User login
A lump in the umbilicus
A 60-year-old man presented to the emergency department with abdominal pain. The pain was dull and constant, with no radiation and no aggravating or relieving factors. He also reported decreased appetite, weight loss, and constipation over the past 3 months.
He had no history of significant medical problems and was not taking any medications. He had no fever and no evidence of gastrointestinal bleeding.
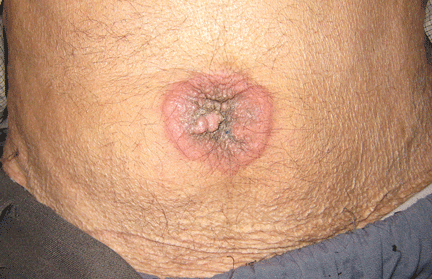
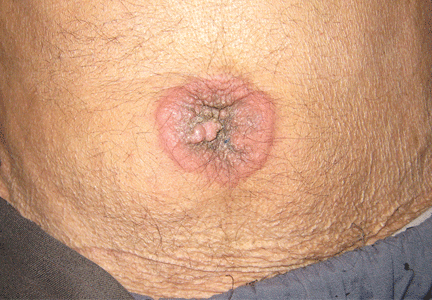
Physical examination showed mild tenderness around the umbilicus and a painless, small nodule (15 mm by 6 mm) protruding through the umbilicus with surrounding erythematous discoloration (Figure 1). A digital rectal examination was normal. Laboratory studies showed only mild normocytic anemia.
The patient underwent abdominal ultrasonography, which showed free fluid in the abdominopelvic cavity. This was followed by computed tomography of the abdominopelvic cavity, which revealed ascites and a small mass in the umbilicus. Punch biopsy of the umbilical lesion was performed, and histologic study indicated a diagnosis of adenocarcinoma.
Based on the biopsy results and the patient’s history of gastrointestinal symptoms, colonoscopy was performed, which showed an exophytic tumor of the transverse colon. The tumor was biopsied, and pathologic evaluation confirmed adenocarcinoma. A diagnosis of metastatic colon cancer was made. The patient received chemotherapy and underwent surgery to relieve the bowel obstruction.
SISTER MARY JOSEPH NODULE
A periumbilical nodule representing metastatic cancer, also known as Sister Mary Joseph nodule,1 is typically associated with intra-abdominal malignancy. An estimated 1% to 3% of patients with abdominopelvic malignancy present with this nodule,2 most often from gastrointestinal cancer but also from gynecologic malignancies. In about 15% to 30% of cases, no origin is identified.3
How these cancers spread to the umbilicus is not known. Proposed mechanisms include direct transperitoneal, lymphatic, or hematogenous spread, and even iatrogenic spread during laparotomy.4,5
The differential diagnosis includes umbilical hernia, cutaneous endometriosis, lymphangioma, melanoma, pilonidal sinus, and pyogenic granuloma. It is usually described as a painful nodule with irregular margins and a mean diameter of 2 to 3 cm.2 The condition is always a sign of metastatic cancer. Although it can be useful for diagnosing advanced disease, whether this would lead to earlier diagnosis is doubtful. Palliative treatment is generally most appropriate.
- Albano EA, Kanter J. Images in clinical medicine. Sister Mary Joseph’s nodule. N Engl J Med 2005; 352:1913.
- Iavazzo C, Madhuri K, Essapen S, Akrivos N, Tailor A, Butler-Manuel S. Sister Mary Joseph’s nodule as a first manifestation of primary peritoneal cancer. Case Rep Obstet Gynecol 2012; 2012:467240.
- Gabriele R, Borghese M, Conte M, Basso L. Sister Mary Joseph’s nodule as a first sign of cancer of the cecum: report of a case. Dis Colon Rectum 2004; 47:115–117.
- Dar IH, Kamili MA, Dar SH, Kuchaai FA. Sister Mary Joseph nodule—a case report with review of literature. J Res Med Sci 2009; 14:385–387.
- Martínez-Palones JM, Gil-Moreno A, Pérez-Benavente MA, Garcia-Giménez A, Xercavins J. Umbilical metastasis after laparoscopic retroperitoneal paraaortic lymphadenectomy for cervical cancer: a true port-site metastasis? Gynecol Oncol 2005; 97:292–295.
A 60-year-old man presented to the emergency department with abdominal pain. The pain was dull and constant, with no radiation and no aggravating or relieving factors. He also reported decreased appetite, weight loss, and constipation over the past 3 months.
He had no history of significant medical problems and was not taking any medications. He had no fever and no evidence of gastrointestinal bleeding.
Physical examination showed mild tenderness around the umbilicus and a painless, small nodule (15 mm by 6 mm) protruding through the umbilicus with surrounding erythematous discoloration (Figure 1). A digital rectal examination was normal. Laboratory studies showed only mild normocytic anemia.
The patient underwent abdominal ultrasonography, which showed free fluid in the abdominopelvic cavity. This was followed by computed tomography of the abdominopelvic cavity, which revealed ascites and a small mass in the umbilicus. Punch biopsy of the umbilical lesion was performed, and histologic study indicated a diagnosis of adenocarcinoma.
Based on the biopsy results and the patient’s history of gastrointestinal symptoms, colonoscopy was performed, which showed an exophytic tumor of the transverse colon. The tumor was biopsied, and pathologic evaluation confirmed adenocarcinoma. A diagnosis of metastatic colon cancer was made. The patient received chemotherapy and underwent surgery to relieve the bowel obstruction.
SISTER MARY JOSEPH NODULE
A periumbilical nodule representing metastatic cancer, also known as Sister Mary Joseph nodule,1 is typically associated with intra-abdominal malignancy. An estimated 1% to 3% of patients with abdominopelvic malignancy present with this nodule,2 most often from gastrointestinal cancer but also from gynecologic malignancies. In about 15% to 30% of cases, no origin is identified.3
How these cancers spread to the umbilicus is not known. Proposed mechanisms include direct transperitoneal, lymphatic, or hematogenous spread, and even iatrogenic spread during laparotomy.4,5
The differential diagnosis includes umbilical hernia, cutaneous endometriosis, lymphangioma, melanoma, pilonidal sinus, and pyogenic granuloma. It is usually described as a painful nodule with irregular margins and a mean diameter of 2 to 3 cm.2 The condition is always a sign of metastatic cancer. Although it can be useful for diagnosing advanced disease, whether this would lead to earlier diagnosis is doubtful. Palliative treatment is generally most appropriate.
A 60-year-old man presented to the emergency department with abdominal pain. The pain was dull and constant, with no radiation and no aggravating or relieving factors. He also reported decreased appetite, weight loss, and constipation over the past 3 months.
He had no history of significant medical problems and was not taking any medications. He had no fever and no evidence of gastrointestinal bleeding.
Physical examination showed mild tenderness around the umbilicus and a painless, small nodule (15 mm by 6 mm) protruding through the umbilicus with surrounding erythematous discoloration (Figure 1). A digital rectal examination was normal. Laboratory studies showed only mild normocytic anemia.
The patient underwent abdominal ultrasonography, which showed free fluid in the abdominopelvic cavity. This was followed by computed tomography of the abdominopelvic cavity, which revealed ascites and a small mass in the umbilicus. Punch biopsy of the umbilical lesion was performed, and histologic study indicated a diagnosis of adenocarcinoma.
Based on the biopsy results and the patient’s history of gastrointestinal symptoms, colonoscopy was performed, which showed an exophytic tumor of the transverse colon. The tumor was biopsied, and pathologic evaluation confirmed adenocarcinoma. A diagnosis of metastatic colon cancer was made. The patient received chemotherapy and underwent surgery to relieve the bowel obstruction.
SISTER MARY JOSEPH NODULE
A periumbilical nodule representing metastatic cancer, also known as Sister Mary Joseph nodule,1 is typically associated with intra-abdominal malignancy. An estimated 1% to 3% of patients with abdominopelvic malignancy present with this nodule,2 most often from gastrointestinal cancer but also from gynecologic malignancies. In about 15% to 30% of cases, no origin is identified.3
How these cancers spread to the umbilicus is not known. Proposed mechanisms include direct transperitoneal, lymphatic, or hematogenous spread, and even iatrogenic spread during laparotomy.4,5
The differential diagnosis includes umbilical hernia, cutaneous endometriosis, lymphangioma, melanoma, pilonidal sinus, and pyogenic granuloma. It is usually described as a painful nodule with irregular margins and a mean diameter of 2 to 3 cm.2 The condition is always a sign of metastatic cancer. Although it can be useful for diagnosing advanced disease, whether this would lead to earlier diagnosis is doubtful. Palliative treatment is generally most appropriate.
- Albano EA, Kanter J. Images in clinical medicine. Sister Mary Joseph’s nodule. N Engl J Med 2005; 352:1913.
- Iavazzo C, Madhuri K, Essapen S, Akrivos N, Tailor A, Butler-Manuel S. Sister Mary Joseph’s nodule as a first manifestation of primary peritoneal cancer. Case Rep Obstet Gynecol 2012; 2012:467240.
- Gabriele R, Borghese M, Conte M, Basso L. Sister Mary Joseph’s nodule as a first sign of cancer of the cecum: report of a case. Dis Colon Rectum 2004; 47:115–117.
- Dar IH, Kamili MA, Dar SH, Kuchaai FA. Sister Mary Joseph nodule—a case report with review of literature. J Res Med Sci 2009; 14:385–387.
- Martínez-Palones JM, Gil-Moreno A, Pérez-Benavente MA, Garcia-Giménez A, Xercavins J. Umbilical metastasis after laparoscopic retroperitoneal paraaortic lymphadenectomy for cervical cancer: a true port-site metastasis? Gynecol Oncol 2005; 97:292–295.
- Albano EA, Kanter J. Images in clinical medicine. Sister Mary Joseph’s nodule. N Engl J Med 2005; 352:1913.
- Iavazzo C, Madhuri K, Essapen S, Akrivos N, Tailor A, Butler-Manuel S. Sister Mary Joseph’s nodule as a first manifestation of primary peritoneal cancer. Case Rep Obstet Gynecol 2012; 2012:467240.
- Gabriele R, Borghese M, Conte M, Basso L. Sister Mary Joseph’s nodule as a first sign of cancer of the cecum: report of a case. Dis Colon Rectum 2004; 47:115–117.
- Dar IH, Kamili MA, Dar SH, Kuchaai FA. Sister Mary Joseph nodule—a case report with review of literature. J Res Med Sci 2009; 14:385–387.
- Martínez-Palones JM, Gil-Moreno A, Pérez-Benavente MA, Garcia-Giménez A, Xercavins J. Umbilical metastasis after laparoscopic retroperitoneal paraaortic lymphadenectomy for cervical cancer: a true port-site metastasis? Gynecol Oncol 2005; 97:292–295.
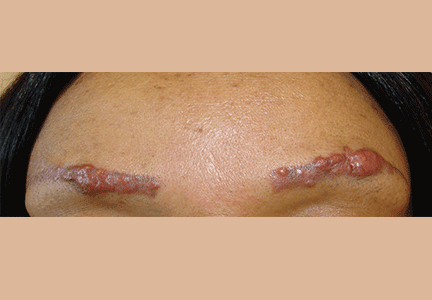
Corkscrew hairs
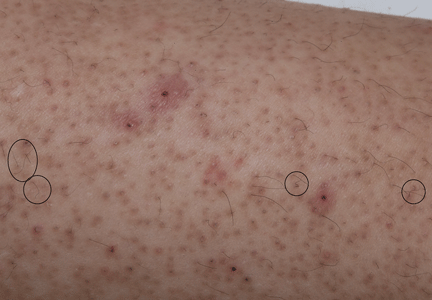
A 22-year-old woman with a 1-year history of mild systemic lupus erythematosus presented with disproportionately severe constitutional symptoms of fatigue and malaise. Physical examination showed multiple follicular-based hyperkeratotic papules with coiled “corkscrew” hairs on the outer surface of the arms and on the front of the legs (Figure 1). The patient reported a diet consisting mainly of white meat and processed foods. Although levels of serum folate, ferritin, zinc, and vitamins A, B1, B2, B6, B12, D, and E were within normal limits, the serum ascorbic acid level was low at 0.2 mg/dL (reference range 0.6–2.0 mg/dL). Ascorbic acid supplementation and dietary modification were recommended.
Ascorbic acid deficiency, or scurvy, is often considered a disease primarily of historical significance, with occurrences today limited to malnutrition or poverty.1 However, 18% of adults in the United States consume less than the recommend daily allowance of ascorbic acid.2 Ascorbic acid is minimally stored in the body,3 and scurvy can develop after 60 to 90 days of a diet free of ascorbic acid.4
Initial symptoms of fatigue, mood changes, and other constitutional symptoms are nonspecific, leading to a delay in diagnosis. Cutaneous manifestations include follicular hyperkeratosis associated with coiled or “corkscrew” hairs. Fragility of cutaneous blood vessels leads to perifollicular hemorrhages, petechiae, purpura, and ecchymoses. Extracutaneous manifestations are diverse and include oral involvement and intramuscular or intra-articular hemorrhage.1 Clinicians should have a high index of suspicion in socially isolated adults, elderly patients, those with alcoholism, mental illness, or chronic illness, and those with restrictive dietary preferences, particularly with predominant intake of processed foods.5
- Nguyen RT, Cowley DM, Muir JB. Scurvy: a cutaneous clinical diagnosis. Australas J Dermatol 2003; 44:48–51.
- Hampl JS, Taylor CA, Johnston CS. Vitamin C deficiency and depletion in the United States: the Third National Health and Nutrition Examination Survey, 1988 to 1994. Am J Public Health 2004; 94:870–875.
- Kluesner NH, Miller DG. Scurvy: malnourishment in the land of plenty. J Emerg Med 2014; 46:530–532.
- Popovich D, McAlhany A, Adewumi AO, Barnes MM. Scurvy: forgotten but definitely not gone. J Pediatr Health Care 2009; 23:405–415.
Velandia B, Centor RM, McConnell V, Shah M. Scurvy is still present in developed countries. J Gen Intern Med 2008; 23:1281–1284.
A 22-year-old woman with a 1-year history of mild systemic lupus erythematosus presented with disproportionately severe constitutional symptoms of fatigue and malaise. Physical examination showed multiple follicular-based hyperkeratotic papules with coiled “corkscrew” hairs on the outer surface of the arms and on the front of the legs (Figure 1). The patient reported a diet consisting mainly of white meat and processed foods. Although levels of serum folate, ferritin, zinc, and vitamins A, B1, B2, B6, B12, D, and E were within normal limits, the serum ascorbic acid level was low at 0.2 mg/dL (reference range 0.6–2.0 mg/dL). Ascorbic acid supplementation and dietary modification were recommended.
Ascorbic acid deficiency, or scurvy, is often considered a disease primarily of historical significance, with occurrences today limited to malnutrition or poverty.1 However, 18% of adults in the United States consume less than the recommend daily allowance of ascorbic acid.2 Ascorbic acid is minimally stored in the body,3 and scurvy can develop after 60 to 90 days of a diet free of ascorbic acid.4
Initial symptoms of fatigue, mood changes, and other constitutional symptoms are nonspecific, leading to a delay in diagnosis. Cutaneous manifestations include follicular hyperkeratosis associated with coiled or “corkscrew” hairs. Fragility of cutaneous blood vessels leads to perifollicular hemorrhages, petechiae, purpura, and ecchymoses. Extracutaneous manifestations are diverse and include oral involvement and intramuscular or intra-articular hemorrhage.1 Clinicians should have a high index of suspicion in socially isolated adults, elderly patients, those with alcoholism, mental illness, or chronic illness, and those with restrictive dietary preferences, particularly with predominant intake of processed foods.5
A 22-year-old woman with a 1-year history of mild systemic lupus erythematosus presented with disproportionately severe constitutional symptoms of fatigue and malaise. Physical examination showed multiple follicular-based hyperkeratotic papules with coiled “corkscrew” hairs on the outer surface of the arms and on the front of the legs (Figure 1). The patient reported a diet consisting mainly of white meat and processed foods. Although levels of serum folate, ferritin, zinc, and vitamins A, B1, B2, B6, B12, D, and E were within normal limits, the serum ascorbic acid level was low at 0.2 mg/dL (reference range 0.6–2.0 mg/dL). Ascorbic acid supplementation and dietary modification were recommended.
Ascorbic acid deficiency, or scurvy, is often considered a disease primarily of historical significance, with occurrences today limited to malnutrition or poverty.1 However, 18% of adults in the United States consume less than the recommend daily allowance of ascorbic acid.2 Ascorbic acid is minimally stored in the body,3 and scurvy can develop after 60 to 90 days of a diet free of ascorbic acid.4
Initial symptoms of fatigue, mood changes, and other constitutional symptoms are nonspecific, leading to a delay in diagnosis. Cutaneous manifestations include follicular hyperkeratosis associated with coiled or “corkscrew” hairs. Fragility of cutaneous blood vessels leads to perifollicular hemorrhages, petechiae, purpura, and ecchymoses. Extracutaneous manifestations are diverse and include oral involvement and intramuscular or intra-articular hemorrhage.1 Clinicians should have a high index of suspicion in socially isolated adults, elderly patients, those with alcoholism, mental illness, or chronic illness, and those with restrictive dietary preferences, particularly with predominant intake of processed foods.5
- Nguyen RT, Cowley DM, Muir JB. Scurvy: a cutaneous clinical diagnosis. Australas J Dermatol 2003; 44:48–51.
- Hampl JS, Taylor CA, Johnston CS. Vitamin C deficiency and depletion in the United States: the Third National Health and Nutrition Examination Survey, 1988 to 1994. Am J Public Health 2004; 94:870–875.
- Kluesner NH, Miller DG. Scurvy: malnourishment in the land of plenty. J Emerg Med 2014; 46:530–532.
- Popovich D, McAlhany A, Adewumi AO, Barnes MM. Scurvy: forgotten but definitely not gone. J Pediatr Health Care 2009; 23:405–415.
