User login
Bringing CME to the Bedside
Hospitalists, and physicians in general, recognize the need for continuing medical education (CME) to update their knowledge and skills to provide the best possible care for patients. Interactive and personalized learning activities provide the most effective approaches for maintaining or improving physician competency.[1, 2] Despite guidelines that recommend a shift of CME from the traditional large lecture format to case‐based and highly interactive learning techniques,[3] this has been challenging to achieve in practice.
In this issue of the Journal of Hospital Medicine, Sehgal and collaborators at the University of California, San Francisco (UCSF) report innovative and highly appealing CME activity that provides a short, focused experience for the practicing hospitalist seeking to update his or her skills.[4] The UCSF Hospitalist Mini‐College (UHMC) embraced the principles for creation of effective CME by conducting needs assessment from community hospitalists and constructing a program that provides focused, interactive, small‐group, intensive experiences and then evaluating the experience to improve subsequent iterations of the Mini‐College. The UHMC immerses participants in a relatively intense experience that includes close interaction with prominent faculty, hands‐on bedside experiences, practical skills, and attendance at sessions (resident report, morbidity and mortality conferences) that are part of every resident trainee's experience. Participants would be linked to their previous learning activities. As the authors point out, there may be a powerful stimulus to learning when practicing physicians return to the milieu of training environments. This observation deserves further investigation.
The report does not provide evidence that participation in the Mini‐College improved patient outcomes or physician performance in practice; these outcome measures remain elusive and an aspirational goal in medical education research. However, experienced clinician educators have come to recognize and adopt effective interventions that simply make sense in the same fashion that it makes sense to use a parachute when jumping out of an airplane in flight.[5] The UHMC makes sense. The medical education literature is replete with articles describing educational innovations and their 1‐ to 2‐year outcomes, leaving the reader wondering about sustainability. It is reassuring that Sehgal et al. report 5 years of experience with the UHMC, and that the program has consistently had a waiting list of hospitalists who want to participate despite the expense. Although it requires patience on the part of educational innovators, this report helps set a standard for reporting enduring innovation in the education arena.
The article provides a description that is sufficiently detailed for other academic medical centers to replicate the intervention or to effectively adapt the principles of the intervention for the needs of their local hospitalist community. The authors should be congratulated for sharing the details of their program and for sharing powerful comments by participants. For hospitalist medical educators interested in sharing details of effective and sustained innovations, publication of this article emphasizes the Journal of Hospital Medicine's interest in disseminating these important projects.
In summary, the report on the UHMC model challenges all of us in academic hospital medicine to think creatively about how to provide effective, engaging, and exciting learning opportunities beyond the years of medical school and residency training.
- . Effects of continuing medical education on improving physician clinical care and patient health: a review of systematic reviews. Int J Technol Assess Health Care. 2005;21:380–385.
- , . Continuing medical education: the link between physician learning and health care outcomes. Acad Med. 2011;86:1339.
- , , . Continuing medical education: AMEE Education Guide No. 35. Med Teach. 2008;30:652–666.
- , , . Bringing continuing medical education to the bedside: The University of California, San Francisco hospitalist mini‐college. J HospMed. 2014;9:129–134.
- , . Parachute use to prevent death and major trauma related to gravitational challenge: systematic review of randomised controlled trials. BMJ. 2003;327:1459–1461.
Hospitalists, and physicians in general, recognize the need for continuing medical education (CME) to update their knowledge and skills to provide the best possible care for patients. Interactive and personalized learning activities provide the most effective approaches for maintaining or improving physician competency.[1, 2] Despite guidelines that recommend a shift of CME from the traditional large lecture format to case‐based and highly interactive learning techniques,[3] this has been challenging to achieve in practice.
In this issue of the Journal of Hospital Medicine, Sehgal and collaborators at the University of California, San Francisco (UCSF) report innovative and highly appealing CME activity that provides a short, focused experience for the practicing hospitalist seeking to update his or her skills.[4] The UCSF Hospitalist Mini‐College (UHMC) embraced the principles for creation of effective CME by conducting needs assessment from community hospitalists and constructing a program that provides focused, interactive, small‐group, intensive experiences and then evaluating the experience to improve subsequent iterations of the Mini‐College. The UHMC immerses participants in a relatively intense experience that includes close interaction with prominent faculty, hands‐on bedside experiences, practical skills, and attendance at sessions (resident report, morbidity and mortality conferences) that are part of every resident trainee's experience. Participants would be linked to their previous learning activities. As the authors point out, there may be a powerful stimulus to learning when practicing physicians return to the milieu of training environments. This observation deserves further investigation.
The report does not provide evidence that participation in the Mini‐College improved patient outcomes or physician performance in practice; these outcome measures remain elusive and an aspirational goal in medical education research. However, experienced clinician educators have come to recognize and adopt effective interventions that simply make sense in the same fashion that it makes sense to use a parachute when jumping out of an airplane in flight.[5] The UHMC makes sense. The medical education literature is replete with articles describing educational innovations and their 1‐ to 2‐year outcomes, leaving the reader wondering about sustainability. It is reassuring that Sehgal et al. report 5 years of experience with the UHMC, and that the program has consistently had a waiting list of hospitalists who want to participate despite the expense. Although it requires patience on the part of educational innovators, this report helps set a standard for reporting enduring innovation in the education arena.
The article provides a description that is sufficiently detailed for other academic medical centers to replicate the intervention or to effectively adapt the principles of the intervention for the needs of their local hospitalist community. The authors should be congratulated for sharing the details of their program and for sharing powerful comments by participants. For hospitalist medical educators interested in sharing details of effective and sustained innovations, publication of this article emphasizes the Journal of Hospital Medicine's interest in disseminating these important projects.
In summary, the report on the UHMC model challenges all of us in academic hospital medicine to think creatively about how to provide effective, engaging, and exciting learning opportunities beyond the years of medical school and residency training.
Hospitalists, and physicians in general, recognize the need for continuing medical education (CME) to update their knowledge and skills to provide the best possible care for patients. Interactive and personalized learning activities provide the most effective approaches for maintaining or improving physician competency.[1, 2] Despite guidelines that recommend a shift of CME from the traditional large lecture format to case‐based and highly interactive learning techniques,[3] this has been challenging to achieve in practice.
In this issue of the Journal of Hospital Medicine, Sehgal and collaborators at the University of California, San Francisco (UCSF) report innovative and highly appealing CME activity that provides a short, focused experience for the practicing hospitalist seeking to update his or her skills.[4] The UCSF Hospitalist Mini‐College (UHMC) embraced the principles for creation of effective CME by conducting needs assessment from community hospitalists and constructing a program that provides focused, interactive, small‐group, intensive experiences and then evaluating the experience to improve subsequent iterations of the Mini‐College. The UHMC immerses participants in a relatively intense experience that includes close interaction with prominent faculty, hands‐on bedside experiences, practical skills, and attendance at sessions (resident report, morbidity and mortality conferences) that are part of every resident trainee's experience. Participants would be linked to their previous learning activities. As the authors point out, there may be a powerful stimulus to learning when practicing physicians return to the milieu of training environments. This observation deserves further investigation.
The report does not provide evidence that participation in the Mini‐College improved patient outcomes or physician performance in practice; these outcome measures remain elusive and an aspirational goal in medical education research. However, experienced clinician educators have come to recognize and adopt effective interventions that simply make sense in the same fashion that it makes sense to use a parachute when jumping out of an airplane in flight.[5] The UHMC makes sense. The medical education literature is replete with articles describing educational innovations and their 1‐ to 2‐year outcomes, leaving the reader wondering about sustainability. It is reassuring that Sehgal et al. report 5 years of experience with the UHMC, and that the program has consistently had a waiting list of hospitalists who want to participate despite the expense. Although it requires patience on the part of educational innovators, this report helps set a standard for reporting enduring innovation in the education arena.
The article provides a description that is sufficiently detailed for other academic medical centers to replicate the intervention or to effectively adapt the principles of the intervention for the needs of their local hospitalist community. The authors should be congratulated for sharing the details of their program and for sharing powerful comments by participants. For hospitalist medical educators interested in sharing details of effective and sustained innovations, publication of this article emphasizes the Journal of Hospital Medicine's interest in disseminating these important projects.
In summary, the report on the UHMC model challenges all of us in academic hospital medicine to think creatively about how to provide effective, engaging, and exciting learning opportunities beyond the years of medical school and residency training.
- . Effects of continuing medical education on improving physician clinical care and patient health: a review of systematic reviews. Int J Technol Assess Health Care. 2005;21:380–385.
- , . Continuing medical education: the link between physician learning and health care outcomes. Acad Med. 2011;86:1339.
- , , . Continuing medical education: AMEE Education Guide No. 35. Med Teach. 2008;30:652–666.
- , , . Bringing continuing medical education to the bedside: The University of California, San Francisco hospitalist mini‐college. J HospMed. 2014;9:129–134.
- , . Parachute use to prevent death and major trauma related to gravitational challenge: systematic review of randomised controlled trials. BMJ. 2003;327:1459–1461.
- . Effects of continuing medical education on improving physician clinical care and patient health: a review of systematic reviews. Int J Technol Assess Health Care. 2005;21:380–385.
- , . Continuing medical education: the link between physician learning and health care outcomes. Acad Med. 2011;86:1339.
- , , . Continuing medical education: AMEE Education Guide No. 35. Med Teach. 2008;30:652–666.
- , , . Bringing continuing medical education to the bedside: The University of California, San Francisco hospitalist mini‐college. J HospMed. 2014;9:129–134.
- , . Parachute use to prevent death and major trauma related to gravitational challenge: systematic review of randomised controlled trials. BMJ. 2003;327:1459–1461.
Perioral dermatitis and diet
Could it be the carbs?
In my practice, I have observed consistent improvements in recalcitrant perioral dermatitis when patients switch to low-carbohydrate diets. Several of my patients with perioral dermatitis that responded poorly to oral doxycycline, topical metronidazole, and topical tacrolimus – or recurred upon cessation of therapy – have proven to have gluten sensitivity or intolerance. Their skin condition improves when they go on a gluten-free diet. But I have also seen considerable improvements after patients undertake low-carbohydrate, high-protein diets, even if those patients have no diagnosed gluten sensitivity. These improvements have occurred with minimal oral and topical treatments, and these patients have not experienced recurrences.
There have been no well-controlled studies, or even case reports to my knowledge, linking carbohydrate or gluten intake to perioral dermatitis. Could the improvement be serendipitous, or is there some basis for carbohydrates contributing to inflammatory status in the oral and gastrointestinal mucosa?
Alcohol, spicy foods, and chocolate have been linked to exacerbation of erythemogenic and papulopustular rosacea. However, the precipitating ingredients in these foods have not been identified. Could the common link simply be an abundance of carbohydrates?
More studies are needed to better define the role of diet in perioral dermatitis. In the meantime, I am seeing good results with low-carb/carb-free diets and will continue to suggest them to prevent recurrences in my patients with perioral dermatitis.
Dr. Talakoub is in private practice in McLean, Va.
Could it be the carbs?
In my practice, I have observed consistent improvements in recalcitrant perioral dermatitis when patients switch to low-carbohydrate diets. Several of my patients with perioral dermatitis that responded poorly to oral doxycycline, topical metronidazole, and topical tacrolimus – or recurred upon cessation of therapy – have proven to have gluten sensitivity or intolerance. Their skin condition improves when they go on a gluten-free diet. But I have also seen considerable improvements after patients undertake low-carbohydrate, high-protein diets, even if those patients have no diagnosed gluten sensitivity. These improvements have occurred with minimal oral and topical treatments, and these patients have not experienced recurrences.
There have been no well-controlled studies, or even case reports to my knowledge, linking carbohydrate or gluten intake to perioral dermatitis. Could the improvement be serendipitous, or is there some basis for carbohydrates contributing to inflammatory status in the oral and gastrointestinal mucosa?
Alcohol, spicy foods, and chocolate have been linked to exacerbation of erythemogenic and papulopustular rosacea. However, the precipitating ingredients in these foods have not been identified. Could the common link simply be an abundance of carbohydrates?
