User login
“Something Abnormal” on a Chest X-ray
ANSWER
The radiograph demonstrates a fairly large (4 x 6 cm) right paratracheal mass of unclear etiology. This type of finding warrants further evaluation with contrasted CT.
Fortunately for this patient, a subsequent study demonstrated a slightly enlarged thyroid gland. This correlated with the radiographic
finding.

ANSWER
The radiograph demonstrates a fairly large (4 x 6 cm) right paratracheal mass of unclear etiology. This type of finding warrants further evaluation with contrasted CT.
Fortunately for this patient, a subsequent study demonstrated a slightly enlarged thyroid gland. This correlated with the radiographic
finding.
ANSWER
The radiograph demonstrates a fairly large (4 x 6 cm) right paratracheal mass of unclear etiology. This type of finding warrants further evaluation with contrasted CT.
Fortunately for this patient, a subsequent study demonstrated a slightly enlarged thyroid gland. This correlated with the radiographic
finding.
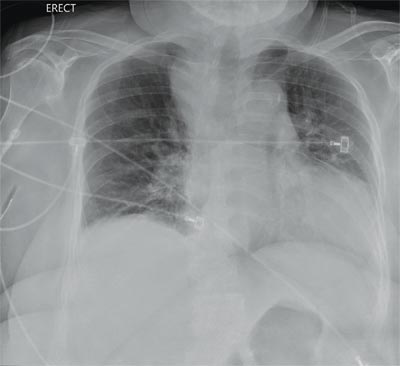
You are doing preoperative orders on a patient scheduled for surgery tomorrow morning. The patient is a 75-year-old woman who was admitted with an acute left subdural hematoma after sustaining a ground-level fall. Her medical history is significant for hypertension and diabetes. Social history is unremarkable. She is neurologically intact except for occasional confusion and aphasia. She moves all her extremities well. As you review her lab results, one of the nurses mentions that the radiology department called about “something abnormal” on the patient’s chest radiograph. You pull up the patient’s portable chest radiograph on the computer to review. What is your impression?
Itchy Lesion Heralds Pervasive Problem
ANSWER
The correct answer is pityriasis rosea (choice “b”), a common and very distinctive eruption related to human herpesvirus 6 and 7.
Allergic reaction to methotrexate (choice “a”), while far from unknown, does not resemble pityriasis rosea. It also would not be limited to such a relatively small area.
Pityriasis rosea is often designated as “fungal infection” (choice “c”) by the uninitiated. However, the lesions of dermatophytosis would be round, with a leading scaly edge, and unlikely to be found in this distribution.
Secondary syphilis (choice “d”) is a major item in the pityriasis rosea differential, but it almost always involves the palms and soles and the lesions would be round (not oval) scaly brown papules. Furthermore, assuming we have an honest patient, we’re also missing a source for sexually transmitted infection.
DISCUSSION
One could hardly ask for a more classic case of pityriasis rosea (PR), which primarily affects patients ages 14 to 40. Alas, that being said, one cannot depend on seeing all these clues in every PR patient.
For example, the herald patch (also known as the mother patch) is missing in at least half of cases. In others, the lesions are smaller, sparser, and more papular (especially in young black patients). The condition may even be confined to intertriginous areas (eg, the groin and/or axillae); this is known as inverse PR.
While salmon-colored scaly lesions are considered a classic presentation, PR can present with darker ovoid macules that have minimal scale and, rarely, become bullous. Involvement above the neck is rare.
What is consistent and dependable among signs of PR is the centripetal scale, seen even in the smallest lesions. This scale is so fine that the old dermatology texts called it “cigarette paper” scale or “scurf.”
After decades of speculation, researchers finally provided strong evidence of the probable cause of PR: replication of human herpesvirus 6 and 7, present in mono-
nuclear cells of lesional skin. Though universally acquired in childhood, these viruses are thought to remain latent until reactivated, leading to viremia.
Itching can be moderately severe in a minority of cases. Most patients, such as this one, are not bothered much by the condition once they understand its self-limited nature. They usually are not happy, however, to learn that it could persist for nine weeks or more, whether treated or not.
UV light exposure can be helpful in hastening PR’s departure, and topical corticosteroids (class III or IV; eg, triamcinolone 0.1% cream) can help control the itching. Neither oral nor topical antihistamines will help, since PR is not a histamine-mediated problem.
If the diagnosis is in doubt, a punch biopsy could at least rule out the more serious items in the differential, which include syphilis, drug rash, and psoriasis. In cases in which fungal origin is a possibility, a quick KOH prep will settle the issue. However, it must be remembered that one doesn’t just “get” a fungal infection. There has to be a source (animal, child), and that source is usually identified with minimal history taking.
ANSWER
The correct answer is pityriasis rosea (choice “b”), a common and very distinctive eruption related to human herpesvirus 6 and 7.
Allergic reaction to methotrexate (choice “a”), while far from unknown, does not resemble pityriasis rosea. It also would not be limited to such a relatively small area.
Pityriasis rosea is often designated as “fungal infection” (choice “c”) by the uninitiated. However, the lesions of dermatophytosis would be round, with a leading scaly edge, and unlikely to be found in this distribution.
Secondary syphilis (choice “d”) is a major item in the pityriasis rosea differential, but it almost always involves the palms and soles and the lesions would be round (not oval) scaly brown papules. Furthermore, assuming we have an honest patient, we’re also missing a source for sexually transmitted infection.
DISCUSSION
One could hardly ask for a more classic case of pityriasis rosea (PR), which primarily affects patients ages 14 to 40. Alas, that being said, one cannot depend on seeing all these clues in every PR patient.
For example, the herald patch (also known as the mother patch) is missing in at least half of cases. In others, the lesions are smaller, sparser, and more papular (especially in young black patients). The condition may even be confined to intertriginous areas (eg, the groin and/or axillae); this is known as inverse PR.
While salmon-colored scaly lesions are considered a classic presentation, PR can present with darker ovoid macules that have minimal scale and, rarely, become bullous. Involvement above the neck is rare.
What is consistent and dependable among signs of PR is the centripetal scale, seen even in the smallest lesions. This scale is so fine that the old dermatology texts called it “cigarette paper” scale or “scurf.”
After decades of speculation, researchers finally provided strong evidence of the probable cause of PR: replication of human herpesvirus 6 and 7, present in mono-
nuclear cells of lesional skin. Though universally acquired in childhood, these viruses are thought to remain latent until reactivated, leading to viremia.
Itching can be moderately severe in a minority of cases. Most patients, such as this one, are not bothered much by the condition once they understand its self-limited nature. They usually are not happy, however, to learn that it could persist for nine weeks or more, whether treated or not.
UV light exposure can be helpful in hastening PR’s departure, and topical corticosteroids (class III or IV; eg, triamcinolone 0.1% cream) can help control the itching. Neither oral nor topical antihistamines will help, since PR is not a histamine-mediated problem.
If the diagnosis is in doubt, a punch biopsy could at least rule out the more serious items in the differential, which include syphilis, drug rash, and psoriasis. In cases in which fungal origin is a possibility, a quick KOH prep will settle the issue. However, it must be remembered that one doesn’t just “get” a fungal infection. There has to be a source (animal, child), and that source is usually identified with minimal history taking.
ANSWER
The correct answer is pityriasis rosea (choice “b”), a common and very distinctive eruption related to human herpesvirus 6 and 7.
Allergic reaction to methotrexate (choice “a”), while far from unknown, does not resemble pityriasis rosea. It also would not be limited to such a relatively small area.
Pityriasis rosea is often designated as “fungal infection” (choice “c”) by the uninitiated. However, the lesions of dermatophytosis would be round, with a leading scaly edge, and unlikely to be found in this distribution.
Secondary syphilis (choice “d”) is a major item in the pityriasis rosea differential, but it almost always involves the palms and soles and the lesions would be round (not oval) scaly brown papules. Furthermore, assuming we have an honest patient, we’re also missing a source for sexually transmitted infection.
DISCUSSION
One could hardly ask for a more classic case of pityriasis rosea (PR), which primarily affects patients ages 14 to 40. Alas, that being said, one cannot depend on seeing all these clues in every PR patient.
For example, the herald patch (also known as the mother patch) is missing in at least half of cases. In others, the lesions are smaller, sparser, and more papular (especially in young black patients). The condition may even be confined to intertriginous areas (eg, the groin and/or axillae); this is known as inverse PR.
While salmon-colored scaly lesions are considered a classic presentation, PR can present with darker ovoid macules that have minimal scale and, rarely, become bullous. Involvement above the neck is rare.
What is consistent and dependable among signs of PR is the centripetal scale, seen even in the smallest lesions. This scale is so fine that the old dermatology texts called it “cigarette paper” scale or “scurf.”
After decades of speculation, researchers finally provided strong evidence of the probable cause of PR: replication of human herpesvirus 6 and 7, present in mono-
nuclear cells of lesional skin. Though universally acquired in childhood, these viruses are thought to remain latent until reactivated, leading to viremia.
Itching can be moderately severe in a minority of cases. Most patients, such as this one, are not bothered much by the condition once they understand its self-limited nature. They usually are not happy, however, to learn that it could persist for nine weeks or more, whether treated or not.
UV light exposure can be helpful in hastening PR’s departure, and topical corticosteroids (class III or IV; eg, triamcinolone 0.1% cream) can help control the itching. Neither oral nor topical antihistamines will help, since PR is not a histamine-mediated problem.
If the diagnosis is in doubt, a punch biopsy could at least rule out the more serious items in the differential, which include syphilis, drug rash, and psoriasis. In cases in which fungal origin is a possibility, a quick KOH prep will settle the issue. However, it must be remembered that one doesn’t just “get” a fungal infection. There has to be a source (animal, child), and that source is usually identified with minimal history taking.
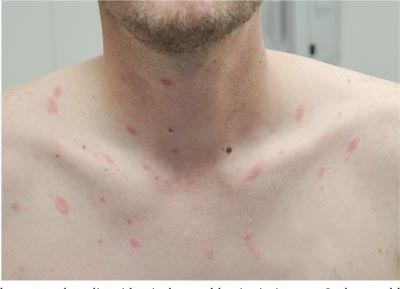
Two weeks ago, an itchy rash appeared on a man’s back before spreading to his chest and neck. He has never experienced anything like it before, and no one else in his household is similarly affected. He denies night sweats, fever, and malaise but reports that he was recently diagnosed with rheumatoid arthritis. His rheumatologist started him on methotrexate (12.5 mg/wk) after extensive labwork (complete blood count, complete metabolic panel, and hepatitis profile) was performed. He denies any history of high-risk sexual behavior or exposure, exposure to animals or children, or history of foreign travel. The patient, who appears well, is afebrile and in no distress. The original lesion, on his upper left back, is distinctly pinkish brown and round, with an odd fine scale around its inner rim, and measures about 3 cm in diameter. Elsewhere, examination reveals about 15 more lesions. All are oval but similarly pinkish brown, averaging about 2 cm in their long axis. These smaller lesions form a necklace-like configuration, paralleling the natural skin lines of the neck. Each lesion has central scaling identical to that of the original back lesion. Examination of the patient’s palms and soles fails to reveal any cutaneous abnormalities. Likewise, examination of the oral cavity is normal. No palpable nodes are felt around the neck, in the axillae, or in the groin.