Velandia B, Centor RM, McConnell V, Shah M. Scurvy is still present in developed countries. J Gen Intern Med 2008; 23:1281–1284.
- Nguyen RT, Cowley DM, Muir JB. Scurvy: a cutaneous clinical diagnosis. Australas J Dermatol 2003; 44:48–51.
- Hampl JS, Taylor CA, Johnston CS. Vitamin C deficiency and depletion in the United States: the Third National Health and Nutrition Examination Survey, 1988 to 1994. Am J Public Health 2004; 94:870–875.
- Kluesner NH, Miller DG. Scurvy: malnourishment in the land of plenty. J Emerg Med 2014; 46:530–532.
- Popovich D, McAlhany A, Adewumi AO, Barnes MM. Scurvy: forgotten but definitely not gone. J Pediatr Health Care 2009; 23:405–415.
Velandia B, Centor RM, McConnell V, Shah M. Scurvy is still present in developed countries. J Gen Intern Med 2008; 23:1281–1284.
A nonhealing oral ulcer in a man with HIV
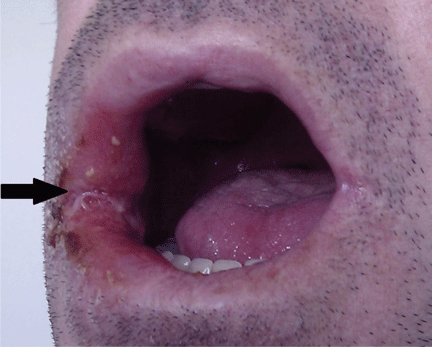
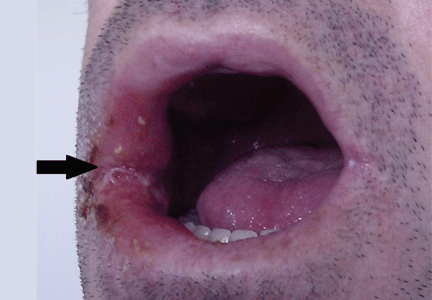
A 44-year-old man presented with a 3-week history of a painless, nonhealing ulcer affecting the mucosa in the right angle of the mouth. He reported no fever, weight loss, or systemic symptoms of chronic disease. His medical history included chronic hepatitis C infection and human immunodeficiency virus (HIV) infection; his antiretroviral regimen for HIV was abacavir, lamivudine, and darunavir. His CD4+ T-cell count was 153 cells/mm3, and his viral load was 154.88 copies/mL. He formerly used injected drugs, and he currently smoked 20 cigarettes a day. He had no history of periodontal disease.
Oral examination showed poor oral hygiene and a solitary ulcerated lesion with an infiltrated base and indurated borders in the right oral commissure (Figure 1). There was also soft-tissue induration in the ipsilateral cheek. He had no other oral lesions or signs of neck lymphadenopathy.
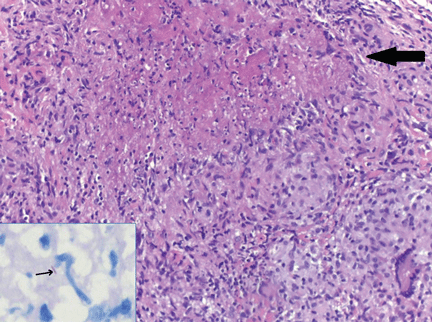
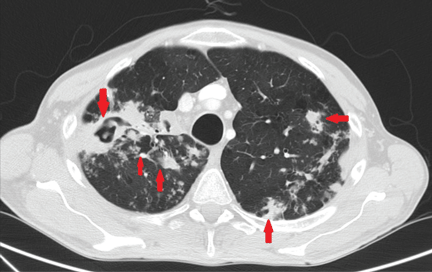
Based on the clinical examination, a provisional diagnosis of malignancy was suspected, and an incisional biopsy of the ulcer was done. The findings on histopathologic study of the biopsy specimen (below; Figure 2) led to additional evaluation with chest radiography and thoracic computed tomography (Figure 3).
FURTHER WORKUP
Histologic study of the biopsy specimen showed ulcerated mucosa with extensive granulomatous inflammation and with caseation necrosis. Ziehl-Neelsen staining demonstrated a few acid-fast bacilli. The patient was then evaluated for pulmonary tuberculosis.
Chest radiography showed pulmonary infiltrates in both bases and the upper right lobe and cavitation in the upper left lobe. Thoracic computed tomography confirmed the presence of multiple cavitated lesions in both left and right lung fields. Sputum cultures were positive for Mycobacterium tuberculosis, an organism sensitive to several agents. Laboratory investigations that included blood cell counts, biochemical tests, and liver and kidney function tests were normal, with the exception of a low lymphocyte count.
ORAL TUBERCULOSIS SECONDARY TO ACTIVE PULMONARY TUBERCULOSIS
Even though the incidence of extrapulmonary tuberculosis has been increasing worldwide in recent years, cutaneous manifestations are uncommon (2% to 10%) and are seen mainly in immunosuppressed patients with coexisting tuberculosis infection of internal organs.1 Oral manifestations of tuberculosis are extremely rare, accounting for 2% of cases of cutaneous manifestations.1,2 For this reason and because of its clinical heterogeneity, oral tuberculosis is often not considered in the differential diagnosis of oral lesions.
The differential diagnosis of oral ulcers in patients with HIV includes adverse drug reactions (eg, nevirapine-induced Stevens-Johnson syndrome); oral ulcers and necrotizing gingivitis related to HIV-associated neutropenia; aphthous ulcers; oral ulcers in reactive arthritis; malignancies such as lymphoma, Kaposi sarcoma, and squamous cell carcinoma; and infections such as candidiasis, herpes simplex virus, cytomegalovirus, primary syphilis, mucosal leishmaniasis, histoplasmosis, and periorificial tuberculosis. Periorificial tuberculosis is more commonly seen in HIV patients, in whom extrapulmonary forms of tuberculosis are frequent.3
Oral tuberculosis is often mistaken for a malignancy such as squamous cell carcinoma; in fact, carcinoma coexists with tubercular ulcer in up to 3% of patients.2 The typical clinical presentation of oral tuberculosis is one or more painful or painless ulcers with irregular borders, usually localized on the tongue (nearly half of cases4), and less frequently affecting the lip, buccal mucosa, gingiva, soft palate, and extraction sockets. It can also present as a nodule, fissure, vesicle, tuberculoma, tubercular papilloma, or periapical granuloma.2,5
Oral tuberculosis may be primary but is more commonly secondary to internal-organ tuberculosis. Secondary disease is seen in patients with pulmonary forms (affecting 0.05% to 1.5% of patients with pulmonary tuberculosis1,2) because of autoinoculation by infected sputum or hematogenous spread.1,4,5
Clinicians should consider oral tuberculosis in the differential diagnosis of a nonhealing ulcer of the mouth, particularly in an immunosuppressed patient such as ours. It is also important to highlight the necessity of searching for a primary site of infection, more frequently in the lungs.
- Kiliç A, Gül U, Gönül M, Soylu S, Cakmak SK, Demiriz M. Orificial tuberculosis of the lip: a case report and review of the literature. Int J Dermatol 2009; 48:178–180.
- Ram H, Kumar S, Mehrotra S, Mohommad S. Tubercular ulcer: mimicking squamous cell carcinoma of buccal mucosa. J Maxillofac Oral Surg 2012; 11:105–108.
- Frezzini C, Leao JC, Porter S. Current trends of HIV disease of the mouth. J Oral Pathol Med 2005; 34:513–531.
- Chauhan V, Mahesh DM, Panda P, Mahajan S, Thakur S. Tuberculosis cutis orificialis (TBCO): a rare manifestation of tuberculosis. J Assoc Physicians India 2012; 60:126–127.
- Kannan S, Thakkar P, Dcruz AK. Tuberculosis masquerading as oral malignancy. Indian J Med Paediatr Oncol 2011; 32:180–182.
A 44-year-old man presented with a 3-week history of a painless, nonhealing ulcer affecting the mucosa in the right angle of the mouth. He reported no fever, weight loss, or systemic symptoms of chronic disease. His medical history included chronic hepatitis C infection and human immunodeficiency virus (HIV) infection; his antiretroviral regimen for HIV was abacavir, lamivudine, and darunavir. His CD4+ T-cell count was 153 cells/mm3, and his viral load was 154.88 copies/mL. He formerly used injected drugs, and he currently smoked 20 cigarettes a day. He had no history of periodontal disease.
Oral examination showed poor oral hygiene and a solitary ulcerated lesion with an infiltrated base and indurated borders in the right oral commissure (Figure 1). There was also soft-tissue induration in the ipsilateral cheek. He had no other oral lesions or signs of neck lymphadenopathy.
Based on the clinical examination, a provisional diagnosis of malignancy was suspected, and an incisional biopsy of the ulcer was done. The findings on histopathologic study of the biopsy specimen (below; Figure 2) led to additional evaluation with chest radiography and thoracic computed tomography (Figure 3).
FURTHER WORKUP
Histologic study of the biopsy specimen showed ulcerated mucosa with extensive granulomatous inflammation and with caseation necrosis. Ziehl-Neelsen staining demonstrated a few acid-fast bacilli. The patient was then evaluated for pulmonary tuberculosis.
Chest radiography showed pulmonary infiltrates in both bases and the upper right lobe and cavitation in the upper left lobe. Thoracic computed tomography confirmed the presence of multiple cavitated lesions in both left and right lung fields. Sputum cultures were positive for Mycobacterium tuberculosis, an organism sensitive to several agents. Laboratory investigations that included blood cell counts, biochemical tests, and liver and kidney function tests were normal, with the exception of a low lymphocyte count.
ORAL TUBERCULOSIS SECONDARY TO ACTIVE PULMONARY TUBERCULOSIS
Even though the incidence of extrapulmonary tuberculosis has been increasing worldwide in recent years, cutaneous manifestations are uncommon (2% to 10%) and are seen mainly in immunosuppressed patients with coexisting tuberculosis infection of internal organs.1 Oral manifestations of tuberculosis are extremely rare, accounting for 2% of cases of cutaneous manifestations.1,2 For this reason and because of its clinical heterogeneity, oral tuberculosis is often not considered in the differential diagnosis of oral lesions.
The differential diagnosis of oral ulcers in patients with HIV includes adverse drug reactions (eg, nevirapine-induced Stevens-Johnson syndrome); oral ulcers and necrotizing gingivitis related to HIV-associated neutropenia; aphthous ulcers; oral ulcers in reactive arthritis; malignancies such as lymphoma, Kaposi sarcoma, and squamous cell carcinoma; and infections such as candidiasis, herpes simplex virus, cytomegalovirus, primary syphilis, mucosal leishmaniasis, histoplasmosis, and periorificial tuberculosis. Periorificial tuberculosis is more commonly seen in HIV patients, in whom extrapulmonary forms of tuberculosis are frequent.3
Oral tuberculosis is often mistaken for a malignancy such as squamous cell carcinoma; in fact, carcinoma coexists with tubercular ulcer in up to 3% of patients.2 The typical clinical presentation of oral tuberculosis is one or more painful or painless ulcers with irregular borders, usually localized on the tongue (nearly half of cases4), and less frequently affecting the lip, buccal mucosa, gingiva, soft palate, and extraction sockets. It can also present as a nodule, fissure, vesicle, tuberculoma, tubercular papilloma, or periapical granuloma.2,5
Oral tuberculosis may be primary but is more commonly secondary to internal-organ tuberculosis. Secondary disease is seen in patients with pulmonary forms (affecting 0.05% to 1.5% of patients with pulmonary tuberculosis1,2) because of autoinoculation by infected sputum or hematogenous spread.1,4,5
Clinicians should consider oral tuberculosis in the differential diagnosis of a nonhealing ulcer of the mouth, particularly in an immunosuppressed patient such as ours. It is also important to highlight the necessity of searching for a primary site of infection, more frequently in the lungs.
A 44-year-old man presented with a 3-week history of a painless, nonhealing ulcer affecting the mucosa in the right angle of the mouth. He reported no fever, weight loss, or systemic symptoms of chronic disease. His medical history included chronic hepatitis C infection and human immunodeficiency virus (HIV) infection; his antiretroviral regimen for HIV was abacavir, lamivudine, and darunavir. His CD4+ T-cell count was 153 cells/mm3, and his viral load was 154.88 copies/mL. He formerly used injected drugs, and he currently smoked 20 cigarettes a day. He had no history of periodontal disease.
Oral examination showed poor oral hygiene and a solitary ulcerated lesion with an infiltrated base and indurated borders in the right oral commissure (Figure 1). There was also soft-tissue induration in the ipsilateral cheek. He had no other oral lesions or signs of neck lymphadenopathy.
Based on the clinical examination, a provisional diagnosis of malignancy was suspected, and an incisional biopsy of the ulcer was done. The findings on histopathologic study of the biopsy specimen (below; Figure 2) led to additional evaluation with chest radiography and thoracic computed tomography (Figure 3).
FURTHER WORKUP
Histologic study of the biopsy specimen showed ulcerated mucosa with extensive granulomatous inflammation and with caseation necrosis. Ziehl-Neelsen staining demonstrated a few acid-fast bacilli. The patient was then evaluated for pulmonary tuberculosis.
Chest radiography showed pulmonary infiltrates in both bases and the upper right lobe and cavitation in the upper left lobe. Thoracic computed tomography confirmed the presence of multiple cavitated lesions in both left and right lung fields. Sputum cultures were positive for Mycobacterium tuberculosis, an organism sensitive to several agents. Laboratory investigations that included blood cell counts, biochemical tests, and liver and kidney function tests were normal, with the exception of a low lymphocyte count.
ORAL TUBERCULOSIS SECONDARY TO ACTIVE PULMONARY TUBERCULOSIS
Even though the incidence of extrapulmonary tuberculosis has been increasing worldwide in recent years, cutaneous manifestations are uncommon (2% to 10%) and are seen mainly in immunosuppressed patients with coexisting tuberculosis infection of internal organs.1 Oral manifestations of tuberculosis are extremely rare, accounting for 2% of cases of cutaneous manifestations.1,2 For this reason and because of its clinical heterogeneity, oral tuberculosis is often not considered in the differential diagnosis of oral lesions.
The differential diagnosis of oral ulcers in patients with HIV includes adverse drug reactions (eg, nevirapine-induced Stevens-Johnson syndrome); oral ulcers and necrotizing gingivitis related to HIV-associated neutropenia; aphthous ulcers; oral ulcers in reactive arthritis; malignancies such as lymphoma, Kaposi sarcoma, and squamous cell carcinoma; and infections such as candidiasis, herpes simplex virus, cytomegalovirus, primary syphilis, mucosal leishmaniasis, histoplasmosis, and periorificial tuberculosis. Periorificial tuberculosis is more commonly seen in HIV patients, in whom extrapulmonary forms of tuberculosis are frequent.3
Oral tuberculosis is often mistaken for a malignancy such as squamous cell carcinoma; in fact, carcinoma coexists with tubercular ulcer in up to 3% of patients.2 The typical clinical presentation of oral tuberculosis is one or more painful or painless ulcers with irregular borders, usually localized on the tongue (nearly half of cases4), and less frequently affecting the lip, buccal mucosa, gingiva, soft palate, and extraction sockets. It can also present as a nodule, fissure, vesicle, tuberculoma, tubercular papilloma, or periapical granuloma.2,5
Oral tuberculosis may be primary but is more commonly secondary to internal-organ tuberculosis. Secondary disease is seen in patients with pulmonary forms (affecting 0.05% to 1.5% of patients with pulmonary tuberculosis1,2) because of autoinoculation by infected sputum or hematogenous spread.1,4,5
Clinicians should consider oral tuberculosis in the differential diagnosis of a nonhealing ulcer of the mouth, particularly in an immunosuppressed patient such as ours. It is also important to highlight the necessity of searching for a primary site of infection, more frequently in the lungs.
- Kiliç A, Gül U, Gönül M, Soylu S, Cakmak SK, Demiriz M. Orificial tuberculosis of the lip: a case report and review of the literature. Int J Dermatol 2009; 48:178–180.
- Ram H, Kumar S, Mehrotra S, Mohommad S. Tubercular ulcer: mimicking squamous cell carcinoma of buccal mucosa. J Maxillofac Oral Surg 2012; 11:105–108.
- Frezzini C, Leao JC, Porter S. Current trends of HIV disease of the mouth. J Oral Pathol Med 2005; 34:513–531.
- Chauhan V, Mahesh DM, Panda P, Mahajan S, Thakur S. Tuberculosis cutis orificialis (TBCO): a rare manifestation of tuberculosis. J Assoc Physicians India 2012; 60:126–127.
- Kannan S, Thakkar P, Dcruz AK. Tuberculosis masquerading as oral malignancy. Indian J Med Paediatr Oncol 2011; 32:180–182.
- Kiliç A, Gül U, Gönül M, Soylu S, Cakmak SK, Demiriz M. Orificial tuberculosis of the lip: a case report and review of the literature. Int J Dermatol 2009; 48:178–180.
- Ram H, Kumar S, Mehrotra S, Mohommad S. Tubercular ulcer: mimicking squamous cell carcinoma of buccal mucosa. J Maxillofac Oral Surg 2012; 11:105–108.
- Frezzini C, Leao JC, Porter S. Current trends of HIV disease of the mouth. J Oral Pathol Med 2005; 34:513–531.
- Chauhan V, Mahesh DM, Panda P, Mahajan S, Thakur S. Tuberculosis cutis orificialis (TBCO): a rare manifestation of tuberculosis. J Assoc Physicians India 2012; 60:126–127.
- Kannan S, Thakkar P, Dcruz AK. Tuberculosis masquerading as oral malignancy. Indian J Med Paediatr Oncol 2011; 32:180–182.