More studies are needed to better define the role of diet in perioral dermatitis. In the meantime, I am seeing good results with low-carb/carb-free diets and will continue to suggest them to prevent recurrences in my patients with perioral dermatitis.
Dr. Talakoub is in private practice in McLean, Va.
Could it be the carbs?
In my practice, I have observed consistent improvements in recalcitrant perioral dermatitis when patients switch to low-carbohydrate diets. Several of my patients with perioral dermatitis that responded poorly to oral doxycycline, topical metronidazole, and topical tacrolimus – or recurred upon cessation of therapy – have proven to have gluten sensitivity or intolerance. Their skin condition improves when they go on a gluten-free diet. But I have also seen considerable improvements after patients undertake low-carbohydrate, high-protein diets, even if those patients have no diagnosed gluten sensitivity. These improvements have occurred with minimal oral and topical treatments, and these patients have not experienced recurrences.
There have been no well-controlled studies, or even case reports to my knowledge, linking carbohydrate or gluten intake to perioral dermatitis. Could the improvement be serendipitous, or is there some basis for carbohydrates contributing to inflammatory status in the oral and gastrointestinal mucosa?
Alcohol, spicy foods, and chocolate have been linked to exacerbation of erythemogenic and papulopustular rosacea. However, the precipitating ingredients in these foods have not been identified. Could the common link simply be an abundance of carbohydrates?
More studies are needed to better define the role of diet in perioral dermatitis. In the meantime, I am seeing good results with low-carb/carb-free diets and will continue to suggest them to prevent recurrences in my patients with perioral dermatitis.
Dr. Talakoub is in private practice in McLean, Va.

First-in-man bioengineered graft proves enduring for vascular access
DALLAS – An investigational tissue-engineered vascular graft has enduring potential for vascular access for hemodialysis in patients with end-stage renal disease, based on early clinical results.
Moreover, other potential uses are on the horizon. The big picture involves subsequent extrapolation of this technology from the large-diameter, high-flow bioengineered vessels required for hemodialysis to the creation of small-diameter, low-flow vessels for coronary artery and peripheral arterial graft surgery, Dr. Jeffrey H. Lawson explained at the American Heart Association scientific sessions.

"Our goal is to make a tissue-engineered conduit that could be used widely throughout the body," said Dr. Lawson, professor of surgery and of pathology at Duke University Medical Center, Durham, N.C.
He presented the results from the first-in-man, ongoing phase I clinical experience with the Humacyte graft, which to date has been implanted to provide vascular access for hemodialysis in 28 patients, with 6-month patency as the primary study endpoint. This was a challenging study population, with an average of 4.1 previous access procedure failures per patient. The presentation at the AHA was the first public disclosure of the results of a project Dr. Lawson has been working on for more than 15 years. His surgical colleagues from Poland, who have done the implantations in patients with end-stage renal disease, were in attendance.
The overall 6-month patency was 100%, with no infections, no sign of an immune response, and no aneurysms or other indication of structural degeneration, he said.
Of the 28 patients, 20 had no further interventions, yielding a primary unassisted 6-month patency rate of 71%. Eight patients collectively underwent 10 interventions to maintain patency: eight had thrombectomies for graft- or surgically related thrombosis and two had venous anastomoses. Flow rates have remained suitable for dialysis in all patients, and the grafts are being used for dialysis three times per week. Dr. Lawson described the grafts as easy to cannulate via standard techniques.
He characterized these initial results as "quite remarkable" compared with the outcomes in two large studies of the current benchmark technologies, which are synthetic grafts made of PTFE (polytetrafluoroethyline). In those studies, the primary patency rate at 6 months was less than 50%, with a secondary patency rate of 77% and a 10% infection rate. In other studies, 30%-40% of PTFE grafts are abandoned within 12 months due to loss of patency.
The process of creating the bioengineered grafts begins with harvesting human aortic vascular smooth muscle cells, seeding them on a biodegradable matrix, then culturing them under pulsatile conditions. When the biodegradable matrix melts away, what remains is a tube comprised of vascular smooth muscle cells and extracellular matrix. This is then decellularized, yielding a tube of extracellular matrix that can be shipped off the shelf and around the world.
In primate models, the implanted bioengineered graft has been shown to repopulate with the host’s own vascular smooth muscle cells lined intimally by endothelium.
"Where we implanted an acellular structure, it appears to now be a living tissue, suggesting [the graft] has become their tissue, not ours," Dr. Lawson said.
To date, none of the bioengineered grafts implanted in patients has been explanted, so it’s unknown whether the favorable histologic changes seen in primates’ grafts also occur in humans. Larger clinical trials with longer follow-up are planned in order to assess the bioengineered graft’s durability.
Dr. Lawson’s study is funded by a Department of Defense research grant and by Humacyte. He serves as a consultant to the company.
This work is exciting. The early patency, thrombosis, and infection rates are encouraging.
The unmet clinical need for better ways to provide vascular access for hemodialysis is huge. There are 450,000 U.S. patients with end-stage renal disease on long-term hemodialysis. In this population, hemodialysis access morbidity costs more than $1 billion per year. Although the preferred means of vascular access is an arteriovenous fistula, many hemodialysis patients don’t have suitable veins. And 60% of fistulas become unusable within 6 months.

|
|
We’ve got a conundrum where PTFE grafts have their problems and fistulas have their own problems. We don’t have a good clinical armamentarium.
Synthetic grafts most often lose patency because of venous outflow tract stenosis due to intimal hyperplasia. Balloon angioplasty of the stenotic anastomosis has been the conventional treatment to restore patency, but a landmark randomized trial carried out several years ago (N. Engl. J. Med. 2010;362:494-503) showed the patency rate was a mere 23%, significantly worse than the 51% patency rate with a PTFE-covered stent graft – and even that 51% patency rate, is abysmal.
Dr. Sanjay Misra is professor of radiology at the Mayo Clinic in Rochester, Minn. He was the invited discussant of the paper at the meeting and declared having no relevant financial disclosures.
This work is exciting. The early patency, thrombosis, and infection rates are encouraging.
The unmet clinical need for better ways to provide vascular access for hemodialysis is huge. There are 450,000 U.S. patients with end-stage renal disease on long-term hemodialysis. In this population, hemodialysis access morbidity costs more than $1 billion per year. Although the preferred means of vascular access is an arteriovenous fistula, many hemodialysis patients don’t have suitable veins. And 60% of fistulas become unusable within 6 months.
|
|
|
We’ve got a conundrum where PTFE grafts have their problems and fistulas have their own problems. We don’t have a good clinical armamentarium.
Synthetic grafts most often lose patency because of venous outflow tract stenosis due to intimal hyperplasia. Balloon angioplasty of the stenotic anastomosis has been the conventional treatment to restore patency, but a landmark randomized trial carried out several years ago (N. Engl. J. Med. 2010;362:494-503) showed the patency rate was a mere 23%, significantly worse than the 51% patency rate with a PTFE-covered stent graft – and even that 51% patency rate, is abysmal.
Dr. Sanjay Misra is professor of radiology at the Mayo Clinic in Rochester, Minn. He was the invited discussant of the paper at the meeting and declared having no relevant financial disclosures.
This work is exciting. The early patency, thrombosis, and infection rates are encouraging.
The unmet clinical need for better ways to provide vascular access for hemodialysis is huge. There are 450,000 U.S. patients with end-stage renal disease on long-term hemodialysis. In this population, hemodialysis access morbidity costs more than $1 billion per year. Although the preferred means of vascular access is an arteriovenous fistula, many hemodialysis patients don’t have suitable veins. And 60% of fistulas become unusable within 6 months.
|
|
|
We’ve got a conundrum where PTFE grafts have their problems and fistulas have their own problems. We don’t have a good clinical armamentarium.
Synthetic grafts most often lose patency because of venous outflow tract stenosis due to intimal hyperplasia. Balloon angioplasty of the stenotic anastomosis has been the conventional treatment to restore patency, but a landmark randomized trial carried out several years ago (N. Engl. J. Med. 2010;362:494-503) showed the patency rate was a mere 23%, significantly worse than the 51% patency rate with a PTFE-covered stent graft – and even that 51% patency rate, is abysmal.
Dr. Sanjay Misra is professor of radiology at the Mayo Clinic in Rochester, Minn. He was the invited discussant of the paper at the meeting and declared having no relevant financial disclosures.
DALLAS – An investigational tissue-engineered vascular graft has enduring potential for vascular access for hemodialysis in patients with end-stage renal disease, based on early clinical results.
Moreover, other potential uses are on the horizon. The big picture involves subsequent extrapolation of this technology from the large-diameter, high-flow bioengineered vessels required for hemodialysis to the creation of small-diameter, low-flow vessels for coronary artery and peripheral arterial graft surgery, Dr. Jeffrey H. Lawson explained at the American Heart Association scientific sessions.
"Our goal is to make a tissue-engineered conduit that could be used widely throughout the body," said Dr. Lawson, professor of surgery and of pathology at Duke University Medical Center, Durham, N.C.
He presented the results from the first-in-man, ongoing phase I clinical experience with the Humacyte graft, which to date has been implanted to provide vascular access for hemodialysis in 28 patients, with 6-month patency as the primary study endpoint. This was a challenging study population, with an average of 4.1 previous access procedure failures per patient. The presentation at the AHA was the first public disclosure of the results of a project Dr. Lawson has been working on for more than 15 years. His surgical colleagues from Poland, who have done the implantations in patients with end-stage renal disease, were in attendance.
The overall 6-month patency was 100%, with no infections, no sign of an immune response, and no aneurysms or other indication of structural degeneration, he said.
Of the 28 patients, 20 had no further interventions, yielding a primary unassisted 6-month patency rate of 71%. Eight patients collectively underwent 10 interventions to maintain patency: eight had thrombectomies for graft- or surgically related thrombosis and two had venous anastomoses. Flow rates have remained suitable for dialysis in all patients, and the grafts are being used for dialysis three times per week. Dr. Lawson described the grafts as easy to cannulate via standard techniques.
He characterized these initial results as "quite remarkable" compared with the outcomes in two large studies of the current benchmark technologies, which are synthetic grafts made of PTFE (polytetrafluoroethyline). In those studies, the primary patency rate at 6 months was less than 50%, with a secondary patency rate of 77% and a 10% infection rate. In other studies, 30%-40% of PTFE grafts are abandoned within 12 months due to loss of patency.
The process of creating the bioengineered grafts begins with harvesting human aortic vascular smooth muscle cells, seeding them on a biodegradable matrix, then culturing them under pulsatile conditions. When the biodegradable matrix melts away, what remains is a tube comprised of vascular smooth muscle cells and extracellular matrix. This is then decellularized, yielding a tube of extracellular matrix that can be shipped off the shelf and around the world.
In primate models, the implanted bioengineered graft has been shown to repopulate with the host’s own vascular smooth muscle cells lined intimally by endothelium.
"Where we implanted an acellular structure, it appears to now be a living tissue, suggesting [the graft] has become their tissue, not ours," Dr. Lawson said.
To date, none of the bioengineered grafts implanted in patients has been explanted, so it’s unknown whether the favorable histologic changes seen in primates’ grafts also occur in humans. Larger clinical trials with longer follow-up are planned in order to assess the bioengineered graft’s durability.
Dr. Lawson’s study is funded by a Department of Defense research grant and by Humacyte. He serves as a consultant to the company.
DALLAS – An investigational tissue-engineered vascular graft has enduring potential for vascular access for hemodialysis in patients with end-stage renal disease, based on early clinical results.
Moreover, other potential uses are on the horizon. The big picture involves subsequent extrapolation of this technology from the large-diameter, high-flow bioengineered vessels required for hemodialysis to the creation of small-diameter, low-flow vessels for coronary artery and peripheral arterial graft surgery, Dr. Jeffrey H. Lawson explained at the American Heart Association scientific sessions.
