User login
Rash from waist to below knees
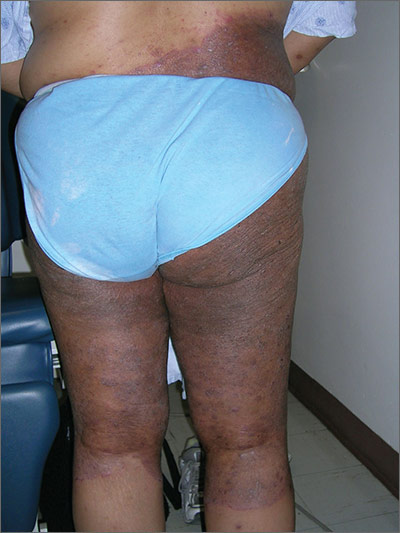
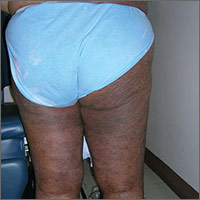
The FP suspected a fungal infection that was worsened by topical triamcinolone. He performed a potassium hydroxide (KOH) preparation and it was positive for branching septate hyphae, confirming the diagnosis of tinea corporis (See video on how to perform a KOH preparation here).
Knowing something about fungal epidemiology, he realized that this was most likely Trichophyton rubrum. He looked at the patient’s feet as well, but she did not have evidence of tinea pedis or onychomycosis. This patient’s diagnosis could also be called tinea incognito because a topical steroid was applied to the area, potentially changing the appearance of the infection. Since the infection was also in the groin, the term tinea cruris would describe that part of the rash. The hyperpigmentation seen is one form of post-inflammatory hyperpigmentation and may or may not resolve after the tinea is eradicated.
The FP knew that a topical antifungal cream wouldn’t be able to cure this massive case of tinea corporis, so he prescribed oral terbinafine 250 mg/d for 3 weeks. He also advised the patient to discontinue the hydroxyzine and the triamcinolone. The patient returned for a follow-up visit 3 weeks later and was delighted by the results. She had been able to sleep well—without incessant itching—for the first time in a year.
One month later, she returned to the FP with regrowth of fungus in the involved area. It wasn’t as bad as when she first presented, but the KOH preparation was again positive. The FP reassessed the situation and realized that large tinea infections may require more than the recommended duration of therapy. As she had no risk factors for liver disease and previous liver function tests were normal, the FP gave the patient an additional 4 weeks of oral terbinafine 250 mg/d. A 2-month follow-up appointment was arranged, at which time there was no further evidence of an active fungal infection. The post-inflammatory hyperpigmentation was lighter, but not fully gone.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R, Jimenez A. Tinea corporis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill;2013:788-794.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP suspected a fungal infection that was worsened by topical triamcinolone. He performed a potassium hydroxide (KOH) preparation and it was positive for branching septate hyphae, confirming the diagnosis of tinea corporis (See video on how to perform a KOH preparation here).
Knowing something about fungal epidemiology, he realized that this was most likely Trichophyton rubrum. He looked at the patient’s feet as well, but she did not have evidence of tinea pedis or onychomycosis. This patient’s diagnosis could also be called tinea incognito because a topical steroid was applied to the area, potentially changing the appearance of the infection. Since the infection was also in the groin, the term tinea cruris would describe that part of the rash. The hyperpigmentation seen is one form of post-inflammatory hyperpigmentation and may or may not resolve after the tinea is eradicated.
The FP knew that a topical antifungal cream wouldn’t be able to cure this massive case of tinea corporis, so he prescribed oral terbinafine 250 mg/d for 3 weeks. He also advised the patient to discontinue the hydroxyzine and the triamcinolone. The patient returned for a follow-up visit 3 weeks later and was delighted by the results. She had been able to sleep well—without incessant itching—for the first time in a year.
One month later, she returned to the FP with regrowth of fungus in the involved area. It wasn’t as bad as when she first presented, but the KOH preparation was again positive. The FP reassessed the situation and realized that large tinea infections may require more than the recommended duration of therapy. As she had no risk factors for liver disease and previous liver function tests were normal, the FP gave the patient an additional 4 weeks of oral terbinafine 250 mg/d. A 2-month follow-up appointment was arranged, at which time there was no further evidence of an active fungal infection. The post-inflammatory hyperpigmentation was lighter, but not fully gone.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R, Jimenez A. Tinea corporis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill;2013:788-794.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
The FP suspected a fungal infection that was worsened by topical triamcinolone. He performed a potassium hydroxide (KOH) preparation and it was positive for branching septate hyphae, confirming the diagnosis of tinea corporis (See video on how to perform a KOH preparation here).
Knowing something about fungal epidemiology, he realized that this was most likely Trichophyton rubrum. He looked at the patient’s feet as well, but she did not have evidence of tinea pedis or onychomycosis. This patient’s diagnosis could also be called tinea incognito because a topical steroid was applied to the area, potentially changing the appearance of the infection. Since the infection was also in the groin, the term tinea cruris would describe that part of the rash. The hyperpigmentation seen is one form of post-inflammatory hyperpigmentation and may or may not resolve after the tinea is eradicated.
The FP knew that a topical antifungal cream wouldn’t be able to cure this massive case of tinea corporis, so he prescribed oral terbinafine 250 mg/d for 3 weeks. He also advised the patient to discontinue the hydroxyzine and the triamcinolone. The patient returned for a follow-up visit 3 weeks later and was delighted by the results. She had been able to sleep well—without incessant itching—for the first time in a year.
One month later, she returned to the FP with regrowth of fungus in the involved area. It wasn’t as bad as when she first presented, but the KOH preparation was again positive. The FP reassessed the situation and realized that large tinea infections may require more than the recommended duration of therapy. As she had no risk factors for liver disease and previous liver function tests were normal, the FP gave the patient an additional 4 weeks of oral terbinafine 250 mg/d. A 2-month follow-up appointment was arranged, at which time there was no further evidence of an active fungal infection. The post-inflammatory hyperpigmentation was lighter, but not fully gone.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Usatine R, Jimenez A. Tinea corporis. In: Usatine R, Smith M, Mayeaux EJ, et al, eds. Color Atlas of Family Medicine. 2nd ed. New York, NY: McGraw-Hill;2013:788-794.
To learn more about the Color Atlas of Family Medicine, see: www.amazon.com/Color-Family-Medicine-Richard-Usatine/dp/0071769641/
You can now get the second edition of the Color Atlas of Family Medicine as an app by clicking on this link: usatinemedia.com
A Creepy Crawly Anomaly
ANSWER
The correct answer is dermatofibroma (choice “c”), based on the classic histologic picture and the lack of supportive findings for other items in the differential. These include seborrheic keratosis, granular cell tumor, basal cell carcinoma, and sweat duct cancer.
DISCUSSION
This is a perfect example of one of the most common benign tumors, seen daily in dermatology offices worldwide. Alternately referred to as superficial benign fibrous histiocytomas, dermatofibromas (DFs) typically manifest on lower extremities, are about twice as common in women as in men, and usually affect patients in their early 40s.
DFs appear most commonly as low, firm, round to ovoid, pinkish brown papules that dimple with lateral digital pressure. Though not pathognomic, the “dimple sign” is highly suggestive of this diagnosis. DF lesions can also manifest as firm, convex papules or nodules (as in this case) with the same coloring but without the dimple sign.
For years, DFs were believed to be a reaction to trauma (eg, bug bite). While this theory still has its adherents, more recent studies suggest these are true tumors composed of skin fibroblasts. Their ability to occur internally, even in bone, provides further evidence against their putatively reactive nature.
Histologically, the typical DF shows whorling fascicles of a fibroblastic spindle cell proliferation in the dermis. By contrast, the most dangerous item in the differential, the rare but greatly feared malignant dermatofibrosarcoma protuberans, is characterized by a storiform (cartwheel-shaped) pattern of spindle cells.
DFs are often subject to trauma from shaving and therefore surgically removed. However, since this was not the case for this patient, she chose to leave her lesion in place.
ANSWER
The correct answer is dermatofibroma (choice “c”), based on the classic histologic picture and the lack of supportive findings for other items in the differential. These include seborrheic keratosis, granular cell tumor, basal cell carcinoma, and sweat duct cancer.
DISCUSSION
This is a perfect example of one of the most common benign tumors, seen daily in dermatology offices worldwide. Alternately referred to as superficial benign fibrous histiocytomas, dermatofibromas (DFs) typically manifest on lower extremities, are about twice as common in women as in men, and usually affect patients in their early 40s.
DFs appear most commonly as low, firm, round to ovoid, pinkish brown papules that dimple with lateral digital pressure. Though not pathognomic, the “dimple sign” is highly suggestive of this diagnosis. DF lesions can also manifest as firm, convex papules or nodules (as in this case) with the same coloring but without the dimple sign.
For years, DFs were believed to be a reaction to trauma (eg, bug bite). While this theory still has its adherents, more recent studies suggest these are true tumors composed of skin fibroblasts. Their ability to occur internally, even in bone, provides further evidence against their putatively reactive nature.
Histologically, the typical DF shows whorling fascicles of a fibroblastic spindle cell proliferation in the dermis. By contrast, the most dangerous item in the differential, the rare but greatly feared malignant dermatofibrosarcoma protuberans, is characterized by a storiform (cartwheel-shaped) pattern of spindle cells.
DFs are often subject to trauma from shaving and therefore surgically removed. However, since this was not the case for this patient, she chose to leave her lesion in place.
ANSWER
The correct answer is dermatofibroma (choice “c”), based on the classic histologic picture and the lack of supportive findings for other items in the differential. These include seborrheic keratosis, granular cell tumor, basal cell carcinoma, and sweat duct cancer.
DISCUSSION
This is a perfect example of one of the most common benign tumors, seen daily in dermatology offices worldwide. Alternately referred to as superficial benign fibrous histiocytomas, dermatofibromas (DFs) typically manifest on lower extremities, are about twice as common in women as in men, and usually affect patients in their early 40s.
DFs appear most commonly as low, firm, round to ovoid, pinkish brown papules that dimple with lateral digital pressure. Though not pathognomic, the “dimple sign” is highly suggestive of this diagnosis. DF lesions can also manifest as firm, convex papules or nodules (as in this case) with the same coloring but without the dimple sign.
For years, DFs were believed to be a reaction to trauma (eg, bug bite). While this theory still has its adherents, more recent studies suggest these are true tumors composed of skin fibroblasts. Their ability to occur internally, even in bone, provides further evidence against their putatively reactive nature.
