User login
Denosumab boosts bone strength in glucocorticoid users
Bone strength and microarchitecture remained stronger at 24 months after treatment with denosumab compared to risedronate, in a study of 110 adults using glucocorticoids.
Patients using glucocorticoids are at increased risk for vertebral and nonvertebral fractures at both the start of treatment or as treatment continues, wrote Piet Geusens, MD, of Maastricht University, the Netherlands, and colleagues.

Imaging data collected via high-resolution peripheral quantitative computed tomography (HR-pQCT) allow for the assessment of bone microarchitecture and strength, but specific data comparing the impact of bone treatment in patients using glucocorticoids are lacking, they said.
In a study published in the Journal of Bone and Mineral Research, the researchers identified a subset of 56 patients randomized to denosumab and 54 to risedronate patients out of a total of 590 patients who were enrolled in a phase 3 randomized, controlled trial of denosumab vs. risedronate for bone mineral density. The main results of the larger trial – presented at EULAR 2018 – showed greater increases in bone strength with denosumab over risedronate in patients receiving glucocorticoids.
In the current study, the researchers reviewed HR-pQCT scans of the distal radius and tibia at baseline, 12 months, and 24 months. Bone strength and microarchitecture were defined in terms of failure load (FL) as a primary outcome. Patients also were divided into subpopulations of those initiating glucocorticoid treatment (GC-I) and continuing treatment (GC-C).
Baseline characteristics were mainly balanced among the treatment groups within the GC-I and GC-C categories.
Among the GC-I patients, in the denosumab group, FL increased significantly from baseline to 12 months at the radius at tibia (1.8% and 1.7%, respectively) but did not change significantly in the risedronate group, which translated to a significant treatment difference between the drugs of 3.3% for radius and 2.5% for tibia.
At 24 months, the radius measure of FL was unchanged from baseline in denosumab patients but significantly decreased in risedronate patients, with a difference of –4.1%, which translated to a significant between-treatment difference at the radius of 5.6% (P < .001). Changes at the tibia were not significantly different between the groups at 24 months.
Among the GC-C patients, FL was unchanged from baseline to 12 months for both the denosumab and risedronate groups. However, FL significantly increased with denosumab (4.3%) and remained unchanged in the risedronate group.
The researchers also found significant differences between denosumab and risedronate in percentage changes in cortical bone mineral density, and less prominent changes and differences in trabecular bone mineral density.
The study findings were limited by several factors including the use of the HR-pQCT scanner, which limits the measurement of trabecular microarchitecture, and the use of only standard HR-pQCT parameters, which do not allow insight into endosteal changes, and the inability to correct for multiplicity of data, the researchers noted.
However, the results support the superiority of denosumab over risedronate for preventing FL and total bone mineral density loss at the radius and tibia in new glucocorticoid users, and for increasing FL and total bone mineral density at the radius in long-term glucocorticoid users, they said.
Denosumab therefore could be a useful therapeutic option and could inform decision-making in patients initiating GC-therapy or on long-term GC-therapy, they concluded.
The study was supported by Amgen. Dr. Geusens disclosed grants from Amgen, Celgene, Lilly, Merck, Pfizer, Roche, UCB, Fresenius, Mylan, and Sandoz, and grants and other funding from AbbVie, outside the current study.
Bone strength and microarchitecture remained stronger at 24 months after treatment with denosumab compared to risedronate, in a study of 110 adults using glucocorticoids.
Patients using glucocorticoids are at increased risk for vertebral and nonvertebral fractures at both the start of treatment or as treatment continues, wrote Piet Geusens, MD, of Maastricht University, the Netherlands, and colleagues.
Imaging data collected via high-resolution peripheral quantitative computed tomography (HR-pQCT) allow for the assessment of bone microarchitecture and strength, but specific data comparing the impact of bone treatment in patients using glucocorticoids are lacking, they said.
In a study published in the Journal of Bone and Mineral Research, the researchers identified a subset of 56 patients randomized to denosumab and 54 to risedronate patients out of a total of 590 patients who were enrolled in a phase 3 randomized, controlled trial of denosumab vs. risedronate for bone mineral density. The main results of the larger trial – presented at EULAR 2018 – showed greater increases in bone strength with denosumab over risedronate in patients receiving glucocorticoids.
In the current study, the researchers reviewed HR-pQCT scans of the distal radius and tibia at baseline, 12 months, and 24 months. Bone strength and microarchitecture were defined in terms of failure load (FL) as a primary outcome. Patients also were divided into subpopulations of those initiating glucocorticoid treatment (GC-I) and continuing treatment (GC-C).
Baseline characteristics were mainly balanced among the treatment groups within the GC-I and GC-C categories.
Among the GC-I patients, in the denosumab group, FL increased significantly from baseline to 12 months at the radius at tibia (1.8% and 1.7%, respectively) but did not change significantly in the risedronate group, which translated to a significant treatment difference between the drugs of 3.3% for radius and 2.5% for tibia.
At 24 months, the radius measure of FL was unchanged from baseline in denosumab patients but significantly decreased in risedronate patients, with a difference of –4.1%, which translated to a significant between-treatment difference at the radius of 5.6% (P < .001). Changes at the tibia were not significantly different between the groups at 24 months.
Among the GC-C patients, FL was unchanged from baseline to 12 months for both the denosumab and risedronate groups. However, FL significantly increased with denosumab (4.3%) and remained unchanged in the risedronate group.
The researchers also found significant differences between denosumab and risedronate in percentage changes in cortical bone mineral density, and less prominent changes and differences in trabecular bone mineral density.
The study findings were limited by several factors including the use of the HR-pQCT scanner, which limits the measurement of trabecular microarchitecture, and the use of only standard HR-pQCT parameters, which do not allow insight into endosteal changes, and the inability to correct for multiplicity of data, the researchers noted.
However, the results support the superiority of denosumab over risedronate for preventing FL and total bone mineral density loss at the radius and tibia in new glucocorticoid users, and for increasing FL and total bone mineral density at the radius in long-term glucocorticoid users, they said.
Denosumab therefore could be a useful therapeutic option and could inform decision-making in patients initiating GC-therapy or on long-term GC-therapy, they concluded.
The study was supported by Amgen. Dr. Geusens disclosed grants from Amgen, Celgene, Lilly, Merck, Pfizer, Roche, UCB, Fresenius, Mylan, and Sandoz, and grants and other funding from AbbVie, outside the current study.
Bone strength and microarchitecture remained stronger at 24 months after treatment with denosumab compared to risedronate, in a study of 110 adults using glucocorticoids.
Patients using glucocorticoids are at increased risk for vertebral and nonvertebral fractures at both the start of treatment or as treatment continues, wrote Piet Geusens, MD, of Maastricht University, the Netherlands, and colleagues.
Imaging data collected via high-resolution peripheral quantitative computed tomography (HR-pQCT) allow for the assessment of bone microarchitecture and strength, but specific data comparing the impact of bone treatment in patients using glucocorticoids are lacking, they said.
In a study published in the Journal of Bone and Mineral Research, the researchers identified a subset of 56 patients randomized to denosumab and 54 to risedronate patients out of a total of 590 patients who were enrolled in a phase 3 randomized, controlled trial of denosumab vs. risedronate for bone mineral density. The main results of the larger trial – presented at EULAR 2018 – showed greater increases in bone strength with denosumab over risedronate in patients receiving glucocorticoids.
In the current study, the researchers reviewed HR-pQCT scans of the distal radius and tibia at baseline, 12 months, and 24 months. Bone strength and microarchitecture were defined in terms of failure load (FL) as a primary outcome. Patients also were divided into subpopulations of those initiating glucocorticoid treatment (GC-I) and continuing treatment (GC-C).
Baseline characteristics were mainly balanced among the treatment groups within the GC-I and GC-C categories.
Among the GC-I patients, in the denosumab group, FL increased significantly from baseline to 12 months at the radius at tibia (1.8% and 1.7%, respectively) but did not change significantly in the risedronate group, which translated to a significant treatment difference between the drugs of 3.3% for radius and 2.5% for tibia.
At 24 months, the radius measure of FL was unchanged from baseline in denosumab patients but significantly decreased in risedronate patients, with a difference of –4.1%, which translated to a significant between-treatment difference at the radius of 5.6% (P < .001). Changes at the tibia were not significantly different between the groups at 24 months.
Among the GC-C patients, FL was unchanged from baseline to 12 months for both the denosumab and risedronate groups. However, FL significantly increased with denosumab (4.3%) and remained unchanged in the risedronate group.
The researchers also found significant differences between denosumab and risedronate in percentage changes in cortical bone mineral density, and less prominent changes and differences in trabecular bone mineral density.
The study findings were limited by several factors including the use of the HR-pQCT scanner, which limits the measurement of trabecular microarchitecture, and the use of only standard HR-pQCT parameters, which do not allow insight into endosteal changes, and the inability to correct for multiplicity of data, the researchers noted.
However, the results support the superiority of denosumab over risedronate for preventing FL and total bone mineral density loss at the radius and tibia in new glucocorticoid users, and for increasing FL and total bone mineral density at the radius in long-term glucocorticoid users, they said.
Denosumab therefore could be a useful therapeutic option and could inform decision-making in patients initiating GC-therapy or on long-term GC-therapy, they concluded.
The study was supported by Amgen. Dr. Geusens disclosed grants from Amgen, Celgene, Lilly, Merck, Pfizer, Roche, UCB, Fresenius, Mylan, and Sandoz, and grants and other funding from AbbVie, outside the current study.
FROM THE JOURNAL OF BONE AND MINERAL RESEARCH
FDA okays first sublingual med for agitation in serious mental illness
This is the first FDA-approved, orally dissolving, self-administered sublingual treatment for this indication. With a demonstrated onset of action as early as 20 minutes, it shows a high response rate in patients at both 120-mcg and 180-mcg doses.
An estimated 7.3 million individuals in the United States are diagnosed with schizophrenia or bipolar disorders, and up to one-quarter of them experience episodes of agitation that can occur 10-17 times annually. These episodes represent a significant burden for patients, caregivers, and the health care system.
“There are large numbers of patients who experience agitation associated with schizophrenia and bipolar disorders, and this condition has been a long-standing challenge for health care professionals to treat,” said John Krystal, MD, the Robert L. McNeil Jr. Professor of Translational Research and chair of the department of psychiatry at Yale University, New Haven, Conn.
“The approval of Igalmi, a self-administered film with a desirable onset of action, represents a milestone moment. It provides health care teams with an innovative tool to help control agitation. As clinicians, we welcome this much-needed new oral treatment option,” he added.
“Igalmi is the first new acute treatment for schizophrenia or bipolar disorder–associated agitation in nearly a decade and represents a differentiated approach to helping patients manage this difficult and debilitating symptom,” said Vimal Mehta, PhD, CEO of BioXcel Therapeutics.
The FDA approval of Igalmi is based on data from two pivotal randomized, double-blinded, placebo-controlled, parallel-group, phase 3 trials that evaluated Igalmi for the acute treatment of agitation associated with schizophrenia (SERENITY I) or bipolar I or II disorder (SERENITY II).
The most common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were somnolence, paresthesia or oral hypoesthesia, dizziness, dry mouth, hypotension, and orthostatic hypotension. All adverse drug reactions were mild to moderate in severity. While Igalmi was not associated with any treatment-related serious adverse effects in phase 3 studies, it may cause notable side effects, including hypotension, orthostatic hypotension, bradycardia, QT interval prolongation, and somnolence.
As previously reported by this news organization, data from the phase 3 SERENITY II trial that evaluated Igalmi in bipolar disorders were published in JAMA.
A version of this article first appeared on Medscape.com.
This is the first FDA-approved, orally dissolving, self-administered sublingual treatment for this indication. With a demonstrated onset of action as early as 20 minutes, it shows a high response rate in patients at both 120-mcg and 180-mcg doses.
An estimated 7.3 million individuals in the United States are diagnosed with schizophrenia or bipolar disorders, and up to one-quarter of them experience episodes of agitation that can occur 10-17 times annually. These episodes represent a significant burden for patients, caregivers, and the health care system.
“There are large numbers of patients who experience agitation associated with schizophrenia and bipolar disorders, and this condition has been a long-standing challenge for health care professionals to treat,” said John Krystal, MD, the Robert L. McNeil Jr. Professor of Translational Research and chair of the department of psychiatry at Yale University, New Haven, Conn.
