User login
High-intensity treadmill workouts preserved motor function in early-stage Parkinson’s
Six months of high-intensity treadmill walking largely preserved motor function in patients with recently diagnosed Parkinson’s disease, a phase 2 study has determined.
Patients randomized to the high-intensity exercise arm stayed very close to their baseline motor scores on the Unified Parkinson’s Disease Rating Scale (UPDRS), while the scores of those randomized to a wait list increased a mean of 3 points over 24 weeks. The scores of patients randomized to an intermediate-intensity treadmill workout increased 2 points over the study, which was not significantly different than the increase seen in the control group, Margaret Schenkman, PhD, and her colleagues wrote Dec. 11 in JAMA Neurology.
“A larger efficacy trial is warranted to determine whether exercising at 80%-85% maximum heart rate produces meaningful clinical benefits in de novo Parkinson disease,” wrote Dr. Schenkman and her colleagues. “Meanwhile, clinicians may safely prescribe exercise at this intensity level for this population.”
The Study in Parkinson Disease of Exercise (SPARX) randomized 128 patients with newly diagnosed Parkinson’s to 30-minute treadmill workouts, four times weekly, at either 80%-85% or 60%-65% maximum heart rate. They were compared against a control group of wait-listed patients.
Subjects were a mean of 64 years old and had been diagnosed for about 3-4 months before enrolling. Their mean total UPDRS score was 23.
Both active arms hit the gym a mean of 3 days/week and were able to exercise at their target heart rates, confirming the feasibility of treadmill workouts for this patient population.
The mean change in UPDRS motor score in the high-intensity group was an increase of 0.3 at 24 weeks, compared with an increase of 3.2 in the usual care group – a statistically significant difference. The mean change in the moderate-intensity group was an increase of 2.0, a nonsignificant difference.
Adverse events consisted largely of falls and musculoskeletal pain. There were 6 falls in the high-intensity group, 5 in the moderate-intensity group, and 11 in the control group. Of these falls, one in the high-intensity and one in the moderate-intensity group were considered serious.
SPARX was largely funded by a grant from the National Institute of Neurological Disorders and Stroke. None of the investigators reported having any financial conflicts.
SOURCE: Schenkman M et al. JAMA Neurol. 2017 Dec 11. doi: 10.1001/jamaneurol.2017.3517
Six months of high-intensity treadmill walking largely preserved motor function in patients with recently diagnosed Parkinson’s disease, a phase 2 study has determined.
Patients randomized to the high-intensity exercise arm stayed very close to their baseline motor scores on the Unified Parkinson’s Disease Rating Scale (UPDRS), while the scores of those randomized to a wait list increased a mean of 3 points over 24 weeks. The scores of patients randomized to an intermediate-intensity treadmill workout increased 2 points over the study, which was not significantly different than the increase seen in the control group, Margaret Schenkman, PhD, and her colleagues wrote Dec. 11 in JAMA Neurology.
“A larger efficacy trial is warranted to determine whether exercising at 80%-85% maximum heart rate produces meaningful clinical benefits in de novo Parkinson disease,” wrote Dr. Schenkman and her colleagues. “Meanwhile, clinicians may safely prescribe exercise at this intensity level for this population.”
The Study in Parkinson Disease of Exercise (SPARX) randomized 128 patients with newly diagnosed Parkinson’s to 30-minute treadmill workouts, four times weekly, at either 80%-85% or 60%-65% maximum heart rate. They were compared against a control group of wait-listed patients.
Subjects were a mean of 64 years old and had been diagnosed for about 3-4 months before enrolling. Their mean total UPDRS score was 23.
Both active arms hit the gym a mean of 3 days/week and were able to exercise at their target heart rates, confirming the feasibility of treadmill workouts for this patient population.
The mean change in UPDRS motor score in the high-intensity group was an increase of 0.3 at 24 weeks, compared with an increase of 3.2 in the usual care group – a statistically significant difference. The mean change in the moderate-intensity group was an increase of 2.0, a nonsignificant difference.
Adverse events consisted largely of falls and musculoskeletal pain. There were 6 falls in the high-intensity group, 5 in the moderate-intensity group, and 11 in the control group. Of these falls, one in the high-intensity and one in the moderate-intensity group were considered serious.
SPARX was largely funded by a grant from the National Institute of Neurological Disorders and Stroke. None of the investigators reported having any financial conflicts.
SOURCE: Schenkman M et al. JAMA Neurol. 2017 Dec 11. doi: 10.1001/jamaneurol.2017.3517
Six months of high-intensity treadmill walking largely preserved motor function in patients with recently diagnosed Parkinson’s disease, a phase 2 study has determined.
Patients randomized to the high-intensity exercise arm stayed very close to their baseline motor scores on the Unified Parkinson’s Disease Rating Scale (UPDRS), while the scores of those randomized to a wait list increased a mean of 3 points over 24 weeks. The scores of patients randomized to an intermediate-intensity treadmill workout increased 2 points over the study, which was not significantly different than the increase seen in the control group, Margaret Schenkman, PhD, and her colleagues wrote Dec. 11 in JAMA Neurology.
“A larger efficacy trial is warranted to determine whether exercising at 80%-85% maximum heart rate produces meaningful clinical benefits in de novo Parkinson disease,” wrote Dr. Schenkman and her colleagues. “Meanwhile, clinicians may safely prescribe exercise at this intensity level for this population.”
The Study in Parkinson Disease of Exercise (SPARX) randomized 128 patients with newly diagnosed Parkinson’s to 30-minute treadmill workouts, four times weekly, at either 80%-85% or 60%-65% maximum heart rate. They were compared against a control group of wait-listed patients.
Subjects were a mean of 64 years old and had been diagnosed for about 3-4 months before enrolling. Their mean total UPDRS score was 23.
Both active arms hit the gym a mean of 3 days/week and were able to exercise at their target heart rates, confirming the feasibility of treadmill workouts for this patient population.
The mean change in UPDRS motor score in the high-intensity group was an increase of 0.3 at 24 weeks, compared with an increase of 3.2 in the usual care group – a statistically significant difference. The mean change in the moderate-intensity group was an increase of 2.0, a nonsignificant difference.
Adverse events consisted largely of falls and musculoskeletal pain. There were 6 falls in the high-intensity group, 5 in the moderate-intensity group, and 11 in the control group. Of these falls, one in the high-intensity and one in the moderate-intensity group were considered serious.
SPARX was largely funded by a grant from the National Institute of Neurological Disorders and Stroke. None of the investigators reported having any financial conflicts.
SOURCE: Schenkman M et al. JAMA Neurol. 2017 Dec 11. doi: 10.1001/jamaneurol.2017.3517
FROM JAMA NEUROLOGY
Key clinical point:
Major finding: The high-intensity exercisers had a mean increase of 0.3 points on the UPDRS motor scale, compared with a 3.2-point increase in the control group.
Study details: The study randomized 128 patients to high-intensity or moderate-intensity treadmill exercise or to a wait list.
Disclosures: SPARX was largely funded by a grant from the National Institute of Neurological Disorders and Stroke. None of the investigators reported having any financial conflicts.
Source: Schenkman M et al. JAMA Neurol. 2017 Dec 11. doi: 10.1001/jamaneurol.2017.3517
Interleukin-23 inhibition for psoriasis shows ‘wow’ factor
GENEVA – The merits of addressing interleukin-23 as a novel therapeutic target in moderate to severe plaque psoriasis were abundantly displayed in 2-year outcomes data for two anti–IL-23 monoclonal antibodies – guselkumab and tildrakizumab – in studies presented back to back at the annual congress of the European Academy of Dermatology and Venereology.
These long-term, open-label extensions of previously reported phase 3, randomized, double-blind clinical trials provided evidence of multiple advantages for IL-23 inhibition. The story was similar for both agents: After 2 years of use in the extension studies, the two biologics demonstrated stellar treatment response rates that would have been unimaginable only a few years ago, maintenance of efficacy without drop-off over time, exceedingly low dropout rates, and a safety picture that remains reassuring as experience accumulates. Also, the subcutaneously administered IL-23 inhibitors are attractive from a patient convenience standpoint in that maintenance guselkumab is dosed at 100 mg once every 8 weeks, and tildrakizumab is given once every 12 weeks.
Still, there are differences between the two drugs, most notably in apparent effectiveness. While more than half of guselkumab-treated patients had a Psoriasis Area Severity Index (PASI) 100 response – that is, totally clear skin – at 2 years, that was the case for only one-quarter to one-third of patients on tildrakizumab.
Guselkumab (Tremfya) was approved by the Food and Drug Administration in July 2017 for treatment of adults with moderate to severe plaque psoriasis. Tildrakizumab remains investigational.
Guselkumab
The 2-year, open-label extension of the phase 3 VOYAGE 1 trial included 735 patients who were either on guselkumab continuously, crossed from adalimumab (Humira) to guselkumab after 48 weeks, or switched from placebo after 16 weeks.

PASI 100 rates at 2 years were 49%-55% in the three patient groups. Of the patients in these groups, 54%-59% achieved an Investigator’s Global Assessment (IGA) score of 0. IGA scores of 0 or 1, meaning clear or almost clear skin, were present in 82%-85% of patients at 2 years.
“Dropout rate is an important consideration in long-term studies,” observed Dr. Blauvelt, a dermatologist and president of Oregon Medical Research Center in Portland. “For patients on continuous guselkumab there was a 6% dropout rate in the first year and 6% in the second year, so 88% of patients that started guselkumab were still on guselkumab 2 years later. That’s impressive. In the other two groups, the dropout rate was 2% per year.”
A Dermatology Life Quality Index (DLQI) score of 0 or 1, meaning no disease effects on quality of life, was recorded in 62.5% of the continuous guselkumab group at 48 weeks and 71.1% at 2 years.
“The interesting thing here is that, even though the efficacy numbers are fairly constant between year 1 and year 2, the DLQI goes up and up. Surprising? Maybe not. I think it shows patients are getting happier and happier over time with their disease control,” Dr. Blauvelt continued.
Rates of serious adverse events remained low and stable, with no negative surprises during year 2. The serious infection rate was 1.02 cases/100 patient-years in year 1 and 0.84 cases/100 patient-years in year 2. No cases of tuberculosis, opportunistic infections, or serious hypersensitivity reactions occurred during 2 years of treatment.
Tildrakizumab
Two-year results from the ongoing 5-year extension of the phase 3 reSURFACE 1 and reSURFACE 2 trials were presented by Kim A. Papp, MD, PhD, president of Probity Medical Research, Waterloo, Ont. This presentation of 2-year outcomes for 1,237 study participants was a feat, considering that the 12-week results of the trials had been published less than 3 months earlier (Lancet. 2017 Jul 15;390[10091]:276-88).

“I think these data are very compelling that the loss of response over time is minimal,” according to the dermatologist. “We’ve also seen that safety over 2 years has no surprises; in fact, it’s remarkably quiet. The rate of severe infections, which is important to look at for any treatment suppressing the immune system, is low and occurs almost independent of dose, which is very hopeful. It’s a promising sign.”
Indeed, the serious infection rate was 0.8 cases/100 patient-years regardless of whether subjects were on tildrakizumab at 100 mg or 200 mg.
Controversy over how to report long-term outcomes
A hot topic among clinical trialists in dermatology concerns how to report study results. The traditional method in studies funded by pharmaceutical companies is known as the “last observation carried forward” analysis. It casts the study drug results in the most favorable possible light because, when a subject drops out of a trial for any reason, their last measured value for response to treatment is carried forward as though the patient completed the study. Thus, psoriasis patients who drop out because they couldn’t tolerate a therapy or developed a serious side effect dictating discontinuation will be scored on the basis of their last PASI response, creating a bias in favor of active treatment.
A more conservative analytic method is known as the “nonresponder imputation” analysis. By this method, a patient who drops out of a trial is automatically categorized as a treatment failure, even if the reason was that the patient moved and could no longer make visits to the study center.
The prespecified guselkumab analysis presented by Dr. Blauvelt involved nonresponder imputation through year 1 and imputation based on the reason for discontinuation in the second year. In contrast, the 2-year tildrakizumab analysis presented by Dr. Papp used the far more common last observation carried forward method.
To help the audience appreciate the importance of looking at the analytic methods used in a studies and help them understand the clinical significance of the results, Dr. Blauvelt provided a reanalysis of the 2-year guselkumab data using the last observation carried forward method. Across the board, the numbers became more favorable. For example, the PASI 75 rate of 95.7% using the prespecified nonresponder imputation analysis crept up to 96.8% under the last observation carried forward method; for comparison, the PASI 75 rates were 81%-84% in the tildrakizumab analysis.
“If you wanted to compare apples to apples with some other drugs, you would use these numbers – the as-observed analysis numbers used by most other companies with other drugs. If you wanted to determine what the true-life numbers are, they’d probably be something between the nonresponder imputation and as-observed numbers,” said to Dr. Blauvelt.
Dr. Papp was untroubled by the use of the last observation carried forward method in the particular case of the tildrakizumab long-term extension study.
“There is reason to believe the as-observed analysis doesn’t affect the integrity of the data because the dropout rate is extraordinarily low,” he said.
The guselkumab analysis was sponsored by Janssen Pharmaceutica; the tildrakizumab analysis was sponsored by Merck and by Sun Pharma. Dr. Blauvelt and Dr. Papp were paid investigators in both studies and serve as scientific advisers to virtually all companies invested in the psoriasis therapy developmental pipeline.
GENEVA – The merits of addressing interleukin-23 as a novel therapeutic target in moderate to severe plaque psoriasis were abundantly displayed in 2-year outcomes data for two anti–IL-23 monoclonal antibodies – guselkumab and tildrakizumab – in studies presented back to back at the annual congress of the European Academy of Dermatology and Venereology.
These long-term, open-label extensions of previously reported phase 3, randomized, double-blind clinical trials provided evidence of multiple advantages for IL-23 inhibition. The story was similar for both agents: After 2 years of use in the extension studies, the two biologics demonstrated stellar treatment response rates that would have been unimaginable only a few years ago, maintenance of efficacy without drop-off over time, exceedingly low dropout rates, and a safety picture that remains reassuring as experience accumulates. Also, the subcutaneously administered IL-23 inhibitors are attractive from a patient convenience standpoint in that maintenance guselkumab is dosed at 100 mg once every 8 weeks, and tildrakizumab is given once every 12 weeks.
Still, there are differences between the two drugs, most notably in apparent effectiveness. While more than half of guselkumab-treated patients had a Psoriasis Area Severity Index (PASI) 100 response – that is, totally clear skin – at 2 years, that was the case for only one-quarter to one-third of patients on tildrakizumab.
Guselkumab (Tremfya) was approved by the Food and Drug Administration in July 2017 for treatment of adults with moderate to severe plaque psoriasis. Tildrakizumab remains investigational.
Guselkumab
The 2-year, open-label extension of the phase 3 VOYAGE 1 trial included 735 patients who were either on guselkumab continuously, crossed from adalimumab (Humira) to guselkumab after 48 weeks, or switched from placebo after 16 weeks.
PASI 100 rates at 2 years were 49%-55% in the three patient groups. Of the patients in these groups, 54%-59% achieved an Investigator’s Global Assessment (IGA) score of 0. IGA scores of 0 or 1, meaning clear or almost clear skin, were present in 82%-85% of patients at 2 years.
“Dropout rate is an important consideration in long-term studies,” observed Dr. Blauvelt, a dermatologist and president of Oregon Medical Research Center in Portland. “For patients on continuous guselkumab there was a 6% dropout rate in the first year and 6% in the second year, so 88% of patients that started guselkumab were still on guselkumab 2 years later. That’s impressive. In the other two groups, the dropout rate was 2% per year.”
A Dermatology Life Quality Index (DLQI) score of 0 or 1, meaning no disease effects on quality of life, was recorded in 62.5% of the continuous guselkumab group at 48 weeks and 71.1% at 2 years.
“The interesting thing here is that, even though the efficacy numbers are fairly constant between year 1 and year 2, the DLQI goes up and up. Surprising? Maybe not. I think it shows patients are getting happier and happier over time with their disease control,” Dr. Blauvelt continued.
Rates of serious adverse events remained low and stable, with no negative surprises during year 2. The serious infection rate was 1.02 cases/100 patient-years in year 1 and 0.84 cases/100 patient-years in year 2. No cases of tuberculosis, opportunistic infections, or serious hypersensitivity reactions occurred during 2 years of treatment.
Tildrakizumab
Two-year results from the ongoing 5-year extension of the phase 3 reSURFACE 1 and reSURFACE 2 trials were presented by Kim A. Papp, MD, PhD, president of Probity Medical Research, Waterloo, Ont. This presentation of 2-year outcomes for 1,237 study participants was a feat, considering that the 12-week results of the trials had been published less than 3 months earlier (Lancet. 2017 Jul 15;390[10091]:276-88).
“I think these data are very compelling that the loss of response over time is minimal,” according to the dermatologist. “We’ve also seen that safety over 2 years has no surprises; in fact, it’s remarkably quiet. The rate of severe infections, which is important to look at for any treatment suppressing the immune system, is low and occurs almost independent of dose, which is very hopeful. It’s a promising sign.”
Indeed, the serious infection rate was 0.8 cases/100 patient-years regardless of whether subjects were on tildrakizumab at 100 mg or 200 mg.
Controversy over how to report long-term outcomes
A hot topic among clinical trialists in dermatology concerns how to report study results. The traditional method in studies funded by pharmaceutical companies is known as the “last observation carried forward” analysis. It casts the study drug results in the most favorable possible light because, when a subject drops out of a trial for any reason, their last measured value for response to treatment is carried forward as though the patient completed the study. Thus, psoriasis patients who drop out because they couldn’t tolerate a therapy or developed a serious side effect dictating discontinuation will be scored on the basis of their last PASI response, creating a bias in favor of active treatment.
A more conservative analytic method is known as the “nonresponder imputation” analysis. By this method, a patient who drops out of a trial is automatically categorized as a treatment failure, even if the reason was that the patient moved and could no longer make visits to the study center.
The prespecified guselkumab analysis presented by Dr. Blauvelt involved nonresponder imputation through year 1 and imputation based on the reason for discontinuation in the second year. In contrast, the 2-year tildrakizumab analysis presented by Dr. Papp used the far more common last observation carried forward method.
To help the audience appreciate the importance of looking at the analytic methods used in a studies and help them understand the clinical significance of the results, Dr. Blauvelt provided a reanalysis of the 2-year guselkumab data using the last observation carried forward method. Across the board, the numbers became more favorable. For example, the PASI 75 rate of 95.7% using the prespecified nonresponder imputation analysis crept up to 96.8% under the last observation carried forward method; for comparison, the PASI 75 rates were 81%-84% in the tildrakizumab analysis.
“If you wanted to compare apples to apples with some other drugs, you would use these numbers – the as-observed analysis numbers used by most other companies with other drugs. If you wanted to determine what the true-life numbers are, they’d probably be something between the nonresponder imputation and as-observed numbers,” said to Dr. Blauvelt.
Dr. Papp was untroubled by the use of the last observation carried forward method in the particular case of the tildrakizumab long-term extension study.
“There is reason to believe the as-observed analysis doesn’t affect the integrity of the data because the dropout rate is extraordinarily low,” he said.
The guselkumab analysis was sponsored by Janssen Pharmaceutica; the tildrakizumab analysis was sponsored by Merck and by Sun Pharma. Dr. Blauvelt and Dr. Papp were paid investigators in both studies and serve as scientific advisers to virtually all companies invested in the psoriasis therapy developmental pipeline.
