User login
President to nominate oncologist to lead FDA
Stephen M. Hahn, MD, a radiation oncologist and researcher, may soon take the reins of the Food and Drug Administration.

President Trump indicated his intent to nominate Dr. Hahn as FDA Commissioner in a brief Nov.1 statement that outlined Dr. Hahn’s background. Dr. Hahn currently serves as chief medical executive at MD Anderson Cancer Center, Houston, where he heads the radiology oncology division.
Dr. Hahn specializes in treating lung cancer and sarcoma and has authored 220 peer-reviewed original research articles, according to his biography. He was previously chair of the department of radiology oncology at the University of Pennsylvania, Philadelphia, and also served as a senior investigator at the National Cancer Institute.
Dr. Hahn completed his residency in radiation oncology at NCI and his residency in internal medicine at the University of California, San Francisco.
Margaret Foti, PhD, chief executive officer for the American Association for Cancer Research called Dr. Hahn a renowned expert in radiation oncology and research, an experienced and highly effective administrator, and an innovative leader.
“I have seen firsthand Dr. Hahn’s extraordinary dedication and commitment to cancer patients, and the AACR is extremely confident that he will be an outstanding leader for the FDA,” Dr. Foti said in a statement. “Dr. Hahn, who is board certified in both radiation and medical oncology, is esteemed for the breadth and depth of his scientific knowledge and expertise, and he has consistently advocated for a drug review process at the FDA that is both science-directed and patient-focused.”
The American Society of Clinical Oncology also congratulated Dr. Hahn on the upcoming nomination, noting that he has a strong grasp of the drug development process and understands the realities of working in a complex clinical care environment.
“The role of FDA commissioner requires a strong commitment to advancing the agency’s mission to protect public health across the United States, and an understanding of how to help speed innovations to get new treatments to patients, while also ensuring the safety and efficacy of the medical products that millions of Americans rely on to manage, treat, and cure their cancer,” the society stated. “ASCO has a long and productive history of collaborating with FDA, including with current acting Commissioner Norman E. “Ned” Sharpless, MD, in support of the agency’s important role in reducing cancer incidence, advancing treatment options, and improving the lives of individuals with cancer. We look forward to continuing our close collaboration to make it possible for every American with cancer to have access to medical products that are safe and effective.”
Dr. Sharpless will return to his position as NCI director; he served as interim FDA commissioner from the April departure of then-FDA commissioner, Scott Gottlieb, MD.
“As one of the nation’s leading oncologists who has devoted his entire professional career to helping patients in the fight against cancer, Ned is returning home to NCI to continue this work and we look forward to working closely with him once again,” Francis S. Collins, MD, director of the National Institutes of Health, said in a statement. “I want to thank Dr. Doug Lowy, principal deputy director of NCI, for having stepped in, once again, to take the helm at NCI and lead the institute so skillfully while Ned was at FDA.”
At press time, neither Dr. Hahn nor MD Anderson Cancer Center had returned messages seeking comment about his nomination.
Stephen M. Hahn, MD, a radiation oncologist and researcher, may soon take the reins of the Food and Drug Administration.
President Trump indicated his intent to nominate Dr. Hahn as FDA Commissioner in a brief Nov.1 statement that outlined Dr. Hahn’s background. Dr. Hahn currently serves as chief medical executive at MD Anderson Cancer Center, Houston, where he heads the radiology oncology division.
Dr. Hahn specializes in treating lung cancer and sarcoma and has authored 220 peer-reviewed original research articles, according to his biography. He was previously chair of the department of radiology oncology at the University of Pennsylvania, Philadelphia, and also served as a senior investigator at the National Cancer Institute.
Dr. Hahn completed his residency in radiation oncology at NCI and his residency in internal medicine at the University of California, San Francisco.
Margaret Foti, PhD, chief executive officer for the American Association for Cancer Research called Dr. Hahn a renowned expert in radiation oncology and research, an experienced and highly effective administrator, and an innovative leader.
“I have seen firsthand Dr. Hahn’s extraordinary dedication and commitment to cancer patients, and the AACR is extremely confident that he will be an outstanding leader for the FDA,” Dr. Foti said in a statement. “Dr. Hahn, who is board certified in both radiation and medical oncology, is esteemed for the breadth and depth of his scientific knowledge and expertise, and he has consistently advocated for a drug review process at the FDA that is both science-directed and patient-focused.”
The American Society of Clinical Oncology also congratulated Dr. Hahn on the upcoming nomination, noting that he has a strong grasp of the drug development process and understands the realities of working in a complex clinical care environment.
“The role of FDA commissioner requires a strong commitment to advancing the agency’s mission to protect public health across the United States, and an understanding of how to help speed innovations to get new treatments to patients, while also ensuring the safety and efficacy of the medical products that millions of Americans rely on to manage, treat, and cure their cancer,” the society stated. “ASCO has a long and productive history of collaborating with FDA, including with current acting Commissioner Norman E. “Ned” Sharpless, MD, in support of the agency’s important role in reducing cancer incidence, advancing treatment options, and improving the lives of individuals with cancer. We look forward to continuing our close collaboration to make it possible for every American with cancer to have access to medical products that are safe and effective.”
Dr. Sharpless will return to his position as NCI director; he served as interim FDA commissioner from the April departure of then-FDA commissioner, Scott Gottlieb, MD.
“As one of the nation’s leading oncologists who has devoted his entire professional career to helping patients in the fight against cancer, Ned is returning home to NCI to continue this work and we look forward to working closely with him once again,” Francis S. Collins, MD, director of the National Institutes of Health, said in a statement. “I want to thank Dr. Doug Lowy, principal deputy director of NCI, for having stepped in, once again, to take the helm at NCI and lead the institute so skillfully while Ned was at FDA.”
At press time, neither Dr. Hahn nor MD Anderson Cancer Center had returned messages seeking comment about his nomination.
Stephen M. Hahn, MD, a radiation oncologist and researcher, may soon take the reins of the Food and Drug Administration.
President Trump indicated his intent to nominate Dr. Hahn as FDA Commissioner in a brief Nov.1 statement that outlined Dr. Hahn’s background. Dr. Hahn currently serves as chief medical executive at MD Anderson Cancer Center, Houston, where he heads the radiology oncology division.
Dr. Hahn specializes in treating lung cancer and sarcoma and has authored 220 peer-reviewed original research articles, according to his biography. He was previously chair of the department of radiology oncology at the University of Pennsylvania, Philadelphia, and also served as a senior investigator at the National Cancer Institute.
Dr. Hahn completed his residency in radiation oncology at NCI and his residency in internal medicine at the University of California, San Francisco.
Margaret Foti, PhD, chief executive officer for the American Association for Cancer Research called Dr. Hahn a renowned expert in radiation oncology and research, an experienced and highly effective administrator, and an innovative leader.
“I have seen firsthand Dr. Hahn’s extraordinary dedication and commitment to cancer patients, and the AACR is extremely confident that he will be an outstanding leader for the FDA,” Dr. Foti said in a statement. “Dr. Hahn, who is board certified in both radiation and medical oncology, is esteemed for the breadth and depth of his scientific knowledge and expertise, and he has consistently advocated for a drug review process at the FDA that is both science-directed and patient-focused.”
The American Society of Clinical Oncology also congratulated Dr. Hahn on the upcoming nomination, noting that he has a strong grasp of the drug development process and understands the realities of working in a complex clinical care environment.
“The role of FDA commissioner requires a strong commitment to advancing the agency’s mission to protect public health across the United States, and an understanding of how to help speed innovations to get new treatments to patients, while also ensuring the safety and efficacy of the medical products that millions of Americans rely on to manage, treat, and cure their cancer,” the society stated. “ASCO has a long and productive history of collaborating with FDA, including with current acting Commissioner Norman E. “Ned” Sharpless, MD, in support of the agency’s important role in reducing cancer incidence, advancing treatment options, and improving the lives of individuals with cancer. We look forward to continuing our close collaboration to make it possible for every American with cancer to have access to medical products that are safe and effective.”
Dr. Sharpless will return to his position as NCI director; he served as interim FDA commissioner from the April departure of then-FDA commissioner, Scott Gottlieb, MD.
“As one of the nation’s leading oncologists who has devoted his entire professional career to helping patients in the fight against cancer, Ned is returning home to NCI to continue this work and we look forward to working closely with him once again,” Francis S. Collins, MD, director of the National Institutes of Health, said in a statement. “I want to thank Dr. Doug Lowy, principal deputy director of NCI, for having stepped in, once again, to take the helm at NCI and lead the institute so skillfully while Ned was at FDA.”
At press time, neither Dr. Hahn nor MD Anderson Cancer Center had returned messages seeking comment about his nomination.