Eikenella corrodens Septic Hip Arthritis in a Healthy Adult Treated With Arthroscopic Irrigation and Debridement
A summary of the new ACOG report on neonatal brachial plexus palsy. Part 1: Can it be predicted?
Neonatal brachial plexus palsy (NBPP) after a delivery involving shoulder dystocia is not only a clinical disaster—it constitutes the second largest category of litigation in obstetrics.1
Lawsuits that center on NBPP often feature plaintiff expert witnesses who claim that the only way a permanent brachial plexus injury can occur is by a clinician applying “excessive” traction on the fetal head during delivery. The same experts often claim that the mother had multiple risk factors for shoulder dystocia and should never have been allowed a trial of labor in the first place.
The jury is left suspecting that the NBPP was a disaster waiting to happen, with warning signs that were ignored by the clinician. Jurors also may be convinced that, when the dystocia occurred, the defendant handled it badly, causing a severe, lifelong injury to the beautiful child whose images they are shown in the courtroom.
But this scenario is far from accurate.
ACOG publishes new guidance on NBPPThe American College of Obstetricians and Gynecologists (ACOG) periodically issues practice bulletins on the subject of shoulder dystocia, the most recent one written in 2002 and reaffirmed in 2013.2 These bulletins are, of necessity, relatively brief summaries of current thinking about the causes, pathophysiology, treatment, and preventability of shoulder dystocia and associated brachial plexus injuries.
In 2011, James Breeden, MD, then president-elect of ACOG, called for formation of a task force on NBPP. The task force’s report, Neonatal Brachial Plexus Palsy,3 was published earlier this year and represents ACOG’s official position on the important—but still controversial—subjects of shoulder dystocia and NBPP. This report should serve not only to help clinicians better understand and manage these entities but also as a foundational document in the prolific and complex medicolegal suits involving them.
Given the length of this report, however, a concise summary of the key takeaways is in order.
NBPP and shoulder dystocia are not always linked
Early in the report, ACOG presents three very important statements, all of which challenge claims that are frequently made by plaintiffs in brachial plexus injury cases:
- NBPP can occur without concomitant, clinically recognizable shoulder dystocia, although it often is associated with shoulder dystocia.
- In the presence of shoulder dystocia, all ancillary maneuvers necessarily increase strain on the brachial plexus, no matter how expertly the maneuvers are performed.
- Recent multidisciplinary research now indicates that the existence of NBPP after birth does not prove that exogenous forces are the sole cause of this injury.
These findings raise a number of questions, including:
- Can NBPP be predicted and prevented?
- What is the pathophysiologic mechanism for NBPP with and without shoulder dystocia?
- Are there specific interventions that may reduce the frequency of NBPP?
In Part 1 of this article, I summarize ACOG data on whether and how NBPP might be predicted. Part 2, to follow in October 2014, will discuss the pathophysiologic mechanism for NBPP and discuss potential interventions.
The data on NBPP without shoulder dystocia
The results of 12 reports published between 1990 and 2011 describe NBPP (temporary and persistent) that occurred without concomitant shoulder dystocia. These reports indicate that 46% of NBPP cases occurred without documented shoulder dystocia (0.9 cases/1,000 births).
Persistent NBPP. Two of these reports provide data on persistent NBPP without shoulder dystocia. Even when injury to the brachial plexus was documented as lasting more than 1 year, 26% of cases occurred in the absence of documented shoulder dystocia.
NBPP sometimes can occur during cesarean delivery. Four studies evaluated more than 240,000 births and found a rate of NBPP with cesarean delivery ranging from 0.3 to 1.5 cases per 1,000 live births.
All of these studies are described in the ACOG report.
When NBPP is related to shoulder dystocia
Shoulder dystocia may occur when there is a lack of fit of the transverse diameter of the fetal shoulders through the different pelvic diameters the shoulders encounter as they descend through the pelvis during the course of labor and delivery. This lack of fit can be related to excessive size of the fetal shoulders, inadequacy of pelvic dimensions to allow passage of a given fetus, or both. Abnormalities of fetal anatomy, fetal presentation, and soft tissue obstruction are rarely the cause of shoulder dystocia.
The difference between anterior shoulder obstruction behind the symphysis pubis and posterior shoulder obstruction from arrest at the level of the sacral promontory also is discussed in the ACOG report. In both cases, it is this obstruction of the affected shoulder while the long axis of the body continues to be pushed downward that widens the angle between the neck and impacted shoulder and stretches the brachial plexus.
The ACOG report acknowledges that may cases of NBPP do occur in conjunction with shoulder dystocia and that the same biomechanical factors that predispose a fetus to develop NBPP are associated with shoulder dystocia as well. However, the report takes pains to point out that the frequent conjunction of these two entities—NBPP and shoulder dystocia—may lead to an “erroneous retrospective inference of causation.”
Risk and predictive factors
The ACOG report states: “Various risk factors have been described in association with NBPP. Overall, however, these risk factors have not been shown to be statistically reliable or clinically useful predictors for...NBPP.”
For example, fetal macrosomia, defined as a birth weight of 4,000 g or more, has been reported as a risk factor for NBPP either alone or in conjunction with maternal diabetes. Although NBPP does occur more frequently as birth weight increases, seven studies over the past 20 years have shown that most cases of NBPP occur in infants of mothers without diabetes and in infants who weigh less than 4,000 g.
Other studies have shown that, if cesarean delivery were performed in cases of suspected macrosomia, it would have only a limited effect on reducing the incidence of NBPP. Specifically, in women with diabetes who have an estimated fetal weight of more than 4,500 g, the positive predictive value for NBPP is only 5%. Without maternal diabetes, that figure is less than 2%.
Estimating fetal weight by ultrasound does not significantly enhance our ability to predict NBPP. Ultrasound estimates of birth weight usually fall within 15% to 20% of actual birth weight, and the sensitivity of ultrasound in detecting birth weights more than 4,500 g is only 40%.
Therefore, ultrasound estimates of birth weight are of limited utility for contemporaneous clinical management. Furthermore, no data exist to support the claim that estimated fetal weight can be used prophylactically to reduce the incidence of NBPP.
Recurrent shoulder dystocia may be predictive of future NBPP
Whether studied alone or with NBPP, risk factors for shoulder dystocia are not reliable predictors of its occurrence. This is not the case, however, for recurrent shoulder dystocia, where the risk of neonatal brachial plexus palsy can be as high as 4.5%, compared with 1% to 2% for a first episode of shoulder dystocia.
NBPP is a rare phenomenon
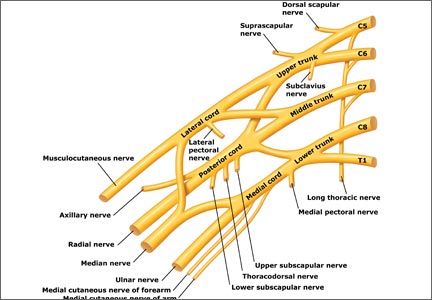
The frequency of NBPP is “rare,” according to the ACOG report, which cites a rate of 1.5 cases for every 1,000 births. Favorable outcomes with complete recovery are estimated to range from 50% to 80%.3
Brachial plexus injuries are classically defined as Erb’s palsy—involving C5 and C6 nerve roots—or Klumpke’s palsy, in which there is damage to the C8 and T1 nerve roots.
Erb’s palsy is recognizable by the characteristic “waiter’s tip” position of the hand, which is caused by muscle imbalance in the shoulder and upper arm. Most NBPP injuries are Erb’s palsy, which affect 1.2 infants in every 1,000 births.
Klumpke’s palsy results in weakness of the hand and medial forearm muscles. It affects 0.05 infants in every 1,000 births. The remaining cases involve a combination of the two types of palsy.
These injuries can be temporary, resolving by 12 months after birth, or permanent. The rate of persistence of NBPP at 12 months ranges from 3% to 33%.
Can clinician maneuvers increase the likelihood of NBPP?
The ACOG report addresses the direction and angle of clinician traction at delivery. The report confirms what clinicians generally have been taught: The application of fundal pressure during a delivery in which shoulder dystocia is recognized can exacerbate shoulder impaction and can lead to an increased risk of NBPP.
Traction applied by the clinician and lateral bending of the fetal neck often are implicated as causative factors of NBPP. However, ACOG presents evidence that NBPP can occur entirely unrelated to clinician traction. The report cites studies involving both transient and persistent NBPP in fetuses delivered vaginally without evident shoulder dystocia. The same types of injury are sometimes seen in fetuses delivered by cesarean, as has been mentioned.
The report goes on to state:
Recommendations for practice
At the close of its second chapter (“Risk and predictive factors”), the ACOG report offers the same official recommendations that appear in its current practice bulletin on shoulder dystocia. It notes that there are three clinical situations in which it may be prudent to alter usual obstetric management, with an aim of reducing the risk of shoulder dystocia and NBPP:
- when fetal macrosomia is suspected, with fetal weight estimated to exceed 5,000 g in a woman without diabetes or 4,500 g in a woman with diabetes
- when the mother has a history of recognized shoulder dystocia, especially when neonatal injury was severe
- when midpelvic operative vaginal delivery is contemplated with a fetus estimated to weigh more than 4,000 g.
It is interesting to note that these recommendations are made, according to the report, “notwithstanding the unreliability of specific risk factors to predict NBPP or clinically apparent shoulder dystocia in a specific case.” The report further adds:
More to come
For ACOG’s conclusions on the pathophysiology and causation of NBPP, with a view toward formulating specific protective interventions, see Part 2 of this article, which will appear in the October 2014 issue of OBG Management.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Physician Insurers Association of America. http://www.piaa.us. Accessed August 21, 2014.
2. American College of Obstetricians and Gynecologists. Practice Bulletin #40: shoulder dystocia. Obstet Gynecol. 2002;100(5 pt 1):1045–1050.
3. American College of Obstetricians and Gynecologists. Executive summary: neonatal brachial plexus palsy. Report of the American College of Obstetricians and Gynecologists’ Task Force on neonatal brachial plexus palsy. Obstet Gynecol. 2014;123(4):902–904.

Henry M. Lerner, MD
Dr. Lerner is Assistant Clinical Professor of Obstetrics and Gynecology at Harvard Medical School in Boston, Massachusetts.
Dr. Lerner reports that he has been a consultant to the Sullivan Group, which provides patient safety services to labor and delivery units and obstetric practices.
Henry M. Lerner, MD
Dr. Lerner is Assistant Clinical Professor of Obstetrics and Gynecology at Harvard Medical School in Boston, Massachusetts.
Dr. Lerner reports that he has been a consultant to the Sullivan Group, which provides patient safety services to labor and delivery units and obstetric practices.
