User login
Gene therapy normalized or near-normalized factor VIII in hemophilia A
ATLANTA – A single infusion of valoctocogene roxaparvovec normalized or nearly normalized factor VIII levels in 11 of 13 adults with severe hemophilia A, eliminated spontaneous bleeds and the need for factor VIII infusions, showed durable effects for up to 72 weeks of follow-up, K. John Pasi, MD, said at the annual meeting of the American Society of Hematology.

Nicknamed valrox and designated as a breakthrough therapy by the Food and Drug Administration in October 2017, valoctocogene roxaparvovec uses an adenoviral vector to deliver a functional copy of the factor VIII gene to patients with hemophilia A, said Dr. Pasi of Barts and The London School of Medicine and Dentistry.
Gene therapy has long been the “holy grail” for managing hemophilia because it is a single-gene disorder with a clear relationship between clotting factor level and bleeding severity, Dr. Pasi said. In a mouse model of hemophilia A, valrox restored factor VIII plasma concentrations to levels thought to be adequate to support normal clotting in humans.
Accordingly, the phase 2/3 enrolled 13 patients with severe hemophilia A whose baseline factor VIII levels were less than 1 IU/dL. Patients started at the lowest dose of gene therapy (4 x 1013 vector genomes/kg) and then received a higher dose ( 6 x 1013 VG/kg) if their factor VIII level remained under 5 IU/dL at week 3. Six patients received the lower dose and seven received the higher dose.
At 78 weeks, median factor VIII level in the higher-dose cohort was 90 IU/mL, as Dr. Pasi and his associates reported simultaneously in the New England Journal of Medicine (2017 Dec 9. doi: 10. 1056/NEJMoa1708483).
Before undergoing gene therapy, study participants had endured up to 41 breakthrough bleeds per year despite often receiving more than 150 infusions of factor VIII annually. Median annualized bleeding rates, which at baseline were 16.5 in the higher dose group and 8 in the lower dose group, zeroed out in both groups after factor VIII activity rose above 5%. Quality of life was evaluated in five patients, who reported substantial improvements across all domains.
All patients began producing factor VIII several weeks after infusion. Median levels plateaued within normal range by 20 weeks in the higher-dose group. At the lower dose, median levels rose steadily to a median of 34 IU/dL by 20 weeks. Additionally, three recipients of the lower dose who were followed for 32 weeks achieved factor VIII levels within normal range (median 51 IU/dL). Levels of factor VIII remained within normal range for up to 78 weeks of posttreatment follow-up, Dr. Pasi said.
No patients developed inhibitors or signs of immune-related adverse effects, nor were adverse events qualitatively different between dose groups, Dr. Pasi said. The most common adverse effects were transient increases in alanine transaminase (ALT), which peaked between 44 IU/L and 141 IU/L and lasted anywhere from several days to 15 weeks. Patients whose ALT rose 1.5-fold above baseline received short-term corticosteroids with no adverse effects. All but one was tapered off. There were two serious adverse events – one elective knee surgery and one case of transient fever, headache, and myalgia at time of infusion.
So far, valrox appears to be long lasting, but “durability is a huge question for any gene therapy approach,” Dr. Pasi said. “The only way to answer it is to follow patients through.”
In hemophilia B, studies indicate that some patients continue expressing factor IX years after a single infusion of gene therapy (N Engl J Med. 2017 Dec 7;377:2215-27).
Two phase 3 trials will further evaluate safety and optimal dosing of valrox, Dr. Pasi said. The GENEr8-1 trial will use the 6 x 1013 VG/kg dose and the GENEr8-2 trial will use the 4 x 1013 VG/kg dose. Like the pilot study, these trials will exclude patients with inhibitors, but they may include patients with comorbidities such as liver disease, he said.
Valrox was previously known as BMN 270.
The study was sponsored by BioMarin. Dr. Pasi disclosed research funding, consultancy fees, and speaker and advisory relationships with BioMarin. He disclosed ties to many other companies that develop hemophilia therapies.
SOURCE: Pasi KJ et al. ASH 2017 Abstract 603
ATLANTA – A single infusion of valoctocogene roxaparvovec normalized or nearly normalized factor VIII levels in 11 of 13 adults with severe hemophilia A, eliminated spontaneous bleeds and the need for factor VIII infusions, showed durable effects for up to 72 weeks of follow-up, K. John Pasi, MD, said at the annual meeting of the American Society of Hematology.
Nicknamed valrox and designated as a breakthrough therapy by the Food and Drug Administration in October 2017, valoctocogene roxaparvovec uses an adenoviral vector to deliver a functional copy of the factor VIII gene to patients with hemophilia A, said Dr. Pasi of Barts and The London School of Medicine and Dentistry.
Gene therapy has long been the “holy grail” for managing hemophilia because it is a single-gene disorder with a clear relationship between clotting factor level and bleeding severity, Dr. Pasi said. In a mouse model of hemophilia A, valrox restored factor VIII plasma concentrations to levels thought to be adequate to support normal clotting in humans.
Accordingly, the phase 2/3 enrolled 13 patients with severe hemophilia A whose baseline factor VIII levels were less than 1 IU/dL. Patients started at the lowest dose of gene therapy (4 x 1013 vector genomes/kg) and then received a higher dose ( 6 x 1013 VG/kg) if their factor VIII level remained under 5 IU/dL at week 3. Six patients received the lower dose and seven received the higher dose.
At 78 weeks, median factor VIII level in the higher-dose cohort was 90 IU/mL, as Dr. Pasi and his associates reported simultaneously in the New England Journal of Medicine (2017 Dec 9. doi: 10. 1056/NEJMoa1708483).
Before undergoing gene therapy, study participants had endured up to 41 breakthrough bleeds per year despite often receiving more than 150 infusions of factor VIII annually. Median annualized bleeding rates, which at baseline were 16.5 in the higher dose group and 8 in the lower dose group, zeroed out in both groups after factor VIII activity rose above 5%. Quality of life was evaluated in five patients, who reported substantial improvements across all domains.
All patients began producing factor VIII several weeks after infusion. Median levels plateaued within normal range by 20 weeks in the higher-dose group. At the lower dose, median levels rose steadily to a median of 34 IU/dL by 20 weeks. Additionally, three recipients of the lower dose who were followed for 32 weeks achieved factor VIII levels within normal range (median 51 IU/dL). Levels of factor VIII remained within normal range for up to 78 weeks of posttreatment follow-up, Dr. Pasi said.
No patients developed inhibitors or signs of immune-related adverse effects, nor were adverse events qualitatively different between dose groups, Dr. Pasi said. The most common adverse effects were transient increases in alanine transaminase (ALT), which peaked between 44 IU/L and 141 IU/L and lasted anywhere from several days to 15 weeks. Patients whose ALT rose 1.5-fold above baseline received short-term corticosteroids with no adverse effects. All but one was tapered off. There were two serious adverse events – one elective knee surgery and one case of transient fever, headache, and myalgia at time of infusion.
So far, valrox appears to be long lasting, but “durability is a huge question for any gene therapy approach,” Dr. Pasi said. “The only way to answer it is to follow patients through.”
In hemophilia B, studies indicate that some patients continue expressing factor IX years after a single infusion of gene therapy (N Engl J Med. 2017 Dec 7;377:2215-27).
Two phase 3 trials will further evaluate safety and optimal dosing of valrox, Dr. Pasi said. The GENEr8-1 trial will use the 6 x 1013 VG/kg dose and the GENEr8-2 trial will use the 4 x 1013 VG/kg dose. Like the pilot study, these trials will exclude patients with inhibitors, but they may include patients with comorbidities such as liver disease, he said.
Valrox was previously known as BMN 270.
The study was sponsored by BioMarin. Dr. Pasi disclosed research funding, consultancy fees, and speaker and advisory relationships with BioMarin. He disclosed ties to many other companies that develop hemophilia therapies.
SOURCE: Pasi KJ et al. ASH 2017 Abstract 603
ATLANTA – A single infusion of valoctocogene roxaparvovec normalized or nearly normalized factor VIII levels in 11 of 13 adults with severe hemophilia A, eliminated spontaneous bleeds and the need for factor VIII infusions, showed durable effects for up to 72 weeks of follow-up, K. John Pasi, MD, said at the annual meeting of the American Society of Hematology.
Nicknamed valrox and designated as a breakthrough therapy by the Food and Drug Administration in October 2017, valoctocogene roxaparvovec uses an adenoviral vector to deliver a functional copy of the factor VIII gene to patients with hemophilia A, said Dr. Pasi of Barts and The London School of Medicine and Dentistry.
Gene therapy has long been the “holy grail” for managing hemophilia because it is a single-gene disorder with a clear relationship between clotting factor level and bleeding severity, Dr. Pasi said. In a mouse model of hemophilia A, valrox restored factor VIII plasma concentrations to levels thought to be adequate to support normal clotting in humans.
Accordingly, the phase 2/3 enrolled 13 patients with severe hemophilia A whose baseline factor VIII levels were less than 1 IU/dL. Patients started at the lowest dose of gene therapy (4 x 1013 vector genomes/kg) and then received a higher dose ( 6 x 1013 VG/kg) if their factor VIII level remained under 5 IU/dL at week 3. Six patients received the lower dose and seven received the higher dose.
At 78 weeks, median factor VIII level in the higher-dose cohort was 90 IU/mL, as Dr. Pasi and his associates reported simultaneously in the New England Journal of Medicine (2017 Dec 9. doi: 10. 1056/NEJMoa1708483).
Before undergoing gene therapy, study participants had endured up to 41 breakthrough bleeds per year despite often receiving more than 150 infusions of factor VIII annually. Median annualized bleeding rates, which at baseline were 16.5 in the higher dose group and 8 in the lower dose group, zeroed out in both groups after factor VIII activity rose above 5%. Quality of life was evaluated in five patients, who reported substantial improvements across all domains.
All patients began producing factor VIII several weeks after infusion. Median levels plateaued within normal range by 20 weeks in the higher-dose group. At the lower dose, median levels rose steadily to a median of 34 IU/dL by 20 weeks. Additionally, three recipients of the lower dose who were followed for 32 weeks achieved factor VIII levels within normal range (median 51 IU/dL). Levels of factor VIII remained within normal range for up to 78 weeks of posttreatment follow-up, Dr. Pasi said.
No patients developed inhibitors or signs of immune-related adverse effects, nor were adverse events qualitatively different between dose groups, Dr. Pasi said. The most common adverse effects were transient increases in alanine transaminase (ALT), which peaked between 44 IU/L and 141 IU/L and lasted anywhere from several days to 15 weeks. Patients whose ALT rose 1.5-fold above baseline received short-term corticosteroids with no adverse effects. All but one was tapered off. There were two serious adverse events – one elective knee surgery and one case of transient fever, headache, and myalgia at time of infusion.
So far, valrox appears to be long lasting, but “durability is a huge question for any gene therapy approach,” Dr. Pasi said. “The only way to answer it is to follow patients through.”
In hemophilia B, studies indicate that some patients continue expressing factor IX years after a single infusion of gene therapy (N Engl J Med. 2017 Dec 7;377:2215-27).
Two phase 3 trials will further evaluate safety and optimal dosing of valrox, Dr. Pasi said. The GENEr8-1 trial will use the 6 x 1013 VG/kg dose and the GENEr8-2 trial will use the 4 x 1013 VG/kg dose. Like the pilot study, these trials will exclude patients with inhibitors, but they may include patients with comorbidities such as liver disease, he said.
Valrox was previously known as BMN 270.
The study was sponsored by BioMarin. Dr. Pasi disclosed research funding, consultancy fees, and speaker and advisory relationships with BioMarin. He disclosed ties to many other companies that develop hemophilia therapies.
SOURCE: Pasi KJ et al. ASH 2017 Abstract 603
REPORTING FROM ASH 2017
Key clinical point: The investigational gene therapy BMN 270 (valoctocogene roxaparvovec; valrox) eliminated spontaneous bleeds and the need for factor VIII infusions in patients with hemophilia A, and the effects persisted for up to 78 weeks.
Major finding: Median FVIII level was 90 IU/dL at 78 weeks in the higher (6 x 1013 VG/kg) dose cohort.
Data source: A phase I/II, first-in-human study of adenoassociated viral factor VIII gene transfer in 15 patients with severe hemophilia A without inhibitors.
Disclosures: The study was sponsored by BioMarin. Dr. Pasi disclosed research funding, consultancy fees, and speaker and advisory relationships with BioMarin. He disclosed ties to many other companies that develop hemophilia therapies.
Source: Pasi KJ et al. ASH 2017 Abstract 603
Hallmark tumor metabolism becomes a validated therapeutic target
Altered cell metabolism has long been recognized as a distinctive feature of malignant cells but, until recently, research efforts had focused on a single aspect. It has become increasingly evident that many metabolic pathways are altered in cancer cells. Improved understanding has yielded the first regulatory approval in this new class of drugs. Here, we discuss the latest developments in the therapeutic targeting of the cancer metabolism hallmark.
A cancer cell’s sweet tooth
The metabolism of cancer cells differs from that of normal cells, an observation that has spawned a dedicated field of research and new targeted drug development. The German physiologist Otto Warburg is credited as the father of the field with his observations about the way in which cancer cells derive energy from glucose.1
In normal cells, glucose is converted into pyruvate in the cytoplasm, which is then, most often, fed to the mitochondria that use oxidative phosphorylation to produce energy in the form of adenosine triphosphate (ATP). Cancer cells seem instead to favor using the pyruvate to produce lactate through glycolysis (Figure 1).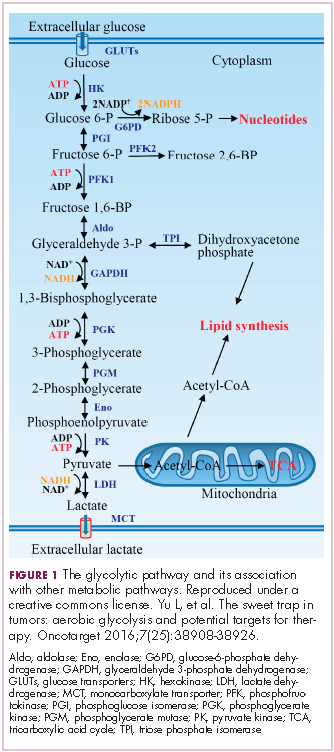
Glycolysis is usually reserved for conditions of poor oxygen availability, but although the tumor microenvironment is often hypoxic, cancer cells have been shown to use glycolysis even when oxygen is plentiful. As a result, the phenomenon is known as aerobic glycolysis, although it is most often referred to as the Warburg effect.2
Glycolysis is much less efficient than oxidative phosphorylation at producing energy, yielding only 2 ATP. In order to meet their energy demands in this way, cancer cells ramp up their glucose intake, an effect that has been exploited for the detection of cancer with positron-emission tomography.
Warburg postulated that this metabolic shift was a result of mitochondrial damage and defective oxidative phosphorylation, even going so far as to suggest that cancer was a mitochondrial disease. It has subsequently been shown that the mitochondria are mostly intact in cancer cells and that oxidative phosphorylation can still occur.3
The Warburg effect has been the subject of significant investigative efforts as researchers have attempted to better understand how this phenomenon comes about. Studies have shown that it is driven in large part by the transcription factors hypoxia inducible factor 1 alpha (HIF-1α) and c-Myc. In addition, numerous other signaling pathways, including the phosphatidylinositol 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) pathway, and the activation of oncogenes and inactivation of tumor suppressors, are thought to play a central role.
HIF-1α is an oxygen-sensing transcription factor that coordinates cellular responses to reduced oxygen levels by binding to specific regions, known as hypoxia response elements, on target genes in the nucleus and regulating their subsequent expression. Oxygen levels and metabolism are tightly linked, and HIF-1α sits at the intersection of the 2 since many of its target genes are involved in metabolic pathways, including many glycolytic enzymes, but it also directly inhibits oxidative phosphorylation by suppressing key enzymes in this metabolic pathway.
The expression of HIF-1α and numerous glycolytic enzymes, including lactate dehydrogenase (LDH), phosphofructokinase (PFK), hexokinase II (HKII), and pyruvate dehydrogenase kinase (PDK) is increased in many tumor types. Other molecules that are associated with glucose uptake and metabolism are also dysregulated, such as the GLUT-1 glucose transporter.2,4-6
Targeting glycolysis and glucose uptake
According to one study, glucose transporters and glycolytic enzymes are overexpressed in 24 different types of cancer, representing more than 70% of all cancer cases.7 This enables cancer cells to respond metabolically as though they are experiencing hypoxia, even when oxygen is plentiful and, indeed, when hypoxia is a concern, to mount a faster response. It also provides a tempting avenue for anticancer drug design by exploiting the dependency of cancer cells on glycolysis to survive and thrive.
Inhibitors of HKII, LDH, PFK, PDK, and GLUT-1 have been and continue to be developed. For example, 2-deoxy-D-glucose is a glucose molecule in which the 2-hydroxyl group has been replaced by hydrogen, preventing further glycolysis; it acts as a competitive inhibitor of HKII. Dichloroacetate (DCA) activates the pyruvate dehydrogenase complex and inhibits the actions of the PDKs. Although development of DCA itself was unsuccessful, DCA derivatives continue to be pursued. WZB117 and STF-31 are novel small-molecule inhibitors of GLUT-1-mediated glucose transport. To date, where inhibitors of glycolysis have progressed into clinical trials, they have not proved successful, often limited by off-target effects and low potency.8-11
A variety of cell signaling pathways are implicated in metabolism by tightly regulating the ability of cells to gain access to and use nutrients. Through aberrations in these pathways, cancer cells can essentially go rogue, ignoring regulatory signals and taking up nutrients in an autonomous manner. One of the most frequently altered signaling pathways in human cancer, the PI3K-Akt-mTOR pathway, is also an important regulator of metabolism, coordinating the uptake of multiple nutrients, including glucose.
