User login
ACOG issues guidelines for managing listeriosis during pregnancy
The first medical management guidelines for treating the bacteria listeria during pregnancy were recently released by the American College of Obstetricians and Gynecologists (ACOG).1
The new guidelines were developed in response to recent reports of recalls of listeria-contaminated food and concern because data show the incidence of listeriosis among pregnant women is approximately 13 times higher than in the general population.1 Maternal listeriosis can cause significant fetal and perinatal complications, including miscarriage, preterm labor, stillbirth, as well as neonatal listeriosis and neonatal death.2
“It is essential for ObGyns not only to be aware of the best ways to manage the care of a pregnant patient who has been exposed to listeria bacteria, but, just as important, to counsel pregnant women regarding how to avoid potential exposure,” said Jeffrey L. Ecker, MD, chair of the ACOG Committee on Obstetric Practice.2
Symptoms of listeriosis are similar to a flu-like infection and can include fever, muscle pain, backache, headache, and gastrointestinal symptoms, including diarrhea.2
The Committee Opinion offers management recommendations for three scenarios1:
- Asymptomatic women who have been exposed to listeria: No testing is necessary unless symptoms develop within 2 months of exposure. Listeriosis-related fetal surveillance is unnecessary.
- No fever, with mild symptoms consistent with listeriosis: A pregnant woman who has been exposed to a listeria-containing food and is displaying mild symptoms, but does not have a fever, does not require culture testing (although it may be done). If her blood is tested, the laboratory should be informed about the listeriosis threat so that the bacteria is not confused with a contaminant.
- Fever, with or without symptoms consistent with listeriosis: A pregnant woman who has been exposed and has a fever exceeding 100.6°F should undergo blood culture testing. Because results will not be available for several days, the Committee Opinion recommends simultaneous listeriosis treatment for the patient and surveillance of the fetus.
Preventive measures to avoid listeria exposure include not eating the following foods1:
- hot dogs, lunch meats, cold cuts served cold or heated to less than 165°F
- refrigerated pate and meat spreads
- refrigerated smoked seafood
- raw (unpasteurized) milk
- unpasteurized soft cheese (feta, queso blanco, Brie, blue-veined cheeses)
- unwashed raw produce (when eating raw fruits and vegetables, skin should be washed thoroughly in running tap water, even if it will be peeled or cut).
Share your thoughts on this news! Send your Letter to the Editor to [email protected]. Please include your name, and the city and state in which you practice.
- American College of Obstetricians and Gynecologists. Committee Opinion. Management of pregnant women with presumptive exposure to Listeria monocytogenes [published online ahead of print August 5, 2014]. http://www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Obstetric-Practice/Management-of-Pregnant-Women-With-Presumptive-Exposure-to-Listeria-monocytogenes. Accessed September 29, 2014.
- American College of Obstetricians and Gynecologists. Ob-Gyns address management of listeria during pregnancy [press release]. http://www.acog.org/About-ACOG/News-Room/News-Releases/2014/Ob-Gyns-Address-Management-of-Listeria-During-Pregnancy. Published August 6, 2014. Accessed September 29, 2014.
The first medical management guidelines for treating the bacteria listeria during pregnancy were recently released by the American College of Obstetricians and Gynecologists (ACOG).1
The new guidelines were developed in response to recent reports of recalls of listeria-contaminated food and concern because data show the incidence of listeriosis among pregnant women is approximately 13 times higher than in the general population.1 Maternal listeriosis can cause significant fetal and perinatal complications, including miscarriage, preterm labor, stillbirth, as well as neonatal listeriosis and neonatal death.2
“It is essential for ObGyns not only to be aware of the best ways to manage the care of a pregnant patient who has been exposed to listeria bacteria, but, just as important, to counsel pregnant women regarding how to avoid potential exposure,” said Jeffrey L. Ecker, MD, chair of the ACOG Committee on Obstetric Practice.2
Symptoms of listeriosis are similar to a flu-like infection and can include fever, muscle pain, backache, headache, and gastrointestinal symptoms, including diarrhea.2
The Committee Opinion offers management recommendations for three scenarios1:
- Asymptomatic women who have been exposed to listeria: No testing is necessary unless symptoms develop within 2 months of exposure. Listeriosis-related fetal surveillance is unnecessary.
- No fever, with mild symptoms consistent with listeriosis: A pregnant woman who has been exposed to a listeria-containing food and is displaying mild symptoms, but does not have a fever, does not require culture testing (although it may be done). If her blood is tested, the laboratory should be informed about the listeriosis threat so that the bacteria is not confused with a contaminant.
- Fever, with or without symptoms consistent with listeriosis: A pregnant woman who has been exposed and has a fever exceeding 100.6°F should undergo blood culture testing. Because results will not be available for several days, the Committee Opinion recommends simultaneous listeriosis treatment for the patient and surveillance of the fetus.
Preventive measures to avoid listeria exposure include not eating the following foods1:
- hot dogs, lunch meats, cold cuts served cold or heated to less than 165°F
- refrigerated pate and meat spreads
- refrigerated smoked seafood
- raw (unpasteurized) milk
- unpasteurized soft cheese (feta, queso blanco, Brie, blue-veined cheeses)
- unwashed raw produce (when eating raw fruits and vegetables, skin should be washed thoroughly in running tap water, even if it will be peeled or cut).
Share your thoughts on this news! Send your Letter to the Editor to [email protected]. Please include your name, and the city and state in which you practice.
The first medical management guidelines for treating the bacteria listeria during pregnancy were recently released by the American College of Obstetricians and Gynecologists (ACOG).1
The new guidelines were developed in response to recent reports of recalls of listeria-contaminated food and concern because data show the incidence of listeriosis among pregnant women is approximately 13 times higher than in the general population.1 Maternal listeriosis can cause significant fetal and perinatal complications, including miscarriage, preterm labor, stillbirth, as well as neonatal listeriosis and neonatal death.2
“It is essential for ObGyns not only to be aware of the best ways to manage the care of a pregnant patient who has been exposed to listeria bacteria, but, just as important, to counsel pregnant women regarding how to avoid potential exposure,” said Jeffrey L. Ecker, MD, chair of the ACOG Committee on Obstetric Practice.2
Symptoms of listeriosis are similar to a flu-like infection and can include fever, muscle pain, backache, headache, and gastrointestinal symptoms, including diarrhea.2
The Committee Opinion offers management recommendations for three scenarios1:
- Asymptomatic women who have been exposed to listeria: No testing is necessary unless symptoms develop within 2 months of exposure. Listeriosis-related fetal surveillance is unnecessary.
- No fever, with mild symptoms consistent with listeriosis: A pregnant woman who has been exposed to a listeria-containing food and is displaying mild symptoms, but does not have a fever, does not require culture testing (although it may be done). If her blood is tested, the laboratory should be informed about the listeriosis threat so that the bacteria is not confused with a contaminant.
- Fever, with or without symptoms consistent with listeriosis: A pregnant woman who has been exposed and has a fever exceeding 100.6°F should undergo blood culture testing. Because results will not be available for several days, the Committee Opinion recommends simultaneous listeriosis treatment for the patient and surveillance of the fetus.
Preventive measures to avoid listeria exposure include not eating the following foods1:
- hot dogs, lunch meats, cold cuts served cold or heated to less than 165°F
- refrigerated pate and meat spreads
- refrigerated smoked seafood
- raw (unpasteurized) milk
- unpasteurized soft cheese (feta, queso blanco, Brie, blue-veined cheeses)
- unwashed raw produce (when eating raw fruits and vegetables, skin should be washed thoroughly in running tap water, even if it will be peeled or cut).
Share your thoughts on this news! Send your Letter to the Editor to [email protected]. Please include your name, and the city and state in which you practice.
- American College of Obstetricians and Gynecologists. Committee Opinion. Management of pregnant women with presumptive exposure to Listeria monocytogenes [published online ahead of print August 5, 2014]. http://www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Obstetric-Practice/Management-of-Pregnant-Women-With-Presumptive-Exposure-to-Listeria-monocytogenes. Accessed September 29, 2014.
- American College of Obstetricians and Gynecologists. Ob-Gyns address management of listeria during pregnancy [press release]. http://www.acog.org/About-ACOG/News-Room/News-Releases/2014/Ob-Gyns-Address-Management-of-Listeria-During-Pregnancy. Published August 6, 2014. Accessed September 29, 2014.
- American College of Obstetricians and Gynecologists. Committee Opinion. Management of pregnant women with presumptive exposure to Listeria monocytogenes [published online ahead of print August 5, 2014]. http://www.acog.org/Resources-And-Publications/Committee-Opinions/Committee-on-Obstetric-Practice/Management-of-Pregnant-Women-With-Presumptive-Exposure-to-Listeria-monocytogenes. Accessed September 29, 2014.
- American College of Obstetricians and Gynecologists. Ob-Gyns address management of listeria during pregnancy [press release]. http://www.acog.org/About-ACOG/News-Room/News-Releases/2014/Ob-Gyns-Address-Management-of-Listeria-During-Pregnancy. Published August 6, 2014. Accessed September 29, 2014.
E-cigarettes: What to tell your patients

EHRs: Something’s gotta give

Clicking on a checklist of symptoms seldom provides sufficient information about the patient’s illness. “Hypertension” and “type 2 diabetes” are not assessments; they are diagnoses that do not tell the person reading the electronic health record (EHR) how the patient is doing. A diagnosis of “abdominal pain” without a prioritized differential is inadequate, especially in court.
Why is visit documentation too often inadequate? I am convinced it is rarely due to clinician incompetence, laziness, or lack of knowledge, but nearly always due to a combination of inadequate EHR formats and billing documentation requirements that encourage quantity rather than quality. Documentation is no longer driven by the essential need to record the care provided.
I’m sure a lot of you are nodding your heads in agreement. You all know what those EHR notes look like—cluttered with cut-and-pasted information drawn from prior encounters that document no end of details regarding medical, family, and social histories, facts that are often completely irrelevant to the reason the patient is in the office today. And unless one meticulously updates those other elements of the patient’s record, this information pulled into the note may be inaccurate.
I am not the only one complaining. The American Medical Association just published Improving Care: Priorities to Improve Electronic Health Record Usability,1 which outlines 8 priorities for EHR improvement. The first is to “Enhance physicians’ ability to provide high-quality patient care.” I could not agree more. I think today’s EHRs are like an old-fashioned crank phone and what we really need is an iPhone.
Something has got to change.
So tell me: Have any of you figured out how to use your EHR to enhance the quality of your documentation?
Reference
1. American Medical Association. Improving Care: Priorities to Improve Electronic Health Record Usability. American Medical Association Web site. Available at: https://download.ama-assn.org/resources/doc/ps2/x-pub/ehr-priorities.pdf. Accessed September 18, 2014.
Clicking on a checklist of symptoms seldom provides sufficient information about the patient’s illness. “Hypertension” and “type 2 diabetes” are not assessments; they are diagnoses that do not tell the person reading the electronic health record (EHR) how the patient is doing. A diagnosis of “abdominal pain” without a prioritized differential is inadequate, especially in court.
Why is visit documentation too often inadequate? I am convinced it is rarely due to clinician incompetence, laziness, or lack of knowledge, but nearly always due to a combination of inadequate EHR formats and billing documentation requirements that encourage quantity rather than quality. Documentation is no longer driven by the essential need to record the care provided.
I’m sure a lot of you are nodding your heads in agreement. You all know what those EHR notes look like—cluttered with cut-and-pasted information drawn from prior encounters that document no end of details regarding medical, family, and social histories, facts that are often completely irrelevant to the reason the patient is in the office today. And unless one meticulously updates those other elements of the patient’s record, this information pulled into the note may be inaccurate.
I am not the only one complaining. The American Medical Association just published Improving Care: Priorities to Improve Electronic Health Record Usability,1 which outlines 8 priorities for EHR improvement. The first is to “Enhance physicians’ ability to provide high-quality patient care.” I could not agree more. I think today’s EHRs are like an old-fashioned crank phone and what we really need is an iPhone.
Something has got to change.
So tell me: Have any of you figured out how to use your EHR to enhance the quality of your documentation?
Clicking on a checklist of symptoms seldom provides sufficient information about the patient’s illness. “Hypertension” and “type 2 diabetes” are not assessments; they are diagnoses that do not tell the person reading the electronic health record (EHR) how the patient is doing. A diagnosis of “abdominal pain” without a prioritized differential is inadequate, especially in court.
Why is visit documentation too often inadequate? I am convinced it is rarely due to clinician incompetence, laziness, or lack of knowledge, but nearly always due to a combination of inadequate EHR formats and billing documentation requirements that encourage quantity rather than quality. Documentation is no longer driven by the essential need to record the care provided.
I’m sure a lot of you are nodding your heads in agreement. You all know what those EHR notes look like—cluttered with cut-and-pasted information drawn from prior encounters that document no end of details regarding medical, family, and social histories, facts that are often completely irrelevant to the reason the patient is in the office today. And unless one meticulously updates those other elements of the patient’s record, this information pulled into the note may be inaccurate.
I am not the only one complaining. The American Medical Association just published Improving Care: Priorities to Improve Electronic Health Record Usability,1 which outlines 8 priorities for EHR improvement. The first is to “Enhance physicians’ ability to provide high-quality patient care.” I could not agree more. I think today’s EHRs are like an old-fashioned crank phone and what we really need is an iPhone.
Something has got to change.
So tell me: Have any of you figured out how to use your EHR to enhance the quality of your documentation?
Reference
1. American Medical Association. Improving Care: Priorities to Improve Electronic Health Record Usability. American Medical Association Web site. Available at: https://download.ama-assn.org/resources/doc/ps2/x-pub/ehr-priorities.pdf. Accessed September 18, 2014.
Reference
1. American Medical Association. Improving Care: Priorities to Improve Electronic Health Record Usability. American Medical Association Web site. Available at: https://download.ama-assn.org/resources/doc/ps2/x-pub/ehr-priorities.pdf. Accessed September 18, 2014.

Uterine rupture, child stillborn: $3.8M net award
Uterine rupture, child stillborn: $3.8M net award
At 35 weeks' gestation, a woman went to the emergency department (ED) with abdominal pain, fast heartbeat, and irregular contractions. Her history included three cesarean deliveries, including one with a vertical incision. She was admitted, and a cesarean delivery was planned for the next day. After 8 hours, during which the patient’s condition worsened, an emergency cesarean delivery was undertaken. A full rupture of the uterus was found; the baby’s body had extruded into the mother’s abdomen. The child was stillborn.
PARENTS’ CLAIM The stillbirth could have been avoided if the nurses had communicated the mother’s worsening condition to the physicians.
DEFENDANTS’ DEFENSE After the hospital and physicians settled prior to trial, the case continued against the nurse in charge of the mother’s care and the nurse-staffing group. Negligence was denied; all protocols were followed.
VERDICT A $2.9 million Illinois verdict was returned. With a $900,000 settlement from the hospital and physicians, the net award was $3.8 million.
_______________
Where did rare strep A infection come from?
A 36-year-old woman reported heavy vaginal bleeding to her ObGyn. She underwent endometrial ablation in her physician’s office.
The next day, the woman called the office to report abdominal pain. She was told to stop the medication she was taking, and if the pain continued to the next day, to go to an ED. The next day, the patient went to the ED and was found to be in septic shock. During emergency laparotomy, 50 mL of purulent fluid were drained and an emergency hysterectomy was performed. Three days later, the patient died from pulmonary arrest caused by toxic shock syndrome. An autopsy revealed that the patient’s sepsis was caused by group A streptococci (GAS) infection.
ESTATE’S CLAIM The patient was not a proper candidate for endometrial ablation because of her history of chronic cervical infection. The ObGyn perforated the cervix during the procedure and tried to conceal it. At autopsy, bone wax was found in the rectal lumen that had been used to cover up damage to the cervix. The ObGyn introduced GAS bacteria into the patient’s system. The ObGyn’s staff failed to ask the proper questions when she called the day after the procedure. She should have been told to go directly to the ED.
DEFENDANTS’ DEFENSE The ObGyn did not perforate the cervix or uterus during the procedure. GAS infection is so rare that it would have been difficult to foresee or diagnose. Potentially, the patient had a chronic
cervical infection before ablation.
VERDICT A Texas defense verdict was returned.
_______________
DURING INSERTION, IUD PERFORATES UTERINE WALL; LATER FOUND BELOW LIVER
On July 21, a 46-year-old woman went to an ObGyn for placement of an intrauterine device (IUD). Shortly after the ObGyn inserted the levonorgestrel-releasing intrauterine system (Mirena, Bayer HealthCare), the patient reported severe pelvic and abdominal pain. On July 26, the patient underwent surgical removal of the IUD.
She was discharged on July 29 but continued to report pain. She was readmitted to the hospital the next day and treated for pain. She was bed ridden for 3 weeks after IUD-removal surgery, and had a 3-month recovery before feeling pain free.
PATIENT’S CLAIM The ObGyn was negligent in perforating the patient’s uterine wall during IUD insertion, causing the device to ultimately migrate under the patient’s liver.
DEFENDANTS’ DEFENSE Uterine perforation is a known complication of IUD insertion. The IUD escaped from the patient’s uterus at a later time and not during the insertion procedure.
VERDICT A Florida verdict of $208,839 was returned; the amount was reduced to $161,058 because the medical expenses were written off by the health-care providers.
_______________
Was travel appropriate for this pregnant woman?
