User login
No longer a death sentence, HIV diagnosis still hits hard
Veronica Brady and her team at the University of Texas Health Science Center, Houston, sat down with 37 people diagnosed with HIV or AIDS to ask them what that felt like.
“The results were really eye-opening and sad,” says Brady, PhD, RN, from the Cizik School of Nursing with UTHealth, Houston.
Many of the people Dr. Brady and her team spoke with were diagnosed through routine or random testing. They ranged in age from 21 years to 65 and said they did not know how they had been infected and felt shocked, freaked out, scared, and in a state of disbelief.
Their conversations about being diagnosed with HIV, presented at the annual meeting of the Association of Nurses in AIDS Care in New Orleans, also described how symptoms of the disease or side effects from treatment can have a huge impact on the daily lives of those affected.
Jesse Milan Jr., president of AIDS United, an HIV advocacy organization based in Washington, D.C., says he recognizes all of these feelings from his own experience with HIV after being diagnosed more than 40 years ago.
“All of those have come up over the years,” he says. “They are all relevant and important at different times.”
For Mr. Milan, less was known about the virus at the time of his diagnosis, and he watched loved ones die. He lived to see the introduction of antiretroviral therapies and receive treatment when his partner and many of his friends did not.
Effective treatments
There is a marked difference between the reaction of people diagnosed with HIV years ago and those diagnosed more recently, Dr. Brady explains. Those diagnosed before much was known about the virus and before there were effective treatments were more frightened, she says, whereas people hearing the news recently are much less worried and understand that if they take their medication, they will be fine.
Still, Mr. Milan says when he talks to people diagnosed now, they seem to experience more shame and embarrassment than before. Because it is long known how to prevent HIV infection, they often worry what people will think if they disclose their status. “It makes things harder for people diagnosed today,” says Mr. Milan. “There is a different level of embarrassment tinged with, ‘Why was I so stupid?’ ”
Diagnosis can also be hard on health care professionals, says Dr. Brady. “You never want to tell anyone they’re sick with a chronic disease, especially younger people,” she adds. “You know you’re adding a burden to someone’s life.”
Symptoms and side effects of treatment also had an important impact on the people in this report, with most aspects of their lives affected, including work, relationships, mood, and daily activities.
Clinicians should be supportive and spend some time sitting with patients as they come to terms with the diagnosis and its implications. They should help them understand what to expect and talk about how – or whether – to talk about their status with family and friends. “You need to show you care about the person and that they are not alone,” Dr. Brady says.
And most of all, clinicians need to explain that patients can live a long and healthy life and go on to become whoever they want to be. “Twenty years ago, we wouldn’t have as hopeful a message as we do now,” she says.
Hope is the most important thing for doctors and nurses to communicate to their patients. “There are medications available, and it will be okay. You don’t have to die,” Mr. Milan says. “That’s the core message that everyone needs to hear, whether they were diagnosed 30 years ago or 30 minutes ago.”
A version of this article appeared on Medscape.com.
Veronica Brady and her team at the University of Texas Health Science Center, Houston, sat down with 37 people diagnosed with HIV or AIDS to ask them what that felt like.
“The results were really eye-opening and sad,” says Brady, PhD, RN, from the Cizik School of Nursing with UTHealth, Houston.
Many of the people Dr. Brady and her team spoke with were diagnosed through routine or random testing. They ranged in age from 21 years to 65 and said they did not know how they had been infected and felt shocked, freaked out, scared, and in a state of disbelief.
Their conversations about being diagnosed with HIV, presented at the annual meeting of the Association of Nurses in AIDS Care in New Orleans, also described how symptoms of the disease or side effects from treatment can have a huge impact on the daily lives of those affected.
Jesse Milan Jr., president of AIDS United, an HIV advocacy organization based in Washington, D.C., says he recognizes all of these feelings from his own experience with HIV after being diagnosed more than 40 years ago.
“All of those have come up over the years,” he says. “They are all relevant and important at different times.”
For Mr. Milan, less was known about the virus at the time of his diagnosis, and he watched loved ones die. He lived to see the introduction of antiretroviral therapies and receive treatment when his partner and many of his friends did not.
Effective treatments
There is a marked difference between the reaction of people diagnosed with HIV years ago and those diagnosed more recently, Dr. Brady explains. Those diagnosed before much was known about the virus and before there were effective treatments were more frightened, she says, whereas people hearing the news recently are much less worried and understand that if they take their medication, they will be fine.
Still, Mr. Milan says when he talks to people diagnosed now, they seem to experience more shame and embarrassment than before. Because it is long known how to prevent HIV infection, they often worry what people will think if they disclose their status. “It makes things harder for people diagnosed today,” says Mr. Milan. “There is a different level of embarrassment tinged with, ‘Why was I so stupid?’ ”
Diagnosis can also be hard on health care professionals, says Dr. Brady. “You never want to tell anyone they’re sick with a chronic disease, especially younger people,” she adds. “You know you’re adding a burden to someone’s life.”
Symptoms and side effects of treatment also had an important impact on the people in this report, with most aspects of their lives affected, including work, relationships, mood, and daily activities.
Clinicians should be supportive and spend some time sitting with patients as they come to terms with the diagnosis and its implications. They should help them understand what to expect and talk about how – or whether – to talk about their status with family and friends. “You need to show you care about the person and that they are not alone,” Dr. Brady says.
And most of all, clinicians need to explain that patients can live a long and healthy life and go on to become whoever they want to be. “Twenty years ago, we wouldn’t have as hopeful a message as we do now,” she says.
Hope is the most important thing for doctors and nurses to communicate to their patients. “There are medications available, and it will be okay. You don’t have to die,” Mr. Milan says. “That’s the core message that everyone needs to hear, whether they were diagnosed 30 years ago or 30 minutes ago.”
A version of this article appeared on Medscape.com.
Veronica Brady and her team at the University of Texas Health Science Center, Houston, sat down with 37 people diagnosed with HIV or AIDS to ask them what that felt like.
“The results were really eye-opening and sad,” says Brady, PhD, RN, from the Cizik School of Nursing with UTHealth, Houston.
Many of the people Dr. Brady and her team spoke with were diagnosed through routine or random testing. They ranged in age from 21 years to 65 and said they did not know how they had been infected and felt shocked, freaked out, scared, and in a state of disbelief.
Their conversations about being diagnosed with HIV, presented at the annual meeting of the Association of Nurses in AIDS Care in New Orleans, also described how symptoms of the disease or side effects from treatment can have a huge impact on the daily lives of those affected.
Jesse Milan Jr., president of AIDS United, an HIV advocacy organization based in Washington, D.C., says he recognizes all of these feelings from his own experience with HIV after being diagnosed more than 40 years ago.
“All of those have come up over the years,” he says. “They are all relevant and important at different times.”
For Mr. Milan, less was known about the virus at the time of his diagnosis, and he watched loved ones die. He lived to see the introduction of antiretroviral therapies and receive treatment when his partner and many of his friends did not.
Effective treatments
There is a marked difference between the reaction of people diagnosed with HIV years ago and those diagnosed more recently, Dr. Brady explains. Those diagnosed before much was known about the virus and before there were effective treatments were more frightened, she says, whereas people hearing the news recently are much less worried and understand that if they take their medication, they will be fine.
Still, Mr. Milan says when he talks to people diagnosed now, they seem to experience more shame and embarrassment than before. Because it is long known how to prevent HIV infection, they often worry what people will think if they disclose their status. “It makes things harder for people diagnosed today,” says Mr. Milan. “There is a different level of embarrassment tinged with, ‘Why was I so stupid?’ ”
Diagnosis can also be hard on health care professionals, says Dr. Brady. “You never want to tell anyone they’re sick with a chronic disease, especially younger people,” she adds. “You know you’re adding a burden to someone’s life.”
Symptoms and side effects of treatment also had an important impact on the people in this report, with most aspects of their lives affected, including work, relationships, mood, and daily activities.
Clinicians should be supportive and spend some time sitting with patients as they come to terms with the diagnosis and its implications. They should help them understand what to expect and talk about how – or whether – to talk about their status with family and friends. “You need to show you care about the person and that they are not alone,” Dr. Brady says.
And most of all, clinicians need to explain that patients can live a long and healthy life and go on to become whoever they want to be. “Twenty years ago, we wouldn’t have as hopeful a message as we do now,” she says.
Hope is the most important thing for doctors and nurses to communicate to their patients. “There are medications available, and it will be okay. You don’t have to die,” Mr. Milan says. “That’s the core message that everyone needs to hear, whether they were diagnosed 30 years ago or 30 minutes ago.”
A version of this article appeared on Medscape.com.
How can we improve our approach to cancer-related fatigue?
MADRID – These were the messages delivered by speakers at the annual meeting of the European Society for Medical Oncology during a session titled “The Multiple Faces of Fatigue in the Cancer Ecosystem.”
Cancer-related fatigue is said to affect 40% of patients at the time of cancer diagnosis, 65% of patients during active or maintenance treatment, 21%-52% of patients in the 5 years following cancer diagnosis, and even one quarter of patients who are between 5 and 30 years post diagnosis, said Florian Scotté, MD, PhD, head of the interdisciplinary department for the Organization of Patient Pathways at Gustave Roussy Institute in Villejuif, France.
However, he underlines that “up to 50% of cancer survivors report never having discussed their cancer-related fatigue or received advice or support on how to manage it.”
What exactly is this fatigue? According to the definition set out in the ESMO 2020 recommendations and repeated word for word in the latest recommendations issued by the National Comprehensive Cancer Network published on Oct. 6, cancer-related fatigue is “a distressing, persistent, subjective sense of physical, emotional, and/or cognitive tiredness or exhaustion related to cancer or cancer treatment that is not proportional to recent activity and interferes with usual functioning.”
Mechanisms at play
The mechanisms at play in cancer-related fatigue are clinical, molecular, and psychological, stated Dr. Scotté.
In terms of the clinical factors responsible for patients’ fatigue, comorbidities such as anemia, diabetes, heart disease, and even psychological conditions are significant elements. In addition, taking medicinal products such as antidepressants or beta-blockers can also cause fatigue. Furthermore, cancer treatment itself has many possible side effects, such as anemia, hypothyroidism, insomnia, pain, and hypopituitarism.
In terms of molecular and physiologic factors, central nervous system dysfunction (inflammation, hypothalamic-pituitary-adrenal axis) leads to perceived reduced physical and mental capacity with no clear motor or cognitive deficiencies. Changes in the peripheral nervous system also cause reduced energy metabolism, which hampers the response of muscles to stimuli, possibly even limiting endurance. Finally, several studies have shown that systemic inflammation is involved in the onset of fatigue.
Dr. Scotté also highlighted the importance of psychological factors, citing depression, psychosocial stress before treatment, negative attention to symptoms, and fear of relapse as key features in the development of cancer-related fatigue.
Among the risk factors for developing cancer-related fatigue, the speaker mentioned a combination of genetic, psychological, and biobehavioral factors (such as preexisting risk factors, depression, sleep disorders, physical inactivity, BMI, smoking, alcohol consumption, and adaptability).
Screen and diagnose
“Cancer-related fatigue is one of the most underestimated and least researched side effects,” said Christina Ruhlmann, MD, PhD, an oncology consultant at Odense (Denmark) University Hospital. “It is important to screen for fatigue in cancer patients.”
There are several tools available to enable this screening, she noted. The EORTC Core Quality of Life Questionnaire (EORTC QLQ-C30) is a three-item subscale evaluating the symptoms of fatigue, weakness, and lack of energy. The MD Anderson Symptom Inventory (13 items) assesses fatigue, sleep disorders, and drowsiness. The numeric rating scale (NRS) for fatigue is an 11-point visual self-assessment scale comprising a single element, with 0 representing no fatigue and 10 representing intense fatigue.
When screening for cancer-related fatigue, whenever a score of 4 or more is obtained on the NRS, a diagnostic assessment is needed based on clinical history-taking, fatigue assessment, and evaluation of comorbidities.
When taking the clinical history, information should be obtained on the type of condition, its stage, any relapse or progression, metastases, the date of diagnosis, length of treatment, any cancer or surgical treatments carried out, other treatments administered, and the risk for drug interactions.
In addition, to assess fatigue, the diagnostic process consists of documenting the start, type, and duration of the fatigue, as well as the presence of attenuating factors and interference with activities of daily living and leisure activities.
Seeking information regarding environmental factors such as availability of a support network of family and friends or financial resources is also paramount, said Dr. Ruhlmann.
Finally, contributory factors that may require treatment must be assessed. They include pain, emotional distress, anemia, sleep disorders, nutritional deficiencies, inactivity, smoking and alcohol consumption, and comorbidities (such as cardiac, endocrine, gastrointestinal, hepatic, infectious, and renal conditions).
The following two simple questions can be used to screen for symptoms of depression quickly:
- Over the past month, have you often felt despondent, sad, depressed, or in despair?
- Over the past month, have you found less pleasure than usual in doing the things you normally enjoy doing?
How to treat?
“All of the elements associated with fatigue that can be taken into account ought to be,” stressed Dr. Ruhlmann before insisting on the key role played by physical activity in combating the feeling of exhaustion.
The ESMO recommendations indicate that, according to the results of randomized clinical trials and systematic literature reviews, physical exercise can be recommended in patients with cancer who do not have cachexia (level of evidence I, B).
The type of physical activity recommended is moderate, aerobic, and functional strength exercises (I, B). Walking, aerobic exercises at home, and strength exercises are recommended to improve cancer-related fatigue and quality of life (II, B). “They help with fatigue and also with side effects such as depression, anxiety, pain, and muscle strength,” said Dr. Ruhlmann.
Alongside exercise, and with a lower level of evidence, pharmacologic treatments can sometimes be used (II, B; II, D). Short-term use of dexamethasone or methylprednisolone is recommended for managing fatigue linked to metastatic cancer except during the course of immunotherapy (II, B).
The ESMO expert group did not reach a consensus on the use of methylphenidate, dexmethylphenidate, slow-release methylphenidate, and dexamphetamine.
Modafinil and armodafinil, antidepressants (especially paroxetine), donepezil and eszopiclone, megestrol acetate, and melatonin are not recommended (II, D).
No consensus could be reached on nutraceuticals, and they are not recommended, said Dr. Ruhlmann (II, C; II, D).
Finally, psychosocial interventions in the form of information, advice, psychoeducation, and cognitive-behavioral therapy are useful tools (II, B).
Another area being explored is the gut microbiota. “Research into the microbiota and its role in systemic inflammation is underway and could pave the way for future strategies for managing cancer-related fatigue,” said Dr. Ruhlmann. “Fatigue is a subjective experience, unlike other symptoms. It’s what those people suffering from it say it is!”
This article was translated from the Medscape French edition.
MADRID – These were the messages delivered by speakers at the annual meeting of the European Society for Medical Oncology during a session titled “The Multiple Faces of Fatigue in the Cancer Ecosystem.”
Cancer-related fatigue is said to affect 40% of patients at the time of cancer diagnosis, 65% of patients during active or maintenance treatment, 21%-52% of patients in the 5 years following cancer diagnosis, and even one quarter of patients who are between 5 and 30 years post diagnosis, said Florian Scotté, MD, PhD, head of the interdisciplinary department for the Organization of Patient Pathways at Gustave Roussy Institute in Villejuif, France.
However, he underlines that “up to 50% of cancer survivors report never having discussed their cancer-related fatigue or received advice or support on how to manage it.”
What exactly is this fatigue? According to the definition set out in the ESMO 2020 recommendations and repeated word for word in the latest recommendations issued by the National Comprehensive Cancer Network published on Oct. 6, cancer-related fatigue is “a distressing, persistent, subjective sense of physical, emotional, and/or cognitive tiredness or exhaustion related to cancer or cancer treatment that is not proportional to recent activity and interferes with usual functioning.”
Mechanisms at play
The mechanisms at play in cancer-related fatigue are clinical, molecular, and psychological, stated Dr. Scotté.
In terms of the clinical factors responsible for patients’ fatigue, comorbidities such as anemia, diabetes, heart disease, and even psychological conditions are significant elements. In addition, taking medicinal products such as antidepressants or beta-blockers can also cause fatigue. Furthermore, cancer treatment itself has many possible side effects, such as anemia, hypothyroidism, insomnia, pain, and hypopituitarism.
In terms of molecular and physiologic factors, central nervous system dysfunction (inflammation, hypothalamic-pituitary-adrenal axis) leads to perceived reduced physical and mental capacity with no clear motor or cognitive deficiencies. Changes in the peripheral nervous system also cause reduced energy metabolism, which hampers the response of muscles to stimuli, possibly even limiting endurance. Finally, several studies have shown that systemic inflammation is involved in the onset of fatigue.
Dr. Scotté also highlighted the importance of psychological factors, citing depression, psychosocial stress before treatment, negative attention to symptoms, and fear of relapse as key features in the development of cancer-related fatigue.
Among the risk factors for developing cancer-related fatigue, the speaker mentioned a combination of genetic, psychological, and biobehavioral factors (such as preexisting risk factors, depression, sleep disorders, physical inactivity, BMI, smoking, alcohol consumption, and adaptability).
Screen and diagnose
“Cancer-related fatigue is one of the most underestimated and least researched side effects,” said Christina Ruhlmann, MD, PhD, an oncology consultant at Odense (Denmark) University Hospital. “It is important to screen for fatigue in cancer patients.”
There are several tools available to enable this screening, she noted. The EORTC Core Quality of Life Questionnaire (EORTC QLQ-C30) is a three-item subscale evaluating the symptoms of fatigue, weakness, and lack of energy. The MD Anderson Symptom Inventory (13 items) assesses fatigue, sleep disorders, and drowsiness. The numeric rating scale (NRS) for fatigue is an 11-point visual self-assessment scale comprising a single element, with 0 representing no fatigue and 10 representing intense fatigue.
When screening for cancer-related fatigue, whenever a score of 4 or more is obtained on the NRS, a diagnostic assessment is needed based on clinical history-taking, fatigue assessment, and evaluation of comorbidities.
When taking the clinical history, information should be obtained on the type of condition, its stage, any relapse or progression, metastases, the date of diagnosis, length of treatment, any cancer or surgical treatments carried out, other treatments administered, and the risk for drug interactions.
In addition, to assess fatigue, the diagnostic process consists of documenting the start, type, and duration of the fatigue, as well as the presence of attenuating factors and interference with activities of daily living and leisure activities.
Seeking information regarding environmental factors such as availability of a support network of family and friends or financial resources is also paramount, said Dr. Ruhlmann.
Finally, contributory factors that may require treatment must be assessed. They include pain, emotional distress, anemia, sleep disorders, nutritional deficiencies, inactivity, smoking and alcohol consumption, and comorbidities (such as cardiac, endocrine, gastrointestinal, hepatic, infectious, and renal conditions).
The following two simple questions can be used to screen for symptoms of depression quickly:
- Over the past month, have you often felt despondent, sad, depressed, or in despair?
- Over the past month, have you found less pleasure than usual in doing the things you normally enjoy doing?
How to treat?
“All of the elements associated with fatigue that can be taken into account ought to be,” stressed Dr. Ruhlmann before insisting on the key role played by physical activity in combating the feeling of exhaustion.
The ESMO recommendations indicate that, according to the results of randomized clinical trials and systematic literature reviews, physical exercise can be recommended in patients with cancer who do not have cachexia (level of evidence I, B).
The type of physical activity recommended is moderate, aerobic, and functional strength exercises (I, B). Walking, aerobic exercises at home, and strength exercises are recommended to improve cancer-related fatigue and quality of life (II, B). “They help with fatigue and also with side effects such as depression, anxiety, pain, and muscle strength,” said Dr. Ruhlmann.
Alongside exercise, and with a lower level of evidence, pharmacologic treatments can sometimes be used (II, B; II, D). Short-term use of dexamethasone or methylprednisolone is recommended for managing fatigue linked to metastatic cancer except during the course of immunotherapy (II, B).
The ESMO expert group did not reach a consensus on the use of methylphenidate, dexmethylphenidate, slow-release methylphenidate, and dexamphetamine.
Modafinil and armodafinil, antidepressants (especially paroxetine), donepezil and eszopiclone, megestrol acetate, and melatonin are not recommended (II, D).
No consensus could be reached on nutraceuticals, and they are not recommended, said Dr. Ruhlmann (II, C; II, D).
Finally, psychosocial interventions in the form of information, advice, psychoeducation, and cognitive-behavioral therapy are useful tools (II, B).
Another area being explored is the gut microbiota. “Research into the microbiota and its role in systemic inflammation is underway and could pave the way for future strategies for managing cancer-related fatigue,” said Dr. Ruhlmann. “Fatigue is a subjective experience, unlike other symptoms. It’s what those people suffering from it say it is!”
This article was translated from the Medscape French edition.
MADRID – These were the messages delivered by speakers at the annual meeting of the European Society for Medical Oncology during a session titled “The Multiple Faces of Fatigue in the Cancer Ecosystem.”
Cancer-related fatigue is said to affect 40% of patients at the time of cancer diagnosis, 65% of patients during active or maintenance treatment, 21%-52% of patients in the 5 years following cancer diagnosis, and even one quarter of patients who are between 5 and 30 years post diagnosis, said Florian Scotté, MD, PhD, head of the interdisciplinary department for the Organization of Patient Pathways at Gustave Roussy Institute in Villejuif, France.
However, he underlines that “up to 50% of cancer survivors report never having discussed their cancer-related fatigue or received advice or support on how to manage it.”
What exactly is this fatigue? According to the definition set out in the ESMO 2020 recommendations and repeated word for word in the latest recommendations issued by the National Comprehensive Cancer Network published on Oct. 6, cancer-related fatigue is “a distressing, persistent, subjective sense of physical, emotional, and/or cognitive tiredness or exhaustion related to cancer or cancer treatment that is not proportional to recent activity and interferes with usual functioning.”
Mechanisms at play
The mechanisms at play in cancer-related fatigue are clinical, molecular, and psychological, stated Dr. Scotté.
In terms of the clinical factors responsible for patients’ fatigue, comorbidities such as anemia, diabetes, heart disease, and even psychological conditions are significant elements. In addition, taking medicinal products such as antidepressants or beta-blockers can also cause fatigue. Furthermore, cancer treatment itself has many possible side effects, such as anemia, hypothyroidism, insomnia, pain, and hypopituitarism.
In terms of molecular and physiologic factors, central nervous system dysfunction (inflammation, hypothalamic-pituitary-adrenal axis) leads to perceived reduced physical and mental capacity with no clear motor or cognitive deficiencies. Changes in the peripheral nervous system also cause reduced energy metabolism, which hampers the response of muscles to stimuli, possibly even limiting endurance. Finally, several studies have shown that systemic inflammation is involved in the onset of fatigue.
Dr. Scotté also highlighted the importance of psychological factors, citing depression, psychosocial stress before treatment, negative attention to symptoms, and fear of relapse as key features in the development of cancer-related fatigue.
Among the risk factors for developing cancer-related fatigue, the speaker mentioned a combination of genetic, psychological, and biobehavioral factors (such as preexisting risk factors, depression, sleep disorders, physical inactivity, BMI, smoking, alcohol consumption, and adaptability).
Screen and diagnose
“Cancer-related fatigue is one of the most underestimated and least researched side effects,” said Christina Ruhlmann, MD, PhD, an oncology consultant at Odense (Denmark) University Hospital. “It is important to screen for fatigue in cancer patients.”
There are several tools available to enable this screening, she noted. The EORTC Core Quality of Life Questionnaire (EORTC QLQ-C30) is a three-item subscale evaluating the symptoms of fatigue, weakness, and lack of energy. The MD Anderson Symptom Inventory (13 items) assesses fatigue, sleep disorders, and drowsiness. The numeric rating scale (NRS) for fatigue is an 11-point visual self-assessment scale comprising a single element, with 0 representing no fatigue and 10 representing intense fatigue.
When screening for cancer-related fatigue, whenever a score of 4 or more is obtained on the NRS, a diagnostic assessment is needed based on clinical history-taking, fatigue assessment, and evaluation of comorbidities.
When taking the clinical history, information should be obtained on the type of condition, its stage, any relapse or progression, metastases, the date of diagnosis, length of treatment, any cancer or surgical treatments carried out, other treatments administered, and the risk for drug interactions.
In addition, to assess fatigue, the diagnostic process consists of documenting the start, type, and duration of the fatigue, as well as the presence of attenuating factors and interference with activities of daily living and leisure activities.
Seeking information regarding environmental factors such as availability of a support network of family and friends or financial resources is also paramount, said Dr. Ruhlmann.
Finally, contributory factors that may require treatment must be assessed. They include pain, emotional distress, anemia, sleep disorders, nutritional deficiencies, inactivity, smoking and alcohol consumption, and comorbidities (such as cardiac, endocrine, gastrointestinal, hepatic, infectious, and renal conditions).
The following two simple questions can be used to screen for symptoms of depression quickly:
- Over the past month, have you often felt despondent, sad, depressed, or in despair?
- Over the past month, have you found less pleasure than usual in doing the things you normally enjoy doing?
How to treat?
“All of the elements associated with fatigue that can be taken into account ought to be,” stressed Dr. Ruhlmann before insisting on the key role played by physical activity in combating the feeling of exhaustion.
The ESMO recommendations indicate that, according to the results of randomized clinical trials and systematic literature reviews, physical exercise can be recommended in patients with cancer who do not have cachexia (level of evidence I, B).