“The approval of Igalmi, a self-administered film with a desirable onset of action, represents a milestone moment. It provides health care teams with an innovative tool to help control agitation. As clinicians, we welcome this much-needed new oral treatment option,” he added.
“Igalmi is the first new acute treatment for schizophrenia or bipolar disorder–associated agitation in nearly a decade and represents a differentiated approach to helping patients manage this difficult and debilitating symptom,” said Vimal Mehta, PhD, CEO of BioXcel Therapeutics.
The FDA approval of Igalmi is based on data from two pivotal randomized, double-blinded, placebo-controlled, parallel-group, phase 3 trials that evaluated Igalmi for the acute treatment of agitation associated with schizophrenia (SERENITY I) or bipolar I or II disorder (SERENITY II).
The most common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were somnolence, paresthesia or oral hypoesthesia, dizziness, dry mouth, hypotension, and orthostatic hypotension. All adverse drug reactions were mild to moderate in severity. While Igalmi was not associated with any treatment-related serious adverse effects in phase 3 studies, it may cause notable side effects, including hypotension, orthostatic hypotension, bradycardia, QT interval prolongation, and somnolence.
As previously reported by this news organization, data from the phase 3 SERENITY II trial that evaluated Igalmi in bipolar disorders were published in JAMA.
A version of this article first appeared on Medscape.com.
This is the first FDA-approved, orally dissolving, self-administered sublingual treatment for this indication. With a demonstrated onset of action as early as 20 minutes, it shows a high response rate in patients at both 120-mcg and 180-mcg doses.
An estimated 7.3 million individuals in the United States are diagnosed with schizophrenia or bipolar disorders, and up to one-quarter of them experience episodes of agitation that can occur 10-17 times annually. These episodes represent a significant burden for patients, caregivers, and the health care system.
“There are large numbers of patients who experience agitation associated with schizophrenia and bipolar disorders, and this condition has been a long-standing challenge for health care professionals to treat,” said John Krystal, MD, the Robert L. McNeil Jr. Professor of Translational Research and chair of the department of psychiatry at Yale University, New Haven, Conn.
“The approval of Igalmi, a self-administered film with a desirable onset of action, represents a milestone moment. It provides health care teams with an innovative tool to help control agitation. As clinicians, we welcome this much-needed new oral treatment option,” he added.
“Igalmi is the first new acute treatment for schizophrenia or bipolar disorder–associated agitation in nearly a decade and represents a differentiated approach to helping patients manage this difficult and debilitating symptom,” said Vimal Mehta, PhD, CEO of BioXcel Therapeutics.
The FDA approval of Igalmi is based on data from two pivotal randomized, double-blinded, placebo-controlled, parallel-group, phase 3 trials that evaluated Igalmi for the acute treatment of agitation associated with schizophrenia (SERENITY I) or bipolar I or II disorder (SERENITY II).
The most common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were somnolence, paresthesia or oral hypoesthesia, dizziness, dry mouth, hypotension, and orthostatic hypotension. All adverse drug reactions were mild to moderate in severity. While Igalmi was not associated with any treatment-related serious adverse effects in phase 3 studies, it may cause notable side effects, including hypotension, orthostatic hypotension, bradycardia, QT interval prolongation, and somnolence.
As previously reported by this news organization, data from the phase 3 SERENITY II trial that evaluated Igalmi in bipolar disorders were published in JAMA.
A version of this article first appeared on Medscape.com.
Incorporation of Clinical Staff Pharmacists in the Emergency Department Sepsis Response at a Single Institution
Sepsis is life-threatening organ dysfunction caused by dysregulated host response to an infection that can progress to shock. Sepsis is a major cause of death in the United States, with > 1 million people developing sepsis and > 250,000 people dying from sepsis annually.1 The Surviving Sepsis Campaign (SSC) guidelines recommend treating sepsis as an emergency with timely administration of fluids and antibiotics, as administering antibiotics within the first hour has been found to reduce mortality and disease progression. In addition, empiric antibiotic regimens should be chosen to target the most probable pathogens and dosing should be optimized. To achieve this, the SSC guidelines recommend that hospitals develop quality improvement (QI) programs developed by a multidisciplinary group to improve sepsis recognition and response using a protocolized approach.2
There are several studies describing efforts to improve the sepsis response at facilities, some of which have evaluated the addition of a pharmacist into the sepsis response, particularly in the emergency department (ED). Some studies found improved selection and decreased time to antibiotic administration with the addition of an ED pharmacist.3-7 Despite this, ED pharmacists are not present in all hospitals, with a 2015 national survey reporting the presence of an ED pharmacist in 68.7% of respondents at 187 facilities. Even facilities with ED pharmacists often have limited hours of coverage, with at least 8 hours of coverage in 49.4% of facilities with an ED pharmacist and no weekend coverage at 34.8% of these facilities.8
While many hospitals do not routinely employ ED pharmacists, most hospitals have clinical staff pharmacists (CSPs), and many inpatient hospital pharmacies are staffed with CSPs 24 hours per day, 7 days per week. A 2017 survey conducted by the American Society of Health-System Pharmacists (ASHP) found 43% of all hospital pharmacy departments were staffed by a CSP around the clock, with the prevalence increasing to 56.7 to 100% in hospitals with > 100 beds.9 As a result, CSPs may be a useful resource to assist with the management of patients with sepsis in hospitals without an ED pharmacist.
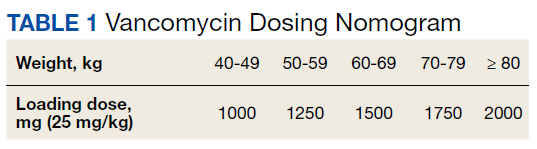
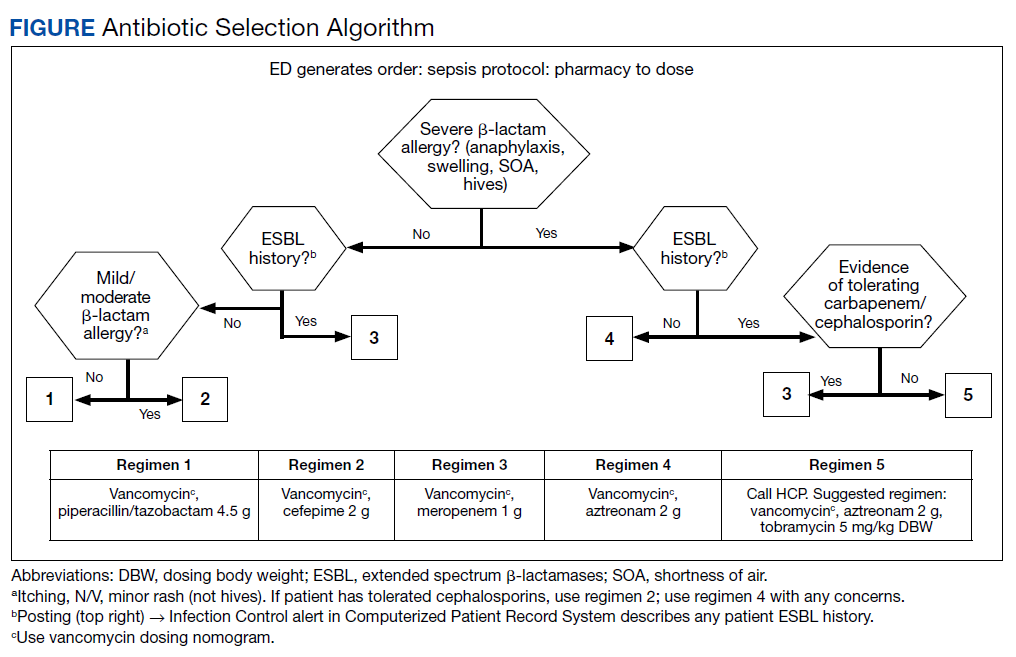
At the Lexington Veterans Affairs Health Care System (LVAHCS) in Kentucky, the inpatient pharmacy department is staffed with a CSP 24/7 but does not have an ED pharmacist. Therefore, when an interdisciplinary group developed an ED sepsis bundle as part of a QI initiative on sepsis recognition and response, the group took a unique approach of incorporating CSPs into the response team to assist with antimicrobial selection and dosing. An antibiotic selection algorithm and vancomycin dosing nomogram were developed to aid CSPs to select and dose antibiotics (Figure, Table 1). We describe the implementation of this process and evaluate CSPs’ accuracy in antimicrobial selection and vancomycin dosing.
Methods
Lexington VAHCS is a 94-bed hospital that provides services to veterans, including an ED, inpatient medical services, surgical services, acute mental health, progressive care, and intensive care units. This facility has 1 antimicrobial stewardship clinical pharmacy specialist, 2 critical care clinical pharmacy specialists, and 16 full-time CSPs with 24-hour CSP coverage. The annual ED volume at the time of this study was approximately 21,000 patients.
Consistent with the SSC guideline recommendation to develop multidisciplinary QI initiatives on sepsis recognition and response, an Interdisciplinary Sepsis Committee (ISC) was created in 2018 comprised of ED, pulmonary, critical care, and infectious diseases licensed independent practitioners (LIPs), ED nurses, and pharmacists. The ISC developed a comprehensive set of sepsis tools that included a sepsis screening tool used by ED triage nurses to provide early detection of sepsis and an updated electronic order set to decrease time to appropriate treatment. This order set included automatic orders for blood cultures and serum lactate, the initiation of IV crystalloids, as well as a Sepsis Alert order placed by ED LIPs which alerted CSPs to a patient with sepsis in the ED.
To ensure a protocol-based approach by the CSPs responding to the sepsis alert, an antibiotic algorithm and vancomycin dosing nomogram were developed by the ISC based on current guideline recommendations and the local antibiogram. These were subsequently approved by ED practitioners, the pharmacy and therapeutics committee, and the critical care committee. The antibiotic algorithm prompts CSPs to perform a chart review to identify β-lactam allergies, evaluate the severity of the allergy and which agents the patient has tolerated in the past, as well as determine whether the patient has a history of extended spectrum β-lactamase (ESBL)–producing organisms from previous cultures. A decision tree then guides CSPs toward the selection of 1 of 5 empiric antibiotic regimens to cover all likely pathogens. The medication orders are then entered by the CSPs as a telephone order from the ED LIP per protocol. Unless patients had a true vancomycin allergy, all patients received vancomycin as the empiric gram-positive backbone of the regimen. The vancomycin dosing nomogram was created to ensure an appropriate and consistent vancomycin weight-based loading dose was administered.
Prior to implementation, the antimicrobial stewardship pharmacist educated CSPs on the use of these tools, including simulated orders for mock sepsis alerts to ensure competency. A copy of the algorithm and nomogram were emailed to all CSPs and posted in a prominent location in the pharmacy.
As part of continuous performance improvement efforts of the ISC, a retrospective cohort study was conducted through chart review on patients at the Lexington VAHCS with an order for a sepsis alert in the ED from December 3, 2018 to May 31, 2020 to assess the accuracy of the CSPs’ antibiotic selection and dosing. Patients were excluded if they had a vancomycin allergy or if the ED practitioner ordered antibiotics prior to the CSPs placing orders. Patients could be included more than once in the study if they had sepsis alerts placed on different dates.
The primary outcomes were CSPs’ accuracy in antimicrobial selection with the antibiotic selection algorithm and vancomycin dosing nomogram. The antibiotic selection was deemed accurate if the appropriate antibiotic regimen was selected based on allergy status and previous cultures as directed in the algorithm. The vancomycin dose was considered accurate if the dose chosen was appropriate based on the patient’s weight at the time of ED presentation. Secondary outcomes included time to administration of antibiotics from ED presentation as well as time to antibiotics administration from sepsis alert initiation. Time of administration was considered the time the antibiotics were scanned in the bar code medication administration (BCMA) system.
Descriptive statistics were used with data presented as percentages for nominal data and median as IQR for continuous data. In accordance with our facility’s project assessment process, this project was determined not to constitute human subjects research; therefore, this QI project did not require review by the institutional review board.
