User login
45-year-old man • fever • generalized rash • recent history of calcaneal osteomyelitis • Dx?
THE CASE
A 45-year-old man was admitted to the hospital with a fever and generalized rash. For the previous 2 weeks, he had been treated at a skilled nursing facility with IV vancomycin and cefepime for left calcaneal osteomyelitis. He reported that the rash was pruritic and started 2 days prior to hospital admission.
His past medical history was significant for type 2 diabetes mellitus and polysubstance drug abuse. Medical and travel history were otherwise unremarkable. The patient was taking the following medications at the time of presentation: hydrocodone-acetaminophen, cyclobenzaprine, melatonin, and metformin.
Initial vital signs included a temperature of 102.9°F; respiratory rate, 22 breaths/min; heart rate, 97 beats/min; and blood pressure, 89/50 mm Hg. Physical exam was notable for left anterior cervical and axillary lymphadenopathy. The patient had no facial edema, but he did have a diffuse, morbilliform rash on his bilateral upper and lower extremities, encompassing about 54% of his body surface area (FIGURE 1).

Laboratory studies revealed a white blood cell count of 4.7/mcL, with 3.4% eosinophils and 10.9% monocytes; an erythrocyte sedimentation rate of 60 mm/h; and a C-reactive protein level of 1 mg/dL. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels were both elevated (AST: 95 U/L [normal range, 8 - 48 U/L]; ALT: 115 U/L [normal range: 7 - 55 U/L]). A chest x-ray was obtained and showed new lung infiltrates (FIGURE 2).

Linezolid and meropenem were initiated for a presumed health care–associated pneumonia, and a sepsis work-up was initiated.
THE DIAGNOSIS
The patient’s rash and pruritus worsened after meropenem was introduced. A hepatitis panel was nonreactive except for prior hepatitis A exposure. Ultrasound of the liver and spleen was normal. Investigation of pneumonia pathogens including Legionella, Streptococcus, Mycoplasma, and Chlamydia psittaci did not reveal any causative agents. A skin biopsy revealed perivascular neutrophilic dermatitis with dyskeratosis.
The patient was diagnosed with DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome based on his fever, worsening morbilliform rash, lymphadenopathy, and elevated liver transaminase levels. Although he did not have marked eosinophilia, atypical lymphocytes were present. Serologies for human herpesvirus (HHV), Epstein-Barr virus (EBV), and cytomegalovirus (CMV) were all unremarkable.
Continue to: During discussions...
During discussions with an infectious disease specialist, it was concluded that the patient’s DRESS syndrome was likely secondary to beta-lactam antibiotics. The patient had been receiving cefepime prior to hospitalization. Meropenem was discontinued and aztreonam was started, with continued linezolid. This patient did not have a reactivation of a herpesvirus (HHV-6, HHV-7, EBV, or CMV), which has been previously reported in cases of DRESS syndrome.
DISCUSSION
DRESS syndrome is a challenging diagnosis to make due to the multiplicity of presenting symptoms. Skin rash, lymphadenopathy, hepatic involvement, and hypereosinophilia are characteristic findings.1 Accurate diagnosis reduces fatal disease outcomes, which are estimated to occur in 5%-10% of cases.1,2
Causative agents. DRESS syndrome typically occurs 2 to 6 weeks after the introduction of the causative agent, commonly an aromatic anticonvulsant or antibiotic.3 The incidence of DRESS syndrome in patients using carbamazepine and phenytoin is estimated to be 1 to 5 per 10,000 patients. The incidence of DRESS syndrome in patients using antibiotics is unknown. Frequently, the inducing antibiotic is a beta-lactam, as in this case.4,5
The pathogenesis of DRESS syndrome is not well understood, although there appears to be an immune-mediated reaction that occurs in certain patients after viral reactivation, particularly with herpesviruses. In vitro studies have demonstrated that the culprit drug is able to induce viral reactivation leading to T-lymphocyte response and systemic inflammation, which occurs in multiple organs.6,7 Reported long-term sequelae of DRESS syndrome include immune-mediated diseases such as thyroiditis and type 1 diabetes. In addition, it is hypothesized that there is a genetic predisposition involving human leukocyte antigens that increases the likelihood that individuals will develop DRESS syndrome.5,8
Diagnosis. The

Continue to: Treatment
Treatment is aimed at stopping the causative agent and starting moderate- to high-dose systemic corticosteroids (from 0.5 to 2 mg/kg/d). If symptoms continue to progress, cyclosporine can be used. N-acetylcysteine may also be beneficial due to its ability to neutralize drug metabolites that can stimulate T-cell response.7 There has not been sufficient evidence to suggest that antiviral medication should be initiated.1,7
Our patient was treated with 2 mg/kg/d of prednisone, along with triamcinolone cream, diphenhydramine, and N-acetylcysteine. His rash improved dramatically during his hospital stay and at the subsequent 1-month follow-up was completely resolved.
THE TAKEAWAY
DRESS syndrome should be suspected in patients presenting with fever, rash, lymphadenopathy, pulmonary infiltrates, and liver involvement after initiation of drugs commonly associated with this syndrome. Our case reinforces previous clinical evidence that beta-lactam antibiotics are a common cause of DRESS syndrome; patients taking these medications should be closely monitored. Cross-reactions are frequent, and it is imperative that patients avoid related drugs to prevent recurrence. Although glucocorticoids are the mainstay of treatment, further studies are needed to assess the benefits of N-acetylcysteine.
CORRESPONDENCE
W. Jacob Cobb, MD, JPS Health Network, 1500 South Main Street, Fort Worth, TX, 76104; [email protected]
1. Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124:588-597.
2. Chen Y, Chiu H, Chu C. Drug reaction with eosinophilia and systemic symptoms: a retrospective study of 60 cases. Arch Dermatol. 2010;146:1373-1379.
3. Jeung Y-J, Lee J-Y, Oh M-J, et al. Comparison of the causes and clinical features of drug rash with eosinophilia and systemic symptoms and Stevens-Johnson syndrome. Allergy Asthma Immunol Res. 2010;2:123–126.
4. Shiohara T, Iijima M, Ikezawa Z, et al. The diagnosis of a DRESS syndrome has been sufficiently established on the basis of typical clinical features and viral reactivations [commentary]. Br J Dermatol. 2006;156:1083-1084.
5. Ben-Said B, Arnaud-Butel S, Rozières A, et al. Allergic delayed drug hypersensitivity is more frequently diagnosed in drug reaction, eosinophilia and systemic symptoms (DRESS) syndrome than in exanthema induced by beta lactam antibiotics. J Dermatol Sci. 2015;80:71-74.
6. Schrijvers R, Gilissen L, Chiriac AM, et al. Pathogenesis and diagnosis of delayed-type drug hypersensitivity reactions, from bedside to bench and back. Clin Transl Allergy. 2015;5:31.
7. Moling O, Tappeiner L, Piccin A, et al. Treatment of DIHS/DRESS syndrome with combined N-acetylcysteine, prednisone and valganciclovir—a hypothesis. Med Sci Monit. 2012;18:CS57-CS62.
8. Cardoso CS, Vieira AM, Oliveira AP. DRESS syndrome: a case report and literature review. BMJ Case Rep. 2011;2011:bcr0220113898.
9. Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080.
10. Bernard L, Eichenfield L. Drug-associated rashes. In: Zaoutis L, Chiang V, eds. Comprehensive Pediatric Hospital Medicine. Philadelphia, PA: Elsevier; 2010: 1005-1011.
11. Grover S. Severe cutaneous adverse reactions. Indian J Dermatol Venereol Leprol. 2011;77:3-6.
THE CASE
A 45-year-old man was admitted to the hospital with a fever and generalized rash. For the previous 2 weeks, he had been treated at a skilled nursing facility with IV vancomycin and cefepime for left calcaneal osteomyelitis. He reported that the rash was pruritic and started 2 days prior to hospital admission.
His past medical history was significant for type 2 diabetes mellitus and polysubstance drug abuse. Medical and travel history were otherwise unremarkable. The patient was taking the following medications at the time of presentation: hydrocodone-acetaminophen, cyclobenzaprine, melatonin, and metformin.
Initial vital signs included a temperature of 102.9°F; respiratory rate, 22 breaths/min; heart rate, 97 beats/min; and blood pressure, 89/50 mm Hg. Physical exam was notable for left anterior cervical and axillary lymphadenopathy. The patient had no facial edema, but he did have a diffuse, morbilliform rash on his bilateral upper and lower extremities, encompassing about 54% of his body surface area (FIGURE 1).
Laboratory studies revealed a white blood cell count of 4.7/mcL, with 3.4% eosinophils and 10.9% monocytes; an erythrocyte sedimentation rate of 60 mm/h; and a C-reactive protein level of 1 mg/dL. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels were both elevated (AST: 95 U/L [normal range, 8 - 48 U/L]; ALT: 115 U/L [normal range: 7 - 55 U/L]). A chest x-ray was obtained and showed new lung infiltrates (FIGURE 2).
Linezolid and meropenem were initiated for a presumed health care–associated pneumonia, and a sepsis work-up was initiated.
THE DIAGNOSIS
The patient’s rash and pruritus worsened after meropenem was introduced. A hepatitis panel was nonreactive except for prior hepatitis A exposure. Ultrasound of the liver and spleen was normal. Investigation of pneumonia pathogens including Legionella, Streptococcus, Mycoplasma, and Chlamydia psittaci did not reveal any causative agents. A skin biopsy revealed perivascular neutrophilic dermatitis with dyskeratosis.
The patient was diagnosed with DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome based on his fever, worsening morbilliform rash, lymphadenopathy, and elevated liver transaminase levels. Although he did not have marked eosinophilia, atypical lymphocytes were present. Serologies for human herpesvirus (HHV), Epstein-Barr virus (EBV), and cytomegalovirus (CMV) were all unremarkable.
Continue to: During discussions...
During discussions with an infectious disease specialist, it was concluded that the patient’s DRESS syndrome was likely secondary to beta-lactam antibiotics. The patient had been receiving cefepime prior to hospitalization. Meropenem was discontinued and aztreonam was started, with continued linezolid. This patient did not have a reactivation of a herpesvirus (HHV-6, HHV-7, EBV, or CMV), which has been previously reported in cases of DRESS syndrome.
DISCUSSION
DRESS syndrome is a challenging diagnosis to make due to the multiplicity of presenting symptoms. Skin rash, lymphadenopathy, hepatic involvement, and hypereosinophilia are characteristic findings.1 Accurate diagnosis reduces fatal disease outcomes, which are estimated to occur in 5%-10% of cases.1,2
Causative agents. DRESS syndrome typically occurs 2 to 6 weeks after the introduction of the causative agent, commonly an aromatic anticonvulsant or antibiotic.3 The incidence of DRESS syndrome in patients using carbamazepine and phenytoin is estimated to be 1 to 5 per 10,000 patients. The incidence of DRESS syndrome in patients using antibiotics is unknown. Frequently, the inducing antibiotic is a beta-lactam, as in this case.4,5
The pathogenesis of DRESS syndrome is not well understood, although there appears to be an immune-mediated reaction that occurs in certain patients after viral reactivation, particularly with herpesviruses. In vitro studies have demonstrated that the culprit drug is able to induce viral reactivation leading to T-lymphocyte response and systemic inflammation, which occurs in multiple organs.6,7 Reported long-term sequelae of DRESS syndrome include immune-mediated diseases such as thyroiditis and type 1 diabetes. In addition, it is hypothesized that there is a genetic predisposition involving human leukocyte antigens that increases the likelihood that individuals will develop DRESS syndrome.5,8
Diagnosis. The
Continue to: Treatment
Treatment is aimed at stopping the causative agent and starting moderate- to high-dose systemic corticosteroids (from 0.5 to 2 mg/kg/d). If symptoms continue to progress, cyclosporine can be used. N-acetylcysteine may also be beneficial due to its ability to neutralize drug metabolites that can stimulate T-cell response.7 There has not been sufficient evidence to suggest that antiviral medication should be initiated.1,7
Our patient was treated with 2 mg/kg/d of prednisone, along with triamcinolone cream, diphenhydramine, and N-acetylcysteine. His rash improved dramatically during his hospital stay and at the subsequent 1-month follow-up was completely resolved.
THE TAKEAWAY
DRESS syndrome should be suspected in patients presenting with fever, rash, lymphadenopathy, pulmonary infiltrates, and liver involvement after initiation of drugs commonly associated with this syndrome. Our case reinforces previous clinical evidence that beta-lactam antibiotics are a common cause of DRESS syndrome; patients taking these medications should be closely monitored. Cross-reactions are frequent, and it is imperative that patients avoid related drugs to prevent recurrence. Although glucocorticoids are the mainstay of treatment, further studies are needed to assess the benefits of N-acetylcysteine.
CORRESPONDENCE
W. Jacob Cobb, MD, JPS Health Network, 1500 South Main Street, Fort Worth, TX, 76104; [email protected]
THE CASE
A 45-year-old man was admitted to the hospital with a fever and generalized rash. For the previous 2 weeks, he had been treated at a skilled nursing facility with IV vancomycin and cefepime for left calcaneal osteomyelitis. He reported that the rash was pruritic and started 2 days prior to hospital admission.
His past medical history was significant for type 2 diabetes mellitus and polysubstance drug abuse. Medical and travel history were otherwise unremarkable. The patient was taking the following medications at the time of presentation: hydrocodone-acetaminophen, cyclobenzaprine, melatonin, and metformin.
Initial vital signs included a temperature of 102.9°F; respiratory rate, 22 breaths/min; heart rate, 97 beats/min; and blood pressure, 89/50 mm Hg. Physical exam was notable for left anterior cervical and axillary lymphadenopathy. The patient had no facial edema, but he did have a diffuse, morbilliform rash on his bilateral upper and lower extremities, encompassing about 54% of his body surface area (FIGURE 1).
Laboratory studies revealed a white blood cell count of 4.7/mcL, with 3.4% eosinophils and 10.9% monocytes; an erythrocyte sedimentation rate of 60 mm/h; and a C-reactive protein level of 1 mg/dL. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels were both elevated (AST: 95 U/L [normal range, 8 - 48 U/L]; ALT: 115 U/L [normal range: 7 - 55 U/L]). A chest x-ray was obtained and showed new lung infiltrates (FIGURE 2).
Linezolid and meropenem were initiated for a presumed health care–associated pneumonia, and a sepsis work-up was initiated.
THE DIAGNOSIS
The patient’s rash and pruritus worsened after meropenem was introduced. A hepatitis panel was nonreactive except for prior hepatitis A exposure. Ultrasound of the liver and spleen was normal. Investigation of pneumonia pathogens including Legionella, Streptococcus, Mycoplasma, and Chlamydia psittaci did not reveal any causative agents. A skin biopsy revealed perivascular neutrophilic dermatitis with dyskeratosis.
The patient was diagnosed with DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome based on his fever, worsening morbilliform rash, lymphadenopathy, and elevated liver transaminase levels. Although he did not have marked eosinophilia, atypical lymphocytes were present. Serologies for human herpesvirus (HHV), Epstein-Barr virus (EBV), and cytomegalovirus (CMV) were all unremarkable.
Continue to: During discussions...
During discussions with an infectious disease specialist, it was concluded that the patient’s DRESS syndrome was likely secondary to beta-lactam antibiotics. The patient had been receiving cefepime prior to hospitalization. Meropenem was discontinued and aztreonam was started, with continued linezolid. This patient did not have a reactivation of a herpesvirus (HHV-6, HHV-7, EBV, or CMV), which has been previously reported in cases of DRESS syndrome.
DISCUSSION
DRESS syndrome is a challenging diagnosis to make due to the multiplicity of presenting symptoms. Skin rash, lymphadenopathy, hepatic involvement, and hypereosinophilia are characteristic findings.1 Accurate diagnosis reduces fatal disease outcomes, which are estimated to occur in 5%-10% of cases.1,2
Causative agents. DRESS syndrome typically occurs 2 to 6 weeks after the introduction of the causative agent, commonly an aromatic anticonvulsant or antibiotic.3 The incidence of DRESS syndrome in patients using carbamazepine and phenytoin is estimated to be 1 to 5 per 10,000 patients. The incidence of DRESS syndrome in patients using antibiotics is unknown. Frequently, the inducing antibiotic is a beta-lactam, as in this case.4,5
The pathogenesis of DRESS syndrome is not well understood, although there appears to be an immune-mediated reaction that occurs in certain patients after viral reactivation, particularly with herpesviruses. In vitro studies have demonstrated that the culprit drug is able to induce viral reactivation leading to T-lymphocyte response and systemic inflammation, which occurs in multiple organs.6,7 Reported long-term sequelae of DRESS syndrome include immune-mediated diseases such as thyroiditis and type 1 diabetes. In addition, it is hypothesized that there is a genetic predisposition involving human leukocyte antigens that increases the likelihood that individuals will develop DRESS syndrome.5,8
Diagnosis. The
Continue to: Treatment
Treatment is aimed at stopping the causative agent and starting moderate- to high-dose systemic corticosteroids (from 0.5 to 2 mg/kg/d). If symptoms continue to progress, cyclosporine can be used. N-acetylcysteine may also be beneficial due to its ability to neutralize drug metabolites that can stimulate T-cell response.7 There has not been sufficient evidence to suggest that antiviral medication should be initiated.1,7
Our patient was treated with 2 mg/kg/d of prednisone, along with triamcinolone cream, diphenhydramine, and N-acetylcysteine. His rash improved dramatically during his hospital stay and at the subsequent 1-month follow-up was completely resolved.
THE TAKEAWAY
DRESS syndrome should be suspected in patients presenting with fever, rash, lymphadenopathy, pulmonary infiltrates, and liver involvement after initiation of drugs commonly associated with this syndrome. Our case reinforces previous clinical evidence that beta-lactam antibiotics are a common cause of DRESS syndrome; patients taking these medications should be closely monitored. Cross-reactions are frequent, and it is imperative that patients avoid related drugs to prevent recurrence. Although glucocorticoids are the mainstay of treatment, further studies are needed to assess the benefits of N-acetylcysteine.
CORRESPONDENCE
W. Jacob Cobb, MD, JPS Health Network, 1500 South Main Street, Fort Worth, TX, 76104; [email protected]
1. Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124:588-597.
2. Chen Y, Chiu H, Chu C. Drug reaction with eosinophilia and systemic symptoms: a retrospective study of 60 cases. Arch Dermatol. 2010;146:1373-1379.
3. Jeung Y-J, Lee J-Y, Oh M-J, et al. Comparison of the causes and clinical features of drug rash with eosinophilia and systemic symptoms and Stevens-Johnson syndrome. Allergy Asthma Immunol Res. 2010;2:123–126.
4. Shiohara T, Iijima M, Ikezawa Z, et al. The diagnosis of a DRESS syndrome has been sufficiently established on the basis of typical clinical features and viral reactivations [commentary]. Br J Dermatol. 2006;156:1083-1084.
5. Ben-Said B, Arnaud-Butel S, Rozières A, et al. Allergic delayed drug hypersensitivity is more frequently diagnosed in drug reaction, eosinophilia and systemic symptoms (DRESS) syndrome than in exanthema induced by beta lactam antibiotics. J Dermatol Sci. 2015;80:71-74.
6. Schrijvers R, Gilissen L, Chiriac AM, et al. Pathogenesis and diagnosis of delayed-type drug hypersensitivity reactions, from bedside to bench and back. Clin Transl Allergy. 2015;5:31.
7. Moling O, Tappeiner L, Piccin A, et al. Treatment of DIHS/DRESS syndrome with combined N-acetylcysteine, prednisone and valganciclovir—a hypothesis. Med Sci Monit. 2012;18:CS57-CS62.
8. Cardoso CS, Vieira AM, Oliveira AP. DRESS syndrome: a case report and literature review. BMJ Case Rep. 2011;2011:bcr0220113898.
9. Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080.
10. Bernard L, Eichenfield L. Drug-associated rashes. In: Zaoutis L, Chiang V, eds. Comprehensive Pediatric Hospital Medicine. Philadelphia, PA: Elsevier; 2010: 1005-1011.
11. Grover S. Severe cutaneous adverse reactions. Indian J Dermatol Venereol Leprol. 2011;77:3-6.
1. Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med. 2011;124:588-597.
2. Chen Y, Chiu H, Chu C. Drug reaction with eosinophilia and systemic symptoms: a retrospective study of 60 cases. Arch Dermatol. 2010;146:1373-1379.
3. Jeung Y-J, Lee J-Y, Oh M-J, et al. Comparison of the causes and clinical features of drug rash with eosinophilia and systemic symptoms and Stevens-Johnson syndrome. Allergy Asthma Immunol Res. 2010;2:123–126.
4. Shiohara T, Iijima M, Ikezawa Z, et al. The diagnosis of a DRESS syndrome has been sufficiently established on the basis of typical clinical features and viral reactivations [commentary]. Br J Dermatol. 2006;156:1083-1084.
5. Ben-Said B, Arnaud-Butel S, Rozières A, et al. Allergic delayed drug hypersensitivity is more frequently diagnosed in drug reaction, eosinophilia and systemic symptoms (DRESS) syndrome than in exanthema induced by beta lactam antibiotics. J Dermatol Sci. 2015;80:71-74.
6. Schrijvers R, Gilissen L, Chiriac AM, et al. Pathogenesis and diagnosis of delayed-type drug hypersensitivity reactions, from bedside to bench and back. Clin Transl Allergy. 2015;5:31.
7. Moling O, Tappeiner L, Piccin A, et al. Treatment of DIHS/DRESS syndrome with combined N-acetylcysteine, prednisone and valganciclovir—a hypothesis. Med Sci Monit. 2012;18:CS57-CS62.
8. Cardoso CS, Vieira AM, Oliveira AP. DRESS syndrome: a case report and literature review. BMJ Case Rep. 2011;2011:bcr0220113898.
9. Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080.
10. Bernard L, Eichenfield L. Drug-associated rashes. In: Zaoutis L, Chiang V, eds. Comprehensive Pediatric Hospital Medicine. Philadelphia, PA: Elsevier; 2010: 1005-1011.
11. Grover S. Severe cutaneous adverse reactions. Indian J Dermatol Venereol Leprol. 2011;77:3-6.
An Atypical Long-Term Thiamine Treatment Regimen for Wernicke Encephalopathy
Wernicke-Korsakoff syndrome is a cluster of symptoms attributed to a disorder of vitamin B1 (thiamine) deficiency, manifesting as a combined presentation of alcohol-induced Wernicke encephalopathy (WE) and Korsakoff syndrome (KS).1 While there is consensus on the characteristic presentation and symptoms of WE, there is a lack of agreement on the exact definition of KS. The classic triad describing WE consists of ataxia, ophthalmoplegia, and confusion; however, reports now suggest that a majority of patients exhibit only 1 or 2 of the elements of the triad. KS is often seen as a condition of chronic thiamine deficiency manifesting as memory impairment alongside a cognitive and behavioral decline, with no clear consensus on the sequence of appearance of symptoms. The typical relationship is thought to be a progression of WE to KS if untreated.
From a mental health perspective, WE presents with delirium and confusion whereas KS manifests with irreversible dementia and a cognitive deterioration. Though it is commonly taught that KS-induced memory loss is permanent due to neuronal damage (classically identified as damage to the mammillary bodies - though other structures have been implicated as well), more recent research suggest otherwise.2 A review published in 2018, for example, gathered several case reports and case series that suggest significant improvement in memory and cognition attributed to behavioral and pharmacologic interventions, indicating this as an area deserving of further study.3 About 20% of patients diagnosed with WE by autopsy exhibited none of the classical triad symptoms prior to death.4 Hence, these conditions are surmised to be significantly underdiagnosed and misdiagnosed.
Though consensus regarding the appropriate treatment regimen is lacking for WE, a common protocol consists of high-dose parenteral thiamine for 4 to 7 days.5 This is usually followed by daily oral thiamine repletion until the patient either achieves complete abstinence from alcohol (ideal) or decreases consumption. The goal is to allow thiamine stores to replete and maintain at minimum required body levels moving forward. In this case report, we highlight the utilization of a long-term, unconventional intramuscular (IM) thiamine repletion regimen to ensure maintenance of a patient’s mental status, highlighting discrepancies in our understanding of the mechanisms at play in WE and its treatment.
Case Presentation
A 65-year-old male patient with a more than 3-decade history of daily hard liquor intake, multiple psychiatric hospitalizations for WE, and a prior suicide attempt, presented to the emergency department (ED) with increased frequency of falls, poor oral intake, confabulation, and diminished verbal communication. A chart review revealed memory impairment alongside the diagnoses of schizoaffective disorder and WE, and confusion that was responsive to thiamine administration as well as a history of hypertension, hyperlipidemia, osteoarthritis, and urinary retention secondary to benign prostatic hyperplasia (BPH).
On examination the patient was found to be disoriented with a clouded sensorium. While the history of heavy daily alcohol use was clear in the chart and confirmed by other sources, it appeared unlikely that the patient had been using alcohol in the preceding month due to restricted access in his most recent living environment (a shared apartment with daily nursing assistance). He reported no lightheadedness, dizziness, palpitations, numbness, tingling, or any head trauma. He also negated the presence of active mood symptoms, auditory or visual hallucinations or suicidal ideation (SI)
The patient was admitted to the Internal Medicine Service and received a workup for the causes of delirium, including consideration of normal pressure hydrocephalus (NPH) and other neurologic conditions. Laboratory tests including a comprehensive metabolic panel, thyroid stimulating hormone, urinalysis, urine toxicology screen, and vitamin B12 and folate levels were in normal ranges. Although brain imaging revealed enlarged ventricles, NPH was considered unlikely because of the absence of ophthalmologic abnormalities, like gaze nystagmus, and urinary incontinence; conversely, there was some presence of urinary retention attributed to BPH and required an admission a few months prior. Moreover, magnetic resonance images showed that the ventricles were enlarged slightly out of proportion to the sulci, which can be seen with predominantly central volume loss compared with the pattern typically seen in NPH.
In light of concern for WE and the patient's history, treatment with IV thiamine and IV fluids was initiated and the Liaison Psychiatry Service was consulted for cognitive disability and treatment of his mood. Administration of IV thiamine rapidly restored his sensorium, but he became abruptly disorganized as the IV regimen graduated to an oral thiamine dose of 200 mg 3 times daily. Simultaneously, as medical stabilization was achieved, the patient was transferred to the inpatient psychiatry unit to address the nonresolving cognitive impairment and behavioral disorganization. This specifically involved newly emerging, impulsive, self-harming behaviors like throwing himself on the ground and banging his head on the floor. Such behaviors along with paucity of speech and decreased oral intake, ultimately warranted constant observation, which led to a decrease in self-harming activity. All this behavior was noted even though the patient was adherent to oral administration of thiamine. Throughout this time, the patient underwent several transfers back and forth between the Psychiatry and Internal Medicine services due to ongoing concern for the possibility of delirium or WE. However, the Neurology and Internal Medicine services did not feel that WE would explain the patient’s mental and behavioral status, in part due to his ongoing adherence with daily oral thiamine dosing that was not associated with improvement in mental status.
Recollecting the patient’s improvement with the parenteral thiamine regimen (IV and IM), the psychiatry unit tried a thiamine regimen of 200 mg IM and 100 mg oral 2 times daily. After about 2 weeks on this regimen, the patient subsequently achieved remarkable improvement in his cognitive and behavioral status, with resolution of selfharming behaviors. The patient was noted to be calmer, more linear, and more oriented, though he remained incompletely oriented throughout his hospitalization. As improvement in sensorium was established and the patient’s hospital stay prolonged (Figure), his mood symptoms began manifesting as guilt, low energy, decreased appetite, withdrawal, and passive SI. This was followed by a trial of lithium that was discontinued due to elevated creatine levels. As the patient continued to report depression, a multidrug regimen of divalproex, fluoxetine, and quetiapine was administered, which lead to remarkable improvement.
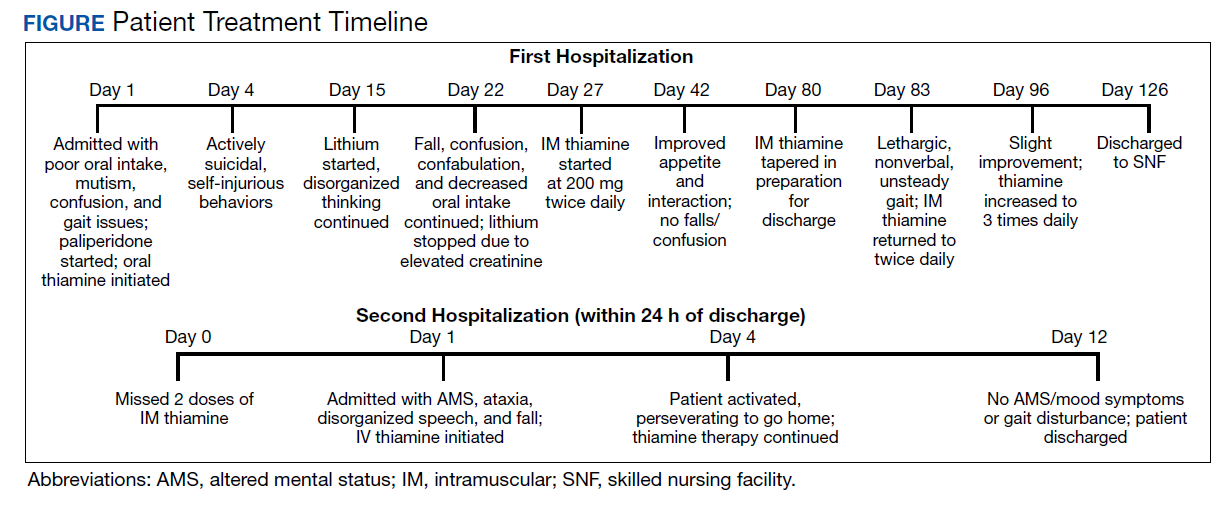
At this time, it was concluded that the stores of thiamine in the patient’s body may have been replenished, the alcohol intake completely ceased and that he needed to be weaned off of thiamine. The next step taken was reduction of the twice daily 200 mg IM thiamine dose to a once daily regimen, and oral thiamine was put on hold. Over the next 48 hours, the patient became less verbal, more withdrawn, incontinent of urine, and delirious. The twice daily IM 200 mg thiamine was restarted, but this time the patient demonstrated very slow improvement. After 2 weeks, the IM thiamine 200 mg was increased to 3 times daily, and the patient showed marked improvement in recall, mood, and effect.
Several attempts were made to reduce the IM thiamine burden on the patient and/ or transition to an exclusively oral regimen. However, he rapidly decompensated within hours of each attempt to taper the IM dose and required immediate reinstation. On the IM thiamine regimen, he eventually appeared to reach a stable cognitive and affective baseline marked by incomplete orientation but pleasant affect, he reported no mood complaints, behavioral stability, and an ability to comply with care needs and have simple conversations. Some speech content remained disorganized particularly if engaged beyond simple exchanges.
The patient was discharged to a skilled nursing facility after a month of 3 times daily IM administration of thiamine. Within the next 24 hours, the patient returned to the ED with the originally reported symptoms of ataxia, agitation, and confusion. On inquiry, it was revealed that the ordered vials of IM thiamine for injection had not arrived with him at the nursing facility and he had missed 2 doses. The blood laboratory results, scans, and all other parameters were otherwise found to be normal and the patient was adherent to his prescribed antipsychotics and antidepressants. As anticipated, restoration of the IM thiamine regimen revived his baseline within hours. While confusion and delirium resolved completely with treatment, the memory impairments persisted. This patient has been administered a 3 times daily IM dose of 200 mg thiamine for more than 2 years with a stable cognitive clinical picture.
Discussion
According to data from the 2016 National Survey on Drug Use and Health, 16 million individuals in the US aged ≥ 12 years reported heavy alcohol use, which is defined as binge drinking on ≥ 5 days in the past month.6,7 Thiamine deficiency is an alcoholrelated disorder that is frequently encountered in hospital settings. This deficiency can also occur in the context of malabsorption, malnutrition, a prolonged course of vomiting, and bariatric surgery.8,9
The deficiency in thiamine, which is sometimes known as WE, manifests rarely with all 3 of the classic triad of gait disturbances, abnormal eye movements, and mental status changes, with only 16.5% of patients displaying all of the triad.4 Moreover, there may be additional symptoms not listed in this triad, such as memory impairment, bilateral sixth nerve palsy, ptosis, hypotension, and hypothermia.10.11 This inconsistent presentation makes the diagnosis challenging and therefore requires a higher threshold for suspicion. If undiagnosed and/or untreated, WE can lead to chronic thiamine deficiency causing permanent brain damage in the guise of KS. This further increases the importance of timely diagnosis and treatment.
Our case highlights the utilization of an unconventional thiamine regimen that appeared to be temporally associated with mental status improvement. The patient’s clouded sensorium and confusion could not be attributed to metabolic, encephalopathic, or infectious pathologies due to the absence of supportive laboratory evidence. He responded to IV and IM doses of thiamine, but repeated attempts to taper the IM doses with the objective of transitioning to oral thiamine supplementation were followed by immediate decompensations in mental status. This was atypical of WE as the patient seemed adequately replete with thiamine, and missing a few doses should not be enough to deplete his stores. Thus, reflecting a unique case of thiamine-dependent chronically set WE when even a single missed dose of thiamine adversely affected the patient’s cognitive baseline. Interesting to note is this patient’s memory issue, as evident by clinical examination and dating back at least 5 years as per chart review. This premature amnestic component of his presentation indicates a likely parallel running KS component of his presentation. Conversely, the patient’s long history of alcohol use disorder, prior episodes of WE, and ideal response achieved only on parenteral thiamine repletion further supported the diagnosis of WE and our impression of the scenario.
Even though this patient had prior episodes of WE, there remained diagnostic uncertainty regarding his altered mental status for some time before the nonoral thiamine repletion treatment was implemented. Particularly in this admission, the patient’s mental status frequently waxed and waned and there was the additional confusion of whether a potential psychiatric etiology contributed to some of the elements of his presentation, such as his impulsive self-harm behaviors. This behavior led to recurrent transfers among the Psychiatry Service, Internal Medicine Service, and the ED.
The patient’s presentation did not reflect the classical triad of WE, and while this is consistent with the majority of clinical manifestations, various services were reluctant to attribute his symptoms to WE. Once the threshold of suspicion of thiamine deficiency was lowered and the deficit treated more aggressively, the patient seemed to improve tremendously. Presence of memory problems and confabulation, both of which this patient exhibited, are suggestive of KS and are not expected to recover with treatment, yet for this patient there did seem to be some improvement—though not complete resolution. This is consistent with newer evidence suggesting that some recovery from the deficits seen in KS is possible.3
Once diagnosed, the treatment objective is the replenishment of thiamine stores and optimization of the metabolic scenario of the body to prevent recurrence. For acute WE symptoms, many regimens call for 250 to 500 mg of IV thiamine supplementation 2 to 3 times daily for 3 to 5 days. High dose IV thiamine (≥ 500 mg daily) has been proposed to be efficacious and free of considerable adverse effects.12 A study conducted at the University of North Carolina described thiamine prescribing practices in a large academic hospital, analyzing data with the objective of assessing outcomes of ordering high-dose IV thiamine (HDIV, ≥ 200 mg IV twice daily) to patients with encephalopathy. 13 The researchers concluded that HDIV, even though rarely prescribed, was associated with decreased inpatient mortality in bivariable models. However, in multivariable analyses this decrease was found to be clinically insignificant. Our patient benefitted from both IV and IM delivery.
Ideally, after the initial IV thiamine dose, oral administration of thiamine 250 to 1,000 mg is continued until a reduction, if not abstinence, from alcohol use is achieved.5 Many patients are discharged on an oral maintenance dose of thiamine 100 mg. Oral thiamine is poorly absorbed and less effective in both prophylaxis and treatment of newly diagnosed WE; therefore, it is typically used only after IM or IV replenishment. It remains unclear why this patient required IM thiamine multiple times per day to maintain his mental status, and why he would present with selfinjurious behaviors after missing doses. The patient’s response can be attributed to late-onset defects in oral thiamine absorption at the carrier protein level of the brush border and basolateral membranes of his jejunum; however, an invasive procedure like a jejunal biopsy to establish the definitive etiology was neither necessary nor practical once treatment response was observed. 14 Other possible explanations include rapid thiamine metabolism, poor gastrointestinal absorption and a late-onset deficit in the thiamine diffusion mechanisms, and active transport systems (thiamine utilization depends on active transport in low availability states and passive transport when readily available). The nature of these mechanisms deserves further study. Less data have been reported on the administration and utility of IM thiamine for chronic WE; hence, our case report is one of the first illustrating the role of this method for sustained repletion.
Conclusions
This case presented a clinical dilemma because the conventional treatment regimen for WE didn’t yield the desired outcome until the mode and duration of thiamine administration were adjusted. It illustrates the utility of a sustained intensive thiamine regimen irrespective of sobriety status, as opposed to the traditional regimen of parenteral (primarily IV) thiamine for 3 to 7 days, followed by oral repletion until the patient achieves sustained abstinence. In this patient’s case, access to nursing care postdischarge facilitated his continued adherence to IM thiamine therapy.
The longitudinal time course of this case suggests a relationship between this route of administration and improvement in symptom burden and indicates that this patient may have a long-term need for IM thiamine to maintain his baseline mental status. Of great benefit in such patients would be the availability of a long-acting IM thiamine therapy. Risk of overdose is unlikely due to the water solubility of B group vitamins.
This case report highlights the importance of setting a high clinical suspicion for WE due to its ever-increasing incidence in these times. We also wish to direct researchers to consider other out-of-the-box treatment options in case of failure of the conventional regime. In documenting this patient report, we invite more medical providers to investigate and explore other therapeutic options for WE treatment with the aim of decreasing both morbidity and mortality secondary to the condition.
1. Lough ME. Wernicke’s encephalopathy: expanding the diagnostic toolbox. Neuropsychol Rev. 2012;22(2):181-194. doi:10.1007/s11065-012-9200-7
2. Arts NJ, Walvoort SJ, Kessels RP. Korsakoff’s syndrome: a critical review. Neuropsychiatr Dis Treat. 2017;13:2875- 2890. Published 2017 Nov 27. doi:10.2147/NDT.S130078
3. Johnson JM, Fox V. Beyond thiamine: treatment for cognitive impairment in Korsakoff’s syndrome. Psychosomatics. 2018;59(4):311-317. doi:10.1016/j.psym.2018.03.011
4. Harper CG, Giles M, Finlay-Jones R. Clinical signs in the Wernicke-Korsakoff complex: a retrospective analysis of 131 cases diagnosed at necropsy. J Neurol Neurosurg Psychiatry. 1986;49(4):341-345. doi:10.1136/ jnnp.49.4.341
5. Xiong GL, Kenedl, CA. Wernicke-Korsakoff syndrome. https://emedicine.medscape.com/article/288379-overview. Updated May 16, 2018, Accessed July 24, 2020.
6. Ahrnsbrak R, Bose J, Hedden SL, Lipari RN, Park-Lee E. Results from the 2016 National Survey on Drug Use and Health. https://www.samhsa.gov/data/sites/default/files /NSDUH-FFR1-2016/NSDUH-FFR1-2016.htm. Accessed July 22, 2020.
7. National Institute on Alcohol Abuse and Alcoholism. Drinking Levels Defined. https://www.niaaa.nih.gov /alcohol-health/overview-alcohol-consumption/moderate -binge-drinking Accessed July 24, 2020.
