User login
HHS Secretary resigns amid flight criticism
U.S. Department of Health & Human Services Secretary Tom Price, MD, has resigned from his post following furor over his use of private planes for government business paid for by taxpayers.
In a Sept. 29 press statement, White House press secretary Sarah Huckabee Sanders said Dr. Price offered his resignation early Sept. 29 and that President Trump accepted. Don J. Wright, deputy assistant secretary for health and director of the office of disease prevention and health promotion, will serve as Acting Secretary effective Sept. 30, according to the statement.

Dr. Price’s resignation comes after widespread criticism over his alleged repeated use of taxpayer-funded charter flights. Politico first broke the story, reporting that he has taken at least 24 flights on private charter planes at taxpayers’ expense since early May, costing an estimated $400,000-$500,000. After the reports, Dr. Price announced that he planned to write a personal check to the government for $60,000 to cover the cost of the flights.
During a press briefing on Sept. 29, President Trump said he was “disappointed” with Dr. Price’s actions, and that the White House was looking into the accusations.
“I felt very badly because Secretary Price is a good man, but we are looking into it, and we’re looking into it very strongly,” President Trump said at the briefing.
In his resignation letter, posted on Twitter, Dr. Price expressed remorse for having created a distraction for the administration and said he was grateful for the opportunity to have served as HHS Secretary.
“I have spent 40 years both as a doctor and public servant putting people first,” Dr. Price said in his resignation letter. “I regret that the recent events have created a distraction from these important objectives. Success on these issues is more important than any one person. In order for you to move forward without further disruption, I am officially tendering my resignation.”
[email protected]
On Twitter @legal_med
U.S. Department of Health & Human Services Secretary Tom Price, MD, has resigned from his post following furor over his use of private planes for government business paid for by taxpayers.
In a Sept. 29 press statement, White House press secretary Sarah Huckabee Sanders said Dr. Price offered his resignation early Sept. 29 and that President Trump accepted. Don J. Wright, deputy assistant secretary for health and director of the office of disease prevention and health promotion, will serve as Acting Secretary effective Sept. 30, according to the statement.
Dr. Price’s resignation comes after widespread criticism over his alleged repeated use of taxpayer-funded charter flights. Politico first broke the story, reporting that he has taken at least 24 flights on private charter planes at taxpayers’ expense since early May, costing an estimated $400,000-$500,000. After the reports, Dr. Price announced that he planned to write a personal check to the government for $60,000 to cover the cost of the flights.
During a press briefing on Sept. 29, President Trump said he was “disappointed” with Dr. Price’s actions, and that the White House was looking into the accusations.
“I felt very badly because Secretary Price is a good man, but we are looking into it, and we’re looking into it very strongly,” President Trump said at the briefing.
In his resignation letter, posted on Twitter, Dr. Price expressed remorse for having created a distraction for the administration and said he was grateful for the opportunity to have served as HHS Secretary.
“I have spent 40 years both as a doctor and public servant putting people first,” Dr. Price said in his resignation letter. “I regret that the recent events have created a distraction from these important objectives. Success on these issues is more important than any one person. In order for you to move forward without further disruption, I am officially tendering my resignation.”
[email protected]
On Twitter @legal_med
U.S. Department of Health & Human Services Secretary Tom Price, MD, has resigned from his post following furor over his use of private planes for government business paid for by taxpayers.
In a Sept. 29 press statement, White House press secretary Sarah Huckabee Sanders said Dr. Price offered his resignation early Sept. 29 and that President Trump accepted. Don J. Wright, deputy assistant secretary for health and director of the office of disease prevention and health promotion, will serve as Acting Secretary effective Sept. 30, according to the statement.
Dr. Price’s resignation comes after widespread criticism over his alleged repeated use of taxpayer-funded charter flights. Politico first broke the story, reporting that he has taken at least 24 flights on private charter planes at taxpayers’ expense since early May, costing an estimated $400,000-$500,000. After the reports, Dr. Price announced that he planned to write a personal check to the government for $60,000 to cover the cost of the flights.
During a press briefing on Sept. 29, President Trump said he was “disappointed” with Dr. Price’s actions, and that the White House was looking into the accusations.
“I felt very badly because Secretary Price is a good man, but we are looking into it, and we’re looking into it very strongly,” President Trump said at the briefing.
In his resignation letter, posted on Twitter, Dr. Price expressed remorse for having created a distraction for the administration and said he was grateful for the opportunity to have served as HHS Secretary.
“I have spent 40 years both as a doctor and public servant putting people first,” Dr. Price said in his resignation letter. “I regret that the recent events have created a distraction from these important objectives. Success on these issues is more important than any one person. In order for you to move forward without further disruption, I am officially tendering my resignation.”
[email protected]
On Twitter @legal_med
Japan approves product for hemophilia A

Japan’s Ministry of Health, Labor and Welfare has approved lonoctocog alfa (AFSTYLA®), a recombinant single-chain coagulation factor VIII product, for use in patients with hemophilia A.
The product is approved for use as routine prophylaxis to prevent or reduce the frequency of bleeding episodes, for on-demand treatment and control of bleeding, and for perioperative management.
Lonoctocog alfa is the first and only single-chain recombinant factor VIII product specifically designed to treat hemophilia A.
According to CSL Behring, the company developing lonoctocog alfa, the product was designed to provide greater molecular stability and longer duration of action. Lonoctocog alfa uses a covalent bond to form one structural entity, a single polypeptide chain, to improve the stability of factor VIII and provide factor VIII activity with the option of twice-weekly dosing.
Lonoctocog alfa is also approved in the European Union, US, Canada, Switzerland, and Australia.
AFFINITY trials
Japan’s approval of lonoctocog alfa is based on results from the AFFINITY clinical development program, which includes a trial of children (n=84) and a trial of adolescents and adults (n=175).
Among patients who received lonoctocog alfa prophylactically in these trials, the median annualized bleeding rate was 1.14 in the adults/adolescents and 3.69 in children younger than 12.
In all, there were 1195 bleeding events—848 in the adults/adolescents and 347 in the children.
Ninety-four percent of bleeds in adults/adolescents and 96% of bleeds in pediatric patients were effectively controlled with no more than 2 infusions of lonoctocog alfa weekly.
Eighty-one percent of bleeds in adults/adolescents and 86% of bleeds in pediatric patients were controlled by a single infusion.
Researchers assessed safety in 258 patients from both studies. Adverse reactions occurred in 14 patients and included hypersensitivity (n=4), dizziness (n=2), paresthesia (n=1), rash (n=1), erythema (n=1), pruritus (n=1), pyrexia (n=1), injection-site pain (n=1), chills (n=1), and feeling hot (n=1).
One patient withdrew from treatment due to hypersensitivity.
None of the patients developed neutralizing antibodies to factor VIII or antibodies to host cell proteins. There were no reports of anaphylaxis or thrombosis.
Results from the trial of adolescents/adults were published in Blood in August 2016. Results from the trial of children were published in the Journal of Thrombosis and Haemostasis in March 2017. ![]()
Japan’s Ministry of Health, Labor and Welfare has approved lonoctocog alfa (AFSTYLA®), a recombinant single-chain coagulation factor VIII product, for use in patients with hemophilia A.
The product is approved for use as routine prophylaxis to prevent or reduce the frequency of bleeding episodes, for on-demand treatment and control of bleeding, and for perioperative management.
Lonoctocog alfa is the first and only single-chain recombinant factor VIII product specifically designed to treat hemophilia A.
According to CSL Behring, the company developing lonoctocog alfa, the product was designed to provide greater molecular stability and longer duration of action. Lonoctocog alfa uses a covalent bond to form one structural entity, a single polypeptide chain, to improve the stability of factor VIII and provide factor VIII activity with the option of twice-weekly dosing.
Lonoctocog alfa is also approved in the European Union, US, Canada, Switzerland, and Australia.
AFFINITY trials
Japan’s approval of lonoctocog alfa is based on results from the AFFINITY clinical development program, which includes a trial of children (n=84) and a trial of adolescents and adults (n=175).
Among patients who received lonoctocog alfa prophylactically in these trials, the median annualized bleeding rate was 1.14 in the adults/adolescents and 3.69 in children younger than 12.
In all, there were 1195 bleeding events—848 in the adults/adolescents and 347 in the children.
Ninety-four percent of bleeds in adults/adolescents and 96% of bleeds in pediatric patients were effectively controlled with no more than 2 infusions of lonoctocog alfa weekly.
Eighty-one percent of bleeds in adults/adolescents and 86% of bleeds in pediatric patients were controlled by a single infusion.
Researchers assessed safety in 258 patients from both studies. Adverse reactions occurred in 14 patients and included hypersensitivity (n=4), dizziness (n=2), paresthesia (n=1), rash (n=1), erythema (n=1), pruritus (n=1), pyrexia (n=1), injection-site pain (n=1), chills (n=1), and feeling hot (n=1).
One patient withdrew from treatment due to hypersensitivity.
None of the patients developed neutralizing antibodies to factor VIII or antibodies to host cell proteins. There were no reports of anaphylaxis or thrombosis.
Results from the trial of adolescents/adults were published in Blood in August 2016. Results from the trial of children were published in the Journal of Thrombosis and Haemostasis in March 2017. ![]()
Japan’s Ministry of Health, Labor and Welfare has approved lonoctocog alfa (AFSTYLA®), a recombinant single-chain coagulation factor VIII product, for use in patients with hemophilia A.