The type of physical activity recommended is moderate, aerobic, and functional strength exercises (I, B). Walking, aerobic exercises at home, and strength exercises are recommended to improve cancer-related fatigue and quality of life (II, B). “They help with fatigue and also with side effects such as depression, anxiety, pain, and muscle strength,” said Dr. Ruhlmann.
Alongside exercise, and with a lower level of evidence, pharmacologic treatments can sometimes be used (II, B; II, D). Short-term use of dexamethasone or methylprednisolone is recommended for managing fatigue linked to metastatic cancer except during the course of immunotherapy (II, B).
The ESMO expert group did not reach a consensus on the use of methylphenidate, dexmethylphenidate, slow-release methylphenidate, and dexamphetamine.
Modafinil and armodafinil, antidepressants (especially paroxetine), donepezil and eszopiclone, megestrol acetate, and melatonin are not recommended (II, D).
No consensus could be reached on nutraceuticals, and they are not recommended, said Dr. Ruhlmann (II, C; II, D).
Finally, psychosocial interventions in the form of information, advice, psychoeducation, and cognitive-behavioral therapy are useful tools (II, B).
Another area being explored is the gut microbiota. “Research into the microbiota and its role in systemic inflammation is underway and could pave the way for future strategies for managing cancer-related fatigue,” said Dr. Ruhlmann. “Fatigue is a subjective experience, unlike other symptoms. It’s what those people suffering from it say it is!”
This article was translated from the Medscape French edition.
AT ESMO 2023
Forgetfulness and mood fluctuations
This patient's symptoms go beyond just memory problems: She has difficulty with daily tasks, shows behavioral changes, and has significant communication difficulties — symptoms not found in mild cognitive impairment. While the patient has some behavioral changes, she does not exhibit the pronounced personality changes typical of frontotemporal dementia. Finally, the patient's cognitive decline is gradual and consistent without the stepwise progression typical of vascular dementia. Given the comprehensive presentation of the patient's symptoms and the results of her clinical investigations, middle-stage Alzheimer's disease is the most fitting diagnosis.
Alzheimer's disease is a progressive and irreversible brain disorder that affects memory, behavior, and cognitive skills. This condition causes the degeneration and death of brain cells, leading to various cognitive issues. Alzheimer's disease is the most common cause of dementia and accounts for 60%-80% of dementia cases. Although the exact cause is unknown, it is believed to result from genetic, lifestyle, and environmental factors. Alzheimer's disease progresses through stages — mild (early stage), moderate (middle stage), and severe (late stage) — and each stage has different signs and symptoms.
Alzheimer's disease is commonly observed in individuals 65 years or older, as age is the most significant risk factor. Another risk factor for Alzheimer's disease is family history; individuals who have parents or siblings with Alzheimer's disease are more likely to develop the disease. The risk increases with the number of family members diagnosed with the disease. Genetics also contribute to the development of Alzheimer's disease. Genes for developing Alzheimer's disease have been classified as deterministic and risk genes, which imply that they can cause the disease or increase the risk of developing it; however, the deterministic gene, which almost guarantees the occurrence of Alzheimer's, is rare and is found in less than 1% of cases. Experiencing a head injury is also a possible risk factor for Alzheimer's disease.
Accurate diagnosis of Alzheimer's disease requires a thorough history and physical examination. Gathering information from the patient's family and caregivers is important because some patients may not be aware of their condition. It is common for Alzheimer's disease patients to experience "sundowning," which causes confusion, agitation, and behavioral issues in the evening. A comprehensive physical examination, including a detailed neurologic and mental status exam, is necessary to determine the stage of the disease and rule out other conditions. Typically, the neurologic exam of Alzheimer's disease patients is normal.
Volumetric MRI is a recent technique that allows precise measurement of changes in brain volume. In Alzheimer's disease, shrinkage in the medial temporal lobe is visible through volumetric MRI. However, hippocampal atrophy is also a normal part of age-related memory decline, which raises doubts about the appropriateness of using volumetric MRI for early detection of Alzheimer's disease. The full potential of volumetric MRI in aiding the diagnosis of Alzheimer's disease is yet to be fully established.
Alzheimer's disease has no known cure, and treatment options are limited to addressing symptoms. Currently, three types of drugs are approved for treating the moderate or severe stages of the disease: cholinesterase inhibitors, partial N-methyl D-aspartate (NMDA) antagonists, and amyloid-directed antibodies. Cholinesterase inhibitors increase acetylcholine levels, a chemical crucial for cognitive functions such as memory and learning. NMDA antagonists (memantine) blocks NMDA receptors whose overactivation is implicated in Alzheimer's disease and related to synaptic dysfunction. Antiamyloid monoclonal antibodies bind to and promote the clearance of amyloid-beta peptides, thereby reducing amyloid plaques in the brain, which are associated with Alzheimer's disease.
Jasvinder Chawla, MD, Professor of Neurology, Loyola University Medical Center, Maywood; Director, Clinical Neurophysiology Lab, Department of Neurology, Hines VA Hospital, Hines, IL.
Jasvinder Chawla, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's symptoms go beyond just memory problems: She has difficulty with daily tasks, shows behavioral changes, and has significant communication difficulties — symptoms not found in mild cognitive impairment. While the patient has some behavioral changes, she does not exhibit the pronounced personality changes typical of frontotemporal dementia. Finally, the patient's cognitive decline is gradual and consistent without the stepwise progression typical of vascular dementia. Given the comprehensive presentation of the patient's symptoms and the results of her clinical investigations, middle-stage Alzheimer's disease is the most fitting diagnosis.
Alzheimer's disease is a progressive and irreversible brain disorder that affects memory, behavior, and cognitive skills. This condition causes the degeneration and death of brain cells, leading to various cognitive issues. Alzheimer's disease is the most common cause of dementia and accounts for 60%-80% of dementia cases. Although the exact cause is unknown, it is believed to result from genetic, lifestyle, and environmental factors. Alzheimer's disease progresses through stages — mild (early stage), moderate (middle stage), and severe (late stage) — and each stage has different signs and symptoms.
Alzheimer's disease is commonly observed in individuals 65 years or older, as age is the most significant risk factor. Another risk factor for Alzheimer's disease is family history; individuals who have parents or siblings with Alzheimer's disease are more likely to develop the disease. The risk increases with the number of family members diagnosed with the disease. Genetics also contribute to the development of Alzheimer's disease. Genes for developing Alzheimer's disease have been classified as deterministic and risk genes, which imply that they can cause the disease or increase the risk of developing it; however, the deterministic gene, which almost guarantees the occurrence of Alzheimer's, is rare and is found in less than 1% of cases. Experiencing a head injury is also a possible risk factor for Alzheimer's disease.
Accurate diagnosis of Alzheimer's disease requires a thorough history and physical examination. Gathering information from the patient's family and caregivers is important because some patients may not be aware of their condition. It is common for Alzheimer's disease patients to experience "sundowning," which causes confusion, agitation, and behavioral issues in the evening. A comprehensive physical examination, including a detailed neurologic and mental status exam, is necessary to determine the stage of the disease and rule out other conditions. Typically, the neurologic exam of Alzheimer's disease patients is normal.
Volumetric MRI is a recent technique that allows precise measurement of changes in brain volume. In Alzheimer's disease, shrinkage in the medial temporal lobe is visible through volumetric MRI. However, hippocampal atrophy is also a normal part of age-related memory decline, which raises doubts about the appropriateness of using volumetric MRI for early detection of Alzheimer's disease. The full potential of volumetric MRI in aiding the diagnosis of Alzheimer's disease is yet to be fully established.
Alzheimer's disease has no known cure, and treatment options are limited to addressing symptoms. Currently, three types of drugs are approved for treating the moderate or severe stages of the disease: cholinesterase inhibitors, partial N-methyl D-aspartate (NMDA) antagonists, and amyloid-directed antibodies. Cholinesterase inhibitors increase acetylcholine levels, a chemical crucial for cognitive functions such as memory and learning. NMDA antagonists (memantine) blocks NMDA receptors whose overactivation is implicated in Alzheimer's disease and related to synaptic dysfunction. Antiamyloid monoclonal antibodies bind to and promote the clearance of amyloid-beta peptides, thereby reducing amyloid plaques in the brain, which are associated with Alzheimer's disease.
Jasvinder Chawla, MD, Professor of Neurology, Loyola University Medical Center, Maywood; Director, Clinical Neurophysiology Lab, Department of Neurology, Hines VA Hospital, Hines, IL.
Jasvinder Chawla, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's symptoms go beyond just memory problems: She has difficulty with daily tasks, shows behavioral changes, and has significant communication difficulties — symptoms not found in mild cognitive impairment. While the patient has some behavioral changes, she does not exhibit the pronounced personality changes typical of frontotemporal dementia. Finally, the patient's cognitive decline is gradual and consistent without the stepwise progression typical of vascular dementia. Given the comprehensive presentation of the patient's symptoms and the results of her clinical investigations, middle-stage Alzheimer's disease is the most fitting diagnosis.
Alzheimer's disease is a progressive and irreversible brain disorder that affects memory, behavior, and cognitive skills. This condition causes the degeneration and death of brain cells, leading to various cognitive issues. Alzheimer's disease is the most common cause of dementia and accounts for 60%-80% of dementia cases. Although the exact cause is unknown, it is believed to result from genetic, lifestyle, and environmental factors. Alzheimer's disease progresses through stages — mild (early stage), moderate (middle stage), and severe (late stage) — and each stage has different signs and symptoms.
Alzheimer's disease is commonly observed in individuals 65 years or older, as age is the most significant risk factor. Another risk factor for Alzheimer's disease is family history; individuals who have parents or siblings with Alzheimer's disease are more likely to develop the disease. The risk increases with the number of family members diagnosed with the disease. Genetics also contribute to the development of Alzheimer's disease. Genes for developing Alzheimer's disease have been classified as deterministic and risk genes, which imply that they can cause the disease or increase the risk of developing it; however, the deterministic gene, which almost guarantees the occurrence of Alzheimer's, is rare and is found in less than 1% of cases. Experiencing a head injury is also a possible risk factor for Alzheimer's disease.
Accurate diagnosis of Alzheimer's disease requires a thorough history and physical examination. Gathering information from the patient's family and caregivers is important because some patients may not be aware of their condition. It is common for Alzheimer's disease patients to experience "sundowning," which causes confusion, agitation, and behavioral issues in the evening. A comprehensive physical examination, including a detailed neurologic and mental status exam, is necessary to determine the stage of the disease and rule out other conditions. Typically, the neurologic exam of Alzheimer's disease patients is normal.
Volumetric MRI is a recent technique that allows precise measurement of changes in brain volume. In Alzheimer's disease, shrinkage in the medial temporal lobe is visible through volumetric MRI. However, hippocampal atrophy is also a normal part of age-related memory decline, which raises doubts about the appropriateness of using volumetric MRI for early detection of Alzheimer's disease. The full potential of volumetric MRI in aiding the diagnosis of Alzheimer's disease is yet to be fully established.
Alzheimer's disease has no known cure, and treatment options are limited to addressing symptoms. Currently, three types of drugs are approved for treating the moderate or severe stages of the disease: cholinesterase inhibitors, partial N-methyl D-aspartate (NMDA) antagonists, and amyloid-directed antibodies. Cholinesterase inhibitors increase acetylcholine levels, a chemical crucial for cognitive functions such as memory and learning. NMDA antagonists (memantine) blocks NMDA receptors whose overactivation is implicated in Alzheimer's disease and related to synaptic dysfunction. Antiamyloid monoclonal antibodies bind to and promote the clearance of amyloid-beta peptides, thereby reducing amyloid plaques in the brain, which are associated with Alzheimer's disease.
Jasvinder Chawla, MD, Professor of Neurology, Loyola University Medical Center, Maywood; Director, Clinical Neurophysiology Lab, Department of Neurology, Hines VA Hospital, Hines, IL.
Jasvinder Chawla, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
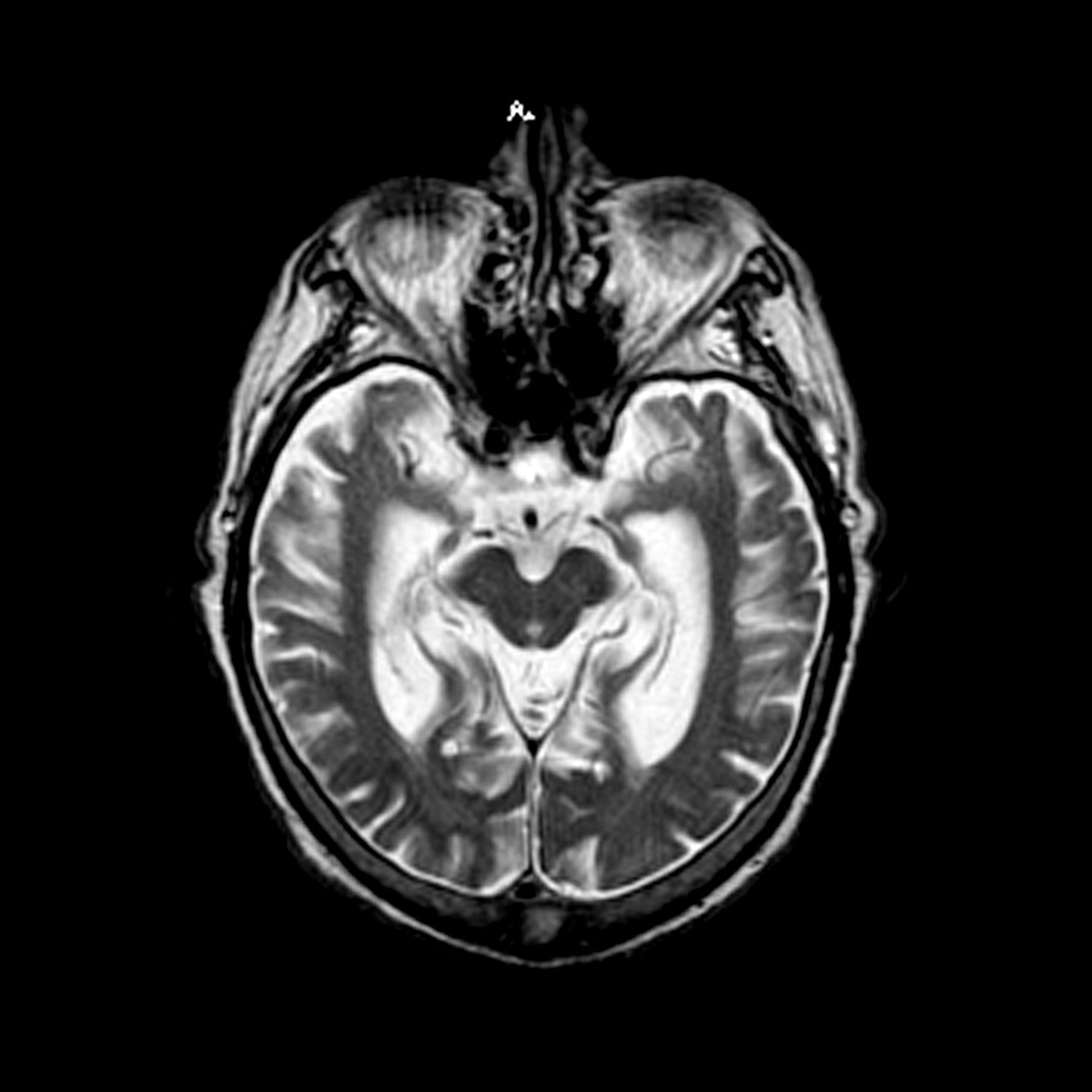
The patient is a 72-year-old retired schoolteacher accompanied by her daughter. Over the past year, her family has become increasingly concerned about her forgetfulness, mood fluctuations, and challenges in performing daily activities. The patient often forgets her grandchildren's names and struggles to recall significant recent events. She frequently misplaces household items and has missed several appointments. During her consultation, she has difficulty finding the right words, often repeats herself, and seems to lose track of the conversation. Her daughter shared concerning incidents, such as the patient wearing heavy sweaters during hot summer days and falling victim to a phone scam, which was uncharacteristic of her previous discerning nature. Additionally, the patient has become more reclusive, avoiding the social gatherings she once loved. She occasionally exhibits signs of agitation, especially in the evening. She has also stopped cooking as a result of instances of forgetting to turn off the stove and has had challenges managing her finances, leading to unpaid bills. A thorough neurologic exam is performed and is normal. Coronal T1-weighted MRI reveals hippocampal atrophy, particularly on the right side.

The challenges of managing CMV infection during pregnancy
CASE Anomalous findings on fetal anatomic survey
A 27-year-old previously healthy primigravid woman is at 18 weeks’ gestation. She is a first-grade schoolteacher. On her fetal anatomic survey, the estimated fetal weight was in the eighth percentile. Echogenic bowel and a small amount of ascitic fluid were noted in the fetal abdomen. The lateral and third ventricles were mildly dilated, the head circumference was 2 standard deviations below normal, and the placenta was slightly thickened and edematous.
What is the most likely diagnosis?
What diagnostic tests are indicated?
What management options are available for this patient?
Cytomegalovirus (CMV) is the most common of the perinatally transmitted infections, affecting 1% to 4% of all pregnancies. Although the virus typically causes either asymptomatic infection or only mild illness in immunocompetent individuals, it can cause life-threatening disease in immunocompromised persons and in the developing fetus. In this article, we review the virology and epidemiology of CMV infection and then focus on the key methods to diagnose infection in the mother and fetus. We conclude by considering measures that may be of at least modest value in treating CMV in pregnancy.
Virology of CMV infection
Cytomegalovirus is a double-stranded DNA virus in the Herpesviridae family. This ubiquitous virus is present in virtually all secretions and excretions of an infected host, including blood, urine, saliva, breast milk, genital secretions, and tissues and organs used for donation. Infection is transmitted through direct contact with any of the substances listed; contact with infected urine or saliva is the most common mode of transmission. Disease occurrence does not show seasonal variation.
After exposure, an incubation period of 28 to 60 days ensues, followed by development of viremia and clinical symptoms. In the majority of exposed individuals, CMV establishes a lifelong latent infection, and recurrent episodes of illness can occur as a result of reactivation of latent virus (also known as secondary infection) or, more rarely, infection with a new viral strain. In fact, most CMV illness episodes in pregnancy represent a reactivation of a previous infection rather than a new infection.
Following initial infection, both IgM (immunoglobulin M) and IgG (immunoglobulin G) antibodies develop rapidly and can be detected in blood within 1 to 2 weeks. IgM levels typically wane within 30 to 60 days, although persistence for several months is not unusual, and levels also can increase with viral reactivation (secondary infection). IgG antibodies typically persist for many years after a primary infection.
Intrauterine CMV infection occurs through hematogenous transplacental passage during maternal viremia. The risk of transmission and severity of fetal effects depend on whether or not the infection is primary or secondary in nature as well as the gestational age at fetal exposure.1,2
Additionally, postnatal vertical transmission can occur through exposure to viral particles in genital secretions as well as breast milk. CMV acquired in the postnatal period rarely produces severe sequelae in a healthy term neonate, but it has been associated with an increased rate of complications in very low birth weight and premature newborns.3
Continue to: Who is at risk...
Who is at risk
Congenital CMV, which occurs in 2.1 to 7.7 per 10,000 live births in the United States, is both the most common congenital infection and the leading cause of nonhereditary congenital hearing loss in children.4,5 The main reservoir of CMV in the United States is young children in day care settings, with approximately 50% of this population showing evidence of viral shedding in saliva.1 Adult populations in North America have a high prevalence of CMV IgG antibodies indicative of prior infection, with rates reaching 50% to 80%. Among seronegative individuals aged 12 to 49, the rate of seroconversion is approximately 1 in 60 annually.6 Significant racial disparities have been noted in rates of seroprevalence and seroconversion, with higher rates of infection in non-Hispanic Black and Mexican American individuals.6 Overall, the rate of new CMV infection among pregnant women in the United States is 0.7% to 4%.7
Clinical manifestations
Manifestations of infection differ depending on whether or not infection is primary or recurrent (secondary) and whether or not the host is immunocompetent or has a compromised immune system. Unique manifestations develop in the fetus.
CMV infection in children and adults. Among individuals with a normal immune response, the typical course of CMV is either no symptoms or a mononucleosis-like illness. In symptomatic patients, the most common symptoms include malaise, fever, and night sweats, and the most common associated laboratory abnormalities are elevation in liver function tests and a decreased white blood cell count, with a predominance of lymphocytes.8
Immunocompromised individuals are at risk for significant morbidity and mortality resulting from CMV. Illness may be the result of reactivation of latent infection due to decreased immune function or may be acquired as a result of treatment such as transplantation of CMV-positive organs or tissues, including bone marrow. Virtually any organ system can be affected, with potential for permanent organ damage and death. Severe systemic infection also can occur.
CMV infection in the fetus and neonate. As noted previously, fetal infection develops as a result of transplacental passage coincident with maternal infection. The risk of CMV transmission to the fetus and the severity of fetal injury vary based on gestational age at fetal infection and whether or not maternal infection is primary or secondary.
In most studies, primary maternal infections are associated with higher rates of fetal infection and more severe fetal and neonatal disease manifestations.2,7,9,10 Primary infections carry an overall 30% to 40% risk of transmission to the fetus.7,11 The risk of fetal transmission is much lower with a recurrent infection and is usually less than 2%.11 Due to their greater overall incidence, secondary infections account for the majority of cases of fetal and neonatal CMV disease.7 Importantly, although secondary infections generally have been regarded as having a lower risk and lower severity of fetal and neonatal disease, several recent studies have demonstrated rates of complications similar to, and even exceeding, those of primary infections.12-15 The TABLE provides a summary of the risks of fetal transmission and symptomatic fetal infection based on trimester of pregnancy.2,11,16-18
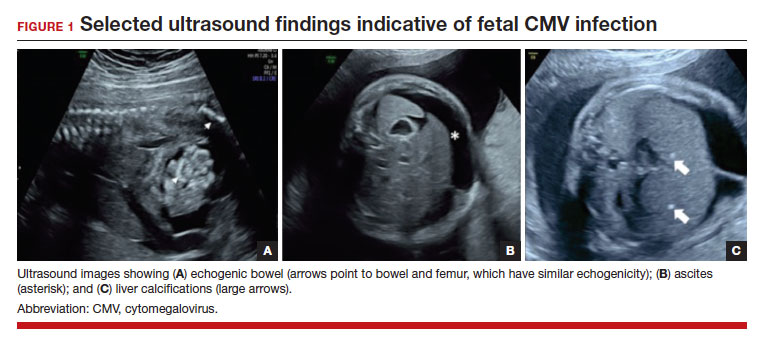
In the fetus, CMV may affect multiple organ systems. Among sonographic and magnetic resonance imaging (MRI) findings, central nervous system (CNS) anomalies are the most common.19,20 These can include microcephaly, ventriculomegaly, and periventricular calcifications. The gastrointestinal system also is frequently affected, and findings include echogenic bowel, hepatosplenomegaly, and liver calcifications. Lastly, isolated effusions, placentomegaly, fetal growth restriction, and even frank hydrops can develop. More favorable neurologic outcomes have been demonstrated in infants with no prenatal brain imaging abnormalities.20,21 However, the role of MRI in prenatal prognosis currently is not well defined.
FIGURE 1 illustrates selected sonographic findings associated with fetal CMV infection.
About 85% to 90% of infants with congenital CMV that results from primary maternal infection have no symptoms at birth. Among the 10% to 15% of infants that do have symptoms, petechial rash, jaundice, and hepatosplenomegaly are the most common manifestations (“blueberry muffin baby”). Approximately 10% to 20% of infants in this group have evidence of chorioretinitis on ophthalmologic examination, and 50% show either microcephaly or low birth weight.22Among survivors of symptomatic congenital CMV, more than 50% have long-term neurologic morbidities that may include sensorineural hearing loss, seizures, vision impairment, and developmental disabilities. Note that even when neonates appear asymptomatic at birth (regardless of whether infection is primary or secondary), 5% may develop microcephaly and motor deficits, 10% go on to develop sensorineural hearing loss, and the overall rate of neurologic morbidity reaches 13% to 15%.12,23 Some of the observed deficits manifest at several years of age, and, currently, no models exist for prediction of outcome.
Continue to: Diagnosing CMV infection...
Diagnosing CMV infection
Maternal infection
If maternal CMV infection is suspected based on a symptomatic illness or an abnormal fetal ultrasound exam, the first diagnostic test should be an assessment of IgM and IgG serology. If the former test results are positive and the latter negative, the diagnosis of acute CMV infection is confirmed. A positive serum CMV DNA polymerase chain reaction (PCR) test adds additional assurance that the diagnosis is correct. Primary infection, as noted above, poses the greatest risk of serious injury to the fetus.1
A frequent diagnostic dilemma arises when both the IgM and IgG antibody are positive. Remember that CMV IgM antibody can remain positive for 9 to 12 months after a primary infection and can reappear in the maternal serum in the face of a recurrent or reactivated infection. When confronted by both a positive IgM and positive IgG result, the clinician should then order IgG avidity testing. If the avidity is low to moderate, which reflects poor binding of antibody to the virus, the patient likely has an acute infection. If the avidity is high, which reflects enhanced binding of antibody to virus, the patient probably has a recurrent or reactivated infection; this scenario poses less danger to the developing fetus. The presence of CMV DNA in serum is also more consistent with acute infection, although viremia still can occur with recurrent infection. FIGURE 2 presents a suggested algorithm for the diagnosis of CMV in the pregnant patient.1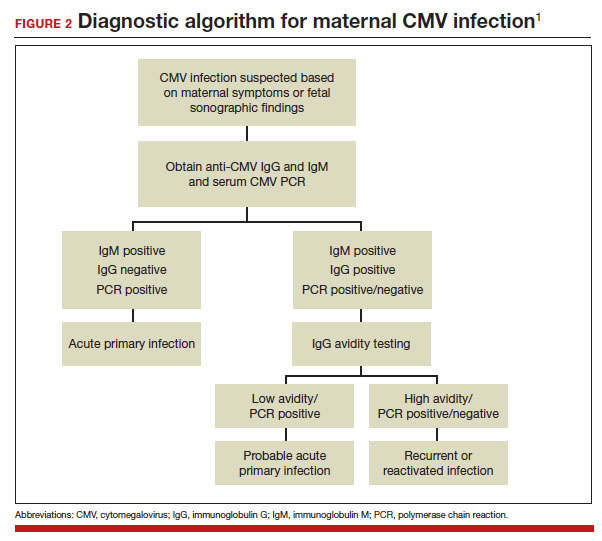
If a diagnosis of maternal CMV infection is confirmed, liver function tests should be obtained to determine if CMV hepatitis is present. If the liver function tests are abnormal, a coagulation profile also should be performed to identify the mother who might be at risk for peripartum hemorrhage.