A woman with a history of two premature deliveries and one miscarriage became pregnant again. She received prenatal care at an Army hospital. She traveled to Spain, where the baby was born at 31 weeks’ gestation. The baby required treatment in a neonatal intensive care unit (NICU) for 17 days. The child has cerebral palsy, with tetraplegia of all four extremities. She cannot walk without assistance and suffers severe cognitive and vision impairment.
PARENTS’ CLAIM The ObGyn at the Army hospital should not have approved the mother’s request for travel; he did so, despite knowing that the mother was at high risk for premature birth. The military medical hospital to which she was assigned in Spain could not manage a high-risk pregnancy, didn’t have a NICU, and didn’t have specialists to treat premature infants.
DEFENDANTS’ DEFENSE The ObGyn argued that he did not have access to the medical records showing the mother’s history. The patient countered that the ObGyn did indeed have the patient’s records, as he had discussed them with her.
VERDICT A $10,409,700 California verdict was returned against the ObGyn and the government facility.
_______________
Triple-negative BrCa not diagnosed until metastasized: $5.2M
After finding lumps in both breasts, a woman in her 30s saw a nurse practitioner (NP) at an Army hospital. A radiologist reported no mass in the right breast and multiple benign-appearing anechoic lesions in the left breast after bilateral mammography and ultrasonography (US) in July 2008. The Chief of Mammography Services recommended referral to a breast surgeon, but the patient never received the letter. It was placed in her mammography file, not in the treatment file.
In November 2008, the patient returned to the clinic. Bilateral diagnostic mammography and US were ordered, but for unknown reasons, cancelled. US of the left breast was interpreted as benign in January 2009.
After imaging in March 2010, followed by a needle biopsy of the right breast, a radiologist reported finding intermediate-grade infiltrating ductal carcinoma.
The patient sought care outside the military medical system at a large university hospital. In April 2010, stage 3 triple-negative invasive ductal carcinoma (IDC) was identified. The patient underwent chemotherapy, a double mastectomy, removal of 21 lymph nodes, and breast reconstruction. She was given a 60% chance of recurrence in 5–7 years.
PATIENT’S CLAIM It was negligent to not inform her of imaging results. Biopsy should have been performed in 2008, when the IDC was likely at stage 1; treatment would have been far less aggressive. Electronic medical records showed that the 2008 mammography and US results had been “signed off” by an NP at the clinic.
DEFENDANTS’ DEFENSE While unable to concede liability, the government agency did not contest the point.
VERDICT A $5.2 million Tennessee federal court bench verdict was returned, citing failures in communication, poor and improper record keeping and retention, failure to follow-up, and an unexplained cancellation of a medical order.
_______________
Woman dies from cervical cancer: $2.3M
In 2001, a 41-year-old woman had abnormal Pap smear results but her gynecologist did not order more testing. The patient was told to return in 3 months, but she did not return until 2007—reporting abnormal bleeding, vaginal discharge, and pain. Her Pap results were normal, however, and the gynecologist did not order further testing. In 2009, the patient was found to have advanced cervical cancer. She died 2 years later.
ESTATE’S CLAIM Further testing should have been ordered in 2001, which would have likely revealed dysplasia, which can lead to cancer. The laboratory incorrectly interpreted the 2007 Pap test; if the results had been properly reported, additional testing could have been ordered.
DEFENDANTS’ DEFENSE The laboratory and patient’s estate settled for a confidential amount before trial. The gynecologist denied negligence.
VERDICT A New Jersey jury found the gynecologist 40% at fault for his actions in 2007. The jury found the laboratory 50% at fault, and the patient 10% at fault. A gross verdict of $2.33 million was returned.
_______________
Bowel injury after cesarean delivery; mother dies of sepsis
At 40 4/7 weeks' gestation, a 37-year-old woman gave birth to a healthy child by cesarean delivery. The next day, the patient had an elevated white blood cell (WBC) count with a left shift, her abdomen was tympanic but soft, and she was passing flatus and belching. The ObGyn ordered a Fleet enema; only flatus was released. A covering ObGyn ordered an abdominal radiograph, which the radiologist reported as showing postoperative ileus and mild constipation. The patient was given a second Fleet enema the next day, resulting in watery stool. She vomited 300 mL of dark green fluid.
After a rectal tube was placed 2 days later, one hard brown stool and several brown, pasty, loose, and liquid stools were returned. She vomited several times that day, and was found to have hypoactive bowel sounds with continued tympanic quality in the upper quadrants. Laboratory testing revealed continued elevated WBC count with left shift. The next day, she had hypoactive bowel sounds with brown liquid stools. Later that morning, she was able to tolerate clear liquids. The ObGyn decided to discharge her home with instructions to continue on a clear liquid diet for 2 more days before advancing her diet.
The day after discharge, she was found unresponsive at home. She was taken to the hospital, but resuscitation attempts failed. She died. An autopsy revealed that the cause of death was sepsis.
ESTATE’S CLAIM The ObGyn was negligent in failing to diagnose and treat a postoperative intra-abdominal infection caused by bowel perforation. A surgical consult should have been obtained. The woman was prematurely discharged. The radiologist failed to report the presence of free air on the abdominal x-ray.
DEFENDANTS’ DEFENSE The case was settled during trial.
VERDICT A $1 million Maryland settlement was reached.
_______________
Right ureter injury detected and repaired
During laparoscopic-assisted vaginal hysterectomy, the ObGyn detected and repaired an injury to the right ureter. The patient’s recovery was delayed by the injury.
PATIENT’S CLAIM The ObGyn was negligent in using a Kleppinger bipolar cauterizing instrument to cauterize the vaginal cuff. Thermal overspray from the instrument or the instrument itself damaged the ureter. The ObGyn was also negligent in not performing diagnostic cystoscopy to confirm patency of the ureter after the repair was made.
PHYSICIAN’S DEFENSE Ureter injury is a known risk of the procedure. All procedures were performed according to protocol.
VERDICT A Florida defense verdict was returned.
_______________
Failure to detect inflammatory BrCa; woman dies
A 42-year-old woman underwent mammography in February 2002 after reporting pain, discoloration, inflammation, and swelling in her left breast. The radiologist who interpreted the mammography suggested a biopsy for a differential diagnosis of mastitis or inflammatory carcinoma. The biopsy results were negative.
The patient’s symptoms persisted, and she underwent US in late May 2002. Another radiologist interpreted the US, noting that the patient could not tolerate compression, which led to less than optimal evaluation. The radiologist suggested that mastitis was the likely cause of the patient’s symptoms.
The patient then consulted a surgeon, who ordered mammography and magnetic resonance imaging (MRI) followed by biopsy, which indicated cancer. The patient underwent a mastectomy but metastasis had already occurred. She died at age 50 prior to the trial.
ESTATE’S CLAIM If the cancer had been diagnosed earlier, the outcome would have been better. Both radiologists misinterpreted the mammographies.
DEFENDANTS’ DEFENSE The mammographies had been properly interpreted. Any missed diagnosis would not have impacted the outcome due to the type of cancer. The scans had been released to the patient, but were subsequently lost; an adverse interference instruction was given to the jury.
VERDICT A New York defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Uterine rupture, child stillborn: $3.8M net award
At 35 weeks' gestation, a woman went to the emergency department (ED) with abdominal pain, fast heartbeat, and irregular contractions. Her history included three cesarean deliveries, including one with a vertical incision. She was admitted, and a cesarean delivery was planned for the next day. After 8 hours, during which the patient’s condition worsened, an emergency cesarean delivery was undertaken. A full rupture of the uterus was found; the baby’s body had extruded into the mother’s abdomen. The child was stillborn.
PARENTS’ CLAIM The stillbirth could have been avoided if the nurses had communicated the mother’s worsening condition to the physicians.
DEFENDANTS’ DEFENSE After the hospital and physicians settled prior to trial, the case continued against the nurse in charge of the mother’s care and the nurse-staffing group. Negligence was denied; all protocols were followed.
VERDICT A $2.9 million Illinois verdict was returned. With a $900,000 settlement from the hospital and physicians, the net award was $3.8 million.
_______________
Where did rare strep A infection come from?
A 36-year-old woman reported heavy vaginal bleeding to her ObGyn. She underwent endometrial ablation in her physician’s office.
The next day, the woman called the office to report abdominal pain. She was told to stop the medication she was taking, and if the pain continued to the next day, to go to an ED. The next day, the patient went to the ED and was found to be in septic shock. During emergency laparotomy, 50 mL of purulent fluid were drained and an emergency hysterectomy was performed. Three days later, the patient died from pulmonary arrest caused by toxic shock syndrome. An autopsy revealed that the patient’s sepsis was caused by group A streptococci (GAS) infection.
ESTATE’S CLAIM The patient was not a proper candidate for endometrial ablation because of her history of chronic cervical infection. The ObGyn perforated the cervix during the procedure and tried to conceal it. At autopsy, bone wax was found in the rectal lumen that had been used to cover up damage to the cervix. The ObGyn introduced GAS bacteria into the patient’s system. The ObGyn’s staff failed to ask the proper questions when she called the day after the procedure. She should have been told to go directly to the ED.
DEFENDANTS’ DEFENSE The ObGyn did not perforate the cervix or uterus during the procedure. GAS infection is so rare that it would have been difficult to foresee or diagnose. Potentially, the patient had a chronic
cervical infection before ablation.
VERDICT A Texas defense verdict was returned.
_______________
DURING INSERTION, IUD PERFORATES UTERINE WALL; LATER FOUND BELOW LIVER
On July 21, a 46-year-old woman went to an ObGyn for placement of an intrauterine device (IUD). Shortly after the ObGyn inserted the levonorgestrel-releasing intrauterine system (Mirena, Bayer HealthCare), the patient reported severe pelvic and abdominal pain. On July 26, the patient underwent surgical removal of the IUD.
She was discharged on July 29 but continued to report pain. She was readmitted to the hospital the next day and treated for pain. She was bed ridden for 3 weeks after IUD-removal surgery, and had a 3-month recovery before feeling pain free.
PATIENT’S CLAIM The ObGyn was negligent in perforating the patient’s uterine wall during IUD insertion, causing the device to ultimately migrate under the patient’s liver.
DEFENDANTS’ DEFENSE Uterine perforation is a known complication of IUD insertion. The IUD escaped from the patient’s uterus at a later time and not during the insertion procedure.
VERDICT A Florida verdict of $208,839 was returned; the amount was reduced to $161,058 because the medical expenses were written off by the health-care providers.
_______________
Was travel appropriate for this pregnant woman?
A woman with a history of two premature deliveries and one miscarriage became pregnant again. She received prenatal care at an Army hospital. She traveled to Spain, where the baby was born at 31 weeks’ gestation. The baby required treatment in a neonatal intensive care unit (NICU) for 17 days. The child has cerebral palsy, with tetraplegia of all four extremities. She cannot walk without assistance and suffers severe cognitive and vision impairment.
PARENTS’ CLAIM The ObGyn at the Army hospital should not have approved the mother’s request for travel; he did so, despite knowing that the mother was at high risk for premature birth. The military medical hospital to which she was assigned in Spain could not manage a high-risk pregnancy, didn’t have a NICU, and didn’t have specialists to treat premature infants.
DEFENDANTS’ DEFENSE The ObGyn argued that he did not have access to the medical records showing the mother’s history. The patient countered that the ObGyn did indeed have the patient’s records, as he had discussed them with her.
VERDICT A $10,409,700 California verdict was returned against the ObGyn and the government facility.
_______________
Triple-negative BrCa not diagnosed until metastasized: $5.2M
After finding lumps in both breasts, a woman in her 30s saw a nurse practitioner (NP) at an Army hospital. A radiologist reported no mass in the right breast and multiple benign-appearing anechoic lesions in the left breast after bilateral mammography and ultrasonography (US) in July 2008. The Chief of Mammography Services recommended referral to a breast surgeon, but the patient never received the letter. It was placed in her mammography file, not in the treatment file.
In November 2008, the patient returned to the clinic. Bilateral diagnostic mammography and US were ordered, but for unknown reasons, cancelled. US of the left breast was interpreted as benign in January 2009.
After imaging in March 2010, followed by a needle biopsy of the right breast, a radiologist reported finding intermediate-grade infiltrating ductal carcinoma.
The patient sought care outside the military medical system at a large university hospital. In April 2010, stage 3 triple-negative invasive ductal carcinoma (IDC) was identified. The patient underwent chemotherapy, a double mastectomy, removal of 21 lymph nodes, and breast reconstruction. She was given a 60% chance of recurrence in 5–7 years.
PATIENT’S CLAIM It was negligent to not inform her of imaging results. Biopsy should have been performed in 2008, when the IDC was likely at stage 1; treatment would have been far less aggressive. Electronic medical records showed that the 2008 mammography and US results had been “signed off” by an NP at the clinic.
DEFENDANTS’ DEFENSE While unable to concede liability, the government agency did not contest the point.
VERDICT A $5.2 million Tennessee federal court bench verdict was returned, citing failures in communication, poor and improper record keeping and retention, failure to follow-up, and an unexplained cancellation of a medical order.
_______________
Woman dies from cervical cancer: $2.3M
In 2001, a 41-year-old woman had abnormal Pap smear results but her gynecologist did not order more testing. The patient was told to return in 3 months, but she did not return until 2007—reporting abnormal bleeding, vaginal discharge, and pain. Her Pap results were normal, however, and the gynecologist did not order further testing. In 2009, the patient was found to have advanced cervical cancer. She died 2 years later.
ESTATE’S CLAIM Further testing should have been ordered in 2001, which would have likely revealed dysplasia, which can lead to cancer. The laboratory incorrectly interpreted the 2007 Pap test; if the results had been properly reported, additional testing could have been ordered.
DEFENDANTS’ DEFENSE The laboratory and patient’s estate settled for a confidential amount before trial. The gynecologist denied negligence.
VERDICT A New Jersey jury found the gynecologist 40% at fault for his actions in 2007. The jury found the laboratory 50% at fault, and the patient 10% at fault. A gross verdict of $2.33 million was returned.
_______________
Bowel injury after cesarean delivery; mother dies of sepsis
At 40 4/7 weeks' gestation, a 37-year-old woman gave birth to a healthy child by cesarean delivery. The next day, the patient had an elevated white blood cell (WBC) count with a left shift, her abdomen was tympanic but soft, and she was passing flatus and belching. The ObGyn ordered a Fleet enema; only flatus was released. A covering ObGyn ordered an abdominal radiograph, which the radiologist reported as showing postoperative ileus and mild constipation. The patient was given a second Fleet enema the next day, resulting in watery stool. She vomited 300 mL of dark green fluid.
After a rectal tube was placed 2 days later, one hard brown stool and several brown, pasty, loose, and liquid stools were returned. She vomited several times that day, and was found to have hypoactive bowel sounds with continued tympanic quality in the upper quadrants. Laboratory testing revealed continued elevated WBC count with left shift. The next day, she had hypoactive bowel sounds with brown liquid stools. Later that morning, she was able to tolerate clear liquids. The ObGyn decided to discharge her home with instructions to continue on a clear liquid diet for 2 more days before advancing her diet.
The day after discharge, she was found unresponsive at home. She was taken to the hospital, but resuscitation attempts failed. She died. An autopsy revealed that the cause of death was sepsis.
ESTATE’S CLAIM The ObGyn was negligent in failing to diagnose and treat a postoperative intra-abdominal infection caused by bowel perforation. A surgical consult should have been obtained. The woman was prematurely discharged. The radiologist failed to report the presence of free air on the abdominal x-ray.
DEFENDANTS’ DEFENSE The case was settled during trial.
VERDICT A $1 million Maryland settlement was reached.
_______________
Right ureter injury detected and repaired
During laparoscopic-assisted vaginal hysterectomy, the ObGyn detected and repaired an injury to the right ureter. The patient’s recovery was delayed by the injury.
PATIENT’S CLAIM The ObGyn was negligent in using a Kleppinger bipolar cauterizing instrument to cauterize the vaginal cuff. Thermal overspray from the instrument or the instrument itself damaged the ureter. The ObGyn was also negligent in not performing diagnostic cystoscopy to confirm patency of the ureter after the repair was made.
PHYSICIAN’S DEFENSE Ureter injury is a known risk of the procedure. All procedures were performed according to protocol.
VERDICT A Florida defense verdict was returned.
_______________
Failure to detect inflammatory BrCa; woman dies
A 42-year-old woman underwent mammography in February 2002 after reporting pain, discoloration, inflammation, and swelling in her left breast. The radiologist who interpreted the mammography suggested a biopsy for a differential diagnosis of mastitis or inflammatory carcinoma. The biopsy results were negative.
The patient’s symptoms persisted, and she underwent US in late May 2002. Another radiologist interpreted the US, noting that the patient could not tolerate compression, which led to less than optimal evaluation. The radiologist suggested that mastitis was the likely cause of the patient’s symptoms.
The patient then consulted a surgeon, who ordered mammography and magnetic resonance imaging (MRI) followed by biopsy, which indicated cancer. The patient underwent a mastectomy but metastasis had already occurred. She died at age 50 prior to the trial.
ESTATE’S CLAIM If the cancer had been diagnosed earlier, the outcome would have been better. Both radiologists misinterpreted the mammographies.
DEFENDANTS’ DEFENSE The mammographies had been properly interpreted. Any missed diagnosis would not have impacted the outcome due to the type of cancer. The scans had been released to the patient, but were subsequently lost; an adverse interference instruction was given to the jury.
