User login
Is It Possible To Treat Patients You Dislike?
This transcript has been edited for clarity.
What do we do if we don’t like patients? We take the Hippocratic Oath as young students in Glasgow. We do that just before our graduation ceremony; we hold our hands up and repeat the Hippocratic Oath: “First, do no harm,” and so on.
I was thinking back over a long career. I’ve been a cancer doctor for 40 years and I quite like saying that.
I can only think genuinely over a couple of times in which I’ve acted reflexively when a patient has done something awful. The couple of times it happened, it was just terrible racist comments to junior doctors who were with me. Extraordinarily dreadful things such as, “I don’t want to be touched by ...” or something of that sort.
Without really thinking about it, you react as a normal citizen and say, “That’s absolutely awful. Apologize immediately or leave the consultation room, and never ever come back again.”
I remember that it happened once in Glasgow and once when I was a young professor in Birmingham, and it’s just an automatic gut reaction. The patient got a fright, and I immediately apologized and groveled around. In that relationship, we hold all the power, don’t we? Rather than being gentle about it, I was genuinely angry because of these ridiculous comments.
Otherwise, I think most of the doctor-patient relationships are predicated on nonromantic love. I think patients want us to love them as one would a son, mother, father, or daughter, because if we do, then we will do better for them and we’ll pull out all the stops. “Placebo” means “I will please.” I think in the vast majority of cases, at least in our National Health Service (NHS), patients come with trust and a sense of wanting to build that relationship. That may be changing, but not for me.
What about putting the boot on the other foot? What if the patients don’t like us rather than vice versa? As part of our accreditation appraisal process, from time to time we have to take patient surveys as to whether the patients felt that, after they had been seen in a consultation, they were treated with dignity, the quality of information given was appropriate, and they were treated with kindness.
It’s an excellent exercise. Without bragging about it, patients objectively, according to these measures, appreciate the service that I give. It’s like getting five-star reviews on Trustpilot, or whatever these things are, that allow you to review car salesmen and so on. I have always had five-star reviews across the board.
That, again, I thought was just a feature of that relationship, of patients wanting to please. These are patients who had been treated, who were in the outpatient department, who were in the midst of battle. Still, the scores are very high. I speak to my colleagues and that’s not uniformly the case. Patients actually do use these feedback forms, I think in a positive rather than negative way, reflecting back on the way that they were treated.
It has caused some of my colleagues to think quite hard about their personal style and approach to patients. That sense of feedback is important.
What about losing trust? If that’s at the heart of everything that we do, then what would be an objective measure of losing trust? Again, in our healthcare system, it has been exceedingly unusual for a patient to request a second opinion. Now, that’s changing. The government is trying to change it. Leaders of the NHS are trying to change it so that patients feel assured that they can seek second opinions.
Again, in all the years I’ve been a cancer doctor, it has been incredibly infrequent that somebody has sought a second opinion after I’ve said something. That may be a measure of trust. Again, I’ve lived through an NHS in which seeking second opinions was something of a rarity.
I’d be really interested to see what you think. In your own sphere of healthcare practice, is it possible for us to look after patients that we don’t like, or should we be honest and say, “I don’t like you. Our relationship has broken down. I want you to be seen by a colleague,” or “I want you to be nursed by somebody else”?
Has that happened? Is that something that you think is common or may become more common? What about when trust breaks down the other way? Can you think of instances in which the relationship, for whatever reason, just didn’t work and the patient had to move on because of that loss of trust and what underpinned it? I’d be really interested to know.
I seek to be informed rather than the other way around. Can we truly look after patients that we don’t like or can we rise above it as Hippocrates might have done?
Thanks for listening, as always. For the time being, over and out.
Dr. Kerr, Professor, Nuffield Department of Clinical Laboratory Science, University of Oxford; Professor of Cancer Medicine, Oxford Cancer Centre, Oxford, United Kingdom, disclosed ties with Celleron Therapeutics, Oxford Cancer Biomarkers, Afrox, GlaxoSmithKline, Bayer HealthCare Pharmaceuticals, Genomic Health, Merck Serono, and Roche.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
What do we do if we don’t like patients? We take the Hippocratic Oath as young students in Glasgow. We do that just before our graduation ceremony; we hold our hands up and repeat the Hippocratic Oath: “First, do no harm,” and so on.
I was thinking back over a long career. I’ve been a cancer doctor for 40 years and I quite like saying that.
I can only think genuinely over a couple of times in which I’ve acted reflexively when a patient has done something awful. The couple of times it happened, it was just terrible racist comments to junior doctors who were with me. Extraordinarily dreadful things such as, “I don’t want to be touched by ...” or something of that sort.
Without really thinking about it, you react as a normal citizen and say, “That’s absolutely awful. Apologize immediately or leave the consultation room, and never ever come back again.”
I remember that it happened once in Glasgow and once when I was a young professor in Birmingham, and it’s just an automatic gut reaction. The patient got a fright, and I immediately apologized and groveled around. In that relationship, we hold all the power, don’t we? Rather than being gentle about it, I was genuinely angry because of these ridiculous comments.
Otherwise, I think most of the doctor-patient relationships are predicated on nonromantic love. I think patients want us to love them as one would a son, mother, father, or daughter, because if we do, then we will do better for them and we’ll pull out all the stops. “Placebo” means “I will please.” I think in the vast majority of cases, at least in our National Health Service (NHS), patients come with trust and a sense of wanting to build that relationship. That may be changing, but not for me.
What about putting the boot on the other foot? What if the patients don’t like us rather than vice versa? As part of our accreditation appraisal process, from time to time we have to take patient surveys as to whether the patients felt that, after they had been seen in a consultation, they were treated with dignity, the quality of information given was appropriate, and they were treated with kindness.
It’s an excellent exercise. Without bragging about it, patients objectively, according to these measures, appreciate the service that I give. It’s like getting five-star reviews on Trustpilot, or whatever these things are, that allow you to review car salesmen and so on. I have always had five-star reviews across the board.
That, again, I thought was just a feature of that relationship, of patients wanting to please. These are patients who had been treated, who were in the outpatient department, who were in the midst of battle. Still, the scores are very high. I speak to my colleagues and that’s not uniformly the case. Patients actually do use these feedback forms, I think in a positive rather than negative way, reflecting back on the way that they were treated.
It has caused some of my colleagues to think quite hard about their personal style and approach to patients. That sense of feedback is important.
What about losing trust? If that’s at the heart of everything that we do, then what would be an objective measure of losing trust? Again, in our healthcare system, it has been exceedingly unusual for a patient to request a second opinion. Now, that’s changing. The government is trying to change it. Leaders of the NHS are trying to change it so that patients feel assured that they can seek second opinions.
Again, in all the years I’ve been a cancer doctor, it has been incredibly infrequent that somebody has sought a second opinion after I’ve said something. That may be a measure of trust. Again, I’ve lived through an NHS in which seeking second opinions was something of a rarity.
I’d be really interested to see what you think. In your own sphere of healthcare practice, is it possible for us to look after patients that we don’t like, or should we be honest and say, “I don’t like you. Our relationship has broken down. I want you to be seen by a colleague,” or “I want you to be nursed by somebody else”?
Has that happened? Is that something that you think is common or may become more common? What about when trust breaks down the other way? Can you think of instances in which the relationship, for whatever reason, just didn’t work and the patient had to move on because of that loss of trust and what underpinned it? I’d be really interested to know.
I seek to be informed rather than the other way around. Can we truly look after patients that we don’t like or can we rise above it as Hippocrates might have done?
Thanks for listening, as always. For the time being, over and out.
Dr. Kerr, Professor, Nuffield Department of Clinical Laboratory Science, University of Oxford; Professor of Cancer Medicine, Oxford Cancer Centre, Oxford, United Kingdom, disclosed ties with Celleron Therapeutics, Oxford Cancer Biomarkers, Afrox, GlaxoSmithKline, Bayer HealthCare Pharmaceuticals, Genomic Health, Merck Serono, and Roche.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
What do we do if we don’t like patients? We take the Hippocratic Oath as young students in Glasgow. We do that just before our graduation ceremony; we hold our hands up and repeat the Hippocratic Oath: “First, do no harm,” and so on.
I was thinking back over a long career. I’ve been a cancer doctor for 40 years and I quite like saying that.
I can only think genuinely over a couple of times in which I’ve acted reflexively when a patient has done something awful. The couple of times it happened, it was just terrible racist comments to junior doctors who were with me. Extraordinarily dreadful things such as, “I don’t want to be touched by ...” or something of that sort.
Without really thinking about it, you react as a normal citizen and say, “That’s absolutely awful. Apologize immediately or leave the consultation room, and never ever come back again.”
I remember that it happened once in Glasgow and once when I was a young professor in Birmingham, and it’s just an automatic gut reaction. The patient got a fright, and I immediately apologized and groveled around. In that relationship, we hold all the power, don’t we? Rather than being gentle about it, I was genuinely angry because of these ridiculous comments.
Otherwise, I think most of the doctor-patient relationships are predicated on nonromantic love. I think patients want us to love them as one would a son, mother, father, or daughter, because if we do, then we will do better for them and we’ll pull out all the stops. “Placebo” means “I will please.” I think in the vast majority of cases, at least in our National Health Service (NHS), patients come with trust and a sense of wanting to build that relationship. That may be changing, but not for me.
What about putting the boot on the other foot? What if the patients don’t like us rather than vice versa? As part of our accreditation appraisal process, from time to time we have to take patient surveys as to whether the patients felt that, after they had been seen in a consultation, they were treated with dignity, the quality of information given was appropriate, and they were treated with kindness.
It’s an excellent exercise. Without bragging about it, patients objectively, according to these measures, appreciate the service that I give. It’s like getting five-star reviews on Trustpilot, or whatever these things are, that allow you to review car salesmen and so on. I have always had five-star reviews across the board.
That, again, I thought was just a feature of that relationship, of patients wanting to please. These are patients who had been treated, who were in the outpatient department, who were in the midst of battle. Still, the scores are very high. I speak to my colleagues and that’s not uniformly the case. Patients actually do use these feedback forms, I think in a positive rather than negative way, reflecting back on the way that they were treated.
It has caused some of my colleagues to think quite hard about their personal style and approach to patients. That sense of feedback is important.
What about losing trust? If that’s at the heart of everything that we do, then what would be an objective measure of losing trust? Again, in our healthcare system, it has been exceedingly unusual for a patient to request a second opinion. Now, that’s changing. The government is trying to change it. Leaders of the NHS are trying to change it so that patients feel assured that they can seek second opinions.
Again, in all the years I’ve been a cancer doctor, it has been incredibly infrequent that somebody has sought a second opinion after I’ve said something. That may be a measure of trust. Again, I’ve lived through an NHS in which seeking second opinions was something of a rarity.
I’d be really interested to see what you think. In your own sphere of healthcare practice, is it possible for us to look after patients that we don’t like, or should we be honest and say, “I don’t like you. Our relationship has broken down. I want you to be seen by a colleague,” or “I want you to be nursed by somebody else”?
Has that happened? Is that something that you think is common or may become more common? What about when trust breaks down the other way? Can you think of instances in which the relationship, for whatever reason, just didn’t work and the patient had to move on because of that loss of trust and what underpinned it? I’d be really interested to know.
I seek to be informed rather than the other way around. Can we truly look after patients that we don’t like or can we rise above it as Hippocrates might have done?
Thanks for listening, as always. For the time being, over and out.
Dr. Kerr, Professor, Nuffield Department of Clinical Laboratory Science, University of Oxford; Professor of Cancer Medicine, Oxford Cancer Centre, Oxford, United Kingdom, disclosed ties with Celleron Therapeutics, Oxford Cancer Biomarkers, Afrox, GlaxoSmithKline, Bayer HealthCare Pharmaceuticals, Genomic Health, Merck Serono, and Roche.
A version of this article appeared on Medscape.com.
Ultraprocessed Foods and CVD: Myths vs Facts
I’d like to talk with you about ultraprocessed foods (UPFs) and risk for cardiovascular disease (CVD) and try to separate some of the facts from the myths. I’d like to discuss a recent report in The Lancet Regional Health that looks at this topic comprehensively and in detail.
This report includes three large-scale prospective cohort studies of US female and male health professionals, more than 200,000 participants in total. It also includes a meta-analysis of 22 international cohorts with about 1.2 million participants. I’d like to acknowledge that I’m a co-author of this study.
What are UPFs, and why are they important? Why do we care, and what are the knowledge gaps? UPFs are generally packaged foods that contain ingredients to extend shelf life and improve taste and palatability. It’s important because 60%-70% of the US diet, if not more, is made up of UPFs. So, the relationship between UPFs and CVD and other health outcomes is actually very important.
And the research to date on this subject has been quite limited.
In other studies, these UPFs have been linked to weight gain and dyslipidemia; some tissue glycation has been found, and some changes in the microbiome. Some studies have linked higher UPF intake with type 2 diabetes. A few have looked at certain selected UPF foods and found a higher risk for CVD, but a really comprehensive look at this question hasn’t been done.
So, that’s what we did in this paper and in the meta-analysis with the 22 cohorts, and we saw a very clear and distinct significant increase in coronary heart disease by 23%, total CVD by 17%, and stroke by 9% when comparing the highest vs the lowest category [of UPF intake]. When we drilled down deeply into the types of UPFs in the US health professional cohorts, we saw that there were some major differences in the relationship with CVD depending on the type of UPF.
In comparing the highest quintile vs the lowest quintile [of total UPF intake], we saw that some of the UPFs were associated with significant elevations in risk for CVD. These included sugar-sweetened beverages and processed meats. But some UPFs were linked with a lower risk for CVD. These included breakfast cereals, yogurt, some dairy desserts, and whole grains.
Overall, it seemed that UPFs are actually quite diverse in their association with health. It’s not one size fits all. They’re not all created equal, and some of these differences matter. Although overall we would recommend that our diets be focused on whole foods, primarily plant based, lots of fruits and vegetables, whole grains, fish, and other whole foods, it seems from this report and the meta-analysis that certain types of UPFs can be incorporated into a healthy diet and don’t need to be avoided entirely.
Dr. Manson is Professor of Medicine and the Michael and Lee Bell Professor of Women’s Health, Harvard Medical School, and Chief of the Division of Preventive Medicine, Brigham and Women’s Hospital, both in Boston, Massachusetts. She reported receiving donations and infrastructure support from Mars Symbioscience.
A version of this article first appeared on Medscape.com.
I’d like to talk with you about ultraprocessed foods (UPFs) and risk for cardiovascular disease (CVD) and try to separate some of the facts from the myths. I’d like to discuss a recent report in The Lancet Regional Health that looks at this topic comprehensively and in detail.
This report includes three large-scale prospective cohort studies of US female and male health professionals, more than 200,000 participants in total. It also includes a meta-analysis of 22 international cohorts with about 1.2 million participants. I’d like to acknowledge that I’m a co-author of this study.
What are UPFs, and why are they important? Why do we care, and what are the knowledge gaps? UPFs are generally packaged foods that contain ingredients to extend shelf life and improve taste and palatability. It’s important because 60%-70% of the US diet, if not more, is made up of UPFs. So, the relationship between UPFs and CVD and other health outcomes is actually very important.
And the research to date on this subject has been quite limited.
In other studies, these UPFs have been linked to weight gain and dyslipidemia; some tissue glycation has been found, and some changes in the microbiome. Some studies have linked higher UPF intake with type 2 diabetes. A few have looked at certain selected UPF foods and found a higher risk for CVD, but a really comprehensive look at this question hasn’t been done.
So, that’s what we did in this paper and in the meta-analysis with the 22 cohorts, and we saw a very clear and distinct significant increase in coronary heart disease by 23%, total CVD by 17%, and stroke by 9% when comparing the highest vs the lowest category [of UPF intake]. When we drilled down deeply into the types of UPFs in the US health professional cohorts, we saw that there were some major differences in the relationship with CVD depending on the type of UPF.
In comparing the highest quintile vs the lowest quintile [of total UPF intake], we saw that some of the UPFs were associated with significant elevations in risk for CVD. These included sugar-sweetened beverages and processed meats. But some UPFs were linked with a lower risk for CVD. These included breakfast cereals, yogurt, some dairy desserts, and whole grains.
Overall, it seemed that UPFs are actually quite diverse in their association with health. It’s not one size fits all. They’re not all created equal, and some of these differences matter. Although overall we would recommend that our diets be focused on whole foods, primarily plant based, lots of fruits and vegetables, whole grains, fish, and other whole foods, it seems from this report and the meta-analysis that certain types of UPFs can be incorporated into a healthy diet and don’t need to be avoided entirely.
Dr. Manson is Professor of Medicine and the Michael and Lee Bell Professor of Women’s Health, Harvard Medical School, and Chief of the Division of Preventive Medicine, Brigham and Women’s Hospital, both in Boston, Massachusetts. She reported receiving donations and infrastructure support from Mars Symbioscience.
A version of this article first appeared on Medscape.com.
I’d like to talk with you about ultraprocessed foods (UPFs) and risk for cardiovascular disease (CVD) and try to separate some of the facts from the myths. I’d like to discuss a recent report in The Lancet Regional Health that looks at this topic comprehensively and in detail.
This report includes three large-scale prospective cohort studies of US female and male health professionals, more than 200,000 participants in total. It also includes a meta-analysis of 22 international cohorts with about 1.2 million participants. I’d like to acknowledge that I’m a co-author of this study.
What are UPFs, and why are they important? Why do we care, and what are the knowledge gaps? UPFs are generally packaged foods that contain ingredients to extend shelf life and improve taste and palatability. It’s important because 60%-70% of the US diet, if not more, is made up of UPFs. So, the relationship between UPFs and CVD and other health outcomes is actually very important.
And the research to date on this subject has been quite limited.
In other studies, these UPFs have been linked to weight gain and dyslipidemia; some tissue glycation has been found, and some changes in the microbiome. Some studies have linked higher UPF intake with type 2 diabetes. A few have looked at certain selected UPF foods and found a higher risk for CVD, but a really comprehensive look at this question hasn’t been done.
So, that’s what we did in this paper and in the meta-analysis with the 22 cohorts, and we saw a very clear and distinct significant increase in coronary heart disease by 23%, total CVD by 17%, and stroke by 9% when comparing the highest vs the lowest category [of UPF intake]. When we drilled down deeply into the types of UPFs in the US health professional cohorts, we saw that there were some major differences in the relationship with CVD depending on the type of UPF.
In comparing the highest quintile vs the lowest quintile [of total UPF intake], we saw that some of the UPFs were associated with significant elevations in risk for CVD. These included sugar-sweetened beverages and processed meats. But some UPFs were linked with a lower risk for CVD. These included breakfast cereals, yogurt, some dairy desserts, and whole grains.
Overall, it seemed that UPFs are actually quite diverse in their association with health. It’s not one size fits all. They’re not all created equal, and some of these differences matter. Although overall we would recommend that our diets be focused on whole foods, primarily plant based, lots of fruits and vegetables, whole grains, fish, and other whole foods, it seems from this report and the meta-analysis that certain types of UPFs can be incorporated into a healthy diet and don’t need to be avoided entirely.
