User login
No Benefits to Therapeutic Hypothermia for Severe Bacterial Meningitis
Clinical question
Can therapeutic hypothermia improve functional outcomes in comatose patients with severe bacterial meningitis?
Bottom line
For critically ill patients with severe bacterial meningitis, induced hypothermia using intravascular cooling or other cooling techniques does not improve outcomes and may lead to increased mortality. This trial was stopped early and thus lacked the statistical power to make definitive conclusions about the potential harmful effects of this intervention. (LOE = 1b-)
Reference
Mourvillier B, Tubach F, van de Beek D, et al. Induced hypothermia in severe bacterial meningitis: A randomized clinical trial. JAMA 2013;310(20):2174-2183.
Study design
Randomized controlled trial (nonblinded)
Funding source
Industry + govt
Allocation
Concealed
Setting
Inpatient (ICU only)
Synopsis
Adult patients with suspected or confirmed bacterial meningitis who had a Glasgow Coma Scale (GCS) score of less than 8 for fewer than 12 hours were randomized, using concealed allocation, into the induced hypothermia group or to usual care. All patients received appropriate antimicrobial therapy. In the hypothermia group, intravascular cooling was achieved by a loading dose of 1500 mL 40C saline over 30 minutes, and additional 500 mL boluses over 10 minutes as needed, to achieve a temperature of 33.50C or lower. Other cooling techniques, including ice packs, cooling air, and cooling pads, were also used. Temperatures were maintained between 32C and 34C for 48 hours, and the rewarming phase was passive. Baseline characteristics in the intervention group and control group were similar: mean age was 59 years, median GCS score was 7, all patients were mechanically ventilated, and the causative organism was identified as Streptococcus pneumoniae in the majority of patients. Analysis was by intention to treat. The primary outcome was the score on the Glasgow Outcome Scale. A favorable outcome was a score of 5, indicating mild or no disability; an unfavorable outcome was any score 1 through 4, with 1 indicating death. At 3 months, there was a trend toward unfavorable outcomes in the hypothermia group (86% vs 73% in the control group; relative risk = 1.17; 0.95-1.43; P = .13), as well as a trend toward increased mortality (hazard ratio = 1.76; 0.89-3.45; P = .10). The trial was stopped early because of the higher mortality in the hypothermia group.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question
Can therapeutic hypothermia improve functional outcomes in comatose patients with severe bacterial meningitis?
Bottom line
For critically ill patients with severe bacterial meningitis, induced hypothermia using intravascular cooling or other cooling techniques does not improve outcomes and may lead to increased mortality. This trial was stopped early and thus lacked the statistical power to make definitive conclusions about the potential harmful effects of this intervention. (LOE = 1b-)
Reference
Mourvillier B, Tubach F, van de Beek D, et al. Induced hypothermia in severe bacterial meningitis: A randomized clinical trial. JAMA 2013;310(20):2174-2183.
Study design
Randomized controlled trial (nonblinded)
Funding source
Industry + govt
Allocation
Concealed
Setting
Inpatient (ICU only)
Synopsis
Adult patients with suspected or confirmed bacterial meningitis who had a Glasgow Coma Scale (GCS) score of less than 8 for fewer than 12 hours were randomized, using concealed allocation, into the induced hypothermia group or to usual care. All patients received appropriate antimicrobial therapy. In the hypothermia group, intravascular cooling was achieved by a loading dose of 1500 mL 40C saline over 30 minutes, and additional 500 mL boluses over 10 minutes as needed, to achieve a temperature of 33.50C or lower. Other cooling techniques, including ice packs, cooling air, and cooling pads, were also used. Temperatures were maintained between 32C and 34C for 48 hours, and the rewarming phase was passive. Baseline characteristics in the intervention group and control group were similar: mean age was 59 years, median GCS score was 7, all patients were mechanically ventilated, and the causative organism was identified as Streptococcus pneumoniae in the majority of patients. Analysis was by intention to treat. The primary outcome was the score on the Glasgow Outcome Scale. A favorable outcome was a score of 5, indicating mild or no disability; an unfavorable outcome was any score 1 through 4, with 1 indicating death. At 3 months, there was a trend toward unfavorable outcomes in the hypothermia group (86% vs 73% in the control group; relative risk = 1.17; 0.95-1.43; P = .13), as well as a trend toward increased mortality (hazard ratio = 1.76; 0.89-3.45; P = .10). The trial was stopped early because of the higher mortality in the hypothermia group.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question
Can therapeutic hypothermia improve functional outcomes in comatose patients with severe bacterial meningitis?
Bottom line
For critically ill patients with severe bacterial meningitis, induced hypothermia using intravascular cooling or other cooling techniques does not improve outcomes and may lead to increased mortality. This trial was stopped early and thus lacked the statistical power to make definitive conclusions about the potential harmful effects of this intervention. (LOE = 1b-)
Reference
Mourvillier B, Tubach F, van de Beek D, et al. Induced hypothermia in severe bacterial meningitis: A randomized clinical trial. JAMA 2013;310(20):2174-2183.
Study design
Randomized controlled trial (nonblinded)
Funding source
Industry + govt
Allocation
Concealed
Setting
Inpatient (ICU only)
Synopsis
Adult patients with suspected or confirmed bacterial meningitis who had a Glasgow Coma Scale (GCS) score of less than 8 for fewer than 12 hours were randomized, using concealed allocation, into the induced hypothermia group or to usual care. All patients received appropriate antimicrobial therapy. In the hypothermia group, intravascular cooling was achieved by a loading dose of 1500 mL 40C saline over 30 minutes, and additional 500 mL boluses over 10 minutes as needed, to achieve a temperature of 33.50C or lower. Other cooling techniques, including ice packs, cooling air, and cooling pads, were also used. Temperatures were maintained between 32C and 34C for 48 hours, and the rewarming phase was passive. Baseline characteristics in the intervention group and control group were similar: mean age was 59 years, median GCS score was 7, all patients were mechanically ventilated, and the causative organism was identified as Streptococcus pneumoniae in the majority of patients. Analysis was by intention to treat. The primary outcome was the score on the Glasgow Outcome Scale. A favorable outcome was a score of 5, indicating mild or no disability; an unfavorable outcome was any score 1 through 4, with 1 indicating death. At 3 months, there was a trend toward unfavorable outcomes in the hypothermia group (86% vs 73% in the control group; relative risk = 1.17; 0.95-1.43; P = .13), as well as a trend toward increased mortality (hazard ratio = 1.76; 0.89-3.45; P = .10). The trial was stopped early because of the higher mortality in the hypothermia group.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
ACP Guidelines on Treatment of Anemia in Patients With Heart Disease
Clinical question
How should anemia and iron deficiency be treated in adults with heart disease?
Bottom line
For hospitalized patients with anemia and coronary heart disease, the American College of Physicians recommends a restrictive transfusion strategy and a trigger hemoglobin of 7 g/dL to 8 g/dL. Furthermore, erythropoiesis-stimulating agents (ESAs) should be avoided in patients with coronary heart disease or congestive heart failure and mild to moderate anemia. Evidence regarding intravenous iron for this patient population is inconclusive. (LOE = 1a)
Reference
Kansagara D, Dyer E, Englander H, Fu R, Freeman M, Kagen D. Treatment of anemia in patients with heart disease: A systematic review. Ann Intern Med 2013;159(11):746-757. Qaseem A, Humphrey LL, Fitterman N, Starkey M, Shekelle P; Clinical Guidelines Committee of the American College of Physicians. Treatment of anemia in patients with heart disease: A clinical practice guideline from the American College of Physicians. Ann Intern Med 2013;159(11):770-779.
Study design
Practice guideline
Funding source
Government
Allocation
Uncertain
Setting
Various (meta-analysis)
Synopsis
The American College of Physicians developed this guideline based on a systematic review of the literature that evaluated the benefits and harms of anemia treatment in adults with heart disease. The authors searched multiple databases including MEDLINE and the Cochrane Library, to identify trials that studied the effects of blood transfusions, ESAs, and iron in patients with anemia and congestive heart failure or coronary heart disease. Observational transfusion studies were also included. Two reviewers independently assessed studies for inclusion, extracted data, and assessed study quality. Data was combined for meta-analysis when possible. Although it was low-quality evidence, liberal transfusion strategies as compared with restrictive strategies in treating anemia showed no effect on mortality for patients with heart disease. Moderate-strength to high-strength evidence from the ESA studies also showed no benefit, but did show a potential for harm, including an increased risk of venous thromboembolism. Finally, although few studies evaluated intravenous iron therapy, one good-quality study showed that it increased short-term exercise tolerance and quality of life in patients with heart failure. Based on these findings, the American College of Physicians guideline committee makes the following recommendations: (1) Use a restrictive red blood cell transfusion strategy with a hemoglobin threshold of 7 g/dL to 8 g/dL in hospitalized patients with coronary heart disease; and (2) avoid ESAs in patients with mild to moderate anemia and congestive heart failure or coronary heart disease. Because of lack of evidence regarding long-term outcomes and possible harms, as well as limited overall data, there was no recommendation made regarding the use of intravenous iron.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question
How should anemia and iron deficiency be treated in adults with heart disease?
Bottom line
For hospitalized patients with anemia and coronary heart disease, the American College of Physicians recommends a restrictive transfusion strategy and a trigger hemoglobin of 7 g/dL to 8 g/dL. Furthermore, erythropoiesis-stimulating agents (ESAs) should be avoided in patients with coronary heart disease or congestive heart failure and mild to moderate anemia. Evidence regarding intravenous iron for this patient population is inconclusive. (LOE = 1a)
Reference
Kansagara D, Dyer E, Englander H, Fu R, Freeman M, Kagen D. Treatment of anemia in patients with heart disease: A systematic review. Ann Intern Med 2013;159(11):746-757. Qaseem A, Humphrey LL, Fitterman N, Starkey M, Shekelle P; Clinical Guidelines Committee of the American College of Physicians. Treatment of anemia in patients with heart disease: A clinical practice guideline from the American College of Physicians. Ann Intern Med 2013;159(11):770-779.
Study design
Practice guideline
Funding source
Government
Allocation
Uncertain
Setting
Various (meta-analysis)
Synopsis
The American College of Physicians developed this guideline based on a systematic review of the literature that evaluated the benefits and harms of anemia treatment in adults with heart disease. The authors searched multiple databases including MEDLINE and the Cochrane Library, to identify trials that studied the effects of blood transfusions, ESAs, and iron in patients with anemia and congestive heart failure or coronary heart disease. Observational transfusion studies were also included. Two reviewers independently assessed studies for inclusion, extracted data, and assessed study quality. Data was combined for meta-analysis when possible. Although it was low-quality evidence, liberal transfusion strategies as compared with restrictive strategies in treating anemia showed no effect on mortality for patients with heart disease. Moderate-strength to high-strength evidence from the ESA studies also showed no benefit, but did show a potential for harm, including an increased risk of venous thromboembolism. Finally, although few studies evaluated intravenous iron therapy, one good-quality study showed that it increased short-term exercise tolerance and quality of life in patients with heart failure. Based on these findings, the American College of Physicians guideline committee makes the following recommendations: (1) Use a restrictive red blood cell transfusion strategy with a hemoglobin threshold of 7 g/dL to 8 g/dL in hospitalized patients with coronary heart disease; and (2) avoid ESAs in patients with mild to moderate anemia and congestive heart failure or coronary heart disease. Because of lack of evidence regarding long-term outcomes and possible harms, as well as limited overall data, there was no recommendation made regarding the use of intravenous iron.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
Clinical question
How should anemia and iron deficiency be treated in adults with heart disease?
Bottom line
For hospitalized patients with anemia and coronary heart disease, the American College of Physicians recommends a restrictive transfusion strategy and a trigger hemoglobin of 7 g/dL to 8 g/dL. Furthermore, erythropoiesis-stimulating agents (ESAs) should be avoided in patients with coronary heart disease or congestive heart failure and mild to moderate anemia. Evidence regarding intravenous iron for this patient population is inconclusive. (LOE = 1a)
Reference
Kansagara D, Dyer E, Englander H, Fu R, Freeman M, Kagen D. Treatment of anemia in patients with heart disease: A systematic review. Ann Intern Med 2013;159(11):746-757. Qaseem A, Humphrey LL, Fitterman N, Starkey M, Shekelle P; Clinical Guidelines Committee of the American College of Physicians. Treatment of anemia in patients with heart disease: A clinical practice guideline from the American College of Physicians. Ann Intern Med 2013;159(11):770-779.
Study design
Practice guideline
Funding source
Government
Allocation
Uncertain
Setting
Various (meta-analysis)
Synopsis
The American College of Physicians developed this guideline based on a systematic review of the literature that evaluated the benefits and harms of anemia treatment in adults with heart disease. The authors searched multiple databases including MEDLINE and the Cochrane Library, to identify trials that studied the effects of blood transfusions, ESAs, and iron in patients with anemia and congestive heart failure or coronary heart disease. Observational transfusion studies were also included. Two reviewers independently assessed studies for inclusion, extracted data, and assessed study quality. Data was combined for meta-analysis when possible. Although it was low-quality evidence, liberal transfusion strategies as compared with restrictive strategies in treating anemia showed no effect on mortality for patients with heart disease. Moderate-strength to high-strength evidence from the ESA studies also showed no benefit, but did show a potential for harm, including an increased risk of venous thromboembolism. Finally, although few studies evaluated intravenous iron therapy, one good-quality study showed that it increased short-term exercise tolerance and quality of life in patients with heart failure. Based on these findings, the American College of Physicians guideline committee makes the following recommendations: (1) Use a restrictive red blood cell transfusion strategy with a hemoglobin threshold of 7 g/dL to 8 g/dL in hospitalized patients with coronary heart disease; and (2) avoid ESAs in patients with mild to moderate anemia and congestive heart failure or coronary heart disease. Because of lack of evidence regarding long-term outcomes and possible harms, as well as limited overall data, there was no recommendation made regarding the use of intravenous iron.
Dr. Kulkarni is an assistant professor of hospital medicine at Northwestern University in Chicago.
2014 Update on abnormal uterine bleeding
As recently defined by the International Federation of Gynecology and Obstetrics (FIGO)—and endorsed by the American College of Obstetricians and Gynecologists—the term “abnormal uterine bleeding” (AUB) now describes any departure from normal menstrual bleeding.1 To determine the most appropriate intervention for this widespread problem, FIGO proposed that clinicians consider potential contributors to the clinical problem by investigating and categorizing patients according to the following system:
- Polyp
- Adenomyosis
- Leiomyoma
- Malignancy and hyperplasia
- Coagulopathy
- Ovulatory disorders
- Endometrial dysfunction
- Iatrogenic
- Not otherwise classified.
A given individual may be found to have one or more of these features, but not all of the features may contribute to the AUB. To facilitate their use, these nine causes are more commonly identified using the acronym PALM-COEIN.
In this article, I focus on three of these categories, presenting recent data on AUB associated with leiomyomata (AUB-L) or adenomyosis (AUB-A), and AUB of an iatrogenic nature (AUB-I).
AUB-L: SATISFACTION RATES ARE SIMILAR 5 YEARS AFTER FIBROID TREATMENT BY SURGERY OR UTERINE ARTERY EMBOLIZATION
Gupta JK, Sinha A, Lumsden MA, Hickey M. Uterine artery embolization for symptomatic uterine fibroids. Cochrane Database Syst Rev. 2012;5:CD005073. doi:10.1002/14651858.CD005073.pub3.
Women who undergo uterine artery embolization (UAE) for the treatment of symptomatic uterine fibroids are just as satisfied with the outcome as women treated with hysterectomy or myomectomy, according to this 2012 review from the Cochrane Database.
Gupta and colleagues found similar patient-satisfaction rates at 5 years (odds ratio [OR] 0.9; 95% confidence interval [CI], 0.45–1.8), although women undergoing UAE were more likely to require additional interventions within 2 years (56 additional interventions per 1,000 women for surgery vs 250 per 1,000 women for UAE; OR, 5.64).
Details and general findings
Gupta and colleagues selected randomized, controlled trials comparing UAE with surgery:
- three trials of UAE versus abdominal hysterectomy (n = 291)
- one trial of UAE versus hysterectomy or myomectomy (the specific surgery was determined by patient preference) (n = 157)
- one trial of UAE versus myomectomy in women desiring future childbearing (n = 121).
In these trials, UAE was bilateral and involved the use of permanent embolic material.
Among the findings:
- Costs were lower with UAE, as assessed by measuring the duration of the procedure, length of hospitalization, and time to resumption of normal activities.
- Ovarian-failure rates were comparable between women in the UAE and surgery groups. Ovarian function was assessed by measuring follicle-stimulating hormone (FSH), although FSH thresholds varied in some of the studies.
- Pregnancy was less likely after UAE than after myomectomy. In the trial comparing UAE with myomectomy, 26 women later tried to conceive after UAE versus 40 after myomectomy. Significantly fewer women became pregnant after UAE (OR, 0.29; 95% CI, 0.10–0.85).
Related Article: Update on Fertility G. David Adamson, MD; Mary E. Abusief, MD (February 2014)
Bleeding outcomes were not measured
Strengths of this systematic review are its inclusion of high-quality, randomized, controlled trials and its assessment of ovarian-failure rates. However, a major weakness is the fact that its design does not allow for discrete evaluation of bleeding outcomes. Nor can its findings be broken down by the type of leiomyoma being treated.
WHAT THIS EVIDENCE MEANS FOR PRACTICE
This review demonstrates that women are satisfied with outcomes five years after UAE and that ovarian failure is not more common after UAE than after surgery. Although the available evidence demonstrates that pregnancy following UAE is possible, women requiring a surgical procedure for AUB-L who are uncertain about their childbearing plans or who are hoping to conceive should be encouraged to select myomectomy as their intervention of choice.
AUB-A: FOR ADENOMYOSIS-ASSOCIATED AUB, CONSIDER THE LNG-IUS AS AN ALTERNATIVE TO HYSTERECTOMY
Ozdegirmenci O, Kayikcioglu F, Akgul MA, et al. Comparison of levonorgestrel intrauterine system versus hysterectomy on efficacy and quality of life in patients with adenomyosis. Fertil Steril. 2011;95(2):497–502.
In a small randomized, controlled trial of the levonorgestrel-releasing intrauterine system (LNG-IUS; Mirena) versus hysterectomy for adenomyosis-associated AUB, women allocated to the LNG-IUS experienced a reduction in bleeding and comparable gains in hemoglobin values during the first year of use. Both the LNG-IUS and hysterectomy improved health-related quality of life, but the LNG-IUS was associated with superior improvements in measures of psychological and social functioning.