Histologically, the typical DF shows whorling fascicles of a fibroblastic spindle cell proliferation in the dermis. By contrast, the most dangerous item in the differential, the rare but greatly feared malignant dermatofibrosarcoma protuberans, is characterized by a storiform (cartwheel-shaped) pattern of spindle cells.
DFs are often subject to trauma from shaving and therefore surgically removed. However, since this was not the case for this patient, she chose to leave her lesion in place.
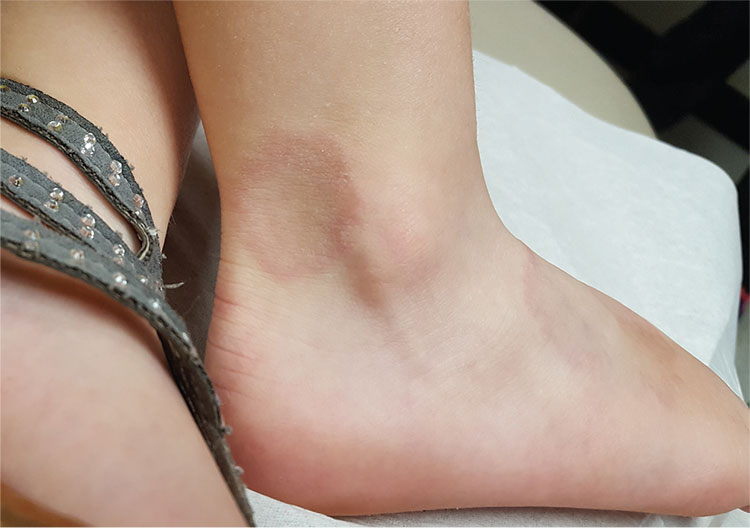
A 48-year-old woman is referred to dermatology for examination of a lesion on her ankle. Although the patient is certain that it has remained unchanged for years, she takes her primary care provider’s recommendation and agrees to be seen. She says the lesion occasionally itches and has a “crawly” feeling to it, but its size remains consistent.
Located on the lateral aspect of her left ankle is a brownish red, firm, round, intradermal nodule. It measures 8.5 mm and has a faint brown macular halo around it. Biopsy shows multiple round fascicles of spindle cells proliferating in the dermis. Special stains rule out the possibility of dermatofibrosarcoma protuberans, the most serious item in the differential.
The patient’s medical history includes breast cancer and a significant family history of skin cancer.

Suicide risk factors differ for women in military than in civilian population
WASHINGTON – Women service personnel face different suicide risks from their civilian counterparts, according to a Department of Defense appointee.
Data are few about suicide among women in the military – in part because not much research has been conducted over the years into service women’s health outcomes – according to Jacqueline Garrick, but insights gleaned from the reports of military women, both active duty and veterans, who survived suicide attempts, shed light on what to look for as risk factors. Ms. Garrick, special assistant, Manpower and Reserve Affairs in the Department of Defense, made her comments during a panel discussion at the American Psychiatric Association’s Institute on Psychiatric Services.*
One of the most salient of suicide risks can emerge when a service woman’s intimate relationship ends. This loss is compounded by the absence of social support that results from the military’s inherently masculine environment where “fitting in is definitely harder for women,” according to Ms. Garrick, a licensed clinical social worker, U.S. Army veteran, and policy analyst.
Deployment and combat zone traumas, whether physical, mental, or both, are other risk factors. Horrors witnessed in war can have psychological implications for men and women personnel. But for women, who also possibly face additional concerns of sexual assault and lack of social support, the traumas can become debilitating and lead to risk of suicide, Ms. Garrick said.
Women in the military overlap with civilians in their suicide risk factors where mental health history, abuse, and exposure to suicide are concerned, but where the two cohorts particularly diverge, Ms. Garrick said, is access to lethal means, particularly among women veterans. Civilian women who attempt suicide are more likely to cut themselves or overdose on drugs, whereas, “Military women have firearms, and they know how to use them,” Ms. Garrick said. “So, if you’re screening [for suicide in this population], pay close attention to whether there are weapons in the home.”
Traumatic brain injury is another area in which risks for suicide in military women could exist, but not enough is known at this point, Ms. Garrick said.
A suicide risk intervention called “safety planning” is one that Ms. Garrick said she has been developing in her work with the DOD. This includes asking these women what makes them feel “safe” at home, determining what their families know about the whereabouts and the safety features of their firearms, and learning what level of peer support exists for them and how to build it if it is lacking. Building resilience is another area, including finding military women opportunities to use their experiences in positive ways, such as through mentoring others.
For more information on suicide prevention for these women, Ms. Garrick referred clinicians to the suicide risk assessment and prevention clinical guidelines issued by the DOD and the Department of Veterans Affairs.
For patients at acute risk, Ms. Garrick said, “I recommend sitting with them as you watch them put this number into their phone: 800-273-8255. That’s the lifeline number that will connect you directly with the VA if you press 1.”
Because there has been a historic lack of interest on behalf of the military in women’s health outcomes related to their service compared with that of men, there is a need to create a database going forward to better inform DOD health and disability policies for women in the military, Ms. Garrick said. This places the onus on psychiatrists who evaluate this cohort to “tease out any potential psychological stressors that might not be obvious from their personnel file.” Some women have been exposed to the same levels of traumatic combat experiences as their male colleagues, even though it wasn’t until earlier this year that women became eligible for the same combat roles as men.
“If you look in their files, they might not have the same awards and titles as men, but they might have seen the same people being killed or the same number of dead bodies,” she said.
Ms. Garrick’s views are her own and do not represent those of the Department of Defense.
*Correction 10/14/16: An earlier version of this story misstated Ms. Garrick's position.
[email protected]
On Twitter @whitneymcknight
WASHINGTON – Women service personnel face different suicide risks from their civilian counterparts, according to a Department of Defense appointee.
Data are few about suicide among women in the military – in part because not much research has been conducted over the years into service women’s health outcomes – according to Jacqueline Garrick, but insights gleaned from the reports of military women, both active duty and veterans, who survived suicide attempts, shed light on what to look for as risk factors. Ms. Garrick, special assistant, Manpower and Reserve Affairs in the Department of Defense, made her comments during a panel discussion at the American Psychiatric Association’s Institute on Psychiatric Services.*
One of the most salient of suicide risks can emerge when a service woman’s intimate relationship ends. This loss is compounded by the absence of social support that results from the military’s inherently masculine environment where “fitting in is definitely harder for women,” according to Ms. Garrick, a licensed clinical social worker, U.S. Army veteran, and policy analyst.
Deployment and combat zone traumas, whether physical, mental, or both, are other risk factors. Horrors witnessed in war can have psychological implications for men and women personnel. But for women, who also possibly face additional concerns of sexual assault and lack of social support, the traumas can become debilitating and lead to risk of suicide, Ms. Garrick said.
Women in the military overlap with civilians in their suicide risk factors where mental health history, abuse, and exposure to suicide are concerned, but where the two cohorts particularly diverge, Ms. Garrick said, is access to lethal means, particularly among women veterans. Civilian women who attempt suicide are more likely to cut themselves or overdose on drugs, whereas, “Military women have firearms, and they know how to use them,” Ms. Garrick said. “So, if you’re screening [for suicide in this population], pay close attention to whether there are weapons in the home.”
Traumatic brain injury is another area in which risks for suicide in military women could exist, but not enough is known at this point, Ms. Garrick said.
A suicide risk intervention called “safety planning” is one that Ms. Garrick said she has been developing in her work with the DOD. This includes asking these women what makes them feel “safe” at home, determining what their families know about the whereabouts and the safety features of their firearms, and learning what level of peer support exists for them and how to build it if it is lacking. Building resilience is another area, including finding military women opportunities to use their experiences in positive ways, such as through mentoring others.
For more information on suicide prevention for these women, Ms. Garrick referred clinicians to the suicide risk assessment and prevention clinical guidelines issued by the DOD and the Department of Veterans Affairs.
For patients at acute risk, Ms. Garrick said, “I recommend sitting with them as you watch them put this number into their phone: 800-273-8255. That’s the lifeline number that will connect you directly with the VA if you press 1.”
Because there has been a historic lack of interest on behalf of the military in women’s health outcomes related to their service compared with that of men, there is a need to create a database going forward to better inform DOD health and disability policies for women in the military, Ms. Garrick said. This places the onus on psychiatrists who evaluate this cohort to “tease out any potential psychological stressors that might not be obvious from their personnel file.” Some women have been exposed to the same levels of traumatic combat experiences as their male colleagues, even though it wasn’t until earlier this year that women became eligible for the same combat roles as men.
“If you look in their files, they might not have the same awards and titles as men, but they might have seen the same people being killed or the same number of dead bodies,” she said.
Ms. Garrick’s views are her own and do not represent those of the Department of Defense.
*Correction 10/14/16: An earlier version of this story misstated Ms. Garrick's position.
[email protected]
On Twitter @whitneymcknight
WASHINGTON – Women service personnel face different suicide risks from their civilian counterparts, according to a Department of Defense appointee.
Data are few about suicide among women in the military – in part because not much research has been conducted over the years into service women’s health outcomes – according to Jacqueline Garrick, but insights gleaned from the reports of military women, both active duty and veterans, who survived suicide attempts, shed light on what to look for as risk factors. Ms. Garrick, special assistant, Manpower and Reserve Affairs in the Department of Defense, made her comments during a panel discussion at the American Psychiatric Association’s Institute on Psychiatric Services.*
One of the most salient of suicide risks can emerge when a service woman’s intimate relationship ends. This loss is compounded by the absence of social support that results from the military’s inherently masculine environment where “fitting in is definitely harder for women,” according to Ms. Garrick, a licensed clinical social worker, U.S. Army veteran, and policy analyst.
Deployment and combat zone traumas, whether physical, mental, or both, are other risk factors. Horrors witnessed in war can have psychological implications for men and women personnel. But for women, who also possibly face additional concerns of sexual assault and lack of social support, the traumas can become debilitating and lead to risk of suicide, Ms. Garrick said.
Women in the military overlap with civilians in their suicide risk factors where mental health history, abuse, and exposure to suicide are concerned, but where the two cohorts particularly diverge, Ms. Garrick said, is access to lethal means, particularly among women veterans. Civilian women who attempt suicide are more likely to cut themselves or overdose on drugs, whereas, “Military women have firearms, and they know how to use them,” Ms. Garrick said. “So, if you’re screening [for suicide in this population], pay close attention to whether there are weapons in the home.”
Traumatic brain injury is another area in which risks for suicide in military women could exist, but not enough is known at this point, Ms. Garrick said.
A suicide risk intervention called “safety planning” is one that Ms. Garrick said she has been developing in her work with the DOD. This includes asking these women what makes them feel “safe” at home, determining what their families know about the whereabouts and the safety features of their firearms, and learning what level of peer support exists for them and how to build it if it is lacking. Building resilience is another area, including finding military women opportunities to use their experiences in positive ways, such as through mentoring others.