Dr. Manson is Professor of Medicine and the Michael and Lee Bell Professor of Women’s Health, Harvard Medical School, and Chief of the Division of Preventive Medicine, Brigham and Women’s Hospital, both in Boston, Massachusetts. She reported receiving donations and infrastructure support from Mars Symbioscience.
A version of this article first appeared on Medscape.com.
The New Cancer Stats Might Look Like a Death Sentence. They Aren’t.
Cancer is becoming more common in younger generations. Data show that people under 50 are experiencing higher rates of cancer than any generation before them. As a genetic counselor, I hoped these upward trends in early-onset malignancies would slow with a better understanding of risk factors and prevention strategies. Unfortunately, the opposite is happening. Recent findings from the American Cancer Society reveal that the incidence of at least 17 of 34 cancer types is rising among GenX and Millennials.
These statistics are alarming. I appreciate how easy it is for patients to get lost in the headlines about cancer, which may shape how they approach their healthcare. Each year, millions of Americans miss critical cancer screenings, with many citing fear of a positive test result as a leading reason. Others believe, despite the statistics, that cancer is not something they need to worry about until they are older. And then, of course, getting screened is not as easy as it should be.
In my work, I meet with people from both younger and older generations who have either faced cancer themselves or witnessed a loved one experience the disease. One of the most common sentiments I hear from these patients is the desire to catch cancer earlier. My answer is always this: The first and most important step everyone can take is understanding their risk.
For some, knowing they are at increased risk for cancer means starting screenings earlier — sometimes as early as age 25 — or getting screened with a more sensitive test.
This proactive approach is the right one. It also significantly reduces the burden of total and cancer-specific healthcare costs. While screening may carry some potential risks, clinicians can minimize these risks by adhering to evidence-based guidelines, such as those from the American Cancer Society, and ensuring there is appropriate discussion of treatment options when a diagnosis is made.
Normalizing Cancer Risk Assessment and Screening
A detailed cancer risk assessment and education about signs and symptoms should be part of every preventive care visit, regardless of someone’s age. Further, that cancer risk assessment should lead to clear recommendations and support for taking the next steps.
This is where care advocacy and patient navigation come in. Care advocacy can improve outcomes at every stage of the cancer journey, from increasing screening rates to improving quality of life for survivors. I’ve seen first-hand how care advocates help patients overcome hurdles like long wait times for appointments they need, making both screening and diagnostic care easier to access.
Now, with the finalization of a new rule from the Centers for Medicare & Medicaid Services, providers can bill for oncology navigation services that occur under their supervision. This formal recognition of care navigation affirms the value of these services not just clinically but financially as well. It will be through methods like care navigation, targeted outreach, and engaging educational resources — built into and covered by health plans — that patients will feel more in control over their health and have tools to help minimize the effects of cancer on the rest of their lives.
These services benefit healthcare providers as well. Care navigation supports clinical care teams, from primary care providers to oncologists, by ensuring patients are seen before their cancer progresses to a more advanced stage. And even if patients follow screening recommendations for the rest of their lives and never get a positive result, they’ve still gained something invaluable: peace of mind, knowing they’ve taken an active role in their health.
Fighting Fear With Routine
Treating cancer as a normal part of young people’s healthcare means helping them envision the disease as a condition that can be treated, much like a diagnosis of diabetes or high cholesterol. This mindset shift means quickly following up on a concerning symptom or screening result and reducing the time to start treatment if needed. And with treatment options and success rates for some cancers being better than ever, survivorship support must be built into every treatment plan from the start. Before treatment begins, healthcare providers should make time to talk about sometimes-overlooked key topics, such as reproductive options for people whose fertility may be affected by their cancer treatment, about plans for returning to work during or after treatment, and finding the right mental health support.
Where we can’t prevent cancer, both primary care providers and oncologists can work together to help patients receive the right diagnosis and treatment as quickly as possible. Knowing insurance coverage has a direct effect on how early cancer is caught, for example, younger people need support in understanding and accessing benefits and resources that may be available through their existing healthcare channels, like some employer-sponsored health plans. Even if getting treated for cancer is inevitable for some, taking immediate action to get screened when it’s appropriate is the best thing we can do to lessen the impact of these rising cancer incidences across the country. At the end of the day, being afraid of cancer doesn’t decrease the chances of getting sick or dying from it. Proactive screening and early detection do.
Brockman, Genetic Counselor, Color Health, Buffalo, New York, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
Cancer is becoming more common in younger generations. Data show that people under 50 are experiencing higher rates of cancer than any generation before them. As a genetic counselor, I hoped these upward trends in early-onset malignancies would slow with a better understanding of risk factors and prevention strategies. Unfortunately, the opposite is happening. Recent findings from the American Cancer Society reveal that the incidence of at least 17 of 34 cancer types is rising among GenX and Millennials.
These statistics are alarming. I appreciate how easy it is for patients to get lost in the headlines about cancer, which may shape how they approach their healthcare. Each year, millions of Americans miss critical cancer screenings, with many citing fear of a positive test result as a leading reason. Others believe, despite the statistics, that cancer is not something they need to worry about until they are older. And then, of course, getting screened is not as easy as it should be.
In my work, I meet with people from both younger and older generations who have either faced cancer themselves or witnessed a loved one experience the disease. One of the most common sentiments I hear from these patients is the desire to catch cancer earlier. My answer is always this: The first and most important step everyone can take is understanding their risk.
For some, knowing they are at increased risk for cancer means starting screenings earlier — sometimes as early as age 25 — or getting screened with a more sensitive test.
This proactive approach is the right one. It also significantly reduces the burden of total and cancer-specific healthcare costs. While screening may carry some potential risks, clinicians can minimize these risks by adhering to evidence-based guidelines, such as those from the American Cancer Society, and ensuring there is appropriate discussion of treatment options when a diagnosis is made.
Normalizing Cancer Risk Assessment and Screening
A detailed cancer risk assessment and education about signs and symptoms should be part of every preventive care visit, regardless of someone’s age. Further, that cancer risk assessment should lead to clear recommendations and support for taking the next steps.
This is where care advocacy and patient navigation come in. Care advocacy can improve outcomes at every stage of the cancer journey, from increasing screening rates to improving quality of life for survivors. I’ve seen first-hand how care advocates help patients overcome hurdles like long wait times for appointments they need, making both screening and diagnostic care easier to access.
Now, with the finalization of a new rule from the Centers for Medicare & Medicaid Services, providers can bill for oncology navigation services that occur under their supervision. This formal recognition of care navigation affirms the value of these services not just clinically but financially as well. It will be through methods like care navigation, targeted outreach, and engaging educational resources — built into and covered by health plans — that patients will feel more in control over their health and have tools to help minimize the effects of cancer on the rest of their lives.
These services benefit healthcare providers as well. Care navigation supports clinical care teams, from primary care providers to oncologists, by ensuring patients are seen before their cancer progresses to a more advanced stage. And even if patients follow screening recommendations for the rest of their lives and never get a positive result, they’ve still gained something invaluable: peace of mind, knowing they’ve taken an active role in their health.
Fighting Fear With Routine
Treating cancer as a normal part of young people’s healthcare means helping them envision the disease as a condition that can be treated, much like a diagnosis of diabetes or high cholesterol. This mindset shift means quickly following up on a concerning symptom or screening result and reducing the time to start treatment if needed. And with treatment options and success rates for some cancers being better than ever, survivorship support must be built into every treatment plan from the start. Before treatment begins, healthcare providers should make time to talk about sometimes-overlooked key topics, such as reproductive options for people whose fertility may be affected by their cancer treatment, about plans for returning to work during or after treatment, and finding the right mental health support.
Where we can’t prevent cancer, both primary care providers and oncologists can work together to help patients receive the right diagnosis and treatment as quickly as possible. Knowing insurance coverage has a direct effect on how early cancer is caught, for example, younger people need support in understanding and accessing benefits and resources that may be available through their existing healthcare channels, like some employer-sponsored health plans. Even if getting treated for cancer is inevitable for some, taking immediate action to get screened when it’s appropriate is the best thing we can do to lessen the impact of these rising cancer incidences across the country. At the end of the day, being afraid of cancer doesn’t decrease the chances of getting sick or dying from it. Proactive screening and early detection do.
Brockman, Genetic Counselor, Color Health, Buffalo, New York, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
Cancer is becoming more common in younger generations. Data show that people under 50 are experiencing higher rates of cancer than any generation before them. As a genetic counselor, I hoped these upward trends in early-onset malignancies would slow with a better understanding of risk factors and prevention strategies. Unfortunately, the opposite is happening. Recent findings from the American Cancer Society reveal that the incidence of at least 17 of 34 cancer types is rising among GenX and Millennials.
These statistics are alarming. I appreciate how easy it is for patients to get lost in the headlines about cancer, which may shape how they approach their healthcare. Each year, millions of Americans miss critical cancer screenings, with many citing fear of a positive test result as a leading reason. Others believe, despite the statistics, that cancer is not something they need to worry about until they are older. And then, of course, getting screened is not as easy as it should be.
In my work, I meet with people from both younger and older generations who have either faced cancer themselves or witnessed a loved one experience the disease. One of the most common sentiments I hear from these patients is the desire to catch cancer earlier. My answer is always this: The first and most important step everyone can take is understanding their risk.
For some, knowing they are at increased risk for cancer means starting screenings earlier — sometimes as early as age 25 — or getting screened with a more sensitive test.
This proactive approach is the right one. It also significantly reduces the burden of total and cancer-specific healthcare costs. While screening may carry some potential risks, clinicians can minimize these risks by adhering to evidence-based guidelines, such as those from the American Cancer Society, and ensuring there is appropriate discussion of treatment options when a diagnosis is made.
Normalizing Cancer Risk Assessment and Screening
A detailed cancer risk assessment and education about signs and symptoms should be part of every preventive care visit, regardless of someone’s age. Further, that cancer risk assessment should lead to clear recommendations and support for taking the next steps.
This is where care advocacy and patient navigation come in. Care advocacy can improve outcomes at every stage of the cancer journey, from increasing screening rates to improving quality of life for survivors. I’ve seen first-hand how care advocates help patients overcome hurdles like long wait times for appointments they need, making both screening and diagnostic care easier to access.
Now, with the finalization of a new rule from the Centers for Medicare & Medicaid Services, providers can bill for oncology navigation services that occur under their supervision. This formal recognition of care navigation affirms the value of these services not just clinically but financially as well. It will be through methods like care navigation, targeted outreach, and engaging educational resources — built into and covered by health plans — that patients will feel more in control over their health and have tools to help minimize the effects of cancer on the rest of their lives.
These services benefit healthcare providers as well. Care navigation supports clinical care teams, from primary care providers to oncologists, by ensuring patients are seen before their cancer progresses to a more advanced stage. And even if patients follow screening recommendations for the rest of their lives and never get a positive result, they’ve still gained something invaluable: peace of mind, knowing they’ve taken an active role in their health.
Fighting Fear With Routine
Treating cancer as a normal part of young people’s healthcare means helping them envision the disease as a condition that can be treated, much like a diagnosis of diabetes or high cholesterol. This mindset shift means quickly following up on a concerning symptom or screening result and reducing the time to start treatment if needed. And with treatment options and success rates for some cancers being better than ever, survivorship support must be built into every treatment plan from the start. Before treatment begins, healthcare providers should make time to talk about sometimes-overlooked key topics, such as reproductive options for people whose fertility may be affected by their cancer treatment, about plans for returning to work during or after treatment, and finding the right mental health support.
Where we can’t prevent cancer, both primary care providers and oncologists can work together to help patients receive the right diagnosis and treatment as quickly as possible. Knowing insurance coverage has a direct effect on how early cancer is caught, for example, younger people need support in understanding and accessing benefits and resources that may be available through their existing healthcare channels, like some employer-sponsored health plans. Even if getting treated for cancer is inevitable for some, taking immediate action to get screened when it’s appropriate is the best thing we can do to lessen the impact of these rising cancer incidences across the country. At the end of the day, being afraid of cancer doesn’t decrease the chances of getting sick or dying from it. Proactive screening and early detection do.
Brockman, Genetic Counselor, Color Health, Buffalo, New York, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
Unseen Cost of Weight Loss and Aging: Tackling Sarcopenia
Losses of muscle and strength are inescapable effects of the aging process. Left unchecked, these progressive losses will start to impair physical function.
Once a certain level of impairment occurs, an individual can be diagnosed with sarcopenia, which comes from the Greek words “sarco” (flesh) and “penia” (poverty).
Muscle mass losses generally occur with weight loss, and the increasing use of glucagon-like peptide 1 (GLP-1) medications may lead to greater incidence and prevalence of sarcopenia in the years to come.
A recent meta-analysis of 56 studies (mean participant age, 50 years) found a twofold greater risk for mortality in those with sarcopenia vs those without. Despite its health consequences, sarcopenia tends to be underdiagnosed and, consequently, undertreated at a population and individual level. Part of the reason probably stems from the lack of health insurance reimbursement for individual clinicians and hospital systems to perform sarcopenia screening assessments.
In aging and obesity, it appears justified to include and emphasize a recommendation for sarcopenia screening in medical society guidelines; however, individual patients and clinicians do not need to wait for updated guidelines to implement sarcopenia screening, treatment, and prevention strategies in their own lives and/or clinical practice.
Simple Prevention and Treatment Strategy
Much can be done to help prevent sarcopenia. The primary strategy, unsurprisingly, is engaging in frequent strength training. But that doesn’t mean hours in the gym every week.
With just one session per week over 10 weeks, lean body mass (LBM), a common proxy for muscle mass, increased by 0.33 kg, according to a study which evaluated LBM improvements across different strength training frequencies. Adding a second weekly session was significantly better. In the twice-weekly group, LBM increased by 1.4 kg over 10 weeks, resulting in an increase in LBM more than four times greater than the once-a-week group. (There was no greater improvement in LBM by adding a third weekly session vs two weekly sessions.)
Although that particular study didn’t identify greater benefit at three times a week, compared with twice a week, the specific training routines and lack of a protein consumption assessment may have played a role in that finding.
Underlying the diminishing benefits, a different study found a marginally greater benefit in favor of performing ≥ five sets per major muscle group per week, compared with < five sets per week for increasing muscle in the legs, arms, back, chest, and shoulders.
Expensive gym memberships and fancy equipment are not necessary. While the use of strength training machines and free weights have been viewed by many as the optimal approach, a recent systematic review and meta-analysis found that comparable improvements to strength can be achieved with workouts using resistance bands. For those who struggle to find the time to go to a gym, or for whom gym fees are not financially affordable, resistance bands are a cheaper and more convenient alternative.
Lucas, Assistant Professor of Clinical Medicine, Comprehensive Weight Control Center, Weill Cornell Medicine, New York City, disclosed ties with Measured (Better Health Labs).
A version of this article appeared on Medscape.com.
Losses of muscle and strength are inescapable effects of the aging process. Left unchecked, these progressive losses will start to impair physical function.
Once a certain level of impairment occurs, an individual can be diagnosed with sarcopenia, which comes from the Greek words “sarco” (flesh) and “penia” (poverty).
Muscle mass losses generally occur with weight loss, and the increasing use of glucagon-like peptide 1 (GLP-1) medications may lead to greater incidence and prevalence of sarcopenia in the years to come.
A recent meta-analysis of 56 studies (mean participant age, 50 years) found a twofold greater risk for mortality in those with sarcopenia vs those without. Despite its health consequences, sarcopenia tends to be underdiagnosed and, consequently, undertreated at a population and individual level. Part of the reason probably stems from the lack of health insurance reimbursement for individual clinicians and hospital systems to perform sarcopenia screening assessments.
In aging and obesity, it appears justified to include and emphasize a recommendation for sarcopenia screening in medical society guidelines; however, individual patients and clinicians do not need to wait for updated guidelines to implement sarcopenia screening, treatment, and prevention strategies in their own lives and/or clinical practice.
Simple Prevention and Treatment Strategy
Much can be done to help prevent sarcopenia. The primary strategy, unsurprisingly, is engaging in frequent strength training. But that doesn’t mean hours in the gym every week.
With just one session per week over 10 weeks, lean body mass (LBM), a common proxy for muscle mass, increased by 0.33 kg, according to a study which evaluated LBM improvements across different strength training frequencies. Adding a second weekly session was significantly better. In the twice-weekly group, LBM increased by 1.4 kg over 10 weeks, resulting in an increase in LBM more than four times greater than the once-a-week group. (There was no greater improvement in LBM by adding a third weekly session vs two weekly sessions.)
Although that particular study didn’t identify greater benefit at three times a week, compared with twice a week, the specific training routines and lack of a protein consumption assessment may have played a role in that finding.
Underlying the diminishing benefits, a different study found a marginally greater benefit in favor of performing ≥ five sets per major muscle group per week, compared with < five sets per week for increasing muscle in the legs, arms, back, chest, and shoulders.
Expensive gym memberships and fancy equipment are not necessary. While the use of strength training machines and free weights have been viewed by many as the optimal approach, a recent systematic review and meta-analysis found that comparable improvements to strength can be achieved with workouts using resistance bands. For those who struggle to find the time to go to a gym, or for whom gym fees are not financially affordable, resistance bands are a cheaper and more convenient alternative.
Lucas, Assistant Professor of Clinical Medicine, Comprehensive Weight Control Center, Weill Cornell Medicine, New York City, disclosed ties with Measured (Better Health Labs).
A version of this article appeared on Medscape.com.
Losses of muscle and strength are inescapable effects of the aging process. Left unchecked, these progressive losses will start to impair physical function.
Once a certain level of impairment occurs, an individual can be diagnosed with sarcopenia, which comes from the Greek words “sarco” (flesh) and “penia” (poverty).
Muscle mass losses generally occur with weight loss, and the increasing use of glucagon-like peptide 1 (GLP-1) medications may lead to greater incidence and prevalence of sarcopenia in the years to come.
A recent meta-analysis of 56 studies (mean participant age, 50 years) found a twofold greater risk for mortality in those with sarcopenia vs those without. Despite its health consequences, sarcopenia tends to be underdiagnosed and, consequently, undertreated at a population and individual level. Part of the reason probably stems from the lack of health insurance reimbursement for individual clinicians and hospital systems to perform sarcopenia screening assessments.
In aging and obesity, it appears justified to include and emphasize a recommendation for sarcopenia screening in medical society guidelines; however, individual patients and clinicians do not need to wait for updated guidelines to implement sarcopenia screening, treatment, and prevention strategies in their own lives and/or clinical practice.
Simple Prevention and Treatment Strategy
Much can be done to help prevent sarcopenia. The primary strategy, unsurprisingly, is engaging in frequent strength training. But that doesn’t mean hours in the gym every week.
With just one session per week over 10 weeks, lean body mass (LBM), a common proxy for muscle mass, increased by 0.33 kg, according to a study which evaluated LBM improvements across different strength training frequencies. Adding a second weekly session was significantly better. In the twice-weekly group, LBM increased by 1.4 kg over 10 weeks, resulting in an increase in LBM more than four times greater than the once-a-week group. (There was no greater improvement in LBM by adding a third weekly session vs two weekly sessions.)
Although that particular study didn’t identify greater benefit at three times a week, compared with twice a week, the specific training routines and lack of a protein consumption assessment may have played a role in that finding.