GENEVA – The merits of addressing interleukin-23 as a novel therapeutic target in moderate to severe plaque psoriasis were abundantly displayed in 2-year outcomes data for two anti–IL-23 monoclonal antibodies – guselkumab and tildrakizumab – in studies presented back to back at the annual congress of the European Academy of Dermatology and Venereology.
These long-term, open-label extensions of previously reported phase 3, randomized, double-blind clinical trials provided evidence of multiple advantages for IL-23 inhibition. The story was similar for both agents: After 2 years of use in the extension studies, the two biologics demonstrated stellar treatment response rates that would have been unimaginable only a few years ago, maintenance of efficacy without drop-off over time, exceedingly low dropout rates, and a safety picture that remains reassuring as experience accumulates. Also, the subcutaneously administered IL-23 inhibitors are attractive from a patient convenience standpoint in that maintenance guselkumab is dosed at 100 mg once every 8 weeks, and tildrakizumab is given once every 12 weeks.
Still, there are differences between the two drugs, most notably in apparent effectiveness. While more than half of guselkumab-treated patients had a Psoriasis Area Severity Index (PASI) 100 response – that is, totally clear skin – at 2 years, that was the case for only one-quarter to one-third of patients on tildrakizumab.
Guselkumab (Tremfya) was approved by the Food and Drug Administration in July 2017 for treatment of adults with moderate to severe plaque psoriasis. Tildrakizumab remains investigational.
Guselkumab
The 2-year, open-label extension of the phase 3 VOYAGE 1 trial included 735 patients who were either on guselkumab continuously, crossed from adalimumab (Humira) to guselkumab after 48 weeks, or switched from placebo after 16 weeks.
PASI 100 rates at 2 years were 49%-55% in the three patient groups. Of the patients in these groups, 54%-59% achieved an Investigator’s Global Assessment (IGA) score of 0. IGA scores of 0 or 1, meaning clear or almost clear skin, were present in 82%-85% of patients at 2 years.
“Dropout rate is an important consideration in long-term studies,” observed Dr. Blauvelt, a dermatologist and president of Oregon Medical Research Center in Portland. “For patients on continuous guselkumab there was a 6% dropout rate in the first year and 6% in the second year, so 88% of patients that started guselkumab were still on guselkumab 2 years later. That’s impressive. In the other two groups, the dropout rate was 2% per year.”
A Dermatology Life Quality Index (DLQI) score of 0 or 1, meaning no disease effects on quality of life, was recorded in 62.5% of the continuous guselkumab group at 48 weeks and 71.1% at 2 years.
“The interesting thing here is that, even though the efficacy numbers are fairly constant between year 1 and year 2, the DLQI goes up and up. Surprising? Maybe not. I think it shows patients are getting happier and happier over time with their disease control,” Dr. Blauvelt continued.
Rates of serious adverse events remained low and stable, with no negative surprises during year 2. The serious infection rate was 1.02 cases/100 patient-years in year 1 and 0.84 cases/100 patient-years in year 2. No cases of tuberculosis, opportunistic infections, or serious hypersensitivity reactions occurred during 2 years of treatment.
Tildrakizumab
Two-year results from the ongoing 5-year extension of the phase 3 reSURFACE 1 and reSURFACE 2 trials were presented by Kim A. Papp, MD, PhD, president of Probity Medical Research, Waterloo, Ont. This presentation of 2-year outcomes for 1,237 study participants was a feat, considering that the 12-week results of the trials had been published less than 3 months earlier (Lancet. 2017 Jul 15;390[10091]:276-88).
“I think these data are very compelling that the loss of response over time is minimal,” according to the dermatologist. “We’ve also seen that safety over 2 years has no surprises; in fact, it’s remarkably quiet. The rate of severe infections, which is important to look at for any treatment suppressing the immune system, is low and occurs almost independent of dose, which is very hopeful. It’s a promising sign.”
Indeed, the serious infection rate was 0.8 cases/100 patient-years regardless of whether subjects were on tildrakizumab at 100 mg or 200 mg.
Controversy over how to report long-term outcomes
A hot topic among clinical trialists in dermatology concerns how to report study results. The traditional method in studies funded by pharmaceutical companies is known as the “last observation carried forward” analysis. It casts the study drug results in the most favorable possible light because, when a subject drops out of a trial for any reason, their last measured value for response to treatment is carried forward as though the patient completed the study. Thus, psoriasis patients who drop out because they couldn’t tolerate a therapy or developed a serious side effect dictating discontinuation will be scored on the basis of their last PASI response, creating a bias in favor of active treatment.
A more conservative analytic method is known as the “nonresponder imputation” analysis. By this method, a patient who drops out of a trial is automatically categorized as a treatment failure, even if the reason was that the patient moved and could no longer make visits to the study center.
The prespecified guselkumab analysis presented by Dr. Blauvelt involved nonresponder imputation through year 1 and imputation based on the reason for discontinuation in the second year. In contrast, the 2-year tildrakizumab analysis presented by Dr. Papp used the far more common last observation carried forward method.
To help the audience appreciate the importance of looking at the analytic methods used in a studies and help them understand the clinical significance of the results, Dr. Blauvelt provided a reanalysis of the 2-year guselkumab data using the last observation carried forward method. Across the board, the numbers became more favorable. For example, the PASI 75 rate of 95.7% using the prespecified nonresponder imputation analysis crept up to 96.8% under the last observation carried forward method; for comparison, the PASI 75 rates were 81%-84% in the tildrakizumab analysis.
“If you wanted to compare apples to apples with some other drugs, you would use these numbers – the as-observed analysis numbers used by most other companies with other drugs. If you wanted to determine what the true-life numbers are, they’d probably be something between the nonresponder imputation and as-observed numbers,” said to Dr. Blauvelt.
Dr. Papp was untroubled by the use of the last observation carried forward method in the particular case of the tildrakizumab long-term extension study.
“There is reason to believe the as-observed analysis doesn’t affect the integrity of the data because the dropout rate is extraordinarily low,” he said.
The guselkumab analysis was sponsored by Janssen Pharmaceutica; the tildrakizumab analysis was sponsored by Merck and by Sun Pharma. Dr. Blauvelt and Dr. Papp were paid investigators in both studies and serve as scientific advisers to virtually all companies invested in the psoriasis therapy developmental pipeline.
expert analysis from tHE EADV CONGRESS
VIDEO: CAR T cell axi-cel drives B-cell lymphomas into remission
ATLANTA – In the ZUMA-1 trial, more than one-third of patients with refractory large B-cell lymphomas treated with the chimeric antigen receptor (CAR) T-cell product axicabtagene ciloleucel (Yescarta; axi-cel) had durable responses, with some patients having complete responses lasting more than 1 year after a single infusion.
Updated combined phase 1 and 2 results in 108 patients with diffuse large B-cell lymphoma, primary mediastinal B-cell lymphoma, or transformed follicular lymphoma showed an objective response rate of 82% of patients – including 58% showing complete responses – after a median follow-up of 15.4 months.
In a video interview at the annual meeting of the American Society of Hematology, Sattva S. Neelapu, MD, from the University of Texas MD Anderson Cancer Center in Houston discusses the use of CAR T cells directed against the CD19 antigen in patients with relapsed/refractory B-cell lymphomas and describes efforts to improve responses while managing adverse events common to CAR T-cell therapies, notably cytokine release syndrome.
ZUMA-1 is supported by Kite Pharma, which developed axicabtagene ciloleucel, and the Leukemia & Lymphoma Society’s Therapy Acceleration Program. Dr. Neelapu reported receiving advisory board fees from the company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ATLANTA – In the ZUMA-1 trial, more than one-third of patients with refractory large B-cell lymphomas treated with the chimeric antigen receptor (CAR) T-cell product axicabtagene ciloleucel (Yescarta; axi-cel) had durable responses, with some patients having complete responses lasting more than 1 year after a single infusion.
Updated combined phase 1 and 2 results in 108 patients with diffuse large B-cell lymphoma, primary mediastinal B-cell lymphoma, or transformed follicular lymphoma showed an objective response rate of 82% of patients – including 58% showing complete responses – after a median follow-up of 15.4 months.
In a video interview at the annual meeting of the American Society of Hematology, Sattva S. Neelapu, MD, from the University of Texas MD Anderson Cancer Center in Houston discusses the use of CAR T cells directed against the CD19 antigen in patients with relapsed/refractory B-cell lymphomas and describes efforts to improve responses while managing adverse events common to CAR T-cell therapies, notably cytokine release syndrome.
ZUMA-1 is supported by Kite Pharma, which developed axicabtagene ciloleucel, and the Leukemia & Lymphoma Society’s Therapy Acceleration Program. Dr. Neelapu reported receiving advisory board fees from the company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ATLANTA – In the ZUMA-1 trial, more than one-third of patients with refractory large B-cell lymphomas treated with the chimeric antigen receptor (CAR) T-cell product axicabtagene ciloleucel (Yescarta; axi-cel) had durable responses, with some patients having complete responses lasting more than 1 year after a single infusion.
Updated combined phase 1 and 2 results in 108 patients with diffuse large B-cell lymphoma, primary mediastinal B-cell lymphoma, or transformed follicular lymphoma showed an objective response rate of 82% of patients – including 58% showing complete responses – after a median follow-up of 15.4 months.
In a video interview at the annual meeting of the American Society of Hematology, Sattva S. Neelapu, MD, from the University of Texas MD Anderson Cancer Center in Houston discusses the use of CAR T cells directed against the CD19 antigen in patients with relapsed/refractory B-cell lymphomas and describes efforts to improve responses while managing adverse events common to CAR T-cell therapies, notably cytokine release syndrome.
ZUMA-1 is supported by Kite Pharma, which developed axicabtagene ciloleucel, and the Leukemia & Lymphoma Society’s Therapy Acceleration Program. Dr. Neelapu reported receiving advisory board fees from the company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
REPORTING FROM ASH 2017
Consider ‘impactibility’ to prevent hospital readmissions
With the goal of reducing 28-day or 30-day readmissions, some health care teams are turning to predictive models to identify patients at high risk for readmission and to efficiently focus resource-intensive prevention strategies. Recently, there’s been a rapid multiplying of these models.
Many of these models do accurately predict readmission risk, according to a recent BMJ editorial. “Among the 14 published models that target all unplanned readmissions (rather than readmissions for specific patient groups), the ‘C statistic’ ranges from 0.55 to 0.80, meaning that, when presented with two patients, these models correctly identify the higher risk individual between 55% and 80% of the time,” the authors wrote.
But, the authors suggested, the real value is not in simply making predictions but in using predictive models in ways that improve outcomes for patients.
“This will require linking predictive models to actionable opportunities for improving care,” they wrote. “Such linkages will most likely be identified through close collaboration between analytical teams, health care practitioners, and patients.” Being at high risk of readmission is not the only consideration; the patient must also be able to benefit from interventions being considered – they must be “impactible.”
“The distinction between predictive risk and impactibility might explain why practitioners tend to identify quite different patients for intervention than predictive risk models,” the authors wrote.
But together, predictive models and clinicians might produce more effective decisions than either does alone. “One of the strengths of predictive models is that they produce objective and consistent judgments regarding readmission risk, whereas clinical judgment can be affected by personal attitudes or attentiveness. Predictive risk models can also be operationalised across whole populations, and might therefore identify needs that would otherwise be missed by clinical teams (e.g., among more socioeconomically deprived neighbourhoods or groups with inadequate primary care). On the other hand, clinicians have access to a much wider range of information regarding patients than predictive risk models, which is essential to judge impactibility.”
The authors conclude, “The predictive modelling enterprise would benefit enormously from such collaboration because the real goal of this activity lies not in predicting the risk of readmission but in identifying patients at risk for preventable readmissions and ‘impactible’ by available interventions.”
Reference
Steventon A et al. Preventing hospital readmissions: The importance of considering ‘impactibility,’ not just predicted risk. BMJ Qual Saf. 2017 Oct;26(10):782-5. Accessed Oct. 9, 2017.
With the goal of reducing 28-day or 30-day readmissions, some health care teams are turning to predictive models to identify patients at high risk for readmission and to efficiently focus resource-intensive prevention strategies. Recently, there’s been a rapid multiplying of these models.
Many of these models do accurately predict readmission risk, according to a recent BMJ editorial. “Among the 14 published models that target all unplanned readmissions (rather than readmissions for specific patient groups), the ‘C statistic’ ranges from 0.55 to 0.80, meaning that, when presented with two patients, these models correctly identify the higher risk individual between 55% and 80% of the time,” the authors wrote.
But, the authors suggested, the real value is not in simply making predictions but in using predictive models in ways that improve outcomes for patients.
“This will require linking predictive models to actionable opportunities for improving care,” they wrote. “Such linkages will most likely be identified through close collaboration between analytical teams, health care practitioners, and patients.” Being at high risk of readmission is not the only consideration; the patient must also be able to benefit from interventions being considered – they must be “impactible.”
“The distinction between predictive risk and impactibility might explain why practitioners tend to identify quite different patients for intervention than predictive risk models,” the authors wrote.
But together, predictive models and clinicians might produce more effective decisions than either does alone. “One of the strengths of predictive models is that they produce objective and consistent judgments regarding readmission risk, whereas clinical judgment can be affected by personal attitudes or attentiveness. Predictive risk models can also be operationalised across whole populations, and might therefore identify needs that would otherwise be missed by clinical teams (e.g., among more socioeconomically deprived neighbourhoods or groups with inadequate primary care). On the other hand, clinicians have access to a much wider range of information regarding patients than predictive risk models, which is essential to judge impactibility.”
The authors conclude, “The predictive modelling enterprise would benefit enormously from such collaboration because the real goal of this activity lies not in predicting the risk of readmission but in identifying patients at risk for preventable readmissions and ‘impactible’ by available interventions.”
Reference
Steventon A et al. Preventing hospital readmissions: The importance of considering ‘impactibility,’ not just predicted risk. BMJ Qual Saf. 2017 Oct;26(10):782-5. Accessed Oct. 9, 2017.
With the goal of reducing 28-day or 30-day readmissions, some health care teams are turning to predictive models to identify patients at high risk for readmission and to efficiently focus resource-intensive prevention strategies. Recently, there’s been a rapid multiplying of these models.
Many of these models do accurately predict readmission risk, according to a recent BMJ editorial. “Among the 14 published models that target all unplanned readmissions (rather than readmissions for specific patient groups), the ‘C statistic’ ranges from 0.55 to 0.80, meaning that, when presented with two patients, these models correctly identify the higher risk individual between 55% and 80% of the time,” the authors wrote.
But, the authors suggested, the real value is not in simply making predictions but in using predictive models in ways that improve outcomes for patients.
“This will require linking predictive models to actionable opportunities for improving care,” they wrote. “Such linkages will most likely be identified through close collaboration between analytical teams, health care practitioners, and patients.” Being at high risk of readmission is not the only consideration; the patient must also be able to benefit from interventions being considered – they must be “impactible.”
“The distinction between predictive risk and impactibility might explain why practitioners tend to identify quite different patients for intervention than predictive risk models,” the authors wrote.
But together, predictive models and clinicians might produce more effective decisions than either does alone. “One of the strengths of predictive models is that they produce objective and consistent judgments regarding readmission risk, whereas clinical judgment can be affected by personal attitudes or attentiveness. Predictive risk models can also be operationalised across whole populations, and might therefore identify needs that would otherwise be missed by clinical teams (e.g., among more socioeconomically deprived neighbourhoods or groups with inadequate primary care). On the other hand, clinicians have access to a much wider range of information regarding patients than predictive risk models, which is essential to judge impactibility.”
The authors conclude, “The predictive modelling enterprise would benefit enormously from such collaboration because the real goal of this activity lies not in predicting the risk of readmission but in identifying patients at risk for preventable readmissions and ‘impactible’ by available interventions.”
Reference
Steventon A et al. Preventing hospital readmissions: The importance of considering ‘impactibility,’ not just predicted risk. BMJ Qual Saf. 2017 Oct;26(10):782-5. Accessed Oct. 9, 2017.
Three-month response to CAR T-cells looks durable in DLBCL
ATLANTA – Responses 3 months after chimeric antigen receptor (CAR) T-cell therapy look durable in adults with transplant-ineligible relapsed/refractory diffuse large B-cell lymphoma (DLBCL), according to updated results from the single-arm, global, phase 2 JULIET trial.
Fully 95% of patients who had a complete response to CTL019 (tisagenlecleucel; Kymriah) at 3 months maintained that complete response at 6 months, Stephen J. Schuster, MD, said at the annual meeting of the American Society of Hematology.

Patients with relapsed/refractory DLBCL tend to face a very poor prognosis, noted Dr. Schuster of Perelman School of Medicine and Abramson Cancer Center, University of Pennsylvania, Philadelphia. High-dose chemotherapy followed by autologous stem cell transplantation “is capable of long-term survival, but in very few patients,” he said. A dismal 8% of patients completely respond to salvage treatment and only about one in five partially respond. Both levels of response are short-lived, with a median survival of about 4 months.
Meanwhile, CTL019 therapy has produced durable complete remissions in children with lymphoblastic leukemia and in adults with chronic lymphocytic leukemia, Dr. Schuster and his associates wrote in an article simultaneously published in the New England Journal of Medicine (2017 Dec 10. doi: 10.1056/NEJMoa1708566).
To test the CAR T-cell therapy in relapsed/refractory DLBCL, they enrolled affected adults who had received at least two prior lines of antineoplastic treatment and who were not candidates for autologous stem cell transplantation.
Treatment consisted of a single CTL019 infusion (median dose, 3.1 × 108 cells; range, 0.1 × 108 to 6.0 × 108 cells), usually after lymphodepleting chemotherapy. Previously, patients had received a median of three lines of therapy, and about half had undergone autologous stem cell transplantation.
Median time from infusion to data cutoff in March 2017 was 5.6 months. Among 81 patients followed for at least 3 months before data cutoff, best overall response rate was 53% and 40% had a complete response. Overall response rates were 38% at 3 months and 37% at 6 months. Rates of complete response as confirmed by 18F-fluorodeoxyglucose–positron-emission tomography (PET) were 32% at 3 months and 30% at 6 months.These findings highlight the predictive power of 3-month response to CTL019 therapy in relapsed/refractory DLBCL, Dr. Schuster said. Among all responders, 74% remained relapse free at 6 months, meaning that median duration of response and median overall survival were not reached at data cutoff.
Dr. Schuster also reported that 26% of patients were infused as outpatients, which he called “easy to do” and appropriate as long as patients who become febrile are admitted and monitored for cytokine release syndrome. Three-quarters of patients who were infused as outpatients were able to remain home for at least 3 days afterward, he said.
Adverse events typified those of CAR T-cell therapy, including cytokine release syndrome (all grades: 58%; grade 3-4: 23%) and neurological toxicities (all grades: 21%; grade 3-4: 12%). The current labeling for CTL019 in children and young adults with acute lymphoblastic leukemia also includes a boxed warning for these toxicities.Tisagenlecleucel, the first-ever approved CAR T-cell therapy, is made by using a lentiviral vector to genetically engineer a patient’s own T-cells to express a CAR for the pan-B-cell CD19 antigen. These anti-CD19 CAR T-cells are then expanded in the laboratory, frozen for shipping purposes, and infused back into patients. In October 2017, Novartis submitted a biologics license application to the Food and Drug Administration to expand the label for CTL019 to include transplant-ineligible relapsed/refractory DLBCL.