For more information on suicide prevention for these women, Ms. Garrick referred clinicians to the suicide risk assessment and prevention clinical guidelines issued by the DOD and the Department of Veterans Affairs.
For patients at acute risk, Ms. Garrick said, “I recommend sitting with them as you watch them put this number into their phone: 800-273-8255. That’s the lifeline number that will connect you directly with the VA if you press 1.”
Because there has been a historic lack of interest on behalf of the military in women’s health outcomes related to their service compared with that of men, there is a need to create a database going forward to better inform DOD health and disability policies for women in the military, Ms. Garrick said. This places the onus on psychiatrists who evaluate this cohort to “tease out any potential psychological stressors that might not be obvious from their personnel file.” Some women have been exposed to the same levels of traumatic combat experiences as their male colleagues, even though it wasn’t until earlier this year that women became eligible for the same combat roles as men.
“If you look in their files, they might not have the same awards and titles as men, but they might have seen the same people being killed or the same number of dead bodies,” she said.
Ms. Garrick’s views are her own and do not represent those of the Department of Defense.
*Correction 10/14/16: An earlier version of this story misstated Ms. Garrick's position.
[email protected]
On Twitter @whitneymcknight
Survival benefit maintained long term with ipilimumab for high-risk melanoma
COPENHAGEN – Five years on, patients with high-risk stage III melanoma treated with the checkpoint inhibitor ipilimumab, following complete resection, continue to have significantly better overall, recurrence-free, and distant metastasis–free survival, compared with patients treated with placebo, reported investigators in a phase III trial.
Five-year overall survival among patients who received a 10-mg/kg dose of ipilimumab (Yervoy) in the EORTC 18071 trial was 65.4%, compared with 54.4% for patients who received placebo. This difference translated into a hazard ratio for death with ipilimumab of 0.72 (P = .001), reported Alexander M.M. Eggermont, MD, from the Gustave Roussy Cancer Center in Villejuif, France.

The survival benefit with ipilimumab “was consistent across all survival endpoints,” he said at a briefing prior to his presentation of the data in a symposium at the European Society for Medical Oncology Congress.
He noted, however, that the 10-mg/kg dose of ipilimumab selected in phase II trials is associated with significant toxicities.
“Ipilimumab is not an easy drug to handle. My recommendation is to keep it in [cancer] centers,” he said.
In the trial, 951 patients with high-risk, stage III, completely resected melanoma were randomly assigned to receive induction therapy with ipilimumab 10 mg/kg every 3 weeks for four cycles or placebo, followed by maintenance with the assigned therapy every 12 weeks for up to 3 years.
The investigators previously reported that, at a median follow-up of 2.7 years, ipilimumab was associated with significantly prolonged overall survival (the primary endpoint), with a hazard ratio vs. placebo of 0.75 (P = .001).
At ESMO 2016, Dr. Eggermont reported final survival results from the trial, at a median follow-up of 5.3 years.
The rate of 5-year overall survival for the 475 patients assigned to ipilimumab was 65.4%, compared with 54.4% among the 476 patients assigned to placebo (HR, 0.72, P = .001).
At 5 years, 41% of patients assigned to ipilimumab were free of recurrences, compared with 30% of patients on placebo, The median recurrence-free survival was 27.6 months vs. 17.1 months, respectively (HR, 0.76, P = .0008).
Similarly, the rate of distant metastasis-free survival at 5 years was 48.3% for patients assigned to ipilimumab, vs. 38.9% in the placebo group (HR for death or distant metastasis, 0.76; P = .002).
The safety analysis showed that twice as many patients assigned to ipilimumab had grade 3 or 4 adverse events (54.1% vs. 26.2%). Grade 3 or 4 adverse immune events occurred in 41.6% vs. 2.7%, respectively.
Five patients assigned to ipilimumab died from immune-related causes: three from colitis (two of whom had intestinal perforations), one from myocarditis, and one from multiorgan failure associated with Guillain-Barré syndrome.

“The final analysis shows, for the first time, that checkpoint blockade is effective in the adjuvant setting,” he said.
The data suggest, however, that the benefit appears to be concentrated in patients with higher-risk features, such as involvement of four or more lymph nodes, microscopic nodal disease, or ulceration, he said.
The discussant also agreed with Dr. Eggermont’s assertions that the decision to treat patients with ipilimumab should factor in toxicity, and that treatment should be administered only in centers experienced in using the drug.
The trial was sponsored by Bristol-Myers Squibb. Dr. Eggermont disclosed serving on an advisory board for Bristol-Myers Squibb and Merck. Dr. Michielin disclosed consulting and/or honoraria from Amgen, Bristol-Myers Squibb, Roche, Merck Sharp & Dohme, Novartis, and GlaxoSmithKline.
COPENHAGEN – Five years on, patients with high-risk stage III melanoma treated with the checkpoint inhibitor ipilimumab, following complete resection, continue to have significantly better overall, recurrence-free, and distant metastasis–free survival, compared with patients treated with placebo, reported investigators in a phase III trial.
Five-year overall survival among patients who received a 10-mg/kg dose of ipilimumab (Yervoy) in the EORTC 18071 trial was 65.4%, compared with 54.4% for patients who received placebo. This difference translated into a hazard ratio for death with ipilimumab of 0.72 (P = .001), reported Alexander M.M. Eggermont, MD, from the Gustave Roussy Cancer Center in Villejuif, France.
The survival benefit with ipilimumab “was consistent across all survival endpoints,” he said at a briefing prior to his presentation of the data in a symposium at the European Society for Medical Oncology Congress.
He noted, however, that the 10-mg/kg dose of ipilimumab selected in phase II trials is associated with significant toxicities.
“Ipilimumab is not an easy drug to handle. My recommendation is to keep it in [cancer] centers,” he said.
In the trial, 951 patients with high-risk, stage III, completely resected melanoma were randomly assigned to receive induction therapy with ipilimumab 10 mg/kg every 3 weeks for four cycles or placebo, followed by maintenance with the assigned therapy every 12 weeks for up to 3 years.
The investigators previously reported that, at a median follow-up of 2.7 years, ipilimumab was associated with significantly prolonged overall survival (the primary endpoint), with a hazard ratio vs. placebo of 0.75 (P = .001).
At ESMO 2016, Dr. Eggermont reported final survival results from the trial, at a median follow-up of 5.3 years.
The rate of 5-year overall survival for the 475 patients assigned to ipilimumab was 65.4%, compared with 54.4% among the 476 patients assigned to placebo (HR, 0.72, P = .001).
At 5 years, 41% of patients assigned to ipilimumab were free of recurrences, compared with 30% of patients on placebo, The median recurrence-free survival was 27.6 months vs. 17.1 months, respectively (HR, 0.76, P = .0008).
Similarly, the rate of distant metastasis-free survival at 5 years was 48.3% for patients assigned to ipilimumab, vs. 38.9% in the placebo group (HR for death or distant metastasis, 0.76; P = .002).
The safety analysis showed that twice as many patients assigned to ipilimumab had grade 3 or 4 adverse events (54.1% vs. 26.2%). Grade 3 or 4 adverse immune events occurred in 41.6% vs. 2.7%, respectively.
Five patients assigned to ipilimumab died from immune-related causes: three from colitis (two of whom had intestinal perforations), one from myocarditis, and one from multiorgan failure associated with Guillain-Barré syndrome.
“The final analysis shows, for the first time, that checkpoint blockade is effective in the adjuvant setting,” he said.
The data suggest, however, that the benefit appears to be concentrated in patients with higher-risk features, such as involvement of four or more lymph nodes, microscopic nodal disease, or ulceration, he said.
The discussant also agreed with Dr. Eggermont’s assertions that the decision to treat patients with ipilimumab should factor in toxicity, and that treatment should be administered only in centers experienced in using the drug.
The trial was sponsored by Bristol-Myers Squibb. Dr. Eggermont disclosed serving on an advisory board for Bristol-Myers Squibb and Merck. Dr. Michielin disclosed consulting and/or honoraria from Amgen, Bristol-Myers Squibb, Roche, Merck Sharp & Dohme, Novartis, and GlaxoSmithKline.
COPENHAGEN – Five years on, patients with high-risk stage III melanoma treated with the checkpoint inhibitor ipilimumab, following complete resection, continue to have significantly better overall, recurrence-free, and distant metastasis–free survival, compared with patients treated with placebo, reported investigators in a phase III trial.
Five-year overall survival among patients who received a 10-mg/kg dose of ipilimumab (Yervoy) in the EORTC 18071 trial was 65.4%, compared with 54.4% for patients who received placebo. This difference translated into a hazard ratio for death with ipilimumab of 0.72 (P = .001), reported Alexander M.M. Eggermont, MD, from the Gustave Roussy Cancer Center in Villejuif, France.
The survival benefit with ipilimumab “was consistent across all survival endpoints,” he said at a briefing prior to his presentation of the data in a symposium at the European Society for Medical Oncology Congress.
He noted, however, that the 10-mg/kg dose of ipilimumab selected in phase II trials is associated with significant toxicities.
“Ipilimumab is not an easy drug to handle. My recommendation is to keep it in [cancer] centers,” he said.
In the trial, 951 patients with high-risk, stage III, completely resected melanoma were randomly assigned to receive induction therapy with ipilimumab 10 mg/kg every 3 weeks for four cycles or placebo, followed by maintenance with the assigned therapy every 12 weeks for up to 3 years.
The investigators previously reported that, at a median follow-up of 2.7 years, ipilimumab was associated with significantly prolonged overall survival (the primary endpoint), with a hazard ratio vs. placebo of 0.75 (P = .001).
At ESMO 2016, Dr. Eggermont reported final survival results from the trial, at a median follow-up of 5.3 years.
The rate of 5-year overall survival for the 475 patients assigned to ipilimumab was 65.4%, compared with 54.4% among the 476 patients assigned to placebo (HR, 0.72, P = .001).
At 5 years, 41% of patients assigned to ipilimumab were free of recurrences, compared with 30% of patients on placebo, The median recurrence-free survival was 27.6 months vs. 17.1 months, respectively (HR, 0.76, P = .0008).
Similarly, the rate of distant metastasis-free survival at 5 years was 48.3% for patients assigned to ipilimumab, vs. 38.9% in the placebo group (HR for death or distant metastasis, 0.76; P = .002).
The safety analysis showed that twice as many patients assigned to ipilimumab had grade 3 or 4 adverse events (54.1% vs. 26.2%). Grade 3 or 4 adverse immune events occurred in 41.6% vs. 2.7%, respectively.
