User login
In the Face of It All
A 59 year‐old man was sent from urgent care clinic to the emergency room for further evaluation because of 1 month of diarrhea and an acute elevation in his serum creatinine.
Whereas acute diarrhea is commonly due to a self‐limited and often unspecified infection, diarrhea that extends beyond 23 weeks (chronic) warrants consideration of malabsorptive, inflammatory, infectious, and malignant processes. The acute renal failure likely is a consequence of dehydration, but the possibility of simultaneous gastrointestinal and renal involvement from a systemic process (eg, vasculitis) must be considered.
The patient's diarrhea began 1 month prior, shortly after having a milkshake at a fast food restaurant. The diarrhea was initially watery, occurred 8‐10 times per day, occasionally awakened him at night, and was associated with nausea. There was no mucus, blood, or steatorrhea until 1 day prior to presentation, when he developed epigastric pain and bloody stools. He denied any recent travel outside of Northern California and had no sick contacts. He had lost 10 pounds over the preceding month. He denied fevers, chills, vomiting, or jaundice, and had not taken antibiotics recently.
In the setting of chronic diarrhea, unintentional weight loss is an alarm feature but does not narrow the diagnostic possibilities significantly. The appearance of blood and pain on a single day after 1 month of symptoms renders their diagnostic value uncertain. For instance, rectal or hemorrhoidal bleeding would be a common occurrence after 1 month of frequent defecation. Sustained bloody stools might be seen in any form of erosive luminal disease, such as infection, inflammatory bowel disease, or neoplasm. Pain is compatible with inflammatory bowel disease, obstructing neoplasms, infections, or ischemia (eg, vasculitis). There are no fever or chills to support infection, and common gram‐negative enteric pathogens (such as Salmonella, Campylobacter, and Yersinia) usually do not produce symptoms for such an extended period. He has not taken antibiotics, which would predispose him to infection with Clostridum difficile, and he has no obvious exposure to parasites such as Entamoeba.
The patient had diabetes mellitus with microalbuminuria, chronic obstructive pulmonary disease, hypertension, hyperlipidemia, chronic low back pain, and gastritis, and had undergone a Billroth II procedure for a perforated gastric ulcer in the remote past. His medications included omeprazole, insulin glargine, simvastatin, lisinopril, amlodipine, and albuterol and beclomethasone metered‐dose inhalers. He had been married for 31 years, lived at home with his wife, was a former rigger in a shipyard and was on disability for chronic low back pain. He denied alcohol or intravenous drug use but had quit tobacco 5 years prior after more than 40 pack‐years of smoking. He had three healthy adult children and there was no family history of cancer, liver disease, or inflammatory bowel disease. There was no history of sexually transmitted diseases or unprotected sexual intercourse.
Bacterial overgrowth in the blind loop following a Billroth II operation can lead to malabsorption, but the diarrhea would not begin so abruptly this long after surgery. Medications are common causes of diarrhea. Proton‐pump inhibitors, by reducing gastric acidity, confer an increased risk of bacterial enteritis; they also are a risk factor for C difficile. Lisinopril may cause bowel angioedema months or years after initiation. Occult laxative use is a well‐recognized cause of chronic diarrhea and should also be considered. The most relevant element of his social history is the prolonged smoking and the attendant risk of cancer, although diarrhea is a rare paraneoplastic phenomenon.
On exam, temperature was 36.6C, blood pressure 125/78, pulse 88, respiratory rate 16 per minute, and oxygen saturation 97% while breathing room air. There was temporal wasting and mild scleral icterus, but no jaundice. Lungs were clear to auscultation and heart was regular in rate and rhythm without murmurs or gallops. There was no jugular venous distention. A large abdominal midline scar was present, bowel sounds were normoactive, and the abdomen was soft, nontender, and nondistended. The hard was regular in rate and rhythm the liver edge was 6 cm below costal margin; there was no splenomegaly. The patient was alert and oriented, with a normal neurologic exam.
The liver generally enlarges because of acute inflammation, congestion, or infiltration. Infiltration can be due to tumors, infections, hemochromatosis, amyloidosis, or sarcoidosis. A normal cardiac exam argues against hepatic congestion from right‐sided heart failure or pericardial disease.
The key elements of the case are diarrhea and hepatomegaly. Inflammatory bowel disease can be accompanied by sclerosing cholangitis, but this should not enlarge the liver. Mycobacterial infections and syphilis can infiltrate the liver and intestinal mucosa, causing diarrhea, but he lacks typical risk factors.
Malignancy is an increasing concern. Colon cancer commonly metastasizes to the liver and can occasionally be intensely secretory. Pancreatic cancer could account for these symptoms, especially if pancreatic exocrine insufficiency caused malabsorption. Various rare neuroendocrine tumors that arise in the pancreas can cause secretory diarrheas and liver metastases, such as carcinoid, VIPoma, and Zollinger‐Ellison syndrome.
Laboratory results revealed a serum sodium of 143 mmol/L, potassium 4.7 mmol/L, chloride 110 mmol/L, bicarbonate 25 mmol/L, urea nitrogen 24 mg/dL, and creatinine 2.5 mg/dL (baseline had been 1.2 mg/dL 2 months previously). Serum glucose was 108 mg/dL and calcium was 8.8 mg/dL. The total white blood cell count was 9300 per mm3 with a normal differential, hemoglobin was 14.4 g/dL, mean corpuscular volume was 87 fL, and the platelet count was normal. Total bilirubin was 3.7 mg/dL, and direct bilirubin was 3.1 mg/dL. Aspartate aminotransferase (AST) was 122 U/L (normal range, 831), alanine aminotransferase (ALT) 79 U/L (normal range, 731), alkaline phosphatase 1591 U/L (normal range, 39117), and gamma‐glutamyltransferase (GGT) 980 U/L (normal range, 57). Serum albumin was 2.5 mg/dL, prothrombin time was 16.4 seconds, and international normalized ratio (INR) was 1.6.
Urinalysis was normal except for trace hemoglobin, small bilirubin, and 70 mg/dL of protein; specific gravity was 1.007. Urine microscopy demonstrated no cells or casts. The ratio of protein to creatinine on a spot urine sample was less than 1. Chest x‐ray was normal. The electrocardiogram demonstrated sinus rhythm with an old right bundle branch block and normal QRS voltages.
The disproportionate elevation in alkaline phosphatase points to an infiltrative hepatopathy from a cancer originating in the gastrointestinal tract or infection. Other infiltrative processes such as sarcoidosis or amyloidosis usually have evidence of disease elsewhere before hepatic disease becomes apparent.
Mild proteinuria may be explained by diabetes. The specific gravity of 1.007 is atypical for dehydration and could suggest ischemic tubular injury. Although intrinsic renal diseases must continue to be entertained, hypovolemia (compounded by angiotensin‐converting enzyme [ACE] inhibitor use) is the leading explanation in light of the nondiagnostic renal studies. The preserved hemoglobin may simply indicate dehydration, but otherwise is somewhat reassuring in the context of bloody diarrhea.
The patient was admitted to the hospital. Three stool samples returned negative for C difficile toxin. No white cells were detected in the stool, and no ova or parasites were detected. Stool culture was negative for routine bacterial pathogens and for E coli O157. Tests for HIV and antinuclear antibodies (ANAs) and serologies for hepatitis A, B, and C were negative. Abdominal ultrasound demonstrated no intra‐ or extrahepatic bile duct dilatation; no hepatic masses were seen. Kidneys were normal in size and appearance without hydronephrosis. Computed tomography (CT) of the abdomen without intravenous contrast revealed normal‐appearing liver (with a 12‐cm span), spleen, biliary ducts, and pancreas, and there was no intra‐abdominal adenopathy.
The stool studies point away from infectious colitis. Infiltrative processes of the liver, including metastases, lymphoma, tuberculosis, syphilis, amyloidosis, and sarcoidosis, can be microscopic and therefore evade detection by ultrasound and CT scan. In conditions such as these, endoscopic retrograde cholangiopanccreatography/magnetic resonance cholangiopancreatography (ERCP/MRCP) or liver biopsy may be required. The CT is limited without contrast but does not suggest extrahepatic disease in the abdomen.
MRCP was performed, but was a technically suboptimal study due to the presence of ascites. The serum creatinine improved to 1.4 mg/dL over the next 4 days, and the patient's diarrhea decreased to two bowel movements daily with the use of loperamide. The patient was discharged home with outpatient gastroenterology follow‐up planned to discuss further evaluation of the abnormal liver enzymes.
Prior to being seen in the Gastroenterology Clinic, the patient's nonbloody diarrhea worsened. He felt weaker and continued to lose weight. He also noted new onset of bilateral lower face numbness and burning, which was followed by swelling of his lower lip 12 hours later. He returned to the hospital.
On examination, he was afebrile. His lower lip was markedly swollen and was drooping from his face. He could not move the lip to close his mouth. The upper lip and tongue were normal size and moved without restriction. Facial sensation was intact, but there was weakness when he attempted to wrinkle both of his brows and close his eyelids. The rest of his physical examination was unchanged.
The serum creatinine had risen to 3.6 mg/dL, and the complete blood count remained normal. Serum total bilirubin was 4.6 mg/dL, AST 87 U/L, ALT 76 U/L, and alkaline phosphatase 1910 U/L. The 24‐hour urine protein measurement was 86 mg.
Lip swelling suggests angioedema. ACE inhibitors are frequent offenders, and it would be important to know whether his lisinopril was restarted at discharge. ACE‐inhibitor angioedema can also affect the intestine, causing abdominal pain and diarrhea, but does not cause a systemic wasting illness or infiltrative hepatopathy. The difficulty moving the lip may reflect the physical effects of swelling, but generalized facial weakness supports a cranial neuropathy. Basilar meningitis may produce multiple cranial neuropathies, the etiologies of which are quite similar to the previously mentioned causes of infiltrative liver disease: sarcoidosis, syphilis, tuberculosis, or lymphoma.
The patient had not resumed lisinopril since his prior hospitalization. The lower lip swelling and paralysis persisted, and new sensory paresthesias developed over the right side of his chin. A consulting neurologist found normal language and speech and moderate dysarthria. Cranial nerve exam was normal except bilateral lower motor neuron facial nerve palsy was noted with bilateral facial droop, reduced strength of eyelid closure, and diminished forehead movement bilaterally; facial sensation was normal. Extremity motor exam revealed proximal iliopsoas muscle weakness bilaterally rated as 4/5 and was otherwise normal. Sensation to pinprick was diminished in a stocking/glove distribution. Deep‐tendon reflexes were normal and plantar response was down‐going bilaterally. Coordination was intact, Romberg was negative, and gait was slowed due to weakness.
Over the next several days, the patient continued to have diarrhea and facial symptoms. The serum total bilirubin increased to 14 mg/dL, alkaline phosphatase rose above 2,000 U/L, and serum creatinine increased to 5.5 mg/dL. Noncontrast CT scan of the head was normal.
Along with a mild peripheral sensory neuropathy, the exam indicates bilateral palsies of the facial nerve. Lyme disease is a frequent etiology, but this patient is not from an endemic area. I am most suspicious of bilateral infiltration of cranial nerve VII. I am thinking analogically to the numb chin syndrome, wherein lymphoma or breast cancer infiltration along the mental branch of V3 causes sensory loss, and perhaps these disorders can produce infiltrative facial neuropathy. At this point I am most concerned about lymphomatous meningitis with cranial nerve involvement. Cerebrospinal fluid (CSF) analysis (including cytology) would be informative.
Lumbar puncture demonstrated clear CSF with one white blood cell per mm3 and no red blood cells. Glucose was normal, and protein was 95.5 (normal range, 15‐45 mg/dL). Gram stain and culture for bacteria were negative, as were polymerase chain reaction (PCR) testing for herpes simplex virus, mycobacterial and fungal stains and cultures, and cytology. Transthoracic echocardiogram demonstrated severe concentric left ventricular (LV) hypertrophy, normal LV systolic function, and impaired LV relaxation. CT scan of the chest identified no adenopathy or other abnormality.
The CSF analysis does not support basilar meningitis, although the cytoalbuminologic dissociation makes me wonder whether there is some intrathecal antibody production or an autoimmune process we have yet to uncover. The absence of lymphadenopathy anywhere in the body and the negative CSF cytology now point away from lymphoma. As the case for lymphoma or an infection diminishes, systemic amyloidosis rises to the top of possibilities in this afebrile man who is losing weight, has infiltrative liver and nerve abnormalities, renal failure, cardiac enlargement, and suspected gastrointestinal luminal abnormality. Although the echocardiographic findings are most likely explained by hypertension, they are compatible with amyloid infiltration. A tissue specimen is needed, and either colonoscopy or liver biopsy should be suitable.
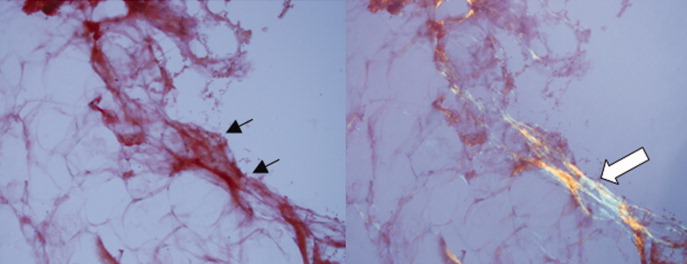
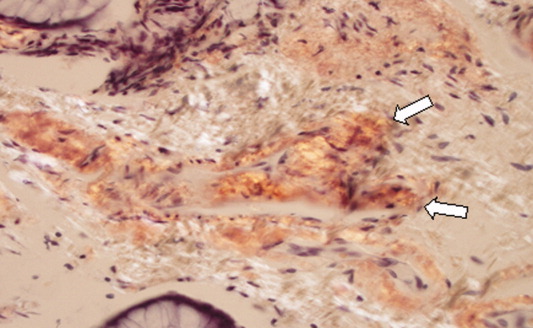
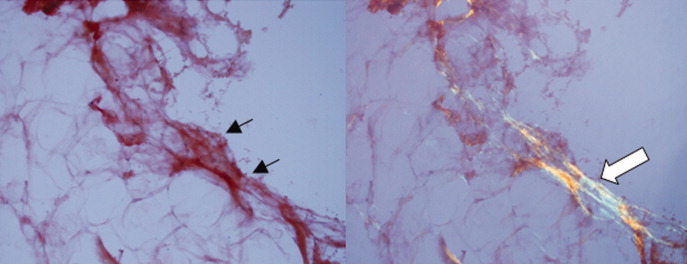
A pathologist performed a fat pad biopsy that demonstrated scant congophilic and birefringent material associated with blood vessels, suggestive of amyloid (Fig. 1). Colonoscopy demonstrated normal mucosa, and a rectal biopsy revealed congophilic material within the blood vessels consistent with amyloid (Fig. 2). No monoclonal band was present on serum protein electrophoresis. Urine protein electrophoresis identified a homogenous band in the gamma region, and urine kappa and lambda free light chains were increased: kappa was 10.7 mg/dL (normal range, 2), and lambda was 4.25 mg/dL (normal range, 2).
After extensive discussion among the patient, his wife, and a palliative care physician, the patient declined chemotherapy and elected to go home. Two days after discharge (7 weeks after his initial admission for diarrhea) he died in his sleep at home. Permission for a postmortem examination was not granted.
Discussion
Amyloidosis refers to abnormal extracellular deposition of fibril. There are many types of amyloidosis including primary amyloidosis (AL amyloidosis), secondary amyloidosis (AA amyloidosis), and hereditary causes. Systemic AL amyloidosis is a rare plasma cell disorder characterized by misfolding of insoluble extracellular fibrillar proteins derived from immunoglobulin light chains. These insoluble proteins typically deposit in the kidney, heart, and nervous system.1 Although the mechanism of organ dysfunction is debated, deposition of these proteins may disrupt the tissue architecture by interacting with local receptors and causing apoptosis.1
Table 1 indicates the most common findings in patients with AL amyloidosis.2 While our patient ultimately developed many common findings of AL amyloidosis, several features were atypical, including the marked hyperbilirubinemia, profound diarrhea, and bilateral facial diplegia.
| Organ Involvement | Incidence of Organ Involvement (%) | Symptoms | Signs | Laboratory/Test Finding |
|---|---|---|---|---|
| ||||
| General | Malaise, weight loss | |||
| Renal | 33 | Fatigue | Peripheral edema | Proteinuria with or without renal insufficiency, pleural effusion, hypercholesterolemia |
| Cardiac | 20 | Palpitations, dyspnea | Elevated jugular venous pressure, S3, peripheral edema, hepatomegaly | Low‐voltage or atrial fibrillation on electrocardiogram; echocardiogram: thickened ventricles, dilated atria |
| Neurological | 20 | Paresthesias, numbness, weakness, autonomic insufficiency | Carpal tunnel syndrome, postural hypotension | |
| Gastrointestinal and Hepatic | 16 | Diarrhea, nausea, weight loss | Macroglossia, hepatomegaly | Elevated alkaline phosphatase |
| Hematology | Rare | Bleeding | Periorbital purpura (raccoon eyes) | Prolonged prothrombin time, Factor X deficiency |
Up to 70% of patients with amyloidosis will have detectable liver deposits, typically involving portal vessels, portal stroma, central vein, and sinusoidal parenchyma.3 Clinically overt hepatic dysfunction from amyloid is less frequent,4 and the most characteristic findings are hepatomegaly with a markedly elevated serum alkaline phosphatase concentration; jaundice is rare. Palpable hepatic enlargement without abnormal liver enzymes should be interpreted with caution. The finding of a palpable liver edge correlates poorly with frank hepatomegaly, with a positive likelihood ratio of just 1.7.5 In the patient under discussion, suspected hepatomegaly was not confirmed on a subsequent CT scan. Nonetheless, the elevated alkaline phosphatase represented an important clue to potential infiltrative liver disease. In a series of amyloidosis patients from the Mayo Clinic, 81% had hepatomegaly on physical exam, and the mean alkaline phosphatase level was 1,029 U/L (normal, 250 U/L), while the mean serum bilirubin and AST levels were only modestly elevated, at 3.2 mg/dL and 72 U/L respectively. The prothrombin time was prolonged in 35% of patients.
Upper gastrointestinal tract involvement by AL amyloid may be found in up to a third of cases at autopsy, but clinically significant gastrointestinal features are seen in fewer than 5% of patients.6 Predominant intestinal manifestations are unintentional weight loss (average 7 kg) and diarrhea, nonspecific features that result in delayed diagnosis for a median of 7 months after symptom onset.7 Diarrhea in AL amyloid may stem from several mechanisms: small intestine mucosal infiltration, steatorrhea from pancreatic insufficiency, autonomic neuropathy leading to pseudo‐obstruction and bacterial overgrowth, bile acid malabsorption, or rapid transit time. Diarrhea in AL amyloid is often resistant to treatment and may be the primary cause of death.7
Systemic amyloidosis commonly produces peripheral neuropathies. Involvement of small unmyelinated fibers causes paresthesias and progressive sensory loss in a pattern that is usually distal, symmetric, and progressive.6, 9 Our patient presented with bilateral sensory paresthesias of the chin, suggesting the numb chin syndrome (NCS). NCS is characterized by facial numbness along the distribution of the mental branch of the trigeminal nerve. While dental disorders and infiltration from malignant tumors (mostly lung and breast cancer) account for most cases, amyloidosis and other infiltrative disorders are known to cause NCS as well.10, 11 Our patient's sensory paresthesias may have represented amyloid infiltration of peripheral nerves.
With the exception of carpal tunnel syndrome, motor or cranial neuropathy is uncommon in amyloid, and when present usually heralds advanced disease.12 Descriptions of bilateral facial weakness, also known as facial diplegia, from amyloidosis are limited to case reports.1315 Other causes of this rare finding include sarcoidosis, Guillain‐Barr syndrome, and Lyme disease.16
The diagnosis of primary amyloidosis requires histologic evidence of amyloid from a tissue biopsy specimen (demonstrating positive Congo red staining and pathognomonic green birefringence under cross‐polarized light microscopy), and the presence of a clonal plasma cell disorder. While biopsy of an affected organ is diagnostic, more easily obtained samples such as fat pad biopsy and rectal biopsy yield positive results in up to 80% of cases.2 Serum and urine protein electrophoresis with immunofixation identify an underlying plasma cell disorder in 90% of cases of primary amyloidosis. When these tests are inconclusive, serum or urine free light chain assays or bone marrow aspirate and biopsy are useful aids to detect underlying plasma cell dyscrasia.2 AL amyloidosis is a progressive disease with median survival of about 12 years.8 Poorer prognosis is associated with substantial echocardiographic findings, autonomic neuropathy, and liver involvement.2 Hyperbilirubinemia is associated with a poor prognosis, with a median survival of 8.5 months.4 Proteinuria or peripheral neuropathy portends a less ominous course.6
Treatment goals include reducing production and deposition of fibril proteins and contending with organ dysfunction (eg, congestive heart failure [CHF] management). Selected patients with AL amyloidosis may be candidates for high‐dose melphalan and autologous stem cell transplantation.
It would not be reasonable for clinicians to suspect amyloidosis in cases of diarrhea until two conditions are met: 1) the absence of evidence for the typical etiologies of diarrhea; and 2) the evolving picture of an infiltrative disorder. The latter was heralded by the elevated alkaline phosphatase, and was supported by the subsequent multiorgan involvement. Conceptualizing the disease as infiltrative still required a diligent exclusion of infection and invasive tumor cells, which invade disparate organs far more commonly than amyloidosis. Their absence and the organ pattern that is typical of AL amyloidosis (heart, kidney, and peripheral nerve involvement) allowed the discussant to reason by analogy that amyloidosis was also responsible for the most symptomatic phenomena, namely, the diarrhea and facial diplegia (and numb chin syndrome).
Key Teaching Points
-
Hospitalists should consider systemic amyloidosis in cases of unexplained diarrhea when other clinical features of AL amyloidosis are present, including nephrotic syndrome with or without renal insufficiency, cardiomyopathy, peripheral neuropathy, and hepatomegaly.
-
Hepatic amyloidosis should be suspected when weight loss, hepatomegaly, and elevated alkaline phosphatase are present. Although jaundice is rare in amyloidosis, liver involvement and hyperbilirubinemia portend a poorer prognosis.
-
Numb chin syndrome and bilateral facial diplegia are rare manifestations of AL amyloid deposition in peripheral nerves.
- ,.Molecular mechanisms of amyloidosis.N Engl J Med.2003;349(6):583–596.
- Guidelines Working Group of UK Myeloma Forum; British Committee for Standards in Haematology, British Society for Haematology.Guidelines on the diagnosis and management of AL amyloidosis.Br J Haematol.2004;125:681–700.
- ,.Hepatic amyloidosis: morphologic differences between systemic AL and AA types.Hum Pathol.1991;22(9):904–907.
- ,,, et al.Primary (AL) hepatic amyloidosis clinical features and natural history in 98 patients.Medicine.2003;82(5):291–298.
- .Evidence‐Based Physical Diagnosis.Philadelphia, PA:WB Saunders;2001:595–599.
- ,,, et al.Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis.Am J Hematol.2005;79:319–328.
- .Primary (AL) amyloidosis with gastrointestinal involvement.Scand J Gastroenterol.2009;44(6):708–711.
- ,.Gastrointestinal manifestations of amyloid.Am J Gastroenterol.2008;103:776–787.
- ,.Primary systemic amyloidosis: clinical and laboratory features in 474 cases.Semin Hematol.1995;32:45–59.
- ,,,,.Chin numbness: a symptom that should not be underestimated: a review of 12 cases.Am J Med Sci.2009;337:407–410.
- .Numb chin syndrome: a possible clue to serious illness.Hosp Physician.2000;54–56.
- .Autonomic peripheral neuropathy.Neurol Clin.2007;25:277–301.
- ,.Facial diplegia due to amyloidosis.South Med J.1986;79(11):1458–1459.
- ,,,,.Familial amyloidosis with cranial neuropathy and corneal lattice dystrophy.Neurology.1986;36:432–435.
- ,,.Crainal neuropathy associated with primary amyloidosis.Ann Neurol.1991;29:451–454.
- .Bilateral seventh nerve palsy: analysis of 43 cases and review of the literature.Neurology.1994;44:1198–202.
A 59 year‐old man was sent from urgent care clinic to the emergency room for further evaluation because of 1 month of diarrhea and an acute elevation in his serum creatinine.
Whereas acute diarrhea is commonly due to a self‐limited and often unspecified infection, diarrhea that extends beyond 23 weeks (chronic) warrants consideration of malabsorptive, inflammatory, infectious, and malignant processes. The acute renal failure likely is a consequence of dehydration, but the possibility of simultaneous gastrointestinal and renal involvement from a systemic process (eg, vasculitis) must be considered.
The patient's diarrhea began 1 month prior, shortly after having a milkshake at a fast food restaurant. The diarrhea was initially watery, occurred 8‐10 times per day, occasionally awakened him at night, and was associated with nausea. There was no mucus, blood, or steatorrhea until 1 day prior to presentation, when he developed epigastric pain and bloody stools. He denied any recent travel outside of Northern California and had no sick contacts. He had lost 10 pounds over the preceding month. He denied fevers, chills, vomiting, or jaundice, and had not taken antibiotics recently.
In the setting of chronic diarrhea, unintentional weight loss is an alarm feature but does not narrow the diagnostic possibilities significantly. The appearance of blood and pain on a single day after 1 month of symptoms renders their diagnostic value uncertain. For instance, rectal or hemorrhoidal bleeding would be a common occurrence after 1 month of frequent defecation. Sustained bloody stools might be seen in any form of erosive luminal disease, such as infection, inflammatory bowel disease, or neoplasm. Pain is compatible with inflammatory bowel disease, obstructing neoplasms, infections, or ischemia (eg, vasculitis). There are no fever or chills to support infection, and common gram‐negative enteric pathogens (such as Salmonella, Campylobacter, and Yersinia) usually do not produce symptoms for such an extended period. He has not taken antibiotics, which would predispose him to infection with Clostridum difficile, and he has no obvious exposure to parasites such as Entamoeba.
The patient had diabetes mellitus with microalbuminuria, chronic obstructive pulmonary disease, hypertension, hyperlipidemia, chronic low back pain, and gastritis, and had undergone a Billroth II procedure for a perforated gastric ulcer in the remote past. His medications included omeprazole, insulin glargine, simvastatin, lisinopril, amlodipine, and albuterol and beclomethasone metered‐dose inhalers. He had been married for 31 years, lived at home with his wife, was a former rigger in a shipyard and was on disability for chronic low back pain. He denied alcohol or intravenous drug use but had quit tobacco 5 years prior after more than 40 pack‐years of smoking. He had three healthy adult children and there was no family history of cancer, liver disease, or inflammatory bowel disease. There was no history of sexually transmitted diseases or unprotected sexual intercourse.
Bacterial overgrowth in the blind loop following a Billroth II operation can lead to malabsorption, but the diarrhea would not begin so abruptly this long after surgery. Medications are common causes of diarrhea. Proton‐pump inhibitors, by reducing gastric acidity, confer an increased risk of bacterial enteritis; they also are a risk factor for C difficile. Lisinopril may cause bowel angioedema months or years after initiation. Occult laxative use is a well‐recognized cause of chronic diarrhea and should also be considered. The most relevant element of his social history is the prolonged smoking and the attendant risk of cancer, although diarrhea is a rare paraneoplastic phenomenon.
On exam, temperature was 36.6C, blood pressure 125/78, pulse 88, respiratory rate 16 per minute, and oxygen saturation 97% while breathing room air. There was temporal wasting and mild scleral icterus, but no jaundice. Lungs were clear to auscultation and heart was regular in rate and rhythm without murmurs or gallops. There was no jugular venous distention. A large abdominal midline scar was present, bowel sounds were normoactive, and the abdomen was soft, nontender, and nondistended. The hard was regular in rate and rhythm the liver edge was 6 cm below costal margin; there was no splenomegaly. The patient was alert and oriented, with a normal neurologic exam.
The liver generally enlarges because of acute inflammation, congestion, or infiltration. Infiltration can be due to tumors, infections, hemochromatosis, amyloidosis, or sarcoidosis. A normal cardiac exam argues against hepatic congestion from right‐sided heart failure or pericardial disease.
The key elements of the case are diarrhea and hepatomegaly. Inflammatory bowel disease can be accompanied by sclerosing cholangitis, but this should not enlarge the liver. Mycobacterial infections and syphilis can infiltrate the liver and intestinal mucosa, causing diarrhea, but he lacks typical risk factors.
Malignancy is an increasing concern. Colon cancer commonly metastasizes to the liver and can occasionally be intensely secretory. Pancreatic cancer could account for these symptoms, especially if pancreatic exocrine insufficiency caused malabsorption. Various rare neuroendocrine tumors that arise in the pancreas can cause secretory diarrheas and liver metastases, such as carcinoid, VIPoma, and Zollinger‐Ellison syndrome.
Laboratory results revealed a serum sodium of 143 mmol/L, potassium 4.7 mmol/L, chloride 110 mmol/L, bicarbonate 25 mmol/L, urea nitrogen 24 mg/dL, and creatinine 2.5 mg/dL (baseline had been 1.2 mg/dL 2 months previously). Serum glucose was 108 mg/dL and calcium was 8.8 mg/dL. The total white blood cell count was 9300 per mm3 with a normal differential, hemoglobin was 14.4 g/dL, mean corpuscular volume was 87 fL, and the platelet count was normal. Total bilirubin was 3.7 mg/dL, and direct bilirubin was 3.1 mg/dL. Aspartate aminotransferase (AST) was 122 U/L (normal range, 831), alanine aminotransferase (ALT) 79 U/L (normal range, 731), alkaline phosphatase 1591 U/L (normal range, 39117), and gamma‐glutamyltransferase (GGT) 980 U/L (normal range, 57). Serum albumin was 2.5 mg/dL, prothrombin time was 16.4 seconds, and international normalized ratio (INR) was 1.6.
Urinalysis was normal except for trace hemoglobin, small bilirubin, and 70 mg/dL of protein; specific gravity was 1.007. Urine microscopy demonstrated no cells or casts. The ratio of protein to creatinine on a spot urine sample was less than 1. Chest x‐ray was normal. The electrocardiogram demonstrated sinus rhythm with an old right bundle branch block and normal QRS voltages.
The disproportionate elevation in alkaline phosphatase points to an infiltrative hepatopathy from a cancer originating in the gastrointestinal tract or infection. Other infiltrative processes such as sarcoidosis or amyloidosis usually have evidence of disease elsewhere before hepatic disease becomes apparent.
Mild proteinuria may be explained by diabetes. The specific gravity of 1.007 is atypical for dehydration and could suggest ischemic tubular injury. Although intrinsic renal diseases must continue to be entertained, hypovolemia (compounded by angiotensin‐converting enzyme [ACE] inhibitor use) is the leading explanation in light of the nondiagnostic renal studies. The preserved hemoglobin may simply indicate dehydration, but otherwise is somewhat reassuring in the context of bloody diarrhea.
The patient was admitted to the hospital. Three stool samples returned negative for C difficile toxin. No white cells were detected in the stool, and no ova or parasites were detected. Stool culture was negative for routine bacterial pathogens and for E coli O157. Tests for HIV and antinuclear antibodies (ANAs) and serologies for hepatitis A, B, and C were negative. Abdominal ultrasound demonstrated no intra‐ or extrahepatic bile duct dilatation; no hepatic masses were seen. Kidneys were normal in size and appearance without hydronephrosis. Computed tomography (CT) of the abdomen without intravenous contrast revealed normal‐appearing liver (with a 12‐cm span), spleen, biliary ducts, and pancreas, and there was no intra‐abdominal adenopathy.
The stool studies point away from infectious colitis. Infiltrative processes of the liver, including metastases, lymphoma, tuberculosis, syphilis, amyloidosis, and sarcoidosis, can be microscopic and therefore evade detection by ultrasound and CT scan. In conditions such as these, endoscopic retrograde cholangiopanccreatography/magnetic resonance cholangiopancreatography (ERCP/MRCP) or liver biopsy may be required. The CT is limited without contrast but does not suggest extrahepatic disease in the abdomen.
MRCP was performed, but was a technically suboptimal study due to the presence of ascites. The serum creatinine improved to 1.4 mg/dL over the next 4 days, and the patient's diarrhea decreased to two bowel movements daily with the use of loperamide. The patient was discharged home with outpatient gastroenterology follow‐up planned to discuss further evaluation of the abnormal liver enzymes.
Prior to being seen in the Gastroenterology Clinic, the patient's nonbloody diarrhea worsened. He felt weaker and continued to lose weight. He also noted new onset of bilateral lower face numbness and burning, which was followed by swelling of his lower lip 12 hours later. He returned to the hospital.
On examination, he was afebrile. His lower lip was markedly swollen and was drooping from his face. He could not move the lip to close his mouth. The upper lip and tongue were normal size and moved without restriction. Facial sensation was intact, but there was weakness when he attempted to wrinkle both of his brows and close his eyelids. The rest of his physical examination was unchanged.
The serum creatinine had risen to 3.6 mg/dL, and the complete blood count remained normal. Serum total bilirubin was 4.6 mg/dL, AST 87 U/L, ALT 76 U/L, and alkaline phosphatase 1910 U/L. The 24‐hour urine protein measurement was 86 mg.
Lip swelling suggests angioedema. ACE inhibitors are frequent offenders, and it would be important to know whether his lisinopril was restarted at discharge. ACE‐inhibitor angioedema can also affect the intestine, causing abdominal pain and diarrhea, but does not cause a systemic wasting illness or infiltrative hepatopathy. The difficulty moving the lip may reflect the physical effects of swelling, but generalized facial weakness supports a cranial neuropathy. Basilar meningitis may produce multiple cranial neuropathies, the etiologies of which are quite similar to the previously mentioned causes of infiltrative liver disease: sarcoidosis, syphilis, tuberculosis, or lymphoma.
The patient had not resumed lisinopril since his prior hospitalization. The lower lip swelling and paralysis persisted, and new sensory paresthesias developed over the right side of his chin. A consulting neurologist found normal language and speech and moderate dysarthria. Cranial nerve exam was normal except bilateral lower motor neuron facial nerve palsy was noted with bilateral facial droop, reduced strength of eyelid closure, and diminished forehead movement bilaterally; facial sensation was normal. Extremity motor exam revealed proximal iliopsoas muscle weakness bilaterally rated as 4/5 and was otherwise normal. Sensation to pinprick was diminished in a stocking/glove distribution. Deep‐tendon reflexes were normal and plantar response was down‐going bilaterally. Coordination was intact, Romberg was negative, and gait was slowed due to weakness.
Over the next several days, the patient continued to have diarrhea and facial symptoms. The serum total bilirubin increased to 14 mg/dL, alkaline phosphatase rose above 2,000 U/L, and serum creatinine increased to 5.5 mg/dL. Noncontrast CT scan of the head was normal.