Novartis Pharmaceuticals anticipates large-scale production in 2018, Dr. Schuster said. Manufacturing time has been cut to 22 days from the 30-day turnaround used in the trial, he reported.
Dr. Schuster also said that he sees no point in retreating patients whose relapsed/refractory DLBCL doesn’t respond to tisagenlecleucel, and that JULIET did not test this approach. “If someone fails therapy and you retreat, you don’t see success, in my experience,” he said. “If patients respond and then fail later, then you retreat and you may succeed.”
Novartis Pharmaceuticals sponsored JULIET. Dr. Schuster disclosed consultancy and research funding from Novartis and ties to Celgene, Gilead, Genentech, and several other pharmaceutical companies.
SOURCE: Schuster S et al. ASH 2017 Abstract 577.
ATLANTA – Responses 3 months after chimeric antigen receptor (CAR) T-cell therapy look durable in adults with transplant-ineligible relapsed/refractory diffuse large B-cell lymphoma (DLBCL), according to updated results from the single-arm, global, phase 2 JULIET trial.
Fully 95% of patients who had a complete response to CTL019 (tisagenlecleucel; Kymriah) at 3 months maintained that complete response at 6 months, Stephen J. Schuster, MD, said at the annual meeting of the American Society of Hematology.
Patients with relapsed/refractory DLBCL tend to face a very poor prognosis, noted Dr. Schuster of Perelman School of Medicine and Abramson Cancer Center, University of Pennsylvania, Philadelphia. High-dose chemotherapy followed by autologous stem cell transplantation “is capable of long-term survival, but in very few patients,” he said. A dismal 8% of patients completely respond to salvage treatment and only about one in five partially respond. Both levels of response are short-lived, with a median survival of about 4 months.
Meanwhile, CTL019 therapy has produced durable complete remissions in children with lymphoblastic leukemia and in adults with chronic lymphocytic leukemia, Dr. Schuster and his associates wrote in an article simultaneously published in the New England Journal of Medicine (2017 Dec 10. doi: 10.1056/NEJMoa1708566).
To test the CAR T-cell therapy in relapsed/refractory DLBCL, they enrolled affected adults who had received at least two prior lines of antineoplastic treatment and who were not candidates for autologous stem cell transplantation.
Treatment consisted of a single CTL019 infusion (median dose, 3.1 × 108 cells; range, 0.1 × 108 to 6.0 × 108 cells), usually after lymphodepleting chemotherapy. Previously, patients had received a median of three lines of therapy, and about half had undergone autologous stem cell transplantation.
Median time from infusion to data cutoff in March 2017 was 5.6 months. Among 81 patients followed for at least 3 months before data cutoff, best overall response rate was 53% and 40% had a complete response. Overall response rates were 38% at 3 months and 37% at 6 months. Rates of complete response as confirmed by 18F-fluorodeoxyglucose–positron-emission tomography (PET) were 32% at 3 months and 30% at 6 months.These findings highlight the predictive power of 3-month response to CTL019 therapy in relapsed/refractory DLBCL, Dr. Schuster said. Among all responders, 74% remained relapse free at 6 months, meaning that median duration of response and median overall survival were not reached at data cutoff.
Dr. Schuster also reported that 26% of patients were infused as outpatients, which he called “easy to do” and appropriate as long as patients who become febrile are admitted and monitored for cytokine release syndrome. Three-quarters of patients who were infused as outpatients were able to remain home for at least 3 days afterward, he said.
Adverse events typified those of CAR T-cell therapy, including cytokine release syndrome (all grades: 58%; grade 3-4: 23%) and neurological toxicities (all grades: 21%; grade 3-4: 12%). The current labeling for CTL019 in children and young adults with acute lymphoblastic leukemia also includes a boxed warning for these toxicities.Tisagenlecleucel, the first-ever approved CAR T-cell therapy, is made by using a lentiviral vector to genetically engineer a patient’s own T-cells to express a CAR for the pan-B-cell CD19 antigen. These anti-CD19 CAR T-cells are then expanded in the laboratory, frozen for shipping purposes, and infused back into patients. In October 2017, Novartis submitted a biologics license application to the Food and Drug Administration to expand the label for CTL019 to include transplant-ineligible relapsed/refractory DLBCL.
Novartis Pharmaceuticals anticipates large-scale production in 2018, Dr. Schuster said. Manufacturing time has been cut to 22 days from the 30-day turnaround used in the trial, he reported.
Dr. Schuster also said that he sees no point in retreating patients whose relapsed/refractory DLBCL doesn’t respond to tisagenlecleucel, and that JULIET did not test this approach. “If someone fails therapy and you retreat, you don’t see success, in my experience,” he said. “If patients respond and then fail later, then you retreat and you may succeed.”
Novartis Pharmaceuticals sponsored JULIET. Dr. Schuster disclosed consultancy and research funding from Novartis and ties to Celgene, Gilead, Genentech, and several other pharmaceutical companies.
SOURCE: Schuster S et al. ASH 2017 Abstract 577.
ATLANTA – Responses 3 months after chimeric antigen receptor (CAR) T-cell therapy look durable in adults with transplant-ineligible relapsed/refractory diffuse large B-cell lymphoma (DLBCL), according to updated results from the single-arm, global, phase 2 JULIET trial.
Fully 95% of patients who had a complete response to CTL019 (tisagenlecleucel; Kymriah) at 3 months maintained that complete response at 6 months, Stephen J. Schuster, MD, said at the annual meeting of the American Society of Hematology.
Patients with relapsed/refractory DLBCL tend to face a very poor prognosis, noted Dr. Schuster of Perelman School of Medicine and Abramson Cancer Center, University of Pennsylvania, Philadelphia. High-dose chemotherapy followed by autologous stem cell transplantation “is capable of long-term survival, but in very few patients,” he said. A dismal 8% of patients completely respond to salvage treatment and only about one in five partially respond. Both levels of response are short-lived, with a median survival of about 4 months.
Meanwhile, CTL019 therapy has produced durable complete remissions in children with lymphoblastic leukemia and in adults with chronic lymphocytic leukemia, Dr. Schuster and his associates wrote in an article simultaneously published in the New England Journal of Medicine (2017 Dec 10. doi: 10.1056/NEJMoa1708566).
To test the CAR T-cell therapy in relapsed/refractory DLBCL, they enrolled affected adults who had received at least two prior lines of antineoplastic treatment and who were not candidates for autologous stem cell transplantation.
Treatment consisted of a single CTL019 infusion (median dose, 3.1 × 108 cells; range, 0.1 × 108 to 6.0 × 108 cells), usually after lymphodepleting chemotherapy. Previously, patients had received a median of three lines of therapy, and about half had undergone autologous stem cell transplantation.
Median time from infusion to data cutoff in March 2017 was 5.6 months. Among 81 patients followed for at least 3 months before data cutoff, best overall response rate was 53% and 40% had a complete response. Overall response rates were 38% at 3 months and 37% at 6 months. Rates of complete response as confirmed by 18F-fluorodeoxyglucose–positron-emission tomography (PET) were 32% at 3 months and 30% at 6 months.These findings highlight the predictive power of 3-month response to CTL019 therapy in relapsed/refractory DLBCL, Dr. Schuster said. Among all responders, 74% remained relapse free at 6 months, meaning that median duration of response and median overall survival were not reached at data cutoff.
Dr. Schuster also reported that 26% of patients were infused as outpatients, which he called “easy to do” and appropriate as long as patients who become febrile are admitted and monitored for cytokine release syndrome. Three-quarters of patients who were infused as outpatients were able to remain home for at least 3 days afterward, he said.
Adverse events typified those of CAR T-cell therapy, including cytokine release syndrome (all grades: 58%; grade 3-4: 23%) and neurological toxicities (all grades: 21%; grade 3-4: 12%). The current labeling for CTL019 in children and young adults with acute lymphoblastic leukemia also includes a boxed warning for these toxicities.Tisagenlecleucel, the first-ever approved CAR T-cell therapy, is made by using a lentiviral vector to genetically engineer a patient’s own T-cells to express a CAR for the pan-B-cell CD19 antigen. These anti-CD19 CAR T-cells are then expanded in the laboratory, frozen for shipping purposes, and infused back into patients. In October 2017, Novartis submitted a biologics license application to the Food and Drug Administration to expand the label for CTL019 to include transplant-ineligible relapsed/refractory DLBCL.
Novartis Pharmaceuticals anticipates large-scale production in 2018, Dr. Schuster said. Manufacturing time has been cut to 22 days from the 30-day turnaround used in the trial, he reported.
Dr. Schuster also said that he sees no point in retreating patients whose relapsed/refractory DLBCL doesn’t respond to tisagenlecleucel, and that JULIET did not test this approach. “If someone fails therapy and you retreat, you don’t see success, in my experience,” he said. “If patients respond and then fail later, then you retreat and you may succeed.”
Novartis Pharmaceuticals sponsored JULIET. Dr. Schuster disclosed consultancy and research funding from Novartis and ties to Celgene, Gilead, Genentech, and several other pharmaceutical companies.
SOURCE: Schuster S et al. ASH 2017 Abstract 577.
REPORTING FROM ASH 2017
Key clinical point:
Major finding: Among 81 patients with at least 3 months of follow-up, best overall response rate was 53% (95% CI, 42%-64%; P less than .0001) and rates of complete response were 32% at 3 months and 30% at 6 months.
Study details: JULIET is an international, single-arm, phase 2 study of adults with relapsed/refractory DLBCL.
Disclosures: Novartis Pharmaceuticals sponsored JULIET. Dr. Schuster reported consultancy and research funding from Novartis and ties to Celgene, Gilead, Genentech, and several other pharmaceutical companies.
Source: Schuster S et al. ASH 2017 Abstract 577.
Shift to long-term ‘maternal care’ needs boost
WASHINGTON – George R. Saade, MD, wants to see a Time magazine cover story like the 2010 feature titled, “How the first 9 months shape the rest of your life” – except this new story would read “How the first 9 months shape the rest of the mother’s life.”
It’s time, he says, that pregnancy truly be appreciated as a “window to future health” for the mother as well as for the baby, and that the term “maternal care” replaces prenatal care. “How many of your [primary care] providers have asked you if you’ve had any pregnancies and if any of your pregnancies were complicated by hypertension, preterm delivery, growth restriction, or gestational diabetes?” Dr. Saade asked at the biennial meeting of the Diabetes in Pregnancy Study Group of North America.
“It’s better than before, but not good enough ... Checking with patients 6 weeks postpartum is not enough” to prevent long-term metabolic and cardiovascular disorders, he said. “We need regular screening of women.”
The relationship between gestational diabetes (GDM) and subsequent type 2 diabetes, demonstrated several decades ago, offered the “first evidence that pregnancy is a window to future health,” and evidence of the relationship continues to grow. “We know today that there is no other predictive marker of type 2 diabetes that is better and stronger than gestational diabetes,” said Dr. Saade, chief of obstetrics and maternal-fetal medicine at the University of Texas Medical Branch, Galveston.
GDM has also recently been shown to elevate cardiovascular risk independent of its association with type 2 diabetes and metabolic disease.
And similarly, there is now an incontrovertible body of evidence that women who have had preeclampsia are at significantly higher risk of developing hypertension, stroke, and ischemic heart disease later in life than are women who have not have preeclampsia, Dr. Saade said.
Layered evidence
The study that first caught Dr. Saade’s attention was a large Norwegian population-based study published in 2001 that looked at maternal mortality up to 25 years after pregnancy. Women who had preeclampsia had a 1.2-fold higher long-term risk of death from cardiovascular diseases, cancer, and stroke – and women with a history of both preeclampsia and a preterm delivery had a 2.71-fold higher risk – than that of women without such history.
Looking at cardiovascular causes of death specifically, the risk among women with both preeclampsia and preterm delivery was 8.12-fold higher than in women who did not have preeclampsia.
Since then, studies and reviews conducted in the United States and Europe have shown that a history of preeclampsia doubles the risk of developing cardiovascular disease, more than triples the risk of later hypertension, and also increases the risk of stroke, though more moderately.
Recently, Dr. Saade said, researchers have also begun reporting subclinical cardiac abnormalities in women with a history of preeclampsia. A study of 107 women with preeclampsia and 41 women with uneventful pregnancies found that the prevalence of subclinical heart failure (heart failure Stage B) was approximately 3.5% higher in the short term in the preeclampsia group. The women underwent regular cardiac ultrasound and other cardiovascular risk assessment tests 4-10 years postpartum (Ultrasound Obstet Gynecol. 2017;49[1]:143-9).
Preterm delivery and small for gestational age have also been associated with increased risk of ischemic heart disease and other cardiovascular events later in life. And gestational hypertension, research has shown, is a clear risk factor for later hypertension. “We always think of preeclampsia as a different disease, but as far as long-term health is concerned, it doesn’t matter if a woman had preeclampsia or gestational hypertension; she’s still at [greater] risk for hypertension later,” Dr. Saade said.
“And we don’t have to wait 30 years to see evidence” of the association between pregnancy complications and adverse cardiovascular outcomes, he emphasized. A recent retrospective cohort study of more than 300,000 women in Florida showed that women who experienced a maternal placental syndrome during their first pregnancy were at higher risk of subsequent cardiovascular disease during just 5 years of follow-up (Am J Obstet Gynecol. 2016;215[4]:484.e1-14).
Into practice
Regular screening of women whose pregnancies were complicated by conditions associated with long-term health risks “need not be that sophisticated,” Dr. Saade said. Measurement of blood pressure, waist circumference, fasting lipid profile, and fasting glucose is often enough for basic maternal health surveillance, he said.
Dr. Saade said he and some other experts in the field are recommending yearly follow-up for patients who’ve had preeclampsia and other complications. Among the other experts urging early heart disease risk screening is Graeme N. Smith, MD, PhD, an ob.gyn in Ontario who has developed surveillance protocols, follow-up forms, and risk prediction tools for use in a maternal health clinic he established at Kingston General Hospital, Queens University. At the meeting, Dr. Saade encouraged the audience to access Dr. Smith’s resources.
The American Heart Association, in its 2011 guidelines for cardiovascular disease prevention in women, includes pregnancy risks factors (specifically a history of preeclampsia, gestational diabetes, or pregnancy-induced hypertension) as part of its list of major factors for use in risk assessment (Circulation. 2011;123:1243-62).
But as Erica P. Gunderson, PhD, MPH, of Kaiser Permanente Northern California’s division of research, pointed out during another presentation at the meeting, there is more work to be done. Reproductive history is not included in existing disease prediction or risk stratification models for atherosclerotic cardiovascular disease, making it hard at this point to devise specific screening protocols and schedules, especially when it comes to a history of GDM, she said.
“We need more coordinated systems, surveillance in younger women, and some prediction models so that we can know who is at highest risk and needs more surveillance,” she said.
The AHA’s inclusion of GDM history is based on its strong link to overt diabetes, but recent evidence has shown that a history of GDM can independently elevate cardiovascular risk, she noted. Research from the Nurses Health Study II cohort, for instance, found a 30% higher relative risk of cardiovascular events (myocardial infarction and stroke) in women with a history of GDM without progression to diabetes, compared with women who did not have GDM or diabetes (JAMA Intern Med. 2017;177[12]:1735-42).
Dr. Gunderson has also found through analyses of the Coronary Artery Risk Development in Young Adults study data that women who had GDM but did not go on to develop overt diabetes or impaired glycemia had greater carotid intima media thickness many years post delivery than did women without a history of gestational diabetes (J Am Heart Assoc. 2014;3[2]:e000490).
In some models, she has written, this difference in carotid intima media thickness could represent 3-5 years of greater vascular aging for women with previous gestational diabetes and no apparent metabolic dysfunction outside of pregnancy (JAMA Intern Med. 2017;177[12]:1742-4).
There’s a question, she and Dr. Saade both noted, of whether pregnancies unmask previous dispositions to cardiovascular disease or whether pregnancy complications more directly drive adverse long-term outcomes. There is evidence that disorders such as GDM, Dr. Gunderson said, are superimposed on already altered metabolism. But at this time, Dr. Saade said, it appears that “the answer is both.”
According to Dr. Saade, at least three studies are currently following women prospectively to learn more about pregnancy as a window to future cardiovascular health. One of them is the National Institute of Child Health and Human Development’s Nulliparous Pregnancy Outcomes Study–Monitoring Mothers-to-Be Heart Health Study; some data from this study will be presented soon, he said.
Both Dr. Saade and Dr. Gunderson reported in their presentations that they had no disclosures.
WASHINGTON – George R. Saade, MD, wants to see a Time magazine cover story like the 2010 feature titled, “How the first 9 months shape the rest of your life” – except this new story would read “How the first 9 months shape the rest of the mother’s life.”
It’s time, he says, that pregnancy truly be appreciated as a “window to future health” for the mother as well as for the baby, and that the term “maternal care” replaces prenatal care. “How many of your [primary care] providers have asked you if you’ve had any pregnancies and if any of your pregnancies were complicated by hypertension, preterm delivery, growth restriction, or gestational diabetes?” Dr. Saade asked at the biennial meeting of the Diabetes in Pregnancy Study Group of North America.
“It’s better than before, but not good enough ... Checking with patients 6 weeks postpartum is not enough” to prevent long-term metabolic and cardiovascular disorders, he said. “We need regular screening of women.”
The relationship between gestational diabetes (GDM) and subsequent type 2 diabetes, demonstrated several decades ago, offered the “first evidence that pregnancy is a window to future health,” and evidence of the relationship continues to grow. “We know today that there is no other predictive marker of type 2 diabetes that is better and stronger than gestational diabetes,” said Dr. Saade, chief of obstetrics and maternal-fetal medicine at the University of Texas Medical Branch, Galveston.
GDM has also recently been shown to elevate cardiovascular risk independent of its association with type 2 diabetes and metabolic disease.
And similarly, there is now an incontrovertible body of evidence that women who have had preeclampsia are at significantly higher risk of developing hypertension, stroke, and ischemic heart disease later in life than are women who have not have preeclampsia, Dr. Saade said.
Layered evidence
The study that first caught Dr. Saade’s attention was a large Norwegian population-based study published in 2001 that looked at maternal mortality up to 25 years after pregnancy. Women who had preeclampsia had a 1.2-fold higher long-term risk of death from cardiovascular diseases, cancer, and stroke – and women with a history of both preeclampsia and a preterm delivery had a 2.71-fold higher risk – than that of women without such history.
Looking at cardiovascular causes of death specifically, the risk among women with both preeclampsia and preterm delivery was 8.12-fold higher than in women who did not have preeclampsia.
Since then, studies and reviews conducted in the United States and Europe have shown that a history of preeclampsia doubles the risk of developing cardiovascular disease, more than triples the risk of later hypertension, and also increases the risk of stroke, though more moderately.
Recently, Dr. Saade said, researchers have also begun reporting subclinical cardiac abnormalities in women with a history of preeclampsia. A study of 107 women with preeclampsia and 41 women with uneventful pregnancies found that the prevalence of subclinical heart failure (heart failure Stage B) was approximately 3.5% higher in the short term in the preeclampsia group. The women underwent regular cardiac ultrasound and other cardiovascular risk assessment tests 4-10 years postpartum (Ultrasound Obstet Gynecol. 2017;49[1]:143-9).
