User login
Extramedullary plasmacytoma of the thyroid, refractory to radiation therapy and treated with bortezomib
Plasma cell neoplasms involving tissues other than the bone marrow are known as extramedullary plasmacytoma (EMP).1 EMPs mostly involve the head and neck region.2 Solitary EMP involving only the thyroid gland is very rare.3,4 Because of the limited knowledge about this condition and its rarity, its management can be challenging and is often extrapolated from plasma cell myeloma.5,6 In general, surgery or radiation are considered as front-line therapy.3,5 EMPs usually respond well to radiotherapy with almost complete remission. No definite guidelines outlining the treatment of radio-resistant EMP of the thyroid have yet been published. Data supporting the use of chemotherapy is particularly limited.4,7,8
Here, we describe the case of a 53-year-old woman with a long history of thyroiditis who presented with rapidly worsening symptomatic thyroid enlargement. She was diagnosed with EMP of the thyroid gland that was not amenable to surgery and was refractory to radiotherapy but responded to adjuvant chemotherapy with bortezomib. This report highlights 2 unique aspects of this condition: it focuses on a rare case of EMP and, as far as we know, it reports for the first time on EMP that was resistant to radiotherapy. It also highlights the need for guidelines for the treatment of EMPs.
Case presentation and summary
A 53-year-old woman presented to the emergency department with complaints of difficulty swallowing, hoarseness, and neck pain during the previous 1 month. She had a known history of Hashimoto’s thyroiditis, and an ultrasound scan of her neck 6 years previously had demonstrated diffuse thyromegaly without discrete nodules. On presentation, the patient’s vitals were stable, and a neck examination revealed a firm and enlarged thyroid without any cervical adenopathy. Laboratory investigations revealed a normal complete blood count and comprehensive metabolic panel. She had an elevated thyroid-stimulating hormone level of 13.40 mIU/L (reference range, 0.47-4.68 mIU/L) and normal thyroxine level of 4.5 pmol/L (reference range, 4.5-12.0 pmol/L). A computerized tomography (CT) scan of the neck revealed an enlarged thyroid gland (right lobe length, 10.3 cm; isthmus, 2 cm; left lobe, 8 cm) with a focal area of increased echogenicity in the midpole of the left lobe measuring 9.5 mm × 5.5 mm. The patient was discharged to home with pain medications, and urgent follow-up with an otolaryngologist was arranged. A flexible laryngoscopy was done in the otolaryngology clinic, which revealed retropharyngeal bulging that correlated with the thyromegaly evident on the CT scan.
Because of the patient’s significant symptoms, we decided to proceed with surgery with a clinical diagnosis of likely thyroiditis. A left subtotal thyroidectomy with extension to the superior mediastinum was performed, but a right thyroidectomy could not be done safely. On gross examination, a well-capsulated left lobe with a tan-white, lobulated, soft cut surface was seen. Microscopic examination revealed replacement of thyroid parenchyma with sheets of mature-appearing plasma cells with eccentric round nuclei, abundant eosinophilic cytoplasm without atypia, and few scattered thyroid follicles with lymphoepithelial lesions (Figure 1A). Immunohistochemistry confirmed plasma cells with expression of CD138 (Figure 1B).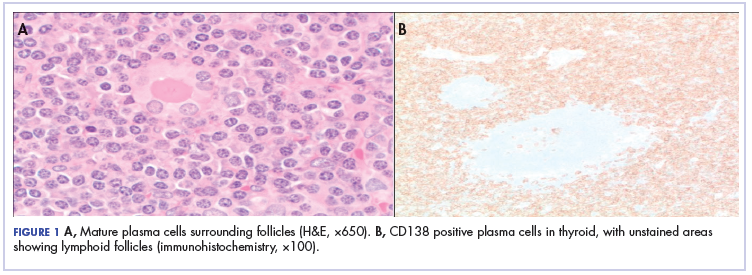
Fluorescence in situ hybridization (FISH) showed that the neoplastic plasma cells contained monotypic kappa immunoglobulin light chain messenger RNA. Clonal immunoglobulin gene rearrangement was detected on polymerase chain reaction. A diagnosis of plasmacytoma of the thyroid gland in a background of thyroiditis was made on the basis of the aforementioned observations.
After that diagnosis, we performed an extensive work-up for plasma cell myeloma. Bone marrow biopsy showed normal maturing trilineage hematopoiesis with scattered mature-appearing plasma cells Figure 2A. Flow cytometry showed a rare (0.2%) population of polytypic plasma cells and was confirmed by CD138 immunohistochemistry. FISH showed proportionate distribution (2-5:1) of kappa and lambda light chains in plasma cells (Figure 2B).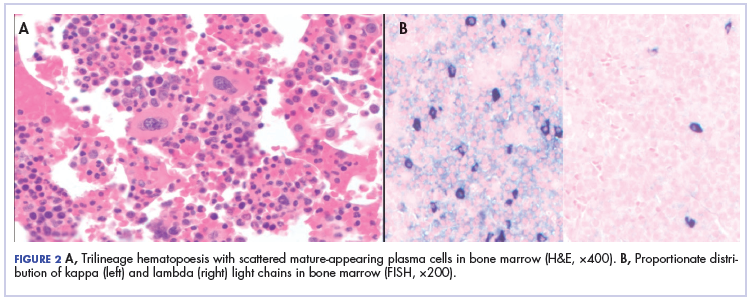
Serum protein electrophoresis showed normal levels of serum proteins with no M spike. Serum total protein was 7.9 g/dL, albumin 5.0 g/dL, α1-globulin 0.3 g/dL, α2-globulin 0.8 g/dL, β-globulin 0.7 g/dL, and γ-globulin 1.6 g/dL, with an albumin–globulin ratio of 1.47. Calcium and β2-microglobulin were also in the normal ranges. Serum-free kappa light chain was found to be elevated (20.9 mg/L; reference range, 3.3-19.4 mg/L). The immunoglobulin G level was also elevated at 3,104 mg/dL (reference range, 700-1,600 mg/dL).
A positron-emission tomographic (PET) scan done 1 month after the surgery showed no other sites of disease except the thyroid. No lytic bone lesions were present. The patient was treated with 50.4 Gy of radiation by external beam radiotherapy to the thyroid in 28 fractions as definitive therapy. Despite treatment with surgery and radiation, she continued to have pain around the neck, and a repeat PET scan 3 months after completion of radiation showed persistent uptake in the thyroid. Because of her refractoriness to radiotherapy, she was started on systemic therapy with a weekly regimen of bortezomib and dexamethasone for 9 cycles. Her symptoms began to resolve, and a repeat PET scan done after completion of chemotherapy showed no evidence of uptake, suggesting adequate response to chemotherapy, and chemotherapy was therefore stopped. The patient was scheduled a regular follow-up in 3 months. Because of continued local symptoms in this follow-up period, the decision was made to perform surgical gland removal, and she underwent completion of
Discussion
Plasma cells are well-differentiated B-lymphocytes that secrete antibodies and provide protective immunity to the human body.9 Plasma cell neoplasms are clonal proliferation of plasma cells, producing monoclonal immunoglobulins. They are of the following different types: plasma cell myeloma, monoclonal gammopathy of unknown significance, immunoglobulin deposition disease, POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes) syndrome, and plasmacytomas, which are divided into 2 types – solitary plasmacytoma of the bone, and extramedullary plasmacytoma (EMP).10 EMP is a rare condition and encompasses 3% to 5% of all plasma cell neoplasms, depending on the study.1,2,5 It is more common in men than in women (2.6:1, respectively), with equal incidence among black and white patients. Median age at diagnosis is 62 years, and it is more common among those aged 40 to 70 years.2,11 The most common sites of occurrence are the respiratory tract, the mouth, and the pharynx, but other sites such as the eyes, brain, skin, and lymph nodes may also be involved.2
EMP involving the thyroid gland is a very rare occurrence, but plasma cell myeloma has been shown to secondarily involve the thyroid.4 Similar to our report, EMP of the thyroid in the setting of thyroiditis has been reported by other authors.3,4 The incidence of EMP occurring in the thyroid varies according to different authors. Wiltshaw found 7 cases involving the thyroid out of 272 cases of EMP.1 Galieni and colleagues reported only 1 case that involved the thyroid out of 46 described cases of EMP.12
El- Siemińska and colleagues showed that levels of interleukin (IL)-6 are elevated in thyroiditis.13 IL-6 promotes monoclonal as well as polyclonal proliferation of plasma cells. Kovalchuk and colleagues showed an increase in EMP in IL-6 transgenic mice, suggesting a pathophysiologic explanation.14
The diagnostic requirements of EMP include the following: histology showing monoclonal plasma cell infiltration in tissue; bone marrow biopsy with normal plasma cell aspirate and biopsy (plasma cells, <5%); no lytic lesions on skeletal survey; no anemia, renal impairment, or hypercalcemia; and absent or low serum M protein.12
Our case fulfilled those criteria.
The treatment options for EMP include surgery, radiotherapy, or a combined approach including both. Usually, EMPs are very sensitive to radiotherapy, and complete remission can be achieved by radiotherapy alone in 80% to 100% of cases.6,11,15 Surgery is considered if the tumor is diffuse or is causing symptoms secondary to pressure on surrounding structures. A combined approach is recommended in cases with incomplete surgical margin or lymph node involvement.5,6
There is limited evidence about and experience with the use of chemotherapy in the treatment of EMP. It has been recommended that chemotherapy be considered in patients with refractory or relapsed disease using the same regimen used in plasma cell myeloma.5 Katodritou and colleagues have reported using bortezomib and dexamethasone without surgery in a solitary gastric plasmacytoma to avoid the toxicity of gastrointestinal irradiation.7 Wei and colleagues treated a patient with EMP in the pancreas with bortezomib and achieved a near-complete remission.8 To our knowledge, there is no documented literature about the treatment of EMP of the thyroid with chemotherapy. In our patient, despite the treatment with surgery and radiation, there was persistent uptake on the PET scan, so we treated her with bortezomib and dexamethasone. Because there is an 11% to 30% risk of progression to multiple myeloma, long-term follow-up is recommended in EMP.11
Conclusions
Solitary EMP of the thyroid gland is a rare condition. Plasma cell myeloma must be ruled out to make a diagnosis. Data on the incidence of EMP and its clinicopathological features are sparse, and literature describing proper guidelines on treatment is limited. It can be treated with radiotherapy, surgery, or a combined approach. There is limited data on the role of chemotherapy; our case adds to the available literature on using myeloma-based therapy in refractory disease and, to our knowledge, is the only case report using this in the literature on cases of EMP of the thyroid. Regular follow-up, even after the disease is in remission, is necessary because of the high risk of progression to plasma cell myeloma.
1. Wiltshaw E. The natural history of extramedullary plasmacytoma and its relation to solitary myeloma of bone and myelomatosis. Medicine (Baltimore). 1976;55(3):217-238.
2. Dores GM, Landgren O, McGlynn KA, Curtis RE, Linet MS, Devesa SS. Plasmacytoma of bone, extramedullary plasmacytoma, and multiple myeloma: incidence and survival in the United States, 1992-2004. Br J Haematol. 2009;144(1):86-94.
3. Kovacs CS, Mant MJ, Nguyen GK, Ginsberg J. Plasma cell lesions of the thyroid: report of a case of solitary plasmacytoma and a review of the literature. Thyroid. 1994;4(1):65-71.
4. Avila A, Villalpando A, Montoya G, Luna MA. Clinical features and differential diagnoses of solitary extramedullary plasmacytoma of the thyroid: a case report. Ann Diagn Pathol. 2009;13(2):119-123.
5. Hughes M, Soutar R, Lucraft H, Owen R, Bird J. Guidelines on the diagnosis and management of solitary plasmacytoma of bone, extramedullary plasmacytoma and multiple solitary plasmacytomas: 2009 update. London, United Kingdom: British Committee for Standards in Haematology; 2009.
6. Weber DM. Solitary bone and extramedullary plasmacytoma. Hematology Am Soc Hematol Educ Program. 2005;373-376.
7. Katodritou E, Kartsios C, Gastari V, et al. Successful treatment of extramedullary gastric plasmacytoma with the combination of bortezomib and dexamethasone: first reported case. Leuk Res. 2008;32(2):339-341.
8. Wei JY, Tong HY, Zhu WF, et al. Bortezomib in treatment of extramedullary plasmacytoma of the pancreas. Hepatobiliary Pancreat Dis Int. 2009;8(3):329-331.
9. Roth K, Oehme L, Zehentmeier S, Zhang Y, Niesner R, Hauser AE. Tracking plasma cell differentiation and survival. Cytometry A. 2014;85(1):15-24.
10. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: International Agency for Research on Cancer; 2008.
11. Alexiou C, Kau RJ, Dietzfelbinger H, et al. Extramedullary plasmacytoma: tumor occurrence and therapeutic concepts. Cancer. 1999;85(11):2305-2314.
12. Galieni P, Cavo M, Pulsoni A, et al. Clinical outcome of extramedullary plasmacytoma. Haematologica. 2000;85(1):47-51.
13. Siemińska L, Wojciechowska C, Kos-Kudła B, et al. Serum concentrations of leptin, adiponectin, and interleukin-6 in postmenopausal women with Hashimoto’s thyroiditis. Endokrynol Pol. 2010;61(1):112-116.
14. Kovalchuk AL, Kim JS, Park SS, et al. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc Natl Acad Sci US
15. Chao MW, Gibbs P, Wirth A, Quong G, Guiney MJ, Liew KH. Radiotherapy in the management of solitary extramedullary plasmacytoma. Intern Med J. 2005;35(4):211-215.
Plasma cell neoplasms involving tissues other than the bone marrow are known as extramedullary plasmacytoma (EMP).1 EMPs mostly involve the head and neck region.2 Solitary EMP involving only the thyroid gland is very rare.3,4 Because of the limited knowledge about this condition and its rarity, its management can be challenging and is often extrapolated from plasma cell myeloma.5,6 In general, surgery or radiation are considered as front-line therapy.3,5 EMPs usually respond well to radiotherapy with almost complete remission. No definite guidelines outlining the treatment of radio-resistant EMP of the thyroid have yet been published. Data supporting the use of chemotherapy is particularly limited.4,7,8
Here, we describe the case of a 53-year-old woman with a long history of thyroiditis who presented with rapidly worsening symptomatic thyroid enlargement. She was diagnosed with EMP of the thyroid gland that was not amenable to surgery and was refractory to radiotherapy but responded to adjuvant chemotherapy with bortezomib. This report highlights 2 unique aspects of this condition: it focuses on a rare case of EMP and, as far as we know, it reports for the first time on EMP that was resistant to radiotherapy. It also highlights the need for guidelines for the treatment of EMPs.
Case presentation and summary
A 53-year-old woman presented to the emergency department with complaints of difficulty swallowing, hoarseness, and neck pain during the previous 1 month. She had a known history of Hashimoto’s thyroiditis, and an ultrasound scan of her neck 6 years previously had demonstrated diffuse thyromegaly without discrete nodules. On presentation, the patient’s vitals were stable, and a neck examination revealed a firm and enlarged thyroid without any cervical adenopathy. Laboratory investigations revealed a normal complete blood count and comprehensive metabolic panel. She had an elevated thyroid-stimulating hormone level of 13.40 mIU/L (reference range, 0.47-4.68 mIU/L) and normal thyroxine level of 4.5 pmol/L (reference range, 4.5-12.0 pmol/L). A computerized tomography (CT) scan of the neck revealed an enlarged thyroid gland (right lobe length, 10.3 cm; isthmus, 2 cm; left lobe, 8 cm) with a focal area of increased echogenicity in the midpole of the left lobe measuring 9.5 mm × 5.5 mm. The patient was discharged to home with pain medications, and urgent follow-up with an otolaryngologist was arranged. A flexible laryngoscopy was done in the otolaryngology clinic, which revealed retropharyngeal bulging that correlated with the thyromegaly evident on the CT scan.
Because of the patient’s significant symptoms, we decided to proceed with surgery with a clinical diagnosis of likely thyroiditis. A left subtotal thyroidectomy with extension to the superior mediastinum was performed, but a right thyroidectomy could not be done safely. On gross examination, a well-capsulated left lobe with a tan-white, lobulated, soft cut surface was seen. Microscopic examination revealed replacement of thyroid parenchyma with sheets of mature-appearing plasma cells with eccentric round nuclei, abundant eosinophilic cytoplasm without atypia, and few scattered thyroid follicles with lymphoepithelial lesions (Figure 1A). Immunohistochemistry confirmed plasma cells with expression of CD138 (Figure 1B).
Fluorescence in situ hybridization (FISH) showed that the neoplastic plasma cells contained monotypic kappa immunoglobulin light chain messenger RNA. Clonal immunoglobulin gene rearrangement was detected on polymerase chain reaction. A diagnosis of plasmacytoma of the thyroid gland in a background of thyroiditis was made on the basis of the aforementioned observations.
After that diagnosis, we performed an extensive work-up for plasma cell myeloma. Bone marrow biopsy showed normal maturing trilineage hematopoiesis with scattered mature-appearing plasma cells Figure 2A. Flow cytometry showed a rare (0.2%) population of polytypic plasma cells and was confirmed by CD138 immunohistochemistry. FISH showed proportionate distribution (2-5:1) of kappa and lambda light chains in plasma cells (Figure 2B).
Serum protein electrophoresis showed normal levels of serum proteins with no M spike. Serum total protein was 7.9 g/dL, albumin 5.0 g/dL, α1-globulin 0.3 g/dL, α2-globulin 0.8 g/dL, β-globulin 0.7 g/dL, and γ-globulin 1.6 g/dL, with an albumin–globulin ratio of 1.47. Calcium and β2-microglobulin were also in the normal ranges. Serum-free kappa light chain was found to be elevated (20.9 mg/L; reference range, 3.3-19.4 mg/L). The immunoglobulin G level was also elevated at 3,104 mg/dL (reference range, 700-1,600 mg/dL).
A positron-emission tomographic (PET) scan done 1 month after the surgery showed no other sites of disease except the thyroid. No lytic bone lesions were present. The patient was treated with 50.4 Gy of radiation by external beam radiotherapy to the thyroid in 28 fractions as definitive therapy. Despite treatment with surgery and radiation, she continued to have pain around the neck, and a repeat PET scan 3 months after completion of radiation showed persistent uptake in the thyroid. Because of her refractoriness to radiotherapy, she was started on systemic therapy with a weekly regimen of bortezomib and dexamethasone for 9 cycles. Her symptoms began to resolve, and a repeat PET scan done after completion of chemotherapy showed no evidence of uptake, suggesting adequate response to chemotherapy, and chemotherapy was therefore stopped. The patient was scheduled a regular follow-up in 3 months. Because of continued local symptoms in this follow-up period, the decision was made to perform surgical gland removal, and she underwent completion of
Discussion
Plasma cells are well-differentiated B-lymphocytes that secrete antibodies and provide protective immunity to the human body.9 Plasma cell neoplasms are clonal proliferation of plasma cells, producing monoclonal immunoglobulins. They are of the following different types: plasma cell myeloma, monoclonal gammopathy of unknown significance, immunoglobulin deposition disease, POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes) syndrome, and plasmacytomas, which are divided into 2 types – solitary plasmacytoma of the bone, and extramedullary plasmacytoma (EMP).10 EMP is a rare condition and encompasses 3% to 5% of all plasma cell neoplasms, depending on the study.1,2,5 It is more common in men than in women (2.6:1, respectively), with equal incidence among black and white patients. Median age at diagnosis is 62 years, and it is more common among those aged 40 to 70 years.2,11 The most common sites of occurrence are the respiratory tract, the mouth, and the pharynx, but other sites such as the eyes, brain, skin, and lymph nodes may also be involved.2
EMP involving the thyroid gland is a very rare occurrence, but plasma cell myeloma has been shown to secondarily involve the thyroid.4 Similar to our report, EMP of the thyroid in the setting of thyroiditis has been reported by other authors.3,4 The incidence of EMP occurring in the thyroid varies according to different authors. Wiltshaw found 7 cases involving the thyroid out of 272 cases of EMP.1 Galieni and colleagues reported only 1 case that involved the thyroid out of 46 described cases of EMP.12
El- Siemińska and colleagues showed that levels of interleukin (IL)-6 are elevated in thyroiditis.13 IL-6 promotes monoclonal as well as polyclonal proliferation of plasma cells. Kovalchuk and colleagues showed an increase in EMP in IL-6 transgenic mice, suggesting a pathophysiologic explanation.14
The diagnostic requirements of EMP include the following: histology showing monoclonal plasma cell infiltration in tissue; bone marrow biopsy with normal plasma cell aspirate and biopsy (plasma cells, <5%); no lytic lesions on skeletal survey; no anemia, renal impairment, or hypercalcemia; and absent or low serum M protein.12
Our case fulfilled those criteria.
The treatment options for EMP include surgery, radiotherapy, or a combined approach including both. Usually, EMPs are very sensitive to radiotherapy, and complete remission can be achieved by radiotherapy alone in 80% to 100% of cases.6,11,15 Surgery is considered if the tumor is diffuse or is causing symptoms secondary to pressure on surrounding structures. A combined approach is recommended in cases with incomplete surgical margin or lymph node involvement.5,6
There is limited evidence about and experience with the use of chemotherapy in the treatment of EMP. It has been recommended that chemotherapy be considered in patients with refractory or relapsed disease using the same regimen used in plasma cell myeloma.5 Katodritou and colleagues have reported using bortezomib and dexamethasone without surgery in a solitary gastric plasmacytoma to avoid the toxicity of gastrointestinal irradiation.7 Wei and colleagues treated a patient with EMP in the pancreas with bortezomib and achieved a near-complete remission.8 To our knowledge, there is no documented literature about the treatment of EMP of the thyroid with chemotherapy. In our patient, despite the treatment with surgery and radiation, there was persistent uptake on the PET scan, so we treated her with bortezomib and dexamethasone. Because there is an 11% to 30% risk of progression to multiple myeloma, long-term follow-up is recommended in EMP.11
Conclusions
Solitary EMP of the thyroid gland is a rare condition. Plasma cell myeloma must be ruled out to make a diagnosis. Data on the incidence of EMP and its clinicopathological features are sparse, and literature describing proper guidelines on treatment is limited. It can be treated with radiotherapy, surgery, or a combined approach. There is limited data on the role of chemotherapy; our case adds to the available literature on using myeloma-based therapy in refractory disease and, to our knowledge, is the only case report using this in the literature on cases of EMP of the thyroid. Regular follow-up, even after the disease is in remission, is necessary because of the high risk of progression to plasma cell myeloma.
Plasma cell neoplasms involving tissues other than the bone marrow are known as extramedullary plasmacytoma (EMP).1 EMPs mostly involve the head and neck region.2 Solitary EMP involving only the thyroid gland is very rare.3,4 Because of the limited knowledge about this condition and its rarity, its management can be challenging and is often extrapolated from plasma cell myeloma.5,6 In general, surgery or radiation are considered as front-line therapy.3,5 EMPs usually respond well to radiotherapy with almost complete remission. No definite guidelines outlining the treatment of radio-resistant EMP of the thyroid have yet been published. Data supporting the use of chemotherapy is particularly limited.4,7,8
Here, we describe the case of a 53-year-old woman with a long history of thyroiditis who presented with rapidly worsening symptomatic thyroid enlargement. She was diagnosed with EMP of the thyroid gland that was not amenable to surgery and was refractory to radiotherapy but responded to adjuvant chemotherapy with bortezomib. This report highlights 2 unique aspects of this condition: it focuses on a rare case of EMP and, as far as we know, it reports for the first time on EMP that was resistant to radiotherapy. It also highlights the need for guidelines for the treatment of EMPs.
Case presentation and summary
A 53-year-old woman presented to the emergency department with complaints of difficulty swallowing, hoarseness, and neck pain during the previous 1 month. She had a known history of Hashimoto’s thyroiditis, and an ultrasound scan of her neck 6 years previously had demonstrated diffuse thyromegaly without discrete nodules. On presentation, the patient’s vitals were stable, and a neck examination revealed a firm and enlarged thyroid without any cervical adenopathy. Laboratory investigations revealed a normal complete blood count and comprehensive metabolic panel. She had an elevated thyroid-stimulating hormone level of 13.40 mIU/L (reference range, 0.47-4.68 mIU/L) and normal thyroxine level of 4.5 pmol/L (reference range, 4.5-12.0 pmol/L). A computerized tomography (CT) scan of the neck revealed an enlarged thyroid gland (right lobe length, 10.3 cm; isthmus, 2 cm; left lobe, 8 cm) with a focal area of increased echogenicity in the midpole of the left lobe measuring 9.5 mm × 5.5 mm. The patient was discharged to home with pain medications, and urgent follow-up with an otolaryngologist was arranged. A flexible laryngoscopy was done in the otolaryngology clinic, which revealed retropharyngeal bulging that correlated with the thyromegaly evident on the CT scan.
Because of the patient’s significant symptoms, we decided to proceed with surgery with a clinical diagnosis of likely thyroiditis. A left subtotal thyroidectomy with extension to the superior mediastinum was performed, but a right thyroidectomy could not be done safely. On gross examination, a well-capsulated left lobe with a tan-white, lobulated, soft cut surface was seen. Microscopic examination revealed replacement of thyroid parenchyma with sheets of mature-appearing plasma cells with eccentric round nuclei, abundant eosinophilic cytoplasm without atypia, and few scattered thyroid follicles with lymphoepithelial lesions (Figure 1A). Immunohistochemistry confirmed plasma cells with expression of CD138 (Figure 1B).
Fluorescence in situ hybridization (FISH) showed that the neoplastic plasma cells contained monotypic kappa immunoglobulin light chain messenger RNA. Clonal immunoglobulin gene rearrangement was detected on polymerase chain reaction. A diagnosis of plasmacytoma of the thyroid gland in a background of thyroiditis was made on the basis of the aforementioned observations.
After that diagnosis, we performed an extensive work-up for plasma cell myeloma. Bone marrow biopsy showed normal maturing trilineage hematopoiesis with scattered mature-appearing plasma cells Figure 2A. Flow cytometry showed a rare (0.2%) population of polytypic plasma cells and was confirmed by CD138 immunohistochemistry. FISH showed proportionate distribution (2-5:1) of kappa and lambda light chains in plasma cells (Figure 2B).
Serum protein electrophoresis showed normal levels of serum proteins with no M spike. Serum total protein was 7.9 g/dL, albumin 5.0 g/dL, α1-globulin 0.3 g/dL, α2-globulin 0.8 g/dL, β-globulin 0.7 g/dL, and γ-globulin 1.6 g/dL, with an albumin–globulin ratio of 1.47. Calcium and β2-microglobulin were also in the normal ranges. Serum-free kappa light chain was found to be elevated (20.9 mg/L; reference range, 3.3-19.4 mg/L). The immunoglobulin G level was also elevated at 3,104 mg/dL (reference range, 700-1,600 mg/dL).
A positron-emission tomographic (PET) scan done 1 month after the surgery showed no other sites of disease except the thyroid. No lytic bone lesions were present. The patient was treated with 50.4 Gy of radiation by external beam radiotherapy to the thyroid in 28 fractions as definitive therapy. Despite treatment with surgery and radiation, she continued to have pain around the neck, and a repeat PET scan 3 months after completion of radiation showed persistent uptake in the thyroid. Because of her refractoriness to radiotherapy, she was started on systemic therapy with a weekly regimen of bortezomib and dexamethasone for 9 cycles. Her symptoms began to resolve, and a repeat PET scan done after completion of chemotherapy showed no evidence of uptake, suggesting adequate response to chemotherapy, and chemotherapy was therefore stopped. The patient was scheduled a regular follow-up in 3 months. Because of continued local symptoms in this follow-up period, the decision was made to perform surgical gland removal, and she underwent completion of
Discussion
Plasma cells are well-differentiated B-lymphocytes that secrete antibodies and provide protective immunity to the human body.9 Plasma cell neoplasms are clonal proliferation of plasma cells, producing monoclonal immunoglobulins. They are of the following different types: plasma cell myeloma, monoclonal gammopathy of unknown significance, immunoglobulin deposition disease, POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes) syndrome, and plasmacytomas, which are divided into 2 types – solitary plasmacytoma of the bone, and extramedullary plasmacytoma (EMP).10 EMP is a rare condition and encompasses 3% to 5% of all plasma cell neoplasms, depending on the study.1,2,5 It is more common in men than in women (2.6:1, respectively), with equal incidence among black and white patients. Median age at diagnosis is 62 years, and it is more common among those aged 40 to 70 years.2,11 The most common sites of occurrence are the respiratory tract, the mouth, and the pharynx, but other sites such as the eyes, brain, skin, and lymph nodes may also be involved.2
EMP involving the thyroid gland is a very rare occurrence, but plasma cell myeloma has been shown to secondarily involve the thyroid.4 Similar to our report, EMP of the thyroid in the setting of thyroiditis has been reported by other authors.3,4 The incidence of EMP occurring in the thyroid varies according to different authors. Wiltshaw found 7 cases involving the thyroid out of 272 cases of EMP.1 Galieni and colleagues reported only 1 case that involved the thyroid out of 46 described cases of EMP.12
El- Siemińska and colleagues showed that levels of interleukin (IL)-6 are elevated in thyroiditis.13 IL-6 promotes monoclonal as well as polyclonal proliferation of plasma cells. Kovalchuk and colleagues showed an increase in EMP in IL-6 transgenic mice, suggesting a pathophysiologic explanation.14
The diagnostic requirements of EMP include the following: histology showing monoclonal plasma cell infiltration in tissue; bone marrow biopsy with normal plasma cell aspirate and biopsy (plasma cells, <5%); no lytic lesions on skeletal survey; no anemia, renal impairment, or hypercalcemia; and absent or low serum M protein.12
Our case fulfilled those criteria.
The treatment options for EMP include surgery, radiotherapy, or a combined approach including both. Usually, EMPs are very sensitive to radiotherapy, and complete remission can be achieved by radiotherapy alone in 80% to 100% of cases.6,11,15 Surgery is considered if the tumor is diffuse or is causing symptoms secondary to pressure on surrounding structures. A combined approach is recommended in cases with incomplete surgical margin or lymph node involvement.5,6
There is limited evidence about and experience with the use of chemotherapy in the treatment of EMP. It has been recommended that chemotherapy be considered in patients with refractory or relapsed disease using the same regimen used in plasma cell myeloma.5 Katodritou and colleagues have reported using bortezomib and dexamethasone without surgery in a solitary gastric plasmacytoma to avoid the toxicity of gastrointestinal irradiation.7 Wei and colleagues treated a patient with EMP in the pancreas with bortezomib and achieved a near-complete remission.8 To our knowledge, there is no documented literature about the treatment of EMP of the thyroid with chemotherapy. In our patient, despite the treatment with surgery and radiation, there was persistent uptake on the PET scan, so we treated her with bortezomib and dexamethasone. Because there is an 11% to 30% risk of progression to multiple myeloma, long-term follow-up is recommended in EMP.11
Conclusions
Solitary EMP of the thyroid gland is a rare condition. Plasma cell myeloma must be ruled out to make a diagnosis. Data on the incidence of EMP and its clinicopathological features are sparse, and literature describing proper guidelines on treatment is limited. It can be treated with radiotherapy, surgery, or a combined approach. There is limited data on the role of chemotherapy; our case adds to the available literature on using myeloma-based therapy in refractory disease and, to our knowledge, is the only case report using this in the literature on cases of EMP of the thyroid. Regular follow-up, even after the disease is in remission, is necessary because of the high risk of progression to plasma cell myeloma.
1. Wiltshaw E. The natural history of extramedullary plasmacytoma and its relation to solitary myeloma of bone and myelomatosis. Medicine (Baltimore). 1976;55(3):217-238.
2. Dores GM, Landgren O, McGlynn KA, Curtis RE, Linet MS, Devesa SS. Plasmacytoma of bone, extramedullary plasmacytoma, and multiple myeloma: incidence and survival in the United States, 1992-2004. Br J Haematol. 2009;144(1):86-94.
3. Kovacs CS, Mant MJ, Nguyen GK, Ginsberg J. Plasma cell lesions of the thyroid: report of a case of solitary plasmacytoma and a review of the literature. Thyroid. 1994;4(1):65-71.
4. Avila A, Villalpando A, Montoya G, Luna MA. Clinical features and differential diagnoses of solitary extramedullary plasmacytoma of the thyroid: a case report. Ann Diagn Pathol. 2009;13(2):119-123.
5. Hughes M, Soutar R, Lucraft H, Owen R, Bird J. Guidelines on the diagnosis and management of solitary plasmacytoma of bone, extramedullary plasmacytoma and multiple solitary plasmacytomas: 2009 update. London, United Kingdom: British Committee for Standards in Haematology; 2009.