Henry M. Lerner, MD
Dr. Lerner is Assistant Clinical Professor of Obstetrics and Gynecology at Harvard Medical School in Boston, Massachusetts.
Dr. Lerner reports that he has been a consultant to the Sullivan Group, which provides patient safety services to labor and delivery units and obstetric practices.
Neonatal brachial plexus palsy (NBPP) after a delivery involving shoulder dystocia is not only a clinical disaster—it constitutes the second largest category of litigation in obstetrics.1
Lawsuits that center on NBPP often feature plaintiff expert witnesses who claim that the only way a permanent brachial plexus injury can occur is by a clinician applying “excessive” traction on the fetal head during delivery. The same experts often claim that the mother had multiple risk factors for shoulder dystocia and should never have been allowed a trial of labor in the first place.
The jury is left suspecting that the NBPP was a disaster waiting to happen, with warning signs that were ignored by the clinician. Jurors also may be convinced that, when the dystocia occurred, the defendant handled it badly, causing a severe, lifelong injury to the beautiful child whose images they are shown in the courtroom.
But this scenario is far from accurate.
ACOG publishes new guidance on NBPPThe American College of Obstetricians and Gynecologists (ACOG) periodically issues practice bulletins on the subject of shoulder dystocia, the most recent one written in 2002 and reaffirmed in 2013.2 These bulletins are, of necessity, relatively brief summaries of current thinking about the causes, pathophysiology, treatment, and preventability of shoulder dystocia and associated brachial plexus injuries.
In 2011, James Breeden, MD, then president-elect of ACOG, called for formation of a task force on NBPP. The task force’s report, Neonatal Brachial Plexus Palsy,3 was published earlier this year and represents ACOG’s official position on the important—but still controversial—subjects of shoulder dystocia and NBPP. This report should serve not only to help clinicians better understand and manage these entities but also as a foundational document in the prolific and complex medicolegal suits involving them.
Given the length of this report, however, a concise summary of the key takeaways is in order.
NBPP and shoulder dystocia are not always linked
Early in the report, ACOG presents three very important statements, all of which challenge claims that are frequently made by plaintiffs in brachial plexus injury cases:
- NBPP can occur without concomitant, clinically recognizable shoulder dystocia, although it often is associated with shoulder dystocia.
- In the presence of shoulder dystocia, all ancillary maneuvers necessarily increase strain on the brachial plexus, no matter how expertly the maneuvers are performed.
- Recent multidisciplinary research now indicates that the existence of NBPP after birth does not prove that exogenous forces are the sole cause of this injury.
These findings raise a number of questions, including:
- Can NBPP be predicted and prevented?
- What is the pathophysiologic mechanism for NBPP with and without shoulder dystocia?
- Are there specific interventions that may reduce the frequency of NBPP?
In Part 1 of this article, I summarize ACOG data on whether and how NBPP might be predicted. Part 2, to follow in October 2014, will discuss the pathophysiologic mechanism for NBPP and discuss potential interventions.
The data on NBPP without shoulder dystocia
The results of 12 reports published between 1990 and 2011 describe NBPP (temporary and persistent) that occurred without concomitant shoulder dystocia. These reports indicate that 46% of NBPP cases occurred without documented shoulder dystocia (0.9 cases/1,000 births).
Persistent NBPP. Two of these reports provide data on persistent NBPP without shoulder dystocia. Even when injury to the brachial plexus was documented as lasting more than 1 year, 26% of cases occurred in the absence of documented shoulder dystocia.
NBPP sometimes can occur during cesarean delivery. Four studies evaluated more than 240,000 births and found a rate of NBPP with cesarean delivery ranging from 0.3 to 1.5 cases per 1,000 live births.
All of these studies are described in the ACOG report.
When NBPP is related to shoulder dystocia
Shoulder dystocia may occur when there is a lack of fit of the transverse diameter of the fetal shoulders through the different pelvic diameters the shoulders encounter as they descend through the pelvis during the course of labor and delivery. This lack of fit can be related to excessive size of the fetal shoulders, inadequacy of pelvic dimensions to allow passage of a given fetus, or both. Abnormalities of fetal anatomy, fetal presentation, and soft tissue obstruction are rarely the cause of shoulder dystocia.
The difference between anterior shoulder obstruction behind the symphysis pubis and posterior shoulder obstruction from arrest at the level of the sacral promontory also is discussed in the ACOG report. In both cases, it is this obstruction of the affected shoulder while the long axis of the body continues to be pushed downward that widens the angle between the neck and impacted shoulder and stretches the brachial plexus.
The ACOG report acknowledges that may cases of NBPP do occur in conjunction with shoulder dystocia and that the same biomechanical factors that predispose a fetus to develop NBPP are associated with shoulder dystocia as well. However, the report takes pains to point out that the frequent conjunction of these two entities—NBPP and shoulder dystocia—may lead to an “erroneous retrospective inference of causation.”
Risk and predictive factors
The ACOG report states: “Various risk factors have been described in association with NBPP. Overall, however, these risk factors have not been shown to be statistically reliable or clinically useful predictors for...NBPP.”
For example, fetal macrosomia, defined as a birth weight of 4,000 g or more, has been reported as a risk factor for NBPP either alone or in conjunction with maternal diabetes. Although NBPP does occur more frequently as birth weight increases, seven studies over the past 20 years have shown that most cases of NBPP occur in infants of mothers without diabetes and in infants who weigh less than 4,000 g.
Other studies have shown that, if cesarean delivery were performed in cases of suspected macrosomia, it would have only a limited effect on reducing the incidence of NBPP. Specifically, in women with diabetes who have an estimated fetal weight of more than 4,500 g, the positive predictive value for NBPP is only 5%. Without maternal diabetes, that figure is less than 2%.
Estimating fetal weight by ultrasound does not significantly enhance our ability to predict NBPP. Ultrasound estimates of birth weight usually fall within 15% to 20% of actual birth weight, and the sensitivity of ultrasound in detecting birth weights more than 4,500 g is only 40%.
Therefore, ultrasound estimates of birth weight are of limited utility for contemporaneous clinical management. Furthermore, no data exist to support the claim that estimated fetal weight can be used prophylactically to reduce the incidence of NBPP.
Recurrent shoulder dystocia may be predictive of future NBPP
Whether studied alone or with NBPP, risk factors for shoulder dystocia are not reliable predictors of its occurrence. This is not the case, however, for recurrent shoulder dystocia, where the risk of neonatal brachial plexus palsy can be as high as 4.5%, compared with 1% to 2% for a first episode of shoulder dystocia.
NBPP is a rare phenomenon
The frequency of NBPP is “rare,” according to the ACOG report, which cites a rate of 1.5 cases for every 1,000 births. Favorable outcomes with complete recovery are estimated to range from 50% to 80%.3
Brachial plexus injuries are classically defined as Erb’s palsy—involving C5 and C6 nerve roots—or Klumpke’s palsy, in which there is damage to the C8 and T1 nerve roots.
Erb’s palsy is recognizable by the characteristic “waiter’s tip” position of the hand, which is caused by muscle imbalance in the shoulder and upper arm. Most NBPP injuries are Erb’s palsy, which affect 1.2 infants in every 1,000 births.
Klumpke’s palsy results in weakness of the hand and medial forearm muscles. It affects 0.05 infants in every 1,000 births. The remaining cases involve a combination of the two types of palsy.
These injuries can be temporary, resolving by 12 months after birth, or permanent. The rate of persistence of NBPP at 12 months ranges from 3% to 33%.
Can clinician maneuvers increase the likelihood of NBPP?
The ACOG report addresses the direction and angle of clinician traction at delivery. The report confirms what clinicians generally have been taught: The application of fundal pressure during a delivery in which shoulder dystocia is recognized can exacerbate shoulder impaction and can lead to an increased risk of NBPP.
Traction applied by the clinician and lateral bending of the fetal neck often are implicated as causative factors of NBPP. However, ACOG presents evidence that NBPP can occur entirely unrelated to clinician traction. The report cites studies involving both transient and persistent NBPP in fetuses delivered vaginally without evident shoulder dystocia. The same types of injury are sometimes seen in fetuses delivered by cesarean, as has been mentioned.
The report goes on to state:
Recommendations for practice
At the close of its second chapter (“Risk and predictive factors”), the ACOG report offers the same official recommendations that appear in its current practice bulletin on shoulder dystocia. It notes that there are three clinical situations in which it may be prudent to alter usual obstetric management, with an aim of reducing the risk of shoulder dystocia and NBPP:
- when fetal macrosomia is suspected, with fetal weight estimated to exceed 5,000 g in a woman without diabetes or 4,500 g in a woman with diabetes
- when the mother has a history of recognized shoulder dystocia, especially when neonatal injury was severe
- when midpelvic operative vaginal delivery is contemplated with a fetus estimated to weigh more than 4,000 g.
It is interesting to note that these recommendations are made, according to the report, “notwithstanding the unreliability of specific risk factors to predict NBPP or clinically apparent shoulder dystocia in a specific case.” The report further adds:
More to come
For ACOG’s conclusions on the pathophysiology and causation of NBPP, with a view toward formulating specific protective interventions, see Part 2 of this article, which will appear in the October 2014 issue of OBG Management.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Neonatal brachial plexus palsy (NBPP) after a delivery involving shoulder dystocia is not only a clinical disaster—it constitutes the second largest category of litigation in obstetrics.1
Lawsuits that center on NBPP often feature plaintiff expert witnesses who claim that the only way a permanent brachial plexus injury can occur is by a clinician applying “excessive” traction on the fetal head during delivery. The same experts often claim that the mother had multiple risk factors for shoulder dystocia and should never have been allowed a trial of labor in the first place.
The jury is left suspecting that the NBPP was a disaster waiting to happen, with warning signs that were ignored by the clinician. Jurors also may be convinced that, when the dystocia occurred, the defendant handled it badly, causing a severe, lifelong injury to the beautiful child whose images they are shown in the courtroom.
But this scenario is far from accurate.
ACOG publishes new guidance on NBPPThe American College of Obstetricians and Gynecologists (ACOG) periodically issues practice bulletins on the subject of shoulder dystocia, the most recent one written in 2002 and reaffirmed in 2013.2 These bulletins are, of necessity, relatively brief summaries of current thinking about the causes, pathophysiology, treatment, and preventability of shoulder dystocia and associated brachial plexus injuries.
In 2011, James Breeden, MD, then president-elect of ACOG, called for formation of a task force on NBPP. The task force’s report, Neonatal Brachial Plexus Palsy,3 was published earlier this year and represents ACOG’s official position on the important—but still controversial—subjects of shoulder dystocia and NBPP. This report should serve not only to help clinicians better understand and manage these entities but also as a foundational document in the prolific and complex medicolegal suits involving them.
Given the length of this report, however, a concise summary of the key takeaways is in order.
NBPP and shoulder dystocia are not always linked
Early in the report, ACOG presents three very important statements, all of which challenge claims that are frequently made by plaintiffs in brachial plexus injury cases:
- NBPP can occur without concomitant, clinically recognizable shoulder dystocia, although it often is associated with shoulder dystocia.