Related Article: Update: Minimally invasive gynecology Amy Garcia, MD (April 2013)
Details and general findings of the trial
Eighty-six women were enrolled in the trial after exclusion of endometrial pathology as a cause of their heavy menstrual bleeding and after transvaginal ultrasound and magnetic resonance imaging findings were consistent with the diagnosis of adenomyosis. Participants then were randomly assigned to undergo hysterectomy or insertion of an LNG-IUS (43 women in each group). At baseline, the mean (SD) age was 44.28 (4.36) years among women in the LNG-IUS group versus 46.38 (3.76) years among women undergoing hysterectomy (P = .032), a statistical difference that I suspect is not clinically significant.
Menstrual bleeding, hemoglobin levels, and quality of life were assessed prior to insertion or surgery, and again at 6- and 12-month follow-up. Eleven women in the hysterectomy group were lost to follow-up.
General findings of the trial include:
- Women in the LNG-IUS group had a mean reduction in the volume of menstrual bleeding—as measured by the number of pads used—from two pads to one pad at 6 months, remaining at that level until 12 months. Serum hemoglobin levels increased from a median of just over
11 g/dL at the time of insertion to 13 g/dL at 6 months and slightly higher at 12 months. In the five self-reported quality-of-life domains assessed (physical, psychological, social, environmental, and a national environmental domain), women using the LNG-IUS demonstrated improvement in all five. - Women in the hysterectomy group were treated using an abdominal surgical approach, with one patient experiencing postoperative wound infection that required secondary suture. Postoperative pathologic analysis found that 21 of these women (65.6%) had adenomyosis, six women (18.8%) had myomas, three women (9.4%) had both adenomyosis and a myoma, and two women (6.2%) had a normal uterus. Serum hemoglobin levels increased from a median of roughly 10.5 g/dL at the time of treatment to 13 g/dL at 6 months and slightly higher at 12 months. (There were no statistically significant differences in hemoglobin values between the LNG-IUS and hysterectomy groups at any point in the study.) Quality of life improved in three of the five domains assessed (physical and both environmental domains).
Although 11 women were lost to follow-up, this trial appeared to have an adequate sample size to examine the selected outcomes, and the population was well defined.
Two weaknesses were the limited follow-up (only 12 months) and the use of quality-of-life measures designed for a Turkish population (the trial was conducted in Turkey), which may or may not be fully applicable to a US population.
WHAT THIS EVIDENCE MEANS FOR PRACTICE
The relationship of adenomyosis to gynecologic symptoms, including heavy menstrual bleeding and dysmenorrhea, needs further study. However, this trial confirmed that transvaginal ultrasound is helpful in the nonsurgical diagnosis of adenomyosis and suggests that the LNG-IUS may be as effective at 1 year as hysterectomy for the treatment of adenomyosis-associated heavy menstrual bleeding (AUB-A).
Clinicians who perform office-based ultrasound to assess AUB should familiarize themselves with the criteria for ultrasonic diagnosis of adenomyosis. These criteria include the presence of heterogeneous myometrial echogenicity, a loss of clarity of the endo-myometrial interface, typically radially oriented linear striations, the appearance of myometrial cysts, and an overall globular enlarged uterus characterized by asymmetric thickening of the myometrium.2
In patients with heavy menstrual bleeding who have these findings, particularly if there is coexistent dysmenorrhea and uterine tenderness, it behooves the clinician to consider the LNG-IUS as first-line therapy, especially for women who wish to preserve fertility, but also for women for whom fertility is not an issue.
There is some evidence that the therapeutic effect of the LNG-IUS containing 20 µg of levonorgestrel may start to fade at 2 or 3 years, a possibility that should be shared with patients.3 Other features, such as cavity size, thickness of the myometrium, and the coexistence of clinically relevant leiomyomas, have not been evaluated but may have an impact on the clinical response.
AUB-I: LOW-DOSE DOXYCYCLINE REDUCES THE TIME TO AMENORRHEA IN USERS OF CONTINUOUS ORAL CONTRACEPTIVES
Kaneshiro B, Edelman A, Carlson NE, Nichols M, Forbes MM, Jensen J. A randomized controlled trial of subantimicrobial-dose doxycycline to prevent unscheduled bleeding with continuous oral contraceptive pill use. Contraception. 2012;85(4):351–358.
Unscheduled bleeding is the most common complaint among women who use continuous combination oral contraceptives (OCs). Because unscheduled bleeding has been correlated with the upregulation of matrix metalloprotineases (MMPs), Kaneshiro and colleagues conducted a randomized, controlled trial of doxycycline (an MMP inhibitor) versus placebo among users of continuous OCs. The addition of doxycycline to the OC regimen did not significantly reduce unscheduled bleeding during the first 84 days of use, but it did shorten the time required to achieve amenorrhea (mean of 61.7 days for doxycycline vs 85.2 days for placebo; standard error [SE], 7.7 vs 6.7, respectively; P = .03).
Related Article: Big step forward and downward: An OC with 10 μg of estrogen Robert L. Barbieri, MD (Editorial, May 2011)
Details and general findings of the trial
Participants (n = 65) were healthy women aged 18 to 45 years who had no contraindications to continuous use of combination OCs. Prior to enrollment, they all had used cyclic combination contraception (pill, patch, or ring) without unscheduled bleeding, thereby avoiding the “transition bleeding” that often occurs when continuous OCs are initiated.
All women in the trial were started on continuous OCs (20 µg ethinyl estradiol with 100 µg levonorgestrel; Aviane) and then randomly assigned to receive one of the following for 84 days in addition to the OC:
- doxycycline 40 mg daily (controlled-release Oracea), a subantimicrobial dose
- placebo.
After 84 days, doxycycline was discontinued, and participants were observed for an additional 28 days on the OC regimen alone for the documentation of bleeding patterns.
General findings:
- The number of bleeding and spotting days decreased in both groups over the course of the study.
- During the first 84 days of the trial, bleeding and spotting occurred among a median of 11 and 17 women in the doxycycline and placebo groups, respectively, and bleeding alone (without spotting) occurred in a median 3 and 4 women in the doxycycline and placebo groups, respectively.
- During the 28-day observation period, bleeding and spotting occurred among a median of 0 and 6 women in the doxycycline and placebo groups, respectively. Bleeding alone (without spotting) was absent in both groups.
- Women in the doxycycline group were significantly less likely to report side effects such as headache, depressed mood, and abdominal cramping. However, they were more likely to prefer continuous OCs without doxycycline, compared with women receiving placebo (16.1% vs 10.7%).
WHAT THIS EVIDENCE MEANS FOR PRACTICE
This trial increases our insight into AUB associated with the use of progestins and suggests that concomitant doxycycline may reduce unscheduled bleeding and spotting in women using continuous combination OCs. The trial was of adequate sample size for the primary outcomes, lending credence to its findings, although longer-term data would be helpful.
I have included this trial for two reasons:
It offers useful information regarding the mechanisms and potential prevention or reduction of AUB-I in users of continuous combined estrogen-progestin contraception.
Doxycycline is one of the agents covered in a Cochrane review of high-quality research into AUB-I in women using progestin-only products, including injectables, implantables, intrauterine systems, and oral agents.4 Estrogens have been shown to have some value in reducing breakthrough bleeding associated with depot medroxyprogesterone acetate, and individual use of tranexamic acid or doxycycline has shown value in terminating an episode of breakthrough bleeding in women using progestin-only contraceptives.
TELL US WHAT YOU THINK!
Share your thoughts on this article or on any topic relevant to ObGyns and women’s health practitioners. Tell us which topics you’d like to see covered in future issues, and what challenges you face in daily practice. We will consider publishing your letter and in a future issue.
Send your letter to: [email protected] Please include the city and state in which you practice.
Stay in touch! Your feedback is important to us!
- Munro MG, Critchley HO, Broder MS, Fraser IS; FIGO Working Group on Menstrual Disorders. The FIGO classification for causes of abnormal bleeding in the reproductive years. Fertil Steril. 2011;95(7):2204–2208.
- Champaneria R, Abedin P, Daniels J, Balogun M, Khan KS. Ultrasound scan and magnetic resonance imaging for the diagnosis of adenomyosis: Systematic review comparing test accuracy. Acta Obstet Gynecol Scand. 2010;89(11):1374–1384.
- Cho S, Nam A, Kim H, et al. Clinical effects of the levonorgestrel-releasing intrauterine device in patients with adenomyosis. Am J Obstet Gynecol. 2008;198(4):373.e1–e7.
- Abdel-Aleem H, d’Arcangues C, Vogelsong KM, Gaffield ML, Gulmezoglu AM. Treatment of vaginal bleeding irregularities induced by progestin-only contraceptives. Cochrane Database Syst Rev. 2013;10:CD003449.

Malcolm G. Munro, MD, is Professor of Obstetrics and Gynecology at the David Geffen School of Medicine at UCLA and Director of Gynecologic Services at Kaiser Permanente, Los Angeles Medical Center, in Los Angeles, California
The author reports that he is a consultant to Bayer HealthCare.
Malcolm G. Munro, MD, is Professor of Obstetrics and Gynecology at the David Geffen School of Medicine at UCLA and Director of Gynecologic Services at Kaiser Permanente, Los Angeles Medical Center, in Los Angeles, California
The author reports that he is a consultant to Bayer HealthCare.
Malcolm G. Munro, MD, is Professor of Obstetrics and Gynecology at the David Geffen School of Medicine at UCLA and Director of Gynecologic Services at Kaiser Permanente, Los Angeles Medical Center, in Los Angeles, California
The author reports that he is a consultant to Bayer HealthCare.
As recently defined by the International Federation of Gynecology and Obstetrics (FIGO)—and endorsed by the American College of Obstetricians and Gynecologists—the term “abnormal uterine bleeding” (AUB) now describes any departure from normal menstrual bleeding.1 To determine the most appropriate intervention for this widespread problem, FIGO proposed that clinicians consider potential contributors to the clinical problem by investigating and categorizing patients according to the following system:
- Polyp
- Adenomyosis
- Leiomyoma
- Malignancy and hyperplasia
- Coagulopathy
- Ovulatory disorders
- Endometrial dysfunction
- Iatrogenic
- Not otherwise classified.
A given individual may be found to have one or more of these features, but not all of the features may contribute to the AUB. To facilitate their use, these nine causes are more commonly identified using the acronym PALM-COEIN.
In this article, I focus on three of these categories, presenting recent data on AUB associated with leiomyomata (AUB-L) or adenomyosis (AUB-A), and AUB of an iatrogenic nature (AUB-I).
AUB-L: SATISFACTION RATES ARE SIMILAR 5 YEARS AFTER FIBROID TREATMENT BY SURGERY OR UTERINE ARTERY EMBOLIZATION
Gupta JK, Sinha A, Lumsden MA, Hickey M. Uterine artery embolization for symptomatic uterine fibroids. Cochrane Database Syst Rev. 2012;5:CD005073. doi:10.1002/14651858.CD005073.pub3.
Women who undergo uterine artery embolization (UAE) for the treatment of symptomatic uterine fibroids are just as satisfied with the outcome as women treated with hysterectomy or myomectomy, according to this 2012 review from the Cochrane Database.
Gupta and colleagues found similar patient-satisfaction rates at 5 years (odds ratio [OR] 0.9; 95% confidence interval [CI], 0.45–1.8), although women undergoing UAE were more likely to require additional interventions within 2 years (56 additional interventions per 1,000 women for surgery vs 250 per 1,000 women for UAE; OR, 5.64).
Details and general findings
Gupta and colleagues selected randomized, controlled trials comparing UAE with surgery:
- three trials of UAE versus abdominal hysterectomy (n = 291)
- one trial of UAE versus hysterectomy or myomectomy (the specific surgery was determined by patient preference) (n = 157)
- one trial of UAE versus myomectomy in women desiring future childbearing (n = 121).
In these trials, UAE was bilateral and involved the use of permanent embolic material.
Among the findings:
- Costs were lower with UAE, as assessed by measuring the duration of the procedure, length of hospitalization, and time to resumption of normal activities.
- Ovarian-failure rates were comparable between women in the UAE and surgery groups. Ovarian function was assessed by measuring follicle-stimulating hormone (FSH), although FSH thresholds varied in some of the studies.
- Pregnancy was less likely after UAE than after myomectomy. In the trial comparing UAE with myomectomy, 26 women later tried to conceive after UAE versus 40 after myomectomy. Significantly fewer women became pregnant after UAE (OR, 0.29; 95% CI, 0.10–0.85).
Related Article: Update on Fertility G. David Adamson, MD; Mary E. Abusief, MD (February 2014)
Bleeding outcomes were not measured
Strengths of this systematic review are its inclusion of high-quality, randomized, controlled trials and its assessment of ovarian-failure rates. However, a major weakness is the fact that its design does not allow for discrete evaluation of bleeding outcomes. Nor can its findings be broken down by the type of leiomyoma being treated.
WHAT THIS EVIDENCE MEANS FOR PRACTICE
This review demonstrates that women are satisfied with outcomes five years after UAE and that ovarian failure is not more common after UAE than after surgery. Although the available evidence demonstrates that pregnancy following UAE is possible, women requiring a surgical procedure for AUB-L who are uncertain about their childbearing plans or who are hoping to conceive should be encouraged to select myomectomy as their intervention of choice.
AUB-A: FOR ADENOMYOSIS-ASSOCIATED AUB, CONSIDER THE LNG-IUS AS AN ALTERNATIVE TO HYSTERECTOMY
Ozdegirmenci O, Kayikcioglu F, Akgul MA, et al. Comparison of levonorgestrel intrauterine system versus hysterectomy on efficacy and quality of life in patients with adenomyosis. Fertil Steril. 2011;95(2):497–502.
In a small randomized, controlled trial of the levonorgestrel-releasing intrauterine system (LNG-IUS; Mirena) versus hysterectomy for adenomyosis-associated AUB, women allocated to the LNG-IUS experienced a reduction in bleeding and comparable gains in hemoglobin values during the first year of use. Both the LNG-IUS and hysterectomy improved health-related quality of life, but the LNG-IUS was associated with superior improvements in measures of psychological and social functioning.
Related Article: Update: Minimally invasive gynecology Amy Garcia, MD (April 2013)
Details and general findings of the trial
Eighty-six women were enrolled in the trial after exclusion of endometrial pathology as a cause of their heavy menstrual bleeding and after transvaginal ultrasound and magnetic resonance imaging findings were consistent with the diagnosis of adenomyosis. Participants then were randomly assigned to undergo hysterectomy or insertion of an LNG-IUS (43 women in each group). At baseline, the mean (SD) age was 44.28 (4.36) years among women in the LNG-IUS group versus 46.38 (3.76) years among women undergoing hysterectomy (P = .032), a statistical difference that I suspect is not clinically significant.
Menstrual bleeding, hemoglobin levels, and quality of life were assessed prior to insertion or surgery, and again at 6- and 12-month follow-up. Eleven women in the hysterectomy group were lost to follow-up.
General findings of the trial include:
- Women in the LNG-IUS group had a mean reduction in the volume of menstrual bleeding—as measured by the number of pads used—from two pads to one pad at 6 months, remaining at that level until 12 months. Serum hemoglobin levels increased from a median of just over
11 g/dL at the time of insertion to 13 g/dL at 6 months and slightly higher at 12 months. In the five self-reported quality-of-life domains assessed (physical, psychological, social, environmental, and a national environmental domain), women using the LNG-IUS demonstrated improvement in all five. - Women in the hysterectomy group were treated using an abdominal surgical approach, with one patient experiencing postoperative wound infection that required secondary suture. Postoperative pathologic analysis found that 21 of these women (65.6%) had adenomyosis, six women (18.8%) had myomas, three women (9.4%) had both adenomyosis and a myoma, and two women (6.2%) had a normal uterus. Serum hemoglobin levels increased from a median of roughly 10.5 g/dL at the time of treatment to 13 g/dL at 6 months and slightly higher at 12 months. (There were no statistically significant differences in hemoglobin values between the LNG-IUS and hysterectomy groups at any point in the study.) Quality of life improved in three of the five domains assessed (physical and both environmental domains).
Although 11 women were lost to follow-up, this trial appeared to have an adequate sample size to examine the selected outcomes, and the population was well defined.
Two weaknesses were the limited follow-up (only 12 months) and the use of quality-of-life measures designed for a Turkish population (the trial was conducted in Turkey), which may or may not be fully applicable to a US population.
WHAT THIS EVIDENCE MEANS FOR PRACTICE
The relationship of adenomyosis to gynecologic symptoms, including heavy menstrual bleeding and dysmenorrhea, needs further study. However, this trial confirmed that transvaginal ultrasound is helpful in the nonsurgical diagnosis of adenomyosis and suggests that the LNG-IUS may be as effective at 1 year as hysterectomy for the treatment of adenomyosis-associated heavy menstrual bleeding (AUB-A).
Clinicians who perform office-based ultrasound to assess AUB should familiarize themselves with the criteria for ultrasonic diagnosis of adenomyosis. These criteria include the presence of heterogeneous myometrial echogenicity, a loss of clarity of the endo-myometrial interface, typically radially oriented linear striations, the appearance of myometrial cysts, and an overall globular enlarged uterus characterized by asymmetric thickening of the myometrium.2
In patients with heavy menstrual bleeding who have these findings, particularly if there is coexistent dysmenorrhea and uterine tenderness, it behooves the clinician to consider the LNG-IUS as first-line therapy, especially for women who wish to preserve fertility, but also for women for whom fertility is not an issue.
There is some evidence that the therapeutic effect of the LNG-IUS containing 20 µg of levonorgestrel may start to fade at 2 or 3 years, a possibility that should be shared with patients.3 Other features, such as cavity size, thickness of the myometrium, and the coexistence of clinically relevant leiomyomas, have not been evaluated but may have an impact on the clinical response.
AUB-I: LOW-DOSE DOXYCYCLINE REDUCES THE TIME TO AMENORRHEA IN USERS OF CONTINUOUS ORAL CONTRACEPTIVES
Kaneshiro B, Edelman A, Carlson NE, Nichols M, Forbes MM, Jensen J. A randomized controlled trial of subantimicrobial-dose doxycycline to prevent unscheduled bleeding with continuous oral contraceptive pill use. Contraception. 2012;85(4):351–358.
Unscheduled bleeding is the most common complaint among women who use continuous combination oral contraceptives (OCs). Because unscheduled bleeding has been correlated with the upregulation of matrix metalloprotineases (MMPs), Kaneshiro and colleagues conducted a randomized, controlled trial of doxycycline (an MMP inhibitor) versus placebo among users of continuous OCs. The addition of doxycycline to the OC regimen did not significantly reduce unscheduled bleeding during the first 84 days of use, but it did shorten the time required to achieve amenorrhea (mean of 61.7 days for doxycycline vs 85.2 days for placebo; standard error [SE], 7.7 vs 6.7, respectively; P = .03).
