User login
Recliner Butt
Recliner Butt
DIAGNOSIS
Senile gluteal dermatosis (SGD). SGD is a friction- related skin injury, also known as recliner butt, chronic tissue injury, or grandfather’s disease.1-4 The hallmarks include blanchable erythematous plaques and/or purplish discoloration of the fleshy part of the buttocks or posterior thighs, with little to no change over months to years. Additional findings may include skin erosions, lichenification, and ridging. SGD is most commonly seen in older adults with impaired mobility who spend prolonged periods in a reclined position, particularly those who slide down in a chair, “scoot,” or drag the buttocks during transfers or repositioning.
The pathogenesis of SGD is thought to involve microischemia associated with prolonged sitting.4 Histopathologic findings are nonspecific and may include hyperkeratosis, psoriasiform epidermal hyperplasia, vascular dilatation or proliferation in the superficial dermis, and reactive lymphohistiocytic perivascular infiltrate.4 The condition is poorly recognized and is likely underreported. Treatment involves reducing frictional injury by avoiding the reclined position, minimizing sliding during transfers, and frequent repositioning. Petroleum-based ointments may be applied to reduce friction and protect the skin barrier. Heat-dissipating chair cushions can be used to offload pressure and improve the local microclimate. Friction-related skin injuries need to be differentiated from pressure injuries, in which pressure and shear are the driving forces, and lesions are located over bony prominences.
Unlike SGD, chronic lichen sclerosus typically occurs in the anogenital area, including the scrotum and vulva, and is typically intensely pruritic, with white, atrophic plaques.
A stage 2 pressure injury is characterized by an area of partial-thickness skin loss with exposed dermis, usually overlying a bony prominence. Although friction-related skin injuries may contain erosions, they are often maroon or purple and are not located over a bony prominence.
Deep tissue injury (DTI) is characterized by nonblanchable dark red or purple skin discoloration, with intact or nonintact skin. While friction injuries may mimic DTIs, they lack the characteristic anatomic location over a bony prominence and the predictable evolution pattern seen in DTIs.
Incontinence-associated dermatitis (IAD) results from prolonged exposure to urine and/or feces and presents with erythema, inflammation, and epidermal erosion. Although IAD can look similar or coincide with SGD, the affected area is typically red, not purple. Skin ridging and lichenification are also not seen in IAD cases.
Sedentary behavior is prevalent among older adults, with nearly 60% spending > 4 hours per day sitting.5 Prolonged sitting puts them at risk for friction-related skin injuries. Even though friction-related skin injuries are typically nonprogressive, these patients are also at risk for pressure injuries that are typically acquired in a sitting position (eg, ischial and sacrococcygeal). Therefore, it is imperative that clinicians not only address SGD but also implement a pressure injury prevention plan.
- Berke CT. Pathology and clinical presentation of friction injuries case series and literature review. J Wound Ostomy Continence Nurs. 2105;42:47-61. doi:10.1097/WON.0000000000000087
- Mahoney MF, Rozenboom BJ. Definition and characteristics of chronic tissue injury: a unique form of skin damage. J Wound Ostomy Continence Nurs. 2019;46:187-191. doi:10.1097/WON.0000000000000527
- Kelechi, TJ. Commentary: chronic tissue injury. Making the case for a new form of skin damage. J Wound Ostomy Continence Nurs. 2019;46:192-193. doi:10.1097/WON.0000000000000533
- Majid I, Jairam D, Baheti K, et al. Senile guletal dermatosis: update on etiopathogenesis, Diagnostic Criteria, and Management. Dermatol Ther. 2024;37:e5556190.
- Harvey JA, Chastin SF, Skelton DA. Prevalence of sedentary behavior in older adults: a systematic review. Int J Environ Res Public Health. 2013;10:6645-6661. doi:10.3390/ijerph10126645
DIAGNOSIS
Senile gluteal dermatosis (SGD). SGD is a friction- related skin injury, also known as recliner butt, chronic tissue injury, or grandfather’s disease.1-4 The hallmarks include blanchable erythematous plaques and/or purplish discoloration of the fleshy part of the buttocks or posterior thighs, with little to no change over months to years. Additional findings may include skin erosions, lichenification, and ridging. SGD is most commonly seen in older adults with impaired mobility who spend prolonged periods in a reclined position, particularly those who slide down in a chair, “scoot,” or drag the buttocks during transfers or repositioning.
The pathogenesis of SGD is thought to involve microischemia associated with prolonged sitting.4 Histopathologic findings are nonspecific and may include hyperkeratosis, psoriasiform epidermal hyperplasia, vascular dilatation or proliferation in the superficial dermis, and reactive lymphohistiocytic perivascular infiltrate.4 The condition is poorly recognized and is likely underreported. Treatment involves reducing frictional injury by avoiding the reclined position, minimizing sliding during transfers, and frequent repositioning. Petroleum-based ointments may be applied to reduce friction and protect the skin barrier. Heat-dissipating chair cushions can be used to offload pressure and improve the local microclimate. Friction-related skin injuries need to be differentiated from pressure injuries, in which pressure and shear are the driving forces, and lesions are located over bony prominences.
Unlike SGD, chronic lichen sclerosus typically occurs in the anogenital area, including the scrotum and vulva, and is typically intensely pruritic, with white, atrophic plaques.
A stage 2 pressure injury is characterized by an area of partial-thickness skin loss with exposed dermis, usually overlying a bony prominence. Although friction-related skin injuries may contain erosions, they are often maroon or purple and are not located over a bony prominence.
Deep tissue injury (DTI) is characterized by nonblanchable dark red or purple skin discoloration, with intact or nonintact skin. While friction injuries may mimic DTIs, they lack the characteristic anatomic location over a bony prominence and the predictable evolution pattern seen in DTIs.
Incontinence-associated dermatitis (IAD) results from prolonged exposure to urine and/or feces and presents with erythema, inflammation, and epidermal erosion. Although IAD can look similar or coincide with SGD, the affected area is typically red, not purple. Skin ridging and lichenification are also not seen in IAD cases.
Sedentary behavior is prevalent among older adults, with nearly 60% spending > 4 hours per day sitting.5 Prolonged sitting puts them at risk for friction-related skin injuries. Even though friction-related skin injuries are typically nonprogressive, these patients are also at risk for pressure injuries that are typically acquired in a sitting position (eg, ischial and sacrococcygeal). Therefore, it is imperative that clinicians not only address SGD but also implement a pressure injury prevention plan.
DIAGNOSIS
Senile gluteal dermatosis (SGD). SGD is a friction- related skin injury, also known as recliner butt, chronic tissue injury, or grandfather’s disease.1-4 The hallmarks include blanchable erythematous plaques and/or purplish discoloration of the fleshy part of the buttocks or posterior thighs, with little to no change over months to years. Additional findings may include skin erosions, lichenification, and ridging. SGD is most commonly seen in older adults with impaired mobility who spend prolonged periods in a reclined position, particularly those who slide down in a chair, “scoot,” or drag the buttocks during transfers or repositioning.
The pathogenesis of SGD is thought to involve microischemia associated with prolonged sitting.4 Histopathologic findings are nonspecific and may include hyperkeratosis, psoriasiform epidermal hyperplasia, vascular dilatation or proliferation in the superficial dermis, and reactive lymphohistiocytic perivascular infiltrate.4 The condition is poorly recognized and is likely underreported. Treatment involves reducing frictional injury by avoiding the reclined position, minimizing sliding during transfers, and frequent repositioning. Petroleum-based ointments may be applied to reduce friction and protect the skin barrier. Heat-dissipating chair cushions can be used to offload pressure and improve the local microclimate. Friction-related skin injuries need to be differentiated from pressure injuries, in which pressure and shear are the driving forces, and lesions are located over bony prominences.
Unlike SGD, chronic lichen sclerosus typically occurs in the anogenital area, including the scrotum and vulva, and is typically intensely pruritic, with white, atrophic plaques.
A stage 2 pressure injury is characterized by an area of partial-thickness skin loss with exposed dermis, usually overlying a bony prominence. Although friction-related skin injuries may contain erosions, they are often maroon or purple and are not located over a bony prominence.
Deep tissue injury (DTI) is characterized by nonblanchable dark red or purple skin discoloration, with intact or nonintact skin. While friction injuries may mimic DTIs, they lack the characteristic anatomic location over a bony prominence and the predictable evolution pattern seen in DTIs.
Incontinence-associated dermatitis (IAD) results from prolonged exposure to urine and/or feces and presents with erythema, inflammation, and epidermal erosion. Although IAD can look similar or coincide with SGD, the affected area is typically red, not purple. Skin ridging and lichenification are also not seen in IAD cases.
Sedentary behavior is prevalent among older adults, with nearly 60% spending > 4 hours per day sitting.5 Prolonged sitting puts them at risk for friction-related skin injuries. Even though friction-related skin injuries are typically nonprogressive, these patients are also at risk for pressure injuries that are typically acquired in a sitting position (eg, ischial and sacrococcygeal). Therefore, it is imperative that clinicians not only address SGD but also implement a pressure injury prevention plan.
- Berke CT. Pathology and clinical presentation of friction injuries case series and literature review. J Wound Ostomy Continence Nurs. 2105;42:47-61. doi:10.1097/WON.0000000000000087
- Mahoney MF, Rozenboom BJ. Definition and characteristics of chronic tissue injury: a unique form of skin damage. J Wound Ostomy Continence Nurs. 2019;46:187-191. doi:10.1097/WON.0000000000000527
- Kelechi, TJ. Commentary: chronic tissue injury. Making the case for a new form of skin damage. J Wound Ostomy Continence Nurs. 2019;46:192-193. doi:10.1097/WON.0000000000000533
- Majid I, Jairam D, Baheti K, et al. Senile guletal dermatosis: update on etiopathogenesis, Diagnostic Criteria, and Management. Dermatol Ther. 2024;37:e5556190.
- Harvey JA, Chastin SF, Skelton DA. Prevalence of sedentary behavior in older adults: a systematic review. Int J Environ Res Public Health. 2013;10:6645-6661. doi:10.3390/ijerph10126645
- Berke CT. Pathology and clinical presentation of friction injuries case series and literature review. J Wound Ostomy Continence Nurs. 2105;42:47-61. doi:10.1097/WON.0000000000000087
- Mahoney MF, Rozenboom BJ. Definition and characteristics of chronic tissue injury: a unique form of skin damage. J Wound Ostomy Continence Nurs. 2019;46:187-191. doi:10.1097/WON.0000000000000527
- Kelechi, TJ. Commentary: chronic tissue injury. Making the case for a new form of skin damage. J Wound Ostomy Continence Nurs. 2019;46:192-193. doi:10.1097/WON.0000000000000533
- Majid I, Jairam D, Baheti K, et al. Senile guletal dermatosis: update on etiopathogenesis, Diagnostic Criteria, and Management. Dermatol Ther. 2024;37:e5556190.
- Harvey JA, Chastin SF, Skelton DA. Prevalence of sedentary behavior in older adults: a systematic review. Int J Environ Res Public Health. 2013;10:6645-6661. doi:10.3390/ijerph10126645
Recliner Butt
Recliner Butt
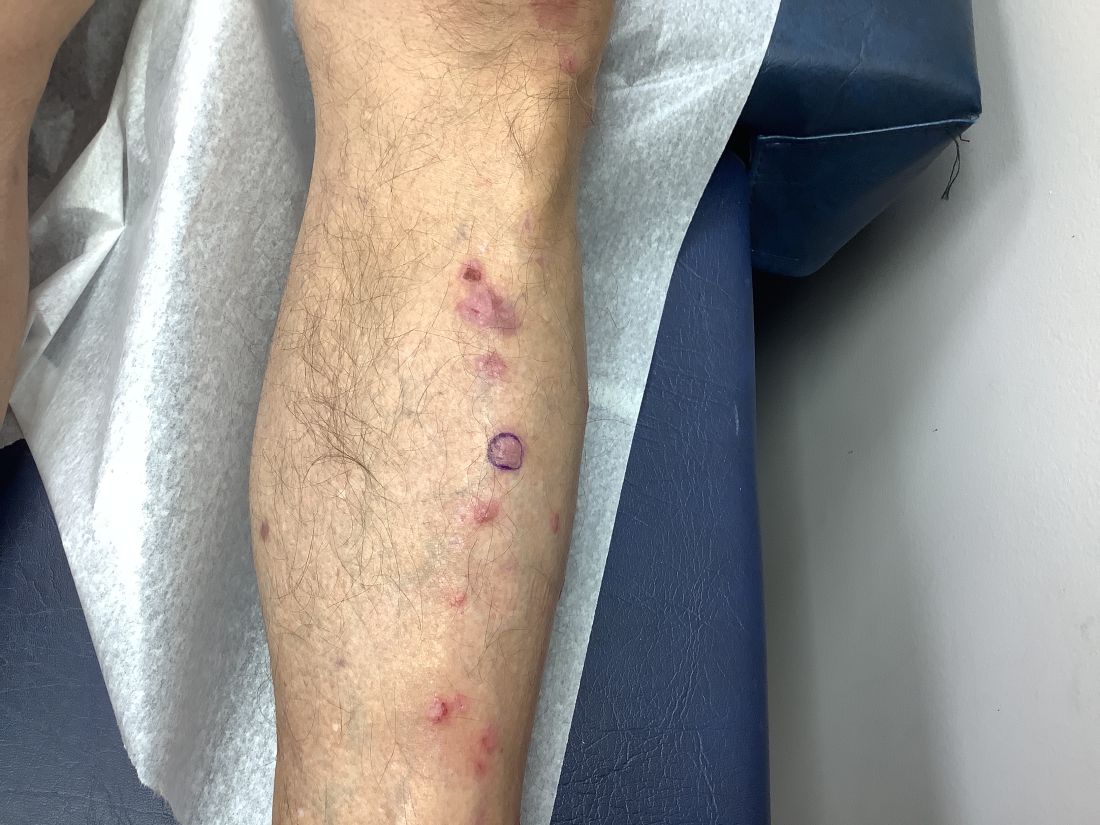
An 89-year-old male veteran with a history of obesity (body mass index, 33), osteoarthritis, anemia, pulmonary embolism, and urinary incontinence presented for evaluation of gluteal skin lesions (Figure). The patient had poor mobility and spent most of the day in a recliner chair. He also slept in the recliner due to chronic dyspnea and orthopnea.
The gluteal region demonstrated purplish discoloration with blanchable erythema and superficial ulcerations. The affected area was not pruritic and had remained unchanged for 3 months.
A punch biopsy of the discolored gluteal area was performed. Histopathologic examination revealed hyperkeratosis, orthokeratosis, irregular acanthosis, and mild spongiosis. Vascular proliferation and papillary dermal edema were also noted.
Thoracic Intramedullary Mass Causing Neurologic Weakness
Thoracic Intramedullary Mass Causing Neurologic Weakness
Discussion
A diagnosis of dural arteriovenous fistula (dAVF) was made. Lesions involving the spinal cord are traditionally classified by location as extradural, intradural/extramedullary, or intramedullary. Intramedullary spinal cord abnormalities pose considerable diagnostic and management challenges because of the risks of biopsy in this location and the added potential for morbidity and mortality from improperly treated lesions. Although MRI is the preferred imaging modality, PET/CT and magnetic resonance angiography (MRA) may also help narrow the differential diagnosis and potentially avoid complications from an invasive biopsy.1 This patient’s intramedullary lesion, which represented a dAVF, posed a diagnostic challenge; after diagnosis, it was successfully managed conservatively with dexamethasone and physical therapy.
Intradural tumors account for 2% to 4% of all primary central nervous system (CNS) tumors.2 Ependymomas account for 50% to 60% of intramedullary tumors in adults, while astrocytomas account for about 60% of all lesions in children and adolescents.3,4 The differential diagnosis for intramedullary tumors also includes hemangioblastoma, metastases, primary CNS lymphoma, germ cell tumors, and gangliogliomas.5,6
Intramedullary metastases remain rare, although the incidence is rising with improvements in oncologic and supportive treatments. Autopsy studies conducted decades ago demonstrated that about 0.9% to 2.1% of patients with systemic cancer have intramedullary metastases at death.7,8 In patients with an established history of malignancy, a metastatic intramedullary tumor should be placed higher on the differential diagnosis. Intramedullary metastases most often occur in the setting of widespread metastatic disease. A systematic review of the literature on patients with lung cancer (small cell and non-small cell lung carcinomas) and ≥ 1 intramedullary spinal cord metastasis demonstrated that 55.8% of patients had concurrent brain metastases, 20.0% had leptomeningeal carcinomatosis, and 19.5% had vertebral metastases.9 While about half of all intramedullary metastases are associated with lung cancer, other common malignancies that metastasize to this area include colorectal, breast, and renal cell carcinoma, as well as lymphoma and melanoma primaries.10,11
On imaging, intramedullary metastases often appear as several short, studded segments with surrounding edema, typically out of proportion to the size of the lesion.1 By contrast, astrocytomas and ependymomas often span multiple segments, and enhancement patterns can vary depending on the subtype and grade. Glioblastoma multiforme, or grade 4 IDH wild-type astrocytomas, demonstrate an irregular, heterogeneous pattern of enhancement. Hemangioblastomas vary in size and are classically hypointense to isointense on T1-weighted sequences, isointense to hyperintense on T2-weighted sequences, and demonstrate avid enhancement on T1- postcontrast images. In large hemangioblastomas, flow voids due to prominent vasculature may be visualized.
Numerous nonneoplastic tumor mimics can obscure the differential diagnosis. Vascular malformations, including cavernomas and dAVFs, can also present with enhancement and edema. dAVFs are the most common type of spinal vascular malformation, accounting for about 70% of cases.12 They are supplied by the radiculomeningeal arteries, whereas pial arteriovenous malformations (AVMs) are supplied by the radiculomedullary and radiculopial arteries. On MRI, dAVFs usually have venous congestion with intramedullary edema, which appears as an ill-defined centromedullary hyperintensity on T2-weighted imaging over multiple segments. The spinal cord may appear swollen with atrophic changes in chronic cases. Spinal cord AVMs are rarer and have an intramedullary nidus. They usually demonstrate mixed heterogeneous signal on T1- and T2-weighted imaging due to blood products, while the nidus demonstrates a variable degree of enhancement. Serpiginous flow voids are seen both within the nidus and at the cord surface.