Preterm delivery and small for gestational age have also been associated with increased risk of ischemic heart disease and other cardiovascular events later in life. And gestational hypertension, research has shown, is a clear risk factor for later hypertension. “We always think of preeclampsia as a different disease, but as far as long-term health is concerned, it doesn’t matter if a woman had preeclampsia or gestational hypertension; she’s still at [greater] risk for hypertension later,” Dr. Saade said.
“And we don’t have to wait 30 years to see evidence” of the association between pregnancy complications and adverse cardiovascular outcomes, he emphasized. A recent retrospective cohort study of more than 300,000 women in Florida showed that women who experienced a maternal placental syndrome during their first pregnancy were at higher risk of subsequent cardiovascular disease during just 5 years of follow-up (Am J Obstet Gynecol. 2016;215[4]:484.e1-14).
Into practice
Regular screening of women whose pregnancies were complicated by conditions associated with long-term health risks “need not be that sophisticated,” Dr. Saade said. Measurement of blood pressure, waist circumference, fasting lipid profile, and fasting glucose is often enough for basic maternal health surveillance, he said.
Dr. Saade said he and some other experts in the field are recommending yearly follow-up for patients who’ve had preeclampsia and other complications. Among the other experts urging early heart disease risk screening is Graeme N. Smith, MD, PhD, an ob.gyn in Ontario who has developed surveillance protocols, follow-up forms, and risk prediction tools for use in a maternal health clinic he established at Kingston General Hospital, Queens University. At the meeting, Dr. Saade encouraged the audience to access Dr. Smith’s resources.
The American Heart Association, in its 2011 guidelines for cardiovascular disease prevention in women, includes pregnancy risks factors (specifically a history of preeclampsia, gestational diabetes, or pregnancy-induced hypertension) as part of its list of major factors for use in risk assessment (Circulation. 2011;123:1243-62).
But as Erica P. Gunderson, PhD, MPH, of Kaiser Permanente Northern California’s division of research, pointed out during another presentation at the meeting, there is more work to be done. Reproductive history is not included in existing disease prediction or risk stratification models for atherosclerotic cardiovascular disease, making it hard at this point to devise specific screening protocols and schedules, especially when it comes to a history of GDM, she said.
“We need more coordinated systems, surveillance in younger women, and some prediction models so that we can know who is at highest risk and needs more surveillance,” she said.
The AHA’s inclusion of GDM history is based on its strong link to overt diabetes, but recent evidence has shown that a history of GDM can independently elevate cardiovascular risk, she noted. Research from the Nurses Health Study II cohort, for instance, found a 30% higher relative risk of cardiovascular events (myocardial infarction and stroke) in women with a history of GDM without progression to diabetes, compared with women who did not have GDM or diabetes (JAMA Intern Med. 2017;177[12]:1735-42).
Dr. Gunderson has also found through analyses of the Coronary Artery Risk Development in Young Adults study data that women who had GDM but did not go on to develop overt diabetes or impaired glycemia had greater carotid intima media thickness many years post delivery than did women without a history of gestational diabetes (J Am Heart Assoc. 2014;3[2]:e000490).
In some models, she has written, this difference in carotid intima media thickness could represent 3-5 years of greater vascular aging for women with previous gestational diabetes and no apparent metabolic dysfunction outside of pregnancy (JAMA Intern Med. 2017;177[12]:1742-4).
There’s a question, she and Dr. Saade both noted, of whether pregnancies unmask previous dispositions to cardiovascular disease or whether pregnancy complications more directly drive adverse long-term outcomes. There is evidence that disorders such as GDM, Dr. Gunderson said, are superimposed on already altered metabolism. But at this time, Dr. Saade said, it appears that “the answer is both.”
According to Dr. Saade, at least three studies are currently following women prospectively to learn more about pregnancy as a window to future cardiovascular health. One of them is the National Institute of Child Health and Human Development’s Nulliparous Pregnancy Outcomes Study–Monitoring Mothers-to-Be Heart Health Study; some data from this study will be presented soon, he said.
Both Dr. Saade and Dr. Gunderson reported in their presentations that they had no disclosures.
WASHINGTON – George R. Saade, MD, wants to see a Time magazine cover story like the 2010 feature titled, “How the first 9 months shape the rest of your life” – except this new story would read “How the first 9 months shape the rest of the mother’s life.”
It’s time, he says, that pregnancy truly be appreciated as a “window to future health” for the mother as well as for the baby, and that the term “maternal care” replaces prenatal care. “How many of your [primary care] providers have asked you if you’ve had any pregnancies and if any of your pregnancies were complicated by hypertension, preterm delivery, growth restriction, or gestational diabetes?” Dr. Saade asked at the biennial meeting of the Diabetes in Pregnancy Study Group of North America.
“It’s better than before, but not good enough ... Checking with patients 6 weeks postpartum is not enough” to prevent long-term metabolic and cardiovascular disorders, he said. “We need regular screening of women.”
The relationship between gestational diabetes (GDM) and subsequent type 2 diabetes, demonstrated several decades ago, offered the “first evidence that pregnancy is a window to future health,” and evidence of the relationship continues to grow. “We know today that there is no other predictive marker of type 2 diabetes that is better and stronger than gestational diabetes,” said Dr. Saade, chief of obstetrics and maternal-fetal medicine at the University of Texas Medical Branch, Galveston.
GDM has also recently been shown to elevate cardiovascular risk independent of its association with type 2 diabetes and metabolic disease.
And similarly, there is now an incontrovertible body of evidence that women who have had preeclampsia are at significantly higher risk of developing hypertension, stroke, and ischemic heart disease later in life than are women who have not have preeclampsia, Dr. Saade said.
Layered evidence
The study that first caught Dr. Saade’s attention was a large Norwegian population-based study published in 2001 that looked at maternal mortality up to 25 years after pregnancy. Women who had preeclampsia had a 1.2-fold higher long-term risk of death from cardiovascular diseases, cancer, and stroke – and women with a history of both preeclampsia and a preterm delivery had a 2.71-fold higher risk – than that of women without such history.
Looking at cardiovascular causes of death specifically, the risk among women with both preeclampsia and preterm delivery was 8.12-fold higher than in women who did not have preeclampsia.
Since then, studies and reviews conducted in the United States and Europe have shown that a history of preeclampsia doubles the risk of developing cardiovascular disease, more than triples the risk of later hypertension, and also increases the risk of stroke, though more moderately.
Recently, Dr. Saade said, researchers have also begun reporting subclinical cardiac abnormalities in women with a history of preeclampsia. A study of 107 women with preeclampsia and 41 women with uneventful pregnancies found that the prevalence of subclinical heart failure (heart failure Stage B) was approximately 3.5% higher in the short term in the preeclampsia group. The women underwent regular cardiac ultrasound and other cardiovascular risk assessment tests 4-10 years postpartum (Ultrasound Obstet Gynecol. 2017;49[1]:143-9).
Preterm delivery and small for gestational age have also been associated with increased risk of ischemic heart disease and other cardiovascular events later in life. And gestational hypertension, research has shown, is a clear risk factor for later hypertension. “We always think of preeclampsia as a different disease, but as far as long-term health is concerned, it doesn’t matter if a woman had preeclampsia or gestational hypertension; she’s still at [greater] risk for hypertension later,” Dr. Saade said.
“And we don’t have to wait 30 years to see evidence” of the association between pregnancy complications and adverse cardiovascular outcomes, he emphasized. A recent retrospective cohort study of more than 300,000 women in Florida showed that women who experienced a maternal placental syndrome during their first pregnancy were at higher risk of subsequent cardiovascular disease during just 5 years of follow-up (Am J Obstet Gynecol. 2016;215[4]:484.e1-14).
Into practice
Regular screening of women whose pregnancies were complicated by conditions associated with long-term health risks “need not be that sophisticated,” Dr. Saade said. Measurement of blood pressure, waist circumference, fasting lipid profile, and fasting glucose is often enough for basic maternal health surveillance, he said.
Dr. Saade said he and some other experts in the field are recommending yearly follow-up for patients who’ve had preeclampsia and other complications. Among the other experts urging early heart disease risk screening is Graeme N. Smith, MD, PhD, an ob.gyn in Ontario who has developed surveillance protocols, follow-up forms, and risk prediction tools for use in a maternal health clinic he established at Kingston General Hospital, Queens University. At the meeting, Dr. Saade encouraged the audience to access Dr. Smith’s resources.
The American Heart Association, in its 2011 guidelines for cardiovascular disease prevention in women, includes pregnancy risks factors (specifically a history of preeclampsia, gestational diabetes, or pregnancy-induced hypertension) as part of its list of major factors for use in risk assessment (Circulation. 2011;123:1243-62).
But as Erica P. Gunderson, PhD, MPH, of Kaiser Permanente Northern California’s division of research, pointed out during another presentation at the meeting, there is more work to be done. Reproductive history is not included in existing disease prediction or risk stratification models for atherosclerotic cardiovascular disease, making it hard at this point to devise specific screening protocols and schedules, especially when it comes to a history of GDM, she said.
“We need more coordinated systems, surveillance in younger women, and some prediction models so that we can know who is at highest risk and needs more surveillance,” she said.
The AHA’s inclusion of GDM history is based on its strong link to overt diabetes, but recent evidence has shown that a history of GDM can independently elevate cardiovascular risk, she noted. Research from the Nurses Health Study II cohort, for instance, found a 30% higher relative risk of cardiovascular events (myocardial infarction and stroke) in women with a history of GDM without progression to diabetes, compared with women who did not have GDM or diabetes (JAMA Intern Med. 2017;177[12]:1735-42).
Dr. Gunderson has also found through analyses of the Coronary Artery Risk Development in Young Adults study data that women who had GDM but did not go on to develop overt diabetes or impaired glycemia had greater carotid intima media thickness many years post delivery than did women without a history of gestational diabetes (J Am Heart Assoc. 2014;3[2]:e000490).
In some models, she has written, this difference in carotid intima media thickness could represent 3-5 years of greater vascular aging for women with previous gestational diabetes and no apparent metabolic dysfunction outside of pregnancy (JAMA Intern Med. 2017;177[12]:1742-4).
There’s a question, she and Dr. Saade both noted, of whether pregnancies unmask previous dispositions to cardiovascular disease or whether pregnancy complications more directly drive adverse long-term outcomes. There is evidence that disorders such as GDM, Dr. Gunderson said, are superimposed on already altered metabolism. But at this time, Dr. Saade said, it appears that “the answer is both.”
According to Dr. Saade, at least three studies are currently following women prospectively to learn more about pregnancy as a window to future cardiovascular health. One of them is the National Institute of Child Health and Human Development’s Nulliparous Pregnancy Outcomes Study–Monitoring Mothers-to-Be Heart Health Study; some data from this study will be presented soon, he said.
Both Dr. Saade and Dr. Gunderson reported in their presentations that they had no disclosures.
EXPERT ANALYSIS FROM DPSG-NA 2017
Minimally Invasive Anatomical Reconstruction of Posteromedial Corner of Knee: A Cadaveric Study
Take-Home Points
- Injuries to the medial knee are the most common knee ligament injuries, and often occur in the athletic population.
- Complete posteromedial corner injuries require surgical treatment to restore joint stability and biomechanics.
- Biomechanical evidence has demonstrated an important load-sharing distribution between the sMCL and the POL.
- Valgus instability caused by a medial side injury, can lead to both ACL/posterior cruciate ligament reconstruction graft failure if the medial sided injury is not concurrently repaired or reconstructed.
- Anatomic posteromedial corner reconstruction yields excellent biomechanical and patient-reported outcomes.
Most injuries of the medial structures of the knee are treated conservatively.1-3 In severe acute injuries and chronic symptomatic instabilities, however, surgical treatment is needed to restore knee stability and to prevent degenerative changes secondary to instability.4 Three structures involved in medial stability are the superficial medial collateral ligament (sMCL), which is the primary valgus restraint; the posterior oblique ligament (POL), which is the primary restraint to internal rotation and the secondary valgus restraint; and the semimembranosus.5,6
Surgical techniques for posteromedial knee reconstruction include direct repair,7 repair with augmentation,8,9 advancement of the tibial insertion of the sMCL,10 and transfer of the pes anserine tendons.11 In anatomical reconstruction of the posteromedial corner, which has been described before, the sMCL and the POL are reconstructed to reproduce the native motion and stability of the knee.12 Clinically, repair and reconstruction have similar patient-reported outcomes and medial opening evaluations over the short term.
These approaches require large incisions and extensive dissection of soft tissue on the medial aspect of the knee.5 Given these drawbacks, it is reasonable to consider less invasive options. Minimally invasive surgery has the advantages of reduced scarring and blood loss, less disruption of surrounding tissue, faster recovery, and improved aesthetics.4
We conducted a study of a minimally invasive technique for reconstructing the posteromedial structures of the knee. We compared medial compartment stability measured on valgus stress radiographs in intact, sectioned, and reconstructed states in cadaveric knees. We hypothesized that a minimally invasive technique using autogenous hamstring graft in the appropriate anatomical location would return valgus stability to its nearly native state.
Materials and Methods
This study was conducted at the Buenos Aires British Hospital in Buenos Aires, Argentina, and at the University of Colorado Hospital in Aurora. Ten fresh-frozen cadaveric knees with no evidence of ligamentous injuries, osteoarthritis, or previous surgery were used. Mean donor age was 69.4 years (range, 45-87 years). Each specimen was maintained at room temperature for 24 hours before use. The femur was sectioned 20 cm proximal to the knee joint. The tibia was sectioned 12.5 cm distal to the knee joint.
Identification and Sectioning of Posteromedial Structures
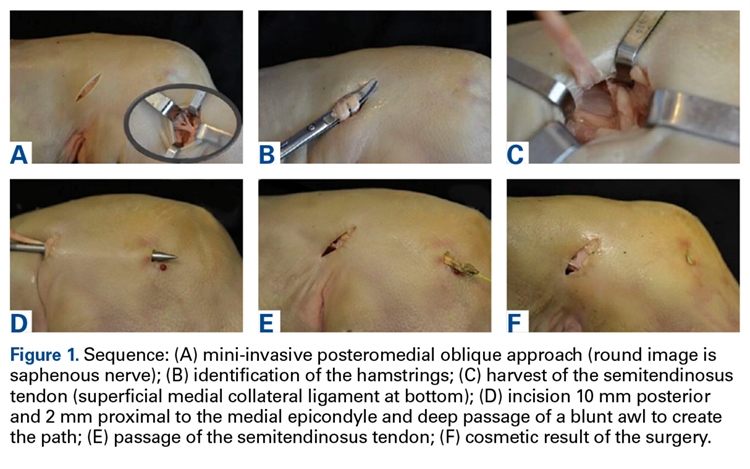
After intact-state evaluation, each knee’s sMCL, dMCL, and POL were sectioned at their tibial insertion. Valgus stress radiograph was repeated and medial compartment gap was remeasured for comparison of the sectioned state with the intact and reconstructed states.
Anatomical Reconstruction With Mini-Invasive Technique
After sectioning of medial stabilizing structures, minimally invasive reconstruction was performed through 2 small incisions on the medial aspect of each of the 10 knees, as follows. First, the semitendinosus tendon was identified through the oblique incision that had been used for sectioning. Then, an open-ended tendon stripper was placed around the circumference of the semitendinosus and was passed proximomedially, transecting the tendon at its musculotendinous junction. While the tendon stripper was being passed, care was taken to maintain the nearby tibial insertion of the sartorius fascia (Figures 1D-1F).
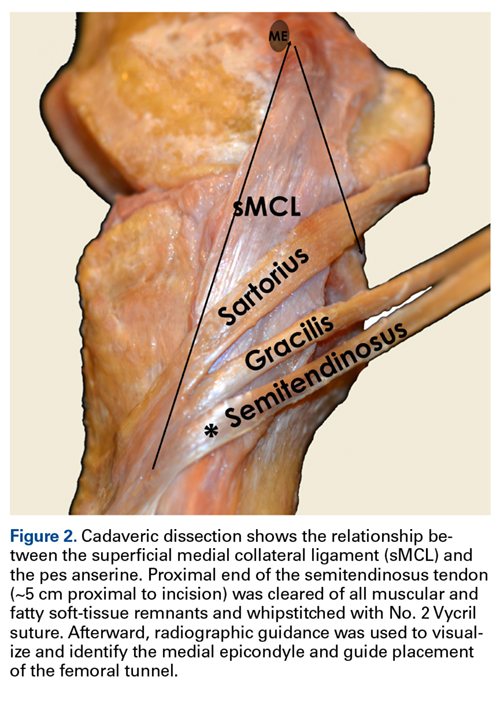
With the semitendinosus tendon looped around the wire, isometricity was tested by pulling the suture within the tendon and moving the knee through a full range of motion. The isometric point was confirmed by tendon migration of <2 mm.13 Migration was measured by marking the graft 2 mm from its insertion; the graft was then pulled to ensure correct isometric point position. An 18-mm cannulated spiked screw and washer (Arthrex) were then passed over the wire and partially secured to the femur—the attachment point for the proximal sMCL portion of the semitendinosus graft. The semitendinosus tendon was then secured beneath the spiked washer with the knee in 20° of flexion with neutral rotation, recreating the sMCL.
Posteriorly, the distal insertion site of the POL was identified at the posteromedial aspect of the tibia through the oblique incision previously described. A 7-mm tunnel was drilled starting posteromedial (10 mm under tibial articular surface) and exiting just distal and medial to the Gerdy tubercle.
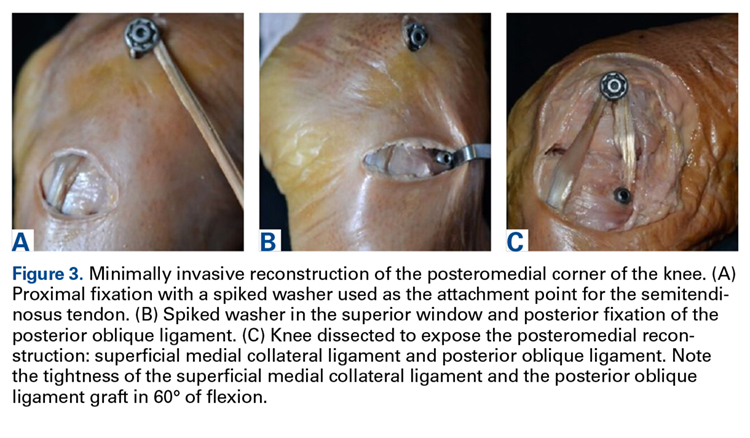
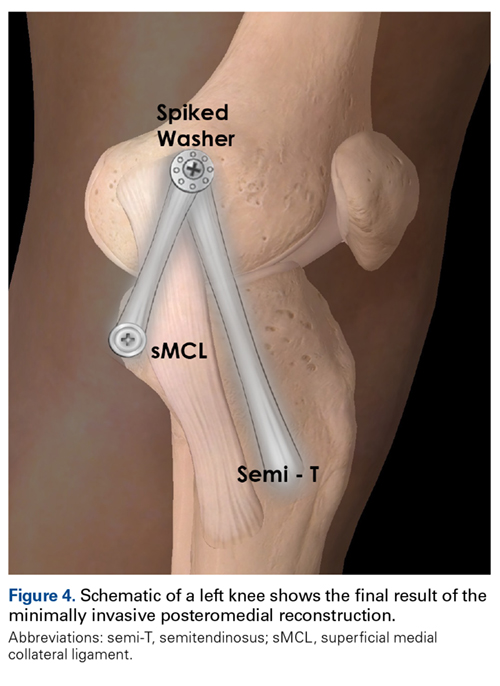
After final fixation, the medial knee was openly dissected to assess the inverted-V ligament reconstruction for anatomical placement and avoidance of crucial structures.