"Our goal is to make a tissue-engineered conduit that could be used widely throughout the body," said Dr. Lawson, professor of surgery and of pathology at Duke University Medical Center, Durham, N.C.
He presented the results from the first-in-man, ongoing phase I clinical experience with the Humacyte graft, which to date has been implanted to provide vascular access for hemodialysis in 28 patients, with 6-month patency as the primary study endpoint. This was a challenging study population, with an average of 4.1 previous access procedure failures per patient. The presentation at the AHA was the first public disclosure of the results of a project Dr. Lawson has been working on for more than 15 years. His surgical colleagues from Poland, who have done the implantations in patients with end-stage renal disease, were in attendance.
The overall 6-month patency was 100%, with no infections, no sign of an immune response, and no aneurysms or other indication of structural degeneration, he said.
Of the 28 patients, 20 had no further interventions, yielding a primary unassisted 6-month patency rate of 71%. Eight patients collectively underwent 10 interventions to maintain patency: eight had thrombectomies for graft- or surgically related thrombosis and two had venous anastomoses. Flow rates have remained suitable for dialysis in all patients, and the grafts are being used for dialysis three times per week. Dr. Lawson described the grafts as easy to cannulate via standard techniques.
He characterized these initial results as "quite remarkable" compared with the outcomes in two large studies of the current benchmark technologies, which are synthetic grafts made of PTFE (polytetrafluoroethyline). In those studies, the primary patency rate at 6 months was less than 50%, with a secondary patency rate of 77% and a 10% infection rate. In other studies, 30%-40% of PTFE grafts are abandoned within 12 months due to loss of patency.
The process of creating the bioengineered grafts begins with harvesting human aortic vascular smooth muscle cells, seeding them on a biodegradable matrix, then culturing them under pulsatile conditions. When the biodegradable matrix melts away, what remains is a tube comprised of vascular smooth muscle cells and extracellular matrix. This is then decellularized, yielding a tube of extracellular matrix that can be shipped off the shelf and around the world.
In primate models, the implanted bioengineered graft has been shown to repopulate with the host’s own vascular smooth muscle cells lined intimally by endothelium.
"Where we implanted an acellular structure, it appears to now be a living tissue, suggesting [the graft] has become their tissue, not ours," Dr. Lawson said.
To date, none of the bioengineered grafts implanted in patients has been explanted, so it’s unknown whether the favorable histologic changes seen in primates’ grafts also occur in humans. Larger clinical trials with longer follow-up are planned in order to assess the bioengineered graft’s durability.
Dr. Lawson’s study is funded by a Department of Defense research grant and by Humacyte. He serves as a consultant to the company.
AT THE AHA SCIENTIFIC SESSIONS
Major finding: The 6-month enduring patency rate of an investigational tissue-engineered vascular graft for hemodialysis access was 100%, markedly better than rates achievable with synthetic PTFE grafts, the current benchmark technology.
Data source: An initial report from an ongoing prospective first-in-man study in which, to date, 28 patients with end-stage renal disease have been implanted with a novel tissue-engineered vascular graft for use as a hemodialysis access.
Disclosures: The study was funded by the Department of Defense and Humacyte. The presenter is a consultant to the company.

Is Spreading Pain Due to Injury?
Answer
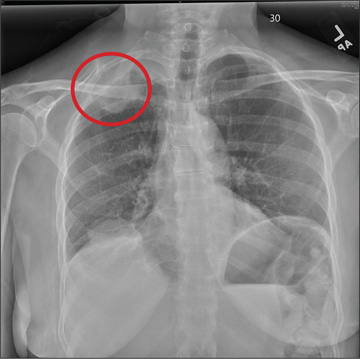
The radiograph shows a right apical mass. This clinical and radiographic presentation is strongly suggestive of a Pancoast tumor. Such lung masses (typically non–small cell carcinomas) can cause brachial plexus compression when they progress, which results in thoracic outlet obstruction and symptoms similar to those seen in this patient.
The patient was admitted by a hospitalist service, and further imaging did confirm the presence of a lung mass, as well as extension to the chest wall and cervicothoracic portion of the spinal canal. CT-guided biopsy of the mass is pending.
Answer
The radiograph shows a right apical mass. This clinical and radiographic presentation is strongly suggestive of a Pancoast tumor. Such lung masses (typically non–small cell carcinomas) can cause brachial plexus compression when they progress, which results in thoracic outlet obstruction and symptoms similar to those seen in this patient.
The patient was admitted by a hospitalist service, and further imaging did confirm the presence of a lung mass, as well as extension to the chest wall and cervicothoracic portion of the spinal canal. CT-guided biopsy of the mass is pending.
Answer
The radiograph shows a right apical mass. This clinical and radiographic presentation is strongly suggestive of a Pancoast tumor. Such lung masses (typically non–small cell carcinomas) can cause brachial plexus compression when they progress, which results in thoracic outlet obstruction and symptoms similar to those seen in this patient.
The patient was admitted by a hospitalist service, and further imaging did confirm the presence of a lung mass, as well as extension to the chest wall and cervicothoracic portion of the spinal canal. CT-guided biopsy of the mass is pending.
A 53-year-old woman presents with complaints of right-side chest wall, neck, and shoulder pain. Her symptoms started two months ago, when she says she injured herself while doing yard work. She initially self-treated but subsequently went to various emergency departments and walk-in clinics on several occasions; no definitive diagnosis was established. Recently, she has noticed increasing weakness in her right arm and hand as well. Medical history is significant for hypertension. Family history is remarkable for non-Hodgkin’s lymphoma (mother). Social history reveals that the patient is a smoker, with a pack-a-day habit for at least 40 years. On physical exam, you note normal vital signs. The patient has good range of motion in her extremities; however, the strength in her right upper extremity is significantly diminished. Her deltoid, biceps, triceps, and hand grip are all about 2/5. She also notes a paresthesia along her right anterior chest wall, although sensation is intact. Chest radiograph is ordered (shown). What is your impression?
Man, 45, With Greasy Rash and Deformed Nails
A 45-year-old man presented to the dermatology office complaining of a pruritic rash on his neck, chest, abdomen, and upper back. The rash had been present since the patient was 20, intermittently flaring and causing severe pruritus. For the past two weeks, it had become increasingly bothersome.
The patient described the rash as “greasy” brown plaques diffusely scattered on his body. The rash on his neck was the most bothersome, and the patient felt an uncontrollable need to scratch that area.
Since it first developed 25 years ago, he had used OTC hydrocortisone cream as needed to treat the rash. Although effective for past flares, the cream provided only minimal relief during the current episode.
The patient’s medical history included brittle nails with a worsening of nail quality in recent years. The family history revealed that the patient’s father and sister were affected by the same type of rash, which developed in adolescence for each of them, as well as brittle nails.
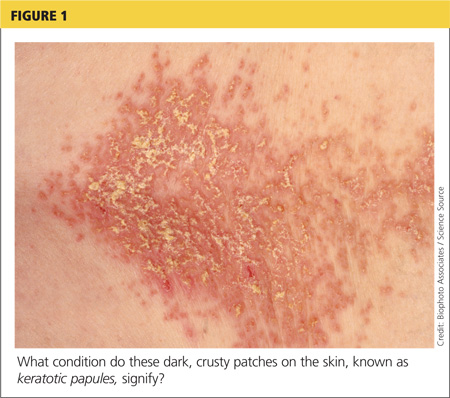
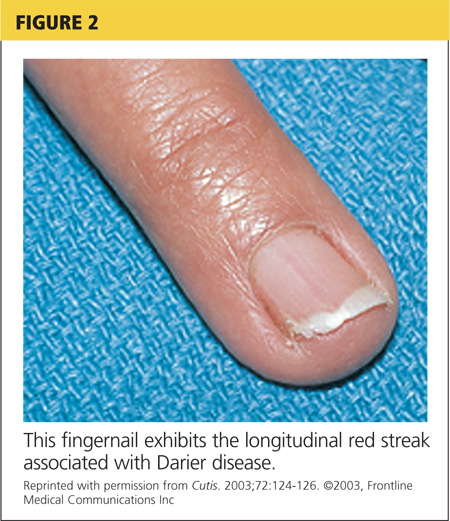
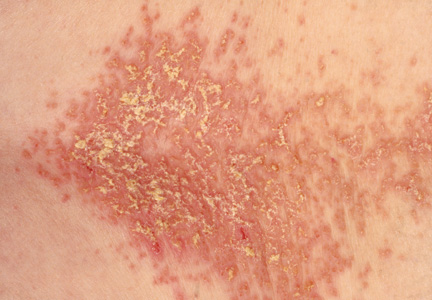
On physical examination, the skin was warm and moist to the touch. Flat, slightly elevated, greasy brown papules were scattered on the chest, abdomen, and upper back, with mild surrounding erythema (see Figure 1). Excoriated lesions were noted on the anterior surface of the neck, with pinpoint bleeding resulting from constant irritation. The patient’s fingernails were deformed, with longitudinal ridges and v-shaped notching of the free margin. The remainder of the physical exam was unremarkable, and review of systems was negative.
This patient’s symptoms could result from a variety of causes. Seborrheic dermatitis is a common skin condition that presents with brown plaques similar to those on the patient’s trunk. Another possible diagnosis is Grover’s disease, a rare disorder also known as transient acantholytic dermatosis, in which keratotic plaques appear on the torso and are thought to occur from trauma to sun-damaged skin. An additional consideration is Hailey-Hailey disease, a rare genetic disorder also known as benign familial pemphigus, which is characterized by red-brown plaques located predominantly on flexure surfaces.1 Skin biopsy should be performed for a definitive diagnosis.
Given the family history of a similar rash occurring in first-degree relatives and the distinct physical exam findings, the most likely diagnosis for this patient is keratosis follicularis, also known as Darier disease (DD) or Darier-White disease.
DISCUSSION
Named after Ferdinand-Jean Darier, who discovered this rare genodermatosis, DD is a rare genetic skin disorder caused by mutations of the ATP2A2 gene, located on the long arm of chromosome 12 at position 24,11.1,2 The mutation disrupts the encoding of the enzyme sarco/endoplasmic reticulum calcium-ATPase 2 (SERCA2). This enzyme is important in the transport of calcium ions across the cell membrane, and insufficient amounts lead to a defect in intracellular calcium signaling.2,3
This genetic mutation is inherited as an autosomal dominant trait with complete penetrance. DD affects men and women equally, with progressive skin signs of interfamilial and intrafamilial variability.4 Skin manifestations occur from late childhood to early adulthood and are typical during adolescence.4 Acute flare-ups can be triggered by heat, perspiration, sunlight, ultraviolet B exposure, stress, or certain medications (in particular, lithium).2 DD is not contagious.2
CLINICAL PRESENTATION
The characteristics of DD include yellow or brown, rough, firm papules that are frequently crusted. The papules often appear in seborrheic areas of the body, such as the chest, back, ears, nasolabial fold, forehead, scalp, and groin.4 The severity of expression varies from mild, with few lesions, to severe, in which the entire body is covered with disfiguring, macerated plaques emitting a strong odor. On biopsy, the histopathologic findings are typical of dyskeratosis and acantholysis.4
Fingernails (and occasionally toenails) display broad, white or red, somewhat translucent, longitudinal bands accompanied by v-shaped notching1,4,5 (see Figure 2). Such nail changes are diagnostic and occur in 92% to 95% of patients with DD.6 They may, in fact, occur in the absence of cutaneous disease. All nails may be affected, but usually only two to three are involved.6
Although uncommon in DD, white, umbilicated, or cobblestone plaques may be found on intraoral mucous membranes (ie, tongue, buccal mucosa, palate, epiglottis, pharyngeal wall, and esophagus); due to confluence, papules may mimic leukoplakia.7 Lesions may also appear on the vulva or rectum.1,5 In severe cases, the salivary glands can become blocked, and the gums can hypertrophy.5
Since epidermal and brain tissue both derive from ectoderm, pathologic processes that affect one organ system may also affect the other.8 Indeed, among patients with DD, neuropsychiatric problems—including epilepsy, learning difficulties, and schizoaffective disorder—are commonly reported.1 To confirm an association between DD and ATP2A2 mutations, Jacobsen and colleagues performed an analysis of 19 unrelated DD patients with neuropsychiatric phenotypes. They discovered evidence to support the gene’s pleiotropic effects in the brain and hypothesized that mutations in the enzyme SERCA2 correlate with these phenotypes, most specifically for mood disorders.9
TREATMENT AND MANAGEMENT
Although no cure is currently available for DD, both short- and long-term treatment options are available; the choice should be based on the severity of an individual patient’s signs and symptoms. For mild cases, topical therapy, such as general emollients, corticosteroid ointments, and high sun protection factor sunscreen, is sufficient.1
For moderate cases, topical retinoids, including tretinoin cream, adapalene gel or cream, and tazarotene gel, may be necessary.4 Keratolytics, including salicylic acid in propylene glycol gel, may be used to regulate hyperkeratosis.4 Celecoxib, a COX-2 inhibitor, is another option that may restore the down regulation of SERCA2. This can prevent progression of the disease.10
Long-term management includes use of oral retinoid therapy (eg, acitretin), which might reduce the frequency of inflammatory flares.1 Systemic adverse effects from long-term use of oral retinoids are cause for concern, however. Close monitoring along with patient education can limit the occurrence of complications.11
If DD is uncontrolled with medication, dermabrasion and erbium:YAG laser ablation have been used to successfully treat chronic cases.12 Although these treatment options may remove existing lesions, it is important to inform patients that the disease has not been cured, that remission is difficult to attain, and that lesions may recur.