- In the presence of shoulder dystocia, all ancillary maneuvers necessarily increase strain on the brachial plexus, no matter how expertly the maneuvers are performed.
- Recent multidisciplinary research now indicates that the existence of NBPP after birth does not prove that exogenous forces are the sole cause of this injury.
These findings raise a number of questions, including:
- Can NBPP be predicted and prevented?
- What is the pathophysiologic mechanism for NBPP with and without shoulder dystocia?
- Are there specific interventions that may reduce the frequency of NBPP?
In Part 1 of this article, I summarize ACOG data on whether and how NBPP might be predicted. Part 2, to follow in October 2014, will discuss the pathophysiologic mechanism for NBPP and discuss potential interventions.
The data on NBPP without shoulder dystocia
The results of 12 reports published between 1990 and 2011 describe NBPP (temporary and persistent) that occurred without concomitant shoulder dystocia. These reports indicate that 46% of NBPP cases occurred without documented shoulder dystocia (0.9 cases/1,000 births).
Persistent NBPP. Two of these reports provide data on persistent NBPP without shoulder dystocia. Even when injury to the brachial plexus was documented as lasting more than 1 year, 26% of cases occurred in the absence of documented shoulder dystocia.
NBPP sometimes can occur during cesarean delivery. Four studies evaluated more than 240,000 births and found a rate of NBPP with cesarean delivery ranging from 0.3 to 1.5 cases per 1,000 live births.
All of these studies are described in the ACOG report.
When NBPP is related to shoulder dystocia
Shoulder dystocia may occur when there is a lack of fit of the transverse diameter of the fetal shoulders through the different pelvic diameters the shoulders encounter as they descend through the pelvis during the course of labor and delivery. This lack of fit can be related to excessive size of the fetal shoulders, inadequacy of pelvic dimensions to allow passage of a given fetus, or both. Abnormalities of fetal anatomy, fetal presentation, and soft tissue obstruction are rarely the cause of shoulder dystocia.
The difference between anterior shoulder obstruction behind the symphysis pubis and posterior shoulder obstruction from arrest at the level of the sacral promontory also is discussed in the ACOG report. In both cases, it is this obstruction of the affected shoulder while the long axis of the body continues to be pushed downward that widens the angle between the neck and impacted shoulder and stretches the brachial plexus.
The ACOG report acknowledges that may cases of NBPP do occur in conjunction with shoulder dystocia and that the same biomechanical factors that predispose a fetus to develop NBPP are associated with shoulder dystocia as well. However, the report takes pains to point out that the frequent conjunction of these two entities—NBPP and shoulder dystocia—may lead to an “erroneous retrospective inference of causation.”
Risk and predictive factors
The ACOG report states: “Various risk factors have been described in association with NBPP. Overall, however, these risk factors have not been shown to be statistically reliable or clinically useful predictors for...NBPP.”
For example, fetal macrosomia, defined as a birth weight of 4,000 g or more, has been reported as a risk factor for NBPP either alone or in conjunction with maternal diabetes. Although NBPP does occur more frequently as birth weight increases, seven studies over the past 20 years have shown that most cases of NBPP occur in infants of mothers without diabetes and in infants who weigh less than 4,000 g.
Other studies have shown that, if cesarean delivery were performed in cases of suspected macrosomia, it would have only a limited effect on reducing the incidence of NBPP. Specifically, in women with diabetes who have an estimated fetal weight of more than 4,500 g, the positive predictive value for NBPP is only 5%. Without maternal diabetes, that figure is less than 2%.
Estimating fetal weight by ultrasound does not significantly enhance our ability to predict NBPP. Ultrasound estimates of birth weight usually fall within 15% to 20% of actual birth weight, and the sensitivity of ultrasound in detecting birth weights more than 4,500 g is only 40%.
Therefore, ultrasound estimates of birth weight are of limited utility for contemporaneous clinical management. Furthermore, no data exist to support the claim that estimated fetal weight can be used prophylactically to reduce the incidence of NBPP.
Recurrent shoulder dystocia may be predictive of future NBPP
Whether studied alone or with NBPP, risk factors for shoulder dystocia are not reliable predictors of its occurrence. This is not the case, however, for recurrent shoulder dystocia, where the risk of neonatal brachial plexus palsy can be as high as 4.5%, compared with 1% to 2% for a first episode of shoulder dystocia.
NBPP is a rare phenomenon
The frequency of NBPP is “rare,” according to the ACOG report, which cites a rate of 1.5 cases for every 1,000 births. Favorable outcomes with complete recovery are estimated to range from 50% to 80%.3
Brachial plexus injuries are classically defined as Erb’s palsy—involving C5 and C6 nerve roots—or Klumpke’s palsy, in which there is damage to the C8 and T1 nerve roots.
Erb’s palsy is recognizable by the characteristic “waiter’s tip” position of the hand, which is caused by muscle imbalance in the shoulder and upper arm. Most NBPP injuries are Erb’s palsy, which affect 1.2 infants in every 1,000 births.
Klumpke’s palsy results in weakness of the hand and medial forearm muscles. It affects 0.05 infants in every 1,000 births. The remaining cases involve a combination of the two types of palsy.
These injuries can be temporary, resolving by 12 months after birth, or permanent. The rate of persistence of NBPP at 12 months ranges from 3% to 33%.
Can clinician maneuvers increase the likelihood of NBPP?
The ACOG report addresses the direction and angle of clinician traction at delivery. The report confirms what clinicians generally have been taught: The application of fundal pressure during a delivery in which shoulder dystocia is recognized can exacerbate shoulder impaction and can lead to an increased risk of NBPP.
Traction applied by the clinician and lateral bending of the fetal neck often are implicated as causative factors of NBPP. However, ACOG presents evidence that NBPP can occur entirely unrelated to clinician traction. The report cites studies involving both transient and persistent NBPP in fetuses delivered vaginally without evident shoulder dystocia. The same types of injury are sometimes seen in fetuses delivered by cesarean, as has been mentioned.
The report goes on to state:
Recommendations for practice
At the close of its second chapter (“Risk and predictive factors”), the ACOG report offers the same official recommendations that appear in its current practice bulletin on shoulder dystocia. It notes that there are three clinical situations in which it may be prudent to alter usual obstetric management, with an aim of reducing the risk of shoulder dystocia and NBPP:
- when fetal macrosomia is suspected, with fetal weight estimated to exceed 5,000 g in a woman without diabetes or 4,500 g in a woman with diabetes
- when the mother has a history of recognized shoulder dystocia, especially when neonatal injury was severe
- when midpelvic operative vaginal delivery is contemplated with a fetus estimated to weigh more than 4,000 g.
It is interesting to note that these recommendations are made, according to the report, “notwithstanding the unreliability of specific risk factors to predict NBPP or clinically apparent shoulder dystocia in a specific case.” The report further adds:
More to come
For ACOG’s conclusions on the pathophysiology and causation of NBPP, with a view toward formulating specific protective interventions, see Part 2 of this article, which will appear in the October 2014 issue of OBG Management.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Physician Insurers Association of America. http://www.piaa.us. Accessed August 21, 2014.
2. American College of Obstetricians and Gynecologists. Practice Bulletin #40: shoulder dystocia. Obstet Gynecol. 2002;100(5 pt 1):1045–1050.
3. American College of Obstetricians and Gynecologists. Executive summary: neonatal brachial plexus palsy. Report of the American College of Obstetricians and Gynecologists’ Task Force on neonatal brachial plexus palsy. Obstet Gynecol. 2014;123(4):902–904.
1. Physician Insurers Association of America. http://www.piaa.us. Accessed August 21, 2014.
2. American College of Obstetricians and Gynecologists. Practice Bulletin #40: shoulder dystocia. Obstet Gynecol. 2002;100(5 pt 1):1045–1050.
3. American College of Obstetricians and Gynecologists. Executive summary: neonatal brachial plexus palsy. Report of the American College of Obstetricians and Gynecologists’ Task Force on neonatal brachial plexus palsy. Obstet Gynecol. 2014;123(4):902–904.
Novel anticoagulants given to 60% of newly diagnosed AF patients
Novel oral anticoagulants introduced since October 2010 have been adopted into clinical practice rapidly, and within 2.5 years were prescribed for more than 60% of patients with newly diagnosed atrial fibrillation, according to a report published online in the American Journal of Medicine.
Further, the new drugs are being prescribed for a different patient population from that indicated by the clinical trials on which Food and Drug Administration (FDA) approval was based. Specifically, dabigatran, rivaroxaban, and apixaban are selectively prescribed for younger, healthier men who have high incomes and reside in wealthier communities, reported Dr. Nihar R. Desai of the division of pharmacoepidemiology and pharmacoeconomics, Brigham and Women’s Hospital and Harvard Medical School, Boston, and his associates.
In what they described as the first study to evaluate real-world use of all novel anticoagulants, researchers found that the rapid uptake of the drugs as first-line therapy for atrial fibrillation (AF) was accompanied by a marked decline in the use of warfarin. The difference in total costs between the generic warfarin and the proprietary dabigatran, rivaroxaban, or apixaban totaled $900 per patient during the first 6 months alone, which "translates into billions of dollars at the national level."
This has important economic implications for patients, payers, and the health care system. The impending FDA approval of new factor Xa inhibitors such as edoxaban and betrixaban will likely further complicate the picture, the researchers said.
The researchers analyzed nationwide medical and prescription claims data for 6,893 adults covered by Aetna who had newly diagnosed nonvalvular AF and were prescribed an oral anticoagulant between October 2010 and June 2013. The direct thrombin inhibitor dabigatran was approved in October 2010, and the factor Xa inhibitors rivaroxaban and apixaban were approved in November 2011 and December 2012.
During the study period, these patients filled 45,472 prescriptions for oral anticoagulants: 57.7% for warfarin, 32.8% for dabigatran, 9.3% for rivaroxaban, and 0.1% for apixaban. However, these figures don’t reflect the trend over time in which prescriptions for the newer agents rapidly displaced those for warfarin. Within 1 year of appearing on the market, dabigatran was equally likely to be prescribed as warfarin was for new AF patients. Its use as a first-line therapy dropped considerably a year later, after reports of excess rates of myocardial infarction and serious and fatal bleeding events in patients taking dabigatran. But at that point rivaroxaban had been introduced, and it soon overtook both dabigatran and warfarin as first-line therapy for AF. (Apixaban accounted for 2% of new anticoagulant prescriptions as of 6 months after it was approved, which is the most recent date for which such statistics were available.)
Simultaneously, the costs of oral anticoagulants rose dramatically, with the new agents accounting for 98% of that escalation. It is estimated that insurers spend $5.82 million every month for all agents combined, and that warfarin accounts for only $0.43 million of that. Similarly, patient out-of-pocket spending for all oral anticoagulants combined was estimated to be $1.3 million per month, with warfarin accounting for $3,844 of that total.