Demyelinating lesions of the spine may be seen in neuroinflammatory conditions such as multiple sclerosis, neuromyelitis optica spectrum disorder, acute transverse myelitis, and acute disseminated encephalomyelitis. In multiple sclerosis, lesions typically extend ≤ 2 vertebral segments in length, cover less than half of the vertebral cross-sectional area, and have a dorsolateral predilection.13 Active lesions may demonstrate enhancement along the rim or in a patchy pattern. In the presence of demyelinating lesions, there may occasionally appear to be an expansile mass with a syrinx.14
Infections such as tuberculosis and neurosarcoidosis should also remain on the differential diagnosis. On MRI, tuberculosis usually involves the thoracic cord and is typically rim-enhancing.15 If there are caseating granulomas, T2-weighted images may also demonstrate rim enhancement.16 Spinal sarcoidosis is unusual without intracranial involvement, and its appearance may include leptomeningeal enhancement, cord expansion, and hyperintense signal on T2- weighted imaging.17
Finally, iatrogenic causes are also possible, including radiation myelopathy and mechanical spinal cord injury. For radiation myelopathy, it is important to ascertain whether a patient has undergone prior radiotherapy in the region and to obtain the pertinent dosimetry. Spinal cord injury may cause a focal signal abnormality within the cord, with T2 hyperintensity; these foci may or may not present with enhancement, edema, or hematoma and therefore may resemble tumors.13
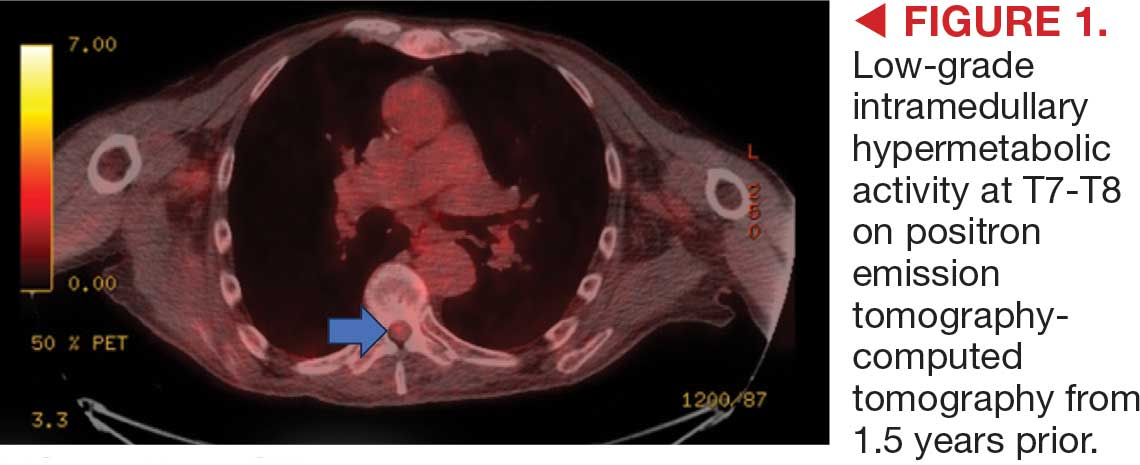
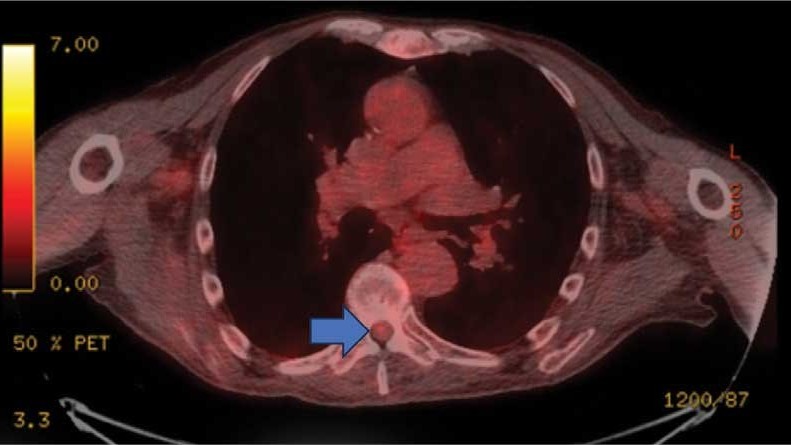
This patient presented with progressive right-sided lower extremity weakness and hypoesthesia and a history of a low-grade right renal/pelvic ureteral tumor. The immediate impression was that the thoracic intramedullary lesion represented a metastatic lesion. However, in the absence of any systemic or intracranial metastases, this progression was much less likely. An extensive interdisciplinary workup was conducted that included medical oncology, neurology, neuroradiology, neuro-oncology, neurosurgery, nuclear medicine, and radiation oncology. Neuroradiology and nuclear medicine identified a slightly hypermetabolic focus on the PET/CT from 1.5 years prior that correlated exactly with the same location as the lesion on the recent spinal MRI. This finding, along with the MRA, confirmed the diagnosis of a dAVF, which was successfully managed conservatively with dexamethasone and physical therapy, rather than through oncologic treatments such as radiotherapy
There remains debate regarding the utility of steroids in treating patients with dAVF. Although there are some case reports documenting that the edema associated with the dAVF responds to steroids, other case series have found that steroids may worsen outcomes in patients with dAVF, possibly due to increased venous hydrostatic pressure.
This case demonstrates the importance of an interdisciplinary workup when evaluating an intramedullary lesion, as well as maintaining a wide differential diagnosis, particularly in the absence of a history of polymetastatic cancer. All the clues (such as the slightly hypermetabolic focus on a PET/CT from 1.5 years prior) need to be obtained to comfortably reach a diagnosis in the absence of pathologic confirmation. These cases can be especially challenging due to the lack of pathologic confirmation, but by understanding the main differentiating features among the various etiologies and obtaining all available information, a correct diagnosis can be made without unnecessary interventions.
- Moghaddam SM, Bhatt AA. Location, length, and enhancement: systematic approach to differentiating intramedullary spinal cord lesions. Insights Imaging. 2018;9:511-526. doi:10.1007/s13244-018-0608-3
- Grimm S, Chamberlain MC. Adult primary spinal cord tumors. Expert Rev Neurother. 2009;9:1487-1495. doi:10.1586/ern.09.101
- Miller DJ, McCutcheon IE. Hemangioblastomas and other uncommon intramedullary tumors. J Neurooncol. 2000;47:253- 270. doi:10.1023/a:1006403500801
- Mottl H, Koutecky J. Treatment of spinal cord tumors in children. Med Pediatr Oncol. 1997;29:293-295.
- Kandemirli SG, Reddy A, Hitchon P, et al. Intramedullary tumours and tumour mimics. Clin Radiol. 2020;75:876.e17-876. e32. doi:10.1016/j.crad.2020.05.010
- Tobin MK, Geraghty JR, Engelhard HH, et al. Intramedullary spinal cord tumors: a review of current and future treatment strategies. Neurosurg Focus. 2015;39:E14. doi:10.3171/2015.5.FOCUS15158
- Chason JL, Walker FB, Landers JW. Metastatic carcinoma in the central nervous system and dorsal root ganglia. A prospective autopsy study. Cancer. 1963;16:781-787.
- Costigan DA, Winkelman MD. Intramedullary spinal cord metastasis. A clinicopathological study of 13 cases. J Neurosurg. 1985;62:227-233.
- Wu L, Wang L, Yang J, et al. Clinical features, treatments, and prognosis of intramedullary spinal cord metastases from lung cancer: a case series and systematic review. Neurospine. 2022;19:65-76. doi:10.14245/ns.2142910.455
- Lv J, Liu B, Quan X, et al. Intramedullary spinal cord metastasis in malignancies: an institutional analysis and review. Onco Targets Ther. 2019;12:4741-4753. doi:10.2147/OTT.S193235
- Goyal A, Yolcu Y, Kerezoudis P, et al. Intramedullary spinal cord metastases: an institutional review of survival and outcomes. J Neurooncol. 2019;142:347-354. doi:10.1007/s11060-019-03105-2
- Krings T. Vascular malformations of the spine and spinal cord: anatomy, classification, treatment. Clin Neuroradiol. 2010;20:5-24. doi:10.1007/s00062-010-9036-6
- Maj E, Wojtowicz K, Aleksandra PP, et al. Intramedullary spinal tumor-like lesions. Acta Radiol. 2019;60:994-1010. doi:10.1177/0284185118809540
- Waziri A, Vonsattel JP, Kaiser MG, et al. Expansile, enhancing cervical cord lesion with an associated syrinx secondary to demyelination. Case report and review of the literature. J Neurosurg Spine. 2007;6:52-56. doi:10.3171/spi.2007.6.1.52
- Nussbaum ES, Rockswold GL, Bergman TA, et al. Spinal tuberculosis: a diagnostic and management challenge. J Neurosurg. 1995;83:243-247. doi:10.3171/jns.1995.83.2.0243
- Lu M. Imaging diagnosis of spinal intramedullary tuberculoma: case reports and literature review. J Spinal Cord Med. 2010;33:159-162. doi:10.1080/10790268.2010.11689691
- Do-Dai DD, Brooks MK, Goldkamp A, et al. Magnetic resonance imaging of intramedullary spinal cord lesions: a pictorial review. Curr Probl Diagn Radiol. 2010;39:160-185. doi:10.1067/j.cpradiol.2009.05.004
Discussion
A diagnosis of dural arteriovenous fistula (dAVF) was made. Lesions involving the spinal cord are traditionally classified by location as extradural, intradural/extramedullary, or intramedullary. Intramedullary spinal cord abnormalities pose considerable diagnostic and management challenges because of the risks of biopsy in this location and the added potential for morbidity and mortality from improperly treated lesions. Although MRI is the preferred imaging modality, PET/CT and magnetic resonance angiography (MRA) may also help narrow the differential diagnosis and potentially avoid complications from an invasive biopsy.1 This patient’s intramedullary lesion, which represented a dAVF, posed a diagnostic challenge; after diagnosis, it was successfully managed conservatively with dexamethasone and physical therapy.
Intradural tumors account for 2% to 4% of all primary central nervous system (CNS) tumors.2 Ependymomas account for 50% to 60% of intramedullary tumors in adults, while astrocytomas account for about 60% of all lesions in children and adolescents.3,4 The differential diagnosis for intramedullary tumors also includes hemangioblastoma, metastases, primary CNS lymphoma, germ cell tumors, and gangliogliomas.5,6
Intramedullary metastases remain rare, although the incidence is rising with improvements in oncologic and supportive treatments. Autopsy studies conducted decades ago demonstrated that about 0.9% to 2.1% of patients with systemic cancer have intramedullary metastases at death.7,8 In patients with an established history of malignancy, a metastatic intramedullary tumor should be placed higher on the differential diagnosis. Intramedullary metastases most often occur in the setting of widespread metastatic disease. A systematic review of the literature on patients with lung cancer (small cell and non-small cell lung carcinomas) and ≥ 1 intramedullary spinal cord metastasis demonstrated that 55.8% of patients had concurrent brain metastases, 20.0% had leptomeningeal carcinomatosis, and 19.5% had vertebral metastases.9 While about half of all intramedullary metastases are associated with lung cancer, other common malignancies that metastasize to this area include colorectal, breast, and renal cell carcinoma, as well as lymphoma and melanoma primaries.10,11
On imaging, intramedullary metastases often appear as several short, studded segments with surrounding edema, typically out of proportion to the size of the lesion.1 By contrast, astrocytomas and ependymomas often span multiple segments, and enhancement patterns can vary depending on the subtype and grade. Glioblastoma multiforme, or grade 4 IDH wild-type astrocytomas, demonstrate an irregular, heterogeneous pattern of enhancement. Hemangioblastomas vary in size and are classically hypointense to isointense on T1-weighted sequences, isointense to hyperintense on T2-weighted sequences, and demonstrate avid enhancement on T1- postcontrast images. In large hemangioblastomas, flow voids due to prominent vasculature may be visualized.
Numerous nonneoplastic tumor mimics can obscure the differential diagnosis. Vascular malformations, including cavernomas and dAVFs, can also present with enhancement and edema. dAVFs are the most common type of spinal vascular malformation, accounting for about 70% of cases.12 They are supplied by the radiculomeningeal arteries, whereas pial arteriovenous malformations (AVMs) are supplied by the radiculomedullary and radiculopial arteries. On MRI, dAVFs usually have venous congestion with intramedullary edema, which appears as an ill-defined centromedullary hyperintensity on T2-weighted imaging over multiple segments. The spinal cord may appear swollen with atrophic changes in chronic cases. Spinal cord AVMs are rarer and have an intramedullary nidus. They usually demonstrate mixed heterogeneous signal on T1- and T2-weighted imaging due to blood products, while the nidus demonstrates a variable degree of enhancement. Serpiginous flow voids are seen both within the nidus and at the cord surface.
Demyelinating lesions of the spine may be seen in neuroinflammatory conditions such as multiple sclerosis, neuromyelitis optica spectrum disorder, acute transverse myelitis, and acute disseminated encephalomyelitis. In multiple sclerosis, lesions typically extend ≤ 2 vertebral segments in length, cover less than half of the vertebral cross-sectional area, and have a dorsolateral predilection.13 Active lesions may demonstrate enhancement along the rim or in a patchy pattern. In the presence of demyelinating lesions, there may occasionally appear to be an expansile mass with a syrinx.14
Infections such as tuberculosis and neurosarcoidosis should also remain on the differential diagnosis. On MRI, tuberculosis usually involves the thoracic cord and is typically rim-enhancing.15 If there are caseating granulomas, T2-weighted images may also demonstrate rim enhancement.16 Spinal sarcoidosis is unusual without intracranial involvement, and its appearance may include leptomeningeal enhancement, cord expansion, and hyperintense signal on T2- weighted imaging.17
Finally, iatrogenic causes are also possible, including radiation myelopathy and mechanical spinal cord injury. For radiation myelopathy, it is important to ascertain whether a patient has undergone prior radiotherapy in the region and to obtain the pertinent dosimetry. Spinal cord injury may cause a focal signal abnormality within the cord, with T2 hyperintensity; these foci may or may not present with enhancement, edema, or hematoma and therefore may resemble tumors.13
This patient presented with progressive right-sided lower extremity weakness and hypoesthesia and a history of a low-grade right renal/pelvic ureteral tumor. The immediate impression was that the thoracic intramedullary lesion represented a metastatic lesion. However, in the absence of any systemic or intracranial metastases, this progression was much less likely. An extensive interdisciplinary workup was conducted that included medical oncology, neurology, neuroradiology, neuro-oncology, neurosurgery, nuclear medicine, and radiation oncology. Neuroradiology and nuclear medicine identified a slightly hypermetabolic focus on the PET/CT from 1.5 years prior that correlated exactly with the same location as the lesion on the recent spinal MRI. This finding, along with the MRA, confirmed the diagnosis of a dAVF, which was successfully managed conservatively with dexamethasone and physical therapy, rather than through oncologic treatments such as radiotherapy
There remains debate regarding the utility of steroids in treating patients with dAVF. Although there are some case reports documenting that the edema associated with the dAVF responds to steroids, other case series have found that steroids may worsen outcomes in patients with dAVF, possibly due to increased venous hydrostatic pressure.
This case demonstrates the importance of an interdisciplinary workup when evaluating an intramedullary lesion, as well as maintaining a wide differential diagnosis, particularly in the absence of a history of polymetastatic cancer. All the clues (such as the slightly hypermetabolic focus on a PET/CT from 1.5 years prior) need to be obtained to comfortably reach a diagnosis in the absence of pathologic confirmation. These cases can be especially challenging due to the lack of pathologic confirmation, but by understanding the main differentiating features among the various etiologies and obtaining all available information, a correct diagnosis can be made without unnecessary interventions.
Discussion
A diagnosis of dural arteriovenous fistula (dAVF) was made. Lesions involving the spinal cord are traditionally classified by location as extradural, intradural/extramedullary, or intramedullary. Intramedullary spinal cord abnormalities pose considerable diagnostic and management challenges because of the risks of biopsy in this location and the added potential for morbidity and mortality from improperly treated lesions. Although MRI is the preferred imaging modality, PET/CT and magnetic resonance angiography (MRA) may also help narrow the differential diagnosis and potentially avoid complications from an invasive biopsy.1 This patient’s intramedullary lesion, which represented a dAVF, posed a diagnostic challenge; after diagnosis, it was successfully managed conservatively with dexamethasone and physical therapy.
Intradural tumors account for 2% to 4% of all primary central nervous system (CNS) tumors.2 Ependymomas account for 50% to 60% of intramedullary tumors in adults, while astrocytomas account for about 60% of all lesions in children and adolescents.3,4 The differential diagnosis for intramedullary tumors also includes hemangioblastoma, metastases, primary CNS lymphoma, germ cell tumors, and gangliogliomas.5,6
Intramedullary metastases remain rare, although the incidence is rising with improvements in oncologic and supportive treatments. Autopsy studies conducted decades ago demonstrated that about 0.9% to 2.1% of patients with systemic cancer have intramedullary metastases at death.7,8 In patients with an established history of malignancy, a metastatic intramedullary tumor should be placed higher on the differential diagnosis. Intramedullary metastases most often occur in the setting of widespread metastatic disease. A systematic review of the literature on patients with lung cancer (small cell and non-small cell lung carcinomas) and ≥ 1 intramedullary spinal cord metastasis demonstrated that 55.8% of patients had concurrent brain metastases, 20.0% had leptomeningeal carcinomatosis, and 19.5% had vertebral metastases.9 While about half of all intramedullary metastases are associated with lung cancer, other common malignancies that metastasize to this area include colorectal, breast, and renal cell carcinoma, as well as lymphoma and melanoma primaries.10,11
On imaging, intramedullary metastases often appear as several short, studded segments with surrounding edema, typically out of proportion to the size of the lesion.1 By contrast, astrocytomas and ependymomas often span multiple segments, and enhancement patterns can vary depending on the subtype and grade. Glioblastoma multiforme, or grade 4 IDH wild-type astrocytomas, demonstrate an irregular, heterogeneous pattern of enhancement. Hemangioblastomas vary in size and are classically hypointense to isointense on T1-weighted sequences, isointense to hyperintense on T2-weighted sequences, and demonstrate avid enhancement on T1- postcontrast images. In large hemangioblastomas, flow voids due to prominent vasculature may be visualized.
Numerous nonneoplastic tumor mimics can obscure the differential diagnosis. Vascular malformations, including cavernomas and dAVFs, can also present with enhancement and edema. dAVFs are the most common type of spinal vascular malformation, accounting for about 70% of cases.12 They are supplied by the radiculomeningeal arteries, whereas pial arteriovenous malformations (AVMs) are supplied by the radiculomedullary and radiculopial arteries. On MRI, dAVFs usually have venous congestion with intramedullary edema, which appears as an ill-defined centromedullary hyperintensity on T2-weighted imaging over multiple segments. The spinal cord may appear swollen with atrophic changes in chronic cases. Spinal cord AVMs are rarer and have an intramedullary nidus. They usually demonstrate mixed heterogeneous signal on T1- and T2-weighted imaging due to blood products, while the nidus demonstrates a variable degree of enhancement. Serpiginous flow voids are seen both within the nidus and at the cord surface.