Fetal infection
The single best test for confirmation of congenital CMV infection is detection of viral DNA and quantitation of viral load in the amniotic fluid by PCR. If the amniocentesis is performed prior to 20 weeks’ gestation and is negative, the test should be repeated in approximately 4 weeks.1,19,24
Detection of viral DNA indicates congenital infection. The ultimate task, however, is to determine if the infection has injured the fetus. Detailed ultrasound examination is the key to identifying fetal injury. As noted previously, the principal ultrasonographic findings that suggest congenital CMV infection include2,19,20,21,25:
- hydropic placenta
- fetal growth restriction
- microcephaly (head circumference more than 3 standard deviations below the mean)
- periventricular calcifications
- enlarged liver
- echogenic bowel
- ascites
- fetal hydrops.
Management: Evidence on CMV hyperimmune globulin, valacyclovir
If the immunocompetent mother has clinical manifestations of infection, she should receive symptomatic treatment. She should be encouraged to rest as much as possible, stay well hydrated, and use acetaminophen (1,000 mg every 6 to 8 hours) as needed for malaise and fever.
However, if the mother is immunocompromised and has signs of serious complications, such as chorioretinitis, hepatitis, or pneumonia, more aggressive therapy is indicated. Drugs used in this setting include foscarnet and ganciclovir and are best prescribed in consultation with a medical infectious disease specialist.
At this time, no consistently effective therapy for congenital infection is available. Therefore, if a patient has primary CMV infection in the first half of pregnancy, particularly in the first trimester, she should be counseled that the risk of fetal infection is approximately 40% and that approximately 5% to 15% of infants will be severely affected at birth. Given this information, some patients may opt for pregnancy termination.
In 2005, a report from Nigro and colleagues stimulated great hope that CMV-specific hyperimmune globulin (CytoGam) might be of value for both treatment and prophylaxis for congenital infection.26 These authors studied 157 women with confirmed primary CMV infection. One-hundred forty-eight women were asymptomatic and were identified by routine serologic screening, 8 had symptomatic infection, and 1 was identified because of abnormal fetal ultrasound findings. Forty-five women had CMV detected in amniotic fluid by PCR or culture more than 6 weeks before study enrollment. Thirty-one of these women were treated with intravenous hyperimmune globulin (200 U or 200 mg/kg maternal body weight); 14 declined treatment. Seven of the latter women had infants who were acutely symptomatic at the time of delivery; only 1 of the 31 treated women had an affected neonate (adjusted odds ratio [OR], 0.02; P<.001). In this same study, 84 women did not have a diagnostic amniocentesis because their infection occurred within 6 weeks of enrollment, their gestational age was less than 20 weeks, or they declined the procedure. Thirty-seven of these women received hyperimmune globulin (100 U or 100 mg/kg) every month until delivery, and 47 declined treatment. Six of the treated women delivered infected infants compared with 19 of the untreated women (adjusted OR, 0.32; P<.04).
Although these results were quite encouraging, several problems existed with the study’s design, as noted in an editorial that accompanied the study’s publication.27 First, the study was not randomized or placebo controlled. Second, patients were not stratified based on the severity of fetal ultrasound abnormalities. Third, the dosing of hyperimmune globulin varied; 9 of the 31 patients in the treatment group received additional infusions of drug into either the amniotic fluid or fetal umbilical vein. Moreover, patients in the prophylaxis group actually received a higher cumulative dose of hyperimmune globulin than patients in the treatment group.
Two subsequent investigations that were better designed were unable to verify the effectiveness of hyperimmune globulin. In 2014, Revello and colleagues reported the results of a prospective, randomized, placebo-controlled, double-blinded study of 124 women at 5 to 26 weeks’ gestation with confirmed primary CMV infection.28 The rate of congenital infection was 30% in the group treated with hyperimmune globulin and 44% in the placebo group (P=.13). There also was no significant difference in the concentration of serum CMV DNA in treated versus untreated mothers. Moreover, the number of adverse obstetric events (preterm delivery, fetal growth restriction, intrahepatic cholestasis of pregnancy, and postpartum preeclampsia) in the treatment group was higher than in the placebo group, 13% versus 2%.
In 2021, Hughes and colleagues published the results of a multicenter, double-blind trial in 399 women who had a diagnosis of primary CMV infection before 23 weeks’ gestation.29 The primary outcome was defined as a composite of congenital CMV infection or fetal/neonatal death. An adverse primary outcome occurred in 22.7% of the patients who received hyperimmune globulin and 19.4% of those who received placebo (relative risk, 1.17; 95% confidence interval [CI], 0.80–1.72; P=.42).
Continue to: Jacquemard and colleagues...
Jacquemard and colleagues then proposed a different approach.30 In a small pilot study of 20 patients, these authors used high doses of oral valacylovir (2 g 4 times daily) and documented therapeutic drug concentrations and a decline in CMV viral load in fetal serum. Patients were not stratified by severity of fetal injury at onset of treatment, so the authors were unable to define which fetuses were most likely to benefit from treatment.
In a follow-up investigation, Leruez-Ville and colleagues reported another small series in which high-dose oral valacyclovir (8 g daily) was used for treatment.31 They excluded fetuses with severe brain anomalies and fetuses with no sonographic evidence of injury. The median gestational age at diagnosis was 26 weeks. Thirty-four of 43 treated fetuses were free of injury at birth. In addition, the viral load in the neonate’s serum decreased significantly after treatment, and the platelet count increased. The authors then compared these outcomes to a historical cohort and confirmed that treatment increased the proportion of asymptomatic neonates from 43% without treatment to 82% with treatment (P<.05 with no overlapping confidence intervals).
We conclude from these investigations that hyperimmune globulin is unlikely to be of value in treating congenital CMV infection, especially if the fetus already has sonographic findings of severe injury. High-dose oral valacyclovir also is unlikely to be of value in severely affected fetuses, particularly those with evidence of CNS injury. However, antiviral therapy may be of modest value in situations when the fetus is less severely injured.
Preventive measures
Since no definitive treatment is available for congenital CMV infection, our efforts as clinicians should focus on measures that may prevent transmission of infection to the pregnant patient. These measures include:
- Encouraging patients to use careful handwashing techniques when handling infant diapers and toys.
- Encouraging patients to adopt safe sexual practices if not already engaged in a mutually faithful, monogamous relationship.
- Using CMV-negative blood when transfusing a pregnant woman or a fetus.
At the present time, unfortunately, a readily available and highly effective therapy for prevention of CMV infection is not available.
CASE Congenital infection diagnosed
The ultrasound findings are most consistent with congenital CMV infection, especially given the patient’s work as an elementary schoolteacher. The diagnosis of maternal infection is best established by conventional serology (positive IgM, negative IgM) and detection of viral DNA in maternal blood by PCR testing. The diagnosis of congenital infection is best confirmed by documentation of viral DNA in the amniotic fluid by PCR testing. Given that this fetus already has evidence of moderate to severe injury, no treatment is likely to be effective in reversing the abnormal ultrasound findings. Pregnancy termination may be an option, depending upon the patient’s desires and the legal restrictions prevalent in the patient’s geographic area. ●
- Cytomegalovirus infection is the most common of the perinatally transmitted infections.
- Maternal infection is often asymptomatic. When symptoms are present, they resemble those of an influenza-like illness. In immunocompromised persons, however, CMV may cause serious complications, including pneumonia, hepatitis, and chorioretinitis.
- The virus is transmitted by contact with contaminated body fluids, such as saliva, urine, blood, and genital secretions.
- The greatest risk of severe fetal injury results from primary maternal infection in the first trimester of pregnancy.
- Manifestations of severe congenital CMV infection include growth restriction, microcephaly, ventriculomegaly, hepatosplenomegaly, ascites, chorioretinitis, thrombocytopenia, purpura, and hydrops (“blueberry muffin baby”).
- Late manifestations of infection, which usually follow recurrent maternal infection, may appear as a child enters elementary school and include visual and auditory deficits, developmental delays, and learning disabilities.
- The diagnosis of maternal infection is confirmed by serology and detection of viral DNA in the serum by PCR testing.
- The diagnosis of fetal infection is best made by a combination of abnormal ultrasound findings and detection of CMV DNA in amniotic fluid. The characteristic ultrasound findings include placentomegaly, microcephaly, ventriculomegaly, growth restriction, echogenic bowel, and serous effusions/hydrops.
- Treatment of the mother with antiviral medications such as valacyclovir may be of modest value in reducing placental edema, decreasing viral load in the fetus, and hastening the resolution of some ultrasound findings, such as echogenic bowel.
- While initial studies seemed promising, the use of hyperimmune globulin has not proven to be consistently effective in treating congenital infection.
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TR, et al, eds. Creasy and Resnik’s Maternal Fetal Medicine: Principles and Practice. 8th ed. 2019:888-890.
- Chatzakis C, Ville Y, Makrydimas G, et al. Timing of primary maternal cytomegalovirus infection and rates of vertical transmission and fetal consequences. Am J Obstet Gynecol. 2020;223:870-883.e11. doi:10.1016/j.ajog.2020.05.038
- Kelly MS, Benjamin DK, Puopolo KM, et al. Postnatal cytomegalovirus infection and the risk for bronchopulmonary dysplasia. JAMA Pediatr. 2015;169:e153785. doi:10.1001 /jamapediatrics.2015.3785
- Messinger CJ, Lipsitch M, Bateman BT, et al. Association between congenital cytomegalovirus and the prevalence at birth of microcephaly in the United States. JAMA Pediatr. 2020;174:1159-1167. doi:10.1001/jamapediatrics.2020.3009
- De Cuyper E, Acke F, Keymeulen A, et al. Risk factors for hearing loss at birth in newborns with congenital cytomegalovirus infection. JAMA Otolaryngol Head Neck Surg. 2023;149:122-130. doi:10.1001/jamaoto.2022.4109
- Colugnati FA, Staras SA, Dollard SC, et al. Incidence of cytomegalovirus infection among the general population and pregnant women in the United States. BMC Infect Dis. 2007;7:71. doi:10.1186/1471-2334-7-71
- Stagno S, Pass RF, Cloud G, et al. Primary cytomegalovirus infection in pregnancy. Incidence, transmission to fetus, and clinical outcome. JAMA. 1986;256:1904-1908.
- Wreghitt TG, Teare EL, Sule O, et al. Cytomegalovirus infection in immunocompetent patients. Clin Infect Dis. 2003;37:1603-1606. doi:10.1086/379711
- Fowler KB, Stagno S, Pass RF, et al. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N Engl J Med. 1992;326:663-667. doi:10.1056 /NEJM199203053261003
- Faure-Bardon V, Magny JF, Parodi M, et al. Sequelae of congenital cytomegalovirus following maternal primary infections are limited to those acquired in the first trimester of pregnancy. Clin Infect Dis. 2019;69:1526-1532. doi:10.1093/ cid/ciy1128
- Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol. 2007;17:253-276. doi:10.1002/ rmv.535
- Boppana SB, Pass RF, Britt WJ, et al. Symptomatic congenital cytomegalovirus infection: neonatal morbidity and mortality. Pediatr Infect Dis J. 1992;11:93-99. doi:10.1097/00006454-199202000-00007
- Ross SA, Fowler KB, Ashrith G, et al. Hearing loss in children with congenital cytomegalovirus infection born to mothers with preexisting immunity. J Pediatr. 2006;148:332-336. doi:10.1016/j.jpeds.2005.09.003
- Zalel Y, Gilboa Y, Berkenshtat M, et al. Secondary cytomegalovirus infection can cause severe fetal sequelae despite maternal preconceptional immunity. Ultrasound Obstet Gynecol. 31:417-420. doi:10.1002/uog.5255
- Scaramuzzino F, Di Pastena M, Chiurchiu S, et al. Secondary cytomegalovirus infections: how much do we still not know? Comparison of children with symptomatic congenital cytomegalovirus born to mothers with primary and secondary infection. Front Pediatr. 2022;10:885926. doi:10.3389/fped.2022.885926
- Gindes L, Teperberg-Oikawa M, Sherman D, et al. Congenital cytomegalovirus infection following primary maternal infection in the third trimester. BJOG. 2008;115:830-835. doi:10.1111/j.1471-0528.2007.01651.x
- Hadar E, Dorfman E, Bardin R, et al. Symptomatic congenital cytomegalovirus disease following non-primary maternal infection: a retrospective cohort study. BMC Infect Dis. 2017;17:31. doi:10.1186/s12879-016-2161-3
- Elkan Miller T, Weisz B, Yinon Y, et al. Congenital cytomegalovirus infection following second and third trimester maternal infection is associated with mild childhood adverse outcome not predicted by prenatal imaging. J Pediatric Infect Dis Soc. 2021;10:562-568. doi:10.1093/jpids/ piaa154
- Lipitz S, Yinon Y, Malinger G, et al. Risk of cytomegalovirusassociated sequelae in relation to time of infection and findings on prenatal imaging. Ultrasound Obstet Gynecol. 2013;41:508-514. doi:10.1002/uog.12377
- Lipitz S, Elkan Miller T, Yinon Y, et al. Revisiting short- and long-term outcome after fetal first-trimester primary cytomegalovirus infection in relation to prenatal imaging findings. Ultrasound Obstet Gynecol. 2020;56:572-578. doi:10.1002/uog.21946
- Buca D, Di Mascio D, Rizzo G, et al. Outcome of fetuses with congenital cytomegalovirus infection and normal ultrasound at diagnosis: systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2021;57:551-559. doi:10.1002/uog.23143
- Boppana SB, Ross SA, Fowler KB. Congenital cytomegalovirus infection: clinical outcome. Clin Infect Dis. 2013;57 (suppl 4):S178-S181. doi:10.1093/cid/cit629
- Dollard SC, Grosse SD, Ross DS. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol. 2007;17:355-363. doi:10.1002/rmv.544
- Hughes BL, Gyamfi-Bannerman C. Diagnosis and antenatal management of congenital cytomegalovirus infection. Am J Obstet Gynecol. 2016;214:B5-11. doi:10.1016 /j.ajog.2016.02.042
- Rouse DJ, Fette LM, Hughes BL, et al. Noninvasive prediction of congenital cytomegalovirus infection after maternal primary infection. Obstet Gynecol. 2022;139:400-406. doi:10.1097/AOG.0000000000004691
- Nigro G, Adler SP, La Torre R, et al; Congenital Cytomegalovirus Collaborating Group. Passive immunization during pregnancy for congenital cytomegalovirus infection. N Engl J Med. 2005;353:1350-1362. doi:10.1056/NEJMoa043337
- Duff P. Immunotherapy for congenital cytomegalovirus infection. N Engl J Med. 2005;355:1402-1404. doi:10.1056 /NEJMe058172
- Revello MG, Lazzarotto T, Guerra B, et al. A randomized trial of hyperimmune globulin to prevent congenital cytomegalovirus. N Engl J Med. 2014;370:1316-1326. doi:10.1056/NEJMoa1310214
- Hughes BL, Clifton RG, Rouse DJ, et al. A trial of hyperimmune globulin to prevent congenital cytomegalovirus infection. N Engl J Med. 2021;385:436-444. doi:10.1056/NEJMoa1913569
- Jacquemard F, Yamamoto M, Costa JM, et al. Maternal administration of valaciclovir in symptomatic intrauterine cytomegalovirus infection. BJOG. 2007;114:1113-1121. doi:10.1111/j.1471-0528.2007.01308.x
- Leruez-Ville M, Ghout I, Bussières L, et al. In utero treatment of congenital cytomegalovirus infection with valacyclovir in a multicenter, open-label, phase II study. Am J Obstet Gynecol. 2016;215:462.e1-462.e10. doi:10.1016/j.ajog.2016.04.003

Dr. Berwick is a first-year Maternal-Fetal Medicine Fellow, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.

Dr. Duff is Professor, Division of MaternalFetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
Dr. Berwick is a first-year Maternal-Fetal Medicine Fellow, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
Dr. Duff is Professor, Division of MaternalFetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
Dr. Berwick is a first-year Maternal-Fetal Medicine Fellow, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
Dr. Duff is Professor, Division of MaternalFetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
CASE Anomalous findings on fetal anatomic survey
A 27-year-old previously healthy primigravid woman is at 18 weeks’ gestation. She is a first-grade schoolteacher. On her fetal anatomic survey, the estimated fetal weight was in the eighth percentile. Echogenic bowel and a small amount of ascitic fluid were noted in the fetal abdomen. The lateral and third ventricles were mildly dilated, the head circumference was 2 standard deviations below normal, and the placenta was slightly thickened and edematous.
What is the most likely diagnosis?
What diagnostic tests are indicated?
What management options are available for this patient?
Cytomegalovirus (CMV) is the most common of the perinatally transmitted infections, affecting 1% to 4% of all pregnancies. Although the virus typically causes either asymptomatic infection or only mild illness in immunocompetent individuals, it can cause life-threatening disease in immunocompromised persons and in the developing fetus. In this article, we review the virology and epidemiology of CMV infection and then focus on the key methods to diagnose infection in the mother and fetus. We conclude by considering measures that may be of at least modest value in treating CMV in pregnancy.
Virology of CMV infection
Cytomegalovirus is a double-stranded DNA virus in the Herpesviridae family. This ubiquitous virus is present in virtually all secretions and excretions of an infected host, including blood, urine, saliva, breast milk, genital secretions, and tissues and organs used for donation. Infection is transmitted through direct contact with any of the substances listed; contact with infected urine or saliva is the most common mode of transmission. Disease occurrence does not show seasonal variation.
After exposure, an incubation period of 28 to 60 days ensues, followed by development of viremia and clinical symptoms. In the majority of exposed individuals, CMV establishes a lifelong latent infection, and recurrent episodes of illness can occur as a result of reactivation of latent virus (also known as secondary infection) or, more rarely, infection with a new viral strain. In fact, most CMV illness episodes in pregnancy represent a reactivation of a previous infection rather than a new infection.
Following initial infection, both IgM (immunoglobulin M) and IgG (immunoglobulin G) antibodies develop rapidly and can be detected in blood within 1 to 2 weeks. IgM levels typically wane within 30 to 60 days, although persistence for several months is not unusual, and levels also can increase with viral reactivation (secondary infection). IgG antibodies typically persist for many years after a primary infection.
Intrauterine CMV infection occurs through hematogenous transplacental passage during maternal viremia. The risk of transmission and severity of fetal effects depend on whether or not the infection is primary or secondary in nature as well as the gestational age at fetal exposure.1,2
Additionally, postnatal vertical transmission can occur through exposure to viral particles in genital secretions as well as breast milk. CMV acquired in the postnatal period rarely produces severe sequelae in a healthy term neonate, but it has been associated with an increased rate of complications in very low birth weight and premature newborns.3
Continue to: Who is at risk...
Who is at risk
Congenital CMV, which occurs in 2.1 to 7.7 per 10,000 live births in the United States, is both the most common congenital infection and the leading cause of nonhereditary congenital hearing loss in children.4,5 The main reservoir of CMV in the United States is young children in day care settings, with approximately 50% of this population showing evidence of viral shedding in saliva.1 Adult populations in North America have a high prevalence of CMV IgG antibodies indicative of prior infection, with rates reaching 50% to 80%. Among seronegative individuals aged 12 to 49, the rate of seroconversion is approximately 1 in 60 annually.6 Significant racial disparities have been noted in rates of seroprevalence and seroconversion, with higher rates of infection in non-Hispanic Black and Mexican American individuals.6 Overall, the rate of new CMV infection among pregnant women in the United States is 0.7% to 4%.7
Clinical manifestations
Manifestations of infection differ depending on whether or not infection is primary or recurrent (secondary) and whether or not the host is immunocompetent or has a compromised immune system. Unique manifestations develop in the fetus.
CMV infection in children and adults. Among individuals with a normal immune response, the typical course of CMV is either no symptoms or a mononucleosis-like illness. In symptomatic patients, the most common symptoms include malaise, fever, and night sweats, and the most common associated laboratory abnormalities are elevation in liver function tests and a decreased white blood cell count, with a predominance of lymphocytes.8
Immunocompromised individuals are at risk for significant morbidity and mortality resulting from CMV. Illness may be the result of reactivation of latent infection due to decreased immune function or may be acquired as a result of treatment such as transplantation of CMV-positive organs or tissues, including bone marrow. Virtually any organ system can be affected, with potential for permanent organ damage and death. Severe systemic infection also can occur.
CMV infection in the fetus and neonate. As noted previously, fetal infection develops as a result of transplacental passage coincident with maternal infection. The risk of CMV transmission to the fetus and the severity of fetal injury vary based on gestational age at fetal infection and whether or not maternal infection is primary or secondary.
In most studies, primary maternal infections are associated with higher rates of fetal infection and more severe fetal and neonatal disease manifestations.2,7,9,10 Primary infections carry an overall 30% to 40% risk of transmission to the fetus.7,11 The risk of fetal transmission is much lower with a recurrent infection and is usually less than 2%.11 Due to their greater overall incidence, secondary infections account for the majority of cases of fetal and neonatal CMV disease.7 Importantly, although secondary infections generally have been regarded as having a lower risk and lower severity of fetal and neonatal disease, several recent studies have demonstrated rates of complications similar to, and even exceeding, those of primary infections.12-15 The TABLE provides a summary of the risks of fetal transmission and symptomatic fetal infection based on trimester of pregnancy.2,11,16-18
In the fetus, CMV may affect multiple organ systems. Among sonographic and magnetic resonance imaging (MRI) findings, central nervous system (CNS) anomalies are the most common.19,20 These can include microcephaly, ventriculomegaly, and periventricular calcifications. The gastrointestinal system also is frequently affected, and findings include echogenic bowel, hepatosplenomegaly, and liver calcifications. Lastly, isolated effusions, placentomegaly, fetal growth restriction, and even frank hydrops can develop. More favorable neurologic outcomes have been demonstrated in infants with no prenatal brain imaging abnormalities.20,21 However, the role of MRI in prenatal prognosis currently is not well defined.
FIGURE 1 illustrates selected sonographic findings associated with fetal CMV infection.
About 85% to 90% of infants with congenital CMV that results from primary maternal infection have no symptoms at birth. Among the 10% to 15% of infants that do have symptoms, petechial rash, jaundice, and hepatosplenomegaly are the most common manifestations (“blueberry muffin baby”). Approximately 10% to 20% of infants in this group have evidence of chorioretinitis on ophthalmologic examination, and 50% show either microcephaly or low birth weight.22Among survivors of symptomatic congenital CMV, more than 50% have long-term neurologic morbidities that may include sensorineural hearing loss, seizures, vision impairment, and developmental disabilities. Note that even when neonates appear asymptomatic at birth (regardless of whether infection is primary or secondary), 5% may develop microcephaly and motor deficits, 10% go on to develop sensorineural hearing loss, and the overall rate of neurologic morbidity reaches 13% to 15%.12,23 Some of the observed deficits manifest at several years of age, and, currently, no models exist for prediction of outcome.
Continue to: Diagnosing CMV infection...
Diagnosing CMV infection
Maternal infection
If maternal CMV infection is suspected based on a symptomatic illness or an abnormal fetal ultrasound exam, the first diagnostic test should be an assessment of IgM and IgG serology. If the former test results are positive and the latter negative, the diagnosis of acute CMV infection is confirmed. A positive serum CMV DNA polymerase chain reaction (PCR) test adds additional assurance that the diagnosis is correct. Primary infection, as noted above, poses the greatest risk of serious injury to the fetus.1
A frequent diagnostic dilemma arises when both the IgM and IgG antibody are positive. Remember that CMV IgM antibody can remain positive for 9 to 12 months after a primary infection and can reappear in the maternal serum in the face of a recurrent or reactivated infection. When confronted by both a positive IgM and positive IgG result, the clinician should then order IgG avidity testing. If the avidity is low to moderate, which reflects poor binding of antibody to the virus, the patient likely has an acute infection. If the avidity is high, which reflects enhanced binding of antibody to virus, the patient probably has a recurrent or reactivated infection; this scenario poses less danger to the developing fetus. The presence of CMV DNA in serum is also more consistent with acute infection, although viremia still can occur with recurrent infection. FIGURE 2 presents a suggested algorithm for the diagnosis of CMV in the pregnant patient.1
If a diagnosis of maternal CMV infection is confirmed, liver function tests should be obtained to determine if CMV hepatitis is present. If the liver function tests are abnormal, a coagulation profile also should be performed to identify the mother who might be at risk for peripartum hemorrhage.