Underlying the diminishing benefits, a different study found a marginally greater benefit in favor of performing ≥ five sets per major muscle group per week, compared with < five sets per week for increasing muscle in the legs, arms, back, chest, and shoulders.
Expensive gym memberships and fancy equipment are not necessary. While the use of strength training machines and free weights have been viewed by many as the optimal approach, a recent systematic review and meta-analysis found that comparable improvements to strength can be achieved with workouts using resistance bands. For those who struggle to find the time to go to a gym, or for whom gym fees are not financially affordable, resistance bands are a cheaper and more convenient alternative.
Lucas, Assistant Professor of Clinical Medicine, Comprehensive Weight Control Center, Weill Cornell Medicine, New York City, disclosed ties with Measured (Better Health Labs).
A version of this article appeared on Medscape.com.
Heard of ApoB Testing? New Guidelines
This transcript has been edited for clarity.
I've been hearing a lot about apolipoprotein B (apoB) lately. It keeps popping up, but I've not been sure where it fits in or what I should do about it. The new Expert Clinical Consensus from the National Lipid Association now finally gives us clear guidance.
ApoB is the main protein that is found on all atherogenic lipoproteins. It is found on low-density lipoprotein (LDL) but also on other atherogenic lipoprotein particles. Because it is a part of all atherogenic particles, it predicts cardiovascular (CV) risk more accurately than does LDL cholesterol (LDL-C).
ApoB and LDL-C tend to run together, but not always. While they are correlated fairly well on a population level, for a given individual they can diverge; and when they do, apoB is the better predictor of future CV outcomes. This divergence occurs frequently, and it can occur even more frequently after treatment with statins. When LDL decreases to reach the LDL threshold for treatment, but apoB remains elevated, there is the potential for misclassification of CV risk and essentially the risk for undertreatment of someone whose CV risk is actually higher than it appears to be if we only look at their LDL-C. The consensus statement says, "Where there is discordance between apoB and LDL-C, risk follows apoB."
This understanding leads to the places where measurement of apoB may be helpful:
In patients with borderline atherosclerotic cardiovascular disease risk in whom a shared decision about statin therapy is being determined and the patient prefers not to start a statin, apoB can be useful for further risk stratification. If apoB suggests low risk, then statin therapy could be withheld, and if apoB is high, that would favor starting statin therapy. Certain common conditions, such as obesity and insulin resistance, can lead to smaller cholesterol-depleted LDL particles that result in lower LDL-C, but elevated apoB levels in this circumstance may drive the decision to treat with a statin.
In patients already treated with statins, but a decision must be made about whether treatment intensification is warranted. If the LDL-C is to goal and apoB is above threshold, treatment intensification may be considered. In patients who are not yet to goal, based on an elevated apoB, the first step is intensification of statin therapy. After that, intensification would be the same as has already been addressed in my review of the 2022 ACC Expert Consensus Decision Pathway on the Role of Nonstatin Therapies for LDL-Cholesterol Lowering.
After clarifying the importance of apoB in providing additional discrimination of CV risk, the consensus statement clarifies the treatment thresholds, or goals for treatment, for apoB that correlate with established LDL-C thresholds, as shown in this table:
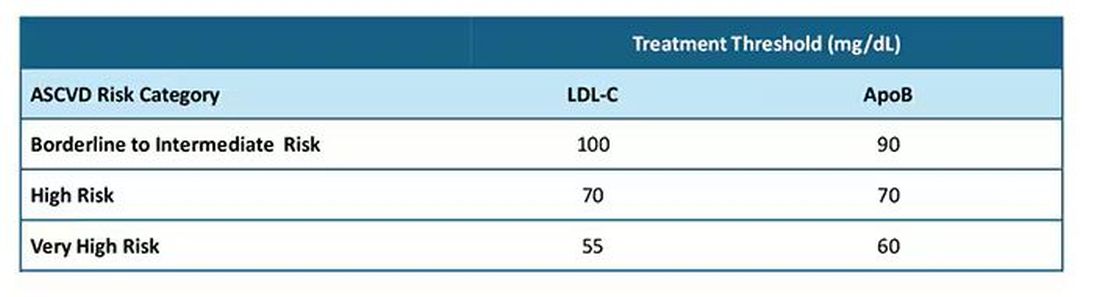
Let me be really clear: The consensus statement does not say that we need to measure apoB in all patients or that such measurement is the standard of care. It is not. It says, and I'll quote, "At present, the use of apoB to assess the effectiveness of lipid-lowering therapies remains a matter of clinical judgment." This guideline is helpful in pointing out the patients most likely to benefit from this additional measurement, including those with hypertriglyceridemia, diabetes, visceral adiposity, insulin resistance/metabolic syndrome, low HDL-C, or very low LDL-C levels.
In summary, measurement of apoB can be helpful for further risk stratification in patients with borderline or intermediate LDL-C levels, and for deciding whether further intensification of lipid-lowering therapy may be warranted when the LDL threshold has been reached.
Lipid management is something that we do every day in the office. This is new information, or at least clarifying information, for most of us. Hopefully it is helpful. I'm interested in your thoughts on this topic, including whether and how you plan to use apoB measurements.
Dr. Skolnik, Professor, Department of Family Medicine, Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia; Associate Director, Department of Family Medicine, Abington Jefferson Health, Abington, Pennsylvania, disclosed ties with AstraZeneca, Teva, Eli Lilly, Boehringer Ingelheim, Sanofi, Sanofi Pasteur, GlaxoSmithKline, Merck, and Bayer.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
I've been hearing a lot about apolipoprotein B (apoB) lately. It keeps popping up, but I've not been sure where it fits in or what I should do about it. The new Expert Clinical Consensus from the National Lipid Association now finally gives us clear guidance.
ApoB is the main protein that is found on all atherogenic lipoproteins. It is found on low-density lipoprotein (LDL) but also on other atherogenic lipoprotein particles. Because it is a part of all atherogenic particles, it predicts cardiovascular (CV) risk more accurately than does LDL cholesterol (LDL-C).
ApoB and LDL-C tend to run together, but not always. While they are correlated fairly well on a population level, for a given individual they can diverge; and when they do, apoB is the better predictor of future CV outcomes. This divergence occurs frequently, and it can occur even more frequently after treatment with statins. When LDL decreases to reach the LDL threshold for treatment, but apoB remains elevated, there is the potential for misclassification of CV risk and essentially the risk for undertreatment of someone whose CV risk is actually higher than it appears to be if we only look at their LDL-C. The consensus statement says, "Where there is discordance between apoB and LDL-C, risk follows apoB."
This understanding leads to the places where measurement of apoB may be helpful:
In patients with borderline atherosclerotic cardiovascular disease risk in whom a shared decision about statin therapy is being determined and the patient prefers not to start a statin, apoB can be useful for further risk stratification. If apoB suggests low risk, then statin therapy could be withheld, and if apoB is high, that would favor starting statin therapy. Certain common conditions, such as obesity and insulin resistance, can lead to smaller cholesterol-depleted LDL particles that result in lower LDL-C, but elevated apoB levels in this circumstance may drive the decision to treat with a statin.
In patients already treated with statins, but a decision must be made about whether treatment intensification is warranted. If the LDL-C is to goal and apoB is above threshold, treatment intensification may be considered. In patients who are not yet to goal, based on an elevated apoB, the first step is intensification of statin therapy. After that, intensification would be the same as has already been addressed in my review of the 2022 ACC Expert Consensus Decision Pathway on the Role of Nonstatin Therapies for LDL-Cholesterol Lowering.
After clarifying the importance of apoB in providing additional discrimination of CV risk, the consensus statement clarifies the treatment thresholds, or goals for treatment, for apoB that correlate with established LDL-C thresholds, as shown in this table:
Let me be really clear: The consensus statement does not say that we need to measure apoB in all patients or that such measurement is the standard of care. It is not. It says, and I'll quote, "At present, the use of apoB to assess the effectiveness of lipid-lowering therapies remains a matter of clinical judgment." This guideline is helpful in pointing out the patients most likely to benefit from this additional measurement, including those with hypertriglyceridemia, diabetes, visceral adiposity, insulin resistance/metabolic syndrome, low HDL-C, or very low LDL-C levels.
In summary, measurement of apoB can be helpful for further risk stratification in patients with borderline or intermediate LDL-C levels, and for deciding whether further intensification of lipid-lowering therapy may be warranted when the LDL threshold has been reached.
Lipid management is something that we do every day in the office. This is new information, or at least clarifying information, for most of us. Hopefully it is helpful. I'm interested in your thoughts on this topic, including whether and how you plan to use apoB measurements.
Dr. Skolnik, Professor, Department of Family Medicine, Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia; Associate Director, Department of Family Medicine, Abington Jefferson Health, Abington, Pennsylvania, disclosed ties with AstraZeneca, Teva, Eli Lilly, Boehringer Ingelheim, Sanofi, Sanofi Pasteur, GlaxoSmithKline, Merck, and Bayer.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
I've been hearing a lot about apolipoprotein B (apoB) lately. It keeps popping up, but I've not been sure where it fits in or what I should do about it. The new Expert Clinical Consensus from the National Lipid Association now finally gives us clear guidance.
ApoB is the main protein that is found on all atherogenic lipoproteins. It is found on low-density lipoprotein (LDL) but also on other atherogenic lipoprotein particles. Because it is a part of all atherogenic particles, it predicts cardiovascular (CV) risk more accurately than does LDL cholesterol (LDL-C).
ApoB and LDL-C tend to run together, but not always. While they are correlated fairly well on a population level, for a given individual they can diverge; and when they do, apoB is the better predictor of future CV outcomes. This divergence occurs frequently, and it can occur even more frequently after treatment with statins. When LDL decreases to reach the LDL threshold for treatment, but apoB remains elevated, there is the potential for misclassification of CV risk and essentially the risk for undertreatment of someone whose CV risk is actually higher than it appears to be if we only look at their LDL-C. The consensus statement says, "Where there is discordance between apoB and LDL-C, risk follows apoB."
This understanding leads to the places where measurement of apoB may be helpful:
In patients with borderline atherosclerotic cardiovascular disease risk in whom a shared decision about statin therapy is being determined and the patient prefers not to start a statin, apoB can be useful for further risk stratification. If apoB suggests low risk, then statin therapy could be withheld, and if apoB is high, that would favor starting statin therapy. Certain common conditions, such as obesity and insulin resistance, can lead to smaller cholesterol-depleted LDL particles that result in lower LDL-C, but elevated apoB levels in this circumstance may drive the decision to treat with a statin.
In patients already treated with statins, but a decision must be made about whether treatment intensification is warranted. If the LDL-C is to goal and apoB is above threshold, treatment intensification may be considered. In patients who are not yet to goal, based on an elevated apoB, the first step is intensification of statin therapy. After that, intensification would be the same as has already been addressed in my review of the 2022 ACC Expert Consensus Decision Pathway on the Role of Nonstatin Therapies for LDL-Cholesterol Lowering.
After clarifying the importance of apoB in providing additional discrimination of CV risk, the consensus statement clarifies the treatment thresholds, or goals for treatment, for apoB that correlate with established LDL-C thresholds, as shown in this table:
Let me be really clear: The consensus statement does not say that we need to measure apoB in all patients or that such measurement is the standard of care. It is not. It says, and I'll quote, "At present, the use of apoB to assess the effectiveness of lipid-lowering therapies remains a matter of clinical judgment." This guideline is helpful in pointing out the patients most likely to benefit from this additional measurement, including those with hypertriglyceridemia, diabetes, visceral adiposity, insulin resistance/metabolic syndrome, low HDL-C, or very low LDL-C levels.
In summary, measurement of apoB can be helpful for further risk stratification in patients with borderline or intermediate LDL-C levels, and for deciding whether further intensification of lipid-lowering therapy may be warranted when the LDL threshold has been reached.
Lipid management is something that we do every day in the office. This is new information, or at least clarifying information, for most of us. Hopefully it is helpful. I'm interested in your thoughts on this topic, including whether and how you plan to use apoB measurements.
Dr. Skolnik, Professor, Department of Family Medicine, Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia; Associate Director, Department of Family Medicine, Abington Jefferson Health, Abington, Pennsylvania, disclosed ties with AstraZeneca, Teva, Eli Lilly, Boehringer Ingelheim, Sanofi, Sanofi Pasteur, GlaxoSmithKline, Merck, and Bayer.
A version of this article first appeared on Medscape.com.
What Should You Do When a Patient Asks for a PSA Test?
Many patients ask us to request a prostate-specific antigen (PSA) test. According to the Brazilian Ministry of Health, prostate cancer is the second most common type of cancer in the male population in all regions of our country. It is the second-leading cause of cancer death in the male population, reaffirming its epidemiologic importance in Brazil. On the other hand, a Ministry of Health technical paper recommends against population-based screening for prostate cancer. So, what should we do?
First, it is important to distinguish early diagnosis from screening. Early diagnosis is the identification of cancer in early stages in people with signs and symptoms. Screening is characterized by the systematic application of exams — digital rectal exam and PSA test — in asymptomatic people, with the aim of identifying cancer in an early stage.
A recent European epidemiologic study reinforced this thesis and helps guide us.
The study included men aged 35-84 years from 26 European countries. Data on cancer incidence and mortality were collected between 1980 and 2017. The data suggested overdiagnosis of prostate cancer, which varied over time and among populations. The findings supported previous recommendations that any implementation of prostate cancer screening should be carefully designed, with an emphasis on minimizing the harms of overdiagnosis.
The clinical evolution of prostate cancer is still not well understood. Increasing age is associated with increased mortality. Many men with less aggressive disease tend to die with cancer rather than die of cancer. However, it is not always possible at the time of diagnosis to determine which tumors will be aggressive and which will grow slowly.
On the other hand, with screening, many of these indolent cancers are unnecessarily detected, generating excessive exams and treatments with negative repercussions (eg, pain, bleeding, infections, stress, and urinary and sexual dysfunction).
So, how should we as clinicians proceed regarding screening?
We should request the PSA test and emphasize the importance of digital rectal exam by a urologist for those at high risk for prostatic neoplasia (ie, those with family history) or those with urinary symptoms that may be associated with prostate cancer.
In general, we should draw attention to the possible risks and benefits of testing and adopt a shared decision-making approach with asymptomatic men or those at low risk who wish to have the screening exam. But achieving a shared decision is not a simple task.
I always have a thorough conversation with patients, but I confess that I request the exam in most cases.
Dr. Wajngarten is a professor of cardiology, Faculty of Medicine, at the University of São Paulo in Brazil. Dr. Wajngarten reported no conflicts of interest.
This story was translated from the Medscape Portuguese edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Many patients ask us to request a prostate-specific antigen (PSA) test. According to the Brazilian Ministry of Health, prostate cancer is the second most common type of cancer in the male population in all regions of our country. It is the second-leading cause of cancer death in the male population, reaffirming its epidemiologic importance in Brazil. On the other hand, a Ministry of Health technical paper recommends against population-based screening for prostate cancer. So, what should we do?
First, it is important to distinguish early diagnosis from screening. Early diagnosis is the identification of cancer in early stages in people with signs and symptoms. Screening is characterized by the systematic application of exams — digital rectal exam and PSA test — in asymptomatic people, with the aim of identifying cancer in an early stage.
A recent European epidemiologic study reinforced this thesis and helps guide us.
The study included men aged 35-84 years from 26 European countries. Data on cancer incidence and mortality were collected between 1980 and 2017. The data suggested overdiagnosis of prostate cancer, which varied over time and among populations. The findings supported previous recommendations that any implementation of prostate cancer screening should be carefully designed, with an emphasis on minimizing the harms of overdiagnosis.
The clinical evolution of prostate cancer is still not well understood. Increasing age is associated with increased mortality. Many men with less aggressive disease tend to die with cancer rather than die of cancer. However, it is not always possible at the time of diagnosis to determine which tumors will be aggressive and which will grow slowly.
On the other hand, with screening, many of these indolent cancers are unnecessarily detected, generating excessive exams and treatments with negative repercussions (eg, pain, bleeding, infections, stress, and urinary and sexual dysfunction).
So, how should we as clinicians proceed regarding screening?
We should request the PSA test and emphasize the importance of digital rectal exam by a urologist for those at high risk for prostatic neoplasia (ie, those with family history) or those with urinary symptoms that may be associated with prostate cancer.
In general, we should draw attention to the possible risks and benefits of testing and adopt a shared decision-making approach with asymptomatic men or those at low risk who wish to have the screening exam. But achieving a shared decision is not a simple task.
I always have a thorough conversation with patients, but I confess that I request the exam in most cases.
Dr. Wajngarten is a professor of cardiology, Faculty of Medicine, at the University of São Paulo in Brazil. Dr. Wajngarten reported no conflicts of interest.
This story was translated from the Medscape Portuguese edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Many patients ask us to request a prostate-specific antigen (PSA) test. According to the Brazilian Ministry of Health, prostate cancer is the second most common type of cancer in the male population in all regions of our country. It is the second-leading cause of cancer death in the male population, reaffirming its epidemiologic importance in Brazil. On the other hand, a Ministry of Health technical paper recommends against population-based screening for prostate cancer. So, what should we do?
First, it is important to distinguish early diagnosis from screening. Early diagnosis is the identification of cancer in early stages in people with signs and symptoms. Screening is characterized by the systematic application of exams — digital rectal exam and PSA test — in asymptomatic people, with the aim of identifying cancer in an early stage.
A recent European epidemiologic study reinforced this thesis and helps guide us.
The study included men aged 35-84 years from 26 European countries. Data on cancer incidence and mortality were collected between 1980 and 2017. The data suggested overdiagnosis of prostate cancer, which varied over time and among populations. The findings supported previous recommendations that any implementation of prostate cancer screening should be carefully designed, with an emphasis on minimizing the harms of overdiagnosis.
The clinical evolution of prostate cancer is still not well understood. Increasing age is associated with increased mortality. Many men with less aggressive disease tend to die with cancer rather than die of cancer. However, it is not always possible at the time of diagnosis to determine which tumors will be aggressive and which will grow slowly.
On the other hand, with screening, many of these indolent cancers are unnecessarily detected, generating excessive exams and treatments with negative repercussions (eg, pain, bleeding, infections, stress, and urinary and sexual dysfunction).
So, how should we as clinicians proceed regarding screening?
We should request the PSA test and emphasize the importance of digital rectal exam by a urologist for those at high risk for prostatic neoplasia (ie, those with family history) or those with urinary symptoms that may be associated with prostate cancer.
In general, we should draw attention to the possible risks and benefits of testing and adopt a shared decision-making approach with asymptomatic men or those at low risk who wish to have the screening exam. But achieving a shared decision is not a simple task.
I always have a thorough conversation with patients, but I confess that I request the exam in most cases.
Dr. Wajngarten is a professor of cardiology, Faculty of Medicine, at the University of São Paulo in Brazil. Dr. Wajngarten reported no conflicts of interest.
This story was translated from the Medscape Portuguese edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
Maternal Immunization to Prevent Serious Respiratory Illness
Editor’s Note: Sadly, this is the last column in the Master Class Obstetrics series. This award-winning column has been part of Ob.Gyn. News for 20 years. The deep discussion of cutting-edge topics in obstetrics by specialists and researchers will be missed as will the leadership and curation of topics by Dr. E. Albert Reece.