Demyelinating lesions of the spine may be seen in neuroinflammatory conditions such as multiple sclerosis, neuromyelitis optica spectrum disorder, acute transverse myelitis, and acute disseminated encephalomyelitis. In multiple sclerosis, lesions typically extend ≤ 2 vertebral segments in length, cover less than half of the vertebral cross-sectional area, and have a dorsolateral predilection.13 Active lesions may demonstrate enhancement along the rim or in a patchy pattern. In the presence of demyelinating lesions, there may occasionally appear to be an expansile mass with a syrinx.14
Infections such as tuberculosis and neurosarcoidosis should also remain on the differential diagnosis. On MRI, tuberculosis usually involves the thoracic cord and is typically rim-enhancing.15 If there are caseating granulomas, T2-weighted images may also demonstrate rim enhancement.16 Spinal sarcoidosis is unusual without intracranial involvement, and its appearance may include leptomeningeal enhancement, cord expansion, and hyperintense signal on T2- weighted imaging.17
Finally, iatrogenic causes are also possible, including radiation myelopathy and mechanical spinal cord injury. For radiation myelopathy, it is important to ascertain whether a patient has undergone prior radiotherapy in the region and to obtain the pertinent dosimetry. Spinal cord injury may cause a focal signal abnormality within the cord, with T2 hyperintensity; these foci may or may not present with enhancement, edema, or hematoma and therefore may resemble tumors.13
This patient presented with progressive right-sided lower extremity weakness and hypoesthesia and a history of a low-grade right renal/pelvic ureteral tumor. The immediate impression was that the thoracic intramedullary lesion represented a metastatic lesion. However, in the absence of any systemic or intracranial metastases, this progression was much less likely. An extensive interdisciplinary workup was conducted that included medical oncology, neurology, neuroradiology, neuro-oncology, neurosurgery, nuclear medicine, and radiation oncology. Neuroradiology and nuclear medicine identified a slightly hypermetabolic focus on the PET/CT from 1.5 years prior that correlated exactly with the same location as the lesion on the recent spinal MRI. This finding, along with the MRA, confirmed the diagnosis of a dAVF, which was successfully managed conservatively with dexamethasone and physical therapy, rather than through oncologic treatments such as radiotherapy
There remains debate regarding the utility of steroids in treating patients with dAVF. Although there are some case reports documenting that the edema associated with the dAVF responds to steroids, other case series have found that steroids may worsen outcomes in patients with dAVF, possibly due to increased venous hydrostatic pressure.
This case demonstrates the importance of an interdisciplinary workup when evaluating an intramedullary lesion, as well as maintaining a wide differential diagnosis, particularly in the absence of a history of polymetastatic cancer. All the clues (such as the slightly hypermetabolic focus on a PET/CT from 1.5 years prior) need to be obtained to comfortably reach a diagnosis in the absence of pathologic confirmation. These cases can be especially challenging due to the lack of pathologic confirmation, but by understanding the main differentiating features among the various etiologies and obtaining all available information, a correct diagnosis can be made without unnecessary interventions.
- Moghaddam SM, Bhatt AA. Location, length, and enhancement: systematic approach to differentiating intramedullary spinal cord lesions. Insights Imaging. 2018;9:511-526. doi:10.1007/s13244-018-0608-3
- Grimm S, Chamberlain MC. Adult primary spinal cord tumors. Expert Rev Neurother. 2009;9:1487-1495. doi:10.1586/ern.09.101
- Miller DJ, McCutcheon IE. Hemangioblastomas and other uncommon intramedullary tumors. J Neurooncol. 2000;47:253- 270. doi:10.1023/a:1006403500801
- Mottl H, Koutecky J. Treatment of spinal cord tumors in children. Med Pediatr Oncol. 1997;29:293-295.
- Kandemirli SG, Reddy A, Hitchon P, et al. Intramedullary tumours and tumour mimics. Clin Radiol. 2020;75:876.e17-876. e32. doi:10.1016/j.crad.2020.05.010
- Tobin MK, Geraghty JR, Engelhard HH, et al. Intramedullary spinal cord tumors: a review of current and future treatment strategies. Neurosurg Focus. 2015;39:E14. doi:10.3171/2015.5.FOCUS15158
- Chason JL, Walker FB, Landers JW. Metastatic carcinoma in the central nervous system and dorsal root ganglia. A prospective autopsy study. Cancer. 1963;16:781-787.
- Costigan DA, Winkelman MD. Intramedullary spinal cord metastasis. A clinicopathological study of 13 cases. J Neurosurg. 1985;62:227-233.
- Wu L, Wang L, Yang J, et al. Clinical features, treatments, and prognosis of intramedullary spinal cord metastases from lung cancer: a case series and systematic review. Neurospine. 2022;19:65-76. doi:10.14245/ns.2142910.455
- Lv J, Liu B, Quan X, et al. Intramedullary spinal cord metastasis in malignancies: an institutional analysis and review. Onco Targets Ther. 2019;12:4741-4753. doi:10.2147/OTT.S193235
- Goyal A, Yolcu Y, Kerezoudis P, et al. Intramedullary spinal cord metastases: an institutional review of survival and outcomes. J Neurooncol. 2019;142:347-354. doi:10.1007/s11060-019-03105-2
- Krings T. Vascular malformations of the spine and spinal cord: anatomy, classification, treatment. Clin Neuroradiol. 2010;20:5-24. doi:10.1007/s00062-010-9036-6
- Maj E, Wojtowicz K, Aleksandra PP, et al. Intramedullary spinal tumor-like lesions. Acta Radiol. 2019;60:994-1010. doi:10.1177/0284185118809540
- Waziri A, Vonsattel JP, Kaiser MG, et al. Expansile, enhancing cervical cord lesion with an associated syrinx secondary to demyelination. Case report and review of the literature. J Neurosurg Spine. 2007;6:52-56. doi:10.3171/spi.2007.6.1.52
- Nussbaum ES, Rockswold GL, Bergman TA, et al. Spinal tuberculosis: a diagnostic and management challenge. J Neurosurg. 1995;83:243-247. doi:10.3171/jns.1995.83.2.0243
- Lu M. Imaging diagnosis of spinal intramedullary tuberculoma: case reports and literature review. J Spinal Cord Med. 2010;33:159-162. doi:10.1080/10790268.2010.11689691
- Do-Dai DD, Brooks MK, Goldkamp A, et al. Magnetic resonance imaging of intramedullary spinal cord lesions: a pictorial review. Curr Probl Diagn Radiol. 2010;39:160-185. doi:10.1067/j.cpradiol.2009.05.004
- Moghaddam SM, Bhatt AA. Location, length, and enhancement: systematic approach to differentiating intramedullary spinal cord lesions. Insights Imaging. 2018;9:511-526. doi:10.1007/s13244-018-0608-3
- Grimm S, Chamberlain MC. Adult primary spinal cord tumors. Expert Rev Neurother. 2009;9:1487-1495. doi:10.1586/ern.09.101
- Miller DJ, McCutcheon IE. Hemangioblastomas and other uncommon intramedullary tumors. J Neurooncol. 2000;47:253- 270. doi:10.1023/a:1006403500801
- Mottl H, Koutecky J. Treatment of spinal cord tumors in children. Med Pediatr Oncol. 1997;29:293-295.
- Kandemirli SG, Reddy A, Hitchon P, et al. Intramedullary tumours and tumour mimics. Clin Radiol. 2020;75:876.e17-876. e32. doi:10.1016/j.crad.2020.05.010
- Tobin MK, Geraghty JR, Engelhard HH, et al. Intramedullary spinal cord tumors: a review of current and future treatment strategies. Neurosurg Focus. 2015;39:E14. doi:10.3171/2015.5.FOCUS15158
- Chason JL, Walker FB, Landers JW. Metastatic carcinoma in the central nervous system and dorsal root ganglia. A prospective autopsy study. Cancer. 1963;16:781-787.
- Costigan DA, Winkelman MD. Intramedullary spinal cord metastasis. A clinicopathological study of 13 cases. J Neurosurg. 1985;62:227-233.
- Wu L, Wang L, Yang J, et al. Clinical features, treatments, and prognosis of intramedullary spinal cord metastases from lung cancer: a case series and systematic review. Neurospine. 2022;19:65-76. doi:10.14245/ns.2142910.455
- Lv J, Liu B, Quan X, et al. Intramedullary spinal cord metastasis in malignancies: an institutional analysis and review. Onco Targets Ther. 2019;12:4741-4753. doi:10.2147/OTT.S193235
- Goyal A, Yolcu Y, Kerezoudis P, et al. Intramedullary spinal cord metastases: an institutional review of survival and outcomes. J Neurooncol. 2019;142:347-354. doi:10.1007/s11060-019-03105-2
- Krings T. Vascular malformations of the spine and spinal cord: anatomy, classification, treatment. Clin Neuroradiol. 2010;20:5-24. doi:10.1007/s00062-010-9036-6
- Maj E, Wojtowicz K, Aleksandra PP, et al. Intramedullary spinal tumor-like lesions. Acta Radiol. 2019;60:994-1010. doi:10.1177/0284185118809540
- Waziri A, Vonsattel JP, Kaiser MG, et al. Expansile, enhancing cervical cord lesion with an associated syrinx secondary to demyelination. Case report and review of the literature. J Neurosurg Spine. 2007;6:52-56. doi:10.3171/spi.2007.6.1.52
- Nussbaum ES, Rockswold GL, Bergman TA, et al. Spinal tuberculosis: a diagnostic and management challenge. J Neurosurg. 1995;83:243-247. doi:10.3171/jns.1995.83.2.0243
- Lu M. Imaging diagnosis of spinal intramedullary tuberculoma: case reports and literature review. J Spinal Cord Med. 2010;33:159-162. doi:10.1080/10790268.2010.11689691
- Do-Dai DD, Brooks MK, Goldkamp A, et al. Magnetic resonance imaging of intramedullary spinal cord lesions: a pictorial review. Curr Probl Diagn Radiol. 2010;39:160-185. doi:10.1067/j.cpradiol.2009.05.004
Thoracic Intramedullary Mass Causing Neurologic Weakness
Thoracic Intramedullary Mass Causing Neurologic Weakness
An 87-year-old man presented to the emergency department reporting a 1-month history of right lower extremity weakness, progressing to an inability to ambulate. The patient had a history of hyperlipidemia, hypertension, benign prostatic hyperplasia, chronic obstructive pulmonary disease, low-grade right urothelial carcinoma status postbiopsy 2 years earlier, and atrial fibrillation following cardioversion 6 years earlier without anticoagulation therapy. He also reported severe right groin pain and increasing urinary obstruction.
On admission, neurology evaluated the patient’s lower extremity strength as 5/5 on his left, 1/5 on his right hip, and 2/5 on his right knee, with hypoesthesia of his right lower extremity. Computed tomography (CT) with contrast of the chest, abdomen, and pelvis demonstrated moderate to severe right-sided hydronephrosis, possibly due to a proximal right ureteric mass; no evidence of systemic metastases was found. He underwent a gadolinium-enhanced magnetic resonance imaging (MRI) of the cervical, thoracic, and lumbar spine, which showed a mass at T7-T8, a mass effect in the central cord, and abnormal spinal cord enhancement from T7 through the conus medullaris. A review of fluorodeoxyglucose- 18 (FDG-18) positron emission tomography (PET)-CT imaging from 1.5 years prior showed a low-grade focus (Figures 1-3). A gadolinium-enhanced brain MRI did not demonstrate any intracranial metastatic disease, acute infarct, hemorrhage, mass effect, or extra-axial fluid collections.


Atrophic Areas on the Axillary and Anogenital Anatomy
Atrophic Areas on the Axillary and Anogenital Anatomy
Discussion
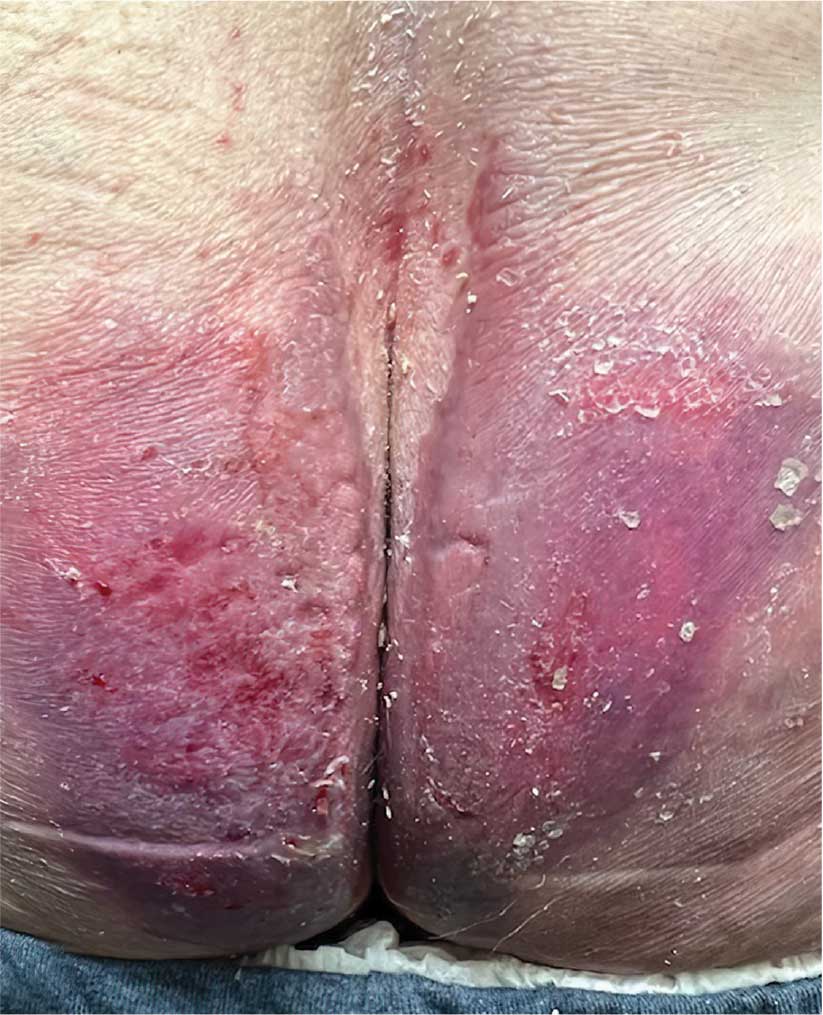
A diagnosis of lichen sclerosus (LS) was made based on clinical and dermoscopic features, followed by confirmation with histology. The patient’s presentation included typical signs and symptoms of LS: itching, burning, intermittent bleeding, perianal hemorrhage, fusion of the clitoral head, and fissures. Other presentations can include dyspareunia, erosions, and excoriations; however, these symptoms and signs were not reported or seen in this patient.
LS typically affects the anogenital region and has 2 peak incidences: in preadolescent teens and during the fifth to sixth decade of life.1 This patient presented with a case of extragenital LS, which is less common than the classic presentation of LS that affects the genitals. This variant’s epidemiology differs, as it is less common in children and more common in postmenopausal women.2 Extragenital LS presents as white, atrophic plaques with a predilection for sites including the trunk, breasts, upper arms, and sites of physical trauma, with symptoms of dryness and pruritus. Over time, the papules can coalesce and form ivory, scar-like papules or plaques with a wrinkled surface. In advanced stages, telangiectasia or follicular plugging can be present, along with flattening of the dermal-epidermal junction. This flat interface is fragile and can result in bullae that may become hemorrhagic.
Cutaneous squamous cell carcinoma (SCC) may infrequently arise from LS, similar to other chronic inflammatory dermatoses.3 Lichen planus is typically not associated with an increased risk of SCC, except in the oral and hypertrophic variants. However, LS may be considered a premalignant process, and many vulvar SCC cases are noted to have adjacent LS lesions.3
Autoimmune and genetic factors contribute to the pathogenesis of LS. Extracellular matrix protein 1 (ECM1) binds molecules of the basement membrane zone and dermis, contributing to the structure and integrity of skin. Autoantibodies against ECM1 and other antigens of the basement membrane zone, including BP180 and BP320, were found in LS.2 HLA-DQ7 major histocompatibility complex class II antigens have been associated with LS.1
On histologic examination, the epidermis of LS is atrophic with hyperkeratosis. The dermis shows homogenization and sclerosis of superficial collagen with a band-like lymphocytic infiltrate below the sclerosis. The basal layer is thickened, showing basal cell vacuolization and hydropic degeneration.4
First-line treatment for genital and extragenital variants of LS is high-potency topical steroids for 3 months or until the skin texture and color resolve (ie, clobetasol 0.05% cream or ointment). The second-line treatment is a topical calcineurin inhibitor. These treatments are used for management. They are not cures for LS, as relapse is possible after the initial treatment course is completed. Adverse effects of high potency topical steroids are skin burning, skin atrophy, and fragility, telangiectasia. The adverse effects of topical calcineurin inhibitors are stinging and burning on application.
Other Diagnostic Considerations
Inverse psoriasis (IP) is a variant of psoriasis that presents as erythematous, well-demarcated plaques with minimal scale in intertriginous areas and flexural surfaces. Localized dermatophyte, candidal, or bacterial infections can trigger IP.5 It occurs in about 3% to 7% of patients with plaque psoriasis and is thought to form due to koebnerization via mechanical friction of flexural zones.6 The patient described in this case did not have IP because IP would be more likely to present as a well-demarcated erythematous plaque rather than a patch.
Histologically, IP shows regular psoriasiform acanthosis and hypogranulosis of the epidermis, Munro microabscess, spongiform pustules of Kogoj, dilated tortuous dermal vessels, and thinning of the suprapapillary plates.5
Lichen planus pigmentosus-inversus (LPPI) is also known as lichen planus pigmentosus—intertriginous variant. This variant of lichen planus pigmentosus presents as multiple gray to dark brown macules and patches with poorly defined borders in a linear distribution limited to intertriginous areas, flexural surfaces, or following the lines of Blaschko.7 About 20% of cases present with frontal fibrosing alopecia. It is most common in individuals with intermediate and darker skin pigmentation, has a higher prevalence in females, and typically occurs within the third and fifth decades of life. Friction is a common trigger of LPPI.7 A diagnosis of LPPI is incorrect because the lesions would present as gray to dark brown macules, as opposed to the shiny white atrophic thin papules with surrounding pink and purple patches seen in this case.
Histologically, while both LS and LPPI share band-like lymphocytic infiltrate and basal cell vacuolization, findings in the dermis differ. LPPI shows melanophages and prominent melanin incontinence, while LS shows homogenization and sclerosis of superficial collagen.1,8 LPPI also shows absence of compensatory keratinocyte proliferation.
Morphea is an inflammatory disease that affects the dermis and subcutaneous fat, resulting in sclerosis that appears scarlike. Its prevalence increases with age and has a 4:1 prevalence in females, with the plaque type being the most common variant. 9 The typical presentation of plaque-type morphea is an insidious onset of asymptomatic, slightly elevated, erythematous or violaceous, slightly edematous plaques with centrifugal expansion. The center of the plaque may become sclerotic and indurated, acquiring a shiny white color with a peripheral “lilac” ring. Trunk and upper extremity involvement is common. Morphea is associated with increased antisingle-stranded DNA, antitopoisomerase IIa, antiphospholipid, antifibrillin-1, and antihistone antibodies. Triggers of morphea are believed to be localized insults to the skin, including mechanical trauma, injections, vaccinations, and irradiation.9 This answer is incorrect because the patient’s lesions were pruritic and had genital involvement, which are not typical of morphea. Morphea can be differentiated with based on symptoms (lack of pruritus, pain, burning), morphology of lesions (induration versus atrophy), dermoscopy (fibrotic beams with less scale and hemorrhage vs keratotic follicular plugs), and histopathology (depth of inflammation in superficial and deep dermis).