Along with a mild peripheral sensory neuropathy, the exam indicates bilateral palsies of the facial nerve. Lyme disease is a frequent etiology, but this patient is not from an endemic area. I am most suspicious of bilateral infiltration of cranial nerve VII. I am thinking analogically to the numb chin syndrome, wherein lymphoma or breast cancer infiltration along the mental branch of V3 causes sensory loss, and perhaps these disorders can produce infiltrative facial neuropathy. At this point I am most concerned about lymphomatous meningitis with cranial nerve involvement. Cerebrospinal fluid (CSF) analysis (including cytology) would be informative.
Lumbar puncture demonstrated clear CSF with one white blood cell per mm3 and no red blood cells. Glucose was normal, and protein was 95.5 (normal range, 15‐45 mg/dL). Gram stain and culture for bacteria were negative, as were polymerase chain reaction (PCR) testing for herpes simplex virus, mycobacterial and fungal stains and cultures, and cytology. Transthoracic echocardiogram demonstrated severe concentric left ventricular (LV) hypertrophy, normal LV systolic function, and impaired LV relaxation. CT scan of the chest identified no adenopathy or other abnormality.
The CSF analysis does not support basilar meningitis, although the cytoalbuminologic dissociation makes me wonder whether there is some intrathecal antibody production or an autoimmune process we have yet to uncover. The absence of lymphadenopathy anywhere in the body and the negative CSF cytology now point away from lymphoma. As the case for lymphoma or an infection diminishes, systemic amyloidosis rises to the top of possibilities in this afebrile man who is losing weight, has infiltrative liver and nerve abnormalities, renal failure, cardiac enlargement, and suspected gastrointestinal luminal abnormality. Although the echocardiographic findings are most likely explained by hypertension, they are compatible with amyloid infiltration. A tissue specimen is needed, and either colonoscopy or liver biopsy should be suitable.
A pathologist performed a fat pad biopsy that demonstrated scant congophilic and birefringent material associated with blood vessels, suggestive of amyloid (Fig. 1). Colonoscopy demonstrated normal mucosa, and a rectal biopsy revealed congophilic material within the blood vessels consistent with amyloid (Fig. 2). No monoclonal band was present on serum protein electrophoresis. Urine protein electrophoresis identified a homogenous band in the gamma region, and urine kappa and lambda free light chains were increased: kappa was 10.7 mg/dL (normal range, 2), and lambda was 4.25 mg/dL (normal range, 2).
After extensive discussion among the patient, his wife, and a palliative care physician, the patient declined chemotherapy and elected to go home. Two days after discharge (7 weeks after his initial admission for diarrhea) he died in his sleep at home. Permission for a postmortem examination was not granted.
Discussion
Amyloidosis refers to abnormal extracellular deposition of fibril. There are many types of amyloidosis including primary amyloidosis (AL amyloidosis), secondary amyloidosis (AA amyloidosis), and hereditary causes. Systemic AL amyloidosis is a rare plasma cell disorder characterized by misfolding of insoluble extracellular fibrillar proteins derived from immunoglobulin light chains. These insoluble proteins typically deposit in the kidney, heart, and nervous system.1 Although the mechanism of organ dysfunction is debated, deposition of these proteins may disrupt the tissue architecture by interacting with local receptors and causing apoptosis.1
Table 1 indicates the most common findings in patients with AL amyloidosis.2 While our patient ultimately developed many common findings of AL amyloidosis, several features were atypical, including the marked hyperbilirubinemia, profound diarrhea, and bilateral facial diplegia.
| Organ Involvement | Incidence of Organ Involvement (%) | Symptoms | Signs | Laboratory/Test Finding |
|---|---|---|---|---|
| ||||
| General | Malaise, weight loss | |||
| Renal | 33 | Fatigue | Peripheral edema | Proteinuria with or without renal insufficiency, pleural effusion, hypercholesterolemia |
| Cardiac | 20 | Palpitations, dyspnea | Elevated jugular venous pressure, S3, peripheral edema, hepatomegaly | Low‐voltage or atrial fibrillation on electrocardiogram; echocardiogram: thickened ventricles, dilated atria |
| Neurological | 20 | Paresthesias, numbness, weakness, autonomic insufficiency | Carpal tunnel syndrome, postural hypotension | |
| Gastrointestinal and Hepatic | 16 | Diarrhea, nausea, weight loss | Macroglossia, hepatomegaly | Elevated alkaline phosphatase |
| Hematology | Rare | Bleeding | Periorbital purpura (raccoon eyes) | Prolonged prothrombin time, Factor X deficiency |
Up to 70% of patients with amyloidosis will have detectable liver deposits, typically involving portal vessels, portal stroma, central vein, and sinusoidal parenchyma.3 Clinically overt hepatic dysfunction from amyloid is less frequent,4 and the most characteristic findings are hepatomegaly with a markedly elevated serum alkaline phosphatase concentration; jaundice is rare. Palpable hepatic enlargement without abnormal liver enzymes should be interpreted with caution. The finding of a palpable liver edge correlates poorly with frank hepatomegaly, with a positive likelihood ratio of just 1.7.5 In the patient under discussion, suspected hepatomegaly was not confirmed on a subsequent CT scan. Nonetheless, the elevated alkaline phosphatase represented an important clue to potential infiltrative liver disease. In a series of amyloidosis patients from the Mayo Clinic, 81% had hepatomegaly on physical exam, and the mean alkaline phosphatase level was 1,029 U/L (normal, 250 U/L), while the mean serum bilirubin and AST levels were only modestly elevated, at 3.2 mg/dL and 72 U/L respectively. The prothrombin time was prolonged in 35% of patients.
Upper gastrointestinal tract involvement by AL amyloid may be found in up to a third of cases at autopsy, but clinically significant gastrointestinal features are seen in fewer than 5% of patients.6 Predominant intestinal manifestations are unintentional weight loss (average 7 kg) and diarrhea, nonspecific features that result in delayed diagnosis for a median of 7 months after symptom onset.7 Diarrhea in AL amyloid may stem from several mechanisms: small intestine mucosal infiltration, steatorrhea from pancreatic insufficiency, autonomic neuropathy leading to pseudo‐obstruction and bacterial overgrowth, bile acid malabsorption, or rapid transit time. Diarrhea in AL amyloid is often resistant to treatment and may be the primary cause of death.7
Systemic amyloidosis commonly produces peripheral neuropathies. Involvement of small unmyelinated fibers causes paresthesias and progressive sensory loss in a pattern that is usually distal, symmetric, and progressive.6, 9 Our patient presented with bilateral sensory paresthesias of the chin, suggesting the numb chin syndrome (NCS). NCS is characterized by facial numbness along the distribution of the mental branch of the trigeminal nerve. While dental disorders and infiltration from malignant tumors (mostly lung and breast cancer) account for most cases, amyloidosis and other infiltrative disorders are known to cause NCS as well.10, 11 Our patient's sensory paresthesias may have represented amyloid infiltration of peripheral nerves.
With the exception of carpal tunnel syndrome, motor or cranial neuropathy is uncommon in amyloid, and when present usually heralds advanced disease.12 Descriptions of bilateral facial weakness, also known as facial diplegia, from amyloidosis are limited to case reports.1315 Other causes of this rare finding include sarcoidosis, Guillain‐Barr syndrome, and Lyme disease.16
The diagnosis of primary amyloidosis requires histologic evidence of amyloid from a tissue biopsy specimen (demonstrating positive Congo red staining and pathognomonic green birefringence under cross‐polarized light microscopy), and the presence of a clonal plasma cell disorder. While biopsy of an affected organ is diagnostic, more easily obtained samples such as fat pad biopsy and rectal biopsy yield positive results in up to 80% of cases.2 Serum and urine protein electrophoresis with immunofixation identify an underlying plasma cell disorder in 90% of cases of primary amyloidosis. When these tests are inconclusive, serum or urine free light chain assays or bone marrow aspirate and biopsy are useful aids to detect underlying plasma cell dyscrasia.2 AL amyloidosis is a progressive disease with median survival of about 12 years.8 Poorer prognosis is associated with substantial echocardiographic findings, autonomic neuropathy, and liver involvement.2 Hyperbilirubinemia is associated with a poor prognosis, with a median survival of 8.5 months.4 Proteinuria or peripheral neuropathy portends a less ominous course.6
Treatment goals include reducing production and deposition of fibril proteins and contending with organ dysfunction (eg, congestive heart failure [CHF] management). Selected patients with AL amyloidosis may be candidates for high‐dose melphalan and autologous stem cell transplantation.
It would not be reasonable for clinicians to suspect amyloidosis in cases of diarrhea until two conditions are met: 1) the absence of evidence for the typical etiologies of diarrhea; and 2) the evolving picture of an infiltrative disorder. The latter was heralded by the elevated alkaline phosphatase, and was supported by the subsequent multiorgan involvement. Conceptualizing the disease as infiltrative still required a diligent exclusion of infection and invasive tumor cells, which invade disparate organs far more commonly than amyloidosis. Their absence and the organ pattern that is typical of AL amyloidosis (heart, kidney, and peripheral nerve involvement) allowed the discussant to reason by analogy that amyloidosis was also responsible for the most symptomatic phenomena, namely, the diarrhea and facial diplegia (and numb chin syndrome).
Key Teaching Points
-
Hospitalists should consider systemic amyloidosis in cases of unexplained diarrhea when other clinical features of AL amyloidosis are present, including nephrotic syndrome with or without renal insufficiency, cardiomyopathy, peripheral neuropathy, and hepatomegaly.
-
Hepatic amyloidosis should be suspected when weight loss, hepatomegaly, and elevated alkaline phosphatase are present. Although jaundice is rare in amyloidosis, liver involvement and hyperbilirubinemia portend a poorer prognosis.
-
Numb chin syndrome and bilateral facial diplegia are rare manifestations of AL amyloid deposition in peripheral nerves.
A 59 year‐old man was sent from urgent care clinic to the emergency room for further evaluation because of 1 month of diarrhea and an acute elevation in his serum creatinine.
Whereas acute diarrhea is commonly due to a self‐limited and often unspecified infection, diarrhea that extends beyond 23 weeks (chronic) warrants consideration of malabsorptive, inflammatory, infectious, and malignant processes. The acute renal failure likely is a consequence of dehydration, but the possibility of simultaneous gastrointestinal and renal involvement from a systemic process (eg, vasculitis) must be considered.
The patient's diarrhea began 1 month prior, shortly after having a milkshake at a fast food restaurant. The diarrhea was initially watery, occurred 8‐10 times per day, occasionally awakened him at night, and was associated with nausea. There was no mucus, blood, or steatorrhea until 1 day prior to presentation, when he developed epigastric pain and bloody stools. He denied any recent travel outside of Northern California and had no sick contacts. He had lost 10 pounds over the preceding month. He denied fevers, chills, vomiting, or jaundice, and had not taken antibiotics recently.
In the setting of chronic diarrhea, unintentional weight loss is an alarm feature but does not narrow the diagnostic possibilities significantly. The appearance of blood and pain on a single day after 1 month of symptoms renders their diagnostic value uncertain. For instance, rectal or hemorrhoidal bleeding would be a common occurrence after 1 month of frequent defecation. Sustained bloody stools might be seen in any form of erosive luminal disease, such as infection, inflammatory bowel disease, or neoplasm. Pain is compatible with inflammatory bowel disease, obstructing neoplasms, infections, or ischemia (eg, vasculitis). There are no fever or chills to support infection, and common gram‐negative enteric pathogens (such as Salmonella, Campylobacter, and Yersinia) usually do not produce symptoms for such an extended period. He has not taken antibiotics, which would predispose him to infection with Clostridum difficile, and he has no obvious exposure to parasites such as Entamoeba.
The patient had diabetes mellitus with microalbuminuria, chronic obstructive pulmonary disease, hypertension, hyperlipidemia, chronic low back pain, and gastritis, and had undergone a Billroth II procedure for a perforated gastric ulcer in the remote past. His medications included omeprazole, insulin glargine, simvastatin, lisinopril, amlodipine, and albuterol and beclomethasone metered‐dose inhalers. He had been married for 31 years, lived at home with his wife, was a former rigger in a shipyard and was on disability for chronic low back pain. He denied alcohol or intravenous drug use but had quit tobacco 5 years prior after more than 40 pack‐years of smoking. He had three healthy adult children and there was no family history of cancer, liver disease, or inflammatory bowel disease. There was no history of sexually transmitted diseases or unprotected sexual intercourse.
Bacterial overgrowth in the blind loop following a Billroth II operation can lead to malabsorption, but the diarrhea would not begin so abruptly this long after surgery. Medications are common causes of diarrhea. Proton‐pump inhibitors, by reducing gastric acidity, confer an increased risk of bacterial enteritis; they also are a risk factor for C difficile. Lisinopril may cause bowel angioedema months or years after initiation. Occult laxative use is a well‐recognized cause of chronic diarrhea and should also be considered. The most relevant element of his social history is the prolonged smoking and the attendant risk of cancer, although diarrhea is a rare paraneoplastic phenomenon.
On exam, temperature was 36.6C, blood pressure 125/78, pulse 88, respiratory rate 16 per minute, and oxygen saturation 97% while breathing room air. There was temporal wasting and mild scleral icterus, but no jaundice. Lungs were clear to auscultation and heart was regular in rate and rhythm without murmurs or gallops. There was no jugular venous distention. A large abdominal midline scar was present, bowel sounds were normoactive, and the abdomen was soft, nontender, and nondistended. The hard was regular in rate and rhythm the liver edge was 6 cm below costal margin; there was no splenomegaly. The patient was alert and oriented, with a normal neurologic exam.
The liver generally enlarges because of acute inflammation, congestion, or infiltration. Infiltration can be due to tumors, infections, hemochromatosis, amyloidosis, or sarcoidosis. A normal cardiac exam argues against hepatic congestion from right‐sided heart failure or pericardial disease.
The key elements of the case are diarrhea and hepatomegaly. Inflammatory bowel disease can be accompanied by sclerosing cholangitis, but this should not enlarge the liver. Mycobacterial infections and syphilis can infiltrate the liver and intestinal mucosa, causing diarrhea, but he lacks typical risk factors.
Malignancy is an increasing concern. Colon cancer commonly metastasizes to the liver and can occasionally be intensely secretory. Pancreatic cancer could account for these symptoms, especially if pancreatic exocrine insufficiency caused malabsorption. Various rare neuroendocrine tumors that arise in the pancreas can cause secretory diarrheas and liver metastases, such as carcinoid, VIPoma, and Zollinger‐Ellison syndrome.
Laboratory results revealed a serum sodium of 143 mmol/L, potassium 4.7 mmol/L, chloride 110 mmol/L, bicarbonate 25 mmol/L, urea nitrogen 24 mg/dL, and creatinine 2.5 mg/dL (baseline had been 1.2 mg/dL 2 months previously). Serum glucose was 108 mg/dL and calcium was 8.8 mg/dL. The total white blood cell count was 9300 per mm3 with a normal differential, hemoglobin was 14.4 g/dL, mean corpuscular volume was 87 fL, and the platelet count was normal. Total bilirubin was 3.7 mg/dL, and direct bilirubin was 3.1 mg/dL. Aspartate aminotransferase (AST) was 122 U/L (normal range, 831), alanine aminotransferase (ALT) 79 U/L (normal range, 731), alkaline phosphatase 1591 U/L (normal range, 39117), and gamma‐glutamyltransferase (GGT) 980 U/L (normal range, 57). Serum albumin was 2.5 mg/dL, prothrombin time was 16.4 seconds, and international normalized ratio (INR) was 1.6.
Urinalysis was normal except for trace hemoglobin, small bilirubin, and 70 mg/dL of protein; specific gravity was 1.007. Urine microscopy demonstrated no cells or casts. The ratio of protein to creatinine on a spot urine sample was less than 1. Chest x‐ray was normal. The electrocardiogram demonstrated sinus rhythm with an old right bundle branch block and normal QRS voltages.
The disproportionate elevation in alkaline phosphatase points to an infiltrative hepatopathy from a cancer originating in the gastrointestinal tract or infection. Other infiltrative processes such as sarcoidosis or amyloidosis usually have evidence of disease elsewhere before hepatic disease becomes apparent.
Mild proteinuria may be explained by diabetes. The specific gravity of 1.007 is atypical for dehydration and could suggest ischemic tubular injury. Although intrinsic renal diseases must continue to be entertained, hypovolemia (compounded by angiotensin‐converting enzyme [ACE] inhibitor use) is the leading explanation in light of the nondiagnostic renal studies. The preserved hemoglobin may simply indicate dehydration, but otherwise is somewhat reassuring in the context of bloody diarrhea.
The patient was admitted to the hospital. Three stool samples returned negative for C difficile toxin. No white cells were detected in the stool, and no ova or parasites were detected. Stool culture was negative for routine bacterial pathogens and for E coli O157. Tests for HIV and antinuclear antibodies (ANAs) and serologies for hepatitis A, B, and C were negative. Abdominal ultrasound demonstrated no intra‐ or extrahepatic bile duct dilatation; no hepatic masses were seen. Kidneys were normal in size and appearance without hydronephrosis. Computed tomography (CT) of the abdomen without intravenous contrast revealed normal‐appearing liver (with a 12‐cm span), spleen, biliary ducts, and pancreas, and there was no intra‐abdominal adenopathy.
The stool studies point away from infectious colitis. Infiltrative processes of the liver, including metastases, lymphoma, tuberculosis, syphilis, amyloidosis, and sarcoidosis, can be microscopic and therefore evade detection by ultrasound and CT scan. In conditions such as these, endoscopic retrograde cholangiopanccreatography/magnetic resonance cholangiopancreatography (ERCP/MRCP) or liver biopsy may be required. The CT is limited without contrast but does not suggest extrahepatic disease in the abdomen.
MRCP was performed, but was a technically suboptimal study due to the presence of ascites. The serum creatinine improved to 1.4 mg/dL over the next 4 days, and the patient's diarrhea decreased to two bowel movements daily with the use of loperamide. The patient was discharged home with outpatient gastroenterology follow‐up planned to discuss further evaluation of the abnormal liver enzymes.
Prior to being seen in the Gastroenterology Clinic, the patient's nonbloody diarrhea worsened. He felt weaker and continued to lose weight. He also noted new onset of bilateral lower face numbness and burning, which was followed by swelling of his lower lip 12 hours later. He returned to the hospital.
On examination, he was afebrile. His lower lip was markedly swollen and was drooping from his face. He could not move the lip to close his mouth. The upper lip and tongue were normal size and moved without restriction. Facial sensation was intact, but there was weakness when he attempted to wrinkle both of his brows and close his eyelids. The rest of his physical examination was unchanged.
The serum creatinine had risen to 3.6 mg/dL, and the complete blood count remained normal. Serum total bilirubin was 4.6 mg/dL, AST 87 U/L, ALT 76 U/L, and alkaline phosphatase 1910 U/L. The 24‐hour urine protein measurement was 86 mg.
Lip swelling suggests angioedema. ACE inhibitors are frequent offenders, and it would be important to know whether his lisinopril was restarted at discharge. ACE‐inhibitor angioedema can also affect the intestine, causing abdominal pain and diarrhea, but does not cause a systemic wasting illness or infiltrative hepatopathy. The difficulty moving the lip may reflect the physical effects of swelling, but generalized facial weakness supports a cranial neuropathy. Basilar meningitis may produce multiple cranial neuropathies, the etiologies of which are quite similar to the previously mentioned causes of infiltrative liver disease: sarcoidosis, syphilis, tuberculosis, or lymphoma.
The patient had not resumed lisinopril since his prior hospitalization. The lower lip swelling and paralysis persisted, and new sensory paresthesias developed over the right side of his chin. A consulting neurologist found normal language and speech and moderate dysarthria. Cranial nerve exam was normal except bilateral lower motor neuron facial nerve palsy was noted with bilateral facial droop, reduced strength of eyelid closure, and diminished forehead movement bilaterally; facial sensation was normal. Extremity motor exam revealed proximal iliopsoas muscle weakness bilaterally rated as 4/5 and was otherwise normal. Sensation to pinprick was diminished in a stocking/glove distribution. Deep‐tendon reflexes were normal and plantar response was down‐going bilaterally. Coordination was intact, Romberg was negative, and gait was slowed due to weakness.
Over the next several days, the patient continued to have diarrhea and facial symptoms. The serum total bilirubin increased to 14 mg/dL, alkaline phosphatase rose above 2,000 U/L, and serum creatinine increased to 5.5 mg/dL. Noncontrast CT scan of the head was normal.
Along with a mild peripheral sensory neuropathy, the exam indicates bilateral palsies of the facial nerve. Lyme disease is a frequent etiology, but this patient is not from an endemic area. I am most suspicious of bilateral infiltration of cranial nerve VII. I am thinking analogically to the numb chin syndrome, wherein lymphoma or breast cancer infiltration along the mental branch of V3 causes sensory loss, and perhaps these disorders can produce infiltrative facial neuropathy. At this point I am most concerned about lymphomatous meningitis with cranial nerve involvement. Cerebrospinal fluid (CSF) analysis (including cytology) would be informative.
Lumbar puncture demonstrated clear CSF with one white blood cell per mm3 and no red blood cells. Glucose was normal, and protein was 95.5 (normal range, 15‐45 mg/dL). Gram stain and culture for bacteria were negative, as were polymerase chain reaction (PCR) testing for herpes simplex virus, mycobacterial and fungal stains and cultures, and cytology. Transthoracic echocardiogram demonstrated severe concentric left ventricular (LV) hypertrophy, normal LV systolic function, and impaired LV relaxation. CT scan of the chest identified no adenopathy or other abnormality.
The CSF analysis does not support basilar meningitis, although the cytoalbuminologic dissociation makes me wonder whether there is some intrathecal antibody production or an autoimmune process we have yet to uncover. The absence of lymphadenopathy anywhere in the body and the negative CSF cytology now point away from lymphoma. As the case for lymphoma or an infection diminishes, systemic amyloidosis rises to the top of possibilities in this afebrile man who is losing weight, has infiltrative liver and nerve abnormalities, renal failure, cardiac enlargement, and suspected gastrointestinal luminal abnormality. Although the echocardiographic findings are most likely explained by hypertension, they are compatible with amyloid infiltration. A tissue specimen is needed, and either colonoscopy or liver biopsy should be suitable.
A pathologist performed a fat pad biopsy that demonstrated scant congophilic and birefringent material associated with blood vessels, suggestive of amyloid (Fig. 1). Colonoscopy demonstrated normal mucosa, and a rectal biopsy revealed congophilic material within the blood vessels consistent with amyloid (Fig. 2). No monoclonal band was present on serum protein electrophoresis. Urine protein electrophoresis identified a homogenous band in the gamma region, and urine kappa and lambda free light chains were increased: kappa was 10.7 mg/dL (normal range, 2), and lambda was 4.25 mg/dL (normal range, 2).
After extensive discussion among the patient, his wife, and a palliative care physician, the patient declined chemotherapy and elected to go home. Two days after discharge (7 weeks after his initial admission for diarrhea) he died in his sleep at home. Permission for a postmortem examination was not granted.
Discussion
Amyloidosis refers to abnormal extracellular deposition of fibril. There are many types of amyloidosis including primary amyloidosis (AL amyloidosis), secondary amyloidosis (AA amyloidosis), and hereditary causes. Systemic AL amyloidosis is a rare plasma cell disorder characterized by misfolding of insoluble extracellular fibrillar proteins derived from immunoglobulin light chains. These insoluble proteins typically deposit in the kidney, heart, and nervous system.1 Although the mechanism of organ dysfunction is debated, deposition of these proteins may disrupt the tissue architecture by interacting with local receptors and causing apoptosis.1
Table 1 indicates the most common findings in patients with AL amyloidosis.2 While our patient ultimately developed many common findings of AL amyloidosis, several features were atypical, including the marked hyperbilirubinemia, profound diarrhea, and bilateral facial diplegia.
| Organ Involvement | Incidence of Organ Involvement (%) | Symptoms | Signs | Laboratory/Test Finding |
|---|---|---|---|---|
| ||||
| General | Malaise, weight loss | |||
| Renal | 33 | Fatigue | Peripheral edema | Proteinuria with or without renal insufficiency, pleural effusion, hypercholesterolemia |
| Cardiac | 20 | Palpitations, dyspnea | Elevated jugular venous pressure, S3, peripheral edema, hepatomegaly | Low‐voltage or atrial fibrillation on electrocardiogram; echocardiogram: thickened ventricles, dilated atria |
| Neurological | 20 | Paresthesias, numbness, weakness, autonomic insufficiency | Carpal tunnel syndrome, postural hypotension | |
| Gastrointestinal and Hepatic | 16 | Diarrhea, nausea, weight loss | Macroglossia, hepatomegaly | Elevated alkaline phosphatase |
| Hematology | Rare | Bleeding | Periorbital purpura (raccoon eyes) | Prolonged prothrombin time, Factor X deficiency |
Up to 70% of patients with amyloidosis will have detectable liver deposits, typically involving portal vessels, portal stroma, central vein, and sinusoidal parenchyma.3 Clinically overt hepatic dysfunction from amyloid is less frequent,4 and the most characteristic findings are hepatomegaly with a markedly elevated serum alkaline phosphatase concentration; jaundice is rare. Palpable hepatic enlargement without abnormal liver enzymes should be interpreted with caution. The finding of a palpable liver edge correlates poorly with frank hepatomegaly, with a positive likelihood ratio of just 1.7.5 In the patient under discussion, suspected hepatomegaly was not confirmed on a subsequent CT scan. Nonetheless, the elevated alkaline phosphatase represented an important clue to potential infiltrative liver disease. In a series of amyloidosis patients from the Mayo Clinic, 81% had hepatomegaly on physical exam, and the mean alkaline phosphatase level was 1,029 U/L (normal, 250 U/L), while the mean serum bilirubin and AST levels were only modestly elevated, at 3.2 mg/dL and 72 U/L respectively. The prothrombin time was prolonged in 35% of patients.
Upper gastrointestinal tract involvement by AL amyloid may be found in up to a third of cases at autopsy, but clinically significant gastrointestinal features are seen in fewer than 5% of patients.6 Predominant intestinal manifestations are unintentional weight loss (average 7 kg) and diarrhea, nonspecific features that result in delayed diagnosis for a median of 7 months after symptom onset.7 Diarrhea in AL amyloid may stem from several mechanisms: small intestine mucosal infiltration, steatorrhea from pancreatic insufficiency, autonomic neuropathy leading to pseudo‐obstruction and bacterial overgrowth, bile acid malabsorption, or rapid transit time. Diarrhea in AL amyloid is often resistant to treatment and may be the primary cause of death.7
Systemic amyloidosis commonly produces peripheral neuropathies. Involvement of small unmyelinated fibers causes paresthesias and progressive sensory loss in a pattern that is usually distal, symmetric, and progressive.6, 9 Our patient presented with bilateral sensory paresthesias of the chin, suggesting the numb chin syndrome (NCS). NCS is characterized by facial numbness along the distribution of the mental branch of the trigeminal nerve. While dental disorders and infiltration from malignant tumors (mostly lung and breast cancer) account for most cases, amyloidosis and other infiltrative disorders are known to cause NCS as well.10, 11 Our patient's sensory paresthesias may have represented amyloid infiltration of peripheral nerves.
With the exception of carpal tunnel syndrome, motor or cranial neuropathy is uncommon in amyloid, and when present usually heralds advanced disease.12 Descriptions of bilateral facial weakness, also known as facial diplegia, from amyloidosis are limited to case reports.1315 Other causes of this rare finding include sarcoidosis, Guillain‐Barr syndrome, and Lyme disease.16
The diagnosis of primary amyloidosis requires histologic evidence of amyloid from a tissue biopsy specimen (demonstrating positive Congo red staining and pathognomonic green birefringence under cross‐polarized light microscopy), and the presence of a clonal plasma cell disorder. While biopsy of an affected organ is diagnostic, more easily obtained samples such as fat pad biopsy and rectal biopsy yield positive results in up to 80% of cases.2 Serum and urine protein electrophoresis with immunofixation identify an underlying plasma cell disorder in 90% of cases of primary amyloidosis. When these tests are inconclusive, serum or urine free light chain assays or bone marrow aspirate and biopsy are useful aids to detect underlying plasma cell dyscrasia.2 AL amyloidosis is a progressive disease with median survival of about 12 years.8 Poorer prognosis is associated with substantial echocardiographic findings, autonomic neuropathy, and liver involvement.2 Hyperbilirubinemia is associated with a poor prognosis, with a median survival of 8.5 months.4 Proteinuria or peripheral neuropathy portends a less ominous course.6
Treatment goals include reducing production and deposition of fibril proteins and contending with organ dysfunction (eg, congestive heart failure [CHF] management). Selected patients with AL amyloidosis may be candidates for high‐dose melphalan and autologous stem cell transplantation.
It would not be reasonable for clinicians to suspect amyloidosis in cases of diarrhea until two conditions are met: 1) the absence of evidence for the typical etiologies of diarrhea; and 2) the evolving picture of an infiltrative disorder. The latter was heralded by the elevated alkaline phosphatase, and was supported by the subsequent multiorgan involvement. Conceptualizing the disease as infiltrative still required a diligent exclusion of infection and invasive tumor cells, which invade disparate organs far more commonly than amyloidosis. Their absence and the organ pattern that is typical of AL amyloidosis (heart, kidney, and peripheral nerve involvement) allowed the discussant to reason by analogy that amyloidosis was also responsible for the most symptomatic phenomena, namely, the diarrhea and facial diplegia (and numb chin syndrome).
Key Teaching Points
-
Hospitalists should consider systemic amyloidosis in cases of unexplained diarrhea when other clinical features of AL amyloidosis are present, including nephrotic syndrome with or without renal insufficiency, cardiomyopathy, peripheral neuropathy, and hepatomegaly.
-
Hepatic amyloidosis should be suspected when weight loss, hepatomegaly, and elevated alkaline phosphatase are present. Although jaundice is rare in amyloidosis, liver involvement and hyperbilirubinemia portend a poorer prognosis.
-
Numb chin syndrome and bilateral facial diplegia are rare manifestations of AL amyloid deposition in peripheral nerves.
- ,.Molecular mechanisms of amyloidosis.N Engl J Med.2003;349(6):583–596.
- Guidelines Working Group of UK Myeloma Forum; British Committee for Standards in Haematology, British Society for Haematology.Guidelines on the diagnosis and management of AL amyloidosis.Br J Haematol.2004;125:681–700.
- ,.Hepatic amyloidosis: morphologic differences between systemic AL and AA types.Hum Pathol.1991;22(9):904–907.
- ,,, et al.Primary (AL) hepatic amyloidosis clinical features and natural history in 98 patients.Medicine.2003;82(5):291–298.
- .Evidence‐Based Physical Diagnosis.Philadelphia, PA:WB Saunders;2001:595–599.
- ,,, et al.Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis.Am J Hematol.2005;79:319–328.
- .Primary (AL) amyloidosis with gastrointestinal involvement.Scand J Gastroenterol.2009;44(6):708–711.
- ,.Gastrointestinal manifestations of amyloid.Am J Gastroenterol.2008;103:776–787.
- ,.Primary systemic amyloidosis: clinical and laboratory features in 474 cases.Semin Hematol.1995;32:45–59.
- ,,,,.Chin numbness: a symptom that should not be underestimated: a review of 12 cases.Am J Med Sci.2009;337:407–410.
- .Numb chin syndrome: a possible clue to serious illness.Hosp Physician.2000;54–56.
- .Autonomic peripheral neuropathy.Neurol Clin.2007;25:277–301.
- ,.Facial diplegia due to amyloidosis.South Med J.1986;79(11):1458–1459.
- ,,,,.Familial amyloidosis with cranial neuropathy and corneal lattice dystrophy.Neurology.1986;36:432–435.
- ,,.Crainal neuropathy associated with primary amyloidosis.Ann Neurol.1991;29:451–454.
- .Bilateral seventh nerve palsy: analysis of 43 cases and review of the literature.Neurology.1994;44:1198–202.
- ,.Molecular mechanisms of amyloidosis.N Engl J Med.2003;349(6):583–596.
- Guidelines Working Group of UK Myeloma Forum; British Committee for Standards in Haematology, British Society for Haematology.Guidelines on the diagnosis and management of AL amyloidosis.Br J Haematol.2004;125:681–700.
- ,.Hepatic amyloidosis: morphologic differences between systemic AL and AA types.Hum Pathol.1991;22(9):904–907.
- ,,, et al.Primary (AL) hepatic amyloidosis clinical features and natural history in 98 patients.Medicine.2003;82(5):291–298.
- .Evidence‐Based Physical Diagnosis.Philadelphia, PA:WB Saunders;2001:595–599.
- ,,, et al.Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis.Am J Hematol.2005;79:319–328.
- .Primary (AL) amyloidosis with gastrointestinal involvement.Scand J Gastroenterol.2009;44(6):708–711.
- ,.Gastrointestinal manifestations of amyloid.Am J Gastroenterol.2008;103:776–787.
- ,.Primary systemic amyloidosis: clinical and laboratory features in 474 cases.Semin Hematol.1995;32:45–59.
- ,,,,.Chin numbness: a symptom that should not be underestimated: a review of 12 cases.Am J Med Sci.2009;337:407–410.
- .Numb chin syndrome: a possible clue to serious illness.Hosp Physician.2000;54–56.
- .Autonomic peripheral neuropathy.Neurol Clin.2007;25:277–301.
- ,.Facial diplegia due to amyloidosis.South Med J.1986;79(11):1458–1459.
- ,,,,.Familial amyloidosis with cranial neuropathy and corneal lattice dystrophy.Neurology.1986;36:432–435.
- ,,.Crainal neuropathy associated with primary amyloidosis.Ann Neurol.1991;29:451–454.
- .Bilateral seventh nerve palsy: analysis of 43 cases and review of the literature.Neurology.1994;44:1198–202.
Hospitalist Down Under
antipodes (n.): 1. two points diametrically opposite on the globe; 2. a nickname for New Zealand.
With that definition in mind, this hospitalist decided to seek a working sabbatical—not so much over dissatisfaction with my job back home, but to see how the other side lives with nationalized healthcare. Of course, moving to the beach in undeniably beautiful New Zealand never hurts, either. When it comes to government involvement and healthcare, the U.S. is in a distinct minority globally; New Zealand’s healthcare delivery system, while not diametrically opposite, offers some fascinating differences.
Last July, my wife and I, along with our two young boys, relocated to Ohope Beach, a quaint resort community on the northeast coast of New Zealand about 100 miles south of the nation's largest and most well-known city, Auckland. I am now six months into a year’s assignment at Whakatane Hospital.
I do not claim to be an expert on the comparative aspects of national healthcare systems, and some of my observations are directly related to my move from an urban to a rural setting. Still, the differences between the health systems in New Zealand and the U.S. are striking. Compared to Americans, New Zealanders spend less money on healthcare, their medications cost a third of what ours do, they undergo less testing, and they spend less time in the ICU, yet they live longer. Additionally, the public sector pays for the health insurance of every single New Zealand resident.