Triple-drug therapy proves effective in CF patients with most common mutation
Reinforcing previous findings, a new study has determined that the next-generation corrector elexacaftor, in combination with tezacaftor and ivacaftor, can effectively treat patients with Phe508del-minimal function genotypes who did not respond to previous cystic fibrosis transmembrane conductance regulator (CFTR) modulator regimens.
“These results provide evidence that , thus addressing the underlying cause of disease in the large majority of patients,” wrote Peter G. Middleton, PhD, of the University of Sydney (Australia) and his coauthors. The study was published in the New England Journal of Medicine.
To further determine if the elexacaftor-tezacaftor-ivacaftor regimen was effective and safe, the researchers launched a randomized, placebo-controlled phase 3 trial of 403 cystic fibrosis patients age 12 or older who had a single Phe508del allele. Patients in the combination group (n = 200) received 200 mg of elexacaftor once daily, 100 mg of tezacaftor once daily, and 150 mg of ivacaftor every 12 hours for 24 weeks. Patients in the other group (n = 203) received matched placebos.
At 14 weeks, patients in the combination group had a change in percentage of predicted forced expiratory volume in 1 second (FEV1) that was 13.8 points higher than the placebo group (95% confidence interval, 12.1-15.4, P less than .001). At 24 weeks, the combination group had a predicted FEV1 difference that was 14.3 percentage points higher (95% confidence interval, 12.7-15.8, P less than .001). The rate of pulmonary exacerbations was 63% lower (rate ratio 0.37; 95% CI, 0.25-0.55, P less than .001) and sweat chloride concentration was 41.8 mmol/L lower (95% CI, –44.4 to –39.3, P less than .001) in the combination group through 24 weeks.
At least one adverse event occurred in 93.1% of patients in the combination group and 96% of patients in the placebo group. Serious adverse events occurred in 28 patients (13.9%) in the combination group and 42 patients (20.9%) in the placebo group. There were no deaths in either group.
The study was funded by Vertex Pharmaceuticals. The authors had disclosures, including receiving personal fees and grants from various pharmaceutical companies and being on the advisory board, owning stock, or being an employee of Vertex Pharmaceuticals.
SOURCE: Middleton PG et al. 2019 Oct 31. N Engl J Med. doi: 10.1056/NEJMoa1908639.
After 30 years, new research from Middleton et al. and others appears to be the breakthrough we’ve been waiting for in treating cystic fibrosis, wrote Francis S. Collins, MD, PhD, of the National Institutes of Health in an accompanying editorial (N Engl J Med. 2019 Oct 31. doi: 10.1056/NEJMe1911602).
As one of the researchers who discovered the cystic fibrosis gene, he acknowledged the 3 decades of work that followed their discovery and the excitement that comes from being able to counter the common Phe508del CFTR mutation that afflicts so many cystic fibrosis patients. “These findings indicate that it may soon be possible to offer safe and effective molecularly targeted therapies to 90% of persons with cystic fibrosis,” he wrote.
“Yet we must not abandon the patients with cystic fibrosis who have null mutations and will not have a response to these drugs,” he added, noting that those challenges remain “substantial” and potentially will involve in vivo somatic-cell gene editing of airway epithelial cells. That said, what once was a dream 30 years ago now appears to be a reality.
Dr. Collins reported being a coinventor of the original patents on the CFTR gene, for which he donated all royalties to the Cystic Fibrosis Foundation.
After 30 years, new research from Middleton et al. and others appears to be the breakthrough we’ve been waiting for in treating cystic fibrosis, wrote Francis S. Collins, MD, PhD, of the National Institutes of Health in an accompanying editorial (N Engl J Med. 2019 Oct 31. doi: 10.1056/NEJMe1911602).
As one of the researchers who discovered the cystic fibrosis gene, he acknowledged the 3 decades of work that followed their discovery and the excitement that comes from being able to counter the common Phe508del CFTR mutation that afflicts so many cystic fibrosis patients. “These findings indicate that it may soon be possible to offer safe and effective molecularly targeted therapies to 90% of persons with cystic fibrosis,” he wrote.
“Yet we must not abandon the patients with cystic fibrosis who have null mutations and will not have a response to these drugs,” he added, noting that those challenges remain “substantial” and potentially will involve in vivo somatic-cell gene editing of airway epithelial cells. That said, what once was a dream 30 years ago now appears to be a reality.
Dr. Collins reported being a coinventor of the original patents on the CFTR gene, for which he donated all royalties to the Cystic Fibrosis Foundation.
After 30 years, new research from Middleton et al. and others appears to be the breakthrough we’ve been waiting for in treating cystic fibrosis, wrote Francis S. Collins, MD, PhD, of the National Institutes of Health in an accompanying editorial (N Engl J Med. 2019 Oct 31. doi: 10.1056/NEJMe1911602).
As one of the researchers who discovered the cystic fibrosis gene, he acknowledged the 3 decades of work that followed their discovery and the excitement that comes from being able to counter the common Phe508del CFTR mutation that afflicts so many cystic fibrosis patients. “These findings indicate that it may soon be possible to offer safe and effective molecularly targeted therapies to 90% of persons with cystic fibrosis,” he wrote.
“Yet we must not abandon the patients with cystic fibrosis who have null mutations and will not have a response to these drugs,” he added, noting that those challenges remain “substantial” and potentially will involve in vivo somatic-cell gene editing of airway epithelial cells. That said, what once was a dream 30 years ago now appears to be a reality.
Dr. Collins reported being a coinventor of the original patents on the CFTR gene, for which he donated all royalties to the Cystic Fibrosis Foundation.
Reinforcing previous findings, a new study has determined that the next-generation corrector elexacaftor, in combination with tezacaftor and ivacaftor, can effectively treat patients with Phe508del-minimal function genotypes who did not respond to previous cystic fibrosis transmembrane conductance regulator (CFTR) modulator regimens.
“These results provide evidence that , thus addressing the underlying cause of disease in the large majority of patients,” wrote Peter G. Middleton, PhD, of the University of Sydney (Australia) and his coauthors. The study was published in the New England Journal of Medicine.
To further determine if the elexacaftor-tezacaftor-ivacaftor regimen was effective and safe, the researchers launched a randomized, placebo-controlled phase 3 trial of 403 cystic fibrosis patients age 12 or older who had a single Phe508del allele. Patients in the combination group (n = 200) received 200 mg of elexacaftor once daily, 100 mg of tezacaftor once daily, and 150 mg of ivacaftor every 12 hours for 24 weeks. Patients in the other group (n = 203) received matched placebos.
At 14 weeks, patients in the combination group had a change in percentage of predicted forced expiratory volume in 1 second (FEV1) that was 13.8 points higher than the placebo group (95% confidence interval, 12.1-15.4, P less than .001). At 24 weeks, the combination group had a predicted FEV1 difference that was 14.3 percentage points higher (95% confidence interval, 12.7-15.8, P less than .001). The rate of pulmonary exacerbations was 63% lower (rate ratio 0.37; 95% CI, 0.25-0.55, P less than .001) and sweat chloride concentration was 41.8 mmol/L lower (95% CI, –44.4 to –39.3, P less than .001) in the combination group through 24 weeks.
At least one adverse event occurred in 93.1% of patients in the combination group and 96% of patients in the placebo group. Serious adverse events occurred in 28 patients (13.9%) in the combination group and 42 patients (20.9%) in the placebo group. There were no deaths in either group.
The study was funded by Vertex Pharmaceuticals. The authors had disclosures, including receiving personal fees and grants from various pharmaceutical companies and being on the advisory board, owning stock, or being an employee of Vertex Pharmaceuticals.
SOURCE: Middleton PG et al. 2019 Oct 31. N Engl J Med. doi: 10.1056/NEJMoa1908639.
Reinforcing previous findings, a new study has determined that the next-generation corrector elexacaftor, in combination with tezacaftor and ivacaftor, can effectively treat patients with Phe508del-minimal function genotypes who did not respond to previous cystic fibrosis transmembrane conductance regulator (CFTR) modulator regimens.
“These results provide evidence that , thus addressing the underlying cause of disease in the large majority of patients,” wrote Peter G. Middleton, PhD, of the University of Sydney (Australia) and his coauthors. The study was published in the New England Journal of Medicine.
To further determine if the elexacaftor-tezacaftor-ivacaftor regimen was effective and safe, the researchers launched a randomized, placebo-controlled phase 3 trial of 403 cystic fibrosis patients age 12 or older who had a single Phe508del allele. Patients in the combination group (n = 200) received 200 mg of elexacaftor once daily, 100 mg of tezacaftor once daily, and 150 mg of ivacaftor every 12 hours for 24 weeks. Patients in the other group (n = 203) received matched placebos.