Viewed from another perspective, the average combined patient and insurer spending for anticoagulants during the first 6 months of therapy for warfarin was $122, dabigatran $1,053, and rivaroxaban $1,084. "This represents a difference of more than $900 per patient," said Dr. Desai, who is also at the Center for Outcomes Research and Evaluation, Yale-New Haven Health Services, and his associates.
The greatest benefit from novel anticoagulants is among patients at the highest risk for stroke or systemic embolization, as measured by higher scores on CHADS (Congestive Heart Failure, Hypertension, Age of 75 years or more, Diabetes Mellitus, and Stroke) and HAS-BLED (Hypertension, Abnormal Renal/Liver Function, Stroke, Bleeding History or Predisposition, Labile INR, Elderly, Drugs/Alcohol) assessments. This is also the patient population targeted in the clinical trials that formed the basis for FDA approval.
But in this study, 46% of patients with low CHADS and HAS-BLED scores were initially prescribed the novel anticoagulants, compared with 26% of those with high scores. "For every 1-point increase in CHADS, patients were 20% less likely to receive a novel anticoagulant. Similarly, for every 1-point increase in HAS-BLED, patients were 18% less likely to receive a novel anticoagulant.
"In addition, women were 24% less likely to be initiated on a novel oral anticoagulant as compared with men. [And] there was a significant, stepwise increase in the likelihood of receiving a novel agent with progressively increasing neighborhood household income, compared with a median household income of $50,000 or less," the investigators said (Amer. J. Med. 2014 May 20 [doi: 10.1016/j.amjmed.2014.05.013]).
"These findings point to the need to conduct ongoing surveillance of the adoption of new agents into clinical practice, as well as the need for robust, real-world comparative-effectiveness analyses of these medications, to enable patients and providers to make informed decisions about their relative benefit, safety, and cost-effectiveness," Dr. Desai and his associates said.
This study was funded by an unrestricted research grant from CVS Caremark. Dr. Desai’s associates reported ties to CVS Caremark and Aetna.
It is not surprising that the novel oral anticoagulants appear to be supplanting warfarin as first-line therapy for nonvalvular AF. The newer drugs are much easier to use because they don’t require frequent monitoring of clotting parameters, require no dietary restrictions, and have simple and straightforward dosing.
I expect the use of these novel anticoagulants – and others soon to be approved – to increase over time, particularly once they become generic and cost gradually becomes less of an issue.
Dr. Joseph S. Alpert is professor of medicine at the University of Arizona, Tucson, and the editor-in-chief of the American Journal of Medicine. Dr. Alpert made these remarks in an editorial (Amer. J. Med. 2014 Aug. 8 [doi: 10.1016/amjmed.2014.07.028]) accompanying Dr. Desai’s report. He reported cochairing the data monitoring committees for two of the large clinical trials that led to FDA approval of rivaroxaban.
It is not surprising that the novel oral anticoagulants appear to be supplanting warfarin as first-line therapy for nonvalvular AF. The newer drugs are much easier to use because they don’t require frequent monitoring of clotting parameters, require no dietary restrictions, and have simple and straightforward dosing.
I expect the use of these novel anticoagulants – and others soon to be approved – to increase over time, particularly once they become generic and cost gradually becomes less of an issue.
Dr. Joseph S. Alpert is professor of medicine at the University of Arizona, Tucson, and the editor-in-chief of the American Journal of Medicine. Dr. Alpert made these remarks in an editorial (Amer. J. Med. 2014 Aug. 8 [doi: 10.1016/amjmed.2014.07.028]) accompanying Dr. Desai’s report. He reported cochairing the data monitoring committees for two of the large clinical trials that led to FDA approval of rivaroxaban.
It is not surprising that the novel oral anticoagulants appear to be supplanting warfarin as first-line therapy for nonvalvular AF. The newer drugs are much easier to use because they don’t require frequent monitoring of clotting parameters, require no dietary restrictions, and have simple and straightforward dosing.
I expect the use of these novel anticoagulants – and others soon to be approved – to increase over time, particularly once they become generic and cost gradually becomes less of an issue.
Dr. Joseph S. Alpert is professor of medicine at the University of Arizona, Tucson, and the editor-in-chief of the American Journal of Medicine. Dr. Alpert made these remarks in an editorial (Amer. J. Med. 2014 Aug. 8 [doi: 10.1016/amjmed.2014.07.028]) accompanying Dr. Desai’s report. He reported cochairing the data monitoring committees for two of the large clinical trials that led to FDA approval of rivaroxaban.
Novel oral anticoagulants introduced since October 2010 have been adopted into clinical practice rapidly, and within 2.5 years were prescribed for more than 60% of patients with newly diagnosed atrial fibrillation, according to a report published online in the American Journal of Medicine.
Further, the new drugs are being prescribed for a different patient population from that indicated by the clinical trials on which Food and Drug Administration (FDA) approval was based. Specifically, dabigatran, rivaroxaban, and apixaban are selectively prescribed for younger, healthier men who have high incomes and reside in wealthier communities, reported Dr. Nihar R. Desai of the division of pharmacoepidemiology and pharmacoeconomics, Brigham and Women’s Hospital and Harvard Medical School, Boston, and his associates.
In what they described as the first study to evaluate real-world use of all novel anticoagulants, researchers found that the rapid uptake of the drugs as first-line therapy for atrial fibrillation (AF) was accompanied by a marked decline in the use of warfarin. The difference in total costs between the generic warfarin and the proprietary dabigatran, rivaroxaban, or apixaban totaled $900 per patient during the first 6 months alone, which "translates into billions of dollars at the national level."
This has important economic implications for patients, payers, and the health care system. The impending FDA approval of new factor Xa inhibitors such as edoxaban and betrixaban will likely further complicate the picture, the researchers said.
The researchers analyzed nationwide medical and prescription claims data for 6,893 adults covered by Aetna who had newly diagnosed nonvalvular AF and were prescribed an oral anticoagulant between October 2010 and June 2013. The direct thrombin inhibitor dabigatran was approved in October 2010, and the factor Xa inhibitors rivaroxaban and apixaban were approved in November 2011 and December 2012.
During the study period, these patients filled 45,472 prescriptions for oral anticoagulants: 57.7% for warfarin, 32.8% for dabigatran, 9.3% for rivaroxaban, and 0.1% for apixaban. However, these figures don’t reflect the trend over time in which prescriptions for the newer agents rapidly displaced those for warfarin. Within 1 year of appearing on the market, dabigatran was equally likely to be prescribed as warfarin was for new AF patients. Its use as a first-line therapy dropped considerably a year later, after reports of excess rates of myocardial infarction and serious and fatal bleeding events in patients taking dabigatran. But at that point rivaroxaban had been introduced, and it soon overtook both dabigatran and warfarin as first-line therapy for AF. (Apixaban accounted for 2% of new anticoagulant prescriptions as of 6 months after it was approved, which is the most recent date for which such statistics were available.)
Simultaneously, the costs of oral anticoagulants rose dramatically, with the new agents accounting for 98% of that escalation. It is estimated that insurers spend $5.82 million every month for all agents combined, and that warfarin accounts for only $0.43 million of that. Similarly, patient out-of-pocket spending for all oral anticoagulants combined was estimated to be $1.3 million per month, with warfarin accounting for $3,844 of that total.
Viewed from another perspective, the average combined patient and insurer spending for anticoagulants during the first 6 months of therapy for warfarin was $122, dabigatran $1,053, and rivaroxaban $1,084. "This represents a difference of more than $900 per patient," said Dr. Desai, who is also at the Center for Outcomes Research and Evaluation, Yale-New Haven Health Services, and his associates.
The greatest benefit from novel anticoagulants is among patients at the highest risk for stroke or systemic embolization, as measured by higher scores on CHADS (Congestive Heart Failure, Hypertension, Age of 75 years or more, Diabetes Mellitus, and Stroke) and HAS-BLED (Hypertension, Abnormal Renal/Liver Function, Stroke, Bleeding History or Predisposition, Labile INR, Elderly, Drugs/Alcohol) assessments. This is also the patient population targeted in the clinical trials that formed the basis for FDA approval.
But in this study, 46% of patients with low CHADS and HAS-BLED scores were initially prescribed the novel anticoagulants, compared with 26% of those with high scores. "For every 1-point increase in CHADS, patients were 20% less likely to receive a novel anticoagulant. Similarly, for every 1-point increase in HAS-BLED, patients were 18% less likely to receive a novel anticoagulant.
"In addition, women were 24% less likely to be initiated on a novel oral anticoagulant as compared with men. [And] there was a significant, stepwise increase in the likelihood of receiving a novel agent with progressively increasing neighborhood household income, compared with a median household income of $50,000 or less," the investigators said (Amer. J. Med. 2014 May 20 [doi: 10.1016/j.amjmed.2014.05.013]).
"These findings point to the need to conduct ongoing surveillance of the adoption of new agents into clinical practice, as well as the need for robust, real-world comparative-effectiveness analyses of these medications, to enable patients and providers to make informed decisions about their relative benefit, safety, and cost-effectiveness," Dr. Desai and his associates said.
This study was funded by an unrestricted research grant from CVS Caremark. Dr. Desai’s associates reported ties to CVS Caremark and Aetna.
Novel oral anticoagulants introduced since October 2010 have been adopted into clinical practice rapidly, and within 2.5 years were prescribed for more than 60% of patients with newly diagnosed atrial fibrillation, according to a report published online in the American Journal of Medicine.
Further, the new drugs are being prescribed for a different patient population from that indicated by the clinical trials on which Food and Drug Administration (FDA) approval was based. Specifically, dabigatran, rivaroxaban, and apixaban are selectively prescribed for younger, healthier men who have high incomes and reside in wealthier communities, reported Dr. Nihar R. Desai of the division of pharmacoepidemiology and pharmacoeconomics, Brigham and Women’s Hospital and Harvard Medical School, Boston, and his associates.
In what they described as the first study to evaluate real-world use of all novel anticoagulants, researchers found that the rapid uptake of the drugs as first-line therapy for atrial fibrillation (AF) was accompanied by a marked decline in the use of warfarin. The difference in total costs between the generic warfarin and the proprietary dabigatran, rivaroxaban, or apixaban totaled $900 per patient during the first 6 months alone, which "translates into billions of dollars at the national level."
This has important economic implications for patients, payers, and the health care system. The impending FDA approval of new factor Xa inhibitors such as edoxaban and betrixaban will likely further complicate the picture, the researchers said.
The researchers analyzed nationwide medical and prescription claims data for 6,893 adults covered by Aetna who had newly diagnosed nonvalvular AF and were prescribed an oral anticoagulant between October 2010 and June 2013. The direct thrombin inhibitor dabigatran was approved in October 2010, and the factor Xa inhibitors rivaroxaban and apixaban were approved in November 2011 and December 2012.