Fetal infection
The single best test for confirmation of congenital CMV infection is detection of viral DNA and quantitation of viral load in the amniotic fluid by PCR. If the amniocentesis is performed prior to 20 weeks’ gestation and is negative, the test should be repeated in approximately 4 weeks.1,19,24
Detection of viral DNA indicates congenital infection. The ultimate task, however, is to determine if the infection has injured the fetus. Detailed ultrasound examination is the key to identifying fetal injury. As noted previously, the principal ultrasonographic findings that suggest congenital CMV infection include2,19,20,21,25:
- hydropic placenta
- fetal growth restriction
- microcephaly (head circumference more than 3 standard deviations below the mean)
- periventricular calcifications
- enlarged liver
- echogenic bowel
- ascites
- fetal hydrops.
Management: Evidence on CMV hyperimmune globulin, valacyclovir
If the immunocompetent mother has clinical manifestations of infection, she should receive symptomatic treatment. She should be encouraged to rest as much as possible, stay well hydrated, and use acetaminophen (1,000 mg every 6 to 8 hours) as needed for malaise and fever.
However, if the mother is immunocompromised and has signs of serious complications, such as chorioretinitis, hepatitis, or pneumonia, more aggressive therapy is indicated. Drugs used in this setting include foscarnet and ganciclovir and are best prescribed in consultation with a medical infectious disease specialist.
At this time, no consistently effective therapy for congenital infection is available. Therefore, if a patient has primary CMV infection in the first half of pregnancy, particularly in the first trimester, she should be counseled that the risk of fetal infection is approximately 40% and that approximately 5% to 15% of infants will be severely affected at birth. Given this information, some patients may opt for pregnancy termination.
In 2005, a report from Nigro and colleagues stimulated great hope that CMV-specific hyperimmune globulin (CytoGam) might be of value for both treatment and prophylaxis for congenital infection.26 These authors studied 157 women with confirmed primary CMV infection. One-hundred forty-eight women were asymptomatic and were identified by routine serologic screening, 8 had symptomatic infection, and 1 was identified because of abnormal fetal ultrasound findings. Forty-five women had CMV detected in amniotic fluid by PCR or culture more than 6 weeks before study enrollment. Thirty-one of these women were treated with intravenous hyperimmune globulin (200 U or 200 mg/kg maternal body weight); 14 declined treatment. Seven of the latter women had infants who were acutely symptomatic at the time of delivery; only 1 of the 31 treated women had an affected neonate (adjusted odds ratio [OR], 0.02; P<.001). In this same study, 84 women did not have a diagnostic amniocentesis because their infection occurred within 6 weeks of enrollment, their gestational age was less than 20 weeks, or they declined the procedure. Thirty-seven of these women received hyperimmune globulin (100 U or 100 mg/kg) every month until delivery, and 47 declined treatment. Six of the treated women delivered infected infants compared with 19 of the untreated women (adjusted OR, 0.32; P<.04).
Although these results were quite encouraging, several problems existed with the study’s design, as noted in an editorial that accompanied the study’s publication.27 First, the study was not randomized or placebo controlled. Second, patients were not stratified based on the severity of fetal ultrasound abnormalities. Third, the dosing of hyperimmune globulin varied; 9 of the 31 patients in the treatment group received additional infusions of drug into either the amniotic fluid or fetal umbilical vein. Moreover, patients in the prophylaxis group actually received a higher cumulative dose of hyperimmune globulin than patients in the treatment group.
Two subsequent investigations that were better designed were unable to verify the effectiveness of hyperimmune globulin. In 2014, Revello and colleagues reported the results of a prospective, randomized, placebo-controlled, double-blinded study of 124 women at 5 to 26 weeks’ gestation with confirmed primary CMV infection.28 The rate of congenital infection was 30% in the group treated with hyperimmune globulin and 44% in the placebo group (P=.13). There also was no significant difference in the concentration of serum CMV DNA in treated versus untreated mothers. Moreover, the number of adverse obstetric events (preterm delivery, fetal growth restriction, intrahepatic cholestasis of pregnancy, and postpartum preeclampsia) in the treatment group was higher than in the placebo group, 13% versus 2%.
In 2021, Hughes and colleagues published the results of a multicenter, double-blind trial in 399 women who had a diagnosis of primary CMV infection before 23 weeks’ gestation.29 The primary outcome was defined as a composite of congenital CMV infection or fetal/neonatal death. An adverse primary outcome occurred in 22.7% of the patients who received hyperimmune globulin and 19.4% of those who received placebo (relative risk, 1.17; 95% confidence interval [CI], 0.80–1.72; P=.42).
Continue to: Jacquemard and colleagues...
Jacquemard and colleagues then proposed a different approach.30 In a small pilot study of 20 patients, these authors used high doses of oral valacylovir (2 g 4 times daily) and documented therapeutic drug concentrations and a decline in CMV viral load in fetal serum. Patients were not stratified by severity of fetal injury at onset of treatment, so the authors were unable to define which fetuses were most likely to benefit from treatment.
In a follow-up investigation, Leruez-Ville and colleagues reported another small series in which high-dose oral valacyclovir (8 g daily) was used for treatment.31 They excluded fetuses with severe brain anomalies and fetuses with no sonographic evidence of injury. The median gestational age at diagnosis was 26 weeks. Thirty-four of 43 treated fetuses were free of injury at birth. In addition, the viral load in the neonate’s serum decreased significantly after treatment, and the platelet count increased. The authors then compared these outcomes to a historical cohort and confirmed that treatment increased the proportion of asymptomatic neonates from 43% without treatment to 82% with treatment (P<.05 with no overlapping confidence intervals).
We conclude from these investigations that hyperimmune globulin is unlikely to be of value in treating congenital CMV infection, especially if the fetus already has sonographic findings of severe injury. High-dose oral valacyclovir also is unlikely to be of value in severely affected fetuses, particularly those with evidence of CNS injury. However, antiviral therapy may be of modest value in situations when the fetus is less severely injured.
Preventive measures
Since no definitive treatment is available for congenital CMV infection, our efforts as clinicians should focus on measures that may prevent transmission of infection to the pregnant patient. These measures include:
- Encouraging patients to use careful handwashing techniques when handling infant diapers and toys.
- Encouraging patients to adopt safe sexual practices if not already engaged in a mutually faithful, monogamous relationship.
- Using CMV-negative blood when transfusing a pregnant woman or a fetus.
At the present time, unfortunately, a readily available and highly effective therapy for prevention of CMV infection is not available.
CASE Congenital infection diagnosed
The ultrasound findings are most consistent with congenital CMV infection, especially given the patient’s work as an elementary schoolteacher. The diagnosis of maternal infection is best established by conventional serology (positive IgM, negative IgM) and detection of viral DNA in maternal blood by PCR testing. The diagnosis of congenital infection is best confirmed by documentation of viral DNA in the amniotic fluid by PCR testing. Given that this fetus already has evidence of moderate to severe injury, no treatment is likely to be effective in reversing the abnormal ultrasound findings. Pregnancy termination may be an option, depending upon the patient’s desires and the legal restrictions prevalent in the patient’s geographic area. ●
- Cytomegalovirus infection is the most common of the perinatally transmitted infections.
- Maternal infection is often asymptomatic. When symptoms are present, they resemble those of an influenza-like illness. In immunocompromised persons, however, CMV may cause serious complications, including pneumonia, hepatitis, and chorioretinitis.
- The virus is transmitted by contact with contaminated body fluids, such as saliva, urine, blood, and genital secretions.
- The greatest risk of severe fetal injury results from primary maternal infection in the first trimester of pregnancy.
- Manifestations of severe congenital CMV infection include growth restriction, microcephaly, ventriculomegaly, hepatosplenomegaly, ascites, chorioretinitis, thrombocytopenia, purpura, and hydrops (“blueberry muffin baby”).
- Late manifestations of infection, which usually follow recurrent maternal infection, may appear as a child enters elementary school and include visual and auditory deficits, developmental delays, and learning disabilities.
- The diagnosis of maternal infection is confirmed by serology and detection of viral DNA in the serum by PCR testing.
- The diagnosis of fetal infection is best made by a combination of abnormal ultrasound findings and detection of CMV DNA in amniotic fluid. The characteristic ultrasound findings include placentomegaly, microcephaly, ventriculomegaly, growth restriction, echogenic bowel, and serous effusions/hydrops.
- Treatment of the mother with antiviral medications such as valacyclovir may be of modest value in reducing placental edema, decreasing viral load in the fetus, and hastening the resolution of some ultrasound findings, such as echogenic bowel.
- While initial studies seemed promising, the use of hyperimmune globulin has not proven to be consistently effective in treating congenital infection.
CASE Anomalous findings on fetal anatomic survey
A 27-year-old previously healthy primigravid woman is at 18 weeks’ gestation. She is a first-grade schoolteacher. On her fetal anatomic survey, the estimated fetal weight was in the eighth percentile. Echogenic bowel and a small amount of ascitic fluid were noted in the fetal abdomen. The lateral and third ventricles were mildly dilated, the head circumference was 2 standard deviations below normal, and the placenta was slightly thickened and edematous.
What is the most likely diagnosis?
What diagnostic tests are indicated?
What management options are available for this patient?
Cytomegalovirus (CMV) is the most common of the perinatally transmitted infections, affecting 1% to 4% of all pregnancies. Although the virus typically causes either asymptomatic infection or only mild illness in immunocompetent individuals, it can cause life-threatening disease in immunocompromised persons and in the developing fetus. In this article, we review the virology and epidemiology of CMV infection and then focus on the key methods to diagnose infection in the mother and fetus. We conclude by considering measures that may be of at least modest value in treating CMV in pregnancy.
Virology of CMV infection
Cytomegalovirus is a double-stranded DNA virus in the Herpesviridae family. This ubiquitous virus is present in virtually all secretions and excretions of an infected host, including blood, urine, saliva, breast milk, genital secretions, and tissues and organs used for donation. Infection is transmitted through direct contact with any of the substances listed; contact with infected urine or saliva is the most common mode of transmission. Disease occurrence does not show seasonal variation.
After exposure, an incubation period of 28 to 60 days ensues, followed by development of viremia and clinical symptoms. In the majority of exposed individuals, CMV establishes a lifelong latent infection, and recurrent episodes of illness can occur as a result of reactivation of latent virus (also known as secondary infection) or, more rarely, infection with a new viral strain. In fact, most CMV illness episodes in pregnancy represent a reactivation of a previous infection rather than a new infection.
Following initial infection, both IgM (immunoglobulin M) and IgG (immunoglobulin G) antibodies develop rapidly and can be detected in blood within 1 to 2 weeks. IgM levels typically wane within 30 to 60 days, although persistence for several months is not unusual, and levels also can increase with viral reactivation (secondary infection). IgG antibodies typically persist for many years after a primary infection.
Intrauterine CMV infection occurs through hematogenous transplacental passage during maternal viremia. The risk of transmission and severity of fetal effects depend on whether or not the infection is primary or secondary in nature as well as the gestational age at fetal exposure.1,2
Additionally, postnatal vertical transmission can occur through exposure to viral particles in genital secretions as well as breast milk. CMV acquired in the postnatal period rarely produces severe sequelae in a healthy term neonate, but it has been associated with an increased rate of complications in very low birth weight and premature newborns.3
Continue to: Who is at risk...
Who is at risk
Congenital CMV, which occurs in 2.1 to 7.7 per 10,000 live births in the United States, is both the most common congenital infection and the leading cause of nonhereditary congenital hearing loss in children.4,5 The main reservoir of CMV in the United States is young children in day care settings, with approximately 50% of this population showing evidence of viral shedding in saliva.1 Adult populations in North America have a high prevalence of CMV IgG antibodies indicative of prior infection, with rates reaching 50% to 80%. Among seronegative individuals aged 12 to 49, the rate of seroconversion is approximately 1 in 60 annually.6 Significant racial disparities have been noted in rates of seroprevalence and seroconversion, with higher rates of infection in non-Hispanic Black and Mexican American individuals.6 Overall, the rate of new CMV infection among pregnant women in the United States is 0.7% to 4%.7
Clinical manifestations
Manifestations of infection differ depending on whether or not infection is primary or recurrent (secondary) and whether or not the host is immunocompetent or has a compromised immune system. Unique manifestations develop in the fetus.
CMV infection in children and adults. Among individuals with a normal immune response, the typical course of CMV is either no symptoms or a mononucleosis-like illness. In symptomatic patients, the most common symptoms include malaise, fever, and night sweats, and the most common associated laboratory abnormalities are elevation in liver function tests and a decreased white blood cell count, with a predominance of lymphocytes.8
Immunocompromised individuals are at risk for significant morbidity and mortality resulting from CMV. Illness may be the result of reactivation of latent infection due to decreased immune function or may be acquired as a result of treatment such as transplantation of CMV-positive organs or tissues, including bone marrow. Virtually any organ system can be affected, with potential for permanent organ damage and death. Severe systemic infection also can occur.
CMV infection in the fetus and neonate. As noted previously, fetal infection develops as a result of transplacental passage coincident with maternal infection. The risk of CMV transmission to the fetus and the severity of fetal injury vary based on gestational age at fetal infection and whether or not maternal infection is primary or secondary.
In most studies, primary maternal infections are associated with higher rates of fetal infection and more severe fetal and neonatal disease manifestations.2,7,9,10 Primary infections carry an overall 30% to 40% risk of transmission to the fetus.7,11 The risk of fetal transmission is much lower with a recurrent infection and is usually less than 2%.11 Due to their greater overall incidence, secondary infections account for the majority of cases of fetal and neonatal CMV disease.7 Importantly, although secondary infections generally have been regarded as having a lower risk and lower severity of fetal and neonatal disease, several recent studies have demonstrated rates of complications similar to, and even exceeding, those of primary infections.12-15 The TABLE provides a summary of the risks of fetal transmission and symptomatic fetal infection based on trimester of pregnancy.2,11,16-18
In the fetus, CMV may affect multiple organ systems. Among sonographic and magnetic resonance imaging (MRI) findings, central nervous system (CNS) anomalies are the most common.19,20 These can include microcephaly, ventriculomegaly, and periventricular calcifications. The gastrointestinal system also is frequently affected, and findings include echogenic bowel, hepatosplenomegaly, and liver calcifications. Lastly, isolated effusions, placentomegaly, fetal growth restriction, and even frank hydrops can develop. More favorable neurologic outcomes have been demonstrated in infants with no prenatal brain imaging abnormalities.20,21 However, the role of MRI in prenatal prognosis currently is not well defined.
FIGURE 1 illustrates selected sonographic findings associated with fetal CMV infection.
About 85% to 90% of infants with congenital CMV that results from primary maternal infection have no symptoms at birth. Among the 10% to 15% of infants that do have symptoms, petechial rash, jaundice, and hepatosplenomegaly are the most common manifestations (“blueberry muffin baby”). Approximately 10% to 20% of infants in this group have evidence of chorioretinitis on ophthalmologic examination, and 50% show either microcephaly or low birth weight.22Among survivors of symptomatic congenital CMV, more than 50% have long-term neurologic morbidities that may include sensorineural hearing loss, seizures, vision impairment, and developmental disabilities. Note that even when neonates appear asymptomatic at birth (regardless of whether infection is primary or secondary), 5% may develop microcephaly and motor deficits, 10% go on to develop sensorineural hearing loss, and the overall rate of neurologic morbidity reaches 13% to 15%.12,23 Some of the observed deficits manifest at several years of age, and, currently, no models exist for prediction of outcome.
Continue to: Diagnosing CMV infection...
Diagnosing CMV infection
Maternal infection
If maternal CMV infection is suspected based on a symptomatic illness or an abnormal fetal ultrasound exam, the first diagnostic test should be an assessment of IgM and IgG serology. If the former test results are positive and the latter negative, the diagnosis of acute CMV infection is confirmed. A positive serum CMV DNA polymerase chain reaction (PCR) test adds additional assurance that the diagnosis is correct. Primary infection, as noted above, poses the greatest risk of serious injury to the fetus.1
A frequent diagnostic dilemma arises when both the IgM and IgG antibody are positive. Remember that CMV IgM antibody can remain positive for 9 to 12 months after a primary infection and can reappear in the maternal serum in the face of a recurrent or reactivated infection. When confronted by both a positive IgM and positive IgG result, the clinician should then order IgG avidity testing. If the avidity is low to moderate, which reflects poor binding of antibody to the virus, the patient likely has an acute infection. If the avidity is high, which reflects enhanced binding of antibody to virus, the patient probably has a recurrent or reactivated infection; this scenario poses less danger to the developing fetus. The presence of CMV DNA in serum is also more consistent with acute infection, although viremia still can occur with recurrent infection. FIGURE 2 presents a suggested algorithm for the diagnosis of CMV in the pregnant patient.1
If a diagnosis of maternal CMV infection is confirmed, liver function tests should be obtained to determine if CMV hepatitis is present. If the liver function tests are abnormal, a coagulation profile also should be performed to identify the mother who might be at risk for peripartum hemorrhage.
Fetal infection
The single best test for confirmation of congenital CMV infection is detection of viral DNA and quantitation of viral load in the amniotic fluid by PCR. If the amniocentesis is performed prior to 20 weeks’ gestation and is negative, the test should be repeated in approximately 4 weeks.1,19,24
Detection of viral DNA indicates congenital infection. The ultimate task, however, is to determine if the infection has injured the fetus. Detailed ultrasound examination is the key to identifying fetal injury. As noted previously, the principal ultrasonographic findings that suggest congenital CMV infection include2,19,20,21,25:
- hydropic placenta
- fetal growth restriction
- microcephaly (head circumference more than 3 standard deviations below the mean)
- periventricular calcifications
- enlarged liver
- echogenic bowel
- ascites
- fetal hydrops.
Management: Evidence on CMV hyperimmune globulin, valacyclovir
If the immunocompetent mother has clinical manifestations of infection, she should receive symptomatic treatment. She should be encouraged to rest as much as possible, stay well hydrated, and use acetaminophen (1,000 mg every 6 to 8 hours) as needed for malaise and fever.
However, if the mother is immunocompromised and has signs of serious complications, such as chorioretinitis, hepatitis, or pneumonia, more aggressive therapy is indicated. Drugs used in this setting include foscarnet and ganciclovir and are best prescribed in consultation with a medical infectious disease specialist.
At this time, no consistently effective therapy for congenital infection is available. Therefore, if a patient has primary CMV infection in the first half of pregnancy, particularly in the first trimester, she should be counseled that the risk of fetal infection is approximately 40% and that approximately 5% to 15% of infants will be severely affected at birth. Given this information, some patients may opt for pregnancy termination.
In 2005, a report from Nigro and colleagues stimulated great hope that CMV-specific hyperimmune globulin (CytoGam) might be of value for both treatment and prophylaxis for congenital infection.26 These authors studied 157 women with confirmed primary CMV infection. One-hundred forty-eight women were asymptomatic and were identified by routine serologic screening, 8 had symptomatic infection, and 1 was identified because of abnormal fetal ultrasound findings. Forty-five women had CMV detected in amniotic fluid by PCR or culture more than 6 weeks before study enrollment. Thirty-one of these women were treated with intravenous hyperimmune globulin (200 U or 200 mg/kg maternal body weight); 14 declined treatment. Seven of the latter women had infants who were acutely symptomatic at the time of delivery; only 1 of the 31 treated women had an affected neonate (adjusted odds ratio [OR], 0.02; P<.001). In this same study, 84 women did not have a diagnostic amniocentesis because their infection occurred within 6 weeks of enrollment, their gestational age was less than 20 weeks, or they declined the procedure. Thirty-seven of these women received hyperimmune globulin (100 U or 100 mg/kg) every month until delivery, and 47 declined treatment. Six of the treated women delivered infected infants compared with 19 of the untreated women (adjusted OR, 0.32; P<.04).
Although these results were quite encouraging, several problems existed with the study’s design, as noted in an editorial that accompanied the study’s publication.27 First, the study was not randomized or placebo controlled. Second, patients were not stratified based on the severity of fetal ultrasound abnormalities. Third, the dosing of hyperimmune globulin varied; 9 of the 31 patients in the treatment group received additional infusions of drug into either the amniotic fluid or fetal umbilical vein. Moreover, patients in the prophylaxis group actually received a higher cumulative dose of hyperimmune globulin than patients in the treatment group.
Two subsequent investigations that were better designed were unable to verify the effectiveness of hyperimmune globulin. In 2014, Revello and colleagues reported the results of a prospective, randomized, placebo-controlled, double-blinded study of 124 women at 5 to 26 weeks’ gestation with confirmed primary CMV infection.28 The rate of congenital infection was 30% in the group treated with hyperimmune globulin and 44% in the placebo group (P=.13). There also was no significant difference in the concentration of serum CMV DNA in treated versus untreated mothers. Moreover, the number of adverse obstetric events (preterm delivery, fetal growth restriction, intrahepatic cholestasis of pregnancy, and postpartum preeclampsia) in the treatment group was higher than in the placebo group, 13% versus 2%.
In 2021, Hughes and colleagues published the results of a multicenter, double-blind trial in 399 women who had a diagnosis of primary CMV infection before 23 weeks’ gestation.29 The primary outcome was defined as a composite of congenital CMV infection or fetal/neonatal death. An adverse primary outcome occurred in 22.7% of the patients who received hyperimmune globulin and 19.4% of those who received placebo (relative risk, 1.17; 95% confidence interval [CI], 0.80–1.72; P=.42).
Continue to: Jacquemard and colleagues...
Jacquemard and colleagues then proposed a different approach.30 In a small pilot study of 20 patients, these authors used high doses of oral valacylovir (2 g 4 times daily) and documented therapeutic drug concentrations and a decline in CMV viral load in fetal serum. Patients were not stratified by severity of fetal injury at onset of treatment, so the authors were unable to define which fetuses were most likely to benefit from treatment.
In a follow-up investigation, Leruez-Ville and colleagues reported another small series in which high-dose oral valacyclovir (8 g daily) was used for treatment.31 They excluded fetuses with severe brain anomalies and fetuses with no sonographic evidence of injury. The median gestational age at diagnosis was 26 weeks. Thirty-four of 43 treated fetuses were free of injury at birth. In addition, the viral load in the neonate’s serum decreased significantly after treatment, and the platelet count increased. The authors then compared these outcomes to a historical cohort and confirmed that treatment increased the proportion of asymptomatic neonates from 43% without treatment to 82% with treatment (P<.05 with no overlapping confidence intervals).
We conclude from these investigations that hyperimmune globulin is unlikely to be of value in treating congenital CMV infection, especially if the fetus already has sonographic findings of severe injury. High-dose oral valacyclovir also is unlikely to be of value in severely affected fetuses, particularly those with evidence of CNS injury. However, antiviral therapy may be of modest value in situations when the fetus is less severely injured.
Preventive measures
Since no definitive treatment is available for congenital CMV infection, our efforts as clinicians should focus on measures that may prevent transmission of infection to the pregnant patient. These measures include:
- Encouraging patients to use careful handwashing techniques when handling infant diapers and toys.
- Encouraging patients to adopt safe sexual practices if not already engaged in a mutually faithful, monogamous relationship.
- Using CMV-negative blood when transfusing a pregnant woman or a fetus.
At the present time, unfortunately, a readily available and highly effective therapy for prevention of CMV infection is not available.
CASE Congenital infection diagnosed
The ultrasound findings are most consistent with congenital CMV infection, especially given the patient’s work as an elementary schoolteacher. The diagnosis of maternal infection is best established by conventional serology (positive IgM, negative IgM) and detection of viral DNA in maternal blood by PCR testing. The diagnosis of congenital infection is best confirmed by documentation of viral DNA in the amniotic fluid by PCR testing. Given that this fetus already has evidence of moderate to severe injury, no treatment is likely to be effective in reversing the abnormal ultrasound findings. Pregnancy termination may be an option, depending upon the patient’s desires and the legal restrictions prevalent in the patient’s geographic area. ●
- Cytomegalovirus infection is the most common of the perinatally transmitted infections.
- Maternal infection is often asymptomatic. When symptoms are present, they resemble those of an influenza-like illness. In immunocompromised persons, however, CMV may cause serious complications, including pneumonia, hepatitis, and chorioretinitis.
- The virus is transmitted by contact with contaminated body fluids, such as saliva, urine, blood, and genital secretions.
- The greatest risk of severe fetal injury results from primary maternal infection in the first trimester of pregnancy.
- Manifestations of severe congenital CMV infection include growth restriction, microcephaly, ventriculomegaly, hepatosplenomegaly, ascites, chorioretinitis, thrombocytopenia, purpura, and hydrops (“blueberry muffin baby”).
- Late manifestations of infection, which usually follow recurrent maternal infection, may appear as a child enters elementary school and include visual and auditory deficits, developmental delays, and learning disabilities.
- The diagnosis of maternal infection is confirmed by serology and detection of viral DNA in the serum by PCR testing.
- The diagnosis of fetal infection is best made by a combination of abnormal ultrasound findings and detection of CMV DNA in amniotic fluid. The characteristic ultrasound findings include placentomegaly, microcephaly, ventriculomegaly, growth restriction, echogenic bowel, and serous effusions/hydrops.
- Treatment of the mother with antiviral medications such as valacyclovir may be of modest value in reducing placental edema, decreasing viral load in the fetus, and hastening the resolution of some ultrasound findings, such as echogenic bowel.
