User login
Smoldering myeloma progressed more rapidly in patients with elevated BMIs
An elevated body mass index appears to be a risk factor for progression of smoldering multiple myeloma, according to Wilson I. Gonsalves, MD, and his colleagues at Mayo Clinic, Rochester, Minn.
The findings, based on median follow up data of 106 months from 306 patients diagnosed with smoldering multiple myeloma from 2000-2010 at the Mayo Clinic, need to be confirmed in larger studies. Nevertheless, the results imply that patient weight is a potentially modifiable risk factor for progression from smoldering disease to multiple myeloma, Dr. Gonsalves and his colleagues wrote.

At initial evaluation, 28% of patients had myeloma defining events, such as a serum free light chain ratio greater than 100 or over 60% clonal bone marrow plasma cells. Myeloma defining events were present in 17% of patients with normal BMIs and 33% of patients with elevated BMIs, a statistically significant difference (P = .011).
When the analysis was limited to the 187 patients without myeloma-defining events at initial evaluation, the 2-year rate of progression to symptomatic multiple myeloma was 15% in those with a normal BMI and 33% in those with an elevated BMI (P = .013).
In a multivariable model, only elevated BMI (P = .004) and increasing clonal bone marrow plasma cells (P = .001) were statistically significant in predicting 2-year progression to multiple myeloma.
At last follow-up, 66% of patients had progressed to symptomatic multiple myeloma.
Dr. Gonsalves had no relationships to disclose.
The impact of body mass index on the risk of early progression of smoldering multiple myeloma to symptomatic myeloma. 2017 ASCO annual meeting. Abstract No: 8032.
[email protected]
On Twitter @maryjodales
An elevated body mass index appears to be a risk factor for progression of smoldering multiple myeloma, according to Wilson I. Gonsalves, MD, and his colleagues at Mayo Clinic, Rochester, Minn.
The findings, based on median follow up data of 106 months from 306 patients diagnosed with smoldering multiple myeloma from 2000-2010 at the Mayo Clinic, need to be confirmed in larger studies. Nevertheless, the results imply that patient weight is a potentially modifiable risk factor for progression from smoldering disease to multiple myeloma, Dr. Gonsalves and his colleagues wrote.
At initial evaluation, 28% of patients had myeloma defining events, such as a serum free light chain ratio greater than 100 or over 60% clonal bone marrow plasma cells. Myeloma defining events were present in 17% of patients with normal BMIs and 33% of patients with elevated BMIs, a statistically significant difference (P = .011).
When the analysis was limited to the 187 patients without myeloma-defining events at initial evaluation, the 2-year rate of progression to symptomatic multiple myeloma was 15% in those with a normal BMI and 33% in those with an elevated BMI (P = .013).
In a multivariable model, only elevated BMI (P = .004) and increasing clonal bone marrow plasma cells (P = .001) were statistically significant in predicting 2-year progression to multiple myeloma.
At last follow-up, 66% of patients had progressed to symptomatic multiple myeloma.
Dr. Gonsalves had no relationships to disclose.
The impact of body mass index on the risk of early progression of smoldering multiple myeloma to symptomatic myeloma. 2017 ASCO annual meeting. Abstract No: 8032.
[email protected]
On Twitter @maryjodales
An elevated body mass index appears to be a risk factor for progression of smoldering multiple myeloma, according to Wilson I. Gonsalves, MD, and his colleagues at Mayo Clinic, Rochester, Minn.
The findings, based on median follow up data of 106 months from 306 patients diagnosed with smoldering multiple myeloma from 2000-2010 at the Mayo Clinic, need to be confirmed in larger studies. Nevertheless, the results imply that patient weight is a potentially modifiable risk factor for progression from smoldering disease to multiple myeloma, Dr. Gonsalves and his colleagues wrote.
At initial evaluation, 28% of patients had myeloma defining events, such as a serum free light chain ratio greater than 100 or over 60% clonal bone marrow plasma cells. Myeloma defining events were present in 17% of patients with normal BMIs and 33% of patients with elevated BMIs, a statistically significant difference (P = .011).
When the analysis was limited to the 187 patients without myeloma-defining events at initial evaluation, the 2-year rate of progression to symptomatic multiple myeloma was 15% in those with a normal BMI and 33% in those with an elevated BMI (P = .013).
In a multivariable model, only elevated BMI (P = .004) and increasing clonal bone marrow plasma cells (P = .001) were statistically significant in predicting 2-year progression to multiple myeloma.
At last follow-up, 66% of patients had progressed to symptomatic multiple myeloma.
Dr. Gonsalves had no relationships to disclose.
The impact of body mass index on the risk of early progression of smoldering multiple myeloma to symptomatic myeloma. 2017 ASCO annual meeting. Abstract No: 8032.
[email protected]
On Twitter @maryjodales
FROM ASCO 2017
Key clinical point:
Major finding: The 2-year rate of progression from smoldering disease to symptomatic multiple myeloma was 16% in patients with a normal BMI and 42% in patients with an elevated BMI (P less than 0.0001).
Data source: Median follow up data of 106 months from 306 patients diagnosed with smoldering multiple myeloma during 2000-2010 at the Mayo Clinic.
Disclosures: Dr. Gonsalves had no relationships to disclose.
Citation: The impact of body mass index on the risk of early progression of smoldering multiple myeloma to symptomatic myeloma. 2017 ASCO annual meeting. Abstract No: 8032.
Postop satisfaction scores not tied to restricted opioid prescribing
Reduced opioid prescribing did not correlate with inpatient pain management scores, a study has shown.
The Centers for Medicare & Medicaid Services announced recently that, as of 2018, pain management will no longer be rated in the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey, citing concerns that patient satisfaction surveys given at the time of postoperative discharge incentivizes clinicians to over-prescribe pain medication. Surgical patients are key contributors to HCAHPS scores, and opioids account for almost 40% of surgical prescriptions, according to the study.
A new study throws some shade on the CMS decision to delete pain management from the HCAHPS survey.
Pain management scores were calculated as the percentage of patients who reported that their pain was “always” well controlled. The pain dimension was calculated from the number of opioid prescriptions and also pain management scores compared to national benchmarks. Hospitals were then grouped into quintiles according to opioid prescriptions measured in oral morphine equivalents. The first quintile has the lowest number of prescriptions.
Unadjusted comparisons showed no significant differences in pain management or pain dimension scores between the first and fifth quintiles of hospitals. For pain management scores that ranked hospital staff as always controlling pain, the first quintile had a mean score of 69.5 (95% confidence interval, 66.7-71.7) out of 100, compared with 69.1 for the fifth quintile (95% CI, 67.2-71.4). On a scale of 1-10, pain dimension scores in the first quintile averaged 1.9 (mean 95% CI, 1.5-2.0), compared with 1.4 in the fifth quintile (mean 95% CI, 0.9-1.9).
So, for these institutions, the number of pain prescriptions was not correlated with HCAHPS scores for pain management. The study suggests that the concern that reducing opioid prescriptions may have a negative impact on patient satisfaction assessments may not be realized.
Other analyses controlling for a variety of comorbidities also showed no correlations between pain management scores and opioid prescribing. Of the surgeries considered – orthopedic, general, gynecologic, cancer, cardiac, and vascular – gynecologic procedures were most likely to be associated with improved pain management and pain dimension scores.
Dr. Brummett disclosed relationships with Tonix and Neuros Medical. He also holds a patent for peripheral perineural dexmedetomidine. Mr. Syrjamaki and Dr. Dupree received support from Blue Cross Blue Shield of Michigan for their respective roles in the Michigan Value Collaborative. Dr. Waljee is an unpaid consultant for 3MHealth.
[email protected]
On Twitter @whitneymcknight
Reduced opioid prescribing did not correlate with inpatient pain management scores, a study has shown.
The Centers for Medicare & Medicaid Services announced recently that, as of 2018, pain management will no longer be rated in the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey, citing concerns that patient satisfaction surveys given at the time of postoperative discharge incentivizes clinicians to over-prescribe pain medication. Surgical patients are key contributors to HCAHPS scores, and opioids account for almost 40% of surgical prescriptions, according to the study.
A new study throws some shade on the CMS decision to delete pain management from the HCAHPS survey.
Pain management scores were calculated as the percentage of patients who reported that their pain was “always” well controlled. The pain dimension was calculated from the number of opioid prescriptions and also pain management scores compared to national benchmarks. Hospitals were then grouped into quintiles according to opioid prescriptions measured in oral morphine equivalents. The first quintile has the lowest number of prescriptions.
Unadjusted comparisons showed no significant differences in pain management or pain dimension scores between the first and fifth quintiles of hospitals. For pain management scores that ranked hospital staff as always controlling pain, the first quintile had a mean score of 69.5 (95% confidence interval, 66.7-71.7) out of 100, compared with 69.1 for the fifth quintile (95% CI, 67.2-71.4). On a scale of 1-10, pain dimension scores in the first quintile averaged 1.9 (mean 95% CI, 1.5-2.0), compared with 1.4 in the fifth quintile (mean 95% CI, 0.9-1.9).
So, for these institutions, the number of pain prescriptions was not correlated with HCAHPS scores for pain management. The study suggests that the concern that reducing opioid prescriptions may have a negative impact on patient satisfaction assessments may not be realized.
Other analyses controlling for a variety of comorbidities also showed no correlations between pain management scores and opioid prescribing. Of the surgeries considered – orthopedic, general, gynecologic, cancer, cardiac, and vascular – gynecologic procedures were most likely to be associated with improved pain management and pain dimension scores.
Dr. Brummett disclosed relationships with Tonix and Neuros Medical. He also holds a patent for peripheral perineural dexmedetomidine. Mr. Syrjamaki and Dr. Dupree received support from Blue Cross Blue Shield of Michigan for their respective roles in the Michigan Value Collaborative. Dr. Waljee is an unpaid consultant for 3MHealth.
[email protected]
On Twitter @whitneymcknight
Reduced opioid prescribing did not correlate with inpatient pain management scores, a study has shown.
The Centers for Medicare & Medicaid Services announced recently that, as of 2018, pain management will no longer be rated in the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey, citing concerns that patient satisfaction surveys given at the time of postoperative discharge incentivizes clinicians to over-prescribe pain medication. Surgical patients are key contributors to HCAHPS scores, and opioids account for almost 40% of surgical prescriptions, according to the study.
A new study throws some shade on the CMS decision to delete pain management from the HCAHPS survey.
Pain management scores were calculated as the percentage of patients who reported that their pain was “always” well controlled. The pain dimension was calculated from the number of opioid prescriptions and also pain management scores compared to national benchmarks. Hospitals were then grouped into quintiles according to opioid prescriptions measured in oral morphine equivalents. The first quintile has the lowest number of prescriptions.
Unadjusted comparisons showed no significant differences in pain management or pain dimension scores between the first and fifth quintiles of hospitals. For pain management scores that ranked hospital staff as always controlling pain, the first quintile had a mean score of 69.5 (95% confidence interval, 66.7-71.7) out of 100, compared with 69.1 for the fifth quintile (95% CI, 67.2-71.4). On a scale of 1-10, pain dimension scores in the first quintile averaged 1.9 (mean 95% CI, 1.5-2.0), compared with 1.4 in the fifth quintile (mean 95% CI, 0.9-1.9).
So, for these institutions, the number of pain prescriptions was not correlated with HCAHPS scores for pain management. The study suggests that the concern that reducing opioid prescriptions may have a negative impact on patient satisfaction assessments may not be realized.
Other analyses controlling for a variety of comorbidities also showed no correlations between pain management scores and opioid prescribing. Of the surgeries considered – orthopedic, general, gynecologic, cancer, cardiac, and vascular – gynecologic procedures were most likely to be associated with improved pain management and pain dimension scores.
Dr. Brummett disclosed relationships with Tonix and Neuros Medical. He also holds a patent for peripheral perineural dexmedetomidine. Mr. Syrjamaki and Dr. Dupree received support from Blue Cross Blue Shield of Michigan for their respective roles in the Michigan Value Collaborative. Dr. Waljee is an unpaid consultant for 3MHealth.
[email protected]
On Twitter @whitneymcknight
Key clinical point:
Major finding: No significant differences between top and bottom quintiles of 47 Michigan hospitals’ opioid prescribing patterns existed when comparing their HCAHPS scores.
Data source: Pharmacy and insurance claims and HCAHPS pain management for 31,481 surgery patients between 2012 and 2014.
Disclosures: Dr. Brummett disclosed relationships with Tonix and Neuros Medical. He also holds a patent for peripheral perineural dexmedetomidine. Mr. Syrjamaki and Dr. Dupree received support from Blue Cross Blue Shield of Michigan for their respective roles in the Michigan Value Collaborative. Dr. Waljee is an unpaid consultant for 3MHealth.

GOLD guidelines for the management of COPD – 2017 update
Chronic obstructive lung disease (COPD) is the third leading cause of death in the United States1 and a major cause of mortality and morbidity around the world. The Global Initiative for Chronic Obstructive Lung Disease (GOLD) released a new “2017 Report”2 with modified recommendations for the diagnosis, management, and prevention of COPD. The report contains several changes that are relevant to the primary care provider that will be outlined below.
Redefining COPD
GOLD’s definition of COPD was changed in its 2017 Report: “COPD is a common, preventable, and treatable disease that is characterized by persistent respiratory symptoms and airflow limitations that are due to airway and/or alveolar abnormalities usually caused by significant exposure to noxious particles or gases.” The report emphasizes that “COPD may be punctuated by periods of acute worsening of respiratory symptoms, called exacerbations.” Note that the terms “emphysema” and “chronic bronchitis” have been removed in favor of a more comprehensive description of the pathophysiology of COPD. Importantly, the report states that cough and sputum production for at least 3 months in each of 2 consecutive years, previously accepted as diagnostic criteria, are present in only a minority of patients. It is noted that chronic respiratory symptoms may exist without spirometric changes and many patients (usually smokers) have structural evidence of COPD without airflow limitation.

Changes to COPD initial assessment
The primary criterion for diagnosis is unchanged: post-bronchodilator forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) less than 0.70. Spirometry remains important to confirm the diagnosis in those with classic symptoms of dyspnea, chronic cough, and/or sputum production with a history of exposure to noxious particles or gases.
The GOLD assessment system previously incorporated spirometry and included an “ABCD” system such that patients in group A are least severe. Spirometry has been progressively deemphasized in favor of symptom-based classification and the 2017 Report, for the first time, dissociates spirometric findings from severity classification.
The new system uses symptom severity and exacerbation risk to classify COPD. Two specific standardized COPD symptom measurement tools, The Modified British Medical Research Council (mMRC) questionnaire and COPD Assessment Test (CAT), are reported by GOLD as the most widely used. Low symptom severity is considered an mMRC less than or equal to 1 or CAT less than or equal to 9, high symptom severity is considered an mMRC greater than or equal to 2 or CAT greater than or equal to 10. Low risk of exacerbation is defined as no more than one exacerbation not resulting in hospital admission in the last 12 months; high risk of exacerbation is defined as at least two exacerbations or any exacerbations resulting in hospital admission in the last 12 months. Symptom severity and exacerbation risk is divided into four quadrants:
• GOLD group A: Low symptom severity, low exacerbation risk.
• GOLD group B: High symptom severity, low exacerbation risk.
• GOLD group C: Low symptom severity, high exacerbation risk.
• GOLD group D: High symptom severity, high exacerbation risk.
Changes to prevention and management of stable COPD
Smoking cessation remains important in the prevention of COPD. The 2017 Report reflects the U.S. Preventive Services Task Force’s guidelines for smoking cessation: Offer nicotine replacement, cessation counseling, and pharmacotherapy (varenicline, bupropion or nortriptyline). There is insufficient evidence to support the use of e-cigarettes. Influenza and pneumococcal vaccinations are recommended. Pulmonary rehabilitation remains important.
The 2017 Report includes an expanded discussion of COPD medications. The role of short-acting bronchodilators (SABD) in COPD remains prominent. Changes include a stronger recommendation to use combination short-acting beta-agonists and short-acting muscarinic antagonists (SABA/SAMA) as these seem to be superior to SABD monotherapy in improving symptoms and FEV1.
There were several changes to the pharmacologic treatment algorithm. For the first time, GOLD proposes escalation strategies. Preference is given to LABA/LAMA (long-acting beta-agonist/long-acting muscarinic antagonists) combinations over LABA/ICS (long-acting beta-agonist/inhaled corticosteroid) combinations as a mainstay of treatment. The rationale for this change is that LABA/LAMAs give greater bronchodilation compared with LABA/ICS, and one study showed a decreased rate of exacerbations compared to LABA/ICS in patients with a history of exacerbations. In addition, patients with COPD who receive ICS appear to have a higher risk of developing pneumonia. GOLD recommendations are:
• Group A: Start with single bronchodilator (short- or long-acting), escalate to alternative class of bronchodilator if necessary.
• Group B: Start with LABA or LAMA, escalate to LABA/LAMA if symptoms persist.
• Group C: Start with LAMA, escalate to LABA/LAMA (preferred) or LABA/ICS if exacerbations continue.
• Group D: Start with LABA/LAMA (preferred) or LAMA monotherapy, escalate to LABA/LAMA/ICS (preferred) or try LABA/ICS before escalating to LAMA/LABA/ICS if symptoms persist or exacerbations continue; roflumilast and/or a macrolide may be considered if further exacerbations occur with LABA/LAMA/ICS.
Bottom line
1. GOLD classification of COPD severity is now based on clinical criteria alone: symptom assessment and risk for exacerbation.
2. SABA/SAMA combination therapy seems to be superior to either SABA or SAMA alone.
3. Patients in group A (milder symptoms, low exacerbation risk) may be initiated on either short- or long-acting bronchodilator therapy.
4. Patients in group B (milder symptoms, increased exacerbation risk) should be initiated on LAMA monotherapy.
5. LABA/LAMA combination therapy seems to be superior to LABA/ICS combination therapy and should be used when long-acting bronchodilator monotherapy fails to control symptoms or reduce exacerbations.
References
1. CDC MMWR 11/23/12
2. Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2017 at http://goldcopd.org (accessed 3/10/2017)
Dr. Skolnik is associate director of the family medicine residency program at Abington (Pa.) Memorial Hospital. Dr. Lent is chief resident in the program.
Chronic obstructive lung disease (COPD) is the third leading cause of death in the United States1 and a major cause of mortality and morbidity around the world. The Global Initiative for Chronic Obstructive Lung Disease (GOLD) released a new “2017 Report”2 with modified recommendations for the diagnosis, management, and prevention of COPD. The report contains several changes that are relevant to the primary care provider that will be outlined below.
Redefining COPD
GOLD’s definition of COPD was changed in its 2017 Report: “COPD is a common, preventable, and treatable disease that is characterized by persistent respiratory symptoms and airflow limitations that are due to airway and/or alveolar abnormalities usually caused by significant exposure to noxious particles or gases.” The report emphasizes that “COPD may be punctuated by periods of acute worsening of respiratory symptoms, called exacerbations.” Note that the terms “emphysema” and “chronic bronchitis” have been removed in favor of a more comprehensive description of the pathophysiology of COPD. Importantly, the report states that cough and sputum production for at least 3 months in each of 2 consecutive years, previously accepted as diagnostic criteria, are present in only a minority of patients. It is noted that chronic respiratory symptoms may exist without spirometric changes and many patients (usually smokers) have structural evidence of COPD without airflow limitation.
Changes to COPD initial assessment
The primary criterion for diagnosis is unchanged: post-bronchodilator forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) less than 0.70. Spirometry remains important to confirm the diagnosis in those with classic symptoms of dyspnea, chronic cough, and/or sputum production with a history of exposure to noxious particles or gases.
The GOLD assessment system previously incorporated spirometry and included an “ABCD” system such that patients in group A are least severe. Spirometry has been progressively deemphasized in favor of symptom-based classification and the 2017 Report, for the first time, dissociates spirometric findings from severity classification.
The new system uses symptom severity and exacerbation risk to classify COPD. Two specific standardized COPD symptom measurement tools, The Modified British Medical Research Council (mMRC) questionnaire and COPD Assessment Test (CAT), are reported by GOLD as the most widely used. Low symptom severity is considered an mMRC less than or equal to 1 or CAT less than or equal to 9, high symptom severity is considered an mMRC greater than or equal to 2 or CAT greater than or equal to 10. Low risk of exacerbation is defined as no more than one exacerbation not resulting in hospital admission in the last 12 months; high risk of exacerbation is defined as at least two exacerbations or any exacerbations resulting in hospital admission in the last 12 months. Symptom severity and exacerbation risk is divided into four quadrants:
• GOLD group A: Low symptom severity, low exacerbation risk.
• GOLD group B: High symptom severity, low exacerbation risk.
• GOLD group C: Low symptom severity, high exacerbation risk.
• GOLD group D: High symptom severity, high exacerbation risk.
Changes to prevention and management of stable COPD
Smoking cessation remains important in the prevention of COPD. The 2017 Report reflects the U.S. Preventive Services Task Force’s guidelines for smoking cessation: Offer nicotine replacement, cessation counseling, and pharmacotherapy (varenicline, bupropion or nortriptyline). There is insufficient evidence to support the use of e-cigarettes. Influenza and pneumococcal vaccinations are recommended. Pulmonary rehabilitation remains important.
The 2017 Report includes an expanded discussion of COPD medications. The role of short-acting bronchodilators (SABD) in COPD remains prominent. Changes include a stronger recommendation to use combination short-acting beta-agonists and short-acting muscarinic antagonists (SABA/SAMA) as these seem to be superior to SABD monotherapy in improving symptoms and FEV1.