8. Heye N, Terstegge K, Sirtl C, McMonagle U, Schreiber K, Meyer-Gessner M. Wernicke’s encephalopathy--causes to consider. Intensive Care Med. 1994;20(4):282-286. doi:10.1007/BF01708966
9. Aasheim ET. Wernicke encephalopathy after bariatric surgery: a systematic review. Ann Surg. 2008;248(5):714-720. doi:10.1097/SLA.0b013e3181884308
10. Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff Syndrome and Related Neurologic Disorders Due to Alcoholism and Malnutrition. Philadelphia, PA: FA Davis; 1989.
11. Thomson AD, Cook CC, Touquet R, Henry JA; Royal College of Physicians, London. The Royal College of Physicians report on alcohol: guidelines for managing Wernicke’s encephalopathy in the accident and Emergency Department [published correction appears in Alcohol Alcohol. 2003 May-Jun;38(3):291]. Alcohol Alcohol. 2002;37(6):513-521. doi:10.1093/alcalc/37.6.513
12. Nishimoto A, Usery J, Winton JC, Twilla J. High-dose parenteral thiamine in treatment of Wernicke’s encephalopathy: case series and review of the literature. In Vivo. 2017;31(1):121-124. doi:10.21873/invivo.11034
13. Nakamura ZM, Tatreau JR, Rosenstein DL, Park EM. Clinical characteristics and outcomes associated with highdose intravenous thiamine administration in patients with encephalopathy. Psychosomatics. 2018;59(4):379-387. doi:10.1016/j.psym.2018.01.004
14. Subramanya SB, Subramanian VS, Said HM. Chronic alcohol consumption and intestinal thiamin absorption: effects on physiological and molecular parameters of the uptake process. Am J Physiol Gastrointest Liver Physiol. 2010;299(1):G23-G31. doi:10.1152/ajpgi.00132.2010
Wernicke-Korsakoff syndrome is a cluster of symptoms attributed to a disorder of vitamin B1 (thiamine) deficiency, manifesting as a combined presentation of alcohol-induced Wernicke encephalopathy (WE) and Korsakoff syndrome (KS).1 While there is consensus on the characteristic presentation and symptoms of WE, there is a lack of agreement on the exact definition of KS. The classic triad describing WE consists of ataxia, ophthalmoplegia, and confusion; however, reports now suggest that a majority of patients exhibit only 1 or 2 of the elements of the triad. KS is often seen as a condition of chronic thiamine deficiency manifesting as memory impairment alongside a cognitive and behavioral decline, with no clear consensus on the sequence of appearance of symptoms. The typical relationship is thought to be a progression of WE to KS if untreated.
From a mental health perspective, WE presents with delirium and confusion whereas KS manifests with irreversible dementia and a cognitive deterioration. Though it is commonly taught that KS-induced memory loss is permanent due to neuronal damage (classically identified as damage to the mammillary bodies - though other structures have been implicated as well), more recent research suggest otherwise.2 A review published in 2018, for example, gathered several case reports and case series that suggest significant improvement in memory and cognition attributed to behavioral and pharmacologic interventions, indicating this as an area deserving of further study.3 About 20% of patients diagnosed with WE by autopsy exhibited none of the classical triad symptoms prior to death.4 Hence, these conditions are surmised to be significantly underdiagnosed and misdiagnosed.
Though consensus regarding the appropriate treatment regimen is lacking for WE, a common protocol consists of high-dose parenteral thiamine for 4 to 7 days.5 This is usually followed by daily oral thiamine repletion until the patient either achieves complete abstinence from alcohol (ideal) or decreases consumption. The goal is to allow thiamine stores to replete and maintain at minimum required body levels moving forward. In this case report, we highlight the utilization of a long-term, unconventional intramuscular (IM) thiamine repletion regimen to ensure maintenance of a patient’s mental status, highlighting discrepancies in our understanding of the mechanisms at play in WE and its treatment.
Case Presentation
A 65-year-old male patient with a more than 3-decade history of daily hard liquor intake, multiple psychiatric hospitalizations for WE, and a prior suicide attempt, presented to the emergency department (ED) with increased frequency of falls, poor oral intake, confabulation, and diminished verbal communication. A chart review revealed memory impairment alongside the diagnoses of schizoaffective disorder and WE, and confusion that was responsive to thiamine administration as well as a history of hypertension, hyperlipidemia, osteoarthritis, and urinary retention secondary to benign prostatic hyperplasia (BPH).
On examination the patient was found to be disoriented with a clouded sensorium. While the history of heavy daily alcohol use was clear in the chart and confirmed by other sources, it appeared unlikely that the patient had been using alcohol in the preceding month due to restricted access in his most recent living environment (a shared apartment with daily nursing assistance). He reported no lightheadedness, dizziness, palpitations, numbness, tingling, or any head trauma. He also negated the presence of active mood symptoms, auditory or visual hallucinations or suicidal ideation (SI)
The patient was admitted to the Internal Medicine Service and received a workup for the causes of delirium, including consideration of normal pressure hydrocephalus (NPH) and other neurologic conditions. Laboratory tests including a comprehensive metabolic panel, thyroid stimulating hormone, urinalysis, urine toxicology screen, and vitamin B12 and folate levels were in normal ranges. Although brain imaging revealed enlarged ventricles, NPH was considered unlikely because of the absence of ophthalmologic abnormalities, like gaze nystagmus, and urinary incontinence; conversely, there was some presence of urinary retention attributed to BPH and required an admission a few months prior. Moreover, magnetic resonance images showed that the ventricles were enlarged slightly out of proportion to the sulci, which can be seen with predominantly central volume loss compared with the pattern typically seen in NPH.
In light of concern for WE and the patient's history, treatment with IV thiamine and IV fluids was initiated and the Liaison Psychiatry Service was consulted for cognitive disability and treatment of his mood. Administration of IV thiamine rapidly restored his sensorium, but he became abruptly disorganized as the IV regimen graduated to an oral thiamine dose of 200 mg 3 times daily. Simultaneously, as medical stabilization was achieved, the patient was transferred to the inpatient psychiatry unit to address the nonresolving cognitive impairment and behavioral disorganization. This specifically involved newly emerging, impulsive, self-harming behaviors like throwing himself on the ground and banging his head on the floor. Such behaviors along with paucity of speech and decreased oral intake, ultimately warranted constant observation, which led to a decrease in self-harming activity. All this behavior was noted even though the patient was adherent to oral administration of thiamine. Throughout this time, the patient underwent several transfers back and forth between the Psychiatry and Internal Medicine services due to ongoing concern for the possibility of delirium or WE. However, the Neurology and Internal Medicine services did not feel that WE would explain the patient’s mental and behavioral status, in part due to his ongoing adherence with daily oral thiamine dosing that was not associated with improvement in mental status.
Recollecting the patient’s improvement with the parenteral thiamine regimen (IV and IM), the psychiatry unit tried a thiamine regimen of 200 mg IM and 100 mg oral 2 times daily. After about 2 weeks on this regimen, the patient subsequently achieved remarkable improvement in his cognitive and behavioral status, with resolution of selfharming behaviors. The patient was noted to be calmer, more linear, and more oriented, though he remained incompletely oriented throughout his hospitalization. As improvement in sensorium was established and the patient’s hospital stay prolonged (Figure), his mood symptoms began manifesting as guilt, low energy, decreased appetite, withdrawal, and passive SI. This was followed by a trial of lithium that was discontinued due to elevated creatine levels. As the patient continued to report depression, a multidrug regimen of divalproex, fluoxetine, and quetiapine was administered, which lead to remarkable improvement.
At this time, it was concluded that the stores of thiamine in the patient’s body may have been replenished, the alcohol intake completely ceased and that he needed to be weaned off of thiamine. The next step taken was reduction of the twice daily 200 mg IM thiamine dose to a once daily regimen, and oral thiamine was put on hold. Over the next 48 hours, the patient became less verbal, more withdrawn, incontinent of urine, and delirious. The twice daily IM 200 mg thiamine was restarted, but this time the patient demonstrated very slow improvement. After 2 weeks, the IM thiamine 200 mg was increased to 3 times daily, and the patient showed marked improvement in recall, mood, and effect.
Several attempts were made to reduce the IM thiamine burden on the patient and/ or transition to an exclusively oral regimen. However, he rapidly decompensated within hours of each attempt to taper the IM dose and required immediate reinstation. On the IM thiamine regimen, he eventually appeared to reach a stable cognitive and affective baseline marked by incomplete orientation but pleasant affect, he reported no mood complaints, behavioral stability, and an ability to comply with care needs and have simple conversations. Some speech content remained disorganized particularly if engaged beyond simple exchanges.
The patient was discharged to a skilled nursing facility after a month of 3 times daily IM administration of thiamine. Within the next 24 hours, the patient returned to the ED with the originally reported symptoms of ataxia, agitation, and confusion. On inquiry, it was revealed that the ordered vials of IM thiamine for injection had not arrived with him at the nursing facility and he had missed 2 doses. The blood laboratory results, scans, and all other parameters were otherwise found to be normal and the patient was adherent to his prescribed antipsychotics and antidepressants. As anticipated, restoration of the IM thiamine regimen revived his baseline within hours. While confusion and delirium resolved completely with treatment, the memory impairments persisted. This patient has been administered a 3 times daily IM dose of 200 mg thiamine for more than 2 years with a stable cognitive clinical picture.
Discussion
According to data from the 2016 National Survey on Drug Use and Health, 16 million individuals in the US aged ≥ 12 years reported heavy alcohol use, which is defined as binge drinking on ≥ 5 days in the past month.6,7 Thiamine deficiency is an alcoholrelated disorder that is frequently encountered in hospital settings. This deficiency can also occur in the context of malabsorption, malnutrition, a prolonged course of vomiting, and bariatric surgery.8,9
The deficiency in thiamine, which is sometimes known as WE, manifests rarely with all 3 of the classic triad of gait disturbances, abnormal eye movements, and mental status changes, with only 16.5% of patients displaying all of the triad.4 Moreover, there may be additional symptoms not listed in this triad, such as memory impairment, bilateral sixth nerve palsy, ptosis, hypotension, and hypothermia.10.11 This inconsistent presentation makes the diagnosis challenging and therefore requires a higher threshold for suspicion. If undiagnosed and/or untreated, WE can lead to chronic thiamine deficiency causing permanent brain damage in the guise of KS. This further increases the importance of timely diagnosis and treatment.
Our case highlights the utilization of an unconventional thiamine regimen that appeared to be temporally associated with mental status improvement. The patient’s clouded sensorium and confusion could not be attributed to metabolic, encephalopathic, or infectious pathologies due to the absence of supportive laboratory evidence. He responded to IV and IM doses of thiamine, but repeated attempts to taper the IM doses with the objective of transitioning to oral thiamine supplementation were followed by immediate decompensations in mental status. This was atypical of WE as the patient seemed adequately replete with thiamine, and missing a few doses should not be enough to deplete his stores. Thus, reflecting a unique case of thiamine-dependent chronically set WE when even a single missed dose of thiamine adversely affected the patient’s cognitive baseline. Interesting to note is this patient’s memory issue, as evident by clinical examination and dating back at least 5 years as per chart review. This premature amnestic component of his presentation indicates a likely parallel running KS component of his presentation. Conversely, the patient’s long history of alcohol use disorder, prior episodes of WE, and ideal response achieved only on parenteral thiamine repletion further supported the diagnosis of WE and our impression of the scenario.
Even though this patient had prior episodes of WE, there remained diagnostic uncertainty regarding his altered mental status for some time before the nonoral thiamine repletion treatment was implemented. Particularly in this admission, the patient’s mental status frequently waxed and waned and there was the additional confusion of whether a potential psychiatric etiology contributed to some of the elements of his presentation, such as his impulsive self-harm behaviors. This behavior led to recurrent transfers among the Psychiatry Service, Internal Medicine Service, and the ED.
The patient’s presentation did not reflect the classical triad of WE, and while this is consistent with the majority of clinical manifestations, various services were reluctant to attribute his symptoms to WE. Once the threshold of suspicion of thiamine deficiency was lowered and the deficit treated more aggressively, the patient seemed to improve tremendously. Presence of memory problems and confabulation, both of which this patient exhibited, are suggestive of KS and are not expected to recover with treatment, yet for this patient there did seem to be some improvement—though not complete resolution. This is consistent with newer evidence suggesting that some recovery from the deficits seen in KS is possible.3
Once diagnosed, the treatment objective is the replenishment of thiamine stores and optimization of the metabolic scenario of the body to prevent recurrence. For acute WE symptoms, many regimens call for 250 to 500 mg of IV thiamine supplementation 2 to 3 times daily for 3 to 5 days. High dose IV thiamine (≥ 500 mg daily) has been proposed to be efficacious and free of considerable adverse effects.12 A study conducted at the University of North Carolina described thiamine prescribing practices in a large academic hospital, analyzing data with the objective of assessing outcomes of ordering high-dose IV thiamine (HDIV, ≥ 200 mg IV twice daily) to patients with encephalopathy. 13 The researchers concluded that HDIV, even though rarely prescribed, was associated with decreased inpatient mortality in bivariable models. However, in multivariable analyses this decrease was found to be clinically insignificant. Our patient benefitted from both IV and IM delivery.
Ideally, after the initial IV thiamine dose, oral administration of thiamine 250 to 1,000 mg is continued until a reduction, if not abstinence, from alcohol use is achieved.5 Many patients are discharged on an oral maintenance dose of thiamine 100 mg. Oral thiamine is poorly absorbed and less effective in both prophylaxis and treatment of newly diagnosed WE; therefore, it is typically used only after IM or IV replenishment. It remains unclear why this patient required IM thiamine multiple times per day to maintain his mental status, and why he would present with selfinjurious behaviors after missing doses. The patient’s response can be attributed to late-onset defects in oral thiamine absorption at the carrier protein level of the brush border and basolateral membranes of his jejunum; however, an invasive procedure like a jejunal biopsy to establish the definitive etiology was neither necessary nor practical once treatment response was observed. 14 Other possible explanations include rapid thiamine metabolism, poor gastrointestinal absorption and a late-onset deficit in the thiamine diffusion mechanisms, and active transport systems (thiamine utilization depends on active transport in low availability states and passive transport when readily available). The nature of these mechanisms deserves further study. Less data have been reported on the administration and utility of IM thiamine for chronic WE; hence, our case report is one of the first illustrating the role of this method for sustained repletion.
Conclusions
This case presented a clinical dilemma because the conventional treatment regimen for WE didn’t yield the desired outcome until the mode and duration of thiamine administration were adjusted. It illustrates the utility of a sustained intensive thiamine regimen irrespective of sobriety status, as opposed to the traditional regimen of parenteral (primarily IV) thiamine for 3 to 7 days, followed by oral repletion until the patient achieves sustained abstinence. In this patient’s case, access to nursing care postdischarge facilitated his continued adherence to IM thiamine therapy.
The longitudinal time course of this case suggests a relationship between this route of administration and improvement in symptom burden and indicates that this patient may have a long-term need for IM thiamine to maintain his baseline mental status. Of great benefit in such patients would be the availability of a long-acting IM thiamine therapy. Risk of overdose is unlikely due to the water solubility of B group vitamins.
This case report highlights the importance of setting a high clinical suspicion for WE due to its ever-increasing incidence in these times. We also wish to direct researchers to consider other out-of-the-box treatment options in case of failure of the conventional regime. In documenting this patient report, we invite more medical providers to investigate and explore other therapeutic options for WE treatment with the aim of decreasing both morbidity and mortality secondary to the condition.
Wernicke-Korsakoff syndrome is a cluster of symptoms attributed to a disorder of vitamin B1 (thiamine) deficiency, manifesting as a combined presentation of alcohol-induced Wernicke encephalopathy (WE) and Korsakoff syndrome (KS).1 While there is consensus on the characteristic presentation and symptoms of WE, there is a lack of agreement on the exact definition of KS. The classic triad describing WE consists of ataxia, ophthalmoplegia, and confusion; however, reports now suggest that a majority of patients exhibit only 1 or 2 of the elements of the triad. KS is often seen as a condition of chronic thiamine deficiency manifesting as memory impairment alongside a cognitive and behavioral decline, with no clear consensus on the sequence of appearance of symptoms. The typical relationship is thought to be a progression of WE to KS if untreated.
From a mental health perspective, WE presents with delirium and confusion whereas KS manifests with irreversible dementia and a cognitive deterioration. Though it is commonly taught that KS-induced memory loss is permanent due to neuronal damage (classically identified as damage to the mammillary bodies - though other structures have been implicated as well), more recent research suggest otherwise.2 A review published in 2018, for example, gathered several case reports and case series that suggest significant improvement in memory and cognition attributed to behavioral and pharmacologic interventions, indicating this as an area deserving of further study.3 About 20% of patients diagnosed with WE by autopsy exhibited none of the classical triad symptoms prior to death.4 Hence, these conditions are surmised to be significantly underdiagnosed and misdiagnosed.
Though consensus regarding the appropriate treatment regimen is lacking for WE, a common protocol consists of high-dose parenteral thiamine for 4 to 7 days.5 This is usually followed by daily oral thiamine repletion until the patient either achieves complete abstinence from alcohol (ideal) or decreases consumption. The goal is to allow thiamine stores to replete and maintain at minimum required body levels moving forward. In this case report, we highlight the utilization of a long-term, unconventional intramuscular (IM) thiamine repletion regimen to ensure maintenance of a patient’s mental status, highlighting discrepancies in our understanding of the mechanisms at play in WE and its treatment.
Case Presentation
A 65-year-old male patient with a more than 3-decade history of daily hard liquor intake, multiple psychiatric hospitalizations for WE, and a prior suicide attempt, presented to the emergency department (ED) with increased frequency of falls, poor oral intake, confabulation, and diminished verbal communication. A chart review revealed memory impairment alongside the diagnoses of schizoaffective disorder and WE, and confusion that was responsive to thiamine administration as well as a history of hypertension, hyperlipidemia, osteoarthritis, and urinary retention secondary to benign prostatic hyperplasia (BPH).
On examination the patient was found to be disoriented with a clouded sensorium. While the history of heavy daily alcohol use was clear in the chart and confirmed by other sources, it appeared unlikely that the patient had been using alcohol in the preceding month due to restricted access in his most recent living environment (a shared apartment with daily nursing assistance). He reported no lightheadedness, dizziness, palpitations, numbness, tingling, or any head trauma. He also negated the presence of active mood symptoms, auditory or visual hallucinations or suicidal ideation (SI)
The patient was admitted to the Internal Medicine Service and received a workup for the causes of delirium, including consideration of normal pressure hydrocephalus (NPH) and other neurologic conditions. Laboratory tests including a comprehensive metabolic panel, thyroid stimulating hormone, urinalysis, urine toxicology screen, and vitamin B12 and folate levels were in normal ranges. Although brain imaging revealed enlarged ventricles, NPH was considered unlikely because of the absence of ophthalmologic abnormalities, like gaze nystagmus, and urinary incontinence; conversely, there was some presence of urinary retention attributed to BPH and required an admission a few months prior. Moreover, magnetic resonance images showed that the ventricles were enlarged slightly out of proportion to the sulci, which can be seen with predominantly central volume loss compared with the pattern typically seen in NPH.
In light of concern for WE and the patient's history, treatment with IV thiamine and IV fluids was initiated and the Liaison Psychiatry Service was consulted for cognitive disability and treatment of his mood. Administration of IV thiamine rapidly restored his sensorium, but he became abruptly disorganized as the IV regimen graduated to an oral thiamine dose of 200 mg 3 times daily. Simultaneously, as medical stabilization was achieved, the patient was transferred to the inpatient psychiatry unit to address the nonresolving cognitive impairment and behavioral disorganization. This specifically involved newly emerging, impulsive, self-harming behaviors like throwing himself on the ground and banging his head on the floor. Such behaviors along with paucity of speech and decreased oral intake, ultimately warranted constant observation, which led to a decrease in self-harming activity. All this behavior was noted even though the patient was adherent to oral administration of thiamine. Throughout this time, the patient underwent several transfers back and forth between the Psychiatry and Internal Medicine services due to ongoing concern for the possibility of delirium or WE. However, the Neurology and Internal Medicine services did not feel that WE would explain the patient’s mental and behavioral status, in part due to his ongoing adherence with daily oral thiamine dosing that was not associated with improvement in mental status.
Recollecting the patient’s improvement with the parenteral thiamine regimen (IV and IM), the psychiatry unit tried a thiamine regimen of 200 mg IM and 100 mg oral 2 times daily. After about 2 weeks on this regimen, the patient subsequently achieved remarkable improvement in his cognitive and behavioral status, with resolution of selfharming behaviors. The patient was noted to be calmer, more linear, and more oriented, though he remained incompletely oriented throughout his hospitalization. As improvement in sensorium was established and the patient’s hospital stay prolonged (Figure), his mood symptoms began manifesting as guilt, low energy, decreased appetite, withdrawal, and passive SI. This was followed by a trial of lithium that was discontinued due to elevated creatine levels. As the patient continued to report depression, a multidrug regimen of divalproex, fluoxetine, and quetiapine was administered, which lead to remarkable improvement.
At this time, it was concluded that the stores of thiamine in the patient’s body may have been replenished, the alcohol intake completely ceased and that he needed to be weaned off of thiamine. The next step taken was reduction of the twice daily 200 mg IM thiamine dose to a once daily regimen, and oral thiamine was put on hold. Over the next 48 hours, the patient became less verbal, more withdrawn, incontinent of urine, and delirious. The twice daily IM 200 mg thiamine was restarted, but this time the patient demonstrated very slow improvement. After 2 weeks, the IM thiamine 200 mg was increased to 3 times daily, and the patient showed marked improvement in recall, mood, and effect.
Several attempts were made to reduce the IM thiamine burden on the patient and/ or transition to an exclusively oral regimen. However, he rapidly decompensated within hours of each attempt to taper the IM dose and required immediate reinstation. On the IM thiamine regimen, he eventually appeared to reach a stable cognitive and affective baseline marked by incomplete orientation but pleasant affect, he reported no mood complaints, behavioral stability, and an ability to comply with care needs and have simple conversations. Some speech content remained disorganized particularly if engaged beyond simple exchanges.
The patient was discharged to a skilled nursing facility after a month of 3 times daily IM administration of thiamine. Within the next 24 hours, the patient returned to the ED with the originally reported symptoms of ataxia, agitation, and confusion. On inquiry, it was revealed that the ordered vials of IM thiamine for injection had not arrived with him at the nursing facility and he had missed 2 doses. The blood laboratory results, scans, and all other parameters were otherwise found to be normal and the patient was adherent to his prescribed antipsychotics and antidepressants. As anticipated, restoration of the IM thiamine regimen revived his baseline within hours. While confusion and delirium resolved completely with treatment, the memory impairments persisted. This patient has been administered a 3 times daily IM dose of 200 mg thiamine for more than 2 years with a stable cognitive clinical picture.
Discussion
According to data from the 2016 National Survey on Drug Use and Health, 16 million individuals in the US aged ≥ 12 years reported heavy alcohol use, which is defined as binge drinking on ≥ 5 days in the past month.6,7 Thiamine deficiency is an alcoholrelated disorder that is frequently encountered in hospital settings. This deficiency can also occur in the context of malabsorption, malnutrition, a prolonged course of vomiting, and bariatric surgery.8,9
The deficiency in thiamine, which is sometimes known as WE, manifests rarely with all 3 of the classic triad of gait disturbances, abnormal eye movements, and mental status changes, with only 16.5% of patients displaying all of the triad.4 Moreover, there may be additional symptoms not listed in this triad, such as memory impairment, bilateral sixth nerve palsy, ptosis, hypotension, and hypothermia.10.11 This inconsistent presentation makes the diagnosis challenging and therefore requires a higher threshold for suspicion. If undiagnosed and/or untreated, WE can lead to chronic thiamine deficiency causing permanent brain damage in the guise of KS. This further increases the importance of timely diagnosis and treatment.
Our case highlights the utilization of an unconventional thiamine regimen that appeared to be temporally associated with mental status improvement. The patient’s clouded sensorium and confusion could not be attributed to metabolic, encephalopathic, or infectious pathologies due to the absence of supportive laboratory evidence. He responded to IV and IM doses of thiamine, but repeated attempts to taper the IM doses with the objective of transitioning to oral thiamine supplementation were followed by immediate decompensations in mental status. This was atypical of WE as the patient seemed adequately replete with thiamine, and missing a few doses should not be enough to deplete his stores. Thus, reflecting a unique case of thiamine-dependent chronically set WE when even a single missed dose of thiamine adversely affected the patient’s cognitive baseline. Interesting to note is this patient’s memory issue, as evident by clinical examination and dating back at least 5 years as per chart review. This premature amnestic component of his presentation indicates a likely parallel running KS component of his presentation. Conversely, the patient’s long history of alcohol use disorder, prior episodes of WE, and ideal response achieved only on parenteral thiamine repletion further supported the diagnosis of WE and our impression of the scenario.
Even though this patient had prior episodes of WE, there remained diagnostic uncertainty regarding his altered mental status for some time before the nonoral thiamine repletion treatment was implemented. Particularly in this admission, the patient’s mental status frequently waxed and waned and there was the additional confusion of whether a potential psychiatric etiology contributed to some of the elements of his presentation, such as his impulsive self-harm behaviors. This behavior led to recurrent transfers among the Psychiatry Service, Internal Medicine Service, and the ED.
The patient’s presentation did not reflect the classical triad of WE, and while this is consistent with the majority of clinical manifestations, various services were reluctant to attribute his symptoms to WE. Once the threshold of suspicion of thiamine deficiency was lowered and the deficit treated more aggressively, the patient seemed to improve tremendously. Presence of memory problems and confabulation, both of which this patient exhibited, are suggestive of KS and are not expected to recover with treatment, yet for this patient there did seem to be some improvement—though not complete resolution. This is consistent with newer evidence suggesting that some recovery from the deficits seen in KS is possible.3
Once diagnosed, the treatment objective is the replenishment of thiamine stores and optimization of the metabolic scenario of the body to prevent recurrence. For acute WE symptoms, many regimens call for 250 to 500 mg of IV thiamine supplementation 2 to 3 times daily for 3 to 5 days. High dose IV thiamine (≥ 500 mg daily) has been proposed to be efficacious and free of considerable adverse effects.12 A study conducted at the University of North Carolina described thiamine prescribing practices in a large academic hospital, analyzing data with the objective of assessing outcomes of ordering high-dose IV thiamine (HDIV, ≥ 200 mg IV twice daily) to patients with encephalopathy. 13 The researchers concluded that HDIV, even though rarely prescribed, was associated with decreased inpatient mortality in bivariable models. However, in multivariable analyses this decrease was found to be clinically insignificant. Our patient benefitted from both IV and IM delivery.
Ideally, after the initial IV thiamine dose, oral administration of thiamine 250 to 1,000 mg is continued until a reduction, if not abstinence, from alcohol use is achieved.5 Many patients are discharged on an oral maintenance dose of thiamine 100 mg. Oral thiamine is poorly absorbed and less effective in both prophylaxis and treatment of newly diagnosed WE; therefore, it is typically used only after IM or IV replenishment. It remains unclear why this patient required IM thiamine multiple times per day to maintain his mental status, and why he would present with selfinjurious behaviors after missing doses. The patient’s response can be attributed to late-onset defects in oral thiamine absorption at the carrier protein level of the brush border and basolateral membranes of his jejunum; however, an invasive procedure like a jejunal biopsy to establish the definitive etiology was neither necessary nor practical once treatment response was observed. 14 Other possible explanations include rapid thiamine metabolism, poor gastrointestinal absorption and a late-onset deficit in the thiamine diffusion mechanisms, and active transport systems (thiamine utilization depends on active transport in low availability states and passive transport when readily available). The nature of these mechanisms deserves further study. Less data have been reported on the administration and utility of IM thiamine for chronic WE; hence, our case report is one of the first illustrating the role of this method for sustained repletion.
Conclusions
This case presented a clinical dilemma because the conventional treatment regimen for WE didn’t yield the desired outcome until the mode and duration of thiamine administration were adjusted. It illustrates the utility of a sustained intensive thiamine regimen irrespective of sobriety status, as opposed to the traditional regimen of parenteral (primarily IV) thiamine for 3 to 7 days, followed by oral repletion until the patient achieves sustained abstinence. In this patient’s case, access to nursing care postdischarge facilitated his continued adherence to IM thiamine therapy.
The longitudinal time course of this case suggests a relationship between this route of administration and improvement in symptom burden and indicates that this patient may have a long-term need for IM thiamine to maintain his baseline mental status. Of great benefit in such patients would be the availability of a long-acting IM thiamine therapy. Risk of overdose is unlikely due to the water solubility of B group vitamins.
This case report highlights the importance of setting a high clinical suspicion for WE due to its ever-increasing incidence in these times. We also wish to direct researchers to consider other out-of-the-box treatment options in case of failure of the conventional regime. In documenting this patient report, we invite more medical providers to investigate and explore other therapeutic options for WE treatment with the aim of decreasing both morbidity and mortality secondary to the condition.
1. Lough ME. Wernicke’s encephalopathy: expanding the diagnostic toolbox. Neuropsychol Rev. 2012;22(2):181-194. doi:10.1007/s11065-012-9200-7
2. Arts NJ, Walvoort SJ, Kessels RP. Korsakoff’s syndrome: a critical review. Neuropsychiatr Dis Treat. 2017;13:2875- 2890. Published 2017 Nov 27. doi:10.2147/NDT.S130078
3. Johnson JM, Fox V. Beyond thiamine: treatment for cognitive impairment in Korsakoff’s syndrome. Psychosomatics. 2018;59(4):311-317. doi:10.1016/j.psym.2018.03.011
4. Harper CG, Giles M, Finlay-Jones R. Clinical signs in the Wernicke-Korsakoff complex: a retrospective analysis of 131 cases diagnosed at necropsy. J Neurol Neurosurg Psychiatry. 1986;49(4):341-345. doi:10.1136/ jnnp.49.4.341
5. Xiong GL, Kenedl, CA. Wernicke-Korsakoff syndrome. https://emedicine.medscape.com/article/288379-overview. Updated May 16, 2018, Accessed July 24, 2020.
6. Ahrnsbrak R, Bose J, Hedden SL, Lipari RN, Park-Lee E. Results from the 2016 National Survey on Drug Use and Health. https://www.samhsa.gov/data/sites/default/files /NSDUH-FFR1-2016/NSDUH-FFR1-2016.htm. Accessed July 22, 2020.
7. National Institute on Alcohol Abuse and Alcoholism. Drinking Levels Defined. https://www.niaaa.nih.gov /alcohol-health/overview-alcohol-consumption/moderate -binge-drinking Accessed July 24, 2020.
8. Heye N, Terstegge K, Sirtl C, McMonagle U, Schreiber K, Meyer-Gessner M. Wernicke’s encephalopathy--causes to consider. Intensive Care Med. 1994;20(4):282-286. doi:10.1007/BF01708966
9. Aasheim ET. Wernicke encephalopathy after bariatric surgery: a systematic review. Ann Surg. 2008;248(5):714-720. doi:10.1097/SLA.0b013e3181884308
10. Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff Syndrome and Related Neurologic Disorders Due to Alcoholism and Malnutrition. Philadelphia, PA: FA Davis; 1989.
11. Thomson AD, Cook CC, Touquet R, Henry JA; Royal College of Physicians, London. The Royal College of Physicians report on alcohol: guidelines for managing Wernicke’s encephalopathy in the accident and Emergency Department [published correction appears in Alcohol Alcohol. 2003 May-Jun;38(3):291]. Alcohol Alcohol. 2002;37(6):513-521. doi:10.1093/alcalc/37.6.513
12. Nishimoto A, Usery J, Winton JC, Twilla J. High-dose parenteral thiamine in treatment of Wernicke’s encephalopathy: case series and review of the literature. In Vivo. 2017;31(1):121-124. doi:10.21873/invivo.11034
13. Nakamura ZM, Tatreau JR, Rosenstein DL, Park EM. Clinical characteristics and outcomes associated with highdose intravenous thiamine administration in patients with encephalopathy. Psychosomatics. 2018;59(4):379-387. doi:10.1016/j.psym.2018.01.004
14. Subramanya SB, Subramanian VS, Said HM. Chronic alcohol consumption and intestinal thiamin absorption: effects on physiological and molecular parameters of the uptake process. Am J Physiol Gastrointest Liver Physiol. 2010;299(1):G23-G31. doi:10.1152/ajpgi.00132.2010
1. Lough ME. Wernicke’s encephalopathy: expanding the diagnostic toolbox. Neuropsychol Rev. 2012;22(2):181-194. doi:10.1007/s11065-012-9200-7
2. Arts NJ, Walvoort SJ, Kessels RP. Korsakoff’s syndrome: a critical review. Neuropsychiatr Dis Treat. 2017;13:2875- 2890. Published 2017 Nov 27. doi:10.2147/NDT.S130078
3. Johnson JM, Fox V. Beyond thiamine: treatment for cognitive impairment in Korsakoff’s syndrome. Psychosomatics. 2018;59(4):311-317. doi:10.1016/j.psym.2018.03.011
4. Harper CG, Giles M, Finlay-Jones R. Clinical signs in the Wernicke-Korsakoff complex: a retrospective analysis of 131 cases diagnosed at necropsy. J Neurol Neurosurg Psychiatry. 1986;49(4):341-345. doi:10.1136/ jnnp.49.4.341
5. Xiong GL, Kenedl, CA. Wernicke-Korsakoff syndrome. https://emedicine.medscape.com/article/288379-overview. Updated May 16, 2018, Accessed July 24, 2020.
6. Ahrnsbrak R, Bose J, Hedden SL, Lipari RN, Park-Lee E. Results from the 2016 National Survey on Drug Use and Health. https://www.samhsa.gov/data/sites/default/files /NSDUH-FFR1-2016/NSDUH-FFR1-2016.htm. Accessed July 22, 2020.
7. National Institute on Alcohol Abuse and Alcoholism. Drinking Levels Defined. https://www.niaaa.nih.gov /alcohol-health/overview-alcohol-consumption/moderate -binge-drinking Accessed July 24, 2020.
8. Heye N, Terstegge K, Sirtl C, McMonagle U, Schreiber K, Meyer-Gessner M. Wernicke’s encephalopathy--causes to consider. Intensive Care Med. 1994;20(4):282-286. doi:10.1007/BF01708966
9. Aasheim ET. Wernicke encephalopathy after bariatric surgery: a systematic review. Ann Surg. 2008;248(5):714-720. doi:10.1097/SLA.0b013e3181884308
10. Victor M, Adams RD, Collins GH. The Wernicke-Korsakoff Syndrome and Related Neurologic Disorders Due to Alcoholism and Malnutrition. Philadelphia, PA: FA Davis; 1989.
11. Thomson AD, Cook CC, Touquet R, Henry JA; Royal College of Physicians, London. The Royal College of Physicians report on alcohol: guidelines for managing Wernicke’s encephalopathy in the accident and Emergency Department [published correction appears in Alcohol Alcohol. 2003 May-Jun;38(3):291]. Alcohol Alcohol. 2002;37(6):513-521. doi:10.1093/alcalc/37.6.513
12. Nishimoto A, Usery J, Winton JC, Twilla J. High-dose parenteral thiamine in treatment of Wernicke’s encephalopathy: case series and review of the literature. In Vivo. 2017;31(1):121-124. doi:10.21873/invivo.11034
13. Nakamura ZM, Tatreau JR, Rosenstein DL, Park EM. Clinical characteristics and outcomes associated with highdose intravenous thiamine administration in patients with encephalopathy. Psychosomatics. 2018;59(4):379-387. doi:10.1016/j.psym.2018.01.004
14. Subramanya SB, Subramanian VS, Said HM. Chronic alcohol consumption and intestinal thiamin absorption: effects on physiological and molecular parameters of the uptake process. Am J Physiol Gastrointest Liver Physiol. 2010;299(1):G23-G31. doi:10.1152/ajpgi.00132.2010
Smallpox Vaccination-Associated Myopericarditis
A renewed effort to vaccinate service members fighting the global war on terrorism has brought new diagnostic challenges. Vaccinations not generally given to the public are routinely given to service members when they deploy to various parts of the world. Examples include anthrax, yellow fever, Japanese encephalitis, rabies, polio, and smallpox. Every vaccination has potential for adverse effects (AEs), which can range from mild to severe life-threatening complications. These AEs often go unrecognized and untreated because physicians are not routinely screening for vaccination administration.
Background
Smallpox (Variola major) was successfully eradicated in 1977 due to worldwide vaccination efforts.1 However, the threat of bioterrorism has renewed mandatory smallpox vaccinations for high-risk individuals, such as active-duty military personnel.1,2 A notable increase in myopericarditis has been reported with the new generation of smallpox vaccination, ACAM2000.3 We present a case of a 27-year-old healthy male who presented with chest pain and diffuse ST segment elevations consistent with myopericarditis after vaccination with ACAM2000.
Case Presentation
A healthy 27-year-old soldier presented to the emergency department with sudden, new onset, sharp-stabbing, substernal chest pain, which was made worse with lying flat and better with leaning forward. Vital signs were unremarkable. He recently enlisted in the US Army and received the smallpox vaccination about 11 days before as part of a routine predeployment checklist. The patient reported he did not have any viral symptoms, such as fever, chills, nausea, vomiting, diarrhea, shortness of breath, sore throat, rhinorrhea, or sputum production. He also reported having no prior illness for the past 3 months, sick contacts at home or work, or recent travel outside the US. He reported no tobacco use, alcohol use, or illicit drug use. The patient’s family history was negative for significant cardiac disease.
A physical examination was unremarkable. The initial laboratory report showed no leukocytosis, anemia, thrombocytopenia, electrolytes derangement, abnormal kidney function, or abnormal liver function tests. Initial troponin was 0.25 ng/mL, erythrocyte sedimentation rate (ESR) was 40 mmol/h and C-reactive protein (CRP) was 120.2 mg/L suggestive of acute inflammation. A urine drug screen was negative. D-dimer was < 0.27. An electrocardiogram (ECG) showed diffuse ST segment elevation (Figure 1). An echocardiogram showed normal left ventricle size, and function with ejection fraction 55 to 60%, normal diastolic dysfunction, and trivial pericardial effusion. Magnetic resonance imaging (MRI) showed increased T2 signal intensity of the myocardium suggestive of myopericarditis (Figure 2). A computed tomography (CT) angiogram of the coronary arteries showed no significant stenosis.
The patient was treated with ibuprofen for 2 weeks and colchicine for 3 months, and his symptoms resolved. He followed up with an appointment in the cardiology clinic 1 month later, and his ESR, CRP, and troponin results were negative. A limited echocardiogram showed ejection fraction 60 to 65%, no regional wall motion abnormalities, normal diastolic function, and resolution of the pericardial effusion.