Introduction: The Need for Increased Vigilance About Maternal Immunization
Viruses are becoming increasingly prevalent in our world and the consequences of viral infections are implicated in a growing number of disease states. It is well established that certain cancers are caused by viruses and it is increasingly evident that viral infections can trigger the development of chronic illness. In pregnant women, viruses such as cytomegalovirus can cause infection in utero and lead to long-term impairments for the baby.
Likewise, it appears that the virulence of viruses is increasing, whether it be the respiratory syncytial virus (RSV) in children or the severe acute respiratory syndrome (SARS) coronaviruses in adults. Clearly, our environment is changing, with increases in population growth and urbanization, for instance, and an intensification of climate change and its effects. Viruses are part of this changing background.

Vaccines are our most powerful tool to protect people of all ages against viral threats, and fortunately, we benefit from increasing expertise in vaccinology. Since 1974, the University of Maryland School of Medicine has a Center for Vaccine Development and Global Health that has conducted research on vaccines to defend against the Zika virus, H1N1, Ebola, and SARS-CoV-2.
We’re not alone. Other vaccinology centers across the country — as well as the National Institutes of Health at the national level, through its National Institute of Allergy and Infectious Diseases — are doing research and developing vaccines to combat viral diseases.
In this column, we are focused on viral diseases in pregnancy and the role that vaccines can play in preventing serious respiratory illness in mothers and their newborns. I have invited Laura E. Riley, MD, the Given Foundation Professor and Chair of Obstetrics and Gynecology at Weill Cornell Medicine, to address the importance of maternal immunization and how we can best counsel our patients and improve immunization rates.
As Dr. Riley explains, we are in a new era, and it behooves us all to be more vigilant about recommending vaccines, combating misperceptions, addressing patients’ knowledge gaps, and administering vaccines whenever possible.
Dr. Reece is the former Dean of Medicine & University Executive VP, and The Distinguished University and Endowed Professor & Director of the Center for Advanced Research Training and Innovation (CARTI) at the University of Maryland School of Medicine, as well as senior scientist at the Center for Birth Defects Research.
The alarming decline in maternal immunization rates that occurred in the wake of the COVID-19 pandemic means that, now more than ever, we must fully embrace our responsibility to recommend immunizations in pregnancy and to communicate what is known about their efficacy and safety. Data show that vaccination rates drop when we do not offer vaccines in our offices, so whenever possible, we should administer them as well.
The ob.gyn. is the patient’s most trusted person in pregnancy. When patients decline or express hesitancy about vaccines, it is incumbent upon us to ask why. Oftentimes, we can identify areas in which patients lack knowledge or have misperceptions and we can successfully educate the patient or change their perspective or misunderstanding concerning the importance of vaccination for themselves and their babies. (See Table 1.) We can also successfully address concerns about safety.
The safety of COVID-19 vaccinations in pregnancy is now backed by several years of data from multiple studies showing no increase in birth defects, preterm delivery, miscarriage, or stillbirth.
Data also show that pregnant patients are more likely than patients who are not pregnant to need hospitalization and intensive care when infected with SARS-CoV-2 and are at risk of having complications that can affect pregnancy and the newborn, including preterm birth and stillbirth. Vaccination has been shown to reduce the risk of severe illness and the risk of such adverse obstetrical outcomes, in addition to providing protection for the infant early on.
Similarly, influenza has long been more likely to be severe in pregnant patients, with an increased risk of poor obstetrical outcomes. Vaccines similarly provide “two for one protection,” protecting both mother and baby, and are, of course, backed by many years of safety and efficacy data.

With the new maternal respiratory syncytial virus (RSV) vaccine, now in its second year of availability, the goal is to protect the baby from RSV-caused serious lower respiratory tract illness. The illness has contributed to tens of thousands of annual hospitalizations and up to several hundred deaths every year in children younger than 5 years — particularly in those under age 6 months.
The RSV monoclonal antibody nirsevimab is available for the newborn as an alternative to maternal immunization but the maternal vaccine is optimal in that it will provide immediate rather than delayed protection for the newborn. The maternal vaccine is recommended during weeks 32-36 of pregnancy in mothers who were not vaccinated during last year’s RSV season. With real-world experience from year one, the available safety data are reassuring.
Counseling About Influenza and COVID-19 Vaccination
The COVID-19 pandemic took a toll on vaccination interest/receptivity broadly in pregnant and nonpregnant people. Among pregnant individuals, influenza vaccination coverage declined from 71% in the 2019-2020 influenza season to 56% in the 2021-2022 season, according to data from the Centers for Disease Control and Prevention’s Vaccine Safety Datalink.4 Coverage for the 2022-2023 and 2023-2024 influenza seasons was even worse: well under 50%.5
Fewer pregnant women have received updated COVID-19 vaccines. Only 13% of pregnant persons overall received the updated 2023-2024 COVID-19 booster vaccine (through March 30, 2024), according to the CDC.6
Maternal immunization for influenza has been recommended in the United States since 2004 (part of the recommendation that everyone over the age of 6 months receive an annual flu vaccine), and flu vaccines have been given to millions of pregnant women, but the H1N1 pandemic of 2009 reinforced its value as a priority for prenatal care. Most of the women who became severely ill from the H1N1 virus were young and healthy, without co-existing conditions known to increase risk.7
It became clearer during the H1N1 pandemic that pregnancy itself — which is associated with physiologic changes such as decreased lung capacity, increased nasal congestion and changes in the immune system – is its own significant risk factor for severe illness from the influenza virus. This increased risk applies to COVID-19 as well.
As COVID-19 has become endemic, with hospitalizations and deaths not reaching the levels of previous surges — and with mask-wearing and other preventive measures having declined — patients understandably have become more complacent. Some patients are vaccine deniers, but in my practice, these patients are a much smaller group than those who believe COVID-19 “is no big deal,” especially if they have had infections recently.
This is why it’s important to actively listen to concerns and to ask patients who decline a vaccination why they are hesitant. Blanket messages about vaccine efficacy and safety are the first step, but individualized, more pointed conversations based on the patient’s personal experiences and beliefs have become increasingly important.
I routinely tell pregnant patients about the risks of COVID-19 and I explain that it has been difficult to predict who will develop severe illness. Sometimes more conversation is needed. For those who are still hesitant or who tell me they feel protected by a recent infection, for instance, I provide more detail on the unique risks of pregnancy — the fact that “pregnancy is different” — and that natural immunity wanes while the protection afforded by immunization is believed to last longer. Many women are also concerned about the safety of the COVID-19 vaccine, so having safety data at your fingertips is helpful. (See Table 2.)
The fact that influenza and COVID-19 vaccination protect the newborn as well as the mother is something that I find is underappreciated by many patients. Explaining that infants likely benefit from the passage of antibodies across the placenta should be part of patient counseling.
Counseling About RSV Vaccination
Importantly, for the 2024-2025 RSV season, the maternal RSV vaccine (Abrysvo, Pfizer) is recommended only for pregnant women who did not receive the vaccine during the 2023-2024 season. When more research is done and more data are obtained showing how long the immune response persists post vaccination, it may be that the US Food and Drug Administration (FDA) will approve the maternal RSV vaccine for use in every pregnancy.
The later timing of the vaccination recommendation — 32-36 weeks’ gestation — reflects a conservative approach taken by the FDA in response to data from one of the pivotal trials showing a numerical trend toward more preterm deliveries among vaccinated compared with unvaccinated patients. This imbalance in the original trial, which administered the vaccine during 24-36 weeks of gestation, was seen only in low-income countries with no temporal association, however.
In our experience at two Weill Cornell Medical College–associated hospitals we did not see this trend. Our cohort study of almost 3000 pregnant patients who delivered at 32 weeks’ gestation or later found no increased risk of preterm birth among the 35% of patients who received the RSV vaccine during the 2023-2024 RSV season. We also did not see any difference in preeclampsia, in contrast with original trial data that showed a signal for increased risk.11
When fewer than 2 weeks have elapsed between maternal vaccination and delivery, the monoclonal antibody nirsevimab is recommended for the newborn — ideally before the newborn leaves the hospital. Nirsevimab is also recommended for newborns of mothers who decline vaccination or were not candidates (e.g. vaccinated in a previous pregnancy), or when there is concern about the adequacy of the maternal immune response to the vaccine (e.g. in cases of immunosuppression).
While there was a limited supply of the monoclonal antibody last year, limitations are not expected this year, especially after October.
The ultimate goal is that patients choose the vaccine or the immunoglobulin, given the severity of RSV disease. Patient preferences should be considered. However, given that it takes 2 weeks after vaccination for protection to build up, I stress to patients that if they’ve vaccinated themselves, their newborn will leave the hospital with protection. If nirsevimab is relied upon, I explain, their newborn may not be protected for some period of time.
Take-home Messages
- When patients decline or are hesitant about vaccines, ask why. Listen actively, and work to correct misperceptions and knowledge gaps.
- Whenever possible, offer vaccines in your practice. Vaccination rates drop when this does not occur.
- COVID-vaccine safety is backed by many studies showing no increase in birth defects, preterm delivery, miscarriage, or stillbirth.
- Pregnant women are more likely to have severe illness from the influenza and SARS-CoV-2 viruses. Vaccines can prevent severe illness and can protect the newborn as well as the mother.
- Recommend/administer the maternal RSV vaccine at 32-36 weeks’ gestation in women who did not receive the vaccine in the 2023-2024 season. If mothers aren’t eligible their babies should be offered nirsevimab.
Dr. Riley is the Given Foundation Professor and Chair of Obstetrics and Gynecology at Weill Cornell Medicine and the obstetrician and gynecologist-in-chief at New York Presbyterian Hospital. She disclosed that she has provided one-time consultations to Pfizer (Abrysvo RSV vaccine) and GSK (cytomegalovirus vaccine), and is providing consultant education on CMV for Moderna. She is chair of ACOG’s task force on immunization and emerging infectious diseases, serves on the medical advisory board for MAVEN, and serves as an editor or editorial board member for several medical publications.
References
1. ACOG Committee Opinion No. 741: Maternal Immunization. Obstet Gynecol. 2018;131(6):e214-e217.
2. Centers for Disease Control and Prevention. COVID-19 Vaccination for People Who are Pregnant or Breastfeeding. https://www.cdc.gov/covid/vaccines/pregnant-or-breastfeeding.html.
3. ACOG Practice Advisory on Maternal Respiratory Syncytial Virus Vaccination, September 2023. (Updated August 2024).4. Irving S et al. Open Forum Infect Dis. 2023;10(Suppl 2):ofad500.1002.
5. Flu Vaccination Dashboard, CDC, National Center for Immunization and Respiratory Diseases.
6. Weekly COVID-19 Vaccination Dashboard, CDC. https://www.cdc.gov/covidvaxview/weekly-dashboard/index.html
7. Louie JK et al. N Engl J Med. 2010;362:27-35. 8. Ciapponi A et al. Vaccine. 2021;39(40):5891-908.
9. Prasad S et al. Nature Communications. 2022;13:2414. 10. Fleming-Dutra KE et al. Obstet Gynecol Clin North Am 2023;50(2):279-97. 11. Mouen S et al. JAMA Network Open 2024;7(7):e2419268.
Editor’s Note: Sadly, this is the last column in the Master Class Obstetrics series. This award-winning column has been part of Ob.Gyn. News for 20 years. The deep discussion of cutting-edge topics in obstetrics by specialists and researchers will be missed as will the leadership and curation of topics by Dr. E. Albert Reece.
Introduction: The Need for Increased Vigilance About Maternal Immunization
Viruses are becoming increasingly prevalent in our world and the consequences of viral infections are implicated in a growing number of disease states. It is well established that certain cancers are caused by viruses and it is increasingly evident that viral infections can trigger the development of chronic illness. In pregnant women, viruses such as cytomegalovirus can cause infection in utero and lead to long-term impairments for the baby.
Likewise, it appears that the virulence of viruses is increasing, whether it be the respiratory syncytial virus (RSV) in children or the severe acute respiratory syndrome (SARS) coronaviruses in adults. Clearly, our environment is changing, with increases in population growth and urbanization, for instance, and an intensification of climate change and its effects. Viruses are part of this changing background.
Vaccines are our most powerful tool to protect people of all ages against viral threats, and fortunately, we benefit from increasing expertise in vaccinology. Since 1974, the University of Maryland School of Medicine has a Center for Vaccine Development and Global Health that has conducted research on vaccines to defend against the Zika virus, H1N1, Ebola, and SARS-CoV-2.
We’re not alone. Other vaccinology centers across the country — as well as the National Institutes of Health at the national level, through its National Institute of Allergy and Infectious Diseases — are doing research and developing vaccines to combat viral diseases.
In this column, we are focused on viral diseases in pregnancy and the role that vaccines can play in preventing serious respiratory illness in mothers and their newborns. I have invited Laura E. Riley, MD, the Given Foundation Professor and Chair of Obstetrics and Gynecology at Weill Cornell Medicine, to address the importance of maternal immunization and how we can best counsel our patients and improve immunization rates.
As Dr. Riley explains, we are in a new era, and it behooves us all to be more vigilant about recommending vaccines, combating misperceptions, addressing patients’ knowledge gaps, and administering vaccines whenever possible.
Dr. Reece is the former Dean of Medicine & University Executive VP, and The Distinguished University and Endowed Professor & Director of the Center for Advanced Research Training and Innovation (CARTI) at the University of Maryland School of Medicine, as well as senior scientist at the Center for Birth Defects Research.
The alarming decline in maternal immunization rates that occurred in the wake of the COVID-19 pandemic means that, now more than ever, we must fully embrace our responsibility to recommend immunizations in pregnancy and to communicate what is known about their efficacy and safety. Data show that vaccination rates drop when we do not offer vaccines in our offices, so whenever possible, we should administer them as well.
The ob.gyn. is the patient’s most trusted person in pregnancy. When patients decline or express hesitancy about vaccines, it is incumbent upon us to ask why. Oftentimes, we can identify areas in which patients lack knowledge or have misperceptions and we can successfully educate the patient or change their perspective or misunderstanding concerning the importance of vaccination for themselves and their babies. (See Table 1.) We can also successfully address concerns about safety.
The safety of COVID-19 vaccinations in pregnancy is now backed by several years of data from multiple studies showing no increase in birth defects, preterm delivery, miscarriage, or stillbirth.
Data also show that pregnant patients are more likely than patients who are not pregnant to need hospitalization and intensive care when infected with SARS-CoV-2 and are at risk of having complications that can affect pregnancy and the newborn, including preterm birth and stillbirth. Vaccination has been shown to reduce the risk of severe illness and the risk of such adverse obstetrical outcomes, in addition to providing protection for the infant early on.
Similarly, influenza has long been more likely to be severe in pregnant patients, with an increased risk of poor obstetrical outcomes. Vaccines similarly provide “two for one protection,” protecting both mother and baby, and are, of course, backed by many years of safety and efficacy data.
With the new maternal respiratory syncytial virus (RSV) vaccine, now in its second year of availability, the goal is to protect the baby from RSV-caused serious lower respiratory tract illness. The illness has contributed to tens of thousands of annual hospitalizations and up to several hundred deaths every year in children younger than 5 years — particularly in those under age 6 months.
The RSV monoclonal antibody nirsevimab is available for the newborn as an alternative to maternal immunization but the maternal vaccine is optimal in that it will provide immediate rather than delayed protection for the newborn. The maternal vaccine is recommended during weeks 32-36 of pregnancy in mothers who were not vaccinated during last year’s RSV season. With real-world experience from year one, the available safety data are reassuring.
Counseling About Influenza and COVID-19 Vaccination
The COVID-19 pandemic took a toll on vaccination interest/receptivity broadly in pregnant and nonpregnant people. Among pregnant individuals, influenza vaccination coverage declined from 71% in the 2019-2020 influenza season to 56% in the 2021-2022 season, according to data from the Centers for Disease Control and Prevention’s Vaccine Safety Datalink.4 Coverage for the 2022-2023 and 2023-2024 influenza seasons was even worse: well under 50%.5
Fewer pregnant women have received updated COVID-19 vaccines. Only 13% of pregnant persons overall received the updated 2023-2024 COVID-19 booster vaccine (through March 30, 2024), according to the CDC.6
Maternal immunization for influenza has been recommended in the United States since 2004 (part of the recommendation that everyone over the age of 6 months receive an annual flu vaccine), and flu vaccines have been given to millions of pregnant women, but the H1N1 pandemic of 2009 reinforced its value as a priority for prenatal care. Most of the women who became severely ill from the H1N1 virus were young and healthy, without co-existing conditions known to increase risk.7
It became clearer during the H1N1 pandemic that pregnancy itself — which is associated with physiologic changes such as decreased lung capacity, increased nasal congestion and changes in the immune system – is its own significant risk factor for severe illness from the influenza virus. This increased risk applies to COVID-19 as well.
As COVID-19 has become endemic, with hospitalizations and deaths not reaching the levels of previous surges — and with mask-wearing and other preventive measures having declined — patients understandably have become more complacent. Some patients are vaccine deniers, but in my practice, these patients are a much smaller group than those who believe COVID-19 “is no big deal,” especially if they have had infections recently.
This is why it’s important to actively listen to concerns and to ask patients who decline a vaccination why they are hesitant. Blanket messages about vaccine efficacy and safety are the first step, but individualized, more pointed conversations based on the patient’s personal experiences and beliefs have become increasingly important.
I routinely tell pregnant patients about the risks of COVID-19 and I explain that it has been difficult to predict who will develop severe illness. Sometimes more conversation is needed. For those who are still hesitant or who tell me they feel protected by a recent infection, for instance, I provide more detail on the unique risks of pregnancy — the fact that “pregnancy is different” — and that natural immunity wanes while the protection afforded by immunization is believed to last longer. Many women are also concerned about the safety of the COVID-19 vaccine, so having safety data at your fingertips is helpful. (See Table 2.)
The fact that influenza and COVID-19 vaccination protect the newborn as well as the mother is something that I find is underappreciated by many patients. Explaining that infants likely benefit from the passage of antibodies across the placenta should be part of patient counseling.
Counseling About RSV Vaccination
Importantly, for the 2024-2025 RSV season, the maternal RSV vaccine (Abrysvo, Pfizer) is recommended only for pregnant women who did not receive the vaccine during the 2023-2024 season. When more research is done and more data are obtained showing how long the immune response persists post vaccination, it may be that the US Food and Drug Administration (FDA) will approve the maternal RSV vaccine for use in every pregnancy.
The later timing of the vaccination recommendation — 32-36 weeks’ gestation — reflects a conservative approach taken by the FDA in response to data from one of the pivotal trials showing a numerical trend toward more preterm deliveries among vaccinated compared with unvaccinated patients. This imbalance in the original trial, which administered the vaccine during 24-36 weeks of gestation, was seen only in low-income countries with no temporal association, however.
In our experience at two Weill Cornell Medical College–associated hospitals we did not see this trend. Our cohort study of almost 3000 pregnant patients who delivered at 32 weeks’ gestation or later found no increased risk of preterm birth among the 35% of patients who received the RSV vaccine during the 2023-2024 RSV season. We also did not see any difference in preeclampsia, in contrast with original trial data that showed a signal for increased risk.11
When fewer than 2 weeks have elapsed between maternal vaccination and delivery, the monoclonal antibody nirsevimab is recommended for the newborn — ideally before the newborn leaves the hospital. Nirsevimab is also recommended for newborns of mothers who decline vaccination or were not candidates (e.g. vaccinated in a previous pregnancy), or when there is concern about the adequacy of the maternal immune response to the vaccine (e.g. in cases of immunosuppression).
