User login
Advance Care Planning Among Patients with Heart Failure: A Review of Challenges and Approaches to Better Communication
From the Rand Corporation and UCLA Fielding School of Public Health, Santa Monica, CA (Dr. Ahluwalia) and University of Southern California, Leonard Davis School of Gerontology, Los Angeles, CA (Dr. Enguidanos).
Abstract
- Objective: To review the relevance of advance care planning to heart failure management, describe key advance care planning challenges, and provide clinicians with actionable guidance for engaging in advance care planning conversations.
- Methods: Review of the literature.
- Results: Although most patients with heart failure prefer to receive thorough and honest information about their health condition and prognosis, the unpredictability of the heart failure trajectory coupled with physician barriers including discomfort with emotionally-laden topics and difficulty identifying the “right” time to engage in advance care planning, and systems barriers such as inadequate clinic time and limited reimbursement, impede timely engagement in advance care planning discussions. Approaches to effective advance care planning communication include using open-ended questions to stimulate patient engagement, evaluating how much information the patient wants to ensure patient-centeredness, and using empathic language to demonstrate support and understanding. While successful models of advance care planning communication have been identified, replication is limited due to the resource intense nature of these approaches.
- Conclusion: Challenges to advance care planning discussions among patients with heart failure may be mitigated through the establishment of communication quality standards as well as guidelines promoting early and ongoing advance care planning discussions, as well as reimbursement for outpatient discussions.
Heart failure, a leading cause of death, disability, and health care costs in the United States, is an incurable and life-limiting illness that is becoming increasingly prevalent due to an aging population and improved life expectancy. Approximately 5.3 million Americans are currently living with heart failure [1], with more than 550,000 new cases diagnosed each year [2]. Heart failure disproportionately affects older adults; about 80% of all cases occur in persons aged 65 years or older [3], and heart failure is the leading cause of hospital admissions among older adults [4]. The burden and impact of heart failure peaks near the end of life; 80% of Medicare beneficiaries with heart failure are hospitalized in the last 6 months of life [5].
The Trajectory of Heart Failure
Patients with heart failure experience a highly variable, nonlinear clinical trajectory marked by progressive deterioration and frequent exacerbations requiring hospitalization [6]. Their prognosis, though uncertain, is poor, with reported 1-year mortality rates following a hospitalization between 30% and 50% and 5-year mortality as high as 75% [7–11], a survival rate worse than that of some cancers [12]. Patients with heart failure caused by ischemic heart disease are at high risk for sudden cardiac death, particularly at earlier stages of the disease, which can confound the ability to appropriately plan for the future [13]. Those who survive to more advanced stages of heart failure face worsening quality of life [14–16], driven by a high prevalence of fatigue, breathlessness, pain, and depression [17–24]. Indeed, patients with heart failure have a similar symptom burden to patients with advanced cancer [25]. Older adults with heart failure also have a high comorbidity burden that further complicates both symptomology and disease trajectory with implications for decision-making about life-prolonging heart failure therapies [26,27].
Advance Care Planning in Heart Failure
The unpredictable nature of heart failure makes it difficult for patients and families to plan and prepare for their future, yet it is this very uncertainty that makes advance care planning (ACP) so critical for heart failure patients. Clear and honest patient-clinician communication about ACP, including an exploration of patient values and goals for care in the context of prognostic information, is essential to patient-centered treatment decision-making [28]. This is particularly relevant in heart failure, where a range of high-intensity, invasive, and costly interventions are increasingly being applied (eg, ventricular assist devices) without equivalent attention to quality of life and patients’ long-term goals for care.
Patients with heart failure and their families face multiple complex treatment decisions along the trajectory of their illness, such as discontinuation of beta blockers among patients with refractory fluid overload or angiotensin-converting enzyme inhibitors in end-stage patients with symptomatic hypotension [29,30]. In end-stage heart failure patients, deactivation of an implantable cardiac defibrillator might be considered to avoid the pain and distress associated with repeated shocks. In contrast, other interventions such as cardiac resynchronization therapy and continuous inotropic infusion have quality of life benefits; continuation of these therapies may be appropriate even when discontinuing other interventions. Such decisions should be guided by a thorough understanding of the patient’s expressed preferences and values, ideally assessed early in the trajectory of the disease and continuously re-evaluated as the diseases progresses.
The American Heart Association supports early and regular patient-provider ACP discussions to guide heart failure patients’ future decision-making [31], and recommends that such discussions be initiated in the outpatient setting, prior to and in anticipation of clinical decline. ACP communication plays a critical role in enhancing patients’ understanding of their diagnosis, treatment, prognosis, and choices in end-of-life care [31]. ACP communication also helps the clinician to better understand the context within which patients and their caregivers might make health care decisions, including their values and preferences for care. Patient-provider discussions about ACP focused on understanding patient values and initiated early in the trajectory of serious illness can support future in-the-moment decision-making, and is likely more effective than asking patients to make specific treatment decisions in advance [32]. A growing body of rigorous research has shown that ACP communication is associated with greater preference-concordant care and congruence in patient-surrogate understanding of patient preferences, lower costs, and less aggressive care at the end of life [33–37].
Patient Preferences for ACP Communication
Most patients with heart failure and their caregivers want honest disclosure regarding prognosis and to receive information about the expected trajectory of their disease [38–41] as early as possible [38] to help them plan and prepare for their future. Patients and their caregivers prefer to have these conversations with their physician [38] or other provider most familiar with the patient and family [39]. Patients also express a preference for support with dealing with the uncertainty inherent to heart failure [39]. Although most patients and caregivers desire to receive clear and honest communication about their disease, it is important to note that patients may vary in the extent of information they prefer to receive about their heart failure, with some individuals preferring not to talk about the end of life and future care needs at all [39,42–44].
Challenges to ACP Communication in Heart Failure
Despite patient and caregiver preferences for ACP communication with their providers, evidence suggests such communication occurs infrequently [40,45] and that heart failure patients may lack important information about their prognosis and treatment options [40,44,46,47]. For example, patients may not recognize the terminal nature of heart failure, and may be unaware of the range of treatment options, including hospice, available to them. Evidence also demonstrates that ACP is infrequently discussed with their health care providers [40], resulting in these conversations being avoided or deferred until an emergent clinical situation [44,48] when hasty questions about treatment choices may yield uncertain and conflicting answers not representative of a patient’s underlying values.
The infrequent, late, and often lack of discussions about ACP are driven by several challenges. First, the uncertain trajectory of heart failure makes communication regarding “what to expect” difficult. Prognostication is an immense challenge in heart failure [40,49–52], making it harder to talk about end-of-life issues and hindering the ability of patients, caregivers and health care providers to plan and prepare for the future. It is often difficult for clinicians, who face the challenge of instilling hope in the face of truthful disclosure [53], to identify the “right time” to initiate such discussions.
Second, a lack of time, particularly during outpatient visits, impedes physician ability to have considered discussions about future care needs and preferences [32,54]. The U.S. health care system currently lacks financial reimbursement for these discussions, which poses a significant barrier to the integration of ACP conversations into routine clinical practice. Moreover, these conversations are lengthy and iterative [53]. ACP discussions that are focused on facilitating patient-centered decision-making ideally begin with a discussion of expected prognosis, followed by an exploration of patient preferences and values for health care, and then a review of treatment options to be considered in the context of those preferences. Often additional time is needed for completing advance directive documents or for charting key outcomes from these discussions. Clinicians today are frequently overloaded with addressing multiple medical issues during outpatient visits that leave little time for non-medical tasks such as ACP discussions. The lack of financial incentives to support in-depth discussions is a critical challenge in improving ACP.
Third, a lack of training in specialized communication skills, particularly focused on empathic and emotionally sensitive disclosure, may further hinder physicians from initiating frank discussions with their patients. ACP conversations are highly sensitive and fraught with emotional complexity, and clinicians understandably experience discomfort with breaking bad news [49,51,55] or with broader issues of decline and death [51,56,57]. Physicians tend to be most comfortable addressing cognitive aspects of communication; addressing the emotional needs of patients is harder. Medical school training teaches detachment in physician practice, perhaps as a way of coping with the sadness they regularly confront and in maintaining their ability to provide clinical care. In fact, physicians describe their most difficult encounters as those with the most negative expressed emotions and miss opportunities to respond with empathy [58–60], a critical skill in effective patient-physician communication that is associated with improved patient satisfaction [61,62]. While patients value good communication skills in their health care encounters, many providers feel they lack the necessary skills to lead effective ACP discussions [49,63].
Finally, information gaps with regards to heart failure contribute to delayed or absent conversations about planning for future care. Many heart failure patients have a limited understanding of their disease [32,40,44,55], particularly an inaccurate perception that heart failure is not a terminal and life-limiting illness [42,49,64]. Compounding this is the fact that even some health care providers are reluctant to acknowledge the terminal nature of heart failure [50,56]. Without frank acknowledgement of the terminal nature of heart failure, the initiation of discussions regarding end-of-life care will remain difficult if not impossible.
Approaches to ACP in Heart Failure

When Is the Right Time?
Given the complexity and unpredictable trajectory of heart failure, indicators of disease progression, including changes in health status and health service use, may serve as useful signals to help clinicians identify the appropriate time to initiate care planning discussions. Repeated hospital admissions for heart failure are strongly associated with increased mortality. In a sample of community heart failure patients [8], median survival after the first, second, and third hospitalization was 2.4, 1.4, and 1.0 years, respectively. In light of this, a patient with 1 or more hospitalizations in a 12-month period may be an appropriate candidate for an ACP conversation. Similarly, comorbidity in patients with heart failure may signal the relevance and need for discussions about future care. In a sample of Medicare beneficiaries with advanced heart failure, an increasing burden of comorbidity was associated with significantly higher mortality, as were certain conditions (COPD, CKD, dementia, depression) and combinations of conditions (eg, CKD and dementia) [26]. Davidson and colleagues [68] suggest a list of clinical indicators signaling the need for an ACP conversation, including any of the following:
- > 1 episodes of exacerbation of heart failure leading to hospital admission
- New York Heart Association Class IV heart failure
- Decline in function and mobility
- Unexplained weight loss
- Resting pulse rate greater than 100 beats/minute
- Raised serum creatinine (> 150 µmol/L)
- Low serum sodium (< 135 mmol/L)
- Low serum albumen (< 33 g/L)
- High dose of loop diuretic (eg, furosemide ≥ 160 mg daily)
Given the considerable complexity and multisystem nature of heart failure, none of these indicators alone can signal certainty about disease progression and consequent outcomes; however, they can serve as a useful heuristic for helping clinicians identify appropriate times to raise the topic of ACP with their patients.
What Do I Say? Structuring the Conversation
Heart failure patients and their caregivers may vary in their preferences for hearing information about their disease; therefore, it is critical to open any conversation about planning for future in the context of their illness by asking what and how much information is desired. This includes evaluating how involved in decision-making the patient wants to be. Previously suggested language includes [69,70]:
- Would you like to consider all the options, or my opinion about the options that fit best with what I know about you?
- Some people like to know everything about their disease and be involved in all decision making. Others do not want all the news and would rather the doctor talk to __________. Which kind of person are you? How involved do you want to be in these decisions?
- Would you like me to tell you the full details of your condition?
- If you prefer not to hear the details, is there someone in your family who you trust to receive this information?
After establishing the patient’s preferences for hearing different types of information and level of involvement in decision-making about their care, the ask-tell-ask model [69,71] provides a useful approach to communicating with patients and their families. The conversation generally begins by asking patients what he or she understands about their illness (eg, “What do you understand about your heart failure?”; “I want to make sure we’re on the same page; what have other doctors told you?”). Building on what the patient already knows, the clinician can then disclose new information, correct misunderstandings, or confirm impressions and expectations the patient might have. In this way, information is tailored to the patient’s understanding and aimed at addressing potential knowledge gaps, all within the context of their preferences. Finally, the clinician asks the patient to describe their new understanding and whether or not they have questions or concerns (eg, “To make sure I did a good job of explaining to you, can you tell me what you now know about your condition from our conversation?”; “I know I’ve covered a lot and I want to make sure I was clear. When you get home, how are you going to explain what I’ve told you to your spouse?”). This approach encourages communication and exchange between patient and physician. Additionally, expressions of concern promote relationship building and bonding between physician and patient.
Keeping the Conversation Going
ACP communication can cover a wide range of topics beyond disease and prognostic disclosure by the provider to the patient. A critical aspect of ACP conversations is an exploration of the patient’s values and preferences, which can be used to help contextualize treatment choices and subsequently guide in-the-moment decision-making [72]. Using open-ended questions throughout the conversation gives the patient an opportunity to reflect on and communicate their wishes and values and allows them to engage in the conversation on their own terms. Examples of discussion-stimulating questions include [69,73]:
- What concerns you most about your illness?
- How is treatment going for you (your family)?
- As you think about your illness, what is the best and worst that might happen?
- What are your greatest hopes about your health?
- What has been most difficult about this illness for you?
- Looking back at your life, what has been important to you?
- At this point, what is most important for you to do?
Language
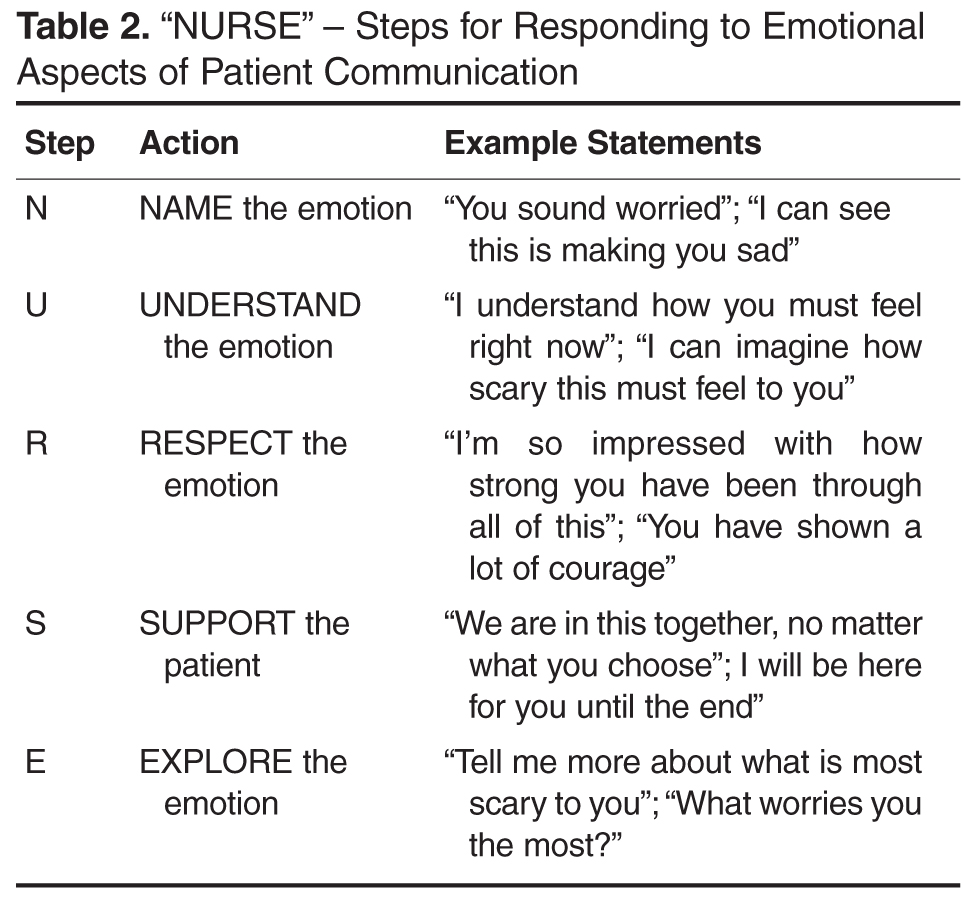
Central to this process is the use of empathic language to demonstrate support and understanding. An expression of empathy is also an appropriate way to acknowledge and share difficult emotions when it becomes hard to know where to take the conversation next. Quill and colleagues [74] suggest the following empathic responses to patients’ emotional expressions:
- I wish for that too
- It's unfortunate that things aren't different
- I am so sorry that this happened to you
- I understand how much you want that
- It must be very hard to accept the seriousness of this illness
Relatedly, the use of medical jargon in ACP conversations can increase the distance between patients and their providers, and may hinder patient understanding. Physicians may use technical language out of habit, or as an unconscious way to emotionally separate themselves from the task of delivering bad news. However, clear communication using layperson terms is the most effective approach to providing information necessary to patient-centered decision making. Explaining medical procedures in simple terms can improve understanding and help to build trust with the physician (eg, “We will perform an angioplasty – a procedure where a special tube with a balloon on the end of it is inserted into your artery to stretch it open. This will improve blood flow and relieve some of the symptoms you are currently experiencing”.)
Cultural Issues in Communication
There are various cultural issues to consider and address when conducting ACP discussions with heart failure patients and their families. Heart failure disproportionately affects certain racial and ethnic groups (eg, African Americans) [77–79], and effective management of heart failure depends on the provision of culturally sensitive information and facilitation of culturally informed self-care behaviors. There is evidence of cultural variation in preferences for information and role in decision-making. For example, most white and African-American patients prefer to be fully informed of their condition [80], whereas other cultures may focus on protecting the patient from difficult information in order to maintain hope [80–86]. Moreover, even in cultures where nondisclosure is preferred, patients may want to be told the truth in an indirect, euphemistic, or even nonverbal manner [80,87–89]. These complexities underscore the importance of taking a patient-centered approach to ACP communication, respecting individuality and autonomy while ultimately facilitating decision-making [90,91].
Are There Effective Training Programs for ACP Communication?
Effective communication skills are a critical component of ACP conversations between clinicians and their patients; however, most clinicians do not receive formal training in ACP communication and believe it to be a difficult task [92]. Strong evidence of the effectiveness of communication skills training has yet to be established, largely due to variation in the approach to training and the specification of relevant outcomes. For example, a systematic review of communication skills training courses found that some courses are effective at improving different types of communication skills related to providing support and gathering information, but these courses lacked effectiveness in improving patient satisfaction or provider burnout and distress [93]. Similarly, a range of approaches to teaching clinicians effective ACP communication skills early in their medical training have been identified [94], but considerable variation in quality preclude any conclusions from being drawn about their effectiveness.
Despite these challenges, there are some studies of communication skills training courses that have demonstrated the ability to increase providers’ use of empathic and facilitative communication (eg, use of open-ended questions) [58,95], and to increase self-efficacy and confidence among providers [96]. One particular teaching model that is increasingly used in cancer care is Oncotalk (http://depts.washington.edu/oncotalk/). Oncotalk has been shown to significantly increase clinical skills in giving bad news and facilitating the transition to palliative care. Building on this success, the program has expanded to training courses focused on the intensive care setting (http://depts.washington.edu/icutalk/) and geriatrics care [97–99]. It is important to note, however, that the considerable time and resource-intensive nature of communication training programs limits widespread implementation of any one approach into routine medical education. More attention to the type and structure of communication skills training programs are needed as well as scalable approaches to assist clinicians in developing effective ACP communication skills.
Policy Implications of ACP and Future Directions
There is growing recognition of the need to improve ACP among patients with seriously illness, including heart failure. In a recent Institute of Medicine (IOM) report, Dying in America [100], the need for clinician-patient communication about ACP was identified as a primary area of improvement. Recommendations include the establishment of communication quality standards as well as guidelines promoting early and ongoing ACP discussions. This is supported by recommendations from medical professional societies for an iterative model of ACP that follows the course of a serious illness [2,101]. At early stages of the illness, ACP might be focused on helping patients clarify their broad health care values and raise awareness of their disease and expected prognosis. As the condition progresses, ACP discussion might focus on exploring disease-specific treatment options within the context of previously expressed preferences, as well as identifying changes in patients’ values over time, particularly as they gain experience with their illness and health status changes [102]. In late stages of the disease, ACP might focus on documenting specific treatment choices (eg, DNR orders) and on exploring options such as palliative care, while also ensuring that patients and caregivers are appropriately prepared for imminent decline and death.
The IOM report also calls for payment reforms to include reimbursement for outpatient ACP discussions [100]. There is burgeoning national support for developing reimbursement models for ACP discussions. The American Medical Association has recently released current procedural terminology (CPT) codes for ACP services, a first step toward urging Medicare to consider reimbursement for ACP discussions with physicians.
Finally, the IOM report calls for improved education and training in ACP communication across all disciplines and specialties providing care to patients with serious illness. These recommendations bring national attention to the current limitations surrounding ACP discussions for those with serious illness, including heart failure. Further research is needed to identify methods and care models to address the gap in communication skills, processes, and policies.
Corresponding author: Sangeeta C. Ahluwalia, Rand Corporation, 1776 Main St., Santa Monica, CA, 90401, [email protected].
Financial disclosures: None.
Author contributions: conception and design, SCA, SE; analysis and interpretation of data, SCA, SE; drafting of article, SCA, SE; critical revision of the article, SCA, SE.
1. American Heart Association. Heart disease and stroke statistics—2008 update. [Internet]. Available at www.americanheart.org/downloadable/heart/1200078608862HS_Stats%202008.final.pdf.
2. Hunt SA, Abraham WT, Chin MH, et al. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult—summary article. Circulation 2005;112:1825–52.
3. Masoudi FA, Havranek EP, Krumholz HM. The burden of chronic congestive heart failure in older persons: magnitude and implications for policy and research. Heart Fail Rev 2002;7:9–16.
4. McMurray JJ PM. Heart failure. Lancet 2005;365:1877–89.
5. Unroe KT, Greiner MA, Hernandez AF, et al. Resource use in the last 6 months of life among medicare beneficiaries with heart failure, 2000-2007. Arch Intern Med 2011;171:196–203.
6. Lunney JR, Lynn J, Foley DJ, et al. Patterns of functional decline at the end of life. JAMA 2003;289:2387–92.
7. Ko D, Alter D, Austin P, et al. Life expectancy after an index hospitalization for patients with heart failure: A population-based study. Am Heart J 2008;155:324–31.
8. Setoguchi S, Stevenson LW, Schneeweiss S. Repeated hospitalizations predict mortality in the community population with heart failure. Am Heart J 2007;154:260–6.
9. Jong P, Vowinskel E, Liu PP, et al. Prognosis and determinants for survival in patients newly hospitalized for heart failure. Arch Intern Med 2002;162:1689-94.
10. Thom T, Haase N, Rodamond W, et al. Heart disease and stroke statistics- 2006 update. Circulation 2006;113:e85-e151.
11. Shahar E, Lee S, Kim J, et al. Hospitalized heart failure: Rates and long-term mortality. J Card Fail 2004;10:374–9.
12. Kirkpatrick JN, Guger CJ, Arnsdorf MF, et al. Advance directives in the cardiac care unit. Am Heart J 2007;154:477–81.
13. Orn S, Dickstein K. How do heart failure patients die? Eur Heart J. 2002;4(suppl D).
14. Juenger J, Schellberg D, Kraemer S, et al. Health related quality of life in patients with congestive heart failure: comparison with other chronic disease and relation to functional variables. Heart 2002;87:235–41.
15. Steptoe A, Mohabir A, Mahon NG, et al. Health related quality of life and psychological wellbeing in patients with dilated cardiomyopathy. Heart 2000;83:645–50.
16. Johansson P, Agnebrink M, Dahlstrom U, et al. Measurement of health-related quality of life in chronic heart failure, form a nursing perspective--a review of the literature. Eur J Cardiovasc Nurs 2004;3:7–20.
17. Levenson J, McCarthy E, Lynn J, et al. The last six months of life for patients with congestive heart failure. J Am Geriatr Soc 2000;48(Suppl 5):S101–S109.
18. Sullivan M, Levy W, Russo J, Spertus J. Depression and health status in patients with advanced heart failure: a prospective study in tertiary care. J Card Fail 2004;10:390–6.
19. Bekelman DB, Havranek EP, Becker DM, et al. Symptoms, depression, and quality of life in patients with heart failure. J Card Fail 2007;13:643–8.
20. Godfrey CM, Harrison MB, Friedberg E, et al. The symptom of pain in individuals recently hospitalized for heart failure. J Cardiovasc Nurs 2007;22:368–74.
21. McCarthy M, Lay M, Addington-Hall J. Dying from heart disease. J R Coll Physicians Lond 1996;30:325–8.
22. Norgren L SS. Symptoms experienced in the last six months of life in patients with end-stage heart failure. Eur J Cardiovasc Nurs 2003;2:213–7.
23. Zambroski CH, Moser DK, Bhat G, et al. Impact of symptom prevalence and symptom burden on quality of life in patients with heart failure. Eur J Cardiovasc Nurs 2005;4:198–206.
24. Walke LM, Byers AL, Tinetti ME, et al. Range and severity of symptoms over time among older adults wih chronic obstructive pulmonary disease and heart failure. Arch Intern Med 2007;167:2503–8.
25. Bekelman DB, Rumsfeld JS, Havranek EP, et al. Symptom burden, depression, and spiritual well-being: a comparison of heart failure and advanced cancer patients. J Gen Intern Med 2009;24:592–8.
26. Ahluwalia SC, Gross CP, Chaudhry SI, et al. Impact of comorbidity on mortality among older persons with advanced heart failure. J Gen Intern Med 2012;27:513–9.
27. Ahluwalia SC, Gross CP, Chaudhry SI, et al. Change in comorbidity prevalence with advancing age among persons with heart failure. J Gen Intern Med 2011;26:1145–51.
28. Corrigan JM, Donaldson MS, Kohn LT, et al. A new health system for the 21st century. crossing the quality chasm. Washington, DC: Institute of Medicine, National Academy of Sciences, National Academies Press; 2001.
29. Kirchhoff KT, Hammes BJ, Kehl KA, et al. Effect of a disease-specific advance care planning intervention on end-of-life care. J Am Geriatr Soc 2012;60:946–50.
30. Kirchhoff KT, Hammes BJ, Kehl KA, et al. Effect of a disease-specific planning intervention on surrogate understanding of patient goals for future medical treatment. J Am Geriatr Soc 2010;58:1233–40.
31. Janssen DJ, Engelberg RA, Wouters EF, Curtis JR. Advance care planning for patients with COPD: past, present and future. Patient Educ Couns 2012;86:19–24.
32. Aldred H, Gott M, Gariballa S. Advanced heart failure: Impact on older patients and informal carers. J Adv Nurs 2005;49:116–24.
33. Zhang B, Wright AA, Huskamp HA, et al. Health care costs in the last week of life: Associations with end-of-life conversations. Arch Intern Med 2009;169:480–8.
34. Wright AA, Zhang B, Ray A, et al. Associations between end-of-life discussions, patient mental health, medical care near death, and caregiver bereavement adjustment. JAMA 2008;300:1665–73.
35. Mack JW, Smith TJ. Reasons why physicians do not have discussions about poor prognosis, why it matters, and what can be improved. J Clin Oncol 2012;30:2715–7.
36. Detering KM, Hancock AD, Reade MC, Silvester W. The impact of advance care planning on end of life care in elderly patients: randomised controlled trial. BMJ 2010;340:c1345.
37. Schwartz CE, Wheeler HB, Hammes B, et al. Early intervention in planning end-of-life care with ambulatory geriatric patients: results of a pilot trial. Arch Intern Med 2002;162:1611–8.
38. Caldwell PH, Arthur HM, Demers C. Preferences of patients with heart failure for prognosis communication. Can J Cardiol 2007;23:791–6.
39. Bekelman DB, Nowels Ct, Retrum JH, et al. Giving voice to patients’ and family caregivers’ needs in chronic heart failure: implications for palliative care programs. J Palliat Med 2011;14:1317–24.
40. Harding R, Selman L, Beynon T, et al. Meeting the communication and information needs of chronic heart failure patients. J Pain Symptom Manage 2008;36:149–56.
41. Strachan PH, Ross H, Dodek PM, et al. Mind the gap: opportunities for improving end-of-life care for patients with advanced heart failure. Can J Cardiol 2009;25:635–40.
42. Ågård A, Hermerén G, Herlitz J. When is a patient with heart failure adequately informed? A study of patients’ knowledge of and attitudes toward medical information. Heart Lung 2004;33:219–26.
43. Gott M, Small N, Barnes S, et al. Older people’s views of a good death in heart failure: implications for palliative care provision. Soc Sci Med 2008;67:1113–21.
44. Murray SA, Boyd K, Kendall M, et al. Dying of lung cancer or cardiac failure: prospective qualitative interview study of patients and their carers in the community. BMJ 2002;325:929.
45. Ahluwalia SC, Levin JR, Lorenz KA, et al. Missed opportunities for advance care planning communication during outpatient clinic visits. J Gen Intern Med 2012;27:445–51.
46. Rodriguez KL, Appelt CJ, Switzer GE, et al. “They diagnosed bad heart”: a qualitative exploration of patients’ knowledge about and experiences with heart failure. Heart Lung. 2008;37:257–65.
47. Remme WJ,McMurray JJ, Rauch B, et al. Public awareness of heart failure in Europe: first results from SHAPE. Eur Heart J 2005;22:2413e21.
48. Golin CE, Wenger NS, Liu H, et al. A prospective study of patient-physician communication about resuscitation. J Am Geriatr Soc 2000;48(5 Suppl):S52–60.
49. Selman L, Harding R, Beynon T, et al. Improving end of life care for patients with chronic heart failure: ‘let’s hope it’ll get better when I know in my heart of hearts it won’t’. Heart 2007;93:963–7.
50. Barnes S, Gott M, Payne S, et al. Communication in heart failure: Perspectives from older people and primary care professionals. Health Soc Care Comm 2006;14:482–90.
51. Brännström M, Ekman I, Norberg A, et al. Living with severe chronic heart failure in palliative advanced home care. Eur J Cardiovasc Nurs 2006;5:295–302.
52. Barclay S, Momen N, Case-Upton S, et al. End-of-life care conversations with heart failure patients: a systematic literature review and narrative synthesis. Br J Gen Pract 2011;61:e49–62.
53. Whitney SN, McCullough LB, Fruge E, et al. Beyond breaking bad news: the roles of hope and hopefulness. Cancer 2008;113:442–5.
54. Tung EE, North F. Advance care planning in the primary care setting: a comparison of attending staff and resident barriers. Am J Hosp Palliat Care 2009;26:456–63.
55. Boyd K, Murray S, Kendall M, et al. Living with advanced heart failure: A prospective, community based study of patients and their carers. Eur J Heart Fail 2004;6:585–91.
56. Borbasi S, Wotton K, Redden M, et al. Letting go: A qualitative study of acute care and community nurses’ perceptions of a ‘good’ versus a ‘bad’ death. Austr Crit Care 2005 2005;18:104–13.
57. Hanratty B, Hibbert D, Mair F, et al. Doctors’ perceptions of palliative care for heart failure: Focus group study. BMJ 2002;325:581–5.
58. Fallowfield L, Jenkins V, Farewell V, et al. Efficacy of a communication skills training model for oncologists: a randomized controlled trial. Lancet 2002;359:650–6.
59. Platt F, Keller V. Empathic communication: a teachable and learnable skill. J Gen Intern Med 1994;9:222–6.
60. Morse D, Edwardsen E, Gordon H. Missed opportunities for interval empathy in lung cancer communication. Arch Intern Med 2008;22;168:1853–8.
61. Epstein R, Hadee T, Carroll J, et al. “Could this be something serious?” reassurance, uncertainty, and empathy in response to patients’ expressions of worry. J Gen Intern Med 2007;22:1731–9.
62. Stewart M. What is a successful doctor-patient interview? A study of interactions and outcomes. Soc Sci Med 1984;19:
167–75.
63. Wotton K, Borbasi S, Redden M. When all else has failed. Nurses’ perception of factors influencing palliative care for patients with end-stage heart failure. J Cardiovasc Nurs 2005;20:18–25.
64. Willems DL, Hak A, Visser F, Van der Wal G. Thoughts of patients with advanced heart failure on dying. Palliat Med 2004;18:564–72.
65. Briggs L, Kirchhoff K, Hammes B, et al. Patient-centered advance care planning in special patient populations: a pilot study. J Prof Nurs 2004;20:47–58.
66. Hammes B, Rooney B. Death and EOL planning in one midwestern community. Arch Intern Med 1998;158:390.
67. Lilly C, DeMeo D, Sonna L, et al. An intensive communication intervention for the critically ill. Am J Med 2000;109:469–75.
68. Davidson P, Macdonald P, Newton P, et al. End stage heart failure patients: Palliative care in general practice. Aust Fam Physician 2010;39:920.
69. Goodlin S, Quill T, Arnold R. Communication and decision-making about prognosis in heart failure care. J Card Fail 2008;14:106–13.
70. Analysis of U.S. hospital palliative care programs 2010 snapshot. Center to Advance Palliative Care (CAPC). Accessed 24 Nov 2014 at www.capc.org/news-and-events/releases/analysis-of-us-hospital-palliative-care-programs-2010-snapshot.pdf.
71. Back AL, Arnold RM, Baile WF, et al. Approaching difficult communication tasks in oncology. CA Cancer J Clin 2005;55:164–77.
72. Sudore RL, Fried TR, Redefining the “planning” in advance care planning: preparing for end-of-life decision making. Ann Intern Med 2010;153:256–61.
73. Lo B, Quill T, Tulsky J. Discussing palliative care with patients. Ann Intern Med 1999;130:744–9.
74. Quill T, Arnold R, Platt F. I wish things were different: Expressing wishes in response to loss, futility, and unrealistic hopes. Ann Intern Med 2001;135:551–5.
75. Pollak K, Arnold R, Jeffreys A, et al. Oncologist communication about emotion during visits with patients with advanced cancer. J Clin Oncol 2007;25:5748–52.
76. Back AL, Anderson WG, Bunch L, et al. Communication about cancer near the end of life. Cancer 2008;113(S7):1897–910.
77. Dries D, Exner D, Gersh B, et al. Racial differences in the outcome of left ventricular dysfunction. N Engl J Med 1999;340:609–16.
78. Alexander M, Grumbach K, Selby J, et al. Hospitalization for congestive heart failure. Explaining racial differences. JAMA 1995;274:1037–42.
79. Afzal A, Ananthasubramaniam K, Sharma N, et al. Racial differences in patients with heart failure. Clin Cardiol 1999;22:791–4.
80. Blackhall L, Murphy S, Frank G, et al. Ethnicity and attitudes toward patient autonomy. JAMA 1995;274:820–5.
81. Huang X, Butow P, Meiser B, et al. Attitudes and information needs of chinese migrant cancer patients and their relatives. Aust N Z J Med 1999;29:207–13.
82. Tan T, Teo F, Wong K, et al. Cancer: to tell or not to tell? Singapore Med J 1993;34:202–3.
83. Gorgaki S, Kalaidopoulou O, Liarmakopoulos I, et al. Nurses’ attitudes toward truthful communication with patients with cancer. A Greek study. Cancer Nurs 2002;25:436–41.
84. Harris J, Shao J, Sugarman J. Disclosure of cancer diagnosis and prognosis in northern Tanzania. Soc Sci Med 2003;56:905–13.
85. Goldstein D, Thewes B, Butow P. Communicating in a multicultural society. II: Greek community attitudes towards cancer in Australia. Intern Med J 2002;32:289–96.
86. Beyene Y. Medical disclosure and refugees. Telling bad news to Ethiopian patients. West J Med 1992;157:328–32.
87. Matsumura S, Bito S, Liu H, et al. Acculturation of attitudes toward end-of-life care: A cross-cultural survey of Japanese Americans and Japanese. J Gen Intern Med 2002;17:531–9.
88. Yick AG, Gupta R. Chinese cultural dimensions of death, dying, and bereavement: Focus group findings. J Cult Divers 2002 Summer;9:32–42.
89. Frank G, Blackhall L, Murphy S, et al. Ambiguity and hope: Disclosure preferences of less acculturated elderly Mexican Americans concerning terminal cancer—A case story. Camb Q Healthc Ethics 2002;11:117–26.
90. Hern HJ, Koenig B, Moore L, et al. The difference that culture can make in end-of-life decisionmaking. Camb Q Healthc Ethics 1998;7:27–48.
91. Kagawa-Singer M, Kassim-Lakha S. A strategy to reduce cross-cultural miscommunication and increase the likelihood of improving health outcomes. Acad Med 2003;78:577–87.
92. Barnett MM, Fisher JD, Cooke H, et al. Breaking bad news: consultants’ experience, previous education, and views on educational format and timing. Med Educ 2007;41:947–56.
93. Moore P, Rivera Mercado S, Grez Artigues M, Lawrie T. Communication skills training for healthcare professionals working with people who have cancer. Cochrane Database Syst Rev 2013;3CD003751.
94. Alelwani S, Ahmed Y. Medical training for communication of bad news: A literature review. J Educ Health Promot 2014;3
95. Delvaux N, Razavi D, Marchal S, et al. Effects of a 105 hour psychological training program on attitudes, communication skills and occupational stress in oncology: a randomised study. Br J Cancer 2004;90:106–14.
96. Baile WF, Lenzi R, Kudelka AP, et al. Improving physician-patient communication in cancer care: Outcome of a workshop for oncologists. J Cancer Educ 1997;12:166–73.
97. Back AL, Arnold RM, Baile WF, et al. Efficacy of communication skills training for giving bad news and discussing transitions to palliative care. Arch Intern Med 2007;167:453–60.
98. Gelfman LP, Lindenberger E, Fernandez H, et al. The effectiveness of the Geritalk communication skills course: a real-time assessment of skill acquisition and deliberate practice. J Pain Sympt Manage 2014;48:738–44.
99. Kelley AS, Back AL, Arnold RM, et al. Geritalk: communication skills training for geriatric and palliative medicine fellows. J Am Geriatr Soc 2012;60:332–7.
100. Institute of Medicine. Dying in America: Improving quality and honoring individual preferences near the end of life. Washington, DC: National Academies Press; 2014.
101. Allen LA, Stevenson LW, Grady KL, et al. Decision making in advanced heart failure: a scientific statement From the American Heart Association. Circulation 2012;125:1928–52.
102. Ditto PH, Jacobson JA, Smucker WD, et al. Context changes choices: a prospective study of the effects of hospitalization on life-sustaining treatment preferences. Med Decis Making 2006;26:313–22.
From the Rand Corporation and UCLA Fielding School of Public Health, Santa Monica, CA (Dr. Ahluwalia) and University of Southern California, Leonard Davis School of Gerontology, Los Angeles, CA (Dr. Enguidanos).
Abstract
- Objective: To review the relevance of advance care planning to heart failure management, describe key advance care planning challenges, and provide clinicians with actionable guidance for engaging in advance care planning conversations.
- Methods: Review of the literature.
- Results: Although most patients with heart failure prefer to receive thorough and honest information about their health condition and prognosis, the unpredictability of the heart failure trajectory coupled with physician barriers including discomfort with emotionally-laden topics and difficulty identifying the “right” time to engage in advance care planning, and systems barriers such as inadequate clinic time and limited reimbursement, impede timely engagement in advance care planning discussions. Approaches to effective advance care planning communication include using open-ended questions to stimulate patient engagement, evaluating how much information the patient wants to ensure patient-centeredness, and using empathic language to demonstrate support and understanding. While successful models of advance care planning communication have been identified, replication is limited due to the resource intense nature of these approaches.
- Conclusion: Challenges to advance care planning discussions among patients with heart failure may be mitigated through the establishment of communication quality standards as well as guidelines promoting early and ongoing advance care planning discussions, as well as reimbursement for outpatient discussions.
Heart failure, a leading cause of death, disability, and health care costs in the United States, is an incurable and life-limiting illness that is becoming increasingly prevalent due to an aging population and improved life expectancy. Approximately 5.3 million Americans are currently living with heart failure [1], with more than 550,000 new cases diagnosed each year [2]. Heart failure disproportionately affects older adults; about 80% of all cases occur in persons aged 65 years or older [3], and heart failure is the leading cause of hospital admissions among older adults [4]. The burden and impact of heart failure peaks near the end of life; 80% of Medicare beneficiaries with heart failure are hospitalized in the last 6 months of life [5].
The Trajectory of Heart Failure
Patients with heart failure experience a highly variable, nonlinear clinical trajectory marked by progressive deterioration and frequent exacerbations requiring hospitalization [6]. Their prognosis, though uncertain, is poor, with reported 1-year mortality rates following a hospitalization between 30% and 50% and 5-year mortality as high as 75% [7–11], a survival rate worse than that of some cancers [12]. Patients with heart failure caused by ischemic heart disease are at high risk for sudden cardiac death, particularly at earlier stages of the disease, which can confound the ability to appropriately plan for the future [13]. Those who survive to more advanced stages of heart failure face worsening quality of life [14–16], driven by a high prevalence of fatigue, breathlessness, pain, and depression [17–24]. Indeed, patients with heart failure have a similar symptom burden to patients with advanced cancer [25]. Older adults with heart failure also have a high comorbidity burden that further complicates both symptomology and disease trajectory with implications for decision-making about life-prolonging heart failure therapies [26,27].
Advance Care Planning in Heart Failure
The unpredictable nature of heart failure makes it difficult for patients and families to plan and prepare for their future, yet it is this very uncertainty that makes advance care planning (ACP) so critical for heart failure patients. Clear and honest patient-clinician communication about ACP, including an exploration of patient values and goals for care in the context of prognostic information, is essential to patient-centered treatment decision-making [28]. This is particularly relevant in heart failure, where a range of high-intensity, invasive, and costly interventions are increasingly being applied (eg, ventricular assist devices) without equivalent attention to quality of life and patients’ long-term goals for care.
Patients with heart failure and their families face multiple complex treatment decisions along the trajectory of their illness, such as discontinuation of beta blockers among patients with refractory fluid overload or angiotensin-converting enzyme inhibitors in end-stage patients with symptomatic hypotension [29,30]. In end-stage heart failure patients, deactivation of an implantable cardiac defibrillator might be considered to avoid the pain and distress associated with repeated shocks. In contrast, other interventions such as cardiac resynchronization therapy and continuous inotropic infusion have quality of life benefits; continuation of these therapies may be appropriate even when discontinuing other interventions. Such decisions should be guided by a thorough understanding of the patient’s expressed preferences and values, ideally assessed early in the trajectory of the disease and continuously re-evaluated as the diseases progresses.
The American Heart Association supports early and regular patient-provider ACP discussions to guide heart failure patients’ future decision-making [31], and recommends that such discussions be initiated in the outpatient setting, prior to and in anticipation of clinical decline. ACP communication plays a critical role in enhancing patients’ understanding of their diagnosis, treatment, prognosis, and choices in end-of-life care [31]. ACP communication also helps the clinician to better understand the context within which patients and their caregivers might make health care decisions, including their values and preferences for care. Patient-provider discussions about ACP focused on understanding patient values and initiated early in the trajectory of serious illness can support future in-the-moment decision-making, and is likely more effective than asking patients to make specific treatment decisions in advance [32]. A growing body of rigorous research has shown that ACP communication is associated with greater preference-concordant care and congruence in patient-surrogate understanding of patient preferences, lower costs, and less aggressive care at the end of life [33–37].
Patient Preferences for ACP Communication
Most patients with heart failure and their caregivers want honest disclosure regarding prognosis and to receive information about the expected trajectory of their disease [38–41] as early as possible [38] to help them plan and prepare for their future. Patients and their caregivers prefer to have these conversations with their physician [38] or other provider most familiar with the patient and family [39]. Patients also express a preference for support with dealing with the uncertainty inherent to heart failure [39]. Although most patients and caregivers desire to receive clear and honest communication about their disease, it is important to note that patients may vary in the extent of information they prefer to receive about their heart failure, with some individuals preferring not to talk about the end of life and future care needs at all [39,42–44].
Challenges to ACP Communication in Heart Failure
Despite patient and caregiver preferences for ACP communication with their providers, evidence suggests such communication occurs infrequently [40,45] and that heart failure patients may lack important information about their prognosis and treatment options [40,44,46,47]. For example, patients may not recognize the terminal nature of heart failure, and may be unaware of the range of treatment options, including hospice, available to them. Evidence also demonstrates that ACP is infrequently discussed with their health care providers [40], resulting in these conversations being avoided or deferred until an emergent clinical situation [44,48] when hasty questions about treatment choices may yield uncertain and conflicting answers not representative of a patient’s underlying values.
The infrequent, late, and often lack of discussions about ACP are driven by several challenges. First, the uncertain trajectory of heart failure makes communication regarding “what to expect” difficult. Prognostication is an immense challenge in heart failure [40,49–52], making it harder to talk about end-of-life issues and hindering the ability of patients, caregivers and health care providers to plan and prepare for the future. It is often difficult for clinicians, who face the challenge of instilling hope in the face of truthful disclosure [53], to identify the “right time” to initiate such discussions.
Second, a lack of time, particularly during outpatient visits, impedes physician ability to have considered discussions about future care needs and preferences [32,54]. The U.S. health care system currently lacks financial reimbursement for these discussions, which poses a significant barrier to the integration of ACP conversations into routine clinical practice. Moreover, these conversations are lengthy and iterative [53]. ACP discussions that are focused on facilitating patient-centered decision-making ideally begin with a discussion of expected prognosis, followed by an exploration of patient preferences and values for health care, and then a review of treatment options to be considered in the context of those preferences. Often additional time is needed for completing advance directive documents or for charting key outcomes from these discussions. Clinicians today are frequently overloaded with addressing multiple medical issues during outpatient visits that leave little time for non-medical tasks such as ACP discussions. The lack of financial incentives to support in-depth discussions is a critical challenge in improving ACP.
Third, a lack of training in specialized communication skills, particularly focused on empathic and emotionally sensitive disclosure, may further hinder physicians from initiating frank discussions with their patients. ACP conversations are highly sensitive and fraught with emotional complexity, and clinicians understandably experience discomfort with breaking bad news [49,51,55] or with broader issues of decline and death [51,56,57]. Physicians tend to be most comfortable addressing cognitive aspects of communication; addressing the emotional needs of patients is harder. Medical school training teaches detachment in physician practice, perhaps as a way of coping with the sadness they regularly confront and in maintaining their ability to provide clinical care. In fact, physicians describe their most difficult encounters as those with the most negative expressed emotions and miss opportunities to respond with empathy [58–60], a critical skill in effective patient-physician communication that is associated with improved patient satisfaction [61,62]. While patients value good communication skills in their health care encounters, many providers feel they lack the necessary skills to lead effective ACP discussions [49,63].
Finally, information gaps with regards to heart failure contribute to delayed or absent conversations about planning for future care. Many heart failure patients have a limited understanding of their disease [32,40,44,55], particularly an inaccurate perception that heart failure is not a terminal and life-limiting illness [42,49,64]. Compounding this is the fact that even some health care providers are reluctant to acknowledge the terminal nature of heart failure [50,56]. Without frank acknowledgement of the terminal nature of heart failure, the initiation of discussions regarding end-of-life care will remain difficult if not impossible.
Approaches to ACP in Heart Failure
When Is the Right Time?
Given the complexity and unpredictable trajectory of heart failure, indicators of disease progression, including changes in health status and health service use, may serve as useful signals to help clinicians identify the appropriate time to initiate care planning discussions. Repeated hospital admissions for heart failure are strongly associated with increased mortality. In a sample of community heart failure patients [8], median survival after the first, second, and third hospitalization was 2.4, 1.4, and 1.0 years, respectively. In light of this, a patient with 1 or more hospitalizations in a 12-month period may be an appropriate candidate for an ACP conversation. Similarly, comorbidity in patients with heart failure may signal the relevance and need for discussions about future care. In a sample of Medicare beneficiaries with advanced heart failure, an increasing burden of comorbidity was associated with significantly higher mortality, as were certain conditions (COPD, CKD, dementia, depression) and combinations of conditions (eg, CKD and dementia) [26]. Davidson and colleagues [68] suggest a list of clinical indicators signaling the need for an ACP conversation, including any of the following:
- > 1 episodes of exacerbation of heart failure leading to hospital admission
- New York Heart Association Class IV heart failure
- Decline in function and mobility
- Unexplained weight loss
- Resting pulse rate greater than 100 beats/minute
- Raised serum creatinine (> 150 µmol/L)
- Low serum sodium (< 135 mmol/L)
- Low serum albumen (< 33 g/L)
- High dose of loop diuretic (eg, furosemide ≥ 160 mg daily)
Given the considerable complexity and multisystem nature of heart failure, none of these indicators alone can signal certainty about disease progression and consequent outcomes; however, they can serve as a useful heuristic for helping clinicians identify appropriate times to raise the topic of ACP with their patients.
What Do I Say? Structuring the Conversation
Heart failure patients and their caregivers may vary in their preferences for hearing information about their disease; therefore, it is critical to open any conversation about planning for future in the context of their illness by asking what and how much information is desired. This includes evaluating how involved in decision-making the patient wants to be. Previously suggested language includes [69,70]:
- Would you like to consider all the options, or my opinion about the options that fit best with what I know about you?
- Some people like to know everything about their disease and be involved in all decision making. Others do not want all the news and would rather the doctor talk to __________. Which kind of person are you? How involved do you want to be in these decisions?
- Would you like me to tell you the full details of your condition?
- If you prefer not to hear the details, is there someone in your family who you trust to receive this information?
After establishing the patient’s preferences for hearing different types of information and level of involvement in decision-making about their care, the ask-tell-ask model [69,71] provides a useful approach to communicating with patients and their families. The conversation generally begins by asking patients what he or she understands about their illness (eg, “What do you understand about your heart failure?”; “I want to make sure we’re on the same page; what have other doctors told you?”). Building on what the patient already knows, the clinician can then disclose new information, correct misunderstandings, or confirm impressions and expectations the patient might have. In this way, information is tailored to the patient’s understanding and aimed at addressing potential knowledge gaps, all within the context of their preferences. Finally, the clinician asks the patient to describe their new understanding and whether or not they have questions or concerns (eg, “To make sure I did a good job of explaining to you, can you tell me what you now know about your condition from our conversation?”; “I know I’ve covered a lot and I want to make sure I was clear. When you get home, how are you going to explain what I’ve told you to your spouse?”). This approach encourages communication and exchange between patient and physician. Additionally, expressions of concern promote relationship building and bonding between physician and patient.
Keeping the Conversation Going
ACP communication can cover a wide range of topics beyond disease and prognostic disclosure by the provider to the patient. A critical aspect of ACP conversations is an exploration of the patient’s values and preferences, which can be used to help contextualize treatment choices and subsequently guide in-the-moment decision-making [72]. Using open-ended questions throughout the conversation gives the patient an opportunity to reflect on and communicate their wishes and values and allows them to engage in the conversation on their own terms. Examples of discussion-stimulating questions include [69,73]:
- What concerns you most about your illness?
- How is treatment going for you (your family)?
- As you think about your illness, what is the best and worst that might happen?
- What are your greatest hopes about your health?
- What has been most difficult about this illness for you?
- Looking back at your life, what has been important to you?
- At this point, what is most important for you to do?
Language
Central to this process is the use of empathic language to demonstrate support and understanding. An expression of empathy is also an appropriate way to acknowledge and share difficult emotions when it becomes hard to know where to take the conversation next. Quill and colleagues [74] suggest the following empathic responses to patients’ emotional expressions:
- I wish for that too
- It's unfortunate that things aren't different
- I am so sorry that this happened to you
- I understand how much you want that
- It must be very hard to accept the seriousness of this illness
Relatedly, the use of medical jargon in ACP conversations can increase the distance between patients and their providers, and may hinder patient understanding. Physicians may use technical language out of habit, or as an unconscious way to emotionally separate themselves from the task of delivering bad news. However, clear communication using layperson terms is the most effective approach to providing information necessary to patient-centered decision making. Explaining medical procedures in simple terms can improve understanding and help to build trust with the physician (eg, “We will perform an angioplasty – a procedure where a special tube with a balloon on the end of it is inserted into your artery to stretch it open. This will improve blood flow and relieve some of the symptoms you are currently experiencing”.)
Cultural Issues in Communication
There are various cultural issues to consider and address when conducting ACP discussions with heart failure patients and their families. Heart failure disproportionately affects certain racial and ethnic groups (eg, African Americans) [77–79], and effective management of heart failure depends on the provision of culturally sensitive information and facilitation of culturally informed self-care behaviors. There is evidence of cultural variation in preferences for information and role in decision-making. For example, most white and African-American patients prefer to be fully informed of their condition [80], whereas other cultures may focus on protecting the patient from difficult information in order to maintain hope [80–86]. Moreover, even in cultures where nondisclosure is preferred, patients may want to be told the truth in an indirect, euphemistic, or even nonverbal manner [80,87–89]. These complexities underscore the importance of taking a patient-centered approach to ACP communication, respecting individuality and autonomy while ultimately facilitating decision-making [90,91].
Are There Effective Training Programs for ACP Communication?
Effective communication skills are a critical component of ACP conversations between clinicians and their patients; however, most clinicians do not receive formal training in ACP communication and believe it to be a difficult task [92]. Strong evidence of the effectiveness of communication skills training has yet to be established, largely due to variation in the approach to training and the specification of relevant outcomes. For example, a systematic review of communication skills training courses found that some courses are effective at improving different types of communication skills related to providing support and gathering information, but these courses lacked effectiveness in improving patient satisfaction or provider burnout and distress [93]. Similarly, a range of approaches to teaching clinicians effective ACP communication skills early in their medical training have been identified [94], but considerable variation in quality preclude any conclusions from being drawn about their effectiveness.
Despite these challenges, there are some studies of communication skills training courses that have demonstrated the ability to increase providers’ use of empathic and facilitative communication (eg, use of open-ended questions) [58,95], and to increase self-efficacy and confidence among providers [96]. One particular teaching model that is increasingly used in cancer care is Oncotalk (http://depts.washington.edu/oncotalk/). Oncotalk has been shown to significantly increase clinical skills in giving bad news and facilitating the transition to palliative care. Building on this success, the program has expanded to training courses focused on the intensive care setting (http://depts.washington.edu/icutalk/) and geriatrics care [97–99]. It is important to note, however, that the considerable time and resource-intensive nature of communication training programs limits widespread implementation of any one approach into routine medical education. More attention to the type and structure of communication skills training programs are needed as well as scalable approaches to assist clinicians in developing effective ACP communication skills.
Policy Implications of ACP and Future Directions
There is growing recognition of the need to improve ACP among patients with seriously illness, including heart failure. In a recent Institute of Medicine (IOM) report, Dying in America [100], the need for clinician-patient communication about ACP was identified as a primary area of improvement. Recommendations include the establishment of communication quality standards as well as guidelines promoting early and ongoing ACP discussions. This is supported by recommendations from medical professional societies for an iterative model of ACP that follows the course of a serious illness [2,101]. At early stages of the illness, ACP might be focused on helping patients clarify their broad health care values and raise awareness of their disease and expected prognosis. As the condition progresses, ACP discussion might focus on exploring disease-specific treatment options within the context of previously expressed preferences, as well as identifying changes in patients’ values over time, particularly as they gain experience with their illness and health status changes [102]. In late stages of the disease, ACP might focus on documenting specific treatment choices (eg, DNR orders) and on exploring options such as palliative care, while also ensuring that patients and caregivers are appropriately prepared for imminent decline and death.
The IOM report also calls for payment reforms to include reimbursement for outpatient ACP discussions [100]. There is burgeoning national support for developing reimbursement models for ACP discussions. The American Medical Association has recently released current procedural terminology (CPT) codes for ACP services, a first step toward urging Medicare to consider reimbursement for ACP discussions with physicians.
Finally, the IOM report calls for improved education and training in ACP communication across all disciplines and specialties providing care to patients with serious illness. These recommendations bring national attention to the current limitations surrounding ACP discussions for those with serious illness, including heart failure. Further research is needed to identify methods and care models to address the gap in communication skills, processes, and policies.
Corresponding author: Sangeeta C. Ahluwalia, Rand Corporation, 1776 Main St., Santa Monica, CA, 90401, [email protected].
Financial disclosures: None.
Author contributions: conception and design, SCA, SE; analysis and interpretation of data, SCA, SE; drafting of article, SCA, SE; critical revision of the article, SCA, SE.
From the Rand Corporation and UCLA Fielding School of Public Health, Santa Monica, CA (Dr. Ahluwalia) and University of Southern California, Leonard Davis School of Gerontology, Los Angeles, CA (Dr. Enguidanos).
Abstract
- Objective: To review the relevance of advance care planning to heart failure management, describe key advance care planning challenges, and provide clinicians with actionable guidance for engaging in advance care planning conversations.
- Methods: Review of the literature.
- Results: Although most patients with heart failure prefer to receive thorough and honest information about their health condition and prognosis, the unpredictability of the heart failure trajectory coupled with physician barriers including discomfort with emotionally-laden topics and difficulty identifying the “right” time to engage in advance care planning, and systems barriers such as inadequate clinic time and limited reimbursement, impede timely engagement in advance care planning discussions. Approaches to effective advance care planning communication include using open-ended questions to stimulate patient engagement, evaluating how much information the patient wants to ensure patient-centeredness, and using empathic language to demonstrate support and understanding. While successful models of advance care planning communication have been identified, replication is limited due to the resource intense nature of these approaches.
- Conclusion: Challenges to advance care planning discussions among patients with heart failure may be mitigated through the establishment of communication quality standards as well as guidelines promoting early and ongoing advance care planning discussions, as well as reimbursement for outpatient discussions.
Heart failure, a leading cause of death, disability, and health care costs in the United States, is an incurable and life-limiting illness that is becoming increasingly prevalent due to an aging population and improved life expectancy. Approximately 5.3 million Americans are currently living with heart failure [1], with more than 550,000 new cases diagnosed each year [2]. Heart failure disproportionately affects older adults; about 80% of all cases occur in persons aged 65 years or older [3], and heart failure is the leading cause of hospital admissions among older adults [4]. The burden and impact of heart failure peaks near the end of life; 80% of Medicare beneficiaries with heart failure are hospitalized in the last 6 months of life [5].
The Trajectory of Heart Failure
Patients with heart failure experience a highly variable, nonlinear clinical trajectory marked by progressive deterioration and frequent exacerbations requiring hospitalization [6]. Their prognosis, though uncertain, is poor, with reported 1-year mortality rates following a hospitalization between 30% and 50% and 5-year mortality as high as 75% [7–11], a survival rate worse than that of some cancers [12]. Patients with heart failure caused by ischemic heart disease are at high risk for sudden cardiac death, particularly at earlier stages of the disease, which can confound the ability to appropriately plan for the future [13]. Those who survive to more advanced stages of heart failure face worsening quality of life [14–16], driven by a high prevalence of fatigue, breathlessness, pain, and depression [17–24]. Indeed, patients with heart failure have a similar symptom burden to patients with advanced cancer [25]. Older adults with heart failure also have a high comorbidity burden that further complicates both symptomology and disease trajectory with implications for decision-making about life-prolonging heart failure therapies [26,27].
Advance Care Planning in Heart Failure
The unpredictable nature of heart failure makes it difficult for patients and families to plan and prepare for their future, yet it is this very uncertainty that makes advance care planning (ACP) so critical for heart failure patients. Clear and honest patient-clinician communication about ACP, including an exploration of patient values and goals for care in the context of prognostic information, is essential to patient-centered treatment decision-making [28]. This is particularly relevant in heart failure, where a range of high-intensity, invasive, and costly interventions are increasingly being applied (eg, ventricular assist devices) without equivalent attention to quality of life and patients’ long-term goals for care.
Patients with heart failure and their families face multiple complex treatment decisions along the trajectory of their illness, such as discontinuation of beta blockers among patients with refractory fluid overload or angiotensin-converting enzyme inhibitors in end-stage patients with symptomatic hypotension [29,30]. In end-stage heart failure patients, deactivation of an implantable cardiac defibrillator might be considered to avoid the pain and distress associated with repeated shocks. In contrast, other interventions such as cardiac resynchronization therapy and continuous inotropic infusion have quality of life benefits; continuation of these therapies may be appropriate even when discontinuing other interventions. Such decisions should be guided by a thorough understanding of the patient’s expressed preferences and values, ideally assessed early in the trajectory of the disease and continuously re-evaluated as the diseases progresses.
The American Heart Association supports early and regular patient-provider ACP discussions to guide heart failure patients’ future decision-making [31], and recommends that such discussions be initiated in the outpatient setting, prior to and in anticipation of clinical decline. ACP communication plays a critical role in enhancing patients’ understanding of their diagnosis, treatment, prognosis, and choices in end-of-life care [31]. ACP communication also helps the clinician to better understand the context within which patients and their caregivers might make health care decisions, including their values and preferences for care. Patient-provider discussions about ACP focused on understanding patient values and initiated early in the trajectory of serious illness can support future in-the-moment decision-making, and is likely more effective than asking patients to make specific treatment decisions in advance [32]. A growing body of rigorous research has shown that ACP communication is associated with greater preference-concordant care and congruence in patient-surrogate understanding of patient preferences, lower costs, and less aggressive care at the end of life [33–37].
Patient Preferences for ACP Communication
Most patients with heart failure and their caregivers want honest disclosure regarding prognosis and to receive information about the expected trajectory of their disease [38–41] as early as possible [38] to help them plan and prepare for their future. Patients and their caregivers prefer to have these conversations with their physician [38] or other provider most familiar with the patient and family [39]. Patients also express a preference for support with dealing with the uncertainty inherent to heart failure [39]. Although most patients and caregivers desire to receive clear and honest communication about their disease, it is important to note that patients may vary in the extent of information they prefer to receive about their heart failure, with some individuals preferring not to talk about the end of life and future care needs at all [39,42–44].
Challenges to ACP Communication in Heart Failure
Despite patient and caregiver preferences for ACP communication with their providers, evidence suggests such communication occurs infrequently [40,45] and that heart failure patients may lack important information about their prognosis and treatment options [40,44,46,47]. For example, patients may not recognize the terminal nature of heart failure, and may be unaware of the range of treatment options, including hospice, available to them. Evidence also demonstrates that ACP is infrequently discussed with their health care providers [40], resulting in these conversations being avoided or deferred until an emergent clinical situation [44,48] when hasty questions about treatment choices may yield uncertain and conflicting answers not representative of a patient’s underlying values.
The infrequent, late, and often lack of discussions about ACP are driven by several challenges. First, the uncertain trajectory of heart failure makes communication regarding “what to expect” difficult. Prognostication is an immense challenge in heart failure [40,49–52], making it harder to talk about end-of-life issues and hindering the ability of patients, caregivers and health care providers to plan and prepare for the future. It is often difficult for clinicians, who face the challenge of instilling hope in the face of truthful disclosure [53], to identify the “right time” to initiate such discussions.
Second, a lack of time, particularly during outpatient visits, impedes physician ability to have considered discussions about future care needs and preferences [32,54]. The U.S. health care system currently lacks financial reimbursement for these discussions, which poses a significant barrier to the integration of ACP conversations into routine clinical practice. Moreover, these conversations are lengthy and iterative [53]. ACP discussions that are focused on facilitating patient-centered decision-making ideally begin with a discussion of expected prognosis, followed by an exploration of patient preferences and values for health care, and then a review of treatment options to be considered in the context of those preferences. Often additional time is needed for completing advance directive documents or for charting key outcomes from these discussions. Clinicians today are frequently overloaded with addressing multiple medical issues during outpatient visits that leave little time for non-medical tasks such as ACP discussions. The lack of financial incentives to support in-depth discussions is a critical challenge in improving ACP.
Third, a lack of training in specialized communication skills, particularly focused on empathic and emotionally sensitive disclosure, may further hinder physicians from initiating frank discussions with their patients. ACP conversations are highly sensitive and fraught with emotional complexity, and clinicians understandably experience discomfort with breaking bad news [49,51,55] or with broader issues of decline and death [51,56,57]. Physicians tend to be most comfortable addressing cognitive aspects of communication; addressing the emotional needs of patients is harder. Medical school training teaches detachment in physician practice, perhaps as a way of coping with the sadness they regularly confront and in maintaining their ability to provide clinical care. In fact, physicians describe their most difficult encounters as those with the most negative expressed emotions and miss opportunities to respond with empathy [58–60], a critical skill in effective patient-physician communication that is associated with improved patient satisfaction [61,62]. While patients value good communication skills in their health care encounters, many providers feel they lack the necessary skills to lead effective ACP discussions [49,63].
Finally, information gaps with regards to heart failure contribute to delayed or absent conversations about planning for future care. Many heart failure patients have a limited understanding of their disease [32,40,44,55], particularly an inaccurate perception that heart failure is not a terminal and life-limiting illness [42,49,64]. Compounding this is the fact that even some health care providers are reluctant to acknowledge the terminal nature of heart failure [50,56]. Without frank acknowledgement of the terminal nature of heart failure, the initiation of discussions regarding end-of-life care will remain difficult if not impossible.
Approaches to ACP in Heart Failure
When Is the Right Time?
Given the complexity and unpredictable trajectory of heart failure, indicators of disease progression, including changes in health status and health service use, may serve as useful signals to help clinicians identify the appropriate time to initiate care planning discussions. Repeated hospital admissions for heart failure are strongly associated with increased mortality. In a sample of community heart failure patients [8], median survival after the first, second, and third hospitalization was 2.4, 1.4, and 1.0 years, respectively. In light of this, a patient with 1 or more hospitalizations in a 12-month period may be an appropriate candidate for an ACP conversation. Similarly, comorbidity in patients with heart failure may signal the relevance and need for discussions about future care. In a sample of Medicare beneficiaries with advanced heart failure, an increasing burden of comorbidity was associated with significantly higher mortality, as were certain conditions (COPD, CKD, dementia, depression) and combinations of conditions (eg, CKD and dementia) [26]. Davidson and colleagues [68] suggest a list of clinical indicators signaling the need for an ACP conversation, including any of the following:
- > 1 episodes of exacerbation of heart failure leading to hospital admission
- New York Heart Association Class IV heart failure
- Decline in function and mobility
- Unexplained weight loss
- Resting pulse rate greater than 100 beats/minute
- Raised serum creatinine (> 150 µmol/L)
- Low serum sodium (< 135 mmol/L)
- Low serum albumen (< 33 g/L)
- High dose of loop diuretic (eg, furosemide ≥ 160 mg daily)
Given the considerable complexity and multisystem nature of heart failure, none of these indicators alone can signal certainty about disease progression and consequent outcomes; however, they can serve as a useful heuristic for helping clinicians identify appropriate times to raise the topic of ACP with their patients.
What Do I Say? Structuring the Conversation
Heart failure patients and their caregivers may vary in their preferences for hearing information about their disease; therefore, it is critical to open any conversation about planning for future in the context of their illness by asking what and how much information is desired. This includes evaluating how involved in decision-making the patient wants to be. Previously suggested language includes [69,70]:
- Would you like to consider all the options, or my opinion about the options that fit best with what I know about you?
- Some people like to know everything about their disease and be involved in all decision making. Others do not want all the news and would rather the doctor talk to __________. Which kind of person are you? How involved do you want to be in these decisions?
- Would you like me to tell you the full details of your condition?
- If you prefer not to hear the details, is there someone in your family who you trust to receive this information?
After establishing the patient’s preferences for hearing different types of information and level of involvement in decision-making about their care, the ask-tell-ask model [69,71] provides a useful approach to communicating with patients and their families. The conversation generally begins by asking patients what he or she understands about their illness (eg, “What do you understand about your heart failure?”; “I want to make sure we’re on the same page; what have other doctors told you?”). Building on what the patient already knows, the clinician can then disclose new information, correct misunderstandings, or confirm impressions and expectations the patient might have. In this way, information is tailored to the patient’s understanding and aimed at addressing potential knowledge gaps, all within the context of their preferences. Finally, the clinician asks the patient to describe their new understanding and whether or not they have questions or concerns (eg, “To make sure I did a good job of explaining to you, can you tell me what you now know about your condition from our conversation?”; “I know I’ve covered a lot and I want to make sure I was clear. When you get home, how are you going to explain what I’ve told you to your spouse?”). This approach encourages communication and exchange between patient and physician. Additionally, expressions of concern promote relationship building and bonding between physician and patient.
Keeping the Conversation Going
ACP communication can cover a wide range of topics beyond disease and prognostic disclosure by the provider to the patient. A critical aspect of ACP conversations is an exploration of the patient’s values and preferences, which can be used to help contextualize treatment choices and subsequently guide in-the-moment decision-making [72]. Using open-ended questions throughout the conversation gives the patient an opportunity to reflect on and communicate their wishes and values and allows them to engage in the conversation on their own terms. Examples of discussion-stimulating questions include [69,73]:
- What concerns you most about your illness?
- How is treatment going for you (your family)?
- As you think about your illness, what is the best and worst that might happen?
- What are your greatest hopes about your health?
- What has been most difficult about this illness for you?
- Looking back at your life, what has been important to you?
- At this point, what is most important for you to do?
Language
Central to this process is the use of empathic language to demonstrate support and understanding. An expression of empathy is also an appropriate way to acknowledge and share difficult emotions when it becomes hard to know where to take the conversation next. Quill and colleagues [74] suggest the following empathic responses to patients’ emotional expressions:
- I wish for that too
- It's unfortunate that things aren't different
- I am so sorry that this happened to you
- I understand how much you want that
- It must be very hard to accept the seriousness of this illness
Relatedly, the use of medical jargon in ACP conversations can increase the distance between patients and their providers, and may hinder patient understanding. Physicians may use technical language out of habit, or as an unconscious way to emotionally separate themselves from the task of delivering bad news. However, clear communication using layperson terms is the most effective approach to providing information necessary to patient-centered decision making. Explaining medical procedures in simple terms can improve understanding and help to build trust with the physician (eg, “We will perform an angioplasty – a procedure where a special tube with a balloon on the end of it is inserted into your artery to stretch it open. This will improve blood flow and relieve some of the symptoms you are currently experiencing”.)
Cultural Issues in Communication
There are various cultural issues to consider and address when conducting ACP discussions with heart failure patients and their families. Heart failure disproportionately affects certain racial and ethnic groups (eg, African Americans) [77–79], and effective management of heart failure depends on the provision of culturally sensitive information and facilitation of culturally informed self-care behaviors. There is evidence of cultural variation in preferences for information and role in decision-making. For example, most white and African-American patients prefer to be fully informed of their condition [80], whereas other cultures may focus on protecting the patient from difficult information in order to maintain hope [80–86]. Moreover, even in cultures where nondisclosure is preferred, patients may want to be told the truth in an indirect, euphemistic, or even nonverbal manner [80,87–89]. These complexities underscore the importance of taking a patient-centered approach to ACP communication, respecting individuality and autonomy while ultimately facilitating decision-making [90,91].
Are There Effective Training Programs for ACP Communication?
Effective communication skills are a critical component of ACP conversations between clinicians and their patients; however, most clinicians do not receive formal training in ACP communication and believe it to be a difficult task [92]. Strong evidence of the effectiveness of communication skills training has yet to be established, largely due to variation in the approach to training and the specification of relevant outcomes. For example, a systematic review of communication skills training courses found that some courses are effective at improving different types of communication skills related to providing support and gathering information, but these courses lacked effectiveness in improving patient satisfaction or provider burnout and distress [93]. Similarly, a range of approaches to teaching clinicians effective ACP communication skills early in their medical training have been identified [94], but considerable variation in quality preclude any conclusions from being drawn about their effectiveness.