The product is approved for use as routine prophylaxis to prevent or reduce the frequency of bleeding episodes, for on-demand treatment and control of bleeding, and for perioperative management.
Lonoctocog alfa is the first and only single-chain recombinant factor VIII product specifically designed to treat hemophilia A.
According to CSL Behring, the company developing lonoctocog alfa, the product was designed to provide greater molecular stability and longer duration of action. Lonoctocog alfa uses a covalent bond to form one structural entity, a single polypeptide chain, to improve the stability of factor VIII and provide factor VIII activity with the option of twice-weekly dosing.
Lonoctocog alfa is also approved in the European Union, US, Canada, Switzerland, and Australia.
AFFINITY trials
Japan’s approval of lonoctocog alfa is based on results from the AFFINITY clinical development program, which includes a trial of children (n=84) and a trial of adolescents and adults (n=175).
Among patients who received lonoctocog alfa prophylactically in these trials, the median annualized bleeding rate was 1.14 in the adults/adolescents and 3.69 in children younger than 12.
In all, there were 1195 bleeding events—848 in the adults/adolescents and 347 in the children.
Ninety-four percent of bleeds in adults/adolescents and 96% of bleeds in pediatric patients were effectively controlled with no more than 2 infusions of lonoctocog alfa weekly.
Eighty-one percent of bleeds in adults/adolescents and 86% of bleeds in pediatric patients were controlled by a single infusion.
Researchers assessed safety in 258 patients from both studies. Adverse reactions occurred in 14 patients and included hypersensitivity (n=4), dizziness (n=2), paresthesia (n=1), rash (n=1), erythema (n=1), pruritus (n=1), pyrexia (n=1), injection-site pain (n=1), chills (n=1), and feeling hot (n=1).
One patient withdrew from treatment due to hypersensitivity.
None of the patients developed neutralizing antibodies to factor VIII or antibodies to host cell proteins. There were no reports of anaphylaxis or thrombosis.
Results from the trial of adolescents/adults were published in Blood in August 2016. Results from the trial of children were published in the Journal of Thrombosis and Haemostasis in March 2017. ![]()
Add aggressiveness to mixed features specifier for major depressive episode
PARIS – Aggressiveness deserves to be incorporated in the next Diagnostic and Statistical Manual of Mental Disorders update as a new clinical criterion triggering application of the “with mixed features” specifier in patients diagnosed with a major depressive episode, Norma Verdolini, MD, said at the annual congress of the European College of Neuropsychopharmacology.
“Aggressiveness might be a trait component of bipolarity and a diagnostic indicator of ‘mixicity’ in patients with a major depressive episode. This has implications for the therapeutic strategy,” said Dr. Verdolini of the bipolar disorders unit at the University of Barcelona Institute of Neurosciences.

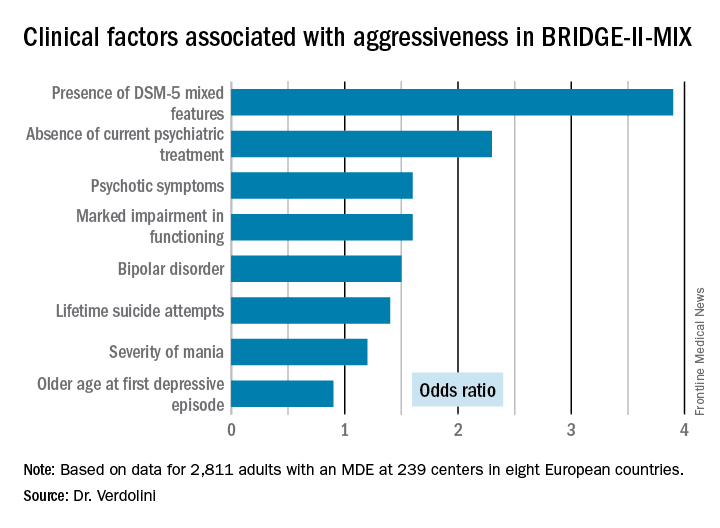
The BRIDGE-II-MIX study was a cross-sectional observational study of 2,811 adults with MDE at 239 centers in eight European countries (J Clin Psychiatry. 2015 Mar;76[3]:e351-8). Three hundred ninety-nine participants (14.2%) met the operational definition of physical or verbal aggressiveness used in Dr. Verdolini’s new post-hoc analysis.
Statistically significant and clinically meaningful differences were found between MDE patients with aggressiveness (MDE-aggro) and MDE without aggressiveness. For example, the MDE-aggro group was twice as likely to meet DSM-IV-TR criteria for bipolar disorder I. Twenty-seven percent of the MDE-aggro group met DSM-5 criteria for a mixed state, meaning both depressed mood and mania in the same episode, compared with just 4% of the MDE-no-aggro group.
The MDE-aggro patients also had a strikingly greater prevalence of comorbid borderline personality disorder, by a margin of 20% versus 4%. They had a younger mean age at their first depressive episode: 29.9 years old, compared with 36.1 in the MDE-no-aggro group. The MDE-aggro patients had more prior mood episodes and a greater number of lifetime suicide attempts. In addition, they had significantly more severe depression, mania, and bipolar disorder scores on the Clinical Global Impression Scale for Bipolar Disorder.
“Our results should prompt reconsideration of the diagnostic criteria for the mixed features specifier. The detection of aggression in MDE could represent a therapeutic target in personalized pharmacological treatment for bipolar disorder,” Dr. Verdolini concluded.
The BRIDGE-II-MIX study was sponsored by Sanofi-Aventis. Dr. Verdolini reported receiving research funding from the company.
PARIS – Aggressiveness deserves to be incorporated in the next Diagnostic and Statistical Manual of Mental Disorders update as a new clinical criterion triggering application of the “with mixed features” specifier in patients diagnosed with a major depressive episode, Norma Verdolini, MD, said at the annual congress of the European College of Neuropsychopharmacology.
“Aggressiveness might be a trait component of bipolarity and a diagnostic indicator of ‘mixicity’ in patients with a major depressive episode. This has implications for the therapeutic strategy,” said Dr. Verdolini of the bipolar disorders unit at the University of Barcelona Institute of Neurosciences.
The BRIDGE-II-MIX study was a cross-sectional observational study of 2,811 adults with MDE at 239 centers in eight European countries (J Clin Psychiatry. 2015 Mar;76[3]:e351-8). Three hundred ninety-nine participants (14.2%) met the operational definition of physical or verbal aggressiveness used in Dr. Verdolini’s new post-hoc analysis.
Statistically significant and clinically meaningful differences were found between MDE patients with aggressiveness (MDE-aggro) and MDE without aggressiveness. For example, the MDE-aggro group was twice as likely to meet DSM-IV-TR criteria for bipolar disorder I. Twenty-seven percent of the MDE-aggro group met DSM-5 criteria for a mixed state, meaning both depressed mood and mania in the same episode, compared with just 4% of the MDE-no-aggro group.
The MDE-aggro patients also had a strikingly greater prevalence of comorbid borderline personality disorder, by a margin of 20% versus 4%. They had a younger mean age at their first depressive episode: 29.9 years old, compared with 36.1 in the MDE-no-aggro group. The MDE-aggro patients had more prior mood episodes and a greater number of lifetime suicide attempts. In addition, they had significantly more severe depression, mania, and bipolar disorder scores on the Clinical Global Impression Scale for Bipolar Disorder.
“Our results should prompt reconsideration of the diagnostic criteria for the mixed features specifier. The detection of aggression in MDE could represent a therapeutic target in personalized pharmacological treatment for bipolar disorder,” Dr. Verdolini concluded.
The BRIDGE-II-MIX study was sponsored by Sanofi-Aventis. Dr. Verdolini reported receiving research funding from the company.
PARIS – Aggressiveness deserves to be incorporated in the next Diagnostic and Statistical Manual of Mental Disorders update as a new clinical criterion triggering application of the “with mixed features” specifier in patients diagnosed with a major depressive episode, Norma Verdolini, MD, said at the annual congress of the European College of Neuropsychopharmacology.
“Aggressiveness might be a trait component of bipolarity and a diagnostic indicator of ‘mixicity’ in patients with a major depressive episode. This has implications for the therapeutic strategy,” said Dr. Verdolini of the bipolar disorders unit at the University of Barcelona Institute of Neurosciences.
The BRIDGE-II-MIX study was a cross-sectional observational study of 2,811 adults with MDE at 239 centers in eight European countries (J Clin Psychiatry. 2015 Mar;76[3]:e351-8). Three hundred ninety-nine participants (14.2%) met the operational definition of physical or verbal aggressiveness used in Dr. Verdolini’s new post-hoc analysis.
Statistically significant and clinically meaningful differences were found between MDE patients with aggressiveness (MDE-aggro) and MDE without aggressiveness. For example, the MDE-aggro group was twice as likely to meet DSM-IV-TR criteria for bipolar disorder I. Twenty-seven percent of the MDE-aggro group met DSM-5 criteria for a mixed state, meaning both depressed mood and mania in the same episode, compared with just 4% of the MDE-no-aggro group.
The MDE-aggro patients also had a strikingly greater prevalence of comorbid borderline personality disorder, by a margin of 20% versus 4%. They had a younger mean age at their first depressive episode: 29.9 years old, compared with 36.1 in the MDE-no-aggro group. The MDE-aggro patients had more prior mood episodes and a greater number of lifetime suicide attempts. In addition, they had significantly more severe depression, mania, and bipolar disorder scores on the Clinical Global Impression Scale for Bipolar Disorder.