At 14 weeks, patients in the combination group had a change in percentage of predicted forced expiratory volume in 1 second (FEV1) that was 13.8 points higher than the placebo group (95% confidence interval, 12.1-15.4, P less than .001). At 24 weeks, the combination group had a predicted FEV1 difference that was 14.3 percentage points higher (95% confidence interval, 12.7-15.8, P less than .001). The rate of pulmonary exacerbations was 63% lower (rate ratio 0.37; 95% CI, 0.25-0.55, P less than .001) and sweat chloride concentration was 41.8 mmol/L lower (95% CI, –44.4 to –39.3, P less than .001) in the combination group through 24 weeks.
At least one adverse event occurred in 93.1% of patients in the combination group and 96% of patients in the placebo group. Serious adverse events occurred in 28 patients (13.9%) in the combination group and 42 patients (20.9%) in the placebo group. There were no deaths in either group.
The study was funded by Vertex Pharmaceuticals. The authors had disclosures, including receiving personal fees and grants from various pharmaceutical companies and being on the advisory board, owning stock, or being an employee of Vertex Pharmaceuticals.
SOURCE: Middleton PG et al. 2019 Oct 31. N Engl J Med. doi: 10.1056/NEJMoa1908639.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Vaping-linked lung injury cases near 1,900
according to the latest update provided by the Centers for Disease Control and Prevention. Thirty-seven deaths have been confirmed.
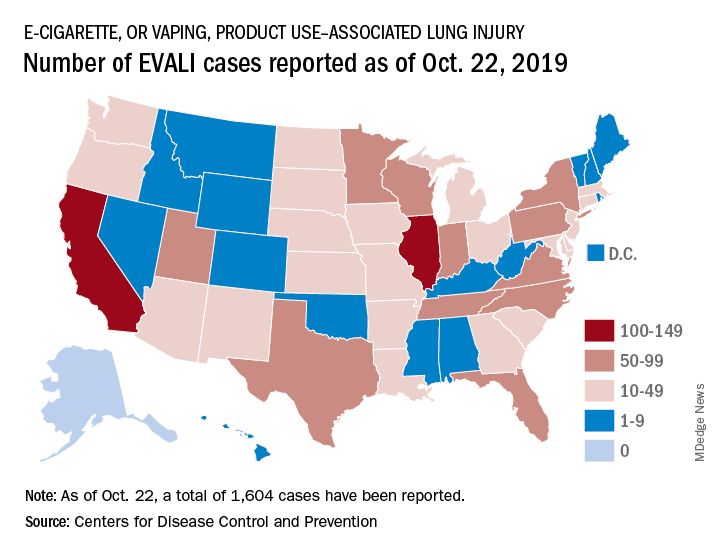
Deaths have occurred in 24 states and the District of Columbia: Alabama, California (3), Connecticut, Delaware, Florida, Georgia (3), Illinois (2), Indiana (3), Kansas (2), Massachusetts, Michigan, Minnesota (3), Mississippi, Missouri, Montana, Nebraska, New Jersey, New York, Oregon (2), Pennsylvania, Tennessee (2), Texas, Utah, and Virginia. As on Oct. 28, the median age of deceased patients was 49 years and ranged from 17 to 75 years.
The CDC is now doing additional testing on available samples for chemical in the bronchoalveolar lavage fluid, blood, or urine, as well as lung biopsy or autopsy specimens. It also is validating methods for aerosol emission testing of case-associated product samples from vaping products and e-liquids.
For more information and resources visit For the Public, For Healthcare Providers, and For State and Local Health Departments pages, as well as the CDC’s Publications and Resources page.
according to the latest update provided by the Centers for Disease Control and Prevention. Thirty-seven deaths have been confirmed.
Deaths have occurred in 24 states and the District of Columbia: Alabama, California (3), Connecticut, Delaware, Florida, Georgia (3), Illinois (2), Indiana (3), Kansas (2), Massachusetts, Michigan, Minnesota (3), Mississippi, Missouri, Montana, Nebraska, New Jersey, New York, Oregon (2), Pennsylvania, Tennessee (2), Texas, Utah, and Virginia. As on Oct. 28, the median age of deceased patients was 49 years and ranged from 17 to 75 years.
The CDC is now doing additional testing on available samples for chemical in the bronchoalveolar lavage fluid, blood, or urine, as well as lung biopsy or autopsy specimens. It also is validating methods for aerosol emission testing of case-associated product samples from vaping products and e-liquids.
For more information and resources visit For the Public, For Healthcare Providers, and For State and Local Health Departments pages, as well as the CDC’s Publications and Resources page.
according to the latest update provided by the Centers for Disease Control and Prevention. Thirty-seven deaths have been confirmed.
Deaths have occurred in 24 states and the District of Columbia: Alabama, California (3), Connecticut, Delaware, Florida, Georgia (3), Illinois (2), Indiana (3), Kansas (2), Massachusetts, Michigan, Minnesota (3), Mississippi, Missouri, Montana, Nebraska, New Jersey, New York, Oregon (2), Pennsylvania, Tennessee (2), Texas, Utah, and Virginia. As on Oct. 28, the median age of deceased patients was 49 years and ranged from 17 to 75 years.
The CDC is now doing additional testing on available samples for chemical in the bronchoalveolar lavage fluid, blood, or urine, as well as lung biopsy or autopsy specimens. It also is validating methods for aerosol emission testing of case-associated product samples from vaping products and e-liquids.
For more information and resources visit For the Public, For Healthcare Providers, and For State and Local Health Departments pages, as well as the CDC’s Publications and Resources page.
2019 at a glance: Hem-onc U.S. drug approvals
The rapid development and identification of novel drugs has translated into innovative therapies in hematology and oncology. The aim of this piece is to present newly approved drugs and expanded indications to serve as a reference guide for practicing clinicians.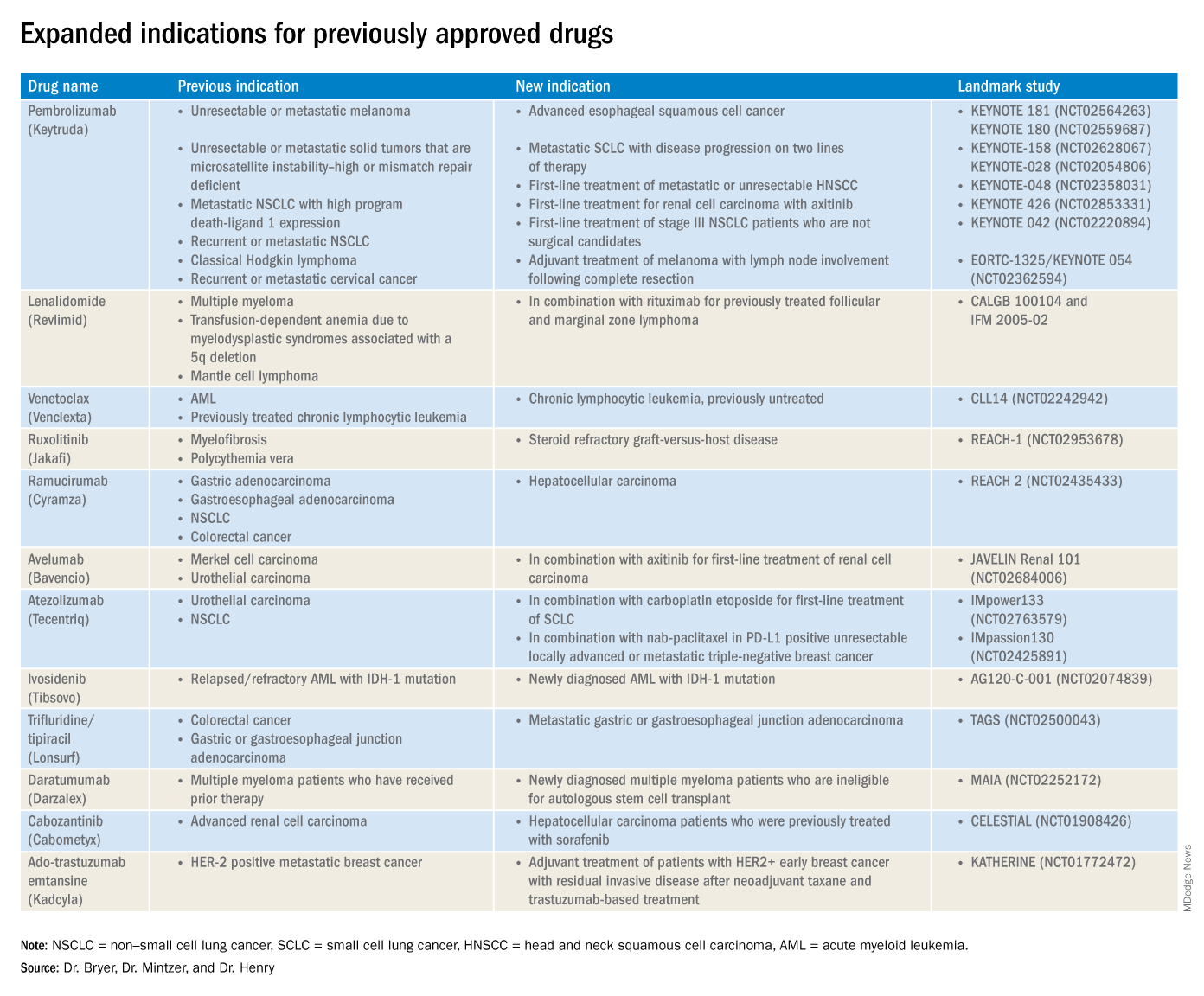
This article reviews therapies that were newly approved so far in 2019, as well as those previously approved whose indications were expanded this past year. The list highlights the most clinically important approvals, as well as adverse events that are unique or especially severe.