There were several changes to the pharmacologic treatment algorithm. For the first time, GOLD proposes escalation strategies. Preference is given to LABA/LAMA (long-acting beta-agonist/long-acting muscarinic antagonists) combinations over LABA/ICS (long-acting beta-agonist/inhaled corticosteroid) combinations as a mainstay of treatment. The rationale for this change is that LABA/LAMAs give greater bronchodilation compared with LABA/ICS, and one study showed a decreased rate of exacerbations compared to LABA/ICS in patients with a history of exacerbations. In addition, patients with COPD who receive ICS appear to have a higher risk of developing pneumonia. GOLD recommendations are:
• Group A: Start with single bronchodilator (short- or long-acting), escalate to alternative class of bronchodilator if necessary.
• Group B: Start with LABA or LAMA, escalate to LABA/LAMA if symptoms persist.
• Group C: Start with LAMA, escalate to LABA/LAMA (preferred) or LABA/ICS if exacerbations continue.
• Group D: Start with LABA/LAMA (preferred) or LAMA monotherapy, escalate to LABA/LAMA/ICS (preferred) or try LABA/ICS before escalating to LAMA/LABA/ICS if symptoms persist or exacerbations continue; roflumilast and/or a macrolide may be considered if further exacerbations occur with LABA/LAMA/ICS.
Bottom line
1. GOLD classification of COPD severity is now based on clinical criteria alone: symptom assessment and risk for exacerbation.
2. SABA/SAMA combination therapy seems to be superior to either SABA or SAMA alone.
3. Patients in group A (milder symptoms, low exacerbation risk) may be initiated on either short- or long-acting bronchodilator therapy.
4. Patients in group B (milder symptoms, increased exacerbation risk) should be initiated on LAMA monotherapy.
5. LABA/LAMA combination therapy seems to be superior to LABA/ICS combination therapy and should be used when long-acting bronchodilator monotherapy fails to control symptoms or reduce exacerbations.
References
1. CDC MMWR 11/23/12
2. Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2017 at http://goldcopd.org (accessed 3/10/2017)
Dr. Skolnik is associate director of the family medicine residency program at Abington (Pa.) Memorial Hospital. Dr. Lent is chief resident in the program.
Chronic obstructive lung disease (COPD) is the third leading cause of death in the United States1 and a major cause of mortality and morbidity around the world. The Global Initiative for Chronic Obstructive Lung Disease (GOLD) released a new “2017 Report”2 with modified recommendations for the diagnosis, management, and prevention of COPD. The report contains several changes that are relevant to the primary care provider that will be outlined below.
Redefining COPD
GOLD’s definition of COPD was changed in its 2017 Report: “COPD is a common, preventable, and treatable disease that is characterized by persistent respiratory symptoms and airflow limitations that are due to airway and/or alveolar abnormalities usually caused by significant exposure to noxious particles or gases.” The report emphasizes that “COPD may be punctuated by periods of acute worsening of respiratory symptoms, called exacerbations.” Note that the terms “emphysema” and “chronic bronchitis” have been removed in favor of a more comprehensive description of the pathophysiology of COPD. Importantly, the report states that cough and sputum production for at least 3 months in each of 2 consecutive years, previously accepted as diagnostic criteria, are present in only a minority of patients. It is noted that chronic respiratory symptoms may exist without spirometric changes and many patients (usually smokers) have structural evidence of COPD without airflow limitation.
Changes to COPD initial assessment
The primary criterion for diagnosis is unchanged: post-bronchodilator forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) less than 0.70. Spirometry remains important to confirm the diagnosis in those with classic symptoms of dyspnea, chronic cough, and/or sputum production with a history of exposure to noxious particles or gases.
The GOLD assessment system previously incorporated spirometry and included an “ABCD” system such that patients in group A are least severe. Spirometry has been progressively deemphasized in favor of symptom-based classification and the 2017 Report, for the first time, dissociates spirometric findings from severity classification.
The new system uses symptom severity and exacerbation risk to classify COPD. Two specific standardized COPD symptom measurement tools, The Modified British Medical Research Council (mMRC) questionnaire and COPD Assessment Test (CAT), are reported by GOLD as the most widely used. Low symptom severity is considered an mMRC less than or equal to 1 or CAT less than or equal to 9, high symptom severity is considered an mMRC greater than or equal to 2 or CAT greater than or equal to 10. Low risk of exacerbation is defined as no more than one exacerbation not resulting in hospital admission in the last 12 months; high risk of exacerbation is defined as at least two exacerbations or any exacerbations resulting in hospital admission in the last 12 months. Symptom severity and exacerbation risk is divided into four quadrants:
• GOLD group A: Low symptom severity, low exacerbation risk.
• GOLD group B: High symptom severity, low exacerbation risk.
• GOLD group C: Low symptom severity, high exacerbation risk.
• GOLD group D: High symptom severity, high exacerbation risk.
Changes to prevention and management of stable COPD
Smoking cessation remains important in the prevention of COPD. The 2017 Report reflects the U.S. Preventive Services Task Force’s guidelines for smoking cessation: Offer nicotine replacement, cessation counseling, and pharmacotherapy (varenicline, bupropion or nortriptyline). There is insufficient evidence to support the use of e-cigarettes. Influenza and pneumococcal vaccinations are recommended. Pulmonary rehabilitation remains important.
The 2017 Report includes an expanded discussion of COPD medications. The role of short-acting bronchodilators (SABD) in COPD remains prominent. Changes include a stronger recommendation to use combination short-acting beta-agonists and short-acting muscarinic antagonists (SABA/SAMA) as these seem to be superior to SABD monotherapy in improving symptoms and FEV1.
There were several changes to the pharmacologic treatment algorithm. For the first time, GOLD proposes escalation strategies. Preference is given to LABA/LAMA (long-acting beta-agonist/long-acting muscarinic antagonists) combinations over LABA/ICS (long-acting beta-agonist/inhaled corticosteroid) combinations as a mainstay of treatment. The rationale for this change is that LABA/LAMAs give greater bronchodilation compared with LABA/ICS, and one study showed a decreased rate of exacerbations compared to LABA/ICS in patients with a history of exacerbations. In addition, patients with COPD who receive ICS appear to have a higher risk of developing pneumonia. GOLD recommendations are:
• Group A: Start with single bronchodilator (short- or long-acting), escalate to alternative class of bronchodilator if necessary.
• Group B: Start with LABA or LAMA, escalate to LABA/LAMA if symptoms persist.
• Group C: Start with LAMA, escalate to LABA/LAMA (preferred) or LABA/ICS if exacerbations continue.
• Group D: Start with LABA/LAMA (preferred) or LAMA monotherapy, escalate to LABA/LAMA/ICS (preferred) or try LABA/ICS before escalating to LAMA/LABA/ICS if symptoms persist or exacerbations continue; roflumilast and/or a macrolide may be considered if further exacerbations occur with LABA/LAMA/ICS.
Bottom line
1. GOLD classification of COPD severity is now based on clinical criteria alone: symptom assessment and risk for exacerbation.
2. SABA/SAMA combination therapy seems to be superior to either SABA or SAMA alone.
3. Patients in group A (milder symptoms, low exacerbation risk) may be initiated on either short- or long-acting bronchodilator therapy.
4. Patients in group B (milder symptoms, increased exacerbation risk) should be initiated on LAMA monotherapy.
5. LABA/LAMA combination therapy seems to be superior to LABA/ICS combination therapy and should be used when long-acting bronchodilator monotherapy fails to control symptoms or reduce exacerbations.
References
1. CDC MMWR 11/23/12
2. Global Strategy for the Diagnosis, Management and Prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2017 at http://goldcopd.org (accessed 3/10/2017)
Dr. Skolnik is associate director of the family medicine residency program at Abington (Pa.) Memorial Hospital. Dr. Lent is chief resident in the program.
ADHD medication may lower risk of motor vehicle crashes
Men with ADHD had a 38% lower risk of motor vehicle crashes (MVCs) when receiving ADHD medication, compared with months off medication. Women had a 42% lower risk, according to the results of a U.S. study.
Estimates suggested that up to 22% of MVCs in patients with ADHD could have been avoided if they had received medication during the whole length of the study, reported Zheng Chang, PhD, of the Karolinska Institutet, Sweden, and his colleagues (JAMA Psychiatry. 2017 May 10. doi: 10.1001/jamapsychiatry.2017.0659)
“This study is the first, to date, to demonstrate a long-term association between receiving ADHD medication and decreased MVCs,” said Dr. Chang and his associates. If this result demonstrates a protective effect, it is possible that continuous ADHD medication use might lead to lower risk of other problems, such as substance abuse disorder, or provide long-term improvements in life functioning for people with ADHD.
This study was supported by grants from the Swedish Research Council and the National Institute of Mental Health, as well as grants to two of the researchers from the Swedish Research Council for Health, Working Life and Welfare, and the National Institute on Drug Abuse. Dr. Chang and the other researchers had no relevant financial disclosures, except for Henrik Larsson, PhD, who received some speaker’s fees and research grants from pharmaceutical companies outside this work.
Prescribing medication to ADHD patients does not guarantee they will take it. Therefore, there is a chance that some of the motor vehicle crashes that occurred during a month when a patient reportedly was on medication may have occurred on a day when the patient had not actually taken medication. Also, using ED visits to measure the number of MVCs has a major drawback: vehicular accidents do not necessarily result in ED visits. Therefore, the study by Chang et al. may not accurately report the benefits of ADHD medication on safe driving.
Management of ADHD is not limited to school or the workplace but extends to other aspects of life, such as driving, which clinicians must consider when prescribing. It also is important to keep in mind, while prescribing, that the progression of ADHD often involves a decrease in hyperactivity during adulthood, while inattention and impulsivity may continue, and that the latter two traits can lead to distracted driving. Another important variable is that MVCs involving individuals with ADHD often happen later in the evening, when their medications may have worn off.
Customizing and improving ADHD pharmacotherapy, while being mindful of effects, is the most sensible way forward.
Vishal Madaan, MD, and Daniel J. Cox, PhD, are at the University of Virginia Health System in Charlottesville. Dr. Madaan reported receiving research support from Forest, Purdue, Aevi Genomic Medicine (formerly Medgenics), Sunovion, and Pfizer, as well as receiving royalties from Taylor & Francis. Dr. Cox reported receiving research support from the National Institutes of Health, Purdue, Johnson & Johnson, and Dexcom. They made these remarks in a commentary accompanying the study by Dr. Chang et al. (JAMA Psychiatry. 2017 May 10. doi: 10.1001/jamapsychiatry.2017.0659).
Prescribing medication to ADHD patients does not guarantee they will take it. Therefore, there is a chance that some of the motor vehicle crashes that occurred during a month when a patient reportedly was on medication may have occurred on a day when the patient had not actually taken medication. Also, using ED visits to measure the number of MVCs has a major drawback: vehicular accidents do not necessarily result in ED visits. Therefore, the study by Chang et al. may not accurately report the benefits of ADHD medication on safe driving.
Management of ADHD is not limited to school or the workplace but extends to other aspects of life, such as driving, which clinicians must consider when prescribing. It also is important to keep in mind, while prescribing, that the progression of ADHD often involves a decrease in hyperactivity during adulthood, while inattention and impulsivity may continue, and that the latter two traits can lead to distracted driving. Another important variable is that MVCs involving individuals with ADHD often happen later in the evening, when their medications may have worn off.
Customizing and improving ADHD pharmacotherapy, while being mindful of effects, is the most sensible way forward.
Vishal Madaan, MD, and Daniel J. Cox, PhD, are at the University of Virginia Health System in Charlottesville. Dr. Madaan reported receiving research support from Forest, Purdue, Aevi Genomic Medicine (formerly Medgenics), Sunovion, and Pfizer, as well as receiving royalties from Taylor & Francis. Dr. Cox reported receiving research support from the National Institutes of Health, Purdue, Johnson & Johnson, and Dexcom. They made these remarks in a commentary accompanying the study by Dr. Chang et al. (JAMA Psychiatry. 2017 May 10. doi: 10.1001/jamapsychiatry.2017.0659).
Prescribing medication to ADHD patients does not guarantee they will take it. Therefore, there is a chance that some of the motor vehicle crashes that occurred during a month when a patient reportedly was on medication may have occurred on a day when the patient had not actually taken medication. Also, using ED visits to measure the number of MVCs has a major drawback: vehicular accidents do not necessarily result in ED visits. Therefore, the study by Chang et al. may not accurately report the benefits of ADHD medication on safe driving.
Management of ADHD is not limited to school or the workplace but extends to other aspects of life, such as driving, which clinicians must consider when prescribing. It also is important to keep in mind, while prescribing, that the progression of ADHD often involves a decrease in hyperactivity during adulthood, while inattention and impulsivity may continue, and that the latter two traits can lead to distracted driving. Another important variable is that MVCs involving individuals with ADHD often happen later in the evening, when their medications may have worn off.
Customizing and improving ADHD pharmacotherapy, while being mindful of effects, is the most sensible way forward.
Vishal Madaan, MD, and Daniel J. Cox, PhD, are at the University of Virginia Health System in Charlottesville. Dr. Madaan reported receiving research support from Forest, Purdue, Aevi Genomic Medicine (formerly Medgenics), Sunovion, and Pfizer, as well as receiving royalties from Taylor & Francis. Dr. Cox reported receiving research support from the National Institutes of Health, Purdue, Johnson & Johnson, and Dexcom. They made these remarks in a commentary accompanying the study by Dr. Chang et al. (JAMA Psychiatry. 2017 May 10. doi: 10.1001/jamapsychiatry.2017.0659).
Men with ADHD had a 38% lower risk of motor vehicle crashes (MVCs) when receiving ADHD medication, compared with months off medication. Women had a 42% lower risk, according to the results of a U.S. study.
Estimates suggested that up to 22% of MVCs in patients with ADHD could have been avoided if they had received medication during the whole length of the study, reported Zheng Chang, PhD, of the Karolinska Institutet, Sweden, and his colleagues (JAMA Psychiatry. 2017 May 10. doi: 10.1001/jamapsychiatry.2017.0659)
“This study is the first, to date, to demonstrate a long-term association between receiving ADHD medication and decreased MVCs,” said Dr. Chang and his associates. If this result demonstrates a protective effect, it is possible that continuous ADHD medication use might lead to lower risk of other problems, such as substance abuse disorder, or provide long-term improvements in life functioning for people with ADHD.
This study was supported by grants from the Swedish Research Council and the National Institute of Mental Health, as well as grants to two of the researchers from the Swedish Research Council for Health, Working Life and Welfare, and the National Institute on Drug Abuse. Dr. Chang and the other researchers had no relevant financial disclosures, except for Henrik Larsson, PhD, who received some speaker’s fees and research grants from pharmaceutical companies outside this work.
Men with ADHD had a 38% lower risk of motor vehicle crashes (MVCs) when receiving ADHD medication, compared with months off medication. Women had a 42% lower risk, according to the results of a U.S. study.
Estimates suggested that up to 22% of MVCs in patients with ADHD could have been avoided if they had received medication during the whole length of the study, reported Zheng Chang, PhD, of the Karolinska Institutet, Sweden, and his colleagues (JAMA Psychiatry. 2017 May 10. doi: 10.1001/jamapsychiatry.2017.0659)
“This study is the first, to date, to demonstrate a long-term association between receiving ADHD medication and decreased MVCs,” said Dr. Chang and his associates. If this result demonstrates a protective effect, it is possible that continuous ADHD medication use might lead to lower risk of other problems, such as substance abuse disorder, or provide long-term improvements in life functioning for people with ADHD.
This study was supported by grants from the Swedish Research Council and the National Institute of Mental Health, as well as grants to two of the researchers from the Swedish Research Council for Health, Working Life and Welfare, and the National Institute on Drug Abuse. Dr. Chang and the other researchers had no relevant financial disclosures, except for Henrik Larsson, PhD, who received some speaker’s fees and research grants from pharmaceutical companies outside this work.
FROM JAMA PSYCHIATRY
Key clinical point:
Major finding: Patients with ADHD have 22% less risk for motor vehicle crashes when they are on medication.
Data source: Data were gathered from commercial insurance claims of a national cohort of 2,319,450 patients with ADHD and ED visits for motor vehicle crashes.
Disclosures: This study was supported by grants from the Swedish Research Council and the National Institute of Mental Health, as well grants to two of the researchers from the Swedish Research Council for Health, Working Life and Welfare, and the National Institute on Drug Abuse. Dr. Chang and the other researchers had no relevant financial disclosures, except for Dr. Larsson who received some speaker’s fees and research grants from pharmaceutical companies outside this work.
Myeloma patients who get solid tumor cancers do as well as other cancer patients
With improved treatment, patients with multiple myeloma are surviving long enough to develop other cancers, Jorge J. Castillo, MD, and Adam J. Olszewski, MD, reported in a poster to be presented at the annual meeting of the American Society of Clinical Oncology.
The good news is that myeloma patients, when diagnosed with a subsequent solid tumor, are just as likely to respond to treatment and do just as well as patients without myeloma, according to Dr. Castillo of Dana-Farber Cancer Institute, Boston, and Dr. Olszewski of Alpert Medical School of Brown University, Providence, R.I.

They based their conclusion on Surveillance, Epidemiology, and End Results (SEER) data for patients diagnosed with six common cancers from 2004-2013.
“Among them, we identified [nearly 1,300] myeloma survivors, and we matched each to 50 randomly sampled controls with the same cancer by age, sex, race, and year of diagnosis. We then compared [cancer specific survival], cumulative incidence function (CIF) for death from the non-myeloma index cancer, and whether patients had surgery for non-metastastic, stage-matched tumors only,” the researchers wrote in their abstract.
They did analyses for breast, lung, prostate, colorectal, melanoma, and bladder cancers. The median time from diagnosis of myeloma to diagnosis of the second ranged from 35 months (bladder [133 myeloma patients] and lung [286 myeloma patients] cancers) to 50 months (melanoma [140 myeloma patients]). The median time after myeloma diagnosis was 40 months for those patients who developed breast, prostate, or colorectal cancers.
In the comparisons, myeloma survivors were significantly older (P less than .001) than patients initially diagnosed with the same respective cancers. In the case-control analysis, breast (P = .002) and lung cancers (P = .003) were more often diagnosed at an early stage among myeloma survivors.
The hazard ratio (HR) for cancer-specific survival for 189 myeloma patients diagnosed with breast cancer as compared to other breast cancer patients, for example, was 0.99, 95% confidence interval (CI) 0.61-1.61. The HR for the cumulative incidence function of cancer death was 0.82, 95% CI 0.50-1.35.
Myeloma patients were no less likely than were case-control subjects to have surgery for their cancers, with the exception of the 330 myeloma patients who developed prostate cancer (odds ratio, 0.59, 95% CI, 0.44-0.81).
Cancer-specific survival significantly differed (P less than .05) only for lung cancer, and was better among the 286 myeloma patients with lung cancer even when stratified by stage (HR 0.64, 95% CI 0.54-0.75). For cumulative incidence function of cancer death for lung cancer, the hazard ratio was 0.52 (95% CI 0.44-0.61). Better outcomes in lung cancer are not fully explained by earlier detection, suggesting a biological difference, the researchers reported.
Cumulative incidence function of cancer death was significantly lower for myeloma patients with lung and colorectal cancers.
Dr. Castillo disclosed honoraria from Celgene and Janssen; a consulting or advisory role with Biogen, Otsuka, and Pharmacyclics; and institutional research funding from Abbvie, Gilead Sciences, Millennium, and Pharmacyclics. Dr. Olszewski disclosed institutional research funding from Genentech, Incyte, and TG Therapeutics.
Citation: Outcomes of secondary cancers among myeloma survivors. 2017 ASCO annual meeting. Abstract No. 8043.
[email protected]
On Twitter @maryjodales
With improved treatment, patients with multiple myeloma are surviving long enough to develop other cancers, Jorge J. Castillo, MD, and Adam J. Olszewski, MD, reported in a poster to be presented at the annual meeting of the American Society of Clinical Oncology.
The good news is that myeloma patients, when diagnosed with a subsequent solid tumor, are just as likely to respond to treatment and do just as well as patients without myeloma, according to Dr. Castillo of Dana-Farber Cancer Institute, Boston, and Dr. Olszewski of Alpert Medical School of Brown University, Providence, R.I.
They based their conclusion on Surveillance, Epidemiology, and End Results (SEER) data for patients diagnosed with six common cancers from 2004-2013.
“Among them, we identified [nearly 1,300] myeloma survivors, and we matched each to 50 randomly sampled controls with the same cancer by age, sex, race, and year of diagnosis. We then compared [cancer specific survival], cumulative incidence function (CIF) for death from the non-myeloma index cancer, and whether patients had surgery for non-metastastic, stage-matched tumors only,” the researchers wrote in their abstract.
They did analyses for breast, lung, prostate, colorectal, melanoma, and bladder cancers. The median time from diagnosis of myeloma to diagnosis of the second ranged from 35 months (bladder [133 myeloma patients] and lung [286 myeloma patients] cancers) to 50 months (melanoma [140 myeloma patients]). The median time after myeloma diagnosis was 40 months for those patients who developed breast, prostate, or colorectal cancers.
In the comparisons, myeloma survivors were significantly older (P less than .001) than patients initially diagnosed with the same respective cancers. In the case-control analysis, breast (P = .002) and lung cancers (P = .003) were more often diagnosed at an early stage among myeloma survivors.
The hazard ratio (HR) for cancer-specific survival for 189 myeloma patients diagnosed with breast cancer as compared to other breast cancer patients, for example, was 0.99, 95% confidence interval (CI) 0.61-1.61. The HR for the cumulative incidence function of cancer death was 0.82, 95% CI 0.50-1.35.