Discussion
Smallpox was a major worldwide cause of mortality; about 30% of those infected died because of smallpox.2,4,5 Due to a worldwide vaccination effort, the World Health Organization declared smallpox was eradicated in 1977.2,4,5 However, despite successful eradication, smallpox is considered a possible bioterrorism target, which prompted a resurgence of mandatory smallpox vaccinations for active-duty personnel.2,5
Dryvax, a freeze-dried calf lymph smallpox vaccine was used extensively from the 1940s to the 1980s but was replaced in 2008 by ACAM2000, a smallpox vaccine cultured in kidney epithelial cells from African green monkeys.3,5 Myopericarditis was rarely associated with the Dryvax, with only 5 cases reported from 1955 to 1986 after millions of doses of vaccines were administered; however, in 230,734 administered ACAM2000 doses, 18 cases of myopericarditis (incidence, 7.8 per 100,000) were reported during a surveillance study in 2002 and 2003.3,5
Myopericarditis presents with a wide variety of symptoms, such as chest pain, palpitations, chills, shortness of breath, and fever.6,7 Mainstay diagnostic criteria include ECG findings consistent with myopericarditis (such as diffuse ST segment elevations) and elevated cardiac biomarkers (elevated troponins).5-7 An echocardiogram can be helpful in diagnosis, as most cases will not have regional wall motion abnormalities (to distinguish against coronary artery disease).5-7 MRI with diffuse enhancement of the myocardium can be helpful in diagnosis.5,6 The gold standard for diagnosis is an endomyocardial biopsy, which carries a significant risk of complications and is not routinely performed to diagnose myopericarditis.5,6 US military smallpox vaccination data showed that the onset of vaccine-associated myopericarditis averaged (SD) 10.4 (3.6) days after vaccination.5
Vaccine-associated myopericarditis treatment is focused on decreasing inflammation.5,6 Nonsteroidal anti-inflammatory drugs are advised for about 2 weeks with cessation of intensive cardiac activities for between 4 and 6 weeks due to risks of congestive heart failure and fatal cardiac arrhythmias.5,6
Conclusions
Since the September 11 attacks, the US needs to be continually prepared for potential terrorism on American soil and abroad. The threat of bioterrorism has renewed efforts to vaccinate or revaccinate American service members deployed to high-risk regions. These vaccinations put them at risk for vaccination-induced complications that can range from mild fever to life-threatening complications.
1. Bruner DI, Butler BS. Smallpox vaccination-associated myopericarditis is more common with the newest smallpox vaccine. J Emerg Med. 2014;46(3):e85-e87. doi:10.1016/j.jemermed.2013.06.001
2. Halsell JS, Riddle JR, Atwood JE, et al. Myopericarditis following smallpox vaccination among vaccinia-naive US military personnel. JAMA. 2003;289(24):3283-3289. doi:10.1001/jama.289.24.3283
3. Nalca A, Zumbrun EE. ACAM2000: the new smallpox vaccine for United States Strategic National Stockpile. Drug Des Devel Ther. 2010;4:71-79. doi:10.2147/dddt.s3687
4. Wollenberg A, Engler R. Smallpox, vaccination and adverse reactions to smallpox vaccine. Curr Opin Allergy Clin Immunol. 2004;4(4):271-275. doi:10.1097/01.all.0000136758.66442.28
5. Cassimatis DC, Atwood JE, Engler RM, Linz PE, Grabenstein JD, Vernalis MN. Smallpox vaccination and myopericarditis: a clinical review. J Am Coll Cardiol. 2004;43(9):1503-1510. doi:10.1016/j.jacc.2003.11.053
6. Sharma U, Tak T. A report of 2 cases of myopericarditis after Vaccinia virus (smallpox) immunization. WMJ. 2011;110(6):291-294.
7. Sarkisian SA, Hand G, Rivera VM, Smith M, Miller JA. A case series of smallpox vaccination-associated myopericarditis: effects on safety and readiness of the active duty soldier. Mil Med. 2019;184(1-2):e280-e283. doi:10.1093/milmed/usy159
A renewed effort to vaccinate service members fighting the global war on terrorism has brought new diagnostic challenges. Vaccinations not generally given to the public are routinely given to service members when they deploy to various parts of the world. Examples include anthrax, yellow fever, Japanese encephalitis, rabies, polio, and smallpox. Every vaccination has potential for adverse effects (AEs), which can range from mild to severe life-threatening complications. These AEs often go unrecognized and untreated because physicians are not routinely screening for vaccination administration.
Background
Smallpox (Variola major) was successfully eradicated in 1977 due to worldwide vaccination efforts.1 However, the threat of bioterrorism has renewed mandatory smallpox vaccinations for high-risk individuals, such as active-duty military personnel.1,2 A notable increase in myopericarditis has been reported with the new generation of smallpox vaccination, ACAM2000.3 We present a case of a 27-year-old healthy male who presented with chest pain and diffuse ST segment elevations consistent with myopericarditis after vaccination with ACAM2000.
Case Presentation
A healthy 27-year-old soldier presented to the emergency department with sudden, new onset, sharp-stabbing, substernal chest pain, which was made worse with lying flat and better with leaning forward. Vital signs were unremarkable. He recently enlisted in the US Army and received the smallpox vaccination about 11 days before as part of a routine predeployment checklist. The patient reported he did not have any viral symptoms, such as fever, chills, nausea, vomiting, diarrhea, shortness of breath, sore throat, rhinorrhea, or sputum production. He also reported having no prior illness for the past 3 months, sick contacts at home or work, or recent travel outside the US. He reported no tobacco use, alcohol use, or illicit drug use. The patient’s family history was negative for significant cardiac disease.
A physical examination was unremarkable. The initial laboratory report showed no leukocytosis, anemia, thrombocytopenia, electrolytes derangement, abnormal kidney function, or abnormal liver function tests. Initial troponin was 0.25 ng/mL, erythrocyte sedimentation rate (ESR) was 40 mmol/h and C-reactive protein (CRP) was 120.2 mg/L suggestive of acute inflammation. A urine drug screen was negative. D-dimer was < 0.27. An electrocardiogram (ECG) showed diffuse ST segment elevation (Figure 1). An echocardiogram showed normal left ventricle size, and function with ejection fraction 55 to 60%, normal diastolic dysfunction, and trivial pericardial effusion. Magnetic resonance imaging (MRI) showed increased T2 signal intensity of the myocardium suggestive of myopericarditis (Figure 2). A computed tomography (CT) angiogram of the coronary arteries showed no significant stenosis.
The patient was treated with ibuprofen for 2 weeks and colchicine for 3 months, and his symptoms resolved. He followed up with an appointment in the cardiology clinic 1 month later, and his ESR, CRP, and troponin results were negative. A limited echocardiogram showed ejection fraction 60 to 65%, no regional wall motion abnormalities, normal diastolic function, and resolution of the pericardial effusion.
Discussion
Smallpox was a major worldwide cause of mortality; about 30% of those infected died because of smallpox.2,4,5 Due to a worldwide vaccination effort, the World Health Organization declared smallpox was eradicated in 1977.2,4,5 However, despite successful eradication, smallpox is considered a possible bioterrorism target, which prompted a resurgence of mandatory smallpox vaccinations for active-duty personnel.2,5
Dryvax, a freeze-dried calf lymph smallpox vaccine was used extensively from the 1940s to the 1980s but was replaced in 2008 by ACAM2000, a smallpox vaccine cultured in kidney epithelial cells from African green monkeys.3,5 Myopericarditis was rarely associated with the Dryvax, with only 5 cases reported from 1955 to 1986 after millions of doses of vaccines were administered; however, in 230,734 administered ACAM2000 doses, 18 cases of myopericarditis (incidence, 7.8 per 100,000) were reported during a surveillance study in 2002 and 2003.3,5
Myopericarditis presents with a wide variety of symptoms, such as chest pain, palpitations, chills, shortness of breath, and fever.6,7 Mainstay diagnostic criteria include ECG findings consistent with myopericarditis (such as diffuse ST segment elevations) and elevated cardiac biomarkers (elevated troponins).5-7 An echocardiogram can be helpful in diagnosis, as most cases will not have regional wall motion abnormalities (to distinguish against coronary artery disease).5-7 MRI with diffuse enhancement of the myocardium can be helpful in diagnosis.5,6 The gold standard for diagnosis is an endomyocardial biopsy, which carries a significant risk of complications and is not routinely performed to diagnose myopericarditis.5,6 US military smallpox vaccination data showed that the onset of vaccine-associated myopericarditis averaged (SD) 10.4 (3.6) days after vaccination.5
Vaccine-associated myopericarditis treatment is focused on decreasing inflammation.5,6 Nonsteroidal anti-inflammatory drugs are advised for about 2 weeks with cessation of intensive cardiac activities for between 4 and 6 weeks due to risks of congestive heart failure and fatal cardiac arrhythmias.5,6
Conclusions
Since the September 11 attacks, the US needs to be continually prepared for potential terrorism on American soil and abroad. The threat of bioterrorism has renewed efforts to vaccinate or revaccinate American service members deployed to high-risk regions. These vaccinations put them at risk for vaccination-induced complications that can range from mild fever to life-threatening complications.
A renewed effort to vaccinate service members fighting the global war on terrorism has brought new diagnostic challenges. Vaccinations not generally given to the public are routinely given to service members when they deploy to various parts of the world. Examples include anthrax, yellow fever, Japanese encephalitis, rabies, polio, and smallpox. Every vaccination has potential for adverse effects (AEs), which can range from mild to severe life-threatening complications. These AEs often go unrecognized and untreated because physicians are not routinely screening for vaccination administration.
Background
Smallpox (Variola major) was successfully eradicated in 1977 due to worldwide vaccination efforts.1 However, the threat of bioterrorism has renewed mandatory smallpox vaccinations for high-risk individuals, such as active-duty military personnel.1,2 A notable increase in myopericarditis has been reported with the new generation of smallpox vaccination, ACAM2000.3 We present a case of a 27-year-old healthy male who presented with chest pain and diffuse ST segment elevations consistent with myopericarditis after vaccination with ACAM2000.
Case Presentation
A healthy 27-year-old soldier presented to the emergency department with sudden, new onset, sharp-stabbing, substernal chest pain, which was made worse with lying flat and better with leaning forward. Vital signs were unremarkable. He recently enlisted in the US Army and received the smallpox vaccination about 11 days before as part of a routine predeployment checklist. The patient reported he did not have any viral symptoms, such as fever, chills, nausea, vomiting, diarrhea, shortness of breath, sore throat, rhinorrhea, or sputum production. He also reported having no prior illness for the past 3 months, sick contacts at home or work, or recent travel outside the US. He reported no tobacco use, alcohol use, or illicit drug use. The patient’s family history was negative for significant cardiac disease.
A physical examination was unremarkable. The initial laboratory report showed no leukocytosis, anemia, thrombocytopenia, electrolytes derangement, abnormal kidney function, or abnormal liver function tests. Initial troponin was 0.25 ng/mL, erythrocyte sedimentation rate (ESR) was 40 mmol/h and C-reactive protein (CRP) was 120.2 mg/L suggestive of acute inflammation. A urine drug screen was negative. D-dimer was < 0.27. An electrocardiogram (ECG) showed diffuse ST segment elevation (Figure 1). An echocardiogram showed normal left ventricle size, and function with ejection fraction 55 to 60%, normal diastolic dysfunction, and trivial pericardial effusion. Magnetic resonance imaging (MRI) showed increased T2 signal intensity of the myocardium suggestive of myopericarditis (Figure 2). A computed tomography (CT) angiogram of the coronary arteries showed no significant stenosis.
The patient was treated with ibuprofen for 2 weeks and colchicine for 3 months, and his symptoms resolved. He followed up with an appointment in the cardiology clinic 1 month later, and his ESR, CRP, and troponin results were negative. A limited echocardiogram showed ejection fraction 60 to 65%, no regional wall motion abnormalities, normal diastolic function, and resolution of the pericardial effusion.
Discussion
Smallpox was a major worldwide cause of mortality; about 30% of those infected died because of smallpox.2,4,5 Due to a worldwide vaccination effort, the World Health Organization declared smallpox was eradicated in 1977.2,4,5 However, despite successful eradication, smallpox is considered a possible bioterrorism target, which prompted a resurgence of mandatory smallpox vaccinations for active-duty personnel.2,5
Dryvax, a freeze-dried calf lymph smallpox vaccine was used extensively from the 1940s to the 1980s but was replaced in 2008 by ACAM2000, a smallpox vaccine cultured in kidney epithelial cells from African green monkeys.3,5 Myopericarditis was rarely associated with the Dryvax, with only 5 cases reported from 1955 to 1986 after millions of doses of vaccines were administered; however, in 230,734 administered ACAM2000 doses, 18 cases of myopericarditis (incidence, 7.8 per 100,000) were reported during a surveillance study in 2002 and 2003.3,5
Myopericarditis presents with a wide variety of symptoms, such as chest pain, palpitations, chills, shortness of breath, and fever.6,7 Mainstay diagnostic criteria include ECG findings consistent with myopericarditis (such as diffuse ST segment elevations) and elevated cardiac biomarkers (elevated troponins).5-7 An echocardiogram can be helpful in diagnosis, as most cases will not have regional wall motion abnormalities (to distinguish against coronary artery disease).5-7 MRI with diffuse enhancement of the myocardium can be helpful in diagnosis.5,6 The gold standard for diagnosis is an endomyocardial biopsy, which carries a significant risk of complications and is not routinely performed to diagnose myopericarditis.5,6 US military smallpox vaccination data showed that the onset of vaccine-associated myopericarditis averaged (SD) 10.4 (3.6) days after vaccination.5
Vaccine-associated myopericarditis treatment is focused on decreasing inflammation.5,6 Nonsteroidal anti-inflammatory drugs are advised for about 2 weeks with cessation of intensive cardiac activities for between 4 and 6 weeks due to risks of congestive heart failure and fatal cardiac arrhythmias.5,6
Conclusions
Since the September 11 attacks, the US needs to be continually prepared for potential terrorism on American soil and abroad. The threat of bioterrorism has renewed efforts to vaccinate or revaccinate American service members deployed to high-risk regions. These vaccinations put them at risk for vaccination-induced complications that can range from mild fever to life-threatening complications.
1. Bruner DI, Butler BS. Smallpox vaccination-associated myopericarditis is more common with the newest smallpox vaccine. J Emerg Med. 2014;46(3):e85-e87. doi:10.1016/j.jemermed.2013.06.001
2. Halsell JS, Riddle JR, Atwood JE, et al. Myopericarditis following smallpox vaccination among vaccinia-naive US military personnel. JAMA. 2003;289(24):3283-3289. doi:10.1001/jama.289.24.3283
3. Nalca A, Zumbrun EE. ACAM2000: the new smallpox vaccine for United States Strategic National Stockpile. Drug Des Devel Ther. 2010;4:71-79. doi:10.2147/dddt.s3687
4. Wollenberg A, Engler R. Smallpox, vaccination and adverse reactions to smallpox vaccine. Curr Opin Allergy Clin Immunol. 2004;4(4):271-275. doi:10.1097/01.all.0000136758.66442.28
5. Cassimatis DC, Atwood JE, Engler RM, Linz PE, Grabenstein JD, Vernalis MN. Smallpox vaccination and myopericarditis: a clinical review. J Am Coll Cardiol. 2004;43(9):1503-1510. doi:10.1016/j.jacc.2003.11.053
6. Sharma U, Tak T. A report of 2 cases of myopericarditis after Vaccinia virus (smallpox) immunization. WMJ. 2011;110(6):291-294.
7. Sarkisian SA, Hand G, Rivera VM, Smith M, Miller JA. A case series of smallpox vaccination-associated myopericarditis: effects on safety and readiness of the active duty soldier. Mil Med. 2019;184(1-2):e280-e283. doi:10.1093/milmed/usy159
1. Bruner DI, Butler BS. Smallpox vaccination-associated myopericarditis is more common with the newest smallpox vaccine. J Emerg Med. 2014;46(3):e85-e87. doi:10.1016/j.jemermed.2013.06.001
2. Halsell JS, Riddle JR, Atwood JE, et al. Myopericarditis following smallpox vaccination among vaccinia-naive US military personnel. JAMA. 2003;289(24):3283-3289. doi:10.1001/jama.289.24.3283
3. Nalca A, Zumbrun EE. ACAM2000: the new smallpox vaccine for United States Strategic National Stockpile. Drug Des Devel Ther. 2010;4:71-79. doi:10.2147/dddt.s3687
4. Wollenberg A, Engler R. Smallpox, vaccination and adverse reactions to smallpox vaccine. Curr Opin Allergy Clin Immunol. 2004;4(4):271-275. doi:10.1097/01.all.0000136758.66442.28
5. Cassimatis DC, Atwood JE, Engler RM, Linz PE, Grabenstein JD, Vernalis MN. Smallpox vaccination and myopericarditis: a clinical review. J Am Coll Cardiol. 2004;43(9):1503-1510. doi:10.1016/j.jacc.2003.11.053
6. Sharma U, Tak T. A report of 2 cases of myopericarditis after Vaccinia virus (smallpox) immunization. WMJ. 2011;110(6):291-294.
7. Sarkisian SA, Hand G, Rivera VM, Smith M, Miller JA. A case series of smallpox vaccination-associated myopericarditis: effects on safety and readiness of the active duty soldier. Mil Med. 2019;184(1-2):e280-e283. doi:10.1093/milmed/usy159
Long-standing Dermatitis Treated With Dupilumab With Subsequent Progression to Cutaneous T-cell Lymphoma
Dupilumab is a novel medication that is approved by the US Food and Drug Administration to treat moderate to severe atopic dermatitis (AD) in patients 6 years and older. Dupilumab is an injectable fully human monoclonal antibody. It provides a giant leap toward a better quality of life for patients with AD. Dupilumab works by binding to the shared α subunit of the IL-4 receptor (IL-4R), thus inhibiting IL-4 and IL-13 from using that signaling pathway. The documented side-effect profile includes injection-site reaction, keratitis, nasopharyngitis, and headache.1
We initiated off-label treatment with dupilumab in 3 adult patients who had a history of long-standing adult-onset dermatitis confirmed by histopathology. The 3 patients received a loading dose of 600 mg subcutaneously, followed by 300 mg every other week. Following treatment, the patients had expansion of their disease, with features consistent with cutaneous T-cell lymphoma (CTCL) on subsequent biopsies. These 3 cases demonstrate the well-known adage that the diagnosis of CTCL often requires multiple biopsies performed over time. Although dupilumab has proved efficacious and safe for treating AD, dermatologists should be cautious before starting this medication in an adult who has new-onset dermatitis and no history of atopy.
Case Reports
Patient 1
A 61-year-old man presented to dermatology after being lost to follow-up for several years and was started on dupilumab for long-standing nonspecific eczematous dermatitis based on histopathology. He had a pruritic rash of 10 years’ duration that had been biopsied multiple times and was found to be consistent with dermatitis and lichen simplex chronicus (Figure 1). He had been treated with triamcinolone ointment 0.1% and narrowband UVB as often as 3 times weekly over many years. The patient also had a history of idiopathic CD4 lymphopenia with consistently negative tests for human immunodeficiency virus.
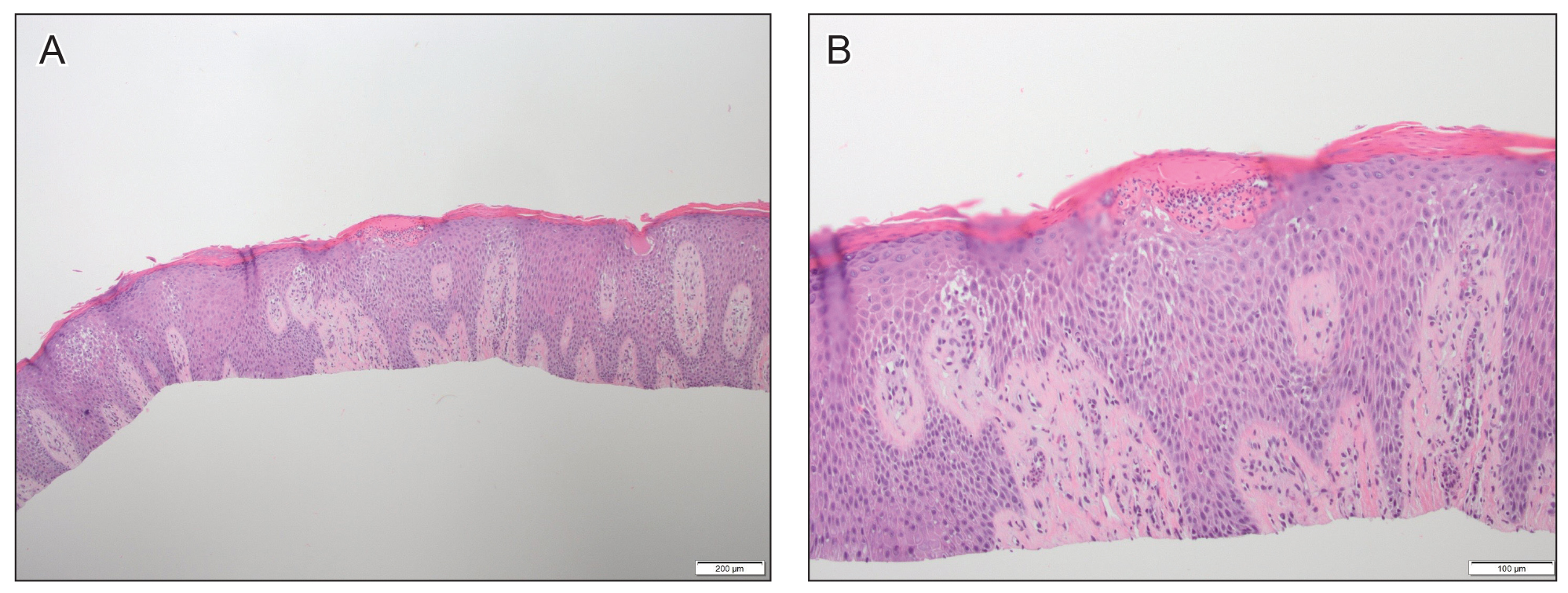
At approximately the same time as dupilumab was initiated, he was started on 60 mg daily of prednisone by his pulmonologist because of a history of restrictive lung disease of unknown cause. While taking prednisone, he experienced notable improvement in his skin condition; however, as he was slowly tapered off prednisone, he noted remarkable worsening of the dermatitis. Dupilumab was discontinued. Two more biopsies were performed; findings on both were consistent with mycosis fungoides (MF)(Figure 2).
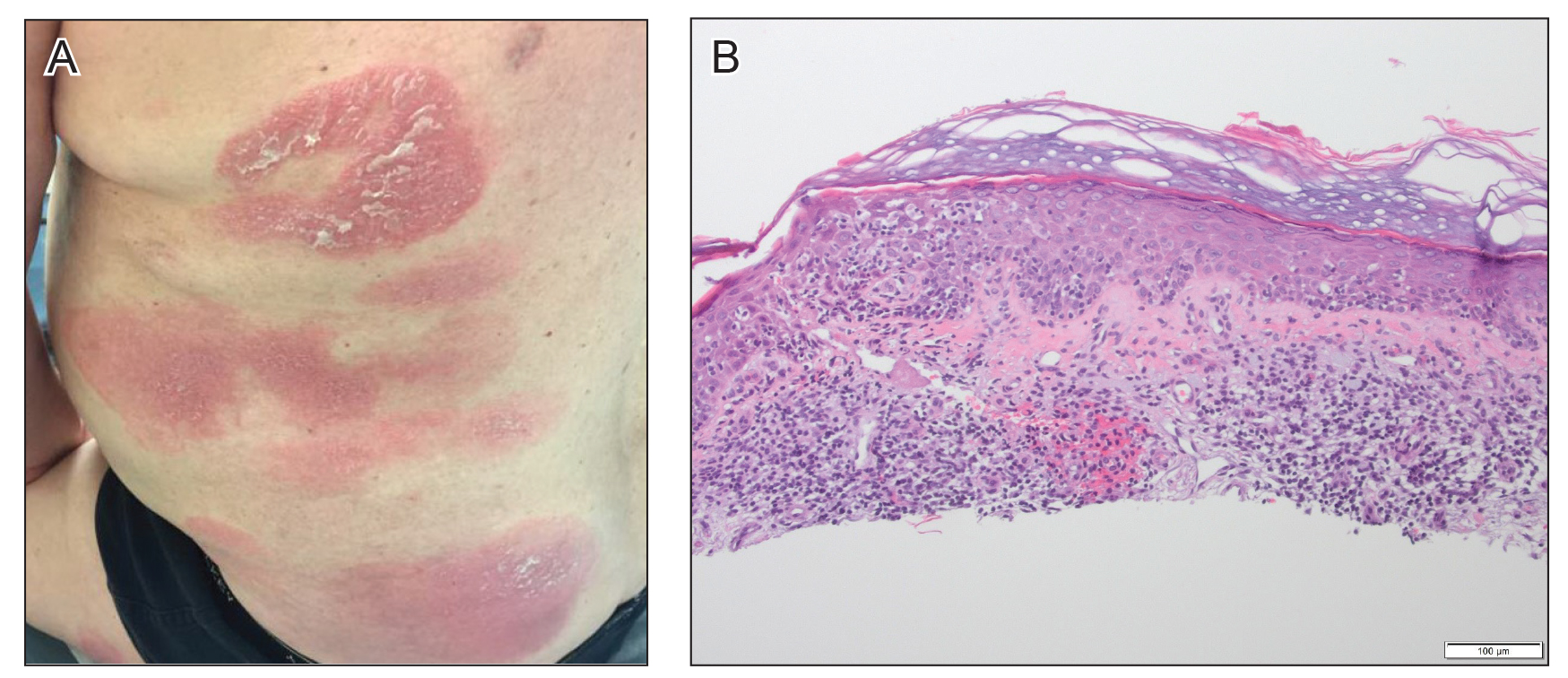
Patient 2
A 52-year-old man presented with indurated, red, scaly plaques on the legs and arms. Initial biopsy was consistent with psoriasiform dermatitis that was thought to be due to a primarily eczematous process. Because of the clinical suspicion of psoriasis, the patient was at first treated with topical betamethasone and eventually was transitioned to multiple injectable biologics without improvement. There was no response to multiple psoriasis treatments, and the original pathology report was re-reviewed. The report noted a substantial eczematous component; therefore, a decision was made to transition him to dupilumab. He also was at first provided with a prednisone taper due to the severity of the cutaneous disease.
Initially, the patient noted 15% to 20% improvement; however, after 6 injections, dupilumab appeared to lose efficacy. Due to a lack of response to multiple biologic medications as well as dupilumab, another biopsy was performed. Findings were consistent with MF.
Patient 3
A 60-year-old woman with diffuse, pruritic, and erythematous dermatitis of 3 years’ duration was referred from an outside dermatology group. Prior biopsies were consistent with eczematous dermatitis. However, because 1 isolated plaque demonstrated findings consistent with psoriasis, she was started on guselkumab, which was discontinued after 12 weeks of therapy for lack of efficacy. The patient also had been treated with a short course of narrowband UVB and topical corticosteroids without benefit.
Upon initial evaluation in our clinic, there was concern for Sézary syndrome; however, peripheral blood studies were normal, and there was no monoclonal spike or irregularity in the patient’s Sézary flow cytometry panel. A biopsy demonstrated lichenoid dermatitis, possibly consistent with drug eruption. All supplements and likely medication culprits were discontinued without improvement.
Prior to follow-up in our clinic, the patient was again evaluated by an outside dermatologist and started on dupilumab. After 3 doses, she discontinued the medication because there was no improvement in the cutaneous symptoms. Findings on repeat biopsy following dupilumab treatment were consistent with MF.
Comment
Mycosis fungoides is a rare chronic T-cell lymphoma that can smolder for decades as nonspecific dermatitis before declaring itself fully on skin biopsy.2 In many cases, MF masquerades as eczema, psoriasis, contact dermatitis, or other dermatitides, and it often responds to the same medications, making diagnosis even more challenging. Treatment options include topical steroids, narrowband UVB, topical nitrogen mustard, topical carmustine, and bexarotene gel for early-stage disease.3 Although it cannot be determined which patients will progress, some do, and therapies must then be upgraded.
We reported 3 patients with adult-onset dermatitis and multiple biopsies demonstrating nondiagnostic findings, which, in retrospect, likely represented early smoldering CTCL. Each of these patients was treated with dupilumab because multiple biopsies demonstrated findings consistent with nondiagnostic dermatitis, along with a lack of response to standard therapies. In all 3 cases, however, the patients had no history of eczema or atopy. After starting dupilumab, each patient had an acute exacerbation of dermatitis; immediately thereafter, biopsies were consistent with CTCL.
These patients most likely had smoldering CTCL that expressed itself fully after dupilumab was started. Biologic medications and their effects on the immune system have been shown to have multiple unanticipated effects on the skin.4-6 We are not insinuating that dupilumab was the cause of our patients having developed CTCL, but we do propose that the underlying interplay of dupilumab with the immune system might have accelerated progression of underlying CTCL, resulting in the lymphoma presenting itself clinically and histopathologically. We also must mention that all 3 cases could represent a “true, true, and unrelated” phenomenon.
A proposed mechanism for how dupilumab might hasten progression of CTCL is based on a functional increase of IL-13 available for binding at the IL-13 receptor (IL-13R) α2 site following blockade of the IL-13Rα1 site by dupilumab (Figure 3). The pathway that is blocked by dupilumab provides improvement in AD by blocking the α subunit of the IL-4R, making it a receptor antagonist for both IL-4 and IL-13. The IL-4R forms a heterodimer with both γ c and separately with IL-13Rα1. As a result, IL-4 and IL-13 cannot bind to their respective targets; thus, downstream signaling that is required for AD is halted.7 IL-13, in addition to IL-4R, also binds to an IL-13Rα2. IL-13 and both of its receptors are upregulated in CTCL, particularly IL-13Rα2.8
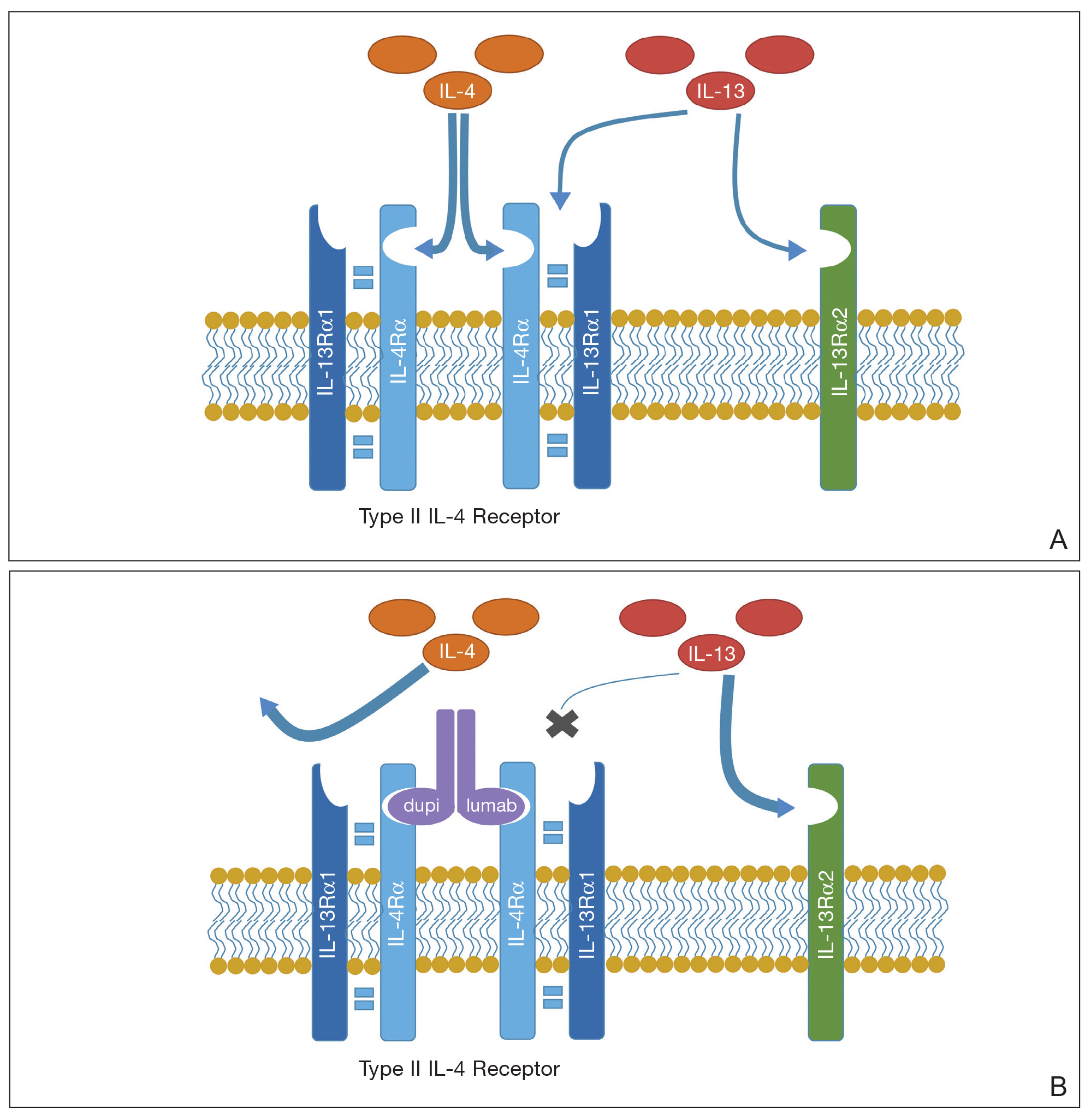
One of the principal ways that CTCL survives is through autocrine signaling, inducing more IL-13 and more IL-13Rα2, which is not seen in normal skin.8 Autocrine signaling plays a critical role in cancer activation and in providing self-sustaining growth signals to tumors.9 In addition, it has been documented that IL-13Rα2 has a higher affinity for IL-13 than the affinity of IL-13Rα1.10 As such, when the dupilumab receptor is blocked, our proposed mechanism of acceleration of CTCL is based on a functional increase in IL-13 available for binding at the IL-13Rα2 site, following indirect blockade of the α1 receptor with dupilumab, which effectively increases available IL-13 to be shunted down the tumorigenic pathway.
We recognize that this proposed mechanism is a theory; additionally, it should be noted that dupilumab is approved only for the treatment of AD and asthma. In our 3 cases, we used dupilumab off label in patients who did not have a clear case of AD or a childhood history of the disease.
When screening patients for the use of dupilumab, it is important to treat only those who have a classic history of moderate to severe AD, including itch, family history, and rash in the classic atopic distribution. We propose that these cases represent potential exacerbation of extant CTCL following exposure to dupilumab.
The manufacturer of dupilumab has reported 1 case of stage IV MF in a 57-year-old man 48 days after the first dose of dupilumab, leading to permanent discontinuation. The patient had ongoing disease at the time of the report, and the manufacturer stated that use of dupilumab was unrelated to disease.11 Studies are needed to explore any potential immunologic link between dupilumab and progression of CTCL.
- Raedler LA. Dupixent (dupilumab) first biologic drug approved for patients with moderate-to-severe atopic dermatitis. Am Health Drug Benefits. 2018;11:58-60.
- Skov AG, Gniadecki R. Delay in the histopathologic diagnosis of mycosis fungoides. Acta Derm Venereol. 2015;95:472-475.
- Ramsay DL, Meller JA, Zackheim HS. Topical treatment of early cutaneous T-cell lymphoma. Hematol Oncol Clin North Am. 1995;9:1031-1056.
- Mazloom SE, Yan D, Hu JZ, et al. TNF-α inhibitor-induced psoriasis: a decade of experience at the Cleveland Clinic [published online December 18, 2018]. J Am Acad Dermatol. doi: 10.1016/j.jaad.2018.12.018.
- Tierney E, Kirthi S, Ramsay B, et al. Ustekinumab-induced subacute cutaneous lupus. JAAD Case Rep. 2019;5:271-273.
- Orrell KA, Murphrey M, Kelm RC, et al. Inflammatory bowel disease events after exposure to interleukin 17 inhibitors secukinumab and ixekizumab: postmarketing analysis from the RADAR (“Research on Adverse Drug events And Reports”) program. J Am Acad Dermatol. 2018;79:777-778.
- Sastre J, Dávila I. Dupilumab: a new paradigm for the treatment of allergic diseases. J Investig Allergol Clin Immunol. 2018;28:139-150.
- Geskin LJ, Viragova S, Stolz DB, et al. Interleukin-13 is over-expressed in cutaneous T-cell lymphoma cells and regulates their proliferation. Blood. 2015;125:2798-2805.
- Barderas R, Bartolomé RA, Fernandez-Aceñero MJ, et al. High expression of IL-13 receptor α2 in colorectal cancer is associated with invasion, liver metastasis, and poor prognosis. Cancer Res. 2012;72:2780-2790.
- Andrews A-L, Holloway JW, Puddicombe SM, et al. Kinetic analysis of the interleukin-13 receptor complex. J Biol Chem. 2002;277:46073-46078.
- Data on file. Tarrytown, NY: Regeneron Pharmaceuticals, Inc; 2017.
Dupilumab is a novel medication that is approved by the US Food and Drug Administration to treat moderate to severe atopic dermatitis (AD) in patients 6 years and older. Dupilumab is an injectable fully human monoclonal antibody. It provides a giant leap toward a better quality of life for patients with AD. Dupilumab works by binding to the shared α subunit of the IL-4 receptor (IL-4R), thus inhibiting IL-4 and IL-13 from using that signaling pathway. The documented side-effect profile includes injection-site reaction, keratitis, nasopharyngitis, and headache.1
We initiated off-label treatment with dupilumab in 3 adult patients who had a history of long-standing adult-onset dermatitis confirmed by histopathology. The 3 patients received a loading dose of 600 mg subcutaneously, followed by 300 mg every other week. Following treatment, the patients had expansion of their disease, with features consistent with cutaneous T-cell lymphoma (CTCL) on subsequent biopsies. These 3 cases demonstrate the well-known adage that the diagnosis of CTCL often requires multiple biopsies performed over time. Although dupilumab has proved efficacious and safe for treating AD, dermatologists should be cautious before starting this medication in an adult who has new-onset dermatitis and no history of atopy.
Case Reports
Patient 1
A 61-year-old man presented to dermatology after being lost to follow-up for several years and was started on dupilumab for long-standing nonspecific eczematous dermatitis based on histopathology. He had a pruritic rash of 10 years’ duration that had been biopsied multiple times and was found to be consistent with dermatitis and lichen simplex chronicus (Figure 1). He had been treated with triamcinolone ointment 0.1% and narrowband UVB as often as 3 times weekly over many years. The patient also had a history of idiopathic CD4 lymphopenia with consistently negative tests for human immunodeficiency virus.
At approximately the same time as dupilumab was initiated, he was started on 60 mg daily of prednisone by his pulmonologist because of a history of restrictive lung disease of unknown cause. While taking prednisone, he experienced notable improvement in his skin condition; however, as he was slowly tapered off prednisone, he noted remarkable worsening of the dermatitis. Dupilumab was discontinued. Two more biopsies were performed; findings on both were consistent with mycosis fungoides (MF)(Figure 2).
Patient 2
A 52-year-old man presented with indurated, red, scaly plaques on the legs and arms. Initial biopsy was consistent with psoriasiform dermatitis that was thought to be due to a primarily eczematous process. Because of the clinical suspicion of psoriasis, the patient was at first treated with topical betamethasone and eventually was transitioned to multiple injectable biologics without improvement. There was no response to multiple psoriasis treatments, and the original pathology report was re-reviewed. The report noted a substantial eczematous component; therefore, a decision was made to transition him to dupilumab. He also was at first provided with a prednisone taper due to the severity of the cutaneous disease.