“Our results should prompt reconsideration of the diagnostic criteria for the mixed features specifier. The detection of aggression in MDE could represent a therapeutic target in personalized pharmacological treatment for bipolar disorder,” Dr. Verdolini concluded.
The BRIDGE-II-MIX study was sponsored by Sanofi-Aventis. Dr. Verdolini reported receiving research funding from the company.
AT THE ECNP CONGRESS
Key clinical point:
Major finding: Patients who fulfilled the DSM-5 criteria for a major depressive episode with mixed features were 3.9-fold more likely to meet investigators’ operational definition of aggressiveness.
Data source: This was a post-hoc analysis of the BRIDGE-II-MIX study, an observational cross-sectional study of 2,811 adults experiencing a major depressive episode.
Disclosures: The BRIDGE-II-MIX study was sponsored by Sanofi-Aventis. The presenter reported receiving research funding from the company.
Laugier-Hunziker Syndrome
To the Editor:
A 55-year-old man presented with hyperpigmented brown macules on the lips, hands, and fingertips of 6 years’ duration. The spots were persistent, asymptomatic, and had not changed in size. The patient denied a history of alopecia or dystrophic nails. He also denied a family history of similar skin findings. He had no personal history of cancer and a colonoscopy performed 5 years prior revealed no notable abnormalities. His medications included amlodipine and hydrocodone-acetaminophen. His mother died of “abdominal bleeding” at 74 years of age and his father died of a brain tumor at 64 years of age. Physical examination demonstrated numerous well-defined, dark brown macules of variable size distributed on the lower and upper mucosal lips (Figure 1A), buccal mucosa, hard palate, and gingiva, as well as the dorsal aspect of the fingers (Figure 1B) and volar aspect of the fingertips (Figure 1C).

A shave biopsy of a dark brown macule from the lower lip (Figure 2) was performed. Histopathologic examination revealed pigmentation of the basal layer of the epidermis with pigment-laden cells in the dermis immediately deep to the surface epithelium. Immunoperoxidase stains showed a normal number and distribution of melanocytes.
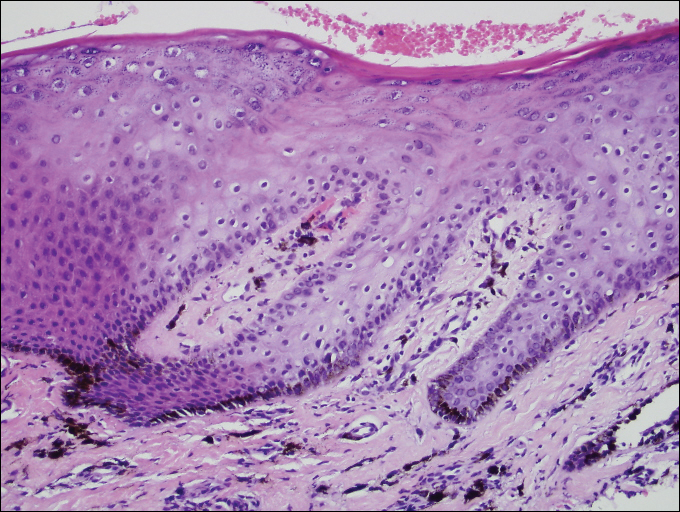
A diagnosis of Laugier-Hunziker syndrome (LHS) was made given the age of onset; distribution of pigmentation; and lack of pathologic colonoscopic findings, personal history of cancer, or gastrointestinal tract symptoms.
Benign hyperpigmentation of the lips and fingers has been reported.1 The average age of onset of LHS is 52 years, and it typically is diagnosed in white adults.1,2 In LHS, pigmentation is most commonly distributed on the lips, especially the lower lips and oral mucosa.2 Pigmentation of the nails in the form of longitudinal melanonychia is present in approximately half of cases.2,3 There also may be pigmentation of the neck; thorax; abdomen; and acral surfaces, especially the fingertips.1-3 Rarely, pigmented macules can occur on the genitalia or sclera.1,2 Unlike Peutz-Jeghers syndrome, the diagnosis of LHS does not result from a germline mutation and carries no risk of gastrointestinal polyposis or internal malignancy.3,4 The histopathology of a pigmented macule of LHS shows a normal number and morphology of melanocytes. Epidermal basement membrane pigmentation is common, with pigment-laden macrophages evident in the papillary dermis.3
RELATED ARTICLE: Asymptomatic Lower Lip Hyperpigmentation From Laugier-Hunziker Syndrome
The differential diagnosis of multiple lentigines is broad and includes Peutz-Jeghers syndrome; LEOPARD (lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, deafness) syndrome; Carney complexes, including LAMB (lentigines, atrial myxoma, mucocutaneous myxoma, blue nevi) and NAME (nevi, atrial myxoma, myxoid neurofibroma, ephelide) syndromes5; primary adrenocortical insufficiency (Addison disease); and idiopathic melanoplakia.2 Peutz-Jeghers syndrome, an autosomal-dominant syndrome with mucocutaneous lentigines, has a similar clinical appearance to LHS; therefore, it is necessary to exclude this diagnosis due to its association with intestinal hamartomatous polyps and internal malignancies (Table).3,6,7
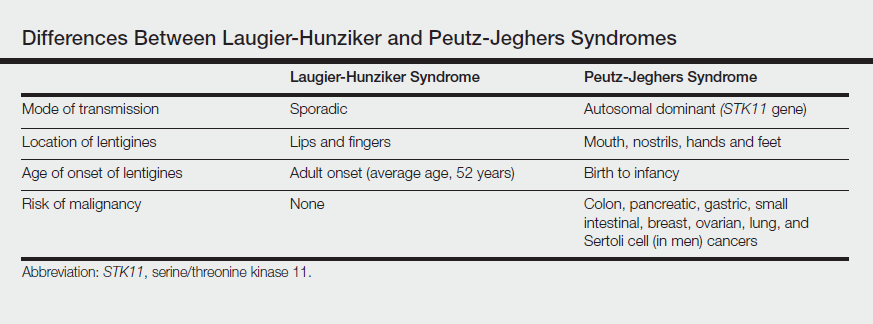
Peutz-Jeghers syndrome is characterized by mucocutaneous hyperpigmentation and intestinal hamartomatous polyposis and is associated with internal malignancies of the colon, breast, pancreas, stomach, small intestines, ovaries, lung, and Sertoli cells in men.6,7 Associated gastrointestinal tract malignancies in descending order of frequency are colon (39%), pancreatic (36%), gastric (29%), and small intestine (13%).1 It is caused by a germ line mutation of the serine/threonine kinase 11 gene, STK11. Although the appearance and distribution of the mucocutaneous lentigines is similar to individuals with LHS, by contrast the lentiginosis in individuals with Peutz-Jeghers syndrome is present from birth or develops during infancy.6 Aggressive cancer screening guidelines aid in early detection and begin at 8 years of age with a baseline colonoscopy and esophagogastroduodenoscopy; future screening is dictated by the presence or absence of polyps. If no polyps are detected at 8 years of age, a colonoscopy and esophagogastroduodenoscopy are repeated at 18 years of age and then every 3 years until 50 years of age.8
In an adult patient, the diagnosis of LHS can be made clinically and a correct diagnosis prevents frequent and unpleasant gastrointestinal tract cancer screening examinations. Lampe et al2 described a man with LHS who was incorrectly diagnosed with Peutz-Jeghers syndrome and experienced a colonic perforation as a complication of a screening colonoscopy. Their case report underscores the importance of making the correct diagnosis of LHS to avoid undertaking unnecessary aggressive cancer screening regimens.2
Although LHS is a benign condition that does not require treatment, Q-switched alexandrite or erbium:YAG laser therapy has been shown to improve the pigmentary findings associated with LHS.9,10 It has been suggested that LHS should be renamed Laugier-Hunziker pigmentation2 or mucocutaneous lentiginosis of Laugier and Hunziker1 to differentiate LHS as simply a disorder of pigmentation rather than a potentially morbid genetic defect, as in Peutz-Jeghers syndrome.
- Moore RT, Chae KA, Rhodes AR. Laugier and Hunziker pigmentation: a lentiginous proliferation of melanocytes. J Am Acad Dermatol. 2004;50(5 suppl):S70-S74.
- Lampe AK, Hampton PJ, Woodford-Richens K, et al. Laugier-Hunziker Syndrome: an important differential diagnosis for Peutz-Jeghers Syndrome. J Med Genet. 2003;40:E77.
- Baran R. Longitudinal melanotic streaks as a clue for Laugier-Hunziker syndrome. Arch Dermatol. 1979;115:1148-1149.
- Grimes P, Nordlund JJ, Pandya AG, et al. Increasing our understanding of pigmentary disorders. J Am Acad Dermatol. 2006;54(5 suppl 2):S255-S261.
- Bertherat J. Carney complex (CNC). Orphanet J Rare Dis. 2006;1:21.
- Giardiello FM, Brensinger JD, Tersemette AC, et al. Very high risk of cancer in Peutz-Jeghers Syndrome. Gastroenterology. 2000;119:1447-1453.
- Brosens LA, van Hattem WA, Jansen M, et al. Gastrointestinal polyposis syndromes. Curr Mol Med. 2007;7:29-46.
- Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59:975-986.
- Zuo YG, Ma DL, Jin HZ, et al. Treatment of Laugier-Hunziker syndrome with the Q-switched alexandrite laser in 22 Chinese patients. Arch Dermatol Res. 2010;302:125-130.
- Ergun S, Saruhanog˘lu A, Migliari DA, et al. Refractory pigmentation associated with Laugier-Hunziker syndrome following Er:YAG laser treatment [published online December 3, 2013]. Case Rep Dent. 2013;2013:561040.
To the Editor:
A 55-year-old man presented with hyperpigmented brown macules on the lips, hands, and fingertips of 6 years’ duration. The spots were persistent, asymptomatic, and had not changed in size. The patient denied a history of alopecia or dystrophic nails. He also denied a family history of similar skin findings. He had no personal history of cancer and a colonoscopy performed 5 years prior revealed no notable abnormalities. His medications included amlodipine and hydrocodone-acetaminophen. His mother died of “abdominal bleeding” at 74 years of age and his father died of a brain tumor at 64 years of age. Physical examination demonstrated numerous well-defined, dark brown macules of variable size distributed on the lower and upper mucosal lips (Figure 1A), buccal mucosa, hard palate, and gingiva, as well as the dorsal aspect of the fingers (Figure 1B) and volar aspect of the fingertips (Figure 1C).
A shave biopsy of a dark brown macule from the lower lip (Figure 2) was performed. Histopathologic examination revealed pigmentation of the basal layer of the epidermis with pigment-laden cells in the dermis immediately deep to the surface epithelium. Immunoperoxidase stains showed a normal number and distribution of melanocytes.
A diagnosis of Laugier-Hunziker syndrome (LHS) was made given the age of onset; distribution of pigmentation; and lack of pathologic colonoscopic findings, personal history of cancer, or gastrointestinal tract symptoms.
Benign hyperpigmentation of the lips and fingers has been reported.1 The average age of onset of LHS is 52 years, and it typically is diagnosed in white adults.1,2 In LHS, pigmentation is most commonly distributed on the lips, especially the lower lips and oral mucosa.2 Pigmentation of the nails in the form of longitudinal melanonychia is present in approximately half of cases.2,3 There also may be pigmentation of the neck; thorax; abdomen; and acral surfaces, especially the fingertips.1-3 Rarely, pigmented macules can occur on the genitalia or sclera.1,2 Unlike Peutz-Jeghers syndrome, the diagnosis of LHS does not result from a germline mutation and carries no risk of gastrointestinal polyposis or internal malignancy.3,4 The histopathology of a pigmented macule of LHS shows a normal number and morphology of melanocytes. Epidermal basement membrane pigmentation is common, with pigment-laden macrophages evident in the papillary dermis.3
RELATED ARTICLE: Asymptomatic Lower Lip Hyperpigmentation From Laugier-Hunziker Syndrome
The differential diagnosis of multiple lentigines is broad and includes Peutz-Jeghers syndrome; LEOPARD (lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, deafness) syndrome; Carney complexes, including LAMB (lentigines, atrial myxoma, mucocutaneous myxoma, blue nevi) and NAME (nevi, atrial myxoma, myxoid neurofibroma, ephelide) syndromes5; primary adrenocortical insufficiency (Addison disease); and idiopathic melanoplakia.2 Peutz-Jeghers syndrome, an autosomal-dominant syndrome with mucocutaneous lentigines, has a similar clinical appearance to LHS; therefore, it is necessary to exclude this diagnosis due to its association with intestinal hamartomatous polyps and internal malignancies (Table).3,6,7
Peutz-Jeghers syndrome is characterized by mucocutaneous hyperpigmentation and intestinal hamartomatous polyposis and is associated with internal malignancies of the colon, breast, pancreas, stomach, small intestines, ovaries, lung, and Sertoli cells in men.6,7 Associated gastrointestinal tract malignancies in descending order of frequency are colon (39%), pancreatic (36%), gastric (29%), and small intestine (13%).1 It is caused by a germ line mutation of the serine/threonine kinase 11 gene, STK11. Although the appearance and distribution of the mucocutaneous lentigines is similar to individuals with LHS, by contrast the lentiginosis in individuals with Peutz-Jeghers syndrome is present from birth or develops during infancy.6 Aggressive cancer screening guidelines aid in early detection and begin at 8 years of age with a baseline colonoscopy and esophagogastroduodenoscopy; future screening is dictated by the presence or absence of polyps. If no polyps are detected at 8 years of age, a colonoscopy and esophagogastroduodenoscopy are repeated at 18 years of age and then every 3 years until 50 years of age.8
In an adult patient, the diagnosis of LHS can be made clinically and a correct diagnosis prevents frequent and unpleasant gastrointestinal tract cancer screening examinations. Lampe et al2 described a man with LHS who was incorrectly diagnosed with Peutz-Jeghers syndrome and experienced a colonic perforation as a complication of a screening colonoscopy. Their case report underscores the importance of making the correct diagnosis of LHS to avoid undertaking unnecessary aggressive cancer screening regimens.2
Although LHS is a benign condition that does not require treatment, Q-switched alexandrite or erbium:YAG laser therapy has been shown to improve the pigmentary findings associated with LHS.9,10 It has been suggested that LHS should be renamed Laugier-Hunziker pigmentation2 or mucocutaneous lentiginosis of Laugier and Hunziker1 to differentiate LHS as simply a disorder of pigmentation rather than a potentially morbid genetic defect, as in Peutz-Jeghers syndrome.
To the Editor:
A 55-year-old man presented with hyperpigmented brown macules on the lips, hands, and fingertips of 6 years’ duration. The spots were persistent, asymptomatic, and had not changed in size. The patient denied a history of alopecia or dystrophic nails. He also denied a family history of similar skin findings. He had no personal history of cancer and a colonoscopy performed 5 years prior revealed no notable abnormalities. His medications included amlodipine and hydrocodone-acetaminophen. His mother died of “abdominal bleeding” at 74 years of age and his father died of a brain tumor at 64 years of age. Physical examination demonstrated numerous well-defined, dark brown macules of variable size distributed on the lower and upper mucosal lips (Figure 1A), buccal mucosa, hard palate, and gingiva, as well as the dorsal aspect of the fingers (Figure 1B) and volar aspect of the fingertips (Figure 1C).
A shave biopsy of a dark brown macule from the lower lip (Figure 2) was performed. Histopathologic examination revealed pigmentation of the basal layer of the epidermis with pigment-laden cells in the dermis immediately deep to the surface epithelium. Immunoperoxidase stains showed a normal number and distribution of melanocytes.
A diagnosis of Laugier-Hunziker syndrome (LHS) was made given the age of onset; distribution of pigmentation; and lack of pathologic colonoscopic findings, personal history of cancer, or gastrointestinal tract symptoms.
Benign hyperpigmentation of the lips and fingers has been reported.1 The average age of onset of LHS is 52 years, and it typically is diagnosed in white adults.1,2 In LHS, pigmentation is most commonly distributed on the lips, especially the lower lips and oral mucosa.2 Pigmentation of the nails in the form of longitudinal melanonychia is present in approximately half of cases.2,3 There also may be pigmentation of the neck; thorax; abdomen; and acral surfaces, especially the fingertips.1-3 Rarely, pigmented macules can occur on the genitalia or sclera.1,2 Unlike Peutz-Jeghers syndrome, the diagnosis of LHS does not result from a germline mutation and carries no risk of gastrointestinal polyposis or internal malignancy.3,4 The histopathology of a pigmented macule of LHS shows a normal number and morphology of melanocytes. Epidermal basement membrane pigmentation is common, with pigment-laden macrophages evident in the papillary dermis.3
RELATED ARTICLE: Asymptomatic Lower Lip Hyperpigmentation From Laugier-Hunziker Syndrome
The differential diagnosis of multiple lentigines is broad and includes Peutz-Jeghers syndrome; LEOPARD (lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, deafness) syndrome; Carney complexes, including LAMB (lentigines, atrial myxoma, mucocutaneous myxoma, blue nevi) and NAME (nevi, atrial myxoma, myxoid neurofibroma, ephelide) syndromes5; primary adrenocortical insufficiency (Addison disease); and idiopathic melanoplakia.2 Peutz-Jeghers syndrome, an autosomal-dominant syndrome with mucocutaneous lentigines, has a similar clinical appearance to LHS; therefore, it is necessary to exclude this diagnosis due to its association with intestinal hamartomatous polyps and internal malignancies (Table).3,6,7
Peutz-Jeghers syndrome is characterized by mucocutaneous hyperpigmentation and intestinal hamartomatous polyposis and is associated with internal malignancies of the colon, breast, pancreas, stomach, small intestines, ovaries, lung, and Sertoli cells in men.6,7 Associated gastrointestinal tract malignancies in descending order of frequency are colon (39%), pancreatic (36%), gastric (29%), and small intestine (13%).1 It is caused by a germ line mutation of the serine/threonine kinase 11 gene, STK11. Although the appearance and distribution of the mucocutaneous lentigines is similar to individuals with LHS, by contrast the lentiginosis in individuals with Peutz-Jeghers syndrome is present from birth or develops during infancy.6 Aggressive cancer screening guidelines aid in early detection and begin at 8 years of age with a baseline colonoscopy and esophagogastroduodenoscopy; future screening is dictated by the presence or absence of polyps. If no polyps are detected at 8 years of age, a colonoscopy and esophagogastroduodenoscopy are repeated at 18 years of age and then every 3 years until 50 years of age.8
In an adult patient, the diagnosis of LHS can be made clinically and a correct diagnosis prevents frequent and unpleasant gastrointestinal tract cancer screening examinations. Lampe et al2 described a man with LHS who was incorrectly diagnosed with Peutz-Jeghers syndrome and experienced a colonic perforation as a complication of a screening colonoscopy. Their case report underscores the importance of making the correct diagnosis of LHS to avoid undertaking unnecessary aggressive cancer screening regimens.2
Although LHS is a benign condition that does not require treatment, Q-switched alexandrite or erbium:YAG laser therapy has been shown to improve the pigmentary findings associated with LHS.9,10 It has been suggested that LHS should be renamed Laugier-Hunziker pigmentation2 or mucocutaneous lentiginosis of Laugier and Hunziker1 to differentiate LHS as simply a disorder of pigmentation rather than a potentially morbid genetic defect, as in Peutz-Jeghers syndrome.