Results
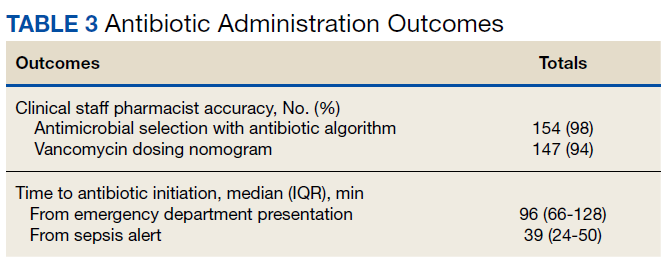
Between December 3, 2018 and May 31, 2020, 160 sepsis alerts were ordered by ED practitioners. Of the 160 patients, 157 were included in the final data analysis. Two patients were excluded due to vancomycin allergy, and 1 patient because the physician ordered antibiotics prior to pharmacist order entry. The population was largely composed of male patients (98%) with a median age of 72 years (Table 2).
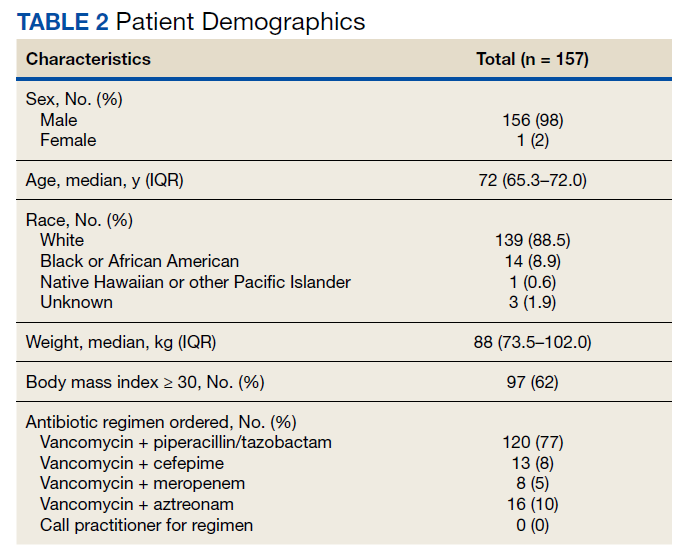
Of 157 sepsis alerts, the antibiotic selection algorithm was used appropriately in 154 (98%) instances (Table 3). Chart reviews were performed in instances of antimicrobial selection different from the algorithm. Of the 3 patients who received antibiotics not consistent with the algorithm, 1 patient without a history of ESBL-producing organisms in their culture history received meropenem instead of piperacillin/tazobactam. Another patient without a penicillin allergy received cefepime (plus metronidazole ordered separately from the ED practitioner) instead of piperacillin/tazobactam, and the third patient received piperacillin/tazobactam instead of meropenem despite a culture history of ESBL-producing organisms. Vancomycin dose was appropriate according to the weight-based nomogram in 147 cases (94%). The median time to administration of first dose antibiotics was 39 minutes after the sepsis alert order was placed and 96 minutes after initial ED presentation.
Discussion
This study found extremely high rates of accuracy among CSPs for both the antibiotic selection algorithm (98%) and the vancomycin dosing nomogram (94%). Moreover, analysis of the 3 patients who received antibiotics that were inconsistent with the algorithm revealed that 2 of these patients arguably still received adequate empiric coverage, increasing the percentage of patients receiving appropriate empiric antibiotics to 99.4%. Similarly, chart review of 10 patients who received vancomycin doses that deviated from the nomogram revealed that in at least 3 cases, patients were likely given correct vancomycin doses based on the patient’s last known weight. However, when actual current weights were recorded soon after admission, the updated weights rendered the initial vancomycin loading dose incorrect when this analysis was performed. Thus, the adherence to the vancomycin dosing nomogram is higher than it appears.
Median time to antibiotic administration from the sepsis alert was 39 minutes—well within SSC recommendations (60 minutes).2 Previous internal analyses at Lexington VAHCS demonstrated the mean time to first dose of antibiotics in the ED has been 39 minutes since about 2015. Thus, this initiative did not necessarily make this process quicker; however it did remove 1 responsibility from LIPs so that they could focus their efforts on other components of sepsis management.
Further studies are needed to evaluate the effects of this initiative on other aspects of the sepsis bundle, such as volume of fluid administered and appropriateness of laboratory tests. It was noted that while the time to first-dose antibiotic administration was < 1 hour from order placement, the median time from ED presentation to antibiotic administration was 96 minutes. This suggests that another focus of the sepsis workgroup should be on speeding recognition of sepsis, triggering the sepsis alert even sooner, and evaluating the feasibility of storing first doses of antibiotics in the automatic dispensing cabinets in the ED.
Limitations
This descriptive study evaluating CSPs’ ability to accurately use the newly developed antibiotic selection algorithm and vancomycin dosing nomogram had no control group for outcome comparison. This study was not designed to evaluate clinical outcomes, such as mortality, so the impact of these interventions need to be further studied. In addition, as veterans receive most of their care at our facility, with their allergies and previous cultures readily available in our electronic health record, this process may not be feasible at other facilities where patients' care is divided among multiple facilities/systems.
Moreover, as the veteran population studied was predominately male patients aged > 60 years, implementation at other hospitals may require the dosing nomograms and treatment algorithms to be adapted for a broader population, such as children and pregnant women. In particular, the ISC chose to implement an algorithm that did not differentiate between suspected source of infections and included anti-Pseudomonal coverage in all regimens based on the most encountered diseases among our veteran population and our local antibiogram; implementation at other facilities would require a thoughtful evaluation of the most appropriate site-specific regimen. Finally, many of the CSPs at our facility are board certified and/or residency trained, so more staff development may be required prior to implementation at other facilities, depending on the experience and comfort level of the CSPs.
Strengths
This study describes an example of a protocolized and multidisciplinary approach to improve sepsis recognition and standardize the response, consistent with SSC guideline recommendations. To the best of our knowledge, this is the first study to demonstrate the incorporation of CSPs into the interdisciplinary sepsis response. This allows for CSPs to practice at the top of their license and contributes to their professional development. Although it was not formally assessed, anecdotally CSPs reported that this process presented a negligible addition to their workload (< 5 minutes was the most reported time requirement), and they expressed satisfaction with their involvement in the sepsis response. Overall, this presents a possible solution to improve the sepsis response in hospitals without a dedicated ED pharmacist.
Conclusions
This study describes the successful incorporation of CSPs into the sepsis response in the ED. As CSPs are more likely than ED pharmacists to be present at a facility, they are arguably an underused resource whose clinical skills can be used to optimize the treatment of patients with sepsis.
1. Centers for Disease Control and Prevention. Sepsis. Accessed March 8, 2022. https://www.cdc.gov/sepsis/what-is-sepsis.html
2. Rhodes A, Evans LE, Alhazzani W, et al. Surviving Sepsis Campaign: international guidelines for management of sepsis and septic shock: 2016. Crit Care Med. 2017 Mar;45(3):486-552. doi:10.1097/CCM.0000000000002255
3. Denny KJ, Gartside JG, Alcorn K, et al. Appropriateness of antibiotic prescribing in the emergency department. J Antimicrob Chemother. 2019 Feb 1;74(2):515-520. doi:10.1093/jac/dky447
4. Laine ME, Flynn JD, Flannery AH. Impact of pharmacist intervention on selection and timing of appropriate antimicrobial therapy in septic shock. J Pharm Pract. 2018 Feb;31(1):46-51. doi:10.1177/0897190017696953
5. Weant KA, Baker SN. Emergency medicine pharmacists and sepsis management. J Pharm Pract. 2013 Aug;26(4):401-5. doi:10.1177/0897190012467211
6. Farmer BM, Hayes BD, Rao R, et al. The role of clinical pharmacists in the emergency department. J Med Toxicol. 2018 Mar;14(1):114-116. doi:10.1007/s13181-017-0634-4
7. Yarbrough N, Bloxam M, Priano J, Louzon Lynch P, Hunt LN, Elfman J. Pharmacist impact on sepsis bundle compliance through participation on an emergency department sepsis alert team. Am J Emerg Med. 2019;37(4):762-763. doi:10.1016/j.ajem.2018.08.00
8. Thomas MC, Acquisto NM, Shirk MB, et al. A national survey of emergency pharmacy practice in the United States. Am J Health Syst Pharm. 2016 Mar 15;73(6):386-94. doi:10.2146/ajhp150321
9. Schneider PJ, Pedersen CA, Scheckelhoff DJ. ASHP national survey of pharmacy practice in hospital settings: dispensing and administration-2017. Am J Health Syst Pharm. 2018;75(16):1203-1226. doi:10.2146/ajhp180151
Sepsis is life-threatening organ dysfunction caused by dysregulated host response to an infection that can progress to shock. Sepsis is a major cause of death in the United States, with > 1 million people developing sepsis and > 250,000 people dying from sepsis annually.1 The Surviving Sepsis Campaign (SSC) guidelines recommend treating sepsis as an emergency with timely administration of fluids and antibiotics, as administering antibiotics within the first hour has been found to reduce mortality and disease progression. In addition, empiric antibiotic regimens should be chosen to target the most probable pathogens and dosing should be optimized. To achieve this, the SSC guidelines recommend that hospitals develop quality improvement (QI) programs developed by a multidisciplinary group to improve sepsis recognition and response using a protocolized approach.2
There are several studies describing efforts to improve the sepsis response at facilities, some of which have evaluated the addition of a pharmacist into the sepsis response, particularly in the emergency department (ED). Some studies found improved selection and decreased time to antibiotic administration with the addition of an ED pharmacist.3-7 Despite this, ED pharmacists are not present in all hospitals, with a 2015 national survey reporting the presence of an ED pharmacist in 68.7% of respondents at 187 facilities. Even facilities with ED pharmacists often have limited hours of coverage, with at least 8 hours of coverage in 49.4% of facilities with an ED pharmacist and no weekend coverage at 34.8% of these facilities.8
While many hospitals do not routinely employ ED pharmacists, most hospitals have clinical staff pharmacists (CSPs), and many inpatient hospital pharmacies are staffed with CSPs 24 hours per day, 7 days per week. A 2017 survey conducted by the American Society of Health-System Pharmacists (ASHP) found 43% of all hospital pharmacy departments were staffed by a CSP around the clock, with the prevalence increasing to 56.7 to 100% in hospitals with > 100 beds.9 As a result, CSPs may be a useful resource to assist with the management of patients with sepsis in hospitals without an ED pharmacist.
At the Lexington Veterans Affairs Health Care System (LVAHCS) in Kentucky, the inpatient pharmacy department is staffed with a CSP 24/7 but does not have an ED pharmacist. Therefore, when an interdisciplinary group developed an ED sepsis bundle as part of a QI initiative on sepsis recognition and response, the group took a unique approach of incorporating CSPs into the response team to assist with antimicrobial selection and dosing. An antibiotic selection algorithm and vancomycin dosing nomogram were developed to aid CSPs to select and dose antibiotics (Figure, Table 1). We describe the implementation of this process and evaluate CSPs’ accuracy in antimicrobial selection and vancomycin dosing.
Methods
Lexington VAHCS is a 94-bed hospital that provides services to veterans, including an ED, inpatient medical services, surgical services, acute mental health, progressive care, and intensive care units. This facility has 1 antimicrobial stewardship clinical pharmacy specialist, 2 critical care clinical pharmacy specialists, and 16 full-time CSPs with 24-hour CSP coverage. The annual ED volume at the time of this study was approximately 21,000 patients.
Consistent with the SSC guideline recommendation to develop multidisciplinary QI initiatives on sepsis recognition and response, an Interdisciplinary Sepsis Committee (ISC) was created in 2018 comprised of ED, pulmonary, critical care, and infectious diseases licensed independent practitioners (LIPs), ED nurses, and pharmacists. The ISC developed a comprehensive set of sepsis tools that included a sepsis screening tool used by ED triage nurses to provide early detection of sepsis and an updated electronic order set to decrease time to appropriate treatment. This order set included automatic orders for blood cultures and serum lactate, the initiation of IV crystalloids, as well as a Sepsis Alert order placed by ED LIPs which alerted CSPs to a patient with sepsis in the ED.
To ensure a protocol-based approach by the CSPs responding to the sepsis alert, an antibiotic algorithm and vancomycin dosing nomogram were developed by the ISC based on current guideline recommendations and the local antibiogram. These were subsequently approved by ED practitioners, the pharmacy and therapeutics committee, and the critical care committee. The antibiotic algorithm prompts CSPs to perform a chart review to identify β-lactam allergies, evaluate the severity of the allergy and which agents the patient has tolerated in the past, as well as determine whether the patient has a history of extended spectrum β-lactamase (ESBL)–producing organisms from previous cultures. A decision tree then guides CSPs toward the selection of 1 of 5 empiric antibiotic regimens to cover all likely pathogens. The medication orders are then entered by the CSPs as a telephone order from the ED LIP per protocol. Unless patients had a true vancomycin allergy, all patients received vancomycin as the empiric gram-positive backbone of the regimen. The vancomycin dosing nomogram was created to ensure an appropriate and consistent vancomycin weight-based loading dose was administered.