Initially, the patient noted 15% to 20% improvement; however, after 6 injections, dupilumab appeared to lose efficacy. Due to a lack of response to multiple biologic medications as well as dupilumab, another biopsy was performed. Findings were consistent with MF.
Patient 3
A 60-year-old woman with diffuse, pruritic, and erythematous dermatitis of 3 years’ duration was referred from an outside dermatology group. Prior biopsies were consistent with eczematous dermatitis. However, because 1 isolated plaque demonstrated findings consistent with psoriasis, she was started on guselkumab, which was discontinued after 12 weeks of therapy for lack of efficacy. The patient also had been treated with a short course of narrowband UVB and topical corticosteroids without benefit.
Upon initial evaluation in our clinic, there was concern for Sézary syndrome; however, peripheral blood studies were normal, and there was no monoclonal spike or irregularity in the patient’s Sézary flow cytometry panel. A biopsy demonstrated lichenoid dermatitis, possibly consistent with drug eruption. All supplements and likely medication culprits were discontinued without improvement.
Prior to follow-up in our clinic, the patient was again evaluated by an outside dermatologist and started on dupilumab. After 3 doses, she discontinued the medication because there was no improvement in the cutaneous symptoms. Findings on repeat biopsy following dupilumab treatment were consistent with MF.
Comment
Mycosis fungoides is a rare chronic T-cell lymphoma that can smolder for decades as nonspecific dermatitis before declaring itself fully on skin biopsy.2 In many cases, MF masquerades as eczema, psoriasis, contact dermatitis, or other dermatitides, and it often responds to the same medications, making diagnosis even more challenging. Treatment options include topical steroids, narrowband UVB, topical nitrogen mustard, topical carmustine, and bexarotene gel for early-stage disease.3 Although it cannot be determined which patients will progress, some do, and therapies must then be upgraded.
We reported 3 patients with adult-onset dermatitis and multiple biopsies demonstrating nondiagnostic findings, which, in retrospect, likely represented early smoldering CTCL. Each of these patients was treated with dupilumab because multiple biopsies demonstrated findings consistent with nondiagnostic dermatitis, along with a lack of response to standard therapies. In all 3 cases, however, the patients had no history of eczema or atopy. After starting dupilumab, each patient had an acute exacerbation of dermatitis; immediately thereafter, biopsies were consistent with CTCL.
These patients most likely had smoldering CTCL that expressed itself fully after dupilumab was started. Biologic medications and their effects on the immune system have been shown to have multiple unanticipated effects on the skin.4-6 We are not insinuating that dupilumab was the cause of our patients having developed CTCL, but we do propose that the underlying interplay of dupilumab with the immune system might have accelerated progression of underlying CTCL, resulting in the lymphoma presenting itself clinically and histopathologically. We also must mention that all 3 cases could represent a “true, true, and unrelated” phenomenon.
A proposed mechanism for how dupilumab might hasten progression of CTCL is based on a functional increase of IL-13 available for binding at the IL-13 receptor (IL-13R) α2 site following blockade of the IL-13Rα1 site by dupilumab (Figure 3). The pathway that is blocked by dupilumab provides improvement in AD by blocking the α subunit of the IL-4R, making it a receptor antagonist for both IL-4 and IL-13. The IL-4R forms a heterodimer with both γ c and separately with IL-13Rα1. As a result, IL-4 and IL-13 cannot bind to their respective targets; thus, downstream signaling that is required for AD is halted.7 IL-13, in addition to IL-4R, also binds to an IL-13Rα2. IL-13 and both of its receptors are upregulated in CTCL, particularly IL-13Rα2.8
One of the principal ways that CTCL survives is through autocrine signaling, inducing more IL-13 and more IL-13Rα2, which is not seen in normal skin.8 Autocrine signaling plays a critical role in cancer activation and in providing self-sustaining growth signals to tumors.9 In addition, it has been documented that IL-13Rα2 has a higher affinity for IL-13 than the affinity of IL-13Rα1.10 As such, when the dupilumab receptor is blocked, our proposed mechanism of acceleration of CTCL is based on a functional increase in IL-13 available for binding at the IL-13Rα2 site, following indirect blockade of the α1 receptor with dupilumab, which effectively increases available IL-13 to be shunted down the tumorigenic pathway.
We recognize that this proposed mechanism is a theory; additionally, it should be noted that dupilumab is approved only for the treatment of AD and asthma. In our 3 cases, we used dupilumab off label in patients who did not have a clear case of AD or a childhood history of the disease.
When screening patients for the use of dupilumab, it is important to treat only those who have a classic history of moderate to severe AD, including itch, family history, and rash in the classic atopic distribution. We propose that these cases represent potential exacerbation of extant CTCL following exposure to dupilumab.
The manufacturer of dupilumab has reported 1 case of stage IV MF in a 57-year-old man 48 days after the first dose of dupilumab, leading to permanent discontinuation. The patient had ongoing disease at the time of the report, and the manufacturer stated that use of dupilumab was unrelated to disease.11 Studies are needed to explore any potential immunologic link between dupilumab and progression of CTCL.
Dupilumab is a novel medication that is approved by the US Food and Drug Administration to treat moderate to severe atopic dermatitis (AD) in patients 6 years and older. Dupilumab is an injectable fully human monoclonal antibody. It provides a giant leap toward a better quality of life for patients with AD. Dupilumab works by binding to the shared α subunit of the IL-4 receptor (IL-4R), thus inhibiting IL-4 and IL-13 from using that signaling pathway. The documented side-effect profile includes injection-site reaction, keratitis, nasopharyngitis, and headache.1
We initiated off-label treatment with dupilumab in 3 adult patients who had a history of long-standing adult-onset dermatitis confirmed by histopathology. The 3 patients received a loading dose of 600 mg subcutaneously, followed by 300 mg every other week. Following treatment, the patients had expansion of their disease, with features consistent with cutaneous T-cell lymphoma (CTCL) on subsequent biopsies. These 3 cases demonstrate the well-known adage that the diagnosis of CTCL often requires multiple biopsies performed over time. Although dupilumab has proved efficacious and safe for treating AD, dermatologists should be cautious before starting this medication in an adult who has new-onset dermatitis and no history of atopy.
Case Reports
Patient 1
A 61-year-old man presented to dermatology after being lost to follow-up for several years and was started on dupilumab for long-standing nonspecific eczematous dermatitis based on histopathology. He had a pruritic rash of 10 years’ duration that had been biopsied multiple times and was found to be consistent with dermatitis and lichen simplex chronicus (Figure 1). He had been treated with triamcinolone ointment 0.1% and narrowband UVB as often as 3 times weekly over many years. The patient also had a history of idiopathic CD4 lymphopenia with consistently negative tests for human immunodeficiency virus.
At approximately the same time as dupilumab was initiated, he was started on 60 mg daily of prednisone by his pulmonologist because of a history of restrictive lung disease of unknown cause. While taking prednisone, he experienced notable improvement in his skin condition; however, as he was slowly tapered off prednisone, he noted remarkable worsening of the dermatitis. Dupilumab was discontinued. Two more biopsies were performed; findings on both were consistent with mycosis fungoides (MF)(Figure 2).
Patient 2
A 52-year-old man presented with indurated, red, scaly plaques on the legs and arms. Initial biopsy was consistent with psoriasiform dermatitis that was thought to be due to a primarily eczematous process. Because of the clinical suspicion of psoriasis, the patient was at first treated with topical betamethasone and eventually was transitioned to multiple injectable biologics without improvement. There was no response to multiple psoriasis treatments, and the original pathology report was re-reviewed. The report noted a substantial eczematous component; therefore, a decision was made to transition him to dupilumab. He also was at first provided with a prednisone taper due to the severity of the cutaneous disease.
Initially, the patient noted 15% to 20% improvement; however, after 6 injections, dupilumab appeared to lose efficacy. Due to a lack of response to multiple biologic medications as well as dupilumab, another biopsy was performed. Findings were consistent with MF.
Patient 3
A 60-year-old woman with diffuse, pruritic, and erythematous dermatitis of 3 years’ duration was referred from an outside dermatology group. Prior biopsies were consistent with eczematous dermatitis. However, because 1 isolated plaque demonstrated findings consistent with psoriasis, she was started on guselkumab, which was discontinued after 12 weeks of therapy for lack of efficacy. The patient also had been treated with a short course of narrowband UVB and topical corticosteroids without benefit.
Upon initial evaluation in our clinic, there was concern for Sézary syndrome; however, peripheral blood studies were normal, and there was no monoclonal spike or irregularity in the patient’s Sézary flow cytometry panel. A biopsy demonstrated lichenoid dermatitis, possibly consistent with drug eruption. All supplements and likely medication culprits were discontinued without improvement.
Prior to follow-up in our clinic, the patient was again evaluated by an outside dermatologist and started on dupilumab. After 3 doses, she discontinued the medication because there was no improvement in the cutaneous symptoms. Findings on repeat biopsy following dupilumab treatment were consistent with MF.
Comment
Mycosis fungoides is a rare chronic T-cell lymphoma that can smolder for decades as nonspecific dermatitis before declaring itself fully on skin biopsy.2 In many cases, MF masquerades as eczema, psoriasis, contact dermatitis, or other dermatitides, and it often responds to the same medications, making diagnosis even more challenging. Treatment options include topical steroids, narrowband UVB, topical nitrogen mustard, topical carmustine, and bexarotene gel for early-stage disease.3 Although it cannot be determined which patients will progress, some do, and therapies must then be upgraded.
We reported 3 patients with adult-onset dermatitis and multiple biopsies demonstrating nondiagnostic findings, which, in retrospect, likely represented early smoldering CTCL. Each of these patients was treated with dupilumab because multiple biopsies demonstrated findings consistent with nondiagnostic dermatitis, along with a lack of response to standard therapies. In all 3 cases, however, the patients had no history of eczema or atopy. After starting dupilumab, each patient had an acute exacerbation of dermatitis; immediately thereafter, biopsies were consistent with CTCL.
These patients most likely had smoldering CTCL that expressed itself fully after dupilumab was started. Biologic medications and their effects on the immune system have been shown to have multiple unanticipated effects on the skin.4-6 We are not insinuating that dupilumab was the cause of our patients having developed CTCL, but we do propose that the underlying interplay of dupilumab with the immune system might have accelerated progression of underlying CTCL, resulting in the lymphoma presenting itself clinically and histopathologically. We also must mention that all 3 cases could represent a “true, true, and unrelated” phenomenon.
A proposed mechanism for how dupilumab might hasten progression of CTCL is based on a functional increase of IL-13 available for binding at the IL-13 receptor (IL-13R) α2 site following blockade of the IL-13Rα1 site by dupilumab (Figure 3). The pathway that is blocked by dupilumab provides improvement in AD by blocking the α subunit of the IL-4R, making it a receptor antagonist for both IL-4 and IL-13. The IL-4R forms a heterodimer with both γ c and separately with IL-13Rα1. As a result, IL-4 and IL-13 cannot bind to their respective targets; thus, downstream signaling that is required for AD is halted.7 IL-13, in addition to IL-4R, also binds to an IL-13Rα2. IL-13 and both of its receptors are upregulated in CTCL, particularly IL-13Rα2.8
One of the principal ways that CTCL survives is through autocrine signaling, inducing more IL-13 and more IL-13Rα2, which is not seen in normal skin.8 Autocrine signaling plays a critical role in cancer activation and in providing self-sustaining growth signals to tumors.9 In addition, it has been documented that IL-13Rα2 has a higher affinity for IL-13 than the affinity of IL-13Rα1.10 As such, when the dupilumab receptor is blocked, our proposed mechanism of acceleration of CTCL is based on a functional increase in IL-13 available for binding at the IL-13Rα2 site, following indirect blockade of the α1 receptor with dupilumab, which effectively increases available IL-13 to be shunted down the tumorigenic pathway.
We recognize that this proposed mechanism is a theory; additionally, it should be noted that dupilumab is approved only for the treatment of AD and asthma. In our 3 cases, we used dupilumab off label in patients who did not have a clear case of AD or a childhood history of the disease.
When screening patients for the use of dupilumab, it is important to treat only those who have a classic history of moderate to severe AD, including itch, family history, and rash in the classic atopic distribution. We propose that these cases represent potential exacerbation of extant CTCL following exposure to dupilumab.
The manufacturer of dupilumab has reported 1 case of stage IV MF in a 57-year-old man 48 days after the first dose of dupilumab, leading to permanent discontinuation. The patient had ongoing disease at the time of the report, and the manufacturer stated that use of dupilumab was unrelated to disease.11 Studies are needed to explore any potential immunologic link between dupilumab and progression of CTCL.
- Raedler LA. Dupixent (dupilumab) first biologic drug approved for patients with moderate-to-severe atopic dermatitis. Am Health Drug Benefits. 2018;11:58-60.
- Skov AG, Gniadecki R. Delay in the histopathologic diagnosis of mycosis fungoides. Acta Derm Venereol. 2015;95:472-475.
- Ramsay DL, Meller JA, Zackheim HS. Topical treatment of early cutaneous T-cell lymphoma. Hematol Oncol Clin North Am. 1995;9:1031-1056.
- Mazloom SE, Yan D, Hu JZ, et al. TNF-α inhibitor-induced psoriasis: a decade of experience at the Cleveland Clinic [published online December 18, 2018]. J Am Acad Dermatol. doi: 10.1016/j.jaad.2018.12.018.
- Tierney E, Kirthi S, Ramsay B, et al. Ustekinumab-induced subacute cutaneous lupus. JAAD Case Rep. 2019;5:271-273.
- Orrell KA, Murphrey M, Kelm RC, et al. Inflammatory bowel disease events after exposure to interleukin 17 inhibitors secukinumab and ixekizumab: postmarketing analysis from the RADAR (“Research on Adverse Drug events And Reports”) program. J Am Acad Dermatol. 2018;79:777-778.
- Sastre J, Dávila I. Dupilumab: a new paradigm for the treatment of allergic diseases. J Investig Allergol Clin Immunol. 2018;28:139-150.
- Geskin LJ, Viragova S, Stolz DB, et al. Interleukin-13 is over-expressed in cutaneous T-cell lymphoma cells and regulates their proliferation. Blood. 2015;125:2798-2805.
- Barderas R, Bartolomé RA, Fernandez-Aceñero MJ, et al. High expression of IL-13 receptor α2 in colorectal cancer is associated with invasion, liver metastasis, and poor prognosis. Cancer Res. 2012;72:2780-2790.
- Andrews A-L, Holloway JW, Puddicombe SM, et al. Kinetic analysis of the interleukin-13 receptor complex. J Biol Chem. 2002;277:46073-46078.
- Data on file. Tarrytown, NY: Regeneron Pharmaceuticals, Inc; 2017.
- Raedler LA. Dupixent (dupilumab) first biologic drug approved for patients with moderate-to-severe atopic dermatitis. Am Health Drug Benefits. 2018;11:58-60.
- Skov AG, Gniadecki R. Delay in the histopathologic diagnosis of mycosis fungoides. Acta Derm Venereol. 2015;95:472-475.
- Ramsay DL, Meller JA, Zackheim HS. Topical treatment of early cutaneous T-cell lymphoma. Hematol Oncol Clin North Am. 1995;9:1031-1056.
- Mazloom SE, Yan D, Hu JZ, et al. TNF-α inhibitor-induced psoriasis: a decade of experience at the Cleveland Clinic [published online December 18, 2018]. J Am Acad Dermatol. doi: 10.1016/j.jaad.2018.12.018.
- Tierney E, Kirthi S, Ramsay B, et al. Ustekinumab-induced subacute cutaneous lupus. JAAD Case Rep. 2019;5:271-273.
- Orrell KA, Murphrey M, Kelm RC, et al. Inflammatory bowel disease events after exposure to interleukin 17 inhibitors secukinumab and ixekizumab: postmarketing analysis from the RADAR (“Research on Adverse Drug events And Reports”) program. J Am Acad Dermatol. 2018;79:777-778.
- Sastre J, Dávila I. Dupilumab: a new paradigm for the treatment of allergic diseases. J Investig Allergol Clin Immunol. 2018;28:139-150.
- Geskin LJ, Viragova S, Stolz DB, et al. Interleukin-13 is over-expressed in cutaneous T-cell lymphoma cells and regulates their proliferation. Blood. 2015;125:2798-2805.
- Barderas R, Bartolomé RA, Fernandez-Aceñero MJ, et al. High expression of IL-13 receptor α2 in colorectal cancer is associated with invasion, liver metastasis, and poor prognosis. Cancer Res. 2012;72:2780-2790.
- Andrews A-L, Holloway JW, Puddicombe SM, et al. Kinetic analysis of the interleukin-13 receptor complex. J Biol Chem. 2002;277:46073-46078.
- Data on file. Tarrytown, NY: Regeneron Pharmaceuticals, Inc; 2017.
Practice Points
- Dupilumab is a safe and effective treatment for atopic dermatitis (AD) in both children and adults.
- Prior to starting treatment for presumed adult-onset AD, consider smoldering cutaneous T-cell lymphoma (CTCL).
- Dupilumab may interact with the cutaneous immune system, leading to an expedited presentation of CTCL in patients with chronic adult-onset AD.
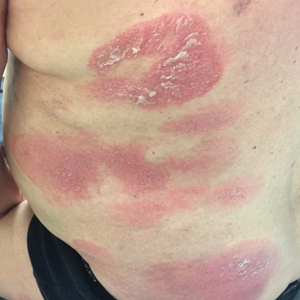
Valproate-Induced Lower Extremity Swelling
Bilateral lower extremity edema is a common condition with a broad differential diagnosis. New, severe peripheral edema implies a more nefarious underlying etiology than chronic venous insufficiency and should prompt a thorough evaluation for underlying conditions, such as congestive heart failure (CHF), cirrhosis, nephrotic syndrome, hypoalbuminemia, or lymphatic or venous obstruction. We present a case of a patient with sudden onset new bilateral lower extremity edema due to a rare adverse drug reaction (ADR) from valproate.
Case Presentation
A 63-year-old male with a history of seizures, bipolar disorder type I, and memory impairment due to traumatic brain injury (TBI) from a gunshot wound 24 years prior presented to the emergency department for witnessed seizure activity in the community. The patient had been incarcerated for the past 20 years, throughout which he had been taking the antiepileptic drugs (AEDs) phenytoin and divalproex and did not have any seizure activity. No records prior to his incarceration were available for review.
The patient recently had been released from prison and was nonadherent with his AEDs, leading to a witnessed seizure. This episode was described as preceded by an electric sensation, followed by rhythmic shaking of the right upper extremity without loss of consciousness. His regimen prior to admission included divalproex 1,000 mg daily and phenytoin 200 mg daily. His only other medication was folic acid.
Neurology was consulted on admission. An awake and asleep 4-hour electroencephalogram showed intermittent focal slowing of the right frontocentral region and frequent epileptiform discharges in the right prefrontal region during sleep, corresponding to areas of chronic right anterior frontal and temporal encephalomalacia seen on brain imaging. His seizures were thought likely to be secondary to prior head trauma. While the described seizure activity involving the right upper extremity was not consistent with the location of his prior TBI, neurology considered that he might have simple partial seizures with multiple foci or that his seizure event prior to admission was not accurately described. The neurology consult recommended switching from phenytoin 200 mg daily to lacosamide 100 mg twice daily on admission. His prior dose of divalproex 1,000 mg daily also was resumed for its antiepileptic effect and the added benefit of mood stabilization, as the patient reported elevated mood and decreased need for sleep on admission.
Eight days after changing his AED regimen, the patient was found to have new onset bilateral grade 1+ pitting edema to the level of his shins. He had no history of dyspnea, orthopnea, paroxysmal nocturnal dyspnea, dysuria, or changes in his urination. Although medical records from his incarceration were not available for review, the patient reported that he had never had peripheral edema.
On physical examination, the patient had no periorbital edema, jugular venous pressure of 8 cm H2O, negative hepatojugular reflex, unremarkable cardiac and lung examination, and grade 2+ posterior tibial and dorsalis pedis pulses bilaterally. He underwent extensive laboratory evaluation for potential underlying causes, including nephrotic syndrome, cirrhosis, hypothyroidism, and CHF (Table). Valproate levels were initially subtherapeutic on admission (< 10 µg/mL, reference range 50-125 µg/mL) then rose to within therapeutic range (54 µg/mL-80 µg/mL throughout admission) after neurology recommended increasing the dose from 1,000 mg daily to 1,500 mg daily. His measured valproate levels were never supratherapeutic.
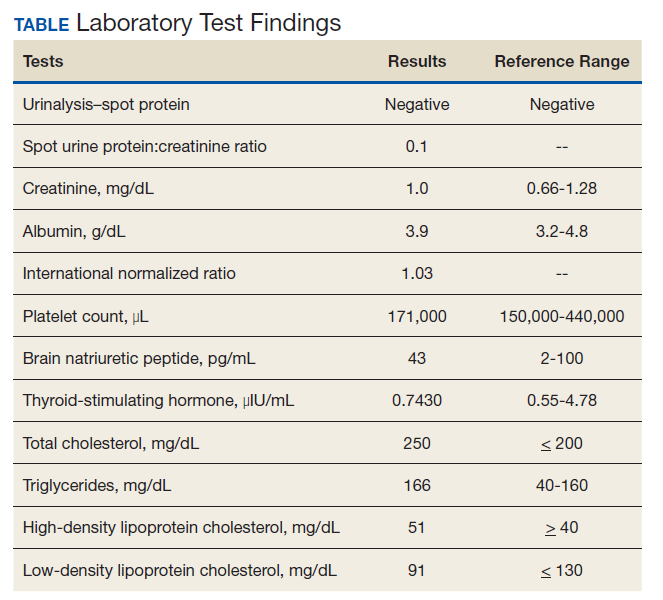
An electrocardiogram showed normal sinus rhythm unchanged from admission. Transthoracic echocardiogram showed normal left ventricular (LV) size and estimated LV ejection fraction of 55 to 60%. Abdominal ultrasound showed no evidence of cirrhosis and normal portal vein flow. Ultrasound of the lower extremities showed no deep venous thrombosis or valvular insufficiency. The patient was prescribed compression stockings. However, due to memory impairment, he was relatively nonadherent, and his lower extremity edema worsened to grade 3+ over several days. Due to the progressive swelling with no identified cause, a computed tomographic venogram of the abdomen and pelvis was performed to determine whether an inferior vena cava (IVC) thrombus was present. This study was unremarkable and did not show any external IVC compression.
After extensive evaluation did not reveal any other cause, the temporal course of events suggested an association between the patient’s peripheral edema and resumption of divalproex. His swelling remained stable. Discontinuation of divalproex was considered, but the patient’s mood remained euthymic, and he had no further seizure activity while on this medication, so the benefit of continuation was felt to outweigh any risks of switching to another agent.
Discussion
Valproate and its related forms, such as divalproex, often are used in the treatment of generalized or partial seizures, psychiatric disorders, and the prophylaxis of migraine headaches. Common ADRs include gastrointestinal symptoms, sedation, and dose-related thrombocytopenia, among many others. Rare ADRs include fulminant hepatitis, pancreatitis, hyperammonemia, and peripheral edema.1 There have been case reports of valproate-induced peripheral edema, which seems to be an idiosyncratic ADR that occurs after long-term administration of the medication.2,3 Early studies reported valproate-related edema in the context of valproate-induced hepatic injury.4 However, in more recent case reports, valproate-related edema has been found in patients without hepatotoxicity or supratherapeutic drug levels.1,2
The exact mechanism by which valproate causes peripheral edema is unknown. It has been reported that medications affecting the γ-aminobutyric acid (GABA) system such as benzodiazepines, for example, can cause this rare ADR.5 Unlike benzodiazepines, valproate has an indirect effect on the GABA system, through increasing availability of GABA.6 GABA receptors have been identified on peripheral tissues, suggesting that GABAergic medications also may have an effect on regional vascular resistance.7 This mechanism was proposed by prior case reports but has yet to be proven in studies.2
In this case, initiation of lacosamide temporally coinciding with development of the patient’s edema leads one to question whether lacosamide may have caused this ADR. Other medications commonly used in seizure management (such as benzodiazepines and gabapentin) have been reported to cause new onset peripheral edema.5,8 To date, however, there are no reported cases of peripheral edema due to lacosamide. While there are known interactions between various AEDs that may impact drug levels of valproate, there are no reported drug-drug interactions between lacosamide and valproate.9
Conclusions
Our case adds to the small but growing body of literature that suggests peripheral edema is a rare but clinically significant ADR of valproate. With its broad differential diagnosis, new onset peripheral edema is a concern that often warrants an extensive evaluation for underlying causes. Clinicians should be aware of this ADR as use of valproate becomes increasingly common so that an extensive workup is not always performed on patients with peripheral edema.
1. Prajapati H, Kansal D, Negi R. Magnesium valproate-induced pedal edema on chronic therapy: a rare adverse drug reaction. Indian J Pharmacol. 2017;49(5):399. doi:10.4103/ijp.IJP_239_17
2. Lin ST, Chen CS, Yen CF, Tsei JH, Wang SY. Valproate-related peripheral oedema: a manageable but probably neglected condition. Int J Neuropsychopharmacol. 2009;12(7):991-993. doi:10.1017/S1461145709000509
3. Ettinger A, Moshe S, Shinnar S. Edema associated with long‐term valproate therapy. Epilepsia. 1990;31(2):211-213. doi:10.1111/j.1528-1167.1990.tb06308.x
4. Zimmerman HJ, Ishak KG. Valproate‐induced hepatic injury: analyses of 23 fatal cases. Hepatology. 1982;2(5):591S-597S. doi:10.1002/hep.1840020513
5. Mathew T, D’Souza D, Nadimpally US, Nadig R. Clobazam‐induced pedal edema: “an unrecognized side effect of a common antiepileptic drug.” Epilepsia. 2016;57(3): 524-525. doi:10.1111/epi.13316
6. Bourin M, Chenu F, Hascoët M. The role of sodium channels in the mechanism of action of antidepressants and mood stabilizers. Curr Drug Targets. 2009;10(11):1052-1060. doi:10.2174/138945009789735138
7. Takemoto Y. Effects of gamma‐aminobutyric acid on regional vascular resistances of conscious spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. 1995;22(suppl):S102-Sl04. doi:10.1111/j.1440-1681.1995.tb02839.x
8. Bidaki R, Sadeghi Z, Shafizadegan S, et al. Gabapentin induces edema, hyperesthesia and scaling in a depressed patient; a diagnostic challenge. Adv Biomed Res. 2016;5:1. doi:10.4103/2277-9175.174955
9. Cawello W, Nickel B, Eggert‐Formella A. No pharmacokinetic interaction between lacosamide and carbamazepine in healthy volunteers. J Clin Pharmacol. 2010;50(4):459-471. doi:10.1177/0091270009347675
Bilateral lower extremity edema is a common condition with a broad differential diagnosis. New, severe peripheral edema implies a more nefarious underlying etiology than chronic venous insufficiency and should prompt a thorough evaluation for underlying conditions, such as congestive heart failure (CHF), cirrhosis, nephrotic syndrome, hypoalbuminemia, or lymphatic or venous obstruction. We present a case of a patient with sudden onset new bilateral lower extremity edema due to a rare adverse drug reaction (ADR) from valproate.
Case Presentation
A 63-year-old male with a history of seizures, bipolar disorder type I, and memory impairment due to traumatic brain injury (TBI) from a gunshot wound 24 years prior presented to the emergency department for witnessed seizure activity in the community. The patient had been incarcerated for the past 20 years, throughout which he had been taking the antiepileptic drugs (AEDs) phenytoin and divalproex and did not have any seizure activity. No records prior to his incarceration were available for review.
The patient recently had been released from prison and was nonadherent with his AEDs, leading to a witnessed seizure. This episode was described as preceded by an electric sensation, followed by rhythmic shaking of the right upper extremity without loss of consciousness. His regimen prior to admission included divalproex 1,000 mg daily and phenytoin 200 mg daily. His only other medication was folic acid.
Neurology was consulted on admission. An awake and asleep 4-hour electroencephalogram showed intermittent focal slowing of the right frontocentral region and frequent epileptiform discharges in the right prefrontal region during sleep, corresponding to areas of chronic right anterior frontal and temporal encephalomalacia seen on brain imaging. His seizures were thought likely to be secondary to prior head trauma. While the described seizure activity involving the right upper extremity was not consistent with the location of his prior TBI, neurology considered that he might have simple partial seizures with multiple foci or that his seizure event prior to admission was not accurately described. The neurology consult recommended switching from phenytoin 200 mg daily to lacosamide 100 mg twice daily on admission. His prior dose of divalproex 1,000 mg daily also was resumed for its antiepileptic effect and the added benefit of mood stabilization, as the patient reported elevated mood and decreased need for sleep on admission.
Eight days after changing his AED regimen, the patient was found to have new onset bilateral grade 1+ pitting edema to the level of his shins. He had no history of dyspnea, orthopnea, paroxysmal nocturnal dyspnea, dysuria, or changes in his urination. Although medical records from his incarceration were not available for review, the patient reported that he had never had peripheral edema.
On physical examination, the patient had no periorbital edema, jugular venous pressure of 8 cm H2O, negative hepatojugular reflex, unremarkable cardiac and lung examination, and grade 2+ posterior tibial and dorsalis pedis pulses bilaterally. He underwent extensive laboratory evaluation for potential underlying causes, including nephrotic syndrome, cirrhosis, hypothyroidism, and CHF (Table). Valproate levels were initially subtherapeutic on admission (< 10 µg/mL, reference range 50-125 µg/mL) then rose to within therapeutic range (54 µg/mL-80 µg/mL throughout admission) after neurology recommended increasing the dose from 1,000 mg daily to 1,500 mg daily. His measured valproate levels were never supratherapeutic.
An electrocardiogram showed normal sinus rhythm unchanged from admission. Transthoracic echocardiogram showed normal left ventricular (LV) size and estimated LV ejection fraction of 55 to 60%. Abdominal ultrasound showed no evidence of cirrhosis and normal portal vein flow. Ultrasound of the lower extremities showed no deep venous thrombosis or valvular insufficiency. The patient was prescribed compression stockings. However, due to memory impairment, he was relatively nonadherent, and his lower extremity edema worsened to grade 3+ over several days. Due to the progressive swelling with no identified cause, a computed tomographic venogram of the abdomen and pelvis was performed to determine whether an inferior vena cava (IVC) thrombus was present. This study was unremarkable and did not show any external IVC compression.
After extensive evaluation did not reveal any other cause, the temporal course of events suggested an association between the patient’s peripheral edema and resumption of divalproex. His swelling remained stable. Discontinuation of divalproex was considered, but the patient’s mood remained euthymic, and he had no further seizure activity while on this medication, so the benefit of continuation was felt to outweigh any risks of switching to another agent.
Discussion
Valproate and its related forms, such as divalproex, often are used in the treatment of generalized or partial seizures, psychiatric disorders, and the prophylaxis of migraine headaches. Common ADRs include gastrointestinal symptoms, sedation, and dose-related thrombocytopenia, among many others. Rare ADRs include fulminant hepatitis, pancreatitis, hyperammonemia, and peripheral edema.1 There have been case reports of valproate-induced peripheral edema, which seems to be an idiosyncratic ADR that occurs after long-term administration of the medication.2,3 Early studies reported valproate-related edema in the context of valproate-induced hepatic injury.4 However, in more recent case reports, valproate-related edema has been found in patients without hepatotoxicity or supratherapeutic drug levels.1,2
The exact mechanism by which valproate causes peripheral edema is unknown. It has been reported that medications affecting the γ-aminobutyric acid (GABA) system such as benzodiazepines, for example, can cause this rare ADR.5 Unlike benzodiazepines, valproate has an indirect effect on the GABA system, through increasing availability of GABA.6 GABA receptors have been identified on peripheral tissues, suggesting that GABAergic medications also may have an effect on regional vascular resistance.7 This mechanism was proposed by prior case reports but has yet to be proven in studies.2
In this case, initiation of lacosamide temporally coinciding with development of the patient’s edema leads one to question whether lacosamide may have caused this ADR. Other medications commonly used in seizure management (such as benzodiazepines and gabapentin) have been reported to cause new onset peripheral edema.5,8 To date, however, there are no reported cases of peripheral edema due to lacosamide. While there are known interactions between various AEDs that may impact drug levels of valproate, there are no reported drug-drug interactions between lacosamide and valproate.9
Conclusions
Our case adds to the small but growing body of literature that suggests peripheral edema is a rare but clinically significant ADR of valproate. With its broad differential diagnosis, new onset peripheral edema is a concern that often warrants an extensive evaluation for underlying causes. Clinicians should be aware of this ADR as use of valproate becomes increasingly common so that an extensive workup is not always performed on patients with peripheral edema.
Bilateral lower extremity edema is a common condition with a broad differential diagnosis. New, severe peripheral edema implies a more nefarious underlying etiology than chronic venous insufficiency and should prompt a thorough evaluation for underlying conditions, such as congestive heart failure (CHF), cirrhosis, nephrotic syndrome, hypoalbuminemia, or lymphatic or venous obstruction. We present a case of a patient with sudden onset new bilateral lower extremity edema due to a rare adverse drug reaction (ADR) from valproate.
Case Presentation
A 63-year-old male with a history of seizures, bipolar disorder type I, and memory impairment due to traumatic brain injury (TBI) from a gunshot wound 24 years prior presented to the emergency department for witnessed seizure activity in the community. The patient had been incarcerated for the past 20 years, throughout which he had been taking the antiepileptic drugs (AEDs) phenytoin and divalproex and did not have any seizure activity. No records prior to his incarceration were available for review.
The patient recently had been released from prison and was nonadherent with his AEDs, leading to a witnessed seizure. This episode was described as preceded by an electric sensation, followed by rhythmic shaking of the right upper extremity without loss of consciousness. His regimen prior to admission included divalproex 1,000 mg daily and phenytoin 200 mg daily. His only other medication was folic acid.
Neurology was consulted on admission. An awake and asleep 4-hour electroencephalogram showed intermittent focal slowing of the right frontocentral region and frequent epileptiform discharges in the right prefrontal region during sleep, corresponding to areas of chronic right anterior frontal and temporal encephalomalacia seen on brain imaging. His seizures were thought likely to be secondary to prior head trauma. While the described seizure activity involving the right upper extremity was not consistent with the location of his prior TBI, neurology considered that he might have simple partial seizures with multiple foci or that his seizure event prior to admission was not accurately described. The neurology consult recommended switching from phenytoin 200 mg daily to lacosamide 100 mg twice daily on admission. His prior dose of divalproex 1,000 mg daily also was resumed for its antiepileptic effect and the added benefit of mood stabilization, as the patient reported elevated mood and decreased need for sleep on admission.
Eight days after changing his AED regimen, the patient was found to have new onset bilateral grade 1+ pitting edema to the level of his shins. He had no history of dyspnea, orthopnea, paroxysmal nocturnal dyspnea, dysuria, or changes in his urination. Although medical records from his incarceration were not available for review, the patient reported that he had never had peripheral edema.
On physical examination, the patient had no periorbital edema, jugular venous pressure of 8 cm H2O, negative hepatojugular reflex, unremarkable cardiac and lung examination, and grade 2+ posterior tibial and dorsalis pedis pulses bilaterally. He underwent extensive laboratory evaluation for potential underlying causes, including nephrotic syndrome, cirrhosis, hypothyroidism, and CHF (Table). Valproate levels were initially subtherapeutic on admission (< 10 µg/mL, reference range 50-125 µg/mL) then rose to within therapeutic range (54 µg/mL-80 µg/mL throughout admission) after neurology recommended increasing the dose from 1,000 mg daily to 1,500 mg daily. His measured valproate levels were never supratherapeutic.
An electrocardiogram showed normal sinus rhythm unchanged from admission. Transthoracic echocardiogram showed normal left ventricular (LV) size and estimated LV ejection fraction of 55 to 60%. Abdominal ultrasound showed no evidence of cirrhosis and normal portal vein flow. Ultrasound of the lower extremities showed no deep venous thrombosis or valvular insufficiency. The patient was prescribed compression stockings. However, due to memory impairment, he was relatively nonadherent, and his lower extremity edema worsened to grade 3+ over several days. Due to the progressive swelling with no identified cause, a computed tomographic venogram of the abdomen and pelvis was performed to determine whether an inferior vena cava (IVC) thrombus was present. This study was unremarkable and did not show any external IVC compression.
After extensive evaluation did not reveal any other cause, the temporal course of events suggested an association between the patient’s peripheral edema and resumption of divalproex. His swelling remained stable. Discontinuation of divalproex was considered, but the patient’s mood remained euthymic, and he had no further seizure activity while on this medication, so the benefit of continuation was felt to outweigh any risks of switching to another agent.
Discussion
Valproate and its related forms, such as divalproex, often are used in the treatment of generalized or partial seizures, psychiatric disorders, and the prophylaxis of migraine headaches. Common ADRs include gastrointestinal symptoms, sedation, and dose-related thrombocytopenia, among many others. Rare ADRs include fulminant hepatitis, pancreatitis, hyperammonemia, and peripheral edema.1 There have been case reports of valproate-induced peripheral edema, which seems to be an idiosyncratic ADR that occurs after long-term administration of the medication.2,3 Early studies reported valproate-related edema in the context of valproate-induced hepatic injury.4 However, in more recent case reports, valproate-related edema has been found in patients without hepatotoxicity or supratherapeutic drug levels.1,2
The exact mechanism by which valproate causes peripheral edema is unknown. It has been reported that medications affecting the γ-aminobutyric acid (GABA) system such as benzodiazepines, for example, can cause this rare ADR.5 Unlike benzodiazepines, valproate has an indirect effect on the GABA system, through increasing availability of GABA.6 GABA receptors have been identified on peripheral tissues, suggesting that GABAergic medications also may have an effect on regional vascular resistance.7 This mechanism was proposed by prior case reports but has yet to be proven in studies.2
In this case, initiation of lacosamide temporally coinciding with development of the patient’s edema leads one to question whether lacosamide may have caused this ADR. Other medications commonly used in seizure management (such as benzodiazepines and gabapentin) have been reported to cause new onset peripheral edema.5,8 To date, however, there are no reported cases of peripheral edema due to lacosamide. While there are known interactions between various AEDs that may impact drug levels of valproate, there are no reported drug-drug interactions between lacosamide and valproate.9
Conclusions
Our case adds to the small but growing body of literature that suggests peripheral edema is a rare but clinically significant ADR of valproate. With its broad differential diagnosis, new onset peripheral edema is a concern that often warrants an extensive evaluation for underlying causes. Clinicians should be aware of this ADR as use of valproate becomes increasingly common so that an extensive workup is not always performed on patients with peripheral edema.