Related Article: Big step forward and downward: An OC with 10 μg of estrogen Robert L. Barbieri, MD (Editorial, May 2011)
Details and general findings of the trial
Participants (n = 65) were healthy women aged 18 to 45 years who had no contraindications to continuous use of combination OCs. Prior to enrollment, they all had used cyclic combination contraception (pill, patch, or ring) without unscheduled bleeding, thereby avoiding the “transition bleeding” that often occurs when continuous OCs are initiated.
All women in the trial were started on continuous OCs (20 µg ethinyl estradiol with 100 µg levonorgestrel; Aviane) and then randomly assigned to receive one of the following for 84 days in addition to the OC:
- doxycycline 40 mg daily (controlled-release Oracea), a subantimicrobial dose
- placebo.
After 84 days, doxycycline was discontinued, and participants were observed for an additional 28 days on the OC regimen alone for the documentation of bleeding patterns.
General findings:
- The number of bleeding and spotting days decreased in both groups over the course of the study.
- During the first 84 days of the trial, bleeding and spotting occurred among a median of 11 and 17 women in the doxycycline and placebo groups, respectively, and bleeding alone (without spotting) occurred in a median 3 and 4 women in the doxycycline and placebo groups, respectively.
- During the 28-day observation period, bleeding and spotting occurred among a median of 0 and 6 women in the doxycycline and placebo groups, respectively. Bleeding alone (without spotting) was absent in both groups.
- Women in the doxycycline group were significantly less likely to report side effects such as headache, depressed mood, and abdominal cramping. However, they were more likely to prefer continuous OCs without doxycycline, compared with women receiving placebo (16.1% vs 10.7%).
WHAT THIS EVIDENCE MEANS FOR PRACTICE
This trial increases our insight into AUB associated with the use of progestins and suggests that concomitant doxycycline may reduce unscheduled bleeding and spotting in women using continuous combination OCs. The trial was of adequate sample size for the primary outcomes, lending credence to its findings, although longer-term data would be helpful.
I have included this trial for two reasons:
It offers useful information regarding the mechanisms and potential prevention or reduction of AUB-I in users of continuous combined estrogen-progestin contraception.
Doxycycline is one of the agents covered in a Cochrane review of high-quality research into AUB-I in women using progestin-only products, including injectables, implantables, intrauterine systems, and oral agents.4 Estrogens have been shown to have some value in reducing breakthrough bleeding associated with depot medroxyprogesterone acetate, and individual use of tranexamic acid or doxycycline has shown value in terminating an episode of breakthrough bleeding in women using progestin-only contraceptives.
TELL US WHAT YOU THINK!
Share your thoughts on this article or on any topic relevant to ObGyns and women’s health practitioners. Tell us which topics you’d like to see covered in future issues, and what challenges you face in daily practice. We will consider publishing your letter and in a future issue.
Send your letter to: [email protected] Please include the city and state in which you practice.
Stay in touch! Your feedback is important to us!
As recently defined by the International Federation of Gynecology and Obstetrics (FIGO)—and endorsed by the American College of Obstetricians and Gynecologists—the term “abnormal uterine bleeding” (AUB) now describes any departure from normal menstrual bleeding.1 To determine the most appropriate intervention for this widespread problem, FIGO proposed that clinicians consider potential contributors to the clinical problem by investigating and categorizing patients according to the following system:
- Polyp
- Adenomyosis
- Leiomyoma
- Malignancy and hyperplasia
- Coagulopathy
- Ovulatory disorders
- Endometrial dysfunction
- Iatrogenic
- Not otherwise classified.
A given individual may be found to have one or more of these features, but not all of the features may contribute to the AUB. To facilitate their use, these nine causes are more commonly identified using the acronym PALM-COEIN.
In this article, I focus on three of these categories, presenting recent data on AUB associated with leiomyomata (AUB-L) or adenomyosis (AUB-A), and AUB of an iatrogenic nature (AUB-I).
AUB-L: SATISFACTION RATES ARE SIMILAR 5 YEARS AFTER FIBROID TREATMENT BY SURGERY OR UTERINE ARTERY EMBOLIZATION
Gupta JK, Sinha A, Lumsden MA, Hickey M. Uterine artery embolization for symptomatic uterine fibroids. Cochrane Database Syst Rev. 2012;5:CD005073. doi:10.1002/14651858.CD005073.pub3.
Women who undergo uterine artery embolization (UAE) for the treatment of symptomatic uterine fibroids are just as satisfied with the outcome as women treated with hysterectomy or myomectomy, according to this 2012 review from the Cochrane Database.
Gupta and colleagues found similar patient-satisfaction rates at 5 years (odds ratio [OR] 0.9; 95% confidence interval [CI], 0.45–1.8), although women undergoing UAE were more likely to require additional interventions within 2 years (56 additional interventions per 1,000 women for surgery vs 250 per 1,000 women for UAE; OR, 5.64).
Details and general findings
Gupta and colleagues selected randomized, controlled trials comparing UAE with surgery:
- three trials of UAE versus abdominal hysterectomy (n = 291)
- one trial of UAE versus hysterectomy or myomectomy (the specific surgery was determined by patient preference) (n = 157)
- one trial of UAE versus myomectomy in women desiring future childbearing (n = 121).
In these trials, UAE was bilateral and involved the use of permanent embolic material.
Among the findings:
- Costs were lower with UAE, as assessed by measuring the duration of the procedure, length of hospitalization, and time to resumption of normal activities.
- Ovarian-failure rates were comparable between women in the UAE and surgery groups. Ovarian function was assessed by measuring follicle-stimulating hormone (FSH), although FSH thresholds varied in some of the studies.
- Pregnancy was less likely after UAE than after myomectomy. In the trial comparing UAE with myomectomy, 26 women later tried to conceive after UAE versus 40 after myomectomy. Significantly fewer women became pregnant after UAE (OR, 0.29; 95% CI, 0.10–0.85).
Related Article: Update on Fertility G. David Adamson, MD; Mary E. Abusief, MD (February 2014)
Bleeding outcomes were not measured
Strengths of this systematic review are its inclusion of high-quality, randomized, controlled trials and its assessment of ovarian-failure rates. However, a major weakness is the fact that its design does not allow for discrete evaluation of bleeding outcomes. Nor can its findings be broken down by the type of leiomyoma being treated.
WHAT THIS EVIDENCE MEANS FOR PRACTICE
This review demonstrates that women are satisfied with outcomes five years after UAE and that ovarian failure is not more common after UAE than after surgery. Although the available evidence demonstrates that pregnancy following UAE is possible, women requiring a surgical procedure for AUB-L who are uncertain about their childbearing plans or who are hoping to conceive should be encouraged to select myomectomy as their intervention of choice.
AUB-A: FOR ADENOMYOSIS-ASSOCIATED AUB, CONSIDER THE LNG-IUS AS AN ALTERNATIVE TO HYSTERECTOMY
Ozdegirmenci O, Kayikcioglu F, Akgul MA, et al. Comparison of levonorgestrel intrauterine system versus hysterectomy on efficacy and quality of life in patients with adenomyosis. Fertil Steril. 2011;95(2):497–502.
In a small randomized, controlled trial of the levonorgestrel-releasing intrauterine system (LNG-IUS; Mirena) versus hysterectomy for adenomyosis-associated AUB, women allocated to the LNG-IUS experienced a reduction in bleeding and comparable gains in hemoglobin values during the first year of use. Both the LNG-IUS and hysterectomy improved health-related quality of life, but the LNG-IUS was associated with superior improvements in measures of psychological and social functioning.
Related Article: Update: Minimally invasive gynecology Amy Garcia, MD (April 2013)
Details and general findings of the trial
Eighty-six women were enrolled in the trial after exclusion of endometrial pathology as a cause of their heavy menstrual bleeding and after transvaginal ultrasound and magnetic resonance imaging findings were consistent with the diagnosis of adenomyosis. Participants then were randomly assigned to undergo hysterectomy or insertion of an LNG-IUS (43 women in each group). At baseline, the mean (SD) age was 44.28 (4.36) years among women in the LNG-IUS group versus 46.38 (3.76) years among women undergoing hysterectomy (P = .032), a statistical difference that I suspect is not clinically significant.
Menstrual bleeding, hemoglobin levels, and quality of life were assessed prior to insertion or surgery, and again at 6- and 12-month follow-up. Eleven women in the hysterectomy group were lost to follow-up.
General findings of the trial include:
- Women in the LNG-IUS group had a mean reduction in the volume of menstrual bleeding—as measured by the number of pads used—from two pads to one pad at 6 months, remaining at that level until 12 months. Serum hemoglobin levels increased from a median of just over
11 g/dL at the time of insertion to 13 g/dL at 6 months and slightly higher at 12 months. In the five self-reported quality-of-life domains assessed (physical, psychological, social, environmental, and a national environmental domain), women using the LNG-IUS demonstrated improvement in all five. - Women in the hysterectomy group were treated using an abdominal surgical approach, with one patient experiencing postoperative wound infection that required secondary suture. Postoperative pathologic analysis found that 21 of these women (65.6%) had adenomyosis, six women (18.8%) had myomas, three women (9.4%) had both adenomyosis and a myoma, and two women (6.2%) had a normal uterus. Serum hemoglobin levels increased from a median of roughly 10.5 g/dL at the time of treatment to 13 g/dL at 6 months and slightly higher at 12 months. (There were no statistically significant differences in hemoglobin values between the LNG-IUS and hysterectomy groups at any point in the study.) Quality of life improved in three of the five domains assessed (physical and both environmental domains).
Although 11 women were lost to follow-up, this trial appeared to have an adequate sample size to examine the selected outcomes, and the population was well defined.
Two weaknesses were the limited follow-up (only 12 months) and the use of quality-of-life measures designed for a Turkish population (the trial was conducted in Turkey), which may or may not be fully applicable to a US population.
WHAT THIS EVIDENCE MEANS FOR PRACTICE
The relationship of adenomyosis to gynecologic symptoms, including heavy menstrual bleeding and dysmenorrhea, needs further study. However, this trial confirmed that transvaginal ultrasound is helpful in the nonsurgical diagnosis of adenomyosis and suggests that the LNG-IUS may be as effective at 1 year as hysterectomy for the treatment of adenomyosis-associated heavy menstrual bleeding (AUB-A).
Clinicians who perform office-based ultrasound to assess AUB should familiarize themselves with the criteria for ultrasonic diagnosis of adenomyosis. These criteria include the presence of heterogeneous myometrial echogenicity, a loss of clarity of the endo-myometrial interface, typically radially oriented linear striations, the appearance of myometrial cysts, and an overall globular enlarged uterus characterized by asymmetric thickening of the myometrium.2
In patients with heavy menstrual bleeding who have these findings, particularly if there is coexistent dysmenorrhea and uterine tenderness, it behooves the clinician to consider the LNG-IUS as first-line therapy, especially for women who wish to preserve fertility, but also for women for whom fertility is not an issue.
There is some evidence that the therapeutic effect of the LNG-IUS containing 20 µg of levonorgestrel may start to fade at 2 or 3 years, a possibility that should be shared with patients.3 Other features, such as cavity size, thickness of the myometrium, and the coexistence of clinically relevant leiomyomas, have not been evaluated but may have an impact on the clinical response.
AUB-I: LOW-DOSE DOXYCYCLINE REDUCES THE TIME TO AMENORRHEA IN USERS OF CONTINUOUS ORAL CONTRACEPTIVES
Kaneshiro B, Edelman A, Carlson NE, Nichols M, Forbes MM, Jensen J. A randomized controlled trial of subantimicrobial-dose doxycycline to prevent unscheduled bleeding with continuous oral contraceptive pill use. Contraception. 2012;85(4):351–358.
Unscheduled bleeding is the most common complaint among women who use continuous combination oral contraceptives (OCs). Because unscheduled bleeding has been correlated with the upregulation of matrix metalloprotineases (MMPs), Kaneshiro and colleagues conducted a randomized, controlled trial of doxycycline (an MMP inhibitor) versus placebo among users of continuous OCs. The addition of doxycycline to the OC regimen did not significantly reduce unscheduled bleeding during the first 84 days of use, but it did shorten the time required to achieve amenorrhea (mean of 61.7 days for doxycycline vs 85.2 days for placebo; standard error [SE], 7.7 vs 6.7, respectively; P = .03).
Related Article: Big step forward and downward: An OC with 10 μg of estrogen Robert L. Barbieri, MD (Editorial, May 2011)
Details and general findings of the trial
Participants (n = 65) were healthy women aged 18 to 45 years who had no contraindications to continuous use of combination OCs. Prior to enrollment, they all had used cyclic combination contraception (pill, patch, or ring) without unscheduled bleeding, thereby avoiding the “transition bleeding” that often occurs when continuous OCs are initiated.
All women in the trial were started on continuous OCs (20 µg ethinyl estradiol with 100 µg levonorgestrel; Aviane) and then randomly assigned to receive one of the following for 84 days in addition to the OC:
- doxycycline 40 mg daily (controlled-release Oracea), a subantimicrobial dose
- placebo.
After 84 days, doxycycline was discontinued, and participants were observed for an additional 28 days on the OC regimen alone for the documentation of bleeding patterns.
General findings:
- The number of bleeding and spotting days decreased in both groups over the course of the study.
- During the first 84 days of the trial, bleeding and spotting occurred among a median of 11 and 17 women in the doxycycline and placebo groups, respectively, and bleeding alone (without spotting) occurred in a median 3 and 4 women in the doxycycline and placebo groups, respectively.
- During the 28-day observation period, bleeding and spotting occurred among a median of 0 and 6 women in the doxycycline and placebo groups, respectively. Bleeding alone (without spotting) was absent in both groups.
- Women in the doxycycline group were significantly less likely to report side effects such as headache, depressed mood, and abdominal cramping. However, they were more likely to prefer continuous OCs without doxycycline, compared with women receiving placebo (16.1% vs 10.7%).
WHAT THIS EVIDENCE MEANS FOR PRACTICE
This trial increases our insight into AUB associated with the use of progestins and suggests that concomitant doxycycline may reduce unscheduled bleeding and spotting in women using continuous combination OCs. The trial was of adequate sample size for the primary outcomes, lending credence to its findings, although longer-term data would be helpful.
I have included this trial for two reasons:
It offers useful information regarding the mechanisms and potential prevention or reduction of AUB-I in users of continuous combined estrogen-progestin contraception.
Doxycycline is one of the agents covered in a Cochrane review of high-quality research into AUB-I in women using progestin-only products, including injectables, implantables, intrauterine systems, and oral agents.4 Estrogens have been shown to have some value in reducing breakthrough bleeding associated with depot medroxyprogesterone acetate, and individual use of tranexamic acid or doxycycline has shown value in terminating an episode of breakthrough bleeding in women using progestin-only contraceptives.
TELL US WHAT YOU THINK!
Share your thoughts on this article or on any topic relevant to ObGyns and women’s health practitioners. Tell us which topics you’d like to see covered in future issues, and what challenges you face in daily practice. We will consider publishing your letter and in a future issue.
Send your letter to: [email protected] Please include the city and state in which you practice.
Stay in touch! Your feedback is important to us!
- Munro MG, Critchley HO, Broder MS, Fraser IS; FIGO Working Group on Menstrual Disorders. The FIGO classification for causes of abnormal bleeding in the reproductive years. Fertil Steril. 2011;95(7):2204–2208.
- Champaneria R, Abedin P, Daniels J, Balogun M, Khan KS. Ultrasound scan and magnetic resonance imaging for the diagnosis of adenomyosis: Systematic review comparing test accuracy. Acta Obstet Gynecol Scand. 2010;89(11):1374–1384.
- Cho S, Nam A, Kim H, et al. Clinical effects of the levonorgestrel-releasing intrauterine device in patients with adenomyosis. Am J Obstet Gynecol. 2008;198(4):373.e1–e7.
- Abdel-Aleem H, d’Arcangues C, Vogelsong KM, Gaffield ML, Gulmezoglu AM. Treatment of vaginal bleeding irregularities induced by progestin-only contraceptives. Cochrane Database Syst Rev. 2013;10:CD003449.
- Munro MG, Critchley HO, Broder MS, Fraser IS; FIGO Working Group on Menstrual Disorders. The FIGO classification for causes of abnormal bleeding in the reproductive years. Fertil Steril. 2011;95(7):2204–2208.
- Champaneria R, Abedin P, Daniels J, Balogun M, Khan KS. Ultrasound scan and magnetic resonance imaging for the diagnosis of adenomyosis: Systematic review comparing test accuracy. Acta Obstet Gynecol Scand. 2010;89(11):1374–1384.
- Cho S, Nam A, Kim H, et al. Clinical effects of the levonorgestrel-releasing intrauterine device in patients with adenomyosis. Am J Obstet Gynecol. 2008;198(4):373.e1–e7.
- Abdel-Aleem H, d’Arcangues C, Vogelsong KM, Gaffield ML, Gulmezoglu AM. Treatment of vaginal bleeding irregularities induced by progestin-only contraceptives. Cochrane Database Syst Rev. 2013;10:CD003449.

Targeting pathways can override resistance in ALL
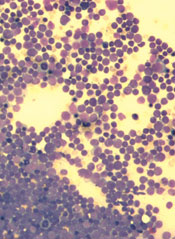
Inhibiting two biosynthesis pathways can override treatment resistance in acute lymphoblastic leukemia (ALL), preclinical research suggests.
Researchers found that inhibiting only one pathway did not effectively kill ALL cells. The cells simply used another pathway to replicate their DNA and continue dividing.
But inhibiting both pathways induced apoptosis in human leukemia cells and reduced tumor burden in mouse models of T- and B-cell ALL.
“This new, dual-targeting approach shows that we can overcome the redundancy in DNA synthesis in ALL cells and identifies a potential target for metabolic intervention in ALL, and possibly in other hematological cancers,” said study author Caius Radu, MD, of the University of California, Los Angeles.
He and his colleagues described this approach in The Journal of Experimental Medicine.
The research began with the knowledge that deoxyribonucleotide triphosphates, including deoxycytidine triphosphate (dCTP), are required for DNA replication, which is necessary for cell division. And dCTP can be produced by the de novo pathway or the nucleoside salvage pathway.
Dr Radu and his colleagues discovered that inhibiting the de novo pathway with the compound thymidine caused leukemia cells to switch to the nucleoside salvage pathway for dCTP production.
However, inhibiting both the de novo and nucleoside salvage pathways prevented dCTP production and proved lethal for leukemia cells.