Stability Testing
Per International Knee Documentation Committee guidelines for stressing the medial compartment,14 valgus stress radiographs were obtained for all specimens at 0° and 20° of flexion in intact, sectioned, and reconstructed states.
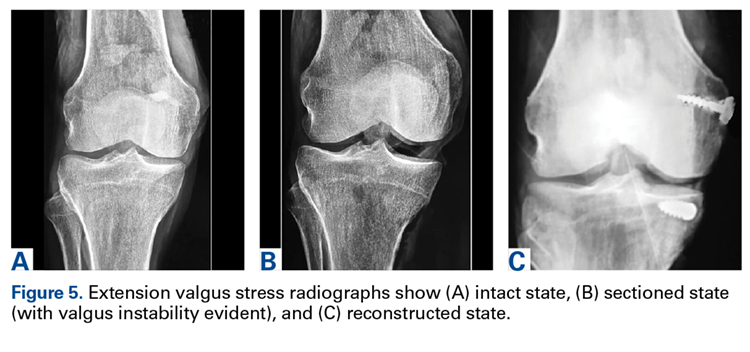
The medial gap formed by the femoral condyle and its corresponding tibial plateau (at site of maximal separation) was tested in all 3 state conditions (intact, sectioned, reconstructed). Distances were digitally measured with a picture archiving and communication system viewer (Imagecast; IDX Systems Corporation). Medial gap was measured by taking the shortest distance between the subchondral bone surface of the most distal aspect of the medial femoral condyle and the corresponding medial tibial plateau. Three independent examiners took all the measurements; each examiner was blinded to the others’ measurements.
Statistics
Paired Student t tests were used to compare the 3 conditions, and the Shapiro-Wilk test was used to check for a normally distributed population. Statistical significance was set at P < .05. Statistical analyses were performed with GraphPad software.
Results
In all 10 specimens, the sMCL, the dMCL, and the POL were successfully identified and sectioned through a medial oblique incision over the distal insertion of the structures.
During all valgus testing states, there was no loss of graft fixation, and there was no gross graft slippage. In addition, all grafts remained in continuity with no evidence of failure, and there were no failures or breakages of the proximal or distal screw.
After posteromedial sectioning, mean medial gap was statistically significantly larger (P = .0002) at full extension (11 mm vs 3.3 mm) and at 20° of flexion (12.6 mm vs 3.8 mm). There was no statistically significant difference between the value of the intact state and the value after minimally invasive reconstruction at 0° (P = .56) or 20° (P = .102) of flexion.
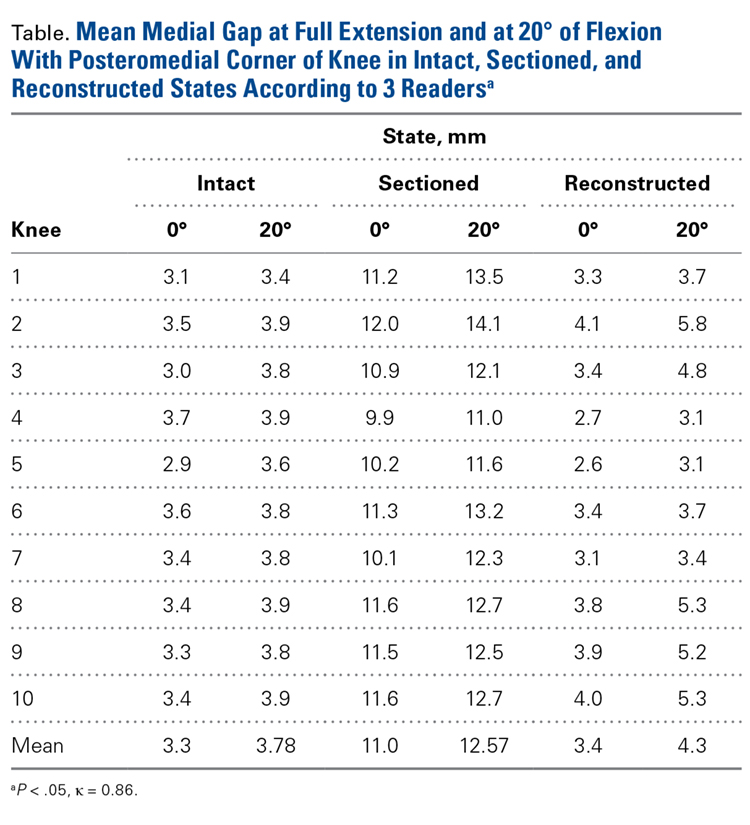
Discussion
In this article, we describe a minimally invasive technique for anatomical posteromedial reconstruction of the knee in a cadaveric model. This technique restores the knee’s native valgus stability without causing extensive damage to the surrounding soft tissues and thereby potentially prevents scar formation and reduces blood loss.
Superficial MCL injury, one of the most common knee ligament injuries, is often associated with POL injury.7 Although most sMCL injuries are treated nonoperatively, with good results,3 surgical treatment is needed for severe (grade III) instabilities, symptomatic chronic instabilities, and knee dislocations.12,17 Most posteromedial reconstruction techniques require an extensive approach that causes damage to surrounding soft tissue,6,7,9,10 which in turn may compromise healing and positive patient outcomes. Surgical techniques include direct repair with sutures or anchors,18 capsular procedures,19 augmentations,9 internal bracing,6 and complete reconstruction of the posteromedial corner.20
LaPrade and Wijdicks12 have previously described anatomical reconstruction of the posteromedial corner. In their technique, a split semitendinosus autograft is used to reconstruct the sMCL and the POL separately, using 4 implants and reproducing each ligament’s anatomical attachment site. In this proposed technique, the distal attachment of the semitendinosus insertion is left intact, and uses 1 attachment point on the distal femur and 1 on the proximal tibia, allowing use of only 2 implants. In addition, it is performed with a minimally invasive approach, reduces cost, limits surgical exposure, and with experience may shorten operative time. To reduce the graft failure rate, the technique of LaPrade and Wijdicks12 positions the sMCL tibial attachment as posterior as possible, which can be performed with this minimally invasive approach as well.
To reduce the graft failure rate, the technique of LaPrade and Wijdicks12 positions the sMCL as posterior as possible. Despite the potential for increased graft stress with an anterior position, as in our modified technique, our group of 10 knees had no graft fixation failures in isolated valgus stress testing in either extension or flexion. Our minimally invasive posteromedial knee reconstruction significantly improved knee stability over the sectioned state as well as medial compartment gapping with valgus stress. There was no significant difference in medial compartment gapping between the intact and reconstructed states.
Our technique was built on open procedures (described by Kim and colleagues13) that carefully identify the isometric point of the graft. In addition, it adopted the modification (proposed by Lind and colleagues21) in which a fixation point is added at the distal insertion of the POL instead of being sutured to the direct arm of the semitendinosus tendon.
Furthermore, our technique, despite being similar to those described by Dong and colleagues22 and Borden and colleagues,23 has the advantages of minimally invasive surgery and reduced disruption of soft tissues. Dong and colleagues22 reported on 64 patients with a mean follow-up of 34 months; patients’ medial opening measurements were significantly decreased at follow-up and fell within the normal range.
The present study had several limitations. First, the age of our specimens was higher than the mean age of patients with knee ligament injury, potentially leading to firmer or more fibrotic tendons less susceptible to elongation. Second, we did not evaluate the knees’ rotational stability, and anterior cruciate ligaments (ACLs) were intact. As most posteromedial injuries co-occur with ACL injuries, a more realistic situation would have been reproduced by assessing rotational stability while performing both ACL reconstruction and the proposed posteromedial reconstruction. Third, static specimen measurements do not reflect the dynamic function of the posteromedial corner. Prospective clinical studies are needed to assess the true effectiveness of the posteromedial corner in the clinical scenario.
Knowledge of the anatomy of the medial aspect of the knee is vital to reconstruction of the medial side of the knee. Our results suggest that a minimally invasive technique can restore valgus stability without the need for extensive dissection and disruption of surrounding soft tissues. More research is needed to determine the results of this technique in vivo.
1. Ellsasser JC, Reynolds FC, Omohundro JR. The non-operative treatment of collateral ligament injuries of the knee in professional football players. An analysis of seventy-four injuries treated non-operatively and twenty-four injuries treated surgically. J Bone Joint Surg Am. 1974;56(6):1185-1190.
2. Indelicato PA. Non-operative treatment of complete tears of the medial collateral ligament of the knee. J Bone Joint Surg Am. 1983;65(3):323-329.
3. Indelicato PA, Hermansdorfer J, Huegel M. Nonoperative management of complete tears of the medial collateral ligament of the knee in intercollegiate football players. Clin Orthop Rel Res. 1990;(256):174-177.
4. Jeng CL, Bluman EM, Myerson MS. Minimally invasive deltoid ligament reconstruction for stage IV flatfoot deformity. Foot Ankle Int. 2011;32(1):21-30.
5. Coobs BR, Wijdicks CA, Armitage BM, et al. An in vitro analysis of an anatomical medial knee reconstruction. Am J Sports Med. 2010;38(2):339-347.
6. Lubowitz JH, MacKay G, Gilmer B. Knee medial collateral ligament and posteromedial corner anatomic repair with internal bracing. Arthrosc Tech. 2014;3(4):e505-e508.
7. Hughston JC, Eilers AF. The role of the posterior oblique ligament in repairs of acute medial (collateral) ligament tears of the knee. J Bone Joint Surg Am. 1973;55(5):923-940.
8. Gorin S, Paul DD, Wilkinson EJ. An anterior cruciate ligament and medial collateral ligament tear in a skeletally immature patient: a new technique to augment primary repair of the medial collateral ligament and an allograft reconstruction of the anterior cruciate ligament. Arthroscopy. 2003;19(10):E21-E26.
Take-Home Points
- Injuries to the medial knee are the most common knee ligament injuries, and often occur in the athletic population.
- Complete posteromedial corner injuries require surgical treatment to restore joint stability and biomechanics.
- Biomechanical evidence has demonstrated an important load-sharing distribution between the sMCL and the POL.
- Valgus instability caused by a medial side injury, can lead to both ACL/posterior cruciate ligament reconstruction graft failure if the medial sided injury is not concurrently repaired or reconstructed.
- Anatomic posteromedial corner reconstruction yields excellent biomechanical and patient-reported outcomes.
Most injuries of the medial structures of the knee are treated conservatively.1-3 In severe acute injuries and chronic symptomatic instabilities, however, surgical treatment is needed to restore knee stability and to prevent degenerative changes secondary to instability.4 Three structures involved in medial stability are the superficial medial collateral ligament (sMCL), which is the primary valgus restraint; the posterior oblique ligament (POL), which is the primary restraint to internal rotation and the secondary valgus restraint; and the semimembranosus.5,6
Surgical techniques for posteromedial knee reconstruction include direct repair,7 repair with augmentation,8,9 advancement of the tibial insertion of the sMCL,10 and transfer of the pes anserine tendons.11 In anatomical reconstruction of the posteromedial corner, which has been described before, the sMCL and the POL are reconstructed to reproduce the native motion and stability of the knee.12 Clinically, repair and reconstruction have similar patient-reported outcomes and medial opening evaluations over the short term.
These approaches require large incisions and extensive dissection of soft tissue on the medial aspect of the knee.5 Given these drawbacks, it is reasonable to consider less invasive options. Minimally invasive surgery has the advantages of reduced scarring and blood loss, less disruption of surrounding tissue, faster recovery, and improved aesthetics.4
We conducted a study of a minimally invasive technique for reconstructing the posteromedial structures of the knee. We compared medial compartment stability measured on valgus stress radiographs in intact, sectioned, and reconstructed states in cadaveric knees. We hypothesized that a minimally invasive technique using autogenous hamstring graft in the appropriate anatomical location would return valgus stability to its nearly native state.
Materials and Methods
This study was conducted at the Buenos Aires British Hospital in Buenos Aires, Argentina, and at the University of Colorado Hospital in Aurora. Ten fresh-frozen cadaveric knees with no evidence of ligamentous injuries, osteoarthritis, or previous surgery were used. Mean donor age was 69.4 years (range, 45-87 years). Each specimen was maintained at room temperature for 24 hours before use. The femur was sectioned 20 cm proximal to the knee joint. The tibia was sectioned 12.5 cm distal to the knee joint.
Identification and Sectioning of Posteromedial Structures
After intact-state evaluation, each knee’s sMCL, dMCL, and POL were sectioned at their tibial insertion. Valgus stress radiograph was repeated and medial compartment gap was remeasured for comparison of the sectioned state with the intact and reconstructed states.
Anatomical Reconstruction With Mini-Invasive Technique
After sectioning of medial stabilizing structures, minimally invasive reconstruction was performed through 2 small incisions on the medial aspect of each of the 10 knees, as follows. First, the semitendinosus tendon was identified through the oblique incision that had been used for sectioning. Then, an open-ended tendon stripper was placed around the circumference of the semitendinosus and was passed proximomedially, transecting the tendon at its musculotendinous junction. While the tendon stripper was being passed, care was taken to maintain the nearby tibial insertion of the sartorius fascia (Figures 1D-1F).
With the semitendinosus tendon looped around the wire, isometricity was tested by pulling the suture within the tendon and moving the knee through a full range of motion. The isometric point was confirmed by tendon migration of <2 mm.13 Migration was measured by marking the graft 2 mm from its insertion; the graft was then pulled to ensure correct isometric point position. An 18-mm cannulated spiked screw and washer (Arthrex) were then passed over the wire and partially secured to the femur—the attachment point for the proximal sMCL portion of the semitendinosus graft. The semitendinosus tendon was then secured beneath the spiked washer with the knee in 20° of flexion with neutral rotation, recreating the sMCL.
Posteriorly, the distal insertion site of the POL was identified at the posteromedial aspect of the tibia through the oblique incision previously described. A 7-mm tunnel was drilled starting posteromedial (10 mm under tibial articular surface) and exiting just distal and medial to the Gerdy tubercle.
After final fixation, the medial knee was openly dissected to assess the inverted-V ligament reconstruction for anatomical placement and avoidance of crucial structures.
Stability Testing
Per International Knee Documentation Committee guidelines for stressing the medial compartment,14 valgus stress radiographs were obtained for all specimens at 0° and 20° of flexion in intact, sectioned, and reconstructed states.
The medial gap formed by the femoral condyle and its corresponding tibial plateau (at site of maximal separation) was tested in all 3 state conditions (intact, sectioned, reconstructed). Distances were digitally measured with a picture archiving and communication system viewer (Imagecast; IDX Systems Corporation). Medial gap was measured by taking the shortest distance between the subchondral bone surface of the most distal aspect of the medial femoral condyle and the corresponding medial tibial plateau. Three independent examiners took all the measurements; each examiner was blinded to the others’ measurements.
Statistics
Paired Student t tests were used to compare the 3 conditions, and the Shapiro-Wilk test was used to check for a normally distributed population. Statistical significance was set at P < .05. Statistical analyses were performed with GraphPad software.
Results
In all 10 specimens, the sMCL, the dMCL, and the POL were successfully identified and sectioned through a medial oblique incision over the distal insertion of the structures.
During all valgus testing states, there was no loss of graft fixation, and there was no gross graft slippage. In addition, all grafts remained in continuity with no evidence of failure, and there were no failures or breakages of the proximal or distal screw.
After posteromedial sectioning, mean medial gap was statistically significantly larger (P = .0002) at full extension (11 mm vs 3.3 mm) and at 20° of flexion (12.6 mm vs 3.8 mm). There was no statistically significant difference between the value of the intact state and the value after minimally invasive reconstruction at 0° (P = .56) or 20° (P = .102) of flexion.
Discussion
In this article, we describe a minimally invasive technique for anatomical posteromedial reconstruction of the knee in a cadaveric model. This technique restores the knee’s native valgus stability without causing extensive damage to the surrounding soft tissues and thereby potentially prevents scar formation and reduces blood loss.
Superficial MCL injury, one of the most common knee ligament injuries, is often associated with POL injury.7 Although most sMCL injuries are treated nonoperatively, with good results,3 surgical treatment is needed for severe (grade III) instabilities, symptomatic chronic instabilities, and knee dislocations.12,17 Most posteromedial reconstruction techniques require an extensive approach that causes damage to surrounding soft tissue,6,7,9,10 which in turn may compromise healing and positive patient outcomes. Surgical techniques include direct repair with sutures or anchors,18 capsular procedures,19 augmentations,9 internal bracing,6 and complete reconstruction of the posteromedial corner.20
LaPrade and Wijdicks12 have previously described anatomical reconstruction of the posteromedial corner. In their technique, a split semitendinosus autograft is used to reconstruct the sMCL and the POL separately, using 4 implants and reproducing each ligament’s anatomical attachment site. In this proposed technique, the distal attachment of the semitendinosus insertion is left intact, and uses 1 attachment point on the distal femur and 1 on the proximal tibia, allowing use of only 2 implants. In addition, it is performed with a minimally invasive approach, reduces cost, limits surgical exposure, and with experience may shorten operative time. To reduce the graft failure rate, the technique of LaPrade and Wijdicks12 positions the sMCL tibial attachment as posterior as possible, which can be performed with this minimally invasive approach as well.
To reduce the graft failure rate, the technique of LaPrade and Wijdicks12 positions the sMCL as posterior as possible. Despite the potential for increased graft stress with an anterior position, as in our modified technique, our group of 10 knees had no graft fixation failures in isolated valgus stress testing in either extension or flexion. Our minimally invasive posteromedial knee reconstruction significantly improved knee stability over the sectioned state as well as medial compartment gapping with valgus stress. There was no significant difference in medial compartment gapping between the intact and reconstructed states.
Our technique was built on open procedures (described by Kim and colleagues13) that carefully identify the isometric point of the graft. In addition, it adopted the modification (proposed by Lind and colleagues21) in which a fixation point is added at the distal insertion of the POL instead of being sutured to the direct arm of the semitendinosus tendon.
Furthermore, our technique, despite being similar to those described by Dong and colleagues22 and Borden and colleagues,23 has the advantages of minimally invasive surgery and reduced disruption of soft tissues. Dong and colleagues22 reported on 64 patients with a mean follow-up of 34 months; patients’ medial opening measurements were significantly decreased at follow-up and fell within the normal range.
The present study had several limitations. First, the age of our specimens was higher than the mean age of patients with knee ligament injury, potentially leading to firmer or more fibrotic tendons less susceptible to elongation. Second, we did not evaluate the knees’ rotational stability, and anterior cruciate ligaments (ACLs) were intact. As most posteromedial injuries co-occur with ACL injuries, a more realistic situation would have been reproduced by assessing rotational stability while performing both ACL reconstruction and the proposed posteromedial reconstruction. Third, static specimen measurements do not reflect the dynamic function of the posteromedial corner. Prospective clinical studies are needed to assess the true effectiveness of the posteromedial corner in the clinical scenario.
Knowledge of the anatomy of the medial aspect of the knee is vital to reconstruction of the medial side of the knee. Our results suggest that a minimally invasive technique can restore valgus stability without the need for extensive dissection and disruption of surrounding soft tissues. More research is needed to determine the results of this technique in vivo.
Take-Home Points
- Injuries to the medial knee are the most common knee ligament injuries, and often occur in the athletic population.
- Complete posteromedial corner injuries require surgical treatment to restore joint stability and biomechanics.