1. Prajapati H, Kansal D, Negi R. Magnesium valproate-induced pedal edema on chronic therapy: a rare adverse drug reaction. Indian J Pharmacol. 2017;49(5):399. doi:10.4103/ijp.IJP_239_17
2. Lin ST, Chen CS, Yen CF, Tsei JH, Wang SY. Valproate-related peripheral oedema: a manageable but probably neglected condition. Int J Neuropsychopharmacol. 2009;12(7):991-993. doi:10.1017/S1461145709000509
3. Ettinger A, Moshe S, Shinnar S. Edema associated with long‐term valproate therapy. Epilepsia. 1990;31(2):211-213. doi:10.1111/j.1528-1167.1990.tb06308.x
4. Zimmerman HJ, Ishak KG. Valproate‐induced hepatic injury: analyses of 23 fatal cases. Hepatology. 1982;2(5):591S-597S. doi:10.1002/hep.1840020513
5. Mathew T, D’Souza D, Nadimpally US, Nadig R. Clobazam‐induced pedal edema: “an unrecognized side effect of a common antiepileptic drug.” Epilepsia. 2016;57(3): 524-525. doi:10.1111/epi.13316
6. Bourin M, Chenu F, Hascoët M. The role of sodium channels in the mechanism of action of antidepressants and mood stabilizers. Curr Drug Targets. 2009;10(11):1052-1060. doi:10.2174/138945009789735138
7. Takemoto Y. Effects of gamma‐aminobutyric acid on regional vascular resistances of conscious spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. 1995;22(suppl):S102-Sl04. doi:10.1111/j.1440-1681.1995.tb02839.x
8. Bidaki R, Sadeghi Z, Shafizadegan S, et al. Gabapentin induces edema, hyperesthesia and scaling in a depressed patient; a diagnostic challenge. Adv Biomed Res. 2016;5:1. doi:10.4103/2277-9175.174955
9. Cawello W, Nickel B, Eggert‐Formella A. No pharmacokinetic interaction between lacosamide and carbamazepine in healthy volunteers. J Clin Pharmacol. 2010;50(4):459-471. doi:10.1177/0091270009347675
1. Prajapati H, Kansal D, Negi R. Magnesium valproate-induced pedal edema on chronic therapy: a rare adverse drug reaction. Indian J Pharmacol. 2017;49(5):399. doi:10.4103/ijp.IJP_239_17
2. Lin ST, Chen CS, Yen CF, Tsei JH, Wang SY. Valproate-related peripheral oedema: a manageable but probably neglected condition. Int J Neuropsychopharmacol. 2009;12(7):991-993. doi:10.1017/S1461145709000509
3. Ettinger A, Moshe S, Shinnar S. Edema associated with long‐term valproate therapy. Epilepsia. 1990;31(2):211-213. doi:10.1111/j.1528-1167.1990.tb06308.x
4. Zimmerman HJ, Ishak KG. Valproate‐induced hepatic injury: analyses of 23 fatal cases. Hepatology. 1982;2(5):591S-597S. doi:10.1002/hep.1840020513
5. Mathew T, D’Souza D, Nadimpally US, Nadig R. Clobazam‐induced pedal edema: “an unrecognized side effect of a common antiepileptic drug.” Epilepsia. 2016;57(3): 524-525. doi:10.1111/epi.13316
6. Bourin M, Chenu F, Hascoët M. The role of sodium channels in the mechanism of action of antidepressants and mood stabilizers. Curr Drug Targets. 2009;10(11):1052-1060. doi:10.2174/138945009789735138
7. Takemoto Y. Effects of gamma‐aminobutyric acid on regional vascular resistances of conscious spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. 1995;22(suppl):S102-Sl04. doi:10.1111/j.1440-1681.1995.tb02839.x
8. Bidaki R, Sadeghi Z, Shafizadegan S, et al. Gabapentin induces edema, hyperesthesia and scaling in a depressed patient; a diagnostic challenge. Adv Biomed Res. 2016;5:1. doi:10.4103/2277-9175.174955
9. Cawello W, Nickel B, Eggert‐Formella A. No pharmacokinetic interaction between lacosamide and carbamazepine in healthy volunteers. J Clin Pharmacol. 2010;50(4):459-471. doi:10.1177/0091270009347675
Chronic Microaspiration and Frailty: A Geriatric Smoking Gun?
Frailty is a highly prevalent syndrome in nursing homes, occurring in at least 50% of patients.1 The frailty phenotype has been described by Fried and colleagues as impairment in ≥ 3 of 5 domains: unintentional weight loss, self-reported exhaustion, muscle weakness, slow gait speed, and low physical activity. By this definition, frailty is highly associated with poor quality of life and mortality.2,3
In recent years, there has been evolving evidence of a relationship between frailty and chronic systemic inflammation.4-6 Some degree of chronic inflammation is likely inherent to the aging process and increases the risk of frailty (so-called inflammaging) but is seen to a greater degree in many pathologic conditions in nursing homes, including cancer, organ failure, and chronic infection.4,6-8
Dysphagia also is highly prevalent in nursing homes, affecting up to 60% of patients and is a strong predictor of hospital utilization and of mortality.9,10 Overt aspiration pneumonitis and pneumonia are perhaps the best studied sequelae, but chronic occult microaspiration also is prevalent in this population.11 Just as normal systemic inflammatory changes in aging may increase vulnerability to frailty with additional illness burden, normal aging changes in swallowing function may increase vulnerability to dysphagia and to microaspiration with additional illness burden.12,13 In older adults, important risk factors for microaspiration include not only overt dysphagia, dementia, and other neurologic illnesses, but also general debility, weakness, and immobility.14
Matsuse and colleagues have described diffuse aspiration bronchiolitis (DAB) in patients with chronic microaspiration.14 DAB often goes undiagnosed.14-16 As in frailty, weight loss and chronic anemia may be seen, and many of these patients are bedridden.14,17 Episodes of macroaspiration and overt lobar pneumonia also may occur.14 Lung biopsy or autopsy reveals chronic bronchiolar inflammation and sometimes pulmonary fibrosis, but to date there have been no reports suggesting chronic systemic inflammation or elevated proinflammatory cytokines.14,15,17 We present 3 patients with progressive weight loss, functional decline, and frailty in whom chronic microaspiration likely played a significant role.
Case 1 Presentation
A 68-year-old man with a 6-year history of rapidly progressive Parkinson disease was admitted to the Haley’s Cove Community Living Center (CLC) on the James A. Haley Veterans’ Hospital campus in Tampa, Florida for long-term care. The patient’s medical history also was significant for bipolar illness and for small cell carcinoma of the lung in sustained remission.
Medications included levodopa/carbidopa 50 mg/200 mg 4 times daily, entacapone 200 mg 4 times daily, lithium carbonate 600 mg every night at bedtime, lamotrigine 150 mg daily, quetiapine 200 mg every night at bedtime, pravastatin 40 mg every night at bedtime, omeprazole 20 mg daily, tamsulosin 0.4 mg every night at bedtime, and aspirin 81 mg daily. He initially did well, but after 6 months the nursing staff began to notice the patient coughing during and after meals. Speech pathology evaluation revealed moderate oropharyngeal dysphagia, and his diet was downgraded to nectar-thickened liquids.
Over the subsequent 10 months, he became progressively weaker in physical therapy and more inactive, with about a 20-lb weight loss and mild hypoalbuminemia of 3.0 gm/dL. He had developed 3 episodes of aspiration pneumonia during this period; a repeat swallow evaluation after the last episode revealed worsened dysphagia, and his physician suggested nil per os (NPO) status and an alternative feeding route. His guardian declined placement of a percutaneous endoscopic gastrostomy (PEG) tube, he was transferred to the inpatient hospice unit, and died 2 weeks later. An autopsy was declined.
Case 2 Presentation
A 66-year-old man with a medical history of multiple traumatic brain injuries (TBIs) was admitted to the CLC for long-term care. Sequelae of the TBIs included moderate dementia, spastic paraparesis with multiple pressure injuries, a well-controlled seizure disorder, and severe oropharyngeal dysphagia with NPO status and a percutaneous endoscopic gastrostomy (PEG) tube. His medical history included TBIs and hepatitis C virus infection; medications included levetiracetam 1,000 mg twice daily, lamotrigine 25 mg twice daily, and cholecalciferol 2,000 U daily. He had multiple stage III pressure injuries and an ischial stage IV injury at the time of admission.
His 11-month stay in the CLC was characterized by progressively worsening weakness and inactivity, with a 25-lb weight loss in spite of adequate tube feeding. Serum albumin remained in the 2.0 to 2.5 gm/dL range, hemoglobin in the 7 to 9 gm/dL range without any obvious source of anemia. Most of the pressure injuries worsened during his stay in spite of aggressive wound care, and he developed a second stage IV sacral wound. A single C-reactive protein (CRP) level 2 months prior to his death was markedly elevated at 19.5 mg/dL. In spite of maintaining NPO status, he developed 3 episodes of aspiration pneumonia, all of which responded well to treatment. Ultimately, he was found pulseless and apneic and resuscitation was unsuccessful. An autopsy revealed purulent material in the small airways.
Case 3 Presentation
A 65-year-old man with a long history of paranoid schizophrenia and severe gastroesophageal reflux disease had resided in the CLC for about 10 years. Medications included risperidone microspheres 37.5 mg every 2 weeks, valproic acid 500 mg 3 times daily and 1,000 mg every night at bedtime, lansoprazole 30 mg twice daily, ranitidine 150 mg every night at bedtime, sucralfate 1,000 mg 3 times daily, simvastatin 20 mg every night at bedtime, and tamsulosin 0.4 mg every night at bedtime. He had done well for many years but developed some drooling and a modest resting tremor (but no other signs of pseudoparkinsonism) about 8 years after admission.
There had been no changes to his risperidone dosage. He also lost about 20 lb over a period of 1 year and became increasingly weak and dependent in gait, serum albumin dropped as low as 1.6 gm/dL, hemoglobin dropped to the 7 to 8 gm/dL range (without any other obvious source of anemia), and he developed a gradually worsening right-sided pleural effusion. CRP was chronically elevated at this point, in the 6 to 15 mg/dL range and as high as 17.2 mg/dL. Ultimately, he developed 3 episodes of aspiration pneumonia over a period of 2 months. Swallowing evaluation at that time revealed severe oropharyngeal dysphagia and a PEG tube was placed. Due to concerns for possible antipsychotic-induced dysphagia, risperidone was discontinued, and quetiapine 400 mg a day was substituted. He did well over the subsequent year with no further pneumonia and advancement back to a regular diet. He regained all of the lost weight and began independent ambulation. Albumin improved to the 3 gm/dL range, hemoglobin to the 12 to 13 gm/dL range, and CRP had decreased to 0.7 mg/dL. The pleural effusion (believed to have been a parapneumonic effusion) had resolved.
Discussion
All 3 patients met the Fried criteria for frailty, although there were several confounding issues.2 All 3 patients lost between 20 and 25 lb; all had clearly become weaker according to nursing and rehabilitation staff (although none were formally assessed for grip strength); and all had clear declines in their activity level. Patient 3 had a clear decrement in gait speed, but patient 1 had severe gait impairment due to Parkinson disease (although his gait in therapy had clearly worsened). Patient 2 was paraparetic and unable to ambulate. There also was evidence of limited biomarkers of systemic inflammation; all 3 patients’ albumin had decreased, and patients 2 and 3 had significant decrease in hemoglobin; but these commonplace clinical biomarkers are obviously multifactorially determined. We have limited data on our patients’ CRP levels; serial levels would have been more specific for systemic inflammation but were infrequently performed on the patients.
Multimorbidity and medical complexity are more the rule than the exception in frail geriatric patients,and it is difficult to separate the role of microaspiration from other confounding conditions that might have contributed to these patients’ evolving systemic inflammation and frailty.18 It might be argued that the decline for patient 1 was related to the underlying Parkinson disease (a progressive neurologic illness in which systemic inflammation has been reported), or that the decline of patient 2 was related to the worsening pressure injuries rather than to covert microaspiration.19 However, the TBIs for patient 2 and the schizophrenia for patient 3 would not be expected to be associated with frailty or with systemic inflammation. Furthermore, the frailty symptoms of patient 3 and inflammatory biomarkers improved after the risperidone, which was likely responsible for his microaspiration, was discontinued. All 3 patients were at risk for oropharyngeal dysphagia (antipsychotic medication is clearly associated with dysphagia20); patient 2 demonstrated pathologic evidence of DAB at autopsy.
There is evolving evidence that chronic systemic inflammation and immune activation are key mechanisms in the pathogenesis of frailty.4-6 It is known that elevated serum levels of proinflammatory cytokines, including tumor necrosis factor-α, interleukin-6, and CRP are directly associated with frailty and are inversely associated with levels of albumin, hemoglobin, insulin-like growth factor-1, and several micronutrients in frail individuals.4-7,21,22 Chronic inflammation contributes to the pathophysiology of frailty through detrimental effects on a broad range of systems, including the musculoskeletal, endocrine, and hematopoietic systems and through nutritional dysregulation.2,4,23 These changes may lead to further deleterious effects, creating a downward spiral of worsening frailty. For example, it seems likely that our patients’ progressive weakness further compromised airway protection, creating a vicious cycle of worsening microaspiration and chronic inflammation.
Conclusions
To date, the role of chronic microaspiration and DAB in chronic systemic inflammation or in frailty has not been explored. Given the prevalence of microaspiration in nursing home residents and the devastating consequences of frailty, though, this seems to be a crucial area of investigation. It is equally crucial for long-term care staff, both providers and nursing staff, to have a heightened awareness of covert microaspiration and a low threshold for referral to speech pathology for further investigation. Staff also should be aware of the utility of the Fried criteria to improve identification of frailty in general. It is probable that covert microaspiration will prove to be an important part of the differential diagnosis of frailty.
1. Kojima G. Prevalence of frailty in nursing homes: a systematic review and meta-analysis. J Am Med Dir Assoc. 2015;16(11):940-945. doi:10.1016/j.jamda.2015.06.025
2. Fried LP, Tangen CM, Walston J, et al. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci. 2001;56(3):M146-M157. doi:10.1093/gerona/56.3.m146
3. Morley JE, Vellas B, van Kan GA, et al. Frailty consensus: a call to action. J Am Med Dir Assoc. 2013;14(6):392-397. doi:10.1016/j.jamda.2013.03.022
4. Chen X, Mao G, Leng SX. Frailty syndrome: an overview. Clin Interv Aging. 2014;9:433-441. doi:10.2147/CIA.S45300.
5. Soysal P, Stubbs B, Lucato P, et al. Inflammation and frailty in the elderly: a systematic review and meta-analysis. Ageing Res Rev. 2016;31:1-8. doi:10.1016/j.arr.2016.08.006
6. Langmann GA, Perera S, Ferchak MA, Nace DA, Resnick NM, Greenspan SL. Inflammatory markers and frailty in long-term care residents. J Am Geriatr Soc. 2017;65(8):1777-1783. doi:10.1111/jgs.14876
7. Michaud M, Balardy L, Moulis G, et al. Proinflammatory cytokines, aging, and age-related diseases. J Am Med Dir Assoc. 2013;14(12):877-882. doi:10.1016/j.jamda.2013.05.009
8. Fougere B, Boulanger E, Nourhashemi F, Guyonnet S, Cesari M. Chronic inflammation: accelerator of biological aging. J Gerontol A Biol Sci Med Sci. 2017;72(9):1218-1225. doi:10.1093/gerona/glw240
9. Shanley C, O’Loughlin G. Dysphagia among nursing home residents: an assessment and management protocol. J Gerontol Nurs. 2000;26(8):35-48. doi:10.3928/0098-9134-20000801-09
10. Altman KW, Yu GP, Schaefer SD. Consequences of dysphagia in the hospitalized patient: impact on prognosis and hospital resources. Arch Otolaryngol Head Neck Surg. 2010;136(8):784-789. doi:10.1001/archoto.2010.129
11. Sakai K, Hirano H, Watanabe Y, et al. An examination of factors related to aspiration and silent aspiration in older adults requiring long-term care in rural Japan. J Oral Rehabil. 2016;43(2):103-110. doi:10.1111/joor.12349
12. Nilsson H, Ekberg O, Olsson R, Hindfelt B. Quantitative aspects of swallowing in an elderly nondysphagic population. Dysphagia. 1996;11(3):180-184. doi:10.1007/BF00366381
13. Daggett A, Logemann J, Rademaker A, Pauloski B. Laryngeal penetration during deglutition in normal subjects of various ages. Dysphagia. 2006;21(4):270-274. doi:10.1007/s00455-006-9051-6
14. Matsuse T, Oka T, Kida K, Fukuchi Y. Importance of diffuse aspiration bronchiolitis caused by chronic occult aspiration in the elderly. Chest. 1996;110(5):1289-1293. doi:10.1378/chest.110.5.1289
15. Cardasis JJ, MacMahon H, Husain AN. The spectrum of lung disease due to chronic occult aspiration. Ann Am Thorac Soc. 2014;11(6):865-873. doi:10.1513/AnnalsATS.201310-360OC
16. Pereira-Silva JL, Silva CIS, Araujo Neto CA, Andrade TL, Muller NL. Chronic pulmonary microaspiration: high-resolution computed tomographic findings in 13 patients. J Thorac Imaging. 2014;29(5):298-303. doi:10.1097/RTI.0000000000000091
17. Hu X, Lee JS, Pianosi PT, Ryu JH. Aspiration-related pulmonary syndromes. Chest. 2015;147(3):815-823. doi:10.1378/chest.14-1049
18. Yarnall AJ, Sayer AA, Clegg A, Rockwood K, Parker S, Hindle JV. New horizons in multimorbidity in older adults. Age Aging. 2017;46(6):882-888. doi:10.1093/ageing/afx150
19. Calabrese V, Santoro A, Monti D, et al. Aging and Parkinson’s disease: inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic Biol Med. 2018;115:80-91. doi:10.1016/j.freeradbiomed.2017.10.379
20. Kulkarni DP, Kamath VD, Stewart JT. Swallowing disorders in schizophrenia. Dysphagia. 2017;32(4):467-471. doi:10.1007/s00455-017-9802-6
21. Velissaris D, Pantzaris N, Koniari I, et al. C-reactive protein and frailty in the elderly: a literature review. J Clin Med Res. 2017;9(6):461-465. doi:10.14740/jocmr2959w
22. Hubbard RE, O’Mahoney MS, Savva GM, Calver BL, Woodhouse KW. Inflammation and frailty measures in older people. J Cell Mol Med. 2009;13(9B):3103-3109. doi:10.1111/j.1582-4934.2009.00733.x
23. Argiles JM, Busquets S, Stemmler B, Lotez-Soriano FJ. Cachexia and sarcopenia: mechanisms and potential targets for intervention. Curr Opin Pharmacol. 2015;22:100-106. doi:10.1016/j.coph.2015.04.003
Frailty is a highly prevalent syndrome in nursing homes, occurring in at least 50% of patients.1 The frailty phenotype has been described by Fried and colleagues as impairment in ≥ 3 of 5 domains: unintentional weight loss, self-reported exhaustion, muscle weakness, slow gait speed, and low physical activity. By this definition, frailty is highly associated with poor quality of life and mortality.2,3
In recent years, there has been evolving evidence of a relationship between frailty and chronic systemic inflammation.4-6 Some degree of chronic inflammation is likely inherent to the aging process and increases the risk of frailty (so-called inflammaging) but is seen to a greater degree in many pathologic conditions in nursing homes, including cancer, organ failure, and chronic infection.4,6-8
Dysphagia also is highly prevalent in nursing homes, affecting up to 60% of patients and is a strong predictor of hospital utilization and of mortality.9,10 Overt aspiration pneumonitis and pneumonia are perhaps the best studied sequelae, but chronic occult microaspiration also is prevalent in this population.11 Just as normal systemic inflammatory changes in aging may increase vulnerability to frailty with additional illness burden, normal aging changes in swallowing function may increase vulnerability to dysphagia and to microaspiration with additional illness burden.12,13 In older adults, important risk factors for microaspiration include not only overt dysphagia, dementia, and other neurologic illnesses, but also general debility, weakness, and immobility.14
Matsuse and colleagues have described diffuse aspiration bronchiolitis (DAB) in patients with chronic microaspiration.14 DAB often goes undiagnosed.14-16 As in frailty, weight loss and chronic anemia may be seen, and many of these patients are bedridden.14,17 Episodes of macroaspiration and overt lobar pneumonia also may occur.14 Lung biopsy or autopsy reveals chronic bronchiolar inflammation and sometimes pulmonary fibrosis, but to date there have been no reports suggesting chronic systemic inflammation or elevated proinflammatory cytokines.14,15,17 We present 3 patients with progressive weight loss, functional decline, and frailty in whom chronic microaspiration likely played a significant role.
Case 1 Presentation
A 68-year-old man with a 6-year history of rapidly progressive Parkinson disease was admitted to the Haley’s Cove Community Living Center (CLC) on the James A. Haley Veterans’ Hospital campus in Tampa, Florida for long-term care. The patient’s medical history also was significant for bipolar illness and for small cell carcinoma of the lung in sustained remission.
Medications included levodopa/carbidopa 50 mg/200 mg 4 times daily, entacapone 200 mg 4 times daily, lithium carbonate 600 mg every night at bedtime, lamotrigine 150 mg daily, quetiapine 200 mg every night at bedtime, pravastatin 40 mg every night at bedtime, omeprazole 20 mg daily, tamsulosin 0.4 mg every night at bedtime, and aspirin 81 mg daily. He initially did well, but after 6 months the nursing staff began to notice the patient coughing during and after meals. Speech pathology evaluation revealed moderate oropharyngeal dysphagia, and his diet was downgraded to nectar-thickened liquids.
Over the subsequent 10 months, he became progressively weaker in physical therapy and more inactive, with about a 20-lb weight loss and mild hypoalbuminemia of 3.0 gm/dL. He had developed 3 episodes of aspiration pneumonia during this period; a repeat swallow evaluation after the last episode revealed worsened dysphagia, and his physician suggested nil per os (NPO) status and an alternative feeding route. His guardian declined placement of a percutaneous endoscopic gastrostomy (PEG) tube, he was transferred to the inpatient hospice unit, and died 2 weeks later. An autopsy was declined.
Case 2 Presentation
A 66-year-old man with a medical history of multiple traumatic brain injuries (TBIs) was admitted to the CLC for long-term care. Sequelae of the TBIs included moderate dementia, spastic paraparesis with multiple pressure injuries, a well-controlled seizure disorder, and severe oropharyngeal dysphagia with NPO status and a percutaneous endoscopic gastrostomy (PEG) tube. His medical history included TBIs and hepatitis C virus infection; medications included levetiracetam 1,000 mg twice daily, lamotrigine 25 mg twice daily, and cholecalciferol 2,000 U daily. He had multiple stage III pressure injuries and an ischial stage IV injury at the time of admission.
His 11-month stay in the CLC was characterized by progressively worsening weakness and inactivity, with a 25-lb weight loss in spite of adequate tube feeding. Serum albumin remained in the 2.0 to 2.5 gm/dL range, hemoglobin in the 7 to 9 gm/dL range without any obvious source of anemia. Most of the pressure injuries worsened during his stay in spite of aggressive wound care, and he developed a second stage IV sacral wound. A single C-reactive protein (CRP) level 2 months prior to his death was markedly elevated at 19.5 mg/dL. In spite of maintaining NPO status, he developed 3 episodes of aspiration pneumonia, all of which responded well to treatment. Ultimately, he was found pulseless and apneic and resuscitation was unsuccessful. An autopsy revealed purulent material in the small airways.
Case 3 Presentation
A 65-year-old man with a long history of paranoid schizophrenia and severe gastroesophageal reflux disease had resided in the CLC for about 10 years. Medications included risperidone microspheres 37.5 mg every 2 weeks, valproic acid 500 mg 3 times daily and 1,000 mg every night at bedtime, lansoprazole 30 mg twice daily, ranitidine 150 mg every night at bedtime, sucralfate 1,000 mg 3 times daily, simvastatin 20 mg every night at bedtime, and tamsulosin 0.4 mg every night at bedtime. He had done well for many years but developed some drooling and a modest resting tremor (but no other signs of pseudoparkinsonism) about 8 years after admission.
There had been no changes to his risperidone dosage. He also lost about 20 lb over a period of 1 year and became increasingly weak and dependent in gait, serum albumin dropped as low as 1.6 gm/dL, hemoglobin dropped to the 7 to 8 gm/dL range (without any other obvious source of anemia), and he developed a gradually worsening right-sided pleural effusion. CRP was chronically elevated at this point, in the 6 to 15 mg/dL range and as high as 17.2 mg/dL. Ultimately, he developed 3 episodes of aspiration pneumonia over a period of 2 months. Swallowing evaluation at that time revealed severe oropharyngeal dysphagia and a PEG tube was placed. Due to concerns for possible antipsychotic-induced dysphagia, risperidone was discontinued, and quetiapine 400 mg a day was substituted. He did well over the subsequent year with no further pneumonia and advancement back to a regular diet. He regained all of the lost weight and began independent ambulation. Albumin improved to the 3 gm/dL range, hemoglobin to the 12 to 13 gm/dL range, and CRP had decreased to 0.7 mg/dL. The pleural effusion (believed to have been a parapneumonic effusion) had resolved.
Discussion
All 3 patients met the Fried criteria for frailty, although there were several confounding issues.2 All 3 patients lost between 20 and 25 lb; all had clearly become weaker according to nursing and rehabilitation staff (although none were formally assessed for grip strength); and all had clear declines in their activity level. Patient 3 had a clear decrement in gait speed, but patient 1 had severe gait impairment due to Parkinson disease (although his gait in therapy had clearly worsened). Patient 2 was paraparetic and unable to ambulate. There also was evidence of limited biomarkers of systemic inflammation; all 3 patients’ albumin had decreased, and patients 2 and 3 had significant decrease in hemoglobin; but these commonplace clinical biomarkers are obviously multifactorially determined. We have limited data on our patients’ CRP levels; serial levels would have been more specific for systemic inflammation but were infrequently performed on the patients.
Multimorbidity and medical complexity are more the rule than the exception in frail geriatric patients,and it is difficult to separate the role of microaspiration from other confounding conditions that might have contributed to these patients’ evolving systemic inflammation and frailty.18 It might be argued that the decline for patient 1 was related to the underlying Parkinson disease (a progressive neurologic illness in which systemic inflammation has been reported), or that the decline of patient 2 was related to the worsening pressure injuries rather than to covert microaspiration.19 However, the TBIs for patient 2 and the schizophrenia for patient 3 would not be expected to be associated with frailty or with systemic inflammation. Furthermore, the frailty symptoms of patient 3 and inflammatory biomarkers improved after the risperidone, which was likely responsible for his microaspiration, was discontinued. All 3 patients were at risk for oropharyngeal dysphagia (antipsychotic medication is clearly associated with dysphagia20); patient 2 demonstrated pathologic evidence of DAB at autopsy.
There is evolving evidence that chronic systemic inflammation and immune activation are key mechanisms in the pathogenesis of frailty.4-6 It is known that elevated serum levels of proinflammatory cytokines, including tumor necrosis factor-α, interleukin-6, and CRP are directly associated with frailty and are inversely associated with levels of albumin, hemoglobin, insulin-like growth factor-1, and several micronutrients in frail individuals.4-7,21,22 Chronic inflammation contributes to the pathophysiology of frailty through detrimental effects on a broad range of systems, including the musculoskeletal, endocrine, and hematopoietic systems and through nutritional dysregulation.2,4,23 These changes may lead to further deleterious effects, creating a downward spiral of worsening frailty. For example, it seems likely that our patients’ progressive weakness further compromised airway protection, creating a vicious cycle of worsening microaspiration and chronic inflammation.
Conclusions
To date, the role of chronic microaspiration and DAB in chronic systemic inflammation or in frailty has not been explored. Given the prevalence of microaspiration in nursing home residents and the devastating consequences of frailty, though, this seems to be a crucial area of investigation. It is equally crucial for long-term care staff, both providers and nursing staff, to have a heightened awareness of covert microaspiration and a low threshold for referral to speech pathology for further investigation. Staff also should be aware of the utility of the Fried criteria to improve identification of frailty in general. It is probable that covert microaspiration will prove to be an important part of the differential diagnosis of frailty.
Frailty is a highly prevalent syndrome in nursing homes, occurring in at least 50% of patients.1 The frailty phenotype has been described by Fried and colleagues as impairment in ≥ 3 of 5 domains: unintentional weight loss, self-reported exhaustion, muscle weakness, slow gait speed, and low physical activity. By this definition, frailty is highly associated with poor quality of life and mortality.2,3
In recent years, there has been evolving evidence of a relationship between frailty and chronic systemic inflammation.4-6 Some degree of chronic inflammation is likely inherent to the aging process and increases the risk of frailty (so-called inflammaging) but is seen to a greater degree in many pathologic conditions in nursing homes, including cancer, organ failure, and chronic infection.4,6-8
Dysphagia also is highly prevalent in nursing homes, affecting up to 60% of patients and is a strong predictor of hospital utilization and of mortality.9,10 Overt aspiration pneumonitis and pneumonia are perhaps the best studied sequelae, but chronic occult microaspiration also is prevalent in this population.11 Just as normal systemic inflammatory changes in aging may increase vulnerability to frailty with additional illness burden, normal aging changes in swallowing function may increase vulnerability to dysphagia and to microaspiration with additional illness burden.12,13 In older adults, important risk factors for microaspiration include not only overt dysphagia, dementia, and other neurologic illnesses, but also general debility, weakness, and immobility.14
Matsuse and colleagues have described diffuse aspiration bronchiolitis (DAB) in patients with chronic microaspiration.14 DAB often goes undiagnosed.14-16 As in frailty, weight loss and chronic anemia may be seen, and many of these patients are bedridden.14,17 Episodes of macroaspiration and overt lobar pneumonia also may occur.14 Lung biopsy or autopsy reveals chronic bronchiolar inflammation and sometimes pulmonary fibrosis, but to date there have been no reports suggesting chronic systemic inflammation or elevated proinflammatory cytokines.14,15,17 We present 3 patients with progressive weight loss, functional decline, and frailty in whom chronic microaspiration likely played a significant role.
Case 1 Presentation
A 68-year-old man with a 6-year history of rapidly progressive Parkinson disease was admitted to the Haley’s Cove Community Living Center (CLC) on the James A. Haley Veterans’ Hospital campus in Tampa, Florida for long-term care. The patient’s medical history also was significant for bipolar illness and for small cell carcinoma of the lung in sustained remission.
Medications included levodopa/carbidopa 50 mg/200 mg 4 times daily, entacapone 200 mg 4 times daily, lithium carbonate 600 mg every night at bedtime, lamotrigine 150 mg daily, quetiapine 200 mg every night at bedtime, pravastatin 40 mg every night at bedtime, omeprazole 20 mg daily, tamsulosin 0.4 mg every night at bedtime, and aspirin 81 mg daily. He initially did well, but after 6 months the nursing staff began to notice the patient coughing during and after meals. Speech pathology evaluation revealed moderate oropharyngeal dysphagia, and his diet was downgraded to nectar-thickened liquids.
Over the subsequent 10 months, he became progressively weaker in physical therapy and more inactive, with about a 20-lb weight loss and mild hypoalbuminemia of 3.0 gm/dL. He had developed 3 episodes of aspiration pneumonia during this period; a repeat swallow evaluation after the last episode revealed worsened dysphagia, and his physician suggested nil per os (NPO) status and an alternative feeding route. His guardian declined placement of a percutaneous endoscopic gastrostomy (PEG) tube, he was transferred to the inpatient hospice unit, and died 2 weeks later. An autopsy was declined.
Case 2 Presentation
A 66-year-old man with a medical history of multiple traumatic brain injuries (TBIs) was admitted to the CLC for long-term care. Sequelae of the TBIs included moderate dementia, spastic paraparesis with multiple pressure injuries, a well-controlled seizure disorder, and severe oropharyngeal dysphagia with NPO status and a percutaneous endoscopic gastrostomy (PEG) tube. His medical history included TBIs and hepatitis C virus infection; medications included levetiracetam 1,000 mg twice daily, lamotrigine 25 mg twice daily, and cholecalciferol 2,000 U daily. He had multiple stage III pressure injuries and an ischial stage IV injury at the time of admission.
His 11-month stay in the CLC was characterized by progressively worsening weakness and inactivity, with a 25-lb weight loss in spite of adequate tube feeding. Serum albumin remained in the 2.0 to 2.5 gm/dL range, hemoglobin in the 7 to 9 gm/dL range without any obvious source of anemia. Most of the pressure injuries worsened during his stay in spite of aggressive wound care, and he developed a second stage IV sacral wound. A single C-reactive protein (CRP) level 2 months prior to his death was markedly elevated at 19.5 mg/dL. In spite of maintaining NPO status, he developed 3 episodes of aspiration pneumonia, all of which responded well to treatment. Ultimately, he was found pulseless and apneic and resuscitation was unsuccessful. An autopsy revealed purulent material in the small airways.
Case 3 Presentation
A 65-year-old man with a long history of paranoid schizophrenia and severe gastroesophageal reflux disease had resided in the CLC for about 10 years. Medications included risperidone microspheres 37.5 mg every 2 weeks, valproic acid 500 mg 3 times daily and 1,000 mg every night at bedtime, lansoprazole 30 mg twice daily, ranitidine 150 mg every night at bedtime, sucralfate 1,000 mg 3 times daily, simvastatin 20 mg every night at bedtime, and tamsulosin 0.4 mg every night at bedtime. He had done well for many years but developed some drooling and a modest resting tremor (but no other signs of pseudoparkinsonism) about 8 years after admission.
There had been no changes to his risperidone dosage. He also lost about 20 lb over a period of 1 year and became increasingly weak and dependent in gait, serum albumin dropped as low as 1.6 gm/dL, hemoglobin dropped to the 7 to 8 gm/dL range (without any other obvious source of anemia), and he developed a gradually worsening right-sided pleural effusion. CRP was chronically elevated at this point, in the 6 to 15 mg/dL range and as high as 17.2 mg/dL. Ultimately, he developed 3 episodes of aspiration pneumonia over a period of 2 months. Swallowing evaluation at that time revealed severe oropharyngeal dysphagia and a PEG tube was placed. Due to concerns for possible antipsychotic-induced dysphagia, risperidone was discontinued, and quetiapine 400 mg a day was substituted. He did well over the subsequent year with no further pneumonia and advancement back to a regular diet. He regained all of the lost weight and began independent ambulation. Albumin improved to the 3 gm/dL range, hemoglobin to the 12 to 13 gm/dL range, and CRP had decreased to 0.7 mg/dL. The pleural effusion (believed to have been a parapneumonic effusion) had resolved.
Discussion
All 3 patients met the Fried criteria for frailty, although there were several confounding issues.2 All 3 patients lost between 20 and 25 lb; all had clearly become weaker according to nursing and rehabilitation staff (although none were formally assessed for grip strength); and all had clear declines in their activity level. Patient 3 had a clear decrement in gait speed, but patient 1 had severe gait impairment due to Parkinson disease (although his gait in therapy had clearly worsened). Patient 2 was paraparetic and unable to ambulate. There also was evidence of limited biomarkers of systemic inflammation; all 3 patients’ albumin had decreased, and patients 2 and 3 had significant decrease in hemoglobin; but these commonplace clinical biomarkers are obviously multifactorially determined. We have limited data on our patients’ CRP levels; serial levels would have been more specific for systemic inflammation but were infrequently performed on the patients.
Multimorbidity and medical complexity are more the rule than the exception in frail geriatric patients,and it is difficult to separate the role of microaspiration from other confounding conditions that might have contributed to these patients’ evolving systemic inflammation and frailty.18 It might be argued that the decline for patient 1 was related to the underlying Parkinson disease (a progressive neurologic illness in which systemic inflammation has been reported), or that the decline of patient 2 was related to the worsening pressure injuries rather than to covert microaspiration.19 However, the TBIs for patient 2 and the schizophrenia for patient 3 would not be expected to be associated with frailty or with systemic inflammation. Furthermore, the frailty symptoms of patient 3 and inflammatory biomarkers improved after the risperidone, which was likely responsible for his microaspiration, was discontinued. All 3 patients were at risk for oropharyngeal dysphagia (antipsychotic medication is clearly associated with dysphagia20); patient 2 demonstrated pathologic evidence of DAB at autopsy.
There is evolving evidence that chronic systemic inflammation and immune activation are key mechanisms in the pathogenesis of frailty.4-6 It is known that elevated serum levels of proinflammatory cytokines, including tumor necrosis factor-α, interleukin-6, and CRP are directly associated with frailty and are inversely associated with levels of albumin, hemoglobin, insulin-like growth factor-1, and several micronutrients in frail individuals.4-7,21,22 Chronic inflammation contributes to the pathophysiology of frailty through detrimental effects on a broad range of systems, including the musculoskeletal, endocrine, and hematopoietic systems and through nutritional dysregulation.2,4,23 These changes may lead to further deleterious effects, creating a downward spiral of worsening frailty. For example, it seems likely that our patients’ progressive weakness further compromised airway protection, creating a vicious cycle of worsening microaspiration and chronic inflammation.
Conclusions
To date, the role of chronic microaspiration and DAB in chronic systemic inflammation or in frailty has not been explored. Given the prevalence of microaspiration in nursing home residents and the devastating consequences of frailty, though, this seems to be a crucial area of investigation. It is equally crucial for long-term care staff, both providers and nursing staff, to have a heightened awareness of covert microaspiration and a low threshold for referral to speech pathology for further investigation. Staff also should be aware of the utility of the Fried criteria to improve identification of frailty in general. It is probable that covert microaspiration will prove to be an important part of the differential diagnosis of frailty.