Myeloma patients were no less likely than were case-control subjects to have surgery for their cancers, with the exception of the 330 myeloma patients who developed prostate cancer (odds ratio, 0.59, 95% CI, 0.44-0.81).
Cancer-specific survival significantly differed (P less than .05) only for lung cancer, and was better among the 286 myeloma patients with lung cancer even when stratified by stage (HR 0.64, 95% CI 0.54-0.75). For cumulative incidence function of cancer death for lung cancer, the hazard ratio was 0.52 (95% CI 0.44-0.61). Better outcomes in lung cancer are not fully explained by earlier detection, suggesting a biological difference, the researchers reported.
Cumulative incidence function of cancer death was significantly lower for myeloma patients with lung and colorectal cancers.
Dr. Castillo disclosed honoraria from Celgene and Janssen; a consulting or advisory role with Biogen, Otsuka, and Pharmacyclics; and institutional research funding from Abbvie, Gilead Sciences, Millennium, and Pharmacyclics. Dr. Olszewski disclosed institutional research funding from Genentech, Incyte, and TG Therapeutics.
Citation: Outcomes of secondary cancers among myeloma survivors. 2017 ASCO annual meeting. Abstract No. 8043.
[email protected]
On Twitter @maryjodales
With improved treatment, patients with multiple myeloma are surviving long enough to develop other cancers, Jorge J. Castillo, MD, and Adam J. Olszewski, MD, reported in a poster to be presented at the annual meeting of the American Society of Clinical Oncology.
The good news is that myeloma patients, when diagnosed with a subsequent solid tumor, are just as likely to respond to treatment and do just as well as patients without myeloma, according to Dr. Castillo of Dana-Farber Cancer Institute, Boston, and Dr. Olszewski of Alpert Medical School of Brown University, Providence, R.I.
They based their conclusion on Surveillance, Epidemiology, and End Results (SEER) data for patients diagnosed with six common cancers from 2004-2013.
“Among them, we identified [nearly 1,300] myeloma survivors, and we matched each to 50 randomly sampled controls with the same cancer by age, sex, race, and year of diagnosis. We then compared [cancer specific survival], cumulative incidence function (CIF) for death from the non-myeloma index cancer, and whether patients had surgery for non-metastastic, stage-matched tumors only,” the researchers wrote in their abstract.
They did analyses for breast, lung, prostate, colorectal, melanoma, and bladder cancers. The median time from diagnosis of myeloma to diagnosis of the second ranged from 35 months (bladder [133 myeloma patients] and lung [286 myeloma patients] cancers) to 50 months (melanoma [140 myeloma patients]). The median time after myeloma diagnosis was 40 months for those patients who developed breast, prostate, or colorectal cancers.
In the comparisons, myeloma survivors were significantly older (P less than .001) than patients initially diagnosed with the same respective cancers. In the case-control analysis, breast (P = .002) and lung cancers (P = .003) were more often diagnosed at an early stage among myeloma survivors.
The hazard ratio (HR) for cancer-specific survival for 189 myeloma patients diagnosed with breast cancer as compared to other breast cancer patients, for example, was 0.99, 95% confidence interval (CI) 0.61-1.61. The HR for the cumulative incidence function of cancer death was 0.82, 95% CI 0.50-1.35.
Myeloma patients were no less likely than were case-control subjects to have surgery for their cancers, with the exception of the 330 myeloma patients who developed prostate cancer (odds ratio, 0.59, 95% CI, 0.44-0.81).
Cancer-specific survival significantly differed (P less than .05) only for lung cancer, and was better among the 286 myeloma patients with lung cancer even when stratified by stage (HR 0.64, 95% CI 0.54-0.75). For cumulative incidence function of cancer death for lung cancer, the hazard ratio was 0.52 (95% CI 0.44-0.61). Better outcomes in lung cancer are not fully explained by earlier detection, suggesting a biological difference, the researchers reported.
Cumulative incidence function of cancer death was significantly lower for myeloma patients with lung and colorectal cancers.
Dr. Castillo disclosed honoraria from Celgene and Janssen; a consulting or advisory role with Biogen, Otsuka, and Pharmacyclics; and institutional research funding from Abbvie, Gilead Sciences, Millennium, and Pharmacyclics. Dr. Olszewski disclosed institutional research funding from Genentech, Incyte, and TG Therapeutics.
Citation: Outcomes of secondary cancers among myeloma survivors. 2017 ASCO annual meeting. Abstract No. 8043.
[email protected]
On Twitter @maryjodales
FROM 2017 ASCO ANNUAL MEETING
Key clinical point:
Major finding: Cancer-specific survival significantly differed (P less than .05) only for lung cancer, and was better among the 286 myeloma patients with lung cancer even when stratified by stage (HR, 0.64, 95% CI 0.54-0.75).
Data source: Surveillance, Epidemiology, and End Results (SEER) data for patients diagnosed with six common cancers from 2004-2013.
Disclosures: Dr. Castillo disclosed honoraria from Celgene and Janssen; a consulting or advisory role with Biogen, Otsuka, and Pharmacyclics; and institutional research funding from Abbvie, Gilead Sciences, Millennium, and Pharmacyclics. Dr. Olszewski disclosed institutional research funding from Genentech, Incyte, and TG Therapeutics.
Citation: Outcomes of secondary cancers among myeloma survivors. 2017 ASCO annual meeting. Abstract No. 8043.
HM17 session summary: Focus on POCUS – Introduction to Point-of-Care Ultrasound for pediatric hospitalists
Presenters
Nilam Soni, MD, FHM; Thomas Conlon, MD; Ria Dancel, MD, FAAP, FHM; Daniel Schnobrich, MD
Summary
Point-of-care ultrasound (POCUS) is rapidly gaining acceptance in the medical community as a goal-directed examination that answers a specific diagnostic question or guides a bedside invasive procedure. Adoption by pediatric hospitalists is increasing, aided by multiple training pathways, opportunities for scholarship, and organization development.
The use of POCUS is increasing among nonradiologist physicians due to the expectation for perfection, desire for improved patient experience, and increased availability of ultrasound machines. POCUS is rapid and safe, and can be used serially to monitor, provide procedural guidance, and lead to initiation of appropriate therapies.

Training in POCUS in limited applications is possible in short periods of time. One recent study showed that approximately 40% of POCUS cases led to new findings or alteration of treatment. However, POCUS requires training, monitoring for competence, transparency of training/competence, and a QA process that supports the training. One solution at Children’s Hospital of Philadelphia was to use American College of Emergency Physician guidelines for POCUS training.
Pediatric applications include guidance of bladder catheterization, identifying occult abscesses, diagnosis of pneumonia and associated parapneumonic effusion, and IV placement. More advanced applications include diagnosis of appendicitis, intussusception, and increased intracranial pressure. Novel applications conceived by nonradiologist physicians have included sinus ultrasound.
Initial training can be provided by “in-house experts,” such as pediatric ED physicians and PICU physicians. Alternatively, an on-site commercial course can be arranged for larger groups. Consideration should be given to mentorship, with comparison to formal imaging and/or clinical progression. Relationships with traditional imagers should be cultivated, as POCUS can potentially be misunderstood. In fact, formal US utilization has been found to increase once clinicals begin to use POCUS.
Key takeaways for HM
- Point-of-care ultrasound (POCUS) is rapidly being adopted by pediatric hospitalists.
- Pediatric applications are still being developed, but include guidance of bladder catheterization, identifying occult abscesses, diagnosis of pneumonia/associated effusions, and IV placement.
- Initial training can be provided by pediatric ED physicians/PICU physicians or an on-site commercial course can be arranged for larger groups.
- Relationships with radiologists should be established at the outset to avoid misunderstanding of POCUS.
Dr. Chang is a pediatric hospitalist at Baystate Children’s Hospital and is the pediatric editor of The Hospitalist.
Presenters
Nilam Soni, MD, FHM; Thomas Conlon, MD; Ria Dancel, MD, FAAP, FHM; Daniel Schnobrich, MD
Summary
Point-of-care ultrasound (POCUS) is rapidly gaining acceptance in the medical community as a goal-directed examination that answers a specific diagnostic question or guides a bedside invasive procedure. Adoption by pediatric hospitalists is increasing, aided by multiple training pathways, opportunities for scholarship, and organization development.
The use of POCUS is increasing among nonradiologist physicians due to the expectation for perfection, desire for improved patient experience, and increased availability of ultrasound machines. POCUS is rapid and safe, and can be used serially to monitor, provide procedural guidance, and lead to initiation of appropriate therapies.
Training in POCUS in limited applications is possible in short periods of time. One recent study showed that approximately 40% of POCUS cases led to new findings or alteration of treatment. However, POCUS requires training, monitoring for competence, transparency of training/competence, and a QA process that supports the training. One solution at Children’s Hospital of Philadelphia was to use American College of Emergency Physician guidelines for POCUS training.
Pediatric applications include guidance of bladder catheterization, identifying occult abscesses, diagnosis of pneumonia and associated parapneumonic effusion, and IV placement. More advanced applications include diagnosis of appendicitis, intussusception, and increased intracranial pressure. Novel applications conceived by nonradiologist physicians have included sinus ultrasound.
Initial training can be provided by “in-house experts,” such as pediatric ED physicians and PICU physicians. Alternatively, an on-site commercial course can be arranged for larger groups. Consideration should be given to mentorship, with comparison to formal imaging and/or clinical progression. Relationships with traditional imagers should be cultivated, as POCUS can potentially be misunderstood. In fact, formal US utilization has been found to increase once clinicals begin to use POCUS.
Key takeaways for HM
- Point-of-care ultrasound (POCUS) is rapidly being adopted by pediatric hospitalists.
- Pediatric applications are still being developed, but include guidance of bladder catheterization, identifying occult abscesses, diagnosis of pneumonia/associated effusions, and IV placement.
- Initial training can be provided by pediatric ED physicians/PICU physicians or an on-site commercial course can be arranged for larger groups.
- Relationships with radiologists should be established at the outset to avoid misunderstanding of POCUS.
Dr. Chang is a pediatric hospitalist at Baystate Children’s Hospital and is the pediatric editor of The Hospitalist.
Presenters
Nilam Soni, MD, FHM; Thomas Conlon, MD; Ria Dancel, MD, FAAP, FHM; Daniel Schnobrich, MD
Summary
Point-of-care ultrasound (POCUS) is rapidly gaining acceptance in the medical community as a goal-directed examination that answers a specific diagnostic question or guides a bedside invasive procedure. Adoption by pediatric hospitalists is increasing, aided by multiple training pathways, opportunities for scholarship, and organization development.
The use of POCUS is increasing among nonradiologist physicians due to the expectation for perfection, desire for improved patient experience, and increased availability of ultrasound machines. POCUS is rapid and safe, and can be used serially to monitor, provide procedural guidance, and lead to initiation of appropriate therapies.
Training in POCUS in limited applications is possible in short periods of time. One recent study showed that approximately 40% of POCUS cases led to new findings or alteration of treatment. However, POCUS requires training, monitoring for competence, transparency of training/competence, and a QA process that supports the training. One solution at Children’s Hospital of Philadelphia was to use American College of Emergency Physician guidelines for POCUS training.
Pediatric applications include guidance of bladder catheterization, identifying occult abscesses, diagnosis of pneumonia and associated parapneumonic effusion, and IV placement. More advanced applications include diagnosis of appendicitis, intussusception, and increased intracranial pressure. Novel applications conceived by nonradiologist physicians have included sinus ultrasound.
Initial training can be provided by “in-house experts,” such as pediatric ED physicians and PICU physicians. Alternatively, an on-site commercial course can be arranged for larger groups. Consideration should be given to mentorship, with comparison to formal imaging and/or clinical progression. Relationships with traditional imagers should be cultivated, as POCUS can potentially be misunderstood. In fact, formal US utilization has been found to increase once clinicals begin to use POCUS.
Key takeaways for HM
- Point-of-care ultrasound (POCUS) is rapidly being adopted by pediatric hospitalists.
- Pediatric applications are still being developed, but include guidance of bladder catheterization, identifying occult abscesses, diagnosis of pneumonia/associated effusions, and IV placement.
- Initial training can be provided by pediatric ED physicians/PICU physicians or an on-site commercial course can be arranged for larger groups.
- Relationships with radiologists should be established at the outset to avoid misunderstanding of POCUS.
Dr. Chang is a pediatric hospitalist at Baystate Children’s Hospital and is the pediatric editor of The Hospitalist.
Omitting ALND in some breast cancer patients may be the right choice
PHILADELPHIA – The safety of sentinel lymph node biopsy (SLNB) alone without axillary lymph node dissection (ALND) has been established for patients with cT1-2N0 cancer that are found to have one or two metastatic sentinel lymph nodes who undergo breast conservation therapy, but questions regarding the role of regional radiation have persisted.
This issue is addressed by the results of a large, prospective, 5+ year study at Memorial Sloan Kettering Cancer Center which confirmed the safety of omitting axillary lymph node dissection and suggested that regional radiation provides minimal benefit.

Dr. Morrow explained that, in August 2010, the breast surgery service at MSKCC adopted the guidelines that arose from the American College of Surgeons Oncology Group’s multicenter Z0011 trial and abandoned routine use of ALND in eligible patients. The goal of the study, she reported, was to determine how frequently axillary dissection was avoided in a consecutive, otherwise unselected, series of patients and to determine the incidence of local regional recurrence after SLNB alone in a population treated with known radiotherapy fields.
Eligible subjects had T1 or T2 node-negative breast cancer, were undergoing breast-conserving surgery with planned whole-breast irradiation, and were found to have hematoxylin-eosin-detected sentinel node metastases. Patients receiving neoadjuvant chemotherapy or requiring conversion to mastectomy, or those in whom partial breast irradiation or no radiotherapy was planned, were ineligible. Axillary imaging was not used in select patients. Criteria for axillary dissection were metastases in three or more sentinel nodes or the presence of matted nodes identified intraoperatively. The researchers did not use the MSKCC nomogram to predict the likelihood of non–sentinel node metastases.
Median patient age was 58 years and median tumor size 1.7 cm. With regard to tumor pathology, 87% had infiltrating ductal tumors, 94% had grade 2 or 3 disease, and the most common subtype was HR+, HER2– disease in 84%. “In this node-positive cohort of patients, 98% received adjuvant systemic therapy, most commonly both chemotherapy and endocrine therapy (received by 65%), and 93% completed radiotherapy,” Dr. Morrow said.
In the entire patient cohort, 84% (663) were treated with SLNB alone, Dr. Morrow said. Among the 130 patients requiring ALND, 68% (88) had metastases in three or more nodes, 26% (34) were found to have had matted nodes intraoperatively, and 6% (8) were eligible for SLNB alone but opted for ALND or had it recommended by their surgeon. “All of these occurred early in our experience, and this has not been repeated since,” Dr. Morrow said.
Among the SLNB-only patients, the 5-year event-free survival was 93%. “There were no isolated axillary recurrences,” Dr. Morrow said. The study reported four combined breast and axillary recurrences, three in nonradiated patients, and four combined nodal and distant recurrences, only one of which involved the axillary nodes. “The median time to any nodal recurrence was 25 months,” Dr. Morrow added. Among 484 patients who had 1 year or more of follow-up, 58% (280) received conventional supine breast tangents, 21% were treated prone – “meaning their axilla received essentially no radiotherapy,” Dr. Morrow said – and 21% had node field irradiation.
“If we compare patient characteristics based on radiotherapy fields treated, it’s clear that the patients who received nodal irradiation were a higher-risk group,” Dr. Morrow said. While all three groups had a median of one positive sentinel node, that “skewed towards two” in the nodal irradiation group, she said. This group also had higher rates of lymphovascular invasion (72% vs. 56% and 49% in the supine and prone groups, respectively) and extracapsular extension (41% vs. 31% and 25%).
The rates of nodal relapse were not statistically significant among the three groups: 1% in the prone group, 1.4% in the supine group, and 0% in the node irradiation group.
“Factors associated with a higher risk of distant metastases, such as young patient age, estrogen receptor negativity, or HER2 over-expression, were not associated with the need for axillary dissection and should not be used as priority selection criteria,” Dr. Morrow said. “Nodal recurrence was uncommon in the absence of routine nodal radiation therapy, and no isolated nodal failures were observed.
In his comments, Armando Giuliano, MD, of Cedars Sinai Medical Center in Los Angeles, principal investigator of the Z0011 trial, said the MSKCC study “extends and informs” the Z0011 findings. He noted that the prone treatment group in the MSKCC trial had a low rate of axillary recurrence. “Can you speculate how such excellent results are achieved without resection or irradiation?” he asked Dr. Morrow. “To me it appears that nodal irradiation provides very little benefit to this selected group of patients.”
The patients in the prone group were in the lowest-risk category of the study, Dr. Morrow said, but the fact that not all nodal disease becomes clinically evident, even in patients who do not receive radiotherapy or systemic therapy, along with the high use of systemic therapy in this group, may explain the low rates of axillary recurrence. “What I think we still need to find out, though, is whether or not failure to irradiate the nodes at all is in any way associated with decreased survival, as would be suggested in the MA.20 trial,” she said. “I think we will find that out from ongoing trials looking at no axillary dissection in mastectomy patients.”
Dr. Morrow and Dr. Giuliano reported no financial disclosures.
The complete manuscript of this study and its presentation at the American Surgical Association’s 137th Annual Meeting, April 2017, in Philadelphia, Pennsylvania, is to be published in Annals of Surgery pending editorial review.
PHILADELPHIA – The safety of sentinel lymph node biopsy (SLNB) alone without axillary lymph node dissection (ALND) has been established for patients with cT1-2N0 cancer that are found to have one or two metastatic sentinel lymph nodes who undergo breast conservation therapy, but questions regarding the role of regional radiation have persisted.
This issue is addressed by the results of a large, prospective, 5+ year study at Memorial Sloan Kettering Cancer Center which confirmed the safety of omitting axillary lymph node dissection and suggested that regional radiation provides minimal benefit.
Dr. Morrow explained that, in August 2010, the breast surgery service at MSKCC adopted the guidelines that arose from the American College of Surgeons Oncology Group’s multicenter Z0011 trial and abandoned routine use of ALND in eligible patients. The goal of the study, she reported, was to determine how frequently axillary dissection was avoided in a consecutive, otherwise unselected, series of patients and to determine the incidence of local regional recurrence after SLNB alone in a population treated with known radiotherapy fields.
Eligible subjects had T1 or T2 node-negative breast cancer, were undergoing breast-conserving surgery with planned whole-breast irradiation, and were found to have hematoxylin-eosin-detected sentinel node metastases. Patients receiving neoadjuvant chemotherapy or requiring conversion to mastectomy, or those in whom partial breast irradiation or no radiotherapy was planned, were ineligible. Axillary imaging was not used in select patients. Criteria for axillary dissection were metastases in three or more sentinel nodes or the presence of matted nodes identified intraoperatively. The researchers did not use the MSKCC nomogram to predict the likelihood of non–sentinel node metastases.
Median patient age was 58 years and median tumor size 1.7 cm. With regard to tumor pathology, 87% had infiltrating ductal tumors, 94% had grade 2 or 3 disease, and the most common subtype was HR+, HER2– disease in 84%. “In this node-positive cohort of patients, 98% received adjuvant systemic therapy, most commonly both chemotherapy and endocrine therapy (received by 65%), and 93% completed radiotherapy,” Dr. Morrow said.
In the entire patient cohort, 84% (663) were treated with SLNB alone, Dr. Morrow said. Among the 130 patients requiring ALND, 68% (88) had metastases in three or more nodes, 26% (34) were found to have had matted nodes intraoperatively, and 6% (8) were eligible for SLNB alone but opted for ALND or had it recommended by their surgeon. “All of these occurred early in our experience, and this has not been repeated since,” Dr. Morrow said.
Among the SLNB-only patients, the 5-year event-free survival was 93%. “There were no isolated axillary recurrences,” Dr. Morrow said. The study reported four combined breast and axillary recurrences, three in nonradiated patients, and four combined nodal and distant recurrences, only one of which involved the axillary nodes. “The median time to any nodal recurrence was 25 months,” Dr. Morrow added. Among 484 patients who had 1 year or more of follow-up, 58% (280) received conventional supine breast tangents, 21% were treated prone – “meaning their axilla received essentially no radiotherapy,” Dr. Morrow said – and 21% had node field irradiation.
“If we compare patient characteristics based on radiotherapy fields treated, it’s clear that the patients who received nodal irradiation were a higher-risk group,” Dr. Morrow said. While all three groups had a median of one positive sentinel node, that “skewed towards two” in the nodal irradiation group, she said. This group also had higher rates of lymphovascular invasion (72% vs. 56% and 49% in the supine and prone groups, respectively) and extracapsular extension (41% vs. 31% and 25%).
The rates of nodal relapse were not statistically significant among the three groups: 1% in the prone group, 1.4% in the supine group, and 0% in the node irradiation group.
“Factors associated with a higher risk of distant metastases, such as young patient age, estrogen receptor negativity, or HER2 over-expression, were not associated with the need for axillary dissection and should not be used as priority selection criteria,” Dr. Morrow said. “Nodal recurrence was uncommon in the absence of routine nodal radiation therapy, and no isolated nodal failures were observed.
In his comments, Armando Giuliano, MD, of Cedars Sinai Medical Center in Los Angeles, principal investigator of the Z0011 trial, said the MSKCC study “extends and informs” the Z0011 findings. He noted that the prone treatment group in the MSKCC trial had a low rate of axillary recurrence. “Can you speculate how such excellent results are achieved without resection or irradiation?” he asked Dr. Morrow. “To me it appears that nodal irradiation provides very little benefit to this selected group of patients.”