A number of experiments elicited these results. In one, the researchers knocked down the deoxycytidine kinase (dCK) in human T-ALL cells to inhibit the nucleoside salvage pathway. Then, they administered thymidine to inhibit the de novo pathway. This resulted in dCTP depletion, stalled DNA replication, replication stress, DNA damage, and apoptosis.
The researchers also used the small-molecule inhibitor DI-39 to target dCK. They found that co-administration of DI-39 and thymidine induced replication stress and apoptosis in several leukemia cell lines: CEM, Jurkat, MOLT-4, NALM-6 and RS4;116.
The team then tested DI-39 and thymidine in mice bearing CEM tumors. They found the combination reduced tumor growth in mice bearing established, subcutaneous CEM xenografts and in mice with systemic T-ALL.
In the systemic T-ALL model, treatment with thymidine alone resulted in a 7-fold reduction in tumor burden compared to vehicle control or DI-39 alone. But when thymidine and DI-39 were administered together, mice saw a 100-fold reduction in tumor burden compared to thymidine alone.
The thymidine-DI-39 combination also proved effective against B-ALL cells and in a mouse model of B-ALL. However, the effects were not as great as those observed in T-ALL.
Finally, the researchers evaluated the effects of thymidine and DI-39 in hematopoietic progenitor cells. They looked at the Lineage- Sca-1+ c-Kit+ hematopoietic stem cell population, as well as short-term, long-term, and multipotent progenitor hematopoietic progenitor cells.
There was a minor decrease in the percentage of Lineage- Sca-1+ c-Kit+ cells after thymidine treatment. However, there were no other significant changes in progenitor cells between the treatment and control groups. Why leukemic cells and normal hematopoietic progenitors respond so differently to this treatment requires further investigation, the researchers said.
But they also noted that this study advances our understanding of DNA synthesis in leukemic cells and suggests that targeted metabolic intervention could be a new therapeutic approach in ALL. ![]()
Inhibiting two biosynthesis pathways can override treatment resistance in acute lymphoblastic leukemia (ALL), preclinical research suggests.
Researchers found that inhibiting only one pathway did not effectively kill ALL cells. The cells simply used another pathway to replicate their DNA and continue dividing.
But inhibiting both pathways induced apoptosis in human leukemia cells and reduced tumor burden in mouse models of T- and B-cell ALL.
“This new, dual-targeting approach shows that we can overcome the redundancy in DNA synthesis in ALL cells and identifies a potential target for metabolic intervention in ALL, and possibly in other hematological cancers,” said study author Caius Radu, MD, of the University of California, Los Angeles.
He and his colleagues described this approach in The Journal of Experimental Medicine.
The research began with the knowledge that deoxyribonucleotide triphosphates, including deoxycytidine triphosphate (dCTP), are required for DNA replication, which is necessary for cell division. And dCTP can be produced by the de novo pathway or the nucleoside salvage pathway.
Dr Radu and his colleagues discovered that inhibiting the de novo pathway with the compound thymidine caused leukemia cells to switch to the nucleoside salvage pathway for dCTP production.
However, inhibiting both the de novo and nucleoside salvage pathways prevented dCTP production and proved lethal for leukemia cells.
A number of experiments elicited these results. In one, the researchers knocked down the deoxycytidine kinase (dCK) in human T-ALL cells to inhibit the nucleoside salvage pathway. Then, they administered thymidine to inhibit the de novo pathway. This resulted in dCTP depletion, stalled DNA replication, replication stress, DNA damage, and apoptosis.
The researchers also used the small-molecule inhibitor DI-39 to target dCK. They found that co-administration of DI-39 and thymidine induced replication stress and apoptosis in several leukemia cell lines: CEM, Jurkat, MOLT-4, NALM-6 and RS4;116.
The team then tested DI-39 and thymidine in mice bearing CEM tumors. They found the combination reduced tumor growth in mice bearing established, subcutaneous CEM xenografts and in mice with systemic T-ALL.
In the systemic T-ALL model, treatment with thymidine alone resulted in a 7-fold reduction in tumor burden compared to vehicle control or DI-39 alone. But when thymidine and DI-39 were administered together, mice saw a 100-fold reduction in tumor burden compared to thymidine alone.
The thymidine-DI-39 combination also proved effective against B-ALL cells and in a mouse model of B-ALL. However, the effects were not as great as those observed in T-ALL.
Finally, the researchers evaluated the effects of thymidine and DI-39 in hematopoietic progenitor cells. They looked at the Lineage- Sca-1+ c-Kit+ hematopoietic stem cell population, as well as short-term, long-term, and multipotent progenitor hematopoietic progenitor cells.
There was a minor decrease in the percentage of Lineage- Sca-1+ c-Kit+ cells after thymidine treatment. However, there were no other significant changes in progenitor cells between the treatment and control groups. Why leukemic cells and normal hematopoietic progenitors respond so differently to this treatment requires further investigation, the researchers said.
But they also noted that this study advances our understanding of DNA synthesis in leukemic cells and suggests that targeted metabolic intervention could be a new therapeutic approach in ALL. ![]()
Inhibiting two biosynthesis pathways can override treatment resistance in acute lymphoblastic leukemia (ALL), preclinical research suggests.
Researchers found that inhibiting only one pathway did not effectively kill ALL cells. The cells simply used another pathway to replicate their DNA and continue dividing.
But inhibiting both pathways induced apoptosis in human leukemia cells and reduced tumor burden in mouse models of T- and B-cell ALL.
“This new, dual-targeting approach shows that we can overcome the redundancy in DNA synthesis in ALL cells and identifies a potential target for metabolic intervention in ALL, and possibly in other hematological cancers,” said study author Caius Radu, MD, of the University of California, Los Angeles.
He and his colleagues described this approach in The Journal of Experimental Medicine.
The research began with the knowledge that deoxyribonucleotide triphosphates, including deoxycytidine triphosphate (dCTP), are required for DNA replication, which is necessary for cell division. And dCTP can be produced by the de novo pathway or the nucleoside salvage pathway.
Dr Radu and his colleagues discovered that inhibiting the de novo pathway with the compound thymidine caused leukemia cells to switch to the nucleoside salvage pathway for dCTP production.
However, inhibiting both the de novo and nucleoside salvage pathways prevented dCTP production and proved lethal for leukemia cells.
A number of experiments elicited these results. In one, the researchers knocked down the deoxycytidine kinase (dCK) in human T-ALL cells to inhibit the nucleoside salvage pathway. Then, they administered thymidine to inhibit the de novo pathway. This resulted in dCTP depletion, stalled DNA replication, replication stress, DNA damage, and apoptosis.
The researchers also used the small-molecule inhibitor DI-39 to target dCK. They found that co-administration of DI-39 and thymidine induced replication stress and apoptosis in several leukemia cell lines: CEM, Jurkat, MOLT-4, NALM-6 and RS4;116.
The team then tested DI-39 and thymidine in mice bearing CEM tumors. They found the combination reduced tumor growth in mice bearing established, subcutaneous CEM xenografts and in mice with systemic T-ALL.
In the systemic T-ALL model, treatment with thymidine alone resulted in a 7-fold reduction in tumor burden compared to vehicle control or DI-39 alone. But when thymidine and DI-39 were administered together, mice saw a 100-fold reduction in tumor burden compared to thymidine alone.
The thymidine-DI-39 combination also proved effective against B-ALL cells and in a mouse model of B-ALL. However, the effects were not as great as those observed in T-ALL.
Finally, the researchers evaluated the effects of thymidine and DI-39 in hematopoietic progenitor cells. They looked at the Lineage- Sca-1+ c-Kit+ hematopoietic stem cell population, as well as short-term, long-term, and multipotent progenitor hematopoietic progenitor cells.
There was a minor decrease in the percentage of Lineage- Sca-1+ c-Kit+ cells after thymidine treatment. However, there were no other significant changes in progenitor cells between the treatment and control groups. Why leukemic cells and normal hematopoietic progenitors respond so differently to this treatment requires further investigation, the researchers said.
But they also noted that this study advances our understanding of DNA synthesis in leukemic cells and suggests that targeted metabolic intervention could be a new therapeutic approach in ALL. ![]()
Malaria parasite originated in Africa, team says

David Morgan & Crickette Sanz
Investigators have found evidence suggesting the malaria parasite Plasmodium vivax originated in Africa.
Until recently, the closest genetic relatives of human P vivax were found only in Asian macaques, leading researchers to believe that P vivax originated in Asia.
The current study, published in Nature Communications, showed that wild apes in central Africa are widely infected with parasites that are, genetically, nearly identical to human P vivax.
This finding overturns the dogma that P vivax originated in Asia, despite being most prevalent in humans there now, and also solves other questions about P vivax infection.
For example, it explains why the Duffy-null phenotype, which confers resistance to P vivax, is common among people indigenous to Africa. And it explains how travelers returning from regions where most people are Duffy-negative can be infected with P vivax.
Paul Sharp, PhD, of the University of Edinburgh in the UK, and his colleagues conducted this research, testing more than 5000 ape fecal samples from dozens of field stations and sanctuaries in Africa for P vivax DNA.
They found P vivax-like sequences in chimpanzees, western gorillas, and eastern gorillas, but not in bonobos. Ape P vivax was highly prevalent in wild communities, exhibiting infection rates consistent with stable transmission of the parasite within the wild apes.
To examine the evolutionary relationships between ape and human parasites, the researchers generated parasite DNA sequences from wild and sanctuary apes, as well as from a global sampling of human P vivax infections.
They constructed a family tree of the sequences and found that ape and human parasites were very closely related. But ape parasites were more diverse than the human parasites and did not group according to their host species. The human parasites formed a single lineage that fell within the branches of ape parasite sequences.
From these evolutionary relationships, the investigators concluded that P vivax is of African—not Asian—origin and that all existing human P vivax parasites evolved from a single ancestor that spread out of Africa.
The high prevalence of P vivax in wild apes, along with the recent finding of ape P vivax in a European traveler, indicates the existence of a substantial natural reservoir of P vivax in Africa.
Resolving the Duffy-negative paradox
Of the 5 Plasmodium species known to cause malaria in humans, P vivax is the most widespread. Although highly prevalent in Asia and Latin America, P vivax was thought to be absent from west and central Africa due to a mutation that causes the Duffy-negative phenotype in most indigenous African people.
P vivax parasites enter human red blood cells via the Duffy protein receptor. Because the absence of the receptor on the surface of these cells confers protection against P vivax malaria, this parasite has long been suspected to be the agent that selected for this mutation. However, this hypothesis had been difficult to reconcile with the belief that P vivax originated in Asia.
“Our finding that wild-living apes in central Africa show widespread infection with diverse strains of P vivax provides new insight into the evolutionary history of human P vivax and resolves the paradox that a mutation conferring resistance to P vivax occurs with high frequency in the very region where this parasite is absent in humans,” said study author Beatrice Hahn, MD, of the University of Pennsylvania in Philadelphia.
“One interpretation of the relationships that we observed is that a single host switch from apes gave rise to human P vivax, analogous to the origin of human P falciparum,” Dr Sharp added. “However, this seems unlikely in this case, since ape P vivax does not divide into gorilla- and chimpanzee-specific lineages.”
A more plausible scenario, according to the researchers, is that an ancestral P vivax stock was able to infect humans, gorillas, and chimpanzees in Africa until the Duffy-negative mutation started to spread—around 30,000 years ago—and eliminated P vivax from humans there.
Under this scenario, existing human-infecting P vivax is a parasite that survived after spreading out of Africa.
“The existence of a P vivax reservoir within the forests of central Africa has public health implications,” said study author Martine Peeters, PhD, of the Institut de Recherche pour le Développement and the University of Montpellier in France.
“First, it solves the mystery of P vivax infections in travelers returning from regions where 99% of the human population is Duffy-negative. It also raises the possibility that Duffy-positive humans whose work may bring them in close proximity to chimpanzees and gorillas may become infected by ape P vivax. This has already happened once and may happen again, with unknown consequences.”
The investigators are also concerned about the possibility that ape P vivax may spread via international travel to countries where human P vivax is actively transmitted. Since ape P vivax is more genetically diverse than human P vivax, it may have more versatility to escape treatment and prevention measures, especially if human and ape parasites were able to recombine.
Given what biologists know about P vivax’s ability to switch hosts, the researchers suggest it is important to screen Duffy-positive and Duffy-negative humans in west central Africa, as well as transmitting mosquito vectors, for the presence of ape P vivax. The team believes this information is necessary to inform malaria control and eradication efforts of the propensity of ape P vivax to cross over to humans.
The investigators are also planning to compare and contrast the molecular and biological properties of human and ape parasites to identify host-specific interactions and transmission requirements, thereby uncovering vulnerabilities that can be exploited to combat human malaria. ![]()
David Morgan & Crickette Sanz
Investigators have found evidence suggesting the malaria parasite Plasmodium vivax originated in Africa.
Until recently, the closest genetic relatives of human P vivax were found only in Asian macaques, leading researchers to believe that P vivax originated in Asia.
The current study, published in Nature Communications, showed that wild apes in central Africa are widely infected with parasites that are, genetically, nearly identical to human P vivax.
This finding overturns the dogma that P vivax originated in Asia, despite being most prevalent in humans there now, and also solves other questions about P vivax infection.
For example, it explains why the Duffy-null phenotype, which confers resistance to P vivax, is common among people indigenous to Africa. And it explains how travelers returning from regions where most people are Duffy-negative can be infected with P vivax.
Paul Sharp, PhD, of the University of Edinburgh in the UK, and his colleagues conducted this research, testing more than 5000 ape fecal samples from dozens of field stations and sanctuaries in Africa for P vivax DNA.
They found P vivax-like sequences in chimpanzees, western gorillas, and eastern gorillas, but not in bonobos. Ape P vivax was highly prevalent in wild communities, exhibiting infection rates consistent with stable transmission of the parasite within the wild apes.
To examine the evolutionary relationships between ape and human parasites, the researchers generated parasite DNA sequences from wild and sanctuary apes, as well as from a global sampling of human P vivax infections.
They constructed a family tree of the sequences and found that ape and human parasites were very closely related. But ape parasites were more diverse than the human parasites and did not group according to their host species. The human parasites formed a single lineage that fell within the branches of ape parasite sequences.
From these evolutionary relationships, the investigators concluded that P vivax is of African—not Asian—origin and that all existing human P vivax parasites evolved from a single ancestor that spread out of Africa.
The high prevalence of P vivax in wild apes, along with the recent finding of ape P vivax in a European traveler, indicates the existence of a substantial natural reservoir of P vivax in Africa.
Resolving the Duffy-negative paradox
Of the 5 Plasmodium species known to cause malaria in humans, P vivax is the most widespread. Although highly prevalent in Asia and Latin America, P vivax was thought to be absent from west and central Africa due to a mutation that causes the Duffy-negative phenotype in most indigenous African people.
P vivax parasites enter human red blood cells via the Duffy protein receptor. Because the absence of the receptor on the surface of these cells confers protection against P vivax malaria, this parasite has long been suspected to be the agent that selected for this mutation. However, this hypothesis had been difficult to reconcile with the belief that P vivax originated in Asia.
“Our finding that wild-living apes in central Africa show widespread infection with diverse strains of P vivax provides new insight into the evolutionary history of human P vivax and resolves the paradox that a mutation conferring resistance to P vivax occurs with high frequency in the very region where this parasite is absent in humans,” said study author Beatrice Hahn, MD, of the University of Pennsylvania in Philadelphia.
“One interpretation of the relationships that we observed is that a single host switch from apes gave rise to human P vivax, analogous to the origin of human P falciparum,” Dr Sharp added. “However, this seems unlikely in this case, since ape P vivax does not divide into gorilla- and chimpanzee-specific lineages.”
A more plausible scenario, according to the researchers, is that an ancestral P vivax stock was able to infect humans, gorillas, and chimpanzees in Africa until the Duffy-negative mutation started to spread—around 30,000 years ago—and eliminated P vivax from humans there.
Under this scenario, existing human-infecting P vivax is a parasite that survived after spreading out of Africa.
“The existence of a P vivax reservoir within the forests of central Africa has public health implications,” said study author Martine Peeters, PhD, of the Institut de Recherche pour le Développement and the University of Montpellier in France.
“First, it solves the mystery of P vivax infections in travelers returning from regions where 99% of the human population is Duffy-negative. It also raises the possibility that Duffy-positive humans whose work may bring them in close proximity to chimpanzees and gorillas may become infected by ape P vivax. This has already happened once and may happen again, with unknown consequences.”
The investigators are also concerned about the possibility that ape P vivax may spread via international travel to countries where human P vivax is actively transmitted. Since ape P vivax is more genetically diverse than human P vivax, it may have more versatility to escape treatment and prevention measures, especially if human and ape parasites were able to recombine.
Given what biologists know about P vivax’s ability to switch hosts, the researchers suggest it is important to screen Duffy-positive and Duffy-negative humans in west central Africa, as well as transmitting mosquito vectors, for the presence of ape P vivax. The team believes this information is necessary to inform malaria control and eradication efforts of the propensity of ape P vivax to cross over to humans.
The investigators are also planning to compare and contrast the molecular and biological properties of human and ape parasites to identify host-specific interactions and transmission requirements, thereby uncovering vulnerabilities that can be exploited to combat human malaria. ![]()
David Morgan & Crickette Sanz
Investigators have found evidence suggesting the malaria parasite Plasmodium vivax originated in Africa.
Until recently, the closest genetic relatives of human P vivax were found only in Asian macaques, leading researchers to believe that P vivax originated in Asia.
The current study, published in Nature Communications, showed that wild apes in central Africa are widely infected with parasites that are, genetically, nearly identical to human P vivax.
This finding overturns the dogma that P vivax originated in Asia, despite being most prevalent in humans there now, and also solves other questions about P vivax infection.
For example, it explains why the Duffy-null phenotype, which confers resistance to P vivax, is common among people indigenous to Africa. And it explains how travelers returning from regions where most people are Duffy-negative can be infected with P vivax.
Paul Sharp, PhD, of the University of Edinburgh in the UK, and his colleagues conducted this research, testing more than 5000 ape fecal samples from dozens of field stations and sanctuaries in Africa for P vivax DNA.
They found P vivax-like sequences in chimpanzees, western gorillas, and eastern gorillas, but not in bonobos. Ape P vivax was highly prevalent in wild communities, exhibiting infection rates consistent with stable transmission of the parasite within the wild apes.
To examine the evolutionary relationships between ape and human parasites, the researchers generated parasite DNA sequences from wild and sanctuary apes, as well as from a global sampling of human P vivax infections.