- Moore RT, Chae KA, Rhodes AR. Laugier and Hunziker pigmentation: a lentiginous proliferation of melanocytes. J Am Acad Dermatol. 2004;50(5 suppl):S70-S74.
- Lampe AK, Hampton PJ, Woodford-Richens K, et al. Laugier-Hunziker Syndrome: an important differential diagnosis for Peutz-Jeghers Syndrome. J Med Genet. 2003;40:E77.
- Baran R. Longitudinal melanotic streaks as a clue for Laugier-Hunziker syndrome. Arch Dermatol. 1979;115:1148-1149.
- Grimes P, Nordlund JJ, Pandya AG, et al. Increasing our understanding of pigmentary disorders. J Am Acad Dermatol. 2006;54(5 suppl 2):S255-S261.
- Bertherat J. Carney complex (CNC). Orphanet J Rare Dis. 2006;1:21.
- Giardiello FM, Brensinger JD, Tersemette AC, et al. Very high risk of cancer in Peutz-Jeghers Syndrome. Gastroenterology. 2000;119:1447-1453.
- Brosens LA, van Hattem WA, Jansen M, et al. Gastrointestinal polyposis syndromes. Curr Mol Med. 2007;7:29-46.
- Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59:975-986.
- Zuo YG, Ma DL, Jin HZ, et al. Treatment of Laugier-Hunziker syndrome with the Q-switched alexandrite laser in 22 Chinese patients. Arch Dermatol Res. 2010;302:125-130.
- Ergun S, Saruhanog˘lu A, Migliari DA, et al. Refractory pigmentation associated with Laugier-Hunziker syndrome following Er:YAG laser treatment [published online December 3, 2013]. Case Rep Dent. 2013;2013:561040.
- Moore RT, Chae KA, Rhodes AR. Laugier and Hunziker pigmentation: a lentiginous proliferation of melanocytes. J Am Acad Dermatol. 2004;50(5 suppl):S70-S74.
- Lampe AK, Hampton PJ, Woodford-Richens K, et al. Laugier-Hunziker Syndrome: an important differential diagnosis for Peutz-Jeghers Syndrome. J Med Genet. 2003;40:E77.
- Baran R. Longitudinal melanotic streaks as a clue for Laugier-Hunziker syndrome. Arch Dermatol. 1979;115:1148-1149.
- Grimes P, Nordlund JJ, Pandya AG, et al. Increasing our understanding of pigmentary disorders. J Am Acad Dermatol. 2006;54(5 suppl 2):S255-S261.
- Bertherat J. Carney complex (CNC). Orphanet J Rare Dis. 2006;1:21.
- Giardiello FM, Brensinger JD, Tersemette AC, et al. Very high risk of cancer in Peutz-Jeghers Syndrome. Gastroenterology. 2000;119:1447-1453.
- Brosens LA, van Hattem WA, Jansen M, et al. Gastrointestinal polyposis syndromes. Curr Mol Med. 2007;7:29-46.
- Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59:975-986.
- Zuo YG, Ma DL, Jin HZ, et al. Treatment of Laugier-Hunziker syndrome with the Q-switched alexandrite laser in 22 Chinese patients. Arch Dermatol Res. 2010;302:125-130.
- Ergun S, Saruhanog˘lu A, Migliari DA, et al. Refractory pigmentation associated with Laugier-Hunziker syndrome following Er:YAG laser treatment [published online December 3, 2013]. Case Rep Dent. 2013;2013:561040.
Practice Points
- Laugier-Hunziker syndrome (LHS) comprises benign mucosal pigmentation in the absence of gastrointestinal pathology.
- Differentiating LHS from Peutz-Jeghers syndrome can prevent unnecessary aggressive cancer screening protocols.
- The average age of onset of LHS is 52 years and typically occurs in white adults.
- Pigmentation in LHS is most commonly distributed on the lower lips and oral mucosa.
Robot-assisted abdominoperineal resection outperforms open or laparoscopic surgery for rectal cancers
MADRID – The automaton uprising continues: Robot-assisted abdominoperineal resection (APR) can be safely performed in patients with rectal cancers within 5 cm of the anal verge, with surgical results equivalent to those seen with open or laparoscopic APR, investigators say.
Robot-assisted procedures were associated with a significantly lower rate of postoperative complications and with faster functional recovery than either laparoscopic or open surgery in a randomized trial, reported Jianmin Xu, MD, PhD., from Fudan University in Shanghai, China, and colleagues in a scientific poster presented at the European Society for Medical Oncology Congress.
“Retrospective studies have showed that robotic surgery was better than laparoscopic surgery in ensuring radical resection, reducing complications, and promoting recovery. However, there is no clinical trial reported for robotic surgery for rectal cancer. Thus, we conduct this randomized controlled trial to compare the safety and efficacy of robotic, laparoscopic, and open surgery for low rectal cancer,” they wrote.
Dr. Xu and colleagues enrolled 506 patients from 18 to 75 years of age with clinical stage T1 to T3 cancers within 5 cm of the anal verge and no distant metastases and randomly assigned them to resection with either a robot-assisted, laparoscopic, or open APR technique. Three of the 506 patients randomized did not undergo resection, leaving 503 for the per-protocol analysis presented here.
For the primary endpoint of complications rates within 30 days following surgery, the investigators found that patients assigned to robotic-assisted surgery (173 patients) had a total complication rate of 10.4%, compared with 18.8% for 176 patients assigned to laparoscopy (P = .027), and 26% for 154 assigned to open APR (P less than .001). The latter group included four patients assigned to laparoscopy whose procedures were converted to open surgery.
Among patients without complications, robot-assisted procedures were also associated with faster recovery, as measured by days to first flatus, at a median of 1 vs. 2 for laparoscopy and 3 for open procedures (P less than .001 for robots vs. each other surgery type). The robotic surgery was also significantly associated with fewer days to first automatic urination (median 2 vs. 4 for each of the other procedures; P less than .001), and with fewer days to discharge (median 5 vs. 7 for the other procedures; P less than .001).

"These are excellent postoperative complication rates reported, especially for the robotic treatment group, commented Thomas Gruenberger, MD, an oncologic surgeon at Rudolf Hospital in Vienna, in a poster discussion session.
“We all are now favoring laparoscopic surgery for these kinds of patients. The robot is a nice thing to have; however, we cannot use it in every hospital because it’s still quite expensive,” he said.
“We require – and this is a secondary endpoint of the study – long-term follow-up for local and distant outcomes,” he added,
The investigators did not report a funding source. All authors declared having no conflicts of interest. Dr. Gruenberger disclosed research funding, speakers bureau participation, and/or advisory roles with several companies.
MADRID – The automaton uprising continues: Robot-assisted abdominoperineal resection (APR) can be safely performed in patients with rectal cancers within 5 cm of the anal verge, with surgical results equivalent to those seen with open or laparoscopic APR, investigators say.
Robot-assisted procedures were associated with a significantly lower rate of postoperative complications and with faster functional recovery than either laparoscopic or open surgery in a randomized trial, reported Jianmin Xu, MD, PhD., from Fudan University in Shanghai, China, and colleagues in a scientific poster presented at the European Society for Medical Oncology Congress.
“Retrospective studies have showed that robotic surgery was better than laparoscopic surgery in ensuring radical resection, reducing complications, and promoting recovery. However, there is no clinical trial reported for robotic surgery for rectal cancer. Thus, we conduct this randomized controlled trial to compare the safety and efficacy of robotic, laparoscopic, and open surgery for low rectal cancer,” they wrote.
Dr. Xu and colleagues enrolled 506 patients from 18 to 75 years of age with clinical stage T1 to T3 cancers within 5 cm of the anal verge and no distant metastases and randomly assigned them to resection with either a robot-assisted, laparoscopic, or open APR technique. Three of the 506 patients randomized did not undergo resection, leaving 503 for the per-protocol analysis presented here.
For the primary endpoint of complications rates within 30 days following surgery, the investigators found that patients assigned to robotic-assisted surgery (173 patients) had a total complication rate of 10.4%, compared with 18.8% for 176 patients assigned to laparoscopy (P = .027), and 26% for 154 assigned to open APR (P less than .001). The latter group included four patients assigned to laparoscopy whose procedures were converted to open surgery.