Akt in particular is thought to have a critical role in glucose metabolism and increased Akt pathway signaling has been shown to correlate with increased rates of glycolysis in cancer cells. Thus, Akt inhibitors could double as glycolytic or glucose transport inhibitors.12,13
A number of Akt inhibitors are being evaluated in clinical trials (Table) and results from the phase 2 LOTUS trial of ipatasertib (GDC-0068) were recently published.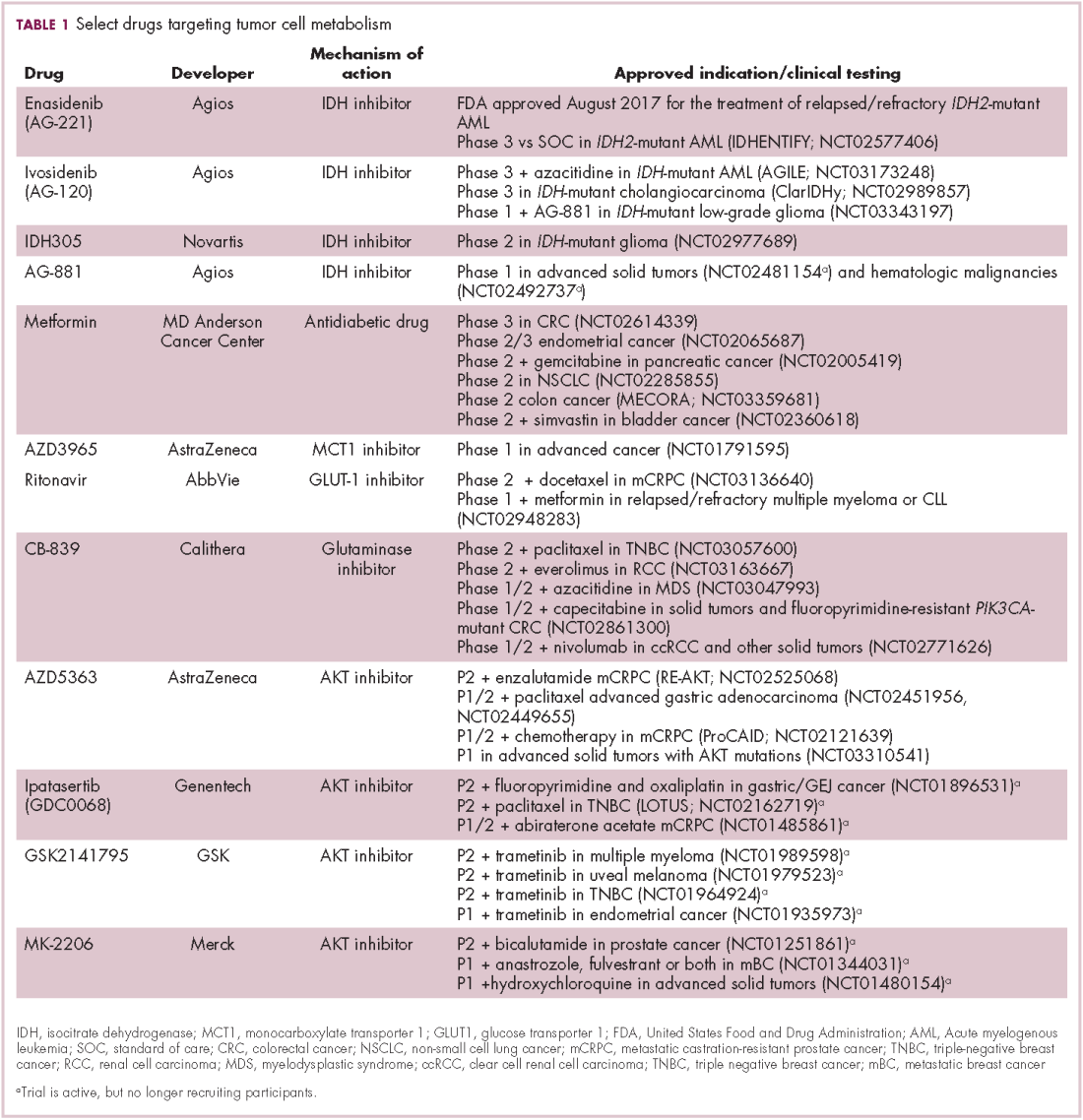
Among 124 patients randomly assigned to paclitaxel in combination with either ipatasertib or placebo, there was a modest improvement in progression-free survival (PFS) in the ipatasertib arm in patients with triple-negative breast cancer (TNBC; 6 months vs 4.2 months, respectively; hazard ratio [HR], 0.60; P = .037). The effect was more pronounced, though not statistically significant, in patients with phosphatase and tensin homolog (PTEN)-low tumors (6.2 months vs 3.7 months; HR, 0.59; P = .18). The most common grade 3 and higher adverse events (AEs) were diarrhea, reduced neutrophil count, and neutropenia.14
The Warburg paradox
Although the molecular mechanisms underlying the Warburg effect have been revealed to some extent, why cancer cells would choose to use such an energy-inefficient process when they have such high energy demands, remains something of a paradox. It’s still not entirely clear, but several explanations that are not necessarily mutually exclusive have been proposed and relate to the inherent benefits of glycolysis and might explain why cancer cells favor this pathway despite its poor energy yield. First, ATP is produced much more rapidly through glycolysis than oxidative phosphorylation, up to 100 times faster. Thus, using glycolysis is a trade-off, between making less energy and making it more quickly.
Second, cancer cells require more than just ATP to meet their metabolic demands. They need amino acids for protein synthesis; nucleotides for DNA replication; lipids for cell membrane synthesis; nicotinamide adenine dinucleotide phosphate (NADPH), which helps the cancer cell deal with oxidative stress; and various other metabolites. Glycolysis branches off into other metabolic pathways that generate many of these metabolites. Among these branched pathways is the pentose phosphate pathway (PPP), which is required for the generation of ribonucleotides and is a major source for NADPH. Cancer cells have been shown to upregulate the flux of glucose into the PPP to meet their anabolic demands and counter oxidative stress.
Third, the lactic acid produced through glycolysis is actively exported from tumor cells by monocarboxylate transporters (MCTs). This creates a highly acidic tumor microenvironment, which can promote several cancer-related processes and also plays a role in tumor-induced immunosuppression, by inhibiting the activity of tumor-infiltrating T cells, reducing dendritic cell maturation, and promoting the transformation of macrophages to a protumorigenic form.2,4,6
Beyond the Warburg effect
Although the focus has been on glucose metabolism and glycolysis, it has been increasingly recognized that many different metabolic pathways are altered. Fundamental changes to the metabolism of all 4 major classes of macromolecules – carbohydrates, lipids, proteins, and nucleic acids – have been observed, encompassing all aspects of cellular metabolism and enabling cancer cells to meet their complete metabolic requirements. There is also evidence that cancer cells are able to switch between different metabolic pathways depending on the availability of oxygen, their energetic needs, environmental stresses, and many other factors. Certainly, there is significant heterogeneity in the metabolic changes that occur in tumors, which vary from tumor to tumor and even within the same tumor and across the lifespan of a tumor as it progresses from an early stage to more advanced or metastatic disease.
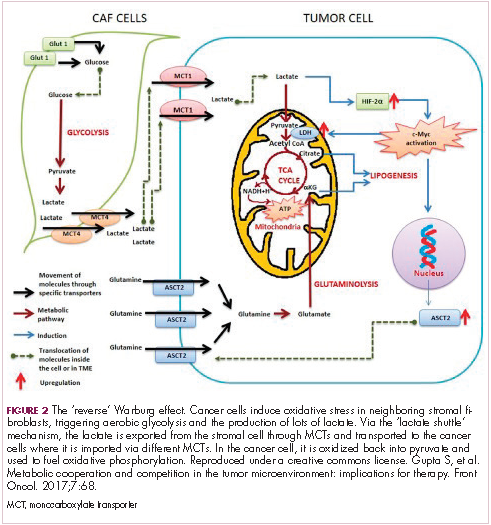
The notion of the Warburg effect as a universal phenomenon in cancer cells is now being widely disregarded. Many tumors continue to use oxidative phosphorylation, particularly slower growing tumors, to meet their energy needs. More recently a “reverse” Warburg effect was described, whereby cancer cells are thought to influence the metabolism of the surrounding stromal fibroblasts and essentially outsource aerobic glycolysis to these cells, while performing energy-efficient oxidative phosphorylation themselves (Figure 2).5,15,16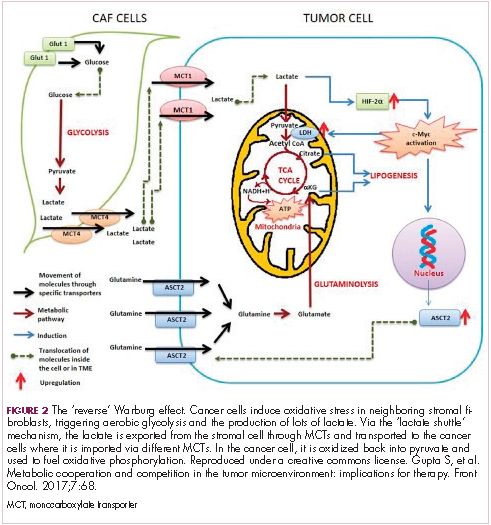
There is thought to be a “lactate shuttle” between the stromal and cancer cells. The stromal cells express high levels of efflux MCTs so that they can remove the subsequently high levels of lactate from the cytoplasm and avoid pickling themselves. The lactate is then shuttled to the cancer cells that have MCTs on their surface that are involved in lactate uptake. The cancer cells oxidize the lactate back into pyruvate, which can then be used in the tricarboxylic acid (TCA) cycle to feed oxidative phosphorylation for efficient ATP production. This hypothesis reflects a broader appreciation of the role of the microenvironment in contributing to cancer metabolism.17,18
An improved holistic understanding of cancer cell metabolism has led to the recognition of altered cancer metabolism as one of the hallmark abilities required for transformation of a normal cell into a cancerous one. It is categorized as “deregulation of bioenergetics” in the most up to date review of the cancer hallmarks.19 It has also begun to shape the therapeutic landscape as new drug targets have emerged.
IDH inhibitors first to market
A number of new metabolically-targeted treatment strategies are being developed. Most promising are small molecule inhibitors of the isocitrate dehydrogenase (IDH) enzymes. These enzymes play an essential role in the TCA cycle, catalyzing the conversion of isocitrate to alpha-ketoglutarate, generating carbon dioxide and NADPH. Recurrent mutations in the IDH1 and IDH2 genes have been observed in several different types of cancer, including glioma, acute myeloid leukemia (AML), and cholangiocarcinoma.
IDH mutations are known as neomorphic mutations because they confer a new function on the altered gene product. In this case, the mutant IDH enzyme converts alpha-ketoglutarate further into D-2-hydroxyglutarate (D-2HG). This molecule has a number of different effects that promote tumorigenesis, including fostering defective DNA repair (Figure 3).20,21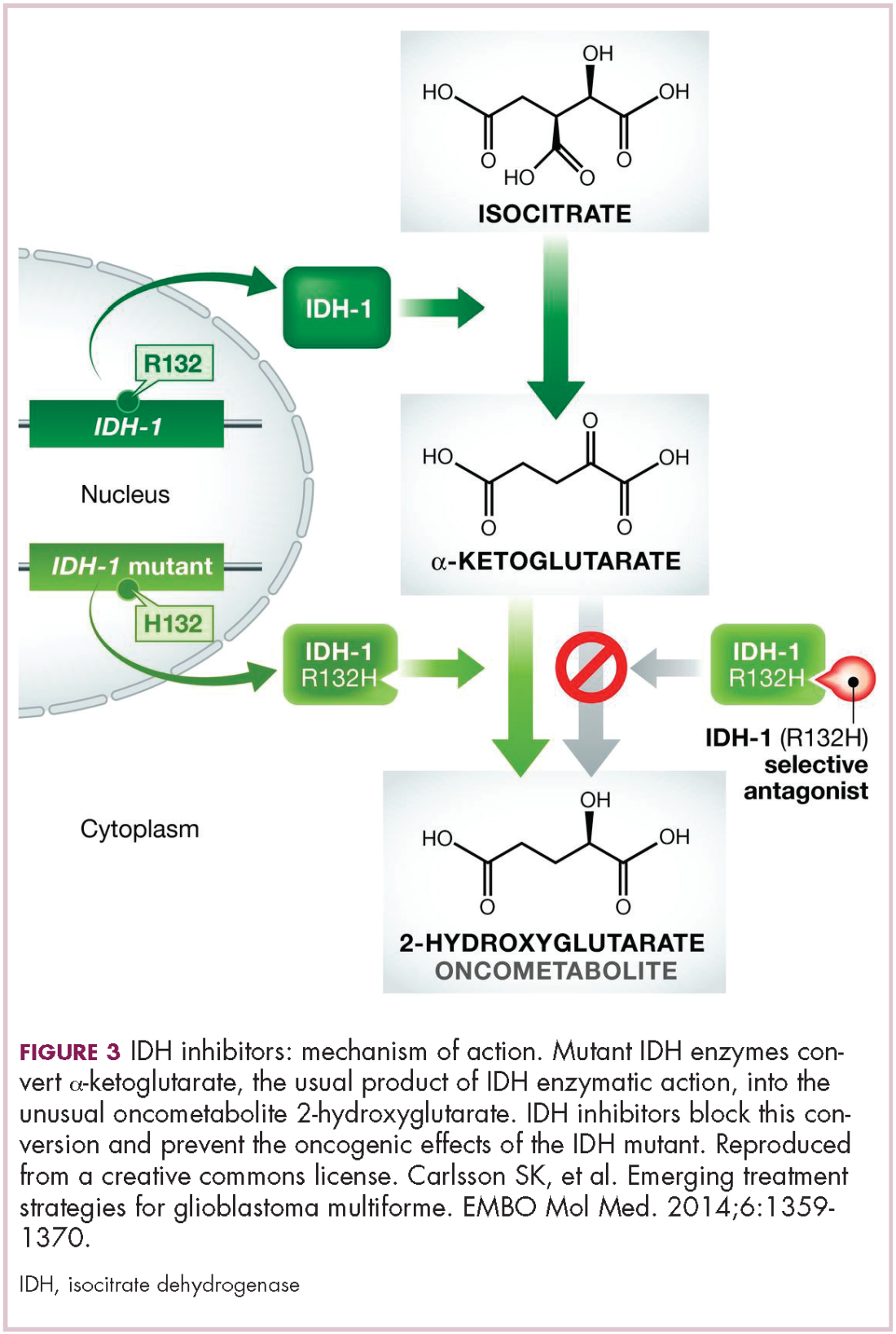
Intriguing research presented at the American Association of Cancer Research Annual Meeting revealed that IDH mutations may make cancer cells more vulnerable to poly (ADP-ribose) polymerase (PARP) inhibition, likely as a result of defects in homologous recombination pathways of DNA repair.22The pursuit of IDH as a potential therapeutic target has yielded the first regulatory approval for a metabolically targeted anticancer therapy. In August 2017, the United States Food and Drug Administration (FDA) approved enasidenib, an IDH2 inhibitor, for the treatment of relapsed or refractory AML with an IDH2 mutation. It was approved in combination with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect IDH2 mutations.
The approval was based on a single-arm trial in which responses occurred in almost a quarter of the 199 patients treated with 100 mg oral enasidenib daily. After a median follow-up of 6.6 months, 23% of the patients experienced a complete response or a complete response with partial hematologic recovery lasting a median of 8.2 months. The most common AEs were nausea, vomiting, diarrhea, elevated bilirubin levels, and reduced appetite.23
Several other IDH inhibitors are also showing encouraging efficacy. Ivosidenib is an IDH1 inhibitor and the results of a phase 1 study in patients with cholangiocarcinoma were recently presented at a leading conference. Escalating doses of ivosidenib (100 mg twice daily to 1,200 mg once daily) were administered to 73 patients (as of December 2016). The confirmed partial response (PR) rate was 6%, the rate of stable disease was 56%, and PFS at 6 months was 40%. There were no dose-limiting toxicities (DLTs) and treatment-emergent AEs included fatigue, nausea, vomiting, diarrhea, decreased appetite, dysgeusia, and QT prolongation.24
Another study of ivosidenib was presented at the 2017 annual meeting of the Society for Neuro-Oncology. In that study, patients with glioma received daily doses of ivosidenib ranging from 300 mg to 900 mg. Two patients had a minor response, 83% had stable disease, and the median PFS was 13 months. There were no DLTs and most AEs were mild to moderate and included, most commonly, headache, nausea, diarrhea, and vomiting.25
Pursuing alternative targets and repurposing drugs
Other metabolic targets that are being pursued include glutaminase, given the observation of significantly enhanced glutamine uptake in cancer cells. CB-839 is a glutaminase inhibitor that is currently being evaluated in phase 1 and 2 clinical trials. Updated clinical trial data from a phase 1 trial of CB-839 in combination with paclitaxel in patients with advanced/metastatic TNBC were presented at the San Antonio Breast Cancer Symposium last year.26
As of October 2017, 49 patients had been treated with 400 mg, 600 mg, or 800 mg CB-839 twice daily in combination with 80 mg/m2 intravenous paclitaxel weekly. Among the 44 patients evaluable for response, the rate of PR was 22% and of disease control, 59%. The one DLT was grade 3 neutropenia at the 400 mg dose. Overall AEs were mostly low grade and reversible.
In recent years, lactate has emerged as more than just a by-product of altered cancer cell metabolism. It is responsible, at least in part, for the highly acidic tumor microenvironment that fosters many of the other hallmarks of cancer. In addition, lactate promotes angiogenesis by upregulating HIF-1α in endothelial cells. Depriving tumor cells of the ability to export lactate is a potentially promising therapeutic strategy. An MCT-1 inhibitor, AZD3965, is being evaluated in early stage clinical trials.
Finally, several drugs that are renowned for their use in other disease settings are being repurposed for cancer therapy because of their potential effects on cancer cell metabolism. Ritonavir, an antiretroviral drug used in the treatment of HIV, is an inhibitor of GLUT-1 and is being evaluated in phase 1 and 2 clinical trials. Meanwhile, long-term studies of metformin, a drug that has revolutionized the treatment of diabetes, have revealed a reduction in the emergence of new cancers in diabetic patients treated who are treated with it, and the drug has been shown to improve breast cancer survival rates. Its precise anticancer effects are somewhat unclear, but it is thought to act in part by inhibiting oxidative phosphorylation. Numerous clinical trials of metformin in different types of cancer are ongoing.27,2
1. Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269-270.
2. Yu L, Chen X, Wang L, Chen S. The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget. 2016;7(25):38908-38926.
3. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309-314.