VERDICT A New York defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Uterine rupture, child stillborn: $3.8M net award
At 35 weeks' gestation, a woman went to the emergency department (ED) with abdominal pain, fast heartbeat, and irregular contractions. Her history included three cesarean deliveries, including one with a vertical incision. She was admitted, and a cesarean delivery was planned for the next day. After 8 hours, during which the patient’s condition worsened, an emergency cesarean delivery was undertaken. A full rupture of the uterus was found; the baby’s body had extruded into the mother’s abdomen. The child was stillborn.
PARENTS’ CLAIM The stillbirth could have been avoided if the nurses had communicated the mother’s worsening condition to the physicians.
DEFENDANTS’ DEFENSE After the hospital and physicians settled prior to trial, the case continued against the nurse in charge of the mother’s care and the nurse-staffing group. Negligence was denied; all protocols were followed.
VERDICT A $2.9 million Illinois verdict was returned. With a $900,000 settlement from the hospital and physicians, the net award was $3.8 million.
_______________
Where did rare strep A infection come from?
A 36-year-old woman reported heavy vaginal bleeding to her ObGyn. She underwent endometrial ablation in her physician’s office.
The next day, the woman called the office to report abdominal pain. She was told to stop the medication she was taking, and if the pain continued to the next day, to go to an ED. The next day, the patient went to the ED and was found to be in septic shock. During emergency laparotomy, 50 mL of purulent fluid were drained and an emergency hysterectomy was performed. Three days later, the patient died from pulmonary arrest caused by toxic shock syndrome. An autopsy revealed that the patient’s sepsis was caused by group A streptococci (GAS) infection.
ESTATE’S CLAIM The patient was not a proper candidate for endometrial ablation because of her history of chronic cervical infection. The ObGyn perforated the cervix during the procedure and tried to conceal it. At autopsy, bone wax was found in the rectal lumen that had been used to cover up damage to the cervix. The ObGyn introduced GAS bacteria into the patient’s system. The ObGyn’s staff failed to ask the proper questions when she called the day after the procedure. She should have been told to go directly to the ED.
DEFENDANTS’ DEFENSE The ObGyn did not perforate the cervix or uterus during the procedure. GAS infection is so rare that it would have been difficult to foresee or diagnose. Potentially, the patient had a chronic
cervical infection before ablation.
VERDICT A Texas defense verdict was returned.
_______________
DURING INSERTION, IUD PERFORATES UTERINE WALL; LATER FOUND BELOW LIVER
On July 21, a 46-year-old woman went to an ObGyn for placement of an intrauterine device (IUD). Shortly after the ObGyn inserted the levonorgestrel-releasing intrauterine system (Mirena, Bayer HealthCare), the patient reported severe pelvic and abdominal pain. On July 26, the patient underwent surgical removal of the IUD.
She was discharged on July 29 but continued to report pain. She was readmitted to the hospital the next day and treated for pain. She was bed ridden for 3 weeks after IUD-removal surgery, and had a 3-month recovery before feeling pain free.
PATIENT’S CLAIM The ObGyn was negligent in perforating the patient’s uterine wall during IUD insertion, causing the device to ultimately migrate under the patient’s liver.
DEFENDANTS’ DEFENSE Uterine perforation is a known complication of IUD insertion. The IUD escaped from the patient’s uterus at a later time and not during the insertion procedure.
VERDICT A Florida verdict of $208,839 was returned; the amount was reduced to $161,058 because the medical expenses were written off by the health-care providers.
_______________
Was travel appropriate for this pregnant woman?
A woman with a history of two premature deliveries and one miscarriage became pregnant again. She received prenatal care at an Army hospital. She traveled to Spain, where the baby was born at 31 weeks’ gestation. The baby required treatment in a neonatal intensive care unit (NICU) for 17 days. The child has cerebral palsy, with tetraplegia of all four extremities. She cannot walk without assistance and suffers severe cognitive and vision impairment.
PARENTS’ CLAIM The ObGyn at the Army hospital should not have approved the mother’s request for travel; he did so, despite knowing that the mother was at high risk for premature birth. The military medical hospital to which she was assigned in Spain could not manage a high-risk pregnancy, didn’t have a NICU, and didn’t have specialists to treat premature infants.
DEFENDANTS’ DEFENSE The ObGyn argued that he did not have access to the medical records showing the mother’s history. The patient countered that the ObGyn did indeed have the patient’s records, as he had discussed them with her.
VERDICT A $10,409,700 California verdict was returned against the ObGyn and the government facility.
_______________
Triple-negative BrCa not diagnosed until metastasized: $5.2M
After finding lumps in both breasts, a woman in her 30s saw a nurse practitioner (NP) at an Army hospital. A radiologist reported no mass in the right breast and multiple benign-appearing anechoic lesions in the left breast after bilateral mammography and ultrasonography (US) in July 2008. The Chief of Mammography Services recommended referral to a breast surgeon, but the patient never received the letter. It was placed in her mammography file, not in the treatment file.
In November 2008, the patient returned to the clinic. Bilateral diagnostic mammography and US were ordered, but for unknown reasons, cancelled. US of the left breast was interpreted as benign in January 2009.
After imaging in March 2010, followed by a needle biopsy of the right breast, a radiologist reported finding intermediate-grade infiltrating ductal carcinoma.
The patient sought care outside the military medical system at a large university hospital. In April 2010, stage 3 triple-negative invasive ductal carcinoma (IDC) was identified. The patient underwent chemotherapy, a double mastectomy, removal of 21 lymph nodes, and breast reconstruction. She was given a 60% chance of recurrence in 5–7 years.
PATIENT’S CLAIM It was negligent to not inform her of imaging results. Biopsy should have been performed in 2008, when the IDC was likely at stage 1; treatment would have been far less aggressive. Electronic medical records showed that the 2008 mammography and US results had been “signed off” by an NP at the clinic.
DEFENDANTS’ DEFENSE While unable to concede liability, the government agency did not contest the point.
VERDICT A $5.2 million Tennessee federal court bench verdict was returned, citing failures in communication, poor and improper record keeping and retention, failure to follow-up, and an unexplained cancellation of a medical order.
_______________
Woman dies from cervical cancer: $2.3M
In 2001, a 41-year-old woman had abnormal Pap smear results but her gynecologist did not order more testing. The patient was told to return in 3 months, but she did not return until 2007—reporting abnormal bleeding, vaginal discharge, and pain. Her Pap results were normal, however, and the gynecologist did not order further testing. In 2009, the patient was found to have advanced cervical cancer. She died 2 years later.
ESTATE’S CLAIM Further testing should have been ordered in 2001, which would have likely revealed dysplasia, which can lead to cancer. The laboratory incorrectly interpreted the 2007 Pap test; if the results had been properly reported, additional testing could have been ordered.
DEFENDANTS’ DEFENSE The laboratory and patient’s estate settled for a confidential amount before trial. The gynecologist denied negligence.
VERDICT A New Jersey jury found the gynecologist 40% at fault for his actions in 2007. The jury found the laboratory 50% at fault, and the patient 10% at fault. A gross verdict of $2.33 million was returned.
_______________
Bowel injury after cesarean delivery; mother dies of sepsis
At 40 4/7 weeks' gestation, a 37-year-old woman gave birth to a healthy child by cesarean delivery. The next day, the patient had an elevated white blood cell (WBC) count with a left shift, her abdomen was tympanic but soft, and she was passing flatus and belching. The ObGyn ordered a Fleet enema; only flatus was released. A covering ObGyn ordered an abdominal radiograph, which the radiologist reported as showing postoperative ileus and mild constipation. The patient was given a second Fleet enema the next day, resulting in watery stool. She vomited 300 mL of dark green fluid.
After a rectal tube was placed 2 days later, one hard brown stool and several brown, pasty, loose, and liquid stools were returned. She vomited several times that day, and was found to have hypoactive bowel sounds with continued tympanic quality in the upper quadrants. Laboratory testing revealed continued elevated WBC count with left shift. The next day, she had hypoactive bowel sounds with brown liquid stools. Later that morning, she was able to tolerate clear liquids. The ObGyn decided to discharge her home with instructions to continue on a clear liquid diet for 2 more days before advancing her diet.
The day after discharge, she was found unresponsive at home. She was taken to the hospital, but resuscitation attempts failed. She died. An autopsy revealed that the cause of death was sepsis.
ESTATE’S CLAIM The ObGyn was negligent in failing to diagnose and treat a postoperative intra-abdominal infection caused by bowel perforation. A surgical consult should have been obtained. The woman was prematurely discharged. The radiologist failed to report the presence of free air on the abdominal x-ray.
DEFENDANTS’ DEFENSE The case was settled during trial.
VERDICT A $1 million Maryland settlement was reached.
_______________
Right ureter injury detected and repaired
During laparoscopic-assisted vaginal hysterectomy, the ObGyn detected and repaired an injury to the right ureter. The patient’s recovery was delayed by the injury.
PATIENT’S CLAIM The ObGyn was negligent in using a Kleppinger bipolar cauterizing instrument to cauterize the vaginal cuff. Thermal overspray from the instrument or the instrument itself damaged the ureter. The ObGyn was also negligent in not performing diagnostic cystoscopy to confirm patency of the ureter after the repair was made.
PHYSICIAN’S DEFENSE Ureter injury is a known risk of the procedure. All procedures were performed according to protocol.
VERDICT A Florida defense verdict was returned.
_______________
Failure to detect inflammatory BrCa; woman dies
A 42-year-old woman underwent mammography in February 2002 after reporting pain, discoloration, inflammation, and swelling in her left breast. The radiologist who interpreted the mammography suggested a biopsy for a differential diagnosis of mastitis or inflammatory carcinoma. The biopsy results were negative.
The patient’s symptoms persisted, and she underwent US in late May 2002. Another radiologist interpreted the US, noting that the patient could not tolerate compression, which led to less than optimal evaluation. The radiologist suggested that mastitis was the likely cause of the patient’s symptoms.
The patient then consulted a surgeon, who ordered mammography and magnetic resonance imaging (MRI) followed by biopsy, which indicated cancer. The patient underwent a mastectomy but metastasis had already occurred. She died at age 50 prior to the trial.
ESTATE’S CLAIM If the cancer had been diagnosed earlier, the outcome would have been better. Both radiologists misinterpreted the mammographies.
DEFENDANTS’ DEFENSE The mammographies had been properly interpreted. Any missed diagnosis would not have impacted the outcome due to the type of cancer. The scans had been released to the patient, but were subsequently lost; an adverse interference instruction was given to the jury.
VERDICT A New York defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
More inclusions:
- Where did rare strep A infection come from?
- During insertion, IUD perforates uterine wall; Later found below liver
- Was travel appropriate for this pregnant woman?
- Triple-negative BrCa not diagnosed until metastasized: $5.2M
- Woman dies from cervical cancer: $2.3M
- Bowel injury after cesarean delivery; mother dies of sepsis
- Right ureter injury detected and repaired
- Failure to detect inflammatory BrCa; woman dies
What treatments relieve arthritis and fatigue associated with systemic lupus erythematosus?
EVIDENCE-BASED ANSWER:
Hydroxychloroquine and chloroquine improve the arthritis associated with mild systemic lupus erythematosus (SLE)—producing a 50% reduction in arthritis flares and articular involvement—and have few adverse effects (strength of recommendation [SOR]: A, systematic review of randomized controlled trials [RCTs]).
Methotrexate reduces arthralgias by as much as 79%, but produces adverse effects in up to 70% of patients (SOR: B, systematic review of RCTs with limited patient-oriented evidence).
Nonsteroidal anti-inflammatory drugs (NSAIDs) and corticosteroids are often used for SLE joint pain (SOR: C, expert opinion).
Omega-3 fatty acids may reduce arthritis symptoms by about 35% (SOR: B, RCTs with inconsistent evidence).
Abatacept and dehydroepiandrosterone don’t produce clinically meaningful improvements in fatigue associated with SLE, and abatacept causes significant adverse effects (SOR: B, posthoc analysis of a single RCT).
Aerobic exercise may help fatigue (SOR: B, systematic review with inconsistent evidence).
EVIDENCE SUMMARY
A systematic review of pharmacotherapy for joint pain in patients with SLE found 4 poor-quality RCTs that evaluated hydroxychloroquine, chloroquine, and methotrexate.1 Of the 2 studies that examined the effect of hydroxychloroquine, one (47 patients) showed a statistically significant 50% reduction in SLE flares (including arthritis, pleuritis, and cutaneous symptoms) over 24 weeks in patients treated with hydroxychloroquine compared with placebo (TABLE1-8). The second study (71 subjects) found a nonquantified decrease in self-reported pain when hydroxychloroquine was compared with placebo, although some of the patients were also taking prednisone (10 mg/d).
An RCT that evaluated the effect of chloroquine showed a statistically significant reduction in unspecified “articular involvement” compared with placebo.
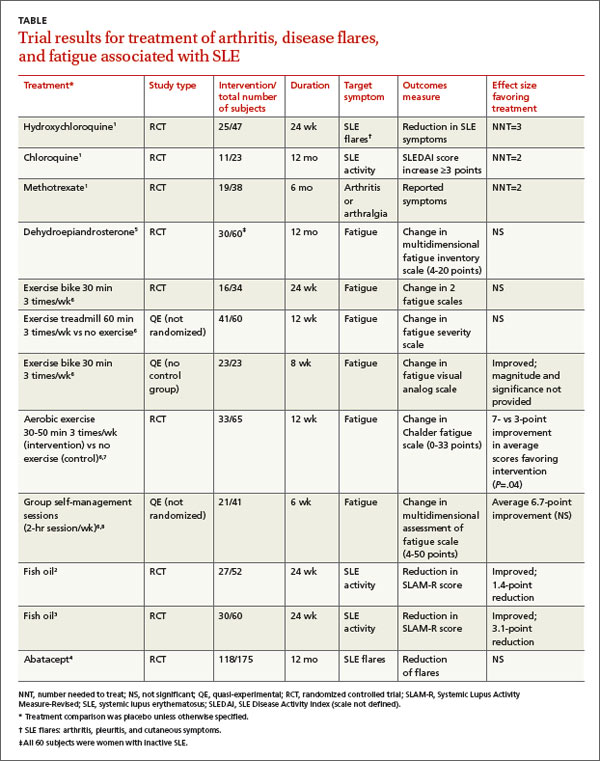
The fourth RCT, assessing methotrexate, found a statistically significant reduction by as much as 79% in patients with residual arthritis or arthralgia at 6 months compared with placebo, although 70% of patients taking methotrexate developed significant adverse effects, including infections, gastrointestinal symptoms, and elevated transaminases compared with 14% on placebo (number needed to harm [NNH]=2).
The authors of the review noted that consensus opinion holds that oral corticosteroids and NSAIDs reduce SLE-associated joint pain, but they found no studies that objectively evaluated either of these interventions.1
Fish oil also helps arthritis
Two RCTs on the effects of 3 g/d of omega-3 polyunsaturated fatty acids (fish oil) for 24 weeks in SLE patients with mild disease found a reduction in Systemic Lupus Activity Measure-Revised (SLAM-R) scores.2,3 SLAM-R is a validated measure of SLE disease activity, rated on a scale from 0 to 81, including 23 clinical and 7 laboratory manifestations of disease.
In the first study (52 subjects), disease activity decreased from an average SLAM-R score of 6.1 at baseline to 4.7 (P<.05). The second study (60 subjects) found a similar reduction in mean SLAM-R scores from 9.4 to 6.3 (P<.001) and joint pain scores from 1.27 to 0.83 (P=.047).
Drug treatments don’t significantly relieve fatigue
An industry-sponsored RCT that compared abatacept with placebo found improvements in fatigue that weren’t clinically meaningful in posthoc analysis (-9.45 points difference on a self-reported 0-to-100 visual analog scale; 95% confidence interval, -17.65 to -1.25, with a 10-point reduction considered to be clinically meaningful). Abatacept also had a high rate of serious adverse events, including facial edema, polyneuropathy, and serious infections (24/121 with abatacept vs 4/59 placebo; NNH=8).4
Another RCT found no effect of dehydroepiandrosterone on fatigue in women with inactive SLE.5
Nondrug treatments for fatigue produce mixed results
Studies of nondrug treatment of SLE-associated fatigue show inconsistent results. A systematic review of nonpharmacologic interventions for fatigue in several chronic diseases found 2 RCTs and 4 quasi-experimental studies that included 324 patients with SLE.6 Of 4 studies that evaluated the effect of exercise, 2 showed improvement and 2 didn’t. Neither group self-management nor relaxation therapy and telephone counseling significantly relieved fatigue.6-8 A small RCT (24 patients) found no benefit for acupuncture over sham needling in treating pain and fatigue in SLE.9
RECOMMENDATIONS
The American College of Rheumatology guideline for referral and management of SLE states that “NSAIDs are sometimes helpful for control of fever, arthritis, and mild serositis. Antimalarial agents (eg, hydroxychloroquine) are useful for skin and joint manifestations of SLE, for preventing flares, and for other constitutional symptoms of the disease. They may also reduce fatigue.”10
The European League Against Rheumatism recommends antimalarials or glucocorticoids to treat patients with SLE without major organ manifestations. They also say clinicians may try NSAIDs for limited periods of time in patients at low risk for the drugs’ complications.11
1. Madhok R, Wu O. Systemic lupus erythematosus. Clin Evid. 2009;7:1123.
2. Duffy EM, Meenagh GK, McMillan SA, et al. The clinical effect of dietary supplementation with omega-3 fish oils and/ or copper in systemic lupus erythematosus. J Rheumatol. 2004;31:1551-1556.
3. Wright SA, O’Prey FM, McHenry MT, et al. A randomised interventional trial of omega-3-polyunsaturated fatty acids on endothelial function and disease activity in systemic lupus erythematosus. Ann Rheum Dis. 2008;67:841-848.