- While initial studies seemed promising, the use of hyperimmune globulin has not proven to be consistently effective in treating congenital infection.
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TR, et al, eds. Creasy and Resnik’s Maternal Fetal Medicine: Principles and Practice. 8th ed. 2019:888-890.
- Chatzakis C, Ville Y, Makrydimas G, et al. Timing of primary maternal cytomegalovirus infection and rates of vertical transmission and fetal consequences. Am J Obstet Gynecol. 2020;223:870-883.e11. doi:10.1016/j.ajog.2020.05.038
- Kelly MS, Benjamin DK, Puopolo KM, et al. Postnatal cytomegalovirus infection and the risk for bronchopulmonary dysplasia. JAMA Pediatr. 2015;169:e153785. doi:10.1001 /jamapediatrics.2015.3785
- Messinger CJ, Lipsitch M, Bateman BT, et al. Association between congenital cytomegalovirus and the prevalence at birth of microcephaly in the United States. JAMA Pediatr. 2020;174:1159-1167. doi:10.1001/jamapediatrics.2020.3009
- De Cuyper E, Acke F, Keymeulen A, et al. Risk factors for hearing loss at birth in newborns with congenital cytomegalovirus infection. JAMA Otolaryngol Head Neck Surg. 2023;149:122-130. doi:10.1001/jamaoto.2022.4109
- Colugnati FA, Staras SA, Dollard SC, et al. Incidence of cytomegalovirus infection among the general population and pregnant women in the United States. BMC Infect Dis. 2007;7:71. doi:10.1186/1471-2334-7-71
- Stagno S, Pass RF, Cloud G, et al. Primary cytomegalovirus infection in pregnancy. Incidence, transmission to fetus, and clinical outcome. JAMA. 1986;256:1904-1908.
- Wreghitt TG, Teare EL, Sule O, et al. Cytomegalovirus infection in immunocompetent patients. Clin Infect Dis. 2003;37:1603-1606. doi:10.1086/379711
- Fowler KB, Stagno S, Pass RF, et al. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N Engl J Med. 1992;326:663-667. doi:10.1056 /NEJM199203053261003
- Faure-Bardon V, Magny JF, Parodi M, et al. Sequelae of congenital cytomegalovirus following maternal primary infections are limited to those acquired in the first trimester of pregnancy. Clin Infect Dis. 2019;69:1526-1532. doi:10.1093/ cid/ciy1128
- Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol. 2007;17:253-276. doi:10.1002/ rmv.535
- Boppana SB, Pass RF, Britt WJ, et al. Symptomatic congenital cytomegalovirus infection: neonatal morbidity and mortality. Pediatr Infect Dis J. 1992;11:93-99. doi:10.1097/00006454-199202000-00007
- Ross SA, Fowler KB, Ashrith G, et al. Hearing loss in children with congenital cytomegalovirus infection born to mothers with preexisting immunity. J Pediatr. 2006;148:332-336. doi:10.1016/j.jpeds.2005.09.003
- Zalel Y, Gilboa Y, Berkenshtat M, et al. Secondary cytomegalovirus infection can cause severe fetal sequelae despite maternal preconceptional immunity. Ultrasound Obstet Gynecol. 31:417-420. doi:10.1002/uog.5255
- Scaramuzzino F, Di Pastena M, Chiurchiu S, et al. Secondary cytomegalovirus infections: how much do we still not know? Comparison of children with symptomatic congenital cytomegalovirus born to mothers with primary and secondary infection. Front Pediatr. 2022;10:885926. doi:10.3389/fped.2022.885926
- Gindes L, Teperberg-Oikawa M, Sherman D, et al. Congenital cytomegalovirus infection following primary maternal infection in the third trimester. BJOG. 2008;115:830-835. doi:10.1111/j.1471-0528.2007.01651.x
- Hadar E, Dorfman E, Bardin R, et al. Symptomatic congenital cytomegalovirus disease following non-primary maternal infection: a retrospective cohort study. BMC Infect Dis. 2017;17:31. doi:10.1186/s12879-016-2161-3
- Elkan Miller T, Weisz B, Yinon Y, et al. Congenital cytomegalovirus infection following second and third trimester maternal infection is associated with mild childhood adverse outcome not predicted by prenatal imaging. J Pediatric Infect Dis Soc. 2021;10:562-568. doi:10.1093/jpids/ piaa154
- Lipitz S, Yinon Y, Malinger G, et al. Risk of cytomegalovirusassociated sequelae in relation to time of infection and findings on prenatal imaging. Ultrasound Obstet Gynecol. 2013;41:508-514. doi:10.1002/uog.12377
- Lipitz S, Elkan Miller T, Yinon Y, et al. Revisiting short- and long-term outcome after fetal first-trimester primary cytomegalovirus infection in relation to prenatal imaging findings. Ultrasound Obstet Gynecol. 2020;56:572-578. doi:10.1002/uog.21946
- Buca D, Di Mascio D, Rizzo G, et al. Outcome of fetuses with congenital cytomegalovirus infection and normal ultrasound at diagnosis: systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2021;57:551-559. doi:10.1002/uog.23143
- Boppana SB, Ross SA, Fowler KB. Congenital cytomegalovirus infection: clinical outcome. Clin Infect Dis. 2013;57 (suppl 4):S178-S181. doi:10.1093/cid/cit629
- Dollard SC, Grosse SD, Ross DS. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol. 2007;17:355-363. doi:10.1002/rmv.544
- Hughes BL, Gyamfi-Bannerman C. Diagnosis and antenatal management of congenital cytomegalovirus infection. Am J Obstet Gynecol. 2016;214:B5-11. doi:10.1016 /j.ajog.2016.02.042
- Rouse DJ, Fette LM, Hughes BL, et al. Noninvasive prediction of congenital cytomegalovirus infection after maternal primary infection. Obstet Gynecol. 2022;139:400-406. doi:10.1097/AOG.0000000000004691
- Nigro G, Adler SP, La Torre R, et al; Congenital Cytomegalovirus Collaborating Group. Passive immunization during pregnancy for congenital cytomegalovirus infection. N Engl J Med. 2005;353:1350-1362. doi:10.1056/NEJMoa043337
- Duff P. Immunotherapy for congenital cytomegalovirus infection. N Engl J Med. 2005;355:1402-1404. doi:10.1056 /NEJMe058172
- Revello MG, Lazzarotto T, Guerra B, et al. A randomized trial of hyperimmune globulin to prevent congenital cytomegalovirus. N Engl J Med. 2014;370:1316-1326. doi:10.1056/NEJMoa1310214
- Hughes BL, Clifton RG, Rouse DJ, et al. A trial of hyperimmune globulin to prevent congenital cytomegalovirus infection. N Engl J Med. 2021;385:436-444. doi:10.1056/NEJMoa1913569
- Jacquemard F, Yamamoto M, Costa JM, et al. Maternal administration of valaciclovir in symptomatic intrauterine cytomegalovirus infection. BJOG. 2007;114:1113-1121. doi:10.1111/j.1471-0528.2007.01308.x
- Leruez-Ville M, Ghout I, Bussières L, et al. In utero treatment of congenital cytomegalovirus infection with valacyclovir in a multicenter, open-label, phase II study. Am J Obstet Gynecol. 2016;215:462.e1-462.e10. doi:10.1016/j.ajog.2016.04.003
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TR, et al, eds. Creasy and Resnik’s Maternal Fetal Medicine: Principles and Practice. 8th ed. 2019:888-890.
- Chatzakis C, Ville Y, Makrydimas G, et al. Timing of primary maternal cytomegalovirus infection and rates of vertical transmission and fetal consequences. Am J Obstet Gynecol. 2020;223:870-883.e11. doi:10.1016/j.ajog.2020.05.038
- Kelly MS, Benjamin DK, Puopolo KM, et al. Postnatal cytomegalovirus infection and the risk for bronchopulmonary dysplasia. JAMA Pediatr. 2015;169:e153785. doi:10.1001 /jamapediatrics.2015.3785
- Messinger CJ, Lipsitch M, Bateman BT, et al. Association between congenital cytomegalovirus and the prevalence at birth of microcephaly in the United States. JAMA Pediatr. 2020;174:1159-1167. doi:10.1001/jamapediatrics.2020.3009
- De Cuyper E, Acke F, Keymeulen A, et al. Risk factors for hearing loss at birth in newborns with congenital cytomegalovirus infection. JAMA Otolaryngol Head Neck Surg. 2023;149:122-130. doi:10.1001/jamaoto.2022.4109
- Colugnati FA, Staras SA, Dollard SC, et al. Incidence of cytomegalovirus infection among the general population and pregnant women in the United States. BMC Infect Dis. 2007;7:71. doi:10.1186/1471-2334-7-71
- Stagno S, Pass RF, Cloud G, et al. Primary cytomegalovirus infection in pregnancy. Incidence, transmission to fetus, and clinical outcome. JAMA. 1986;256:1904-1908.
- Wreghitt TG, Teare EL, Sule O, et al. Cytomegalovirus infection in immunocompetent patients. Clin Infect Dis. 2003;37:1603-1606. doi:10.1086/379711
- Fowler KB, Stagno S, Pass RF, et al. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N Engl J Med. 1992;326:663-667. doi:10.1056 /NEJM199203053261003
- Faure-Bardon V, Magny JF, Parodi M, et al. Sequelae of congenital cytomegalovirus following maternal primary infections are limited to those acquired in the first trimester of pregnancy. Clin Infect Dis. 2019;69:1526-1532. doi:10.1093/ cid/ciy1128
- Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol. 2007;17:253-276. doi:10.1002/ rmv.535
- Boppana SB, Pass RF, Britt WJ, et al. Symptomatic congenital cytomegalovirus infection: neonatal morbidity and mortality. Pediatr Infect Dis J. 1992;11:93-99. doi:10.1097/00006454-199202000-00007
- Ross SA, Fowler KB, Ashrith G, et al. Hearing loss in children with congenital cytomegalovirus infection born to mothers with preexisting immunity. J Pediatr. 2006;148:332-336. doi:10.1016/j.jpeds.2005.09.003
- Zalel Y, Gilboa Y, Berkenshtat M, et al. Secondary cytomegalovirus infection can cause severe fetal sequelae despite maternal preconceptional immunity. Ultrasound Obstet Gynecol. 31:417-420. doi:10.1002/uog.5255
- Scaramuzzino F, Di Pastena M, Chiurchiu S, et al. Secondary cytomegalovirus infections: how much do we still not know? Comparison of children with symptomatic congenital cytomegalovirus born to mothers with primary and secondary infection. Front Pediatr. 2022;10:885926. doi:10.3389/fped.2022.885926
- Gindes L, Teperberg-Oikawa M, Sherman D, et al. Congenital cytomegalovirus infection following primary maternal infection in the third trimester. BJOG. 2008;115:830-835. doi:10.1111/j.1471-0528.2007.01651.x
- Hadar E, Dorfman E, Bardin R, et al. Symptomatic congenital cytomegalovirus disease following non-primary maternal infection: a retrospective cohort study. BMC Infect Dis. 2017;17:31. doi:10.1186/s12879-016-2161-3
- Elkan Miller T, Weisz B, Yinon Y, et al. Congenital cytomegalovirus infection following second and third trimester maternal infection is associated with mild childhood adverse outcome not predicted by prenatal imaging. J Pediatric Infect Dis Soc. 2021;10:562-568. doi:10.1093/jpids/ piaa154
- Lipitz S, Yinon Y, Malinger G, et al. Risk of cytomegalovirusassociated sequelae in relation to time of infection and findings on prenatal imaging. Ultrasound Obstet Gynecol. 2013;41:508-514. doi:10.1002/uog.12377
- Lipitz S, Elkan Miller T, Yinon Y, et al. Revisiting short- and long-term outcome after fetal first-trimester primary cytomegalovirus infection in relation to prenatal imaging findings. Ultrasound Obstet Gynecol. 2020;56:572-578. doi:10.1002/uog.21946
- Buca D, Di Mascio D, Rizzo G, et al. Outcome of fetuses with congenital cytomegalovirus infection and normal ultrasound at diagnosis: systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2021;57:551-559. doi:10.1002/uog.23143
- Boppana SB, Ross SA, Fowler KB. Congenital cytomegalovirus infection: clinical outcome. Clin Infect Dis. 2013;57 (suppl 4):S178-S181. doi:10.1093/cid/cit629
- Dollard SC, Grosse SD, Ross DS. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol. 2007;17:355-363. doi:10.1002/rmv.544
- Hughes BL, Gyamfi-Bannerman C. Diagnosis and antenatal management of congenital cytomegalovirus infection. Am J Obstet Gynecol. 2016;214:B5-11. doi:10.1016 /j.ajog.2016.02.042
- Rouse DJ, Fette LM, Hughes BL, et al. Noninvasive prediction of congenital cytomegalovirus infection after maternal primary infection. Obstet Gynecol. 2022;139:400-406. doi:10.1097/AOG.0000000000004691
- Nigro G, Adler SP, La Torre R, et al; Congenital Cytomegalovirus Collaborating Group. Passive immunization during pregnancy for congenital cytomegalovirus infection. N Engl J Med. 2005;353:1350-1362. doi:10.1056/NEJMoa043337
- Duff P. Immunotherapy for congenital cytomegalovirus infection. N Engl J Med. 2005;355:1402-1404. doi:10.1056 /NEJMe058172
- Revello MG, Lazzarotto T, Guerra B, et al. A randomized trial of hyperimmune globulin to prevent congenital cytomegalovirus. N Engl J Med. 2014;370:1316-1326. doi:10.1056/NEJMoa1310214
- Hughes BL, Clifton RG, Rouse DJ, et al. A trial of hyperimmune globulin to prevent congenital cytomegalovirus infection. N Engl J Med. 2021;385:436-444. doi:10.1056/NEJMoa1913569
- Jacquemard F, Yamamoto M, Costa JM, et al. Maternal administration of valaciclovir in symptomatic intrauterine cytomegalovirus infection. BJOG. 2007;114:1113-1121. doi:10.1111/j.1471-0528.2007.01308.x
- Leruez-Ville M, Ghout I, Bussières L, et al. In utero treatment of congenital cytomegalovirus infection with valacyclovir in a multicenter, open-label, phase II study. Am J Obstet Gynecol. 2016;215:462.e1-462.e10. doi:10.1016/j.ajog.2016.04.003
Duchenne muscular dystrophy gene therapy safe, effective at 4 years
PHOENIX – compared with untreated patients who showed significant decline over the same time period, new research shows.
“Functional assessments demonstrated long-term sustained stabilization of motor function that was clinically meaningful, at ages where functional decline would be expected based on natural history,” the investigators noted in their abstract. Furthermore, the treatment, known as delandistrogene moxeparvovec-rokl (SRP-9001), was well tolerated 4 years post treatment.
The study was presented at the annual meeting of the American Association of Neuromuscular Electrodiagnostic Medicine.
Severe type of DMD
Considered one of the most severe forms of muscular dystrophy, DMD causes progressive muscle wasting stemming from the root genetic cause of missing dystrophin in muscle cells. Often referred to as a molecular “shock absorber,” dystrophin stabilizes the sarcolemma during muscle contractions to prevent degeneration.
SRP-9001, a single-dose recombinant gene therapy administered as an intravenous infusion, was designed to deliver a trimmed down form of dystrophin to compensate for the deficit.
In July, the adeno-associated virus vector (AAV)–based SRP-9001 gene therapy was granted accelerated approval by the Food and Drug Administration for the treatment of ambulatory pediatric patients aged 4-5 years with DMD with a confirmed mutation in the DMD gene.
The therapy is administered over 1-2 hours at a dose of 133 trillion vector genomes per kilogram of body weight.
For Study 101, one of several evaluating the novel therapy, a research team led by senior investigator Jerry Mendell, MD, an attending neurologist at Nationwide Children’s Hospital and professor of pediatrics and neurology at Ohio State University, both in Columbus,evaluated data on four ambulatory male patients aged 4-8 years who received a single IV infusion of the therapy.
All patients also received prednisone 1 mg/kg, 1 day preinfusion and 30 days post infusion.
At 4 years post treatment, there were no new safety events. All treatment-related adverse events occurred mainly within the first 70 days, and all resolved.
The most commonly reported adverse reactions of the gene therapy include vomiting, nausea, increases in liver enzymes, pyrexia (fever), and thrombocytopenia, all of which occurred within 90 days of infusion and been manageable.
Risk mitigation strategies for hepatotoxicity or acute liver injury include pre- and postinfusion monitoring of liver enzymes, the authors noted.
No serious abnormalities were observed in hematologic or chemistry panels, and while three patients had elevated gamma-glutamyl transpeptidase in the first 3 months post treatment, those cases resolved with oral steroid treatment.
Significant improvements in function were observed, with a mean improvement in North Star Ambulatory Assessment (NSAA) scores from baseline of 7.0 points (range, 4-11).
Exploratory analyses further showed that, compared with a propensity score–weighted external control cohort of 21 patients with DMD who did not receive the therapy, those receiving SRP-9001 had a statistically significant difference of 9.4 points in least-squares mean change from baseline to 4 years on the NSAA score (P = .0125).
Similar trends were observed in improvement from baseline in key measures of time to rise, 4-stair climb, and 10- and 100-meter walk/run function tests.
Other reported adverse events include acute serious liver injury, immune-mediated myositis, and myocarditis. Because of the latter risk, the therapy is contraindicated in patients with any deletion in exon 8 and/or exon 9 in the DMD gene.
The current 4-year update on SRP-9001 adds to clinical trial results that have been reported on more than 80 patients treated to date, with favorable results and consistent safety profiles reported at other time points.
Continued FDA approval for the therapy will be contingent upon verification of a clinical benefit in the confirmatory trials, including the EMBARK trial.
Increased function, long-term stability
Discussing the research at the meeting, Craig McDonald, MD, professor and chair of physical medicine & rehabilitation, a professor of pediatrics and study chair of the CINRG Duchenne Natural History Study at University of California Davis Health, noted that top-line results from the ongoing, confirmatory phase 3 EMBARK trial show functional benefits of SRP-9001 not only in 4- to 5-year-olds but also in other older age groups.
“What’s really striking, and in my mind the most impressive, is that when you follow these patients out 3 or 4 years ... you see there is this bump in function followed by long-term stability, whereas the external control cohort predictably shows really quite significant declines in their [NSAA] functional values,” he said in his presentation.
“When you look at each individually treated patient versus their own predicted trajectory using their baseline values on the time function test, each of the patients actually has a really quite impressive stabilization of function over their predicted disease trajectory,” he added.
A caveat that SRP-9001 shares with other gene therapies is the issue of cost – reported in the range of $2 million–$3 million.
In the context of racial and socioeconomic disparities in access to diagnosis and care reported in DMD, Emma Ciafaloni, MD, a professor of neurology and pediatrics at the University of Rochester (N.Y.) Medical Center, underscored the need to consider approval versus access to gene therapies and how to optimize access to the novel treatments.
“We need to consider what the cost is, how it’s going to be accessed, and whether there is a sustainable model,” said Ciafaloni, who was not associated with the study. “There will need to be institutional readiness and support for specialized multidisciplinary clinics for gene therapy.”
She also noted “we need to consider how we can do better on a broader level, because this is not a provider problem or a manufacturer problem — it’s a society problem.”
The study was funded by Sarepta Therapeutics. McDonald reported consulting work for Sarepta Therapeutics and has been an investigator in SRP-9001 research. Ciafaloni reported serving on advisory boards or other relationships with Alexion, Argenx, Biogen, Amicus, Momenta, Medscape, Pfizer, Sanofi/Genzyme, Sarepta, Jansen, NS Pharma, CureSMA, Orphazyme, the Patient-Centered Outcomes Research Institute, PPMD, PTC Therapeutics, and Santhera.
A version of this article first appeared on Medscape.com.
PHOENIX – compared with untreated patients who showed significant decline over the same time period, new research shows.
“Functional assessments demonstrated long-term sustained stabilization of motor function that was clinically meaningful, at ages where functional decline would be expected based on natural history,” the investigators noted in their abstract. Furthermore, the treatment, known as delandistrogene moxeparvovec-rokl (SRP-9001), was well tolerated 4 years post treatment.
The study was presented at the annual meeting of the American Association of Neuromuscular Electrodiagnostic Medicine.
Severe type of DMD
Considered one of the most severe forms of muscular dystrophy, DMD causes progressive muscle wasting stemming from the root genetic cause of missing dystrophin in muscle cells. Often referred to as a molecular “shock absorber,” dystrophin stabilizes the sarcolemma during muscle contractions to prevent degeneration.
SRP-9001, a single-dose recombinant gene therapy administered as an intravenous infusion, was designed to deliver a trimmed down form of dystrophin to compensate for the deficit.
In July, the adeno-associated virus vector (AAV)–based SRP-9001 gene therapy was granted accelerated approval by the Food and Drug Administration for the treatment of ambulatory pediatric patients aged 4-5 years with DMD with a confirmed mutation in the DMD gene.
The therapy is administered over 1-2 hours at a dose of 133 trillion vector genomes per kilogram of body weight.
For Study 101, one of several evaluating the novel therapy, a research team led by senior investigator Jerry Mendell, MD, an attending neurologist at Nationwide Children’s Hospital and professor of pediatrics and neurology at Ohio State University, both in Columbus,evaluated data on four ambulatory male patients aged 4-8 years who received a single IV infusion of the therapy.
All patients also received prednisone 1 mg/kg, 1 day preinfusion and 30 days post infusion.
At 4 years post treatment, there were no new safety events. All treatment-related adverse events occurred mainly within the first 70 days, and all resolved.
The most commonly reported adverse reactions of the gene therapy include vomiting, nausea, increases in liver enzymes, pyrexia (fever), and thrombocytopenia, all of which occurred within 90 days of infusion and been manageable.
Risk mitigation strategies for hepatotoxicity or acute liver injury include pre- and postinfusion monitoring of liver enzymes, the authors noted.
No serious abnormalities were observed in hematologic or chemistry panels, and while three patients had elevated gamma-glutamyl transpeptidase in the first 3 months post treatment, those cases resolved with oral steroid treatment.
Significant improvements in function were observed, with a mean improvement in North Star Ambulatory Assessment (NSAA) scores from baseline of 7.0 points (range, 4-11).
Exploratory analyses further showed that, compared with a propensity score–weighted external control cohort of 21 patients with DMD who did not receive the therapy, those receiving SRP-9001 had a statistically significant difference of 9.4 points in least-squares mean change from baseline to 4 years on the NSAA score (P = .0125).
Similar trends were observed in improvement from baseline in key measures of time to rise, 4-stair climb, and 10- and 100-meter walk/run function tests.
Other reported adverse events include acute serious liver injury, immune-mediated myositis, and myocarditis. Because of the latter risk, the therapy is contraindicated in patients with any deletion in exon 8 and/or exon 9 in the DMD gene.
The current 4-year update on SRP-9001 adds to clinical trial results that have been reported on more than 80 patients treated to date, with favorable results and consistent safety profiles reported at other time points.
Continued FDA approval for the therapy will be contingent upon verification of a clinical benefit in the confirmatory trials, including the EMBARK trial.
Increased function, long-term stability
Discussing the research at the meeting, Craig McDonald, MD, professor and chair of physical medicine & rehabilitation, a professor of pediatrics and study chair of the CINRG Duchenne Natural History Study at University of California Davis Health, noted that top-line results from the ongoing, confirmatory phase 3 EMBARK trial show functional benefits of SRP-9001 not only in 4- to 5-year-olds but also in other older age groups.
“What’s really striking, and in my mind the most impressive, is that when you follow these patients out 3 or 4 years ... you see there is this bump in function followed by long-term stability, whereas the external control cohort predictably shows really quite significant declines in their [NSAA] functional values,” he said in his presentation.
“When you look at each individually treated patient versus their own predicted trajectory using their baseline values on the time function test, each of the patients actually has a really quite impressive stabilization of function over their predicted disease trajectory,” he added.
A caveat that SRP-9001 shares with other gene therapies is the issue of cost – reported in the range of $2 million–$3 million.
In the context of racial and socioeconomic disparities in access to diagnosis and care reported in DMD, Emma Ciafaloni, MD, a professor of neurology and pediatrics at the University of Rochester (N.Y.) Medical Center, underscored the need to consider approval versus access to gene therapies and how to optimize access to the novel treatments.
“We need to consider what the cost is, how it’s going to be accessed, and whether there is a sustainable model,” said Ciafaloni, who was not associated with the study. “There will need to be institutional readiness and support for specialized multidisciplinary clinics for gene therapy.”
She also noted “we need to consider how we can do better on a broader level, because this is not a provider problem or a manufacturer problem — it’s a society problem.”
The study was funded by Sarepta Therapeutics. McDonald reported consulting work for Sarepta Therapeutics and has been an investigator in SRP-9001 research. Ciafaloni reported serving on advisory boards or other relationships with Alexion, Argenx, Biogen, Amicus, Momenta, Medscape, Pfizer, Sanofi/Genzyme, Sarepta, Jansen, NS Pharma, CureSMA, Orphazyme, the Patient-Centered Outcomes Research Institute, PPMD, PTC Therapeutics, and Santhera.
A version of this article first appeared on Medscape.com.
PHOENIX – compared with untreated patients who showed significant decline over the same time period, new research shows.
“Functional assessments demonstrated long-term sustained stabilization of motor function that was clinically meaningful, at ages where functional decline would be expected based on natural history,” the investigators noted in their abstract. Furthermore, the treatment, known as delandistrogene moxeparvovec-rokl (SRP-9001), was well tolerated 4 years post treatment.