New approvals
Fedratinib (Inrebic)
Class: JAK2 and FLT3 selective kinase inhibitor.
Disease: Intermediate or high-risk primary or secondary (postpolycythemia vera or postessential thrombocythemia) myelofibrosis.
Dose: 400 mg orally once daily, with or without food.
Adverse events (AEs): Black box warning: Fatal encephalopathy, including Wernicke’s (thiamine level monitoring suggested).
Trials: In JAKARTA (NCT01437787), 37% of patients achieved a 35% or greater reduction in spleen volume and 40% received a 50% or greater reduction in myelofibrosis-related symptoms. In Jakarta-2, there was a 55% spleen response in patients resistant or intolerant to ruxolitinib.
Entrectinib (Rozlytrek)
Class: Tropomyosin receptor tyrosine kinase inhibitor.
Disease: Solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion and for ROS-1 positive non–small cell lung cancer (NSCLC).
Dose: 600 mg orally once daily.
AEs: Heart failure, QT prolongation, skeletal fractures, hepatotoxicity, central nervous system effects, and hyperuricemia.
Trial: ALKA, STARTRK-1 (NCT02097810) and STARTRK-2 (NCT02568267): Overall response rate of 57% for NTRK positive patients; response rate of 77% in ROS-1 positive NSCLC.
Pexidartinib (Turalio)
Class: Small molecule tyrosine kinase inhibitor targeting CSF1R.
Disease: Symptomatic tenosynovial giant cell tumor.
Dose: 400 mg orally twice daily without food.
AEs: Black box warning on hepatotoxicity.
Trial: ENLIVEN (NCT02371369): Overall response rate of 38% at 25 weeks, with a 15% complete response rate and a 23% partial response rate.
Darolutamide (Nubeqa)
Class: Androgen receptor inhibitor.
Disease: Nonmetastatic castration-resistant prostate cancer.
Dose: 600 mg orally twice daily with food with concomitant androgen deprivation therapy.
AEs: Fatigue, extremity pain, and rash.
Trial: ARAMIS (NCT02200614): Median metastasis free survival was 40.4 months for patients with darolutamide, compared with 18.4 months for controls.
Selinexor (Xpovio)
Class: Reversible inhibitor of nuclear export of tumor suppressor proteins, growth regulators, and mRNAs of oncogenic proteins.
Disease: Relapsed or refractory multiple myeloma. Indicated for patients who have received at least four prior therapies, including at least two immunomodulatory agents and an anti-CD38 monoclonal antibody.
Dose: 80 mg orally in combination with oral dexamethasone on days 1 and 3 of each week.
AEs: Thrombocytopenia, fatigue, pancytopenia, and hyponatremia.
Trial: STORM (NCT02336815): Overall response rate 25.3% with a median time to first response of 4 weeks and 3.8-month median duration of response.
Polatuzumab vedotin-piiq (Polivy)
Class: CD79b-directed antibody-drug conjugate.
Disease: Relapsed or refractory diffuse large B-cell lymphoma. Indicated for patients who have had at least two prior therapies.
Dose: 1.8 mg/kg intravenous infusion every 21 days for six cycles in combination with bendamustine and a rituximab product.
AEs: Pancytopenia, peripheral neuropathy.
Trial: GO29365 (NCT02257567): Complete response rate was 40% for polatuzumab vedotin-piiq plus bendamustine/rituximab, compared with 18% with bendamustine/rituximab alone.*
Caplacizumab-yhdp (Cablivi)
Class: Monoclonal antibody fragment directed against von Willebrand factor.
Disease: Thrombotic thrombocytopenic purpura.
Dose: 11 mg IV initially, then daily subcutaneously; in combination with plasma exchange and immunosuppressive therapy.
AEs: Epistaxis, headache, and gingival bleeding.
Trial: Hercules trial (NCT02553317): More rapid normalization of platelets, lower incidence of composite TTP-related death, and lower rate of recurrence when added to plasma exchange and steroids.
Alpelisib (Piqray)
Class: Phosphatidylinositol-3-kinase (PI3K) inhibitor.
Disease: Hormone receptor positive HER2-negative PIK3CA-mutated, advanced or metastatic breast cancer.
Dose: 300 mg orally once daily with food with concomitant fulvestrant.
AEs: Hyperglycemia, pancytopenia.
Trial: SOLAR-1 (NCT02437318): 11-month progression-free survival among patients treated with alpelisib and fulvestrant, compared with 5.7 months in fulvestrant alone control arm; overall response rate of 36% versus 16%, respectively.
Erdafitinib (Balversa)
Class: Fibroblast growth factor receptor kinase inhibitor.
Disease: Locally advanced or metastatic urothelial carcinoma with FGFR3 or FGFR2 mutations.
Dose: 8 mg orally once daily, with or without food.
AEs: Ocular disorders including retinopathy or retinal detachment.
Trial: BLC2001 (NCT02365597): Objective response rate of 32.2%, with a complete response in 2.3% of patients and partial response in 29.9% of patients.
Biosimilar approvals
Trastuzumab and hyaluronidase-oysk (Herceptin Hylecta)
Biosimilar to: Trastuzumab.
Indication: HER2-overexpressing breast cancer.
Dr. Bryer is a resident in the department of internal medicine at the University of Pennsylvania, Philadelphia. Dr. Mintzer is chief of hematology-oncology at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania. Dr. Henry is a hematologist-oncologist at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania.
*Correction, 11/7/2019: An earlier version of this article misstated the drug combination in the GO29365 trial.
The rapid development and identification of novel drugs has translated into innovative therapies in hematology and oncology. The aim of this piece is to present newly approved drugs and expanded indications to serve as a reference guide for practicing clinicians.
This article reviews therapies that were newly approved so far in 2019, as well as those previously approved whose indications were expanded this past year. The list highlights the most clinically important approvals, as well as adverse events that are unique or especially severe.
New approvals
Fedratinib (Inrebic)
Class: JAK2 and FLT3 selective kinase inhibitor.
Disease: Intermediate or high-risk primary or secondary (postpolycythemia vera or postessential thrombocythemia) myelofibrosis.
Dose: 400 mg orally once daily, with or without food.
Adverse events (AEs): Black box warning: Fatal encephalopathy, including Wernicke’s (thiamine level monitoring suggested).
Trials: In JAKARTA (NCT01437787), 37% of patients achieved a 35% or greater reduction in spleen volume and 40% received a 50% or greater reduction in myelofibrosis-related symptoms. In Jakarta-2, there was a 55% spleen response in patients resistant or intolerant to ruxolitinib.
Entrectinib (Rozlytrek)
Class: Tropomyosin receptor tyrosine kinase inhibitor.
Disease: Solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion and for ROS-1 positive non–small cell lung cancer (NSCLC).
Dose: 600 mg orally once daily.
AEs: Heart failure, QT prolongation, skeletal fractures, hepatotoxicity, central nervous system effects, and hyperuricemia.
Trial: ALKA, STARTRK-1 (NCT02097810) and STARTRK-2 (NCT02568267): Overall response rate of 57% for NTRK positive patients; response rate of 77% in ROS-1 positive NSCLC.
Pexidartinib (Turalio)
Class: Small molecule tyrosine kinase inhibitor targeting CSF1R.
Disease: Symptomatic tenosynovial giant cell tumor.
Dose: 400 mg orally twice daily without food.
AEs: Black box warning on hepatotoxicity.
Trial: ENLIVEN (NCT02371369): Overall response rate of 38% at 25 weeks, with a 15% complete response rate and a 23% partial response rate.
Darolutamide (Nubeqa)
Class: Androgen receptor inhibitor.
Disease: Nonmetastatic castration-resistant prostate cancer.
Dose: 600 mg orally twice daily with food with concomitant androgen deprivation therapy.
AEs: Fatigue, extremity pain, and rash.