During the study period, these patients filled 45,472 prescriptions for oral anticoagulants: 57.7% for warfarin, 32.8% for dabigatran, 9.3% for rivaroxaban, and 0.1% for apixaban. However, these figures don’t reflect the trend over time in which prescriptions for the newer agents rapidly displaced those for warfarin. Within 1 year of appearing on the market, dabigatran was equally likely to be prescribed as warfarin was for new AF patients. Its use as a first-line therapy dropped considerably a year later, after reports of excess rates of myocardial infarction and serious and fatal bleeding events in patients taking dabigatran. But at that point rivaroxaban had been introduced, and it soon overtook both dabigatran and warfarin as first-line therapy for AF. (Apixaban accounted for 2% of new anticoagulant prescriptions as of 6 months after it was approved, which is the most recent date for which such statistics were available.)
Simultaneously, the costs of oral anticoagulants rose dramatically, with the new agents accounting for 98% of that escalation. It is estimated that insurers spend $5.82 million every month for all agents combined, and that warfarin accounts for only $0.43 million of that. Similarly, patient out-of-pocket spending for all oral anticoagulants combined was estimated to be $1.3 million per month, with warfarin accounting for $3,844 of that total.
Viewed from another perspective, the average combined patient and insurer spending for anticoagulants during the first 6 months of therapy for warfarin was $122, dabigatran $1,053, and rivaroxaban $1,084. "This represents a difference of more than $900 per patient," said Dr. Desai, who is also at the Center for Outcomes Research and Evaluation, Yale-New Haven Health Services, and his associates.
The greatest benefit from novel anticoagulants is among patients at the highest risk for stroke or systemic embolization, as measured by higher scores on CHADS (Congestive Heart Failure, Hypertension, Age of 75 years or more, Diabetes Mellitus, and Stroke) and HAS-BLED (Hypertension, Abnormal Renal/Liver Function, Stroke, Bleeding History or Predisposition, Labile INR, Elderly, Drugs/Alcohol) assessments. This is also the patient population targeted in the clinical trials that formed the basis for FDA approval.
But in this study, 46% of patients with low CHADS and HAS-BLED scores were initially prescribed the novel anticoagulants, compared with 26% of those with high scores. "For every 1-point increase in CHADS, patients were 20% less likely to receive a novel anticoagulant. Similarly, for every 1-point increase in HAS-BLED, patients were 18% less likely to receive a novel anticoagulant.
"In addition, women were 24% less likely to be initiated on a novel oral anticoagulant as compared with men. [And] there was a significant, stepwise increase in the likelihood of receiving a novel agent with progressively increasing neighborhood household income, compared with a median household income of $50,000 or less," the investigators said (Amer. J. Med. 2014 May 20 [doi: 10.1016/j.amjmed.2014.05.013]).
"These findings point to the need to conduct ongoing surveillance of the adoption of new agents into clinical practice, as well as the need for robust, real-world comparative-effectiveness analyses of these medications, to enable patients and providers to make informed decisions about their relative benefit, safety, and cost-effectiveness," Dr. Desai and his associates said.
This study was funded by an unrestricted research grant from CVS Caremark. Dr. Desai’s associates reported ties to CVS Caremark and Aetna.
FROM THE AMERICAN JOURNAL OF MEDICINE
Key clinical point: Novel anticoagulants are being prescribed for younger men, a different patient population from that indicated by the clinical trials on which FDA approval was based.
Major finding: The average combined patient and insurer spending for oral anticoagulants during the first 6 months of therapy was $122 for warfarin, $1,053 for dabigatran, and $1,084 for rivaroxaban.
Data source: A retrospective, longitudinal analysis of nationwide Aetna prescription claims data for 6,893 adults with nonvalvular AF who initiated oral anticoagulants in 2010-2013.
Disclosures: This study was funded by an unrestricted research grant from CVS Caremark. Dr. Desai’s associates reported ties to CVS Caremark and Aetna.
Drug can treat inflammation-induced anemia
An experimental drug designed to help regulate the blood’s iron supply may be a viable treatment option for inflammation-induced anemia, according to a study published in Blood.
The only current treatment strategy for this type of anemia involves targeting the underlying disease or infection.
However, recent research has sought to explore additional options for patients whose inflammation is difficult to control or when the cause of inflammation is unknown.
A hepcidin inhibitor called lexaptepid pegol (lexaptepid) has demonstrated efficacy in treating inflammation-induced anemia in animal studies. Lexaptepid inactivates hepcidin, thereby maintaining the transport of iron to the bloodstream.
To evaluate lexaptepid’s potential in humans, investigators caused inflammation-induced anemia in 24 healthy male adults and randomized them to receive lexaptepid or placebo.
Subjects received a low dose of Escherichia coli endotoxin to induce controlled inflammation and received either lexaptepid or placebo 30 minutes later.
After 9 hours, serum iron had decreased by 8.3±9.0 μmol/L in controls but increased by 15.9±9.8 μmol/L in lexaptepid-treated subjects (P<0.0001).
In addition to evaluating whether lexaptepid interfered with hepcidin production, the researchers also sought to determine whether the drug influenced the immune response.
Results suggested it did not. Treated subjects and controls alike experienced flu-like symptoms, increased body temperature and white blood cell counts, and higher concentrations of inflammatory and signaling proteins.
“It is quite encouraging that lexaptepid helped maintain appropriate levels of iron in the bloodstream of healthy volunteers without compromising the immune response,” said lead study author Lucas van Eijk, MD, of Radboud University Medical Center in Nijmegen, The Netherlands.
“We are hopeful that, with further study, this first-of-its-kind therapy could significantly improve quality of life for patients suffering from chronic illnesses.”
Results of a phase 2 study testing lexaptepid in anemic cancer patients were presented at the AACR Annual Meeting 2014. ![]()
An experimental drug designed to help regulate the blood’s iron supply may be a viable treatment option for inflammation-induced anemia, according to a study published in Blood.
The only current treatment strategy for this type of anemia involves targeting the underlying disease or infection.
However, recent research has sought to explore additional options for patients whose inflammation is difficult to control or when the cause of inflammation is unknown.
A hepcidin inhibitor called lexaptepid pegol (lexaptepid) has demonstrated efficacy in treating inflammation-induced anemia in animal studies. Lexaptepid inactivates hepcidin, thereby maintaining the transport of iron to the bloodstream.
To evaluate lexaptepid’s potential in humans, investigators caused inflammation-induced anemia in 24 healthy male adults and randomized them to receive lexaptepid or placebo.
Subjects received a low dose of Escherichia coli endotoxin to induce controlled inflammation and received either lexaptepid or placebo 30 minutes later.
After 9 hours, serum iron had decreased by 8.3±9.0 μmol/L in controls but increased by 15.9±9.8 μmol/L in lexaptepid-treated subjects (P<0.0001).
In addition to evaluating whether lexaptepid interfered with hepcidin production, the researchers also sought to determine whether the drug influenced the immune response.
Results suggested it did not. Treated subjects and controls alike experienced flu-like symptoms, increased body temperature and white blood cell counts, and higher concentrations of inflammatory and signaling proteins.
“It is quite encouraging that lexaptepid helped maintain appropriate levels of iron in the bloodstream of healthy volunteers without compromising the immune response,” said lead study author Lucas van Eijk, MD, of Radboud University Medical Center in Nijmegen, The Netherlands.
“We are hopeful that, with further study, this first-of-its-kind therapy could significantly improve quality of life for patients suffering from chronic illnesses.”
Results of a phase 2 study testing lexaptepid in anemic cancer patients were presented at the AACR Annual Meeting 2014. ![]()
An experimental drug designed to help regulate the blood’s iron supply may be a viable treatment option for inflammation-induced anemia, according to a study published in Blood.
The only current treatment strategy for this type of anemia involves targeting the underlying disease or infection.
However, recent research has sought to explore additional options for patients whose inflammation is difficult to control or when the cause of inflammation is unknown.
A hepcidin inhibitor called lexaptepid pegol (lexaptepid) has demonstrated efficacy in treating inflammation-induced anemia in animal studies. Lexaptepid inactivates hepcidin, thereby maintaining the transport of iron to the bloodstream.
To evaluate lexaptepid’s potential in humans, investigators caused inflammation-induced anemia in 24 healthy male adults and randomized them to receive lexaptepid or placebo.
Subjects received a low dose of Escherichia coli endotoxin to induce controlled inflammation and received either lexaptepid or placebo 30 minutes later.
After 9 hours, serum iron had decreased by 8.3±9.0 μmol/L in controls but increased by 15.9±9.8 μmol/L in lexaptepid-treated subjects (P<0.0001).
In addition to evaluating whether lexaptepid interfered with hepcidin production, the researchers also sought to determine whether the drug influenced the immune response.
Results suggested it did not. Treated subjects and controls alike experienced flu-like symptoms, increased body temperature and white blood cell counts, and higher concentrations of inflammatory and signaling proteins.
“It is quite encouraging that lexaptepid helped maintain appropriate levels of iron in the bloodstream of healthy volunteers without compromising the immune response,” said lead study author Lucas van Eijk, MD, of Radboud University Medical Center in Nijmegen, The Netherlands.
“We are hopeful that, with further study, this first-of-its-kind therapy could significantly improve quality of life for patients suffering from chronic illnesses.”
Results of a phase 2 study testing lexaptepid in anemic cancer patients were presented at the AACR Annual Meeting 2014. ![]()
T cells play role in clinical tolerance of malaria, team says

Credit: Malayaka house
Children repeatedly infected with malaria have been known to become asymptomatic, and researchers have found evidence suggesting a subset of γδ T cells play a role in this phenomenon.
Studying young children in Uganda, the team discovered that repeated malaria infection was associated with the loss and dysfunction of Vδ2+ γδ T cells.
This appeared to facilitate immunological tolerance of the malaria parasite and, therefore, a reduction in clinical symptoms.
“These inflammatory immune cells are depleted in children with repeated malaria exposure, and those that remain behave differently than the same cell types in children who have not previously been infected,” said Prasanna Jagannathan, MD, of the University of California, San Francisco (UCSF).
He and his colleagues reported these findings in Science Translational Medicine.
The researchers collected data on malaria infections, disease symptoms, and immune responses in 78 children who were monitored from infancy as part of a research collaboration between UCSF and Makarere University in Kampala, Africa. The study was conducted in Tororo, Uganda.
At 1 year of age, all children showed clinical symptoms of malaria with each infection. At 4 years, fewer than 10% were symptom-free upon infection. But 1 year later, more than 20% were symptom-free when infected.
The researchers found that repeated malaria infection was associated with the loss and dysfunction of Vδ2+ γδ T cells. They observed a decrease in cell proliferation and in the production of inflammatory cytokines (IFN-γ and TNF-α) in response to malaria antigens.
Repeated malaria infection was also associated with the upregulation of immunoregulatory pathways—increased expression of genes such as HAVCR2, FCRL6, LYN, BATF, and B3GAT1—that dampen the immune response.
Children with these characteristics were less likely than their peers to exhibit clinical symptoms upon subsequent malaria infection.
So it seems the depletion of Vδ2+ γδ T cells is beneficial in some ways and detrimental in others, said Margaret Feeney, MD, of UCSF. Individuals may no longer suffer symptoms, but they might not clear the parasite and could remain infectious.