They constructed a family tree of the sequences and found that ape and human parasites were very closely related. But ape parasites were more diverse than the human parasites and did not group according to their host species. The human parasites formed a single lineage that fell within the branches of ape parasite sequences.
From these evolutionary relationships, the investigators concluded that P vivax is of African—not Asian—origin and that all existing human P vivax parasites evolved from a single ancestor that spread out of Africa.
The high prevalence of P vivax in wild apes, along with the recent finding of ape P vivax in a European traveler, indicates the existence of a substantial natural reservoir of P vivax in Africa.
Resolving the Duffy-negative paradox
Of the 5 Plasmodium species known to cause malaria in humans, P vivax is the most widespread. Although highly prevalent in Asia and Latin America, P vivax was thought to be absent from west and central Africa due to a mutation that causes the Duffy-negative phenotype in most indigenous African people.
P vivax parasites enter human red blood cells via the Duffy protein receptor. Because the absence of the receptor on the surface of these cells confers protection against P vivax malaria, this parasite has long been suspected to be the agent that selected for this mutation. However, this hypothesis had been difficult to reconcile with the belief that P vivax originated in Asia.
“Our finding that wild-living apes in central Africa show widespread infection with diverse strains of P vivax provides new insight into the evolutionary history of human P vivax and resolves the paradox that a mutation conferring resistance to P vivax occurs with high frequency in the very region where this parasite is absent in humans,” said study author Beatrice Hahn, MD, of the University of Pennsylvania in Philadelphia.
“One interpretation of the relationships that we observed is that a single host switch from apes gave rise to human P vivax, analogous to the origin of human P falciparum,” Dr Sharp added. “However, this seems unlikely in this case, since ape P vivax does not divide into gorilla- and chimpanzee-specific lineages.”
A more plausible scenario, according to the researchers, is that an ancestral P vivax stock was able to infect humans, gorillas, and chimpanzees in Africa until the Duffy-negative mutation started to spread—around 30,000 years ago—and eliminated P vivax from humans there.
Under this scenario, existing human-infecting P vivax is a parasite that survived after spreading out of Africa.
“The existence of a P vivax reservoir within the forests of central Africa has public health implications,” said study author Martine Peeters, PhD, of the Institut de Recherche pour le Développement and the University of Montpellier in France.
“First, it solves the mystery of P vivax infections in travelers returning from regions where 99% of the human population is Duffy-negative. It also raises the possibility that Duffy-positive humans whose work may bring them in close proximity to chimpanzees and gorillas may become infected by ape P vivax. This has already happened once and may happen again, with unknown consequences.”
The investigators are also concerned about the possibility that ape P vivax may spread via international travel to countries where human P vivax is actively transmitted. Since ape P vivax is more genetically diverse than human P vivax, it may have more versatility to escape treatment and prevention measures, especially if human and ape parasites were able to recombine.
Given what biologists know about P vivax’s ability to switch hosts, the researchers suggest it is important to screen Duffy-positive and Duffy-negative humans in west central Africa, as well as transmitting mosquito vectors, for the presence of ape P vivax. The team believes this information is necessary to inform malaria control and eradication efforts of the propensity of ape P vivax to cross over to humans.
The investigators are also planning to compare and contrast the molecular and biological properties of human and ape parasites to identify host-specific interactions and transmission requirements, thereby uncovering vulnerabilities that can be exploited to combat human malaria. ![]()
Hospital to Home Transitions
Hospital readmissions, which account for a substantial proportion of healthcare expenditures, have increasingly become a focus for hospitals and health systems. Hospitals now assume greater responsibility for population health, and face financial penalties by federal and state agencies that consider readmissions a key measure of the quality of care provided during hospitalization. Consequently, there is broad interest in identifying approaches to reduce hospital reutilization, including emergency department (ED) revisits and hospital readmissions. In this issue of the Journal of Hospital Medicine, Auger et al.[1] report the results of a systematic review, which evaluates the effect of discharge interventions on hospital reutilization among children.
As Auger et al. note, the transition from hospital to home is a vulnerable time for children and their families, with 1 in 5 parents reporting major challenges with such transitions.[2] Auger and colleagues identified 14 studies spanning 3 pediatric disease processes that addressed this issue. The authors concluded that several interventions were potentially effective, but individual studies frequently used multifactorial interventions, precluding determination of discrete elements essential to success. The larger body of care transitions literature in adult populations provides insights for interventions that may benefit pediatric patients, as well as informs future research and quality improvement priorities.
The authors identified some distinct interventions that may successfully decrease hospital reutilization, which share common themes from the adult literature. The first is the use of a dedicated transition coordinator (eg, nurse) or coordinating center to assist with the patient's transition home after discharge. In adult studies, this bridging strategy[3, 4] (ie, use of a dedicated transition coordinator or provider) is initiated during the hospitalization and continues postdischarge in the form of phone calls or home visits. The second theme illustrated in both this pediatric review[1] and adult reviews[3, 4, 5] focuses on enhanced or individualized patient education. Most studies have used a combination of these strategies. For example, the Care Transitions Intervention (one of the best validated adult discharge approaches) uses a transition coach to aid the patient in medication self‐management, creation of a patient‐centered record, scheduling follow‐up appointments, and understanding signs and symptoms of a worsening condition.[6] In a randomized study, this intervention demonstrated a reduction in readmissions within 90 days to 16.7% in the intervention group, compared with 22.5% in the control group.[6] One of the pediatric studies highlighted in the review by Auger et al. achieved a decrease in 14‐day ED revisits from 8% prior to implementation of the program to 2.7% following implementation of the program.[7] This program was for patients discharged from the neonatal intensive care unit and involved a nurse coordinator (similar to a transition coach) who worked closely with families and ensured adequate resources prior to discharge as well as a home visitation program.[7]
Although Auger et al. identify some effective approaches to reducing hospital reutilization after discharge in children, their review and the complementary adult literature bring to light 4 main unresolved questions for hospitalists seeking to improve care transitions: (1) how to dissect diverse and heterogeneous interventions to determine the key driver of success, (2) how to interpret and generally apply interventions from single centers where they may have been tailored to a specific healthcare environment, (3) how to generalize the findings of many disease‐specific interventions to other populations, and (4) how to evaluate the cost and assess the costbenefit of implementing many of the more resource intensive interventions. An example of a heterogeneous intervention addressed in this pediatric systematic review was described by Ng et al.,[8] in which the intervention group received a combination of an enhanced discharge education session, disease‐specific nurse evaluation, an animated education booklet, and postdischarge telephone follow‐up, whereas the control group received a shorter discharge education session, a disease‐specific nurse evaluation only if referred by a physician, a written education booklet, and no telephone follow‐up. Investigators found that intervention patients were less likely to be readmitted or revisit the ED as compared with controls. A similarly multifaceted intervention introduced by Taggart et al.[9] was unable to detect a difference in readmissions or ED revisits. It is unclear whether or not the differences in outcomes were related to differences in the intervention bundle itself or institutional or local contextual factors, thus limiting application to other hospitals. Generalizability of interventions is similarly complicated in adults.
The studies presented in this pediatric review article are specific to 3 disease processes: cancer, asthma, and neonatal intensive care (ie, premature) populations. Beyond these populations, there were no other pediatric conditions that met inclusion criteria, thus limiting the generalizability of the findings. As described by Rennke et al.,[3] adult systematic reviews that have focused only on disease‐specific interventions to reduce hospital reutilization are also difficult to generalize to broader populations. Two of the 3 recent adult transition intervention systematic reviews excluded disease‐specific interventions in an attempt to find more broadly applicable interventions but struggled with the same heterogeneity discussed in this review by Auger et al.[3, 4] Although disease‐specific interventions were included in the third adult systematic review and the evaluation was restricted to randomized controlled trials, the authors still grappled with finding 1 or 2 common, successful intervention components.[5] The fourth unresolved question involves understanding the financial burden of implementing more resource‐intensive interventions such as postdischarge home nurse visits. For example, it may be difficult to justify the business case for hiring a transition coach or initiating home nurse visits when the cost and financial implications are unclear. Neither the pediatric nor adult literature describes this well.
Some of the challenges in identifying effective interventions differ between adult and pediatric populations. Adults tend to have multiple comorbid conditions, making them more medically complex and at greater risk for adverse outcomes, medication errors, and hospital utilization.[10] Although a small subset of the pediatric population with complex chronic medical conditions accounts for a majority of hospital reutilization and cost,[11] most hospitalized pediatric patients are otherwise healthy with acute illnesses.[12] Additionally, pediatric patients have lower overall hospital reutilization rates when compared with adults. Adult 30‐day readmission rates are approximately 20%[13] compared with pediatric patients whose mean 30‐day readmission rate is 6.5%.[14] With readmission being an outcome upon which studies are basing intervention success or failure, the relatively low readmission rates in the pediatric population make shifting that outcome more challenging.
There is also controversy about whether policymakers should be focusing on decreasing 30‐day readmission rates as a measure of success. We believe that efforts should focus on identifying more meaningful outcomes, especially outcomes important to patients and their families. No single metric is likely to be an adequate measure of the quality of care transitions, but a combination of outcome measures could potentially be more informative both for patients and clinicians. Patient satisfaction with the discharge process is measured as part of standard patient experience surveys, and the 3‐question Care Transitions Measure[15] has been validated and endorsed as a measure of patient perception of discharge safety in adult populations. There is a growing consensus that 30‐day readmission rates are lacking as a measure of discharge quality, and therefore, measuring shorter‐term7‐ or 14‐dayreadmission rates along with short‐term ED utilization after discharge would likely be more helpful for identifying care transitions problems. Attention should also be paid to measuring rates of specific adverse events in the postdischarge period, such as adverse drug events or failure to follow up on pending test results, as these failures are often implicated in reutilization.
In reflecting upon the published data on adult and pediatric transitions of care interventions and the lingering unanswered questions, we propose a few considerations for future direction of the field. First, engagement of the primary care provider may be beneficial. In many interventions describing a care transition coordinator, nursing fulfilled this role; however, there are opportunities for the primary care provider to play a greater role in this arena. Second, the use of factorial design in future studies may help elucidate which specific parts of each intervention may be the most crucial.[16] Finally, readmission rates are a controversial quality measure in adults. Pediatric readmissions are relatively uncommon, making it difficult to track measurements and show improvement. Clinicians, patients, and policymakers should prioritize outcome measures that are most meaningful to patients and their families that occur at a much higher rate than that of readmissions.
- , , , . Pediatric hospital discharge interventions to reduce subsequent utilization: a systematic review. J Hosp Med. 2014;9(0):000–000.
- , , , , . Are hospital characteristics associated with parental views of pediatric inpatient care quality? Pediatrics. 2003;111(2):308–314.
- , , , , , . Hospital‐initiated transitional care interventions as a patient safety strategy: a systematic review. Ann Intern Med. 2013;158(5 pt 2):433–440.
- , , , , . Interventions to reduce 30‐day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520–528.
- , , , et al. Improving patient handovers from hospital to primary care: a systematic review. Ann Intern Med. 2012;157(6):417–428.
- , , , . The care transitions intervention: results of a randomized controlled trial. Arch Intern Med. 2006;166(17):1822–1828.
- , , , , . Description and evaluation of a program for the early discharge of infants from a neonatal intensive care unit. J Pediatr. 1995;127(2):285–290.
- , , , , , . Effect of a structured asthma education program on hospitalized asthmatic children: a randomized controlled study. Pediatr Int. 2006;48(2):158–162.
- , , , et al. You Can Control Asthma: evaluation of an asthma education‐program for hospitalized inner‐city children. Patient Educ Couns. 1991;17(1):35–47.
- , , , et al. Adverse events among medical patients after discharge from hospital. CMAJ. 2004;170(3):345–349.
- , , , et al. Hospital utilization and characteristics of patients experiencing recurrent readmissions within children's hospitals. JAMA. 2011;305(7):682–690.
- , , , et al. Prioritization of comparative effectiveness research topics in hospital pediatrics. Arch Pediatr Adolesc Med. 2012;166(12):1155–1164.
- , , . Thirty‐day readmission rates for Medicare beneficiaries by race and site of care. JAMA. 2011;305(7):675–681.
- , , , et al. Pediatric readmission prevalence and variability across hospitals. JAMA. 2013;309(4):372–380.
- , , , . Assessing the quality of transitional care: further applications of the care transitions measure. Med Care. 2008;46(3):317–322.
- , , . Quality Improvement Through Planned Experimentation. 2nd ed. New York, NY: McGraw‐Hill; 1999.
Hospital readmissions, which account for a substantial proportion of healthcare expenditures, have increasingly become a focus for hospitals and health systems. Hospitals now assume greater responsibility for population health, and face financial penalties by federal and state agencies that consider readmissions a key measure of the quality of care provided during hospitalization. Consequently, there is broad interest in identifying approaches to reduce hospital reutilization, including emergency department (ED) revisits and hospital readmissions. In this issue of the Journal of Hospital Medicine, Auger et al.[1] report the results of a systematic review, which evaluates the effect of discharge interventions on hospital reutilization among children.
As Auger et al. note, the transition from hospital to home is a vulnerable time for children and their families, with 1 in 5 parents reporting major challenges with such transitions.[2] Auger and colleagues identified 14 studies spanning 3 pediatric disease processes that addressed this issue. The authors concluded that several interventions were potentially effective, but individual studies frequently used multifactorial interventions, precluding determination of discrete elements essential to success. The larger body of care transitions literature in adult populations provides insights for interventions that may benefit pediatric patients, as well as informs future research and quality improvement priorities.
The authors identified some distinct interventions that may successfully decrease hospital reutilization, which share common themes from the adult literature. The first is the use of a dedicated transition coordinator (eg, nurse) or coordinating center to assist with the patient's transition home after discharge. In adult studies, this bridging strategy[3, 4] (ie, use of a dedicated transition coordinator or provider) is initiated during the hospitalization and continues postdischarge in the form of phone calls or home visits. The second theme illustrated in both this pediatric review[1] and adult reviews[3, 4, 5] focuses on enhanced or individualized patient education. Most studies have used a combination of these strategies. For example, the Care Transitions Intervention (one of the best validated adult discharge approaches) uses a transition coach to aid the patient in medication self‐management, creation of a patient‐centered record, scheduling follow‐up appointments, and understanding signs and symptoms of a worsening condition.[6] In a randomized study, this intervention demonstrated a reduction in readmissions within 90 days to 16.7% in the intervention group, compared with 22.5% in the control group.[6] One of the pediatric studies highlighted in the review by Auger et al. achieved a decrease in 14‐day ED revisits from 8% prior to implementation of the program to 2.7% following implementation of the program.[7] This program was for patients discharged from the neonatal intensive care unit and involved a nurse coordinator (similar to a transition coach) who worked closely with families and ensured adequate resources prior to discharge as well as a home visitation program.[7]
Although Auger et al. identify some effective approaches to reducing hospital reutilization after discharge in children, their review and the complementary adult literature bring to light 4 main unresolved questions for hospitalists seeking to improve care transitions: (1) how to dissect diverse and heterogeneous interventions to determine the key driver of success, (2) how to interpret and generally apply interventions from single centers where they may have been tailored to a specific healthcare environment, (3) how to generalize the findings of many disease‐specific interventions to other populations, and (4) how to evaluate the cost and assess the costbenefit of implementing many of the more resource intensive interventions. An example of a heterogeneous intervention addressed in this pediatric systematic review was described by Ng et al.,[8] in which the intervention group received a combination of an enhanced discharge education session, disease‐specific nurse evaluation, an animated education booklet, and postdischarge telephone follow‐up, whereas the control group received a shorter discharge education session, a disease‐specific nurse evaluation only if referred by a physician, a written education booklet, and no telephone follow‐up. Investigators found that intervention patients were less likely to be readmitted or revisit the ED as compared with controls. A similarly multifaceted intervention introduced by Taggart et al.[9] was unable to detect a difference in readmissions or ED revisits. It is unclear whether or not the differences in outcomes were related to differences in the intervention bundle itself or institutional or local contextual factors, thus limiting application to other hospitals. Generalizability of interventions is similarly complicated in adults.
The studies presented in this pediatric review article are specific to 3 disease processes: cancer, asthma, and neonatal intensive care (ie, premature) populations. Beyond these populations, there were no other pediatric conditions that met inclusion criteria, thus limiting the generalizability of the findings. As described by Rennke et al.,[3] adult systematic reviews that have focused only on disease‐specific interventions to reduce hospital reutilization are also difficult to generalize to broader populations. Two of the 3 recent adult transition intervention systematic reviews excluded disease‐specific interventions in an attempt to find more broadly applicable interventions but struggled with the same heterogeneity discussed in this review by Auger et al.[3, 4] Although disease‐specific interventions were included in the third adult systematic review and the evaluation was restricted to randomized controlled trials, the authors still grappled with finding 1 or 2 common, successful intervention components.[5] The fourth unresolved question involves understanding the financial burden of implementing more resource‐intensive interventions such as postdischarge home nurse visits. For example, it may be difficult to justify the business case for hiring a transition coach or initiating home nurse visits when the cost and financial implications are unclear. Neither the pediatric nor adult literature describes this well.
Some of the challenges in identifying effective interventions differ between adult and pediatric populations. Adults tend to have multiple comorbid conditions, making them more medically complex and at greater risk for adverse outcomes, medication errors, and hospital utilization.[10] Although a small subset of the pediatric population with complex chronic medical conditions accounts for a majority of hospital reutilization and cost,[11] most hospitalized pediatric patients are otherwise healthy with acute illnesses.[12] Additionally, pediatric patients have lower overall hospital reutilization rates when compared with adults. Adult 30‐day readmission rates are approximately 20%[13] compared with pediatric patients whose mean 30‐day readmission rate is 6.5%.[14] With readmission being an outcome upon which studies are basing intervention success or failure, the relatively low readmission rates in the pediatric population make shifting that outcome more challenging.
There is also controversy about whether policymakers should be focusing on decreasing 30‐day readmission rates as a measure of success. We believe that efforts should focus on identifying more meaningful outcomes, especially outcomes important to patients and their families. No single metric is likely to be an adequate measure of the quality of care transitions, but a combination of outcome measures could potentially be more informative both for patients and clinicians. Patient satisfaction with the discharge process is measured as part of standard patient experience surveys, and the 3‐question Care Transitions Measure[15] has been validated and endorsed as a measure of patient perception of discharge safety in adult populations. There is a growing consensus that 30‐day readmission rates are lacking as a measure of discharge quality, and therefore, measuring shorter‐term7‐ or 14‐dayreadmission rates along with short‐term ED utilization after discharge would likely be more helpful for identifying care transitions problems. Attention should also be paid to measuring rates of specific adverse events in the postdischarge period, such as adverse drug events or failure to follow up on pending test results, as these failures are often implicated in reutilization.