Trial: ARAMIS (NCT02200614): Median metastasis free survival was 40.4 months for patients with darolutamide, compared with 18.4 months for controls.
Selinexor (Xpovio)
Class: Reversible inhibitor of nuclear export of tumor suppressor proteins, growth regulators, and mRNAs of oncogenic proteins.
Disease: Relapsed or refractory multiple myeloma. Indicated for patients who have received at least four prior therapies, including at least two immunomodulatory agents and an anti-CD38 monoclonal antibody.
Dose: 80 mg orally in combination with oral dexamethasone on days 1 and 3 of each week.
AEs: Thrombocytopenia, fatigue, pancytopenia, and hyponatremia.
Trial: STORM (NCT02336815): Overall response rate 25.3% with a median time to first response of 4 weeks and 3.8-month median duration of response.
Polatuzumab vedotin-piiq (Polivy)
Class: CD79b-directed antibody-drug conjugate.
Disease: Relapsed or refractory diffuse large B-cell lymphoma. Indicated for patients who have had at least two prior therapies.
Dose: 1.8 mg/kg intravenous infusion every 21 days for six cycles in combination with bendamustine and a rituximab product.
AEs: Pancytopenia, peripheral neuropathy.
Trial: GO29365 (NCT02257567): Complete response rate was 40% for polatuzumab vedotin-piiq plus bendamustine/rituximab, compared with 18% with bendamustine/rituximab alone.*
Caplacizumab-yhdp (Cablivi)
Class: Monoclonal antibody fragment directed against von Willebrand factor.
Disease: Thrombotic thrombocytopenic purpura.
Dose: 11 mg IV initially, then daily subcutaneously; in combination with plasma exchange and immunosuppressive therapy.
AEs: Epistaxis, headache, and gingival bleeding.
Trial: Hercules trial (NCT02553317): More rapid normalization of platelets, lower incidence of composite TTP-related death, and lower rate of recurrence when added to plasma exchange and steroids.
Alpelisib (Piqray)
Class: Phosphatidylinositol-3-kinase (PI3K) inhibitor.
Disease: Hormone receptor positive HER2-negative PIK3CA-mutated, advanced or metastatic breast cancer.
Dose: 300 mg orally once daily with food with concomitant fulvestrant.
AEs: Hyperglycemia, pancytopenia.
Trial: SOLAR-1 (NCT02437318): 11-month progression-free survival among patients treated with alpelisib and fulvestrant, compared with 5.7 months in fulvestrant alone control arm; overall response rate of 36% versus 16%, respectively.
Erdafitinib (Balversa)
Class: Fibroblast growth factor receptor kinase inhibitor.
Disease: Locally advanced or metastatic urothelial carcinoma with FGFR3 or FGFR2 mutations.
Dose: 8 mg orally once daily, with or without food.
AEs: Ocular disorders including retinopathy or retinal detachment.
Trial: BLC2001 (NCT02365597): Objective response rate of 32.2%, with a complete response in 2.3% of patients and partial response in 29.9% of patients.
Biosimilar approvals
Trastuzumab and hyaluronidase-oysk (Herceptin Hylecta)
Biosimilar to: Trastuzumab.
Indication: HER2-overexpressing breast cancer.
Dr. Bryer is a resident in the department of internal medicine at the University of Pennsylvania, Philadelphia. Dr. Mintzer is chief of hematology-oncology at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania. Dr. Henry is a hematologist-oncologist at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania.
*Correction, 11/7/2019: An earlier version of this article misstated the drug combination in the GO29365 trial.
The rapid development and identification of novel drugs has translated into innovative therapies in hematology and oncology. The aim of this piece is to present newly approved drugs and expanded indications to serve as a reference guide for practicing clinicians.
This article reviews therapies that were newly approved so far in 2019, as well as those previously approved whose indications were expanded this past year. The list highlights the most clinically important approvals, as well as adverse events that are unique or especially severe.
New approvals
Fedratinib (Inrebic)
Class: JAK2 and FLT3 selective kinase inhibitor.
Disease: Intermediate or high-risk primary or secondary (postpolycythemia vera or postessential thrombocythemia) myelofibrosis.
Dose: 400 mg orally once daily, with or without food.
Adverse events (AEs): Black box warning: Fatal encephalopathy, including Wernicke’s (thiamine level monitoring suggested).
Trials: In JAKARTA (NCT01437787), 37% of patients achieved a 35% or greater reduction in spleen volume and 40% received a 50% or greater reduction in myelofibrosis-related symptoms. In Jakarta-2, there was a 55% spleen response in patients resistant or intolerant to ruxolitinib.
Entrectinib (Rozlytrek)
Class: Tropomyosin receptor tyrosine kinase inhibitor.
Disease: Solid tumors that have a neurotrophic tyrosine receptor kinase (NTRK) gene fusion and for ROS-1 positive non–small cell lung cancer (NSCLC).
Dose: 600 mg orally once daily.
AEs: Heart failure, QT prolongation, skeletal fractures, hepatotoxicity, central nervous system effects, and hyperuricemia.
Trial: ALKA, STARTRK-1 (NCT02097810) and STARTRK-2 (NCT02568267): Overall response rate of 57% for NTRK positive patients; response rate of 77% in ROS-1 positive NSCLC.
Pexidartinib (Turalio)
Class: Small molecule tyrosine kinase inhibitor targeting CSF1R.
Disease: Symptomatic tenosynovial giant cell tumor.
Dose: 400 mg orally twice daily without food.
AEs: Black box warning on hepatotoxicity.
Trial: ENLIVEN (NCT02371369): Overall response rate of 38% at 25 weeks, with a 15% complete response rate and a 23% partial response rate.
Darolutamide (Nubeqa)
Class: Androgen receptor inhibitor.
Disease: Nonmetastatic castration-resistant prostate cancer.
Dose: 600 mg orally twice daily with food with concomitant androgen deprivation therapy.
AEs: Fatigue, extremity pain, and rash.
Trial: ARAMIS (NCT02200614): Median metastasis free survival was 40.4 months for patients with darolutamide, compared with 18.4 months for controls.
Selinexor (Xpovio)
Class: Reversible inhibitor of nuclear export of tumor suppressor proteins, growth regulators, and mRNAs of oncogenic proteins.
Disease: Relapsed or refractory multiple myeloma. Indicated for patients who have received at least four prior therapies, including at least two immunomodulatory agents and an anti-CD38 monoclonal antibody.
Dose: 80 mg orally in combination with oral dexamethasone on days 1 and 3 of each week.
AEs: Thrombocytopenia, fatigue, pancytopenia, and hyponatremia.
Trial: STORM (NCT02336815): Overall response rate 25.3% with a median time to first response of 4 weeks and 3.8-month median duration of response.
Polatuzumab vedotin-piiq (Polivy)
Class: CD79b-directed antibody-drug conjugate.
Disease: Relapsed or refractory diffuse large B-cell lymphoma. Indicated for patients who have had at least two prior therapies.
Dose: 1.8 mg/kg intravenous infusion every 21 days for six cycles in combination with bendamustine and a rituximab product.
AEs: Pancytopenia, peripheral neuropathy.
Trial: GO29365 (NCT02257567): Complete response rate was 40% for polatuzumab vedotin-piiq plus bendamustine/rituximab, compared with 18% with bendamustine/rituximab alone.*
Caplacizumab-yhdp (Cablivi)
Class: Monoclonal antibody fragment directed against von Willebrand factor.
Disease: Thrombotic thrombocytopenic purpura.
Dose: 11 mg IV initially, then daily subcutaneously; in combination with plasma exchange and immunosuppressive therapy.
AEs: Epistaxis, headache, and gingival bleeding.
Trial: Hercules trial (NCT02553317): More rapid normalization of platelets, lower incidence of composite TTP-related death, and lower rate of recurrence when added to plasma exchange and steroids.
Alpelisib (Piqray)
Class: Phosphatidylinositol-3-kinase (PI3K) inhibitor.
Disease: Hormone receptor positive HER2-negative PIK3CA-mutated, advanced or metastatic breast cancer.
Dose: 300 mg orally once daily with food with concomitant fulvestrant.
AEs: Hyperglycemia, pancytopenia.
Trial: SOLAR-1 (NCT02437318): 11-month progression-free survival among patients treated with alpelisib and fulvestrant, compared with 5.7 months in fulvestrant alone control arm; overall response rate of 36% versus 16%, respectively.
Erdafitinib (Balversa)
Class: Fibroblast growth factor receptor kinase inhibitor.
Disease: Locally advanced or metastatic urothelial carcinoma with FGFR3 or FGFR2 mutations.
Dose: 8 mg orally once daily, with or without food.
AEs: Ocular disorders including retinopathy or retinal detachment.