The patients in the prone group were in the lowest-risk category of the study, Dr. Morrow said, but the fact that not all nodal disease becomes clinically evident, even in patients who do not receive radiotherapy or systemic therapy, along with the high use of systemic therapy in this group, may explain the low rates of axillary recurrence. “What I think we still need to find out, though, is whether or not failure to irradiate the nodes at all is in any way associated with decreased survival, as would be suggested in the MA.20 trial,” she said. “I think we will find that out from ongoing trials looking at no axillary dissection in mastectomy patients.”
Dr. Morrow and Dr. Giuliano reported no financial disclosures.
The complete manuscript of this study and its presentation at the American Surgical Association’s 137th Annual Meeting, April 2017, in Philadelphia, Pennsylvania, is to be published in Annals of Surgery pending editorial review.
PHILADELPHIA – The safety of sentinel lymph node biopsy (SLNB) alone without axillary lymph node dissection (ALND) has been established for patients with cT1-2N0 cancer that are found to have one or two metastatic sentinel lymph nodes who undergo breast conservation therapy, but questions regarding the role of regional radiation have persisted.
This issue is addressed by the results of a large, prospective, 5+ year study at Memorial Sloan Kettering Cancer Center which confirmed the safety of omitting axillary lymph node dissection and suggested that regional radiation provides minimal benefit.
Dr. Morrow explained that, in August 2010, the breast surgery service at MSKCC adopted the guidelines that arose from the American College of Surgeons Oncology Group’s multicenter Z0011 trial and abandoned routine use of ALND in eligible patients. The goal of the study, she reported, was to determine how frequently axillary dissection was avoided in a consecutive, otherwise unselected, series of patients and to determine the incidence of local regional recurrence after SLNB alone in a population treated with known radiotherapy fields.
Eligible subjects had T1 or T2 node-negative breast cancer, were undergoing breast-conserving surgery with planned whole-breast irradiation, and were found to have hematoxylin-eosin-detected sentinel node metastases. Patients receiving neoadjuvant chemotherapy or requiring conversion to mastectomy, or those in whom partial breast irradiation or no radiotherapy was planned, were ineligible. Axillary imaging was not used in select patients. Criteria for axillary dissection were metastases in three or more sentinel nodes or the presence of matted nodes identified intraoperatively. The researchers did not use the MSKCC nomogram to predict the likelihood of non–sentinel node metastases.
Median patient age was 58 years and median tumor size 1.7 cm. With regard to tumor pathology, 87% had infiltrating ductal tumors, 94% had grade 2 or 3 disease, and the most common subtype was HR+, HER2– disease in 84%. “In this node-positive cohort of patients, 98% received adjuvant systemic therapy, most commonly both chemotherapy and endocrine therapy (received by 65%), and 93% completed radiotherapy,” Dr. Morrow said.
In the entire patient cohort, 84% (663) were treated with SLNB alone, Dr. Morrow said. Among the 130 patients requiring ALND, 68% (88) had metastases in three or more nodes, 26% (34) were found to have had matted nodes intraoperatively, and 6% (8) were eligible for SLNB alone but opted for ALND or had it recommended by their surgeon. “All of these occurred early in our experience, and this has not been repeated since,” Dr. Morrow said.
Among the SLNB-only patients, the 5-year event-free survival was 93%. “There were no isolated axillary recurrences,” Dr. Morrow said. The study reported four combined breast and axillary recurrences, three in nonradiated patients, and four combined nodal and distant recurrences, only one of which involved the axillary nodes. “The median time to any nodal recurrence was 25 months,” Dr. Morrow added. Among 484 patients who had 1 year or more of follow-up, 58% (280) received conventional supine breast tangents, 21% were treated prone – “meaning their axilla received essentially no radiotherapy,” Dr. Morrow said – and 21% had node field irradiation.
“If we compare patient characteristics based on radiotherapy fields treated, it’s clear that the patients who received nodal irradiation were a higher-risk group,” Dr. Morrow said. While all three groups had a median of one positive sentinel node, that “skewed towards two” in the nodal irradiation group, she said. This group also had higher rates of lymphovascular invasion (72% vs. 56% and 49% in the supine and prone groups, respectively) and extracapsular extension (41% vs. 31% and 25%).
The rates of nodal relapse were not statistically significant among the three groups: 1% in the prone group, 1.4% in the supine group, and 0% in the node irradiation group.
“Factors associated with a higher risk of distant metastases, such as young patient age, estrogen receptor negativity, or HER2 over-expression, were not associated with the need for axillary dissection and should not be used as priority selection criteria,” Dr. Morrow said. “Nodal recurrence was uncommon in the absence of routine nodal radiation therapy, and no isolated nodal failures were observed.
In his comments, Armando Giuliano, MD, of Cedars Sinai Medical Center in Los Angeles, principal investigator of the Z0011 trial, said the MSKCC study “extends and informs” the Z0011 findings. He noted that the prone treatment group in the MSKCC trial had a low rate of axillary recurrence. “Can you speculate how such excellent results are achieved without resection or irradiation?” he asked Dr. Morrow. “To me it appears that nodal irradiation provides very little benefit to this selected group of patients.”
The patients in the prone group were in the lowest-risk category of the study, Dr. Morrow said, but the fact that not all nodal disease becomes clinically evident, even in patients who do not receive radiotherapy or systemic therapy, along with the high use of systemic therapy in this group, may explain the low rates of axillary recurrence. “What I think we still need to find out, though, is whether or not failure to irradiate the nodes at all is in any way associated with decreased survival, as would be suggested in the MA.20 trial,” she said. “I think we will find that out from ongoing trials looking at no axillary dissection in mastectomy patients.”
Dr. Morrow and Dr. Giuliano reported no financial disclosures.
The complete manuscript of this study and its presentation at the American Surgical Association’s 137th Annual Meeting, April 2017, in Philadelphia, Pennsylvania, is to be published in Annals of Surgery pending editorial review.
AT THE ANNUAL ASA MEETING
Rates, predictors and variability of interhospital transfers: A national evaluation
Interhospital transfer (IHT) is defined as the transfer of hospitalized patients between acute care hospitals. Although cited reasons for transfer include providing patients access to unique specialty services,1 patterns and practices of IHT remain largely unstudied. Interhospital transfer is known to be common in certain patient populations, including selected patients presenting to the intensive care unit2 and those with acute myocardial infarction (AMI),3-5 but no recent studies have looked at frequency of IHT among a broader group of hospitalized patients nationally. Little is known about which patients are selected for transfer and why.6 Limited evidence suggests poor concordance between cited reason for transfer among patients, transferring physicians, and receiving physicians,7 indicating ambiguity in this care process.
Interhospital transfer exposes patients to the potential risks associated with discontinuity of care. Communication is particularly vulnerable to error during times of transition.8-10 Patients transferred between acute care hospitals are especially vulnerable, given the severity of illness in this patient population,11 and the absence of other factors to fill in gaps in communication, such as common electronic health records. Limited existing literature suggests transferred patients use more resources 12-13 and experience worse outcomes compared to nontransferred patients,11 although these data involved limited patient populations, and adjustment for illness severity and other factors was variably addressed.14-16
To improve the quality and safety of IHT, therefore, it is necessary to understand which patients benefit from IHT and identify best practices in the IHT process.17 A fundamental first step is to study patterns and practices of IHT, in particular with an eye towards identifying unwarranted variation.18 This is important to understand the prevalence of the issue, provide possible evidence of lack of standardization, and natural experiments with which to identify best practices.
To address this, we conducted a foundational study examining a national sample of Medicare patients to determine the nationwide frequency of IHT among elderly patients, patient and hospital-level predictors of transfer, and hospital variability in IHT practices.
METHODS
We performed a cross-sectional analysis using 2 nationally representative datasets: (1) Center for Medicare and Medicaid Services (CMS) 2013 100% Master Beneficiary Summary and Inpatient claims files, which contains data on all fee-for-service program Medicare enrollees’ demographic information, date of death, and hospitalization claims, including ICD-9 codes for diagnoses, diagnosis-related group (DRG), and dates of service; merged with (2) 2013 American Hospital Association (AHA) data,19 which contains hospital-level characteristics for all acute care hospitals in the U.S. Our study protocol was approved by the Partners Healthcare Human Subjects Review Committee.
Beneficiaries were eligible for inclusion if they were 65 years or older, continuously enrolled in Medicare A and B, with an acute care hospitalization claim in 2013, excluding Medicare managed care and end-stage renal disease (ESRD) beneficiaries. We additionally excluded beneficiaries hospitalized at federal or nonacute care hospitals, or critical access hospitals given their mission to stabilize and transfer patients to referral hospitals.20
Transferred patients were defined as: (1) beneficiaries with a “transfer out” claim and a corresponding “transfer in” claim at a different hospital; as well as (2) beneficiaries with a “transfer out” claim and a corresponding date of admission to another hospital within 1 day following the date of claim; and (3) beneficiaries with a “transfer in” claim and a corresponding date of discharge from another hospital within 1 day preceding the date of claim. Beneficiaries transferred to the same hospital, or cared for at hospitals with “outlier” transfer in rates equal to 100% or transfer out rates greater than 35%, were excluded from analysis given the suggestion of nonstandard claims practices. Beneficiaries with greater than 1 transfer within the same hospitalization were additionally excluded.
Patient Characteristics
Patient characteristics were obtained from the CMS data files and included: demographics (age, sex, race); DRG-weight, categorized into quartiles; primary diagnosis for the index hospitalization using ICD-9 codes; patient comorbidity using ICD-9 codes compiled into a CMS-Hierarchical Condition Category (HCC) risk score;21 presence of Medicaid co-insurance; number of hospitalizations in the past 12 months, categorized into 0, 1, 2-3, and 4 or more; season, defined as calendar quarters; and median income per household by census tract. These characteristics were chosen a priori given expert opinion in combination with prior research demonstrating association with IHT.11,22
Hospital Characteristics
Hospital characteristics were obtained from AHA data files and included hospitals’ size, categorized into small, medium, and large (less than 100, 100 to 399, 400 or more beds); geographic location; ownership; teaching status; setting (urban vs. rural); case mix index (CMI) for all patients cared for at the hospital; and presence of selected specialty services, including certified trauma center, medical intensive care unit, cardiac intensive care unit, cardiac surgery services, adult interventional cardiac catheterization, adult cardiac electrophysiology, and composite score of presence of 55 other specialty services (complete list in Appendix A). All characteristics were chosen a priori given expert opinion or relationship of characteristics with IHT, and prior research utilizing AHA data.23-24
Analysis
Descriptive statistics were used to evaluate the frequency of IHT, characteristics of transferred patients, and number of days to transfer. Patient and hospital characteristics of transferred vs. nontransferred patients were compared using chi-square analyses.
To analyze the effects of each patient and hospital characteristic on the odds of transfer, we used logistic regression models incorporating all patient and hospital characteristics, accounting for fixed effects for diagnosis, and utilizing generalized estimating equations (the GENMOD procedure in SAS statistical software, v 9.4; SAS Institute Inc., Cary, North Carolina) to account for the clustering of patients within hospitals.25 Indicator variables were created for missing covariate data and included in analyses when missing data accounted for greater than 10% of the total cohort.
To measure the variability in transfer rates between hospitals, we used a sequence of random effects logistic regression models. We first ran a model with no covariates, representing the unadjusted differences in transfer rates between hospitals. We then added patient characteristics to see if the unadjusted differences in IHT rates were explained by differences in patient characteristics between hospitals. Lastly, we added hospital characteristics to determine if these explained the remaining differences in transfer rates. Each of the 3 models provided a measure of between-hospital variability, reflecting the degree to which IHT rates differed between hospitals. Additionally, we used the intercept from the unadjusted model and the measure of between-hospital variability from each model to calculate the 95% confidence intervals, illustrating the range of IHT rates spanning 95% of all hospitals. We used those same numbers to calculate the 25th and 75th percentiles, illustrating the range of IHT rates for the middle half of hospitals.

RESULTS
Among 28 million eligible beneficiaries, 6.6 million had an acute care hospitalization to nonfederal, noncritical access hospitals, and 107,741 met our defined criteria for IHT. An additional 3790 beneficiaries were excluded for being transferred to the same facility, 416 beneficiaries (115 transferred, 301 nontransferred) were excluded as they were cared for at 1 of the 11 hospitals with “outlier” transfer in/out rates, and 2329 were excluded because they had more than 1 transfer during hospitalization. Thus, the final cohort consisted of 101,507 transferred (1.5%) and 6,625,474 nontransferred beneficiaries (Figure 1). Of the 101,507 transferred beneficiaries, 2799 (2.8%) were included more than once (ie, experienced more than 1 IHT on separate hospitalizations throughout the study period; the vast majority of these had 2 separate hospitalizations resulting in IHT). Characteristics of transferred and nontransferred beneficiaries are shown (Table 1).

Among transferred patients, the top 5 primary diagnoses at time of transfer included AMI (12.2%), congestive heart failure (CHF) (7.2%), sepsis (6.6%), arrhythmia (6.6%), and pneumonia (3.4%). Comorbid conditions most commonly present in transferred patients included CHF (52.6%), renal failure (51.8%), arrhythmia (49.8%), and chronic obstructive pulmonary disease (COPD; 37.0%). The most common day of transfer was day after admission (hospital day 2, 24.7%), with 75% of transferred patients transferred before hospital day 6 (Appendix B).
After adjusting for all other patient and hospital characteristics and clustering by hospital, the following variables were associated with greater odds of transfer: older age, male sex, nonblack race, non-Medicaid co-insurance, higher comorbidity (HCC score), lower DRG-weight, and fewer hospitalizations in the prior 12 months. Beneficiaries also had greater odds of transfer if initially hospitalized at smaller hospitals, nonteaching hospitals, public hospitals, at hospitals in the Northeast, those with fewer specialty services, and those with a low CMI (Table 2).
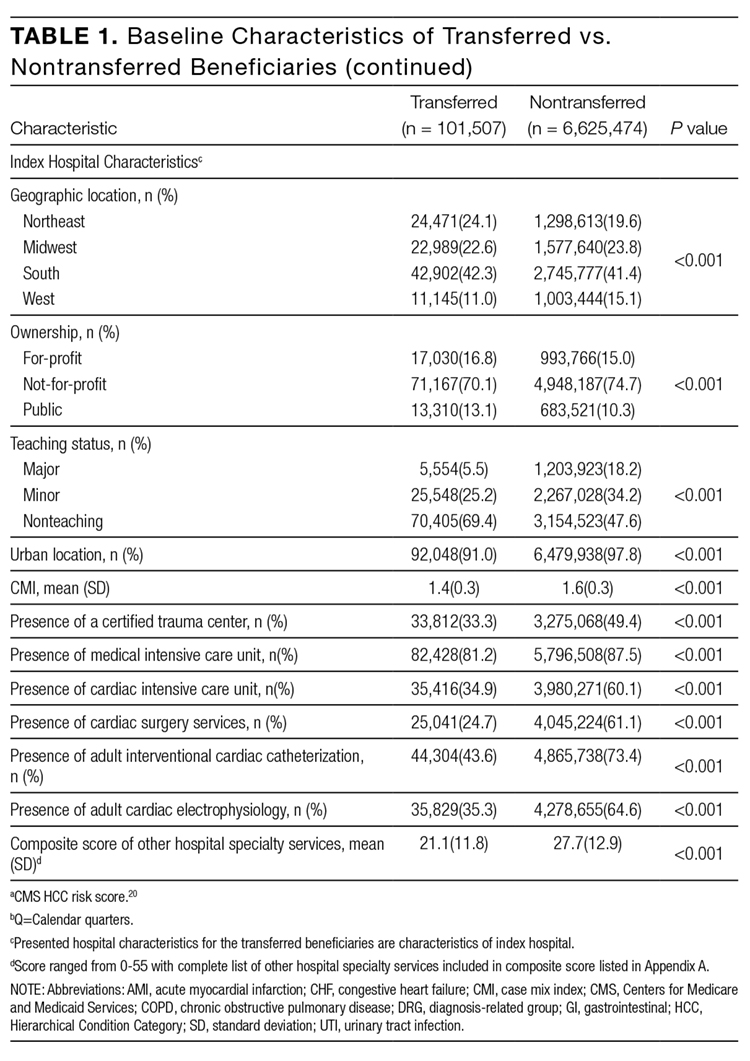
DISCUSSION
In this nationally representative study of 6.6 million Medicare beneficiaries, we found that 1.5% of patients were transferred between acute care facilities and were most often transferred prior to hospital day 6. Older age, male sex, nonblack race, higher medical comorbidity, lower DRG weight, and fewer recent hospitalizations were associated with greater odds of transfer. Initial hospitalization at smaller, nonteaching, public hospitals, with fewer specialty services were associated with greater odds of transfer, while higher CMI was associated with a lower odds of transfer. The most common comorbid conditions among transferred patients included CHF, renal failure, arrhythmia, and COPD; particularly notable was the very high prevalence of these conditions among transferred as compared with nontransferred patients. Importantly, we found significant variation in IHT by region and a large variation in transfer practices by hospital, with significant variability in transfer rates even after accounting for known patient and hospital characteristics.
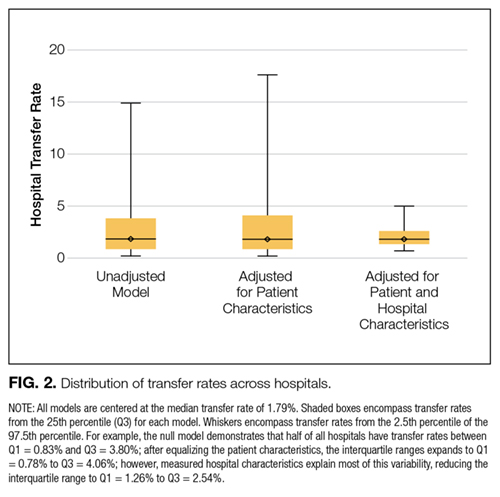
Among our examined population, we found that a sizable number of patients undergo IHT—more than 100,000 per year. Primary diagnoses at time of transfer consist of common inpatient conditions, including AMI, CHF, sepsis, arrhythmia, and pneumonia. Limited prior data support our findings, with up to 50% of AMI patients reportedly undergoing IHT,3-5 and severe sepsis and respiratory illness reported as common diagnoses at transfer.11 Although knowledge of these primary diagnoses does not directly confer an understanding of reason for transfer, one can speculate based on our findings. For example, research demonstrates the majority of AMI patients who undergo IHT had further intervention, including stress testing, cardiac catheterization, and/or coronary artery bypass graft surgery.5,26 Thus, it is reasonable to presume that many of the beneficiaries
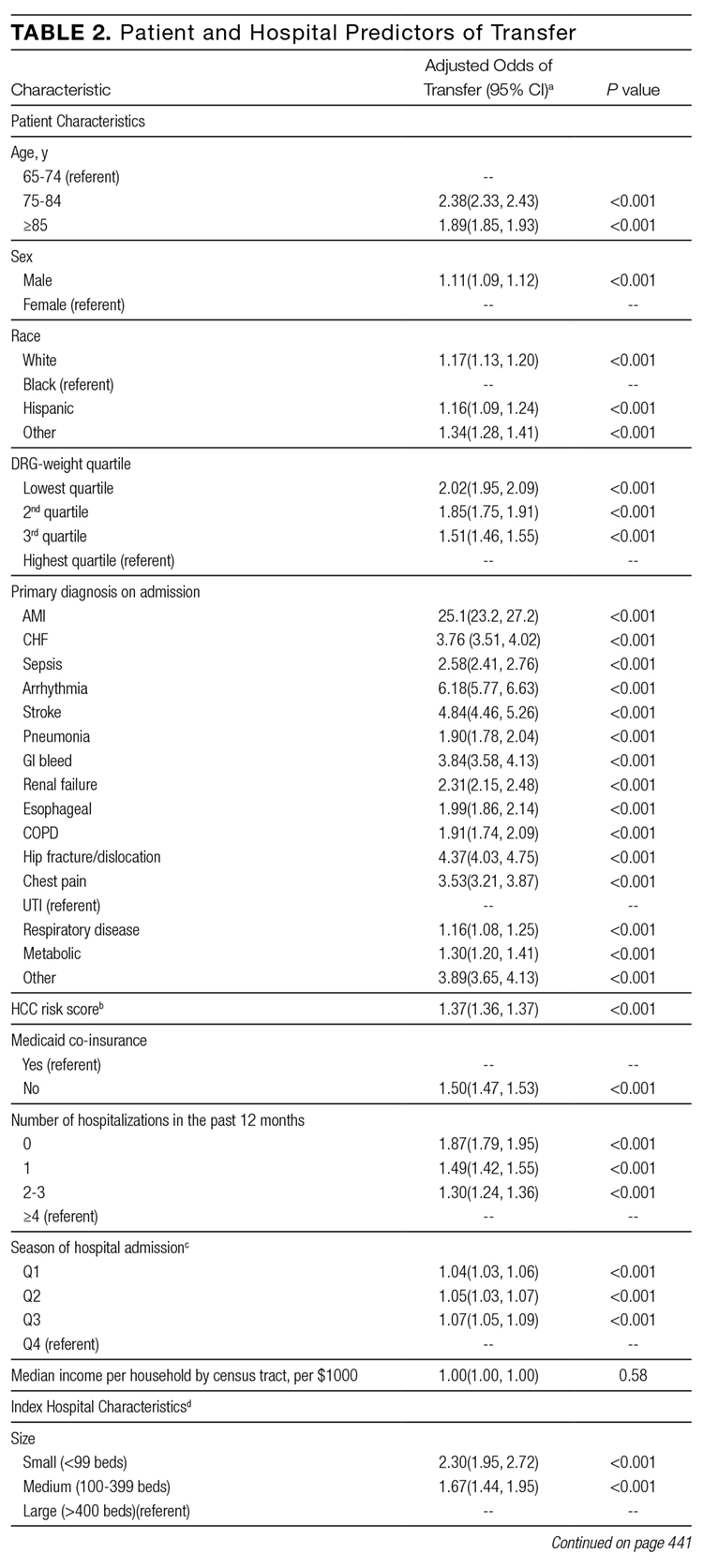
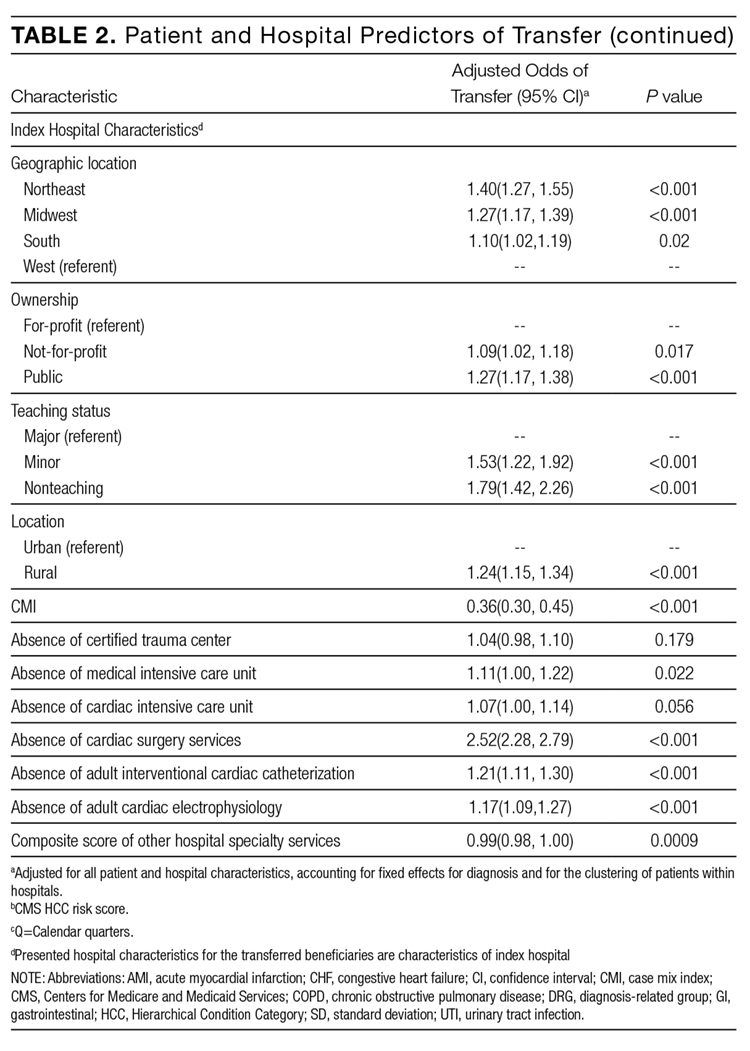
We additionally found that certain patient characteristics were associated with greater odds of transfer. Research suggests that transferred patients are “sicker” than nontransferred patients.1,11 Although our findings in part confirm these data, we paradoxically found that higher DRG-weight and 4 or more hospitalizations in the past year were actually associated with lower odds of transfer. In addition, the oldest patients in our cohort (85 years or older) were actually less likely to be transferred than their slightly younger counterparts (75 to 84 years). These variables may reflect extreme illness or frailty,27 and providers consciously (or subconsciously) may factor this in to their decision to transfer, considering a threshold past which transfer would confer more risk than benefit (eg, a patient may be “too sick” for transfer). Indeed, in a secondary analysis without hospital characteristics or comorbidities, and with fixed effects by hospital, we found the highest rates of IHT in patients in the middle 2 quartiles of DRG-weight, supporting this threshold hypothesis. It is also possible that patients with numerous hospitalizations may be less likely to be transferred because of familiarity and a strong sense of responsibility to continue to care for those patients (although we cannot confirm that those prior hospitalizations were all with the same index hospital).