Prior to implementation, the antimicrobial stewardship pharmacist educated CSPs on the use of these tools, including simulated orders for mock sepsis alerts to ensure competency. A copy of the algorithm and nomogram were emailed to all CSPs and posted in a prominent location in the pharmacy.
As part of continuous performance improvement efforts of the ISC, a retrospective cohort study was conducted through chart review on patients at the Lexington VAHCS with an order for a sepsis alert in the ED from December 3, 2018 to May 31, 2020 to assess the accuracy of the CSPs’ antibiotic selection and dosing. Patients were excluded if they had a vancomycin allergy or if the ED practitioner ordered antibiotics prior to the CSPs placing orders. Patients could be included more than once in the study if they had sepsis alerts placed on different dates.
The primary outcomes were CSPs’ accuracy in antimicrobial selection with the antibiotic selection algorithm and vancomycin dosing nomogram. The antibiotic selection was deemed accurate if the appropriate antibiotic regimen was selected based on allergy status and previous cultures as directed in the algorithm. The vancomycin dose was considered accurate if the dose chosen was appropriate based on the patient’s weight at the time of ED presentation. Secondary outcomes included time to administration of antibiotics from ED presentation as well as time to antibiotics administration from sepsis alert initiation. Time of administration was considered the time the antibiotics were scanned in the bar code medication administration (BCMA) system.
Descriptive statistics were used with data presented as percentages for nominal data and median as IQR for continuous data. In accordance with our facility’s project assessment process, this project was determined not to constitute human subjects research; therefore, this QI project did not require review by the institutional review board.
Results
Between December 3, 2018 and May 31, 2020, 160 sepsis alerts were ordered by ED practitioners. Of the 160 patients, 157 were included in the final data analysis. Two patients were excluded due to vancomycin allergy, and 1 patient because the physician ordered antibiotics prior to pharmacist order entry. The population was largely composed of male patients (98%) with a median age of 72 years (Table 2).
Of 157 sepsis alerts, the antibiotic selection algorithm was used appropriately in 154 (98%) instances (Table 3). Chart reviews were performed in instances of antimicrobial selection different from the algorithm. Of the 3 patients who received antibiotics not consistent with the algorithm, 1 patient without a history of ESBL-producing organisms in their culture history received meropenem instead of piperacillin/tazobactam. Another patient without a penicillin allergy received cefepime (plus metronidazole ordered separately from the ED practitioner) instead of piperacillin/tazobactam, and the third patient received piperacillin/tazobactam instead of meropenem despite a culture history of ESBL-producing organisms. Vancomycin dose was appropriate according to the weight-based nomogram in 147 cases (94%). The median time to administration of first dose antibiotics was 39 minutes after the sepsis alert order was placed and 96 minutes after initial ED presentation.
Discussion
This study found extremely high rates of accuracy among CSPs for both the antibiotic selection algorithm (98%) and the vancomycin dosing nomogram (94%). Moreover, analysis of the 3 patients who received antibiotics that were inconsistent with the algorithm revealed that 2 of these patients arguably still received adequate empiric coverage, increasing the percentage of patients receiving appropriate empiric antibiotics to 99.4%. Similarly, chart review of 10 patients who received vancomycin doses that deviated from the nomogram revealed that in at least 3 cases, patients were likely given correct vancomycin doses based on the patient’s last known weight. However, when actual current weights were recorded soon after admission, the updated weights rendered the initial vancomycin loading dose incorrect when this analysis was performed. Thus, the adherence to the vancomycin dosing nomogram is higher than it appears.
Median time to antibiotic administration from the sepsis alert was 39 minutes—well within SSC recommendations (60 minutes).2 Previous internal analyses at Lexington VAHCS demonstrated the mean time to first dose of antibiotics in the ED has been 39 minutes since about 2015. Thus, this initiative did not necessarily make this process quicker; however it did remove 1 responsibility from LIPs so that they could focus their efforts on other components of sepsis management.
Further studies are needed to evaluate the effects of this initiative on other aspects of the sepsis bundle, such as volume of fluid administered and appropriateness of laboratory tests. It was noted that while the time to first-dose antibiotic administration was < 1 hour from order placement, the median time from ED presentation to antibiotic administration was 96 minutes. This suggests that another focus of the sepsis workgroup should be on speeding recognition of sepsis, triggering the sepsis alert even sooner, and evaluating the feasibility of storing first doses of antibiotics in the automatic dispensing cabinets in the ED.
Limitations
This descriptive study evaluating CSPs’ ability to accurately use the newly developed antibiotic selection algorithm and vancomycin dosing nomogram had no control group for outcome comparison. This study was not designed to evaluate clinical outcomes, such as mortality, so the impact of these interventions need to be further studied. In addition, as veterans receive most of their care at our facility, with their allergies and previous cultures readily available in our electronic health record, this process may not be feasible at other facilities where patients' care is divided among multiple facilities/systems.
Moreover, as the veteran population studied was predominately male patients aged > 60 years, implementation at other hospitals may require the dosing nomograms and treatment algorithms to be adapted for a broader population, such as children and pregnant women. In particular, the ISC chose to implement an algorithm that did not differentiate between suspected source of infections and included anti-Pseudomonal coverage in all regimens based on the most encountered diseases among our veteran population and our local antibiogram; implementation at other facilities would require a thoughtful evaluation of the most appropriate site-specific regimen. Finally, many of the CSPs at our facility are board certified and/or residency trained, so more staff development may be required prior to implementation at other facilities, depending on the experience and comfort level of the CSPs.
Strengths
This study describes an example of a protocolized and multidisciplinary approach to improve sepsis recognition and standardize the response, consistent with SSC guideline recommendations. To the best of our knowledge, this is the first study to demonstrate the incorporation of CSPs into the interdisciplinary sepsis response. This allows for CSPs to practice at the top of their license and contributes to their professional development. Although it was not formally assessed, anecdotally CSPs reported that this process presented a negligible addition to their workload (< 5 minutes was the most reported time requirement), and they expressed satisfaction with their involvement in the sepsis response. Overall, this presents a possible solution to improve the sepsis response in hospitals without a dedicated ED pharmacist.
Conclusions
This study describes the successful incorporation of CSPs into the sepsis response in the ED. As CSPs are more likely than ED pharmacists to be present at a facility, they are arguably an underused resource whose clinical skills can be used to optimize the treatment of patients with sepsis.
Sepsis is life-threatening organ dysfunction caused by dysregulated host response to an infection that can progress to shock. Sepsis is a major cause of death in the United States, with > 1 million people developing sepsis and > 250,000 people dying from sepsis annually.1 The Surviving Sepsis Campaign (SSC) guidelines recommend treating sepsis as an emergency with timely administration of fluids and antibiotics, as administering antibiotics within the first hour has been found to reduce mortality and disease progression. In addition, empiric antibiotic regimens should be chosen to target the most probable pathogens and dosing should be optimized. To achieve this, the SSC guidelines recommend that hospitals develop quality improvement (QI) programs developed by a multidisciplinary group to improve sepsis recognition and response using a protocolized approach.2
There are several studies describing efforts to improve the sepsis response at facilities, some of which have evaluated the addition of a pharmacist into the sepsis response, particularly in the emergency department (ED). Some studies found improved selection and decreased time to antibiotic administration with the addition of an ED pharmacist.3-7 Despite this, ED pharmacists are not present in all hospitals, with a 2015 national survey reporting the presence of an ED pharmacist in 68.7% of respondents at 187 facilities. Even facilities with ED pharmacists often have limited hours of coverage, with at least 8 hours of coverage in 49.4% of facilities with an ED pharmacist and no weekend coverage at 34.8% of these facilities.8
While many hospitals do not routinely employ ED pharmacists, most hospitals have clinical staff pharmacists (CSPs), and many inpatient hospital pharmacies are staffed with CSPs 24 hours per day, 7 days per week. A 2017 survey conducted by the American Society of Health-System Pharmacists (ASHP) found 43% of all hospital pharmacy departments were staffed by a CSP around the clock, with the prevalence increasing to 56.7 to 100% in hospitals with > 100 beds.9 As a result, CSPs may be a useful resource to assist with the management of patients with sepsis in hospitals without an ED pharmacist.
At the Lexington Veterans Affairs Health Care System (LVAHCS) in Kentucky, the inpatient pharmacy department is staffed with a CSP 24/7 but does not have an ED pharmacist. Therefore, when an interdisciplinary group developed an ED sepsis bundle as part of a QI initiative on sepsis recognition and response, the group took a unique approach of incorporating CSPs into the response team to assist with antimicrobial selection and dosing. An antibiotic selection algorithm and vancomycin dosing nomogram were developed to aid CSPs to select and dose antibiotics (Figure, Table 1). We describe the implementation of this process and evaluate CSPs’ accuracy in antimicrobial selection and vancomycin dosing.
Methods
Lexington VAHCS is a 94-bed hospital that provides services to veterans, including an ED, inpatient medical services, surgical services, acute mental health, progressive care, and intensive care units. This facility has 1 antimicrobial stewardship clinical pharmacy specialist, 2 critical care clinical pharmacy specialists, and 16 full-time CSPs with 24-hour CSP coverage. The annual ED volume at the time of this study was approximately 21,000 patients.
Consistent with the SSC guideline recommendation to develop multidisciplinary QI initiatives on sepsis recognition and response, an Interdisciplinary Sepsis Committee (ISC) was created in 2018 comprised of ED, pulmonary, critical care, and infectious diseases licensed independent practitioners (LIPs), ED nurses, and pharmacists. The ISC developed a comprehensive set of sepsis tools that included a sepsis screening tool used by ED triage nurses to provide early detection of sepsis and an updated electronic order set to decrease time to appropriate treatment. This order set included automatic orders for blood cultures and serum lactate, the initiation of IV crystalloids, as well as a Sepsis Alert order placed by ED LIPs which alerted CSPs to a patient with sepsis in the ED.
To ensure a protocol-based approach by the CSPs responding to the sepsis alert, an antibiotic algorithm and vancomycin dosing nomogram were developed by the ISC based on current guideline recommendations and the local antibiogram. These were subsequently approved by ED practitioners, the pharmacy and therapeutics committee, and the critical care committee. The antibiotic algorithm prompts CSPs to perform a chart review to identify β-lactam allergies, evaluate the severity of the allergy and which agents the patient has tolerated in the past, as well as determine whether the patient has a history of extended spectrum β-lactamase (ESBL)–producing organisms from previous cultures. A decision tree then guides CSPs toward the selection of 1 of 5 empiric antibiotic regimens to cover all likely pathogens. The medication orders are then entered by the CSPs as a telephone order from the ED LIP per protocol. Unless patients had a true vancomycin allergy, all patients received vancomycin as the empiric gram-positive backbone of the regimen. The vancomycin dosing nomogram was created to ensure an appropriate and consistent vancomycin weight-based loading dose was administered.
Prior to implementation, the antimicrobial stewardship pharmacist educated CSPs on the use of these tools, including simulated orders for mock sepsis alerts to ensure competency. A copy of the algorithm and nomogram were emailed to all CSPs and posted in a prominent location in the pharmacy.
As part of continuous performance improvement efforts of the ISC, a retrospective cohort study was conducted through chart review on patients at the Lexington VAHCS with an order for a sepsis alert in the ED from December 3, 2018 to May 31, 2020 to assess the accuracy of the CSPs’ antibiotic selection and dosing. Patients were excluded if they had a vancomycin allergy or if the ED practitioner ordered antibiotics prior to the CSPs placing orders. Patients could be included more than once in the study if they had sepsis alerts placed on different dates.
The primary outcomes were CSPs’ accuracy in antimicrobial selection with the antibiotic selection algorithm and vancomycin dosing nomogram. The antibiotic selection was deemed accurate if the appropriate antibiotic regimen was selected based on allergy status and previous cultures as directed in the algorithm. The vancomycin dose was considered accurate if the dose chosen was appropriate based on the patient’s weight at the time of ED presentation. Secondary outcomes included time to administration of antibiotics from ED presentation as well as time to antibiotics administration from sepsis alert initiation. Time of administration was considered the time the antibiotics were scanned in the bar code medication administration (BCMA) system.