Histology of morphea can differ based on the stage, whether the lesion is sampled in the inflammatory margin or central sclerosis, and the depth of affected skin. At the inflammatory margin, vascular changes, including endothelial swelling and edema, are present, as well as CD4+ T cells, eosinophils, plasma cells, and mast cells surrounding smaller blood vessels. In late stages, the inflammatory infiltrate is no longer present, the epidermis appears regular, and there is a flattened dermal-epidermal junction. Distinct features include homogenous collagen bundles that replace many dermal structures, with atrophic eccrine glands that appear “trapped” in the thickened dermis, and homogenized and hyalinized subcutis.9
Mycosis fungoides (MF) is the most common type of cutaneous T-cell lymphoma and presents as annular, erythematous or hypopigmented patches and plaques with fine scale and tumors on the buttocks and sun-protected areas of the limbs and trunk. Lesions can appear with prominent poikiloderma or atrophic or lichenified skin.10 It is most common in males of African descent aged 50 to 55 years. The etiology is largely unknown but believed to be multifactorial. This answer is incorrect because the lesions in this patient appeared more atrophic, were less well demarcated, and lacked the scale that would be present in MF.
On histology, both LS and MF show band-like lymphocytic infiltrate, however MF lacks the homogenization and sclerosis of superficial collagen that is present in the dermis of LS. Also, MF demonstrates epidermotropism of atypical lymphocytes forming Pautrier microabscess.10
Primary Care Role
Primary care physicians can diagnose and treat LS. Referral to dermatology is not mandatory. Note that topical steroids can be used daily for up to 12 weeks. In LS, early treatment is associated with improved outcomes and minimizes the risk of irreversible skin changes.11 Follow-up during the treatment period is recommended to monitor subjective and objective response to treatment. Follow-up after the initial treatment is recommended since LS is typically chronic, can relapse, and SCC can infrequently arise from LS lesions.11
- Tran DA, Tan X, Macri CJ, Goldstein AT, Fu SW. Lichen sclerosus: an autoimmunopathogenic and genomic enigma with emerging genetic and immune targets. Int J Biol Sci. 2019;15:1429-1439. doi:10.7150/ijbs.34613
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389/fmed.2023.1106318
- Kuraitis D, Murina A. Squamous cell carcinoma arising in chronic inflammatory dermatoses. Cutis. 2024;113:29-34. doi:10.12788/cutis.0914
- Gaertner E, Elstein W. Lichen planus pigmentosus-inversus: case report and review of an unusual entity. Dermatol Online J. 2012;18:11.
- Micali G, Verzì AE, Giuffrida G, et al. Inverse psoriasis: from diagnosis to current treatment options. Clin Cosmet Investig Dermatol. 2019;12:953-959. doi:10.2147/CCID.S189000
- Syed ZU, Khachemoune A. Inverse psoriasis: case presentation and review. Am J Clin Dermatol. 2011;12:143-146. doi:10.2165/11532060-000000000-00000
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514. doi:10.1111/ijd.13806
- Vinay K, Kumar S, Bishnoi A, et al. A clinico-demographic study of 344 patients with lichen planus pigmentosus seen in a tertiary care center in India over an 8-year period. Int J Dermatol. 2020;59:245-252. doi:10.1111/ijd.14540
- Papara C, De Luca DA, Bieber K, et al. Morphea: the 2023 update. Front Med (Lausanne). 2023;10:1108623. doi:10.3389/fmed.2023.1108623
- Zinzani PL, Ferreri AJ, Cerroni L. Mycosis fungoides. Cri t Rev Oncol Hematol. 2008;65:172-182. doi:10.1016/j.critrevonc.2007.08.004
- Lee A, Bradford J, Fischer G. Long-term management of adult vulvar lichen sclerosus: a prospective cohort study of 507 women. JAMA Dermatol. 2015;151(10):1061-1067. doi:10.1001/jamadermatol.2015.0643
Discussion
A diagnosis of lichen sclerosus (LS) was made based on clinical and dermoscopic features, followed by confirmation with histology. The patient’s presentation included typical signs and symptoms of LS: itching, burning, intermittent bleeding, perianal hemorrhage, fusion of the clitoral head, and fissures. Other presentations can include dyspareunia, erosions, and excoriations; however, these symptoms and signs were not reported or seen in this patient.
LS typically affects the anogenital region and has 2 peak incidences: in preadolescent teens and during the fifth to sixth decade of life.1 This patient presented with a case of extragenital LS, which is less common than the classic presentation of LS that affects the genitals. This variant’s epidemiology differs, as it is less common in children and more common in postmenopausal women.2 Extragenital LS presents as white, atrophic plaques with a predilection for sites including the trunk, breasts, upper arms, and sites of physical trauma, with symptoms of dryness and pruritus. Over time, the papules can coalesce and form ivory, scar-like papules or plaques with a wrinkled surface. In advanced stages, telangiectasia or follicular plugging can be present, along with flattening of the dermal-epidermal junction. This flat interface is fragile and can result in bullae that may become hemorrhagic.
Cutaneous squamous cell carcinoma (SCC) may infrequently arise from LS, similar to other chronic inflammatory dermatoses.3 Lichen planus is typically not associated with an increased risk of SCC, except in the oral and hypertrophic variants. However, LS may be considered a premalignant process, and many vulvar SCC cases are noted to have adjacent LS lesions.3
Autoimmune and genetic factors contribute to the pathogenesis of LS. Extracellular matrix protein 1 (ECM1) binds molecules of the basement membrane zone and dermis, contributing to the structure and integrity of skin. Autoantibodies against ECM1 and other antigens of the basement membrane zone, including BP180 and BP320, were found in LS.2 HLA-DQ7 major histocompatibility complex class II antigens have been associated with LS.1
On histologic examination, the epidermis of LS is atrophic with hyperkeratosis. The dermis shows homogenization and sclerosis of superficial collagen with a band-like lymphocytic infiltrate below the sclerosis. The basal layer is thickened, showing basal cell vacuolization and hydropic degeneration.4
First-line treatment for genital and extragenital variants of LS is high-potency topical steroids for 3 months or until the skin texture and color resolve (ie, clobetasol 0.05% cream or ointment). The second-line treatment is a topical calcineurin inhibitor. These treatments are used for management. They are not cures for LS, as relapse is possible after the initial treatment course is completed. Adverse effects of high potency topical steroids are skin burning, skin atrophy, and fragility, telangiectasia. The adverse effects of topical calcineurin inhibitors are stinging and burning on application.
Other Diagnostic Considerations
Inverse psoriasis (IP) is a variant of psoriasis that presents as erythematous, well-demarcated plaques with minimal scale in intertriginous areas and flexural surfaces. Localized dermatophyte, candidal, or bacterial infections can trigger IP.5 It occurs in about 3% to 7% of patients with plaque psoriasis and is thought to form due to koebnerization via mechanical friction of flexural zones.6 The patient described in this case did not have IP because IP would be more likely to present as a well-demarcated erythematous plaque rather than a patch.
Histologically, IP shows regular psoriasiform acanthosis and hypogranulosis of the epidermis, Munro microabscess, spongiform pustules of Kogoj, dilated tortuous dermal vessels, and thinning of the suprapapillary plates.5
Lichen planus pigmentosus-inversus (LPPI) is also known as lichen planus pigmentosus—intertriginous variant. This variant of lichen planus pigmentosus presents as multiple gray to dark brown macules and patches with poorly defined borders in a linear distribution limited to intertriginous areas, flexural surfaces, or following the lines of Blaschko.7 About 20% of cases present with frontal fibrosing alopecia. It is most common in individuals with intermediate and darker skin pigmentation, has a higher prevalence in females, and typically occurs within the third and fifth decades of life. Friction is a common trigger of LPPI.7 A diagnosis of LPPI is incorrect because the lesions would present as gray to dark brown macules, as opposed to the shiny white atrophic thin papules with surrounding pink and purple patches seen in this case.
Histologically, while both LS and LPPI share band-like lymphocytic infiltrate and basal cell vacuolization, findings in the dermis differ. LPPI shows melanophages and prominent melanin incontinence, while LS shows homogenization and sclerosis of superficial collagen.1,8 LPPI also shows absence of compensatory keratinocyte proliferation.
Morphea is an inflammatory disease that affects the dermis and subcutaneous fat, resulting in sclerosis that appears scarlike. Its prevalence increases with age and has a 4:1 prevalence in females, with the plaque type being the most common variant. 9 The typical presentation of plaque-type morphea is an insidious onset of asymptomatic, slightly elevated, erythematous or violaceous, slightly edematous plaques with centrifugal expansion. The center of the plaque may become sclerotic and indurated, acquiring a shiny white color with a peripheral “lilac” ring. Trunk and upper extremity involvement is common. Morphea is associated with increased antisingle-stranded DNA, antitopoisomerase IIa, antiphospholipid, antifibrillin-1, and antihistone antibodies. Triggers of morphea are believed to be localized insults to the skin, including mechanical trauma, injections, vaccinations, and irradiation.9 This answer is incorrect because the patient’s lesions were pruritic and had genital involvement, which are not typical of morphea. Morphea can be differentiated with based on symptoms (lack of pruritus, pain, burning), morphology of lesions (induration versus atrophy), dermoscopy (fibrotic beams with less scale and hemorrhage vs keratotic follicular plugs), and histopathology (depth of inflammation in superficial and deep dermis).
Histology of morphea can differ based on the stage, whether the lesion is sampled in the inflammatory margin or central sclerosis, and the depth of affected skin. At the inflammatory margin, vascular changes, including endothelial swelling and edema, are present, as well as CD4+ T cells, eosinophils, plasma cells, and mast cells surrounding smaller blood vessels. In late stages, the inflammatory infiltrate is no longer present, the epidermis appears regular, and there is a flattened dermal-epidermal junction. Distinct features include homogenous collagen bundles that replace many dermal structures, with atrophic eccrine glands that appear “trapped” in the thickened dermis, and homogenized and hyalinized subcutis.9
Mycosis fungoides (MF) is the most common type of cutaneous T-cell lymphoma and presents as annular, erythematous or hypopigmented patches and plaques with fine scale and tumors on the buttocks and sun-protected areas of the limbs and trunk. Lesions can appear with prominent poikiloderma or atrophic or lichenified skin.10 It is most common in males of African descent aged 50 to 55 years. The etiology is largely unknown but believed to be multifactorial. This answer is incorrect because the lesions in this patient appeared more atrophic, were less well demarcated, and lacked the scale that would be present in MF.
On histology, both LS and MF show band-like lymphocytic infiltrate, however MF lacks the homogenization and sclerosis of superficial collagen that is present in the dermis of LS. Also, MF demonstrates epidermotropism of atypical lymphocytes forming Pautrier microabscess.10
Primary Care Role
Primary care physicians can diagnose and treat LS. Referral to dermatology is not mandatory. Note that topical steroids can be used daily for up to 12 weeks. In LS, early treatment is associated with improved outcomes and minimizes the risk of irreversible skin changes.11 Follow-up during the treatment period is recommended to monitor subjective and objective response to treatment. Follow-up after the initial treatment is recommended since LS is typically chronic, can relapse, and SCC can infrequently arise from LS lesions.11
Discussion
A diagnosis of lichen sclerosus (LS) was made based on clinical and dermoscopic features, followed by confirmation with histology. The patient’s presentation included typical signs and symptoms of LS: itching, burning, intermittent bleeding, perianal hemorrhage, fusion of the clitoral head, and fissures. Other presentations can include dyspareunia, erosions, and excoriations; however, these symptoms and signs were not reported or seen in this patient.
LS typically affects the anogenital region and has 2 peak incidences: in preadolescent teens and during the fifth to sixth decade of life.1 This patient presented with a case of extragenital LS, which is less common than the classic presentation of LS that affects the genitals. This variant’s epidemiology differs, as it is less common in children and more common in postmenopausal women.2 Extragenital LS presents as white, atrophic plaques with a predilection for sites including the trunk, breasts, upper arms, and sites of physical trauma, with symptoms of dryness and pruritus. Over time, the papules can coalesce and form ivory, scar-like papules or plaques with a wrinkled surface. In advanced stages, telangiectasia or follicular plugging can be present, along with flattening of the dermal-epidermal junction. This flat interface is fragile and can result in bullae that may become hemorrhagic.
Cutaneous squamous cell carcinoma (SCC) may infrequently arise from LS, similar to other chronic inflammatory dermatoses.3 Lichen planus is typically not associated with an increased risk of SCC, except in the oral and hypertrophic variants. However, LS may be considered a premalignant process, and many vulvar SCC cases are noted to have adjacent LS lesions.3
Autoimmune and genetic factors contribute to the pathogenesis of LS. Extracellular matrix protein 1 (ECM1) binds molecules of the basement membrane zone and dermis, contributing to the structure and integrity of skin. Autoantibodies against ECM1 and other antigens of the basement membrane zone, including BP180 and BP320, were found in LS.2 HLA-DQ7 major histocompatibility complex class II antigens have been associated with LS.1
On histologic examination, the epidermis of LS is atrophic with hyperkeratosis. The dermis shows homogenization and sclerosis of superficial collagen with a band-like lymphocytic infiltrate below the sclerosis. The basal layer is thickened, showing basal cell vacuolization and hydropic degeneration.4
First-line treatment for genital and extragenital variants of LS is high-potency topical steroids for 3 months or until the skin texture and color resolve (ie, clobetasol 0.05% cream or ointment). The second-line treatment is a topical calcineurin inhibitor. These treatments are used for management. They are not cures for LS, as relapse is possible after the initial treatment course is completed. Adverse effects of high potency topical steroids are skin burning, skin atrophy, and fragility, telangiectasia. The adverse effects of topical calcineurin inhibitors are stinging and burning on application.
Other Diagnostic Considerations
Inverse psoriasis (IP) is a variant of psoriasis that presents as erythematous, well-demarcated plaques with minimal scale in intertriginous areas and flexural surfaces. Localized dermatophyte, candidal, or bacterial infections can trigger IP.5 It occurs in about 3% to 7% of patients with plaque psoriasis and is thought to form due to koebnerization via mechanical friction of flexural zones.6 The patient described in this case did not have IP because IP would be more likely to present as a well-demarcated erythematous plaque rather than a patch.
Histologically, IP shows regular psoriasiform acanthosis and hypogranulosis of the epidermis, Munro microabscess, spongiform pustules of Kogoj, dilated tortuous dermal vessels, and thinning of the suprapapillary plates.5
Lichen planus pigmentosus-inversus (LPPI) is also known as lichen planus pigmentosus—intertriginous variant. This variant of lichen planus pigmentosus presents as multiple gray to dark brown macules and patches with poorly defined borders in a linear distribution limited to intertriginous areas, flexural surfaces, or following the lines of Blaschko.7 About 20% of cases present with frontal fibrosing alopecia. It is most common in individuals with intermediate and darker skin pigmentation, has a higher prevalence in females, and typically occurs within the third and fifth decades of life. Friction is a common trigger of LPPI.7 A diagnosis of LPPI is incorrect because the lesions would present as gray to dark brown macules, as opposed to the shiny white atrophic thin papules with surrounding pink and purple patches seen in this case.
Histologically, while both LS and LPPI share band-like lymphocytic infiltrate and basal cell vacuolization, findings in the dermis differ. LPPI shows melanophages and prominent melanin incontinence, while LS shows homogenization and sclerosis of superficial collagen.1,8 LPPI also shows absence of compensatory keratinocyte proliferation.
Morphea is an inflammatory disease that affects the dermis and subcutaneous fat, resulting in sclerosis that appears scarlike. Its prevalence increases with age and has a 4:1 prevalence in females, with the plaque type being the most common variant. 9 The typical presentation of plaque-type morphea is an insidious onset of asymptomatic, slightly elevated, erythematous or violaceous, slightly edematous plaques with centrifugal expansion. The center of the plaque may become sclerotic and indurated, acquiring a shiny white color with a peripheral “lilac” ring. Trunk and upper extremity involvement is common. Morphea is associated with increased antisingle-stranded DNA, antitopoisomerase IIa, antiphospholipid, antifibrillin-1, and antihistone antibodies. Triggers of morphea are believed to be localized insults to the skin, including mechanical trauma, injections, vaccinations, and irradiation.9 This answer is incorrect because the patient’s lesions were pruritic and had genital involvement, which are not typical of morphea. Morphea can be differentiated with based on symptoms (lack of pruritus, pain, burning), morphology of lesions (induration versus atrophy), dermoscopy (fibrotic beams with less scale and hemorrhage vs keratotic follicular plugs), and histopathology (depth of inflammation in superficial and deep dermis).
Histology of morphea can differ based on the stage, whether the lesion is sampled in the inflammatory margin or central sclerosis, and the depth of affected skin. At the inflammatory margin, vascular changes, including endothelial swelling and edema, are present, as well as CD4+ T cells, eosinophils, plasma cells, and mast cells surrounding smaller blood vessels. In late stages, the inflammatory infiltrate is no longer present, the epidermis appears regular, and there is a flattened dermal-epidermal junction. Distinct features include homogenous collagen bundles that replace many dermal structures, with atrophic eccrine glands that appear “trapped” in the thickened dermis, and homogenized and hyalinized subcutis.9
Mycosis fungoides (MF) is the most common type of cutaneous T-cell lymphoma and presents as annular, erythematous or hypopigmented patches and plaques with fine scale and tumors on the buttocks and sun-protected areas of the limbs and trunk. Lesions can appear with prominent poikiloderma or atrophic or lichenified skin.10 It is most common in males of African descent aged 50 to 55 years. The etiology is largely unknown but believed to be multifactorial. This answer is incorrect because the lesions in this patient appeared more atrophic, were less well demarcated, and lacked the scale that would be present in MF.
On histology, both LS and MF show band-like lymphocytic infiltrate, however MF lacks the homogenization and sclerosis of superficial collagen that is present in the dermis of LS. Also, MF demonstrates epidermotropism of atypical lymphocytes forming Pautrier microabscess.10
Primary Care Role
Primary care physicians can diagnose and treat LS. Referral to dermatology is not mandatory. Note that topical steroids can be used daily for up to 12 weeks. In LS, early treatment is associated with improved outcomes and minimizes the risk of irreversible skin changes.11 Follow-up during the treatment period is recommended to monitor subjective and objective response to treatment. Follow-up after the initial treatment is recommended since LS is typically chronic, can relapse, and SCC can infrequently arise from LS lesions.11
- Tran DA, Tan X, Macri CJ, Goldstein AT, Fu SW. Lichen sclerosus: an autoimmunopathogenic and genomic enigma with emerging genetic and immune targets. Int J Biol Sci. 2019;15:1429-1439. doi:10.7150/ijbs.34613
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389/fmed.2023.1106318
- Kuraitis D, Murina A. Squamous cell carcinoma arising in chronic inflammatory dermatoses. Cutis. 2024;113:29-34. doi:10.12788/cutis.0914
- Gaertner E, Elstein W. Lichen planus pigmentosus-inversus: case report and review of an unusual entity. Dermatol Online J. 2012;18:11.