Trial: BLC2001 (NCT02365597): Objective response rate of 32.2%, with a complete response in 2.3% of patients and partial response in 29.9% of patients.
Biosimilar approvals
Trastuzumab and hyaluronidase-oysk (Herceptin Hylecta)
Biosimilar to: Trastuzumab.
Indication: HER2-overexpressing breast cancer.
Dr. Bryer is a resident in the department of internal medicine at the University of Pennsylvania, Philadelphia. Dr. Mintzer is chief of hematology-oncology at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania. Dr. Henry is a hematologist-oncologist at Pennsylvania Hospital and professor of medicine at the University of Pennsylvania.
*Correction, 11/7/2019: An earlier version of this article misstated the drug combination in the GO29365 trial.
FDA approves diroximel fumarate for relapsing MS
The Food and Drug Administration has approved diroximel fumarate (Vumerity) for the treatment of relapsing forms of multiple sclerosis (MS) in adults, including clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, according to an Oct. 30 announcement from its developers, Biogen and Alkermes.

The approval is based on pharmacokinetic studies that established the bioequivalence of diroximel fumarate and dimethyl fumarate (Tecfidera), and it relied in part on the safety and efficacy data for dimethyl fumarate, which was approved in 2013. Diroximel fumarate rapidly converts to monomethyl fumarate, the same active metabolite as dimethyl fumarate.
Diroximel fumarate may be better tolerated than dimethyl fumarate. A trial found that the newer drug has significantly better gastrointestinal tolerability, the developers of the drug announced in July. In addition, the drug application for diroximel fumarate included interim data from EVOLVE-MS-1, an ongoing, open-label, 2-year safety study evaluating diroximel fumarate in patients with relapsing-remitting MS. Researchers found a 6.3% rate of treatment discontinuation attributable to adverse events. Less than 1% of patients discontinued treatment because of gastrointestinal adverse events.
Serious side effects of diroximel fumarate may include allergic reaction, progressive multifocal leukoencephalopathy, decreases in white blood cell count, and liver problems. Flushing and stomach problems are the most common side effects, which may decrease over time.
Biogen plans to make diroximel fumarate available in the United States in the near future, the company said. Prescribing information is available online.
The Food and Drug Administration has approved diroximel fumarate (Vumerity) for the treatment of relapsing forms of multiple sclerosis (MS) in adults, including clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, according to an Oct. 30 announcement from its developers, Biogen and Alkermes.
The approval is based on pharmacokinetic studies that established the bioequivalence of diroximel fumarate and dimethyl fumarate (Tecfidera), and it relied in part on the safety and efficacy data for dimethyl fumarate, which was approved in 2013. Diroximel fumarate rapidly converts to monomethyl fumarate, the same active metabolite as dimethyl fumarate.
Diroximel fumarate may be better tolerated than dimethyl fumarate. A trial found that the newer drug has significantly better gastrointestinal tolerability, the developers of the drug announced in July. In addition, the drug application for diroximel fumarate included interim data from EVOLVE-MS-1, an ongoing, open-label, 2-year safety study evaluating diroximel fumarate in patients with relapsing-remitting MS. Researchers found a 6.3% rate of treatment discontinuation attributable to adverse events. Less than 1% of patients discontinued treatment because of gastrointestinal adverse events.
Serious side effects of diroximel fumarate may include allergic reaction, progressive multifocal leukoencephalopathy, decreases in white blood cell count, and liver problems. Flushing and stomach problems are the most common side effects, which may decrease over time.
Biogen plans to make diroximel fumarate available in the United States in the near future, the company said. Prescribing information is available online.
The Food and Drug Administration has approved diroximel fumarate (Vumerity) for the treatment of relapsing forms of multiple sclerosis (MS) in adults, including clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, according to an Oct. 30 announcement from its developers, Biogen and Alkermes.
The approval is based on pharmacokinetic studies that established the bioequivalence of diroximel fumarate and dimethyl fumarate (Tecfidera), and it relied in part on the safety and efficacy data for dimethyl fumarate, which was approved in 2013. Diroximel fumarate rapidly converts to monomethyl fumarate, the same active metabolite as dimethyl fumarate.
Diroximel fumarate may be better tolerated than dimethyl fumarate. A trial found that the newer drug has significantly better gastrointestinal tolerability, the developers of the drug announced in July. In addition, the drug application for diroximel fumarate included interim data from EVOLVE-MS-1, an ongoing, open-label, 2-year safety study evaluating diroximel fumarate in patients with relapsing-remitting MS. Researchers found a 6.3% rate of treatment discontinuation attributable to adverse events. Less than 1% of patients discontinued treatment because of gastrointestinal adverse events.
Serious side effects of diroximel fumarate may include allergic reaction, progressive multifocal leukoencephalopathy, decreases in white blood cell count, and liver problems. Flushing and stomach problems are the most common side effects, which may decrease over time.
Biogen plans to make diroximel fumarate available in the United States in the near future, the company said. Prescribing information is available online.
Three companies issue recall for ranitidine because of NDMA impurities
The Food and Drug Administration has issued an alert to health care providers and patients about voluntary recalls of ranitidine (Zantac) from three separate companies because of the potential of N-nitrosodimethylamine (NDMA) in the medicine.

According to the FDA alert, Perrigo is recalling over-the-counter ranitidine tablets of all sizes, Novitium Pharma is recalling all unexpired quantities and lots of ranitidine hydrochloride capsules, and Lannett is recalling all unexpired lots of prescription ranitidine syrup (ranitidine oral solution (15 mg/mL).
Patients who are using over-the-counter ranitidine should consider switching to an alternative, such as famotidine, cimetidine, esomeprazole, lansoprazole, and omeprazole, the FDA noted. None of these medications have shown evidence of containing NDMA.
The alert is the fifth update on ranitidine since the initial FDA announcement that NDMA had been found in ranitidine on Sept. 13, 2019.
The recent FDA safety alerts on ranitidine might be causing concern among your patients about their heartburn treatment. AGA offers key points that you can share with your patients at www.gastro.org/news/talking-to-your-patients-about-ranitidine.
The Food and Drug Administration has issued an alert to health care providers and patients about voluntary recalls of ranitidine (Zantac) from three separate companies because of the potential of N-nitrosodimethylamine (NDMA) in the medicine.
According to the FDA alert, Perrigo is recalling over-the-counter ranitidine tablets of all sizes, Novitium Pharma is recalling all unexpired quantities and lots of ranitidine hydrochloride capsules, and Lannett is recalling all unexpired lots of prescription ranitidine syrup (ranitidine oral solution (15 mg/mL).
Patients who are using over-the-counter ranitidine should consider switching to an alternative, such as famotidine, cimetidine, esomeprazole, lansoprazole, and omeprazole, the FDA noted. None of these medications have shown evidence of containing NDMA.
The alert is the fifth update on ranitidine since the initial FDA announcement that NDMA had been found in ranitidine on Sept. 13, 2019.
The recent FDA safety alerts on ranitidine might be causing concern among your patients about their heartburn treatment. AGA offers key points that you can share with your patients at www.gastro.org/news/talking-to-your-patients-about-ranitidine.
The Food and Drug Administration has issued an alert to health care providers and patients about voluntary recalls of ranitidine (Zantac) from three separate companies because of the potential of N-nitrosodimethylamine (NDMA) in the medicine.
According to the FDA alert, Perrigo is recalling over-the-counter ranitidine tablets of all sizes, Novitium Pharma is recalling all unexpired quantities and lots of ranitidine hydrochloride capsules, and Lannett is recalling all unexpired lots of prescription ranitidine syrup (ranitidine oral solution (15 mg/mL).
Patients who are using over-the-counter ranitidine should consider switching to an alternative, such as famotidine, cimetidine, esomeprazole, lansoprazole, and omeprazole, the FDA noted. None of these medications have shown evidence of containing NDMA.
The alert is the fifth update on ranitidine since the initial FDA announcement that NDMA had been found in ranitidine on Sept. 13, 2019.
The recent FDA safety alerts on ranitidine might be causing concern among your patients about their heartburn treatment. AGA offers key points that you can share with your patients at www.gastro.org/news/talking-to-your-patients-about-ranitidine.
THC use reported in majority of vaping-related illnesses
(EVALI), according to the Centers for Disease Control and Prevention.