- Biomechanical evidence has demonstrated an important load-sharing distribution between the sMCL and the POL.
- Valgus instability caused by a medial side injury, can lead to both ACL/posterior cruciate ligament reconstruction graft failure if the medial sided injury is not concurrently repaired or reconstructed.
- Anatomic posteromedial corner reconstruction yields excellent biomechanical and patient-reported outcomes.
Most injuries of the medial structures of the knee are treated conservatively.1-3 In severe acute injuries and chronic symptomatic instabilities, however, surgical treatment is needed to restore knee stability and to prevent degenerative changes secondary to instability.4 Three structures involved in medial stability are the superficial medial collateral ligament (sMCL), which is the primary valgus restraint; the posterior oblique ligament (POL), which is the primary restraint to internal rotation and the secondary valgus restraint; and the semimembranosus.5,6
Surgical techniques for posteromedial knee reconstruction include direct repair,7 repair with augmentation,8,9 advancement of the tibial insertion of the sMCL,10 and transfer of the pes anserine tendons.11 In anatomical reconstruction of the posteromedial corner, which has been described before, the sMCL and the POL are reconstructed to reproduce the native motion and stability of the knee.12 Clinically, repair and reconstruction have similar patient-reported outcomes and medial opening evaluations over the short term.
These approaches require large incisions and extensive dissection of soft tissue on the medial aspect of the knee.5 Given these drawbacks, it is reasonable to consider less invasive options. Minimally invasive surgery has the advantages of reduced scarring and blood loss, less disruption of surrounding tissue, faster recovery, and improved aesthetics.4
We conducted a study of a minimally invasive technique for reconstructing the posteromedial structures of the knee. We compared medial compartment stability measured on valgus stress radiographs in intact, sectioned, and reconstructed states in cadaveric knees. We hypothesized that a minimally invasive technique using autogenous hamstring graft in the appropriate anatomical location would return valgus stability to its nearly native state.
Materials and Methods
This study was conducted at the Buenos Aires British Hospital in Buenos Aires, Argentina, and at the University of Colorado Hospital in Aurora. Ten fresh-frozen cadaveric knees with no evidence of ligamentous injuries, osteoarthritis, or previous surgery were used. Mean donor age was 69.4 years (range, 45-87 years). Each specimen was maintained at room temperature for 24 hours before use. The femur was sectioned 20 cm proximal to the knee joint. The tibia was sectioned 12.5 cm distal to the knee joint.
Identification and Sectioning of Posteromedial Structures
After intact-state evaluation, each knee’s sMCL, dMCL, and POL were sectioned at their tibial insertion. Valgus stress radiograph was repeated and medial compartment gap was remeasured for comparison of the sectioned state with the intact and reconstructed states.
Anatomical Reconstruction With Mini-Invasive Technique
After sectioning of medial stabilizing structures, minimally invasive reconstruction was performed through 2 small incisions on the medial aspect of each of the 10 knees, as follows. First, the semitendinosus tendon was identified through the oblique incision that had been used for sectioning. Then, an open-ended tendon stripper was placed around the circumference of the semitendinosus and was passed proximomedially, transecting the tendon at its musculotendinous junction. While the tendon stripper was being passed, care was taken to maintain the nearby tibial insertion of the sartorius fascia (Figures 1D-1F).
With the semitendinosus tendon looped around the wire, isometricity was tested by pulling the suture within the tendon and moving the knee through a full range of motion. The isometric point was confirmed by tendon migration of <2 mm.13 Migration was measured by marking the graft 2 mm from its insertion; the graft was then pulled to ensure correct isometric point position. An 18-mm cannulated spiked screw and washer (Arthrex) were then passed over the wire and partially secured to the femur—the attachment point for the proximal sMCL portion of the semitendinosus graft. The semitendinosus tendon was then secured beneath the spiked washer with the knee in 20° of flexion with neutral rotation, recreating the sMCL.
Posteriorly, the distal insertion site of the POL was identified at the posteromedial aspect of the tibia through the oblique incision previously described. A 7-mm tunnel was drilled starting posteromedial (10 mm under tibial articular surface) and exiting just distal and medial to the Gerdy tubercle.
After final fixation, the medial knee was openly dissected to assess the inverted-V ligament reconstruction for anatomical placement and avoidance of crucial structures.
Stability Testing
Per International Knee Documentation Committee guidelines for stressing the medial compartment,14 valgus stress radiographs were obtained for all specimens at 0° and 20° of flexion in intact, sectioned, and reconstructed states.
The medial gap formed by the femoral condyle and its corresponding tibial plateau (at site of maximal separation) was tested in all 3 state conditions (intact, sectioned, reconstructed). Distances were digitally measured with a picture archiving and communication system viewer (Imagecast; IDX Systems Corporation). Medial gap was measured by taking the shortest distance between the subchondral bone surface of the most distal aspect of the medial femoral condyle and the corresponding medial tibial plateau. Three independent examiners took all the measurements; each examiner was blinded to the others’ measurements.
Statistics
Paired Student t tests were used to compare the 3 conditions, and the Shapiro-Wilk test was used to check for a normally distributed population. Statistical significance was set at P < .05. Statistical analyses were performed with GraphPad software.
Results
In all 10 specimens, the sMCL, the dMCL, and the POL were successfully identified and sectioned through a medial oblique incision over the distal insertion of the structures.
During all valgus testing states, there was no loss of graft fixation, and there was no gross graft slippage. In addition, all grafts remained in continuity with no evidence of failure, and there were no failures or breakages of the proximal or distal screw.
After posteromedial sectioning, mean medial gap was statistically significantly larger (P = .0002) at full extension (11 mm vs 3.3 mm) and at 20° of flexion (12.6 mm vs 3.8 mm). There was no statistically significant difference between the value of the intact state and the value after minimally invasive reconstruction at 0° (P = .56) or 20° (P = .102) of flexion.
Discussion
In this article, we describe a minimally invasive technique for anatomical posteromedial reconstruction of the knee in a cadaveric model. This technique restores the knee’s native valgus stability without causing extensive damage to the surrounding soft tissues and thereby potentially prevents scar formation and reduces blood loss.
Superficial MCL injury, one of the most common knee ligament injuries, is often associated with POL injury.7 Although most sMCL injuries are treated nonoperatively, with good results,3 surgical treatment is needed for severe (grade III) instabilities, symptomatic chronic instabilities, and knee dislocations.12,17 Most posteromedial reconstruction techniques require an extensive approach that causes damage to surrounding soft tissue,6,7,9,10 which in turn may compromise healing and positive patient outcomes. Surgical techniques include direct repair with sutures or anchors,18 capsular procedures,19 augmentations,9 internal bracing,6 and complete reconstruction of the posteromedial corner.20
LaPrade and Wijdicks12 have previously described anatomical reconstruction of the posteromedial corner. In their technique, a split semitendinosus autograft is used to reconstruct the sMCL and the POL separately, using 4 implants and reproducing each ligament’s anatomical attachment site. In this proposed technique, the distal attachment of the semitendinosus insertion is left intact, and uses 1 attachment point on the distal femur and 1 on the proximal tibia, allowing use of only 2 implants. In addition, it is performed with a minimally invasive approach, reduces cost, limits surgical exposure, and with experience may shorten operative time. To reduce the graft failure rate, the technique of LaPrade and Wijdicks12 positions the sMCL tibial attachment as posterior as possible, which can be performed with this minimally invasive approach as well.
To reduce the graft failure rate, the technique of LaPrade and Wijdicks12 positions the sMCL as posterior as possible. Despite the potential for increased graft stress with an anterior position, as in our modified technique, our group of 10 knees had no graft fixation failures in isolated valgus stress testing in either extension or flexion. Our minimally invasive posteromedial knee reconstruction significantly improved knee stability over the sectioned state as well as medial compartment gapping with valgus stress. There was no significant difference in medial compartment gapping between the intact and reconstructed states.
Our technique was built on open procedures (described by Kim and colleagues13) that carefully identify the isometric point of the graft. In addition, it adopted the modification (proposed by Lind and colleagues21) in which a fixation point is added at the distal insertion of the POL instead of being sutured to the direct arm of the semitendinosus tendon.
Furthermore, our technique, despite being similar to those described by Dong and colleagues22 and Borden and colleagues,23 has the advantages of minimally invasive surgery and reduced disruption of soft tissues. Dong and colleagues22 reported on 64 patients with a mean follow-up of 34 months; patients’ medial opening measurements were significantly decreased at follow-up and fell within the normal range.
The present study had several limitations. First, the age of our specimens was higher than the mean age of patients with knee ligament injury, potentially leading to firmer or more fibrotic tendons less susceptible to elongation. Second, we did not evaluate the knees’ rotational stability, and anterior cruciate ligaments (ACLs) were intact. As most posteromedial injuries co-occur with ACL injuries, a more realistic situation would have been reproduced by assessing rotational stability while performing both ACL reconstruction and the proposed posteromedial reconstruction. Third, static specimen measurements do not reflect the dynamic function of the posteromedial corner. Prospective clinical studies are needed to assess the true effectiveness of the posteromedial corner in the clinical scenario.
Knowledge of the anatomy of the medial aspect of the knee is vital to reconstruction of the medial side of the knee. Our results suggest that a minimally invasive technique can restore valgus stability without the need for extensive dissection and disruption of surrounding soft tissues. More research is needed to determine the results of this technique in vivo.
1. Ellsasser JC, Reynolds FC, Omohundro JR. The non-operative treatment of collateral ligament injuries of the knee in professional football players. An analysis of seventy-four injuries treated non-operatively and twenty-four injuries treated surgically. J Bone Joint Surg Am. 1974;56(6):1185-1190.
2. Indelicato PA. Non-operative treatment of complete tears of the medial collateral ligament of the knee. J Bone Joint Surg Am. 1983;65(3):323-329.
3. Indelicato PA, Hermansdorfer J, Huegel M. Nonoperative management of complete tears of the medial collateral ligament of the knee in intercollegiate football players. Clin Orthop Rel Res. 1990;(256):174-177.
4. Jeng CL, Bluman EM, Myerson MS. Minimally invasive deltoid ligament reconstruction for stage IV flatfoot deformity. Foot Ankle Int. 2011;32(1):21-30.
5. Coobs BR, Wijdicks CA, Armitage BM, et al. An in vitro analysis of an anatomical medial knee reconstruction. Am J Sports Med. 2010;38(2):339-347.
6. Lubowitz JH, MacKay G, Gilmer B. Knee medial collateral ligament and posteromedial corner anatomic repair with internal bracing. Arthrosc Tech. 2014;3(4):e505-e508.
7. Hughston JC, Eilers AF. The role of the posterior oblique ligament in repairs of acute medial (collateral) ligament tears of the knee. J Bone Joint Surg Am. 1973;55(5):923-940.
8. Gorin S, Paul DD, Wilkinson EJ. An anterior cruciate ligament and medial collateral ligament tear in a skeletally immature patient: a new technique to augment primary repair of the medial collateral ligament and an allograft reconstruction of the anterior cruciate ligament. Arthroscopy. 2003;19(10):E21-E26.
1. Ellsasser JC, Reynolds FC, Omohundro JR. The non-operative treatment of collateral ligament injuries of the knee in professional football players. An analysis of seventy-four injuries treated non-operatively and twenty-four injuries treated surgically. J Bone Joint Surg Am. 1974;56(6):1185-1190.
2. Indelicato PA. Non-operative treatment of complete tears of the medial collateral ligament of the knee. J Bone Joint Surg Am. 1983;65(3):323-329.
3. Indelicato PA, Hermansdorfer J, Huegel M. Nonoperative management of complete tears of the medial collateral ligament of the knee in intercollegiate football players. Clin Orthop Rel Res. 1990;(256):174-177.
4. Jeng CL, Bluman EM, Myerson MS. Minimally invasive deltoid ligament reconstruction for stage IV flatfoot deformity. Foot Ankle Int. 2011;32(1):21-30.
5. Coobs BR, Wijdicks CA, Armitage BM, et al. An in vitro analysis of an anatomical medial knee reconstruction. Am J Sports Med. 2010;38(2):339-347.
6. Lubowitz JH, MacKay G, Gilmer B. Knee medial collateral ligament and posteromedial corner anatomic repair with internal bracing. Arthrosc Tech. 2014;3(4):e505-e508.
7. Hughston JC, Eilers AF. The role of the posterior oblique ligament in repairs of acute medial (collateral) ligament tears of the knee. J Bone Joint Surg Am. 1973;55(5):923-940.
8. Gorin S, Paul DD, Wilkinson EJ. An anterior cruciate ligament and medial collateral ligament tear in a skeletally immature patient: a new technique to augment primary repair of the medial collateral ligament and an allograft reconstruction of the anterior cruciate ligament. Arthroscopy. 2003;19(10):E21-E26.
Ozanimod Is Superior to Interferon in Relapsing-Remitting MS
PARIS—Ozanimod is superior to interferon beta-1a on clinical and MRI measures in patients with relapsing-remitting multiple sclerosis (MS), according to the results of two studies presented at the Seventh Joint ECTRIMS–ACTRIMS Meeting. The drug could become a safe and effective oral medication for this population, said the investigators.
Scientists at the Scripps Research Institute developed ozanimod, a selective sphingosine 1-phosphate 1 and 5 receptor modulator. “Receptor specificity for subtypes 1 and 5 is anticipated to preserve efficacy, but to lessen off-target treatment effects, as compared to nonselective sphingosine 1-phosphate receptor modulators,” said Jeffrey Cohen, MD, a neurologist at the Cleveland Clinic and an investigator on both of the studies.

The SUNBEAM Study
SUNBEAM was the first of the two trials of ozanimod to be presented at the meeting. Eligible participants were between ages 18 and 55, had relapsing-remitting MS, had had at least one documented relapse in the previous year, had not had a relapse in the previous 30 days, and had no history of cardiac conditions. The trial randomized participants to 0.5 mg/day of oral ozanimod, 1 mg/day of oral ozanimod, or weekly intramuscular injections of interferon beta-1a (30 µg). Patients in the ozanimod groups underwent a seven-day dose escalation as an initiation.
The trial’s primary end point was annualized relapse rate. Among the secondary end points were new and enlarging T2 lesions from baseline to month 12 and T1, gadolinium-enhancing lesions at month 12. The investigators carried out a prespecified evaluation of disability progression, based on Expanded Disability Status Scale (EDSS), using a pooled analysis of SUNBEAM and RADIANCE, the other phase III study.
The researchers enrolled 1,346 patients in 20 countries. The treatment groups were well balanced at baseline. Participants’ mean age was 36, and 66% of the population was female. Mean EDSS score at baseline was 2.6, mean number of relapses in the prior year was 1.3, and 47% of participants had gadolinium-enhancing lesions. In addition, 31% of participants previously had been treated with disease-modifying therapy.
The mean treatment duration in SUNBEAM was 13.6 months. The annualized relapse rate was reduced by 31% with 0.5 mg/day of ozanimod and by 48% with 1 mg/day of ozanimod, compared with interferon beta-1a. Overall, ozanimod reduced MRI activity by 63%, compared with interferon beta-1a. The number of new or enlarging T2 lesions and the adjusted mean number of gadolinium-enhancing lesions at month 12 were significantly reduced in the ozanimod groups, compared with the interferon group. The investigators observed a consistent dose response to ozanimod for all efficacy end points.
The majority of treatment-emergent adverse events were mild, and serious adverse events were uncommon. The rate of adverse events was similar for all treatment groups. The rate of discontinuation due to adverse events also was low and similar between treatment groups. The researchers observed no first-dose, clinically relevant cases of bradycardia.
The RADIANCE Trial
The eligibility criteria for the RADIANCE trial were the same as those of the SUNBEAM study. Participants were randomized to 0.5-mg/day or 1-mg/day doses of ozanimod or to 30 µg/week of interferon beta-1a. The primary end point in RADIANCE was annualized relapse rate at two years. Secondary end points included measures of MRI lesion activity, brain volume loss over two years, and a pooled analysis of confirmed disability worsening. Disability was assessed during a clinical evaluation every three months and at the time of potential relapse.
The investigators enrolled 1,313 patients into the study, and their baseline characteristics were similar across treatment groups. Participants’ mean age was 36, and 67% of the population was female. Mean EDSS score at baseline was 2.5, and mean number of relapses in the prior year was 1.3. About 43% of patients had gadolinium-enhancing lesions at baseline, and 29% previously had received disease-modifying therapy. Overall, 86% of the participants completed the trial, and the discontinuation rate was similar in all treatment arms.
The 0.5-mg dose of ozanimod was associated with a 21% reduction in annualized relapse rate, compared with interferon, and the 1-mg dose of ozanimod was associated with a 38% reduction in this measure. The adjusted mean number of new or enlarging T2 lesions per scan was reduced by 42% with ozanimod 1 mg and by 35% with 0.5 mg of ozanimod, compared with interferon. The adjusted mean number of gadolinium-enhancing lesions at 24 months also was reduced by 53% with 1 mg of ozanimod and by 47% with 0.5 mg of ozanimod, compared with interferon.
In addition, the study demonstrated an approximately 25% reduction in whole-brain volume loss in the two ozanimod arms, compared with the interferon group. Ozanimod also reduced segmented cortical gray volume loss and segmented thalamic volume loss to a similar extent, compared with interferon.
In the pooled analysis of both trials, the rate of confirmed disability worsening was low. The investigators did not observe any difference in this outcome between the three treatment arms.
Ozanimod was well tolerated. Liver enzyme elevations associated with the drug tended to be mild and resolved despite continuation of study drug. The drug was associated with a minimal change in heart rate, and the researchers observed no cases of second-degree or higher heart block. “It appears that the first-dose escalation protocol effectively mitigated cardiac effects,” said Dr. Cohen.
Celgene, the company developing the drug, expects to submit the compound for FDA approval and introduce it to the market by the end of 2018, according to a press release.
—Erik Greb
Suggested Reading
Cohen JA, Arnold DL, Comi G, et al. Safety and efficacy of the selective sphingosine 1-phosphate receptor modulator ozanimod in relapsing multiple sclerosis (RADIANCE): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(4):373-381.
Sørensen PS. Ozanimod: a better or just another S1P receptor modulator? Lancet Neurol. 2016;15(4):345-347.
PARIS—Ozanimod is superior to interferon beta-1a on clinical and MRI measures in patients with relapsing-remitting multiple sclerosis (MS), according to the results of two studies presented at the Seventh Joint ECTRIMS–ACTRIMS Meeting. The drug could become a safe and effective oral medication for this population, said the investigators.
Scientists at the Scripps Research Institute developed ozanimod, a selective sphingosine 1-phosphate 1 and 5 receptor modulator. “Receptor specificity for subtypes 1 and 5 is anticipated to preserve efficacy, but to lessen off-target treatment effects, as compared to nonselective sphingosine 1-phosphate receptor modulators,” said Jeffrey Cohen, MD, a neurologist at the Cleveland Clinic and an investigator on both of the studies.
The SUNBEAM Study
SUNBEAM was the first of the two trials of ozanimod to be presented at the meeting. Eligible participants were between ages 18 and 55, had relapsing-remitting MS, had had at least one documented relapse in the previous year, had not had a relapse in the previous 30 days, and had no history of cardiac conditions. The trial randomized participants to 0.5 mg/day of oral ozanimod, 1 mg/day of oral ozanimod, or weekly intramuscular injections of interferon beta-1a (30 µg). Patients in the ozanimod groups underwent a seven-day dose escalation as an initiation.