Among patients without complications, robot-assisted procedures were also associated with faster recovery, as measured by days to first flatus, at a median of 1 vs. 2 for laparoscopy and 3 for open procedures (P less than .001 for robots vs. each other surgery type). The robotic surgery was also significantly associated with fewer days to first automatic urination (median 2 vs. 4 for each of the other procedures; P less than .001), and with fewer days to discharge (median 5 vs. 7 for the other procedures; P less than .001).
"These are excellent postoperative complication rates reported, especially for the robotic treatment group, commented Thomas Gruenberger, MD, an oncologic surgeon at Rudolf Hospital in Vienna, in a poster discussion session.
“We all are now favoring laparoscopic surgery for these kinds of patients. The robot is a nice thing to have; however, we cannot use it in every hospital because it’s still quite expensive,” he said.
“We require – and this is a secondary endpoint of the study – long-term follow-up for local and distant outcomes,” he added,
The investigators did not report a funding source. All authors declared having no conflicts of interest. Dr. Gruenberger disclosed research funding, speakers bureau participation, and/or advisory roles with several companies.
MADRID – The automaton uprising continues: Robot-assisted abdominoperineal resection (APR) can be safely performed in patients with rectal cancers within 5 cm of the anal verge, with surgical results equivalent to those seen with open or laparoscopic APR, investigators say.
Robot-assisted procedures were associated with a significantly lower rate of postoperative complications and with faster functional recovery than either laparoscopic or open surgery in a randomized trial, reported Jianmin Xu, MD, PhD., from Fudan University in Shanghai, China, and colleagues in a scientific poster presented at the European Society for Medical Oncology Congress.
“Retrospective studies have showed that robotic surgery was better than laparoscopic surgery in ensuring radical resection, reducing complications, and promoting recovery. However, there is no clinical trial reported for robotic surgery for rectal cancer. Thus, we conduct this randomized controlled trial to compare the safety and efficacy of robotic, laparoscopic, and open surgery for low rectal cancer,” they wrote.
Dr. Xu and colleagues enrolled 506 patients from 18 to 75 years of age with clinical stage T1 to T3 cancers within 5 cm of the anal verge and no distant metastases and randomly assigned them to resection with either a robot-assisted, laparoscopic, or open APR technique. Three of the 506 patients randomized did not undergo resection, leaving 503 for the per-protocol analysis presented here.
For the primary endpoint of complications rates within 30 days following surgery, the investigators found that patients assigned to robotic-assisted surgery (173 patients) had a total complication rate of 10.4%, compared with 18.8% for 176 patients assigned to laparoscopy (P = .027), and 26% for 154 assigned to open APR (P less than .001). The latter group included four patients assigned to laparoscopy whose procedures were converted to open surgery.
Among patients without complications, robot-assisted procedures were also associated with faster recovery, as measured by days to first flatus, at a median of 1 vs. 2 for laparoscopy and 3 for open procedures (P less than .001 for robots vs. each other surgery type). The robotic surgery was also significantly associated with fewer days to first automatic urination (median 2 vs. 4 for each of the other procedures; P less than .001), and with fewer days to discharge (median 5 vs. 7 for the other procedures; P less than .001).
"These are excellent postoperative complication rates reported, especially for the robotic treatment group, commented Thomas Gruenberger, MD, an oncologic surgeon at Rudolf Hospital in Vienna, in a poster discussion session.
“We all are now favoring laparoscopic surgery for these kinds of patients. The robot is a nice thing to have; however, we cannot use it in every hospital because it’s still quite expensive,” he said.
“We require – and this is a secondary endpoint of the study – long-term follow-up for local and distant outcomes,” he added,
The investigators did not report a funding source. All authors declared having no conflicts of interest. Dr. Gruenberger disclosed research funding, speakers bureau participation, and/or advisory roles with several companies.
AT ESMO 2017

Launch of adalimumab biosimilar Amjevita postponed
Amgen, maker of the adalimumab biosimilar Amjevita (adalimumab-atto) has reached an agreement with AbbVie, manufacturer of the originator adalimumab Humira, that halts marketing of Amjevita in the United States until 2023 and in Europe until 2018, according to a company statement.
The deal between the two manufacturers settles a patent infringement lawsuit that AbbVie brought against Amgen after it received Food and Drug Administration approval in September 2016 for seven of Humira’s nine indications: rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, plaque psoriasis, and polyarticular juvenile idiopathic arthritis. Amjevita is not approved for two of Humira’s indications, hidradenitis suppurativa and uveitis.
Amgen said in its statement that AbbVie will grant patent licenses for the use and sale of Amjevita worldwide, on a country-by-country basis, with current expectations that marketing will begin in Europe on Oct. 16, 2018, and in the United States on Jan. 31, 2023. Amjevita is named Amgevita in Europe.
Amgen, maker of the adalimumab biosimilar Amjevita (adalimumab-atto) has reached an agreement with AbbVie, manufacturer of the originator adalimumab Humira, that halts marketing of Amjevita in the United States until 2023 and in Europe until 2018, according to a company statement.
The deal between the two manufacturers settles a patent infringement lawsuit that AbbVie brought against Amgen after it received Food and Drug Administration approval in September 2016 for seven of Humira’s nine indications: rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, plaque psoriasis, and polyarticular juvenile idiopathic arthritis. Amjevita is not approved for two of Humira’s indications, hidradenitis suppurativa and uveitis.
Amgen said in its statement that AbbVie will grant patent licenses for the use and sale of Amjevita worldwide, on a country-by-country basis, with current expectations that marketing will begin in Europe on Oct. 16, 2018, and in the United States on Jan. 31, 2023. Amjevita is named Amgevita in Europe.
Amgen, maker of the adalimumab biosimilar Amjevita (adalimumab-atto) has reached an agreement with AbbVie, manufacturer of the originator adalimumab Humira, that halts marketing of Amjevita in the United States until 2023 and in Europe until 2018, according to a company statement.
The deal between the two manufacturers settles a patent infringement lawsuit that AbbVie brought against Amgen after it received Food and Drug Administration approval in September 2016 for seven of Humira’s nine indications: rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, plaque psoriasis, and polyarticular juvenile idiopathic arthritis. Amjevita is not approved for two of Humira’s indications, hidradenitis suppurativa and uveitis.
Amgen said in its statement that AbbVie will grant patent licenses for the use and sale of Amjevita worldwide, on a country-by-country basis, with current expectations that marketing will begin in Europe on Oct. 16, 2018, and in the United States on Jan. 31, 2023. Amjevita is named Amgevita in Europe.
Under our noses
If you graduated from medical school after 1990, you may be surprised to learn that there was a time when the typical general pediatrician could go through an entire day of seeing patients and not write a single prescription for a stimulant medication. In fact, he or she could go for several months without writing for any controlled substance.
ADHD is a modern phenomenon. There always have been children with “ants in their pants” who couldn’t sit still. And there always were “daydreamers” who didn’t pay attention in school. But in the 1970s, the number of children who might now be labeled as having ADHD was nowhere near the 11% often quoted for the prevalence in the current pediatric population.
Could there be some genetic selection process that is favoring the birth and survival of hyperactive and distractible children? In the last decade or two, biologists have discovered evolutionary changes in some animals occurring at pace far faster than had been previously imagined. However, a Darwinian explanation seems unlikely in the case of the emergence of ADHD.
Could it be a diet laced with high fructose sugars or artificial dyes and food coloring? While there continues to be a significant number of parents whose anecdotal observations point to a relationship between diet and behavior, to date controlled studies have not supported a dietary cause for the ADHD phenomenon.
Within a few years of beginning my dual careers as parent and pediatrician, I began to notice that children who were sleep deprived often were distractible and inattentive. Some also were hyperactive, an observation that initially seemed counterintuitive. Over the ensuing four decades, I have become more convinced that a substantial driver of the emergence of the ADHD phenomenon is the fact that the North American lifestyle places sleep so far down on its priority list that a significant percentage of both the pediatric and adult populations are sleep deprived.
I freely admit that my initial anecdotal observations have evolved to the point of an obsession. Of course, I look at the data that show that children are getting less sleep than they did a century ago and suspect that this decline must somehow be reflected in their behavior (“Never enough sleep: A brief history of sleep recommendations for children” by Matricciani et al. Pediatrics. 2012 Mar;129[3]:548-56). And, of course, I wonder whether the success and popularity of stimulant medication to treat ADHD is just chance or whether it simply could be waking up a bunch of children who aren’t getting enough sleep.
At times, it has been a lonely several decades, trying to convince parents and other pediatricians that sleep may be the answer. I can’t point to my own research because I have been too busy doing general pediatrics. I can only point to the observations of others that fit into my construct.
You can imagine the warm glow that swept over me when I came across an article in the Washington Post titled “Could some ADHD be a type of sleep disorder? That would fundamentally change how we treat it” (A.E. Cha, Sep. 20, 2017). The studies referred to in the article are not terribly earth shaking. But it was nice to read some quotes in a national newspaper from scientists who share my suspicions about sleep deprivation as a major contributor to the ADHD phenomenon. I instantly felt less lonely.
Unfortunately, it is still a long way from this token recognition in the Washington Post to convincing parents and pediatricians to do the heavy lifting that will be required to undo decades of our society’s sleep-unfriendly norms. It’s so much easier to pull out a prescription pad and write for a stimulant.

Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.”
Email him at [email protected].
If you graduated from medical school after 1990, you may be surprised to learn that there was a time when the typical general pediatrician could go through an entire day of seeing patients and not write a single prescription for a stimulant medication. In fact, he or she could go for several months without writing for any controlled substance.