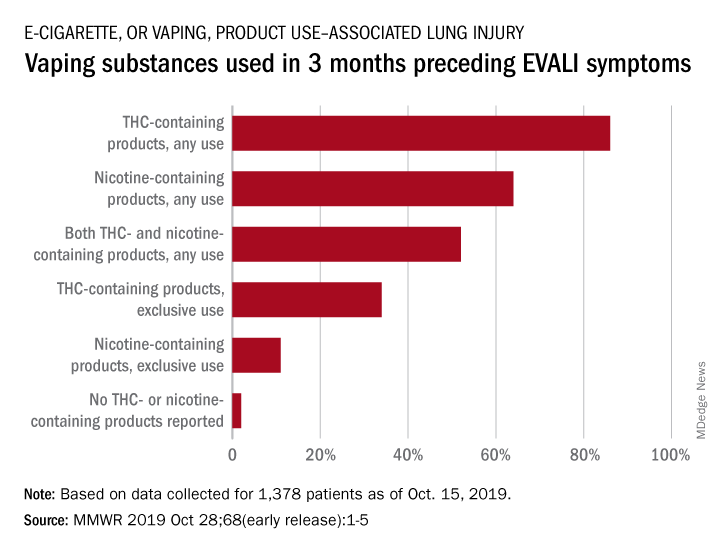
In the largest analysis to date, exclusive use of THC-containing products was reported for 34% of the 1,378 patients with confirmed or probable EVALI as of Oct. 15, 2019. Among those who died, 63% had been using THC exclusively during the 3 months preceding symptom onset, Erin D. Moritz, PhD, and associates said Oct. 28 in the Morbidity and Mortality Weekly Report.
Almost two-thirds (64%) of all EVALI patients had used nicotine-containing products at some time in the 3 months before symptom onset, and nicotine use was exclusive for 11%. Any nicotine use was reported for 37% of EVALI-related deaths, with exclusive use at 16%, the investigators reported.
“The data presented here suggest that THC-containing products are playing an important role in this outbreak,” they wrote, but “to date, no single compound or ingredient has emerged as the cause of EVALI, and there might be more than one cause.”
Dr. Moritz and associates also noted that many “patients likely did not know the content of the e-cigarette, or vaping, products they used,” which may have led to misclassification of substances.
SOURCE: Moritz ED et al. MMWR. Morbidity and mortality weekly report 2019 Oct 28;68(early release):1-4.
(EVALI), according to the Centers for Disease Control and Prevention.
In the largest analysis to date, exclusive use of THC-containing products was reported for 34% of the 1,378 patients with confirmed or probable EVALI as of Oct. 15, 2019. Among those who died, 63% had been using THC exclusively during the 3 months preceding symptom onset, Erin D. Moritz, PhD, and associates said Oct. 28 in the Morbidity and Mortality Weekly Report.
Almost two-thirds (64%) of all EVALI patients had used nicotine-containing products at some time in the 3 months before symptom onset, and nicotine use was exclusive for 11%. Any nicotine use was reported for 37% of EVALI-related deaths, with exclusive use at 16%, the investigators reported.
“The data presented here suggest that THC-containing products are playing an important role in this outbreak,” they wrote, but “to date, no single compound or ingredient has emerged as the cause of EVALI, and there might be more than one cause.”
Dr. Moritz and associates also noted that many “patients likely did not know the content of the e-cigarette, or vaping, products they used,” which may have led to misclassification of substances.
SOURCE: Moritz ED et al. MMWR. Morbidity and mortality weekly report 2019 Oct 28;68(early release):1-4.
(EVALI), according to the Centers for Disease Control and Prevention.
In the largest analysis to date, exclusive use of THC-containing products was reported for 34% of the 1,378 patients with confirmed or probable EVALI as of Oct. 15, 2019. Among those who died, 63% had been using THC exclusively during the 3 months preceding symptom onset, Erin D. Moritz, PhD, and associates said Oct. 28 in the Morbidity and Mortality Weekly Report.
Almost two-thirds (64%) of all EVALI patients had used nicotine-containing products at some time in the 3 months before symptom onset, and nicotine use was exclusive for 11%. Any nicotine use was reported for 37% of EVALI-related deaths, with exclusive use at 16%, the investigators reported.
“The data presented here suggest that THC-containing products are playing an important role in this outbreak,” they wrote, but “to date, no single compound or ingredient has emerged as the cause of EVALI, and there might be more than one cause.”
Dr. Moritz and associates also noted that many “patients likely did not know the content of the e-cigarette, or vaping, products they used,” which may have led to misclassification of substances.
SOURCE: Moritz ED et al. MMWR. Morbidity and mortality weekly report 2019 Oct 28;68(early release):1-4.
FROM MMWR
ICD-10 codes for EVALI released
The Centers for Disease Control and Prevention has issued coding guidance to help track e-cigarette, or vaping, product use–associated lung injury (EVALI).

The purpose of the coding guidelines “is to provide official diagnosis coding guidance for healthcare encounters related to the 2019 health care encounters and deaths related to” EVALI, CDC stated in a document detailing the coding update. The document was posted on the CDC website. The guidance is consistent with current clinical knowledge about e-cigarette, or vaping, related disorders.
CDC noted in the document that the guidance “is intended to be used in conjunction with current ICD-10-CM classification,” and the codes provided “are intended to provide e-cigarette, or vaping, product use coding guidance only.”
The codes are intended to track a number of areas related to EVALI, including lung-related complications, poisoning and toxicity, and substance use, abuse, and dependence.
The following conditions associated with EVALI are covered in the new coding guidance:
- Bronchitis and pneumonitis caused by chemicals, gases, and fumes.
- Bronchitis and pneumonitis caused by chemicals, gases, fumes, and vapors; includes chemical pneumonitis.
- Pneumonitis caused by inhalation of oils and essences; includes lipoid pneumonia.
- Acute respiratory distress syndrome.
- Pulmonary eosinophilia, not elsewhere classified.
- Acute interstitial pneumonitis.
The document notes that the coding guidance has been approved by the National Center for Health Statistics, the American Health Information Management Association, the American Hospital Association, and the Centers for Medicare & Medicaid Services.
The Centers for Disease Control and Prevention has issued coding guidance to help track e-cigarette, or vaping, product use–associated lung injury (EVALI).
The purpose of the coding guidelines “is to provide official diagnosis coding guidance for healthcare encounters related to the 2019 health care encounters and deaths related to” EVALI, CDC stated in a document detailing the coding update. The document was posted on the CDC website. The guidance is consistent with current clinical knowledge about e-cigarette, or vaping, related disorders.
CDC noted in the document that the guidance “is intended to be used in conjunction with current ICD-10-CM classification,” and the codes provided “are intended to provide e-cigarette, or vaping, product use coding guidance only.”
The codes are intended to track a number of areas related to EVALI, including lung-related complications, poisoning and toxicity, and substance use, abuse, and dependence.
The following conditions associated with EVALI are covered in the new coding guidance:
- Bronchitis and pneumonitis caused by chemicals, gases, and fumes.
- Bronchitis and pneumonitis caused by chemicals, gases, fumes, and vapors; includes chemical pneumonitis.
- Pneumonitis caused by inhalation of oils and essences; includes lipoid pneumonia.
- Acute respiratory distress syndrome.
- Pulmonary eosinophilia, not elsewhere classified.
- Acute interstitial pneumonitis.
The document notes that the coding guidance has been approved by the National Center for Health Statistics, the American Health Information Management Association, the American Hospital Association, and the Centers for Medicare & Medicaid Services.
The Centers for Disease Control and Prevention has issued coding guidance to help track e-cigarette, or vaping, product use–associated lung injury (EVALI).
The purpose of the coding guidelines “is to provide official diagnosis coding guidance for healthcare encounters related to the 2019 health care encounters and deaths related to” EVALI, CDC stated in a document detailing the coding update. The document was posted on the CDC website. The guidance is consistent with current clinical knowledge about e-cigarette, or vaping, related disorders.
CDC noted in the document that the guidance “is intended to be used in conjunction with current ICD-10-CM classification,” and the codes provided “are intended to provide e-cigarette, or vaping, product use coding guidance only.”
The codes are intended to track a number of areas related to EVALI, including lung-related complications, poisoning and toxicity, and substance use, abuse, and dependence.
The following conditions associated with EVALI are covered in the new coding guidance:
- Bronchitis and pneumonitis caused by chemicals, gases, and fumes.
- Bronchitis and pneumonitis caused by chemicals, gases, fumes, and vapors; includes chemical pneumonitis.
- Pneumonitis caused by inhalation of oils and essences; includes lipoid pneumonia.
- Acute respiratory distress syndrome.
- Pulmonary eosinophilia, not elsewhere classified.
- Acute interstitial pneumonitis.
The document notes that the coding guidance has been approved by the National Center for Health Statistics, the American Health Information Management Association, the American Hospital Association, and the Centers for Medicare & Medicaid Services.
Suicide deaths rising in children aged 10-19 years
according to the National Center for Health Statistics.