6. Weber DM. Solitary bone and extramedullary plasmacytoma. Hematology Am Soc Hematol Educ Program. 2005;373-376.
7. Katodritou E, Kartsios C, Gastari V, et al. Successful treatment of extramedullary gastric plasmacytoma with the combination of bortezomib and dexamethasone: first reported case. Leuk Res. 2008;32(2):339-341.
8. Wei JY, Tong HY, Zhu WF, et al. Bortezomib in treatment of extramedullary plasmacytoma of the pancreas. Hepatobiliary Pancreat Dis Int. 2009;8(3):329-331.
9. Roth K, Oehme L, Zehentmeier S, Zhang Y, Niesner R, Hauser AE. Tracking plasma cell differentiation and survival. Cytometry A. 2014;85(1):15-24.
10. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: International Agency for Research on Cancer; 2008.
11. Alexiou C, Kau RJ, Dietzfelbinger H, et al. Extramedullary plasmacytoma: tumor occurrence and therapeutic concepts. Cancer. 1999;85(11):2305-2314.
12. Galieni P, Cavo M, Pulsoni A, et al. Clinical outcome of extramedullary plasmacytoma. Haematologica. 2000;85(1):47-51.
13. Siemińska L, Wojciechowska C, Kos-Kudła B, et al. Serum concentrations of leptin, adiponectin, and interleukin-6 in postmenopausal women with Hashimoto’s thyroiditis. Endokrynol Pol. 2010;61(1):112-116.
14. Kovalchuk AL, Kim JS, Park SS, et al. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc Natl Acad Sci US
15. Chao MW, Gibbs P, Wirth A, Quong G, Guiney MJ, Liew KH. Radiotherapy in the management of solitary extramedullary plasmacytoma. Intern Med J. 2005;35(4):211-215.
1. Wiltshaw E. The natural history of extramedullary plasmacytoma and its relation to solitary myeloma of bone and myelomatosis. Medicine (Baltimore). 1976;55(3):217-238.
2. Dores GM, Landgren O, McGlynn KA, Curtis RE, Linet MS, Devesa SS. Plasmacytoma of bone, extramedullary plasmacytoma, and multiple myeloma: incidence and survival in the United States, 1992-2004. Br J Haematol. 2009;144(1):86-94.
3. Kovacs CS, Mant MJ, Nguyen GK, Ginsberg J. Plasma cell lesions of the thyroid: report of a case of solitary plasmacytoma and a review of the literature. Thyroid. 1994;4(1):65-71.
4. Avila A, Villalpando A, Montoya G, Luna MA. Clinical features and differential diagnoses of solitary extramedullary plasmacytoma of the thyroid: a case report. Ann Diagn Pathol. 2009;13(2):119-123.
5. Hughes M, Soutar R, Lucraft H, Owen R, Bird J. Guidelines on the diagnosis and management of solitary plasmacytoma of bone, extramedullary plasmacytoma and multiple solitary plasmacytomas: 2009 update. London, United Kingdom: British Committee for Standards in Haematology; 2009.
6. Weber DM. Solitary bone and extramedullary plasmacytoma. Hematology Am Soc Hematol Educ Program. 2005;373-376.
7. Katodritou E, Kartsios C, Gastari V, et al. Successful treatment of extramedullary gastric plasmacytoma with the combination of bortezomib and dexamethasone: first reported case. Leuk Res. 2008;32(2):339-341.
8. Wei JY, Tong HY, Zhu WF, et al. Bortezomib in treatment of extramedullary plasmacytoma of the pancreas. Hepatobiliary Pancreat Dis Int. 2009;8(3):329-331.
9. Roth K, Oehme L, Zehentmeier S, Zhang Y, Niesner R, Hauser AE. Tracking plasma cell differentiation and survival. Cytometry A. 2014;85(1):15-24.
10. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: International Agency for Research on Cancer; 2008.
11. Alexiou C, Kau RJ, Dietzfelbinger H, et al. Extramedullary plasmacytoma: tumor occurrence and therapeutic concepts. Cancer. 1999;85(11):2305-2314.
12. Galieni P, Cavo M, Pulsoni A, et al. Clinical outcome of extramedullary plasmacytoma. Haematologica. 2000;85(1):47-51.
13. Siemińska L, Wojciechowska C, Kos-Kudła B, et al. Serum concentrations of leptin, adiponectin, and interleukin-6 in postmenopausal women with Hashimoto’s thyroiditis. Endokrynol Pol. 2010;61(1):112-116.
14. Kovalchuk AL, Kim JS, Park SS, et al. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc Natl Acad Sci US
15. Chao MW, Gibbs P, Wirth A, Quong G, Guiney MJ, Liew KH. Radiotherapy in the management of solitary extramedullary plasmacytoma. Intern Med J. 2005;35(4):211-215.
Salivary ductal adenocarcinoma with complete response to androgen blockade
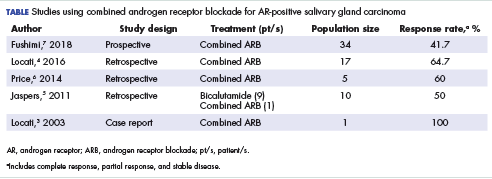
Salivary ductal adenocarcinomas make up about 9% of malignant salivary gland tumors and occur mostly in men older than 50 years, with a peak incidence in the sixth and seventh decades. It is the most aggressive of salivary gland tumors and is histologically similar to high-grade, invasive ductal carcinoma of the breast. In all, 65% of patients will die of the disease, and most will experience skin ulceration and nerve palsy.1 With such an aggressive clinical picture, the temptation for many oncologists and patients is to use aggressive cytotoxic chemotherapies. Considering the lack of large trials exploring treatment options in this less-common subtype of salivary gland carcinoma, practice guidelines also recommend the use of aggressive chemotherapies. Unlike other types of malignant cancers of the salivary glands, 70% to 90% of ductal adenocarcinomas express the androgen receptor (AR) by immunohistochemistry.2 There are reported cases of androgen deprivation therapy (ADT) as a successful treatment for salivary ductal adenocarcinomas that express the AR (Table).In 2003, Locati and colleagues reported the case of a man with salivary ductal adenocarcinomas who had a complete response with ADT.3 In 2016, the same group of authors published a retrospective analysis of 17 patients with recurrent or metastatic AR-positive salivary gland cancers who were treated with ADT and reported a 64.7% overall response rate among the patients.4 A 10-patient case series in the Netherlands demonstrated a 50% response rate to ADT plus bicalutamide, including a palliative effect in the form of pain relief.5 A retrospective analysis by Price and colleagues of 5 patients with AR-positive metastatic salivary duct adenocarcinoma showed a 60% response rate to a combination of leuprolide and bicalutamide.6
Case presentation and summary
A 91-year-old man was diagnosed with salivary ductal adenocarcinoma of the left parotid gland in September 2013 and underwent left parotidectomy and lymph node dissection, which revealed AJCC stage IVA (pT2 pN3 M0) disease. The following year, in December 2014, he had an enlarging left neck mass that was pathologically confirmed to be recurrent disease, and he underwent left level V neck dissection in February 2015. Five months after surgery, in July 2015, he presented with left neck fullness and new skin nodules, and the results of a biopsy confirmed recurrent disease. Given his relatively asymptomatic state and advanced age, the oncology care team decided to follow the patient without any pharmacologic therapy.
The patient felt relatively well for 11 months but slowly developed increasing pain in the left neck in June 2016. The skin nodules also began to spread inferiorly from his left neck to his upper chest with the development of open sores that wept serous fluid with scab formation (Figure 1). He and his wife lived independently and managed all their own instrumental activities of daily living (IADL). Eventually, the pain in his neck became so severe that it began to interfere with his ability to drive. He declined radiation therapy because of side effects and transportation issues, but he desired something to alleviate the burden of the disease. During a multidisciplinary cancer conference, the staff pathologist and oncologist discussed AR immunohistochemistry to assist with management. In June 2016, the patient’s tumor was found to have AR immunostaining (nuclear pattern) in 100% of cells, and he was treated with combined androgen blockade, consisting of monthly 3.6 mg goserelin injections and daily bicalutamide 50 mg orally.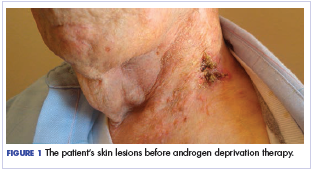
Within a week, the patient noticed that the skin lesions stopped weeping fluid. Within 2 weeks, the pain had begun to resolve. At his formal follow-up visit 11 weeks after starting treatment, he was not taking any pain medications and reported no pain. In addition, his visually apparent disease had almost completely resolved (Figure 2). He was fully able to manage his own IADL and reported a marked increase in satisfaction with the quality of his life.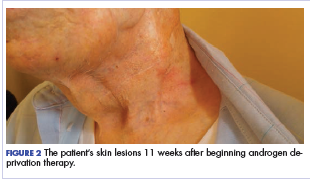
Discussion
The oncology care team clearly defined the goal of care for this patient as palliative and conveyed as such to the patient. The team considered the risks and side effects of cytotoxic chemotherapy agents to be contrary to the patient’s stated primary goal of independence. We selected the combined androgen blockade because it has a low toxicity rate and thus met the primary goals of therapy.
The European Organization for Research and Treatment of Cancer is presently conducting a trial in which cytotoxic chemotherapy is being compared with ADT in AR-positive salivary duct tumors. Findings from a recent prospective, phase-2 trial conducted in Japan suggested that combined AR blockade has similar efficacy and less toxicity than conventional cytotoxic chemotherapy for recurrent and/or metastatic and unresectable locally advanced AR-positive salivary gland carcinoma.7 As more data become available from other studies, it is possible that practice guidelines will be revised to recommend this treatment approach for these cancers.
1. Eveson JW, Thompson LDR. Malignant neoplasms of the salivary glands. In: Thompson LDR, ed. Head and neck pathology. 2nd ed. Philadelphia, PA: Elsevier Inc; 2013:304-305.
2. Luk PP, Weston JD, Yu B, et al. Salivary duct carcinoma: clinicopathologic features, morphologic spectrum, and somatic mutations. Head Neck. 2016;38(suppl 1):E1838-E1847.
3. Locati LD, Quattrone P, Bossi P, Marchianò AV, Cantù G, Licitra L. A complete remission with androgen-deprivation therapy in a recurrent androgen receptor-expressing adenocarcinoma of the parotid gland. Ann Oncol. 2003;14(8):1327-1328.
4. Locati LD, Perrone F, Cortelazzi B, et al. Clinical activity of androgen deprivation therapy in patients with metastatic/relapsed androgen receptor-positive salivary gland cancers. Head Neck. 2016;38(5):724-731.
5. Jaspers HC, Verbist BM, Schoffelen R, et al. Androgen receptor-positive salivary duct carcinoma: a disease entity with promising new treatment options. J Clin Oncol. 2011;29(16):e473-e476.
6. Price KAR, Okuno SH, Molina JR, Garcia JJ. Treatment of metastatic salivary duct carcinoma with combined androgen blockade (CAB) with leuprolide acetate and bicalutamide. Int J Radiat Oncol Biol Phys. 2014;88(2):521-522.
7. Fushimi C, Tada Y, Takahashi H, et al. A prospective phase II study of combined androgen blockade in patients with androgen receptor-positive metastatic or locally advanced unresectable salivary gland carcinoma. Ann Oncol. 2018;29(4):979-984.
Salivary ductal adenocarcinomas make up about 9% of malignant salivary gland tumors and occur mostly in men older than 50 years, with a peak incidence in the sixth and seventh decades. It is the most aggressive of salivary gland tumors and is histologically similar to high-grade, invasive ductal carcinoma of the breast. In all, 65% of patients will die of the disease, and most will experience skin ulceration and nerve palsy.1 With such an aggressive clinical picture, the temptation for many oncologists and patients is to use aggressive cytotoxic chemotherapies. Considering the lack of large trials exploring treatment options in this less-common subtype of salivary gland carcinoma, practice guidelines also recommend the use of aggressive chemotherapies. Unlike other types of malignant cancers of the salivary glands, 70% to 90% of ductal adenocarcinomas express the androgen receptor (AR) by immunohistochemistry.2 There are reported cases of androgen deprivation therapy (ADT) as a successful treatment for salivary ductal adenocarcinomas that express the AR (Table).In 2003, Locati and colleagues reported the case of a man with salivary ductal adenocarcinomas who had a complete response with ADT.3 In 2016, the same group of authors published a retrospective analysis of 17 patients with recurrent or metastatic AR-positive salivary gland cancers who were treated with ADT and reported a 64.7% overall response rate among the patients.4 A 10-patient case series in the Netherlands demonstrated a 50% response rate to ADT plus bicalutamide, including a palliative effect in the form of pain relief.5 A retrospective analysis by Price and colleagues of 5 patients with AR-positive metastatic salivary duct adenocarcinoma showed a 60% response rate to a combination of leuprolide and bicalutamide.6
Case presentation and summary
A 91-year-old man was diagnosed with salivary ductal adenocarcinoma of the left parotid gland in September 2013 and underwent left parotidectomy and lymph node dissection, which revealed AJCC stage IVA (pT2 pN3 M0) disease. The following year, in December 2014, he had an enlarging left neck mass that was pathologically confirmed to be recurrent disease, and he underwent left level V neck dissection in February 2015. Five months after surgery, in July 2015, he presented with left neck fullness and new skin nodules, and the results of a biopsy confirmed recurrent disease. Given his relatively asymptomatic state and advanced age, the oncology care team decided to follow the patient without any pharmacologic therapy.
The patient felt relatively well for 11 months but slowly developed increasing pain in the left neck in June 2016. The skin nodules also began to spread inferiorly from his left neck to his upper chest with the development of open sores that wept serous fluid with scab formation (Figure 1). He and his wife lived independently and managed all their own instrumental activities of daily living (IADL). Eventually, the pain in his neck became so severe that it began to interfere with his ability to drive. He declined radiation therapy because of side effects and transportation issues, but he desired something to alleviate the burden of the disease. During a multidisciplinary cancer conference, the staff pathologist and oncologist discussed AR immunohistochemistry to assist with management. In June 2016, the patient’s tumor was found to have AR immunostaining (nuclear pattern) in 100% of cells, and he was treated with combined androgen blockade, consisting of monthly 3.6 mg goserelin injections and daily bicalutamide 50 mg orally.
Within a week, the patient noticed that the skin lesions stopped weeping fluid. Within 2 weeks, the pain had begun to resolve. At his formal follow-up visit 11 weeks after starting treatment, he was not taking any pain medications and reported no pain. In addition, his visually apparent disease had almost completely resolved (Figure 2). He was fully able to manage his own IADL and reported a marked increase in satisfaction with the quality of his life.
Discussion
The oncology care team clearly defined the goal of care for this patient as palliative and conveyed as such to the patient. The team considered the risks and side effects of cytotoxic chemotherapy agents to be contrary to the patient’s stated primary goal of independence. We selected the combined androgen blockade because it has a low toxicity rate and thus met the primary goals of therapy.
The European Organization for Research and Treatment of Cancer is presently conducting a trial in which cytotoxic chemotherapy is being compared with ADT in AR-positive salivary duct tumors. Findings from a recent prospective, phase-2 trial conducted in Japan suggested that combined AR blockade has similar efficacy and less toxicity than conventional cytotoxic chemotherapy for recurrent and/or metastatic and unresectable locally advanced AR-positive salivary gland carcinoma.7 As more data become available from other studies, it is possible that practice guidelines will be revised to recommend this treatment approach for these cancers.
Salivary ductal adenocarcinomas make up about 9% of malignant salivary gland tumors and occur mostly in men older than 50 years, with a peak incidence in the sixth and seventh decades. It is the most aggressive of salivary gland tumors and is histologically similar to high-grade, invasive ductal carcinoma of the breast. In all, 65% of patients will die of the disease, and most will experience skin ulceration and nerve palsy.1 With such an aggressive clinical picture, the temptation for many oncologists and patients is to use aggressive cytotoxic chemotherapies. Considering the lack of large trials exploring treatment options in this less-common subtype of salivary gland carcinoma, practice guidelines also recommend the use of aggressive chemotherapies. Unlike other types of malignant cancers of the salivary glands, 70% to 90% of ductal adenocarcinomas express the androgen receptor (AR) by immunohistochemistry.2 There are reported cases of androgen deprivation therapy (ADT) as a successful treatment for salivary ductal adenocarcinomas that express the AR (Table).In 2003, Locati and colleagues reported the case of a man with salivary ductal adenocarcinomas who had a complete response with ADT.3 In 2016, the same group of authors published a retrospective analysis of 17 patients with recurrent or metastatic AR-positive salivary gland cancers who were treated with ADT and reported a 64.7% overall response rate among the patients.4 A 10-patient case series in the Netherlands demonstrated a 50% response rate to ADT plus bicalutamide, including a palliative effect in the form of pain relief.5 A retrospective analysis by Price and colleagues of 5 patients with AR-positive metastatic salivary duct adenocarcinoma showed a 60% response rate to a combination of leuprolide and bicalutamide.6
Case presentation and summary
A 91-year-old man was diagnosed with salivary ductal adenocarcinoma of the left parotid gland in September 2013 and underwent left parotidectomy and lymph node dissection, which revealed AJCC stage IVA (pT2 pN3 M0) disease. The following year, in December 2014, he had an enlarging left neck mass that was pathologically confirmed to be recurrent disease, and he underwent left level V neck dissection in February 2015. Five months after surgery, in July 2015, he presented with left neck fullness and new skin nodules, and the results of a biopsy confirmed recurrent disease. Given his relatively asymptomatic state and advanced age, the oncology care team decided to follow the patient without any pharmacologic therapy.
The patient felt relatively well for 11 months but slowly developed increasing pain in the left neck in June 2016. The skin nodules also began to spread inferiorly from his left neck to his upper chest with the development of open sores that wept serous fluid with scab formation (Figure 1). He and his wife lived independently and managed all their own instrumental activities of daily living (IADL). Eventually, the pain in his neck became so severe that it began to interfere with his ability to drive. He declined radiation therapy because of side effects and transportation issues, but he desired something to alleviate the burden of the disease. During a multidisciplinary cancer conference, the staff pathologist and oncologist discussed AR immunohistochemistry to assist with management. In June 2016, the patient’s tumor was found to have AR immunostaining (nuclear pattern) in 100% of cells, and he was treated with combined androgen blockade, consisting of monthly 3.6 mg goserelin injections and daily bicalutamide 50 mg orally.
Within a week, the patient noticed that the skin lesions stopped weeping fluid. Within 2 weeks, the pain had begun to resolve. At his formal follow-up visit 11 weeks after starting treatment, he was not taking any pain medications and reported no pain. In addition, his visually apparent disease had almost completely resolved (Figure 2). He was fully able to manage his own IADL and reported a marked increase in satisfaction with the quality of his life.
Discussion
The oncology care team clearly defined the goal of care for this patient as palliative and conveyed as such to the patient. The team considered the risks and side effects of cytotoxic chemotherapy agents to be contrary to the patient’s stated primary goal of independence. We selected the combined androgen blockade because it has a low toxicity rate and thus met the primary goals of therapy.
The European Organization for Research and Treatment of Cancer is presently conducting a trial in which cytotoxic chemotherapy is being compared with ADT in AR-positive salivary duct tumors. Findings from a recent prospective, phase-2 trial conducted in Japan suggested that combined AR blockade has similar efficacy and less toxicity than conventional cytotoxic chemotherapy for recurrent and/or metastatic and unresectable locally advanced AR-positive salivary gland carcinoma.7 As more data become available from other studies, it is possible that practice guidelines will be revised to recommend this treatment approach for these cancers.
1. Eveson JW, Thompson LDR. Malignant neoplasms of the salivary glands. In: Thompson LDR, ed. Head and neck pathology. 2nd ed. Philadelphia, PA: Elsevier Inc; 2013:304-305.
2. Luk PP, Weston JD, Yu B, et al. Salivary duct carcinoma: clinicopathologic features, morphologic spectrum, and somatic mutations. Head Neck. 2016;38(suppl 1):E1838-E1847.
3. Locati LD, Quattrone P, Bossi P, Marchianò AV, Cantù G, Licitra L. A complete remission with androgen-deprivation therapy in a recurrent androgen receptor-expressing adenocarcinoma of the parotid gland. Ann Oncol. 2003;14(8):1327-1328.
4. Locati LD, Perrone F, Cortelazzi B, et al. Clinical activity of androgen deprivation therapy in patients with metastatic/relapsed androgen receptor-positive salivary gland cancers. Head Neck. 2016;38(5):724-731.
5. Jaspers HC, Verbist BM, Schoffelen R, et al. Androgen receptor-positive salivary duct carcinoma: a disease entity with promising new treatment options. J Clin Oncol. 2011;29(16):e473-e476.
6. Price KAR, Okuno SH, Molina JR, Garcia JJ. Treatment of metastatic salivary duct carcinoma with combined androgen blockade (CAB) with leuprolide acetate and bicalutamide. Int J Radiat Oncol Biol Phys. 2014;88(2):521-522.
7. Fushimi C, Tada Y, Takahashi H, et al. A prospective phase II study of combined androgen blockade in patients with androgen receptor-positive metastatic or locally advanced unresectable salivary gland carcinoma. Ann Oncol. 2018;29(4):979-984.
1. Eveson JW, Thompson LDR. Malignant neoplasms of the salivary glands. In: Thompson LDR, ed. Head and neck pathology. 2nd ed. Philadelphia, PA: Elsevier Inc; 2013:304-305.
2. Luk PP, Weston JD, Yu B, et al. Salivary duct carcinoma: clinicopathologic features, morphologic spectrum, and somatic mutations. Head Neck. 2016;38(suppl 1):E1838-E1847.
3. Locati LD, Quattrone P, Bossi P, Marchianò AV, Cantù G, Licitra L. A complete remission with androgen-deprivation therapy in a recurrent androgen receptor-expressing adenocarcinoma of the parotid gland. Ann Oncol. 2003;14(8):1327-1328.
4. Locati LD, Perrone F, Cortelazzi B, et al. Clinical activity of androgen deprivation therapy in patients with metastatic/relapsed androgen receptor-positive salivary gland cancers. Head Neck. 2016;38(5):724-731.
5. Jaspers HC, Verbist BM, Schoffelen R, et al. Androgen receptor-positive salivary duct carcinoma: a disease entity with promising new treatment options. J Clin Oncol. 2011;29(16):e473-e476.
6. Price KAR, Okuno SH, Molina JR, Garcia JJ. Treatment of metastatic salivary duct carcinoma with combined androgen blockade (CAB) with leuprolide acetate and bicalutamide. Int J Radiat Oncol Biol Phys. 2014;88(2):521-522.
7. Fushimi C, Tada Y, Takahashi H, et al. A prospective phase II study of combined androgen blockade in patients with androgen receptor-positive metastatic or locally advanced unresectable salivary gland carcinoma. Ann Oncol. 2018;29(4):979-984.
Metastatic Vulvovaginal Crohn Disease in the Setting of Well-Controlled Intestinal Disease
The cutaneous manifestations of Crohn disease (CD) are varied, including pyoderma gangrenosum, erythema nodosum, and metastatic CD (MCD). First described by Parks et al,1 MCD is defined as the occurrence of granulomatous lesions at a skin site distant from the gastrointestinal tract.1-20 Metastatic CD presents a diagnostic challenge because it is a rare component in the spectrum of inflammatory bowel disease complications, and many physicians are unaware of its existence. It may precede, coincide with, or develop after the diagnosis of intestinal disease.2-5 Vulvoperineal involvement is particularly problematic because a multitude of other, more likely disease processes are considered first. Typically it is initially diagnosed as a presumed infection prompting reflexive treatment with antivirals, antifungals, and antibiotics. Patients may experience symptoms for years prior to correct diagnosis and institution of proper therapy. A variety of clinical presentations have been described, including nonspecific pain and swelling, erythematous papules and plaques, and nonhealing ulcers. Skin biopsy characteristically confirms the diagnosis and reveals dermal noncaseating granulomas. Multiple oral and parenteral therapies are available, with surgical intervention reserved for resistant cases. We present a case of vulvovaginal MCD in the setting of well-controlled intestinal disease. We also provide a review of the literature regarding genital CD and emphasize the need to keep MCD in the differential of vulvoperineal pathology.
Case Report
A 29-year-old woman was referred to the dermatology clinic with vulvar pain, swelling, and pruritus of 14 months’ duration. Her medical history was remarkable for CD following a colectomy with colostomy. Prior therapies included methotrexate with infliximab for 5 years followed by a 2-year regimen with adalimumab, which induced remission of the intestinal disease.
The patient previously had taken a variety of topical and oral antimicrobials based on treatment from a primary care physician because fungal, bacterial, and viral infections initially were suspected; however, the vulvar disease persisted, and drug-induced immunosuppression was considered to be an underlying factor. Thus, adalimumab was discontinued. Despite elimination of the biologic, the vulvar disease progressed, which prompted referral to the dermatology clinic.
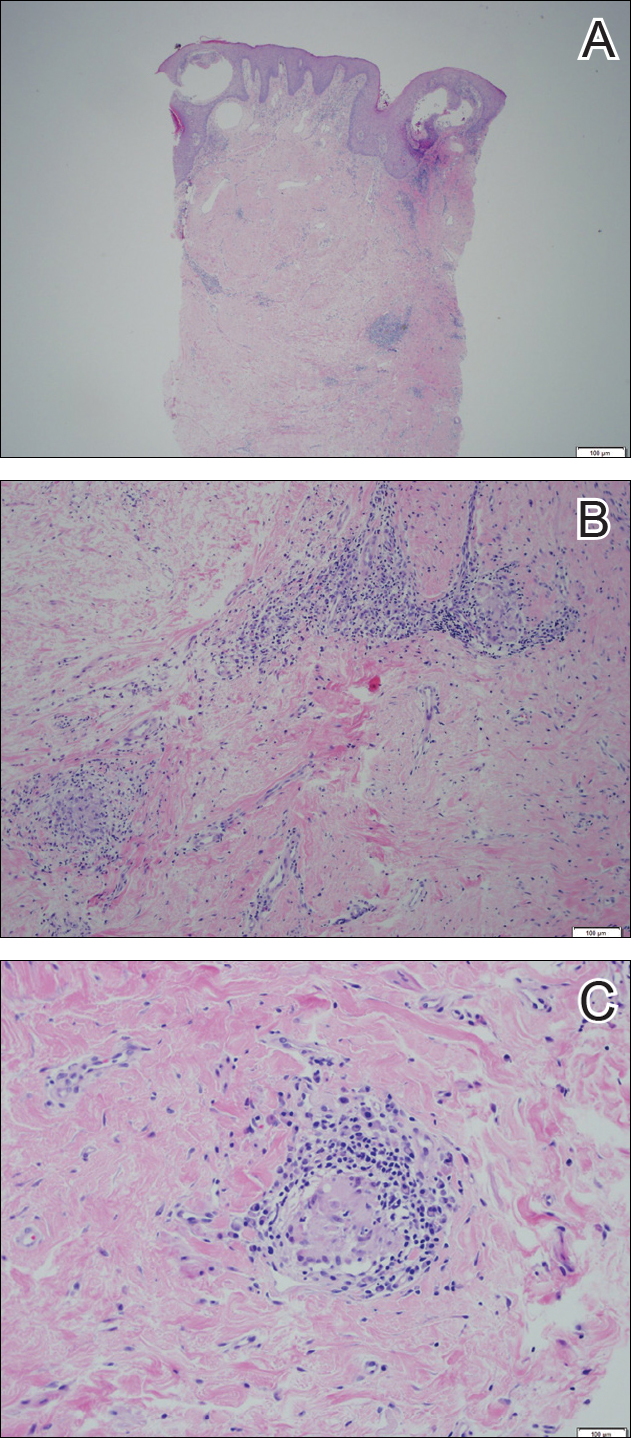
Physical examination revealed diffuse vulvar edema with overlying erythema and scale (Figure 1A). Upon closer inspection, scattered violaceous papules atop a backdrop of lichenification were evident, imparting a cobblestone appearance (Figure 1B). Additionally, a fissure was present on the gluteal cleft. Biopsy from the left labia majora demonstrated well-formed granulomas within a fibrotic reticular dermis (Figures 2A and 2B). The granulomas consisted of both mononucleated and multinucleated histiocytes, rimmed peripherally by lymphocytes and plasma cells (Figure 2C). Periodic acid–Schiff–diastase and acid-fast bacilli stains as well as polarizing microscopy were negative.
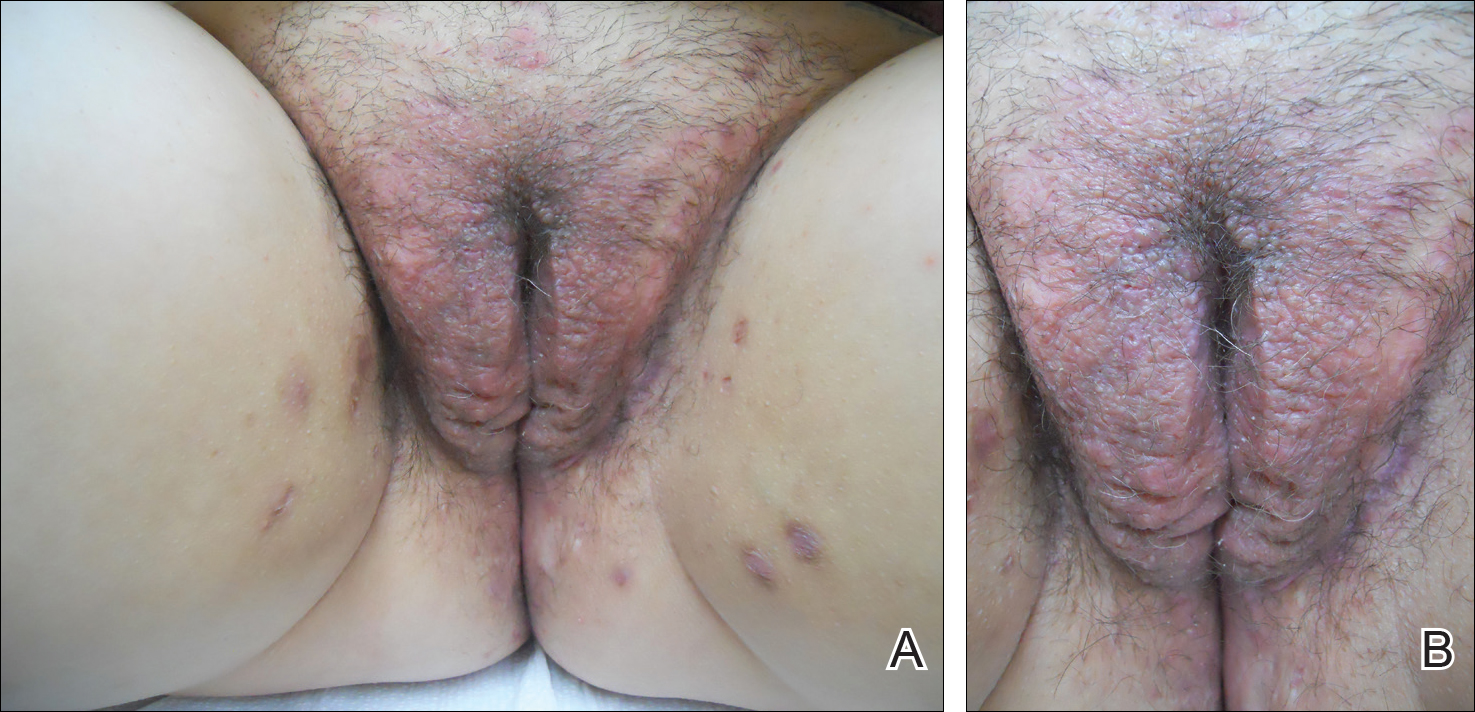
Given the patient’s history, a diagnosis of vulvoperineal MCD was rendered. The patient was started on oral metronidazole 250 mg 3 times daily with topical fluocinonide and tacrolimus. She responded well to this treatment regimen and was referred back to the gastroenterologist for management of the intestinal disease.