- Micali G, Verzì AE, Giuffrida G, et al. Inverse psoriasis: from diagnosis to current treatment options. Clin Cosmet Investig Dermatol. 2019;12:953-959. doi:10.2147/CCID.S189000
- Syed ZU, Khachemoune A. Inverse psoriasis: case presentation and review. Am J Clin Dermatol. 2011;12:143-146. doi:10.2165/11532060-000000000-00000
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514. doi:10.1111/ijd.13806
- Vinay K, Kumar S, Bishnoi A, et al. A clinico-demographic study of 344 patients with lichen planus pigmentosus seen in a tertiary care center in India over an 8-year period. Int J Dermatol. 2020;59:245-252. doi:10.1111/ijd.14540
- Papara C, De Luca DA, Bieber K, et al. Morphea: the 2023 update. Front Med (Lausanne). 2023;10:1108623. doi:10.3389/fmed.2023.1108623
- Zinzani PL, Ferreri AJ, Cerroni L. Mycosis fungoides. Cri t Rev Oncol Hematol. 2008;65:172-182. doi:10.1016/j.critrevonc.2007.08.004
- Lee A, Bradford J, Fischer G. Long-term management of adult vulvar lichen sclerosus: a prospective cohort study of 507 women. JAMA Dermatol. 2015;151(10):1061-1067. doi:10.1001/jamadermatol.2015.0643
- Tran DA, Tan X, Macri CJ, Goldstein AT, Fu SW. Lichen sclerosus: an autoimmunopathogenic and genomic enigma with emerging genetic and immune targets. Int J Biol Sci. 2019;15:1429-1439. doi:10.7150/ijbs.34613
- De Luca DA, Papara C, Vorobyev A, et al. Lichen sclerosus: the 2023 update. Front Med (Lausanne). 2023;10:1106318. doi:10.3389/fmed.2023.1106318
- Kuraitis D, Murina A. Squamous cell carcinoma arising in chronic inflammatory dermatoses. Cutis. 2024;113:29-34. doi:10.12788/cutis.0914
- Gaertner E, Elstein W. Lichen planus pigmentosus-inversus: case report and review of an unusual entity. Dermatol Online J. 2012;18:11.
- Micali G, Verzì AE, Giuffrida G, et al. Inverse psoriasis: from diagnosis to current treatment options. Clin Cosmet Investig Dermatol. 2019;12:953-959. doi:10.2147/CCID.S189000
- Syed ZU, Khachemoune A. Inverse psoriasis: case presentation and review. Am J Clin Dermatol. 2011;12:143-146. doi:10.2165/11532060-000000000-00000
- Robles-Méndez JC, Rizo-Frías P, Herz-Ruelas ME, et al. Lichen planus pigmentosus and its variants: review and update. Int J Dermatol. 2018;57:505-514. doi:10.1111/ijd.13806
- Vinay K, Kumar S, Bishnoi A, et al. A clinico-demographic study of 344 patients with lichen planus pigmentosus seen in a tertiary care center in India over an 8-year period. Int J Dermatol. 2020;59:245-252. doi:10.1111/ijd.14540
- Papara C, De Luca DA, Bieber K, et al. Morphea: the 2023 update. Front Med (Lausanne). 2023;10:1108623. doi:10.3389/fmed.2023.1108623
- Zinzani PL, Ferreri AJ, Cerroni L. Mycosis fungoides. Cri t Rev Oncol Hematol. 2008;65:172-182. doi:10.1016/j.critrevonc.2007.08.004
- Lee A, Bradford J, Fischer G. Long-term management of adult vulvar lichen sclerosus: a prospective cohort study of 507 women. JAMA Dermatol. 2015;151(10):1061-1067. doi:10.1001/jamadermatol.2015.0643
Atrophic Areas on the Axillary and Anogenital Anatomy
Atrophic Areas on the Axillary and Anogenital Anatomy
A 62-year-old woman presented for a fullbody skin examination and was found to have a rash in her axillae and inframammary regions. The rash was intermittently pruritic, and the patient felt that the inframammary rash had started from contact with brassiere underwires. She had no oral lesions but noted intermittent burning and itching of the vaginal folds and intermittent bleeding near her anus. Physical examination revealed confluent, shiny, white, atrophic, thin papules with surrounding pink and purple patches on bilateral axillae, bilateral inframammary folds, bilateral inner thighs, and on the clitoral hood and labia minora. There was also an hourglass-shaped erythematous patch involving the vagina and anus. A small fissure was noted perianally, and small hemorrhage was noted on the clitoral head, with fusion of the clitoral head and superior labia minora (Figures 1 and 2).
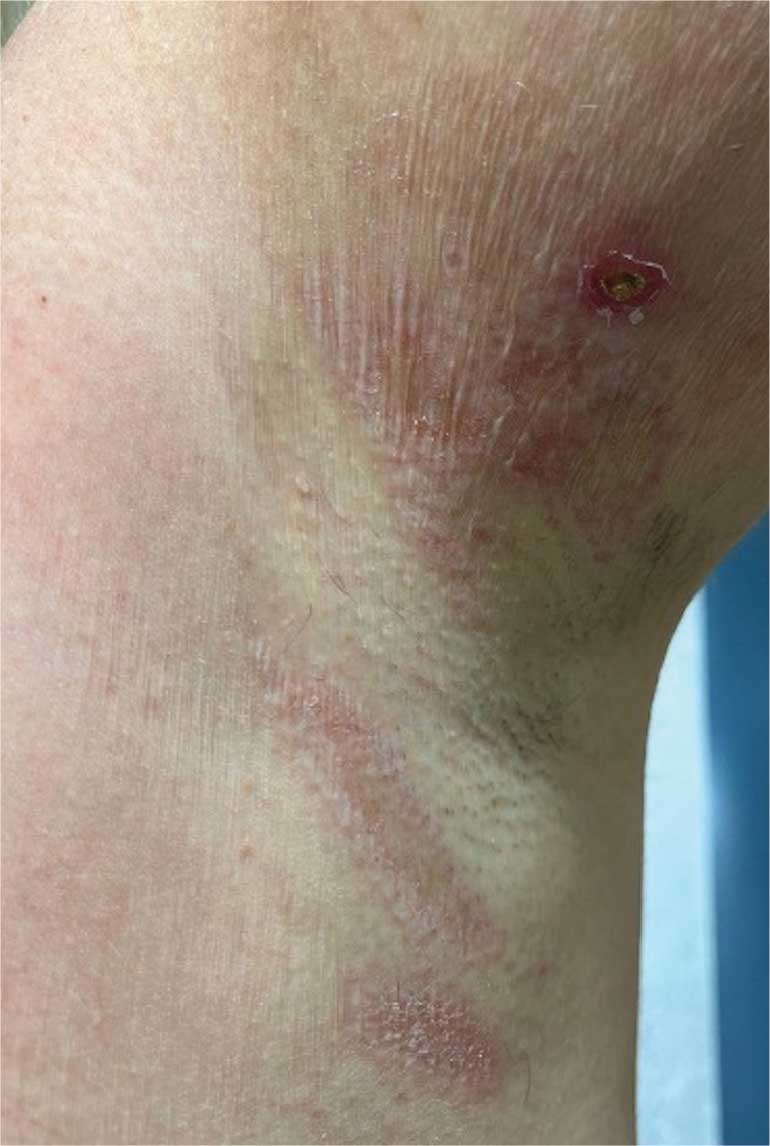
lesion from punch biopsy of the patient’s left axilla.
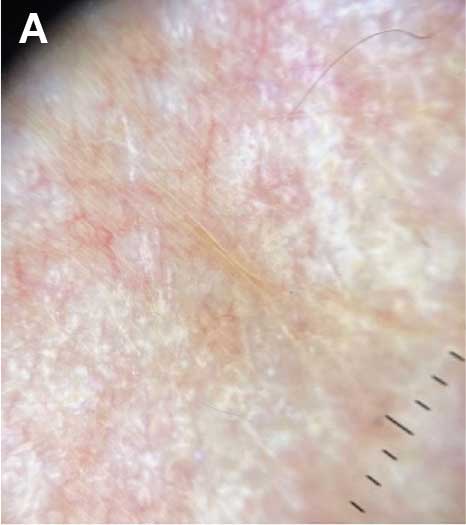
sclerosus plaque showing bright white grouped dots
on a pink background with follicular plugging and linear
branching vessels.
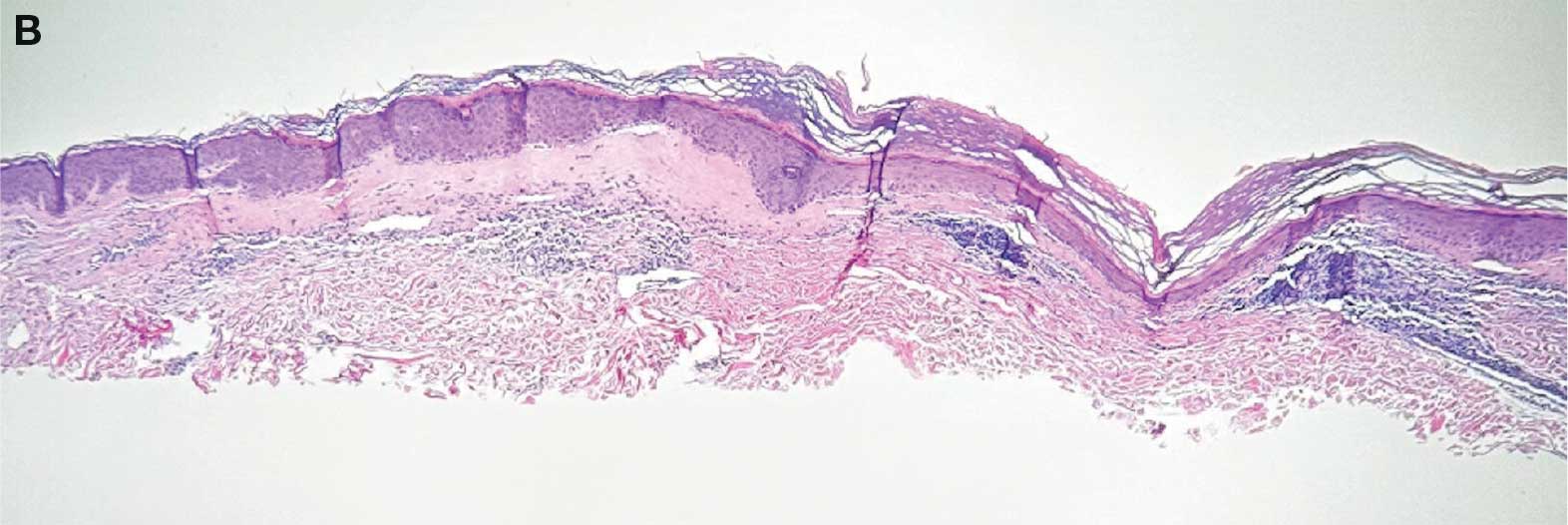
showing a compact corneal layer with a pale papillary
dermis and an underlying lymphocytic infiltrate. These
findings give the “red, white, and blue” appearance.
Low power 20× magnification.
nsbp;
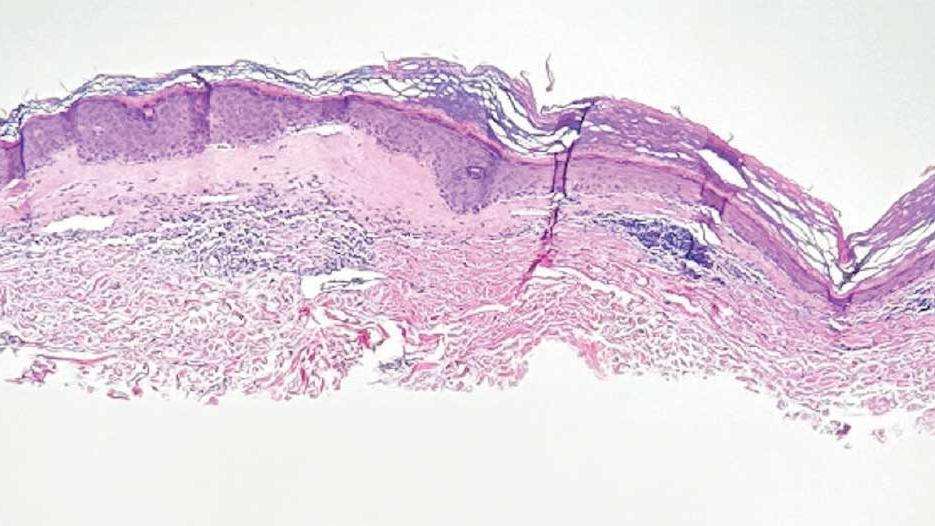
An 81-Year-Old White Woman Presented With a 2-Week History of a Painful Lesion on Her Left Calf
Calciphylaxis, also known as calcific uremic arteriolopathy, is a rare condition most commonly observed in patients with end-stage renal disease (ESRD). Because of the non-healing nature of the wounds and need for frequent hospitalizations, there is a significant risk of sepsis with a 1-year mortality rate greater than 50%.
Beyond ESRD, calciphylaxis is also associated with obesity, diabetes, hypoalbuminemia, autoimmune conditions, hepatic disease, malignancies, and dialysis. Rates in patients on dialysis have been increasing, ranging from 1% to 4%. Certain medications have also been implicated in the development of calciphylaxis, including warfarin, steroids, calcium-based phosphate binders, vitamin D, and iron. There is also an association with White individuals and more cases have been reported in females.
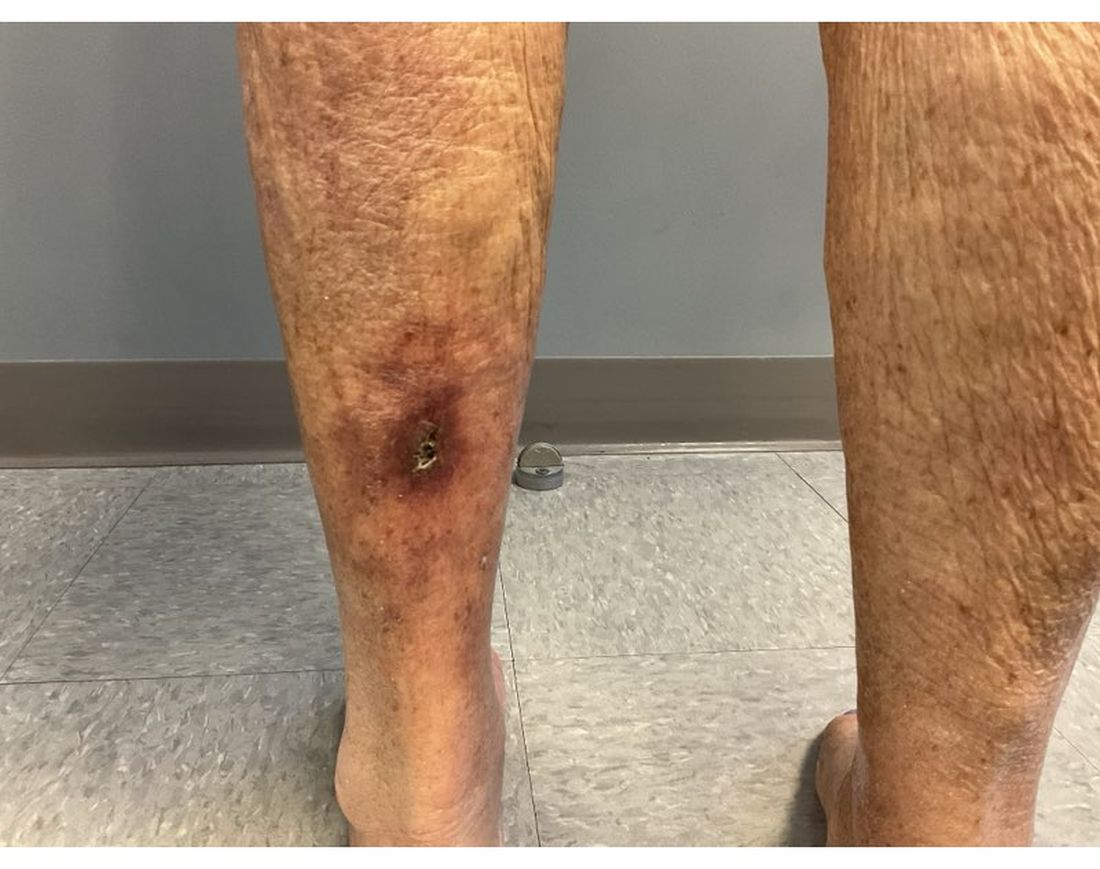
Pathophysiology of this condition includes calcification of the medial layer of arterioles and small arteries near the skin. Damage to vessel endothelium and formation of microthrombi contribute to the ischemia, which results in necrosis and ulceration of the skin. Elevated calcium and phosphate have been associated with these findings; however, these lab abnormalities alone are typically not enough to cause calciphylaxis. Vascular calcification inhibitors such as fetuin-A, osteoprotegerin, and matrix G1a protein may play a role in pathogenesis, with individuals lacking these factors potentially being at a greater risk. Specifically, matrix G1a protein is dependent on vitamin K dependent carboxylation, which may elucidate why warfarin has been implicated in the development of calciphylaxis because of interference with this pathway.
Upon presentation, patients will have painful ischemic plaques on the skin or painful subcutaneous nodules. Long-standing lesions may have a necrotic eschar or secondary infection, or may be associated with livedo reticularis. Areas with a greater concentration of adipose tissue such as the abdomen, thighs, and buttocks are most commonly affected, but lesions may appear anywhere. A biopsy may be done, but a clinical diagnosis is often sufficient as biopsies carry risks of prolonged healing and infection.

The differential diagnosis includes warfarin skin necrosis, cholesterol embolization, vasculitis, antiphospholipid syndrome, and cellulitis. Although this is a cutaneous manifestation, calciphylaxis is indicative of a systemic problem and requires multidisciplinary intervention.
Patients who present with calciphylaxis require a complete metabolic panel, liver function tests, coagulation studies, and albumin tests. Depending on the presentation, imaging studies such as nuclear medicine scans may be used if extensive soft tissue involvement is suspected.
Clinical management includes carefully avoiding electrolyte imbalances, initiating dialysis if necessary, discontinuing potentially offending supplements and medications, and administering proper wound care and pain management. Debridement of necrotic tissue may be necessary and should be initiated early as this has been associated with a 6-month increase in survival. Physicians should have a low threshold for starting antibiotics if secondary infection is suspected, but prophylaxis is not recommended. Sodium thiosulfate has been used off-label, but the mechanism of action is unknown and some meta-analyses indicate this treatment is not significantly associated with improvement of skin lesions. Interventions such as hyperbaric oxygen have also been used, but there is still more research to be done on these modalities.
The case and photo were submitted by Lucas Shapiro, BS, Nova Southeastern University College of Osteopathic Medicine, and Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, Fort Lauderdale, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Kodumudi V et al. Adv Ther. 2020 Dec;37(12):4797-4807. doi: 10.1007/s12325-020-01504-w.
Seethapathy H et al. Adv Chronic Kidney Dis. 2019 Nov;26(6):484-490. doi: 10.1053/j.ackd.2019.09.005.
Turek M et al. Am J Case Rep. 2021 Jun 7:22:e930026. doi: 10.12659/AJCR.930026.
Wen W at al. JAMA Netw Open. 2023;6(4):e2310068. doi:10.1001/jamanetworkopen.2023.10068.
Westphal SG, Plumb T. Calciphylaxis. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: www.ncbi.nlm.nih.gov/books/NBK519020/.
Calciphylaxis, also known as calcific uremic arteriolopathy, is a rare condition most commonly observed in patients with end-stage renal disease (ESRD). Because of the non-healing nature of the wounds and need for frequent hospitalizations, there is a significant risk of sepsis with a 1-year mortality rate greater than 50%.