It is also notable that odds of transfer differed by race, with black patients 17% less likely to undergo transfer compared to whites, similar to findings in other IHT studies.11 This finding, in combination with our demonstration that Medicaid patients also have lower odds of transfer, warrants further investigation to ensure the process of IHT does not bias against these populations, as with other well-documented health disparities.28-30
The hospital predictors of transfer were largely expected. However, interestingly, when we controlled for all other patient and hospital characteristics, regional variation persisted, with highest odds of transfer with hospitalization in the Northeast, indicating variability by region not explained by other factors, and findings supported by other limited data.31 This variability was further elucidated in our examination of change in variance estimates accounting for patient, then hospital, characteristics. Although we expected and found marked variability in hospital transfer rates in our null model (without accounting for any patient or hospital characteristics), we interestingly found that variability increased upon adjusting for patient characteristics. This result is presumably due to the fact that patients who are more likely to be transferred (ie, “sick” patients) are more often already at hospitals less likely to transfer patients, supported by our findings that hospital CMI is inversely associated with odds of transfer (in other words, hospitals that care for a less sick patient population are more likely to transfer their patients, and hospitals that care for a sicker patient population [higher CMI] are less likely to transfer). Adjusting solely for patient characteristics effectively equalizes these patients across hospitals, which would lead to even increased variability in transfer rates. Conversely, when we then adjusted for hospital characteristics, variability in hospital transfer rates decreased by 83% (in other words, hospital characteristics, rather than patient characteristics, explained much of the variability in transfer rates), although significant unexplained variability remained. We should note that although the observed reduction in variability was explained by the patient and hospital characteristics included in the model, these characteristics do not necessarily justify the variability they accounted for; although patients’ race or hospitals’ location may explain some of the observed variability, this does not reasonably justify it.
This observed variability in transfer practices is not surprising given the absence of standardization and clear guidelines to direct clinical IHT practice.17 Selection of patients that may benefit from transfer is often ambiguous and subjective.6 The Emergency Medical Treatment and Active Labor Act laws dictate that hospitals transfer patients requiring a more specialized service, or when “medical benefits ... outweigh the increased risks to the individual...,” although in practice this provides little guidance to practitioners.1 Thus, clearer guidelines may be necessary to achieve less variable practices.
Our study is subject to several limitations. First, although nationally representative, the Medicare population is not reflective of all hospitalized patients nationwide. Additionally, we excluded patients transferred from the emergency room. Thus, the total number of patients who undergo IHT nationally is expected to be much higher than reflected in our analysis. We also excluded patients who were transferred more than once during a given hospitalization. This enabled us to focus on the initial transfer decision but does not allow us to look at patients who are transferred to a referral center and then transferred back. Second, given the criteria we used to define transfer, it is possible that we included nontransferred patients within our transferred cohort if they were discharged from one hospital and admitted to a different hospital within 1 day. However, on quality assurance analyses where we limited our cohort to only those beneficiaries with corresponding “transfer in” and “transfer out” claims (87% of the total cohort), we found no marked differences in our results. Additionally, although we assume that patient transfer status was coded correctly within the Medicare dataset, we could not confirm by individually examining each patient we defined as “transferred.” However, on additional quality assurance analyses where we examined randomly selected excluded patients with greater than 1 transfer during hospitalization, we found differing provider numbers with each transfer, suggesting validity of the coding. Third, because there are likely many unmeasured patient confounders, we cannot be sure how much of the between-hospital variation is due to incomplete adjustment for patient characteristics. However, since adjusting for patient characteristics actually increased variability in hospital transfer rates, it is unlikely that residual patient confounders fully explain our observed results. Despite this, other variables that are not available within the CMS or AHA datasets may further elucidate hospital transfer practices, including variables reflective of the transfer process (eg, time of day of patient transfer, time delay between initiation of transfer and patient arrival at accepting hospital, accepting service on transfer, etc.); other markers of illness severity (eg, clinical service at the time of index admission, acute physiology score, utilization of critical care services on arrival at receiving hospital); and other hospital system variables (ie, membership in an accountable care organization and/or regional care network, the density of nearby tertiary referral centers (indicating possible supply-induced demand), other variables reflective of the “transfer culture” (such as the transfer rate at the hospital or region where the attending physician trained, etc.). Lastly, though our examination provides important foundational information regarding IHT nationally, this study did not examine patient outcomes in transferred and nontransferred patients, which may help to determine which patients benefit (or do not benefit) from transfer and why. Further investigation is needed to study these outcomes.
CONCLUSION
In this national study of IHT, we found that a sizable number of patients admitted to the hospital undergo transfer to another acute care facility. Patients are transferred with common medical conditions, including those requiring specialized care such as AMI, and a high rate of comorbid clinical conditions, and certain patient and hospital characteristics are associated with greater odds of transfer. Although many of the observed associations between characteristics and odds of transfer were expected based on limited existing literature, we found several unexpected findings, eg, suggesting the possibility of a threshold beyond which sicker patients are not transferred. Additionally, we found that black and Medicaid patients had lower odds of transfer, which warrants further investigation for potential health care disparity. Importantly, we found much variability in the practice of IHT, as evidenced by the inexplicable differences in transfer by hospital region, and by residual unexplained variability in hospital transfer rates after accounting for patient and hospital characteristics, which may be due to lack of standard guidelines to direct IHT practices. In conclusion, this study of hospitalized Medicare patients provides important foundational information regarding rates and predictors of IHT nationally, as well as unexplained variability that exists within this complex care transition. Further investigation will be essential to understand reasons for, processes related to, and outcomes of transferred patients, to help guide standardization in best practices in care.
Disclosure
Nothing to report.
1. Iwashyna TJ. The incomplete infrastructure for interhospital patient transfer. Crit Care Med. 2012;40(8):2470-2478. PubMed
2. Iwashyna TJ, Christie JD, Moody J, Kahn JM, Asch DA. The structure of critical care transfer networks. Med Care. 2009;47(7):787-793. PubMed
3. Mehta RH, Stalhandske EJ, McCargar PA, Ruane TJ, Eagle KA. Elderly patients at highest risk with acute myocardial infarction are more frequently transferred from community hospitals to tertiary centers: reality or myth? Am Heart J. 1999;138(4 Pt 1):688-695. PubMed
4. Iwashyna TJ, Kahn JM, Hayward RA, Nallamothu BK. Interhospital transfers among Medicare beneficiaries admitted for acute myocardial infarction at nonrevascularization hospitals. Circ Cardiovasc Qual Outcomes. 2010;3(5):468-475. PubMed
5. Roe MT, Chen AY, Delong ER, Boden WE, Calvin JE Jr, Cairns CB, et al. Patterns of transfer for patients with non-ST-segment elevation acute coronary syndrome from community to tertiary care hospitals. Am Heart J. 2008;156(1):185-192. PubMed
6. Bosk EA, Veinot T, Iwashyna TJ. Which patients and where: a qualitative study of patient transfers from community hospitals. Med Care. 2011;49(6):592-598. PubMed
7. Wagner J, Iwashyna TJ, Kahn JM. Reasons underlying interhospital transfers to an academic medical intensive care unit. J Crit Care. 2013;28(2):202-208. PubMed
8. Cohen MD, Hilligoss PB. The published literature on handoffs in hospitals: deficiencies identified in an extensive review. Qual Saf Health Care. 2010;19(6):493-497. PubMed
9. Riesenberg LA, Leitzsch J, Massucci JL, et al. Residents’ and attending physicians’ handoffs: a systematic review of the literature. Acad Med. 2009;84(12):1775-1787. PubMed
10. Arora V, Johnson J, Lovinger D, Humphrey HJ, Meltzer DO. Communication failures in patient sign-out and suggestions for improvement: a critical incident analysis. Qual Saf Health Care. 2005;14(6):401-407. PubMed
11. Sokol-Hessner L, White AA, Davis KF, Herzig SJ, Hohmann SF. Interhospital transfer patients discharged by academic hospitalists and general internists: characteristics and outcomes. J Hosp Med. 2016;11(4):245-250. PubMed
12. Bernard AM, Hayward RA, Rosevear J, Chun H, McMahon LF. Comparing the hospitalizations of transfer and non-transfer patients in an academic medical center. Acad Med. 1996;71(3):262-266. PubMed
13. Golestanian E, Scruggs JE, Gangnon RE, Mak RP, Wood KE. Effect of interhospital transfer on resource utilization and outcomes at a tertiary care referral center. Crit Care Med. 2007;35(6):1470-1476. PubMed
14. Durairaj L, Will JG, Torner JC, Doebbeling BN. Prognostic factors for mortality following interhospital transfers to the medical intensive care unit of a tertiary referral center. Crit Care Med. 2003;31(7):1981-1986. PubMed
15. Kerr HD, Byrd JC. Community hospital transfers to a VA Medical Center. JAMA. 1989;262(1):70-73. PubMed
16. Dragsted L, Jörgensen J, Jensen NH, et al. Interhospital comparisons of patient outcome from intensive care: importance of lead-time bias. Crit Care Med. 1989;17(5):418-422. PubMed
17. Gupta K, Mueller SK. Interhospital transfers: the need for standards. J Hosp Med. 2015;10(6):415-417. PubMed
18. The Dartmouth Atlas of Health Care: Understanding of the Efficiency and Effectiveness of the Health Care System. The Dartmouth Institute for Health Practice and Clinical Policy, Lebanon, NH. http://www.dartmouthatlas.org/. Accessed November 1, 2016.
19. American Hospital Association Annual Survey Database. American Hospital Association, Chicago, IL. http://www.ahadataviewer.com/book-cd-products/AHA-Survey/. Accessed July 1, 2013.
20. U.S. Department of Health and Human Services (HRSA): What are critical access hospitals (CAH)? http://www.hrsa.gov/healthit/toolbox/RuralHealthITtoolbox/Introduction/critical.html. Accessed June 9, 2016.
21. Li P, Kim MM, Doshi JA. Comparison of the performance of the CMS Hierarchical Condition Category (CMS-HCC) risk adjuster with the Charlson and Elixhauser comorbidity measures in predicting mortality. BMC Health Serv Res. 2010;10:245. PubMed
22. Hernandez-Boussard T, Davies S, McDonald K, Wang NE. Interhospital facility transfers in the United States: a nationwide outcomes study. J Patient Saf. Nov 13 2014. PubMed
23. Landon BE, Normand SL, Lessler A, et al. Quality of care for the treatment of acute medical conditions in US hospitals. Arch Intern Med. 2006;166(22):2511-2517. PubMed
24. Mueller SK, Lipsitz S, Hicks LS. Impact of hospital teaching intensity on quality of care and patient outcomes. Med Care.2013;51(7):567-574. PubMed
25. Lopez L, Hicks LS, Cohen AP, McKean S, Weissman JS. Hospitalists and the quality of care in hospitals. Arch Intern Med. 2009;169(15):1389-1394. PubMed
26. Barreto-Filho JA, Wang Y, Rathore SS, et al. Transfer rates from nonprocedure hospitals after initial admission and outcomes among elderly patients with acute myocardial infarction. JAMA Intern Med. 2014;174(2):213-222. PubMed
27. Carlson JE, Zocchi KA, Bettencourt DM, et al. Measuring frailty in the hospitalized elderly: concept of functional homeostasis. Am J Phys Med Rehabil. 1998;77(3):252-257. PubMed
28. Ward E, Jemal A, Cokkinides V, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin. 2004;54(2):78-93. PubMed
29. Iribarren C, Tolstykh I, Somkin CP, et al. Sex and racial/ethnic disparities in outcomes after acute myocardial infarction: a cohort study among members of a large integrated health care delivery system in northern California. Arch Intern Med. 2005;165(18):2105-2113. PubMed
30. Kawachi I, Daniels N, Robinson DE. Health disparities by race and class: why both matter. Health Aff (Millwood). 2005;24(2):343-352. PubMed
31. Herrigel DJ, Carroll M, Fanning C, Steinberg MB, Parikh A, Usher M. Interhospital transfer handoff practices among US tertiary care centers: a descriptive survey. J Hosp Med. 2016;11(6):413-417. PubMed
Interhospital transfer (IHT) is defined as the transfer of hospitalized patients between acute care hospitals. Although cited reasons for transfer include providing patients access to unique specialty services,1 patterns and practices of IHT remain largely unstudied. Interhospital transfer is known to be common in certain patient populations, including selected patients presenting to the intensive care unit2 and those with acute myocardial infarction (AMI),3-5 but no recent studies have looked at frequency of IHT among a broader group of hospitalized patients nationally. Little is known about which patients are selected for transfer and why.6 Limited evidence suggests poor concordance between cited reason for transfer among patients, transferring physicians, and receiving physicians,7 indicating ambiguity in this care process.
Interhospital transfer exposes patients to the potential risks associated with discontinuity of care. Communication is particularly vulnerable to error during times of transition.8-10 Patients transferred between acute care hospitals are especially vulnerable, given the severity of illness in this patient population,11 and the absence of other factors to fill in gaps in communication, such as common electronic health records. Limited existing literature suggests transferred patients use more resources 12-13 and experience worse outcomes compared to nontransferred patients,11 although these data involved limited patient populations, and adjustment for illness severity and other factors was variably addressed.14-16
To improve the quality and safety of IHT, therefore, it is necessary to understand which patients benefit from IHT and identify best practices in the IHT process.17 A fundamental first step is to study patterns and practices of IHT, in particular with an eye towards identifying unwarranted variation.18 This is important to understand the prevalence of the issue, provide possible evidence of lack of standardization, and natural experiments with which to identify best practices.
To address this, we conducted a foundational study examining a national sample of Medicare patients to determine the nationwide frequency of IHT among elderly patients, patient and hospital-level predictors of transfer, and hospital variability in IHT practices.
METHODS
We performed a cross-sectional analysis using 2 nationally representative datasets: (1) Center for Medicare and Medicaid Services (CMS) 2013 100% Master Beneficiary Summary and Inpatient claims files, which contains data on all fee-for-service program Medicare enrollees’ demographic information, date of death, and hospitalization claims, including ICD-9 codes for diagnoses, diagnosis-related group (DRG), and dates of service; merged with (2) 2013 American Hospital Association (AHA) data,19 which contains hospital-level characteristics for all acute care hospitals in the U.S. Our study protocol was approved by the Partners Healthcare Human Subjects Review Committee.
Beneficiaries were eligible for inclusion if they were 65 years or older, continuously enrolled in Medicare A and B, with an acute care hospitalization claim in 2013, excluding Medicare managed care and end-stage renal disease (ESRD) beneficiaries. We additionally excluded beneficiaries hospitalized at federal or nonacute care hospitals, or critical access hospitals given their mission to stabilize and transfer patients to referral hospitals.20
Transferred patients were defined as: (1) beneficiaries with a “transfer out” claim and a corresponding “transfer in” claim at a different hospital; as well as (2) beneficiaries with a “transfer out” claim and a corresponding date of admission to another hospital within 1 day following the date of claim; and (3) beneficiaries with a “transfer in” claim and a corresponding date of discharge from another hospital within 1 day preceding the date of claim. Beneficiaries transferred to the same hospital, or cared for at hospitals with “outlier” transfer in rates equal to 100% or transfer out rates greater than 35%, were excluded from analysis given the suggestion of nonstandard claims practices. Beneficiaries with greater than 1 transfer within the same hospitalization were additionally excluded.
Patient Characteristics
Patient characteristics were obtained from the CMS data files and included: demographics (age, sex, race); DRG-weight, categorized into quartiles; primary diagnosis for the index hospitalization using ICD-9 codes; patient comorbidity using ICD-9 codes compiled into a CMS-Hierarchical Condition Category (HCC) risk score;21 presence of Medicaid co-insurance; number of hospitalizations in the past 12 months, categorized into 0, 1, 2-3, and 4 or more; season, defined as calendar quarters; and median income per household by census tract. These characteristics were chosen a priori given expert opinion in combination with prior research demonstrating association with IHT.11,22
Hospital Characteristics
Hospital characteristics were obtained from AHA data files and included hospitals’ size, categorized into small, medium, and large (less than 100, 100 to 399, 400 or more beds); geographic location; ownership; teaching status; setting (urban vs. rural); case mix index (CMI) for all patients cared for at the hospital; and presence of selected specialty services, including certified trauma center, medical intensive care unit, cardiac intensive care unit, cardiac surgery services, adult interventional cardiac catheterization, adult cardiac electrophysiology, and composite score of presence of 55 other specialty services (complete list in Appendix A). All characteristics were chosen a priori given expert opinion or relationship of characteristics with IHT, and prior research utilizing AHA data.23-24
Analysis
Descriptive statistics were used to evaluate the frequency of IHT, characteristics of transferred patients, and number of days to transfer. Patient and hospital characteristics of transferred vs. nontransferred patients were compared using chi-square analyses.
To analyze the effects of each patient and hospital characteristic on the odds of transfer, we used logistic regression models incorporating all patient and hospital characteristics, accounting for fixed effects for diagnosis, and utilizing generalized estimating equations (the GENMOD procedure in SAS statistical software, v 9.4; SAS Institute Inc., Cary, North Carolina) to account for the clustering of patients within hospitals.25 Indicator variables were created for missing covariate data and included in analyses when missing data accounted for greater than 10% of the total cohort.
To measure the variability in transfer rates between hospitals, we used a sequence of random effects logistic regression models. We first ran a model with no covariates, representing the unadjusted differences in transfer rates between hospitals. We then added patient characteristics to see if the unadjusted differences in IHT rates were explained by differences in patient characteristics between hospitals. Lastly, we added hospital characteristics to determine if these explained the remaining differences in transfer rates. Each of the 3 models provided a measure of between-hospital variability, reflecting the degree to which IHT rates differed between hospitals. Additionally, we used the intercept from the unadjusted model and the measure of between-hospital variability from each model to calculate the 95% confidence intervals, illustrating the range of IHT rates spanning 95% of all hospitals. We used those same numbers to calculate the 25th and 75th percentiles, illustrating the range of IHT rates for the middle half of hospitals.