Because viral, bacterial, and fungal superinfections are common and may exacerbate the disease, be sure to check for signs of infection while examining the patient.4 Patients should be advised to avoid hot environments, and if that is not possible, to dress in cool cotton clothing to allow for proper ventilation and avoid the build-up of perspiration. Excessive perspiration along with poor hygiene can contribute to the formation of infections as well as trigger a flare-up. If an infection develops, patients should consult a health care provider.
Keeping the skin well moisturized can alleviate the constant pruritus that many patients experience. Daily sunscreen use is essential to avoid skin irritation caused by the sun, which can trigger an acute flare-up. Patients should be advised to avoid the long-term use of corticosteroid ointment. They should also contact their health care provider before using OTC treatments such as Burow’s solution.
CONCLUSION
A thorough history and physical exam are crucial in the diagnosis of DD. In this particular case, inquiry into family history was the key to proper diagnosis. That information, paired with a thorough physical exam, led to the correct diagnosis of this rare genetic skin disorder. A skin biopsy provided definitive confirmation.
This patient had a mild-to-moderate manifestation of DD. He was prescribed retinoid therapy, and routine follow-up visits were recommended to monitor the efficacy of medical therapy and to screen for secondary infections or neuropsychiatric disorders.
This case illustrates the importance of taking a full history and performing an in-depth physical exam when a patient presents with an unfamiliar complaint. Being thorough reduces the risk of missing a crucial element that can guide the diagnostic process.
REFERENCES
1. Creamer D, Barker J, Kerdel FA. Papular and papulosquamous dermatoses. In: Acute Adult Dermatology: Diagnosis and Management (A Colour Handbook). London, UK: Manson Publishing Ltd; 2011:48.
2. Kelly EB. Darier disease (DAR). In: Encyclopedia of Human Genetics and Disease. Santa Barbara, CA: ABC-CLIO; 2013:186-187.
3. Klausegger A, Laimer M, Bauer JW. Darier disease. [In German.] Hautarzt. 2013;64:22-25.
4. Ringpfeil F. Dermatologic disorders. In: NORD Guide to Rare Disorders. Philadelphia, PA: Lippincott Williams & Wilkins; 2003:101.
5. Disorders of keratinization. In: Ostler HB, Maibach HI, Hoke AW, Schwab IR, eds. Diseases of the Eye and Skin: A Color Atlas. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:23-34.
6. Baran R, de Berker D, Holzberg M, Thomas L, eds. Baran & Dawber’s Diseases of the Nails and their Management. 4th ed. West Sussex, UK: John Wiley & Sons, Ltd; 2012:295-296.
7. Thiagarajan MK, Narasimhan M, Sankarasubramanian A. Darier disease with oral and esophageal involvement: a case report. Indian J Dent Res. 2011;22:843-846.
8. Medansky RS, Woloshin AA. Darier’s disease: an evaluation of its neuropsychiatric component. Arch Dermatol. 1961;84:482-484.
9. Jacobsen NJ, Lyons I, Hoogendoorn B, et al. ATP2A2 mutations in Darier’s disease and their relationship to neuropsychiatric phenotypes. Hum Mol Genet. 1999;8:1631-1636.
10. Kamijo M, Nishiyama C, Takagi A, et al. Cyclooxygenase-2 inhibition restores ultraviolet B-induced downregulation of ATP2A2/SERCA2 in keratinocytes: possible therapeutic approach of cyclooxygenase-2 inhibition for treatment of Darier disease. Br J Dermatol. 2012;166: 1017-1022.
11. Brecher AR, Orlow SJ. Oral retinoid therapy for dermatologic conditions in children and adolescents. J Am Acad Dermatol. 2003;49:171-182.
12. Beier C, Kaufmann R. Efficacy of erbium:YAG laser ablation in Darier disease and Hailey-Hailey disease. Arch Dermatol. 1999;35:423-427.
A 45-year-old man presented to the dermatology office complaining of a pruritic rash on his neck, chest, abdomen, and upper back. The rash had been present since the patient was 20, intermittently flaring and causing severe pruritus. For the past two weeks, it had become increasingly bothersome.
The patient described the rash as “greasy” brown plaques diffusely scattered on his body. The rash on his neck was the most bothersome, and the patient felt an uncontrollable need to scratch that area.
Since it first developed 25 years ago, he had used OTC hydrocortisone cream as needed to treat the rash. Although effective for past flares, the cream provided only minimal relief during the current episode.
The patient’s medical history included brittle nails with a worsening of nail quality in recent years. The family history revealed that the patient’s father and sister were affected by the same type of rash, which developed in adolescence for each of them, as well as brittle nails.
On physical examination, the skin was warm and moist to the touch. Flat, slightly elevated, greasy brown papules were scattered on the chest, abdomen, and upper back, with mild surrounding erythema (see Figure 1). Excoriated lesions were noted on the anterior surface of the neck, with pinpoint bleeding resulting from constant irritation. The patient’s fingernails were deformed, with longitudinal ridges and v-shaped notching of the free margin. The remainder of the physical exam was unremarkable, and review of systems was negative.
This patient’s symptoms could result from a variety of causes. Seborrheic dermatitis is a common skin condition that presents with brown plaques similar to those on the patient’s trunk. Another possible diagnosis is Grover’s disease, a rare disorder also known as transient acantholytic dermatosis, in which keratotic plaques appear on the torso and are thought to occur from trauma to sun-damaged skin. An additional consideration is Hailey-Hailey disease, a rare genetic disorder also known as benign familial pemphigus, which is characterized by red-brown plaques located predominantly on flexure surfaces.1 Skin biopsy should be performed for a definitive diagnosis.
Given the family history of a similar rash occurring in first-degree relatives and the distinct physical exam findings, the most likely diagnosis for this patient is keratosis follicularis, also known as Darier disease (DD) or Darier-White disease.
DISCUSSION
Named after Ferdinand-Jean Darier, who discovered this rare genodermatosis, DD is a rare genetic skin disorder caused by mutations of the ATP2A2 gene, located on the long arm of chromosome 12 at position 24,11.1,2 The mutation disrupts the encoding of the enzyme sarco/endoplasmic reticulum calcium-ATPase 2 (SERCA2). This enzyme is important in the transport of calcium ions across the cell membrane, and insufficient amounts lead to a defect in intracellular calcium signaling.2,3
This genetic mutation is inherited as an autosomal dominant trait with complete penetrance. DD affects men and women equally, with progressive skin signs of interfamilial and intrafamilial variability.4 Skin manifestations occur from late childhood to early adulthood and are typical during adolescence.4 Acute flare-ups can be triggered by heat, perspiration, sunlight, ultraviolet B exposure, stress, or certain medications (in particular, lithium).2 DD is not contagious.2
CLINICAL PRESENTATION
The characteristics of DD include yellow or brown, rough, firm papules that are frequently crusted. The papules often appear in seborrheic areas of the body, such as the chest, back, ears, nasolabial fold, forehead, scalp, and groin.4 The severity of expression varies from mild, with few lesions, to severe, in which the entire body is covered with disfiguring, macerated plaques emitting a strong odor. On biopsy, the histopathologic findings are typical of dyskeratosis and acantholysis.4
Fingernails (and occasionally toenails) display broad, white or red, somewhat translucent, longitudinal bands accompanied by v-shaped notching1,4,5 (see Figure 2). Such nail changes are diagnostic and occur in 92% to 95% of patients with DD.6 They may, in fact, occur in the absence of cutaneous disease. All nails may be affected, but usually only two to three are involved.6
Although uncommon in DD, white, umbilicated, or cobblestone plaques may be found on intraoral mucous membranes (ie, tongue, buccal mucosa, palate, epiglottis, pharyngeal wall, and esophagus); due to confluence, papules may mimic leukoplakia.7 Lesions may also appear on the vulva or rectum.1,5 In severe cases, the salivary glands can become blocked, and the gums can hypertrophy.5
Since epidermal and brain tissue both derive from ectoderm, pathologic processes that affect one organ system may also affect the other.8 Indeed, among patients with DD, neuropsychiatric problems—including epilepsy, learning difficulties, and schizoaffective disorder—are commonly reported.1 To confirm an association between DD and ATP2A2 mutations, Jacobsen and colleagues performed an analysis of 19 unrelated DD patients with neuropsychiatric phenotypes. They discovered evidence to support the gene’s pleiotropic effects in the brain and hypothesized that mutations in the enzyme SERCA2 correlate with these phenotypes, most specifically for mood disorders.9
TREATMENT AND MANAGEMENT
Although no cure is currently available for DD, both short- and long-term treatment options are available; the choice should be based on the severity of an individual patient’s signs and symptoms. For mild cases, topical therapy, such as general emollients, corticosteroid ointments, and high sun protection factor sunscreen, is sufficient.1
For moderate cases, topical retinoids, including tretinoin cream, adapalene gel or cream, and tazarotene gel, may be necessary.4 Keratolytics, including salicylic acid in propylene glycol gel, may be used to regulate hyperkeratosis.4 Celecoxib, a COX-2 inhibitor, is another option that may restore the down regulation of SERCA2. This can prevent progression of the disease.10
Long-term management includes use of oral retinoid therapy (eg, acitretin), which might reduce the frequency of inflammatory flares.1 Systemic adverse effects from long-term use of oral retinoids are cause for concern, however. Close monitoring along with patient education can limit the occurrence of complications.11
If DD is uncontrolled with medication, dermabrasion and erbium:YAG laser ablation have been used to successfully treat chronic cases.12 Although these treatment options may remove existing lesions, it is important to inform patients that the disease has not been cured, that remission is difficult to attain, and that lesions may recur.
Because viral, bacterial, and fungal superinfections are common and may exacerbate the disease, be sure to check for signs of infection while examining the patient.4 Patients should be advised to avoid hot environments, and if that is not possible, to dress in cool cotton clothing to allow for proper ventilation and avoid the build-up of perspiration. Excessive perspiration along with poor hygiene can contribute to the formation of infections as well as trigger a flare-up. If an infection develops, patients should consult a health care provider.
Keeping the skin well moisturized can alleviate the constant pruritus that many patients experience. Daily sunscreen use is essential to avoid skin irritation caused by the sun, which can trigger an acute flare-up. Patients should be advised to avoid the long-term use of corticosteroid ointment. They should also contact their health care provider before using OTC treatments such as Burow’s solution.