The study was presented at the annual meeting of the American Association of Neuromuscular Electrodiagnostic Medicine.
Severe type of DMD
Considered one of the most severe forms of muscular dystrophy, DMD causes progressive muscle wasting stemming from the root genetic cause of missing dystrophin in muscle cells. Often referred to as a molecular “shock absorber,” dystrophin stabilizes the sarcolemma during muscle contractions to prevent degeneration.
SRP-9001, a single-dose recombinant gene therapy administered as an intravenous infusion, was designed to deliver a trimmed down form of dystrophin to compensate for the deficit.
In July, the adeno-associated virus vector (AAV)–based SRP-9001 gene therapy was granted accelerated approval by the Food and Drug Administration for the treatment of ambulatory pediatric patients aged 4-5 years with DMD with a confirmed mutation in the DMD gene.
The therapy is administered over 1-2 hours at a dose of 133 trillion vector genomes per kilogram of body weight.
For Study 101, one of several evaluating the novel therapy, a research team led by senior investigator Jerry Mendell, MD, an attending neurologist at Nationwide Children’s Hospital and professor of pediatrics and neurology at Ohio State University, both in Columbus,evaluated data on four ambulatory male patients aged 4-8 years who received a single IV infusion of the therapy.
All patients also received prednisone 1 mg/kg, 1 day preinfusion and 30 days post infusion.
At 4 years post treatment, there were no new safety events. All treatment-related adverse events occurred mainly within the first 70 days, and all resolved.
The most commonly reported adverse reactions of the gene therapy include vomiting, nausea, increases in liver enzymes, pyrexia (fever), and thrombocytopenia, all of which occurred within 90 days of infusion and been manageable.
Risk mitigation strategies for hepatotoxicity or acute liver injury include pre- and postinfusion monitoring of liver enzymes, the authors noted.
No serious abnormalities were observed in hematologic or chemistry panels, and while three patients had elevated gamma-glutamyl transpeptidase in the first 3 months post treatment, those cases resolved with oral steroid treatment.
Significant improvements in function were observed, with a mean improvement in North Star Ambulatory Assessment (NSAA) scores from baseline of 7.0 points (range, 4-11).
Exploratory analyses further showed that, compared with a propensity score–weighted external control cohort of 21 patients with DMD who did not receive the therapy, those receiving SRP-9001 had a statistically significant difference of 9.4 points in least-squares mean change from baseline to 4 years on the NSAA score (P = .0125).
Similar trends were observed in improvement from baseline in key measures of time to rise, 4-stair climb, and 10- and 100-meter walk/run function tests.
Other reported adverse events include acute serious liver injury, immune-mediated myositis, and myocarditis. Because of the latter risk, the therapy is contraindicated in patients with any deletion in exon 8 and/or exon 9 in the DMD gene.
The current 4-year update on SRP-9001 adds to clinical trial results that have been reported on more than 80 patients treated to date, with favorable results and consistent safety profiles reported at other time points.
Continued FDA approval for the therapy will be contingent upon verification of a clinical benefit in the confirmatory trials, including the EMBARK trial.
Increased function, long-term stability
Discussing the research at the meeting, Craig McDonald, MD, professor and chair of physical medicine & rehabilitation, a professor of pediatrics and study chair of the CINRG Duchenne Natural History Study at University of California Davis Health, noted that top-line results from the ongoing, confirmatory phase 3 EMBARK trial show functional benefits of SRP-9001 not only in 4- to 5-year-olds but also in other older age groups.
“What’s really striking, and in my mind the most impressive, is that when you follow these patients out 3 or 4 years ... you see there is this bump in function followed by long-term stability, whereas the external control cohort predictably shows really quite significant declines in their [NSAA] functional values,” he said in his presentation.
“When you look at each individually treated patient versus their own predicted trajectory using their baseline values on the time function test, each of the patients actually has a really quite impressive stabilization of function over their predicted disease trajectory,” he added.
A caveat that SRP-9001 shares with other gene therapies is the issue of cost – reported in the range of $2 million–$3 million.
In the context of racial and socioeconomic disparities in access to diagnosis and care reported in DMD, Emma Ciafaloni, MD, a professor of neurology and pediatrics at the University of Rochester (N.Y.) Medical Center, underscored the need to consider approval versus access to gene therapies and how to optimize access to the novel treatments.
“We need to consider what the cost is, how it’s going to be accessed, and whether there is a sustainable model,” said Ciafaloni, who was not associated with the study. “There will need to be institutional readiness and support for specialized multidisciplinary clinics for gene therapy.”
She also noted “we need to consider how we can do better on a broader level, because this is not a provider problem or a manufacturer problem — it’s a society problem.”
The study was funded by Sarepta Therapeutics. McDonald reported consulting work for Sarepta Therapeutics and has been an investigator in SRP-9001 research. Ciafaloni reported serving on advisory boards or other relationships with Alexion, Argenx, Biogen, Amicus, Momenta, Medscape, Pfizer, Sanofi/Genzyme, Sarepta, Jansen, NS Pharma, CureSMA, Orphazyme, the Patient-Centered Outcomes Research Institute, PPMD, PTC Therapeutics, and Santhera.
A version of this article first appeared on Medscape.com.
AT AANEM 2023
Five hours or less of sleep per night tied to subsequent depression
TOPLINE:
, new research shows.
METHODOLOGY:
- The analysis included participants in the English Longitudinal Study of Ageing (ELSA), a prospective cohort study of a representative U.K. sample (mean age, 65 years) that is assessed biennially.
- Researchers collected data on sleep duration and depression through nursing home visits and computer-assisted personal interviews and used combined ELSA waves from 2004 to 2008, when collection of genetic data began.
- Using genome-wide association studies from the U.K. Biobank, the authors constructed polygenic scores (PGSs) to predict an individual’s genetic risk over an average of 8 years for a disease or outcome, overall sleep duration, short sleep (≤ 5 hours nightly), long sleep (≥ 9 hours of sleep nightly), and depression.
- The analysis included two analytic samples; one involved 6,521 persons to determine the role of baseline sleep on depression (assessed using the Center for Epidemiologic Studies Depression Scale) at follow-up, and the other involved 6,070 persons to determine the role of baseline depression on suboptimal sleep at follow-up.
TAKEAWAY:
- After adjustments, including for age and sex, a 1–standard deviation increase in PGS for short sleep was associated with an increase of 14% in odds of developing depression during the follow-up period (odds ratio, 1.14; P = .008).
- There was no significant association of the PGS for sleep duration (P = .053) or long sleep (P = .544) with the onset of depression.
- There were no significant associations between PGS for depression and future overall sleep duration, short sleep, and long sleep by the end of the follow-up, suggesting that different mechanisms underlie the relationship between depression and subsequent onset of suboptimal sleep in older adults.
- Several sensitivity analyses – including additional adjustment for socioeconomic, environmental, and behavioral factors – upheld the findings of the main analysis, highlighting the robustness of the results.
IN PRACTICE:
The study showed that common genetic markers for short sleep play an important role in the incidence of depression in older adults, the authors note, adding that the new findings “support a growing view that short-sleep is more salient to the experience of depression than long sleep” across the lifespan.
SOURCE:
The study was led by Odessa S. Hamilton, department of behavioral science and health, University College London. It was published online in Translational Psychiatry.
LIMITATIONS:
There are probably intraindividual differences in sleep duration that were not assessed in the study. The depression scale used may be indicative of subclinical depression and not major depressive disorder. The phenotypic sensitivity analyses did not account for comorbidities or medications that can affect sleep duration and depression.
DISCLOSURES:
ELSA is funded by the National Institute on Aging and by a consortium of U.K. government departments coordinated by the National Institute for Health and Care Research. The authors report no relevant conflicts of interests.
A version of this article first appeared on Medscape.com.
TOPLINE:
, new research shows.
METHODOLOGY:
- The analysis included participants in the English Longitudinal Study of Ageing (ELSA), a prospective cohort study of a representative U.K. sample (mean age, 65 years) that is assessed biennially.
- Researchers collected data on sleep duration and depression through nursing home visits and computer-assisted personal interviews and used combined ELSA waves from 2004 to 2008, when collection of genetic data began.
- Using genome-wide association studies from the U.K. Biobank, the authors constructed polygenic scores (PGSs) to predict an individual’s genetic risk over an average of 8 years for a disease or outcome, overall sleep duration, short sleep (≤ 5 hours nightly), long sleep (≥ 9 hours of sleep nightly), and depression.
- The analysis included two analytic samples; one involved 6,521 persons to determine the role of baseline sleep on depression (assessed using the Center for Epidemiologic Studies Depression Scale) at follow-up, and the other involved 6,070 persons to determine the role of baseline depression on suboptimal sleep at follow-up.
TAKEAWAY:
- After adjustments, including for age and sex, a 1–standard deviation increase in PGS for short sleep was associated with an increase of 14% in odds of developing depression during the follow-up period (odds ratio, 1.14; P = .008).
- There was no significant association of the PGS for sleep duration (P = .053) or long sleep (P = .544) with the onset of depression.
- There were no significant associations between PGS for depression and future overall sleep duration, short sleep, and long sleep by the end of the follow-up, suggesting that different mechanisms underlie the relationship between depression and subsequent onset of suboptimal sleep in older adults.
- Several sensitivity analyses – including additional adjustment for socioeconomic, environmental, and behavioral factors – upheld the findings of the main analysis, highlighting the robustness of the results.
IN PRACTICE:
The study showed that common genetic markers for short sleep play an important role in the incidence of depression in older adults, the authors note, adding that the new findings “support a growing view that short-sleep is more salient to the experience of depression than long sleep” across the lifespan.
SOURCE:
The study was led by Odessa S. Hamilton, department of behavioral science and health, University College London. It was published online in Translational Psychiatry.
LIMITATIONS:
There are probably intraindividual differences in sleep duration that were not assessed in the study. The depression scale used may be indicative of subclinical depression and not major depressive disorder. The phenotypic sensitivity analyses did not account for comorbidities or medications that can affect sleep duration and depression.
DISCLOSURES:
ELSA is funded by the National Institute on Aging and by a consortium of U.K. government departments coordinated by the National Institute for Health and Care Research. The authors report no relevant conflicts of interests.
A version of this article first appeared on Medscape.com.
TOPLINE:
, new research shows.
METHODOLOGY:
- The analysis included participants in the English Longitudinal Study of Ageing (ELSA), a prospective cohort study of a representative U.K. sample (mean age, 65 years) that is assessed biennially.
- Researchers collected data on sleep duration and depression through nursing home visits and computer-assisted personal interviews and used combined ELSA waves from 2004 to 2008, when collection of genetic data began.
- Using genome-wide association studies from the U.K. Biobank, the authors constructed polygenic scores (PGSs) to predict an individual’s genetic risk over an average of 8 years for a disease or outcome, overall sleep duration, short sleep (≤ 5 hours nightly), long sleep (≥ 9 hours of sleep nightly), and depression.
- The analysis included two analytic samples; one involved 6,521 persons to determine the role of baseline sleep on depression (assessed using the Center for Epidemiologic Studies Depression Scale) at follow-up, and the other involved 6,070 persons to determine the role of baseline depression on suboptimal sleep at follow-up.
TAKEAWAY:
- After adjustments, including for age and sex, a 1–standard deviation increase in PGS for short sleep was associated with an increase of 14% in odds of developing depression during the follow-up period (odds ratio, 1.14; P = .008).
- There was no significant association of the PGS for sleep duration (P = .053) or long sleep (P = .544) with the onset of depression.
- There were no significant associations between PGS for depression and future overall sleep duration, short sleep, and long sleep by the end of the follow-up, suggesting that different mechanisms underlie the relationship between depression and subsequent onset of suboptimal sleep in older adults.
- Several sensitivity analyses – including additional adjustment for socioeconomic, environmental, and behavioral factors – upheld the findings of the main analysis, highlighting the robustness of the results.
IN PRACTICE:
The study showed that common genetic markers for short sleep play an important role in the incidence of depression in older adults, the authors note, adding that the new findings “support a growing view that short-sleep is more salient to the experience of depression than long sleep” across the lifespan.
SOURCE:
The study was led by Odessa S. Hamilton, department of behavioral science and health, University College London. It was published online in Translational Psychiatry.
LIMITATIONS:
There are probably intraindividual differences in sleep duration that were not assessed in the study. The depression scale used may be indicative of subclinical depression and not major depressive disorder. The phenotypic sensitivity analyses did not account for comorbidities or medications that can affect sleep duration and depression.
DISCLOSURES:
ELSA is funded by the National Institute on Aging and by a consortium of U.K. government departments coordinated by the National Institute for Health and Care Research. The authors report no relevant conflicts of interests.
A version of this article first appeared on Medscape.com.
FROM TRANSLATIONAL PSYCHIATRY
Psychological safety in cardiology training
Training in medicine has long been thought of as a tough process, but the issue of creating a psychologically safe environment for young doctors is now being highlighted as an important way of providing an improved learning environment, which will ultimately lead to better patient care. And cardiology is one field that needs to work harder on this.
“We all remember attendings who made our training experience memorable, who made us excited to come to work and learn, and who inspired us to become better,” Vivek Kulkarni, MD, wrote in a recent commentary. “Unfortunately, we also all remember the learning environments where we were terrified, where thriving took a backseat to surviving, and where learning was an afterthought.”
Writing in an article in the Journal of the American College of Cardiology, Dr. Kulkarni asked the question: “Why are some learning environments better than others, and what can we do to improve the learning environment for our trainees?”
Dr. Kulkarni, director of the training program for cardiology fellows at Cooper University Hospital, Camden, New Jersey, said “There may be some people in some institutions that do pay attention to this but as wider field we could do better.”
Dr. Kulkarni explained that psychological safety is the comfort to engage with others genuinely, with honesty and without fear.
It has been defined as a “willingness to take interpersonal risks at work, whether to admit error, ask a question, seek help, or simply say ‘I don’t know,’ ” or as “the perception that a working environment is safe for team members to express a concern, ask a question, or acknowledge a mistake without fear of humiliation, retaliation, blame, or being ignored.”
“In the medical environment we usually work in teams: older doctors, younger doctors, nurses, other staff,” Dr. Kulkarni said in an interview. “A psychologically safe environment would be one where a trainee feels comfortable so that they can ask a question about something that they don’t understand. That comfort comes from the idea that it is okay to get something wrong or to not know something and to ask for help.
“The flip side of that is an environment in which people are so afraid to make a mistake out of fear of retribution or punishment that they don’t take risks, or they don’t openly acknowledge when they might need help with something,” he said. “That would be a psychologically unsafe environment.”
What exactly this looks like varies in different environments and culture of the group, he noted, “but in general, you can tell if you are part of a psychologically safe environment because you are excited to come to work and feel comfortable at work.”
Dr. Kulkarni added that a growing body of literature now shows that psychological safety is critical for optimal learning but that cardiovascular fellowship training poses unique barriers to psychological safety.
‘Arrogant, unkind, and unwelcoming’
First, he said that the “high-stakes” nature of cardiology, in which decisions often must be made quickly and can have life-or-death consequences, can create fear about making mistakes and that some trainees may be so afraid that they cannot speak up and ask for help when struggling or cannot incorporate feedback in real time.
Second, in medicine at large, there is a stereotype that cardiologists can be “arrogant, unkind, and unwelcoming,” which may discourage new fellows from honest interaction.
Third, cardiology involves many different technical skills that fellows have little to no previous experience with; this may contribute to a perceived sense of being judged when making mistakes or asking for help.
Finally, demographics may be a factor, with only one in eight cardiologists in the United States being women and only 7.5% of cardiologists being from traditionally underrepresented racial and ethnic minority groups, which Dr. Kulkarni said may lead to a lack of psychological safety because of “bias, microaggressions, or even just a lack of mentors of similar backgrounds.”
But he believes that the cardiology training culture is improving.
“I think it is getting better. Even the fact that I can publish this article is a positive sign. I think there’s an audience for this type of thing now.”
He believes that part of the reason for this is the availability of research and evidence showing there are better ways to teach than the old traditional approaches.
He noted that some teaching physicians receive training on how to teach and some don’t, and this is an area that could be improved.
“I think the knowledge of how to produce psychologically safe environments is already there,” he said. “It just has to be standardized and publicized. That would make the learning environment better.”
“Nothing about this is groundbreaking,” he added. “We all know psychologically unsafe environments exist. The novelty is just that it is now starting to be discussed. It’s one of those things that we can likely improve the ways our trainees learn and the kind of doctors we produce just by thinking a little bit more carefully about the way we interact with each other.”
Dr. Kulkarni said trainees often drop out because they have had a negative experience of feeling psychologically unsafe. “They may drop out of medicine all together or they may choose to pursue a career in a different part of medicine, where they perceive a more psychologically safe environment.”
He also suggested that this issue can affect patient care.
“If the medical team does not provide a psychologically safe environment for trainees, it is very likely that that team is not operating as effectively as it could, and it is very likely that patients being taken care of by that team may have missed opportunities for better care,” he concluded. Examples could include trainees recognizing errors and bringing things that might not be right to the attention of their superiors. “That is something that requires some degree of psychological safety.”
Action for improvement
Dr. Kulkarni suggested several strategies to promote psychological safety in cardiology training.
As a first step, institutions should investigate the culture of learning within their fellowship programs and gather feedback from anonymous surveys of fellows. They can then implement policies to address gaps.
He noted that, at Cooper University Hospital, standardized documents have been created that explicitly outline policies for attendings on teaching services, which establish expectations for all team members, encourage fellows to ask for help, set guidelines for feedback conversations with fellows, and delineate situations when calling the attending is expected.
Dr. Kulkarni also suggested that cardiologists involved in teaching fellows can try several strategies to promote psychological safety. These include setting clear expectations on their tasks and graded autonomy, inviting participation in decisions, acknowledging that gaps in knowledge are not a personal failure but rather a normal part of the growth process, encouraging fellows to seek help when they need it, fostering collegial relationships with fellows, acknowledging your own uncertainty in difficult situations, checking in about emotions after challenging situations, and seeking feedback on your own performance.
He added that changes on a larger scale are also needed, such as training for cardiology program directors including more on this issue as well as developing best practices.
“If we as a community could come together and agree on the things needed to create a psychologically safe environment for training, that would be a big improvement.”
Addressing the challenges of different generations
In a response to Dr. Kulkarni’s article, Margo Vassar, MD, The Queen’s Medical Center, Honolulu, and Sandra Lewis, MD, Legacy Health System, Portland, Ore., make the case that to succeed in providing psychological safety, the cardiovascular community also needs to address intergenerational cultural challenges.
“Twenty years ago, to have raised the idea of psychological safety in any phase of training would likely have been met with intergenerational pushback and complete disregard,” they say, adding that: “Asking senior Baby Boomer cardiologists to develop skills to implement psychological safety, with just a list of action items, to suddenly create safe environments, belies the challenges inherent in intergenerational understanding and collaboration.”
In an interview, Dr. Lewis elaborated: “Many cardiology training program directors are Baby Boomers, but there is a whole new group of younger people moving in, and the way they deal with things and communicate is quite different.”
Dr. Lewis gave an example of when she was in training the attending was the “be all and end all,” and it was not expected that fellows would ask questions. “I think there is more communication now and a willingness to take risks and ask questions.”
But she said because everyone is so busy now, building relationships within a team can be difficult.
“We don’t have the doctors’ lounge anymore. We don’t sit and have lunch together. Computers are taking over now, no one actually talks to each other anymore,” she said. “We need to try to get to know each other and become colleagues. It’s easy when you don’t know somebody to be abrupt or brusque; it’s harder when you’re friends.”
She noted that the Mayo Clinic is one institution that is doing a lot of work on this, arranging for groups of doctors to go out for dinner together to get to know each other.
“This bringing people together socially happens in a lot of workplaces, and it can happen in medicine.”
Dr. Lewis, who has some leadership positions at the American College of Cardiology, said the organization is focusing on “intergenerational opportunities and challenges” to help improve psychological safety for trainees.
Noting that a recent survey of medical residents found that “contemporary residents were more likely than their predecessors to agree with negative perceptions of cardiology,” Lewis said the ACC is also reaching out to medical residents who may think that cardiology is an unwelcoming environment to enter and to minority groups of medical residents such as women and ethnic minorities to try and attract them to become cardiology fellows.
“If fellows find in hard to speak up because they are in this hierarchical learning situation, that can be even more difficult if you feel you’re in a minority group. ... We need to create a culture of colleagues rather than perpetuating a culture of us and them, to provide a safe and thriving cardiovascular community,” she added.
A version of this article first appeared on Medscape.com.
Training in medicine has long been thought of as a tough process, but the issue of creating a psychologically safe environment for young doctors is now being highlighted as an important way of providing an improved learning environment, which will ultimately lead to better patient care. And cardiology is one field that needs to work harder on this.
“We all remember attendings who made our training experience memorable, who made us excited to come to work and learn, and who inspired us to become better,” Vivek Kulkarni, MD, wrote in a recent commentary. “Unfortunately, we also all remember the learning environments where we were terrified, where thriving took a backseat to surviving, and where learning was an afterthought.”
Writing in an article in the Journal of the American College of Cardiology, Dr. Kulkarni asked the question: “Why are some learning environments better than others, and what can we do to improve the learning environment for our trainees?”
Dr. Kulkarni, director of the training program for cardiology fellows at Cooper University Hospital, Camden, New Jersey, said “There may be some people in some institutions that do pay attention to this but as wider field we could do better.”
Dr. Kulkarni explained that psychological safety is the comfort to engage with others genuinely, with honesty and without fear.
It has been defined as a “willingness to take interpersonal risks at work, whether to admit error, ask a question, seek help, or simply say ‘I don’t know,’ ” or as “the perception that a working environment is safe for team members to express a concern, ask a question, or acknowledge a mistake without fear of humiliation, retaliation, blame, or being ignored.”
“In the medical environment we usually work in teams: older doctors, younger doctors, nurses, other staff,” Dr. Kulkarni said in an interview. “A psychologically safe environment would be one where a trainee feels comfortable so that they can ask a question about something that they don’t understand. That comfort comes from the idea that it is okay to get something wrong or to not know something and to ask for help.
“The flip side of that is an environment in which people are so afraid to make a mistake out of fear of retribution or punishment that they don’t take risks, or they don’t openly acknowledge when they might need help with something,” he said. “That would be a psychologically unsafe environment.”
What exactly this looks like varies in different environments and culture of the group, he noted, “but in general, you can tell if you are part of a psychologically safe environment because you are excited to come to work and feel comfortable at work.”
Dr. Kulkarni added that a growing body of literature now shows that psychological safety is critical for optimal learning but that cardiovascular fellowship training poses unique barriers to psychological safety.
‘Arrogant, unkind, and unwelcoming’
First, he said that the “high-stakes” nature of cardiology, in which decisions often must be made quickly and can have life-or-death consequences, can create fear about making mistakes and that some trainees may be so afraid that they cannot speak up and ask for help when struggling or cannot incorporate feedback in real time.
Second, in medicine at large, there is a stereotype that cardiologists can be “arrogant, unkind, and unwelcoming,” which may discourage new fellows from honest interaction.
Third, cardiology involves many different technical skills that fellows have little to no previous experience with; this may contribute to a perceived sense of being judged when making mistakes or asking for help.
Finally, demographics may be a factor, with only one in eight cardiologists in the United States being women and only 7.5% of cardiologists being from traditionally underrepresented racial and ethnic minority groups, which Dr. Kulkarni said may lead to a lack of psychological safety because of “bias, microaggressions, or even just a lack of mentors of similar backgrounds.”
But he believes that the cardiology training culture is improving.
“I think it is getting better. Even the fact that I can publish this article is a positive sign. I think there’s an audience for this type of thing now.”
He believes that part of the reason for this is the availability of research and evidence showing there are better ways to teach than the old traditional approaches.
He noted that some teaching physicians receive training on how to teach and some don’t, and this is an area that could be improved.
“I think the knowledge of how to produce psychologically safe environments is already there,” he said. “It just has to be standardized and publicized. That would make the learning environment better.”
“Nothing about this is groundbreaking,” he added. “We all know psychologically unsafe environments exist. The novelty is just that it is now starting to be discussed. It’s one of those things that we can likely improve the ways our trainees learn and the kind of doctors we produce just by thinking a little bit more carefully about the way we interact with each other.”
Dr. Kulkarni said trainees often drop out because they have had a negative experience of feeling psychologically unsafe. “They may drop out of medicine all together or they may choose to pursue a career in a different part of medicine, where they perceive a more psychologically safe environment.”
He also suggested that this issue can affect patient care.
“If the medical team does not provide a psychologically safe environment for trainees, it is very likely that that team is not operating as effectively as it could, and it is very likely that patients being taken care of by that team may have missed opportunities for better care,” he concluded. Examples could include trainees recognizing errors and bringing things that might not be right to the attention of their superiors. “That is something that requires some degree of psychological safety.”
Action for improvement
Dr. Kulkarni suggested several strategies to promote psychological safety in cardiology training.
As a first step, institutions should investigate the culture of learning within their fellowship programs and gather feedback from anonymous surveys of fellows. They can then implement policies to address gaps.
He noted that, at Cooper University Hospital, standardized documents have been created that explicitly outline policies for attendings on teaching services, which establish expectations for all team members, encourage fellows to ask for help, set guidelines for feedback conversations with fellows, and delineate situations when calling the attending is expected.