RESULTS
Among 28 million eligible beneficiaries, 6.6 million had an acute care hospitalization to nonfederal, noncritical access hospitals, and 107,741 met our defined criteria for IHT. An additional 3790 beneficiaries were excluded for being transferred to the same facility, 416 beneficiaries (115 transferred, 301 nontransferred) were excluded as they were cared for at 1 of the 11 hospitals with “outlier” transfer in/out rates, and 2329 were excluded because they had more than 1 transfer during hospitalization. Thus, the final cohort consisted of 101,507 transferred (1.5%) and 6,625,474 nontransferred beneficiaries (Figure 1). Of the 101,507 transferred beneficiaries, 2799 (2.8%) were included more than once (ie, experienced more than 1 IHT on separate hospitalizations throughout the study period; the vast majority of these had 2 separate hospitalizations resulting in IHT). Characteristics of transferred and nontransferred beneficiaries are shown (Table 1).
Among transferred patients, the top 5 primary diagnoses at time of transfer included AMI (12.2%), congestive heart failure (CHF) (7.2%), sepsis (6.6%), arrhythmia (6.6%), and pneumonia (3.4%). Comorbid conditions most commonly present in transferred patients included CHF (52.6%), renal failure (51.8%), arrhythmia (49.8%), and chronic obstructive pulmonary disease (COPD; 37.0%). The most common day of transfer was day after admission (hospital day 2, 24.7%), with 75% of transferred patients transferred before hospital day 6 (Appendix B).
After adjusting for all other patient and hospital characteristics and clustering by hospital, the following variables were associated with greater odds of transfer: older age, male sex, nonblack race, non-Medicaid co-insurance, higher comorbidity (HCC score), lower DRG-weight, and fewer hospitalizations in the prior 12 months. Beneficiaries also had greater odds of transfer if initially hospitalized at smaller hospitals, nonteaching hospitals, public hospitals, at hospitals in the Northeast, those with fewer specialty services, and those with a low CMI (Table 2).
DISCUSSION
In this nationally representative study of 6.6 million Medicare beneficiaries, we found that 1.5% of patients were transferred between acute care facilities and were most often transferred prior to hospital day 6. Older age, male sex, nonblack race, higher medical comorbidity, lower DRG weight, and fewer recent hospitalizations were associated with greater odds of transfer. Initial hospitalization at smaller, nonteaching, public hospitals, with fewer specialty services were associated with greater odds of transfer, while higher CMI was associated with a lower odds of transfer. The most common comorbid conditions among transferred patients included CHF, renal failure, arrhythmia, and COPD; particularly notable was the very high prevalence of these conditions among transferred as compared with nontransferred patients. Importantly, we found significant variation in IHT by region and a large variation in transfer practices by hospital, with significant variability in transfer rates even after accounting for known patient and hospital characteristics.
Among our examined population, we found that a sizable number of patients undergo IHT—more than 100,000 per year. Primary diagnoses at time of transfer consist of common inpatient conditions, including AMI, CHF, sepsis, arrhythmia, and pneumonia. Limited prior data support our findings, with up to 50% of AMI patients reportedly undergoing IHT,3-5 and severe sepsis and respiratory illness reported as common diagnoses at transfer.11 Although knowledge of these primary diagnoses does not directly confer an understanding of reason for transfer, one can speculate based on our findings. For example, research demonstrates the majority of AMI patients who undergo IHT had further intervention, including stress testing, cardiac catheterization, and/or coronary artery bypass graft surgery.5,26 Thus, it is reasonable to presume that many of the beneficiaries
We additionally found that certain patient characteristics were associated with greater odds of transfer. Research suggests that transferred patients are “sicker” than nontransferred patients.1,11 Although our findings in part confirm these data, we paradoxically found that higher DRG-weight and 4 or more hospitalizations in the past year were actually associated with lower odds of transfer. In addition, the oldest patients in our cohort (85 years or older) were actually less likely to be transferred than their slightly younger counterparts (75 to 84 years). These variables may reflect extreme illness or frailty,27 and providers consciously (or subconsciously) may factor this in to their decision to transfer, considering a threshold past which transfer would confer more risk than benefit (eg, a patient may be “too sick” for transfer). Indeed, in a secondary analysis without hospital characteristics or comorbidities, and with fixed effects by hospital, we found the highest rates of IHT in patients in the middle 2 quartiles of DRG-weight, supporting this threshold hypothesis. It is also possible that patients with numerous hospitalizations may be less likely to be transferred because of familiarity and a strong sense of responsibility to continue to care for those patients (although we cannot confirm that those prior hospitalizations were all with the same index hospital).
It is also notable that odds of transfer differed by race, with black patients 17% less likely to undergo transfer compared to whites, similar to findings in other IHT studies.11 This finding, in combination with our demonstration that Medicaid patients also have lower odds of transfer, warrants further investigation to ensure the process of IHT does not bias against these populations, as with other well-documented health disparities.28-30
The hospital predictors of transfer were largely expected. However, interestingly, when we controlled for all other patient and hospital characteristics, regional variation persisted, with highest odds of transfer with hospitalization in the Northeast, indicating variability by region not explained by other factors, and findings supported by other limited data.31 This variability was further elucidated in our examination of change in variance estimates accounting for patient, then hospital, characteristics. Although we expected and found marked variability in hospital transfer rates in our null model (without accounting for any patient or hospital characteristics), we interestingly found that variability increased upon adjusting for patient characteristics. This result is presumably due to the fact that patients who are more likely to be transferred (ie, “sick” patients) are more often already at hospitals less likely to transfer patients, supported by our findings that hospital CMI is inversely associated with odds of transfer (in other words, hospitals that care for a less sick patient population are more likely to transfer their patients, and hospitals that care for a sicker patient population [higher CMI] are less likely to transfer). Adjusting solely for patient characteristics effectively equalizes these patients across hospitals, which would lead to even increased variability in transfer rates. Conversely, when we then adjusted for hospital characteristics, variability in hospital transfer rates decreased by 83% (in other words, hospital characteristics, rather than patient characteristics, explained much of the variability in transfer rates), although significant unexplained variability remained. We should note that although the observed reduction in variability was explained by the patient and hospital characteristics included in the model, these characteristics do not necessarily justify the variability they accounted for; although patients’ race or hospitals’ location may explain some of the observed variability, this does not reasonably justify it.
This observed variability in transfer practices is not surprising given the absence of standardization and clear guidelines to direct clinical IHT practice.17 Selection of patients that may benefit from transfer is often ambiguous and subjective.6 The Emergency Medical Treatment and Active Labor Act laws dictate that hospitals transfer patients requiring a more specialized service, or when “medical benefits ... outweigh the increased risks to the individual...,” although in practice this provides little guidance to practitioners.1 Thus, clearer guidelines may be necessary to achieve less variable practices.
Our study is subject to several limitations. First, although nationally representative, the Medicare population is not reflective of all hospitalized patients nationwide. Additionally, we excluded patients transferred from the emergency room. Thus, the total number of patients who undergo IHT nationally is expected to be much higher than reflected in our analysis. We also excluded patients who were transferred more than once during a given hospitalization. This enabled us to focus on the initial transfer decision but does not allow us to look at patients who are transferred to a referral center and then transferred back. Second, given the criteria we used to define transfer, it is possible that we included nontransferred patients within our transferred cohort if they were discharged from one hospital and admitted to a different hospital within 1 day. However, on quality assurance analyses where we limited our cohort to only those beneficiaries with corresponding “transfer in” and “transfer out” claims (87% of the total cohort), we found no marked differences in our results. Additionally, although we assume that patient transfer status was coded correctly within the Medicare dataset, we could not confirm by individually examining each patient we defined as “transferred.” However, on additional quality assurance analyses where we examined randomly selected excluded patients with greater than 1 transfer during hospitalization, we found differing provider numbers with each transfer, suggesting validity of the coding. Third, because there are likely many unmeasured patient confounders, we cannot be sure how much of the between-hospital variation is due to incomplete adjustment for patient characteristics. However, since adjusting for patient characteristics actually increased variability in hospital transfer rates, it is unlikely that residual patient confounders fully explain our observed results. Despite this, other variables that are not available within the CMS or AHA datasets may further elucidate hospital transfer practices, including variables reflective of the transfer process (eg, time of day of patient transfer, time delay between initiation of transfer and patient arrival at accepting hospital, accepting service on transfer, etc.); other markers of illness severity (eg, clinical service at the time of index admission, acute physiology score, utilization of critical care services on arrival at receiving hospital); and other hospital system variables (ie, membership in an accountable care organization and/or regional care network, the density of nearby tertiary referral centers (indicating possible supply-induced demand), other variables reflective of the “transfer culture” (such as the transfer rate at the hospital or region where the attending physician trained, etc.). Lastly, though our examination provides important foundational information regarding IHT nationally, this study did not examine patient outcomes in transferred and nontransferred patients, which may help to determine which patients benefit (or do not benefit) from transfer and why. Further investigation is needed to study these outcomes.
CONCLUSION
In this national study of IHT, we found that a sizable number of patients admitted to the hospital undergo transfer to another acute care facility. Patients are transferred with common medical conditions, including those requiring specialized care such as AMI, and a high rate of comorbid clinical conditions, and certain patient and hospital characteristics are associated with greater odds of transfer. Although many of the observed associations between characteristics and odds of transfer were expected based on limited existing literature, we found several unexpected findings, eg, suggesting the possibility of a threshold beyond which sicker patients are not transferred. Additionally, we found that black and Medicaid patients had lower odds of transfer, which warrants further investigation for potential health care disparity. Importantly, we found much variability in the practice of IHT, as evidenced by the inexplicable differences in transfer by hospital region, and by residual unexplained variability in hospital transfer rates after accounting for patient and hospital characteristics, which may be due to lack of standard guidelines to direct IHT practices. In conclusion, this study of hospitalized Medicare patients provides important foundational information regarding rates and predictors of IHT nationally, as well as unexplained variability that exists within this complex care transition. Further investigation will be essential to understand reasons for, processes related to, and outcomes of transferred patients, to help guide standardization in best practices in care.
Disclosure
Nothing to report.
Interhospital transfer (IHT) is defined as the transfer of hospitalized patients between acute care hospitals. Although cited reasons for transfer include providing patients access to unique specialty services,1 patterns and practices of IHT remain largely unstudied. Interhospital transfer is known to be common in certain patient populations, including selected patients presenting to the intensive care unit2 and those with acute myocardial infarction (AMI),3-5 but no recent studies have looked at frequency of IHT among a broader group of hospitalized patients nationally. Little is known about which patients are selected for transfer and why.6 Limited evidence suggests poor concordance between cited reason for transfer among patients, transferring physicians, and receiving physicians,7 indicating ambiguity in this care process.
Interhospital transfer exposes patients to the potential risks associated with discontinuity of care. Communication is particularly vulnerable to error during times of transition.8-10 Patients transferred between acute care hospitals are especially vulnerable, given the severity of illness in this patient population,11 and the absence of other factors to fill in gaps in communication, such as common electronic health records. Limited existing literature suggests transferred patients use more resources 12-13 and experience worse outcomes compared to nontransferred patients,11 although these data involved limited patient populations, and adjustment for illness severity and other factors was variably addressed.14-16
To improve the quality and safety of IHT, therefore, it is necessary to understand which patients benefit from IHT and identify best practices in the IHT process.17 A fundamental first step is to study patterns and practices of IHT, in particular with an eye towards identifying unwarranted variation.18 This is important to understand the prevalence of the issue, provide possible evidence of lack of standardization, and natural experiments with which to identify best practices.
To address this, we conducted a foundational study examining a national sample of Medicare patients to determine the nationwide frequency of IHT among elderly patients, patient and hospital-level predictors of transfer, and hospital variability in IHT practices.
METHODS
We performed a cross-sectional analysis using 2 nationally representative datasets: (1) Center for Medicare and Medicaid Services (CMS) 2013 100% Master Beneficiary Summary and Inpatient claims files, which contains data on all fee-for-service program Medicare enrollees’ demographic information, date of death, and hospitalization claims, including ICD-9 codes for diagnoses, diagnosis-related group (DRG), and dates of service; merged with (2) 2013 American Hospital Association (AHA) data,19 which contains hospital-level characteristics for all acute care hospitals in the U.S. Our study protocol was approved by the Partners Healthcare Human Subjects Review Committee.
Beneficiaries were eligible for inclusion if they were 65 years or older, continuously enrolled in Medicare A and B, with an acute care hospitalization claim in 2013, excluding Medicare managed care and end-stage renal disease (ESRD) beneficiaries. We additionally excluded beneficiaries hospitalized at federal or nonacute care hospitals, or critical access hospitals given their mission to stabilize and transfer patients to referral hospitals.20
Transferred patients were defined as: (1) beneficiaries with a “transfer out” claim and a corresponding “transfer in” claim at a different hospital; as well as (2) beneficiaries with a “transfer out” claim and a corresponding date of admission to another hospital within 1 day following the date of claim; and (3) beneficiaries with a “transfer in” claim and a corresponding date of discharge from another hospital within 1 day preceding the date of claim. Beneficiaries transferred to the same hospital, or cared for at hospitals with “outlier” transfer in rates equal to 100% or transfer out rates greater than 35%, were excluded from analysis given the suggestion of nonstandard claims practices. Beneficiaries with greater than 1 transfer within the same hospitalization were additionally excluded.
Patient Characteristics
Patient characteristics were obtained from the CMS data files and included: demographics (age, sex, race); DRG-weight, categorized into quartiles; primary diagnosis for the index hospitalization using ICD-9 codes; patient comorbidity using ICD-9 codes compiled into a CMS-Hierarchical Condition Category (HCC) risk score;21 presence of Medicaid co-insurance; number of hospitalizations in the past 12 months, categorized into 0, 1, 2-3, and 4 or more; season, defined as calendar quarters; and median income per household by census tract. These characteristics were chosen a priori given expert opinion in combination with prior research demonstrating association with IHT.11,22
Hospital Characteristics
Hospital characteristics were obtained from AHA data files and included hospitals’ size, categorized into small, medium, and large (less than 100, 100 to 399, 400 or more beds); geographic location; ownership; teaching status; setting (urban vs. rural); case mix index (CMI) for all patients cared for at the hospital; and presence of selected specialty services, including certified trauma center, medical intensive care unit, cardiac intensive care unit, cardiac surgery services, adult interventional cardiac catheterization, adult cardiac electrophysiology, and composite score of presence of 55 other specialty services (complete list in Appendix A). All characteristics were chosen a priori given expert opinion or relationship of characteristics with IHT, and prior research utilizing AHA data.23-24
Analysis
Descriptive statistics were used to evaluate the frequency of IHT, characteristics of transferred patients, and number of days to transfer. Patient and hospital characteristics of transferred vs. nontransferred patients were compared using chi-square analyses.
To analyze the effects of each patient and hospital characteristic on the odds of transfer, we used logistic regression models incorporating all patient and hospital characteristics, accounting for fixed effects for diagnosis, and utilizing generalized estimating equations (the GENMOD procedure in SAS statistical software, v 9.4; SAS Institute Inc., Cary, North Carolina) to account for the clustering of patients within hospitals.25 Indicator variables were created for missing covariate data and included in analyses when missing data accounted for greater than 10% of the total cohort.
To measure the variability in transfer rates between hospitals, we used a sequence of random effects logistic regression models. We first ran a model with no covariates, representing the unadjusted differences in transfer rates between hospitals. We then added patient characteristics to see if the unadjusted differences in IHT rates were explained by differences in patient characteristics between hospitals. Lastly, we added hospital characteristics to determine if these explained the remaining differences in transfer rates. Each of the 3 models provided a measure of between-hospital variability, reflecting the degree to which IHT rates differed between hospitals. Additionally, we used the intercept from the unadjusted model and the measure of between-hospital variability from each model to calculate the 95% confidence intervals, illustrating the range of IHT rates spanning 95% of all hospitals. We used those same numbers to calculate the 25th and 75th percentiles, illustrating the range of IHT rates for the middle half of hospitals.
RESULTS
Among 28 million eligible beneficiaries, 6.6 million had an acute care hospitalization to nonfederal, noncritical access hospitals, and 107,741 met our defined criteria for IHT. An additional 3790 beneficiaries were excluded for being transferred to the same facility, 416 beneficiaries (115 transferred, 301 nontransferred) were excluded as they were cared for at 1 of the 11 hospitals with “outlier” transfer in/out rates, and 2329 were excluded because they had more than 1 transfer during hospitalization. Thus, the final cohort consisted of 101,507 transferred (1.5%) and 6,625,474 nontransferred beneficiaries (Figure 1). Of the 101,507 transferred beneficiaries, 2799 (2.8%) were included more than once (ie, experienced more than 1 IHT on separate hospitalizations throughout the study period; the vast majority of these had 2 separate hospitalizations resulting in IHT). Characteristics of transferred and nontransferred beneficiaries are shown (Table 1).
Among transferred patients, the top 5 primary diagnoses at time of transfer included AMI (12.2%), congestive heart failure (CHF) (7.2%), sepsis (6.6%), arrhythmia (6.6%), and pneumonia (3.4%). Comorbid conditions most commonly present in transferred patients included CHF (52.6%), renal failure (51.8%), arrhythmia (49.8%), and chronic obstructive pulmonary disease (COPD; 37.0%). The most common day of transfer was day after admission (hospital day 2, 24.7%), with 75% of transferred patients transferred before hospital day 6 (Appendix B).
After adjusting for all other patient and hospital characteristics and clustering by hospital, the following variables were associated with greater odds of transfer: older age, male sex, nonblack race, non-Medicaid co-insurance, higher comorbidity (HCC score), lower DRG-weight, and fewer hospitalizations in the prior 12 months. Beneficiaries also had greater odds of transfer if initially hospitalized at smaller hospitals, nonteaching hospitals, public hospitals, at hospitals in the Northeast, those with fewer specialty services, and those with a low CMI (Table 2).
DISCUSSION
In this nationally representative study of 6.6 million Medicare beneficiaries, we found that 1.5% of patients were transferred between acute care facilities and were most often transferred prior to hospital day 6. Older age, male sex, nonblack race, higher medical comorbidity, lower DRG weight, and fewer recent hospitalizations were associated with greater odds of transfer. Initial hospitalization at smaller, nonteaching, public hospitals, with fewer specialty services were associated with greater odds of transfer, while higher CMI was associated with a lower odds of transfer. The most common comorbid conditions among transferred patients included CHF, renal failure, arrhythmia, and COPD; particularly notable was the very high prevalence of these conditions among transferred as compared with nontransferred patients. Importantly, we found significant variation in IHT by region and a large variation in transfer practices by hospital, with significant variability in transfer rates even after accounting for known patient and hospital characteristics.
Among our examined population, we found that a sizable number of patients undergo IHT—more than 100,000 per year. Primary diagnoses at time of transfer consist of common inpatient conditions, including AMI, CHF, sepsis, arrhythmia, and pneumonia. Limited prior data support our findings, with up to 50% of AMI patients reportedly undergoing IHT,3-5 and severe sepsis and respiratory illness reported as common diagnoses at transfer.11 Although knowledge of these primary diagnoses does not directly confer an understanding of reason for transfer, one can speculate based on our findings. For example, research demonstrates the majority of AMI patients who undergo IHT had further intervention, including stress testing, cardiac catheterization, and/or coronary artery bypass graft surgery.5,26 Thus, it is reasonable to presume that many of the beneficiaries
We additionally found that certain patient characteristics were associated with greater odds of transfer. Research suggests that transferred patients are “sicker” than nontransferred patients.1,11 Although our findings in part confirm these data, we paradoxically found that higher DRG-weight and 4 or more hospitalizations in the past year were actually associated with lower odds of transfer. In addition, the oldest patients in our cohort (85 years or older) were actually less likely to be transferred than their slightly younger counterparts (75 to 84 years). These variables may reflect extreme illness or frailty,27 and providers consciously (or subconsciously) may factor this in to their decision to transfer, considering a threshold past which transfer would confer more risk than benefit (eg, a patient may be “too sick” for transfer). Indeed, in a secondary analysis without hospital characteristics or comorbidities, and with fixed effects by hospital, we found the highest rates of IHT in patients in the middle 2 quartiles of DRG-weight, supporting this threshold hypothesis. It is also possible that patients with numerous hospitalizations may be less likely to be transferred because of familiarity and a strong sense of responsibility to continue to care for those patients (although we cannot confirm that those prior hospitalizations were all with the same index hospital).
It is also notable that odds of transfer differed by race, with black patients 17% less likely to undergo transfer compared to whites, similar to findings in other IHT studies.11 This finding, in combination with our demonstration that Medicaid patients also have lower odds of transfer, warrants further investigation to ensure the process of IHT does not bias against these populations, as with other well-documented health disparities.28-30
The hospital predictors of transfer were largely expected. However, interestingly, when we controlled for all other patient and hospital characteristics, regional variation persisted, with highest odds of transfer with hospitalization in the Northeast, indicating variability by region not explained by other factors, and findings supported by other limited data.31 This variability was further elucidated in our examination of change in variance estimates accounting for patient, then hospital, characteristics. Although we expected and found marked variability in hospital transfer rates in our null model (without accounting for any patient or hospital characteristics), we interestingly found that variability increased upon adjusting for patient characteristics. This result is presumably due to the fact that patients who are more likely to be transferred (ie, “sick” patients) are more often already at hospitals less likely to transfer patients, supported by our findings that hospital CMI is inversely associated with odds of transfer (in other words, hospitals that care for a less sick patient population are more likely to transfer their patients, and hospitals that care for a sicker patient population [higher CMI] are less likely to transfer). Adjusting solely for patient characteristics effectively equalizes these patients across hospitals, which would lead to even increased variability in transfer rates. Conversely, when we then adjusted for hospital characteristics, variability in hospital transfer rates decreased by 83% (in other words, hospital characteristics, rather than patient characteristics, explained much of the variability in transfer rates), although significant unexplained variability remained. We should note that although the observed reduction in variability was explained by the patient and hospital characteristics included in the model, these characteristics do not necessarily justify the variability they accounted for; although patients’ race or hospitals’ location may explain some of the observed variability, this does not reasonably justify it.