Beyond ESRD, calciphylaxis is also associated with obesity, diabetes, hypoalbuminemia, autoimmune conditions, hepatic disease, malignancies, and dialysis. Rates in patients on dialysis have been increasing, ranging from 1% to 4%. Certain medications have also been implicated in the development of calciphylaxis, including warfarin, steroids, calcium-based phosphate binders, vitamin D, and iron. There is also an association with White individuals and more cases have been reported in females.
Pathophysiology of this condition includes calcification of the medial layer of arterioles and small arteries near the skin. Damage to vessel endothelium and formation of microthrombi contribute to the ischemia, which results in necrosis and ulceration of the skin. Elevated calcium and phosphate have been associated with these findings; however, these lab abnormalities alone are typically not enough to cause calciphylaxis. Vascular calcification inhibitors such as fetuin-A, osteoprotegerin, and matrix G1a protein may play a role in pathogenesis, with individuals lacking these factors potentially being at a greater risk. Specifically, matrix G1a protein is dependent on vitamin K dependent carboxylation, which may elucidate why warfarin has been implicated in the development of calciphylaxis because of interference with this pathway.
Upon presentation, patients will have painful ischemic plaques on the skin or painful subcutaneous nodules. Long-standing lesions may have a necrotic eschar or secondary infection, or may be associated with livedo reticularis. Areas with a greater concentration of adipose tissue such as the abdomen, thighs, and buttocks are most commonly affected, but lesions may appear anywhere. A biopsy may be done, but a clinical diagnosis is often sufficient as biopsies carry risks of prolonged healing and infection.
The differential diagnosis includes warfarin skin necrosis, cholesterol embolization, vasculitis, antiphospholipid syndrome, and cellulitis. Although this is a cutaneous manifestation, calciphylaxis is indicative of a systemic problem and requires multidisciplinary intervention.
Patients who present with calciphylaxis require a complete metabolic panel, liver function tests, coagulation studies, and albumin tests. Depending on the presentation, imaging studies such as nuclear medicine scans may be used if extensive soft tissue involvement is suspected.
Clinical management includes carefully avoiding electrolyte imbalances, initiating dialysis if necessary, discontinuing potentially offending supplements and medications, and administering proper wound care and pain management. Debridement of necrotic tissue may be necessary and should be initiated early as this has been associated with a 6-month increase in survival. Physicians should have a low threshold for starting antibiotics if secondary infection is suspected, but prophylaxis is not recommended. Sodium thiosulfate has been used off-label, but the mechanism of action is unknown and some meta-analyses indicate this treatment is not significantly associated with improvement of skin lesions. Interventions such as hyperbaric oxygen have also been used, but there is still more research to be done on these modalities.
The case and photo were submitted by Lucas Shapiro, BS, Nova Southeastern University College of Osteopathic Medicine, and Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, Fort Lauderdale, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Kodumudi V et al. Adv Ther. 2020 Dec;37(12):4797-4807. doi: 10.1007/s12325-020-01504-w.
Seethapathy H et al. Adv Chronic Kidney Dis. 2019 Nov;26(6):484-490. doi: 10.1053/j.ackd.2019.09.005.
Turek M et al. Am J Case Rep. 2021 Jun 7:22:e930026. doi: 10.12659/AJCR.930026.
Wen W at al. JAMA Netw Open. 2023;6(4):e2310068. doi:10.1001/jamanetworkopen.2023.10068.
Westphal SG, Plumb T. Calciphylaxis. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: www.ncbi.nlm.nih.gov/books/NBK519020/.
Calciphylaxis, also known as calcific uremic arteriolopathy, is a rare condition most commonly observed in patients with end-stage renal disease (ESRD). Because of the non-healing nature of the wounds and need for frequent hospitalizations, there is a significant risk of sepsis with a 1-year mortality rate greater than 50%.
Beyond ESRD, calciphylaxis is also associated with obesity, diabetes, hypoalbuminemia, autoimmune conditions, hepatic disease, malignancies, and dialysis. Rates in patients on dialysis have been increasing, ranging from 1% to 4%. Certain medications have also been implicated in the development of calciphylaxis, including warfarin, steroids, calcium-based phosphate binders, vitamin D, and iron. There is also an association with White individuals and more cases have been reported in females.
Pathophysiology of this condition includes calcification of the medial layer of arterioles and small arteries near the skin. Damage to vessel endothelium and formation of microthrombi contribute to the ischemia, which results in necrosis and ulceration of the skin. Elevated calcium and phosphate have been associated with these findings; however, these lab abnormalities alone are typically not enough to cause calciphylaxis. Vascular calcification inhibitors such as fetuin-A, osteoprotegerin, and matrix G1a protein may play a role in pathogenesis, with individuals lacking these factors potentially being at a greater risk. Specifically, matrix G1a protein is dependent on vitamin K dependent carboxylation, which may elucidate why warfarin has been implicated in the development of calciphylaxis because of interference with this pathway.
Upon presentation, patients will have painful ischemic plaques on the skin or painful subcutaneous nodules. Long-standing lesions may have a necrotic eschar or secondary infection, or may be associated with livedo reticularis. Areas with a greater concentration of adipose tissue such as the abdomen, thighs, and buttocks are most commonly affected, but lesions may appear anywhere. A biopsy may be done, but a clinical diagnosis is often sufficient as biopsies carry risks of prolonged healing and infection.
The differential diagnosis includes warfarin skin necrosis, cholesterol embolization, vasculitis, antiphospholipid syndrome, and cellulitis. Although this is a cutaneous manifestation, calciphylaxis is indicative of a systemic problem and requires multidisciplinary intervention.
Patients who present with calciphylaxis require a complete metabolic panel, liver function tests, coagulation studies, and albumin tests. Depending on the presentation, imaging studies such as nuclear medicine scans may be used if extensive soft tissue involvement is suspected.
Clinical management includes carefully avoiding electrolyte imbalances, initiating dialysis if necessary, discontinuing potentially offending supplements and medications, and administering proper wound care and pain management. Debridement of necrotic tissue may be necessary and should be initiated early as this has been associated with a 6-month increase in survival. Physicians should have a low threshold for starting antibiotics if secondary infection is suspected, but prophylaxis is not recommended. Sodium thiosulfate has been used off-label, but the mechanism of action is unknown and some meta-analyses indicate this treatment is not significantly associated with improvement of skin lesions. Interventions such as hyperbaric oxygen have also been used, but there is still more research to be done on these modalities.
The case and photo were submitted by Lucas Shapiro, BS, Nova Southeastern University College of Osteopathic Medicine, and Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, Fort Lauderdale, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Kodumudi V et al. Adv Ther. 2020 Dec;37(12):4797-4807. doi: 10.1007/s12325-020-01504-w.
Seethapathy H et al. Adv Chronic Kidney Dis. 2019 Nov;26(6):484-490. doi: 10.1053/j.ackd.2019.09.005.
Turek M et al. Am J Case Rep. 2021 Jun 7:22:e930026. doi: 10.12659/AJCR.930026.
Wen W at al. JAMA Netw Open. 2023;6(4):e2310068. doi:10.1001/jamanetworkopen.2023.10068.
Westphal SG, Plumb T. Calciphylaxis. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: www.ncbi.nlm.nih.gov/books/NBK519020/.
An 81-year-old White woman with a medical history significant for end stage renal disease (ESRD) on dialysis, diabetes, and a cerebrovascular accident presented with a 2-week history of a very painful lesion on her left calf. Upon physical exam, she was also noted to have tender subcutaneous nodules on her left anterolateral thigh that had been present for several weeks.
What's your diagnosis?
A 58-year-old White male presented with lesions on his index and middle finger for 3 months
Syphilis
Two biopsies by punch technique were performed; one for pathology and one for tissue culture (fungal and atypical mycobacteria). Tissue cultures showed no growth at 4 and 6 weeks, respectively. The lesions were swabbed for bacterial and viral cultures. Bacterial culture was positive for methicillin-resistant Staphylococcus aureus (MRSA), Pseudomonas aeruginosa, and group C Streptococcus. Viral culture for herpes simplex virus (HSV) and varicella zoster virus (VZV) was negative. Histopathology confirmed the diagnosis of syphilis. Immunoperoxidase stain was positive for Treponema pallidum, and negative for HSV-1, HSV-2, and VZV. Special stains for PAS, GMS, Fite, and AFB were negative for organisms.
Syphilis, also known as Lues disease, is a contagious, sexually acquired disease caused by the spirochete T pallidum. The skin and mucous membranes are primarily infected. There are primary, secondary, and tertiary stages. In the primary or initial stage of syphilis, a chancre appears, usually 3-4 weeks after infection. The chancre is a painless papule or erosion that progresses to a firm ulceration. Lymphadenopathy may be present. Less often, multiple chancres may be present. Primary chancre on the finger has been reported in the literature, although it is far less common to have extragenital primary syphilis. The incidence ranges from 2% to 10%. Other extragenital areas that can be affected include lips, intraoral lesions, and the anus. Atypical chancres can be formed when other microbial agents are also present. Generally, an untreated chancre will heal spontaneously within a few months.
The patient referred to the department of health for treatment with penicillin G and further workup of sexually transmitted diseases. He was also seen by infectious disease for treatment of the superimposed bacterial infections and treated with an antibiotic regimen.
The case and photo were submitted by Dr. Bilu Martin.
Dr Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Ramoni S et al. Sex Transm Dis. 2010 Jul;37(7):468. doi: 10.1097/OLQ.0b013e3181e2cfac.
Starzycki Z. Br J Vener Dis. 1983 Jun;59(3):169-71. doi: 10.1136/sti.59.3.169.
Syphilis
Two biopsies by punch technique were performed; one for pathology and one for tissue culture (fungal and atypical mycobacteria). Tissue cultures showed no growth at 4 and 6 weeks, respectively. The lesions were swabbed for bacterial and viral cultures. Bacterial culture was positive for methicillin-resistant Staphylococcus aureus (MRSA), Pseudomonas aeruginosa, and group C Streptococcus. Viral culture for herpes simplex virus (HSV) and varicella zoster virus (VZV) was negative. Histopathology confirmed the diagnosis of syphilis. Immunoperoxidase stain was positive for Treponema pallidum, and negative for HSV-1, HSV-2, and VZV. Special stains for PAS, GMS, Fite, and AFB were negative for organisms.
Syphilis, also known as Lues disease, is a contagious, sexually acquired disease caused by the spirochete T pallidum. The skin and mucous membranes are primarily infected. There are primary, secondary, and tertiary stages. In the primary or initial stage of syphilis, a chancre appears, usually 3-4 weeks after infection. The chancre is a painless papule or erosion that progresses to a firm ulceration. Lymphadenopathy may be present. Less often, multiple chancres may be present. Primary chancre on the finger has been reported in the literature, although it is far less common to have extragenital primary syphilis. The incidence ranges from 2% to 10%. Other extragenital areas that can be affected include lips, intraoral lesions, and the anus. Atypical chancres can be formed when other microbial agents are also present. Generally, an untreated chancre will heal spontaneously within a few months.
The patient referred to the department of health for treatment with penicillin G and further workup of sexually transmitted diseases. He was also seen by infectious disease for treatment of the superimposed bacterial infections and treated with an antibiotic regimen.
The case and photo were submitted by Dr. Bilu Martin.
Dr Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Ramoni S et al. Sex Transm Dis. 2010 Jul;37(7):468. doi: 10.1097/OLQ.0b013e3181e2cfac.
Starzycki Z. Br J Vener Dis. 1983 Jun;59(3):169-71. doi: 10.1136/sti.59.3.169.
Syphilis
Two biopsies by punch technique were performed; one for pathology and one for tissue culture (fungal and atypical mycobacteria). Tissue cultures showed no growth at 4 and 6 weeks, respectively. The lesions were swabbed for bacterial and viral cultures. Bacterial culture was positive for methicillin-resistant Staphylococcus aureus (MRSA), Pseudomonas aeruginosa, and group C Streptococcus. Viral culture for herpes simplex virus (HSV) and varicella zoster virus (VZV) was negative. Histopathology confirmed the diagnosis of syphilis. Immunoperoxidase stain was positive for Treponema pallidum, and negative for HSV-1, HSV-2, and VZV. Special stains for PAS, GMS, Fite, and AFB were negative for organisms.
Syphilis, also known as Lues disease, is a contagious, sexually acquired disease caused by the spirochete T pallidum. The skin and mucous membranes are primarily infected. There are primary, secondary, and tertiary stages. In the primary or initial stage of syphilis, a chancre appears, usually 3-4 weeks after infection. The chancre is a painless papule or erosion that progresses to a firm ulceration. Lymphadenopathy may be present. Less often, multiple chancres may be present. Primary chancre on the finger has been reported in the literature, although it is far less common to have extragenital primary syphilis. The incidence ranges from 2% to 10%. Other extragenital areas that can be affected include lips, intraoral lesions, and the anus. Atypical chancres can be formed when other microbial agents are also present. Generally, an untreated chancre will heal spontaneously within a few months.
The patient referred to the department of health for treatment with penicillin G and further workup of sexually transmitted diseases. He was also seen by infectious disease for treatment of the superimposed bacterial infections and treated with an antibiotic regimen.
The case and photo were submitted by Dr. Bilu Martin.
Dr Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Ramoni S et al. Sex Transm Dis. 2010 Jul;37(7):468. doi: 10.1097/OLQ.0b013e3181e2cfac.
Starzycki Z. Br J Vener Dis. 1983 Jun;59(3):169-71. doi: 10.1136/sti.59.3.169.
A 58-year-old White male with no significant past medical history presented with lesions on his right index and middle fingers, which had been present for 3 months. The lesions were painless. The patient has a history of hand dermatitis. Upon questioning, the patient said he had not fished or cleaned fish tanks. He did garden occasionally (no roses). He has been using Neosporin on the lesions. He denied any fever or systemic symptoms and had no lymphadenopathy.
What's your diagnosis?
A 51-year-old woman presented for a routine full body skin exam after vacationing in Hawaii.
Primary adrenal insufficiency (Addison’s disease) results from a dysfunction of the adrenal glands, which may be secondary to autoimmune diseases, genetic conditions, infections, and vasculopathies,or may be drug-induced (e.g. checkpoint inhibitors), among others . In contrast, secondary adrenal insufficiency results from pituitary dysfunction of low adrenocorticotropic hormone (ACTH). The most common cause of primary adrenal insufficiency in developed countries is autoimmune adrenalitis, which accounts for upwards of 90% of cases. Typically, 21-hydroxylase autoantibodies are identified and account for destruction of the adrenal cortex through cell-mediated and humoral immune responses.
Palmar creases, subungual surfaces, sites of trauma, and joint spaces (including the knees, spine, elbows, and shoulders) are commonly affected. Hair depletes in the pubic area and axillary vaults. Nevi may also appear darker. In patients with autoimmune adrenalitis, vitiligo may be seen secondary to autoimmune destruction of melanocytes.
Diagnosis may be difficult in the early stages, but historical findings of fatigue and clinical findings of hyperpigmentation in classic areas may prompt appropriate lab screening workup. It is essential to determine whether adrenal insufficiency is primary or secondary. Evaluation of decreased cortisol production, determination of whether production is ACTH-dependent or -independent, and evaluation for the underlying causes of adrenal dysfunction are important. Lab screening includes morning serum cortisol, morning ACTH (cosyntropin) stimulation test, fasting CBC with differential, and CMP to evaluate for normocytic normochromic anemia, hyponatremia, hyperkalemia, hypoglycemia, plasma renin/aldosterone ratio, and 21-hydroxylase autoantibodies.
Management strategies of primary adrenal insufficiency require corticosteroid supplementation and multidisciplinary collaboration with endocrinology. If untreated, primary adrenal insufficiency can be fatal. Adrenal crisis is a critical condition following a precipitating event, such as GI infection, fever, acute stress, and/or untreated adrenal or pituitary disorders. Clinical findings include acute shock with hypotension, nausea, vomiting, abdominal pain, back or leg pain, and a change in mental status. In this scenario, increasing the dose of corticosteroid supplementation is essential for reducing mortality.
Upon examining this patient’s new skin findings of hyperpigmentation and discussing her fatigue, primary adrenal insufficiency was suspected. With further prompting, the patient reported an ICU hospitalization several months prior because of sepsis originating from a peritonsillar abscess. With these clinical and historical findings, preliminary workup was conducted by dermatology, which included morning cortisol level, ACTH, CBC with differential, CMP, plasma renin-aldosterone ratio, and 21-hydroxylase autoantibodies. Work up demonstrated a low morning cortisol level of 1.3 mcg/dL, an elevated ACTH of 2,739 pg/mL, and positive 21-hydroxylase autoantibodies. The patient was urgently referred to endocrinology and started on oral hydrocortisone. Her fatigue immediately improved, and at 1-year follow-up with dermatology, her mucocutaneous hyperpigmentation had subsided dramatically.
Dermatologists can play a major role in the early diagnosis of primary adrenal insufficiency, which is essential for reducing patient morbidity and mortality. Skin findings on full body skin exams can clue in dermatologists for ordering preliminary workup to expedite care for these patients.
The case and photos were submitted by Dr. Akhiyat, Scripps Clinic Medical Group, La Jolla, California. Donna Bilu Martin, MD, edited the column.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
J Am Acad Dermatol. 2014 May;70(5):Supplement 1AB118. doi: 10.1016/j.jaad.2014.01.491.
Michels A, Michels N. Am Fam Physician. 2014 Apr 1;89(7):563-568.
Kauzman A et al. J Can Dent Assoc. 2004 Nov;70(10):682-683.
Primary adrenal insufficiency (Addison’s disease) results from a dysfunction of the adrenal glands, which may be secondary to autoimmune diseases, genetic conditions, infections, and vasculopathies,or may be drug-induced (e.g. checkpoint inhibitors), among others . In contrast, secondary adrenal insufficiency results from pituitary dysfunction of low adrenocorticotropic hormone (ACTH). The most common cause of primary adrenal insufficiency in developed countries is autoimmune adrenalitis, which accounts for upwards of 90% of cases. Typically, 21-hydroxylase autoantibodies are identified and account for destruction of the adrenal cortex through cell-mediated and humoral immune responses.
Palmar creases, subungual surfaces, sites of trauma, and joint spaces (including the knees, spine, elbows, and shoulders) are commonly affected. Hair depletes in the pubic area and axillary vaults. Nevi may also appear darker. In patients with autoimmune adrenalitis, vitiligo may be seen secondary to autoimmune destruction of melanocytes.
Diagnosis may be difficult in the early stages, but historical findings of fatigue and clinical findings of hyperpigmentation in classic areas may prompt appropriate lab screening workup. It is essential to determine whether adrenal insufficiency is primary or secondary. Evaluation of decreased cortisol production, determination of whether production is ACTH-dependent or -independent, and evaluation for the underlying causes of adrenal dysfunction are important. Lab screening includes morning serum cortisol, morning ACTH (cosyntropin) stimulation test, fasting CBC with differential, and CMP to evaluate for normocytic normochromic anemia, hyponatremia, hyperkalemia, hypoglycemia, plasma renin/aldosterone ratio, and 21-hydroxylase autoantibodies.