Dr. Kulkarni also suggested that cardiologists involved in teaching fellows can try several strategies to promote psychological safety. These include setting clear expectations on their tasks and graded autonomy, inviting participation in decisions, acknowledging that gaps in knowledge are not a personal failure but rather a normal part of the growth process, encouraging fellows to seek help when they need it, fostering collegial relationships with fellows, acknowledging your own uncertainty in difficult situations, checking in about emotions after challenging situations, and seeking feedback on your own performance.
He added that changes on a larger scale are also needed, such as training for cardiology program directors including more on this issue as well as developing best practices.
“If we as a community could come together and agree on the things needed to create a psychologically safe environment for training, that would be a big improvement.”
Addressing the challenges of different generations
In a response to Dr. Kulkarni’s article, Margo Vassar, MD, The Queen’s Medical Center, Honolulu, and Sandra Lewis, MD, Legacy Health System, Portland, Ore., make the case that to succeed in providing psychological safety, the cardiovascular community also needs to address intergenerational cultural challenges.
“Twenty years ago, to have raised the idea of psychological safety in any phase of training would likely have been met with intergenerational pushback and complete disregard,” they say, adding that: “Asking senior Baby Boomer cardiologists to develop skills to implement psychological safety, with just a list of action items, to suddenly create safe environments, belies the challenges inherent in intergenerational understanding and collaboration.”
In an interview, Dr. Lewis elaborated: “Many cardiology training program directors are Baby Boomers, but there is a whole new group of younger people moving in, and the way they deal with things and communicate is quite different.”
Dr. Lewis gave an example of when she was in training the attending was the “be all and end all,” and it was not expected that fellows would ask questions. “I think there is more communication now and a willingness to take risks and ask questions.”
But she said because everyone is so busy now, building relationships within a team can be difficult.
“We don’t have the doctors’ lounge anymore. We don’t sit and have lunch together. Computers are taking over now, no one actually talks to each other anymore,” she said. “We need to try to get to know each other and become colleagues. It’s easy when you don’t know somebody to be abrupt or brusque; it’s harder when you’re friends.”
She noted that the Mayo Clinic is one institution that is doing a lot of work on this, arranging for groups of doctors to go out for dinner together to get to know each other.
“This bringing people together socially happens in a lot of workplaces, and it can happen in medicine.”
Dr. Lewis, who has some leadership positions at the American College of Cardiology, said the organization is focusing on “intergenerational opportunities and challenges” to help improve psychological safety for trainees.
Noting that a recent survey of medical residents found that “contemporary residents were more likely than their predecessors to agree with negative perceptions of cardiology,” Lewis said the ACC is also reaching out to medical residents who may think that cardiology is an unwelcoming environment to enter and to minority groups of medical residents such as women and ethnic minorities to try and attract them to become cardiology fellows.
“If fellows find in hard to speak up because they are in this hierarchical learning situation, that can be even more difficult if you feel you’re in a minority group. ... We need to create a culture of colleagues rather than perpetuating a culture of us and them, to provide a safe and thriving cardiovascular community,” she added.
A version of this article first appeared on Medscape.com.
Training in medicine has long been thought of as a tough process, but the issue of creating a psychologically safe environment for young doctors is now being highlighted as an important way of providing an improved learning environment, which will ultimately lead to better patient care. And cardiology is one field that needs to work harder on this.
“We all remember attendings who made our training experience memorable, who made us excited to come to work and learn, and who inspired us to become better,” Vivek Kulkarni, MD, wrote in a recent commentary. “Unfortunately, we also all remember the learning environments where we were terrified, where thriving took a backseat to surviving, and where learning was an afterthought.”
Writing in an article in the Journal of the American College of Cardiology, Dr. Kulkarni asked the question: “Why are some learning environments better than others, and what can we do to improve the learning environment for our trainees?”
Dr. Kulkarni, director of the training program for cardiology fellows at Cooper University Hospital, Camden, New Jersey, said “There may be some people in some institutions that do pay attention to this but as wider field we could do better.”
Dr. Kulkarni explained that psychological safety is the comfort to engage with others genuinely, with honesty and without fear.
It has been defined as a “willingness to take interpersonal risks at work, whether to admit error, ask a question, seek help, or simply say ‘I don’t know,’ ” or as “the perception that a working environment is safe for team members to express a concern, ask a question, or acknowledge a mistake without fear of humiliation, retaliation, blame, or being ignored.”
“In the medical environment we usually work in teams: older doctors, younger doctors, nurses, other staff,” Dr. Kulkarni said in an interview. “A psychologically safe environment would be one where a trainee feels comfortable so that they can ask a question about something that they don’t understand. That comfort comes from the idea that it is okay to get something wrong or to not know something and to ask for help.
“The flip side of that is an environment in which people are so afraid to make a mistake out of fear of retribution or punishment that they don’t take risks, or they don’t openly acknowledge when they might need help with something,” he said. “That would be a psychologically unsafe environment.”
What exactly this looks like varies in different environments and culture of the group, he noted, “but in general, you can tell if you are part of a psychologically safe environment because you are excited to come to work and feel comfortable at work.”
Dr. Kulkarni added that a growing body of literature now shows that psychological safety is critical for optimal learning but that cardiovascular fellowship training poses unique barriers to psychological safety.
‘Arrogant, unkind, and unwelcoming’
First, he said that the “high-stakes” nature of cardiology, in which decisions often must be made quickly and can have life-or-death consequences, can create fear about making mistakes and that some trainees may be so afraid that they cannot speak up and ask for help when struggling or cannot incorporate feedback in real time.
Second, in medicine at large, there is a stereotype that cardiologists can be “arrogant, unkind, and unwelcoming,” which may discourage new fellows from honest interaction.
Third, cardiology involves many different technical skills that fellows have little to no previous experience with; this may contribute to a perceived sense of being judged when making mistakes or asking for help.
Finally, demographics may be a factor, with only one in eight cardiologists in the United States being women and only 7.5% of cardiologists being from traditionally underrepresented racial and ethnic minority groups, which Dr. Kulkarni said may lead to a lack of psychological safety because of “bias, microaggressions, or even just a lack of mentors of similar backgrounds.”
But he believes that the cardiology training culture is improving.
“I think it is getting better. Even the fact that I can publish this article is a positive sign. I think there’s an audience for this type of thing now.”
He believes that part of the reason for this is the availability of research and evidence showing there are better ways to teach than the old traditional approaches.
He noted that some teaching physicians receive training on how to teach and some don’t, and this is an area that could be improved.
“I think the knowledge of how to produce psychologically safe environments is already there,” he said. “It just has to be standardized and publicized. That would make the learning environment better.”
“Nothing about this is groundbreaking,” he added. “We all know psychologically unsafe environments exist. The novelty is just that it is now starting to be discussed. It’s one of those things that we can likely improve the ways our trainees learn and the kind of doctors we produce just by thinking a little bit more carefully about the way we interact with each other.”
Dr. Kulkarni said trainees often drop out because they have had a negative experience of feeling psychologically unsafe. “They may drop out of medicine all together or they may choose to pursue a career in a different part of medicine, where they perceive a more psychologically safe environment.”
He also suggested that this issue can affect patient care.
“If the medical team does not provide a psychologically safe environment for trainees, it is very likely that that team is not operating as effectively as it could, and it is very likely that patients being taken care of by that team may have missed opportunities for better care,” he concluded. Examples could include trainees recognizing errors and bringing things that might not be right to the attention of their superiors. “That is something that requires some degree of psychological safety.”
Action for improvement
Dr. Kulkarni suggested several strategies to promote psychological safety in cardiology training.
As a first step, institutions should investigate the culture of learning within their fellowship programs and gather feedback from anonymous surveys of fellows. They can then implement policies to address gaps.
He noted that, at Cooper University Hospital, standardized documents have been created that explicitly outline policies for attendings on teaching services, which establish expectations for all team members, encourage fellows to ask for help, set guidelines for feedback conversations with fellows, and delineate situations when calling the attending is expected.
Dr. Kulkarni also suggested that cardiologists involved in teaching fellows can try several strategies to promote psychological safety. These include setting clear expectations on their tasks and graded autonomy, inviting participation in decisions, acknowledging that gaps in knowledge are not a personal failure but rather a normal part of the growth process, encouraging fellows to seek help when they need it, fostering collegial relationships with fellows, acknowledging your own uncertainty in difficult situations, checking in about emotions after challenging situations, and seeking feedback on your own performance.
He added that changes on a larger scale are also needed, such as training for cardiology program directors including more on this issue as well as developing best practices.
“If we as a community could come together and agree on the things needed to create a psychologically safe environment for training, that would be a big improvement.”
Addressing the challenges of different generations
In a response to Dr. Kulkarni’s article, Margo Vassar, MD, The Queen’s Medical Center, Honolulu, and Sandra Lewis, MD, Legacy Health System, Portland, Ore., make the case that to succeed in providing psychological safety, the cardiovascular community also needs to address intergenerational cultural challenges.
“Twenty years ago, to have raised the idea of psychological safety in any phase of training would likely have been met with intergenerational pushback and complete disregard,” they say, adding that: “Asking senior Baby Boomer cardiologists to develop skills to implement psychological safety, with just a list of action items, to suddenly create safe environments, belies the challenges inherent in intergenerational understanding and collaboration.”
In an interview, Dr. Lewis elaborated: “Many cardiology training program directors are Baby Boomers, but there is a whole new group of younger people moving in, and the way they deal with things and communicate is quite different.”
Dr. Lewis gave an example of when she was in training the attending was the “be all and end all,” and it was not expected that fellows would ask questions. “I think there is more communication now and a willingness to take risks and ask questions.”
But she said because everyone is so busy now, building relationships within a team can be difficult.
“We don’t have the doctors’ lounge anymore. We don’t sit and have lunch together. Computers are taking over now, no one actually talks to each other anymore,” she said. “We need to try to get to know each other and become colleagues. It’s easy when you don’t know somebody to be abrupt or brusque; it’s harder when you’re friends.”
She noted that the Mayo Clinic is one institution that is doing a lot of work on this, arranging for groups of doctors to go out for dinner together to get to know each other.
“This bringing people together socially happens in a lot of workplaces, and it can happen in medicine.”
Dr. Lewis, who has some leadership positions at the American College of Cardiology, said the organization is focusing on “intergenerational opportunities and challenges” to help improve psychological safety for trainees.
Noting that a recent survey of medical residents found that “contemporary residents were more likely than their predecessors to agree with negative perceptions of cardiology,” Lewis said the ACC is also reaching out to medical residents who may think that cardiology is an unwelcoming environment to enter and to minority groups of medical residents such as women and ethnic minorities to try and attract them to become cardiology fellows.
“If fellows find in hard to speak up because they are in this hierarchical learning situation, that can be even more difficult if you feel you’re in a minority group. ... We need to create a culture of colleagues rather than perpetuating a culture of us and them, to provide a safe and thriving cardiovascular community,” she added.
A version of this article first appeared on Medscape.com.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
Black psychiatric inpatients more likely to be restrained and for longer
TOPLINE:
, new research suggests.
METHODOLOGY:
- The study, part of a larger retrospective chart review of inpatient psychiatric electronic medical records (EMRs), included 29,739 adolescents (aged 12-17 years) and adults admitted because of severe and disruptive psychiatric illness or concerns about self-harm.
- A restraint event was defined as a physician-ordered physical or mechanical hold in which patients are unable to move their limbs, body, or head or are given medication to restrict their movement.
- Researchers used scores on the Dynamic Appraisal of Situational Aggression (DASA) at admission to assess risk for aggression among high-risk psychiatric inpatients (scores ranged from a low of 0 to a high of 7).
- From restraint event data extracted from the EMR, researchers investigated whether restraint frequency or duration was affected by “adultification,” a form of racial bias in which adolescents are perceived as being older than their actual age and are treated accordingly.
TAKEAWAY:
- Of the entire cohort, 1867 (6.3%) experienced a restraint event, and 27,872 (93.7%) did not.
- Compared with White patients, restraint was 85% more likely among Black patients (adjusted odds ratio, 1.85; P < .001) and 36% more likely among multiracial patients (aOR, 1.36; P = .006), which researchers suggest may reflect systemic racism within psychiatry and medicine, as well as an implicit or learned perception that people of color are more aggressive and dangerous.
- Lower DASA score at admission (P = .001), shorter length of stay (P < .001), adult age (P = .001), female sex (P = .042), and Black race, compared with White race (P = .001), were significantly associated with longer restraint duration, which may serve as a proxy for psychiatric symptom severity.
- Neither age group alone (adolescent or adult) nor the interaction of race and age group was significantly associated with experiencing a restraint event, suggesting no evidence of “adultification.”
IN PRACTICE:
It’s important to raise awareness about racial differences in restraint events in inpatient psychiatric settings, the authors write, adding that addressing overcrowding and investing in bias assessment and restraint education may reduce bias in the care of agitated patients and the use of restraints.
SOURCE:
The study was carried out by Sonali Singal, BS, Institute of Behavioral Science, Feinstein Institutes for Medical Research, Manhasset, N.Y., and colleagues. It was published online in Psychiatric Services.
LIMITATIONS:
The variables analyzed in the study were limited by the retrospective chart review and by the available EMR data, which may have contained entry errors. Although the investigators used DASA scores to control for differences in aggression, they could not control for symptom severity. The study could not examine the impact of race on seclusion (involuntary confinement), a variable often examined in tandem with restraint, because there were too few such events. The analysis also did not control for substance use disorder, which can influence a patient’s behavior and be related to restraint use.
DISCLOSURES:
Ms. Singal reported no relevant financial relationships. The original article has disclosures of other authors.
A version of this article first appeared on Medscape.com.
TOPLINE:
, new research suggests.
METHODOLOGY:
- The study, part of a larger retrospective chart review of inpatient psychiatric electronic medical records (EMRs), included 29,739 adolescents (aged 12-17 years) and adults admitted because of severe and disruptive psychiatric illness or concerns about self-harm.
- A restraint event was defined as a physician-ordered physical or mechanical hold in which patients are unable to move their limbs, body, or head or are given medication to restrict their movement.
- Researchers used scores on the Dynamic Appraisal of Situational Aggression (DASA) at admission to assess risk for aggression among high-risk psychiatric inpatients (scores ranged from a low of 0 to a high of 7).
- From restraint event data extracted from the EMR, researchers investigated whether restraint frequency or duration was affected by “adultification,” a form of racial bias in which adolescents are perceived as being older than their actual age and are treated accordingly.
TAKEAWAY:
- Of the entire cohort, 1867 (6.3%) experienced a restraint event, and 27,872 (93.7%) did not.
- Compared with White patients, restraint was 85% more likely among Black patients (adjusted odds ratio, 1.85; P < .001) and 36% more likely among multiracial patients (aOR, 1.36; P = .006), which researchers suggest may reflect systemic racism within psychiatry and medicine, as well as an implicit or learned perception that people of color are more aggressive and dangerous.
- Lower DASA score at admission (P = .001), shorter length of stay (P < .001), adult age (P = .001), female sex (P = .042), and Black race, compared with White race (P = .001), were significantly associated with longer restraint duration, which may serve as a proxy for psychiatric symptom severity.
- Neither age group alone (adolescent or adult) nor the interaction of race and age group was significantly associated with experiencing a restraint event, suggesting no evidence of “adultification.”
IN PRACTICE:
It’s important to raise awareness about racial differences in restraint events in inpatient psychiatric settings, the authors write, adding that addressing overcrowding and investing in bias assessment and restraint education may reduce bias in the care of agitated patients and the use of restraints.
SOURCE:
The study was carried out by Sonali Singal, BS, Institute of Behavioral Science, Feinstein Institutes for Medical Research, Manhasset, N.Y., and colleagues. It was published online in Psychiatric Services.
LIMITATIONS:
The variables analyzed in the study were limited by the retrospective chart review and by the available EMR data, which may have contained entry errors. Although the investigators used DASA scores to control for differences in aggression, they could not control for symptom severity. The study could not examine the impact of race on seclusion (involuntary confinement), a variable often examined in tandem with restraint, because there were too few such events. The analysis also did not control for substance use disorder, which can influence a patient’s behavior and be related to restraint use.
DISCLOSURES:
Ms. Singal reported no relevant financial relationships. The original article has disclosures of other authors.
A version of this article first appeared on Medscape.com.
TOPLINE:
, new research suggests.
METHODOLOGY:
- The study, part of a larger retrospective chart review of inpatient psychiatric electronic medical records (EMRs), included 29,739 adolescents (aged 12-17 years) and adults admitted because of severe and disruptive psychiatric illness or concerns about self-harm.
- A restraint event was defined as a physician-ordered physical or mechanical hold in which patients are unable to move their limbs, body, or head or are given medication to restrict their movement.
- Researchers used scores on the Dynamic Appraisal of Situational Aggression (DASA) at admission to assess risk for aggression among high-risk psychiatric inpatients (scores ranged from a low of 0 to a high of 7).
- From restraint event data extracted from the EMR, researchers investigated whether restraint frequency or duration was affected by “adultification,” a form of racial bias in which adolescents are perceived as being older than their actual age and are treated accordingly.
TAKEAWAY:
- Of the entire cohort, 1867 (6.3%) experienced a restraint event, and 27,872 (93.7%) did not.
- Compared with White patients, restraint was 85% more likely among Black patients (adjusted odds ratio, 1.85; P < .001) and 36% more likely among multiracial patients (aOR, 1.36; P = .006), which researchers suggest may reflect systemic racism within psychiatry and medicine, as well as an implicit or learned perception that people of color are more aggressive and dangerous.
- Lower DASA score at admission (P = .001), shorter length of stay (P < .001), adult age (P = .001), female sex (P = .042), and Black race, compared with White race (P = .001), were significantly associated with longer restraint duration, which may serve as a proxy for psychiatric symptom severity.
- Neither age group alone (adolescent or adult) nor the interaction of race and age group was significantly associated with experiencing a restraint event, suggesting no evidence of “adultification.”
IN PRACTICE:
It’s important to raise awareness about racial differences in restraint events in inpatient psychiatric settings, the authors write, adding that addressing overcrowding and investing in bias assessment and restraint education may reduce bias in the care of agitated patients and the use of restraints.
SOURCE:
The study was carried out by Sonali Singal, BS, Institute of Behavioral Science, Feinstein Institutes for Medical Research, Manhasset, N.Y., and colleagues. It was published online in Psychiatric Services.
LIMITATIONS:
The variables analyzed in the study were limited by the retrospective chart review and by the available EMR data, which may have contained entry errors. Although the investigators used DASA scores to control for differences in aggression, they could not control for symptom severity. The study could not examine the impact of race on seclusion (involuntary confinement), a variable often examined in tandem with restraint, because there were too few such events. The analysis also did not control for substance use disorder, which can influence a patient’s behavior and be related to restraint use.
DISCLOSURES:
Ms. Singal reported no relevant financial relationships. The original article has disclosures of other authors.
A version of this article first appeared on Medscape.com.
FROM PSYCHIATRIC SERVICES
Aprocitentan reduces resistant hypertension in CKD
PHILADELPHIA – (CKD). The results come from a prespecified subgroup analysis of data collected in the drug’s pivotal trial, PRECISION.
The findings provide support for potentially using aprocitentan, if approved for U.S. marketing in 2024, in patients with blood pressure that remains elevated despite treatment with three established antihypertensive drug classes and with stage 3 CKD with an estimated glomerular filtration rate of 30-59 mL/min per 1.73 m2. This is a key group of patients because “chronic kidney disease is the most common comorbidity in patients with resistant hypertension,” said George Bakris, MD, who presented the subgroup analysis at Kidney Week 2023, organized by the American Society of Nephrology.

The CKD subgroup analysis showed “good evidence for safety and evidence in stage 3 CKD,” a subgroup of 141 patients among the total 730 enrolled in PRECISION, said Dr. Bakris. Professor and director of the Comprehensive Hypertension Center at the University of Chicago, he acknowledged that while the results also showed a signal for safety and efficacy in the 21 enrolled patients with stage 4 hypertension, 15-29 mL/min per 1.73m2, this number of stage 4 patients was too small to allow definitive conclusions.
Nephrologist Nishigandha Pradhan, MD, who cochaired the session with this report, agreed. “Resistant hypertension is a particularly intractable problem in patients with CKD, and the risk is greatest with stage 4 CKD. If studies could show that aprocitentan is safe in people with stage 4 CKD, that would be a big plus, but we need more data,” commented Dr. Pradhan in an interview.
Incremental blood pressure reductions
The parallel-group, phase 3 PRECISION trial investigated the safety and short-term antihypertensive effect of aprocitentan in patients with resistant hypertension. The study’s primary efficacy endpoint was blood pressure reduction from baseline in 730 randomized people with persistent systolic hypertension despite treatment with three established antihypertensive agents including a diuretic. The study ran during June 2018–April 2022 at 191 sites in 22 countries.
The primary outcome after 4 weeks on treatment was a least-square mean reduction in office-measured systolic blood pressure, compared with placebo, of 3.8 mm Hg with a 12.5-mg daily oral dose of aprocitentan and 3.7 mm Hg with a 25-mg daily oral dose. Both significant differences were first reported in 2022. Twenty-four–hour ambulatory systolic blood pressures after 4 weeks of treatment fell by an average of 4.2 mm Hg on the lower dose compared with placebo and by an average of 5.9 mm Hg on the higher daily dose, compared with placebo.
Consistent blood pressure reductions occurred in the CKD subgroups. Among people with stage 3 CKD, daytime ambulatory blood pressure at 4 weeks fell by about 10 mm Hg on both the 12.5-mg daily and 25-mg daily doses, compared with placebo.
Among the small number of people with stage 4 CKD, the incremental nighttime systolic blood pressure on aprocitentan, compared with placebo, was even greater, with about a 15–mm Hg incremental reduction on 12.5 mg daily and about a 17–mm Hg incremental reduction on the higher dose.
“This is the first evidence for a change in nocturnal blood pressure in people with stage 4 CKD [and treatment-resistant hypertension], but it was just 21 patients so not yet a big deal,” Dr. Bakris noted.
Increased rates of fluid retention
Although aprocitentan was generally well tolerated, the most common adverse effect was edema or fluid retention, mainly during the first 4 weeks of treatment. In the full PRECISION cohort, this adverse event occurred in 2.1% of people treated with placebo, 9.1% of those on the 12.5-mg daily dose, and in 18.4% of those on the higher dose during the initial 4-week phase of treatment.
Among all stage 3 and 4 CKD patients on aprocitentan, edema or fluid retention occurred in 21% during the first 4 weeks, and in 27% during an additional 32 weeks of treatment with 25 mg aprocitentan daily. A majority of these patients started a diuretic to address their excess fluid, with only two discontinuing aprocitentan treatment.
“Fluid retention is an issue with aprocitentan,” Dr. Bakris acknowledged. But he also highlighted than only 6 of the 162 patients with CKD required hospitalization for heart failure during the study, and one of these cases had placebo treatment. Among the five with acute heart failure while on aprocitentan, none had to stop their treatment, and two had a clear prior history of heart failure.
The companies developing aprocitentan, Janssen and Idorsia, used the PRECISION results as the centerpiece in filing for a new drug approval to the FDA, with a March 2024 goal for the FDA‘s decision. Dr. Bakris called the application “a solid case for approval.” But he added that approval will likely require that all treatment candidates first undergo testing of their heart function or fluid volume, such as a measure of their blood level of N-terminal pro-B-type natriuretic peptide, with treatment withheld when the level is too high.
The upside of aprocitentan compared with current drug options for treating resistant hypertension is that it has not appeared to cause any increase in blood potassium levels, which is an issue with the current top agent for resistant hypertension, spironolactone.
“The problem with spironolactone is the risk for hyperkalemia, which keeps us looking for something with lower risk,” commented Dr. Pradhan, a nephrologist with University Hospitals in Cleveland. Hyperkalemia is an even greater risk for people with CKD. Although the PRECISION trial identified the issue of fluid retention with aprocitentan, titrating an effective dose of a loop diuretic for treated patients may effectively blunt the edema risk, Dr. Pradhan said.
Endothelin has a potent vasoconstrictive effect and is “implicated in the pathogenesis of hypertension,” Dr. Bakris explained. Aprocitentan antagonizes both the endothelin A and B receptors. The subgroup analyses also showed that in people with CKD, treatment with aprocitentan led to roughly a halving of the baseline level of urine albumin-to-creatinine ratio, a small and stable decrease in estimated glomerular filtration rate, and a modest and stable increase in blood levels of N-terminal pro-B-type natriuretic hormone.
The PRECISION trial was sponsored by Janssen Pharmaceuticals and Idorsia Pharmaceuticals, the companies jointly developing aprocitentan. Dr. Bakris has been a consultant to Janssen, and also a consultant to or honoraria recipient of Alnylam, AstraZeneca, Bayer, Dia Medica Therapeutics, Ionis, inREGEN, KBP Biosciences, Merck, Novo Nordisk, and Quantum Genomics. Dr. Pradhan had no disclosures.
PHILADELPHIA – (CKD). The results come from a prespecified subgroup analysis of data collected in the drug’s pivotal trial, PRECISION.
The findings provide support for potentially using aprocitentan, if approved for U.S. marketing in 2024, in patients with blood pressure that remains elevated despite treatment with three established antihypertensive drug classes and with stage 3 CKD with an estimated glomerular filtration rate of 30-59 mL/min per 1.73 m2. This is a key group of patients because “chronic kidney disease is the most common comorbidity in patients with resistant hypertension,” said George Bakris, MD, who presented the subgroup analysis at Kidney Week 2023, organized by the American Society of Nephrology.