4. Chen XS, Li LY, Guan YD, Yang JM, Cheng Y. Anticancer strategies based on the metabolic profile of tumor cells: therapeutic targeting of the Warburg effect. Acta Pharmacol Sin. 2016;37(8):1013-1019.
5. Gupta S, Roy A, Dwarakanath BS. Metabolic cooperation and competition in the tumor microenvironment: implications for therapy. Front Oncol. 2017;7:68.
6. Marchiq I, Pouyssegur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J Mol Med (Berl). 2016;94(2):155-171.
7. Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84(6):1014-1020.
8. Yu L, Chen X, Sun X, Wang L, Chen S. The glycolytic switch in tumors: how many players are involved? J Cancer. 2017;8(17):3430-3440.
9. Zhang W, Zhang SL, Hu X, Tam KY. Targeting tumor metabolism for cancer treatment: is pyruvate dehydrogenase kinases (PDKs) a viable anticancer target? Int J Biol Sci. 2015;11(12):1390-1400.
10. Talekar M, Boreddy SR, Singh A, Amiji M. Tumor aerobic glycolysis: new insights into therapeutic strategies with targeted delivery. Expert Opin Biol Ther. 2014;14(8):1145-1159.
11. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152.
12. Lien EC, Lyssiotis CA, Cantley LC. Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer. In: Cramer T, Schmitt CA, eds. Metabolism in Cancer. Cham, Switzerland: Springer International Publishing; 2016:39-72.
13. Simons AL, Orcutt KP, Madsen JM, Scarbrough PM, Spitz DR. The role of Akt pathway signaling in glucose metabolism and metabolic oxidative stress. In: Spitz DR, Dornfeld KJ, Krishnan K, Gius D (eds). Oxidative stress in cancer biology and therapy. Humana Press (copyright holder, Springer Science+Business Media, LLC); 2012:21-46.
14. Kim S-B, Dent R, Im S-A, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18(10):1360-1372.
15. Fu Y, Liu S, Yin S, et al. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget. 2017;8(34):57813-57825.
16. Wilde L, Roche M, Domingo-Vidal M, et al. Metabolic coupling and the reverse Warburg effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol. 2017;44(3):198-203.
17. Brooks GA. Cell–cell and intracellular lactate shuttles. Journal Physiol. 2009;587(23):5591-5600.
18. Chiarugi P, Cirri P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016;380(1):272-280.
19. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674.
20. Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 2016;21(117):373-380.
21. Carlsson SK, Brothers SP, Wahlestedt C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol Med. 2014;6(11):1359-1370.
22. Lu Y, Kwintkiewicz J, Liu Y, et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. 2017;77(7):1709-1718.
23. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731.
24. Lowery MA, Abou-Alfa GK, Burris HA, et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: results from the cholangiocarcinoma dose escalation and expansion cohorts. J Clin Oncol. 2017;35(15_suppl):4015-4015.
25. Mellinghoff IK, Touat M, Maher E, et al. ACTR-46. AG-120, a first-in-class mutant IDH1 inhibitor in patients with recurrent or progressive IDH1 mutant glioma: updated results from the phase 1 non-enhancing glioma population. Neuro Oncol. 2017;19(suppl_6):vi10-vi11.
26. Kalinsky K, Harding J, DeMichele A, et al. Phase 1 study of CB-839, a first-in-class oral inhibitor of glutaminase, in combination with paclitaxel in patients with advanced triple negative breast cancer. Paper presented at San Antonio Breast Cancer Symposium; December 5-9, 2017; San Antonio, Texas.
27. Hatoum D, McGowan EM. Recent advances in the use of metformin: can treating diabetes prevent breast cancer? Biomed Res Int. 2015;2015:548436.
28. Leone A, Di Gennaro E, Bruzzese F, Avallone A, Budillon A. New perspective for an old antidiabetic drug: metformin as anticancer agent. Cancer Treat Res. 2014;159:355-376.
Altered cell metabolism has long been recognized as a distinctive feature of malignant cells but, until recently, research efforts had focused on a single aspect. It has become increasingly evident that many metabolic pathways are altered in cancer cells. Improved understanding has yielded the first regulatory approval in this new class of drugs. Here, we discuss the latest developments in the therapeutic targeting of the cancer metabolism hallmark.
A cancer cell’s sweet tooth
The metabolism of cancer cells differs from that of normal cells, an observation that has spawned a dedicated field of research and new targeted drug development. The German physiologist Otto Warburg is credited as the father of the field with his observations about the way in which cancer cells derive energy from glucose.1
In normal cells, glucose is converted into pyruvate in the cytoplasm, which is then, most often, fed to the mitochondria that use oxidative phosphorylation to produce energy in the form of adenosine triphosphate (ATP). Cancer cells seem instead to favor using the pyruvate to produce lactate through glycolysis (Figure 1).
Glycolysis is usually reserved for conditions of poor oxygen availability, but although the tumor microenvironment is often hypoxic, cancer cells have been shown to use glycolysis even when oxygen is plentiful. As a result, the phenomenon is known as aerobic glycolysis, although it is most often referred to as the Warburg effect.2
Glycolysis is much less efficient than oxidative phosphorylation at producing energy, yielding only 2 ATP. In order to meet their energy demands in this way, cancer cells ramp up their glucose intake, an effect that has been exploited for the detection of cancer with positron-emission tomography.
Warburg postulated that this metabolic shift was a result of mitochondrial damage and defective oxidative phosphorylation, even going so far as to suggest that cancer was a mitochondrial disease. It has subsequently been shown that the mitochondria are mostly intact in cancer cells and that oxidative phosphorylation can still occur.3
The Warburg effect has been the subject of significant investigative efforts as researchers have attempted to better understand how this phenomenon comes about. Studies have shown that it is driven in large part by the transcription factors hypoxia inducible factor 1 alpha (HIF-1α) and c-Myc. In addition, numerous other signaling pathways, including the phosphatidylinositol 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) pathway, and the activation of oncogenes and inactivation of tumor suppressors, are thought to play a central role.
HIF-1α is an oxygen-sensing transcription factor that coordinates cellular responses to reduced oxygen levels by binding to specific regions, known as hypoxia response elements, on target genes in the nucleus and regulating their subsequent expression. Oxygen levels and metabolism are tightly linked, and HIF-1α sits at the intersection of the 2 since many of its target genes are involved in metabolic pathways, including many glycolytic enzymes, but it also directly inhibits oxidative phosphorylation by suppressing key enzymes in this metabolic pathway.
The expression of HIF-1α and numerous glycolytic enzymes, including lactate dehydrogenase (LDH), phosphofructokinase (PFK), hexokinase II (HKII), and pyruvate dehydrogenase kinase (PDK) is increased in many tumor types. Other molecules that are associated with glucose uptake and metabolism are also dysregulated, such as the GLUT-1 glucose transporter.2,4-6
Targeting glycolysis and glucose uptake
According to one study, glucose transporters and glycolytic enzymes are overexpressed in 24 different types of cancer, representing more than 70% of all cancer cases.7 This enables cancer cells to respond metabolically as though they are experiencing hypoxia, even when oxygen is plentiful and, indeed, when hypoxia is a concern, to mount a faster response. It also provides a tempting avenue for anticancer drug design by exploiting the dependency of cancer cells on glycolysis to survive and thrive.
Inhibitors of HKII, LDH, PFK, PDK, and GLUT-1 have been and continue to be developed. For example, 2-deoxy-D-glucose is a glucose molecule in which the 2-hydroxyl group has been replaced by hydrogen, preventing further glycolysis; it acts as a competitive inhibitor of HKII. Dichloroacetate (DCA) activates the pyruvate dehydrogenase complex and inhibits the actions of the PDKs. Although development of DCA itself was unsuccessful, DCA derivatives continue to be pursued. WZB117 and STF-31 are novel small-molecule inhibitors of GLUT-1-mediated glucose transport. To date, where inhibitors of glycolysis have progressed into clinical trials, they have not proved successful, often limited by off-target effects and low potency.8-11
A variety of cell signaling pathways are implicated in metabolism by tightly regulating the ability of cells to gain access to and use nutrients. Through aberrations in these pathways, cancer cells can essentially go rogue, ignoring regulatory signals and taking up nutrients in an autonomous manner. One of the most frequently altered signaling pathways in human cancer, the PI3K-Akt-mTOR pathway, is also an important regulator of metabolism, coordinating the uptake of multiple nutrients, including glucose.
Akt in particular is thought to have a critical role in glucose metabolism and increased Akt pathway signaling has been shown to correlate with increased rates of glycolysis in cancer cells. Thus, Akt inhibitors could double as glycolytic or glucose transport inhibitors.12,13
A number of Akt inhibitors are being evaluated in clinical trials (Table) and results from the phase 2 LOTUS trial of ipatasertib (GDC-0068) were recently published.
Among 124 patients randomly assigned to paclitaxel in combination with either ipatasertib or placebo, there was a modest improvement in progression-free survival (PFS) in the ipatasertib arm in patients with triple-negative breast cancer (TNBC; 6 months vs 4.2 months, respectively; hazard ratio [HR], 0.60; P = .037). The effect was more pronounced, though not statistically significant, in patients with phosphatase and tensin homolog (PTEN)-low tumors (6.2 months vs 3.7 months; HR, 0.59; P = .18). The most common grade 3 and higher adverse events (AEs) were diarrhea, reduced neutrophil count, and neutropenia.14
The Warburg paradox
Although the molecular mechanisms underlying the Warburg effect have been revealed to some extent, why cancer cells would choose to use such an energy-inefficient process when they have such high energy demands, remains something of a paradox. It’s still not entirely clear, but several explanations that are not necessarily mutually exclusive have been proposed and relate to the inherent benefits of glycolysis and might explain why cancer cells favor this pathway despite its poor energy yield. First, ATP is produced much more rapidly through glycolysis than oxidative phosphorylation, up to 100 times faster. Thus, using glycolysis is a trade-off, between making less energy and making it more quickly.
Second, cancer cells require more than just ATP to meet their metabolic demands. They need amino acids for protein synthesis; nucleotides for DNA replication; lipids for cell membrane synthesis; nicotinamide adenine dinucleotide phosphate (NADPH), which helps the cancer cell deal with oxidative stress; and various other metabolites. Glycolysis branches off into other metabolic pathways that generate many of these metabolites. Among these branched pathways is the pentose phosphate pathway (PPP), which is required for the generation of ribonucleotides and is a major source for NADPH. Cancer cells have been shown to upregulate the flux of glucose into the PPP to meet their anabolic demands and counter oxidative stress.
Third, the lactic acid produced through glycolysis is actively exported from tumor cells by monocarboxylate transporters (MCTs). This creates a highly acidic tumor microenvironment, which can promote several cancer-related processes and also plays a role in tumor-induced immunosuppression, by inhibiting the activity of tumor-infiltrating T cells, reducing dendritic cell maturation, and promoting the transformation of macrophages to a protumorigenic form.2,4,6
Beyond the Warburg effect
Although the focus has been on glucose metabolism and glycolysis, it has been increasingly recognized that many different metabolic pathways are altered. Fundamental changes to the metabolism of all 4 major classes of macromolecules – carbohydrates, lipids, proteins, and nucleic acids – have been observed, encompassing all aspects of cellular metabolism and enabling cancer cells to meet their complete metabolic requirements. There is also evidence that cancer cells are able to switch between different metabolic pathways depending on the availability of oxygen, their energetic needs, environmental stresses, and many other factors. Certainly, there is significant heterogeneity in the metabolic changes that occur in tumors, which vary from tumor to tumor and even within the same tumor and across the lifespan of a tumor as it progresses from an early stage to more advanced or metastatic disease.
The notion of the Warburg effect as a universal phenomenon in cancer cells is now being widely disregarded. Many tumors continue to use oxidative phosphorylation, particularly slower growing tumors, to meet their energy needs. More recently a “reverse” Warburg effect was described, whereby cancer cells are thought to influence the metabolism of the surrounding stromal fibroblasts and essentially outsource aerobic glycolysis to these cells, while performing energy-efficient oxidative phosphorylation themselves (Figure 2).5,15,16
There is thought to be a “lactate shuttle” between the stromal and cancer cells. The stromal cells express high levels of efflux MCTs so that they can remove the subsequently high levels of lactate from the cytoplasm and avoid pickling themselves. The lactate is then shuttled to the cancer cells that have MCTs on their surface that are involved in lactate uptake. The cancer cells oxidize the lactate back into pyruvate, which can then be used in the tricarboxylic acid (TCA) cycle to feed oxidative phosphorylation for efficient ATP production. This hypothesis reflects a broader appreciation of the role of the microenvironment in contributing to cancer metabolism.17,18
An improved holistic understanding of cancer cell metabolism has led to the recognition of altered cancer metabolism as one of the hallmark abilities required for transformation of a normal cell into a cancerous one. It is categorized as “deregulation of bioenergetics” in the most up to date review of the cancer hallmarks.19 It has also begun to shape the therapeutic landscape as new drug targets have emerged.
IDH inhibitors first to market
A number of new metabolically-targeted treatment strategies are being developed. Most promising are small molecule inhibitors of the isocitrate dehydrogenase (IDH) enzymes. These enzymes play an essential role in the TCA cycle, catalyzing the conversion of isocitrate to alpha-ketoglutarate, generating carbon dioxide and NADPH. Recurrent mutations in the IDH1 and IDH2 genes have been observed in several different types of cancer, including glioma, acute myeloid leukemia (AML), and cholangiocarcinoma.
IDH mutations are known as neomorphic mutations because they confer a new function on the altered gene product. In this case, the mutant IDH enzyme converts alpha-ketoglutarate further into D-2-hydroxyglutarate (D-2HG). This molecule has a number of different effects that promote tumorigenesis, including fostering defective DNA repair (Figure 3).20,21
Intriguing research presented at the American Association of Cancer Research Annual Meeting revealed that IDH mutations may make cancer cells more vulnerable to poly (ADP-ribose) polymerase (PARP) inhibition, likely as a result of defects in homologous recombination pathways of DNA repair.22The pursuit of IDH as a potential therapeutic target has yielded the first regulatory approval for a metabolically targeted anticancer therapy. In August 2017, the United States Food and Drug Administration (FDA) approved enasidenib, an IDH2 inhibitor, for the treatment of relapsed or refractory AML with an IDH2 mutation. It was approved in combination with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect IDH2 mutations.
The approval was based on a single-arm trial in which responses occurred in almost a quarter of the 199 patients treated with 100 mg oral enasidenib daily. After a median follow-up of 6.6 months, 23% of the patients experienced a complete response or a complete response with partial hematologic recovery lasting a median of 8.2 months. The most common AEs were nausea, vomiting, diarrhea, elevated bilirubin levels, and reduced appetite.23
Several other IDH inhibitors are also showing encouraging efficacy. Ivosidenib is an IDH1 inhibitor and the results of a phase 1 study in patients with cholangiocarcinoma were recently presented at a leading conference. Escalating doses of ivosidenib (100 mg twice daily to 1,200 mg once daily) were administered to 73 patients (as of December 2016). The confirmed partial response (PR) rate was 6%, the rate of stable disease was 56%, and PFS at 6 months was 40%. There were no dose-limiting toxicities (DLTs) and treatment-emergent AEs included fatigue, nausea, vomiting, diarrhea, decreased appetite, dysgeusia, and QT prolongation.24
Another study of ivosidenib was presented at the 2017 annual meeting of the Society for Neuro-Oncology. In that study, patients with glioma received daily doses of ivosidenib ranging from 300 mg to 900 mg. Two patients had a minor response, 83% had stable disease, and the median PFS was 13 months. There were no DLTs and most AEs were mild to moderate and included, most commonly, headache, nausea, diarrhea, and vomiting.25
Pursuing alternative targets and repurposing drugs
Other metabolic targets that are being pursued include glutaminase, given the observation of significantly enhanced glutamine uptake in cancer cells. CB-839 is a glutaminase inhibitor that is currently being evaluated in phase 1 and 2 clinical trials. Updated clinical trial data from a phase 1 trial of CB-839 in combination with paclitaxel in patients with advanced/metastatic TNBC were presented at the San Antonio Breast Cancer Symposium last year.26
As of October 2017, 49 patients had been treated with 400 mg, 600 mg, or 800 mg CB-839 twice daily in combination with 80 mg/m2 intravenous paclitaxel weekly. Among the 44 patients evaluable for response, the rate of PR was 22% and of disease control, 59%. The one DLT was grade 3 neutropenia at the 400 mg dose. Overall AEs were mostly low grade and reversible.
In recent years, lactate has emerged as more than just a by-product of altered cancer cell metabolism. It is responsible, at least in part, for the highly acidic tumor microenvironment that fosters many of the other hallmarks of cancer. In addition, lactate promotes angiogenesis by upregulating HIF-1α in endothelial cells. Depriving tumor cells of the ability to export lactate is a potentially promising therapeutic strategy. An MCT-1 inhibitor, AZD3965, is being evaluated in early stage clinical trials.
Finally, several drugs that are renowned for their use in other disease settings are being repurposed for cancer therapy because of their potential effects on cancer cell metabolism. Ritonavir, an antiretroviral drug used in the treatment of HIV, is an inhibitor of GLUT-1 and is being evaluated in phase 1 and 2 clinical trials. Meanwhile, long-term studies of metformin, a drug that has revolutionized the treatment of diabetes, have revealed a reduction in the emergence of new cancers in diabetic patients treated who are treated with it, and the drug has been shown to improve breast cancer survival rates. Its precise anticancer effects are somewhat unclear, but it is thought to act in part by inhibiting oxidative phosphorylation. Numerous clinical trials of metformin in different types of cancer are ongoing.27,2
Altered cell metabolism has long been recognized as a distinctive feature of malignant cells but, until recently, research efforts had focused on a single aspect. It has become increasingly evident that many metabolic pathways are altered in cancer cells. Improved understanding has yielded the first regulatory approval in this new class of drugs. Here, we discuss the latest developments in the therapeutic targeting of the cancer metabolism hallmark.
A cancer cell’s sweet tooth
The metabolism of cancer cells differs from that of normal cells, an observation that has spawned a dedicated field of research and new targeted drug development. The German physiologist Otto Warburg is credited as the father of the field with his observations about the way in which cancer cells derive energy from glucose.1
In normal cells, glucose is converted into pyruvate in the cytoplasm, which is then, most often, fed to the mitochondria that use oxidative phosphorylation to produce energy in the form of adenosine triphosphate (ATP). Cancer cells seem instead to favor using the pyruvate to produce lactate through glycolysis (Figure 1).