Comment
Crohn disease is an idiopathic chronic inflammatory condition that primarily affects the gastrointestinal tract, anywhere from the mouth to the anus. It is characterized by transmural inflammation and fissures that can extend beyond the muscularis propria.4,6 Extraintestinal manifestations are common.3
Cutaneous CD often presents as perianal, perifistular, or peristomal inflammation or ulceration.7 Other skin manifestations include pyoderma gangrenosum, erythema nodosum, erythema multiforme, epidermolysis bullosa acquisita, and palmar erythema.7 Metastatic CD involves skin noncontiguous with the gastrointestinal tract1-20 and may involve any portion of the cutis. Although rare, MCD is the typical etiology underlying vulvar CD.8
Approximately 20% of MCD patients have cutaneous lesions without a history of gastrointestinal disease. More than half of cases in adults and approximately two-thirds in children involve the genitalia. Although more common in adults, vulvar involvement has been reported in children as young as 6 years of age.2 Diagnosis is especially challenging when bowel symptoms are absent; those patients should be evaluated and followed for subsequent intestinal involvement.6
Clinically, symptoms may include general discomfort, pain, pruritus, and dyspareunia. Psychosocial and sexual dysfunction are prevalent and also should be addressed.9 Depending on the stage of the disease, physical examination may reveal erythema, edema, papules, pustules, nodules, condylomatous lesions, abscesses, fissures, fistulas, ulceration, acrochordons, and scarring.2-6,10,11
A host of infections (ie, mycobacterial, actinomycosis, deep fungal, sexually transmitted, schistosomiasis), inflammatory conditions (ie, sarcoid, hidradenitis suppurativa), foreign body reactions, Melkersson-Rosenthal syndrome, and sexual abuse should be included in the differential diagnosis.2,6,10-12 Once infection, sarcoid, and foreign body reaction have been ruled out, noncaseating granulomas in skin are highly suggestive of CD.7
Histopathologic findings of MCD reveal myriad morphological reaction patterns,5,13 including high-grade dysplasia and carcinoma of the vulva; therefore, it may be imprudent to withhold diagnosis based on the absence of the historically pathognomonic noncaseating granulomas.5
The etiopathogenesis of MCD remains an enigma. Dermatopathologic examinations consistently reveal a vascular injury syndrome,13 implicating a possible circulatory system contribution via deposition of immune complexes or antigens in skin.7 Bacterial infection has been implicated in the intestinal manifestations of CD; however, failure to detect microbial ribosomal RNA in MCD biopsies refutes theories of hematogenous spread of microbes.13 Another plausible explanation is that antibodies are formed to conserved microbial epitopes following loss of tolerance to gut flora, which results in an excessive immunologic response at distinct sites in susceptible individuals.13 A T-lymphocyte–mediated type IV hypersensitivity reaction also has been proposed via cross-reactivity of lymphocytes, with skin antigens precipitating extraintestinal granuloma formation and vascular injury.3 Clearly, further investigation is needed.
Magnetic resonanance imaging can identify the extent and anatomy of intestinal and pelvic disease and can assist in the diagnosis of vulvar CD.10,11,14 For these reasons, some experts propose that imaging should be instituted prior to therapy,12,15,16 especially when direct extension is suspected.17
Treatment is challenging and often involves collaboration among several specialties.12 Many treatment options exist because therapeutic responses vary and genital MCD is frequently recalcitrant to therapy.4 Medical therapy includes antibiotics such as metronidazole, corticosteroids (ie, topical, intralesional, systemic), and immune modulators (eg, azathioprine, 6-mercaptopurine, cyclosporine, methotrexate, mycophenolate mofetil, tumor necrosis factor α inhibitors).2,3,6,10,16,18 Thalidomide has been used for refractory cases.19 These treatments can be used alone or in combination. Patients should be monitored for side effects and informed that many treatment regimens may be required before a sustained response is achieved.4,16,18 Surgery is reserved for the most resistant cases. Extensive radical excision of the involved area is the best approach, as limited local excision often is followed by recurrence.20
Conclusion
Our case highlights that vulvar CD can develop in the setting of well-controlled intestinal disease. Vulvoperineal CD should be considered in the differential diagnosis of chronic vulvar pain, swelling, and pruritus, especially in cases resistant to standard therapies and regardless of whether or not gastrointestinal tract symptoms are present. Physicians must be cognizant that vulvar signs and symptoms may precede, coincide with, or follow the diagnosis of intestinal CD. Increased awareness of this entity may facilitate its early recognition and prompt more timely treatment among women with vulvar disease caused by MCD.
- Parks AG, Morson BC, Pegum JS. Crohn’s disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Ploysangam T, Heubi JE, Eisen D, et al. Cutaneous Crohn’s disease in children. J Am Acad Dermatol. 1997;36:697-704.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn’s disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Leu S, Sun PK, Collyer J, et al. Clinical spectrum of vulvar metastatic Crohn’s disease. Dig Dis Sci. 2009;54:1565-1571.
- Foo WC, Papalas JA, Robboy SJ, et al. Vulvar manifestations of Crohn’s disease. Am J Dermatopathol. 2001;33:588-593.
- Urbanek M, Neill SM, McKee PH. Vulval Crohn’s disease: difficulties in diagnosis. Clin Exp Dermatol. 1996;21:211-214.
- Burgdorf W. Cutaneous manifestations of Crohn’s disease. J Am Acad Dermatol. 1981;5:689-695.
- Andreani SM, Ratnasingham K, Dang HH, et al. Crohn’s disease of the vulva. Int J Surg. 2010;8:2-5.
- Feller E, Ribaudo S, Jackson N. Gynecologic aspects of Crohn’s disease. Am Fam Physician. 2001;64:1725-1728.
- Corbett SL, Walsh CM, Spitzer RF, et al. Vulvar inflammation as the only clinical manifestation of Crohn disease in an 8-year-old girl [published online May 10, 2010]. Pediatrics. 2010;125:E1518-E1522.
- Tonolini M, Villa C, Campari A, et al. Common and unusual urogenital Crohn’s disease complications: spectrum of cross-sectional imaging findings. Abdom Imaging. 2013;38:32-41.
- Bhaduri S, Jenkinson S, Lewis F. Vulval Crohn’s disease—a multi-specialty approach. Int J STD AIDS. 2005;16:512-514.
- Crowson AN, Nuovo GJ, Mihm MC Jr, et al. Cutaneous manifestations of Crohn’s disease, its spectrum, and its pathogenesis: intracellular consensus bacterial 16S rRNA is associated with the gastrointestinal but not the cutaneous manifestations of Crohn’s disease. Hum Pathol. 2003;34:1185-1192.
- Pai D, Dillman JR, Mahani MG, et al. MRI of vulvar Crohn disease. Pediatr Radiol. 2011;41:537-541.
- Madnani NA, Desai D, Gandhi N, et al. Isolated Crohn’s disease of the vulva. Indian J Dermatol Venereol Leprol. 2011;77:342-344.
- Makhija S, Trotter M, Wagner E, et al. Refractory Crohn’s disease of the vulva treated with infliximab: a case report. Can J Gastroenterol. 2007;21:835-837.
- Fahmy N, Kalidindi M, Khan R. Direct colo-labial Crohn’s abscess mimicking bartholinitis. Am J Obstret Gynecol. 2010;30:741-742.
- Preston PW, Hudson N, Lewis FM. Treatment of vulval Crohn’s disease with infliximab. Clin Exp Derm. 2006;31:378-380.
- Kolivras A, De Maubeuge J, André J, et al. Thalidomide in refractory vulvar ulcerations associated with Crohn’s disease. Dermatology. 2003;206:381-383.
- Kao MS, Paulson JD, Askin FB. Crohn’s disease of the vulva. Obstet Gynecol. 1975;46:329-333.
The cutaneous manifestations of Crohn disease (CD) are varied, including pyoderma gangrenosum, erythema nodosum, and metastatic CD (MCD). First described by Parks et al,1 MCD is defined as the occurrence of granulomatous lesions at a skin site distant from the gastrointestinal tract.1-20 Metastatic CD presents a diagnostic challenge because it is a rare component in the spectrum of inflammatory bowel disease complications, and many physicians are unaware of its existence. It may precede, coincide with, or develop after the diagnosis of intestinal disease.2-5 Vulvoperineal involvement is particularly problematic because a multitude of other, more likely disease processes are considered first. Typically it is initially diagnosed as a presumed infection prompting reflexive treatment with antivirals, antifungals, and antibiotics. Patients may experience symptoms for years prior to correct diagnosis and institution of proper therapy. A variety of clinical presentations have been described, including nonspecific pain and swelling, erythematous papules and plaques, and nonhealing ulcers. Skin biopsy characteristically confirms the diagnosis and reveals dermal noncaseating granulomas. Multiple oral and parenteral therapies are available, with surgical intervention reserved for resistant cases. We present a case of vulvovaginal MCD in the setting of well-controlled intestinal disease. We also provide a review of the literature regarding genital CD and emphasize the need to keep MCD in the differential of vulvoperineal pathology.
Case Report
A 29-year-old woman was referred to the dermatology clinic with vulvar pain, swelling, and pruritus of 14 months’ duration. Her medical history was remarkable for CD following a colectomy with colostomy. Prior therapies included methotrexate with infliximab for 5 years followed by a 2-year regimen with adalimumab, which induced remission of the intestinal disease.
The patient previously had taken a variety of topical and oral antimicrobials based on treatment from a primary care physician because fungal, bacterial, and viral infections initially were suspected; however, the vulvar disease persisted, and drug-induced immunosuppression was considered to be an underlying factor. Thus, adalimumab was discontinued. Despite elimination of the biologic, the vulvar disease progressed, which prompted referral to the dermatology clinic.
Physical examination revealed diffuse vulvar edema with overlying erythema and scale (Figure 1A). Upon closer inspection, scattered violaceous papules atop a backdrop of lichenification were evident, imparting a cobblestone appearance (Figure 1B). Additionally, a fissure was present on the gluteal cleft. Biopsy from the left labia majora demonstrated well-formed granulomas within a fibrotic reticular dermis (Figures 2A and 2B). The granulomas consisted of both mononucleated and multinucleated histiocytes, rimmed peripherally by lymphocytes and plasma cells (Figure 2C). Periodic acid–Schiff–diastase and acid-fast bacilli stains as well as polarizing microscopy were negative.
Given the patient’s history, a diagnosis of vulvoperineal MCD was rendered. The patient was started on oral metronidazole 250 mg 3 times daily with topical fluocinonide and tacrolimus. She responded well to this treatment regimen and was referred back to the gastroenterologist for management of the intestinal disease.
Comment
Crohn disease is an idiopathic chronic inflammatory condition that primarily affects the gastrointestinal tract, anywhere from the mouth to the anus. It is characterized by transmural inflammation and fissures that can extend beyond the muscularis propria.4,6 Extraintestinal manifestations are common.3
Cutaneous CD often presents as perianal, perifistular, or peristomal inflammation or ulceration.7 Other skin manifestations include pyoderma gangrenosum, erythema nodosum, erythema multiforme, epidermolysis bullosa acquisita, and palmar erythema.7 Metastatic CD involves skin noncontiguous with the gastrointestinal tract1-20 and may involve any portion of the cutis. Although rare, MCD is the typical etiology underlying vulvar CD.8
Approximately 20% of MCD patients have cutaneous lesions without a history of gastrointestinal disease. More than half of cases in adults and approximately two-thirds in children involve the genitalia. Although more common in adults, vulvar involvement has been reported in children as young as 6 years of age.2 Diagnosis is especially challenging when bowel symptoms are absent; those patients should be evaluated and followed for subsequent intestinal involvement.6
Clinically, symptoms may include general discomfort, pain, pruritus, and dyspareunia. Psychosocial and sexual dysfunction are prevalent and also should be addressed.9 Depending on the stage of the disease, physical examination may reveal erythema, edema, papules, pustules, nodules, condylomatous lesions, abscesses, fissures, fistulas, ulceration, acrochordons, and scarring.2-6,10,11
A host of infections (ie, mycobacterial, actinomycosis, deep fungal, sexually transmitted, schistosomiasis), inflammatory conditions (ie, sarcoid, hidradenitis suppurativa), foreign body reactions, Melkersson-Rosenthal syndrome, and sexual abuse should be included in the differential diagnosis.2,6,10-12 Once infection, sarcoid, and foreign body reaction have been ruled out, noncaseating granulomas in skin are highly suggestive of CD.7
Histopathologic findings of MCD reveal myriad morphological reaction patterns,5,13 including high-grade dysplasia and carcinoma of the vulva; therefore, it may be imprudent to withhold diagnosis based on the absence of the historically pathognomonic noncaseating granulomas.5
The etiopathogenesis of MCD remains an enigma. Dermatopathologic examinations consistently reveal a vascular injury syndrome,13 implicating a possible circulatory system contribution via deposition of immune complexes or antigens in skin.7 Bacterial infection has been implicated in the intestinal manifestations of CD; however, failure to detect microbial ribosomal RNA in MCD biopsies refutes theories of hematogenous spread of microbes.13 Another plausible explanation is that antibodies are formed to conserved microbial epitopes following loss of tolerance to gut flora, which results in an excessive immunologic response at distinct sites in susceptible individuals.13 A T-lymphocyte–mediated type IV hypersensitivity reaction also has been proposed via cross-reactivity of lymphocytes, with skin antigens precipitating extraintestinal granuloma formation and vascular injury.3 Clearly, further investigation is needed.
Magnetic resonanance imaging can identify the extent and anatomy of intestinal and pelvic disease and can assist in the diagnosis of vulvar CD.10,11,14 For these reasons, some experts propose that imaging should be instituted prior to therapy,12,15,16 especially when direct extension is suspected.17
Treatment is challenging and often involves collaboration among several specialties.12 Many treatment options exist because therapeutic responses vary and genital MCD is frequently recalcitrant to therapy.4 Medical therapy includes antibiotics such as metronidazole, corticosteroids (ie, topical, intralesional, systemic), and immune modulators (eg, azathioprine, 6-mercaptopurine, cyclosporine, methotrexate, mycophenolate mofetil, tumor necrosis factor α inhibitors).2,3,6,10,16,18 Thalidomide has been used for refractory cases.19 These treatments can be used alone or in combination. Patients should be monitored for side effects and informed that many treatment regimens may be required before a sustained response is achieved.4,16,18 Surgery is reserved for the most resistant cases. Extensive radical excision of the involved area is the best approach, as limited local excision often is followed by recurrence.20
Conclusion
Our case highlights that vulvar CD can develop in the setting of well-controlled intestinal disease. Vulvoperineal CD should be considered in the differential diagnosis of chronic vulvar pain, swelling, and pruritus, especially in cases resistant to standard therapies and regardless of whether or not gastrointestinal tract symptoms are present. Physicians must be cognizant that vulvar signs and symptoms may precede, coincide with, or follow the diagnosis of intestinal CD. Increased awareness of this entity may facilitate its early recognition and prompt more timely treatment among women with vulvar disease caused by MCD.
The cutaneous manifestations of Crohn disease (CD) are varied, including pyoderma gangrenosum, erythema nodosum, and metastatic CD (MCD). First described by Parks et al,1 MCD is defined as the occurrence of granulomatous lesions at a skin site distant from the gastrointestinal tract.1-20 Metastatic CD presents a diagnostic challenge because it is a rare component in the spectrum of inflammatory bowel disease complications, and many physicians are unaware of its existence. It may precede, coincide with, or develop after the diagnosis of intestinal disease.2-5 Vulvoperineal involvement is particularly problematic because a multitude of other, more likely disease processes are considered first. Typically it is initially diagnosed as a presumed infection prompting reflexive treatment with antivirals, antifungals, and antibiotics. Patients may experience symptoms for years prior to correct diagnosis and institution of proper therapy. A variety of clinical presentations have been described, including nonspecific pain and swelling, erythematous papules and plaques, and nonhealing ulcers. Skin biopsy characteristically confirms the diagnosis and reveals dermal noncaseating granulomas. Multiple oral and parenteral therapies are available, with surgical intervention reserved for resistant cases. We present a case of vulvovaginal MCD in the setting of well-controlled intestinal disease. We also provide a review of the literature regarding genital CD and emphasize the need to keep MCD in the differential of vulvoperineal pathology.
Case Report
A 29-year-old woman was referred to the dermatology clinic with vulvar pain, swelling, and pruritus of 14 months’ duration. Her medical history was remarkable for CD following a colectomy with colostomy. Prior therapies included methotrexate with infliximab for 5 years followed by a 2-year regimen with adalimumab, which induced remission of the intestinal disease.
The patient previously had taken a variety of topical and oral antimicrobials based on treatment from a primary care physician because fungal, bacterial, and viral infections initially were suspected; however, the vulvar disease persisted, and drug-induced immunosuppression was considered to be an underlying factor. Thus, adalimumab was discontinued. Despite elimination of the biologic, the vulvar disease progressed, which prompted referral to the dermatology clinic.
Physical examination revealed diffuse vulvar edema with overlying erythema and scale (Figure 1A). Upon closer inspection, scattered violaceous papules atop a backdrop of lichenification were evident, imparting a cobblestone appearance (Figure 1B). Additionally, a fissure was present on the gluteal cleft. Biopsy from the left labia majora demonstrated well-formed granulomas within a fibrotic reticular dermis (Figures 2A and 2B). The granulomas consisted of both mononucleated and multinucleated histiocytes, rimmed peripherally by lymphocytes and plasma cells (Figure 2C). Periodic acid–Schiff–diastase and acid-fast bacilli stains as well as polarizing microscopy were negative.
Given the patient’s history, a diagnosis of vulvoperineal MCD was rendered. The patient was started on oral metronidazole 250 mg 3 times daily with topical fluocinonide and tacrolimus. She responded well to this treatment regimen and was referred back to the gastroenterologist for management of the intestinal disease.
Comment
Crohn disease is an idiopathic chronic inflammatory condition that primarily affects the gastrointestinal tract, anywhere from the mouth to the anus. It is characterized by transmural inflammation and fissures that can extend beyond the muscularis propria.4,6 Extraintestinal manifestations are common.3
Cutaneous CD often presents as perianal, perifistular, or peristomal inflammation or ulceration.7 Other skin manifestations include pyoderma gangrenosum, erythema nodosum, erythema multiforme, epidermolysis bullosa acquisita, and palmar erythema.7 Metastatic CD involves skin noncontiguous with the gastrointestinal tract1-20 and may involve any portion of the cutis. Although rare, MCD is the typical etiology underlying vulvar CD.8
Approximately 20% of MCD patients have cutaneous lesions without a history of gastrointestinal disease. More than half of cases in adults and approximately two-thirds in children involve the genitalia. Although more common in adults, vulvar involvement has been reported in children as young as 6 years of age.2 Diagnosis is especially challenging when bowel symptoms are absent; those patients should be evaluated and followed for subsequent intestinal involvement.6
Clinically, symptoms may include general discomfort, pain, pruritus, and dyspareunia. Psychosocial and sexual dysfunction are prevalent and also should be addressed.9 Depending on the stage of the disease, physical examination may reveal erythema, edema, papules, pustules, nodules, condylomatous lesions, abscesses, fissures, fistulas, ulceration, acrochordons, and scarring.2-6,10,11
A host of infections (ie, mycobacterial, actinomycosis, deep fungal, sexually transmitted, schistosomiasis), inflammatory conditions (ie, sarcoid, hidradenitis suppurativa), foreign body reactions, Melkersson-Rosenthal syndrome, and sexual abuse should be included in the differential diagnosis.2,6,10-12 Once infection, sarcoid, and foreign body reaction have been ruled out, noncaseating granulomas in skin are highly suggestive of CD.7
Histopathologic findings of MCD reveal myriad morphological reaction patterns,5,13 including high-grade dysplasia and carcinoma of the vulva; therefore, it may be imprudent to withhold diagnosis based on the absence of the historically pathognomonic noncaseating granulomas.5
The etiopathogenesis of MCD remains an enigma. Dermatopathologic examinations consistently reveal a vascular injury syndrome,13 implicating a possible circulatory system contribution via deposition of immune complexes or antigens in skin.7 Bacterial infection has been implicated in the intestinal manifestations of CD; however, failure to detect microbial ribosomal RNA in MCD biopsies refutes theories of hematogenous spread of microbes.13 Another plausible explanation is that antibodies are formed to conserved microbial epitopes following loss of tolerance to gut flora, which results in an excessive immunologic response at distinct sites in susceptible individuals.13 A T-lymphocyte–mediated type IV hypersensitivity reaction also has been proposed via cross-reactivity of lymphocytes, with skin antigens precipitating extraintestinal granuloma formation and vascular injury.3 Clearly, further investigation is needed.
Magnetic resonanance imaging can identify the extent and anatomy of intestinal and pelvic disease and can assist in the diagnosis of vulvar CD.10,11,14 For these reasons, some experts propose that imaging should be instituted prior to therapy,12,15,16 especially when direct extension is suspected.17
Treatment is challenging and often involves collaboration among several specialties.12 Many treatment options exist because therapeutic responses vary and genital MCD is frequently recalcitrant to therapy.4 Medical therapy includes antibiotics such as metronidazole, corticosteroids (ie, topical, intralesional, systemic), and immune modulators (eg, azathioprine, 6-mercaptopurine, cyclosporine, methotrexate, mycophenolate mofetil, tumor necrosis factor α inhibitors).2,3,6,10,16,18 Thalidomide has been used for refractory cases.19 These treatments can be used alone or in combination. Patients should be monitored for side effects and informed that many treatment regimens may be required before a sustained response is achieved.4,16,18 Surgery is reserved for the most resistant cases. Extensive radical excision of the involved area is the best approach, as limited local excision often is followed by recurrence.20
Conclusion
Our case highlights that vulvar CD can develop in the setting of well-controlled intestinal disease. Vulvoperineal CD should be considered in the differential diagnosis of chronic vulvar pain, swelling, and pruritus, especially in cases resistant to standard therapies and regardless of whether or not gastrointestinal tract symptoms are present. Physicians must be cognizant that vulvar signs and symptoms may precede, coincide with, or follow the diagnosis of intestinal CD. Increased awareness of this entity may facilitate its early recognition and prompt more timely treatment among women with vulvar disease caused by MCD.
- Parks AG, Morson BC, Pegum JS. Crohn’s disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Ploysangam T, Heubi JE, Eisen D, et al. Cutaneous Crohn’s disease in children. J Am Acad Dermatol. 1997;36:697-704.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn’s disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Leu S, Sun PK, Collyer J, et al. Clinical spectrum of vulvar metastatic Crohn’s disease. Dig Dis Sci. 2009;54:1565-1571.
- Foo WC, Papalas JA, Robboy SJ, et al. Vulvar manifestations of Crohn’s disease. Am J Dermatopathol. 2001;33:588-593.
- Urbanek M, Neill SM, McKee PH. Vulval Crohn’s disease: difficulties in diagnosis. Clin Exp Dermatol. 1996;21:211-214.
- Burgdorf W. Cutaneous manifestations of Crohn’s disease. J Am Acad Dermatol. 1981;5:689-695.
- Andreani SM, Ratnasingham K, Dang HH, et al. Crohn’s disease of the vulva. Int J Surg. 2010;8:2-5.
- Feller E, Ribaudo S, Jackson N. Gynecologic aspects of Crohn’s disease. Am Fam Physician. 2001;64:1725-1728.
- Corbett SL, Walsh CM, Spitzer RF, et al. Vulvar inflammation as the only clinical manifestation of Crohn disease in an 8-year-old girl [published online May 10, 2010]. Pediatrics. 2010;125:E1518-E1522.
- Tonolini M, Villa C, Campari A, et al. Common and unusual urogenital Crohn’s disease complications: spectrum of cross-sectional imaging findings. Abdom Imaging. 2013;38:32-41.
- Bhaduri S, Jenkinson S, Lewis F. Vulval Crohn’s disease—a multi-specialty approach. Int J STD AIDS. 2005;16:512-514.
- Crowson AN, Nuovo GJ, Mihm MC Jr, et al. Cutaneous manifestations of Crohn’s disease, its spectrum, and its pathogenesis: intracellular consensus bacterial 16S rRNA is associated with the gastrointestinal but not the cutaneous manifestations of Crohn’s disease. Hum Pathol. 2003;34:1185-1192.
- Pai D, Dillman JR, Mahani MG, et al. MRI of vulvar Crohn disease. Pediatr Radiol. 2011;41:537-541.
- Madnani NA, Desai D, Gandhi N, et al. Isolated Crohn’s disease of the vulva. Indian J Dermatol Venereol Leprol. 2011;77:342-344.
- Makhija S, Trotter M, Wagner E, et al. Refractory Crohn’s disease of the vulva treated with infliximab: a case report. Can J Gastroenterol. 2007;21:835-837.
- Fahmy N, Kalidindi M, Khan R. Direct colo-labial Crohn’s abscess mimicking bartholinitis. Am J Obstret Gynecol. 2010;30:741-742.
- Preston PW, Hudson N, Lewis FM. Treatment of vulval Crohn’s disease with infliximab. Clin Exp Derm. 2006;31:378-380.
- Kolivras A, De Maubeuge J, André J, et al. Thalidomide in refractory vulvar ulcerations associated with Crohn’s disease. Dermatology. 2003;206:381-383.
- Kao MS, Paulson JD, Askin FB. Crohn’s disease of the vulva. Obstet Gynecol. 1975;46:329-333.
- Parks AG, Morson BC, Pegum JS. Crohn’s disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Ploysangam T, Heubi JE, Eisen D, et al. Cutaneous Crohn’s disease in children. J Am Acad Dermatol. 1997;36:697-704.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn’s disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Leu S, Sun PK, Collyer J, et al. Clinical spectrum of vulvar metastatic Crohn’s disease. Dig Dis Sci. 2009;54:1565-1571.
- Foo WC, Papalas JA, Robboy SJ, et al. Vulvar manifestations of Crohn’s disease. Am J Dermatopathol. 2001;33:588-593.
- Urbanek M, Neill SM, McKee PH. Vulval Crohn’s disease: difficulties in diagnosis. Clin Exp Dermatol. 1996;21:211-214.
- Burgdorf W. Cutaneous manifestations of Crohn’s disease. J Am Acad Dermatol. 1981;5:689-695.
- Andreani SM, Ratnasingham K, Dang HH, et al. Crohn’s disease of the vulva. Int J Surg. 2010;8:2-5.
- Feller E, Ribaudo S, Jackson N. Gynecologic aspects of Crohn’s disease. Am Fam Physician. 2001;64:1725-1728.
- Corbett SL, Walsh CM, Spitzer RF, et al. Vulvar inflammation as the only clinical manifestation of Crohn disease in an 8-year-old girl [published online May 10, 2010]. Pediatrics. 2010;125:E1518-E1522.
- Tonolini M, Villa C, Campari A, et al. Common and unusual urogenital Crohn’s disease complications: spectrum of cross-sectional imaging findings. Abdom Imaging. 2013;38:32-41.
- Bhaduri S, Jenkinson S, Lewis F. Vulval Crohn’s disease—a multi-specialty approach. Int J STD AIDS. 2005;16:512-514.
- Crowson AN, Nuovo GJ, Mihm MC Jr, et al. Cutaneous manifestations of Crohn’s disease, its spectrum, and its pathogenesis: intracellular consensus bacterial 16S rRNA is associated with the gastrointestinal but not the cutaneous manifestations of Crohn’s disease. Hum Pathol. 2003;34:1185-1192.
- Pai D, Dillman JR, Mahani MG, et al. MRI of vulvar Crohn disease. Pediatr Radiol. 2011;41:537-541.
- Madnani NA, Desai D, Gandhi N, et al. Isolated Crohn’s disease of the vulva. Indian J Dermatol Venereol Leprol. 2011;77:342-344.
- Makhija S, Trotter M, Wagner E, et al. Refractory Crohn’s disease of the vulva treated with infliximab: a case report. Can J Gastroenterol. 2007;21:835-837.
- Fahmy N, Kalidindi M, Khan R. Direct colo-labial Crohn’s abscess mimicking bartholinitis. Am J Obstret Gynecol. 2010;30:741-742.
- Preston PW, Hudson N, Lewis FM. Treatment of vulval Crohn’s disease with infliximab. Clin Exp Derm. 2006;31:378-380.
- Kolivras A, De Maubeuge J, André J, et al. Thalidomide in refractory vulvar ulcerations associated with Crohn’s disease. Dermatology. 2003;206:381-383.
- Kao MS, Paulson JD, Askin FB. Crohn’s disease of the vulva. Obstet Gynecol. 1975;46:329-333.

Carcinoma of the colon in a child
Colon cancer is not common in childhood even though cases have been reported in children and adolescents.1,2 Although it is sporadic, it can arise in the setting of predisposing illnesses such as familial polyposis syndrome or inflammatory bowel disease.2-5 Only 1 or 2 cases per million children are reported globally each year, but the incidence has been noted to be on the rise.2 The nonspecific gastrointestinal symptoms and anemia as features of the disease could also be seen in other common childhood ailments, such as helminthiasis in our region in West Africa. As a result, unless there is a high index of suspicion at the outset, there is a risk that colon cancer will be diagnosed at a late stage, especially in children with no apparent predisposing factor.
In this case, an 11-year-old girl presented to our institution with abdominal pain, melena, abdominal swelling, and iron deficiency anemia. A positive family history of colon cancer in the mother and a brain tumor in an elder sibling prompted a search for and subsequent diagnosis of colon cancer. Her case highlights the importance of a high index of suspicion in making an early diagnosis to achieve the best possible outcomes. This case is being reported in line with the SCARE guidelines.6
Case summary and presentation
An 11-year-old girl presented to our facilty with recurrent abdominal pain of 8 months duration, a 4-month history of progressive paleness of the palms, and a month-long fever. There was an associated change in bowel habit to about 2-3 times per day, weight loss despite a preserved appetite, and black, tarry stools. A month before she presented, she developed low-grade pyrexia, dysuria, and pica. She was treated for iron deficiency anemia at a peripheral hospital where she first sought for care with oral iron, folic acid, and vitamin C, but with no improvement in symptoms.
She was the youngest of 8 children born to parents who were first cousins. Her father had died in a car accident when she was a year old, and her mother had died 6 years later after being diagnosed with and treated for colon cancer. An elder sibling died of a brain tumor at the age of 9 years.
On admission to our institution, the girl looked acutely ill. She was severely pale, but afebrile and anicteric. She had no petechial or purpuric skin rashes, but had glossitis with areas of papules on the anterior two-thirds of the dorsum of the tongue. She had no gingival hypertrophy, but had significant peripheral lymphadenopathy and weighed 67% of the weight for her age. In addition, she had generalized abdominal pain and a soft, well-circumscribed tender mass located at the right iliac fossa was palpated and estimated to be 8 cm x 6 cm.
A full blood count showed severe hypochromic microcytic anemia, with a red blood cell count of 2.53 x 1012/L, packed cell volume of 9%, white blood cell count 9.4 x109/L, platelet cell count of 453 x 109/L, mean corpuscular volume of 48.6 fl, and a red cell distribution width of 23.7%. Iron studies could not be done because we lacked the facilities, but a bone marrow aspiration biopsy showed reduced bone marrow iron stores. A fecal occult blood test was positive for blood, but negative for culture, ova, or cysts. An abdominopelvic ultrasound showed the well-circumscribed mass at the right iliac fossa, and that was confirmed by a computed-tomographic scan (Figure 1).
An upper endoscopy revealed fundal and prepyloric erosions and reflux eosophagitis. Although findings from a sigmoidoscopy were normal, a histology of biopsied tissues showed features of chronic inflammation.
There was a delay in arriving at the final diagnosis because the patient’s family faced financial difficulties and some of the imaging procedures were not available at our institution. Other diagnoses that were entertained and managed in this case were iron deficiency anemia from peptic ulcer disease. Six weeks after her initial presentation to our institution, the patient had an exploratory laparotomy. The findings intra-operatively were those of a huge tumor involving the ascending colon measuring 16 x14 cm and extending to involve the cecum and mesenteric lymph nodes (Figure 2).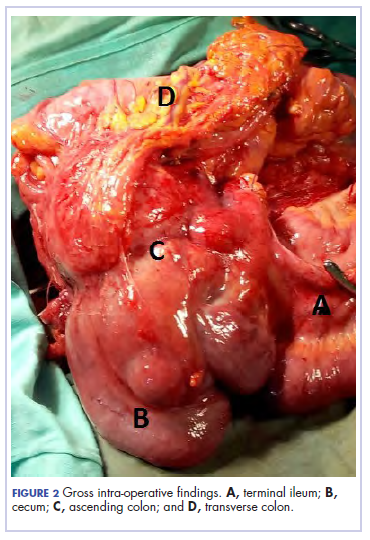
Kidneys, liver and spleen were macroscopically normal. An assessment of Duke’s stage 3C colon cancer was made and she had an extended radical hemicolectomy with anastomosis.