1. Kojima G. Prevalence of frailty in nursing homes: a systematic review and meta-analysis. J Am Med Dir Assoc. 2015;16(11):940-945. doi:10.1016/j.jamda.2015.06.025
2. Fried LP, Tangen CM, Walston J, et al. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci. 2001;56(3):M146-M157. doi:10.1093/gerona/56.3.m146
3. Morley JE, Vellas B, van Kan GA, et al. Frailty consensus: a call to action. J Am Med Dir Assoc. 2013;14(6):392-397. doi:10.1016/j.jamda.2013.03.022
4. Chen X, Mao G, Leng SX. Frailty syndrome: an overview. Clin Interv Aging. 2014;9:433-441. doi:10.2147/CIA.S45300.
5. Soysal P, Stubbs B, Lucato P, et al. Inflammation and frailty in the elderly: a systematic review and meta-analysis. Ageing Res Rev. 2016;31:1-8. doi:10.1016/j.arr.2016.08.006
6. Langmann GA, Perera S, Ferchak MA, Nace DA, Resnick NM, Greenspan SL. Inflammatory markers and frailty in long-term care residents. J Am Geriatr Soc. 2017;65(8):1777-1783. doi:10.1111/jgs.14876
7. Michaud M, Balardy L, Moulis G, et al. Proinflammatory cytokines, aging, and age-related diseases. J Am Med Dir Assoc. 2013;14(12):877-882. doi:10.1016/j.jamda.2013.05.009
8. Fougere B, Boulanger E, Nourhashemi F, Guyonnet S, Cesari M. Chronic inflammation: accelerator of biological aging. J Gerontol A Biol Sci Med Sci. 2017;72(9):1218-1225. doi:10.1093/gerona/glw240
9. Shanley C, O’Loughlin G. Dysphagia among nursing home residents: an assessment and management protocol. J Gerontol Nurs. 2000;26(8):35-48. doi:10.3928/0098-9134-20000801-09
10. Altman KW, Yu GP, Schaefer SD. Consequences of dysphagia in the hospitalized patient: impact on prognosis and hospital resources. Arch Otolaryngol Head Neck Surg. 2010;136(8):784-789. doi:10.1001/archoto.2010.129
11. Sakai K, Hirano H, Watanabe Y, et al. An examination of factors related to aspiration and silent aspiration in older adults requiring long-term care in rural Japan. J Oral Rehabil. 2016;43(2):103-110. doi:10.1111/joor.12349
12. Nilsson H, Ekberg O, Olsson R, Hindfelt B. Quantitative aspects of swallowing in an elderly nondysphagic population. Dysphagia. 1996;11(3):180-184. doi:10.1007/BF00366381
13. Daggett A, Logemann J, Rademaker A, Pauloski B. Laryngeal penetration during deglutition in normal subjects of various ages. Dysphagia. 2006;21(4):270-274. doi:10.1007/s00455-006-9051-6
14. Matsuse T, Oka T, Kida K, Fukuchi Y. Importance of diffuse aspiration bronchiolitis caused by chronic occult aspiration in the elderly. Chest. 1996;110(5):1289-1293. doi:10.1378/chest.110.5.1289
15. Cardasis JJ, MacMahon H, Husain AN. The spectrum of lung disease due to chronic occult aspiration. Ann Am Thorac Soc. 2014;11(6):865-873. doi:10.1513/AnnalsATS.201310-360OC
16. Pereira-Silva JL, Silva CIS, Araujo Neto CA, Andrade TL, Muller NL. Chronic pulmonary microaspiration: high-resolution computed tomographic findings in 13 patients. J Thorac Imaging. 2014;29(5):298-303. doi:10.1097/RTI.0000000000000091
17. Hu X, Lee JS, Pianosi PT, Ryu JH. Aspiration-related pulmonary syndromes. Chest. 2015;147(3):815-823. doi:10.1378/chest.14-1049
18. Yarnall AJ, Sayer AA, Clegg A, Rockwood K, Parker S, Hindle JV. New horizons in multimorbidity in older adults. Age Aging. 2017;46(6):882-888. doi:10.1093/ageing/afx150
19. Calabrese V, Santoro A, Monti D, et al. Aging and Parkinson’s disease: inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic Biol Med. 2018;115:80-91. doi:10.1016/j.freeradbiomed.2017.10.379
20. Kulkarni DP, Kamath VD, Stewart JT. Swallowing disorders in schizophrenia. Dysphagia. 2017;32(4):467-471. doi:10.1007/s00455-017-9802-6
21. Velissaris D, Pantzaris N, Koniari I, et al. C-reactive protein and frailty in the elderly: a literature review. J Clin Med Res. 2017;9(6):461-465. doi:10.14740/jocmr2959w
22. Hubbard RE, O’Mahoney MS, Savva GM, Calver BL, Woodhouse KW. Inflammation and frailty measures in older people. J Cell Mol Med. 2009;13(9B):3103-3109. doi:10.1111/j.1582-4934.2009.00733.x
23. Argiles JM, Busquets S, Stemmler B, Lotez-Soriano FJ. Cachexia and sarcopenia: mechanisms and potential targets for intervention. Curr Opin Pharmacol. 2015;22:100-106. doi:10.1016/j.coph.2015.04.003
1. Kojima G. Prevalence of frailty in nursing homes: a systematic review and meta-analysis. J Am Med Dir Assoc. 2015;16(11):940-945. doi:10.1016/j.jamda.2015.06.025
2. Fried LP, Tangen CM, Walston J, et al. Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci. 2001;56(3):M146-M157. doi:10.1093/gerona/56.3.m146
3. Morley JE, Vellas B, van Kan GA, et al. Frailty consensus: a call to action. J Am Med Dir Assoc. 2013;14(6):392-397. doi:10.1016/j.jamda.2013.03.022
4. Chen X, Mao G, Leng SX. Frailty syndrome: an overview. Clin Interv Aging. 2014;9:433-441. doi:10.2147/CIA.S45300.
5. Soysal P, Stubbs B, Lucato P, et al. Inflammation and frailty in the elderly: a systematic review and meta-analysis. Ageing Res Rev. 2016;31:1-8. doi:10.1016/j.arr.2016.08.006
6. Langmann GA, Perera S, Ferchak MA, Nace DA, Resnick NM, Greenspan SL. Inflammatory markers and frailty in long-term care residents. J Am Geriatr Soc. 2017;65(8):1777-1783. doi:10.1111/jgs.14876
7. Michaud M, Balardy L, Moulis G, et al. Proinflammatory cytokines, aging, and age-related diseases. J Am Med Dir Assoc. 2013;14(12):877-882. doi:10.1016/j.jamda.2013.05.009
8. Fougere B, Boulanger E, Nourhashemi F, Guyonnet S, Cesari M. Chronic inflammation: accelerator of biological aging. J Gerontol A Biol Sci Med Sci. 2017;72(9):1218-1225. doi:10.1093/gerona/glw240
9. Shanley C, O’Loughlin G. Dysphagia among nursing home residents: an assessment and management protocol. J Gerontol Nurs. 2000;26(8):35-48. doi:10.3928/0098-9134-20000801-09
10. Altman KW, Yu GP, Schaefer SD. Consequences of dysphagia in the hospitalized patient: impact on prognosis and hospital resources. Arch Otolaryngol Head Neck Surg. 2010;136(8):784-789. doi:10.1001/archoto.2010.129
11. Sakai K, Hirano H, Watanabe Y, et al. An examination of factors related to aspiration and silent aspiration in older adults requiring long-term care in rural Japan. J Oral Rehabil. 2016;43(2):103-110. doi:10.1111/joor.12349
12. Nilsson H, Ekberg O, Olsson R, Hindfelt B. Quantitative aspects of swallowing in an elderly nondysphagic population. Dysphagia. 1996;11(3):180-184. doi:10.1007/BF00366381
13. Daggett A, Logemann J, Rademaker A, Pauloski B. Laryngeal penetration during deglutition in normal subjects of various ages. Dysphagia. 2006;21(4):270-274. doi:10.1007/s00455-006-9051-6
14. Matsuse T, Oka T, Kida K, Fukuchi Y. Importance of diffuse aspiration bronchiolitis caused by chronic occult aspiration in the elderly. Chest. 1996;110(5):1289-1293. doi:10.1378/chest.110.5.1289
15. Cardasis JJ, MacMahon H, Husain AN. The spectrum of lung disease due to chronic occult aspiration. Ann Am Thorac Soc. 2014;11(6):865-873. doi:10.1513/AnnalsATS.201310-360OC
16. Pereira-Silva JL, Silva CIS, Araujo Neto CA, Andrade TL, Muller NL. Chronic pulmonary microaspiration: high-resolution computed tomographic findings in 13 patients. J Thorac Imaging. 2014;29(5):298-303. doi:10.1097/RTI.0000000000000091
17. Hu X, Lee JS, Pianosi PT, Ryu JH. Aspiration-related pulmonary syndromes. Chest. 2015;147(3):815-823. doi:10.1378/chest.14-1049
18. Yarnall AJ, Sayer AA, Clegg A, Rockwood K, Parker S, Hindle JV. New horizons in multimorbidity in older adults. Age Aging. 2017;46(6):882-888. doi:10.1093/ageing/afx150
19. Calabrese V, Santoro A, Monti D, et al. Aging and Parkinson’s disease: inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic Biol Med. 2018;115:80-91. doi:10.1016/j.freeradbiomed.2017.10.379
20. Kulkarni DP, Kamath VD, Stewart JT. Swallowing disorders in schizophrenia. Dysphagia. 2017;32(4):467-471. doi:10.1007/s00455-017-9802-6
21. Velissaris D, Pantzaris N, Koniari I, et al. C-reactive protein and frailty in the elderly: a literature review. J Clin Med Res. 2017;9(6):461-465. doi:10.14740/jocmr2959w
22. Hubbard RE, O’Mahoney MS, Savva GM, Calver BL, Woodhouse KW. Inflammation and frailty measures in older people. J Cell Mol Med. 2009;13(9B):3103-3109. doi:10.1111/j.1582-4934.2009.00733.x
23. Argiles JM, Busquets S, Stemmler B, Lotez-Soriano FJ. Cachexia and sarcopenia: mechanisms and potential targets for intervention. Curr Opin Pharmacol. 2015;22:100-106. doi:10.1016/j.coph.2015.04.003
Pan-Pseudothrombocytopenia in COVID-19: A Harbinger for Lethal Arterial Thrombosis?
In late 2019 a new pandemic started in Wuhan, China, caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) due to its similarities with the virus responsible for the SARS outbreak of 2003. The disease manifestations are named coronavirus disease 2019 (COVID-19).1
Pseudothrombocytopenia, or platelet clumping, visualized on the peripheral blood smear, is a common cause for artificial thrombocytopenia laboratory reporting and is frequently attributed to laboratory artifact. In this case presentation, a critically ill patient with COVID-19 developed pan-pseudothrombocytopenia (ethylenediaminetetraacetic acid [EDTA], sodium citrate, and heparin tubes) just prior to his death from a ST-segment elevation myocardial infarction (STEMI) in the setting of therapeutic anticoagulation during a prolonged hospitalization. This case raises the possibility that pseudothrombocytopenia in the setting of COVID-19 critical illness may represent an ominous feature of COVID-19-associated coagulopathy (CAC). Furthermore, it prompts the question whether pseudothrombocytopenia in this setting is representative of increased platelet aggregation activity in vivo.
Case Presentation
A 50-year-old African American man who was diagnosed with COVID-19 3 days prior to admission presented to the emergency department of the W.G. (Bill) Hefner VA Medical Center in Salisbury, North Carolina, with worsening dyspnea and fever. His primary chronic medical problems included obesity (body mass index, 33), type 2 diabetes mellitus (hemoglobin A1c 2 months prior of 6.6%), migraine headaches, and obstructive sleep apnea. Shortly after presentation, his respiratory status declined, requiring intubation. He was admitted to the medical intensive care unit for further management.
Notable findings at admission included > 20 mcg/mL FEU D-dimer (normal range, 0-0.56 mcg/mL FEU), 20.4 mg/dL C-reactive protein (normal range, < 1 mg/dL), 30 mm/h erythrocyte sedimentation rate (normal range, 0-25 mm/h), and 3.56 ng/mL procalcitonin (normal range, 0.05-1.99 ng/mL). Patient’s hemoglobin and platelet counts were normal. Empiric antimicrobial therapy was initiated with ceftriaxone (2 g IV daily) and doxycycline (100 mg IV twice daily) due to concern of superimposed infection in the setting of an elevated procalcitonin.
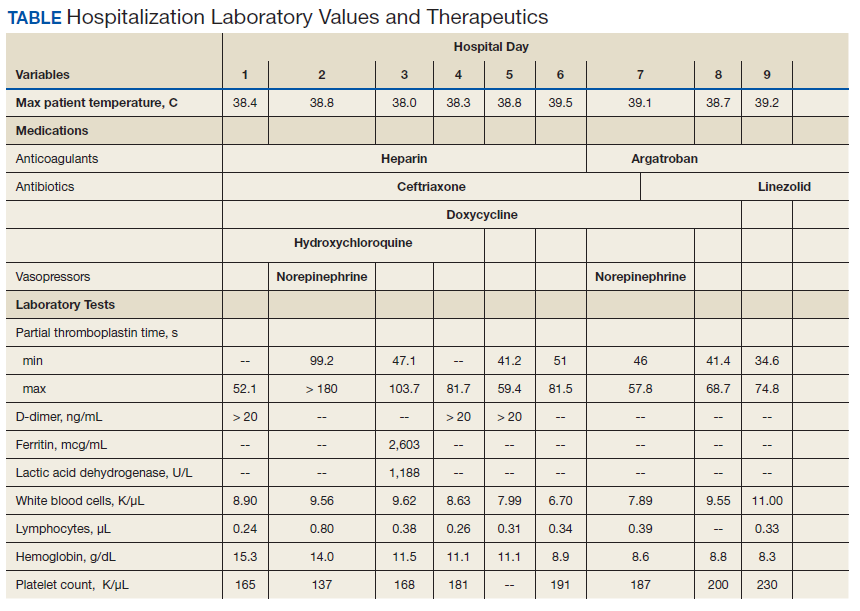
A heparin infusion was initiated (5,000 U IV bolus followed by continuous infusion with goal partial thromboplastin time [PTT] of 1.5x the upper limit of normal) on admission to treat CAC. Renal function worsened requiring intermittent renal replacement therapy on day 3. His lactate dehydrogenase was elevated to 1,188 U/L (normal range: 100-240 U/L) and ferritin was elevated to 2,603 ng/mL (normal range: 25-350 ng/mL) (Table). Initial neuromuscular blockade and prone positioning maneuvers were instituted to optimize oxygenation based on the latest literature for respiratory distress in the COVID-19 management.2
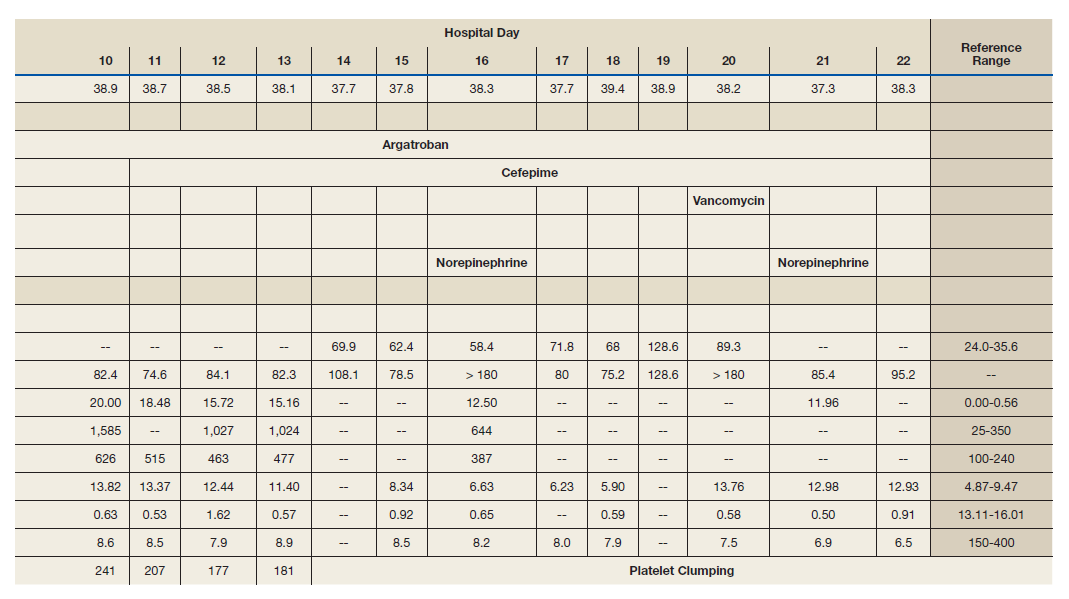
Intermittent norepinephrine infusion (5 mcg/min with a 2 mcg/min titration every 5 minutes as needed to maintain mean arterial pressure of > 65 mm Hg) was required for hemodynamic support throughout the patient’s course. Several therapies for COVID-19 were considered and were a reflection of the rapidly evolving literature during the care of patients with this disease. The patient originally received hydroxychloroquine (200 mg by mouth twice daily) in accordance with the US Department of Veterans Affairs (VA) institutional protocol between day 2 and day 4; however, hydroxychloroquine was stopped due to concerns of QTc prolongation. The patient also received 1 unit of convalescent plasma on day 6 after being enrolled in the expanded access program.3 The patient was not a candidate for remdesivir due to his unstable renal function and need for vasopressors. Finally, interleukin-6 inhibitors also were considered; however, the risk of superimposed infection precluded its use.
On day 7 antimicrobial therapy was transitioned to linezolid (600 mg IV twice daily) due to the persistence of fever and a portable chest radiograph revealing diffuse infiltrates throughout the bilateral lungs, worse compared with prior radiograph on day 5, suggesting a worsening of pneumonia. On day 12, the patient was transitioned to cefepime (1 gram IV daily) to broaden antimicrobial coverage and was continued thereafter. Blood cultures were negative throughout his hospitalization.
Given his worsening clinical scenario there was a question about whether or not the patient was still shedding virus for prognostic and therapeutic implications. Therefore, his SARS-CoV-2 test by polymerase chain reaction nasopharyngeal was positive again on day 18. On day 20, the patient developed leukocytosis, his fever persisted, and a portable chest radiograph revealed extensive bilateral pulmonary opacities with focal worsening in left lower base. Due to this constellation of findings, a vancomycin IV (1,500 mg once) was started for empirical treatment of hospital-acquired pneumonia. Sputum samples obtained on day 20 revealed Staphylococcus aureus on subsequent days.
From a hematologic perspective, on day 9 due to challenges to maintain a therapeutic level of anticoagulation with heparin infusion thought to be related to antithrombin deficiency, anticoagulation was changed to argatroban infusion (0.5 mcg/kg/min targeting a PTT of 70-105 seconds) for ongoing management of CAC. Although D-dimer was > 20 mcg/mL FEU on admission and on days 4 and 5, D-dimer trended down to 12.5 mcg/mL FEU on day 16.
Throughout the patient’s hospital stay, no significant bleeding was seen. Hemoglobin was 15.2 g/dL on admission, but anemia developed with a nadir of 6.5 g/dL, warranting transfusion of red blood cells on day 22. Platelet count was 165,000 per microliter on admission and remained within normal limits until platelet clumping was noted on day 15 laboratory collection.
Hematology was consulted on day 20 to obtain an accurate platelet count. A peripheral blood smear from a sodium citrate containing tube was remarkable for prominent platelet clumping, particularly at the periphery of the slide (Figure 1). Platelet clumping was reproduced in samples containing EDTA and heparin. Other features of the peripheral blood smear included the presence of echinocytes with rare schistocytes. To investigate for presence of disseminated intravascular coagulation on day 22, fibrinogen was found to be mildly elevated at 538 mg/dL (normal range: 243-517 mg/dL) and a D-dimer value of 11.96 mcg/mL FEU.
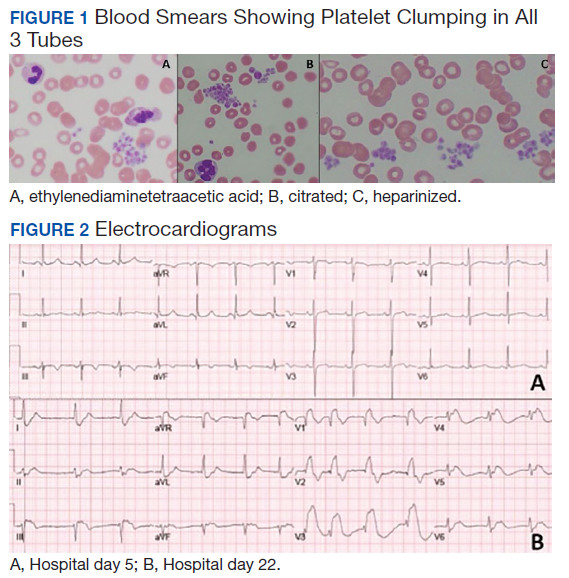
On day 22, the patient’s ventilator requirements escalated to requiring 100% FiO2 and 10 cm H20 of positive end-expiratory pressure with mean arterial pressures in the 50 to 60 mm Hg range. Within 30 minutes an electrocardiogram (EKG) obtained revealed a STEMI (Figure 2). Troponin was measured at 0.65 ng/mL (normal range: 0.02-0.06 ng/mL). Just after an EKG was performed, the patient developed a ventricular fibrillation arrest and was unable to obtain return of spontaneous circulation. The patient was pronounced dead. The family declined an autopsy.
Discussion
Pseudothrombocytopenia, or platelet clumping (agglutination), is estimated to be present in up to 2% of hospitalized patients.4 Pseudothrombocytopenia was found to be the root cause of thrombocytopenia hematology consultations in up to 4% of hospitalized patients.5 The etiology is commonly ascribed to EDTA inducing a conformational change in the GpIIb-IIIa platelet complex, rendering it susceptible to binding of autoantibodies, which cause subsequent platelet agglutination.6 In most cases (83%), the use of a non-EDTA anticoagulant, such as sodium citrate, resolves the platelet agglutination and allows for accurate platelet count reporting.4 Pseudothrombocytopenia in most cases is considered an in vitro finding without clinical relevance.7 However, in this patient’s case, his pan-pseudothrombocytopenia was temporally associated with an arterial occlusive event (STEMI) leading to his demise despite therapeutic anticoagulation in the setting of CAC. This temporal association raises the possibility that pseudothrombocytopenia seen on the peripheral blood smear is an accurate representation of in vivo activity.
Pseudothrombocytopenia has been associated with sepsis from bacterial and viral causes as well as autoimmune and medication effect.4,8-10 Li and colleagues reported transient EDTA-dependent pseudothrombocytopenia in a patient with COVID-19 infection; however, platelet clumping resolved with use of a citrate tube, and the EDTA-dependent pseudothrombocytopenia phenomenon resolved with patient recovery.11 The frequency of COVID-19-related pseudothrombocytopenia is currently unknown.
Although the understanding of COVID-19-associated CAC continues to evolve, it seems that initial reports support the idea that hemostatic dysfunction tends to more thrombosis than to bleeding.12 Rather than overt disseminated intravascular coagulation with reduced fibrinogen and bleeding, CAC is more closely associated with blood clotting, as demonstrated by autopsy studies revealing microvascular thrombosis in the lungs.13 The D-dimer test has been identified as the most useful biomarker by the International Society of Thrombosis and Hemostasis to screen for CAC and stratify patients who warrant admission or closer monitoring.12 Other identified features of CAC include prolonged prothrombin time and thrombocytopenia.12
There have been varying clinical approaches to CAC management. A retrospective review found that prophylactic heparin doses were associated with improved mortality in those with elevated D-dimer > 3.0 mg/L.14 There continues to be a diversity of varying clinical approaches with many medical centers advocating for an intensified prophylactic twice daily low molecular-weight heparin compared with others advocating for full therapeutic dose anticoagulation for patients with elevated D-dimer.15 This patient was treated aggressively with full-dose anticoagulation, and despite his having a down-trend in D-dimer, he suffered a lethal arterial thrombosis in the form of a STEMI.
Varatharajah and Rajah
Conclusions
This patient’s case highlights the presence of pan-pseudothrombocytopenia despite the use of a sodium citrate and heparin containing tube in a COVID-19 infection with multiorgan dysfunction. This developed 1 week prior to the patient suffering a STEMI despite therapeutic anticoagulation. Although the exact nature of CAC remains to be worked out, it is possible that platelet agglutination/clumping seen on the peripheral blood smear is representative of in vivo activity and serves as a harbinger for worsening thrombosis. The frequency of such phenomenon and efficacy of further interventions has yet to be explored.
1. World Health Organization. Naming the coronavirus disease (COVID-19) and the virus that causes it. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/naming-the-coronavirus-disease-(COVID-2019)-and-the-virus-that-causes-it. Accessed July 15, 2020.
2. Ghelichkhani P, Esmaeili M. Prone position in management of COVID-19 patients; a commentary. Arch Acad Emerg Med. 2020;8(1):e48. Published 2020 April 11.
3. National Library of Medicine, Clinicaltrials.gov. Expanded access to convalescent plasma for the treatment of patients with COVID-19. NCT04338360. https://clinicaltrials.gov/ct2/show/nct04338360. Update April 20, 2020. Accessed July 15, 2020.
4. Tan GC, Stalling M, Dennis G, Nunez M, Kahwash SB. Pseudothrombocytopenia due to platelet clumping: a case report and brief review of the literature. Case Rep Hematol. 2016;2016:3036476. doi:10.1155/2016/3036476
5. Boxer M, Biuso TJ. Etiologies of thrombocytopenia in the community hospital: the experience of 1 hematologist. Am J Med. 2020;133(5):e183-e186. doi:10.1016/j.amjmed.2019.10.027
6. Fiorin F, Steffan A, Pradella P, Bizzaro N, Potenza R, De Angelis V. IgG platelet antibodies in EDTA-dependent pseudothrombocytopenia bind to platelet membrane glycoprotein IIb. Am J Clin Pathol. 1998;110(2):178-183. doi:10.1093/ajcp/110.2.178
7. Nagler M, Keller P, Siegrist S, Alberio L. A case of EDTA-Dependent pseudothrombocytopenia: simple recognition of an underdiagnosed and misleading phenomenon. BMC Clin Pathol. 2014;14:19. doi:10.1186/1472-6890-14-19
8. Mori M, Kudo H, Yoshitake S, Ito K, Shinguu C, Noguchi T. Transient EDTA-dependent pseudothrombocytopenia in a patient with sepsis. Intensive Care Med. 2000;26(2):218-220. doi:10.1007/s001340050050.
9. Choe W-H, Cho Y-U, Chae J-D, Kim S-H. 2013. Pseudothrombocytopenia or platelet clumping as a possible cause of low platelet count in patients with viral infection: a case series from single institution focusing on hepatitis A virus infection. Int J Lab Hematol. 2013;35(1):70-76. doi:10.1111/j.1751-553x.2012.01466.
10. Hsieh AT, Chao TY, Chen YC. Pseudothrombocytopenia associated with infectious mononucleosis. Arch Pathol Lab Med. 2003;127(1):e17-e18. doi:10.1043/0003-9985(2003)1272.0.CO;2
11. Li H, Wang B, Ning L, Luo Y, Xiang S. Transient appearance of EDTA dependent pseudothrombocytopenia in a patient with 2019 novel coronavirus pneumonia [published online ahead of print, 2020 May 5]. Platelets. 2020;1-2. doi:10.1080/09537104.2020.1760231
12. Thachil J, Tang N, Gando S, et al. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J Thromb Haemost. 2020;18(5):1023-1026. doi:10.1111/jth.14810
13. Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1-13. doi:10.1016/j.trsl.2020.04.007
14. Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020;18(5):1094-1099. doi:10.1111/jth.14817
15. Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;125(23):2033-2040. doi.org/10.1182/blood.2020006000.
16. Varatharajah N, Rajah S. Microthrombotic complications of COVID-19 are likely due to embolism of circulating endothelial derived ultralarge von Willebrand factor (eULVWF) Decorated-Platelet Strings. Fed Pract. 2020;37(6):258-259. doi:10.12788/fp.0001
17. Bernardo A, Ball C, Nolasco L, Choi H, Moake JL, Dong JF. Platelets adhered to endothelial cell-bound ultra-large von Willebrand factor strings support leukocyte tethering and rolling under high shear stress. J Thromb Haemost. 2005;3(3):562-570. doi:10.1111/j.1538-7836.2005.01122.x
In late 2019 a new pandemic started in Wuhan, China, caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) due to its similarities with the virus responsible for the SARS outbreak of 2003. The disease manifestations are named coronavirus disease 2019 (COVID-19).1
Pseudothrombocytopenia, or platelet clumping, visualized on the peripheral blood smear, is a common cause for artificial thrombocytopenia laboratory reporting and is frequently attributed to laboratory artifact. In this case presentation, a critically ill patient with COVID-19 developed pan-pseudothrombocytopenia (ethylenediaminetetraacetic acid [EDTA], sodium citrate, and heparin tubes) just prior to his death from a ST-segment elevation myocardial infarction (STEMI) in the setting of therapeutic anticoagulation during a prolonged hospitalization. This case raises the possibility that pseudothrombocytopenia in the setting of COVID-19 critical illness may represent an ominous feature of COVID-19-associated coagulopathy (CAC). Furthermore, it prompts the question whether pseudothrombocytopenia in this setting is representative of increased platelet aggregation activity in vivo.
Case Presentation
A 50-year-old African American man who was diagnosed with COVID-19 3 days prior to admission presented to the emergency department of the W.G. (Bill) Hefner VA Medical Center in Salisbury, North Carolina, with worsening dyspnea and fever. His primary chronic medical problems included obesity (body mass index, 33), type 2 diabetes mellitus (hemoglobin A1c 2 months prior of 6.6%), migraine headaches, and obstructive sleep apnea. Shortly after presentation, his respiratory status declined, requiring intubation. He was admitted to the medical intensive care unit for further management.
Notable findings at admission included > 20 mcg/mL FEU D-dimer (normal range, 0-0.56 mcg/mL FEU), 20.4 mg/dL C-reactive protein (normal range, < 1 mg/dL), 30 mm/h erythrocyte sedimentation rate (normal range, 0-25 mm/h), and 3.56 ng/mL procalcitonin (normal range, 0.05-1.99 ng/mL). Patient’s hemoglobin and platelet counts were normal. Empiric antimicrobial therapy was initiated with ceftriaxone (2 g IV daily) and doxycycline (100 mg IV twice daily) due to concern of superimposed infection in the setting of an elevated procalcitonin.
A heparin infusion was initiated (5,000 U IV bolus followed by continuous infusion with goal partial thromboplastin time [PTT] of 1.5x the upper limit of normal) on admission to treat CAC. Renal function worsened requiring intermittent renal replacement therapy on day 3. His lactate dehydrogenase was elevated to 1,188 U/L (normal range: 100-240 U/L) and ferritin was elevated to 2,603 ng/mL (normal range: 25-350 ng/mL) (Table). Initial neuromuscular blockade and prone positioning maneuvers were instituted to optimize oxygenation based on the latest literature for respiratory distress in the COVID-19 management.2
Intermittent norepinephrine infusion (5 mcg/min with a 2 mcg/min titration every 5 minutes as needed to maintain mean arterial pressure of > 65 mm Hg) was required for hemodynamic support throughout the patient’s course. Several therapies for COVID-19 were considered and were a reflection of the rapidly evolving literature during the care of patients with this disease. The patient originally received hydroxychloroquine (200 mg by mouth twice daily) in accordance with the US Department of Veterans Affairs (VA) institutional protocol between day 2 and day 4; however, hydroxychloroquine was stopped due to concerns of QTc prolongation. The patient also received 1 unit of convalescent plasma on day 6 after being enrolled in the expanded access program.3 The patient was not a candidate for remdesivir due to his unstable renal function and need for vasopressors. Finally, interleukin-6 inhibitors also were considered; however, the risk of superimposed infection precluded its use.
On day 7 antimicrobial therapy was transitioned to linezolid (600 mg IV twice daily) due to the persistence of fever and a portable chest radiograph revealing diffuse infiltrates throughout the bilateral lungs, worse compared with prior radiograph on day 5, suggesting a worsening of pneumonia. On day 12, the patient was transitioned to cefepime (1 gram IV daily) to broaden antimicrobial coverage and was continued thereafter. Blood cultures were negative throughout his hospitalization.
Given his worsening clinical scenario there was a question about whether or not the patient was still shedding virus for prognostic and therapeutic implications. Therefore, his SARS-CoV-2 test by polymerase chain reaction nasopharyngeal was positive again on day 18. On day 20, the patient developed leukocytosis, his fever persisted, and a portable chest radiograph revealed extensive bilateral pulmonary opacities with focal worsening in left lower base. Due to this constellation of findings, a vancomycin IV (1,500 mg once) was started for empirical treatment of hospital-acquired pneumonia. Sputum samples obtained on day 20 revealed Staphylococcus aureus on subsequent days.
From a hematologic perspective, on day 9 due to challenges to maintain a therapeutic level of anticoagulation with heparin infusion thought to be related to antithrombin deficiency, anticoagulation was changed to argatroban infusion (0.5 mcg/kg/min targeting a PTT of 70-105 seconds) for ongoing management of CAC. Although D-dimer was > 20 mcg/mL FEU on admission and on days 4 and 5, D-dimer trended down to 12.5 mcg/mL FEU on day 16.
Throughout the patient’s hospital stay, no significant bleeding was seen. Hemoglobin was 15.2 g/dL on admission, but anemia developed with a nadir of 6.5 g/dL, warranting transfusion of red blood cells on day 22. Platelet count was 165,000 per microliter on admission and remained within normal limits until platelet clumping was noted on day 15 laboratory collection.
Hematology was consulted on day 20 to obtain an accurate platelet count. A peripheral blood smear from a sodium citrate containing tube was remarkable for prominent platelet clumping, particularly at the periphery of the slide (Figure 1). Platelet clumping was reproduced in samples containing EDTA and heparin. Other features of the peripheral blood smear included the presence of echinocytes with rare schistocytes. To investigate for presence of disseminated intravascular coagulation on day 22, fibrinogen was found to be mildly elevated at 538 mg/dL (normal range: 243-517 mg/dL) and a D-dimer value of 11.96 mcg/mL FEU.
On day 22, the patient’s ventilator requirements escalated to requiring 100% FiO2 and 10 cm H20 of positive end-expiratory pressure with mean arterial pressures in the 50 to 60 mm Hg range. Within 30 minutes an electrocardiogram (EKG) obtained revealed a STEMI (Figure 2). Troponin was measured at 0.65 ng/mL (normal range: 0.02-0.06 ng/mL). Just after an EKG was performed, the patient developed a ventricular fibrillation arrest and was unable to obtain return of spontaneous circulation. The patient was pronounced dead. The family declined an autopsy.
Discussion
Pseudothrombocytopenia, or platelet clumping (agglutination), is estimated to be present in up to 2% of hospitalized patients.4 Pseudothrombocytopenia was found to be the root cause of thrombocytopenia hematology consultations in up to 4% of hospitalized patients.5 The etiology is commonly ascribed to EDTA inducing a conformational change in the GpIIb-IIIa platelet complex, rendering it susceptible to binding of autoantibodies, which cause subsequent platelet agglutination.6 In most cases (83%), the use of a non-EDTA anticoagulant, such as sodium citrate, resolves the platelet agglutination and allows for accurate platelet count reporting.4 Pseudothrombocytopenia in most cases is considered an in vitro finding without clinical relevance.7 However, in this patient’s case, his pan-pseudothrombocytopenia was temporally associated with an arterial occlusive event (STEMI) leading to his demise despite therapeutic anticoagulation in the setting of CAC. This temporal association raises the possibility that pseudothrombocytopenia seen on the peripheral blood smear is an accurate representation of in vivo activity.
Pseudothrombocytopenia has been associated with sepsis from bacterial and viral causes as well as autoimmune and medication effect.4,8-10 Li and colleagues reported transient EDTA-dependent pseudothrombocytopenia in a patient with COVID-19 infection; however, platelet clumping resolved with use of a citrate tube, and the EDTA-dependent pseudothrombocytopenia phenomenon resolved with patient recovery.11 The frequency of COVID-19-related pseudothrombocytopenia is currently unknown.
Although the understanding of COVID-19-associated CAC continues to evolve, it seems that initial reports support the idea that hemostatic dysfunction tends to more thrombosis than to bleeding.12 Rather than overt disseminated intravascular coagulation with reduced fibrinogen and bleeding, CAC is more closely associated with blood clotting, as demonstrated by autopsy studies revealing microvascular thrombosis in the lungs.13 The D-dimer test has been identified as the most useful biomarker by the International Society of Thrombosis and Hemostasis to screen for CAC and stratify patients who warrant admission or closer monitoring.12 Other identified features of CAC include prolonged prothrombin time and thrombocytopenia.12
There have been varying clinical approaches to CAC management. A retrospective review found that prophylactic heparin doses were associated with improved mortality in those with elevated D-dimer > 3.0 mg/L.14 There continues to be a diversity of varying clinical approaches with many medical centers advocating for an intensified prophylactic twice daily low molecular-weight heparin compared with others advocating for full therapeutic dose anticoagulation for patients with elevated D-dimer.15 This patient was treated aggressively with full-dose anticoagulation, and despite his having a down-trend in D-dimer, he suffered a lethal arterial thrombosis in the form of a STEMI.
Varatharajah and Rajah
Conclusions
This patient’s case highlights the presence of pan-pseudothrombocytopenia despite the use of a sodium citrate and heparin containing tube in a COVID-19 infection with multiorgan dysfunction. This developed 1 week prior to the patient suffering a STEMI despite therapeutic anticoagulation. Although the exact nature of CAC remains to be worked out, it is possible that platelet agglutination/clumping seen on the peripheral blood smear is representative of in vivo activity and serves as a harbinger for worsening thrombosis. The frequency of such phenomenon and efficacy of further interventions has yet to be explored.
In late 2019 a new pandemic started in Wuhan, China, caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) due to its similarities with the virus responsible for the SARS outbreak of 2003. The disease manifestations are named coronavirus disease 2019 (COVID-19).1
Pseudothrombocytopenia, or platelet clumping, visualized on the peripheral blood smear, is a common cause for artificial thrombocytopenia laboratory reporting and is frequently attributed to laboratory artifact. In this case presentation, a critically ill patient with COVID-19 developed pan-pseudothrombocytopenia (ethylenediaminetetraacetic acid [EDTA], sodium citrate, and heparin tubes) just prior to his death from a ST-segment elevation myocardial infarction (STEMI) in the setting of therapeutic anticoagulation during a prolonged hospitalization. This case raises the possibility that pseudothrombocytopenia in the setting of COVID-19 critical illness may represent an ominous feature of COVID-19-associated coagulopathy (CAC). Furthermore, it prompts the question whether pseudothrombocytopenia in this setting is representative of increased platelet aggregation activity in vivo.
Case Presentation
A 50-year-old African American man who was diagnosed with COVID-19 3 days prior to admission presented to the emergency department of the W.G. (Bill) Hefner VA Medical Center in Salisbury, North Carolina, with worsening dyspnea and fever. His primary chronic medical problems included obesity (body mass index, 33), type 2 diabetes mellitus (hemoglobin A1c 2 months prior of 6.6%), migraine headaches, and obstructive sleep apnea. Shortly after presentation, his respiratory status declined, requiring intubation. He was admitted to the medical intensive care unit for further management.
Notable findings at admission included > 20 mcg/mL FEU D-dimer (normal range, 0-0.56 mcg/mL FEU), 20.4 mg/dL C-reactive protein (normal range, < 1 mg/dL), 30 mm/h erythrocyte sedimentation rate (normal range, 0-25 mm/h), and 3.56 ng/mL procalcitonin (normal range, 0.05-1.99 ng/mL). Patient’s hemoglobin and platelet counts were normal. Empiric antimicrobial therapy was initiated with ceftriaxone (2 g IV daily) and doxycycline (100 mg IV twice daily) due to concern of superimposed infection in the setting of an elevated procalcitonin.
A heparin infusion was initiated (5,000 U IV bolus followed by continuous infusion with goal partial thromboplastin time [PTT] of 1.5x the upper limit of normal) on admission to treat CAC. Renal function worsened requiring intermittent renal replacement therapy on day 3. His lactate dehydrogenase was elevated to 1,188 U/L (normal range: 100-240 U/L) and ferritin was elevated to 2,603 ng/mL (normal range: 25-350 ng/mL) (Table). Initial neuromuscular blockade and prone positioning maneuvers were instituted to optimize oxygenation based on the latest literature for respiratory distress in the COVID-19 management.2
Intermittent norepinephrine infusion (5 mcg/min with a 2 mcg/min titration every 5 minutes as needed to maintain mean arterial pressure of > 65 mm Hg) was required for hemodynamic support throughout the patient’s course. Several therapies for COVID-19 were considered and were a reflection of the rapidly evolving literature during the care of patients with this disease. The patient originally received hydroxychloroquine (200 mg by mouth twice daily) in accordance with the US Department of Veterans Affairs (VA) institutional protocol between day 2 and day 4; however, hydroxychloroquine was stopped due to concerns of QTc prolongation. The patient also received 1 unit of convalescent plasma on day 6 after being enrolled in the expanded access program.3 The patient was not a candidate for remdesivir due to his unstable renal function and need for vasopressors. Finally, interleukin-6 inhibitors also were considered; however, the risk of superimposed infection precluded its use.