While there was a limited supply of the monoclonal antibody last year, limitations are not expected this year, especially after October.
The ultimate goal is that patients choose the vaccine or the immunoglobulin, given the severity of RSV disease. Patient preferences should be considered. However, given that it takes 2 weeks after vaccination for protection to build up, I stress to patients that if they’ve vaccinated themselves, their newborn will leave the hospital with protection. If nirsevimab is relied upon, I explain, their newborn may not be protected for some period of time.
Take-home Messages
- When patients decline or are hesitant about vaccines, ask why. Listen actively, and work to correct misperceptions and knowledge gaps.
- Whenever possible, offer vaccines in your practice. Vaccination rates drop when this does not occur.
- COVID-vaccine safety is backed by many studies showing no increase in birth defects, preterm delivery, miscarriage, or stillbirth.
- Pregnant women are more likely to have severe illness from the influenza and SARS-CoV-2 viruses. Vaccines can prevent severe illness and can protect the newborn as well as the mother.
- Recommend/administer the maternal RSV vaccine at 32-36 weeks’ gestation in women who did not receive the vaccine in the 2023-2024 season. If mothers aren’t eligible their babies should be offered nirsevimab.
Dr. Riley is the Given Foundation Professor and Chair of Obstetrics and Gynecology at Weill Cornell Medicine and the obstetrician and gynecologist-in-chief at New York Presbyterian Hospital. She disclosed that she has provided one-time consultations to Pfizer (Abrysvo RSV vaccine) and GSK (cytomegalovirus vaccine), and is providing consultant education on CMV for Moderna. She is chair of ACOG’s task force on immunization and emerging infectious diseases, serves on the medical advisory board for MAVEN, and serves as an editor or editorial board member for several medical publications.
References
1. ACOG Committee Opinion No. 741: Maternal Immunization. Obstet Gynecol. 2018;131(6):e214-e217.
2. Centers for Disease Control and Prevention. COVID-19 Vaccination for People Who are Pregnant or Breastfeeding. https://www.cdc.gov/covid/vaccines/pregnant-or-breastfeeding.html.
3. ACOG Practice Advisory on Maternal Respiratory Syncytial Virus Vaccination, September 2023. (Updated August 2024).4. Irving S et al. Open Forum Infect Dis. 2023;10(Suppl 2):ofad500.1002.
5. Flu Vaccination Dashboard, CDC, National Center for Immunization and Respiratory Diseases.
6. Weekly COVID-19 Vaccination Dashboard, CDC. https://www.cdc.gov/covidvaxview/weekly-dashboard/index.html
7. Louie JK et al. N Engl J Med. 2010;362:27-35. 8. Ciapponi A et al. Vaccine. 2021;39(40):5891-908.
9. Prasad S et al. Nature Communications. 2022;13:2414. 10. Fleming-Dutra KE et al. Obstet Gynecol Clin North Am 2023;50(2):279-97. 11. Mouen S et al. JAMA Network Open 2024;7(7):e2419268.
Editor’s Note: Sadly, this is the last column in the Master Class Obstetrics series. This award-winning column has been part of Ob.Gyn. News for 20 years. The deep discussion of cutting-edge topics in obstetrics by specialists and researchers will be missed as will the leadership and curation of topics by Dr. E. Albert Reece.
Introduction: The Need for Increased Vigilance About Maternal Immunization
Viruses are becoming increasingly prevalent in our world and the consequences of viral infections are implicated in a growing number of disease states. It is well established that certain cancers are caused by viruses and it is increasingly evident that viral infections can trigger the development of chronic illness. In pregnant women, viruses such as cytomegalovirus can cause infection in utero and lead to long-term impairments for the baby.
Likewise, it appears that the virulence of viruses is increasing, whether it be the respiratory syncytial virus (RSV) in children or the severe acute respiratory syndrome (SARS) coronaviruses in adults. Clearly, our environment is changing, with increases in population growth and urbanization, for instance, and an intensification of climate change and its effects. Viruses are part of this changing background.
Vaccines are our most powerful tool to protect people of all ages against viral threats, and fortunately, we benefit from increasing expertise in vaccinology. Since 1974, the University of Maryland School of Medicine has a Center for Vaccine Development and Global Health that has conducted research on vaccines to defend against the Zika virus, H1N1, Ebola, and SARS-CoV-2.
We’re not alone. Other vaccinology centers across the country — as well as the National Institutes of Health at the national level, through its National Institute of Allergy and Infectious Diseases — are doing research and developing vaccines to combat viral diseases.
In this column, we are focused on viral diseases in pregnancy and the role that vaccines can play in preventing serious respiratory illness in mothers and their newborns. I have invited Laura E. Riley, MD, the Given Foundation Professor and Chair of Obstetrics and Gynecology at Weill Cornell Medicine, to address the importance of maternal immunization and how we can best counsel our patients and improve immunization rates.
As Dr. Riley explains, we are in a new era, and it behooves us all to be more vigilant about recommending vaccines, combating misperceptions, addressing patients’ knowledge gaps, and administering vaccines whenever possible.
Dr. Reece is the former Dean of Medicine & University Executive VP, and The Distinguished University and Endowed Professor & Director of the Center for Advanced Research Training and Innovation (CARTI) at the University of Maryland School of Medicine, as well as senior scientist at the Center for Birth Defects Research.
The alarming decline in maternal immunization rates that occurred in the wake of the COVID-19 pandemic means that, now more than ever, we must fully embrace our responsibility to recommend immunizations in pregnancy and to communicate what is known about their efficacy and safety. Data show that vaccination rates drop when we do not offer vaccines in our offices, so whenever possible, we should administer them as well.
The ob.gyn. is the patient’s most trusted person in pregnancy. When patients decline or express hesitancy about vaccines, it is incumbent upon us to ask why. Oftentimes, we can identify areas in which patients lack knowledge or have misperceptions and we can successfully educate the patient or change their perspective or misunderstanding concerning the importance of vaccination for themselves and their babies. (See Table 1.) We can also successfully address concerns about safety.
The safety of COVID-19 vaccinations in pregnancy is now backed by several years of data from multiple studies showing no increase in birth defects, preterm delivery, miscarriage, or stillbirth.
Data also show that pregnant patients are more likely than patients who are not pregnant to need hospitalization and intensive care when infected with SARS-CoV-2 and are at risk of having complications that can affect pregnancy and the newborn, including preterm birth and stillbirth. Vaccination has been shown to reduce the risk of severe illness and the risk of such adverse obstetrical outcomes, in addition to providing protection for the infant early on.
Similarly, influenza has long been more likely to be severe in pregnant patients, with an increased risk of poor obstetrical outcomes. Vaccines similarly provide “two for one protection,” protecting both mother and baby, and are, of course, backed by many years of safety and efficacy data.
With the new maternal respiratory syncytial virus (RSV) vaccine, now in its second year of availability, the goal is to protect the baby from RSV-caused serious lower respiratory tract illness. The illness has contributed to tens of thousands of annual hospitalizations and up to several hundred deaths every year in children younger than 5 years — particularly in those under age 6 months.
The RSV monoclonal antibody nirsevimab is available for the newborn as an alternative to maternal immunization but the maternal vaccine is optimal in that it will provide immediate rather than delayed protection for the newborn. The maternal vaccine is recommended during weeks 32-36 of pregnancy in mothers who were not vaccinated during last year’s RSV season. With real-world experience from year one, the available safety data are reassuring.
Counseling About Influenza and COVID-19 Vaccination
The COVID-19 pandemic took a toll on vaccination interest/receptivity broadly in pregnant and nonpregnant people. Among pregnant individuals, influenza vaccination coverage declined from 71% in the 2019-2020 influenza season to 56% in the 2021-2022 season, according to data from the Centers for Disease Control and Prevention’s Vaccine Safety Datalink.4 Coverage for the 2022-2023 and 2023-2024 influenza seasons was even worse: well under 50%.5
Fewer pregnant women have received updated COVID-19 vaccines. Only 13% of pregnant persons overall received the updated 2023-2024 COVID-19 booster vaccine (through March 30, 2024), according to the CDC.6
Maternal immunization for influenza has been recommended in the United States since 2004 (part of the recommendation that everyone over the age of 6 months receive an annual flu vaccine), and flu vaccines have been given to millions of pregnant women, but the H1N1 pandemic of 2009 reinforced its value as a priority for prenatal care. Most of the women who became severely ill from the H1N1 virus were young and healthy, without co-existing conditions known to increase risk.7
It became clearer during the H1N1 pandemic that pregnancy itself — which is associated with physiologic changes such as decreased lung capacity, increased nasal congestion and changes in the immune system – is its own significant risk factor for severe illness from the influenza virus. This increased risk applies to COVID-19 as well.
As COVID-19 has become endemic, with hospitalizations and deaths not reaching the levels of previous surges — and with mask-wearing and other preventive measures having declined — patients understandably have become more complacent. Some patients are vaccine deniers, but in my practice, these patients are a much smaller group than those who believe COVID-19 “is no big deal,” especially if they have had infections recently.
This is why it’s important to actively listen to concerns and to ask patients who decline a vaccination why they are hesitant. Blanket messages about vaccine efficacy and safety are the first step, but individualized, more pointed conversations based on the patient’s personal experiences and beliefs have become increasingly important.
I routinely tell pregnant patients about the risks of COVID-19 and I explain that it has been difficult to predict who will develop severe illness. Sometimes more conversation is needed. For those who are still hesitant or who tell me they feel protected by a recent infection, for instance, I provide more detail on the unique risks of pregnancy — the fact that “pregnancy is different” — and that natural immunity wanes while the protection afforded by immunization is believed to last longer. Many women are also concerned about the safety of the COVID-19 vaccine, so having safety data at your fingertips is helpful. (See Table 2.)
The fact that influenza and COVID-19 vaccination protect the newborn as well as the mother is something that I find is underappreciated by many patients. Explaining that infants likely benefit from the passage of antibodies across the placenta should be part of patient counseling.
Counseling About RSV Vaccination
Importantly, for the 2024-2025 RSV season, the maternal RSV vaccine (Abrysvo, Pfizer) is recommended only for pregnant women who did not receive the vaccine during the 2023-2024 season. When more research is done and more data are obtained showing how long the immune response persists post vaccination, it may be that the US Food and Drug Administration (FDA) will approve the maternal RSV vaccine for use in every pregnancy.
The later timing of the vaccination recommendation — 32-36 weeks’ gestation — reflects a conservative approach taken by the FDA in response to data from one of the pivotal trials showing a numerical trend toward more preterm deliveries among vaccinated compared with unvaccinated patients. This imbalance in the original trial, which administered the vaccine during 24-36 weeks of gestation, was seen only in low-income countries with no temporal association, however.
In our experience at two Weill Cornell Medical College–associated hospitals we did not see this trend. Our cohort study of almost 3000 pregnant patients who delivered at 32 weeks’ gestation or later found no increased risk of preterm birth among the 35% of patients who received the RSV vaccine during the 2023-2024 RSV season. We also did not see any difference in preeclampsia, in contrast with original trial data that showed a signal for increased risk.11
When fewer than 2 weeks have elapsed between maternal vaccination and delivery, the monoclonal antibody nirsevimab is recommended for the newborn — ideally before the newborn leaves the hospital. Nirsevimab is also recommended for newborns of mothers who decline vaccination or were not candidates (e.g. vaccinated in a previous pregnancy), or when there is concern about the adequacy of the maternal immune response to the vaccine (e.g. in cases of immunosuppression).
While there was a limited supply of the monoclonal antibody last year, limitations are not expected this year, especially after October.
The ultimate goal is that patients choose the vaccine or the immunoglobulin, given the severity of RSV disease. Patient preferences should be considered. However, given that it takes 2 weeks after vaccination for protection to build up, I stress to patients that if they’ve vaccinated themselves, their newborn will leave the hospital with protection. If nirsevimab is relied upon, I explain, their newborn may not be protected for some period of time.
Take-home Messages
- When patients decline or are hesitant about vaccines, ask why. Listen actively, and work to correct misperceptions and knowledge gaps.
- Whenever possible, offer vaccines in your practice. Vaccination rates drop when this does not occur.
- COVID-vaccine safety is backed by many studies showing no increase in birth defects, preterm delivery, miscarriage, or stillbirth.
- Pregnant women are more likely to have severe illness from the influenza and SARS-CoV-2 viruses. Vaccines can prevent severe illness and can protect the newborn as well as the mother.
- Recommend/administer the maternal RSV vaccine at 32-36 weeks’ gestation in women who did not receive the vaccine in the 2023-2024 season. If mothers aren’t eligible their babies should be offered nirsevimab.
Dr. Riley is the Given Foundation Professor and Chair of Obstetrics and Gynecology at Weill Cornell Medicine and the obstetrician and gynecologist-in-chief at New York Presbyterian Hospital. She disclosed that she has provided one-time consultations to Pfizer (Abrysvo RSV vaccine) and GSK (cytomegalovirus vaccine), and is providing consultant education on CMV for Moderna. She is chair of ACOG’s task force on immunization and emerging infectious diseases, serves on the medical advisory board for MAVEN, and serves as an editor or editorial board member for several medical publications.
References
1. ACOG Committee Opinion No. 741: Maternal Immunization. Obstet Gynecol. 2018;131(6):e214-e217.
2. Centers for Disease Control and Prevention. COVID-19 Vaccination for People Who are Pregnant or Breastfeeding. https://www.cdc.gov/covid/vaccines/pregnant-or-breastfeeding.html.
3. ACOG Practice Advisory on Maternal Respiratory Syncytial Virus Vaccination, September 2023. (Updated August 2024).4. Irving S et al. Open Forum Infect Dis. 2023;10(Suppl 2):ofad500.1002.
5. Flu Vaccination Dashboard, CDC, National Center for Immunization and Respiratory Diseases.
6. Weekly COVID-19 Vaccination Dashboard, CDC. https://www.cdc.gov/covidvaxview/weekly-dashboard/index.html
7. Louie JK et al. N Engl J Med. 2010;362:27-35. 8. Ciapponi A et al. Vaccine. 2021;39(40):5891-908.
9. Prasad S et al. Nature Communications. 2022;13:2414. 10. Fleming-Dutra KE et al. Obstet Gynecol Clin North Am 2023;50(2):279-97. 11. Mouen S et al. JAMA Network Open 2024;7(7):e2419268.
Are Targeted Drugs the Future in Colorectal Cancer?
This transcript has been edited for clarity.
Welcome back, everybody, from the European Society for Medical Oncology (ESMO) Congress in the wonderful city of Barcelona in Spain. I was coming from ESMO drenched in huge amounts of new data.
They looked at molecularly targeted drugs, some early-stage and a later-stage study in which there’s some evidence of promise.
She talked a little about the preliminary results from three trials suggesting some benefits, pretty marginal, of cetuximab plus irinotecan in patients who’d already had epidermal growth factor receptor (EGFR) receptor inhibitory treatment.
Amivantamab plus FOLFOX or FOLFIRI was also discussed. This is a bispecific antibody against EGFR and MET. Again, very early, but there are some potential marginal benefits coming through. She also discussed the results of a larger phase 3 randomized trial with an old friend, ramucirumab, the anti-angiogenic agent, in which the ramucirumab in combination with trifluridine-tipiracil failed to meet its primary endpoint of improving overall survival.
There were some interesting post hoc subgroup analyses showing potential benefits for women, left-sided tumors, and so on. She made an excellent presentation, which she summarized by saying that the future of colorectal cancer treatment lies in further defining molecularly targeted treatment.
Nobody would disagree with that. What is interesting, though, is that, if I were to use the analogy of mining, the more deeply we mine, perhaps the lower marginal the benefits are becoming. There’s no doubt that we’re understanding better the exquisite machinery of cell signaling. We understand that there’s redundancy, there’s repeatability, and the possibility of emergence of resistance can come quite quickly.
Although we can develop ever more precise molecularly targeted drugs, it does seem as if the clinical benefits of these, in some cases, are marginally small. I’d like to suggest that, in addition to Sara’s call for more molecularly targeted drugs, we should think about cellular targets.
We did a large amount of work (as have many others, of course) looking at the immune tumor microenvironment and trying to, in a way, separate and understand the contribution of the individual component cells — of which there are many, including cancer-associated fibroblasts, natural killer (NK) cells, whole hosts of different types of T-cell subsets, B cells, tumor-associated neutrophils, and so on — and how these interact together and of interact with the epithelial colorectal cancer cells.
We are collaborating with Patrick Soon-Shiong, a clever chap, who believes in combination immunotherapy, dissecting and understanding the individual role of these different cells, and coming up with cellular therapies or targeted therapies that either inhibit or stimulate some of the different cell components to be the way ahead for an immunologically cold tumor such as microsatellite-stable colorectal cancer.
For example, we’re looking at combinations of our histone deacetylase (HDAC) inhibitor, which switches on the machinery of antigen presentation, up-regulating major histocompatibility complex (MHC) class 1 and class 2, and some other of the molecules involved in antigen chopping and presentation; it’s like turning a microsatellite-stable immunologically cold tumor hot; an interleukin-15 superagonist that stimulates NK cells; and we’ve found a way to manipulate and reduce the number of Treg cells.
We have various approaches to reducing the microenvironment transforming growth factor beta and some of the downstream elements from that. We can look at combinatorial immunotherapy, but thinking at a cellular level and developing anticancer agents that either activate or inhibit these different cell components. I’d bring the two together.
Of course, the future has got to be better molecularly targeted drugs, but let’s think at a macro level as to how we can look at the different cellular interactions within the tumor microenvironment, and perhaps through that, come up with synergistic immunotherapeutic combinations.
Dr. Kerr is Professor, Nuffield Department of Clinical Laboratory Science, University of Oxford, and Professor of Cancer Medicine, Oxford Cancer Centre, both in England. He reported conflicts of interest with Celleron Therapeutics, Oxford Cancer Biomarkers, Afrox, GlaxoSmithKline, Genomic Health, and Merck Serono.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
Welcome back, everybody, from the European Society for Medical Oncology (ESMO) Congress in the wonderful city of Barcelona in Spain. I was coming from ESMO drenched in huge amounts of new data.
They looked at molecularly targeted drugs, some early-stage and a later-stage study in which there’s some evidence of promise.
She talked a little about the preliminary results from three trials suggesting some benefits, pretty marginal, of cetuximab plus irinotecan in patients who’d already had epidermal growth factor receptor (EGFR) receptor inhibitory treatment.
Amivantamab plus FOLFOX or FOLFIRI was also discussed. This is a bispecific antibody against EGFR and MET. Again, very early, but there are some potential marginal benefits coming through. She also discussed the results of a larger phase 3 randomized trial with an old friend, ramucirumab, the anti-angiogenic agent, in which the ramucirumab in combination with trifluridine-tipiracil failed to meet its primary endpoint of improving overall survival.