Eruptive xanthoma
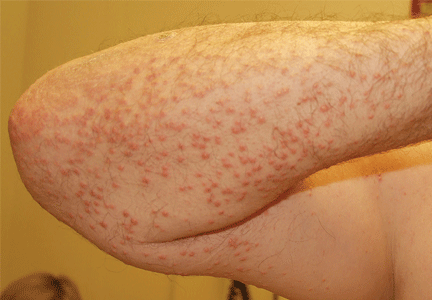
An obese 50-year-old man with hypertension, hyperlipidemia, recently diagnosed diabetes, and a history of grand mal seizures presented to the emergency room complaining of skin rash for 1 week. He denied having fever, chills, myalgia, abdominal pain, visual changes, recent changes in medications, or contact with anyone with similar symptoms.
He was a smoker, with a history of 20 pack-years; he denied abusing alcohol and taking illicit drugs.
He had no family history of diabetes, peripheral vascular disease, or coronary artery disease. His medications included lisinopril, simvastatin, niacin, metformin, and phenytoin.
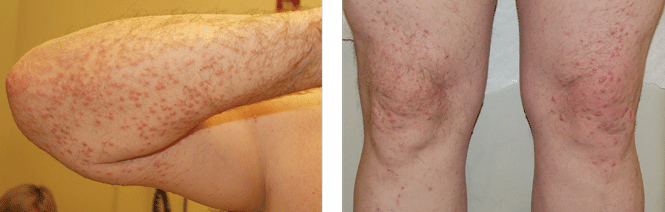
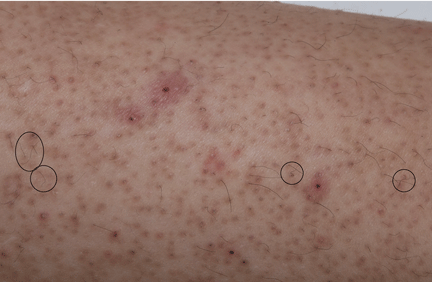
On physical examination, the lesions were small, reddish-yellow, nonpruritic tender papules covering the extensor surfaces of the knees, the forearms, the abdomen, and the back (Figure 1). Laboratory test results:
- Total cholesterol 1,045 mg/dL (reference range 100–199)
- Triglycerides 7,855 mg/dL (30–149)
- Thyroid-stimulating hormone 0.52 mIU/L (0.4–5.5)
- Fasting blood glucose 441 mg/dL (65–100)
- Hemoglobin A1c 12.6% (4.0–6.0)
- Total protein 7.2 g/dL (6.0–8.4)
- Albumin 4 g/dL (3.5–5.0)
- Creatinine 1 mg/dL (0.70–1.40)
- Glomerular filtration rate 79 mL/min/1.73 m2 (> 60)
- Urinalysis showed no proteinuria.
Histologic analysis of a lesion-biopsy specimen showed dermal foamy macrophages and loose lipids, which confirmed the suspicion of eruptive xanthoma.

The patient was started on strict glycemic and lipid control. Metformin and statin doses were increased and insulin was added. Three months later, laboratory results showed total cholesterol 128 mg/dL, triglycerides 164 mg/dL, fasting blood glucose 88 mg/dL, and hemoglobin A1c 5.5%. This was accompanied by marked improvement of the skin lesions (Figure 2).
CAUSES AND DIFFERENTIAL DIAGNOSIS
Eruptive xanthoma is a cutaneous disease most commonly arising over the extensor surfaces of the extremities and on the buttocks and shoulders, and it can be caused by high levels of serum triglycerides and uncontrolled diabetes mellitus.1 Hypothyroidism, end-stage renal disease, and nephrotic syndrome can cause secondary hypertriglyceridemia,2 which can cause eruptive xanthoma in severe cases. Patients with eruptive xanthoma may also have ophthalmologic and gastrointestinal involvement, such as lipemia retinalis (salmon-colored retina with creamy-white retinal vessels), abdominal pain, and hepatosplenomegaly.3
Other types of xanthoma associated with dyslipidemia include tuberous, tendinous, and plane xanthoma. Tuberous xanthoma is a firm, painless, deeper, red-yellow, larger nodular lesion, and the size may vary.4 Tendinous xanthoma is a slowly enlarging subcutaneous nodule typically located near tendons or ligaments in the hands, feet, and the Achilles tendon. Plane xanthoma is a flat papule or patch that can occur anywhere on the body.
The differential diagnosis includes disseminated granuloma annulare, non-Langerhans cell histiocytosis (xanthoma disseminatum, micronodular form of juvenile xanthogranuloma), and generalized eruptive histiocytoma. Eruptive xanthoma is differentiated from disseminated granuloma annulare by the abundance of perivascular histiocytes and xanthomized histiocytes, the presence of lipid deposits, and the deposition of hyaluronic acid on the edges.5 Xanthoma disseminatum consists of numerous, small, red-brown papules that are evenly spread on the face, skin-folds, trunk, and proximal extremities.6 Juvenile xanthogranuloma occurs mostly in children and is characterized by discrete orange-yellow nodules, which commonly appear on the scalp, face, and upper trunk. It is in most cases a solitary lesion, but multiple lesions may occur.7 Lesions of generalized eruptive histiocytoma are firm, erythematous or brownish papules that appear in successive crops over the face, trunk, and proximal surfaces of the limbs.
TREATMENT
Treatment of eruptive xanthoma involves dietary restriction, exercise, and drug therapy to control the hyperlipidemia and the diabetes.2 Early recognition and proper control of hypertriglyceridemia can prevent sequelae such as acute pancreatitis.3
- Durrington P. Dyslipidaemia. Lancet 2003; 362:717–731.
- Brunzell JD. Clinical practice. Hypertriglyceridemia. N Engl J Med 2007; 357:1009–1017.
- Leaf DA. Chylomicronemia and the chylomicronemia syndrome: a practical approach to management. Am J Med 2008; 121:10–12.
- Siddi GM, Pes GM, Errigo A, Corraduzza G, Ena P. Multiple tuberous xanthomas as the first manifestation of autosomal recessive hypercholesterolemia. J Eur Acad Dermatol Venereol 2006; 20:1376–1378.
- Cooper PH. Eruptive xanthoma: a microscopic simulant of granuloma annulare. J Cutan Pathol 1986; 13:207–215.
- Rupec RA, Schaller M. Xanthoma disseminatum. Int J Dermatol 2002; 41:911–913.
- Ferrari F, Masurel A, Olivier-Faivre L, Vabres P. Juvenile xanthogranuloma and nevus anemicus in the diagnosis of neurofibromatosis type 1. JAMA Dermatol 2014; 150:42–46.
An obese 50-year-old man with hypertension, hyperlipidemia, recently diagnosed diabetes, and a history of grand mal seizures presented to the emergency room complaining of skin rash for 1 week. He denied having fever, chills, myalgia, abdominal pain, visual changes, recent changes in medications, or contact with anyone with similar symptoms.
He was a smoker, with a history of 20 pack-years; he denied abusing alcohol and taking illicit drugs.
He had no family history of diabetes, peripheral vascular disease, or coronary artery disease. His medications included lisinopril, simvastatin, niacin, metformin, and phenytoin.
On physical examination, the lesions were small, reddish-yellow, nonpruritic tender papules covering the extensor surfaces of the knees, the forearms, the abdomen, and the back (Figure 1). Laboratory test results:
- Total cholesterol 1,045 mg/dL (reference range 100–199)
- Triglycerides 7,855 mg/dL (30–149)
- Thyroid-stimulating hormone 0.52 mIU/L (0.4–5.5)
- Fasting blood glucose 441 mg/dL (65–100)
- Hemoglobin A1c 12.6% (4.0–6.0)
- Total protein 7.2 g/dL (6.0–8.4)
- Albumin 4 g/dL (3.5–5.0)
- Creatinine 1 mg/dL (0.70–1.40)
- Glomerular filtration rate 79 mL/min/1.73 m2 (> 60)
- Urinalysis showed no proteinuria.
Histologic analysis of a lesion-biopsy specimen showed dermal foamy macrophages and loose lipids, which confirmed the suspicion of eruptive xanthoma.
The patient was started on strict glycemic and lipid control. Metformin and statin doses were increased and insulin was added. Three months later, laboratory results showed total cholesterol 128 mg/dL, triglycerides 164 mg/dL, fasting blood glucose 88 mg/dL, and hemoglobin A1c 5.5%. This was accompanied by marked improvement of the skin lesions (Figure 2).
CAUSES AND DIFFERENTIAL DIAGNOSIS
Eruptive xanthoma is a cutaneous disease most commonly arising over the extensor surfaces of the extremities and on the buttocks and shoulders, and it can be caused by high levels of serum triglycerides and uncontrolled diabetes mellitus.1 Hypothyroidism, end-stage renal disease, and nephrotic syndrome can cause secondary hypertriglyceridemia,2 which can cause eruptive xanthoma in severe cases. Patients with eruptive xanthoma may also have ophthalmologic and gastrointestinal involvement, such as lipemia retinalis (salmon-colored retina with creamy-white retinal vessels), abdominal pain, and hepatosplenomegaly.3
Other types of xanthoma associated with dyslipidemia include tuberous, tendinous, and plane xanthoma. Tuberous xanthoma is a firm, painless, deeper, red-yellow, larger nodular lesion, and the size may vary.4 Tendinous xanthoma is a slowly enlarging subcutaneous nodule typically located near tendons or ligaments in the hands, feet, and the Achilles tendon. Plane xanthoma is a flat papule or patch that can occur anywhere on the body.
The differential diagnosis includes disseminated granuloma annulare, non-Langerhans cell histiocytosis (xanthoma disseminatum, micronodular form of juvenile xanthogranuloma), and generalized eruptive histiocytoma. Eruptive xanthoma is differentiated from disseminated granuloma annulare by the abundance of perivascular histiocytes and xanthomized histiocytes, the presence of lipid deposits, and the deposition of hyaluronic acid on the edges.5 Xanthoma disseminatum consists of numerous, small, red-brown papules that are evenly spread on the face, skin-folds, trunk, and proximal extremities.6 Juvenile xanthogranuloma occurs mostly in children and is characterized by discrete orange-yellow nodules, which commonly appear on the scalp, face, and upper trunk. It is in most cases a solitary lesion, but multiple lesions may occur.7 Lesions of generalized eruptive histiocytoma are firm, erythematous or brownish papules that appear in successive crops over the face, trunk, and proximal surfaces of the limbs.
TREATMENT
Treatment of eruptive xanthoma involves dietary restriction, exercise, and drug therapy to control the hyperlipidemia and the diabetes.2 Early recognition and proper control of hypertriglyceridemia can prevent sequelae such as acute pancreatitis.3
An obese 50-year-old man with hypertension, hyperlipidemia, recently diagnosed diabetes, and a history of grand mal seizures presented to the emergency room complaining of skin rash for 1 week. He denied having fever, chills, myalgia, abdominal pain, visual changes, recent changes in medications, or contact with anyone with similar symptoms.
He was a smoker, with a history of 20 pack-years; he denied abusing alcohol and taking illicit drugs.
He had no family history of diabetes, peripheral vascular disease, or coronary artery disease. His medications included lisinopril, simvastatin, niacin, metformin, and phenytoin.
On physical examination, the lesions were small, reddish-yellow, nonpruritic tender papules covering the extensor surfaces of the knees, the forearms, the abdomen, and the back (Figure 1). Laboratory test results:
- Total cholesterol 1,045 mg/dL (reference range 100–199)
- Triglycerides 7,855 mg/dL (30–149)
- Thyroid-stimulating hormone 0.52 mIU/L (0.4–5.5)
- Fasting blood glucose 441 mg/dL (65–100)
- Hemoglobin A1c 12.6% (4.0–6.0)
- Total protein 7.2 g/dL (6.0–8.4)
- Albumin 4 g/dL (3.5–5.0)
- Creatinine 1 mg/dL (0.70–1.40)
- Glomerular filtration rate 79 mL/min/1.73 m2 (> 60)
- Urinalysis showed no proteinuria.
Histologic analysis of a lesion-biopsy specimen showed dermal foamy macrophages and loose lipids, which confirmed the suspicion of eruptive xanthoma.
The patient was started on strict glycemic and lipid control. Metformin and statin doses were increased and insulin was added. Three months later, laboratory results showed total cholesterol 128 mg/dL, triglycerides 164 mg/dL, fasting blood glucose 88 mg/dL, and hemoglobin A1c 5.5%. This was accompanied by marked improvement of the skin lesions (Figure 2).
CAUSES AND DIFFERENTIAL DIAGNOSIS
Eruptive xanthoma is a cutaneous disease most commonly arising over the extensor surfaces of the extremities and on the buttocks and shoulders, and it can be caused by high levels of serum triglycerides and uncontrolled diabetes mellitus.1 Hypothyroidism, end-stage renal disease, and nephrotic syndrome can cause secondary hypertriglyceridemia,2 which can cause eruptive xanthoma in severe cases. Patients with eruptive xanthoma may also have ophthalmologic and gastrointestinal involvement, such as lipemia retinalis (salmon-colored retina with creamy-white retinal vessels), abdominal pain, and hepatosplenomegaly.3
Other types of xanthoma associated with dyslipidemia include tuberous, tendinous, and plane xanthoma. Tuberous xanthoma is a firm, painless, deeper, red-yellow, larger nodular lesion, and the size may vary.4 Tendinous xanthoma is a slowly enlarging subcutaneous nodule typically located near tendons or ligaments in the hands, feet, and the Achilles tendon. Plane xanthoma is a flat papule or patch that can occur anywhere on the body.
The differential diagnosis includes disseminated granuloma annulare, non-Langerhans cell histiocytosis (xanthoma disseminatum, micronodular form of juvenile xanthogranuloma), and generalized eruptive histiocytoma. Eruptive xanthoma is differentiated from disseminated granuloma annulare by the abundance of perivascular histiocytes and xanthomized histiocytes, the presence of lipid deposits, and the deposition of hyaluronic acid on the edges.5 Xanthoma disseminatum consists of numerous, small, red-brown papules that are evenly spread on the face, skin-folds, trunk, and proximal extremities.6 Juvenile xanthogranuloma occurs mostly in children and is characterized by discrete orange-yellow nodules, which commonly appear on the scalp, face, and upper trunk. It is in most cases a solitary lesion, but multiple lesions may occur.7 Lesions of generalized eruptive histiocytoma are firm, erythematous or brownish papules that appear in successive crops over the face, trunk, and proximal surfaces of the limbs.
TREATMENT
Treatment of eruptive xanthoma involves dietary restriction, exercise, and drug therapy to control the hyperlipidemia and the diabetes.2 Early recognition and proper control of hypertriglyceridemia can prevent sequelae such as acute pancreatitis.3
- Durrington P. Dyslipidaemia. Lancet 2003; 362:717–731.
- Brunzell JD. Clinical practice. Hypertriglyceridemia. N Engl J Med 2007; 357:1009–1017.
- Leaf DA. Chylomicronemia and the chylomicronemia syndrome: a practical approach to management. Am J Med 2008; 121:10–12.
- Siddi GM, Pes GM, Errigo A, Corraduzza G, Ena P. Multiple tuberous xanthomas as the first manifestation of autosomal recessive hypercholesterolemia. J Eur Acad Dermatol Venereol 2006; 20:1376–1378.
- Cooper PH. Eruptive xanthoma: a microscopic simulant of granuloma annulare. J Cutan Pathol 1986; 13:207–215.
- Rupec RA, Schaller M. Xanthoma disseminatum. Int J Dermatol 2002; 41:911–913.
- Ferrari F, Masurel A, Olivier-Faivre L, Vabres P. Juvenile xanthogranuloma and nevus anemicus in the diagnosis of neurofibromatosis type 1. JAMA Dermatol 2014; 150:42–46.
- Durrington P. Dyslipidaemia. Lancet 2003; 362:717–731.
- Brunzell JD. Clinical practice. Hypertriglyceridemia. N Engl J Med 2007; 357:1009–1017.
- Leaf DA. Chylomicronemia and the chylomicronemia syndrome: a practical approach to management. Am J Med 2008; 121:10–12.
- Siddi GM, Pes GM, Errigo A, Corraduzza G, Ena P. Multiple tuberous xanthomas as the first manifestation of autosomal recessive hypercholesterolemia. J Eur Acad Dermatol Venereol 2006; 20:1376–1378.
- Cooper PH. Eruptive xanthoma: a microscopic simulant of granuloma annulare. J Cutan Pathol 1986; 13:207–215.
- Rupec RA, Schaller M. Xanthoma disseminatum. Int J Dermatol 2002; 41:911–913.
- Ferrari F, Masurel A, Olivier-Faivre L, Vabres P. Juvenile xanthogranuloma and nevus anemicus in the diagnosis of neurofibromatosis type 1. JAMA Dermatol 2014; 150:42–46.
Heart on the right may sometimes be ‘right’
A 76-year-old man presented to the emergency department with right-sided exertional chest pain radiating to the right shoulder and arm associated with shortness of breath. His vital signs were normal. On clinical examination, the cardiac apex was palpated on the right side, 9 cm from the midsternal line in the fifth intercostal space.