A 44.5-cm long right hemicolectomy segment comprising a 17-cm ileal segment, a 6-cm cecum, 21.5-cm ascending colon, and an 8-cm appendix was removed. The tumor was located in the ascending colon at 7.5 cm from the distal resection margin and extending 1 cm into the cecum. It had a circumference of 27 cm with fibrinous exudates on its peritoneal surface. Dissection revealed uneven circumferential thickening of the bowel wall, luminal dilatation, marked mucosal ulcerations, and liquid content made up of fecal material and necrotic debris. The tumor cut surface was solid white. We also removed 4 lymph nodes. Other uninvolved areas showed focal mucosal hyperemia, but no polyps were observed. Histology showed moderately differentiated adenocarcinoma (pT4) with ¼ nodal involvement (Figure 3).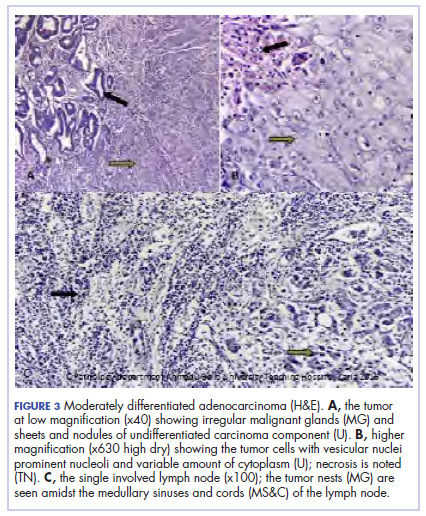
The patient’s postoperative course was uneventful, and she had adjuvant chemotherapy with oral capecitabine and intravenous oxaliplatin. She completed the 8-cycle protocol with excellent clinical response and minimal adverse events were recorded. A repeat abdominal CT scan showed no residual tumor (Figure 4), and her full blood count showed normal hematological profile with no evidence of iron deficiency.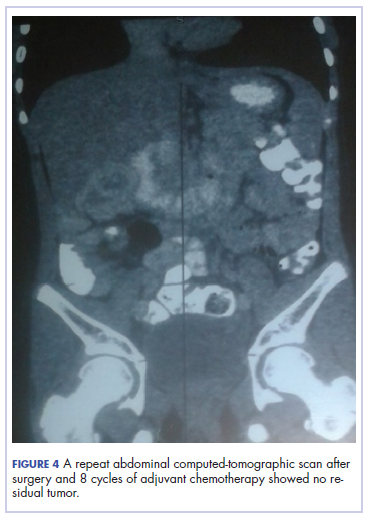
She is presently on follow up 2 years after confirmation of the diagnosis. (Her histological diagnosis was made June 2016, and her last clinic follow-up was March 2018.
Discussion
Our patient presented with symptoms of abdominal pain, dysuria, melena, and pallor as in other case reports.7-10 A diagnosis of iron deficiency anemia was initially entertained in view of the hematologic profile, and for which management was instituted. The findings of gastric and duodenal erosions on endoscopy further supported the assumption for and treatment of peptic ulcer disease. Iron deficiency in this patient was owing to chronic blood loss from a tumour located at the upper parts of the. Vague and nonspecific symptoms are associated with delayed diagnosis and poor prognosis.1-5,11 Nonspecificity of symptoms is typical feature of colon cancer as reported in other studies.1,11-13 However, the strong family history of colon cancer heightened suspicion in this case, otherwise the diagnosis of an ascending colon tumor could have been delayed until much later and with graver consequences.
The diagnosis of colon cancer in this child was made about a year after her initial symptoms, and 3 months after her presentation to us. Ascending and transverse colon cancers are usually diagnosed late because the symptoms of intestinal obstruction – frank bleeding – will not present until the illness is substantially advanced. Ameh and Nmadu reported a case series of 8 patients from our facility with rectosigmoid tumor, of whom 6 had mucinous adenocarcinaoma and 5 of those 6 had stage 3C disease. Although the patient in the present case had an advanced disease at diagnosis, she had a moderately differentiated histology in contrast to the 6 previously reported cases, who had mucinous histology.14
Previous studies have shown that colorectal carcinoma is a rare disease worldwide, with an annual age-adjusted incidence of 0.38 people/million.1,2 When it occurs in the young, familial or hereditary predisposition should be highly suspected.1-3 To date, there is scant literature on children younger than 16 years in Nigeria.15 Various studies have found a relationship between patients with early-stage colon cancer and inherited genetic predisposition to the disease.2,5 Familial adenomatous polyposis syndrome is an autosomal dominant disorder characterized by the development of polyps during the first decade of life, extensive polyposis in the second decade, and transformation into frank carcinoma in early adulthood.1-5
Although our patient’s mother was diagnosed with and died of colon cancer, the type of which could not be ascertained because her records could not be traced. However, the operative and histological findings in this patient did not suggest the presence of polyposis. The clinical phenotype for the autosomal recessive mismatch repair deficiency includes susceptibity to glioma, leukemia, lymphoma, and colorectal carcinoma in children and young adults.1,5 Screening for genetic markers in the child in the present case might have identified the genetic abnormalities involved and would have been invaluable in the evaluation of her 6 surviving siblings and further management of this family. In conclusion. A high index of suspicion should prompt inclusion of colon cancer in the differential diagnosis of nonspecific gastrointestinal symptoms associated with colon cancer in children.
Acknowledg
The authors obtained written informed consent from the patient and her elder sibling before writing this report. In addition, the authors thank all the staff involved in the management of this child in the pediatric medical and surgical wards.
1. Sultan I, Rodriguez-Galindo C, El-Taani H, Pastore G, Casanova M, Gallino G, Ferrari A. Distinct features of colorectal cancer in children and adolescents. A population-based study of 159 cases. Cancer. 2010;1;116(3):758-65.
2. Ferrari A. Intestinal carcinomas. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. 1st ed. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 32.
3. Hill DA, Furman WL, Bilups CA, Riedly SE, Cain AM, Rao BN. Colorectal carcinoma in childhood and adolescence: a clinicopathological review. J Clin Oncol. 2007;25(36):5808-5814.
4. Saab OKR, Furman WL. Epidemiology and management options for colorectal cancer in children. Paediatr Drugs. 2008;10(3):177-192.
5. Bertario L, Signoroni S. Gastrointestinal cancer predisposition syndromes. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 30.
6. Agha RA, Fowler AJ, Saetta A, et al, for the SCARE Group. The SCARE Statement: consensus-based surgical case report guidelines. Int J Surg. 2016;34:180-186.
7. Tricoli JV, Seibel NL, Blair DG, Albritton K, Hayes-Lattin B. Unique characteristics of adolescent and young adult acute lymphoblastic leukemia, breast cancer, and colon cancer. J Natl Cancer Inst. 2011;103(8):628-635.
8. Begum M, Khan ZJ, Hassan K, Karim S. Carcinoma colon of a child presenting with abdominal pain. Bangaladesh J Child Health. 2014;38(1):44-47.
9. Woods R, Larkin JO, Muldoon C, Kennedy MJ, Mehigan B, McCormick P. Metastatic paediatric colorectal carcinoma. Ir Med J. 2012;105(3):88-89.
10. Bjoernsen LP, Lindsay MB. An unusual case of pediatric abdominal pain. CJEM. 2011;13(2):133-138.
11. Takalkar UV, Asegaonkar SB, Kulkarni U, Jadhav A, Advani S, Reddy DN. Carcinoma of colon in an adolescent: a case report with review of literature. Int J Sci Rep 2015;1(2):151-3.
12. Zamir N, Ahmad S, Akhtar J. Mucinous adenocarcinoma of colon. APSP J Case Rep. 2010;1(2):20.
13. Al-Tonbary Y, Darwish A, El-Hussein A, Fouda A. Adenocarcinoma of the colon in children: case series and mini-review of the literature. Hematol Oncol Stem Cell Ther. 2013;6(1):29-33.
14. Ameh EA, Nmadu PT. Colorectal adenocarcinoma in children and adolescents: a report of 8 patients from Zaria, Nigeria. West Afr J Med. 2000;19(4):273-276.
15. Ibrahim, AE, Afolayan KA, Adeniji OM, Buhari KB. Colorectal carcinoma in children and young adults in Ilorin, Nigeria. West Afr J Med. 2011;30(3):202-205.
Colon cancer is not common in childhood even though cases have been reported in children and adolescents.1,2 Although it is sporadic, it can arise in the setting of predisposing illnesses such as familial polyposis syndrome or inflammatory bowel disease.2-5 Only 1 or 2 cases per million children are reported globally each year, but the incidence has been noted to be on the rise.2 The nonspecific gastrointestinal symptoms and anemia as features of the disease could also be seen in other common childhood ailments, such as helminthiasis in our region in West Africa. As a result, unless there is a high index of suspicion at the outset, there is a risk that colon cancer will be diagnosed at a late stage, especially in children with no apparent predisposing factor.
In this case, an 11-year-old girl presented to our institution with abdominal pain, melena, abdominal swelling, and iron deficiency anemia. A positive family history of colon cancer in the mother and a brain tumor in an elder sibling prompted a search for and subsequent diagnosis of colon cancer. Her case highlights the importance of a high index of suspicion in making an early diagnosis to achieve the best possible outcomes. This case is being reported in line with the SCARE guidelines.6
Case summary and presentation
An 11-year-old girl presented to our facilty with recurrent abdominal pain of 8 months duration, a 4-month history of progressive paleness of the palms, and a month-long fever. There was an associated change in bowel habit to about 2-3 times per day, weight loss despite a preserved appetite, and black, tarry stools. A month before she presented, she developed low-grade pyrexia, dysuria, and pica. She was treated for iron deficiency anemia at a peripheral hospital where she first sought for care with oral iron, folic acid, and vitamin C, but with no improvement in symptoms.
She was the youngest of 8 children born to parents who were first cousins. Her father had died in a car accident when she was a year old, and her mother had died 6 years later after being diagnosed with and treated for colon cancer. An elder sibling died of a brain tumor at the age of 9 years.
On admission to our institution, the girl looked acutely ill. She was severely pale, but afebrile and anicteric. She had no petechial or purpuric skin rashes, but had glossitis with areas of papules on the anterior two-thirds of the dorsum of the tongue. She had no gingival hypertrophy, but had significant peripheral lymphadenopathy and weighed 67% of the weight for her age. In addition, she had generalized abdominal pain and a soft, well-circumscribed tender mass located at the right iliac fossa was palpated and estimated to be 8 cm x 6 cm.
A full blood count showed severe hypochromic microcytic anemia, with a red blood cell count of 2.53 x 1012/L, packed cell volume of 9%, white blood cell count 9.4 x109/L, platelet cell count of 453 x 109/L, mean corpuscular volume of 48.6 fl, and a red cell distribution width of 23.7%. Iron studies could not be done because we lacked the facilities, but a bone marrow aspiration biopsy showed reduced bone marrow iron stores. A fecal occult blood test was positive for blood, but negative for culture, ova, or cysts. An abdominopelvic ultrasound showed the well-circumscribed mass at the right iliac fossa, and that was confirmed by a computed-tomographic scan (Figure 1).
An upper endoscopy revealed fundal and prepyloric erosions and reflux eosophagitis. Although findings from a sigmoidoscopy were normal, a histology of biopsied tissues showed features of chronic inflammation.
There was a delay in arriving at the final diagnosis because the patient’s family faced financial difficulties and some of the imaging procedures were not available at our institution. Other diagnoses that were entertained and managed in this case were iron deficiency anemia from peptic ulcer disease. Six weeks after her initial presentation to our institution, the patient had an exploratory laparotomy. The findings intra-operatively were those of a huge tumor involving the ascending colon measuring 16 x14 cm and extending to involve the cecum and mesenteric lymph nodes (Figure 2).
Kidneys, liver and spleen were macroscopically normal. An assessment of Duke’s stage 3C colon cancer was made and she had an extended radical hemicolectomy with anastomosis.
A 44.5-cm long right hemicolectomy segment comprising a 17-cm ileal segment, a 6-cm cecum, 21.5-cm ascending colon, and an 8-cm appendix was removed. The tumor was located in the ascending colon at 7.5 cm from the distal resection margin and extending 1 cm into the cecum. It had a circumference of 27 cm with fibrinous exudates on its peritoneal surface. Dissection revealed uneven circumferential thickening of the bowel wall, luminal dilatation, marked mucosal ulcerations, and liquid content made up of fecal material and necrotic debris. The tumor cut surface was solid white. We also removed 4 lymph nodes. Other uninvolved areas showed focal mucosal hyperemia, but no polyps were observed. Histology showed moderately differentiated adenocarcinoma (pT4) with ¼ nodal involvement (Figure 3).
The patient’s postoperative course was uneventful, and she had adjuvant chemotherapy with oral capecitabine and intravenous oxaliplatin. She completed the 8-cycle protocol with excellent clinical response and minimal adverse events were recorded. A repeat abdominal CT scan showed no residual tumor (Figure 4), and her full blood count showed normal hematological profile with no evidence of iron deficiency.
She is presently on follow up 2 years after confirmation of the diagnosis. (Her histological diagnosis was made June 2016, and her last clinic follow-up was March 2018.
Discussion
Our patient presented with symptoms of abdominal pain, dysuria, melena, and pallor as in other case reports.7-10 A diagnosis of iron deficiency anemia was initially entertained in view of the hematologic profile, and for which management was instituted. The findings of gastric and duodenal erosions on endoscopy further supported the assumption for and treatment of peptic ulcer disease. Iron deficiency in this patient was owing to chronic blood loss from a tumour located at the upper parts of the. Vague and nonspecific symptoms are associated with delayed diagnosis and poor prognosis.1-5,11 Nonspecificity of symptoms is typical feature of colon cancer as reported in other studies.1,11-13 However, the strong family history of colon cancer heightened suspicion in this case, otherwise the diagnosis of an ascending colon tumor could have been delayed until much later and with graver consequences.
The diagnosis of colon cancer in this child was made about a year after her initial symptoms, and 3 months after her presentation to us. Ascending and transverse colon cancers are usually diagnosed late because the symptoms of intestinal obstruction – frank bleeding – will not present until the illness is substantially advanced. Ameh and Nmadu reported a case series of 8 patients from our facility with rectosigmoid tumor, of whom 6 had mucinous adenocarcinaoma and 5 of those 6 had stage 3C disease. Although the patient in the present case had an advanced disease at diagnosis, she had a moderately differentiated histology in contrast to the 6 previously reported cases, who had mucinous histology.14
Previous studies have shown that colorectal carcinoma is a rare disease worldwide, with an annual age-adjusted incidence of 0.38 people/million.1,2 When it occurs in the young, familial or hereditary predisposition should be highly suspected.1-3 To date, there is scant literature on children younger than 16 years in Nigeria.15 Various studies have found a relationship between patients with early-stage colon cancer and inherited genetic predisposition to the disease.2,5 Familial adenomatous polyposis syndrome is an autosomal dominant disorder characterized by the development of polyps during the first decade of life, extensive polyposis in the second decade, and transformation into frank carcinoma in early adulthood.1-5
Although our patient’s mother was diagnosed with and died of colon cancer, the type of which could not be ascertained because her records could not be traced. However, the operative and histological findings in this patient did not suggest the presence of polyposis. The clinical phenotype for the autosomal recessive mismatch repair deficiency includes susceptibity to glioma, leukemia, lymphoma, and colorectal carcinoma in children and young adults.1,5 Screening for genetic markers in the child in the present case might have identified the genetic abnormalities involved and would have been invaluable in the evaluation of her 6 surviving siblings and further management of this family. In conclusion. A high index of suspicion should prompt inclusion of colon cancer in the differential diagnosis of nonspecific gastrointestinal symptoms associated with colon cancer in children.
Acknowledg
The authors obtained written informed consent from the patient and her elder sibling before writing this report. In addition, the authors thank all the staff involved in the management of this child in the pediatric medical and surgical wards.
Colon cancer is not common in childhood even though cases have been reported in children and adolescents.1,2 Although it is sporadic, it can arise in the setting of predisposing illnesses such as familial polyposis syndrome or inflammatory bowel disease.2-5 Only 1 or 2 cases per million children are reported globally each year, but the incidence has been noted to be on the rise.2 The nonspecific gastrointestinal symptoms and anemia as features of the disease could also be seen in other common childhood ailments, such as helminthiasis in our region in West Africa. As a result, unless there is a high index of suspicion at the outset, there is a risk that colon cancer will be diagnosed at a late stage, especially in children with no apparent predisposing factor.
In this case, an 11-year-old girl presented to our institution with abdominal pain, melena, abdominal swelling, and iron deficiency anemia. A positive family history of colon cancer in the mother and a brain tumor in an elder sibling prompted a search for and subsequent diagnosis of colon cancer. Her case highlights the importance of a high index of suspicion in making an early diagnosis to achieve the best possible outcomes. This case is being reported in line with the SCARE guidelines.6
Case summary and presentation
An 11-year-old girl presented to our facilty with recurrent abdominal pain of 8 months duration, a 4-month history of progressive paleness of the palms, and a month-long fever. There was an associated change in bowel habit to about 2-3 times per day, weight loss despite a preserved appetite, and black, tarry stools. A month before she presented, she developed low-grade pyrexia, dysuria, and pica. She was treated for iron deficiency anemia at a peripheral hospital where she first sought for care with oral iron, folic acid, and vitamin C, but with no improvement in symptoms.
She was the youngest of 8 children born to parents who were first cousins. Her father had died in a car accident when she was a year old, and her mother had died 6 years later after being diagnosed with and treated for colon cancer. An elder sibling died of a brain tumor at the age of 9 years.
On admission to our institution, the girl looked acutely ill. She was severely pale, but afebrile and anicteric. She had no petechial or purpuric skin rashes, but had glossitis with areas of papules on the anterior two-thirds of the dorsum of the tongue. She had no gingival hypertrophy, but had significant peripheral lymphadenopathy and weighed 67% of the weight for her age. In addition, she had generalized abdominal pain and a soft, well-circumscribed tender mass located at the right iliac fossa was palpated and estimated to be 8 cm x 6 cm.
A full blood count showed severe hypochromic microcytic anemia, with a red blood cell count of 2.53 x 1012/L, packed cell volume of 9%, white blood cell count 9.4 x109/L, platelet cell count of 453 x 109/L, mean corpuscular volume of 48.6 fl, and a red cell distribution width of 23.7%. Iron studies could not be done because we lacked the facilities, but a bone marrow aspiration biopsy showed reduced bone marrow iron stores. A fecal occult blood test was positive for blood, but negative for culture, ova, or cysts. An abdominopelvic ultrasound showed the well-circumscribed mass at the right iliac fossa, and that was confirmed by a computed-tomographic scan (Figure 1).
An upper endoscopy revealed fundal and prepyloric erosions and reflux eosophagitis. Although findings from a sigmoidoscopy were normal, a histology of biopsied tissues showed features of chronic inflammation.
There was a delay in arriving at the final diagnosis because the patient’s family faced financial difficulties and some of the imaging procedures were not available at our institution. Other diagnoses that were entertained and managed in this case were iron deficiency anemia from peptic ulcer disease. Six weeks after her initial presentation to our institution, the patient had an exploratory laparotomy. The findings intra-operatively were those of a huge tumor involving the ascending colon measuring 16 x14 cm and extending to involve the cecum and mesenteric lymph nodes (Figure 2).
Kidneys, liver and spleen were macroscopically normal. An assessment of Duke’s stage 3C colon cancer was made and she had an extended radical hemicolectomy with anastomosis.
A 44.5-cm long right hemicolectomy segment comprising a 17-cm ileal segment, a 6-cm cecum, 21.5-cm ascending colon, and an 8-cm appendix was removed. The tumor was located in the ascending colon at 7.5 cm from the distal resection margin and extending 1 cm into the cecum. It had a circumference of 27 cm with fibrinous exudates on its peritoneal surface. Dissection revealed uneven circumferential thickening of the bowel wall, luminal dilatation, marked mucosal ulcerations, and liquid content made up of fecal material and necrotic debris. The tumor cut surface was solid white. We also removed 4 lymph nodes. Other uninvolved areas showed focal mucosal hyperemia, but no polyps were observed. Histology showed moderately differentiated adenocarcinoma (pT4) with ¼ nodal involvement (Figure 3).
The patient’s postoperative course was uneventful, and she had adjuvant chemotherapy with oral capecitabine and intravenous oxaliplatin. She completed the 8-cycle protocol with excellent clinical response and minimal adverse events were recorded. A repeat abdominal CT scan showed no residual tumor (Figure 4), and her full blood count showed normal hematological profile with no evidence of iron deficiency.
She is presently on follow up 2 years after confirmation of the diagnosis. (Her histological diagnosis was made June 2016, and her last clinic follow-up was March 2018.
Discussion
Our patient presented with symptoms of abdominal pain, dysuria, melena, and pallor as in other case reports.7-10 A diagnosis of iron deficiency anemia was initially entertained in view of the hematologic profile, and for which management was instituted. The findings of gastric and duodenal erosions on endoscopy further supported the assumption for and treatment of peptic ulcer disease. Iron deficiency in this patient was owing to chronic blood loss from a tumour located at the upper parts of the. Vague and nonspecific symptoms are associated with delayed diagnosis and poor prognosis.1-5,11 Nonspecificity of symptoms is typical feature of colon cancer as reported in other studies.1,11-13 However, the strong family history of colon cancer heightened suspicion in this case, otherwise the diagnosis of an ascending colon tumor could have been delayed until much later and with graver consequences.
The diagnosis of colon cancer in this child was made about a year after her initial symptoms, and 3 months after her presentation to us. Ascending and transverse colon cancers are usually diagnosed late because the symptoms of intestinal obstruction – frank bleeding – will not present until the illness is substantially advanced. Ameh and Nmadu reported a case series of 8 patients from our facility with rectosigmoid tumor, of whom 6 had mucinous adenocarcinaoma and 5 of those 6 had stage 3C disease. Although the patient in the present case had an advanced disease at diagnosis, she had a moderately differentiated histology in contrast to the 6 previously reported cases, who had mucinous histology.14
Previous studies have shown that colorectal carcinoma is a rare disease worldwide, with an annual age-adjusted incidence of 0.38 people/million.1,2 When it occurs in the young, familial or hereditary predisposition should be highly suspected.1-3 To date, there is scant literature on children younger than 16 years in Nigeria.15 Various studies have found a relationship between patients with early-stage colon cancer and inherited genetic predisposition to the disease.2,5 Familial adenomatous polyposis syndrome is an autosomal dominant disorder characterized by the development of polyps during the first decade of life, extensive polyposis in the second decade, and transformation into frank carcinoma in early adulthood.1-5
Although our patient’s mother was diagnosed with and died of colon cancer, the type of which could not be ascertained because her records could not be traced. However, the operative and histological findings in this patient did not suggest the presence of polyposis. The clinical phenotype for the autosomal recessive mismatch repair deficiency includes susceptibity to glioma, leukemia, lymphoma, and colorectal carcinoma in children and young adults.1,5 Screening for genetic markers in the child in the present case might have identified the genetic abnormalities involved and would have been invaluable in the evaluation of her 6 surviving siblings and further management of this family. In conclusion. A high index of suspicion should prompt inclusion of colon cancer in the differential diagnosis of nonspecific gastrointestinal symptoms associated with colon cancer in children.
Acknowledg
The authors obtained written informed consent from the patient and her elder sibling before writing this report. In addition, the authors thank all the staff involved in the management of this child in the pediatric medical and surgical wards.
1. Sultan I, Rodriguez-Galindo C, El-Taani H, Pastore G, Casanova M, Gallino G, Ferrari A. Distinct features of colorectal cancer in children and adolescents. A population-based study of 159 cases. Cancer. 2010;1;116(3):758-65.
2. Ferrari A. Intestinal carcinomas. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. 1st ed. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 32.
3. Hill DA, Furman WL, Bilups CA, Riedly SE, Cain AM, Rao BN. Colorectal carcinoma in childhood and adolescence: a clinicopathological review. J Clin Oncol. 2007;25(36):5808-5814.
4. Saab OKR, Furman WL. Epidemiology and management options for colorectal cancer in children. Paediatr Drugs. 2008;10(3):177-192.
5. Bertario L, Signoroni S. Gastrointestinal cancer predisposition syndromes. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 30.
6. Agha RA, Fowler AJ, Saetta A, et al, for the SCARE Group. The SCARE Statement: consensus-based surgical case report guidelines. Int J Surg. 2016;34:180-186.
7. Tricoli JV, Seibel NL, Blair DG, Albritton K, Hayes-Lattin B. Unique characteristics of adolescent and young adult acute lymphoblastic leukemia, breast cancer, and colon cancer. J Natl Cancer Inst. 2011;103(8):628-635.
8. Begum M, Khan ZJ, Hassan K, Karim S. Carcinoma colon of a child presenting with abdominal pain. Bangaladesh J Child Health. 2014;38(1):44-47.
9. Woods R, Larkin JO, Muldoon C, Kennedy MJ, Mehigan B, McCormick P. Metastatic paediatric colorectal carcinoma. Ir Med J. 2012;105(3):88-89.
10. Bjoernsen LP, Lindsay MB. An unusual case of pediatric abdominal pain. CJEM. 2011;13(2):133-138.
11. Takalkar UV, Asegaonkar SB, Kulkarni U, Jadhav A, Advani S, Reddy DN. Carcinoma of colon in an adolescent: a case report with review of literature. Int J Sci Rep 2015;1(2):151-3.
12. Zamir N, Ahmad S, Akhtar J. Mucinous adenocarcinoma of colon. APSP J Case Rep. 2010;1(2):20.
13. Al-Tonbary Y, Darwish A, El-Hussein A, Fouda A. Adenocarcinoma of the colon in children: case series and mini-review of the literature. Hematol Oncol Stem Cell Ther. 2013;6(1):29-33.
14. Ameh EA, Nmadu PT. Colorectal adenocarcinoma in children and adolescents: a report of 8 patients from Zaria, Nigeria. West Afr J Med. 2000;19(4):273-276.
15. Ibrahim, AE, Afolayan KA, Adeniji OM, Buhari KB. Colorectal carcinoma in children and young adults in Ilorin, Nigeria. West Afr J Med. 2011;30(3):202-205.
1. Sultan I, Rodriguez-Galindo C, El-Taani H, Pastore G, Casanova M, Gallino G, Ferrari A. Distinct features of colorectal cancer in children and adolescents. A population-based study of 159 cases. Cancer. 2010;1;116(3):758-65.
2. Ferrari A. Intestinal carcinomas. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. 1st ed. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 32.
3. Hill DA, Furman WL, Bilups CA, Riedly SE, Cain AM, Rao BN. Colorectal carcinoma in childhood and adolescence: a clinicopathological review. J Clin Oncol. 2007;25(36):5808-5814.
4. Saab OKR, Furman WL. Epidemiology and management options for colorectal cancer in children. Paediatr Drugs. 2008;10(3):177-192.
5. Bertario L, Signoroni S. Gastrointestinal cancer predisposition syndromes. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 30.
6. Agha RA, Fowler AJ, Saetta A, et al, for the SCARE Group. The SCARE Statement: consensus-based surgical case report guidelines. Int J Surg. 2016;34:180-186.
7. Tricoli JV, Seibel NL, Blair DG, Albritton K, Hayes-Lattin B. Unique characteristics of adolescent and young adult acute lymphoblastic leukemia, breast cancer, and colon cancer. J Natl Cancer Inst. 2011;103(8):628-635.
8. Begum M, Khan ZJ, Hassan K, Karim S. Carcinoma colon of a child presenting with abdominal pain. Bangaladesh J Child Health. 2014;38(1):44-47.
9. Woods R, Larkin JO, Muldoon C, Kennedy MJ, Mehigan B, McCormick P. Metastatic paediatric colorectal carcinoma. Ir Med J. 2012;105(3):88-89.
10. Bjoernsen LP, Lindsay MB. An unusual case of pediatric abdominal pain. CJEM. 2011;13(2):133-138.
11. Takalkar UV, Asegaonkar SB, Kulkarni U, Jadhav A, Advani S, Reddy DN. Carcinoma of colon in an adolescent: a case report with review of literature. Int J Sci Rep 2015;1(2):151-3.
12. Zamir N, Ahmad S, Akhtar J. Mucinous adenocarcinoma of colon. APSP J Case Rep. 2010;1(2):20.
13. Al-Tonbary Y, Darwish A, El-Hussein A, Fouda A. Adenocarcinoma of the colon in children: case series and mini-review of the literature. Hematol Oncol Stem Cell Ther. 2013;6(1):29-33.
14. Ameh EA, Nmadu PT. Colorectal adenocarcinoma in children and adolescents: a report of 8 patients from Zaria, Nigeria. West Afr J Med. 2000;19(4):273-276.
15. Ibrahim, AE, Afolayan KA, Adeniji OM, Buhari KB. Colorectal carcinoma in children and young adults in Ilorin, Nigeria. West Afr J Med. 2011;30(3):202-205.
Acute Aortic Occlusion With Spinal Cord Infarction
Acute aortic occlusion (AAO) is a relatively rare vascular emergency. The actual incidence of AAO is unknown but has been variously reported to be 1% to 4%, and the incidence of AAO secondary to infrarenal abdominal aortic aneurysm is reported to be about 2%.1 Acute aortic occlusion may present with acute onset of neurologic deficits as a consequence of spinal cord ischemia from thrombotic or embolic etiology. Risk factors for thrombosis include hypertension, tobacco smoking, and diabetes mellitus; heart disease and female gender are associated with embolism.2 Spinal cord infarction accounts for only 1% to 2% of all strokes and is characterized by acute onset of paralysis, bowel and bladder dysfunction, and loss of pain and temperature perception. Proprioception and vibratory sense are typically preserved.3 The authors present the case of a patient with acute onset of lower limb paralysis and urinary incontinence who was later found to have AAO due to thrombosis and consequent spinal cord infarction.
Case Presentation
An 80-year-old white woman presented to the emergency department of Jefferson Regional Medical Center in Pine Bluff, Arkansas, with the sudden onset of severe lower back pain, bilateral leg paralysis and paresthesia, and urinary incontinence. The patient stated that she had been watching television when her legs began to tingle and feel numb. Within 10 to 20 minutes she was unable to move her legs and became incontinent of urine. She reported no injury or previous history of back pain. Her medical history was significant for “irregular heartbeat my whole life,” hypertension, hyperlipidemia, and bladder cancer. She was not receiving systemic anticoagulation therapy. She reported a previous 15 pack-year smoking history but reported that she had quit cigarette smoking and usually drank 1 glass of wine daily. She had previously completed 3 rounds of chemotherapy for bladder cancer and received her first radiation treatment earlier that day. The symptoms began about 8 hours later that evening.
On examination the patient was noted to be in acute distress due to pain. Her vital signs in triage were blood pressure (BP) 122/71 mm Hg, pulse 54 beats per min (BPM), respirations 18 breathes per min, temperature 98° F, and pulse oximetry 90% on 2 L/min oxygen via nasal cannula. Laboratory evaluation was remarkable only for serum sodium 132 mEq/L, potassium 2.8 mEq/L, and thrombocytosis with platelets 697 × 103/μL. A neurologic examination showed normal motor function, strength of the upper extremities, and paralysis of the lower extremities, which were insensate to blunt or sharp touch and with decreased skin temperature from the groin distally. Pedal pulses were absent bilaterally. She was incontinent of urine and had anal sphincter laxity.
Magnetic resonance imaging showed bulging lumbar intervertebral discs and foraminal narrowing, which the consulting neurosurgeon did not feel explained her presentation and suggested that a vascular etiology was more likely. A contrast-enhanced computed tomography (CT) scan of the abdomen and pelvis showed occlusion of the infrarenal abdominal aorta, bilateral common, and external iliac arteries. The proximal inferior mesenteric artery was occluded, and there was 90% stenosis of the proximal superior mesenteric artery with noncalcified plaque. There was no abdominal aortic aneurysm or dissection demonstrated. A chest CT was unremarkable.
The patient was started on IV heparin 800 U/h and transferred via ambulance to the University of Arkansas for Medical Sciences in Little Rock, Arkansas, for vascular surgery. On arrival her vital signs were BP 108/69 mm Hg, pulse 123 BPM, respirations 18 breathes per min, temperature 98° F, and pulse oximetry 94% on 2 L/min oxygen via nasal cannula. The patient’s electrocardiogram demonstrated atrial fibrillation with rapid ventricular response with premature ventricular complexes. On examination the bilateral lower extremities were cyanotic and cold to the touch. The pedal pulses were nonpalpable, and decreased distal sensation with dense paralysis was noted. 
The patient was taken emergently to the operating room (OR) for left axillary bifemoral bypass. Severe atherosclerotic disease was noted at surgery. She was transferred to the surgical intensive care unit (SICU) for postoperative hemodynamic monitoring. Her clinical course became complicated by mesenteric ischemia from chronic superior mesenteric artery (SMA) occlusion. On postoperative day 2, she became progressively more hypotensive, and she was placed on vasopressin 0.04 U/min and amiodarone 0.5 mg/min infusions.
Bedside echocardiography showed diffuse ventricular hypokinesis and a left ventricular ejection fraction (LVEF) of about 30% but no mural thrombus. The patient developed altered mental status and respiratory distress, and her serum lactate increased to 6.1 mg/dL. She was emergently intubated and taken to the OR to attempt recanalization of the SMA occlusion, which was unsuccessful. She was returned to the SICU for continued resuscitation and monitoring. She continued to decline with hypotensive pressures, increasing serum creatinine and lactate, and worsening metabolic acidosis. Management options and goals of care were discussed with the family, and it was decided to honor her do not resuscitate status and pursue comfort care. She was extubated and expired a short time after this was done.