There were some interesting post hoc subgroup analyses showing potential benefits for women, left-sided tumors, and so on. She made an excellent presentation, which she summarized by saying that the future of colorectal cancer treatment lies in further defining molecularly targeted treatment.
Nobody would disagree with that. What is interesting, though, is that, if I were to use the analogy of mining, the more deeply we mine, perhaps the lower marginal the benefits are becoming. There’s no doubt that we’re understanding better the exquisite machinery of cell signaling. We understand that there’s redundancy, there’s repeatability, and the possibility of emergence of resistance can come quite quickly.
Although we can develop ever more precise molecularly targeted drugs, it does seem as if the clinical benefits of these, in some cases, are marginally small. I’d like to suggest that, in addition to Sara’s call for more molecularly targeted drugs, we should think about cellular targets.
We did a large amount of work (as have many others, of course) looking at the immune tumor microenvironment and trying to, in a way, separate and understand the contribution of the individual component cells — of which there are many, including cancer-associated fibroblasts, natural killer (NK) cells, whole hosts of different types of T-cell subsets, B cells, tumor-associated neutrophils, and so on — and how these interact together and of interact with the epithelial colorectal cancer cells.
We are collaborating with Patrick Soon-Shiong, a clever chap, who believes in combination immunotherapy, dissecting and understanding the individual role of these different cells, and coming up with cellular therapies or targeted therapies that either inhibit or stimulate some of the different cell components to be the way ahead for an immunologically cold tumor such as microsatellite-stable colorectal cancer.
For example, we’re looking at combinations of our histone deacetylase (HDAC) inhibitor, which switches on the machinery of antigen presentation, up-regulating major histocompatibility complex (MHC) class 1 and class 2, and some other of the molecules involved in antigen chopping and presentation; it’s like turning a microsatellite-stable immunologically cold tumor hot; an interleukin-15 superagonist that stimulates NK cells; and we’ve found a way to manipulate and reduce the number of Treg cells.
We have various approaches to reducing the microenvironment transforming growth factor beta and some of the downstream elements from that. We can look at combinatorial immunotherapy, but thinking at a cellular level and developing anticancer agents that either activate or inhibit these different cell components. I’d bring the two together.
Of course, the future has got to be better molecularly targeted drugs, but let’s think at a macro level as to how we can look at the different cellular interactions within the tumor microenvironment, and perhaps through that, come up with synergistic immunotherapeutic combinations.
Dr. Kerr is Professor, Nuffield Department of Clinical Laboratory Science, University of Oxford, and Professor of Cancer Medicine, Oxford Cancer Centre, both in England. He reported conflicts of interest with Celleron Therapeutics, Oxford Cancer Biomarkers, Afrox, GlaxoSmithKline, Genomic Health, and Merck Serono.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
Welcome back, everybody, from the European Society for Medical Oncology (ESMO) Congress in the wonderful city of Barcelona in Spain. I was coming from ESMO drenched in huge amounts of new data.
They looked at molecularly targeted drugs, some early-stage and a later-stage study in which there’s some evidence of promise.
She talked a little about the preliminary results from three trials suggesting some benefits, pretty marginal, of cetuximab plus irinotecan in patients who’d already had epidermal growth factor receptor (EGFR) receptor inhibitory treatment.
Amivantamab plus FOLFOX or FOLFIRI was also discussed. This is a bispecific antibody against EGFR and MET. Again, very early, but there are some potential marginal benefits coming through. She also discussed the results of a larger phase 3 randomized trial with an old friend, ramucirumab, the anti-angiogenic agent, in which the ramucirumab in combination with trifluridine-tipiracil failed to meet its primary endpoint of improving overall survival.
There were some interesting post hoc subgroup analyses showing potential benefits for women, left-sided tumors, and so on. She made an excellent presentation, which she summarized by saying that the future of colorectal cancer treatment lies in further defining molecularly targeted treatment.
Nobody would disagree with that. What is interesting, though, is that, if I were to use the analogy of mining, the more deeply we mine, perhaps the lower marginal the benefits are becoming. There’s no doubt that we’re understanding better the exquisite machinery of cell signaling. We understand that there’s redundancy, there’s repeatability, and the possibility of emergence of resistance can come quite quickly.
Although we can develop ever more precise molecularly targeted drugs, it does seem as if the clinical benefits of these, in some cases, are marginally small. I’d like to suggest that, in addition to Sara’s call for more molecularly targeted drugs, we should think about cellular targets.
We did a large amount of work (as have many others, of course) looking at the immune tumor microenvironment and trying to, in a way, separate and understand the contribution of the individual component cells — of which there are many, including cancer-associated fibroblasts, natural killer (NK) cells, whole hosts of different types of T-cell subsets, B cells, tumor-associated neutrophils, and so on — and how these interact together and of interact with the epithelial colorectal cancer cells.
We are collaborating with Patrick Soon-Shiong, a clever chap, who believes in combination immunotherapy, dissecting and understanding the individual role of these different cells, and coming up with cellular therapies or targeted therapies that either inhibit or stimulate some of the different cell components to be the way ahead for an immunologically cold tumor such as microsatellite-stable colorectal cancer.
For example, we’re looking at combinations of our histone deacetylase (HDAC) inhibitor, which switches on the machinery of antigen presentation, up-regulating major histocompatibility complex (MHC) class 1 and class 2, and some other of the molecules involved in antigen chopping and presentation; it’s like turning a microsatellite-stable immunologically cold tumor hot; an interleukin-15 superagonist that stimulates NK cells; and we’ve found a way to manipulate and reduce the number of Treg cells.
We have various approaches to reducing the microenvironment transforming growth factor beta and some of the downstream elements from that. We can look at combinatorial immunotherapy, but thinking at a cellular level and developing anticancer agents that either activate or inhibit these different cell components. I’d bring the two together.
Of course, the future has got to be better molecularly targeted drugs, but let’s think at a macro level as to how we can look at the different cellular interactions within the tumor microenvironment, and perhaps through that, come up with synergistic immunotherapeutic combinations.
Dr. Kerr is Professor, Nuffield Department of Clinical Laboratory Science, University of Oxford, and Professor of Cancer Medicine, Oxford Cancer Centre, both in England. He reported conflicts of interest with Celleron Therapeutics, Oxford Cancer Biomarkers, Afrox, GlaxoSmithKline, Genomic Health, and Merck Serono.
A version of this article first appeared on Medscape.com.
Nonalcoholic Beer and Underage Drinking
Several months ago in a letter about healthcare providers and the decision to use alcohol and other mind-altering substances on the job, I waxed enthusiastically about the new wave of no alcohol (NA) and zero (00) alcohol beers that have come on the market. In the last 2 years our local grocery store’s cooler space for nonalcoholic beer has grown from less than 24 inches to something approaching the height of the average sixth grader.
In a bold act of chivalry at the beginning of the pandemic I accepted the mantle of designated grocery shopper and over the last 3 years have become uncommonly proud of my ability to bring home the groceries efficiently and cost effectively, without catching COVID in the process. I have developed a sixth sense of choosing which human checker/bagger combination is fastest or whether the self-checkout is the way to go.

For obvious reasons the human checkers don’t ask for my ID when I am buying adult beverages. However, the self-check register freezes up instantly when I scan my 12-pack of Run Wild nonalcoholic. This necessitates a search for the MIA store person assigned to patrol the self-check corral, ever on the lookout for shoplifters, underage drinkers, and other generally shifty looking characters.
When I find one of the grocery store detectives (who is likely to have been a former patient), I say: “You know, this doesn’t have any alcohol in it.” They invariably reply with a shrug. “I know. But, the rules are the rules.” Occasionally, they may add: “It doesn’t make sense, does it?”
At first blush checking IDs for a nonalcoholic beverage may sound dumb, certainly to someone who is just a few years on either side of the legal drinking age. Why are we trying to protect some crazy teenager from the futility of getting high on a six-pack of something that at worst will make him spend most of the next couple of hours peeing?
But, there is concern in some corners that nonalcoholic drinks pose a significant threat to teenagers. Two PhDs at Stanford University have recently published a paper in which they worry that the dramatic rise in US sales of nonalcoholic drinks from 15% to 30% since 2018 may be socializing “users of alcohol drinking experiences by exposing them to the taste, look, and even brands of alcoholic beverages”.
Is there evidence to support their concern? I could only find one brief report in the Japanese literature that states that among young people “who experienced the nonalcoholic beverage intake, interest in or motivation for drinking alcoholic beverages, and/or smoking is higher than [among] those who did not.” The study didn’t appear to clearly separate the exposure in a family setting from the actual intake.
Beer is an acquired taste. If someone offered you your first taste of beer after a hot-weather set of tennis most of you would reject it and ask for water or lemonade. I can recall my first taste of beer. For some reason my father thought at age 11 or 12 I might like to try some from his glass. I’m not sure of his motivation, but he tried the same thing with oysters. I didn’t drink beer again until I was 16, motivated at that time by a group dynamic. The oyster trial, however, backfired on him and from then on he had to share his coveted dozen with me. Alcohol, unless heavily disguised by a mixer, is also not a taste that most young people find appealing.
It is unlikely that the average thrill-seeking teenager is going to ask his older-appearing buddy with a fake ID to buy him some nonalcoholic beer. Nor would he go to the effort or risk of acquiring his own fake ID just to see how it tastes. It just doesn’t compute, especially to a self-check corral patroller.
I guess one could envision a scenario in which a teenager wanting to fit in with the fast crowd would ask a trusted adult (or clueless parent) to buy him some nonalcoholic beer to bring to a party. He is running a serious risk of being laughed at by his friends if they find he’s drinking the fake stuff. It also seems unlikely that a parent would buy nonalcoholic beer to introduce his teenager to the taste of beer.
So,
Although it runs counter to my usual commitment to evidence-based decisions, making it difficult for adolescents to buy nonalcoholic beverages feels like the right think to do. As long as alcoholic and nonalcoholic beverages share the same display space and are packaged in nearly identical containers, there is ample opportunity for confusion. Recent evidence suggesting that even small amounts of alcohol increases some health risks should strengthen our resolve to minimize that confusion.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at [email protected].
Several months ago in a letter about healthcare providers and the decision to use alcohol and other mind-altering substances on the job, I waxed enthusiastically about the new wave of no alcohol (NA) and zero (00) alcohol beers that have come on the market. In the last 2 years our local grocery store’s cooler space for nonalcoholic beer has grown from less than 24 inches to something approaching the height of the average sixth grader.
In a bold act of chivalry at the beginning of the pandemic I accepted the mantle of designated grocery shopper and over the last 3 years have become uncommonly proud of my ability to bring home the groceries efficiently and cost effectively, without catching COVID in the process. I have developed a sixth sense of choosing which human checker/bagger combination is fastest or whether the self-checkout is the way to go.
For obvious reasons the human checkers don’t ask for my ID when I am buying adult beverages. However, the self-check register freezes up instantly when I scan my 12-pack of Run Wild nonalcoholic. This necessitates a search for the MIA store person assigned to patrol the self-check corral, ever on the lookout for shoplifters, underage drinkers, and other generally shifty looking characters.
When I find one of the grocery store detectives (who is likely to have been a former patient), I say: “You know, this doesn’t have any alcohol in it.” They invariably reply with a shrug. “I know. But, the rules are the rules.” Occasionally, they may add: “It doesn’t make sense, does it?”
At first blush checking IDs for a nonalcoholic beverage may sound dumb, certainly to someone who is just a few years on either side of the legal drinking age. Why are we trying to protect some crazy teenager from the futility of getting high on a six-pack of something that at worst will make him spend most of the next couple of hours peeing?
But, there is concern in some corners that nonalcoholic drinks pose a significant threat to teenagers. Two PhDs at Stanford University have recently published a paper in which they worry that the dramatic rise in US sales of nonalcoholic drinks from 15% to 30% since 2018 may be socializing “users of alcohol drinking experiences by exposing them to the taste, look, and even brands of alcoholic beverages”.
Is there evidence to support their concern? I could only find one brief report in the Japanese literature that states that among young people “who experienced the nonalcoholic beverage intake, interest in or motivation for drinking alcoholic beverages, and/or smoking is higher than [among] those who did not.” The study didn’t appear to clearly separate the exposure in a family setting from the actual intake.
Beer is an acquired taste. If someone offered you your first taste of beer after a hot-weather set of tennis most of you would reject it and ask for water or lemonade. I can recall my first taste of beer. For some reason my father thought at age 11 or 12 I might like to try some from his glass. I’m not sure of his motivation, but he tried the same thing with oysters. I didn’t drink beer again until I was 16, motivated at that time by a group dynamic. The oyster trial, however, backfired on him and from then on he had to share his coveted dozen with me. Alcohol, unless heavily disguised by a mixer, is also not a taste that most young people find appealing.
It is unlikely that the average thrill-seeking teenager is going to ask his older-appearing buddy with a fake ID to buy him some nonalcoholic beer. Nor would he go to the effort or risk of acquiring his own fake ID just to see how it tastes. It just doesn’t compute, especially to a self-check corral patroller.
I guess one could envision a scenario in which a teenager wanting to fit in with the fast crowd would ask a trusted adult (or clueless parent) to buy him some nonalcoholic beer to bring to a party. He is running a serious risk of being laughed at by his friends if they find he’s drinking the fake stuff. It also seems unlikely that a parent would buy nonalcoholic beer to introduce his teenager to the taste of beer.
So,
Although it runs counter to my usual commitment to evidence-based decisions, making it difficult for adolescents to buy nonalcoholic beverages feels like the right think to do. As long as alcoholic and nonalcoholic beverages share the same display space and are packaged in nearly identical containers, there is ample opportunity for confusion. Recent evidence suggesting that even small amounts of alcohol increases some health risks should strengthen our resolve to minimize that confusion.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at [email protected].
Several months ago in a letter about healthcare providers and the decision to use alcohol and other mind-altering substances on the job, I waxed enthusiastically about the new wave of no alcohol (NA) and zero (00) alcohol beers that have come on the market. In the last 2 years our local grocery store’s cooler space for nonalcoholic beer has grown from less than 24 inches to something approaching the height of the average sixth grader.
In a bold act of chivalry at the beginning of the pandemic I accepted the mantle of designated grocery shopper and over the last 3 years have become uncommonly proud of my ability to bring home the groceries efficiently and cost effectively, without catching COVID in the process. I have developed a sixth sense of choosing which human checker/bagger combination is fastest or whether the self-checkout is the way to go.
For obvious reasons the human checkers don’t ask for my ID when I am buying adult beverages. However, the self-check register freezes up instantly when I scan my 12-pack of Run Wild nonalcoholic. This necessitates a search for the MIA store person assigned to patrol the self-check corral, ever on the lookout for shoplifters, underage drinkers, and other generally shifty looking characters.
When I find one of the grocery store detectives (who is likely to have been a former patient), I say: “You know, this doesn’t have any alcohol in it.” They invariably reply with a shrug. “I know. But, the rules are the rules.” Occasionally, they may add: “It doesn’t make sense, does it?”
At first blush checking IDs for a nonalcoholic beverage may sound dumb, certainly to someone who is just a few years on either side of the legal drinking age. Why are we trying to protect some crazy teenager from the futility of getting high on a six-pack of something that at worst will make him spend most of the next couple of hours peeing?
But, there is concern in some corners that nonalcoholic drinks pose a significant threat to teenagers. Two PhDs at Stanford University have recently published a paper in which they worry that the dramatic rise in US sales of nonalcoholic drinks from 15% to 30% since 2018 may be socializing “users of alcohol drinking experiences by exposing them to the taste, look, and even brands of alcoholic beverages”.
Is there evidence to support their concern? I could only find one brief report in the Japanese literature that states that among young people “who experienced the nonalcoholic beverage intake, interest in or motivation for drinking alcoholic beverages, and/or smoking is higher than [among] those who did not.” The study didn’t appear to clearly separate the exposure in a family setting from the actual intake.
Beer is an acquired taste. If someone offered you your first taste of beer after a hot-weather set of tennis most of you would reject it and ask for water or lemonade. I can recall my first taste of beer. For some reason my father thought at age 11 or 12 I might like to try some from his glass. I’m not sure of his motivation, but he tried the same thing with oysters. I didn’t drink beer again until I was 16, motivated at that time by a group dynamic. The oyster trial, however, backfired on him and from then on he had to share his coveted dozen with me. Alcohol, unless heavily disguised by a mixer, is also not a taste that most young people find appealing.
It is unlikely that the average thrill-seeking teenager is going to ask his older-appearing buddy with a fake ID to buy him some nonalcoholic beer. Nor would he go to the effort or risk of acquiring his own fake ID just to see how it tastes. It just doesn’t compute, especially to a self-check corral patroller.
I guess one could envision a scenario in which a teenager wanting to fit in with the fast crowd would ask a trusted adult (or clueless parent) to buy him some nonalcoholic beer to bring to a party. He is running a serious risk of being laughed at by his friends if they find he’s drinking the fake stuff. It also seems unlikely that a parent would buy nonalcoholic beer to introduce his teenager to the taste of beer.
So,
Although it runs counter to my usual commitment to evidence-based decisions, making it difficult for adolescents to buy nonalcoholic beverages feels like the right think to do. As long as alcoholic and nonalcoholic beverages share the same display space and are packaged in nearly identical containers, there is ample opportunity for confusion. Recent evidence suggesting that even small amounts of alcohol increases some health risks should strengthen our resolve to minimize that confusion.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Other than a Littman stethoscope he accepted as a first-year medical student in 1966, Dr. Wilkoff reports having nothing to disclose. Email him at [email protected].
Age-Friendly Health Systems Transformation: A Whole Person Approach to Support the Well-Being of Older Adults
The COVID-19 pandemic established a new normal for health care delivery, with leaders rethinking core practices to survive and thrive in a changing environment and improve the health and well-being of patients. The Veterans Health Administration (VHA) is embracing a shift in focus from “what is the matter” to “what really matters” to address pre- and postpandemic challenges through a whole health approach.1 Initially conceptualized by the VHA in 2011, whole health “is an approach to health care that empowers and equips people to take charge of their health and well-being so that they can live their life to the fullest.”1 Whole health integrates evidence-based complementary and integrative health (CIH) therapies to manage pain; this includes acupuncture, meditation, tai chi, yoga, massage therapy, guided imagery, biofeedback, and clinical hypnosis.1 The VHA now recognizes well-being as a core value, helping clinicians respond to emerging challenges related to the social determinants of health (eg, access to health care, physical activity, and healthy foods) and guiding health care decision making.1,2
Well-being through empowerment—elements of whole health and Age-Friendly Health Systems (AFHS)—encourages health care institutions to work with employees, patients, and other stakeholders to address global challenges, clinician burnout, and social issues faced by their communities. This approach focuses on life’s purpose and meaning for individuals and inspires leaders to engage with patients, staff, and communities in new, impactful ways by focusing on wellbeing and wholeness rather than illness and disease. Having a higher sense of purpose is associated with lower all-cause mortality, reduced risk of specific diseases, better health behaviors, greater use of preventive services, and fewer hospital days of care.3
This article describes how AFHS supports the well-being of older adults and aligns with the whole health model of care. It also outlines the VHA investment to transform health care to be more person-centered by documenting what matters in the electronic health record (EHR).