CONCLUSION
A thorough history and physical exam are crucial in the diagnosis of DD. In this particular case, inquiry into family history was the key to proper diagnosis. That information, paired with a thorough physical exam, led to the correct diagnosis of this rare genetic skin disorder. A skin biopsy provided definitive confirmation.
This patient had a mild-to-moderate manifestation of DD. He was prescribed retinoid therapy, and routine follow-up visits were recommended to monitor the efficacy of medical therapy and to screen for secondary infections or neuropsychiatric disorders.
This case illustrates the importance of taking a full history and performing an in-depth physical exam when a patient presents with an unfamiliar complaint. Being thorough reduces the risk of missing a crucial element that can guide the diagnostic process.
REFERENCES
1. Creamer D, Barker J, Kerdel FA. Papular and papulosquamous dermatoses. In: Acute Adult Dermatology: Diagnosis and Management (A Colour Handbook). London, UK: Manson Publishing Ltd; 2011:48.
2. Kelly EB. Darier disease (DAR). In: Encyclopedia of Human Genetics and Disease. Santa Barbara, CA: ABC-CLIO; 2013:186-187.
3. Klausegger A, Laimer M, Bauer JW. Darier disease. [In German.] Hautarzt. 2013;64:22-25.
4. Ringpfeil F. Dermatologic disorders. In: NORD Guide to Rare Disorders. Philadelphia, PA: Lippincott Williams & Wilkins; 2003:101.
5. Disorders of keratinization. In: Ostler HB, Maibach HI, Hoke AW, Schwab IR, eds. Diseases of the Eye and Skin: A Color Atlas. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:23-34.
6. Baran R, de Berker D, Holzberg M, Thomas L, eds. Baran & Dawber’s Diseases of the Nails and their Management. 4th ed. West Sussex, UK: John Wiley & Sons, Ltd; 2012:295-296.
7. Thiagarajan MK, Narasimhan M, Sankarasubramanian A. Darier disease with oral and esophageal involvement: a case report. Indian J Dent Res. 2011;22:843-846.
8. Medansky RS, Woloshin AA. Darier’s disease: an evaluation of its neuropsychiatric component. Arch Dermatol. 1961;84:482-484.
9. Jacobsen NJ, Lyons I, Hoogendoorn B, et al. ATP2A2 mutations in Darier’s disease and their relationship to neuropsychiatric phenotypes. Hum Mol Genet. 1999;8:1631-1636.
10. Kamijo M, Nishiyama C, Takagi A, et al. Cyclooxygenase-2 inhibition restores ultraviolet B-induced downregulation of ATP2A2/SERCA2 in keratinocytes: possible therapeutic approach of cyclooxygenase-2 inhibition for treatment of Darier disease. Br J Dermatol. 2012;166: 1017-1022.
11. Brecher AR, Orlow SJ. Oral retinoid therapy for dermatologic conditions in children and adolescents. J Am Acad Dermatol. 2003;49:171-182.
12. Beier C, Kaufmann R. Efficacy of erbium:YAG laser ablation in Darier disease and Hailey-Hailey disease. Arch Dermatol. 1999;35:423-427.
A 45-year-old man presented to the dermatology office complaining of a pruritic rash on his neck, chest, abdomen, and upper back. The rash had been present since the patient was 20, intermittently flaring and causing severe pruritus. For the past two weeks, it had become increasingly bothersome.
The patient described the rash as “greasy” brown plaques diffusely scattered on his body. The rash on his neck was the most bothersome, and the patient felt an uncontrollable need to scratch that area.
Since it first developed 25 years ago, he had used OTC hydrocortisone cream as needed to treat the rash. Although effective for past flares, the cream provided only minimal relief during the current episode.
The patient’s medical history included brittle nails with a worsening of nail quality in recent years. The family history revealed that the patient’s father and sister were affected by the same type of rash, which developed in adolescence for each of them, as well as brittle nails.
On physical examination, the skin was warm and moist to the touch. Flat, slightly elevated, greasy brown papules were scattered on the chest, abdomen, and upper back, with mild surrounding erythema (see Figure 1). Excoriated lesions were noted on the anterior surface of the neck, with pinpoint bleeding resulting from constant irritation. The patient’s fingernails were deformed, with longitudinal ridges and v-shaped notching of the free margin. The remainder of the physical exam was unremarkable, and review of systems was negative.
This patient’s symptoms could result from a variety of causes. Seborrheic dermatitis is a common skin condition that presents with brown plaques similar to those on the patient’s trunk. Another possible diagnosis is Grover’s disease, a rare disorder also known as transient acantholytic dermatosis, in which keratotic plaques appear on the torso and are thought to occur from trauma to sun-damaged skin. An additional consideration is Hailey-Hailey disease, a rare genetic disorder also known as benign familial pemphigus, which is characterized by red-brown plaques located predominantly on flexure surfaces.1 Skin biopsy should be performed for a definitive diagnosis.
Given the family history of a similar rash occurring in first-degree relatives and the distinct physical exam findings, the most likely diagnosis for this patient is keratosis follicularis, also known as Darier disease (DD) or Darier-White disease.
DISCUSSION
Named after Ferdinand-Jean Darier, who discovered this rare genodermatosis, DD is a rare genetic skin disorder caused by mutations of the ATP2A2 gene, located on the long arm of chromosome 12 at position 24,11.1,2 The mutation disrupts the encoding of the enzyme sarco/endoplasmic reticulum calcium-ATPase 2 (SERCA2). This enzyme is important in the transport of calcium ions across the cell membrane, and insufficient amounts lead to a defect in intracellular calcium signaling.2,3
This genetic mutation is inherited as an autosomal dominant trait with complete penetrance. DD affects men and women equally, with progressive skin signs of interfamilial and intrafamilial variability.4 Skin manifestations occur from late childhood to early adulthood and are typical during adolescence.4 Acute flare-ups can be triggered by heat, perspiration, sunlight, ultraviolet B exposure, stress, or certain medications (in particular, lithium).2 DD is not contagious.2
CLINICAL PRESENTATION
The characteristics of DD include yellow or brown, rough, firm papules that are frequently crusted. The papules often appear in seborrheic areas of the body, such as the chest, back, ears, nasolabial fold, forehead, scalp, and groin.4 The severity of expression varies from mild, with few lesions, to severe, in which the entire body is covered with disfiguring, macerated plaques emitting a strong odor. On biopsy, the histopathologic findings are typical of dyskeratosis and acantholysis.4
Fingernails (and occasionally toenails) display broad, white or red, somewhat translucent, longitudinal bands accompanied by v-shaped notching1,4,5 (see Figure 2). Such nail changes are diagnostic and occur in 92% to 95% of patients with DD.6 They may, in fact, occur in the absence of cutaneous disease. All nails may be affected, but usually only two to three are involved.6
Although uncommon in DD, white, umbilicated, or cobblestone plaques may be found on intraoral mucous membranes (ie, tongue, buccal mucosa, palate, epiglottis, pharyngeal wall, and esophagus); due to confluence, papules may mimic leukoplakia.7 Lesions may also appear on the vulva or rectum.1,5 In severe cases, the salivary glands can become blocked, and the gums can hypertrophy.5
Since epidermal and brain tissue both derive from ectoderm, pathologic processes that affect one organ system may also affect the other.8 Indeed, among patients with DD, neuropsychiatric problems—including epilepsy, learning difficulties, and schizoaffective disorder—are commonly reported.1 To confirm an association between DD and ATP2A2 mutations, Jacobsen and colleagues performed an analysis of 19 unrelated DD patients with neuropsychiatric phenotypes. They discovered evidence to support the gene’s pleiotropic effects in the brain and hypothesized that mutations in the enzyme SERCA2 correlate with these phenotypes, most specifically for mood disorders.9
TREATMENT AND MANAGEMENT
Although no cure is currently available for DD, both short- and long-term treatment options are available; the choice should be based on the severity of an individual patient’s signs and symptoms. For mild cases, topical therapy, such as general emollients, corticosteroid ointments, and high sun protection factor sunscreen, is sufficient.1
For moderate cases, topical retinoids, including tretinoin cream, adapalene gel or cream, and tazarotene gel, may be necessary.4 Keratolytics, including salicylic acid in propylene glycol gel, may be used to regulate hyperkeratosis.4 Celecoxib, a COX-2 inhibitor, is another option that may restore the down regulation of SERCA2. This can prevent progression of the disease.10
Long-term management includes use of oral retinoid therapy (eg, acitretin), which might reduce the frequency of inflammatory flares.1 Systemic adverse effects from long-term use of oral retinoids are cause for concern, however. Close monitoring along with patient education can limit the occurrence of complications.11
If DD is uncontrolled with medication, dermabrasion and erbium:YAG laser ablation have been used to successfully treat chronic cases.12 Although these treatment options may remove existing lesions, it is important to inform patients that the disease has not been cured, that remission is difficult to attain, and that lesions may recur.
Because viral, bacterial, and fungal superinfections are common and may exacerbate the disease, be sure to check for signs of infection while examining the patient.4 Patients should be advised to avoid hot environments, and if that is not possible, to dress in cool cotton clothing to allow for proper ventilation and avoid the build-up of perspiration. Excessive perspiration along with poor hygiene can contribute to the formation of infections as well as trigger a flare-up. If an infection develops, patients should consult a health care provider.
Keeping the skin well moisturized can alleviate the constant pruritus that many patients experience. Daily sunscreen use is essential to avoid skin irritation caused by the sun, which can trigger an acute flare-up. Patients should be advised to avoid the long-term use of corticosteroid ointment. They should also contact their health care provider before using OTC treatments such as Burow’s solution.
CONCLUSION
A thorough history and physical exam are crucial in the diagnosis of DD. In this particular case, inquiry into family history was the key to proper diagnosis. That information, paired with a thorough physical exam, led to the correct diagnosis of this rare genetic skin disorder. A skin biopsy provided definitive confirmation.
This patient had a mild-to-moderate manifestation of DD. He was prescribed retinoid therapy, and routine follow-up visits were recommended to monitor the efficacy of medical therapy and to screen for secondary infections or neuropsychiatric disorders.
This case illustrates the importance of taking a full history and performing an in-depth physical exam when a patient presents with an unfamiliar complaint. Being thorough reduces the risk of missing a crucial element that can guide the diagnostic process.
REFERENCES
1. Creamer D, Barker J, Kerdel FA. Papular and papulosquamous dermatoses. In: Acute Adult Dermatology: Diagnosis and Management (A Colour Handbook). London, UK: Manson Publishing Ltd; 2011:48.
2. Kelly EB. Darier disease (DAR). In: Encyclopedia of Human Genetics and Disease. Santa Barbara, CA: ABC-CLIO; 2013:186-187.
3. Klausegger A, Laimer M, Bauer JW. Darier disease. [In German.] Hautarzt. 2013;64:22-25.
4. Ringpfeil F. Dermatologic disorders. In: NORD Guide to Rare Disorders. Philadelphia, PA: Lippincott Williams & Wilkins; 2003:101.
5. Disorders of keratinization. In: Ostler HB, Maibach HI, Hoke AW, Schwab IR, eds. Diseases of the Eye and Skin: A Color Atlas. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:23-34.
6. Baran R, de Berker D, Holzberg M, Thomas L, eds. Baran & Dawber’s Diseases of the Nails and their Management. 4th ed. West Sussex, UK: John Wiley & Sons, Ltd; 2012:295-296.
7. Thiagarajan MK, Narasimhan M, Sankarasubramanian A. Darier disease with oral and esophageal involvement: a case report. Indian J Dent Res. 2011;22:843-846.
8. Medansky RS, Woloshin AA. Darier’s disease: an evaluation of its neuropsychiatric component. Arch Dermatol. 1961;84:482-484.
9. Jacobsen NJ, Lyons I, Hoogendoorn B, et al. ATP2A2 mutations in Darier’s disease and their relationship to neuropsychiatric phenotypes. Hum Mol Genet. 1999;8:1631-1636.