Despite these challenges, there are some studies of communication skills training courses that have demonstrated the ability to increase providers’ use of empathic and facilitative communication (eg, use of open-ended questions) [58,95], and to increase self-efficacy and confidence among providers [96]. One particular teaching model that is increasingly used in cancer care is Oncotalk (http://depts.washington.edu/oncotalk/). Oncotalk has been shown to significantly increase clinical skills in giving bad news and facilitating the transition to palliative care. Building on this success, the program has expanded to training courses focused on the intensive care setting (http://depts.washington.edu/icutalk/) and geriatrics care [97–99]. It is important to note, however, that the considerable time and resource-intensive nature of communication training programs limits widespread implementation of any one approach into routine medical education. More attention to the type and structure of communication skills training programs are needed as well as scalable approaches to assist clinicians in developing effective ACP communication skills.
Policy Implications of ACP and Future Directions
There is growing recognition of the need to improve ACP among patients with seriously illness, including heart failure. In a recent Institute of Medicine (IOM) report, Dying in America [100], the need for clinician-patient communication about ACP was identified as a primary area of improvement. Recommendations include the establishment of communication quality standards as well as guidelines promoting early and ongoing ACP discussions. This is supported by recommendations from medical professional societies for an iterative model of ACP that follows the course of a serious illness [2,101]. At early stages of the illness, ACP might be focused on helping patients clarify their broad health care values and raise awareness of their disease and expected prognosis. As the condition progresses, ACP discussion might focus on exploring disease-specific treatment options within the context of previously expressed preferences, as well as identifying changes in patients’ values over time, particularly as they gain experience with their illness and health status changes [102]. In late stages of the disease, ACP might focus on documenting specific treatment choices (eg, DNR orders) and on exploring options such as palliative care, while also ensuring that patients and caregivers are appropriately prepared for imminent decline and death.
The IOM report also calls for payment reforms to include reimbursement for outpatient ACP discussions [100]. There is burgeoning national support for developing reimbursement models for ACP discussions. The American Medical Association has recently released current procedural terminology (CPT) codes for ACP services, a first step toward urging Medicare to consider reimbursement for ACP discussions with physicians.
Finally, the IOM report calls for improved education and training in ACP communication across all disciplines and specialties providing care to patients with serious illness. These recommendations bring national attention to the current limitations surrounding ACP discussions for those with serious illness, including heart failure. Further research is needed to identify methods and care models to address the gap in communication skills, processes, and policies.
Corresponding author: Sangeeta C. Ahluwalia, Rand Corporation, 1776 Main St., Santa Monica, CA, 90401, [email protected].
Financial disclosures: None.
Author contributions: conception and design, SCA, SE; analysis and interpretation of data, SCA, SE; drafting of article, SCA, SE; critical revision of the article, SCA, SE.
1. American Heart Association. Heart disease and stroke statistics—2008 update. [Internet]. Available at www.americanheart.org/downloadable/heart/1200078608862HS_Stats%202008.final.pdf.
2. Hunt SA, Abraham WT, Chin MH, et al. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult—summary article. Circulation 2005;112:1825–52.
3. Masoudi FA, Havranek EP, Krumholz HM. The burden of chronic congestive heart failure in older persons: magnitude and implications for policy and research. Heart Fail Rev 2002;7:9–16.
4. McMurray JJ PM. Heart failure. Lancet 2005;365:1877–89.
5. Unroe KT, Greiner MA, Hernandez AF, et al. Resource use in the last 6 months of life among medicare beneficiaries with heart failure, 2000-2007. Arch Intern Med 2011;171:196–203.
6. Lunney JR, Lynn J, Foley DJ, et al. Patterns of functional decline at the end of life. JAMA 2003;289:2387–92.
7. Ko D, Alter D, Austin P, et al. Life expectancy after an index hospitalization for patients with heart failure: A population-based study. Am Heart J 2008;155:324–31.
8. Setoguchi S, Stevenson LW, Schneeweiss S. Repeated hospitalizations predict mortality in the community population with heart failure. Am Heart J 2007;154:260–6.
9. Jong P, Vowinskel E, Liu PP, et al. Prognosis and determinants for survival in patients newly hospitalized for heart failure. Arch Intern Med 2002;162:1689-94.
10. Thom T, Haase N, Rodamond W, et al. Heart disease and stroke statistics- 2006 update. Circulation 2006;113:e85-e151.
11. Shahar E, Lee S, Kim J, et al. Hospitalized heart failure: Rates and long-term mortality. J Card Fail 2004;10:374–9.
12. Kirkpatrick JN, Guger CJ, Arnsdorf MF, et al. Advance directives in the cardiac care unit. Am Heart J 2007;154:477–81.
13. Orn S, Dickstein K. How do heart failure patients die? Eur Heart J. 2002;4(suppl D).
14. Juenger J, Schellberg D, Kraemer S, et al. Health related quality of life in patients with congestive heart failure: comparison with other chronic disease and relation to functional variables. Heart 2002;87:235–41.
15. Steptoe A, Mohabir A, Mahon NG, et al. Health related quality of life and psychological wellbeing in patients with dilated cardiomyopathy. Heart 2000;83:645–50.
16. Johansson P, Agnebrink M, Dahlstrom U, et al. Measurement of health-related quality of life in chronic heart failure, form a nursing perspective--a review of the literature. Eur J Cardiovasc Nurs 2004;3:7–20.
17. Levenson J, McCarthy E, Lynn J, et al. The last six months of life for patients with congestive heart failure. J Am Geriatr Soc 2000;48(Suppl 5):S101–S109.
18. Sullivan M, Levy W, Russo J, Spertus J. Depression and health status in patients with advanced heart failure: a prospective study in tertiary care. J Card Fail 2004;10:390–6.
19. Bekelman DB, Havranek EP, Becker DM, et al. Symptoms, depression, and quality of life in patients with heart failure. J Card Fail 2007;13:643–8.
20. Godfrey CM, Harrison MB, Friedberg E, et al. The symptom of pain in individuals recently hospitalized for heart failure. J Cardiovasc Nurs 2007;22:368–74.
21. McCarthy M, Lay M, Addington-Hall J. Dying from heart disease. J R Coll Physicians Lond 1996;30:325–8.
22. Norgren L SS. Symptoms experienced in the last six months of life in patients with end-stage heart failure. Eur J Cardiovasc Nurs 2003;2:213–7.
23. Zambroski CH, Moser DK, Bhat G, et al. Impact of symptom prevalence and symptom burden on quality of life in patients with heart failure. Eur J Cardiovasc Nurs 2005;4:198–206.
24. Walke LM, Byers AL, Tinetti ME, et al. Range and severity of symptoms over time among older adults wih chronic obstructive pulmonary disease and heart failure. Arch Intern Med 2007;167:2503–8.
25. Bekelman DB, Rumsfeld JS, Havranek EP, et al. Symptom burden, depression, and spiritual well-being: a comparison of heart failure and advanced cancer patients. J Gen Intern Med 2009;24:592–8.
26. Ahluwalia SC, Gross CP, Chaudhry SI, et al. Impact of comorbidity on mortality among older persons with advanced heart failure. J Gen Intern Med 2012;27:513–9.
27. Ahluwalia SC, Gross CP, Chaudhry SI, et al. Change in comorbidity prevalence with advancing age among persons with heart failure. J Gen Intern Med 2011;26:1145–51.
28. Corrigan JM, Donaldson MS, Kohn LT, et al. A new health system for the 21st century. crossing the quality chasm. Washington, DC: Institute of Medicine, National Academy of Sciences, National Academies Press; 2001.
29. Kirchhoff KT, Hammes BJ, Kehl KA, et al. Effect of a disease-specific advance care planning intervention on end-of-life care. J Am Geriatr Soc 2012;60:946–50.
30. Kirchhoff KT, Hammes BJ, Kehl KA, et al. Effect of a disease-specific planning intervention on surrogate understanding of patient goals for future medical treatment. J Am Geriatr Soc 2010;58:1233–40.
31. Janssen DJ, Engelberg RA, Wouters EF, Curtis JR. Advance care planning for patients with COPD: past, present and future. Patient Educ Couns 2012;86:19–24.
32. Aldred H, Gott M, Gariballa S. Advanced heart failure: Impact on older patients and informal carers. J Adv Nurs 2005;49:116–24.
33. Zhang B, Wright AA, Huskamp HA, et al. Health care costs in the last week of life: Associations with end-of-life conversations. Arch Intern Med 2009;169:480–8.
34. Wright AA, Zhang B, Ray A, et al. Associations between end-of-life discussions, patient mental health, medical care near death, and caregiver bereavement adjustment. JAMA 2008;300:1665–73.
35. Mack JW, Smith TJ. Reasons why physicians do not have discussions about poor prognosis, why it matters, and what can be improved. J Clin Oncol 2012;30:2715–7.
36. Detering KM, Hancock AD, Reade MC, Silvester W. The impact of advance care planning on end of life care in elderly patients: randomised controlled trial. BMJ 2010;340:c1345.
37. Schwartz CE, Wheeler HB, Hammes B, et al. Early intervention in planning end-of-life care with ambulatory geriatric patients: results of a pilot trial. Arch Intern Med 2002;162:1611–8.
38. Caldwell PH, Arthur HM, Demers C. Preferences of patients with heart failure for prognosis communication. Can J Cardiol 2007;23:791–6.
39. Bekelman DB, Nowels Ct, Retrum JH, et al. Giving voice to patients’ and family caregivers’ needs in chronic heart failure: implications for palliative care programs. J Palliat Med 2011;14:1317–24.
40. Harding R, Selman L, Beynon T, et al. Meeting the communication and information needs of chronic heart failure patients. J Pain Symptom Manage 2008;36:149–56.
41. Strachan PH, Ross H, Dodek PM, et al. Mind the gap: opportunities for improving end-of-life care for patients with advanced heart failure. Can J Cardiol 2009;25:635–40.
42. Ågård A, Hermerén G, Herlitz J. When is a patient with heart failure adequately informed? A study of patients’ knowledge of and attitudes toward medical information. Heart Lung 2004;33:219–26.
43. Gott M, Small N, Barnes S, et al. Older people’s views of a good death in heart failure: implications for palliative care provision. Soc Sci Med 2008;67:1113–21.
44. Murray SA, Boyd K, Kendall M, et al. Dying of lung cancer or cardiac failure: prospective qualitative interview study of patients and their carers in the community. BMJ 2002;325:929.
45. Ahluwalia SC, Levin JR, Lorenz KA, et al. Missed opportunities for advance care planning communication during outpatient clinic visits. J Gen Intern Med 2012;27:445–51.
46. Rodriguez KL, Appelt CJ, Switzer GE, et al. “They diagnosed bad heart”: a qualitative exploration of patients’ knowledge about and experiences with heart failure. Heart Lung. 2008;37:257–65.
47. Remme WJ,McMurray JJ, Rauch B, et al. Public awareness of heart failure in Europe: first results from SHAPE. Eur Heart J 2005;22:2413e21.
48. Golin CE, Wenger NS, Liu H, et al. A prospective study of patient-physician communication about resuscitation. J Am Geriatr Soc 2000;48(5 Suppl):S52–60.
49. Selman L, Harding R, Beynon T, et al. Improving end of life care for patients with chronic heart failure: ‘let’s hope it’ll get better when I know in my heart of hearts it won’t’. Heart 2007;93:963–7.
50. Barnes S, Gott M, Payne S, et al. Communication in heart failure: Perspectives from older people and primary care professionals. Health Soc Care Comm 2006;14:482–90.
51. Brännström M, Ekman I, Norberg A, et al. Living with severe chronic heart failure in palliative advanced home care. Eur J Cardiovasc Nurs 2006;5:295–302.
52. Barclay S, Momen N, Case-Upton S, et al. End-of-life care conversations with heart failure patients: a systematic literature review and narrative synthesis. Br J Gen Pract 2011;61:e49–62.
53. Whitney SN, McCullough LB, Fruge E, et al. Beyond breaking bad news: the roles of hope and hopefulness. Cancer 2008;113:442–5.
54. Tung EE, North F. Advance care planning in the primary care setting: a comparison of attending staff and resident barriers. Am J Hosp Palliat Care 2009;26:456–63.
55. Boyd K, Murray S, Kendall M, et al. Living with advanced heart failure: A prospective, community based study of patients and their carers. Eur J Heart Fail 2004;6:585–91.
56. Borbasi S, Wotton K, Redden M, et al. Letting go: A qualitative study of acute care and community nurses’ perceptions of a ‘good’ versus a ‘bad’ death. Austr Crit Care 2005 2005;18:104–13.
57. Hanratty B, Hibbert D, Mair F, et al. Doctors’ perceptions of palliative care for heart failure: Focus group study. BMJ 2002;325:581–5.
58. Fallowfield L, Jenkins V, Farewell V, et al. Efficacy of a communication skills training model for oncologists: a randomized controlled trial. Lancet 2002;359:650–6.
59. Platt F, Keller V. Empathic communication: a teachable and learnable skill. J Gen Intern Med 1994;9:222–6.
60. Morse D, Edwardsen E, Gordon H. Missed opportunities for interval empathy in lung cancer communication. Arch Intern Med 2008;22;168:1853–8.
61. Epstein R, Hadee T, Carroll J, et al. “Could this be something serious?” reassurance, uncertainty, and empathy in response to patients’ expressions of worry. J Gen Intern Med 2007;22:1731–9.
62. Stewart M. What is a successful doctor-patient interview? A study of interactions and outcomes. Soc Sci Med 1984;19:
167–75.
63. Wotton K, Borbasi S, Redden M. When all else has failed. Nurses’ perception of factors influencing palliative care for patients with end-stage heart failure. J Cardiovasc Nurs 2005;20:18–25.
64. Willems DL, Hak A, Visser F, Van der Wal G. Thoughts of patients with advanced heart failure on dying. Palliat Med 2004;18:564–72.
65. Briggs L, Kirchhoff K, Hammes B, et al. Patient-centered advance care planning in special patient populations: a pilot study. J Prof Nurs 2004;20:47–58.
66. Hammes B, Rooney B. Death and EOL planning in one midwestern community. Arch Intern Med 1998;158:390.
67. Lilly C, DeMeo D, Sonna L, et al. An intensive communication intervention for the critically ill. Am J Med 2000;109:469–75.
68. Davidson P, Macdonald P, Newton P, et al. End stage heart failure patients: Palliative care in general practice. Aust Fam Physician 2010;39:920.
69. Goodlin S, Quill T, Arnold R. Communication and decision-making about prognosis in heart failure care. J Card Fail 2008;14:106–13.
70. Analysis of U.S. hospital palliative care programs 2010 snapshot. Center to Advance Palliative Care (CAPC). Accessed 24 Nov 2014 at www.capc.org/news-and-events/releases/analysis-of-us-hospital-palliative-care-programs-2010-snapshot.pdf.
71. Back AL, Arnold RM, Baile WF, et al. Approaching difficult communication tasks in oncology. CA Cancer J Clin 2005;55:164–77.
72. Sudore RL, Fried TR, Redefining the “planning” in advance care planning: preparing for end-of-life decision making. Ann Intern Med 2010;153:256–61.
73. Lo B, Quill T, Tulsky J. Discussing palliative care with patients. Ann Intern Med 1999;130:744–9.
74. Quill T, Arnold R, Platt F. I wish things were different: Expressing wishes in response to loss, futility, and unrealistic hopes. Ann Intern Med 2001;135:551–5.
75. Pollak K, Arnold R, Jeffreys A, et al. Oncologist communication about emotion during visits with patients with advanced cancer. J Clin Oncol 2007;25:5748–52.
76. Back AL, Anderson WG, Bunch L, et al. Communication about cancer near the end of life. Cancer 2008;113(S7):1897–910.
77. Dries D, Exner D, Gersh B, et al. Racial differences in the outcome of left ventricular dysfunction. N Engl J Med 1999;340:609–16.
78. Alexander M, Grumbach K, Selby J, et al. Hospitalization for congestive heart failure. Explaining racial differences. JAMA 1995;274:1037–42.
79. Afzal A, Ananthasubramaniam K, Sharma N, et al. Racial differences in patients with heart failure. Clin Cardiol 1999;22:791–4.
80. Blackhall L, Murphy S, Frank G, et al. Ethnicity and attitudes toward patient autonomy. JAMA 1995;274:820–5.
81. Huang X, Butow P, Meiser B, et al. Attitudes and information needs of chinese migrant cancer patients and their relatives. Aust N Z J Med 1999;29:207–13.
82. Tan T, Teo F, Wong K, et al. Cancer: to tell or not to tell? Singapore Med J 1993;34:202–3.
83. Gorgaki S, Kalaidopoulou O, Liarmakopoulos I, et al. Nurses’ attitudes toward truthful communication with patients with cancer. A Greek study. Cancer Nurs 2002;25:436–41.
84. Harris J, Shao J, Sugarman J. Disclosure of cancer diagnosis and prognosis in northern Tanzania. Soc Sci Med 2003;56:905–13.
85. Goldstein D, Thewes B, Butow P. Communicating in a multicultural society. II: Greek community attitudes towards cancer in Australia. Intern Med J 2002;32:289–96.
86. Beyene Y. Medical disclosure and refugees. Telling bad news to Ethiopian patients. West J Med 1992;157:328–32.
87. Matsumura S, Bito S, Liu H, et al. Acculturation of attitudes toward end-of-life care: A cross-cultural survey of Japanese Americans and Japanese. J Gen Intern Med 2002;17:531–9.
88. Yick AG, Gupta R. Chinese cultural dimensions of death, dying, and bereavement: Focus group findings. J Cult Divers 2002 Summer;9:32–42.
89. Frank G, Blackhall L, Murphy S, et al. Ambiguity and hope: Disclosure preferences of less acculturated elderly Mexican Americans concerning terminal cancer—A case story. Camb Q Healthc Ethics 2002;11:117–26.
90. Hern HJ, Koenig B, Moore L, et al. The difference that culture can make in end-of-life decisionmaking. Camb Q Healthc Ethics 1998;7:27–48.
91. Kagawa-Singer M, Kassim-Lakha S. A strategy to reduce cross-cultural miscommunication and increase the likelihood of improving health outcomes. Acad Med 2003;78:577–87.
92. Barnett MM, Fisher JD, Cooke H, et al. Breaking bad news: consultants’ experience, previous education, and views on educational format and timing. Med Educ 2007;41:947–56.
93. Moore P, Rivera Mercado S, Grez Artigues M, Lawrie T. Communication skills training for healthcare professionals working with people who have cancer. Cochrane Database Syst Rev 2013;3CD003751.
94. Alelwani S, Ahmed Y. Medical training for communication of bad news: A literature review. J Educ Health Promot 2014;3
95. Delvaux N, Razavi D, Marchal S, et al. Effects of a 105 hour psychological training program on attitudes, communication skills and occupational stress in oncology: a randomised study. Br J Cancer 2004;90:106–14.
96. Baile WF, Lenzi R, Kudelka AP, et al. Improving physician-patient communication in cancer care: Outcome of a workshop for oncologists. J Cancer Educ 1997;12:166–73.
97. Back AL, Arnold RM, Baile WF, et al. Efficacy of communication skills training for giving bad news and discussing transitions to palliative care. Arch Intern Med 2007;167:453–60.
98. Gelfman LP, Lindenberger E, Fernandez H, et al. The effectiveness of the Geritalk communication skills course: a real-time assessment of skill acquisition and deliberate practice. J Pain Sympt Manage 2014;48:738–44.
99. Kelley AS, Back AL, Arnold RM, et al. Geritalk: communication skills training for geriatric and palliative medicine fellows. J Am Geriatr Soc 2012;60:332–7.
100. Institute of Medicine. Dying in America: Improving quality and honoring individual preferences near the end of life. Washington, DC: National Academies Press; 2014.
101. Allen LA, Stevenson LW, Grady KL, et al. Decision making in advanced heart failure: a scientific statement From the American Heart Association. Circulation 2012;125:1928–52.
102. Ditto PH, Jacobson JA, Smucker WD, et al. Context changes choices: a prospective study of the effects of hospitalization on life-sustaining treatment preferences. Med Decis Making 2006;26:313–22.
1. American Heart Association. Heart disease and stroke statistics—2008 update. [Internet]. Available at www.americanheart.org/downloadable/heart/1200078608862HS_Stats%202008.final.pdf.
2. Hunt SA, Abraham WT, Chin MH, et al. ACC/AHA 2005 guideline update for the diagnosis and management of chronic heart failure in the adult—summary article. Circulation 2005;112:1825–52.
3. Masoudi FA, Havranek EP, Krumholz HM. The burden of chronic congestive heart failure in older persons: magnitude and implications for policy and research. Heart Fail Rev 2002;7:9–16.
4. McMurray JJ PM. Heart failure. Lancet 2005;365:1877–89.
5. Unroe KT, Greiner MA, Hernandez AF, et al. Resource use in the last 6 months of life among medicare beneficiaries with heart failure, 2000-2007. Arch Intern Med 2011;171:196–203.
6. Lunney JR, Lynn J, Foley DJ, et al. Patterns of functional decline at the end of life. JAMA 2003;289:2387–92.
7. Ko D, Alter D, Austin P, et al. Life expectancy after an index hospitalization for patients with heart failure: A population-based study. Am Heart J 2008;155:324–31.
8. Setoguchi S, Stevenson LW, Schneeweiss S. Repeated hospitalizations predict mortality in the community population with heart failure. Am Heart J 2007;154:260–6.
9. Jong P, Vowinskel E, Liu PP, et al. Prognosis and determinants for survival in patients newly hospitalized for heart failure. Arch Intern Med 2002;162:1689-94.
10. Thom T, Haase N, Rodamond W, et al. Heart disease and stroke statistics- 2006 update. Circulation 2006;113:e85-e151.
11. Shahar E, Lee S, Kim J, et al. Hospitalized heart failure: Rates and long-term mortality. J Card Fail 2004;10:374–9.
12. Kirkpatrick JN, Guger CJ, Arnsdorf MF, et al. Advance directives in the cardiac care unit. Am Heart J 2007;154:477–81.
13. Orn S, Dickstein K. How do heart failure patients die? Eur Heart J. 2002;4(suppl D).
14. Juenger J, Schellberg D, Kraemer S, et al. Health related quality of life in patients with congestive heart failure: comparison with other chronic disease and relation to functional variables. Heart 2002;87:235–41.
15. Steptoe A, Mohabir A, Mahon NG, et al. Health related quality of life and psychological wellbeing in patients with dilated cardiomyopathy. Heart 2000;83:645–50.
16. Johansson P, Agnebrink M, Dahlstrom U, et al. Measurement of health-related quality of life in chronic heart failure, form a nursing perspective--a review of the literature. Eur J Cardiovasc Nurs 2004;3:7–20.
17. Levenson J, McCarthy E, Lynn J, et al. The last six months of life for patients with congestive heart failure. J Am Geriatr Soc 2000;48(Suppl 5):S101–S109.
18. Sullivan M, Levy W, Russo J, Spertus J. Depression and health status in patients with advanced heart failure: a prospective study in tertiary care. J Card Fail 2004;10:390–6.
19. Bekelman DB, Havranek EP, Becker DM, et al. Symptoms, depression, and quality of life in patients with heart failure. J Card Fail 2007;13:643–8.
20. Godfrey CM, Harrison MB, Friedberg E, et al. The symptom of pain in individuals recently hospitalized for heart failure. J Cardiovasc Nurs 2007;22:368–74.
21. McCarthy M, Lay M, Addington-Hall J. Dying from heart disease. J R Coll Physicians Lond 1996;30:325–8.
22. Norgren L SS. Symptoms experienced in the last six months of life in patients with end-stage heart failure. Eur J Cardiovasc Nurs 2003;2:213–7.
23. Zambroski CH, Moser DK, Bhat G, et al. Impact of symptom prevalence and symptom burden on quality of life in patients with heart failure. Eur J Cardiovasc Nurs 2005;4:198–206.
24. Walke LM, Byers AL, Tinetti ME, et al. Range and severity of symptoms over time among older adults wih chronic obstructive pulmonary disease and heart failure. Arch Intern Med 2007;167:2503–8.
25. Bekelman DB, Rumsfeld JS, Havranek EP, et al. Symptom burden, depression, and spiritual well-being: a comparison of heart failure and advanced cancer patients. J Gen Intern Med 2009;24:592–8.
26. Ahluwalia SC, Gross CP, Chaudhry SI, et al. Impact of comorbidity on mortality among older persons with advanced heart failure. J Gen Intern Med 2012;27:513–9.
27. Ahluwalia SC, Gross CP, Chaudhry SI, et al. Change in comorbidity prevalence with advancing age among persons with heart failure. J Gen Intern Med 2011;26:1145–51.
28. Corrigan JM, Donaldson MS, Kohn LT, et al. A new health system for the 21st century. crossing the quality chasm. Washington, DC: Institute of Medicine, National Academy of Sciences, National Academies Press; 2001.
29. Kirchhoff KT, Hammes BJ, Kehl KA, et al. Effect of a disease-specific advance care planning intervention on end-of-life care. J Am Geriatr Soc 2012;60:946–50.
30. Kirchhoff KT, Hammes BJ, Kehl KA, et al. Effect of a disease-specific planning intervention on surrogate understanding of patient goals for future medical treatment. J Am Geriatr Soc 2010;58:1233–40.
31. Janssen DJ, Engelberg RA, Wouters EF, Curtis JR. Advance care planning for patients with COPD: past, present and future. Patient Educ Couns 2012;86:19–24.
32. Aldred H, Gott M, Gariballa S. Advanced heart failure: Impact on older patients and informal carers. J Adv Nurs 2005;49:116–24.
33. Zhang B, Wright AA, Huskamp HA, et al. Health care costs in the last week of life: Associations with end-of-life conversations. Arch Intern Med 2009;169:480–8.
34. Wright AA, Zhang B, Ray A, et al. Associations between end-of-life discussions, patient mental health, medical care near death, and caregiver bereavement adjustment. JAMA 2008;300:1665–73.
35. Mack JW, Smith TJ. Reasons why physicians do not have discussions about poor prognosis, why it matters, and what can be improved. J Clin Oncol 2012;30:2715–7.
36. Detering KM, Hancock AD, Reade MC, Silvester W. The impact of advance care planning on end of life care in elderly patients: randomised controlled trial. BMJ 2010;340:c1345.
37. Schwartz CE, Wheeler HB, Hammes B, et al. Early intervention in planning end-of-life care with ambulatory geriatric patients: results of a pilot trial. Arch Intern Med 2002;162:1611–8.
38. Caldwell PH, Arthur HM, Demers C. Preferences of patients with heart failure for prognosis communication. Can J Cardiol 2007;23:791–6.
39. Bekelman DB, Nowels Ct, Retrum JH, et al. Giving voice to patients’ and family caregivers’ needs in chronic heart failure: implications for palliative care programs. J Palliat Med 2011;14:1317–24.
40. Harding R, Selman L, Beynon T, et al. Meeting the communication and information needs of chronic heart failure patients. J Pain Symptom Manage 2008;36:149–56.
41. Strachan PH, Ross H, Dodek PM, et al. Mind the gap: opportunities for improving end-of-life care for patients with advanced heart failure. Can J Cardiol 2009;25:635–40.
42. Ågård A, Hermerén G, Herlitz J. When is a patient with heart failure adequately informed? A study of patients’ knowledge of and attitudes toward medical information. Heart Lung 2004;33:219–26.
43. Gott M, Small N, Barnes S, et al. Older people’s views of a good death in heart failure: implications for palliative care provision. Soc Sci Med 2008;67:1113–21.
44. Murray SA, Boyd K, Kendall M, et al. Dying of lung cancer or cardiac failure: prospective qualitative interview study of patients and their carers in the community. BMJ 2002;325:929.
45. Ahluwalia SC, Levin JR, Lorenz KA, et al. Missed opportunities for advance care planning communication during outpatient clinic visits. J Gen Intern Med 2012;27:445–51.
46. Rodriguez KL, Appelt CJ, Switzer GE, et al. “They diagnosed bad heart”: a qualitative exploration of patients’ knowledge about and experiences with heart failure. Heart Lung. 2008;37:257–65.
47. Remme WJ,McMurray JJ, Rauch B, et al. Public awareness of heart failure in Europe: first results from SHAPE. Eur Heart J 2005;22:2413e21.
48. Golin CE, Wenger NS, Liu H, et al. A prospective study of patient-physician communication about resuscitation. J Am Geriatr Soc 2000;48(5 Suppl):S52–60.
49. Selman L, Harding R, Beynon T, et al. Improving end of life care for patients with chronic heart failure: ‘let’s hope it’ll get better when I know in my heart of hearts it won’t’. Heart 2007;93:963–7.
50. Barnes S, Gott M, Payne S, et al. Communication in heart failure: Perspectives from older people and primary care professionals. Health Soc Care Comm 2006;14:482–90.
51. Brännström M, Ekman I, Norberg A, et al. Living with severe chronic heart failure in palliative advanced home care. Eur J Cardiovasc Nurs 2006;5:295–302.
52. Barclay S, Momen N, Case-Upton S, et al. End-of-life care conversations with heart failure patients: a systematic literature review and narrative synthesis. Br J Gen Pract 2011;61:e49–62.
53. Whitney SN, McCullough LB, Fruge E, et al. Beyond breaking bad news: the roles of hope and hopefulness. Cancer 2008;113:442–5.
54. Tung EE, North F. Advance care planning in the primary care setting: a comparison of attending staff and resident barriers. Am J Hosp Palliat Care 2009;26:456–63.
55. Boyd K, Murray S, Kendall M, et al. Living with advanced heart failure: A prospective, community based study of patients and their carers. Eur J Heart Fail 2004;6:585–91.
56. Borbasi S, Wotton K, Redden M, et al. Letting go: A qualitative study of acute care and community nurses’ perceptions of a ‘good’ versus a ‘bad’ death. Austr Crit Care 2005 2005;18:104–13.
57. Hanratty B, Hibbert D, Mair F, et al. Doctors’ perceptions of palliative care for heart failure: Focus group study. BMJ 2002;325:581–5.
58. Fallowfield L, Jenkins V, Farewell V, et al. Efficacy of a communication skills training model for oncologists: a randomized controlled trial. Lancet 2002;359:650–6.
59. Platt F, Keller V. Empathic communication: a teachable and learnable skill. J Gen Intern Med 1994;9:222–6.
60. Morse D, Edwardsen E, Gordon H. Missed opportunities for interval empathy in lung cancer communication. Arch Intern Med 2008;22;168:1853–8.
61. Epstein R, Hadee T, Carroll J, et al. “Could this be something serious?” reassurance, uncertainty, and empathy in response to patients’ expressions of worry. J Gen Intern Med 2007;22:1731–9.
62. Stewart M. What is a successful doctor-patient interview? A study of interactions and outcomes. Soc Sci Med 1984;19:
167–75.
63. Wotton K, Borbasi S, Redden M. When all else has failed. Nurses’ perception of factors influencing palliative care for patients with end-stage heart failure. J Cardiovasc Nurs 2005;20:18–25.
64. Willems DL, Hak A, Visser F, Van der Wal G. Thoughts of patients with advanced heart failure on dying. Palliat Med 2004;18:564–72.
65. Briggs L, Kirchhoff K, Hammes B, et al. Patient-centered advance care planning in special patient populations: a pilot study. J Prof Nurs 2004;20:47–58.
66. Hammes B, Rooney B. Death and EOL planning in one midwestern community. Arch Intern Med 1998;158:390.
67. Lilly C, DeMeo D, Sonna L, et al. An intensive communication intervention for the critically ill. Am J Med 2000;109:469–75.
68. Davidson P, Macdonald P, Newton P, et al. End stage heart failure patients: Palliative care in general practice. Aust Fam Physician 2010;39:920.
69. Goodlin S, Quill T, Arnold R. Communication and decision-making about prognosis in heart failure care. J Card Fail 2008;14:106–13.
70. Analysis of U.S. hospital palliative care programs 2010 snapshot. Center to Advance Palliative Care (CAPC). Accessed 24 Nov 2014 at www.capc.org/news-and-events/releases/analysis-of-us-hospital-palliative-care-programs-2010-snapshot.pdf.
71. Back AL, Arnold RM, Baile WF, et al. Approaching difficult communication tasks in oncology. CA Cancer J Clin 2005;55:164–77.
72. Sudore RL, Fried TR, Redefining the “planning” in advance care planning: preparing for end-of-life decision making. Ann Intern Med 2010;153:256–61.
73. Lo B, Quill T, Tulsky J. Discussing palliative care with patients. Ann Intern Med 1999;130:744–9.
74. Quill T, Arnold R, Platt F. I wish things were different: Expressing wishes in response to loss, futility, and unrealistic hopes. Ann Intern Med 2001;135:551–5.
75. Pollak K, Arnold R, Jeffreys A, et al. Oncologist communication about emotion during visits with patients with advanced cancer. J Clin Oncol 2007;25:5748–52.
76. Back AL, Anderson WG, Bunch L, et al. Communication about cancer near the end of life. Cancer 2008;113(S7):1897–910.
77. Dries D, Exner D, Gersh B, et al. Racial differences in the outcome of left ventricular dysfunction. N Engl J Med 1999;340:609–16.
78. Alexander M, Grumbach K, Selby J, et al. Hospitalization for congestive heart failure. Explaining racial differences. JAMA 1995;274:1037–42.
79. Afzal A, Ananthasubramaniam K, Sharma N, et al. Racial differences in patients with heart failure. Clin Cardiol 1999;22:791–4.
80. Blackhall L, Murphy S, Frank G, et al. Ethnicity and attitudes toward patient autonomy. JAMA 1995;274:820–5.
81. Huang X, Butow P, Meiser B, et al. Attitudes and information needs of chinese migrant cancer patients and their relatives. Aust N Z J Med 1999;29:207–13.
82. Tan T, Teo F, Wong K, et al. Cancer: to tell or not to tell? Singapore Med J 1993;34:202–3.
83. Gorgaki S, Kalaidopoulou O, Liarmakopoulos I, et al. Nurses’ attitudes toward truthful communication with patients with cancer. A Greek study. Cancer Nurs 2002;25:436–41.
84. Harris J, Shao J, Sugarman J. Disclosure of cancer diagnosis and prognosis in northern Tanzania. Soc Sci Med 2003;56:905–13.
85. Goldstein D, Thewes B, Butow P. Communicating in a multicultural society. II: Greek community attitudes towards cancer in Australia. Intern Med J 2002;32:289–96.
86. Beyene Y. Medical disclosure and refugees. Telling bad news to Ethiopian patients. West J Med 1992;157:328–32.
87. Matsumura S, Bito S, Liu H, et al. Acculturation of attitudes toward end-of-life care: A cross-cultural survey of Japanese Americans and Japanese. J Gen Intern Med 2002;17:531–9.
88. Yick AG, Gupta R. Chinese cultural dimensions of death, dying, and bereavement: Focus group findings. J Cult Divers 2002 Summer;9:32–42.
89. Frank G, Blackhall L, Murphy S, et al. Ambiguity and hope: Disclosure preferences of less acculturated elderly Mexican Americans concerning terminal cancer—A case story. Camb Q Healthc Ethics 2002;11:117–26.
90. Hern HJ, Koenig B, Moore L, et al. The difference that culture can make in end-of-life decisionmaking. Camb Q Healthc Ethics 1998;7:27–48.
91. Kagawa-Singer M, Kassim-Lakha S. A strategy to reduce cross-cultural miscommunication and increase the likelihood of improving health outcomes. Acad Med 2003;78:577–87.
92. Barnett MM, Fisher JD, Cooke H, et al. Breaking bad news: consultants’ experience, previous education, and views on educational format and timing. Med Educ 2007;41:947–56.
93. Moore P, Rivera Mercado S, Grez Artigues M, Lawrie T. Communication skills training for healthcare professionals working with people who have cancer. Cochrane Database Syst Rev 2013;3CD003751.
94. Alelwani S, Ahmed Y. Medical training for communication of bad news: A literature review. J Educ Health Promot 2014;3
95. Delvaux N, Razavi D, Marchal S, et al. Effects of a 105 hour psychological training program on attitudes, communication skills and occupational stress in oncology: a randomised study. Br J Cancer 2004;90:106–14.
96. Baile WF, Lenzi R, Kudelka AP, et al. Improving physician-patient communication in cancer care: Outcome of a workshop for oncologists. J Cancer Educ 1997;12:166–73.
97. Back AL, Arnold RM, Baile WF, et al. Efficacy of communication skills training for giving bad news and discussing transitions to palliative care. Arch Intern Med 2007;167:453–60.
98. Gelfman LP, Lindenberger E, Fernandez H, et al. The effectiveness of the Geritalk communication skills course: a real-time assessment of skill acquisition and deliberate practice. J Pain Sympt Manage 2014;48:738–44.
99. Kelley AS, Back AL, Arnold RM, et al. Geritalk: communication skills training for geriatric and palliative medicine fellows. J Am Geriatr Soc 2012;60:332–7.
100. Institute of Medicine. Dying in America: Improving quality and honoring individual preferences near the end of life. Washington, DC: National Academies Press; 2014.
101. Allen LA, Stevenson LW, Grady KL, et al. Decision making in advanced heart failure: a scientific statement From the American Heart Association. Circulation 2012;125:1928–52.
102. Ditto PH, Jacobson JA, Smucker WD, et al. Context changes choices: a prospective study of the effects of hospitalization on life-sustaining treatment preferences. Med Decis Making 2006;26:313–22.
Helping Patients Set Goals for Better Health
It has long been recognized that setting realistic and achievable short-term goals can help people improve their health long-term. But does goal setting work for older adults with multiple health issues or cognitive impairment? Researchers from the University of Pittsburgh in Pennsylvania conducted a study to find out.
They asked 27 patients from the University of Pittsburgh Medical Center Benedum Geriatric Center, a multidisciplinary outpatient geriatric clinic, to use goal attainment scaling (GAS) to set 2 to 4 activity-based goals to work toward. Goal attainment scaling is a tool for setting quantifiable patient-centered goals, measuring improvement toward the goals, and facilitating communication of shared priorities between patient and provider, the researchers say. They note that although originally designed for use in mental health, GAS has been used effectively as an outcome measure with older adults in multiple settings.
Related: Infusing Gerontologic Practice Into PACT
In a guided hour-by-hour review of their typical day, patients were directed to choose activities from 3 major domains: self-care, productivity, and leisure. Using the GAS 5-point scale, the physicians helped patients specify goals and determine the criteria for attaining them. Physicians were asked to rate on a 5-point scale whether their patients’ goals were realistic. The physicians felt that 100% of the goals were realistic, and 93% of the goals were achievable.
The researchers also measured cognitive function, physical function, and disability. About half the patients had ≥ 10 medical diagnoses, and most had both medical and psychiatric diagnoses. Fourteen patients reported some form of caregiver support. Fifteen were evaluated as having cognitive impairment.
Related: Development and Evaluation of a Geriatric Mood Management Program
At baseline, 25 participants demonstrated the ability to set a minimum of 2 goals. One patient chose not to, and another, who had dementia, could not. In all, the participants set 60 goals, half of them addressing leisure activity, such as getting exercise, playing cards, and attending church. Thirty percent of goals reflected patients’ desires to improve their personal care, for example, improving functional mobility. The final 20% of goals addressed productivity, such as managing a budget and finances.
At the 8-week follow-up, patients evaluated their current performance of the goal activities in relation to the indicators for levels of goal attainment they had established at baseline. They were also asked to rate their satisfaction with the process on a 5-point scale.
At the follow-up, 21 participants reported gains in ≥ 1 of their goal areas, reflected in significant changes in their GAS scores (P < .001). The patients were also highly satisfied with the process and their progress. Interesting, and a bit surprising, the patients who had no caregiver support were more likely to achieve their goals.
Related: Mobility in the Elderly: Speed Counts Most
The researchers note that because they did not include any additional intervention beyond goal setting, their findings support existing research that suggests the process of personal goal setting is a strategy in and of itself for increasing motivation toward achieving goals.
Primary care has become the preferred setting for chronic disease management, the researchers say. A patient-centered model could help solve the “impending crisis” of a growing population with many chronic conditions. They suggest that using GAS might be a way to enhance patient engagement, motivation, and satisfaction; improve provider-patient communication; and foster collaborative or shared priorities between patients and health care providers.
Source
Toto PE, Skidmore ER, Terhorst L, Rosen J, Weiner DK. Arch Gerontol Geriatr. 2015;60(1):16-21.
doi: 10.1016/j.archger.2014.10.022.
It has long been recognized that setting realistic and achievable short-term goals can help people improve their health long-term. But does goal setting work for older adults with multiple health issues or cognitive impairment? Researchers from the University of Pittsburgh in Pennsylvania conducted a study to find out.
They asked 27 patients from the University of Pittsburgh Medical Center Benedum Geriatric Center, a multidisciplinary outpatient geriatric clinic, to use goal attainment scaling (GAS) to set 2 to 4 activity-based goals to work toward. Goal attainment scaling is a tool for setting quantifiable patient-centered goals, measuring improvement toward the goals, and facilitating communication of shared priorities between patient and provider, the researchers say. They note that although originally designed for use in mental health, GAS has been used effectively as an outcome measure with older adults in multiple settings.
Related: Infusing Gerontologic Practice Into PACT
In a guided hour-by-hour review of their typical day, patients were directed to choose activities from 3 major domains: self-care, productivity, and leisure. Using the GAS 5-point scale, the physicians helped patients specify goals and determine the criteria for attaining them. Physicians were asked to rate on a 5-point scale whether their patients’ goals were realistic. The physicians felt that 100% of the goals were realistic, and 93% of the goals were achievable.
The researchers also measured cognitive function, physical function, and disability. About half the patients had ≥ 10 medical diagnoses, and most had both medical and psychiatric diagnoses. Fourteen patients reported some form of caregiver support. Fifteen were evaluated as having cognitive impairment.
Related: Development and Evaluation of a Geriatric Mood Management Program
At baseline, 25 participants demonstrated the ability to set a minimum of 2 goals. One patient chose not to, and another, who had dementia, could not. In all, the participants set 60 goals, half of them addressing leisure activity, such as getting exercise, playing cards, and attending church. Thirty percent of goals reflected patients’ desires to improve their personal care, for example, improving functional mobility. The final 20% of goals addressed productivity, such as managing a budget and finances.
At the 8-week follow-up, patients evaluated their current performance of the goal activities in relation to the indicators for levels of goal attainment they had established at baseline. They were also asked to rate their satisfaction with the process on a 5-point scale.
At the follow-up, 21 participants reported gains in ≥ 1 of their goal areas, reflected in significant changes in their GAS scores (P < .001). The patients were also highly satisfied with the process and their progress. Interesting, and a bit surprising, the patients who had no caregiver support were more likely to achieve their goals.
Related: Mobility in the Elderly: Speed Counts Most
The researchers note that because they did not include any additional intervention beyond goal setting, their findings support existing research that suggests the process of personal goal setting is a strategy in and of itself for increasing motivation toward achieving goals.
Primary care has become the preferred setting for chronic disease management, the researchers say. A patient-centered model could help solve the “impending crisis” of a growing population with many chronic conditions. They suggest that using GAS might be a way to enhance patient engagement, motivation, and satisfaction; improve provider-patient communication; and foster collaborative or shared priorities between patients and health care providers.
Source
Toto PE, Skidmore ER, Terhorst L, Rosen J, Weiner DK. Arch Gerontol Geriatr. 2015;60(1):16-21.
doi: 10.1016/j.archger.2014.10.022.
It has long been recognized that setting realistic and achievable short-term goals can help people improve their health long-term. But does goal setting work for older adults with multiple health issues or cognitive impairment? Researchers from the University of Pittsburgh in Pennsylvania conducted a study to find out.
They asked 27 patients from the University of Pittsburgh Medical Center Benedum Geriatric Center, a multidisciplinary outpatient geriatric clinic, to use goal attainment scaling (GAS) to set 2 to 4 activity-based goals to work toward. Goal attainment scaling is a tool for setting quantifiable patient-centered goals, measuring improvement toward the goals, and facilitating communication of shared priorities between patient and provider, the researchers say. They note that although originally designed for use in mental health, GAS has been used effectively as an outcome measure with older adults in multiple settings.
Related: Infusing Gerontologic Practice Into PACT
In a guided hour-by-hour review of their typical day, patients were directed to choose activities from 3 major domains: self-care, productivity, and leisure. Using the GAS 5-point scale, the physicians helped patients specify goals and determine the criteria for attaining them. Physicians were asked to rate on a 5-point scale whether their patients’ goals were realistic. The physicians felt that 100% of the goals were realistic, and 93% of the goals were achievable.
The researchers also measured cognitive function, physical function, and disability. About half the patients had ≥ 10 medical diagnoses, and most had both medical and psychiatric diagnoses. Fourteen patients reported some form of caregiver support. Fifteen were evaluated as having cognitive impairment.
Related: Development and Evaluation of a Geriatric Mood Management Program
At baseline, 25 participants demonstrated the ability to set a minimum of 2 goals. One patient chose not to, and another, who had dementia, could not. In all, the participants set 60 goals, half of them addressing leisure activity, such as getting exercise, playing cards, and attending church. Thirty percent of goals reflected patients’ desires to improve their personal care, for example, improving functional mobility. The final 20% of goals addressed productivity, such as managing a budget and finances.
At the 8-week follow-up, patients evaluated their current performance of the goal activities in relation to the indicators for levels of goal attainment they had established at baseline. They were also asked to rate their satisfaction with the process on a 5-point scale.
At the follow-up, 21 participants reported gains in ≥ 1 of their goal areas, reflected in significant changes in their GAS scores (P < .001). The patients were also highly satisfied with the process and their progress. Interesting, and a bit surprising, the patients who had no caregiver support were more likely to achieve their goals.
Related: Mobility in the Elderly: Speed Counts Most
The researchers note that because they did not include any additional intervention beyond goal setting, their findings support existing research that suggests the process of personal goal setting is a strategy in and of itself for increasing motivation toward achieving goals.
Primary care has become the preferred setting for chronic disease management, the researchers say. A patient-centered model could help solve the “impending crisis” of a growing population with many chronic conditions. They suggest that using GAS might be a way to enhance patient engagement, motivation, and satisfaction; improve provider-patient communication; and foster collaborative or shared priorities between patients and health care providers.
Source
Toto PE, Skidmore ER, Terhorst L, Rosen J, Weiner DK. Arch Gerontol Geriatr. 2015;60(1):16-21.
doi: 10.1016/j.archger.2014.10.022.
How to Avoid the Mistakes That Everyone Makes
In his book, Better: A Surgeon’s Notes on Performance, Atul Gawande asks the question, “What does it take to be good at something in which failure is so easy, so effortless?”1 Consider this statement for just a moment. Every day, over 355,000 patents seek care in this nation’s EDs.2 These visits have a wide-range of significance, from the low acuity and low impact self-limited problems to the cases in which every decision and every second counts. Reflect on the 1999 Institute of Medicine (IOM) report, “To Err is Human.” At that time, 16 years ago, this seminal work estimated that up to 98,000 people die each year (268 each day) as a result of errors made in US hospitals.3 Variability in documentation, for many reasons, is a plausible factor in underestimation of accurate numbers. Since 2001, the worrisome number of deaths reported by the IOM has been re-evaluated a number of times, with each successive “deep dive” looking more ominous than the last.
In 2013, John James published a more recent estimate of preventable adverse events in the Journal of Patient Safety.4 He applied a literature review method to target the Global Trigger Tool from the Institute for Healthcare Improvement as the litmus test to estimate preventable error. In this limited review, James found that between 210,000 and over 400,000 premature deaths per year (575-1,095 deaths per day) are associated with harm that is preventable in hospitals. This number accounts for approximately 17% of the annual US population mortality and exceeds the national death toll from chronic lower respiratory tract infections, strokes, and accidents.5 Estimates of serious harm events (ie, morbidity) appear to be significantly greater than mortality. The adoption of the electronic medical record has not eliminated inaccuracies due to variation in documentation, reluctance of providers to report known errors, and lack of patient perspective in the recounting of their medical stories. The enormous magnitude of public-health consequences due to medical errors thus seems clear.
We become doctors and nurses primarily to help people, and not to cause harm to anyone. When harm occurs as the result of medical errors, the gut-wrenching guilt and self-deprecation that follows for most of us, and the doubt cast on our abilities as physicians, raise the question of why errors happen, and why more is not done to prevent them or to mitigate the consequences.
An awareness of some of the circumstances that lead to error can be a tremendous help in its prevention. High reliability organizations recognize that humans are fallible and that variation in human factors contributes to error, while also focusing on building safer environments designed to create layers of defense against error and mitigate their impact. Blame, shame, and accusatory approaches fail to improve any type of error. Environmental and situational hazards such as ED overcrowding, understaffing, high-patient volumes, rigid throughput demands, lack of equipment/subspecialty services and support in the system are highly contributory and must be addressed. Systemic issues aside, there are compelling individual factors that can lead anyone to make a mistake. Although lessons learned from mistakes are paramount to improvement, an understanding and awareness of the science of error and the necessity of “mindful medicine” can help protect individuals from the personal tolls of making a mistake.
Cognitive Biases
There are five significant cognitive biases that can result in preventable errors: availability bias, anchoring bias, framing bias, confirmation bias, and premature closure. Availability bias favors the common diagnosis without proving its validity. Anchoring bias occurs when a prior diagnosis or opinion is favored and misleads one from the correct current diagnosis. Framing bias can occur when it is not recognized that the data fail to fit the diagnostic presumptions. Confirmation bias can result when information is selectively interpreted to confirm a belief. Premature closure can lead hastily to an incorrect, rushed diagnostic conclusion. The following case scenarios illustrate examples of each of these biases.
Case Scenario 1: The Most Common Diagnosis Versus Looking for the Needle in the Haystack
On a busy night in the ED, the fifth patient of an emergency physician’s (EP) fourth consecutive shift was a quiet young lady home from college during the flu season. With a temperature of 102.6˚F, a heart rate of 110 beats/minute, blood pressure of 105/68 mm Hg, respiratory rate of 16 breaths/minute, and an oxygen saturation of 100% on room air, the EP was confident she would see another case of influenza. With the patient’s body aches, fever, and cough, she clearly appeared to be suffering from the flu, just like so many others this particular week. After treating the patient with fluids, antipyretics, and reassurance, she was sent home to rest in her own bed, with care instructions for influenza.
Because influenza was prevalent in the community, and a myriad of patients with the virus were seen this particular week, the EP was assured that this young lady had the flu—until she returned the next day with a petechial rash and sepsis from bacterial meningitis. This case illustrates the influence of “availability bias” in decision making. Treating a myriad of patients with the same symptoms, some with positive influenza screens and others with negative screens, led the physician to believe that the correct diagnosis, influenza for this patient, was the most common, some might say logical, diagnosis, while discounting other, and more serious possibilities as improbable.
By referring to the disease process that comes to mind most easily, and basing a diagnosis on previous patient experiences with similar symptoms, availability bias confounded the ability to look deeper into other possible causes. A more thorough neck examination, and careful skin and neurological examinations, coupled with the knowledge of a negative rapid influenza test, might have provided enough information—or doubt—to change the physician’s frame of reference and initially establish the correct diagnosis.
“Availability Bias” Mitigating Strategy: Always take a moment to consider diagnoses other than the most common in the differential and prove why the common diagnosis is valid and why other diagnoses could not be the case. If unable to do so, go back and re-evaluate.
Case Scenario 2: It Is Not Always ‘What It Is’
The next patient, a woman in her late 20s, presented to the ED less than 48 hours after discharge from the trauma service at another hospital. She had been admitted after a motor vehicle accident that resulted in an isolated traumatic subarachnoid hemorrhage. After observation, with no surgical intervention, she was discharged in good condition and was able to resume her normal activities with supportive care for a persistent headache postinjury. However, the patient returned after 2 days to an ED closer to her home, because she felt “foggy” and more irritable than usual.
As is customary, this busy unit employs nursing preemptive (ie, standing) orders, and the patient was triaged and laboratory tests were drawn including a basic chemistry panel. Upon evaluation by the attending EP, a concern for re-bleed led to a request for a noncontrast computed tomography (CT) scan of the head, which was interpreted as stable with no new bleed. The case was discussed with the trauma service from the initial hospital and follow-up was arranged.
Prior to the follow-up appointment, the patient returned to the ED because of a further deterioration in mental status. A third head CT was taken and interpreted as stable; however, her serum sodium level was 114 mmol/L. This patient suffered from posttraumatic syndrome of inappropriate antidiuretic hormone secretion (SIADH), and a retrospective review of the laboratory values from the prior ED visit showed the sodium level to be abnormally low at 121 mmol/L.
This case is a good illustration of “anchoring bias,” in which the existing diagnosis of traumatic subarachnoid hemorrhage was maintained as the etiology of the patient’s symptoms, despite a new piece of significant information (ie, the low sodium level) that was not integrated into the differential of possible etiologies for continued deterioration of mental status.
“Anchoring Bias” Mitigating Strategy: Awareness of the power of a prior diagnosis or opinion to mislead is paramount; be sure to carefully review all available data and account for anything that does not lie within the range expected for your diagnosis whenever a patient returns to the ED.
Case Scenario 3: The Search for the Right Piece of the Puzzle
A merchant marine in his mid-40s had fever, jaundice, vomiting, and right upper-quadrant pain for 2 days. He had been airlifted off his ship in the mid-Atlantic Ocean because the medical crew on the ship was concerned that he had a life-threatening illness and they had no surgical facilities available to care for acute cholecystitis with ascending cholangitis.

This patient was otherwise healthy and had all current immunizations. Upon arrival in the ED, he was given intravenous (IV) fluids, antiemetics, and medication for pain control while the workup was underway. He was somnolent and critically ill-appearing. As he spoke only German, a Red Cross interpreter was engaged in an attempt to obtain further information, but the patient was unable to provide additional history. The physician was able to elicit the travel history of the ship by connecting the interpreter with a crewmember on board and learned that the ship was on a return voyage from Haiti, a country endemic with Plasmodium falciparum. It was further determined that this patient was not taking malaria prophylaxis; his blood smear turned out to be positive for the disease.
This case emphasizes the impact of “framing bias” in the thought process that led to the initial diagnosis of ascending cholangitis. Charcot’s Triad of fever, jaundice, and right upper-quadrant pain was the “frame” in which the suspected—yet uncommon—diagnosis was made. The frame of reference, however, changed significantly when the additional information of P. falciparum malaria exposure was elucidated, allowing the correct diagnosis to come into view.
I “Framing Bias” Mitigation Strategy: Confirm that the points of data align properly and fit within the diagnostic possibilities. If they do not connect, seek further information and widen the frame of reference.
Case Scenario 4: Mother Knows Best
The parents of an 11-year-old boy brought their child to the ED for abdominal pain, nausea, and vomiting that started 10 hours prior to presentation. The boy had no medical or surgical history. During the examination, his mother expressed concern for appendicitis, a common concern of parents whose children have abdominal pain.
Although the boy was uncomfortable, he had no right lower-quadrant tenderness; his bowel sounds were normal, and there was no tenderness to heel tap or Rovsing’s sign. He did have periumbilical tenderness and distractible guarding. His white blood-cell (WBC) count was 12.5 K/uL with 78% segmented neutrophils and urinalysis showed 15 WBCs/hpf with no bacteria. With IV hydration, opioid pain medication, and antiemetic medication, the patient felt much better, and the abdominal examination improved with no tenderness.
After a clear liquid trial was successful, careful counseling of the parents made it clear that appendicitis was possible, but unlikely given the improvement in symptoms, and the boy was discharged with the diagnosis of acute gastroenteritis. Six hours later, the boy was brought back to the ED and then taken to the operating room for acute appendicitis.
This case illustrates the effect of “confirmation bias,” in which the preference to favor a particular diagnosis outweighs the clinical clues that suggested the correct diagnosis. Neither possible opioid effect in masking pain nor the lack of history or complaint of diarrhea was effectively taken into consideration before making the diagnosis of “gastroenteritis.”
I “Confirmation Bias” Mitigating Strategy: Be aware that selective filtering of information to confirm a belief is wrought with danger. Seek data and information that could weaken or negate the belief and give it serious consideration.
Case Scenario 5: The “Frequent Flyer”
This case involved an extremely difficult-to-manage patient who had presented to the ED many times in the past with “the worst headache of his life.” During these visits, he was typically disruptive, problematic to discharge safely, and a particular behavioral issue for the nurses. As a result, when he presented to the ED, the goal was to evaluate him quickly, treat him humanely, and diagnose and discharge him as soon as possible. He was never accompanied by family or friends. His personal medical history was positive for uncontrolled hypertension and chronic alcoholism.
Attempts to coordinate his care with social services and community mental health were unsuccessful. On examination, his blood pressure was 210/105 mm Hg and a severe headache was once again his chief complaint. A head CT scan performed less than 12 hours prior to this presentation revealed no new findings compared with many prior CTs. However, there was a slight slurring of his speech on this visit that wasn’t part of his prior presentations. After he was given lorazepam and a meal tray, the patient felt a little better and was discharged. The patient never returned to the ED, and the staff has since become concerned that on this last ED visit, their “frequent flyer” had something real.
Although it is not known what exactly happened to this patient, the manner in which he was treated represents the effect of premature closure on the medical decision-making process. When a clinician jumps to conclusions without seeking more information, the possibility of an error in judgment exists.
I “Premature Closure” Mitigating Strategies: Never stop thinking even after a conclusion has been reached. Instead, stop and think again about what else could be happening.

Conclusion
In all of the case scenarios presented, any or all of the patients might have survived the errors made without any adverse outcome, but the errors might also have resulted in mild to severe permanent disability or death. It is important to remember that errors can happen regardless of good intentions and acceptable practice, and that although an error may have no consequence in one set of circumstances, the same error could be deadly in others.
On the systems level, approaches to assess and control the work environment are necessary to mitigate risk to individuals, create a culture of safety, and encourage collective learning and continuous improvement. Recognition of the frequent incidence of error and awareness of the overlapping cognitive biases (Table), while always being mindful to take a moment to think further, can help avoid—but never eliminate--these types of mistakes.
Dr McCammon is an assistant professor, department of emergency medicine, Eastern Virginia Medical School, Norfolk.
- Gawande A. Better: A Surgeon’s Notes on Performance. New York, NY: Picador; 2007.
- National Hospital Ambulatory Medical Care Survey: 2010 Emergency Department Summary
- Tables. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2010_ed_web_tables.pdf. Accessed January 26, 2015.
- Committee on Quality of Health Care in America; Institute of Medicine. To err is human: building a safer health system. https://www.iom.edu/~/media/Files/Report%20Files/1999/To-Err-is-Human/To%20Err%20is%20Human%201999%20%20report%20brief.pdf. Published November 1999. Accessed January 26, 2015.
- James, JT. A new, evidence-based estimate of patient harms associated with hospital care. J Pat Saf. 2013;9(3):122-128.
- Hoyert DL, Xu J. Deaths: preliminary data for 2011. Natl Vital Stat Rep. 2012;61(6):1-51. http://www.cdc.gov/nchs/data/nvsr/nvsr61/nvsr61_06.pdf. Accessed January 26, 2015.
- Wellbury C. Flaws in clinical reasoning: a common cause of diagnostic error. Am Fam Physician. 2011; 84(9):1042-1048
In his book, Better: A Surgeon’s Notes on Performance, Atul Gawande asks the question, “What does it take to be good at something in which failure is so easy, so effortless?”1 Consider this statement for just a moment. Every day, over 355,000 patents seek care in this nation’s EDs.2 These visits have a wide-range of significance, from the low acuity and low impact self-limited problems to the cases in which every decision and every second counts. Reflect on the 1999 Institute of Medicine (IOM) report, “To Err is Human.” At that time, 16 years ago, this seminal work estimated that up to 98,000 people die each year (268 each day) as a result of errors made in US hospitals.3 Variability in documentation, for many reasons, is a plausible factor in underestimation of accurate numbers. Since 2001, the worrisome number of deaths reported by the IOM has been re-evaluated a number of times, with each successive “deep dive” looking more ominous than the last.
In 2013, John James published a more recent estimate of preventable adverse events in the Journal of Patient Safety.4 He applied a literature review method to target the Global Trigger Tool from the Institute for Healthcare Improvement as the litmus test to estimate preventable error. In this limited review, James found that between 210,000 and over 400,000 premature deaths per year (575-1,095 deaths per day) are associated with harm that is preventable in hospitals. This number accounts for approximately 17% of the annual US population mortality and exceeds the national death toll from chronic lower respiratory tract infections, strokes, and accidents.5 Estimates of serious harm events (ie, morbidity) appear to be significantly greater than mortality. The adoption of the electronic medical record has not eliminated inaccuracies due to variation in documentation, reluctance of providers to report known errors, and lack of patient perspective in the recounting of their medical stories. The enormous magnitude of public-health consequences due to medical errors thus seems clear.
We become doctors and nurses primarily to help people, and not to cause harm to anyone. When harm occurs as the result of medical errors, the gut-wrenching guilt and self-deprecation that follows for most of us, and the doubt cast on our abilities as physicians, raise the question of why errors happen, and why more is not done to prevent them or to mitigate the consequences.
An awareness of some of the circumstances that lead to error can be a tremendous help in its prevention. High reliability organizations recognize that humans are fallible and that variation in human factors contributes to error, while also focusing on building safer environments designed to create layers of defense against error and mitigate their impact. Blame, shame, and accusatory approaches fail to improve any type of error. Environmental and situational hazards such as ED overcrowding, understaffing, high-patient volumes, rigid throughput demands, lack of equipment/subspecialty services and support in the system are highly contributory and must be addressed. Systemic issues aside, there are compelling individual factors that can lead anyone to make a mistake. Although lessons learned from mistakes are paramount to improvement, an understanding and awareness of the science of error and the necessity of “mindful medicine” can help protect individuals from the personal tolls of making a mistake.
Cognitive Biases
There are five significant cognitive biases that can result in preventable errors: availability bias, anchoring bias, framing bias, confirmation bias, and premature closure. Availability bias favors the common diagnosis without proving its validity. Anchoring bias occurs when a prior diagnosis or opinion is favored and misleads one from the correct current diagnosis. Framing bias can occur when it is not recognized that the data fail to fit the diagnostic presumptions. Confirmation bias can result when information is selectively interpreted to confirm a belief. Premature closure can lead hastily to an incorrect, rushed diagnostic conclusion. The following case scenarios illustrate examples of each of these biases.
Case Scenario 1: The Most Common Diagnosis Versus Looking for the Needle in the Haystack
On a busy night in the ED, the fifth patient of an emergency physician’s (EP) fourth consecutive shift was a quiet young lady home from college during the flu season. With a temperature of 102.6˚F, a heart rate of 110 beats/minute, blood pressure of 105/68 mm Hg, respiratory rate of 16 breaths/minute, and an oxygen saturation of 100% on room air, the EP was confident she would see another case of influenza. With the patient’s body aches, fever, and cough, she clearly appeared to be suffering from the flu, just like so many others this particular week. After treating the patient with fluids, antipyretics, and reassurance, she was sent home to rest in her own bed, with care instructions for influenza.
Because influenza was prevalent in the community, and a myriad of patients with the virus were seen this particular week, the EP was assured that this young lady had the flu—until she returned the next day with a petechial rash and sepsis from bacterial meningitis. This case illustrates the influence of “availability bias” in decision making. Treating a myriad of patients with the same symptoms, some with positive influenza screens and others with negative screens, led the physician to believe that the correct diagnosis, influenza for this patient, was the most common, some might say logical, diagnosis, while discounting other, and more serious possibilities as improbable.
By referring to the disease process that comes to mind most easily, and basing a diagnosis on previous patient experiences with similar symptoms, availability bias confounded the ability to look deeper into other possible causes. A more thorough neck examination, and careful skin and neurological examinations, coupled with the knowledge of a negative rapid influenza test, might have provided enough information—or doubt—to change the physician’s frame of reference and initially establish the correct diagnosis.
“Availability Bias” Mitigating Strategy: Always take a moment to consider diagnoses other than the most common in the differential and prove why the common diagnosis is valid and why other diagnoses could not be the case. If unable to do so, go back and re-evaluate.
Case Scenario 2: It Is Not Always ‘What It Is’
The next patient, a woman in her late 20s, presented to the ED less than 48 hours after discharge from the trauma service at another hospital. She had been admitted after a motor vehicle accident that resulted in an isolated traumatic subarachnoid hemorrhage. After observation, with no surgical intervention, she was discharged in good condition and was able to resume her normal activities with supportive care for a persistent headache postinjury. However, the patient returned after 2 days to an ED closer to her home, because she felt “foggy” and more irritable than usual.
As is customary, this busy unit employs nursing preemptive (ie, standing) orders, and the patient was triaged and laboratory tests were drawn including a basic chemistry panel. Upon evaluation by the attending EP, a concern for re-bleed led to a request for a noncontrast computed tomography (CT) scan of the head, which was interpreted as stable with no new bleed. The case was discussed with the trauma service from the initial hospital and follow-up was arranged.
Prior to the follow-up appointment, the patient returned to the ED because of a further deterioration in mental status. A third head CT was taken and interpreted as stable; however, her serum sodium level was 114 mmol/L. This patient suffered from posttraumatic syndrome of inappropriate antidiuretic hormone secretion (SIADH), and a retrospective review of the laboratory values from the prior ED visit showed the sodium level to be abnormally low at 121 mmol/L.
This case is a good illustration of “anchoring bias,” in which the existing diagnosis of traumatic subarachnoid hemorrhage was maintained as the etiology of the patient’s symptoms, despite a new piece of significant information (ie, the low sodium level) that was not integrated into the differential of possible etiologies for continued deterioration of mental status.
“Anchoring Bias” Mitigating Strategy: Awareness of the power of a prior diagnosis or opinion to mislead is paramount; be sure to carefully review all available data and account for anything that does not lie within the range expected for your diagnosis whenever a patient returns to the ED.
Case Scenario 3: The Search for the Right Piece of the Puzzle
A merchant marine in his mid-40s had fever, jaundice, vomiting, and right upper-quadrant pain for 2 days. He had been airlifted off his ship in the mid-Atlantic Ocean because the medical crew on the ship was concerned that he had a life-threatening illness and they had no surgical facilities available to care for acute cholecystitis with ascending cholangitis.
This patient was otherwise healthy and had all current immunizations. Upon arrival in the ED, he was given intravenous (IV) fluids, antiemetics, and medication for pain control while the workup was underway. He was somnolent and critically ill-appearing. As he spoke only German, a Red Cross interpreter was engaged in an attempt to obtain further information, but the patient was unable to provide additional history. The physician was able to elicit the travel history of the ship by connecting the interpreter with a crewmember on board and learned that the ship was on a return voyage from Haiti, a country endemic with Plasmodium falciparum. It was further determined that this patient was not taking malaria prophylaxis; his blood smear turned out to be positive for the disease.
This case emphasizes the impact of “framing bias” in the thought process that led to the initial diagnosis of ascending cholangitis. Charcot’s Triad of fever, jaundice, and right upper-quadrant pain was the “frame” in which the suspected—yet uncommon—diagnosis was made. The frame of reference, however, changed significantly when the additional information of P. falciparum malaria exposure was elucidated, allowing the correct diagnosis to come into view.
I “Framing Bias” Mitigation Strategy: Confirm that the points of data align properly and fit within the diagnostic possibilities. If they do not connect, seek further information and widen the frame of reference.
Case Scenario 4: Mother Knows Best
The parents of an 11-year-old boy brought their child to the ED for abdominal pain, nausea, and vomiting that started 10 hours prior to presentation. The boy had no medical or surgical history. During the examination, his mother expressed concern for appendicitis, a common concern of parents whose children have abdominal pain.
Although the boy was uncomfortable, he had no right lower-quadrant tenderness; his bowel sounds were normal, and there was no tenderness to heel tap or Rovsing’s sign. He did have periumbilical tenderness and distractible guarding. His white blood-cell (WBC) count was 12.5 K/uL with 78% segmented neutrophils and urinalysis showed 15 WBCs/hpf with no bacteria. With IV hydration, opioid pain medication, and antiemetic medication, the patient felt much better, and the abdominal examination improved with no tenderness.
After a clear liquid trial was successful, careful counseling of the parents made it clear that appendicitis was possible, but unlikely given the improvement in symptoms, and the boy was discharged with the diagnosis of acute gastroenteritis. Six hours later, the boy was brought back to the ED and then taken to the operating room for acute appendicitis.
This case illustrates the effect of “confirmation bias,” in which the preference to favor a particular diagnosis outweighs the clinical clues that suggested the correct diagnosis. Neither possible opioid effect in masking pain nor the lack of history or complaint of diarrhea was effectively taken into consideration before making the diagnosis of “gastroenteritis.”
I “Confirmation Bias” Mitigating Strategy: Be aware that selective filtering of information to confirm a belief is wrought with danger. Seek data and information that could weaken or negate the belief and give it serious consideration.
Case Scenario 5: The “Frequent Flyer”
This case involved an extremely difficult-to-manage patient who had presented to the ED many times in the past with “the worst headache of his life.” During these visits, he was typically disruptive, problematic to discharge safely, and a particular behavioral issue for the nurses. As a result, when he presented to the ED, the goal was to evaluate him quickly, treat him humanely, and diagnose and discharge him as soon as possible. He was never accompanied by family or friends. His personal medical history was positive for uncontrolled hypertension and chronic alcoholism.
Attempts to coordinate his care with social services and community mental health were unsuccessful. On examination, his blood pressure was 210/105 mm Hg and a severe headache was once again his chief complaint. A head CT scan performed less than 12 hours prior to this presentation revealed no new findings compared with many prior CTs. However, there was a slight slurring of his speech on this visit that wasn’t part of his prior presentations. After he was given lorazepam and a meal tray, the patient felt a little better and was discharged. The patient never returned to the ED, and the staff has since become concerned that on this last ED visit, their “frequent flyer” had something real.
Although it is not known what exactly happened to this patient, the manner in which he was treated represents the effect of premature closure on the medical decision-making process. When a clinician jumps to conclusions without seeking more information, the possibility of an error in judgment exists.
I “Premature Closure” Mitigating Strategies: Never stop thinking even after a conclusion has been reached. Instead, stop and think again about what else could be happening.
Conclusion
In all of the case scenarios presented, any or all of the patients might have survived the errors made without any adverse outcome, but the errors might also have resulted in mild to severe permanent disability or death. It is important to remember that errors can happen regardless of good intentions and acceptable practice, and that although an error may have no consequence in one set of circumstances, the same error could be deadly in others.
On the systems level, approaches to assess and control the work environment are necessary to mitigate risk to individuals, create a culture of safety, and encourage collective learning and continuous improvement. Recognition of the frequent incidence of error and awareness of the overlapping cognitive biases (Table), while always being mindful to take a moment to think further, can help avoid—but never eliminate--these types of mistakes.
Dr McCammon is an assistant professor, department of emergency medicine, Eastern Virginia Medical School, Norfolk.
In his book, Better: A Surgeon’s Notes on Performance, Atul Gawande asks the question, “What does it take to be good at something in which failure is so easy, so effortless?”1 Consider this statement for just a moment. Every day, over 355,000 patents seek care in this nation’s EDs.2 These visits have a wide-range of significance, from the low acuity and low impact self-limited problems to the cases in which every decision and every second counts. Reflect on the 1999 Institute of Medicine (IOM) report, “To Err is Human.” At that time, 16 years ago, this seminal work estimated that up to 98,000 people die each year (268 each day) as a result of errors made in US hospitals.3 Variability in documentation, for many reasons, is a plausible factor in underestimation of accurate numbers. Since 2001, the worrisome number of deaths reported by the IOM has been re-evaluated a number of times, with each successive “deep dive” looking more ominous than the last.