AGE-FRIENDLY CARE
Given that nearly half of veterans enrolled in the VHA are aged ≥ 65 years, there is an increased need to identify models of care to support this aging population.4 This is especially critical because older veterans often have multiple chronic conditions and complex care needs that benefit from a whole person approach. The AFHS movement aims to provide evidence-based care aligned with what matters to older adults and provides a mechanism for transforming care to meet the needs of older veterans. This includes addressing age-related health concerns while promoting optimal health outcomes and quality of life. AFHS follows the 4Ms framework: what matters, medication, mentation, and mobility.5 The 4Ms serve as a guide for the health care of older adults in any setting, where each “M” is assessed and acted on to support what matters.5 Since 2020, > 390 teams have developed a plan to implement the 4Ms at 156 VHA facilities, demonstrating the VHA commitment to transforming health care for veterans.6
When VHA teams join the AFHS movement, they may also engage older veterans in a whole health system (WHS) (Figure). While AFHS is designed to improve care for patients aged ≥ 65 years, it also complements whole health, a person-centered approach available to all veterans enrolled in the VHA. Through the WHS and AFHS, veterans are empowered and equipped to take charge of their health and well-being through conversations about their unique goals, preferences, and health priorities.4 Clinicians are challenged to assess what matters by asking questions like, “What brings you joy?” and, “How can we help you meet your health goals?”1,5 These questions shift the conversation from disease-based treatment and enable clinicians to better understand the veteran as a person.1,5
For whole health and AFHS, conversations about what matters are anchored in the veteran’s goals and preferences, especially those facing a significant health change (ie, a new diagnosis or treatment decision).5,7 Together, the veteran’s goals and priorities serve as the foundation for developing person-centered care plans that often go beyond conventional medical treatments to address the physical, mental, emotional, and social aspects of health.
SYSTEM-WIDE DIRECTIVE
The WHS enhances AFHS discussions about what matters to veterans by adding a system-level lens for conceptualizing health care delivery by leveraging the 3 components of WHS: the “pathway,” well-being programs, and whole health clinical care.
The Pathway
Discovering what matters, or the veteran’s “mission, aspiration, and purpose,” begins with the WHS pathway. When stepping into the pathway, veterans begin completing a personal health inventory, or “walking the circle of health,” which encourages self-reflection that focuses on components of their life that can influence health and well-being.1,8 The circle of health offers a visual representation of the 4 most important aspects of health and well-being: First, “Me” at the center as an individual who is the expert on their life, values, goals, and priorities. Only the individual can know what really matters through mindful awareness and what works for their life. Second, self-care consists of 8 areas that impact health and wellbeing: working your body; surroundings; personal development; food and drink; recharge; family, friends, and coworkers; spirit and soul; and power of the mind. Third, professional care consists of prevention, conventional care, and complementary care. Finally, the community that supports the individual.
Well-Being Programs
VHA provides WHS programs that support veterans in building self-care skills and improving their quality of life, often through integrative care clinics that offer coaching and CIH therapies. For example, a veteran who prioritizes mobility when seeking care at an integrative care clinic will not only receive conventional medical treatment for their physical symptoms but may also be offered CIH therapies depending on their goals. The veteran may set a daily mobility goal with their care team that supports what matters, incorporating CIH approaches, such as yoga and tai chi into the care plan.5 These holistic approaches for moving the body can help alleviate physical symptoms, reduce stress, improve mindful awareness, and provide opportunities for self-discovery and growth, thus promote overall well-being
Whole Health Clinical Care
AFHS and the 4Ms embody the clinical care component of the WHS. Because what matters is the driver of the 4Ms, every action taken by the care team supports wellbeing and quality of life by promoting independence, connection, and support, and addressing external factors, such as social determinants of health. At a minimum, well-being includes “functioning well: the experience of positive emotions such as happiness and contentment as well as the development of one’s potential, having some control over one’s life, having a sense of purpose, and experiencing positive relationships.”9 From a system perspective, the VHA has begun to normalize focusing on what matters to veterans, using an interprofessional approach, one of the first steps to implementing AFHS.
As the programs expand, AFHS teams can learn from whole health well-being programs and increase the capacity for self-care in older veterans. Learning about the key elements included in the circle of health helps clinicians understand each veteran’s perceived strengths and weaknesses to support their self-care. From there, teams can act on the 4Ms and connect older veterans with the most appropriate programs and services at their facility, ensuring continuum of care.
DOCUMENTATION
The VHA leverages several tools and evidence-based practices to assess and act on what matters for veterans of all ages (Table).5,10-16 The VHA EHR and associated dashboards contain a wealth of information about whole health and AFHS implementation, scale up, and spread. A national AFHS 4Ms note template contains standardized data elements called health factors, which provide a mechanism for monitoring 4Ms care via its related dashboard. This template was developed by an interprofessional workgroup of VHA staff and underwent a thorough human factors engineering review and testing process prior to its release. Although teams continue to personalize care based on what matters to the veteran, data from the standardized 4Ms note template and dashboard provide a way to establish consistent, equitable care across multiple care settings.17
Between January 2022 and December 2023, > 612,000 participants aged ≥ 65 years identified what matters to them through 1.35 million assessments. During that period, > 36,000 veterans aged ≥ 65 years participated in AFHS and had what matters conversations documented. A personalized health plan was completed by 585,270 veterans for a total of 1.1 million assessments.11 Whole health coaching has been documented for > 57,000 veterans with > 200,000 assessments completed.13 In fiscal year 2023, a total of 1,802,131 veterans participated in whole health.
When teams share information about what matters to the veteran in a clinicianfacing format in the EHR, this helps ensure that the VHA honors veteran preferences throughout transitions of care and across all phases of health care. Although the EHR captures data on what matters, measurement of the overall impact on veteran and health system outcomes is essential. Further evaluation and ongoing education are needed to ensure clinicians are accurately and efficiently capturing the care provided by completing the appropriate EHR. Additional challenges include identifying ways to balance the documentation burden, while ensuring notes include valuable patient-centered information to guide care. EHR tools and templates have helped to unlock important insights on health care delivery in the VHA; however, health systems must consider how these clinical practices support the overall well-being of patients. How leaders empower frontline clinicians in any care setting to use these data to drive meaningful change is also important.
TRANSFORMING VHA CARE DELIVERY
In Achieving Whole Health: A New Approach for Veterans and the Nation, the National Academy of Science proposes a framework for the transformation of health care institutions to provide better whole health to veterans.3 Transformation requires change in entire systems and leaders who mobilize people “for participation in the process of change, encouraging a sense of collective identity and collective efficacy, which in turn brings stronger feelings of self-worth and self-efficacy,” and an enhanced sense of meaningfulness in their work and lives.18
Shifting health care approaches to equipping and empowering veterans and employees with whole health and AFHS resources is transformational and requires radically different assumptions and approaches that cannot be realized through traditional approaches. This change requires robust and multifaceted cultural transformation spanning all levels of the organization. Whole health and AFHS are facilitating this transformation by supporting documentation and data needs, tracking outcomes across settings, and accelerating spread to new facilities and care settings nationwide to support older veterans in improving their health and well-being.
Whole health and AFHS are complementary approaches to care that can work to empower veterans (as well as caregivers and clinicians) to align services with what matters most to veterans. Lessons such as standardizing person-centered assessments of what matters, creating supportive structures to better align care with veterans’ priorities, and identifying meaningful veteran and system-level outcomes to help sustain transformational change can be applied from whole health to AFHS. Together these programs have the potential to enhance overall health outcomes and quality of life for veterans.
- Kligler B, Hyde J, Gantt C, Bokhour B. The Whole Health transformation at the Veterans Health Administration: moving from “what’s the matter with you?” to “what matters to you?” Med Care. 2022;60(5):387-391. doi:10.1097/MLR.0000000000001706
- Centers for Disease Control and Prevention. Social determinants of health (SDOH) at CDC. January 17, 2024. Accessed September 12, 2024. https://www.cdc.gov/public-health-gateway/php/about/social-determinants-of-health.html
- National Academies of Sciences, Engineering, and Medicine. Achieving Whole Health: A New Approach for Veterans and the Nation. The National Academies Press; 2023. Accessed September 9, 2024. doi:10.17226/26854
- Church K, Munro S, Shaughnessy M, Clancy C. Age-friendly health systems: improving care for older adults in the Veterans Health Administration. Health Serv Res. 2023;58 Suppl 1(Suppl 1):5-8. doi:10.1111/1475-6773.14110
- Laderman M, Jackson C, Little K, Duong T, Pelton L. “What Matters” to older adults? A toolkit for health systems to design better care with older adults. Institute for Healthcare Improvement; 2019. Accessed September 9, 2024. https://www.ihi.org/Engage/Initiatives/Age-Friendly-Health-Systems/Documents/IHI_Age_Friendly_What_Matters_to_Older_Adults_Toolkit.pdf
- U.S. Department of Veterans Affairs. Age-Friendly Health Systems. Updated September 4, 2024. Accessed September 9, 2024. https://marketplace.va.gov/innovations/age-friendly-health-systems
- Brown TT, Hurley VB, Rodriguez HP, et al. Shared dec i s i o n - m a k i n g l o w e r s m e d i c a l e x p e n d i t u re s a n d the effect is amplified in racially-ethnically concordant relationships. Med Care. 2023;61(8):528-535. doi:10.1097/MLR.0000000000001881
- Kligler B. Whole Health in the Veterans Health Administration. Glob Adv Health Med. 2022;11:2164957X221077214.
- Ruggeri K, Garcia-Garzon E, Maguire Á, Matz S, Huppert FA. Well-being is more than happiness and life satisfaction: a multidimensional analysis of 21 countries. Health Qual Life Outcomes. 2020;18(1):192. doi:10.1186/s12955-020-01423-y
- U.S. Department of Veterans Affairs. Personal Health Inventory. Updated May 2022. Accessed September 9, 2024. https://www.va.gov/WHOLEHEALTH/docs/PHI-long-May22-fillable-508.pdf doi:10.1177/2164957X221077214
- Veterans Health Administration. Personal Health Plan. Updated March 2019. Accessed September 9, 2024. https:// www.va.gov/WHOLEHEALTH/docs/PersonalHealthPlan_508_03-2019.pdf
- Veterans Health Administration. Whole Health: My Life, My Story. Updated March 20, 2024. Accessed September 9, 2024. https://www.va.gov/WHOLEHEALTH/mylifemystory/index.asp
- U.S. Department of Veterans Affairs. Whole Health Library: Whole Health for Skill Building. Updated April 17, 2024. Accessed September 9, 2024. https://www.va.gov/WHOLEHEALTHLIBRARY/courses/whole-health-skill-building.asp
- U.S. Department of Veterans Affairs. Making Decisions: Current Care Planning. Updated May 21, 2024. Accessed September 9, 2024. https://www.va.gov/geriatrics/pages/making_decisions.asp
- U.S. Department of Veterans Affairs. Life-Sustaining Treatment Decisions Initiative (LSTDI). Updated March 2024. Accessed September 12, 2024. https://marketplace.va.gov/innovations/life-sustaining-treatment-decisions-initiative
- U.S. Department of Veterans Affairs. Center for Health Equity Research and Promotion: Surgical Pause Saving Veterans Lives. Updated September 22, 2021. Accessed September 9, 2024. https://www.cherp.research.va.gov/features/Surgical_Pause_Saving_Veterans_Lives.asp
- Munro S, Church K, Berner C, et al. Implementation of an agefriendly template in the Veterans Health Administration electronic health record. J Inform Nurs. 2023;8(3):6-11.
- Burns JM. Transforming Leadership: A New Pursuit of Happiness. Grove Press; 2003.
- US Department of Veterans Affairs, Veterans Health Administration. Whole Health: Circle of Health Overview. Updated May 20, 2024. Accessed September 12, 2024. https://www.va.gov/WHOLEHEALTH/circle-of-health/index.asp
The COVID-19 pandemic established a new normal for health care delivery, with leaders rethinking core practices to survive and thrive in a changing environment and improve the health and well-being of patients. The Veterans Health Administration (VHA) is embracing a shift in focus from “what is the matter” to “what really matters” to address pre- and postpandemic challenges through a whole health approach.1 Initially conceptualized by the VHA in 2011, whole health “is an approach to health care that empowers and equips people to take charge of their health and well-being so that they can live their life to the fullest.”1 Whole health integrates evidence-based complementary and integrative health (CIH) therapies to manage pain; this includes acupuncture, meditation, tai chi, yoga, massage therapy, guided imagery, biofeedback, and clinical hypnosis.1 The VHA now recognizes well-being as a core value, helping clinicians respond to emerging challenges related to the social determinants of health (eg, access to health care, physical activity, and healthy foods) and guiding health care decision making.1,2
Well-being through empowerment—elements of whole health and Age-Friendly Health Systems (AFHS)—encourages health care institutions to work with employees, patients, and other stakeholders to address global challenges, clinician burnout, and social issues faced by their communities. This approach focuses on life’s purpose and meaning for individuals and inspires leaders to engage with patients, staff, and communities in new, impactful ways by focusing on wellbeing and wholeness rather than illness and disease. Having a higher sense of purpose is associated with lower all-cause mortality, reduced risk of specific diseases, better health behaviors, greater use of preventive services, and fewer hospital days of care.3
This article describes how AFHS supports the well-being of older adults and aligns with the whole health model of care. It also outlines the VHA investment to transform health care to be more person-centered by documenting what matters in the electronic health record (EHR).
AGE-FRIENDLY CARE
Given that nearly half of veterans enrolled in the VHA are aged ≥ 65 years, there is an increased need to identify models of care to support this aging population.4 This is especially critical because older veterans often have multiple chronic conditions and complex care needs that benefit from a whole person approach. The AFHS movement aims to provide evidence-based care aligned with what matters to older adults and provides a mechanism for transforming care to meet the needs of older veterans. This includes addressing age-related health concerns while promoting optimal health outcomes and quality of life. AFHS follows the 4Ms framework: what matters, medication, mentation, and mobility.5 The 4Ms serve as a guide for the health care of older adults in any setting, where each “M” is assessed and acted on to support what matters.5 Since 2020, > 390 teams have developed a plan to implement the 4Ms at 156 VHA facilities, demonstrating the VHA commitment to transforming health care for veterans.6
When VHA teams join the AFHS movement, they may also engage older veterans in a whole health system (WHS) (Figure). While AFHS is designed to improve care for patients aged ≥ 65 years, it also complements whole health, a person-centered approach available to all veterans enrolled in the VHA. Through the WHS and AFHS, veterans are empowered and equipped to take charge of their health and well-being through conversations about their unique goals, preferences, and health priorities.4 Clinicians are challenged to assess what matters by asking questions like, “What brings you joy?” and, “How can we help you meet your health goals?”1,5 These questions shift the conversation from disease-based treatment and enable clinicians to better understand the veteran as a person.1,5
For whole health and AFHS, conversations about what matters are anchored in the veteran’s goals and preferences, especially those facing a significant health change (ie, a new diagnosis or treatment decision).5,7 Together, the veteran’s goals and priorities serve as the foundation for developing person-centered care plans that often go beyond conventional medical treatments to address the physical, mental, emotional, and social aspects of health.
SYSTEM-WIDE DIRECTIVE
The WHS enhances AFHS discussions about what matters to veterans by adding a system-level lens for conceptualizing health care delivery by leveraging the 3 components of WHS: the “pathway,” well-being programs, and whole health clinical care.
The Pathway
Discovering what matters, or the veteran’s “mission, aspiration, and purpose,” begins with the WHS pathway. When stepping into the pathway, veterans begin completing a personal health inventory, or “walking the circle of health,” which encourages self-reflection that focuses on components of their life that can influence health and well-being.1,8 The circle of health offers a visual representation of the 4 most important aspects of health and well-being: First, “Me” at the center as an individual who is the expert on their life, values, goals, and priorities. Only the individual can know what really matters through mindful awareness and what works for their life. Second, self-care consists of 8 areas that impact health and wellbeing: working your body; surroundings; personal development; food and drink; recharge; family, friends, and coworkers; spirit and soul; and power of the mind. Third, professional care consists of prevention, conventional care, and complementary care. Finally, the community that supports the individual.
Well-Being Programs
VHA provides WHS programs that support veterans in building self-care skills and improving their quality of life, often through integrative care clinics that offer coaching and CIH therapies. For example, a veteran who prioritizes mobility when seeking care at an integrative care clinic will not only receive conventional medical treatment for their physical symptoms but may also be offered CIH therapies depending on their goals. The veteran may set a daily mobility goal with their care team that supports what matters, incorporating CIH approaches, such as yoga and tai chi into the care plan.5 These holistic approaches for moving the body can help alleviate physical symptoms, reduce stress, improve mindful awareness, and provide opportunities for self-discovery and growth, thus promote overall well-being
Whole Health Clinical Care
AFHS and the 4Ms embody the clinical care component of the WHS. Because what matters is the driver of the 4Ms, every action taken by the care team supports wellbeing and quality of life by promoting independence, connection, and support, and addressing external factors, such as social determinants of health. At a minimum, well-being includes “functioning well: the experience of positive emotions such as happiness and contentment as well as the development of one’s potential, having some control over one’s life, having a sense of purpose, and experiencing positive relationships.”9 From a system perspective, the VHA has begun to normalize focusing on what matters to veterans, using an interprofessional approach, one of the first steps to implementing AFHS.
As the programs expand, AFHS teams can learn from whole health well-being programs and increase the capacity for self-care in older veterans. Learning about the key elements included in the circle of health helps clinicians understand each veteran’s perceived strengths and weaknesses to support their self-care. From there, teams can act on the 4Ms and connect older veterans with the most appropriate programs and services at their facility, ensuring continuum of care.
DOCUMENTATION
The VHA leverages several tools and evidence-based practices to assess and act on what matters for veterans of all ages (Table).5,10-16 The VHA EHR and associated dashboards contain a wealth of information about whole health and AFHS implementation, scale up, and spread. A national AFHS 4Ms note template contains standardized data elements called health factors, which provide a mechanism for monitoring 4Ms care via its related dashboard. This template was developed by an interprofessional workgroup of VHA staff and underwent a thorough human factors engineering review and testing process prior to its release. Although teams continue to personalize care based on what matters to the veteran, data from the standardized 4Ms note template and dashboard provide a way to establish consistent, equitable care across multiple care settings.17
Between January 2022 and December 2023, > 612,000 participants aged ≥ 65 years identified what matters to them through 1.35 million assessments. During that period, > 36,000 veterans aged ≥ 65 years participated in AFHS and had what matters conversations documented. A personalized health plan was completed by 585,270 veterans for a total of 1.1 million assessments.11 Whole health coaching has been documented for > 57,000 veterans with > 200,000 assessments completed.13 In fiscal year 2023, a total of 1,802,131 veterans participated in whole health.
When teams share information about what matters to the veteran in a clinicianfacing format in the EHR, this helps ensure that the VHA honors veteran preferences throughout transitions of care and across all phases of health care. Although the EHR captures data on what matters, measurement of the overall impact on veteran and health system outcomes is essential. Further evaluation and ongoing education are needed to ensure clinicians are accurately and efficiently capturing the care provided by completing the appropriate EHR. Additional challenges include identifying ways to balance the documentation burden, while ensuring notes include valuable patient-centered information to guide care. EHR tools and templates have helped to unlock important insights on health care delivery in the VHA; however, health systems must consider how these clinical practices support the overall well-being of patients. How leaders empower frontline clinicians in any care setting to use these data to drive meaningful change is also important.