On day 7 antimicrobial therapy was transitioned to linezolid (600 mg IV twice daily) due to the persistence of fever and a portable chest radiograph revealing diffuse infiltrates throughout the bilateral lungs, worse compared with prior radiograph on day 5, suggesting a worsening of pneumonia. On day 12, the patient was transitioned to cefepime (1 gram IV daily) to broaden antimicrobial coverage and was continued thereafter. Blood cultures were negative throughout his hospitalization.
Given his worsening clinical scenario there was a question about whether or not the patient was still shedding virus for prognostic and therapeutic implications. Therefore, his SARS-CoV-2 test by polymerase chain reaction nasopharyngeal was positive again on day 18. On day 20, the patient developed leukocytosis, his fever persisted, and a portable chest radiograph revealed extensive bilateral pulmonary opacities with focal worsening in left lower base. Due to this constellation of findings, a vancomycin IV (1,500 mg once) was started for empirical treatment of hospital-acquired pneumonia. Sputum samples obtained on day 20 revealed Staphylococcus aureus on subsequent days.
From a hematologic perspective, on day 9 due to challenges to maintain a therapeutic level of anticoagulation with heparin infusion thought to be related to antithrombin deficiency, anticoagulation was changed to argatroban infusion (0.5 mcg/kg/min targeting a PTT of 70-105 seconds) for ongoing management of CAC. Although D-dimer was > 20 mcg/mL FEU on admission and on days 4 and 5, D-dimer trended down to 12.5 mcg/mL FEU on day 16.
Throughout the patient’s hospital stay, no significant bleeding was seen. Hemoglobin was 15.2 g/dL on admission, but anemia developed with a nadir of 6.5 g/dL, warranting transfusion of red blood cells on day 22. Platelet count was 165,000 per microliter on admission and remained within normal limits until platelet clumping was noted on day 15 laboratory collection.
Hematology was consulted on day 20 to obtain an accurate platelet count. A peripheral blood smear from a sodium citrate containing tube was remarkable for prominent platelet clumping, particularly at the periphery of the slide (Figure 1). Platelet clumping was reproduced in samples containing EDTA and heparin. Other features of the peripheral blood smear included the presence of echinocytes with rare schistocytes. To investigate for presence of disseminated intravascular coagulation on day 22, fibrinogen was found to be mildly elevated at 538 mg/dL (normal range: 243-517 mg/dL) and a D-dimer value of 11.96 mcg/mL FEU.
On day 22, the patient’s ventilator requirements escalated to requiring 100% FiO2 and 10 cm H20 of positive end-expiratory pressure with mean arterial pressures in the 50 to 60 mm Hg range. Within 30 minutes an electrocardiogram (EKG) obtained revealed a STEMI (Figure 2). Troponin was measured at 0.65 ng/mL (normal range: 0.02-0.06 ng/mL). Just after an EKG was performed, the patient developed a ventricular fibrillation arrest and was unable to obtain return of spontaneous circulation. The patient was pronounced dead. The family declined an autopsy.
Discussion
Pseudothrombocytopenia, or platelet clumping (agglutination), is estimated to be present in up to 2% of hospitalized patients.4 Pseudothrombocytopenia was found to be the root cause of thrombocytopenia hematology consultations in up to 4% of hospitalized patients.5 The etiology is commonly ascribed to EDTA inducing a conformational change in the GpIIb-IIIa platelet complex, rendering it susceptible to binding of autoantibodies, which cause subsequent platelet agglutination.6 In most cases (83%), the use of a non-EDTA anticoagulant, such as sodium citrate, resolves the platelet agglutination and allows for accurate platelet count reporting.4 Pseudothrombocytopenia in most cases is considered an in vitro finding without clinical relevance.7 However, in this patient’s case, his pan-pseudothrombocytopenia was temporally associated with an arterial occlusive event (STEMI) leading to his demise despite therapeutic anticoagulation in the setting of CAC. This temporal association raises the possibility that pseudothrombocytopenia seen on the peripheral blood smear is an accurate representation of in vivo activity.
Pseudothrombocytopenia has been associated with sepsis from bacterial and viral causes as well as autoimmune and medication effect.4,8-10 Li and colleagues reported transient EDTA-dependent pseudothrombocytopenia in a patient with COVID-19 infection; however, platelet clumping resolved with use of a citrate tube, and the EDTA-dependent pseudothrombocytopenia phenomenon resolved with patient recovery.11 The frequency of COVID-19-related pseudothrombocytopenia is currently unknown.
Although the understanding of COVID-19-associated CAC continues to evolve, it seems that initial reports support the idea that hemostatic dysfunction tends to more thrombosis than to bleeding.12 Rather than overt disseminated intravascular coagulation with reduced fibrinogen and bleeding, CAC is more closely associated with blood clotting, as demonstrated by autopsy studies revealing microvascular thrombosis in the lungs.13 The D-dimer test has been identified as the most useful biomarker by the International Society of Thrombosis and Hemostasis to screen for CAC and stratify patients who warrant admission or closer monitoring.12 Other identified features of CAC include prolonged prothrombin time and thrombocytopenia.12
There have been varying clinical approaches to CAC management. A retrospective review found that prophylactic heparin doses were associated with improved mortality in those with elevated D-dimer > 3.0 mg/L.14 There continues to be a diversity of varying clinical approaches with many medical centers advocating for an intensified prophylactic twice daily low molecular-weight heparin compared with others advocating for full therapeutic dose anticoagulation for patients with elevated D-dimer.15 This patient was treated aggressively with full-dose anticoagulation, and despite his having a down-trend in D-dimer, he suffered a lethal arterial thrombosis in the form of a STEMI.
Varatharajah and Rajah
Conclusions
This patient’s case highlights the presence of pan-pseudothrombocytopenia despite the use of a sodium citrate and heparin containing tube in a COVID-19 infection with multiorgan dysfunction. This developed 1 week prior to the patient suffering a STEMI despite therapeutic anticoagulation. Although the exact nature of CAC remains to be worked out, it is possible that platelet agglutination/clumping seen on the peripheral blood smear is representative of in vivo activity and serves as a harbinger for worsening thrombosis. The frequency of such phenomenon and efficacy of further interventions has yet to be explored.
1. World Health Organization. Naming the coronavirus disease (COVID-19) and the virus that causes it. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/naming-the-coronavirus-disease-(COVID-2019)-and-the-virus-that-causes-it. Accessed July 15, 2020.
2. Ghelichkhani P, Esmaeili M. Prone position in management of COVID-19 patients; a commentary. Arch Acad Emerg Med. 2020;8(1):e48. Published 2020 April 11.
3. National Library of Medicine, Clinicaltrials.gov. Expanded access to convalescent plasma for the treatment of patients with COVID-19. NCT04338360. https://clinicaltrials.gov/ct2/show/nct04338360. Update April 20, 2020. Accessed July 15, 2020.
4. Tan GC, Stalling M, Dennis G, Nunez M, Kahwash SB. Pseudothrombocytopenia due to platelet clumping: a case report and brief review of the literature. Case Rep Hematol. 2016;2016:3036476. doi:10.1155/2016/3036476
5. Boxer M, Biuso TJ. Etiologies of thrombocytopenia in the community hospital: the experience of 1 hematologist. Am J Med. 2020;133(5):e183-e186. doi:10.1016/j.amjmed.2019.10.027
6. Fiorin F, Steffan A, Pradella P, Bizzaro N, Potenza R, De Angelis V. IgG platelet antibodies in EDTA-dependent pseudothrombocytopenia bind to platelet membrane glycoprotein IIb. Am J Clin Pathol. 1998;110(2):178-183. doi:10.1093/ajcp/110.2.178
7. Nagler M, Keller P, Siegrist S, Alberio L. A case of EDTA-Dependent pseudothrombocytopenia: simple recognition of an underdiagnosed and misleading phenomenon. BMC Clin Pathol. 2014;14:19. doi:10.1186/1472-6890-14-19
8. Mori M, Kudo H, Yoshitake S, Ito K, Shinguu C, Noguchi T. Transient EDTA-dependent pseudothrombocytopenia in a patient with sepsis. Intensive Care Med. 2000;26(2):218-220. doi:10.1007/s001340050050.
9. Choe W-H, Cho Y-U, Chae J-D, Kim S-H. 2013. Pseudothrombocytopenia or platelet clumping as a possible cause of low platelet count in patients with viral infection: a case series from single institution focusing on hepatitis A virus infection. Int J Lab Hematol. 2013;35(1):70-76. doi:10.1111/j.1751-553x.2012.01466.
10. Hsieh AT, Chao TY, Chen YC. Pseudothrombocytopenia associated with infectious mononucleosis. Arch Pathol Lab Med. 2003;127(1):e17-e18. doi:10.1043/0003-9985(2003)1272.0.CO;2
11. Li H, Wang B, Ning L, Luo Y, Xiang S. Transient appearance of EDTA dependent pseudothrombocytopenia in a patient with 2019 novel coronavirus pneumonia [published online ahead of print, 2020 May 5]. Platelets. 2020;1-2. doi:10.1080/09537104.2020.1760231
12. Thachil J, Tang N, Gando S, et al. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J Thromb Haemost. 2020;18(5):1023-1026. doi:10.1111/jth.14810
13. Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1-13. doi:10.1016/j.trsl.2020.04.007
14. Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020;18(5):1094-1099. doi:10.1111/jth.14817
15. Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;125(23):2033-2040. doi.org/10.1182/blood.2020006000.
16. Varatharajah N, Rajah S. Microthrombotic complications of COVID-19 are likely due to embolism of circulating endothelial derived ultralarge von Willebrand factor (eULVWF) Decorated-Platelet Strings. Fed Pract. 2020;37(6):258-259. doi:10.12788/fp.0001
17. Bernardo A, Ball C, Nolasco L, Choi H, Moake JL, Dong JF. Platelets adhered to endothelial cell-bound ultra-large von Willebrand factor strings support leukocyte tethering and rolling under high shear stress. J Thromb Haemost. 2005;3(3):562-570. doi:10.1111/j.1538-7836.2005.01122.x
1. World Health Organization. Naming the coronavirus disease (COVID-19) and the virus that causes it. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/naming-the-coronavirus-disease-(COVID-2019)-and-the-virus-that-causes-it. Accessed July 15, 2020.
2. Ghelichkhani P, Esmaeili M. Prone position in management of COVID-19 patients; a commentary. Arch Acad Emerg Med. 2020;8(1):e48. Published 2020 April 11.
3. National Library of Medicine, Clinicaltrials.gov. Expanded access to convalescent plasma for the treatment of patients with COVID-19. NCT04338360. https://clinicaltrials.gov/ct2/show/nct04338360. Update April 20, 2020. Accessed July 15, 2020.
4. Tan GC, Stalling M, Dennis G, Nunez M, Kahwash SB. Pseudothrombocytopenia due to platelet clumping: a case report and brief review of the literature. Case Rep Hematol. 2016;2016:3036476. doi:10.1155/2016/3036476
5. Boxer M, Biuso TJ. Etiologies of thrombocytopenia in the community hospital: the experience of 1 hematologist. Am J Med. 2020;133(5):e183-e186. doi:10.1016/j.amjmed.2019.10.027
6. Fiorin F, Steffan A, Pradella P, Bizzaro N, Potenza R, De Angelis V. IgG platelet antibodies in EDTA-dependent pseudothrombocytopenia bind to platelet membrane glycoprotein IIb. Am J Clin Pathol. 1998;110(2):178-183. doi:10.1093/ajcp/110.2.178
7. Nagler M, Keller P, Siegrist S, Alberio L. A case of EDTA-Dependent pseudothrombocytopenia: simple recognition of an underdiagnosed and misleading phenomenon. BMC Clin Pathol. 2014;14:19. doi:10.1186/1472-6890-14-19
8. Mori M, Kudo H, Yoshitake S, Ito K, Shinguu C, Noguchi T. Transient EDTA-dependent pseudothrombocytopenia in a patient with sepsis. Intensive Care Med. 2000;26(2):218-220. doi:10.1007/s001340050050.
9. Choe W-H, Cho Y-U, Chae J-D, Kim S-H. 2013. Pseudothrombocytopenia or platelet clumping as a possible cause of low platelet count in patients with viral infection: a case series from single institution focusing on hepatitis A virus infection. Int J Lab Hematol. 2013;35(1):70-76. doi:10.1111/j.1751-553x.2012.01466.
10. Hsieh AT, Chao TY, Chen YC. Pseudothrombocytopenia associated with infectious mononucleosis. Arch Pathol Lab Med. 2003;127(1):e17-e18. doi:10.1043/0003-9985(2003)1272.0.CO;2
11. Li H, Wang B, Ning L, Luo Y, Xiang S. Transient appearance of EDTA dependent pseudothrombocytopenia in a patient with 2019 novel coronavirus pneumonia [published online ahead of print, 2020 May 5]. Platelets. 2020;1-2. doi:10.1080/09537104.2020.1760231
12. Thachil J, Tang N, Gando S, et al. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J Thromb Haemost. 2020;18(5):1023-1026. doi:10.1111/jth.14810
13. Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1-13. doi:10.1016/j.trsl.2020.04.007
14. Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. 2020;18(5):1094-1099. doi:10.1111/jth.14817
15. Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;125(23):2033-2040. doi.org/10.1182/blood.2020006000.
16. Varatharajah N, Rajah S. Microthrombotic complications of COVID-19 are likely due to embolism of circulating endothelial derived ultralarge von Willebrand factor (eULVWF) Decorated-Platelet Strings. Fed Pract. 2020;37(6):258-259. doi:10.12788/fp.0001
17. Bernardo A, Ball C, Nolasco L, Choi H, Moake JL, Dong JF. Platelets adhered to endothelial cell-bound ultra-large von Willebrand factor strings support leukocyte tethering and rolling under high shear stress. J Thromb Haemost. 2005;3(3):562-570. doi:10.1111/j.1538-7836.2005.01122.x
Psoriasis in Patients of Color: Differences in Morphology, Clinical Presentation, and Treatment
Psoriasis is a chronic inflammatory skin disease that affects 2% to 3% of individuals worldwide.1 Despite extensive research, the majority of clinical data are in white patients with limited data in patients of color, yet a number of differences are known. The prevalence of psoriasis differs among racial and ethnic groups, with lower prevalence in racial minorities.2 A cross-sectional American study using data from 2009 through 2010 showed the prevalence for psoriasis was 3.6% in white patients, 1.9% in black patients, 1.6% in Hispanic patients, and 1.4% in other racial groups.3 Psoriasis presents differently in patients of color, both in morphology and severity. Cultural differences and stigma may contribute to the differences seen in severity but also to the psychological impact and treatment choices in patients of color compared to white patients.4 It has even been theorized that treatment efficacy could differ because of potential genetic differences.5 Psoriasis in patients of color is an emerging clinical issue that requires further attention so that dermatologists can learn about, diagnose, and treat them.
We report 3 cases of patients of color with psoriasis who presented to an urban and racially diverse dermatology clinic affiliated with Scarborough General Hospital in Toronto, Ontario, Canada. A retrospective chart review was performed on these high-yield representative cases to demonstrate differences in color and morphology, disease severity, and treatment in patients of various races seen at our clinic. After informed consent was obtained, photographs were taken of patient cutaneous findings to illustrate these differences. Discussion with these selected patients yielded supplementary qualitative data, highlighting individual perspectives of their disease.
Case Series
Patient 1
A 53-year-old black man from Grenada presented to our clinic with a history of psoriasis for a number of years that presented as violaceous plaques throughout large portions of the body (Figure 1). He previously had achieved inadequate results while using topical therapies, methotrexate, acitretin, apremilast, ustekinumab, ixekizumab, and guselkumab at adequate or even maximum doses. His disease affected 30% of the body surface area, with a psoriasis area and severity index score of 27 and a dermatology life quality index score of 23. The patient’s life was quite affected by psoriasis, with emphasis on choice of clothing worn and effect on body image. He also discussed the stigma psoriasis may have in black patients, stating that he has been told multiple times that “black people do not get psoriasis.”
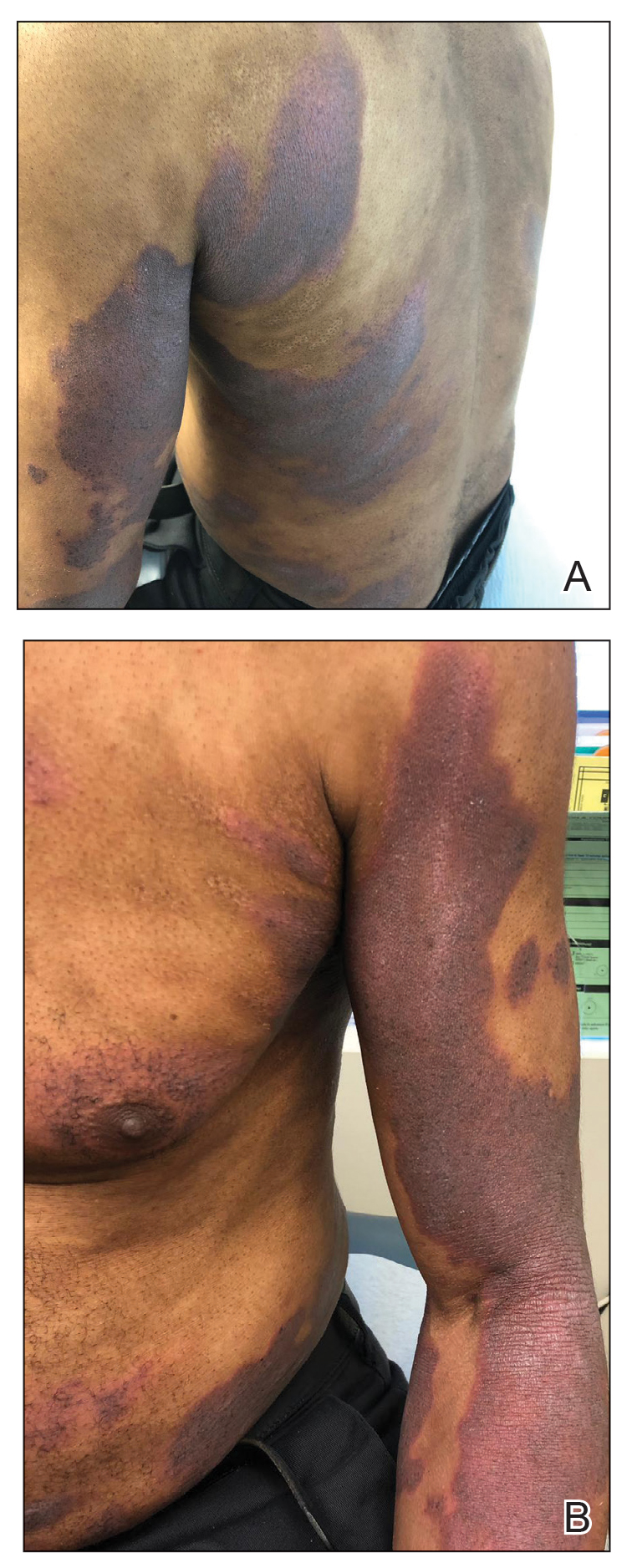
Patient 2
A 27-year-old man from India presented with guttate psoriasis (Figure 2). He was treated with methotrexate 2 years prior and currently is on maintenance therapy with topical treatments alone. His main concerns pertained to the persistent dyschromia that occurred secondary to the psoriatic lesions. Through discussion, the patient stated that he “would do anything to get rid of it.”
Patient 3
A 49-year-old man from the Philippines presented to our clinic with plaque psoriasis that predominantly affected the trunk and scalp (Figure 3). He had been treated with methotrexate and phototherapy with suboptimal efficacy and was planning for biologic therapy. Although he had active plaques on the trunk, the patient stated, “I am most bothered by my scalp,” particularly referring to the itch and scale and their effects on hair and hairstyling.
Comment
Clinical differences in patients of color with psoriasis affect the management of the disease. Special consideration should be given to variances in morphology, presentation, treatment, and psychosocial factors in the management of psoriasis for these patient populations, as summarized in the eTable.
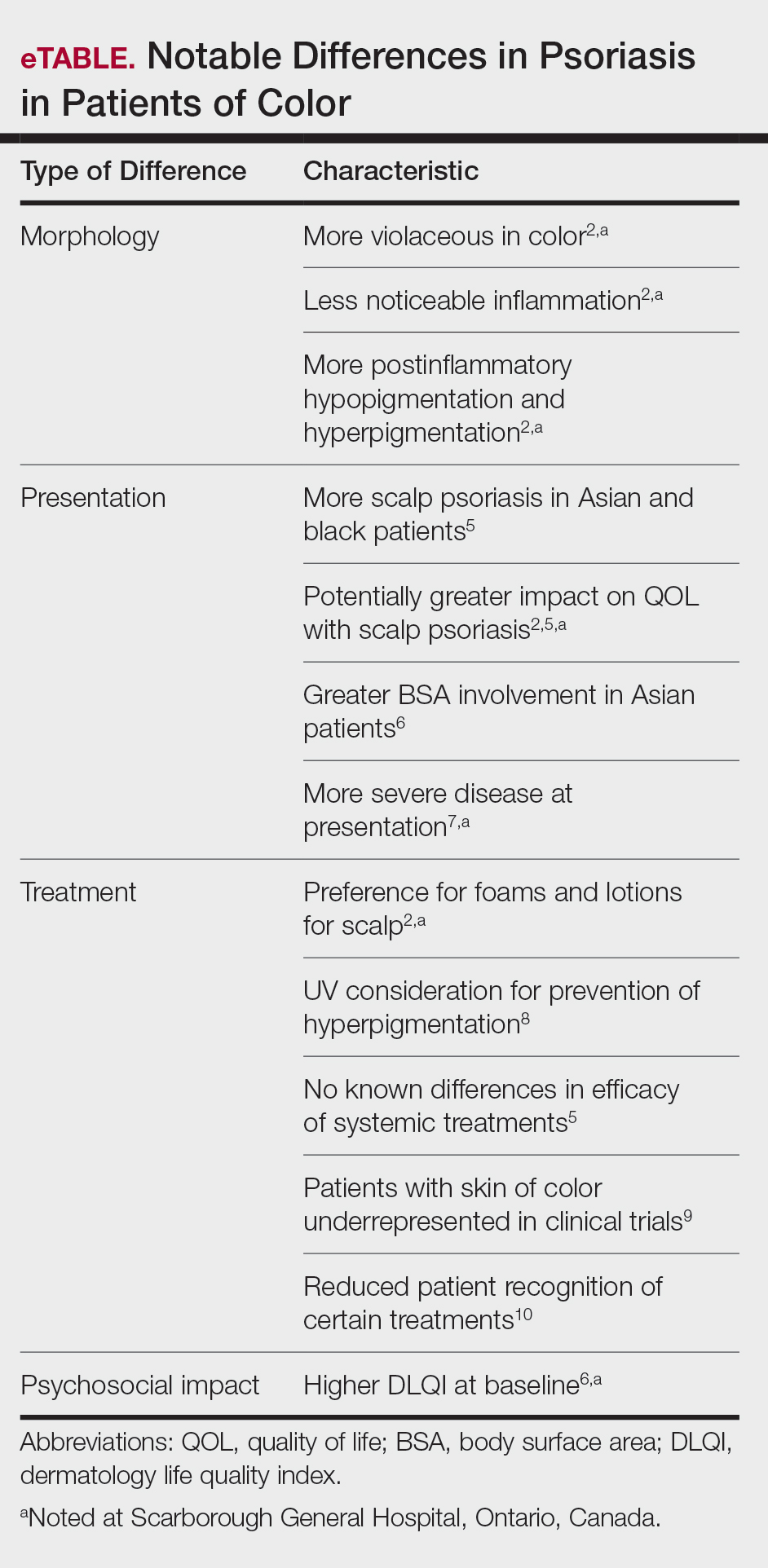
Morphology
At our clinic, patients of color have been found to have differences in morphology, including lesions that are more violaceous in color, as seen in patient 1; less noticeable inflammation; and more postinflammatory hypopigmentation and hyperpigmentation changes, as seen in patient 2. These changes are supported by the literature and differ from typical psoriasis plaques, which are pink-red and have more overlying scale. The varied morphology also may affect the differential, and other mimickers may be considered, such as lichen planus, cutaneous lupus erythematosus, and sarcoidosis.2
Presentation
There are differences in presentation among patients of color, particularly in distribution, type of psoriasis, and severity. As seen in patient 3, Asian and black patients are more likely to present with scalp psoriasis.2,5 Hairstyling and hair care practices can differ considerably between racial groups. Given the differences in hairstyling, scalp psoriasis also may have a greater impact on patient quality of life (QOL).
Racial differences affect the type of psoriasis seen. Asian patients are more likely to present with pustular and erythrodermic psoriasis and less likely to present with inverse psoriasis compared to white patients. Hispanic patients are more likely to present with pustular psoriasis.11 Black patients have been reported to have lower frequencies of psoriatic arthritis compared to white patients.12 Recognition of these differences may help guide initial choice for therapeutics.
Notably, patients of color may present with much more severe psoriasis, particularly Asian and Hispanic patients.7 One retrospective study looking at patients with psoriasis treated with etanercept found that Asian patients were more likely to have greater baseline body surface area involvement.6 An American cross-sectional study reported higher psoriasis area and severity index scores in black patients compared to white patients,12 possibly because patients of color do not normalize the experience of having psoriasis and feel stigmatized, which can cause delays in seeking medical attention and worsen disease burden. For patient 1, the stigma of black patients having psoriasis affected his body image and may have led to a delay in seeking medical attention due to him not believing it was possible for people of his skin color to have psoriasis. Increased disease severity may contribute to treatment resistance or numerous trials of topicals or biologics before the disease improves. Patient education in the community as well as patient support groups are paramount, and increased awareness of psoriasis can help improve disease management.
Treatment
Topical therapies are the first-line treatment of psoriasis. Although there is no evidence showing differences in topical treatment efficacy, patient preference for different topical treatments may vary based on race. For example, patients with Afro-textured hair may prefer foams and lotions and would avoid shampoo therapies, as frequent hair washing may not be feasible with certain hairstyles and may cause hair breakage or dryness.2
UV therapy can be an effective treatment modality for patients with psoriasis. The strength of therapy tends to be dictated by the Fitzpatrick skin phototype rather than race. Darker-skinned individuals may have an increased risk for hyperpigmentation, so caution should be taken to prevent burning during therapy. Suberythemogenic dosing—70% of minimal erythema dose—of narrowband UVB treatments has shown the same efficacy as using minimal erythema dose in patients with darker skin types in addition to fair-skinned patients.8
Although we found poor efficacy of systemic treatments in patient 1, to our knowledge, studies examining the efficacy of systemic therapeutic options have not shown differences in patients of color.6,13 Studies show similar efficacy in treatments among races, particularly biologic therapies.5 However, patients with skin of color historically have been underrepresented in clinical trials,9 which may contribute to these patients, particularly black patients, being less familiar with biologics as a treatment option for psoriasis, as reported by Takeshita et al.10 Therefore, patient-centered discussions regarding treatment choices are important to ensure patients understand all options available to manage their disease.
Psychosocial Impact
Because of its chronic remitting course, psoriasis has a notable psychosocial impact on the lives of all patients, though the literature suggests there may be more of an impact on QOL in patients of color. Higher baseline dermatology life quality index scores have been reported in patients of color compared to white patients.6 Kerr et al12 reported significantly greater psoriasis area and severity index scores (P=.06) and greater psychological impact in black patients compared to white patients. Stress also was more likely to be reported as a trigger for psoriasis in patients of Hispanic background compared to white patients.14 Many patients report body image issues with large physical lesions; however, the difference may lie in personal and cultural views about psoriasis, as one of our patients stated, “black people do not get psoriasis.” In addition to the cosmetic challenges that patients face with active lesions, postinflammatory pigmentary changes can be equally as burdensome to patients, as one of our patients stated he “would do anything to get rid of it.” Increased rates of depression and anxiety in patients of color can worsen their outlook on the condition.15,16 The increased stigma and burden of psoriasis in patients of color calls for clinicians to counsel and address psoriasis in a holistic way and refer patients to psoriasis support groups when appropriate. Although the burden of psoriasis is clear, more studies can be carried out to investigate the impact on QOL in different ethnic populations.
Dermatology Education
Although differences have been found in patients of color with psoriasis, dissemination of this knowledge continues to be a challenge. In dermatology residency programs, the majority of teaching is provided with examples of skin diseases in white patients, which can complicate pattern recognition and diagnostic ability for trainees. Although dermatologists recognize that ethnic skin has unique dermatologic considerations, there is a persistent need for increasing skin of color education within dermatology residency programs.17,18 Implementing more educational programs on skin of color has been proposed, and these programs will continue to be in demand as our population increasingly diversifies.19
Conclusion
Psoriasis in patients of color carries unique challenges when compared to psoriasis in white patients. Differences in morphology and presentation can make the disease difficult to accurately diagnose. These differences in addition to cultural differences may contribute to a greater impact on QOL and psychological health. Although treatment preferences and recognition may differ, treatment efficacy has so far been similar, albeit with a low proportion of patients with skin of color included in clinical trials.
Further focus should now lie within knowledge translation of these differences, which would normalize the condition for patients, support them seeking medical attention sooner, and inform them of all treatment options possible. For clinicians, more attention on the differences would help make earlier diagnoses, personalize physician-patient conversations, and advocate for further education on this issue in residency training programs.
- National Psoriasis Foundation. Statistics. https://www.psoriasis.org/content/statistics. Accessed July 14, 2020.
- Alexis AF, Blackcloud P. Psoriasis in skin of color: epidemiology, genetics, clinical presentation, and treatment nuances. J Clin Aesthet Dermatol. 2014;7:16-24.
- Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol. 2014;70:512-516.
- Goff KL, Karimkhani C, Boyers LN, et al. The global burden of psoriatic skin disease. Br J Dermatol. 2015;172:1665-1668.
- Kaufman BP, Alexis AF. Psoriasis in skin of color: insights into the epidemiology, clinical presentation, genetics, quality-of-life impact, and treatment of psoriasis in non-white racial/ethnic groups. Am J Clin Dermatol. 2018;19:405-423.
- Shah SK, Arthur A, Yang YC, et al. A retrospective study to investigate racial and ethnic variations in the treatment of psoriasis with etanercept. J Drugs Dermatol. 2011;10:866-872.
- Abrouk M, Lee K, Brodsky M, et al. Ethnicity affects the presenting severity of psoriasis. J Am Acad Dermatol. 2017;77:180-182.
- Youssef RM, Mahgoub D, Mashaly HM, et al. Different narrowband UVB dosage regimens in dark skinned psoriatics: a preliminary study. Photodermatol Photoimmunol Photomed. 2008;24:256-259.
- Charrow A, Xia F Di, Joyce C, et al. Diversity in dermatology clinical trials: a systematic review. JAMA Dermatol. 2017;153:193-198.
- Takeshita J, Eriksen WT, Raziano VT, et al. Racial differences in perceptions of psoriasis therapies: implications for racial disparities in psoriasis treatment. J Invest Dermatol. 2019;139:1672-1679.
- Yan D, Afifi L, Jeon C, et al. A cross-sectional study of the distribution of psoriasis subtypes in different ethno-racial groups. Dermatol Online J. 2018;24. pii:13030/qt5z21q4k2.
- Kerr GS, Qaiyumi S, Richards J, et al. Psoriasis and psoriatic arthritis in African-American patients—the need to measure disease burden. Clin Rheumatol. 2015;34:1753-1759.
- Edson-Heredia E, Sterling KL, Alatorre CI, et al. Heterogeneity of response to biologic treatment: perspective for psoriasis. J Invest Dermatol. 2014;134:18-23.
- Yan D, Afifi L, Jeon C, et al. A cross-sectional study of psoriasis triggers among different ethno-racial groups. J Am Acad Dermatol. 2017;77:756-758.
- Bailey RK, Mokonogho J, Kumar A. Racial and ethnic differences in depression: current perspectives. Neuropsychiatr Dis Treat. 2019;15:603-609.
- Jackson C, Maibach H. Ethnic and socioeconomic disparities in dermatology. J Dermatolog Treat. 2016;27:290-291.
- Salam A, Dadzie OE. Dermatology training in the U.K.: does it reflect the changing demographics of our population? Br J Dermatol. 2013;169:1360-1362.
- Nijhawan RI, Jacob SE, Woolery-Lloyd H. Skin of color education in dermatology residency programs: does residency training reflect the changing demographics of the United States? J Am Acad Dermatol. 2008;59:615-618.
- Ogunyemi B, Miller-Monthrope Y. The state of ethnic dermatology in Canada. J Cutan Med Surg. 2017;21:464-466.
Psoriasis is a chronic inflammatory skin disease that affects 2% to 3% of individuals worldwide.1 Despite extensive research, the majority of clinical data are in white patients with limited data in patients of color, yet a number of differences are known. The prevalence of psoriasis differs among racial and ethnic groups, with lower prevalence in racial minorities.2 A cross-sectional American study using data from 2009 through 2010 showed the prevalence for psoriasis was 3.6% in white patients, 1.9% in black patients, 1.6% in Hispanic patients, and 1.4% in other racial groups.3 Psoriasis presents differently in patients of color, both in morphology and severity. Cultural differences and stigma may contribute to the differences seen in severity but also to the psychological impact and treatment choices in patients of color compared to white patients.4 It has even been theorized that treatment efficacy could differ because of potential genetic differences.5 Psoriasis in patients of color is an emerging clinical issue that requires further attention so that dermatologists can learn about, diagnose, and treat them.
We report 3 cases of patients of color with psoriasis who presented to an urban and racially diverse dermatology clinic affiliated with Scarborough General Hospital in Toronto, Ontario, Canada. A retrospective chart review was performed on these high-yield representative cases to demonstrate differences in color and morphology, disease severity, and treatment in patients of various races seen at our clinic. After informed consent was obtained, photographs were taken of patient cutaneous findings to illustrate these differences. Discussion with these selected patients yielded supplementary qualitative data, highlighting individual perspectives of their disease.
Case Series
Patient 1
A 53-year-old black man from Grenada presented to our clinic with a history of psoriasis for a number of years that presented as violaceous plaques throughout large portions of the body (Figure 1). He previously had achieved inadequate results while using topical therapies, methotrexate, acitretin, apremilast, ustekinumab, ixekizumab, and guselkumab at adequate or even maximum doses. His disease affected 30% of the body surface area, with a psoriasis area and severity index score of 27 and a dermatology life quality index score of 23. The patient’s life was quite affected by psoriasis, with emphasis on choice of clothing worn and effect on body image. He also discussed the stigma psoriasis may have in black patients, stating that he has been told multiple times that “black people do not get psoriasis.”
Patient 2
A 27-year-old man from India presented with guttate psoriasis (Figure 2). He was treated with methotrexate 2 years prior and currently is on maintenance therapy with topical treatments alone. His main concerns pertained to the persistent dyschromia that occurred secondary to the psoriatic lesions. Through discussion, the patient stated that he “would do anything to get rid of it.”
Patient 3
A 49-year-old man from the Philippines presented to our clinic with plaque psoriasis that predominantly affected the trunk and scalp (Figure 3). He had been treated with methotrexate and phototherapy with suboptimal efficacy and was planning for biologic therapy. Although he had active plaques on the trunk, the patient stated, “I am most bothered by my scalp,” particularly referring to the itch and scale and their effects on hair and hairstyling.
Comment
Clinical differences in patients of color with psoriasis affect the management of the disease. Special consideration should be given to variances in morphology, presentation, treatment, and psychosocial factors in the management of psoriasis for these patient populations, as summarized in the eTable.
Morphology
At our clinic, patients of color have been found to have differences in morphology, including lesions that are more violaceous in color, as seen in patient 1; less noticeable inflammation; and more postinflammatory hypopigmentation and hyperpigmentation changes, as seen in patient 2. These changes are supported by the literature and differ from typical psoriasis plaques, which are pink-red and have more overlying scale. The varied morphology also may affect the differential, and other mimickers may be considered, such as lichen planus, cutaneous lupus erythematosus, and sarcoidosis.2
Presentation
There are differences in presentation among patients of color, particularly in distribution, type of psoriasis, and severity. As seen in patient 3, Asian and black patients are more likely to present with scalp psoriasis.2,5 Hairstyling and hair care practices can differ considerably between racial groups. Given the differences in hairstyling, scalp psoriasis also may have a greater impact on patient quality of life (QOL).
Racial differences affect the type of psoriasis seen. Asian patients are more likely to present with pustular and erythrodermic psoriasis and less likely to present with inverse psoriasis compared to white patients. Hispanic patients are more likely to present with pustular psoriasis.11 Black patients have been reported to have lower frequencies of psoriatic arthritis compared to white patients.12 Recognition of these differences may help guide initial choice for therapeutics.
Notably, patients of color may present with much more severe psoriasis, particularly Asian and Hispanic patients.7 One retrospective study looking at patients with psoriasis treated with etanercept found that Asian patients were more likely to have greater baseline body surface area involvement.6 An American cross-sectional study reported higher psoriasis area and severity index scores in black patients compared to white patients,12 possibly because patients of color do not normalize the experience of having psoriasis and feel stigmatized, which can cause delays in seeking medical attention and worsen disease burden. For patient 1, the stigma of black patients having psoriasis affected his body image and may have led to a delay in seeking medical attention due to him not believing it was possible for people of his skin color to have psoriasis. Increased disease severity may contribute to treatment resistance or numerous trials of topicals or biologics before the disease improves. Patient education in the community as well as patient support groups are paramount, and increased awareness of psoriasis can help improve disease management.
Treatment
Topical therapies are the first-line treatment of psoriasis. Although there is no evidence showing differences in topical treatment efficacy, patient preference for different topical treatments may vary based on race. For example, patients with Afro-textured hair may prefer foams and lotions and would avoid shampoo therapies, as frequent hair washing may not be feasible with certain hairstyles and may cause hair breakage or dryness.2
UV therapy can be an effective treatment modality for patients with psoriasis. The strength of therapy tends to be dictated by the Fitzpatrick skin phototype rather than race. Darker-skinned individuals may have an increased risk for hyperpigmentation, so caution should be taken to prevent burning during therapy. Suberythemogenic dosing—70% of minimal erythema dose—of narrowband UVB treatments has shown the same efficacy as using minimal erythema dose in patients with darker skin types in addition to fair-skinned patients.8
Although we found poor efficacy of systemic treatments in patient 1, to our knowledge, studies examining the efficacy of systemic therapeutic options have not shown differences in patients of color.6,13 Studies show similar efficacy in treatments among races, particularly biologic therapies.5 However, patients with skin of color historically have been underrepresented in clinical trials,9 which may contribute to these patients, particularly black patients, being less familiar with biologics as a treatment option for psoriasis, as reported by Takeshita et al.10 Therefore, patient-centered discussions regarding treatment choices are important to ensure patients understand all options available to manage their disease.