Glycolysis is usually reserved for conditions of poor oxygen availability, but although the tumor microenvironment is often hypoxic, cancer cells have been shown to use glycolysis even when oxygen is plentiful. As a result, the phenomenon is known as aerobic glycolysis, although it is most often referred to as the Warburg effect.2
Glycolysis is much less efficient than oxidative phosphorylation at producing energy, yielding only 2 ATP. In order to meet their energy demands in this way, cancer cells ramp up their glucose intake, an effect that has been exploited for the detection of cancer with positron-emission tomography.
Warburg postulated that this metabolic shift was a result of mitochondrial damage and defective oxidative phosphorylation, even going so far as to suggest that cancer was a mitochondrial disease. It has subsequently been shown that the mitochondria are mostly intact in cancer cells and that oxidative phosphorylation can still occur.3
The Warburg effect has been the subject of significant investigative efforts as researchers have attempted to better understand how this phenomenon comes about. Studies have shown that it is driven in large part by the transcription factors hypoxia inducible factor 1 alpha (HIF-1α) and c-Myc. In addition, numerous other signaling pathways, including the phosphatidylinositol 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) pathway, and the activation of oncogenes and inactivation of tumor suppressors, are thought to play a central role.
HIF-1α is an oxygen-sensing transcription factor that coordinates cellular responses to reduced oxygen levels by binding to specific regions, known as hypoxia response elements, on target genes in the nucleus and regulating their subsequent expression. Oxygen levels and metabolism are tightly linked, and HIF-1α sits at the intersection of the 2 since many of its target genes are involved in metabolic pathways, including many glycolytic enzymes, but it also directly inhibits oxidative phosphorylation by suppressing key enzymes in this metabolic pathway.
The expression of HIF-1α and numerous glycolytic enzymes, including lactate dehydrogenase (LDH), phosphofructokinase (PFK), hexokinase II (HKII), and pyruvate dehydrogenase kinase (PDK) is increased in many tumor types. Other molecules that are associated with glucose uptake and metabolism are also dysregulated, such as the GLUT-1 glucose transporter.2,4-6
Targeting glycolysis and glucose uptake
According to one study, glucose transporters and glycolytic enzymes are overexpressed in 24 different types of cancer, representing more than 70% of all cancer cases.7 This enables cancer cells to respond metabolically as though they are experiencing hypoxia, even when oxygen is plentiful and, indeed, when hypoxia is a concern, to mount a faster response. It also provides a tempting avenue for anticancer drug design by exploiting the dependency of cancer cells on glycolysis to survive and thrive.
Inhibitors of HKII, LDH, PFK, PDK, and GLUT-1 have been and continue to be developed. For example, 2-deoxy-D-glucose is a glucose molecule in which the 2-hydroxyl group has been replaced by hydrogen, preventing further glycolysis; it acts as a competitive inhibitor of HKII. Dichloroacetate (DCA) activates the pyruvate dehydrogenase complex and inhibits the actions of the PDKs. Although development of DCA itself was unsuccessful, DCA derivatives continue to be pursued. WZB117 and STF-31 are novel small-molecule inhibitors of GLUT-1-mediated glucose transport. To date, where inhibitors of glycolysis have progressed into clinical trials, they have not proved successful, often limited by off-target effects and low potency.8-11
A variety of cell signaling pathways are implicated in metabolism by tightly regulating the ability of cells to gain access to and use nutrients. Through aberrations in these pathways, cancer cells can essentially go rogue, ignoring regulatory signals and taking up nutrients in an autonomous manner. One of the most frequently altered signaling pathways in human cancer, the PI3K-Akt-mTOR pathway, is also an important regulator of metabolism, coordinating the uptake of multiple nutrients, including glucose.
Akt in particular is thought to have a critical role in glucose metabolism and increased Akt pathway signaling has been shown to correlate with increased rates of glycolysis in cancer cells. Thus, Akt inhibitors could double as glycolytic or glucose transport inhibitors.12,13
A number of Akt inhibitors are being evaluated in clinical trials (Table) and results from the phase 2 LOTUS trial of ipatasertib (GDC-0068) were recently published.
Among 124 patients randomly assigned to paclitaxel in combination with either ipatasertib or placebo, there was a modest improvement in progression-free survival (PFS) in the ipatasertib arm in patients with triple-negative breast cancer (TNBC; 6 months vs 4.2 months, respectively; hazard ratio [HR], 0.60; P = .037). The effect was more pronounced, though not statistically significant, in patients with phosphatase and tensin homolog (PTEN)-low tumors (6.2 months vs 3.7 months; HR, 0.59; P = .18). The most common grade 3 and higher adverse events (AEs) were diarrhea, reduced neutrophil count, and neutropenia.14
The Warburg paradox
Although the molecular mechanisms underlying the Warburg effect have been revealed to some extent, why cancer cells would choose to use such an energy-inefficient process when they have such high energy demands, remains something of a paradox. It’s still not entirely clear, but several explanations that are not necessarily mutually exclusive have been proposed and relate to the inherent benefits of glycolysis and might explain why cancer cells favor this pathway despite its poor energy yield. First, ATP is produced much more rapidly through glycolysis than oxidative phosphorylation, up to 100 times faster. Thus, using glycolysis is a trade-off, between making less energy and making it more quickly.
Second, cancer cells require more than just ATP to meet their metabolic demands. They need amino acids for protein synthesis; nucleotides for DNA replication; lipids for cell membrane synthesis; nicotinamide adenine dinucleotide phosphate (NADPH), which helps the cancer cell deal with oxidative stress; and various other metabolites. Glycolysis branches off into other metabolic pathways that generate many of these metabolites. Among these branched pathways is the pentose phosphate pathway (PPP), which is required for the generation of ribonucleotides and is a major source for NADPH. Cancer cells have been shown to upregulate the flux of glucose into the PPP to meet their anabolic demands and counter oxidative stress.
Third, the lactic acid produced through glycolysis is actively exported from tumor cells by monocarboxylate transporters (MCTs). This creates a highly acidic tumor microenvironment, which can promote several cancer-related processes and also plays a role in tumor-induced immunosuppression, by inhibiting the activity of tumor-infiltrating T cells, reducing dendritic cell maturation, and promoting the transformation of macrophages to a protumorigenic form.2,4,6
Beyond the Warburg effect
Although the focus has been on glucose metabolism and glycolysis, it has been increasingly recognized that many different metabolic pathways are altered. Fundamental changes to the metabolism of all 4 major classes of macromolecules – carbohydrates, lipids, proteins, and nucleic acids – have been observed, encompassing all aspects of cellular metabolism and enabling cancer cells to meet their complete metabolic requirements. There is also evidence that cancer cells are able to switch between different metabolic pathways depending on the availability of oxygen, their energetic needs, environmental stresses, and many other factors. Certainly, there is significant heterogeneity in the metabolic changes that occur in tumors, which vary from tumor to tumor and even within the same tumor and across the lifespan of a tumor as it progresses from an early stage to more advanced or metastatic disease.
The notion of the Warburg effect as a universal phenomenon in cancer cells is now being widely disregarded. Many tumors continue to use oxidative phosphorylation, particularly slower growing tumors, to meet their energy needs. More recently a “reverse” Warburg effect was described, whereby cancer cells are thought to influence the metabolism of the surrounding stromal fibroblasts and essentially outsource aerobic glycolysis to these cells, while performing energy-efficient oxidative phosphorylation themselves (Figure 2).5,15,16
There is thought to be a “lactate shuttle” between the stromal and cancer cells. The stromal cells express high levels of efflux MCTs so that they can remove the subsequently high levels of lactate from the cytoplasm and avoid pickling themselves. The lactate is then shuttled to the cancer cells that have MCTs on their surface that are involved in lactate uptake. The cancer cells oxidize the lactate back into pyruvate, which can then be used in the tricarboxylic acid (TCA) cycle to feed oxidative phosphorylation for efficient ATP production. This hypothesis reflects a broader appreciation of the role of the microenvironment in contributing to cancer metabolism.17,18
An improved holistic understanding of cancer cell metabolism has led to the recognition of altered cancer metabolism as one of the hallmark abilities required for transformation of a normal cell into a cancerous one. It is categorized as “deregulation of bioenergetics” in the most up to date review of the cancer hallmarks.19 It has also begun to shape the therapeutic landscape as new drug targets have emerged.
IDH inhibitors first to market
A number of new metabolically-targeted treatment strategies are being developed. Most promising are small molecule inhibitors of the isocitrate dehydrogenase (IDH) enzymes. These enzymes play an essential role in the TCA cycle, catalyzing the conversion of isocitrate to alpha-ketoglutarate, generating carbon dioxide and NADPH. Recurrent mutations in the IDH1 and IDH2 genes have been observed in several different types of cancer, including glioma, acute myeloid leukemia (AML), and cholangiocarcinoma.
IDH mutations are known as neomorphic mutations because they confer a new function on the altered gene product. In this case, the mutant IDH enzyme converts alpha-ketoglutarate further into D-2-hydroxyglutarate (D-2HG). This molecule has a number of different effects that promote tumorigenesis, including fostering defective DNA repair (Figure 3).20,21
Intriguing research presented at the American Association of Cancer Research Annual Meeting revealed that IDH mutations may make cancer cells more vulnerable to poly (ADP-ribose) polymerase (PARP) inhibition, likely as a result of defects in homologous recombination pathways of DNA repair.22The pursuit of IDH as a potential therapeutic target has yielded the first regulatory approval for a metabolically targeted anticancer therapy. In August 2017, the United States Food and Drug Administration (FDA) approved enasidenib, an IDH2 inhibitor, for the treatment of relapsed or refractory AML with an IDH2 mutation. It was approved in combination with a companion diagnostic, the RealTime IDH2 Assay, which is used to detect IDH2 mutations.
The approval was based on a single-arm trial in which responses occurred in almost a quarter of the 199 patients treated with 100 mg oral enasidenib daily. After a median follow-up of 6.6 months, 23% of the patients experienced a complete response or a complete response with partial hematologic recovery lasting a median of 8.2 months. The most common AEs were nausea, vomiting, diarrhea, elevated bilirubin levels, and reduced appetite.23
Several other IDH inhibitors are also showing encouraging efficacy. Ivosidenib is an IDH1 inhibitor and the results of a phase 1 study in patients with cholangiocarcinoma were recently presented at a leading conference. Escalating doses of ivosidenib (100 mg twice daily to 1,200 mg once daily) were administered to 73 patients (as of December 2016). The confirmed partial response (PR) rate was 6%, the rate of stable disease was 56%, and PFS at 6 months was 40%. There were no dose-limiting toxicities (DLTs) and treatment-emergent AEs included fatigue, nausea, vomiting, diarrhea, decreased appetite, dysgeusia, and QT prolongation.24
Another study of ivosidenib was presented at the 2017 annual meeting of the Society for Neuro-Oncology. In that study, patients with glioma received daily doses of ivosidenib ranging from 300 mg to 900 mg. Two patients had a minor response, 83% had stable disease, and the median PFS was 13 months. There were no DLTs and most AEs were mild to moderate and included, most commonly, headache, nausea, diarrhea, and vomiting.25
Pursuing alternative targets and repurposing drugs
Other metabolic targets that are being pursued include glutaminase, given the observation of significantly enhanced glutamine uptake in cancer cells. CB-839 is a glutaminase inhibitor that is currently being evaluated in phase 1 and 2 clinical trials. Updated clinical trial data from a phase 1 trial of CB-839 in combination with paclitaxel in patients with advanced/metastatic TNBC were presented at the San Antonio Breast Cancer Symposium last year.26
As of October 2017, 49 patients had been treated with 400 mg, 600 mg, or 800 mg CB-839 twice daily in combination with 80 mg/m2 intravenous paclitaxel weekly. Among the 44 patients evaluable for response, the rate of PR was 22% and of disease control, 59%. The one DLT was grade 3 neutropenia at the 400 mg dose. Overall AEs were mostly low grade and reversible.
In recent years, lactate has emerged as more than just a by-product of altered cancer cell metabolism. It is responsible, at least in part, for the highly acidic tumor microenvironment that fosters many of the other hallmarks of cancer. In addition, lactate promotes angiogenesis by upregulating HIF-1α in endothelial cells. Depriving tumor cells of the ability to export lactate is a potentially promising therapeutic strategy. An MCT-1 inhibitor, AZD3965, is being evaluated in early stage clinical trials.
Finally, several drugs that are renowned for their use in other disease settings are being repurposed for cancer therapy because of their potential effects on cancer cell metabolism. Ritonavir, an antiretroviral drug used in the treatment of HIV, is an inhibitor of GLUT-1 and is being evaluated in phase 1 and 2 clinical trials. Meanwhile, long-term studies of metformin, a drug that has revolutionized the treatment of diabetes, have revealed a reduction in the emergence of new cancers in diabetic patients treated who are treated with it, and the drug has been shown to improve breast cancer survival rates. Its precise anticancer effects are somewhat unclear, but it is thought to act in part by inhibiting oxidative phosphorylation. Numerous clinical trials of metformin in different types of cancer are ongoing.27,2
1. Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269-270.
2. Yu L, Chen X, Wang L, Chen S. The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget. 2016;7(25):38908-38926.
3. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309-314.
4. Chen XS, Li LY, Guan YD, Yang JM, Cheng Y. Anticancer strategies based on the metabolic profile of tumor cells: therapeutic targeting of the Warburg effect. Acta Pharmacol Sin. 2016;37(8):1013-1019.
5. Gupta S, Roy A, Dwarakanath BS. Metabolic cooperation and competition in the tumor microenvironment: implications for therapy. Front Oncol. 2017;7:68.
6. Marchiq I, Pouyssegur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J Mol Med (Berl). 2016;94(2):155-171.
7. Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84(6):1014-1020.
8. Yu L, Chen X, Sun X, Wang L, Chen S. The glycolytic switch in tumors: how many players are involved? J Cancer. 2017;8(17):3430-3440.
9. Zhang W, Zhang SL, Hu X, Tam KY. Targeting tumor metabolism for cancer treatment: is pyruvate dehydrogenase kinases (PDKs) a viable anticancer target? Int J Biol Sci. 2015;11(12):1390-1400.
10. Talekar M, Boreddy SR, Singh A, Amiji M. Tumor aerobic glycolysis: new insights into therapeutic strategies with targeted delivery. Expert Opin Biol Ther. 2014;14(8):1145-1159.
11. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152.
12. Lien EC, Lyssiotis CA, Cantley LC. Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer. In: Cramer T, Schmitt CA, eds. Metabolism in Cancer. Cham, Switzerland: Springer International Publishing; 2016:39-72.
13. Simons AL, Orcutt KP, Madsen JM, Scarbrough PM, Spitz DR. The role of Akt pathway signaling in glucose metabolism and metabolic oxidative stress. In: Spitz DR, Dornfeld KJ, Krishnan K, Gius D (eds). Oxidative stress in cancer biology and therapy. Humana Press (copyright holder, Springer Science+Business Media, LLC); 2012:21-46.
14. Kim S-B, Dent R, Im S-A, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18(10):1360-1372.
15. Fu Y, Liu S, Yin S, et al. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget. 2017;8(34):57813-57825.
16. Wilde L, Roche M, Domingo-Vidal M, et al. Metabolic coupling and the reverse Warburg effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol. 2017;44(3):198-203.
17. Brooks GA. Cell–cell and intracellular lactate shuttles. Journal Physiol. 2009;587(23):5591-5600.
18. Chiarugi P, Cirri P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016;380(1):272-280.
19. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674.
20. Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 2016;21(117):373-380.
21. Carlsson SK, Brothers SP, Wahlestedt C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol Med. 2014;6(11):1359-1370.
22. Lu Y, Kwintkiewicz J, Liu Y, et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. 2017;77(7):1709-1718.
23. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731.
24. Lowery MA, Abou-Alfa GK, Burris HA, et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: results from the cholangiocarcinoma dose escalation and expansion cohorts. J Clin Oncol. 2017;35(15_suppl):4015-4015.
25. Mellinghoff IK, Touat M, Maher E, et al. ACTR-46. AG-120, a first-in-class mutant IDH1 inhibitor in patients with recurrent or progressive IDH1 mutant glioma: updated results from the phase 1 non-enhancing glioma population. Neuro Oncol. 2017;19(suppl_6):vi10-vi11.
26. Kalinsky K, Harding J, DeMichele A, et al. Phase 1 study of CB-839, a first-in-class oral inhibitor of glutaminase, in combination with paclitaxel in patients with advanced triple negative breast cancer. Paper presented at San Antonio Breast Cancer Symposium; December 5-9, 2017; San Antonio, Texas.
27. Hatoum D, McGowan EM. Recent advances in the use of metformin: can treating diabetes prevent breast cancer? Biomed Res Int. 2015;2015:548436.
28. Leone A, Di Gennaro E, Bruzzese F, Avallone A, Budillon A. New perspective for an old antidiabetic drug: metformin as anticancer agent. Cancer Treat Res. 2014;159:355-376.
1. Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269-270.
2. Yu L, Chen X, Wang L, Chen S. The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget. 2016;7(25):38908-38926.
3. Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309-314.
4. Chen XS, Li LY, Guan YD, Yang JM, Cheng Y. Anticancer strategies based on the metabolic profile of tumor cells: therapeutic targeting of the Warburg effect. Acta Pharmacol Sin. 2016;37(8):1013-1019.
5. Gupta S, Roy A, Dwarakanath BS. Metabolic cooperation and competition in the tumor microenvironment: implications for therapy. Front Oncol. 2017;7:68.
6. Marchiq I, Pouyssegur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J Mol Med (Berl). 2016;94(2):155-171.
7. Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84(6):1014-1020.
8. Yu L, Chen X, Sun X, Wang L, Chen S. The glycolytic switch in tumors: how many players are involved? J Cancer. 2017;8(17):3430-3440.
9. Zhang W, Zhang SL, Hu X, Tam KY. Targeting tumor metabolism for cancer treatment: is pyruvate dehydrogenase kinases (PDKs) a viable anticancer target? Int J Biol Sci. 2015;11(12):1390-1400.
10. Talekar M, Boreddy SR, Singh A, Amiji M. Tumor aerobic glycolysis: new insights into therapeutic strategies with targeted delivery. Expert Opin Biol Ther. 2014;14(8):1145-1159.
11. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152.
12. Lien EC, Lyssiotis CA, Cantley LC. Metabolic reprogramming by the PI3K-Akt-mTOR pathway in cancer. In: Cramer T, Schmitt CA, eds. Metabolism in Cancer. Cham, Switzerland: Springer International Publishing; 2016:39-72.
13. Simons AL, Orcutt KP, Madsen JM, Scarbrough PM, Spitz DR. The role of Akt pathway signaling in glucose metabolism and metabolic oxidative stress. In: Spitz DR, Dornfeld KJ, Krishnan K, Gius D (eds). Oxidative stress in cancer biology and therapy. Humana Press (copyright holder, Springer Science+Business Media, LLC); 2012:21-46.
14. Kim S-B, Dent R, Im S-A, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017;18(10):1360-1372.
15. Fu Y, Liu S, Yin S, et al. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget. 2017;8(34):57813-57825.
16. Wilde L, Roche M, Domingo-Vidal M, et al. Metabolic coupling and the reverse Warburg effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol. 2017;44(3):198-203.
17. Brooks GA. Cell–cell and intracellular lactate shuttles. Journal Physiol. 2009;587(23):5591-5600.
18. Chiarugi P, Cirri P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016;380(1):272-280.
19. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674.
20. Fujii T, Khawaja MR, DiNardo CD, Atkins JT, Janku F. Targeting isocitrate dehydrogenase (IDH) in cancer. Discov Med. 2016;21(117):373-380.
21. Carlsson SK, Brothers SP, Wahlestedt C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol Med. 2014;6(11):1359-1370.
22. Lu Y, Kwintkiewicz J, Liu Y, et al. Chemosensitivity of IDH1-mutated gliomas due to an impairment in PARP1-mediated DNA repair. Cancer Res. 2017;77(7):1709-1718.
23. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731.
24. Lowery MA, Abou-Alfa GK, Burris HA, et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: results from the cholangiocarcinoma dose escalation and expansion cohorts. J Clin Oncol. 2017;35(15_suppl):4015-4015.
25. Mellinghoff IK, Touat M, Maher E, et al. ACTR-46. AG-120, a first-in-class mutant IDH1 inhibitor in patients with recurrent or progressive IDH1 mutant glioma: updated results from the phase 1 non-enhancing glioma population. Neuro Oncol. 2017;19(suppl_6):vi10-vi11.
26. Kalinsky K, Harding J, DeMichele A, et al. Phase 1 study of CB-839, a first-in-class oral inhibitor of glutaminase, in combination with paclitaxel in patients with advanced triple negative breast cancer. Paper presented at San Antonio Breast Cancer Symposium; December 5-9, 2017; San Antonio, Texas.
27. Hatoum D, McGowan EM. Recent advances in the use of metformin: can treating diabetes prevent breast cancer? Biomed Res Int. 2015;2015:548436.
28. Leone A, Di Gennaro E, Bruzzese F, Avallone A, Budillon A. New perspective for an old antidiabetic drug: metformin as anticancer agent. Cancer Treat Res. 2014;159:355-376.
Gene therapy regimen for XSCID shows rapid results in newly diagnosed infants
ATLANTA – A new approach to gene therapy produced rapid T cell reconstruction and normal markers of B-cell and natural killer (NK)-cell function in newly diagnosed infants with X-linked Severe Combined Immunodeficiency (XSCID), according to initial results from a phase 1/2 trial.
Researchers used a regimen of safety-modified lentiviral vector plus reduced-exposure busulfan conditioning in seven newly diagnosed infants and found that it was well tolerated in all patients and quickly improved T-cell immunity in five of seven patients.

Initial gene therapy trials used a mouse vector without busulfan conditioning. These trials produced T-cell correction but were unable to restore B-cell function, NK-cell function, or myeloid-cell function. As a result, patients continued to experience viral infections and required monthly, life-long intravenous immunoglobulin infusions, Dr. Mamcarz said.
The phase 1/2 multicenter safety and efficacy study tested the gene therapy regimen for the first time in seven newly diagnosed infants. The regimen involved transducing purified bone marrow CD34+ cells with the lentiviral vector, which was generated by a stable producer cell line and then cryopreserved. The busulfan was given as two single daily doses, which were tailored based on patient age and weight.
In total, seven patients have been treated and all tolerated the low-dose busulfan chemotherapy well. Five patients had normal numbers of previously defective cells (T cells, B cells, and NK cells) and were taken off protective isolation and prophylactic medications. Those infants appear to have functioning immune systems, Dr. Mamcarz said. One infant has stopped monthly intravenous immunoglobulin infusions and has received normal pediatric vaccines, but responses have not yet been tested.
For the two infants who did not show responses, one is early in the trial and there is not yet adequate follow-up data to assess the immune system reconstitution, Dr. Mamcarz said.
The other infant had high levels of maternal T cell engraftment, severe neutropenia, and ongoing cytomegalovirus infection, resulting in delayed and partial T-cell reconstitution. The researchers sought to boost the patient’s immunity through an infusion of corrected cells – without busulfan – at 1 year after receiving the initial therapy. Three months after the second treatment, the infant has normal functioning of T cells and NK cells, but he is not yet engrafting B cells, Dr. Mamcarz said.
Overall, there was no evidence of vector-mediated effects on blood formation, Dr. Mamcarz reported.
The ultimate evaluation of the efficacy of the trial would be to assess vaccine responses in the infants treated with gene therapy, she said.
The study was supported by the Assisi Foundation of Memphis and the California Institute of Regenerative Medicine. Dr. Mamcarz reported having no relevant financial disclosures. Her coauthors reported financial relationships with InsightRX, UpToDate, Invitae, and Homology Medicines.
[email protected]
SOURCE: Mamcarz E et al. Abstract 523.
ATLANTA – A new approach to gene therapy produced rapid T cell reconstruction and normal markers of B-cell and natural killer (NK)-cell function in newly diagnosed infants with X-linked Severe Combined Immunodeficiency (XSCID), according to initial results from a phase 1/2 trial.
Researchers used a regimen of safety-modified lentiviral vector plus reduced-exposure busulfan conditioning in seven newly diagnosed infants and found that it was well tolerated in all patients and quickly improved T-cell immunity in five of seven patients.
Initial gene therapy trials used a mouse vector without busulfan conditioning. These trials produced T-cell correction but were unable to restore B-cell function, NK-cell function, or myeloid-cell function. As a result, patients continued to experience viral infections and required monthly, life-long intravenous immunoglobulin infusions, Dr. Mamcarz said.
The phase 1/2 multicenter safety and efficacy study tested the gene therapy regimen for the first time in seven newly diagnosed infants. The regimen involved transducing purified bone marrow CD34+ cells with the lentiviral vector, which was generated by a stable producer cell line and then cryopreserved. The busulfan was given as two single daily doses, which were tailored based on patient age and weight.
In total, seven patients have been treated and all tolerated the low-dose busulfan chemotherapy well. Five patients had normal numbers of previously defective cells (T cells, B cells, and NK cells) and were taken off protective isolation and prophylactic medications. Those infants appear to have functioning immune systems, Dr. Mamcarz said. One infant has stopped monthly intravenous immunoglobulin infusions and has received normal pediatric vaccines, but responses have not yet been tested.
For the two infants who did not show responses, one is early in the trial and there is not yet adequate follow-up data to assess the immune system reconstitution, Dr. Mamcarz said.
The other infant had high levels of maternal T cell engraftment, severe neutropenia, and ongoing cytomegalovirus infection, resulting in delayed and partial T-cell reconstitution. The researchers sought to boost the patient’s immunity through an infusion of corrected cells – without busulfan – at 1 year after receiving the initial therapy. Three months after the second treatment, the infant has normal functioning of T cells and NK cells, but he is not yet engrafting B cells, Dr. Mamcarz said.
Overall, there was no evidence of vector-mediated effects on blood formation, Dr. Mamcarz reported.
The ultimate evaluation of the efficacy of the trial would be to assess vaccine responses in the infants treated with gene therapy, she said.
The study was supported by the Assisi Foundation of Memphis and the California Institute of Regenerative Medicine. Dr. Mamcarz reported having no relevant financial disclosures. Her coauthors reported financial relationships with InsightRX, UpToDate, Invitae, and Homology Medicines.
[email protected]
SOURCE: Mamcarz E et al. Abstract 523.
ATLANTA – A new approach to gene therapy produced rapid T cell reconstruction and normal markers of B-cell and natural killer (NK)-cell function in newly diagnosed infants with X-linked Severe Combined Immunodeficiency (XSCID), according to initial results from a phase 1/2 trial.
Researchers used a regimen of safety-modified lentiviral vector plus reduced-exposure busulfan conditioning in seven newly diagnosed infants and found that it was well tolerated in all patients and quickly improved T-cell immunity in five of seven patients.
Initial gene therapy trials used a mouse vector without busulfan conditioning. These trials produced T-cell correction but were unable to restore B-cell function, NK-cell function, or myeloid-cell function. As a result, patients continued to experience viral infections and required monthly, life-long intravenous immunoglobulin infusions, Dr. Mamcarz said.
The phase 1/2 multicenter safety and efficacy study tested the gene therapy regimen for the first time in seven newly diagnosed infants. The regimen involved transducing purified bone marrow CD34+ cells with the lentiviral vector, which was generated by a stable producer cell line and then cryopreserved. The busulfan was given as two single daily doses, which were tailored based on patient age and weight.
In total, seven patients have been treated and all tolerated the low-dose busulfan chemotherapy well. Five patients had normal numbers of previously defective cells (T cells, B cells, and NK cells) and were taken off protective isolation and prophylactic medications. Those infants appear to have functioning immune systems, Dr. Mamcarz said. One infant has stopped monthly intravenous immunoglobulin infusions and has received normal pediatric vaccines, but responses have not yet been tested.
For the two infants who did not show responses, one is early in the trial and there is not yet adequate follow-up data to assess the immune system reconstitution, Dr. Mamcarz said.
The other infant had high levels of maternal T cell engraftment, severe neutropenia, and ongoing cytomegalovirus infection, resulting in delayed and partial T-cell reconstitution. The researchers sought to boost the patient’s immunity through an infusion of corrected cells – without busulfan – at 1 year after receiving the initial therapy. Three months after the second treatment, the infant has normal functioning of T cells and NK cells, but he is not yet engrafting B cells, Dr. Mamcarz said.
Overall, there was no evidence of vector-mediated effects on blood formation, Dr. Mamcarz reported.
The ultimate evaluation of the efficacy of the trial would be to assess vaccine responses in the infants treated with gene therapy, she said.
The study was supported by the Assisi Foundation of Memphis and the California Institute of Regenerative Medicine. Dr. Mamcarz reported having no relevant financial disclosures. Her coauthors reported financial relationships with InsightRX, UpToDate, Invitae, and Homology Medicines.
[email protected]
SOURCE: Mamcarz E et al. Abstract 523.
REPORTING FROM ASH 2017
Key clinical point:
Major finding: In total, five of seven infants with XSCID had normal functioning of T cells, B cells, and NK cells.
Study details: A phase 1/2 multicenter study of seven infants with newly diagnosed XSCID.
Disclosures: The study was supported by the Assisi Foundation of Memphis and the California Institute of Regenerative Medicine. Dr. Mamcarz reported having no relevant financial disclosures. Her coauthors reported financial relationships with InsightRX, UpToDate, Invitae, and Homology Medicines.
Source: Mamcarz E et al. Abstract 523.
HAVEN 2: Emicizumab represents new standard of care for hemophilia A with inhibitors
ATLANTA – The bispecific humanized monoclonal antibody emicizumab safely prevents or substantially reduces bleeds in children with hemophilia A with inhibitors that render standard factor VIII replacement therapies ineffective, according to an updated interim analysis of data from the open-label phase 3 HAVEN 2 study.
Guy Young, MD, said during a press briefing at the annual meeting of the American Society of Hematology.

Of 57 patients under age 12 years receiving weekly subcutaneous emicizumab (Hemlibra) for bleeding prophylaxis at the data cutoff, 54 (94.7%) had zero treated bleeds, said Dr. Young of the University of Southern California, Los Angeles.
“That’s, honestly, truly remarkable. I mean, most of the time, these patients are bleeding at least on a monthly basis, sometimes more often than that,” he said.
Further, of 23 patients who were treated for at least 12 weeks and had a median efficacy period of 38.1 weeks, 87% had no bleeding events whatsoever (annual bleed rate of 0.2 events vs. a usual annual bleed rate of 1.3 events in patients without inhibitors). The annual treated joint bleed and treated target joint bleed rates each were 0.1, he said.
Overall, only three treated bleeds occurred in the 57 patients, and only one was a spontaneous bleed; one occurred in a joint, one occurred in a muscle, and one occurred in the hip and was classified as “other,” he noted.
The findings expand upon those from a previously reported interim analysis of data on 20 children from the study, which also showed that once-weekly subcutaneous emicizumab prophylaxis prevented or reduced bleeds, and provided clinically meaningful reductions in the annualized bleed rate when compared with prior bypassing agent treatment, Dr Young said.
In that prior analysis of participants aged 2-12 years (or up to 17 years with weight below 40 kg), which was presented in July at the annual meeting of the International Society on Thrombosis and Haemostasis, the data cutoff was Oct. 28, 2016. The current analysis had a May 8, 2017 cutoff, and therefore included about 6 additional months of data.
The treatment, which is given at a dose of 3 mg/kg per week for 4 weeks and then 1.5 mg/kg per week thereafter, was well tolerated. Ten nonserious, treatment-related adverse events occurred, and mainly involved injection site reactions. Serious adverse events occurred in six patients but were mainly related to the disease or to the use of catheters, or were otherwise unrelated to emicizumab. There were no thromboembolic or thrombotic microangiopathy events reported. Mean steady state trough concentrations (approximately 50 mcg/mL) were consistent with the pharmacokinetic profile seen in prior studies in the adolescent/adult population.
Health-related quality of life improvements were considerable, and in many cases, dramatic, Dr. Young said, noting in an interview that, for some patients, the effects were life changing both for the patients and their families. He described one young patient who had experienced scores of debilitating ankle bleeds, but had had no bleeds since he began receiving treatment as part of this study.
Emicizumab, which recently received Food and Drug Administration approval for use in patients of all ages with hemophilia A with inhibitors, bridges factor IXa and factor X to “bring them into proper alignment, which is really what the function of factor VIII is,” he said.
“So, it’s essentially replacing, or mimicking, the function of factor VIII, but without being factor VIII,” he said, explaining that it can be used to treat patients with inhibitors, because the antibodies they develop are specifically to factor VIII.
“This is not factor VIII, it’s just doing the job of factor VIII,” he added.
Studies of emicizumab for hemophilia A without inhibitors are ongoing (HAVEN 3), and could result in expanded indications, he noted.
The primary efficacy results of HAVEN 2 will be reported 52 weeks after the last patient is enrolled. These interim findings, however, demonstrate the potential for weekly subcutaneous injections of emicizumab to safely and conveniently reduce overall disease burden, as well as overall treatment burden in patients with hemophilia A with inhibitors, as these patients previously had to undergo intravenous treatment multiple times per week – and sometimes multiple times per day, he said.
The HAVEN 2 study is sponsored by Hoffman-La Roche. Dr. Young has received honoraria and/or consulting fees from Alnylam, Bayer, Bioverativ, CSL Behring, Genentech/Roche, Kedrion, Novo Nordisk, and Shire.
SOURCE: Young G et al. ASH 2017 Abstract 85.
ATLANTA – The bispecific humanized monoclonal antibody emicizumab safely prevents or substantially reduces bleeds in children with hemophilia A with inhibitors that render standard factor VIII replacement therapies ineffective, according to an updated interim analysis of data from the open-label phase 3 HAVEN 2 study.
Guy Young, MD, said during a press briefing at the annual meeting of the American Society of Hematology.
Of 57 patients under age 12 years receiving weekly subcutaneous emicizumab (Hemlibra) for bleeding prophylaxis at the data cutoff, 54 (94.7%) had zero treated bleeds, said Dr. Young of the University of Southern California, Los Angeles.
“That’s, honestly, truly remarkable. I mean, most of the time, these patients are bleeding at least on a monthly basis, sometimes more often than that,” he said.
Further, of 23 patients who were treated for at least 12 weeks and had a median efficacy period of 38.1 weeks, 87% had no bleeding events whatsoever (annual bleed rate of 0.2 events vs. a usual annual bleed rate of 1.3 events in patients without inhibitors). The annual treated joint bleed and treated target joint bleed rates each were 0.1, he said.
Overall, only three treated bleeds occurred in the 57 patients, and only one was a spontaneous bleed; one occurred in a joint, one occurred in a muscle, and one occurred in the hip and was classified as “other,” he noted.
The findings expand upon those from a previously reported interim analysis of data on 20 children from the study, which also showed that once-weekly subcutaneous emicizumab prophylaxis prevented or reduced bleeds, and provided clinically meaningful reductions in the annualized bleed rate when compared with prior bypassing agent treatment, Dr Young said.
In that prior analysis of participants aged 2-12 years (or up to 17 years with weight below 40 kg), which was presented in July at the annual meeting of the International Society on Thrombosis and Haemostasis, the data cutoff was Oct. 28, 2016. The current analysis had a May 8, 2017 cutoff, and therefore included about 6 additional months of data.
The treatment, which is given at a dose of 3 mg/kg per week for 4 weeks and then 1.5 mg/kg per week thereafter, was well tolerated. Ten nonserious, treatment-related adverse events occurred, and mainly involved injection site reactions. Serious adverse events occurred in six patients but were mainly related to the disease or to the use of catheters, or were otherwise unrelated to emicizumab. There were no thromboembolic or thrombotic microangiopathy events reported. Mean steady state trough concentrations (approximately 50 mcg/mL) were consistent with the pharmacokinetic profile seen in prior studies in the adolescent/adult population.
Health-related quality of life improvements were considerable, and in many cases, dramatic, Dr. Young said, noting in an interview that, for some patients, the effects were life changing both for the patients and their families. He described one young patient who had experienced scores of debilitating ankle bleeds, but had had no bleeds since he began receiving treatment as part of this study.
Emicizumab, which recently received Food and Drug Administration approval for use in patients of all ages with hemophilia A with inhibitors, bridges factor IXa and factor X to “bring them into proper alignment, which is really what the function of factor VIII is,” he said.