Descriptive statistics were used with data presented as percentages for nominal data and median as IQR for continuous data. In accordance with our facility’s project assessment process, this project was determined not to constitute human subjects research; therefore, this QI project did not require review by the institutional review board.
Results
Between December 3, 2018 and May 31, 2020, 160 sepsis alerts were ordered by ED practitioners. Of the 160 patients, 157 were included in the final data analysis. Two patients were excluded due to vancomycin allergy, and 1 patient because the physician ordered antibiotics prior to pharmacist order entry. The population was largely composed of male patients (98%) with a median age of 72 years (Table 2).
Of 157 sepsis alerts, the antibiotic selection algorithm was used appropriately in 154 (98%) instances (Table 3). Chart reviews were performed in instances of antimicrobial selection different from the algorithm. Of the 3 patients who received antibiotics not consistent with the algorithm, 1 patient without a history of ESBL-producing organisms in their culture history received meropenem instead of piperacillin/tazobactam. Another patient without a penicillin allergy received cefepime (plus metronidazole ordered separately from the ED practitioner) instead of piperacillin/tazobactam, and the third patient received piperacillin/tazobactam instead of meropenem despite a culture history of ESBL-producing organisms. Vancomycin dose was appropriate according to the weight-based nomogram in 147 cases (94%). The median time to administration of first dose antibiotics was 39 minutes after the sepsis alert order was placed and 96 minutes after initial ED presentation.
Discussion
This study found extremely high rates of accuracy among CSPs for both the antibiotic selection algorithm (98%) and the vancomycin dosing nomogram (94%). Moreover, analysis of the 3 patients who received antibiotics that were inconsistent with the algorithm revealed that 2 of these patients arguably still received adequate empiric coverage, increasing the percentage of patients receiving appropriate empiric antibiotics to 99.4%. Similarly, chart review of 10 patients who received vancomycin doses that deviated from the nomogram revealed that in at least 3 cases, patients were likely given correct vancomycin doses based on the patient’s last known weight. However, when actual current weights were recorded soon after admission, the updated weights rendered the initial vancomycin loading dose incorrect when this analysis was performed. Thus, the adherence to the vancomycin dosing nomogram is higher than it appears.
Median time to antibiotic administration from the sepsis alert was 39 minutes—well within SSC recommendations (60 minutes).2 Previous internal analyses at Lexington VAHCS demonstrated the mean time to first dose of antibiotics in the ED has been 39 minutes since about 2015. Thus, this initiative did not necessarily make this process quicker; however it did remove 1 responsibility from LIPs so that they could focus their efforts on other components of sepsis management.
Further studies are needed to evaluate the effects of this initiative on other aspects of the sepsis bundle, such as volume of fluid administered and appropriateness of laboratory tests. It was noted that while the time to first-dose antibiotic administration was < 1 hour from order placement, the median time from ED presentation to antibiotic administration was 96 minutes. This suggests that another focus of the sepsis workgroup should be on speeding recognition of sepsis, triggering the sepsis alert even sooner, and evaluating the feasibility of storing first doses of antibiotics in the automatic dispensing cabinets in the ED.
Limitations
This descriptive study evaluating CSPs’ ability to accurately use the newly developed antibiotic selection algorithm and vancomycin dosing nomogram had no control group for outcome comparison. This study was not designed to evaluate clinical outcomes, such as mortality, so the impact of these interventions need to be further studied. In addition, as veterans receive most of their care at our facility, with their allergies and previous cultures readily available in our electronic health record, this process may not be feasible at other facilities where patients' care is divided among multiple facilities/systems.
Moreover, as the veteran population studied was predominately male patients aged > 60 years, implementation at other hospitals may require the dosing nomograms and treatment algorithms to be adapted for a broader population, such as children and pregnant women. In particular, the ISC chose to implement an algorithm that did not differentiate between suspected source of infections and included anti-Pseudomonal coverage in all regimens based on the most encountered diseases among our veteran population and our local antibiogram; implementation at other facilities would require a thoughtful evaluation of the most appropriate site-specific regimen. Finally, many of the CSPs at our facility are board certified and/or residency trained, so more staff development may be required prior to implementation at other facilities, depending on the experience and comfort level of the CSPs.
Strengths
This study describes an example of a protocolized and multidisciplinary approach to improve sepsis recognition and standardize the response, consistent with SSC guideline recommendations. To the best of our knowledge, this is the first study to demonstrate the incorporation of CSPs into the interdisciplinary sepsis response. This allows for CSPs to practice at the top of their license and contributes to their professional development. Although it was not formally assessed, anecdotally CSPs reported that this process presented a negligible addition to their workload (< 5 minutes was the most reported time requirement), and they expressed satisfaction with their involvement in the sepsis response. Overall, this presents a possible solution to improve the sepsis response in hospitals without a dedicated ED pharmacist.
Conclusions
This study describes the successful incorporation of CSPs into the sepsis response in the ED. As CSPs are more likely than ED pharmacists to be present at a facility, they are arguably an underused resource whose clinical skills can be used to optimize the treatment of patients with sepsis.
1. Centers for Disease Control and Prevention. Sepsis. Accessed March 8, 2022. https://www.cdc.gov/sepsis/what-is-sepsis.html
2. Rhodes A, Evans LE, Alhazzani W, et al. Surviving Sepsis Campaign: international guidelines for management of sepsis and septic shock: 2016. Crit Care Med. 2017 Mar;45(3):486-552. doi:10.1097/CCM.0000000000002255
3. Denny KJ, Gartside JG, Alcorn K, et al. Appropriateness of antibiotic prescribing in the emergency department. J Antimicrob Chemother. 2019 Feb 1;74(2):515-520. doi:10.1093/jac/dky447
4. Laine ME, Flynn JD, Flannery AH. Impact of pharmacist intervention on selection and timing of appropriate antimicrobial therapy in septic shock. J Pharm Pract. 2018 Feb;31(1):46-51. doi:10.1177/0897190017696953
5. Weant KA, Baker SN. Emergency medicine pharmacists and sepsis management. J Pharm Pract. 2013 Aug;26(4):401-5. doi:10.1177/0897190012467211
6. Farmer BM, Hayes BD, Rao R, et al. The role of clinical pharmacists in the emergency department. J Med Toxicol. 2018 Mar;14(1):114-116. doi:10.1007/s13181-017-0634-4
7. Yarbrough N, Bloxam M, Priano J, Louzon Lynch P, Hunt LN, Elfman J. Pharmacist impact on sepsis bundle compliance through participation on an emergency department sepsis alert team. Am J Emerg Med. 2019;37(4):762-763. doi:10.1016/j.ajem.2018.08.00
8. Thomas MC, Acquisto NM, Shirk MB, et al. A national survey of emergency pharmacy practice in the United States. Am J Health Syst Pharm. 2016 Mar 15;73(6):386-94. doi:10.2146/ajhp150321
9. Schneider PJ, Pedersen CA, Scheckelhoff DJ. ASHP national survey of pharmacy practice in hospital settings: dispensing and administration-2017. Am J Health Syst Pharm. 2018;75(16):1203-1226. doi:10.2146/ajhp180151
1. Centers for Disease Control and Prevention. Sepsis. Accessed March 8, 2022. https://www.cdc.gov/sepsis/what-is-sepsis.html
2. Rhodes A, Evans LE, Alhazzani W, et al. Surviving Sepsis Campaign: international guidelines for management of sepsis and septic shock: 2016. Crit Care Med. 2017 Mar;45(3):486-552. doi:10.1097/CCM.0000000000002255
3. Denny KJ, Gartside JG, Alcorn K, et al. Appropriateness of antibiotic prescribing in the emergency department. J Antimicrob Chemother. 2019 Feb 1;74(2):515-520. doi:10.1093/jac/dky447
4. Laine ME, Flynn JD, Flannery AH. Impact of pharmacist intervention on selection and timing of appropriate antimicrobial therapy in septic shock. J Pharm Pract. 2018 Feb;31(1):46-51. doi:10.1177/0897190017696953
5. Weant KA, Baker SN. Emergency medicine pharmacists and sepsis management. J Pharm Pract. 2013 Aug;26(4):401-5. doi:10.1177/0897190012467211
6. Farmer BM, Hayes BD, Rao R, et al. The role of clinical pharmacists in the emergency department. J Med Toxicol. 2018 Mar;14(1):114-116. doi:10.1007/s13181-017-0634-4
7. Yarbrough N, Bloxam M, Priano J, Louzon Lynch P, Hunt LN, Elfman J. Pharmacist impact on sepsis bundle compliance through participation on an emergency department sepsis alert team. Am J Emerg Med. 2019;37(4):762-763. doi:10.1016/j.ajem.2018.08.00
8. Thomas MC, Acquisto NM, Shirk MB, et al. A national survey of emergency pharmacy practice in the United States. Am J Health Syst Pharm. 2016 Mar 15;73(6):386-94. doi:10.2146/ajhp150321
9. Schneider PJ, Pedersen CA, Scheckelhoff DJ. ASHP national survey of pharmacy practice in hospital settings: dispensing and administration-2017. Am J Health Syst Pharm. 2018;75(16):1203-1226. doi:10.2146/ajhp180151
U.S. pulls COVID drug as Omicron subvariant spreads
F, the Omicron subvariant that now accounts for most new cases in the United States, The Associated Press reports.
The Food and Drug Administration announced that the antibody drug sotrovimab is no longer authorized to treat patients in U.S. states or territories. The decision was expected, as the FDA restricted the drug’s use across the country throughout March as BA.2 became dominant in certain regions, the AP reported.
The BA.2 subvariant now accounts for 72% of new COVID-19 cases sequenced by health authorities, according to the latest CDC data updated April 5. The FDA cited the CDC data in its reason for pulling back on the authorization of the drug.
The GlaxoSmithKline drug is the latest antibody medication to be pulled due to coronavirus mutations. In January, the FDA halted the use of antibody drugs from Regeneron and Eli Lilly because they didn’t work against the Omicron variant.
The FDA’s decision means that one antibody drug is still authorized for use against routine COVID-19 cases, the AP reported. A different Eli Lilly drug – bebtelovimab – still appears to work against BA.2.
Doctors can also prescribe antiviral pills, which typically affect the coronavirus spike protein and aren’t affected by mutations, to treat mild to moderate COVID-19, the AP reported. The authorized pills from Pfizer and Merck – Paxlovid and Lagevrio – have been shipped to pharmacy chains and medical clinics in hopes of getting them to patients early enough to work.
The federal government purchased nearly $2 billion worth of the GlaxoSmithKline drug and shipped more than 900,000 doses to states last fall, the AP reported. In March, the company announced that it was studying a higher dose that could be effective against BA.2, which would require FDA approval before resuming use in the United States.
The antibody drugs mimic the virus-blocking proteins found in the human body, the AP reported. They’re designed to attack a specific virus and need to be updated as the coronavirus mutates.
A version of this article first appeared on WebMD.com.
F, the Omicron subvariant that now accounts for most new cases in the United States, The Associated Press reports.
The Food and Drug Administration announced that the antibody drug sotrovimab is no longer authorized to treat patients in U.S. states or territories. The decision was expected, as the FDA restricted the drug’s use across the country throughout March as BA.2 became dominant in certain regions, the AP reported.
The BA.2 subvariant now accounts for 72% of new COVID-19 cases sequenced by health authorities, according to the latest CDC data updated April 5. The FDA cited the CDC data in its reason for pulling back on the authorization of the drug.
The GlaxoSmithKline drug is the latest antibody medication to be pulled due to coronavirus mutations. In January, the FDA halted the use of antibody drugs from Regeneron and Eli Lilly because they didn’t work against the Omicron variant.
The FDA’s decision means that one antibody drug is still authorized for use against routine COVID-19 cases, the AP reported. A different Eli Lilly drug – bebtelovimab – still appears to work against BA.2.
Doctors can also prescribe antiviral pills, which typically affect the coronavirus spike protein and aren’t affected by mutations, to treat mild to moderate COVID-19, the AP reported. The authorized pills from Pfizer and Merck – Paxlovid and Lagevrio – have been shipped to pharmacy chains and medical clinics in hopes of getting them to patients early enough to work.