The trial’s primary end point was annualized relapse rate. Among the secondary end points were new and enlarging T2 lesions from baseline to month 12 and T1, gadolinium-enhancing lesions at month 12. The investigators carried out a prespecified evaluation of disability progression, based on Expanded Disability Status Scale (EDSS), using a pooled analysis of SUNBEAM and RADIANCE, the other phase III study.
The researchers enrolled 1,346 patients in 20 countries. The treatment groups were well balanced at baseline. Participants’ mean age was 36, and 66% of the population was female. Mean EDSS score at baseline was 2.6, mean number of relapses in the prior year was 1.3, and 47% of participants had gadolinium-enhancing lesions. In addition, 31% of participants previously had been treated with disease-modifying therapy.
The mean treatment duration in SUNBEAM was 13.6 months. The annualized relapse rate was reduced by 31% with 0.5 mg/day of ozanimod and by 48% with 1 mg/day of ozanimod, compared with interferon beta-1a. Overall, ozanimod reduced MRI activity by 63%, compared with interferon beta-1a. The number of new or enlarging T2 lesions and the adjusted mean number of gadolinium-enhancing lesions at month 12 were significantly reduced in the ozanimod groups, compared with the interferon group. The investigators observed a consistent dose response to ozanimod for all efficacy end points.
The majority of treatment-emergent adverse events were mild, and serious adverse events were uncommon. The rate of adverse events was similar for all treatment groups. The rate of discontinuation due to adverse events also was low and similar between treatment groups. The researchers observed no first-dose, clinically relevant cases of bradycardia.
The RADIANCE Trial
The eligibility criteria for the RADIANCE trial were the same as those of the SUNBEAM study. Participants were randomized to 0.5-mg/day or 1-mg/day doses of ozanimod or to 30 µg/week of interferon beta-1a. The primary end point in RADIANCE was annualized relapse rate at two years. Secondary end points included measures of MRI lesion activity, brain volume loss over two years, and a pooled analysis of confirmed disability worsening. Disability was assessed during a clinical evaluation every three months and at the time of potential relapse.
The investigators enrolled 1,313 patients into the study, and their baseline characteristics were similar across treatment groups. Participants’ mean age was 36, and 67% of the population was female. Mean EDSS score at baseline was 2.5, and mean number of relapses in the prior year was 1.3. About 43% of patients had gadolinium-enhancing lesions at baseline, and 29% previously had received disease-modifying therapy. Overall, 86% of the participants completed the trial, and the discontinuation rate was similar in all treatment arms.
The 0.5-mg dose of ozanimod was associated with a 21% reduction in annualized relapse rate, compared with interferon, and the 1-mg dose of ozanimod was associated with a 38% reduction in this measure. The adjusted mean number of new or enlarging T2 lesions per scan was reduced by 42% with ozanimod 1 mg and by 35% with 0.5 mg of ozanimod, compared with interferon. The adjusted mean number of gadolinium-enhancing lesions at 24 months also was reduced by 53% with 1 mg of ozanimod and by 47% with 0.5 mg of ozanimod, compared with interferon.
In addition, the study demonstrated an approximately 25% reduction in whole-brain volume loss in the two ozanimod arms, compared with the interferon group. Ozanimod also reduced segmented cortical gray volume loss and segmented thalamic volume loss to a similar extent, compared with interferon.
In the pooled analysis of both trials, the rate of confirmed disability worsening was low. The investigators did not observe any difference in this outcome between the three treatment arms.
Ozanimod was well tolerated. Liver enzyme elevations associated with the drug tended to be mild and resolved despite continuation of study drug. The drug was associated with a minimal change in heart rate, and the researchers observed no cases of second-degree or higher heart block. “It appears that the first-dose escalation protocol effectively mitigated cardiac effects,” said Dr. Cohen.
Celgene, the company developing the drug, expects to submit the compound for FDA approval and introduce it to the market by the end of 2018, according to a press release.
—Erik Greb
Suggested Reading
Cohen JA, Arnold DL, Comi G, et al. Safety and efficacy of the selective sphingosine 1-phosphate receptor modulator ozanimod in relapsing multiple sclerosis (RADIANCE): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(4):373-381.
Sørensen PS. Ozanimod: a better or just another S1P receptor modulator? Lancet Neurol. 2016;15(4):345-347.
PARIS—Ozanimod is superior to interferon beta-1a on clinical and MRI measures in patients with relapsing-remitting multiple sclerosis (MS), according to the results of two studies presented at the Seventh Joint ECTRIMS–ACTRIMS Meeting. The drug could become a safe and effective oral medication for this population, said the investigators.
Scientists at the Scripps Research Institute developed ozanimod, a selective sphingosine 1-phosphate 1 and 5 receptor modulator. “Receptor specificity for subtypes 1 and 5 is anticipated to preserve efficacy, but to lessen off-target treatment effects, as compared to nonselective sphingosine 1-phosphate receptor modulators,” said Jeffrey Cohen, MD, a neurologist at the Cleveland Clinic and an investigator on both of the studies.
The SUNBEAM Study
SUNBEAM was the first of the two trials of ozanimod to be presented at the meeting. Eligible participants were between ages 18 and 55, had relapsing-remitting MS, had had at least one documented relapse in the previous year, had not had a relapse in the previous 30 days, and had no history of cardiac conditions. The trial randomized participants to 0.5 mg/day of oral ozanimod, 1 mg/day of oral ozanimod, or weekly intramuscular injections of interferon beta-1a (30 µg). Patients in the ozanimod groups underwent a seven-day dose escalation as an initiation.
The trial’s primary end point was annualized relapse rate. Among the secondary end points were new and enlarging T2 lesions from baseline to month 12 and T1, gadolinium-enhancing lesions at month 12. The investigators carried out a prespecified evaluation of disability progression, based on Expanded Disability Status Scale (EDSS), using a pooled analysis of SUNBEAM and RADIANCE, the other phase III study.
The researchers enrolled 1,346 patients in 20 countries. The treatment groups were well balanced at baseline. Participants’ mean age was 36, and 66% of the population was female. Mean EDSS score at baseline was 2.6, mean number of relapses in the prior year was 1.3, and 47% of participants had gadolinium-enhancing lesions. In addition, 31% of participants previously had been treated with disease-modifying therapy.
The mean treatment duration in SUNBEAM was 13.6 months. The annualized relapse rate was reduced by 31% with 0.5 mg/day of ozanimod and by 48% with 1 mg/day of ozanimod, compared with interferon beta-1a. Overall, ozanimod reduced MRI activity by 63%, compared with interferon beta-1a. The number of new or enlarging T2 lesions and the adjusted mean number of gadolinium-enhancing lesions at month 12 were significantly reduced in the ozanimod groups, compared with the interferon group. The investigators observed a consistent dose response to ozanimod for all efficacy end points.
The majority of treatment-emergent adverse events were mild, and serious adverse events were uncommon. The rate of adverse events was similar for all treatment groups. The rate of discontinuation due to adverse events also was low and similar between treatment groups. The researchers observed no first-dose, clinically relevant cases of bradycardia.
The RADIANCE Trial
The eligibility criteria for the RADIANCE trial were the same as those of the SUNBEAM study. Participants were randomized to 0.5-mg/day or 1-mg/day doses of ozanimod or to 30 µg/week of interferon beta-1a. The primary end point in RADIANCE was annualized relapse rate at two years. Secondary end points included measures of MRI lesion activity, brain volume loss over two years, and a pooled analysis of confirmed disability worsening. Disability was assessed during a clinical evaluation every three months and at the time of potential relapse.
The investigators enrolled 1,313 patients into the study, and their baseline characteristics were similar across treatment groups. Participants’ mean age was 36, and 67% of the population was female. Mean EDSS score at baseline was 2.5, and mean number of relapses in the prior year was 1.3. About 43% of patients had gadolinium-enhancing lesions at baseline, and 29% previously had received disease-modifying therapy. Overall, 86% of the participants completed the trial, and the discontinuation rate was similar in all treatment arms.
The 0.5-mg dose of ozanimod was associated with a 21% reduction in annualized relapse rate, compared with interferon, and the 1-mg dose of ozanimod was associated with a 38% reduction in this measure. The adjusted mean number of new or enlarging T2 lesions per scan was reduced by 42% with ozanimod 1 mg and by 35% with 0.5 mg of ozanimod, compared with interferon. The adjusted mean number of gadolinium-enhancing lesions at 24 months also was reduced by 53% with 1 mg of ozanimod and by 47% with 0.5 mg of ozanimod, compared with interferon.
In addition, the study demonstrated an approximately 25% reduction in whole-brain volume loss in the two ozanimod arms, compared with the interferon group. Ozanimod also reduced segmented cortical gray volume loss and segmented thalamic volume loss to a similar extent, compared with interferon.
In the pooled analysis of both trials, the rate of confirmed disability worsening was low. The investigators did not observe any difference in this outcome between the three treatment arms.
Ozanimod was well tolerated. Liver enzyme elevations associated with the drug tended to be mild and resolved despite continuation of study drug. The drug was associated with a minimal change in heart rate, and the researchers observed no cases of second-degree or higher heart block. “It appears that the first-dose escalation protocol effectively mitigated cardiac effects,” said Dr. Cohen.
Celgene, the company developing the drug, expects to submit the compound for FDA approval and introduce it to the market by the end of 2018, according to a press release.
—Erik Greb
Suggested Reading
Cohen JA, Arnold DL, Comi G, et al. Safety and efficacy of the selective sphingosine 1-phosphate receptor modulator ozanimod in relapsing multiple sclerosis (RADIANCE): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(4):373-381.
Sørensen PS. Ozanimod: a better or just another S1P receptor modulator? Lancet Neurol. 2016;15(4):345-347.
Gene Therapy May Benefit Patients With Cerebral ALD
KANSAS CITY, MO—Lentiviral gene therapy halts inflammation and demyelination in patients with cerebral adrenoleukodystrophy (ALD), according to research presented at the 46th Annual Meeting of the Child Neurology Society and published in the New England Journal of Medicine. The treatment appears to stabilize neurologic function and to have an acceptable safety profile.
ALD is an X-linked genetic disease caused by a defect in ABCD1, which encodes the peroxisomal ABC half-transporter ALD protein. The defect results in abnormal breakdown of very-long-chain fatty acids, which build up and affect tissues in the nervous system. The most common phenotype of ALD is adrenomyeloneuropathy, which involves axonal degeneration in the spinal cord. A more severe phenotype is childhood cerebral adenopathy, which manifests as an inflammatory demyelination in the white matter.

Allogeneic bone marrow transplantation is the only other treatment with proven efficacy in childhood cerebral ALD. It halts disease progression and improves survival in presymptomatic patients. Finding a donor with identical human leukocyte antigen often is difficult, however. In addition, the treatment entails risks of graft failure and graft-versus-host disease, and the rate of treatment-related mortality is greater than 10%. Gene therapy “may be an alternative to allogeneic bone marrow transplantation, particularly for patients who do not have a matched sibling donor who are at risk for graft failure and graft-versus-host [disease],” said Florian Eichler, MD, Director of the Leukodystrophy Service at Massachusetts General Hospital for Children in Boston.
Researchers Transduced Stem Cells Ex Vivo
In the first trial of its kind, Dr. Eichler and colleagues investigated the safety and efficacy of gene therapy with autologous hematopoietic stem cells for cerebral ALD. The enrollment criteria for the phase II–III, single-arm, open-label study were identical to those for conventional bone marrow transplantation, said Dr. Eichler. Eligible participants were age 17 or younger, had a defect in ABCD1, and had evidence of an active, inflammatory lesion on brain MRI. Children with sibling donors for bone marrow transplantation were excluded from the study.
The investigators obtained CD34+ cells from participants through apheresis and transduced them ex vivo with a lentiviral vector containing normal ABCD1. After undergoing conditioning with busulfan and cyclophosphamide, the patients received an infusion of the transduced CD34+ cells. Patients were assessed regularly for graft-versus-host disease, death, and major functional disabilities. Dr. Eichler and colleagues also monitored participants for changes in neurologic function and increases in MRI lesions. At the end of a two-year follow-up period, patients were offered enrollment in a 13-year long-term follow-up study. The main study’s primary end point was being alive and having no major functional disability at 24 months.
Lesion Progression Stabilized in Most Patients
Dr. Eichler and colleagues treated 17 patients with a mean age at enrollment of 6. Participants’ median Loes score at baseline was 2.0, which indicated a low level of neurologic progression. All patients had an ALD neurologic function scale score of 0 at baseline, indicating an absence of clinical signs of cerebral disease.
The vector copy number in peripheral blood ranged from 0.10 to 1.55 in the 14 patients assessed at month 24. At month 36, the number ranged from 0.36 to 1.83 in the three patients assessed. The number had generally stabilized by two months after infusion. The investigators did not observe preferential integration in or near genes that have previously been associated with serious adverse events related to gene therapy (eg, MDS1, EVI1, and LMO2). All participants had expression of ALD protein in peripheral-blood leukocytes at the most recent follow-up (median follow-up time was 29.4 months). The median percentage of CD14+ cells that expressed ALD protein was 19% at 24 months.
Of the 17 patients, 15 were alive at 24 months and maintained an ALD neurologic function scale score of 0 or 1. In untreated patients, ALD neurologic function scale score usually increases significantly after the first symptoms appear. One patient in the study was progressing rapidly at the time of enrollment and subsequently died of disease progression. Another patient with disease progression on MRI withdrew from the study after the infusion and subsequently died from the complications of an allogeneic transplantation
An exploratory analysis indicated that participants with higher vector copy number in their peripheral blood tended to have better neurologic outcomes, while participants with lower vector copy number tended to have poor neurologic outcomes.
Lesion progression, as measured with the Loes score, had stabilized in 12 of the 17 patients (71%) at the time of the interim analysis. “Half the patients stabilized immediately in their Loes scores after treatment. The other half tended to progress within the first 12 months and then stabilize over time,” said Dr. Eichler.
Gadolinium enhancement resolved in 16 of 17 patients by six months. It reappeared in some patients after treatment, although it was much fainter and more poorly circumscribed, compared with the time of screening, said Dr. Eichler.
The investigators did not observe graft failure or graft-versus-host disease in the population. Most adverse events associated with the treatment were consistent with those associated with myeloablative chemotherapy and occurred during conditioning or during the first two weeks after the infusion. One serious adverse event that possibly was related to treatment was hemorrhagic cystitis associated with BK virus, which resolved with conservative measures.
The results have implications for leukodystrophies in general, according to Dr. Eichler. “Specific phenotypes, even within the same individual leukodystrophy, require different approaches” such as ex vivo or in vivo gene therapy, he added. Overall, the safety data are reassuring, and gene therapy appears to have encouraging efficacy in ALD, he concluded.
—Erik Greb
Suggested Reading
Eichler F, Duncan C, Musolino PL, et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med. 2017;377(17):1630-1638.
Engelen M. Optimizing treatment for cerebral adrenoleukodystrophy in the era of gene therapy. N Engl J Med. 2017;377(17):1682-1684.
KANSAS CITY, MO—Lentiviral gene therapy halts inflammation and demyelination in patients with cerebral adrenoleukodystrophy (ALD), according to research presented at the 46th Annual Meeting of the Child Neurology Society and published in the New England Journal of Medicine. The treatment appears to stabilize neurologic function and to have an acceptable safety profile.
ALD is an X-linked genetic disease caused by a defect in ABCD1, which encodes the peroxisomal ABC half-transporter ALD protein. The defect results in abnormal breakdown of very-long-chain fatty acids, which build up and affect tissues in the nervous system. The most common phenotype of ALD is adrenomyeloneuropathy, which involves axonal degeneration in the spinal cord. A more severe phenotype is childhood cerebral adenopathy, which manifests as an inflammatory demyelination in the white matter.
Allogeneic bone marrow transplantation is the only other treatment with proven efficacy in childhood cerebral ALD. It halts disease progression and improves survival in presymptomatic patients. Finding a donor with identical human leukocyte antigen often is difficult, however. In addition, the treatment entails risks of graft failure and graft-versus-host disease, and the rate of treatment-related mortality is greater than 10%. Gene therapy “may be an alternative to allogeneic bone marrow transplantation, particularly for patients who do not have a matched sibling donor who are at risk for graft failure and graft-versus-host [disease],” said Florian Eichler, MD, Director of the Leukodystrophy Service at Massachusetts General Hospital for Children in Boston.
Researchers Transduced Stem Cells Ex Vivo
In the first trial of its kind, Dr. Eichler and colleagues investigated the safety and efficacy of gene therapy with autologous hematopoietic stem cells for cerebral ALD. The enrollment criteria for the phase II–III, single-arm, open-label study were identical to those for conventional bone marrow transplantation, said Dr. Eichler. Eligible participants were age 17 or younger, had a defect in ABCD1, and had evidence of an active, inflammatory lesion on brain MRI. Children with sibling donors for bone marrow transplantation were excluded from the study.
The investigators obtained CD34+ cells from participants through apheresis and transduced them ex vivo with a lentiviral vector containing normal ABCD1. After undergoing conditioning with busulfan and cyclophosphamide, the patients received an infusion of the transduced CD34+ cells. Patients were assessed regularly for graft-versus-host disease, death, and major functional disabilities. Dr. Eichler and colleagues also monitored participants for changes in neurologic function and increases in MRI lesions. At the end of a two-year follow-up period, patients were offered enrollment in a 13-year long-term follow-up study. The main study’s primary end point was being alive and having no major functional disability at 24 months.
Lesion Progression Stabilized in Most Patients
Dr. Eichler and colleagues treated 17 patients with a mean age at enrollment of 6. Participants’ median Loes score at baseline was 2.0, which indicated a low level of neurologic progression. All patients had an ALD neurologic function scale score of 0 at baseline, indicating an absence of clinical signs of cerebral disease.
The vector copy number in peripheral blood ranged from 0.10 to 1.55 in the 14 patients assessed at month 24. At month 36, the number ranged from 0.36 to 1.83 in the three patients assessed. The number had generally stabilized by two months after infusion. The investigators did not observe preferential integration in or near genes that have previously been associated with serious adverse events related to gene therapy (eg, MDS1, EVI1, and LMO2). All participants had expression of ALD protein in peripheral-blood leukocytes at the most recent follow-up (median follow-up time was 29.4 months). The median percentage of CD14+ cells that expressed ALD protein was 19% at 24 months.
Of the 17 patients, 15 were alive at 24 months and maintained an ALD neurologic function scale score of 0 or 1. In untreated patients, ALD neurologic function scale score usually increases significantly after the first symptoms appear. One patient in the study was progressing rapidly at the time of enrollment and subsequently died of disease progression. Another patient with disease progression on MRI withdrew from the study after the infusion and subsequently died from the complications of an allogeneic transplantation
An exploratory analysis indicated that participants with higher vector copy number in their peripheral blood tended to have better neurologic outcomes, while participants with lower vector copy number tended to have poor neurologic outcomes.
Lesion progression, as measured with the Loes score, had stabilized in 12 of the 17 patients (71%) at the time of the interim analysis. “Half the patients stabilized immediately in their Loes scores after treatment. The other half tended to progress within the first 12 months and then stabilize over time,” said Dr. Eichler.
Gadolinium enhancement resolved in 16 of 17 patients by six months. It reappeared in some patients after treatment, although it was much fainter and more poorly circumscribed, compared with the time of screening, said Dr. Eichler.