Let’s dispense with a few dry facts:
- Healthcare spending as a percentage of GDP: in the U.S., 16%; in New Zealand, 8%;1
- Per capita spending on healthcare in U.S. dollars: $7,500 for Americans; $2,700 for New Zealanders;
- Percent of healthcare spending by public sector: U.S., 45%; New Zealand, 80%1;
- Average life expectancy in the U.S.: 78.2 years (38th in the world); average in New Zealand: 80.2 years (13th in the world);2 and
- World ranking in infant mortality: U.S., 33rd; New Zealand, 27th.2
How are those figures possible? Does New Zealand employ death panels? No. Is there something in the water (e.g. statins and ACEs)? No. Is everyone a non-GMO, fair-trade, shade-grown, sustainably harvested vegan monk triathlete? Hmm.
Anyway, here are a few observations—from a hospitalist’s point of view.
Hospitalist by Another Name
My job title here at 100-bed Whakatane Hospital is “consultant physician.” The term “hospitalist” is not common, but it is exactly the job the consultant physician performs. Inpatient ward rounds make up the majority of the consultant’s role, and the only outpatient responsibility is a twice-weekly clinic to see patients referred from primary-care physicians (still called GPs here) for clinical questions. Because it’s a public hospital, we have house officers and junior physicians as well. Roughly, that means that the consultant examines the patient and formulates the plan, the junior physician does the new intake, and the house officer does all the writing.
As for the team, it’s a bit like working at the United Nations. So far, I have met physicians from New Zealand, Australia, Ireland, Sri Lanka, Spain, Jordan, Iraq, the United Kingdom, India, Zambia, South Africa, and, of course, America. Thank goodness we—and the patients—all speak varying degrees of English. It’s certainly a bit odd to find yourself misunderstood by someone else speaking your (and their) native tongue.
The work schedule is quite reasonable, or, as I have come to call it, “civilized.” It is a 40-hour workweek, 8 a.m. to 4 p.m. daily, with at least 12 hours of each week dedicated to nonclinical time in the form of reading, research, and teaching. The daily patient census per team is about 12, and the call (or “take”) ratio is about 1:4. With no nights and rare weekends, the whole experience is a welcome change of pace. For the Kiwis, this pace is perfectly accepted and expected, with little pressure to work harder, longer, or faster. Teatime is 10 in the morning, and breaking for a cuppa is just part of the job.
In switching from the private sector to the public sector, I braced myself for a significant cut in pay. As it turns out, the compensation is actually quite reasonable when matched to the schedule. Based on the current exchange rate of $1 USD equal to $0.75 NZD, the annual salary is about $165,000 USD. You also receive six weeks of vacation, two weeks of CME leave, and a $12,000 CME allowance. All in all, it’s a nice package. (Unfortunately, the pay in Australia is higher, and many Kiwi physicians choose to practice over there.) The resulting shortage of locally educated and trained physicians in New Zealand explains the ongoing need for overseas physicians.

Fewer Resources, So Choose Wisely
The hospital is part of a larger network called a district health board (DHB). The DHBs receive government funding based on population, then must decide how to spend the money. For that reason, there are only a few hospitals that include subspecialties. Smaller sites, such as Whakatane, have no subspecialists at all.
That also means we have no MRI machine, no cardiac catheter lab, no echocardiography, no hemodialysis. So, to do one of these procedures, it involves speaking to a specialist at a tertiary site and requesting a transfer, which, in turn, forces you to explain and justify your request. All of a sudden, the history and the physical exam regain their proper importance rather than being subverted to ordering a confirmatory imaging study or specialist consultation. It’s much different than back home, where the indication to perform coronary angiography can be the presence of a cardiologist and a groin in the same room.
In addition, the community GPs cannot order such testing as exercise treadmills or CT scans without going through a consultant first. Although this inevitably leads to delays, it also means some thought is introduced into the process.
Dialysis is another example. In New Zealand, more than 40% of patients receive peritoneal dialysis, and many of those who get hemodialysis do so at home. The average cost per patient per year is $25,000. By contrast, 93% of U.S. dialysis patients are on hemodialysis at a cost of $67,000 per patient per year.3 The outcomes for the two treatments are generally accepted to be the same; one just costs a whole lot more.

This Is the Only Formulary
U.S. physicians are all too familiar with MRSA, VRE, C. diff, and their ilk. Soon we may have to deal with NDM-1. Over and over again, we hear about how we should curtail our antibiotic use, yet little is actually done. New Zealand has strict antibiotic guidelines in accordance with a patient’s diagnosis. Ordering antibiotics outside the protocol is not accepted, unless you can convince the infectious-disease physician otherwise. It is an eminently reasonable practice that has simply never gained broad acceptance in America. What is the result of the New Zealand practice mandate? The hospital where I work has experienced only rare MRSA cases (9% of all S. aureus vs. 35% in the U.S.), no VRE, and zero cases of C. diff in the past nine months.
New Zealand has one drug-purchasing entity: Pharmac. The entity negotiates prices with suppliers and writes the formulary. If a drug is not on the national formulary, it is not available. Period.
Most drugs are fully subsidized; some drugs are partially subsidized. If it is available and subsidized, a 90-day prescription costs $3. All in all, Kiwi pharmaceutical costs are about 30% of the costs in the U.S. The choices are fewer, yet patients still receive appropriate pharmacologic treatment.
Every hospitalist has worked with an electronic health record (EHR) in one form or another, but one of the most frustrating things in the States is the lack of interface between EHR in different health systems. Such disconnect leads to an extraordinary amount of effort not to duplicate tests for patients who frequent more than one hospital. Often, these patients are high consumers of healthcare at baseline, and duplicating tests only makes it worse.
New Zealand has one computer system that allows the GPs and the hospitalists to view all the labs and imaging results together, in the same system. No matter who orders the test, it all comes back to the same place. It might seem small, but it makes a tremendous difference to the management of the patient. One could argue that defensive medicine is good practice, but we’ve all seen the extra study get ordered somewhere along the line. I won’t get into the details of malpractice insurance (the editors said stop at 2,000 words, and I could go on for 200,000), but back home in Colorado, I paid about $20,000 a year for malpractice insurance. Here, it’s $1,200.
New Zealand has a no-fault compensation system in which injured patients apply for government-funded compensation and thus give up the right to litigation. Most claims are processed within weeks, and all decisions are final within nine months. The patients here know that they have recourse for a bad outcome, and the physicians practice in a manner according to clinical judgment, rather than trying to avoid being sued.

Confusion and Delay
You might get the sense that practicing medicine in New Zealand has been a refreshing change of pace, and you would be right. My physical exam skills (what was left of them) are starting to return. I think long and hard before ordering an imaging study or requesting consultation. I order antibiotics according to the guidelines. I only prescribe medicines that are on the formulary. The care given to the patients by the staff and the knowledge exhibited by our physicians are quite good.
Still, this system is by no means perfect. The delays in care can be excruciating in both the urgent and routine settings. For example, my first patient on my first day of work had an acute myocardial infarction. The ED physician admitted him to the hospital at midnight, but no one on our team had met the patient when I arrived for work the next day. At 8 a.m., he had dynamic EKG changes, a positive troponin, ongoing chest pain, and a systolic BP of about 90 mm/HG. Back home, I never would have met the man, as he would have gone pretty much straight to the cath lab—with the cardiologists—within 60 minutes, even at midnight. Here, we had to call the hospital that does interventions, arrange a bed (hoping one was available), then put him in an ambulance for a four-hour drive on a two-lane road to receive a stent for his 100% coronary lesion.
The average wait for transfer for acute coronary angiography is about five days. The average wait for routine outpatient echocardiography is two years. Yes, two years.
Ultimately, the right thing gets done, but sometimes way too slowly. Thus, there is a smaller, parallel private insurance system, but people view it as optional and only use it when they want to speed up the schedule.
A Few Things I Have No Answer For
I have yet to admit someone with alcohol withdrawal—this in a country with a higher per capita alcohol consumption. I have yet to order a PCA pump for a medical patient. Low-back pain is managed by surgery, and a pneumothorax goes to the medical service. There is no Pyxis equivalent, just an open cupboard in the ICU where the meds are kept. There is no separate page for physician orders; the staff is expected to read the notes. In an old physical plant such as Whakatane, there are no private bathrooms for patients—it’s down the hall. Oh, and it’s four patients to a room, with no television, no Internet, and no telephone. There are plans for a new hospital in a few years’ time.
Culturally, the patients and their families are more accepting of the fragile nature of old age, with a strong desire to avoid unnecessary interventions at the end of life. There is a robust community hospice program that helps patients remain comfortable at home. As a local colleague of mine explains, the Kiwis are uniformly grateful for the care they receive, even if it means sharing a room with another patient (or three).

Final Observations
Healthcare delivery in New Zealand is different from what we see in the U.S. Obviously, the vast majority of healthcare delivered in New Zealand is through the public sector. No matter which direction U.S. healthcare reform goes next, it’s highly improbable it will ever resemble New Zealand’s system.
One quote that resonates comes from the medical practice handbook, which is given to all newly registered physicians in New Zealand: “Doctors have a responsibility to the community to foster the proper use of resources—in particular, by making efforts to use resources efficiently, consistent with good patient care.”4 It’s sound advice.
Changes are coming, and hospitalists everywhere are in a unique position to gain knowledge and lead the change. In the U.S. or in New Zealand, as a hospitalist or a physician consultant, medicine is a fascinating field of practice.
I hope you enjoyed reading about one hospitalist’s observations from New Zealand. Time for tea. TH
M-A Williams has been a practicing hospitalist in Denver since 1999. He worked with the same company (Inpatient Services, which then merged with Sound Physicians) for 11 years until departing for New Zealand. He is a member of Team Hospitalist.
References
- Organisation for Economic Cooperation and Development website. Available at: www.oecd.org. Accessed Dec. 31, 2010.
- The United Nations Statistical Division website. Available at: http://unstats.un.org/unsd/default.htm. Accessed Jan. 4, 2011.
- Ashton T, Marshall RM. The organization and financing of dialysis and kidney transplantation services in New Zealand. Int J Healthcare Finance and Econ. 2007;7:233-252.
- Cole’s Medical Practice in New Zealand (2009). Ed., Ian St. George. Medical Council of New Zealand: Wellington:18.
New Zealand Fast Facts

Demographics
- New Zealand pop.: 4.3 million
- Capital: Wellington
- Largest city: Auckland, 1.4 million pop.
- Whakatane District pop.: 32,814
- Ohope Beach pop.: 2,760
Information
- Whakatane District in the Bay of Plenty features 34 miles of sandy beaches.
- Ohope Beach is a popular vacation spot. White Island, an active volcano offshore, is a world-renowned tourism attraction.
- The kiwi, NZ’s national emblem, is a flightless bird with hair-like feathers and a long, slender bill, which it uses to pull worms and insects out of the ground.
- New Zealanders often refer to themselves as Kiwis, and the term is also used as a short form for the famous kiwi fruit. The NZ dollar is referred to as the kiwi.
- Whakatane summer temperatures average in the 80s, and winter months in the 50s. The district records New Zealand’s high temperature about 55 days a year.
- New Zealand’s seasons are the reverse of the Northern Hemisphere.
Health Stats
- Spending as a percentage of GDP: 8%
- Per capita spending on healthcare: $2,700
- Average life expectancy: 80.2 years (13th in the world)
antipodes (n.): 1. two points diametrically opposite on the globe; 2. a nickname for New Zealand.
With that definition in mind, this hospitalist decided to seek a working sabbatical—not so much over dissatisfaction with my job back home, but to see how the other side lives with nationalized healthcare. Of course, moving to the beach in undeniably beautiful New Zealand never hurts, either. When it comes to government involvement and healthcare, the U.S. is in a distinct minority globally; New Zealand’s healthcare delivery system, while not diametrically opposite, offers some fascinating differences.
Last July, my wife and I, along with our two young boys, relocated to Ohope Beach, a quaint resort community on the northeast coast of New Zealand about 100 miles south of the nation's largest and most well-known city, Auckland. I am now six months into a year’s assignment at Whakatane Hospital.
I do not claim to be an expert on the comparative aspects of national healthcare systems, and some of my observations are directly related to my move from an urban to a rural setting. Still, the differences between the health systems in New Zealand and the U.S. are striking. Compared to Americans, New Zealanders spend less money on healthcare, their medications cost a third of what ours do, they undergo less testing, and they spend less time in the ICU, yet they live longer. Additionally, the public sector pays for the health insurance of every single New Zealand resident.
Let’s dispense with a few dry facts:
- Healthcare spending as a percentage of GDP: in the U.S., 16%; in New Zealand, 8%;1
- Per capita spending on healthcare in U.S. dollars: $7,500 for Americans; $2,700 for New Zealanders;
- Percent of healthcare spending by public sector: U.S., 45%; New Zealand, 80%1;
- Average life expectancy in the U.S.: 78.2 years (38th in the world); average in New Zealand: 80.2 years (13th in the world);2 and
- World ranking in infant mortality: U.S., 33rd; New Zealand, 27th.2
How are those figures possible? Does New Zealand employ death panels? No. Is there something in the water (e.g. statins and ACEs)? No. Is everyone a non-GMO, fair-trade, shade-grown, sustainably harvested vegan monk triathlete? Hmm.
Anyway, here are a few observations—from a hospitalist’s point of view.
Hospitalist by Another Name
My job title here at 100-bed Whakatane Hospital is “consultant physician.” The term “hospitalist” is not common, but it is exactly the job the consultant physician performs. Inpatient ward rounds make up the majority of the consultant’s role, and the only outpatient responsibility is a twice-weekly clinic to see patients referred from primary-care physicians (still called GPs here) for clinical questions. Because it’s a public hospital, we have house officers and junior physicians as well. Roughly, that means that the consultant examines the patient and formulates the plan, the junior physician does the new intake, and the house officer does all the writing.
As for the team, it’s a bit like working at the United Nations. So far, I have met physicians from New Zealand, Australia, Ireland, Sri Lanka, Spain, Jordan, Iraq, the United Kingdom, India, Zambia, South Africa, and, of course, America. Thank goodness we—and the patients—all speak varying degrees of English. It’s certainly a bit odd to find yourself misunderstood by someone else speaking your (and their) native tongue.
The work schedule is quite reasonable, or, as I have come to call it, “civilized.” It is a 40-hour workweek, 8 a.m. to 4 p.m. daily, with at least 12 hours of each week dedicated to nonclinical time in the form of reading, research, and teaching. The daily patient census per team is about 12, and the call (or “take”) ratio is about 1:4. With no nights and rare weekends, the whole experience is a welcome change of pace. For the Kiwis, this pace is perfectly accepted and expected, with little pressure to work harder, longer, or faster. Teatime is 10 in the morning, and breaking for a cuppa is just part of the job.
In switching from the private sector to the public sector, I braced myself for a significant cut in pay. As it turns out, the compensation is actually quite reasonable when matched to the schedule. Based on the current exchange rate of $1 USD equal to $0.75 NZD, the annual salary is about $165,000 USD. You also receive six weeks of vacation, two weeks of CME leave, and a $12,000 CME allowance. All in all, it’s a nice package. (Unfortunately, the pay in Australia is higher, and many Kiwi physicians choose to practice over there.) The resulting shortage of locally educated and trained physicians in New Zealand explains the ongoing need for overseas physicians.
Fewer Resources, So Choose Wisely
The hospital is part of a larger network called a district health board (DHB). The DHBs receive government funding based on population, then must decide how to spend the money. For that reason, there are only a few hospitals that include subspecialties. Smaller sites, such as Whakatane, have no subspecialists at all.
That also means we have no MRI machine, no cardiac catheter lab, no echocardiography, no hemodialysis. So, to do one of these procedures, it involves speaking to a specialist at a tertiary site and requesting a transfer, which, in turn, forces you to explain and justify your request. All of a sudden, the history and the physical exam regain their proper importance rather than being subverted to ordering a confirmatory imaging study or specialist consultation. It’s much different than back home, where the indication to perform coronary angiography can be the presence of a cardiologist and a groin in the same room.
In addition, the community GPs cannot order such testing as exercise treadmills or CT scans without going through a consultant first. Although this inevitably leads to delays, it also means some thought is introduced into the process.
Dialysis is another example. In New Zealand, more than 40% of patients receive peritoneal dialysis, and many of those who get hemodialysis do so at home. The average cost per patient per year is $25,000. By contrast, 93% of U.S. dialysis patients are on hemodialysis at a cost of $67,000 per patient per year.3 The outcomes for the two treatments are generally accepted to be the same; one just costs a whole lot more.
This Is the Only Formulary
U.S. physicians are all too familiar with MRSA, VRE, C. diff, and their ilk. Soon we may have to deal with NDM-1. Over and over again, we hear about how we should curtail our antibiotic use, yet little is actually done. New Zealand has strict antibiotic guidelines in accordance with a patient’s diagnosis. Ordering antibiotics outside the protocol is not accepted, unless you can convince the infectious-disease physician otherwise. It is an eminently reasonable practice that has simply never gained broad acceptance in America. What is the result of the New Zealand practice mandate? The hospital where I work has experienced only rare MRSA cases (9% of all S. aureus vs. 35% in the U.S.), no VRE, and zero cases of C. diff in the past nine months.
New Zealand has one drug-purchasing entity: Pharmac. The entity negotiates prices with suppliers and writes the formulary. If a drug is not on the national formulary, it is not available. Period.
Most drugs are fully subsidized; some drugs are partially subsidized. If it is available and subsidized, a 90-day prescription costs $3. All in all, Kiwi pharmaceutical costs are about 30% of the costs in the U.S. The choices are fewer, yet patients still receive appropriate pharmacologic treatment.
Every hospitalist has worked with an electronic health record (EHR) in one form or another, but one of the most frustrating things in the States is the lack of interface between EHR in different health systems. Such disconnect leads to an extraordinary amount of effort not to duplicate tests for patients who frequent more than one hospital. Often, these patients are high consumers of healthcare at baseline, and duplicating tests only makes it worse.
New Zealand has one computer system that allows the GPs and the hospitalists to view all the labs and imaging results together, in the same system. No matter who orders the test, it all comes back to the same place. It might seem small, but it makes a tremendous difference to the management of the patient. One could argue that defensive medicine is good practice, but we’ve all seen the extra study get ordered somewhere along the line. I won’t get into the details of malpractice insurance (the editors said stop at 2,000 words, and I could go on for 200,000), but back home in Colorado, I paid about $20,000 a year for malpractice insurance. Here, it’s $1,200.
New Zealand has a no-fault compensation system in which injured patients apply for government-funded compensation and thus give up the right to litigation. Most claims are processed within weeks, and all decisions are final within nine months. The patients here know that they have recourse for a bad outcome, and the physicians practice in a manner according to clinical judgment, rather than trying to avoid being sued.
Confusion and Delay
You might get the sense that practicing medicine in New Zealand has been a refreshing change of pace, and you would be right. My physical exam skills (what was left of them) are starting to return. I think long and hard before ordering an imaging study or requesting consultation. I order antibiotics according to the guidelines. I only prescribe medicines that are on the formulary. The care given to the patients by the staff and the knowledge exhibited by our physicians are quite good.
Still, this system is by no means perfect. The delays in care can be excruciating in both the urgent and routine settings. For example, my first patient on my first day of work had an acute myocardial infarction. The ED physician admitted him to the hospital at midnight, but no one on our team had met the patient when I arrived for work the next day. At 8 a.m., he had dynamic EKG changes, a positive troponin, ongoing chest pain, and a systolic BP of about 90 mm/HG. Back home, I never would have met the man, as he would have gone pretty much straight to the cath lab—with the cardiologists—within 60 minutes, even at midnight. Here, we had to call the hospital that does interventions, arrange a bed (hoping one was available), then put him in an ambulance for a four-hour drive on a two-lane road to receive a stent for his 100% coronary lesion.
The average wait for transfer for acute coronary angiography is about five days. The average wait for routine outpatient echocardiography is two years. Yes, two years.
Ultimately, the right thing gets done, but sometimes way too slowly. Thus, there is a smaller, parallel private insurance system, but people view it as optional and only use it when they want to speed up the schedule.
A Few Things I Have No Answer For
I have yet to admit someone with alcohol withdrawal—this in a country with a higher per capita alcohol consumption. I have yet to order a PCA pump for a medical patient. Low-back pain is managed by surgery, and a pneumothorax goes to the medical service. There is no Pyxis equivalent, just an open cupboard in the ICU where the meds are kept. There is no separate page for physician orders; the staff is expected to read the notes. In an old physical plant such as Whakatane, there are no private bathrooms for patients—it’s down the hall. Oh, and it’s four patients to a room, with no television, no Internet, and no telephone. There are plans for a new hospital in a few years’ time.
Culturally, the patients and their families are more accepting of the fragile nature of old age, with a strong desire to avoid unnecessary interventions at the end of life. There is a robust community hospice program that helps patients remain comfortable at home. As a local colleague of mine explains, the Kiwis are uniformly grateful for the care they receive, even if it means sharing a room with another patient (or three).
Final Observations
Healthcare delivery in New Zealand is different from what we see in the U.S. Obviously, the vast majority of healthcare delivered in New Zealand is through the public sector. No matter which direction U.S. healthcare reform goes next, it’s highly improbable it will ever resemble New Zealand’s system.
One quote that resonates comes from the medical practice handbook, which is given to all newly registered physicians in New Zealand: “Doctors have a responsibility to the community to foster the proper use of resources—in particular, by making efforts to use resources efficiently, consistent with good patient care.”4 It’s sound advice.
Changes are coming, and hospitalists everywhere are in a unique position to gain knowledge and lead the change. In the U.S. or in New Zealand, as a hospitalist or a physician consultant, medicine is a fascinating field of practice.
I hope you enjoyed reading about one hospitalist’s observations from New Zealand. Time for tea. TH
M-A Williams has been a practicing hospitalist in Denver since 1999. He worked with the same company (Inpatient Services, which then merged with Sound Physicians) for 11 years until departing for New Zealand. He is a member of Team Hospitalist.
References
- Organisation for Economic Cooperation and Development website. Available at: www.oecd.org. Accessed Dec. 31, 2010.
- The United Nations Statistical Division website. Available at: http://unstats.un.org/unsd/default.htm. Accessed Jan. 4, 2011.
- Ashton T, Marshall RM. The organization and financing of dialysis and kidney transplantation services in New Zealand. Int J Healthcare Finance and Econ. 2007;7:233-252.
- Cole’s Medical Practice in New Zealand (2009). Ed., Ian St. George. Medical Council of New Zealand: Wellington:18.
New Zealand Fast Facts
Demographics
- New Zealand pop.: 4.3 million
- Capital: Wellington
- Largest city: Auckland, 1.4 million pop.
- Whakatane District pop.: 32,814
- Ohope Beach pop.: 2,760
Information
- Whakatane District in the Bay of Plenty features 34 miles of sandy beaches.
- Ohope Beach is a popular vacation spot. White Island, an active volcano offshore, is a world-renowned tourism attraction.
- The kiwi, NZ’s national emblem, is a flightless bird with hair-like feathers and a long, slender bill, which it uses to pull worms and insects out of the ground.
- New Zealanders often refer to themselves as Kiwis, and the term is also used as a short form for the famous kiwi fruit. The NZ dollar is referred to as the kiwi.
- Whakatane summer temperatures average in the 80s, and winter months in the 50s. The district records New Zealand’s high temperature about 55 days a year.
- New Zealand’s seasons are the reverse of the Northern Hemisphere.
Health Stats
- Spending as a percentage of GDP: 8%
- Per capita spending on healthcare: $2,700
- Average life expectancy: 80.2 years (13th in the world)
antipodes (n.): 1. two points diametrically opposite on the globe; 2. a nickname for New Zealand.
With that definition in mind, this hospitalist decided to seek a working sabbatical—not so much over dissatisfaction with my job back home, but to see how the other side lives with nationalized healthcare. Of course, moving to the beach in undeniably beautiful New Zealand never hurts, either. When it comes to government involvement and healthcare, the U.S. is in a distinct minority globally; New Zealand’s healthcare delivery system, while not diametrically opposite, offers some fascinating differences.
Last July, my wife and I, along with our two young boys, relocated to Ohope Beach, a quaint resort community on the northeast coast of New Zealand about 100 miles south of the nation's largest and most well-known city, Auckland. I am now six months into a year’s assignment at Whakatane Hospital.
I do not claim to be an expert on the comparative aspects of national healthcare systems, and some of my observations are directly related to my move from an urban to a rural setting. Still, the differences between the health systems in New Zealand and the U.S. are striking. Compared to Americans, New Zealanders spend less money on healthcare, their medications cost a third of what ours do, they undergo less testing, and they spend less time in the ICU, yet they live longer. Additionally, the public sector pays for the health insurance of every single New Zealand resident.
Let’s dispense with a few dry facts:
- Healthcare spending as a percentage of GDP: in the U.S., 16%; in New Zealand, 8%;1
- Per capita spending on healthcare in U.S. dollars: $7,500 for Americans; $2,700 for New Zealanders;
- Percent of healthcare spending by public sector: U.S., 45%; New Zealand, 80%1;
- Average life expectancy in the U.S.: 78.2 years (38th in the world); average in New Zealand: 80.2 years (13th in the world);2 and
- World ranking in infant mortality: U.S., 33rd; New Zealand, 27th.2
How are those figures possible? Does New Zealand employ death panels? No. Is there something in the water (e.g. statins and ACEs)? No. Is everyone a non-GMO, fair-trade, shade-grown, sustainably harvested vegan monk triathlete? Hmm.
Anyway, here are a few observations—from a hospitalist’s point of view.
Hospitalist by Another Name
My job title here at 100-bed Whakatane Hospital is “consultant physician.” The term “hospitalist” is not common, but it is exactly the job the consultant physician performs. Inpatient ward rounds make up the majority of the consultant’s role, and the only outpatient responsibility is a twice-weekly clinic to see patients referred from primary-care physicians (still called GPs here) for clinical questions. Because it’s a public hospital, we have house officers and junior physicians as well. Roughly, that means that the consultant examines the patient and formulates the plan, the junior physician does the new intake, and the house officer does all the writing.
As for the team, it’s a bit like working at the United Nations. So far, I have met physicians from New Zealand, Australia, Ireland, Sri Lanka, Spain, Jordan, Iraq, the United Kingdom, India, Zambia, South Africa, and, of course, America. Thank goodness we—and the patients—all speak varying degrees of English. It’s certainly a bit odd to find yourself misunderstood by someone else speaking your (and their) native tongue.
The work schedule is quite reasonable, or, as I have come to call it, “civilized.” It is a 40-hour workweek, 8 a.m. to 4 p.m. daily, with at least 12 hours of each week dedicated to nonclinical time in the form of reading, research, and teaching. The daily patient census per team is about 12, and the call (or “take”) ratio is about 1:4. With no nights and rare weekends, the whole experience is a welcome change of pace. For the Kiwis, this pace is perfectly accepted and expected, with little pressure to work harder, longer, or faster. Teatime is 10 in the morning, and breaking for a cuppa is just part of the job.
In switching from the private sector to the public sector, I braced myself for a significant cut in pay. As it turns out, the compensation is actually quite reasonable when matched to the schedule. Based on the current exchange rate of $1 USD equal to $0.75 NZD, the annual salary is about $165,000 USD. You also receive six weeks of vacation, two weeks of CME leave, and a $12,000 CME allowance. All in all, it’s a nice package. (Unfortunately, the pay in Australia is higher, and many Kiwi physicians choose to practice over there.) The resulting shortage of locally educated and trained physicians in New Zealand explains the ongoing need for overseas physicians.
Fewer Resources, So Choose Wisely
The hospital is part of a larger network called a district health board (DHB). The DHBs receive government funding based on population, then must decide how to spend the money. For that reason, there are only a few hospitals that include subspecialties. Smaller sites, such as Whakatane, have no subspecialists at all.
That also means we have no MRI machine, no cardiac catheter lab, no echocardiography, no hemodialysis. So, to do one of these procedures, it involves speaking to a specialist at a tertiary site and requesting a transfer, which, in turn, forces you to explain and justify your request. All of a sudden, the history and the physical exam regain their proper importance rather than being subverted to ordering a confirmatory imaging study or specialist consultation. It’s much different than back home, where the indication to perform coronary angiography can be the presence of a cardiologist and a groin in the same room.
In addition, the community GPs cannot order such testing as exercise treadmills or CT scans without going through a consultant first. Although this inevitably leads to delays, it also means some thought is introduced into the process.
Dialysis is another example. In New Zealand, more than 40% of patients receive peritoneal dialysis, and many of those who get hemodialysis do so at home. The average cost per patient per year is $25,000. By contrast, 93% of U.S. dialysis patients are on hemodialysis at a cost of $67,000 per patient per year.3 The outcomes for the two treatments are generally accepted to be the same; one just costs a whole lot more.
This Is the Only Formulary
U.S. physicians are all too familiar with MRSA, VRE, C. diff, and their ilk. Soon we may have to deal with NDM-1. Over and over again, we hear about how we should curtail our antibiotic use, yet little is actually done. New Zealand has strict antibiotic guidelines in accordance with a patient’s diagnosis. Ordering antibiotics outside the protocol is not accepted, unless you can convince the infectious-disease physician otherwise. It is an eminently reasonable practice that has simply never gained broad acceptance in America. What is the result of the New Zealand practice mandate? The hospital where I work has experienced only rare MRSA cases (9% of all S. aureus vs. 35% in the U.S.), no VRE, and zero cases of C. diff in the past nine months.
New Zealand has one drug-purchasing entity: Pharmac. The entity negotiates prices with suppliers and writes the formulary. If a drug is not on the national formulary, it is not available. Period.
Most drugs are fully subsidized; some drugs are partially subsidized. If it is available and subsidized, a 90-day prescription costs $3. All in all, Kiwi pharmaceutical costs are about 30% of the costs in the U.S. The choices are fewer, yet patients still receive appropriate pharmacologic treatment.
Every hospitalist has worked with an electronic health record (EHR) in one form or another, but one of the most frustrating things in the States is the lack of interface between EHR in different health systems. Such disconnect leads to an extraordinary amount of effort not to duplicate tests for patients who frequent more than one hospital. Often, these patients are high consumers of healthcare at baseline, and duplicating tests only makes it worse.
New Zealand has one computer system that allows the GPs and the hospitalists to view all the labs and imaging results together, in the same system. No matter who orders the test, it all comes back to the same place. It might seem small, but it makes a tremendous difference to the management of the patient. One could argue that defensive medicine is good practice, but we’ve all seen the extra study get ordered somewhere along the line. I won’t get into the details of malpractice insurance (the editors said stop at 2,000 words, and I could go on for 200,000), but back home in Colorado, I paid about $20,000 a year for malpractice insurance. Here, it’s $1,200.
New Zealand has a no-fault compensation system in which injured patients apply for government-funded compensation and thus give up the right to litigation. Most claims are processed within weeks, and all decisions are final within nine months. The patients here know that they have recourse for a bad outcome, and the physicians practice in a manner according to clinical judgment, rather than trying to avoid being sued.
Confusion and Delay
You might get the sense that practicing medicine in New Zealand has been a refreshing change of pace, and you would be right. My physical exam skills (what was left of them) are starting to return. I think long and hard before ordering an imaging study or requesting consultation. I order antibiotics according to the guidelines. I only prescribe medicines that are on the formulary. The care given to the patients by the staff and the knowledge exhibited by our physicians are quite good.
Still, this system is by no means perfect. The delays in care can be excruciating in both the urgent and routine settings. For example, my first patient on my first day of work had an acute myocardial infarction. The ED physician admitted him to the hospital at midnight, but no one on our team had met the patient when I arrived for work the next day. At 8 a.m., he had dynamic EKG changes, a positive troponin, ongoing chest pain, and a systolic BP of about 90 mm/HG. Back home, I never would have met the man, as he would have gone pretty much straight to the cath lab—with the cardiologists—within 60 minutes, even at midnight. Here, we had to call the hospital that does interventions, arrange a bed (hoping one was available), then put him in an ambulance for a four-hour drive on a two-lane road to receive a stent for his 100% coronary lesion.
The average wait for transfer for acute coronary angiography is about five days. The average wait for routine outpatient echocardiography is two years. Yes, two years.
Ultimately, the right thing gets done, but sometimes way too slowly. Thus, there is a smaller, parallel private insurance system, but people view it as optional and only use it when they want to speed up the schedule.
A Few Things I Have No Answer For
I have yet to admit someone with alcohol withdrawal—this in a country with a higher per capita alcohol consumption. I have yet to order a PCA pump for a medical patient. Low-back pain is managed by surgery, and a pneumothorax goes to the medical service. There is no Pyxis equivalent, just an open cupboard in the ICU where the meds are kept. There is no separate page for physician orders; the staff is expected to read the notes. In an old physical plant such as Whakatane, there are no private bathrooms for patients—it’s down the hall. Oh, and it’s four patients to a room, with no television, no Internet, and no telephone. There are plans for a new hospital in a few years’ time.
Culturally, the patients and their families are more accepting of the fragile nature of old age, with a strong desire to avoid unnecessary interventions at the end of life. There is a robust community hospice program that helps patients remain comfortable at home. As a local colleague of mine explains, the Kiwis are uniformly grateful for the care they receive, even if it means sharing a room with another patient (or three).
Final Observations
Healthcare delivery in New Zealand is different from what we see in the U.S. Obviously, the vast majority of healthcare delivered in New Zealand is through the public sector. No matter which direction U.S. healthcare reform goes next, it’s highly improbable it will ever resemble New Zealand’s system.
One quote that resonates comes from the medical practice handbook, which is given to all newly registered physicians in New Zealand: “Doctors have a responsibility to the community to foster the proper use of resources—in particular, by making efforts to use resources efficiently, consistent with good patient care.”4 It’s sound advice.
Changes are coming, and hospitalists everywhere are in a unique position to gain knowledge and lead the change. In the U.S. or in New Zealand, as a hospitalist or a physician consultant, medicine is a fascinating field of practice.
I hope you enjoyed reading about one hospitalist’s observations from New Zealand. Time for tea. TH
M-A Williams has been a practicing hospitalist in Denver since 1999. He worked with the same company (Inpatient Services, which then merged with Sound Physicians) for 11 years until departing for New Zealand. He is a member of Team Hospitalist.
References
- Organisation for Economic Cooperation and Development website. Available at: www.oecd.org. Accessed Dec. 31, 2010.
- The United Nations Statistical Division website. Available at: http://unstats.un.org/unsd/default.htm. Accessed Jan. 4, 2011.
- Ashton T, Marshall RM. The organization and financing of dialysis and kidney transplantation services in New Zealand. Int J Healthcare Finance and Econ. 2007;7:233-252.
- Cole’s Medical Practice in New Zealand (2009). Ed., Ian St. George. Medical Council of New Zealand: Wellington:18.