Five patients assigned to ipilimumab died from immune-related causes: three from colitis (two of whom had intestinal perforations), one from myocarditis, and one from multiorgan failure associated with Guillain-Barré syndrome.
“The final analysis shows, for the first time, that checkpoint blockade is effective in the adjuvant setting,” he said.
The data suggest, however, that the benefit appears to be concentrated in patients with higher-risk features, such as involvement of four or more lymph nodes, microscopic nodal disease, or ulceration, he said.
The discussant also agreed with Dr. Eggermont’s assertions that the decision to treat patients with ipilimumab should factor in toxicity, and that treatment should be administered only in centers experienced in using the drug.
The trial was sponsored by Bristol-Myers Squibb. Dr. Eggermont disclosed serving on an advisory board for Bristol-Myers Squibb and Merck. Dr. Michielin disclosed consulting and/or honoraria from Amgen, Bristol-Myers Squibb, Roche, Merck Sharp & Dohme, Novartis, and GlaxoSmithKline.
Key clinical point: The CTLA-4 checkpoint inhibitor ipilimumab offers survival benefit, compared with placebo in patients with malignant melanoma.
Major finding: The hazard ratio for death with ipilimumab vs. placebo was 0.72 (P = .001).
Data source: Randomized, controlled, phase III trial in 951 patients with high-risk stage III malignant melanoma following complete resection.
Disclosures: The trial was sponsored by Bristol-Myers Squibb. Dr. Eggermont disclosed serving on an advisory board for Bristol-Myers Squibb and Merck. Dr. Michielin disclosed consulting and/or honoraria from Amgen, Bristol-Myers Squibb, Roche, Merck Sharp & Dohme, Novartis, and GlaxoSmithKline.
‘Thank you, EMR!’
“Thank you, EMR!” Said no doctor. Ever.
At least up until now.
For years, we have had to put up with these machines in our exam rooms, distracting data entry devices that offer insignificant contributions to the work we do. That’s starting to change.
Recently, I led a workshop at the annual Kaiser Permanente internal medicine conference in Southern California. I gave one of my more popular sessions on the art of diagnosis and therapy (inspired and borrowed from Dr. Irwin M. Braverman’s marvelous lectures on learning dermatology through art).

What’s more, the listed steroids change automatically based on the current formulary. This ensures the lowest cost to the patient and minimizes rework of having to go back and pick another when the patient balks at unjustifiably high prices. The clinician has only to click and sign to place the order. Now a primary care physician – or even a dermatologist! – needs only to estimate the potency of the therapy, pick a vehicle (cream, ointment, gel, solution), and the EMR guides him or her to prescribe the right medication. It is easy to use, active at the point of care, and helpful to both clinician and patient.
This SRX program was developed by our local physicians in conjunction with pharmacists and the informatics team. It has enormous potential, providing more point of care clinical decision support based on best practice, formulary, and even personalized information automatically gleaned from that patient’s chart. As of now, we can customize our order entry such that if I want to order labs to look for connective tissue disease, I have to type only .CTD, and my personal picks for a lupus workup come up. It saves me time. Yes, I did just say that in reference to my EMR. And it helps ensure high-quality care. Whenever new diagnostics or new treatments become best practice, I can put them on my preference list, thereby making the best thing to do the easy thing to do.
The internal medicine physicians were appreciative for my lecture and loved learning through art. However, the big hit was the SRX DERM. “This will make it so much easier,” said one hospitalist, “thanks for doing this!”
I had nothing to do with it though. Thank you, EMR.
Dr. Benabio is a partner physician in the department of dermatology of the Southern California Permanente Group in San Diego. He is @Dermdoc on Twitter. Write to him at [email protected].
“Thank you, EMR!” Said no doctor. Ever.
At least up until now.
For years, we have had to put up with these machines in our exam rooms, distracting data entry devices that offer insignificant contributions to the work we do. That’s starting to change.
Recently, I led a workshop at the annual Kaiser Permanente internal medicine conference in Southern California. I gave one of my more popular sessions on the art of diagnosis and therapy (inspired and borrowed from Dr. Irwin M. Braverman’s marvelous lectures on learning dermatology through art).
What’s more, the listed steroids change automatically based on the current formulary. This ensures the lowest cost to the patient and minimizes rework of having to go back and pick another when the patient balks at unjustifiably high prices. The clinician has only to click and sign to place the order. Now a primary care physician – or even a dermatologist! – needs only to estimate the potency of the therapy, pick a vehicle (cream, ointment, gel, solution), and the EMR guides him or her to prescribe the right medication. It is easy to use, active at the point of care, and helpful to both clinician and patient.
This SRX program was developed by our local physicians in conjunction with pharmacists and the informatics team. It has enormous potential, providing more point of care clinical decision support based on best practice, formulary, and even personalized information automatically gleaned from that patient’s chart. As of now, we can customize our order entry such that if I want to order labs to look for connective tissue disease, I have to type only .CTD, and my personal picks for a lupus workup come up. It saves me time. Yes, I did just say that in reference to my EMR. And it helps ensure high-quality care. Whenever new diagnostics or new treatments become best practice, I can put them on my preference list, thereby making the best thing to do the easy thing to do.
The internal medicine physicians were appreciative for my lecture and loved learning through art. However, the big hit was the SRX DERM. “This will make it so much easier,” said one hospitalist, “thanks for doing this!”
I had nothing to do with it though. Thank you, EMR.
Dr. Benabio is a partner physician in the department of dermatology of the Southern California Permanente Group in San Diego. He is @Dermdoc on Twitter. Write to him at [email protected].
“Thank you, EMR!” Said no doctor. Ever.
At least up until now.
For years, we have had to put up with these machines in our exam rooms, distracting data entry devices that offer insignificant contributions to the work we do. That’s starting to change.
Recently, I led a workshop at the annual Kaiser Permanente internal medicine conference in Southern California. I gave one of my more popular sessions on the art of diagnosis and therapy (inspired and borrowed from Dr. Irwin M. Braverman’s marvelous lectures on learning dermatology through art).
What’s more, the listed steroids change automatically based on the current formulary. This ensures the lowest cost to the patient and minimizes rework of having to go back and pick another when the patient balks at unjustifiably high prices. The clinician has only to click and sign to place the order. Now a primary care physician – or even a dermatologist! – needs only to estimate the potency of the therapy, pick a vehicle (cream, ointment, gel, solution), and the EMR guides him or her to prescribe the right medication. It is easy to use, active at the point of care, and helpful to both clinician and patient.
This SRX program was developed by our local physicians in conjunction with pharmacists and the informatics team. It has enormous potential, providing more point of care clinical decision support based on best practice, formulary, and even personalized information automatically gleaned from that patient’s chart. As of now, we can customize our order entry such that if I want to order labs to look for connective tissue disease, I have to type only .CTD, and my personal picks for a lupus workup come up. It saves me time. Yes, I did just say that in reference to my EMR. And it helps ensure high-quality care. Whenever new diagnostics or new treatments become best practice, I can put them on my preference list, thereby making the best thing to do the easy thing to do.
The internal medicine physicians were appreciative for my lecture and loved learning through art. However, the big hit was the SRX DERM. “This will make it so much easier,” said one hospitalist, “thanks for doing this!”
I had nothing to do with it though. Thank you, EMR.
Dr. Benabio is a partner physician in the department of dermatology of the Southern California Permanente Group in San Diego. He is @Dermdoc on Twitter. Write to him at [email protected].
Ebola research update: September 2016
The struggle to defeat Ebola virus disease continues globally, although it may not always make the headlines. To catch up on what you may have missed, here are some notable news items and journal articles published over the past few weeks that are worth a second look.
, according to a study of EVD patients treated in the United States.
An analysis of the 2014 Ebola virus disease outbreak in Nigeria found that early detection of cases, an efficacious incident management system, and rapid case management with on-site mobilization and training of local professionals were important to better outcomes, prompt containment, and no infection among EVD care-providers.
Viral genome sequence data uniquely reveals the 2013-2016 epidemic of Ebola virus disease in West Africa to be “a heterogeneous and spatially dissociated collection of transmission clusters of that were of varying size, duration, and connectivity,” according to a recent study.
A case study of Lassa fever involved the development of a mathematical framework which was applied to try to determine how much of disease transmission was from animal to human and how much was from human to human. This knowledge can be used to “infer human disease risk based on knowledge of infection patterns in the animal reservoir host and the contact mechanisms required for transmission to humans.”
A decontamination protocol that relies on the use of both peracetic acid and hydrogen peroxide fumigation was proposed for a biosafety level 3 field laboratory as a part of an Ebola treatment center in Guinea. Inoculated stainless steel disks of bioindicators containing spores of Geobacillus stearothermophilus were used to control the protocol.
A survey in New Zealand indicated that a future Ebola outbreak would have “large social and economic consequences” because judging from survey responses, a large percentage of the population would avoid social contact, such as going to work, school, and social events, to protect their health, according to a study in Disaster Medicine and Public Health Preparedness. Survey respondents also indicated a willingness to receive a vaccine.
Investigators identified contact tracing as an important determinant of the 2014-2015 Ebola epidemic’s behavior in Guinea. Also, early availability of Ebola treatment unit beds was key in limiting the number of Ebola cases.
The WHO Ebola Response Team said that empirical and modeling studies performed during the West African Ebola virus disease epidemic have demonstrated that large epidemics of EVD can be prevented, that “a rapid response can interrupt transmission and restrict the size of outbreaks, even in densely populated cities.”
In 2015, the first nationwide semen testing and counseling program for male Ebola survivors, the Men’s Health Screening Program, was established in Liberia, according to a report in MMWR. Researchers said involvement with the survivor community, communication, and flexibility were key to the program’s success.
A $13 million NIH grant to study how the Ebola virus replicates has been awarded to a team at Washington University, St. Louis.
A recent study found that vector delivery of two antibody components of the ZMapp product works in mice against systemic and airway challenge with a mouse-adapted strain of Ebola virus. The authors say this platform “provides a generic manufacturing solution and overcomes some of the delivery challenges associated with repeated administration of the protective protein.”
U.S. Army researchers based at Fort Detrick, Md., are developing relationships with Ebola survivors in Uganda, who the researchers believe may hold the key to a vaccine or treatment for the infection because many Ugandans have survived the epidemic.
A qualitative study in PLOS Current Outbreaks found that the preparedness of U.S. health care volunteers in the West Africa Ebola deployment was inadequate. The authors said effective policies and practices must be developed and implemented to properly protect the health and well-being of volunteers.
Investigators hypothesized that cannabidiol, based on its pharmacological effects and favorable safety profile, should be considered as a treatment for individuals with post-Ebola sequelae because it can reduce pain and inflammation.