In 2013, John James published a more recent estimate of preventable adverse events in the Journal of Patient Safety.4 He applied a literature review method to target the Global Trigger Tool from the Institute for Healthcare Improvement as the litmus test to estimate preventable error. In this limited review, James found that between 210,000 and over 400,000 premature deaths per year (575-1,095 deaths per day) are associated with harm that is preventable in hospitals. This number accounts for approximately 17% of the annual US population mortality and exceeds the national death toll from chronic lower respiratory tract infections, strokes, and accidents.5 Estimates of serious harm events (ie, morbidity) appear to be significantly greater than mortality. The adoption of the electronic medical record has not eliminated inaccuracies due to variation in documentation, reluctance of providers to report known errors, and lack of patient perspective in the recounting of their medical stories. The enormous magnitude of public-health consequences due to medical errors thus seems clear.
We become doctors and nurses primarily to help people, and not to cause harm to anyone. When harm occurs as the result of medical errors, the gut-wrenching guilt and self-deprecation that follows for most of us, and the doubt cast on our abilities as physicians, raise the question of why errors happen, and why more is not done to prevent them or to mitigate the consequences.
An awareness of some of the circumstances that lead to error can be a tremendous help in its prevention. High reliability organizations recognize that humans are fallible and that variation in human factors contributes to error, while also focusing on building safer environments designed to create layers of defense against error and mitigate their impact. Blame, shame, and accusatory approaches fail to improve any type of error. Environmental and situational hazards such as ED overcrowding, understaffing, high-patient volumes, rigid throughput demands, lack of equipment/subspecialty services and support in the system are highly contributory and must be addressed. Systemic issues aside, there are compelling individual factors that can lead anyone to make a mistake. Although lessons learned from mistakes are paramount to improvement, an understanding and awareness of the science of error and the necessity of “mindful medicine” can help protect individuals from the personal tolls of making a mistake.
Cognitive Biases
There are five significant cognitive biases that can result in preventable errors: availability bias, anchoring bias, framing bias, confirmation bias, and premature closure. Availability bias favors the common diagnosis without proving its validity. Anchoring bias occurs when a prior diagnosis or opinion is favored and misleads one from the correct current diagnosis. Framing bias can occur when it is not recognized that the data fail to fit the diagnostic presumptions. Confirmation bias can result when information is selectively interpreted to confirm a belief. Premature closure can lead hastily to an incorrect, rushed diagnostic conclusion. The following case scenarios illustrate examples of each of these biases.
Case Scenario 1: The Most Common Diagnosis Versus Looking for the Needle in the Haystack
On a busy night in the ED, the fifth patient of an emergency physician’s (EP) fourth consecutive shift was a quiet young lady home from college during the flu season. With a temperature of 102.6˚F, a heart rate of 110 beats/minute, blood pressure of 105/68 mm Hg, respiratory rate of 16 breaths/minute, and an oxygen saturation of 100% on room air, the EP was confident she would see another case of influenza. With the patient’s body aches, fever, and cough, she clearly appeared to be suffering from the flu, just like so many others this particular week. After treating the patient with fluids, antipyretics, and reassurance, she was sent home to rest in her own bed, with care instructions for influenza.
Because influenza was prevalent in the community, and a myriad of patients with the virus were seen this particular week, the EP was assured that this young lady had the flu—until she returned the next day with a petechial rash and sepsis from bacterial meningitis. This case illustrates the influence of “availability bias” in decision making. Treating a myriad of patients with the same symptoms, some with positive influenza screens and others with negative screens, led the physician to believe that the correct diagnosis, influenza for this patient, was the most common, some might say logical, diagnosis, while discounting other, and more serious possibilities as improbable.
By referring to the disease process that comes to mind most easily, and basing a diagnosis on previous patient experiences with similar symptoms, availability bias confounded the ability to look deeper into other possible causes. A more thorough neck examination, and careful skin and neurological examinations, coupled with the knowledge of a negative rapid influenza test, might have provided enough information—or doubt—to change the physician’s frame of reference and initially establish the correct diagnosis.
“Availability Bias” Mitigating Strategy: Always take a moment to consider diagnoses other than the most common in the differential and prove why the common diagnosis is valid and why other diagnoses could not be the case. If unable to do so, go back and re-evaluate.
Case Scenario 2: It Is Not Always ‘What It Is’
The next patient, a woman in her late 20s, presented to the ED less than 48 hours after discharge from the trauma service at another hospital. She had been admitted after a motor vehicle accident that resulted in an isolated traumatic subarachnoid hemorrhage. After observation, with no surgical intervention, she was discharged in good condition and was able to resume her normal activities with supportive care for a persistent headache postinjury. However, the patient returned after 2 days to an ED closer to her home, because she felt “foggy” and more irritable than usual.
As is customary, this busy unit employs nursing preemptive (ie, standing) orders, and the patient was triaged and laboratory tests were drawn including a basic chemistry panel. Upon evaluation by the attending EP, a concern for re-bleed led to a request for a noncontrast computed tomography (CT) scan of the head, which was interpreted as stable with no new bleed. The case was discussed with the trauma service from the initial hospital and follow-up was arranged.
Prior to the follow-up appointment, the patient returned to the ED because of a further deterioration in mental status. A third head CT was taken and interpreted as stable; however, her serum sodium level was 114 mmol/L. This patient suffered from posttraumatic syndrome of inappropriate antidiuretic hormone secretion (SIADH), and a retrospective review of the laboratory values from the prior ED visit showed the sodium level to be abnormally low at 121 mmol/L.
This case is a good illustration of “anchoring bias,” in which the existing diagnosis of traumatic subarachnoid hemorrhage was maintained as the etiology of the patient’s symptoms, despite a new piece of significant information (ie, the low sodium level) that was not integrated into the differential of possible etiologies for continued deterioration of mental status.
“Anchoring Bias” Mitigating Strategy: Awareness of the power of a prior diagnosis or opinion to mislead is paramount; be sure to carefully review all available data and account for anything that does not lie within the range expected for your diagnosis whenever a patient returns to the ED.
Case Scenario 3: The Search for the Right Piece of the Puzzle
A merchant marine in his mid-40s had fever, jaundice, vomiting, and right upper-quadrant pain for 2 days. He had been airlifted off his ship in the mid-Atlantic Ocean because the medical crew on the ship was concerned that he had a life-threatening illness and they had no surgical facilities available to care for acute cholecystitis with ascending cholangitis.
This patient was otherwise healthy and had all current immunizations. Upon arrival in the ED, he was given intravenous (IV) fluids, antiemetics, and medication for pain control while the workup was underway. He was somnolent and critically ill-appearing. As he spoke only German, a Red Cross interpreter was engaged in an attempt to obtain further information, but the patient was unable to provide additional history. The physician was able to elicit the travel history of the ship by connecting the interpreter with a crewmember on board and learned that the ship was on a return voyage from Haiti, a country endemic with Plasmodium falciparum. It was further determined that this patient was not taking malaria prophylaxis; his blood smear turned out to be positive for the disease.
This case emphasizes the impact of “framing bias” in the thought process that led to the initial diagnosis of ascending cholangitis. Charcot’s Triad of fever, jaundice, and right upper-quadrant pain was the “frame” in which the suspected—yet uncommon—diagnosis was made. The frame of reference, however, changed significantly when the additional information of P. falciparum malaria exposure was elucidated, allowing the correct diagnosis to come into view.
I “Framing Bias” Mitigation Strategy: Confirm that the points of data align properly and fit within the diagnostic possibilities. If they do not connect, seek further information and widen the frame of reference.
Case Scenario 4: Mother Knows Best
The parents of an 11-year-old boy brought their child to the ED for abdominal pain, nausea, and vomiting that started 10 hours prior to presentation. The boy had no medical or surgical history. During the examination, his mother expressed concern for appendicitis, a common concern of parents whose children have abdominal pain.
Although the boy was uncomfortable, he had no right lower-quadrant tenderness; his bowel sounds were normal, and there was no tenderness to heel tap or Rovsing’s sign. He did have periumbilical tenderness and distractible guarding. His white blood-cell (WBC) count was 12.5 K/uL with 78% segmented neutrophils and urinalysis showed 15 WBCs/hpf with no bacteria. With IV hydration, opioid pain medication, and antiemetic medication, the patient felt much better, and the abdominal examination improved with no tenderness.
After a clear liquid trial was successful, careful counseling of the parents made it clear that appendicitis was possible, but unlikely given the improvement in symptoms, and the boy was discharged with the diagnosis of acute gastroenteritis. Six hours later, the boy was brought back to the ED and then taken to the operating room for acute appendicitis.
This case illustrates the effect of “confirmation bias,” in which the preference to favor a particular diagnosis outweighs the clinical clues that suggested the correct diagnosis. Neither possible opioid effect in masking pain nor the lack of history or complaint of diarrhea was effectively taken into consideration before making the diagnosis of “gastroenteritis.”
I “Confirmation Bias” Mitigating Strategy: Be aware that selective filtering of information to confirm a belief is wrought with danger. Seek data and information that could weaken or negate the belief and give it serious consideration.
Case Scenario 5: The “Frequent Flyer”
This case involved an extremely difficult-to-manage patient who had presented to the ED many times in the past with “the worst headache of his life.” During these visits, he was typically disruptive, problematic to discharge safely, and a particular behavioral issue for the nurses. As a result, when he presented to the ED, the goal was to evaluate him quickly, treat him humanely, and diagnose and discharge him as soon as possible. He was never accompanied by family or friends. His personal medical history was positive for uncontrolled hypertension and chronic alcoholism.
Attempts to coordinate his care with social services and community mental health were unsuccessful. On examination, his blood pressure was 210/105 mm Hg and a severe headache was once again his chief complaint. A head CT scan performed less than 12 hours prior to this presentation revealed no new findings compared with many prior CTs. However, there was a slight slurring of his speech on this visit that wasn’t part of his prior presentations. After he was given lorazepam and a meal tray, the patient felt a little better and was discharged. The patient never returned to the ED, and the staff has since become concerned that on this last ED visit, their “frequent flyer” had something real.
Although it is not known what exactly happened to this patient, the manner in which he was treated represents the effect of premature closure on the medical decision-making process. When a clinician jumps to conclusions without seeking more information, the possibility of an error in judgment exists.
I “Premature Closure” Mitigating Strategies: Never stop thinking even after a conclusion has been reached. Instead, stop and think again about what else could be happening.
Conclusion
In all of the case scenarios presented, any or all of the patients might have survived the errors made without any adverse outcome, but the errors might also have resulted in mild to severe permanent disability or death. It is important to remember that errors can happen regardless of good intentions and acceptable practice, and that although an error may have no consequence in one set of circumstances, the same error could be deadly in others.
On the systems level, approaches to assess and control the work environment are necessary to mitigate risk to individuals, create a culture of safety, and encourage collective learning and continuous improvement. Recognition of the frequent incidence of error and awareness of the overlapping cognitive biases (Table), while always being mindful to take a moment to think further, can help avoid—but never eliminate--these types of mistakes.
Dr McCammon is an assistant professor, department of emergency medicine, Eastern Virginia Medical School, Norfolk.
- Gawande A. Better: A Surgeon’s Notes on Performance. New York, NY: Picador; 2007.
- National Hospital Ambulatory Medical Care Survey: 2010 Emergency Department Summary
- Tables. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2010_ed_web_tables.pdf. Accessed January 26, 2015.
- Committee on Quality of Health Care in America; Institute of Medicine. To err is human: building a safer health system. https://www.iom.edu/~/media/Files/Report%20Files/1999/To-Err-is-Human/To%20Err%20is%20Human%201999%20%20report%20brief.pdf. Published November 1999. Accessed January 26, 2015.
- James, JT. A new, evidence-based estimate of patient harms associated with hospital care. J Pat Saf. 2013;9(3):122-128.
- Hoyert DL, Xu J. Deaths: preliminary data for 2011. Natl Vital Stat Rep. 2012;61(6):1-51. http://www.cdc.gov/nchs/data/nvsr/nvsr61/nvsr61_06.pdf. Accessed January 26, 2015.
- Wellbury C. Flaws in clinical reasoning: a common cause of diagnostic error. Am Fam Physician. 2011; 84(9):1042-1048
- Gawande A. Better: A Surgeon’s Notes on Performance. New York, NY: Picador; 2007.
- National Hospital Ambulatory Medical Care Survey: 2010 Emergency Department Summary
- Tables. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/nchs/data/ahcd/nhamcs_emergency/2010_ed_web_tables.pdf. Accessed January 26, 2015.
- Committee on Quality of Health Care in America; Institute of Medicine. To err is human: building a safer health system. https://www.iom.edu/~/media/Files/Report%20Files/1999/To-Err-is-Human/To%20Err%20is%20Human%201999%20%20report%20brief.pdf. Published November 1999. Accessed January 26, 2015.
- James, JT. A new, evidence-based estimate of patient harms associated with hospital care. J Pat Saf. 2013;9(3):122-128.
- Hoyert DL, Xu J. Deaths: preliminary data for 2011. Natl Vital Stat Rep. 2012;61(6):1-51. http://www.cdc.gov/nchs/data/nvsr/nvsr61/nvsr61_06.pdf. Accessed January 26, 2015.
- Wellbury C. Flaws in clinical reasoning: a common cause of diagnostic error. Am Fam Physician. 2011; 84(9):1042-1048

2015 Update on fertility
The first human birth from a frozen oocyte was reported in 1986.1 Nearly 3 decades later, mature oocyte cryopreservation has emerged as a meaningful technology to preserve reproductive potential in women of reproductive age. In 2013, the American Society for Reproductive Medicine (ASRM) removed the “experimental” label from egg freezing but cautioned that more data on safety and efficacy were needed prior to widespread adoption of the technique.2
In this Update, we present the current protocols for oocyte cryopreservation, how we arrived at them, and the questions regarding outcomes that still remain. In addition, we discuss the ethical dilemmas egg freezing presents, according to the varying rhetoric within the media and our own profession. Finally, we consider what preliminary data suggest as to the live-birth rate using frozen eggs from women of varying ages and what the costs are associated with using oocyte cryopreservation as the approach to fertility treatment.

Vitrification and slow freezing: How did we get here and how effective are they?
Fertility preservation is a rapidly advancing area of reproductive medicine. Cryopreservation is the cooling of cells to subzero temperatures to halt biologic activity and preserve the cells for future use. Clinically, oocyte cryopreservation requires a patient to undergo in vitro fertilization (IVF). After egg retrieval, the oocytes are cryopreserved for use at a later time.
The prefix “cryo” originated from the Greek word “kryos,” meaning icy cold or frost. Cryopreservation is not a new science. In 1776, the Italian priest and scientist Lazzaro Spallanzani reported that sperm became motionless when cooled by snow. A pivotal discovery in the field came in 1949, when Christopher Polge, an English scientist, showed that glycerol, a permeating solute, could provide protection to cells at low temperatures.3 Progress in sperm cryopreservation advanced quickly, partly due to the ease of observing sperm motility as a marker of postthaw function.4
The ongoing evolution of cryopreservation science led to landmark achievements, including the first birth using human cryopreserved sperm in the 1950s, and the first human birth after embryo thaw in 1983. Since that time cryopreservation has become a cornerstone in the field of reproductive medicine.
Initial problems encountered with egg freezing
Although the first birth after thaw of a human oocyte occurred in 1986, oocyte cryopreservation was fraught with technical difficulties. Oocytes (vs sperm and embryos) proved challenging to successfully cryopreserve. The problem lay in the damage caused by water crystals forming ice and rising concentrations of intracellular solutes as cells were cooled to freezing temperatures.5 The large size and high water content of the human oocyte made it particularly vulnerable to the detrimental effects of freezing. In addition, freeze−thaw hardening of the zone pellucida led to decreased postthaw fertilization. The delicate meiotic spindle within the oocyte was prone to injury from ice crystals.6
Use of cryoprotectants, such as ethylene glycol, glycerol, and dimethylsulfoxide (DMSO), can prevent ice crystal formation, but high concentrations are theoretically toxic. The fine balance between protection and toxicity led to the development of diverse egg freezing protocols using various types and concentrations of cryoprotectants. Inconsistent results and lack of reproducibility among labs, together with concerns about postthaw oocyte function and safety, slowed the progression of oocyte freezing. By the end of the 1980s, clinical oocyte cryopreservation had been effectively halted and the field was confined to small groups of researchers who continued laboratory experiments with limited success.5
In 1997, clinical work with frozen oocytes resumed with a Bologna team reporting postthawing oocyte survival rates of up to 80% using propanediol as the primary cryoprotectant, and viable pregnancies with the use of intracytoplasmic sperm injection (ICSI) for fertilization.7,8 Since the late 1990s, further modifications in freezing technologies have resulted in greater success. And currently, both slow freezing and vitrification methods are used to preserve oocytes.
Slow freezing
Slow freezing involves a low rate of oocyte temperature decline with a simultaneous gradual increase in the concentration of cryoprotectants. As the metabolic activity of the oocyte decreases, the concentration of cryoprotectant can be increased to prevent ice crystal formation. Once solidification of the oocyte is achieved, the oocyte can be exposed to freezing at colder temperatures. Results of a meta-analysis of 26 studies revealed that, compared with using fresh oocytes, eggs thawed after slow-freezing yielded significantly lower rates of fertilization (61.0% [1,346/2,217] vs 76.7% [2,788/3,637]), clinical pregnancy rate per transfer (27.1% [95/351] vs 68.5% [272/397]), and live birth per transfer (21.6% [76/351] vs 32.4% [24/72]).9
Vitrification
Vitrification involves the rapid cooling of cells to extremely low temperatures. During vitrification, oocytes are exposed to high concentrations of cryoprotectants and, after a short equilibration time, rapidly cooled. The rate of cooling is dramatic, up to 20,000°C per minute—so fast that ice does not have time to form and a glass-like state is achieved within the oocyte. Studies suggest that the use of vitrification improves oocyte survival and function compared with slow freezing.9-11 A prospective randomized controlled trial comparing frozen/thawed with vitrified/warmed oocytes demonstrated superior oocyte function in the vitrification group, with higher oocyte survival (81% for vitrification/warming vs 67% for slow freezing/thawing); higher rates of fertilization, cleavage, and embryo morphology; as well as higher clinical pregnancy rates (38% for vitrified/warmed vs 13% for frozen/thawed).10
The Practice Committee of ASRM published a guideline for mature cryopreservation in 2013.2 The committee reviewed the literature on efficacy and safety of mature oocyte cryopreservation. Although data are limited, studies comparing outcomes of IVF using cryopreserved versus fresh oocytes, including four randomized controlled trials and a meta-analysis, provide evidence that previously vitrified/thawed eggs result in similar fertilization and pregnancy rates as IVF/ICSI with fresh oocytes. Similar to data from fresh IVF cycles, decreased success with oocyte vitrification is seen in women with advanced age, and delivery rates, not unexpectedly, are inversely correlated with maternal age.12
Safety outcomes data are limited but reassuring
Two major factors limit our current understanding of egg cryopreservation outcomes. First, many women who have cryopreserved their eggs have not yet used them and, second, babies born after use of cryopreserved oocytes have not reached ages in which safety of the technique can be fully evaluated. Despite this important gap in our knowledge, to date, results of studies examining safety outcomes of the procedure have been reassuring.
For instance, chromosomal analysis via fluorescence in-situ hybridization of embryos created with thawed oocytes versus controls revealed no difference in the incidence of chromosomal abnormalities, decreasing concerns about damage to the oocyte spindle secondary to freezing.13
Data from a review of 900 live births resulting from embryos created from thawed oocytes frozen via the slow freeze technique revealed no increase in the risk of congenital anomalies.14 Similarly, no increased risk of congenital anomalies or difference in birth weights was noted in a study of 200 live births after transfers with embryos derived from vitrified oocytes compared with fresh oocytes.15
In a study of 954 clinical pregnancies occurring in 855 couples with cryopreserved oocytes after assisted reproductive technology, the outcomes of 197 pregnancies from frozen/thawed oocytes were compared with 757 obtained from fresh sibling oocyte cycles. A significantly higher rate of spontaneous abortions at 12 weeks or less was observed in the frozen/thawed oocyte group. No statistically significant differences were noted in gestational age at delivery or in the incidence of major congenital anomalies at birth, but mean birth weights were significantly lower in fresh oocyte pregnancies. Interestingly, in the group of 63 women who had pregnancies derived from both fresh and thawed oocytes, no differences were noted in the abortion rate or mean birth weight.16
We can freeze eggs, but when should we?
Based on media presentations and professional perspectives, it appears that many people differentiate between “medical” and “social” egg freezing.
Medical versus social freezing
Medical egg freezing is done when there is an immediate medical need to preserve fertility, such as before cancer treatment when the woman can’t reproduce now and will have reduced or no capacity later. Social freezing, on the other hand, occurs when there is no immediate need, such as when there is a desire to delay parenthood so that educational, professional, or other goals can be met. The difference is important because medical freezing is usually seen as a “need” and is therefore acceptable, whereas social freezing is elective or a “wish” and therefore is questionable.17
The labels are important for both ethical and political reasons because most people would consider medical freezing to be ethically acceptable and also worthy of societal support, perhaps even financial coverage, while some might consider social freezing to be neither ethically acceptable nor worthy of coverage.
What’s the difference?
But is the difference really all that clear? If a woman has a mother and a sister who have undergone premature menopause in their 30s and she now has signs of diminishing ovarian reserve in her late 20s, would a desire to freeze eggs be medical or social? She has no immediate need for treatment but a reasonable expectation of need later. One could argue that she should go to a sperm bank now if she has no partner, or change her life plans—but is this a reasonable expectation? If a woman is perfectly healthy but her husband has severe sperm problems and she elects IVF to treat male-factor infertility, is it medical or social? There are many situations in which it is unclear whether the reason for egg freezing would be medical or social.
Does it matter?
In any event, are social reasons to freeze eggs not legitimate? Many would argue that medical services should be used to treat diseases, not social causes. Yet we use medicine all the time to treat problems caused by social factors (obesity, depression, anxiety).
Some would argue that it is a personal decision to delay reproduction, and that health problems caused by personal decisions do not merit medical intervention. However, it is common to provide medical services to people who require the services only because of personal decisions—for instance, professional and amateur athletes who injure themselves pursuing activities for compensation or pleasure, or smokers or persons with alcoholism.
Others have argued that social freezing is inappropriate because it is only being done to avoid the consequences of aging, and that its need could be avoided by not waiting too long to get pregnant. But we treat many conditions that occur primarily as a result of aging (hypertension, dementia, poor eyesight).
Because it has become generally accepted to treat older women with diminished ovarian reserve and infertility, why is it inappropriate to treat women—when they are younger—with egg freezing to mitigate the impact of aging on reproductive performance that we know will occur later? If we could prevent or limit the impact of aging on the cardiovascular or neurologic systems by interventions earlier in a person’s life, would we not provide that medical service? Do we not provide statins and other medications to delay or limit the sequelae of aging? What is the difference with egg freezing?
Therefore, could it be discriminatory not to consider egg freezing ethical and acceptable, even if the reason for the procedure is considered social? Why should egg freezing for social reasons not be acceptable and widely available?
Who should pay for egg freezing?
Even if egg freezing performed because of social reasons is considered ethical and is supported by society, it does not necessarily follow that it will or should be paid for by society. The creation of policies determining coverage for health-care services is a complex process and is based on overall societal needs, economic capabilities, and relative social value of the services. Because infertility carries such a large personal burden and childbearing is so essential to any society, one can argue that infertility, per se, should be covered by society and, in the United States, its surrogate employers and insurance companies. This is often not the case, however. So, while it can be argued that egg freezing should be covered by insurance for both medical and social reasons, even the success of that argument does not mean it will be so in the current US health-care system.
Because egg freezing involves two major steps: (1) ovarian stimulation, egg retrieval, egg freezing, and egg storage followed at a later date by (2) egg thawing, fertilization, embryo culture, and embryo replacement in the uterus, what would be socially justified coverage of egg freezing? Society could cover just the first step or just the second, or both. Such decisions would depend on an assessment of the social benefit from coverage of these services. Such analysis is not yet available because of limited experience.
Is the cost worth it?
A major issue for women considering egg freezing for social reasons is whether a sufficient number of eggs will be retrieved to provide a reasonable chance for pregnancy later when they are used. The FIGURE illustrates the probability of a live birth after egg freezing. It should be noted that while most, but not all, eggs survive thawing after vitrification, not all eggs will become fertilized. Only about half of the fertilized eggs will grow to a day 3 embryo, and not all of those embryos will be viable. Therefore, constant reproductive loss occurs after the eggs are retrieved.

Furthermore, even after embryo replacement, pregnancy does not occur in every case, and some pregnancies are lost to miscarriage as well as other complications of pregnancy and childbirth. The FIGURE shows that a 25-year-old woman with 12 eggs frozen would have an estimated pregnancy rate much greater than 50%. However, the numbers also indicate that egg freezing is not very successful for older women who, at this time, constitute many of those considering the procedure.18
Another consideration is that a significant, but currently unknown, number of women who freeze their eggs will never use them for a variety of reasons. This is especially true of younger women, for whom many of the factors determining their eventual reproductive life might well change. They may eventually decide not to have children or they might become pregnant naturally or after fertility treatments that are cheaper than using the frozen eggs.
A $200,000 price tag?
Let’s consider the near 20% estimated pregnancy rate for age 35 in the FIGURE. If only half of the women aged 35 who freeze 6 eggs eventually use them (but, again, only about 20% have a baby), it means that only one of every 10 women who freeze their eggs eventually will have a baby as a result of the procedure. The number needed to treat (NNT) is therefore 10, and if the cost is $20,000 for the egg freezing procedure and storage over 5 to 10 years, the overall cost per baby born is about $200,000. If 12 eggs are frozen, the cost is $100,000. This clearly is a significant cost, and a greater cost than most other fertility treatments to achieve a baby, even in the older population. Therefore, the cost-effectiveness of social egg freezing is yet to be determined.
What should we do as we move forward?
Abandon the medical versus social rhetoric
It is difficult to argue against egg freezing for medical reasons, and the distinction between medical and social freezing is largely an artificial construct. In general, therefore, the differentiation between medical and social egg freezing should be abandoned, and egg freezing to preserve future fertility should be considered ethical for whatever reasons.
Consider the ideal time frame for health insurance coverage of egg freezing
That does not mean that egg freezing should always be reimbursed. The decision for coverage by employers, insurers, and other payers should be based on a cost–benefit analysis of the social benefit, individual benefit, biological chances of success, probability that the frozen eggs will be used, medical risks/sequelae, and the financial costs. Therefore, whether or not egg freezing for fertility preservation is covered will vary among countries and even within countries and among different individuals. Such an approach to coverage should apply to all medical interventions, including both medical and social egg freezing.
This approach could possibly result in findings and resulting policies that do not cover egg freezing before age 30 because too few women will return to use their eggs, or after age 38 because the chances of success are too low. Other instances of freezing should not be forbidden but would not be reimbursed by public or payer money.17
Many considerations must go into the development of social, professional, and payment policies. Policies that are seen as family-friendly that promote childbearing, especially at an earlier age, can be seen as limiting women’s academic and career opportunities and therefore women-unfriendly. Policies supporting women’s reproductive autonomy and ability to delay childbearing can be seen as women- but not family-friendly. Therefore, reproductive policies affect not only the individual woman but also society, its demographics, politics, and economics.17
The future
The new technology of egg freezing is a wonderful advance for many people. We are learning innovative ways to apply this technology for both infertile and noninfertile people. Research, better evidence, public education, informed consent, ethical practice of medicine, societal support for reproductive rights, and consideration of patient autonomy and social justice will enable us to optimize egg freezing as a treatment intervention.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Chen C. Pregnancy after human oocyte cryopreservation. Lancet. 1986;1(8486):884–886.
2. Practice Committees of the American Society of Reproductive Medicine; Society for Assisted Reproductive Technology. Mature oocyte cryopreservation: a guideline. Fertil Steril. 2013;99(1):37–43.
3. Polge C, Smith AU, Parkes AS. Revival of spermatozoa after vitrification and dehydration at low temperatures. Nature. 1949;164(4172):666.
4. Gook D. History of oocyte cryopreservation. Reprod Biomed Online. 2011;23(3):281−289.
5. Gosden R. Cryopreservation: a cold look at technology for fertility preservation. Fertil Steril. 2011;96(2):264−268.
6. Van der Elst J. Oocyte freezing: here to stay? Hum Reprod Update. 2003;9(5):463–470.
7. Porcu E, Fabbri R, Seracchioli R, Ciotti PM, Magrini O, Flamigni C. Birth of a healthy female after intracytoplasmic sperm injection of cryopreserved human oocytes. Fertil Steril. 1997;68(4):724–726.
8. Fabbri R, Porcu E, Marsella T, Rocchetta G, Venturoli S, Flamigni C. Human oocyte cryopreservation: new perspectives regarding oocyte survival. Hum Reprod. 2001;16(3):411–416.
9. Oktay K, Cil AP, Bang H. Efficiency of oocyte cryopreservation: a meta-analysis. Fertil Steril. 2006;86(1):70–80.
10. Smith GD, Serafini PC, Fioravanti J, et al. Prospective randomized comparison of human oocyte cryopreservationwith slow-rate freezing or vitrification. Fertil Steril. 2010;94(6):2088–2095.
11. Gook DA, Edgar DH. Human oocyte cryopreservation. Hum Reprod Update. 2007;13(6):591–605.
12. Rienzi L, Cobo A, Paffoni A, et al. Consistent and predictable delivery rates after oocyte vitrification: an observational longitudinal cohort multicentric study. Hum Reprod. 2012;27(6):1606–1612.
13. Cobo A, Rubio C, Gerli S, Ruiz A, Pellicer A, Remohi J. Use of fluorescence in situ hybridization to assess the chromosomal status of embryos obtained from cryopreserved oocytes. Fertil Steril. 2001;75(2):354–360.
14. Noyes N, Porcu E, Borini A. Over 900 oocyte cryopreservation babies born with no apparent increase in congenital anomalies. Reprod Biomed Online. 2009;18(6):769–776.
15. Chian RC, Huang JY, Tan SL, et al. Obstetric and perinatal outcome in 200 infants conceived from vitrified oocytes. Reprod Biomed Online. 2008;16(5):608–610.
16. Levi Setti P, Albani E, Morenghi E, et al. Comparative analysis of fetal and neonatal outcomes of pregnancies from fresh and cryopreserved/thawed oocytes in the same group of patients. Fertil Steril. 2013;100(2):396–401.
17. Pennings G. Ethical aspects of social freezing. Gynecol Obstet Fertil. 2013;41(9):521–523.
18. Cil AP, Bang H, Oktay K. Age-specific probability of live birth with oocyte cryopreservation: an individual patient data meta-analysis. Fertil Steril. 2013;100(2):492–499.
Mary E. Abusief, MD and G. David Adamson, MD

Dr. Abusief is a Board-Certified Specialist in Reproductive Endocrinology and Infertility at Palo Alto Medical Foundation Fertility Physicians of Northern California.

Dr. Adamson is Founder/CEO of Advanced Reproductive Care, Inc; Adjunct Clinical Professor at Stanford University; and Associate Clinical Professor at the University of California, San Francisco. He is also Medical Director, Assisted Reproductive Technologies Program, Palo Alto Medical Foundation Fertility Physicians of Northern California in Palo Alto and San Jose, California.
Dr. Abusief reports no financial relationships relevant to this article. Dr. Adamson reports that he receives grant or research support from Auxogyn, LabCorp, Schering Plough, and IBSA, is a consultant to Auxogyn, Bayer HealthCare, Ferring, LabCorp, Ziva Medical, and has other financial relationships with Advanced Reproductive Care.
Mary E. Abusief, MD and G. David Adamson, MD
Dr. Abusief is a Board-Certified Specialist in Reproductive Endocrinology and Infertility at Palo Alto Medical Foundation Fertility Physicians of Northern California.
Dr. Adamson is Founder/CEO of Advanced Reproductive Care, Inc; Adjunct Clinical Professor at Stanford University; and Associate Clinical Professor at the University of California, San Francisco. He is also Medical Director, Assisted Reproductive Technologies Program, Palo Alto Medical Foundation Fertility Physicians of Northern California in Palo Alto and San Jose, California.
Dr. Abusief reports no financial relationships relevant to this article. Dr. Adamson reports that he receives grant or research support from Auxogyn, LabCorp, Schering Plough, and IBSA, is a consultant to Auxogyn, Bayer HealthCare, Ferring, LabCorp, Ziva Medical, and has other financial relationships with Advanced Reproductive Care.
Mary E. Abusief, MD and G. David Adamson, MD
Dr. Abusief is a Board-Certified Specialist in Reproductive Endocrinology and Infertility at Palo Alto Medical Foundation Fertility Physicians of Northern California.
Dr. Adamson is Founder/CEO of Advanced Reproductive Care, Inc; Adjunct Clinical Professor at Stanford University; and Associate Clinical Professor at the University of California, San Francisco. He is also Medical Director, Assisted Reproductive Technologies Program, Palo Alto Medical Foundation Fertility Physicians of Northern California in Palo Alto and San Jose, California.
Dr. Abusief reports no financial relationships relevant to this article. Dr. Adamson reports that he receives grant or research support from Auxogyn, LabCorp, Schering Plough, and IBSA, is a consultant to Auxogyn, Bayer HealthCare, Ferring, LabCorp, Ziva Medical, and has other financial relationships with Advanced Reproductive Care.
The first human birth from a frozen oocyte was reported in 1986.1 Nearly 3 decades later, mature oocyte cryopreservation has emerged as a meaningful technology to preserve reproductive potential in women of reproductive age. In 2013, the American Society for Reproductive Medicine (ASRM) removed the “experimental” label from egg freezing but cautioned that more data on safety and efficacy were needed prior to widespread adoption of the technique.2
In this Update, we present the current protocols for oocyte cryopreservation, how we arrived at them, and the questions regarding outcomes that still remain. In addition, we discuss the ethical dilemmas egg freezing presents, according to the varying rhetoric within the media and our own profession. Finally, we consider what preliminary data suggest as to the live-birth rate using frozen eggs from women of varying ages and what the costs are associated with using oocyte cryopreservation as the approach to fertility treatment.
Vitrification and slow freezing: How did we get here and how effective are they?
Fertility preservation is a rapidly advancing area of reproductive medicine. Cryopreservation is the cooling of cells to subzero temperatures to halt biologic activity and preserve the cells for future use. Clinically, oocyte cryopreservation requires a patient to undergo in vitro fertilization (IVF). After egg retrieval, the oocytes are cryopreserved for use at a later time.
The prefix “cryo” originated from the Greek word “kryos,” meaning icy cold or frost. Cryopreservation is not a new science. In 1776, the Italian priest and scientist Lazzaro Spallanzani reported that sperm became motionless when cooled by snow. A pivotal discovery in the field came in 1949, when Christopher Polge, an English scientist, showed that glycerol, a permeating solute, could provide protection to cells at low temperatures.3 Progress in sperm cryopreservation advanced quickly, partly due to the ease of observing sperm motility as a marker of postthaw function.4
The ongoing evolution of cryopreservation science led to landmark achievements, including the first birth using human cryopreserved sperm in the 1950s, and the first human birth after embryo thaw in 1983. Since that time cryopreservation has become a cornerstone in the field of reproductive medicine.
Initial problems encountered with egg freezing
Although the first birth after thaw of a human oocyte occurred in 1986, oocyte cryopreservation was fraught with technical difficulties. Oocytes (vs sperm and embryos) proved challenging to successfully cryopreserve. The problem lay in the damage caused by water crystals forming ice and rising concentrations of intracellular solutes as cells were cooled to freezing temperatures.5 The large size and high water content of the human oocyte made it particularly vulnerable to the detrimental effects of freezing. In addition, freeze−thaw hardening of the zone pellucida led to decreased postthaw fertilization. The delicate meiotic spindle within the oocyte was prone to injury from ice crystals.6
Use of cryoprotectants, such as ethylene glycol, glycerol, and dimethylsulfoxide (DMSO), can prevent ice crystal formation, but high concentrations are theoretically toxic. The fine balance between protection and toxicity led to the development of diverse egg freezing protocols using various types and concentrations of cryoprotectants. Inconsistent results and lack of reproducibility among labs, together with concerns about postthaw oocyte function and safety, slowed the progression of oocyte freezing. By the end of the 1980s, clinical oocyte cryopreservation had been effectively halted and the field was confined to small groups of researchers who continued laboratory experiments with limited success.5
In 1997, clinical work with frozen oocytes resumed with a Bologna team reporting postthawing oocyte survival rates of up to 80% using propanediol as the primary cryoprotectant, and viable pregnancies with the use of intracytoplasmic sperm injection (ICSI) for fertilization.7,8 Since the late 1990s, further modifications in freezing technologies have resulted in greater success. And currently, both slow freezing and vitrification methods are used to preserve oocytes.
Slow freezing
Slow freezing involves a low rate of oocyte temperature decline with a simultaneous gradual increase in the concentration of cryoprotectants. As the metabolic activity of the oocyte decreases, the concentration of cryoprotectant can be increased to prevent ice crystal formation. Once solidification of the oocyte is achieved, the oocyte can be exposed to freezing at colder temperatures. Results of a meta-analysis of 26 studies revealed that, compared with using fresh oocytes, eggs thawed after slow-freezing yielded significantly lower rates of fertilization (61.0% [1,346/2,217] vs 76.7% [2,788/3,637]), clinical pregnancy rate per transfer (27.1% [95/351] vs 68.5% [272/397]), and live birth per transfer (21.6% [76/351] vs 32.4% [24/72]).9
Vitrification
Vitrification involves the rapid cooling of cells to extremely low temperatures. During vitrification, oocytes are exposed to high concentrations of cryoprotectants and, after a short equilibration time, rapidly cooled. The rate of cooling is dramatic, up to 20,000°C per minute—so fast that ice does not have time to form and a glass-like state is achieved within the oocyte. Studies suggest that the use of vitrification improves oocyte survival and function compared with slow freezing.9-11 A prospective randomized controlled trial comparing frozen/thawed with vitrified/warmed oocytes demonstrated superior oocyte function in the vitrification group, with higher oocyte survival (81% for vitrification/warming vs 67% for slow freezing/thawing); higher rates of fertilization, cleavage, and embryo morphology; as well as higher clinical pregnancy rates (38% for vitrified/warmed vs 13% for frozen/thawed).10
The Practice Committee of ASRM published a guideline for mature cryopreservation in 2013.2 The committee reviewed the literature on efficacy and safety of mature oocyte cryopreservation. Although data are limited, studies comparing outcomes of IVF using cryopreserved versus fresh oocytes, including four randomized controlled trials and a meta-analysis, provide evidence that previously vitrified/thawed eggs result in similar fertilization and pregnancy rates as IVF/ICSI with fresh oocytes. Similar to data from fresh IVF cycles, decreased success with oocyte vitrification is seen in women with advanced age, and delivery rates, not unexpectedly, are inversely correlated with maternal age.12
Safety outcomes data are limited but reassuring
Two major factors limit our current understanding of egg cryopreservation outcomes. First, many women who have cryopreserved their eggs have not yet used them and, second, babies born after use of cryopreserved oocytes have not reached ages in which safety of the technique can be fully evaluated. Despite this important gap in our knowledge, to date, results of studies examining safety outcomes of the procedure have been reassuring.
For instance, chromosomal analysis via fluorescence in-situ hybridization of embryos created with thawed oocytes versus controls revealed no difference in the incidence of chromosomal abnormalities, decreasing concerns about damage to the oocyte spindle secondary to freezing.13
Data from a review of 900 live births resulting from embryos created from thawed oocytes frozen via the slow freeze technique revealed no increase in the risk of congenital anomalies.14 Similarly, no increased risk of congenital anomalies or difference in birth weights was noted in a study of 200 live births after transfers with embryos derived from vitrified oocytes compared with fresh oocytes.15
In a study of 954 clinical pregnancies occurring in 855 couples with cryopreserved oocytes after assisted reproductive technology, the outcomes of 197 pregnancies from frozen/thawed oocytes were compared with 757 obtained from fresh sibling oocyte cycles. A significantly higher rate of spontaneous abortions at 12 weeks or less was observed in the frozen/thawed oocyte group. No statistically significant differences were noted in gestational age at delivery or in the incidence of major congenital anomalies at birth, but mean birth weights were significantly lower in fresh oocyte pregnancies. Interestingly, in the group of 63 women who had pregnancies derived from both fresh and thawed oocytes, no differences were noted in the abortion rate or mean birth weight.16
We can freeze eggs, but when should we?
Based on media presentations and professional perspectives, it appears that many people differentiate between “medical” and “social” egg freezing.
Medical versus social freezing
Medical egg freezing is done when there is an immediate medical need to preserve fertility, such as before cancer treatment when the woman can’t reproduce now and will have reduced or no capacity later. Social freezing, on the other hand, occurs when there is no immediate need, such as when there is a desire to delay parenthood so that educational, professional, or other goals can be met. The difference is important because medical freezing is usually seen as a “need” and is therefore acceptable, whereas social freezing is elective or a “wish” and therefore is questionable.17
The labels are important for both ethical and political reasons because most people would consider medical freezing to be ethically acceptable and also worthy of societal support, perhaps even financial coverage, while some might consider social freezing to be neither ethically acceptable nor worthy of coverage.
What’s the difference?
But is the difference really all that clear? If a woman has a mother and a sister who have undergone premature menopause in their 30s and she now has signs of diminishing ovarian reserve in her late 20s, would a desire to freeze eggs be medical or social? She has no immediate need for treatment but a reasonable expectation of need later. One could argue that she should go to a sperm bank now if she has no partner, or change her life plans—but is this a reasonable expectation? If a woman is perfectly healthy but her husband has severe sperm problems and she elects IVF to treat male-factor infertility, is it medical or social? There are many situations in which it is unclear whether the reason for egg freezing would be medical or social.
Does it matter?
In any event, are social reasons to freeze eggs not legitimate? Many would argue that medical services should be used to treat diseases, not social causes. Yet we use medicine all the time to treat problems caused by social factors (obesity, depression, anxiety).
Some would argue that it is a personal decision to delay reproduction, and that health problems caused by personal decisions do not merit medical intervention. However, it is common to provide medical services to people who require the services only because of personal decisions—for instance, professional and amateur athletes who injure themselves pursuing activities for compensation or pleasure, or smokers or persons with alcoholism.
Others have argued that social freezing is inappropriate because it is only being done to avoid the consequences of aging, and that its need could be avoided by not waiting too long to get pregnant. But we treat many conditions that occur primarily as a result of aging (hypertension, dementia, poor eyesight).
Because it has become generally accepted to treat older women with diminished ovarian reserve and infertility, why is it inappropriate to treat women—when they are younger—with egg freezing to mitigate the impact of aging on reproductive performance that we know will occur later? If we could prevent or limit the impact of aging on the cardiovascular or neurologic systems by interventions earlier in a person’s life, would we not provide that medical service? Do we not provide statins and other medications to delay or limit the sequelae of aging? What is the difference with egg freezing?
Therefore, could it be discriminatory not to consider egg freezing ethical and acceptable, even if the reason for the procedure is considered social? Why should egg freezing for social reasons not be acceptable and widely available?
Who should pay for egg freezing?
Even if egg freezing performed because of social reasons is considered ethical and is supported by society, it does not necessarily follow that it will or should be paid for by society. The creation of policies determining coverage for health-care services is a complex process and is based on overall societal needs, economic capabilities, and relative social value of the services. Because infertility carries such a large personal burden and childbearing is so essential to any society, one can argue that infertility, per se, should be covered by society and, in the United States, its surrogate employers and insurance companies. This is often not the case, however. So, while it can be argued that egg freezing should be covered by insurance for both medical and social reasons, even the success of that argument does not mean it will be so in the current US health-care system.
Because egg freezing involves two major steps: (1) ovarian stimulation, egg retrieval, egg freezing, and egg storage followed at a later date by (2) egg thawing, fertilization, embryo culture, and embryo replacement in the uterus, what would be socially justified coverage of egg freezing? Society could cover just the first step or just the second, or both. Such decisions would depend on an assessment of the social benefit from coverage of these services. Such analysis is not yet available because of limited experience.
Is the cost worth it?
A major issue for women considering egg freezing for social reasons is whether a sufficient number of eggs will be retrieved to provide a reasonable chance for pregnancy later when they are used. The FIGURE illustrates the probability of a live birth after egg freezing. It should be noted that while most, but not all, eggs survive thawing after vitrification, not all eggs will become fertilized. Only about half of the fertilized eggs will grow to a day 3 embryo, and not all of those embryos will be viable. Therefore, constant reproductive loss occurs after the eggs are retrieved.
Furthermore, even after embryo replacement, pregnancy does not occur in every case, and some pregnancies are lost to miscarriage as well as other complications of pregnancy and childbirth. The FIGURE shows that a 25-year-old woman with 12 eggs frozen would have an estimated pregnancy rate much greater than 50%. However, the numbers also indicate that egg freezing is not very successful for older women who, at this time, constitute many of those considering the procedure.18
Another consideration is that a significant, but currently unknown, number of women who freeze their eggs will never use them for a variety of reasons. This is especially true of younger women, for whom many of the factors determining their eventual reproductive life might well change. They may eventually decide not to have children or they might become pregnant naturally or after fertility treatments that are cheaper than using the frozen eggs.
A $200,000 price tag?
Let’s consider the near 20% estimated pregnancy rate for age 35 in the FIGURE. If only half of the women aged 35 who freeze 6 eggs eventually use them (but, again, only about 20% have a baby), it means that only one of every 10 women who freeze their eggs eventually will have a baby as a result of the procedure. The number needed to treat (NNT) is therefore 10, and if the cost is $20,000 for the egg freezing procedure and storage over 5 to 10 years, the overall cost per baby born is about $200,000. If 12 eggs are frozen, the cost is $100,000. This clearly is a significant cost, and a greater cost than most other fertility treatments to achieve a baby, even in the older population. Therefore, the cost-effectiveness of social egg freezing is yet to be determined.
What should we do as we move forward?
Abandon the medical versus social rhetoric
It is difficult to argue against egg freezing for medical reasons, and the distinction between medical and social freezing is largely an artificial construct. In general, therefore, the differentiation between medical and social egg freezing should be abandoned, and egg freezing to preserve future fertility should be considered ethical for whatever reasons.
Consider the ideal time frame for health insurance coverage of egg freezing
That does not mean that egg freezing should always be reimbursed. The decision for coverage by employers, insurers, and other payers should be based on a cost–benefit analysis of the social benefit, individual benefit, biological chances of success, probability that the frozen eggs will be used, medical risks/sequelae, and the financial costs. Therefore, whether or not egg freezing for fertility preservation is covered will vary among countries and even within countries and among different individuals. Such an approach to coverage should apply to all medical interventions, including both medical and social egg freezing.
This approach could possibly result in findings and resulting policies that do not cover egg freezing before age 30 because too few women will return to use their eggs, or after age 38 because the chances of success are too low. Other instances of freezing should not be forbidden but would not be reimbursed by public or payer money.17
Many considerations must go into the development of social, professional, and payment policies. Policies that are seen as family-friendly that promote childbearing, especially at an earlier age, can be seen as limiting women’s academic and career opportunities and therefore women-unfriendly. Policies supporting women’s reproductive autonomy and ability to delay childbearing can be seen as women- but not family-friendly. Therefore, reproductive policies affect not only the individual woman but also society, its demographics, politics, and economics.17
The future
The new technology of egg freezing is a wonderful advance for many people. We are learning innovative ways to apply this technology for both infertile and noninfertile people. Research, better evidence, public education, informed consent, ethical practice of medicine, societal support for reproductive rights, and consideration of patient autonomy and social justice will enable us to optimize egg freezing as a treatment intervention.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
The first human birth from a frozen oocyte was reported in 1986.1 Nearly 3 decades later, mature oocyte cryopreservation has emerged as a meaningful technology to preserve reproductive potential in women of reproductive age. In 2013, the American Society for Reproductive Medicine (ASRM) removed the “experimental” label from egg freezing but cautioned that more data on safety and efficacy were needed prior to widespread adoption of the technique.2
In this Update, we present the current protocols for oocyte cryopreservation, how we arrived at them, and the questions regarding outcomes that still remain. In addition, we discuss the ethical dilemmas egg freezing presents, according to the varying rhetoric within the media and our own profession. Finally, we consider what preliminary data suggest as to the live-birth rate using frozen eggs from women of varying ages and what the costs are associated with using oocyte cryopreservation as the approach to fertility treatment.
Vitrification and slow freezing: How did we get here and how effective are they?
Fertility preservation is a rapidly advancing area of reproductive medicine. Cryopreservation is the cooling of cells to subzero temperatures to halt biologic activity and preserve the cells for future use. Clinically, oocyte cryopreservation requires a patient to undergo in vitro fertilization (IVF). After egg retrieval, the oocytes are cryopreserved for use at a later time.
The prefix “cryo” originated from the Greek word “kryos,” meaning icy cold or frost. Cryopreservation is not a new science. In 1776, the Italian priest and scientist Lazzaro Spallanzani reported that sperm became motionless when cooled by snow. A pivotal discovery in the field came in 1949, when Christopher Polge, an English scientist, showed that glycerol, a permeating solute, could provide protection to cells at low temperatures.3 Progress in sperm cryopreservation advanced quickly, partly due to the ease of observing sperm motility as a marker of postthaw function.4
The ongoing evolution of cryopreservation science led to landmark achievements, including the first birth using human cryopreserved sperm in the 1950s, and the first human birth after embryo thaw in 1983. Since that time cryopreservation has become a cornerstone in the field of reproductive medicine.
Initial problems encountered with egg freezing
Although the first birth after thaw of a human oocyte occurred in 1986, oocyte cryopreservation was fraught with technical difficulties. Oocytes (vs sperm and embryos) proved challenging to successfully cryopreserve. The problem lay in the damage caused by water crystals forming ice and rising concentrations of intracellular solutes as cells were cooled to freezing temperatures.5 The large size and high water content of the human oocyte made it particularly vulnerable to the detrimental effects of freezing. In addition, freeze−thaw hardening of the zone pellucida led to decreased postthaw fertilization. The delicate meiotic spindle within the oocyte was prone to injury from ice crystals.6
Use of cryoprotectants, such as ethylene glycol, glycerol, and dimethylsulfoxide (DMSO), can prevent ice crystal formation, but high concentrations are theoretically toxic. The fine balance between protection and toxicity led to the development of diverse egg freezing protocols using various types and concentrations of cryoprotectants. Inconsistent results and lack of reproducibility among labs, together with concerns about postthaw oocyte function and safety, slowed the progression of oocyte freezing. By the end of the 1980s, clinical oocyte cryopreservation had been effectively halted and the field was confined to small groups of researchers who continued laboratory experiments with limited success.5
In 1997, clinical work with frozen oocytes resumed with a Bologna team reporting postthawing oocyte survival rates of up to 80% using propanediol as the primary cryoprotectant, and viable pregnancies with the use of intracytoplasmic sperm injection (ICSI) for fertilization.7,8 Since the late 1990s, further modifications in freezing technologies have resulted in greater success. And currently, both slow freezing and vitrification methods are used to preserve oocytes.
Slow freezing
Slow freezing involves a low rate of oocyte temperature decline with a simultaneous gradual increase in the concentration of cryoprotectants. As the metabolic activity of the oocyte decreases, the concentration of cryoprotectant can be increased to prevent ice crystal formation. Once solidification of the oocyte is achieved, the oocyte can be exposed to freezing at colder temperatures. Results of a meta-analysis of 26 studies revealed that, compared with using fresh oocytes, eggs thawed after slow-freezing yielded significantly lower rates of fertilization (61.0% [1,346/2,217] vs 76.7% [2,788/3,637]), clinical pregnancy rate per transfer (27.1% [95/351] vs 68.5% [272/397]), and live birth per transfer (21.6% [76/351] vs 32.4% [24/72]).9
Vitrification
Vitrification involves the rapid cooling of cells to extremely low temperatures. During vitrification, oocytes are exposed to high concentrations of cryoprotectants and, after a short equilibration time, rapidly cooled. The rate of cooling is dramatic, up to 20,000°C per minute—so fast that ice does not have time to form and a glass-like state is achieved within the oocyte. Studies suggest that the use of vitrification improves oocyte survival and function compared with slow freezing.9-11 A prospective randomized controlled trial comparing frozen/thawed with vitrified/warmed oocytes demonstrated superior oocyte function in the vitrification group, with higher oocyte survival (81% for vitrification/warming vs 67% for slow freezing/thawing); higher rates of fertilization, cleavage, and embryo morphology; as well as higher clinical pregnancy rates (38% for vitrified/warmed vs 13% for frozen/thawed).10
The Practice Committee of ASRM published a guideline for mature cryopreservation in 2013.2 The committee reviewed the literature on efficacy and safety of mature oocyte cryopreservation. Although data are limited, studies comparing outcomes of IVF using cryopreserved versus fresh oocytes, including four randomized controlled trials and a meta-analysis, provide evidence that previously vitrified/thawed eggs result in similar fertilization and pregnancy rates as IVF/ICSI with fresh oocytes. Similar to data from fresh IVF cycles, decreased success with oocyte vitrification is seen in women with advanced age, and delivery rates, not unexpectedly, are inversely correlated with maternal age.12
Safety outcomes data are limited but reassuring
Two major factors limit our current understanding of egg cryopreservation outcomes. First, many women who have cryopreserved their eggs have not yet used them and, second, babies born after use of cryopreserved oocytes have not reached ages in which safety of the technique can be fully evaluated. Despite this important gap in our knowledge, to date, results of studies examining safety outcomes of the procedure have been reassuring.
For instance, chromosomal analysis via fluorescence in-situ hybridization of embryos created with thawed oocytes versus controls revealed no difference in the incidence of chromosomal abnormalities, decreasing concerns about damage to the oocyte spindle secondary to freezing.13
Data from a review of 900 live births resulting from embryos created from thawed oocytes frozen via the slow freeze technique revealed no increase in the risk of congenital anomalies.14 Similarly, no increased risk of congenital anomalies or difference in birth weights was noted in a study of 200 live births after transfers with embryos derived from vitrified oocytes compared with fresh oocytes.15
In a study of 954 clinical pregnancies occurring in 855 couples with cryopreserved oocytes after assisted reproductive technology, the outcomes of 197 pregnancies from frozen/thawed oocytes were compared with 757 obtained from fresh sibling oocyte cycles. A significantly higher rate of spontaneous abortions at 12 weeks or less was observed in the frozen/thawed oocyte group. No statistically significant differences were noted in gestational age at delivery or in the incidence of major congenital anomalies at birth, but mean birth weights were significantly lower in fresh oocyte pregnancies. Interestingly, in the group of 63 women who had pregnancies derived from both fresh and thawed oocytes, no differences were noted in the abortion rate or mean birth weight.16
We can freeze eggs, but when should we?
Based on media presentations and professional perspectives, it appears that many people differentiate between “medical” and “social” egg freezing.
Medical versus social freezing
Medical egg freezing is done when there is an immediate medical need to preserve fertility, such as before cancer treatment when the woman can’t reproduce now and will have reduced or no capacity later. Social freezing, on the other hand, occurs when there is no immediate need, such as when there is a desire to delay parenthood so that educational, professional, or other goals can be met. The difference is important because medical freezing is usually seen as a “need” and is therefore acceptable, whereas social freezing is elective or a “wish” and therefore is questionable.17
The labels are important for both ethical and political reasons because most people would consider medical freezing to be ethically acceptable and also worthy of societal support, perhaps even financial coverage, while some might consider social freezing to be neither ethically acceptable nor worthy of coverage.
What’s the difference?
But is the difference really all that clear? If a woman has a mother and a sister who have undergone premature menopause in their 30s and she now has signs of diminishing ovarian reserve in her late 20s, would a desire to freeze eggs be medical or social? She has no immediate need for treatment but a reasonable expectation of need later. One could argue that she should go to a sperm bank now if she has no partner, or change her life plans—but is this a reasonable expectation? If a woman is perfectly healthy but her husband has severe sperm problems and she elects IVF to treat male-factor infertility, is it medical or social? There are many situations in which it is unclear whether the reason for egg freezing would be medical or social.
Does it matter?
In any event, are social reasons to freeze eggs not legitimate? Many would argue that medical services should be used to treat diseases, not social causes. Yet we use medicine all the time to treat problems caused by social factors (obesity, depression, anxiety).
Some would argue that it is a personal decision to delay reproduction, and that health problems caused by personal decisions do not merit medical intervention. However, it is common to provide medical services to people who require the services only because of personal decisions—for instance, professional and amateur athletes who injure themselves pursuing activities for compensation or pleasure, or smokers or persons with alcoholism.
Others have argued that social freezing is inappropriate because it is only being done to avoid the consequences of aging, and that its need could be avoided by not waiting too long to get pregnant. But we treat many conditions that occur primarily as a result of aging (hypertension, dementia, poor eyesight).
Because it has become generally accepted to treat older women with diminished ovarian reserve and infertility, why is it inappropriate to treat women—when they are younger—with egg freezing to mitigate the impact of aging on reproductive performance that we know will occur later? If we could prevent or limit the impact of aging on the cardiovascular or neurologic systems by interventions earlier in a person’s life, would we not provide that medical service? Do we not provide statins and other medications to delay or limit the sequelae of aging? What is the difference with egg freezing?
Therefore, could it be discriminatory not to consider egg freezing ethical and acceptable, even if the reason for the procedure is considered social? Why should egg freezing for social reasons not be acceptable and widely available?
Who should pay for egg freezing?
Even if egg freezing performed because of social reasons is considered ethical and is supported by society, it does not necessarily follow that it will or should be paid for by society. The creation of policies determining coverage for health-care services is a complex process and is based on overall societal needs, economic capabilities, and relative social value of the services. Because infertility carries such a large personal burden and childbearing is so essential to any society, one can argue that infertility, per se, should be covered by society and, in the United States, its surrogate employers and insurance companies. This is often not the case, however. So, while it can be argued that egg freezing should be covered by insurance for both medical and social reasons, even the success of that argument does not mean it will be so in the current US health-care system.
Because egg freezing involves two major steps: (1) ovarian stimulation, egg retrieval, egg freezing, and egg storage followed at a later date by (2) egg thawing, fertilization, embryo culture, and embryo replacement in the uterus, what would be socially justified coverage of egg freezing? Society could cover just the first step or just the second, or both. Such decisions would depend on an assessment of the social benefit from coverage of these services. Such analysis is not yet available because of limited experience.
Is the cost worth it?
A major issue for women considering egg freezing for social reasons is whether a sufficient number of eggs will be retrieved to provide a reasonable chance for pregnancy later when they are used. The FIGURE illustrates the probability of a live birth after egg freezing. It should be noted that while most, but not all, eggs survive thawing after vitrification, not all eggs will become fertilized. Only about half of the fertilized eggs will grow to a day 3 embryo, and not all of those embryos will be viable. Therefore, constant reproductive loss occurs after the eggs are retrieved.
Furthermore, even after embryo replacement, pregnancy does not occur in every case, and some pregnancies are lost to miscarriage as well as other complications of pregnancy and childbirth. The FIGURE shows that a 25-year-old woman with 12 eggs frozen would have an estimated pregnancy rate much greater than 50%. However, the numbers also indicate that egg freezing is not very successful for older women who, at this time, constitute many of those considering the procedure.18
Another consideration is that a significant, but currently unknown, number of women who freeze their eggs will never use them for a variety of reasons. This is especially true of younger women, for whom many of the factors determining their eventual reproductive life might well change. They may eventually decide not to have children or they might become pregnant naturally or after fertility treatments that are cheaper than using the frozen eggs.
A $200,000 price tag?
Let’s consider the near 20% estimated pregnancy rate for age 35 in the FIGURE. If only half of the women aged 35 who freeze 6 eggs eventually use them (but, again, only about 20% have a baby), it means that only one of every 10 women who freeze their eggs eventually will have a baby as a result of the procedure. The number needed to treat (NNT) is therefore 10, and if the cost is $20,000 for the egg freezing procedure and storage over 5 to 10 years, the overall cost per baby born is about $200,000. If 12 eggs are frozen, the cost is $100,000. This clearly is a significant cost, and a greater cost than most other fertility treatments to achieve a baby, even in the older population. Therefore, the cost-effectiveness of social egg freezing is yet to be determined.
What should we do as we move forward?
Abandon the medical versus social rhetoric
It is difficult to argue against egg freezing for medical reasons, and the distinction between medical and social freezing is largely an artificial construct. In general, therefore, the differentiation between medical and social egg freezing should be abandoned, and egg freezing to preserve future fertility should be considered ethical for whatever reasons.
Consider the ideal time frame for health insurance coverage of egg freezing
That does not mean that egg freezing should always be reimbursed. The decision for coverage by employers, insurers, and other payers should be based on a cost–benefit analysis of the social benefit, individual benefit, biological chances of success, probability that the frozen eggs will be used, medical risks/sequelae, and the financial costs. Therefore, whether or not egg freezing for fertility preservation is covered will vary among countries and even within countries and among different individuals. Such an approach to coverage should apply to all medical interventions, including both medical and social egg freezing.
This approach could possibly result in findings and resulting policies that do not cover egg freezing before age 30 because too few women will return to use their eggs, or after age 38 because the chances of success are too low. Other instances of freezing should not be forbidden but would not be reimbursed by public or payer money.17
Many considerations must go into the development of social, professional, and payment policies. Policies that are seen as family-friendly that promote childbearing, especially at an earlier age, can be seen as limiting women’s academic and career opportunities and therefore women-unfriendly. Policies supporting women’s reproductive autonomy and ability to delay childbearing can be seen as women- but not family-friendly. Therefore, reproductive policies affect not only the individual woman but also society, its demographics, politics, and economics.17
The future
The new technology of egg freezing is a wonderful advance for many people. We are learning innovative ways to apply this technology for both infertile and noninfertile people. Research, better evidence, public education, informed consent, ethical practice of medicine, societal support for reproductive rights, and consideration of patient autonomy and social justice will enable us to optimize egg freezing as a treatment intervention.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Chen C. Pregnancy after human oocyte cryopreservation. Lancet. 1986;1(8486):884–886.
2. Practice Committees of the American Society of Reproductive Medicine; Society for Assisted Reproductive Technology. Mature oocyte cryopreservation: a guideline. Fertil Steril. 2013;99(1):37–43.
3. Polge C, Smith AU, Parkes AS. Revival of spermatozoa after vitrification and dehydration at low temperatures. Nature. 1949;164(4172):666.
4. Gook D. History of oocyte cryopreservation. Reprod Biomed Online. 2011;23(3):281−289.
5. Gosden R. Cryopreservation: a cold look at technology for fertility preservation. Fertil Steril. 2011;96(2):264−268.
6. Van der Elst J. Oocyte freezing: here to stay? Hum Reprod Update. 2003;9(5):463–470.
7. Porcu E, Fabbri R, Seracchioli R, Ciotti PM, Magrini O, Flamigni C. Birth of a healthy female after intracytoplasmic sperm injection of cryopreserved human oocytes. Fertil Steril. 1997;68(4):724–726.
8. Fabbri R, Porcu E, Marsella T, Rocchetta G, Venturoli S, Flamigni C. Human oocyte cryopreservation: new perspectives regarding oocyte survival. Hum Reprod. 2001;16(3):411–416.
9. Oktay K, Cil AP, Bang H. Efficiency of oocyte cryopreservation: a meta-analysis. Fertil Steril. 2006;86(1):70–80.
10. Smith GD, Serafini PC, Fioravanti J, et al. Prospective randomized comparison of human oocyte cryopreservationwith slow-rate freezing or vitrification. Fertil Steril. 2010;94(6):2088–2095.
11. Gook DA, Edgar DH. Human oocyte cryopreservation. Hum Reprod Update. 2007;13(6):591–605.
12. Rienzi L, Cobo A, Paffoni A, et al. Consistent and predictable delivery rates after oocyte vitrification: an observational longitudinal cohort multicentric study. Hum Reprod. 2012;27(6):1606–1612.
13. Cobo A, Rubio C, Gerli S, Ruiz A, Pellicer A, Remohi J. Use of fluorescence in situ hybridization to assess the chromosomal status of embryos obtained from cryopreserved oocytes. Fertil Steril. 2001;75(2):354–360.
14. Noyes N, Porcu E, Borini A. Over 900 oocyte cryopreservation babies born with no apparent increase in congenital anomalies. Reprod Biomed Online. 2009;18(6):769–776.
15. Chian RC, Huang JY, Tan SL, et al. Obstetric and perinatal outcome in 200 infants conceived from vitrified oocytes. Reprod Biomed Online. 2008;16(5):608–610.
16. Levi Setti P, Albani E, Morenghi E, et al. Comparative analysis of fetal and neonatal outcomes of pregnancies from fresh and cryopreserved/thawed oocytes in the same group of patients. Fertil Steril. 2013;100(2):396–401.
17. Pennings G. Ethical aspects of social freezing. Gynecol Obstet Fertil. 2013;41(9):521–523.
18. Cil AP, Bang H, Oktay K. Age-specific probability of live birth with oocyte cryopreservation: an individual patient data meta-analysis. Fertil Steril. 2013;100(2):492–499.
1. Chen C. Pregnancy after human oocyte cryopreservation. Lancet. 1986;1(8486):884–886.
2. Practice Committees of the American Society of Reproductive Medicine; Society for Assisted Reproductive Technology. Mature oocyte cryopreservation: a guideline. Fertil Steril. 2013;99(1):37–43.
3. Polge C, Smith AU, Parkes AS. Revival of spermatozoa after vitrification and dehydration at low temperatures. Nature. 1949;164(4172):666.
4. Gook D. History of oocyte cryopreservation. Reprod Biomed Online. 2011;23(3):281−289.
5. Gosden R. Cryopreservation: a cold look at technology for fertility preservation. Fertil Steril. 2011;96(2):264−268.
6. Van der Elst J. Oocyte freezing: here to stay? Hum Reprod Update. 2003;9(5):463–470.
7. Porcu E, Fabbri R, Seracchioli R, Ciotti PM, Magrini O, Flamigni C. Birth of a healthy female after intracytoplasmic sperm injection of cryopreserved human oocytes. Fertil Steril. 1997;68(4):724–726.
8. Fabbri R, Porcu E, Marsella T, Rocchetta G, Venturoli S, Flamigni C. Human oocyte cryopreservation: new perspectives regarding oocyte survival. Hum Reprod. 2001;16(3):411–416.
9. Oktay K, Cil AP, Bang H. Efficiency of oocyte cryopreservation: a meta-analysis. Fertil Steril. 2006;86(1):70–80.
10. Smith GD, Serafini PC, Fioravanti J, et al. Prospective randomized comparison of human oocyte cryopreservationwith slow-rate freezing or vitrification. Fertil Steril. 2010;94(6):2088–2095.
11. Gook DA, Edgar DH. Human oocyte cryopreservation. Hum Reprod Update. 2007;13(6):591–605.
12. Rienzi L, Cobo A, Paffoni A, et al. Consistent and predictable delivery rates after oocyte vitrification: an observational longitudinal cohort multicentric study. Hum Reprod. 2012;27(6):1606–1612.
13. Cobo A, Rubio C, Gerli S, Ruiz A, Pellicer A, Remohi J. Use of fluorescence in situ hybridization to assess the chromosomal status of embryos obtained from cryopreserved oocytes. Fertil Steril. 2001;75(2):354–360.
14. Noyes N, Porcu E, Borini A. Over 900 oocyte cryopreservation babies born with no apparent increase in congenital anomalies. Reprod Biomed Online. 2009;18(6):769–776.
15. Chian RC, Huang JY, Tan SL, et al. Obstetric and perinatal outcome in 200 infants conceived from vitrified oocytes. Reprod Biomed Online. 2008;16(5):608–610.
16. Levi Setti P, Albani E, Morenghi E, et al. Comparative analysis of fetal and neonatal outcomes of pregnancies from fresh and cryopreserved/thawed oocytes in the same group of patients. Fertil Steril. 2013;100(2):396–401.
17. Pennings G. Ethical aspects of social freezing. Gynecol Obstet Fertil. 2013;41(9):521–523.
18. Cil AP, Bang H, Oktay K. Age-specific probability of live birth with oocyte cryopreservation: an individual patient data meta-analysis. Fertil Steril. 2013;100(2):492–499.
IN THIS ARTICLE
-Vitrification and slow freezing: How did we get here and how effective are they?
-Safety outcomes data are limited but reassuring
-We can freeze eggs, but when should we?
-Who should pay for egg freezing?
-What should we do as we move forward?

75-Year-Old Woman With Elevated Liver Enzymes
A 75-year-old woman, Gladys, was brought to the psychiatric clinic in a manic state by her concerned sister. The patient was disheveled, dehydrated, and having difficulty expressing her thoughts. Vital signs included a blood pressure of 200/94 mm Hg; pulse, 88 beats/min; temperature, 98.4°F; and respiratory rate, 20 breaths/min. Psychiatric history included a diagnosis of schizoaffective disorder with inconsistent adherence to treatment regimens, particularly mood stabilizers; and attention-deficit/hyperactivity disorder, for which she took methylphenidate regularly. Medical history was significant for asthma, osteoporosis, hypertension, type 2 diabetes, and hypothyroidism.
Gladys tended to become involved in personal relationships that adversely affected her mental health. This, in fact, had just happened: A “friend” had taken advantage of her kindness and then abruptly moved away, triggering the patient’s current decompensation. She was referred for admission for psychiatric evaluation and treatment.
During the three-week hospitalization, Gladys was diagnosed with bipolar I disorder. She agreed to take mood-stabilizing medication primarily to alleviate her insomnia during manic episodes. She was discharged on a multidrug regimen for her coexisting conditions (see Table 1). Of note, her blood pressure at discharge was 148/66 mm Hg.
At outpatient follow-up five days later, the patient reported feeling better and stronger. However, five weeks after discharge, Gladys returned with complaints of tiredness during the day (though sleeping well at night), severe dry mouth, aching joints, and poor appetite. Her blood pressure was 100/50 mm Hg. She denied abdominal pain or change in the color of her urine or stool. She also denied use of alcohol, illicit drugs, or OTC medications. Laboratory results revealed elevated levels of several liver enzymes (see Table 2), all of which had been normal when she was admitted to the hospital two months earlier.
Continue for discussion >>
DISCUSSION
Elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels may result from a variety of factors. Mild elevations are commonly caused by alcohol consumption, hemochromatosis, medications, nonalcoholic fatty liver disease, and viral hepatitis (with which elevations may range from mild to marked).1 Moderate to marked elevations of ALT and AST are commonly seen with acute biliary obstruction, alcoholic hepatitis, toxic injury, and ischemic injury.2
Abnormal liver enzyme levels are common with use of psychotropic drugs, such as antipsychotics and mood stabilizers.3 In a systematic review that examined the effects of antipsychotics on liver function tests, a median 4% of patients experienced elevated ALT, AST, or gamma-glutamyl transferase (GGT) levels (defined as more than triple the normal level) or alkaline phosphatase (ALP) level (defined as more than twice the normal level).3 Of the studies reviewed, five noted an interval of one to six weeks between initiation of antipsychotic drugs and detection of liver function test abnormalities. None of the included studies reported severe or fatal hepatic injury.