Discussion
Acute aortic occlusion is a rare vascular emergency with a mortality rate that approaches 75%.4-6 It results from numerous etiologies, including saddle embolism at the aortic bifurcation, acute thrombus formation, subsequent to aortic dissection, or other causes related to severe atherosclerotic disease or hypercoagulable states.4,7
A recent retrospective series of 29 cases of AAO found that thrombosis was the cause for 76% of cases, and > 40% of patients had a hypercoagulable state either because of antiphospholipid antibody syndrome (17%) or malignancy (24%).6 The most common presentation of AAO is the abrupt onset of painful bilateral paresis or paraplegia.5,6 While some studies have suggested that the major determinant of mortality is time elapsed until revascularization,7 other studies have reported that the neurologic status of the extremities is more closely related with mortality.2
The anterior spinal artery is the major independent provider of blood flow to the anterior two-thirds of the spinal cord, including the anterior horns, the anterior commissure, the anterior funiculi, and to a variable extent, the lateral funiculi. The largest segmental posterior radicular branch of the anterior spinal artery is the artery of Adamkiewicz, which arises from the T9 to T12 level on the left in 75% of cases and provides perfusion to the lumbar spinal cord and the conus medullaris. Obstruction of blood flow in this region has been implicated in the clinical picture of anterior cord syndrome characterized by abrupt onset of radicular pain, flaccid paresis or paralysis, sphincter dysfunction with urinary and fecal incontinence, and decreased pain and temperature sensation below a sensory level with spared proprioception and vibratory sensation.3, 8-10
Aortography is the gold standard procedure for diagnosis of AAO, but it is a time-consuming procedure, and preoperative testing is controversial. Contrast-enhanced CT is useful for evaluation as it can be quickly accomplished and is more available in general hospitals. Moreover, CT scanning may reveal aortic dissections or aneurysms as the cause of occlusion. Deep Doppler ultrasonography also has demonstrated utility as a noninvasive and rapidly performed diagnostic procedure. Magnetic resonance angiography or CT should be performed for all cases unless the patient’s clinical condition prevents this evaluation. Imaging not only confirms diagnosis, but also is valuable for assessment and planning management.11-14
Once the diagnosis of AAO is made, management with IV fluid hydration, heparin administration, and optimizing cardiac function are essential. However, conservative management with anticoagulation alone is associated with high mortality, and unless the ischemia is irreversible or unless the patient is in a dying state, surgery is appropriate.5,7 Depending on the etiology of the AAO, anatomic considerations, and other patient factors, urgent revascularization with thrombo-embolectomy, direct aortic reconstruction, or anatomic or extra-anatomic bypass procedures may be employed. Aortic reconstruction has been advocated for all patients with infrarenal aortic occlusion given the concern for propagation of thrombosis at the distal aorta proximally to the renal and mesenteric arteries.7 Axillary-bifemoral bypass has been advocated as a rapid revascularization strategy with good patency and less physiologic strain for critically ill AAO patients.6
The patient in this study had a constellation of risk factors for developing AAO due to thrombosis and consequently sustaining spinal cord infarction. Echocardiography ruled out embolism of a mural thrombus. She had cardiac dysfunction due to atrial fibrillation and left ventricular failure causing a low-flow state (LVEF 30%). She also had a hypercoagulable state due to bladder malignancy in addition to severe atherosclerotic disease. She was not on systemic anticoagulation therapy because of her high fall risk. Hence, her risk for thrombosis was quite high. Despite expedient revascularization surgery, her postoperative course was complicated as a result of severe mesenteric ischemia due to chronic SMA occlusion, which caused her death.
Conclusion
Acute aortic occlusion is a rare vascular emergency. The patient presenting with the abrupt onset of bilateral leg pain, neurologic deficits of paresis/paralysis, sensory disturbance, and/or sphincter dysfunction, and stigmata of vascular compromise with lower extremity mottling should alert the physician to AAO. Acute aortic occlusion continues to have high morbidity and mortality, and prompt recognition and appropriate transfer for surgical intervention are essential for improving outcomes.
1. de Varona Frolov SR, Acosta Silva MP, Volvo Pérez G, Fiuza Pérez MD. Outcomes after treatment of acute aortic occlusion. [Article in English, Spanish] Cir Esp. 2015;93(9):573-579.
2. Dossa CD, Shepard AD, Reddy DJ, et al. Acute aortic occlusion: a 40-year experience. Arch Surg. 1994;129(6):603-608.
3. Sandson TA, Friedman JH. Spinal cord infarction. Report of 8 cases and review of the literature. Medicine (Baltimore). 1989;68(5):282-292.
4. Yamamoto H, Yamamoto F, Tanaka F, et al. Acute occlusion of the abdominal aorta with concomitant internal iliac artery occlusion. Ann Thorac Cardiovasc Surg. 2011;17(4):422-427.
5. Zainal AA, Oommen G, Chew LG, Yusha AW. Acute aortic occlusion: the need to be aware. Med J Malaysia. 2000;55(1):29-32.
6. Crawford JD, Perrone KH, Wong VW, et al. A modern series of acute aortic occlusion. J Vasc Surg. 2014;59(4):1044-1050.
7. Babu SC, Shah PM, Nitahara J. Acute aortic occlusion—factors that influence outcome. J Vasc Surg. 1995;21(4):567-572.
8. Cheshire WP, Santos CC, Massey EW, Howard JF Jr. Spinal cord infarction: etiology and outcome. Neurology. 1996;47(2):321-330.
9. Rosenthal D. Spinal cord ischemia after abdominal aortic operation: is it preventable? J Vasc Surg. 1999;30(3):391-397.
10. Triantafyllopoulos GK, Athanassacopoulos M, Maltezos C, Pneumaticos SG. Acute infrarenal aortic thrombosis presenting as flaccid paraplegia. Spine (Phila Pa 1976). 2011;36(15):E1042-E1045.
11. Nienaber CA. The role of imaging in acute aortic syndromes. Eur Heart J Cardiovasc Imaging. 2013;14(1):15-23.
12. Bollinger B, Strandberg C, Baekgaard N, Mantoni M, Helweg-Larsen S. Diagnosis of acute aortic occlusion by computer tomography. Vasa. 1995;24(2):199-201.
13. Bertucci B, Rotundo A, Perri G, Sessa E, Tamburrini O. Acute thrombotic occlusion of the infrarenal abdominal aorta: its diagnosis with spiral computed tomography in a case [Article in Italian]. Radiol Med. 1997;94(5):541-543.
14. Battaglia S, Danesino GM, Danesino V, Castellani S. Color doppler ultrasonography of the abdominal aorta. J Ultrasound. 2010;13(3):107-117.
Acute aortic occlusion (AAO) is a relatively rare vascular emergency. The actual incidence of AAO is unknown but has been variously reported to be 1% to 4%, and the incidence of AAO secondary to infrarenal abdominal aortic aneurysm is reported to be about 2%.1 Acute aortic occlusion may present with acute onset of neurologic deficits as a consequence of spinal cord ischemia from thrombotic or embolic etiology. Risk factors for thrombosis include hypertension, tobacco smoking, and diabetes mellitus; heart disease and female gender are associated with embolism.2 Spinal cord infarction accounts for only 1% to 2% of all strokes and is characterized by acute onset of paralysis, bowel and bladder dysfunction, and loss of pain and temperature perception. Proprioception and vibratory sense are typically preserved.3 The authors present the case of a patient with acute onset of lower limb paralysis and urinary incontinence who was later found to have AAO due to thrombosis and consequent spinal cord infarction.
Case Presentation
An 80-year-old white woman presented to the emergency department of Jefferson Regional Medical Center in Pine Bluff, Arkansas, with the sudden onset of severe lower back pain, bilateral leg paralysis and paresthesia, and urinary incontinence. The patient stated that she had been watching television when her legs began to tingle and feel numb. Within 10 to 20 minutes she was unable to move her legs and became incontinent of urine. She reported no injury or previous history of back pain. Her medical history was significant for “irregular heartbeat my whole life,” hypertension, hyperlipidemia, and bladder cancer. She was not receiving systemic anticoagulation therapy. She reported a previous 15 pack-year smoking history but reported that she had quit cigarette smoking and usually drank 1 glass of wine daily. She had previously completed 3 rounds of chemotherapy for bladder cancer and received her first radiation treatment earlier that day. The symptoms began about 8 hours later that evening.
On examination the patient was noted to be in acute distress due to pain. Her vital signs in triage were blood pressure (BP) 122/71 mm Hg, pulse 54 beats per min (BPM), respirations 18 breathes per min, temperature 98° F, and pulse oximetry 90% on 2 L/min oxygen via nasal cannula. Laboratory evaluation was remarkable only for serum sodium 132 mEq/L, potassium 2.8 mEq/L, and thrombocytosis with platelets 697 × 103/μL. A neurologic examination showed normal motor function, strength of the upper extremities, and paralysis of the lower extremities, which were insensate to blunt or sharp touch and with decreased skin temperature from the groin distally. Pedal pulses were absent bilaterally. She was incontinent of urine and had anal sphincter laxity.
Magnetic resonance imaging showed bulging lumbar intervertebral discs and foraminal narrowing, which the consulting neurosurgeon did not feel explained her presentation and suggested that a vascular etiology was more likely. A contrast-enhanced computed tomography (CT) scan of the abdomen and pelvis showed occlusion of the infrarenal abdominal aorta, bilateral common, and external iliac arteries. The proximal inferior mesenteric artery was occluded, and there was 90% stenosis of the proximal superior mesenteric artery with noncalcified plaque. There was no abdominal aortic aneurysm or dissection demonstrated. A chest CT was unremarkable.
The patient was started on IV heparin 800 U/h and transferred via ambulance to the University of Arkansas for Medical Sciences in Little Rock, Arkansas, for vascular surgery. On arrival her vital signs were BP 108/69 mm Hg, pulse 123 BPM, respirations 18 breathes per min, temperature 98° F, and pulse oximetry 94% on 2 L/min oxygen via nasal cannula. The patient’s electrocardiogram demonstrated atrial fibrillation with rapid ventricular response with premature ventricular complexes. On examination the bilateral lower extremities were cyanotic and cold to the touch. The pedal pulses were nonpalpable, and decreased distal sensation with dense paralysis was noted.
The patient was taken emergently to the operating room (OR) for left axillary bifemoral bypass. Severe atherosclerotic disease was noted at surgery. She was transferred to the surgical intensive care unit (SICU) for postoperative hemodynamic monitoring. Her clinical course became complicated by mesenteric ischemia from chronic superior mesenteric artery (SMA) occlusion. On postoperative day 2, she became progressively more hypotensive, and she was placed on vasopressin 0.04 U/min and amiodarone 0.5 mg/min infusions.
Bedside echocardiography showed diffuse ventricular hypokinesis and a left ventricular ejection fraction (LVEF) of about 30% but no mural thrombus. The patient developed altered mental status and respiratory distress, and her serum lactate increased to 6.1 mg/dL. She was emergently intubated and taken to the OR to attempt recanalization of the SMA occlusion, which was unsuccessful. She was returned to the SICU for continued resuscitation and monitoring. She continued to decline with hypotensive pressures, increasing serum creatinine and lactate, and worsening metabolic acidosis. Management options and goals of care were discussed with the family, and it was decided to honor her do not resuscitate status and pursue comfort care. She was extubated and expired a short time after this was done.
Discussion
Acute aortic occlusion is a rare vascular emergency with a mortality rate that approaches 75%.4-6 It results from numerous etiologies, including saddle embolism at the aortic bifurcation, acute thrombus formation, subsequent to aortic dissection, or other causes related to severe atherosclerotic disease or hypercoagulable states.4,7
A recent retrospective series of 29 cases of AAO found that thrombosis was the cause for 76% of cases, and > 40% of patients had a hypercoagulable state either because of antiphospholipid antibody syndrome (17%) or malignancy (24%).6 The most common presentation of AAO is the abrupt onset of painful bilateral paresis or paraplegia.5,6 While some studies have suggested that the major determinant of mortality is time elapsed until revascularization,7 other studies have reported that the neurologic status of the extremities is more closely related with mortality.2
The anterior spinal artery is the major independent provider of blood flow to the anterior two-thirds of the spinal cord, including the anterior horns, the anterior commissure, the anterior funiculi, and to a variable extent, the lateral funiculi. The largest segmental posterior radicular branch of the anterior spinal artery is the artery of Adamkiewicz, which arises from the T9 to T12 level on the left in 75% of cases and provides perfusion to the lumbar spinal cord and the conus medullaris. Obstruction of blood flow in this region has been implicated in the clinical picture of anterior cord syndrome characterized by abrupt onset of radicular pain, flaccid paresis or paralysis, sphincter dysfunction with urinary and fecal incontinence, and decreased pain and temperature sensation below a sensory level with spared proprioception and vibratory sensation.3, 8-10
Aortography is the gold standard procedure for diagnosis of AAO, but it is a time-consuming procedure, and preoperative testing is controversial. Contrast-enhanced CT is useful for evaluation as it can be quickly accomplished and is more available in general hospitals. Moreover, CT scanning may reveal aortic dissections or aneurysms as the cause of occlusion. Deep Doppler ultrasonography also has demonstrated utility as a noninvasive and rapidly performed diagnostic procedure. Magnetic resonance angiography or CT should be performed for all cases unless the patient’s clinical condition prevents this evaluation. Imaging not only confirms diagnosis, but also is valuable for assessment and planning management.11-14
Once the diagnosis of AAO is made, management with IV fluid hydration, heparin administration, and optimizing cardiac function are essential. However, conservative management with anticoagulation alone is associated with high mortality, and unless the ischemia is irreversible or unless the patient is in a dying state, surgery is appropriate.5,7 Depending on the etiology of the AAO, anatomic considerations, and other patient factors, urgent revascularization with thrombo-embolectomy, direct aortic reconstruction, or anatomic or extra-anatomic bypass procedures may be employed. Aortic reconstruction has been advocated for all patients with infrarenal aortic occlusion given the concern for propagation of thrombosis at the distal aorta proximally to the renal and mesenteric arteries.7 Axillary-bifemoral bypass has been advocated as a rapid revascularization strategy with good patency and less physiologic strain for critically ill AAO patients.6
The patient in this study had a constellation of risk factors for developing AAO due to thrombosis and consequently sustaining spinal cord infarction. Echocardiography ruled out embolism of a mural thrombus. She had cardiac dysfunction due to atrial fibrillation and left ventricular failure causing a low-flow state (LVEF 30%). She also had a hypercoagulable state due to bladder malignancy in addition to severe atherosclerotic disease. She was not on systemic anticoagulation therapy because of her high fall risk. Hence, her risk for thrombosis was quite high. Despite expedient revascularization surgery, her postoperative course was complicated as a result of severe mesenteric ischemia due to chronic SMA occlusion, which caused her death.
Conclusion
Acute aortic occlusion is a rare vascular emergency. The patient presenting with the abrupt onset of bilateral leg pain, neurologic deficits of paresis/paralysis, sensory disturbance, and/or sphincter dysfunction, and stigmata of vascular compromise with lower extremity mottling should alert the physician to AAO. Acute aortic occlusion continues to have high morbidity and mortality, and prompt recognition and appropriate transfer for surgical intervention are essential for improving outcomes.
Acute aortic occlusion (AAO) is a relatively rare vascular emergency. The actual incidence of AAO is unknown but has been variously reported to be 1% to 4%, and the incidence of AAO secondary to infrarenal abdominal aortic aneurysm is reported to be about 2%.1 Acute aortic occlusion may present with acute onset of neurologic deficits as a consequence of spinal cord ischemia from thrombotic or embolic etiology. Risk factors for thrombosis include hypertension, tobacco smoking, and diabetes mellitus; heart disease and female gender are associated with embolism.2 Spinal cord infarction accounts for only 1% to 2% of all strokes and is characterized by acute onset of paralysis, bowel and bladder dysfunction, and loss of pain and temperature perception. Proprioception and vibratory sense are typically preserved.3 The authors present the case of a patient with acute onset of lower limb paralysis and urinary incontinence who was later found to have AAO due to thrombosis and consequent spinal cord infarction.
Case Presentation
An 80-year-old white woman presented to the emergency department of Jefferson Regional Medical Center in Pine Bluff, Arkansas, with the sudden onset of severe lower back pain, bilateral leg paralysis and paresthesia, and urinary incontinence. The patient stated that she had been watching television when her legs began to tingle and feel numb. Within 10 to 20 minutes she was unable to move her legs and became incontinent of urine. She reported no injury or previous history of back pain. Her medical history was significant for “irregular heartbeat my whole life,” hypertension, hyperlipidemia, and bladder cancer. She was not receiving systemic anticoagulation therapy. She reported a previous 15 pack-year smoking history but reported that she had quit cigarette smoking and usually drank 1 glass of wine daily. She had previously completed 3 rounds of chemotherapy for bladder cancer and received her first radiation treatment earlier that day. The symptoms began about 8 hours later that evening.
On examination the patient was noted to be in acute distress due to pain. Her vital signs in triage were blood pressure (BP) 122/71 mm Hg, pulse 54 beats per min (BPM), respirations 18 breathes per min, temperature 98° F, and pulse oximetry 90% on 2 L/min oxygen via nasal cannula. Laboratory evaluation was remarkable only for serum sodium 132 mEq/L, potassium 2.8 mEq/L, and thrombocytosis with platelets 697 × 103/μL. A neurologic examination showed normal motor function, strength of the upper extremities, and paralysis of the lower extremities, which were insensate to blunt or sharp touch and with decreased skin temperature from the groin distally. Pedal pulses were absent bilaterally. She was incontinent of urine and had anal sphincter laxity.
Magnetic resonance imaging showed bulging lumbar intervertebral discs and foraminal narrowing, which the consulting neurosurgeon did not feel explained her presentation and suggested that a vascular etiology was more likely. A contrast-enhanced computed tomography (CT) scan of the abdomen and pelvis showed occlusion of the infrarenal abdominal aorta, bilateral common, and external iliac arteries. The proximal inferior mesenteric artery was occluded, and there was 90% stenosis of the proximal superior mesenteric artery with noncalcified plaque. There was no abdominal aortic aneurysm or dissection demonstrated. A chest CT was unremarkable.
The patient was started on IV heparin 800 U/h and transferred via ambulance to the University of Arkansas for Medical Sciences in Little Rock, Arkansas, for vascular surgery. On arrival her vital signs were BP 108/69 mm Hg, pulse 123 BPM, respirations 18 breathes per min, temperature 98° F, and pulse oximetry 94% on 2 L/min oxygen via nasal cannula. The patient’s electrocardiogram demonstrated atrial fibrillation with rapid ventricular response with premature ventricular complexes. On examination the bilateral lower extremities were cyanotic and cold to the touch. The pedal pulses were nonpalpable, and decreased distal sensation with dense paralysis was noted.
The patient was taken emergently to the operating room (OR) for left axillary bifemoral bypass. Severe atherosclerotic disease was noted at surgery. She was transferred to the surgical intensive care unit (SICU) for postoperative hemodynamic monitoring. Her clinical course became complicated by mesenteric ischemia from chronic superior mesenteric artery (SMA) occlusion. On postoperative day 2, she became progressively more hypotensive, and she was placed on vasopressin 0.04 U/min and amiodarone 0.5 mg/min infusions.
Bedside echocardiography showed diffuse ventricular hypokinesis and a left ventricular ejection fraction (LVEF) of about 30% but no mural thrombus. The patient developed altered mental status and respiratory distress, and her serum lactate increased to 6.1 mg/dL. She was emergently intubated and taken to the OR to attempt recanalization of the SMA occlusion, which was unsuccessful. She was returned to the SICU for continued resuscitation and monitoring. She continued to decline with hypotensive pressures, increasing serum creatinine and lactate, and worsening metabolic acidosis. Management options and goals of care were discussed with the family, and it was decided to honor her do not resuscitate status and pursue comfort care. She was extubated and expired a short time after this was done.
Discussion
Acute aortic occlusion is a rare vascular emergency with a mortality rate that approaches 75%.4-6 It results from numerous etiologies, including saddle embolism at the aortic bifurcation, acute thrombus formation, subsequent to aortic dissection, or other causes related to severe atherosclerotic disease or hypercoagulable states.4,7
A recent retrospective series of 29 cases of AAO found that thrombosis was the cause for 76% of cases, and > 40% of patients had a hypercoagulable state either because of antiphospholipid antibody syndrome (17%) or malignancy (24%).6 The most common presentation of AAO is the abrupt onset of painful bilateral paresis or paraplegia.5,6 While some studies have suggested that the major determinant of mortality is time elapsed until revascularization,7 other studies have reported that the neurologic status of the extremities is more closely related with mortality.2
The anterior spinal artery is the major independent provider of blood flow to the anterior two-thirds of the spinal cord, including the anterior horns, the anterior commissure, the anterior funiculi, and to a variable extent, the lateral funiculi. The largest segmental posterior radicular branch of the anterior spinal artery is the artery of Adamkiewicz, which arises from the T9 to T12 level on the left in 75% of cases and provides perfusion to the lumbar spinal cord and the conus medullaris. Obstruction of blood flow in this region has been implicated in the clinical picture of anterior cord syndrome characterized by abrupt onset of radicular pain, flaccid paresis or paralysis, sphincter dysfunction with urinary and fecal incontinence, and decreased pain and temperature sensation below a sensory level with spared proprioception and vibratory sensation.3, 8-10
Aortography is the gold standard procedure for diagnosis of AAO, but it is a time-consuming procedure, and preoperative testing is controversial. Contrast-enhanced CT is useful for evaluation as it can be quickly accomplished and is more available in general hospitals. Moreover, CT scanning may reveal aortic dissections or aneurysms as the cause of occlusion. Deep Doppler ultrasonography also has demonstrated utility as a noninvasive and rapidly performed diagnostic procedure. Magnetic resonance angiography or CT should be performed for all cases unless the patient’s clinical condition prevents this evaluation. Imaging not only confirms diagnosis, but also is valuable for assessment and planning management.11-14
Once the diagnosis of AAO is made, management with IV fluid hydration, heparin administration, and optimizing cardiac function are essential. However, conservative management with anticoagulation alone is associated with high mortality, and unless the ischemia is irreversible or unless the patient is in a dying state, surgery is appropriate.5,7 Depending on the etiology of the AAO, anatomic considerations, and other patient factors, urgent revascularization with thrombo-embolectomy, direct aortic reconstruction, or anatomic or extra-anatomic bypass procedures may be employed. Aortic reconstruction has been advocated for all patients with infrarenal aortic occlusion given the concern for propagation of thrombosis at the distal aorta proximally to the renal and mesenteric arteries.7 Axillary-bifemoral bypass has been advocated as a rapid revascularization strategy with good patency and less physiologic strain for critically ill AAO patients.6
The patient in this study had a constellation of risk factors for developing AAO due to thrombosis and consequently sustaining spinal cord infarction. Echocardiography ruled out embolism of a mural thrombus. She had cardiac dysfunction due to atrial fibrillation and left ventricular failure causing a low-flow state (LVEF 30%). She also had a hypercoagulable state due to bladder malignancy in addition to severe atherosclerotic disease. She was not on systemic anticoagulation therapy because of her high fall risk. Hence, her risk for thrombosis was quite high. Despite expedient revascularization surgery, her postoperative course was complicated as a result of severe mesenteric ischemia due to chronic SMA occlusion, which caused her death.
Conclusion
Acute aortic occlusion is a rare vascular emergency. The patient presenting with the abrupt onset of bilateral leg pain, neurologic deficits of paresis/paralysis, sensory disturbance, and/or sphincter dysfunction, and stigmata of vascular compromise with lower extremity mottling should alert the physician to AAO. Acute aortic occlusion continues to have high morbidity and mortality, and prompt recognition and appropriate transfer for surgical intervention are essential for improving outcomes.
1. de Varona Frolov SR, Acosta Silva MP, Volvo Pérez G, Fiuza Pérez MD. Outcomes after treatment of acute aortic occlusion. [Article in English, Spanish] Cir Esp. 2015;93(9):573-579.
2. Dossa CD, Shepard AD, Reddy DJ, et al. Acute aortic occlusion: a 40-year experience. Arch Surg. 1994;129(6):603-608.
3. Sandson TA, Friedman JH. Spinal cord infarction. Report of 8 cases and review of the literature. Medicine (Baltimore). 1989;68(5):282-292.
4. Yamamoto H, Yamamoto F, Tanaka F, et al. Acute occlusion of the abdominal aorta with concomitant internal iliac artery occlusion. Ann Thorac Cardiovasc Surg. 2011;17(4):422-427.
5. Zainal AA, Oommen G, Chew LG, Yusha AW. Acute aortic occlusion: the need to be aware. Med J Malaysia. 2000;55(1):29-32.
6. Crawford JD, Perrone KH, Wong VW, et al. A modern series of acute aortic occlusion. J Vasc Surg. 2014;59(4):1044-1050.
7. Babu SC, Shah PM, Nitahara J. Acute aortic occlusion—factors that influence outcome. J Vasc Surg. 1995;21(4):567-572.
8. Cheshire WP, Santos CC, Massey EW, Howard JF Jr. Spinal cord infarction: etiology and outcome. Neurology. 1996;47(2):321-330.
9. Rosenthal D. Spinal cord ischemia after abdominal aortic operation: is it preventable? J Vasc Surg. 1999;30(3):391-397.
10. Triantafyllopoulos GK, Athanassacopoulos M, Maltezos C, Pneumaticos SG. Acute infrarenal aortic thrombosis presenting as flaccid paraplegia. Spine (Phila Pa 1976). 2011;36(15):E1042-E1045.
11. Nienaber CA. The role of imaging in acute aortic syndromes. Eur Heart J Cardiovasc Imaging. 2013;14(1):15-23.
12. Bollinger B, Strandberg C, Baekgaard N, Mantoni M, Helweg-Larsen S. Diagnosis of acute aortic occlusion by computer tomography. Vasa. 1995;24(2):199-201.
13. Bertucci B, Rotundo A, Perri G, Sessa E, Tamburrini O. Acute thrombotic occlusion of the infrarenal abdominal aorta: its diagnosis with spiral computed tomography in a case [Article in Italian]. Radiol Med. 1997;94(5):541-543.
14. Battaglia S, Danesino GM, Danesino V, Castellani S. Color doppler ultrasonography of the abdominal aorta. J Ultrasound. 2010;13(3):107-117.
1. de Varona Frolov SR, Acosta Silva MP, Volvo Pérez G, Fiuza Pérez MD. Outcomes after treatment of acute aortic occlusion. [Article in English, Spanish] Cir Esp. 2015;93(9):573-579.
2. Dossa CD, Shepard AD, Reddy DJ, et al. Acute aortic occlusion: a 40-year experience. Arch Surg. 1994;129(6):603-608.
3. Sandson TA, Friedman JH. Spinal cord infarction. Report of 8 cases and review of the literature. Medicine (Baltimore). 1989;68(5):282-292.
4. Yamamoto H, Yamamoto F, Tanaka F, et al. Acute occlusion of the abdominal aorta with concomitant internal iliac artery occlusion. Ann Thorac Cardiovasc Surg. 2011;17(4):422-427.
5. Zainal AA, Oommen G, Chew LG, Yusha AW. Acute aortic occlusion: the need to be aware. Med J Malaysia. 2000;55(1):29-32.
6. Crawford JD, Perrone KH, Wong VW, et al. A modern series of acute aortic occlusion. J Vasc Surg. 2014;59(4):1044-1050.
7. Babu SC, Shah PM, Nitahara J. Acute aortic occlusion—factors that influence outcome. J Vasc Surg. 1995;21(4):567-572.
8. Cheshire WP, Santos CC, Massey EW, Howard JF Jr. Spinal cord infarction: etiology and outcome. Neurology. 1996;47(2):321-330.
9. Rosenthal D. Spinal cord ischemia after abdominal aortic operation: is it preventable? J Vasc Surg. 1999;30(3):391-397.
10. Triantafyllopoulos GK, Athanassacopoulos M, Maltezos C, Pneumaticos SG. Acute infrarenal aortic thrombosis presenting as flaccid paraplegia. Spine (Phila Pa 1976). 2011;36(15):E1042-E1045.
11. Nienaber CA. The role of imaging in acute aortic syndromes. Eur Heart J Cardiovasc Imaging. 2013;14(1):15-23.
12. Bollinger B, Strandberg C, Baekgaard N, Mantoni M, Helweg-Larsen S. Diagnosis of acute aortic occlusion by computer tomography. Vasa. 1995;24(2):199-201.
13. Bertucci B, Rotundo A, Perri G, Sessa E, Tamburrini O. Acute thrombotic occlusion of the infrarenal abdominal aorta: its diagnosis with spiral computed tomography in a case [Article in Italian]. Radiol Med. 1997;94(5):541-543.
14. Battaglia S, Danesino GM, Danesino V, Castellani S. Color doppler ultrasonography of the abdominal aorta. J Ultrasound. 2010;13(3):107-117.
An unusual case of primary cardiac prosthetic valve-associated lymphoma
Primary cardiac tumors are extremely rare neoplasms with an incidence of less than 0.4%.1-3 Primary cardiac lymphoma (PCL), the majority of which is non-Hodgkin lymphoma, accounts for around 2% of cardiac tumors and less than 0.5% of extranodal lymphomas.1,4-6 Primary lymphoma involving cardiac valves has been described in few case reports and small case series owing to its rarity.7-10 Most cases of PCL present with manifestations of congestive heart failure or cardiac arrhythmias,11 whereas primary valve-associated lymphoma (PV-AL) is usually diagnosed incidentally during valve repair or replacement. The pathophysiology remains unclear, but a few cases have been associated with Epstein Barr virus (EBV).7 Cases previously described in the literature carried an overall poor prognosis and to date there is no standardized treatment approach. We provide here an unusual case of primary prosthetic valve-associated cardiac large B-cell lymphoma, which was successfully treated with adjuvant chemotherapy after valve repair and which resulted in an excellent long-term outcome.
Case presentation and summary
The patient presented in 2012 as a 65-year-old man with a history of ascending aortic aneurysm with secondary aortic insufficiency who in 2004 had undergone composite valve replacement of the aortic valve (AV) root and ascending aorta with a St Jude Toronto root. In June 2011, he was found to have a right parietal intraparenchymal hemorrhage that was thought to be a thromboembolic hemorrhagic ischemic stroke. In March 2012, he had routine follow-up brain magnetic resonance imaging that incidentally showed a left frontal ischemic stroke with hemorrhagic conversion. In June 2012, he was found to have first degree atrioventricular block with episodic runs of supraventricular tachycardia.
In September 2012, transthoracic echocardiography was done for further evaluation of possible recurrent cryptogenic strokes. The results showed a hypo-echogenic mass within the proximal ascending aortic root, but this was not confirmed on transesophageal echocardiography. A chest computed-tomography (CT) scan was therefore performed, and it showed aneurysmal dilatation of the aortic root with an irregular marginal filling defect just above the AV suggestive of intraluminal thrombus. The patient was placed on full anticoagulation with warfarin and referred for cardiothoracic surgery to consider graft and valve replacement. However, 3 weeks later and before the surgery, the patient developed a third thromboembolic ischemic event (transient ischemic attack). The recurrent strokes were attributed to thromboembolic events secondary to prosthetic AV thrombosis.
A repeat transthoracic echocardiography was significant for an abnormal AV bioprosthesis with associated thrombus extending to the ascending aorta. Surgical excision and replacement of the AV conduit explant were performed in November 2012. The final pathology was consistent with EBV-associated large B-cell lymphoma (Figure). The initial staging evaluation, including a CT and positron-emission tomography scan and bone marrow biopsy, was negative for any systemic disease. The patient received 4 cycles of R-CHOP-21 (rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2 , vincristine 2 mg, and prednisone 100 mg) every 3 weeks in an “adjuvant” setting (because patient had no evidence of disease when given the systemic chemotherapy). The patient tolerated chemotherapy well without significant complications, and he is now over 36 months post-treatment without evidence of recurrent disease.