New Zealand Fast Facts
Demographics
- New Zealand pop.: 4.3 million
- Capital: Wellington
- Largest city: Auckland, 1.4 million pop.
- Whakatane District pop.: 32,814
- Ohope Beach pop.: 2,760
Information
- Whakatane District in the Bay of Plenty features 34 miles of sandy beaches.
- Ohope Beach is a popular vacation spot. White Island, an active volcano offshore, is a world-renowned tourism attraction.
- The kiwi, NZ’s national emblem, is a flightless bird with hair-like feathers and a long, slender bill, which it uses to pull worms and insects out of the ground.
- New Zealanders often refer to themselves as Kiwis, and the term is also used as a short form for the famous kiwi fruit. The NZ dollar is referred to as the kiwi.
- Whakatane summer temperatures average in the 80s, and winter months in the 50s. The district records New Zealand’s high temperature about 55 days a year.
- New Zealand’s seasons are the reverse of the Northern Hemisphere.
Health Stats
- Spending as a percentage of GDP: 8%
- Per capita spending on healthcare: $2,700
- Average life expectancy: 80.2 years (13th in the world)
All Aboard
Ten years ago, Stephen Jencks, MD, MPH, was hospitalized after taking a nasty spill and rupturing a kidney, breaking two ribs, and fracturing two transverse processes. The independent healthcare safety and quality consultant based in Baltimore still laughs ruefully at what happened next.
Dr. Jencks was stabilized and given OxyContin to treat his considerable pain, and then he was discharged—without his wife or another caregiver present, with a prescription for nothing more than Tylenol, and without any instructions on what to do if his condition worsened. Twelve hours after returning home, his pain re-emerged with such a vengeance that he experienced severe muscle spasms.
Dr. Jencks suspects his doctor was so focused on his ruptured kidney that pain management and follow-up fell by the wayside. “I am not an unassertive individual, so why didn’t I say something?” he asks. “The simple answer is that, at least for me, if I’m taking OxyContin, there are no problems. People tend not to be at the very top of their game when they’re on opioids and traumatized.”
He made it through the night at home and received better pain medication in the morning, but his experience, he says, “beautifully illustrates” the chronic problem of less-than-graceful transfers of care that can lead to unnecessary hospital readmissions. If it nearly happened to him, it can happen to anyone.
And, based on his research, it often does. In an influential 2009 New England Journal of Medicine study coauthored with Mark Williams, MD, FACP, FHM, professor and chief of the division of hospital medicine at Northwestern University’s Feinberg School of Medicine in Chicago, and principal investigator of SHM’s Project BOOST, and Eric Coleman, MD, MPH, FACP, associate professor of medicine and director of the care transitions program at the University of Colorado Denver, Dr. Jencks helped uncover some startling statistics: During a 15-month period from 2003 to 2004, nearly 20% of the roughly 12 million Medicare beneficiaries discharged from hospitals were readmitted within 30 days (see “State-by-State Breakdown of 30-Day Rehospitalizations of Medicare Beneficiaries,” p. 7).1 Of those patients discharged to the community and then rehospitalized, half had not seen their own primary-care physician (PCP) in the interim. In all, the authors estimated Medicare’s financial toll from unplanned rehospitalizations at $17.4 billion for 2004 alone.

Surprisingly, Dr. Jencks’ study and a 2007 Medicare Payment Advisory Commission report to Congress provided the first estimates of the overall burden of rehospitalization in nearly a quarter-century. Since then, however, the topic has been a mainstay in conversations about the kinds of interventions that could yield major improvements in healthcare.
“The thing that has propelled this to the front is the recognition that we really can do better,” Dr. Jencks says. “What had tended to be seen as just an evitable consequence of people being sick is now increasingly seen as often being the consequence of not having done as good a job as we should have.”
Beyond the potential for poor patient outcomes and wasted money, healthcare experts say excessive readmissions have the potential to undermine the reputations of hospitalists just as they are moving to center stage in national quality-improvement (QI) efforts.
“I see, basically every day, patients that come back to the hospital because the discharge process is broken,” says Eric Howell, MD, SFHM, director of the hospitalist division at Johns Hopkins Bayview Medical Center in Baltimore. Dr. Howell says communication difficulties between the hospital and a nursing home have plagued one “revolving door” case involving a patient with a stomach ulcer that requires surgical resection. Hospital surgeons have repeatedly arranged to see her as an outpatient and schedule the surgery, but before the surgery can take place, the patient vomits up blood and is rehospitalized.
Another contributing factor, Dr. Howell argues, is the lack of incentives for both hospitals and hospitalists to work hard at preventing the next readmission. Although Dr. Jencks’ study suggests readmissions might not always be profitable, Dr. Howell and others say the sizeable contribution of rehospitalizations to overall admission numbers and the single-digit profit margins of most hospitals offer little motivation to change the status quo. “I think there are good people who want to fix it,” says Dr. Howell, an SHM board member and Project BOOST mentor. But changing the reimbursement system so that hospitalists can better focus on reducing readmissions, he adds, “will really go a long way.”
A New Landscape
Change is in the air. As part of the federal Affordable Care Act of 2010, the Centers for Medicare & Medicaid Services (CMS) is expanding a pilot project on bundling payments to doctors and hospitals around episodes of care. Starting Jan. 1, 2013, the bundling pilot will define “episodes” as all medical services administered three days before a hospital admission until 30 days after discharge. A rehospitalization within that timeframe would net reduced reimbursements.
CMS also has begun accepting applications for what’s known as the Community-Based Care Transitions Program, with $500 million over five years authorized by the healthcare reform act to fund collaborative, readmission-reducing efforts between hospitals and community-based organizations. Linda Magno, CMS director of the Medicare Demonstrations Program Group (www.cms.gov/CMSLeadership/19_Office_ORDI.asp), says program participants will form a learning network so the agency can quickly deliver information about who’s doing well and what approaches are working better than others. The participating organizations, she says, can then help teach best practices to other hospitals around the country.
CMS has adopted public reporting requirements as another tactic. The “Hospital Compare” website (www.hospitalcompare.hhs.gov/) set up by CMS, for example, uses discharge data to publish rehospitalization rates for heart failure, acute myocardial infarction, and pneumonia. More published rates will be added soon. More importantly, Medicare will begin penalizing poorly performing institutions in October 2012 by withholding a percentage of their payments, starting at 1% and rising to 3% within three years, as part of the value-based purchasing initiative.
For hospitals, the looming deadline has prompted widespread concern about the potential financial impact. With a growing number of models and projects springing up around the country, however, hospitalists and other healthcare providers are finding encouraging signs that even relatively simple interventions might help profoundly change the trajectory of care transitions.
Rachel George, MD, MBA, FHM, regional medical director and vice president of operations for West Cogent Healthcare Inc., says Cogent has found success with one tactic—ensuring that all patients are called after being discharged. The call helps to verify that prescribed medications have been picked up and that other care-related questions have been answered. Even before discharge, Dr. George says, Cogent also tries to ensure that a follow-up appointment with every patient’s PCP is on the calendar.
Debbie White, project coordinator for the Little Rock, Ark.-based National Transitions of Care Coalition (NTOCC), says it helps to frame the entire process as a transition plan rather than a discharge. White says patients—and often their family caregivers—are the one constant in every transition. “Some older Americans, including the baby boomers, came from a culture where you don’t question your physician or even an RN,” White says. “So they’ve had a hard time speaking up and learning to ask for a list of their medications, or who’s going to make their next follow-up appointment.” Among its tools, NTOCC offers resources to teach patients how to take more responsibility for their own care (see “Patient Interaction,” p. 5).

On the other side of the equation, the most downloaded tool on the coalition’s website is an evaluation and implementation plan that helps healthcare professionals find the gaps in care transitions. Other tools, including case scenarios and checklists, help healthcare providers consider specific steps, and a compendium of evidence offers a look at successful models and projects.
Dr. Bradley M. Sherman, MD, FHM, chairman of the department of medicine at Glen Cove Hospital/North Shore-LIJ University Health System in New York, led one such project, sponsored by the Greater New York Hospital Association. Dr. Sherman targeted heart failure, the condition with the highest readmission rate for both Glen Cove Hospital and the North Shore/LIJ system. By placing special emphasis on medication compliance, dietary adherence, and physician follow-up, Dr. Sherman says, the hospital cut its readmission rates by more than half, to well below the national average.
Another effort led by Johns Hopkins’ Dr. Howell, known as Safe and Successful Transition of Elderly Patients (Safe STEP), used a collaborative staff approach in general medicine wards overseen by hospitalists to reduce 30-day readmission rates from 22% to 14%. The encouraging results, first reported at SHM’s annual meeting in 2008, provided the impetus for a project called Better Outcomes for Older Adults through Safe Transitions, or Project BOOST (www.hospitalmedicine.org/BOOST).
Developed by SHM, BOOST features a yearlong mentoring program to help sites implement the QI project. It began at six hospitals and has since spread to 62 active mentor sites. Enrollment may swell to between 100 and 120 sites by the end of 2011, according to project director Tina Budnitz, MPH. Data from the first phase revealed a 21% reduction in 30-day readmission rates at the six pilot sites, to 11.2% from 14.2%. Follow-up data from the larger cohort are expected this spring.
Eric Siegal, MD, SFHM, an SHM board member, past chair of SHM’s Public Policy Committee, and a clinical assistant professor of medicine at the University of Wisconsin School of Medicine and Public Health, says BOOST has benefited from being solidly in place at the right time, gaining momentum and garnering significant national attention as the focus on better care transitions has intensified.

“If BOOST demonstrates substantial and reproducible decreases in rehospitalizations, improvements in quality, and presumed projected cost reductions, I think that it’s going to go off like a bomb,” he says, “in a good way.”
Lakshmi Halasyamani, MD, SFHM, vice president for medical affairs for the Saint Joseph Mercy Health System in Michigan and an SHM board member, says BOOST encourages hospitalists to think about ways in which a discharge might fail. “And then we need to actively mitigate those risks,” she says.
National Collaborations
CMS has tapped a network of technical assistance and QI contractors in all 50 states, known as quality-improvement organizations (QIOs), for its own project addressing rehospitalizations. In 2008, these QIOs began working with communities in 14 states to implement what’s known as the Care Transitions Program.
The program has helped community leaders highlight three root causes of high readmission rates: patients’ lack of knowledge and understanding about their chronic conditions, lack of communication among providers, and the healthcare system’s lack of known standards.
The 14 communities, 70 hospitals, and 1.25 million Medicare beneficiaries being followed to date suggest that 30-day readmission rates can be significantly decreased, says Paul McGann, MD, CMS deputy chief medical officer. Preliminary data based on the number of readmissions per 1,000 Medicare beneficiaries, he says, show that participating communities have improved by an average of 4.7% over the first two years of the project, with the top performer improving 14% (for more information, visit www.cfmc.org/caretransitions).
Dr. Halasyamani says no single program has necessarily found the “secret sauce” to improve readmission rates across the board. “And we definitely haven’t figured out how to implement that in as cost-effective a way as possible,” she says.
But optimism is clearly building. With the initial focus on coaching low-performing institutions to improve their rates, Medicare could tap programs that demonstrate early promise as the main go-to teaching aids.
More importantly, hospitals around the country are finding what it takes to help their own patients.
“The question isn’t, ‘Is our number better than St. Elsewhere’s down the street?’ ” Dr. Jencks concludes. “The real question is, ‘Are there things we could reasonably have done for this patient and could do for the next patient that will keep this from happening to them?’ ” TH
Bryn Nelson is a freelance medical writer based in Seattle.
Reference
- Jencks SJ, Williams MV, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428.
Ten years ago, Stephen Jencks, MD, MPH, was hospitalized after taking a nasty spill and rupturing a kidney, breaking two ribs, and fracturing two transverse processes. The independent healthcare safety and quality consultant based in Baltimore still laughs ruefully at what happened next.
Dr. Jencks was stabilized and given OxyContin to treat his considerable pain, and then he was discharged—without his wife or another caregiver present, with a prescription for nothing more than Tylenol, and without any instructions on what to do if his condition worsened. Twelve hours after returning home, his pain re-emerged with such a vengeance that he experienced severe muscle spasms.
Dr. Jencks suspects his doctor was so focused on his ruptured kidney that pain management and follow-up fell by the wayside. “I am not an unassertive individual, so why didn’t I say something?” he asks. “The simple answer is that, at least for me, if I’m taking OxyContin, there are no problems. People tend not to be at the very top of their game when they’re on opioids and traumatized.”
He made it through the night at home and received better pain medication in the morning, but his experience, he says, “beautifully illustrates” the chronic problem of less-than-graceful transfers of care that can lead to unnecessary hospital readmissions. If it nearly happened to him, it can happen to anyone.
And, based on his research, it often does. In an influential 2009 New England Journal of Medicine study coauthored with Mark Williams, MD, FACP, FHM, professor and chief of the division of hospital medicine at Northwestern University’s Feinberg School of Medicine in Chicago, and principal investigator of SHM’s Project BOOST, and Eric Coleman, MD, MPH, FACP, associate professor of medicine and director of the care transitions program at the University of Colorado Denver, Dr. Jencks helped uncover some startling statistics: During a 15-month period from 2003 to 2004, nearly 20% of the roughly 12 million Medicare beneficiaries discharged from hospitals were readmitted within 30 days (see “State-by-State Breakdown of 30-Day Rehospitalizations of Medicare Beneficiaries,” p. 7).1 Of those patients discharged to the community and then rehospitalized, half had not seen their own primary-care physician (PCP) in the interim. In all, the authors estimated Medicare’s financial toll from unplanned rehospitalizations at $17.4 billion for 2004 alone.
Surprisingly, Dr. Jencks’ study and a 2007 Medicare Payment Advisory Commission report to Congress provided the first estimates of the overall burden of rehospitalization in nearly a quarter-century. Since then, however, the topic has been a mainstay in conversations about the kinds of interventions that could yield major improvements in healthcare.
“The thing that has propelled this to the front is the recognition that we really can do better,” Dr. Jencks says. “What had tended to be seen as just an evitable consequence of people being sick is now increasingly seen as often being the consequence of not having done as good a job as we should have.”
Beyond the potential for poor patient outcomes and wasted money, healthcare experts say excessive readmissions have the potential to undermine the reputations of hospitalists just as they are moving to center stage in national quality-improvement (QI) efforts.
“I see, basically every day, patients that come back to the hospital because the discharge process is broken,” says Eric Howell, MD, SFHM, director of the hospitalist division at Johns Hopkins Bayview Medical Center in Baltimore. Dr. Howell says communication difficulties between the hospital and a nursing home have plagued one “revolving door” case involving a patient with a stomach ulcer that requires surgical resection. Hospital surgeons have repeatedly arranged to see her as an outpatient and schedule the surgery, but before the surgery can take place, the patient vomits up blood and is rehospitalized.
Another contributing factor, Dr. Howell argues, is the lack of incentives for both hospitals and hospitalists to work hard at preventing the next readmission. Although Dr. Jencks’ study suggests readmissions might not always be profitable, Dr. Howell and others say the sizeable contribution of rehospitalizations to overall admission numbers and the single-digit profit margins of most hospitals offer little motivation to change the status quo. “I think there are good people who want to fix it,” says Dr. Howell, an SHM board member and Project BOOST mentor. But changing the reimbursement system so that hospitalists can better focus on reducing readmissions, he adds, “will really go a long way.”
A New Landscape
Change is in the air. As part of the federal Affordable Care Act of 2010, the Centers for Medicare & Medicaid Services (CMS) is expanding a pilot project on bundling payments to doctors and hospitals around episodes of care. Starting Jan. 1, 2013, the bundling pilot will define “episodes” as all medical services administered three days before a hospital admission until 30 days after discharge. A rehospitalization within that timeframe would net reduced reimbursements.
CMS also has begun accepting applications for what’s known as the Community-Based Care Transitions Program, with $500 million over five years authorized by the healthcare reform act to fund collaborative, readmission-reducing efforts between hospitals and community-based organizations. Linda Magno, CMS director of the Medicare Demonstrations Program Group (www.cms.gov/CMSLeadership/19_Office_ORDI.asp), says program participants will form a learning network so the agency can quickly deliver information about who’s doing well and what approaches are working better than others. The participating organizations, she says, can then help teach best practices to other hospitals around the country.
CMS has adopted public reporting requirements as another tactic. The “Hospital Compare” website (www.hospitalcompare.hhs.gov/) set up by CMS, for example, uses discharge data to publish rehospitalization rates for heart failure, acute myocardial infarction, and pneumonia. More published rates will be added soon. More importantly, Medicare will begin penalizing poorly performing institutions in October 2012 by withholding a percentage of their payments, starting at 1% and rising to 3% within three years, as part of the value-based purchasing initiative.
For hospitals, the looming deadline has prompted widespread concern about the potential financial impact. With a growing number of models and projects springing up around the country, however, hospitalists and other healthcare providers are finding encouraging signs that even relatively simple interventions might help profoundly change the trajectory of care transitions.
Rachel George, MD, MBA, FHM, regional medical director and vice president of operations for West Cogent Healthcare Inc., says Cogent has found success with one tactic—ensuring that all patients are called after being discharged. The call helps to verify that prescribed medications have been picked up and that other care-related questions have been answered. Even before discharge, Dr. George says, Cogent also tries to ensure that a follow-up appointment with every patient’s PCP is on the calendar.
Debbie White, project coordinator for the Little Rock, Ark.-based National Transitions of Care Coalition (NTOCC), says it helps to frame the entire process as a transition plan rather than a discharge. White says patients—and often their family caregivers—are the one constant in every transition. “Some older Americans, including the baby boomers, came from a culture where you don’t question your physician or even an RN,” White says. “So they’ve had a hard time speaking up and learning to ask for a list of their medications, or who’s going to make their next follow-up appointment.” Among its tools, NTOCC offers resources to teach patients how to take more responsibility for their own care (see “Patient Interaction,” p. 5).
On the other side of the equation, the most downloaded tool on the coalition’s website is an evaluation and implementation plan that helps healthcare professionals find the gaps in care transitions. Other tools, including case scenarios and checklists, help healthcare providers consider specific steps, and a compendium of evidence offers a look at successful models and projects.
Dr. Bradley M. Sherman, MD, FHM, chairman of the department of medicine at Glen Cove Hospital/North Shore-LIJ University Health System in New York, led one such project, sponsored by the Greater New York Hospital Association. Dr. Sherman targeted heart failure, the condition with the highest readmission rate for both Glen Cove Hospital and the North Shore/LIJ system. By placing special emphasis on medication compliance, dietary adherence, and physician follow-up, Dr. Sherman says, the hospital cut its readmission rates by more than half, to well below the national average.
Another effort led by Johns Hopkins’ Dr. Howell, known as Safe and Successful Transition of Elderly Patients (Safe STEP), used a collaborative staff approach in general medicine wards overseen by hospitalists to reduce 30-day readmission rates from 22% to 14%. The encouraging results, first reported at SHM’s annual meeting in 2008, provided the impetus for a project called Better Outcomes for Older Adults through Safe Transitions, or Project BOOST (www.hospitalmedicine.org/BOOST).
Developed by SHM, BOOST features a yearlong mentoring program to help sites implement the QI project. It began at six hospitals and has since spread to 62 active mentor sites. Enrollment may swell to between 100 and 120 sites by the end of 2011, according to project director Tina Budnitz, MPH. Data from the first phase revealed a 21% reduction in 30-day readmission rates at the six pilot sites, to 11.2% from 14.2%. Follow-up data from the larger cohort are expected this spring.
Eric Siegal, MD, SFHM, an SHM board member, past chair of SHM’s Public Policy Committee, and a clinical assistant professor of medicine at the University of Wisconsin School of Medicine and Public Health, says BOOST has benefited from being solidly in place at the right time, gaining momentum and garnering significant national attention as the focus on better care transitions has intensified.
“If BOOST demonstrates substantial and reproducible decreases in rehospitalizations, improvements in quality, and presumed projected cost reductions, I think that it’s going to go off like a bomb,” he says, “in a good way.”
Lakshmi Halasyamani, MD, SFHM, vice president for medical affairs for the Saint Joseph Mercy Health System in Michigan and an SHM board member, says BOOST encourages hospitalists to think about ways in which a discharge might fail. “And then we need to actively mitigate those risks,” she says.
National Collaborations
CMS has tapped a network of technical assistance and QI contractors in all 50 states, known as quality-improvement organizations (QIOs), for its own project addressing rehospitalizations. In 2008, these QIOs began working with communities in 14 states to implement what’s known as the Care Transitions Program.
The program has helped community leaders highlight three root causes of high readmission rates: patients’ lack of knowledge and understanding about their chronic conditions, lack of communication among providers, and the healthcare system’s lack of known standards.
The 14 communities, 70 hospitals, and 1.25 million Medicare beneficiaries being followed to date suggest that 30-day readmission rates can be significantly decreased, says Paul McGann, MD, CMS deputy chief medical officer. Preliminary data based on the number of readmissions per 1,000 Medicare beneficiaries, he says, show that participating communities have improved by an average of 4.7% over the first two years of the project, with the top performer improving 14% (for more information, visit www.cfmc.org/caretransitions).
Dr. Halasyamani says no single program has necessarily found the “secret sauce” to improve readmission rates across the board. “And we definitely haven’t figured out how to implement that in as cost-effective a way as possible,” she says.
But optimism is clearly building. With the initial focus on coaching low-performing institutions to improve their rates, Medicare could tap programs that demonstrate early promise as the main go-to teaching aids.
More importantly, hospitals around the country are finding what it takes to help their own patients.
“The question isn’t, ‘Is our number better than St. Elsewhere’s down the street?’ ” Dr. Jencks concludes. “The real question is, ‘Are there things we could reasonably have done for this patient and could do for the next patient that will keep this from happening to them?’ ” TH
Bryn Nelson is a freelance medical writer based in Seattle.
Reference
- Jencks SJ, Williams MV, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428.
Ten years ago, Stephen Jencks, MD, MPH, was hospitalized after taking a nasty spill and rupturing a kidney, breaking two ribs, and fracturing two transverse processes. The independent healthcare safety and quality consultant based in Baltimore still laughs ruefully at what happened next.
Dr. Jencks was stabilized and given OxyContin to treat his considerable pain, and then he was discharged—without his wife or another caregiver present, with a prescription for nothing more than Tylenol, and without any instructions on what to do if his condition worsened. Twelve hours after returning home, his pain re-emerged with such a vengeance that he experienced severe muscle spasms.
Dr. Jencks suspects his doctor was so focused on his ruptured kidney that pain management and follow-up fell by the wayside. “I am not an unassertive individual, so why didn’t I say something?” he asks. “The simple answer is that, at least for me, if I’m taking OxyContin, there are no problems. People tend not to be at the very top of their game when they’re on opioids and traumatized.”
He made it through the night at home and received better pain medication in the morning, but his experience, he says, “beautifully illustrates” the chronic problem of less-than-graceful transfers of care that can lead to unnecessary hospital readmissions. If it nearly happened to him, it can happen to anyone.
And, based on his research, it often does. In an influential 2009 New England Journal of Medicine study coauthored with Mark Williams, MD, FACP, FHM, professor and chief of the division of hospital medicine at Northwestern University’s Feinberg School of Medicine in Chicago, and principal investigator of SHM’s Project BOOST, and Eric Coleman, MD, MPH, FACP, associate professor of medicine and director of the care transitions program at the University of Colorado Denver, Dr. Jencks helped uncover some startling statistics: During a 15-month period from 2003 to 2004, nearly 20% of the roughly 12 million Medicare beneficiaries discharged from hospitals were readmitted within 30 days (see “State-by-State Breakdown of 30-Day Rehospitalizations of Medicare Beneficiaries,” p. 7).1 Of those patients discharged to the community and then rehospitalized, half had not seen their own primary-care physician (PCP) in the interim. In all, the authors estimated Medicare’s financial toll from unplanned rehospitalizations at $17.4 billion for 2004 alone.
Surprisingly, Dr. Jencks’ study and a 2007 Medicare Payment Advisory Commission report to Congress provided the first estimates of the overall burden of rehospitalization in nearly a quarter-century. Since then, however, the topic has been a mainstay in conversations about the kinds of interventions that could yield major improvements in healthcare.
“The thing that has propelled this to the front is the recognition that we really can do better,” Dr. Jencks says. “What had tended to be seen as just an evitable consequence of people being sick is now increasingly seen as often being the consequence of not having done as good a job as we should have.”
Beyond the potential for poor patient outcomes and wasted money, healthcare experts say excessive readmissions have the potential to undermine the reputations of hospitalists just as they are moving to center stage in national quality-improvement (QI) efforts.
“I see, basically every day, patients that come back to the hospital because the discharge process is broken,” says Eric Howell, MD, SFHM, director of the hospitalist division at Johns Hopkins Bayview Medical Center in Baltimore. Dr. Howell says communication difficulties between the hospital and a nursing home have plagued one “revolving door” case involving a patient with a stomach ulcer that requires surgical resection. Hospital surgeons have repeatedly arranged to see her as an outpatient and schedule the surgery, but before the surgery can take place, the patient vomits up blood and is rehospitalized.
Another contributing factor, Dr. Howell argues, is the lack of incentives for both hospitals and hospitalists to work hard at preventing the next readmission. Although Dr. Jencks’ study suggests readmissions might not always be profitable, Dr. Howell and others say the sizeable contribution of rehospitalizations to overall admission numbers and the single-digit profit margins of most hospitals offer little motivation to change the status quo. “I think there are good people who want to fix it,” says Dr. Howell, an SHM board member and Project BOOST mentor. But changing the reimbursement system so that hospitalists can better focus on reducing readmissions, he adds, “will really go a long way.”
A New Landscape
Change is in the air. As part of the federal Affordable Care Act of 2010, the Centers for Medicare & Medicaid Services (CMS) is expanding a pilot project on bundling payments to doctors and hospitals around episodes of care. Starting Jan. 1, 2013, the bundling pilot will define “episodes” as all medical services administered three days before a hospital admission until 30 days after discharge. A rehospitalization within that timeframe would net reduced reimbursements.
CMS also has begun accepting applications for what’s known as the Community-Based Care Transitions Program, with $500 million over five years authorized by the healthcare reform act to fund collaborative, readmission-reducing efforts between hospitals and community-based organizations. Linda Magno, CMS director of the Medicare Demonstrations Program Group (www.cms.gov/CMSLeadership/19_Office_ORDI.asp), says program participants will form a learning network so the agency can quickly deliver information about who’s doing well and what approaches are working better than others. The participating organizations, she says, can then help teach best practices to other hospitals around the country.
CMS has adopted public reporting requirements as another tactic. The “Hospital Compare” website (www.hospitalcompare.hhs.gov/) set up by CMS, for example, uses discharge data to publish rehospitalization rates for heart failure, acute myocardial infarction, and pneumonia. More published rates will be added soon. More importantly, Medicare will begin penalizing poorly performing institutions in October 2012 by withholding a percentage of their payments, starting at 1% and rising to 3% within three years, as part of the value-based purchasing initiative.
For hospitals, the looming deadline has prompted widespread concern about the potential financial impact. With a growing number of models and projects springing up around the country, however, hospitalists and other healthcare providers are finding encouraging signs that even relatively simple interventions might help profoundly change the trajectory of care transitions.
Rachel George, MD, MBA, FHM, regional medical director and vice president of operations for West Cogent Healthcare Inc., says Cogent has found success with one tactic—ensuring that all patients are called after being discharged. The call helps to verify that prescribed medications have been picked up and that other care-related questions have been answered. Even before discharge, Dr. George says, Cogent also tries to ensure that a follow-up appointment with every patient’s PCP is on the calendar.
Debbie White, project coordinator for the Little Rock, Ark.-based National Transitions of Care Coalition (NTOCC), says it helps to frame the entire process as a transition plan rather than a discharge. White says patients—and often their family caregivers—are the one constant in every transition. “Some older Americans, including the baby boomers, came from a culture where you don’t question your physician or even an RN,” White says. “So they’ve had a hard time speaking up and learning to ask for a list of their medications, or who’s going to make their next follow-up appointment.” Among its tools, NTOCC offers resources to teach patients how to take more responsibility for their own care (see “Patient Interaction,” p. 5).
On the other side of the equation, the most downloaded tool on the coalition’s website is an evaluation and implementation plan that helps healthcare professionals find the gaps in care transitions. Other tools, including case scenarios and checklists, help healthcare providers consider specific steps, and a compendium of evidence offers a look at successful models and projects.
Dr. Bradley M. Sherman, MD, FHM, chairman of the department of medicine at Glen Cove Hospital/North Shore-LIJ University Health System in New York, led one such project, sponsored by the Greater New York Hospital Association. Dr. Sherman targeted heart failure, the condition with the highest readmission rate for both Glen Cove Hospital and the North Shore/LIJ system. By placing special emphasis on medication compliance, dietary adherence, and physician follow-up, Dr. Sherman says, the hospital cut its readmission rates by more than half, to well below the national average.
Another effort led by Johns Hopkins’ Dr. Howell, known as Safe and Successful Transition of Elderly Patients (Safe STEP), used a collaborative staff approach in general medicine wards overseen by hospitalists to reduce 30-day readmission rates from 22% to 14%. The encouraging results, first reported at SHM’s annual meeting in 2008, provided the impetus for a project called Better Outcomes for Older Adults through Safe Transitions, or Project BOOST (www.hospitalmedicine.org/BOOST).
Developed by SHM, BOOST features a yearlong mentoring program to help sites implement the QI project. It began at six hospitals and has since spread to 62 active mentor sites. Enrollment may swell to between 100 and 120 sites by the end of 2011, according to project director Tina Budnitz, MPH. Data from the first phase revealed a 21% reduction in 30-day readmission rates at the six pilot sites, to 11.2% from 14.2%. Follow-up data from the larger cohort are expected this spring.
Eric Siegal, MD, SFHM, an SHM board member, past chair of SHM’s Public Policy Committee, and a clinical assistant professor of medicine at the University of Wisconsin School of Medicine and Public Health, says BOOST has benefited from being solidly in place at the right time, gaining momentum and garnering significant national attention as the focus on better care transitions has intensified.
“If BOOST demonstrates substantial and reproducible decreases in rehospitalizations, improvements in quality, and presumed projected cost reductions, I think that it’s going to go off like a bomb,” he says, “in a good way.”
Lakshmi Halasyamani, MD, SFHM, vice president for medical affairs for the Saint Joseph Mercy Health System in Michigan and an SHM board member, says BOOST encourages hospitalists to think about ways in which a discharge might fail. “And then we need to actively mitigate those risks,” she says.
National Collaborations
CMS has tapped a network of technical assistance and QI contractors in all 50 states, known as quality-improvement organizations (QIOs), for its own project addressing rehospitalizations. In 2008, these QIOs began working with communities in 14 states to implement what’s known as the Care Transitions Program.
The program has helped community leaders highlight three root causes of high readmission rates: patients’ lack of knowledge and understanding about their chronic conditions, lack of communication among providers, and the healthcare system’s lack of known standards.
The 14 communities, 70 hospitals, and 1.25 million Medicare beneficiaries being followed to date suggest that 30-day readmission rates can be significantly decreased, says Paul McGann, MD, CMS deputy chief medical officer. Preliminary data based on the number of readmissions per 1,000 Medicare beneficiaries, he says, show that participating communities have improved by an average of 4.7% over the first two years of the project, with the top performer improving 14% (for more information, visit www.cfmc.org/caretransitions).
Dr. Halasyamani says no single program has necessarily found the “secret sauce” to improve readmission rates across the board. “And we definitely haven’t figured out how to implement that in as cost-effective a way as possible,” she says.
But optimism is clearly building. With the initial focus on coaching low-performing institutions to improve their rates, Medicare could tap programs that demonstrate early promise as the main go-to teaching aids.
More importantly, hospitals around the country are finding what it takes to help their own patients.
“The question isn’t, ‘Is our number better than St. Elsewhere’s down the street?’ ” Dr. Jencks concludes. “The real question is, ‘Are there things we could reasonably have done for this patient and could do for the next patient that will keep this from happening to them?’ ” TH
Bryn Nelson is a freelance medical writer based in Seattle.
Reference
- Jencks SJ, Williams MV, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428.
POLICY CORNER: An inside look at the most pressing policy issues
This year, hospitalists will begin to see health reform affect the way they work, and SHM is bringing the best perspective and access to its members.
With the proposed rules anticipated to have been in effect by the end of January, the definition and development of accountable care organizations (ACOs) will answer two long-awaited questions: How will these organizations impact the practice of hospital medicine … and when? Additionally, the Community-Based Care Transitions Program available to hospitals identified as having high readmission rates is scheduled to begin in early 2011.
So how can hospitalists get the information they need to prepare for, and succeed under, all of these new rules? Launched in mid-January, our new Advocacy & Public Policy portal at www.hospitalmedicine.org provides summaries and background material for relevant reform provisions, educational resources, headlines, and coming events—along with an easy way to reach out to elected officials through our Legislative Action Center.
Specifically outlined are SHM’s top priority issues (hospital value-based purchasing [HVBP], bundled payments, and reducing readmissions/improving care transitions), identified by the Public Policy Committee. The summaries also include SHM’s position statement so hospitalists know where SHM stands and what we’re doing to help hospitalists best position themselves to succeed.
In addition to provisions of the Affordable Care Act (ACA) of 2010, we’ve devoted a section to health information technology and updated the Physician Quality Reporting System to reflect ACA changes (including Maintenance of Certification [MOC] and the Physician Compare website).
In January, Patrick Conway, MD, and Patrick Torcson, MD, MMM, FACP, SFHM, chairmen of the Public Policy Committee and Performance & Standards Committee, respectively, presented the “Health Reform: Highlights and Practical Implications for Hospitalists” webinar, which explored ACOs, readmissions, HVBP, and the Centers for Medicare & Medicaid Services’ role in the implementation process. If you missed the presentation, it is available on demand at www.hospitalmedicine.org/webinars.
HM11, which is May 10-13 in Grapevine, Texas, will feature a session on the latest reform news: “The Biggest Changes in Healthcare Reform: What We Know Now.” Though the final presentation likely will change in the days leading up to the meeting, the panel plans to review how other ACA provisions will set hospitalists up to succeed under the new ACO model.
Now is the time for hospitalists to get up to speed. TH
Find all this and more by visiting www.hospitalmedicine.org/advocacy and let us know what you think by e-mailing [email protected].
This year, hospitalists will begin to see health reform affect the way they work, and SHM is bringing the best perspective and access to its members.
With the proposed rules anticipated to have been in effect by the end of January, the definition and development of accountable care organizations (ACOs) will answer two long-awaited questions: How will these organizations impact the practice of hospital medicine … and when? Additionally, the Community-Based Care Transitions Program available to hospitals identified as having high readmission rates is scheduled to begin in early 2011.