Although this discovery has not provided a disease-fighting strategy as of yet, it does point to further avenues of study, according to Dr Feeney.
“We want to understand whether this is a generalizable phenomenon that also occurs among those who are first exposed to malaria as adults and in regions where malaria incidence is lower,” she said.
Dr Feeney speculates that malaria infection, by reshaping immune responses, might influence a person’s susceptibility to, and protection from, other infectious diseases. ![]()
Credit: Malayaka house
Children repeatedly infected with malaria have been known to become asymptomatic, and researchers have found evidence suggesting a subset of γδ T cells play a role in this phenomenon.
Studying young children in Uganda, the team discovered that repeated malaria infection was associated with the loss and dysfunction of Vδ2+ γδ T cells.
This appeared to facilitate immunological tolerance of the malaria parasite and, therefore, a reduction in clinical symptoms.
“These inflammatory immune cells are depleted in children with repeated malaria exposure, and those that remain behave differently than the same cell types in children who have not previously been infected,” said Prasanna Jagannathan, MD, of the University of California, San Francisco (UCSF).
He and his colleagues reported these findings in Science Translational Medicine.
The researchers collected data on malaria infections, disease symptoms, and immune responses in 78 children who were monitored from infancy as part of a research collaboration between UCSF and Makarere University in Kampala, Africa. The study was conducted in Tororo, Uganda.
At 1 year of age, all children showed clinical symptoms of malaria with each infection. At 4 years, fewer than 10% were symptom-free upon infection. But 1 year later, more than 20% were symptom-free when infected.
The researchers found that repeated malaria infection was associated with the loss and dysfunction of Vδ2+ γδ T cells. They observed a decrease in cell proliferation and in the production of inflammatory cytokines (IFN-γ and TNF-α) in response to malaria antigens.
Repeated malaria infection was also associated with the upregulation of immunoregulatory pathways—increased expression of genes such as HAVCR2, FCRL6, LYN, BATF, and B3GAT1—that dampen the immune response.
Children with these characteristics were less likely than their peers to exhibit clinical symptoms upon subsequent malaria infection.
So it seems the depletion of Vδ2+ γδ T cells is beneficial in some ways and detrimental in others, said Margaret Feeney, MD, of UCSF. Individuals may no longer suffer symptoms, but they might not clear the parasite and could remain infectious.
Although this discovery has not provided a disease-fighting strategy as of yet, it does point to further avenues of study, according to Dr Feeney.
“We want to understand whether this is a generalizable phenomenon that also occurs among those who are first exposed to malaria as adults and in regions where malaria incidence is lower,” she said.
Dr Feeney speculates that malaria infection, by reshaping immune responses, might influence a person’s susceptibility to, and protection from, other infectious diseases. ![]()
Credit: Malayaka house
Children repeatedly infected with malaria have been known to become asymptomatic, and researchers have found evidence suggesting a subset of γδ T cells play a role in this phenomenon.
Studying young children in Uganda, the team discovered that repeated malaria infection was associated with the loss and dysfunction of Vδ2+ γδ T cells.
This appeared to facilitate immunological tolerance of the malaria parasite and, therefore, a reduction in clinical symptoms.
“These inflammatory immune cells are depleted in children with repeated malaria exposure, and those that remain behave differently than the same cell types in children who have not previously been infected,” said Prasanna Jagannathan, MD, of the University of California, San Francisco (UCSF).
He and his colleagues reported these findings in Science Translational Medicine.
The researchers collected data on malaria infections, disease symptoms, and immune responses in 78 children who were monitored from infancy as part of a research collaboration between UCSF and Makarere University in Kampala, Africa. The study was conducted in Tororo, Uganda.
At 1 year of age, all children showed clinical symptoms of malaria with each infection. At 4 years, fewer than 10% were symptom-free upon infection. But 1 year later, more than 20% were symptom-free when infected.
The researchers found that repeated malaria infection was associated with the loss and dysfunction of Vδ2+ γδ T cells. They observed a decrease in cell proliferation and in the production of inflammatory cytokines (IFN-γ and TNF-α) in response to malaria antigens.
Repeated malaria infection was also associated with the upregulation of immunoregulatory pathways—increased expression of genes such as HAVCR2, FCRL6, LYN, BATF, and B3GAT1—that dampen the immune response.
Children with these characteristics were less likely than their peers to exhibit clinical symptoms upon subsequent malaria infection.
So it seems the depletion of Vδ2+ γδ T cells is beneficial in some ways and detrimental in others, said Margaret Feeney, MD, of UCSF. Individuals may no longer suffer symptoms, but they might not clear the parasite and could remain infectious.
Although this discovery has not provided a disease-fighting strategy as of yet, it does point to further avenues of study, according to Dr Feeney.
“We want to understand whether this is a generalizable phenomenon that also occurs among those who are first exposed to malaria as adults and in regions where malaria incidence is lower,” she said.
Dr Feeney speculates that malaria infection, by reshaping immune responses, might influence a person’s susceptibility to, and protection from, other infectious diseases. ![]()
Nanoparticle may have multiple cancer applications

Credit: PNAS
A new type of nanoparticle (NP) could aid the diagnosis and treatment of cancers, according to research published in Nature Communications.
Built on an easy-to-make polymer, these particles can be used as contrast agents to light up tumors for MRI and PET scans or to deliver chemotherapy and other treatments to cancer cells.
Furthermore, in vivo experiments showed the particles are biocompatible and elicit minimal side effects.
“These are amazingly useful particles,” said study author Yuanpei Li, PhD, of the UC Davis Comprehensive Cancer Center in Sacramento, California.
“As a contrast agent, they make tumors easier to see on MRI and other scans. We can also use them as vehicles to deliver chemotherapy directly to tumors, apply light to make the nanoparticles release singlet oxygen (photodynamic therapy), or use a laser to heat them (photothermal therapy)—all proven ways to destroy tumors.”
These NPs are built on a porphyrin/cholic acid polymer. Porphyrins are common organic compounds, and cholic acid is produced by the liver.
To further stabilize the particles, the researchers added the amino acid cysteine (creating CNPs), which prevents them from prematurely releasing their therapeutic payload when exposed to blood proteins and other barriers.
Therapeutic applications
The researchers tested the CNPs, both in vitro and in vivo, for a wide range of tasks. On the therapeutic side, the particles effectively transported anticancer drugs, such as doxorubicin.
CNPs carrying doxorubicin provided excellent cancer control in animals, with minimal side effects.
Even when kept in the blood for many hours, CNPs only released small amounts of the drug. However, when exposed to light or agents such as glutathione, they readily released their payloads.
The researchers showed that, when exposed to a single wavelength of light, the CNPs could generate heat or produce singlet oxygen to destroy tumor cells.
Imaging applications
CNPs offer a number of advantages to enhance imaging, according to the researchers. The particles readily chelate imaging agents and can remain in the body for long periods.
In animal studies, CNPs largely accumulated in tumors, rather than in normal tissue. So they dramatically enhanced tumor contrast for MRI and may also be promising for PET-MRI scans, the researchers said.
“These particles can combine imaging and therapeutics,” Dr Li noted. “We could potentially use them to simultaneously deliver treatment and monitor treatment efficacy.”
The researchers are now conducting additional preclinical studies with the CNPs. If all goes well, they will proceed to human trials. In the meantime, the team is excited about these capabilities.
“This is the first nanoparticle to perform so many different jobs,” Dr Li said. “From delivering chemo, photodynamic, and photothermal therapies, to enhancing diagnostic imaging, it’s the complete package.” ![]()
Credit: PNAS
A new type of nanoparticle (NP) could aid the diagnosis and treatment of cancers, according to research published in Nature Communications.
Built on an easy-to-make polymer, these particles can be used as contrast agents to light up tumors for MRI and PET scans or to deliver chemotherapy and other treatments to cancer cells.
Furthermore, in vivo experiments showed the particles are biocompatible and elicit minimal side effects.
“These are amazingly useful particles,” said study author Yuanpei Li, PhD, of the UC Davis Comprehensive Cancer Center in Sacramento, California.
“As a contrast agent, they make tumors easier to see on MRI and other scans. We can also use them as vehicles to deliver chemotherapy directly to tumors, apply light to make the nanoparticles release singlet oxygen (photodynamic therapy), or use a laser to heat them (photothermal therapy)—all proven ways to destroy tumors.”
These NPs are built on a porphyrin/cholic acid polymer. Porphyrins are common organic compounds, and cholic acid is produced by the liver.
To further stabilize the particles, the researchers added the amino acid cysteine (creating CNPs), which prevents them from prematurely releasing their therapeutic payload when exposed to blood proteins and other barriers.
Therapeutic applications
The researchers tested the CNPs, both in vitro and in vivo, for a wide range of tasks. On the therapeutic side, the particles effectively transported anticancer drugs, such as doxorubicin.
CNPs carrying doxorubicin provided excellent cancer control in animals, with minimal side effects.
Even when kept in the blood for many hours, CNPs only released small amounts of the drug. However, when exposed to light or agents such as glutathione, they readily released their payloads.
The researchers showed that, when exposed to a single wavelength of light, the CNPs could generate heat or produce singlet oxygen to destroy tumor cells.
Imaging applications
CNPs offer a number of advantages to enhance imaging, according to the researchers. The particles readily chelate imaging agents and can remain in the body for long periods.
In animal studies, CNPs largely accumulated in tumors, rather than in normal tissue. So they dramatically enhanced tumor contrast for MRI and may also be promising for PET-MRI scans, the researchers said.
“These particles can combine imaging and therapeutics,” Dr Li noted. “We could potentially use them to simultaneously deliver treatment and monitor treatment efficacy.”
The researchers are now conducting additional preclinical studies with the CNPs. If all goes well, they will proceed to human trials. In the meantime, the team is excited about these capabilities.
“This is the first nanoparticle to perform so many different jobs,” Dr Li said. “From delivering chemo, photodynamic, and photothermal therapies, to enhancing diagnostic imaging, it’s the complete package.” ![]()
Credit: PNAS
A new type of nanoparticle (NP) could aid the diagnosis and treatment of cancers, according to research published in Nature Communications.
Built on an easy-to-make polymer, these particles can be used as contrast agents to light up tumors for MRI and PET scans or to deliver chemotherapy and other treatments to cancer cells.
Furthermore, in vivo experiments showed the particles are biocompatible and elicit minimal side effects.
“These are amazingly useful particles,” said study author Yuanpei Li, PhD, of the UC Davis Comprehensive Cancer Center in Sacramento, California.
“As a contrast agent, they make tumors easier to see on MRI and other scans. We can also use them as vehicles to deliver chemotherapy directly to tumors, apply light to make the nanoparticles release singlet oxygen (photodynamic therapy), or use a laser to heat them (photothermal therapy)—all proven ways to destroy tumors.”
These NPs are built on a porphyrin/cholic acid polymer. Porphyrins are common organic compounds, and cholic acid is produced by the liver.