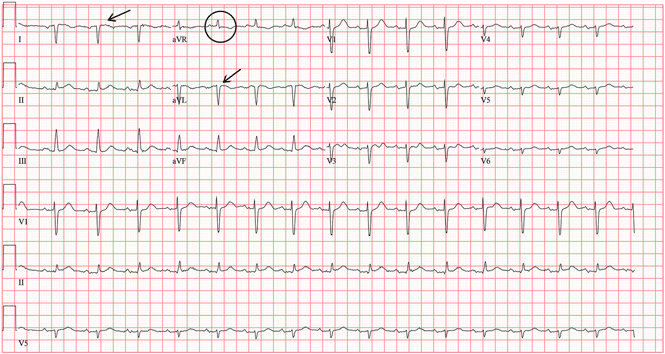
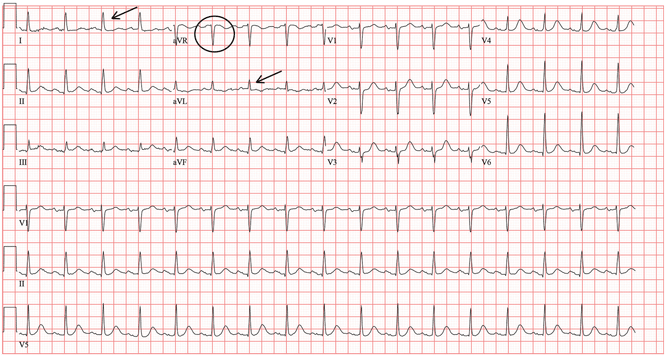
A standard left-sided 12-lead electrocardiogram (ECG) showed right-axis deviation and inverted P, QRS, and T waves in leads I and aVL (Figure 1). Although these changes are also seen when the right and left arm electrode wires are transposed, the precordial lead morphology in such a situation would usually be normal. In our patient, the precordial leads showed the absence or even slight reversal of R-wave progression, a feature indicative of dextrocardia.1,2
In patients with dextrocardia, right-sided hookup of the electrodes is usually necessary for proper interpretation of the ECG. When this was done in our patient, the ECG showed a normal cardiac axis, a negative QRS complex in lead aVR, a positive P wave and other complexes in lead I, and normal R-wave progression in the precordial leads—findings suggestive of dextrocardia (Figure 2).
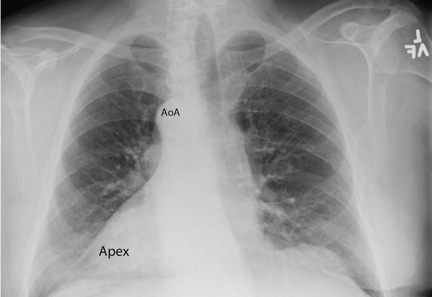
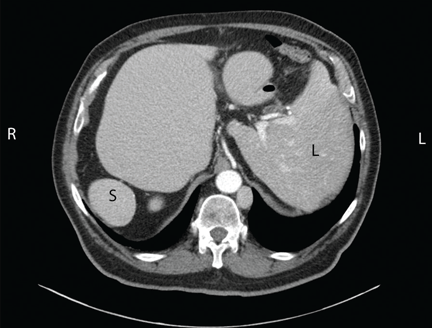
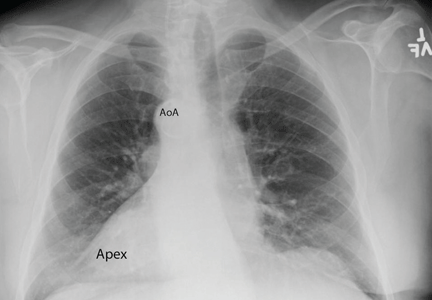
Chest radiography showed a right-sided cardiac silhouette (Figure 3), and computed tomography of the abdomen (Figure 4) revealed the liver positioned on the left side and the spleen on the right, confirming the diagnosis of situs inversus totalis. The ECG showed dextrocardia, but no other abnormalities. The patient eventually underwent coronary angiography, which showed nonobstructive coronary artery disease.
DEXTROCARDIA, OTHER CONGENITAL CARDIOVASCULAR MALFORMATIONS
Dextrocardia was first described in early 17th century.1 Situs solitus is the normal position of the heart and viscera, whereas situs inversus is a mirror-image anatomic arrangement of the organs. Situs inversus with dextrocardia, also called situs inversus totalis, is a rare condition (with a prevalence of 1 in 8,000) in which the heart and descending aorta are on the right and the thoracic and abdominal viscera are usually mirror images of the normal morphology.1,3,4 A mirror-image sinus node lies at the junction of the left superior vena cava and the left-sided (morphologic right) atrium.1 People with situs inversus with dextrocardia are usually asymptomatic and have a normal life expectancy.1,2 Situs inversus with levocardia is a rare condition in which the heart is in the normal position but the viscera are in the dextro-position. This anomaly has a prevalence of 1 in 22,000.5
Atrial situs almost always corresponds to visceral situs. However, when the alignment of the atria and viscera is inconsistent and situs cannot be determined clearly because of the malpositioning of organs, the condition is called “situs ambiguous.” This is very rare, with a prevalence of 1 in 40,000.6
Risk factors
The cause of congenital cardiovascular malformations such as these is not known, but risk factors include positive family history, maternal diabetes, and cocaine use in the first trimester.7
The prevalence of congenital heart disease in patients with situs inversus with dextrocardia is low and ranges from 2% to 5%. This is in contrast to situs solitus with dextrocardia (isolated dextrocardia), which is almost always associated with cardiovascular anomalies.2,4 Kartagener syndrome—the triad of situs inversus, sinusitis, and bronchiectasis—occurs in 25% of people with situs inversus with dextrocardia.4 Situs inversus with levocardia is also frequently associated with cardiac anomalies.5
The major features of dextrocardia on ECG are:
- Negative P wave, QRS complex, and T wave in lead I
- Positive QRS complex in aVR
- Right-axis deviation
- Reversal of R-wave progression in the precordial leads.
Ventricular activation and repolarization are reversed, resulting in a negative QRS complex and an inverted T wave in lead I. The absence of R-wave progression in the precordial leads helps differentiate mirror-image dextrocardia from erroneously reversed limb-electrode placement, which shows normal R-wave progression from V1 to V6 while showing similar features to those seen in dextrocardia in the limb leads.2 In right-sided hookup, the limb electrodes are reversed, and the chest electrodes are recorded from the right precordium.
CORONARY INTERVENTIONS REQUIRE SPECIAL CONSIDERATION
In patients with dextrocardia, coronary interventions can be challenging because of the mirror-image position of the coronary ostia and the aortic arch.8 These patients also need careful imaging, consideration of other associated congenital cardiac abnormalities, and detailed planning before cardiac surgery, including coronary artery bypass grafting.9
Patients with dextrocardia may present with cardiac symptoms localized to the right side of the body and have confusing clinical and diagnostic findings. Keeping dextrocardia and other such anomalies in mind can prevent delay in appropriately directed interventions. In a patient such as ours, the heart on the right side of the chest may indeed be “right.” Still, diagnostic tests to look for disorders encountered with dextrocardia may be necessary.
- Perloff JK. The cardiac malpositions. Am J Cardiol 2011; 108:1352–1361.
- Tanawuttiwat T, Vasaiwala S, Dia M. ECG image of the month. Mirror mirror. Am J Med 2010; 123:34–36.
- Douard R, Feldman A, Bargy F, Loric S, Delmas V. Anomalies of lateralization in man: a case of total situs in-versus. Surg Radiol Anat 2000; 22:293–297.
- Maldjian PD, Saric M. Approach to dextrocardia in adults: review. AJR Am J Roentgenol 2007; 188(suppl 6):S39–S49.
- Gindes L, Hegesh J, Barkai G, Jacobson JM, Achiron R. Isolated levocardia: prenatal diagnosis, clinical im-portance, and literature review. J Ultrasound Med 2007; 26:361–365.
- Abut E, Arman A, Güveli H, et al. Malposition of internal organs: a case of situs ambiguous anomaly in an adult. Turk J Gastroenterol 2003; 14:151–155.
- Kuehl KS, Loffredo C. Risk factors for heart disease associated with abnormal sidedness. Teratology 2002; 66:242–248.
- Aksoy S, Cam N, Gurkan U, Altay S, Bozbay M, Agirbasli M. Primary percutaneous intervention: for acute myo-cardial infarction in a patient with dextrocardia and situs inversus. Tex Heart Inst J 2012; 39:140–141.
- Murtuza B, Gupta P, Goli G, Lall KS. Coronary revascularization in adults with dextrocardia: surgical implications of the anatomic variants. Tex Heart Inst J 2010; 37:633–640.
A 76-year-old man presented to the emergency department with right-sided exertional chest pain radiating to the right shoulder and arm associated with shortness of breath. His vital signs were normal. On clinical examination, the cardiac apex was palpated on the right side, 9 cm from the midsternal line in the fifth intercostal space.
A standard left-sided 12-lead electrocardiogram (ECG) showed right-axis deviation and inverted P, QRS, and T waves in leads I and aVL (Figure 1). Although these changes are also seen when the right and left arm electrode wires are transposed, the precordial lead morphology in such a situation would usually be normal. In our patient, the precordial leads showed the absence or even slight reversal of R-wave progression, a feature indicative of dextrocardia.1,2
In patients with dextrocardia, right-sided hookup of the electrodes is usually necessary for proper interpretation of the ECG. When this was done in our patient, the ECG showed a normal cardiac axis, a negative QRS complex in lead aVR, a positive P wave and other complexes in lead I, and normal R-wave progression in the precordial leads—findings suggestive of dextrocardia (Figure 2).
Chest radiography showed a right-sided cardiac silhouette (Figure 3), and computed tomography of the abdomen (Figure 4) revealed the liver positioned on the left side and the spleen on the right, confirming the diagnosis of situs inversus totalis. The ECG showed dextrocardia, but no other abnormalities. The patient eventually underwent coronary angiography, which showed nonobstructive coronary artery disease.
DEXTROCARDIA, OTHER CONGENITAL CARDIOVASCULAR MALFORMATIONS
Dextrocardia was first described in early 17th century.1 Situs solitus is the normal position of the heart and viscera, whereas situs inversus is a mirror-image anatomic arrangement of the organs. Situs inversus with dextrocardia, also called situs inversus totalis, is a rare condition (with a prevalence of 1 in 8,000) in which the heart and descending aorta are on the right and the thoracic and abdominal viscera are usually mirror images of the normal morphology.1,3,4 A mirror-image sinus node lies at the junction of the left superior vena cava and the left-sided (morphologic right) atrium.1 People with situs inversus with dextrocardia are usually asymptomatic and have a normal life expectancy.1,2 Situs inversus with levocardia is a rare condition in which the heart is in the normal position but the viscera are in the dextro-position. This anomaly has a prevalence of 1 in 22,000.5
Atrial situs almost always corresponds to visceral situs. However, when the alignment of the atria and viscera is inconsistent and situs cannot be determined clearly because of the malpositioning of organs, the condition is called “situs ambiguous.” This is very rare, with a prevalence of 1 in 40,000.6
Risk factors
The cause of congenital cardiovascular malformations such as these is not known, but risk factors include positive family history, maternal diabetes, and cocaine use in the first trimester.7
The prevalence of congenital heart disease in patients with situs inversus with dextrocardia is low and ranges from 2% to 5%. This is in contrast to situs solitus with dextrocardia (isolated dextrocardia), which is almost always associated with cardiovascular anomalies.2,4 Kartagener syndrome—the triad of situs inversus, sinusitis, and bronchiectasis—occurs in 25% of people with situs inversus with dextrocardia.4 Situs inversus with levocardia is also frequently associated with cardiac anomalies.5
The major features of dextrocardia on ECG are:
- Negative P wave, QRS complex, and T wave in lead I
- Positive QRS complex in aVR
- Right-axis deviation
- Reversal of R-wave progression in the precordial leads.
Ventricular activation and repolarization are reversed, resulting in a negative QRS complex and an inverted T wave in lead I. The absence of R-wave progression in the precordial leads helps differentiate mirror-image dextrocardia from erroneously reversed limb-electrode placement, which shows normal R-wave progression from V1 to V6 while showing similar features to those seen in dextrocardia in the limb leads.2 In right-sided hookup, the limb electrodes are reversed, and the chest electrodes are recorded from the right precordium.
CORONARY INTERVENTIONS REQUIRE SPECIAL CONSIDERATION
In patients with dextrocardia, coronary interventions can be challenging because of the mirror-image position of the coronary ostia and the aortic arch.8 These patients also need careful imaging, consideration of other associated congenital cardiac abnormalities, and detailed planning before cardiac surgery, including coronary artery bypass grafting.9
Patients with dextrocardia may present with cardiac symptoms localized to the right side of the body and have confusing clinical and diagnostic findings. Keeping dextrocardia and other such anomalies in mind can prevent delay in appropriately directed interventions. In a patient such as ours, the heart on the right side of the chest may indeed be “right.” Still, diagnostic tests to look for disorders encountered with dextrocardia may be necessary.
A 76-year-old man presented to the emergency department with right-sided exertional chest pain radiating to the right shoulder and arm associated with shortness of breath. His vital signs were normal. On clinical examination, the cardiac apex was palpated on the right side, 9 cm from the midsternal line in the fifth intercostal space.
A standard left-sided 12-lead electrocardiogram (ECG) showed right-axis deviation and inverted P, QRS, and T waves in leads I and aVL (Figure 1). Although these changes are also seen when the right and left arm electrode wires are transposed, the precordial lead morphology in such a situation would usually be normal. In our patient, the precordial leads showed the absence or even slight reversal of R-wave progression, a feature indicative of dextrocardia.1,2
In patients with dextrocardia, right-sided hookup of the electrodes is usually necessary for proper interpretation of the ECG. When this was done in our patient, the ECG showed a normal cardiac axis, a negative QRS complex in lead aVR, a positive P wave and other complexes in lead I, and normal R-wave progression in the precordial leads—findings suggestive of dextrocardia (Figure 2).
Chest radiography showed a right-sided cardiac silhouette (Figure 3), and computed tomography of the abdomen (Figure 4) revealed the liver positioned on the left side and the spleen on the right, confirming the diagnosis of situs inversus totalis. The ECG showed dextrocardia, but no other abnormalities. The patient eventually underwent coronary angiography, which showed nonobstructive coronary artery disease.
DEXTROCARDIA, OTHER CONGENITAL CARDIOVASCULAR MALFORMATIONS
Dextrocardia was first described in early 17th century.1 Situs solitus is the normal position of the heart and viscera, whereas situs inversus is a mirror-image anatomic arrangement of the organs. Situs inversus with dextrocardia, also called situs inversus totalis, is a rare condition (with a prevalence of 1 in 8,000) in which the heart and descending aorta are on the right and the thoracic and abdominal viscera are usually mirror images of the normal morphology.1,3,4 A mirror-image sinus node lies at the junction of the left superior vena cava and the left-sided (morphologic right) atrium.1 People with situs inversus with dextrocardia are usually asymptomatic and have a normal life expectancy.1,2 Situs inversus with levocardia is a rare condition in which the heart is in the normal position but the viscera are in the dextro-position. This anomaly has a prevalence of 1 in 22,000.5
Atrial situs almost always corresponds to visceral situs. However, when the alignment of the atria and viscera is inconsistent and situs cannot be determined clearly because of the malpositioning of organs, the condition is called “situs ambiguous.” This is very rare, with a prevalence of 1 in 40,000.6
Risk factors
The cause of congenital cardiovascular malformations such as these is not known, but risk factors include positive family history, maternal diabetes, and cocaine use in the first trimester.7
The prevalence of congenital heart disease in patients with situs inversus with dextrocardia is low and ranges from 2% to 5%. This is in contrast to situs solitus with dextrocardia (isolated dextrocardia), which is almost always associated with cardiovascular anomalies.2,4 Kartagener syndrome—the triad of situs inversus, sinusitis, and bronchiectasis—occurs in 25% of people with situs inversus with dextrocardia.4 Situs inversus with levocardia is also frequently associated with cardiac anomalies.5
The major features of dextrocardia on ECG are:
- Negative P wave, QRS complex, and T wave in lead I
- Positive QRS complex in aVR
- Right-axis deviation
- Reversal of R-wave progression in the precordial leads.
Ventricular activation and repolarization are reversed, resulting in a negative QRS complex and an inverted T wave in lead I. The absence of R-wave progression in the precordial leads helps differentiate mirror-image dextrocardia from erroneously reversed limb-electrode placement, which shows normal R-wave progression from V1 to V6 while showing similar features to those seen in dextrocardia in the limb leads.2 In right-sided hookup, the limb electrodes are reversed, and the chest electrodes are recorded from the right precordium.
CORONARY INTERVENTIONS REQUIRE SPECIAL CONSIDERATION
In patients with dextrocardia, coronary interventions can be challenging because of the mirror-image position of the coronary ostia and the aortic arch.8 These patients also need careful imaging, consideration of other associated congenital cardiac abnormalities, and detailed planning before cardiac surgery, including coronary artery bypass grafting.9
Patients with dextrocardia may present with cardiac symptoms localized to the right side of the body and have confusing clinical and diagnostic findings. Keeping dextrocardia and other such anomalies in mind can prevent delay in appropriately directed interventions. In a patient such as ours, the heart on the right side of the chest may indeed be “right.” Still, diagnostic tests to look for disorders encountered with dextrocardia may be necessary.
- Perloff JK. The cardiac malpositions. Am J Cardiol 2011; 108:1352–1361.
- Tanawuttiwat T, Vasaiwala S, Dia M. ECG image of the month. Mirror mirror. Am J Med 2010; 123:34–36.
- Douard R, Feldman A, Bargy F, Loric S, Delmas V. Anomalies of lateralization in man: a case of total situs in-versus. Surg Radiol Anat 2000; 22:293–297.
- Maldjian PD, Saric M. Approach to dextrocardia in adults: review. AJR Am J Roentgenol 2007; 188(suppl 6):S39–S49.
- Gindes L, Hegesh J, Barkai G, Jacobson JM, Achiron R. Isolated levocardia: prenatal diagnosis, clinical im-portance, and literature review. J Ultrasound Med 2007; 26:361–365.
- Abut E, Arman A, Güveli H, et al. Malposition of internal organs: a case of situs ambiguous anomaly in an adult. Turk J Gastroenterol 2003; 14:151–155.
- Kuehl KS, Loffredo C. Risk factors for heart disease associated with abnormal sidedness. Teratology 2002; 66:242–248.
- Aksoy S, Cam N, Gurkan U, Altay S, Bozbay M, Agirbasli M. Primary percutaneous intervention: for acute myo-cardial infarction in a patient with dextrocardia and situs inversus. Tex Heart Inst J 2012; 39:140–141.