“So, it’s essentially replacing, or mimicking, the function of factor VIII, but without being factor VIII,” he said, explaining that it can be used to treat patients with inhibitors, because the antibodies they develop are specifically to factor VIII.
“This is not factor VIII, it’s just doing the job of factor VIII,” he added.
Studies of emicizumab for hemophilia A without inhibitors are ongoing (HAVEN 3), and could result in expanded indications, he noted.
The primary efficacy results of HAVEN 2 will be reported 52 weeks after the last patient is enrolled. These interim findings, however, demonstrate the potential for weekly subcutaneous injections of emicizumab to safely and conveniently reduce overall disease burden, as well as overall treatment burden in patients with hemophilia A with inhibitors, as these patients previously had to undergo intravenous treatment multiple times per week – and sometimes multiple times per day, he said.
The HAVEN 2 study is sponsored by Hoffman-La Roche. Dr. Young has received honoraria and/or consulting fees from Alnylam, Bayer, Bioverativ, CSL Behring, Genentech/Roche, Kedrion, Novo Nordisk, and Shire.
SOURCE: Young G et al. ASH 2017 Abstract 85.
ATLANTA – The bispecific humanized monoclonal antibody emicizumab safely prevents or substantially reduces bleeds in children with hemophilia A with inhibitors that render standard factor VIII replacement therapies ineffective, according to an updated interim analysis of data from the open-label phase 3 HAVEN 2 study.
Guy Young, MD, said during a press briefing at the annual meeting of the American Society of Hematology.
Of 57 patients under age 12 years receiving weekly subcutaneous emicizumab (Hemlibra) for bleeding prophylaxis at the data cutoff, 54 (94.7%) had zero treated bleeds, said Dr. Young of the University of Southern California, Los Angeles.
“That’s, honestly, truly remarkable. I mean, most of the time, these patients are bleeding at least on a monthly basis, sometimes more often than that,” he said.
Further, of 23 patients who were treated for at least 12 weeks and had a median efficacy period of 38.1 weeks, 87% had no bleeding events whatsoever (annual bleed rate of 0.2 events vs. a usual annual bleed rate of 1.3 events in patients without inhibitors). The annual treated joint bleed and treated target joint bleed rates each were 0.1, he said.
Overall, only three treated bleeds occurred in the 57 patients, and only one was a spontaneous bleed; one occurred in a joint, one occurred in a muscle, and one occurred in the hip and was classified as “other,” he noted.
The findings expand upon those from a previously reported interim analysis of data on 20 children from the study, which also showed that once-weekly subcutaneous emicizumab prophylaxis prevented or reduced bleeds, and provided clinically meaningful reductions in the annualized bleed rate when compared with prior bypassing agent treatment, Dr Young said.
In that prior analysis of participants aged 2-12 years (or up to 17 years with weight below 40 kg), which was presented in July at the annual meeting of the International Society on Thrombosis and Haemostasis, the data cutoff was Oct. 28, 2016. The current analysis had a May 8, 2017 cutoff, and therefore included about 6 additional months of data.
The treatment, which is given at a dose of 3 mg/kg per week for 4 weeks and then 1.5 mg/kg per week thereafter, was well tolerated. Ten nonserious, treatment-related adverse events occurred, and mainly involved injection site reactions. Serious adverse events occurred in six patients but were mainly related to the disease or to the use of catheters, or were otherwise unrelated to emicizumab. There were no thromboembolic or thrombotic microangiopathy events reported. Mean steady state trough concentrations (approximately 50 mcg/mL) were consistent with the pharmacokinetic profile seen in prior studies in the adolescent/adult population.
Health-related quality of life improvements were considerable, and in many cases, dramatic, Dr. Young said, noting in an interview that, for some patients, the effects were life changing both for the patients and their families. He described one young patient who had experienced scores of debilitating ankle bleeds, but had had no bleeds since he began receiving treatment as part of this study.
Emicizumab, which recently received Food and Drug Administration approval for use in patients of all ages with hemophilia A with inhibitors, bridges factor IXa and factor X to “bring them into proper alignment, which is really what the function of factor VIII is,” he said.
“So, it’s essentially replacing, or mimicking, the function of factor VIII, but without being factor VIII,” he said, explaining that it can be used to treat patients with inhibitors, because the antibodies they develop are specifically to factor VIII.
“This is not factor VIII, it’s just doing the job of factor VIII,” he added.
Studies of emicizumab for hemophilia A without inhibitors are ongoing (HAVEN 3), and could result in expanded indications, he noted.
The primary efficacy results of HAVEN 2 will be reported 52 weeks after the last patient is enrolled. These interim findings, however, demonstrate the potential for weekly subcutaneous injections of emicizumab to safely and conveniently reduce overall disease burden, as well as overall treatment burden in patients with hemophilia A with inhibitors, as these patients previously had to undergo intravenous treatment multiple times per week – and sometimes multiple times per day, he said.
The HAVEN 2 study is sponsored by Hoffman-La Roche. Dr. Young has received honoraria and/or consulting fees from Alnylam, Bayer, Bioverativ, CSL Behring, Genentech/Roche, Kedrion, Novo Nordisk, and Shire.
SOURCE: Young G et al. ASH 2017 Abstract 85.
REPORTING FROM ASH 2017
Key clinical point: Emicizumab prevents/substantially reduces bleeds in children with hemophilia A with inhibitors
Major finding: 94.7% of 57 patients had zero bleeds.
Study details: An interim analysis of data from 57 patients in the open-label phase 3 HAVEN 2 study.
Disclosures: The HAVEN 2 study is sponsored by Hoffman-La Roche. Dr. Young has received honoraria and/or consulting fees from Alnylam, Bayer, Bioverativ, CSL Behring, Genentech/Roche, Kedrion, Novo Nordisk, and Shire.
Source: Young G et al. ASH 2017 Abstract 85.
Polymerization inhibitor improves hemoglobin levels in adolescents with SCD
ATLANTA – Voxelotor, an investigational inhibitor of hemoglobin polymerization, demonstrated consistent and sustained improvements in hemoglobin levels in adolescents with sickle cell disease (SCD), according to first results of a phase 2b study.
A hemoglobin response of at least 1 g/dL was achieved in 55% of patients (6 of 11) at 16 weeks, in results of the ongoing open-label, 24-week study presented at the annual meeting of the American Society of Hematology.

The treatment was well tolerated, associated with mostly grade 1-2 adverse events, and also improved patient-reported symptoms, according to Carolyn C. Hoppe, MD, of University of California, San Francisco, Benioff Children’s Hospital.
at a press conference. “We need therapies to treat this disease and prevent the complications.”
Dr. Hoppe presented data on the first 12 patients in a study designed to assess the safety, pharmacokinetics, and efficacy of voxelotor in adolescent patients with homozygous hemoglobin SS or hemoglobin S beta-0 thalassemia.
All patients except one were already receiving standard care with hydroxyurea, suggesting that the two drugs may provide additive effects, according to investigators.
Treatment with voxelotor was associated with improvements in hemoglobin at 16 weeks, along with improvements in clinical measures of hemolysis – a 34% median reductions in reticulocytes and a 27% reduction in indirect bilirubin, Dr. Hoppe reported.
Patient-reported symptoms improved in 10 of 12 patients, with a median reduction of 94% in total symptom scores (TSS) at week 16, including 5 subjects with a mean TSS of 0.
Voxelotor is designed to address the cause of sickle cell disease, according to Dr. Hoppe, by modulating hemoglobin’s affinity for oxygen, which in turn inhibits hemoglobin polymerization.
“If we can interrupt that early on, we can hopefully disrupt or interrupt the biologic pathways that lead to the consequences of the disease,” she said.
The study is ongoing, as is a phase 3 clinical trial evaluating voxelotor in patients aged 12 years and older with SCD.
Voxelotor was previously known as GBT440
The study was sponsored by Global Blood Therapeutics. Dr. Hoppe reported research funding from and advisory relationships with Global Blood Therapeutics. Several coauthors are employees of the company.
SOURCE: Hoppe C et al. ASH 2017. Abstract 689
ATLANTA – Voxelotor, an investigational inhibitor of hemoglobin polymerization, demonstrated consistent and sustained improvements in hemoglobin levels in adolescents with sickle cell disease (SCD), according to first results of a phase 2b study.
A hemoglobin response of at least 1 g/dL was achieved in 55% of patients (6 of 11) at 16 weeks, in results of the ongoing open-label, 24-week study presented at the annual meeting of the American Society of Hematology.
The treatment was well tolerated, associated with mostly grade 1-2 adverse events, and also improved patient-reported symptoms, according to Carolyn C. Hoppe, MD, of University of California, San Francisco, Benioff Children’s Hospital.
at a press conference. “We need therapies to treat this disease and prevent the complications.”
Dr. Hoppe presented data on the first 12 patients in a study designed to assess the safety, pharmacokinetics, and efficacy of voxelotor in adolescent patients with homozygous hemoglobin SS or hemoglobin S beta-0 thalassemia.
All patients except one were already receiving standard care with hydroxyurea, suggesting that the two drugs may provide additive effects, according to investigators.
Treatment with voxelotor was associated with improvements in hemoglobin at 16 weeks, along with improvements in clinical measures of hemolysis – a 34% median reductions in reticulocytes and a 27% reduction in indirect bilirubin, Dr. Hoppe reported.
Patient-reported symptoms improved in 10 of 12 patients, with a median reduction of 94% in total symptom scores (TSS) at week 16, including 5 subjects with a mean TSS of 0.
Voxelotor is designed to address the cause of sickle cell disease, according to Dr. Hoppe, by modulating hemoglobin’s affinity for oxygen, which in turn inhibits hemoglobin polymerization.
“If we can interrupt that early on, we can hopefully disrupt or interrupt the biologic pathways that lead to the consequences of the disease,” she said.
The study is ongoing, as is a phase 3 clinical trial evaluating voxelotor in patients aged 12 years and older with SCD.
Voxelotor was previously known as GBT440
The study was sponsored by Global Blood Therapeutics. Dr. Hoppe reported research funding from and advisory relationships with Global Blood Therapeutics. Several coauthors are employees of the company.
SOURCE: Hoppe C et al. ASH 2017. Abstract 689
ATLANTA – Voxelotor, an investigational inhibitor of hemoglobin polymerization, demonstrated consistent and sustained improvements in hemoglobin levels in adolescents with sickle cell disease (SCD), according to first results of a phase 2b study.
A hemoglobin response of at least 1 g/dL was achieved in 55% of patients (6 of 11) at 16 weeks, in results of the ongoing open-label, 24-week study presented at the annual meeting of the American Society of Hematology.
The treatment was well tolerated, associated with mostly grade 1-2 adverse events, and also improved patient-reported symptoms, according to Carolyn C. Hoppe, MD, of University of California, San Francisco, Benioff Children’s Hospital.
at a press conference. “We need therapies to treat this disease and prevent the complications.”
Dr. Hoppe presented data on the first 12 patients in a study designed to assess the safety, pharmacokinetics, and efficacy of voxelotor in adolescent patients with homozygous hemoglobin SS or hemoglobin S beta-0 thalassemia.
All patients except one were already receiving standard care with hydroxyurea, suggesting that the two drugs may provide additive effects, according to investigators.
Treatment with voxelotor was associated with improvements in hemoglobin at 16 weeks, along with improvements in clinical measures of hemolysis – a 34% median reductions in reticulocytes and a 27% reduction in indirect bilirubin, Dr. Hoppe reported.
Patient-reported symptoms improved in 10 of 12 patients, with a median reduction of 94% in total symptom scores (TSS) at week 16, including 5 subjects with a mean TSS of 0.
Voxelotor is designed to address the cause of sickle cell disease, according to Dr. Hoppe, by modulating hemoglobin’s affinity for oxygen, which in turn inhibits hemoglobin polymerization.
“If we can interrupt that early on, we can hopefully disrupt or interrupt the biologic pathways that lead to the consequences of the disease,” she said.
The study is ongoing, as is a phase 3 clinical trial evaluating voxelotor in patients aged 12 years and older with SCD.
Voxelotor was previously known as GBT440
The study was sponsored by Global Blood Therapeutics. Dr. Hoppe reported research funding from and advisory relationships with Global Blood Therapeutics. Several coauthors are employees of the company.
SOURCE: Hoppe C et al. ASH 2017. Abstract 689
REPORTING FROM ASH 2017
Key clinical point: Voxelotor was well tolerated and demonstrated consistent and sustained improvements in hemoglobin levels in adolescents with sickle cell disease (SCD).
Major finding: A hemoglobin response of at least 1 g/dL was achieved in 55% of subjects at 16 weeks.
Data source: A phase 2a open-label, interventional study of 12 adolescent patients with sickle cell disease.
Disclosures: The study was sponsored by Global Blood Therapeutics. Dr. Hoppe reported research funding from and advisory relationships with Global Blood Therapeutics. Several coauthors are employees of the company.
Source: Hoppe C et al. ASH 2017. Abstract #689
Expert discusses the role of salt and fructose in diabetes
LOS ANGELES – Sugar consumption has been implicated as a risk factor for the development of diabetes since at least the 1920s, but high salt intake may also increase the risk for obesity and prediabetes by stimulating fructose production in the liver.
“We think about high-salt diets as being associated with hypertension, but if you put people on a high-salt diet, you can induce insulin resistance within 5 or 10 days,” Richard J. Johnson, MD, said at the World Congress on Insulin Resistance, Diabetes & Cardiovascular Disease. “There is a fair amount of published data suggesting that a high salt intake, defined as greater than 150 mmol per day, is associated with hypertension, insulin resistance, obesity, and metabolic syndrome. In fact, it’s been reported that obese people are slightly hyperosmolar and tend to have elevated vasopressin levels. Interestingly, sugar stimulates vasopressin production.”

Furthermore, the mice on high-salt diets became insulin resistant. “Their fasting glucose went up, their fasting insulin went up, and they developed marked fatty liver, obesity, abdominal fat, and hypertension,” Dr. Johnson added. The discovery supports the notion that osmolality is the mechanism by which salt drives blood pressure, not volume expansion. “And I think that’s going to turn out to be important in obesity and metabolic syndrome as well,” he said.
Dr. Johnson, a professor of medicine in the department of renal diseases and hypertension at the University of Colorado at Denver, Aurora, discussed the role of role of sugar and fructose in the development of diabetes as well. He noted that annual sugar consumption in the United States rose from about 4 pounds per person in 1700 to about 150 pounds per person today. About one-third of current sugar intake comes from soft drinks.
“The reason why we think that fructose is a good candidate for playing a role in the diabetes epidemic is because, when you give an animal water drinking combined with fructose, they will rapidly start increasing their energy intake,” he said. “They become lethargic and less active, and they gain weight.”
He and his colleagues have demonstrated that when rats are fed fructose over time, they become leptin resistant (Am J Physiol. 2008 Nov;295:R1370-5). “Not only that, it’s been shown in animals and humans that, if you feed people fructose over time, fructose will decrease resting energy expenditure,” he said. “Our work and that of others has shown that fructose stimulates weight gain by stimulating energy intake. It does so by inducing leptin resistance. It also works in the brain to stimulate dopamine and to drive food intake that way as well.”
Fructose also impairs fatty acid oxidation and reduces energy expenditure, he continued. In one human trial, in which men consumed 200g of fructose for 2 weeks, Dr. Johnson and his associates found that 25% of them developed features of metabolic syndrome (Int J Obes. 2010;34[3]:454-61). “Just think about how long it takes to get obese or metabolic syndrome,” he said. “We think in terms of years, but this was a 2-week study! This means there’s something special about fructose that seems to drive metabolic syndrome.”
A key player in the process appears to be an enzyme in the liver known as fructokinase, which metabolizes fructose so rapidly that it causes ATP depletion. “Normally, when glucose is metabolized, ATP levels stay normal in the cell because if you start to consume too much ATP in the initial phosphorylation, there’s a feedback mechanism,” Dr. Johnson said. “But not so for fructose; it’s a runaway train, and it activates a nucleotide degradation pathway, which we call the energy depletion pathway. This seems to be what is critical for fructose effects.”
Further evaluation of that pathway led him and his associates to discover that lowering uric acid reduced fatty liver formation in fructose-fed rats (PLOS One 2012 Oct. 24. doi:10.1371/journal.pone.0047948). “We started looking at how this worked, and we found that, when we put uric acid on liver cells, that they actually stimulated fat accumulation,” he said. “We showed that in an in vitro system, and we found that both fructose and uric acid stimulate oxidative stress in the mitochondria. It’s very specific. You can actually block the production of oxidative stress with allopurinol, a drug that lowers uric acid. We’ve been building a case that this pathway is involved in a lot of mechanisms that lead to obesity, insulin resistance, and hypertension.”
Dr. Johnson concluded by noting that not all calories are created equal. “Some additives, like salt, might be playing a role in metabolic syndrome, obesity, and diabetes,” he said. “We think that sugar and high fructose corn sugar are the major causes driving metabolic syndrome. High-glycemic carbs are working primarily through fructose to induce [insulin resistance]. We think that salt may accelerate this pathway as well.”
He disclosed that he holds patents and patent applications related to this work and that he has launched a start-up company trying to develop inhibitors of fructose metabolism.
Dr. Johnson reported having no conflicts of interest related to this article.
LOS ANGELES – Sugar consumption has been implicated as a risk factor for the development of diabetes since at least the 1920s, but high salt intake may also increase the risk for obesity and prediabetes by stimulating fructose production in the liver.
“We think about high-salt diets as being associated with hypertension, but if you put people on a high-salt diet, you can induce insulin resistance within 5 or 10 days,” Richard J. Johnson, MD, said at the World Congress on Insulin Resistance, Diabetes & Cardiovascular Disease. “There is a fair amount of published data suggesting that a high salt intake, defined as greater than 150 mmol per day, is associated with hypertension, insulin resistance, obesity, and metabolic syndrome. In fact, it’s been reported that obese people are slightly hyperosmolar and tend to have elevated vasopressin levels. Interestingly, sugar stimulates vasopressin production.”
Furthermore, the mice on high-salt diets became insulin resistant. “Their fasting glucose went up, their fasting insulin went up, and they developed marked fatty liver, obesity, abdominal fat, and hypertension,” Dr. Johnson added. The discovery supports the notion that osmolality is the mechanism by which salt drives blood pressure, not volume expansion. “And I think that’s going to turn out to be important in obesity and metabolic syndrome as well,” he said.