ADHD is a modern phenomenon. There always have been children with “ants in their pants” who couldn’t sit still. And there always were “daydreamers” who didn’t pay attention in school. But in the 1970s, the number of children who might now be labeled as having ADHD was nowhere near the 11% often quoted for the prevalence in the current pediatric population.
Could there be some genetic selection process that is favoring the birth and survival of hyperactive and distractible children? In the last decade or two, biologists have discovered evolutionary changes in some animals occurring at pace far faster than had been previously imagined. However, a Darwinian explanation seems unlikely in the case of the emergence of ADHD.
Could it be a diet laced with high fructose sugars or artificial dyes and food coloring? While there continues to be a significant number of parents whose anecdotal observations point to a relationship between diet and behavior, to date controlled studies have not supported a dietary cause for the ADHD phenomenon.
Within a few years of beginning my dual careers as parent and pediatrician, I began to notice that children who were sleep deprived often were distractible and inattentive. Some also were hyperactive, an observation that initially seemed counterintuitive. Over the ensuing four decades, I have become more convinced that a substantial driver of the emergence of the ADHD phenomenon is the fact that the North American lifestyle places sleep so far down on its priority list that a significant percentage of both the pediatric and adult populations are sleep deprived.
I freely admit that my initial anecdotal observations have evolved to the point of an obsession. Of course, I look at the data that show that children are getting less sleep than they did a century ago and suspect that this decline must somehow be reflected in their behavior (“Never enough sleep: A brief history of sleep recommendations for children” by Matricciani et al. Pediatrics. 2012 Mar;129[3]:548-56). And, of course, I wonder whether the success and popularity of stimulant medication to treat ADHD is just chance or whether it simply could be waking up a bunch of children who aren’t getting enough sleep.
At times, it has been a lonely several decades, trying to convince parents and other pediatricians that sleep may be the answer. I can’t point to my own research because I have been too busy doing general pediatrics. I can only point to the observations of others that fit into my construct.
You can imagine the warm glow that swept over me when I came across an article in the Washington Post titled “Could some ADHD be a type of sleep disorder? That would fundamentally change how we treat it” (A.E. Cha, Sep. 20, 2017). The studies referred to in the article are not terribly earth shaking. But it was nice to read some quotes in a national newspaper from scientists who share my suspicions about sleep deprivation as a major contributor to the ADHD phenomenon. I instantly felt less lonely.
Unfortunately, it is still a long way from this token recognition in the Washington Post to convincing parents and pediatricians to do the heavy lifting that will be required to undo decades of our society’s sleep-unfriendly norms. It’s so much easier to pull out a prescription pad and write for a stimulant.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.”
Email him at [email protected].
If you graduated from medical school after 1990, you may be surprised to learn that there was a time when the typical general pediatrician could go through an entire day of seeing patients and not write a single prescription for a stimulant medication. In fact, he or she could go for several months without writing for any controlled substance.
ADHD is a modern phenomenon. There always have been children with “ants in their pants” who couldn’t sit still. And there always were “daydreamers” who didn’t pay attention in school. But in the 1970s, the number of children who might now be labeled as having ADHD was nowhere near the 11% often quoted for the prevalence in the current pediatric population.
Could there be some genetic selection process that is favoring the birth and survival of hyperactive and distractible children? In the last decade or two, biologists have discovered evolutionary changes in some animals occurring at pace far faster than had been previously imagined. However, a Darwinian explanation seems unlikely in the case of the emergence of ADHD.
Could it be a diet laced with high fructose sugars or artificial dyes and food coloring? While there continues to be a significant number of parents whose anecdotal observations point to a relationship between diet and behavior, to date controlled studies have not supported a dietary cause for the ADHD phenomenon.
Within a few years of beginning my dual careers as parent and pediatrician, I began to notice that children who were sleep deprived often were distractible and inattentive. Some also were hyperactive, an observation that initially seemed counterintuitive. Over the ensuing four decades, I have become more convinced that a substantial driver of the emergence of the ADHD phenomenon is the fact that the North American lifestyle places sleep so far down on its priority list that a significant percentage of both the pediatric and adult populations are sleep deprived.
I freely admit that my initial anecdotal observations have evolved to the point of an obsession. Of course, I look at the data that show that children are getting less sleep than they did a century ago and suspect that this decline must somehow be reflected in their behavior (“Never enough sleep: A brief history of sleep recommendations for children” by Matricciani et al. Pediatrics. 2012 Mar;129[3]:548-56). And, of course, I wonder whether the success and popularity of stimulant medication to treat ADHD is just chance or whether it simply could be waking up a bunch of children who aren’t getting enough sleep.
At times, it has been a lonely several decades, trying to convince parents and other pediatricians that sleep may be the answer. I can’t point to my own research because I have been too busy doing general pediatrics. I can only point to the observations of others that fit into my construct.
You can imagine the warm glow that swept over me when I came across an article in the Washington Post titled “Could some ADHD be a type of sleep disorder? That would fundamentally change how we treat it” (A.E. Cha, Sep. 20, 2017). The studies referred to in the article are not terribly earth shaking. But it was nice to read some quotes in a national newspaper from scientists who share my suspicions about sleep deprivation as a major contributor to the ADHD phenomenon. I instantly felt less lonely.
Unfortunately, it is still a long way from this token recognition in the Washington Post to convincing parents and pediatricians to do the heavy lifting that will be required to undo decades of our society’s sleep-unfriendly norms. It’s so much easier to pull out a prescription pad and write for a stimulant.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.”
Email him at [email protected].
Transoral robotic surgery assessed for oral lesions
A single-arm study is being conducted at the Ohio State University Comprehensive Cancer Center, Columbus, to assess transoral robotic surgery (TORS) for oral and laryngopharyngeal benign and malignant lesions using the da Vinci Robotic Surgical System.
The study started in 2007, and the estimated completion date is December 2020. Investigators hope to enroll 360 adults.
Patients are scheduled for regular postop visits to assess quality of life and other matters. Those unable to return to Ohio State University are contacted by phone or provided with the questionnaire by mail.
A single-arm study is being conducted at the Ohio State University Comprehensive Cancer Center, Columbus, to assess transoral robotic surgery (TORS) for oral and laryngopharyngeal benign and malignant lesions using the da Vinci Robotic Surgical System.
The study started in 2007, and the estimated completion date is December 2020. Investigators hope to enroll 360 adults.
Patients are scheduled for regular postop visits to assess quality of life and other matters. Those unable to return to Ohio State University are contacted by phone or provided with the questionnaire by mail.
A single-arm study is being conducted at the Ohio State University Comprehensive Cancer Center, Columbus, to assess transoral robotic surgery (TORS) for oral and laryngopharyngeal benign and malignant lesions using the da Vinci Robotic Surgical System.
The study started in 2007, and the estimated completion date is December 2020. Investigators hope to enroll 360 adults.
Patients are scheduled for regular postop visits to assess quality of life and other matters. Those unable to return to Ohio State University are contacted by phone or provided with the questionnaire by mail.
‘Without clinical prodrome’
For the most part pediatricians are insulated from death. Our little patients are surprisingly resilient. Once past that anxiety-provoking transition from placental dependence to air breathing, children will thrive in an environment that includes immunizations, potable water, and adequate nutrition. But pediatric deaths do occur infrequently in North America, and they are particularly unsettling to us because we are so unaccustomed to processing the emotions that swirl around the end of life. Did I miss something at the last health maintenance visit? Should I have taken more seriously that call last week about what sounded like a simple viral prodrome? Should I have asked that mother to make an appointment?

Their approach, which has been labeled the Robert’s Program, is particularly appealing because it is careful to address the families’ concerns about their surviving and future children. I found the inclusion of the dead child’s pediatrician and the office of the chief medical examiner in the summation of the investigation especially appealing.
However, I have trouble envisioning how this novel approach, funded by several philanthropic organizations, could be rolled out on a larger scale. Here in Maine and in many other smaller cash-strapped communities, the medical examiner’s office is overburdened with opioid overdoses and traumatic deaths. The police and sheriffs’ departments may lack sufficient training and experience to do careful scene investigations.
In reviewing the summary of the 17 deaths included in the article, I was struck by the inclusion of 3 cases in which the final cause of death was meningitis or encephalitis “without clinical prodrome.”
While a thorough investigation did eventually unearth the cause of death in these three cases, it is in that devilish prodrome that the seeds of guilt can continue to germinate. Parents and physicians will continue to wonder whether someone else with more sensitive antennae might have picked up those early signs of impending disaster. The answer is that there probably wasn’t anyone with better antennae, but there may have been someone with better luck.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.”
For the most part pediatricians are insulated from death. Our little patients are surprisingly resilient. Once past that anxiety-provoking transition from placental dependence to air breathing, children will thrive in an environment that includes immunizations, potable water, and adequate nutrition. But pediatric deaths do occur infrequently in North America, and they are particularly unsettling to us because we are so unaccustomed to processing the emotions that swirl around the end of life. Did I miss something at the last health maintenance visit? Should I have taken more seriously that call last week about what sounded like a simple viral prodrome? Should I have asked that mother to make an appointment?
Their approach, which has been labeled the Robert’s Program, is particularly appealing because it is careful to address the families’ concerns about their surviving and future children. I found the inclusion of the dead child’s pediatrician and the office of the chief medical examiner in the summation of the investigation especially appealing.