[email protected]
On Twitter @richpizzi
The struggle to defeat Ebola virus disease continues globally, although it may not always make the headlines. To catch up on what you may have missed, here are some notable news items and journal articles published over the past few weeks that are worth a second look.
, according to a study of EVD patients treated in the United States.
An analysis of the 2014 Ebola virus disease outbreak in Nigeria found that early detection of cases, an efficacious incident management system, and rapid case management with on-site mobilization and training of local professionals were important to better outcomes, prompt containment, and no infection among EVD care-providers.
Viral genome sequence data uniquely reveals the 2013-2016 epidemic of Ebola virus disease in West Africa to be “a heterogeneous and spatially dissociated collection of transmission clusters of that were of varying size, duration, and connectivity,” according to a recent study.
A case study of Lassa fever involved the development of a mathematical framework which was applied to try to determine how much of disease transmission was from animal to human and how much was from human to human. This knowledge can be used to “infer human disease risk based on knowledge of infection patterns in the animal reservoir host and the contact mechanisms required for transmission to humans.”
A decontamination protocol that relies on the use of both peracetic acid and hydrogen peroxide fumigation was proposed for a biosafety level 3 field laboratory as a part of an Ebola treatment center in Guinea. Inoculated stainless steel disks of bioindicators containing spores of Geobacillus stearothermophilus were used to control the protocol.
A survey in New Zealand indicated that a future Ebola outbreak would have “large social and economic consequences” because judging from survey responses, a large percentage of the population would avoid social contact, such as going to work, school, and social events, to protect their health, according to a study in Disaster Medicine and Public Health Preparedness. Survey respondents also indicated a willingness to receive a vaccine.
Investigators identified contact tracing as an important determinant of the 2014-2015 Ebola epidemic’s behavior in Guinea. Also, early availability of Ebola treatment unit beds was key in limiting the number of Ebola cases.
The WHO Ebola Response Team said that empirical and modeling studies performed during the West African Ebola virus disease epidemic have demonstrated that large epidemics of EVD can be prevented, that “a rapid response can interrupt transmission and restrict the size of outbreaks, even in densely populated cities.”
In 2015, the first nationwide semen testing and counseling program for male Ebola survivors, the Men’s Health Screening Program, was established in Liberia, according to a report in MMWR. Researchers said involvement with the survivor community, communication, and flexibility were key to the program’s success.
A $13 million NIH grant to study how the Ebola virus replicates has been awarded to a team at Washington University, St. Louis.
A recent study found that vector delivery of two antibody components of the ZMapp product works in mice against systemic and airway challenge with a mouse-adapted strain of Ebola virus. The authors say this platform “provides a generic manufacturing solution and overcomes some of the delivery challenges associated with repeated administration of the protective protein.”
U.S. Army researchers based at Fort Detrick, Md., are developing relationships with Ebola survivors in Uganda, who the researchers believe may hold the key to a vaccine or treatment for the infection because many Ugandans have survived the epidemic.
A qualitative study in PLOS Current Outbreaks found that the preparedness of U.S. health care volunteers in the West Africa Ebola deployment was inadequate. The authors said effective policies and practices must be developed and implemented to properly protect the health and well-being of volunteers.
Investigators hypothesized that cannabidiol, based on its pharmacological effects and favorable safety profile, should be considered as a treatment for individuals with post-Ebola sequelae because it can reduce pain and inflammation.
[email protected]
On Twitter @richpizzi
The struggle to defeat Ebola virus disease continues globally, although it may not always make the headlines. To catch up on what you may have missed, here are some notable news items and journal articles published over the past few weeks that are worth a second look.
, according to a study of EVD patients treated in the United States.
An analysis of the 2014 Ebola virus disease outbreak in Nigeria found that early detection of cases, an efficacious incident management system, and rapid case management with on-site mobilization and training of local professionals were important to better outcomes, prompt containment, and no infection among EVD care-providers.
Viral genome sequence data uniquely reveals the 2013-2016 epidemic of Ebola virus disease in West Africa to be “a heterogeneous and spatially dissociated collection of transmission clusters of that were of varying size, duration, and connectivity,” according to a recent study.
A case study of Lassa fever involved the development of a mathematical framework which was applied to try to determine how much of disease transmission was from animal to human and how much was from human to human. This knowledge can be used to “infer human disease risk based on knowledge of infection patterns in the animal reservoir host and the contact mechanisms required for transmission to humans.”
A decontamination protocol that relies on the use of both peracetic acid and hydrogen peroxide fumigation was proposed for a biosafety level 3 field laboratory as a part of an Ebola treatment center in Guinea. Inoculated stainless steel disks of bioindicators containing spores of Geobacillus stearothermophilus were used to control the protocol.
A survey in New Zealand indicated that a future Ebola outbreak would have “large social and economic consequences” because judging from survey responses, a large percentage of the population would avoid social contact, such as going to work, school, and social events, to protect their health, according to a study in Disaster Medicine and Public Health Preparedness. Survey respondents also indicated a willingness to receive a vaccine.
Investigators identified contact tracing as an important determinant of the 2014-2015 Ebola epidemic’s behavior in Guinea. Also, early availability of Ebola treatment unit beds was key in limiting the number of Ebola cases.
The WHO Ebola Response Team said that empirical and modeling studies performed during the West African Ebola virus disease epidemic have demonstrated that large epidemics of EVD can be prevented, that “a rapid response can interrupt transmission and restrict the size of outbreaks, even in densely populated cities.”
In 2015, the first nationwide semen testing and counseling program for male Ebola survivors, the Men’s Health Screening Program, was established in Liberia, according to a report in MMWR. Researchers said involvement with the survivor community, communication, and flexibility were key to the program’s success.
A $13 million NIH grant to study how the Ebola virus replicates has been awarded to a team at Washington University, St. Louis.
A recent study found that vector delivery of two antibody components of the ZMapp product works in mice against systemic and airway challenge with a mouse-adapted strain of Ebola virus. The authors say this platform “provides a generic manufacturing solution and overcomes some of the delivery challenges associated with repeated administration of the protective protein.”
U.S. Army researchers based at Fort Detrick, Md., are developing relationships with Ebola survivors in Uganda, who the researchers believe may hold the key to a vaccine or treatment for the infection because many Ugandans have survived the epidemic.
A qualitative study in PLOS Current Outbreaks found that the preparedness of U.S. health care volunteers in the West Africa Ebola deployment was inadequate. The authors said effective policies and practices must be developed and implemented to properly protect the health and well-being of volunteers.
Investigators hypothesized that cannabidiol, based on its pharmacological effects and favorable safety profile, should be considered as a treatment for individuals with post-Ebola sequelae because it can reduce pain and inflammation.
[email protected]
On Twitter @richpizzi
PARP inhibitor prolongs PFS in ovarian cancer patients with and without BRCA mutations
COPENHAGEN – Women with platinum-sensitive, recurrent ovarian cancer treated with the PARP 1/2 inhibitor niraparib had significantly longer progression-free survival than did women who received a placebo, regardless of their BRCA mutational status, according to results of a phase III trial.
Median progression-free survival (PFS) among women with germline BRCA mutations who received niraparib was 21 months, compared with 5.5 months for women with germline BRCA mutations who received placebo (P less than .001).
For the overall population of women with no germline mutations, median PFS was 9.3 months for those who received niraparib, vs. 3.9 months for placebo-treated controls. Among women in this group whose tumors tested positive for homologous recombination deficiency (HRD), median PFS was 12.9 months, vs. 3.8 months without HRD.

Dr. Mirza reported results of the The NGOT-OV16/NOVA trial, the first phase III trial with a PARP inhibitor, at the European Society for Medical Oncology Congress. Results of the trial were simultaneously published online in The New England Journal of Medicine.
Niraparib is a selective, oral inhibitor of poly(adenosine diphosphate-ribose) polymerase (PARP) 1 and 2 that was previously shown to have efficacy against ovarian cancer in a phase I dose-escalation trial.
In the NGOT-OV16 NOVA trial, patients with platinum-sensitive high grade serous ovarian cancer first underwent chemotherapy with 4-6 cycles of a platinum-based regimen, and those who responded to platinum treatment were then stratified by the presence or absence of germline BRCA mutations, and then randomized on a 2:1 basis to either niraparib 300 mg once daily or placebo until disease progression.
A total of 203 patients with germline BRCA mutations and 350 with no mutations were enrolled in the trial.
As noted before, patients treated with niraparib in both trials arms had significantly longer PFS than did controls. The hazard ratio (HR) for niraparib in patients with germline BRCA mutations was 0.27. For the overall non–germline mutation population, the HR was 0.45, and for HRD-positive and HRD-negative subgroups, the HRs were 0.38 and 0.56, respectively (the latter is an exploratory endpoint, however; P less than .001 for the first three HRs shown).
Secondary efficacy endpoints, including chemotherapy-free interval and time to first subsequent treatment, also favored niraparib in patients with and without BRCA mutations.
Overall survival data for the trial are not sufficiently mature for reporting, however.
The safety profile of the drug was in line with that seen in other studies of PARP inhibitors, Dr. Mirza said. Grade 3/4 adverse events occurring in 5% or more of patients included thrombocytopenias in 33.8% of niraparib recipients vs. 0.6% of controls, anemia in 25.3% vs. 0%, neutropenia in 19.6% vs. 1.7%, fatigue in 8.2% vs. 0.6%, and hypertension in 8.2% vs. 2.2%.
Five of the 367 patients who received niraparib (1.4%) developed myelodysplasia or acute myeloid leukemia, compared with 2 of 179 patients (1.1%) treated with placebo.
Patient-reported outcomes measured via the Functional Assessment of Cancer Therapy – Ovarian Symptom Index and EQ (EuroQol) 5D-5L instrument showed high compliance rates and patient-reported symptom rates that were similar between niraparib and placebo groups.

The results also demonstrated that HRD testing can be used to identify patients without germline mutations in BRCA who may benefit from a PARP inhibitor, he said.
Tesaro funded the study. Dr. Mirza disclosed serving on the board of directors of the pharmaceutical companies, and consultant or advisory roles with multiple other companies. Dr. Pignata disclosed consulting, honoraria, and or research funding from several companies, but reported no relationship with Tesaro
COPENHAGEN – Women with platinum-sensitive, recurrent ovarian cancer treated with the PARP 1/2 inhibitor niraparib had significantly longer progression-free survival than did women who received a placebo, regardless of their BRCA mutational status, according to results of a phase III trial.
Median progression-free survival (PFS) among women with germline BRCA mutations who received niraparib was 21 months, compared with 5.5 months for women with germline BRCA mutations who received placebo (P less than .001).