4. Merrill JT, Burgos-Vargas R, Westhovens R, et al. The efficacy and safety of abatacept in patients with non-life-threatening manifestations of systemic lupus erythematosus: results of a twelve-month, multicenter, exploratory, phase IIb, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2010;62:3077-3087.
5. Hartkamp A, Geenen R, Godaert GL, et al. Effects of dehydroepiandrosterone on fatigue and well-being in women with quiescent systemic lupus erythematosus: a randomized controlled trial. Ann Rheum Dis. 2010;69:1144-1147.
6. Neill J, Belan I, Reid K. Effectiveness of non-pharmacological interventions for fatigue in adults with multiple sclerosis, rheumatoid arthritis, or systemic lupus erythematosis: a systematic review. J Adv Nurs. 2006;56:617-635.
7. Tench CM, McCarthy J, McCurdie I, et al. Fatigue in systemic lupus erythematosus: a randomized controlled trial of exercise. Rheumatology (Oxford). 2003;42:1050-1054.
8. Sohng KY. Effects of a self-management course for patients with systemic lupus erythematosus. J Adv Nurs. 2003;42:479-486.
9. Greco CM, Kao AH, Maksimowicz-McKinnon K, et al. Acupuncture for systemic lupus erythematosus: a pilot RCT feasibility and safety study. Lupus. 2008;17:1108-1116.
10. American College of Rheumatology Ad Hoc Committee on Systemic Lupus Erythematosus Guidelines. Guidelines for referral and management of systemic lupus erythematosus in adults. Arthritis Rheum. 1999;42:1785-1796.
11. Bertsias G, Ioannidis JP, Boletis J, et al; Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR recommendations for the management of systemic lupus erythematosus. Report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics. Ann Rheum Dis. 2008;67:195-205.
EVIDENCE-BASED ANSWER:
Hydroxychloroquine and chloroquine improve the arthritis associated with mild systemic lupus erythematosus (SLE)—producing a 50% reduction in arthritis flares and articular involvement—and have few adverse effects (strength of recommendation [SOR]: A, systematic review of randomized controlled trials [RCTs]).
Methotrexate reduces arthralgias by as much as 79%, but produces adverse effects in up to 70% of patients (SOR: B, systematic review of RCTs with limited patient-oriented evidence).
Nonsteroidal anti-inflammatory drugs (NSAIDs) and corticosteroids are often used for SLE joint pain (SOR: C, expert opinion).
Omega-3 fatty acids may reduce arthritis symptoms by about 35% (SOR: B, RCTs with inconsistent evidence).
Abatacept and dehydroepiandrosterone don’t produce clinically meaningful improvements in fatigue associated with SLE, and abatacept causes significant adverse effects (SOR: B, posthoc analysis of a single RCT).
Aerobic exercise may help fatigue (SOR: B, systematic review with inconsistent evidence).
EVIDENCE SUMMARY
A systematic review of pharmacotherapy for joint pain in patients with SLE found 4 poor-quality RCTs that evaluated hydroxychloroquine, chloroquine, and methotrexate.1 Of the 2 studies that examined the effect of hydroxychloroquine, one (47 patients) showed a statistically significant 50% reduction in SLE flares (including arthritis, pleuritis, and cutaneous symptoms) over 24 weeks in patients treated with hydroxychloroquine compared with placebo (TABLE1-8). The second study (71 subjects) found a nonquantified decrease in self-reported pain when hydroxychloroquine was compared with placebo, although some of the patients were also taking prednisone (10 mg/d).
An RCT that evaluated the effect of chloroquine showed a statistically significant reduction in unspecified “articular involvement” compared with placebo.
The fourth RCT, assessing methotrexate, found a statistically significant reduction by as much as 79% in patients with residual arthritis or arthralgia at 6 months compared with placebo, although 70% of patients taking methotrexate developed significant adverse effects, including infections, gastrointestinal symptoms, and elevated transaminases compared with 14% on placebo (number needed to harm [NNH]=2).
The authors of the review noted that consensus opinion holds that oral corticosteroids and NSAIDs reduce SLE-associated joint pain, but they found no studies that objectively evaluated either of these interventions.1
Fish oil also helps arthritis
Two RCTs on the effects of 3 g/d of omega-3 polyunsaturated fatty acids (fish oil) for 24 weeks in SLE patients with mild disease found a reduction in Systemic Lupus Activity Measure-Revised (SLAM-R) scores.2,3 SLAM-R is a validated measure of SLE disease activity, rated on a scale from 0 to 81, including 23 clinical and 7 laboratory manifestations of disease.
In the first study (52 subjects), disease activity decreased from an average SLAM-R score of 6.1 at baseline to 4.7 (P<.05). The second study (60 subjects) found a similar reduction in mean SLAM-R scores from 9.4 to 6.3 (P<.001) and joint pain scores from 1.27 to 0.83 (P=.047).
Drug treatments don’t significantly relieve fatigue
An industry-sponsored RCT that compared abatacept with placebo found improvements in fatigue that weren’t clinically meaningful in posthoc analysis (-9.45 points difference on a self-reported 0-to-100 visual analog scale; 95% confidence interval, -17.65 to -1.25, with a 10-point reduction considered to be clinically meaningful). Abatacept also had a high rate of serious adverse events, including facial edema, polyneuropathy, and serious infections (24/121 with abatacept vs 4/59 placebo; NNH=8).4
Another RCT found no effect of dehydroepiandrosterone on fatigue in women with inactive SLE.5
Nondrug treatments for fatigue produce mixed results
Studies of nondrug treatment of SLE-associated fatigue show inconsistent results. A systematic review of nonpharmacologic interventions for fatigue in several chronic diseases found 2 RCTs and 4 quasi-experimental studies that included 324 patients with SLE.6 Of 4 studies that evaluated the effect of exercise, 2 showed improvement and 2 didn’t. Neither group self-management nor relaxation therapy and telephone counseling significantly relieved fatigue.6-8 A small RCT (24 patients) found no benefit for acupuncture over sham needling in treating pain and fatigue in SLE.9
RECOMMENDATIONS
The American College of Rheumatology guideline for referral and management of SLE states that “NSAIDs are sometimes helpful for control of fever, arthritis, and mild serositis. Antimalarial agents (eg, hydroxychloroquine) are useful for skin and joint manifestations of SLE, for preventing flares, and for other constitutional symptoms of the disease. They may also reduce fatigue.”10
The European League Against Rheumatism recommends antimalarials or glucocorticoids to treat patients with SLE without major organ manifestations. They also say clinicians may try NSAIDs for limited periods of time in patients at low risk for the drugs’ complications.11
EVIDENCE-BASED ANSWER:
Hydroxychloroquine and chloroquine improve the arthritis associated with mild systemic lupus erythematosus (SLE)—producing a 50% reduction in arthritis flares and articular involvement—and have few adverse effects (strength of recommendation [SOR]: A, systematic review of randomized controlled trials [RCTs]).
Methotrexate reduces arthralgias by as much as 79%, but produces adverse effects in up to 70% of patients (SOR: B, systematic review of RCTs with limited patient-oriented evidence).
Nonsteroidal anti-inflammatory drugs (NSAIDs) and corticosteroids are often used for SLE joint pain (SOR: C, expert opinion).
Omega-3 fatty acids may reduce arthritis symptoms by about 35% (SOR: B, RCTs with inconsistent evidence).
Abatacept and dehydroepiandrosterone don’t produce clinically meaningful improvements in fatigue associated with SLE, and abatacept causes significant adverse effects (SOR: B, posthoc analysis of a single RCT).
Aerobic exercise may help fatigue (SOR: B, systematic review with inconsistent evidence).
EVIDENCE SUMMARY
A systematic review of pharmacotherapy for joint pain in patients with SLE found 4 poor-quality RCTs that evaluated hydroxychloroquine, chloroquine, and methotrexate.1 Of the 2 studies that examined the effect of hydroxychloroquine, one (47 patients) showed a statistically significant 50% reduction in SLE flares (including arthritis, pleuritis, and cutaneous symptoms) over 24 weeks in patients treated with hydroxychloroquine compared with placebo (TABLE1-8). The second study (71 subjects) found a nonquantified decrease in self-reported pain when hydroxychloroquine was compared with placebo, although some of the patients were also taking prednisone (10 mg/d).
An RCT that evaluated the effect of chloroquine showed a statistically significant reduction in unspecified “articular involvement” compared with placebo.
The fourth RCT, assessing methotrexate, found a statistically significant reduction by as much as 79% in patients with residual arthritis or arthralgia at 6 months compared with placebo, although 70% of patients taking methotrexate developed significant adverse effects, including infections, gastrointestinal symptoms, and elevated transaminases compared with 14% on placebo (number needed to harm [NNH]=2).
The authors of the review noted that consensus opinion holds that oral corticosteroids and NSAIDs reduce SLE-associated joint pain, but they found no studies that objectively evaluated either of these interventions.1
Fish oil also helps arthritis
Two RCTs on the effects of 3 g/d of omega-3 polyunsaturated fatty acids (fish oil) for 24 weeks in SLE patients with mild disease found a reduction in Systemic Lupus Activity Measure-Revised (SLAM-R) scores.2,3 SLAM-R is a validated measure of SLE disease activity, rated on a scale from 0 to 81, including 23 clinical and 7 laboratory manifestations of disease.
In the first study (52 subjects), disease activity decreased from an average SLAM-R score of 6.1 at baseline to 4.7 (P<.05). The second study (60 subjects) found a similar reduction in mean SLAM-R scores from 9.4 to 6.3 (P<.001) and joint pain scores from 1.27 to 0.83 (P=.047).
Drug treatments don’t significantly relieve fatigue
An industry-sponsored RCT that compared abatacept with placebo found improvements in fatigue that weren’t clinically meaningful in posthoc analysis (-9.45 points difference on a self-reported 0-to-100 visual analog scale; 95% confidence interval, -17.65 to -1.25, with a 10-point reduction considered to be clinically meaningful). Abatacept also had a high rate of serious adverse events, including facial edema, polyneuropathy, and serious infections (24/121 with abatacept vs 4/59 placebo; NNH=8).4
Another RCT found no effect of dehydroepiandrosterone on fatigue in women with inactive SLE.5
Nondrug treatments for fatigue produce mixed results
Studies of nondrug treatment of SLE-associated fatigue show inconsistent results. A systematic review of nonpharmacologic interventions for fatigue in several chronic diseases found 2 RCTs and 4 quasi-experimental studies that included 324 patients with SLE.6 Of 4 studies that evaluated the effect of exercise, 2 showed improvement and 2 didn’t. Neither group self-management nor relaxation therapy and telephone counseling significantly relieved fatigue.6-8 A small RCT (24 patients) found no benefit for acupuncture over sham needling in treating pain and fatigue in SLE.9
RECOMMENDATIONS
The American College of Rheumatology guideline for referral and management of SLE states that “NSAIDs are sometimes helpful for control of fever, arthritis, and mild serositis. Antimalarial agents (eg, hydroxychloroquine) are useful for skin and joint manifestations of SLE, for preventing flares, and for other constitutional symptoms of the disease. They may also reduce fatigue.”10
The European League Against Rheumatism recommends antimalarials or glucocorticoids to treat patients with SLE without major organ manifestations. They also say clinicians may try NSAIDs for limited periods of time in patients at low risk for the drugs’ complications.11
1. Madhok R, Wu O. Systemic lupus erythematosus. Clin Evid. 2009;7:1123.
2. Duffy EM, Meenagh GK, McMillan SA, et al. The clinical effect of dietary supplementation with omega-3 fish oils and/ or copper in systemic lupus erythematosus. J Rheumatol. 2004;31:1551-1556.
3. Wright SA, O’Prey FM, McHenry MT, et al. A randomised interventional trial of omega-3-polyunsaturated fatty acids on endothelial function and disease activity in systemic lupus erythematosus. Ann Rheum Dis. 2008;67:841-848.
4. Merrill JT, Burgos-Vargas R, Westhovens R, et al. The efficacy and safety of abatacept in patients with non-life-threatening manifestations of systemic lupus erythematosus: results of a twelve-month, multicenter, exploratory, phase IIb, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2010;62:3077-3087.
5. Hartkamp A, Geenen R, Godaert GL, et al. Effects of dehydroepiandrosterone on fatigue and well-being in women with quiescent systemic lupus erythematosus: a randomized controlled trial. Ann Rheum Dis. 2010;69:1144-1147.
6. Neill J, Belan I, Reid K. Effectiveness of non-pharmacological interventions for fatigue in adults with multiple sclerosis, rheumatoid arthritis, or systemic lupus erythematosis: a systematic review. J Adv Nurs. 2006;56:617-635.
7. Tench CM, McCarthy J, McCurdie I, et al. Fatigue in systemic lupus erythematosus: a randomized controlled trial of exercise. Rheumatology (Oxford). 2003;42:1050-1054.
8. Sohng KY. Effects of a self-management course for patients with systemic lupus erythematosus. J Adv Nurs. 2003;42:479-486.
9. Greco CM, Kao AH, Maksimowicz-McKinnon K, et al. Acupuncture for systemic lupus erythematosus: a pilot RCT feasibility and safety study. Lupus. 2008;17:1108-1116.
10. American College of Rheumatology Ad Hoc Committee on Systemic Lupus Erythematosus Guidelines. Guidelines for referral and management of systemic lupus erythematosus in adults. Arthritis Rheum. 1999;42:1785-1796.
11. Bertsias G, Ioannidis JP, Boletis J, et al; Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR recommendations for the management of systemic lupus erythematosus. Report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics. Ann Rheum Dis. 2008;67:195-205.
1. Madhok R, Wu O. Systemic lupus erythematosus. Clin Evid. 2009;7:1123.
2. Duffy EM, Meenagh GK, McMillan SA, et al. The clinical effect of dietary supplementation with omega-3 fish oils and/ or copper in systemic lupus erythematosus. J Rheumatol. 2004;31:1551-1556.
3. Wright SA, O’Prey FM, McHenry MT, et al. A randomised interventional trial of omega-3-polyunsaturated fatty acids on endothelial function and disease activity in systemic lupus erythematosus. Ann Rheum Dis. 2008;67:841-848.
4. Merrill JT, Burgos-Vargas R, Westhovens R, et al. The efficacy and safety of abatacept in patients with non-life-threatening manifestations of systemic lupus erythematosus: results of a twelve-month, multicenter, exploratory, phase IIb, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2010;62:3077-3087.
5. Hartkamp A, Geenen R, Godaert GL, et al. Effects of dehydroepiandrosterone on fatigue and well-being in women with quiescent systemic lupus erythematosus: a randomized controlled trial. Ann Rheum Dis. 2010;69:1144-1147.
6. Neill J, Belan I, Reid K. Effectiveness of non-pharmacological interventions for fatigue in adults with multiple sclerosis, rheumatoid arthritis, or systemic lupus erythematosis: a systematic review. J Adv Nurs. 2006;56:617-635.
7. Tench CM, McCarthy J, McCurdie I, et al. Fatigue in systemic lupus erythematosus: a randomized controlled trial of exercise. Rheumatology (Oxford). 2003;42:1050-1054.
8. Sohng KY. Effects of a self-management course for patients with systemic lupus erythematosus. J Adv Nurs. 2003;42:479-486.
9. Greco CM, Kao AH, Maksimowicz-McKinnon K, et al. Acupuncture for systemic lupus erythematosus: a pilot RCT feasibility and safety study. Lupus. 2008;17:1108-1116.
10. American College of Rheumatology Ad Hoc Committee on Systemic Lupus Erythematosus Guidelines. Guidelines for referral and management of systemic lupus erythematosus in adults. Arthritis Rheum. 1999;42:1785-1796.
11. Bertsias G, Ioannidis JP, Boletis J, et al; Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR recommendations for the management of systemic lupus erythematosus. Report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics. Ann Rheum Dis. 2008;67:195-205.
Evidence-based answers from the Family Physicians Inquiries Network
Lack of energy, petechiae, elevated PSA level—Dx?
THE CASE
A 57-year-old Hispanic man sought treatment because he had been feeling tired for a few weeks. He had not seen a physician for 15 years. When he came in, his temperature was 98.8°F, blood pressure was 132/82 mm Hg, pulse was 82 beats/min, respiration rate was 16 breaths/min, and oxygen saturation was 93% on room air. Examination of the head, neck, and respiratory and cardiovascular systems was normal. Skin examination showed petechiae and bruising on his abdomen, left ankle, right thigh, and bilateral shin area. His abdomen was nontender with no organomegaly. There was no focal neurological finding or spinal tenderness. Our patient had no chills, chest pains, shortness of breath, headache, dizziness, or loss of consciousness. There was no hematemesis, melena, hematuria, edema, or weight loss. He had no medical or surgical history and denied substance abuse or taking any medications recently; he did use alcohol previously.
Results of some initial lab tests were abnormal, including a decreased white blood cell count (5.82/mcL), platelet count (29 x 103/mcL), hemoglobin (8.6 g/dL), and hematocrit (27%) (TABLE). A peripheral blood smear showed decreased normocytic red blood cells and scattered schistocytes. His prostate-specific antigen (PSA) level was elevated at 212 ng/mL.
The patient’s coagulation profile was normal, and his von Willebrand factor (vWF) protease (ADAMTS-13) level was within normal limits (13.83). Antineutrophil cytoplasmic antibody and antinuclear antibody tests were negative. Testing for pulmonary embolism was negative, as was testing for human immunodeficiency virus. An abdominal ultrasound was normal, as well.