In reflecting upon the published data on adult and pediatric transitions of care interventions and the lingering unanswered questions, we propose a few considerations for future direction of the field. First, engagement of the primary care provider may be beneficial. In many interventions describing a care transition coordinator, nursing fulfilled this role; however, there are opportunities for the primary care provider to play a greater role in this arena. Second, the use of factorial design in future studies may help elucidate which specific parts of each intervention may be the most crucial.[16] Finally, readmission rates are a controversial quality measure in adults. Pediatric readmissions are relatively uncommon, making it difficult to track measurements and show improvement. Clinicians, patients, and policymakers should prioritize outcome measures that are most meaningful to patients and their families that occur at a much higher rate than that of readmissions.
Hospital readmissions, which account for a substantial proportion of healthcare expenditures, have increasingly become a focus for hospitals and health systems. Hospitals now assume greater responsibility for population health, and face financial penalties by federal and state agencies that consider readmissions a key measure of the quality of care provided during hospitalization. Consequently, there is broad interest in identifying approaches to reduce hospital reutilization, including emergency department (ED) revisits and hospital readmissions. In this issue of the Journal of Hospital Medicine, Auger et al.[1] report the results of a systematic review, which evaluates the effect of discharge interventions on hospital reutilization among children.
As Auger et al. note, the transition from hospital to home is a vulnerable time for children and their families, with 1 in 5 parents reporting major challenges with such transitions.[2] Auger and colleagues identified 14 studies spanning 3 pediatric disease processes that addressed this issue. The authors concluded that several interventions were potentially effective, but individual studies frequently used multifactorial interventions, precluding determination of discrete elements essential to success. The larger body of care transitions literature in adult populations provides insights for interventions that may benefit pediatric patients, as well as informs future research and quality improvement priorities.
The authors identified some distinct interventions that may successfully decrease hospital reutilization, which share common themes from the adult literature. The first is the use of a dedicated transition coordinator (eg, nurse) or coordinating center to assist with the patient's transition home after discharge. In adult studies, this bridging strategy[3, 4] (ie, use of a dedicated transition coordinator or provider) is initiated during the hospitalization and continues postdischarge in the form of phone calls or home visits. The second theme illustrated in both this pediatric review[1] and adult reviews[3, 4, 5] focuses on enhanced or individualized patient education. Most studies have used a combination of these strategies. For example, the Care Transitions Intervention (one of the best validated adult discharge approaches) uses a transition coach to aid the patient in medication self‐management, creation of a patient‐centered record, scheduling follow‐up appointments, and understanding signs and symptoms of a worsening condition.[6] In a randomized study, this intervention demonstrated a reduction in readmissions within 90 days to 16.7% in the intervention group, compared with 22.5% in the control group.[6] One of the pediatric studies highlighted in the review by Auger et al. achieved a decrease in 14‐day ED revisits from 8% prior to implementation of the program to 2.7% following implementation of the program.[7] This program was for patients discharged from the neonatal intensive care unit and involved a nurse coordinator (similar to a transition coach) who worked closely with families and ensured adequate resources prior to discharge as well as a home visitation program.[7]
Although Auger et al. identify some effective approaches to reducing hospital reutilization after discharge in children, their review and the complementary adult literature bring to light 4 main unresolved questions for hospitalists seeking to improve care transitions: (1) how to dissect diverse and heterogeneous interventions to determine the key driver of success, (2) how to interpret and generally apply interventions from single centers where they may have been tailored to a specific healthcare environment, (3) how to generalize the findings of many disease‐specific interventions to other populations, and (4) how to evaluate the cost and assess the costbenefit of implementing many of the more resource intensive interventions. An example of a heterogeneous intervention addressed in this pediatric systematic review was described by Ng et al.,[8] in which the intervention group received a combination of an enhanced discharge education session, disease‐specific nurse evaluation, an animated education booklet, and postdischarge telephone follow‐up, whereas the control group received a shorter discharge education session, a disease‐specific nurse evaluation only if referred by a physician, a written education booklet, and no telephone follow‐up. Investigators found that intervention patients were less likely to be readmitted or revisit the ED as compared with controls. A similarly multifaceted intervention introduced by Taggart et al.[9] was unable to detect a difference in readmissions or ED revisits. It is unclear whether or not the differences in outcomes were related to differences in the intervention bundle itself or institutional or local contextual factors, thus limiting application to other hospitals. Generalizability of interventions is similarly complicated in adults.
The studies presented in this pediatric review article are specific to 3 disease processes: cancer, asthma, and neonatal intensive care (ie, premature) populations. Beyond these populations, there were no other pediatric conditions that met inclusion criteria, thus limiting the generalizability of the findings. As described by Rennke et al.,[3] adult systematic reviews that have focused only on disease‐specific interventions to reduce hospital reutilization are also difficult to generalize to broader populations. Two of the 3 recent adult transition intervention systematic reviews excluded disease‐specific interventions in an attempt to find more broadly applicable interventions but struggled with the same heterogeneity discussed in this review by Auger et al.[3, 4] Although disease‐specific interventions were included in the third adult systematic review and the evaluation was restricted to randomized controlled trials, the authors still grappled with finding 1 or 2 common, successful intervention components.[5] The fourth unresolved question involves understanding the financial burden of implementing more resource‐intensive interventions such as postdischarge home nurse visits. For example, it may be difficult to justify the business case for hiring a transition coach or initiating home nurse visits when the cost and financial implications are unclear. Neither the pediatric nor adult literature describes this well.
Some of the challenges in identifying effective interventions differ between adult and pediatric populations. Adults tend to have multiple comorbid conditions, making them more medically complex and at greater risk for adverse outcomes, medication errors, and hospital utilization.[10] Although a small subset of the pediatric population with complex chronic medical conditions accounts for a majority of hospital reutilization and cost,[11] most hospitalized pediatric patients are otherwise healthy with acute illnesses.[12] Additionally, pediatric patients have lower overall hospital reutilization rates when compared with adults. Adult 30‐day readmission rates are approximately 20%[13] compared with pediatric patients whose mean 30‐day readmission rate is 6.5%.[14] With readmission being an outcome upon which studies are basing intervention success or failure, the relatively low readmission rates in the pediatric population make shifting that outcome more challenging.
There is also controversy about whether policymakers should be focusing on decreasing 30‐day readmission rates as a measure of success. We believe that efforts should focus on identifying more meaningful outcomes, especially outcomes important to patients and their families. No single metric is likely to be an adequate measure of the quality of care transitions, but a combination of outcome measures could potentially be more informative both for patients and clinicians. Patient satisfaction with the discharge process is measured as part of standard patient experience surveys, and the 3‐question Care Transitions Measure[15] has been validated and endorsed as a measure of patient perception of discharge safety in adult populations. There is a growing consensus that 30‐day readmission rates are lacking as a measure of discharge quality, and therefore, measuring shorter‐term7‐ or 14‐dayreadmission rates along with short‐term ED utilization after discharge would likely be more helpful for identifying care transitions problems. Attention should also be paid to measuring rates of specific adverse events in the postdischarge period, such as adverse drug events or failure to follow up on pending test results, as these failures are often implicated in reutilization.
In reflecting upon the published data on adult and pediatric transitions of care interventions and the lingering unanswered questions, we propose a few considerations for future direction of the field. First, engagement of the primary care provider may be beneficial. In many interventions describing a care transition coordinator, nursing fulfilled this role; however, there are opportunities for the primary care provider to play a greater role in this arena. Second, the use of factorial design in future studies may help elucidate which specific parts of each intervention may be the most crucial.[16] Finally, readmission rates are a controversial quality measure in adults. Pediatric readmissions are relatively uncommon, making it difficult to track measurements and show improvement. Clinicians, patients, and policymakers should prioritize outcome measures that are most meaningful to patients and their families that occur at a much higher rate than that of readmissions.
- , , , . Pediatric hospital discharge interventions to reduce subsequent utilization: a systematic review. J Hosp Med. 2014;9(0):000–000.
- , , , , . Are hospital characteristics associated with parental views of pediatric inpatient care quality? Pediatrics. 2003;111(2):308–314.
- , , , , , . Hospital‐initiated transitional care interventions as a patient safety strategy: a systematic review. Ann Intern Med. 2013;158(5 pt 2):433–440.
- , , , , . Interventions to reduce 30‐day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520–528.
- , , , et al. Improving patient handovers from hospital to primary care: a systematic review. Ann Intern Med. 2012;157(6):417–428.
- , , , . The care transitions intervention: results of a randomized controlled trial. Arch Intern Med. 2006;166(17):1822–1828.
- , , , , . Description and evaluation of a program for the early discharge of infants from a neonatal intensive care unit. J Pediatr. 1995;127(2):285–290.
- , , , , , . Effect of a structured asthma education program on hospitalized asthmatic children: a randomized controlled study. Pediatr Int. 2006;48(2):158–162.
- , , , et al. You Can Control Asthma: evaluation of an asthma education‐program for hospitalized inner‐city children. Patient Educ Couns. 1991;17(1):35–47.
- , , , et al. Adverse events among medical patients after discharge from hospital. CMAJ. 2004;170(3):345–349.
- , , , et al. Hospital utilization and characteristics of patients experiencing recurrent readmissions within children's hospitals. JAMA. 2011;305(7):682–690.
- , , , et al. Prioritization of comparative effectiveness research topics in hospital pediatrics. Arch Pediatr Adolesc Med. 2012;166(12):1155–1164.
- , , . Thirty‐day readmission rates for Medicare beneficiaries by race and site of care. JAMA. 2011;305(7):675–681.
- , , , et al. Pediatric readmission prevalence and variability across hospitals. JAMA. 2013;309(4):372–380.
- , , , . Assessing the quality of transitional care: further applications of the care transitions measure. Med Care. 2008;46(3):317–322.
- , , . Quality Improvement Through Planned Experimentation. 2nd ed. New York, NY: McGraw‐Hill; 1999.
- , , , . Pediatric hospital discharge interventions to reduce subsequent utilization: a systematic review. J Hosp Med. 2014;9(0):000–000.
- , , , , . Are hospital characteristics associated with parental views of pediatric inpatient care quality? Pediatrics. 2003;111(2):308–314.
- , , , , , . Hospital‐initiated transitional care interventions as a patient safety strategy: a systematic review. Ann Intern Med. 2013;158(5 pt 2):433–440.
- , , , , . Interventions to reduce 30‐day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520–528.
- , , , et al. Improving patient handovers from hospital to primary care: a systematic review. Ann Intern Med. 2012;157(6):417–428.
- , , , . The care transitions intervention: results of a randomized controlled trial. Arch Intern Med. 2006;166(17):1822–1828.
- , , , , . Description and evaluation of a program for the early discharge of infants from a neonatal intensive care unit. J Pediatr. 1995;127(2):285–290.
- , , , , , . Effect of a structured asthma education program on hospitalized asthmatic children: a randomized controlled study. Pediatr Int. 2006;48(2):158–162.
- , , , et al. You Can Control Asthma: evaluation of an asthma education‐program for hospitalized inner‐city children. Patient Educ Couns. 1991;17(1):35–47.
- , , , et al. Adverse events among medical patients after discharge from hospital. CMAJ. 2004;170(3):345–349.
- , , , et al. Hospital utilization and characteristics of patients experiencing recurrent readmissions within children's hospitals. JAMA. 2011;305(7):682–690.
- , , , et al. Prioritization of comparative effectiveness research topics in hospital pediatrics. Arch Pediatr Adolesc Med. 2012;166(12):1155–1164.
- , , . Thirty‐day readmission rates for Medicare beneficiaries by race and site of care. JAMA. 2011;305(7):675–681.
- , , , et al. Pediatric readmission prevalence and variability across hospitals. JAMA. 2013;309(4):372–380.
- , , , . Assessing the quality of transitional care: further applications of the care transitions measure. Med Care. 2008;46(3):317–322.
- , , . Quality Improvement Through Planned Experimentation. 2nd ed. New York, NY: McGraw‐Hill; 1999.
AAN issues nonvalvular atrial fibrillation stroke prevention guideline
A new evidence-based guideline on how to identify and treat patients with nonvalvular atrial fibrillation to prevent cardioembolic stroke from the American Academy of Neurology suggests when to conduct cardiac rhythm monitoring and offer anticoagulation, including newer agents in place of warfarin.
But the guideline might already be outdated in not considering the results of the recent CRYSTAL-AF study, in which long-term cardiac rhythm monitoring of patients with a previous cryptogenic stroke detected asymptomatic patients at a significantly higher rate than did standard monitoring methods.

The guideline also extends the routine use of anticoagulation for patients with nonvalvular atrial fibrillation (NVAF) who are generally undertreated or whose health was thought a possible barrier to their use, such as those aged 75 years or older, those with mild dementia, and those at moderate risk of falls.
"Cognizant of the global reach of the AAN [American Academy of Neurology], the guideline also examines the evidence base for a treatment alternative to warfarin or its analogues for patients in developing countries who may not have access to the new oral anticoagulants," said lead author Dr. Antonio Culebras in an interview.
"The World Health Organization has determined that atrial fibrillation has reached near-epidemic proportions," observed Dr. Culebras of the State University of New York, Syracuse. "Approximately 1 in 20 individuals with AF will have a stroke unless treated appropriately."
The risk for stroke among patients with NVAF is highest in those with a history of transient ischemic attack (TIA) or prior stroke, at an absolute value of around 10% per year. Patients with "lone NVAF," meaning they have no additional risk factors, have less than a 2% increased risk of stroke per year.
The AAN issued a practice parameter on this topic in 1998 (Neurology 1998;51:671-3). At the time, warfarin, adjusted to an international normalized ratio (INR) of 2.0, was, and largely remains, the recommended standard for patients at risk for cardioembolic stroke. Aspirin was the only recommended alterative for those unable to receive the vitamin K antagonist or who were deemed to be at low risk of stroke, although the evidence was scanty.
Since then, several new oral anticoagulant agents have become available, including the direct thrombin inhibitor dabigatran (Pradaxa), and two factor Xa inhibitors – rivaroxaban (Xarelto) and apixaban (Eliquis) – which have been shown to be at least as effective as, if not more effective than, warfarin. Cardiac rhythm monitoring via a variety of methods has also been introduced as a means to try to detect NVAF in asymptomatic patients.
The aim of the AAN guideline (Neurology 2014;82:716-24) was therefore to look at the latest evidence on the detection of AF using new technologies, as well as the use of treatments to reduce the risk of stroke without increasing the risk of hemorrhage versus the long-standing standard of therapy, warfarin. Data published from 1998 to March 2013 were considered in the preparation of the guideline.
Cardiac rhythm monitoring for NVAF
Seventeen studies were found that examined the use of cardiac monitoring technologies to detect new cases of NVAF. The most common methods used were 24-hour Holter monitoring and serial electrocardiograms, but some emerging evidence on newer technologies was included. The proportion of patients identified with NVAF ranged from 0% to 23%, with the average detection rate 10.7% in all of the studies included.
"The guideline addresses the question of long-term monitoring of patients with NVAF," Dr. Culebras said. "It recommends that clinicians ‘might’ [level C evidence] obtain outpatient cardiac rhythm studies in patients with cryptogenic stroke without known NVAF to identify patients with occult NVAF." He added that the guideline also recommends that monitoring might be needed for prolonged periods of 1 or more weeks rather than for shorter periods, such as 24 hours.
However, at the time the guideline was being prepared, recent data from the CRYSTAL-AF study were not available, and this means the guideline is already outdated, Dr. Richard A. Bernstein, professor of neurology at Northwestern University, Chicago, said in an interview. He was not a guideline author.
Dr. Bernstein was on the steering committee for the CRYSTAL-AF trial, which assessed the performance of Medtronic’s Reveal XT Insertable Cardiac Monitor and found that the implanted device could detect NVAF better than serial ECGs or Holter monitoring (8.6% vs. 1.4%; P = .0006); most (74%) cases of NVAF found were asymptomatic.*
"CRYSTAL-AF represents the state of the art for cardiac monitoring in cryptogenic stroke patients and makes the AAN guidelines obsolete," Dr. Berstein said. "[The study] shows that even intermediate-term monitoring (less than 1 month) will miss the majority of AF in this population, and that most of the AF we find with long-term (greater than 1 year) monitoring is likely to be clinically significant."
With regard to the AAN guideline, he added: "There is no discussion of truly long-term monitoring in the guideline, which is unfortunate." That said, "anything that gets neurologists thinking about long-term cardiac monitoring is likely to be beneficial."
Anticoagulation for stroke prevention
The AAN guideline also provides general recommendations on the use of novel oral anticoagulant agents (NOACs) as alternatives to warfarin. Specifically, it notes that in comparison with warfarin, these NOACs are probably at least as effective (rivaroxaban) or more effective (dabigatran and apixaban). Additionally, while apixaban is also likely to be more effective than aspirin, it is associated with a similar risk for bleeding. NOACs have the following advantages over warfarin: an overall lower risk of intracranial hemorrhage and no need for routine anticoagulant monitoring.
From a practical perspective, the AAN guideline suggests that clinicians have the following options available: warfarin to reach an INR of 2.0-3.0, dabigatran 150 mg twice daily, rivaroxaban 15-20 mg/dL, apixaban 2.5-5 mg twice a day, and triflusal 600 mg plus acenocoumarol to reach an INR target of 1.25-2.0. If a patient is already taking warfarin and is well controlled, then they should remain on that therapy and not switch to a newer oral anticoagulant.
The guideline also notes that clopidogrel plus aspirin is probably less effective than warfarin, but the combination is probably better than aspirin alone. However, the risk of hemorrhage is higher.
Where used, triflusal plus acenocoumarol is "likely more effective" than acenocoumarol alone. Triflusal is an antiplatelet drug related to aspirin, used in Europe, Latin America, and Southeast Asia. Acenocoumarol is mostly used in European countries.
Dr. Culebras explained that the guideline was not intended to dictate which treatment to use. "The guideline leaves room on purpose for clinicians to use their judgment," he said. "The overall objective of the guideline is to reduce therapeutic uncertainty and not to issue commandments for treatment."
Although Dr. Bernstein was critical of the guidelines for not advocating the use of anticoagulants strongly enough, he said that the recommendations on anticoagulant choice are "reasonable in that they impute potential clinical profiles of patients who might particularly benefit from one NOAC over another, without making a claim that these recommendations are based on solid data. This reflects how doctors make decisions when we don’t have direct comparative studies, and I think that is helpful."
The guideline was developed with financial support from the American Academy of Neurology. None of the authors received reimbursement, honoraria, or stipends for their participation in the development of the guideline.