For the overall population of women with no germline mutations, median PFS was 9.3 months for those who received niraparib, vs. 3.9 months for placebo-treated controls. Among women in this group whose tumors tested positive for homologous recombination deficiency (HRD), median PFS was 12.9 months, vs. 3.8 months without HRD.
Dr. Mirza reported results of the The NGOT-OV16/NOVA trial, the first phase III trial with a PARP inhibitor, at the European Society for Medical Oncology Congress. Results of the trial were simultaneously published online in The New England Journal of Medicine.
Niraparib is a selective, oral inhibitor of poly(adenosine diphosphate-ribose) polymerase (PARP) 1 and 2 that was previously shown to have efficacy against ovarian cancer in a phase I dose-escalation trial.
In the NGOT-OV16 NOVA trial, patients with platinum-sensitive high grade serous ovarian cancer first underwent chemotherapy with 4-6 cycles of a platinum-based regimen, and those who responded to platinum treatment were then stratified by the presence or absence of germline BRCA mutations, and then randomized on a 2:1 basis to either niraparib 300 mg once daily or placebo until disease progression.
A total of 203 patients with germline BRCA mutations and 350 with no mutations were enrolled in the trial.
As noted before, patients treated with niraparib in both trials arms had significantly longer PFS than did controls. The hazard ratio (HR) for niraparib in patients with germline BRCA mutations was 0.27. For the overall non–germline mutation population, the HR was 0.45, and for HRD-positive and HRD-negative subgroups, the HRs were 0.38 and 0.56, respectively (the latter is an exploratory endpoint, however; P less than .001 for the first three HRs shown).
Secondary efficacy endpoints, including chemotherapy-free interval and time to first subsequent treatment, also favored niraparib in patients with and without BRCA mutations.
Overall survival data for the trial are not sufficiently mature for reporting, however.
The safety profile of the drug was in line with that seen in other studies of PARP inhibitors, Dr. Mirza said. Grade 3/4 adverse events occurring in 5% or more of patients included thrombocytopenias in 33.8% of niraparib recipients vs. 0.6% of controls, anemia in 25.3% vs. 0%, neutropenia in 19.6% vs. 1.7%, fatigue in 8.2% vs. 0.6%, and hypertension in 8.2% vs. 2.2%.
Five of the 367 patients who received niraparib (1.4%) developed myelodysplasia or acute myeloid leukemia, compared with 2 of 179 patients (1.1%) treated with placebo.
Patient-reported outcomes measured via the Functional Assessment of Cancer Therapy – Ovarian Symptom Index and EQ (EuroQol) 5D-5L instrument showed high compliance rates and patient-reported symptom rates that were similar between niraparib and placebo groups.
The results also demonstrated that HRD testing can be used to identify patients without germline mutations in BRCA who may benefit from a PARP inhibitor, he said.
Tesaro funded the study. Dr. Mirza disclosed serving on the board of directors of the pharmaceutical companies, and consultant or advisory roles with multiple other companies. Dr. Pignata disclosed consulting, honoraria, and or research funding from several companies, but reported no relationship with Tesaro
COPENHAGEN – Women with platinum-sensitive, recurrent ovarian cancer treated with the PARP 1/2 inhibitor niraparib had significantly longer progression-free survival than did women who received a placebo, regardless of their BRCA mutational status, according to results of a phase III trial.
Median progression-free survival (PFS) among women with germline BRCA mutations who received niraparib was 21 months, compared with 5.5 months for women with germline BRCA mutations who received placebo (P less than .001).
For the overall population of women with no germline mutations, median PFS was 9.3 months for those who received niraparib, vs. 3.9 months for placebo-treated controls. Among women in this group whose tumors tested positive for homologous recombination deficiency (HRD), median PFS was 12.9 months, vs. 3.8 months without HRD.
Dr. Mirza reported results of the The NGOT-OV16/NOVA trial, the first phase III trial with a PARP inhibitor, at the European Society for Medical Oncology Congress. Results of the trial were simultaneously published online in The New England Journal of Medicine.
Niraparib is a selective, oral inhibitor of poly(adenosine diphosphate-ribose) polymerase (PARP) 1 and 2 that was previously shown to have efficacy against ovarian cancer in a phase I dose-escalation trial.
In the NGOT-OV16 NOVA trial, patients with platinum-sensitive high grade serous ovarian cancer first underwent chemotherapy with 4-6 cycles of a platinum-based regimen, and those who responded to platinum treatment were then stratified by the presence or absence of germline BRCA mutations, and then randomized on a 2:1 basis to either niraparib 300 mg once daily or placebo until disease progression.
A total of 203 patients with germline BRCA mutations and 350 with no mutations were enrolled in the trial.
As noted before, patients treated with niraparib in both trials arms had significantly longer PFS than did controls. The hazard ratio (HR) for niraparib in patients with germline BRCA mutations was 0.27. For the overall non–germline mutation population, the HR was 0.45, and for HRD-positive and HRD-negative subgroups, the HRs were 0.38 and 0.56, respectively (the latter is an exploratory endpoint, however; P less than .001 for the first three HRs shown).
Secondary efficacy endpoints, including chemotherapy-free interval and time to first subsequent treatment, also favored niraparib in patients with and without BRCA mutations.
Overall survival data for the trial are not sufficiently mature for reporting, however.
The safety profile of the drug was in line with that seen in other studies of PARP inhibitors, Dr. Mirza said. Grade 3/4 adverse events occurring in 5% or more of patients included thrombocytopenias in 33.8% of niraparib recipients vs. 0.6% of controls, anemia in 25.3% vs. 0%, neutropenia in 19.6% vs. 1.7%, fatigue in 8.2% vs. 0.6%, and hypertension in 8.2% vs. 2.2%.
Five of the 367 patients who received niraparib (1.4%) developed myelodysplasia or acute myeloid leukemia, compared with 2 of 179 patients (1.1%) treated with placebo.
Patient-reported outcomes measured via the Functional Assessment of Cancer Therapy – Ovarian Symptom Index and EQ (EuroQol) 5D-5L instrument showed high compliance rates and patient-reported symptom rates that were similar between niraparib and placebo groups.
The results also demonstrated that HRD testing can be used to identify patients without germline mutations in BRCA who may benefit from a PARP inhibitor, he said.
Tesaro funded the study. Dr. Mirza disclosed serving on the board of directors of the pharmaceutical companies, and consultant or advisory roles with multiple other companies. Dr. Pignata disclosed consulting, honoraria, and or research funding from several companies, but reported no relationship with Tesaro
Key clinical point: The poly ADP ribose polymerase (PARP) 1/2 inhibitor niraparib improved PFS in patients with ovarian cancer compared with those on placebo.
Major finding: Median PFS in women with germline BRCA mutations who received niraparib was 21 months, compared with 5.5 months for those on placebo.
Data source: Randomized double-blind phase III trial of 553 women with platinum-sensitive high grade serous ovarian cancer.
Disclosures: Tesaro funded the study. Dr. Mirza disclosed serving on the board of directors of the pharmaceutical companies, and consultant or advisory roles with multiple other companies. Dr. Pignata disclosed consulting, honoraria, and or research funding from several companies, but reported no relationship with Tesaro.
Cow’s milk allergy appears to affect more U.S. infants than thought
MONTREAL – The incidence of cow’s milk protein allergy during the first few months of life may be much more common than suggested by published studies, based on what was found is a prospective study with 700 infants seen regularly at a single, general pediatrics practice in suburban Massachusetts.
Among the 700 infants enrolled in this series, 105 (15%) were diagnosed with cow’s milk protein allergy (CMPA) when they were 5-163 days old, with a median age at diagnosis of 33 days, Victoria J. Martin, MD, said at the World Congress of Pediatric Gastroenterology, Hepatology, and Nutrition. She and her associates confirmed that all these infants had true CMPA episodes of proctocolitis by requiring detection of blood in the stool of affected children.

The study results also suggested a protective effect against CMPA when infants received some amount of early breastfeeding, and a pilot substudy run in 47 of the enrolled infants also suggested a link between development of CMPA and abnormalities in the microbiome composition of affected infants, she reported.
While the 15% incidence rate was unexpectedly high, it “absolutely feels like what we see in routine clinical practice,” Dr. Martin said in an interview. She chalked up the much-lower figure cited in the pediatric literature as relying on strict follow-up confirmation by rechallenge of the child with cow’s milk, a step often not taken by busy clinicians. Deferring formal confirmation also often means delayed reintroduction of cow’s milk into the infant’s diet, with restriction often continuing for perhaps a year following the index episode of CMPA. Although such unnecessarily long delays in milk reintroduction have largely been considered benign, recent findings from the Learning Early About Peanut Allergy (LEAP) trial that withholding peanut exposure can increase development of peanut allergies suggests that children also might receive long-term benefit from quicker reintroduction of milk in terms of better development of the immune system and microbiome, she said.
“If we rechallenged all these infants after 1 month, I think we’d find a CMPA rate closer to 3%. Leaving infants on a mild restricted diet for 12 months is a mistake,” she added.
The Gastrointestinal Microbiome & Proctocolitis (GMAP) study enrolled 700 infants seen at a single general practice pediatric practice in suburban Massachusetts at the time of their first well-baby visit, at a median age of 8 days. During 2 years of follow-up, the researchers collected stool specimens from the enrolled children at each of up to five scheduled visits during the first 4 months. They also kept track of when children received a CMPA diagnosis confirmed by at least one bloody stool.
Analysis of CMPA correlates showed that, among infants who developed it, 17% had not received any breastfeeding soon after birth, while among infants who did not develop CMPA, 8% did not undergo early breastfeeding. The incidence of CMPA was roughly similar among infants who received an early combination of breast milk and formula and in those who received exclusively breast milk during the first days of life, showing that even partial breastfeeding is better than no breastfeeding, Dr. Martin noted.
Her analysis also includes initial results from microbial assessment of the collected serial stool specimens from a subgroup of 24 infants who developed CMPA and 23 who did not, with a total of 223 total specimens evaluated. These studies showed that the infants who developed CMPA significantly lagged in their colonization with Bifidobacteria, had significantly higher colonization levels with Enterobacteriaceae, and that in infants who did develop CMPA, their gut level of Clostridia significantly increased as their proctocolitis resolved.
Dr. Martin had no relevant financial disclosures.
[email protected]
On Twitter @mitchelzoler
MONTREAL – The incidence of cow’s milk protein allergy during the first few months of life may be much more common than suggested by published studies, based on what was found is a prospective study with 700 infants seen regularly at a single, general pediatrics practice in suburban Massachusetts.