The investigators did not observe graft failure or graft-versus-host disease in the population. Most adverse events associated with the treatment were consistent with those associated with myeloablative chemotherapy and occurred during conditioning or during the first two weeks after the infusion. One serious adverse event that possibly was related to treatment was hemorrhagic cystitis associated with BK virus, which resolved with conservative measures.
The results have implications for leukodystrophies in general, according to Dr. Eichler. “Specific phenotypes, even within the same individual leukodystrophy, require different approaches” such as ex vivo or in vivo gene therapy, he added. Overall, the safety data are reassuring, and gene therapy appears to have encouraging efficacy in ALD, he concluded.
—Erik Greb
Suggested Reading
Eichler F, Duncan C, Musolino PL, et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med. 2017;377(17):1630-1638.
Engelen M. Optimizing treatment for cerebral adrenoleukodystrophy in the era of gene therapy. N Engl J Med. 2017;377(17):1682-1684.
KANSAS CITY, MO—Lentiviral gene therapy halts inflammation and demyelination in patients with cerebral adrenoleukodystrophy (ALD), according to research presented at the 46th Annual Meeting of the Child Neurology Society and published in the New England Journal of Medicine. The treatment appears to stabilize neurologic function and to have an acceptable safety profile.
ALD is an X-linked genetic disease caused by a defect in ABCD1, which encodes the peroxisomal ABC half-transporter ALD protein. The defect results in abnormal breakdown of very-long-chain fatty acids, which build up and affect tissues in the nervous system. The most common phenotype of ALD is adrenomyeloneuropathy, which involves axonal degeneration in the spinal cord. A more severe phenotype is childhood cerebral adenopathy, which manifests as an inflammatory demyelination in the white matter.
Allogeneic bone marrow transplantation is the only other treatment with proven efficacy in childhood cerebral ALD. It halts disease progression and improves survival in presymptomatic patients. Finding a donor with identical human leukocyte antigen often is difficult, however. In addition, the treatment entails risks of graft failure and graft-versus-host disease, and the rate of treatment-related mortality is greater than 10%. Gene therapy “may be an alternative to allogeneic bone marrow transplantation, particularly for patients who do not have a matched sibling donor who are at risk for graft failure and graft-versus-host [disease],” said Florian Eichler, MD, Director of the Leukodystrophy Service at Massachusetts General Hospital for Children in Boston.
Researchers Transduced Stem Cells Ex Vivo
In the first trial of its kind, Dr. Eichler and colleagues investigated the safety and efficacy of gene therapy with autologous hematopoietic stem cells for cerebral ALD. The enrollment criteria for the phase II–III, single-arm, open-label study were identical to those for conventional bone marrow transplantation, said Dr. Eichler. Eligible participants were age 17 or younger, had a defect in ABCD1, and had evidence of an active, inflammatory lesion on brain MRI. Children with sibling donors for bone marrow transplantation were excluded from the study.
The investigators obtained CD34+ cells from participants through apheresis and transduced them ex vivo with a lentiviral vector containing normal ABCD1. After undergoing conditioning with busulfan and cyclophosphamide, the patients received an infusion of the transduced CD34+ cells. Patients were assessed regularly for graft-versus-host disease, death, and major functional disabilities. Dr. Eichler and colleagues also monitored participants for changes in neurologic function and increases in MRI lesions. At the end of a two-year follow-up period, patients were offered enrollment in a 13-year long-term follow-up study. The main study’s primary end point was being alive and having no major functional disability at 24 months.
Lesion Progression Stabilized in Most Patients
Dr. Eichler and colleagues treated 17 patients with a mean age at enrollment of 6. Participants’ median Loes score at baseline was 2.0, which indicated a low level of neurologic progression. All patients had an ALD neurologic function scale score of 0 at baseline, indicating an absence of clinical signs of cerebral disease.
The vector copy number in peripheral blood ranged from 0.10 to 1.55 in the 14 patients assessed at month 24. At month 36, the number ranged from 0.36 to 1.83 in the three patients assessed. The number had generally stabilized by two months after infusion. The investigators did not observe preferential integration in or near genes that have previously been associated with serious adverse events related to gene therapy (eg, MDS1, EVI1, and LMO2). All participants had expression of ALD protein in peripheral-blood leukocytes at the most recent follow-up (median follow-up time was 29.4 months). The median percentage of CD14+ cells that expressed ALD protein was 19% at 24 months.
Of the 17 patients, 15 were alive at 24 months and maintained an ALD neurologic function scale score of 0 or 1. In untreated patients, ALD neurologic function scale score usually increases significantly after the first symptoms appear. One patient in the study was progressing rapidly at the time of enrollment and subsequently died of disease progression. Another patient with disease progression on MRI withdrew from the study after the infusion and subsequently died from the complications of an allogeneic transplantation
An exploratory analysis indicated that participants with higher vector copy number in their peripheral blood tended to have better neurologic outcomes, while participants with lower vector copy number tended to have poor neurologic outcomes.
Lesion progression, as measured with the Loes score, had stabilized in 12 of the 17 patients (71%) at the time of the interim analysis. “Half the patients stabilized immediately in their Loes scores after treatment. The other half tended to progress within the first 12 months and then stabilize over time,” said Dr. Eichler.
Gadolinium enhancement resolved in 16 of 17 patients by six months. It reappeared in some patients after treatment, although it was much fainter and more poorly circumscribed, compared with the time of screening, said Dr. Eichler.
The investigators did not observe graft failure or graft-versus-host disease in the population. Most adverse events associated with the treatment were consistent with those associated with myeloablative chemotherapy and occurred during conditioning or during the first two weeks after the infusion. One serious adverse event that possibly was related to treatment was hemorrhagic cystitis associated with BK virus, which resolved with conservative measures.
The results have implications for leukodystrophies in general, according to Dr. Eichler. “Specific phenotypes, even within the same individual leukodystrophy, require different approaches” such as ex vivo or in vivo gene therapy, he added. Overall, the safety data are reassuring, and gene therapy appears to have encouraging efficacy in ALD, he concluded.
—Erik Greb
Suggested Reading
Eichler F, Duncan C, Musolino PL, et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med. 2017;377(17):1630-1638.
Engelen M. Optimizing treatment for cerebral adrenoleukodystrophy in the era of gene therapy. N Engl J Med. 2017;377(17):1682-1684.
CLL drug combinations induce MRD negativity
Atlanta – The potential of two- and three-drug combinations to induce deep, minimal residual disease (MRD)–negative responses in patients with chronic lymphocytic leukemia (CLL) was demonstrated in multiple studies presented at the annual meeting of the American Society of Hematology.
Previous studies have suggested that, in contrast to single-agent kinase inhibitor treatment, combinations of treatments, namely kinase or B-cell lyphoma 2 inhibitors combined with anti-CD20 antibodies, can induce MRD negativity at a high rate. For example, in a report on the CLL14 trial, 11 of 12 previously untreated patients were MRD negative after treatment with the BCL-2 inhibitor venetoclax and anti-CD20 antibody obinutuzumab (Blood. 2017;129:2702-5. doi:10.1182/blood-2017-01-761973).
This new batch of studies presented at ASH 2017 provided additional evidence for the venetoclax and obinutuzumab combination, as well as other combinations that appear to provide favorable rates of MRD-negative CLL, including obinutuzumab and venetoclax plus ibrutinib and, notably, a combination that included venetoclax and ibrutinib but no anti-CD20 antibody.
A phase II trial presented by Nitin Jain, MD, of MD Anderson Cancer Center, Houston, looked at the combination of venetoclax and ibrutinib for patients with previously untreated high-risk CLL or relapsed/refractory CLL. The regimen under evaluation starts with daily ibrutinib monotherapy for 3 months before venetoclax is added.
For the first line cohort of 36 patients, the bone marrow MRD-negativity proportion was 0% after 3 months of ibrutinib and 21% after 3 months of ibrutinib plus venetoclax, then 45%, 80%, and 100%, respectively, after 6, 9, and 12 months of combination therapy, Dr. Jain reported. In the relapsed/refractory cohort, MRD negativity was 8% after 3 months of combination therapy and 40% after 12 months. “Responses continue to improve with time, with many patients achieving bone marrow MRD-negative remission,” Dr. Jain said.
The study showed “very impressive MRD rates,” commented Ian W. Flinn, MD, of Sarah Cannon Research Institute, Nashville, Tenn. “I think it’s still up for further discussion and research to find which is the best combination.”
Dr. Flinn presented results from a phase Ib GP28331 study of venetoclax and obinutuzumab in patients with previously untreated CLL in which MRD was assessed in peripheral blood and bone marrow.
All 32 patients achieved peripheral blood negativity at some point on study, Dr. Flinn said, and 75% achieved bone marrow negativity. The complete response rate was 72% (23/32), but notably, high rates of undetectable bone marrow MRD were seen irrespective of response status, Dr. Flinn said.
All patients had at least one adverse event, with grade 3-4 neutropenia, febrile neutropenia, and thrombocytopenia reported most commonly.
Preliminary progression-free survival data (PFS) suggested “durable clinical outcomes” for the combination, with an estimated 18-month PFS of 90.5%, he said.
A third study assessed the combination of obinutuzumab, ibrutinib, and venetoclax, finding that the combination induced MRD negativity in 14 of 24 (58%) treatment-naive CLL patients, according to Kerry A. Rogers, MD, of Ohio State University, Columbus.
The combination had a 96% response rate and is “extremely effective at eliminating detectable CLL,” Dr. Rogers noted.
Most adverse events were hematologic, and high-grade adverse events were rare, she said.
Results for the study’s primary endpoint, rate of MRD-negative complete remission, are expected by May 2018, Dr. Rogers said, adding that further follow-up will be needed to determine PFS for the combination.
Session attendees asked presenters whether they felt there was scientific justification for inclusion of the anti-CD20 antibody rituximab in future trials designed in part to assess MRD negativity.
“I think that obinutuzumab has greater efficacy in patients with CLL, compared to rituximab, and so our hypothesis is that it is a superior antibody to combine with venetoclax,” Dr. Flinn said.
AbbVie provided funding for the study on venetoclax and ibrutinib. Dr. Jain reported disclosures from venetoclax makers AbbVie and Genentech, ibrutinib makers Janssen and Pharmacyclics, and others.
Genentech and AbbVie provided support for the study on venetoclax and obinutuzumab. Dr. Flinn reported disclosures related to both companies and others.
Dr. Rogers reported no conflicts related to the study on obinutuzumab, ibrutinib, and venetoclax. One of her associates reported disclosures related to ibrutinib makers Novartis and Pharmacylics, among others.
SOURCE: Jain N et al. ASH 2017 Abstract 429; Flinn I et al. ASH 2017 Abstract 430; Rogers K et al. ASH 2017 Abstract 431.
Atlanta – The potential of two- and three-drug combinations to induce deep, minimal residual disease (MRD)–negative responses in patients with chronic lymphocytic leukemia (CLL) was demonstrated in multiple studies presented at the annual meeting of the American Society of Hematology.
Previous studies have suggested that, in contrast to single-agent kinase inhibitor treatment, combinations of treatments, namely kinase or B-cell lyphoma 2 inhibitors combined with anti-CD20 antibodies, can induce MRD negativity at a high rate. For example, in a report on the CLL14 trial, 11 of 12 previously untreated patients were MRD negative after treatment with the BCL-2 inhibitor venetoclax and anti-CD20 antibody obinutuzumab (Blood. 2017;129:2702-5. doi:10.1182/blood-2017-01-761973).
This new batch of studies presented at ASH 2017 provided additional evidence for the venetoclax and obinutuzumab combination, as well as other combinations that appear to provide favorable rates of MRD-negative CLL, including obinutuzumab and venetoclax plus ibrutinib and, notably, a combination that included venetoclax and ibrutinib but no anti-CD20 antibody.
A phase II trial presented by Nitin Jain, MD, of MD Anderson Cancer Center, Houston, looked at the combination of venetoclax and ibrutinib for patients with previously untreated high-risk CLL or relapsed/refractory CLL. The regimen under evaluation starts with daily ibrutinib monotherapy for 3 months before venetoclax is added.
For the first line cohort of 36 patients, the bone marrow MRD-negativity proportion was 0% after 3 months of ibrutinib and 21% after 3 months of ibrutinib plus venetoclax, then 45%, 80%, and 100%, respectively, after 6, 9, and 12 months of combination therapy, Dr. Jain reported. In the relapsed/refractory cohort, MRD negativity was 8% after 3 months of combination therapy and 40% after 12 months. “Responses continue to improve with time, with many patients achieving bone marrow MRD-negative remission,” Dr. Jain said.
The study showed “very impressive MRD rates,” commented Ian W. Flinn, MD, of Sarah Cannon Research Institute, Nashville, Tenn. “I think it’s still up for further discussion and research to find which is the best combination.”
Dr. Flinn presented results from a phase Ib GP28331 study of venetoclax and obinutuzumab in patients with previously untreated CLL in which MRD was assessed in peripheral blood and bone marrow.
All 32 patients achieved peripheral blood negativity at some point on study, Dr. Flinn said, and 75% achieved bone marrow negativity. The complete response rate was 72% (23/32), but notably, high rates of undetectable bone marrow MRD were seen irrespective of response status, Dr. Flinn said.
All patients had at least one adverse event, with grade 3-4 neutropenia, febrile neutropenia, and thrombocytopenia reported most commonly.
Preliminary progression-free survival data (PFS) suggested “durable clinical outcomes” for the combination, with an estimated 18-month PFS of 90.5%, he said.
A third study assessed the combination of obinutuzumab, ibrutinib, and venetoclax, finding that the combination induced MRD negativity in 14 of 24 (58%) treatment-naive CLL patients, according to Kerry A. Rogers, MD, of Ohio State University, Columbus.
The combination had a 96% response rate and is “extremely effective at eliminating detectable CLL,” Dr. Rogers noted.
Most adverse events were hematologic, and high-grade adverse events were rare, she said.
Results for the study’s primary endpoint, rate of MRD-negative complete remission, are expected by May 2018, Dr. Rogers said, adding that further follow-up will be needed to determine PFS for the combination.
Session attendees asked presenters whether they felt there was scientific justification for inclusion of the anti-CD20 antibody rituximab in future trials designed in part to assess MRD negativity.
“I think that obinutuzumab has greater efficacy in patients with CLL, compared to rituximab, and so our hypothesis is that it is a superior antibody to combine with venetoclax,” Dr. Flinn said.
AbbVie provided funding for the study on venetoclax and ibrutinib. Dr. Jain reported disclosures from venetoclax makers AbbVie and Genentech, ibrutinib makers Janssen and Pharmacyclics, and others.
Genentech and AbbVie provided support for the study on venetoclax and obinutuzumab. Dr. Flinn reported disclosures related to both companies and others.
Dr. Rogers reported no conflicts related to the study on obinutuzumab, ibrutinib, and venetoclax. One of her associates reported disclosures related to ibrutinib makers Novartis and Pharmacylics, among others.
SOURCE: Jain N et al. ASH 2017 Abstract 429; Flinn I et al. ASH 2017 Abstract 430; Rogers K et al. ASH 2017 Abstract 431.
Atlanta – The potential of two- and three-drug combinations to induce deep, minimal residual disease (MRD)–negative responses in patients with chronic lymphocytic leukemia (CLL) was demonstrated in multiple studies presented at the annual meeting of the American Society of Hematology.
Previous studies have suggested that, in contrast to single-agent kinase inhibitor treatment, combinations of treatments, namely kinase or B-cell lyphoma 2 inhibitors combined with anti-CD20 antibodies, can induce MRD negativity at a high rate. For example, in a report on the CLL14 trial, 11 of 12 previously untreated patients were MRD negative after treatment with the BCL-2 inhibitor venetoclax and anti-CD20 antibody obinutuzumab (Blood. 2017;129:2702-5. doi:10.1182/blood-2017-01-761973).
This new batch of studies presented at ASH 2017 provided additional evidence for the venetoclax and obinutuzumab combination, as well as other combinations that appear to provide favorable rates of MRD-negative CLL, including obinutuzumab and venetoclax plus ibrutinib and, notably, a combination that included venetoclax and ibrutinib but no anti-CD20 antibody.
A phase II trial presented by Nitin Jain, MD, of MD Anderson Cancer Center, Houston, looked at the combination of venetoclax and ibrutinib for patients with previously untreated high-risk CLL or relapsed/refractory CLL. The regimen under evaluation starts with daily ibrutinib monotherapy for 3 months before venetoclax is added.
For the first line cohort of 36 patients, the bone marrow MRD-negativity proportion was 0% after 3 months of ibrutinib and 21% after 3 months of ibrutinib plus venetoclax, then 45%, 80%, and 100%, respectively, after 6, 9, and 12 months of combination therapy, Dr. Jain reported. In the relapsed/refractory cohort, MRD negativity was 8% after 3 months of combination therapy and 40% after 12 months. “Responses continue to improve with time, with many patients achieving bone marrow MRD-negative remission,” Dr. Jain said.
The study showed “very impressive MRD rates,” commented Ian W. Flinn, MD, of Sarah Cannon Research Institute, Nashville, Tenn. “I think it’s still up for further discussion and research to find which is the best combination.”
Dr. Flinn presented results from a phase Ib GP28331 study of venetoclax and obinutuzumab in patients with previously untreated CLL in which MRD was assessed in peripheral blood and bone marrow.
All 32 patients achieved peripheral blood negativity at some point on study, Dr. Flinn said, and 75% achieved bone marrow negativity. The complete response rate was 72% (23/32), but notably, high rates of undetectable bone marrow MRD were seen irrespective of response status, Dr. Flinn said.
All patients had at least one adverse event, with grade 3-4 neutropenia, febrile neutropenia, and thrombocytopenia reported most commonly.
Preliminary progression-free survival data (PFS) suggested “durable clinical outcomes” for the combination, with an estimated 18-month PFS of 90.5%, he said.
A third study assessed the combination of obinutuzumab, ibrutinib, and venetoclax, finding that the combination induced MRD negativity in 14 of 24 (58%) treatment-naive CLL patients, according to Kerry A. Rogers, MD, of Ohio State University, Columbus.
The combination had a 96% response rate and is “extremely effective at eliminating detectable CLL,” Dr. Rogers noted.
Most adverse events were hematologic, and high-grade adverse events were rare, she said.
Results for the study’s primary endpoint, rate of MRD-negative complete remission, are expected by May 2018, Dr. Rogers said, adding that further follow-up will be needed to determine PFS for the combination.
Session attendees asked presenters whether they felt there was scientific justification for inclusion of the anti-CD20 antibody rituximab in future trials designed in part to assess MRD negativity.
“I think that obinutuzumab has greater efficacy in patients with CLL, compared to rituximab, and so our hypothesis is that it is a superior antibody to combine with venetoclax,” Dr. Flinn said.
AbbVie provided funding for the study on venetoclax and ibrutinib. Dr. Jain reported disclosures from venetoclax makers AbbVie and Genentech, ibrutinib makers Janssen and Pharmacyclics, and others.
Genentech and AbbVie provided support for the study on venetoclax and obinutuzumab. Dr. Flinn reported disclosures related to both companies and others.
Dr. Rogers reported no conflicts related to the study on obinutuzumab, ibrutinib, and venetoclax. One of her associates reported disclosures related to ibrutinib makers Novartis and Pharmacylics, among others.
SOURCE: Jain N et al. ASH 2017 Abstract 429; Flinn I et al. ASH 2017 Abstract 430; Rogers K et al. ASH 2017 Abstract 431.
REPORTING FROM ASH 2017