- Murtuza B, Gupta P, Goli G, Lall KS. Coronary revascularization in adults with dextrocardia: surgical implications of the anatomic variants. Tex Heart Inst J 2010; 37:633–640.
- Perloff JK. The cardiac malpositions. Am J Cardiol 2011; 108:1352–1361.
- Tanawuttiwat T, Vasaiwala S, Dia M. ECG image of the month. Mirror mirror. Am J Med 2010; 123:34–36.
- Douard R, Feldman A, Bargy F, Loric S, Delmas V. Anomalies of lateralization in man: a case of total situs in-versus. Surg Radiol Anat 2000; 22:293–297.
- Maldjian PD, Saric M. Approach to dextrocardia in adults: review. AJR Am J Roentgenol 2007; 188(suppl 6):S39–S49.
- Gindes L, Hegesh J, Barkai G, Jacobson JM, Achiron R. Isolated levocardia: prenatal diagnosis, clinical im-portance, and literature review. J Ultrasound Med 2007; 26:361–365.
- Abut E, Arman A, Güveli H, et al. Malposition of internal organs: a case of situs ambiguous anomaly in an adult. Turk J Gastroenterol 2003; 14:151–155.
- Kuehl KS, Loffredo C. Risk factors for heart disease associated with abnormal sidedness. Teratology 2002; 66:242–248.
- Aksoy S, Cam N, Gurkan U, Altay S, Bozbay M, Agirbasli M. Primary percutaneous intervention: for acute myo-cardial infarction in a patient with dextrocardia and situs inversus. Tex Heart Inst J 2012; 39:140–141.
- Murtuza B, Gupta P, Goli G, Lall KS. Coronary revascularization in adults with dextrocardia: surgical implications of the anatomic variants. Tex Heart Inst J 2010; 37:633–640.
Pneumatosis cystoides intestinalis: Is surgery always indicated?
A 57-year-old man with long-standing systemic sclerosis presented with worsening diffuse abdominal pain associated with several episodes of nonbloody emesis for 5 days. He had been hospitalized numerous times over the past 2 years for similar symptoms. In those instances, abdominal radiography and computed tomography (CT) had revealed nonspecific intestinal pseudo-obstruction that had resolved within a few days with bowel rest, antibiotics for small-intestinal bacterial overgrowth, and supportive care.
At the time of this presentation, physical examination showed stable vital signs, a tympanic, distended abdomen with diffuse tenderness, and diminished bowel sounds with no sign of peritonitis. Complete blood cell counts, renal function testing, and serum lactate levels were unremarkable.
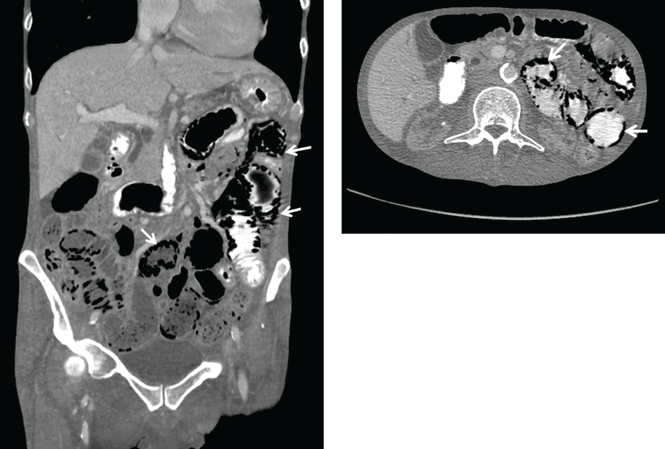
Abdominal radiography showed mildly dilated loops of small bowel with multiple fluid levels, raising concern for intestinal obstruction. Interestingly, abdominal CT revealed extensive pneumatosis cystoides intestinalis of the entire small bowel with sparing of the colon, which raised concern for acute bowel ischemia (Figure 1). However, given the patient’s underlying systemic sclerosis and current stable condition, the general surgeon recommended conservative management with bowel rest, rifaximin to treat the small-intestinal bacterial overgrowth, and intravenous fluids, which resulted in significant clinical improvement. A liquid diet was initiated and advanced as tolerated to a soft diet before he was discharged home after 8 days of hospitalization.
A RARE, USUALLY BENIGN COMPLICATION OF SYSTEMIC SCLEROSIS
Pneumatosis cystoides intestinalis is a rare gastrointestinal complication of systemic sclerosis characterized by intramural accumulation of gas within thin-walled cysts. It is postulated to result either from excess hydrogen gas produced by intraluminal bacterial fermentation and altered partial pressure of nitrogen within the intestinal wall (the bacterial theory),1 or from the transgression of gas cysts through the layers of bowel wall as a result of high luminal pressure from intestinal obstruction (the mechanical theory).2
The more widespread use of diagnostic CT in recent years has led to increased recognition of this condition, a finding that also often raises concern for intestinal necrosis or perforation.3 Meticulous correlation of the clinical presentation with corroborative laboratory testing should determine whether a conservative medical approach or emergency surgical exploration is appropriate.4
Pneumatosis cystoides intestinalis in patients with systemic sclerosis is a benign condition that generally resolves with bowel rest, antibiotics, inhalational oxygen therapy, and supportive care.5 An elevated venous oxygen concentration from high-flow oxygen therapy is believed to attenuate the gaseous cysts by decreasing the partial pressure of the nitrogenous gases and by being toxic to the anaerobic gut bacteria.
About 3% of patients with pneumatosis cystoides intestinalis develop complications such as pneumoperitoneum, intestinal volvulus, obstruction, or hemorrhage. Evidence of pneumoperitoneum or bowel infarction—such as the presence of portomesenteric venous gas, a decreased arterial pH, or an elevated lactic acid or amylase level—warrants immediate surgical intervention. Overall, early recognition and watchful monitoring for bowel necrosis or perforation are preferred over reflexive surgical exploration.
- Levitt MD, Olsson S. Pneumatosis cystoides intestinalis and high breath H2 excretion: insights into the role of H2 in this condition. Gastroenterology 1995; 108:1560–1565.
- Pieterse AS, Leong AS, Rowland R. The mucosal changes and pathogenesis of pneumatosis cystoides intestinalis. Hum Pathol 1985; 16:683–688.
- Ho LM, Paulson EK, Thompson WM. Pneumatosis intestinalis in the adult: benign to life-threatening causes. AJR Am J Roentgenol 2007; 188:1604–1613.
- Khalil PN, Huber-Wagner S, Ladurner R, et al. Natural history, clinical pattern, and surgical considerations of pneumatosis intestinalis. Eur J Med Res 2009; 14:231–239.
- Vischio J, Matlyuk-Urman Z, Lakshminarayanan S. Benign spontaneous pneumoperitoneum in systemic sclerosis. J Clin Rheumatol 2010; 16:379–381.
A 57-year-old man with long-standing systemic sclerosis presented with worsening diffuse abdominal pain associated with several episodes of nonbloody emesis for 5 days. He had been hospitalized numerous times over the past 2 years for similar symptoms. In those instances, abdominal radiography and computed tomography (CT) had revealed nonspecific intestinal pseudo-obstruction that had resolved within a few days with bowel rest, antibiotics for small-intestinal bacterial overgrowth, and supportive care.
At the time of this presentation, physical examination showed stable vital signs, a tympanic, distended abdomen with diffuse tenderness, and diminished bowel sounds with no sign of peritonitis. Complete blood cell counts, renal function testing, and serum lactate levels were unremarkable.
Abdominal radiography showed mildly dilated loops of small bowel with multiple fluid levels, raising concern for intestinal obstruction. Interestingly, abdominal CT revealed extensive pneumatosis cystoides intestinalis of the entire small bowel with sparing of the colon, which raised concern for acute bowel ischemia (Figure 1). However, given the patient’s underlying systemic sclerosis and current stable condition, the general surgeon recommended conservative management with bowel rest, rifaximin to treat the small-intestinal bacterial overgrowth, and intravenous fluids, which resulted in significant clinical improvement. A liquid diet was initiated and advanced as tolerated to a soft diet before he was discharged home after 8 days of hospitalization.
A RARE, USUALLY BENIGN COMPLICATION OF SYSTEMIC SCLEROSIS
Pneumatosis cystoides intestinalis is a rare gastrointestinal complication of systemic sclerosis characterized by intramural accumulation of gas within thin-walled cysts. It is postulated to result either from excess hydrogen gas produced by intraluminal bacterial fermentation and altered partial pressure of nitrogen within the intestinal wall (the bacterial theory),1 or from the transgression of gas cysts through the layers of bowel wall as a result of high luminal pressure from intestinal obstruction (the mechanical theory).2
The more widespread use of diagnostic CT in recent years has led to increased recognition of this condition, a finding that also often raises concern for intestinal necrosis or perforation.3 Meticulous correlation of the clinical presentation with corroborative laboratory testing should determine whether a conservative medical approach or emergency surgical exploration is appropriate.4
Pneumatosis cystoides intestinalis in patients with systemic sclerosis is a benign condition that generally resolves with bowel rest, antibiotics, inhalational oxygen therapy, and supportive care.5 An elevated venous oxygen concentration from high-flow oxygen therapy is believed to attenuate the gaseous cysts by decreasing the partial pressure of the nitrogenous gases and by being toxic to the anaerobic gut bacteria.
About 3% of patients with pneumatosis cystoides intestinalis develop complications such as pneumoperitoneum, intestinal volvulus, obstruction, or hemorrhage. Evidence of pneumoperitoneum or bowel infarction—such as the presence of portomesenteric venous gas, a decreased arterial pH, or an elevated lactic acid or amylase level—warrants immediate surgical intervention. Overall, early recognition and watchful monitoring for bowel necrosis or perforation are preferred over reflexive surgical exploration.
A 57-year-old man with long-standing systemic sclerosis presented with worsening diffuse abdominal pain associated with several episodes of nonbloody emesis for 5 days. He had been hospitalized numerous times over the past 2 years for similar symptoms. In those instances, abdominal radiography and computed tomography (CT) had revealed nonspecific intestinal pseudo-obstruction that had resolved within a few days with bowel rest, antibiotics for small-intestinal bacterial overgrowth, and supportive care.
At the time of this presentation, physical examination showed stable vital signs, a tympanic, distended abdomen with diffuse tenderness, and diminished bowel sounds with no sign of peritonitis. Complete blood cell counts, renal function testing, and serum lactate levels were unremarkable.
Abdominal radiography showed mildly dilated loops of small bowel with multiple fluid levels, raising concern for intestinal obstruction. Interestingly, abdominal CT revealed extensive pneumatosis cystoides intestinalis of the entire small bowel with sparing of the colon, which raised concern for acute bowel ischemia (Figure 1). However, given the patient’s underlying systemic sclerosis and current stable condition, the general surgeon recommended conservative management with bowel rest, rifaximin to treat the small-intestinal bacterial overgrowth, and intravenous fluids, which resulted in significant clinical improvement. A liquid diet was initiated and advanced as tolerated to a soft diet before he was discharged home after 8 days of hospitalization.
A RARE, USUALLY BENIGN COMPLICATION OF SYSTEMIC SCLEROSIS
Pneumatosis cystoides intestinalis is a rare gastrointestinal complication of systemic sclerosis characterized by intramural accumulation of gas within thin-walled cysts. It is postulated to result either from excess hydrogen gas produced by intraluminal bacterial fermentation and altered partial pressure of nitrogen within the intestinal wall (the bacterial theory),1 or from the transgression of gas cysts through the layers of bowel wall as a result of high luminal pressure from intestinal obstruction (the mechanical theory).2
The more widespread use of diagnostic CT in recent years has led to increased recognition of this condition, a finding that also often raises concern for intestinal necrosis or perforation.3 Meticulous correlation of the clinical presentation with corroborative laboratory testing should determine whether a conservative medical approach or emergency surgical exploration is appropriate.4
Pneumatosis cystoides intestinalis in patients with systemic sclerosis is a benign condition that generally resolves with bowel rest, antibiotics, inhalational oxygen therapy, and supportive care.5 An elevated venous oxygen concentration from high-flow oxygen therapy is believed to attenuate the gaseous cysts by decreasing the partial pressure of the nitrogenous gases and by being toxic to the anaerobic gut bacteria.
About 3% of patients with pneumatosis cystoides intestinalis develop complications such as pneumoperitoneum, intestinal volvulus, obstruction, or hemorrhage. Evidence of pneumoperitoneum or bowel infarction—such as the presence of portomesenteric venous gas, a decreased arterial pH, or an elevated lactic acid or amylase level—warrants immediate surgical intervention. Overall, early recognition and watchful monitoring for bowel necrosis or perforation are preferred over reflexive surgical exploration.
- Levitt MD, Olsson S. Pneumatosis cystoides intestinalis and high breath H2 excretion: insights into the role of H2 in this condition. Gastroenterology 1995; 108:1560–1565.
- Pieterse AS, Leong AS, Rowland R. The mucosal changes and pathogenesis of pneumatosis cystoides intestinalis. Hum Pathol 1985; 16:683–688.
- Ho LM, Paulson EK, Thompson WM. Pneumatosis intestinalis in the adult: benign to life-threatening causes. AJR Am J Roentgenol 2007; 188:1604–1613.
- Khalil PN, Huber-Wagner S, Ladurner R, et al. Natural history, clinical pattern, and surgical considerations of pneumatosis intestinalis. Eur J Med Res 2009; 14:231–239.
- Vischio J, Matlyuk-Urman Z, Lakshminarayanan S. Benign spontaneous pneumoperitoneum in systemic sclerosis. J Clin Rheumatol 2010; 16:379–381.
- Levitt MD, Olsson S. Pneumatosis cystoides intestinalis and high breath H2 excretion: insights into the role of H2 in this condition. Gastroenterology 1995; 108:1560–1565.
- Pieterse AS, Leong AS, Rowland R. The mucosal changes and pathogenesis of pneumatosis cystoides intestinalis. Hum Pathol 1985; 16:683–688.
- Ho LM, Paulson EK, Thompson WM. Pneumatosis intestinalis in the adult: benign to life-threatening causes. AJR Am J Roentgenol 2007; 188:1604–1613.
- Khalil PN, Huber-Wagner S, Ladurner R, et al. Natural history, clinical pattern, and surgical considerations of pneumatosis intestinalis. Eur J Med Res 2009; 14:231–239.
- Vischio J, Matlyuk-Urman Z, Lakshminarayanan S. Benign spontaneous pneumoperitoneum in systemic sclerosis. J Clin Rheumatol 2010; 16:379–381.

Syncope from a twiddled ICD
A 61-year-old man with ischemic cardiomyopathy underwent implantation of a cardioverter-defibrillator (ICD) in a left subpectoral pocket. Placement was confirmed with chest radiography (Figure 1). Six weeks later, he presented to the emergency department reporting two episodes of syncope within the previous 24 hours.
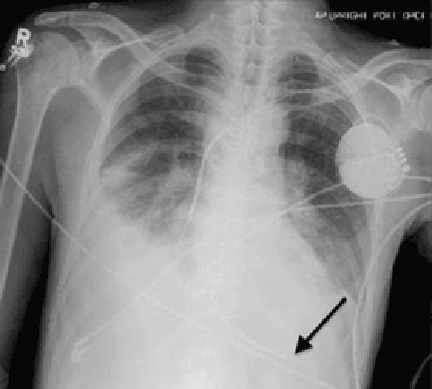
Electrocardiography at the time of presentation showed a normal sinus rhythm with inappropriate ICD discharges (Figure 2). This finding prompted chest radiography, which showed that the ICD had rotated 90 degrees from its original position, and that the lead had completely wrapped around the generator and thus was no longer correctly positioned (Figure 3). The patient admitted to frequently rubbing (ie, twiddling) the area, which led to twisting and dislocation of the device, a complication known as twiddler syndrome.
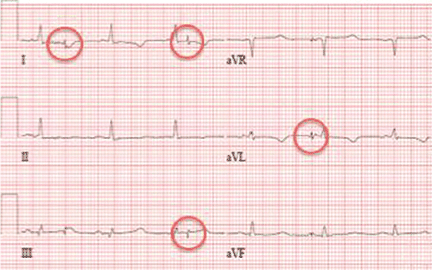
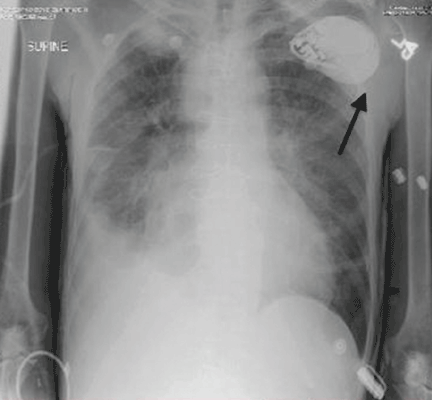
Twiddler syndrome is a rare complication of ICD placement, occurring in only about 0.1% of cases.1 It is usually a result of intentional manipulation (ie, twiddling) of the device by the patient, causing the ICD and the leads to dislodge, break, or retract. It is seen more often in women and in patients with cognitive dysfunction or psychiatric illness, in obese patients, and in patients with laxity of subcutaneous tissues, such as elderly people or people with accelerated weight loss. Placement of the ICD in an inappropriately large pocket predisposes to device rotation.2,3
Twiddler syndrome may cause mild discomfort or may even go unnoticed by the patient.4 In rare cases, it may lead to stimulation of the phrenic nerve, causing diaphragmatic pacing, or to stimulation of the brachial plexus, causing muscle twitching.2,3 However, malfunction of an ICD is life-threatening and requires immediate repair and replacement of the device. Educating the patient about the risks of twiddling the device site and the importance of periodic device checks is imperative to prevent this syndrome and ensure its early identification if it should occur. The patient should understand that dislodging the ICD can make it unable to sense abnormal rhythms and cause the device to deliver inappropriate shocks2,3,5 or stop delivering shocks altogether.