For the atypical antipsychotic quetiapine, elevations in ALT and AST occurred in about 5% and 3% of patients, respectively, in clinical trials of the drug as monotherapy for schizophrenia or bipolar mania.4 These elevations were usually transient, occurring within the first three weeks of treatment initiation and subsiding with continued treatment.
There are rare published reports, however, of serious and even fatal hepatotoxicity induced by quetiapine. One 59-year-old woman developed fulminant hepatic failure (FHF) six weeks after she began taking quetiapine in addition to carbidopa/levodopa for Parkinson disease. She reported nausea, vomiting, poor appetite, and abdominal pain and required a six-week hospitalization, with multidrug treatment that continued after discharge. Liver biopsy identified acute hepatitis with confluent bridging necrosis, a sign that the liver injury was drug-induced. The authors concluded that, because drug-induced hepatotoxicity is the most common cause of FHF in many parts of the world, clinicians should evaluate a patient’s medications for a potential cause.5
In another case report, elevated liver enzymes were identified one month after a 58-year-old woman taking several other medications began treatment with quetiapine (100 mg/d). She developed liver failure and died after a three-week hospitalization. The authors concluded that liver failure was caused by an idiosyncratic reaction to a relatively low dose of quetiapine. This case supports the advisability of close monitoring of liver enzyme levels during quetiapine treatment.6
Naharci et al reported a case of a 77-year-old woman treated with quetiapine (12.5 mg bid for nine days). She developed acute hepatic failure leading to multi-organ system failure and died eight days later. Liver failure was attributed to an idiosyncratic reaction to low-dose quetiapine. The authors concluded that liver function monitoring is essential with quetiapine administration, especially in elderly or fragile patients.7
The initial recommended dosage of quetiapine for elderly patients (defined as age 65 or older) is 50 mg/d, with the dose increased in increments of 50 mg/d, based on clinical response and tolerability. In clinical trials, the mean plasma clearance of quetiapine was reduced by 30% to 50% in the elderly, so dosing adjustments may be necessary in this age-group.4 Gareri et al recommended that atypical antipsychotics be prescribed for elderly patients for the shortest necessary duration and at the lowest effective dose.8
For hepatically impaired patients, recommended initial dosing is 25 mg/d, with increases of 25 to 50 mg/d until an effective and tolerable dose is reached.4 Further, because quetiapine is primarily metabolized via the cytochrome P450 liver enzymes CYP3A4 and CYP2D6,9 when the clinician prescribes a potent CYP3A4 inhibitor (eg, ketoconazole) to a patient taking quetiapine, the quetiapine dosage needs to be reduced. Conversely, when prescribing a CYP3A4 inducer (eg, phenytoin), the quetiapine dosage should be adjusted upward.4
Even when an apparently well-tolerated, effective quetiapine dosage has been reached, clinicians and patients should remain alert to the warning signs of potentially serious events. Adverse effects of atypical antipsychotics, including quetiapine, were summarized by Gareri et al and rated on a scale ranging from no effect to severe effect.8 The most severe adverse effects for quetiapine were hypotension and prolonged QTc interval. Weight gain was identified as a moderate effect, and sedation, gastrointestinal problems (nausea, vomiting, and constipation), and anticholinergic effects as mild. Some effects—tardive dyskinesia, seizures, and hepatic—were deemed “uncertain”; this rating suggests the need for careful monitoring of patients (who should be informed of signs and symptoms that should be reported to the clinician).8
Atasoy et al reviewed the records of 110 patients to assess the effect of atypical antipsychotics on liver function tests. The patients’ records included both baseline liver function tests and repeat testing at six months. Forty-eight patients received quetiapine; 33 patients, olanzapine; and 29 patients, risperidone. Liver enzymes were elevated in 27.1% of the quetiapine group, 30.3% of the olanzapine group, and 27.6% of the risperidone group. In two patients taking olanzapine, liver enzyme levels reached three to four times normal but returned to normal when treatment was stopped. The authors concluded that baseline liver enzyme studies should be done prior to initiation of treatment with atypical antipsychotics, as well as periodically thereafter, especially for patients with preexisting hepatic disorders, those being treated with other potentially hepatotoxic drugs, or those who exhibit signs or symptoms of hepatic impairment.10
Continue for patient outcome >>
PATIENT OUTCOME
Gladys’s ALT and AST levels were mildly elevated. One of the more common causes for this pattern is medication. In addition, her ALP level of more than twice the upper limit of normal further pointed to a viral, alcohol-related, or drug toxicity cause. Since her hepatitis panel was negative and she did not use alcohol, it was determined that elevated liver enzymes were related to the recent addition of quetiapine, which was discontinued. In addition, in light of Gladys’s hypotension (which is also a potential adverse effect of quetiapine8), her dose of lisinopril/hydrochlorothiazide was decreased by half.
One week later, liver enzyme levels were returning to normal. Gladys, however, began having more difficulty sleeping and more trouble remaining focused and getting things done, despite taking methylphenidate. In place of quetiapine, risperidone (0.5 mg at bedtime) was initiated. At first, Gladys was concerned with her continuing dry mouth symptoms, but when she skipped doses of risperidone, she noticed that she functioned less well. Further, her liver enzyme levels were being closely monitored and were normal. With this reassurance, Gladys remained adherent to risperidone for mood stabilization.
CONCLUSION
Atypical antipsychotic drugs such as quetiapine can cause elevated liver enzyme levels, especially in the elderly, patients with hepatic impairment, or patients on polypharmacotherapy. Rarely, quetiapine has been reported to cause serious hepatotoxicity and even death. Patients taking these drugs should be informed of possible symptoms of liver toxicity, including fatigue, nausea, vomiting, abdominal pain, and change in color of urine or stools. Particularly in more vulnerable patients, liver enzyme levels should be monitored carefully to confirm the continued safety of antipsychotic treatment.
REFERENCES
1. Oh RC, Hustead TR. Causes and evaluation of mildly elevated liver transaminase levels. Am Fam Physician. 2011;84(9):1003-1008.
2. Giannini EG, Testa R, Savarino V. Liver enzyme elevation: a guide for clinicians. CMAJ. 2005;172(3):367-379.
3. Marwick KFM, Taylor M, Walker SW. Antipsychotics and abnormal liver function tests: Systematic review. Clin Neuropharmacol. 2012;35(5):244-253.
4. Seroquel [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2013.
5. Al Mutairi F, Dwivedi G, Al Ameel T. Fulminant hepatic failure in association with quetiapine: A case report. J Med Case Rep. 2012;6:418.
6. El Hajj L, Sharara A, Rockey, DC. Subfulminant liver failure associated with quetiapine. Eur J Gastroenterol Hepatol. 2004;16(12):1415-1418.
7. Naharci MI, Karadurmus N, Demir O, et al. Fatal hepatotoxicity in an elderly patient receiving low-dose quetiapine. Am J Psychiatry. 2011;168(2):212-213.
8. Gareri P, Segura-Garcia C, Manfredi VG, et al. Use of atypical antipsychotics in the elderly: a clinical review. Clin Interv Aging. 2014;16(9):1363-1373.
9. Lin S, Chang Y, Moody DE, Foltz RL. A liquid chromatographic-electrospray-tandem mass spectrometric method for quanititation of quetiapine in human plasma and liver microsomes: application to a study of in vitro metabolism. J Anal Toxicol. 2004;28(6):443-446.
10. Atasoy N, Erdogan A, Yalug I, et al. A review of liver function tests during treatment with atypical antipsychotic drugs: a chart review study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1255-1260.
A 75-year-old woman, Gladys, was brought to the psychiatric clinic in a manic state by her concerned sister. The patient was disheveled, dehydrated, and having difficulty expressing her thoughts. Vital signs included a blood pressure of 200/94 mm Hg; pulse, 88 beats/min; temperature, 98.4°F; and respiratory rate, 20 breaths/min. Psychiatric history included a diagnosis of schizoaffective disorder with inconsistent adherence to treatment regimens, particularly mood stabilizers; and attention-deficit/hyperactivity disorder, for which she took methylphenidate regularly. Medical history was significant for asthma, osteoporosis, hypertension, type 2 diabetes, and hypothyroidism.
Gladys tended to become involved in personal relationships that adversely affected her mental health. This, in fact, had just happened: A “friend” had taken advantage of her kindness and then abruptly moved away, triggering the patient’s current decompensation. She was referred for admission for psychiatric evaluation and treatment.
During the three-week hospitalization, Gladys was diagnosed with bipolar I disorder. She agreed to take mood-stabilizing medication primarily to alleviate her insomnia during manic episodes. She was discharged on a multidrug regimen for her coexisting conditions (see Table 1). Of note, her blood pressure at discharge was 148/66 mm Hg.
At outpatient follow-up five days later, the patient reported feeling better and stronger. However, five weeks after discharge, Gladys returned with complaints of tiredness during the day (though sleeping well at night), severe dry mouth, aching joints, and poor appetite. Her blood pressure was 100/50 mm Hg. She denied abdominal pain or change in the color of her urine or stool. She also denied use of alcohol, illicit drugs, or OTC medications. Laboratory results revealed elevated levels of several liver enzymes (see Table 2), all of which had been normal when she was admitted to the hospital two months earlier.
Continue for discussion >>
DISCUSSION
Elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels may result from a variety of factors. Mild elevations are commonly caused by alcohol consumption, hemochromatosis, medications, nonalcoholic fatty liver disease, and viral hepatitis (with which elevations may range from mild to marked).1 Moderate to marked elevations of ALT and AST are commonly seen with acute biliary obstruction, alcoholic hepatitis, toxic injury, and ischemic injury.2
Abnormal liver enzyme levels are common with use of psychotropic drugs, such as antipsychotics and mood stabilizers.3 In a systematic review that examined the effects of antipsychotics on liver function tests, a median 4% of patients experienced elevated ALT, AST, or gamma-glutamyl transferase (GGT) levels (defined as more than triple the normal level) or alkaline phosphatase (ALP) level (defined as more than twice the normal level).3 Of the studies reviewed, five noted an interval of one to six weeks between initiation of antipsychotic drugs and detection of liver function test abnormalities. None of the included studies reported severe or fatal hepatic injury.
For the atypical antipsychotic quetiapine, elevations in ALT and AST occurred in about 5% and 3% of patients, respectively, in clinical trials of the drug as monotherapy for schizophrenia or bipolar mania.4 These elevations were usually transient, occurring within the first three weeks of treatment initiation and subsiding with continued treatment.
There are rare published reports, however, of serious and even fatal hepatotoxicity induced by quetiapine. One 59-year-old woman developed fulminant hepatic failure (FHF) six weeks after she began taking quetiapine in addition to carbidopa/levodopa for Parkinson disease. She reported nausea, vomiting, poor appetite, and abdominal pain and required a six-week hospitalization, with multidrug treatment that continued after discharge. Liver biopsy identified acute hepatitis with confluent bridging necrosis, a sign that the liver injury was drug-induced. The authors concluded that, because drug-induced hepatotoxicity is the most common cause of FHF in many parts of the world, clinicians should evaluate a patient’s medications for a potential cause.5
In another case report, elevated liver enzymes were identified one month after a 58-year-old woman taking several other medications began treatment with quetiapine (100 mg/d). She developed liver failure and died after a three-week hospitalization. The authors concluded that liver failure was caused by an idiosyncratic reaction to a relatively low dose of quetiapine. This case supports the advisability of close monitoring of liver enzyme levels during quetiapine treatment.6
Naharci et al reported a case of a 77-year-old woman treated with quetiapine (12.5 mg bid for nine days). She developed acute hepatic failure leading to multi-organ system failure and died eight days later. Liver failure was attributed to an idiosyncratic reaction to low-dose quetiapine. The authors concluded that liver function monitoring is essential with quetiapine administration, especially in elderly or fragile patients.7
The initial recommended dosage of quetiapine for elderly patients (defined as age 65 or older) is 50 mg/d, with the dose increased in increments of 50 mg/d, based on clinical response and tolerability. In clinical trials, the mean plasma clearance of quetiapine was reduced by 30% to 50% in the elderly, so dosing adjustments may be necessary in this age-group.4 Gareri et al recommended that atypical antipsychotics be prescribed for elderly patients for the shortest necessary duration and at the lowest effective dose.8
For hepatically impaired patients, recommended initial dosing is 25 mg/d, with increases of 25 to 50 mg/d until an effective and tolerable dose is reached.4 Further, because quetiapine is primarily metabolized via the cytochrome P450 liver enzymes CYP3A4 and CYP2D6,9 when the clinician prescribes a potent CYP3A4 inhibitor (eg, ketoconazole) to a patient taking quetiapine, the quetiapine dosage needs to be reduced. Conversely, when prescribing a CYP3A4 inducer (eg, phenytoin), the quetiapine dosage should be adjusted upward.4
Even when an apparently well-tolerated, effective quetiapine dosage has been reached, clinicians and patients should remain alert to the warning signs of potentially serious events. Adverse effects of atypical antipsychotics, including quetiapine, were summarized by Gareri et al and rated on a scale ranging from no effect to severe effect.8 The most severe adverse effects for quetiapine were hypotension and prolonged QTc interval. Weight gain was identified as a moderate effect, and sedation, gastrointestinal problems (nausea, vomiting, and constipation), and anticholinergic effects as mild. Some effects—tardive dyskinesia, seizures, and hepatic—were deemed “uncertain”; this rating suggests the need for careful monitoring of patients (who should be informed of signs and symptoms that should be reported to the clinician).8
Atasoy et al reviewed the records of 110 patients to assess the effect of atypical antipsychotics on liver function tests. The patients’ records included both baseline liver function tests and repeat testing at six months. Forty-eight patients received quetiapine; 33 patients, olanzapine; and 29 patients, risperidone. Liver enzymes were elevated in 27.1% of the quetiapine group, 30.3% of the olanzapine group, and 27.6% of the risperidone group. In two patients taking olanzapine, liver enzyme levels reached three to four times normal but returned to normal when treatment was stopped. The authors concluded that baseline liver enzyme studies should be done prior to initiation of treatment with atypical antipsychotics, as well as periodically thereafter, especially for patients with preexisting hepatic disorders, those being treated with other potentially hepatotoxic drugs, or those who exhibit signs or symptoms of hepatic impairment.10
Continue for patient outcome >>
PATIENT OUTCOME
Gladys’s ALT and AST levels were mildly elevated. One of the more common causes for this pattern is medication. In addition, her ALP level of more than twice the upper limit of normal further pointed to a viral, alcohol-related, or drug toxicity cause. Since her hepatitis panel was negative and she did not use alcohol, it was determined that elevated liver enzymes were related to the recent addition of quetiapine, which was discontinued. In addition, in light of Gladys’s hypotension (which is also a potential adverse effect of quetiapine8), her dose of lisinopril/hydrochlorothiazide was decreased by half.
One week later, liver enzyme levels were returning to normal. Gladys, however, began having more difficulty sleeping and more trouble remaining focused and getting things done, despite taking methylphenidate. In place of quetiapine, risperidone (0.5 mg at bedtime) was initiated. At first, Gladys was concerned with her continuing dry mouth symptoms, but when she skipped doses of risperidone, she noticed that she functioned less well. Further, her liver enzyme levels were being closely monitored and were normal. With this reassurance, Gladys remained adherent to risperidone for mood stabilization.
CONCLUSION
Atypical antipsychotic drugs such as quetiapine can cause elevated liver enzyme levels, especially in the elderly, patients with hepatic impairment, or patients on polypharmacotherapy. Rarely, quetiapine has been reported to cause serious hepatotoxicity and even death. Patients taking these drugs should be informed of possible symptoms of liver toxicity, including fatigue, nausea, vomiting, abdominal pain, and change in color of urine or stools. Particularly in more vulnerable patients, liver enzyme levels should be monitored carefully to confirm the continued safety of antipsychotic treatment.
REFERENCES
1. Oh RC, Hustead TR. Causes and evaluation of mildly elevated liver transaminase levels. Am Fam Physician. 2011;84(9):1003-1008.
2. Giannini EG, Testa R, Savarino V. Liver enzyme elevation: a guide for clinicians. CMAJ. 2005;172(3):367-379.
3. Marwick KFM, Taylor M, Walker SW. Antipsychotics and abnormal liver function tests: Systematic review. Clin Neuropharmacol. 2012;35(5):244-253.
4. Seroquel [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2013.
5. Al Mutairi F, Dwivedi G, Al Ameel T. Fulminant hepatic failure in association with quetiapine: A case report. J Med Case Rep. 2012;6:418.
6. El Hajj L, Sharara A, Rockey, DC. Subfulminant liver failure associated with quetiapine. Eur J Gastroenterol Hepatol. 2004;16(12):1415-1418.
7. Naharci MI, Karadurmus N, Demir O, et al. Fatal hepatotoxicity in an elderly patient receiving low-dose quetiapine. Am J Psychiatry. 2011;168(2):212-213.
8. Gareri P, Segura-Garcia C, Manfredi VG, et al. Use of atypical antipsychotics in the elderly: a clinical review. Clin Interv Aging. 2014;16(9):1363-1373.
9. Lin S, Chang Y, Moody DE, Foltz RL. A liquid chromatographic-electrospray-tandem mass spectrometric method for quanititation of quetiapine in human plasma and liver microsomes: application to a study of in vitro metabolism. J Anal Toxicol. 2004;28(6):443-446.
10. Atasoy N, Erdogan A, Yalug I, et al. A review of liver function tests during treatment with atypical antipsychotic drugs: a chart review study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1255-1260.
A 75-year-old woman, Gladys, was brought to the psychiatric clinic in a manic state by her concerned sister. The patient was disheveled, dehydrated, and having difficulty expressing her thoughts. Vital signs included a blood pressure of 200/94 mm Hg; pulse, 88 beats/min; temperature, 98.4°F; and respiratory rate, 20 breaths/min. Psychiatric history included a diagnosis of schizoaffective disorder with inconsistent adherence to treatment regimens, particularly mood stabilizers; and attention-deficit/hyperactivity disorder, for which she took methylphenidate regularly. Medical history was significant for asthma, osteoporosis, hypertension, type 2 diabetes, and hypothyroidism.
Gladys tended to become involved in personal relationships that adversely affected her mental health. This, in fact, had just happened: A “friend” had taken advantage of her kindness and then abruptly moved away, triggering the patient’s current decompensation. She was referred for admission for psychiatric evaluation and treatment.
During the three-week hospitalization, Gladys was diagnosed with bipolar I disorder. She agreed to take mood-stabilizing medication primarily to alleviate her insomnia during manic episodes. She was discharged on a multidrug regimen for her coexisting conditions (see Table 1). Of note, her blood pressure at discharge was 148/66 mm Hg.
At outpatient follow-up five days later, the patient reported feeling better and stronger. However, five weeks after discharge, Gladys returned with complaints of tiredness during the day (though sleeping well at night), severe dry mouth, aching joints, and poor appetite. Her blood pressure was 100/50 mm Hg. She denied abdominal pain or change in the color of her urine or stool. She also denied use of alcohol, illicit drugs, or OTC medications. Laboratory results revealed elevated levels of several liver enzymes (see Table 2), all of which had been normal when she was admitted to the hospital two months earlier.
Continue for discussion >>
DISCUSSION
Elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels may result from a variety of factors. Mild elevations are commonly caused by alcohol consumption, hemochromatosis, medications, nonalcoholic fatty liver disease, and viral hepatitis (with which elevations may range from mild to marked).1 Moderate to marked elevations of ALT and AST are commonly seen with acute biliary obstruction, alcoholic hepatitis, toxic injury, and ischemic injury.2
Abnormal liver enzyme levels are common with use of psychotropic drugs, such as antipsychotics and mood stabilizers.3 In a systematic review that examined the effects of antipsychotics on liver function tests, a median 4% of patients experienced elevated ALT, AST, or gamma-glutamyl transferase (GGT) levels (defined as more than triple the normal level) or alkaline phosphatase (ALP) level (defined as more than twice the normal level).3 Of the studies reviewed, five noted an interval of one to six weeks between initiation of antipsychotic drugs and detection of liver function test abnormalities. None of the included studies reported severe or fatal hepatic injury.
For the atypical antipsychotic quetiapine, elevations in ALT and AST occurred in about 5% and 3% of patients, respectively, in clinical trials of the drug as monotherapy for schizophrenia or bipolar mania.4 These elevations were usually transient, occurring within the first three weeks of treatment initiation and subsiding with continued treatment.
There are rare published reports, however, of serious and even fatal hepatotoxicity induced by quetiapine. One 59-year-old woman developed fulminant hepatic failure (FHF) six weeks after she began taking quetiapine in addition to carbidopa/levodopa for Parkinson disease. She reported nausea, vomiting, poor appetite, and abdominal pain and required a six-week hospitalization, with multidrug treatment that continued after discharge. Liver biopsy identified acute hepatitis with confluent bridging necrosis, a sign that the liver injury was drug-induced. The authors concluded that, because drug-induced hepatotoxicity is the most common cause of FHF in many parts of the world, clinicians should evaluate a patient’s medications for a potential cause.5
In another case report, elevated liver enzymes were identified one month after a 58-year-old woman taking several other medications began treatment with quetiapine (100 mg/d). She developed liver failure and died after a three-week hospitalization. The authors concluded that liver failure was caused by an idiosyncratic reaction to a relatively low dose of quetiapine. This case supports the advisability of close monitoring of liver enzyme levels during quetiapine treatment.6
Naharci et al reported a case of a 77-year-old woman treated with quetiapine (12.5 mg bid for nine days). She developed acute hepatic failure leading to multi-organ system failure and died eight days later. Liver failure was attributed to an idiosyncratic reaction to low-dose quetiapine. The authors concluded that liver function monitoring is essential with quetiapine administration, especially in elderly or fragile patients.7
The initial recommended dosage of quetiapine for elderly patients (defined as age 65 or older) is 50 mg/d, with the dose increased in increments of 50 mg/d, based on clinical response and tolerability. In clinical trials, the mean plasma clearance of quetiapine was reduced by 30% to 50% in the elderly, so dosing adjustments may be necessary in this age-group.4 Gareri et al recommended that atypical antipsychotics be prescribed for elderly patients for the shortest necessary duration and at the lowest effective dose.8
For hepatically impaired patients, recommended initial dosing is 25 mg/d, with increases of 25 to 50 mg/d until an effective and tolerable dose is reached.4 Further, because quetiapine is primarily metabolized via the cytochrome P450 liver enzymes CYP3A4 and CYP2D6,9 when the clinician prescribes a potent CYP3A4 inhibitor (eg, ketoconazole) to a patient taking quetiapine, the quetiapine dosage needs to be reduced. Conversely, when prescribing a CYP3A4 inducer (eg, phenytoin), the quetiapine dosage should be adjusted upward.4
Even when an apparently well-tolerated, effective quetiapine dosage has been reached, clinicians and patients should remain alert to the warning signs of potentially serious events. Adverse effects of atypical antipsychotics, including quetiapine, were summarized by Gareri et al and rated on a scale ranging from no effect to severe effect.8 The most severe adverse effects for quetiapine were hypotension and prolonged QTc interval. Weight gain was identified as a moderate effect, and sedation, gastrointestinal problems (nausea, vomiting, and constipation), and anticholinergic effects as mild. Some effects—tardive dyskinesia, seizures, and hepatic—were deemed “uncertain”; this rating suggests the need for careful monitoring of patients (who should be informed of signs and symptoms that should be reported to the clinician).8
Atasoy et al reviewed the records of 110 patients to assess the effect of atypical antipsychotics on liver function tests. The patients’ records included both baseline liver function tests and repeat testing at six months. Forty-eight patients received quetiapine; 33 patients, olanzapine; and 29 patients, risperidone. Liver enzymes were elevated in 27.1% of the quetiapine group, 30.3% of the olanzapine group, and 27.6% of the risperidone group. In two patients taking olanzapine, liver enzyme levels reached three to four times normal but returned to normal when treatment was stopped. The authors concluded that baseline liver enzyme studies should be done prior to initiation of treatment with atypical antipsychotics, as well as periodically thereafter, especially for patients with preexisting hepatic disorders, those being treated with other potentially hepatotoxic drugs, or those who exhibit signs or symptoms of hepatic impairment.10
Continue for patient outcome >>
PATIENT OUTCOME
Gladys’s ALT and AST levels were mildly elevated. One of the more common causes for this pattern is medication. In addition, her ALP level of more than twice the upper limit of normal further pointed to a viral, alcohol-related, or drug toxicity cause. Since her hepatitis panel was negative and she did not use alcohol, it was determined that elevated liver enzymes were related to the recent addition of quetiapine, which was discontinued. In addition, in light of Gladys’s hypotension (which is also a potential adverse effect of quetiapine8), her dose of lisinopril/hydrochlorothiazide was decreased by half.
One week later, liver enzyme levels were returning to normal. Gladys, however, began having more difficulty sleeping and more trouble remaining focused and getting things done, despite taking methylphenidate. In place of quetiapine, risperidone (0.5 mg at bedtime) was initiated. At first, Gladys was concerned with her continuing dry mouth symptoms, but when she skipped doses of risperidone, she noticed that she functioned less well. Further, her liver enzyme levels were being closely monitored and were normal. With this reassurance, Gladys remained adherent to risperidone for mood stabilization.
CONCLUSION
Atypical antipsychotic drugs such as quetiapine can cause elevated liver enzyme levels, especially in the elderly, patients with hepatic impairment, or patients on polypharmacotherapy. Rarely, quetiapine has been reported to cause serious hepatotoxicity and even death. Patients taking these drugs should be informed of possible symptoms of liver toxicity, including fatigue, nausea, vomiting, abdominal pain, and change in color of urine or stools. Particularly in more vulnerable patients, liver enzyme levels should be monitored carefully to confirm the continued safety of antipsychotic treatment.
REFERENCES
1. Oh RC, Hustead TR. Causes and evaluation of mildly elevated liver transaminase levels. Am Fam Physician. 2011;84(9):1003-1008.
2. Giannini EG, Testa R, Savarino V. Liver enzyme elevation: a guide for clinicians. CMAJ. 2005;172(3):367-379.
3. Marwick KFM, Taylor M, Walker SW. Antipsychotics and abnormal liver function tests: Systematic review. Clin Neuropharmacol. 2012;35(5):244-253.
4. Seroquel [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2013.
5. Al Mutairi F, Dwivedi G, Al Ameel T. Fulminant hepatic failure in association with quetiapine: A case report. J Med Case Rep. 2012;6:418.
6. El Hajj L, Sharara A, Rockey, DC. Subfulminant liver failure associated with quetiapine. Eur J Gastroenterol Hepatol. 2004;16(12):1415-1418.
7. Naharci MI, Karadurmus N, Demir O, et al. Fatal hepatotoxicity in an elderly patient receiving low-dose quetiapine. Am J Psychiatry. 2011;168(2):212-213.
8. Gareri P, Segura-Garcia C, Manfredi VG, et al. Use of atypical antipsychotics in the elderly: a clinical review. Clin Interv Aging. 2014;16(9):1363-1373.
9. Lin S, Chang Y, Moody DE, Foltz RL. A liquid chromatographic-electrospray-tandem mass spectrometric method for quanititation of quetiapine in human plasma and liver microsomes: application to a study of in vitro metabolism. J Anal Toxicol. 2004;28(6):443-446.
10. Atasoy N, Erdogan A, Yalug I, et al. A review of liver function tests during treatment with atypical antipsychotic drugs: a chart review study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1255-1260.
“Difficult” Patient? Or Is It a Personality Disorder?

If Bob were your patient, how would you proceed?
Personality disorders (PDs) are patterns of inflexible and maladaptive personality traits and behaviors that cause subjective distress and significant social or occupational impairment.1 An individual with a PD tends to have a limited repertoire of responses to the rough-and-tumble of life, with coping mechanisms that often perpetuate difficulty and distress. Examples include distrust and suspiciousness of others’ motives (paranoid PD); disregard and violation of the rights of others (antisocial PD); instability in interpersonal relationships, self-image, and affect (borderline PD); and social inhibition, feelings of inadequacy, and hypersensitivity to negative evaluation (avoidant PD).1
FPPs may view patients with PDs as “difficult patients” because of their frequent crises and the interpersonal problems they bring into the clinician-patient relationship.2,3 Help, of course, can come in the way of a referral to a psychotherapist who specializes in treating PDs. But you can also make use of some evidence-based psychotherapy techniques to improve your patients’ lives and the quality of the clinician-patient relationship. This article focuses on identifying and managing PDs in family practice, using practical strategies drawn from empirically supported therapies.
Next page: How common are PDs?
PDs ARE MORE COMMON THAN YOU MIGHT SUSPECT
The overall prevalence of PD in the community ranges from 4.4% to 14.8%, with no consistent pattern of sex differences.4 Between 31.4% and 45.5% of psychiatric outpatients and up to 24% of primary care patients likely meet criteria for at least one PD.5-7 PDs impede recovery from other mental disorders,8 increase the risk for suicide,9 and are associated with substance abuse, impulsivity, and violence.10,11 Personality pathology also is associated with greater incidence of serious medical illness12,13 and reduced social functioning.14 Not surprisingly, patients with PDs frequently use medical and social services.15
PDs tend to be underdiagnosed, perhaps partly because of concern about stigmatization, but also due to difficulties in identifying and classifying these disorders. Published in 2013, the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, 5th edition (DSM-5) originally was to include a major revision of PDs—reflecting concern about the limitations of PD categories—but ultimately the existing categories were retained (see Table 1).1 There is considerable overlap among PD categories; many patients meet the criteria for more than one PD, but it is unlikely that they actually have several distinct PDs. Other patients—perhaps even the majority—are best diagnosed with “unspecified personality disorder” because they do not neatly fit into one of these categories.
What to do if you suspect your patient has a PD >>
SUSPECT YOUR PATIENT HAS A PD?
Evaluate these two areas
Identifying patients who have PDs in primary care is useful for two reasons: to explore the option of specialty treatment for patients who may be amenable to it, and to improve management of the patient’s complaints in the primary care setting, including a smoother clinician-patient interaction. In either case, determining the specific DSM-5 diagnosis is less important than recognizing core personality impairment: an ingrained disturbance in one’s perceptions of self and others. This can be done by paying attention to how the patient adapts to life’s challenges and if he or she has problematic interpersonal tendencies, including difficulties in the clinician-patient relationship.
Unfortunately, assessing and diagnosing PDs in the primary care setting can be challenging. Limited time doesn’t allow for extensive, personality-focused interviews. Self-report screening tools are limited, because patients may underreport key interpersonal problems such as lack of empathy. Furthermore, very few patients seek help from their FPP in addressing personality dysfunction; PDs typically are identified while investigating other complaints.
The most reliable and useful areas to evaluate in a patient you suspect may have a PD are identity (one’s sense of who one is and can be) and interpersonal relationships, including the capacity for empathy and intimacy.16,17 These should be considered longitudinally and in the context of the individual’s stage of development. For example, identity is generally less stable among adolescents compared to middle-aged adults.
A cohesive sense of identity allows one to embrace life’s tasks and challenges, to develop and strive toward personal goals, and to handle setbacks and disappointments. A person with a stable identity may develop a depressive reaction to difficult life circumstances, but with some assistance can generally bounce back and re-engage in his or her personal goals. By contrast, an individual with an unstable sense of self may feel chronically insecure and empty, with limited capacity to constructively deal with life’s ups and downs. Patients with borderline PD, for example, try to manage a fragmented identity by frantically clinging to others, while narcissistic patients tend to suppress a fragile sense of self by putting forth an arrogant and entitled attitude.
How does the patient interact with others? As is the case with identity, an individual’s capacity for interpersonal functioning is developed early in life, through interactions with primary caregivers. Mental maps of who we are and what we can expect from others are formed and reinforced in attachment relationships, such as those with our parents; traumatic attachments, including abuse or neglect by a caregiver or loved one, are strongly associated with PD.18,19 The resulting belief structures guide subsequent interpersonal functioning, and become interactively reinforced. For example, a person whose internal map of relationships includes others abandoning him might behave in a clingy manner, which may ultimately induce others to reject him, thus creating a self-fulfilling prophecy.
Distorted interpersonal expectations can impair a person’s capacity for sustained intimate connections (a troubled relationship history is characteristic of PDs) and limit empathic functioning.20 Other people’s actions may be interpreted according to the patient’s belief structures rather than with an open mind about the other person’s experience.
Focus on the clinician-patient relationship
The interpersonal dysfunction of patients with PDs will often surface in the clinician-patient relationship, serving as a clue to broader interpersonal dysfunction. An FPP’s relatively innocuous oversight, for example, might be taken as proof of suspected incompetence in the eyes of a patient with paranoid or narcissistic tendencies. Or a patient with a recurrent complaint who repeatedly rejects the clinician’s interventions probably oscillates between seeking and rejecting nurturance in other relationships, as well. A patient who tends to make sarcastic remarks regarding the clinician’s earnest efforts likely holds negative views of others and sabotages potentially positive interactions.
So what strategies are best for managing these types of scenarios? Bringing up a potential diagnosis of PD may be a delicate matter for the FPP; patients might experience this as a jarring diagnosis in the absence of a thorough psychiatric evaluation. If the FPP decides to explore whether the patient is open to discussing the relationship between moods, behaviors, and personality features, he or she can begin this conversation by noting that, as with physical health, we all have our vulnerabilities and that these vulnerabilities may be strengthened through specialist consultation and support. In this way, the patient can view a referral as an opportunity to explore herself with professional support. If a psychiatrist or psychotherapist colleague does become involved, it is important to clarify the roles of treatment providers and to communicate with one another, should difficulties arise.
Continue for forms of psychotherapy >>
EVIDENCE SUPPORTS TWO FORMS OF PSYCHOTHERAPY
Treatment for PDs has seen considerable growth over the past decade, largely due to research on therapies that target the troubling self-injurious and suicidal features of borderline PD. Considerable evidence shows that specialized psychotherapy can significantly reduce suffering and improve functioning among these patients. The two major evidence-based treatments for patients with borderline PD are dialectical behavior therapy (DBT) and psychodynamic therapy.
DBT is an intensive cognitive-behavioral approach that teaches patients how to regulate their emotions and develop an accepting, mindful attitude toward their mental experience.21 Several randomized controlled trials (RCTs) have demonstrated the effectiveness of DBT in reducing hospitalizations and self-injurious and suicidal behavior in patients with borderline PD.22
Psychodynamic therapy, which focuses on helping patients discover how unconscious conflicts influence their present moods and behaviors, has also been validated by multiple RCTs for patients with borderline PD.23-25 Like DBT, empirically supported psychodynamic therapy tends to be structured, long-term (> 12 months), and often intensively delivered in multiple sessions per week. However, a recent study found that a less-intensive, general psychodynamic therapy, along with occasional medication management, was equivalent to intensive DBT.26
Although the research has focused primarily on borderline PD, these approaches can be applied to other PDs. These therapies focus on understanding one’s emotional and behavioral patterns, developing a healthy self-concept, and improving interpersonal relationships—areas that are relevant treatment targets across all PD types.
Indeed, studies of day treatment programs that explicitly welcome patients with a range of PD types have had promising findings.27 Day treatment involves an intensive array of therapies, mostly in a group format; patients work together to support and embolden one another to make positive changes. Unfortunately, FPPs may be challenged to find appropriate services for patients who are amenable to psychotherapy; public mental health resources tend to lag far behind best practices in the case of PD.
MEDICATION MIGHT IMPROVE SYMPTOMS, NOT PERSONALITY DEFICITS
Most research on pharmacotherapy for PDs has focused on borderline PD; findings have been mixed and fairly limited.28 Medication cannot address underlying identity and relational deficits and will not result in remission of PD. Nonetheless, judicious, circumscribed use of medications to target specific symptoms may be helpful for some patients. Selective serotonin reuptake inhibitors can reduce anger and impulsive aggression in patients with borderline PD.28,29
Atypical antipsychotics may help reduce impulsive aggression or transient psychotic symptoms.28-30 For example, olanzapine and aripiprazole can reduce anxiety, anger/aggression, paranoia, and interpersonal sensitivity in borderline PD.31,32 Mood stabilizers such as valproate, lamotrigine, and topiramate may also help some borderline patients, although they do so by reducing impulsivity and aggression rather than improving core unstable identity and affect.28,29
Carefully obtained informed consent is necessary because of the danger of adverse effects with many of these medications; for example, antipsychotics have been associated with metabolic syndrome and weight gain that can threaten a patient’s already fragile self-image.33 Polypharmacy is also a potential problem: Well-intentioned clinicians may be prompted to offer multiple medications in response to patients’ unremitting complaints of distress, when a psychotherapeutic approach may need to be the primary treatment. The bottom line is that medications do not resolve personality dysfunction and are best used symptomatically as adjuncts to psychotherapy.28,30
Next page: Steps to take during the office visit >>
STEPS YOU CAN TAKE DURING THE OFFICE VISIT
Although it is not feasible for most FPPs to provide comprehensive treatment for PD, key elements from specialized therapies can be integrated into your management of these patients. Steps you can take include using validation, promoting mentalization, and managing countertransference.
Validation, which is a component of DBT, is providing the expressed acknowledgement that the patient is entitled to her feelings. This is not the same as agreeing with a position the patient has taken on an issue, but rather conveying the sense that one sees how the patient might feel the way she does. A study of women with borderline PD and substance abuse found a validation intervention by itself was significantly helpful.34 Validation can contribute to a “corrective emotional experience.” For instance, your supportive acknowledgement of a patient with a history of abuse or neglect may counter the patient’s expectation of being invalidated, and over time this can reduce the patient’s defensive rigidity.
Mentalization. Psychodynamic treatment involves a similar tack; clinicians empathize with the patient’s emotional state while also demonstrating a degree of separateness from the emotion.23-25 This promotes mentalization in the patient—the ability to contemplate one’s own and others’ subjective mental states.18 Mentalization is often impaired in PD patients, who presume to “know” what others are thinking. A patient, for instance, “just knows” that her friend secretly hates her, based on a vaguely worded text message.
You can help patients with mentalization by taking an inquisitive “not knowing” stance and by emphasizing a collaborative and reflective approach toward a given problem—to examine the issue together, from all sides. You can point out that while a patient is entitled to feel whatever he is feeling, it may not be in his best interest to act on the feelings without adequately considering the potential consequences of the action. This helps the patient to distinguish thoughts, feelings, and impulses from behavior. It also teaches the value of anticipatory thinking, impulse control, and affect regulation.
Countertransference. Managing your emotional reactions to a patient with PD is a well-documented challenge.35 Your feelings about the patient, known as countertransference, can range from considerable concern and sympathy to severe frustration, bewilderment, and frank hostility. A common reaction is the sense that one must “do something” to respond to the patient’s emotional distress or interpersonal pressure. This may trigger an impulse to give advice or offer tests or medications despite knowing that these are unlikely to be helpful. A more useful response may be to tolerate such feelings and listen empathically to the patient’s frustration. Recognizing subtle countertransference can guard against extreme reactions and maintain an appropriate clinical focus. Discussion with a trusted colleague can be helpful.
Psychodynamic approaches consider managing countertransference to be a therapeutic intervention, even when psychotherapy is not explicitly being carried out. Strong emotional responses may reflect something that the patient needs the clinician to experience, as the patient cannot bear to experience it himself. The patient needs to see—and learn from—the clinician’s handling of unbearable (for the patient) feelings. This occurs at a level of unconscious communication and may be repeated over time. Although not discussed with the patient, a clinician’s capacity for self-containment and provision of undisrupted, good medical care is in itself a psychotherapeutic accomplishment.
Based on Bob’s history of interpersonal conflicts and perceived persecution by coworkers, the FPP consults with a psychotherapist colleague, who says Bob’s chronic mistrust and social isolation suggest he may have a severe identity disturbance and unspecified PD with paranoid and schizoid features. Because Bob refuses to see a therapist, his FPP decides to focus on promoting small improvements in Bob’s interpersonal interactions and reducing absenteeism at work.
The FPP validates Bob’s feelings (“it can be very stressful to constantly feel like others are at odds with you”) and tries to promote mentalizing (“I want to understand more about what you think regarding your work situation and your coworkers. Let’s try to look at this from all perspectives—maybe we can come up with some new ideas.”)
Despite wanting to help his patient, the FPP feels uneasy and reluctant to engage with Bob, who likely evokes such feelings to keep others at a distance. The FPP tactfully seeks to remain Bob’s ally without endorsing his distorted interpretation of events. Given Bob’s paranoid rejection of therapy, the FPP refrains from making further such recommendations. The FPP’s interventions, however, may help Bob warm to the idea of further help over time, and the FPP’s supportive stance will help to ameliorate the patient’s distress. (Additional examples of how to use the strategies described in this article can be found in Table 2.)

References on following page >>
REFERENCES
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
2. Hahn SR, Thompson KS, Wills TA, et al. The difficult clinician-patient relationship: somatization, personality and psychopathology. J Clin Epidemiol. 1994:47:647-657.
3. Schafer S, Nowlis DP. Personality disorders among difficult patients. Arch Fam Med. 1998;7:126-129.
4. Paris J. Estimating the prevalence of personality disorders in the community. J Pers Disord. 2010;24:405-411.
5. Newton-Howes G, Tyrer P, Anagnostakis K, et al. The prevalence of personality disorder, its comorbidity with mental state disorders, and its clinical significance in community mental health teams. Soc Psychiatry Psychiatr Epidemiol. 2010;45:453-460.
6. Zimmerman M, Rothschild L, Chelminski I. The prevalence of DSM-IV personality disorders in psychiatric outpatients. Am J Psychiatry. 2005;162:1911-1918.
7. Moran P, Jenkins R, Tylee A, et al. The prevalence of personality disorder among UK primary care attenders. Acta Psychiatr Scand. 2000;102:52-57.
8. Newton-Howes G, Tyrer P, Johnson T. Personality disorder and the outcome of depression: Meta-analysis of published studies. Br J Psychiatry. 2006;188:13-20.
9. Blasco-Fontecilla H, Baca-Garcia E, Dervic K, et al. Severity of personality disorders and suicide attempt. Acta Psychiatr Scand. 2009;119:149-155.
10. Colpaert K, Vanderplasschen W, De Maeyer J, et al. Prevalence and determinants of personality disorders in a clinical sample of alcohol-, drug-, and dual-dependent patients. Subst Use Misuse. 2012;47:649-661.
11. Yu R, Geddes JR, Fazel S. Personality disorders, violence, and antisocial behavior: A systematic review and meta-regression analysis. J Pers Disord. 2012;26:775-792.
12. Frankenburg FR, Zanarini MC. The association between borderline personality disorder and chronic medical illnesses, poor health-related lifestyle choices, and costly forms of health care utilization. J Clin Psychiatry. 2004;65:1660-1665.
13. Lee HB, Bienvenu OJ, Cho SJ, et al. Personality disorders and traits as predictors of incident cardiovascular disease: Findings from the 23-year follow-up of the Baltimore ECA Study. Psychosomatics. 2010;51:289-296.
14. Skodol AE, Gunderson JG, McGlashan TH, et al. Functional impairment in patients with schizotypal, borderline, avoidant, or obsessive-compulsive personality disorder. Am J Psychiatry. 2002;159:276-283.
15. Bender DS, Dolan RT, Skodol AE, et al. Treatment utilization by patients with personality disorders. Am J Psychiatry. 2001;158:295-302.
16. Livesley WJ. An empirically-based classification of personality disorder.
J Pers Disord. 2011;25:397-420.
17. Bender DS, Morey LC, Skodol AE. Toward a model for assessing personality functioning in DSM-5, part I: a review of theory and methods. J Pers Assess. 2011;93:332-346.
18. Fonagy P, Gergely G, Jurist EL, et al. Affect Regulation, Mentalization, and the Development of the Self. New York, NY: Other Press; 2002.
19. Yen S, Shea MT, Battle CL, et al. Traumatic exposure and posttraumatic stress disorder in borderline, schizotypal, avoidant, and obsessive-compulsive personality disorders: findings from the collaborative longitudinal personality disorders study. J Nerv Ment Dis. 2002;190:510-518.
20. Morey LC, Stagner BH. Narcissistic pathology as core personality dysfunction: comparing DSM-IV and the DSM-5 proposal for narcissistic personality disorder. J Clin Psychol. 2012;68:908-921.
21. Lynch TR, Chapman AL, Rosenthal MZ, et al. Mechanisms of change in dialectical behaviour therapy: theoretical and empirical observations.
J Clin Psychol. 2006;62:459-480.
22. Kliem S, Kröger C, Kosfelder J. Dialectical behavior therapy for borderline personality disorder: a meta-analysis using mixed-effects modeling.
J Consult Clin Psychol. 2010;78:936-951.
23. Clarkin JF, Levy KN, Lenzenweger MF, et al. Evaluating three treatments for borderline personality disorder: a multiwave study. Am J Psychiatry. 2007;164:922-928.
24. Gregory RJ, DeLucia-Deranja E, Mogle JA. Dynamic deconstructive psychotherapy versus optimized community care for borderline personality disorder co-occurring with alcohol use disorders: a 30-month follow-up. J Nerv Ment Dis. 2010;198:292-298.
25. Bateman A, Fonagy P. Randomized controlled trial of outpatient mentalization-based treatment versus structured clinical management for borderline personality disorder. Am J Psychiatry. 2009;166:1355-1364.
26. McMain SF, Links PS, Gnam WH, et al. A randomized trial of dialectical behavior therapy versus general psychiatric management for borderline personality disorder. Am J Psychiatry. 2009;166:1365-1374.
27. Ogrodniczuk JS, Piper WE. Day treatment for personality disorders: a review of research findings. Harv Rev Psychiatry. 2001;9:105-117.
28. Paris J. Pharmacological treatments for personality disorders. Int Rev Psychiatry. 2011;23:303-309.
29. Ripoll LH, Triebwasser J, Siever LJ. Evidence-based pharmacotherapy for personality disorders. Int J Neuropsychopharmacol. 2011;14:1257-1288.
30. Steinberg PI. The use of low-dose neuroleptics in the treatment of patients with severe personality disorder: An adjunct to psychotherapy. BCMJ. 2007;49:306-310.
31. Zanarini MC, Frankenburg FR. Olanzapine treatment of female borderline personality disorder patients: a double-blind, placebo controlled pilot study. J Clin Psychiatry. 2001;62:849-854.
32. Nickel MK, Loew TH, Pedrosa Gil F. Aripiprazole in treatment of borderline patients, part II: an 18-month follow up. Psychopharmacology (Berl). 2007;191:1023-1026.
33. Silk KR. The process of managing medications in patients with borderline personality disorder. J Psychiatr Pract. 2011;17:311-319.
34. Linehan MM, Dimeff LA, Reynolds SK, et al. Dialectal behavior therapy versus comprehensive validation therapy plus 12-step for the treatment of opioid dependent women meeting criteria for borderline personality disorder. Drug Alcohol Depend. 2002;67:13-26.
35. Rossberg JI, Karterud S, Pedersen G, et al. An empirical study of countertransference reactions toward patients with personality disorders. Compr Psychiatry. 2007;48:225-230.
If Bob were your patient, how would you proceed?
Personality disorders (PDs) are patterns of inflexible and maladaptive personality traits and behaviors that cause subjective distress and significant social or occupational impairment.1 An individual with a PD tends to have a limited repertoire of responses to the rough-and-tumble of life, with coping mechanisms that often perpetuate difficulty and distress. Examples include distrust and suspiciousness of others’ motives (paranoid PD); disregard and violation of the rights of others (antisocial PD); instability in interpersonal relationships, self-image, and affect (borderline PD); and social inhibition, feelings of inadequacy, and hypersensitivity to negative evaluation (avoidant PD).1
FPPs may view patients with PDs as “difficult patients” because of their frequent crises and the interpersonal problems they bring into the clinician-patient relationship.2,3 Help, of course, can come in the way of a referral to a psychotherapist who specializes in treating PDs. But you can also make use of some evidence-based psychotherapy techniques to improve your patients’ lives and the quality of the clinician-patient relationship. This article focuses on identifying and managing PDs in family practice, using practical strategies drawn from empirically supported therapies.
Next page: How common are PDs?
PDs ARE MORE COMMON THAN YOU MIGHT SUSPECT
The overall prevalence of PD in the community ranges from 4.4% to 14.8%, with no consistent pattern of sex differences.4 Between 31.4% and 45.5% of psychiatric outpatients and up to 24% of primary care patients likely meet criteria for at least one PD.5-7 PDs impede recovery from other mental disorders,8 increase the risk for suicide,9 and are associated with substance abuse, impulsivity, and violence.10,11 Personality pathology also is associated with greater incidence of serious medical illness12,13 and reduced social functioning.14 Not surprisingly, patients with PDs frequently use medical and social services.15
PDs tend to be underdiagnosed, perhaps partly because of concern about stigmatization, but also due to difficulties in identifying and classifying these disorders. Published in 2013, the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, 5th edition (DSM-5) originally was to include a major revision of PDs—reflecting concern about the limitations of PD categories—but ultimately the existing categories were retained (see Table 1).1 There is considerable overlap among PD categories; many patients meet the criteria for more than one PD, but it is unlikely that they actually have several distinct PDs. Other patients—perhaps even the majority—are best diagnosed with “unspecified personality disorder” because they do not neatly fit into one of these categories.
What to do if you suspect your patient has a PD >>
SUSPECT YOUR PATIENT HAS A PD?
Evaluate these two areas
Identifying patients who have PDs in primary care is useful for two reasons: to explore the option of specialty treatment for patients who may be amenable to it, and to improve management of the patient’s complaints in the primary care setting, including a smoother clinician-patient interaction. In either case, determining the specific DSM-5 diagnosis is less important than recognizing core personality impairment: an ingrained disturbance in one’s perceptions of self and others. This can be done by paying attention to how the patient adapts to life’s challenges and if he or she has problematic interpersonal tendencies, including difficulties in the clinician-patient relationship.
Unfortunately, assessing and diagnosing PDs in the primary care setting can be challenging. Limited time doesn’t allow for extensive, personality-focused interviews. Self-report screening tools are limited, because patients may underreport key interpersonal problems such as lack of empathy. Furthermore, very few patients seek help from their FPP in addressing personality dysfunction; PDs typically are identified while investigating other complaints.
The most reliable and useful areas to evaluate in a patient you suspect may have a PD are identity (one’s sense of who one is and can be) and interpersonal relationships, including the capacity for empathy and intimacy.16,17 These should be considered longitudinally and in the context of the individual’s stage of development. For example, identity is generally less stable among adolescents compared to middle-aged adults.
A cohesive sense of identity allows one to embrace life’s tasks and challenges, to develop and strive toward personal goals, and to handle setbacks and disappointments. A person with a stable identity may develop a depressive reaction to difficult life circumstances, but with some assistance can generally bounce back and re-engage in his or her personal goals. By contrast, an individual with an unstable sense of self may feel chronically insecure and empty, with limited capacity to constructively deal with life’s ups and downs. Patients with borderline PD, for example, try to manage a fragmented identity by frantically clinging to others, while narcissistic patients tend to suppress a fragile sense of self by putting forth an arrogant and entitled attitude.
How does the patient interact with others? As is the case with identity, an individual’s capacity for interpersonal functioning is developed early in life, through interactions with primary caregivers. Mental maps of who we are and what we can expect from others are formed and reinforced in attachment relationships, such as those with our parents; traumatic attachments, including abuse or neglect by a caregiver or loved one, are strongly associated with PD.18,19 The resulting belief structures guide subsequent interpersonal functioning, and become interactively reinforced. For example, a person whose internal map of relationships includes others abandoning him might behave in a clingy manner, which may ultimately induce others to reject him, thus creating a self-fulfilling prophecy.
Distorted interpersonal expectations can impair a person’s capacity for sustained intimate connections (a troubled relationship history is characteristic of PDs) and limit empathic functioning.20 Other people’s actions may be interpreted according to the patient’s belief structures rather than with an open mind about the other person’s experience.
Focus on the clinician-patient relationship
The interpersonal dysfunction of patients with PDs will often surface in the clinician-patient relationship, serving as a clue to broader interpersonal dysfunction. An FPP’s relatively innocuous oversight, for example, might be taken as proof of suspected incompetence in the eyes of a patient with paranoid or narcissistic tendencies. Or a patient with a recurrent complaint who repeatedly rejects the clinician’s interventions probably oscillates between seeking and rejecting nurturance in other relationships, as well. A patient who tends to make sarcastic remarks regarding the clinician’s earnest efforts likely holds negative views of others and sabotages potentially positive interactions.
So what strategies are best for managing these types of scenarios? Bringing up a potential diagnosis of PD may be a delicate matter for the FPP; patients might experience this as a jarring diagnosis in the absence of a thorough psychiatric evaluation. If the FPP decides to explore whether the patient is open to discussing the relationship between moods, behaviors, and personality features, he or she can begin this conversation by noting that, as with physical health, we all have our vulnerabilities and that these vulnerabilities may be strengthened through specialist consultation and support. In this way, the patient can view a referral as an opportunity to explore herself with professional support. If a psychiatrist or psychotherapist colleague does become involved, it is important to clarify the roles of treatment providers and to communicate with one another, should difficulties arise.
Continue for forms of psychotherapy >>
EVIDENCE SUPPORTS TWO FORMS OF PSYCHOTHERAPY
Treatment for PDs has seen considerable growth over the past decade, largely due to research on therapies that target the troubling self-injurious and suicidal features of borderline PD. Considerable evidence shows that specialized psychotherapy can significantly reduce suffering and improve functioning among these patients. The two major evidence-based treatments for patients with borderline PD are dialectical behavior therapy (DBT) and psychodynamic therapy.
DBT is an intensive cognitive-behavioral approach that teaches patients how to regulate their emotions and develop an accepting, mindful attitude toward their mental experience.21 Several randomized controlled trials (RCTs) have demonstrated the effectiveness of DBT in reducing hospitalizations and self-injurious and suicidal behavior in patients with borderline PD.22
Psychodynamic therapy, which focuses on helping patients discover how unconscious conflicts influence their present moods and behaviors, has also been validated by multiple RCTs for patients with borderline PD.23-25 Like DBT, empirically supported psychodynamic therapy tends to be structured, long-term (> 12 months), and often intensively delivered in multiple sessions per week. However, a recent study found that a less-intensive, general psychodynamic therapy, along with occasional medication management, was equivalent to intensive DBT.26
Although the research has focused primarily on borderline PD, these approaches can be applied to other PDs. These therapies focus on understanding one’s emotional and behavioral patterns, developing a healthy self-concept, and improving interpersonal relationships—areas that are relevant treatment targets across all PD types.
Indeed, studies of day treatment programs that explicitly welcome patients with a range of PD types have had promising findings.27 Day treatment involves an intensive array of therapies, mostly in a group format; patients work together to support and embolden one another to make positive changes. Unfortunately, FPPs may be challenged to find appropriate services for patients who are amenable to psychotherapy; public mental health resources tend to lag far behind best practices in the case of PD.
MEDICATION MIGHT IMPROVE SYMPTOMS, NOT PERSONALITY DEFICITS
Most research on pharmacotherapy for PDs has focused on borderline PD; findings have been mixed and fairly limited.28 Medication cannot address underlying identity and relational deficits and will not result in remission of PD. Nonetheless, judicious, circumscribed use of medications to target specific symptoms may be helpful for some patients. Selective serotonin reuptake inhibitors can reduce anger and impulsive aggression in patients with borderline PD.28,29
Atypical antipsychotics may help reduce impulsive aggression or transient psychotic symptoms.28-30 For example, olanzapine and aripiprazole can reduce anxiety, anger/aggression, paranoia, and interpersonal sensitivity in borderline PD.31,32 Mood stabilizers such as valproate, lamotrigine, and topiramate may also help some borderline patients, although they do so by reducing impulsivity and aggression rather than improving core unstable identity and affect.28,29
Carefully obtained informed consent is necessary because of the danger of adverse effects with many of these medications; for example, antipsychotics have been associated with metabolic syndrome and weight gain that can threaten a patient’s already fragile self-image.33 Polypharmacy is also a potential problem: Well-intentioned clinicians may be prompted to offer multiple medications in response to patients’ unremitting complaints of distress, when a psychotherapeutic approach may need to be the primary treatment. The bottom line is that medications do not resolve personality dysfunction and are best used symptomatically as adjuncts to psychotherapy.28,30
Next page: Steps to take during the office visit >>
STEPS YOU CAN TAKE DURING THE OFFICE VISIT
Although it is not feasible for most FPPs to provide comprehensive treatment for PD, key elements from specialized therapies can be integrated into your management of these patients. Steps you can take include using validation, promoting mentalization, and managing countertransference.
Validation, which is a component of DBT, is providing the expressed acknowledgement that the patient is entitled to her feelings. This is not the same as agreeing with a position the patient has taken on an issue, but rather conveying the sense that one sees how the patient might feel the way she does. A study of women with borderline PD and substance abuse found a validation intervention by itself was significantly helpful.34 Validation can contribute to a “corrective emotional experience.” For instance, your supportive acknowledgement of a patient with a history of abuse or neglect may counter the patient’s expectation of being invalidated, and over time this can reduce the patient’s defensive rigidity.
Mentalization. Psychodynamic treatment involves a similar tack; clinicians empathize with the patient’s emotional state while also demonstrating a degree of separateness from the emotion.23-25 This promotes mentalization in the patient—the ability to contemplate one’s own and others’ subjective mental states.18 Mentalization is often impaired in PD patients, who presume to “know” what others are thinking. A patient, for instance, “just knows” that her friend secretly hates her, based on a vaguely worded text message.
You can help patients with mentalization by taking an inquisitive “not knowing” stance and by emphasizing a collaborative and reflective approach toward a given problem—to examine the issue together, from all sides. You can point out that while a patient is entitled to feel whatever he is feeling, it may not be in his best interest to act on the feelings without adequately considering the potential consequences of the action. This helps the patient to distinguish thoughts, feelings, and impulses from behavior. It also teaches the value of anticipatory thinking, impulse control, and affect regulation.
Countertransference. Managing your emotional reactions to a patient with PD is a well-documented challenge.35 Your feelings about the patient, known as countertransference, can range from considerable concern and sympathy to severe frustration, bewilderment, and frank hostility. A common reaction is the sense that one must “do something” to respond to the patient’s emotional distress or interpersonal pressure. This may trigger an impulse to give advice or offer tests or medications despite knowing that these are unlikely to be helpful. A more useful response may be to tolerate such feelings and listen empathically to the patient’s frustration. Recognizing subtle countertransference can guard against extreme reactions and maintain an appropriate clinical focus. Discussion with a trusted colleague can be helpful.
Psychodynamic approaches consider managing countertransference to be a therapeutic intervention, even when psychotherapy is not explicitly being carried out. Strong emotional responses may reflect something that the patient needs the clinician to experience, as the patient cannot bear to experience it himself. The patient needs to see—and learn from—the clinician’s handling of unbearable (for the patient) feelings. This occurs at a level of unconscious communication and may be repeated over time. Although not discussed with the patient, a clinician’s capacity for self-containment and provision of undisrupted, good medical care is in itself a psychotherapeutic accomplishment.
Based on Bob’s history of interpersonal conflicts and perceived persecution by coworkers, the FPP consults with a psychotherapist colleague, who says Bob’s chronic mistrust and social isolation suggest he may have a severe identity disturbance and unspecified PD with paranoid and schizoid features. Because Bob refuses to see a therapist, his FPP decides to focus on promoting small improvements in Bob’s interpersonal interactions and reducing absenteeism at work.
The FPP validates Bob’s feelings (“it can be very stressful to constantly feel like others are at odds with you”) and tries to promote mentalizing (“I want to understand more about what you think regarding your work situation and your coworkers. Let’s try to look at this from all perspectives—maybe we can come up with some new ideas.”)
Despite wanting to help his patient, the FPP feels uneasy and reluctant to engage with Bob, who likely evokes such feelings to keep others at a distance. The FPP tactfully seeks to remain Bob’s ally without endorsing his distorted interpretation of events. Given Bob’s paranoid rejection of therapy, the FPP refrains from making further such recommendations. The FPP’s interventions, however, may help Bob warm to the idea of further help over time, and the FPP’s supportive stance will help to ameliorate the patient’s distress. (Additional examples of how to use the strategies described in this article can be found in Table 2.)
References on following page >>
REFERENCES
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
2. Hahn SR, Thompson KS, Wills TA, et al. The difficult clinician-patient relationship: somatization, personality and psychopathology. J Clin Epidemiol. 1994:47:647-657.
3. Schafer S, Nowlis DP. Personality disorders among difficult patients. Arch Fam Med. 1998;7:126-129.
4. Paris J. Estimating the prevalence of personality disorders in the community. J Pers Disord. 2010;24:405-411.
5. Newton-Howes G, Tyrer P, Anagnostakis K, et al. The prevalence of personality disorder, its comorbidity with mental state disorders, and its clinical significance in community mental health teams. Soc Psychiatry Psychiatr Epidemiol. 2010;45:453-460.
6. Zimmerman M, Rothschild L, Chelminski I. The prevalence of DSM-IV personality disorders in psychiatric outpatients. Am J Psychiatry. 2005;162:1911-1918.
7. Moran P, Jenkins R, Tylee A, et al. The prevalence of personality disorder among UK primary care attenders. Acta Psychiatr Scand. 2000;102:52-57.
8. Newton-Howes G, Tyrer P, Johnson T. Personality disorder and the outcome of depression: Meta-analysis of published studies. Br J Psychiatry. 2006;188:13-20.
9. Blasco-Fontecilla H, Baca-Garcia E, Dervic K, et al. Severity of personality disorders and suicide attempt. Acta Psychiatr Scand. 2009;119:149-155.
10. Colpaert K, Vanderplasschen W, De Maeyer J, et al. Prevalence and determinants of personality disorders in a clinical sample of alcohol-, drug-, and dual-dependent patients. Subst Use Misuse. 2012;47:649-661.
11. Yu R, Geddes JR, Fazel S. Personality disorders, violence, and antisocial behavior: A systematic review and meta-regression analysis. J Pers Disord. 2012;26:775-792.
12. Frankenburg FR, Zanarini MC. The association between borderline personality disorder and chronic medical illnesses, poor health-related lifestyle choices, and costly forms of health care utilization. J Clin Psychiatry. 2004;65:1660-1665.
13. Lee HB, Bienvenu OJ, Cho SJ, et al. Personality disorders and traits as predictors of incident cardiovascular disease: Findings from the 23-year follow-up of the Baltimore ECA Study. Psychosomatics. 2010;51:289-296.
14. Skodol AE, Gunderson JG, McGlashan TH, et al. Functional impairment in patients with schizotypal, borderline, avoidant, or obsessive-compulsive personality disorder. Am J Psychiatry. 2002;159:276-283.
15. Bender DS, Dolan RT, Skodol AE, et al. Treatment utilization by patients with personality disorders. Am J Psychiatry. 2001;158:295-302.
16. Livesley WJ. An empirically-based classification of personality disorder.
J Pers Disord. 2011;25:397-420.
17. Bender DS, Morey LC, Skodol AE. Toward a model for assessing personality functioning in DSM-5, part I: a review of theory and methods. J Pers Assess. 2011;93:332-346.
18. Fonagy P, Gergely G, Jurist EL, et al. Affect Regulation, Mentalization, and the Development of the Self. New York, NY: Other Press; 2002.
19. Yen S, Shea MT, Battle CL, et al. Traumatic exposure and posttraumatic stress disorder in borderline, schizotypal, avoidant, and obsessive-compulsive personality disorders: findings from the collaborative longitudinal personality disorders study. J Nerv Ment Dis. 2002;190:510-518.
20. Morey LC, Stagner BH. Narcissistic pathology as core personality dysfunction: comparing DSM-IV and the DSM-5 proposal for narcissistic personality disorder. J Clin Psychol. 2012;68:908-921.
21. Lynch TR, Chapman AL, Rosenthal MZ, et al. Mechanisms of change in dialectical behaviour therapy: theoretical and empirical observations.
J Clin Psychol. 2006;62:459-480.
22. Kliem S, Kröger C, Kosfelder J. Dialectical behavior therapy for borderline personality disorder: a meta-analysis using mixed-effects modeling.
J Consult Clin Psychol. 2010;78:936-951.
23. Clarkin JF, Levy KN, Lenzenweger MF, et al. Evaluating three treatments for borderline personality disorder: a multiwave study. Am J Psychiatry. 2007;164:922-928.
24. Gregory RJ, DeLucia-Deranja E, Mogle JA. Dynamic deconstructive psychotherapy versus optimized community care for borderline personality disorder co-occurring with alcohol use disorders: a 30-month follow-up. J Nerv Ment Dis. 2010;198:292-298.
25. Bateman A, Fonagy P. Randomized controlled trial of outpatient mentalization-based treatment versus structured clinical management for borderline personality disorder. Am J Psychiatry. 2009;166:1355-1364.
26. McMain SF, Links PS, Gnam WH, et al. A randomized trial of dialectical behavior therapy versus general psychiatric management for borderline personality disorder. Am J Psychiatry. 2009;166:1365-1374.
27. Ogrodniczuk JS, Piper WE. Day treatment for personality disorders: a review of research findings. Harv Rev Psychiatry. 2001;9:105-117.
28. Paris J. Pharmacological treatments for personality disorders. Int Rev Psychiatry. 2011;23:303-309.
29. Ripoll LH, Triebwasser J, Siever LJ. Evidence-based pharmacotherapy for personality disorders. Int J Neuropsychopharmacol. 2011;14:1257-1288.
30. Steinberg PI. The use of low-dose neuroleptics in the treatment of patients with severe personality disorder: An adjunct to psychotherapy. BCMJ. 2007;49:306-310.
31. Zanarini MC, Frankenburg FR. Olanzapine treatment of female borderline personality disorder patients: a double-blind, placebo controlled pilot study. J Clin Psychiatry. 2001;62:849-854.
32. Nickel MK, Loew TH, Pedrosa Gil F. Aripiprazole in treatment of borderline patients, part II: an 18-month follow up. Psychopharmacology (Berl). 2007;191:1023-1026.
33. Silk KR. The process of managing medications in patients with borderline personality disorder. J Psychiatr Pract. 2011;17:311-319.
34. Linehan MM, Dimeff LA, Reynolds SK, et al. Dialectal behavior therapy versus comprehensive validation therapy plus 12-step for the treatment of opioid dependent women meeting criteria for borderline personality disorder. Drug Alcohol Depend. 2002;67:13-26.
35. Rossberg JI, Karterud S, Pedersen G, et al. An empirical study of countertransference reactions toward patients with personality disorders. Compr Psychiatry. 2007;48:225-230.
If Bob were your patient, how would you proceed?
Personality disorders (PDs) are patterns of inflexible and maladaptive personality traits and behaviors that cause subjective distress and significant social or occupational impairment.1 An individual with a PD tends to have a limited repertoire of responses to the rough-and-tumble of life, with coping mechanisms that often perpetuate difficulty and distress. Examples include distrust and suspiciousness of others’ motives (paranoid PD); disregard and violation of the rights of others (antisocial PD); instability in interpersonal relationships, self-image, and affect (borderline PD); and social inhibition, feelings of inadequacy, and hypersensitivity to negative evaluation (avoidant PD).1
FPPs may view patients with PDs as “difficult patients” because of their frequent crises and the interpersonal problems they bring into the clinician-patient relationship.2,3 Help, of course, can come in the way of a referral to a psychotherapist who specializes in treating PDs. But you can also make use of some evidence-based psychotherapy techniques to improve your patients’ lives and the quality of the clinician-patient relationship. This article focuses on identifying and managing PDs in family practice, using practical strategies drawn from empirically supported therapies.
Next page: How common are PDs?
PDs ARE MORE COMMON THAN YOU MIGHT SUSPECT
The overall prevalence of PD in the community ranges from 4.4% to 14.8%, with no consistent pattern of sex differences.4 Between 31.4% and 45.5% of psychiatric outpatients and up to 24% of primary care patients likely meet criteria for at least one PD.5-7 PDs impede recovery from other mental disorders,8 increase the risk for suicide,9 and are associated with substance abuse, impulsivity, and violence.10,11 Personality pathology also is associated with greater incidence of serious medical illness12,13 and reduced social functioning.14 Not surprisingly, patients with PDs frequently use medical and social services.15
PDs tend to be underdiagnosed, perhaps partly because of concern about stigmatization, but also due to difficulties in identifying and classifying these disorders. Published in 2013, the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, 5th edition (DSM-5) originally was to include a major revision of PDs—reflecting concern about the limitations of PD categories—but ultimately the existing categories were retained (see Table 1).1 There is considerable overlap among PD categories; many patients meet the criteria for more than one PD, but it is unlikely that they actually have several distinct PDs. Other patients—perhaps even the majority—are best diagnosed with “unspecified personality disorder” because they do not neatly fit into one of these categories.
What to do if you suspect your patient has a PD >>
SUSPECT YOUR PATIENT HAS A PD?
Evaluate these two areas
Identifying patients who have PDs in primary care is useful for two reasons: to explore the option of specialty treatment for patients who may be amenable to it, and to improve management of the patient’s complaints in the primary care setting, including a smoother clinician-patient interaction. In either case, determining the specific DSM-5 diagnosis is less important than recognizing core personality impairment: an ingrained disturbance in one’s perceptions of self and others. This can be done by paying attention to how the patient adapts to life’s challenges and if he or she has problematic interpersonal tendencies, including difficulties in the clinician-patient relationship.
Unfortunately, assessing and diagnosing PDs in the primary care setting can be challenging. Limited time doesn’t allow for extensive, personality-focused interviews. Self-report screening tools are limited, because patients may underreport key interpersonal problems such as lack of empathy. Furthermore, very few patients seek help from their FPP in addressing personality dysfunction; PDs typically are identified while investigating other complaints.
The most reliable and useful areas to evaluate in a patient you suspect may have a PD are identity (one’s sense of who one is and can be) and interpersonal relationships, including the capacity for empathy and intimacy.16,17 These should be considered longitudinally and in the context of the individual’s stage of development. For example, identity is generally less stable among adolescents compared to middle-aged adults.
A cohesive sense of identity allows one to embrace life’s tasks and challenges, to develop and strive toward personal goals, and to handle setbacks and disappointments. A person with a stable identity may develop a depressive reaction to difficult life circumstances, but with some assistance can generally bounce back and re-engage in his or her personal goals. By contrast, an individual with an unstable sense of self may feel chronically insecure and empty, with limited capacity to constructively deal with life’s ups and downs. Patients with borderline PD, for example, try to manage a fragmented identity by frantically clinging to others, while narcissistic patients tend to suppress a fragile sense of self by putting forth an arrogant and entitled attitude.
How does the patient interact with others? As is the case with identity, an individual’s capacity for interpersonal functioning is developed early in life, through interactions with primary caregivers. Mental maps of who we are and what we can expect from others are formed and reinforced in attachment relationships, such as those with our parents; traumatic attachments, including abuse or neglect by a caregiver or loved one, are strongly associated with PD.18,19 The resulting belief structures guide subsequent interpersonal functioning, and become interactively reinforced. For example, a person whose internal map of relationships includes others abandoning him might behave in a clingy manner, which may ultimately induce others to reject him, thus creating a self-fulfilling prophecy.
Distorted interpersonal expectations can impair a person’s capacity for sustained intimate connections (a troubled relationship history is characteristic of PDs) and limit empathic functioning.20 Other people’s actions may be interpreted according to the patient’s belief structures rather than with an open mind about the other person’s experience.