So how can hospitalists get the information they need to prepare for, and succeed under, all of these new rules? Launched in mid-January, our new Advocacy & Public Policy portal at www.hospitalmedicine.org provides summaries and background material for relevant reform provisions, educational resources, headlines, and coming events—along with an easy way to reach out to elected officials through our Legislative Action Center.
Specifically outlined are SHM’s top priority issues (hospital value-based purchasing [HVBP], bundled payments, and reducing readmissions/improving care transitions), identified by the Public Policy Committee. The summaries also include SHM’s position statement so hospitalists know where SHM stands and what we’re doing to help hospitalists best position themselves to succeed.
In addition to provisions of the Affordable Care Act (ACA) of 2010, we’ve devoted a section to health information technology and updated the Physician Quality Reporting System to reflect ACA changes (including Maintenance of Certification [MOC] and the Physician Compare website).
In January, Patrick Conway, MD, and Patrick Torcson, MD, MMM, FACP, SFHM, chairmen of the Public Policy Committee and Performance & Standards Committee, respectively, presented the “Health Reform: Highlights and Practical Implications for Hospitalists” webinar, which explored ACOs, readmissions, HVBP, and the Centers for Medicare & Medicaid Services’ role in the implementation process. If you missed the presentation, it is available on demand at www.hospitalmedicine.org/webinars.
HM11, which is May 10-13 in Grapevine, Texas, will feature a session on the latest reform news: “The Biggest Changes in Healthcare Reform: What We Know Now.” Though the final presentation likely will change in the days leading up to the meeting, the panel plans to review how other ACA provisions will set hospitalists up to succeed under the new ACO model.
Now is the time for hospitalists to get up to speed. TH
Find all this and more by visiting www.hospitalmedicine.org/advocacy and let us know what you think by e-mailing [email protected].
This year, hospitalists will begin to see health reform affect the way they work, and SHM is bringing the best perspective and access to its members.
With the proposed rules anticipated to have been in effect by the end of January, the definition and development of accountable care organizations (ACOs) will answer two long-awaited questions: How will these organizations impact the practice of hospital medicine … and when? Additionally, the Community-Based Care Transitions Program available to hospitals identified as having high readmission rates is scheduled to begin in early 2011.
So how can hospitalists get the information they need to prepare for, and succeed under, all of these new rules? Launched in mid-January, our new Advocacy & Public Policy portal at www.hospitalmedicine.org provides summaries and background material for relevant reform provisions, educational resources, headlines, and coming events—along with an easy way to reach out to elected officials through our Legislative Action Center.
Specifically outlined are SHM’s top priority issues (hospital value-based purchasing [HVBP], bundled payments, and reducing readmissions/improving care transitions), identified by the Public Policy Committee. The summaries also include SHM’s position statement so hospitalists know where SHM stands and what we’re doing to help hospitalists best position themselves to succeed.
In addition to provisions of the Affordable Care Act (ACA) of 2010, we’ve devoted a section to health information technology and updated the Physician Quality Reporting System to reflect ACA changes (including Maintenance of Certification [MOC] and the Physician Compare website).
In January, Patrick Conway, MD, and Patrick Torcson, MD, MMM, FACP, SFHM, chairmen of the Public Policy Committee and Performance & Standards Committee, respectively, presented the “Health Reform: Highlights and Practical Implications for Hospitalists” webinar, which explored ACOs, readmissions, HVBP, and the Centers for Medicare & Medicaid Services’ role in the implementation process. If you missed the presentation, it is available on demand at www.hospitalmedicine.org/webinars.
HM11, which is May 10-13 in Grapevine, Texas, will feature a session on the latest reform news: “The Biggest Changes in Healthcare Reform: What We Know Now.” Though the final presentation likely will change in the days leading up to the meeting, the panel plans to review how other ACA provisions will set hospitalists up to succeed under the new ACO model.
Now is the time for hospitalists to get up to speed. TH
Find all this and more by visiting www.hospitalmedicine.org/advocacy and let us know what you think by e-mailing [email protected].
HM11: 70-Plus Sessions of Best Practices in Hospital Medicine
Hospitalists of all stripes—educators, practice leaders, clinicians, and others—will find the courses and specialty leaders that can help them advance their careers at HM11, May 10-13 at the Gaylord Texan Resort and Convention Center in Grapevine, Texas.
HM11 sessions offer an unprecedented diversity of topics in hospital medicine, all presented in nine easy-to-follow tracks based on areas of interest:
- Academics and Research: A track specifically designed for hospitalists in academic medical centers or who are interested in research.
- Clinical 1: This track focuses on the essential topics in adult clinical medicine and emphasizes recent advances in HM that should be incorporated into a hospitalist’s approach to clinical-care delivery.
- Clinical 2: This track presents issues and solutions for additional clinical topics beyond the essentials presented in the Clinical 1 track.
- Evidence-Based Rapid Fire: This track gives participants “rapid bursts” of information and addresses 10 of the most pressing clinical issues in HM, based on the highest level of medical evidence available.
- Palliative Care: The track features presentations by experts with insight into the issues of dealing with death with dignity, advanced pain-management strategies, and issues that go beyond pain management.
- Pediatric: This track will focus on pediatric hospitalist practice management, as well as current clinical issues encountered by pediatric hospitalists.
- Practice Management: This year’s topics will focus on understanding key healthcare reform, financial drivers, employment models, improving patient satisfaction, ED throughput, mitigating legal risks, and the results of the first NPP hospitalist survey.
- Quality: Given the importance of quality and patient safety in the delivery of healthcare, this track will address the imperatives around development and implementation of QI efforts in the hospital.
- Workshops: Back by popular demand, workshop topics were submitted by members, underwent a peer review, and were selected based on their relevancy to hospitalists.
To find out more or register for HM11, visit www.hospitalmedicine2011.org.
Hospitalists of all stripes—educators, practice leaders, clinicians, and others—will find the courses and specialty leaders that can help them advance their careers at HM11, May 10-13 at the Gaylord Texan Resort and Convention Center in Grapevine, Texas.
HM11 sessions offer an unprecedented diversity of topics in hospital medicine, all presented in nine easy-to-follow tracks based on areas of interest:
- Academics and Research: A track specifically designed for hospitalists in academic medical centers or who are interested in research.
- Clinical 1: This track focuses on the essential topics in adult clinical medicine and emphasizes recent advances in HM that should be incorporated into a hospitalist’s approach to clinical-care delivery.
- Clinical 2: This track presents issues and solutions for additional clinical topics beyond the essentials presented in the Clinical 1 track.
- Evidence-Based Rapid Fire: This track gives participants “rapid bursts” of information and addresses 10 of the most pressing clinical issues in HM, based on the highest level of medical evidence available.
- Palliative Care: The track features presentations by experts with insight into the issues of dealing with death with dignity, advanced pain-management strategies, and issues that go beyond pain management.
- Pediatric: This track will focus on pediatric hospitalist practice management, as well as current clinical issues encountered by pediatric hospitalists.
- Practice Management: This year’s topics will focus on understanding key healthcare reform, financial drivers, employment models, improving patient satisfaction, ED throughput, mitigating legal risks, and the results of the first NPP hospitalist survey.
- Quality: Given the importance of quality and patient safety in the delivery of healthcare, this track will address the imperatives around development and implementation of QI efforts in the hospital.
- Workshops: Back by popular demand, workshop topics were submitted by members, underwent a peer review, and were selected based on their relevancy to hospitalists.
To find out more or register for HM11, visit www.hospitalmedicine2011.org.
Hospitalists of all stripes—educators, practice leaders, clinicians, and others—will find the courses and specialty leaders that can help them advance their careers at HM11, May 10-13 at the Gaylord Texan Resort and Convention Center in Grapevine, Texas.
HM11 sessions offer an unprecedented diversity of topics in hospital medicine, all presented in nine easy-to-follow tracks based on areas of interest:
- Academics and Research: A track specifically designed for hospitalists in academic medical centers or who are interested in research.
- Clinical 1: This track focuses on the essential topics in adult clinical medicine and emphasizes recent advances in HM that should be incorporated into a hospitalist’s approach to clinical-care delivery.
- Clinical 2: This track presents issues and solutions for additional clinical topics beyond the essentials presented in the Clinical 1 track.
- Evidence-Based Rapid Fire: This track gives participants “rapid bursts” of information and addresses 10 of the most pressing clinical issues in HM, based on the highest level of medical evidence available.
- Palliative Care: The track features presentations by experts with insight into the issues of dealing with death with dignity, advanced pain-management strategies, and issues that go beyond pain management.
- Pediatric: This track will focus on pediatric hospitalist practice management, as well as current clinical issues encountered by pediatric hospitalists.
- Practice Management: This year’s topics will focus on understanding key healthcare reform, financial drivers, employment models, improving patient satisfaction, ED throughput, mitigating legal risks, and the results of the first NPP hospitalist survey.
- Quality: Given the importance of quality and patient safety in the delivery of healthcare, this track will address the imperatives around development and implementation of QI efforts in the hospital.
- Workshops: Back by popular demand, workshop topics were submitted by members, underwent a peer review, and were selected based on their relevancy to hospitalists.
To find out more or register for HM11, visit www.hospitalmedicine2011.org.
The Cutting Edge
Since the beginning of formal medical education, one of the biggest challenges in treating patients has been learning from mistakes. How do providers balance the potentially grave consequences of medical mistakes with the possibilities of improving patient care?
While the conundrum is far from solved, hospitalists at HM11’s “Advanced Interactive Critical Care” pre-course will get hands-on experience in the newest techniques in patient care without affecting real patients. The pre-course will use simulators to replicate real-life situations with critical-care patients.
For the annual meeting’s course director, the simulators are the next step in training hospitalists. “This is really exciting,” says Daniel D. Dressler, MD, MSc, FHM, associate professor and director of internal-medicine teaching services at Emory University Hospital in Atlanta. “As an educator, it’s really something unique. We learn the best by actually doing and sometimes making mistakes; it’s better to do that in simulations with expert advisors offering immediate feedback.
“Not only will pre-course attendees get high-quality didactic information, but they will also participate in the simulation of critical-care events with very experienced faculty,” he says.

Pre-course participants will have the option of having more hands-on time with the simulators in lieu of lectures. Dr. Dressler, an SHM board member, calls the opportunity to work on cutting-edge technology “exceedingly unique.”
The primary simulator will look, feel, and sound like a real person with actual physiological parameters. It breathes and has a pulse—or, in some cases, doesn’t have a pulse. Dressler says participants will be in front of a simulated patient who is going through septic shock or having airway or ventilator problems. While interacting with colleagues and instructors, participants will diagnose and treat the simulated patient. The simulator can replicate real-life complications that can result from treatments.
The pre-course materials state that after completing the course, participants will be able to:
- Explain basic and advanced mechanical ventilator physiology and strategies for complex situations, including acute respiratory distress syndrome (ARDS) and troubleshooting ventilator problems;
- Integrate physiology with treatment of common and less common forms of shock; and
- Apply appropriate sedation and analgesia strategies to minimize delirium in the ICU, and optimize ventilator weaning.
Additionally, the pre-course will present, model, and practice the latest in evidence-based, critical-care practice. Though the pre-course is called “advanced,” it is open to any hospitalist looking to improve their critical-care skills.
“We were getting feedback that people were ready for the next level,” Dr. Dressler says. Along with the basics of critical care (e.g. sepsis, sedation), the pre-course will cover more advanced issues. “We understand that many people have been practicing critical care for a number of years. This course will benefit any hospitalist, regardless of prior critical care experience.”
Led by instructors Kevin Felner, MD, and Brian Kaufman, MD, of New York University Medical Center, “Advanced Interactive Critical Care” begins at 8:50 a.m. May 10. However, the first simulation session begins at 7:20 a.m., and pre-registration is required.
Another new HM11 pre-course will focus solely on using handheld ultrasound devices. The appeal of ultrasounds for use in hospitalists’ rounds is increasing; this course will train hospitalists to use them to look at patients’ vessels, heart, and abdomen.
Hospitalists who take the portable ultrasound pre-course in conjunction with the “Medical Procedures for the Hospitalist” pre-course will receive a full day of the best training in the practice and tools used in medical procedures. The procedures course runs from 8 a.m. to noon May 10. The portable ultrasound course runs from 1:30 to 5:30 p.m. Both pre-courses are taught by Bradley T. Rosen, MD, MBA, FHM, of Cedars-Sinai Medical Center in Los Angeles and Sally Wang, MD, FHM, of Brigham and Women’s Hospital in Boston. TH
Brendon Shank is SHM’s vice president of communications.
Since the beginning of formal medical education, one of the biggest challenges in treating patients has been learning from mistakes. How do providers balance the potentially grave consequences of medical mistakes with the possibilities of improving patient care?
While the conundrum is far from solved, hospitalists at HM11’s “Advanced Interactive Critical Care” pre-course will get hands-on experience in the newest techniques in patient care without affecting real patients. The pre-course will use simulators to replicate real-life situations with critical-care patients.
For the annual meeting’s course director, the simulators are the next step in training hospitalists. “This is really exciting,” says Daniel D. Dressler, MD, MSc, FHM, associate professor and director of internal-medicine teaching services at Emory University Hospital in Atlanta. “As an educator, it’s really something unique. We learn the best by actually doing and sometimes making mistakes; it’s better to do that in simulations with expert advisors offering immediate feedback.
“Not only will pre-course attendees get high-quality didactic information, but they will also participate in the simulation of critical-care events with very experienced faculty,” he says.
Pre-course participants will have the option of having more hands-on time with the simulators in lieu of lectures. Dr. Dressler, an SHM board member, calls the opportunity to work on cutting-edge technology “exceedingly unique.”
The primary simulator will look, feel, and sound like a real person with actual physiological parameters. It breathes and has a pulse—or, in some cases, doesn’t have a pulse. Dressler says participants will be in front of a simulated patient who is going through septic shock or having airway or ventilator problems. While interacting with colleagues and instructors, participants will diagnose and treat the simulated patient. The simulator can replicate real-life complications that can result from treatments.
The pre-course materials state that after completing the course, participants will be able to:
- Explain basic and advanced mechanical ventilator physiology and strategies for complex situations, including acute respiratory distress syndrome (ARDS) and troubleshooting ventilator problems;
- Integrate physiology with treatment of common and less common forms of shock; and
- Apply appropriate sedation and analgesia strategies to minimize delirium in the ICU, and optimize ventilator weaning.
Additionally, the pre-course will present, model, and practice the latest in evidence-based, critical-care practice. Though the pre-course is called “advanced,” it is open to any hospitalist looking to improve their critical-care skills.
“We were getting feedback that people were ready for the next level,” Dr. Dressler says. Along with the basics of critical care (e.g. sepsis, sedation), the pre-course will cover more advanced issues. “We understand that many people have been practicing critical care for a number of years. This course will benefit any hospitalist, regardless of prior critical care experience.”
Led by instructors Kevin Felner, MD, and Brian Kaufman, MD, of New York University Medical Center, “Advanced Interactive Critical Care” begins at 8:50 a.m. May 10. However, the first simulation session begins at 7:20 a.m., and pre-registration is required.
Another new HM11 pre-course will focus solely on using handheld ultrasound devices. The appeal of ultrasounds for use in hospitalists’ rounds is increasing; this course will train hospitalists to use them to look at patients’ vessels, heart, and abdomen.
Hospitalists who take the portable ultrasound pre-course in conjunction with the “Medical Procedures for the Hospitalist” pre-course will receive a full day of the best training in the practice and tools used in medical procedures. The procedures course runs from 8 a.m. to noon May 10. The portable ultrasound course runs from 1:30 to 5:30 p.m. Both pre-courses are taught by Bradley T. Rosen, MD, MBA, FHM, of Cedars-Sinai Medical Center in Los Angeles and Sally Wang, MD, FHM, of Brigham and Women’s Hospital in Boston. TH
Brendon Shank is SHM’s vice president of communications.
Since the beginning of formal medical education, one of the biggest challenges in treating patients has been learning from mistakes. How do providers balance the potentially grave consequences of medical mistakes with the possibilities of improving patient care?
While the conundrum is far from solved, hospitalists at HM11’s “Advanced Interactive Critical Care” pre-course will get hands-on experience in the newest techniques in patient care without affecting real patients. The pre-course will use simulators to replicate real-life situations with critical-care patients.
For the annual meeting’s course director, the simulators are the next step in training hospitalists. “This is really exciting,” says Daniel D. Dressler, MD, MSc, FHM, associate professor and director of internal-medicine teaching services at Emory University Hospital in Atlanta. “As an educator, it’s really something unique. We learn the best by actually doing and sometimes making mistakes; it’s better to do that in simulations with expert advisors offering immediate feedback.
“Not only will pre-course attendees get high-quality didactic information, but they will also participate in the simulation of critical-care events with very experienced faculty,” he says.
Pre-course participants will have the option of having more hands-on time with the simulators in lieu of lectures. Dr. Dressler, an SHM board member, calls the opportunity to work on cutting-edge technology “exceedingly unique.”
The primary simulator will look, feel, and sound like a real person with actual physiological parameters. It breathes and has a pulse—or, in some cases, doesn’t have a pulse. Dressler says participants will be in front of a simulated patient who is going through septic shock or having airway or ventilator problems. While interacting with colleagues and instructors, participants will diagnose and treat the simulated patient. The simulator can replicate real-life complications that can result from treatments.
The pre-course materials state that after completing the course, participants will be able to:
- Explain basic and advanced mechanical ventilator physiology and strategies for complex situations, including acute respiratory distress syndrome (ARDS) and troubleshooting ventilator problems;
- Integrate physiology with treatment of common and less common forms of shock; and
- Apply appropriate sedation and analgesia strategies to minimize delirium in the ICU, and optimize ventilator weaning.
Additionally, the pre-course will present, model, and practice the latest in evidence-based, critical-care practice. Though the pre-course is called “advanced,” it is open to any hospitalist looking to improve their critical-care skills.
“We were getting feedback that people were ready for the next level,” Dr. Dressler says. Along with the basics of critical care (e.g. sepsis, sedation), the pre-course will cover more advanced issues. “We understand that many people have been practicing critical care for a number of years. This course will benefit any hospitalist, regardless of prior critical care experience.”
Led by instructors Kevin Felner, MD, and Brian Kaufman, MD, of New York University Medical Center, “Advanced Interactive Critical Care” begins at 8:50 a.m. May 10. However, the first simulation session begins at 7:20 a.m., and pre-registration is required.
Another new HM11 pre-course will focus solely on using handheld ultrasound devices. The appeal of ultrasounds for use in hospitalists’ rounds is increasing; this course will train hospitalists to use them to look at patients’ vessels, heart, and abdomen.
Hospitalists who take the portable ultrasound pre-course in conjunction with the “Medical Procedures for the Hospitalist” pre-course will receive a full day of the best training in the practice and tools used in medical procedures. The procedures course runs from 8 a.m. to noon May 10. The portable ultrasound course runs from 1:30 to 5:30 p.m. Both pre-courses are taught by Bradley T. Rosen, MD, MBA, FHM, of Cedars-Sinai Medical Center in Los Angeles and Sally Wang, MD, FHM, of Brigham and Women’s Hospital in Boston. TH
Brendon Shank is SHM’s vice president of communications.
In the Literature: HM-Related Research You Need to Know
In This Edition
Literature at a Glance
A guide to this month’s studies
- Effects of extended VTE prophylaxis in medical patients
- Outcomes with and without preprocedural statins
- Association of subclinical hypothyroidism and CHD
- BP treatment after intracerebral hemorrhage
- Outcomes of ICD therapy in the elderly
- Systems delays and outcomes of STEMI
- D-dimer to predict recurrent VTE
- Stocking height and risk of post-stroke DVT
Extending Anticoagulant Prophylaxis after Medical Hospitalization Decreases VTE, Increases Major Bleeding
Clinical question: For patients with acute medical illness, does extending low-molecular-weight heparin (LMWH) administration for up to 28 days after discharge reduce the incidence of venous thromboembolism (VTE)?
Background: DVT and pulmonary embolism (PE) are common hospital-acquired complications. LMWH has been shown to reduce VTE for medical and surgical patients, and extended-duration LMWH reduces VTE in high-risk surgical patients. Whether extending anticoagulant prophylaxis after discharge for acutely ill medical patients with reduced mobility improves outcomes is unknown.
Study design: Randomized, placebo-controlled trial.
Setting: Three hundred seventy hospitals in 20 countries.
Synopsis: Eligible patients were >40 years old, hospitalized with acute medical illness, and had reduced mobility for ≥3 days. Patients received enoxaparin 40 mg SC daily prophylaxis while hospitalized and were then randomized to an additional 28±4 days of enoxaparin or placebo. Patients received a screening ultrasound to assess for asymptomatic DVT. The primary outcome was a composite of asymptomatic proximal DVT, symptomatic DVT or PE, or fatal PE during the period of extended prophylaxis.
An interim analysis indicated that extended prophylaxis was ineffective; at that time, the protocol was amended to target patients with severe immobility or with moderate immobility plus an additional risk factor (e.g. cancer, prior VTE, or age >75).
The study found that extended prophylaxis decreased the composite VTE outcome (2.5% vs 4.0%, P<0.05) and symptomatic VTE (0.2% vs 1.0%, P<0.05). The incidence of major bleeding was increased in the extended prophylaxis group (0.8% vs 0.3%, P<0.05). There was no difference in mortality.
The unplanned, midstudy protocol amendment to target higher-risk patients is a concern, though the final analyses included patients pre- and post-amendment.
Bottom line: Extending LMWH beyond hospitalization for patients admitted with acute medical illness and decreased mobility decreases VTE, but increases major bleeding.
Citation: Hull RD, Schellong SM, Tapson VF, et al. Extended-duration venous thromboembolism prophylaxis in acutely ill medical patients with recently reduced mobility: a randomized trial. Ann Intern Med. 2010;153(1);8-18.
Preprocedural Statin Therapy Reduces Postprocedural Myocardial Infarction
Clinical question: Does statin therapy reduce periprocedural cardiovascular events?
Background: Myocardial infarction (MI) and death are inherent risks of invasive procedures. Reduction of these risks in certain patient populations has been shown with the use of a beta blockade. Statins have shown promise during acute coronary syndrome. Questions remain about the role of statin therapy before invasive procedures in reducing adverse cardiovascular events.
Study design: Meta-analysis of randomized controlled trials.
Setting: Twenty-one studies involving 4,805 patients, published from inception of MEDLINE, Cochrane, and Clinicaltrials to February 2010.
Synopsis: The use of statins one to seven days preprocedure significantly reduced post-procedural MI in percutaneous coronary interventions (PCI) (P<0.0001). Statins given approximately four weeks in advance of noncardiac surgical procedures also significantly reduced postprocedural MI (P=0.004). An absolute risk reduction of 5.8% for postprocedural MI was found after PCI and 4.1% in noncardiac surgical procedures.
Statins did not show a significant reduction in postprocedural MI (P=0.40) or all-cause mortality (P=0.15) in coronary artery bypass graft surgery (CABG). However, statins did reduce post-CABG atrial fibrillation (P<0.0001).
The 21 studies used a variety of drugs and doses. However, the PCI studies favored atorvastatin 40 mg; more than half the CABG studies used atorvastatin 20 mg; and 91% of the noncardiac surgical studies used fluvastatin 80 mg. Dedicated trials are needed to demonstrate optimal statin agent, dose, and timing of therapy.
Bottom line: Preprocedural statin therapy reduces postprocedural MI after both PCI and noncardiac procedures but not after CABG.
Citation: Winchester DE, Wen X, Xie L, Bavry AA. Evidence of pre-procedural statin therapy: a meta-analysis of randomized trials. J Am Coll Cardiol. 2010;56(19); 1099-1109.
Subclinical Hypothyroidism Increases the Risk of Coronary Heart Disease and Mortality
Clinical question: What are the risks of coronary heart disease (CHD) and mortality among adults with subclinical hypothyroidism?
Background: Subclinical hypo-thyroidism is defined as an elevated serum thyroid stimulating hormone (TSH) level with a normal T4 concentration. Controversy exists regarding the treatment of subclinical hypothyroidism. Because of the association with hyperlipidemia and atherosclerosis, treatment of subclinical hypothyroidism is thought to be beneficial. Previous data from large prospective cohort studies regarding this association are conflicting.
Study design: Study-level meta-analysis of prospective cohort studies.
Setting: Eleven prospective cohorts in the U.S., Europe, Australia, Brazil, and Japan from 1972 to 2007.
Synopsis: Among 55,287 adults, 3,450 (6.2%) had subclinical hypothyroidism and 51,837 were euthyroid. Using Cox proportional hazard models, the association of subclinical hypothyroidism with CHD and mortality were determined for each cohort.
The risk of CHD events and CHD mortality increased with higher TSH concentrations.
In age- and sex-adjusted analyses, the hazard ratio (HR) for CHD events were as follows: HR=1.0 (TSH=4.5-6.9 mIU/L); HR=1.17 (TSH=7-9.9 mIU/L), and HR=1.89 (TSH=10-19.9 mIU/L). Similarly, HRs for CHD mortality showed an increasing trend: 1.09, 1.42, and 1.58, respectively.
Although the association is clearly established here, randomized controlled trials are needed to address whether thyroxine replacement can prevent CHD and the TSH threshold that will provide the most clinical benefit.
Bottom line: Subclinical hypo-thyroidism is associated with an increased risk for CHD events and mortality, primarily in patients with TSH concentrations of 10 mIU/L or higher.
Citation: Rodondi N, den Elzen WP, Bauer DC, et al. Subclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA. 2010;304(12): 1365-1374.
Reduction in Hematoma Growth after Acute Intracerebral Hemorrhage Associated with Lower Blood Pressure
Clinical question: Does intensive systolic blood pressure (SBP) <140 mmHg within one hour reduce hematoma growth after acute intracerebral bleeding?
Background: Early elevation of blood pressure after an acute intracerebral bleed is strongly associated with hematoma growth and worse outcomes. The pilot phase of the Intensive Blood Pressure Reduction in Acute Cerebral Hemorrhage Trial (INTERACT) showed that early intensive blood pressure lowering reduced hematoma growth within six hours after onset of intracerebral hemorrhage.
Study design: Randomized controlled trial in which patients received an early intensive blood-pressure-lowering treatment (goal SBP<140 mmHg within one hour) or the AHA recommended best practice guidelines (goal SBP <180 mmHg), which were published in 1999.
Setting: Network of hospitals in China, South Korea, and Australia.
Synopsis: From 2005 to 2007, the study examined 404 patients with computed-tomography (CT) evidence of intracerebral hemorrhage, elevated SBP (150 mmHg-220 mmHg), and ability to commence BP lowering treatment within six hours of onset. Hematoma volumes were measured in the intensive treatment and guideline groups based on CT scans done at baseline and after 24 hours.
No significant association was found between the baseline SBP and the absolute or proportional growth of the hematoma. However, in the intensively treated patients who achieved target SBP within the first 24 hours, a significant association was found with the absolute and proportional hematoma growth.
Maximum reduction of hematoma growth occurred in the group with a median SBP of 135 mmHg.
This single-observational study did not include patients with severe intracranial bleeding who died or required surgical evacuation within the first 24 hours.
Hematoma size reduction did not improve survival or outcomes.
Bottom line: Intensive SBP lowering from 140 mmHg to 130 mmHg within one hour reduces hematoma growth after an intracranial hemorrhage.
Citation: Arima H, Anderson CS, Wang JG, et al. Lower treatment blood pressure is associated with the greatest reduction in hematoma growth after acute intracerebral hemorrhage. Hypertension. 2010;56(5):852-858.
Implantable Cardioverter-Defibrillator (ICD) Therapy for Primary Prevention of Sudden Cardiac Death Might Not Provide Survival Benefit to Elderly
Clinical question: Does ICD therapy for primary prevention of sudden cardiac death in individuals with severe left ventricular dysfunction improve survival in elderly patients?
Background: Several clinical trials of select individuals with severe left ventricular dysfunction (EF <40%) have demonstrated that ICD therapy is associated with a reduction in overall mortality. Given the costs and risks associated with ICD placement, it is important to assess how this therapy affects survival in younger versus older individuals.
Study design: Meta-analysis of five randomized controlled trials.
Setting: Five trials (MADIT-II, DEFINITE, DINAMIT, SCDHeFT, and IRIS).
Synopsis: All included studies compared ICD therapy to standard medical care in the primary prevention of sudden cardiac death in individuals with severe left ventricular dysfunction. Elderly patients comprised 44% of the 5,783 patients included in the study; elderly patients were defined as >65 in three studies, and >60 in two studies. Pooled analysis of the three trials examining ICD use for primary prevention found a nonsignificant reduction in all-cause mortality compared with medical therapy in elderly patients (HR 0.81 [95% CI 0.62 to 1.05], P=0.11). The two remaining studies involving post-MI patients showed no statistically significant reduction in mortality in elderly patients.
In contrast, pooled data from younger patients in the five trials showed that prophylactic ICD therapy reduced all-cause mortality.
None of the selected trials demonstrated evidence of selection, performance, detection, or attrition bias. Only a small number of studies were included, and four potentially relevant trials were not included because mortality data by age group were not available.
Trials including cardiac resynchronization therapy (CRT) were excluded from this meta-analysis, and elderly patients meeting criteria for CRT comprise an important group that must be considered separately.
Bottom line: There is no definitive reduction in mortality with prophylactic ICD therapy in elderly patients with severe left ventricular dysfunction.
Citation: Santangeli P, Di Biase L, Dello Russo A, et al. Meta-analysis: age and effectiveness of prophylactic implantable cardioverter-defibrillators. Ann Intern Med. 2010;153(9):592-599.
System Delay Is Mortality Marker in STEMI Patients Treated with Primary PCI
Clinical question: What is the relationship between system delays to reperfusion therapy and mortality in patients with ST-segment-elevation myocardial infarction (STEMI)?
Background: In patients with STEMI, an early reperfusion strategy is often sought, and several studies have focused on the association between door-to-balloon delay and outcomes. Focusing more broadly on the time from first contact with the healthcare system to the initiation of reperfusion therapy (system delay) might be a more relevant approach.
Study design: Historical follow-up study.
Setting: Three high-volume PCI centers in Denmark.
Synopsis: Using population-based medical registries of 6,209 Danish patients treated for STEMI with percutaneous coronary intervention (PCI), the authors examined the association between delays in reperfusion therapy and mortality. System delay encompassed the entire time from first contact with the healthcare system to the initiation of reperfusion therapy.
Overall, shorter system delay was associated with significantly decreased mortality, as were the individual components of system delay (prehospital delay and door-to-balloon delay). The shortest delays (0-60 minutes) corresponded to a mortality rate of 15%, and the longest delays (181-360 minutes) corresponded to a mortality rate of 31%. Patients were followed for a median of 3.4 years.
This study is unique, as it is the first to look at the association between system delay and outcomes in patients with STEMI transported by EMS and treated with primary PCI. The study highlights the harmful impact of longer system delays on mortality. Limitations of this study include possible underestimation of system delay and challenges surrounding the right marker for reperfusion.
Bottom line: System delay might serve as a broad, comprehensive marker for predicting mortality in patients with STEMI treated with primary PCI.
Citation: Terkelsen CJ, Sorensen JT, Maeng M, et al. System delay and mortality among patients with STEMI treated with primary percutaneous coronary intervention. JAMA. 2010;304(7): 763-771.
D-Dimer Is Effective in Determining the Risk of VTE Recurrence after First Unprovoked Event
Clinical question: Does the timing, patient age, or cut point level affect the predictive value of a D-dimer in predicting VTE recurrence after a first unprovoked event?
Background: Anticoagulation duration after a first unprovoked VTE is at least three months, but it can be indefinite in patients with stable anticoagulation and low bleeding risk. Measuring a D-dimer level after discontinuation of anticoagulation is helpful in determining which patients might benefit from prolonged anticoagulation. However, several unanswered questions remain regarding D-dimer testing.
Study design: Patient-level meta-analysis.
Setting: Pooled patient-level data from seven prospective studies.
Synopsis: Patient-level data were obtained for all patients enrolled using post-treatment D-dimer measurement to predict recurrent VTE in patients with a first unprovoked VTE who had completed at least three months of anticoagulation therapy. The mean length of follow-up was 30 months. Patients with a positive D-dimer had recurrent VTE at a rate of 8.8 per 100 patient-years while those with a negative D-dimer had a rate of 3.7 per 100 patient-years.
Univariate analysis revealed an HR of 2.59 for patients with a positive versus a negative test result. The analysis also showed that the timing of the test, the age of the patient, and the actual cut points used for the various D-dimer tests did not affect the analysis significantly.
These studies’ strength is their large sample sizes and the use of prospective studies. The weaknesses include a mostly white patient population and incomplete data on all patients.
Bottom line: D-dimer testing is useful in predicting VTE recurrence after treatment for a first unprovoked event regardless of patient age, post-treatment timing, or the assay cut point used.
Citation: Douketis J, Tosetto A, Marcucci M, et al. Patient-level meta-analysis: effect of measurement timing, threshold, and patient age on ability of D-dimer testing to assess recurrence risk after unprovoked venous thromboembolism. Ann Intern Med. 2010;153(8): 523-531.
Thigh-High Stockings Are Better than Knee-High Stockings for Post-Stroke DVT Prophylaxis
Clinical question: Are thigh-high compression stockings better then knee-high stockings in immobilized acute-stroke patients?
Background: DVT is common in hospitalized stroke patients with immobility. Graduated compression stockings are often used for DVT prophylaxis, but the CLOTS-1 trial recently found that thigh-high stockings were ineffective after acute stroke. It is unclear if the more commonly used knee-high stockings are more effective than thigh-high stockings.
Study design: Parallel-group trial (the CLOTS-2 trial).
Setting: One hundred twelve hospitals in nine countries.
Synopsis: More than 3,100 patients with acute stroke and immobilization were recruited from January 2002 to May 2009. Patients were randomized to receive thigh-high or knee-high stockings. Patients also received usual care, including anticoagulants and a screening ultrasound for asymptomatic proximal DVT at seven to 10 days. Approximately 640 patients in each group also underwent ultrasound at 25-30 days.
Overall, 6.3% of patients in the thigh-high group had DVT, compared with 8.8% in the knee-high group (P=0.007). There were no significant differences in the secondary outcomes of pulmonary embolism or death. The thigh-high stockings had a higher number of adverse skin events. Enrollment was stopped early when the CLOTS-1 trial showed no difference in DVT rates between thigh-high stockings and no stockings.
Bottom line: Knee-high graduated compression stockings lead to worse outcomes than thigh-high stockings for DVT prophylaxis in immobilized acute-stroke patients.
Citation: CLOTS (Clots in Legs Or sTockings after Stroke) Trial Collaboration. Thigh-length versus below-knee stockings for deep venous thrombosis prophylaxis after stroke: a randomized trial. Ann Intern Med. 2010;153(9):553-562. TH
Pediatric HM Literature
Co-Infection with Pertussis in Infants Hospitalized for Bronchiolitis

Clinical question: How often are infants admitted with bronchiolitis co-infected with Bordatella pertussis?