10. Kamijo M, Nishiyama C, Takagi A, et al. Cyclooxygenase-2 inhibition restores ultraviolet B-induced downregulation of ATP2A2/SERCA2 in keratinocytes: possible therapeutic approach of cyclooxygenase-2 inhibition for treatment of Darier disease. Br J Dermatol. 2012;166: 1017-1022.
11. Brecher AR, Orlow SJ. Oral retinoid therapy for dermatologic conditions in children and adolescents. J Am Acad Dermatol. 2003;49:171-182.
12. Beier C, Kaufmann R. Efficacy of erbium:YAG laser ablation in Darier disease and Hailey-Hailey disease. Arch Dermatol. 1999;35:423-427.
Former Farmer Is Short of Breath
ANSWER
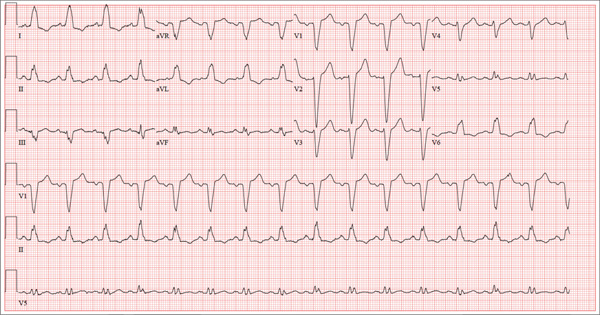
The correct interpretation of this ECG includes normal sinus rhythm with left atrial enlargement and a left bundle branch block (LBBB). Normal sinus rhythm is evidenced by a P wave associated with each QRS complex with a consistent PR interval.
Left atrial enlargement is evidenced by a P-wave duration ≥ 120 ms in lead II, a notched P wave in the limb leads with a peak duration ≥ 4 ms, and a terminal P-wave negativity in lead V1 with a duration ≥ 4 ms and a depth ≥ 1 mm.
An LBBB is illustrated by the QRS duration ≥ 120 ms, a dominant S wave in lead V1, broad monophasic R waves in the lateral leads (including I, aVL, V5, and V6), and R-wave peak times of > 60 ms in leads V5 and V6.
Further work-up revealed elevated left end-diastolic filling pressures, volume overload, and pulmonary edema consistent with diastolic heart failure. Given the unclear etiology of the LBBB, cardiac catheterization was performed. It revealed no significant coronary artery disease.
ANSWER
The correct interpretation of this ECG includes normal sinus rhythm with left atrial enlargement and a left bundle branch block (LBBB). Normal sinus rhythm is evidenced by a P wave associated with each QRS complex with a consistent PR interval.
Left atrial enlargement is evidenced by a P-wave duration ≥ 120 ms in lead II, a notched P wave in the limb leads with a peak duration ≥ 4 ms, and a terminal P-wave negativity in lead V1 with a duration ≥ 4 ms and a depth ≥ 1 mm.
An LBBB is illustrated by the QRS duration ≥ 120 ms, a dominant S wave in lead V1, broad monophasic R waves in the lateral leads (including I, aVL, V5, and V6), and R-wave peak times of > 60 ms in leads V5 and V6.
Further work-up revealed elevated left end-diastolic filling pressures, volume overload, and pulmonary edema consistent with diastolic heart failure. Given the unclear etiology of the LBBB, cardiac catheterization was performed. It revealed no significant coronary artery disease.
ANSWER
The correct interpretation of this ECG includes normal sinus rhythm with left atrial enlargement and a left bundle branch block (LBBB). Normal sinus rhythm is evidenced by a P wave associated with each QRS complex with a consistent PR interval.
Left atrial enlargement is evidenced by a P-wave duration ≥ 120 ms in lead II, a notched P wave in the limb leads with a peak duration ≥ 4 ms, and a terminal P-wave negativity in lead V1 with a duration ≥ 4 ms and a depth ≥ 1 mm.
An LBBB is illustrated by the QRS duration ≥ 120 ms, a dominant S wave in lead V1, broad monophasic R waves in the lateral leads (including I, aVL, V5, and V6), and R-wave peak times of > 60 ms in leads V5 and V6.
Further work-up revealed elevated left end-diastolic filling pressures, volume overload, and pulmonary edema consistent with diastolic heart failure. Given the unclear etiology of the LBBB, cardiac catheterization was performed. It revealed no significant coronary artery disease.
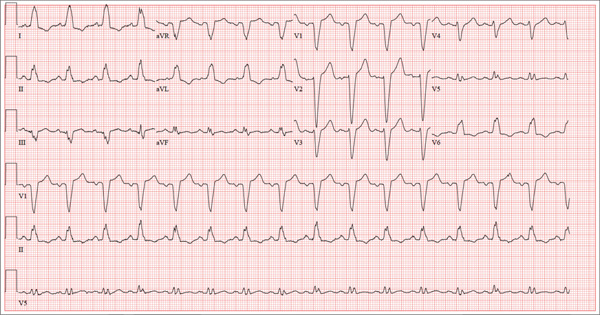
A 67-year-old man has a history of chronic dyspnea. He is a retired farmer who says he “never had time” to seek medical help for anything other than cuts or broken bones. In the past two months, he’s noticed that his dyspnea has progressively worsened. When questioned, he admits that his legs began swelling around that time as well. Two days ago, he awoke from sleep unable to catch his breath. This morning, while walking to his mailbox, he became profoundly short of breath. He sat down by the side of the road and called 911. When the ambulance arrived, he felt much better but agreed to be taken to the emergency department, since his wife is away and he’s home alone. When questioned by the paramedics, he denied having chest pain, palpitations, productive or nonproductive cough, polyuria, polydipsia, nausea, or vomiting. Medical history is positive for hypertension, gastroesophageal reflux disease (GERD), and hypertension. He has had several fractures in his right ankle and left femur, which are well healed. Surgical history is remarkable for a cholecystectomy and multiple laceration repairs on his arms and hands (also well healed). His current medications include one aspirin per day and “a handful” of calcium carbonate tablets. Although he was prescribed “several heart pills” for hypertension, he hasn’t taken them or refilled the prescriptions for at least five years. He is allergic to penicillin and sulfa. He denies recreational or homeopathic drug use. He has never smoked, and he drinks one or two shots of bourbon on weekends. Family history includes a father who died in a farming accident and a mother who died of cervical cancer at age 85. He has seven siblings, all of whom are alive and well. The review of systems is remarkable only for GERD. Physical exam reveals a well-developed, obese male with a height of 6 ft 4 in and a weight of 278 lb. Vital signs include a blood pressure of 184/98 mm Hg; pulse, 90 beats/min; and respiratory rate, 20 breaths/min-1. He is afebrile. The HEENT exam is remarkable for atrophic glossitis. The neck shows no evidence of thyromegaly, and there are no carotid bruits or jugular venous distention. The chest is remarkable for diffuse wheezing and crackles in all lung bases. The cardiac exam reveals a regular rate of 90 beats/min, with no evidence of murmurs, rubs, or gallops. The abdomen is obese. There is no evidence of ascites or masses. Evidence of 2+ pitting edema to the midcalf is present bilaterally. The neurologic exam is grossly intact, and the psychiatric exam reveals the patient to be alert and oriented, with a bright affect. The working diagnosis in the emergency department is acute or chronic heart failure. A chest x-ray reveals moderate-to-severe pulmonary edema, cardiomegaly, and small bilateral effusions. Pertinent laboratory data include a serum glucose of 200 mg/dL and a B-type natriuretic peptide level of 590 pg/mL. All other lab values are within normal limits. An ECG reveals the following: a ventricular rate of 93 beats/min; PR interval, 168 ms; QRS duration, 156 ms; QT/QTc interval, 430/534 ms; P axis, 52°; R axis, 9°; and T axis, 171°. What is your interpretation of this ECG?
Hair Loss at a Very Young Age
ANSWER
The correct answer is trichotillomania (choice “c”). See Discussion for more information.
Alopecia mucinosa (choice “a”) is a rare cause of focal hair loss that can occur in children. However, it usually presents with papules or plaques, unlike the smooth skin surface seen here.
Alopecia areata (choice “b”), common in children, typically entails complete hair loss in a given area—or, as hair regrows, with hairs of equal length. The uneven hairs seen in trichotillomania help a great deal in distinguishing it from alopecia areata.
Traction alopecia (choice “d”) is focal hair loss caused by chronic tension related to hairstyling. Most common in African-American women, and typically affecting the frontal periphery of the scalp, it is an unlikely explanation for hair loss in a 10-year-old boy.
DISCUSSION
Trichotillomania (TT) means, literally, “hair-pulling madness.” But in reality, there’s little actual plucking of hairs in this common condition. Instead, patients habitually manipulate hair by twirling and tugging, which weakens the shafts and follicles and renders them more susceptible to everyday wear and tear. In some cases, individual hairs speed through their growth phases and others break off in mid-shaft. All of this contributes to the classic “uneven” look of TT.
Patients with TT tend to be in the 4-to-17 age range, and most have issues with unresolved anxiety that manifest in part with manipulation of the hair. Officially considered an impulse control disorder, TT in most cases belongs to the psychiatrist’s domain.
In this case, it was enormously helpful to have corroboration from the patient and his mother regarding his role in creating and perpetuating the problem. Had that not been the case—or in the event of other doubts as to the correct diagnosis—biopsy could have been performed to rule out most of the other items in the differential, particularly alopecia areata.
Interestingly enough, studies have shown that the more sharply defined the area of hair loss, the more likely the patient is to admit his/her role in its creation. However, as is often the case with scientific research, contradictory findings have also been made.
TREATMENT
Treatment of TT is problematic, since no medications have proven to be completely helpful. Psychiatrists use a combination of medication, cognitive behavioral therapy, and other behavior modifications that are designed to overcome the habitual component of the problem. Most cases of TT resolve on their own, but in severe cases that persist for years, permanent hair loss can result.
In this case, there was enough insight and motivation on the part of the patient and his family to stop the offending behavior and allow the hair to regrow.
ANSWER
The correct answer is trichotillomania (choice “c”). See Discussion for more information.
Alopecia mucinosa (choice “a”) is a rare cause of focal hair loss that can occur in children. However, it usually presents with papules or plaques, unlike the smooth skin surface seen here.
Alopecia areata (choice “b”), common in children, typically entails complete hair loss in a given area—or, as hair regrows, with hairs of equal length. The uneven hairs seen in trichotillomania help a great deal in distinguishing it from alopecia areata.
Traction alopecia (choice “d”) is focal hair loss caused by chronic tension related to hairstyling. Most common in African-American women, and typically affecting the frontal periphery of the scalp, it is an unlikely explanation for hair loss in a 10-year-old boy.
DISCUSSION
Trichotillomania (TT) means, literally, “hair-pulling madness.” But in reality, there’s little actual plucking of hairs in this common condition. Instead, patients habitually manipulate hair by twirling and tugging, which weakens the shafts and follicles and renders them more susceptible to everyday wear and tear. In some cases, individual hairs speed through their growth phases and others break off in mid-shaft. All of this contributes to the classic “uneven” look of TT.
Patients with TT tend to be in the 4-to-17 age range, and most have issues with unresolved anxiety that manifest in part with manipulation of the hair. Officially considered an impulse control disorder, TT in most cases belongs to the psychiatrist’s domain.
In this case, it was enormously helpful to have corroboration from the patient and his mother regarding his role in creating and perpetuating the problem. Had that not been the case—or in the event of other doubts as to the correct diagnosis—biopsy could have been performed to rule out most of the other items in the differential, particularly alopecia areata.
Interestingly enough, studies have shown that the more sharply defined the area of hair loss, the more likely the patient is to admit his/her role in its creation. However, as is often the case with scientific research, contradictory findings have also been made.
TREATMENT
Treatment of TT is problematic, since no medications have proven to be completely helpful. Psychiatrists use a combination of medication, cognitive behavioral therapy, and other behavior modifications that are designed to overcome the habitual component of the problem. Most cases of TT resolve on their own, but in severe cases that persist for years, permanent hair loss can result.
In this case, there was enough insight and motivation on the part of the patient and his family to stop the offending behavior and allow the hair to regrow.