The federal government purchased nearly $2 billion worth of the GlaxoSmithKline drug and shipped more than 900,000 doses to states last fall, the AP reported. In March, the company announced that it was studying a higher dose that could be effective against BA.2, which would require FDA approval before resuming use in the United States.
The antibody drugs mimic the virus-blocking proteins found in the human body, the AP reported. They’re designed to attack a specific virus and need to be updated as the coronavirus mutates.
A version of this article first appeared on WebMD.com.
F, the Omicron subvariant that now accounts for most new cases in the United States, The Associated Press reports.
The Food and Drug Administration announced that the antibody drug sotrovimab is no longer authorized to treat patients in U.S. states or territories. The decision was expected, as the FDA restricted the drug’s use across the country throughout March as BA.2 became dominant in certain regions, the AP reported.
The BA.2 subvariant now accounts for 72% of new COVID-19 cases sequenced by health authorities, according to the latest CDC data updated April 5. The FDA cited the CDC data in its reason for pulling back on the authorization of the drug.
The GlaxoSmithKline drug is the latest antibody medication to be pulled due to coronavirus mutations. In January, the FDA halted the use of antibody drugs from Regeneron and Eli Lilly because they didn’t work against the Omicron variant.
The FDA’s decision means that one antibody drug is still authorized for use against routine COVID-19 cases, the AP reported. A different Eli Lilly drug – bebtelovimab – still appears to work against BA.2.
Doctors can also prescribe antiviral pills, which typically affect the coronavirus spike protein and aren’t affected by mutations, to treat mild to moderate COVID-19, the AP reported. The authorized pills from Pfizer and Merck – Paxlovid and Lagevrio – have been shipped to pharmacy chains and medical clinics in hopes of getting them to patients early enough to work.
The federal government purchased nearly $2 billion worth of the GlaxoSmithKline drug and shipped more than 900,000 doses to states last fall, the AP reported. In March, the company announced that it was studying a higher dose that could be effective against BA.2, which would require FDA approval before resuming use in the United States.
The antibody drugs mimic the virus-blocking proteins found in the human body, the AP reported. They’re designed to attack a specific virus and need to be updated as the coronavirus mutates.
A version of this article first appeared on WebMD.com.
Novel medication tied to better quality of life in major depression
DENVER –
In a phase 3 trial that included more than 500 adult patients with MDD, those who received zuranolone for 14 days showed greater improvement at day 15 across numerous QoL outcomes, compared with their counterparts in the placebo group.

In addition, combined analysis of four zuranolone clinical trials showed “mental well-being and functioning improved to near general population norm levels” for the active-treatment group, reported the researchers, led by Anita H. Clayton, MD, chair and professor of psychiatry, University of Virginia, Charlottesville.
“Based on these integrated analyses, the benefit of treatment with zuranolone may extend beyond reduction in depressive symptoms to include potential improvement in quality of life and overall health, as perceived by patients,” they add.
The findings were presented as part of the Anxiety and Depression Association of America Anxiety & Depression conference.
First oral formulation
Zuranolone represents the second entry in the new class of neuroactive steroid drugs, which modulate GABA-A receptor activity – but it would be the first to have an oral formulation. Brexanolone, which was approved by the Food and Drug Administration in 2019 for postpartum depression, is administered through continuous IV infusion over 60 hours.
As previously reported by this news organization, zuranolone improved depressive symptoms as early as day 3, achieving the primary endpoint of significantly greater reduction in scores on the 17-item Hamilton Rating Scale for Depression from baseline to day 15 versus placebo (P = .014).
In the new analysis, patient-reported measures of functional health and well-being were assessed in the WATERFALL trial. It included 266 patients with MDD who were treated with zuranolone 50 mg daily for 2 weeks and 268 patients with MDD who were treated with placebo.
The study used the Short Form–36 (SF-36v2), which covers a wide range of patient-reported measures, including physical function, bodily pain, general health, vitality, social function, and “role-emotional” symptoms.
Results showed that although the treatment and placebo groups had similar baseline SF-36v2 scores, those receiving zuranolone reported significantly greater improvements at day 15 in almost all of the assessment’s domains, including physical function (treatment difference, 0.8), general health (1.0), vitality (3.1), social functioning (1.1), and role-emotional symptoms (1.5; for all comparisons, P < .05). The only exceptions were in role-physical symptoms and bodily pain.
In measures that included physical function, bodily pain, and general health, the patients achieved improvements at day 15 that were consistent with normal levels, with the improvement in vitality considered clinically meaningful versus placebo.
Integrated data
In further analysis of integrated data from four zuranolone clinical trials in the NEST and LANDSCAPE programs for patients with MDD and postpartum depression, results showed similar improvements at day 15 for zuranolone in QoL and overall health across all of the SF-36v2 functioning and well-being domains (P <.05), with the exceptions of physical measure and bodily pain.
By day 42, all of the domains showed significantly greater improvement with zuranolone versus placebo (all, P <.05).
Among the strongest score improvements in the integrated trials were measures in social functioning, which improved from baseline scores of 29.66 to 42.82 on day 15 and to 43.59 on day 42.
Emotional domain scores improved from 24.43 at baseline to 39.13 on day 15 and to 39.82 on day 42. For mental health, the integrated scores for the zuranolone group improved from 27.13 at baseline to 42.40 on day 15 and 42.62 on day 42.
Of note, the baseline scores for mental health represented just 54.3% of those in the normal population; with the increase at day 15, the level was 84.8% of the normal population.
“Across four completed placebo-controlled NEST and LANDSCAPE clinical trials, patient reports of functional health and well-being as assessed by the SF-36v2 indicated substantial impairment at baseline compared to the population norm,” the researchers reported.
The improvements are especially important in light of the fact that in some patients with MDD, functional improvement is a top priority.
“Patients have often prioritized returning to their usual level of functioning over reduction in depressive symptoms, and functional recovery has been associated with better prognosis of depression,” the investigators wrote.
Zuranolone trials have shown that treatment-emergent adverse events (AEs) occur among about 60% of patients, versus about 44% with placebo. The most common AEs are somnolence, dizziness, headache, sedation, and diarrhea, with no increases in suicidal ideation or withdrawal.
The rates of severe AEs are low, and they are observed in about 3% of patients, versus 1.1% with placebo, the researchers noted.
Further, as opposed to serotonergic antidepressants such as SNRIs and SSRIs, zuranolone does not appear to have the undesirable side effects of decreased libido and sexual dysfunction, they added.
Clinically meaningful?
Andrew J. Cutler, MD, clinical associate professor of psychiatry at State University of New York, Syracuse, said the data are “very significant” for a number of reasons.
“We need more options to treat depression, especially ones with novel mechanisms of action and faster onset of efficacy, such as zuranolone,” said Dr. Cutler, who was not involved in the current study. He has coauthored other studies on zuranolone.
Regarding the study’s QoL outcomes, “while improvement in depressive symptoms is very important, what really matters to patients is improvement in function and quality of life,” Dr. Cutler noted.
Also commenting on the study, Jonathan E. Alpert, MD, PhD, chair of the department of psychiatry and behavioral sciences and professor of psychiatry, neuroscience, and pediatrics at Albert Einstein College of Medicine, New York, said the investigational drug could represent an important addition to the armamentarium for treating depression.
“Zuranolone has good oral bioavailability and would represent the first neuroactive steroid antidepressant available in oral form and, indeed, the first non–monoamine-based antidepressant available in oral form,” he said in an interview.

Dr. Alpert was not involved in the research and has no relationship with the drug’s development.
He noted that although there are modest differences between the patients who received zuranolone and those who received placebo in the trials, “this may have been related to high placebo response rates, which often complicate antidepressant trials.
“Further research is needed to determine whether differences between zuranolone and placebo are clinically meaningful, though the separation between drug and placebo on the primary endpoint, as well as some other measures, such as quality of life measures, is promising,” Dr. Alpert said.
However, he added that comparisons with other active antidepressants in terms of efficacy and tolerability remain to be seen.
“Given the large number of individuals with major depressive disorder who have incomplete response to or do not tolerate monoaminergic antidepressants, the development of agents that leverage novel nonmonoaminergic mechanisms is important,” Dr. Alpert concluded.
The study was funded by Sage Therapeutics and Biogen. Dr. Cutler has been involved in research of zuranolone for Sage Therapeutics. Dr. Alpert has reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
DENVER –
In a phase 3 trial that included more than 500 adult patients with MDD, those who received zuranolone for 14 days showed greater improvement at day 15 across numerous QoL outcomes, compared with their counterparts in the placebo group.
In addition, combined analysis of four zuranolone clinical trials showed “mental well-being and functioning improved to near general population norm levels” for the active-treatment group, reported the researchers, led by Anita H. Clayton, MD, chair and professor of psychiatry, University of Virginia, Charlottesville.
“Based on these integrated analyses, the benefit of treatment with zuranolone may extend beyond reduction in depressive symptoms to include potential improvement in quality of life and overall health, as perceived by patients,” they add.
The findings were presented as part of the Anxiety and Depression Association of America Anxiety & Depression conference.
First oral formulation
Zuranolone represents the second entry in the new class of neuroactive steroid drugs, which modulate GABA-A receptor activity – but it would be the first to have an oral formulation. Brexanolone, which was approved by the Food and Drug Administration in 2019 for postpartum depression, is administered through continuous IV infusion over 60 hours.
As previously reported by this news organization, zuranolone improved depressive symptoms as early as day 3, achieving the primary endpoint of significantly greater reduction in scores on the 17-item Hamilton Rating Scale for Depression from baseline to day 15 versus placebo (P = .014).
In the new analysis, patient-reported measures of functional health and well-being were assessed in the WATERFALL trial. It included 266 patients with MDD who were treated with zuranolone 50 mg daily for 2 weeks and 268 patients with MDD who were treated with placebo.
The study used the Short Form–36 (SF-36v2), which covers a wide range of patient-reported measures, including physical function, bodily pain, general health, vitality, social function, and “role-emotional” symptoms.
Results showed that although the treatment and placebo groups had similar baseline SF-36v2 scores, those receiving zuranolone reported significantly greater improvements at day 15 in almost all of the assessment’s domains, including physical function (treatment difference, 0.8), general health (1.0), vitality (3.1), social functioning (1.1), and role-emotional symptoms (1.5; for all comparisons, P < .05). The only exceptions were in role-physical symptoms and bodily pain.
In measures that included physical function, bodily pain, and general health, the patients achieved improvements at day 15 that were consistent with normal levels, with the improvement in vitality considered clinically meaningful versus placebo.
Integrated data
In further analysis of integrated data from four zuranolone clinical trials in the NEST and LANDSCAPE programs for patients with MDD and postpartum depression, results showed similar improvements at day 15 for zuranolone in QoL and overall health across all of the SF-36v2 functioning and well-being domains (P <.05), with the exceptions of physical measure and bodily pain.
By day 42, all of the domains showed significantly greater improvement with zuranolone versus placebo (all, P <.05).
Among the strongest score improvements in the integrated trials were measures in social functioning, which improved from baseline scores of 29.66 to 42.82 on day 15 and to 43.59 on day 42.
Emotional domain scores improved from 24.43 at baseline to 39.13 on day 15 and to 39.82 on day 42. For mental health, the integrated scores for the zuranolone group improved from 27.13 at baseline to 42.40 on day 15 and 42.62 on day 42.
Of note, the baseline scores for mental health represented just 54.3% of those in the normal population; with the increase at day 15, the level was 84.8% of the normal population.
“Across four completed placebo-controlled NEST and LANDSCAPE clinical trials, patient reports of functional health and well-being as assessed by the SF-36v2 indicated substantial impairment at baseline compared to the population norm,” the researchers reported.
The improvements are especially important in light of the fact that in some patients with MDD, functional improvement is a top priority.
“Patients have often prioritized returning to their usual level of functioning over reduction in depressive symptoms, and functional recovery has been associated with better prognosis of depression,” the investigators wrote.
Zuranolone trials have shown that treatment-emergent adverse events (AEs) occur among about 60% of patients, versus about 44% with placebo. The most common AEs are somnolence, dizziness, headache, sedation, and diarrhea, with no increases in suicidal ideation or withdrawal.
The rates of severe AEs are low, and they are observed in about 3% of patients, versus 1.1% with placebo, the researchers noted.
Further, as opposed to serotonergic antidepressants such as SNRIs and SSRIs, zuranolone does not appear to have the undesirable side effects of decreased libido and sexual dysfunction, they added.