Focus on the clinician-patient relationship
The interpersonal dysfunction of patients with PDs will often surface in the clinician-patient relationship, serving as a clue to broader interpersonal dysfunction. An FPP’s relatively innocuous oversight, for example, might be taken as proof of suspected incompetence in the eyes of a patient with paranoid or narcissistic tendencies. Or a patient with a recurrent complaint who repeatedly rejects the clinician’s interventions probably oscillates between seeking and rejecting nurturance in other relationships, as well. A patient who tends to make sarcastic remarks regarding the clinician’s earnest efforts likely holds negative views of others and sabotages potentially positive interactions.
So what strategies are best for managing these types of scenarios? Bringing up a potential diagnosis of PD may be a delicate matter for the FPP; patients might experience this as a jarring diagnosis in the absence of a thorough psychiatric evaluation. If the FPP decides to explore whether the patient is open to discussing the relationship between moods, behaviors, and personality features, he or she can begin this conversation by noting that, as with physical health, we all have our vulnerabilities and that these vulnerabilities may be strengthened through specialist consultation and support. In this way, the patient can view a referral as an opportunity to explore herself with professional support. If a psychiatrist or psychotherapist colleague does become involved, it is important to clarify the roles of treatment providers and to communicate with one another, should difficulties arise.
Continue for forms of psychotherapy >>
EVIDENCE SUPPORTS TWO FORMS OF PSYCHOTHERAPY
Treatment for PDs has seen considerable growth over the past decade, largely due to research on therapies that target the troubling self-injurious and suicidal features of borderline PD. Considerable evidence shows that specialized psychotherapy can significantly reduce suffering and improve functioning among these patients. The two major evidence-based treatments for patients with borderline PD are dialectical behavior therapy (DBT) and psychodynamic therapy.
DBT is an intensive cognitive-behavioral approach that teaches patients how to regulate their emotions and develop an accepting, mindful attitude toward their mental experience.21 Several randomized controlled trials (RCTs) have demonstrated the effectiveness of DBT in reducing hospitalizations and self-injurious and suicidal behavior in patients with borderline PD.22
Psychodynamic therapy, which focuses on helping patients discover how unconscious conflicts influence their present moods and behaviors, has also been validated by multiple RCTs for patients with borderline PD.23-25 Like DBT, empirically supported psychodynamic therapy tends to be structured, long-term (> 12 months), and often intensively delivered in multiple sessions per week. However, a recent study found that a less-intensive, general psychodynamic therapy, along with occasional medication management, was equivalent to intensive DBT.26
Although the research has focused primarily on borderline PD, these approaches can be applied to other PDs. These therapies focus on understanding one’s emotional and behavioral patterns, developing a healthy self-concept, and improving interpersonal relationships—areas that are relevant treatment targets across all PD types.
Indeed, studies of day treatment programs that explicitly welcome patients with a range of PD types have had promising findings.27 Day treatment involves an intensive array of therapies, mostly in a group format; patients work together to support and embolden one another to make positive changes. Unfortunately, FPPs may be challenged to find appropriate services for patients who are amenable to psychotherapy; public mental health resources tend to lag far behind best practices in the case of PD.
MEDICATION MIGHT IMPROVE SYMPTOMS, NOT PERSONALITY DEFICITS
Most research on pharmacotherapy for PDs has focused on borderline PD; findings have been mixed and fairly limited.28 Medication cannot address underlying identity and relational deficits and will not result in remission of PD. Nonetheless, judicious, circumscribed use of medications to target specific symptoms may be helpful for some patients. Selective serotonin reuptake inhibitors can reduce anger and impulsive aggression in patients with borderline PD.28,29
Atypical antipsychotics may help reduce impulsive aggression or transient psychotic symptoms.28-30 For example, olanzapine and aripiprazole can reduce anxiety, anger/aggression, paranoia, and interpersonal sensitivity in borderline PD.31,32 Mood stabilizers such as valproate, lamotrigine, and topiramate may also help some borderline patients, although they do so by reducing impulsivity and aggression rather than improving core unstable identity and affect.28,29
Carefully obtained informed consent is necessary because of the danger of adverse effects with many of these medications; for example, antipsychotics have been associated with metabolic syndrome and weight gain that can threaten a patient’s already fragile self-image.33 Polypharmacy is also a potential problem: Well-intentioned clinicians may be prompted to offer multiple medications in response to patients’ unremitting complaints of distress, when a psychotherapeutic approach may need to be the primary treatment. The bottom line is that medications do not resolve personality dysfunction and are best used symptomatically as adjuncts to psychotherapy.28,30
Next page: Steps to take during the office visit >>
STEPS YOU CAN TAKE DURING THE OFFICE VISIT
Although it is not feasible for most FPPs to provide comprehensive treatment for PD, key elements from specialized therapies can be integrated into your management of these patients. Steps you can take include using validation, promoting mentalization, and managing countertransference.
Validation, which is a component of DBT, is providing the expressed acknowledgement that the patient is entitled to her feelings. This is not the same as agreeing with a position the patient has taken on an issue, but rather conveying the sense that one sees how the patient might feel the way she does. A study of women with borderline PD and substance abuse found a validation intervention by itself was significantly helpful.34 Validation can contribute to a “corrective emotional experience.” For instance, your supportive acknowledgement of a patient with a history of abuse or neglect may counter the patient’s expectation of being invalidated, and over time this can reduce the patient’s defensive rigidity.
Mentalization. Psychodynamic treatment involves a similar tack; clinicians empathize with the patient’s emotional state while also demonstrating a degree of separateness from the emotion.23-25 This promotes mentalization in the patient—the ability to contemplate one’s own and others’ subjective mental states.18 Mentalization is often impaired in PD patients, who presume to “know” what others are thinking. A patient, for instance, “just knows” that her friend secretly hates her, based on a vaguely worded text message.
You can help patients with mentalization by taking an inquisitive “not knowing” stance and by emphasizing a collaborative and reflective approach toward a given problem—to examine the issue together, from all sides. You can point out that while a patient is entitled to feel whatever he is feeling, it may not be in his best interest to act on the feelings without adequately considering the potential consequences of the action. This helps the patient to distinguish thoughts, feelings, and impulses from behavior. It also teaches the value of anticipatory thinking, impulse control, and affect regulation.
Countertransference. Managing your emotional reactions to a patient with PD is a well-documented challenge.35 Your feelings about the patient, known as countertransference, can range from considerable concern and sympathy to severe frustration, bewilderment, and frank hostility. A common reaction is the sense that one must “do something” to respond to the patient’s emotional distress or interpersonal pressure. This may trigger an impulse to give advice or offer tests or medications despite knowing that these are unlikely to be helpful. A more useful response may be to tolerate such feelings and listen empathically to the patient’s frustration. Recognizing subtle countertransference can guard against extreme reactions and maintain an appropriate clinical focus. Discussion with a trusted colleague can be helpful.
Psychodynamic approaches consider managing countertransference to be a therapeutic intervention, even when psychotherapy is not explicitly being carried out. Strong emotional responses may reflect something that the patient needs the clinician to experience, as the patient cannot bear to experience it himself. The patient needs to see—and learn from—the clinician’s handling of unbearable (for the patient) feelings. This occurs at a level of unconscious communication and may be repeated over time. Although not discussed with the patient, a clinician’s capacity for self-containment and provision of undisrupted, good medical care is in itself a psychotherapeutic accomplishment.
Based on Bob’s history of interpersonal conflicts and perceived persecution by coworkers, the FPP consults with a psychotherapist colleague, who says Bob’s chronic mistrust and social isolation suggest he may have a severe identity disturbance and unspecified PD with paranoid and schizoid features. Because Bob refuses to see a therapist, his FPP decides to focus on promoting small improvements in Bob’s interpersonal interactions and reducing absenteeism at work.
The FPP validates Bob’s feelings (“it can be very stressful to constantly feel like others are at odds with you”) and tries to promote mentalizing (“I want to understand more about what you think regarding your work situation and your coworkers. Let’s try to look at this from all perspectives—maybe we can come up with some new ideas.”)
Despite wanting to help his patient, the FPP feels uneasy and reluctant to engage with Bob, who likely evokes such feelings to keep others at a distance. The FPP tactfully seeks to remain Bob’s ally without endorsing his distorted interpretation of events. Given Bob’s paranoid rejection of therapy, the FPP refrains from making further such recommendations. The FPP’s interventions, however, may help Bob warm to the idea of further help over time, and the FPP’s supportive stance will help to ameliorate the patient’s distress. (Additional examples of how to use the strategies described in this article can be found in Table 2.)
References on following page >>
REFERENCES
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
2. Hahn SR, Thompson KS, Wills TA, et al. The difficult clinician-patient relationship: somatization, personality and psychopathology. J Clin Epidemiol. 1994:47:647-657.
3. Schafer S, Nowlis DP. Personality disorders among difficult patients. Arch Fam Med. 1998;7:126-129.
4. Paris J. Estimating the prevalence of personality disorders in the community. J Pers Disord. 2010;24:405-411.
5. Newton-Howes G, Tyrer P, Anagnostakis K, et al. The prevalence of personality disorder, its comorbidity with mental state disorders, and its clinical significance in community mental health teams. Soc Psychiatry Psychiatr Epidemiol. 2010;45:453-460.
6. Zimmerman M, Rothschild L, Chelminski I. The prevalence of DSM-IV personality disorders in psychiatric outpatients. Am J Psychiatry. 2005;162:1911-1918.
7. Moran P, Jenkins R, Tylee A, et al. The prevalence of personality disorder among UK primary care attenders. Acta Psychiatr Scand. 2000;102:52-57.
8. Newton-Howes G, Tyrer P, Johnson T. Personality disorder and the outcome of depression: Meta-analysis of published studies. Br J Psychiatry. 2006;188:13-20.
9. Blasco-Fontecilla H, Baca-Garcia E, Dervic K, et al. Severity of personality disorders and suicide attempt. Acta Psychiatr Scand. 2009;119:149-155.
10. Colpaert K, Vanderplasschen W, De Maeyer J, et al. Prevalence and determinants of personality disorders in a clinical sample of alcohol-, drug-, and dual-dependent patients. Subst Use Misuse. 2012;47:649-661.
11. Yu R, Geddes JR, Fazel S. Personality disorders, violence, and antisocial behavior: A systematic review and meta-regression analysis. J Pers Disord. 2012;26:775-792.
12. Frankenburg FR, Zanarini MC. The association between borderline personality disorder and chronic medical illnesses, poor health-related lifestyle choices, and costly forms of health care utilization. J Clin Psychiatry. 2004;65:1660-1665.
13. Lee HB, Bienvenu OJ, Cho SJ, et al. Personality disorders and traits as predictors of incident cardiovascular disease: Findings from the 23-year follow-up of the Baltimore ECA Study. Psychosomatics. 2010;51:289-296.
14. Skodol AE, Gunderson JG, McGlashan TH, et al. Functional impairment in patients with schizotypal, borderline, avoidant, or obsessive-compulsive personality disorder. Am J Psychiatry. 2002;159:276-283.
15. Bender DS, Dolan RT, Skodol AE, et al. Treatment utilization by patients with personality disorders. Am J Psychiatry. 2001;158:295-302.
16. Livesley WJ. An empirically-based classification of personality disorder.
J Pers Disord. 2011;25:397-420.
17. Bender DS, Morey LC, Skodol AE. Toward a model for assessing personality functioning in DSM-5, part I: a review of theory and methods. J Pers Assess. 2011;93:332-346.
18. Fonagy P, Gergely G, Jurist EL, et al. Affect Regulation, Mentalization, and the Development of the Self. New York, NY: Other Press; 2002.
19. Yen S, Shea MT, Battle CL, et al. Traumatic exposure and posttraumatic stress disorder in borderline, schizotypal, avoidant, and obsessive-compulsive personality disorders: findings from the collaborative longitudinal personality disorders study. J Nerv Ment Dis. 2002;190:510-518.
20. Morey LC, Stagner BH. Narcissistic pathology as core personality dysfunction: comparing DSM-IV and the DSM-5 proposal for narcissistic personality disorder. J Clin Psychol. 2012;68:908-921.
21. Lynch TR, Chapman AL, Rosenthal MZ, et al. Mechanisms of change in dialectical behaviour therapy: theoretical and empirical observations.
J Clin Psychol. 2006;62:459-480.
22. Kliem S, Kröger C, Kosfelder J. Dialectical behavior therapy for borderline personality disorder: a meta-analysis using mixed-effects modeling.
J Consult Clin Psychol. 2010;78:936-951.
23. Clarkin JF, Levy KN, Lenzenweger MF, et al. Evaluating three treatments for borderline personality disorder: a multiwave study. Am J Psychiatry. 2007;164:922-928.
24. Gregory RJ, DeLucia-Deranja E, Mogle JA. Dynamic deconstructive psychotherapy versus optimized community care for borderline personality disorder co-occurring with alcohol use disorders: a 30-month follow-up. J Nerv Ment Dis. 2010;198:292-298.
25. Bateman A, Fonagy P. Randomized controlled trial of outpatient mentalization-based treatment versus structured clinical management for borderline personality disorder. Am J Psychiatry. 2009;166:1355-1364.
26. McMain SF, Links PS, Gnam WH, et al. A randomized trial of dialectical behavior therapy versus general psychiatric management for borderline personality disorder. Am J Psychiatry. 2009;166:1365-1374.
27. Ogrodniczuk JS, Piper WE. Day treatment for personality disorders: a review of research findings. Harv Rev Psychiatry. 2001;9:105-117.
28. Paris J. Pharmacological treatments for personality disorders. Int Rev Psychiatry. 2011;23:303-309.
29. Ripoll LH, Triebwasser J, Siever LJ. Evidence-based pharmacotherapy for personality disorders. Int J Neuropsychopharmacol. 2011;14:1257-1288.
30. Steinberg PI. The use of low-dose neuroleptics in the treatment of patients with severe personality disorder: An adjunct to psychotherapy. BCMJ. 2007;49:306-310.
31. Zanarini MC, Frankenburg FR. Olanzapine treatment of female borderline personality disorder patients: a double-blind, placebo controlled pilot study. J Clin Psychiatry. 2001;62:849-854.
32. Nickel MK, Loew TH, Pedrosa Gil F. Aripiprazole in treatment of borderline patients, part II: an 18-month follow up. Psychopharmacology (Berl). 2007;191:1023-1026.
33. Silk KR. The process of managing medications in patients with borderline personality disorder. J Psychiatr Pract. 2011;17:311-319.
34. Linehan MM, Dimeff LA, Reynolds SK, et al. Dialectal behavior therapy versus comprehensive validation therapy plus 12-step for the treatment of opioid dependent women meeting criteria for borderline personality disorder. Drug Alcohol Depend. 2002;67:13-26.
35. Rossberg JI, Karterud S, Pedersen G, et al. An empirical study of countertransference reactions toward patients with personality disorders. Compr Psychiatry. 2007;48:225-230.
Asphyxiation by Cake: An Unusual Case of Dyspnea
A 58-year-old man presented to the emergency department (ED) via emergency medical services (EMS) with shortness of breath, lightheadedness, and nausea. Upon arrival to the ED, most of his symptoms had resolved. The patient reported that he had taken a two-hour flight into town the previous day and had spent an uneventful evening at a local hotel. He said that he began experiencing shortness of breath and lightheadedness soon after entering his rental vehicle an hour prior to presentation, explaining that he felt as if he “could not get any air.”
He denied chest pain, leg pain or swelling, abdominal pain, or recent illness. Medical history was significant for hypertension, for which he was taking losartan and amlodipine. He had no drug allergies, surgical history, or smoking history. Of note, when the hotel clerk got in the same rental vehicle to move it, he developed symptoms similar to those of the patient. As with the patient, the clerk’s symptoms quickly resolved after he got out of the vehicle.
The patient’s vital signs at examination included an oral temperature of 97.5°F; pulse, 62 beats/min; respiratory rate (RR), 18 breaths/min; blood pressure, 133/83 mm Hg; and O2 saturation, 100% on room air. He was alert and oriented, in no distress, easily conversational, and without diaphoresis. The lungs were clear to auscultation bilaterally, and there was no calf swelling, tenderness, or palpable cords. The remainder of the physical exam was normal.
Ancillary studies included a normal chest X-ray. An ECG demonstrated sinus bradycardia with a rate of 56 beats/min but no evidence of ischemia or right heart strain. Complete blood count, troponin I, D-dimer, and creatine phosphokinase (CPK) with MB fraction levels were all within normal limits. A serum chemistry panel was also within normal limits, except for a serum glucose level of 181 mg/dL. Venous co-oximetry showed a carboxyhemoglobin level of 0.0, and methemoglobin level of 0.5 gm% (normal range, 0.4-1.5).
Since both the patient’s and hotel clerk’s symptoms started when each was in the rental car, the patient was questioned about the vehicle and its contents. The car was a late-model rental in good condition per report. The patient informed the treating emergency physician that he worked as a decorative cake salesman and had brought cake samples with him to display at a trade show. He further stated that he had left these samples in the car overnight, packed in dry ice.
What's the solution to this unusual case?
Upon learning this information, EMS was contacted and instructed to return to the hotel and rental vehicle. The hotel room was noted to have normal levels of O2 and carbon monoxide (CO) on measurement. Investigation of the car revealed normal levels of CO but O2 levels too low to read on the sensor. The emergency team concluded that the dry ice (the solid form of carbon dioxide [CO2]) sublimed to CO2 gas overnight. This displaced the O2 in the vehicle, resulting in severe hypoxia and the symptoms of both the patient and hotel clerk.
The patient was initially placed on 15 L of O2 via a nonrebreather mask, then switched to 2 L of O2 via nasal cannula. He was observed for a total of four hours after arrival; as he remained symptom-free, he was discharged home. Postdischarge follow-up information was not obtainable.
DISCUSSION
Carbon dioxide is prevalent in everyday life, from an agent in fire extinguishers and carbonation in beverages to byproducts of cellular metabolism. Similar to CO, it is a colorless and odorless gas.
Carbon dioxide is commonly used in the food industry as dry ice to keep items cold. In its solid state, CO2 can cause severe frostbite with direct contact, similar to a burn. However, when dry ice is warmed and sublimated to a gaseous state, large amounts of CO2 are generated, and this heavy gas can accumulate and displace air (ie, atmospheric O2), especially in confined spaces. In low concentrations, gaseous CO2 appears to have minimal toxicologic effects, but at higher concentrations it can cause tachycardia, tachypnea, dyspnea, visual disturbances, arrhythmias, impaired levels of consciousness, and death.
Carbon dioxide primarily acts as a simple asphyxiant, but it also dissolves in serum as carbonic acid, resulting in a metabolic acidosis. Compensation for this acidosis is accomplished by an increased RR (ie, respiratory alkalosis), which further worsens the intake of CO2.1,2
The normal concentration of CO2 in the atmosphere is approximately 0.04% (396 ppm). The Occupational Safety and Health Administration (OSHA) has set a maximum safe exposure level of CO2 at 0.5% (5,000 ppm) over an eight-hour day.3 Concentrations as low as 1% (10,000 ppm) may cause drowsiness. Exposure to concentrations of 7% to 10% for several minutes to an hour results in headache, tachycardia, dyspnea, and hyperventilation. At levels of 10% to 15%, dizziness, severe muscle twitching, and loss of consciousness can occur after only a few minutes. Death occurs within minutes at concentrations greater than 30%.2
Carbon dioxide also acts as a potent cerebral vasodilator, which may explain symptoms such as headache and dizziness.2 The severity of symptoms is dependent on the concentration of CO2, the length of the exposure, and the underlying health of the patient. Elevated concentrations of CO2 can occur in areas where there is limited or poor ventilation, such as in a mine (where it is known as blackdamp, stythe, or choke damp),4 submarine, grain silo, or a sealed building without mechanical ventilation.
Continue for other case presentations >>
Other case presentations
Similar cases have been described in the literature. In one case, following Hurricane Ivan, a 34-year-old man placed four 25-pound blocks of dry ice wrapped in paper in the front seat of his truck with the windows closed.5 After driving less than one-quarter of a mile, he developed dyspnea and telephoned for help before losing consciousness. Fortunately, he was found in time and recovered soon after the doors to his truck were opened.5
In another case, a 59-year-old man entered a walk-in freezer that contained dry ice wrapped loosely in plastic. He was found inside the freezer 20 minutes later in cardiac arrest; resuscitation efforts were unsuccessful. Investigation of the freezer found an initial O2 concentration of 13% (normal level, 20.93%) and an estimated CO2 level of 40%.5
Similarly, a 35-year-old woman was inadvertently locked in a bank vault while storing receipts. In a bid for help, she pulled the fire alarm, which triggered a CO2-based fire-extinguishing system. The fire department responded and found the woman dead in the vault 30 minutes later. The cause of death was labeled as CO2 intoxication.6
Natural phenomena
There have also been documented cases of CO2 toxicity associated with volcanic eruption and other natural phenomena; for example, the Lake Nyos, Cameroon, West Africa incident in 1986. In this event, a magma pocket underlying the lake saturated the water with CO2 stored as carbonic acid in the water. When a landslide hit the lake, it caused the carbonic acid stored in the depths of the lake to be upheaved to the surface, where it turned back into CO2 and was released into the atmosphere. Since CO2 is heavier than O2, it displaced the O2 near the ground, resulting in the suffocation and death of 1,700 people in the surrounding villages.2
Next page: Differential diagnosis >>
Differential diagnosis
When CO2 toxicity is suspected, other conditions should be considered, as there may be more than one process involved. For example, other causes of coma or dyspnea should be investigated, including trauma, hypoglycemia, CO, methemoglobinemia, or other metabolic processes. In addition, a patient may have a pre-existing condition, such as a trauma or an altered mental status due to drugs or alcohol, all of which can increase his or her susceptibility to the effects of CO2.
Evaluation and treatment
Useful laboratory testing includes arterial blood gas, venous co-oximetry for carboxyhemoglobin, chemistry panels, ethanol testing, and radiographs or CT, as indicated.
Initial management of suspected CO2 toxicity entails first removing the patient from the source of the gas. Rescuers must exercise caution so as to prevent a mass-casualty incident. Once out of the dangerous environment, as long as the patient is conscious and spontaneously breathing, supportive measures are generally all that are necessary. Oxygen should be applied, after which the spontaneously breathing patient without underlying lung disease should rapidly return to normal.
If there is marked decrease in mental status or poor respiratory drive despite O2 administration, intubation with mechanical ventilation may be required. A higher-than-normal RR will help remove excessive CO2 in this instance.
If a respiratory acidosis is present, IV sodium bicarbonate should be avoided, as this may increase the level of serum CO2. IV fluids and other supportive measures, including treatment for any concurrent conditions, may be indicated.
REFERENCES
1. Nelson LS, Odujebe OA. Simple asphyxiants and pulmonary irritants. In: Nelson LS, Lewin NA, Howland MA, et al, eds. Goldfrank’s Toxicologic Emergencies, 9th ed. New York, NY: McGraw-Hill; 2011:1644-1645.
2. Langford NJ. Carbon dioxide poisoning. Toxicol Rev. 2005;24(4):229-235.
3. Occupational Health and Safety Standards. Table Z-1, Limits for air contaminants. Occupational Safety and Health Administration Web site. www.osha.gov/pls/oshaweb/owadisp.show_document?p_table=standards&p_id=9992. Accessed January 19, 2015.
4. Hedlund FH. The extreme carbon dioxide outburst at the Menzengraben potash mine 7 July 1953. Safety Sci. 2012;50(3):537-553.
5. Dunford JV, Lucas J, Vent N, et al. Asphyxiation due to dry ice in a walk-in freezer. J Emerg Med. 2009;36(4):353-356.
6. Gill JR, Ely SF, Hua Z. Environmental gas displacement: three accidental deaths in the workplace. Am J Forensic Med Pathol. 2002;23(1):26-30.
A 58-year-old man presented to the emergency department (ED) via emergency medical services (EMS) with shortness of breath, lightheadedness, and nausea. Upon arrival to the ED, most of his symptoms had resolved. The patient reported that he had taken a two-hour flight into town the previous day and had spent an uneventful evening at a local hotel. He said that he began experiencing shortness of breath and lightheadedness soon after entering his rental vehicle an hour prior to presentation, explaining that he felt as if he “could not get any air.”
He denied chest pain, leg pain or swelling, abdominal pain, or recent illness. Medical history was significant for hypertension, for which he was taking losartan and amlodipine. He had no drug allergies, surgical history, or smoking history. Of note, when the hotel clerk got in the same rental vehicle to move it, he developed symptoms similar to those of the patient. As with the patient, the clerk’s symptoms quickly resolved after he got out of the vehicle.
The patient’s vital signs at examination included an oral temperature of 97.5°F; pulse, 62 beats/min; respiratory rate (RR), 18 breaths/min; blood pressure, 133/83 mm Hg; and O2 saturation, 100% on room air. He was alert and oriented, in no distress, easily conversational, and without diaphoresis. The lungs were clear to auscultation bilaterally, and there was no calf swelling, tenderness, or palpable cords. The remainder of the physical exam was normal.
Ancillary studies included a normal chest X-ray. An ECG demonstrated sinus bradycardia with a rate of 56 beats/min but no evidence of ischemia or right heart strain. Complete blood count, troponin I, D-dimer, and creatine phosphokinase (CPK) with MB fraction levels were all within normal limits. A serum chemistry panel was also within normal limits, except for a serum glucose level of 181 mg/dL. Venous co-oximetry showed a carboxyhemoglobin level of 0.0, and methemoglobin level of 0.5 gm% (normal range, 0.4-1.5).
Since both the patient’s and hotel clerk’s symptoms started when each was in the rental car, the patient was questioned about the vehicle and its contents. The car was a late-model rental in good condition per report. The patient informed the treating emergency physician that he worked as a decorative cake salesman and had brought cake samples with him to display at a trade show. He further stated that he had left these samples in the car overnight, packed in dry ice.
What's the solution to this unusual case?
Upon learning this information, EMS was contacted and instructed to return to the hotel and rental vehicle. The hotel room was noted to have normal levels of O2 and carbon monoxide (CO) on measurement. Investigation of the car revealed normal levels of CO but O2 levels too low to read on the sensor. The emergency team concluded that the dry ice (the solid form of carbon dioxide [CO2]) sublimed to CO2 gas overnight. This displaced the O2 in the vehicle, resulting in severe hypoxia and the symptoms of both the patient and hotel clerk.
The patient was initially placed on 15 L of O2 via a nonrebreather mask, then switched to 2 L of O2 via nasal cannula. He was observed for a total of four hours after arrival; as he remained symptom-free, he was discharged home. Postdischarge follow-up information was not obtainable.
DISCUSSION
Carbon dioxide is prevalent in everyday life, from an agent in fire extinguishers and carbonation in beverages to byproducts of cellular metabolism. Similar to CO, it is a colorless and odorless gas.
Carbon dioxide is commonly used in the food industry as dry ice to keep items cold. In its solid state, CO2 can cause severe frostbite with direct contact, similar to a burn. However, when dry ice is warmed and sublimated to a gaseous state, large amounts of CO2 are generated, and this heavy gas can accumulate and displace air (ie, atmospheric O2), especially in confined spaces. In low concentrations, gaseous CO2 appears to have minimal toxicologic effects, but at higher concentrations it can cause tachycardia, tachypnea, dyspnea, visual disturbances, arrhythmias, impaired levels of consciousness, and death.
Carbon dioxide primarily acts as a simple asphyxiant, but it also dissolves in serum as carbonic acid, resulting in a metabolic acidosis. Compensation for this acidosis is accomplished by an increased RR (ie, respiratory alkalosis), which further worsens the intake of CO2.1,2
The normal concentration of CO2 in the atmosphere is approximately 0.04% (396 ppm). The Occupational Safety and Health Administration (OSHA) has set a maximum safe exposure level of CO2 at 0.5% (5,000 ppm) over an eight-hour day.3 Concentrations as low as 1% (10,000 ppm) may cause drowsiness. Exposure to concentrations of 7% to 10% for several minutes to an hour results in headache, tachycardia, dyspnea, and hyperventilation. At levels of 10% to 15%, dizziness, severe muscle twitching, and loss of consciousness can occur after only a few minutes. Death occurs within minutes at concentrations greater than 30%.2
Carbon dioxide also acts as a potent cerebral vasodilator, which may explain symptoms such as headache and dizziness.2 The severity of symptoms is dependent on the concentration of CO2, the length of the exposure, and the underlying health of the patient. Elevated concentrations of CO2 can occur in areas where there is limited or poor ventilation, such as in a mine (where it is known as blackdamp, stythe, or choke damp),4 submarine, grain silo, or a sealed building without mechanical ventilation.
Continue for other case presentations >>
Other case presentations
Similar cases have been described in the literature. In one case, following Hurricane Ivan, a 34-year-old man placed four 25-pound blocks of dry ice wrapped in paper in the front seat of his truck with the windows closed.5 After driving less than one-quarter of a mile, he developed dyspnea and telephoned for help before losing consciousness. Fortunately, he was found in time and recovered soon after the doors to his truck were opened.5
In another case, a 59-year-old man entered a walk-in freezer that contained dry ice wrapped loosely in plastic. He was found inside the freezer 20 minutes later in cardiac arrest; resuscitation efforts were unsuccessful. Investigation of the freezer found an initial O2 concentration of 13% (normal level, 20.93%) and an estimated CO2 level of 40%.5
Similarly, a 35-year-old woman was inadvertently locked in a bank vault while storing receipts. In a bid for help, she pulled the fire alarm, which triggered a CO2-based fire-extinguishing system. The fire department responded and found the woman dead in the vault 30 minutes later. The cause of death was labeled as CO2 intoxication.6
Natural phenomena
There have also been documented cases of CO2 toxicity associated with volcanic eruption and other natural phenomena; for example, the Lake Nyos, Cameroon, West Africa incident in 1986. In this event, a magma pocket underlying the lake saturated the water with CO2 stored as carbonic acid in the water. When a landslide hit the lake, it caused the carbonic acid stored in the depths of the lake to be upheaved to the surface, where it turned back into CO2 and was released into the atmosphere. Since CO2 is heavier than O2, it displaced the O2 near the ground, resulting in the suffocation and death of 1,700 people in the surrounding villages.2
Next page: Differential diagnosis >>
Differential diagnosis
When CO2 toxicity is suspected, other conditions should be considered, as there may be more than one process involved. For example, other causes of coma or dyspnea should be investigated, including trauma, hypoglycemia, CO, methemoglobinemia, or other metabolic processes. In addition, a patient may have a pre-existing condition, such as a trauma or an altered mental status due to drugs or alcohol, all of which can increase his or her susceptibility to the effects of CO2.
Evaluation and treatment
Useful laboratory testing includes arterial blood gas, venous co-oximetry for carboxyhemoglobin, chemistry panels, ethanol testing, and radiographs or CT, as indicated.
Initial management of suspected CO2 toxicity entails first removing the patient from the source of the gas. Rescuers must exercise caution so as to prevent a mass-casualty incident. Once out of the dangerous environment, as long as the patient is conscious and spontaneously breathing, supportive measures are generally all that are necessary. Oxygen should be applied, after which the spontaneously breathing patient without underlying lung disease should rapidly return to normal.
If there is marked decrease in mental status or poor respiratory drive despite O2 administration, intubation with mechanical ventilation may be required. A higher-than-normal RR will help remove excessive CO2 in this instance.
If a respiratory acidosis is present, IV sodium bicarbonate should be avoided, as this may increase the level of serum CO2. IV fluids and other supportive measures, including treatment for any concurrent conditions, may be indicated.
REFERENCES
1. Nelson LS, Odujebe OA. Simple asphyxiants and pulmonary irritants. In: Nelson LS, Lewin NA, Howland MA, et al, eds. Goldfrank’s Toxicologic Emergencies, 9th ed. New York, NY: McGraw-Hill; 2011:1644-1645.
2. Langford NJ. Carbon dioxide poisoning. Toxicol Rev. 2005;24(4):229-235.
3. Occupational Health and Safety Standards. Table Z-1, Limits for air contaminants. Occupational Safety and Health Administration Web site. www.osha.gov/pls/oshaweb/owadisp.show_document?p_table=standards&p_id=9992. Accessed January 19, 2015.
4. Hedlund FH. The extreme carbon dioxide outburst at the Menzengraben potash mine 7 July 1953. Safety Sci. 2012;50(3):537-553.
5. Dunford JV, Lucas J, Vent N, et al. Asphyxiation due to dry ice in a walk-in freezer. J Emerg Med. 2009;36(4):353-356.
6. Gill JR, Ely SF, Hua Z. Environmental gas displacement: three accidental deaths in the workplace. Am J Forensic Med Pathol. 2002;23(1):26-30.
A 58-year-old man presented to the emergency department (ED) via emergency medical services (EMS) with shortness of breath, lightheadedness, and nausea. Upon arrival to the ED, most of his symptoms had resolved. The patient reported that he had taken a two-hour flight into town the previous day and had spent an uneventful evening at a local hotel. He said that he began experiencing shortness of breath and lightheadedness soon after entering his rental vehicle an hour prior to presentation, explaining that he felt as if he “could not get any air.”
He denied chest pain, leg pain or swelling, abdominal pain, or recent illness. Medical history was significant for hypertension, for which he was taking losartan and amlodipine. He had no drug allergies, surgical history, or smoking history. Of note, when the hotel clerk got in the same rental vehicle to move it, he developed symptoms similar to those of the patient. As with the patient, the clerk’s symptoms quickly resolved after he got out of the vehicle.
The patient’s vital signs at examination included an oral temperature of 97.5°F; pulse, 62 beats/min; respiratory rate (RR), 18 breaths/min; blood pressure, 133/83 mm Hg; and O2 saturation, 100% on room air. He was alert and oriented, in no distress, easily conversational, and without diaphoresis. The lungs were clear to auscultation bilaterally, and there was no calf swelling, tenderness, or palpable cords. The remainder of the physical exam was normal.
Ancillary studies included a normal chest X-ray. An ECG demonstrated sinus bradycardia with a rate of 56 beats/min but no evidence of ischemia or right heart strain. Complete blood count, troponin I, D-dimer, and creatine phosphokinase (CPK) with MB fraction levels were all within normal limits. A serum chemistry panel was also within normal limits, except for a serum glucose level of 181 mg/dL. Venous co-oximetry showed a carboxyhemoglobin level of 0.0, and methemoglobin level of 0.5 gm% (normal range, 0.4-1.5).
Since both the patient’s and hotel clerk’s symptoms started when each was in the rental car, the patient was questioned about the vehicle and its contents. The car was a late-model rental in good condition per report. The patient informed the treating emergency physician that he worked as a decorative cake salesman and had brought cake samples with him to display at a trade show. He further stated that he had left these samples in the car overnight, packed in dry ice.
What's the solution to this unusual case?
Upon learning this information, EMS was contacted and instructed to return to the hotel and rental vehicle. The hotel room was noted to have normal levels of O2 and carbon monoxide (CO) on measurement. Investigation of the car revealed normal levels of CO but O2 levels too low to read on the sensor. The emergency team concluded that the dry ice (the solid form of carbon dioxide [CO2]) sublimed to CO2 gas overnight. This displaced the O2 in the vehicle, resulting in severe hypoxia and the symptoms of both the patient and hotel clerk.
The patient was initially placed on 15 L of O2 via a nonrebreather mask, then switched to 2 L of O2 via nasal cannula. He was observed for a total of four hours after arrival; as he remained symptom-free, he was discharged home. Postdischarge follow-up information was not obtainable.
DISCUSSION
Carbon dioxide is prevalent in everyday life, from an agent in fire extinguishers and carbonation in beverages to byproducts of cellular metabolism. Similar to CO, it is a colorless and odorless gas.
Carbon dioxide is commonly used in the food industry as dry ice to keep items cold. In its solid state, CO2 can cause severe frostbite with direct contact, similar to a burn. However, when dry ice is warmed and sublimated to a gaseous state, large amounts of CO2 are generated, and this heavy gas can accumulate and displace air (ie, atmospheric O2), especially in confined spaces. In low concentrations, gaseous CO2 appears to have minimal toxicologic effects, but at higher concentrations it can cause tachycardia, tachypnea, dyspnea, visual disturbances, arrhythmias, impaired levels of consciousness, and death.
Carbon dioxide primarily acts as a simple asphyxiant, but it also dissolves in serum as carbonic acid, resulting in a metabolic acidosis. Compensation for this acidosis is accomplished by an increased RR (ie, respiratory alkalosis), which further worsens the intake of CO2.1,2
The normal concentration of CO2 in the atmosphere is approximately 0.04% (396 ppm). The Occupational Safety and Health Administration (OSHA) has set a maximum safe exposure level of CO2 at 0.5% (5,000 ppm) over an eight-hour day.3 Concentrations as low as 1% (10,000 ppm) may cause drowsiness. Exposure to concentrations of 7% to 10% for several minutes to an hour results in headache, tachycardia, dyspnea, and hyperventilation. At levels of 10% to 15%, dizziness, severe muscle twitching, and loss of consciousness can occur after only a few minutes. Death occurs within minutes at concentrations greater than 30%.2
Carbon dioxide also acts as a potent cerebral vasodilator, which may explain symptoms such as headache and dizziness.2 The severity of symptoms is dependent on the concentration of CO2, the length of the exposure, and the underlying health of the patient. Elevated concentrations of CO2 can occur in areas where there is limited or poor ventilation, such as in a mine (where it is known as blackdamp, stythe, or choke damp),4 submarine, grain silo, or a sealed building without mechanical ventilation.
Continue for other case presentations >>
Other case presentations
Similar cases have been described in the literature. In one case, following Hurricane Ivan, a 34-year-old man placed four 25-pound blocks of dry ice wrapped in paper in the front seat of his truck with the windows closed.5 After driving less than one-quarter of a mile, he developed dyspnea and telephoned for help before losing consciousness. Fortunately, he was found in time and recovered soon after the doors to his truck were opened.5
In another case, a 59-year-old man entered a walk-in freezer that contained dry ice wrapped loosely in plastic. He was found inside the freezer 20 minutes later in cardiac arrest; resuscitation efforts were unsuccessful. Investigation of the freezer found an initial O2 concentration of 13% (normal level, 20.93%) and an estimated CO2 level of 40%.5
Similarly, a 35-year-old woman was inadvertently locked in a bank vault while storing receipts. In a bid for help, she pulled the fire alarm, which triggered a CO2-based fire-extinguishing system. The fire department responded and found the woman dead in the vault 30 minutes later. The cause of death was labeled as CO2 intoxication.6
Natural phenomena
There have also been documented cases of CO2 toxicity associated with volcanic eruption and other natural phenomena; for example, the Lake Nyos, Cameroon, West Africa incident in 1986. In this event, a magma pocket underlying the lake saturated the water with CO2 stored as carbonic acid in the water. When a landslide hit the lake, it caused the carbonic acid stored in the depths of the lake to be upheaved to the surface, where it turned back into CO2 and was released into the atmosphere. Since CO2 is heavier than O2, it displaced the O2 near the ground, resulting in the suffocation and death of 1,700 people in the surrounding villages.2
Next page: Differential diagnosis >>
Differential diagnosis
When CO2 toxicity is suspected, other conditions should be considered, as there may be more than one process involved. For example, other causes of coma or dyspnea should be investigated, including trauma, hypoglycemia, CO, methemoglobinemia, or other metabolic processes. In addition, a patient may have a pre-existing condition, such as a trauma or an altered mental status due to drugs or alcohol, all of which can increase his or her susceptibility to the effects of CO2.
Evaluation and treatment
Useful laboratory testing includes arterial blood gas, venous co-oximetry for carboxyhemoglobin, chemistry panels, ethanol testing, and radiographs or CT, as indicated.
Initial management of suspected CO2 toxicity entails first removing the patient from the source of the gas. Rescuers must exercise caution so as to prevent a mass-casualty incident. Once out of the dangerous environment, as long as the patient is conscious and spontaneously breathing, supportive measures are generally all that are necessary. Oxygen should be applied, after which the spontaneously breathing patient without underlying lung disease should rapidly return to normal.
If there is marked decrease in mental status or poor respiratory drive despite O2 administration, intubation with mechanical ventilation may be required. A higher-than-normal RR will help remove excessive CO2 in this instance.
If a respiratory acidosis is present, IV sodium bicarbonate should be avoided, as this may increase the level of serum CO2. IV fluids and other supportive measures, including treatment for any concurrent conditions, may be indicated.
REFERENCES
1. Nelson LS, Odujebe OA. Simple asphyxiants and pulmonary irritants. In: Nelson LS, Lewin NA, Howland MA, et al, eds. Goldfrank’s Toxicologic Emergencies, 9th ed. New York, NY: McGraw-Hill; 2011:1644-1645.
2. Langford NJ. Carbon dioxide poisoning. Toxicol Rev. 2005;24(4):229-235.
3. Occupational Health and Safety Standards. Table Z-1, Limits for air contaminants. Occupational Safety and Health Administration Web site. www.osha.gov/pls/oshaweb/owadisp.show_document?p_table=standards&p_id=9992. Accessed January 19, 2015.
4. Hedlund FH. The extreme carbon dioxide outburst at the Menzengraben potash mine 7 July 1953. Safety Sci. 2012;50(3):537-553.
5. Dunford JV, Lucas J, Vent N, et al. Asphyxiation due to dry ice in a walk-in freezer. J Emerg Med. 2009;36(4):353-356.
6. Gill JR, Ely SF, Hua Z. Environmental gas displacement: three accidental deaths in the workplace. Am J Forensic Med Pathol. 2002;23(1):26-30.
Factors Affecting Bone Growth
Differences in bone size are established early in life, before puberty and perhaps even in utero.1 Bone begins to form when mesenchymal cells form condensations—clusters of cells that adhere through expression of adhesion molecules2 (Figure 1). Bone must be stiff, flexible enough to change shape to absorb energy, and light enough to allow mobility.1,3 Longitudinal bone growth is detrimental to bone stability, but this effect is counteracted by concomitant bone growth in width.4 Bone growth in width has not been studied as extensively, despite its paramount role in skeletal development.5
Bone growth and development are products of the complex interactions of genetic and environmental factors, including diet, hormones, and mechanical stimuli.6-9 Longitudinal bone growth is controlled by systemic and local hormones and local mechanical factors. Two models for control of bone growth in width have been suggested—the mechanostat theory (mechanical requirements regulate periosteal apposition) and the sizostat hypothesis (a master gene or set of genes regulates bone growth in width so bone reaches a preprogrammed size, independent of mechanical requirements).5
In this article, we review the most recent data regarding bone growth from the embryonic age and analyze the factors that control bone growth. An understanding of this complex system is important in identifying metabolic and developmental bone diseases10 and fracture risk.11,12
Growth Plate
The growth plate consists mainly of collagen fibrils, proteoglycans, and water, arranged to form a sort of sponge with very small pores.13 The growth plate is located between epiphyseal and metaphyseal bone at the distal end of long bones14 and is strain-rate–dependent,15,16 which means it is hard when squeezed rapidly but soft when deformed slowly. The growth plate becomes ossified after puberty and epiphyseal fusion.17
Histologically, the growth plate consists of horizontal zones of chondrocytes at different stages of differentiation.4 The germinal zone, at the epiphyseal end of the growth plate, contains resting chondrocytes, which seem crucial in orienting the underlying columns of chondrocytes and, therefore, in unidirectional bone growth, probably by secretion of a growth plate–orienting factor.14,18 Next is the proliferative zone, a matrix-rich zone in which flattened chondrocytes undergo longitudinal cell division and orient themselves in typical column-wise fashion. At some point, proliferating chondrocytes lose their capacity to divide; they start to differentiate and become prehypertrophic, coinciding with a size increase.4 Proliferating chondrocytes are located in the transition (maturation or prehypertrophic) zone. In the hypertrophic zone, round chondrocytes secrete matrix proteins in large amounts.14 This stage is characterized by an increase in intracellular calcium concentration, which is essential in the production of matrix vesicles. These vesicles, small membrane-enclosed particles, are released from chondrocytes19,20 and secrete calcium phosphates, hydroxyapatite, and matrix metalloproteinases, resulting in mineralization of the vesicles and their surrounding matrix.4 The chondrocytes in this mineralized zone eventually undergo programmed cell death (apoptosis), leaving a scaffold for new bone formation.
Longitudinal Bone Growth
Generally, bones increase in length as long as new material is being squeezed between the reserve zone of the growth plate and the zone of provisional calcification.4
Postnatal linear growth occurs in 3 phases. Phase 1 is characterized by a high rate of growth at the beginning of fetal life, and then rapid deceleration up to about 3 years; phase 2, by a lower, slowly decelerating growth rate up to puberty; and phase 3, by an increased rate of longitudinal growth until a peak is reached.14,21,22
In 1964, Park23 proposed that the structure of the epiphyseal cartilage may determine the pattern of the growing bone shaft. The changes within the hypertrophic zone are directly related to matrix mineralization, vascular invasion, and subsequent development.24 Intracellular calcium concentration increases in the hypertrophic chondrocytes in the hypertrophic zone of growth plate cartilage; at some point, these chondrocytes begin to mineralize the longitudinal septa in the surrounding matrix25 (Figure 2). At the growth cartilage junction, mononuclear cells of undetermined origin resorb the unmineralized horizontal septa of the growth cartilage. These cells are called septoclasts or chondroclasts.25,26 Blood vessels invade the area and pave the way for bone cell precursors.27 Eighty percent of the longitudinal septa of the growth cartilage is rapidly resorbed in the metaphyseal zone immediately behind the invading blood vessels, paving the way for bone cell precursors.28 Fazzalari and colleagues28 reported that about 40% of mineralized septa serves as scaffold for the formation of primary bone trabeculae; the other 60% is absorbed by chondroclasts (osteoclasts) near the vascular invasion front.
Regulation of Longitudinal Bone Growth
Longitudinal bone growth is regulated by genetic, hormonal, growth, and environment factors17,29-31 (Table). It must be controlled on at least 3 different levels.4 Level 1 is systemic control by factors such as growth hormone (GH), sex hormones, and glucocorticoids. The major systemic hormones that control longitudinal bone growth during childhood are GH, insulin-like growth factor 1 (IGF-1), the thyroid hormones triiodothyronine (T3) and thyroxine (T4), and glucocorticoids; during puberty, the sex steroids play the most significant role.14 Level 2 is local control by factors such as Indian hedgehog (Inh), parathyroid hormone–related peptide (PTHrP), and fibroblast growth factors (FGFs).14,31 Level 3 is mechanical control.4
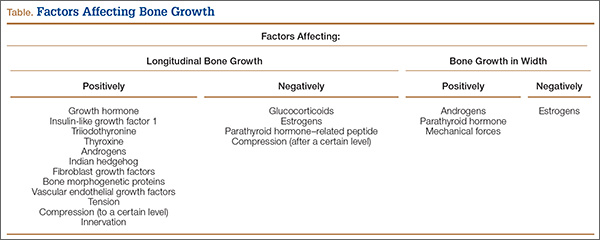
Systemic Regulation. After birth, GH becomes an important modulator of longitudinal growth and appears to be, together with IGF-1, the central player in the hypothalamus–pituitary–growth plate axis.14 According to the original somatomedin hypothesis,32 GH stimulates hepatic production of IGF-1, which in turn promotes growth directly at the epiphyseal plate.17 GH acts on resting zone chondrocytes and is responsible for local IGF-1 production, which stimulates clonal expansion of proliferating chondrocytes in an autocrine/paracrine manner.33 Infusion of GH or IGF-1 shortens stem- and proliferating-cell cycle times in the growth plate of hypophysectomized rats and decreases the duration of the hypertrophic differentiation phase, with GH being more effective.17 According to the experimental study of Hunziker and colleagues,34 GH or IGF-1 treatment restores mean cell volume and height, but the growth rate is not normalized by either hormone.
Thyroid hormones also play a vital role in bone growth. T3 and, to a lesser extent, T4 are crucial in normal bone maturation.30,35 Childhood hypothyroidism causes growth failure; growth failure may develop insidiously, but, once established, it is severe.17 On the other hand, hyperthyroidism increases the growth rate in children but also leads to premature growth plate fusion and short stature.36,37 T3 seems to stimulate recruitment of cells from the germinal zone to the proliferating zone and facilitates differentiation of growth plate chondrocytes.38-40 Its precursor, T4, increases the number of [3H]methylthymidine-labeled chondrocyte nuclei and [35S]incorporation in Snell dwarf mice growth plates, suggesting a stimulatory role in chondrocyte proliferation and differentiation.41
Glucocorticoids suppress growth by modifying the GH/IGF-1 pathway at different levels.17 Silvestrini and colleagues42 localized the glucocorticoid receptor in rat bone cells, including chondrocytes. The glucocorticoid receptor was also localized by Abu and colleagues43 in human growth plates, especially in hypertrophic chondrocytes, suggesting direct effects of glucocorticoids on the growth plate. An excess of glucocorticoids enhances bone resorption, inhibits osteoblast activity, and reduces bone matrix production to retard growth in children.44,45 Excess glucocorticoids also induce apoptosis of osteoblasts and osteocytes in rabbit trabecular bone46 and osteoblasts in rat long bones,47 resulting in an almost complete absence of new bone formation.17 In addition, glucocorticoids induce sex hormone deficiency and alter vitamin D metabolism, leading to deleterious effects on growth and skeletal integrity.48 Excess glucocorticoids modify the GH/IGF-1 pathway at different levels, suppressing growth.17 In contrast, low levels of glucocorticoids, as in familial glucocorticoid deficiency, are associated with tall stature.49
Longitudinal bone growth is also based on sex hormones, especially during puberty.17 In rats, estrogen depletion stimulates longitudinal growth, whereas estrogen administration inhibits longitudinal growth.50-52 Nilsson and colleagues53 studied ovariectomized immature rabbits treated with either estrogen or the selective estrogen receptor modulator raloxifene and found reduced chondrocyte proliferation and growth plate height as well as accelerated growth plate senescence. Many experimental studies have concluded that estrogen can inhibit longitudinal growth in the absence of GH.51,54,55
Androgens can directly influence growth plate function and may account for some skeletal differences between males and females.56-58 Unlike estrogens, androgens stimulate longitudinal growth, as shown in several studies that assessed the effect of administering nonaromatizable androgens on longitudinal growth in boys with constitutionally delayed growth.59,60
Local Regulation. Inh, a master regulator of bone development, coordinates chondrocyte proliferation, chondrocyte differentiation, and osteoblast differentiation.31 Inh belongs to the hedgehog protein family, which plays a crucial role in embryonic patterning and development.4 The proliferative effect of Inh is likely to be direct action on chondrocytes.31 In 1996, Vortkamp and colleagues61 reported that misexpression of Inh in chicken long bones blocked chondrocyte differentiation. More recently, St-Jacques and colleagues62 studied Inh-null mutant mice and found failure of both chondrocyte differentiation and osteoblast development. Inh is now thought to coordinate endochondral ossification, regulating chondrocyte proliferation and differentiation and osteoblast differentiation and coupling chondrogenesis and osteogenesis.62,63
PTHrP acts primarily to keep proliferating chondrocytes in the proliferative pool.31 Mice that did not express PTHrP showed accelerated chondrocyte differentiation leading to dwarfism.64 On the other hand, ectopic expression of PTHrP in the growth plate inhibited chondrocyte differentiation, resulting in a smaller cartilaginous skeleton compared with wild-type mice.65 PTHrP appears to regulate the rate of programmed chondrocyte differentiation in developing endochondral bone and at the level of the growth plate.64,66-69
The family of FGFs, which are major regulators of embryonic bone development, has at least 22 members.70,71 Achondroplasia, the most common type of dwarfism, is caused by an activating mutation in FGF receptor 3 (FGFR3).72-74 FGF18 deficiency also leads to delayed ossification and decreased expression of osteogenic markers.75
Bone morphogenetic proteins (BMPs) are recognized as important regulators of growth, differentiation, and morphogenesis during embryology.76 In 2001, Minina and colleagues77 showed that normal chondrocyte proliferation requires parallel signaling of both Inh and BMPs and that BMPs can inhibit chondrocyte differentiation independently of the Inh/PTHrP pathway.
Vascular endothelial growth factor (VEGF), a chemoattractant for endothelial cells, is one of the most important growth factors for endothelial cells.78 VEGF is a key player in the actions that occur during the end stage of endochondral bone formation; these actions include terminal differentiation of chondrocytes, vascular invasion, chondrocyte apoptosis, and replacement of chondrocytes with bone.27,79,80 When Gerber and colleagues27 inactivated VEGF in 24-day-old mice, they noticed suppressed blood vessel invasion and trabecular bone formation concomitant with an increased width of the hypertrophic zone.
Mechanical Regulation. Mechanical forces influence bone formation and adaptation.81 Growth rates from early infancy through late adolescence were found to be strongly correlated between an appropriate measure of mechanical loading (body size, or body weight–bone length) and bone strength (assessed by section modulus).82 The observation that compression inhibits bone growth was well known to the ancient Romans.83 In the 19th century, the Hueter-Volkmann law was proclaimed. This law is well known to pediatric orthopedic surgeons and is the basis of growth modulation for correcting angular deformities of the lower extremities and spinal deformities.4,84
If compression always inhibited bone growth, as it was believed, growth plates would be extremely unstable, as any slight deviation from the straight alignment of the long bones of the lower extremities would induce a vicious circle of positive feedback and result in catastrophic deformities.4 Mild compression leads to increased, not decreased, growth. Nevertheless, when compression on one side of the growth plate exceeds a certain level, growth is indeed suppressed, and the lesion begins to worsen.4
In 1997, Frost85 proposed using a single graph that combines the clinical observation of mechanical forces affecting longitudinal bone growth. Both mild tension and mild compression induce bone growth, whereas heavy compression inhibits growth (Figure 3).

Three rules describe bone adaptation in mathematical terms. First, bone adaptation is driven by dynamic, not static, loading. Second, only a short period of mechanical loading is needed to initiate an adaptive response (extending the loading period has a diminishing effect on further bone adaptation). Third, bone cells accommodate to a customary mechanical loading environment, making them less responsive to routine loading signals.81
Also playing a significant role in bone physiology is the nervous system, with leptin-dependent central control of bone formation via the sympathetic system.86 Several investigators have tried to determine the effect of muscle activity on bone growth in length.87 Pottorf88 in 1916 and Allison and Brooks89 in 1921 were among the first to study this correlation; they concluded that long bones grow less after denervation. On the other hand, Ring90 in 1961 reported that, despite innervation, longitudinal bone growth was increased. Investigators in more recent studies have advanced the idea that the nervous system plays a negative role in bone physiology. Dysart and colleagues87 showed that muscle pull affects periosteal tension and, consequently, bone form and growth in length. In a clinical study involving 32 children with neonatal brachial plexus injury,91 the ratio of skewness between the affected humeral head and the contralateral normal head was calculated. Skewness was determined by dividing the anterior area of the humeral head by the posterior area. There was a significant preoperative difference between the 2 sides, but the skewness ratio was significantly improved after surgery.
Bone Growth in Width
Bone growth in width has not received as much attention as longitudinal bone growth. Several studies have indicated that body mass and muscle strength have important influences on long bone strength in children and adolescents.92-97 As bone width changes only slowly after the growth period, bone growth in width is one of the most important determinants of bone strength throughout life.4 It is clear that, if bones grew in length without increasing in width, they would become unstable and break.4
Histologically, osteoblasts add mineralized tissue to the outer (periosteal) bone surface. This process is periosteal apposition.98 The periosteum has an outer layer, composed mainly of fibrous tissue, and an inner layer, the cambium, which harbors osteogenic cells.4 In children, bone formation is continuous, which is the hallmark of modeling99,100; in adults, periosteal bone may undergo cyclical resorption and formation, which are characteristic of remodeling.101,102
Macroscopically, bone grows rapidly during early life; then, growth continuously slows down until reaching a nadir during early school age.4 It is clear that wider bones must have higher midshaft periosteal apposition rates, as this is how they become wider.4,103
Regulation of Bone Growth in Width (Table)
Systemic Regulation. Periosteal apposition at diaphyseal bone sites is stimulated by androgen and GH and inhibited by estrogens.104-106 In an experimental study, Turner and colleagues104 found that androgen treatment stimulated bone formation in orchiectomized rats and suppressed bone formation in ovariectomized rats. A large dose of diethylstilbestrol also suppressed bone formation in ovariectomized rats. Parathyroid hormone is associated with faster periosteal expansion in adults, according to Parfitt.107 In addition, nutrition with high calcium intake has the same effects on children, especially those with high levels of physical activity.108
Local Regulation. Given that periosteal bone development is site-specific, whereas systemic hormones and nutrition are blind to structure,4 it is clear that local regulation is key to bone growth in width. Genetic heritage seems to have an overwhelming effect on periosteal bone development. Volkman and colleagues,109 who experimented with various genetic markers in rats, concluded that genetic control of cortical bone geometry is complex and that femoral size and shape may be influenced by different but overlapping groups of polymorphic loci.
Mechanical Regulation. Mechanical forces seem to be very important in determining bone width. For example, the difference in width between femur and humerus can be explained by the different mechanical forces acting on each bone. This perspective is supported by Ruff,82 who showed that the correlation of body size (body weight–bone length) and bone strength is stronger in the femur than in the humerus.
The vital role of mechanical forces in bone growth in width is also supported by results of a study by Goodship and colleagues,110 who overloaded the radius of young pigs by partially removing the ulna. They showed that the radius was strengthened by rapid periosteal apposition. This effect has also been noticed in the clinical setting, when the tibia is replaced with the fibula, which quickly hypertrophies in order to resemble the tibia.111
Conclusion
Longitudinal bone growth has been extensively studied. Systemic and local hormonal pathways control bone growth in a complicated regulation system. Mechanical loading is also strongly correlated with longitudinal bone growth. Bone growth in width has received less attention. Despite its importance in bone stability, periosteal development—and periosteal apposition and resorption more specifically—has not received enough attention. Researchers need to clarify the role of genetic factors affecting periosteal development.
1. Seeman E. Structural basis of growth-related gain and age-related loss of bone strength. Rheumatology. 2008;47(suppl 4):iv2-8.
2. Hall BK, Miyake T. All for one and one for all: condensations and the initiation of skeletal development. Bioessays. 2000;22(2):138-147.
3. Currey JD. Bones: Structure and Mechanics. Princeton, NJ: Princeton University Press; 2002.
4. Rauch F. Bone growth in length and width: the yin and yang of bone stability. J Musculoskelet Neuronal Interact. 2005;5(3):194-201.
5. Seeman E. Periosteal bone formation—a neglected determinant of bone strength. N Engl J Med. 2003;349(4):320-323.
6. Arden NK Spector TD. Genetic influences on muscle strength and bone mineral density: a twin study. J Bone Miner Res. 1997;12(12):2076-2081.
7. Biewener AA, Bertram JEA. Mechanical loading and bone growth in vivo. In: Hall BK, ed. Bone, Vol 7: Bone Growth—B. Boca Raton, FL: CRC Press; 1993:1-36.
8. McGuigan FE, Murray L, Gallagher A, et al. Genetic and environmental determinants of peak bone mass in young men and women. J Bone Miner Res. 2002;17(7):1273-1279.
9. Slemenda CW, Reister TK, Hui SL, Miller JZ, Christian JC, Johnston CC Jr. Influences on skeletal mineralization in children and adolescents: evidence for varying effects of sexual maturation and physical activity. J Pediatr. 1994;125(2):201-207.
10. Schoenau E, Neu CM, Beck B, Manz F, Rauch F. Bone mineral content per muscle cross-sectional area as an index of the functional muscle–bone unit. J Bone Miner Res. 2002;17(6):1095-1101.
11. Rauch F, Neu C, Manz F, Schoenau E. The development of metaphyseal cortex—implications for distal radius fractures during growth. J Bone Miner Res. 2001;16(8):1547-1555.
12. Skaggs DL, Loro ML, Pitukcheewanont P, Tolo V, Gilsanz V. Increased body weight and decreased radial cross-sectional dimensions in girls with forearm fractures. J Bone Miner Res. 2001;16(7):1337-1342.
13. Allen DM, Mao JJ. Heterogenous nanostructural and nanoelastic properties of pericellular and interterritorial matrices of chondrocytes by atomic force microscopy. J Struct Biol. 2004;145(3):196-204.
14. van der Eerden BC, Karperien M, Wit JM. Systemic and local regulation of the growth plate. Endocr Rev. 2003;24(6):782-801.
15. Li LP, Herzog W. Strain-rate dependence of cartilage stiffness in unconfined compression: the role of fibril reinforcement versus tissue volume change in fluid pressurization. J Biomech. 2004;37(3):375-382.
16. Cohen B, Chorney GS, Phillips DP, Dick HM, Mow VC. Compressive stress-relaxation behavior of bovine growth plate may be described by the non-linear biphasic theory. J Orthop Res. 1994;12(6):804-813.
17. Robson H, Siebler T, Shalet SM, Williams GR. Interactions between GH, IGF-Ι, glucocorticoids and thyroid hormones during skeletal growth. Pediatr Res. 2002;52(2):137-147.
18. Abad V, Meyers JL, Weise M, et al. The role of the resting zone in growth plate chondrogenesis. Endocrinology. 2002;143(5):1851-1857.
19. Wang W, Kirsch T. Retinoic acid stimulates annexin-mediated growth plate chondrocyte mineralization. J Cell Biol. 2002;157(6):1061-1069.
20. Anderson HC. Matrix vesicles and calcification. Curr Rheumatol Rep. 2003;5(3):222-226.
21. Tanner JM. The adolescent spurt in animals. In: Thomas CC, ed. Growth at Adolescence. Oxford, UK: Blackwell; 1962:223-239.
22. Drop SL, De Waal WJ, De Muinck Keizer-Schrama SM. Sex steroid treatment of constitutionally tall stature. Endocr Rev. 1998;19(5):540-558.
23. Park EA. The imprinting of nutritional disturbances on the growing bone. Pediatrics. 1964;33(suppl):815-862.
24. Buckwalter JA, Mower D, Unqar R, Schaeffer J, Ginsberg B. Morphometric analysis of chondrocyte hypertrophy. J Bone Joint Surg Am. 1986;68(2):243-255.
25. Sawae Y, Sahara T, Sasaki T. Osteoclast differentiation at the growth plate cartilage–trabecular bone junction in newborn rat femur. J Electron Microsc. 2003;52(6):493-502.
26. Lee ER, Lamplugh L, Shepard NL, Mort JS. The septoclast, a cathepsin B–rich cell involved in the resorption of growth plate cartilage. J Histochem Cytochem. 1995;43(5):525-536.
27. Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5(6):623-628.
28. Fazzalari NL, Moore AJ, Byers S, Byard RW. Quantitative analysis of trabecular morphogenesis in the human costochondral junction during the postnatal period in normal subjects. Anat Rec. 1997;248(1):1-12.
29. Cancedda R, Descalzi Cancedda F, Castagnola P. Chondrocyte differentiation. Int Rev Cytol. 1995;159:265-358.
30. Stevens DA, Williams GR. Hormone regulation of chondrocyte differentiation and endochondral bone formation. Mol Cell Endocrinol. 1999;151(1-2):195-204.
31. Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423(6937):332-336.
32. Daughaday WH, Hall K, Raben MS, Salmon WD Jr, van den Brande JL, van Wyk JJ. Somatomedin: proposed designation for sulphation factor. Nature. 1972;235(5333):107.
33. Isaksson OG, Lindahl A, Nilsson A, Isqaard J. Mechanism of the stimulatory effect of the growth hormone on longitudinal bone growth. Endocr Rev. 1987;8(4):426-438.
34. Hunziker EB, Wagner J, Zapf J. Differential effects of insulin-like growth factor I and growth hormone on developmental stages of rat growth plate chondrocytes in vivo. J Clin Invest. 1994;93(3):1078-1086.
35. Underwood LE, van Wijk JJ. Normal and aberrant growth. In: Wilson JD, Foster DW, eds. Textbook of Endocrinology. Philadelphia, PA: Saunders; 1992:1079-1138.
36. Rivkees SA, Bode HH, Crawford JD. Long-term growth in juvenile acquired hypothyroidism: the failure to achieve normal adult stature. N Engl J Med. 1988;318(10):599-602.
37. Segni M, Leonardi E, Mazzoncini B, Pucarelli I, Pasquino AM. Special features of Graves’ disease in early childhood. Thyroid. 1999;9(9):871-877.
38. Burch WM, Van Wyk JJ. Triiodothyronine stimulates cartilage growth and maturation by different mechanisms. Am J Physiol. 1987;252(2, pt 1):E176-E182.
39. Lewinson D, Bialik GM, Hochberg Z. Differential effects of hypothyroidism on the cartilage and the osteogenic process in the mandipular condyle: recovery by growth hormone and thyroxine. Endocrinology. 1994;135(4):1504-1510.
40. Wakita R, Izumi T, Itoman M. Thyroid hormone–induced chondrocyte terminal differentiation in rat femur organ culture. Cell Tissue Res. 1998;293(2):357-364.
41. Smeets T, van Buul-Offers S. Influence of growth hormone and thyroxine on cell kinetics in the proximal tibial growth plate of Snell dwarf mice. Cell Tissue Kinet. 1986;19(2):161-170.
42. Silvestrini G, Mocetti P, Ballanti P, Di Grezia R, Bonucci E. Cytochemical demonstration of the glucocorticoid receptor in skeletal cells of the rat. Endocr Res. 1999;25(1):117-128.
43. Abu EO, Horner A, Kusec V, Triffitt JT, Compston JE. The localization of the functional glucocorticoid receptor alpha in human bone. J Clin Endocrinol Metab. 2000;85(2):883-889.
44. Magiakou MA, Mastorakos G, Chrousos GP. Final stature in patients with endogenous Cushing’s syndrome. J Clin Endocrinol Metab. 1994;79(4):1082-1085.
45. Avioli LV. Glucocorticoid effects on statural growth. Br J Rheumatol. 1993;32(suppl 2):27-30.
46. Eberhardt AW, Yeager-Jones A, Blair HC. Regional trabecular bone matrix degeneration and osteocyte death in femora if glucocorticoid-treated rabbits. Endocrinology. 2001;142(3):1333-1340.
47. Silvestrini G, Ballanti P, Patacchioli FR, et al. Evaluation of apoptosis and the glucocorticoid receptor in the cartilage growth plate and metaphyseal bone cells of rats after high-dose treatment with corticosterone. Bone. 2000;26(1):33-42.
48. Montecucco C, Caporali R, Caprotti P, Caprotti M, Notario A. Sex hormones and bone metabolism in postmenopausal rheumatoid arthritis treated with two different glucocorticoids. J Rheumatol. 1992;19(12):1895-1900.
49. Bello CE, Garrett SD. Therapeutic issues in oral glucocorticoid use. Lippincotts Prim Care Pract. 1999;3(3):333-341.
50. Turner RT, Riggs BL, Speisberg TC. Skeletal effects of estrogen. Endocr Rev. 1994;15(3):275-300.
51. Gevers EF, Wit JM, Robinson IC. Effect of gonadectomy on growth and GH responsiveness in dwarf rats. J Endocrinol. 1995;145(1):69-79.
52. van der Eerden BC, Emons J, Ahmed S, et al. Evidence for genomic and non-genomic actions of estrogens in growth plate regulation in female and male rats at the onset of sexual maturation. J Endocrinol. 2002;175(2):277-288.
53. Nilsson O, Falk J, Ritzen EM, Baron J, Savendahl L. Raloxifene acts as an estrogen agonist on the rabbit growth plate. Endocrinology. 2003;144(4):1481-1485.
54. Strickland AL, Sprinz H. Studies of the influence of estradiol and growth hormone on the hypophysectomized immature rat epiphyseal cartilage growth plate. Am J Obstet Gynecol. 1973;115(4):471-477.
55. Jansson JO, Eden S, Isaksson O. Sites of action of testosterone and estradiol on longitudinal bone growth. Am J Physiol. 1983;244(2):E135-E140.
56. Abu EO, Horner A, Kusec V, Triffitt JT, Compston JE. The localization of androgen receptors in human bone. J Clin Endocrinol Metab. 1997;82(10):3493-3497.
57. Noble B, Routledge J, Stevens H, Hughes I, Jacobson W. Androgen receptors in bone-forming tissue. Horm Res. 1999;51(1):31-36.
58. van der Eerden BC, van Til NP, Brinkmann AO, Lowik CW, Wit JM, Karperien M. Sex differences in the expression of the androgen receptor in the tibial growth plate and metaphyseal bone of the rat. Bone. 2002;30(6):891-896.
59. Cassorla FG, Skerda MC, Valk IM, Hung W, Cutler GB Jr, Loriaux DL. The effects of sex steroids on ulnar growth during adolescence. J Clin Endocrinol Metab. 1984;58(4):717-720.
60. Zung A, Phillip M, Chalew SA, Palese T, Kowarski AA, Zadik Z. Testosterone effect on growth and growth mediators of the GF–IGF-I axis in the liver and epiphyseal growth plate of juvenile rats. J Mol Endocrinol. 1999;23(2):209-221.
61. Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273(5275):613-622.
62. St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and essential for bone formation. Genes Dev. 1999;13(16):2072-2086.
63. Karp SJ Schipani E, St-Jacques B, Hunzelman J, Kronenberg H, Mcmahon AP. Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone related-protein–dependent and –independent pathways. Development. 2000;127(3):543-548.
64. Karaplis AC, Luz A, Glowacki J, et al. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone–related peptide gene. Genes Dev. 1994;8(3):277-289.
65. Weir EC, Philbrick WM, Amling M, Neff LA, Baron R, Broadus AE. Targeted overexpression of parathyroid hormone–related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc Natl Acad Sci U S A. 1996;93(19):10240-10245.
66. Erlebacher A, Filvaroff EH, Gitelman SE, Derynk R. Toward a molecular understanding of skeletal development. Cell. 1995;80(3):371-378.
67. Iwamoto M, Jikko A, Murakami H, et al. Changes in parathyroid hormone receptors during chondrocyte cytodifferentiation. J Biol Chem. 1994;269(25):17245-17251.
68. Henderson JE, Amizuka N, Warshawsky H, et al. Nucleolar localization of parathyroid hormone–related peptide enhances survival of chondrocytes under conditions that promote apoptotic cell death. Mol Cell Biol. 1995;15(8):4064-4075.
69. Amizuka N, Warshawsky H, Henderson JE, Goltzman D, Karaplis AC. Parathyroid hormone–related peptide-depleted mice show abnormal epiphyseal cartilage development and altered endochondral bone formation. J Cell Biol. 1994;126(6):1611-1623.
70. Szebenyi G, Fallon JF. Fibroblast growth factors as multifunctional signaling factors. Int Rev Cytol. 1999;185:45-106.
71. Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16(12):1446-1465.
72. Shiang R. Thompson LM, Zhu YZ, et al. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78(2):335-342.
73. Rousseau F, Bonaventure J, Legeai-Mallet L, et al. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371(6494):252-254.
74. Vajo Z, Francomano CA, Wilkin DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias. Endocr Rev. 2000;21(1):23-39.
75. Liu Z, Xu J, Colvin JS, Ornitz DM. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 2002;16(7):859-869.
76. Reddi AH. Bone morphogenetic proteins: from basic science to clinical applications. J Bone Joint Surg Am. 2001;83(suppl 1, pt 1):S1-S6.
77. Minina E, Wenzel HM, Kreschel C, et al. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development. 2001;128(22):4523-4534.
78. Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18(1):4-25.