The CKD subgroup analysis showed “good evidence for safety and evidence in stage 3 CKD,” a subgroup of 141 patients among the total 730 enrolled in PRECISION, said Dr. Bakris. Professor and director of the Comprehensive Hypertension Center at the University of Chicago, he acknowledged that while the results also showed a signal for safety and efficacy in the 21 enrolled patients with stage 4 hypertension, 15-29 mL/min per 1.73m2, this number of stage 4 patients was too small to allow definitive conclusions.
Nephrologist Nishigandha Pradhan, MD, who cochaired the session with this report, agreed. “Resistant hypertension is a particularly intractable problem in patients with CKD, and the risk is greatest with stage 4 CKD. If studies could show that aprocitentan is safe in people with stage 4 CKD, that would be a big plus, but we need more data,” commented Dr. Pradhan in an interview.
Incremental blood pressure reductions
The parallel-group, phase 3 PRECISION trial investigated the safety and short-term antihypertensive effect of aprocitentan in patients with resistant hypertension. The study’s primary efficacy endpoint was blood pressure reduction from baseline in 730 randomized people with persistent systolic hypertension despite treatment with three established antihypertensive agents including a diuretic. The study ran during June 2018–April 2022 at 191 sites in 22 countries.
The primary outcome after 4 weeks on treatment was a least-square mean reduction in office-measured systolic blood pressure, compared with placebo, of 3.8 mm Hg with a 12.5-mg daily oral dose of aprocitentan and 3.7 mm Hg with a 25-mg daily oral dose. Both significant differences were first reported in 2022. Twenty-four–hour ambulatory systolic blood pressures after 4 weeks of treatment fell by an average of 4.2 mm Hg on the lower dose compared with placebo and by an average of 5.9 mm Hg on the higher daily dose, compared with placebo.
Consistent blood pressure reductions occurred in the CKD subgroups. Among people with stage 3 CKD, daytime ambulatory blood pressure at 4 weeks fell by about 10 mm Hg on both the 12.5-mg daily and 25-mg daily doses, compared with placebo.
Among the small number of people with stage 4 CKD, the incremental nighttime systolic blood pressure on aprocitentan, compared with placebo, was even greater, with about a 15–mm Hg incremental reduction on 12.5 mg daily and about a 17–mm Hg incremental reduction on the higher dose.
“This is the first evidence for a change in nocturnal blood pressure in people with stage 4 CKD [and treatment-resistant hypertension], but it was just 21 patients so not yet a big deal,” Dr. Bakris noted.
Increased rates of fluid retention
Although aprocitentan was generally well tolerated, the most common adverse effect was edema or fluid retention, mainly during the first 4 weeks of treatment. In the full PRECISION cohort, this adverse event occurred in 2.1% of people treated with placebo, 9.1% of those on the 12.5-mg daily dose, and in 18.4% of those on the higher dose during the initial 4-week phase of treatment.
Among all stage 3 and 4 CKD patients on aprocitentan, edema or fluid retention occurred in 21% during the first 4 weeks, and in 27% during an additional 32 weeks of treatment with 25 mg aprocitentan daily. A majority of these patients started a diuretic to address their excess fluid, with only two discontinuing aprocitentan treatment.
“Fluid retention is an issue with aprocitentan,” Dr. Bakris acknowledged. But he also highlighted than only 6 of the 162 patients with CKD required hospitalization for heart failure during the study, and one of these cases had placebo treatment. Among the five with acute heart failure while on aprocitentan, none had to stop their treatment, and two had a clear prior history of heart failure.
The companies developing aprocitentan, Janssen and Idorsia, used the PRECISION results as the centerpiece in filing for a new drug approval to the FDA, with a March 2024 goal for the FDA‘s decision. Dr. Bakris called the application “a solid case for approval.” But he added that approval will likely require that all treatment candidates first undergo testing of their heart function or fluid volume, such as a measure of their blood level of N-terminal pro-B-type natriuretic peptide, with treatment withheld when the level is too high.
The upside of aprocitentan compared with current drug options for treating resistant hypertension is that it has not appeared to cause any increase in blood potassium levels, which is an issue with the current top agent for resistant hypertension, spironolactone.
“The problem with spironolactone is the risk for hyperkalemia, which keeps us looking for something with lower risk,” commented Dr. Pradhan, a nephrologist with University Hospitals in Cleveland. Hyperkalemia is an even greater risk for people with CKD. Although the PRECISION trial identified the issue of fluid retention with aprocitentan, titrating an effective dose of a loop diuretic for treated patients may effectively blunt the edema risk, Dr. Pradhan said.
Endothelin has a potent vasoconstrictive effect and is “implicated in the pathogenesis of hypertension,” Dr. Bakris explained. Aprocitentan antagonizes both the endothelin A and B receptors. The subgroup analyses also showed that in people with CKD, treatment with aprocitentan led to roughly a halving of the baseline level of urine albumin-to-creatinine ratio, a small and stable decrease in estimated glomerular filtration rate, and a modest and stable increase in blood levels of N-terminal pro-B-type natriuretic hormone.
The PRECISION trial was sponsored by Janssen Pharmaceuticals and Idorsia Pharmaceuticals, the companies jointly developing aprocitentan. Dr. Bakris has been a consultant to Janssen, and also a consultant to or honoraria recipient of Alnylam, AstraZeneca, Bayer, Dia Medica Therapeutics, Ionis, inREGEN, KBP Biosciences, Merck, Novo Nordisk, and Quantum Genomics. Dr. Pradhan had no disclosures.
PHILADELPHIA – (CKD). The results come from a prespecified subgroup analysis of data collected in the drug’s pivotal trial, PRECISION.
The findings provide support for potentially using aprocitentan, if approved for U.S. marketing in 2024, in patients with blood pressure that remains elevated despite treatment with three established antihypertensive drug classes and with stage 3 CKD with an estimated glomerular filtration rate of 30-59 mL/min per 1.73 m2. This is a key group of patients because “chronic kidney disease is the most common comorbidity in patients with resistant hypertension,” said George Bakris, MD, who presented the subgroup analysis at Kidney Week 2023, organized by the American Society of Nephrology.
The CKD subgroup analysis showed “good evidence for safety and evidence in stage 3 CKD,” a subgroup of 141 patients among the total 730 enrolled in PRECISION, said Dr. Bakris. Professor and director of the Comprehensive Hypertension Center at the University of Chicago, he acknowledged that while the results also showed a signal for safety and efficacy in the 21 enrolled patients with stage 4 hypertension, 15-29 mL/min per 1.73m2, this number of stage 4 patients was too small to allow definitive conclusions.
Nephrologist Nishigandha Pradhan, MD, who cochaired the session with this report, agreed. “Resistant hypertension is a particularly intractable problem in patients with CKD, and the risk is greatest with stage 4 CKD. If studies could show that aprocitentan is safe in people with stage 4 CKD, that would be a big plus, but we need more data,” commented Dr. Pradhan in an interview.
Incremental blood pressure reductions
The parallel-group, phase 3 PRECISION trial investigated the safety and short-term antihypertensive effect of aprocitentan in patients with resistant hypertension. The study’s primary efficacy endpoint was blood pressure reduction from baseline in 730 randomized people with persistent systolic hypertension despite treatment with three established antihypertensive agents including a diuretic. The study ran during June 2018–April 2022 at 191 sites in 22 countries.
The primary outcome after 4 weeks on treatment was a least-square mean reduction in office-measured systolic blood pressure, compared with placebo, of 3.8 mm Hg with a 12.5-mg daily oral dose of aprocitentan and 3.7 mm Hg with a 25-mg daily oral dose. Both significant differences were first reported in 2022. Twenty-four–hour ambulatory systolic blood pressures after 4 weeks of treatment fell by an average of 4.2 mm Hg on the lower dose compared with placebo and by an average of 5.9 mm Hg on the higher daily dose, compared with placebo.
Consistent blood pressure reductions occurred in the CKD subgroups. Among people with stage 3 CKD, daytime ambulatory blood pressure at 4 weeks fell by about 10 mm Hg on both the 12.5-mg daily and 25-mg daily doses, compared with placebo.
Among the small number of people with stage 4 CKD, the incremental nighttime systolic blood pressure on aprocitentan, compared with placebo, was even greater, with about a 15–mm Hg incremental reduction on 12.5 mg daily and about a 17–mm Hg incremental reduction on the higher dose.
“This is the first evidence for a change in nocturnal blood pressure in people with stage 4 CKD [and treatment-resistant hypertension], but it was just 21 patients so not yet a big deal,” Dr. Bakris noted.
Increased rates of fluid retention
Although aprocitentan was generally well tolerated, the most common adverse effect was edema or fluid retention, mainly during the first 4 weeks of treatment. In the full PRECISION cohort, this adverse event occurred in 2.1% of people treated with placebo, 9.1% of those on the 12.5-mg daily dose, and in 18.4% of those on the higher dose during the initial 4-week phase of treatment.
Among all stage 3 and 4 CKD patients on aprocitentan, edema or fluid retention occurred in 21% during the first 4 weeks, and in 27% during an additional 32 weeks of treatment with 25 mg aprocitentan daily. A majority of these patients started a diuretic to address their excess fluid, with only two discontinuing aprocitentan treatment.
“Fluid retention is an issue with aprocitentan,” Dr. Bakris acknowledged. But he also highlighted than only 6 of the 162 patients with CKD required hospitalization for heart failure during the study, and one of these cases had placebo treatment. Among the five with acute heart failure while on aprocitentan, none had to stop their treatment, and two had a clear prior history of heart failure.
The companies developing aprocitentan, Janssen and Idorsia, used the PRECISION results as the centerpiece in filing for a new drug approval to the FDA, with a March 2024 goal for the FDA‘s decision. Dr. Bakris called the application “a solid case for approval.” But he added that approval will likely require that all treatment candidates first undergo testing of their heart function or fluid volume, such as a measure of their blood level of N-terminal pro-B-type natriuretic peptide, with treatment withheld when the level is too high.
The upside of aprocitentan compared with current drug options for treating resistant hypertension is that it has not appeared to cause any increase in blood potassium levels, which is an issue with the current top agent for resistant hypertension, spironolactone.
“The problem with spironolactone is the risk for hyperkalemia, which keeps us looking for something with lower risk,” commented Dr. Pradhan, a nephrologist with University Hospitals in Cleveland. Hyperkalemia is an even greater risk for people with CKD. Although the PRECISION trial identified the issue of fluid retention with aprocitentan, titrating an effective dose of a loop diuretic for treated patients may effectively blunt the edema risk, Dr. Pradhan said.
Endothelin has a potent vasoconstrictive effect and is “implicated in the pathogenesis of hypertension,” Dr. Bakris explained. Aprocitentan antagonizes both the endothelin A and B receptors. The subgroup analyses also showed that in people with CKD, treatment with aprocitentan led to roughly a halving of the baseline level of urine albumin-to-creatinine ratio, a small and stable decrease in estimated glomerular filtration rate, and a modest and stable increase in blood levels of N-terminal pro-B-type natriuretic hormone.
The PRECISION trial was sponsored by Janssen Pharmaceuticals and Idorsia Pharmaceuticals, the companies jointly developing aprocitentan. Dr. Bakris has been a consultant to Janssen, and also a consultant to or honoraria recipient of Alnylam, AstraZeneca, Bayer, Dia Medica Therapeutics, Ionis, inREGEN, KBP Biosciences, Merck, Novo Nordisk, and Quantum Genomics. Dr. Pradhan had no disclosures.
AT KIDNEY WEEK 2023
Veterans Get $6 billion in Hearing Loss Settlement
Hearing loss and tinnitus are the top and third most common service-connected disabilities among veterans. According to a Veterans Benefits Administration report, as of fiscal year 2020, more than 1.3 million veterans were receiving disability compensation for hearing loss and more than 2.3 million for tinnitus. Not surprisingly, the US Department of Veterans Affairs (VA) is the largest employer of audiologists and speech-language pathologists in the US.
On the bright side, military hearing losses are at stable levels—but “it’s not improving,” said US Army Lt Col Michael Murphy, chief of the studies and analysis section and Army audiology liaison at the Defense Health Agency Hearing Center of Excellence (HCE), in an interview for Department of Defense news.
Hearing protection is critical to reduce injury. Exposure to firearms, explosives, and other “continuous hazardous noise” puts service members and US Department of Defense (DoD) civilians at risk of permanent hearing loss, said Theresa Schulz, PhD, chief of the HCE prevention and surveillance section. “Good hearing is a key to mission success.”
Hearing protectors, which Shulz calls “the last line of defense from noise-induced hearing loss,” work best when they fit right: protecting against noise and, when necessary, not muffling voices, alarms, and other important sounds. That is why the DoD has updated its requirements for fit testing. All DoD personnel who are exposed to continuous and intermittent noise ≥ 85 decibels (in an 8-hour average) or impulse noise sound pressure ≥ 140 decibels (for ≥ 1 day per year) must be enrolled in a hearing conservation program. Additional criteria are expected for release by December 2023. According to HCE, each service may have more stringent requirements for hearing protector fit testing that better meets the needs of their hearing conservation program.
The question of proper fit was at the root of a recent lawsuit charging 3M with knowingly selling defective earplugs to the US military. The 3M dual-ended Combat Arms Earplug (CAEv2) was designed to eliminate the need for soldiers to carry 2 different sets of earplugs. Worn one way, it was intended to block sound like traditional earplugs; worn in reverse, it would block only certain types of loud battlefield noise while allowing the wearer to hear softer, closer sounds.
However, no 2 ears are the same—even on the same person. According to the HCE, during hearing protection testing, there is a < 2 mm difference in insertion depth between left and right ears for 85% of subjects. A 2016 whistleblower lawsuit accused 3M of not disclosing that the CAEv2 was too short for proper insertion into users’ ears and that it could loosen imperceptibly and fail to form the protective seal.
In 2018, 3M agreed to pay $9.1 million to the Department of Justice to resolve the allegations without admitting liability. That case led to the largest mass tort multidistrict litigation in US history. Last February, Veterans of Foreign Wars (VFW) filed an amicus curiae brief to the Seventh Circuit Court of Appeals in support of claimants seeking relief from 3M for defective ear protection. Approximately 240,000 veterans filed lawsuits against 3M. In September the parties reached a $6 billion settlement—nearly half of 3M’s worth. According to John Muckelbauer, a veteran and general counsel for the VFW in a military.com opinion piece, the settlement achieves balance: not pushing the already financially strapped 3M into bankruptcy, but sending “a strong signal that the safety of our service members can never be compromised.”
Crucially, Muckelbauer notes, the VA says participating in the lawsuit will not result in the loss of health or disability benefits, nor will it adversely affect disability ratings. VA facilities are also barred from recovering any portion of a plaintiff’s award as part of a medical lien.
3M has not admitted responsibility in this settlement either, frustrating the veteran claimants. An admission of guilt was never on the table, says Ronald Miller, Jr., writing for the Lawsuit Information Center, which posts updates on class action lawsuits. “Admitting responsibility would open the door for everyone to opt out and move forward on that admission… Admitting guilt would also be harmful to 3M’s reputation. They have long vigorously denied responsibility, so the optics would be terrible.”
A new twist cropped up almost immediately when claimants began getting cold calls from scammers impersonating employees of Archer Systems LLC, the company designated to administer the settlement. The scammers attempted to extract sensitive personal information, including Social Security numbers. Judge M. Casey Rodgers alerted the Federal Bureau of Investigation and warned claimants to safeguard their data vigilantly and report any fraudulent attempts.
The settlement money will be paid out from 2023 to 2029, with $1 billion in the form of 3M stock, 3M said in a statement. (In August 2023, upon news of the settlement, the price of 3M shares had risen nearly 5%.) Miller says the whole $6 billion will be distributed using a point system that awards amounts according to disability, with, for instance, tinnitus without contemporaneous corroboration getting the least and moderate or greater hearing loss getting the most. “This settlement is a tremendous outcome for veterans of Iraq and Afghanistan who put their lives on the line for our freedom,” said Duane Sarmiento, VFW national commander in a statement. “For those who came home with hearing damage due to 3M’s faulty earplugs, this is not only compensation, it’s a statement that their sacrifices won’t be ignored.”
Hearing loss and tinnitus are the top and third most common service-connected disabilities among veterans. According to a Veterans Benefits Administration report, as of fiscal year 2020, more than 1.3 million veterans were receiving disability compensation for hearing loss and more than 2.3 million for tinnitus. Not surprisingly, the US Department of Veterans Affairs (VA) is the largest employer of audiologists and speech-language pathologists in the US.
On the bright side, military hearing losses are at stable levels—but “it’s not improving,” said US Army Lt Col Michael Murphy, chief of the studies and analysis section and Army audiology liaison at the Defense Health Agency Hearing Center of Excellence (HCE), in an interview for Department of Defense news.
Hearing protection is critical to reduce injury. Exposure to firearms, explosives, and other “continuous hazardous noise” puts service members and US Department of Defense (DoD) civilians at risk of permanent hearing loss, said Theresa Schulz, PhD, chief of the HCE prevention and surveillance section. “Good hearing is a key to mission success.”
Hearing protectors, which Shulz calls “the last line of defense from noise-induced hearing loss,” work best when they fit right: protecting against noise and, when necessary, not muffling voices, alarms, and other important sounds. That is why the DoD has updated its requirements for fit testing. All DoD personnel who are exposed to continuous and intermittent noise ≥ 85 decibels (in an 8-hour average) or impulse noise sound pressure ≥ 140 decibels (for ≥ 1 day per year) must be enrolled in a hearing conservation program. Additional criteria are expected for release by December 2023. According to HCE, each service may have more stringent requirements for hearing protector fit testing that better meets the needs of their hearing conservation program.
The question of proper fit was at the root of a recent lawsuit charging 3M with knowingly selling defective earplugs to the US military. The 3M dual-ended Combat Arms Earplug (CAEv2) was designed to eliminate the need for soldiers to carry 2 different sets of earplugs. Worn one way, it was intended to block sound like traditional earplugs; worn in reverse, it would block only certain types of loud battlefield noise while allowing the wearer to hear softer, closer sounds.
However, no 2 ears are the same—even on the same person. According to the HCE, during hearing protection testing, there is a < 2 mm difference in insertion depth between left and right ears for 85% of subjects. A 2016 whistleblower lawsuit accused 3M of not disclosing that the CAEv2 was too short for proper insertion into users’ ears and that it could loosen imperceptibly and fail to form the protective seal.
In 2018, 3M agreed to pay $9.1 million to the Department of Justice to resolve the allegations without admitting liability. That case led to the largest mass tort multidistrict litigation in US history. Last February, Veterans of Foreign Wars (VFW) filed an amicus curiae brief to the Seventh Circuit Court of Appeals in support of claimants seeking relief from 3M for defective ear protection. Approximately 240,000 veterans filed lawsuits against 3M. In September the parties reached a $6 billion settlement—nearly half of 3M’s worth. According to John Muckelbauer, a veteran and general counsel for the VFW in a military.com opinion piece, the settlement achieves balance: not pushing the already financially strapped 3M into bankruptcy, but sending “a strong signal that the safety of our service members can never be compromised.”
Crucially, Muckelbauer notes, the VA says participating in the lawsuit will not result in the loss of health or disability benefits, nor will it adversely affect disability ratings. VA facilities are also barred from recovering any portion of a plaintiff’s award as part of a medical lien.
3M has not admitted responsibility in this settlement either, frustrating the veteran claimants. An admission of guilt was never on the table, says Ronald Miller, Jr., writing for the Lawsuit Information Center, which posts updates on class action lawsuits. “Admitting responsibility would open the door for everyone to opt out and move forward on that admission… Admitting guilt would also be harmful to 3M’s reputation. They have long vigorously denied responsibility, so the optics would be terrible.”
A new twist cropped up almost immediately when claimants began getting cold calls from scammers impersonating employees of Archer Systems LLC, the company designated to administer the settlement. The scammers attempted to extract sensitive personal information, including Social Security numbers. Judge M. Casey Rodgers alerted the Federal Bureau of Investigation and warned claimants to safeguard their data vigilantly and report any fraudulent attempts.
The settlement money will be paid out from 2023 to 2029, with $1 billion in the form of 3M stock, 3M said in a statement. (In August 2023, upon news of the settlement, the price of 3M shares had risen nearly 5%.) Miller says the whole $6 billion will be distributed using a point system that awards amounts according to disability, with, for instance, tinnitus without contemporaneous corroboration getting the least and moderate or greater hearing loss getting the most. “This settlement is a tremendous outcome for veterans of Iraq and Afghanistan who put their lives on the line for our freedom,” said Duane Sarmiento, VFW national commander in a statement. “For those who came home with hearing damage due to 3M’s faulty earplugs, this is not only compensation, it’s a statement that their sacrifices won’t be ignored.”
Hearing loss and tinnitus are the top and third most common service-connected disabilities among veterans. According to a Veterans Benefits Administration report, as of fiscal year 2020, more than 1.3 million veterans were receiving disability compensation for hearing loss and more than 2.3 million for tinnitus. Not surprisingly, the US Department of Veterans Affairs (VA) is the largest employer of audiologists and speech-language pathologists in the US.
On the bright side, military hearing losses are at stable levels—but “it’s not improving,” said US Army Lt Col Michael Murphy, chief of the studies and analysis section and Army audiology liaison at the Defense Health Agency Hearing Center of Excellence (HCE), in an interview for Department of Defense news.
Hearing protection is critical to reduce injury. Exposure to firearms, explosives, and other “continuous hazardous noise” puts service members and US Department of Defense (DoD) civilians at risk of permanent hearing loss, said Theresa Schulz, PhD, chief of the HCE prevention and surveillance section. “Good hearing is a key to mission success.”
Hearing protectors, which Shulz calls “the last line of defense from noise-induced hearing loss,” work best when they fit right: protecting against noise and, when necessary, not muffling voices, alarms, and other important sounds. That is why the DoD has updated its requirements for fit testing. All DoD personnel who are exposed to continuous and intermittent noise ≥ 85 decibels (in an 8-hour average) or impulse noise sound pressure ≥ 140 decibels (for ≥ 1 day per year) must be enrolled in a hearing conservation program. Additional criteria are expected for release by December 2023. According to HCE, each service may have more stringent requirements for hearing protector fit testing that better meets the needs of their hearing conservation program.
The question of proper fit was at the root of a recent lawsuit charging 3M with knowingly selling defective earplugs to the US military. The 3M dual-ended Combat Arms Earplug (CAEv2) was designed to eliminate the need for soldiers to carry 2 different sets of earplugs. Worn one way, it was intended to block sound like traditional earplugs; worn in reverse, it would block only certain types of loud battlefield noise while allowing the wearer to hear softer, closer sounds.
However, no 2 ears are the same—even on the same person. According to the HCE, during hearing protection testing, there is a < 2 mm difference in insertion depth between left and right ears for 85% of subjects. A 2016 whistleblower lawsuit accused 3M of not disclosing that the CAEv2 was too short for proper insertion into users’ ears and that it could loosen imperceptibly and fail to form the protective seal.
In 2018, 3M agreed to pay $9.1 million to the Department of Justice to resolve the allegations without admitting liability. That case led to the largest mass tort multidistrict litigation in US history. Last February, Veterans of Foreign Wars (VFW) filed an amicus curiae brief to the Seventh Circuit Court of Appeals in support of claimants seeking relief from 3M for defective ear protection. Approximately 240,000 veterans filed lawsuits against 3M. In September the parties reached a $6 billion settlement—nearly half of 3M’s worth. According to John Muckelbauer, a veteran and general counsel for the VFW in a military.com opinion piece, the settlement achieves balance: not pushing the already financially strapped 3M into bankruptcy, but sending “a strong signal that the safety of our service members can never be compromised.”
Crucially, Muckelbauer notes, the VA says participating in the lawsuit will not result in the loss of health or disability benefits, nor will it adversely affect disability ratings. VA facilities are also barred from recovering any portion of a plaintiff’s award as part of a medical lien.
3M has not admitted responsibility in this settlement either, frustrating the veteran claimants. An admission of guilt was never on the table, says Ronald Miller, Jr., writing for the Lawsuit Information Center, which posts updates on class action lawsuits. “Admitting responsibility would open the door for everyone to opt out and move forward on that admission… Admitting guilt would also be harmful to 3M’s reputation. They have long vigorously denied responsibility, so the optics would be terrible.”
A new twist cropped up almost immediately when claimants began getting cold calls from scammers impersonating employees of Archer Systems LLC, the company designated to administer the settlement. The scammers attempted to extract sensitive personal information, including Social Security numbers. Judge M. Casey Rodgers alerted the Federal Bureau of Investigation and warned claimants to safeguard their data vigilantly and report any fraudulent attempts.
The settlement money will be paid out from 2023 to 2029, with $1 billion in the form of 3M stock, 3M said in a statement. (In August 2023, upon news of the settlement, the price of 3M shares had risen nearly 5%.) Miller says the whole $6 billion will be distributed using a point system that awards amounts according to disability, with, for instance, tinnitus without contemporaneous corroboration getting the least and moderate or greater hearing loss getting the most. “This settlement is a tremendous outcome for veterans of Iraq and Afghanistan who put their lives on the line for our freedom,” said Duane Sarmiento, VFW national commander in a statement. “For those who came home with hearing damage due to 3M’s faulty earplugs, this is not only compensation, it’s a statement that their sacrifices won’t be ignored.”