HOW IT IS TREATED
Treatment involves replacing the leads and affixing the device with sutures in the existing pocket or in a new pocket. The subpectoral position is preferred as it is more secure,3 although subcutaneous reimplantation has been successful.1 The device may also be placed in a fabric pouch to help lower the risk of migration or manipulation.3,6
In this case, because of the patient’s slender body habitus (body mass index 13 kg/m2), the ICD was removed from the subpectoral pocket and a new ICD was sutured into a subcutaneous pocket. The single lead was secured to the sternum and fascia using nonabsorbable sutures and a tie-down sleeve, making the device less susceptible to dislodgement by twiddling. Electrocardiography after replacement showed a normal sinus rhythm (Figure 4).
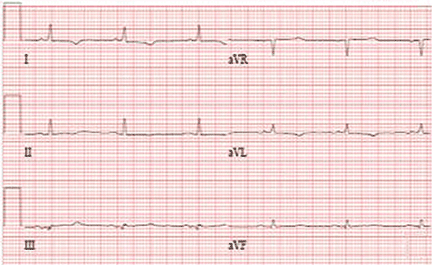
- Constandse J, Smit JJ, Ramdat Misier AR, Elvan A, Delnoy PP. Unusual twiddler syndrome: movement ties the knot. Neth Heart J 2013; 21:253–254.
- Spencker S, Poppelbaum A, Müller D. An unusual cause of oversensing leading to inappropriate ICD discharges. Int J Cardiol 2008; 129:e24–e26.
- Benezet-Mazuecos J, Benezet J, Ortega-Carnicer J. Pacemaker twiddler syndrome. Eur Heart J 2007; 28:2000.
- Chemello D, Subramanian A, Cameron D. Twiddler syndrome with 180 degrees rotation of an implantable cardioverter defibrillator generator resulting in malfunction of one of the shocking coils. Europace 2009; 11:1259.
- Garweg C, Alzand BS, Willems R. Twiddler syndrome causing an inappropriate implantable cardioverter-defibrillator shock. Eur Heart J 2014; 35:516.
- Parsonnet V, Bernstein AD, Neglia D, Omar A. The usefulness of a stretch-polyester pouch to encase implanted pacemakers and defibrillators. Pacing Clin Electrophysiol 1994; 17:2274–2278.
A 61-year-old man with ischemic cardiomyopathy underwent implantation of a cardioverter-defibrillator (ICD) in a left subpectoral pocket. Placement was confirmed with chest radiography (Figure 1). Six weeks later, he presented to the emergency department reporting two episodes of syncope within the previous 24 hours.
Electrocardiography at the time of presentation showed a normal sinus rhythm with inappropriate ICD discharges (Figure 2). This finding prompted chest radiography, which showed that the ICD had rotated 90 degrees from its original position, and that the lead had completely wrapped around the generator and thus was no longer correctly positioned (Figure 3). The patient admitted to frequently rubbing (ie, twiddling) the area, which led to twisting and dislocation of the device, a complication known as twiddler syndrome.
Twiddler syndrome is a rare complication of ICD placement, occurring in only about 0.1% of cases.1 It is usually a result of intentional manipulation (ie, twiddling) of the device by the patient, causing the ICD and the leads to dislodge, break, or retract. It is seen more often in women and in patients with cognitive dysfunction or psychiatric illness, in obese patients, and in patients with laxity of subcutaneous tissues, such as elderly people or people with accelerated weight loss. Placement of the ICD in an inappropriately large pocket predisposes to device rotation.2,3
Twiddler syndrome may cause mild discomfort or may even go unnoticed by the patient.4 In rare cases, it may lead to stimulation of the phrenic nerve, causing diaphragmatic pacing, or to stimulation of the brachial plexus, causing muscle twitching.2,3 However, malfunction of an ICD is life-threatening and requires immediate repair and replacement of the device. Educating the patient about the risks of twiddling the device site and the importance of periodic device checks is imperative to prevent this syndrome and ensure its early identification if it should occur. The patient should understand that dislodging the ICD can make it unable to sense abnormal rhythms and cause the device to deliver inappropriate shocks2,3,5 or stop delivering shocks altogether.
HOW IT IS TREATED
Treatment involves replacing the leads and affixing the device with sutures in the existing pocket or in a new pocket. The subpectoral position is preferred as it is more secure,3 although subcutaneous reimplantation has been successful.1 The device may also be placed in a fabric pouch to help lower the risk of migration or manipulation.3,6
In this case, because of the patient’s slender body habitus (body mass index 13 kg/m2), the ICD was removed from the subpectoral pocket and a new ICD was sutured into a subcutaneous pocket. The single lead was secured to the sternum and fascia using nonabsorbable sutures and a tie-down sleeve, making the device less susceptible to dislodgement by twiddling. Electrocardiography after replacement showed a normal sinus rhythm (Figure 4).
A 61-year-old man with ischemic cardiomyopathy underwent implantation of a cardioverter-defibrillator (ICD) in a left subpectoral pocket. Placement was confirmed with chest radiography (Figure 1). Six weeks later, he presented to the emergency department reporting two episodes of syncope within the previous 24 hours.
Electrocardiography at the time of presentation showed a normal sinus rhythm with inappropriate ICD discharges (Figure 2). This finding prompted chest radiography, which showed that the ICD had rotated 90 degrees from its original position, and that the lead had completely wrapped around the generator and thus was no longer correctly positioned (Figure 3). The patient admitted to frequently rubbing (ie, twiddling) the area, which led to twisting and dislocation of the device, a complication known as twiddler syndrome.
Twiddler syndrome is a rare complication of ICD placement, occurring in only about 0.1% of cases.1 It is usually a result of intentional manipulation (ie, twiddling) of the device by the patient, causing the ICD and the leads to dislodge, break, or retract. It is seen more often in women and in patients with cognitive dysfunction or psychiatric illness, in obese patients, and in patients with laxity of subcutaneous tissues, such as elderly people or people with accelerated weight loss. Placement of the ICD in an inappropriately large pocket predisposes to device rotation.2,3
Twiddler syndrome may cause mild discomfort or may even go unnoticed by the patient.4 In rare cases, it may lead to stimulation of the phrenic nerve, causing diaphragmatic pacing, or to stimulation of the brachial plexus, causing muscle twitching.2,3 However, malfunction of an ICD is life-threatening and requires immediate repair and replacement of the device. Educating the patient about the risks of twiddling the device site and the importance of periodic device checks is imperative to prevent this syndrome and ensure its early identification if it should occur. The patient should understand that dislodging the ICD can make it unable to sense abnormal rhythms and cause the device to deliver inappropriate shocks2,3,5 or stop delivering shocks altogether.
HOW IT IS TREATED
Treatment involves replacing the leads and affixing the device with sutures in the existing pocket or in a new pocket. The subpectoral position is preferred as it is more secure,3 although subcutaneous reimplantation has been successful.1 The device may also be placed in a fabric pouch to help lower the risk of migration or manipulation.3,6
In this case, because of the patient’s slender body habitus (body mass index 13 kg/m2), the ICD was removed from the subpectoral pocket and a new ICD was sutured into a subcutaneous pocket. The single lead was secured to the sternum and fascia using nonabsorbable sutures and a tie-down sleeve, making the device less susceptible to dislodgement by twiddling. Electrocardiography after replacement showed a normal sinus rhythm (Figure 4).
- Constandse J, Smit JJ, Ramdat Misier AR, Elvan A, Delnoy PP. Unusual twiddler syndrome: movement ties the knot. Neth Heart J 2013; 21:253–254.
- Spencker S, Poppelbaum A, Müller D. An unusual cause of oversensing leading to inappropriate ICD discharges. Int J Cardiol 2008; 129:e24–e26.
- Benezet-Mazuecos J, Benezet J, Ortega-Carnicer J. Pacemaker twiddler syndrome. Eur Heart J 2007; 28:2000.
- Chemello D, Subramanian A, Cameron D. Twiddler syndrome with 180 degrees rotation of an implantable cardioverter defibrillator generator resulting in malfunction of one of the shocking coils. Europace 2009; 11:1259.
- Garweg C, Alzand BS, Willems R. Twiddler syndrome causing an inappropriate implantable cardioverter-defibrillator shock. Eur Heart J 2014; 35:516.
- Parsonnet V, Bernstein AD, Neglia D, Omar A. The usefulness of a stretch-polyester pouch to encase implanted pacemakers and defibrillators. Pacing Clin Electrophysiol 1994; 17:2274–2278.
- Constandse J, Smit JJ, Ramdat Misier AR, Elvan A, Delnoy PP. Unusual twiddler syndrome: movement ties the knot. Neth Heart J 2013; 21:253–254.
- Spencker S, Poppelbaum A, Müller D. An unusual cause of oversensing leading to inappropriate ICD discharges. Int J Cardiol 2008; 129:e24–e26.
- Benezet-Mazuecos J, Benezet J, Ortega-Carnicer J. Pacemaker twiddler syndrome. Eur Heart J 2007; 28:2000.
- Chemello D, Subramanian A, Cameron D. Twiddler syndrome with 180 degrees rotation of an implantable cardioverter defibrillator generator resulting in malfunction of one of the shocking coils. Europace 2009; 11:1259.
- Garweg C, Alzand BS, Willems R. Twiddler syndrome causing an inappropriate implantable cardioverter-defibrillator shock. Eur Heart J 2014; 35:516.
- Parsonnet V, Bernstein AD, Neglia D, Omar A. The usefulness of a stretch-polyester pouch to encase implanted pacemakers and defibrillators. Pacing Clin Electrophysiol 1994; 17:2274–2278.
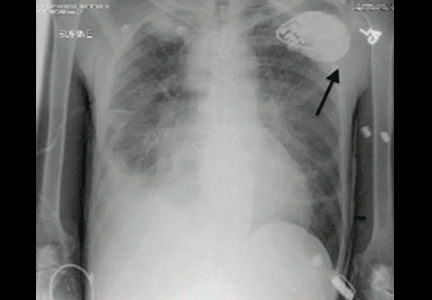
Retroperitoneal cyst hemorrhage in polycystic kidney disease
A 59-year-old man with autosomal dominant polycystic kidney disease (ADPKD), end-stage renal disease on hemodialysis, hypertension, and diverticulosis presented with acute pain in the left lower abdomen. The pain began 4 days previously, was dull and nonradiating, was relieved partially with hydrocodone-acetaminophen, and had no clear exacerbating factors. Two days before presentation, he developed a fever with chills. He reported no recent dysuria, diarrhea, hematuria, hematochezia, or melena. He had not been taking anticoagulants or nonsteroidal anti-inflammatory drugs, and he had no history of heavy lifting or trauma.
His temperature was 38.5˚C (101.3˚F), blood pressure 141/60 mm Hg (normal for this patient). On examination, his left lower quadrant was tender with voluntary guarding. Also present was a reducible ventral hernia, which was not new.
His hemoglobin level was 10.6 g/dL (reference range 13.0–17.0), which had dropped from a previous value of 13.7 g/dL.
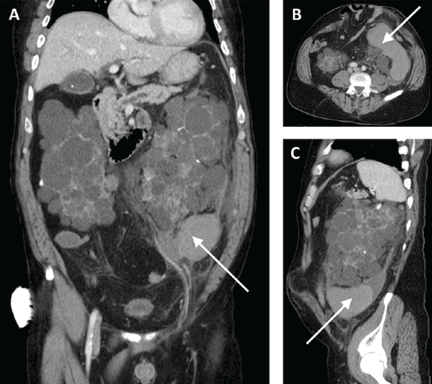
Computed tomography of the abdomen and pelvis revealed a ruptured retroperitoneal hemorrhagic cyst (Figure 1) in the inferior aspect of the left kidney extending into the fascia of Gerota.
Since his vital signs were stable, he was managed supportively during his hospitalization with intravenous fluids, serial hemoglobin checks, and analgesia. He was eventually discharged home in good condition.
CYST HEMORRHAGE IN POLYCYSTIC KIDNEY DISEASE
ADPKD is a relatively common, inherited systemic disease that leads to cyst formation, primarily in the kidneys but also in the liver (94%), seminal vesicles (40%), pancreas (9%), arachnoid membrane (8%), and spinal meningeal area (2%).1
In addition to cyst formation in multiple organs, ADPKD can have extrarenal manifestations such as connective-tissue abnormalities (including mitral valve prolapse) (25%), abdominal hernia (10%), and intracranial aneurysm (8%).1 Management of extrarenal complications of ADPKD is discussed in detail elsewhere.2
The estimated prevalence of ADPKD is 1 of every 400 to 1,000 live births. However, given that ADPKD is often clinically silent, it is diagnosed during the lifetime of fewer than half of people who have it.3
Most ADPKD cases are caused by mutations in either the PKD1 or PKD2 gene.4,5 Although the mechanism of cyst formation in ADPKD is still unclear, it is known that PKD1 and PKD2 encode proteins called polycystin-1 and polycystin-2, respectively. Polycystin-1 is a membrane protein found in renal tubular epithelia, hepatic bile ductules, and pancreatic ducts. Polycystin-2 is involved in cell calcium signaling and has been identified in the renal distal tubules, collecting duct, and thick ascending limb. Mutations in PKD1 and PKD2 are thought to contribute to cyst formation, with PKD1 mutations associated with earlier onset and more severe development of renal and extrarenal cysts.
Cyst hemorrhage
Hemorrhage of renal cysts is a well-known complication, occurring in up to 70% of patients with ADPKD.6 Renal cyst hemorrhage often presents clinically as flank pain with point tenderness or hematuria, or both. Flank pain results from hemorrhage into a cyst with consequent distention of the renal capsule, whereas hematuria results from rupture of a cyst into the collecting system.
Spontaneous nonfatal retroperitoneal cyst hemorrhage, as in our patient, is rare. Indeed, in one series reviewing the abdominal computed tomographic findings of 66 patients with ADPKD, only 2 patients (3%) had perinephric hematomas in the absence of recent trauma.6
Management of cyst hemorrhage is primarily conservative. Pain associated with cyst hemorrhage is managed conservatively with bed rest, intravenous hydration, and analgesics (but not nonsteroidal anti-inflammatory drugs).
Hematuria is also managed conservatively with bedrest and intravenous hydration, and most episodes of hematuria are self-limiting and last 2 to 7 days. However, if excessive bleeding occurs, the patient may be at risk of urinary tract obstruction from clot formation. If obstruction occurs and persists beyond 2 weeks, then ureteral stenting may be necessary. In rare cases of prolonged, severe bleeding with extensive subcapsular or retroperitoneal hematomas, patients require hospitalization, transfusion, or percutaneous transcatheter embolization of the renal artery. If such efforts are not successful, surgery, including nephrectomy, may be required to control the hemorrhage.2
Other causes of abdominal pain
In addition to renal cyst hemorrhage, the differential diagnosis of abdominal pain in a patient with ADPKD includes cyst enlargement causing stretching of the renal capsule or traction on the renal pedicle, cyst infection, nephrolithiasis, pyelonephritis, and rarely, tumors including renal cell carcinoma.
Unlike cyst rupture and hemorrhage, which are associated with point tenderness, cyst infection often manifests as diffuse, usually unilateral flank pain with associated fever, nausea, malaise, and leukocytosis. Our patient had none of these except for fever, which can also occur in cyst hemorrhage.
Nephrolithiasis occurs in up to 35% of patients with ADPKD,7 but no kidney stones were seen on computed tomography in our patient.
Pyelonephritis was unlikely in our patient, given that he had no significant white blood cells in his urinalysis and no leukocytosis.
Abdominal and pelvic imaging did not reveal any tumors in our patient.
- Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 2010; 17:173–180.
- Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant. In: Pagon RA, Adam MP, Bird TD, et al, editors. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993–2014.
- Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 2008; 359:1477–1485.
- Peters DJ, Spruit L, Saris JJ, et al. Chromosome 4 localization of a second gene for autosomal dominant polycystic kidney disease. Nat Genet 1993; 5:359–362.
- Rossetti S, Consugar MB, Chapman AB, et al; CRISP Consortium. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Levine E, Grantham JJ. Perinephric hemorrhage in autosomal dominant polycystic kidney disease: CT and MR findings. J Comput Assist Tomogr 1987; 11:108–111.
- Delaney VB, Adler S, Bruns FJ, Licinia M, Segel DP, Fraley DS. Autosomal dominant polycystic kidney disease: presentation, complications, and prognosis. Am J Kidney Dis 1985; 5:104–111.
A 59-year-old man with autosomal dominant polycystic kidney disease (ADPKD), end-stage renal disease on hemodialysis, hypertension, and diverticulosis presented with acute pain in the left lower abdomen. The pain began 4 days previously, was dull and nonradiating, was relieved partially with hydrocodone-acetaminophen, and had no clear exacerbating factors. Two days before presentation, he developed a fever with chills. He reported no recent dysuria, diarrhea, hematuria, hematochezia, or melena. He had not been taking anticoagulants or nonsteroidal anti-inflammatory drugs, and he had no history of heavy lifting or trauma.
His temperature was 38.5˚C (101.3˚F), blood pressure 141/60 mm Hg (normal for this patient). On examination, his left lower quadrant was tender with voluntary guarding. Also present was a reducible ventral hernia, which was not new.
His hemoglobin level was 10.6 g/dL (reference range 13.0–17.0), which had dropped from a previous value of 13.7 g/dL.
Computed tomography of the abdomen and pelvis revealed a ruptured retroperitoneal hemorrhagic cyst (Figure 1) in the inferior aspect of the left kidney extending into the fascia of Gerota.
Since his vital signs were stable, he was managed supportively during his hospitalization with intravenous fluids, serial hemoglobin checks, and analgesia. He was eventually discharged home in good condition.