THE DIAGNOSIS
Based on our patient’s abnormal blood test results and the presence of petechiae and bruising, we diagnosed thrombotic thrombocytopenic purpura (TTP). The patient’s elevated PSA prompted us to order computed tomography of the chest and abdomen, which showed an enlarged prostate gland and mixed lytic sclerotic lesions in T3 to T5 and T9 vertebrae and in his ribs. A bone marrow biopsy revealed metastatic prostatic adenocarcinoma and a bone scan confirmed multiple metastases in the spine, pelvis, and shoulders.
DISCUSSION
TTP is a rare disorder of increased clotting in small blood vessels throughout the body that can include thrombocytopenia, microangiopathic hemolytic anemia (MAHA), fever, renal dysfunction, and neurological deficits.1 It’s important to maintain a high index of suspicion for TTP because the condition is a hematologic medical emergency that can quickly cause multiorgan failure and death.2
Almost always an acquired condition, TTP can be idiopathic or secondary to another condition, such as collagen vascular diseases, transplants, certain drugs, infections, pregnancy, or cancer.3 In idiopathic TTP, the cause of the condition is believed to be reduced activity of ADAMTS-13, the protease that breaks vWF into smaller pieces—thus preventing the formation of unnecessary blood clots.
In cancer-associated TTP, which could be a complication resulting from chemotherapy or a manifestation of cancer itself,3 ADAMTS-13 level is normal and the condition is likely the result of an increased tumor cell load, which leads to endothelial damage and fragmentation of red blood cells (RBC) as they traverse the injured microvasculature.4 In an analysis of 154 cases of “solid” cancer-related MAHA, Lechner and Obermeier5 found 23 cases were related to prostate cancer, as was the case with our patient.
Treatment for TTP is plasma exchange. The mortality rate of untreated TTP can exceed 90%, but plasma exchange therapy has reduced that rate to <20%.6 It has been suggested that proteolysis of vWF may play a central role in the efficacy of plasma exchange for TTP.7
Our patient was hospitalized and received 2 units of packed RBCs. He also received plasma exchange for 9 days with minimal response. On Day 5, our patient was started on leuprorelin and parenteral steroids. Soon after, his platelet count rose to 33 × 103/mcL and lactate dehydrogenase decreased. He was discharged approximately one week after the steroids were started.
After several months of outpatient treatment with leuprorelin and bicalutamide, the patient’s platelet count normalized to 212 × 103/mcL (from 29 × 103/mcL), alkaline phosphatase decreased to 402 U/L (from 1919 U/L), and PSA levels trended downward to 8.63 ng/mL (from 212 ng/mL). He continued to receive care from our oncology clinic for the next several months and his PSA level continued to decline. However, at his last few visits, his PSA level had trended up, suggesting progression of his prostate cancer. The patient has not followed up with our clinic recently.
THE TAKEAWAY
Suspect TTP in patients who present with unexplained petechiae and bruising, and whose blood work reveals thrombocytopenia and MAHA.2 Patients with TTP who do not respond to plasma exchange should be evaluated for underlying cancer or other potential secondary causes.3 Patients with cancer-associated TTP may respond to steroid therapy.
1. Lichtin AE, Schreiber AD, Hurwitz S, et al. Efficacy of intensive plasmapheresis in thrombotic thrombocytopenic purpura. Archives Intern Med. 1987;147:2122-2126.
2. Blombery P, Scully M. Management of thrombotic thrombocytopenic purpura: current perspectives. J Blood Med. 2014;5:15-23.
3. Chang JC, Naqvi T. Thrombotic thrombocytopenic purpura associated with bone marrow metastasis and secondary myelofibrosis in cancer. Oncologist. 2003;8:375-380.
4. Pirrotta MT, Bucalossi A, Forconi F, et al. Thrombotic thrombocytopenic purpura secondary to an occult adenocarcinoma. Oncologist. 2005;10:299-300.
5. Lechner K, Obermeier HL. Cancer-related microangiopathic hemolytic anemia: clinical and laboratory features in 168 reported cases. Medicine (Baltimore). 2012;91: 195-205.
6. Oberic L, Buffet, M, Scwarzinger M, et al; Reference Center for the Management of Thrombotic Microangiopathies. Cancer awareness in atypical thrombotic microangiopathies. Oncologist. 2009;14:769-779.
7. Zheng X, Chung D, Takayama TK, et al. Structure of von Willebrand factor-cleaving protease (ADAMTS 13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem. 2001;276:41059-41063.
THE CASE
A 57-year-old Hispanic man sought treatment because he had been feeling tired for a few weeks. He had not seen a physician for 15 years. When he came in, his temperature was 98.8°F, blood pressure was 132/82 mm Hg, pulse was 82 beats/min, respiration rate was 16 breaths/min, and oxygen saturation was 93% on room air. Examination of the head, neck, and respiratory and cardiovascular systems was normal. Skin examination showed petechiae and bruising on his abdomen, left ankle, right thigh, and bilateral shin area. His abdomen was nontender with no organomegaly. There was no focal neurological finding or spinal tenderness. Our patient had no chills, chest pains, shortness of breath, headache, dizziness, or loss of consciousness. There was no hematemesis, melena, hematuria, edema, or weight loss. He had no medical or surgical history and denied substance abuse or taking any medications recently; he did use alcohol previously.
Results of some initial lab tests were abnormal, including a decreased white blood cell count (5.82/mcL), platelet count (29 x 103/mcL), hemoglobin (8.6 g/dL), and hematocrit (27%) (TABLE). A peripheral blood smear showed decreased normocytic red blood cells and scattered schistocytes. His prostate-specific antigen (PSA) level was elevated at 212 ng/mL.
The patient’s coagulation profile was normal, and his von Willebrand factor (vWF) protease (ADAMTS-13) level was within normal limits (13.83). Antineutrophil cytoplasmic antibody and antinuclear antibody tests were negative. Testing for pulmonary embolism was negative, as was testing for human immunodeficiency virus. An abdominal ultrasound was normal, as well.
THE DIAGNOSIS
Based on our patient’s abnormal blood test results and the presence of petechiae and bruising, we diagnosed thrombotic thrombocytopenic purpura (TTP). The patient’s elevated PSA prompted us to order computed tomography of the chest and abdomen, which showed an enlarged prostate gland and mixed lytic sclerotic lesions in T3 to T5 and T9 vertebrae and in his ribs. A bone marrow biopsy revealed metastatic prostatic adenocarcinoma and a bone scan confirmed multiple metastases in the spine, pelvis, and shoulders.
DISCUSSION
TTP is a rare disorder of increased clotting in small blood vessels throughout the body that can include thrombocytopenia, microangiopathic hemolytic anemia (MAHA), fever, renal dysfunction, and neurological deficits.1 It’s important to maintain a high index of suspicion for TTP because the condition is a hematologic medical emergency that can quickly cause multiorgan failure and death.2
Almost always an acquired condition, TTP can be idiopathic or secondary to another condition, such as collagen vascular diseases, transplants, certain drugs, infections, pregnancy, or cancer.3 In idiopathic TTP, the cause of the condition is believed to be reduced activity of ADAMTS-13, the protease that breaks vWF into smaller pieces—thus preventing the formation of unnecessary blood clots.
In cancer-associated TTP, which could be a complication resulting from chemotherapy or a manifestation of cancer itself,3 ADAMTS-13 level is normal and the condition is likely the result of an increased tumor cell load, which leads to endothelial damage and fragmentation of red blood cells (RBC) as they traverse the injured microvasculature.4 In an analysis of 154 cases of “solid” cancer-related MAHA, Lechner and Obermeier5 found 23 cases were related to prostate cancer, as was the case with our patient.
Treatment for TTP is plasma exchange. The mortality rate of untreated TTP can exceed 90%, but plasma exchange therapy has reduced that rate to <20%.6 It has been suggested that proteolysis of vWF may play a central role in the efficacy of plasma exchange for TTP.7
Our patient was hospitalized and received 2 units of packed RBCs. He also received plasma exchange for 9 days with minimal response. On Day 5, our patient was started on leuprorelin and parenteral steroids. Soon after, his platelet count rose to 33 × 103/mcL and lactate dehydrogenase decreased. He was discharged approximately one week after the steroids were started.
After several months of outpatient treatment with leuprorelin and bicalutamide, the patient’s platelet count normalized to 212 × 103/mcL (from 29 × 103/mcL), alkaline phosphatase decreased to 402 U/L (from 1919 U/L), and PSA levels trended downward to 8.63 ng/mL (from 212 ng/mL). He continued to receive care from our oncology clinic for the next several months and his PSA level continued to decline. However, at his last few visits, his PSA level had trended up, suggesting progression of his prostate cancer. The patient has not followed up with our clinic recently.
THE TAKEAWAY
Suspect TTP in patients who present with unexplained petechiae and bruising, and whose blood work reveals thrombocytopenia and MAHA.2 Patients with TTP who do not respond to plasma exchange should be evaluated for underlying cancer or other potential secondary causes.3 Patients with cancer-associated TTP may respond to steroid therapy.
THE CASE
A 57-year-old Hispanic man sought treatment because he had been feeling tired for a few weeks. He had not seen a physician for 15 years. When he came in, his temperature was 98.8°F, blood pressure was 132/82 mm Hg, pulse was 82 beats/min, respiration rate was 16 breaths/min, and oxygen saturation was 93% on room air. Examination of the head, neck, and respiratory and cardiovascular systems was normal. Skin examination showed petechiae and bruising on his abdomen, left ankle, right thigh, and bilateral shin area. His abdomen was nontender with no organomegaly. There was no focal neurological finding or spinal tenderness. Our patient had no chills, chest pains, shortness of breath, headache, dizziness, or loss of consciousness. There was no hematemesis, melena, hematuria, edema, or weight loss. He had no medical or surgical history and denied substance abuse or taking any medications recently; he did use alcohol previously.
Results of some initial lab tests were abnormal, including a decreased white blood cell count (5.82/mcL), platelet count (29 x 103/mcL), hemoglobin (8.6 g/dL), and hematocrit (27%) (TABLE). A peripheral blood smear showed decreased normocytic red blood cells and scattered schistocytes. His prostate-specific antigen (PSA) level was elevated at 212 ng/mL.
The patient’s coagulation profile was normal, and his von Willebrand factor (vWF) protease (ADAMTS-13) level was within normal limits (13.83). Antineutrophil cytoplasmic antibody and antinuclear antibody tests were negative. Testing for pulmonary embolism was negative, as was testing for human immunodeficiency virus. An abdominal ultrasound was normal, as well.
THE DIAGNOSIS
Based on our patient’s abnormal blood test results and the presence of petechiae and bruising, we diagnosed thrombotic thrombocytopenic purpura (TTP). The patient’s elevated PSA prompted us to order computed tomography of the chest and abdomen, which showed an enlarged prostate gland and mixed lytic sclerotic lesions in T3 to T5 and T9 vertebrae and in his ribs. A bone marrow biopsy revealed metastatic prostatic adenocarcinoma and a bone scan confirmed multiple metastases in the spine, pelvis, and shoulders.
DISCUSSION
TTP is a rare disorder of increased clotting in small blood vessels throughout the body that can include thrombocytopenia, microangiopathic hemolytic anemia (MAHA), fever, renal dysfunction, and neurological deficits.1 It’s important to maintain a high index of suspicion for TTP because the condition is a hematologic medical emergency that can quickly cause multiorgan failure and death.2
Almost always an acquired condition, TTP can be idiopathic or secondary to another condition, such as collagen vascular diseases, transplants, certain drugs, infections, pregnancy, or cancer.3 In idiopathic TTP, the cause of the condition is believed to be reduced activity of ADAMTS-13, the protease that breaks vWF into smaller pieces—thus preventing the formation of unnecessary blood clots.
In cancer-associated TTP, which could be a complication resulting from chemotherapy or a manifestation of cancer itself,3 ADAMTS-13 level is normal and the condition is likely the result of an increased tumor cell load, which leads to endothelial damage and fragmentation of red blood cells (RBC) as they traverse the injured microvasculature.4 In an analysis of 154 cases of “solid” cancer-related MAHA, Lechner and Obermeier5 found 23 cases were related to prostate cancer, as was the case with our patient.
Treatment for TTP is plasma exchange. The mortality rate of untreated TTP can exceed 90%, but plasma exchange therapy has reduced that rate to <20%.6 It has been suggested that proteolysis of vWF may play a central role in the efficacy of plasma exchange for TTP.7
Our patient was hospitalized and received 2 units of packed RBCs. He also received plasma exchange for 9 days with minimal response. On Day 5, our patient was started on leuprorelin and parenteral steroids. Soon after, his platelet count rose to 33 × 103/mcL and lactate dehydrogenase decreased. He was discharged approximately one week after the steroids were started.
After several months of outpatient treatment with leuprorelin and bicalutamide, the patient’s platelet count normalized to 212 × 103/mcL (from 29 × 103/mcL), alkaline phosphatase decreased to 402 U/L (from 1919 U/L), and PSA levels trended downward to 8.63 ng/mL (from 212 ng/mL). He continued to receive care from our oncology clinic for the next several months and his PSA level continued to decline. However, at his last few visits, his PSA level had trended up, suggesting progression of his prostate cancer. The patient has not followed up with our clinic recently.
THE TAKEAWAY
Suspect TTP in patients who present with unexplained petechiae and bruising, and whose blood work reveals thrombocytopenia and MAHA.2 Patients with TTP who do not respond to plasma exchange should be evaluated for underlying cancer or other potential secondary causes.3 Patients with cancer-associated TTP may respond to steroid therapy.
1. Lichtin AE, Schreiber AD, Hurwitz S, et al. Efficacy of intensive plasmapheresis in thrombotic thrombocytopenic purpura. Archives Intern Med. 1987;147:2122-2126.
2. Blombery P, Scully M. Management of thrombotic thrombocytopenic purpura: current perspectives. J Blood Med. 2014;5:15-23.
3. Chang JC, Naqvi T. Thrombotic thrombocytopenic purpura associated with bone marrow metastasis and secondary myelofibrosis in cancer. Oncologist. 2003;8:375-380.
4. Pirrotta MT, Bucalossi A, Forconi F, et al. Thrombotic thrombocytopenic purpura secondary to an occult adenocarcinoma. Oncologist. 2005;10:299-300.
5. Lechner K, Obermeier HL. Cancer-related microangiopathic hemolytic anemia: clinical and laboratory features in 168 reported cases. Medicine (Baltimore). 2012;91: 195-205.
6. Oberic L, Buffet, M, Scwarzinger M, et al; Reference Center for the Management of Thrombotic Microangiopathies. Cancer awareness in atypical thrombotic microangiopathies. Oncologist. 2009;14:769-779.
7. Zheng X, Chung D, Takayama TK, et al. Structure of von Willebrand factor-cleaving protease (ADAMTS 13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem. 2001;276:41059-41063.
1. Lichtin AE, Schreiber AD, Hurwitz S, et al. Efficacy of intensive plasmapheresis in thrombotic thrombocytopenic purpura. Archives Intern Med. 1987;147:2122-2126.
2. Blombery P, Scully M. Management of thrombotic thrombocytopenic purpura: current perspectives. J Blood Med. 2014;5:15-23.
3. Chang JC, Naqvi T. Thrombotic thrombocytopenic purpura associated with bone marrow metastasis and secondary myelofibrosis in cancer. Oncologist. 2003;8:375-380.
4. Pirrotta MT, Bucalossi A, Forconi F, et al. Thrombotic thrombocytopenic purpura secondary to an occult adenocarcinoma. Oncologist. 2005;10:299-300.
5. Lechner K, Obermeier HL. Cancer-related microangiopathic hemolytic anemia: clinical and laboratory features in 168 reported cases. Medicine (Baltimore). 2012;91: 195-205.
6. Oberic L, Buffet, M, Scwarzinger M, et al; Reference Center for the Management of Thrombotic Microangiopathies. Cancer awareness in atypical thrombotic microangiopathies. Oncologist. 2009;14:769-779.
7. Zheng X, Chung D, Takayama TK, et al. Structure of von Willebrand factor-cleaving protease (ADAMTS 13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem. 2001;276:41059-41063.
Nausea, vomiting, malaise, frequent urination—Dx?
THE CASE
A 63-year-old multiparous woman visited her general practitioner because of nausea, vomiting, and general malaise. A proton pump inhibitor was prescribed, which temporarily relieved her symptoms. Two weeks later, however, her symptoms worsened and she was admitted to the hospital.

The patient’s physical examination on admission was normal, but laboratory findings revealed severe renal failure with a creatinine level of 7.4 mg/dL (normal, 0.6-1.1 mg/dL), potassium level of 7.4 mmol/L (3.5-5 mmol/L), and a sodium level of 123 mmol/L (135-145 mmol/L). A renal ultrasound revealed severe bilateral hydronephrosis with hydroureteronephrosis caused by obstructive uropathy. A radiologist examined the patient and determined that she had a total uterine prolapse; the cervix was 11 cm outside of the vagina (FIGURE 1). Our patient’s untreated pelvic organ prolapse (POP) had caused chronic renal failure. The patient was referred to a urogynecologist.
Previous attempts at treatment. It appeared that our patient had POP for years and there had been a previous attempt to treat it with a pessary. However, because of an unpleasant experience at her initial appointment and because her biggest complaint (until recently) had been the need to urinate frequently, she had not returned for follow-up appointments.