This observed variability in transfer practices is not surprising given the absence of standardization and clear guidelines to direct clinical IHT practice.17 Selection of patients that may benefit from transfer is often ambiguous and subjective.6 The Emergency Medical Treatment and Active Labor Act laws dictate that hospitals transfer patients requiring a more specialized service, or when “medical benefits ... outweigh the increased risks to the individual...,” although in practice this provides little guidance to practitioners.1 Thus, clearer guidelines may be necessary to achieve less variable practices.
Our study is subject to several limitations. First, although nationally representative, the Medicare population is not reflective of all hospitalized patients nationwide. Additionally, we excluded patients transferred from the emergency room. Thus, the total number of patients who undergo IHT nationally is expected to be much higher than reflected in our analysis. We also excluded patients who were transferred more than once during a given hospitalization. This enabled us to focus on the initial transfer decision but does not allow us to look at patients who are transferred to a referral center and then transferred back. Second, given the criteria we used to define transfer, it is possible that we included nontransferred patients within our transferred cohort if they were discharged from one hospital and admitted to a different hospital within 1 day. However, on quality assurance analyses where we limited our cohort to only those beneficiaries with corresponding “transfer in” and “transfer out” claims (87% of the total cohort), we found no marked differences in our results. Additionally, although we assume that patient transfer status was coded correctly within the Medicare dataset, we could not confirm by individually examining each patient we defined as “transferred.” However, on additional quality assurance analyses where we examined randomly selected excluded patients with greater than 1 transfer during hospitalization, we found differing provider numbers with each transfer, suggesting validity of the coding. Third, because there are likely many unmeasured patient confounders, we cannot be sure how much of the between-hospital variation is due to incomplete adjustment for patient characteristics. However, since adjusting for patient characteristics actually increased variability in hospital transfer rates, it is unlikely that residual patient confounders fully explain our observed results. Despite this, other variables that are not available within the CMS or AHA datasets may further elucidate hospital transfer practices, including variables reflective of the transfer process (eg, time of day of patient transfer, time delay between initiation of transfer and patient arrival at accepting hospital, accepting service on transfer, etc.); other markers of illness severity (eg, clinical service at the time of index admission, acute physiology score, utilization of critical care services on arrival at receiving hospital); and other hospital system variables (ie, membership in an accountable care organization and/or regional care network, the density of nearby tertiary referral centers (indicating possible supply-induced demand), other variables reflective of the “transfer culture” (such as the transfer rate at the hospital or region where the attending physician trained, etc.). Lastly, though our examination provides important foundational information regarding IHT nationally, this study did not examine patient outcomes in transferred and nontransferred patients, which may help to determine which patients benefit (or do not benefit) from transfer and why. Further investigation is needed to study these outcomes.
CONCLUSION
In this national study of IHT, we found that a sizable number of patients admitted to the hospital undergo transfer to another acute care facility. Patients are transferred with common medical conditions, including those requiring specialized care such as AMI, and a high rate of comorbid clinical conditions, and certain patient and hospital characteristics are associated with greater odds of transfer. Although many of the observed associations between characteristics and odds of transfer were expected based on limited existing literature, we found several unexpected findings, eg, suggesting the possibility of a threshold beyond which sicker patients are not transferred. Additionally, we found that black and Medicaid patients had lower odds of transfer, which warrants further investigation for potential health care disparity. Importantly, we found much variability in the practice of IHT, as evidenced by the inexplicable differences in transfer by hospital region, and by residual unexplained variability in hospital transfer rates after accounting for patient and hospital characteristics, which may be due to lack of standard guidelines to direct IHT practices. In conclusion, this study of hospitalized Medicare patients provides important foundational information regarding rates and predictors of IHT nationally, as well as unexplained variability that exists within this complex care transition. Further investigation will be essential to understand reasons for, processes related to, and outcomes of transferred patients, to help guide standardization in best practices in care.
Disclosure
Nothing to report.
1. Iwashyna TJ. The incomplete infrastructure for interhospital patient transfer. Crit Care Med. 2012;40(8):2470-2478. PubMed
2. Iwashyna TJ, Christie JD, Moody J, Kahn JM, Asch DA. The structure of critical care transfer networks. Med Care. 2009;47(7):787-793. PubMed
3. Mehta RH, Stalhandske EJ, McCargar PA, Ruane TJ, Eagle KA. Elderly patients at highest risk with acute myocardial infarction are more frequently transferred from community hospitals to tertiary centers: reality or myth? Am Heart J. 1999;138(4 Pt 1):688-695. PubMed
4. Iwashyna TJ, Kahn JM, Hayward RA, Nallamothu BK. Interhospital transfers among Medicare beneficiaries admitted for acute myocardial infarction at nonrevascularization hospitals. Circ Cardiovasc Qual Outcomes. 2010;3(5):468-475. PubMed
5. Roe MT, Chen AY, Delong ER, Boden WE, Calvin JE Jr, Cairns CB, et al. Patterns of transfer for patients with non-ST-segment elevation acute coronary syndrome from community to tertiary care hospitals. Am Heart J. 2008;156(1):185-192. PubMed
6. Bosk EA, Veinot T, Iwashyna TJ. Which patients and where: a qualitative study of patient transfers from community hospitals. Med Care. 2011;49(6):592-598. PubMed
7. Wagner J, Iwashyna TJ, Kahn JM. Reasons underlying interhospital transfers to an academic medical intensive care unit. J Crit Care. 2013;28(2):202-208. PubMed
8. Cohen MD, Hilligoss PB. The published literature on handoffs in hospitals: deficiencies identified in an extensive review. Qual Saf Health Care. 2010;19(6):493-497. PubMed
9. Riesenberg LA, Leitzsch J, Massucci JL, et al. Residents’ and attending physicians’ handoffs: a systematic review of the literature. Acad Med. 2009;84(12):1775-1787. PubMed
10. Arora V, Johnson J, Lovinger D, Humphrey HJ, Meltzer DO. Communication failures in patient sign-out and suggestions for improvement: a critical incident analysis. Qual Saf Health Care. 2005;14(6):401-407. PubMed
11. Sokol-Hessner L, White AA, Davis KF, Herzig SJ, Hohmann SF. Interhospital transfer patients discharged by academic hospitalists and general internists: characteristics and outcomes. J Hosp Med. 2016;11(4):245-250. PubMed
12. Bernard AM, Hayward RA, Rosevear J, Chun H, McMahon LF. Comparing the hospitalizations of transfer and non-transfer patients in an academic medical center. Acad Med. 1996;71(3):262-266. PubMed
13. Golestanian E, Scruggs JE, Gangnon RE, Mak RP, Wood KE. Effect of interhospital transfer on resource utilization and outcomes at a tertiary care referral center. Crit Care Med. 2007;35(6):1470-1476. PubMed
14. Durairaj L, Will JG, Torner JC, Doebbeling BN. Prognostic factors for mortality following interhospital transfers to the medical intensive care unit of a tertiary referral center. Crit Care Med. 2003;31(7):1981-1986. PubMed
15. Kerr HD, Byrd JC. Community hospital transfers to a VA Medical Center. JAMA. 1989;262(1):70-73. PubMed
16. Dragsted L, Jörgensen J, Jensen NH, et al. Interhospital comparisons of patient outcome from intensive care: importance of lead-time bias. Crit Care Med. 1989;17(5):418-422. PubMed
17. Gupta K, Mueller SK. Interhospital transfers: the need for standards. J Hosp Med. 2015;10(6):415-417. PubMed
18. The Dartmouth Atlas of Health Care: Understanding of the Efficiency and Effectiveness of the Health Care System. The Dartmouth Institute for Health Practice and Clinical Policy, Lebanon, NH. http://www.dartmouthatlas.org/. Accessed November 1, 2016.
19. American Hospital Association Annual Survey Database. American Hospital Association, Chicago, IL. http://www.ahadataviewer.com/book-cd-products/AHA-Survey/. Accessed July 1, 2013.
20. U.S. Department of Health and Human Services (HRSA): What are critical access hospitals (CAH)? http://www.hrsa.gov/healthit/toolbox/RuralHealthITtoolbox/Introduction/critical.html. Accessed June 9, 2016.
21. Li P, Kim MM, Doshi JA. Comparison of the performance of the CMS Hierarchical Condition Category (CMS-HCC) risk adjuster with the Charlson and Elixhauser comorbidity measures in predicting mortality. BMC Health Serv Res. 2010;10:245. PubMed
22. Hernandez-Boussard T, Davies S, McDonald K, Wang NE. Interhospital facility transfers in the United States: a nationwide outcomes study. J Patient Saf. Nov 13 2014. PubMed
23. Landon BE, Normand SL, Lessler A, et al. Quality of care for the treatment of acute medical conditions in US hospitals. Arch Intern Med. 2006;166(22):2511-2517. PubMed
24. Mueller SK, Lipsitz S, Hicks LS. Impact of hospital teaching intensity on quality of care and patient outcomes. Med Care.2013;51(7):567-574. PubMed
25. Lopez L, Hicks LS, Cohen AP, McKean S, Weissman JS. Hospitalists and the quality of care in hospitals. Arch Intern Med. 2009;169(15):1389-1394. PubMed
26. Barreto-Filho JA, Wang Y, Rathore SS, et al. Transfer rates from nonprocedure hospitals after initial admission and outcomes among elderly patients with acute myocardial infarction. JAMA Intern Med. 2014;174(2):213-222. PubMed
27. Carlson JE, Zocchi KA, Bettencourt DM, et al. Measuring frailty in the hospitalized elderly: concept of functional homeostasis. Am J Phys Med Rehabil. 1998;77(3):252-257. PubMed
28. Ward E, Jemal A, Cokkinides V, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin. 2004;54(2):78-93. PubMed
29. Iribarren C, Tolstykh I, Somkin CP, et al. Sex and racial/ethnic disparities in outcomes after acute myocardial infarction: a cohort study among members of a large integrated health care delivery system in northern California. Arch Intern Med. 2005;165(18):2105-2113. PubMed
30. Kawachi I, Daniels N, Robinson DE. Health disparities by race and class: why both matter. Health Aff (Millwood). 2005;24(2):343-352. PubMed
31. Herrigel DJ, Carroll M, Fanning C, Steinberg MB, Parikh A, Usher M. Interhospital transfer handoff practices among US tertiary care centers: a descriptive survey. J Hosp Med. 2016;11(6):413-417. PubMed
1. Iwashyna TJ. The incomplete infrastructure for interhospital patient transfer. Crit Care Med. 2012;40(8):2470-2478. PubMed
2. Iwashyna TJ, Christie JD, Moody J, Kahn JM, Asch DA. The structure of critical care transfer networks. Med Care. 2009;47(7):787-793. PubMed
3. Mehta RH, Stalhandske EJ, McCargar PA, Ruane TJ, Eagle KA. Elderly patients at highest risk with acute myocardial infarction are more frequently transferred from community hospitals to tertiary centers: reality or myth? Am Heart J. 1999;138(4 Pt 1):688-695. PubMed
4. Iwashyna TJ, Kahn JM, Hayward RA, Nallamothu BK. Interhospital transfers among Medicare beneficiaries admitted for acute myocardial infarction at nonrevascularization hospitals. Circ Cardiovasc Qual Outcomes. 2010;3(5):468-475. PubMed
5. Roe MT, Chen AY, Delong ER, Boden WE, Calvin JE Jr, Cairns CB, et al. Patterns of transfer for patients with non-ST-segment elevation acute coronary syndrome from community to tertiary care hospitals. Am Heart J. 2008;156(1):185-192. PubMed
6. Bosk EA, Veinot T, Iwashyna TJ. Which patients and where: a qualitative study of patient transfers from community hospitals. Med Care. 2011;49(6):592-598. PubMed
7. Wagner J, Iwashyna TJ, Kahn JM. Reasons underlying interhospital transfers to an academic medical intensive care unit. J Crit Care. 2013;28(2):202-208. PubMed
8. Cohen MD, Hilligoss PB. The published literature on handoffs in hospitals: deficiencies identified in an extensive review. Qual Saf Health Care. 2010;19(6):493-497. PubMed
9. Riesenberg LA, Leitzsch J, Massucci JL, et al. Residents’ and attending physicians’ handoffs: a systematic review of the literature. Acad Med. 2009;84(12):1775-1787. PubMed
10. Arora V, Johnson J, Lovinger D, Humphrey HJ, Meltzer DO. Communication failures in patient sign-out and suggestions for improvement: a critical incident analysis. Qual Saf Health Care. 2005;14(6):401-407. PubMed
11. Sokol-Hessner L, White AA, Davis KF, Herzig SJ, Hohmann SF. Interhospital transfer patients discharged by academic hospitalists and general internists: characteristics and outcomes. J Hosp Med. 2016;11(4):245-250. PubMed
12. Bernard AM, Hayward RA, Rosevear J, Chun H, McMahon LF. Comparing the hospitalizations of transfer and non-transfer patients in an academic medical center. Acad Med. 1996;71(3):262-266. PubMed
13. Golestanian E, Scruggs JE, Gangnon RE, Mak RP, Wood KE. Effect of interhospital transfer on resource utilization and outcomes at a tertiary care referral center. Crit Care Med. 2007;35(6):1470-1476. PubMed
14. Durairaj L, Will JG, Torner JC, Doebbeling BN. Prognostic factors for mortality following interhospital transfers to the medical intensive care unit of a tertiary referral center. Crit Care Med. 2003;31(7):1981-1986. PubMed
15. Kerr HD, Byrd JC. Community hospital transfers to a VA Medical Center. JAMA. 1989;262(1):70-73. PubMed
16. Dragsted L, Jörgensen J, Jensen NH, et al. Interhospital comparisons of patient outcome from intensive care: importance of lead-time bias. Crit Care Med. 1989;17(5):418-422. PubMed
17. Gupta K, Mueller SK. Interhospital transfers: the need for standards. J Hosp Med. 2015;10(6):415-417. PubMed
18. The Dartmouth Atlas of Health Care: Understanding of the Efficiency and Effectiveness of the Health Care System. The Dartmouth Institute for Health Practice and Clinical Policy, Lebanon, NH. http://www.dartmouthatlas.org/. Accessed November 1, 2016.
19. American Hospital Association Annual Survey Database. American Hospital Association, Chicago, IL. http://www.ahadataviewer.com/book-cd-products/AHA-Survey/. Accessed July 1, 2013.
20. U.S. Department of Health and Human Services (HRSA): What are critical access hospitals (CAH)? http://www.hrsa.gov/healthit/toolbox/RuralHealthITtoolbox/Introduction/critical.html. Accessed June 9, 2016.
21. Li P, Kim MM, Doshi JA. Comparison of the performance of the CMS Hierarchical Condition Category (CMS-HCC) risk adjuster with the Charlson and Elixhauser comorbidity measures in predicting mortality. BMC Health Serv Res. 2010;10:245. PubMed
22. Hernandez-Boussard T, Davies S, McDonald K, Wang NE. Interhospital facility transfers in the United States: a nationwide outcomes study. J Patient Saf. Nov 13 2014. PubMed
23. Landon BE, Normand SL, Lessler A, et al. Quality of care for the treatment of acute medical conditions in US hospitals. Arch Intern Med. 2006;166(22):2511-2517. PubMed
24. Mueller SK, Lipsitz S, Hicks LS. Impact of hospital teaching intensity on quality of care and patient outcomes. Med Care.2013;51(7):567-574. PubMed
25. Lopez L, Hicks LS, Cohen AP, McKean S, Weissman JS. Hospitalists and the quality of care in hospitals. Arch Intern Med. 2009;169(15):1389-1394. PubMed
26. Barreto-Filho JA, Wang Y, Rathore SS, et al. Transfer rates from nonprocedure hospitals after initial admission and outcomes among elderly patients with acute myocardial infarction. JAMA Intern Med. 2014;174(2):213-222. PubMed
27. Carlson JE, Zocchi KA, Bettencourt DM, et al. Measuring frailty in the hospitalized elderly: concept of functional homeostasis. Am J Phys Med Rehabil. 1998;77(3):252-257. PubMed
28. Ward E, Jemal A, Cokkinides V, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin. 2004;54(2):78-93. PubMed
29. Iribarren C, Tolstykh I, Somkin CP, et al. Sex and racial/ethnic disparities in outcomes after acute myocardial infarction: a cohort study among members of a large integrated health care delivery system in northern California. Arch Intern Med. 2005;165(18):2105-2113. PubMed
30. Kawachi I, Daniels N, Robinson DE. Health disparities by race and class: why both matter. Health Aff (Millwood). 2005;24(2):343-352. PubMed
31. Herrigel DJ, Carroll M, Fanning C, Steinberg MB, Parikh A, Usher M. Interhospital transfer handoff practices among US tertiary care centers: a descriptive survey. J Hosp Med. 2016;11(6):413-417. PubMed
© 2017 Society of Hospital Medicine
Chemoprevention: Thinking outside the box
WAILEA, HAWAII – Nicotinamide is one of the rare proposed agents for skin cancer chemoprevention distinguished by dirt cheap cost combined with a highly reassuring safety profile plus evidence of efficacy – which, together, make it a reasonable option in high risk patients, according to Daniel M. Siegel, MD.
Other agents that fit into that category include the tropical rainforest fern Polypodium leucotomos and milk thistle, added Dr. Siegel, a dermatologist at the State University of New York, Brooklyn.

“That’s a really interesting one. I don’t know if, 5 years from now, we’ll all be taking low-dose rapamycin as an antiaging drug, but we might, especially if someone figures out the ideal dose,” he said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Research Foundation.
Nicotinamide
In the case of nicotinamide, the efficacy is actually supported by published level 1 evidence in the form of a highly positive 1-year, double-blind, randomized, placebo-controlled phase III clinical trial.
“You can Google ‘nicotinamide’ and find it at places like Costco and Trader Joe’s for less than 6 cents per day. That makes for a really good risk/benefit ratio. A nickel a day: That’s a cheap one. That’s one where I’d say, ‘Why not?’ It seems to be safe,” Dr. Siegel said.
In the phase III ONTRAC trial, Australian investigators randomized 386 patients who averaged roughly eight nonmelanoma skin cancers in the past 5 years to either 500 mg of oral nicotinamide twice daily or matched placebo for 12 months. During the study period, the nicotinamide group had a statistically significant and clinically meaningful 23% reduction in new nonmelanoma skin cancers, compared with the control group. They also had 13% fewer actinic keratoses at 12 months than controls. And the side effect profile mirrored that of placebo (N Engl J Med. 2015 Oct 22;373[17]:1618-26).
“Nicotinamide is vitamin B3. It’s not niacin. It doesn’t cause flushing and other vasodilatory effects. It’s actually pretty innocuous,” Dr. Siegel said.
In laboratory studies, nicotinamide has been shown to enhance DNA repair following UV exposure, as well as curb UV-induced immunosuppression.
Polypodium leucotomos Samambaia
This plant, commonly known as calaguala in the Spanish-speaking tropics and samambaia in Brazil, has a centuries-long tradition of safe medicinal use. It is commercially available over-the-counter (OTC) as a standardized product called Heliocare, designed to avoid the guesswork involved in topical sunscreen application. Each capsule contains 240 mg of an extract of P. leucotomos. Dr. Siegel said he takes it daily when he’s in a sunny locale, such as Hawaii.
Milk thistle
This plant, known as Silybum marianum, has silymarin as its bioactive compound. Dermatologist Haines Ely, MD, of the University of California, Davis, has reported therapeutic success using it in porphyria cutanea tarda and other conditions. It has been shown to inhibit photocarcinogenesis in animal studies.
Dr. Siegel said that, while Dr. Ely has told him his preferred preparation is a German OTC product, milk thistle seeds can be found in health food stores, ground to a powder using a coffee bean grinder, and used as a food supplement. Like Polypodium leucotomos and nicotinamide, milk thistle is nontoxic.
Rapamycin
This macrolide compound is produced by the bacterium Streptomyces hygroscopicus. Rapamycin is an immunosuppressant used to coat coronary stents and prevent rejection of transplanted organs. It is an mechanistic target of rapamycin signaling pathway inhibitor being studied as a cancer prevention and antiaging agent.
Science magazine called the discovery that rapamycin increased the lifespan of mice one of the top scientific breakthroughs of 2009. Subsequent animal studies have established that the extended lifespan wasn’t solely the result of rapamycin’s antineoplastic effects but of across-the-board delayed onset of all the major age-related diseases. Thus, rapamycin could turn out to be a true antiaging agent, in Dr. Siegel’s view.
Studies in humans are underway. Researchers at Novartis have reported that a rapamycin-related compound curbed the typical decline in immune function that accompanies aging as reflected in a 20% enhancement in the response to influenza vaccine in elderly volunteers (Sci Transl Med. 2014 Dec 24;6[268]:268ra179).
Dr. Siegel reported serving as a consultant to Ferndale, which markets Heliocare. The SDEF and this news organization are owned by the same parent company.