Among the 700 infants enrolled in this series, 105 (15%) were diagnosed with cow’s milk protein allergy (CMPA) when they were 5-163 days old, with a median age at diagnosis of 33 days, Victoria J. Martin, MD, said at the World Congress of Pediatric Gastroenterology, Hepatology, and Nutrition. She and her associates confirmed that all these infants had true CMPA episodes of proctocolitis by requiring detection of blood in the stool of affected children.
The study results also suggested a protective effect against CMPA when infants received some amount of early breastfeeding, and a pilot substudy run in 47 of the enrolled infants also suggested a link between development of CMPA and abnormalities in the microbiome composition of affected infants, she reported.
While the 15% incidence rate was unexpectedly high, it “absolutely feels like what we see in routine clinical practice,” Dr. Martin said in an interview. She chalked up the much-lower figure cited in the pediatric literature as relying on strict follow-up confirmation by rechallenge of the child with cow’s milk, a step often not taken by busy clinicians. Deferring formal confirmation also often means delayed reintroduction of cow’s milk into the infant’s diet, with restriction often continuing for perhaps a year following the index episode of CMPA. Although such unnecessarily long delays in milk reintroduction have largely been considered benign, recent findings from the Learning Early About Peanut Allergy (LEAP) trial that withholding peanut exposure can increase development of peanut allergies suggests that children also might receive long-term benefit from quicker reintroduction of milk in terms of better development of the immune system and microbiome, she said.
“If we rechallenged all these infants after 1 month, I think we’d find a CMPA rate closer to 3%. Leaving infants on a mild restricted diet for 12 months is a mistake,” she added.
The Gastrointestinal Microbiome & Proctocolitis (GMAP) study enrolled 700 infants seen at a single general practice pediatric practice in suburban Massachusetts at the time of their first well-baby visit, at a median age of 8 days. During 2 years of follow-up, the researchers collected stool specimens from the enrolled children at each of up to five scheduled visits during the first 4 months. They also kept track of when children received a CMPA diagnosis confirmed by at least one bloody stool.
Analysis of CMPA correlates showed that, among infants who developed it, 17% had not received any breastfeeding soon after birth, while among infants who did not develop CMPA, 8% did not undergo early breastfeeding. The incidence of CMPA was roughly similar among infants who received an early combination of breast milk and formula and in those who received exclusively breast milk during the first days of life, showing that even partial breastfeeding is better than no breastfeeding, Dr. Martin noted.
Her analysis also includes initial results from microbial assessment of the collected serial stool specimens from a subgroup of 24 infants who developed CMPA and 23 who did not, with a total of 223 total specimens evaluated. These studies showed that the infants who developed CMPA significantly lagged in their colonization with Bifidobacteria, had significantly higher colonization levels with Enterobacteriaceae, and that in infants who did develop CMPA, their gut level of Clostridia significantly increased as their proctocolitis resolved.
Dr. Martin had no relevant financial disclosures.
[email protected]
On Twitter @mitchelzoler
MONTREAL – The incidence of cow’s milk protein allergy during the first few months of life may be much more common than suggested by published studies, based on what was found is a prospective study with 700 infants seen regularly at a single, general pediatrics practice in suburban Massachusetts.
Among the 700 infants enrolled in this series, 105 (15%) were diagnosed with cow’s milk protein allergy (CMPA) when they were 5-163 days old, with a median age at diagnosis of 33 days, Victoria J. Martin, MD, said at the World Congress of Pediatric Gastroenterology, Hepatology, and Nutrition. She and her associates confirmed that all these infants had true CMPA episodes of proctocolitis by requiring detection of blood in the stool of affected children.
The study results also suggested a protective effect against CMPA when infants received some amount of early breastfeeding, and a pilot substudy run in 47 of the enrolled infants also suggested a link between development of CMPA and abnormalities in the microbiome composition of affected infants, she reported.
While the 15% incidence rate was unexpectedly high, it “absolutely feels like what we see in routine clinical practice,” Dr. Martin said in an interview. She chalked up the much-lower figure cited in the pediatric literature as relying on strict follow-up confirmation by rechallenge of the child with cow’s milk, a step often not taken by busy clinicians. Deferring formal confirmation also often means delayed reintroduction of cow’s milk into the infant’s diet, with restriction often continuing for perhaps a year following the index episode of CMPA. Although such unnecessarily long delays in milk reintroduction have largely been considered benign, recent findings from the Learning Early About Peanut Allergy (LEAP) trial that withholding peanut exposure can increase development of peanut allergies suggests that children also might receive long-term benefit from quicker reintroduction of milk in terms of better development of the immune system and microbiome, she said.
“If we rechallenged all these infants after 1 month, I think we’d find a CMPA rate closer to 3%. Leaving infants on a mild restricted diet for 12 months is a mistake,” she added.
The Gastrointestinal Microbiome & Proctocolitis (GMAP) study enrolled 700 infants seen at a single general practice pediatric practice in suburban Massachusetts at the time of their first well-baby visit, at a median age of 8 days. During 2 years of follow-up, the researchers collected stool specimens from the enrolled children at each of up to five scheduled visits during the first 4 months. They also kept track of when children received a CMPA diagnosis confirmed by at least one bloody stool.
Analysis of CMPA correlates showed that, among infants who developed it, 17% had not received any breastfeeding soon after birth, while among infants who did not develop CMPA, 8% did not undergo early breastfeeding. The incidence of CMPA was roughly similar among infants who received an early combination of breast milk and formula and in those who received exclusively breast milk during the first days of life, showing that even partial breastfeeding is better than no breastfeeding, Dr. Martin noted.
Her analysis also includes initial results from microbial assessment of the collected serial stool specimens from a subgroup of 24 infants who developed CMPA and 23 who did not, with a total of 223 total specimens evaluated. These studies showed that the infants who developed CMPA significantly lagged in their colonization with Bifidobacteria, had significantly higher colonization levels with Enterobacteriaceae, and that in infants who did develop CMPA, their gut level of Clostridia significantly increased as their proctocolitis resolved.
Dr. Martin had no relevant financial disclosures.
[email protected]
On Twitter @mitchelzoler
AT WCPGHAN 2016
Key clinical point:
Major finding: Among 700 enrolled well infants, aged 5-163 days, 105 (15%) developed proctocolitis linked with cow’s milk.
Data source: Prospective observational study of 700 healthy neonates seen at a single U.S. pediatric practice.
Disclosures: Dr. Martin had no relevant financial disclosures.
Vaccination rates up in U.S. kindergartners in 2015, steady in 19- to 35-month-olds
Vaccination coverage for MMR and DTaP increased for children in kindergarten during the 2015-2016 school year, but remained steady for children aged 19-35 months in 2015, according to reports from the Centers for Disease Control and Prevention.
The median MMR vaccination rate for kindergartners in 2015 was 94.6%, up significantly from 92.6% in 2014. DTaP coverage also increased, rising from 92.4% to 94.2%. A total of 32 states saw an increase in MMR coverage in 2015, with 22 states reporting greater than 95% coverage. Only 3 states and the District of Columbia reported less than 90% coverage, down from 7 states and D.C. in 2014.![]()
While the median vaccination rates were increased in 2015, the median exemption rate also increased by nearly 11% to 1.9% overall. This was caused in part by the addition of reports from Texas and Wyoming, neither of which reported the number of exemptions in 2014, the CDC investigators remarked.
In a second CDC report from Dr. Holly Hill and her associates based on data collected from the National Immunization Survey, the vaccination rate for children aged 19-35 months in 2015 did not increase significantly from the previous year (MMWR. 2016 Oct 6. doi: 10.15585/mmwr.mm639a4). The rate of children who received four or more doses of DTaP and at least one dose of MMR increased by 0.4 percentage points each from 84.2% to 84.6% and from 91.5% to 91.9%, respectively. The largest increase was seen in hepatitis A vaccine, where the rate of vaccination increased from 57.5% to 59.6%.
Healthy People 2020 goals for greater than 90% coverage for children aged 19-35 months were met for four vaccines in 2015: three or more doses of poliovirus vaccine, one or more doses of MMR, three or more doses of hepatitis B vaccine, and one or more doses of varicella vaccine. Vaccine coverage was lower in almost all cases for children living below the poverty level. The largest discrepancies were seen in rotavirus and varicella vaccines. The combined seven-vaccine series rate for children at or above the poverty line was 74.7%, and was 68.7% for children below the poverty line.
“Continued surveillance is needed to monitor coverage, locate pockets of susceptibility, and evaluate the impact of interventions designed to ensure that all children remain adequately protected against vaccine-preventable diseases,” Dr. Hill and her associates noted.
The CDC investigators had no relevant financial disclosures to report.
Vaccination coverage for MMR and DTaP increased for children in kindergarten during the 2015-2016 school year, but remained steady for children aged 19-35 months in 2015, according to reports from the Centers for Disease Control and Prevention.
The median MMR vaccination rate for kindergartners in 2015 was 94.6%, up significantly from 92.6% in 2014. DTaP coverage also increased, rising from 92.4% to 94.2%. A total of 32 states saw an increase in MMR coverage in 2015, with 22 states reporting greater than 95% coverage. Only 3 states and the District of Columbia reported less than 90% coverage, down from 7 states and D.C. in 2014.![]()
While the median vaccination rates were increased in 2015, the median exemption rate also increased by nearly 11% to 1.9% overall. This was caused in part by the addition of reports from Texas and Wyoming, neither of which reported the number of exemptions in 2014, the CDC investigators remarked.
In a second CDC report from Dr. Holly Hill and her associates based on data collected from the National Immunization Survey, the vaccination rate for children aged 19-35 months in 2015 did not increase significantly from the previous year (MMWR. 2016 Oct 6. doi: 10.15585/mmwr.mm639a4). The rate of children who received four or more doses of DTaP and at least one dose of MMR increased by 0.4 percentage points each from 84.2% to 84.6% and from 91.5% to 91.9%, respectively. The largest increase was seen in hepatitis A vaccine, where the rate of vaccination increased from 57.5% to 59.6%.
Healthy People 2020 goals for greater than 90% coverage for children aged 19-35 months were met for four vaccines in 2015: three or more doses of poliovirus vaccine, one or more doses of MMR, three or more doses of hepatitis B vaccine, and one or more doses of varicella vaccine. Vaccine coverage was lower in almost all cases for children living below the poverty level. The largest discrepancies were seen in rotavirus and varicella vaccines. The combined seven-vaccine series rate for children at or above the poverty line was 74.7%, and was 68.7% for children below the poverty line.
“Continued surveillance is needed to monitor coverage, locate pockets of susceptibility, and evaluate the impact of interventions designed to ensure that all children remain adequately protected against vaccine-preventable diseases,” Dr. Hill and her associates noted.