79. Vu TH, Shipley JM, Bergers G, et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93(3):411-422.
80. Gerber HP, Ferrara N. Angiogenesis and bone growth. Trends Cardiovasc Med. 2000;10(5):223-228.
81. Turner CH. Three rules for bone adaptation to mechanical stimuli. Bone. 1998;23(5):399-407.
82. Ruff C. Growth in bone strength, body size, and muscle size in a juvenile longitudinal sample. Bone. 2003;33(3):317-329.
83. Arkin AM, Katz JF. The effects of pressure on epiphyseal growth; the mechanism of plasticity of growing bone. J Bone Joint Surg Am. 1956;38(5):1056-1076.
84. Mehlman CT, Araghi A, Roy DR. Hyphenated history: the Hueter-Volkmann law. Am J Orthop. 1997;26(11):798-800.
85. Frost HM. Biomechanical control of knee alignment: some insights from a new paradigm. Clin Orthop. 1997;(335):335-342.
86. Chenu C. Role of innervation in the control of bone remodeling. J Musculoskelet Neuronal Interact. 2004;4(2):132-134.
87. Dysart PS, Harkness EM, Herbison GP. Growth of the humerus after denervation. An experimental study in the rat. J Anat. 1989;167:147-159.
88. Pottorf JL. An experimental study of bone growth in the dog. Anat Rec. 1916;10:234-235.
89. Allison N, Brooks B. Bone atrophy. An experimental and clinical study of the changes in bone which result from non-use. Surg Gynecol Obstet. 1921;33:250-260.
90. Ring PA. The influence of the nervous system upon the growth of bones. J Bone Joint Surg Br. 1961;43:121-140.
91. Reading BD, Laor T, Salisbury SR, Lippert WC, Cornwall R. Quantification of humeral head deformity following neonatal brachial plexus palsy. J Bone Joint Surg Am. 2012;94(18):e136(1-8).
92. Moro M, van der Meulen MC, Kiratli BJ, Bachrach LK, Carter DR. Body mass is the primary determinant of midfemoral bone acquisition during adolescent growth. Bone. 1996;19(5):519-526.
93. Schoenau E, Neu CM, Mokov E, Wassmer G, Manz F. Influence of puberty on muscle area and cortical bone area and cortical of the forearm in boys and girls. J Clin Endocrinol Metab. 2000;85(3):1095-1098.
94. Schönau E. The development of the skeletal system in children and the influence of muscular strength. Horm Res. 1998;49(1):27-31.
95. Schönau E, Werhahn E, Schiedermaier U, et al. Influence of muscle strength on bone strength during childhood and adolescence. Horm Res. 1996;45(suppl 1):63-66.
96. van der Meulen MC, Ashford MW Jr, Kiratli BJ, Bachrach LK, Carter DR. Determinants of femoral geometry and structure during adolescent growth. J Orthop Res. 1996;14(1):22-29.
97. van der Meulen MC, Moro M, Kiratli BJ, Marcus R, Bachrach LK. Mechanobiology of femoral neck structure during adolescence. J Rehabil Res Dev. 2000;37(2):201-208.
98. Baron R. General principles of bone biology. In: Favus MJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 5th ed. Washington DC: American Society for Bone and Mineral Research; 2003:1-8.
99. Parfitt AM, Travers R, Rauch F, Glorieux FH. Structural and cellular changes during bone growth in healthy children. Bone. 2000;27(4):487-494.
100. Frost HM. Skeletal structural adaptations to mechanical usage (SATMU): 2. Redefining Wolff’s law: the bone modeling problem. Anat Rec. 1990;226(4):414-422.
101. Frost HM. Skeletal structural adaptations to mechanical usage (SATMU): 1. Redefining Wolff’s law: the bone modeling problem. Anat Rec. 1990;226(4):403-413.
102. Balena R, Shih MS, Parfitt AM. Bone resorption and formation on the periosteal envelope of the ilium: a histomorphometric study in healthy women. J Bone Miner Res. 1992;7(12):1475-1482.
103. Tanner JM, Hughes PC, Whitehouse RH. Radiographically determined widths of bone muscle and fat in the upper arm and calf from age 3-18 years. Ann Hum Biol. 1981;8(6):495-517.
104. Turner RT, Wakley GK, Hannon KS. Differential effects of androgens on cortical bone histomorphometry in gonadectomized male and female rats. J Orthop Res. 1990;8(4):612-617.
105. Yeh JK, Chen MM, Aloia JF. Ovariectomy-induced high turnover in cortical bone is dependent on pituitary hormone in rats. Bone. 1996;18(5):443-450.
106. Kim BT, Mosekilde L, Duan Y, et al. The structural and hormonal basis of sex differences in peak appendicular bone strength in rats. J Bone Miner Res. 2003;18(1):150-155.
107. Parfitt AM. Parathyroid hormone and periosteal bone expansion. J Bone Miner Res. 2002;17(10):1741-1743.
108. Specker B, Binkley T. Randomized trial of physical activity and calcium supplementation on bone mineral content in 3- to 5-year-old children. J Bone Miner Res. 2003;18(5):885-892.
109. Volkman SK, Galecki AT, Burke DT, et al. Quantitative trait loci for femoral size and shape in a genetically heterogenous mouse population. J Bone Miner Res. 2003;18(8):1497-1505.
110. Goodship AE, Lanyon LE, McFie H. Functional adaptation of bone to increased stress. An experimental study. J Bone Joint Surg Am. 1979;61(4):539-546.
111. Falder S, Sinclair JS, Rogers CA, Townsend PL. Long-term behavior of the free vascularized fibula following reconstruction of large bony effects. Br J Plast Surg. 2003;56(6):571-584.
Differences in bone size are established early in life, before puberty and perhaps even in utero.1 Bone begins to form when mesenchymal cells form condensations—clusters of cells that adhere through expression of adhesion molecules2 (Figure 1). Bone must be stiff, flexible enough to change shape to absorb energy, and light enough to allow mobility.1,3 Longitudinal bone growth is detrimental to bone stability, but this effect is counteracted by concomitant bone growth in width.4 Bone growth in width has not been studied as extensively, despite its paramount role in skeletal development.5
Bone growth and development are products of the complex interactions of genetic and environmental factors, including diet, hormones, and mechanical stimuli.6-9 Longitudinal bone growth is controlled by systemic and local hormones and local mechanical factors. Two models for control of bone growth in width have been suggested—the mechanostat theory (mechanical requirements regulate periosteal apposition) and the sizostat hypothesis (a master gene or set of genes regulates bone growth in width so bone reaches a preprogrammed size, independent of mechanical requirements).5
In this article, we review the most recent data regarding bone growth from the embryonic age and analyze the factors that control bone growth. An understanding of this complex system is important in identifying metabolic and developmental bone diseases10 and fracture risk.11,12
Growth Plate
The growth plate consists mainly of collagen fibrils, proteoglycans, and water, arranged to form a sort of sponge with very small pores.13 The growth plate is located between epiphyseal and metaphyseal bone at the distal end of long bones14 and is strain-rate–dependent,15,16 which means it is hard when squeezed rapidly but soft when deformed slowly. The growth plate becomes ossified after puberty and epiphyseal fusion.17
Histologically, the growth plate consists of horizontal zones of chondrocytes at different stages of differentiation.4 The germinal zone, at the epiphyseal end of the growth plate, contains resting chondrocytes, which seem crucial in orienting the underlying columns of chondrocytes and, therefore, in unidirectional bone growth, probably by secretion of a growth plate–orienting factor.14,18 Next is the proliferative zone, a matrix-rich zone in which flattened chondrocytes undergo longitudinal cell division and orient themselves in typical column-wise fashion. At some point, proliferating chondrocytes lose their capacity to divide; they start to differentiate and become prehypertrophic, coinciding with a size increase.4 Proliferating chondrocytes are located in the transition (maturation or prehypertrophic) zone. In the hypertrophic zone, round chondrocytes secrete matrix proteins in large amounts.14 This stage is characterized by an increase in intracellular calcium concentration, which is essential in the production of matrix vesicles. These vesicles, small membrane-enclosed particles, are released from chondrocytes19,20 and secrete calcium phosphates, hydroxyapatite, and matrix metalloproteinases, resulting in mineralization of the vesicles and their surrounding matrix.4 The chondrocytes in this mineralized zone eventually undergo programmed cell death (apoptosis), leaving a scaffold for new bone formation.
Longitudinal Bone Growth
Generally, bones increase in length as long as new material is being squeezed between the reserve zone of the growth plate and the zone of provisional calcification.4
Postnatal linear growth occurs in 3 phases. Phase 1 is characterized by a high rate of growth at the beginning of fetal life, and then rapid deceleration up to about 3 years; phase 2, by a lower, slowly decelerating growth rate up to puberty; and phase 3, by an increased rate of longitudinal growth until a peak is reached.14,21,22
In 1964, Park23 proposed that the structure of the epiphyseal cartilage may determine the pattern of the growing bone shaft. The changes within the hypertrophic zone are directly related to matrix mineralization, vascular invasion, and subsequent development.24 Intracellular calcium concentration increases in the hypertrophic chondrocytes in the hypertrophic zone of growth plate cartilage; at some point, these chondrocytes begin to mineralize the longitudinal septa in the surrounding matrix25 (Figure 2). At the growth cartilage junction, mononuclear cells of undetermined origin resorb the unmineralized horizontal septa of the growth cartilage. These cells are called septoclasts or chondroclasts.25,26 Blood vessels invade the area and pave the way for bone cell precursors.27 Eighty percent of the longitudinal septa of the growth cartilage is rapidly resorbed in the metaphyseal zone immediately behind the invading blood vessels, paving the way for bone cell precursors.28 Fazzalari and colleagues28 reported that about 40% of mineralized septa serves as scaffold for the formation of primary bone trabeculae; the other 60% is absorbed by chondroclasts (osteoclasts) near the vascular invasion front.
Regulation of Longitudinal Bone Growth
Longitudinal bone growth is regulated by genetic, hormonal, growth, and environment factors17,29-31 (Table). It must be controlled on at least 3 different levels.4 Level 1 is systemic control by factors such as growth hormone (GH), sex hormones, and glucocorticoids. The major systemic hormones that control longitudinal bone growth during childhood are GH, insulin-like growth factor 1 (IGF-1), the thyroid hormones triiodothyronine (T3) and thyroxine (T4), and glucocorticoids; during puberty, the sex steroids play the most significant role.14 Level 2 is local control by factors such as Indian hedgehog (Inh), parathyroid hormone–related peptide (PTHrP), and fibroblast growth factors (FGFs).14,31 Level 3 is mechanical control.4
Systemic Regulation. After birth, GH becomes an important modulator of longitudinal growth and appears to be, together with IGF-1, the central player in the hypothalamus–pituitary–growth plate axis.14 According to the original somatomedin hypothesis,32 GH stimulates hepatic production of IGF-1, which in turn promotes growth directly at the epiphyseal plate.17 GH acts on resting zone chondrocytes and is responsible for local IGF-1 production, which stimulates clonal expansion of proliferating chondrocytes in an autocrine/paracrine manner.33 Infusion of GH or IGF-1 shortens stem- and proliferating-cell cycle times in the growth plate of hypophysectomized rats and decreases the duration of the hypertrophic differentiation phase, with GH being more effective.17 According to the experimental study of Hunziker and colleagues,34 GH or IGF-1 treatment restores mean cell volume and height, but the growth rate is not normalized by either hormone.
Thyroid hormones also play a vital role in bone growth. T3 and, to a lesser extent, T4 are crucial in normal bone maturation.30,35 Childhood hypothyroidism causes growth failure; growth failure may develop insidiously, but, once established, it is severe.17 On the other hand, hyperthyroidism increases the growth rate in children but also leads to premature growth plate fusion and short stature.36,37 T3 seems to stimulate recruitment of cells from the germinal zone to the proliferating zone and facilitates differentiation of growth plate chondrocytes.38-40 Its precursor, T4, increases the number of [3H]methylthymidine-labeled chondrocyte nuclei and [35S]incorporation in Snell dwarf mice growth plates, suggesting a stimulatory role in chondrocyte proliferation and differentiation.41
Glucocorticoids suppress growth by modifying the GH/IGF-1 pathway at different levels.17 Silvestrini and colleagues42 localized the glucocorticoid receptor in rat bone cells, including chondrocytes. The glucocorticoid receptor was also localized by Abu and colleagues43 in human growth plates, especially in hypertrophic chondrocytes, suggesting direct effects of glucocorticoids on the growth plate. An excess of glucocorticoids enhances bone resorption, inhibits osteoblast activity, and reduces bone matrix production to retard growth in children.44,45 Excess glucocorticoids also induce apoptosis of osteoblasts and osteocytes in rabbit trabecular bone46 and osteoblasts in rat long bones,47 resulting in an almost complete absence of new bone formation.17 In addition, glucocorticoids induce sex hormone deficiency and alter vitamin D metabolism, leading to deleterious effects on growth and skeletal integrity.48 Excess glucocorticoids modify the GH/IGF-1 pathway at different levels, suppressing growth.17 In contrast, low levels of glucocorticoids, as in familial glucocorticoid deficiency, are associated with tall stature.49
Longitudinal bone growth is also based on sex hormones, especially during puberty.17 In rats, estrogen depletion stimulates longitudinal growth, whereas estrogen administration inhibits longitudinal growth.50-52 Nilsson and colleagues53 studied ovariectomized immature rabbits treated with either estrogen or the selective estrogen receptor modulator raloxifene and found reduced chondrocyte proliferation and growth plate height as well as accelerated growth plate senescence. Many experimental studies have concluded that estrogen can inhibit longitudinal growth in the absence of GH.51,54,55
Androgens can directly influence growth plate function and may account for some skeletal differences between males and females.56-58 Unlike estrogens, androgens stimulate longitudinal growth, as shown in several studies that assessed the effect of administering nonaromatizable androgens on longitudinal growth in boys with constitutionally delayed growth.59,60
Local Regulation. Inh, a master regulator of bone development, coordinates chondrocyte proliferation, chondrocyte differentiation, and osteoblast differentiation.31 Inh belongs to the hedgehog protein family, which plays a crucial role in embryonic patterning and development.4 The proliferative effect of Inh is likely to be direct action on chondrocytes.31 In 1996, Vortkamp and colleagues61 reported that misexpression of Inh in chicken long bones blocked chondrocyte differentiation. More recently, St-Jacques and colleagues62 studied Inh-null mutant mice and found failure of both chondrocyte differentiation and osteoblast development. Inh is now thought to coordinate endochondral ossification, regulating chondrocyte proliferation and differentiation and osteoblast differentiation and coupling chondrogenesis and osteogenesis.62,63
PTHrP acts primarily to keep proliferating chondrocytes in the proliferative pool.31 Mice that did not express PTHrP showed accelerated chondrocyte differentiation leading to dwarfism.64 On the other hand, ectopic expression of PTHrP in the growth plate inhibited chondrocyte differentiation, resulting in a smaller cartilaginous skeleton compared with wild-type mice.65 PTHrP appears to regulate the rate of programmed chondrocyte differentiation in developing endochondral bone and at the level of the growth plate.64,66-69
The family of FGFs, which are major regulators of embryonic bone development, has at least 22 members.70,71 Achondroplasia, the most common type of dwarfism, is caused by an activating mutation in FGF receptor 3 (FGFR3).72-74 FGF18 deficiency also leads to delayed ossification and decreased expression of osteogenic markers.75
Bone morphogenetic proteins (BMPs) are recognized as important regulators of growth, differentiation, and morphogenesis during embryology.76 In 2001, Minina and colleagues77 showed that normal chondrocyte proliferation requires parallel signaling of both Inh and BMPs and that BMPs can inhibit chondrocyte differentiation independently of the Inh/PTHrP pathway.
Vascular endothelial growth factor (VEGF), a chemoattractant for endothelial cells, is one of the most important growth factors for endothelial cells.78 VEGF is a key player in the actions that occur during the end stage of endochondral bone formation; these actions include terminal differentiation of chondrocytes, vascular invasion, chondrocyte apoptosis, and replacement of chondrocytes with bone.27,79,80 When Gerber and colleagues27 inactivated VEGF in 24-day-old mice, they noticed suppressed blood vessel invasion and trabecular bone formation concomitant with an increased width of the hypertrophic zone.
Mechanical Regulation. Mechanical forces influence bone formation and adaptation.81 Growth rates from early infancy through late adolescence were found to be strongly correlated between an appropriate measure of mechanical loading (body size, or body weight–bone length) and bone strength (assessed by section modulus).82 The observation that compression inhibits bone growth was well known to the ancient Romans.83 In the 19th century, the Hueter-Volkmann law was proclaimed. This law is well known to pediatric orthopedic surgeons and is the basis of growth modulation for correcting angular deformities of the lower extremities and spinal deformities.4,84
If compression always inhibited bone growth, as it was believed, growth plates would be extremely unstable, as any slight deviation from the straight alignment of the long bones of the lower extremities would induce a vicious circle of positive feedback and result in catastrophic deformities.4 Mild compression leads to increased, not decreased, growth. Nevertheless, when compression on one side of the growth plate exceeds a certain level, growth is indeed suppressed, and the lesion begins to worsen.4
In 1997, Frost85 proposed using a single graph that combines the clinical observation of mechanical forces affecting longitudinal bone growth. Both mild tension and mild compression induce bone growth, whereas heavy compression inhibits growth (Figure 3).
Three rules describe bone adaptation in mathematical terms. First, bone adaptation is driven by dynamic, not static, loading. Second, only a short period of mechanical loading is needed to initiate an adaptive response (extending the loading period has a diminishing effect on further bone adaptation). Third, bone cells accommodate to a customary mechanical loading environment, making them less responsive to routine loading signals.81
Also playing a significant role in bone physiology is the nervous system, with leptin-dependent central control of bone formation via the sympathetic system.86 Several investigators have tried to determine the effect of muscle activity on bone growth in length.87 Pottorf88 in 1916 and Allison and Brooks89 in 1921 were among the first to study this correlation; they concluded that long bones grow less after denervation. On the other hand, Ring90 in 1961 reported that, despite innervation, longitudinal bone growth was increased. Investigators in more recent studies have advanced the idea that the nervous system plays a negative role in bone physiology. Dysart and colleagues87 showed that muscle pull affects periosteal tension and, consequently, bone form and growth in length. In a clinical study involving 32 children with neonatal brachial plexus injury,91 the ratio of skewness between the affected humeral head and the contralateral normal head was calculated. Skewness was determined by dividing the anterior area of the humeral head by the posterior area. There was a significant preoperative difference between the 2 sides, but the skewness ratio was significantly improved after surgery.
Bone Growth in Width
Bone growth in width has not received as much attention as longitudinal bone growth. Several studies have indicated that body mass and muscle strength have important influences on long bone strength in children and adolescents.92-97 As bone width changes only slowly after the growth period, bone growth in width is one of the most important determinants of bone strength throughout life.4 It is clear that, if bones grew in length without increasing in width, they would become unstable and break.4
Histologically, osteoblasts add mineralized tissue to the outer (periosteal) bone surface. This process is periosteal apposition.98 The periosteum has an outer layer, composed mainly of fibrous tissue, and an inner layer, the cambium, which harbors osteogenic cells.4 In children, bone formation is continuous, which is the hallmark of modeling99,100; in adults, periosteal bone may undergo cyclical resorption and formation, which are characteristic of remodeling.101,102
Macroscopically, bone grows rapidly during early life; then, growth continuously slows down until reaching a nadir during early school age.4 It is clear that wider bones must have higher midshaft periosteal apposition rates, as this is how they become wider.4,103
Regulation of Bone Growth in Width (Table)
Systemic Regulation. Periosteal apposition at diaphyseal bone sites is stimulated by androgen and GH and inhibited by estrogens.104-106 In an experimental study, Turner and colleagues104 found that androgen treatment stimulated bone formation in orchiectomized rats and suppressed bone formation in ovariectomized rats. A large dose of diethylstilbestrol also suppressed bone formation in ovariectomized rats. Parathyroid hormone is associated with faster periosteal expansion in adults, according to Parfitt.107 In addition, nutrition with high calcium intake has the same effects on children, especially those with high levels of physical activity.108
Local Regulation. Given that periosteal bone development is site-specific, whereas systemic hormones and nutrition are blind to structure,4 it is clear that local regulation is key to bone growth in width. Genetic heritage seems to have an overwhelming effect on periosteal bone development. Volkman and colleagues,109 who experimented with various genetic markers in rats, concluded that genetic control of cortical bone geometry is complex and that femoral size and shape may be influenced by different but overlapping groups of polymorphic loci.
Mechanical Regulation. Mechanical forces seem to be very important in determining bone width. For example, the difference in width between femur and humerus can be explained by the different mechanical forces acting on each bone. This perspective is supported by Ruff,82 who showed that the correlation of body size (body weight–bone length) and bone strength is stronger in the femur than in the humerus.
The vital role of mechanical forces in bone growth in width is also supported by results of a study by Goodship and colleagues,110 who overloaded the radius of young pigs by partially removing the ulna. They showed that the radius was strengthened by rapid periosteal apposition. This effect has also been noticed in the clinical setting, when the tibia is replaced with the fibula, which quickly hypertrophies in order to resemble the tibia.111
Conclusion
Longitudinal bone growth has been extensively studied. Systemic and local hormonal pathways control bone growth in a complicated regulation system. Mechanical loading is also strongly correlated with longitudinal bone growth. Bone growth in width has received less attention. Despite its importance in bone stability, periosteal development—and periosteal apposition and resorption more specifically—has not received enough attention. Researchers need to clarify the role of genetic factors affecting periosteal development.
Differences in bone size are established early in life, before puberty and perhaps even in utero.1 Bone begins to form when mesenchymal cells form condensations—clusters of cells that adhere through expression of adhesion molecules2 (Figure 1). Bone must be stiff, flexible enough to change shape to absorb energy, and light enough to allow mobility.1,3 Longitudinal bone growth is detrimental to bone stability, but this effect is counteracted by concomitant bone growth in width.4 Bone growth in width has not been studied as extensively, despite its paramount role in skeletal development.5
Bone growth and development are products of the complex interactions of genetic and environmental factors, including diet, hormones, and mechanical stimuli.6-9 Longitudinal bone growth is controlled by systemic and local hormones and local mechanical factors. Two models for control of bone growth in width have been suggested—the mechanostat theory (mechanical requirements regulate periosteal apposition) and the sizostat hypothesis (a master gene or set of genes regulates bone growth in width so bone reaches a preprogrammed size, independent of mechanical requirements).5
In this article, we review the most recent data regarding bone growth from the embryonic age and analyze the factors that control bone growth. An understanding of this complex system is important in identifying metabolic and developmental bone diseases10 and fracture risk.11,12
Growth Plate
The growth plate consists mainly of collagen fibrils, proteoglycans, and water, arranged to form a sort of sponge with very small pores.13 The growth plate is located between epiphyseal and metaphyseal bone at the distal end of long bones14 and is strain-rate–dependent,15,16 which means it is hard when squeezed rapidly but soft when deformed slowly. The growth plate becomes ossified after puberty and epiphyseal fusion.17
Histologically, the growth plate consists of horizontal zones of chondrocytes at different stages of differentiation.4 The germinal zone, at the epiphyseal end of the growth plate, contains resting chondrocytes, which seem crucial in orienting the underlying columns of chondrocytes and, therefore, in unidirectional bone growth, probably by secretion of a growth plate–orienting factor.14,18 Next is the proliferative zone, a matrix-rich zone in which flattened chondrocytes undergo longitudinal cell division and orient themselves in typical column-wise fashion. At some point, proliferating chondrocytes lose their capacity to divide; they start to differentiate and become prehypertrophic, coinciding with a size increase.4 Proliferating chondrocytes are located in the transition (maturation or prehypertrophic) zone. In the hypertrophic zone, round chondrocytes secrete matrix proteins in large amounts.14 This stage is characterized by an increase in intracellular calcium concentration, which is essential in the production of matrix vesicles. These vesicles, small membrane-enclosed particles, are released from chondrocytes19,20 and secrete calcium phosphates, hydroxyapatite, and matrix metalloproteinases, resulting in mineralization of the vesicles and their surrounding matrix.4 The chondrocytes in this mineralized zone eventually undergo programmed cell death (apoptosis), leaving a scaffold for new bone formation.
Longitudinal Bone Growth
Generally, bones increase in length as long as new material is being squeezed between the reserve zone of the growth plate and the zone of provisional calcification.4
Postnatal linear growth occurs in 3 phases. Phase 1 is characterized by a high rate of growth at the beginning of fetal life, and then rapid deceleration up to about 3 years; phase 2, by a lower, slowly decelerating growth rate up to puberty; and phase 3, by an increased rate of longitudinal growth until a peak is reached.14,21,22
In 1964, Park23 proposed that the structure of the epiphyseal cartilage may determine the pattern of the growing bone shaft. The changes within the hypertrophic zone are directly related to matrix mineralization, vascular invasion, and subsequent development.24 Intracellular calcium concentration increases in the hypertrophic chondrocytes in the hypertrophic zone of growth plate cartilage; at some point, these chondrocytes begin to mineralize the longitudinal septa in the surrounding matrix25 (Figure 2). At the growth cartilage junction, mononuclear cells of undetermined origin resorb the unmineralized horizontal septa of the growth cartilage. These cells are called septoclasts or chondroclasts.25,26 Blood vessels invade the area and pave the way for bone cell precursors.27 Eighty percent of the longitudinal septa of the growth cartilage is rapidly resorbed in the metaphyseal zone immediately behind the invading blood vessels, paving the way for bone cell precursors.28 Fazzalari and colleagues28 reported that about 40% of mineralized septa serves as scaffold for the formation of primary bone trabeculae; the other 60% is absorbed by chondroclasts (osteoclasts) near the vascular invasion front.
Regulation of Longitudinal Bone Growth
Longitudinal bone growth is regulated by genetic, hormonal, growth, and environment factors17,29-31 (Table). It must be controlled on at least 3 different levels.4 Level 1 is systemic control by factors such as growth hormone (GH), sex hormones, and glucocorticoids. The major systemic hormones that control longitudinal bone growth during childhood are GH, insulin-like growth factor 1 (IGF-1), the thyroid hormones triiodothyronine (T3) and thyroxine (T4), and glucocorticoids; during puberty, the sex steroids play the most significant role.14 Level 2 is local control by factors such as Indian hedgehog (Inh), parathyroid hormone–related peptide (PTHrP), and fibroblast growth factors (FGFs).14,31 Level 3 is mechanical control.4
Systemic Regulation. After birth, GH becomes an important modulator of longitudinal growth and appears to be, together with IGF-1, the central player in the hypothalamus–pituitary–growth plate axis.14 According to the original somatomedin hypothesis,32 GH stimulates hepatic production of IGF-1, which in turn promotes growth directly at the epiphyseal plate.17 GH acts on resting zone chondrocytes and is responsible for local IGF-1 production, which stimulates clonal expansion of proliferating chondrocytes in an autocrine/paracrine manner.33 Infusion of GH or IGF-1 shortens stem- and proliferating-cell cycle times in the growth plate of hypophysectomized rats and decreases the duration of the hypertrophic differentiation phase, with GH being more effective.17 According to the experimental study of Hunziker and colleagues,34 GH or IGF-1 treatment restores mean cell volume and height, but the growth rate is not normalized by either hormone.
Thyroid hormones also play a vital role in bone growth. T3 and, to a lesser extent, T4 are crucial in normal bone maturation.30,35 Childhood hypothyroidism causes growth failure; growth failure may develop insidiously, but, once established, it is severe.17 On the other hand, hyperthyroidism increases the growth rate in children but also leads to premature growth plate fusion and short stature.36,37 T3 seems to stimulate recruitment of cells from the germinal zone to the proliferating zone and facilitates differentiation of growth plate chondrocytes.38-40 Its precursor, T4, increases the number of [3H]methylthymidine-labeled chondrocyte nuclei and [35S]incorporation in Snell dwarf mice growth plates, suggesting a stimulatory role in chondrocyte proliferation and differentiation.41
Glucocorticoids suppress growth by modifying the GH/IGF-1 pathway at different levels.17 Silvestrini and colleagues42 localized the glucocorticoid receptor in rat bone cells, including chondrocytes. The glucocorticoid receptor was also localized by Abu and colleagues43 in human growth plates, especially in hypertrophic chondrocytes, suggesting direct effects of glucocorticoids on the growth plate. An excess of glucocorticoids enhances bone resorption, inhibits osteoblast activity, and reduces bone matrix production to retard growth in children.44,45 Excess glucocorticoids also induce apoptosis of osteoblasts and osteocytes in rabbit trabecular bone46 and osteoblasts in rat long bones,47 resulting in an almost complete absence of new bone formation.17 In addition, glucocorticoids induce sex hormone deficiency and alter vitamin D metabolism, leading to deleterious effects on growth and skeletal integrity.48 Excess glucocorticoids modify the GH/IGF-1 pathway at different levels, suppressing growth.17 In contrast, low levels of glucocorticoids, as in familial glucocorticoid deficiency, are associated with tall stature.49
Longitudinal bone growth is also based on sex hormones, especially during puberty.17 In rats, estrogen depletion stimulates longitudinal growth, whereas estrogen administration inhibits longitudinal growth.50-52 Nilsson and colleagues53 studied ovariectomized immature rabbits treated with either estrogen or the selective estrogen receptor modulator raloxifene and found reduced chondrocyte proliferation and growth plate height as well as accelerated growth plate senescence. Many experimental studies have concluded that estrogen can inhibit longitudinal growth in the absence of GH.51,54,55
Androgens can directly influence growth plate function and may account for some skeletal differences between males and females.56-58 Unlike estrogens, androgens stimulate longitudinal growth, as shown in several studies that assessed the effect of administering nonaromatizable androgens on longitudinal growth in boys with constitutionally delayed growth.59,60
Local Regulation. Inh, a master regulator of bone development, coordinates chondrocyte proliferation, chondrocyte differentiation, and osteoblast differentiation.31 Inh belongs to the hedgehog protein family, which plays a crucial role in embryonic patterning and development.4 The proliferative effect of Inh is likely to be direct action on chondrocytes.31 In 1996, Vortkamp and colleagues61 reported that misexpression of Inh in chicken long bones blocked chondrocyte differentiation. More recently, St-Jacques and colleagues62 studied Inh-null mutant mice and found failure of both chondrocyte differentiation and osteoblast development. Inh is now thought to coordinate endochondral ossification, regulating chondrocyte proliferation and differentiation and osteoblast differentiation and coupling chondrogenesis and osteogenesis.62,63
PTHrP acts primarily to keep proliferating chondrocytes in the proliferative pool.31 Mice that did not express PTHrP showed accelerated chondrocyte differentiation leading to dwarfism.64 On the other hand, ectopic expression of PTHrP in the growth plate inhibited chondrocyte differentiation, resulting in a smaller cartilaginous skeleton compared with wild-type mice.65 PTHrP appears to regulate the rate of programmed chondrocyte differentiation in developing endochondral bone and at the level of the growth plate.64,66-69
The family of FGFs, which are major regulators of embryonic bone development, has at least 22 members.70,71 Achondroplasia, the most common type of dwarfism, is caused by an activating mutation in FGF receptor 3 (FGFR3).72-74 FGF18 deficiency also leads to delayed ossification and decreased expression of osteogenic markers.75
Bone morphogenetic proteins (BMPs) are recognized as important regulators of growth, differentiation, and morphogenesis during embryology.76 In 2001, Minina and colleagues77 showed that normal chondrocyte proliferation requires parallel signaling of both Inh and BMPs and that BMPs can inhibit chondrocyte differentiation independently of the Inh/PTHrP pathway.
Vascular endothelial growth factor (VEGF), a chemoattractant for endothelial cells, is one of the most important growth factors for endothelial cells.78 VEGF is a key player in the actions that occur during the end stage of endochondral bone formation; these actions include terminal differentiation of chondrocytes, vascular invasion, chondrocyte apoptosis, and replacement of chondrocytes with bone.27,79,80 When Gerber and colleagues27 inactivated VEGF in 24-day-old mice, they noticed suppressed blood vessel invasion and trabecular bone formation concomitant with an increased width of the hypertrophic zone.
Mechanical Regulation. Mechanical forces influence bone formation and adaptation.81 Growth rates from early infancy through late adolescence were found to be strongly correlated between an appropriate measure of mechanical loading (body size, or body weight–bone length) and bone strength (assessed by section modulus).82 The observation that compression inhibits bone growth was well known to the ancient Romans.83 In the 19th century, the Hueter-Volkmann law was proclaimed. This law is well known to pediatric orthopedic surgeons and is the basis of growth modulation for correcting angular deformities of the lower extremities and spinal deformities.4,84
If compression always inhibited bone growth, as it was believed, growth plates would be extremely unstable, as any slight deviation from the straight alignment of the long bones of the lower extremities would induce a vicious circle of positive feedback and result in catastrophic deformities.4 Mild compression leads to increased, not decreased, growth. Nevertheless, when compression on one side of the growth plate exceeds a certain level, growth is indeed suppressed, and the lesion begins to worsen.4
In 1997, Frost85 proposed using a single graph that combines the clinical observation of mechanical forces affecting longitudinal bone growth. Both mild tension and mild compression induce bone growth, whereas heavy compression inhibits growth (Figure 3).
Three rules describe bone adaptation in mathematical terms. First, bone adaptation is driven by dynamic, not static, loading. Second, only a short period of mechanical loading is needed to initiate an adaptive response (extending the loading period has a diminishing effect on further bone adaptation). Third, bone cells accommodate to a customary mechanical loading environment, making them less responsive to routine loading signals.81
Also playing a significant role in bone physiology is the nervous system, with leptin-dependent central control of bone formation via the sympathetic system.86 Several investigators have tried to determine the effect of muscle activity on bone growth in length.87 Pottorf88 in 1916 and Allison and Brooks89 in 1921 were among the first to study this correlation; they concluded that long bones grow less after denervation. On the other hand, Ring90 in 1961 reported that, despite innervation, longitudinal bone growth was increased. Investigators in more recent studies have advanced the idea that the nervous system plays a negative role in bone physiology. Dysart and colleagues87 showed that muscle pull affects periosteal tension and, consequently, bone form and growth in length. In a clinical study involving 32 children with neonatal brachial plexus injury,91 the ratio of skewness between the affected humeral head and the contralateral normal head was calculated. Skewness was determined by dividing the anterior area of the humeral head by the posterior area. There was a significant preoperative difference between the 2 sides, but the skewness ratio was significantly improved after surgery.
Bone Growth in Width
Bone growth in width has not received as much attention as longitudinal bone growth. Several studies have indicated that body mass and muscle strength have important influences on long bone strength in children and adolescents.92-97 As bone width changes only slowly after the growth period, bone growth in width is one of the most important determinants of bone strength throughout life.4 It is clear that, if bones grew in length without increasing in width, they would become unstable and break.4
Histologically, osteoblasts add mineralized tissue to the outer (periosteal) bone surface. This process is periosteal apposition.98 The periosteum has an outer layer, composed mainly of fibrous tissue, and an inner layer, the cambium, which harbors osteogenic cells.4 In children, bone formation is continuous, which is the hallmark of modeling99,100; in adults, periosteal bone may undergo cyclical resorption and formation, which are characteristic of remodeling.101,102
Macroscopically, bone grows rapidly during early life; then, growth continuously slows down until reaching a nadir during early school age.4 It is clear that wider bones must have higher midshaft periosteal apposition rates, as this is how they become wider.4,103
Regulation of Bone Growth in Width (Table)
Systemic Regulation. Periosteal apposition at diaphyseal bone sites is stimulated by androgen and GH and inhibited by estrogens.104-106 In an experimental study, Turner and colleagues104 found that androgen treatment stimulated bone formation in orchiectomized rats and suppressed bone formation in ovariectomized rats. A large dose of diethylstilbestrol also suppressed bone formation in ovariectomized rats. Parathyroid hormone is associated with faster periosteal expansion in adults, according to Parfitt.107 In addition, nutrition with high calcium intake has the same effects on children, especially those with high levels of physical activity.108
Local Regulation. Given that periosteal bone development is site-specific, whereas systemic hormones and nutrition are blind to structure,4 it is clear that local regulation is key to bone growth in width. Genetic heritage seems to have an overwhelming effect on periosteal bone development. Volkman and colleagues,109 who experimented with various genetic markers in rats, concluded that genetic control of cortical bone geometry is complex and that femoral size and shape may be influenced by different but overlapping groups of polymorphic loci.
Mechanical Regulation. Mechanical forces seem to be very important in determining bone width. For example, the difference in width between femur and humerus can be explained by the different mechanical forces acting on each bone. This perspective is supported by Ruff,82 who showed that the correlation of body size (body weight–bone length) and bone strength is stronger in the femur than in the humerus.
The vital role of mechanical forces in bone growth in width is also supported by results of a study by Goodship and colleagues,110 who overloaded the radius of young pigs by partially removing the ulna. They showed that the radius was strengthened by rapid periosteal apposition. This effect has also been noticed in the clinical setting, when the tibia is replaced with the fibula, which quickly hypertrophies in order to resemble the tibia.111
Conclusion
Longitudinal bone growth has been extensively studied. Systemic and local hormonal pathways control bone growth in a complicated regulation system. Mechanical loading is also strongly correlated with longitudinal bone growth. Bone growth in width has received less attention. Despite its importance in bone stability, periosteal development—and periosteal apposition and resorption more specifically—has not received enough attention. Researchers need to clarify the role of genetic factors affecting periosteal development.
1. Seeman E. Structural basis of growth-related gain and age-related loss of bone strength. Rheumatology. 2008;47(suppl 4):iv2-8.
2. Hall BK, Miyake T. All for one and one for all: condensations and the initiation of skeletal development. Bioessays. 2000;22(2):138-147.
3. Currey JD. Bones: Structure and Mechanics. Princeton, NJ: Princeton University Press; 2002.
4. Rauch F. Bone growth in length and width: the yin and yang of bone stability. J Musculoskelet Neuronal Interact. 2005;5(3):194-201.
5. Seeman E. Periosteal bone formation—a neglected determinant of bone strength. N Engl J Med. 2003;349(4):320-323.
6. Arden NK Spector TD. Genetic influences on muscle strength and bone mineral density: a twin study. J Bone Miner Res. 1997;12(12):2076-2081.
7. Biewener AA, Bertram JEA. Mechanical loading and bone growth in vivo. In: Hall BK, ed. Bone, Vol 7: Bone Growth—B. Boca Raton, FL: CRC Press; 1993:1-36.
8. McGuigan FE, Murray L, Gallagher A, et al. Genetic and environmental determinants of peak bone mass in young men and women. J Bone Miner Res. 2002;17(7):1273-1279.
9. Slemenda CW, Reister TK, Hui SL, Miller JZ, Christian JC, Johnston CC Jr. Influences on skeletal mineralization in children and adolescents: evidence for varying effects of sexual maturation and physical activity. J Pediatr. 1994;125(2):201-207.
10. Schoenau E, Neu CM, Beck B, Manz F, Rauch F. Bone mineral content per muscle cross-sectional area as an index of the functional muscle–bone unit. J Bone Miner Res. 2002;17(6):1095-1101.
11. Rauch F, Neu C, Manz F, Schoenau E. The development of metaphyseal cortex—implications for distal radius fractures during growth. J Bone Miner Res. 2001;16(8):1547-1555.
12. Skaggs DL, Loro ML, Pitukcheewanont P, Tolo V, Gilsanz V. Increased body weight and decreased radial cross-sectional dimensions in girls with forearm fractures. J Bone Miner Res. 2001;16(7):1337-1342.
13. Allen DM, Mao JJ. Heterogenous nanostructural and nanoelastic properties of pericellular and interterritorial matrices of chondrocytes by atomic force microscopy. J Struct Biol. 2004;145(3):196-204.
14. van der Eerden BC, Karperien M, Wit JM. Systemic and local regulation of the growth plate. Endocr Rev. 2003;24(6):782-801.
15. Li LP, Herzog W. Strain-rate dependence of cartilage stiffness in unconfined compression: the role of fibril reinforcement versus tissue volume change in fluid pressurization. J Biomech. 2004;37(3):375-382.
16. Cohen B, Chorney GS, Phillips DP, Dick HM, Mow VC. Compressive stress-relaxation behavior of bovine growth plate may be described by the non-linear biphasic theory. J Orthop Res. 1994;12(6):804-813.
17. Robson H, Siebler T, Shalet SM, Williams GR. Interactions between GH, IGF-Ι, glucocorticoids and thyroid hormones during skeletal growth. Pediatr Res. 2002;52(2):137-147.
18. Abad V, Meyers JL, Weise M, et al. The role of the resting zone in growth plate chondrogenesis. Endocrinology. 2002;143(5):1851-1857.
19. Wang W, Kirsch T. Retinoic acid stimulates annexin-mediated growth plate chondrocyte mineralization. J Cell Biol. 2002;157(6):1061-1069.
20. Anderson HC. Matrix vesicles and calcification. Curr Rheumatol Rep. 2003;5(3):222-226.
21. Tanner JM. The adolescent spurt in animals. In: Thomas CC, ed. Growth at Adolescence. Oxford, UK: Blackwell; 1962:223-239.
22. Drop SL, De Waal WJ, De Muinck Keizer-Schrama SM. Sex steroid treatment of constitutionally tall stature. Endocr Rev. 1998;19(5):540-558.
23. Park EA. The imprinting of nutritional disturbances on the growing bone. Pediatrics. 1964;33(suppl):815-862.
24. Buckwalter JA, Mower D, Unqar R, Schaeffer J, Ginsberg B. Morphometric analysis of chondrocyte hypertrophy. J Bone Joint Surg Am. 1986;68(2):243-255.
25. Sawae Y, Sahara T, Sasaki T. Osteoclast differentiation at the growth plate cartilage–trabecular bone junction in newborn rat femur. J Electron Microsc. 2003;52(6):493-502.
26. Lee ER, Lamplugh L, Shepard NL, Mort JS. The septoclast, a cathepsin B–rich cell involved in the resorption of growth plate cartilage. J Histochem Cytochem. 1995;43(5):525-536.
27. Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5(6):623-628.
28. Fazzalari NL, Moore AJ, Byers S, Byard RW. Quantitative analysis of trabecular morphogenesis in the human costochondral junction during the postnatal period in normal subjects. Anat Rec. 1997;248(1):1-12.
29. Cancedda R, Descalzi Cancedda F, Castagnola P. Chondrocyte differentiation. Int Rev Cytol. 1995;159:265-358.
30. Stevens DA, Williams GR. Hormone regulation of chondrocyte differentiation and endochondral bone formation. Mol Cell Endocrinol. 1999;151(1-2):195-204.
31. Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423(6937):332-336.
32. Daughaday WH, Hall K, Raben MS, Salmon WD Jr, van den Brande JL, van Wyk JJ. Somatomedin: proposed designation for sulphation factor. Nature. 1972;235(5333):107.
33. Isaksson OG, Lindahl A, Nilsson A, Isqaard J. Mechanism of the stimulatory effect of the growth hormone on longitudinal bone growth. Endocr Rev. 1987;8(4):426-438.
34. Hunziker EB, Wagner J, Zapf J. Differential effects of insulin-like growth factor I and growth hormone on developmental stages of rat growth plate chondrocytes in vivo. J Clin Invest. 1994;93(3):1078-1086.
35. Underwood LE, van Wijk JJ. Normal and aberrant growth. In: Wilson JD, Foster DW, eds. Textbook of Endocrinology. Philadelphia, PA: Saunders; 1992:1079-1138.
36. Rivkees SA, Bode HH, Crawford JD. Long-term growth in juvenile acquired hypothyroidism: the failure to achieve normal adult stature. N Engl J Med. 1988;318(10):599-602.
37. Segni M, Leonardi E, Mazzoncini B, Pucarelli I, Pasquino AM. Special features of Graves’ disease in early childhood. Thyroid. 1999;9(9):871-877.
38. Burch WM, Van Wyk JJ. Triiodothyronine stimulates cartilage growth and maturation by different mechanisms. Am J Physiol. 1987;252(2, pt 1):E176-E182.
39. Lewinson D, Bialik GM, Hochberg Z. Differential effects of hypothyroidism on the cartilage and the osteogenic process in the mandipular condyle: recovery by growth hormone and thyroxine. Endocrinology. 1994;135(4):1504-1510.
40. Wakita R, Izumi T, Itoman M. Thyroid hormone–induced chondrocyte terminal differentiation in rat femur organ culture. Cell Tissue Res. 1998;293(2):357-364.
41. Smeets T, van Buul-Offers S. Influence of growth hormone and thyroxine on cell kinetics in the proximal tibial growth plate of Snell dwarf mice. Cell Tissue Kinet. 1986;19(2):161-170.
42. Silvestrini G, Mocetti P, Ballanti P, Di Grezia R, Bonucci E. Cytochemical demonstration of the glucocorticoid receptor in skeletal cells of the rat. Endocr Res. 1999;25(1):117-128.
43. Abu EO, Horner A, Kusec V, Triffitt JT, Compston JE. The localization of the functional glucocorticoid receptor alpha in human bone. J Clin Endocrinol Metab. 2000;85(2):883-889.
44. Magiakou MA, Mastorakos G, Chrousos GP. Final stature in patients with endogenous Cushing’s syndrome. J Clin Endocrinol Metab. 1994;79(4):1082-1085.
45. Avioli LV. Glucocorticoid effects on statural growth. Br J Rheumatol. 1993;32(suppl 2):27-30.
46. Eberhardt AW, Yeager-Jones A, Blair HC. Regional trabecular bone matrix degeneration and osteocyte death in femora if glucocorticoid-treated rabbits. Endocrinology. 2001;142(3):1333-1340.
47. Silvestrini G, Ballanti P, Patacchioli FR, et al. Evaluation of apoptosis and the glucocorticoid receptor in the cartilage growth plate and metaphyseal bone cells of rats after high-dose treatment with corticosterone. Bone. 2000;26(1):33-42.
48. Montecucco C, Caporali R, Caprotti P, Caprotti M, Notario A. Sex hormones and bone metabolism in postmenopausal rheumatoid arthritis treated with two different glucocorticoids. J Rheumatol. 1992;19(12):1895-1900.
49. Bello CE, Garrett SD. Therapeutic issues in oral glucocorticoid use. Lippincotts Prim Care Pract. 1999;3(3):333-341.
50. Turner RT, Riggs BL, Speisberg TC. Skeletal effects of estrogen. Endocr Rev. 1994;15(3):275-300.
51. Gevers EF, Wit JM, Robinson IC. Effect of gonadectomy on growth and GH responsiveness in dwarf rats. J Endocrinol. 1995;145(1):69-79.
52. van der Eerden BC, Emons J, Ahmed S, et al. Evidence for genomic and non-genomic actions of estrogens in growth plate regulation in female and male rats at the onset of sexual maturation. J Endocrinol. 2002;175(2):277-288.
53. Nilsson O, Falk J, Ritzen EM, Baron J, Savendahl L. Raloxifene acts as an estrogen agonist on the rabbit growth plate. Endocrinology. 2003;144(4):1481-1485.
54. Strickland AL, Sprinz H. Studies of the influence of estradiol and growth hormone on the hypophysectomized immature rat epiphyseal cartilage growth plate. Am J Obstet Gynecol. 1973;115(4):471-477.
55. Jansson JO, Eden S, Isaksson O. Sites of action of testosterone and estradiol on longitudinal bone growth. Am J Physiol. 1983;244(2):E135-E140.
56. Abu EO, Horner A, Kusec V, Triffitt JT, Compston JE. The localization of androgen receptors in human bone. J Clin Endocrinol Metab. 1997;82(10):3493-3497.
57. Noble B, Routledge J, Stevens H, Hughes I, Jacobson W. Androgen receptors in bone-forming tissue. Horm Res. 1999;51(1):31-36.
58. van der Eerden BC, van Til NP, Brinkmann AO, Lowik CW, Wit JM, Karperien M. Sex differences in the expression of the androgen receptor in the tibial growth plate and metaphyseal bone of the rat. Bone. 2002;30(6):891-896.
59. Cassorla FG, Skerda MC, Valk IM, Hung W, Cutler GB Jr, Loriaux DL. The effects of sex steroids on ulnar growth during adolescence. J Clin Endocrinol Metab. 1984;58(4):717-720.
60. Zung A, Phillip M, Chalew SA, Palese T, Kowarski AA, Zadik Z. Testosterone effect on growth and growth mediators of the GF–IGF-I axis in the liver and epiphyseal growth plate of juvenile rats. J Mol Endocrinol. 1999;23(2):209-221.
61. Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273(5275):613-622.
62. St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and essential for bone formation. Genes Dev. 1999;13(16):2072-2086.
63. Karp SJ Schipani E, St-Jacques B, Hunzelman J, Kronenberg H, Mcmahon AP. Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone related-protein–dependent and –independent pathways. Development. 2000;127(3):543-548.
64. Karaplis AC, Luz A, Glowacki J, et al. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone–related peptide gene. Genes Dev. 1994;8(3):277-289.
65. Weir EC, Philbrick WM, Amling M, Neff LA, Baron R, Broadus AE. Targeted overexpression of parathyroid hormone–related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc Natl Acad Sci U S A. 1996;93(19):10240-10245.
66. Erlebacher A, Filvaroff EH, Gitelman SE, Derynk R. Toward a molecular understanding of skeletal development. Cell. 1995;80(3):371-378.
67. Iwamoto M, Jikko A, Murakami H, et al. Changes in parathyroid hormone receptors during chondrocyte cytodifferentiation. J Biol Chem. 1994;269(25):17245-17251.
68. Henderson JE, Amizuka N, Warshawsky H, et al. Nucleolar localization of parathyroid hormone–related peptide enhances survival of chondrocytes under conditions that promote apoptotic cell death. Mol Cell Biol. 1995;15(8):4064-4075.
69. Amizuka N, Warshawsky H, Henderson JE, Goltzman D, Karaplis AC. Parathyroid hormone–related peptide-depleted mice show abnormal epiphyseal cartilage development and altered endochondral bone formation. J Cell Biol. 1994;126(6):1611-1623.
70. Szebenyi G, Fallon JF. Fibroblast growth factors as multifunctional signaling factors. Int Rev Cytol. 1999;185:45-106.
71. Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16(12):1446-1465.
72. Shiang R. Thompson LM, Zhu YZ, et al. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78(2):335-342.
73. Rousseau F, Bonaventure J, Legeai-Mallet L, et al. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371(6494):252-254.
74. Vajo Z, Francomano CA, Wilkin DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias. Endocr Rev. 2000;21(1):23-39.
75. Liu Z, Xu J, Colvin JS, Ornitz DM. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 2002;16(7):859-869.
76. Reddi AH. Bone morphogenetic proteins: from basic science to clinical applications. J Bone Joint Surg Am. 2001;83(suppl 1, pt 1):S1-S6.
77. Minina E, Wenzel HM, Kreschel C, et al. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development. 2001;128(22):4523-4534.
78. Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18(1):4-25.
79. Vu TH, Shipley JM, Bergers G, et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93(3):411-422.
80. Gerber HP, Ferrara N. Angiogenesis and bone growth. Trends Cardiovasc Med. 2000;10(5):223-228.
81. Turner CH. Three rules for bone adaptation to mechanical stimuli. Bone. 1998;23(5):399-407.
82. Ruff C. Growth in bone strength, body size, and muscle size in a juvenile longitudinal sample. Bone. 2003;33(3):317-329.
83. Arkin AM, Katz JF. The effects of pressure on epiphyseal growth; the mechanism of plasticity of growing bone. J Bone Joint Surg Am. 1956;38(5):1056-1076.
84. Mehlman CT, Araghi A, Roy DR. Hyphenated history: the Hueter-Volkmann law. Am J Orthop. 1997;26(11):798-800.
85. Frost HM. Biomechanical control of knee alignment: some insights from a new paradigm. Clin Orthop. 1997;(335):335-342.
86. Chenu C. Role of innervation in the control of bone remodeling. J Musculoskelet Neuronal Interact. 2004;4(2):132-134.
87. Dysart PS, Harkness EM, Herbison GP. Growth of the humerus after denervation. An experimental study in the rat. J Anat. 1989;167:147-159.
88. Pottorf JL. An experimental study of bone growth in the dog. Anat Rec. 1916;10:234-235.
89. Allison N, Brooks B. Bone atrophy. An experimental and clinical study of the changes in bone which result from non-use. Surg Gynecol Obstet. 1921;33:250-260.
90. Ring PA. The influence of the nervous system upon the growth of bones. J Bone Joint Surg Br. 1961;43:121-140.
91. Reading BD, Laor T, Salisbury SR, Lippert WC, Cornwall R. Quantification of humeral head deformity following neonatal brachial plexus palsy. J Bone Joint Surg Am. 2012;94(18):e136(1-8).
92. Moro M, van der Meulen MC, Kiratli BJ, Bachrach LK, Carter DR. Body mass is the primary determinant of midfemoral bone acquisition during adolescent growth. Bone. 1996;19(5):519-526.
93. Schoenau E, Neu CM, Mokov E, Wassmer G, Manz F. Influence of puberty on muscle area and cortical bone area and cortical of the forearm in boys and girls. J Clin Endocrinol Metab. 2000;85(3):1095-1098.
94. Schönau E. The development of the skeletal system in children and the influence of muscular strength. Horm Res. 1998;49(1):27-31.
95. Schönau E, Werhahn E, Schiedermaier U, et al. Influence of muscle strength on bone strength during childhood and adolescence. Horm Res. 1996;45(suppl 1):63-66.
96. van der Meulen MC, Ashford MW Jr, Kiratli BJ, Bachrach LK, Carter DR. Determinants of femoral geometry and structure during adolescent growth. J Orthop Res. 1996;14(1):22-29.
97. van der Meulen MC, Moro M, Kiratli BJ, Marcus R, Bachrach LK. Mechanobiology of femoral neck structure during adolescence. J Rehabil Res Dev. 2000;37(2):201-208.
98. Baron R. General principles of bone biology. In: Favus MJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 5th ed. Washington DC: American Society for Bone and Mineral Research; 2003:1-8.
99. Parfitt AM, Travers R, Rauch F, Glorieux FH. Structural and cellular changes during bone growth in healthy children. Bone. 2000;27(4):487-494.
100. Frost HM. Skeletal structural adaptations to mechanical usage (SATMU): 2. Redefining Wolff’s law: the bone modeling problem. Anat Rec. 1990;226(4):414-422.
101. Frost HM. Skeletal structural adaptations to mechanical usage (SATMU): 1. Redefining Wolff’s law: the bone modeling problem. Anat Rec. 1990;226(4):403-413.
102. Balena R, Shih MS, Parfitt AM. Bone resorption and formation on the periosteal envelope of the ilium: a histomorphometric study in healthy women. J Bone Miner Res. 1992;7(12):1475-1482.
103. Tanner JM, Hughes PC, Whitehouse RH. Radiographically determined widths of bone muscle and fat in the upper arm and calf from age 3-18 years. Ann Hum Biol. 1981;8(6):495-517.
104. Turner RT, Wakley GK, Hannon KS. Differential effects of androgens on cortical bone histomorphometry in gonadectomized male and female rats. J Orthop Res. 1990;8(4):612-617.
105. Yeh JK, Chen MM, Aloia JF. Ovariectomy-induced high turnover in cortical bone is dependent on pituitary hormone in rats. Bone. 1996;18(5):443-450.
106. Kim BT, Mosekilde L, Duan Y, et al. The structural and hormonal basis of sex differences in peak appendicular bone strength in rats. J Bone Miner Res. 2003;18(1):150-155.
107. Parfitt AM. Parathyroid hormone and periosteal bone expansion. J Bone Miner Res. 2002;17(10):1741-1743.
108. Specker B, Binkley T. Randomized trial of physical activity and calcium supplementation on bone mineral content in 3- to 5-year-old children. J Bone Miner Res. 2003;18(5):885-892.
109. Volkman SK, Galecki AT, Burke DT, et al. Quantitative trait loci for femoral size and shape in a genetically heterogenous mouse population. J Bone Miner Res. 2003;18(8):1497-1505.
110. Goodship AE, Lanyon LE, McFie H. Functional adaptation of bone to increased stress. An experimental study. J Bone Joint Surg Am. 1979;61(4):539-546.
111. Falder S, Sinclair JS, Rogers CA, Townsend PL. Long-term behavior of the free vascularized fibula following reconstruction of large bony effects. Br J Plast Surg. 2003;56(6):571-584.
1. Seeman E. Structural basis of growth-related gain and age-related loss of bone strength. Rheumatology. 2008;47(suppl 4):iv2-8.
2. Hall BK, Miyake T. All for one and one for all: condensations and the initiation of skeletal development. Bioessays. 2000;22(2):138-147.
3. Currey JD. Bones: Structure and Mechanics. Princeton, NJ: Princeton University Press; 2002.
4. Rauch F. Bone growth in length and width: the yin and yang of bone stability. J Musculoskelet Neuronal Interact. 2005;5(3):194-201.
5. Seeman E. Periosteal bone formation—a neglected determinant of bone strength. N Engl J Med. 2003;349(4):320-323.
6. Arden NK Spector TD. Genetic influences on muscle strength and bone mineral density: a twin study. J Bone Miner Res. 1997;12(12):2076-2081.
7. Biewener AA, Bertram JEA. Mechanical loading and bone growth in vivo. In: Hall BK, ed. Bone, Vol 7: Bone Growth—B. Boca Raton, FL: CRC Press; 1993:1-36.
8. McGuigan FE, Murray L, Gallagher A, et al. Genetic and environmental determinants of peak bone mass in young men and women. J Bone Miner Res. 2002;17(7):1273-1279.
9. Slemenda CW, Reister TK, Hui SL, Miller JZ, Christian JC, Johnston CC Jr. Influences on skeletal mineralization in children and adolescents: evidence for varying effects of sexual maturation and physical activity. J Pediatr. 1994;125(2):201-207.
10. Schoenau E, Neu CM, Beck B, Manz F, Rauch F. Bone mineral content per muscle cross-sectional area as an index of the functional muscle–bone unit. J Bone Miner Res. 2002;17(6):1095-1101.
11. Rauch F, Neu C, Manz F, Schoenau E. The development of metaphyseal cortex—implications for distal radius fractures during growth. J Bone Miner Res. 2001;16(8):1547-1555.
12. Skaggs DL, Loro ML, Pitukcheewanont P, Tolo V, Gilsanz V. Increased body weight and decreased radial cross-sectional dimensions in girls with forearm fractures. J Bone Miner Res. 2001;16(7):1337-1342.
13. Allen DM, Mao JJ. Heterogenous nanostructural and nanoelastic properties of pericellular and interterritorial matrices of chondrocytes by atomic force microscopy. J Struct Biol. 2004;145(3):196-204.
14. van der Eerden BC, Karperien M, Wit JM. Systemic and local regulation of the growth plate. Endocr Rev. 2003;24(6):782-801.
15. Li LP, Herzog W. Strain-rate dependence of cartilage stiffness in unconfined compression: the role of fibril reinforcement versus tissue volume change in fluid pressurization. J Biomech. 2004;37(3):375-382.
16. Cohen B, Chorney GS, Phillips DP, Dick HM, Mow VC. Compressive stress-relaxation behavior of bovine growth plate may be described by the non-linear biphasic theory. J Orthop Res. 1994;12(6):804-813.
17. Robson H, Siebler T, Shalet SM, Williams GR. Interactions between GH, IGF-Ι, glucocorticoids and thyroid hormones during skeletal growth. Pediatr Res. 2002;52(2):137-147.
18. Abad V, Meyers JL, Weise M, et al. The role of the resting zone in growth plate chondrogenesis. Endocrinology. 2002;143(5):1851-1857.
19. Wang W, Kirsch T. Retinoic acid stimulates annexin-mediated growth plate chondrocyte mineralization. J Cell Biol. 2002;157(6):1061-1069.
20. Anderson HC. Matrix vesicles and calcification. Curr Rheumatol Rep. 2003;5(3):222-226.
21. Tanner JM. The adolescent spurt in animals. In: Thomas CC, ed. Growth at Adolescence. Oxford, UK: Blackwell; 1962:223-239.
22. Drop SL, De Waal WJ, De Muinck Keizer-Schrama SM. Sex steroid treatment of constitutionally tall stature. Endocr Rev. 1998;19(5):540-558.
23. Park EA. The imprinting of nutritional disturbances on the growing bone. Pediatrics. 1964;33(suppl):815-862.
24. Buckwalter JA, Mower D, Unqar R, Schaeffer J, Ginsberg B. Morphometric analysis of chondrocyte hypertrophy. J Bone Joint Surg Am. 1986;68(2):243-255.
25. Sawae Y, Sahara T, Sasaki T. Osteoclast differentiation at the growth plate cartilage–trabecular bone junction in newborn rat femur. J Electron Microsc. 2003;52(6):493-502.
26. Lee ER, Lamplugh L, Shepard NL, Mort JS. The septoclast, a cathepsin B–rich cell involved in the resorption of growth plate cartilage. J Histochem Cytochem. 1995;43(5):525-536.
27. Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5(6):623-628.
28. Fazzalari NL, Moore AJ, Byers S, Byard RW. Quantitative analysis of trabecular morphogenesis in the human costochondral junction during the postnatal period in normal subjects. Anat Rec. 1997;248(1):1-12.
29. Cancedda R, Descalzi Cancedda F, Castagnola P. Chondrocyte differentiation. Int Rev Cytol. 1995;159:265-358.
30. Stevens DA, Williams GR. Hormone regulation of chondrocyte differentiation and endochondral bone formation. Mol Cell Endocrinol. 1999;151(1-2):195-204.
31. Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423(6937):332-336.
32. Daughaday WH, Hall K, Raben MS, Salmon WD Jr, van den Brande JL, van Wyk JJ. Somatomedin: proposed designation for sulphation factor. Nature. 1972;235(5333):107.
33. Isaksson OG, Lindahl A, Nilsson A, Isqaard J. Mechanism of the stimulatory effect of the growth hormone on longitudinal bone growth. Endocr Rev. 1987;8(4):426-438.
34. Hunziker EB, Wagner J, Zapf J. Differential effects of insulin-like growth factor I and growth hormone on developmental stages of rat growth plate chondrocytes in vivo. J Clin Invest. 1994;93(3):1078-1086.
35. Underwood LE, van Wijk JJ. Normal and aberrant growth. In: Wilson JD, Foster DW, eds. Textbook of Endocrinology. Philadelphia, PA: Saunders; 1992:1079-1138.
36. Rivkees SA, Bode HH, Crawford JD. Long-term growth in juvenile acquired hypothyroidism: the failure to achieve normal adult stature. N Engl J Med. 1988;318(10):599-602.
37. Segni M, Leonardi E, Mazzoncini B, Pucarelli I, Pasquino AM. Special features of Graves’ disease in early childhood. Thyroid. 1999;9(9):871-877.
38. Burch WM, Van Wyk JJ. Triiodothyronine stimulates cartilage growth and maturation by different mechanisms. Am J Physiol. 1987;252(2, pt 1):E176-E182.
39. Lewinson D, Bialik GM, Hochberg Z. Differential effects of hypothyroidism on the cartilage and the osteogenic process in the mandipular condyle: recovery by growth hormone and thyroxine. Endocrinology. 1994;135(4):1504-1510.
40. Wakita R, Izumi T, Itoman M. Thyroid hormone–induced chondrocyte terminal differentiation in rat femur organ culture. Cell Tissue Res. 1998;293(2):357-364.
41. Smeets T, van Buul-Offers S. Influence of growth hormone and thyroxine on cell kinetics in the proximal tibial growth plate of Snell dwarf mice. Cell Tissue Kinet. 1986;19(2):161-170.
42. Silvestrini G, Mocetti P, Ballanti P, Di Grezia R, Bonucci E. Cytochemical demonstration of the glucocorticoid receptor in skeletal cells of the rat. Endocr Res. 1999;25(1):117-128.
43. Abu EO, Horner A, Kusec V, Triffitt JT, Compston JE. The localization of the functional glucocorticoid receptor alpha in human bone. J Clin Endocrinol Metab. 2000;85(2):883-889.
44. Magiakou MA, Mastorakos G, Chrousos GP. Final stature in patients with endogenous Cushing’s syndrome. J Clin Endocrinol Metab. 1994;79(4):1082-1085.
45. Avioli LV. Glucocorticoid effects on statural growth. Br J Rheumatol. 1993;32(suppl 2):27-30.
46. Eberhardt AW, Yeager-Jones A, Blair HC. Regional trabecular bone matrix degeneration and osteocyte death in femora if glucocorticoid-treated rabbits. Endocrinology. 2001;142(3):1333-1340.
47. Silvestrini G, Ballanti P, Patacchioli FR, et al. Evaluation of apoptosis and the glucocorticoid receptor in the cartilage growth plate and metaphyseal bone cells of rats after high-dose treatment with corticosterone. Bone. 2000;26(1):33-42.
48. Montecucco C, Caporali R, Caprotti P, Caprotti M, Notario A. Sex hormones and bone metabolism in postmenopausal rheumatoid arthritis treated with two different glucocorticoids. J Rheumatol. 1992;19(12):1895-1900.
49. Bello CE, Garrett SD. Therapeutic issues in oral glucocorticoid use. Lippincotts Prim Care Pract. 1999;3(3):333-341.
50. Turner RT, Riggs BL, Speisberg TC. Skeletal effects of estrogen. Endocr Rev. 1994;15(3):275-300.
51. Gevers EF, Wit JM, Robinson IC. Effect of gonadectomy on growth and GH responsiveness in dwarf rats. J Endocrinol. 1995;145(1):69-79.
52. van der Eerden BC, Emons J, Ahmed S, et al. Evidence for genomic and non-genomic actions of estrogens in growth plate regulation in female and male rats at the onset of sexual maturation. J Endocrinol. 2002;175(2):277-288.
53. Nilsson O, Falk J, Ritzen EM, Baron J, Savendahl L. Raloxifene acts as an estrogen agonist on the rabbit growth plate. Endocrinology. 2003;144(4):1481-1485.
54. Strickland AL, Sprinz H. Studies of the influence of estradiol and growth hormone on the hypophysectomized immature rat epiphyseal cartilage growth plate. Am J Obstet Gynecol. 1973;115(4):471-477.
55. Jansson JO, Eden S, Isaksson O. Sites of action of testosterone and estradiol on longitudinal bone growth. Am J Physiol. 1983;244(2):E135-E140.
56. Abu EO, Horner A, Kusec V, Triffitt JT, Compston JE. The localization of androgen receptors in human bone. J Clin Endocrinol Metab. 1997;82(10):3493-3497.
57. Noble B, Routledge J, Stevens H, Hughes I, Jacobson W. Androgen receptors in bone-forming tissue. Horm Res. 1999;51(1):31-36.
58. van der Eerden BC, van Til NP, Brinkmann AO, Lowik CW, Wit JM, Karperien M. Sex differences in the expression of the androgen receptor in the tibial growth plate and metaphyseal bone of the rat. Bone. 2002;30(6):891-896.
59. Cassorla FG, Skerda MC, Valk IM, Hung W, Cutler GB Jr, Loriaux DL. The effects of sex steroids on ulnar growth during adolescence. J Clin Endocrinol Metab. 1984;58(4):717-720.
60. Zung A, Phillip M, Chalew SA, Palese T, Kowarski AA, Zadik Z. Testosterone effect on growth and growth mediators of the GF–IGF-I axis in the liver and epiphyseal growth plate of juvenile rats. J Mol Endocrinol. 1999;23(2):209-221.
61. Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273(5275):613-622.
62. St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and essential for bone formation. Genes Dev. 1999;13(16):2072-2086.
63. Karp SJ Schipani E, St-Jacques B, Hunzelman J, Kronenberg H, Mcmahon AP. Indian hedgehog coordinates endochondral bone growth and morphogenesis via parathyroid hormone related-protein–dependent and –independent pathways. Development. 2000;127(3):543-548.
64. Karaplis AC, Luz A, Glowacki J, et al. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone–related peptide gene. Genes Dev. 1994;8(3):277-289.
65. Weir EC, Philbrick WM, Amling M, Neff LA, Baron R, Broadus AE. Targeted overexpression of parathyroid hormone–related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. Proc Natl Acad Sci U S A. 1996;93(19):10240-10245.
66. Erlebacher A, Filvaroff EH, Gitelman SE, Derynk R. Toward a molecular understanding of skeletal development. Cell. 1995;80(3):371-378.
67. Iwamoto M, Jikko A, Murakami H, et al. Changes in parathyroid hormone receptors during chondrocyte cytodifferentiation. J Biol Chem. 1994;269(25):17245-17251.
68. Henderson JE, Amizuka N, Warshawsky H, et al. Nucleolar localization of parathyroid hormone–related peptide enhances survival of chondrocytes under conditions that promote apoptotic cell death. Mol Cell Biol. 1995;15(8):4064-4075.
69. Amizuka N, Warshawsky H, Henderson JE, Goltzman D, Karaplis AC. Parathyroid hormone–related peptide-depleted mice show abnormal epiphyseal cartilage development and altered endochondral bone formation. J Cell Biol. 1994;126(6):1611-1623.
70. Szebenyi G, Fallon JF. Fibroblast growth factors as multifunctional signaling factors. Int Rev Cytol. 1999;185:45-106.
71. Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16(12):1446-1465.
72. Shiang R. Thompson LM, Zhu YZ, et al. Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell. 1994;78(2):335-342.
73. Rousseau F, Bonaventure J, Legeai-Mallet L, et al. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371(6494):252-254.
74. Vajo Z, Francomano CA, Wilkin DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias. Endocr Rev. 2000;21(1):23-39.
75. Liu Z, Xu J, Colvin JS, Ornitz DM. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 2002;16(7):859-869.
76. Reddi AH. Bone morphogenetic proteins: from basic science to clinical applications. J Bone Joint Surg Am. 2001;83(suppl 1, pt 1):S1-S6.
77. Minina E, Wenzel HM, Kreschel C, et al. BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development. 2001;128(22):4523-4534.
78. Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18(1):4-25.
79. Vu TH, Shipley JM, Bergers G, et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93(3):411-422.
80. Gerber HP, Ferrara N. Angiogenesis and bone growth. Trends Cardiovasc Med. 2000;10(5):223-228.
81. Turner CH. Three rules for bone adaptation to mechanical stimuli. Bone. 1998;23(5):399-407.
82. Ruff C. Growth in bone strength, body size, and muscle size in a juvenile longitudinal sample. Bone. 2003;33(3):317-329.
83. Arkin AM, Katz JF. The effects of pressure on epiphyseal growth; the mechanism of plasticity of growing bone. J Bone Joint Surg Am. 1956;38(5):1056-1076.
84. Mehlman CT, Araghi A, Roy DR. Hyphenated history: the Hueter-Volkmann law. Am J Orthop. 1997;26(11):798-800.
85. Frost HM. Biomechanical control of knee alignment: some insights from a new paradigm. Clin Orthop. 1997;(335):335-342.
86. Chenu C. Role of innervation in the control of bone remodeling. J Musculoskelet Neuronal Interact. 2004;4(2):132-134.
87. Dysart PS, Harkness EM, Herbison GP. Growth of the humerus after denervation. An experimental study in the rat. J Anat. 1989;167:147-159.
88. Pottorf JL. An experimental study of bone growth in the dog. Anat Rec. 1916;10:234-235.
89. Allison N, Brooks B. Bone atrophy. An experimental and clinical study of the changes in bone which result from non-use. Surg Gynecol Obstet. 1921;33:250-260.