Background: Infants admitted for bronchiolitis have previously been reported to have co-infection with B. pertussis. Given the nonspecific symptoms associated with early B. pertussis infection, as well as the epidemiology of ongoing outbreaks, it might be useful to better define the risk of co-infection in the seasonal surge of acute bronchiolitis.
Study design: Retrospective cohort study.
Setting: One university hospital in Finland.
Synopsis: A prior study had generated nasopharyngeal aspirate samples for viral antigen detection from 205 healthy, full-term infants younger than 6 months hospitalized for bronchiolitis from 2001 to 2004. Of these samples, 142 (69%) were of quality sufficient for B. pertussis PCR testing in 2009.
Twelve (8.5%) of the 142 infants admitted with bronchiolitis were found to have B. pertussis. Eleven of the 12 infants were co-infected with at least one other virus (RSV in eight of them). Infants who tested positive for pertussis were more often found to have coughing spells (41.7% vs. 14.6% in those who tested negative).
The reported rate of co-infection in this study is higher than other recent reports from the U.S., likely due to the fact that samples were collected during a time period when the incidence of pertussis was relatively high in Finland.
Nevertheless, given the magnitude of a recent pertussis outbreak in California, it might be useful to consider co-infection in young infants admitted for bronchiolitis in areas with a relatively higher incidence of pertussis.
Although the retrospective nature of this study and the lack of a reported definition of coughing spells make further conclusions difficult to draw, cough might deserve further scrutiny during bronchiolitis season.
Bottom line: Consider co-infection with pertussis in young infants admitted with bronchiolitis.
Citation: Nuolivirta K, Koponen P, He Q, et al. Bordatella pertussis infection is common in nonvaccinated infants admitted for bronchiolitis. Pediatr Infect Dis J. 2010;29(11):1013-1015.
In This Edition
Literature at a Glance
A guide to this month’s studies
- Effects of extended VTE prophylaxis in medical patients
- Outcomes with and without preprocedural statins
- Association of subclinical hypothyroidism and CHD
- BP treatment after intracerebral hemorrhage
- Outcomes of ICD therapy in the elderly
- Systems delays and outcomes of STEMI
- D-dimer to predict recurrent VTE
- Stocking height and risk of post-stroke DVT
Extending Anticoagulant Prophylaxis after Medical Hospitalization Decreases VTE, Increases Major Bleeding
Clinical question: For patients with acute medical illness, does extending low-molecular-weight heparin (LMWH) administration for up to 28 days after discharge reduce the incidence of venous thromboembolism (VTE)?
Background: DVT and pulmonary embolism (PE) are common hospital-acquired complications. LMWH has been shown to reduce VTE for medical and surgical patients, and extended-duration LMWH reduces VTE in high-risk surgical patients. Whether extending anticoagulant prophylaxis after discharge for acutely ill medical patients with reduced mobility improves outcomes is unknown.
Study design: Randomized, placebo-controlled trial.
Setting: Three hundred seventy hospitals in 20 countries.
Synopsis: Eligible patients were >40 years old, hospitalized with acute medical illness, and had reduced mobility for ≥3 days. Patients received enoxaparin 40 mg SC daily prophylaxis while hospitalized and were then randomized to an additional 28±4 days of enoxaparin or placebo. Patients received a screening ultrasound to assess for asymptomatic DVT. The primary outcome was a composite of asymptomatic proximal DVT, symptomatic DVT or PE, or fatal PE during the period of extended prophylaxis.
An interim analysis indicated that extended prophylaxis was ineffective; at that time, the protocol was amended to target patients with severe immobility or with moderate immobility plus an additional risk factor (e.g. cancer, prior VTE, or age >75).
The study found that extended prophylaxis decreased the composite VTE outcome (2.5% vs 4.0%, P<0.05) and symptomatic VTE (0.2% vs 1.0%, P<0.05). The incidence of major bleeding was increased in the extended prophylaxis group (0.8% vs 0.3%, P<0.05). There was no difference in mortality.
The unplanned, midstudy protocol amendment to target higher-risk patients is a concern, though the final analyses included patients pre- and post-amendment.
Bottom line: Extending LMWH beyond hospitalization for patients admitted with acute medical illness and decreased mobility decreases VTE, but increases major bleeding.
Citation: Hull RD, Schellong SM, Tapson VF, et al. Extended-duration venous thromboembolism prophylaxis in acutely ill medical patients with recently reduced mobility: a randomized trial. Ann Intern Med. 2010;153(1);8-18.
Preprocedural Statin Therapy Reduces Postprocedural Myocardial Infarction
Clinical question: Does statin therapy reduce periprocedural cardiovascular events?
Background: Myocardial infarction (MI) and death are inherent risks of invasive procedures. Reduction of these risks in certain patient populations has been shown with the use of a beta blockade. Statins have shown promise during acute coronary syndrome. Questions remain about the role of statin therapy before invasive procedures in reducing adverse cardiovascular events.
Study design: Meta-analysis of randomized controlled trials.
Setting: Twenty-one studies involving 4,805 patients, published from inception of MEDLINE, Cochrane, and Clinicaltrials to February 2010.
Synopsis: The use of statins one to seven days preprocedure significantly reduced post-procedural MI in percutaneous coronary interventions (PCI) (P<0.0001). Statins given approximately four weeks in advance of noncardiac surgical procedures also significantly reduced postprocedural MI (P=0.004). An absolute risk reduction of 5.8% for postprocedural MI was found after PCI and 4.1% in noncardiac surgical procedures.
Statins did not show a significant reduction in postprocedural MI (P=0.40) or all-cause mortality (P=0.15) in coronary artery bypass graft surgery (CABG). However, statins did reduce post-CABG atrial fibrillation (P<0.0001).
The 21 studies used a variety of drugs and doses. However, the PCI studies favored atorvastatin 40 mg; more than half the CABG studies used atorvastatin 20 mg; and 91% of the noncardiac surgical studies used fluvastatin 80 mg. Dedicated trials are needed to demonstrate optimal statin agent, dose, and timing of therapy.
Bottom line: Preprocedural statin therapy reduces postprocedural MI after both PCI and noncardiac procedures but not after CABG.
Citation: Winchester DE, Wen X, Xie L, Bavry AA. Evidence of pre-procedural statin therapy: a meta-analysis of randomized trials. J Am Coll Cardiol. 2010;56(19); 1099-1109.
Subclinical Hypothyroidism Increases the Risk of Coronary Heart Disease and Mortality
Clinical question: What are the risks of coronary heart disease (CHD) and mortality among adults with subclinical hypothyroidism?
Background: Subclinical hypo-thyroidism is defined as an elevated serum thyroid stimulating hormone (TSH) level with a normal T4 concentration. Controversy exists regarding the treatment of subclinical hypothyroidism. Because of the association with hyperlipidemia and atherosclerosis, treatment of subclinical hypothyroidism is thought to be beneficial. Previous data from large prospective cohort studies regarding this association are conflicting.
Study design: Study-level meta-analysis of prospective cohort studies.
Setting: Eleven prospective cohorts in the U.S., Europe, Australia, Brazil, and Japan from 1972 to 2007.
Synopsis: Among 55,287 adults, 3,450 (6.2%) had subclinical hypothyroidism and 51,837 were euthyroid. Using Cox proportional hazard models, the association of subclinical hypothyroidism with CHD and mortality were determined for each cohort.
The risk of CHD events and CHD mortality increased with higher TSH concentrations.
In age- and sex-adjusted analyses, the hazard ratio (HR) for CHD events were as follows: HR=1.0 (TSH=4.5-6.9 mIU/L); HR=1.17 (TSH=7-9.9 mIU/L), and HR=1.89 (TSH=10-19.9 mIU/L). Similarly, HRs for CHD mortality showed an increasing trend: 1.09, 1.42, and 1.58, respectively.
Although the association is clearly established here, randomized controlled trials are needed to address whether thyroxine replacement can prevent CHD and the TSH threshold that will provide the most clinical benefit.
Bottom line: Subclinical hypo-thyroidism is associated with an increased risk for CHD events and mortality, primarily in patients with TSH concentrations of 10 mIU/L or higher.
Citation: Rodondi N, den Elzen WP, Bauer DC, et al. Subclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA. 2010;304(12): 1365-1374.
Reduction in Hematoma Growth after Acute Intracerebral Hemorrhage Associated with Lower Blood Pressure
Clinical question: Does intensive systolic blood pressure (SBP) <140 mmHg within one hour reduce hematoma growth after acute intracerebral bleeding?
Background: Early elevation of blood pressure after an acute intracerebral bleed is strongly associated with hematoma growth and worse outcomes. The pilot phase of the Intensive Blood Pressure Reduction in Acute Cerebral Hemorrhage Trial (INTERACT) showed that early intensive blood pressure lowering reduced hematoma growth within six hours after onset of intracerebral hemorrhage.
Study design: Randomized controlled trial in which patients received an early intensive blood-pressure-lowering treatment (goal SBP<140 mmHg within one hour) or the AHA recommended best practice guidelines (goal SBP <180 mmHg), which were published in 1999.
Setting: Network of hospitals in China, South Korea, and Australia.
Synopsis: From 2005 to 2007, the study examined 404 patients with computed-tomography (CT) evidence of intracerebral hemorrhage, elevated SBP (150 mmHg-220 mmHg), and ability to commence BP lowering treatment within six hours of onset. Hematoma volumes were measured in the intensive treatment and guideline groups based on CT scans done at baseline and after 24 hours.
No significant association was found between the baseline SBP and the absolute or proportional growth of the hematoma. However, in the intensively treated patients who achieved target SBP within the first 24 hours, a significant association was found with the absolute and proportional hematoma growth.
Maximum reduction of hematoma growth occurred in the group with a median SBP of 135 mmHg.
This single-observational study did not include patients with severe intracranial bleeding who died or required surgical evacuation within the first 24 hours.
Hematoma size reduction did not improve survival or outcomes.
Bottom line: Intensive SBP lowering from 140 mmHg to 130 mmHg within one hour reduces hematoma growth after an intracranial hemorrhage.
Citation: Arima H, Anderson CS, Wang JG, et al. Lower treatment blood pressure is associated with the greatest reduction in hematoma growth after acute intracerebral hemorrhage. Hypertension. 2010;56(5):852-858.
Implantable Cardioverter-Defibrillator (ICD) Therapy for Primary Prevention of Sudden Cardiac Death Might Not Provide Survival Benefit to Elderly
Clinical question: Does ICD therapy for primary prevention of sudden cardiac death in individuals with severe left ventricular dysfunction improve survival in elderly patients?
Background: Several clinical trials of select individuals with severe left ventricular dysfunction (EF <40%) have demonstrated that ICD therapy is associated with a reduction in overall mortality. Given the costs and risks associated with ICD placement, it is important to assess how this therapy affects survival in younger versus older individuals.
Study design: Meta-analysis of five randomized controlled trials.
Setting: Five trials (MADIT-II, DEFINITE, DINAMIT, SCDHeFT, and IRIS).
Synopsis: All included studies compared ICD therapy to standard medical care in the primary prevention of sudden cardiac death in individuals with severe left ventricular dysfunction. Elderly patients comprised 44% of the 5,783 patients included in the study; elderly patients were defined as >65 in three studies, and >60 in two studies. Pooled analysis of the three trials examining ICD use for primary prevention found a nonsignificant reduction in all-cause mortality compared with medical therapy in elderly patients (HR 0.81 [95% CI 0.62 to 1.05], P=0.11). The two remaining studies involving post-MI patients showed no statistically significant reduction in mortality in elderly patients.
In contrast, pooled data from younger patients in the five trials showed that prophylactic ICD therapy reduced all-cause mortality.
None of the selected trials demonstrated evidence of selection, performance, detection, or attrition bias. Only a small number of studies were included, and four potentially relevant trials were not included because mortality data by age group were not available.
Trials including cardiac resynchronization therapy (CRT) were excluded from this meta-analysis, and elderly patients meeting criteria for CRT comprise an important group that must be considered separately.
Bottom line: There is no definitive reduction in mortality with prophylactic ICD therapy in elderly patients with severe left ventricular dysfunction.
Citation: Santangeli P, Di Biase L, Dello Russo A, et al. Meta-analysis: age and effectiveness of prophylactic implantable cardioverter-defibrillators. Ann Intern Med. 2010;153(9):592-599.
System Delay Is Mortality Marker in STEMI Patients Treated with Primary PCI
Clinical question: What is the relationship between system delays to reperfusion therapy and mortality in patients with ST-segment-elevation myocardial infarction (STEMI)?
Background: In patients with STEMI, an early reperfusion strategy is often sought, and several studies have focused on the association between door-to-balloon delay and outcomes. Focusing more broadly on the time from first contact with the healthcare system to the initiation of reperfusion therapy (system delay) might be a more relevant approach.
Study design: Historical follow-up study.
Setting: Three high-volume PCI centers in Denmark.
Synopsis: Using population-based medical registries of 6,209 Danish patients treated for STEMI with percutaneous coronary intervention (PCI), the authors examined the association between delays in reperfusion therapy and mortality. System delay encompassed the entire time from first contact with the healthcare system to the initiation of reperfusion therapy.
Overall, shorter system delay was associated with significantly decreased mortality, as were the individual components of system delay (prehospital delay and door-to-balloon delay). The shortest delays (0-60 minutes) corresponded to a mortality rate of 15%, and the longest delays (181-360 minutes) corresponded to a mortality rate of 31%. Patients were followed for a median of 3.4 years.
This study is unique, as it is the first to look at the association between system delay and outcomes in patients with STEMI transported by EMS and treated with primary PCI. The study highlights the harmful impact of longer system delays on mortality. Limitations of this study include possible underestimation of system delay and challenges surrounding the right marker for reperfusion.
Bottom line: System delay might serve as a broad, comprehensive marker for predicting mortality in patients with STEMI treated with primary PCI.
Citation: Terkelsen CJ, Sorensen JT, Maeng M, et al. System delay and mortality among patients with STEMI treated with primary percutaneous coronary intervention. JAMA. 2010;304(7): 763-771.
D-Dimer Is Effective in Determining the Risk of VTE Recurrence after First Unprovoked Event
Clinical question: Does the timing, patient age, or cut point level affect the predictive value of a D-dimer in predicting VTE recurrence after a first unprovoked event?
Background: Anticoagulation duration after a first unprovoked VTE is at least three months, but it can be indefinite in patients with stable anticoagulation and low bleeding risk. Measuring a D-dimer level after discontinuation of anticoagulation is helpful in determining which patients might benefit from prolonged anticoagulation. However, several unanswered questions remain regarding D-dimer testing.
Study design: Patient-level meta-analysis.
Setting: Pooled patient-level data from seven prospective studies.
Synopsis: Patient-level data were obtained for all patients enrolled using post-treatment D-dimer measurement to predict recurrent VTE in patients with a first unprovoked VTE who had completed at least three months of anticoagulation therapy. The mean length of follow-up was 30 months. Patients with a positive D-dimer had recurrent VTE at a rate of 8.8 per 100 patient-years while those with a negative D-dimer had a rate of 3.7 per 100 patient-years.
Univariate analysis revealed an HR of 2.59 for patients with a positive versus a negative test result. The analysis also showed that the timing of the test, the age of the patient, and the actual cut points used for the various D-dimer tests did not affect the analysis significantly.
These studies’ strength is their large sample sizes and the use of prospective studies. The weaknesses include a mostly white patient population and incomplete data on all patients.
Bottom line: D-dimer testing is useful in predicting VTE recurrence after treatment for a first unprovoked event regardless of patient age, post-treatment timing, or the assay cut point used.
Citation: Douketis J, Tosetto A, Marcucci M, et al. Patient-level meta-analysis: effect of measurement timing, threshold, and patient age on ability of D-dimer testing to assess recurrence risk after unprovoked venous thromboembolism. Ann Intern Med. 2010;153(8): 523-531.
Thigh-High Stockings Are Better than Knee-High Stockings for Post-Stroke DVT Prophylaxis
Clinical question: Are thigh-high compression stockings better then knee-high stockings in immobilized acute-stroke patients?
Background: DVT is common in hospitalized stroke patients with immobility. Graduated compression stockings are often used for DVT prophylaxis, but the CLOTS-1 trial recently found that thigh-high stockings were ineffective after acute stroke. It is unclear if the more commonly used knee-high stockings are more effective than thigh-high stockings.
Study design: Parallel-group trial (the CLOTS-2 trial).
Setting: One hundred twelve hospitals in nine countries.
Synopsis: More than 3,100 patients with acute stroke and immobilization were recruited from January 2002 to May 2009. Patients were randomized to receive thigh-high or knee-high stockings. Patients also received usual care, including anticoagulants and a screening ultrasound for asymptomatic proximal DVT at seven to 10 days. Approximately 640 patients in each group also underwent ultrasound at 25-30 days.
Overall, 6.3% of patients in the thigh-high group had DVT, compared with 8.8% in the knee-high group (P=0.007). There were no significant differences in the secondary outcomes of pulmonary embolism or death. The thigh-high stockings had a higher number of adverse skin events. Enrollment was stopped early when the CLOTS-1 trial showed no difference in DVT rates between thigh-high stockings and no stockings.
Bottom line: Knee-high graduated compression stockings lead to worse outcomes than thigh-high stockings for DVT prophylaxis in immobilized acute-stroke patients.
Citation: CLOTS (Clots in Legs Or sTockings after Stroke) Trial Collaboration. Thigh-length versus below-knee stockings for deep venous thrombosis prophylaxis after stroke: a randomized trial. Ann Intern Med. 2010;153(9):553-562. TH
Pediatric HM Literature
Co-Infection with Pertussis in Infants Hospitalized for Bronchiolitis
Clinical question: How often are infants admitted with bronchiolitis co-infected with Bordatella pertussis?
Background: Infants admitted for bronchiolitis have previously been reported to have co-infection with B. pertussis. Given the nonspecific symptoms associated with early B. pertussis infection, as well as the epidemiology of ongoing outbreaks, it might be useful to better define the risk of co-infection in the seasonal surge of acute bronchiolitis.
Study design: Retrospective cohort study.
Setting: One university hospital in Finland.
Synopsis: A prior study had generated nasopharyngeal aspirate samples for viral antigen detection from 205 healthy, full-term infants younger than 6 months hospitalized for bronchiolitis from 2001 to 2004. Of these samples, 142 (69%) were of quality sufficient for B. pertussis PCR testing in 2009.
Twelve (8.5%) of the 142 infants admitted with bronchiolitis were found to have B. pertussis. Eleven of the 12 infants were co-infected with at least one other virus (RSV in eight of them). Infants who tested positive for pertussis were more often found to have coughing spells (41.7% vs. 14.6% in those who tested negative).
The reported rate of co-infection in this study is higher than other recent reports from the U.S., likely due to the fact that samples were collected during a time period when the incidence of pertussis was relatively high in Finland.
Nevertheless, given the magnitude of a recent pertussis outbreak in California, it might be useful to consider co-infection in young infants admitted for bronchiolitis in areas with a relatively higher incidence of pertussis.
Although the retrospective nature of this study and the lack of a reported definition of coughing spells make further conclusions difficult to draw, cough might deserve further scrutiny during bronchiolitis season.
Bottom line: Consider co-infection with pertussis in young infants admitted with bronchiolitis.
Citation: Nuolivirta K, Koponen P, He Q, et al. Bordatella pertussis infection is common in nonvaccinated infants admitted for bronchiolitis. Pediatr Infect Dis J. 2010;29(11):1013-1015.
In This Edition
Literature at a Glance
A guide to this month’s studies
- Effects of extended VTE prophylaxis in medical patients
- Outcomes with and without preprocedural statins
- Association of subclinical hypothyroidism and CHD
- BP treatment after intracerebral hemorrhage
- Outcomes of ICD therapy in the elderly
- Systems delays and outcomes of STEMI
- D-dimer to predict recurrent VTE
- Stocking height and risk of post-stroke DVT
Extending Anticoagulant Prophylaxis after Medical Hospitalization Decreases VTE, Increases Major Bleeding
Clinical question: For patients with acute medical illness, does extending low-molecular-weight heparin (LMWH) administration for up to 28 days after discharge reduce the incidence of venous thromboembolism (VTE)?
Background: DVT and pulmonary embolism (PE) are common hospital-acquired complications. LMWH has been shown to reduce VTE for medical and surgical patients, and extended-duration LMWH reduces VTE in high-risk surgical patients. Whether extending anticoagulant prophylaxis after discharge for acutely ill medical patients with reduced mobility improves outcomes is unknown.
Study design: Randomized, placebo-controlled trial.
Setting: Three hundred seventy hospitals in 20 countries.
Synopsis: Eligible patients were >40 years old, hospitalized with acute medical illness, and had reduced mobility for ≥3 days. Patients received enoxaparin 40 mg SC daily prophylaxis while hospitalized and were then randomized to an additional 28±4 days of enoxaparin or placebo. Patients received a screening ultrasound to assess for asymptomatic DVT. The primary outcome was a composite of asymptomatic proximal DVT, symptomatic DVT or PE, or fatal PE during the period of extended prophylaxis.
An interim analysis indicated that extended prophylaxis was ineffective; at that time, the protocol was amended to target patients with severe immobility or with moderate immobility plus an additional risk factor (e.g. cancer, prior VTE, or age >75).
The study found that extended prophylaxis decreased the composite VTE outcome (2.5% vs 4.0%, P<0.05) and symptomatic VTE (0.2% vs 1.0%, P<0.05). The incidence of major bleeding was increased in the extended prophylaxis group (0.8% vs 0.3%, P<0.05). There was no difference in mortality.
The unplanned, midstudy protocol amendment to target higher-risk patients is a concern, though the final analyses included patients pre- and post-amendment.
Bottom line: Extending LMWH beyond hospitalization for patients admitted with acute medical illness and decreased mobility decreases VTE, but increases major bleeding.
Citation: Hull RD, Schellong SM, Tapson VF, et al. Extended-duration venous thromboembolism prophylaxis in acutely ill medical patients with recently reduced mobility: a randomized trial. Ann Intern Med. 2010;153(1);8-18.
Preprocedural Statin Therapy Reduces Postprocedural Myocardial Infarction
Clinical question: Does statin therapy reduce periprocedural cardiovascular events?
Background: Myocardial infarction (MI) and death are inherent risks of invasive procedures. Reduction of these risks in certain patient populations has been shown with the use of a beta blockade. Statins have shown promise during acute coronary syndrome. Questions remain about the role of statin therapy before invasive procedures in reducing adverse cardiovascular events.
Study design: Meta-analysis of randomized controlled trials.
Setting: Twenty-one studies involving 4,805 patients, published from inception of MEDLINE, Cochrane, and Clinicaltrials to February 2010.
Synopsis: The use of statins one to seven days preprocedure significantly reduced post-procedural MI in percutaneous coronary interventions (PCI) (P<0.0001). Statins given approximately four weeks in advance of noncardiac surgical procedures also significantly reduced postprocedural MI (P=0.004). An absolute risk reduction of 5.8% for postprocedural MI was found after PCI and 4.1% in noncardiac surgical procedures.
Statins did not show a significant reduction in postprocedural MI (P=0.40) or all-cause mortality (P=0.15) in coronary artery bypass graft surgery (CABG). However, statins did reduce post-CABG atrial fibrillation (P<0.0001).
The 21 studies used a variety of drugs and doses. However, the PCI studies favored atorvastatin 40 mg; more than half the CABG studies used atorvastatin 20 mg; and 91% of the noncardiac surgical studies used fluvastatin 80 mg. Dedicated trials are needed to demonstrate optimal statin agent, dose, and timing of therapy.
Bottom line: Preprocedural statin therapy reduces postprocedural MI after both PCI and noncardiac procedures but not after CABG.
Citation: Winchester DE, Wen X, Xie L, Bavry AA. Evidence of pre-procedural statin therapy: a meta-analysis of randomized trials. J Am Coll Cardiol. 2010;56(19); 1099-1109.
Subclinical Hypothyroidism Increases the Risk of Coronary Heart Disease and Mortality
Clinical question: What are the risks of coronary heart disease (CHD) and mortality among adults with subclinical hypothyroidism?
Background: Subclinical hypo-thyroidism is defined as an elevated serum thyroid stimulating hormone (TSH) level with a normal T4 concentration. Controversy exists regarding the treatment of subclinical hypothyroidism. Because of the association with hyperlipidemia and atherosclerosis, treatment of subclinical hypothyroidism is thought to be beneficial. Previous data from large prospective cohort studies regarding this association are conflicting.
Study design: Study-level meta-analysis of prospective cohort studies.
Setting: Eleven prospective cohorts in the U.S., Europe, Australia, Brazil, and Japan from 1972 to 2007.
Synopsis: Among 55,287 adults, 3,450 (6.2%) had subclinical hypothyroidism and 51,837 were euthyroid. Using Cox proportional hazard models, the association of subclinical hypothyroidism with CHD and mortality were determined for each cohort.
The risk of CHD events and CHD mortality increased with higher TSH concentrations.
In age- and sex-adjusted analyses, the hazard ratio (HR) for CHD events were as follows: HR=1.0 (TSH=4.5-6.9 mIU/L); HR=1.17 (TSH=7-9.9 mIU/L), and HR=1.89 (TSH=10-19.9 mIU/L). Similarly, HRs for CHD mortality showed an increasing trend: 1.09, 1.42, and 1.58, respectively.
Although the association is clearly established here, randomized controlled trials are needed to address whether thyroxine replacement can prevent CHD and the TSH threshold that will provide the most clinical benefit.
Bottom line: Subclinical hypo-thyroidism is associated with an increased risk for CHD events and mortality, primarily in patients with TSH concentrations of 10 mIU/L or higher.
Citation: Rodondi N, den Elzen WP, Bauer DC, et al. Subclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA. 2010;304(12): 1365-1374.
Reduction in Hematoma Growth after Acute Intracerebral Hemorrhage Associated with Lower Blood Pressure
Clinical question: Does intensive systolic blood pressure (SBP) <140 mmHg within one hour reduce hematoma growth after acute intracerebral bleeding?
Background: Early elevation of blood pressure after an acute intracerebral bleed is strongly associated with hematoma growth and worse outcomes. The pilot phase of the Intensive Blood Pressure Reduction in Acute Cerebral Hemorrhage Trial (INTERACT) showed that early intensive blood pressure lowering reduced hematoma growth within six hours after onset of intracerebral hemorrhage.
Study design: Randomized controlled trial in which patients received an early intensive blood-pressure-lowering treatment (goal SBP<140 mmHg within one hour) or the AHA recommended best practice guidelines (goal SBP <180 mmHg), which were published in 1999.
Setting: Network of hospitals in China, South Korea, and Australia.
Synopsis: From 2005 to 2007, the study examined 404 patients with computed-tomography (CT) evidence of intracerebral hemorrhage, elevated SBP (150 mmHg-220 mmHg), and ability to commence BP lowering treatment within six hours of onset. Hematoma volumes were measured in the intensive treatment and guideline groups based on CT scans done at baseline and after 24 hours.
No significant association was found between the baseline SBP and the absolute or proportional growth of the hematoma. However, in the intensively treated patients who achieved target SBP within the first 24 hours, a significant association was found with the absolute and proportional hematoma growth.
Maximum reduction of hematoma growth occurred in the group with a median SBP of 135 mmHg.
This single-observational study did not include patients with severe intracranial bleeding who died or required surgical evacuation within the first 24 hours.
Hematoma size reduction did not improve survival or outcomes.
Bottom line: Intensive SBP lowering from 140 mmHg to 130 mmHg within one hour reduces hematoma growth after an intracranial hemorrhage.
Citation: Arima H, Anderson CS, Wang JG, et al. Lower treatment blood pressure is associated with the greatest reduction in hematoma growth after acute intracerebral hemorrhage. Hypertension. 2010;56(5):852-858.
Implantable Cardioverter-Defibrillator (ICD) Therapy for Primary Prevention of Sudden Cardiac Death Might Not Provide Survival Benefit to Elderly
Clinical question: Does ICD therapy for primary prevention of sudden cardiac death in individuals with severe left ventricular dysfunction improve survival in elderly patients?
Background: Several clinical trials of select individuals with severe left ventricular dysfunction (EF <40%) have demonstrated that ICD therapy is associated with a reduction in overall mortality. Given the costs and risks associated with ICD placement, it is important to assess how this therapy affects survival in younger versus older individuals.
Study design: Meta-analysis of five randomized controlled trials.
Setting: Five trials (MADIT-II, DEFINITE, DINAMIT, SCDHeFT, and IRIS).
Synopsis: All included studies compared ICD therapy to standard medical care in the primary prevention of sudden cardiac death in individuals with severe left ventricular dysfunction. Elderly patients comprised 44% of the 5,783 patients included in the study; elderly patients were defined as >65 in three studies, and >60 in two studies. Pooled analysis of the three trials examining ICD use for primary prevention found a nonsignificant reduction in all-cause mortality compared with medical therapy in elderly patients (HR 0.81 [95% CI 0.62 to 1.05], P=0.11). The two remaining studies involving post-MI patients showed no statistically significant reduction in mortality in elderly patients.
In contrast, pooled data from younger patients in the five trials showed that prophylactic ICD therapy reduced all-cause mortality.
None of the selected trials demonstrated evidence of selection, performance, detection, or attrition bias. Only a small number of studies were included, and four potentially relevant trials were not included because mortality data by age group were not available.
Trials including cardiac resynchronization therapy (CRT) were excluded from this meta-analysis, and elderly patients meeting criteria for CRT comprise an important group that must be considered separately.
Bottom line: There is no definitive reduction in mortality with prophylactic ICD therapy in elderly patients with severe left ventricular dysfunction.
Citation: Santangeli P, Di Biase L, Dello Russo A, et al. Meta-analysis: age and effectiveness of prophylactic implantable cardioverter-defibrillators. Ann Intern Med. 2010;153(9):592-599.
System Delay Is Mortality Marker in STEMI Patients Treated with Primary PCI
Clinical question: What is the relationship between system delays to reperfusion therapy and mortality in patients with ST-segment-elevation myocardial infarction (STEMI)?
Background: In patients with STEMI, an early reperfusion strategy is often sought, and several studies have focused on the association between door-to-balloon delay and outcomes. Focusing more broadly on the time from first contact with the healthcare system to the initiation of reperfusion therapy (system delay) might be a more relevant approach.
Study design: Historical follow-up study.
Setting: Three high-volume PCI centers in Denmark.
Synopsis: Using population-based medical registries of 6,209 Danish patients treated for STEMI with percutaneous coronary intervention (PCI), the authors examined the association between delays in reperfusion therapy and mortality. System delay encompassed the entire time from first contact with the healthcare system to the initiation of reperfusion therapy.
Overall, shorter system delay was associated with significantly decreased mortality, as were the individual components of system delay (prehospital delay and door-to-balloon delay). The shortest delays (0-60 minutes) corresponded to a mortality rate of 15%, and the longest delays (181-360 minutes) corresponded to a mortality rate of 31%. Patients were followed for a median of 3.4 years.
This study is unique, as it is the first to look at the association between system delay and outcomes in patients with STEMI transported by EMS and treated with primary PCI. The study highlights the harmful impact of longer system delays on mortality. Limitations of this study include possible underestimation of system delay and challenges surrounding the right marker for reperfusion.
Bottom line: System delay might serve as a broad, comprehensive marker for predicting mortality in patients with STEMI treated with primary PCI.
Citation: Terkelsen CJ, Sorensen JT, Maeng M, et al. System delay and mortality among patients with STEMI treated with primary percutaneous coronary intervention. JAMA. 2010;304(7): 763-771.
D-Dimer Is Effective in Determining the Risk of VTE Recurrence after First Unprovoked Event
Clinical question: Does the timing, patient age, or cut point level affect the predictive value of a D-dimer in predicting VTE recurrence after a first unprovoked event?
Background: Anticoagulation duration after a first unprovoked VTE is at least three months, but it can be indefinite in patients with stable anticoagulation and low bleeding risk. Measuring a D-dimer level after discontinuation of anticoagulation is helpful in determining which patients might benefit from prolonged anticoagulation. However, several unanswered questions remain regarding D-dimer testing.
Study design: Patient-level meta-analysis.
Setting: Pooled patient-level data from seven prospective studies.
Synopsis: Patient-level data were obtained for all patients enrolled using post-treatment D-dimer measurement to predict recurrent VTE in patients with a first unprovoked VTE who had completed at least three months of anticoagulation therapy. The mean length of follow-up was 30 months. Patients with a positive D-dimer had recurrent VTE at a rate of 8.8 per 100 patient-years while those with a negative D-dimer had a rate of 3.7 per 100 patient-years.
Univariate analysis revealed an HR of 2.59 for patients with a positive versus a negative test result. The analysis also showed that the timing of the test, the age of the patient, and the actual cut points used for the various D-dimer tests did not affect the analysis significantly.
These studies’ strength is their large sample sizes and the use of prospective studies. The weaknesses include a mostly white patient population and incomplete data on all patients.
Bottom line: D-dimer testing is useful in predicting VTE recurrence after treatment for a first unprovoked event regardless of patient age, post-treatment timing, or the assay cut point used.
Citation: Douketis J, Tosetto A, Marcucci M, et al. Patient-level meta-analysis: effect of measurement timing, threshold, and patient age on ability of D-dimer testing to assess recurrence risk after unprovoked venous thromboembolism. Ann Intern Med. 2010;153(8): 523-531.
Thigh-High Stockings Are Better than Knee-High Stockings for Post-Stroke DVT Prophylaxis
Clinical question: Are thigh-high compression stockings better then knee-high stockings in immobilized acute-stroke patients?
Background: DVT is common in hospitalized stroke patients with immobility. Graduated compression stockings are often used for DVT prophylaxis, but the CLOTS-1 trial recently found that thigh-high stockings were ineffective after acute stroke. It is unclear if the more commonly used knee-high stockings are more effective than thigh-high stockings.
Study design: Parallel-group trial (the CLOTS-2 trial).
Setting: One hundred twelve hospitals in nine countries.
Synopsis: More than 3,100 patients with acute stroke and immobilization were recruited from January 2002 to May 2009. Patients were randomized to receive thigh-high or knee-high stockings. Patients also received usual care, including anticoagulants and a screening ultrasound for asymptomatic proximal DVT at seven to 10 days. Approximately 640 patients in each group also underwent ultrasound at 25-30 days.
Overall, 6.3% of patients in the thigh-high group had DVT, compared with 8.8% in the knee-high group (P=0.007). There were no significant differences in the secondary outcomes of pulmonary embolism or death. The thigh-high stockings had a higher number of adverse skin events. Enrollment was stopped early when the CLOTS-1 trial showed no difference in DVT rates between thigh-high stockings and no stockings.
Bottom line: Knee-high graduated compression stockings lead to worse outcomes than thigh-high stockings for DVT prophylaxis in immobilized acute-stroke patients.