DISCUSSION
POP is not life-threatening, but the condition lowers the quality of life for 50% of parous women age >50 years.1 It can present as stress urinary incontinence, fecal incontinence, sexual dysfunction, and mechanical problems due to vaginal bulging or pelvic pressure.2 With the exception of vaginal bulging, symptoms are not specific for POP and there is no linear relationship between the severity of the prolapse and the symptoms.3,4
The condition is staged using the POP-Quantification (POP-Q) system5:
1. Stage 0: no prolapse
2. Stage I: the most distal portion of the prolapse is >1 cm above the hymen
3. Stage II: the prolapse is ≤1 cm proximal or distal to the plane of the hymen
4. Stage III: the prolapse is >1 cm below the plane of the hymen, but protrudes no farther
than 2 cm less than the total vaginal length
5. Stage IV: complete eversion of the lower genital tract.
As was the case with our patient, it is possible for a woman with severe total uterine prolapse (Stage IV) to have no major problems with urination or defecation.
The link between POP and hydronephrosis
Hydronephrosis appears to be a frequent finding in women with POP.4 A recent prospective observational study reported an overall prevalence of 10.3% (95% confidence interval, 6%-14%) in women with POP.4 Patients with advanced stages of POP (POP-Q Stage III or IV)4 who also had diabetes mellitus and hypertension were at particularly high risk, with a prevalence of about 20%. An analysis of factors, including age, parity, diabetes, hypertension, and type of prolapse, found that severity of POP was the strongest predictor of hydronephrosis: Patients with a Stage III to IV prolapse are 3.4 times more likely to have hydronephrosis than those with a Stage I or II prolapse.4,6
Possible causes of hydronephrosis in POP patients. Some researchers have proposed that hydronephrosis in patients with uterine prolapse may be due to a kinking of the ureters by the extrinsic compression of the prolapsed uterus. In patients with vaginal vault prolapse, the cause of the hydronephrosis could be a weakening or disintegration of the cardinal ligaments after hysterectomy.4,7
Patients may not complain. When hydronephrosis caused by POP occurs, it may develop slowly, causing little or no discomfort. As time passes, patients may complain of dull pain in the flank, suffer from urinary tract infections, or develop kidney stones before progressive renal dysfunction or renal failure occurs.4
There are 2 other cases in the literature of women who, like our patient, had uterine prolapse that went untreated until they were in renal failure.8,9 The patients noticed only mechanical problems due to the POP; bilateral hydroureteronephrosis and renal failure had developed undetected. In the end, both women needed lifelong hemodialysis.
Treatment options
Treatment options for POP include supervised pelvic floor exercise programs, pessary insertion, or reconstructive pelvic surgery. If POP is treated adequately, an estimated 95% of the hydronephrosis can resolve, regardless of its severity at presentation.4
Our patient was treated with a 95 mm Falk pessary. After 24 hours, renal ultrasonography showed a decrease in both the hydroureteronephrosis and the hydronephrosis (FIGURE 2A and 2B). Four weeks later, her serum creatinine level had decreased to 3.3 mg/dL. Four years later, our patient continues to wear the pessary but has chronic renal failure.
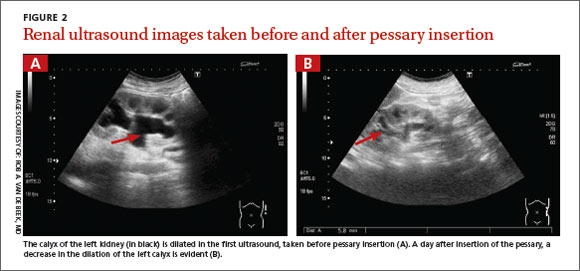
THE TAKEAWAY
POP often is viewed as a minor problem, but it can cause obstructive uropathy with unilateral or bilateral hydronephrosis or renal dysfunction and/or failure. The delay often seen with reporting genital prolapse may be due to the mild symptoms or feelings of shame or fear. Combining screening for cervical pathology in general practice with a screening for genital prolapse could identify these problems.
Monitoring renal function is advised in patients with a Stage III or IV POP and any patients with POP who also have hypertension or diabetes mellitus. Because only minor changes in laboratory findings may be observed in patients with unilateral hydronephrosis, consider renal ultrasonography.
Treatment options for POP includes pelvic floor exercises, pessary insertion, and reconstructive surgery. Early treatment can resolve hydronephrosis and possibly prevent irreversible renal damage.
ACKNOWLEDGEMENTS
The authors thank Wilhelm Van Dorp, MD, Rob A. van de Beek, MD, and Alan Brind for their help with this manuscript.
1. Maher C, Feiner B, Baessler K, et al. Surgical management of pelvic organ prolapse in women. Cochrane Database Syst Rev. 2010;(4):CD004014.
2. Jelovsek JE, Maher C, Barber MD. Pelvic organ prolapse. Lancet. 2007;369:1027-1038.
3. Slieker-ten Hove MC, Pool-Goudzwaard AL, Eijkemans MJ, et al. Symptomatic pelvic organ prolapse and possible risk factors in a general population. Am J Obstet Gynecol. 2009;200:184. e1-184.e7.
4. Hui SY, Chan SC, Lam SY, et al. A prospective study on the prevalence of hydronephrosis in women with pelvic organ prolapse and their outcomes after treatment. Int Urogynecol J. 2011;22:1529-1534.
5. Bump RC, Mattiasson A, Bø K, et al. The standardization of terminology of female pelvic organ prolapse and pelvic floor dysfunction. Am J Obstet Gynecol. 1996;175:10-17.
6. Gemer O, Bergman M, Segal S. Prevalence of hydronephrosis in patients with genital prolapse. Eur J Obstet Gynecol Reprod Biol. 1999;86:11-13.
7. Lieberthal F, Frankenthal L Jr. The mechanism of urethral obstruction in prolapse of the uterus. Surg Gynaecol Obstet. 1941;73:838-842.
8. Sanai T, Yamashiro Y, Nakayama M, et al. End-stage renal failure due to total uterine prolapse. Urology. 2006;67:622. e5-622.e7.
9. Nässberger L, Larsson R. End-stage chronic renal failure due to total uterine prolapse. Acta Obstet Gynecol Scand. 1982;61: 495-497.
THE CASE
A 63-year-old multiparous woman visited her general practitioner because of nausea, vomiting, and general malaise. A proton pump inhibitor was prescribed, which temporarily relieved her symptoms. Two weeks later, however, her symptoms worsened and she was admitted to the hospital.
The patient’s physical examination on admission was normal, but laboratory findings revealed severe renal failure with a creatinine level of 7.4 mg/dL (normal, 0.6-1.1 mg/dL), potassium level of 7.4 mmol/L (3.5-5 mmol/L), and a sodium level of 123 mmol/L (135-145 mmol/L). A renal ultrasound revealed severe bilateral hydronephrosis with hydroureteronephrosis caused by obstructive uropathy. A radiologist examined the patient and determined that she had a total uterine prolapse; the cervix was 11 cm outside of the vagina (FIGURE 1). Our patient’s untreated pelvic organ prolapse (POP) had caused chronic renal failure. The patient was referred to a urogynecologist.
Previous attempts at treatment. It appeared that our patient had POP for years and there had been a previous attempt to treat it with a pessary. However, because of an unpleasant experience at her initial appointment and because her biggest complaint (until recently) had been the need to urinate frequently, she had not returned for follow-up appointments.
DISCUSSION
POP is not life-threatening, but the condition lowers the quality of life for 50% of parous women age >50 years.1 It can present as stress urinary incontinence, fecal incontinence, sexual dysfunction, and mechanical problems due to vaginal bulging or pelvic pressure.2 With the exception of vaginal bulging, symptoms are not specific for POP and there is no linear relationship between the severity of the prolapse and the symptoms.3,4
The condition is staged using the POP-Quantification (POP-Q) system5:
1. Stage 0: no prolapse
2. Stage I: the most distal portion of the prolapse is >1 cm above the hymen
3. Stage II: the prolapse is ≤1 cm proximal or distal to the plane of the hymen
4. Stage III: the prolapse is >1 cm below the plane of the hymen, but protrudes no farther
than 2 cm less than the total vaginal length
5. Stage IV: complete eversion of the lower genital tract.
As was the case with our patient, it is possible for a woman with severe total uterine prolapse (Stage IV) to have no major problems with urination or defecation.
The link between POP and hydronephrosis
Hydronephrosis appears to be a frequent finding in women with POP.4 A recent prospective observational study reported an overall prevalence of 10.3% (95% confidence interval, 6%-14%) in women with POP.4 Patients with advanced stages of POP (POP-Q Stage III or IV)4 who also had diabetes mellitus and hypertension were at particularly high risk, with a prevalence of about 20%. An analysis of factors, including age, parity, diabetes, hypertension, and type of prolapse, found that severity of POP was the strongest predictor of hydronephrosis: Patients with a Stage III to IV prolapse are 3.4 times more likely to have hydronephrosis than those with a Stage I or II prolapse.4,6
Possible causes of hydronephrosis in POP patients. Some researchers have proposed that hydronephrosis in patients with uterine prolapse may be due to a kinking of the ureters by the extrinsic compression of the prolapsed uterus. In patients with vaginal vault prolapse, the cause of the hydronephrosis could be a weakening or disintegration of the cardinal ligaments after hysterectomy.4,7
Patients may not complain. When hydronephrosis caused by POP occurs, it may develop slowly, causing little or no discomfort. As time passes, patients may complain of dull pain in the flank, suffer from urinary tract infections, or develop kidney stones before progressive renal dysfunction or renal failure occurs.4
There are 2 other cases in the literature of women who, like our patient, had uterine prolapse that went untreated until they were in renal failure.8,9 The patients noticed only mechanical problems due to the POP; bilateral hydroureteronephrosis and renal failure had developed undetected. In the end, both women needed lifelong hemodialysis.
Treatment options
Treatment options for POP include supervised pelvic floor exercise programs, pessary insertion, or reconstructive pelvic surgery. If POP is treated adequately, an estimated 95% of the hydronephrosis can resolve, regardless of its severity at presentation.4
Our patient was treated with a 95 mm Falk pessary. After 24 hours, renal ultrasonography showed a decrease in both the hydroureteronephrosis and the hydronephrosis (FIGURE 2A and 2B). Four weeks later, her serum creatinine level had decreased to 3.3 mg/dL. Four years later, our patient continues to wear the pessary but has chronic renal failure.
THE TAKEAWAY
POP often is viewed as a minor problem, but it can cause obstructive uropathy with unilateral or bilateral hydronephrosis or renal dysfunction and/or failure. The delay often seen with reporting genital prolapse may be due to the mild symptoms or feelings of shame or fear. Combining screening for cervical pathology in general practice with a screening for genital prolapse could identify these problems.
Monitoring renal function is advised in patients with a Stage III or IV POP and any patients with POP who also have hypertension or diabetes mellitus. Because only minor changes in laboratory findings may be observed in patients with unilateral hydronephrosis, consider renal ultrasonography.
Treatment options for POP includes pelvic floor exercises, pessary insertion, and reconstructive surgery. Early treatment can resolve hydronephrosis and possibly prevent irreversible renal damage.
ACKNOWLEDGEMENTS
The authors thank Wilhelm Van Dorp, MD, Rob A. van de Beek, MD, and Alan Brind for their help with this manuscript.
THE CASE
A 63-year-old multiparous woman visited her general practitioner because of nausea, vomiting, and general malaise. A proton pump inhibitor was prescribed, which temporarily relieved her symptoms. Two weeks later, however, her symptoms worsened and she was admitted to the hospital.
The patient’s physical examination on admission was normal, but laboratory findings revealed severe renal failure with a creatinine level of 7.4 mg/dL (normal, 0.6-1.1 mg/dL), potassium level of 7.4 mmol/L (3.5-5 mmol/L), and a sodium level of 123 mmol/L (135-145 mmol/L). A renal ultrasound revealed severe bilateral hydronephrosis with hydroureteronephrosis caused by obstructive uropathy. A radiologist examined the patient and determined that she had a total uterine prolapse; the cervix was 11 cm outside of the vagina (FIGURE 1). Our patient’s untreated pelvic organ prolapse (POP) had caused chronic renal failure. The patient was referred to a urogynecologist.
Previous attempts at treatment. It appeared that our patient had POP for years and there had been a previous attempt to treat it with a pessary. However, because of an unpleasant experience at her initial appointment and because her biggest complaint (until recently) had been the need to urinate frequently, she had not returned for follow-up appointments.
DISCUSSION
POP is not life-threatening, but the condition lowers the quality of life for 50% of parous women age >50 years.1 It can present as stress urinary incontinence, fecal incontinence, sexual dysfunction, and mechanical problems due to vaginal bulging or pelvic pressure.2 With the exception of vaginal bulging, symptoms are not specific for POP and there is no linear relationship between the severity of the prolapse and the symptoms.3,4
The condition is staged using the POP-Quantification (POP-Q) system5:
1. Stage 0: no prolapse
2. Stage I: the most distal portion of the prolapse is >1 cm above the hymen
3. Stage II: the prolapse is ≤1 cm proximal or distal to the plane of the hymen
4. Stage III: the prolapse is >1 cm below the plane of the hymen, but protrudes no farther
than 2 cm less than the total vaginal length
5. Stage IV: complete eversion of the lower genital tract.
As was the case with our patient, it is possible for a woman with severe total uterine prolapse (Stage IV) to have no major problems with urination or defecation.
The link between POP and hydronephrosis
Hydronephrosis appears to be a frequent finding in women with POP.4 A recent prospective observational study reported an overall prevalence of 10.3% (95% confidence interval, 6%-14%) in women with POP.4 Patients with advanced stages of POP (POP-Q Stage III or IV)4 who also had diabetes mellitus and hypertension were at particularly high risk, with a prevalence of about 20%. An analysis of factors, including age, parity, diabetes, hypertension, and type of prolapse, found that severity of POP was the strongest predictor of hydronephrosis: Patients with a Stage III to IV prolapse are 3.4 times more likely to have hydronephrosis than those with a Stage I or II prolapse.4,6
Possible causes of hydronephrosis in POP patients. Some researchers have proposed that hydronephrosis in patients with uterine prolapse may be due to a kinking of the ureters by the extrinsic compression of the prolapsed uterus. In patients with vaginal vault prolapse, the cause of the hydronephrosis could be a weakening or disintegration of the cardinal ligaments after hysterectomy.4,7
Patients may not complain. When hydronephrosis caused by POP occurs, it may develop slowly, causing little or no discomfort. As time passes, patients may complain of dull pain in the flank, suffer from urinary tract infections, or develop kidney stones before progressive renal dysfunction or renal failure occurs.4
There are 2 other cases in the literature of women who, like our patient, had uterine prolapse that went untreated until they were in renal failure.8,9 The patients noticed only mechanical problems due to the POP; bilateral hydroureteronephrosis and renal failure had developed undetected. In the end, both women needed lifelong hemodialysis.
Treatment options
Treatment options for POP include supervised pelvic floor exercise programs, pessary insertion, or reconstructive pelvic surgery. If POP is treated adequately, an estimated 95% of the hydronephrosis can resolve, regardless of its severity at presentation.4
Our patient was treated with a 95 mm Falk pessary. After 24 hours, renal ultrasonography showed a decrease in both the hydroureteronephrosis and the hydronephrosis (FIGURE 2A and 2B). Four weeks later, her serum creatinine level had decreased to 3.3 mg/dL. Four years later, our patient continues to wear the pessary but has chronic renal failure.
THE TAKEAWAY
POP often is viewed as a minor problem, but it can cause obstructive uropathy with unilateral or bilateral hydronephrosis or renal dysfunction and/or failure. The delay often seen with reporting genital prolapse may be due to the mild symptoms or feelings of shame or fear. Combining screening for cervical pathology in general practice with a screening for genital prolapse could identify these problems.
Monitoring renal function is advised in patients with a Stage III or IV POP and any patients with POP who also have hypertension or diabetes mellitus. Because only minor changes in laboratory findings may be observed in patients with unilateral hydronephrosis, consider renal ultrasonography.
Treatment options for POP includes pelvic floor exercises, pessary insertion, and reconstructive surgery. Early treatment can resolve hydronephrosis and possibly prevent irreversible renal damage.
ACKNOWLEDGEMENTS
The authors thank Wilhelm Van Dorp, MD, Rob A. van de Beek, MD, and Alan Brind for their help with this manuscript.
1. Maher C, Feiner B, Baessler K, et al. Surgical management of pelvic organ prolapse in women. Cochrane Database Syst Rev. 2010;(4):CD004014.
2. Jelovsek JE, Maher C, Barber MD. Pelvic organ prolapse. Lancet. 2007;369:1027-1038.
3. Slieker-ten Hove MC, Pool-Goudzwaard AL, Eijkemans MJ, et al. Symptomatic pelvic organ prolapse and possible risk factors in a general population. Am J Obstet Gynecol. 2009;200:184. e1-184.e7.
4. Hui SY, Chan SC, Lam SY, et al. A prospective study on the prevalence of hydronephrosis in women with pelvic organ prolapse and their outcomes after treatment. Int Urogynecol J. 2011;22:1529-1534.
5. Bump RC, Mattiasson A, Bø K, et al. The standardization of terminology of female pelvic organ prolapse and pelvic floor dysfunction. Am J Obstet Gynecol. 1996;175:10-17.
6. Gemer O, Bergman M, Segal S. Prevalence of hydronephrosis in patients with genital prolapse. Eur J Obstet Gynecol Reprod Biol. 1999;86:11-13.
7. Lieberthal F, Frankenthal L Jr. The mechanism of urethral obstruction in prolapse of the uterus. Surg Gynaecol Obstet. 1941;73:838-842.