To further stabilize the particles, the researchers added the amino acid cysteine (creating CNPs), which prevents them from prematurely releasing their therapeutic payload when exposed to blood proteins and other barriers.
Therapeutic applications
The researchers tested the CNPs, both in vitro and in vivo, for a wide range of tasks. On the therapeutic side, the particles effectively transported anticancer drugs, such as doxorubicin.
CNPs carrying doxorubicin provided excellent cancer control in animals, with minimal side effects.
Even when kept in the blood for many hours, CNPs only released small amounts of the drug. However, when exposed to light or agents such as glutathione, they readily released their payloads.
The researchers showed that, when exposed to a single wavelength of light, the CNPs could generate heat or produce singlet oxygen to destroy tumor cells.
Imaging applications
CNPs offer a number of advantages to enhance imaging, according to the researchers. The particles readily chelate imaging agents and can remain in the body for long periods.
In animal studies, CNPs largely accumulated in tumors, rather than in normal tissue. So they dramatically enhanced tumor contrast for MRI and may also be promising for PET-MRI scans, the researchers said.
“These particles can combine imaging and therapeutics,” Dr Li noted. “We could potentially use them to simultaneously deliver treatment and monitor treatment efficacy.”
The researchers are now conducting additional preclinical studies with the CNPs. If all goes well, they will proceed to human trials. In the meantime, the team is excited about these capabilities.
“This is the first nanoparticle to perform so many different jobs,” Dr Li said. “From delivering chemo, photodynamic, and photothermal therapies, to enhancing diagnostic imaging, it’s the complete package.” ![]()
RIKEN’s initial STAP cell experiments failed

Investigators at the Japanese research institute RIKEN have failed to create STAP (stimulus-triggered acquisition of pluripotency) cells in the experiments they’ve conducted thus far.
However, the group plans to continue its attempts to replicate the STAP cell phenomenon—inducing pluripotency in somatic cells by exposing them to stress—until next March.
So far, the group has failed to create STAP cells by exposing cells from newborn C57BL/6 mice to a low-pH environment.
Going forward, the researchers plan to conduct their experiments in another mouse strain. They also intend to alter the methods of stressing the cells.
RIKEN investigator Haruko Obokata, PhD, and her colleagues initially reported the STAP cell phenomenon in an article and a letter published in Nature last January.
Not long after the papers were published, members of the scientific community began to question the validity of the research. They voiced concerns about published images, possible plagiarism, and an inability to replicate the experiments described.
So RIKEN launched an investigation, ultimately concluding that Dr Obokata was guilty of misconduct, and some of her colleagues—including the recently deceased Yoshiki Sasai, MD, PhD—were guilty of negligence.
RIKEN also called for the papers to be retracted, and, in July, they were.
Throughout these proceedings, Dr Obokata insisted the STAP cell phenomenon is real. To investigate this claim, RIKEN organized a group of researchers to recreate Dr Obokata’s experiments.
The group has performed 22 experiments using different types of stress and cells from different tissues in C57BL/6 mice, but they have not reproduced the STAP cell phenomenon described in the Nature papers. (A report on these attempts is available in Japanese.)
Still, the investigators are continuing with their experiments and hope to have definitive results by March. ![]()
Investigators at the Japanese research institute RIKEN have failed to create STAP (stimulus-triggered acquisition of pluripotency) cells in the experiments they’ve conducted thus far.
However, the group plans to continue its attempts to replicate the STAP cell phenomenon—inducing pluripotency in somatic cells by exposing them to stress—until next March.
So far, the group has failed to create STAP cells by exposing cells from newborn C57BL/6 mice to a low-pH environment.
Going forward, the researchers plan to conduct their experiments in another mouse strain. They also intend to alter the methods of stressing the cells.
RIKEN investigator Haruko Obokata, PhD, and her colleagues initially reported the STAP cell phenomenon in an article and a letter published in Nature last January.
Not long after the papers were published, members of the scientific community began to question the validity of the research. They voiced concerns about published images, possible plagiarism, and an inability to replicate the experiments described.
So RIKEN launched an investigation, ultimately concluding that Dr Obokata was guilty of misconduct, and some of her colleagues—including the recently deceased Yoshiki Sasai, MD, PhD—were guilty of negligence.
RIKEN also called for the papers to be retracted, and, in July, they were.
Throughout these proceedings, Dr Obokata insisted the STAP cell phenomenon is real. To investigate this claim, RIKEN organized a group of researchers to recreate Dr Obokata’s experiments.
The group has performed 22 experiments using different types of stress and cells from different tissues in C57BL/6 mice, but they have not reproduced the STAP cell phenomenon described in the Nature papers. (A report on these attempts is available in Japanese.)
Still, the investigators are continuing with their experiments and hope to have definitive results by March. ![]()
Investigators at the Japanese research institute RIKEN have failed to create STAP (stimulus-triggered acquisition of pluripotency) cells in the experiments they’ve conducted thus far.
However, the group plans to continue its attempts to replicate the STAP cell phenomenon—inducing pluripotency in somatic cells by exposing them to stress—until next March.
So far, the group has failed to create STAP cells by exposing cells from newborn C57BL/6 mice to a low-pH environment.
Going forward, the researchers plan to conduct their experiments in another mouse strain. They also intend to alter the methods of stressing the cells.
RIKEN investigator Haruko Obokata, PhD, and her colleagues initially reported the STAP cell phenomenon in an article and a letter published in Nature last January.
Not long after the papers were published, members of the scientific community began to question the validity of the research. They voiced concerns about published images, possible plagiarism, and an inability to replicate the experiments described.
So RIKEN launched an investigation, ultimately concluding that Dr Obokata was guilty of misconduct, and some of her colleagues—including the recently deceased Yoshiki Sasai, MD, PhD—were guilty of negligence.
RIKEN also called for the papers to be retracted, and, in July, they were.
Throughout these proceedings, Dr Obokata insisted the STAP cell phenomenon is real. To investigate this claim, RIKEN organized a group of researchers to recreate Dr Obokata’s experiments.
The group has performed 22 experiments using different types of stress and cells from different tissues in C57BL/6 mice, but they have not reproduced the STAP cell phenomenon described in the Nature papers. (A report on these attempts is available in Japanese.)
Still, the investigators are continuing with their experiments and hope to have definitive results by March. ![]()
Acute-Care Surgery Hospitalists: Coming to a Medical Center Near You?
A surgical hospitalist program that's been shown to improve clinical outcomes and reduce inpatient length of stay (LOS) in a non-trauma setting is replicable across the country, says the author of a recent study.
Published in the Journal of the American College of Surgeons, the study reports that a team of surgeons dedicated solely to acute-care surgeries at Sutter Medical Center in Sacramento, Calif., decreased patients' LOS (6.5 days to 5.7 days, P<0.0016), hospital costs ($12,009 to $8306, P<0.0001), and overall complications (21% to 12%, P<0.0001).
Readmissions also "showed a downward trend" but not enough to be statistically significant. The retrospective review looked at the five-year period from 2007 to 2011, which represented the year before the service was initiated and the subsequent four years in which it was practiced.
"By decreasing variation, we improved outcomes and we improved efficiencies," says study author Leon Owens, MD, FACS, president and chief executive officer of the Sacramento-based Surgical Affiliates Management Group, which provided the surgical coverage for Sutter Medical Center. "So we wound up saving money and taking better care of patients. We succeeded because of the uniformity of how we approached the problems and because our team of surgeons is dedicated to doing these surgeries. They do not additionally need to do an elective surgery or deal with other distractions at their office."
Dr. Owens says he believes a well-managed team of acute-care surgeons can improve patient care at any institution that chooses to institute a similar program. "It's not only the brilliance of our doctors but the methodology," he adds. "Our doctors are bright and good, but if you get group-oriented, team-willing, competent surgeons, I believe this is reproducible. We believe this is going to be a common practice across the country. It's just a matter of time to get there." TH
Visit our website for more information on surgical hospitalists.
A surgical hospitalist program that's been shown to improve clinical outcomes and reduce inpatient length of stay (LOS) in a non-trauma setting is replicable across the country, says the author of a recent study.
Published in the Journal of the American College of Surgeons, the study reports that a team of surgeons dedicated solely to acute-care surgeries at Sutter Medical Center in Sacramento, Calif., decreased patients' LOS (6.5 days to 5.7 days, P<0.0016), hospital costs ($12,009 to $8306, P<0.0001), and overall complications (21% to 12%, P<0.0001).
Readmissions also "showed a downward trend" but not enough to be statistically significant. The retrospective review looked at the five-year period from 2007 to 2011, which represented the year before the service was initiated and the subsequent four years in which it was practiced.
"By decreasing variation, we improved outcomes and we improved efficiencies," says study author Leon Owens, MD, FACS, president and chief executive officer of the Sacramento-based Surgical Affiliates Management Group, which provided the surgical coverage for Sutter Medical Center. "So we wound up saving money and taking better care of patients. We succeeded because of the uniformity of how we approached the problems and because our team of surgeons is dedicated to doing these surgeries. They do not additionally need to do an elective surgery or deal with other distractions at their office."
Dr. Owens says he believes a well-managed team of acute-care surgeons can improve patient care at any institution that chooses to institute a similar program. "It's not only the brilliance of our doctors but the methodology," he adds. "Our doctors are bright and good, but if you get group-oriented, team-willing, competent surgeons, I believe this is reproducible. We believe this is going to be a common practice across the country. It's just a matter of time to get there." TH
Visit our website for more information on surgical hospitalists.
A surgical hospitalist program that's been shown to improve clinical outcomes and reduce inpatient length of stay (LOS) in a non-trauma setting is replicable across the country, says the author of a recent study.
Published in the Journal of the American College of Surgeons, the study reports that a team of surgeons dedicated solely to acute-care surgeries at Sutter Medical Center in Sacramento, Calif., decreased patients' LOS (6.5 days to 5.7 days, P<0.0016), hospital costs ($12,009 to $8306, P<0.0001), and overall complications (21% to 12%, P<0.0001).
Readmissions also "showed a downward trend" but not enough to be statistically significant. The retrospective review looked at the five-year period from 2007 to 2011, which represented the year before the service was initiated and the subsequent four years in which it was practiced.
"By decreasing variation, we improved outcomes and we improved efficiencies," says study author Leon Owens, MD, FACS, president and chief executive officer of the Sacramento-based Surgical Affiliates Management Group, which provided the surgical coverage for Sutter Medical Center. "So we wound up saving money and taking better care of patients. We succeeded because of the uniformity of how we approached the problems and because our team of surgeons is dedicated to doing these surgeries. They do not additionally need to do an elective surgery or deal with other distractions at their office."
Dr. Owens says he believes a well-managed team of acute-care surgeons can improve patient care at any institution that chooses to institute a similar program. "It's not only the brilliance of our doctors but the methodology," he adds. "Our doctors are bright and good, but if you get group-oriented, team-willing, competent surgeons, I believe this is reproducible. We believe this is going to be a common practice across the country. It's just a matter of time to get there." TH
Visit our website for more information on surgical hospitalists.