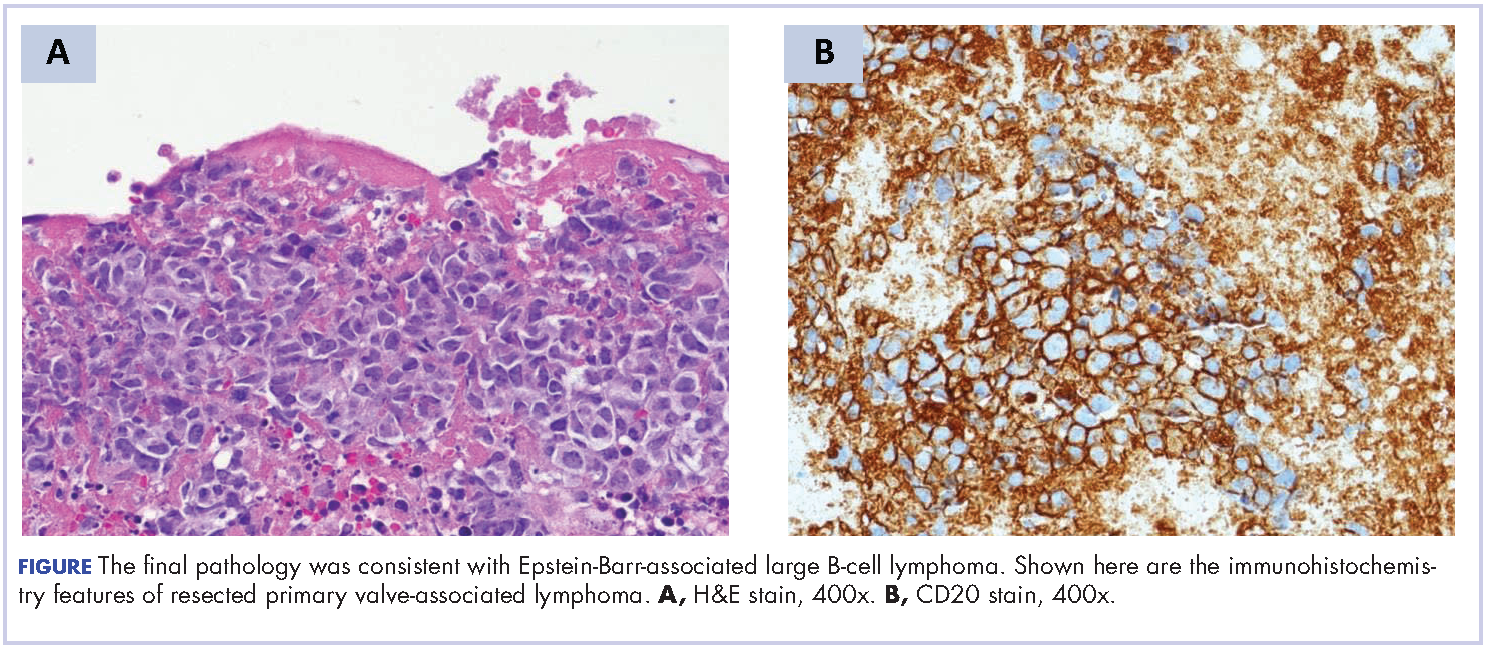
Discussion
Cardiac lymphoma limited only to prosthetic valves is rare, but it has been reported increasingly over the past few years. Until 2010, only six cases of PV-AL had been reported in the literature.7 Including our case, we identified four additional PubMed-indexed cases (using a PubMed search through February 2015). The patient characteristics and treatments received for all identified cases are described in the accompanying Table. The pathology from all of the cases revealed non-Hodgkin lymphoma of large B-cell subtype. PV-AL predominated among men (60%) and older patients with a median age of 62.5 years at diagnosis (range, 48-80 years). Patients had a median duration of 8 years (range, 4-24 years) from date of prosthesis placement to date of lymphoma diagnosis. The three most common presenting manifestations were valvular dysfunction, stroke, and congestive heart failure. All of the patients had surgical intervention on initial presentation. However, management after surgery was not uniform, with only 3 patients reported to have received systemic chemotherapy (Table). None of the patients received adjuvant radiation therapy. Calculated from date of diagnosis, survival duration ranged from less than a month7 to more than 36 months (as reported in our case).
The pathophysiology of PV-AL is not well understood given the rarity of the condition. Similar to other prosthetic-related neoplasms (metallic implants, breast implants),12-14 it has been hypothesized that chronic inflammation and EBV infection may play an essential role in the pathogenesis of this entity. Further, it has been suggested that Dacron, which is used in composite cardiac valve replacements, is carcinogenic and may play a role in some cases.7,15 PV-AL should be highly considered in the differential diagnosis of a suspicious prosthetic valve mass. Various imaging modalities, including echocardiography, CT, and magnetic resonance imaging have been described to have a role in the preoperative evaluation of cardiac tumors by assessing the cardiac function and defining the location and extent of the cardiac tumors.16-19
Given the rarity of this disease entity, there is no standardized approach for treatment. Surgical resection along with repair or replacement of primary involved prosthetic valve is essential for initial treatment. However, there is no consensus about the best approach for subsequent therapy. We cannot be conclusive about the optimum treatment, because of the limited number of published cases, but based on our reading of those cases, it would seem that early surgical intervention and “adjuvant” systemic therapy may have influenced prognosis. We speculate that poor outcomes in the first 6 months were most likely related to primary cardiopulmonary deterioration, whereas later poor outcomes were more likely to be attributable to recurrent lymphoma, particularly for patients who received suboptimal systemic chemotherapy treatment after surgery. All 3 patients who received chemotherapy had no evidence of recurrent disease at last follow-up. Of the 4 patients who received no chemotherapy and survived longer than 6 months (all except 1 died; Table), 2 had recurrent valve lymphoma, 1 had secondary systemic lymphoma, and 1 died of metastatic breast cancer. Those outcomes are in contrast to the 2 out of 3 patients who received adjuvant chemotherapy and who were reported to be alive at 16 and 36 months after diagnosis.
In conclusion, cardiac PV-AL is an increasingly recognized entity that warrants greater awareness among health care providers for early diagnosis and timely surgical intervention. Most of the cases are large B-cell lymphoma. Similar to patients with limited-stage DLBCL, fit patients should be highly considered for “adjuvant” systemic chemotherapy to optimize long-term outcomes. Reporting of similar cases is highly encouraged to better define this rare iatrogenic malignancy.
1. Hudzik B, Miszalski-Jamka K, Glowacki J, et al. Malignant tumors of the heart. Cancer epidemiol. 2015;39(5):665-672.
2. Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, eds. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon, France: IARC Press; 2004.
3. Reynen K. Frequency of primary tumors of the heart. Am J Cardiol. 1996;77(1):107.
4. Neragi-Miandoab S, Kim J, Vlahakes GJ. Malignant tumours of the heart: a review of tumour type, diagnosis and therapy. Clin Oncol. 2007;19(10):748-756.
5. Butany J, Nair V, Naseemuddin A, Nair GM, Catton C, Yau T. Cardiac tumours: diagnosis and management. Lancet Oncol. 2005;6(4):219-228.
6. Burke A, Virmani R. Tumors of the heart and great vessels. In: Atlas of tumor pathology, 3rd Series, Fascicle 16. Washington, DC: Armed Forces Institute of Pathology, 1996.
7. Miller DV, Firchau DJ, McClure RF, Kurtin PJ, Feldman AL. Epstein-Barr virus-associated diffuse large B-cell lymphoma arising on cardiac prostheses. Am J Surg Pathol. 2010;34(3):377-384.
8. Albat B, Messner-Pellenc P, Thevenet A. Surgical treatment for primary lymphoma of the heart simulating prosthetic mitral valve thrombosis. J Thoracic Cardiovasc Surg. 1994;108(1):188-189.
9. Bagwan IN, Desai S, Wotherspoon A, Sheppard MN. Unusual presentation of primary cardiac lymphoma. Interact Cardiovasc Thorac Surg. 2009;9(1):127-129.
10. Durrleman NM, El-Hamamsy I, Demaria RG, Carrier M, Perrault LP, Albat B. Cardiac lymphoma following mitral valve replacement. Ann Thorac Surg. 2005;79(3):1040-1042.
11. Petrich A, Cho SI, Billett H. Primary cardiac lymphoma: an analysis of presentation, treatment, and outcome patterns. Cancer. 2011;117(3):581-589.
12. Cheuk W, Chan AC, Chan JK, Lau GT, Chan VN, Yiu HH. Metallic implant-associated lymphoma: a distinct subgroup of large B-cell lymphoma related to pyothorax-associated lymphoma? Am J Surg Pathol. 2005;29(6):832-836.
13. Roden AC, Macon WR, Keeney GL, Myers JL, Feldman AL, Dogan A. Seroma-associated primary anaplastic large-cell lymphoma adjacent to breast implants: an indolent T-cell lymphoproliferative disorder. Mod Pathol. 2008;21(4):455-463.
14. de Jong D, Vasmel WL, de Boer JP, et al. Anaplastic large-cell lymphoma in women with breast implants. JAMA. 2008;300(17):2030-2035.
15. Durrleman N, El Hamamsy I, Demaria R, Carrier M, Perrault LP, Albat B. Is Dacron carcinogenic? Apropos of a case and review of the literature [In French]. Arch Mal Coeur Vaiss. 2004 Mar;97(3):267-270.16. Peters PJ, Reinhardt S. The echocardiographic evaluation of intracardiac masses: a review. J Am Soc Echocard. 2006;19(2):230-240.
17. Gulati G, Sharma S, Kothari SS, Juneja R, Saxena A, Talwar KK. Comparison of echo and MRI in the imaging evaluation of intracardiac masses. Cardiovasc Intervent Radiol. 2004;27(5):459-469.
18. Krombach GA, Spuentrup E, Buecker A, et al. Heart tumors: magnetic resonance imaging and multislice spiral CT [In German]. RoFo. 2005;177(9):1205-1218.
19. Hoey ET, Mankad K, Puppala S, Gopalan D, Sivananthan MU. MRI and CT appearances of cardiac tumours in adults. Clin Radiol. 2009;64(12):1214-1230.
20. Bonnichsen CR, Dearani JA, Maleszewski JJ, Colgan JP, Williamson EE, Ammash NM. Recurrent Epstein-Barr virus-associated diffuse large B-cell lymphoma in an ascending aorta graft. Circulation. 2013;128(13):1481-1483.
21. Berrio G, Suryadevara A, Singh NK, Wesly OH. Diffuse large B-cell lymphoma in an aortic valve allograft. Tex Heart Inst J. 2010;37(4):492-493.
22. Gruver AM, Huba MA, Dogan A, Hsi ED. Fibrin-associated large B-cell lymphoma: part of the spectrum of cardiac lymphomas. Am J Surg Pathol. 2012;36(10):1527-1537.
23. Farah FJ, Chiles CD. Recurrent primary cardiac lymphoma on aortic valve allograft: implications for therapy. Tex Heart Inst J. 2014;41(5):543-546.
Primary cardiac tumors are extremely rare neoplasms with an incidence of less than 0.4%.1-3 Primary cardiac lymphoma (PCL), the majority of which is non-Hodgkin lymphoma, accounts for around 2% of cardiac tumors and less than 0.5% of extranodal lymphomas.1,4-6 Primary lymphoma involving cardiac valves has been described in few case reports and small case series owing to its rarity.7-10 Most cases of PCL present with manifestations of congestive heart failure or cardiac arrhythmias,11 whereas primary valve-associated lymphoma (PV-AL) is usually diagnosed incidentally during valve repair or replacement. The pathophysiology remains unclear, but a few cases have been associated with Epstein Barr virus (EBV).7 Cases previously described in the literature carried an overall poor prognosis and to date there is no standardized treatment approach. We provide here an unusual case of primary prosthetic valve-associated cardiac large B-cell lymphoma, which was successfully treated with adjuvant chemotherapy after valve repair and which resulted in an excellent long-term outcome.
Case presentation and summary
The patient presented in 2012 as a 65-year-old man with a history of ascending aortic aneurysm with secondary aortic insufficiency who in 2004 had undergone composite valve replacement of the aortic valve (AV) root and ascending aorta with a St Jude Toronto root. In June 2011, he was found to have a right parietal intraparenchymal hemorrhage that was thought to be a thromboembolic hemorrhagic ischemic stroke. In March 2012, he had routine follow-up brain magnetic resonance imaging that incidentally showed a left frontal ischemic stroke with hemorrhagic conversion. In June 2012, he was found to have first degree atrioventricular block with episodic runs of supraventricular tachycardia.
In September 2012, transthoracic echocardiography was done for further evaluation of possible recurrent cryptogenic strokes. The results showed a hypo-echogenic mass within the proximal ascending aortic root, but this was not confirmed on transesophageal echocardiography. A chest computed-tomography (CT) scan was therefore performed, and it showed aneurysmal dilatation of the aortic root with an irregular marginal filling defect just above the AV suggestive of intraluminal thrombus. The patient was placed on full anticoagulation with warfarin and referred for cardiothoracic surgery to consider graft and valve replacement. However, 3 weeks later and before the surgery, the patient developed a third thromboembolic ischemic event (transient ischemic attack). The recurrent strokes were attributed to thromboembolic events secondary to prosthetic AV thrombosis.
A repeat transthoracic echocardiography was significant for an abnormal AV bioprosthesis with associated thrombus extending to the ascending aorta. Surgical excision and replacement of the AV conduit explant were performed in November 2012. The final pathology was consistent with EBV-associated large B-cell lymphoma (Figure). The initial staging evaluation, including a CT and positron-emission tomography scan and bone marrow biopsy, was negative for any systemic disease. The patient received 4 cycles of R-CHOP-21 (rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2 , vincristine 2 mg, and prednisone 100 mg) every 3 weeks in an “adjuvant” setting (because patient had no evidence of disease when given the systemic chemotherapy). The patient tolerated chemotherapy well without significant complications, and he is now over 36 months post-treatment without evidence of recurrent disease.
Discussion
Cardiac lymphoma limited only to prosthetic valves is rare, but it has been reported increasingly over the past few years. Until 2010, only six cases of PV-AL had been reported in the literature.7 Including our case, we identified four additional PubMed-indexed cases (using a PubMed search through February 2015). The patient characteristics and treatments received for all identified cases are described in the accompanying Table. The pathology from all of the cases revealed non-Hodgkin lymphoma of large B-cell subtype. PV-AL predominated among men (60%) and older patients with a median age of 62.5 years at diagnosis (range, 48-80 years). Patients had a median duration of 8 years (range, 4-24 years) from date of prosthesis placement to date of lymphoma diagnosis. The three most common presenting manifestations were valvular dysfunction, stroke, and congestive heart failure. All of the patients had surgical intervention on initial presentation. However, management after surgery was not uniform, with only 3 patients reported to have received systemic chemotherapy (Table). None of the patients received adjuvant radiation therapy. Calculated from date of diagnosis, survival duration ranged from less than a month7 to more than 36 months (as reported in our case).
The pathophysiology of PV-AL is not well understood given the rarity of the condition. Similar to other prosthetic-related neoplasms (metallic implants, breast implants),12-14 it has been hypothesized that chronic inflammation and EBV infection may play an essential role in the pathogenesis of this entity. Further, it has been suggested that Dacron, which is used in composite cardiac valve replacements, is carcinogenic and may play a role in some cases.7,15 PV-AL should be highly considered in the differential diagnosis of a suspicious prosthetic valve mass. Various imaging modalities, including echocardiography, CT, and magnetic resonance imaging have been described to have a role in the preoperative evaluation of cardiac tumors by assessing the cardiac function and defining the location and extent of the cardiac tumors.16-19
Given the rarity of this disease entity, there is no standardized approach for treatment. Surgical resection along with repair or replacement of primary involved prosthetic valve is essential for initial treatment. However, there is no consensus about the best approach for subsequent therapy. We cannot be conclusive about the optimum treatment, because of the limited number of published cases, but based on our reading of those cases, it would seem that early surgical intervention and “adjuvant” systemic therapy may have influenced prognosis. We speculate that poor outcomes in the first 6 months were most likely related to primary cardiopulmonary deterioration, whereas later poor outcomes were more likely to be attributable to recurrent lymphoma, particularly for patients who received suboptimal systemic chemotherapy treatment after surgery. All 3 patients who received chemotherapy had no evidence of recurrent disease at last follow-up. Of the 4 patients who received no chemotherapy and survived longer than 6 months (all except 1 died; Table), 2 had recurrent valve lymphoma, 1 had secondary systemic lymphoma, and 1 died of metastatic breast cancer. Those outcomes are in contrast to the 2 out of 3 patients who received adjuvant chemotherapy and who were reported to be alive at 16 and 36 months after diagnosis.
In conclusion, cardiac PV-AL is an increasingly recognized entity that warrants greater awareness among health care providers for early diagnosis and timely surgical intervention. Most of the cases are large B-cell lymphoma. Similar to patients with limited-stage DLBCL, fit patients should be highly considered for “adjuvant” systemic chemotherapy to optimize long-term outcomes. Reporting of similar cases is highly encouraged to better define this rare iatrogenic malignancy.
Primary cardiac tumors are extremely rare neoplasms with an incidence of less than 0.4%.1-3 Primary cardiac lymphoma (PCL), the majority of which is non-Hodgkin lymphoma, accounts for around 2% of cardiac tumors and less than 0.5% of extranodal lymphomas.1,4-6 Primary lymphoma involving cardiac valves has been described in few case reports and small case series owing to its rarity.7-10 Most cases of PCL present with manifestations of congestive heart failure or cardiac arrhythmias,11 whereas primary valve-associated lymphoma (PV-AL) is usually diagnosed incidentally during valve repair or replacement. The pathophysiology remains unclear, but a few cases have been associated with Epstein Barr virus (EBV).7 Cases previously described in the literature carried an overall poor prognosis and to date there is no standardized treatment approach. We provide here an unusual case of primary prosthetic valve-associated cardiac large B-cell lymphoma, which was successfully treated with adjuvant chemotherapy after valve repair and which resulted in an excellent long-term outcome.
Case presentation and summary
The patient presented in 2012 as a 65-year-old man with a history of ascending aortic aneurysm with secondary aortic insufficiency who in 2004 had undergone composite valve replacement of the aortic valve (AV) root and ascending aorta with a St Jude Toronto root. In June 2011, he was found to have a right parietal intraparenchymal hemorrhage that was thought to be a thromboembolic hemorrhagic ischemic stroke. In March 2012, he had routine follow-up brain magnetic resonance imaging that incidentally showed a left frontal ischemic stroke with hemorrhagic conversion. In June 2012, he was found to have first degree atrioventricular block with episodic runs of supraventricular tachycardia.
In September 2012, transthoracic echocardiography was done for further evaluation of possible recurrent cryptogenic strokes. The results showed a hypo-echogenic mass within the proximal ascending aortic root, but this was not confirmed on transesophageal echocardiography. A chest computed-tomography (CT) scan was therefore performed, and it showed aneurysmal dilatation of the aortic root with an irregular marginal filling defect just above the AV suggestive of intraluminal thrombus. The patient was placed on full anticoagulation with warfarin and referred for cardiothoracic surgery to consider graft and valve replacement. However, 3 weeks later and before the surgery, the patient developed a third thromboembolic ischemic event (transient ischemic attack). The recurrent strokes were attributed to thromboembolic events secondary to prosthetic AV thrombosis.
A repeat transthoracic echocardiography was significant for an abnormal AV bioprosthesis with associated thrombus extending to the ascending aorta. Surgical excision and replacement of the AV conduit explant were performed in November 2012. The final pathology was consistent with EBV-associated large B-cell lymphoma (Figure). The initial staging evaluation, including a CT and positron-emission tomography scan and bone marrow biopsy, was negative for any systemic disease. The patient received 4 cycles of R-CHOP-21 (rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2 , vincristine 2 mg, and prednisone 100 mg) every 3 weeks in an “adjuvant” setting (because patient had no evidence of disease when given the systemic chemotherapy). The patient tolerated chemotherapy well without significant complications, and he is now over 36 months post-treatment without evidence of recurrent disease.
Discussion
Cardiac lymphoma limited only to prosthetic valves is rare, but it has been reported increasingly over the past few years. Until 2010, only six cases of PV-AL had been reported in the literature.7 Including our case, we identified four additional PubMed-indexed cases (using a PubMed search through February 2015). The patient characteristics and treatments received for all identified cases are described in the accompanying Table. The pathology from all of the cases revealed non-Hodgkin lymphoma of large B-cell subtype. PV-AL predominated among men (60%) and older patients with a median age of 62.5 years at diagnosis (range, 48-80 years). Patients had a median duration of 8 years (range, 4-24 years) from date of prosthesis placement to date of lymphoma diagnosis. The three most common presenting manifestations were valvular dysfunction, stroke, and congestive heart failure. All of the patients had surgical intervention on initial presentation. However, management after surgery was not uniform, with only 3 patients reported to have received systemic chemotherapy (Table). None of the patients received adjuvant radiation therapy. Calculated from date of diagnosis, survival duration ranged from less than a month7 to more than 36 months (as reported in our case).
The pathophysiology of PV-AL is not well understood given the rarity of the condition. Similar to other prosthetic-related neoplasms (metallic implants, breast implants),12-14 it has been hypothesized that chronic inflammation and EBV infection may play an essential role in the pathogenesis of this entity. Further, it has been suggested that Dacron, which is used in composite cardiac valve replacements, is carcinogenic and may play a role in some cases.7,15 PV-AL should be highly considered in the differential diagnosis of a suspicious prosthetic valve mass. Various imaging modalities, including echocardiography, CT, and magnetic resonance imaging have been described to have a role in the preoperative evaluation of cardiac tumors by assessing the cardiac function and defining the location and extent of the cardiac tumors.16-19
Given the rarity of this disease entity, there is no standardized approach for treatment. Surgical resection along with repair or replacement of primary involved prosthetic valve is essential for initial treatment. However, there is no consensus about the best approach for subsequent therapy. We cannot be conclusive about the optimum treatment, because of the limited number of published cases, but based on our reading of those cases, it would seem that early surgical intervention and “adjuvant” systemic therapy may have influenced prognosis. We speculate that poor outcomes in the first 6 months were most likely related to primary cardiopulmonary deterioration, whereas later poor outcomes were more likely to be attributable to recurrent lymphoma, particularly for patients who received suboptimal systemic chemotherapy treatment after surgery. All 3 patients who received chemotherapy had no evidence of recurrent disease at last follow-up. Of the 4 patients who received no chemotherapy and survived longer than 6 months (all except 1 died; Table), 2 had recurrent valve lymphoma, 1 had secondary systemic lymphoma, and 1 died of metastatic breast cancer. Those outcomes are in contrast to the 2 out of 3 patients who received adjuvant chemotherapy and who were reported to be alive at 16 and 36 months after diagnosis.
In conclusion, cardiac PV-AL is an increasingly recognized entity that warrants greater awareness among health care providers for early diagnosis and timely surgical intervention. Most of the cases are large B-cell lymphoma. Similar to patients with limited-stage DLBCL, fit patients should be highly considered for “adjuvant” systemic chemotherapy to optimize long-term outcomes. Reporting of similar cases is highly encouraged to better define this rare iatrogenic malignancy.
1. Hudzik B, Miszalski-Jamka K, Glowacki J, et al. Malignant tumors of the heart. Cancer epidemiol. 2015;39(5):665-672.
2. Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, eds. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon, France: IARC Press; 2004.
3. Reynen K. Frequency of primary tumors of the heart. Am J Cardiol. 1996;77(1):107.
4. Neragi-Miandoab S, Kim J, Vlahakes GJ. Malignant tumours of the heart: a review of tumour type, diagnosis and therapy. Clin Oncol. 2007;19(10):748-756.
5. Butany J, Nair V, Naseemuddin A, Nair GM, Catton C, Yau T. Cardiac tumours: diagnosis and management. Lancet Oncol. 2005;6(4):219-228.
6. Burke A, Virmani R. Tumors of the heart and great vessels. In: Atlas of tumor pathology, 3rd Series, Fascicle 16. Washington, DC: Armed Forces Institute of Pathology, 1996.
7. Miller DV, Firchau DJ, McClure RF, Kurtin PJ, Feldman AL. Epstein-Barr virus-associated diffuse large B-cell lymphoma arising on cardiac prostheses. Am J Surg Pathol. 2010;34(3):377-384.
8. Albat B, Messner-Pellenc P, Thevenet A. Surgical treatment for primary lymphoma of the heart simulating prosthetic mitral valve thrombosis. J Thoracic Cardiovasc Surg. 1994;108(1):188-189.
9. Bagwan IN, Desai S, Wotherspoon A, Sheppard MN. Unusual presentation of primary cardiac lymphoma. Interact Cardiovasc Thorac Surg. 2009;9(1):127-129.
10. Durrleman NM, El-Hamamsy I, Demaria RG, Carrier M, Perrault LP, Albat B. Cardiac lymphoma following mitral valve replacement. Ann Thorac Surg. 2005;79(3):1040-1042.
11. Petrich A, Cho SI, Billett H. Primary cardiac lymphoma: an analysis of presentation, treatment, and outcome patterns. Cancer. 2011;117(3):581-589.
12. Cheuk W, Chan AC, Chan JK, Lau GT, Chan VN, Yiu HH. Metallic implant-associated lymphoma: a distinct subgroup of large B-cell lymphoma related to pyothorax-associated lymphoma? Am J Surg Pathol. 2005;29(6):832-836.
13. Roden AC, Macon WR, Keeney GL, Myers JL, Feldman AL, Dogan A. Seroma-associated primary anaplastic large-cell lymphoma adjacent to breast implants: an indolent T-cell lymphoproliferative disorder. Mod Pathol. 2008;21(4):455-463.
14. de Jong D, Vasmel WL, de Boer JP, et al. Anaplastic large-cell lymphoma in women with breast implants. JAMA. 2008;300(17):2030-2035.
15. Durrleman N, El Hamamsy I, Demaria R, Carrier M, Perrault LP, Albat B. Is Dacron carcinogenic? Apropos of a case and review of the literature [In French]. Arch Mal Coeur Vaiss. 2004 Mar;97(3):267-270.16. Peters PJ, Reinhardt S. The echocardiographic evaluation of intracardiac masses: a review. J Am Soc Echocard. 2006;19(2):230-240.
17. Gulati G, Sharma S, Kothari SS, Juneja R, Saxena A, Talwar KK. Comparison of echo and MRI in the imaging evaluation of intracardiac masses. Cardiovasc Intervent Radiol. 2004;27(5):459-469.
18. Krombach GA, Spuentrup E, Buecker A, et al. Heart tumors: magnetic resonance imaging and multislice spiral CT [In German]. RoFo. 2005;177(9):1205-1218.
19. Hoey ET, Mankad K, Puppala S, Gopalan D, Sivananthan MU. MRI and CT appearances of cardiac tumours in adults. Clin Radiol. 2009;64(12):1214-1230.
20. Bonnichsen CR, Dearani JA, Maleszewski JJ, Colgan JP, Williamson EE, Ammash NM. Recurrent Epstein-Barr virus-associated diffuse large B-cell lymphoma in an ascending aorta graft. Circulation. 2013;128(13):1481-1483.
21. Berrio G, Suryadevara A, Singh NK, Wesly OH. Diffuse large B-cell lymphoma in an aortic valve allograft. Tex Heart Inst J. 2010;37(4):492-493.
22. Gruver AM, Huba MA, Dogan A, Hsi ED. Fibrin-associated large B-cell lymphoma: part of the spectrum of cardiac lymphomas. Am J Surg Pathol. 2012;36(10):1527-1537.
23. Farah FJ, Chiles CD. Recurrent primary cardiac lymphoma on aortic valve allograft: implications for therapy. Tex Heart Inst J. 2014;41(5):543-546.
1. Hudzik B, Miszalski-Jamka K, Glowacki J, et al. Malignant tumors of the heart. Cancer epidemiol. 2015;39(5):665-672.
2. Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, eds. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon, France: IARC Press; 2004.
3. Reynen K. Frequency of primary tumors of the heart. Am J Cardiol. 1996;77(1):107.
4. Neragi-Miandoab S, Kim J, Vlahakes GJ. Malignant tumours of the heart: a review of tumour type, diagnosis and therapy. Clin Oncol. 2007;19(10):748-756.
5. Butany J, Nair V, Naseemuddin A, Nair GM, Catton C, Yau T. Cardiac tumours: diagnosis and management. Lancet Oncol. 2005;6(4):219-228.
6. Burke A, Virmani R. Tumors of the heart and great vessels. In: Atlas of tumor pathology, 3rd Series, Fascicle 16. Washington, DC: Armed Forces Institute of Pathology, 1996.
7. Miller DV, Firchau DJ, McClure RF, Kurtin PJ, Feldman AL. Epstein-Barr virus-associated diffuse large B-cell lymphoma arising on cardiac prostheses. Am J Surg Pathol. 2010;34(3):377-384.
8. Albat B, Messner-Pellenc P, Thevenet A. Surgical treatment for primary lymphoma of the heart simulating prosthetic mitral valve thrombosis. J Thoracic Cardiovasc Surg. 1994;108(1):188-189.
9. Bagwan IN, Desai S, Wotherspoon A, Sheppard MN. Unusual presentation of primary cardiac lymphoma. Interact Cardiovasc Thorac Surg. 2009;9(1):127-129.
10. Durrleman NM, El-Hamamsy I, Demaria RG, Carrier M, Perrault LP, Albat B. Cardiac lymphoma following mitral valve replacement. Ann Thorac Surg. 2005;79(3):1040-1042.
11. Petrich A, Cho SI, Billett H. Primary cardiac lymphoma: an analysis of presentation, treatment, and outcome patterns. Cancer. 2011;117(3):581-589.
12. Cheuk W, Chan AC, Chan JK, Lau GT, Chan VN, Yiu HH. Metallic implant-associated lymphoma: a distinct subgroup of large B-cell lymphoma related to pyothorax-associated lymphoma? Am J Surg Pathol. 2005;29(6):832-836.
13. Roden AC, Macon WR, Keeney GL, Myers JL, Feldman AL, Dogan A. Seroma-associated primary anaplastic large-cell lymphoma adjacent to breast implants: an indolent T-cell lymphoproliferative disorder. Mod Pathol. 2008;21(4):455-463.
14. de Jong D, Vasmel WL, de Boer JP, et al. Anaplastic large-cell lymphoma in women with breast implants. JAMA. 2008;300(17):2030-2035.
15. Durrleman N, El Hamamsy I, Demaria R, Carrier M, Perrault LP, Albat B. Is Dacron carcinogenic? Apropos of a case and review of the literature [In French]. Arch Mal Coeur Vaiss. 2004 Mar;97(3):267-270.16. Peters PJ, Reinhardt S. The echocardiographic evaluation of intracardiac masses: a review. J Am Soc Echocard. 2006;19(2):230-240.
17. Gulati G, Sharma S, Kothari SS, Juneja R, Saxena A, Talwar KK. Comparison of echo and MRI in the imaging evaluation of intracardiac masses. Cardiovasc Intervent Radiol. 2004;27(5):459-469.
18. Krombach GA, Spuentrup E, Buecker A, et al. Heart tumors: magnetic resonance imaging and multislice spiral CT [In German]. RoFo. 2005;177(9):1205-1218.
19. Hoey ET, Mankad K, Puppala S, Gopalan D, Sivananthan MU. MRI and CT appearances of cardiac tumours in adults. Clin Radiol. 2009;64(12):1214-1230.
20. Bonnichsen CR, Dearani JA, Maleszewski JJ, Colgan JP, Williamson EE, Ammash NM. Recurrent Epstein-Barr virus-associated diffuse large B-cell lymphoma in an ascending aorta graft. Circulation. 2013;128(13):1481-1483.
21. Berrio G, Suryadevara A, Singh NK, Wesly OH. Diffuse large B-cell lymphoma in an aortic valve allograft. Tex Heart Inst J. 2010;37(4):492-493.
22. Gruver AM, Huba MA, Dogan A, Hsi ED. Fibrin-associated large B-cell lymphoma: part of the spectrum of cardiac lymphomas. Am J Surg Pathol. 2012;36(10):1527-1537.
23. Farah FJ, Chiles CD. Recurrent primary cardiac lymphoma on aortic valve allograft: implications for therapy. Tex Heart Inst J. 2014;41(5):543-546.
Durable response to pralatrexate for aggressive PTCL subtypes
Peripheral T-cell lymphoma (PTCL) is a heterogeneous group of mature T- and natural killer-cell neoplasms that comprise about 10%-15% of all non-Hodgkin lymphomas in the United States.1,2 The development of effective therapies for PTCL has been challenging because of the rare nature and heterogeneity of these lymphomas. Most therapies are a derivative of aggressive B-cell lymphoma therapies, including CHOP (cyclophosphamide, hydroxydaunorubicin, vinicristine, prednisone) and CHOEP (cyclophosphamide, hydroxydaunorubicin, vinicristine, etoposide, prednisone).1 Many centers use autologous or allogeneic stem cell transplant in this setting,1 but outcomes remain poor and progress in developing effective treatments has been slow.
Pralatrexate is the first drug to have been approved by the US Food and Drug Administration specifically for treating patients with relapsed or refractory PTCL.3 As a folate analog metabolic inhibitor, pralatrexate competitively inhibits dihydrofolate reductase and reduces cellular levels of thymidine monophosphate, which prevents the cell from synthesizing genetic material and triggers it to undergo apoptosis.4 The agency’s approval of pralatrexate was based on results from the PROPEL study, which is possibly the largest prospective study conducted in patients with relapsed or refractory PTCL (109 evaluable patients).2 Findings from the study showed an overall response rate (ORR) of 29%, and a median duration of response (DoR) of 10 months.2
Pralatrexate is administered intravenously at 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. It is generally continued until disease progression or an unacceptable level of toxicity.2 Alternative dosing schedules have been described, including 15 mg/m2 once weekly for 3 weeks of a 4-week treatment cycle for cutaneous T-cell lymphomas.5
In this case series, we examine the outcomes of 2 patients with particularly aggressive subtypes of PTCL who were treated with pralatrexate. The significance of this report is in describing the long duration of response and reporting on a PTCL subtype – subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type – that was underrepresented in the PROPEL study and is underreported in the literature.