90. Ring PA. The influence of the nervous system upon the growth of bones. J Bone Joint Surg Br. 1961;43:121-140.
91. Reading BD, Laor T, Salisbury SR, Lippert WC, Cornwall R. Quantification of humeral head deformity following neonatal brachial plexus palsy. J Bone Joint Surg Am. 2012;94(18):e136(1-8).
92. Moro M, van der Meulen MC, Kiratli BJ, Bachrach LK, Carter DR. Body mass is the primary determinant of midfemoral bone acquisition during adolescent growth. Bone. 1996;19(5):519-526.
93. Schoenau E, Neu CM, Mokov E, Wassmer G, Manz F. Influence of puberty on muscle area and cortical bone area and cortical of the forearm in boys and girls. J Clin Endocrinol Metab. 2000;85(3):1095-1098.
94. Schönau E. The development of the skeletal system in children and the influence of muscular strength. Horm Res. 1998;49(1):27-31.
95. Schönau E, Werhahn E, Schiedermaier U, et al. Influence of muscle strength on bone strength during childhood and adolescence. Horm Res. 1996;45(suppl 1):63-66.
96. van der Meulen MC, Ashford MW Jr, Kiratli BJ, Bachrach LK, Carter DR. Determinants of femoral geometry and structure during adolescent growth. J Orthop Res. 1996;14(1):22-29.
97. van der Meulen MC, Moro M, Kiratli BJ, Marcus R, Bachrach LK. Mechanobiology of femoral neck structure during adolescence. J Rehabil Res Dev. 2000;37(2):201-208.
98. Baron R. General principles of bone biology. In: Favus MJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 5th ed. Washington DC: American Society for Bone and Mineral Research; 2003:1-8.
99. Parfitt AM, Travers R, Rauch F, Glorieux FH. Structural and cellular changes during bone growth in healthy children. Bone. 2000;27(4):487-494.
100. Frost HM. Skeletal structural adaptations to mechanical usage (SATMU): 2. Redefining Wolff’s law: the bone modeling problem. Anat Rec. 1990;226(4):414-422.
101. Frost HM. Skeletal structural adaptations to mechanical usage (SATMU): 1. Redefining Wolff’s law: the bone modeling problem. Anat Rec. 1990;226(4):403-413.
102. Balena R, Shih MS, Parfitt AM. Bone resorption and formation on the periosteal envelope of the ilium: a histomorphometric study in healthy women. J Bone Miner Res. 1992;7(12):1475-1482.
103. Tanner JM, Hughes PC, Whitehouse RH. Radiographically determined widths of bone muscle and fat in the upper arm and calf from age 3-18 years. Ann Hum Biol. 1981;8(6):495-517.
104. Turner RT, Wakley GK, Hannon KS. Differential effects of androgens on cortical bone histomorphometry in gonadectomized male and female rats. J Orthop Res. 1990;8(4):612-617.
105. Yeh JK, Chen MM, Aloia JF. Ovariectomy-induced high turnover in cortical bone is dependent on pituitary hormone in rats. Bone. 1996;18(5):443-450.
106. Kim BT, Mosekilde L, Duan Y, et al. The structural and hormonal basis of sex differences in peak appendicular bone strength in rats. J Bone Miner Res. 2003;18(1):150-155.
107. Parfitt AM. Parathyroid hormone and periosteal bone expansion. J Bone Miner Res. 2002;17(10):1741-1743.
108. Specker B, Binkley T. Randomized trial of physical activity and calcium supplementation on bone mineral content in 3- to 5-year-old children. J Bone Miner Res. 2003;18(5):885-892.
109. Volkman SK, Galecki AT, Burke DT, et al. Quantitative trait loci for femoral size and shape in a genetically heterogenous mouse population. J Bone Miner Res. 2003;18(8):1497-1505.
110. Goodship AE, Lanyon LE, McFie H. Functional adaptation of bone to increased stress. An experimental study. J Bone Joint Surg Am. 1979;61(4):539-546.
111. Falder S, Sinclair JS, Rogers CA, Townsend PL. Long-term behavior of the free vascularized fibula following reconstruction of large bony effects. Br J Plast Surg. 2003;56(6):571-584.
Coexisting Frailty, Cognitive Impairment, and Heart Failure: Implications for Clinical Care
From the Nell Hodgson Woodruff School of Nursing, Emory University, Atlanta, GA.
Abstract
- Objective: To review some of the proposed pathways that increase frailty risk in older persons with heart failure and to discuss tools that may be used to assess for changes in physical and cognitive functioning in this population in order to assist with appropriate and timely intervention.
- Methods: Review of the literature.
- Results: Heart failure is the only cardiovascular disease that is increasing by epidemic proportions, largely due to an aging society and therapeutic advances in disease management. Because heart failure is largely a cardiogeriatric syndrome, age-related syndromes such as frailty and cognitive impairment are common in heart failure patients. Compared with age-matched counterparts, older adults with heart failure 4 to 6 times more likely to be frail or cognitively impaired. The reason for the high prevalence of frailty and cognitive impairment in this population is not well known but may likely reflect the synergistic effects of heart failure and aging, which may heighten vulnerability to stressors and accelerate loss of physiologic reserve. Despite the high prevalence of frailty and cognitive impairment in the heart failure population, these conditions are not routinely screened for in clinical practice settings and guidelines on optimal assessment strategies are lacking.
- Conclusion: Persons with heart failure are at an increased risk for frailty, which may worsen symptoms, impair self-management, and lead to worse heart failure outcomes. Early detection of frailty and cognitive impairment may be an opportunity for intervention and a key strategy for improving clinical outcomes in older adults with heart failure.
Approximately 5.7 million persons in the United States are diagnosed with heart failure, and the number of reported new cases is expected to increase to over 700,000 cases annually by the year 2040 [1]. This rising incidence is fueled by an aging population; by the year 2030, 1 in 5 Americans will be over 65 years of age [2]. Heart failure is prevalent among those 65 years of age and older and is the most common reason for hospitalization in this age-group. High readmission rates, approaching 50% over 6 months, are a major contributor to the the escalating economic burden associated with heart failure [3].
Persons with heart failure are more likely to be frail and experience cognitive impairment than their age-matched counterparts without heart failure. The reasons for this are not well known but may be related to hemodynamic, vascular, and inflammatory changes occurring as heart failure progresses. In this paper, we review the link between frailty and cognitive impairment in heart failure, instruments that may be useful for early detection, and interventions such as exercise that may be beneficial for attenuating both conditions.
Frailty in Heart Failure
Epidemiology
Frailty is a heightened vulnerability to stressors in the presence of low physiological reserve [4]. When exposed to stressors, persons who are frail have a much higher probability for disproportionate decompensation, negative events, functional decline, disability, and mortality [5]. Among persons with heart failure, frailty may predispose them to decompensate at a lower threshold, requiring more frequent hospitalizations. Persons with heart failure are more likely to be frail than their age-matched counterparts without heart failure [6,7].
Frailty is a powerful predictor of poor clinical outcomes and mortality in cardiovascular disease [8,9]. Compared with the non-frail, frail persons with heart failure have increased rates of mortality (16.9% vs 4.8%) and increased rates of heart failure hospitalization (20.5% vs 13.3%) [10]. Frailty has also been shown to predict falls, disability, and hospitalization in heart failure patients [6,9,11] and was found to have a negative linear relationship with health-related quality of life [12]. Frail heart failure patients are also more likely to have comorbidities such diabetes mellitus, chronic obstructive pulmonary disease, atrial fibrillation, depression, anemia, and chronic kidney disease [9,13].
Pathophysiology
There is significant overlap in the underlying pathological mechanisms of heart failure and frailty. Symptoms of heart failure, such as dyspnea, fatigue, and muscle loss, mirror components that occur with frailty. Further, cardiac cachexia, a metabolic syndrome in advanced heart failure characterized by a loss of muscle mass, is very similar to the sarcopenia that occurs in frailty.
Frailty, characterized by an increased physiologic vulnerability to stressors, may predispose frail persons with heart failure to exacerbation and worsening of heart failure due to greater susceptibility to the harmful pathophysiologic processes in heart failure, such as inflammation and autonomic dysfunction. Proposed pathophysiologic pathways in frailty include free radicals and oxidative stress, cumulative DNA damage, decreased telomere length, and nuclear fragmentation [14,15]. Frailty has been associated with low-grade chronic inflammation and increased inflammatory cytokines, such as C-reactive protein, tumor necrosis factor–alpha (TNFα), interleukin-6 (IL-6)and fibrinogen [16–18]. Heart failure also is associated with a low-grade and chronic cardiac inflammatory response that is correlated with disease progression [19].
Inflammation. IL-6 is detectable in a higher proportion of persons who are frail compared to non-frail [16] and is the most highly correlated biomarker with frailty. In addition, among those with detectable IL-6 levels, those categorized as frail have higher IL-6 levels compared to those who are non-frail [16,20]. Individuals categorized as frail were found to have significantly higher levels of TNFα than those who were non-frail [16,20]. Increased IL-6 levels are associated with decreased muscle strength, while increased TNFα levels are associated with decreased skeletal muscle protein synthesis [21,22], thus contributing to frailty.
Oxidative stress. Protein carbonyls result from protein oxidation promoted by reactive oxygen species and are markers of oxidative stress. Protein carbonylation is implicated in the pathogenesis of the loss of skeletal muscle mass; high serum protein carbonyls are associated with poor grip strength [23]. 8-OHdG is an oxidized nucleoside indicative of oxidative damage to DNA and a measure of oxidative stress. Accumulation of 8-OHdG in skeletal muscle leads to loss of muscle mass and is associated with decreased hand grip strength in the elderly [24]. Higher serum levels of 8-OHdG are present in older adults who are frail as compared to those who are non-frail [25].
Measurement of Frailty in the Clinical Setting
Frailty has been conceptualized in a number of studies using different models and measures; however, there continues to be a lack of consensus on the definition and operationalization of frailty. Prior research has led to the development of several validated models of frailty that have demonstrated good prediction of adverse outcomes in older adults. Some models, such as the Fried phenotype [6], focus solely on the physical dimension, while other models take a multidimensional approach.Single-item measures (eg, gait speed, 6-minute walk test, handgrip strength) are also commonly used to screen for frailty, but a frailty measure that incorporates more than 1 physical dimension may be more sensitive and reliable. In our opinion, the ideal measure of frailty would consist of a brief assessment that can be serially performed in most clinical practice settings that can identify change in function over time. The incorporation of sensitive physical function measures that can detect frailty early has the potential to slow physical function decline by preserving physiological thresholds.
Cognitive Impairment in Heart Failure
Epidemiology
Cognitive impairment occurs frequently in patients with heart failure, and the presence of cognitive impairment in persons with heart failure has been shown to heighten risk for adverse clinical outcomes, disability, poor quality of life, and mortality [26,27]. Heart failure negatively influences cognitive functioning in most domains [28–32]. The most common domains adversely affected by heart failure and aging are memory and executive function. Deficits in these domains can substantially diminish patient ability to carry out essential self-care behaviors [30,32].
The most common form of cognitive impairment seen in patients with heart failure is mild cognitive impairment (MCI), which is a measurable deficit with memory or another core cognitive domain. Up to 60% of persons with heart failure have been reported to have MCI. Patients with MCI have cognitive deficits that are more pronounced than those seen in normal aging, but lack other symptoms of dementia, such as impaired judgment or reasoning. MCI often will not impede patients’ ability to carry out the activities of daily living (ADLs) independently, but patients may have difficulty in performing some instrumental activities of daily living (IADLs), such as remembering medications, scheduling provider appointments. Dementia, a decline in cognitive ability severe enough to hinder an individual’s ability to perform ADLs or IADLs or engage in social activities or occupational responsibilities, occurs in approximately 25% of persons with heart failure [33].
Persons with heart failure have a fourfold greater likelihood of developing CI than persons without heart failure. Several cohort studies have shown that persons with heart failure had lower performance on cognitive tests than individuals without heart failure [34,35] and were 50% more likely to progress to dementia.
Assessment Tools
Although a comprehensive neurocognitive battery would aid in detecting cognitive impairment in heart failure, few clinical practice settings have the resources to perform such a detailed and time-consuming measurement. Most studies in heart failure have relied on global screening questionnaires such as the Mini-Mental State Examination (MMSE) [36] to assess cognitive functioning in persons with heart failure and in other cardiovascular disorders. Global cognitive measures, however, often lack sensitivity for detecting subtle cognitive deficits such as seen in MCI [28–30]. Screening that measures executive function may be the most beneficial for busy clinical settings, since declines in this domain are well established as contributing to poor outcomes in persons with heart failure.
The Montreal Cognitive Assessment (MoCA) is a rapid screening test designed to detect MCI. It assesses different cognitive domains, including attention, memory, language, and executive function [37]. The MoCA lends itself to use in clinical setting because it is brief, requires little training to administer, and is easy to score. This instrument has been used successfully to assess MCI in persons with heart failure and may be more sensitive than the MMSE in identifying clinically relevant cognitive dysfunction. In 2013 study, Cameron et al [38] administered the MMSE and MoCA to 93 hospitalized heart failure patients and found that the MoCA classified 41% of patients as cognitively impaired that were not classified using the MMSE. For persons with a vascular cognitive deficit, the MMSE has been portrayed as an inadequate screening test due to lack of sensitivity for visuospatial and executive function deficits. Because the MoCA was designed to be more sensitive to such deficits, it may be a superior screening method for persons with heart failure. Although previous studies support the use of the MoCA in persons with heart failure, more research is needed in larger, more diverse heart failure samples with a wide range of cognitive deficits.
A Reasonable Clinical Assessment Approach
Considering the link between heart failure, frailty, and MCI, incorporating simple physical performance measures with cognitive screening may be an effective strategy to identify persons at risk for frailty. Two clinically relevant physical performance-based measures of frailty are proposed: the Fried phenotype (mentioned earlier) and the Short Physical Performance Battery (SBBP). In addition, cognitive screening using the MoCA is recommended as part of the routine examination for determining possible MCI or more severe cognitive deficits. The predictive validity of measuring physical frailty is enhanced when cognitive impairment is included in the assessment [36,39].
The performance-based measures included in this review have previously demonstrated excellent psychometric properties as well as sensitivity for change that is clinically meaningful. Minimal detectable change (MDC), a threshold score that refers to the minimal amount of change outside of error that reflects true change by a patient between 2 time points (rather than variation in measurement), is important for interpreting level of risk for frailty and is included for each instrument [40,41]. If a more brief frailty examination is needed, cut-points for gait speed and handgrip have been used effectively in a number of studies as a threshold for determining frailty, including in older patients with cardiovascular disease and in heart failure [8,42,43].
Fried Frailty Phenotype
The Fried phenotype is an appropriate method of measuring frailty in a clinical setting due to its wide application across diverse populations and consistent identification of adverse outcomes [44]. This model is derived from a frailty model proposed by Fried et al [6] in which a phenotypic cycle exists that includes disease, sarcopenia, decreased walking speed, chronic undernutrition, decreased total energy expenditure, senescent musculoskeletal changes, decreased resting metabolic rate, weight loss and decreased maximal oxygen consumption. Frailty exists when a critical mass of these cycle components are identified in an individual [6].
To validate the model, Fried et al used data from the Cardiovascular Health Study and used the model to show association with a 3-year and 7-year incidence of mobility and ADL disability among 4317 community-dwelling men and women aged 65 years and older, independent of comorbidities. Several studies have directly tested the frailty phenotype model alone and in comparison to other models of frailty in large prospective studies across different populations, such as the Survey of Health, Aging and Retirement in Europe (SHARE) [45], the European Male Aging Study [46], and the Canadian Health Study of Aging [47]. While these studies found the prevalence of frailty to vary across the populations, they all validated the Fried model and found no significant differences in the predictive ability of the Fried model and other models of frailty. The Frailty Consensus conference evaluated the different models of frailty and determined that the Fried model is a validated construct of frailty and is acceptable for use in the identification of individuals who are frail or likely to become frail [48]. Thus, the Fried et al frailty phenotype model is considered to be a standard measure of frailty in older individuals.
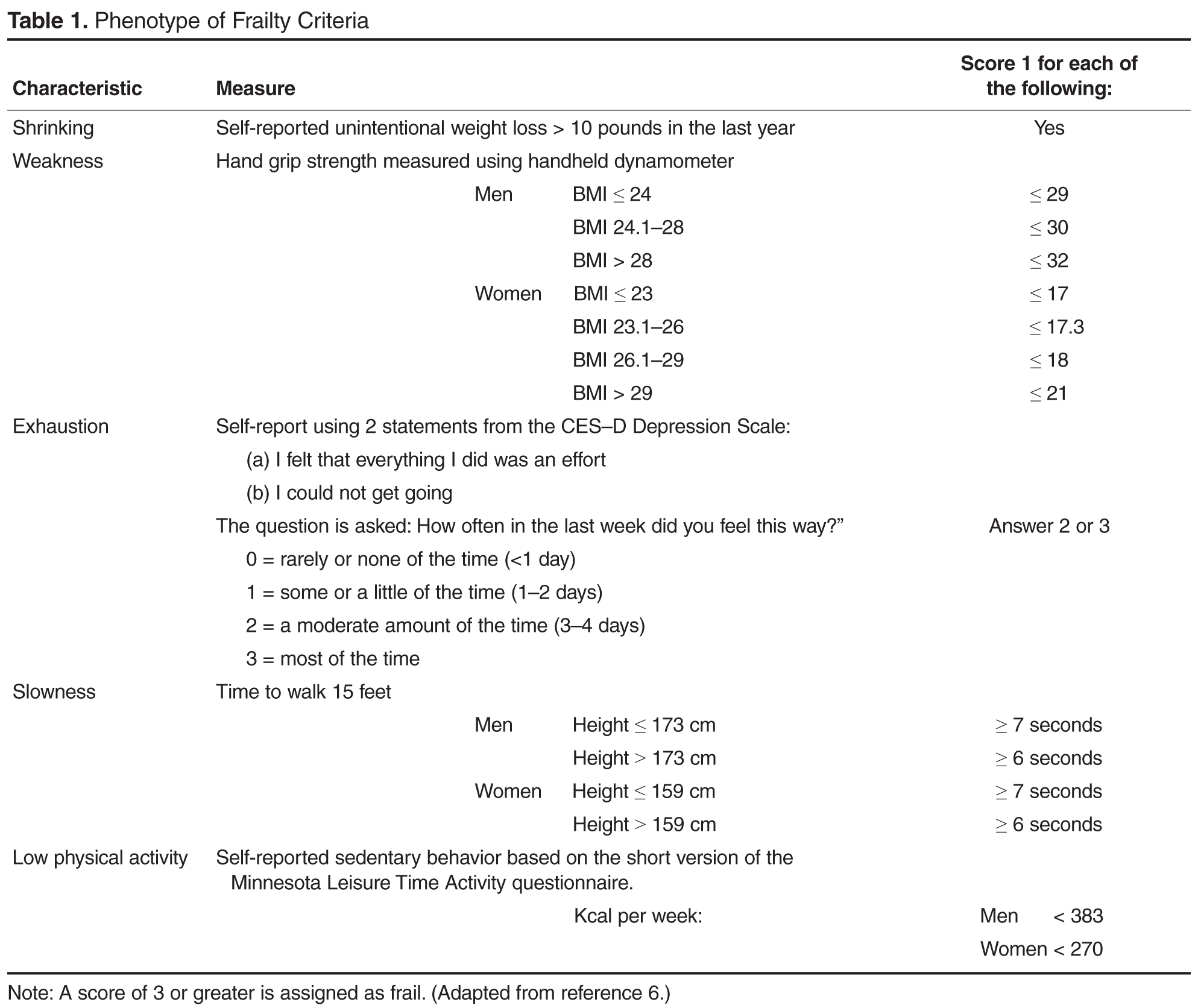
Short Physical Performance Battery
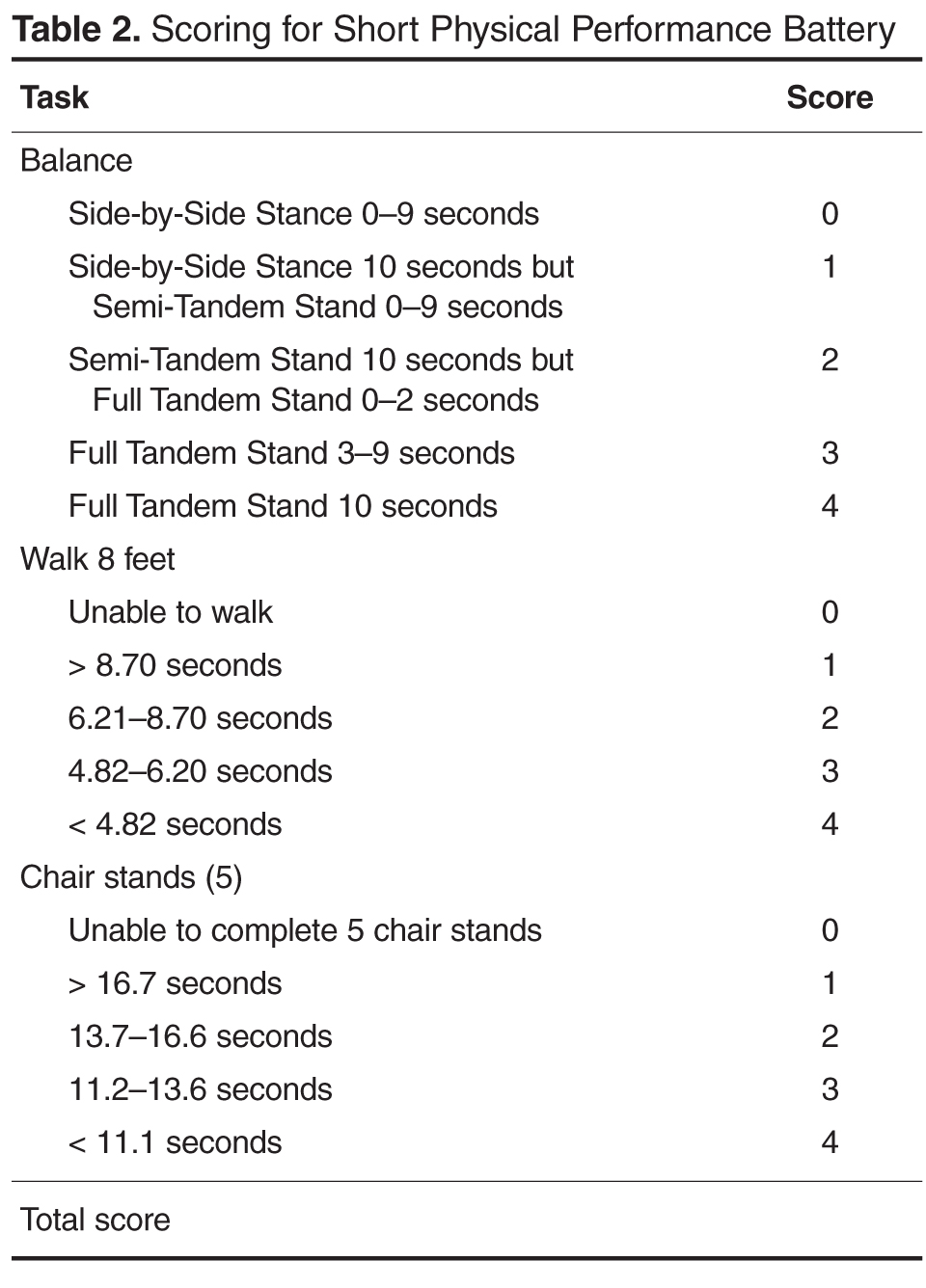
In other chronic illness populations, the SPPB has also been used as a predictor of outcomes before, during, or after hospitalization. Valpato et al [53], for example, used the SPPB to assess older adults (mean age, 78 yr) admitted to the hospital with a diagnosis of heart failure (64%), pneumonia (13%), chronic obstructive pulmonary disease (16%), or minor stroke (6.6%) at admission (baseline) and discharge. Patients with the lowest SPPB quartile scores at hospital discharge had a fivefold greater risk of rehospitalization or mortality compared to the highest quartile. In addition, those who had an early decline in SPPB scores 1 month after hospital discharge had greater limitations in performing activities of daily living and a significantly greater probability of being re-hospitalized or death during the 1-year follow-up period. These studies suggest that the SPPB at the first follow-up outpatient visit following hospital discharge may be beneficial for identifying need for further intervention or the need for more frequent follow-up care. Although the SPPB is not part of the Fried et al phenotype, it may provide additional information concerning risk for falls and lower extremity strength that may be beneficial in the evaluation of some persons with heart failure [54]. The SPPB along with instructions and normative data are available for clinical use at no charge at www.grc.nia.nih.gov/branches/ledb/sppb/index.htm.
Interventions for Frailty in Heart Failure
Interventions to address frailty have included exercise training, comprehensive geriatric assessment and management services, social support systems, nutrition, and drugs; however, few intervention studies have examined frailty in heart failure [8]. Restoration of physical function through aerobic exercise and resistance training has shown benefit in frail older adults [55–57] and in persons with heart failure [58]. Maintaining and/or restoring physical function through aerobic and resistance exercise training may be the key to preventing further decline or potentially reversing frailty in older adults with heart failure.
Aerobic exercise has been shown to be beneficial for both frail older adults and frail persons with heart failure [18]. In a study of community-dwelling frail older adults aged 65 and older, a combined aerobic and resistance exercise intervention, performed over 16 weeks, demonstrated significant improvement in frailty scores during the 1-year follow-up in contrast to worsening frailty measures in the control group [57].
Older adults with heart failure experience a much lower exercise tolerance largely due to a 50% to 75% decrease in aerobic capacity in addition to the well-known alterations in peripheral musculoskeletal performance that contribute to fatigue and greater symptom severity. Aerobic exercise has been shown to be beneficial for most heart failure patients by altering the peripheral and central mechanisms, such as inflammatory cytokines, that contribute to heart failure exacerbations, worsened symptom severity, and poor clinical outcomes [59–62].Lower rates of hospitalization, improved physical function, and enhanced health-related quality of life are reported in heart failure patients who routinely exercise [59]. Resistance training has been shown to improve physical function in frail older adults [55]. Further, the use of TheraBand exercise bands in resistance training demonstrated improvement in physical function among frail older adults [56].
Exercise also appears to exert a positive effect on cognition, particularly executive functioning, and may also have a protective effect against cognitive decline with aging and among those with heart failure. The underlying mechanism for improvement in cognition remains poorly understood but is likely related to improved cardiac function, cerebral perfusion, and oxygenation, although this has not been clearly established. Larson et al (2006) evaluated the frequency of participation in a variety of physical activities (eg, walking, bicycling and swimming) over 6 years in 1740 older adults [63]. Older adults who exercised more than 3 times per week during initial assessment were 34% less likely to be diagnosed with dementia than those who exercised fewer than 3 times per week. Several meta-analyses in recent years have shown a consistent and positive relationship between aerobic exercise and cognition [64,65]. Importantly, findings from meta-analyses have shown a moderate effect size (> 0.5) from aerobic training, which was similar for normal and cognitively impaired adults [64].
Implications for Clinical Care
A systematic assessment performed periodically using physical and cognitive measures that may identify prefrailty may be the best strategy for preventing further functional loss, limitations, and disability in persons with heart failure. Persons with heart failure ideally should be evaluated annually for physical function, since a decline has been consistently shown to be a strong predictor of adverse health outcomes, disability, and death [6,66]. Cognitive function should also be assessed routinely in persons with heart failure, particularly when first diagnosed, when changes in treatment regimen occur, and with worsening disease severity, since these events have been shown to occur before changes in cognition [31]. Incorporating geriatric performance-based measures in heart failure management would allow for more treatment strategies aimed at improving physical function, cognitive outcomes, and quality of life. Further, identifying frailty in heart failure is an important component of clinical decision-making when determining if a patient can tolerate therapies such as implantable defibrillators, cardiac resynchronization therapy, or left ventricular assist device placement.
In older adults, performance measures are well established and commonly used as part of geriatric assessment to evaluate physical and cognitive functioning. Performance-based measures may be particularly beneficial in older adults with heart failure to monitor serial changes in physical function. Performance measures in clinical settings require staff time but little training, space, equipment, or risk. As performance measures become more common in practice settings, MDC thresholds may need to be re-evaluated based on the characteristics of the population [67].
For persons with heart failure whose screening outcomes suggest MCI, more comprehensive neuropsychological testing should be available as well as provision of resources to optimize functional independence. Early identification of impaired cognition may lower risk of poor self-management through simplification of medication regimens or providing resources to help manage other regimens essential for optimal heart failure care. It is also important to recognize that depressive symptoms are common in persons with heart failure and are highly correlated with cognitive impairment in this population. Screening for depressive symptoms therefore, may also enhance identification of persons with heart failure at risk for frailty [4,28].
Conclusion
Effective appraisal and development of effective interventions are essential in older adults with heart failure who are at high risk for frailty and cognitive impairment. This will become increasingly important as the population ages and the incidence of heart failure rises proportionately. Although curative treatments for frailty and cognitive impairment are not available, interdisciplinary interventions such as exercise and comprehensive geriatric assessment may improve outcomes in older persons with heart failure [68]. Information gained from objective, simple, inexpensive physical performance measures, when used in combination with cognitive screening, may enhance the ability to evaluate change that signal onset of frailty or cognitive impairment [54,69,70]. The high morbidity and mortality associated with frailty and cognitive impairment indicate that it should be a priority for future research as a strategy to improve clinical outcomes, enhance quality of life, and lower health care costs in this growing population.
Corresponding author: Rebecca Gary, PhD, RN, Nell Hodgson Woodruff School of Nursing, Emory University, Atlanta, GA 30322, [email protected].
Funding/support: B. Butts was partially funded for this work through National Institutes of Health/National
Institute of Nursing Research Grant #T32NR012715.
1. Velagaleti RS and Vasan RS. Heart failure in the twenty-first century: is it a coronary artery disease or hypertension problem. Cardiol Clin 2007;25:487–95.
2. Vincent GK, Velkoff VA. The next four decades. The older population in the United States: 2010 to 2050. United States Census Bureau Report No: P25-1138. U.S. Department of Commerce; May 2010.
3. Butler J, Kalogeropoulos A. Worsening heart failure hospitalization epidemic we do not know how to prevent and we do not know how to treat. J Am Coll Cardiol 2008;52:435–7.
4. Gary R. Evaluation of frailty in older adults with cardiovascular disease: incorporating physical performance measures. J Cardiovasc Nurs 2012;27:120–131.
5. Shamliyan T, Talley KM, Ramakrishnan R, Kane RL. Association of frailty with survival: a systematic literature review. Age Res Rev 2013;12:719–36.
6. Fried LP, Tangen CM, Walston J, et al. Frailty in older adults: evidence for a phenotype. J Gerontol Med Sci 2001; 56:M146–M156.
7. Newman AB, Gottdiener JS, Mcburnie MA, et al. Associations of subclinical cardiovascular disease with frailty. J Gerontol A Biol Sci Med Sci 2001;56:M158–66.
8. Afilalo J, Karunananthan S, Eisenberg MJ, et al. Role of frailty in patients with cardiovascular disease. Am J Cardiol 2009; 103:1616–21.
9. Cacciatore F, Abete P, Maella F, et al. Frailty predicts long-term mortality in elderly subjects with chronic heart failure. Eur J Clin Invest 2008;35:723–30.
10. Lupón J, González B, Santaeugenia S, et al. Prognostic implication of frailty and depressive symptoms in an outpatient population with heart failure. Rev Españ Cardiol 2008;61:835–42.
11. Rich MW. Heart failure in the oldest patients: the impact of comorbid conditions. Am J Geriatr Cardiol 2007;14:134–41.
12. Buck HG, Riegel B. The impact of frailty on health related quality of life in heart failure. Eur J Cardiovasc Nurs 2011;10:159–66
13. Boxer RS, Shah KB Kenny AM. Frailty and prognosis in advanced heart failure. Curr Opin Supp Pall Care 2014;8:25–9.
14. Afilalo J, Sebag IA, Chalifour LE, et al. Age-related changes in lamin A/C expression in cardiomyocytes. Am J Physiol Heart Circ Physiol 2007;293:H1451–6.
15. Walston J. Frailty—the search for underlying causes. Sci Aging Know Environ 2004;2004:e4.
16. Hubbard RE, O’Mahony MS, Savva GM, et al. Inflammation and frailty measures in older people. J Cell Mol Med 2009; 13:3103–9.
17. Hubbard RE Woodhouse KW. Frailty, inflammation and the elderly. Biogerontol 2010;11:635–41.
18. Baptista G, Dupuy A-M, Jaussent A, et al. Low-grade chronic inflammation and superoxide anion production by NADPH oxidase are the main determinants of physical frailty in older adults. Free Rad Res 2012;46:1108–14.
19. Abbate A. The heart on fire: Inflammasome and cardiomyopathy. Exper Physiol 2013;98:385.
20. Collerton J, Martin-Ruiz C, Davies K, et al. Frailty and the role of inflammation, immunosenescence and cellular ageing in the very old: Cross-sectional findings from the Newcastle 85+ Study. Mech Age Devel 2012;133:456–66.
21. Ferrucci L, Harris TB, Guralnik JM, et al. Serum IL-6 level and the development of disability in older persons. J Am Geriatr Soc 1999;47:639–46.
22. Toth MJ, Matthews DE, Tracy RP and Previs MJ. Age-related differences in skeletal muscle protein synthesis: relation to markers of immune activation. Am j Physiol Endocrin Metab 2005;288:E883–91.
23. Howard C, Ferrucci L, Sun K, et al. Oxidative protein damage is associated with poor grip strength among older women living in the community. J Appl Physiol 2007;103:17–20.
24. Muzembo BA, Nagano Y, Eitoku M, et al. A cross-sectional assessment of oxidative DNA damage and muscle strength among elderly people living in the community. Envir Health Prev Med 2014;19:21–9.
25. Wu I-C, Shiesh S-C, Kuo P-H and Lin X-Z. High oxidative stress is correlated with frailty in elderly Chinese. J Am Geriatr Soc 2009;57:1666–71.
26. Alosco ML, Spitznagel MB, Cohen R, et al. Cognitive impairment is independently associated with reduced instrumental activities of daily living in persons with heart failure. J Cardiovasc Nurs 2012;27:44–50.
27. Feola M, Rosso GL, Peano M, et al. Correlation between cognitive impairment and prognostic parameters in patients with congestive heart failure. Arch Med Res 2007;38:234–9.
28. Pressler SJ, Subramanian U, Kareken D, et al. Cognitive deficits in chronic heart failure. Nurs Res 2010;59:127–39.
29. Pressler SJ, Kim J, Riley P, et al. Memory dysfunction, psychomotor slowing, and decreased executive function predict mortality in patients with heart failure and low ejection fraction. J Cardiac Fail 2010;16:750–60.
30. Pressler SJ, Subramanian U, Kareken D, et al. Cognitive deficits and health-related quality of life in chronic heart failure. J Cardiovasc Nurs 2010;25:189–98.
31. Hajduk AM, Lemon SC, Mcmanus DD, et al. Cognitive impairment and self-care in heart failure. Clin Epidemiol 2013; 24:407–16.
32. Dardiotis E, Giamouzis G, Mastrogiannis D, et al. Cognitive impairment in heart failure. Cardiol Res Prac 2012; 2012:595821.
33. Petersen RC and O’brien J. Mild cognitive impairment should be considered for DSM-V. J Geriatr Psychiatry Neurol 2006; 19:147–54.
34. Hjelm C, Dahl A, Broström A, et al. The influence of heart failure on longitudinal changes in cognition among individuals 80 years of age and older. J Clin Nurs 2012; 21:994–1003.
35. Almeida OP, Garrido GJ, Beer C, et al. Cognitive and brain changes associated with ischaemic heart disease and heart failure. Eur Heart J 2012;33:1769–76.
36. Folstein MF, Folstein SE, McHugh PR. Mini-mental state. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–98.
37. Nasveddine ZS, Phillips NA, Bédirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53:695–699.
38. Cameron J, Worrall-Carter L, Page K, et al. Screening for mild cognitive impairment in patients with heart failure: Montreal cognitive assessment versus mini mental state exam. Eur J Cardiovasc Nurs 2013;12:252–60.
39. Avila-Funes JA, Amieva H, Barberger-Gateau P, et al. Cognitive impairment improves the predictive validity of the phenotype of frailty for adverse health outcomes: the three-city study. J Am Geriatr Soc 57:453–61.
40. Perera S, Mody SH, Woodman RC, Studenski SA. Meaningful change and responsiveness in common physical performance measures in older adults. Am Geriatr Soc 2006;54:743–9.
41. Kwon S, Perera S, Pahor M, et al. What is a meaningful change in physical performance? Findings from a clinical trial in older adults (the LIFE-P study). J Nutr Health Aging 2009;13:538–44.
42. Abellan Van Kan G, Rolland Y, Houles M, et al. The assessment of frailty in older adults. Clin Geriatr Med 2010; 26:275–86.
43. Pahor M, Manini T, Cesari M. Sarcopenia: clinical evaluation, biological markers and other evaluation tools. J Nutr Health Aging 2009;13:724–8.
44. Gary R. Evaluation of frailty in older adults with cardiovascular disease. J Cardiovasc Nurs 2012;27:120–31.
45. Macklai NS, Spagnoli J, Junod J, Santos-Eggimann B. Prospective association of the SHARE-operationalized frailty phenotype with adverse health outcomes: evidence from 620+ community-dwelling Europeans living in 11 countries. BMC Geriatr 2013;13:1–9.
46. Ravinrarajah R, Lee DM, Pye SR, et al. The ability of three different models of frailty to predict all-cause mortality: Results from the European Male Aging Study (EMAS). Arch Gerontol Geriatr 2013;57:360–8.
47. Rockwood K, Andrew M and Mitnitski A. A comparison of two approaches to measuring frailty in elderly people. J Gerontol Med Sci 2007;62:738–43.
48. Morley JE, Vellas B, Van Kan GA, et al. Frailty consensus: a call to action. J Am Med Dir Assoc 2013;14:392–7.
49. Guralnik JM, Simonsick EM, Ferrucci L, et al. A short physical performance battery assessing lower extremity function: association with self-reported disability and prediction of mortality and nursing home admission. J Gerontol 1994;49:M85–94.
50. Guralnik JM, Ferrucci L, Simonsick EM, et al. Lower-extremity function in persons over the age of 70 years as a predictor of subsequent disability. N Engl J Med 1995;332:556–61.
51. Di Bari M, Pozzi C, Cavallini Mc, et al. The diagnosis of heart failure in the community. Comparative validation of four sets of criteria in unselected older adults: the ICARe Dicomano Study. J Am Coll Cardiol 2004;44:1601–08.
52. Chiarantini D, Volpato S, Sioulis F, et al. Lower extremity performance measures predict long-term prognosis in older patients hospitalized for heart failure. J Cardiac Failure 2010; 16:390–5.
53. Volpato S, Cavalieri M, Sioulis F, et al. Predictive value of the short physical performance battery following hospitalization in older patients. J Gerontol A Biol Sci Med Sci 2011;66:89–96.
54. Studenski S, Perera S, Wallace D, et al. Physical performance measures in the clinical setting. J Am Geriatr Soc 2003; 51:314–22.
55. Binder EF, Schechtman KB, Ehsani AA, et al. Effects of exercise training on frailty in community-dwelling older adults: results of a randomized, controlled trial. J Am Geriatr Soc 2012; 50:1921–8.
56. Brown M, Sinacore DR, Ehsani AA, et al. Low-intensity exercise as a modifier of physical frailty in older adults. Arch Phys Med Rehab 2000;81:960–5.
57. Yamada M, Arai H, Sonoda T and Aoyama T. Community-based exercise program is cost-effective by preventing care and disability in Japanese frail older adults. J Am Med Dir Assoc 2012;13:507–11.
58. Gary RA, Cress ME, Higgins MK, et al. A combined aerobic and resistance exercise program improves physical functional performance in patients with heart failure: a pilot study. J Cardiovasc Nurs 2012;27:418–30.
59. De Meirelles L, Matsuura C, Resende AD, et al. Chronic exercise leads to antiaggregant, antioxidant and anti-inflammatory effects in heart failure patients. Eur J Prev Cardiol 2014;21:1225–32.
60. Feiereisen P, Vaillant M, Gilson G, Delagardelle C. Effects of different training modalities on circulating anabolic/catabolic markers in chronic heart failure. J Cardiopulm Rehab Prev 2013;33:303–8.
61. Smart NA, Steele M. The effect of physical training on systemic proinflammatory cytokine expression in heart failure patients: a systematic review. Congest Heart Fail 2011;17:110–4.
62. Nunes RB, Alves JP, Kessler LP, Lago PD. Aerobic exercsie improves the inflammatory profile correlated with cardiac remodeling and function in chronic heart failure rats. Clin Chest Med 2013;68:876–82.
63. Larson EB, Wang L, Bowen JD, et al. Exercise is associated with reduced risk for incident dementia among persons 65 years of age and older. Ann Intern Med 2006;144:73–8.
64. Colcombe S and Kramer AF. Fitness effects on the cognitive function of older adults: a meta-analytic study. Psychol Sci 2003;14:125–30.
65. Heyn P, Abreu BC, Ottenbacher KJ. The effects of exercise training on elderly persons with cognitive impairment and dementia: a meta-analysis. Arch Phys Med Rehab 2004; 85:1694–704.
66. Bautmans I, Vanpuyvelde K, Mets T. Sarcopenia and functional decline: pathophysiology, prevention and therapy. Acta Clinica Belgica 2009;64:303–16.
67. Guralnik JM, Ferrucci L, Pieper CF, et al. Lower extremity function and subsequent disability: Consistency across studies, predictive models, and value of gait speed alone compared with the short physical performance battery. J Gerontol A Biol Sci Med Sci 2000;55A:M221–M231.
68. Kramer AF, Erickson KI. Capitalizing on cortical plasticity: influence of physical activity on cognition and brain function. Trends Cogn Sci 2007;11:342–8.
69. Harkness K, Heckman GA, Mckelvie RS. The older patient with heart failure: high risk for frailty and cognitive impairment. Expert Rev Cardiovasc Ther 2012;10:779–95.
70. Waters DL, Baumgartner RN, Garry PJ, Vellas B. Advantages of dietary, exercise-related, and therapeutic interventions to prevent and treat sarcopenia in adult patients: an update. Clin Interv Aging 2010;5:259–70.
From the Nell Hodgson Woodruff School of Nursing, Emory University, Atlanta, GA.
Abstract
- Objective: To review some of the proposed pathways that increase frailty risk in older persons with heart failure and to discuss tools that may be used to assess for changes in physical and cognitive functioning in this population in order to assist with appropriate and timely intervention.
- Methods: Review of the literature.
- Results: Heart failure is the only cardiovascular disease that is increasing by epidemic proportions, largely due to an aging society and therapeutic advances in disease management. Because heart failure is largely a cardiogeriatric syndrome, age-related syndromes such as frailty and cognitive impairment are common in heart failure patients. Compared with age-matched counterparts, older adults with heart failure 4 to 6 times more likely to be frail or cognitively impaired. The reason for the high prevalence of frailty and cognitive impairment in this population is not well known but may likely reflect the synergistic effects of heart failure and aging, which may heighten vulnerability to stressors and accelerate loss of physiologic reserve. Despite the high prevalence of frailty and cognitive impairment in the heart failure population, these conditions are not routinely screened for in clinical practice settings and guidelines on optimal assessment strategies are lacking.
- Conclusion: Persons with heart failure are at an increased risk for frailty, which may worsen symptoms, impair self-management, and lead to worse heart failure outcomes. Early detection of frailty and cognitive impairment may be an opportunity for intervention and a key strategy for improving clinical outcomes in older adults with heart failure.
Approximately 5.7 million persons in the United States are diagnosed with heart failure, and the number of reported new cases is expected to increase to over 700,000 cases annually by the year 2040 [1]. This rising incidence is fueled by an aging population; by the year 2030, 1 in 5 Americans will be over 65 years of age [2]. Heart failure is prevalent among those 65 years of age and older and is the most common reason for hospitalization in this age-group. High readmission rates, approaching 50% over 6 months, are a major contributor to the the escalating economic burden associated with heart failure [3].
Persons with heart failure are more likely to be frail and experience cognitive impairment than their age-matched counterparts without heart failure. The reasons for this are not well known but may be related to hemodynamic, vascular, and inflammatory changes occurring as heart failure progresses. In this paper, we review the link between frailty and cognitive impairment in heart failure, instruments that may be useful for early detection, and interventions such as exercise that may be beneficial for attenuating both conditions.
Frailty in Heart Failure
Epidemiology
Frailty is a heightened vulnerability to stressors in the presence of low physiological reserve [4]. When exposed to stressors, persons who are frail have a much higher probability for disproportionate decompensation, negative events, functional decline, disability, and mortality [5]. Among persons with heart failure, frailty may predispose them to decompensate at a lower threshold, requiring more frequent hospitalizations. Persons with heart failure are more likely to be frail than their age-matched counterparts without heart failure [6,7].
Frailty is a powerful predictor of poor clinical outcomes and mortality in cardiovascular disease [8,9]. Compared with the non-frail, frail persons with heart failure have increased rates of mortality (16.9% vs 4.8%) and increased rates of heart failure hospitalization (20.5% vs 13.3%) [10]. Frailty has also been shown to predict falls, disability, and hospitalization in heart failure patients [6,9,11] and was found to have a negative linear relationship with health-related quality of life [12]. Frail heart failure patients are also more likely to have comorbidities such diabetes mellitus, chronic obstructive pulmonary disease, atrial fibrillation, depression, anemia, and chronic kidney disease [9,13].
Pathophysiology
There is significant overlap in the underlying pathological mechanisms of heart failure and frailty. Symptoms of heart failure, such as dyspnea, fatigue, and muscle loss, mirror components that occur with frailty. Further, cardiac cachexia, a metabolic syndrome in advanced heart failure characterized by a loss of muscle mass, is very similar to the sarcopenia that occurs in frailty.
Frailty, characterized by an increased physiologic vulnerability to stressors, may predispose frail persons with heart failure to exacerbation and worsening of heart failure due to greater susceptibility to the harmful pathophysiologic processes in heart failure, such as inflammation and autonomic dysfunction. Proposed pathophysiologic pathways in frailty include free radicals and oxidative stress, cumulative DNA damage, decreased telomere length, and nuclear fragmentation [14,15]. Frailty has been associated with low-grade chronic inflammation and increased inflammatory cytokines, such as C-reactive protein, tumor necrosis factor–alpha (TNFα), interleukin-6 (IL-6)and fibrinogen [16–18]. Heart failure also is associated with a low-grade and chronic cardiac inflammatory response that is correlated with disease progression [19].
Inflammation. IL-6 is detectable in a higher proportion of persons who are frail compared to non-frail [16] and is the most highly correlated biomarker with frailty. In addition, among those with detectable IL-6 levels, those categorized as frail have higher IL-6 levels compared to those who are non-frail [16,20]. Individuals categorized as frail were found to have significantly higher levels of TNFα than those who were non-frail [16,20]. Increased IL-6 levels are associated with decreased muscle strength, while increased TNFα levels are associated with decreased skeletal muscle protein synthesis [21,22], thus contributing to frailty.
Oxidative stress. Protein carbonyls result from protein oxidation promoted by reactive oxygen species and are markers of oxidative stress. Protein carbonylation is implicated in the pathogenesis of the loss of skeletal muscle mass; high serum protein carbonyls are associated with poor grip strength [23]. 8-OHdG is an oxidized nucleoside indicative of oxidative damage to DNA and a measure of oxidative stress. Accumulation of 8-OHdG in skeletal muscle leads to loss of muscle mass and is associated with decreased hand grip strength in the elderly [24]. Higher serum levels of 8-OHdG are present in older adults who are frail as compared to those who are non-frail [25].
Measurement of Frailty in the Clinical Setting
Frailty has been conceptualized in a number of studies using different models and measures; however, there continues to be a lack of consensus on the definition and operationalization of frailty. Prior research has led to the development of several validated models of frailty that have demonstrated good prediction of adverse outcomes in older adults. Some models, such as the Fried phenotype [6], focus solely on the physical dimension, while other models take a multidimensional approach.Single-item measures (eg, gait speed, 6-minute walk test, handgrip strength) are also commonly used to screen for frailty, but a frailty measure that incorporates more than 1 physical dimension may be more sensitive and reliable. In our opinion, the ideal measure of frailty would consist of a brief assessment that can be serially performed in most clinical practice settings that can identify change in function over time. The incorporation of sensitive physical function measures that can detect frailty early has the potential to slow physical function decline by preserving physiological thresholds.
Cognitive Impairment in Heart Failure
Epidemiology
Cognitive impairment occurs frequently in patients with heart failure, and the presence of cognitive impairment in persons with heart failure has been shown to heighten risk for adverse clinical outcomes, disability, poor quality of life, and mortality [26,27]. Heart failure negatively influences cognitive functioning in most domains [28–32]. The most common domains adversely affected by heart failure and aging are memory and executive function. Deficits in these domains can substantially diminish patient ability to carry out essential self-care behaviors [30,32].
The most common form of cognitive impairment seen in patients with heart failure is mild cognitive impairment (MCI), which is a measurable deficit with memory or another core cognitive domain. Up to 60% of persons with heart failure have been reported to have MCI. Patients with MCI have cognitive deficits that are more pronounced than those seen in normal aging, but lack other symptoms of dementia, such as impaired judgment or reasoning. MCI often will not impede patients’ ability to carry out the activities of daily living (ADLs) independently, but patients may have difficulty in performing some instrumental activities of daily living (IADLs), such as remembering medications, scheduling provider appointments. Dementia, a decline in cognitive ability severe enough to hinder an individual’s ability to perform ADLs or IADLs or engage in social activities or occupational responsibilities, occurs in approximately 25% of persons with heart failure [33].
Persons with heart failure have a fourfold greater likelihood of developing CI than persons without heart failure. Several cohort studies have shown that persons with heart failure had lower performance on cognitive tests than individuals without heart failure [34,35] and were 50% more likely to progress to dementia.
Assessment Tools
Although a comprehensive neurocognitive battery would aid in detecting cognitive impairment in heart failure, few clinical practice settings have the resources to perform such a detailed and time-consuming measurement. Most studies in heart failure have relied on global screening questionnaires such as the Mini-Mental State Examination (MMSE) [36] to assess cognitive functioning in persons with heart failure and in other cardiovascular disorders. Global cognitive measures, however, often lack sensitivity for detecting subtle cognitive deficits such as seen in MCI [28–30]. Screening that measures executive function may be the most beneficial for busy clinical settings, since declines in this domain are well established as contributing to poor outcomes in persons with heart failure.
The Montreal Cognitive Assessment (MoCA) is a rapid screening test designed to detect MCI. It assesses different cognitive domains, including attention, memory, language, and executive function [37]. The MoCA lends itself to use in clinical setting because it is brief, requires little training to administer, and is easy to score. This instrument has been used successfully to assess MCI in persons with heart failure and may be more sensitive than the MMSE in identifying clinically relevant cognitive dysfunction. In 2013 study, Cameron et al [38] administered the MMSE and MoCA to 93 hospitalized heart failure patients and found that the MoCA classified 41% of patients as cognitively impaired that were not classified using the MMSE. For persons with a vascular cognitive deficit, the MMSE has been portrayed as an inadequate screening test due to lack of sensitivity for visuospatial and executive function deficits. Because the MoCA was designed to be more sensitive to such deficits, it may be a superior screening method for persons with heart failure. Although previous studies support the use of the MoCA in persons with heart failure, more research is needed in larger, more diverse heart failure samples with a wide range of cognitive deficits.
A Reasonable Clinical Assessment Approach
Considering the link between heart failure, frailty, and MCI, incorporating simple physical performance measures with cognitive screening may be an effective strategy to identify persons at risk for frailty. Two clinically relevant physical performance-based measures of frailty are proposed: the Fried phenotype (mentioned earlier) and the Short Physical Performance Battery (SBBP). In addition, cognitive screening using the MoCA is recommended as part of the routine examination for determining possible MCI or more severe cognitive deficits. The predictive validity of measuring physical frailty is enhanced when cognitive impairment is included in the assessment [36,39].
The performance-based measures included in this review have previously demonstrated excellent psychometric properties as well as sensitivity for change that is clinically meaningful. Minimal detectable change (MDC), a threshold score that refers to the minimal amount of change outside of error that reflects true change by a patient between 2 time points (rather than variation in measurement), is important for interpreting level of risk for frailty and is included for each instrument [40,41]. If a more brief frailty examination is needed, cut-points for gait speed and handgrip have been used effectively in a number of studies as a threshold for determining frailty, including in older patients with cardiovascular disease and in heart failure [8,42,43].
Fried Frailty Phenotype
The Fried phenotype is an appropriate method of measuring frailty in a clinical setting due to its wide application across diverse populations and consistent identification of adverse outcomes [44]. This model is derived from a frailty model proposed by Fried et al [6] in which a phenotypic cycle exists that includes disease, sarcopenia, decreased walking speed, chronic undernutrition, decreased total energy expenditure, senescent musculoskeletal changes, decreased resting metabolic rate, weight loss and decreased maximal oxygen consumption. Frailty exists when a critical mass of these cycle components are identified in an individual [6].
To validate the model, Fried et al used data from the Cardiovascular Health Study and used the model to show association with a 3-year and 7-year incidence of mobility and ADL disability among 4317 community-dwelling men and women aged 65 years and older, independent of comorbidities. Several studies have directly tested the frailty phenotype model alone and in comparison to other models of frailty in large prospective studies across different populations, such as the Survey of Health, Aging and Retirement in Europe (SHARE) [45], the European Male Aging Study [46], and the Canadian Health Study of Aging [47]. While these studies found the prevalence of frailty to vary across the populations, they all validated the Fried model and found no significant differences in the predictive ability of the Fried model and other models of frailty. The Frailty Consensus conference evaluated the different models of frailty and determined that the Fried model is a validated construct of frailty and is acceptable for use in the identification of individuals who are frail or likely to become frail [48]. Thus, the Fried et al frailty phenotype model is considered to be a standard measure of frailty in older individuals.
Short Physical Performance Battery
In other chronic illness populations, the SPPB has also been used as a predictor of outcomes before, during, or after hospitalization. Valpato et al [53], for example, used the SPPB to assess older adults (mean age, 78 yr) admitted to the hospital with a diagnosis of heart failure (64%), pneumonia (13%), chronic obstructive pulmonary disease (16%), or minor stroke (6.6%) at admission (baseline) and discharge. Patients with the lowest SPPB quartile scores at hospital discharge had a fivefold greater risk of rehospitalization or mortality compared to the highest quartile. In addition, those who had an early decline in SPPB scores 1 month after hospital discharge had greater limitations in performing activities of daily living and a significantly greater probability of being re-hospitalized or death during the 1-year follow-up period. These studies suggest that the SPPB at the first follow-up outpatient visit following hospital discharge may be beneficial for identifying need for further intervention or the need for more frequent follow-up care. Although the SPPB is not part of the Fried et al phenotype, it may provide additional information concerning risk for falls and lower extremity strength that may be beneficial in the evaluation of some persons with heart failure [54]. The SPPB along with instructions and normative data are available for clinical use at no charge at www.grc.nia.nih.gov/branches/ledb/sppb/index.htm.
Interventions for Frailty in Heart Failure
Interventions to address frailty have included exercise training, comprehensive geriatric assessment and management services, social support systems, nutrition, and drugs; however, few intervention studies have examined frailty in heart failure [8]. Restoration of physical function through aerobic exercise and resistance training has shown benefit in frail older adults [55–57] and in persons with heart failure [58]. Maintaining and/or restoring physical function through aerobic and resistance exercise training may be the key to preventing further decline or potentially reversing frailty in older adults with heart failure.
Aerobic exercise has been shown to be beneficial for both frail older adults and frail persons with heart failure [18]. In a study of community-dwelling frail older adults aged 65 and older, a combined aerobic and resistance exercise intervention, performed over 16 weeks, demonstrated significant improvement in frailty scores during the 1-year follow-up in contrast to worsening frailty measures in the control group [57].
Older adults with heart failure experience a much lower exercise tolerance largely due to a 50% to 75% decrease in aerobic capacity in addition to the well-known alterations in peripheral musculoskeletal performance that contribute to fatigue and greater symptom severity. Aerobic exercise has been shown to be beneficial for most heart failure patients by altering the peripheral and central mechanisms, such as inflammatory cytokines, that contribute to heart failure exacerbations, worsened symptom severity, and poor clinical outcomes [59–62].Lower rates of hospitalization, improved physical function, and enhanced health-related quality of life are reported in heart failure patients who routinely exercise [59]. Resistance training has been shown to improve physical function in frail older adults [55]. Further, the use of TheraBand exercise bands in resistance training demonstrated improvement in physical function among frail older adults [56].
Exercise also appears to exert a positive effect on cognition, particularly executive functioning, and may also have a protective effect against cognitive decline with aging and among those with heart failure. The underlying mechanism for improvement in cognition remains poorly understood but is likely related to improved cardiac function, cerebral perfusion, and oxygenation, although this has not been clearly established. Larson et al (2006) evaluated the frequency of participation in a variety of physical activities (eg, walking, bicycling and swimming) over 6 years in 1740 older adults [63]. Older adults who exercised more than 3 times per week during initial assessment were 34% less likely to be diagnosed with dementia than those who exercised fewer than 3 times per week. Several meta-analyses in recent years have shown a consistent and positive relationship between aerobic exercise and cognition [64,65]. Importantly, findings from meta-analyses have shown a moderate effect size (> 0.5) from aerobic training, which was similar for normal and cognitively impaired adults [64].
Implications for Clinical Care
A systematic assessment performed periodically using physical and cognitive measures that may identify prefrailty may be the best strategy for preventing further functional loss, limitations, and disability in persons with heart failure. Persons with heart failure ideally should be evaluated annually for physical function, since a decline has been consistently shown to be a strong predictor of adverse health outcomes, disability, and death [6,66]. Cognitive function should also be assessed routinely in persons with heart failure, particularly when first diagnosed, when changes in treatment regimen occur, and with worsening disease severity, since these events have been shown to occur before changes in cognition [31]. Incorporating geriatric performance-based measures in heart failure management would allow for more treatment strategies aimed at improving physical function, cognitive outcomes, and quality of life. Further, identifying frailty in heart failure is an important component of clinical decision-making when determining if a patient can tolerate therapies such as implantable defibrillators, cardiac resynchronization therapy, or left ventricular assist device placement.
In older adults, performance measures are well established and commonly used as part of geriatric assessment to evaluate physical and cognitive functioning. Performance-based measures may be particularly beneficial in older adults with heart failure to monitor serial changes in physical function. Performance measures in clinical settings require staff time but little training, space, equipment, or risk. As performance measures become more common in practice settings, MDC thresholds may need to be re-evaluated based on the characteristics of the population [67].
For persons with heart failure whose screening outcomes suggest MCI, more comprehensive neuropsychological testing should be available as well as provision of resources to optimize functional independence. Early identification of impaired cognition may lower risk of poor self-management through simplification of medication regimens or providing resources to help manage other regimens essential for optimal heart failure care. It is also important to recognize that depressive symptoms are common in persons with heart failure and are highly correlated with cognitive impairment in this population. Screening for depressive symptoms therefore, may also enhance identification of persons with heart failure at risk for frailty [4,28].
Conclusion
Effective appraisal and development of effective interventions are essential in older adults with heart failure who are at high risk for frailty and cognitive impairment. This will become increasingly important as the population ages and the incidence of heart failure rises proportionately. Although curative treatments for frailty and cognitive impairment are not available, interdisciplinary interventions such as exercise and comprehensive geriatric assessment may improve outcomes in older persons with heart failure [68]. Information gained from objective, simple, inexpensive physical performance measures, when used in combination with cognitive screening, may enhance the ability to evaluate change that signal onset of frailty or cognitive impairment [54,69,70]. The high morbidity and mortality associated with frailty and cognitive impairment indicate that it should be a priority for future research as a strategy to improve clinical outcomes, enhance quality of life, and lower health care costs in this growing population.
Corresponding author: Rebecca Gary, PhD, RN, Nell Hodgson Woodruff School of Nursing, Emory University, Atlanta, GA 30322, [email protected].
Funding/support: B. Butts was partially funded for this work through National Institutes of Health/National
Institute of Nursing Research Grant #T32NR012715.
From the Nell Hodgson Woodruff School of Nursing, Emory University, Atlanta, GA.
Abstract
- Objective: To review some of the proposed pathways that increase frailty risk in older persons with heart failure and to discuss tools that may be used to assess for changes in physical and cognitive functioning in this population in order to assist with appropriate and timely intervention.
- Methods: Review of the literature.
- Results: Heart failure is the only cardiovascular disease that is increasing by epidemic proportions, largely due to an aging society and therapeutic advances in disease management. Because heart failure is largely a cardiogeriatric syndrome, age-related syndromes such as frailty and cognitive impairment are common in heart failure patients. Compared with age-matched counterparts, older adults with heart failure 4 to 6 times more likely to be frail or cognitively impaired. The reason for the high prevalence of frailty and cognitive impairment in this population is not well known but may likely reflect the synergistic effects of heart failure and aging, which may heighten vulnerability to stressors and accelerate loss of physiologic reserve. Despite the high prevalence of frailty and cognitive impairment in the heart failure population, these conditions are not routinely screened for in clinical practice settings and guidelines on optimal assessment strategies are lacking.
- Conclusion: Persons with heart failure are at an increased risk for frailty, which may worsen symptoms, impair self-management, and lead to worse heart failure outcomes. Early detection of frailty and cognitive impairment may be an opportunity for intervention and a key strategy for improving clinical outcomes in older adults with heart failure.
Approximately 5.7 million persons in the United States are diagnosed with heart failure, and the number of reported new cases is expected to increase to over 700,000 cases annually by the year 2040 [1]. This rising incidence is fueled by an aging population; by the year 2030, 1 in 5 Americans will be over 65 years of age [2]. Heart failure is prevalent among those 65 years of age and older and is the most common reason for hospitalization in this age-group. High readmission rates, approaching 50% over 6 months, are a major contributor to the the escalating economic burden associated with heart failure [3].
Persons with heart failure are more likely to be frail and experience cognitive impairment than their age-matched counterparts without heart failure. The reasons for this are not well known but may be related to hemodynamic, vascular, and inflammatory changes occurring as heart failure progresses. In this paper, we review the link between frailty and cognitive impairment in heart failure, instruments that may be useful for early detection, and interventions such as exercise that may be beneficial for attenuating both conditions.
Frailty in Heart Failure
Epidemiology
Frailty is a heightened vulnerability to stressors in the presence of low physiological reserve [4]. When exposed to stressors, persons who are frail have a much higher probability for disproportionate decompensation, negative events, functional decline, disability, and mortality [5]. Among persons with heart failure, frailty may predispose them to decompensate at a lower threshold, requiring more frequent hospitalizations. Persons with heart failure are more likely to be frail than their age-matched counterparts without heart failure [6,7].
Frailty is a powerful predictor of poor clinical outcomes and mortality in cardiovascular disease [8,9]. Compared with the non-frail, frail persons with heart failure have increased rates of mortality (16.9% vs 4.8%) and increased rates of heart failure hospitalization (20.5% vs 13.3%) [10]. Frailty has also been shown to predict falls, disability, and hospitalization in heart failure patients [6,9,11] and was found to have a negative linear relationship with health-related quality of life [12]. Frail heart failure patients are also more likely to have comorbidities such diabetes mellitus, chronic obstructive pulmonary disease, atrial fibrillation, depression, anemia, and chronic kidney disease [9,13].
Pathophysiology
There is significant overlap in the underlying pathological mechanisms of heart failure and frailty. Symptoms of heart failure, such as dyspnea, fatigue, and muscle loss, mirror components that occur with frailty. Further, cardiac cachexia, a metabolic syndrome in advanced heart failure characterized by a loss of muscle mass, is very similar to the sarcopenia that occurs in frailty.
Frailty, characterized by an increased physiologic vulnerability to stressors, may predispose frail persons with heart failure to exacerbation and worsening of heart failure due to greater susceptibility to the harmful pathophysiologic processes in heart failure, such as inflammation and autonomic dysfunction. Proposed pathophysiologic pathways in frailty include free radicals and oxidative stress, cumulative DNA damage, decreased telomere length, and nuclear fragmentation [14,15]. Frailty has been associated with low-grade chronic inflammation and increased inflammatory cytokines, such as C-reactive protein, tumor necrosis factor–alpha (TNFα), interleukin-6 (IL-6)and fibrinogen [16–18]. Heart failure also is associated with a low-grade and chronic cardiac inflammatory response that is correlated with disease progression [19].
Inflammation. IL-6 is detectable in a higher proportion of persons who are frail compared to non-frail [16] and is the most highly correlated biomarker with frailty. In addition, among those with detectable IL-6 levels, those categorized as frail have higher IL-6 levels compared to those who are non-frail [16,20]. Individuals categorized as frail were found to have significantly higher levels of TNFα than those who were non-frail [16,20]. Increased IL-6 levels are associated with decreased muscle strength, while increased TNFα levels are associated with decreased skeletal muscle protein synthesis [21,22], thus contributing to frailty.
Oxidative stress. Protein carbonyls result from protein oxidation promoted by reactive oxygen species and are markers of oxidative stress. Protein carbonylation is implicated in the pathogenesis of the loss of skeletal muscle mass; high serum protein carbonyls are associated with poor grip strength [23]. 8-OHdG is an oxidized nucleoside indicative of oxidative damage to DNA and a measure of oxidative stress. Accumulation of 8-OHdG in skeletal muscle leads to loss of muscle mass and is associated with decreased hand grip strength in the elderly [24]. Higher serum levels of 8-OHdG are present in older adults who are frail as compared to those who are non-frail [25].
Measurement of Frailty in the Clinical Setting
Frailty has been conceptualized in a number of studies using different models and measures; however, there continues to be a lack of consensus on the definition and operationalization of frailty. Prior research has led to the development of several validated models of frailty that have demonstrated good prediction of adverse outcomes in older adults. Some models, such as the Fried phenotype [6], focus solely on the physical dimension, while other models take a multidimensional approach.Single-item measures (eg, gait speed, 6-minute walk test, handgrip strength) are also commonly used to screen for frailty, but a frailty measure that incorporates more than 1 physical dimension may be more sensitive and reliable. In our opinion, the ideal measure of frailty would consist of a brief assessment that can be serially performed in most clinical practice settings that can identify change in function over time. The incorporation of sensitive physical function measures that can detect frailty early has the potential to slow physical function decline by preserving physiological thresholds.
Cognitive Impairment in Heart Failure
Epidemiology
Cognitive impairment occurs frequently in patients with heart failure, and the presence of cognitive impairment in persons with heart failure has been shown to heighten risk for adverse clinical outcomes, disability, poor quality of life, and mortality [26,27]. Heart failure negatively influences cognitive functioning in most domains [28–32]. The most common domains adversely affected by heart failure and aging are memory and executive function. Deficits in these domains can substantially diminish patient ability to carry out essential self-care behaviors [30,32].
The most common form of cognitive impairment seen in patients with heart failure is mild cognitive impairment (MCI), which is a measurable deficit with memory or another core cognitive domain. Up to 60% of persons with heart failure have been reported to have MCI. Patients with MCI have cognitive deficits that are more pronounced than those seen in normal aging, but lack other symptoms of dementia, such as impaired judgment or reasoning. MCI often will not impede patients’ ability to carry out the activities of daily living (ADLs) independently, but patients may have difficulty in performing some instrumental activities of daily living (IADLs), such as remembering medications, scheduling provider appointments. Dementia, a decline in cognitive ability severe enough to hinder an individual’s ability to perform ADLs or IADLs or engage in social activities or occupational responsibilities, occurs in approximately 25% of persons with heart failure [33].
Persons with heart failure have a fourfold greater likelihood of developing CI than persons without heart failure. Several cohort studies have shown that persons with heart failure had lower performance on cognitive tests than individuals without heart failure [34,35] and were 50% more likely to progress to dementia.
Assessment Tools
Although a comprehensive neurocognitive battery would aid in detecting cognitive impairment in heart failure, few clinical practice settings have the resources to perform such a detailed and time-consuming measurement. Most studies in heart failure have relied on global screening questionnaires such as the Mini-Mental State Examination (MMSE) [36] to assess cognitive functioning in persons with heart failure and in other cardiovascular disorders. Global cognitive measures, however, often lack sensitivity for detecting subtle cognitive deficits such as seen in MCI [28–30]. Screening that measures executive function may be the most beneficial for busy clinical settings, since declines in this domain are well established as contributing to poor outcomes in persons with heart failure.
The Montreal Cognitive Assessment (MoCA) is a rapid screening test designed to detect MCI. It assesses different cognitive domains, including attention, memory, language, and executive function [37]. The MoCA lends itself to use in clinical setting because it is brief, requires little training to administer, and is easy to score. This instrument has been used successfully to assess MCI in persons with heart failure and may be more sensitive than the MMSE in identifying clinically relevant cognitive dysfunction. In 2013 study, Cameron et al [38] administered the MMSE and MoCA to 93 hospitalized heart failure patients and found that the MoCA classified 41% of patients as cognitively impaired that were not classified using the MMSE. For persons with a vascular cognitive deficit, the MMSE has been portrayed as an inadequate screening test due to lack of sensitivity for visuospatial and executive function deficits. Because the MoCA was designed to be more sensitive to such deficits, it may be a superior screening method for persons with heart failure. Although previous studies support the use of the MoCA in persons with heart failure, more research is needed in larger, more diverse heart failure samples with a wide range of cognitive deficits.
A Reasonable Clinical Assessment Approach
Considering the link between heart failure, frailty, and MCI, incorporating simple physical performance measures with cognitive screening may be an effective strategy to identify persons at risk for frailty. Two clinically relevant physical performance-based measures of frailty are proposed: the Fried phenotype (mentioned earlier) and the Short Physical Performance Battery (SBBP). In addition, cognitive screening using the MoCA is recommended as part of the routine examination for determining possible MCI or more severe cognitive deficits. The predictive validity of measuring physical frailty is enhanced when cognitive impairment is included in the assessment [36,39].
The performance-based measures included in this review have previously demonstrated excellent psychometric properties as well as sensitivity for change that is clinically meaningful. Minimal detectable change (MDC), a threshold score that refers to the minimal amount of change outside of error that reflects true change by a patient between 2 time points (rather than variation in measurement), is important for interpreting level of risk for frailty and is included for each instrument [40,41]. If a more brief frailty examination is needed, cut-points for gait speed and handgrip have been used effectively in a number of studies as a threshold for determining frailty, including in older patients with cardiovascular disease and in heart failure [8,42,43].
Fried Frailty Phenotype
The Fried phenotype is an appropriate method of measuring frailty in a clinical setting due to its wide application across diverse populations and consistent identification of adverse outcomes [44]. This model is derived from a frailty model proposed by Fried et al [6] in which a phenotypic cycle exists that includes disease, sarcopenia, decreased walking speed, chronic undernutrition, decreased total energy expenditure, senescent musculoskeletal changes, decreased resting metabolic rate, weight loss and decreased maximal oxygen consumption. Frailty exists when a critical mass of these cycle components are identified in an individual [6].