8. Sanai T, Yamashiro Y, Nakayama M, et al. End-stage renal failure due to total uterine prolapse. Urology. 2006;67:622. e5-622.e7.
9. Nässberger L, Larsson R. End-stage chronic renal failure due to total uterine prolapse. Acta Obstet Gynecol Scand. 1982;61: 495-497.
1. Maher C, Feiner B, Baessler K, et al. Surgical management of pelvic organ prolapse in women. Cochrane Database Syst Rev. 2010;(4):CD004014.
2. Jelovsek JE, Maher C, Barber MD. Pelvic organ prolapse. Lancet. 2007;369:1027-1038.
3. Slieker-ten Hove MC, Pool-Goudzwaard AL, Eijkemans MJ, et al. Symptomatic pelvic organ prolapse and possible risk factors in a general population. Am J Obstet Gynecol. 2009;200:184. e1-184.e7.
4. Hui SY, Chan SC, Lam SY, et al. A prospective study on the prevalence of hydronephrosis in women with pelvic organ prolapse and their outcomes after treatment. Int Urogynecol J. 2011;22:1529-1534.
5. Bump RC, Mattiasson A, Bø K, et al. The standardization of terminology of female pelvic organ prolapse and pelvic floor dysfunction. Am J Obstet Gynecol. 1996;175:10-17.
6. Gemer O, Bergman M, Segal S. Prevalence of hydronephrosis in patients with genital prolapse. Eur J Obstet Gynecol Reprod Biol. 1999;86:11-13.
7. Lieberthal F, Frankenthal L Jr. The mechanism of urethral obstruction in prolapse of the uterus. Surg Gynaecol Obstet. 1941;73:838-842.
8. Sanai T, Yamashiro Y, Nakayama M, et al. End-stage renal failure due to total uterine prolapse. Urology. 2006;67:622. e5-622.e7.
9. Nässberger L, Larsson R. End-stage chronic renal failure due to total uterine prolapse. Acta Obstet Gynecol Scand. 1982;61: 495-497.

Hot topics in vaccines
I recently attended the International Interscience Conference of Infectious Diseases and Vaccines, and I would like to share some of the presentations from the session entitled “Hot Topics in Vaccines.”
CNS complications of varicella-zoster virus infection
Dr. Michelle Science of the Hospital for Sick Children, Toronto, and her associates described the spectrum of CNS complications of varicella-zoster virus (VZV) in children admitted to the hospital during 1999-2012 (J. Pediatr. 2014;165:779-85). Clinical syndromes included 26 cases of acute cerebellar ataxia, 17 of encephalitis, 16 isolated seizures, 10 strokes, 10 cases of meningitis, 2 cases of Guillain-Barré syndrome, 2 cases of acute disseminated encephalomyelitis, and 1 case of Ramsay Hunt syndrome. In children with acute nonstroke complications, neurologic symptoms occurred a median 5 days after the onset of rash, but neurologic symptoms predated the onset of rash in five cases and in two cases there were no exanthems. Time between rash onset and stroke ranged from 2 to 26 weeks (median 16 weeks). There were three deaths among the 17 (18%) children with encephalitis. Among the 39 children with follow-up at 1 year, residual neurologic sequelae occurred in 9 (23%). Only four of the children had received a VZV vaccine. Although an effective vaccine exists, neurologic complications of VZV infection continue to occur.

Timely versus delayed early childhood vaccination and seizures
Dr. Simon J. Hambidge of Denver Health, Colorado, and his associates studied a cohort of 323,247 U.S. children from the Vaccine Safety Datalink born during 2004-2008 for an association between the timing of childhood vaccination and the first occurrence of seizures (Pediatrics 2014;133(6):e1492-9). In the first year, there was no association between the timing of infant vaccination and postvaccination seizures. In the second year, the incidence rate ratio for seizures after receiving the first MMR dose at 12-15 months was 2.7, compared with a rate of 6.5 after an MMR dose at 16-23 months; thus there were more seizures when MMR was delayed. The incidence rate ratio for seizures after receiving the first measles-mumps-rubella-varicella vaccine (MMRV) dose at 12-15 months was 4.95, compared with 9.80 after an MMRV dose at 16-23 months. Again, there were more seizures when MMRV was delayed. These findings suggest that on-time vaccination is as safe with regard to seizures as delayed vaccination in year 1, and that delayed vaccination in year 2 is linked to more postvaccination seizures than on-time vaccination with MMR and that risk is doubled with MMRV.
Effective messages in vaccine promotion: a randomized trial
Brendan Nyhan, Ph.D., of Dartmouth College, Hanover, N.H., and his associates tested the efficacy of various informational messages tailored to reduce misperceptions about vaccines and increase MMR vaccination rates (Pediatrics 2014;133:e835-42). Nearly 1,800 parents were randomly assigned to receive one of four interventions: information explaining the lack of evidence that MMR causes autism from the Centers for Disease Control and Prevention; information about the danger of the diseases prevented by MMR from the Vaccine Information Statement; photos of children with diseases prevented by the MMR vaccine; a dramatic narrative about an infant who almost died of measles from a CDC fact sheet. In addition there was a control group. None of the four interventions increased parents’ intention to vaccinate another child if they had one in the future. Although refuting claims of an MMR/autism link did reduce misperceptions that vaccines cause autism, it decreased intent to vaccinate among parents who had the least favorable attitudes toward vaccines. Also, photos of sick children increased belief in an association between vaccines and autism, and the dramatic narrative about an infant in danger increased belief in serious vaccine side effects. Attempts to rectify misperceptions about vaccines may be counterproductive in some populations, so public health communications about vaccines should be tested before being widely disseminated.
Silent reintroduction of wild-type poliovirus to Israel, 2013
Dr. E. Kaliner of the Israeli Ministry of Health, Jerusalem, and associates, reported that Israel has been certified as polio-free by the World Health Organization for decades and its routine immunization schedule, like the United States, consists of inactivated poliovirus vaccine only (Euro. Surveill. 2014;19:20703). At the end of May 2013, the Israeli Ministry of Health confirmed the reintroduction of wild-type poliovirus 1 into the country. Documented ongoing human-to-human transmission required a thorough risk assessment followed by a supplemental immunization campaign using oral polio vaccine.
Trends in otitis media–related health care use in the United States, 2001-2011
Dr. Tal Marom of the University of Texas, Galveston, and associates studied the trend in otitis media–related health care use in the United States during the pneumococcal conjugate vaccine (PCV) era in 2001-2011 (JAMA Pediatr. 2014;168:68-75). An analysis of an insurance claims database of a large, nationwide managed health care plan was conducted; 7.82 million children aged 6 years and under had 6.21 million primary otitis media (OM) visits. There was an overall downward trend in OM-related health care use across the 10-year study. Recurrent OM rates (defined as greater than or equal to three OM visits within 6 months) decreased at 0.003 per child-year in 2001-2009 and at 0.018 per child-year in 2010-2011. Prior to the pneumococcal conjugate vaccine (PCV-13), there was a stable rate ratio of 1.38 between OM visit rates. During the transition year 2010, the RR decreased significantly to 1.32, and in 2011 the RR decreased further to 1.01. Mastoiditis rates significantly decreased from 61 per 100,000 child-years in 2008 to 37 per 100,000 child-years in 2011. The ventilating tube insertion rate decreased by 19% from 2010 to 2011. Tympanic membrane perforation/otorrhea rates increased gradually and significantly from 3,721 per 100,000 OM child-years in 2001 to 4,542 per 100,000 OM child-years in 2011; the reasons for this are unclear.
Dr. Pichichero, a specialist in pediatric infectious diseases, is director of the Research Institute, Rochester (N.Y.) General Hospital. He is also a pediatrician at Legacy Pediatrics in Rochester. Dr. Pichichero said he had no financial disclosures relevant to this article. To comment, e-mail him at [email protected].
I recently attended the International Interscience Conference of Infectious Diseases and Vaccines, and I would like to share some of the presentations from the session entitled “Hot Topics in Vaccines.”
CNS complications of varicella-zoster virus infection
Dr. Michelle Science of the Hospital for Sick Children, Toronto, and her associates described the spectrum of CNS complications of varicella-zoster virus (VZV) in children admitted to the hospital during 1999-2012 (J. Pediatr. 2014;165:779-85). Clinical syndromes included 26 cases of acute cerebellar ataxia, 17 of encephalitis, 16 isolated seizures, 10 strokes, 10 cases of meningitis, 2 cases of Guillain-Barré syndrome, 2 cases of acute disseminated encephalomyelitis, and 1 case of Ramsay Hunt syndrome. In children with acute nonstroke complications, neurologic symptoms occurred a median 5 days after the onset of rash, but neurologic symptoms predated the onset of rash in five cases and in two cases there were no exanthems. Time between rash onset and stroke ranged from 2 to 26 weeks (median 16 weeks). There were three deaths among the 17 (18%) children with encephalitis. Among the 39 children with follow-up at 1 year, residual neurologic sequelae occurred in 9 (23%). Only four of the children had received a VZV vaccine. Although an effective vaccine exists, neurologic complications of VZV infection continue to occur.
Timely versus delayed early childhood vaccination and seizures
Dr. Simon J. Hambidge of Denver Health, Colorado, and his associates studied a cohort of 323,247 U.S. children from the Vaccine Safety Datalink born during 2004-2008 for an association between the timing of childhood vaccination and the first occurrence of seizures (Pediatrics 2014;133(6):e1492-9). In the first year, there was no association between the timing of infant vaccination and postvaccination seizures. In the second year, the incidence rate ratio for seizures after receiving the first MMR dose at 12-15 months was 2.7, compared with a rate of 6.5 after an MMR dose at 16-23 months; thus there were more seizures when MMR was delayed. The incidence rate ratio for seizures after receiving the first measles-mumps-rubella-varicella vaccine (MMRV) dose at 12-15 months was 4.95, compared with 9.80 after an MMRV dose at 16-23 months. Again, there were more seizures when MMRV was delayed. These findings suggest that on-time vaccination is as safe with regard to seizures as delayed vaccination in year 1, and that delayed vaccination in year 2 is linked to more postvaccination seizures than on-time vaccination with MMR and that risk is doubled with MMRV.
Effective messages in vaccine promotion: a randomized trial
Brendan Nyhan, Ph.D., of Dartmouth College, Hanover, N.H., and his associates tested the efficacy of various informational messages tailored to reduce misperceptions about vaccines and increase MMR vaccination rates (Pediatrics 2014;133:e835-42). Nearly 1,800 parents were randomly assigned to receive one of four interventions: information explaining the lack of evidence that MMR causes autism from the Centers for Disease Control and Prevention; information about the danger of the diseases prevented by MMR from the Vaccine Information Statement; photos of children with diseases prevented by the MMR vaccine; a dramatic narrative about an infant who almost died of measles from a CDC fact sheet. In addition there was a control group. None of the four interventions increased parents’ intention to vaccinate another child if they had one in the future. Although refuting claims of an MMR/autism link did reduce misperceptions that vaccines cause autism, it decreased intent to vaccinate among parents who had the least favorable attitudes toward vaccines. Also, photos of sick children increased belief in an association between vaccines and autism, and the dramatic narrative about an infant in danger increased belief in serious vaccine side effects. Attempts to rectify misperceptions about vaccines may be counterproductive in some populations, so public health communications about vaccines should be tested before being widely disseminated.
Silent reintroduction of wild-type poliovirus to Israel, 2013
Dr. E. Kaliner of the Israeli Ministry of Health, Jerusalem, and associates, reported that Israel has been certified as polio-free by the World Health Organization for decades and its routine immunization schedule, like the United States, consists of inactivated poliovirus vaccine only (Euro. Surveill. 2014;19:20703). At the end of May 2013, the Israeli Ministry of Health confirmed the reintroduction of wild-type poliovirus 1 into the country. Documented ongoing human-to-human transmission required a thorough risk assessment followed by a supplemental immunization campaign using oral polio vaccine.
Trends in otitis media–related health care use in the United States, 2001-2011
Dr. Tal Marom of the University of Texas, Galveston, and associates studied the trend in otitis media–related health care use in the United States during the pneumococcal conjugate vaccine (PCV) era in 2001-2011 (JAMA Pediatr. 2014;168:68-75). An analysis of an insurance claims database of a large, nationwide managed health care plan was conducted; 7.82 million children aged 6 years and under had 6.21 million primary otitis media (OM) visits. There was an overall downward trend in OM-related health care use across the 10-year study. Recurrent OM rates (defined as greater than or equal to three OM visits within 6 months) decreased at 0.003 per child-year in 2001-2009 and at 0.018 per child-year in 2010-2011. Prior to the pneumococcal conjugate vaccine (PCV-13), there was a stable rate ratio of 1.38 between OM visit rates. During the transition year 2010, the RR decreased significantly to 1.32, and in 2011 the RR decreased further to 1.01. Mastoiditis rates significantly decreased from 61 per 100,000 child-years in 2008 to 37 per 100,000 child-years in 2011. The ventilating tube insertion rate decreased by 19% from 2010 to 2011. Tympanic membrane perforation/otorrhea rates increased gradually and significantly from 3,721 per 100,000 OM child-years in 2001 to 4,542 per 100,000 OM child-years in 2011; the reasons for this are unclear.
Dr. Pichichero, a specialist in pediatric infectious diseases, is director of the Research Institute, Rochester (N.Y.) General Hospital. He is also a pediatrician at Legacy Pediatrics in Rochester. Dr. Pichichero said he had no financial disclosures relevant to this article. To comment, e-mail him at [email protected].
I recently attended the International Interscience Conference of Infectious Diseases and Vaccines, and I would like to share some of the presentations from the session entitled “Hot Topics in Vaccines.”
CNS complications of varicella-zoster virus infection
Dr. Michelle Science of the Hospital for Sick Children, Toronto, and her associates described the spectrum of CNS complications of varicella-zoster virus (VZV) in children admitted to the hospital during 1999-2012 (J. Pediatr. 2014;165:779-85). Clinical syndromes included 26 cases of acute cerebellar ataxia, 17 of encephalitis, 16 isolated seizures, 10 strokes, 10 cases of meningitis, 2 cases of Guillain-Barré syndrome, 2 cases of acute disseminated encephalomyelitis, and 1 case of Ramsay Hunt syndrome. In children with acute nonstroke complications, neurologic symptoms occurred a median 5 days after the onset of rash, but neurologic symptoms predated the onset of rash in five cases and in two cases there were no exanthems. Time between rash onset and stroke ranged from 2 to 26 weeks (median 16 weeks). There were three deaths among the 17 (18%) children with encephalitis. Among the 39 children with follow-up at 1 year, residual neurologic sequelae occurred in 9 (23%). Only four of the children had received a VZV vaccine. Although an effective vaccine exists, neurologic complications of VZV infection continue to occur.
Timely versus delayed early childhood vaccination and seizures
Dr. Simon J. Hambidge of Denver Health, Colorado, and his associates studied a cohort of 323,247 U.S. children from the Vaccine Safety Datalink born during 2004-2008 for an association between the timing of childhood vaccination and the first occurrence of seizures (Pediatrics 2014;133(6):e1492-9). In the first year, there was no association between the timing of infant vaccination and postvaccination seizures. In the second year, the incidence rate ratio for seizures after receiving the first MMR dose at 12-15 months was 2.7, compared with a rate of 6.5 after an MMR dose at 16-23 months; thus there were more seizures when MMR was delayed. The incidence rate ratio for seizures after receiving the first measles-mumps-rubella-varicella vaccine (MMRV) dose at 12-15 months was 4.95, compared with 9.80 after an MMRV dose at 16-23 months. Again, there were more seizures when MMRV was delayed. These findings suggest that on-time vaccination is as safe with regard to seizures as delayed vaccination in year 1, and that delayed vaccination in year 2 is linked to more postvaccination seizures than on-time vaccination with MMR and that risk is doubled with MMRV.
Effective messages in vaccine promotion: a randomized trial
Brendan Nyhan, Ph.D., of Dartmouth College, Hanover, N.H., and his associates tested the efficacy of various informational messages tailored to reduce misperceptions about vaccines and increase MMR vaccination rates (Pediatrics 2014;133:e835-42). Nearly 1,800 parents were randomly assigned to receive one of four interventions: information explaining the lack of evidence that MMR causes autism from the Centers for Disease Control and Prevention; information about the danger of the diseases prevented by MMR from the Vaccine Information Statement; photos of children with diseases prevented by the MMR vaccine; a dramatic narrative about an infant who almost died of measles from a CDC fact sheet. In addition there was a control group. None of the four interventions increased parents’ intention to vaccinate another child if they had one in the future. Although refuting claims of an MMR/autism link did reduce misperceptions that vaccines cause autism, it decreased intent to vaccinate among parents who had the least favorable attitudes toward vaccines. Also, photos of sick children increased belief in an association between vaccines and autism, and the dramatic narrative about an infant in danger increased belief in serious vaccine side effects. Attempts to rectify misperceptions about vaccines may be counterproductive in some populations, so public health communications about vaccines should be tested before being widely disseminated.
Silent reintroduction of wild-type poliovirus to Israel, 2013
Dr. E. Kaliner of the Israeli Ministry of Health, Jerusalem, and associates, reported that Israel has been certified as polio-free by the World Health Organization for decades and its routine immunization schedule, like the United States, consists of inactivated poliovirus vaccine only (Euro. Surveill. 2014;19:20703). At the end of May 2013, the Israeli Ministry of Health confirmed the reintroduction of wild-type poliovirus 1 into the country. Documented ongoing human-to-human transmission required a thorough risk assessment followed by a supplemental immunization campaign using oral polio vaccine.