Case presentations and summaries
Case 1
A 23-year-old Asian American man with a medical history of osteogenesis imperfecta presented to Emergency Department at the Hospital of University of Pennsylvania with bilateral lower extremity edema, low-grade fevers, a weight loss of 25 lb, and flat hyperpigmented scaly skin patches across his torso. Symptoms had started manifesting around five months prior to the visit. A punch biopsy of a skin lesion revealed skin tissue with focal infiltrate of small- to medium-sized, atypical lymphocytes infiltrating subcutaneous adipose tissue (panniculitis-like) and adnexa. Immunohistochemical stains showed that the abnormal lymphocytes were positive for CD3, CD8, perforin, granzyme B, TIA-1 (minor subset), and TCR beta; and negative for CD4, CD56, and CD30. Proliferation index (Ki67) was 70%. The findings were consistent with primary subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type (Figure 1). A staging positron-emission tomography–computed tomography (PET–CT) scan demonstrated stage IVB lymphoma with subcutaneous involvement without nodal disease.
He was initially treated with aggressive combination regimens including EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide, hydroxydaunorubicin) and ICE (ifosfamide, carboplatin, etoposide), but he had no response and his disease was primary refractory. Because of his osteogenesis imperfecta, he was not a candidate for allogenic stem cell transplant.
He responded to hyperCVAD B combination therapy (methotrexate and cytarabine), but the course was complicated by cytarabine-induced ataxia and dysarthia. He was then treated with 3 months of intravenous alemtuzumab without response. Intravenous methotrexate (2,000 mg/m2) was then used for 3 cycles, but this exacerbated his previous cytarabine-induced neurological symptoms and resulted in only partial response with persistent fluorine-18-deoxyglucose (FDG) avid lesions on a subsequent PET–CT scan.
At that point, the patient was started on pralatrexate at 15 mg/m2 weekly for 3 weeks on a 4-week cycle schedule. This was his fifth line of therapy and at 16 months from his initial diagnosis. This dosage was continued for 6 months, and he tolerated the therapy well. He reported no exacerbations of his dysarthia, and by the second month, he had achieved clinical and radiographic remission with complete resolution of B symptoms (fevers, night sweats, and weight loss). The dosing was modified to 15 mg/m2 every 2 weeks for 3 months. A whole body PET–CT scan showed resolution of previously FDG avid lesions.
The patient was then continued on 15 mg/m2 pralatrexate every 3 weeks for 1 year and he has been maintained on once-a-month dosing for a second and now third year of therapy. He continues to tolerate the therapy and remains disease free at nearly 2 years since starting pralatrexate.
Case 2
A 64-year-old white man with a medical history of myasthenia gravis (in remission) and invasive thymoma (after thymectomy) presented with diffuse bulky lymphadenopathy and lung lesions to outpatient clinic at the Abramson Cancer Center at the University of Pennsylvania. His LDH was elevated (278 U/L, reference range 98-192 U/L). Excisional biopsy of a left inguinal lymph node revealed sheets of mitotically active large cells with oval to irregular nuclei, clumped chromatin, conspicuous and sometimes multiple nucleoli, and ample eosinophilic cytoplasm. Immunohistochemical staining showed that the neoplastic cells were positive for CD3, CD4, CD30, BCL2 (variable), and MUM1; and negative for ALK 1, CD5, CD8, CD15, CD43, and CD56. Proliferation index (Ki67) was 90% (Figure 2). PET-CT scan showed widespread hypermetabolic lymphoma in the chest, neck, abdomen, and pelvis with pulmonary metastases. Imaging also demonstrated FDG-avid lesions in the gastric and sinus area. The findings were consistent with ALK-negative, anaplastic large cell lymphoma. He was stage IVA; had gastric, lung, and sinus involvement; and disease above and below the diaphragm.
The patient was initially treated with 6 cycles of CHOP and intrathecal methotrexate injections. His post-treatment PET–CT scan showed persistent FDG-avid disease and his LDH level remained elevated. He underwent 1 cycle of ICE and then BCV (busulfan, cyclophosphamide, etoposide) autologous stem cell transplant. Post-transplant PET–CT scan showed improvement from previous 2 scans but still showed several hypermetabolic lymph nodes consistent with persistent disease.
The patient was started on a pralatrexate regimen of 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. After 5 doses, he developed thrombocytopenia and mucositis, which were deemed pralatrexate related. The dosage was reduced to 20 mg/m2 once weekly with variable frequency depending on tolerability. His response assessment with PET–CT scan demonstrated radiographic complete response with resolution of hypermetabolic lesions (Figure 3B).
He then proceeded with pralatrexate for 4 more doses. PET-CT imaging 2 months after the last dose of pralatrexate was consistent with metabolic complete response, and he opted to hold further therapy. His last imaging at 4 years after completion of therapy showed continued remission. At press time, he had been clinically disease free for more than 6 years after his last dose of pralatrexate.
Discussion
PTCL is a rare and heterogeneous lymphoma with poor prognosis. Only 3 agents – pralatrexate, belinostat, and romidespin – have been approved specifically for the treatment of PTCL and all of them have an ORR of less than 30%, based on findings from phase 2 studies.2,6,7 In the PROPEL study, pralatrexate showed an ORR of 29% and a median DoR of 10 months.2 Those results could be considered discouraging, but some PTCL patients may have durable response to pralatrexate monotherapy.
In this case series, each of the patients presented with a particularly aggressive subtype of PTCL, and 1 suffered from a notably rare subtype for which there was scant clinical data to guide treatment. Both patients went through several lines of aggressive treatment that were ineffective and resulted in minimal response. However, both were able to achieve complete resolution of their disease and maintained remission for a significant duration of time after treatment with pralatrexate. In addition, each patient has maintained his remission – one for 6 years after the last dose. These are noteworthy results, and give both patients and clinicians hope that this therapy can be highly effective in some settings.
A better understanding at the molecular level of the oncogenic mechanisms in PTCL patients will be necessary to guide our therapy choices. In these 2 cases, it is likely that the tumor demonstrated superior sensitivity to dihydrofolate reductase inhibition by pralatrexate. In the future, we hope that analysis of the tumor tissue from PTCL patients will allow us to better categorize the tumor sensitivities to particular therapeutic agents. We believe that individualized treatment will lead to better overall outcomes in this challenging group of lymphomas.
1. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
2. O'Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
3. Dondi A, Bari A, Pozzi S, Ferri P, Sacchi S. The potential of pralatrexate as a treatment of peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(5):711-718.
4. Hui J, Przespo E, Elefante A. Pralatrexate: a novel synthetic antifolate for relapsed or refractory peripheral T-cell lymphoma and other potential uses. J Oncol Pharm Pract. 2012;18(2):275-283.
5. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115-4122.
6. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499.
7. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.
Peripheral T-cell lymphoma (PTCL) is a heterogeneous group of mature T- and natural killer-cell neoplasms that comprise about 10%-15% of all non-Hodgkin lymphomas in the United States.1,2 The development of effective therapies for PTCL has been challenging because of the rare nature and heterogeneity of these lymphomas. Most therapies are a derivative of aggressive B-cell lymphoma therapies, including CHOP (cyclophosphamide, hydroxydaunorubicin, vinicristine, prednisone) and CHOEP (cyclophosphamide, hydroxydaunorubicin, vinicristine, etoposide, prednisone).1 Many centers use autologous or allogeneic stem cell transplant in this setting,1 but outcomes remain poor and progress in developing effective treatments has been slow.
Pralatrexate is the first drug to have been approved by the US Food and Drug Administration specifically for treating patients with relapsed or refractory PTCL.3 As a folate analog metabolic inhibitor, pralatrexate competitively inhibits dihydrofolate reductase and reduces cellular levels of thymidine monophosphate, which prevents the cell from synthesizing genetic material and triggers it to undergo apoptosis.4 The agency’s approval of pralatrexate was based on results from the PROPEL study, which is possibly the largest prospective study conducted in patients with relapsed or refractory PTCL (109 evaluable patients).2 Findings from the study showed an overall response rate (ORR) of 29%, and a median duration of response (DoR) of 10 months.2
Pralatrexate is administered intravenously at 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. It is generally continued until disease progression or an unacceptable level of toxicity.2 Alternative dosing schedules have been described, including 15 mg/m2 once weekly for 3 weeks of a 4-week treatment cycle for cutaneous T-cell lymphomas.5
In this case series, we examine the outcomes of 2 patients with particularly aggressive subtypes of PTCL who were treated with pralatrexate. The significance of this report is in describing the long duration of response and reporting on a PTCL subtype – subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type – that was underrepresented in the PROPEL study and is underreported in the literature.
Case presentations and summaries
Case 1
A 23-year-old Asian American man with a medical history of osteogenesis imperfecta presented to Emergency Department at the Hospital of University of Pennsylvania with bilateral lower extremity edema, low-grade fevers, a weight loss of 25 lb, and flat hyperpigmented scaly skin patches across his torso. Symptoms had started manifesting around five months prior to the visit. A punch biopsy of a skin lesion revealed skin tissue with focal infiltrate of small- to medium-sized, atypical lymphocytes infiltrating subcutaneous adipose tissue (panniculitis-like) and adnexa. Immunohistochemical stains showed that the abnormal lymphocytes were positive for CD3, CD8, perforin, granzyme B, TIA-1 (minor subset), and TCR beta; and negative for CD4, CD56, and CD30. Proliferation index (Ki67) was 70%. The findings were consistent with primary subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type (Figure 1). A staging positron-emission tomography–computed tomography (PET–CT) scan demonstrated stage IVB lymphoma with subcutaneous involvement without nodal disease.
He was initially treated with aggressive combination regimens including EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide, hydroxydaunorubicin) and ICE (ifosfamide, carboplatin, etoposide), but he had no response and his disease was primary refractory. Because of his osteogenesis imperfecta, he was not a candidate for allogenic stem cell transplant.
He responded to hyperCVAD B combination therapy (methotrexate and cytarabine), but the course was complicated by cytarabine-induced ataxia and dysarthia. He was then treated with 3 months of intravenous alemtuzumab without response. Intravenous methotrexate (2,000 mg/m2) was then used for 3 cycles, but this exacerbated his previous cytarabine-induced neurological symptoms and resulted in only partial response with persistent fluorine-18-deoxyglucose (FDG) avid lesions on a subsequent PET–CT scan.
At that point, the patient was started on pralatrexate at 15 mg/m2 weekly for 3 weeks on a 4-week cycle schedule. This was his fifth line of therapy and at 16 months from his initial diagnosis. This dosage was continued for 6 months, and he tolerated the therapy well. He reported no exacerbations of his dysarthia, and by the second month, he had achieved clinical and radiographic remission with complete resolution of B symptoms (fevers, night sweats, and weight loss). The dosing was modified to 15 mg/m2 every 2 weeks for 3 months. A whole body PET–CT scan showed resolution of previously FDG avid lesions.
The patient was then continued on 15 mg/m2 pralatrexate every 3 weeks for 1 year and he has been maintained on once-a-month dosing for a second and now third year of therapy. He continues to tolerate the therapy and remains disease free at nearly 2 years since starting pralatrexate.
Case 2
A 64-year-old white man with a medical history of myasthenia gravis (in remission) and invasive thymoma (after thymectomy) presented with diffuse bulky lymphadenopathy and lung lesions to outpatient clinic at the Abramson Cancer Center at the University of Pennsylvania. His LDH was elevated (278 U/L, reference range 98-192 U/L). Excisional biopsy of a left inguinal lymph node revealed sheets of mitotically active large cells with oval to irregular nuclei, clumped chromatin, conspicuous and sometimes multiple nucleoli, and ample eosinophilic cytoplasm. Immunohistochemical staining showed that the neoplastic cells were positive for CD3, CD4, CD30, BCL2 (variable), and MUM1; and negative for ALK 1, CD5, CD8, CD15, CD43, and CD56. Proliferation index (Ki67) was 90% (Figure 2). PET-CT scan showed widespread hypermetabolic lymphoma in the chest, neck, abdomen, and pelvis with pulmonary metastases. Imaging also demonstrated FDG-avid lesions in the gastric and sinus area. The findings were consistent with ALK-negative, anaplastic large cell lymphoma. He was stage IVA; had gastric, lung, and sinus involvement; and disease above and below the diaphragm.
The patient was initially treated with 6 cycles of CHOP and intrathecal methotrexate injections. His post-treatment PET–CT scan showed persistent FDG-avid disease and his LDH level remained elevated. He underwent 1 cycle of ICE and then BCV (busulfan, cyclophosphamide, etoposide) autologous stem cell transplant. Post-transplant PET–CT scan showed improvement from previous 2 scans but still showed several hypermetabolic lymph nodes consistent with persistent disease.
The patient was started on a pralatrexate regimen of 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. After 5 doses, he developed thrombocytopenia and mucositis, which were deemed pralatrexate related. The dosage was reduced to 20 mg/m2 once weekly with variable frequency depending on tolerability. His response assessment with PET–CT scan demonstrated radiographic complete response with resolution of hypermetabolic lesions (Figure 3B).
He then proceeded with pralatrexate for 4 more doses. PET-CT imaging 2 months after the last dose of pralatrexate was consistent with metabolic complete response, and he opted to hold further therapy. His last imaging at 4 years after completion of therapy showed continued remission. At press time, he had been clinically disease free for more than 6 years after his last dose of pralatrexate.
Discussion
PTCL is a rare and heterogeneous lymphoma with poor prognosis. Only 3 agents – pralatrexate, belinostat, and romidespin – have been approved specifically for the treatment of PTCL and all of them have an ORR of less than 30%, based on findings from phase 2 studies.2,6,7 In the PROPEL study, pralatrexate showed an ORR of 29% and a median DoR of 10 months.2 Those results could be considered discouraging, but some PTCL patients may have durable response to pralatrexate monotherapy.
In this case series, each of the patients presented with a particularly aggressive subtype of PTCL, and 1 suffered from a notably rare subtype for which there was scant clinical data to guide treatment. Both patients went through several lines of aggressive treatment that were ineffective and resulted in minimal response. However, both were able to achieve complete resolution of their disease and maintained remission for a significant duration of time after treatment with pralatrexate. In addition, each patient has maintained his remission – one for 6 years after the last dose. These are noteworthy results, and give both patients and clinicians hope that this therapy can be highly effective in some settings.
A better understanding at the molecular level of the oncogenic mechanisms in PTCL patients will be necessary to guide our therapy choices. In these 2 cases, it is likely that the tumor demonstrated superior sensitivity to dihydrofolate reductase inhibition by pralatrexate. In the future, we hope that analysis of the tumor tissue from PTCL patients will allow us to better categorize the tumor sensitivities to particular therapeutic agents. We believe that individualized treatment will lead to better overall outcomes in this challenging group of lymphomas.
Peripheral T-cell lymphoma (PTCL) is a heterogeneous group of mature T- and natural killer-cell neoplasms that comprise about 10%-15% of all non-Hodgkin lymphomas in the United States.1,2 The development of effective therapies for PTCL has been challenging because of the rare nature and heterogeneity of these lymphomas. Most therapies are a derivative of aggressive B-cell lymphoma therapies, including CHOP (cyclophosphamide, hydroxydaunorubicin, vinicristine, prednisone) and CHOEP (cyclophosphamide, hydroxydaunorubicin, vinicristine, etoposide, prednisone).1 Many centers use autologous or allogeneic stem cell transplant in this setting,1 but outcomes remain poor and progress in developing effective treatments has been slow.
Pralatrexate is the first drug to have been approved by the US Food and Drug Administration specifically for treating patients with relapsed or refractory PTCL.3 As a folate analog metabolic inhibitor, pralatrexate competitively inhibits dihydrofolate reductase and reduces cellular levels of thymidine monophosphate, which prevents the cell from synthesizing genetic material and triggers it to undergo apoptosis.4 The agency’s approval of pralatrexate was based on results from the PROPEL study, which is possibly the largest prospective study conducted in patients with relapsed or refractory PTCL (109 evaluable patients).2 Findings from the study showed an overall response rate (ORR) of 29%, and a median duration of response (DoR) of 10 months.2
Pralatrexate is administered intravenously at 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. It is generally continued until disease progression or an unacceptable level of toxicity.2 Alternative dosing schedules have been described, including 15 mg/m2 once weekly for 3 weeks of a 4-week treatment cycle for cutaneous T-cell lymphomas.5
In this case series, we examine the outcomes of 2 patients with particularly aggressive subtypes of PTCL who were treated with pralatrexate. The significance of this report is in describing the long duration of response and reporting on a PTCL subtype – subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type – that was underrepresented in the PROPEL study and is underreported in the literature.
Case presentations and summaries
Case 1
A 23-year-old Asian American man with a medical history of osteogenesis imperfecta presented to Emergency Department at the Hospital of University of Pennsylvania with bilateral lower extremity edema, low-grade fevers, a weight loss of 25 lb, and flat hyperpigmented scaly skin patches across his torso. Symptoms had started manifesting around five months prior to the visit. A punch biopsy of a skin lesion revealed skin tissue with focal infiltrate of small- to medium-sized, atypical lymphocytes infiltrating subcutaneous adipose tissue (panniculitis-like) and adnexa. Immunohistochemical stains showed that the abnormal lymphocytes were positive for CD3, CD8, perforin, granzyme B, TIA-1 (minor subset), and TCR beta; and negative for CD4, CD56, and CD30. Proliferation index (Ki67) was 70%. The findings were consistent with primary subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type (Figure 1). A staging positron-emission tomography–computed tomography (PET–CT) scan demonstrated stage IVB lymphoma with subcutaneous involvement without nodal disease.
He was initially treated with aggressive combination regimens including EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide, hydroxydaunorubicin) and ICE (ifosfamide, carboplatin, etoposide), but he had no response and his disease was primary refractory. Because of his osteogenesis imperfecta, he was not a candidate for allogenic stem cell transplant.
He responded to hyperCVAD B combination therapy (methotrexate and cytarabine), but the course was complicated by cytarabine-induced ataxia and dysarthia. He was then treated with 3 months of intravenous alemtuzumab without response. Intravenous methotrexate (2,000 mg/m2) was then used for 3 cycles, but this exacerbated his previous cytarabine-induced neurological symptoms and resulted in only partial response with persistent fluorine-18-deoxyglucose (FDG) avid lesions on a subsequent PET–CT scan.
At that point, the patient was started on pralatrexate at 15 mg/m2 weekly for 3 weeks on a 4-week cycle schedule. This was his fifth line of therapy and at 16 months from his initial diagnosis. This dosage was continued for 6 months, and he tolerated the therapy well. He reported no exacerbations of his dysarthia, and by the second month, he had achieved clinical and radiographic remission with complete resolution of B symptoms (fevers, night sweats, and weight loss). The dosing was modified to 15 mg/m2 every 2 weeks for 3 months. A whole body PET–CT scan showed resolution of previously FDG avid lesions.
The patient was then continued on 15 mg/m2 pralatrexate every 3 weeks for 1 year and he has been maintained on once-a-month dosing for a second and now third year of therapy. He continues to tolerate the therapy and remains disease free at nearly 2 years since starting pralatrexate.
Case 2
A 64-year-old white man with a medical history of myasthenia gravis (in remission) and invasive thymoma (after thymectomy) presented with diffuse bulky lymphadenopathy and lung lesions to outpatient clinic at the Abramson Cancer Center at the University of Pennsylvania. His LDH was elevated (278 U/L, reference range 98-192 U/L). Excisional biopsy of a left inguinal lymph node revealed sheets of mitotically active large cells with oval to irregular nuclei, clumped chromatin, conspicuous and sometimes multiple nucleoli, and ample eosinophilic cytoplasm. Immunohistochemical staining showed that the neoplastic cells were positive for CD3, CD4, CD30, BCL2 (variable), and MUM1; and negative for ALK 1, CD5, CD8, CD15, CD43, and CD56. Proliferation index (Ki67) was 90% (Figure 2). PET-CT scan showed widespread hypermetabolic lymphoma in the chest, neck, abdomen, and pelvis with pulmonary metastases. Imaging also demonstrated FDG-avid lesions in the gastric and sinus area. The findings were consistent with ALK-negative, anaplastic large cell lymphoma. He was stage IVA; had gastric, lung, and sinus involvement; and disease above and below the diaphragm.
The patient was initially treated with 6 cycles of CHOP and intrathecal methotrexate injections. His post-treatment PET–CT scan showed persistent FDG-avid disease and his LDH level remained elevated. He underwent 1 cycle of ICE and then BCV (busulfan, cyclophosphamide, etoposide) autologous stem cell transplant. Post-transplant PET–CT scan showed improvement from previous 2 scans but still showed several hypermetabolic lymph nodes consistent with persistent disease.
The patient was started on a pralatrexate regimen of 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. After 5 doses, he developed thrombocytopenia and mucositis, which were deemed pralatrexate related. The dosage was reduced to 20 mg/m2 once weekly with variable frequency depending on tolerability. His response assessment with PET–CT scan demonstrated radiographic complete response with resolution of hypermetabolic lesions (Figure 3B).
He then proceeded with pralatrexate for 4 more doses. PET-CT imaging 2 months after the last dose of pralatrexate was consistent with metabolic complete response, and he opted to hold further therapy. His last imaging at 4 years after completion of therapy showed continued remission. At press time, he had been clinically disease free for more than 6 years after his last dose of pralatrexate.
Discussion
PTCL is a rare and heterogeneous lymphoma with poor prognosis. Only 3 agents – pralatrexate, belinostat, and romidespin – have been approved specifically for the treatment of PTCL and all of them have an ORR of less than 30%, based on findings from phase 2 studies.2,6,7 In the PROPEL study, pralatrexate showed an ORR of 29% and a median DoR of 10 months.2 Those results could be considered discouraging, but some PTCL patients may have durable response to pralatrexate monotherapy.
In this case series, each of the patients presented with a particularly aggressive subtype of PTCL, and 1 suffered from a notably rare subtype for which there was scant clinical data to guide treatment. Both patients went through several lines of aggressive treatment that were ineffective and resulted in minimal response. However, both were able to achieve complete resolution of their disease and maintained remission for a significant duration of time after treatment with pralatrexate. In addition, each patient has maintained his remission – one for 6 years after the last dose. These are noteworthy results, and give both patients and clinicians hope that this therapy can be highly effective in some settings.
A better understanding at the molecular level of the oncogenic mechanisms in PTCL patients will be necessary to guide our therapy choices. In these 2 cases, it is likely that the tumor demonstrated superior sensitivity to dihydrofolate reductase inhibition by pralatrexate. In the future, we hope that analysis of the tumor tissue from PTCL patients will allow us to better categorize the tumor sensitivities to particular therapeutic agents. We believe that individualized treatment will lead to better overall outcomes in this challenging group of lymphomas.
1. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
2. O'Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
3. Dondi A, Bari A, Pozzi S, Ferri P, Sacchi S. The potential of pralatrexate as a treatment of peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(5):711-718.
4. Hui J, Przespo E, Elefante A. Pralatrexate: a novel synthetic antifolate for relapsed or refractory peripheral T-cell lymphoma and other potential uses. J Oncol Pharm Pract. 2012;18(2):275-283.
5. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115-4122.
6. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499.
7. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.
1. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
2. O'Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
3. Dondi A, Bari A, Pozzi S, Ferri P, Sacchi S. The potential of pralatrexate as a treatment of peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(5):711-718.
4. Hui J, Przespo E, Elefante A. Pralatrexate: a novel synthetic antifolate for relapsed or refractory peripheral T-cell lymphoma and other potential uses. J Oncol Pharm Pract. 2012;18(2):275-283.
5. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115-4122.
6. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499.
7. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.
A Veteran With Fibromyalgia Presenting With Dyspnea
Case Presentation. A 64-year-old US Army veteran with a history of colorectal cancer, melanoma, and fibrinolytic presented with dyspnea to VA Boston Healthcare System (VABHS). Seven years prior to the current presentation, at the time of her diagnosis of colorectal cancer, the patient was found to be HIV negative but to have a positive purified protein derivative (PPD) test. She was treated with isoniazid (INH) therapy for 9 months. Sputum cultures collected prior to initiation of therapy grew Mycobacterium avium complex (MAC) in 1 of 3 samples, with these results reported several months after initiation of therapy. She was a never smoker with no known travel or exposure. At the time of the current presentation, her medications included bupropion, levothyroxine, capsaicin, cyclobenzaprine, ibuprofen, and acetaminophen.
►Lakshmana Swamy, MD, Chief Medical Resident, VABHS and Boston Medical Center. Dr. Monach, this patient is on a variety of pain medications and has a diagnosis of fibromyalgia. This diagnosis often frustrates doctors and patients alike. Can you tell us about fibromyalgia from the rheumatologist’s perspective and what you think of her current treatment regimen?
►Paul A. Monach, MD, PhD, Chief, Section of Rheumatology, VABHS and Associate Professor of Medicine, Boston University School of Medicine. Fibromyalgia is a syndrome of chronic widespread pain without known pathology in the musculoskeletal system. It is thought to be caused by chronic dysfunction of pain-processing pathways in the central nervous system (CNS). It is often accompanied by other somatic symptoms such as chronic fatigue, irritable bowel syndrome, and bladder pain. It is a common condition, affecting up to 5% of otherwise healthy women. It is particularly common in persons with chronic nonrestorative sleep or posttraumatic stress disorder from a wide range of causes. However, it also is more common in persons with autoimmune inflammatory diseases, such as lupus, Sjögren syndrome, or rheumatoid arthritis. Concern for one of these diseases is the main reason to consider referring a patient for evaluation by a rheumatologist. Often rheumatologists participate in the management of fibromyalgia. A patient should be given appropriate expectations by the referring physician.
Effectiveness of treatment varies widely among patients. Nonpharmacologic approaches such as aerobic exercise, cognitive behavioral therapy, and tai chi have support from clinical trials, and yoga and aquatherapy also are widely used.1,2 The classes of drugs used are the same as for neuropathic pain: tricyclics, including cyclobenzaprine; serotonin and norepinephrine reuptake inhibitors (SNRIs); and gabapentinoids. In contrast, nonsteroidal anti-inflammatory drugs and opioids are ineffective unless there is a superimposed mechanical or inflammatory cause in the periphery. The key point is that continuation of any treatment should be based entirely on the patient’s own assessment of benefit.
►Dr. Swamy. Seven years later, the patient returned to her primary care provider, reporting increased dyspnea on exertion as well as significant fatigue. She was referred to the pulmonary department and had repeat computed tomography (CT) scans of the chest, which indicated persistent right middle lobe (RML) bronchiectasis. She then underwent bronchoscopy with a subsequent bronchoalveolar lavage (BAL) culture growing MAC. Dr. Fine, please interpret the baseline and follow-up CT scans and help us understand the significance of the MAC on sputum and BAL cultures.
►Alan Fine, MD, Section of Pulmonary and Critical Care, VABHS and Professor of Medicine, Boston University School of Medicine. Prior to this presentation, the patient had a pleural-based area of fibrosis with possible associated RML bronchiectasis. This appears to be a postinflammatory process without classic features of malignant or metastatic disease. She then had a sputum, which grew MAC in only 1 of 3 samples and in liquid media only. Importantly, the sputum was not smear positive. All of this suggests a low organism burden. One possibility is that this could reflect colonization with MAC; it is not uncommon for patients with underlying chronic changes in their lung to grow MAC, and it is often difficult to tell whether it is indicative of active disease. Structural lung disease, such as bronchiectasis, predisposes a patient to MAC, but chronic MAC also may cause bronchiectasis. This chicken-and-egg scenario comes up frequently. She may have a MAC infection, but as she is HIV negative and asymptomatic, there is no urgent indication to treat, especially as the burden of therapy is not insignificant.
►Dr. Swamy. Do we need to worry about Mycobacterium tuberculosis (MTB)?
►Dr. Fine. Although she was previously PPD positive, she had already completed 1 year of isoniazid (INH) therapy, making active MTB less likely. From an infection control standpoint, it is important to distinguish MAC from MTB. The former is not contagious, and there is no need for airborne isolation.
►Dr. Swamy. Dr. Fine, where does MAC come from? Does it commonly cause disease?
►Dr. Fine. In the environment, MAC is nearly ubiquitous , especially in water and soil. In one study, 20% of showerheads were positive for MAC; when patients are infected, we may suggest changing/bleaching the showerhead, but there are no definitive recommendations.3 Because MAC is so common in the environment, it is unlikely that measures to target MAC colonization will be clinically meaningful. On the other hand, the incidence of nontuberculous mycobacterial infections is increasing across the US, and it may be a common and frequently underdiagnosed cause of chronic cough, especially in postmenopausal women.
►Dr. Swamy. Four years prior to the current presentation, the patient developed a cough after an upper respiratory tract infection that persisted for more than 2 weeks. Given her history, she underwent a repeat chest CT, which noted a slight increase in nodularity and ground-glass opacity restricted to the RML. She also reported dyspnea on exertion and was referred to the pulmonary medicine department. By the time she arrived, her dyspnea had largely resolved, but she reported persistent fatigue without other systemic symptoms, such as fevers or chills. Dr. Fine, does MAC explain this patient’s dyspnea?
►Dr. Fine. As her pulmonary symptoms resolved in a short period of time with only azithromycin, it is very unlikely that her symptoms were related to her prior disease. The MAC infection is not likely to cause dyspnea on exertion and fatigue and should be worked up more broadly before attributing it to MAC. In view of this, it would not be unreasonable to follow her clinically and see her again in 6 to 8 weeks. In this context, we also should consider the untoward impact of repeated radiation exposure derived from multiple CT scans. When a patient has an abnormality on CT scan, it often leads to further scans even if the symptoms do not match the previous findings, as in this case.

►Dr. Swamy. Given her ongoing fatigue and systemic symptoms (morning stiffness of the shoulders, legs, and thighs, and leg cramps), she was referred to the rheumatology department where the physical examination revealed muscle tenderness in her proximal arms and legs with normal strength, tender points at the elbows and medial side of the bilateral knees, significant tenderness of lower legs, and no synovitis.
Dr. Monach, can you walk us through your approach to this patient? Are we seeing manifestations of fibromyalgia? What diagnoses concerns you and how would you proceed?
►Dr. Monach. The history and exam are most helpful in raising or reducing suspicion for an underlying inflammatory disease. Areas of tenderness described in her case are typical of fibromyalgia, although it can be difficult to interpret symptoms in the hip girdle and shoulder girdle because objective findings are often absent on exam in patients with inflammatory arthritis or bursitis. Similarly, tenderness at sites of tendon insertion (enthuses) without objective abnormalities is common in different forms of spondyloarthritis, so tenderness at the elbow, knee, lateral hip, and low back can be difficult to interpret. What this patient is lacking is prominent subjective or objective findings in the joints most commonly affected in rheumatoid arthritis and lupus: wrists, hands, ankles, and feet.
►Dr. Swamy. Initial laboratory data include an erythrocyte sedimentation rate of 79 with a normal C-reactive protein. A tentative diagnosis of polymyalgia rheumatic is made with consideration of a trial treatment of prednisone.
Dr. Monach, this patient has an indolent infection and is about to be given glucocorticoids. Could you describe the situations in which you feel that glucocorticoids cause a relative immunosuppression?
►Dr. Monach. Glucocorticoids are considered safe in a patient whose infection is not intrinsically dangerous or who has started appropriate antibiotics for that infection. Although all toxicities of glucocorticoids are dose dependent, the long-standing assertion that doses below 10 mg to 15 mg do not increase risk of infection is contradicted by data published in the past 10 to 15 years, with the caveat that these patients were on long-term treatment.
►Dr. Swamy. The patient was started on prednisone 15 mg per day for 15 days. She returned to the clinic after 1 week of prednisone troutment and noted “significant improvement in fatigue, morning stiffness of shoulders, thighs, leg, back is better, leg cramps resolved, shooting pain in many joints resolved.” Further laboratory results were notable for a negative rheumatoid factor, negative antinuclear antibody, and a cyclic citrullinated peptide of 60. A presumptive diagnosis of rheumatoid arthritis (RA) was made and plaquenil 200 mg twice daily was started.
Dr. Monach, can you explain why RA comes up now on serology but was not considered initially? Why does this presentation fit RA, and was her response to treatment typical? How does this fit in with her previous diagnosis of fibromyalgia? Was that just an atypical, indolent presentation of RA?
►Dr. Monach. Though her presentation is atypical for RA, in elderly patients, RA can present with symptoms resembling polymyalgia rheumatica. The question is whether she had RA all along (in which case “elderly onset” would not apply) or had fibromyalgia and developed RA more recently. The response to empiric glucocorticoid therapy is helpful, since fibromyalgia should not improve with prednisone even in a patient with RA unless treatment of RA would allow better sleep and ability to exercise. Rheumatoid arthritis typically responds very well to prednisone in the 5-mg to 15-mg range.