CYST HEMORRHAGE IN POLYCYSTIC KIDNEY DISEASE
ADPKD is a relatively common, inherited systemic disease that leads to cyst formation, primarily in the kidneys but also in the liver (94%), seminal vesicles (40%), pancreas (9%), arachnoid membrane (8%), and spinal meningeal area (2%).1
In addition to cyst formation in multiple organs, ADPKD can have extrarenal manifestations such as connective-tissue abnormalities (including mitral valve prolapse) (25%), abdominal hernia (10%), and intracranial aneurysm (8%).1 Management of extrarenal complications of ADPKD is discussed in detail elsewhere.2
The estimated prevalence of ADPKD is 1 of every 400 to 1,000 live births. However, given that ADPKD is often clinically silent, it is diagnosed during the lifetime of fewer than half of people who have it.3
Most ADPKD cases are caused by mutations in either the PKD1 or PKD2 gene.4,5 Although the mechanism of cyst formation in ADPKD is still unclear, it is known that PKD1 and PKD2 encode proteins called polycystin-1 and polycystin-2, respectively. Polycystin-1 is a membrane protein found in renal tubular epithelia, hepatic bile ductules, and pancreatic ducts. Polycystin-2 is involved in cell calcium signaling and has been identified in the renal distal tubules, collecting duct, and thick ascending limb. Mutations in PKD1 and PKD2 are thought to contribute to cyst formation, with PKD1 mutations associated with earlier onset and more severe development of renal and extrarenal cysts.
Cyst hemorrhage
Hemorrhage of renal cysts is a well-known complication, occurring in up to 70% of patients with ADPKD.6 Renal cyst hemorrhage often presents clinically as flank pain with point tenderness or hematuria, or both. Flank pain results from hemorrhage into a cyst with consequent distention of the renal capsule, whereas hematuria results from rupture of a cyst into the collecting system.
Spontaneous nonfatal retroperitoneal cyst hemorrhage, as in our patient, is rare. Indeed, in one series reviewing the abdominal computed tomographic findings of 66 patients with ADPKD, only 2 patients (3%) had perinephric hematomas in the absence of recent trauma.6
Management of cyst hemorrhage is primarily conservative. Pain associated with cyst hemorrhage is managed conservatively with bed rest, intravenous hydration, and analgesics (but not nonsteroidal anti-inflammatory drugs).
Hematuria is also managed conservatively with bedrest and intravenous hydration, and most episodes of hematuria are self-limiting and last 2 to 7 days. However, if excessive bleeding occurs, the patient may be at risk of urinary tract obstruction from clot formation. If obstruction occurs and persists beyond 2 weeks, then ureteral stenting may be necessary. In rare cases of prolonged, severe bleeding with extensive subcapsular or retroperitoneal hematomas, patients require hospitalization, transfusion, or percutaneous transcatheter embolization of the renal artery. If such efforts are not successful, surgery, including nephrectomy, may be required to control the hemorrhage.2
Other causes of abdominal pain
In addition to renal cyst hemorrhage, the differential diagnosis of abdominal pain in a patient with ADPKD includes cyst enlargement causing stretching of the renal capsule or traction on the renal pedicle, cyst infection, nephrolithiasis, pyelonephritis, and rarely, tumors including renal cell carcinoma.
Unlike cyst rupture and hemorrhage, which are associated with point tenderness, cyst infection often manifests as diffuse, usually unilateral flank pain with associated fever, nausea, malaise, and leukocytosis. Our patient had none of these except for fever, which can also occur in cyst hemorrhage.
Nephrolithiasis occurs in up to 35% of patients with ADPKD,7 but no kidney stones were seen on computed tomography in our patient.
Pyelonephritis was unlikely in our patient, given that he had no significant white blood cells in his urinalysis and no leukocytosis.
Abdominal and pelvic imaging did not reveal any tumors in our patient.
A 59-year-old man with autosomal dominant polycystic kidney disease (ADPKD), end-stage renal disease on hemodialysis, hypertension, and diverticulosis presented with acute pain in the left lower abdomen. The pain began 4 days previously, was dull and nonradiating, was relieved partially with hydrocodone-acetaminophen, and had no clear exacerbating factors. Two days before presentation, he developed a fever with chills. He reported no recent dysuria, diarrhea, hematuria, hematochezia, or melena. He had not been taking anticoagulants or nonsteroidal anti-inflammatory drugs, and he had no history of heavy lifting or trauma.
His temperature was 38.5˚C (101.3˚F), blood pressure 141/60 mm Hg (normal for this patient). On examination, his left lower quadrant was tender with voluntary guarding. Also present was a reducible ventral hernia, which was not new.
His hemoglobin level was 10.6 g/dL (reference range 13.0–17.0), which had dropped from a previous value of 13.7 g/dL.
Computed tomography of the abdomen and pelvis revealed a ruptured retroperitoneal hemorrhagic cyst (Figure 1) in the inferior aspect of the left kidney extending into the fascia of Gerota.
Since his vital signs were stable, he was managed supportively during his hospitalization with intravenous fluids, serial hemoglobin checks, and analgesia. He was eventually discharged home in good condition.
CYST HEMORRHAGE IN POLYCYSTIC KIDNEY DISEASE
ADPKD is a relatively common, inherited systemic disease that leads to cyst formation, primarily in the kidneys but also in the liver (94%), seminal vesicles (40%), pancreas (9%), arachnoid membrane (8%), and spinal meningeal area (2%).1
In addition to cyst formation in multiple organs, ADPKD can have extrarenal manifestations such as connective-tissue abnormalities (including mitral valve prolapse) (25%), abdominal hernia (10%), and intracranial aneurysm (8%).1 Management of extrarenal complications of ADPKD is discussed in detail elsewhere.2
The estimated prevalence of ADPKD is 1 of every 400 to 1,000 live births. However, given that ADPKD is often clinically silent, it is diagnosed during the lifetime of fewer than half of people who have it.3
Most ADPKD cases are caused by mutations in either the PKD1 or PKD2 gene.4,5 Although the mechanism of cyst formation in ADPKD is still unclear, it is known that PKD1 and PKD2 encode proteins called polycystin-1 and polycystin-2, respectively. Polycystin-1 is a membrane protein found in renal tubular epithelia, hepatic bile ductules, and pancreatic ducts. Polycystin-2 is involved in cell calcium signaling and has been identified in the renal distal tubules, collecting duct, and thick ascending limb. Mutations in PKD1 and PKD2 are thought to contribute to cyst formation, with PKD1 mutations associated with earlier onset and more severe development of renal and extrarenal cysts.
Cyst hemorrhage
Hemorrhage of renal cysts is a well-known complication, occurring in up to 70% of patients with ADPKD.6 Renal cyst hemorrhage often presents clinically as flank pain with point tenderness or hematuria, or both. Flank pain results from hemorrhage into a cyst with consequent distention of the renal capsule, whereas hematuria results from rupture of a cyst into the collecting system.
Spontaneous nonfatal retroperitoneal cyst hemorrhage, as in our patient, is rare. Indeed, in one series reviewing the abdominal computed tomographic findings of 66 patients with ADPKD, only 2 patients (3%) had perinephric hematomas in the absence of recent trauma.6
Management of cyst hemorrhage is primarily conservative. Pain associated with cyst hemorrhage is managed conservatively with bed rest, intravenous hydration, and analgesics (but not nonsteroidal anti-inflammatory drugs).
Hematuria is also managed conservatively with bedrest and intravenous hydration, and most episodes of hematuria are self-limiting and last 2 to 7 days. However, if excessive bleeding occurs, the patient may be at risk of urinary tract obstruction from clot formation. If obstruction occurs and persists beyond 2 weeks, then ureteral stenting may be necessary. In rare cases of prolonged, severe bleeding with extensive subcapsular or retroperitoneal hematomas, patients require hospitalization, transfusion, or percutaneous transcatheter embolization of the renal artery. If such efforts are not successful, surgery, including nephrectomy, may be required to control the hemorrhage.2
Other causes of abdominal pain
In addition to renal cyst hemorrhage, the differential diagnosis of abdominal pain in a patient with ADPKD includes cyst enlargement causing stretching of the renal capsule or traction on the renal pedicle, cyst infection, nephrolithiasis, pyelonephritis, and rarely, tumors including renal cell carcinoma.
Unlike cyst rupture and hemorrhage, which are associated with point tenderness, cyst infection often manifests as diffuse, usually unilateral flank pain with associated fever, nausea, malaise, and leukocytosis. Our patient had none of these except for fever, which can also occur in cyst hemorrhage.
Nephrolithiasis occurs in up to 35% of patients with ADPKD,7 but no kidney stones were seen on computed tomography in our patient.
Pyelonephritis was unlikely in our patient, given that he had no significant white blood cells in his urinalysis and no leukocytosis.
Abdominal and pelvic imaging did not reveal any tumors in our patient.
- Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 2010; 17:173–180.
- Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant. In: Pagon RA, Adam MP, Bird TD, et al, editors. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993–2014.
- Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 2008; 359:1477–1485.
- Peters DJ, Spruit L, Saris JJ, et al. Chromosome 4 localization of a second gene for autosomal dominant polycystic kidney disease. Nat Genet 1993; 5:359–362.
- Rossetti S, Consugar MB, Chapman AB, et al; CRISP Consortium. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Levine E, Grantham JJ. Perinephric hemorrhage in autosomal dominant polycystic kidney disease: CT and MR findings. J Comput Assist Tomogr 1987; 11:108–111.
- Delaney VB, Adler S, Bruns FJ, Licinia M, Segel DP, Fraley DS. Autosomal dominant polycystic kidney disease: presentation, complications, and prognosis. Am J Kidney Dis 1985; 5:104–111.
- Pirson Y. Extrarenal manifestations of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis 2010; 17:173–180.
- Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant. In: Pagon RA, Adam MP, Bird TD, et al, editors. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993–2014.
- Grantham JJ. Clinical practice. Autosomal dominant polycystic kidney disease. N Engl J Med 2008; 359:1477–1485.
- Peters DJ, Spruit L, Saris JJ, et al. Chromosome 4 localization of a second gene for autosomal dominant polycystic kidney disease. Nat Genet 1993; 5:359–362.
- Rossetti S, Consugar MB, Chapman AB, et al; CRISP Consortium. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Levine E, Grantham JJ. Perinephric hemorrhage in autosomal dominant polycystic kidney disease: CT and MR findings. J Comput Assist Tomogr 1987; 11:108–111.
- Delaney VB, Adler S, Bruns FJ, Licinia M, Segel DP, Fraley DS. Autosomal dominant polycystic kidney disease: presentation, complications, and prognosis. Am J Kidney Dis 1985; 5:104–111.
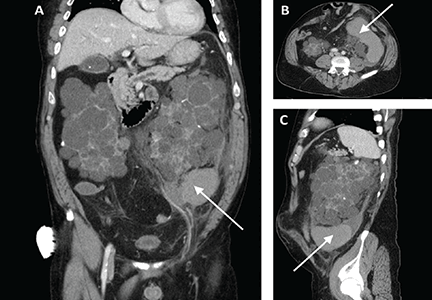
Joint pain in a man with lung cancer
A 46-year-old man with a 50-pack-year smoking history, hepatitis C, and polysubstance abuse presented with worsening joint pain over the past 2 months involving both knees, ankles, wrists, and hands. On examination, along with clubbing of the fingers and toes, he had tenderness and restriction in range of motion in all the affected joints. A fixed, nontender, hard mass was noted in the left axilla extending into the infraclavicular and supraclavicular regions.
Computed tomography (CT) with contrast revealed a mass in the left upper lobe of the lung, eroding the ribs and extending into the axilla, consistent with a suspected diagnosis of lung cancer (Figure 1). Radiography of the hands and feet showed thickening of the periosteal membrane and periosteal formation of new bone (Figure 2). A bone scan revealed diffuse increased uptake in the affected joints with no metastases (Figure 3). A workup for brain and adrenal metastases was negative. CT-guided needle biopsy confirmed adenocarcinoma of the lung.
The final diagnosis was hypertrophic pulmonary osteoarthropathy (HPOA) secondary to stage IIIB: T4N2M0 adenocarcinoma of the lung. Resection of the lung mass was deemed difficult, so the patient was scheduled for neoadjuvant concurrent chemoradiation therapy before resection. Ibuprofen and intravenous pamidronate provided symptom relief.
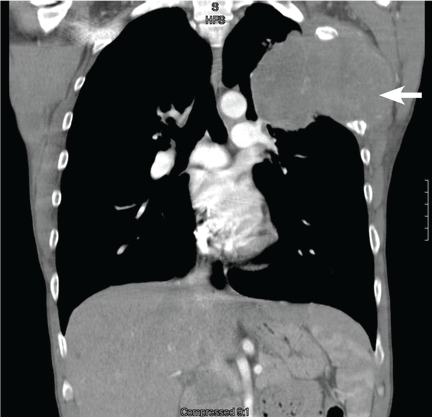
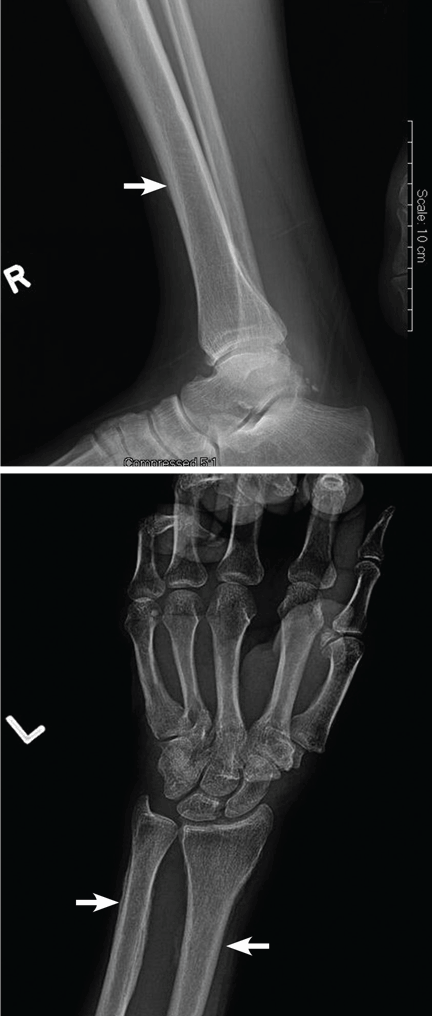
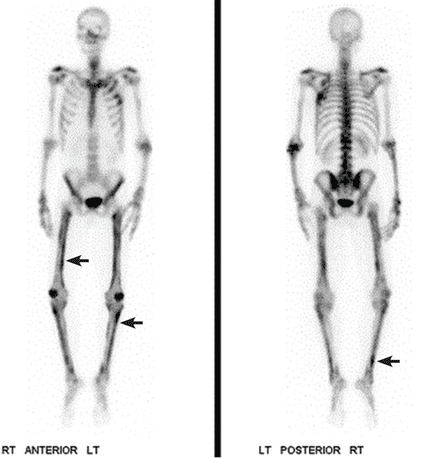
HYPERTROPHIC PULMONARY OSTEOARTHROPATHY
HPOA is characterized by abnormal proliferation of the skin and periosteal formation of new bone at the distal ends of long bones, metatarsals, metacarpals, and proximal phalanges.
Primary HPOA is genetically inherited and accounts for 5% of cases, while secondary HPOA, accounting for the other 95%, is a paraneoplastic syndrome.1 Secondary HPOA is most commonly associated with lung cancer, usually stage III or IV adenocarcinoma.1 However, only 1% of lung cancer patients develop HPOA.1
Other conditions linked to secondary HPOA are mesothelioma, renal cell carcinoma, gastrointestinal cancers, melanoma, thymic cancer, nasopharyngeal cancers, and Hodgkin lymphoma. HPOA is also associated with pulmonary infection, cystic fibrosis, and right-to-left cardiac shunt. Risk factors associated with secondary HPOA are smoking, male sex, and age greater than 55.
Patients often present with unremitting pain, edema, and erythema in the extremities. Radiography reveals periosteal membrane thickening and periosteal new bone formation.1 Findings on bone scintigraphy that confirm the diagnosis include diffuse increased uptake and bracelet-like uptake.
The prognosis and treatment of secondary HPOA are directly related to the underlying disease. Symptoms can be relieved with nonsteroidal anti-inflammatory drugs, bisphosphonates, and octreotide. Unilateral vagotomy is an option in refractory cases.1,2
- Ito T, Goto K, Yoh K, et al. Hypertrophic pulmonary osteoarthropathy as a paraneoplastic manifestation of lung cancer. J Thorac Oncol 2010; 5:976–980.
- Davis MC, Sherry V. Hypertrophic osteoarthropathy as a clinical manifestation of lung cancer. Clin J Oncol Nurs 2011; 15:561–563.
A 46-year-old man with a 50-pack-year smoking history, hepatitis C, and polysubstance abuse presented with worsening joint pain over the past 2 months involving both knees, ankles, wrists, and hands. On examination, along with clubbing of the fingers and toes, he had tenderness and restriction in range of motion in all the affected joints. A fixed, nontender, hard mass was noted in the left axilla extending into the infraclavicular and supraclavicular regions.
Computed tomography (CT) with contrast revealed a mass in the left upper lobe of the lung, eroding the ribs and extending into the axilla, consistent with a suspected diagnosis of lung cancer (Figure 1). Radiography of the hands and feet showed thickening of the periosteal membrane and periosteal formation of new bone (Figure 2). A bone scan revealed diffuse increased uptake in the affected joints with no metastases (Figure 3). A workup for brain and adrenal metastases was negative. CT-guided needle biopsy confirmed adenocarcinoma of the lung.
The final diagnosis was hypertrophic pulmonary osteoarthropathy (HPOA) secondary to stage IIIB: T4N2M0 adenocarcinoma of the lung. Resection of the lung mass was deemed difficult, so the patient was scheduled for neoadjuvant concurrent chemoradiation therapy before resection. Ibuprofen and intravenous pamidronate provided symptom relief.
HYPERTROPHIC PULMONARY OSTEOARTHROPATHY
HPOA is characterized by abnormal proliferation of the skin and periosteal formation of new bone at the distal ends of long bones, metatarsals, metacarpals, and proximal phalanges.