Dr. Johnson, a professor of medicine in the department of renal diseases and hypertension at the University of Colorado at Denver, Aurora, discussed the role of role of sugar and fructose in the development of diabetes as well. He noted that annual sugar consumption in the United States rose from about 4 pounds per person in 1700 to about 150 pounds per person today. About one-third of current sugar intake comes from soft drinks.
“The reason why we think that fructose is a good candidate for playing a role in the diabetes epidemic is because, when you give an animal water drinking combined with fructose, they will rapidly start increasing their energy intake,” he said. “They become lethargic and less active, and they gain weight.”
He and his colleagues have demonstrated that when rats are fed fructose over time, they become leptin resistant (Am J Physiol. 2008 Nov;295:R1370-5). “Not only that, it’s been shown in animals and humans that, if you feed people fructose over time, fructose will decrease resting energy expenditure,” he said. “Our work and that of others has shown that fructose stimulates weight gain by stimulating energy intake. It does so by inducing leptin resistance. It also works in the brain to stimulate dopamine and to drive food intake that way as well.”
Fructose also impairs fatty acid oxidation and reduces energy expenditure, he continued. In one human trial, in which men consumed 200g of fructose for 2 weeks, Dr. Johnson and his associates found that 25% of them developed features of metabolic syndrome (Int J Obes. 2010;34[3]:454-61). “Just think about how long it takes to get obese or metabolic syndrome,” he said. “We think in terms of years, but this was a 2-week study! This means there’s something special about fructose that seems to drive metabolic syndrome.”
A key player in the process appears to be an enzyme in the liver known as fructokinase, which metabolizes fructose so rapidly that it causes ATP depletion. “Normally, when glucose is metabolized, ATP levels stay normal in the cell because if you start to consume too much ATP in the initial phosphorylation, there’s a feedback mechanism,” Dr. Johnson said. “But not so for fructose; it’s a runaway train, and it activates a nucleotide degradation pathway, which we call the energy depletion pathway. This seems to be what is critical for fructose effects.”
Further evaluation of that pathway led him and his associates to discover that lowering uric acid reduced fatty liver formation in fructose-fed rats (PLOS One 2012 Oct. 24. doi:10.1371/journal.pone.0047948). “We started looking at how this worked, and we found that, when we put uric acid on liver cells, that they actually stimulated fat accumulation,” he said. “We showed that in an in vitro system, and we found that both fructose and uric acid stimulate oxidative stress in the mitochondria. It’s very specific. You can actually block the production of oxidative stress with allopurinol, a drug that lowers uric acid. We’ve been building a case that this pathway is involved in a lot of mechanisms that lead to obesity, insulin resistance, and hypertension.”
Dr. Johnson concluded by noting that not all calories are created equal. “Some additives, like salt, might be playing a role in metabolic syndrome, obesity, and diabetes,” he said. “We think that sugar and high fructose corn sugar are the major causes driving metabolic syndrome. High-glycemic carbs are working primarily through fructose to induce [insulin resistance]. We think that salt may accelerate this pathway as well.”
He disclosed that he holds patents and patent applications related to this work and that he has launched a start-up company trying to develop inhibitors of fructose metabolism.
Dr. Johnson reported having no conflicts of interest related to this article.
LOS ANGELES – Sugar consumption has been implicated as a risk factor for the development of diabetes since at least the 1920s, but high salt intake may also increase the risk for obesity and prediabetes by stimulating fructose production in the liver.
“We think about high-salt diets as being associated with hypertension, but if you put people on a high-salt diet, you can induce insulin resistance within 5 or 10 days,” Richard J. Johnson, MD, said at the World Congress on Insulin Resistance, Diabetes & Cardiovascular Disease. “There is a fair amount of published data suggesting that a high salt intake, defined as greater than 150 mmol per day, is associated with hypertension, insulin resistance, obesity, and metabolic syndrome. In fact, it’s been reported that obese people are slightly hyperosmolar and tend to have elevated vasopressin levels. Interestingly, sugar stimulates vasopressin production.”
Furthermore, the mice on high-salt diets became insulin resistant. “Their fasting glucose went up, their fasting insulin went up, and they developed marked fatty liver, obesity, abdominal fat, and hypertension,” Dr. Johnson added. The discovery supports the notion that osmolality is the mechanism by which salt drives blood pressure, not volume expansion. “And I think that’s going to turn out to be important in obesity and metabolic syndrome as well,” he said.
Dr. Johnson, a professor of medicine in the department of renal diseases and hypertension at the University of Colorado at Denver, Aurora, discussed the role of role of sugar and fructose in the development of diabetes as well. He noted that annual sugar consumption in the United States rose from about 4 pounds per person in 1700 to about 150 pounds per person today. About one-third of current sugar intake comes from soft drinks.
“The reason why we think that fructose is a good candidate for playing a role in the diabetes epidemic is because, when you give an animal water drinking combined with fructose, they will rapidly start increasing their energy intake,” he said. “They become lethargic and less active, and they gain weight.”
He and his colleagues have demonstrated that when rats are fed fructose over time, they become leptin resistant (Am J Physiol. 2008 Nov;295:R1370-5). “Not only that, it’s been shown in animals and humans that, if you feed people fructose over time, fructose will decrease resting energy expenditure,” he said. “Our work and that of others has shown that fructose stimulates weight gain by stimulating energy intake. It does so by inducing leptin resistance. It also works in the brain to stimulate dopamine and to drive food intake that way as well.”
Fructose also impairs fatty acid oxidation and reduces energy expenditure, he continued. In one human trial, in which men consumed 200g of fructose for 2 weeks, Dr. Johnson and his associates found that 25% of them developed features of metabolic syndrome (Int J Obes. 2010;34[3]:454-61). “Just think about how long it takes to get obese or metabolic syndrome,” he said. “We think in terms of years, but this was a 2-week study! This means there’s something special about fructose that seems to drive metabolic syndrome.”
A key player in the process appears to be an enzyme in the liver known as fructokinase, which metabolizes fructose so rapidly that it causes ATP depletion. “Normally, when glucose is metabolized, ATP levels stay normal in the cell because if you start to consume too much ATP in the initial phosphorylation, there’s a feedback mechanism,” Dr. Johnson said. “But not so for fructose; it’s a runaway train, and it activates a nucleotide degradation pathway, which we call the energy depletion pathway. This seems to be what is critical for fructose effects.”
Further evaluation of that pathway led him and his associates to discover that lowering uric acid reduced fatty liver formation in fructose-fed rats (PLOS One 2012 Oct. 24. doi:10.1371/journal.pone.0047948). “We started looking at how this worked, and we found that, when we put uric acid on liver cells, that they actually stimulated fat accumulation,” he said. “We showed that in an in vitro system, and we found that both fructose and uric acid stimulate oxidative stress in the mitochondria. It’s very specific. You can actually block the production of oxidative stress with allopurinol, a drug that lowers uric acid. We’ve been building a case that this pathway is involved in a lot of mechanisms that lead to obesity, insulin resistance, and hypertension.”
Dr. Johnson concluded by noting that not all calories are created equal. “Some additives, like salt, might be playing a role in metabolic syndrome, obesity, and diabetes,” he said. “We think that sugar and high fructose corn sugar are the major causes driving metabolic syndrome. High-glycemic carbs are working primarily through fructose to induce [insulin resistance]. We think that salt may accelerate this pathway as well.”
He disclosed that he holds patents and patent applications related to this work and that he has launched a start-up company trying to develop inhibitors of fructose metabolism.
Dr. Johnson reported having no conflicts of interest related to this article.
EXPERT ANALYSIS AT WCIRDC 2017
Nearly all rheumatology slots filled on Specialty Match Day
Once again, rheumatology filled almost every slot offered in the annual Specialty Match.
The specialty had 221 open positions to be filled, with only 3 left open when the 2017 results were announced on Dec. 6.
The number of available positions was up from the 217 slots in 2016, with 7 positions unfilled last year. The number of applicants naming rheumatology as the preferred choice was virtually unchanged year over year, with 313 this year vs. 312 last year.
Of the 313 applicants who identified rheumatology as the preferred choice, 217 were matched to rheumatology, 3 were matched to a different specialty, and 93 did not match.
“Rheumatology did very, very well this year, even better than last year, although it was comparable,” Anne Bass, MD, rheumatology fellowship program director at the Hospital for Special Surgery, New York, said in an interview.
Dr. Bass, who also serves as chair of the American College of Rheumatology Committee on Rheumatology and Training and Workforce Issues, said she expects the remaining open slots to be filled.
“Usually what happens is something called the scramble, which is after the match is over, [the National Resident Matching Program] has a clearinghouse for fellows who haven’t matched and programs that still have slots,” she said in an interview. “It connects them together. I would guess the positions will either be filled or that they are positions that are really targeted, say, for research applicants and there wasn’t anybody appropriate for that position.”
“You do want some excess because not all applicants are necessarily qualified, so you don’t want to have just the right amount of spots if you want to have the best people filling them,” she said, noting that this year, like last, there was around 100 applicants who listed rheumatology as their first choice that did not get assigned as fellowship, about double the number from a few years ago. “At the same time, with the anticipated workforce shortage, what this is telling us is if we created more fellowship slots, we have lots and lots of applicants to fill those slots.”
Dr. Bass said funding issues were the primary stumbling block to training more fellows. She also said more needs to be done to address geographic disparities, as these programs tend to be at major academic institutions in dense urban areas and many fellows practice at sights where they are trained, magnifying shortages in rural areas.
The similarities between last year’s rheumatology fellowship match results and this year’s are in line with what is happening in other specialties.
“Overall, the percentages of programs and positions filled were almost identical to last year,” Mona Singer, president and CEO of the National Resident Matching Program, said. “The same fellowships were competitive this year when compared to last year.”
Specialties filling more than 90% of positions included cardiovascular disease, endocrinology, gastroenterology, hematology/oncology, pulmonary/critical care, and rheumatology.
Ms. Singer noted that infectious diseases and nephrology “have implemented the ‘all-in policy,’ which requires any program participating in the match to place all positions. This was the first year infectious diseases used the policy, and there was a modest improvement in the number of positions filled. Nephrology was unchanged.”
Overall, 4,831 fellowships were available and 4,242 filled. The 87.8% of positions filled is a slight uptick over the 87.5% of positions filled in 2016. A total of 1,249 applicants did not match.
Once again, rheumatology filled almost every slot offered in the annual Specialty Match.
The specialty had 221 open positions to be filled, with only 3 left open when the 2017 results were announced on Dec. 6.
The number of available positions was up from the 217 slots in 2016, with 7 positions unfilled last year. The number of applicants naming rheumatology as the preferred choice was virtually unchanged year over year, with 313 this year vs. 312 last year.
Of the 313 applicants who identified rheumatology as the preferred choice, 217 were matched to rheumatology, 3 were matched to a different specialty, and 93 did not match.
“Rheumatology did very, very well this year, even better than last year, although it was comparable,” Anne Bass, MD, rheumatology fellowship program director at the Hospital for Special Surgery, New York, said in an interview.
Dr. Bass, who also serves as chair of the American College of Rheumatology Committee on Rheumatology and Training and Workforce Issues, said she expects the remaining open slots to be filled.
“Usually what happens is something called the scramble, which is after the match is over, [the National Resident Matching Program] has a clearinghouse for fellows who haven’t matched and programs that still have slots,” she said in an interview. “It connects them together. I would guess the positions will either be filled or that they are positions that are really targeted, say, for research applicants and there wasn’t anybody appropriate for that position.”
“You do want some excess because not all applicants are necessarily qualified, so you don’t want to have just the right amount of spots if you want to have the best people filling them,” she said, noting that this year, like last, there was around 100 applicants who listed rheumatology as their first choice that did not get assigned as fellowship, about double the number from a few years ago. “At the same time, with the anticipated workforce shortage, what this is telling us is if we created more fellowship slots, we have lots and lots of applicants to fill those slots.”
Dr. Bass said funding issues were the primary stumbling block to training more fellows. She also said more needs to be done to address geographic disparities, as these programs tend to be at major academic institutions in dense urban areas and many fellows practice at sights where they are trained, magnifying shortages in rural areas.
The similarities between last year’s rheumatology fellowship match results and this year’s are in line with what is happening in other specialties.
“Overall, the percentages of programs and positions filled were almost identical to last year,” Mona Singer, president and CEO of the National Resident Matching Program, said. “The same fellowships were competitive this year when compared to last year.”
Specialties filling more than 90% of positions included cardiovascular disease, endocrinology, gastroenterology, hematology/oncology, pulmonary/critical care, and rheumatology.
Ms. Singer noted that infectious diseases and nephrology “have implemented the ‘all-in policy,’ which requires any program participating in the match to place all positions. This was the first year infectious diseases used the policy, and there was a modest improvement in the number of positions filled. Nephrology was unchanged.”
Overall, 4,831 fellowships were available and 4,242 filled. The 87.8% of positions filled is a slight uptick over the 87.5% of positions filled in 2016. A total of 1,249 applicants did not match.
Once again, rheumatology filled almost every slot offered in the annual Specialty Match.
The specialty had 221 open positions to be filled, with only 3 left open when the 2017 results were announced on Dec. 6.
The number of available positions was up from the 217 slots in 2016, with 7 positions unfilled last year. The number of applicants naming rheumatology as the preferred choice was virtually unchanged year over year, with 313 this year vs. 312 last year.
Of the 313 applicants who identified rheumatology as the preferred choice, 217 were matched to rheumatology, 3 were matched to a different specialty, and 93 did not match.
“Rheumatology did very, very well this year, even better than last year, although it was comparable,” Anne Bass, MD, rheumatology fellowship program director at the Hospital for Special Surgery, New York, said in an interview.
Dr. Bass, who also serves as chair of the American College of Rheumatology Committee on Rheumatology and Training and Workforce Issues, said she expects the remaining open slots to be filled.
“Usually what happens is something called the scramble, which is after the match is over, [the National Resident Matching Program] has a clearinghouse for fellows who haven’t matched and programs that still have slots,” she said in an interview. “It connects them together. I would guess the positions will either be filled or that they are positions that are really targeted, say, for research applicants and there wasn’t anybody appropriate for that position.”
“You do want some excess because not all applicants are necessarily qualified, so you don’t want to have just the right amount of spots if you want to have the best people filling them,” she said, noting that this year, like last, there was around 100 applicants who listed rheumatology as their first choice that did not get assigned as fellowship, about double the number from a few years ago. “At the same time, with the anticipated workforce shortage, what this is telling us is if we created more fellowship slots, we have lots and lots of applicants to fill those slots.”
Dr. Bass said funding issues were the primary stumbling block to training more fellows. She also said more needs to be done to address geographic disparities, as these programs tend to be at major academic institutions in dense urban areas and many fellows practice at sights where they are trained, magnifying shortages in rural areas.
The similarities between last year’s rheumatology fellowship match results and this year’s are in line with what is happening in other specialties.
“Overall, the percentages of programs and positions filled were almost identical to last year,” Mona Singer, president and CEO of the National Resident Matching Program, said. “The same fellowships were competitive this year when compared to last year.”
Specialties filling more than 90% of positions included cardiovascular disease, endocrinology, gastroenterology, hematology/oncology, pulmonary/critical care, and rheumatology.
Ms. Singer noted that infectious diseases and nephrology “have implemented the ‘all-in policy,’ which requires any program participating in the match to place all positions. This was the first year infectious diseases used the policy, and there was a modest improvement in the number of positions filled. Nephrology was unchanged.”
Overall, 4,831 fellowships were available and 4,242 filled. The 87.8% of positions filled is a slight uptick over the 87.5% of positions filled in 2016. A total of 1,249 applicants did not match.
Sickle cell: Customized stem cells may improve transfusion efficacy, safety
ATLANTA – Matching red cell products to sickle-cell disease (SCD) patients with rare Rh antibodies can be accomplished with a little fancy stem cell footwork and some genetic legerdemain, investigators report.
to produce a renewable source of red cell reagents and potentially universal infusion products for patients with SCD.
“It has been more than 10 years since iPSCs have been available, but we have yet to see applications for blood diseases that improve how we care for our patients,” Dr. Chou said in a statement. “Using this panel, we would be able to more quickly identify what antibodies the patient has made, informing us what kind of blood we have to give them. It means we’ll be able to improve their ability to be transfused safely and reduce delays in their care.”
Up to half of all patients with SCD who require chronic transfusions may develop antibodies to allogeneic blood products, but identifying inactivating antibodies in a given patient can be both challenging and time consuming, due to a paucity of the appropriate reagent red cells, Dr. Chou and her colleagues reported.
The investigators previously reported that despite receiving transfusions from Rh-matched minority donors, patients with SCD have a high prevalence of red blood cell alloimmunization, and that Rh antibodies are the most common type of antibody in SCD patients, with about 33% of Rh antibodies associated with delayed transfusion reactions (Blood. 2013 Aug 8;122:1062-71; doi: 10.1182/blood-2013-03-490623).
It is both costly and time consuming to genotype donated blood for Rh antigens in order to match a low-antigen product to a specific patient. Instead, the investigators identified a workaround involving reprogramming or genetically engineering iPSCs from donors with rare antigen profiles, with the goal of creating a standard panel of red cell reagents that could reliably and quickly identify SCD patients with complex antibodies.
They used CRISPR/Cas9 gene-editing techniques to modify existing iPSCs to include Rh null cells, other cells lacking all or part of certain high-prevalence Rh antigens, and low prevalence novel antigens.
The investigators then showed that they could induce hematopoietic differentiation of their customized iPSCs through a three-step process and demonstrated that, as they had expected, the engineered antibodies were able to more accurately type Rh in gel card assays than did off-the-shelf commercial assays.
“We have designed a panel of customized iPSCs reprogrammed from rare donors or genetically engineered to express rare blood group antigen phenotypes or combinations that are difficult or impossible to find as donor red cells. Any number of combinations not found in natural populations can be produced and generated in quantities sufficient for reagents,” Dr. Chou said.
They also asserted that iPSC-derived red blood cells (iRBCs) produced from their customized iPSCs could be used with standard blood bank assays as a potential means of streamlining and standardizing the antibody identification process in alloimmunized patients with complex antibody specificities.
“In the future, when technology for scale-up is available, Rh null iRBCs could be used as ‘universal’ donor cells for future therapeutic applications,” the reseachers wrote.