Dr. Culebras has received one-time funding for travel from J. Uriach & Co, and he serves on the editorial boards of MedLink, UpToDate.com, and the International Journal of Stroke. He has received royalties from Informa Healthcare and Cambridge University Press, and has held stock in Clinical Stroke Research. Other authors reported current or past ties to companies marketing oral anticoagulants and stroke treatments.
Dr. Bernstein was on the steering committee for the CRYSTAL-AF study and is a paid speaker, researcher, and consultant for Medtronic, Bristol-Myers Squibb, Pfizer, Boehringer Ingelheim, and Lifewatch.
*Correction, 4/8/2014: The article previously misstated what the implantable device was detecting in the CRYSTAL-AF study.

|
|
These guidelines are a missed opportunity to empower neurologists to advocate in favor of anticoagulation to prevent stroke. The biggest public health problem in AF is that only half of patients who need anticoagulation are getting it. This disgraceful state of affairs results in patients having cardioembolic strokes that are fatal or worse and that could have been prevented. We neurologists see these complications of inadequate treatment and should be on the front lines of prevention. These tepid guidelines give as much space to bleeding as they do to ischemic stroke prevention, which is inappropriate, and I fear will make neurologists, who are not terribly assertive under any circumstances, even less willing to push doctors to use anticoagulants.
I would have been happier with a single page that said: "Stop using aspirin. Patients fear major stroke more than they fear bleeding or death, and they are right. Stop undertreating your patients and start preventing strokes."
Dr. Richard A. Bernstein is professor of neurology and director of the stroke program at Northwestern University, Chicago.
|
|
|
These guidelines are a missed opportunity to empower neurologists to advocate in favor of anticoagulation to prevent stroke. The biggest public health problem in AF is that only half of patients who need anticoagulation are getting it. This disgraceful state of affairs results in patients having cardioembolic strokes that are fatal or worse and that could have been prevented. We neurologists see these complications of inadequate treatment and should be on the front lines of prevention. These tepid guidelines give as much space to bleeding as they do to ischemic stroke prevention, which is inappropriate, and I fear will make neurologists, who are not terribly assertive under any circumstances, even less willing to push doctors to use anticoagulants.
I would have been happier with a single page that said: "Stop using aspirin. Patients fear major stroke more than they fear bleeding or death, and they are right. Stop undertreating your patients and start preventing strokes."
Dr. Richard A. Bernstein is professor of neurology and director of the stroke program at Northwestern University, Chicago.
|
|
|
These guidelines are a missed opportunity to empower neurologists to advocate in favor of anticoagulation to prevent stroke. The biggest public health problem in AF is that only half of patients who need anticoagulation are getting it. This disgraceful state of affairs results in patients having cardioembolic strokes that are fatal or worse and that could have been prevented. We neurologists see these complications of inadequate treatment and should be on the front lines of prevention. These tepid guidelines give as much space to bleeding as they do to ischemic stroke prevention, which is inappropriate, and I fear will make neurologists, who are not terribly assertive under any circumstances, even less willing to push doctors to use anticoagulants.
I would have been happier with a single page that said: "Stop using aspirin. Patients fear major stroke more than they fear bleeding or death, and they are right. Stop undertreating your patients and start preventing strokes."
Dr. Richard A. Bernstein is professor of neurology and director of the stroke program at Northwestern University, Chicago.
A new evidence-based guideline on how to identify and treat patients with nonvalvular atrial fibrillation to prevent cardioembolic stroke from the American Academy of Neurology suggests when to conduct cardiac rhythm monitoring and offer anticoagulation, including newer agents in place of warfarin.
But the guideline might already be outdated in not considering the results of the recent CRYSTAL-AF study, in which long-term cardiac rhythm monitoring of patients with a previous cryptogenic stroke detected asymptomatic patients at a significantly higher rate than did standard monitoring methods.
The guideline also extends the routine use of anticoagulation for patients with nonvalvular atrial fibrillation (NVAF) who are generally undertreated or whose health was thought a possible barrier to their use, such as those aged 75 years or older, those with mild dementia, and those at moderate risk of falls.
"Cognizant of the global reach of the AAN [American Academy of Neurology], the guideline also examines the evidence base for a treatment alternative to warfarin or its analogues for patients in developing countries who may not have access to the new oral anticoagulants," said lead author Dr. Antonio Culebras in an interview.
"The World Health Organization has determined that atrial fibrillation has reached near-epidemic proportions," observed Dr. Culebras of the State University of New York, Syracuse. "Approximately 1 in 20 individuals with AF will have a stroke unless treated appropriately."
The risk for stroke among patients with NVAF is highest in those with a history of transient ischemic attack (TIA) or prior stroke, at an absolute value of around 10% per year. Patients with "lone NVAF," meaning they have no additional risk factors, have less than a 2% increased risk of stroke per year.
The AAN issued a practice parameter on this topic in 1998 (Neurology 1998;51:671-3). At the time, warfarin, adjusted to an international normalized ratio (INR) of 2.0, was, and largely remains, the recommended standard for patients at risk for cardioembolic stroke. Aspirin was the only recommended alterative for those unable to receive the vitamin K antagonist or who were deemed to be at low risk of stroke, although the evidence was scanty.
Since then, several new oral anticoagulant agents have become available, including the direct thrombin inhibitor dabigatran (Pradaxa), and two factor Xa inhibitors – rivaroxaban (Xarelto) and apixaban (Eliquis) – which have been shown to be at least as effective as, if not more effective than, warfarin. Cardiac rhythm monitoring via a variety of methods has also been introduced as a means to try to detect NVAF in asymptomatic patients.
The aim of the AAN guideline (Neurology 2014;82:716-24) was therefore to look at the latest evidence on the detection of AF using new technologies, as well as the use of treatments to reduce the risk of stroke without increasing the risk of hemorrhage versus the long-standing standard of therapy, warfarin. Data published from 1998 to March 2013 were considered in the preparation of the guideline.
Cardiac rhythm monitoring for NVAF
Seventeen studies were found that examined the use of cardiac monitoring technologies to detect new cases of NVAF. The most common methods used were 24-hour Holter monitoring and serial electrocardiograms, but some emerging evidence on newer technologies was included. The proportion of patients identified with NVAF ranged from 0% to 23%, with the average detection rate 10.7% in all of the studies included.
"The guideline addresses the question of long-term monitoring of patients with NVAF," Dr. Culebras said. "It recommends that clinicians ‘might’ [level C evidence] obtain outpatient cardiac rhythm studies in patients with cryptogenic stroke without known NVAF to identify patients with occult NVAF." He added that the guideline also recommends that monitoring might be needed for prolonged periods of 1 or more weeks rather than for shorter periods, such as 24 hours.
However, at the time the guideline was being prepared, recent data from the CRYSTAL-AF study were not available, and this means the guideline is already outdated, Dr. Richard A. Bernstein, professor of neurology at Northwestern University, Chicago, said in an interview. He was not a guideline author.
Dr. Bernstein was on the steering committee for the CRYSTAL-AF trial, which assessed the performance of Medtronic’s Reveal XT Insertable Cardiac Monitor and found that the implanted device could detect NVAF better than serial ECGs or Holter monitoring (8.6% vs. 1.4%; P = .0006); most (74%) cases of NVAF found were asymptomatic.*
"CRYSTAL-AF represents the state of the art for cardiac monitoring in cryptogenic stroke patients and makes the AAN guidelines obsolete," Dr. Berstein said. "[The study] shows that even intermediate-term monitoring (less than 1 month) will miss the majority of AF in this population, and that most of the AF we find with long-term (greater than 1 year) monitoring is likely to be clinically significant."
With regard to the AAN guideline, he added: "There is no discussion of truly long-term monitoring in the guideline, which is unfortunate." That said, "anything that gets neurologists thinking about long-term cardiac monitoring is likely to be beneficial."
Anticoagulation for stroke prevention
The AAN guideline also provides general recommendations on the use of novel oral anticoagulant agents (NOACs) as alternatives to warfarin. Specifically, it notes that in comparison with warfarin, these NOACs are probably at least as effective (rivaroxaban) or more effective (dabigatran and apixaban). Additionally, while apixaban is also likely to be more effective than aspirin, it is associated with a similar risk for bleeding. NOACs have the following advantages over warfarin: an overall lower risk of intracranial hemorrhage and no need for routine anticoagulant monitoring.
From a practical perspective, the AAN guideline suggests that clinicians have the following options available: warfarin to reach an INR of 2.0-3.0, dabigatran 150 mg twice daily, rivaroxaban 15-20 mg/dL, apixaban 2.5-5 mg twice a day, and triflusal 600 mg plus acenocoumarol to reach an INR target of 1.25-2.0. If a patient is already taking warfarin and is well controlled, then they should remain on that therapy and not switch to a newer oral anticoagulant.
The guideline also notes that clopidogrel plus aspirin is probably less effective than warfarin, but the combination is probably better than aspirin alone. However, the risk of hemorrhage is higher.
Where used, triflusal plus acenocoumarol is "likely more effective" than acenocoumarol alone. Triflusal is an antiplatelet drug related to aspirin, used in Europe, Latin America, and Southeast Asia. Acenocoumarol is mostly used in European countries.
Dr. Culebras explained that the guideline was not intended to dictate which treatment to use. "The guideline leaves room on purpose for clinicians to use their judgment," he said. "The overall objective of the guideline is to reduce therapeutic uncertainty and not to issue commandments for treatment."
Although Dr. Bernstein was critical of the guidelines for not advocating the use of anticoagulants strongly enough, he said that the recommendations on anticoagulant choice are "reasonable in that they impute potential clinical profiles of patients who might particularly benefit from one NOAC over another, without making a claim that these recommendations are based on solid data. This reflects how doctors make decisions when we don’t have direct comparative studies, and I think that is helpful."
The guideline was developed with financial support from the American Academy of Neurology. None of the authors received reimbursement, honoraria, or stipends for their participation in the development of the guideline.
Dr. Culebras has received one-time funding for travel from J. Uriach & Co, and he serves on the editorial boards of MedLink, UpToDate.com, and the International Journal of Stroke. He has received royalties from Informa Healthcare and Cambridge University Press, and has held stock in Clinical Stroke Research. Other authors reported current or past ties to companies marketing oral anticoagulants and stroke treatments.
Dr. Bernstein was on the steering committee for the CRYSTAL-AF study and is a paid speaker, researcher, and consultant for Medtronic, Bristol-Myers Squibb, Pfizer, Boehringer Ingelheim, and Lifewatch.
*Correction, 4/8/2014: The article previously misstated what the implantable device was detecting in the CRYSTAL-AF study.
A new evidence-based guideline on how to identify and treat patients with nonvalvular atrial fibrillation to prevent cardioembolic stroke from the American Academy of Neurology suggests when to conduct cardiac rhythm monitoring and offer anticoagulation, including newer agents in place of warfarin.
But the guideline might already be outdated in not considering the results of the recent CRYSTAL-AF study, in which long-term cardiac rhythm monitoring of patients with a previous cryptogenic stroke detected asymptomatic patients at a significantly higher rate than did standard monitoring methods.
The guideline also extends the routine use of anticoagulation for patients with nonvalvular atrial fibrillation (NVAF) who are generally undertreated or whose health was thought a possible barrier to their use, such as those aged 75 years or older, those with mild dementia, and those at moderate risk of falls.
"Cognizant of the global reach of the AAN [American Academy of Neurology], the guideline also examines the evidence base for a treatment alternative to warfarin or its analogues for patients in developing countries who may not have access to the new oral anticoagulants," said lead author Dr. Antonio Culebras in an interview.
"The World Health Organization has determined that atrial fibrillation has reached near-epidemic proportions," observed Dr. Culebras of the State University of New York, Syracuse. "Approximately 1 in 20 individuals with AF will have a stroke unless treated appropriately."
The risk for stroke among patients with NVAF is highest in those with a history of transient ischemic attack (TIA) or prior stroke, at an absolute value of around 10% per year. Patients with "lone NVAF," meaning they have no additional risk factors, have less than a 2% increased risk of stroke per year.
The AAN issued a practice parameter on this topic in 1998 (Neurology 1998;51:671-3). At the time, warfarin, adjusted to an international normalized ratio (INR) of 2.0, was, and largely remains, the recommended standard for patients at risk for cardioembolic stroke. Aspirin was the only recommended alterative for those unable to receive the vitamin K antagonist or who were deemed to be at low risk of stroke, although the evidence was scanty.
Since then, several new oral anticoagulant agents have become available, including the direct thrombin inhibitor dabigatran (Pradaxa), and two factor Xa inhibitors – rivaroxaban (Xarelto) and apixaban (Eliquis) – which have been shown to be at least as effective as, if not more effective than, warfarin. Cardiac rhythm monitoring via a variety of methods has also been introduced as a means to try to detect NVAF in asymptomatic patients.
The aim of the AAN guideline (Neurology 2014;82:716-24) was therefore to look at the latest evidence on the detection of AF using new technologies, as well as the use of treatments to reduce the risk of stroke without increasing the risk of hemorrhage versus the long-standing standard of therapy, warfarin. Data published from 1998 to March 2013 were considered in the preparation of the guideline.
Cardiac rhythm monitoring for NVAF
Seventeen studies were found that examined the use of cardiac monitoring technologies to detect new cases of NVAF. The most common methods used were 24-hour Holter monitoring and serial electrocardiograms, but some emerging evidence on newer technologies was included. The proportion of patients identified with NVAF ranged from 0% to 23%, with the average detection rate 10.7% in all of the studies included.
"The guideline addresses the question of long-term monitoring of patients with NVAF," Dr. Culebras said. "It recommends that clinicians ‘might’ [level C evidence] obtain outpatient cardiac rhythm studies in patients with cryptogenic stroke without known NVAF to identify patients with occult NVAF." He added that the guideline also recommends that monitoring might be needed for prolonged periods of 1 or more weeks rather than for shorter periods, such as 24 hours.
However, at the time the guideline was being prepared, recent data from the CRYSTAL-AF study were not available, and this means the guideline is already outdated, Dr. Richard A. Bernstein, professor of neurology at Northwestern University, Chicago, said in an interview. He was not a guideline author.
Dr. Bernstein was on the steering committee for the CRYSTAL-AF trial, which assessed the performance of Medtronic’s Reveal XT Insertable Cardiac Monitor and found that the implanted device could detect NVAF better than serial ECGs or Holter monitoring (8.6% vs. 1.4%; P = .0006); most (74%) cases of NVAF found were asymptomatic.*
"CRYSTAL-AF represents the state of the art for cardiac monitoring in cryptogenic stroke patients and makes the AAN guidelines obsolete," Dr. Berstein said. "[The study] shows that even intermediate-term monitoring (less than 1 month) will miss the majority of AF in this population, and that most of the AF we find with long-term (greater than 1 year) monitoring is likely to be clinically significant."
With regard to the AAN guideline, he added: "There is no discussion of truly long-term monitoring in the guideline, which is unfortunate." That said, "anything that gets neurologists thinking about long-term cardiac monitoring is likely to be beneficial."
Anticoagulation for stroke prevention
The AAN guideline also provides general recommendations on the use of novel oral anticoagulant agents (NOACs) as alternatives to warfarin. Specifically, it notes that in comparison with warfarin, these NOACs are probably at least as effective (rivaroxaban) or more effective (dabigatran and apixaban). Additionally, while apixaban is also likely to be more effective than aspirin, it is associated with a similar risk for bleeding. NOACs have the following advantages over warfarin: an overall lower risk of intracranial hemorrhage and no need for routine anticoagulant monitoring.
From a practical perspective, the AAN guideline suggests that clinicians have the following options available: warfarin to reach an INR of 2.0-3.0, dabigatran 150 mg twice daily, rivaroxaban 15-20 mg/dL, apixaban 2.5-5 mg twice a day, and triflusal 600 mg plus acenocoumarol to reach an INR target of 1.25-2.0. If a patient is already taking warfarin and is well controlled, then they should remain on that therapy and not switch to a newer oral anticoagulant.
The guideline also notes that clopidogrel plus aspirin is probably less effective than warfarin, but the combination is probably better than aspirin alone. However, the risk of hemorrhage is higher.
Where used, triflusal plus acenocoumarol is "likely more effective" than acenocoumarol alone. Triflusal is an antiplatelet drug related to aspirin, used in Europe, Latin America, and Southeast Asia. Acenocoumarol is mostly used in European countries.
Dr. Culebras explained that the guideline was not intended to dictate which treatment to use. "The guideline leaves room on purpose for clinicians to use their judgment," he said. "The overall objective of the guideline is to reduce therapeutic uncertainty and not to issue commandments for treatment."
Although Dr. Bernstein was critical of the guidelines for not advocating the use of anticoagulants strongly enough, he said that the recommendations on anticoagulant choice are "reasonable in that they impute potential clinical profiles of patients who might particularly benefit from one NOAC over another, without making a claim that these recommendations are based on solid data. This reflects how doctors make decisions when we don’t have direct comparative studies, and I think that is helpful."
The guideline was developed with financial support from the American Academy of Neurology. None of the authors received reimbursement, honoraria, or stipends for their participation in the development of the guideline.
Dr. Culebras has received one-time funding for travel from J. Uriach & Co, and he serves on the editorial boards of MedLink, UpToDate.com, and the International Journal of Stroke. He has received royalties from Informa Healthcare and Cambridge University Press, and has held stock in Clinical Stroke Research. Other authors reported current or past ties to companies marketing oral anticoagulants and stroke treatments.
Dr. Bernstein was on the steering committee for the CRYSTAL-AF study and is a paid speaker, researcher, and consultant for Medtronic, Bristol-Myers Squibb, Pfizer, Boehringer Ingelheim, and Lifewatch.
*Correction, 4/8/2014: The article previously misstated what the implantable device was detecting in the CRYSTAL-AF study.
FROM NEUROLOGY

Mammogram data are not to die for
I remember that day like it was yesterday, though it occurred more than a decade ago. I stood leaning over a black entertainment center in my family room, legs wobbly, heart weary – a surreal and solemn snapshot in time. From a speaker streamed a now-favorite Donnie McClurkin song, called "Stand," with its introspective lyrics: "You’ve prayed and you’ve cried ... . After you’ve done all you can, you just stand."
In the next room I could hear her softly gurgling on her secretions. I needed a moment, no, two or three moments, to collect my thoughts and pull myself together before I returned to face the nightmare I was living. My mother was actively dying in my guestroom. Why? I believed then and, today, many years later, believe just as strongly it was because she had not been getting her mammograms.
As a writer, sometimes I struggle with how personal to get in my blogs, but rest assured. I got her permission to share her story while she was still very lucid and competent. You see, she did not want others’ lives to end as hers was ending. She realized, in her final stages of life, that things would have likely been much different had she had her screening mammograms as recommended.