Primary HPOA is genetically inherited and accounts for 5% of cases, while secondary HPOA, accounting for the other 95%, is a paraneoplastic syndrome.1 Secondary HPOA is most commonly associated with lung cancer, usually stage III or IV adenocarcinoma.1 However, only 1% of lung cancer patients develop HPOA.1
Other conditions linked to secondary HPOA are mesothelioma, renal cell carcinoma, gastrointestinal cancers, melanoma, thymic cancer, nasopharyngeal cancers, and Hodgkin lymphoma. HPOA is also associated with pulmonary infection, cystic fibrosis, and right-to-left cardiac shunt. Risk factors associated with secondary HPOA are smoking, male sex, and age greater than 55.
Patients often present with unremitting pain, edema, and erythema in the extremities. Radiography reveals periosteal membrane thickening and periosteal new bone formation.1 Findings on bone scintigraphy that confirm the diagnosis include diffuse increased uptake and bracelet-like uptake.
The prognosis and treatment of secondary HPOA are directly related to the underlying disease. Symptoms can be relieved with nonsteroidal anti-inflammatory drugs, bisphosphonates, and octreotide. Unilateral vagotomy is an option in refractory cases.1,2
A 46-year-old man with a 50-pack-year smoking history, hepatitis C, and polysubstance abuse presented with worsening joint pain over the past 2 months involving both knees, ankles, wrists, and hands. On examination, along with clubbing of the fingers and toes, he had tenderness and restriction in range of motion in all the affected joints. A fixed, nontender, hard mass was noted in the left axilla extending into the infraclavicular and supraclavicular regions.
Computed tomography (CT) with contrast revealed a mass in the left upper lobe of the lung, eroding the ribs and extending into the axilla, consistent with a suspected diagnosis of lung cancer (Figure 1). Radiography of the hands and feet showed thickening of the periosteal membrane and periosteal formation of new bone (Figure 2). A bone scan revealed diffuse increased uptake in the affected joints with no metastases (Figure 3). A workup for brain and adrenal metastases was negative. CT-guided needle biopsy confirmed adenocarcinoma of the lung.
The final diagnosis was hypertrophic pulmonary osteoarthropathy (HPOA) secondary to stage IIIB: T4N2M0 adenocarcinoma of the lung. Resection of the lung mass was deemed difficult, so the patient was scheduled for neoadjuvant concurrent chemoradiation therapy before resection. Ibuprofen and intravenous pamidronate provided symptom relief.
HYPERTROPHIC PULMONARY OSTEOARTHROPATHY
HPOA is characterized by abnormal proliferation of the skin and periosteal formation of new bone at the distal ends of long bones, metatarsals, metacarpals, and proximal phalanges.
Primary HPOA is genetically inherited and accounts for 5% of cases, while secondary HPOA, accounting for the other 95%, is a paraneoplastic syndrome.1 Secondary HPOA is most commonly associated with lung cancer, usually stage III or IV adenocarcinoma.1 However, only 1% of lung cancer patients develop HPOA.1
Other conditions linked to secondary HPOA are mesothelioma, renal cell carcinoma, gastrointestinal cancers, melanoma, thymic cancer, nasopharyngeal cancers, and Hodgkin lymphoma. HPOA is also associated with pulmonary infection, cystic fibrosis, and right-to-left cardiac shunt. Risk factors associated with secondary HPOA are smoking, male sex, and age greater than 55.
Patients often present with unremitting pain, edema, and erythema in the extremities. Radiography reveals periosteal membrane thickening and periosteal new bone formation.1 Findings on bone scintigraphy that confirm the diagnosis include diffuse increased uptake and bracelet-like uptake.
The prognosis and treatment of secondary HPOA are directly related to the underlying disease. Symptoms can be relieved with nonsteroidal anti-inflammatory drugs, bisphosphonates, and octreotide. Unilateral vagotomy is an option in refractory cases.1,2
- Ito T, Goto K, Yoh K, et al. Hypertrophic pulmonary osteoarthropathy as a paraneoplastic manifestation of lung cancer. J Thorac Oncol 2010; 5:976–980.
- Davis MC, Sherry V. Hypertrophic osteoarthropathy as a clinical manifestation of lung cancer. Clin J Oncol Nurs 2011; 15:561–563.
- Ito T, Goto K, Yoh K, et al. Hypertrophic pulmonary osteoarthropathy as a paraneoplastic manifestation of lung cancer. J Thorac Oncol 2010; 5:976–980.
- Davis MC, Sherry V. Hypertrophic osteoarthropathy as a clinical manifestation of lung cancer. Clin J Oncol Nurs 2011; 15:561–563.
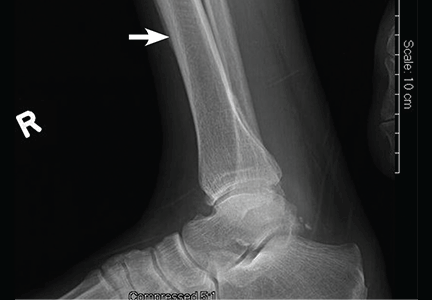
Sarcoidal infiltration of tattoos
A 42-year-old woman presented with painless lesions on her eyebrows that had been progressively growing for the past 3 months. Inspection and palpation revealed reddish papules and nodules on both eyebrows (Figure 1) and on the lips. The remainder of the physical examination was normal.
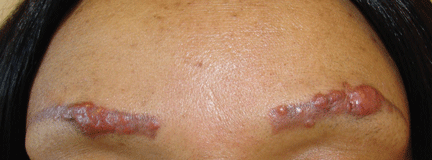
The patient said she had undergone cosmetic tattooing of her eyebrows and lips 10 years previously. She had no other significant medical history.
Her complete blood cell count and biochemistry and immunologic test panels were normal except for a high angiotensin-converting enzyme level.
Suspecting that the lesions represented sarcoidal infiltration of the tattoos, we obtained a biopsy. Histopathologic study showed multiple dermal noncaseating granulomas of epithelioid cells, together with polarizable foreign material and pigmented granules (Figure 2).
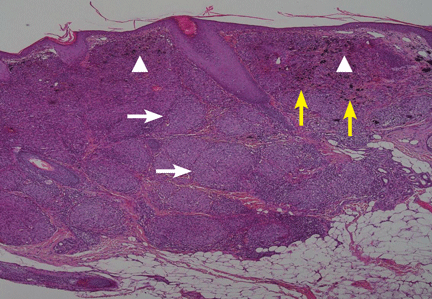
Thoracic multislice computed tomography revealed multiple areas of lymphadenopathy in the mediastinum and both lung hila, with a perilymphatic micronodular interstitial pattern and with thickened and nodular bronchial walls (Figure 3). The patient’s condition was diagnosed as systemic sarcoidosis presenting as sarcoidal infiltration of the tattooed areas.
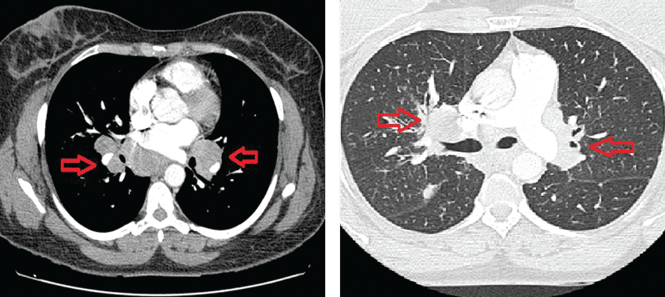
Treatment with oral prednisone 20 mg daily and monthly intralesional injections of triamcinolone 12 ng/mL in the eyebrows and lips resulted in clinical improvement after 3 months (Figure 4).
CUTANEOUS SARCOIDOSIS
Sarcoidosis is a multisystemic granulomatous disease characterized by hyperactivity of the cellular immune system. It usually appears between ages 25 and 35 and is more severe in African Americans.1 About one-third of patients with systemic sarcoidosis develop cutaneous lesions, and these may be the first sign of this disease.
Specific cutaneous lesions contain noncaseating granulomas. They manifest as reddish-brown papules, plaques, and macules and may appear over scars and tattoos.2,3 Other, nonspecific manifestations include erythema nodosum, erythema multiforme, nail clubbing, and Sweet syndrome.4
Diascopy and dermoscopy may be useful in diagnosing cutaneous sarcoidosis. The lesions typically have a characteristic yellowish-brown (“apple-jelly”) color.5
Sarcoidosis is a diagnosis of exclusion. It is suspected clinically, and histologic, radiologic, and analytical tests are indicated. Laboratory evaluation may show an elevated serum angiotensin-converting enzyme level, but this finding alone is not sensitive enough to be useful for diagnosis.6
The treatment and prognosis of skin sarcoidosis depend on the degree of systemic involvement. Localized cutaneous lesions can respond to intralesional corticosteroid injections and tacrolimus 0.1% ointment.7 However, if there is systemic involvement, low-dose prednisolone and weekly methotrexate should be started. This combination has few adverse effects.8 Recently, improvement of cutaneous and systemic sarcoidosis has been observed with agents that block tumor necrosis factor alpha.4
1. English JC 3rd, Patel PJ, Greer KE. Sarcoidosis. J Am Acad Dermatol 2001; 44:725–743.
2. Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol 2005; 141:869–872.
3. Anolik R, Mandal R, Franks AG Jr. Sarcoidal tattoo granuloma. Dermatol Online J 2010; 16:19.
4. Su Ö, Onsun N, Topukçu B, Özçelik HK, Çakıter AU, Büyükpınarbaşılı N. Disseminated scar sarcoidosis may predict pulmonary involvement in sarcoidosis. Acta Dermatovenerol Alp Pannonica Adriat 2013; 22:71–74.
5. Hunt RD, Gonzalez ME, Robinson M, Meehan SA, Franks AG Jr. Ulcerative sarcoidosis. Dermatol Online J 2012; 18:29.
6. Baughman RP, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med 2011; 183:573–581.
7. Landers MC, Skokan M, Law S, Storrs FJ. Cutaneous and pulmonary sarcoidosis in association with tattoos. Cutis 2005; 75:44–48.
8. Nagai S, Yokomatsu T, Tanizawa K, et al. Treatment with methotrexate and low-dose corticosteroids in sarcoidosis patients with cardiac lesions. Intern Med 2014; 53:427–433.
A 42-year-old woman presented with painless lesions on her eyebrows that had been progressively growing for the past 3 months. Inspection and palpation revealed reddish papules and nodules on both eyebrows (Figure 1) and on the lips. The remainder of the physical examination was normal.
The patient said she had undergone cosmetic tattooing of her eyebrows and lips 10 years previously. She had no other significant medical history.
Her complete blood cell count and biochemistry and immunologic test panels were normal except for a high angiotensin-converting enzyme level.
Suspecting that the lesions represented sarcoidal infiltration of the tattoos, we obtained a biopsy. Histopathologic study showed multiple dermal noncaseating granulomas of epithelioid cells, together with polarizable foreign material and pigmented granules (Figure 2).
Thoracic multislice computed tomography revealed multiple areas of lymphadenopathy in the mediastinum and both lung hila, with a perilymphatic micronodular interstitial pattern and with thickened and nodular bronchial walls (Figure 3). The patient’s condition was diagnosed as systemic sarcoidosis presenting as sarcoidal infiltration of the tattooed areas.
Treatment with oral prednisone 20 mg daily and monthly intralesional injections of triamcinolone 12 ng/mL in the eyebrows and lips resulted in clinical improvement after 3 months (Figure 4).
CUTANEOUS SARCOIDOSIS
Sarcoidosis is a multisystemic granulomatous disease characterized by hyperactivity of the cellular immune system. It usually appears between ages 25 and 35 and is more severe in African Americans.1 About one-third of patients with systemic sarcoidosis develop cutaneous lesions, and these may be the first sign of this disease.
Specific cutaneous lesions contain noncaseating granulomas. They manifest as reddish-brown papules, plaques, and macules and may appear over scars and tattoos.2,3 Other, nonspecific manifestations include erythema nodosum, erythema multiforme, nail clubbing, and Sweet syndrome.4
Diascopy and dermoscopy may be useful in diagnosing cutaneous sarcoidosis. The lesions typically have a characteristic yellowish-brown (“apple-jelly”) color.5
Sarcoidosis is a diagnosis of exclusion. It is suspected clinically, and histologic, radiologic, and analytical tests are indicated. Laboratory evaluation may show an elevated serum angiotensin-converting enzyme level, but this finding alone is not sensitive enough to be useful for diagnosis.6
The treatment and prognosis of skin sarcoidosis depend on the degree of systemic involvement. Localized cutaneous lesions can respond to intralesional corticosteroid injections and tacrolimus 0.1% ointment.7 However, if there is systemic involvement, low-dose prednisolone and weekly methotrexate should be started. This combination has few adverse effects.8 Recently, improvement of cutaneous and systemic sarcoidosis has been observed with agents that block tumor necrosis factor alpha.4
A 42-year-old woman presented with painless lesions on her eyebrows that had been progressively growing for the past 3 months. Inspection and palpation revealed reddish papules and nodules on both eyebrows (Figure 1) and on the lips. The remainder of the physical examination was normal.
The patient said she had undergone cosmetic tattooing of her eyebrows and lips 10 years previously. She had no other significant medical history.
Her complete blood cell count and biochemistry and immunologic test panels were normal except for a high angiotensin-converting enzyme level.
Suspecting that the lesions represented sarcoidal infiltration of the tattoos, we obtained a biopsy. Histopathologic study showed multiple dermal noncaseating granulomas of epithelioid cells, together with polarizable foreign material and pigmented granules (Figure 2).
Thoracic multislice computed tomography revealed multiple areas of lymphadenopathy in the mediastinum and both lung hila, with a perilymphatic micronodular interstitial pattern and with thickened and nodular bronchial walls (Figure 3). The patient’s condition was diagnosed as systemic sarcoidosis presenting as sarcoidal infiltration of the tattooed areas.
Treatment with oral prednisone 20 mg daily and monthly intralesional injections of triamcinolone 12 ng/mL in the eyebrows and lips resulted in clinical improvement after 3 months (Figure 4).
CUTANEOUS SARCOIDOSIS
Sarcoidosis is a multisystemic granulomatous disease characterized by hyperactivity of the cellular immune system. It usually appears between ages 25 and 35 and is more severe in African Americans.1 About one-third of patients with systemic sarcoidosis develop cutaneous lesions, and these may be the first sign of this disease.
Specific cutaneous lesions contain noncaseating granulomas. They manifest as reddish-brown papules, plaques, and macules and may appear over scars and tattoos.2,3 Other, nonspecific manifestations include erythema nodosum, erythema multiforme, nail clubbing, and Sweet syndrome.4
Diascopy and dermoscopy may be useful in diagnosing cutaneous sarcoidosis. The lesions typically have a characteristic yellowish-brown (“apple-jelly”) color.5
Sarcoidosis is a diagnosis of exclusion. It is suspected clinically, and histologic, radiologic, and analytical tests are indicated. Laboratory evaluation may show an elevated serum angiotensin-converting enzyme level, but this finding alone is not sensitive enough to be useful for diagnosis.6
The treatment and prognosis of skin sarcoidosis depend on the degree of systemic involvement. Localized cutaneous lesions can respond to intralesional corticosteroid injections and tacrolimus 0.1% ointment.7 However, if there is systemic involvement, low-dose prednisolone and weekly methotrexate should be started. This combination has few adverse effects.8 Recently, improvement of cutaneous and systemic sarcoidosis has been observed with agents that block tumor necrosis factor alpha.4
1. English JC 3rd, Patel PJ, Greer KE. Sarcoidosis. J Am Acad Dermatol 2001; 44:725–743.
2. Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol 2005; 141:869–872.
3. Anolik R, Mandal R, Franks AG Jr. Sarcoidal tattoo granuloma. Dermatol Online J 2010; 16:19.
4. Su Ö, Onsun N, Topukçu B, Özçelik HK, Çakıter AU, Büyükpınarbaşılı N. Disseminated scar sarcoidosis may predict pulmonary involvement in sarcoidosis. Acta Dermatovenerol Alp Pannonica Adriat 2013; 22:71–74.
5. Hunt RD, Gonzalez ME, Robinson M, Meehan SA, Franks AG Jr. Ulcerative sarcoidosis. Dermatol Online J 2012; 18:29.
6. Baughman RP, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med 2011; 183:573–581.
7. Landers MC, Skokan M, Law S, Storrs FJ. Cutaneous and pulmonary sarcoidosis in association with tattoos. Cutis 2005; 75:44–48.
8. Nagai S, Yokomatsu T, Tanizawa K, et al. Treatment with methotrexate and low-dose corticosteroids in sarcoidosis patients with cardiac lesions. Intern Med 2014; 53:427–433.
1. English JC 3rd, Patel PJ, Greer KE. Sarcoidosis. J Am Acad Dermatol 2001; 44:725–743.
2. Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol 2005; 141:869–872.
3. Anolik R, Mandal R, Franks AG Jr. Sarcoidal tattoo granuloma. Dermatol Online J 2010; 16:19.
4. Su Ö, Onsun N, Topukçu B, Özçelik HK, Çakıter AU, Büyükpınarbaşılı N. Disseminated scar sarcoidosis may predict pulmonary involvement in sarcoidosis. Acta Dermatovenerol Alp Pannonica Adriat 2013; 22:71–74.
5. Hunt RD, Gonzalez ME, Robinson M, Meehan SA, Franks AG Jr. Ulcerative sarcoidosis. Dermatol Online J 2012; 18:29.
6. Baughman RP, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med 2011; 183:573–581.
7. Landers MC, Skokan M, Law S, Storrs FJ. Cutaneous and pulmonary sarcoidosis in association with tattoos. Cutis 2005; 75:44–48.
8. Nagai S, Yokomatsu T, Tanizawa K, et al. Treatment with methotrexate and low-dose corticosteroids in sarcoidosis patients with cardiac lesions. Intern Med 2014; 53:427–433.