Trends in otitis media–related health care use in the United States, 2001-2011
Dr. Tal Marom of the University of Texas, Galveston, and associates studied the trend in otitis media–related health care use in the United States during the pneumococcal conjugate vaccine (PCV) era in 2001-2011 (JAMA Pediatr. 2014;168:68-75). An analysis of an insurance claims database of a large, nationwide managed health care plan was conducted; 7.82 million children aged 6 years and under had 6.21 million primary otitis media (OM) visits. There was an overall downward trend in OM-related health care use across the 10-year study. Recurrent OM rates (defined as greater than or equal to three OM visits within 6 months) decreased at 0.003 per child-year in 2001-2009 and at 0.018 per child-year in 2010-2011. Prior to the pneumococcal conjugate vaccine (PCV-13), there was a stable rate ratio of 1.38 between OM visit rates. During the transition year 2010, the RR decreased significantly to 1.32, and in 2011 the RR decreased further to 1.01. Mastoiditis rates significantly decreased from 61 per 100,000 child-years in 2008 to 37 per 100,000 child-years in 2011. The ventilating tube insertion rate decreased by 19% from 2010 to 2011. Tympanic membrane perforation/otorrhea rates increased gradually and significantly from 3,721 per 100,000 OM child-years in 2001 to 4,542 per 100,000 OM child-years in 2011; the reasons for this are unclear.
Dr. Pichichero, a specialist in pediatric infectious diseases, is director of the Research Institute, Rochester (N.Y.) General Hospital. He is also a pediatrician at Legacy Pediatrics in Rochester. Dr. Pichichero said he had no financial disclosures relevant to this article. To comment, e-mail him at [email protected].

Why is metformin contraindicated in chronic kidney disease?
To the Editor: In their article about the care of patients with advanced chronic kidney disease, Sakhuja et al1 mentioned that metformin is contraindicated in chronic kidney disease.
Metformin is a good and useful drug. Not only is it one of the cheapest antidiabetic medications, it is the only one shown to reduce cardiovascular mortality rates in type 2 diabetes mellitus.
Although metformin is thought to increase the risk of lactic acidosis, a Cochrane review2 found that the incidence of lactic acidosis was only 4.3 cases per 100,000 patient-years in patients taking metformin, compared with 5.4 cases per 100,000 patient-years in patients not taking metformin. Furthermore, in a large registry of patients with type 2 diabetes and atherothrombosis,3 the rate of all-cause mortality was 24% lower in metformin users than in nonusers, and in those who had moderate renal impairment (creatinine clearance 30–59 mL/min/1.73 m2) the difference was 36%.3
A trial by Rachmani et al4 raised questions about the standard contraindications to metformin. The authors reviewed 393 patients who had at least one contraindication to metformin but who were receiving it anyway. Their serum creatinine levels ranged from 1.5 to 2.5 mg/dL. There were no cases of lactic acidosis reported. The patients were then randomized either to continue taking metformin or to stop taking it. At 2 years, the group that had stopped taking it had gained more weight, and their glycemic control was worse.
In the Cochrane analysis,2 although individual creatinine levels were not available, 53% of the studies reviewed did not exclude patients with serum creatinine levels higher than 1.5 mg/dL. This equated to 37,360 patient-years of metformin use in studies that included patients with chronic kidney disease, and did not lead to lactic acidosis.
Even though metformin’s US package insert says that it is contraindicated if the serum creatinine level is 1.5 mg/dL or higher in men or 1.4 mg/dL or higher in women or if the creatinine clearance is “abnormal,” in view of the available evidence, many countries (eg, the United Kingdom, Australia, the Netherlands) now allow metformin to be used in patients with glomerular filtration rates as low as 30 mL/min/1.73m2, with lower doses if the glomerular filtration rate is lower than 45.5
The current contraindication to metformin in chronic kidney disease needs to be reviewed. In poor countries like India, this cheap medicine may be the only option available for treating type 2 diabetes mellitus, and it remains the first-line therapy for type 2 diabetes mellitus as recommended by the International Diabetes Federation, the American Diabetes Association, and the European Association for the Study of Diabetes.5
- Sakhuja A, Hyland J, Simon JF. Managing advanced chronic kidney disease: a primary care guide. Cleve Clin J Med 2014; 81:289–299.
- Salpeter SR, Greyber E, Pasternak GA, Salpeter EE. Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitis. Cochrane Database Syst Rev 2010; 4:CD002967.
- Roussel R, Travert F, Pasquet B, et al; Reduction of Atherothrombosis for Continued Health (REACH) Registry Investigators. Metformin use and mortality among patients with diabetes and atherothrombosis. Arch Intern Med 2010; 170:1892–1899.
- Rachmani R, Slavachevski I, Levi Z, Zadok B, Kedar Y, Ravid M. Metformin in patients with type 2 diabetes mellitus: reconsideration of traditional contraindications. Eur J Intern Med 2002; 13:428–433.
- Inzucchi SE, Bergenstal RM, Buse JB, et al; American Diabetes Association (ADA); European Association for the Study of Diabetes (EASD). Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012; 35:1364–1379.
To the Editor: In their article about the care of patients with advanced chronic kidney disease, Sakhuja et al1 mentioned that metformin is contraindicated in chronic kidney disease.
Metformin is a good and useful drug. Not only is it one of the cheapest antidiabetic medications, it is the only one shown to reduce cardiovascular mortality rates in type 2 diabetes mellitus.
Although metformin is thought to increase the risk of lactic acidosis, a Cochrane review2 found that the incidence of lactic acidosis was only 4.3 cases per 100,000 patient-years in patients taking metformin, compared with 5.4 cases per 100,000 patient-years in patients not taking metformin. Furthermore, in a large registry of patients with type 2 diabetes and atherothrombosis,3 the rate of all-cause mortality was 24% lower in metformin users than in nonusers, and in those who had moderate renal impairment (creatinine clearance 30–59 mL/min/1.73 m2) the difference was 36%.3
A trial by Rachmani et al4 raised questions about the standard contraindications to metformin. The authors reviewed 393 patients who had at least one contraindication to metformin but who were receiving it anyway. Their serum creatinine levels ranged from 1.5 to 2.5 mg/dL. There were no cases of lactic acidosis reported. The patients were then randomized either to continue taking metformin or to stop taking it. At 2 years, the group that had stopped taking it had gained more weight, and their glycemic control was worse.
In the Cochrane analysis,2 although individual creatinine levels were not available, 53% of the studies reviewed did not exclude patients with serum creatinine levels higher than 1.5 mg/dL. This equated to 37,360 patient-years of metformin use in studies that included patients with chronic kidney disease, and did not lead to lactic acidosis.
Even though metformin’s US package insert says that it is contraindicated if the serum creatinine level is 1.5 mg/dL or higher in men or 1.4 mg/dL or higher in women or if the creatinine clearance is “abnormal,” in view of the available evidence, many countries (eg, the United Kingdom, Australia, the Netherlands) now allow metformin to be used in patients with glomerular filtration rates as low as 30 mL/min/1.73m2, with lower doses if the glomerular filtration rate is lower than 45.5
The current contraindication to metformin in chronic kidney disease needs to be reviewed. In poor countries like India, this cheap medicine may be the only option available for treating type 2 diabetes mellitus, and it remains the first-line therapy for type 2 diabetes mellitus as recommended by the International Diabetes Federation, the American Diabetes Association, and the European Association for the Study of Diabetes.5
To the Editor: In their article about the care of patients with advanced chronic kidney disease, Sakhuja et al1 mentioned that metformin is contraindicated in chronic kidney disease.
Metformin is a good and useful drug. Not only is it one of the cheapest antidiabetic medications, it is the only one shown to reduce cardiovascular mortality rates in type 2 diabetes mellitus.
Although metformin is thought to increase the risk of lactic acidosis, a Cochrane review2 found that the incidence of lactic acidosis was only 4.3 cases per 100,000 patient-years in patients taking metformin, compared with 5.4 cases per 100,000 patient-years in patients not taking metformin. Furthermore, in a large registry of patients with type 2 diabetes and atherothrombosis,3 the rate of all-cause mortality was 24% lower in metformin users than in nonusers, and in those who had moderate renal impairment (creatinine clearance 30–59 mL/min/1.73 m2) the difference was 36%.3
A trial by Rachmani et al4 raised questions about the standard contraindications to metformin. The authors reviewed 393 patients who had at least one contraindication to metformin but who were receiving it anyway. Their serum creatinine levels ranged from 1.5 to 2.5 mg/dL. There were no cases of lactic acidosis reported. The patients were then randomized either to continue taking metformin or to stop taking it. At 2 years, the group that had stopped taking it had gained more weight, and their glycemic control was worse.
In the Cochrane analysis,2 although individual creatinine levels were not available, 53% of the studies reviewed did not exclude patients with serum creatinine levels higher than 1.5 mg/dL. This equated to 37,360 patient-years of metformin use in studies that included patients with chronic kidney disease, and did not lead to lactic acidosis.
Even though metformin’s US package insert says that it is contraindicated if the serum creatinine level is 1.5 mg/dL or higher in men or 1.4 mg/dL or higher in women or if the creatinine clearance is “abnormal,” in view of the available evidence, many countries (eg, the United Kingdom, Australia, the Netherlands) now allow metformin to be used in patients with glomerular filtration rates as low as 30 mL/min/1.73m2, with lower doses if the glomerular filtration rate is lower than 45.5
The current contraindication to metformin in chronic kidney disease needs to be reviewed. In poor countries like India, this cheap medicine may be the only option available for treating type 2 diabetes mellitus, and it remains the first-line therapy for type 2 diabetes mellitus as recommended by the International Diabetes Federation, the American Diabetes Association, and the European Association for the Study of Diabetes.5
- Sakhuja A, Hyland J, Simon JF. Managing advanced chronic kidney disease: a primary care guide. Cleve Clin J Med 2014; 81:289–299.
- Salpeter SR, Greyber E, Pasternak GA, Salpeter EE. Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitis. Cochrane Database Syst Rev 2010; 4:CD002967.
- Roussel R, Travert F, Pasquet B, et al; Reduction of Atherothrombosis for Continued Health (REACH) Registry Investigators. Metformin use and mortality among patients with diabetes and atherothrombosis. Arch Intern Med 2010; 170:1892–1899.
- Rachmani R, Slavachevski I, Levi Z, Zadok B, Kedar Y, Ravid M. Metformin in patients with type 2 diabetes mellitus: reconsideration of traditional contraindications. Eur J Intern Med 2002; 13:428–433.
- Inzucchi SE, Bergenstal RM, Buse JB, et al; American Diabetes Association (ADA); European Association for the Study of Diabetes (EASD). Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012; 35:1364–1379.
- Sakhuja A, Hyland J, Simon JF. Managing advanced chronic kidney disease: a primary care guide. Cleve Clin J Med 2014; 81:289–299.
- Salpeter SR, Greyber E, Pasternak GA, Salpeter EE. Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitis. Cochrane Database Syst Rev 2010; 4:CD002967.
- Roussel R, Travert F, Pasquet B, et al; Reduction of Atherothrombosis for Continued Health (REACH) Registry Investigators. Metformin use and mortality among patients with diabetes and atherothrombosis. Arch Intern Med 2010; 170:1892–1899.
- Rachmani R, Slavachevski I, Levi Z, Zadok B, Kedar Y, Ravid M. Metformin in patients with type 2 diabetes mellitus: reconsideration of traditional contraindications. Eur J Intern Med 2002; 13:428–433.
- Inzucchi SE, Bergenstal RM, Buse JB, et al; American Diabetes Association (ADA); European Association for the Study of Diabetes (EASD). Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012; 35:1364–1379.
In reply: Why is metformin contraindicated in chronic kidney disease?
In Reply: We appreciate Dr. Imam’s comments regarding using metformin in those with chronic kidney disease.
The US Food and Drug Administration currently lists metformin as contraindicated in those with mild to moderate renal insufficiency, with serum creatinine levels greater than or equal to 1.5 mg/dL in males and greater than or equal to 1.4 mg/dL in females. This contraindication is based on the pharmacokinetics of the medication and, likely, the association of a similar medication, phenformin, with lactic acidosis, which eventually led to its withdrawal from the market. However, lactic acidosis is much less frequent with metformin than with phenformin.1
We agree that metformin is an invaluable medication for diabetes mellitus not requiring insulin. We also agree that lactic acidosis is rare, especially in those with mild renal insufficiency. However, lactic acidosis does occur in patients with chronic kidney disease while on metformin and, however rare, when it does occur it is a life-threatening event.2
The clearance of metformin is strongly dependent on kidney function,3 and therefore guidelines still recommend reducing the dose in those with moderate renal insufficiency and recommend considering stopping the medication in those with severe renal insufficiency—the population we were talking about in our article.4 We are aware of changes to the guidelines that have been made by various groups, and in many circumstances we ourselves take an individualized approach, weighing the risks and benefits of continued therapy with the patient and his or her primary care provider. That being said, we did not believe that such nuanced recommendations were appropriate for our article, especially since they are contrary to marketing restrictions for the drug.
- Bailey CJ, Turner RC. Metformin. N Engl J Med 1996; 334:574–579.
- Lalau JD, Race JM. Lactic acidosis in metformin-treated patients. Prognostic value of arterial lactate levels and plasma metformin concentrations. Drug Saf 1999; 20:377–384.
- Sambol NC, Chiang J, Lin ET, et al. Kidney function and age are both predictors of pharmacokinetics of metformin. J Clin Pharmacol 1995; 35:1094–1102.
- Sakhuja A, Hyland J, Simon JF. Managing advanced chronic kidney disease: a primary care guide. Cleve Clin J Med 2014; 81:289–299.
In Reply: We appreciate Dr. Imam’s comments regarding using metformin in those with chronic kidney disease.
The US Food and Drug Administration currently lists metformin as contraindicated in those with mild to moderate renal insufficiency, with serum creatinine levels greater than or equal to 1.5 mg/dL in males and greater than or equal to 1.4 mg/dL in females. This contraindication is based on the pharmacokinetics of the medication and, likely, the association of a similar medication, phenformin, with lactic acidosis, which eventually led to its withdrawal from the market. However, lactic acidosis is much less frequent with metformin than with phenformin.1
We agree that metformin is an invaluable medication for diabetes mellitus not requiring insulin. We also agree that lactic acidosis is rare, especially in those with mild renal insufficiency. However, lactic acidosis does occur in patients with chronic kidney disease while on metformin and, however rare, when it does occur it is a life-threatening event.2
The clearance of metformin is strongly dependent on kidney function,3 and therefore guidelines still recommend reducing the dose in those with moderate renal insufficiency and recommend considering stopping the medication in those with severe renal insufficiency—the population we were talking about in our article.4 We are aware of changes to the guidelines that have been made by various groups, and in many circumstances we ourselves take an individualized approach, weighing the risks and benefits of continued therapy with the patient and his or her primary care provider. That being said, we did not believe that such nuanced recommendations were appropriate for our article, especially since they are contrary to marketing restrictions for the drug.
In Reply: We appreciate Dr. Imam’s comments regarding using metformin in those with chronic kidney disease.
The US Food and Drug Administration currently lists metformin as contraindicated in those with mild to moderate renal insufficiency, with serum creatinine levels greater than or equal to 1.5 mg/dL in males and greater than or equal to 1.4 mg/dL in females. This contraindication is based on the pharmacokinetics of the medication and, likely, the association of a similar medication, phenformin, with lactic acidosis, which eventually led to its withdrawal from the market. However, lactic acidosis is much less frequent with metformin than with phenformin.1
We agree that metformin is an invaluable medication for diabetes mellitus not requiring insulin. We also agree that lactic acidosis is rare, especially in those with mild renal insufficiency. However, lactic acidosis does occur in patients with chronic kidney disease while on metformin and, however rare, when it does occur it is a life-threatening event.2
The clearance of metformin is strongly dependent on kidney function,3 and therefore guidelines still recommend reducing the dose in those with moderate renal insufficiency and recommend considering stopping the medication in those with severe renal insufficiency—the population we were talking about in our article.4 We are aware of changes to the guidelines that have been made by various groups, and in many circumstances we ourselves take an individualized approach, weighing the risks and benefits of continued therapy with the patient and his or her primary care provider. That being said, we did not believe that such nuanced recommendations were appropriate for our article, especially since they are contrary to marketing restrictions for the drug.
- Bailey CJ, Turner RC. Metformin. N Engl J Med 1996; 334:574–579.
- Lalau JD, Race JM. Lactic acidosis in metformin-treated patients. Prognostic value of arterial lactate levels and plasma metformin concentrations. Drug Saf 1999; 20:377–384.
- Sambol NC, Chiang J, Lin ET, et al. Kidney function and age are both predictors of pharmacokinetics of metformin. J Clin Pharmacol 1995; 35:1094–1102.
- Sakhuja A, Hyland J, Simon JF. Managing advanced chronic kidney disease: a primary care guide. Cleve Clin J Med 2014; 81:289–299.
- Bailey CJ, Turner RC. Metformin. N Engl J Med 1996; 334:574–579.
- Lalau JD, Race JM. Lactic acidosis in metformin-treated patients. Prognostic value of arterial lactate levels and plasma metformin concentrations. Drug Saf 1999; 20:377–384.
- Sambol NC, Chiang J, Lin ET, et al. Kidney function and age are both predictors of pharmacokinetics of metformin. J Clin Pharmacol 1995; 35:1094–1102.
- Sakhuja A, Hyland J, Simon JF. Managing advanced chronic kidney disease: a primary care guide. Cleve Clin J Med 2014; 81:289–299.