Management strategies of primary adrenal insufficiency require corticosteroid supplementation and multidisciplinary collaboration with endocrinology. If untreated, primary adrenal insufficiency can be fatal. Adrenal crisis is a critical condition following a precipitating event, such as GI infection, fever, acute stress, and/or untreated adrenal or pituitary disorders. Clinical findings include acute shock with hypotension, nausea, vomiting, abdominal pain, back or leg pain, and a change in mental status. In this scenario, increasing the dose of corticosteroid supplementation is essential for reducing mortality.
Upon examining this patient’s new skin findings of hyperpigmentation and discussing her fatigue, primary adrenal insufficiency was suspected. With further prompting, the patient reported an ICU hospitalization several months prior because of sepsis originating from a peritonsillar abscess. With these clinical and historical findings, preliminary workup was conducted by dermatology, which included morning cortisol level, ACTH, CBC with differential, CMP, plasma renin-aldosterone ratio, and 21-hydroxylase autoantibodies. Work up demonstrated a low morning cortisol level of 1.3 mcg/dL, an elevated ACTH of 2,739 pg/mL, and positive 21-hydroxylase autoantibodies. The patient was urgently referred to endocrinology and started on oral hydrocortisone. Her fatigue immediately improved, and at 1-year follow-up with dermatology, her mucocutaneous hyperpigmentation had subsided dramatically.
Dermatologists can play a major role in the early diagnosis of primary adrenal insufficiency, which is essential for reducing patient morbidity and mortality. Skin findings on full body skin exams can clue in dermatologists for ordering preliminary workup to expedite care for these patients.
The case and photos were submitted by Dr. Akhiyat, Scripps Clinic Medical Group, La Jolla, California. Donna Bilu Martin, MD, edited the column.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
J Am Acad Dermatol. 2014 May;70(5):Supplement 1AB118. doi: 10.1016/j.jaad.2014.01.491.
Michels A, Michels N. Am Fam Physician. 2014 Apr 1;89(7):563-568.
Kauzman A et al. J Can Dent Assoc. 2004 Nov;70(10):682-683.
Primary adrenal insufficiency (Addison’s disease) results from a dysfunction of the adrenal glands, which may be secondary to autoimmune diseases, genetic conditions, infections, and vasculopathies,or may be drug-induced (e.g. checkpoint inhibitors), among others . In contrast, secondary adrenal insufficiency results from pituitary dysfunction of low adrenocorticotropic hormone (ACTH). The most common cause of primary adrenal insufficiency in developed countries is autoimmune adrenalitis, which accounts for upwards of 90% of cases. Typically, 21-hydroxylase autoantibodies are identified and account for destruction of the adrenal cortex through cell-mediated and humoral immune responses.
Palmar creases, subungual surfaces, sites of trauma, and joint spaces (including the knees, spine, elbows, and shoulders) are commonly affected. Hair depletes in the pubic area and axillary vaults. Nevi may also appear darker. In patients with autoimmune adrenalitis, vitiligo may be seen secondary to autoimmune destruction of melanocytes.
Diagnosis may be difficult in the early stages, but historical findings of fatigue and clinical findings of hyperpigmentation in classic areas may prompt appropriate lab screening workup. It is essential to determine whether adrenal insufficiency is primary or secondary. Evaluation of decreased cortisol production, determination of whether production is ACTH-dependent or -independent, and evaluation for the underlying causes of adrenal dysfunction are important. Lab screening includes morning serum cortisol, morning ACTH (cosyntropin) stimulation test, fasting CBC with differential, and CMP to evaluate for normocytic normochromic anemia, hyponatremia, hyperkalemia, hypoglycemia, plasma renin/aldosterone ratio, and 21-hydroxylase autoantibodies.
Management strategies of primary adrenal insufficiency require corticosteroid supplementation and multidisciplinary collaboration with endocrinology. If untreated, primary adrenal insufficiency can be fatal. Adrenal crisis is a critical condition following a precipitating event, such as GI infection, fever, acute stress, and/or untreated adrenal or pituitary disorders. Clinical findings include acute shock with hypotension, nausea, vomiting, abdominal pain, back or leg pain, and a change in mental status. In this scenario, increasing the dose of corticosteroid supplementation is essential for reducing mortality.
Upon examining this patient’s new skin findings of hyperpigmentation and discussing her fatigue, primary adrenal insufficiency was suspected. With further prompting, the patient reported an ICU hospitalization several months prior because of sepsis originating from a peritonsillar abscess. With these clinical and historical findings, preliminary workup was conducted by dermatology, which included morning cortisol level, ACTH, CBC with differential, CMP, plasma renin-aldosterone ratio, and 21-hydroxylase autoantibodies. Work up demonstrated a low morning cortisol level of 1.3 mcg/dL, an elevated ACTH of 2,739 pg/mL, and positive 21-hydroxylase autoantibodies. The patient was urgently referred to endocrinology and started on oral hydrocortisone. Her fatigue immediately improved, and at 1-year follow-up with dermatology, her mucocutaneous hyperpigmentation had subsided dramatically.
Dermatologists can play a major role in the early diagnosis of primary adrenal insufficiency, which is essential for reducing patient morbidity and mortality. Skin findings on full body skin exams can clue in dermatologists for ordering preliminary workup to expedite care for these patients.
The case and photos were submitted by Dr. Akhiyat, Scripps Clinic Medical Group, La Jolla, California. Donna Bilu Martin, MD, edited the column.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
J Am Acad Dermatol. 2014 May;70(5):Supplement 1AB118. doi: 10.1016/j.jaad.2014.01.491.
Michels A, Michels N. Am Fam Physician. 2014 Apr 1;89(7):563-568.
Kauzman A et al. J Can Dent Assoc. 2004 Nov;70(10):682-683.
A 71-year-old White female developed erosions after hip replacement surgery 2 months prior to presentation
The patient had been diagnosed with pemphigus vulgaris (PV) 1 year prior to presentation with erosions on the axilla. Biopsy at that time revealed intraepithelial acantholytic blistering with areas of suprabasilar and subcorneal clefting. Direct immunofluorescence was positive for linear/granular IgG deposition throughout the epithelial cell surfaces, as well as linear/granular C3 deposits of the lower two thirds of the epithelial strata, consistent for pemphigus vulgaris.
There is likely a genetic predisposition. Medications that may induce pemphigus include penicillamine, nifedipine, or captopril.
Clinically, PV presents with flaccid blistering lesions that may be cutaneous and/or mucosal. Bullae can progress to erosions and crusting, which then heal with pigment alteration but not scarring. The most commonly affected sites are the mouth, intertriginous areas, face, and neck. Mucosal lesions can involve the lips, esophagus, conjunctiva, and genitals.
Biopsy for histology and direct immunofluorescence is important in distinguishing between PV and other blistering disorders. Up to 75% of patients with active disease also have a positive indirect immunofluorescence with circulating IgG.
There are numerous reports in the literature of PV occurring in previous surgical scars, and areas of friction or trauma. This so-called Koebner’s phenomenon is seen more commonly in several dermatologic conditions, such as psoriasis, lichen planus, verruca vulgaris, and vitiligo.
Treatment for PV is generally immunosuppressive. Systemic therapy usually begins with prednisone and then is transitioned to a steroid sparing agent such as mycophenolate mofetil. Other steroid sparing agents include azathioprine, methotrexate, cyclophosphamide, and intravenous immunoglobulin. Secondary infections are possible and should be treated. Topical therapies aimed at reducing pain, especially in mucosal lesions, can be beneficial.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Cerottini JP et al. Eur J Dermatol. 2000 Oct-Nov;10(7):546-7.
Reichert-Penetrat S et al. Eur J Dermatol. 1998 Jan-Feb;8(1):60-2.
Saini P et al. Skinmed. 2020 Aug 1;18(4):252-253.
The patient had been diagnosed with pemphigus vulgaris (PV) 1 year prior to presentation with erosions on the axilla. Biopsy at that time revealed intraepithelial acantholytic blistering with areas of suprabasilar and subcorneal clefting. Direct immunofluorescence was positive for linear/granular IgG deposition throughout the epithelial cell surfaces, as well as linear/granular C3 deposits of the lower two thirds of the epithelial strata, consistent for pemphigus vulgaris.
There is likely a genetic predisposition. Medications that may induce pemphigus include penicillamine, nifedipine, or captopril.
Clinically, PV presents with flaccid blistering lesions that may be cutaneous and/or mucosal. Bullae can progress to erosions and crusting, which then heal with pigment alteration but not scarring. The most commonly affected sites are the mouth, intertriginous areas, face, and neck. Mucosal lesions can involve the lips, esophagus, conjunctiva, and genitals.
Biopsy for histology and direct immunofluorescence is important in distinguishing between PV and other blistering disorders. Up to 75% of patients with active disease also have a positive indirect immunofluorescence with circulating IgG.
There are numerous reports in the literature of PV occurring in previous surgical scars, and areas of friction or trauma. This so-called Koebner’s phenomenon is seen more commonly in several dermatologic conditions, such as psoriasis, lichen planus, verruca vulgaris, and vitiligo.
Treatment for PV is generally immunosuppressive. Systemic therapy usually begins with prednisone and then is transitioned to a steroid sparing agent such as mycophenolate mofetil. Other steroid sparing agents include azathioprine, methotrexate, cyclophosphamide, and intravenous immunoglobulin. Secondary infections are possible and should be treated. Topical therapies aimed at reducing pain, especially in mucosal lesions, can be beneficial.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Cerottini JP et al. Eur J Dermatol. 2000 Oct-Nov;10(7):546-7.
Reichert-Penetrat S et al. Eur J Dermatol. 1998 Jan-Feb;8(1):60-2.
Saini P et al. Skinmed. 2020 Aug 1;18(4):252-253.
The patient had been diagnosed with pemphigus vulgaris (PV) 1 year prior to presentation with erosions on the axilla. Biopsy at that time revealed intraepithelial acantholytic blistering with areas of suprabasilar and subcorneal clefting. Direct immunofluorescence was positive for linear/granular IgG deposition throughout the epithelial cell surfaces, as well as linear/granular C3 deposits of the lower two thirds of the epithelial strata, consistent for pemphigus vulgaris.
There is likely a genetic predisposition. Medications that may induce pemphigus include penicillamine, nifedipine, or captopril.
Clinically, PV presents with flaccid blistering lesions that may be cutaneous and/or mucosal. Bullae can progress to erosions and crusting, which then heal with pigment alteration but not scarring. The most commonly affected sites are the mouth, intertriginous areas, face, and neck. Mucosal lesions can involve the lips, esophagus, conjunctiva, and genitals.
Biopsy for histology and direct immunofluorescence is important in distinguishing between PV and other blistering disorders. Up to 75% of patients with active disease also have a positive indirect immunofluorescence with circulating IgG.
There are numerous reports in the literature of PV occurring in previous surgical scars, and areas of friction or trauma. This so-called Koebner’s phenomenon is seen more commonly in several dermatologic conditions, such as psoriasis, lichen planus, verruca vulgaris, and vitiligo.
Treatment for PV is generally immunosuppressive. Systemic therapy usually begins with prednisone and then is transitioned to a steroid sparing agent such as mycophenolate mofetil. Other steroid sparing agents include azathioprine, methotrexate, cyclophosphamide, and intravenous immunoglobulin. Secondary infections are possible and should be treated. Topical therapies aimed at reducing pain, especially in mucosal lesions, can be beneficial.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Cerottini JP et al. Eur J Dermatol. 2000 Oct-Nov;10(7):546-7.
Reichert-Penetrat S et al. Eur J Dermatol. 1998 Jan-Feb;8(1):60-2.
Saini P et al. Skinmed. 2020 Aug 1;18(4):252-253.
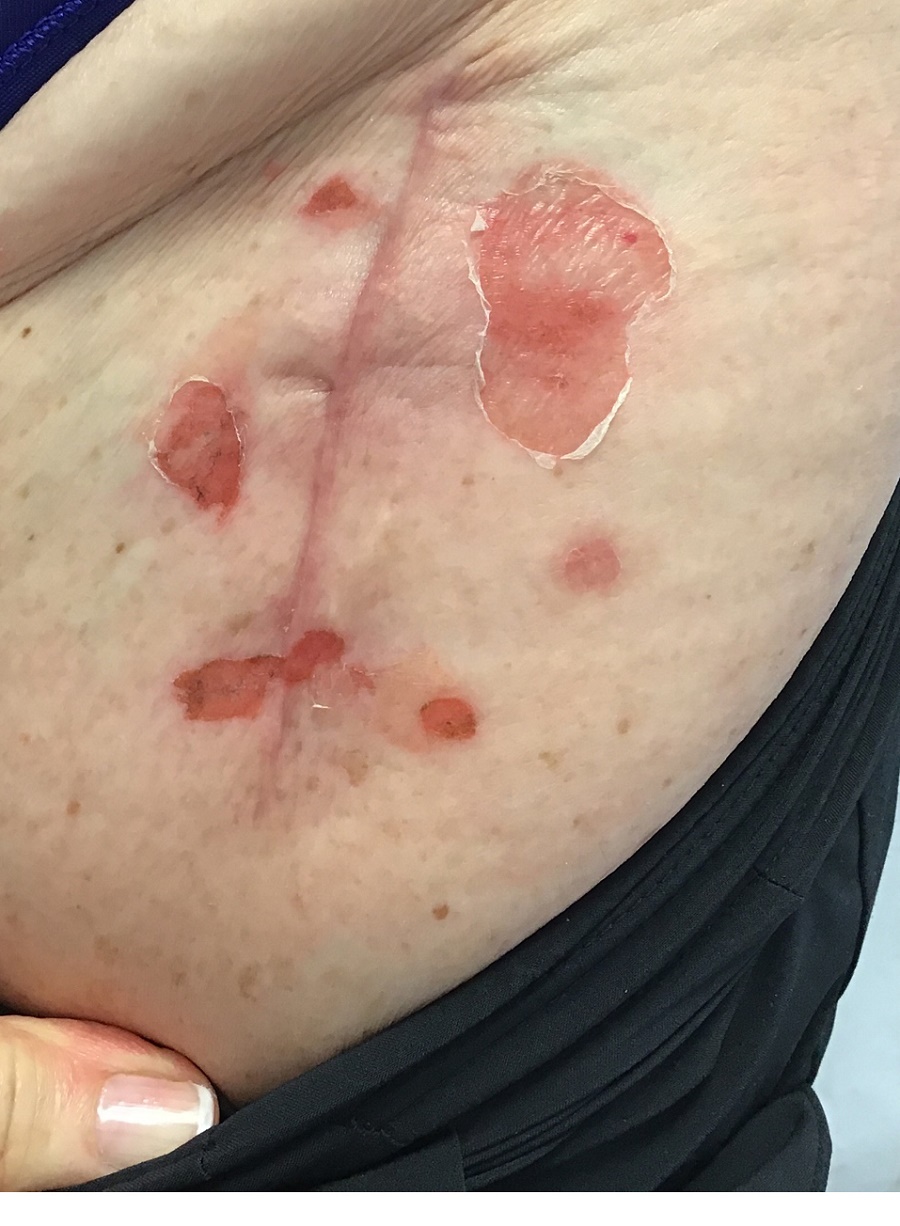
A 62-year-old Black female presented with an epidermal inclusion cyst on her left upper back
This heterogeneous disorder can present with a wide range of clinical manifestations, including dermatological symptoms that may be the first or predominant feature. Systemic amyloidosis is characterized by macroglossia, periorbital purpura, and waxy skin plaques. Lateral scalloping of the tongue may be seen due to impingement of the teeth. Cutaneous amyloidosis occurs when amyloid is deposited in the skin, without internal organ involvement. Variants of cutaneous amyloidosis include macular, lichen, nodular and biphasic.
This condition requires a thorough diagnostic workup, including serum and urine protein electrophoresis and biopsy of the affected tissue. Biopsy of a cutaneous amyloidosis lesion will show fractured, amorphous, eosinophilic material in the dermis. Pigment and epidermal changes are often found with cutaneous amyloidosis, including hyperkeratosis, acanthosis, hypergranulosis, parakeratosis, and epidermal atrophy. Stains that may be used include Congo red showing apple-green birefringence, thioflavin T, and crystal violet.
If untreated, the prognosis is generally poor, related to the extent of organ involvement. Cardiac involvement, a common feature of systemic amyloidosis, can lead to restrictive cardiomyopathy, heart failure, and arrhythmias. Management strategies include steroids, chemotherapy, and stem cell transplantation. Medications include dexamethasone, cyclophosphamide, bortezomib, and melphalan.
This patient went undiagnosed for several years until she began experiencing cardiac issues, including syncope, angina, and restrictive cardiomyopathy with heart failure. A cardiac biopsy confirmed the diagnosis of systemic amyloidosis. This patient is currently awaiting a heart transplant. Early diagnosis of amyloidosis is vital, as it can help prevent severe complications such as heart involvement, significantly impacting the patient’s prognosis and quality of life. When amyloidosis is suspected based on dermatological findings, it is essential to distinguish it from other conditions, such as chronic cutaneous lupus erythematosus, dermatomyositis, scleromyxedema, and lipoid proteinosis. Early identification of characteristic skin lesions and systemic features can lead to timely interventions, more favorable outcomes, and reduction in the risk of advanced organ damage.
The case and photo were submitted by Ms. Cael Aoki and Mr. Shapiro of Nova Southeastern University College of Osteopathic Medicine, Davie, Florida, and Dr. Bartos, of Imperial Dermatology, Hollywood, Florida. The column was edited by Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Brunt EM, Tiniakos DG. Clin Liver Dis. 2004 Nov;8(4):915-30, x. doi: 10.1016/j.cld.2004.06.009.
2. Bolognia JL et al. (2017). Dermatology E-Book. Elsevier Health Sciences.
3. Mehrotra K et al. J Clin Diagn Res. 2017 Aug;11(8):WC01-WC05. doi: 10.7860/JCDR/2017/24273.10334.
4. Banypersad SM et al. J Am Heart Assoc. 2012 Apr;1(2):e000364. doi: 10.1161/JAHA.111.000364.
5. Bustamante JG, Zaidi SRH. Amyloidosis. [Updated 2023 Jul 31]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-.
This heterogeneous disorder can present with a wide range of clinical manifestations, including dermatological symptoms that may be the first or predominant feature. Systemic amyloidosis is characterized by macroglossia, periorbital purpura, and waxy skin plaques. Lateral scalloping of the tongue may be seen due to impingement of the teeth. Cutaneous amyloidosis occurs when amyloid is deposited in the skin, without internal organ involvement. Variants of cutaneous amyloidosis include macular, lichen, nodular and biphasic.
This condition requires a thorough diagnostic workup, including serum and urine protein electrophoresis and biopsy of the affected tissue. Biopsy of a cutaneous amyloidosis lesion will show fractured, amorphous, eosinophilic material in the dermis. Pigment and epidermal changes are often found with cutaneous amyloidosis, including hyperkeratosis, acanthosis, hypergranulosis, parakeratosis, and epidermal atrophy. Stains that may be used include Congo red showing apple-green birefringence, thioflavin T, and crystal violet.