ANSWER
The correct answer is trichotillomania (choice “c”). See Discussion for more information.
Alopecia mucinosa (choice “a”) is a rare cause of focal hair loss that can occur in children. However, it usually presents with papules or plaques, unlike the smooth skin surface seen here.
Alopecia areata (choice “b”), common in children, typically entails complete hair loss in a given area—or, as hair regrows, with hairs of equal length. The uneven hairs seen in trichotillomania help a great deal in distinguishing it from alopecia areata.
Traction alopecia (choice “d”) is focal hair loss caused by chronic tension related to hairstyling. Most common in African-American women, and typically affecting the frontal periphery of the scalp, it is an unlikely explanation for hair loss in a 10-year-old boy.
DISCUSSION
Trichotillomania (TT) means, literally, “hair-pulling madness.” But in reality, there’s little actual plucking of hairs in this common condition. Instead, patients habitually manipulate hair by twirling and tugging, which weakens the shafts and follicles and renders them more susceptible to everyday wear and tear. In some cases, individual hairs speed through their growth phases and others break off in mid-shaft. All of this contributes to the classic “uneven” look of TT.
Patients with TT tend to be in the 4-to-17 age range, and most have issues with unresolved anxiety that manifest in part with manipulation of the hair. Officially considered an impulse control disorder, TT in most cases belongs to the psychiatrist’s domain.
In this case, it was enormously helpful to have corroboration from the patient and his mother regarding his role in creating and perpetuating the problem. Had that not been the case—or in the event of other doubts as to the correct diagnosis—biopsy could have been performed to rule out most of the other items in the differential, particularly alopecia areata.
Interestingly enough, studies have shown that the more sharply defined the area of hair loss, the more likely the patient is to admit his/her role in its creation. However, as is often the case with scientific research, contradictory findings have also been made.
TREATMENT
Treatment of TT is problematic, since no medications have proven to be completely helpful. Psychiatrists use a combination of medication, cognitive behavioral therapy, and other behavior modifications that are designed to overcome the habitual component of the problem. Most cases of TT resolve on their own, but in severe cases that persist for years, permanent hair loss can result.
In this case, there was enough insight and motivation on the part of the patient and his family to stop the offending behavior and allow the hair to regrow.
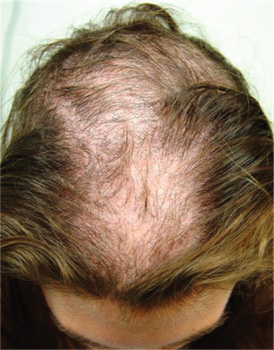
A 10-year-old boy is referred to dermatology with a four-month history of hair loss. The affected area of the vertex is now large enough to alarm his mother, who accompanies him to his appointment. The child’s primary care provider had diagnosed alopecia areata and prescribed triamcinolone 0.1% solution. But after a month of twice-daily application, even more hair has been lost. There is no family history of alopecia areata or other autoimmune disease. The child is otherwise healthy, although he is being treated by a psychiatrist for attention deficit disorder and chronic anxiety (with two medications whose names are unknown). The patient denies any symptoms associated with his hair loss, and his mother denies any skin changes in the affected area. However, she emphasizes that she has seen her son manipulating the area with his hand on several occasions, despite her attempts to make him stop. When pressed, the patient finally admits that throughout the day he twirls and tugs on his hair—although he denies actually pulling out any. On inspection, an 11 x 8–cm oval area of distinct and sharply demarcated hair loss is noted in the vertex scalp. Hairs of different lengths are noted in the central portion of the site; some have obviously been broken off, while others are longer, with thin, tapering ends. There is no disruption (eg, scaling, redness, edema) in the surface of the scalp, but the whole area is darker (brown) than the surrounding, uninvolved scalp. No other areas of hair loss are noted in the scalp or face. No nodes are palpable in the neck.

Lower-dose quizartinib diminishes QT events
NEW ORLEANS – Lower doses of quizartinib reduced worrisome QT-interval prolongation events without a loss of efficacy in patients with FLT3-ITD–positive relapsed or refractory acute myeloid leukemia, a phase II study shows.
In a 76-patient study, grade 2 QT-interval prolongation (QTcF) of more than 480-500 msec occurred in two patients (5%) on oral quizartinib 30 mg/day and in five patients (14%) on 60 mg/day, with no differences between groups in QTcF events of more than 500 msec (5% vs. 3%).
In addition, an increase in QTcF from baseline of more than 60 msec was seen in 19% of patients on the 60-mg dose and in 3% of those on the 30-mg dose, Dr. Jorge Cortes reported at the annual meeting of the American Society of Hematology.
About a third of patients with acute myeloid leukemia (AML) will have FLT3 internal tandem duplications (FLT3-ITD), which are associated with early relapse and poor survival in AML. Quizartinib has shown the highest single-agent activity among FMS-like tyrosine kinase 3 (FLT3)-targeted agents in this population, according to Dr. Cortes.
At last year’s ASH meeting, investigators presented results from a phase II study in which the investigational agent elicited responses in both FLT3-positive and -negative relapsed/refractory AML. The unprecedented results at doses of 90, 135, and 200 mg were partially eclipsed, however, by respective 46%, 39%, and 92% increases in QTcF from baseline of more than 60 msec, noted Dr. Cortes, chair of the AML section, department of leukemia, University of Texas M.D. Anderson Cancer Center, Houston.
The current study randomized 76 patients to quizartinib 30 mg or 60 mg continuous daily dosing for primary AML or AML secondary to myelodysplastic syndrome that relapsed or was refractory to first-line salvage therapy or prior hematopoietic stem cell transplantation. The coprimary endpoints were rate of grade 2 QTc prolongation and the composite complete remission rate, which included complete remission (CR), CR with incomplete platelet recovery, and CR with incomplete hematologic recovery.
In all, 92% of patients had FLT3 internal tandem duplications, and 58 of 60 evaluable patients had intermediate or poor cytogenetic risk. Their mean age was 55 years. Two patients were randomized but not treated.
Treatment with the 30-mg and 60-mg doses resulted in a composite CR rate of 47%, Dr. Cortes said. The median duration of response was 4.1 weeks in the 30-mg group and 20 weeks in the 60-mg group.
Two patients (5%) on the 30-mg dose and 1 patient (3%) on the 60-mg dose achieved CR; 1 patient (3%) in the 60-mg group had a CR with incomplete platelet recovery; and 16 patients (42%) in each arm had a CR with incomplete hematologic recovery.
Partial responses were also seen in 5 patients (13%) in the 30-mg group and 9 (24%) in the 60-mg group.
These results compare favorably with composite CR rates of 47%, 45%, and 42% with the 90-, 135-, and 200-mg doses used in the earlier study, Dr. Cortes observed.
Median overall survival in the current study was 20.7 weeks in the lower-dose group and 25.4 weeks with the 60-mg dose.
Importantly, 34% of patients were successfully bridged to transplant, extending median survival to 31 weeks for those on 30 mg of quizartinib and to 28.1 weeks for those given 60 mg.
"This study demonstrates there is certainly sustained efficacy with these lower doses of quizartinib and a decreased QT signal at doses of 30 and 60 mg compared with the higher doses we’ve tested in the past," Dr. Cortes concluded.
Grade 3/4 adverse events were mainly anemia (39%) in the 30-mg group and febrile neutropenia (36%) in the 60-mg group. Three patients required dose reductions due to QTc prolongation.
A global phase III randomized study of quizartinib in FLT3-ITD–positive patients in first relapse is planned to start in early 2014, he said.
Development of quizartinib has been somewhat rocky, with Astellas Pharma announcing in March 2013 it was ending its collaboration with Ambit Biosciences to develop FLT3 inhibitors including quizartinib.
Dr. Cortes reported research funding from Astellas Pharma, Arog, Novartis, and Ambit Biosciences, which is developing quizartinib, and consulting for Astellas, Arog, and Ambit.
NEW ORLEANS – Lower doses of quizartinib reduced worrisome QT-interval prolongation events without a loss of efficacy in patients with FLT3-ITD–positive relapsed or refractory acute myeloid leukemia, a phase II study shows.
In a 76-patient study, grade 2 QT-interval prolongation (QTcF) of more than 480-500 msec occurred in two patients (5%) on oral quizartinib 30 mg/day and in five patients (14%) on 60 mg/day, with no differences between groups in QTcF events of more than 500 msec (5% vs. 3%).
In addition, an increase in QTcF from baseline of more than 60 msec was seen in 19% of patients on the 60-mg dose and in 3% of those on the 30-mg dose, Dr. Jorge Cortes reported at the annual meeting of the American Society of Hematology.
About a third of patients with acute myeloid leukemia (AML) will have FLT3 internal tandem duplications (FLT3-ITD), which are associated with early relapse and poor survival in AML. Quizartinib has shown the highest single-agent activity among FMS-like tyrosine kinase 3 (FLT3)-targeted agents in this population, according to Dr. Cortes.
At last year’s ASH meeting, investigators presented results from a phase II study in which the investigational agent elicited responses in both FLT3-positive and -negative relapsed/refractory AML. The unprecedented results at doses of 90, 135, and 200 mg were partially eclipsed, however, by respective 46%, 39%, and 92% increases in QTcF from baseline of more than 60 msec, noted Dr. Cortes, chair of the AML section, department of leukemia, University of Texas M.D. Anderson Cancer Center, Houston.
The current study randomized 76 patients to quizartinib 30 mg or 60 mg continuous daily dosing for primary AML or AML secondary to myelodysplastic syndrome that relapsed or was refractory to first-line salvage therapy or prior hematopoietic stem cell transplantation. The coprimary endpoints were rate of grade 2 QTc prolongation and the composite complete remission rate, which included complete remission (CR), CR with incomplete platelet recovery, and CR with incomplete hematologic recovery.
In all, 92% of patients had FLT3 internal tandem duplications, and 58 of 60 evaluable patients had intermediate or poor cytogenetic risk. Their mean age was 55 years. Two patients were randomized but not treated.
Treatment with the 30-mg and 60-mg doses resulted in a composite CR rate of 47%, Dr. Cortes said. The median duration of response was 4.1 weeks in the 30-mg group and 20 weeks in the 60-mg group.
Two patients (5%) on the 30-mg dose and 1 patient (3%) on the 60-mg dose achieved CR; 1 patient (3%) in the 60-mg group had a CR with incomplete platelet recovery; and 16 patients (42%) in each arm had a CR with incomplete hematologic recovery.
Partial responses were also seen in 5 patients (13%) in the 30-mg group and 9 (24%) in the 60-mg group.
These results compare favorably with composite CR rates of 47%, 45%, and 42% with the 90-, 135-, and 200-mg doses used in the earlier study, Dr. Cortes observed.
Median overall survival in the current study was 20.7 weeks in the lower-dose group and 25.4 weeks with the 60-mg dose.
Importantly, 34% of patients were successfully bridged to transplant, extending median survival to 31 weeks for those on 30 mg of quizartinib and to 28.1 weeks for those given 60 mg.
"This study demonstrates there is certainly sustained efficacy with these lower doses of quizartinib and a decreased QT signal at doses of 30 and 60 mg compared with the higher doses we’ve tested in the past," Dr. Cortes concluded.
Grade 3/4 adverse events were mainly anemia (39%) in the 30-mg group and febrile neutropenia (36%) in the 60-mg group. Three patients required dose reductions due to QTc prolongation.
A global phase III randomized study of quizartinib in FLT3-ITD–positive patients in first relapse is planned to start in early 2014, he said.
Development of quizartinib has been somewhat rocky, with Astellas Pharma announcing in March 2013 it was ending its collaboration with Ambit Biosciences to develop FLT3 inhibitors including quizartinib.
Dr. Cortes reported research funding from Astellas Pharma, Arog, Novartis, and Ambit Biosciences, which is developing quizartinib, and consulting for Astellas, Arog, and Ambit.