Citation: CLOTS (Clots in Legs Or sTockings after Stroke) Trial Collaboration. Thigh-length versus below-knee stockings for deep venous thrombosis prophylaxis after stroke: a randomized trial. Ann Intern Med. 2010;153(9):553-562. TH
Pediatric HM Literature
Co-Infection with Pertussis in Infants Hospitalized for Bronchiolitis
Clinical question: How often are infants admitted with bronchiolitis co-infected with Bordatella pertussis?
Background: Infants admitted for bronchiolitis have previously been reported to have co-infection with B. pertussis. Given the nonspecific symptoms associated with early B. pertussis infection, as well as the epidemiology of ongoing outbreaks, it might be useful to better define the risk of co-infection in the seasonal surge of acute bronchiolitis.
Study design: Retrospective cohort study.
Setting: One university hospital in Finland.
Synopsis: A prior study had generated nasopharyngeal aspirate samples for viral antigen detection from 205 healthy, full-term infants younger than 6 months hospitalized for bronchiolitis from 2001 to 2004. Of these samples, 142 (69%) were of quality sufficient for B. pertussis PCR testing in 2009.
Twelve (8.5%) of the 142 infants admitted with bronchiolitis were found to have B. pertussis. Eleven of the 12 infants were co-infected with at least one other virus (RSV in eight of them). Infants who tested positive for pertussis were more often found to have coughing spells (41.7% vs. 14.6% in those who tested negative).
The reported rate of co-infection in this study is higher than other recent reports from the U.S., likely due to the fact that samples were collected during a time period when the incidence of pertussis was relatively high in Finland.
Nevertheless, given the magnitude of a recent pertussis outbreak in California, it might be useful to consider co-infection in young infants admitted for bronchiolitis in areas with a relatively higher incidence of pertussis.
Although the retrospective nature of this study and the lack of a reported definition of coughing spells make further conclusions difficult to draw, cough might deserve further scrutiny during bronchiolitis season.
Bottom line: Consider co-infection with pertussis in young infants admitted with bronchiolitis.
Citation: Nuolivirta K, Koponen P, He Q, et al. Bordatella pertussis infection is common in nonvaccinated infants admitted for bronchiolitis. Pediatr Infect Dis J. 2010;29(11):1013-1015.
What Is the Role of BNP in Diagnosis and Management of Acutely Decompensated Heart Failure?
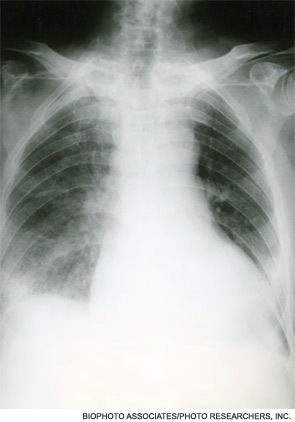
Case
A 76-year-old woman with a history of chronic obstructive pulmonary disease (COPD), congestive heart failure (CHF), and atrial fibrillation presents with shortness of breath. She is tachypneic, her pulse is 105 beats per minute, and her blood pressure is 105/60 mm/Hg. She is obese and has an immeasurable venous pressure with decreased breath sounds in both lung bases, and irregular and distant heart sounds. What is the role of brain (or B-type) natriuretic peptide (BNP) in the diagnosis and management of this patient?
Overview
Each year, more than 1 million patients are admitted to hospitals with acutely decompensated heart failure (ADHF). Although many of these patients carry a pre-admission diagnosis of CHF, their common presenting symptoms are not specific for ADHF, which leads to delays in diagnosis and therapy initiation, and increased diagnostic costs and potentially worse outcomes. Clinical risk scores from NHANES and the Framingham heart study have limited sensitivity, missing nearly 20% of patients.1,2 Moreover, these scores are underused by clinicians who depend heavily on clinical gestalt.3
Once ADHF is diagnosed, ongoing bedside assessment of volume status is a difficult and inexact science. The physiologic goal is achievement of normal left ventricular end diastolic volume; however, surrogate measures of this status, including weight change, venous pressure, and pulmonary and cardiac auscultatory findings, have significant limitations. After discharge, patients have high and heterogeneous risks of readmission, death, and other adverse events. Identifying patients with the highest risk might allow for intensive strategies to improve outcomes.
BNP is a neurohormone released from the ventricular cells in response to increased cardiac filling pressures. Plasma measurements of BNP have been shown to reflect volume status, to predict risk at admission and discharge, and to serve as a treatment guide in a variety of clinical settings.4 This simple laboratory test increasingly has been used to diagnose and manage ADHF; its utility and limitations deserve critical review.
Review of the Data
CHF diagnosis. Since introduction of the rapid BNP assay, several trials have evaluated its clinical utility in determining whether ADHF is the cause of a patient’s dyspnea. The largest of these trials, the Breathing Not Properly Multinational Study, conducted by McCullough et al, enrolled nearly 1,600 patients who presented with the primary complaint of dyspnea.5 After reviewing conventional clinical information, ED physicians were asked to determine the likelihood that ADHF was the etiology of a patient’s dyspnea. These likelihoods were classified as low (<20%), intermediate (20%-80%), or high (>80%). The admission BNP was recorded but was not available for the ED physician decisions.
The “gold standard” was the opinion of two adjudicating cardiologists who reviewed the cases retrospectively and determined whether the dyspnea resulted from ADHF. They were blinded to both the ED physician’s opinion and the BNP results. The accuracy of the ED physician’s initial assessment and the impact of the BNP results were compared with this gold standard.
For the entire cohort, the use of BNP (with a cutoff point of 100 pg/mL) would have improved the ED physician’s assessment from 74% diagnostic accuracy to 81%, which is statistically significant. Most important, in those patients initially given an intermediate likelihood of CHF, BNP results correctly classified 75% of these patients and rarely missed ADHF cases (<10%).
Atrial fibrillation. Since the original trials that established a BNP cutoff of 100 pg/mL for determining the presence of ADHF, several adjustments have been suggested. The presence of atrial fibrillation has been shown to increase BNP values independent of cardiac filling pressures. Breidthardt et al examined patients with atrial fibrillation presenting with dyspnea.4 In their analysis, using a cutoff of 100 pg/mL remained robust in identifying patients without ADHF. However, in the 100 pg/mL-500 pg/mL range, the test was not able to discriminate between atrial fibrillation and ADHF. Values greater than 500 pg/mL proved accurate in supporting the diagnosis of ADHF.
Renal failure. Renal dysfunction also elevates BNP levels independent of filling pressures. McCullough et al re-examined data from their Breathing Not Properly Multinational Study and found that the glomerular filtration rate (GFR) was inversely related to BNP levels.5 They recommend using a cutoff point of 200 pg/mL when the GFR is below 60 mg/dL. Other authors recommend not using BNP levels to diagnose ADHF when the GFR is less than 60 mg/dL due to the lack of data supporting this approach. Until clarified, clinicians should be cautious of interpreting BNP elevations in the setting of kidney disease.

Obesity. Obesity has a negative effect on BNP levels, decreasing the sensitivity of the test in these patients.6 Although no study defines how to adjust for body mass index (BMI), clinicians should be cautious about using a low BNP to rule out ADHF in a dyspneic obese patient.
Historical BNP values. If historical BNP values are available, studies of biological variation have shown that an increase to 123% from 66% from baseline is representative of a clinically meaningful increase in cardiac filling pressures. Less significant changes could merely represent biological variation and should be cautiously interpreted.7
Cost effectiveness. The cost effectiveness of using BNP measurements in dyspneic ED patients has been examined as well. Mueller et al found in a Swiss hospital that BNP testing was associated with a 25% decrease in treatment cost, length of stay (LOS), and ICU usage.8 However, LOS is significantly longer in Switzerland compared with the U.S., and given that much of the cost savings was attributed to reducing LOS, it is not possible to extrapolate these data to the U.S. health system. More evidence is needed to truly evaluate the cost effectiveness of BNP testing.
Serial BNP testing. Once a patient has been diagnosed with ADHF and admitted to the hospital, diuretics are indicated with the goal of achieving euvolemia. The bedside assessment of volume status remains a difficult and inexact science, and failure to appropriately remove fluid is associated with readmissions. Conversely, overdiuresis with a concomitant rise in creatinine has been associated with increased morbidity and mortality.
Several studies have shown that the reduction of volume associated with diuretic administration is coupled with a rapid decrease in BNP levels. Therefore, serial BNP measurement has been evaluated as a tool to guide the daily assessment of volume status in patients admitted with ADHF. Unfortunately, frequent measurements of BNP reveal that a great deal of variance, or “noise,” is present in these repeat measurements. Data do not clearly show how to incorporate serial BNP measurements into daily diuretic management.9
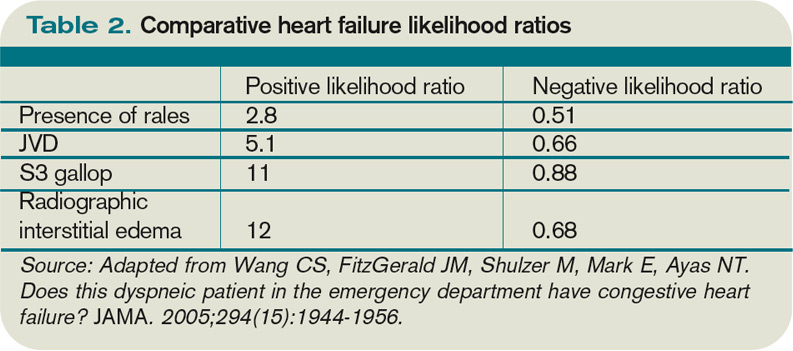
Mortality prediction. Nearly 3.5% of admitted heart failure patients will die during their hospitalization. For perspective, the rate of hospital mortality with acute myocardial infarction is 7%. BNP serves as a powerful and independent predictor of inpatient mortality. The ADHERE (Acute Decompensated Heart Failure National Registry) study showed that when divided into BNP quartiles of <430 pg/mL, 430 pg/mL to 839 pg/mL, 840 pg/mL to 1,729 pg/mL, and >1,730 pg/mL, patients’ risk of inpatient death was accurately predicted as 1.9%, 2.8%, 3.8%, and 6.0%, respectively.10 Even when adjusted for other risk factors, BNP remained a powerful predictor; the mortality rate more than doubled from the lowest to highest quartile.
Different strategies have been proposed to improve the outcomes in these highest-risk patients; however, to date, no evidence-based strategy offers a meaningful way to reduce inpatient mortality beyond the current standard of care.
Readmission and 30-day mortality. The 30-day readmission rate after discharge for ADHF is more than than 25%. A study of Medicare patients showed that more than $17 billion (more than 15% of all Medicare payments to hospitals) was associated with unplanned rehospitalizations.11 As bundling payment trends develop, hospitals have an enormous incentive to identify CHF patients with the highest risk of readmission and attempt to mitigate that risk.
From a patient-centered view, upon hospital discharge a patient with ADHF also realizes a 1 in 10 chance of dying within the first 30 days.
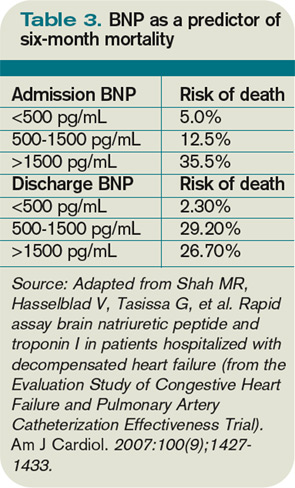
At discharge, BNP serves as a powerful and independent marker of increased risk of readmission, morbidity, and mortality. O’Connor et al developed a discharge risk model in patients with severe left ventricular dysfunction; the ESCAPE risk model and discharge score showed elevated BNP was the single most powerful predictor of six-month mortality.12 For every doubling of the BNP, the odds of death at six months increased by 1.4 times.
After combining discharge BNP with other factors, the ESCAPE discharge score was fairly successful at discriminating between patients who would and would not survive to six months. By identifying these outpatients, intensive management strategies could be focused on individuals with the highest risk. The data support the idea that readmission reductions are significant when outpatients obtain early follow-up. Many healthcare centers struggle to schedule early follow-up for all heart failure patients.
As such, the ability to target individuals with the highest discharge scores for intensive follow-up might improve outcomes. These patients could undergo early evaluation for such advanced therapies as resynchronization, left ventricular assist device implantation, or listing for transplantation. Currently, this strategy is not proven. It also is possible that these high-risk patients might have such advanced diseases that their risk cannot be modified by our current medications and advanced therapies.
Back to the Case
This patient has symptoms and signs that could be caused by ADHF or COPD. Her presentation is consistent with an intermediate probability of ADHF. A rapid BNP reveals a level of 950 pg/mL.
Even considering the higher cutoff required because of her coexistent atrial fibrillation, her BNP is consistent with ADHF. Additionally, her obesity likely has decreased the true value of her BNP. A previous BNP drawn when the patient was not in ADHF was 250 ng/mL, meaning that at least a 70% increase is present.
She was admitted and treated with intravenous diuretics with improvement in her congestion and relief of her symptoms. Daily BNPs were not drawn and her diuretics were titrated based on bedside clinical assessments. Her admission BNP elevation would predict a moderately high risk of short- and intermediate term of morbidity and mortality.
At discharge, a repeat BNP also could add to her risk stratification, though it would not be clear what do with this prognostic information beyond the standard of care.
Bottom Line
BNP measurement in specific situations can complement conventional clinical information in determining the presence of ADHF and also can enhance clinicians’ ability to risk-stratify patients during and after hospitalization. TH
Dr. Wolfe is a hospitalist and assistant professor of medicine at the University of Colorado Denver.
References
- Schocken DD, Arrieta MI, Leaverton PE, Ross EA. Prevalence and mortality of congestive heart failure in the United States. J Am Coll Cardiol. 1992;20(2):301-306.
- McKee PA, Castelli WP, McNamara PM, Kannel WB. The natural history of congestive heart failure: the Framingham study. N Eng J Med. 1971;285(26):1441-1446.
- Wang CS, FitzGerald JM, Schulzer M, Mak E, Ayas NT. Does this dyspneic patient in the emergency department have congestive heart failure? JAMA. 2005;294(15):1944-1956.
- Breidthardt T, Noveanu M, Cayir S, et al. The use of B-type natriuretic peptide in the management of patients with atrial fibrillation and dyspnea. Int J Cardiol. 2009;136(2):193-199.
- McCullough PA, Duc P, Omland T, et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the Breathing Not Properly Multinational Study. Am J Kidney Dis. 2003;41(3):571-579.
- Iwanaga Y, Hihara Y, Nizuma S, et al. BNP in overweight and obese patients with heart failure: an analysis based on the BNP-LV diastolic wall stress relationship. J Card Fail. 2007;13(8):663-667.
- O’Hanlon R, O’Shea P, Ledwidge M. The biologic variability of B-type natriuretic peptide and N-terminal pro-B-type natriuretic peptide in stable heart failure patients. J Card Fail. 2007;13(1):50-55.
- Mueller C, Laule-Kilian K, Schindler C, et al. Cost-effectiveness of B-type natriuretic peptide testing in patients with acute dyspnea. Arch Intern Med. 2006;166(1):1081-1087.
- Wu AH. Serial testing of B-type natriuretic peptide and NTpro-BNP for monitoring therapy of heart failure: the role of biologic variation in the interpretation of results. Am Heart J. 2006;152(5):828-834.
- Fonarow GC, Peacock WF, Phillips CO, et al. ADHERE Scientific Advisory Committee and Investigators. Admission B-type natriuretic peptide levels and in-hospital mortality in acute decompensated heart failure. J Am Coll Cardiol. 2007;48 (19):1943-1950.
- Jencks SF, Williams MC, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428.
- O’Connor CM, Hasselblad V, Mehta RH, et al. Triage after hospitalization with advanced heart failure: the ESCAPE (Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness) risk model and discharge score. J Am Coll Cardiol. 2010;55(9):872-878.
Case
A 76-year-old woman with a history of chronic obstructive pulmonary disease (COPD), congestive heart failure (CHF), and atrial fibrillation presents with shortness of breath. She is tachypneic, her pulse is 105 beats per minute, and her blood pressure is 105/60 mm/Hg. She is obese and has an immeasurable venous pressure with decreased breath sounds in both lung bases, and irregular and distant heart sounds. What is the role of brain (or B-type) natriuretic peptide (BNP) in the diagnosis and management of this patient?
Overview
Each year, more than 1 million patients are admitted to hospitals with acutely decompensated heart failure (ADHF). Although many of these patients carry a pre-admission diagnosis of CHF, their common presenting symptoms are not specific for ADHF, which leads to delays in diagnosis and therapy initiation, and increased diagnostic costs and potentially worse outcomes. Clinical risk scores from NHANES and the Framingham heart study have limited sensitivity, missing nearly 20% of patients.1,2 Moreover, these scores are underused by clinicians who depend heavily on clinical gestalt.3
Once ADHF is diagnosed, ongoing bedside assessment of volume status is a difficult and inexact science. The physiologic goal is achievement of normal left ventricular end diastolic volume; however, surrogate measures of this status, including weight change, venous pressure, and pulmonary and cardiac auscultatory findings, have significant limitations. After discharge, patients have high and heterogeneous risks of readmission, death, and other adverse events. Identifying patients with the highest risk might allow for intensive strategies to improve outcomes.
BNP is a neurohormone released from the ventricular cells in response to increased cardiac filling pressures. Plasma measurements of BNP have been shown to reflect volume status, to predict risk at admission and discharge, and to serve as a treatment guide in a variety of clinical settings.4 This simple laboratory test increasingly has been used to diagnose and manage ADHF; its utility and limitations deserve critical review.
Review of the Data
CHF diagnosis. Since introduction of the rapid BNP assay, several trials have evaluated its clinical utility in determining whether ADHF is the cause of a patient’s dyspnea. The largest of these trials, the Breathing Not Properly Multinational Study, conducted by McCullough et al, enrolled nearly 1,600 patients who presented with the primary complaint of dyspnea.5 After reviewing conventional clinical information, ED physicians were asked to determine the likelihood that ADHF was the etiology of a patient’s dyspnea. These likelihoods were classified as low (<20%), intermediate (20%-80%), or high (>80%). The admission BNP was recorded but was not available for the ED physician decisions.
The “gold standard” was the opinion of two adjudicating cardiologists who reviewed the cases retrospectively and determined whether the dyspnea resulted from ADHF. They were blinded to both the ED physician’s opinion and the BNP results. The accuracy of the ED physician’s initial assessment and the impact of the BNP results were compared with this gold standard.
For the entire cohort, the use of BNP (with a cutoff point of 100 pg/mL) would have improved the ED physician’s assessment from 74% diagnostic accuracy to 81%, which is statistically significant. Most important, in those patients initially given an intermediate likelihood of CHF, BNP results correctly classified 75% of these patients and rarely missed ADHF cases (<10%).
Atrial fibrillation. Since the original trials that established a BNP cutoff of 100 pg/mL for determining the presence of ADHF, several adjustments have been suggested. The presence of atrial fibrillation has been shown to increase BNP values independent of cardiac filling pressures. Breidthardt et al examined patients with atrial fibrillation presenting with dyspnea.4 In their analysis, using a cutoff of 100 pg/mL remained robust in identifying patients without ADHF. However, in the 100 pg/mL-500 pg/mL range, the test was not able to discriminate between atrial fibrillation and ADHF. Values greater than 500 pg/mL proved accurate in supporting the diagnosis of ADHF.
Renal failure. Renal dysfunction also elevates BNP levels independent of filling pressures. McCullough et al re-examined data from their Breathing Not Properly Multinational Study and found that the glomerular filtration rate (GFR) was inversely related to BNP levels.5 They recommend using a cutoff point of 200 pg/mL when the GFR is below 60 mg/dL. Other authors recommend not using BNP levels to diagnose ADHF when the GFR is less than 60 mg/dL due to the lack of data supporting this approach. Until clarified, clinicians should be cautious of interpreting BNP elevations in the setting of kidney disease.
Obesity. Obesity has a negative effect on BNP levels, decreasing the sensitivity of the test in these patients.6 Although no study defines how to adjust for body mass index (BMI), clinicians should be cautious about using a low BNP to rule out ADHF in a dyspneic obese patient.
Historical BNP values. If historical BNP values are available, studies of biological variation have shown that an increase to 123% from 66% from baseline is representative of a clinically meaningful increase in cardiac filling pressures. Less significant changes could merely represent biological variation and should be cautiously interpreted.7
Cost effectiveness. The cost effectiveness of using BNP measurements in dyspneic ED patients has been examined as well. Mueller et al found in a Swiss hospital that BNP testing was associated with a 25% decrease in treatment cost, length of stay (LOS), and ICU usage.8 However, LOS is significantly longer in Switzerland compared with the U.S., and given that much of the cost savings was attributed to reducing LOS, it is not possible to extrapolate these data to the U.S. health system. More evidence is needed to truly evaluate the cost effectiveness of BNP testing.
Serial BNP testing. Once a patient has been diagnosed with ADHF and admitted to the hospital, diuretics are indicated with the goal of achieving euvolemia. The bedside assessment of volume status remains a difficult and inexact science, and failure to appropriately remove fluid is associated with readmissions. Conversely, overdiuresis with a concomitant rise in creatinine has been associated with increased morbidity and mortality.
Several studies have shown that the reduction of volume associated with diuretic administration is coupled with a rapid decrease in BNP levels. Therefore, serial BNP measurement has been evaluated as a tool to guide the daily assessment of volume status in patients admitted with ADHF. Unfortunately, frequent measurements of BNP reveal that a great deal of variance, or “noise,” is present in these repeat measurements. Data do not clearly show how to incorporate serial BNP measurements into daily diuretic management.9
Mortality prediction. Nearly 3.5% of admitted heart failure patients will die during their hospitalization. For perspective, the rate of hospital mortality with acute myocardial infarction is 7%. BNP serves as a powerful and independent predictor of inpatient mortality. The ADHERE (Acute Decompensated Heart Failure National Registry) study showed that when divided into BNP quartiles of <430 pg/mL, 430 pg/mL to 839 pg/mL, 840 pg/mL to 1,729 pg/mL, and >1,730 pg/mL, patients’ risk of inpatient death was accurately predicted as 1.9%, 2.8%, 3.8%, and 6.0%, respectively.10 Even when adjusted for other risk factors, BNP remained a powerful predictor; the mortality rate more than doubled from the lowest to highest quartile.
Different strategies have been proposed to improve the outcomes in these highest-risk patients; however, to date, no evidence-based strategy offers a meaningful way to reduce inpatient mortality beyond the current standard of care.
Readmission and 30-day mortality. The 30-day readmission rate after discharge for ADHF is more than than 25%. A study of Medicare patients showed that more than $17 billion (more than 15% of all Medicare payments to hospitals) was associated with unplanned rehospitalizations.11 As bundling payment trends develop, hospitals have an enormous incentive to identify CHF patients with the highest risk of readmission and attempt to mitigate that risk.
From a patient-centered view, upon hospital discharge a patient with ADHF also realizes a 1 in 10 chance of dying within the first 30 days.
At discharge, BNP serves as a powerful and independent marker of increased risk of readmission, morbidity, and mortality. O’Connor et al developed a discharge risk model in patients with severe left ventricular dysfunction; the ESCAPE risk model and discharge score showed elevated BNP was the single most powerful predictor of six-month mortality.12 For every doubling of the BNP, the odds of death at six months increased by 1.4 times.
After combining discharge BNP with other factors, the ESCAPE discharge score was fairly successful at discriminating between patients who would and would not survive to six months. By identifying these outpatients, intensive management strategies could be focused on individuals with the highest risk. The data support the idea that readmission reductions are significant when outpatients obtain early follow-up. Many healthcare centers struggle to schedule early follow-up for all heart failure patients.
As such, the ability to target individuals with the highest discharge scores for intensive follow-up might improve outcomes. These patients could undergo early evaluation for such advanced therapies as resynchronization, left ventricular assist device implantation, or listing for transplantation. Currently, this strategy is not proven. It also is possible that these high-risk patients might have such advanced diseases that their risk cannot be modified by our current medications and advanced therapies.
Back to the Case
This patient has symptoms and signs that could be caused by ADHF or COPD. Her presentation is consistent with an intermediate probability of ADHF. A rapid BNP reveals a level of 950 pg/mL.
Even considering the higher cutoff required because of her coexistent atrial fibrillation, her BNP is consistent with ADHF. Additionally, her obesity likely has decreased the true value of her BNP. A previous BNP drawn when the patient was not in ADHF was 250 ng/mL, meaning that at least a 70% increase is present.
She was admitted and treated with intravenous diuretics with improvement in her congestion and relief of her symptoms. Daily BNPs were not drawn and her diuretics were titrated based on bedside clinical assessments. Her admission BNP elevation would predict a moderately high risk of short- and intermediate term of morbidity and mortality.
At discharge, a repeat BNP also could add to her risk stratification, though it would not be clear what do with this prognostic information beyond the standard of care.
Bottom Line
BNP measurement in specific situations can complement conventional clinical information in determining the presence of ADHF and also can enhance clinicians’ ability to risk-stratify patients during and after hospitalization. TH
Dr. Wolfe is a hospitalist and assistant professor of medicine at the University of Colorado Denver.
References
- Schocken DD, Arrieta MI, Leaverton PE, Ross EA. Prevalence and mortality of congestive heart failure in the United States. J Am Coll Cardiol. 1992;20(2):301-306.
- McKee PA, Castelli WP, McNamara PM, Kannel WB. The natural history of congestive heart failure: the Framingham study. N Eng J Med. 1971;285(26):1441-1446.
- Wang CS, FitzGerald JM, Schulzer M, Mak E, Ayas NT. Does this dyspneic patient in the emergency department have congestive heart failure? JAMA. 2005;294(15):1944-1956.
- Breidthardt T, Noveanu M, Cayir S, et al. The use of B-type natriuretic peptide in the management of patients with atrial fibrillation and dyspnea. Int J Cardiol. 2009;136(2):193-199.
- McCullough PA, Duc P, Omland T, et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the Breathing Not Properly Multinational Study. Am J Kidney Dis. 2003;41(3):571-579.
- Iwanaga Y, Hihara Y, Nizuma S, et al. BNP in overweight and obese patients with heart failure: an analysis based on the BNP-LV diastolic wall stress relationship. J Card Fail. 2007;13(8):663-667.
- O’Hanlon R, O’Shea P, Ledwidge M. The biologic variability of B-type natriuretic peptide and N-terminal pro-B-type natriuretic peptide in stable heart failure patients. J Card Fail. 2007;13(1):50-55.
- Mueller C, Laule-Kilian K, Schindler C, et al. Cost-effectiveness of B-type natriuretic peptide testing in patients with acute dyspnea. Arch Intern Med. 2006;166(1):1081-1087.
- Wu AH. Serial testing of B-type natriuretic peptide and NTpro-BNP for monitoring therapy of heart failure: the role of biologic variation in the interpretation of results. Am Heart J. 2006;152(5):828-834.
- Fonarow GC, Peacock WF, Phillips CO, et al. ADHERE Scientific Advisory Committee and Investigators. Admission B-type natriuretic peptide levels and in-hospital mortality in acute decompensated heart failure. J Am Coll Cardiol. 2007;48 (19):1943-1950.
- Jencks SF, Williams MC, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428.
- O’Connor CM, Hasselblad V, Mehta RH, et al. Triage after hospitalization with advanced heart failure: the ESCAPE (Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness) risk model and discharge score. J Am Coll Cardiol. 2010;55(9):872-878.
Case
A 76-year-old woman with a history of chronic obstructive pulmonary disease (COPD), congestive heart failure (CHF), and atrial fibrillation presents with shortness of breath. She is tachypneic, her pulse is 105 beats per minute, and her blood pressure is 105/60 mm/Hg. She is obese and has an immeasurable venous pressure with decreased breath sounds in both lung bases, and irregular and distant heart sounds. What is the role of brain (or B-type) natriuretic peptide (BNP) in the diagnosis and management of this patient?
Overview
Each year, more than 1 million patients are admitted to hospitals with acutely decompensated heart failure (ADHF). Although many of these patients carry a pre-admission diagnosis of CHF, their common presenting symptoms are not specific for ADHF, which leads to delays in diagnosis and therapy initiation, and increased diagnostic costs and potentially worse outcomes. Clinical risk scores from NHANES and the Framingham heart study have limited sensitivity, missing nearly 20% of patients.1,2 Moreover, these scores are underused by clinicians who depend heavily on clinical gestalt.3
Once ADHF is diagnosed, ongoing bedside assessment of volume status is a difficult and inexact science. The physiologic goal is achievement of normal left ventricular end diastolic volume; however, surrogate measures of this status, including weight change, venous pressure, and pulmonary and cardiac auscultatory findings, have significant limitations. After discharge, patients have high and heterogeneous risks of readmission, death, and other adverse events. Identifying patients with the highest risk might allow for intensive strategies to improve outcomes.
BNP is a neurohormone released from the ventricular cells in response to increased cardiac filling pressures. Plasma measurements of BNP have been shown to reflect volume status, to predict risk at admission and discharge, and to serve as a treatment guide in a variety of clinical settings.4 This simple laboratory test increasingly has been used to diagnose and manage ADHF; its utility and limitations deserve critical review.
Review of the Data
CHF diagnosis. Since introduction of the rapid BNP assay, several trials have evaluated its clinical utility in determining whether ADHF is the cause of a patient’s dyspnea. The largest of these trials, the Breathing Not Properly Multinational Study, conducted by McCullough et al, enrolled nearly 1,600 patients who presented with the primary complaint of dyspnea.5 After reviewing conventional clinical information, ED physicians were asked to determine the likelihood that ADHF was the etiology of a patient’s dyspnea. These likelihoods were classified as low (<20%), intermediate (20%-80%), or high (>80%). The admission BNP was recorded but was not available for the ED physician decisions.
The “gold standard” was the opinion of two adjudicating cardiologists who reviewed the cases retrospectively and determined whether the dyspnea resulted from ADHF. They were blinded to both the ED physician’s opinion and the BNP results. The accuracy of the ED physician’s initial assessment and the impact of the BNP results were compared with this gold standard.
For the entire cohort, the use of BNP (with a cutoff point of 100 pg/mL) would have improved the ED physician’s assessment from 74% diagnostic accuracy to 81%, which is statistically significant. Most important, in those patients initially given an intermediate likelihood of CHF, BNP results correctly classified 75% of these patients and rarely missed ADHF cases (<10%).
Atrial fibrillation. Since the original trials that established a BNP cutoff of 100 pg/mL for determining the presence of ADHF, several adjustments have been suggested. The presence of atrial fibrillation has been shown to increase BNP values independent of cardiac filling pressures. Breidthardt et al examined patients with atrial fibrillation presenting with dyspnea.4 In their analysis, using a cutoff of 100 pg/mL remained robust in identifying patients without ADHF. However, in the 100 pg/mL-500 pg/mL range, the test was not able to discriminate between atrial fibrillation and ADHF. Values greater than 500 pg/mL proved accurate in supporting the diagnosis of ADHF.
Renal failure. Renal dysfunction also elevates BNP levels independent of filling pressures. McCullough et al re-examined data from their Breathing Not Properly Multinational Study and found that the glomerular filtration rate (GFR) was inversely related to BNP levels.5 They recommend using a cutoff point of 200 pg/mL when the GFR is below 60 mg/dL. Other authors recommend not using BNP levels to diagnose ADHF when the GFR is less than 60 mg/dL due to the lack of data supporting this approach. Until clarified, clinicians should be cautious of interpreting BNP elevations in the setting of kidney disease.
Obesity. Obesity has a negative effect on BNP levels, decreasing the sensitivity of the test in these patients.6 Although no study defines how to adjust for body mass index (BMI), clinicians should be cautious about using a low BNP to rule out ADHF in a dyspneic obese patient.
Historical BNP values. If historical BNP values are available, studies of biological variation have shown that an increase to 123% from 66% from baseline is representative of a clinically meaningful increase in cardiac filling pressures. Less significant changes could merely represent biological variation and should be cautiously interpreted.7
Cost effectiveness. The cost effectiveness of using BNP measurements in dyspneic ED patients has been examined as well. Mueller et al found in a Swiss hospital that BNP testing was associated with a 25% decrease in treatment cost, length of stay (LOS), and ICU usage.8 However, LOS is significantly longer in Switzerland compared with the U.S., and given that much of the cost savings was attributed to reducing LOS, it is not possible to extrapolate these data to the U.S. health system. More evidence is needed to truly evaluate the cost effectiveness of BNP testing.
Serial BNP testing. Once a patient has been diagnosed with ADHF and admitted to the hospital, diuretics are indicated with the goal of achieving euvolemia. The bedside assessment of volume status remains a difficult and inexact science, and failure to appropriately remove fluid is associated with readmissions. Conversely, overdiuresis with a concomitant rise in creatinine has been associated with increased morbidity and mortality.
Several studies have shown that the reduction of volume associated with diuretic administration is coupled with a rapid decrease in BNP levels. Therefore, serial BNP measurement has been evaluated as a tool to guide the daily assessment of volume status in patients admitted with ADHF. Unfortunately, frequent measurements of BNP reveal that a great deal of variance, or “noise,” is present in these repeat measurements. Data do not clearly show how to incorporate serial BNP measurements into daily diuretic management.9
Mortality prediction. Nearly 3.5% of admitted heart failure patients will die during their hospitalization. For perspective, the rate of hospital mortality with acute myocardial infarction is 7%. BNP serves as a powerful and independent predictor of inpatient mortality. The ADHERE (Acute Decompensated Heart Failure National Registry) study showed that when divided into BNP quartiles of <430 pg/mL, 430 pg/mL to 839 pg/mL, 840 pg/mL to 1,729 pg/mL, and >1,730 pg/mL, patients’ risk of inpatient death was accurately predicted as 1.9%, 2.8%, 3.8%, and 6.0%, respectively.10 Even when adjusted for other risk factors, BNP remained a powerful predictor; the mortality rate more than doubled from the lowest to highest quartile.
Different strategies have been proposed to improve the outcomes in these highest-risk patients; however, to date, no evidence-based strategy offers a meaningful way to reduce inpatient mortality beyond the current standard of care.
Readmission and 30-day mortality. The 30-day readmission rate after discharge for ADHF is more than than 25%. A study of Medicare patients showed that more than $17 billion (more than 15% of all Medicare payments to hospitals) was associated with unplanned rehospitalizations.11 As bundling payment trends develop, hospitals have an enormous incentive to identify CHF patients with the highest risk of readmission and attempt to mitigate that risk.
From a patient-centered view, upon hospital discharge a patient with ADHF also realizes a 1 in 10 chance of dying within the first 30 days.
At discharge, BNP serves as a powerful and independent marker of increased risk of readmission, morbidity, and mortality. O’Connor et al developed a discharge risk model in patients with severe left ventricular dysfunction; the ESCAPE risk model and discharge score showed elevated BNP was the single most powerful predictor of six-month mortality.12 For every doubling of the BNP, the odds of death at six months increased by 1.4 times.