►Dr. Swamy. Given the new diagnosis of an inflammatory arthritis requiring immunosuppression, bronchoscopy with BAL is performed to evaluate for the presence of MAC. These cultures were positive for MAC.
Dr. Fine, does the positive BAL culture indicate an active MAC infection?
►Dr. Fine. Yes, based on these updated data, the patient has an active MAC infection. Active infection is defined as symptoms or imaging consistent with the diagnosis, supporting microbiology data (either 2 sputum or 1 BAL sample growing MAC) and the exclusion of other causes. Previously, this patient grew MAC in just one expectorated sputum; this did not meet the microbiologic criteria. Now sputum has grown in the BAL sample; along with the CT imaging, this is enough to diagnosis active MAC infection.
Treatment for MAC must consider the details of each case. First, this is not an emergency; treatment decisions should be made with the rheumatologist to consider the planned immunosuppression. For example, we must consider potential drug interactions. A specific point should be made of the use of tumor necrosis factor (TNF)-α inhibition, which data indicate can reactivate TB and may inhibit mechanisms that restrain mycobacterial disease. Serious cases of MAC infection have been reported in the literature in the setting of TNF-α inhibition.5,6 Despite these concerns, there is not a contraindication to using these therapies from the perspective of the active MAC disease. All of these decisions will impact the need to commit the patient to MAC therapy.
►Dr. Swamy. Dr. Fine, what do you consider prior to initiating MAC therapy?
► Dr. Fine. The decision to pursue MAC therapy should not be taken lightly. Therapy often entails prolonged multidrug regimens, usually spanning more than a year, with frequent adverse effects. Outside of very specific cases, such as TNF-β inhibition, MAC is rarely a life-threatening disease, so the benefit may be limited. Treatment for MAC is certainly unlikely to be fruitful without a diligent and motivated patient able to handle the high and prolonged pill burden. Of note, it is also important to keep this patient up-to-date with influenza and pneumonia vaccination given her structural lung disease.
►Dr. Swamy. The decision is made to treat MAC with azithromycin, rifampin, and ethambutol. The disease is noted to be nonfibrocavitary. The patient underwent monthly liver function test monitoring and visual acuity testing, which were unremarkable. Dr. Fine, can you describe the phenotypes of nontuberculous mycobacterial (NTM) disease?
►Dr. Fine. There are 3 main phenotypes of NTM.3 First, we see the elderly man with preexisting lung disease—usually chronic obstructive pulmonary disease—with fibrocavitary and/or reticulonodular appearance. Second, we see the slim, elderly woman often without any preexisting lung disease presenting with focal bronchiectasis and nodular lesions in right middle lobe and lingula—the Lady Windermere syndrome. This eponym is derived from Oscar Wilde’s play “Lady Windermere’s Fan, a Play About a Good Woman,” and was first associated with this disease in 1992.7 At the time, it was thought that the voluntary suppression of cough led to poorly draining lung regions, vulnerable to engraftment by atypical mycobacteria. Infection with atypical mycobacteria are associated with this population; however, it is no longer thought to be due to the voluntary suppression of cough.7,8 Third, we do occasionally see atypical presentations, such as focal masses and solitary nodules.
►Dr. Swamy. At 1-year follow-up she successfully completed MAC therapy and noted ongoing control of rheumatoid symptoms.
1. Bernardy K, Klose P, Welsch P, Häuser W. Efficacy, acceptability and safety of cognitive behavioural therapies in fibromyalgia syndrome—a systematic review and meta-analysis of randomized controlled trials. Eur J Pain. 2018;22(2):242-260.
2. Geneen LJ, Moore RA, Clarke C, Martin D, Colvin LA, Smith BH. Physical activity and exercise for chronic pain in adults: an overview of Cochrane Reviews. Cochrane Database Syst Rev. 2017;4:CD011279.
3. Aksamit TR, Philley JV, Griffith DE. Nontuberculous mycobacterial (NTM) lung disease: the top ten essentials. Respir Med. 2014;108(3):417-425.
4. Aucott JN. Glucocorticoids and infection. Endocrinol Metab Clin North Am. 1994;23(3):655-670.
5. Curtis JR, Yang S, Patkar NM, et al. Risk of hospitalized bacterial infections associated with biologic treatment among US veterans with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2014;66(7):990-997.
6. Lane MA, McDonald JR, Zeringue AL, et al. TNF-α antagonist use and risk of hospitalization for infection in a national cohort of veterans with rheumatoid arthritis. Medicine (Baltimore). 2011;90(2):139-145.
7. Reich JM, Johnson RE. Mycobacterium avium complex pulmonary disease presenting as an isolated lingular or middle lobe pattern. The Lady Windermere syndrome. Chest. 1992;101(6):1605-1609.
8. Kasthoori JJ, Liam CK, Wastie ML. Lady Windermere syndrome: an inappropriate eponym for an increasingly important condition. Singapore Med J. 2008;49(2):e47-e49.
Case Presentation. A 64-year-old US Army veteran with a history of colorectal cancer, melanoma, and fibrinolytic presented with dyspnea to VA Boston Healthcare System (VABHS). Seven years prior to the current presentation, at the time of her diagnosis of colorectal cancer, the patient was found to be HIV negative but to have a positive purified protein derivative (PPD) test. She was treated with isoniazid (INH) therapy for 9 months. Sputum cultures collected prior to initiation of therapy grew Mycobacterium avium complex (MAC) in 1 of 3 samples, with these results reported several months after initiation of therapy. She was a never smoker with no known travel or exposure. At the time of the current presentation, her medications included bupropion, levothyroxine, capsaicin, cyclobenzaprine, ibuprofen, and acetaminophen.
►Lakshmana Swamy, MD, Chief Medical Resident, VABHS and Boston Medical Center. Dr. Monach, this patient is on a variety of pain medications and has a diagnosis of fibromyalgia. This diagnosis often frustrates doctors and patients alike. Can you tell us about fibromyalgia from the rheumatologist’s perspective and what you think of her current treatment regimen?
►Paul A. Monach, MD, PhD, Chief, Section of Rheumatology, VABHS and Associate Professor of Medicine, Boston University School of Medicine. Fibromyalgia is a syndrome of chronic widespread pain without known pathology in the musculoskeletal system. It is thought to be caused by chronic dysfunction of pain-processing pathways in the central nervous system (CNS). It is often accompanied by other somatic symptoms such as chronic fatigue, irritable bowel syndrome, and bladder pain. It is a common condition, affecting up to 5% of otherwise healthy women. It is particularly common in persons with chronic nonrestorative sleep or posttraumatic stress disorder from a wide range of causes. However, it also is more common in persons with autoimmune inflammatory diseases, such as lupus, Sjögren syndrome, or rheumatoid arthritis. Concern for one of these diseases is the main reason to consider referring a patient for evaluation by a rheumatologist. Often rheumatologists participate in the management of fibromyalgia. A patient should be given appropriate expectations by the referring physician.
Effectiveness of treatment varies widely among patients. Nonpharmacologic approaches such as aerobic exercise, cognitive behavioral therapy, and tai chi have support from clinical trials, and yoga and aquatherapy also are widely used.1,2 The classes of drugs used are the same as for neuropathic pain: tricyclics, including cyclobenzaprine; serotonin and norepinephrine reuptake inhibitors (SNRIs); and gabapentinoids. In contrast, nonsteroidal anti-inflammatory drugs and opioids are ineffective unless there is a superimposed mechanical or inflammatory cause in the periphery. The key point is that continuation of any treatment should be based entirely on the patient’s own assessment of benefit.
►Dr. Swamy. Seven years later, the patient returned to her primary care provider, reporting increased dyspnea on exertion as well as significant fatigue. She was referred to the pulmonary department and had repeat computed tomography (CT) scans of the chest, which indicated persistent right middle lobe (RML) bronchiectasis. She then underwent bronchoscopy with a subsequent bronchoalveolar lavage (BAL) culture growing MAC. Dr. Fine, please interpret the baseline and follow-up CT scans and help us understand the significance of the MAC on sputum and BAL cultures.
►Alan Fine, MD, Section of Pulmonary and Critical Care, VABHS and Professor of Medicine, Boston University School of Medicine. Prior to this presentation, the patient had a pleural-based area of fibrosis with possible associated RML bronchiectasis. This appears to be a postinflammatory process without classic features of malignant or metastatic disease. She then had a sputum, which grew MAC in only 1 of 3 samples and in liquid media only. Importantly, the sputum was not smear positive. All of this suggests a low organism burden. One possibility is that this could reflect colonization with MAC; it is not uncommon for patients with underlying chronic changes in their lung to grow MAC, and it is often difficult to tell whether it is indicative of active disease. Structural lung disease, such as bronchiectasis, predisposes a patient to MAC, but chronic MAC also may cause bronchiectasis. This chicken-and-egg scenario comes up frequently. She may have a MAC infection, but as she is HIV negative and asymptomatic, there is no urgent indication to treat, especially as the burden of therapy is not insignificant.
►Dr. Swamy. Do we need to worry about Mycobacterium tuberculosis (MTB)?
►Dr. Fine. Although she was previously PPD positive, she had already completed 1 year of isoniazid (INH) therapy, making active MTB less likely. From an infection control standpoint, it is important to distinguish MAC from MTB. The former is not contagious, and there is no need for airborne isolation.
►Dr. Swamy. Dr. Fine, where does MAC come from? Does it commonly cause disease?
►Dr. Fine. In the environment, MAC is nearly ubiquitous , especially in water and soil. In one study, 20% of showerheads were positive for MAC; when patients are infected, we may suggest changing/bleaching the showerhead, but there are no definitive recommendations.3 Because MAC is so common in the environment, it is unlikely that measures to target MAC colonization will be clinically meaningful. On the other hand, the incidence of nontuberculous mycobacterial infections is increasing across the US, and it may be a common and frequently underdiagnosed cause of chronic cough, especially in postmenopausal women.
►Dr. Swamy. Four years prior to the current presentation, the patient developed a cough after an upper respiratory tract infection that persisted for more than 2 weeks. Given her history, she underwent a repeat chest CT, which noted a slight increase in nodularity and ground-glass opacity restricted to the RML. She also reported dyspnea on exertion and was referred to the pulmonary medicine department. By the time she arrived, her dyspnea had largely resolved, but she reported persistent fatigue without other systemic symptoms, such as fevers or chills. Dr. Fine, does MAC explain this patient’s dyspnea?
►Dr. Fine. As her pulmonary symptoms resolved in a short period of time with only azithromycin, it is very unlikely that her symptoms were related to her prior disease. The MAC infection is not likely to cause dyspnea on exertion and fatigue and should be worked up more broadly before attributing it to MAC. In view of this, it would not be unreasonable to follow her clinically and see her again in 6 to 8 weeks. In this context, we also should consider the untoward impact of repeated radiation exposure derived from multiple CT scans. When a patient has an abnormality on CT scan, it often leads to further scans even if the symptoms do not match the previous findings, as in this case.
►Dr. Swamy. Given her ongoing fatigue and systemic symptoms (morning stiffness of the shoulders, legs, and thighs, and leg cramps), she was referred to the rheumatology department where the physical examination revealed muscle tenderness in her proximal arms and legs with normal strength, tender points at the elbows and medial side of the bilateral knees, significant tenderness of lower legs, and no synovitis.
Dr. Monach, can you walk us through your approach to this patient? Are we seeing manifestations of fibromyalgia? What diagnoses concerns you and how would you proceed?
►Dr. Monach. The history and exam are most helpful in raising or reducing suspicion for an underlying inflammatory disease. Areas of tenderness described in her case are typical of fibromyalgia, although it can be difficult to interpret symptoms in the hip girdle and shoulder girdle because objective findings are often absent on exam in patients with inflammatory arthritis or bursitis. Similarly, tenderness at sites of tendon insertion (enthuses) without objective abnormalities is common in different forms of spondyloarthritis, so tenderness at the elbow, knee, lateral hip, and low back can be difficult to interpret. What this patient is lacking is prominent subjective or objective findings in the joints most commonly affected in rheumatoid arthritis and lupus: wrists, hands, ankles, and feet.
►Dr. Swamy. Initial laboratory data include an erythrocyte sedimentation rate of 79 with a normal C-reactive protein. A tentative diagnosis of polymyalgia rheumatic is made with consideration of a trial treatment of prednisone.
Dr. Monach, this patient has an indolent infection and is about to be given glucocorticoids. Could you describe the situations in which you feel that glucocorticoids cause a relative immunosuppression?
►Dr. Monach. Glucocorticoids are considered safe in a patient whose infection is not intrinsically dangerous or who has started appropriate antibiotics for that infection. Although all toxicities of glucocorticoids are dose dependent, the long-standing assertion that doses below 10 mg to 15 mg do not increase risk of infection is contradicted by data published in the past 10 to 15 years, with the caveat that these patients were on long-term treatment.
►Dr. Swamy. The patient was started on prednisone 15 mg per day for 15 days. She returned to the clinic after 1 week of prednisone troutment and noted “significant improvement in fatigue, morning stiffness of shoulders, thighs, leg, back is better, leg cramps resolved, shooting pain in many joints resolved.” Further laboratory results were notable for a negative rheumatoid factor, negative antinuclear antibody, and a cyclic citrullinated peptide of 60. A presumptive diagnosis of rheumatoid arthritis (RA) was made and plaquenil 200 mg twice daily was started.
Dr. Monach, can you explain why RA comes up now on serology but was not considered initially? Why does this presentation fit RA, and was her response to treatment typical? How does this fit in with her previous diagnosis of fibromyalgia? Was that just an atypical, indolent presentation of RA?
►Dr. Monach. Though her presentation is atypical for RA, in elderly patients, RA can present with symptoms resembling polymyalgia rheumatica. The question is whether she had RA all along (in which case “elderly onset” would not apply) or had fibromyalgia and developed RA more recently. The response to empiric glucocorticoid therapy is helpful, since fibromyalgia should not improve with prednisone even in a patient with RA unless treatment of RA would allow better sleep and ability to exercise. Rheumatoid arthritis typically responds very well to prednisone in the 5-mg to 15-mg range.
►Dr. Swamy. Given the new diagnosis of an inflammatory arthritis requiring immunosuppression, bronchoscopy with BAL is performed to evaluate for the presence of MAC. These cultures were positive for MAC.
Dr. Fine, does the positive BAL culture indicate an active MAC infection?
►Dr. Fine. Yes, based on these updated data, the patient has an active MAC infection. Active infection is defined as symptoms or imaging consistent with the diagnosis, supporting microbiology data (either 2 sputum or 1 BAL sample growing MAC) and the exclusion of other causes. Previously, this patient grew MAC in just one expectorated sputum; this did not meet the microbiologic criteria. Now sputum has grown in the BAL sample; along with the CT imaging, this is enough to diagnosis active MAC infection.
Treatment for MAC must consider the details of each case. First, this is not an emergency; treatment decisions should be made with the rheumatologist to consider the planned immunosuppression. For example, we must consider potential drug interactions. A specific point should be made of the use of tumor necrosis factor (TNF)-α inhibition, which data indicate can reactivate TB and may inhibit mechanisms that restrain mycobacterial disease. Serious cases of MAC infection have been reported in the literature in the setting of TNF-α inhibition.5,6 Despite these concerns, there is not a contraindication to using these therapies from the perspective of the active MAC disease. All of these decisions will impact the need to commit the patient to MAC therapy.
►Dr. Swamy. Dr. Fine, what do you consider prior to initiating MAC therapy?
► Dr. Fine. The decision to pursue MAC therapy should not be taken lightly. Therapy often entails prolonged multidrug regimens, usually spanning more than a year, with frequent adverse effects. Outside of very specific cases, such as TNF-β inhibition, MAC is rarely a life-threatening disease, so the benefit may be limited. Treatment for MAC is certainly unlikely to be fruitful without a diligent and motivated patient able to handle the high and prolonged pill burden. Of note, it is also important to keep this patient up-to-date with influenza and pneumonia vaccination given her structural lung disease.
►Dr. Swamy. The decision is made to treat MAC with azithromycin, rifampin, and ethambutol. The disease is noted to be nonfibrocavitary. The patient underwent monthly liver function test monitoring and visual acuity testing, which were unremarkable. Dr. Fine, can you describe the phenotypes of nontuberculous mycobacterial (NTM) disease?
►Dr. Fine. There are 3 main phenotypes of NTM.3 First, we see the elderly man with preexisting lung disease—usually chronic obstructive pulmonary disease—with fibrocavitary and/or reticulonodular appearance. Second, we see the slim, elderly woman often without any preexisting lung disease presenting with focal bronchiectasis and nodular lesions in right middle lobe and lingula—the Lady Windermere syndrome. This eponym is derived from Oscar Wilde’s play “Lady Windermere’s Fan, a Play About a Good Woman,” and was first associated with this disease in 1992.7 At the time, it was thought that the voluntary suppression of cough led to poorly draining lung regions, vulnerable to engraftment by atypical mycobacteria. Infection with atypical mycobacteria are associated with this population; however, it is no longer thought to be due to the voluntary suppression of cough.7,8 Third, we do occasionally see atypical presentations, such as focal masses and solitary nodules.
►Dr. Swamy. At 1-year follow-up she successfully completed MAC therapy and noted ongoing control of rheumatoid symptoms.
Case Presentation. A 64-year-old US Army veteran with a history of colorectal cancer, melanoma, and fibrinolytic presented with dyspnea to VA Boston Healthcare System (VABHS). Seven years prior to the current presentation, at the time of her diagnosis of colorectal cancer, the patient was found to be HIV negative but to have a positive purified protein derivative (PPD) test. She was treated with isoniazid (INH) therapy for 9 months. Sputum cultures collected prior to initiation of therapy grew Mycobacterium avium complex (MAC) in 1 of 3 samples, with these results reported several months after initiation of therapy. She was a never smoker with no known travel or exposure. At the time of the current presentation, her medications included bupropion, levothyroxine, capsaicin, cyclobenzaprine, ibuprofen, and acetaminophen.
►Lakshmana Swamy, MD, Chief Medical Resident, VABHS and Boston Medical Center. Dr. Monach, this patient is on a variety of pain medications and has a diagnosis of fibromyalgia. This diagnosis often frustrates doctors and patients alike. Can you tell us about fibromyalgia from the rheumatologist’s perspective and what you think of her current treatment regimen?
►Paul A. Monach, MD, PhD, Chief, Section of Rheumatology, VABHS and Associate Professor of Medicine, Boston University School of Medicine. Fibromyalgia is a syndrome of chronic widespread pain without known pathology in the musculoskeletal system. It is thought to be caused by chronic dysfunction of pain-processing pathways in the central nervous system (CNS). It is often accompanied by other somatic symptoms such as chronic fatigue, irritable bowel syndrome, and bladder pain. It is a common condition, affecting up to 5% of otherwise healthy women. It is particularly common in persons with chronic nonrestorative sleep or posttraumatic stress disorder from a wide range of causes. However, it also is more common in persons with autoimmune inflammatory diseases, such as lupus, Sjögren syndrome, or rheumatoid arthritis. Concern for one of these diseases is the main reason to consider referring a patient for evaluation by a rheumatologist. Often rheumatologists participate in the management of fibromyalgia. A patient should be given appropriate expectations by the referring physician.
Effectiveness of treatment varies widely among patients. Nonpharmacologic approaches such as aerobic exercise, cognitive behavioral therapy, and tai chi have support from clinical trials, and yoga and aquatherapy also are widely used.1,2 The classes of drugs used are the same as for neuropathic pain: tricyclics, including cyclobenzaprine; serotonin and norepinephrine reuptake inhibitors (SNRIs); and gabapentinoids. In contrast, nonsteroidal anti-inflammatory drugs and opioids are ineffective unless there is a superimposed mechanical or inflammatory cause in the periphery. The key point is that continuation of any treatment should be based entirely on the patient’s own assessment of benefit.
►Dr. Swamy. Seven years later, the patient returned to her primary care provider, reporting increased dyspnea on exertion as well as significant fatigue. She was referred to the pulmonary department and had repeat computed tomography (CT) scans of the chest, which indicated persistent right middle lobe (RML) bronchiectasis. She then underwent bronchoscopy with a subsequent bronchoalveolar lavage (BAL) culture growing MAC. Dr. Fine, please interpret the baseline and follow-up CT scans and help us understand the significance of the MAC on sputum and BAL cultures.
►Alan Fine, MD, Section of Pulmonary and Critical Care, VABHS and Professor of Medicine, Boston University School of Medicine. Prior to this presentation, the patient had a pleural-based area of fibrosis with possible associated RML bronchiectasis. This appears to be a postinflammatory process without classic features of malignant or metastatic disease. She then had a sputum, which grew MAC in only 1 of 3 samples and in liquid media only. Importantly, the sputum was not smear positive. All of this suggests a low organism burden. One possibility is that this could reflect colonization with MAC; it is not uncommon for patients with underlying chronic changes in their lung to grow MAC, and it is often difficult to tell whether it is indicative of active disease. Structural lung disease, such as bronchiectasis, predisposes a patient to MAC, but chronic MAC also may cause bronchiectasis. This chicken-and-egg scenario comes up frequently. She may have a MAC infection, but as she is HIV negative and asymptomatic, there is no urgent indication to treat, especially as the burden of therapy is not insignificant.
►Dr. Swamy. Do we need to worry about Mycobacterium tuberculosis (MTB)?
►Dr. Fine. Although she was previously PPD positive, she had already completed 1 year of isoniazid (INH) therapy, making active MTB less likely. From an infection control standpoint, it is important to distinguish MAC from MTB. The former is not contagious, and there is no need for airborne isolation.
►Dr. Swamy. Dr. Fine, where does MAC come from? Does it commonly cause disease?
►Dr. Fine. In the environment, MAC is nearly ubiquitous , especially in water and soil. In one study, 20% of showerheads were positive for MAC; when patients are infected, we may suggest changing/bleaching the showerhead, but there are no definitive recommendations.3 Because MAC is so common in the environment, it is unlikely that measures to target MAC colonization will be clinically meaningful. On the other hand, the incidence of nontuberculous mycobacterial infections is increasing across the US, and it may be a common and frequently underdiagnosed cause of chronic cough, especially in postmenopausal women.
►Dr. Swamy. Four years prior to the current presentation, the patient developed a cough after an upper respiratory tract infection that persisted for more than 2 weeks. Given her history, she underwent a repeat chest CT, which noted a slight increase in nodularity and ground-glass opacity restricted to the RML. She also reported dyspnea on exertion and was referred to the pulmonary medicine department. By the time she arrived, her dyspnea had largely resolved, but she reported persistent fatigue without other systemic symptoms, such as fevers or chills. Dr. Fine, does MAC explain this patient’s dyspnea?
►Dr. Fine. As her pulmonary symptoms resolved in a short period of time with only azithromycin, it is very unlikely that her symptoms were related to her prior disease. The MAC infection is not likely to cause dyspnea on exertion and fatigue and should be worked up more broadly before attributing it to MAC. In view of this, it would not be unreasonable to follow her clinically and see her again in 6 to 8 weeks. In this context, we also should consider the untoward impact of repeated radiation exposure derived from multiple CT scans. When a patient has an abnormality on CT scan, it often leads to further scans even if the symptoms do not match the previous findings, as in this case.
►Dr. Swamy. Given her ongoing fatigue and systemic symptoms (morning stiffness of the shoulders, legs, and thighs, and leg cramps), she was referred to the rheumatology department where the physical examination revealed muscle tenderness in her proximal arms and legs with normal strength, tender points at the elbows and medial side of the bilateral knees, significant tenderness of lower legs, and no synovitis.
Dr. Monach, can you walk us through your approach to this patient? Are we seeing manifestations of fibromyalgia? What diagnoses concerns you and how would you proceed?
►Dr. Monach. The history and exam are most helpful in raising or reducing suspicion for an underlying inflammatory disease. Areas of tenderness described in her case are typical of fibromyalgia, although it can be difficult to interpret symptoms in the hip girdle and shoulder girdle because objective findings are often absent on exam in patients with inflammatory arthritis or bursitis. Similarly, tenderness at sites of tendon insertion (enthuses) without objective abnormalities is common in different forms of spondyloarthritis, so tenderness at the elbow, knee, lateral hip, and low back can be difficult to interpret. What this patient is lacking is prominent subjective or objective findings in the joints most commonly affected in rheumatoid arthritis and lupus: wrists, hands, ankles, and feet.
►Dr. Swamy. Initial laboratory data include an erythrocyte sedimentation rate of 79 with a normal C-reactive protein. A tentative diagnosis of polymyalgia rheumatic is made with consideration of a trial treatment of prednisone.
Dr. Monach, this patient has an indolent infection and is about to be given glucocorticoids. Could you describe the situations in which you feel that glucocorticoids cause a relative immunosuppression?
►Dr. Monach. Glucocorticoids are considered safe in a patient whose infection is not intrinsically dangerous or who has started appropriate antibiotics for that infection. Although all toxicities of glucocorticoids are dose dependent, the long-standing assertion that doses below 10 mg to 15 mg do not increase risk of infection is contradicted by data published in the past 10 to 15 years, with the caveat that these patients were on long-term treatment.
►Dr. Swamy. The patient was started on prednisone 15 mg per day for 15 days. She returned to the clinic after 1 week of prednisone troutment and noted “significant improvement in fatigue, morning stiffness of shoulders, thighs, leg, back is better, leg cramps resolved, shooting pain in many joints resolved.” Further laboratory results were notable for a negative rheumatoid factor, negative antinuclear antibody, and a cyclic citrullinated peptide of 60. A presumptive diagnosis of rheumatoid arthritis (RA) was made and plaquenil 200 mg twice daily was started.
Dr. Monach, can you explain why RA comes up now on serology but was not considered initially? Why does this presentation fit RA, and was her response to treatment typical? How does this fit in with her previous diagnosis of fibromyalgia? Was that just an atypical, indolent presentation of RA?
►Dr. Monach. Though her presentation is atypical for RA, in elderly patients, RA can present with symptoms resembling polymyalgia rheumatica. The question is whether she had RA all along (in which case “elderly onset” would not apply) or had fibromyalgia and developed RA more recently. The response to empiric glucocorticoid therapy is helpful, since fibromyalgia should not improve with prednisone even in a patient with RA unless treatment of RA would allow better sleep and ability to exercise. Rheumatoid arthritis typically responds very well to prednisone in the 5-mg to 15-mg range.
►Dr. Swamy. Given the new diagnosis of an inflammatory arthritis requiring immunosuppression, bronchoscopy with BAL is performed to evaluate for the presence of MAC. These cultures were positive for MAC.
Dr. Fine, does the positive BAL culture indicate an active MAC infection?
►Dr. Fine. Yes, based on these updated data, the patient has an active MAC infection. Active infection is defined as symptoms or imaging consistent with the diagnosis, supporting microbiology data (either 2 sputum or 1 BAL sample growing MAC) and the exclusion of other causes. Previously, this patient grew MAC in just one expectorated sputum; this did not meet the microbiologic criteria. Now sputum has grown in the BAL sample; along with the CT imaging, this is enough to diagnosis active MAC infection.
Treatment for MAC must consider the details of each case. First, this is not an emergency; treatment decisions should be made with the rheumatologist to consider the planned immunosuppression. For example, we must consider potential drug interactions. A specific point should be made of the use of tumor necrosis factor (TNF)-α inhibition, which data indicate can reactivate TB and may inhibit mechanisms that restrain mycobacterial disease. Serious cases of MAC infection have been reported in the literature in the setting of TNF-α inhibition.5,6 Despite these concerns, there is not a contraindication to using these therapies from the perspective of the active MAC disease. All of these decisions will impact the need to commit the patient to MAC therapy.
►Dr. Swamy. Dr. Fine, what do you consider prior to initiating MAC therapy?
► Dr. Fine. The decision to pursue MAC therapy should not be taken lightly. Therapy often entails prolonged multidrug regimens, usually spanning more than a year, with frequent adverse effects. Outside of very specific cases, such as TNF-β inhibition, MAC is rarely a life-threatening disease, so the benefit may be limited. Treatment for MAC is certainly unlikely to be fruitful without a diligent and motivated patient able to handle the high and prolonged pill burden. Of note, it is also important to keep this patient up-to-date with influenza and pneumonia vaccination given her structural lung disease.
►Dr. Swamy. The decision is made to treat MAC with azithromycin, rifampin, and ethambutol. The disease is noted to be nonfibrocavitary. The patient underwent monthly liver function test monitoring and visual acuity testing, which were unremarkable. Dr. Fine, can you describe the phenotypes of nontuberculous mycobacterial (NTM) disease?
►Dr. Fine. There are 3 main phenotypes of NTM.3 First, we see the elderly man with preexisting lung disease—usually chronic obstructive pulmonary disease—with fibrocavitary and/or reticulonodular appearance. Second, we see the slim, elderly woman often without any preexisting lung disease presenting with focal bronchiectasis and nodular lesions in right middle lobe and lingula—the Lady Windermere syndrome. This eponym is derived from Oscar Wilde’s play “Lady Windermere’s Fan, a Play About a Good Woman,” and was first associated with this disease in 1992.7 At the time, it was thought that the voluntary suppression of cough led to poorly draining lung regions, vulnerable to engraftment by atypical mycobacteria. Infection with atypical mycobacteria are associated with this population; however, it is no longer thought to be due to the voluntary suppression of cough.7,8 Third, we do occasionally see atypical presentations, such as focal masses and solitary nodules.
►Dr. Swamy. At 1-year follow-up she successfully completed MAC therapy and noted ongoing control of rheumatoid symptoms.
1. Bernardy K, Klose P, Welsch P, Häuser W. Efficacy, acceptability and safety of cognitive behavioural therapies in fibromyalgia syndrome—a systematic review and meta-analysis of randomized controlled trials. Eur J Pain. 2018;22(2):242-260.
2. Geneen LJ, Moore RA, Clarke C, Martin D, Colvin LA, Smith BH. Physical activity and exercise for chronic pain in adults: an overview of Cochrane Reviews. Cochrane Database Syst Rev. 2017;4:CD011279.
3. Aksamit TR, Philley JV, Griffith DE. Nontuberculous mycobacterial (NTM) lung disease: the top ten essentials. Respir Med. 2014;108(3):417-425.
4. Aucott JN. Glucocorticoids and infection. Endocrinol Metab Clin North Am. 1994;23(3):655-670.
5. Curtis JR, Yang S, Patkar NM, et al. Risk of hospitalized bacterial infections associated with biologic treatment among US veterans with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2014;66(7):990-997.
6. Lane MA, McDonald JR, Zeringue AL, et al. TNF-α antagonist use and risk of hospitalization for infection in a national cohort of veterans with rheumatoid arthritis. Medicine (Baltimore). 2011;90(2):139-145.
7. Reich JM, Johnson RE. Mycobacterium avium complex pulmonary disease presenting as an isolated lingular or middle lobe pattern. The Lady Windermere syndrome. Chest. 1992;101(6):1605-1609.
8. Kasthoori JJ, Liam CK, Wastie ML. Lady Windermere syndrome: an inappropriate eponym for an increasingly important condition. Singapore Med J. 2008;49(2):e47-e49.
1. Bernardy K, Klose P, Welsch P, Häuser W. Efficacy, acceptability and safety of cognitive behavioural therapies in fibromyalgia syndrome—a systematic review and meta-analysis of randomized controlled trials. Eur J Pain. 2018;22(2):242-260.
2. Geneen LJ, Moore RA, Clarke C, Martin D, Colvin LA, Smith BH. Physical activity and exercise for chronic pain in adults: an overview of Cochrane Reviews. Cochrane Database Syst Rev. 2017;4:CD011279.
3. Aksamit TR, Philley JV, Griffith DE. Nontuberculous mycobacterial (NTM) lung disease: the top ten essentials. Respir Med. 2014;108(3):417-425.
4. Aucott JN. Glucocorticoids and infection. Endocrinol Metab Clin North Am. 1994;23(3):655-670.
5. Curtis JR, Yang S, Patkar NM, et al. Risk of hospitalized bacterial infections associated with biologic treatment among US veterans with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2014;66(7):990-997.
6. Lane MA, McDonald JR, Zeringue AL, et al. TNF-α antagonist use and risk of hospitalization for infection in a national cohort of veterans with rheumatoid arthritis. Medicine (Baltimore). 2011;90(2):139-145.
7. Reich JM, Johnson RE. Mycobacterium avium complex pulmonary disease presenting as an isolated lingular or middle lobe pattern. The Lady Windermere syndrome. Chest. 1992;101(6):1605-1609.
8. Kasthoori JJ, Liam CK, Wastie ML. Lady Windermere syndrome: an inappropriate eponym for an increasingly important condition. Singapore Med J. 2008;49(2):e47-e49.