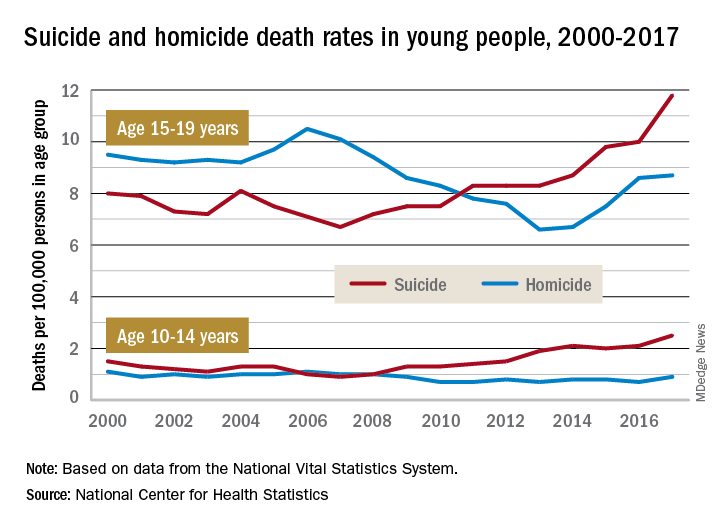
Death rates from suicide for children aged 10-14 years jumped by 178% from 2007 to 2017, while teenagers aged 15-19 years experienced a 76% increase over that period, with both changes reaching significance, the NCHS said in a recent data brief based on data from the National Vital Statistics System.
The actual rate for teens was higher to begin with, however, so in absolute terms the increase is larger for the older group. In 2007, deaths from suicide occurred at a rate of 6.7 per 100,000 persons for persons aged 15-19 years, and by 2017 that rate was up significantly to 11.8 per 100,000. Among children aged 10-14 years, the suicide-related death rate climbed from 0.9 per 100,000 in 2007 to 2.5 in 2014, the NCHS investigators reported.
The news was somewhat better on the other side of the violent death coin. Homicides are down by a significant 18% since 2000 among children aged 10-14 years, as the rate dropped from 1.1 per 100,000 in 2000 to 0.9 in 2017. The homicide rate since 2000 is down slightly for teens aged 15-19 years, but it has risen 32% in recent years, going from 6.6 deaths per 100,000 in 2013 to 8.7 in 2017, they said.
Suicide was the second-leading cause of death in both age groups in 2017, and homicide was third for those aged 15-19 and fifth among 10- to 14-year-olds, the investigators noted.
according to the National Center for Health Statistics.
Death rates from suicide for children aged 10-14 years jumped by 178% from 2007 to 2017, while teenagers aged 15-19 years experienced a 76% increase over that period, with both changes reaching significance, the NCHS said in a recent data brief based on data from the National Vital Statistics System.
The actual rate for teens was higher to begin with, however, so in absolute terms the increase is larger for the older group. In 2007, deaths from suicide occurred at a rate of 6.7 per 100,000 persons for persons aged 15-19 years, and by 2017 that rate was up significantly to 11.8 per 100,000. Among children aged 10-14 years, the suicide-related death rate climbed from 0.9 per 100,000 in 2007 to 2.5 in 2014, the NCHS investigators reported.
The news was somewhat better on the other side of the violent death coin. Homicides are down by a significant 18% since 2000 among children aged 10-14 years, as the rate dropped from 1.1 per 100,000 in 2000 to 0.9 in 2017. The homicide rate since 2000 is down slightly for teens aged 15-19 years, but it has risen 32% in recent years, going from 6.6 deaths per 100,000 in 2013 to 8.7 in 2017, they said.
Suicide was the second-leading cause of death in both age groups in 2017, and homicide was third for those aged 15-19 and fifth among 10- to 14-year-olds, the investigators noted.
according to the National Center for Health Statistics.
Death rates from suicide for children aged 10-14 years jumped by 178% from 2007 to 2017, while teenagers aged 15-19 years experienced a 76% increase over that period, with both changes reaching significance, the NCHS said in a recent data brief based on data from the National Vital Statistics System.
The actual rate for teens was higher to begin with, however, so in absolute terms the increase is larger for the older group. In 2007, deaths from suicide occurred at a rate of 6.7 per 100,000 persons for persons aged 15-19 years, and by 2017 that rate was up significantly to 11.8 per 100,000. Among children aged 10-14 years, the suicide-related death rate climbed from 0.9 per 100,000 in 2007 to 2.5 in 2014, the NCHS investigators reported.
The news was somewhat better on the other side of the violent death coin. Homicides are down by a significant 18% since 2000 among children aged 10-14 years, as the rate dropped from 1.1 per 100,000 in 2000 to 0.9 in 2017. The homicide rate since 2000 is down slightly for teens aged 15-19 years, but it has risen 32% in recent years, going from 6.6 deaths per 100,000 in 2013 to 8.7 in 2017, they said.
Suicide was the second-leading cause of death in both age groups in 2017, and homicide was third for those aged 15-19 and fifth among 10- to 14-year-olds, the investigators noted.
FDA approves onabotulinumtoxinA for pediatric lower limb spasticity
The Food and Drug Administration has approved onabotulinumtoxinA (Botox) for treatment of pediatric lower limb spasticity in patients aged 2-17 years, excluding those in whom it is associated with cerebral palsy, according to an announcement from Allergan.

The approval is based on a phase 3 study evaluating safety and efficacy in more than 300 patients with lower limb spasticity. Although patients with cerebral palsy were included in the study, they’re excluded from this indication. Orphan Drug Exclusivity prevents it from being indicated for lower limb spasticity in cerebral palsy because abobotulinumtoxinA (Dysport) already has marketing exclusivity for the indication. Botox also is indicated for children aged 2-17 years of age with upper limb spasticity, as well as nine other indications.
OnabotulinumtoxinA comes with warnings, including problems of swallowing, speaking, or breathing and even risk of spread of the toxin. It also may cause loss of strength or general muscle weakness, vision problems, or dizziness within hours or weeks of administration. Serious and sometimes immediate allergic reactions have been reported. Patients and health care professionals should discuss various concerns before treatment, including whether the patient has recently received antibiotics by injection, or has taken muscle relaxants, allergy or cold medicine, sleep medicine, and aspirinlike products or blood thinners. It’s important to note that the dose of onabotulinumtoxinA is not the same as that for other botulinum toxin products. The full prescribing information is available on the Allergan website.
The Food and Drug Administration has approved onabotulinumtoxinA (Botox) for treatment of pediatric lower limb spasticity in patients aged 2-17 years, excluding those in whom it is associated with cerebral palsy, according to an announcement from Allergan.
The approval is based on a phase 3 study evaluating safety and efficacy in more than 300 patients with lower limb spasticity. Although patients with cerebral palsy were included in the study, they’re excluded from this indication. Orphan Drug Exclusivity prevents it from being indicated for lower limb spasticity in cerebral palsy because abobotulinumtoxinA (Dysport) already has marketing exclusivity for the indication. Botox also is indicated for children aged 2-17 years of age with upper limb spasticity, as well as nine other indications.
OnabotulinumtoxinA comes with warnings, including problems of swallowing, speaking, or breathing and even risk of spread of the toxin. It also may cause loss of strength or general muscle weakness, vision problems, or dizziness within hours or weeks of administration. Serious and sometimes immediate allergic reactions have been reported. Patients and health care professionals should discuss various concerns before treatment, including whether the patient has recently received antibiotics by injection, or has taken muscle relaxants, allergy or cold medicine, sleep medicine, and aspirinlike products or blood thinners. It’s important to note that the dose of onabotulinumtoxinA is not the same as that for other botulinum toxin products. The full prescribing information is available on the Allergan website.
The Food and Drug Administration has approved onabotulinumtoxinA (Botox) for treatment of pediatric lower limb spasticity in patients aged 2-17 years, excluding those in whom it is associated with cerebral palsy, according to an announcement from Allergan.
The approval is based on a phase 3 study evaluating safety and efficacy in more than 300 patients with lower limb spasticity. Although patients with cerebral palsy were included in the study, they’re excluded from this indication. Orphan Drug Exclusivity prevents it from being indicated for lower limb spasticity in cerebral palsy because abobotulinumtoxinA (Dysport) already has marketing exclusivity for the indication. Botox also is indicated for children aged 2-17 years of age with upper limb spasticity, as well as nine other indications.
OnabotulinumtoxinA comes with warnings, including problems of swallowing, speaking, or breathing and even risk of spread of the toxin. It also may cause loss of strength or general muscle weakness, vision problems, or dizziness within hours or weeks of administration. Serious and sometimes immediate allergic reactions have been reported. Patients and health care professionals should discuss various concerns before treatment, including whether the patient has recently received antibiotics by injection, or has taken muscle relaxants, allergy or cold medicine, sleep medicine, and aspirinlike products or blood thinners. It’s important to note that the dose of onabotulinumtoxinA is not the same as that for other botulinum toxin products. The full prescribing information is available on the Allergan website.