To validate the model, Fried et al used data from the Cardiovascular Health Study and used the model to show association with a 3-year and 7-year incidence of mobility and ADL disability among 4317 community-dwelling men and women aged 65 years and older, independent of comorbidities. Several studies have directly tested the frailty phenotype model alone and in comparison to other models of frailty in large prospective studies across different populations, such as the Survey of Health, Aging and Retirement in Europe (SHARE) [45], the European Male Aging Study [46], and the Canadian Health Study of Aging [47]. While these studies found the prevalence of frailty to vary across the populations, they all validated the Fried model and found no significant differences in the predictive ability of the Fried model and other models of frailty. The Frailty Consensus conference evaluated the different models of frailty and determined that the Fried model is a validated construct of frailty and is acceptable for use in the identification of individuals who are frail or likely to become frail [48]. Thus, the Fried et al frailty phenotype model is considered to be a standard measure of frailty in older individuals.
Short Physical Performance Battery
In other chronic illness populations, the SPPB has also been used as a predictor of outcomes before, during, or after hospitalization. Valpato et al [53], for example, used the SPPB to assess older adults (mean age, 78 yr) admitted to the hospital with a diagnosis of heart failure (64%), pneumonia (13%), chronic obstructive pulmonary disease (16%), or minor stroke (6.6%) at admission (baseline) and discharge. Patients with the lowest SPPB quartile scores at hospital discharge had a fivefold greater risk of rehospitalization or mortality compared to the highest quartile. In addition, those who had an early decline in SPPB scores 1 month after hospital discharge had greater limitations in performing activities of daily living and a significantly greater probability of being re-hospitalized or death during the 1-year follow-up period. These studies suggest that the SPPB at the first follow-up outpatient visit following hospital discharge may be beneficial for identifying need for further intervention or the need for more frequent follow-up care. Although the SPPB is not part of the Fried et al phenotype, it may provide additional information concerning risk for falls and lower extremity strength that may be beneficial in the evaluation of some persons with heart failure [54]. The SPPB along with instructions and normative data are available for clinical use at no charge at www.grc.nia.nih.gov/branches/ledb/sppb/index.htm.
Interventions for Frailty in Heart Failure
Interventions to address frailty have included exercise training, comprehensive geriatric assessment and management services, social support systems, nutrition, and drugs; however, few intervention studies have examined frailty in heart failure [8]. Restoration of physical function through aerobic exercise and resistance training has shown benefit in frail older adults [55–57] and in persons with heart failure [58]. Maintaining and/or restoring physical function through aerobic and resistance exercise training may be the key to preventing further decline or potentially reversing frailty in older adults with heart failure.
Aerobic exercise has been shown to be beneficial for both frail older adults and frail persons with heart failure [18]. In a study of community-dwelling frail older adults aged 65 and older, a combined aerobic and resistance exercise intervention, performed over 16 weeks, demonstrated significant improvement in frailty scores during the 1-year follow-up in contrast to worsening frailty measures in the control group [57].
Older adults with heart failure experience a much lower exercise tolerance largely due to a 50% to 75% decrease in aerobic capacity in addition to the well-known alterations in peripheral musculoskeletal performance that contribute to fatigue and greater symptom severity. Aerobic exercise has been shown to be beneficial for most heart failure patients by altering the peripheral and central mechanisms, such as inflammatory cytokines, that contribute to heart failure exacerbations, worsened symptom severity, and poor clinical outcomes [59–62].Lower rates of hospitalization, improved physical function, and enhanced health-related quality of life are reported in heart failure patients who routinely exercise [59]. Resistance training has been shown to improve physical function in frail older adults [55]. Further, the use of TheraBand exercise bands in resistance training demonstrated improvement in physical function among frail older adults [56].
Exercise also appears to exert a positive effect on cognition, particularly executive functioning, and may also have a protective effect against cognitive decline with aging and among those with heart failure. The underlying mechanism for improvement in cognition remains poorly understood but is likely related to improved cardiac function, cerebral perfusion, and oxygenation, although this has not been clearly established. Larson et al (2006) evaluated the frequency of participation in a variety of physical activities (eg, walking, bicycling and swimming) over 6 years in 1740 older adults [63]. Older adults who exercised more than 3 times per week during initial assessment were 34% less likely to be diagnosed with dementia than those who exercised fewer than 3 times per week. Several meta-analyses in recent years have shown a consistent and positive relationship between aerobic exercise and cognition [64,65]. Importantly, findings from meta-analyses have shown a moderate effect size (> 0.5) from aerobic training, which was similar for normal and cognitively impaired adults [64].
Implications for Clinical Care
A systematic assessment performed periodically using physical and cognitive measures that may identify prefrailty may be the best strategy for preventing further functional loss, limitations, and disability in persons with heart failure. Persons with heart failure ideally should be evaluated annually for physical function, since a decline has been consistently shown to be a strong predictor of adverse health outcomes, disability, and death [6,66]. Cognitive function should also be assessed routinely in persons with heart failure, particularly when first diagnosed, when changes in treatment regimen occur, and with worsening disease severity, since these events have been shown to occur before changes in cognition [31]. Incorporating geriatric performance-based measures in heart failure management would allow for more treatment strategies aimed at improving physical function, cognitive outcomes, and quality of life. Further, identifying frailty in heart failure is an important component of clinical decision-making when determining if a patient can tolerate therapies such as implantable defibrillators, cardiac resynchronization therapy, or left ventricular assist device placement.
In older adults, performance measures are well established and commonly used as part of geriatric assessment to evaluate physical and cognitive functioning. Performance-based measures may be particularly beneficial in older adults with heart failure to monitor serial changes in physical function. Performance measures in clinical settings require staff time but little training, space, equipment, or risk. As performance measures become more common in practice settings, MDC thresholds may need to be re-evaluated based on the characteristics of the population [67].
For persons with heart failure whose screening outcomes suggest MCI, more comprehensive neuropsychological testing should be available as well as provision of resources to optimize functional independence. Early identification of impaired cognition may lower risk of poor self-management through simplification of medication regimens or providing resources to help manage other regimens essential for optimal heart failure care. It is also important to recognize that depressive symptoms are common in persons with heart failure and are highly correlated with cognitive impairment in this population. Screening for depressive symptoms therefore, may also enhance identification of persons with heart failure at risk for frailty [4,28].
Conclusion
Effective appraisal and development of effective interventions are essential in older adults with heart failure who are at high risk for frailty and cognitive impairment. This will become increasingly important as the population ages and the incidence of heart failure rises proportionately. Although curative treatments for frailty and cognitive impairment are not available, interdisciplinary interventions such as exercise and comprehensive geriatric assessment may improve outcomes in older persons with heart failure [68]. Information gained from objective, simple, inexpensive physical performance measures, when used in combination with cognitive screening, may enhance the ability to evaluate change that signal onset of frailty or cognitive impairment [54,69,70]. The high morbidity and mortality associated with frailty and cognitive impairment indicate that it should be a priority for future research as a strategy to improve clinical outcomes, enhance quality of life, and lower health care costs in this growing population.
Corresponding author: Rebecca Gary, PhD, RN, Nell Hodgson Woodruff School of Nursing, Emory University, Atlanta, GA 30322, [email protected].
Funding/support: B. Butts was partially funded for this work through National Institutes of Health/National
Institute of Nursing Research Grant #T32NR012715.
1. Velagaleti RS and Vasan RS. Heart failure in the twenty-first century: is it a coronary artery disease or hypertension problem. Cardiol Clin 2007;25:487–95.
2. Vincent GK, Velkoff VA. The next four decades. The older population in the United States: 2010 to 2050. United States Census Bureau Report No: P25-1138. U.S. Department of Commerce; May 2010.
3. Butler J, Kalogeropoulos A. Worsening heart failure hospitalization epidemic we do not know how to prevent and we do not know how to treat. J Am Coll Cardiol 2008;52:435–7.
4. Gary R. Evaluation of frailty in older adults with cardiovascular disease: incorporating physical performance measures. J Cardiovasc Nurs 2012;27:120–131.
5. Shamliyan T, Talley KM, Ramakrishnan R, Kane RL. Association of frailty with survival: a systematic literature review. Age Res Rev 2013;12:719–36.
6. Fried LP, Tangen CM, Walston J, et al. Frailty in older adults: evidence for a phenotype. J Gerontol Med Sci 2001; 56:M146–M156.
7. Newman AB, Gottdiener JS, Mcburnie MA, et al. Associations of subclinical cardiovascular disease with frailty. J Gerontol A Biol Sci Med Sci 2001;56:M158–66.
8. Afilalo J, Karunananthan S, Eisenberg MJ, et al. Role of frailty in patients with cardiovascular disease. Am J Cardiol 2009; 103:1616–21.
9. Cacciatore F, Abete P, Maella F, et al. Frailty predicts long-term mortality in elderly subjects with chronic heart failure. Eur J Clin Invest 2008;35:723–30.
10. Lupón J, González B, Santaeugenia S, et al. Prognostic implication of frailty and depressive symptoms in an outpatient population with heart failure. Rev Españ Cardiol 2008;61:835–42.
11. Rich MW. Heart failure in the oldest patients: the impact of comorbid conditions. Am J Geriatr Cardiol 2007;14:134–41.
12. Buck HG, Riegel B. The impact of frailty on health related quality of life in heart failure. Eur J Cardiovasc Nurs 2011;10:159–66
13. Boxer RS, Shah KB Kenny AM. Frailty and prognosis in advanced heart failure. Curr Opin Supp Pall Care 2014;8:25–9.
14. Afilalo J, Sebag IA, Chalifour LE, et al. Age-related changes in lamin A/C expression in cardiomyocytes. Am J Physiol Heart Circ Physiol 2007;293:H1451–6.
15. Walston J. Frailty—the search for underlying causes. Sci Aging Know Environ 2004;2004:e4.
16. Hubbard RE, O’Mahony MS, Savva GM, et al. Inflammation and frailty measures in older people. J Cell Mol Med 2009; 13:3103–9.
17. Hubbard RE Woodhouse KW. Frailty, inflammation and the elderly. Biogerontol 2010;11:635–41.
18. Baptista G, Dupuy A-M, Jaussent A, et al. Low-grade chronic inflammation and superoxide anion production by NADPH oxidase are the main determinants of physical frailty in older adults. Free Rad Res 2012;46:1108–14.
19. Abbate A. The heart on fire: Inflammasome and cardiomyopathy. Exper Physiol 2013;98:385.
20. Collerton J, Martin-Ruiz C, Davies K, et al. Frailty and the role of inflammation, immunosenescence and cellular ageing in the very old: Cross-sectional findings from the Newcastle 85+ Study. Mech Age Devel 2012;133:456–66.
21. Ferrucci L, Harris TB, Guralnik JM, et al. Serum IL-6 level and the development of disability in older persons. J Am Geriatr Soc 1999;47:639–46.
22. Toth MJ, Matthews DE, Tracy RP and Previs MJ. Age-related differences in skeletal muscle protein synthesis: relation to markers of immune activation. Am j Physiol Endocrin Metab 2005;288:E883–91.
23. Howard C, Ferrucci L, Sun K, et al. Oxidative protein damage is associated with poor grip strength among older women living in the community. J Appl Physiol 2007;103:17–20.
24. Muzembo BA, Nagano Y, Eitoku M, et al. A cross-sectional assessment of oxidative DNA damage and muscle strength among elderly people living in the community. Envir Health Prev Med 2014;19:21–9.
25. Wu I-C, Shiesh S-C, Kuo P-H and Lin X-Z. High oxidative stress is correlated with frailty in elderly Chinese. J Am Geriatr Soc 2009;57:1666–71.
26. Alosco ML, Spitznagel MB, Cohen R, et al. Cognitive impairment is independently associated with reduced instrumental activities of daily living in persons with heart failure. J Cardiovasc Nurs 2012;27:44–50.
27. Feola M, Rosso GL, Peano M, et al. Correlation between cognitive impairment and prognostic parameters in patients with congestive heart failure. Arch Med Res 2007;38:234–9.
28. Pressler SJ, Subramanian U, Kareken D, et al. Cognitive deficits in chronic heart failure. Nurs Res 2010;59:127–39.
29. Pressler SJ, Kim J, Riley P, et al. Memory dysfunction, psychomotor slowing, and decreased executive function predict mortality in patients with heart failure and low ejection fraction. J Cardiac Fail 2010;16:750–60.
30. Pressler SJ, Subramanian U, Kareken D, et al. Cognitive deficits and health-related quality of life in chronic heart failure. J Cardiovasc Nurs 2010;25:189–98.
31. Hajduk AM, Lemon SC, Mcmanus DD, et al. Cognitive impairment and self-care in heart failure. Clin Epidemiol 2013; 24:407–16.
32. Dardiotis E, Giamouzis G, Mastrogiannis D, et al. Cognitive impairment in heart failure. Cardiol Res Prac 2012; 2012:595821.
33. Petersen RC and O’brien J. Mild cognitive impairment should be considered for DSM-V. J Geriatr Psychiatry Neurol 2006; 19:147–54.
34. Hjelm C, Dahl A, Broström A, et al. The influence of heart failure on longitudinal changes in cognition among individuals 80 years of age and older. J Clin Nurs 2012; 21:994–1003.
35. Almeida OP, Garrido GJ, Beer C, et al. Cognitive and brain changes associated with ischaemic heart disease and heart failure. Eur Heart J 2012;33:1769–76.
36. Folstein MF, Folstein SE, McHugh PR. Mini-mental state. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–98.
37. Nasveddine ZS, Phillips NA, Bédirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53:695–699.
38. Cameron J, Worrall-Carter L, Page K, et al. Screening for mild cognitive impairment in patients with heart failure: Montreal cognitive assessment versus mini mental state exam. Eur J Cardiovasc Nurs 2013;12:252–60.
39. Avila-Funes JA, Amieva H, Barberger-Gateau P, et al. Cognitive impairment improves the predictive validity of the phenotype of frailty for adverse health outcomes: the three-city study. J Am Geriatr Soc 57:453–61.
40. Perera S, Mody SH, Woodman RC, Studenski SA. Meaningful change and responsiveness in common physical performance measures in older adults. Am Geriatr Soc 2006;54:743–9.
41. Kwon S, Perera S, Pahor M, et al. What is a meaningful change in physical performance? Findings from a clinical trial in older adults (the LIFE-P study). J Nutr Health Aging 2009;13:538–44.
42. Abellan Van Kan G, Rolland Y, Houles M, et al. The assessment of frailty in older adults. Clin Geriatr Med 2010; 26:275–86.
43. Pahor M, Manini T, Cesari M. Sarcopenia: clinical evaluation, biological markers and other evaluation tools. J Nutr Health Aging 2009;13:724–8.
44. Gary R. Evaluation of frailty in older adults with cardiovascular disease. J Cardiovasc Nurs 2012;27:120–31.
45. Macklai NS, Spagnoli J, Junod J, Santos-Eggimann B. Prospective association of the SHARE-operationalized frailty phenotype with adverse health outcomes: evidence from 620+ community-dwelling Europeans living in 11 countries. BMC Geriatr 2013;13:1–9.
46. Ravinrarajah R, Lee DM, Pye SR, et al. The ability of three different models of frailty to predict all-cause mortality: Results from the European Male Aging Study (EMAS). Arch Gerontol Geriatr 2013;57:360–8.
47. Rockwood K, Andrew M and Mitnitski A. A comparison of two approaches to measuring frailty in elderly people. J Gerontol Med Sci 2007;62:738–43.
48. Morley JE, Vellas B, Van Kan GA, et al. Frailty consensus: a call to action. J Am Med Dir Assoc 2013;14:392–7.
49. Guralnik JM, Simonsick EM, Ferrucci L, et al. A short physical performance battery assessing lower extremity function: association with self-reported disability and prediction of mortality and nursing home admission. J Gerontol 1994;49:M85–94.
50. Guralnik JM, Ferrucci L, Simonsick EM, et al. Lower-extremity function in persons over the age of 70 years as a predictor of subsequent disability. N Engl J Med 1995;332:556–61.
51. Di Bari M, Pozzi C, Cavallini Mc, et al. The diagnosis of heart failure in the community. Comparative validation of four sets of criteria in unselected older adults: the ICARe Dicomano Study. J Am Coll Cardiol 2004;44:1601–08.
52. Chiarantini D, Volpato S, Sioulis F, et al. Lower extremity performance measures predict long-term prognosis in older patients hospitalized for heart failure. J Cardiac Failure 2010; 16:390–5.
53. Volpato S, Cavalieri M, Sioulis F, et al. Predictive value of the short physical performance battery following hospitalization in older patients. J Gerontol A Biol Sci Med Sci 2011;66:89–96.
54. Studenski S, Perera S, Wallace D, et al. Physical performance measures in the clinical setting. J Am Geriatr Soc 2003; 51:314–22.
55. Binder EF, Schechtman KB, Ehsani AA, et al. Effects of exercise training on frailty in community-dwelling older adults: results of a randomized, controlled trial. J Am Geriatr Soc 2012; 50:1921–8.
56. Brown M, Sinacore DR, Ehsani AA, et al. Low-intensity exercise as a modifier of physical frailty in older adults. Arch Phys Med Rehab 2000;81:960–5.
57. Yamada M, Arai H, Sonoda T and Aoyama T. Community-based exercise program is cost-effective by preventing care and disability in Japanese frail older adults. J Am Med Dir Assoc 2012;13:507–11.
58. Gary RA, Cress ME, Higgins MK, et al. A combined aerobic and resistance exercise program improves physical functional performance in patients with heart failure: a pilot study. J Cardiovasc Nurs 2012;27:418–30.
59. De Meirelles L, Matsuura C, Resende AD, et al. Chronic exercise leads to antiaggregant, antioxidant and anti-inflammatory effects in heart failure patients. Eur J Prev Cardiol 2014;21:1225–32.
60. Feiereisen P, Vaillant M, Gilson G, Delagardelle C. Effects of different training modalities on circulating anabolic/catabolic markers in chronic heart failure. J Cardiopulm Rehab Prev 2013;33:303–8.
61. Smart NA, Steele M. The effect of physical training on systemic proinflammatory cytokine expression in heart failure patients: a systematic review. Congest Heart Fail 2011;17:110–4.
62. Nunes RB, Alves JP, Kessler LP, Lago PD. Aerobic exercsie improves the inflammatory profile correlated with cardiac remodeling and function in chronic heart failure rats. Clin Chest Med 2013;68:876–82.
63. Larson EB, Wang L, Bowen JD, et al. Exercise is associated with reduced risk for incident dementia among persons 65 years of age and older. Ann Intern Med 2006;144:73–8.
64. Colcombe S and Kramer AF. Fitness effects on the cognitive function of older adults: a meta-analytic study. Psychol Sci 2003;14:125–30.
65. Heyn P, Abreu BC, Ottenbacher KJ. The effects of exercise training on elderly persons with cognitive impairment and dementia: a meta-analysis. Arch Phys Med Rehab 2004; 85:1694–704.
66. Bautmans I, Vanpuyvelde K, Mets T. Sarcopenia and functional decline: pathophysiology, prevention and therapy. Acta Clinica Belgica 2009;64:303–16.
67. Guralnik JM, Ferrucci L, Pieper CF, et al. Lower extremity function and subsequent disability: Consistency across studies, predictive models, and value of gait speed alone compared with the short physical performance battery. J Gerontol A Biol Sci Med Sci 2000;55A:M221–M231.
68. Kramer AF, Erickson KI. Capitalizing on cortical plasticity: influence of physical activity on cognition and brain function. Trends Cogn Sci 2007;11:342–8.
69. Harkness K, Heckman GA, Mckelvie RS. The older patient with heart failure: high risk for frailty and cognitive impairment. Expert Rev Cardiovasc Ther 2012;10:779–95.
70. Waters DL, Baumgartner RN, Garry PJ, Vellas B. Advantages of dietary, exercise-related, and therapeutic interventions to prevent and treat sarcopenia in adult patients: an update. Clin Interv Aging 2010;5:259–70.
1. Velagaleti RS and Vasan RS. Heart failure in the twenty-first century: is it a coronary artery disease or hypertension problem. Cardiol Clin 2007;25:487–95.
2. Vincent GK, Velkoff VA. The next four decades. The older population in the United States: 2010 to 2050. United States Census Bureau Report No: P25-1138. U.S. Department of Commerce; May 2010.
3. Butler J, Kalogeropoulos A. Worsening heart failure hospitalization epidemic we do not know how to prevent and we do not know how to treat. J Am Coll Cardiol 2008;52:435–7.
4. Gary R. Evaluation of frailty in older adults with cardiovascular disease: incorporating physical performance measures. J Cardiovasc Nurs 2012;27:120–131.
5. Shamliyan T, Talley KM, Ramakrishnan R, Kane RL. Association of frailty with survival: a systematic literature review. Age Res Rev 2013;12:719–36.
6. Fried LP, Tangen CM, Walston J, et al. Frailty in older adults: evidence for a phenotype. J Gerontol Med Sci 2001; 56:M146–M156.
7. Newman AB, Gottdiener JS, Mcburnie MA, et al. Associations of subclinical cardiovascular disease with frailty. J Gerontol A Biol Sci Med Sci 2001;56:M158–66.
8. Afilalo J, Karunananthan S, Eisenberg MJ, et al. Role of frailty in patients with cardiovascular disease. Am J Cardiol 2009; 103:1616–21.
9. Cacciatore F, Abete P, Maella F, et al. Frailty predicts long-term mortality in elderly subjects with chronic heart failure. Eur J Clin Invest 2008;35:723–30.
10. Lupón J, González B, Santaeugenia S, et al. Prognostic implication of frailty and depressive symptoms in an outpatient population with heart failure. Rev Españ Cardiol 2008;61:835–42.
11. Rich MW. Heart failure in the oldest patients: the impact of comorbid conditions. Am J Geriatr Cardiol 2007;14:134–41.
12. Buck HG, Riegel B. The impact of frailty on health related quality of life in heart failure. Eur J Cardiovasc Nurs 2011;10:159–66
13. Boxer RS, Shah KB Kenny AM. Frailty and prognosis in advanced heart failure. Curr Opin Supp Pall Care 2014;8:25–9.
14. Afilalo J, Sebag IA, Chalifour LE, et al. Age-related changes in lamin A/C expression in cardiomyocytes. Am J Physiol Heart Circ Physiol 2007;293:H1451–6.
15. Walston J. Frailty—the search for underlying causes. Sci Aging Know Environ 2004;2004:e4.
16. Hubbard RE, O’Mahony MS, Savva GM, et al. Inflammation and frailty measures in older people. J Cell Mol Med 2009; 13:3103–9.
17. Hubbard RE Woodhouse KW. Frailty, inflammation and the elderly. Biogerontol 2010;11:635–41.
18. Baptista G, Dupuy A-M, Jaussent A, et al. Low-grade chronic inflammation and superoxide anion production by NADPH oxidase are the main determinants of physical frailty in older adults. Free Rad Res 2012;46:1108–14.
19. Abbate A. The heart on fire: Inflammasome and cardiomyopathy. Exper Physiol 2013;98:385.
20. Collerton J, Martin-Ruiz C, Davies K, et al. Frailty and the role of inflammation, immunosenescence and cellular ageing in the very old: Cross-sectional findings from the Newcastle 85+ Study. Mech Age Devel 2012;133:456–66.
21. Ferrucci L, Harris TB, Guralnik JM, et al. Serum IL-6 level and the development of disability in older persons. J Am Geriatr Soc 1999;47:639–46.
22. Toth MJ, Matthews DE, Tracy RP and Previs MJ. Age-related differences in skeletal muscle protein synthesis: relation to markers of immune activation. Am j Physiol Endocrin Metab 2005;288:E883–91.
23. Howard C, Ferrucci L, Sun K, et al. Oxidative protein damage is associated with poor grip strength among older women living in the community. J Appl Physiol 2007;103:17–20.
24. Muzembo BA, Nagano Y, Eitoku M, et al. A cross-sectional assessment of oxidative DNA damage and muscle strength among elderly people living in the community. Envir Health Prev Med 2014;19:21–9.
25. Wu I-C, Shiesh S-C, Kuo P-H and Lin X-Z. High oxidative stress is correlated with frailty in elderly Chinese. J Am Geriatr Soc 2009;57:1666–71.
26. Alosco ML, Spitznagel MB, Cohen R, et al. Cognitive impairment is independently associated with reduced instrumental activities of daily living in persons with heart failure. J Cardiovasc Nurs 2012;27:44–50.
27. Feola M, Rosso GL, Peano M, et al. Correlation between cognitive impairment and prognostic parameters in patients with congestive heart failure. Arch Med Res 2007;38:234–9.
28. Pressler SJ, Subramanian U, Kareken D, et al. Cognitive deficits in chronic heart failure. Nurs Res 2010;59:127–39.
29. Pressler SJ, Kim J, Riley P, et al. Memory dysfunction, psychomotor slowing, and decreased executive function predict mortality in patients with heart failure and low ejection fraction. J Cardiac Fail 2010;16:750–60.
30. Pressler SJ, Subramanian U, Kareken D, et al. Cognitive deficits and health-related quality of life in chronic heart failure. J Cardiovasc Nurs 2010;25:189–98.
31. Hajduk AM, Lemon SC, Mcmanus DD, et al. Cognitive impairment and self-care in heart failure. Clin Epidemiol 2013; 24:407–16.
32. Dardiotis E, Giamouzis G, Mastrogiannis D, et al. Cognitive impairment in heart failure. Cardiol Res Prac 2012; 2012:595821.
33. Petersen RC and O’brien J. Mild cognitive impairment should be considered for DSM-V. J Geriatr Psychiatry Neurol 2006; 19:147–54.
34. Hjelm C, Dahl A, Broström A, et al. The influence of heart failure on longitudinal changes in cognition among individuals 80 years of age and older. J Clin Nurs 2012; 21:994–1003.
35. Almeida OP, Garrido GJ, Beer C, et al. Cognitive and brain changes associated with ischaemic heart disease and heart failure. Eur Heart J 2012;33:1769–76.
36. Folstein MF, Folstein SE, McHugh PR. Mini-mental state. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–98.
37. Nasveddine ZS, Phillips NA, Bédirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53:695–699.
38. Cameron J, Worrall-Carter L, Page K, et al. Screening for mild cognitive impairment in patients with heart failure: Montreal cognitive assessment versus mini mental state exam. Eur J Cardiovasc Nurs 2013;12:252–60.
39. Avila-Funes JA, Amieva H, Barberger-Gateau P, et al. Cognitive impairment improves the predictive validity of the phenotype of frailty for adverse health outcomes: the three-city study. J Am Geriatr Soc 57:453–61.
40. Perera S, Mody SH, Woodman RC, Studenski SA. Meaningful change and responsiveness in common physical performance measures in older adults. Am Geriatr Soc 2006;54:743–9.
41. Kwon S, Perera S, Pahor M, et al. What is a meaningful change in physical performance? Findings from a clinical trial in older adults (the LIFE-P study). J Nutr Health Aging 2009;13:538–44.
42. Abellan Van Kan G, Rolland Y, Houles M, et al. The assessment of frailty in older adults. Clin Geriatr Med 2010; 26:275–86.
43. Pahor M, Manini T, Cesari M. Sarcopenia: clinical evaluation, biological markers and other evaluation tools. J Nutr Health Aging 2009;13:724–8.
44. Gary R. Evaluation of frailty in older adults with cardiovascular disease. J Cardiovasc Nurs 2012;27:120–31.
45. Macklai NS, Spagnoli J, Junod J, Santos-Eggimann B. Prospective association of the SHARE-operationalized frailty phenotype with adverse health outcomes: evidence from 620+ community-dwelling Europeans living in 11 countries. BMC Geriatr 2013;13:1–9.
46. Ravinrarajah R, Lee DM, Pye SR, et al. The ability of three different models of frailty to predict all-cause mortality: Results from the European Male Aging Study (EMAS). Arch Gerontol Geriatr 2013;57:360–8.
47. Rockwood K, Andrew M and Mitnitski A. A comparison of two approaches to measuring frailty in elderly people. J Gerontol Med Sci 2007;62:738–43.
48. Morley JE, Vellas B, Van Kan GA, et al. Frailty consensus: a call to action. J Am Med Dir Assoc 2013;14:392–7.
49. Guralnik JM, Simonsick EM, Ferrucci L, et al. A short physical performance battery assessing lower extremity function: association with self-reported disability and prediction of mortality and nursing home admission. J Gerontol 1994;49:M85–94.
50. Guralnik JM, Ferrucci L, Simonsick EM, et al. Lower-extremity function in persons over the age of 70 years as a predictor of subsequent disability. N Engl J Med 1995;332:556–61.
51. Di Bari M, Pozzi C, Cavallini Mc, et al. The diagnosis of heart failure in the community. Comparative validation of four sets of criteria in unselected older adults: the ICARe Dicomano Study. J Am Coll Cardiol 2004;44:1601–08.
52. Chiarantini D, Volpato S, Sioulis F, et al. Lower extremity performance measures predict long-term prognosis in older patients hospitalized for heart failure. J Cardiac Failure 2010; 16:390–5.
53. Volpato S, Cavalieri M, Sioulis F, et al. Predictive value of the short physical performance battery following hospitalization in older patients. J Gerontol A Biol Sci Med Sci 2011;66:89–96.
54. Studenski S, Perera S, Wallace D, et al. Physical performance measures in the clinical setting. J Am Geriatr Soc 2003; 51:314–22.
55. Binder EF, Schechtman KB, Ehsani AA, et al. Effects of exercise training on frailty in community-dwelling older adults: results of a randomized, controlled trial. J Am Geriatr Soc 2012; 50:1921–8.
56. Brown M, Sinacore DR, Ehsani AA, et al. Low-intensity exercise as a modifier of physical frailty in older adults. Arch Phys Med Rehab 2000;81:960–5.
57. Yamada M, Arai H, Sonoda T and Aoyama T. Community-based exercise program is cost-effective by preventing care and disability in Japanese frail older adults. J Am Med Dir Assoc 2012;13:507–11.
58. Gary RA, Cress ME, Higgins MK, et al. A combined aerobic and resistance exercise program improves physical functional performance in patients with heart failure: a pilot study. J Cardiovasc Nurs 2012;27:418–30.
59. De Meirelles L, Matsuura C, Resende AD, et al. Chronic exercise leads to antiaggregant, antioxidant and anti-inflammatory effects in heart failure patients. Eur J Prev Cardiol 2014;21:1225–32.
60. Feiereisen P, Vaillant M, Gilson G, Delagardelle C. Effects of different training modalities on circulating anabolic/catabolic markers in chronic heart failure. J Cardiopulm Rehab Prev 2013;33:303–8.
61. Smart NA, Steele M. The effect of physical training on systemic proinflammatory cytokine expression in heart failure patients: a systematic review. Congest Heart Fail 2011;17:110–4.
62. Nunes RB, Alves JP, Kessler LP, Lago PD. Aerobic exercsie improves the inflammatory profile correlated with cardiac remodeling and function in chronic heart failure rats. Clin Chest Med 2013;68:876–82.
63. Larson EB, Wang L, Bowen JD, et al. Exercise is associated with reduced risk for incident dementia among persons 65 years of age and older. Ann Intern Med 2006;144:73–8.
64. Colcombe S and Kramer AF. Fitness effects on the cognitive function of older adults: a meta-analytic study. Psychol Sci 2003;14:125–30.
65. Heyn P, Abreu BC, Ottenbacher KJ. The effects of exercise training on elderly persons with cognitive impairment and dementia: a meta-analysis. Arch Phys Med Rehab 2004; 85:1694–704.
66. Bautmans I, Vanpuyvelde K, Mets T. Sarcopenia and functional decline: pathophysiology, prevention and therapy. Acta Clinica Belgica 2009;64:303–16.
67. Guralnik JM, Ferrucci L, Pieper CF, et al. Lower extremity function and subsequent disability: Consistency across studies, predictive models, and value of gait speed alone compared with the short physical performance battery. J Gerontol A Biol Sci Med Sci 2000;55A:M221–M231.
68. Kramer AF, Erickson KI. Capitalizing on cortical plasticity: influence of physical activity on cognition and brain function. Trends Cogn Sci 2007;11:342–8.
69. Harkness K, Heckman GA, Mckelvie RS. The older patient with heart failure: high risk for frailty and cognitive impairment. Expert Rev Cardiovasc Ther 2012;10:779–95.
70. Waters DL, Baumgartner RN, Garry PJ, Vellas B. Advantages of dietary, exercise-related, and therapeutic interventions to prevent and treat sarcopenia in adult patients: an update. Clin Interv Aging 2010;5:259–70.
2015 Update on obstetrics
Over the past year, much attention has been devoted to labor curves. Is the original Friedman labor curve, which dates to the 1950s, still applicable today? Or do contemporary women labor differently? And if we update our approach to labor management, can we reduce the rate of primary cesarean?
In this Update, we explore these questions, as well as two others:
- How do we minimize infectious morbidity in pregnancy?
- How much prenatal screening is too much?
Is adherence to new labor curves the best way to reduce the rate of primary cesarean?
American College of Obstetricians and Gynecologists. Obstetric Care Consensus No. 1: Safe prevention of the primary cesarean delivery. Obstet Gynecol. 2014;123(3):693–711.
Cohen WR, Friedman EA. Perils of the new labor management guidelines [published online ahead of print September 16, 2014]. Am J Obstet Gynecol. doi:10.1016/j.ajog.2014.09.008.
In 2012, the cesarean delivery rate in the United States remained at 32.8%, a high percentage when one considers the increased risks that major abdominal surgery poses in both the short and long term (blood loss, transfusion, infection, venous thromboembolism, abnormal placentation, hysterectomy).1 The American College of Obstetricians and Gynecologists (ACOG) and the Society for Maternal-Fetal Medicine (SMFM) have made it a priority to reduce the cesarean delivery rate, focusing their efforts on the primary cesarean. In March 2014, they jointly issued guidelines on the “Safe prevention of the primary cesarean delivery,” highlighting labor dystocia as a top cause.
When contemporary data from the Consortium on Safe Labor were applied to the original Friedman labor curve, investigators found that the active phase of labor may be slower than previously thought.2 The maximum slope for the rate of cervical change was not observed until 6 cm of dilation. This finding potentially changes the point at which arrest of the active phase may be declared. The maximum duration of augmentation with oxytocin also has been extended, based on studies that demonstrated increased vaginal delivery rates.
The Consortium on Safe Labor proposed that, by subjecting a contemporary population to decades-old standards, we have been intervening with primary cesarean too early in the treatment of labor dystocia.
What the guidelines say
The new recommendations from ACOG-SMFM suggest that arrest of the active phase of labor can be declared only when the patient is dilated at least 6 cm with ruptured membranes after either 4 hours of adequate uterine contractions or at least 6 hours of oxytocin administration with inadequate uterine contractions or no cervical change.
Although the recommendations state that there is no maximum duration of the second stage of labor, we may increase the vaginal delivery rate by increasing the duration of pushing to 2 hours for a multiparous patient and 3 hours for a nulliparous patient (with an additional hour when an epidural is given).
Are the recommendations ready for prime time?
In response to the recommendations, Cohen and Friedman (author of the original labor curve) published “Perils of the new labor management guidelines,” cited above. In this commentary, they caution against universal acceptance of the guidelines without further validation. They argue that the analytical method used—and not labor itself—has changed, with possible selection biases and unadjusted confounders altering the shape of the dilatation curve. Cohen and Friedman suggest that serial evaluation of the patient is preferable to an arbitrary cutoff of 6 cm.
They also criticize other aspects of the guidelines, focusing on universal use of intrauterine pressure catheters, amniotomy, and a specific duration of pushing without consideration of descent. A “one size fits all” approach may incur risk to both the mother and the fetus without proven benefit, they contend. Clinical judgment and continuous evaluation of the likelihood and safety of vaginal delivery also are encouraged rather than a reliance on labor curves in isolation.
They urge further validation before adoption of the recommendations. “If we direct our clinical and basic science investigations to the goal of practicing obstetrics in a manner that optimizes maternal and newborn outcomes, the ideal cesarean delivery rate, whatever it may be, will follow,” they write.
What this EVIDENCE means for practice
Proceed with caution when applying labor curves to patients. Use clinical judgment in conjunction with any new guidelines.
Be vigilant for infectious threats to your obstetric population
Jamieson DJ, Uyeki TM, Callaghan WM, Meaney-Delman D, Rasmussen SA. What obstetrician-gynecologists should know about Ebola: a perspective from the Centers for Disease Control and Prevention. Obstet Gynecol. 2014;124(5):1005–1010.
American College of Obstetricians and Gynecologists. Committee Opinion No. 614: Management of pregnant women with presumptive exposure to Listeria monocytogenes. Obstet Gynecol. 2014;124(6):1241–1244.
American College of Obstetricians and Gynecologists. Committee Opinion No. 608: Influenza vaccination during pregnancy. Obstet Gynecol. 2014;124(3):648–651.
We no longer consider pregnancy an immunosuppressed state but, rather, a more immune-modulated system. However, there is no question that the unique physiologic state of pregnancy places a woman and her fetus at increased risk for infection. This was devastatingly obvious during the H1N1 epidemic of 2009 and was reemphasized during a 2014 outbreak of Listeria monocytogenes. We are reminded again during the largest Ebola virus outbreak in history in West Africa, where women have been disproportionately affected.
No neonates have survived Ebola
Although Ebola infections in the United States have been very few, vigilance for people at risk of infection and preparedness to act in the case of infection are vitally important.
The Ebola virus is thought to be spread to humans through contact with infected fruit bats or primates. Human-to-human transmission occurs through direct contact with blood or body fluids (urine, feces, sweat, saliva, breast milk, vomit, semen) of an infected person or contaminated objects (needles, syringes). The incubation period is 2 to 21 days (average, 8–10 days).
Infected people become contagious only upon the appearance of fever and symptoms, which include headache, muscle pain, fatigue, weakness, diarrhea, abdominal pain, vomiting, bleeding, and bruising. The differential diagnosis includes malaria, typhoid, Lassa fever, meningococcal disease, influenza, and Marburg virus.
Treatment of Ebola is supportive care and isolation (standard, contact, and droplet precautions). Prevention is through infection-control precautions and isolation and testing of those exposed, with monitoring for 21 days.
Although pregnant women are not thought to be more susceptible to infection, they are at increased risk of severe illness and mortality, as well as spontaneous abortion and pregnancy-related hemorrhage. No neonates of women infected with Ebola have survived to date.
The CDC recommends that physicians screen patients who have traveled to West Africa and those with fevers and implement appropriate isolation and infection-control precautions. Many hospitals have developed Ebola task forces with this in mind.
Updated information is available at www.cdc.gov/vhf/ebola/index.html.
Pregnant women are highly susceptible to Listeriosis
A nationwide food recall in mid-2014 prompted significant media attention to L monocytogenes, particularly its effect on pregnant women, who have an incidence of Listerial infection 13 times higher than the general population. Although maternal illness is relatively mild, ranging from a complete lack of symptoms to febrile diarrhea, there is an increased risk to the fetus or neonate of loss, preterm labor, neonatal sepsis, meningitis, and death. The perinatal mortality rate is 29%.
The mainstay of prevention during pregnancy is improved food safety and handling, as well as counseling of pregnant women to avoid unpasteurized soft cheeses, raw milk, and unwashed fruits and vegetables, and to avoid or heat thoroughly lunch meats and hot dogs.
When a pregnant woman is exposed to Listeria, management depends on the clinical scenario, as outlined by ACOG:
- Asymptomatic pregnant women do not require testing, treatment, or fetal surveillance. Any development of symptoms within 2 months may justify further evaluation, however.
- Pregnant women with mild gastro-intestinal or flulike symptoms but no fever also can be managed expectantly. Blood cultures may be appropriate; if positive, antibiotic therapy should be initiated.
- A febrile pregnant woman should have blood cultures assessed and be started on antibiotics. The preferred regimen is intravenous ampicillin 6 g/day with or without gentamicin for 14 days. If delivery occurs, placental cultures may be assessed. Listeriosis also can be diagnosed by amniocentesis. Stool cultures are not recommended.
Influenza is largely preventable
It is important to remember that one of the most dangerous viruses for pregnant women can be prevented. However, only 38% to 52% of women who should have received the influenza vaccine around the time of pregnancy actually did so between 2009 and 2013, according to the ACOG Committee Opinion cited above. Pregnant and postpartum women are at increased risk of serious illness, prolonged hospitalization, and death from influenza infection.
The vaccine is safe and effective. Not only does it prevent maternal morbidity and mortality, but it reduces neonatal complications. Inactivated vaccine is recommended for all pregnant women at any gestational age during the flu season.
Because many women are hesitant to accept the vaccine, accurate education is essential to dispel misconceptions about it and its components. It has been shown that if an obstetric clinician recommends the vaccine and makes it available, pregnant patients are five to 50 times more likely to receive it. As obstetricians, we are compelled to make this a priority in our practice.
What this EVIDENCE means for practice
Be alert and ready to act if an infectious threat is noted in your obstetric population. Get your flu shot. Give it to your obstetric patients. And don’t forget that ACOG also supports the administration of one dose of the tetanus, diphtheria, and pertussis vaccine during each pregnancy.
How much prenatal screening is too much?
Goetzinger KR, Odibo AO. Screening for abnormal placentation and adverse pregnancy outcomes with maternal serum biomarkers in the second trimester. Prenatal Diagn. 2014;34(7):635–641.
D’Antonio F, Rijo C, Thilaganathan B, et al. Association between first-trimester maternal serum pregnancy associated plasma protein-A and obstetric complications. Prenatal Diagn. 2013;33(9):839–847.
Dugoff L; Society for Maternal-Fetal Medicine. First- and second-trimester maternal serum markers for aneuploidy and adverse obstetric outcomes. Obstet Gynecol. 2010;115(5):1052–1061.
Martin A, Krishna I, Martina B, et al. Can the quantity of cell-free fetal DNA predict preeclampsia: a systematic review. Prenatal Diagn. 2014;34(7): 685–691.
Audibert F, Boucoiran I, An N, et al. Screening for preeclampsia using first-trimester serum markers and uterine artery Doppler in nulliparous women. Am J Obstet Gynecol. 2010;203(4):383.e1–e8.
Myatt L, Clifton RG, Roberts JM, et al. First-trimester prediction of preeclampsia in nulliparous women at low risk. Obstet Gynecol. 2012;119(6):1234–1242.
The placenta of a normal pregnancy secretes small amounts of a variety of biomarkers such as alpha-fetoprotein (AFP), human chorionic gonadotropin, unconjugated estriol, inhibin A, pregnancy-associated placental protein A (PAPP-A), soluble fms-like tyrosine kinase, and placental growth factor.
The association between abnormal maternal serum biomarkers and abnormal pregnancy outcomes has been known since the 1970s, when elevated AFP was noted in pregnancies with fetal open neural tube defects. Shortly thereafter, low levels of AFP were associated with fetuses with trisomy 21.
One theory is that the abnormality in pregnancy leads to abnormal regulation at the level of the fetal-placental interface and over- or under-secretion of the various biomarkers. An offshoot of this theory is the idea that abnormal placentation (ie, preeclampsia, fetal growth restriction, accreta) also may be reflected in elevated or suppressed secretion of placental biomarkers, which could be used to screen for these conditions during pregnancy.
PAPP-A is a placental serum marker that is a component of first-trimester genetic screening. It is a marker of placental function, and low levels have been associated with fetal growth restriction, preterm birth, preeclampsia, and fetal loss. Another first-trimester marker associated with adverse outcomes is cell-free fetal DNA. This DNA, found in the maternal blood, is a product of placental apoptosis, and elevated levels have been demonstrated in women who develop preeclampsia.
Although many of the biomarkers listed here are not available specifically as a clinical screening test in the United States, the link to common genetic screens makes it tempting to try to add prediction of preeclampsia and other information to an existing test. If specific numbers are reported on the genetic screen for the different markers, that information is already there, and some companies may flag abnormally high or low levels.
However, although the association between abnormal pregnancy outcomes and abnormal biomarkers is well established in the literature, the clinical predictive value is not—nor is there always an effective intervention available. One could argue that low-dose aspirin, which is already recommended for patients with a prior delivery before 34 weeks due to preeclampsia, or more than one prior pregnancy with preeclampsia, could be recommended for patients identified on early screens to be at increased risk for preeclampsia. This approach should be tested in randomized clinical trials before universal adoption.
What this EVIDENCE means for practice
Although it is tempting to use associations to predict adverse events, the clinical value of doing so has not yet been proven. Exercise caution before potentially causing concern for both you and your patient.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Martin JA, Hamilton BE, Osterman MJK, Curtin SC, Mathews TJ. Births: final data for 2012. Natl Vital Stat Rep. 2013;62(9):1–67.
2. Zhang J, Landy HJ, Branch DW, et al. Contemporary patterns of spontaneous labor with normal neonatal outcomes. Consortium on Safe Labor. Obstet Gynecol. 2010;116(6):1281–1287.

Dr. Pauli is Assistant Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, Penn State University College of Medicine, and Attending Perinatologist at the Milton S. Hershey Medical Center in Hershey, Pennsylvania.
Dr. Repke is University Professor and Chairman of Obstetrics and Gynecology at Penn State University College of Medicine. He is also Obstetrician-Gynecologist-in-Chief at the Milton S. Hershey Medical Center in Hershey, Pennsylvania. Dr. Repke serves on the OBG Management Board of Editors.
Dr. Pauli reports that she receives research support from the Penn State Department of Obstetrics and Gynecology. Dr. Repke reports no financial relationships relevant to this article.
Dr. Pauli is Assistant Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, Penn State University College of Medicine, and Attending Perinatologist at the Milton S. Hershey Medical Center in Hershey, Pennsylvania.
Dr. Repke is University Professor and Chairman of Obstetrics and Gynecology at Penn State University College of Medicine. He is also Obstetrician-Gynecologist-in-Chief at the Milton S. Hershey Medical Center in Hershey, Pennsylvania. Dr. Repke serves on the OBG Management Board of Editors.
Dr. Pauli reports that she receives research support from the Penn State Department of Obstetrics and Gynecology. Dr. Repke reports no financial relationships relevant to this article.
Dr. Pauli is Assistant Professor, Division of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, Penn State University College of Medicine, and Attending Perinatologist at the Milton S. Hershey Medical Center in Hershey, Pennsylvania.
Dr. Repke is University Professor and Chairman of Obstetrics and Gynecology at Penn State University College of Medicine. He is also Obstetrician-Gynecologist-in-Chief at the Milton S. Hershey Medical Center in Hershey, Pennsylvania. Dr. Repke serves on the OBG Management Board of Editors.
Dr. Pauli reports that she receives research support from the Penn State Department of Obstetrics and Gynecology. Dr. Repke reports no financial relationships relevant to this article.
Over the past year, much attention has been devoted to labor curves. Is the original Friedman labor curve, which dates to the 1950s, still applicable today? Or do contemporary women labor differently? And if we update our approach to labor management, can we reduce the rate of primary cesarean?
In this Update, we explore these questions, as well as two others:
- How do we minimize infectious morbidity in pregnancy?
- How much prenatal screening is too much?
Is adherence to new labor curves the best way to reduce the rate of primary cesarean?
American College of Obstetricians and Gynecologists. Obstetric Care Consensus No. 1: Safe prevention of the primary cesarean delivery. Obstet Gynecol. 2014;123(3):693–711.
Cohen WR, Friedman EA. Perils of the new labor management guidelines [published online ahead of print September 16, 2014]. Am J Obstet Gynecol. doi:10.1016/j.ajog.2014.09.008.
In 2012, the cesarean delivery rate in the United States remained at 32.8%, a high percentage when one considers the increased risks that major abdominal surgery poses in both the short and long term (blood loss, transfusion, infection, venous thromboembolism, abnormal placentation, hysterectomy).1 The American College of Obstetricians and Gynecologists (ACOG) and the Society for Maternal-Fetal Medicine (SMFM) have made it a priority to reduce the cesarean delivery rate, focusing their efforts on the primary cesarean. In March 2014, they jointly issued guidelines on the “Safe prevention of the primary cesarean delivery,” highlighting labor dystocia as a top cause.
When contemporary data from the Consortium on Safe Labor were applied to the original Friedman labor curve, investigators found that the active phase of labor may be slower than previously thought.2 The maximum slope for the rate of cervical change was not observed until 6 cm of dilation. This finding potentially changes the point at which arrest of the active phase may be declared. The maximum duration of augmentation with oxytocin also has been extended, based on studies that demonstrated increased vaginal delivery rates.
The Consortium on Safe Labor proposed that, by subjecting a contemporary population to decades-old standards, we have been intervening with primary cesarean too early in the treatment of labor dystocia.
What the guidelines say
The new recommendations from ACOG-SMFM suggest that arrest of the active phase of labor can be declared only when the patient is dilated at least 6 cm with ruptured membranes after either 4 hours of adequate uterine contractions or at least 6 hours of oxytocin administration with inadequate uterine contractions or no cervical change.
Although the recommendations state that there is no maximum duration of the second stage of labor, we may increase the vaginal delivery rate by increasing the duration of pushing to 2 hours for a multiparous patient and 3 hours for a nulliparous patient (with an additional hour when an epidural is given).
Are the recommendations ready for prime time?
In response to the recommendations, Cohen and Friedman (author of the original labor curve) published “Perils of the new labor management guidelines,” cited above. In this commentary, they caution against universal acceptance of the guidelines without further validation. They argue that the analytical method used—and not labor itself—has changed, with possible selection biases and unadjusted confounders altering the shape of the dilatation curve. Cohen and Friedman suggest that serial evaluation of the patient is preferable to an arbitrary cutoff of 6 cm.
They also criticize other aspects of the guidelines, focusing on universal use of intrauterine pressure catheters, amniotomy, and a specific duration of pushing without consideration of descent. A “one size fits all” approach may incur risk to both the mother and the fetus without proven benefit, they contend. Clinical judgment and continuous evaluation of the likelihood and safety of vaginal delivery also are encouraged rather than a reliance on labor curves in isolation.
They urge further validation before adoption of the recommendations. “If we direct our clinical and basic science investigations to the goal of practicing obstetrics in a manner that optimizes maternal and newborn outcomes, the ideal cesarean delivery rate, whatever it may be, will follow,” they write.
What this EVIDENCE means for practice
Proceed with caution when applying labor curves to patients. Use clinical judgment in conjunction with any new guidelines.
Be vigilant for infectious threats to your obstetric population
Jamieson DJ, Uyeki TM, Callaghan WM, Meaney-Delman D, Rasmussen SA. What obstetrician-gynecologists should know about Ebola: a perspective from the Centers for Disease Control and Prevention. Obstet Gynecol. 2014;124(5):1005–1010.
American College of Obstetricians and Gynecologists. Committee Opinion No. 614: Management of pregnant women with presumptive exposure to Listeria monocytogenes. Obstet Gynecol. 2014;124(6):1241–1244.
American College of Obstetricians and Gynecologists. Committee Opinion No. 608: Influenza vaccination during pregnancy. Obstet Gynecol. 2014;124(3):648–651.
We no longer consider pregnancy an immunosuppressed state but, rather, a more immune-modulated system. However, there is no question that the unique physiologic state of pregnancy places a woman and her fetus at increased risk for infection. This was devastatingly obvious during the H1N1 epidemic of 2009 and was reemphasized during a 2014 outbreak of Listeria monocytogenes. We are reminded again during the largest Ebola virus outbreak in history in West Africa, where women have been disproportionately affected.
No neonates have survived Ebola
Although Ebola infections in the United States have been very few, vigilance for people at risk of infection and preparedness to act in the case of infection are vitally important.
The Ebola virus is thought to be spread to humans through contact with infected fruit bats or primates. Human-to-human transmission occurs through direct contact with blood or body fluids (urine, feces, sweat, saliva, breast milk, vomit, semen) of an infected person or contaminated objects (needles, syringes). The incubation period is 2 to 21 days (average, 8–10 days).
Infected people become contagious only upon the appearance of fever and symptoms, which include headache, muscle pain, fatigue, weakness, diarrhea, abdominal pain, vomiting, bleeding, and bruising. The differential diagnosis includes malaria, typhoid, Lassa fever, meningococcal disease, influenza, and Marburg virus.
Treatment of Ebola is supportive care and isolation (standard, contact, and droplet precautions). Prevention is through infection-control precautions and isolation and testing of those exposed, with monitoring for 21 days.
Although pregnant women are not thought to be more susceptible to infection, they are at increased risk of severe illness and mortality, as well as spontaneous abortion and pregnancy-related hemorrhage. No neonates of women infected with Ebola have survived to date.
The CDC recommends that physicians screen patients who have traveled to West Africa and those with fevers and implement appropriate isolation and infection-control precautions. Many hospitals have developed Ebola task forces with this in mind.
Updated information is available at www.cdc.gov/vhf/ebola/index.html.
Pregnant women are highly susceptible to Listeriosis
A nationwide food recall in mid-2014 prompted significant media attention to L monocytogenes, particularly its effect on pregnant women, who have an incidence of Listerial infection 13 times higher than the general population. Although maternal illness is relatively mild, ranging from a complete lack of symptoms to febrile diarrhea, there is an increased risk to the fetus or neonate of loss, preterm labor, neonatal sepsis, meningitis, and death. The perinatal mortality rate is 29%.
The mainstay of prevention during pregnancy is improved food safety and handling, as well as counseling of pregnant women to avoid unpasteurized soft cheeses, raw milk, and unwashed fruits and vegetables, and to avoid or heat thoroughly lunch meats and hot dogs.
When a pregnant woman is exposed to Listeria, management depends on the clinical scenario, as outlined by ACOG:
- Asymptomatic pregnant women do not require testing, treatment, or fetal surveillance. Any development of symptoms within 2 months may justify further evaluation, however.
- Pregnant women with mild gastro-intestinal or flulike symptoms but no fever also can be managed expectantly. Blood cultures may be appropriate; if positive, antibiotic therapy should be initiated.
- A febrile pregnant woman should have blood cultures assessed and be started on antibiotics. The preferred regimen is intravenous ampicillin 6 g/day with or without gentamicin for 14 days. If delivery occurs, placental cultures may be assessed. Listeriosis also can be diagnosed by amniocentesis. Stool cultures are not recommended.
Influenza is largely preventable
It is important to remember that one of the most dangerous viruses for pregnant women can be prevented. However, only 38% to 52% of women who should have received the influenza vaccine around the time of pregnancy actually did so between 2009 and 2013, according to the ACOG Committee Opinion cited above. Pregnant and postpartum women are at increased risk of serious illness, prolonged hospitalization, and death from influenza infection.
The vaccine is safe and effective. Not only does it prevent maternal morbidity and mortality, but it reduces neonatal complications. Inactivated vaccine is recommended for all pregnant women at any gestational age during the flu season.
Because many women are hesitant to accept the vaccine, accurate education is essential to dispel misconceptions about it and its components. It has been shown that if an obstetric clinician recommends the vaccine and makes it available, pregnant patients are five to 50 times more likely to receive it. As obstetricians, we are compelled to make this a priority in our practice.
What this EVIDENCE means for practice
Be alert and ready to act if an infectious threat is noted in your obstetric population. Get your flu shot. Give it to your obstetric patients. And don’t forget that ACOG also supports the administration of one dose of the tetanus, diphtheria, and pertussis vaccine during each pregnancy.
How much prenatal screening is too much?
Goetzinger KR, Odibo AO. Screening for abnormal placentation and adverse pregnancy outcomes with maternal serum biomarkers in the second trimester. Prenatal Diagn. 2014;34(7):635–641.
D’Antonio F, Rijo C, Thilaganathan B, et al. Association between first-trimester maternal serum pregnancy associated plasma protein-A and obstetric complications. Prenatal Diagn. 2013;33(9):839–847.
Dugoff L; Society for Maternal-Fetal Medicine. First- and second-trimester maternal serum markers for aneuploidy and adverse obstetric outcomes. Obstet Gynecol. 2010;115(5):1052–1061.
Martin A, Krishna I, Martina B, et al. Can the quantity of cell-free fetal DNA predict preeclampsia: a systematic review. Prenatal Diagn. 2014;34(7): 685–691.
Audibert F, Boucoiran I, An N, et al. Screening for preeclampsia using first-trimester serum markers and uterine artery Doppler in nulliparous women. Am J Obstet Gynecol. 2010;203(4):383.e1–e8.
Myatt L, Clifton RG, Roberts JM, et al. First-trimester prediction of preeclampsia in nulliparous women at low risk. Obstet Gynecol. 2012;119(6):1234–1242.
The placenta of a normal pregnancy secretes small amounts of a variety of biomarkers such as alpha-fetoprotein (AFP), human chorionic gonadotropin, unconjugated estriol, inhibin A, pregnancy-associated placental protein A (PAPP-A), soluble fms-like tyrosine kinase, and placental growth factor.
The association between abnormal maternal serum biomarkers and abnormal pregnancy outcomes has been known since the 1970s, when elevated AFP was noted in pregnancies with fetal open neural tube defects. Shortly thereafter, low levels of AFP were associated with fetuses with trisomy 21.
One theory is that the abnormality in pregnancy leads to abnormal regulation at the level of the fetal-placental interface and over- or under-secretion of the various biomarkers. An offshoot of this theory is the idea that abnormal placentation (ie, preeclampsia, fetal growth restriction, accreta) also may be reflected in elevated or suppressed secretion of placental biomarkers, which could be used to screen for these conditions during pregnancy.
PAPP-A is a placental serum marker that is a component of first-trimester genetic screening. It is a marker of placental function, and low levels have been associated with fetal growth restriction, preterm birth, preeclampsia, and fetal loss. Another first-trimester marker associated with adverse outcomes is cell-free fetal DNA. This DNA, found in the maternal blood, is a product of placental apoptosis, and elevated levels have been demonstrated in women who develop preeclampsia.
Although many of the biomarkers listed here are not available specifically as a clinical screening test in the United States, the link to common genetic screens makes it tempting to try to add prediction of preeclampsia and other information to an existing test. If specific numbers are reported on the genetic screen for the different markers, that information is already there, and some companies may flag abnormally high or low levels.
However, although the association between abnormal pregnancy outcomes and abnormal biomarkers is well established in the literature, the clinical predictive value is not—nor is there always an effective intervention available. One could argue that low-dose aspirin, which is already recommended for patients with a prior delivery before 34 weeks due to preeclampsia, or more than one prior pregnancy with preeclampsia, could be recommended for patients identified on early screens to be at increased risk for preeclampsia. This approach should be tested in randomized clinical trials before universal adoption.
What this EVIDENCE means for practice
Although it is tempting to use associations to predict adverse events, the clinical value of doing so has not yet been proven. Exercise caution before potentially causing concern for both you and your patient.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Over the past year, much attention has been devoted to labor curves. Is the original Friedman labor curve, which dates to the 1950s, still applicable today? Or do contemporary women labor differently? And if we update our approach to labor management, can we reduce the rate of primary cesarean?
In this Update, we explore these questions, as well as two others:
- How do we minimize infectious morbidity in pregnancy?
- How much prenatal screening is too much?
Is adherence to new labor curves the best way to reduce the rate of primary cesarean?
American College of Obstetricians and Gynecologists. Obstetric Care Consensus No. 1: Safe prevention of the primary cesarean delivery. Obstet Gynecol. 2014;123(3):693–711.
Cohen WR, Friedman EA. Perils of the new labor management guidelines [published online ahead of print September 16, 2014]. Am J Obstet Gynecol. doi:10.1016/j.ajog.2014.09.008.
In 2012, the cesarean delivery rate in the United States remained at 32.8%, a high percentage when one considers the increased risks that major abdominal surgery poses in both the short and long term (blood loss, transfusion, infection, venous thromboembolism, abnormal placentation, hysterectomy).1 The American College of Obstetricians and Gynecologists (ACOG) and the Society for Maternal-Fetal Medicine (SMFM) have made it a priority to reduce the cesarean delivery rate, focusing their efforts on the primary cesarean. In March 2014, they jointly issued guidelines on the “Safe prevention of the primary cesarean delivery,” highlighting labor dystocia as a top cause.
When contemporary data from the Consortium on Safe Labor were applied to the original Friedman labor curve, investigators found that the active phase of labor may be slower than previously thought.2 The maximum slope for the rate of cervical change was not observed until 6 cm of dilation. This finding potentially changes the point at which arrest of the active phase may be declared. The maximum duration of augmentation with oxytocin also has been extended, based on studies that demonstrated increased vaginal delivery rates.
The Consortium on Safe Labor proposed that, by subjecting a contemporary population to decades-old standards, we have been intervening with primary cesarean too early in the treatment of labor dystocia.
What the guidelines say
The new recommendations from ACOG-SMFM suggest that arrest of the active phase of labor can be declared only when the patient is dilated at least 6 cm with ruptured membranes after either 4 hours of adequate uterine contractions or at least 6 hours of oxytocin administration with inadequate uterine contractions or no cervical change.
Although the recommendations state that there is no maximum duration of the second stage of labor, we may increase the vaginal delivery rate by increasing the duration of pushing to 2 hours for a multiparous patient and 3 hours for a nulliparous patient (with an additional hour when an epidural is given).
Are the recommendations ready for prime time?
In response to the recommendations, Cohen and Friedman (author of the original labor curve) published “Perils of the new labor management guidelines,” cited above. In this commentary, they caution against universal acceptance of the guidelines without further validation. They argue that the analytical method used—and not labor itself—has changed, with possible selection biases and unadjusted confounders altering the shape of the dilatation curve. Cohen and Friedman suggest that serial evaluation of the patient is preferable to an arbitrary cutoff of 6 cm.
They also criticize other aspects of the guidelines, focusing on universal use of intrauterine pressure catheters, amniotomy, and a specific duration of pushing without consideration of descent. A “one size fits all” approach may incur risk to both the mother and the fetus without proven benefit, they contend. Clinical judgment and continuous evaluation of the likelihood and safety of vaginal delivery also are encouraged rather than a reliance on labor curves in isolation.
They urge further validation before adoption of the recommendations. “If we direct our clinical and basic science investigations to the goal of practicing obstetrics in a manner that optimizes maternal and newborn outcomes, the ideal cesarean delivery rate, whatever it may be, will follow,” they write.
What this EVIDENCE means for practice
Proceed with caution when applying labor curves to patients. Use clinical judgment in conjunction with any new guidelines.
Be vigilant for infectious threats to your obstetric population
Jamieson DJ, Uyeki TM, Callaghan WM, Meaney-Delman D, Rasmussen SA. What obstetrician-gynecologists should know about Ebola: a perspective from the Centers for Disease Control and Prevention. Obstet Gynecol. 2014;124(5):1005–1010.
American College of Obstetricians and Gynecologists. Committee Opinion No. 614: Management of pregnant women with presumptive exposure to Listeria monocytogenes. Obstet Gynecol. 2014;124(6):1241–1244.
American College of Obstetricians and Gynecologists. Committee Opinion No. 608: Influenza vaccination during pregnancy. Obstet Gynecol. 2014;124(3):648–651.
We no longer consider pregnancy an immunosuppressed state but, rather, a more immune-modulated system. However, there is no question that the unique physiologic state of pregnancy places a woman and her fetus at increased risk for infection. This was devastatingly obvious during the H1N1 epidemic of 2009 and was reemphasized during a 2014 outbreak of Listeria monocytogenes. We are reminded again during the largest Ebola virus outbreak in history in West Africa, where women have been disproportionately affected.
No neonates have survived Ebola
Although Ebola infections in the United States have been very few, vigilance for people at risk of infection and preparedness to act in the case of infection are vitally important.
The Ebola virus is thought to be spread to humans through contact with infected fruit bats or primates. Human-to-human transmission occurs through direct contact with blood or body fluids (urine, feces, sweat, saliva, breast milk, vomit, semen) of an infected person or contaminated objects (needles, syringes). The incubation period is 2 to 21 days (average, 8–10 days).
Infected people become contagious only upon the appearance of fever and symptoms, which include headache, muscle pain, fatigue, weakness, diarrhea, abdominal pain, vomiting, bleeding, and bruising. The differential diagnosis includes malaria, typhoid, Lassa fever, meningococcal disease, influenza, and Marburg virus.
Treatment of Ebola is supportive care and isolation (standard, contact, and droplet precautions). Prevention is through infection-control precautions and isolation and testing of those exposed, with monitoring for 21 days.
Although pregnant women are not thought to be more susceptible to infection, they are at increased risk of severe illness and mortality, as well as spontaneous abortion and pregnancy-related hemorrhage. No neonates of women infected with Ebola have survived to date.
The CDC recommends that physicians screen patients who have traveled to West Africa and those with fevers and implement appropriate isolation and infection-control precautions. Many hospitals have developed Ebola task forces with this in mind.
Updated information is available at www.cdc.gov/vhf/ebola/index.html.
Pregnant women are highly susceptible to Listeriosis
A nationwide food recall in mid-2014 prompted significant media attention to L monocytogenes, particularly its effect on pregnant women, who have an incidence of Listerial infection 13 times higher than the general population. Although maternal illness is relatively mild, ranging from a complete lack of symptoms to febrile diarrhea, there is an increased risk to the fetus or neonate of loss, preterm labor, neonatal sepsis, meningitis, and death. The perinatal mortality rate is 29%.
The mainstay of prevention during pregnancy is improved food safety and handling, as well as counseling of pregnant women to avoid unpasteurized soft cheeses, raw milk, and unwashed fruits and vegetables, and to avoid or heat thoroughly lunch meats and hot dogs.
When a pregnant woman is exposed to Listeria, management depends on the clinical scenario, as outlined by ACOG:
- Asymptomatic pregnant women do not require testing, treatment, or fetal surveillance. Any development of symptoms within 2 months may justify further evaluation, however.
- Pregnant women with mild gastro-intestinal or flulike symptoms but no fever also can be managed expectantly. Blood cultures may be appropriate; if positive, antibiotic therapy should be initiated.
- A febrile pregnant woman should have blood cultures assessed and be started on antibiotics. The preferred regimen is intravenous ampicillin 6 g/day with or without gentamicin for 14 days. If delivery occurs, placental cultures may be assessed. Listeriosis also can be diagnosed by amniocentesis. Stool cultures are not recommended.
Influenza is largely preventable
It is important to remember that one of the most dangerous viruses for pregnant women can be prevented. However, only 38% to 52% of women who should have received the influenza vaccine around the time of pregnancy actually did so between 2009 and 2013, according to the ACOG Committee Opinion cited above. Pregnant and postpartum women are at increased risk of serious illness, prolonged hospitalization, and death from influenza infection.
The vaccine is safe and effective. Not only does it prevent maternal morbidity and mortality, but it reduces neonatal complications. Inactivated vaccine is recommended for all pregnant women at any gestational age during the flu season.
Because many women are hesitant to accept the vaccine, accurate education is essential to dispel misconceptions about it and its components. It has been shown that if an obstetric clinician recommends the vaccine and makes it available, pregnant patients are five to 50 times more likely to receive it. As obstetricians, we are compelled to make this a priority in our practice.
What this EVIDENCE means for practice
Be alert and ready to act if an infectious threat is noted in your obstetric population. Get your flu shot. Give it to your obstetric patients. And don’t forget that ACOG also supports the administration of one dose of the tetanus, diphtheria, and pertussis vaccine during each pregnancy.
How much prenatal screening is too much?
Goetzinger KR, Odibo AO. Screening for abnormal placentation and adverse pregnancy outcomes with maternal serum biomarkers in the second trimester. Prenatal Diagn. 2014;34(7):635–641.
D’Antonio F, Rijo C, Thilaganathan B, et al. Association between first-trimester maternal serum pregnancy associated plasma protein-A and obstetric complications. Prenatal Diagn. 2013;33(9):839–847.
Dugoff L; Society for Maternal-Fetal Medicine. First- and second-trimester maternal serum markers for aneuploidy and adverse obstetric outcomes. Obstet Gynecol. 2010;115(5):1052–1061.
Martin A, Krishna I, Martina B, et al. Can the quantity of cell-free fetal DNA predict preeclampsia: a systematic review. Prenatal Diagn. 2014;34(7): 685–691.
Audibert F, Boucoiran I, An N, et al. Screening for preeclampsia using first-trimester serum markers and uterine artery Doppler in nulliparous women. Am J Obstet Gynecol. 2010;203(4):383.e1–e8.
Myatt L, Clifton RG, Roberts JM, et al. First-trimester prediction of preeclampsia in nulliparous women at low risk. Obstet Gynecol. 2012;119(6):1234–1242.
The placenta of a normal pregnancy secretes small amounts of a variety of biomarkers such as alpha-fetoprotein (AFP), human chorionic gonadotropin, unconjugated estriol, inhibin A, pregnancy-associated placental protein A (PAPP-A), soluble fms-like tyrosine kinase, and placental growth factor.
The association between abnormal maternal serum biomarkers and abnormal pregnancy outcomes has been known since the 1970s, when elevated AFP was noted in pregnancies with fetal open neural tube defects. Shortly thereafter, low levels of AFP were associated with fetuses with trisomy 21.
One theory is that the abnormality in pregnancy leads to abnormal regulation at the level of the fetal-placental interface and over- or under-secretion of the various biomarkers. An offshoot of this theory is the idea that abnormal placentation (ie, preeclampsia, fetal growth restriction, accreta) also may be reflected in elevated or suppressed secretion of placental biomarkers, which could be used to screen for these conditions during pregnancy.
PAPP-A is a placental serum marker that is a component of first-trimester genetic screening. It is a marker of placental function, and low levels have been associated with fetal growth restriction, preterm birth, preeclampsia, and fetal loss. Another first-trimester marker associated with adverse outcomes is cell-free fetal DNA. This DNA, found in the maternal blood, is a product of placental apoptosis, and elevated levels have been demonstrated in women who develop preeclampsia.
Although many of the biomarkers listed here are not available specifically as a clinical screening test in the United States, the link to common genetic screens makes it tempting to try to add prediction of preeclampsia and other information to an existing test. If specific numbers are reported on the genetic screen for the different markers, that information is already there, and some companies may flag abnormally high or low levels.
However, although the association between abnormal pregnancy outcomes and abnormal biomarkers is well established in the literature, the clinical predictive value is not—nor is there always an effective intervention available. One could argue that low-dose aspirin, which is already recommended for patients with a prior delivery before 34 weeks due to preeclampsia, or more than one prior pregnancy with preeclampsia, could be recommended for patients identified on early screens to be at increased risk for preeclampsia. This approach should be tested in randomized clinical trials before universal adoption.
What this EVIDENCE means for practice
Although it is tempting to use associations to predict adverse events, the clinical value of doing so has not yet been proven. Exercise caution before potentially causing concern for both you and your patient.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
1. Martin JA, Hamilton BE, Osterman MJK, Curtin SC, Mathews TJ. Births: final data for 2012. Natl Vital Stat Rep. 2013;62(9):1–67.
2. Zhang J, Landy HJ, Branch DW, et al. Contemporary patterns of spontaneous labor with normal neonatal outcomes. Consortium on Safe Labor. Obstet Gynecol. 2010;116(6):1281–1287.
1. Martin JA, Hamilton BE, Osterman MJK, Curtin SC, Mathews TJ. Births: final data for 2012. Natl Vital Stat Rep. 2013;62(9):1–67.
2. Zhang J, Landy HJ, Branch DW, et al. Contemporary patterns of spontaneous labor with normal neonatal outcomes. Consortium on Safe Labor. Obstet Gynecol. 2010;116(6):1281–1287.
IN THIS ARTICLE
— Is adherence to new labor curves the best way to reduce the rate of primary cesarean?
— Be vigilant for infectious threats to your obstetric population
— How much prenatal screening is too much?