However, I have trouble envisioning how this novel approach, funded by several philanthropic organizations, could be rolled out on a larger scale. Here in Maine and in many other smaller cash-strapped communities, the medical examiner’s office is overburdened with opioid overdoses and traumatic deaths. The police and sheriffs’ departments may lack sufficient training and experience to do careful scene investigations.
In reviewing the summary of the 17 deaths included in the article, I was struck by the inclusion of 3 cases in which the final cause of death was meningitis or encephalitis “without clinical prodrome.”
While a thorough investigation did eventually unearth the cause of death in these three cases, it is in that devilish prodrome that the seeds of guilt can continue to germinate. Parents and physicians will continue to wonder whether someone else with more sensitive antennae might have picked up those early signs of impending disaster. The answer is that there probably wasn’t anyone with better antennae, but there may have been someone with better luck.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.”
For the most part pediatricians are insulated from death. Our little patients are surprisingly resilient. Once past that anxiety-provoking transition from placental dependence to air breathing, children will thrive in an environment that includes immunizations, potable water, and adequate nutrition. But pediatric deaths do occur infrequently in North America, and they are particularly unsettling to us because we are so unaccustomed to processing the emotions that swirl around the end of life. Did I miss something at the last health maintenance visit? Should I have taken more seriously that call last week about what sounded like a simple viral prodrome? Should I have asked that mother to make an appointment?
Their approach, which has been labeled the Robert’s Program, is particularly appealing because it is careful to address the families’ concerns about their surviving and future children. I found the inclusion of the dead child’s pediatrician and the office of the chief medical examiner in the summation of the investigation especially appealing.
However, I have trouble envisioning how this novel approach, funded by several philanthropic organizations, could be rolled out on a larger scale. Here in Maine and in many other smaller cash-strapped communities, the medical examiner’s office is overburdened with opioid overdoses and traumatic deaths. The police and sheriffs’ departments may lack sufficient training and experience to do careful scene investigations.
In reviewing the summary of the 17 deaths included in the article, I was struck by the inclusion of 3 cases in which the final cause of death was meningitis or encephalitis “without clinical prodrome.”
While a thorough investigation did eventually unearth the cause of death in these three cases, it is in that devilish prodrome that the seeds of guilt can continue to germinate. Parents and physicians will continue to wonder whether someone else with more sensitive antennae might have picked up those early signs of impending disaster. The answer is that there probably wasn’t anyone with better antennae, but there may have been someone with better luck.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.”
Robotic surgery technologies have unique learning curves for trainees
Training surgeons to use robotic technology involves learning curves, and a study has found that robotic technologies have unique learning curve profiles that have implications for the time and number of procedures needed to achieve competence.
Giorgio Mazzon, MD, of the Institute of Urology at University College Hospital, London, and his colleagues reviewed the literature on training surgeons in the use of a variety of technologies for urological procedures. They analyzed learning curves for virtual reality robotic simulators, robot-assisted laparoscopic radical prostatectomy (RALP), robot-assisted radical cystectomy (RARC), and robot-assisted partial nephrectomy (RAPN) (Curr Urol Rep. 2017 Sep 23;18:89).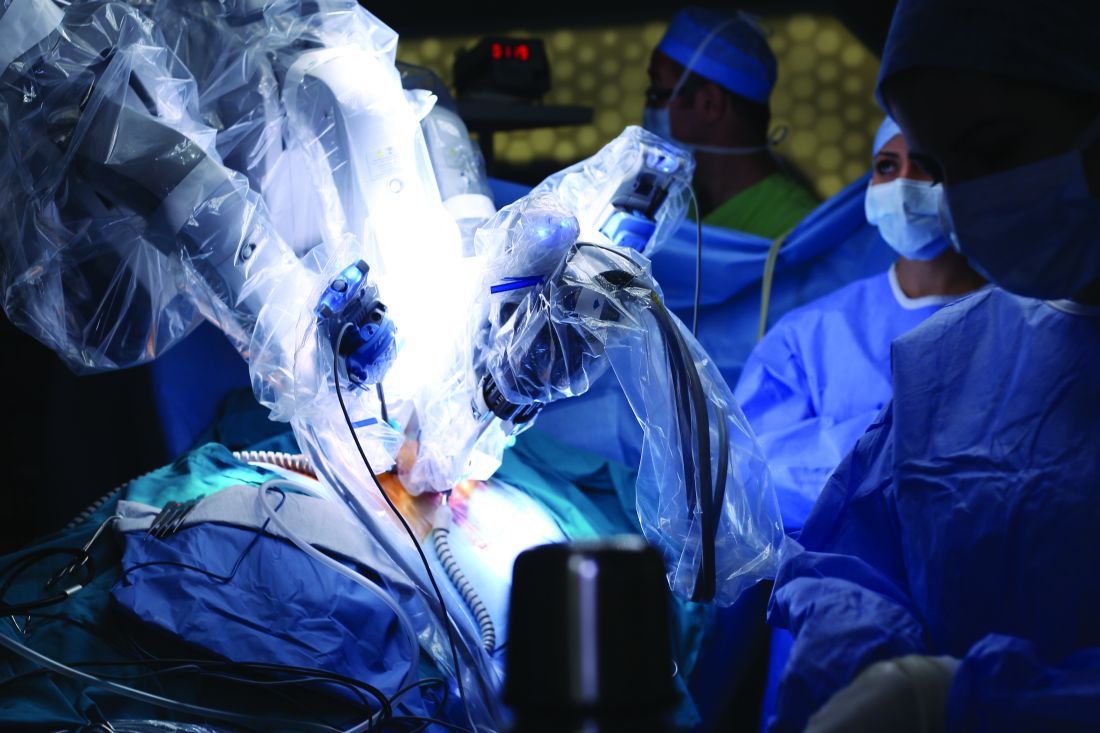
RARC learning curves are more rapid than RALP, but this may be due to the fact that most surgeons practice RALP before learning RARC. It is estimated that it takes 21 procedures for operating time to plateau and 30 patients for proper lymph node yield and positive surgical margins of less than 5% to occur (Eur Urol. 2010 Aug;58[2]:197-202).Safety and competence in RAPN is usually defined by operating times, warm ischemic time, positive surgical margin, and complication rates. It has been reported that RAPN can be safely performed with completion of 25-30 cases (Eur Urol. 2010 Jul;58[1]:127-32).The results of the review “should inform trainers and trainees on what outcomes are expected at a given stage of training,” according to the investigators.
They reported no relevant financial disclosures.
Training surgeons to use robotic technology involves learning curves, and a study has found that robotic technologies have unique learning curve profiles that have implications for the time and number of procedures needed to achieve competence.
Giorgio Mazzon, MD, of the Institute of Urology at University College Hospital, London, and his colleagues reviewed the literature on training surgeons in the use of a variety of technologies for urological procedures. They analyzed learning curves for virtual reality robotic simulators, robot-assisted laparoscopic radical prostatectomy (RALP), robot-assisted radical cystectomy (RARC), and robot-assisted partial nephrectomy (RAPN) (Curr Urol Rep. 2017 Sep 23;18:89).
RARC learning curves are more rapid than RALP, but this may be due to the fact that most surgeons practice RALP before learning RARC. It is estimated that it takes 21 procedures for operating time to plateau and 30 patients for proper lymph node yield and positive surgical margins of less than 5% to occur (Eur Urol. 2010 Aug;58[2]:197-202).Safety and competence in RAPN is usually defined by operating times, warm ischemic time, positive surgical margin, and complication rates. It has been reported that RAPN can be safely performed with completion of 25-30 cases (Eur Urol. 2010 Jul;58[1]:127-32).The results of the review “should inform trainers and trainees on what outcomes are expected at a given stage of training,” according to the investigators.
They reported no relevant financial disclosures.
Training surgeons to use robotic technology involves learning curves, and a study has found that robotic technologies have unique learning curve profiles that have implications for the time and number of procedures needed to achieve competence.
Giorgio Mazzon, MD, of the Institute of Urology at University College Hospital, London, and his colleagues reviewed the literature on training surgeons in the use of a variety of technologies for urological procedures. They analyzed learning curves for virtual reality robotic simulators, robot-assisted laparoscopic radical prostatectomy (RALP), robot-assisted radical cystectomy (RARC), and robot-assisted partial nephrectomy (RAPN) (Curr Urol Rep. 2017 Sep 23;18:89).
RARC learning curves are more rapid than RALP, but this may be due to the fact that most surgeons practice RALP before learning RARC. It is estimated that it takes 21 procedures for operating time to plateau and 30 patients for proper lymph node yield and positive surgical margins of less than 5% to occur (Eur Urol. 2010 Aug;58[2]:197-202).Safety and competence in RAPN is usually defined by operating times, warm ischemic time, positive surgical margin, and complication rates. It has been reported that RAPN can be safely performed with completion of 25-30 cases (Eur Urol. 2010 Jul;58[1]:127-32).The results of the review “should inform trainers and trainees on what outcomes are expected at a given stage of training,” according to the investigators.
They reported no relevant financial disclosures.
FROM CURRENT UROLOGY REPORTS
Key clinical point:
Major finding: For virtual reality training programs, recommendations for achieving safety and competence is between 4 and 12 hours of training in a simulator.
Data source: Review of studies of learning curves in robotic urological surgery.
Disclosures: The researchers reported no financial disclosures.