NEW ORLEANS – Lower doses of quizartinib reduced worrisome QT-interval prolongation events without a loss of efficacy in patients with FLT3-ITD–positive relapsed or refractory acute myeloid leukemia, a phase II study shows.
In a 76-patient study, grade 2 QT-interval prolongation (QTcF) of more than 480-500 msec occurred in two patients (5%) on oral quizartinib 30 mg/day and in five patients (14%) on 60 mg/day, with no differences between groups in QTcF events of more than 500 msec (5% vs. 3%).
In addition, an increase in QTcF from baseline of more than 60 msec was seen in 19% of patients on the 60-mg dose and in 3% of those on the 30-mg dose, Dr. Jorge Cortes reported at the annual meeting of the American Society of Hematology.
About a third of patients with acute myeloid leukemia (AML) will have FLT3 internal tandem duplications (FLT3-ITD), which are associated with early relapse and poor survival in AML. Quizartinib has shown the highest single-agent activity among FMS-like tyrosine kinase 3 (FLT3)-targeted agents in this population, according to Dr. Cortes.
At last year’s ASH meeting, investigators presented results from a phase II study in which the investigational agent elicited responses in both FLT3-positive and -negative relapsed/refractory AML. The unprecedented results at doses of 90, 135, and 200 mg were partially eclipsed, however, by respective 46%, 39%, and 92% increases in QTcF from baseline of more than 60 msec, noted Dr. Cortes, chair of the AML section, department of leukemia, University of Texas M.D. Anderson Cancer Center, Houston.
The current study randomized 76 patients to quizartinib 30 mg or 60 mg continuous daily dosing for primary AML or AML secondary to myelodysplastic syndrome that relapsed or was refractory to first-line salvage therapy or prior hematopoietic stem cell transplantation. The coprimary endpoints were rate of grade 2 QTc prolongation and the composite complete remission rate, which included complete remission (CR), CR with incomplete platelet recovery, and CR with incomplete hematologic recovery.
In all, 92% of patients had FLT3 internal tandem duplications, and 58 of 60 evaluable patients had intermediate or poor cytogenetic risk. Their mean age was 55 years. Two patients were randomized but not treated.
Treatment with the 30-mg and 60-mg doses resulted in a composite CR rate of 47%, Dr. Cortes said. The median duration of response was 4.1 weeks in the 30-mg group and 20 weeks in the 60-mg group.
Two patients (5%) on the 30-mg dose and 1 patient (3%) on the 60-mg dose achieved CR; 1 patient (3%) in the 60-mg group had a CR with incomplete platelet recovery; and 16 patients (42%) in each arm had a CR with incomplete hematologic recovery.
Partial responses were also seen in 5 patients (13%) in the 30-mg group and 9 (24%) in the 60-mg group.
These results compare favorably with composite CR rates of 47%, 45%, and 42% with the 90-, 135-, and 200-mg doses used in the earlier study, Dr. Cortes observed.
Median overall survival in the current study was 20.7 weeks in the lower-dose group and 25.4 weeks with the 60-mg dose.
Importantly, 34% of patients were successfully bridged to transplant, extending median survival to 31 weeks for those on 30 mg of quizartinib and to 28.1 weeks for those given 60 mg.
"This study demonstrates there is certainly sustained efficacy with these lower doses of quizartinib and a decreased QT signal at doses of 30 and 60 mg compared with the higher doses we’ve tested in the past," Dr. Cortes concluded.
Grade 3/4 adverse events were mainly anemia (39%) in the 30-mg group and febrile neutropenia (36%) in the 60-mg group. Three patients required dose reductions due to QTc prolongation.
A global phase III randomized study of quizartinib in FLT3-ITD–positive patients in first relapse is planned to start in early 2014, he said.
Development of quizartinib has been somewhat rocky, with Astellas Pharma announcing in March 2013 it was ending its collaboration with Ambit Biosciences to develop FLT3 inhibitors including quizartinib.
Dr. Cortes reported research funding from Astellas Pharma, Arog, Novartis, and Ambit Biosciences, which is developing quizartinib, and consulting for Astellas, Arog, and Ambit.
AT ASH 2013
Major finding: An increase in QTcF from baseline of more than 60 msec was seen in 19% of patients on quizartinib 60 mg and in 3% of those on 30 mg.
Data source: A prospective phase II study of 76 patients with relapsed/refractory AML.
Disclosures: Dr. Cortes reported research funding from Astellas Pharma, Arog, Novartis, and Ambit Biosciences, which is developing quizartinib, and consulting for Astellas, Arog, and Ambit.
Unexpectedly good results, and no chemotherapy required
Cabozantinib in metastatic prostate cancer1,2
Researchers tested cabozantinib, a tyrosine-kinase inhibitor (TKI) against MET and vascular endothelial growth factor receptor 2 (VEGF), in a large phase 2 randomized discontinuation trial in 9 tumor types. A subset of 171 patients with castrateresistant prostate cancer (CRPC) was reported in this study. Patients were treated on open label for 12 weeks, and then if stable, were randomized to receive the active drug or placebo. The trial was suspended early by the study oversight committee because: a) Cabozantinib was too toxic for the study to continue. b) The prostate-specific antigen (PSA) level fell in most of the treated patients. c) In the initial 121 patients, there was an unexpected improvement in bone scans and decrease in pain in the lead-in stage of the study. d) Unexpected rapid soft tissue progression. Bone scans improved in 78% of patients, and in 12% there was complete remission. After further analysis, the following were true except for: a) Cabozantinib interfered with technetium-99, and thus, the responses were not real, but rather an artifact. b) The PSA did not correlate with improvement in bone pain. c) Markers of bone formation and resorption showed improvement, and there was no correlation with prior bisphosphonate therapy. d) Bone scan improvement correlated with improvement in soft tissue disease.
Key points
The results in patients with prostate cancer were so striking – 72% of patients had regression in soft tissue lesions, and 68% of evaluable patients had improvement on bone scan, including complete resolution in 12% – that the subset analysis was published as a rapid communication.1,2 Because of very high response rates (5% at 12 weeks) and symptomatic improvement in the initial 122 patients who were enrolled, random assignment was discontinued. Bone markers improved in concert with the radiologic and clinical improvement. Answers c, a
Cabozantinib in metastatic prostate cancer1,2
Researchers tested cabozantinib, a tyrosine-kinase inhibitor (TKI) against MET and vascular endothelial growth factor receptor 2 (VEGF), in a large phase 2 randomized discontinuation trial in 9 tumor types. A subset of 171 patients with castrateresistant prostate cancer (CRPC) was reported in this study. Patients were treated on open label for 12 weeks, and then if stable, were randomized to receive the active drug or placebo. The trial was suspended early by the study oversight committee because: a) Cabozantinib was too toxic for the study to continue. b) The prostate-specific antigen (PSA) level fell in most of the treated patients. c) In the initial 121 patients, there was an unexpected improvement in bone scans and decrease in pain in the lead-in stage of the study. d) Unexpected rapid soft tissue progression. Bone scans improved in 78% of patients, and in 12% there was complete remission. After further analysis, the following were true except for: a) Cabozantinib interfered with technetium-99, and thus, the responses were not real, but rather an artifact. b) The PSA did not correlate with improvement in bone pain. c) Markers of bone formation and resorption showed improvement, and there was no correlation with prior bisphosphonate therapy. d) Bone scan improvement correlated with improvement in soft tissue disease.
Key points
The results in patients with prostate cancer were so striking – 72% of patients had regression in soft tissue lesions, and 68% of evaluable patients had improvement on bone scan, including complete resolution in 12% – that the subset analysis was published as a rapid communication.1,2 Because of very high response rates (5% at 12 weeks) and symptomatic improvement in the initial 122 patients who were enrolled, random assignment was discontinued. Bone markers improved in concert with the radiologic and clinical improvement. Answers c, a
Cabozantinib in metastatic prostate cancer1,2
Researchers tested cabozantinib, a tyrosine-kinase inhibitor (TKI) against MET and vascular endothelial growth factor receptor 2 (VEGF), in a large phase 2 randomized discontinuation trial in 9 tumor types. A subset of 171 patients with castrateresistant prostate cancer (CRPC) was reported in this study. Patients were treated on open label for 12 weeks, and then if stable, were randomized to receive the active drug or placebo. The trial was suspended early by the study oversight committee because: a) Cabozantinib was too toxic for the study to continue. b) The prostate-specific antigen (PSA) level fell in most of the treated patients. c) In the initial 121 patients, there was an unexpected improvement in bone scans and decrease in pain in the lead-in stage of the study. d) Unexpected rapid soft tissue progression. Bone scans improved in 78% of patients, and in 12% there was complete remission. After further analysis, the following were true except for: a) Cabozantinib interfered with technetium-99, and thus, the responses were not real, but rather an artifact. b) The PSA did not correlate with improvement in bone pain. c) Markers of bone formation and resorption showed improvement, and there was no correlation with prior bisphosphonate therapy. d) Bone scan improvement correlated with improvement in soft tissue disease.
Key points
The results in patients with prostate cancer were so striking – 72% of patients had regression in soft tissue lesions, and 68% of evaluable patients had improvement on bone scan, including complete resolution in 12% – that the subset analysis was published as a rapid communication.1,2 Because of very high response rates (5% at 12 weeks) and symptomatic improvement in the initial 122 patients who were enrolled, random assignment was discontinued. Bone markers improved in concert with the radiologic and clinical improvement. Answers c, a
BEST PRACTICES IN: Topical Therapy for Actinic Keratosis
Medical Education Library
A Best Practices Supplement to Skin & Allergy News®. This supplement was sponsored by Valeant Pharmaceuticals North America LLC.

- Introduction
- Carac Cream (fluorouracil cream) 0.5%
- Zyclara (imiquimod) Cream 2.5% and 3.75%
- Summary
- IMPORTANT SAFETY INFORMATION
Faculty/Faculty Disclosure
Dr. Harper, MD
Clinical Associate Professor of Dermatology University of Alabama-Birmingham Dermatology and Skin Care Center of Birmingham, P.C.
Birmingham, Alabama
Dr. Harper reported that she is a consultant and speaker for Medicis Pharmaceutical Corporation, a division of Valeant Pharmaceuticals, and received compensation from Valeant for her assistance in developing the content of this article.
LINKS: Click Here for PDF.
Copyright © by Frontline Medical Communications Inc.
Medical Education Library
A Best Practices Supplement to Skin & Allergy News®. This supplement was sponsored by Valeant Pharmaceuticals North America LLC.
- Introduction
- Carac Cream (fluorouracil cream) 0.5%
- Zyclara (imiquimod) Cream 2.5% and 3.75%
- Summary
- IMPORTANT SAFETY INFORMATION
Faculty/Faculty Disclosure
Dr. Harper, MD
Clinical Associate Professor of Dermatology University of Alabama-Birmingham Dermatology and Skin Care Center of Birmingham, P.C.
Birmingham, Alabama
Dr. Harper reported that she is a consultant and speaker for Medicis Pharmaceutical Corporation, a division of Valeant Pharmaceuticals, and received compensation from Valeant for her assistance in developing the content of this article.
LINKS: Click Here for PDF.
Copyright © by Frontline Medical Communications Inc.
Medical Education Library
A Best Practices Supplement to Skin & Allergy News®. This supplement was sponsored by Valeant Pharmaceuticals North America LLC.
- Introduction
- Carac Cream (fluorouracil cream) 0.5%
- Zyclara (imiquimod) Cream 2.5% and 3.75%
- Summary
- IMPORTANT SAFETY INFORMATION
Faculty/Faculty Disclosure
Dr. Harper, MD
Clinical Associate Professor of Dermatology University of Alabama-Birmingham Dermatology and Skin Care Center of Birmingham, P.C.
Birmingham, Alabama
Dr. Harper reported that she is a consultant and speaker for Medicis Pharmaceutical Corporation, a division of Valeant Pharmaceuticals, and received compensation from Valeant for her assistance in developing the content of this article.
LINKS: Click Here for PDF.
Copyright © by Frontline Medical Communications Inc.