By the time of her diagnosis in her early 60s, the cancer had already spread. Would a mammogram in her late 50s have saved her life? I believe so, and I’m not alone. So I take issue with a recent article published in BMJ that downplays the significance of mammography (BMJ 2014;348:g366).
In 1980, Canadian researchers randomized 89,835 women, aged 40-59 to receive five annual mammograms or physical breast examinations. They followed these women over a 25-year period, and concluded that yearly mammography in women aged 40-59 did not decrease breast cancer mortality "beyond that of physical examination or usual care when adjuvant therapy for breast cancer is freely available."
Well, how many of us have taken care of women in their 50s, 40s, and even 30s with terminal breast cancer? How many of us would advise a mother, aunt, sister (or self) not to have routine mammography? Not many, I’m sure. There is the art of medicine and the science of medicine. Sometimes these two clash, but I believe the art of medicine is realizing that the science of medicine really doesn’t matter to dying patients and their family members. Sometimes, we have to act in the best interest of individual patients and not rely too heavily on the "data." Data changes, risk factors emerge, or research findings may prove to be skewed or wrong in hindsight. Explains Dr. Poornima Sharma, an oncologist/hematologist at the University of Maryland Baltimore-Washington Medical Center: "While the methodology, mammographic technique, and equipment used in the Canadian study is being assessed and compared to the mammography standards used in the United States, the standard in this country remains annual mammography starting at age 40."
Still, many women consider themselves at low risk for breast cancer if they have no close relatives with the disease. As erroneous as this assumption may be, this subset of women may be particularly vulnerable to the implication that yearly mammography is not needed.
So do this: Discuss screening mammography with your own family and then use those feelings when a teachable moment presents itself at bedside.
Our patients rely on us to look into their eyes and give them our best advice. Even though I am a hospitalist, there are still those women I feel compelled to counsel about screening mammography, and this study will not lessen my fervor.
Dr. Hester is a hospitalist with Baltimore-Washington Medical Center who has a passion for empowering patients to partner in their health care. She is the creator of the Patient Whiz, a patient-engagement app for iOS.
I remember that day like it was yesterday, though it occurred more than a decade ago. I stood leaning over a black entertainment center in my family room, legs wobbly, heart weary – a surreal and solemn snapshot in time. From a speaker streamed a now-favorite Donnie McClurkin song, called "Stand," with its introspective lyrics: "You’ve prayed and you’ve cried ... . After you’ve done all you can, you just stand."
In the next room I could hear her softly gurgling on her secretions. I needed a moment, no, two or three moments, to collect my thoughts and pull myself together before I returned to face the nightmare I was living. My mother was actively dying in my guestroom. Why? I believed then and, today, many years later, believe just as strongly it was because she had not been getting her mammograms.
As a writer, sometimes I struggle with how personal to get in my blogs, but rest assured. I got her permission to share her story while she was still very lucid and competent. You see, she did not want others’ lives to end as hers was ending. She realized, in her final stages of life, that things would have likely been much different had she had her screening mammograms as recommended.
By the time of her diagnosis in her early 60s, the cancer had already spread. Would a mammogram in her late 50s have saved her life? I believe so, and I’m not alone. So I take issue with a recent article published in BMJ that downplays the significance of mammography (BMJ 2014;348:g366).
In 1980, Canadian researchers randomized 89,835 women, aged 40-59 to receive five annual mammograms or physical breast examinations. They followed these women over a 25-year period, and concluded that yearly mammography in women aged 40-59 did not decrease breast cancer mortality "beyond that of physical examination or usual care when adjuvant therapy for breast cancer is freely available."
Well, how many of us have taken care of women in their 50s, 40s, and even 30s with terminal breast cancer? How many of us would advise a mother, aunt, sister (or self) not to have routine mammography? Not many, I’m sure. There is the art of medicine and the science of medicine. Sometimes these two clash, but I believe the art of medicine is realizing that the science of medicine really doesn’t matter to dying patients and their family members. Sometimes, we have to act in the best interest of individual patients and not rely too heavily on the "data." Data changes, risk factors emerge, or research findings may prove to be skewed or wrong in hindsight. Explains Dr. Poornima Sharma, an oncologist/hematologist at the University of Maryland Baltimore-Washington Medical Center: "While the methodology, mammographic technique, and equipment used in the Canadian study is being assessed and compared to the mammography standards used in the United States, the standard in this country remains annual mammography starting at age 40."
Still, many women consider themselves at low risk for breast cancer if they have no close relatives with the disease. As erroneous as this assumption may be, this subset of women may be particularly vulnerable to the implication that yearly mammography is not needed.
So do this: Discuss screening mammography with your own family and then use those feelings when a teachable moment presents itself at bedside.
Our patients rely on us to look into their eyes and give them our best advice. Even though I am a hospitalist, there are still those women I feel compelled to counsel about screening mammography, and this study will not lessen my fervor.
Dr. Hester is a hospitalist with Baltimore-Washington Medical Center who has a passion for empowering patients to partner in their health care. She is the creator of the Patient Whiz, a patient-engagement app for iOS.
I remember that day like it was yesterday, though it occurred more than a decade ago. I stood leaning over a black entertainment center in my family room, legs wobbly, heart weary – a surreal and solemn snapshot in time. From a speaker streamed a now-favorite Donnie McClurkin song, called "Stand," with its introspective lyrics: "You’ve prayed and you’ve cried ... . After you’ve done all you can, you just stand."
In the next room I could hear her softly gurgling on her secretions. I needed a moment, no, two or three moments, to collect my thoughts and pull myself together before I returned to face the nightmare I was living. My mother was actively dying in my guestroom. Why? I believed then and, today, many years later, believe just as strongly it was because she had not been getting her mammograms.
As a writer, sometimes I struggle with how personal to get in my blogs, but rest assured. I got her permission to share her story while she was still very lucid and competent. You see, she did not want others’ lives to end as hers was ending. She realized, in her final stages of life, that things would have likely been much different had she had her screening mammograms as recommended.
By the time of her diagnosis in her early 60s, the cancer had already spread. Would a mammogram in her late 50s have saved her life? I believe so, and I’m not alone. So I take issue with a recent article published in BMJ that downplays the significance of mammography (BMJ 2014;348:g366).
In 1980, Canadian researchers randomized 89,835 women, aged 40-59 to receive five annual mammograms or physical breast examinations. They followed these women over a 25-year period, and concluded that yearly mammography in women aged 40-59 did not decrease breast cancer mortality "beyond that of physical examination or usual care when adjuvant therapy for breast cancer is freely available."
Well, how many of us have taken care of women in their 50s, 40s, and even 30s with terminal breast cancer? How many of us would advise a mother, aunt, sister (or self) not to have routine mammography? Not many, I’m sure. There is the art of medicine and the science of medicine. Sometimes these two clash, but I believe the art of medicine is realizing that the science of medicine really doesn’t matter to dying patients and their family members. Sometimes, we have to act in the best interest of individual patients and not rely too heavily on the "data." Data changes, risk factors emerge, or research findings may prove to be skewed or wrong in hindsight. Explains Dr. Poornima Sharma, an oncologist/hematologist at the University of Maryland Baltimore-Washington Medical Center: "While the methodology, mammographic technique, and equipment used in the Canadian study is being assessed and compared to the mammography standards used in the United States, the standard in this country remains annual mammography starting at age 40."
Still, many women consider themselves at low risk for breast cancer if they have no close relatives with the disease. As erroneous as this assumption may be, this subset of women may be particularly vulnerable to the implication that yearly mammography is not needed.
So do this: Discuss screening mammography with your own family and then use those feelings when a teachable moment presents itself at bedside.
Our patients rely on us to look into their eyes and give them our best advice. Even though I am a hospitalist, there are still those women I feel compelled to counsel about screening mammography, and this study will not lessen my fervor.
Dr. Hester is a hospitalist with Baltimore-Washington Medical Center who has a passion for empowering patients to partner in their health care. She is the creator of the Patient Whiz, a patient-engagement app for iOS.
Protein appears essential to malaria transmission
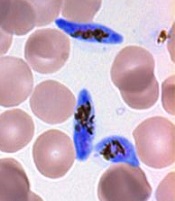
gametocyte stage (blue) and
uninfected red blood cells
Credit: The Llinás lab
Results of 2 new studies suggest that a single regulatory protein acts as a master switch to trigger development of the sexual forms of malaria parasites.
It appears that the protein, AP2-G, is necessary for activating a set of genes that initiate the development of Plasmodium gametocytes, the only forms of the parasite that are infectious to mosquitoes.
This suggests that if researchers can target AP2-G, they can stop sexual parasites from forming.
And if the sexual forms of the parasite never develop in an infected person’s blood, none will enter the mosquito’s gut, and the mosquito will be unable to infect anyone else with malaria.
“Exciting opportunities now lie ahead for finding an effective way to break the chain of malaria transmission by preventing the malaria parasite from completing its full lifecycle,” said Manuel Llinás, PhD, a professor at Pennsylvania State University who was involved in both studies.
The 2 studies, which were published as letters to Nature, had remarkably similar results, despite the fact that the groups worked with 2 different malaria parasites—Plasmodium falciparum and Plasmodium berghei.
In one study, researchers analyzed the whole-genome sequences of 2 P falciparum strains that were unable to produce gametocytes. The only mutated, non-functional gene common to both strains was the AP2-G gene.
In the other study, researchers sequenced P berghei parasites that had lost their ability to make gametocytes. Again, the only common mutated gene in these parasites was AP2-G.
To confirm these observations, both groups of researchers disabled the AP2-G gene in parasites that could generate gametocytes.
As expected, disabling the gene prevented the parasites from producing gametocytes. But the parasites regained their ability to make gametocytes when the mutated gene was repaired.
These results, as well as results of additional experiments, suggest that sexual-stage malaria parasites are produced only when the AP2-G protein is in working order.
“Our research has demonstrated unequivocally that the AP2-G transcription factor protein is essential for flipping the switch that initiates the transformation of malaria parasites in the blood from the asexual stage to the critical sexual stage of their life cycle,” Dr Llinás said.
He and his colleagues believe their discovery is exciting for the future of malaria research. It could spur the development of a sexual-stage vaccine, which would help a person infected with malaria mount an immune response to prevent their parasites from being transmitted to a mosquito, effectively ending the life cycle for that person’s batch of malaria parasites. ![]()
gametocyte stage (blue) and
uninfected red blood cells
Credit: The Llinás lab
Results of 2 new studies suggest that a single regulatory protein acts as a master switch to trigger development of the sexual forms of malaria parasites.
It appears that the protein, AP2-G, is necessary for activating a set of genes that initiate the development of Plasmodium gametocytes, the only forms of the parasite that are infectious to mosquitoes.
This suggests that if researchers can target AP2-G, they can stop sexual parasites from forming.
And if the sexual forms of the parasite never develop in an infected person’s blood, none will enter the mosquito’s gut, and the mosquito will be unable to infect anyone else with malaria.
“Exciting opportunities now lie ahead for finding an effective way to break the chain of malaria transmission by preventing the malaria parasite from completing its full lifecycle,” said Manuel Llinás, PhD, a professor at Pennsylvania State University who was involved in both studies.
The 2 studies, which were published as letters to Nature, had remarkably similar results, despite the fact that the groups worked with 2 different malaria parasites—Plasmodium falciparum and Plasmodium berghei.
In one study, researchers analyzed the whole-genome sequences of 2 P falciparum strains that were unable to produce gametocytes. The only mutated, non-functional gene common to both strains was the AP2-G gene.
In the other study, researchers sequenced P berghei parasites that had lost their ability to make gametocytes. Again, the only common mutated gene in these parasites was AP2-G.
To confirm these observations, both groups of researchers disabled the AP2-G gene in parasites that could generate gametocytes.
As expected, disabling the gene prevented the parasites from producing gametocytes. But the parasites regained their ability to make gametocytes when the mutated gene was repaired.
These results, as well as results of additional experiments, suggest that sexual-stage malaria parasites are produced only when the AP2-G protein is in working order.
“Our research has demonstrated unequivocally that the AP2-G transcription factor protein is essential for flipping the switch that initiates the transformation of malaria parasites in the blood from the asexual stage to the critical sexual stage of their life cycle,” Dr Llinás said.
He and his colleagues believe their discovery is exciting for the future of malaria research. It could spur the development of a sexual-stage vaccine, which would help a person infected with malaria mount an immune response to prevent their parasites from being transmitted to a mosquito, effectively ending the life cycle for that person’s batch of malaria parasites. ![]()
gametocyte stage (blue) and
uninfected red blood cells
Credit: The Llinás lab
Results of 2 new studies suggest that a single regulatory protein acts as a master switch to trigger development of the sexual forms of malaria parasites.
It appears that the protein, AP2-G, is necessary for activating a set of genes that initiate the development of Plasmodium gametocytes, the only forms of the parasite that are infectious to mosquitoes.
This suggests that if researchers can target AP2-G, they can stop sexual parasites from forming.
And if the sexual forms of the parasite never develop in an infected person’s blood, none will enter the mosquito’s gut, and the mosquito will be unable to infect anyone else with malaria.
“Exciting opportunities now lie ahead for finding an effective way to break the chain of malaria transmission by preventing the malaria parasite from completing its full lifecycle,” said Manuel Llinás, PhD, a professor at Pennsylvania State University who was involved in both studies.
The 2 studies, which were published as letters to Nature, had remarkably similar results, despite the fact that the groups worked with 2 different malaria parasites—Plasmodium falciparum and Plasmodium berghei.
In one study, researchers analyzed the whole-genome sequences of 2 P falciparum strains that were unable to produce gametocytes. The only mutated, non-functional gene common to both strains was the AP2-G gene.
In the other study, researchers sequenced P berghei parasites that had lost their ability to make gametocytes. Again, the only common mutated gene in these parasites was AP2-G.
To confirm these observations, both groups of researchers disabled the AP2-G gene in parasites that could generate gametocytes.
As expected, disabling the gene prevented the parasites from producing gametocytes. But the parasites regained their ability to make gametocytes when the mutated gene was repaired.
These results, as well as results of additional experiments, suggest that sexual-stage malaria parasites are produced only when the AP2-G protein is in working order.
“Our research has demonstrated unequivocally that the AP2-G transcription factor protein is essential for flipping the switch that initiates the transformation of malaria parasites in the blood from the asexual stage to the critical sexual stage of their life cycle,” Dr Llinás said.
He and his colleagues believe their discovery is exciting for the future of malaria research. It could spur the development of a sexual-stage vaccine, which would help a person infected with malaria mount an immune response to prevent their parasites from being transmitted to a mosquito, effectively ending the life cycle for that person’s batch of malaria parasites. ![]()
Drug could enhance effects of chemo
Credit: Rhoda Baer
The drug spironolactone could improve the efficacy of platinum-based chemotherapy by preventing tumor cell repair, according to research published in Chemistry & Biology.
The researchers knew that platinum-based chemotherapy drugs bind to cellular DNA to induce damage.
So they theorized that blocking DNA repair mechanisms would help potentiate chemotherapy by reducing cancer cells’ resistance to treatment.
The team focused their efforts on inhibiting nucleotide excision repair (NER), in which a damaged DNA fragment is replaced with an intact fragment.
Frédéric Coin, PhD, of the Institute of Genetics and Molecular and Cellular Biology in Illkirch, France, and his colleagues screened more than 1200 drugs looking for one that would inhibit NER activity.
And they found that spironolactone—a drug already used to treat fluid retention, high blood pressure, and other conditions—affects NER activity.
Specifically, the team found that, when combined with platinum derivatives, spironolactone significantly increased cytotoxicity in ovarian and colon cancer cells.
As platinum-based chemotherapy is used to treat a range of cancers, similar results might occur in other malignancies as well.
The researchers also noted that, because spironolactone is already in use for other purposes, it doesn’t require a new application for marketing authorization. And its side effects are already known.
The team said this suggests that protocols testing spironolactone in combination with platinum-based chemotherapy could be organized rather quickly. ![]()
Credit: Rhoda Baer
The drug spironolactone could improve the efficacy of platinum-based chemotherapy by preventing tumor cell repair, according to research published in Chemistry & Biology.
The researchers knew that platinum-based chemotherapy drugs bind to cellular DNA to induce damage.
So they theorized that blocking DNA repair mechanisms would help potentiate chemotherapy by reducing cancer cells’ resistance to treatment.
The team focused their efforts on inhibiting nucleotide excision repair (NER), in which a damaged DNA fragment is replaced with an intact fragment.
Frédéric Coin, PhD, of the Institute of Genetics and Molecular and Cellular Biology in Illkirch, France, and his colleagues screened more than 1200 drugs looking for one that would inhibit NER activity.
And they found that spironolactone—a drug already used to treat fluid retention, high blood pressure, and other conditions—affects NER activity.
Specifically, the team found that, when combined with platinum derivatives, spironolactone significantly increased cytotoxicity in ovarian and colon cancer cells.
As platinum-based chemotherapy is used to treat a range of cancers, similar results might occur in other malignancies as well.
The researchers also noted that, because spironolactone is already in use for other purposes, it doesn’t require a new application for marketing authorization. And its side effects are already known.
The team said this suggests that protocols testing spironolactone in combination with platinum-based chemotherapy could be organized rather quickly. ![]()
Credit: Rhoda Baer
The drug spironolactone could improve the efficacy of platinum-based chemotherapy by preventing tumor cell repair, according to research published in Chemistry & Biology.
The researchers knew that platinum-based chemotherapy drugs bind to cellular DNA to induce damage.
So they theorized that blocking DNA repair mechanisms would help potentiate chemotherapy by reducing cancer cells’ resistance to treatment.
The team focused their efforts on inhibiting nucleotide excision repair (NER), in which a damaged DNA fragment is replaced with an intact fragment.
Frédéric Coin, PhD, of the Institute of Genetics and Molecular and Cellular Biology in Illkirch, France, and his colleagues screened more than 1200 drugs looking for one that would inhibit NER activity.
And they found that spironolactone—a drug already used to treat fluid retention, high blood pressure, and other conditions—affects NER activity.
Specifically, the team found that, when combined with platinum derivatives, spironolactone significantly increased cytotoxicity in ovarian and colon cancer cells.
As platinum-based chemotherapy is used to treat a range of cancers, similar results might occur in other malignancies as well.
The researchers also noted that, because spironolactone is already in use for other purposes, it doesn’t require a new application for marketing authorization. And its side effects are already known.
The team said this suggests that protocols testing spironolactone in combination with platinum-based chemotherapy could be organized rather quickly. ![]()