WAILEA, HAWAII – Nicotinamide is one of the rare proposed agents for skin cancer chemoprevention distinguished by dirt cheap cost combined with a highly reassuring safety profile plus evidence of efficacy – which, together, make it a reasonable option in high risk patients, according to Daniel M. Siegel, MD.
Other agents that fit into that category include the tropical rainforest fern Polypodium leucotomos and milk thistle, added Dr. Siegel, a dermatologist at the State University of New York, Brooklyn.
“That’s a really interesting one. I don’t know if, 5 years from now, we’ll all be taking low-dose rapamycin as an antiaging drug, but we might, especially if someone figures out the ideal dose,” he said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Research Foundation.
Nicotinamide
In the case of nicotinamide, the efficacy is actually supported by published level 1 evidence in the form of a highly positive 1-year, double-blind, randomized, placebo-controlled phase III clinical trial.
“You can Google ‘nicotinamide’ and find it at places like Costco and Trader Joe’s for less than 6 cents per day. That makes for a really good risk/benefit ratio. A nickel a day: That’s a cheap one. That’s one where I’d say, ‘Why not?’ It seems to be safe,” Dr. Siegel said.
In the phase III ONTRAC trial, Australian investigators randomized 386 patients who averaged roughly eight nonmelanoma skin cancers in the past 5 years to either 500 mg of oral nicotinamide twice daily or matched placebo for 12 months. During the study period, the nicotinamide group had a statistically significant and clinically meaningful 23% reduction in new nonmelanoma skin cancers, compared with the control group. They also had 13% fewer actinic keratoses at 12 months than controls. And the side effect profile mirrored that of placebo (N Engl J Med. 2015 Oct 22;373[17]:1618-26).
“Nicotinamide is vitamin B3. It’s not niacin. It doesn’t cause flushing and other vasodilatory effects. It’s actually pretty innocuous,” Dr. Siegel said.
In laboratory studies, nicotinamide has been shown to enhance DNA repair following UV exposure, as well as curb UV-induced immunosuppression.
Polypodium leucotomos Samambaia
This plant, commonly known as calaguala in the Spanish-speaking tropics and samambaia in Brazil, has a centuries-long tradition of safe medicinal use. It is commercially available over-the-counter (OTC) as a standardized product called Heliocare, designed to avoid the guesswork involved in topical sunscreen application. Each capsule contains 240 mg of an extract of P. leucotomos. Dr. Siegel said he takes it daily when he’s in a sunny locale, such as Hawaii.
Milk thistle
This plant, known as Silybum marianum, has silymarin as its bioactive compound. Dermatologist Haines Ely, MD, of the University of California, Davis, has reported therapeutic success using it in porphyria cutanea tarda and other conditions. It has been shown to inhibit photocarcinogenesis in animal studies.
Dr. Siegel said that, while Dr. Ely has told him his preferred preparation is a German OTC product, milk thistle seeds can be found in health food stores, ground to a powder using a coffee bean grinder, and used as a food supplement. Like Polypodium leucotomos and nicotinamide, milk thistle is nontoxic.
Rapamycin
This macrolide compound is produced by the bacterium Streptomyces hygroscopicus. Rapamycin is an immunosuppressant used to coat coronary stents and prevent rejection of transplanted organs. It is an mechanistic target of rapamycin signaling pathway inhibitor being studied as a cancer prevention and antiaging agent.
Science magazine called the discovery that rapamycin increased the lifespan of mice one of the top scientific breakthroughs of 2009. Subsequent animal studies have established that the extended lifespan wasn’t solely the result of rapamycin’s antineoplastic effects but of across-the-board delayed onset of all the major age-related diseases. Thus, rapamycin could turn out to be a true antiaging agent, in Dr. Siegel’s view.
Studies in humans are underway. Researchers at Novartis have reported that a rapamycin-related compound curbed the typical decline in immune function that accompanies aging as reflected in a 20% enhancement in the response to influenza vaccine in elderly volunteers (Sci Transl Med. 2014 Dec 24;6[268]:268ra179).
Dr. Siegel reported serving as a consultant to Ferndale, which markets Heliocare. The SDEF and this news organization are owned by the same parent company.
WAILEA, HAWAII – Nicotinamide is one of the rare proposed agents for skin cancer chemoprevention distinguished by dirt cheap cost combined with a highly reassuring safety profile plus evidence of efficacy – which, together, make it a reasonable option in high risk patients, according to Daniel M. Siegel, MD.
Other agents that fit into that category include the tropical rainforest fern Polypodium leucotomos and milk thistle, added Dr. Siegel, a dermatologist at the State University of New York, Brooklyn.
“That’s a really interesting one. I don’t know if, 5 years from now, we’ll all be taking low-dose rapamycin as an antiaging drug, but we might, especially if someone figures out the ideal dose,” he said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Research Foundation.
Nicotinamide
In the case of nicotinamide, the efficacy is actually supported by published level 1 evidence in the form of a highly positive 1-year, double-blind, randomized, placebo-controlled phase III clinical trial.
“You can Google ‘nicotinamide’ and find it at places like Costco and Trader Joe’s for less than 6 cents per day. That makes for a really good risk/benefit ratio. A nickel a day: That’s a cheap one. That’s one where I’d say, ‘Why not?’ It seems to be safe,” Dr. Siegel said.
In the phase III ONTRAC trial, Australian investigators randomized 386 patients who averaged roughly eight nonmelanoma skin cancers in the past 5 years to either 500 mg of oral nicotinamide twice daily or matched placebo for 12 months. During the study period, the nicotinamide group had a statistically significant and clinically meaningful 23% reduction in new nonmelanoma skin cancers, compared with the control group. They also had 13% fewer actinic keratoses at 12 months than controls. And the side effect profile mirrored that of placebo (N Engl J Med. 2015 Oct 22;373[17]:1618-26).
“Nicotinamide is vitamin B3. It’s not niacin. It doesn’t cause flushing and other vasodilatory effects. It’s actually pretty innocuous,” Dr. Siegel said.
In laboratory studies, nicotinamide has been shown to enhance DNA repair following UV exposure, as well as curb UV-induced immunosuppression.
Polypodium leucotomos Samambaia
This plant, commonly known as calaguala in the Spanish-speaking tropics and samambaia in Brazil, has a centuries-long tradition of safe medicinal use. It is commercially available over-the-counter (OTC) as a standardized product called Heliocare, designed to avoid the guesswork involved in topical sunscreen application. Each capsule contains 240 mg of an extract of P. leucotomos. Dr. Siegel said he takes it daily when he’s in a sunny locale, such as Hawaii.
Milk thistle
This plant, known as Silybum marianum, has silymarin as its bioactive compound. Dermatologist Haines Ely, MD, of the University of California, Davis, has reported therapeutic success using it in porphyria cutanea tarda and other conditions. It has been shown to inhibit photocarcinogenesis in animal studies.
Dr. Siegel said that, while Dr. Ely has told him his preferred preparation is a German OTC product, milk thistle seeds can be found in health food stores, ground to a powder using a coffee bean grinder, and used as a food supplement. Like Polypodium leucotomos and nicotinamide, milk thistle is nontoxic.
Rapamycin
This macrolide compound is produced by the bacterium Streptomyces hygroscopicus. Rapamycin is an immunosuppressant used to coat coronary stents and prevent rejection of transplanted organs. It is an mechanistic target of rapamycin signaling pathway inhibitor being studied as a cancer prevention and antiaging agent.
Science magazine called the discovery that rapamycin increased the lifespan of mice one of the top scientific breakthroughs of 2009. Subsequent animal studies have established that the extended lifespan wasn’t solely the result of rapamycin’s antineoplastic effects but of across-the-board delayed onset of all the major age-related diseases. Thus, rapamycin could turn out to be a true antiaging agent, in Dr. Siegel’s view.
Studies in humans are underway. Researchers at Novartis have reported that a rapamycin-related compound curbed the typical decline in immune function that accompanies aging as reflected in a 20% enhancement in the response to influenza vaccine in elderly volunteers (Sci Transl Med. 2014 Dec 24;6[268]:268ra179).
Dr. Siegel reported serving as a consultant to Ferndale, which markets Heliocare. The SDEF and this news organization are owned by the same parent company.
EXPERT ANALYSIS FROM SDEF HAWAII DERMATOLOGY SEMINAR
Gefitinib bests chemo as adjuvant therapy for early EGFR-mutant NSCLC
The targeted agent gefitinib is superior to the standard of care chemotherapy for treating resected early non–small cell lung cancer (NSCLC) harboring an epidermal growth factor receptor (EGFR) activating mutation, finds the phase III randomized Chinese ADJUVANT trial.
Gefitinib, an oral tyrosine kinase inhibitor that targets the EGFR kinase among others, is already approved by the Food and Drug Administration for treatment of locally advanced or metastatic disease having mutations in the gene for this receptor.
Trial results reported in a presscast leading up to the annual meeting of the American Society of Clinical Oncology showed that compared with vinorelbine and cisplatin combination chemotherapy, 2 years of gefitinib prolonged the time to recurrence or death by more than 10 months, reducing risk of these events by a significant 40%. Gefitinib also was better tolerated: The rate of grade 3 or worse adverse events with the targeted agent was one-fourth that seen with the chemotherapy.
“Targeted therapy can delay recurrence of intermediate-stage lung cancer after surgery. Two-year treatment duration of gefitinib is efficacious and tolerated well,” said lead study author Yi-Long Wu, MD, director of the Guangdong Lung Cancer Institute, Guangdong General Hospital, Guangzhou, China. “Adjuvant gefitinib should be considered as an important option for stage II to IIIA lung cancer patients with an activating EGFR mutation.”
Clinical implications
The improved disease-free survival seen with gefitinib in ADJUVANT is “encouraging,” according to ASCO President-Elect Bruce E. Johnson, MD, chief clinical research officer and an Institute Physician at the Dana-Farber Cancer Institute in Boston.
Longer follow-up will be needed to obtain a full picture as the horizon for events in the adjuvant setting is more on the order of years, and the disease-free survival curves began converging over time, he noted. “We will ultimately be interested in seeing whether this actually prolongs survival in a longer follow-up study, which Dr. Wu’s group is planning to do.
“I haven’t changed my approach yet for the patients with EGFR-mutant lung cancer,” Dr. Johnson concluded. “But I will be following this [trial] very closely to see what happens to the survival.”
The new data from ADJUVANT will likely have several effects on the clinical management of NSCLC, according to presscast moderator and ASCO Chief Medical Officer Richard L. Schilsky, MD.
“I suspect that many doctors will begin testing these lung cancer tumors right after surgery, to see if they actually have an EGFR mutation. That is not currently standard of care in the U.S.; typically the testing doesn’t take place until the cancer recurs or becomes metastatic,” he said. “So that way, doctors and patients will know whether or not treatment with an EGFR inhibitor is even an option.
“If it is an option, then many factors will likely come into play, and most importantly we will be waiting for the survival data,” said Dr. Schilsky, professor emeritus at the University of Chicago. Another consideration is that the trial compared 12 weeks of chemotherapy with 2 years of continuous gefitinib therapy, the latter of which requires a long-term commitment to adherence by patients and carries much greater cost.
“At the end of the day, I think that once the survival data is known in particular, doctors and patients are going to have to have very thoughtful discussions about what is the magnitude of the survival benefit; what is the burden on the patient to take either cytotoxic chemotherapy for 12 weeks or 2 years of an oral treatment, which, while it is less toxic, is not without toxicity; and what’s the financial burden of that treatment choice going to be for the patient,” he concluded.
Study details
Eligibility for the ADJUVANT trial required completely resected pathological stage II-IIIA (N1-N2) NSCLC with an EGFR-activating mutation. In all, 220 patients were randomized evenly to receive gefitinib (Iressa) once daily for 24 months or vinorelbine plus cisplatin every 3 weeks for 4 cycles.
Results showed that median disease-free survival – the trial’s primary endpoint—was 28.7 months with gefitinib compared with 18.0 months with chemotherapy (hazard ratio, 0.60; P = .005), Dr. Wu reported in the presscast. Corresponding 3-year disease-free survival rates were 34% and 27%.
The rate of grade 3 or worse adverse events was 12.3% in the gefitinib group, compared with 48.3% in the chemotherapy group. Most types of events were less common with the tyrosine kinase inhibitor, with the exception of rash, diarrhea, and elevation of liver enzymes.
Dr. Wu disclosed ties with AstraZeneca, Roche, Merck, Boehringer Ingelheim; Lilly, Pierre Fabre, Pfizer, and Sanofi. The Chinese Thoracic Oncology Group and AstraZeneca Chin funded the trial.
The targeted agent gefitinib is superior to the standard of care chemotherapy for treating resected early non–small cell lung cancer (NSCLC) harboring an epidermal growth factor receptor (EGFR) activating mutation, finds the phase III randomized Chinese ADJUVANT trial.
Gefitinib, an oral tyrosine kinase inhibitor that targets the EGFR kinase among others, is already approved by the Food and Drug Administration for treatment of locally advanced or metastatic disease having mutations in the gene for this receptor.
Trial results reported in a presscast leading up to the annual meeting of the American Society of Clinical Oncology showed that compared with vinorelbine and cisplatin combination chemotherapy, 2 years of gefitinib prolonged the time to recurrence or death by more than 10 months, reducing risk of these events by a significant 40%. Gefitinib also was better tolerated: The rate of grade 3 or worse adverse events with the targeted agent was one-fourth that seen with the chemotherapy.
“Targeted therapy can delay recurrence of intermediate-stage lung cancer after surgery. Two-year treatment duration of gefitinib is efficacious and tolerated well,” said lead study author Yi-Long Wu, MD, director of the Guangdong Lung Cancer Institute, Guangdong General Hospital, Guangzhou, China. “Adjuvant gefitinib should be considered as an important option for stage II to IIIA lung cancer patients with an activating EGFR mutation.”
Clinical implications
The improved disease-free survival seen with gefitinib in ADJUVANT is “encouraging,” according to ASCO President-Elect Bruce E. Johnson, MD, chief clinical research officer and an Institute Physician at the Dana-Farber Cancer Institute in Boston.
Longer follow-up will be needed to obtain a full picture as the horizon for events in the adjuvant setting is more on the order of years, and the disease-free survival curves began converging over time, he noted. “We will ultimately be interested in seeing whether this actually prolongs survival in a longer follow-up study, which Dr. Wu’s group is planning to do.
“I haven’t changed my approach yet for the patients with EGFR-mutant lung cancer,” Dr. Johnson concluded. “But I will be following this [trial] very closely to see what happens to the survival.”
The new data from ADJUVANT will likely have several effects on the clinical management of NSCLC, according to presscast moderator and ASCO Chief Medical Officer Richard L. Schilsky, MD.
“I suspect that many doctors will begin testing these lung cancer tumors right after surgery, to see if they actually have an EGFR mutation. That is not currently standard of care in the U.S.; typically the testing doesn’t take place until the cancer recurs or becomes metastatic,” he said. “So that way, doctors and patients will know whether or not treatment with an EGFR inhibitor is even an option.
“If it is an option, then many factors will likely come into play, and most importantly we will be waiting for the survival data,” said Dr. Schilsky, professor emeritus at the University of Chicago. Another consideration is that the trial compared 12 weeks of chemotherapy with 2 years of continuous gefitinib therapy, the latter of which requires a long-term commitment to adherence by patients and carries much greater cost.
“At the end of the day, I think that once the survival data is known in particular, doctors and patients are going to have to have very thoughtful discussions about what is the magnitude of the survival benefit; what is the burden on the patient to take either cytotoxic chemotherapy for 12 weeks or 2 years of an oral treatment, which, while it is less toxic, is not without toxicity; and what’s the financial burden of that treatment choice going to be for the patient,” he concluded.
Study details
Eligibility for the ADJUVANT trial required completely resected pathological stage II-IIIA (N1-N2) NSCLC with an EGFR-activating mutation. In all, 220 patients were randomized evenly to receive gefitinib (Iressa) once daily for 24 months or vinorelbine plus cisplatin every 3 weeks for 4 cycles.
Results showed that median disease-free survival – the trial’s primary endpoint—was 28.7 months with gefitinib compared with 18.0 months with chemotherapy (hazard ratio, 0.60; P = .005), Dr. Wu reported in the presscast. Corresponding 3-year disease-free survival rates were 34% and 27%.
The rate of grade 3 or worse adverse events was 12.3% in the gefitinib group, compared with 48.3% in the chemotherapy group. Most types of events were less common with the tyrosine kinase inhibitor, with the exception of rash, diarrhea, and elevation of liver enzymes.
Dr. Wu disclosed ties with AstraZeneca, Roche, Merck, Boehringer Ingelheim; Lilly, Pierre Fabre, Pfizer, and Sanofi. The Chinese Thoracic Oncology Group and AstraZeneca Chin funded the trial.
The targeted agent gefitinib is superior to the standard of care chemotherapy for treating resected early non–small cell lung cancer (NSCLC) harboring an epidermal growth factor receptor (EGFR) activating mutation, finds the phase III randomized Chinese ADJUVANT trial.
Gefitinib, an oral tyrosine kinase inhibitor that targets the EGFR kinase among others, is already approved by the Food and Drug Administration for treatment of locally advanced or metastatic disease having mutations in the gene for this receptor.
Trial results reported in a presscast leading up to the annual meeting of the American Society of Clinical Oncology showed that compared with vinorelbine and cisplatin combination chemotherapy, 2 years of gefitinib prolonged the time to recurrence or death by more than 10 months, reducing risk of these events by a significant 40%. Gefitinib also was better tolerated: The rate of grade 3 or worse adverse events with the targeted agent was one-fourth that seen with the chemotherapy.
“Targeted therapy can delay recurrence of intermediate-stage lung cancer after surgery. Two-year treatment duration of gefitinib is efficacious and tolerated well,” said lead study author Yi-Long Wu, MD, director of the Guangdong Lung Cancer Institute, Guangdong General Hospital, Guangzhou, China. “Adjuvant gefitinib should be considered as an important option for stage II to IIIA lung cancer patients with an activating EGFR mutation.”
Clinical implications
The improved disease-free survival seen with gefitinib in ADJUVANT is “encouraging,” according to ASCO President-Elect Bruce E. Johnson, MD, chief clinical research officer and an Institute Physician at the Dana-Farber Cancer Institute in Boston.
Longer follow-up will be needed to obtain a full picture as the horizon for events in the adjuvant setting is more on the order of years, and the disease-free survival curves began converging over time, he noted. “We will ultimately be interested in seeing whether this actually prolongs survival in a longer follow-up study, which Dr. Wu’s group is planning to do.
“I haven’t changed my approach yet for the patients with EGFR-mutant lung cancer,” Dr. Johnson concluded. “But I will be following this [trial] very closely to see what happens to the survival.”
The new data from ADJUVANT will likely have several effects on the clinical management of NSCLC, according to presscast moderator and ASCO Chief Medical Officer Richard L. Schilsky, MD.
“I suspect that many doctors will begin testing these lung cancer tumors right after surgery, to see if they actually have an EGFR mutation. That is not currently standard of care in the U.S.; typically the testing doesn’t take place until the cancer recurs or becomes metastatic,” he said. “So that way, doctors and patients will know whether or not treatment with an EGFR inhibitor is even an option.
“If it is an option, then many factors will likely come into play, and most importantly we will be waiting for the survival data,” said Dr. Schilsky, professor emeritus at the University of Chicago. Another consideration is that the trial compared 12 weeks of chemotherapy with 2 years of continuous gefitinib therapy, the latter of which requires a long-term commitment to adherence by patients and carries much greater cost.
“At the end of the day, I think that once the survival data is known in particular, doctors and patients are going to have to have very thoughtful discussions about what is the magnitude of the survival benefit; what is the burden on the patient to take either cytotoxic chemotherapy for 12 weeks or 2 years of an oral treatment, which, while it is less toxic, is not without toxicity; and what’s the financial burden of that treatment choice going to be for the patient,” he concluded.
Study details
Eligibility for the ADJUVANT trial required completely resected pathological stage II-IIIA (N1-N2) NSCLC with an EGFR-activating mutation. In all, 220 patients were randomized evenly to receive gefitinib (Iressa) once daily for 24 months or vinorelbine plus cisplatin every 3 weeks for 4 cycles.
Results showed that median disease-free survival – the trial’s primary endpoint—was 28.7 months with gefitinib compared with 18.0 months with chemotherapy (hazard ratio, 0.60; P = .005), Dr. Wu reported in the presscast. Corresponding 3-year disease-free survival rates were 34% and 27%.
The rate of grade 3 or worse adverse events was 12.3% in the gefitinib group, compared with 48.3% in the chemotherapy group. Most types of events were less common with the tyrosine kinase inhibitor, with the exception of rash, diarrhea, and elevation of liver enzymes.
Dr. Wu disclosed ties with AstraZeneca, Roche, Merck, Boehringer Ingelheim; Lilly, Pierre Fabre, Pfizer, and Sanofi. The Chinese Thoracic Oncology Group and AstraZeneca Chin funded the trial.
FROM THE 2017 ASCO ANNUAL MEETING
Key clinical point:
Major finding: Patients in the gefitinib group had a sharply reduced risk of recurrence or death relative to peers in the vinorelbine-cisplatin group (hazard ratio, 0.60; P = .005).
Data source: ADJUVANT, a phase III randomized controlled study of 220 patients with completely resected EGFR-mutant pathological stage II-IIIA (N1-N2) NSCLC.
Disclosures: Dr. Wu disclosed ties with AstraZeneca, Roche, Merck, Boehringer Ingelheim; Lilly, Pierre Fabre, Pfizer, and Sanofi. The Chinese Thoracic Oncology Group and AstraZeneca Chin funded the trial.