Psychosocial Impact
Because of its chronic remitting course, psoriasis has a notable psychosocial impact on the lives of all patients, though the literature suggests there may be more of an impact on QOL in patients of color. Higher baseline dermatology life quality index scores have been reported in patients of color compared to white patients.6 Kerr et al12 reported significantly greater psoriasis area and severity index scores (P=.06) and greater psychological impact in black patients compared to white patients. Stress also was more likely to be reported as a trigger for psoriasis in patients of Hispanic background compared to white patients.14 Many patients report body image issues with large physical lesions; however, the difference may lie in personal and cultural views about psoriasis, as one of our patients stated, “black people do not get psoriasis.” In addition to the cosmetic challenges that patients face with active lesions, postinflammatory pigmentary changes can be equally as burdensome to patients, as one of our patients stated he “would do anything to get rid of it.” Increased rates of depression and anxiety in patients of color can worsen their outlook on the condition.15,16 The increased stigma and burden of psoriasis in patients of color calls for clinicians to counsel and address psoriasis in a holistic way and refer patients to psoriasis support groups when appropriate. Although the burden of psoriasis is clear, more studies can be carried out to investigate the impact on QOL in different ethnic populations.
Dermatology Education
Although differences have been found in patients of color with psoriasis, dissemination of this knowledge continues to be a challenge. In dermatology residency programs, the majority of teaching is provided with examples of skin diseases in white patients, which can complicate pattern recognition and diagnostic ability for trainees. Although dermatologists recognize that ethnic skin has unique dermatologic considerations, there is a persistent need for increasing skin of color education within dermatology residency programs.17,18 Implementing more educational programs on skin of color has been proposed, and these programs will continue to be in demand as our population increasingly diversifies.19
Conclusion
Psoriasis in patients of color carries unique challenges when compared to psoriasis in white patients. Differences in morphology and presentation can make the disease difficult to accurately diagnose. These differences in addition to cultural differences may contribute to a greater impact on QOL and psychological health. Although treatment preferences and recognition may differ, treatment efficacy has so far been similar, albeit with a low proportion of patients with skin of color included in clinical trials.
Further focus should now lie within knowledge translation of these differences, which would normalize the condition for patients, support them seeking medical attention sooner, and inform them of all treatment options possible. For clinicians, more attention on the differences would help make earlier diagnoses, personalize physician-patient conversations, and advocate for further education on this issue in residency training programs.
Psoriasis is a chronic inflammatory skin disease that affects 2% to 3% of individuals worldwide.1 Despite extensive research, the majority of clinical data are in white patients with limited data in patients of color, yet a number of differences are known. The prevalence of psoriasis differs among racial and ethnic groups, with lower prevalence in racial minorities.2 A cross-sectional American study using data from 2009 through 2010 showed the prevalence for psoriasis was 3.6% in white patients, 1.9% in black patients, 1.6% in Hispanic patients, and 1.4% in other racial groups.3 Psoriasis presents differently in patients of color, both in morphology and severity. Cultural differences and stigma may contribute to the differences seen in severity but also to the psychological impact and treatment choices in patients of color compared to white patients.4 It has even been theorized that treatment efficacy could differ because of potential genetic differences.5 Psoriasis in patients of color is an emerging clinical issue that requires further attention so that dermatologists can learn about, diagnose, and treat them.
We report 3 cases of patients of color with psoriasis who presented to an urban and racially diverse dermatology clinic affiliated with Scarborough General Hospital in Toronto, Ontario, Canada. A retrospective chart review was performed on these high-yield representative cases to demonstrate differences in color and morphology, disease severity, and treatment in patients of various races seen at our clinic. After informed consent was obtained, photographs were taken of patient cutaneous findings to illustrate these differences. Discussion with these selected patients yielded supplementary qualitative data, highlighting individual perspectives of their disease.
Case Series
Patient 1
A 53-year-old black man from Grenada presented to our clinic with a history of psoriasis for a number of years that presented as violaceous plaques throughout large portions of the body (Figure 1). He previously had achieved inadequate results while using topical therapies, methotrexate, acitretin, apremilast, ustekinumab, ixekizumab, and guselkumab at adequate or even maximum doses. His disease affected 30% of the body surface area, with a psoriasis area and severity index score of 27 and a dermatology life quality index score of 23. The patient’s life was quite affected by psoriasis, with emphasis on choice of clothing worn and effect on body image. He also discussed the stigma psoriasis may have in black patients, stating that he has been told multiple times that “black people do not get psoriasis.”
Patient 2
A 27-year-old man from India presented with guttate psoriasis (Figure 2). He was treated with methotrexate 2 years prior and currently is on maintenance therapy with topical treatments alone. His main concerns pertained to the persistent dyschromia that occurred secondary to the psoriatic lesions. Through discussion, the patient stated that he “would do anything to get rid of it.”
Patient 3
A 49-year-old man from the Philippines presented to our clinic with plaque psoriasis that predominantly affected the trunk and scalp (Figure 3). He had been treated with methotrexate and phototherapy with suboptimal efficacy and was planning for biologic therapy. Although he had active plaques on the trunk, the patient stated, “I am most bothered by my scalp,” particularly referring to the itch and scale and their effects on hair and hairstyling.
Comment
Clinical differences in patients of color with psoriasis affect the management of the disease. Special consideration should be given to variances in morphology, presentation, treatment, and psychosocial factors in the management of psoriasis for these patient populations, as summarized in the eTable.
Morphology
At our clinic, patients of color have been found to have differences in morphology, including lesions that are more violaceous in color, as seen in patient 1; less noticeable inflammation; and more postinflammatory hypopigmentation and hyperpigmentation changes, as seen in patient 2. These changes are supported by the literature and differ from typical psoriasis plaques, which are pink-red and have more overlying scale. The varied morphology also may affect the differential, and other mimickers may be considered, such as lichen planus, cutaneous lupus erythematosus, and sarcoidosis.2
Presentation
There are differences in presentation among patients of color, particularly in distribution, type of psoriasis, and severity. As seen in patient 3, Asian and black patients are more likely to present with scalp psoriasis.2,5 Hairstyling and hair care practices can differ considerably between racial groups. Given the differences in hairstyling, scalp psoriasis also may have a greater impact on patient quality of life (QOL).
Racial differences affect the type of psoriasis seen. Asian patients are more likely to present with pustular and erythrodermic psoriasis and less likely to present with inverse psoriasis compared to white patients. Hispanic patients are more likely to present with pustular psoriasis.11 Black patients have been reported to have lower frequencies of psoriatic arthritis compared to white patients.12 Recognition of these differences may help guide initial choice for therapeutics.
Notably, patients of color may present with much more severe psoriasis, particularly Asian and Hispanic patients.7 One retrospective study looking at patients with psoriasis treated with etanercept found that Asian patients were more likely to have greater baseline body surface area involvement.6 An American cross-sectional study reported higher psoriasis area and severity index scores in black patients compared to white patients,12 possibly because patients of color do not normalize the experience of having psoriasis and feel stigmatized, which can cause delays in seeking medical attention and worsen disease burden. For patient 1, the stigma of black patients having psoriasis affected his body image and may have led to a delay in seeking medical attention due to him not believing it was possible for people of his skin color to have psoriasis. Increased disease severity may contribute to treatment resistance or numerous trials of topicals or biologics before the disease improves. Patient education in the community as well as patient support groups are paramount, and increased awareness of psoriasis can help improve disease management.
Treatment
Topical therapies are the first-line treatment of psoriasis. Although there is no evidence showing differences in topical treatment efficacy, patient preference for different topical treatments may vary based on race. For example, patients with Afro-textured hair may prefer foams and lotions and would avoid shampoo therapies, as frequent hair washing may not be feasible with certain hairstyles and may cause hair breakage or dryness.2
UV therapy can be an effective treatment modality for patients with psoriasis. The strength of therapy tends to be dictated by the Fitzpatrick skin phototype rather than race. Darker-skinned individuals may have an increased risk for hyperpigmentation, so caution should be taken to prevent burning during therapy. Suberythemogenic dosing—70% of minimal erythema dose—of narrowband UVB treatments has shown the same efficacy as using minimal erythema dose in patients with darker skin types in addition to fair-skinned patients.8
Although we found poor efficacy of systemic treatments in patient 1, to our knowledge, studies examining the efficacy of systemic therapeutic options have not shown differences in patients of color.6,13 Studies show similar efficacy in treatments among races, particularly biologic therapies.5 However, patients with skin of color historically have been underrepresented in clinical trials,9 which may contribute to these patients, particularly black patients, being less familiar with biologics as a treatment option for psoriasis, as reported by Takeshita et al.10 Therefore, patient-centered discussions regarding treatment choices are important to ensure patients understand all options available to manage their disease.
Psychosocial Impact
Because of its chronic remitting course, psoriasis has a notable psychosocial impact on the lives of all patients, though the literature suggests there may be more of an impact on QOL in patients of color. Higher baseline dermatology life quality index scores have been reported in patients of color compared to white patients.6 Kerr et al12 reported significantly greater psoriasis area and severity index scores (P=.06) and greater psychological impact in black patients compared to white patients. Stress also was more likely to be reported as a trigger for psoriasis in patients of Hispanic background compared to white patients.14 Many patients report body image issues with large physical lesions; however, the difference may lie in personal and cultural views about psoriasis, as one of our patients stated, “black people do not get psoriasis.” In addition to the cosmetic challenges that patients face with active lesions, postinflammatory pigmentary changes can be equally as burdensome to patients, as one of our patients stated he “would do anything to get rid of it.” Increased rates of depression and anxiety in patients of color can worsen their outlook on the condition.15,16 The increased stigma and burden of psoriasis in patients of color calls for clinicians to counsel and address psoriasis in a holistic way and refer patients to psoriasis support groups when appropriate. Although the burden of psoriasis is clear, more studies can be carried out to investigate the impact on QOL in different ethnic populations.
Dermatology Education
Although differences have been found in patients of color with psoriasis, dissemination of this knowledge continues to be a challenge. In dermatology residency programs, the majority of teaching is provided with examples of skin diseases in white patients, which can complicate pattern recognition and diagnostic ability for trainees. Although dermatologists recognize that ethnic skin has unique dermatologic considerations, there is a persistent need for increasing skin of color education within dermatology residency programs.17,18 Implementing more educational programs on skin of color has been proposed, and these programs will continue to be in demand as our population increasingly diversifies.19
Conclusion
Psoriasis in patients of color carries unique challenges when compared to psoriasis in white patients. Differences in morphology and presentation can make the disease difficult to accurately diagnose. These differences in addition to cultural differences may contribute to a greater impact on QOL and psychological health. Although treatment preferences and recognition may differ, treatment efficacy has so far been similar, albeit with a low proportion of patients with skin of color included in clinical trials.
Further focus should now lie within knowledge translation of these differences, which would normalize the condition for patients, support them seeking medical attention sooner, and inform them of all treatment options possible. For clinicians, more attention on the differences would help make earlier diagnoses, personalize physician-patient conversations, and advocate for further education on this issue in residency training programs.
- National Psoriasis Foundation. Statistics. https://www.psoriasis.org/content/statistics. Accessed July 14, 2020.
- Alexis AF, Blackcloud P. Psoriasis in skin of color: epidemiology, genetics, clinical presentation, and treatment nuances. J Clin Aesthet Dermatol. 2014;7:16-24.
- Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol. 2014;70:512-516.
- Goff KL, Karimkhani C, Boyers LN, et al. The global burden of psoriatic skin disease. Br J Dermatol. 2015;172:1665-1668.
- Kaufman BP, Alexis AF. Psoriasis in skin of color: insights into the epidemiology, clinical presentation, genetics, quality-of-life impact, and treatment of psoriasis in non-white racial/ethnic groups. Am J Clin Dermatol. 2018;19:405-423.
- Shah SK, Arthur A, Yang YC, et al. A retrospective study to investigate racial and ethnic variations in the treatment of psoriasis with etanercept. J Drugs Dermatol. 2011;10:866-872.
- Abrouk M, Lee K, Brodsky M, et al. Ethnicity affects the presenting severity of psoriasis. J Am Acad Dermatol. 2017;77:180-182.
- Youssef RM, Mahgoub D, Mashaly HM, et al. Different narrowband UVB dosage regimens in dark skinned psoriatics: a preliminary study. Photodermatol Photoimmunol Photomed. 2008;24:256-259.
- Charrow A, Xia F Di, Joyce C, et al. Diversity in dermatology clinical trials: a systematic review. JAMA Dermatol. 2017;153:193-198.
- Takeshita J, Eriksen WT, Raziano VT, et al. Racial differences in perceptions of psoriasis therapies: implications for racial disparities in psoriasis treatment. J Invest Dermatol. 2019;139:1672-1679.
- Yan D, Afifi L, Jeon C, et al. A cross-sectional study of the distribution of psoriasis subtypes in different ethno-racial groups. Dermatol Online J. 2018;24. pii:13030/qt5z21q4k2.
- Kerr GS, Qaiyumi S, Richards J, et al. Psoriasis and psoriatic arthritis in African-American patients—the need to measure disease burden. Clin Rheumatol. 2015;34:1753-1759.
- Edson-Heredia E, Sterling KL, Alatorre CI, et al. Heterogeneity of response to biologic treatment: perspective for psoriasis. J Invest Dermatol. 2014;134:18-23.
- Yan D, Afifi L, Jeon C, et al. A cross-sectional study of psoriasis triggers among different ethno-racial groups. J Am Acad Dermatol. 2017;77:756-758.
- Bailey RK, Mokonogho J, Kumar A. Racial and ethnic differences in depression: current perspectives. Neuropsychiatr Dis Treat. 2019;15:603-609.
- Jackson C, Maibach H. Ethnic and socioeconomic disparities in dermatology. J Dermatolog Treat. 2016;27:290-291.
- Salam A, Dadzie OE. Dermatology training in the U.K.: does it reflect the changing demographics of our population? Br J Dermatol. 2013;169:1360-1362.
- Nijhawan RI, Jacob SE, Woolery-Lloyd H. Skin of color education in dermatology residency programs: does residency training reflect the changing demographics of the United States? J Am Acad Dermatol. 2008;59:615-618.
- Ogunyemi B, Miller-Monthrope Y. The state of ethnic dermatology in Canada. J Cutan Med Surg. 2017;21:464-466.
- National Psoriasis Foundation. Statistics. https://www.psoriasis.org/content/statistics. Accessed July 14, 2020.
- Alexis AF, Blackcloud P. Psoriasis in skin of color: epidemiology, genetics, clinical presentation, and treatment nuances. J Clin Aesthet Dermatol. 2014;7:16-24.
- Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol. 2014;70:512-516.
- Goff KL, Karimkhani C, Boyers LN, et al. The global burden of psoriatic skin disease. Br J Dermatol. 2015;172:1665-1668.
- Kaufman BP, Alexis AF. Psoriasis in skin of color: insights into the epidemiology, clinical presentation, genetics, quality-of-life impact, and treatment of psoriasis in non-white racial/ethnic groups. Am J Clin Dermatol. 2018;19:405-423.
- Shah SK, Arthur A, Yang YC, et al. A retrospective study to investigate racial and ethnic variations in the treatment of psoriasis with etanercept. J Drugs Dermatol. 2011;10:866-872.
- Abrouk M, Lee K, Brodsky M, et al. Ethnicity affects the presenting severity of psoriasis. J Am Acad Dermatol. 2017;77:180-182.
- Youssef RM, Mahgoub D, Mashaly HM, et al. Different narrowband UVB dosage regimens in dark skinned psoriatics: a preliminary study. Photodermatol Photoimmunol Photomed. 2008;24:256-259.
- Charrow A, Xia F Di, Joyce C, et al. Diversity in dermatology clinical trials: a systematic review. JAMA Dermatol. 2017;153:193-198.
- Takeshita J, Eriksen WT, Raziano VT, et al. Racial differences in perceptions of psoriasis therapies: implications for racial disparities in psoriasis treatment. J Invest Dermatol. 2019;139:1672-1679.
- Yan D, Afifi L, Jeon C, et al. A cross-sectional study of the distribution of psoriasis subtypes in different ethno-racial groups. Dermatol Online J. 2018;24. pii:13030/qt5z21q4k2.
- Kerr GS, Qaiyumi S, Richards J, et al. Psoriasis and psoriatic arthritis in African-American patients—the need to measure disease burden. Clin Rheumatol. 2015;34:1753-1759.
- Edson-Heredia E, Sterling KL, Alatorre CI, et al. Heterogeneity of response to biologic treatment: perspective for psoriasis. J Invest Dermatol. 2014;134:18-23.
- Yan D, Afifi L, Jeon C, et al. A cross-sectional study of psoriasis triggers among different ethno-racial groups. J Am Acad Dermatol. 2017;77:756-758.
- Bailey RK, Mokonogho J, Kumar A. Racial and ethnic differences in depression: current perspectives. Neuropsychiatr Dis Treat. 2019;15:603-609.
- Jackson C, Maibach H. Ethnic and socioeconomic disparities in dermatology. J Dermatolog Treat. 2016;27:290-291.
- Salam A, Dadzie OE. Dermatology training in the U.K.: does it reflect the changing demographics of our population? Br J Dermatol. 2013;169:1360-1362.
- Nijhawan RI, Jacob SE, Woolery-Lloyd H. Skin of color education in dermatology residency programs: does residency training reflect the changing demographics of the United States? J Am Acad Dermatol. 2008;59:615-618.
- Ogunyemi B, Miller-Monthrope Y. The state of ethnic dermatology in Canada. J Cutan Med Surg. 2017;21:464-466.
Practice Points
- There are key differences in psoriasis in patients with skin of color, including the morphology, clinical presentation, treatment, and psychosocial impact.
- Recognition and awareness of these differences may normalize the condition for patients, support them seeking medical attention sooner, and better inform them of all possible treatment options.
- Advocating further education on these differences in residency training and continuing medical education programs may help physicians make earlier diagnoses and personalize physician-patient conversations.
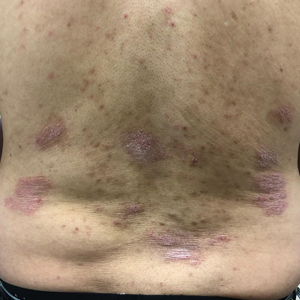
50-year-old man • foot pain • “purple” toe • history of smoking • Dx?
THE CASE
A 50-year-old man presented to the primary care office for evaluation of foot pain. The day before, his left fifth toe had become exquisitely tender. He distinctly remembered that when he awoke, there was no discoloration or pain, but the toe later became “purple.” He denied any trauma. His medical record was notable for an extensive smoking history and a family history of early cardiovascular disease.
The patient appeared well but in obvious distress, secondary to the pain. His vital signs were unremarkable. His head, neck, lung, and cardiac exams revealed no abnormalities. Physical examination revealed a left fifth toe that was dusky purple and warm to the touch. Pain disproportionate to examination was noted on the anterior aspect of the toe, with limited range of motion. The patient walked with a compensated gait. Pulses were palpable on the posterior tibial (PT) and dorsalis pedis (DP) regions.
DIAGNOSIS
Based on our exam findings, we suspected a vascular injury and recommended an emergency consult by Podiatry, for which he was scheduled the following morning. The podiatric evaluation confirmed concern for a vascular injury and prompted a request for an emergent evaluation by Vascular Surgery.
The patient was seen emergently on Day 4 for a vascular surgery evaluation. Examination at that time showed a nearly absent femoral pulse on the left side and diminished and monophasic DP and PT pulses. His left foot demonstrated nonblanchable purpura that was clinically consistent with cholesterol embolization syndrome (CES).
We calculated the patient’s ankle-brachial index, and computed tomography angiography (CTA) was performed. While results were pending, the patient was started on aspirin 81 mg, clopidogrel 75 mg, and atorvastatin 40 mg, for a suspected slowly progressing iliac artery stenosis with a resulting acute atheroembolic event.
The CTA report showed a high-grade stenosis at the bifurcation of the left iliac artery, extending into both external and internal arteries. Of note, mild atherosclerotic disease without significant occlusion and runoff to the foot was observed into the tibial arteries. The stenosis extended into the profonda femoris artery, as well.
DISCUSSION
Atherosclerotic plaques are commonly encountered in patients with atherosclerotic disease; however, there are 2 varieties of emboli that arise from these plaques and one is often overlooked.1-4 The more common of these variants, thromboemboli, originates from an atherosclerotic plaque and can become lodged in a medium or large vessel as a single embolus.
Continue to: By contrast...
By contrast, atheroemboli (commonly known as cholesterol emboli or cholesterol crystal embolization) originate from atherosclerotic plaques in the aorta or another large artery,5 which are prone to embolize if the underlying plaque experiences stress. As the plaque erodes, cholesterol crystals break off and embolize distally. These smaller crystals flood into the circulation, allowing a shower of emboli over time to occlude the arterioles. As occlusion spreads through the arterioles, multiple organ systems are affected. (It was previously thought that procedure-associated cases were common, but a literature review has not borne this out.5)
The shower of emboli often triggers a systemic inflammatory response, causing nondescript abnormalities of laboratory inflammatory markers.6,7 Interestingly, hypereosinophilia is noted in about 80% of patients with CES.8
No disease-specific testing. A confounding factor in validating the diagnosis of CES is the lack of disease-specific testing. However, CES should be considered in a patient with acute kidney injury and hypereosinophilia. Making the diagnosis requires a high degree of clinical suspicion. Any organ can be affected, although the brain, kidneys, gastrointestinal tract, skin, and skeletal muscles of the lower extremities are most frequently involved.9 If left undiagnosed, the results can be devastating: slow and chronic injury to a variety of organ systems over time, which may not be recognized as a harbinger of an insidious underlying process causing end-organ damage.
Technically, definitive diagnosis can be made by biopsy of an affected organ. However, biopsy’s utility is limited due to potential for sampling error, accessibility (as noted, the location of the involved organ[s] may make biopsy nearly impossible without additional surgical risk9), and risk of poor healing to the biopsy site.10
Treatment is two-fold: supportive care for the affected end organ and prevention of subsequent embolic events. The latter entails aggressive risk factor reduction strategies, such as smoking cessation, statin therapy, blood pressure control, and blood sugar control. Warfarin is not recommended for treatment of CES due to the risk of further plaque rupture, hemorrhage, acute and chronic renal failure, and cholesterol microembolization to other organs.11,12
Continue to: Our patient
Our patient. After testing confirmed the diagnosis, the patient underwent an angioplasty. A stent was placed in his left iliac artery. He was continued on antiplatelet and statin therapy and was again counseled regarding smoking cessation.
THE TAKEAWAY
When patients present with symptoms suggestive of a vascular origin, consider CES. Although it can affect a multitude of organs, acute kidney injury and hypereosinophilia are the most common signs. Immediate intervention is required to save the affected organ; strategizing to reduce the risk for further embolic events is also key.
Prompt recognition of vascular emergencies, including those that are harbingers of atherosclerotic disease, is essential. As clinicians, it is imperative that we use all resources to address significant population health burdens. If CES is more prevalent than commonly thought, consideration should be given to increasing education about early detection and treatment of this disorder, including the reinforcement of primary prevention and aggressive treatment of risk factors for atherosclerotic cardiovascular disease.
CORRESPONDENCE
Meagan Vermeulen, MD, FAAFP, Department of Family Medicine, Rowan University School of Osteopathic Medicine, 42 East Laurel Road, Suite 2100A, Stratford, NJ 08084; [email protected]
1. Tunick PA, Kronzon I. Atheromas of the thoracic aorta: clinical and therapeutic update. J Am Coll Cardiol. 2000;35:545-554.
2. Amarenco P, Duyckaerts C, Tzourio C, et al. The prevalence of ulcerated plaques in the aortic arch in patients with stroke. N Engl J Med. 1992;326:221-225.
3. Amarenco P, Cohen A, Tzourio C, et al. Atherosclerotic disease of the aortic arch and the risk of ischemic stroke. N Engl J Med. 1994;331:1474-1479.
4. Amarenco P, Cohen A, et al; French Study of Aortic Plaques in Stroke Group. Atherosclerotic disease of the aortic arch as a risk factor for recurrent ischemic stroke. N Engl J Med. 1996;334:1216-1221.
5. Ong HT, Elmsly WG, Friedlander DH. Cholesterol atheroembolism: an increasingly frequent complication of cardiac catheterisation. Med J Aust. 1991;154:412-414.
6. Kronzon I, Saric M. Cholesterol embolization syndrome. Circulation. 2010;122:631-641.
7. Saric M, Kronzon I. Cholesterol embolization syndrome. Curr Opin Cardiol. 2011;26:472-479.
8. Kasinath BS, Lewis EJ. Eosinophilia as a clue to the diagnosis of atheroembolic renal disease. Arch Intern Med. 1987;147:1384-1385.
9. Quinones A, Saric M. The cholesterol emboli syndrome in atherosclerosis. Curr Atheroscler Rep. 2013;15:315.
10. Jucgla A, Moreso F, Muniesa C, et al. Cholesterol embolism: still an unrecognized entity with a high mortality rate. J Am Acad Dermatol. 2006;55:786-793.
11. Kim H, Zhen DB, Lieske JC, et al. Treatment of cholesterol embolization syndrome in the setting of an acute indication for anticoagulation therapy. J Med Cases. 2014;5:376-379.
12. Igarashi Y, Akimoto T, Kobayashi T, et al. Performing anticoagulation: a puzzling case of cholesterol embolization syndrome. Clin Med Insights Case Rep. 2017;10:1179547616684649. doi:10.1177/1179547616684649.
THE CASE
A 50-year-old man presented to the primary care office for evaluation of foot pain. The day before, his left fifth toe had become exquisitely tender. He distinctly remembered that when he awoke, there was no discoloration or pain, but the toe later became “purple.” He denied any trauma. His medical record was notable for an extensive smoking history and a family history of early cardiovascular disease.
The patient appeared well but in obvious distress, secondary to the pain. His vital signs were unremarkable. His head, neck, lung, and cardiac exams revealed no abnormalities. Physical examination revealed a left fifth toe that was dusky purple and warm to the touch. Pain disproportionate to examination was noted on the anterior aspect of the toe, with limited range of motion. The patient walked with a compensated gait. Pulses were palpable on the posterior tibial (PT) and dorsalis pedis (DP) regions.
DIAGNOSIS
Based on our exam findings, we suspected a vascular injury and recommended an emergency consult by Podiatry, for which he was scheduled the following morning. The podiatric evaluation confirmed concern for a vascular injury and prompted a request for an emergent evaluation by Vascular Surgery.
The patient was seen emergently on Day 4 for a vascular surgery evaluation. Examination at that time showed a nearly absent femoral pulse on the left side and diminished and monophasic DP and PT pulses. His left foot demonstrated nonblanchable purpura that was clinically consistent with cholesterol embolization syndrome (CES).
We calculated the patient’s ankle-brachial index, and computed tomography angiography (CTA) was performed. While results were pending, the patient was started on aspirin 81 mg, clopidogrel 75 mg, and atorvastatin 40 mg, for a suspected slowly progressing iliac artery stenosis with a resulting acute atheroembolic event.
The CTA report showed a high-grade stenosis at the bifurcation of the left iliac artery, extending into both external and internal arteries. Of note, mild atherosclerotic disease without significant occlusion and runoff to the foot was observed into the tibial arteries. The stenosis extended into the profonda femoris artery, as well.
DISCUSSION
Atherosclerotic plaques are commonly encountered in patients with atherosclerotic disease; however, there are 2 varieties of emboli that arise from these plaques and one is often overlooked.1-4 The more common of these variants, thromboemboli, originates from an atherosclerotic plaque and can become lodged in a medium or large vessel as a single embolus.
Continue to: By contrast...
By contrast, atheroemboli (commonly known as cholesterol emboli or cholesterol crystal embolization) originate from atherosclerotic plaques in the aorta or another large artery,5 which are prone to embolize if the underlying plaque experiences stress. As the plaque erodes, cholesterol crystals break off and embolize distally. These smaller crystals flood into the circulation, allowing a shower of emboli over time to occlude the arterioles. As occlusion spreads through the arterioles, multiple organ systems are affected. (It was previously thought that procedure-associated cases were common, but a literature review has not borne this out.5)
The shower of emboli often triggers a systemic inflammatory response, causing nondescript abnormalities of laboratory inflammatory markers.6,7 Interestingly, hypereosinophilia is noted in about 80% of patients with CES.8
No disease-specific testing. A confounding factor in validating the diagnosis of CES is the lack of disease-specific testing. However, CES should be considered in a patient with acute kidney injury and hypereosinophilia. Making the diagnosis requires a high degree of clinical suspicion. Any organ can be affected, although the brain, kidneys, gastrointestinal tract, skin, and skeletal muscles of the lower extremities are most frequently involved.9 If left undiagnosed, the results can be devastating: slow and chronic injury to a variety of organ systems over time, which may not be recognized as a harbinger of an insidious underlying process causing end-organ damage.
Technically, definitive diagnosis can be made by biopsy of an affected organ. However, biopsy’s utility is limited due to potential for sampling error, accessibility (as noted, the location of the involved organ[s] may make biopsy nearly impossible without additional surgical risk9), and risk of poor healing to the biopsy site.10
Treatment is two-fold: supportive care for the affected end organ and prevention of subsequent embolic events. The latter entails aggressive risk factor reduction strategies, such as smoking cessation, statin therapy, blood pressure control, and blood sugar control. Warfarin is not recommended for treatment of CES due to the risk of further plaque rupture, hemorrhage, acute and chronic renal failure, and cholesterol microembolization to other organs.11,12
Continue to: Our patient
Our patient. After testing confirmed the diagnosis, the patient underwent an angioplasty. A stent was placed in his left iliac artery. He was continued on antiplatelet and statin therapy and was again counseled regarding smoking cessation.
THE TAKEAWAY
When patients present with symptoms suggestive of a vascular origin, consider CES. Although it can affect a multitude of organs, acute kidney injury and hypereosinophilia are the most common signs. Immediate intervention is required to save the affected organ; strategizing to reduce the risk for further embolic events is also key.
Prompt recognition of vascular emergencies, including those that are harbingers of atherosclerotic disease, is essential. As clinicians, it is imperative that we use all resources to address significant population health burdens. If CES is more prevalent than commonly thought, consideration should be given to increasing education about early detection and treatment of this disorder, including the reinforcement of primary prevention and aggressive treatment of risk factors for atherosclerotic cardiovascular disease.
CORRESPONDENCE
Meagan Vermeulen, MD, FAAFP, Department of Family Medicine, Rowan University School of Osteopathic Medicine, 42 East Laurel Road, Suite 2100A, Stratford, NJ 08084; [email protected]
THE CASE
A 50-year-old man presented to the primary care office for evaluation of foot pain. The day before, his left fifth toe had become exquisitely tender. He distinctly remembered that when he awoke, there was no discoloration or pain, but the toe later became “purple.” He denied any trauma. His medical record was notable for an extensive smoking history and a family history of early cardiovascular disease.
The patient appeared well but in obvious distress, secondary to the pain. His vital signs were unremarkable. His head, neck, lung, and cardiac exams revealed no abnormalities. Physical examination revealed a left fifth toe that was dusky purple and warm to the touch. Pain disproportionate to examination was noted on the anterior aspect of the toe, with limited range of motion. The patient walked with a compensated gait. Pulses were palpable on the posterior tibial (PT) and dorsalis pedis (DP) regions.
DIAGNOSIS
Based on our exam findings, we suspected a vascular injury and recommended an emergency consult by Podiatry, for which he was scheduled the following morning. The podiatric evaluation confirmed concern for a vascular injury and prompted a request for an emergent evaluation by Vascular Surgery.
The patient was seen emergently on Day 4 for a vascular surgery evaluation. Examination at that time showed a nearly absent femoral pulse on the left side and diminished and monophasic DP and PT pulses. His left foot demonstrated nonblanchable purpura that was clinically consistent with cholesterol embolization syndrome (CES).
We calculated the patient’s ankle-brachial index, and computed tomography angiography (CTA) was performed. While results were pending, the patient was started on aspirin 81 mg, clopidogrel 75 mg, and atorvastatin 40 mg, for a suspected slowly progressing iliac artery stenosis with a resulting acute atheroembolic event.
The CTA report showed a high-grade stenosis at the bifurcation of the left iliac artery, extending into both external and internal arteries. Of note, mild atherosclerotic disease without significant occlusion and runoff to the foot was observed into the tibial arteries. The stenosis extended into the profonda femoris artery, as well.
DISCUSSION
Atherosclerotic plaques are commonly encountered in patients with atherosclerotic disease; however, there are 2 varieties of emboli that arise from these plaques and one is often overlooked.1-4 The more common of these variants, thromboemboli, originates from an atherosclerotic plaque and can become lodged in a medium or large vessel as a single embolus.
Continue to: By contrast...
By contrast, atheroemboli (commonly known as cholesterol emboli or cholesterol crystal embolization) originate from atherosclerotic plaques in the aorta or another large artery,5 which are prone to embolize if the underlying plaque experiences stress. As the plaque erodes, cholesterol crystals break off and embolize distally. These smaller crystals flood into the circulation, allowing a shower of emboli over time to occlude the arterioles. As occlusion spreads through the arterioles, multiple organ systems are affected. (It was previously thought that procedure-associated cases were common, but a literature review has not borne this out.5)
The shower of emboli often triggers a systemic inflammatory response, causing nondescript abnormalities of laboratory inflammatory markers.6,7 Interestingly, hypereosinophilia is noted in about 80% of patients with CES.8
No disease-specific testing. A confounding factor in validating the diagnosis of CES is the lack of disease-specific testing. However, CES should be considered in a patient with acute kidney injury and hypereosinophilia. Making the diagnosis requires a high degree of clinical suspicion. Any organ can be affected, although the brain, kidneys, gastrointestinal tract, skin, and skeletal muscles of the lower extremities are most frequently involved.9 If left undiagnosed, the results can be devastating: slow and chronic injury to a variety of organ systems over time, which may not be recognized as a harbinger of an insidious underlying process causing end-organ damage.
Technically, definitive diagnosis can be made by biopsy of an affected organ. However, biopsy’s utility is limited due to potential for sampling error, accessibility (as noted, the location of the involved organ[s] may make biopsy nearly impossible without additional surgical risk9), and risk of poor healing to the biopsy site.10
Treatment is two-fold: supportive care for the affected end organ and prevention of subsequent embolic events. The latter entails aggressive risk factor reduction strategies, such as smoking cessation, statin therapy, blood pressure control, and blood sugar control. Warfarin is not recommended for treatment of CES due to the risk of further plaque rupture, hemorrhage, acute and chronic renal failure, and cholesterol microembolization to other organs.11,12
Continue to: Our patient
Our patient. After testing confirmed the diagnosis, the patient underwent an angioplasty. A stent was placed in his left iliac artery. He was continued on antiplatelet and statin therapy and was again counseled regarding smoking cessation.
THE TAKEAWAY
When patients present with symptoms suggestive of a vascular origin, consider CES. Although it can affect a multitude of organs, acute kidney injury and hypereosinophilia are the most common signs. Immediate intervention is required to save the affected organ; strategizing to reduce the risk for further embolic events is also key.
Prompt recognition of vascular emergencies, including those that are harbingers of atherosclerotic disease, is essential. As clinicians, it is imperative that we use all resources to address significant population health burdens. If CES is more prevalent than commonly thought, consideration should be given to increasing education about early detection and treatment of this disorder, including the reinforcement of primary prevention and aggressive treatment of risk factors for atherosclerotic cardiovascular disease.
CORRESPONDENCE
Meagan Vermeulen, MD, FAAFP, Department of Family Medicine, Rowan University School of Osteopathic Medicine, 42 East Laurel Road, Suite 2100A, Stratford, NJ 08084; [email protected]
1. Tunick PA, Kronzon I. Atheromas of the thoracic aorta: clinical and therapeutic update. J Am Coll Cardiol. 2000;35:545-554.
2. Amarenco P, Duyckaerts C, Tzourio C, et al. The prevalence of ulcerated plaques in the aortic arch in patients with stroke. N Engl J Med. 1992;326:221-225.
3. Amarenco P, Cohen A, Tzourio C, et al. Atherosclerotic disease of the aortic arch and the risk of ischemic stroke. N Engl J Med. 1994;331:1474-1479.
4. Amarenco P, Cohen A, et al; French Study of Aortic Plaques in Stroke Group. Atherosclerotic disease of the aortic arch as a risk factor for recurrent ischemic stroke. N Engl J Med. 1996;334:1216-1221.
5. Ong HT, Elmsly WG, Friedlander DH. Cholesterol atheroembolism: an increasingly frequent complication of cardiac catheterisation. Med J Aust. 1991;154:412-414.
6. Kronzon I, Saric M. Cholesterol embolization syndrome. Circulation. 2010;122:631-641.
7. Saric M, Kronzon I. Cholesterol embolization syndrome. Curr Opin Cardiol. 2011;26:472-479.
8. Kasinath BS, Lewis EJ. Eosinophilia as a clue to the diagnosis of atheroembolic renal disease. Arch Intern Med. 1987;147:1384-1385.
9. Quinones A, Saric M. The cholesterol emboli syndrome in atherosclerosis. Curr Atheroscler Rep. 2013;15:315.
10. Jucgla A, Moreso F, Muniesa C, et al. Cholesterol embolism: still an unrecognized entity with a high mortality rate. J Am Acad Dermatol. 2006;55:786-793.
11. Kim H, Zhen DB, Lieske JC, et al. Treatment of cholesterol embolization syndrome in the setting of an acute indication for anticoagulation therapy. J Med Cases. 2014;5:376-379.
12. Igarashi Y, Akimoto T, Kobayashi T, et al. Performing anticoagulation: a puzzling case of cholesterol embolization syndrome. Clin Med Insights Case Rep. 2017;10:1179547616684649. doi:10.1177/1179547616684649.
1. Tunick PA, Kronzon I. Atheromas of the thoracic aorta: clinical and therapeutic update. J Am Coll Cardiol. 2000;35:545-554.
2. Amarenco P, Duyckaerts C, Tzourio C, et al. The prevalence of ulcerated plaques in the aortic arch in patients with stroke. N Engl J Med. 1992;326:221-225.
3. Amarenco P, Cohen A, Tzourio C, et al. Atherosclerotic disease of the aortic arch and the risk of ischemic stroke. N Engl J Med. 1994;331:1474-1479.
4. Amarenco P, Cohen A, et al; French Study of Aortic Plaques in Stroke Group. Atherosclerotic disease of the aortic arch as a risk factor for recurrent ischemic stroke. N Engl J Med. 1996;334:1216-1221.
5. Ong HT, Elmsly WG, Friedlander DH. Cholesterol atheroembolism: an increasingly frequent complication of cardiac catheterisation. Med J Aust. 1991;154:412-414.
6. Kronzon I, Saric M. Cholesterol embolization syndrome. Circulation. 2010;122:631-641.
7. Saric M, Kronzon I. Cholesterol embolization syndrome. Curr Opin Cardiol. 2011;26:472-479.
8. Kasinath BS, Lewis EJ. Eosinophilia as a clue to the diagnosis of atheroembolic renal disease. Arch Intern Med. 1987;147:1384-1385.
9. Quinones A, Saric M. The cholesterol emboli syndrome in atherosclerosis. Curr Atheroscler Rep. 2013;15:315.
10. Jucgla A, Moreso F, Muniesa C, et al. Cholesterol embolism: still an unrecognized entity with a high mortality rate. J Am Acad Dermatol. 2006;55:786-793.
11. Kim H, Zhen DB, Lieske JC, et al. Treatment of cholesterol embolization syndrome in the setting of an acute indication for anticoagulation therapy. J Med Cases. 2014;5:376-379.
12. Igarashi Y, Akimoto T, Kobayashi T, et al. Performing anticoagulation: a puzzling case of cholesterol embolization syndrome. Clin Med Insights Case Rep. 2017;10:1179547616684649. doi:10.1177/1179547616684649.