The study was supported by the National Institutes of Health/National Heart Lung and Blood Institute. The authors reported having no conflicts of interest.
SOURCE: Chou S et al. ASH 2017 Abstract 3
ATLANTA – Matching red cell products to sickle-cell disease (SCD) patients with rare Rh antibodies can be accomplished with a little fancy stem cell footwork and some genetic legerdemain, investigators report.
to produce a renewable source of red cell reagents and potentially universal infusion products for patients with SCD.
“It has been more than 10 years since iPSCs have been available, but we have yet to see applications for blood diseases that improve how we care for our patients,” Dr. Chou said in a statement. “Using this panel, we would be able to more quickly identify what antibodies the patient has made, informing us what kind of blood we have to give them. It means we’ll be able to improve their ability to be transfused safely and reduce delays in their care.”
Up to half of all patients with SCD who require chronic transfusions may develop antibodies to allogeneic blood products, but identifying inactivating antibodies in a given patient can be both challenging and time consuming, due to a paucity of the appropriate reagent red cells, Dr. Chou and her colleagues reported.
The investigators previously reported that despite receiving transfusions from Rh-matched minority donors, patients with SCD have a high prevalence of red blood cell alloimmunization, and that Rh antibodies are the most common type of antibody in SCD patients, with about 33% of Rh antibodies associated with delayed transfusion reactions (Blood. 2013 Aug 8;122:1062-71; doi: 10.1182/blood-2013-03-490623).
It is both costly and time consuming to genotype donated blood for Rh antigens in order to match a low-antigen product to a specific patient. Instead, the investigators identified a workaround involving reprogramming or genetically engineering iPSCs from donors with rare antigen profiles, with the goal of creating a standard panel of red cell reagents that could reliably and quickly identify SCD patients with complex antibodies.
They used CRISPR/Cas9 gene-editing techniques to modify existing iPSCs to include Rh null cells, other cells lacking all or part of certain high-prevalence Rh antigens, and low prevalence novel antigens.
The investigators then showed that they could induce hematopoietic differentiation of their customized iPSCs through a three-step process and demonstrated that, as they had expected, the engineered antibodies were able to more accurately type Rh in gel card assays than did off-the-shelf commercial assays.
“We have designed a panel of customized iPSCs reprogrammed from rare donors or genetically engineered to express rare blood group antigen phenotypes or combinations that are difficult or impossible to find as donor red cells. Any number of combinations not found in natural populations can be produced and generated in quantities sufficient for reagents,” Dr. Chou said.
They also asserted that iPSC-derived red blood cells (iRBCs) produced from their customized iPSCs could be used with standard blood bank assays as a potential means of streamlining and standardizing the antibody identification process in alloimmunized patients with complex antibody specificities.
“In the future, when technology for scale-up is available, Rh null iRBCs could be used as ‘universal’ donor cells for future therapeutic applications,” the reseachers wrote.
The study was supported by the National Institutes of Health/National Heart Lung and Blood Institute. The authors reported having no conflicts of interest.
SOURCE: Chou S et al. ASH 2017 Abstract 3
ATLANTA – Matching red cell products to sickle-cell disease (SCD) patients with rare Rh antibodies can be accomplished with a little fancy stem cell footwork and some genetic legerdemain, investigators report.
to produce a renewable source of red cell reagents and potentially universal infusion products for patients with SCD.
“It has been more than 10 years since iPSCs have been available, but we have yet to see applications for blood diseases that improve how we care for our patients,” Dr. Chou said in a statement. “Using this panel, we would be able to more quickly identify what antibodies the patient has made, informing us what kind of blood we have to give them. It means we’ll be able to improve their ability to be transfused safely and reduce delays in their care.”
Up to half of all patients with SCD who require chronic transfusions may develop antibodies to allogeneic blood products, but identifying inactivating antibodies in a given patient can be both challenging and time consuming, due to a paucity of the appropriate reagent red cells, Dr. Chou and her colleagues reported.
The investigators previously reported that despite receiving transfusions from Rh-matched minority donors, patients with SCD have a high prevalence of red blood cell alloimmunization, and that Rh antibodies are the most common type of antibody in SCD patients, with about 33% of Rh antibodies associated with delayed transfusion reactions (Blood. 2013 Aug 8;122:1062-71; doi: 10.1182/blood-2013-03-490623).
It is both costly and time consuming to genotype donated blood for Rh antigens in order to match a low-antigen product to a specific patient. Instead, the investigators identified a workaround involving reprogramming or genetically engineering iPSCs from donors with rare antigen profiles, with the goal of creating a standard panel of red cell reagents that could reliably and quickly identify SCD patients with complex antibodies.
They used CRISPR/Cas9 gene-editing techniques to modify existing iPSCs to include Rh null cells, other cells lacking all or part of certain high-prevalence Rh antigens, and low prevalence novel antigens.
The investigators then showed that they could induce hematopoietic differentiation of their customized iPSCs through a three-step process and demonstrated that, as they had expected, the engineered antibodies were able to more accurately type Rh in gel card assays than did off-the-shelf commercial assays.
“We have designed a panel of customized iPSCs reprogrammed from rare donors or genetically engineered to express rare blood group antigen phenotypes or combinations that are difficult or impossible to find as donor red cells. Any number of combinations not found in natural populations can be produced and generated in quantities sufficient for reagents,” Dr. Chou said.
They also asserted that iPSC-derived red blood cells (iRBCs) produced from their customized iPSCs could be used with standard blood bank assays as a potential means of streamlining and standardizing the antibody identification process in alloimmunized patients with complex antibody specificities.
“In the future, when technology for scale-up is available, Rh null iRBCs could be used as ‘universal’ donor cells for future therapeutic applications,” the reseachers wrote.
The study was supported by the National Institutes of Health/National Heart Lung and Blood Institute. The authors reported having no conflicts of interest.
SOURCE: Chou S et al. ASH 2017 Abstract 3
REPORTING FROM ASH 2017
Key clinical point:. Customized induced pluripotent stem cells (iPSCs) could help to better and more rapidly match sickle-cell disease patients with appropriate transfusion products.
Major finding: Engineered iPSCs accurately typed Rh in iPSC-derived red blood cells in gel card assays.
Data source: Proof of concept in vitro study.
Disclosures: The study was supported by the National Institutes of Health/National Heart Lung and Blood Institute. The authors reported having no conflicts of interest.
Source: Chou S et al. ASH 2017 Abstract 3
Viral failure lower in dolutegravir treated HIV patients
SAN DIEGO – The proportion of viral failure was lower in people living with HIV who took dolutegravir (DTG), compared with patients on other integrase strand transfer inhibitor (INSTI) regimens, according to a study presented at ID Week 2017, an infectious diseases meeting.
Investigators studied records of 6,636 HIV patients who started regimens between August 2013 and August 2016, gathered from the Center for Aids Research (CFAR) Network of Integrated Clinical Systems. The information was collected from eight centers across the United States.
Patients were split among three ART regimen groups: dolutegravir based, other INSTI based, and darunavir (DRV) based. Among the 6,636 people living with HIV studied, those using a DTG-based regimen were less than half as likely to exhibit viral failure as were those in the DRV-based group (hazard ratio, 0.41; 95% confidence interval, 0.37-0.86), according to Heidi Crane, MD, MPH, associate director of CFAR Clinical Epidemiology and Health Services Research at the University of Washington, Seattle.
In a comparison of DTG-based therapy compared to other INSTI-based therapies, patients in the DTG groups showed significantly lower risk for viral failure in a crude hazard ratio (HR, 0.57; 95% CI, 0.46-0.70), although, when adjusted, the investigators found the odds did not hold the same level of significance, despite still being at a lower risk (adjusted HR, 0.82; 95% CI, 0.65-1.03).
In a sensitivity analysis of ART-naive patients, investigators found that patients taking DTG regimens were at nearly one-third the risk of viral failure, compared with those taking a darunavir-based regimen (adjusted HR, 0.32; 95% CI, 0.14-0.75). However, when comparing DTG with other INSTI therapies among the ART-naive population, there was no significant difference reported, Dr. Crane said in her presentation.
Investigators found this difference significant when comparing DTG with DRV and DTG with other INSTIs to be a trend across all testing models, according to Dr. Crane.
“In the various sensitivity models, the association for dolutegravir versus other integrase inhibitors ranged from 0.8 to 0.98 depending on censoring, sometimes significant and sometimes not,” said Dr. Crane. “In contrast, the hazard ratio for the dolutegravir versus the darunavir regimens ranged from 0.4 to 0.5, depending on censoring definitions, and we could not break that binding.”
The changing levels of significance are becuase of the different definitions of censoring for end of follow-up, according to Dr. Crane.
In addition to improved viral failure rates, patients taking DRV-based regimens had a higher proportion of patients lost to follow-up (20%), compared with those assigned DTG regimens (5%), which Dr. Crane hypothesized could be a result of the difference in time on the market between the two drugs.
This study was limited to studying only those patients receiving treatment after 2013,
Funding was provided by ViiV and the National Institutes of Health, which funds the CFAR Network of Integrated Clinical Systems.
[email protected]
SOURCE: Nance R et al. Abstract 1688.
SAN DIEGO – The proportion of viral failure was lower in people living with HIV who took dolutegravir (DTG), compared with patients on other integrase strand transfer inhibitor (INSTI) regimens, according to a study presented at ID Week 2017, an infectious diseases meeting.
Investigators studied records of 6,636 HIV patients who started regimens between August 2013 and August 2016, gathered from the Center for Aids Research (CFAR) Network of Integrated Clinical Systems. The information was collected from eight centers across the United States.
Patients were split among three ART regimen groups: dolutegravir based, other INSTI based, and darunavir (DRV) based. Among the 6,636 people living with HIV studied, those using a DTG-based regimen were less than half as likely to exhibit viral failure as were those in the DRV-based group (hazard ratio, 0.41; 95% confidence interval, 0.37-0.86), according to Heidi Crane, MD, MPH, associate director of CFAR Clinical Epidemiology and Health Services Research at the University of Washington, Seattle.
In a comparison of DTG-based therapy compared to other INSTI-based therapies, patients in the DTG groups showed significantly lower risk for viral failure in a crude hazard ratio (HR, 0.57; 95% CI, 0.46-0.70), although, when adjusted, the investigators found the odds did not hold the same level of significance, despite still being at a lower risk (adjusted HR, 0.82; 95% CI, 0.65-1.03).
In a sensitivity analysis of ART-naive patients, investigators found that patients taking DTG regimens were at nearly one-third the risk of viral failure, compared with those taking a darunavir-based regimen (adjusted HR, 0.32; 95% CI, 0.14-0.75). However, when comparing DTG with other INSTI therapies among the ART-naive population, there was no significant difference reported, Dr. Crane said in her presentation.
Investigators found this difference significant when comparing DTG with DRV and DTG with other INSTIs to be a trend across all testing models, according to Dr. Crane.
“In the various sensitivity models, the association for dolutegravir versus other integrase inhibitors ranged from 0.8 to 0.98 depending on censoring, sometimes significant and sometimes not,” said Dr. Crane. “In contrast, the hazard ratio for the dolutegravir versus the darunavir regimens ranged from 0.4 to 0.5, depending on censoring definitions, and we could not break that binding.”
The changing levels of significance are becuase of the different definitions of censoring for end of follow-up, according to Dr. Crane.
In addition to improved viral failure rates, patients taking DRV-based regimens had a higher proportion of patients lost to follow-up (20%), compared with those assigned DTG regimens (5%), which Dr. Crane hypothesized could be a result of the difference in time on the market between the two drugs.
This study was limited to studying only those patients receiving treatment after 2013,
Funding was provided by ViiV and the National Institutes of Health, which funds the CFAR Network of Integrated Clinical Systems.
[email protected]
SOURCE: Nance R et al. Abstract 1688.
SAN DIEGO – The proportion of viral failure was lower in people living with HIV who took dolutegravir (DTG), compared with patients on other integrase strand transfer inhibitor (INSTI) regimens, according to a study presented at ID Week 2017, an infectious diseases meeting.
Investigators studied records of 6,636 HIV patients who started regimens between August 2013 and August 2016, gathered from the Center for Aids Research (CFAR) Network of Integrated Clinical Systems. The information was collected from eight centers across the United States.
Patients were split among three ART regimen groups: dolutegravir based, other INSTI based, and darunavir (DRV) based. Among the 6,636 people living with HIV studied, those using a DTG-based regimen were less than half as likely to exhibit viral failure as were those in the DRV-based group (hazard ratio, 0.41; 95% confidence interval, 0.37-0.86), according to Heidi Crane, MD, MPH, associate director of CFAR Clinical Epidemiology and Health Services Research at the University of Washington, Seattle.
In a comparison of DTG-based therapy compared to other INSTI-based therapies, patients in the DTG groups showed significantly lower risk for viral failure in a crude hazard ratio (HR, 0.57; 95% CI, 0.46-0.70), although, when adjusted, the investigators found the odds did not hold the same level of significance, despite still being at a lower risk (adjusted HR, 0.82; 95% CI, 0.65-1.03).
In a sensitivity analysis of ART-naive patients, investigators found that patients taking DTG regimens were at nearly one-third the risk of viral failure, compared with those taking a darunavir-based regimen (adjusted HR, 0.32; 95% CI, 0.14-0.75). However, when comparing DTG with other INSTI therapies among the ART-naive population, there was no significant difference reported, Dr. Crane said in her presentation.
Investigators found this difference significant when comparing DTG with DRV and DTG with other INSTIs to be a trend across all testing models, according to Dr. Crane.
“In the various sensitivity models, the association for dolutegravir versus other integrase inhibitors ranged from 0.8 to 0.98 depending on censoring, sometimes significant and sometimes not,” said Dr. Crane. “In contrast, the hazard ratio for the dolutegravir versus the darunavir regimens ranged from 0.4 to 0.5, depending on censoring definitions, and we could not break that binding.”
The changing levels of significance are becuase of the different definitions of censoring for end of follow-up, according to Dr. Crane.
In addition to improved viral failure rates, patients taking DRV-based regimens had a higher proportion of patients lost to follow-up (20%), compared with those assigned DTG regimens (5%), which Dr. Crane hypothesized could be a result of the difference in time on the market between the two drugs.
This study was limited to studying only those patients receiving treatment after 2013,
Funding was provided by ViiV and the National Institutes of Health, which funds the CFAR Network of Integrated Clinical Systems.
[email protected]
SOURCE: Nance R et al. Abstract 1688.
REPORTING FROM ID WEEK 2017
Key clinical point:
Major finding: Patients on DTG-based regiments were half as likely to experience viral failure than those on darunavir-based treatment (HR, 0.41; 95% CI, 0.37-0.86).
Study details: Retrospective study of data of 6,636 HIV patients who started regimens during August 2013-August 2016, gathered from eight CFAR Network of Integrated Clinical Systems centers.
Disclosures: Study funded by ViiV Healthcare, CFAR Network of Integrated Clinical Systems is funded by NIAID/NHLBI R24 A1067039.
Source: Nance R et al. Abstract 1688.
VIDEO: Novel PARP inhibitor boosts PFS in HER2– breast cancer with BRCA mutations
SAN ANTONIO – In women with advanced HER2-negative breast cancer with germline BRCA mutations, talazoparib, an investigational oral PARP inhibitor, was associated with a near doubling in progression-free survival, compared with single-agent chemotherapy in the phase 3 EMBRACA trial.
After a median follow-up of 11.2 months, median progression-free survival by blinded central review was 8.6 months for patients assigned to receive talazoparib, compared with 5.6 months for patients randomized to receive the physician’s choice of either capecitabine, eribulin, gemcitabine, or vinorelbine.
In this video interview from the San Antonio Breast Cancer Symposium, Jennifer K. Litton, MD, from the University of Texas MD Anderson Cancer Center in Houston discusses the comparative efficacy of the drug relative to standard chemotherapy agents in this population, and the association of the PARP inhibitor with improved patient-reported quality of life outcomes.
The EMBRACA study was funded by Pfizer. Dr. Litton has disclosed research funding with EMD Serono, AstraZeneca, Pfizer, Genentech, and GlaxoSmithKline, and serves on advisory boards for Pfizer and AstraZeneca, all uncompensated.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN ANTONIO – In women with advanced HER2-negative breast cancer with germline BRCA mutations, talazoparib, an investigational oral PARP inhibitor, was associated with a near doubling in progression-free survival, compared with single-agent chemotherapy in the phase 3 EMBRACA trial.
After a median follow-up of 11.2 months, median progression-free survival by blinded central review was 8.6 months for patients assigned to receive talazoparib, compared with 5.6 months for patients randomized to receive the physician’s choice of either capecitabine, eribulin, gemcitabine, or vinorelbine.
In this video interview from the San Antonio Breast Cancer Symposium, Jennifer K. Litton, MD, from the University of Texas MD Anderson Cancer Center in Houston discusses the comparative efficacy of the drug relative to standard chemotherapy agents in this population, and the association of the PARP inhibitor with improved patient-reported quality of life outcomes.
The EMBRACA study was funded by Pfizer. Dr. Litton has disclosed research funding with EMD Serono, AstraZeneca, Pfizer, Genentech, and GlaxoSmithKline, and serves on advisory boards for Pfizer and AstraZeneca, all uncompensated.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN ANTONIO – In women with advanced HER2-negative breast cancer with germline BRCA mutations, talazoparib, an investigational oral PARP inhibitor, was associated with a near doubling in progression-free survival, compared with single-agent chemotherapy in the phase 3 EMBRACA trial.
After a median follow-up of 11.2 months, median progression-free survival by blinded central review was 8.6 months for patients assigned to receive talazoparib, compared with 5.6 months for patients randomized to receive the physician’s choice of either capecitabine, eribulin, gemcitabine, or vinorelbine.
In this video interview from the San Antonio Breast Cancer Symposium, Jennifer K. Litton, MD, from the University of Texas MD Anderson Cancer Center in Houston discusses the comparative efficacy of the drug relative to standard chemotherapy agents in this population, and the association of the PARP inhibitor with improved patient-reported quality of life outcomes.
The EMBRACA study was funded by Pfizer. Dr. Litton has disclosed research funding with EMD Serono, AstraZeneca, Pfizer, Genentech, and GlaxoSmithKline, and serves on advisory boards for Pfizer and AstraZeneca, all uncompensated.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
REPORTING FROM SABCS 2017