The CDC investigators had no relevant financial disclosures to report.
Vaccination coverage for MMR and DTaP increased for children in kindergarten during the 2015-2016 school year, but remained steady for children aged 19-35 months in 2015, according to reports from the Centers for Disease Control and Prevention.
The median MMR vaccination rate for kindergartners in 2015 was 94.6%, up significantly from 92.6% in 2014. DTaP coverage also increased, rising from 92.4% to 94.2%. A total of 32 states saw an increase in MMR coverage in 2015, with 22 states reporting greater than 95% coverage. Only 3 states and the District of Columbia reported less than 90% coverage, down from 7 states and D.C. in 2014.![]()
While the median vaccination rates were increased in 2015, the median exemption rate also increased by nearly 11% to 1.9% overall. This was caused in part by the addition of reports from Texas and Wyoming, neither of which reported the number of exemptions in 2014, the CDC investigators remarked.
In a second CDC report from Dr. Holly Hill and her associates based on data collected from the National Immunization Survey, the vaccination rate for children aged 19-35 months in 2015 did not increase significantly from the previous year (MMWR. 2016 Oct 6. doi: 10.15585/mmwr.mm639a4). The rate of children who received four or more doses of DTaP and at least one dose of MMR increased by 0.4 percentage points each from 84.2% to 84.6% and from 91.5% to 91.9%, respectively. The largest increase was seen in hepatitis A vaccine, where the rate of vaccination increased from 57.5% to 59.6%.
Healthy People 2020 goals for greater than 90% coverage for children aged 19-35 months were met for four vaccines in 2015: three or more doses of poliovirus vaccine, one or more doses of MMR, three or more doses of hepatitis B vaccine, and one or more doses of varicella vaccine. Vaccine coverage was lower in almost all cases for children living below the poverty level. The largest discrepancies were seen in rotavirus and varicella vaccines. The combined seven-vaccine series rate for children at or above the poverty line was 74.7%, and was 68.7% for children below the poverty line.
“Continued surveillance is needed to monitor coverage, locate pockets of susceptibility, and evaluate the impact of interventions designed to ensure that all children remain adequately protected against vaccine-preventable diseases,” Dr. Hill and her associates noted.
The CDC investigators had no relevant financial disclosures to report.
Silicone joint arthroplasty for RA shows sustained improvements at 7 years
Rheumatoid arthritis patients who underwent silicone metacarpophalangeal joint replacement maintained significant improvement in ulnar drift and extensor lag after 7 years of follow-up in the prospective, multicenter Silicone Arthroplasty in Rheumatoid Arthritis (SARA) study.
The silicone metacarpophalangeal joint arthroplasty (SMPA) group also showed significantly better metacarpophalangeal (MCP) joint arc of motion, as well as function, aesthetics, and satisfaction scores than did patients in the cohort who chose nonsurgical management.
The practice of replacing deformed MCP joints with a hinged silicone implant has been around for almost half a century, the investigators noted, but despite widespread use, there is a lack of high-quality surgical outcomes data for the procedure. The authors suggested that the scarcity of data may create a barrier for surgical referrals from rheumatologists and could account for the declining rate of surgical intervention for RA joint deformities.
The significant improvement in ulnar drift that occurred in patients who underwent SMPA versus no surgery remained even after adjustment for baseline severity, age, sex, and use of biologics. The adjustment was especially important because the nonsurgical group had better function at baseline, with significantly stronger grip and pinch strength, better Michigan Hand Questionnaire (MHQ) scores, and significantly better ulnar drift, extensor lag, and arc of motion than in the surgical group (Arthritis Care Res. 2016 Oct 1. doi: 10.1002/acr.23105).
The nonsurgical group largely maintained its baseline functional state during the 7-year follow-up period except for a decline in pinch strength.
“When the SMPA cohort was compared with the non-SMPA cohort, the covariate-adjusted difference showed significant benefits associated with SMPA hand outcome as measured by the MHQ function, aesthetics, and satisfaction scores, with no measures showing significantly better outcomes for the non-SMPA group,” the researchers wrote. “Although the average treatment effect estimate (ATT estimate) showed a decline over time for the average benefit from SMPA in those treated, the function score benefit as shown by the ATT estimate remained significant at year 5.”
Patients in the nonsurgical group were also given the option to crossover and receive surgery after 1 year, which two did. Eleven patients in the surgical group also decided to undergo surgery on their other hand.
Researchers saw one mild and three moderate implant-related adverse events during the study, including one patient who experienced ulnar deviation and needed a new joint replacement, one who dislocated the implant of the little finger a few weeks after insertion, and one who experienced sepsis in the joints 6 years after insertion and needed replacements for two implants. The fracture incidence of about 10% fits into the mid-range of previously reported fracture rates with this implant.
The study experienced significant losses to follow-up, particularly in the surgical group in which 7-year data were only available for 43% of participants in this group. The authors suggested this could have been because the surgical patients achieved their goals and were less inclined to the follow-up visits.
The study was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. No conflicts of interest were declared.
Rheumatoid arthritis patients who underwent silicone metacarpophalangeal joint replacement maintained significant improvement in ulnar drift and extensor lag after 7 years of follow-up in the prospective, multicenter Silicone Arthroplasty in Rheumatoid Arthritis (SARA) study.
The silicone metacarpophalangeal joint arthroplasty (SMPA) group also showed significantly better metacarpophalangeal (MCP) joint arc of motion, as well as function, aesthetics, and satisfaction scores than did patients in the cohort who chose nonsurgical management.
The practice of replacing deformed MCP joints with a hinged silicone implant has been around for almost half a century, the investigators noted, but despite widespread use, there is a lack of high-quality surgical outcomes data for the procedure. The authors suggested that the scarcity of data may create a barrier for surgical referrals from rheumatologists and could account for the declining rate of surgical intervention for RA joint deformities.
The significant improvement in ulnar drift that occurred in patients who underwent SMPA versus no surgery remained even after adjustment for baseline severity, age, sex, and use of biologics. The adjustment was especially important because the nonsurgical group had better function at baseline, with significantly stronger grip and pinch strength, better Michigan Hand Questionnaire (MHQ) scores, and significantly better ulnar drift, extensor lag, and arc of motion than in the surgical group (Arthritis Care Res. 2016 Oct 1. doi: 10.1002/acr.23105).
The nonsurgical group largely maintained its baseline functional state during the 7-year follow-up period except for a decline in pinch strength.
“When the SMPA cohort was compared with the non-SMPA cohort, the covariate-adjusted difference showed significant benefits associated with SMPA hand outcome as measured by the MHQ function, aesthetics, and satisfaction scores, with no measures showing significantly better outcomes for the non-SMPA group,” the researchers wrote. “Although the average treatment effect estimate (ATT estimate) showed a decline over time for the average benefit from SMPA in those treated, the function score benefit as shown by the ATT estimate remained significant at year 5.”
Patients in the nonsurgical group were also given the option to crossover and receive surgery after 1 year, which two did. Eleven patients in the surgical group also decided to undergo surgery on their other hand.
Researchers saw one mild and three moderate implant-related adverse events during the study, including one patient who experienced ulnar deviation and needed a new joint replacement, one who dislocated the implant of the little finger a few weeks after insertion, and one who experienced sepsis in the joints 6 years after insertion and needed replacements for two implants. The fracture incidence of about 10% fits into the mid-range of previously reported fracture rates with this implant.
The study experienced significant losses to follow-up, particularly in the surgical group in which 7-year data were only available for 43% of participants in this group. The authors suggested this could have been because the surgical patients achieved their goals and were less inclined to the follow-up visits.
The study was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. No conflicts of interest were declared.
Rheumatoid arthritis patients who underwent silicone metacarpophalangeal joint replacement maintained significant improvement in ulnar drift and extensor lag after 7 years of follow-up in the prospective, multicenter Silicone Arthroplasty in Rheumatoid Arthritis (SARA) study.
The silicone metacarpophalangeal joint arthroplasty (SMPA) group also showed significantly better metacarpophalangeal (MCP) joint arc of motion, as well as function, aesthetics, and satisfaction scores than did patients in the cohort who chose nonsurgical management.
The practice of replacing deformed MCP joints with a hinged silicone implant has been around for almost half a century, the investigators noted, but despite widespread use, there is a lack of high-quality surgical outcomes data for the procedure. The authors suggested that the scarcity of data may create a barrier for surgical referrals from rheumatologists and could account for the declining rate of surgical intervention for RA joint deformities.
The significant improvement in ulnar drift that occurred in patients who underwent SMPA versus no surgery remained even after adjustment for baseline severity, age, sex, and use of biologics. The adjustment was especially important because the nonsurgical group had better function at baseline, with significantly stronger grip and pinch strength, better Michigan Hand Questionnaire (MHQ) scores, and significantly better ulnar drift, extensor lag, and arc of motion than in the surgical group (Arthritis Care Res. 2016 Oct 1. doi: 10.1002/acr.23105).
The nonsurgical group largely maintained its baseline functional state during the 7-year follow-up period except for a decline in pinch strength.
“When the SMPA cohort was compared with the non-SMPA cohort, the covariate-adjusted difference showed significant benefits associated with SMPA hand outcome as measured by the MHQ function, aesthetics, and satisfaction scores, with no measures showing significantly better outcomes for the non-SMPA group,” the researchers wrote. “Although the average treatment effect estimate (ATT estimate) showed a decline over time for the average benefit from SMPA in those treated, the function score benefit as shown by the ATT estimate remained significant at year 5.”
Patients in the nonsurgical group were also given the option to crossover and receive surgery after 1 year, which two did. Eleven patients in the surgical group also decided to undergo surgery on their other hand.
Researchers saw one mild and three moderate implant-related adverse events during the study, including one patient who experienced ulnar deviation and needed a new joint replacement, one who dislocated the implant of the little finger a few weeks after insertion, and one who experienced sepsis in the joints 6 years after insertion and needed replacements for two implants. The fracture incidence of about 10% fits into the mid-range of previously reported fracture rates with this implant.
The study experienced significant losses to follow-up, particularly in the surgical group in which 7-year data were only available for 43% of participants in this group. The authors suggested this could have been because the surgical patients achieved their goals and were less inclined to the follow-up visits.
The study was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. No conflicts of interest were declared.
Key clinical point:
Major finding: Patients who elected to undergo silicone MCP joint replacement showed significant improvements in ulnar drift and extensor lag, as well as in function, aesthetics, and satisfaction scores at 7 years after the procedure.
Data source: Cohort study of 170 patients with rheumatoid arthritis–related severe deformity at the metacarpophalangeal joints.
Disclosures: The study was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. No conflicts of interest were declared.