Clinically meaningful?
Andrew J. Cutler, MD, clinical associate professor of psychiatry at State University of New York, Syracuse, said the data are “very significant” for a number of reasons.
“We need more options to treat depression, especially ones with novel mechanisms of action and faster onset of efficacy, such as zuranolone,” said Dr. Cutler, who was not involved in the current study. He has coauthored other studies on zuranolone.
Regarding the study’s QoL outcomes, “while improvement in depressive symptoms is very important, what really matters to patients is improvement in function and quality of life,” Dr. Cutler noted.
Also commenting on the study, Jonathan E. Alpert, MD, PhD, chair of the department of psychiatry and behavioral sciences and professor of psychiatry, neuroscience, and pediatrics at Albert Einstein College of Medicine, New York, said the investigational drug could represent an important addition to the armamentarium for treating depression.
“Zuranolone has good oral bioavailability and would represent the first neuroactive steroid antidepressant available in oral form and, indeed, the first non–monoamine-based antidepressant available in oral form,” he said in an interview.
Dr. Alpert was not involved in the research and has no relationship with the drug’s development.
He noted that although there are modest differences between the patients who received zuranolone and those who received placebo in the trials, “this may have been related to high placebo response rates, which often complicate antidepressant trials.
“Further research is needed to determine whether differences between zuranolone and placebo are clinically meaningful, though the separation between drug and placebo on the primary endpoint, as well as some other measures, such as quality of life measures, is promising,” Dr. Alpert said.
However, he added that comparisons with other active antidepressants in terms of efficacy and tolerability remain to be seen.
“Given the large number of individuals with major depressive disorder who have incomplete response to or do not tolerate monoaminergic antidepressants, the development of agents that leverage novel nonmonoaminergic mechanisms is important,” Dr. Alpert concluded.
The study was funded by Sage Therapeutics and Biogen. Dr. Cutler has been involved in research of zuranolone for Sage Therapeutics. Dr. Alpert has reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
DENVER –
In a phase 3 trial that included more than 500 adult patients with MDD, those who received zuranolone for 14 days showed greater improvement at day 15 across numerous QoL outcomes, compared with their counterparts in the placebo group.
In addition, combined analysis of four zuranolone clinical trials showed “mental well-being and functioning improved to near general population norm levels” for the active-treatment group, reported the researchers, led by Anita H. Clayton, MD, chair and professor of psychiatry, University of Virginia, Charlottesville.
“Based on these integrated analyses, the benefit of treatment with zuranolone may extend beyond reduction in depressive symptoms to include potential improvement in quality of life and overall health, as perceived by patients,” they add.
The findings were presented as part of the Anxiety and Depression Association of America Anxiety & Depression conference.
First oral formulation
Zuranolone represents the second entry in the new class of neuroactive steroid drugs, which modulate GABA-A receptor activity – but it would be the first to have an oral formulation. Brexanolone, which was approved by the Food and Drug Administration in 2019 for postpartum depression, is administered through continuous IV infusion over 60 hours.
As previously reported by this news organization, zuranolone improved depressive symptoms as early as day 3, achieving the primary endpoint of significantly greater reduction in scores on the 17-item Hamilton Rating Scale for Depression from baseline to day 15 versus placebo (P = .014).
In the new analysis, patient-reported measures of functional health and well-being were assessed in the WATERFALL trial. It included 266 patients with MDD who were treated with zuranolone 50 mg daily for 2 weeks and 268 patients with MDD who were treated with placebo.
The study used the Short Form–36 (SF-36v2), which covers a wide range of patient-reported measures, including physical function, bodily pain, general health, vitality, social function, and “role-emotional” symptoms.
Results showed that although the treatment and placebo groups had similar baseline SF-36v2 scores, those receiving zuranolone reported significantly greater improvements at day 15 in almost all of the assessment’s domains, including physical function (treatment difference, 0.8), general health (1.0), vitality (3.1), social functioning (1.1), and role-emotional symptoms (1.5; for all comparisons, P < .05). The only exceptions were in role-physical symptoms and bodily pain.
In measures that included physical function, bodily pain, and general health, the patients achieved improvements at day 15 that were consistent with normal levels, with the improvement in vitality considered clinically meaningful versus placebo.
Integrated data
In further analysis of integrated data from four zuranolone clinical trials in the NEST and LANDSCAPE programs for patients with MDD and postpartum depression, results showed similar improvements at day 15 for zuranolone in QoL and overall health across all of the SF-36v2 functioning and well-being domains (P <.05), with the exceptions of physical measure and bodily pain.
By day 42, all of the domains showed significantly greater improvement with zuranolone versus placebo (all, P <.05).
Among the strongest score improvements in the integrated trials were measures in social functioning, which improved from baseline scores of 29.66 to 42.82 on day 15 and to 43.59 on day 42.
Emotional domain scores improved from 24.43 at baseline to 39.13 on day 15 and to 39.82 on day 42. For mental health, the integrated scores for the zuranolone group improved from 27.13 at baseline to 42.40 on day 15 and 42.62 on day 42.
Of note, the baseline scores for mental health represented just 54.3% of those in the normal population; with the increase at day 15, the level was 84.8% of the normal population.
“Across four completed placebo-controlled NEST and LANDSCAPE clinical trials, patient reports of functional health and well-being as assessed by the SF-36v2 indicated substantial impairment at baseline compared to the population norm,” the researchers reported.
The improvements are especially important in light of the fact that in some patients with MDD, functional improvement is a top priority.
“Patients have often prioritized returning to their usual level of functioning over reduction in depressive symptoms, and functional recovery has been associated with better prognosis of depression,” the investigators wrote.
Zuranolone trials have shown that treatment-emergent adverse events (AEs) occur among about 60% of patients, versus about 44% with placebo. The most common AEs are somnolence, dizziness, headache, sedation, and diarrhea, with no increases in suicidal ideation or withdrawal.
The rates of severe AEs are low, and they are observed in about 3% of patients, versus 1.1% with placebo, the researchers noted.
Further, as opposed to serotonergic antidepressants such as SNRIs and SSRIs, zuranolone does not appear to have the undesirable side effects of decreased libido and sexual dysfunction, they added.
Clinically meaningful?
Andrew J. Cutler, MD, clinical associate professor of psychiatry at State University of New York, Syracuse, said the data are “very significant” for a number of reasons.
“We need more options to treat depression, especially ones with novel mechanisms of action and faster onset of efficacy, such as zuranolone,” said Dr. Cutler, who was not involved in the current study. He has coauthored other studies on zuranolone.
Regarding the study’s QoL outcomes, “while improvement in depressive symptoms is very important, what really matters to patients is improvement in function and quality of life,” Dr. Cutler noted.
Also commenting on the study, Jonathan E. Alpert, MD, PhD, chair of the department of psychiatry and behavioral sciences and professor of psychiatry, neuroscience, and pediatrics at Albert Einstein College of Medicine, New York, said the investigational drug could represent an important addition to the armamentarium for treating depression.
“Zuranolone has good oral bioavailability and would represent the first neuroactive steroid antidepressant available in oral form and, indeed, the first non–monoamine-based antidepressant available in oral form,” he said in an interview.
Dr. Alpert was not involved in the research and has no relationship with the drug’s development.
He noted that although there are modest differences between the patients who received zuranolone and those who received placebo in the trials, “this may have been related to high placebo response rates, which often complicate antidepressant trials.
“Further research is needed to determine whether differences between zuranolone and placebo are clinically meaningful, though the separation between drug and placebo on the primary endpoint, as well as some other measures, such as quality of life measures, is promising,” Dr. Alpert said.
However, he added that comparisons with other active antidepressants in terms of efficacy and tolerability remain to be seen.
“Given the large number of individuals with major depressive disorder who have incomplete response to or do not tolerate monoaminergic antidepressants, the development of agents that leverage novel nonmonoaminergic mechanisms is important,” Dr. Alpert concluded.
The study was funded by Sage Therapeutics and Biogen. Dr. Cutler has been involved in research of zuranolone for Sage Therapeutics. Dr. Alpert has reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT ADAA 2022
Pembro provides DFS benefit in early NSCLC
Adjuvant pembrolizumab significantly improves disease-free survival (DFS) compared to placebo in patients with early-stage non–small cell lung cancer (NSCLC) who have undergone complete resection, according to findings from the phase 3 PEARLS/KEYNOTE-091 (PEARLS) study.
Patients in the pembrolizumab arm demonstrated median DFS nearly 12 months longer than those in the placebo arm (53.6 vs. 42.0 months). Investigators observed a DFS benefit for patients with any programmed death-ligand 1 (PD-L1) expression.
“We believe that pembrolizumab has the potential to become a new adjuvant treatment option for patient with [stage IB to IIIA] non–small cell lung cancer following complete resection and adjuvant chemotherapy when recommended,” concluded first author Luis Paz-Ares, MD, chair of the clinical research unit at Hospital Universitario 12 de Octubre, CNIO & Universidad Complutense, Madrid. “Pembrolizumab provided a benefit regardless of pathological stage and PD-L1 progression subgroup.”
The findings were presented by Dr. Paz-Ares at the European Society for Medical Oncology (ESMO) March virtual plenary session and published March 17 in Annals of Oncology.
Pembrolizumab is the standard treatment for patients with advanced NSCLC, but its efficacy in early-stage disease remains unclear. To determine whether patients with early-stage disease benefit from pembrolizumab, Dr. Paz-Ares and colleagues randomized 1,177 adults with stage IB, II, or IIIA NSCLC to 200 mg of pembrolizumab (n = 590) or placebo (n = 587) every 3 weeks.
All patients had Eastern Cooperative Oncology Group performance status of 0-1, and any level of PD-L1 expression. Of the study participants, 168 in the pembrolizumab arm and 165 in the placebo arm had PD-L1 expression and a tumor proportion score (TPS) of at least 50%.
Overall, patients receiving pembrolizumab had a DFS of 53.6 months compared to 42.0 months in the placebo arm (hazard ratio [HR], 0.76; P = .0014). The DFS benefit was generally consistent across patients with PD-L1 TPS <1%, 1%-49%, and ≥50%. In the subset of patients with PD-L1 TPS ≥50%, a slightly higher percentage of patients in the pembrolizumab group demonstrated DFS at 18 months (71.7% vs. 70.2%), but the difference did not reach statistical significance (HR, 0.82; P = .14).
Overall survival (OS) at 18 months was 91.7% in the treatment arm and 91.3% in the placebo arm (HR, 0.87; P = .17), but the data were immature.
“The disease-free survival benefit was observed across most prespecified subgroups,” Dr. Paz-Ares said.
No new safety concerns were raised. Grade 3 or greater adverse events occurred in 34.1% of patients in the treatment arm and 25.8% in the placebo arm. Adverse events led to discontinuation in 19.8% of patients receiving pembrolizumab and 5.9% of patients in the placebo group.
Invited discussant Martin Reck, MD, said these findings represent forward progress. “We do see many patients with distant relapse, which indicates that we have to improve our control of the systemic relapse,” said Dr. Reck, head of the department of thoracic oncology and the clinical trial department at the Lungen Clinic Grosshansdorf, Germany.
Prior data provide a rationale for using immune checkpoint inhibition in early-stage NSCLC, and both the PEARLS study and the IMpower010 trial evaluating atezolizumab in a similar setting have demonstrated relevant improvements in DFS.
“I think we are entering the times of perioperative immunotherapies. We are seeing the first signals of efficacy for adjuvant immunotherapy in two large, randomized trials,” Dr. Reck said.
Based on the PEARLS trial results, Dr. Reck said that PD-L1 appears to have predictive and prognostic value but noted that “several other clinical trials say PD-L1 expression is a poor prognostic marker” for sensitivity to immune checkpoint inhibitor. Given this potential inconsistency, Dr. Reck called for further follow-up in this patient population and for studies in larger groups of patients to further delineate the role of PD-L1 as well as EGFR mutations and adjuvant chemotherapy in patients with early NSCLC.
The PEARLS study was funded by Merck Sharp & Dohme Corp. Dr. Paz-Ares and Dr. Reck disclosed numerous relationships with pharmaceutical companies.
Adjuvant pembrolizumab significantly improves disease-free survival (DFS) compared to placebo in patients with early-stage non–small cell lung cancer (NSCLC) who have undergone complete resection, according to findings from the phase 3 PEARLS/KEYNOTE-091 (PEARLS) study.