TRANSFORMING VHA CARE DELIVERY
In Achieving Whole Health: A New Approach for Veterans and the Nation, the National Academy of Science proposes a framework for the transformation of health care institutions to provide better whole health to veterans.3 Transformation requires change in entire systems and leaders who mobilize people “for participation in the process of change, encouraging a sense of collective identity and collective efficacy, which in turn brings stronger feelings of self-worth and self-efficacy,” and an enhanced sense of meaningfulness in their work and lives.18
Shifting health care approaches to equipping and empowering veterans and employees with whole health and AFHS resources is transformational and requires radically different assumptions and approaches that cannot be realized through traditional approaches. This change requires robust and multifaceted cultural transformation spanning all levels of the organization. Whole health and AFHS are facilitating this transformation by supporting documentation and data needs, tracking outcomes across settings, and accelerating spread to new facilities and care settings nationwide to support older veterans in improving their health and well-being.
Whole health and AFHS are complementary approaches to care that can work to empower veterans (as well as caregivers and clinicians) to align services with what matters most to veterans. Lessons such as standardizing person-centered assessments of what matters, creating supportive structures to better align care with veterans’ priorities, and identifying meaningful veteran and system-level outcomes to help sustain transformational change can be applied from whole health to AFHS. Together these programs have the potential to enhance overall health outcomes and quality of life for veterans.
The COVID-19 pandemic established a new normal for health care delivery, with leaders rethinking core practices to survive and thrive in a changing environment and improve the health and well-being of patients. The Veterans Health Administration (VHA) is embracing a shift in focus from “what is the matter” to “what really matters” to address pre- and postpandemic challenges through a whole health approach.1 Initially conceptualized by the VHA in 2011, whole health “is an approach to health care that empowers and equips people to take charge of their health and well-being so that they can live their life to the fullest.”1 Whole health integrates evidence-based complementary and integrative health (CIH) therapies to manage pain; this includes acupuncture, meditation, tai chi, yoga, massage therapy, guided imagery, biofeedback, and clinical hypnosis.1 The VHA now recognizes well-being as a core value, helping clinicians respond to emerging challenges related to the social determinants of health (eg, access to health care, physical activity, and healthy foods) and guiding health care decision making.1,2
Well-being through empowerment—elements of whole health and Age-Friendly Health Systems (AFHS)—encourages health care institutions to work with employees, patients, and other stakeholders to address global challenges, clinician burnout, and social issues faced by their communities. This approach focuses on life’s purpose and meaning for individuals and inspires leaders to engage with patients, staff, and communities in new, impactful ways by focusing on wellbeing and wholeness rather than illness and disease. Having a higher sense of purpose is associated with lower all-cause mortality, reduced risk of specific diseases, better health behaviors, greater use of preventive services, and fewer hospital days of care.3
This article describes how AFHS supports the well-being of older adults and aligns with the whole health model of care. It also outlines the VHA investment to transform health care to be more person-centered by documenting what matters in the electronic health record (EHR).
AGE-FRIENDLY CARE
Given that nearly half of veterans enrolled in the VHA are aged ≥ 65 years, there is an increased need to identify models of care to support this aging population.4 This is especially critical because older veterans often have multiple chronic conditions and complex care needs that benefit from a whole person approach. The AFHS movement aims to provide evidence-based care aligned with what matters to older adults and provides a mechanism for transforming care to meet the needs of older veterans. This includes addressing age-related health concerns while promoting optimal health outcomes and quality of life. AFHS follows the 4Ms framework: what matters, medication, mentation, and mobility.5 The 4Ms serve as a guide for the health care of older adults in any setting, where each “M” is assessed and acted on to support what matters.5 Since 2020, > 390 teams have developed a plan to implement the 4Ms at 156 VHA facilities, demonstrating the VHA commitment to transforming health care for veterans.6
When VHA teams join the AFHS movement, they may also engage older veterans in a whole health system (WHS) (Figure). While AFHS is designed to improve care for patients aged ≥ 65 years, it also complements whole health, a person-centered approach available to all veterans enrolled in the VHA. Through the WHS and AFHS, veterans are empowered and equipped to take charge of their health and well-being through conversations about their unique goals, preferences, and health priorities.4 Clinicians are challenged to assess what matters by asking questions like, “What brings you joy?” and, “How can we help you meet your health goals?”1,5 These questions shift the conversation from disease-based treatment and enable clinicians to better understand the veteran as a person.1,5
For whole health and AFHS, conversations about what matters are anchored in the veteran’s goals and preferences, especially those facing a significant health change (ie, a new diagnosis or treatment decision).5,7 Together, the veteran’s goals and priorities serve as the foundation for developing person-centered care plans that often go beyond conventional medical treatments to address the physical, mental, emotional, and social aspects of health.
SYSTEM-WIDE DIRECTIVE
The WHS enhances AFHS discussions about what matters to veterans by adding a system-level lens for conceptualizing health care delivery by leveraging the 3 components of WHS: the “pathway,” well-being programs, and whole health clinical care.
The Pathway
Discovering what matters, or the veteran’s “mission, aspiration, and purpose,” begins with the WHS pathway. When stepping into the pathway, veterans begin completing a personal health inventory, or “walking the circle of health,” which encourages self-reflection that focuses on components of their life that can influence health and well-being.1,8 The circle of health offers a visual representation of the 4 most important aspects of health and well-being: First, “Me” at the center as an individual who is the expert on their life, values, goals, and priorities. Only the individual can know what really matters through mindful awareness and what works for their life. Second, self-care consists of 8 areas that impact health and wellbeing: working your body; surroundings; personal development; food and drink; recharge; family, friends, and coworkers; spirit and soul; and power of the mind. Third, professional care consists of prevention, conventional care, and complementary care. Finally, the community that supports the individual.
Well-Being Programs
VHA provides WHS programs that support veterans in building self-care skills and improving their quality of life, often through integrative care clinics that offer coaching and CIH therapies. For example, a veteran who prioritizes mobility when seeking care at an integrative care clinic will not only receive conventional medical treatment for their physical symptoms but may also be offered CIH therapies depending on their goals. The veteran may set a daily mobility goal with their care team that supports what matters, incorporating CIH approaches, such as yoga and tai chi into the care plan.5 These holistic approaches for moving the body can help alleviate physical symptoms, reduce stress, improve mindful awareness, and provide opportunities for self-discovery and growth, thus promote overall well-being
Whole Health Clinical Care
AFHS and the 4Ms embody the clinical care component of the WHS. Because what matters is the driver of the 4Ms, every action taken by the care team supports wellbeing and quality of life by promoting independence, connection, and support, and addressing external factors, such as social determinants of health. At a minimum, well-being includes “functioning well: the experience of positive emotions such as happiness and contentment as well as the development of one’s potential, having some control over one’s life, having a sense of purpose, and experiencing positive relationships.”9 From a system perspective, the VHA has begun to normalize focusing on what matters to veterans, using an interprofessional approach, one of the first steps to implementing AFHS.
As the programs expand, AFHS teams can learn from whole health well-being programs and increase the capacity for self-care in older veterans. Learning about the key elements included in the circle of health helps clinicians understand each veteran’s perceived strengths and weaknesses to support their self-care. From there, teams can act on the 4Ms and connect older veterans with the most appropriate programs and services at their facility, ensuring continuum of care.
DOCUMENTATION
The VHA leverages several tools and evidence-based practices to assess and act on what matters for veterans of all ages (Table).5,10-16 The VHA EHR and associated dashboards contain a wealth of information about whole health and AFHS implementation, scale up, and spread. A national AFHS 4Ms note template contains standardized data elements called health factors, which provide a mechanism for monitoring 4Ms care via its related dashboard. This template was developed by an interprofessional workgroup of VHA staff and underwent a thorough human factors engineering review and testing process prior to its release. Although teams continue to personalize care based on what matters to the veteran, data from the standardized 4Ms note template and dashboard provide a way to establish consistent, equitable care across multiple care settings.17
Between January 2022 and December 2023, > 612,000 participants aged ≥ 65 years identified what matters to them through 1.35 million assessments. During that period, > 36,000 veterans aged ≥ 65 years participated in AFHS and had what matters conversations documented. A personalized health plan was completed by 585,270 veterans for a total of 1.1 million assessments.11 Whole health coaching has been documented for > 57,000 veterans with > 200,000 assessments completed.13 In fiscal year 2023, a total of 1,802,131 veterans participated in whole health.
When teams share information about what matters to the veteran in a clinicianfacing format in the EHR, this helps ensure that the VHA honors veteran preferences throughout transitions of care and across all phases of health care. Although the EHR captures data on what matters, measurement of the overall impact on veteran and health system outcomes is essential. Further evaluation and ongoing education are needed to ensure clinicians are accurately and efficiently capturing the care provided by completing the appropriate EHR. Additional challenges include identifying ways to balance the documentation burden, while ensuring notes include valuable patient-centered information to guide care. EHR tools and templates have helped to unlock important insights on health care delivery in the VHA; however, health systems must consider how these clinical practices support the overall well-being of patients. How leaders empower frontline clinicians in any care setting to use these data to drive meaningful change is also important.
TRANSFORMING VHA CARE DELIVERY
In Achieving Whole Health: A New Approach for Veterans and the Nation, the National Academy of Science proposes a framework for the transformation of health care institutions to provide better whole health to veterans.3 Transformation requires change in entire systems and leaders who mobilize people “for participation in the process of change, encouraging a sense of collective identity and collective efficacy, which in turn brings stronger feelings of self-worth and self-efficacy,” and an enhanced sense of meaningfulness in their work and lives.18
Shifting health care approaches to equipping and empowering veterans and employees with whole health and AFHS resources is transformational and requires radically different assumptions and approaches that cannot be realized through traditional approaches. This change requires robust and multifaceted cultural transformation spanning all levels of the organization. Whole health and AFHS are facilitating this transformation by supporting documentation and data needs, tracking outcomes across settings, and accelerating spread to new facilities and care settings nationwide to support older veterans in improving their health and well-being.
Whole health and AFHS are complementary approaches to care that can work to empower veterans (as well as caregivers and clinicians) to align services with what matters most to veterans. Lessons such as standardizing person-centered assessments of what matters, creating supportive structures to better align care with veterans’ priorities, and identifying meaningful veteran and system-level outcomes to help sustain transformational change can be applied from whole health to AFHS. Together these programs have the potential to enhance overall health outcomes and quality of life for veterans.
- Kligler B, Hyde J, Gantt C, Bokhour B. The Whole Health transformation at the Veterans Health Administration: moving from “what’s the matter with you?” to “what matters to you?” Med Care. 2022;60(5):387-391. doi:10.1097/MLR.0000000000001706
- Centers for Disease Control and Prevention. Social determinants of health (SDOH) at CDC. January 17, 2024. Accessed September 12, 2024. https://www.cdc.gov/public-health-gateway/php/about/social-determinants-of-health.html
- National Academies of Sciences, Engineering, and Medicine. Achieving Whole Health: A New Approach for Veterans and the Nation. The National Academies Press; 2023. Accessed September 9, 2024. doi:10.17226/26854
- Church K, Munro S, Shaughnessy M, Clancy C. Age-friendly health systems: improving care for older adults in the Veterans Health Administration. Health Serv Res. 2023;58 Suppl 1(Suppl 1):5-8. doi:10.1111/1475-6773.14110
- Laderman M, Jackson C, Little K, Duong T, Pelton L. “What Matters” to older adults? A toolkit for health systems to design better care with older adults. Institute for Healthcare Improvement; 2019. Accessed September 9, 2024. https://www.ihi.org/Engage/Initiatives/Age-Friendly-Health-Systems/Documents/IHI_Age_Friendly_What_Matters_to_Older_Adults_Toolkit.pdf
- U.S. Department of Veterans Affairs. Age-Friendly Health Systems. Updated September 4, 2024. Accessed September 9, 2024. https://marketplace.va.gov/innovations/age-friendly-health-systems
- Brown TT, Hurley VB, Rodriguez HP, et al. Shared dec i s i o n - m a k i n g l o w e r s m e d i c a l e x p e n d i t u re s a n d the effect is amplified in racially-ethnically concordant relationships. Med Care. 2023;61(8):528-535. doi:10.1097/MLR.0000000000001881
- Kligler B. Whole Health in the Veterans Health Administration. Glob Adv Health Med. 2022;11:2164957X221077214.
- Ruggeri K, Garcia-Garzon E, Maguire Á, Matz S, Huppert FA. Well-being is more than happiness and life satisfaction: a multidimensional analysis of 21 countries. Health Qual Life Outcomes. 2020;18(1):192. doi:10.1186/s12955-020-01423-y
- U.S. Department of Veterans Affairs. Personal Health Inventory. Updated May 2022. Accessed September 9, 2024. https://www.va.gov/WHOLEHEALTH/docs/PHI-long-May22-fillable-508.pdf doi:10.1177/2164957X221077214
- Veterans Health Administration. Personal Health Plan. Updated March 2019. Accessed September 9, 2024. https:// www.va.gov/WHOLEHEALTH/docs/PersonalHealthPlan_508_03-2019.pdf
- Veterans Health Administration. Whole Health: My Life, My Story. Updated March 20, 2024. Accessed September 9, 2024. https://www.va.gov/WHOLEHEALTH/mylifemystory/index.asp
- U.S. Department of Veterans Affairs. Whole Health Library: Whole Health for Skill Building. Updated April 17, 2024. Accessed September 9, 2024. https://www.va.gov/WHOLEHEALTHLIBRARY/courses/whole-health-skill-building.asp
- U.S. Department of Veterans Affairs. Making Decisions: Current Care Planning. Updated May 21, 2024. Accessed September 9, 2024. https://www.va.gov/geriatrics/pages/making_decisions.asp
- U.S. Department of Veterans Affairs. Life-Sustaining Treatment Decisions Initiative (LSTDI). Updated March 2024. Accessed September 12, 2024. https://marketplace.va.gov/innovations/life-sustaining-treatment-decisions-initiative
- U.S. Department of Veterans Affairs. Center for Health Equity Research and Promotion: Surgical Pause Saving Veterans Lives. Updated September 22, 2021. Accessed September 9, 2024. https://www.cherp.research.va.gov/features/Surgical_Pause_Saving_Veterans_Lives.asp
- Munro S, Church K, Berner C, et al. Implementation of an agefriendly template in the Veterans Health Administration electronic health record. J Inform Nurs. 2023;8(3):6-11.
- Burns JM. Transforming Leadership: A New Pursuit of Happiness. Grove Press; 2003.
- US Department of Veterans Affairs, Veterans Health Administration. Whole Health: Circle of Health Overview. Updated May 20, 2024. Accessed September 12, 2024. https://www.va.gov/WHOLEHEALTH/circle-of-health/index.asp
- Kligler B, Hyde J, Gantt C, Bokhour B. The Whole Health transformation at the Veterans Health Administration: moving from “what’s the matter with you?” to “what matters to you?” Med Care. 2022;60(5):387-391. doi:10.1097/MLR.0000000000001706
- Centers for Disease Control and Prevention. Social determinants of health (SDOH) at CDC. January 17, 2024. Accessed September 12, 2024. https://www.cdc.gov/public-health-gateway/php/about/social-determinants-of-health.html
- National Academies of Sciences, Engineering, and Medicine. Achieving Whole Health: A New Approach for Veterans and the Nation. The National Academies Press; 2023. Accessed September 9, 2024. doi:10.17226/26854
- Church K, Munro S, Shaughnessy M, Clancy C. Age-friendly health systems: improving care for older adults in the Veterans Health Administration. Health Serv Res. 2023;58 Suppl 1(Suppl 1):5-8. doi:10.1111/1475-6773.14110
- Laderman M, Jackson C, Little K, Duong T, Pelton L. “What Matters” to older adults? A toolkit for health systems to design better care with older adults. Institute for Healthcare Improvement; 2019. Accessed September 9, 2024. https://www.ihi.org/Engage/Initiatives/Age-Friendly-Health-Systems/Documents/IHI_Age_Friendly_What_Matters_to_Older_Adults_Toolkit.pdf
- U.S. Department of Veterans Affairs. Age-Friendly Health Systems. Updated September 4, 2024. Accessed September 9, 2024. https://marketplace.va.gov/innovations/age-friendly-health-systems
- Brown TT, Hurley VB, Rodriguez HP, et al. Shared dec i s i o n - m a k i n g l o w e r s m e d i c a l e x p e n d i t u re s a n d the effect is amplified in racially-ethnically concordant relationships. Med Care. 2023;61(8):528-535. doi:10.1097/MLR.0000000000001881
- Kligler B. Whole Health in the Veterans Health Administration. Glob Adv Health Med. 2022;11:2164957X221077214.
- Ruggeri K, Garcia-Garzon E, Maguire Á, Matz S, Huppert FA. Well-being is more than happiness and life satisfaction: a multidimensional analysis of 21 countries. Health Qual Life Outcomes. 2020;18(1):192. doi:10.1186/s12955-020-01423-y
- U.S. Department of Veterans Affairs. Personal Health Inventory. Updated May 2022. Accessed September 9, 2024. https://www.va.gov/WHOLEHEALTH/docs/PHI-long-May22-fillable-508.pdf doi:10.1177/2164957X221077214
- Veterans Health Administration. Personal Health Plan. Updated March 2019. Accessed September 9, 2024. https:// www.va.gov/WHOLEHEALTH/docs/PersonalHealthPlan_508_03-2019.pdf
- Veterans Health Administration. Whole Health: My Life, My Story. Updated March 20, 2024. Accessed September 9, 2024. https://www.va.gov/WHOLEHEALTH/mylifemystory/index.asp
- U.S. Department of Veterans Affairs. Whole Health Library: Whole Health for Skill Building. Updated April 17, 2024. Accessed September 9, 2024. https://www.va.gov/WHOLEHEALTHLIBRARY/courses/whole-health-skill-building.asp
- U.S. Department of Veterans Affairs. Making Decisions: Current Care Planning. Updated May 21, 2024. Accessed September 9, 2024. https://www.va.gov/geriatrics/pages/making_decisions.asp
- U.S. Department of Veterans Affairs. Life-Sustaining Treatment Decisions Initiative (LSTDI). Updated March 2024. Accessed September 12, 2024. https://marketplace.va.gov/innovations/life-sustaining-treatment-decisions-initiative
- U.S. Department of Veterans Affairs. Center for Health Equity Research and Promotion: Surgical Pause Saving Veterans Lives. Updated September 22, 2021. Accessed September 9, 2024. https://www.cherp.research.va.gov/features/Surgical_Pause_Saving_Veterans_Lives.asp
- Munro S, Church K, Berner C, et al. Implementation of an agefriendly template in the Veterans Health Administration electronic health record. J Inform Nurs. 2023;8(3):6-11.
- Burns JM. Transforming Leadership: A New Pursuit of Happiness. Grove Press; 2003.
- US Department of Veterans Affairs, Veterans Health Administration. Whole Health: Circle of Health Overview. Updated May 20, 2024. Accessed September 12, 2024. https://www.va.gov/WHOLEHEALTH/circle-of-health/index.asp