After combining discharge BNP with other factors, the ESCAPE discharge score was fairly successful at discriminating between patients who would and would not survive to six months. By identifying these outpatients, intensive management strategies could be focused on individuals with the highest risk. The data support the idea that readmission reductions are significant when outpatients obtain early follow-up. Many healthcare centers struggle to schedule early follow-up for all heart failure patients.
As such, the ability to target individuals with the highest discharge scores for intensive follow-up might improve outcomes. These patients could undergo early evaluation for such advanced therapies as resynchronization, left ventricular assist device implantation, or listing for transplantation. Currently, this strategy is not proven. It also is possible that these high-risk patients might have such advanced diseases that their risk cannot be modified by our current medications and advanced therapies.
Back to the Case
This patient has symptoms and signs that could be caused by ADHF or COPD. Her presentation is consistent with an intermediate probability of ADHF. A rapid BNP reveals a level of 950 pg/mL.
Even considering the higher cutoff required because of her coexistent atrial fibrillation, her BNP is consistent with ADHF. Additionally, her obesity likely has decreased the true value of her BNP. A previous BNP drawn when the patient was not in ADHF was 250 ng/mL, meaning that at least a 70% increase is present.
She was admitted and treated with intravenous diuretics with improvement in her congestion and relief of her symptoms. Daily BNPs were not drawn and her diuretics were titrated based on bedside clinical assessments. Her admission BNP elevation would predict a moderately high risk of short- and intermediate term of morbidity and mortality.
At discharge, a repeat BNP also could add to her risk stratification, though it would not be clear what do with this prognostic information beyond the standard of care.
Bottom Line
BNP measurement in specific situations can complement conventional clinical information in determining the presence of ADHF and also can enhance clinicians’ ability to risk-stratify patients during and after hospitalization. TH
Dr. Wolfe is a hospitalist and assistant professor of medicine at the University of Colorado Denver.
References
- Schocken DD, Arrieta MI, Leaverton PE, Ross EA. Prevalence and mortality of congestive heart failure in the United States. J Am Coll Cardiol. 1992;20(2):301-306.
- McKee PA, Castelli WP, McNamara PM, Kannel WB. The natural history of congestive heart failure: the Framingham study. N Eng J Med. 1971;285(26):1441-1446.
- Wang CS, FitzGerald JM, Schulzer M, Mak E, Ayas NT. Does this dyspneic patient in the emergency department have congestive heart failure? JAMA. 2005;294(15):1944-1956.
- Breidthardt T, Noveanu M, Cayir S, et al. The use of B-type natriuretic peptide in the management of patients with atrial fibrillation and dyspnea. Int J Cardiol. 2009;136(2):193-199.
- McCullough PA, Duc P, Omland T, et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the Breathing Not Properly Multinational Study. Am J Kidney Dis. 2003;41(3):571-579.
- Iwanaga Y, Hihara Y, Nizuma S, et al. BNP in overweight and obese patients with heart failure: an analysis based on the BNP-LV diastolic wall stress relationship. J Card Fail. 2007;13(8):663-667.
- O’Hanlon R, O’Shea P, Ledwidge M. The biologic variability of B-type natriuretic peptide and N-terminal pro-B-type natriuretic peptide in stable heart failure patients. J Card Fail. 2007;13(1):50-55.
- Mueller C, Laule-Kilian K, Schindler C, et al. Cost-effectiveness of B-type natriuretic peptide testing in patients with acute dyspnea. Arch Intern Med. 2006;166(1):1081-1087.
- Wu AH. Serial testing of B-type natriuretic peptide and NTpro-BNP for monitoring therapy of heart failure: the role of biologic variation in the interpretation of results. Am Heart J. 2006;152(5):828-834.
- Fonarow GC, Peacock WF, Phillips CO, et al. ADHERE Scientific Advisory Committee and Investigators. Admission B-type natriuretic peptide levels and in-hospital mortality in acute decompensated heart failure. J Am Coll Cardiol. 2007;48 (19):1943-1950.
- Jencks SF, Williams MC, Coleman EA. Rehospitalizations among patients in the Medicare fee-for-service program. N Engl J Med. 2009;360(14):1418-1428.
- O’Connor CM, Hasselblad V, Mehta RH, et al. Triage after hospitalization with advanced heart failure: the ESCAPE (Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness) risk model and discharge score. J Am Coll Cardiol. 2010;55(9):872-878.
Communication Counts
Put yourself for a moment in your patient’s situation. You are sick enough to have been thrust out of your normal life and admitted to the hospital. You find yourself attached to unfamiliar objects and machines, listening to unfamiliar words, and watching a revolving door of unfamiliar faces stroll in and out of the room to take blood, ask personal questions, touch your body, and monitor equipment. It would be enough to bear if you were well, but you’re not. You are ill and that makes you feel particularly worried and desperate.
Whether the physician succeeds in this scenario largely depends on their communication skills.
“A hospitalist needs to develop an almost immediate relationship with their patients because they are at their most vulnerable,” says Mark Williams, MD, FACP, FHM, professor and chief of the Division of Hospital Medicine at Northwestern University’s Feinberg School of Medicine in Chicago. “It is proven that if a hospitalist can successfully communicate with their patients, the result is much more satisfied patients.”

CAT: The Doctor-Patient Relationship Exam
This is easier said than done, as new research by Dr. Williams and colleagues at Feinberg, Northwestern Memorial Hospital (NMH) in Chicago, and Saint Francis Hospital and Medical Center in Hartford, Conn., has found. As part of the study, which published in the December issue of the Journal of Hospital Medicine, patients who were admitted to NMH between September 2008 and August 2009 and cared for by a hospitalist or hospitalist-led teaching team were interviewed using the Communication Assessment Tool (CAT). The CAT is a 14-item survey designed to measure a patient’s perception of communication with their hospitalist.
The average excellent rating among the 35 hospitalists involved in the study was 59.1% on a scale of 0 to 100 percent. Collectively, the hospitalists scored highest on such items as paying attention to patients (64.1%), talking in terms patients could understand (64.2%), and showing care and concern for patients (63.8%). The hospitalists scored lowest in greeting patients in a way that made them feel comfortable (54.9%), encouraging patients to ask questions (53.2%), and involving patients in decisions as much as they wanted (52.9%).
“There are a lot of factors working against hospitalists. Hospitalists are first meeting their patients when they are at their weakest, they sometimes don’t know the patient’s history, and, of course, there are all the demands on hospitalists’ time,” says Darlene Ferranti, research coordinator at the Feinberg School of Medicine.
What is particularly fascinating about the research is 13% of the patients eligible for the study could not participate because they weren’t able to identify their hospitalist by name or photo, Ferranti says. “If your patient doesn’t know who you are, how can they recall the information you are sharing with them?” she asks.
The study wasn’t designed to test patient communication techniques and their effectiveness, Dr. Williams explains. “We think future research needs to focus on interventions to improve doctor-patient communication,” he says.
However, the study did demonstrate that the CAT survey can be a valuable tool for HM groups interested in learning how their physicians are doing from the patient’s perspective, Dr. Williams notes. Perhaps more importantly, it can also help hospitalists target those communication areas in need of improvement, Ferranti says. For example, each hospitalist in the study was given a report of their individual scores and where they fell in the chart compared to the group as a whole.
“If you want to improve your career, you need to improve your communication with patients,” says Dr. Williams, who notes that hospitalists often don’t know the areas in which they are weak and strong. “It’s a career killer if you have multiple patient complaints against you.”
Risk Reduction
Being an effective communicator can also reduce one’s risk of being sued for malpractice, says Mitchell Wilson, MD, FHM, chief medical officer of Atlanta-based Eagle Hospital Physicians, which manages hospitalist practices for clients in the Southeast and Mid-Atlantic regions of the U.S. Dr. Wilson’s company believes communication is so important that starting with the very first interview of a hospitalist candidate, it considers the candidate’s ability to communicate by taking note of such things as accents, how they present information, and body language, Dr. Wilson says.
“Communication is one of the top three competencies that are essential to hospitalists,” he says.
Certain aspects of hospitalist work make communication exceedingly important, Dr. Wilson says. Hospitalists are coordinators of a patient’s care; they are caring for patients who are out of their comfort zone; many times the patient is in an extreme health situation; and hospitalized patients are of all different ages and backgrounds.
“If a hospitalist is a poor communicator, I would encourage them to seek additional training,” Dr. Wilson says. TH
Lisa Ryan is a freelance writer based in New Jersey.
Put yourself for a moment in your patient’s situation. You are sick enough to have been thrust out of your normal life and admitted to the hospital. You find yourself attached to unfamiliar objects and machines, listening to unfamiliar words, and watching a revolving door of unfamiliar faces stroll in and out of the room to take blood, ask personal questions, touch your body, and monitor equipment. It would be enough to bear if you were well, but you’re not. You are ill and that makes you feel particularly worried and desperate.
Whether the physician succeeds in this scenario largely depends on their communication skills.
“A hospitalist needs to develop an almost immediate relationship with their patients because they are at their most vulnerable,” says Mark Williams, MD, FACP, FHM, professor and chief of the Division of Hospital Medicine at Northwestern University’s Feinberg School of Medicine in Chicago. “It is proven that if a hospitalist can successfully communicate with their patients, the result is much more satisfied patients.”
CAT: The Doctor-Patient Relationship Exam
This is easier said than done, as new research by Dr. Williams and colleagues at Feinberg, Northwestern Memorial Hospital (NMH) in Chicago, and Saint Francis Hospital and Medical Center in Hartford, Conn., has found. As part of the study, which published in the December issue of the Journal of Hospital Medicine, patients who were admitted to NMH between September 2008 and August 2009 and cared for by a hospitalist or hospitalist-led teaching team were interviewed using the Communication Assessment Tool (CAT). The CAT is a 14-item survey designed to measure a patient’s perception of communication with their hospitalist.
The average excellent rating among the 35 hospitalists involved in the study was 59.1% on a scale of 0 to 100 percent. Collectively, the hospitalists scored highest on such items as paying attention to patients (64.1%), talking in terms patients could understand (64.2%), and showing care and concern for patients (63.8%). The hospitalists scored lowest in greeting patients in a way that made them feel comfortable (54.9%), encouraging patients to ask questions (53.2%), and involving patients in decisions as much as they wanted (52.9%).
“There are a lot of factors working against hospitalists. Hospitalists are first meeting their patients when they are at their weakest, they sometimes don’t know the patient’s history, and, of course, there are all the demands on hospitalists’ time,” says Darlene Ferranti, research coordinator at the Feinberg School of Medicine.
What is particularly fascinating about the research is 13% of the patients eligible for the study could not participate because they weren’t able to identify their hospitalist by name or photo, Ferranti says. “If your patient doesn’t know who you are, how can they recall the information you are sharing with them?” she asks.
The study wasn’t designed to test patient communication techniques and their effectiveness, Dr. Williams explains. “We think future research needs to focus on interventions to improve doctor-patient communication,” he says.
However, the study did demonstrate that the CAT survey can be a valuable tool for HM groups interested in learning how their physicians are doing from the patient’s perspective, Dr. Williams notes. Perhaps more importantly, it can also help hospitalists target those communication areas in need of improvement, Ferranti says. For example, each hospitalist in the study was given a report of their individual scores and where they fell in the chart compared to the group as a whole.
“If you want to improve your career, you need to improve your communication with patients,” says Dr. Williams, who notes that hospitalists often don’t know the areas in which they are weak and strong. “It’s a career killer if you have multiple patient complaints against you.”
Risk Reduction
Being an effective communicator can also reduce one’s risk of being sued for malpractice, says Mitchell Wilson, MD, FHM, chief medical officer of Atlanta-based Eagle Hospital Physicians, which manages hospitalist practices for clients in the Southeast and Mid-Atlantic regions of the U.S. Dr. Wilson’s company believes communication is so important that starting with the very first interview of a hospitalist candidate, it considers the candidate’s ability to communicate by taking note of such things as accents, how they present information, and body language, Dr. Wilson says.
“Communication is one of the top three competencies that are essential to hospitalists,” he says.
Certain aspects of hospitalist work make communication exceedingly important, Dr. Wilson says. Hospitalists are coordinators of a patient’s care; they are caring for patients who are out of their comfort zone; many times the patient is in an extreme health situation; and hospitalized patients are of all different ages and backgrounds.
“If a hospitalist is a poor communicator, I would encourage them to seek additional training,” Dr. Wilson says. TH
Lisa Ryan is a freelance writer based in New Jersey.
Put yourself for a moment in your patient’s situation. You are sick enough to have been thrust out of your normal life and admitted to the hospital. You find yourself attached to unfamiliar objects and machines, listening to unfamiliar words, and watching a revolving door of unfamiliar faces stroll in and out of the room to take blood, ask personal questions, touch your body, and monitor equipment. It would be enough to bear if you were well, but you’re not. You are ill and that makes you feel particularly worried and desperate.
Whether the physician succeeds in this scenario largely depends on their communication skills.
“A hospitalist needs to develop an almost immediate relationship with their patients because they are at their most vulnerable,” says Mark Williams, MD, FACP, FHM, professor and chief of the Division of Hospital Medicine at Northwestern University’s Feinberg School of Medicine in Chicago. “It is proven that if a hospitalist can successfully communicate with their patients, the result is much more satisfied patients.”
CAT: The Doctor-Patient Relationship Exam
This is easier said than done, as new research by Dr. Williams and colleagues at Feinberg, Northwestern Memorial Hospital (NMH) in Chicago, and Saint Francis Hospital and Medical Center in Hartford, Conn., has found. As part of the study, which published in the December issue of the Journal of Hospital Medicine, patients who were admitted to NMH between September 2008 and August 2009 and cared for by a hospitalist or hospitalist-led teaching team were interviewed using the Communication Assessment Tool (CAT). The CAT is a 14-item survey designed to measure a patient’s perception of communication with their hospitalist.
The average excellent rating among the 35 hospitalists involved in the study was 59.1% on a scale of 0 to 100 percent. Collectively, the hospitalists scored highest on such items as paying attention to patients (64.1%), talking in terms patients could understand (64.2%), and showing care and concern for patients (63.8%). The hospitalists scored lowest in greeting patients in a way that made them feel comfortable (54.9%), encouraging patients to ask questions (53.2%), and involving patients in decisions as much as they wanted (52.9%).
“There are a lot of factors working against hospitalists. Hospitalists are first meeting their patients when they are at their weakest, they sometimes don’t know the patient’s history, and, of course, there are all the demands on hospitalists’ time,” says Darlene Ferranti, research coordinator at the Feinberg School of Medicine.
What is particularly fascinating about the research is 13% of the patients eligible for the study could not participate because they weren’t able to identify their hospitalist by name or photo, Ferranti says. “If your patient doesn’t know who you are, how can they recall the information you are sharing with them?” she asks.
The study wasn’t designed to test patient communication techniques and their effectiveness, Dr. Williams explains. “We think future research needs to focus on interventions to improve doctor-patient communication,” he says.
However, the study did demonstrate that the CAT survey can be a valuable tool for HM groups interested in learning how their physicians are doing from the patient’s perspective, Dr. Williams notes. Perhaps more importantly, it can also help hospitalists target those communication areas in need of improvement, Ferranti says. For example, each hospitalist in the study was given a report of their individual scores and where they fell in the chart compared to the group as a whole.
“If you want to improve your career, you need to improve your communication with patients,” says Dr. Williams, who notes that hospitalists often don’t know the areas in which they are weak and strong. “It’s a career killer if you have multiple patient complaints against you.”
Risk Reduction
Being an effective communicator can also reduce one’s risk of being sued for malpractice, says Mitchell Wilson, MD, FHM, chief medical officer of Atlanta-based Eagle Hospital Physicians, which manages hospitalist practices for clients in the Southeast and Mid-Atlantic regions of the U.S. Dr. Wilson’s company believes communication is so important that starting with the very first interview of a hospitalist candidate, it considers the candidate’s ability to communicate by taking note of such things as accents, how they present information, and body language, Dr. Wilson says.
“Communication is one of the top three competencies that are essential to hospitalists,” he says.
Certain aspects of hospitalist work make communication exceedingly important, Dr. Wilson says. Hospitalists are coordinators of a patient’s care; they are caring for patients who are out of their comfort zone; many times the patient is in an extreme health situation; and hospitalized patients are of all different ages and backgrounds.
“If a hospitalist is a poor communicator, I would encourage them to seek additional training,” Dr. Wilson says. TH
Lisa Ryan is a freelance writer based in New Jersey.
On the Road Again?
Necessity, as they say, is the mother of invention, and the growing focus on the need for high-quality and cost-effective care is bringing a host of new innovations to light. One that hospitalists are likely to hear about far more about is the evolving role of an “extensivist,” an inpatient provider who ventures to outpatient settings to assist with care transitions.
In many ways, the expanding discussion of what extensivists are and do reflects the success of hospitalists in coordinating inpatient care and improving such metrics as length of stay (LOS). Why should that coordination end upon discharge? healthcare experts have wondered. Instead of a pure hospitalist system, could the experience and training of hospitalists be extended to include transitional or interim settings that provide a safety net between the hospital and a primary-care physician (PCP)? Might that improved inpatient-outpatient coordination help with other metrics, such as reduced rehospitalizations (see “All Aboard,” p. 1)?

One tangible result of those questions has been the growth of high-risk clinics. Hospitalist programs can provide clinic referrals for discharged patients who still require hands-on care, while PCPs can likewise refer some of their more complex patients. The clinic, then, becomes an alternative to hospitalization, or a preventive measure to avoid rehospitalization.
Philip Sanger, MD, founder and former CEO of Houston-based Inpatient Medical Services (now Intercede Health), is credited with one of the first uses of the term “extensivist.” Initially, it only described a hospitalist or other care provider who sees high-risk patients in an outpatient clinic.
Writing in Managed Healthcare Executive in 2002, Dr. Sanger explained: “To move from generally sick to generally well, high-risk patients need something extra—more attention than a busy PCP can offer [and] more individualized care than most protocol-driven disease management programs can provide. A high-risk clinic system is one way to fill this care gap. Also referred to as transitional-care clinics, these outpatient clinics focus on preventing hospital admissions and stabilizing high-risk patients.”1
Measured Improvements
Adam Singer, MD, CEO of North Hollywood, Calif.-based IPC: The Hospitalist Co., says this extensivist model provides a respite for hospitalists, who typically spend a month at a time in these transitional clinics before heading back into the fray of the hospital. As hospitals, independent physician associations, and managed-care organizations try out new models of care, though, the definition of an extensivist has broadened to include providers in a range of outpatient settings, such as skilled nursing facilities, assistant living communities, and even home health services. California’s CareMore Medicare Advantage plan, in particular, has been cited by the Agency for Healthcare Research and Quality (AHRQ) for using hospitalists as extensivists in both outpatient clinics and skilled nursing facilities to reduce hospital readmission rates, LOS, and inpatient resource use.
The CareMore model reduces the caseload of its hospitalists to about six or eight patients per half-day, giving doctors more time to talk to patients and their family members. Based on those conversations, the extensivist works with a case manager to provide needed resources to each patient after discharge. The doctors also spend roughly half of each day in clinics seeing their own recently discharged patients, and one or two days each week in a skilled nursing facility to visit patients transferred from the hospital (for more details, visit www.innovations.ahrq.gov/content.aspx?id=2903).
Average inpatient LOS among the plan’s 44,000 members dipped to 3.2 days, compared with 5.8 days for Medicare fee-for-service providers and 4.5 days for traditional HM programs in the state. Last April, CareMore’s 30-day readmission rate averaged 13.4%, compared with a 19.6% rate for Medicare.
Baltimore-based Bravo Health has begun opening its own transitional advanced-care centers for members of its Medicare Advantage program, offering case management for complex conditions and immediate care when a PCP is unavailable. Another model has been advanced through team approaches practiced by the likes of Kaiser Permanente, though hospitalists aren’t necessarily the ones providing outpatient follow-up care. No matter what the model is called, Dr. Singer says, the main point is the same: “trying to connect the dots so that we get patients continuing to get better along the continuum without having to be readmitted.”
In December, IPC did some more dot-connecting of its own with its announced acquisition of Senior Care of Colorado, which operates more than 200 geriatric-care facilities in the Denver area. Don Murphy, MD, IPC’s practice group leader for Senior Care of Colorado, says the model emphasizes a continuum of care and information flow from hospitalists in the hospital to affiliated providers in skilled nursing facilities and other outpatient settings. “We think that model, where we tie everything together, will be one of the best that we can do,” Dr. Murphy says.
From Dr. Singer’s perspective, the growing opportunities have sprung from efforts to address a persistent challenge. “We write an order of discharge to a skilled nursing facility, and patients go off into the community and we have no idea where they’re going, who they’re going to, what’s the quality of care out there, what’s the capacity of care,” he says. One of the central ideas of healthcare reform—creating true accountability around an episode of care—will require doctors to be linked “not just during what used to be the episode of care in the hospital,” he adds, “but throughout the continuum until that patient is really returned healthy back to wherever they’re going to be living.”
With a new emphasis on avoidable hospitalizations, hospitals will increasingly need to team up with other providers to avoid fragmentation of care. Using extensivists to help avoid gaps might be a good fit for accountable-care organizations, and Dr. Murphy says the process may be easier for big systems, such as Ochsner in New Orleans or the Cleveland Clinic, which are working in a confined geographic area with a defined patient population.
“The real challenge will be those of us out there in larger metropolitan areas where we’re not under the roof of one big conglomerate but still having to work together creatively and effectively to smooth the continuum,” Dr. Singer says.
Emerging Trends
Other trends are making inpatient-outpatient partnerships, whether formal or informal, an increasingly necessary part of providing high-quality healthcare. “We are seeing folks come out of the hospitals who 30 years ago clearly would have been in the hospital for a prolonged stay,” Dr. Murphy says. “A lot of these [patients], instead of going to SNFs, are going back to their homes and to assisted living with additional services; they require a lot of follow-up.”
Dr. Singer says he’s seeing another trend in which PCPs are likewise transitioning to newly created extensivist roles in sub-acute settings such as nursing homes. The position, he says, offers the attraction of a high-impact, longer-term relationship with patients without the high overhead of standalone clinics. The blurring of lines between outpatient and inpatient providers has created questions for hospitalists, too. For example, at what point does a hospitalist working much of the time in an outpatient clinic or skilled nursing facility no longer fit the traditional definition of a hospitalist? Does that detract from the doctor’s hospital duties? TH
Bryn Nelson is a freelance medical writer based in Seattle.
Reference
- Sanger, P. Health plans juggle precarious patients. Managed Healthcare Executive. 2002:40-41.
Necessity, as they say, is the mother of invention, and the growing focus on the need for high-quality and cost-effective care is bringing a host of new innovations to light. One that hospitalists are likely to hear about far more about is the evolving role of an “extensivist,” an inpatient provider who ventures to outpatient settings to assist with care transitions.
In many ways, the expanding discussion of what extensivists are and do reflects the success of hospitalists in coordinating inpatient care and improving such metrics as length of stay (LOS). Why should that coordination end upon discharge? healthcare experts have wondered. Instead of a pure hospitalist system, could the experience and training of hospitalists be extended to include transitional or interim settings that provide a safety net between the hospital and a primary-care physician (PCP)? Might that improved inpatient-outpatient coordination help with other metrics, such as reduced rehospitalizations (see “All Aboard,” p. 1)?
One tangible result of those questions has been the growth of high-risk clinics. Hospitalist programs can provide clinic referrals for discharged patients who still require hands-on care, while PCPs can likewise refer some of their more complex patients. The clinic, then, becomes an alternative to hospitalization, or a preventive measure to avoid rehospitalization.
Philip Sanger, MD, founder and former CEO of Houston-based Inpatient Medical Services (now Intercede Health), is credited with one of the first uses of the term “extensivist.” Initially, it only described a hospitalist or other care provider who sees high-risk patients in an outpatient clinic.
Writing in Managed Healthcare Executive in 2002, Dr. Sanger explained: “To move from generally sick to generally well, high-risk patients need something extra—more attention than a busy PCP can offer [and] more individualized care than most protocol-driven disease management programs can provide. A high-risk clinic system is one way to fill this care gap. Also referred to as transitional-care clinics, these outpatient clinics focus on preventing hospital admissions and stabilizing high-risk patients.”1
Measured Improvements
Adam Singer, MD, CEO of North Hollywood, Calif.-based IPC: The Hospitalist Co., says this extensivist model provides a respite for hospitalists, who typically spend a month at a time in these transitional clinics before heading back into the fray of the hospital. As hospitals, independent physician associations, and managed-care organizations try out new models of care, though, the definition of an extensivist has broadened to include providers in a range of outpatient settings, such as skilled nursing facilities, assistant living communities, and even home health services. California’s CareMore Medicare Advantage plan, in particular, has been cited by the Agency for Healthcare Research and Quality (AHRQ) for using hospitalists as extensivists in both outpatient clinics and skilled nursing facilities to reduce hospital readmission rates, LOS, and inpatient resource use.
The CareMore model reduces the caseload of its hospitalists to about six or eight patients per half-day, giving doctors more time to talk to patients and their family members. Based on those conversations, the extensivist works with a case manager to provide needed resources to each patient after discharge. The doctors also spend roughly half of each day in clinics seeing their own recently discharged patients, and one or two days each week in a skilled nursing facility to visit patients transferred from the hospital (for more details, visit www.innovations.ahrq.gov/content.aspx?id=2903).
Average inpatient LOS among the plan’s 44,000 members dipped to 3.2 days, compared with 5.8 days for Medicare fee-for-service providers and 4.5 days for traditional HM programs in the state. Last April, CareMore’s 30-day readmission rate averaged 13.4%, compared with a 19.6% rate for Medicare.
Baltimore-based Bravo Health has begun opening its own transitional advanced-care centers for members of its Medicare Advantage program, offering case management for complex conditions and immediate care when a PCP is unavailable. Another model has been advanced through team approaches practiced by the likes of Kaiser Permanente, though hospitalists aren’t necessarily the ones providing outpatient follow-up care. No matter what the model is called, Dr. Singer says, the main point is the same: “trying to connect the dots so that we get patients continuing to get better along the continuum without having to be readmitted.”
In December, IPC did some more dot-connecting of its own with its announced acquisition of Senior Care of Colorado, which operates more than 200 geriatric-care facilities in the Denver area. Don Murphy, MD, IPC’s practice group leader for Senior Care of Colorado, says the model emphasizes a continuum of care and information flow from hospitalists in the hospital to affiliated providers in skilled nursing facilities and other outpatient settings. “We think that model, where we tie everything together, will be one of the best that we can do,” Dr. Murphy says.
From Dr. Singer’s perspective, the growing opportunities have sprung from efforts to address a persistent challenge. “We write an order of discharge to a skilled nursing facility, and patients go off into the community and we have no idea where they’re going, who they’re going to, what’s the quality of care out there, what’s the capacity of care,” he says. One of the central ideas of healthcare reform—creating true accountability around an episode of care—will require doctors to be linked “not just during what used to be the episode of care in the hospital,” he adds, “but throughout the continuum until that patient is really returned healthy back to wherever they’re going to be living.”
With a new emphasis on avoidable hospitalizations, hospitals will increasingly need to team up with other providers to avoid fragmentation of care. Using extensivists to help avoid gaps might be a good fit for accountable-care organizations, and Dr. Murphy says the process may be easier for big systems, such as Ochsner in New Orleans or the Cleveland Clinic, which are working in a confined geographic area with a defined patient population.
“The real challenge will be those of us out there in larger metropolitan areas where we’re not under the roof of one big conglomerate but still having to work together creatively and effectively to smooth the continuum,” Dr. Singer says.
Emerging Trends
Other trends are making inpatient-outpatient partnerships, whether formal or informal, an increasingly necessary part of providing high-quality healthcare. “We are seeing folks come out of the hospitals who 30 years ago clearly would have been in the hospital for a prolonged stay,” Dr. Murphy says. “A lot of these [patients], instead of going to SNFs, are going back to their homes and to assisted living with additional services; they require a lot of follow-up.”
Dr. Singer says he’s seeing another trend in which PCPs are likewise transitioning to newly created extensivist roles in sub-acute settings such as nursing homes. The position, he says, offers the attraction of a high-impact, longer-term relationship with patients without the high overhead of standalone clinics. The blurring of lines between outpatient and inpatient providers has created questions for hospitalists, too. For example, at what point does a hospitalist working much of the time in an outpatient clinic or skilled nursing facility no longer fit the traditional definition of a hospitalist? Does that detract from the doctor’s hospital duties? TH
Bryn Nelson is a freelance medical writer based in Seattle.
Reference
- Sanger, P. Health plans juggle precarious patients. Managed Healthcare Executive. 2002:40-41.
Necessity, as they say, is the mother of invention, and the growing focus on the need for high-quality and cost-effective care is bringing a host of new innovations to light. One that hospitalists are likely to hear about far more about is the evolving role of an “extensivist,” an inpatient provider who ventures to outpatient settings to assist with care transitions.
In many ways, the expanding discussion of what extensivists are and do reflects the success of hospitalists in coordinating inpatient care and improving such metrics as length of stay (LOS). Why should that coordination end upon discharge? healthcare experts have wondered. Instead of a pure hospitalist system, could the experience and training of hospitalists be extended to include transitional or interim settings that provide a safety net between the hospital and a primary-care physician (PCP)? Might that improved inpatient-outpatient coordination help with other metrics, such as reduced rehospitalizations (see “All Aboard,” p. 1)?
One tangible result of those questions has been the growth of high-risk clinics. Hospitalist programs can provide clinic referrals for discharged patients who still require hands-on care, while PCPs can likewise refer some of their more complex patients. The clinic, then, becomes an alternative to hospitalization, or a preventive measure to avoid rehospitalization.
Philip Sanger, MD, founder and former CEO of Houston-based Inpatient Medical Services (now Intercede Health), is credited with one of the first uses of the term “extensivist.” Initially, it only described a hospitalist or other care provider who sees high-risk patients in an outpatient clinic.
Writing in Managed Healthcare Executive in 2002, Dr. Sanger explained: “To move from generally sick to generally well, high-risk patients need something extra—more attention than a busy PCP can offer [and] more individualized care than most protocol-driven disease management programs can provide. A high-risk clinic system is one way to fill this care gap. Also referred to as transitional-care clinics, these outpatient clinics focus on preventing hospital admissions and stabilizing high-risk patients.”1
Measured Improvements
Adam Singer, MD, CEO of North Hollywood, Calif.-based IPC: The Hospitalist Co., says this extensivist model provides a respite for hospitalists, who typically spend a month at a time in these transitional clinics before heading back into the fray of the hospital. As hospitals, independent physician associations, and managed-care organizations try out new models of care, though, the definition of an extensivist has broadened to include providers in a range of outpatient settings, such as skilled nursing facilities, assistant living communities, and even home health services. California’s CareMore Medicare Advantage plan, in particular, has been cited by the Agency for Healthcare Research and Quality (AHRQ) for using hospitalists as extensivists in both outpatient clinics and skilled nursing facilities to reduce hospital readmission rates, LOS, and inpatient resource use.
The CareMore model reduces the caseload of its hospitalists to about six or eight patients per half-day, giving doctors more time to talk to patients and their family members. Based on those conversations, the extensivist works with a case manager to provide needed resources to each patient after discharge. The doctors also spend roughly half of each day in clinics seeing their own recently discharged patients, and one or two days each week in a skilled nursing facility to visit patients transferred from the hospital (for more details, visit www.innovations.ahrq.gov/content.aspx?id=2903).
Average inpatient LOS among the plan’s 44,000 members dipped to 3.2 days, compared with 5.8 days for Medicare fee-for-service providers and 4.5 days for traditional HM programs in the state. Last April, CareMore’s 30-day readmission rate averaged 13.4%, compared with a 19.6% rate for Medicare.
Baltimore-based Bravo Health has begun opening its own transitional advanced-care centers for members of its Medicare Advantage program, offering case management for complex conditions and immediate care when a PCP is unavailable. Another model has been advanced through team approaches practiced by the likes of Kaiser Permanente, though hospitalists aren’t necessarily the ones providing outpatient follow-up care. No matter what the model is called, Dr. Singer says, the main point is the same: “trying to connect the dots so that we get patients continuing to get better along the continuum without having to be readmitted.”
In December, IPC did some more dot-connecting of its own with its announced acquisition of Senior Care of Colorado, which operates more than 200 geriatric-care facilities in the Denver area. Don Murphy, MD, IPC’s practice group leader for Senior Care of Colorado, says the model emphasizes a continuum of care and information flow from hospitalists in the hospital to affiliated providers in skilled nursing facilities and other outpatient settings. “We think that model, where we tie everything together, will be one of the best that we can do,” Dr. Murphy says.
From Dr. Singer’s perspective, the growing opportunities have sprung from efforts to address a persistent challenge. “We write an order of discharge to a skilled nursing facility, and patients go off into the community and we have no idea where they’re going, who they’re going to, what’s the quality of care out there, what’s the capacity of care,” he says. One of the central ideas of healthcare reform—creating true accountability around an episode of care—will require doctors to be linked “not just during what used to be the episode of care in the hospital,” he adds, “but throughout the continuum until that patient is really returned healthy back to wherever they’re going to be living.”
With a new emphasis on avoidable hospitalizations, hospitals will increasingly need to team up with other providers to avoid fragmentation of care. Using extensivists to help avoid gaps might be a good fit for accountable-care organizations, and Dr. Murphy says the process may be easier for big systems, such as Ochsner in New Orleans or the Cleveland Clinic, which are working in a confined geographic area with a defined patient population.
“The real challenge will be those of us out there in larger metropolitan areas where we’re not under the roof of one big conglomerate but still having to work together creatively and effectively to smooth the continuum,” Dr. Singer says.
Emerging Trends
Other trends are making inpatient-outpatient partnerships, whether formal or informal, an increasingly necessary part of providing high-quality healthcare. “We are seeing folks come out of the hospitals who 30 years ago clearly would have been in the hospital for a prolonged stay,” Dr. Murphy says. “A lot of these [patients], instead of going to SNFs, are going back to their homes and to assisted living with additional services; they require a lot of follow-up.”
Dr. Singer says he’s seeing another trend in which PCPs are likewise transitioning to newly created extensivist roles in sub-acute settings such as nursing homes. The position, he says, offers the attraction of a high-impact, longer-term relationship with patients without the high overhead of standalone clinics. The blurring of lines between outpatient and inpatient providers has created questions for hospitalists, too. For example, at what point does a hospitalist working much of the time in an outpatient clinic or skilled nursing facility no longer fit the traditional definition of a hospitalist? Does that detract from the doctor’s hospital duties? TH
Bryn Nelson is a freelance medical writer based in Seattle.
Reference
- Sanger, P. Health plans juggle precarious patients. Managed Healthcare Executive. 2002:40-41.