If untreated, the prognosis is generally poor, related to the extent of organ involvement. Cardiac involvement, a common feature of systemic amyloidosis, can lead to restrictive cardiomyopathy, heart failure, and arrhythmias. Management strategies include steroids, chemotherapy, and stem cell transplantation. Medications include dexamethasone, cyclophosphamide, bortezomib, and melphalan.
This patient went undiagnosed for several years until she began experiencing cardiac issues, including syncope, angina, and restrictive cardiomyopathy with heart failure. A cardiac biopsy confirmed the diagnosis of systemic amyloidosis. This patient is currently awaiting a heart transplant. Early diagnosis of amyloidosis is vital, as it can help prevent severe complications such as heart involvement, significantly impacting the patient’s prognosis and quality of life. When amyloidosis is suspected based on dermatological findings, it is essential to distinguish it from other conditions, such as chronic cutaneous lupus erythematosus, dermatomyositis, scleromyxedema, and lipoid proteinosis. Early identification of characteristic skin lesions and systemic features can lead to timely interventions, more favorable outcomes, and reduction in the risk of advanced organ damage.
The case and photo were submitted by Ms. Cael Aoki and Mr. Shapiro of Nova Southeastern University College of Osteopathic Medicine, Davie, Florida, and Dr. Bartos, of Imperial Dermatology, Hollywood, Florida. The column was edited by Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Brunt EM, Tiniakos DG. Clin Liver Dis. 2004 Nov;8(4):915-30, x. doi: 10.1016/j.cld.2004.06.009.
2. Bolognia JL et al. (2017). Dermatology E-Book. Elsevier Health Sciences.
3. Mehrotra K et al. J Clin Diagn Res. 2017 Aug;11(8):WC01-WC05. doi: 10.7860/JCDR/2017/24273.10334.
4. Banypersad SM et al. J Am Heart Assoc. 2012 Apr;1(2):e000364. doi: 10.1161/JAHA.111.000364.
5. Bustamante JG, Zaidi SRH. Amyloidosis. [Updated 2023 Jul 31]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-.
This heterogeneous disorder can present with a wide range of clinical manifestations, including dermatological symptoms that may be the first or predominant feature. Systemic amyloidosis is characterized by macroglossia, periorbital purpura, and waxy skin plaques. Lateral scalloping of the tongue may be seen due to impingement of the teeth. Cutaneous amyloidosis occurs when amyloid is deposited in the skin, without internal organ involvement. Variants of cutaneous amyloidosis include macular, lichen, nodular and biphasic.
This condition requires a thorough diagnostic workup, including serum and urine protein electrophoresis and biopsy of the affected tissue. Biopsy of a cutaneous amyloidosis lesion will show fractured, amorphous, eosinophilic material in the dermis. Pigment and epidermal changes are often found with cutaneous amyloidosis, including hyperkeratosis, acanthosis, hypergranulosis, parakeratosis, and epidermal atrophy. Stains that may be used include Congo red showing apple-green birefringence, thioflavin T, and crystal violet.
If untreated, the prognosis is generally poor, related to the extent of organ involvement. Cardiac involvement, a common feature of systemic amyloidosis, can lead to restrictive cardiomyopathy, heart failure, and arrhythmias. Management strategies include steroids, chemotherapy, and stem cell transplantation. Medications include dexamethasone, cyclophosphamide, bortezomib, and melphalan.
This patient went undiagnosed for several years until she began experiencing cardiac issues, including syncope, angina, and restrictive cardiomyopathy with heart failure. A cardiac biopsy confirmed the diagnosis of systemic amyloidosis. This patient is currently awaiting a heart transplant. Early diagnosis of amyloidosis is vital, as it can help prevent severe complications such as heart involvement, significantly impacting the patient’s prognosis and quality of life. When amyloidosis is suspected based on dermatological findings, it is essential to distinguish it from other conditions, such as chronic cutaneous lupus erythematosus, dermatomyositis, scleromyxedema, and lipoid proteinosis. Early identification of characteristic skin lesions and systemic features can lead to timely interventions, more favorable outcomes, and reduction in the risk of advanced organ damage.
The case and photo were submitted by Ms. Cael Aoki and Mr. Shapiro of Nova Southeastern University College of Osteopathic Medicine, Davie, Florida, and Dr. Bartos, of Imperial Dermatology, Hollywood, Florida. The column was edited by Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Florida. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Brunt EM, Tiniakos DG. Clin Liver Dis. 2004 Nov;8(4):915-30, x. doi: 10.1016/j.cld.2004.06.009.
2. Bolognia JL et al. (2017). Dermatology E-Book. Elsevier Health Sciences.
3. Mehrotra K et al. J Clin Diagn Res. 2017 Aug;11(8):WC01-WC05. doi: 10.7860/JCDR/2017/24273.10334.
4. Banypersad SM et al. J Am Heart Assoc. 2012 Apr;1(2):e000364. doi: 10.1161/JAHA.111.000364.
5. Bustamante JG, Zaidi SRH. Amyloidosis. [Updated 2023 Jul 31]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-.
A young adult with a 1-year history of erythema, papules, and pustules on her cheeks and skin
. It typically presents with a sudden onset of papules, pustules, cysts, painful inflammatory nodules, and erythema on the centrofacial areas. The etiology is unknown but has been speculated to be hormone-related as it is more common in women and can be triggered by acute changes such as stress or medications.
Because of overlapping symptoms with other conditions, an accurate clinical assessment is crucial. Typically, there are no comedones and about half of the patients have a history of acne. Some cases have shown a possible link between pyoderma faciale with inflammatory bowel disease, thyroid disease and liver disease, highlighting the importance of considering these associations in treatment decisions.
Treatment options for pyoderma faciale include isotretinoin, corticosteroids, dapsone, and antibiotics such as doxycycline. Isotretinoin is usually the first-line treatment, with dapsone reserved for cases where other methods have failed. Despite concerns about isotretinoin exacerbating inflammatory bowel disease (IBD), there has been at least one reported case where a patient with ulcerative colitis who had pyoderma faciale that was successfully treated with isotretinoin with no adverse effects.
Isotretinoin has been shown to be effective in treating pyoderma faciale by significantly reducing inflammation and scarring. This is imperative because the scarring from pyoderma faciale can be disfiguring and psychologically harmful for patients. Therefore, an early diagnosis and effective treatment method are essential in preventing these scars and improving patients’ confidence and overall dermatological care.

This patient’s initial bacterial culture was negative. She was treated with a course of low dose isotretinoin. Prednisone was initiated two weeks before starting isotretinoin and then was tapered off during the first month of isotretinoin treatment. The patient was also started on spironolactone. The course of isotretinoin was 9 months. She has remained clear and still takes oral contraceptive pills and low dose spironolactone.
This case and the photos were submitted by Ms. Towe, Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Florida, and Donna Bilu Martin, MD, of Premier Dermatology, MD, Aventura, Florida. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Angileri L et al. J Dermatolog Treat. 2021 Feb;32(1):110-3. doi: 10.1080/09546634.2019.1628175.
Coutinho JC et al. An Bras Dermatol. 2016 Sep-Oct;91(5 suppl 1):151-3. doi: 10.1590/abd1806-4841.20164943.
Rosen T and Unkefer RP. Cutis. 1999 Aug;64(2):107-9.
. It typically presents with a sudden onset of papules, pustules, cysts, painful inflammatory nodules, and erythema on the centrofacial areas. The etiology is unknown but has been speculated to be hormone-related as it is more common in women and can be triggered by acute changes such as stress or medications.
Because of overlapping symptoms with other conditions, an accurate clinical assessment is crucial. Typically, there are no comedones and about half of the patients have a history of acne. Some cases have shown a possible link between pyoderma faciale with inflammatory bowel disease, thyroid disease and liver disease, highlighting the importance of considering these associations in treatment decisions.
Treatment options for pyoderma faciale include isotretinoin, corticosteroids, dapsone, and antibiotics such as doxycycline. Isotretinoin is usually the first-line treatment, with dapsone reserved for cases where other methods have failed. Despite concerns about isotretinoin exacerbating inflammatory bowel disease (IBD), there has been at least one reported case where a patient with ulcerative colitis who had pyoderma faciale that was successfully treated with isotretinoin with no adverse effects.
Isotretinoin has been shown to be effective in treating pyoderma faciale by significantly reducing inflammation and scarring. This is imperative because the scarring from pyoderma faciale can be disfiguring and psychologically harmful for patients. Therefore, an early diagnosis and effective treatment method are essential in preventing these scars and improving patients’ confidence and overall dermatological care.
This patient’s initial bacterial culture was negative. She was treated with a course of low dose isotretinoin. Prednisone was initiated two weeks before starting isotretinoin and then was tapered off during the first month of isotretinoin treatment. The patient was also started on spironolactone. The course of isotretinoin was 9 months. She has remained clear and still takes oral contraceptive pills and low dose spironolactone.
This case and the photos were submitted by Ms. Towe, Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Florida, and Donna Bilu Martin, MD, of Premier Dermatology, MD, Aventura, Florida. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Angileri L et al. J Dermatolog Treat. 2021 Feb;32(1):110-3. doi: 10.1080/09546634.2019.1628175.
Coutinho JC et al. An Bras Dermatol. 2016 Sep-Oct;91(5 suppl 1):151-3. doi: 10.1590/abd1806-4841.20164943.
Rosen T and Unkefer RP. Cutis. 1999 Aug;64(2):107-9.
. It typically presents with a sudden onset of papules, pustules, cysts, painful inflammatory nodules, and erythema on the centrofacial areas. The etiology is unknown but has been speculated to be hormone-related as it is more common in women and can be triggered by acute changes such as stress or medications.
Because of overlapping symptoms with other conditions, an accurate clinical assessment is crucial. Typically, there are no comedones and about half of the patients have a history of acne. Some cases have shown a possible link between pyoderma faciale with inflammatory bowel disease, thyroid disease and liver disease, highlighting the importance of considering these associations in treatment decisions.
Treatment options for pyoderma faciale include isotretinoin, corticosteroids, dapsone, and antibiotics such as doxycycline. Isotretinoin is usually the first-line treatment, with dapsone reserved for cases where other methods have failed. Despite concerns about isotretinoin exacerbating inflammatory bowel disease (IBD), there has been at least one reported case where a patient with ulcerative colitis who had pyoderma faciale that was successfully treated with isotretinoin with no adverse effects.
Isotretinoin has been shown to be effective in treating pyoderma faciale by significantly reducing inflammation and scarring. This is imperative because the scarring from pyoderma faciale can be disfiguring and psychologically harmful for patients. Therefore, an early diagnosis and effective treatment method are essential in preventing these scars and improving patients’ confidence and overall dermatological care.
This patient’s initial bacterial culture was negative. She was treated with a course of low dose isotretinoin. Prednisone was initiated two weeks before starting isotretinoin and then was tapered off during the first month of isotretinoin treatment. The patient was also started on spironolactone. The course of isotretinoin was 9 months. She has remained clear and still takes oral contraceptive pills and low dose spironolactone.
This case and the photos were submitted by Ms. Towe, Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Florida, and Donna Bilu Martin, MD, of Premier Dermatology, MD, Aventura, Florida. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Angileri L et al. J Dermatolog Treat. 2021 Feb;32(1):110-3. doi: 10.1080/09546634.2019.1628175.
Coutinho JC et al. An Bras Dermatol. 2016 Sep-Oct;91(5 suppl 1):151-3. doi: 10.1590/abd1806-4841.20164943.
Rosen T and Unkefer RP. Cutis. 1999 Aug;64(2):107-9.
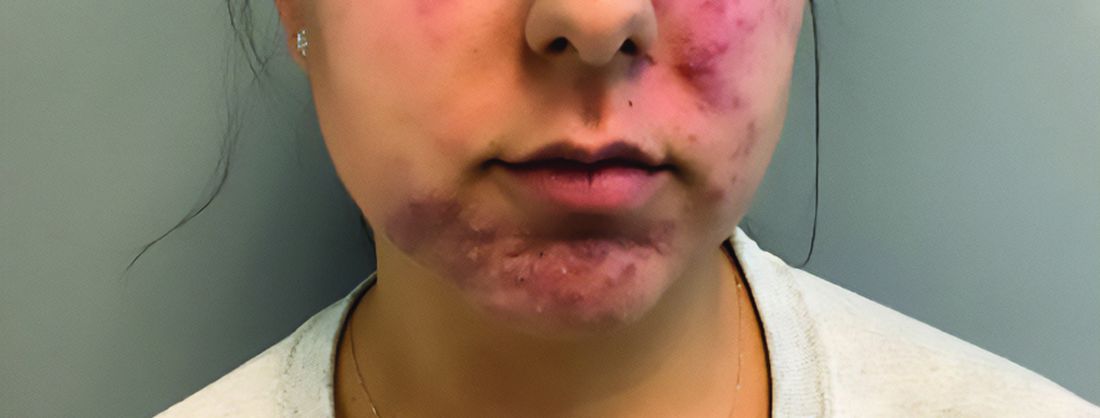
Pruritic, violaceous papules in a patient with renal cell carcinoma
Pembrolizumab (Keytruda) is a programmed cell death protein 1 (PD-1) blocking antibody used to treat different malignancies including melanoma, non–small cell lung cancer, and other advanced solid tumors and hematologic malignancies. and drug rash with eosinophilia and systemic symptoms (DRESS).
Lichen planus-like adverse drug reactions, as seen in this patient, are also referred to as lichenoid drug eruption or drug-induced lichen planus. This cutaneous reaction is one of the more rare side effects of pembrolizumab. It should be noted that in lichenoid reactions, keratinocytes expressing PD-L1 are particularly affected, leading to a dense CD4/CD8 positive lymphocytic infiltration in the basal layer, necrosis of keratinocytes, acanthosis, and hypergranulosis. Subsequently, the cutaneous adverse reaction is a target effect of the PD-1/PD-L1 pathway and not a general hypersensitivity reaction. Clinically, both lichen planus and lichenoid drug eruptions exhibit erythematous papules and plaques. Lichenoid drug eruptions, however, can be scaly, pruritic, and heal with more hyperpigmentation.
A skin biopsy revealed irregular epidermal hyperplasia with jagged rete ridges. Within the dermis, there was a lichenoid inflammatory cell infiltrate obscuring the dermal-epidermal junction. The inflammatory cell infiltrate contained lymphocytes, histiocytes, and eosinophils. A diagnosis of a lichen planus-like adverse drug reaction to pembrolizumab was favored.
If the reaction is mild, topical corticosteroids and oral antihistamines can help with the drug-induced lichen planus. For more severe cases, systemic steroids can be given to help ease the reaction. Physicians should be aware of potential adverse drug effects that can mimic other medical conditions.
The case and photo were submitted by Ms. Towe, Nova Southeastern University College of Osteopathic Medicine, Davie, Florida, and Dr. Berke, Three Rivers Dermatology, Coraopolis, Pennsylvania. The column was edited by Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Bansal A et al. Indian Dermatol Online J. 2023 Apr 4;14(3):391-4. doi: 10.4103/idoj.idoj_377_22.
Sethi A, Raj M. Cureus. 2021 Mar 8;13(3):e13768. doi: 10.7759/cureus.13768.
Pembrolizumab (Keytruda) is a programmed cell death protein 1 (PD-1) blocking antibody used to treat different malignancies including melanoma, non–small cell lung cancer, and other advanced solid tumors and hematologic malignancies. and drug rash with eosinophilia and systemic symptoms (DRESS).
Lichen planus-like adverse drug reactions, as seen in this patient, are also referred to as lichenoid drug eruption or drug-induced lichen planus. This cutaneous reaction is one of the more rare side effects of pembrolizumab. It should be noted that in lichenoid reactions, keratinocytes expressing PD-L1 are particularly affected, leading to a dense CD4/CD8 positive lymphocytic infiltration in the basal layer, necrosis of keratinocytes, acanthosis, and hypergranulosis. Subsequently, the cutaneous adverse reaction is a target effect of the PD-1/PD-L1 pathway and not a general hypersensitivity reaction. Clinically, both lichen planus and lichenoid drug eruptions exhibit erythematous papules and plaques. Lichenoid drug eruptions, however, can be scaly, pruritic, and heal with more hyperpigmentation.
A skin biopsy revealed irregular epidermal hyperplasia with jagged rete ridges. Within the dermis, there was a lichenoid inflammatory cell infiltrate obscuring the dermal-epidermal junction. The inflammatory cell infiltrate contained lymphocytes, histiocytes, and eosinophils. A diagnosis of a lichen planus-like adverse drug reaction to pembrolizumab was favored.
If the reaction is mild, topical corticosteroids and oral antihistamines can help with the drug-induced lichen planus. For more severe cases, systemic steroids can be given to help ease the reaction. Physicians should be aware of potential adverse drug effects that can mimic other medical conditions.
The case and photo were submitted by Ms. Towe, Nova Southeastern University College of Osteopathic Medicine, Davie, Florida, and Dr. Berke, Three Rivers Dermatology, Coraopolis, Pennsylvania. The column was edited by Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Bansal A et al. Indian Dermatol Online J. 2023 Apr 4;14(3):391-4. doi: 10.4103/idoj.idoj_377_22.
Sethi A, Raj M. Cureus. 2021 Mar 8;13(3):e13768. doi: 10.7759/cureus.13768.
Pembrolizumab (Keytruda) is a programmed cell death protein 1 (PD-1) blocking antibody used to treat different malignancies including melanoma, non–small cell lung cancer, and other advanced solid tumors and hematologic malignancies. and drug rash with eosinophilia and systemic symptoms (DRESS).
Lichen planus-like adverse drug reactions, as seen in this patient, are also referred to as lichenoid drug eruption or drug-induced lichen planus. This cutaneous reaction is one of the more rare side effects of pembrolizumab. It should be noted that in lichenoid reactions, keratinocytes expressing PD-L1 are particularly affected, leading to a dense CD4/CD8 positive lymphocytic infiltration in the basal layer, necrosis of keratinocytes, acanthosis, and hypergranulosis. Subsequently, the cutaneous adverse reaction is a target effect of the PD-1/PD-L1 pathway and not a general hypersensitivity reaction. Clinically, both lichen planus and lichenoid drug eruptions exhibit erythematous papules and plaques. Lichenoid drug eruptions, however, can be scaly, pruritic, and heal with more hyperpigmentation.
A skin biopsy revealed irregular epidermal hyperplasia with jagged rete ridges. Within the dermis, there was a lichenoid inflammatory cell infiltrate obscuring the dermal-epidermal junction. The inflammatory cell infiltrate contained lymphocytes, histiocytes, and eosinophils. A diagnosis of a lichen planus-like adverse drug reaction to pembrolizumab was favored.
If the reaction is mild, topical corticosteroids and oral antihistamines can help with the drug-induced lichen planus. For more severe cases, systemic steroids can be given to help ease the reaction. Physicians should be aware of potential adverse drug effects that can mimic other medical conditions.
The case and photo were submitted by Ms. Towe, Nova Southeastern University College of Osteopathic Medicine, Davie, Florida, and Dr. Berke, Three Rivers Dermatology, Coraopolis, Pennsylvania. The column was edited by Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Bansal A et al. Indian Dermatol Online J. 2023 Apr 4;14(3):391-4. doi: 10.4103/idoj.idoj_377_22.
Sethi A, Raj M. Cureus. 2021 Mar 8;13(3):e13768. doi: 10.7759/cureus.13768.