Patients in the pembrolizumab arm demonstrated median DFS nearly 12 months longer than those in the placebo arm (53.6 vs. 42.0 months). Investigators observed a DFS benefit for patients with any programmed death-ligand 1 (PD-L1) expression.
“We believe that pembrolizumab has the potential to become a new adjuvant treatment option for patient with [stage IB to IIIA] non–small cell lung cancer following complete resection and adjuvant chemotherapy when recommended,” concluded first author Luis Paz-Ares, MD, chair of the clinical research unit at Hospital Universitario 12 de Octubre, CNIO & Universidad Complutense, Madrid. “Pembrolizumab provided a benefit regardless of pathological stage and PD-L1 progression subgroup.”
The findings were presented by Dr. Paz-Ares at the European Society for Medical Oncology (ESMO) March virtual plenary session and published March 17 in Annals of Oncology.
Pembrolizumab is the standard treatment for patients with advanced NSCLC, but its efficacy in early-stage disease remains unclear. To determine whether patients with early-stage disease benefit from pembrolizumab, Dr. Paz-Ares and colleagues randomized 1,177 adults with stage IB, II, or IIIA NSCLC to 200 mg of pembrolizumab (n = 590) or placebo (n = 587) every 3 weeks.
All patients had Eastern Cooperative Oncology Group performance status of 0-1, and any level of PD-L1 expression. Of the study participants, 168 in the pembrolizumab arm and 165 in the placebo arm had PD-L1 expression and a tumor proportion score (TPS) of at least 50%.
Overall, patients receiving pembrolizumab had a DFS of 53.6 months compared to 42.0 months in the placebo arm (hazard ratio [HR], 0.76; P = .0014). The DFS benefit was generally consistent across patients with PD-L1 TPS <1%, 1%-49%, and ≥50%. In the subset of patients with PD-L1 TPS ≥50%, a slightly higher percentage of patients in the pembrolizumab group demonstrated DFS at 18 months (71.7% vs. 70.2%), but the difference did not reach statistical significance (HR, 0.82; P = .14).
Overall survival (OS) at 18 months was 91.7% in the treatment arm and 91.3% in the placebo arm (HR, 0.87; P = .17), but the data were immature.
“The disease-free survival benefit was observed across most prespecified subgroups,” Dr. Paz-Ares said.
No new safety concerns were raised. Grade 3 or greater adverse events occurred in 34.1% of patients in the treatment arm and 25.8% in the placebo arm. Adverse events led to discontinuation in 19.8% of patients receiving pembrolizumab and 5.9% of patients in the placebo group.
Invited discussant Martin Reck, MD, said these findings represent forward progress. “We do see many patients with distant relapse, which indicates that we have to improve our control of the systemic relapse,” said Dr. Reck, head of the department of thoracic oncology and the clinical trial department at the Lungen Clinic Grosshansdorf, Germany.
Prior data provide a rationale for using immune checkpoint inhibition in early-stage NSCLC, and both the PEARLS study and the IMpower010 trial evaluating atezolizumab in a similar setting have demonstrated relevant improvements in DFS.
“I think we are entering the times of perioperative immunotherapies. We are seeing the first signals of efficacy for adjuvant immunotherapy in two large, randomized trials,” Dr. Reck said.
Based on the PEARLS trial results, Dr. Reck said that PD-L1 appears to have predictive and prognostic value but noted that “several other clinical trials say PD-L1 expression is a poor prognostic marker” for sensitivity to immune checkpoint inhibitor. Given this potential inconsistency, Dr. Reck called for further follow-up in this patient population and for studies in larger groups of patients to further delineate the role of PD-L1 as well as EGFR mutations and adjuvant chemotherapy in patients with early NSCLC.
The PEARLS study was funded by Merck Sharp & Dohme Corp. Dr. Paz-Ares and Dr. Reck disclosed numerous relationships with pharmaceutical companies.
Adjuvant pembrolizumab significantly improves disease-free survival (DFS) compared to placebo in patients with early-stage non–small cell lung cancer (NSCLC) who have undergone complete resection, according to findings from the phase 3 PEARLS/KEYNOTE-091 (PEARLS) study.
Patients in the pembrolizumab arm demonstrated median DFS nearly 12 months longer than those in the placebo arm (53.6 vs. 42.0 months). Investigators observed a DFS benefit for patients with any programmed death-ligand 1 (PD-L1) expression.
“We believe that pembrolizumab has the potential to become a new adjuvant treatment option for patient with [stage IB to IIIA] non–small cell lung cancer following complete resection and adjuvant chemotherapy when recommended,” concluded first author Luis Paz-Ares, MD, chair of the clinical research unit at Hospital Universitario 12 de Octubre, CNIO & Universidad Complutense, Madrid. “Pembrolizumab provided a benefit regardless of pathological stage and PD-L1 progression subgroup.”
The findings were presented by Dr. Paz-Ares at the European Society for Medical Oncology (ESMO) March virtual plenary session and published March 17 in Annals of Oncology.
Pembrolizumab is the standard treatment for patients with advanced NSCLC, but its efficacy in early-stage disease remains unclear. To determine whether patients with early-stage disease benefit from pembrolizumab, Dr. Paz-Ares and colleagues randomized 1,177 adults with stage IB, II, or IIIA NSCLC to 200 mg of pembrolizumab (n = 590) or placebo (n = 587) every 3 weeks.
All patients had Eastern Cooperative Oncology Group performance status of 0-1, and any level of PD-L1 expression. Of the study participants, 168 in the pembrolizumab arm and 165 in the placebo arm had PD-L1 expression and a tumor proportion score (TPS) of at least 50%.
Overall, patients receiving pembrolizumab had a DFS of 53.6 months compared to 42.0 months in the placebo arm (hazard ratio [HR], 0.76; P = .0014). The DFS benefit was generally consistent across patients with PD-L1 TPS <1%, 1%-49%, and ≥50%. In the subset of patients with PD-L1 TPS ≥50%, a slightly higher percentage of patients in the pembrolizumab group demonstrated DFS at 18 months (71.7% vs. 70.2%), but the difference did not reach statistical significance (HR, 0.82; P = .14).
Overall survival (OS) at 18 months was 91.7% in the treatment arm and 91.3% in the placebo arm (HR, 0.87; P = .17), but the data were immature.
“The disease-free survival benefit was observed across most prespecified subgroups,” Dr. Paz-Ares said.
No new safety concerns were raised. Grade 3 or greater adverse events occurred in 34.1% of patients in the treatment arm and 25.8% in the placebo arm. Adverse events led to discontinuation in 19.8% of patients receiving pembrolizumab and 5.9% of patients in the placebo group.
Invited discussant Martin Reck, MD, said these findings represent forward progress. “We do see many patients with distant relapse, which indicates that we have to improve our control of the systemic relapse,” said Dr. Reck, head of the department of thoracic oncology and the clinical trial department at the Lungen Clinic Grosshansdorf, Germany.
Prior data provide a rationale for using immune checkpoint inhibition in early-stage NSCLC, and both the PEARLS study and the IMpower010 trial evaluating atezolizumab in a similar setting have demonstrated relevant improvements in DFS.
“I think we are entering the times of perioperative immunotherapies. We are seeing the first signals of efficacy for adjuvant immunotherapy in two large, randomized trials,” Dr. Reck said.
Based on the PEARLS trial results, Dr. Reck said that PD-L1 appears to have predictive and prognostic value but noted that “several other clinical trials say PD-L1 expression is a poor prognostic marker” for sensitivity to immune checkpoint inhibitor. Given this potential inconsistency, Dr. Reck called for further follow-up in this patient population and for studies in larger groups of patients to further delineate the role of PD-L1 as well as EGFR mutations and adjuvant chemotherapy in patients with early NSCLC.
The PEARLS study was funded by Merck Sharp & Dohme Corp. Dr. Paz-Ares and Dr. Reck disclosed numerous relationships with pharmaceutical companies.
FROM THE ESMO MARCH PLENARY
Adding immunotherapy to chemo in lung cancer improves patient outcomes, new data show
according to an analysis presented at the annual European Lung Cancer Congress (ELCC) on March 30.
“Overall, it is very clear that chemotherapy plus immunotherapy prolongs the time to symptom deterioration and actually improves symptoms” in this patient population, said study discussant Luis Paz-Ares, MD, PhD, chair of medical oncology at the Hospital Universitario 12 de Octubre, Madrid, who was not involved in the research.
Last September, investigators reported efficacy outcomes from the phase 3 POSEIDON trial, which randomized 1,013 patients with EGFR/ALK wild-type mNSCLC to one of three first-line options: chemotherapy alone, chemotherapy plus the checkpoint inhibitor durvalumab, or chemotherapy plus two check-point inhibitors, durvalumab and tremelimumab. The analysis showed improved progression-free survival in both immunotherapy arms as well as a significant 2.3-month overall survival advantage with dual immunotherapy and a nonsignificant 1.6-month advantage with single agent durvalumab.
At the ELCC meeting, study presenter and lead investigator Edward Garon, MD, reported the latest data on the trial’s secondary endpoints: patient-reported outcomes. Global health status, functioning, and symptom scores were assessed using two questionnaires, the EORTC QLQ-C30 and EORTC QLQ-LC13.
Overall, Dr. Garon and colleagues reported a longer time to deterioration in all three areas – global health status, functioning, and symptoms – for patients who received immunotherapy versus chemotherapy alone, with similar results in both immunotherapy arms.
Time to deterioration in global health status, for instance, was a median of about 8 months on both immunotherapy regimens versus 5.6 months with chemotherapy alone. The positive findings held for many patient-reported treatment side effects, including dyspnea, hemoptysis, nausea/vomiting, and insomnia, but the benefits of adding immunotherapy weren’t always statistically significant.
Adding one or both checkpoint inhibitors to chemotherapy “improved efficacy while delaying deterioration in symptoms, functioning, and [health-related quality of life] versus chemotherapy alone in patients with mNSCLC,” concluded Dr. Garon, a thoracic medical oncologist at the University of California, Los Angeles. Plus, he added, “the pattern was observed across nearly all prespecified symptoms and domains of interest.”
According to study discussant Dr. Paz-Ares, “the data seem to be very consistent with all the trials asking similar questions.” The important thing here is figuring out the ideal candidates for dual inhibitor therapy, he said.
With positive efficacy and patient-reported outcomes for single and dual immunotherapy in this trial, it’s a “relatively straightforward” decision to add immunotherapy to chemotherapy for patients with mNSCLC, Massimo Di Maio, a medical oncologist at the University of Turin, Italy, said in an editorial on the ELCC’s news site.
However, that’s not always the case for every cancer type, which makes patient-reported outcomes “crucial” for determining the right treatment for each patient. Some might opt for a modest survival benefit regardless of the side effects, while others might favor a less toxic approach, even it means not living quite as long, he said.
The problem, he stressed, is that trials often release efficacy data well before patient-reported outcomes, which makes weighing the benefits and risks of a treat-ment option more difficult. The delay between efficacy and patient-reported outcome data was about 6 months in the POSEIDON trial.
“Timing is key when it comes to using [patient reported outcomes] for decision-making in oncology,” Dr. Di Maio said. “In fact, to enable a full assessment of a treatment, results should be published concurrently with the efficacy and safety data. Unfortunately, this is generally not the case.”
POSEIDON was funded by AstraZeneca, which markets durvalumab and is developing tremelimumab. Dr. Garon reported grants from the company. Dr. Paz-Ares reported honoraria and institutional research grants from AstraZeneca. Dr. Di Maio is a consultant for AstraZeneca and reported receiving honoraria and personal fees from the company.
according to an analysis presented at the annual European Lung Cancer Congress (ELCC) on March 30.
“Overall, it is very clear that chemotherapy plus immunotherapy prolongs the time to symptom deterioration and actually improves symptoms” in this patient population, said study discussant Luis Paz-Ares, MD, PhD, chair of medical oncology at the Hospital Universitario 12 de Octubre, Madrid, who was not involved in the research.
Last September, investigators reported efficacy outcomes from the phase 3 POSEIDON trial, which randomized 1,013 patients with EGFR/ALK wild-type mNSCLC to one of three first-line options: chemotherapy alone, chemotherapy plus the checkpoint inhibitor durvalumab, or chemotherapy plus two check-point inhibitors, durvalumab and tremelimumab. The analysis showed improved progression-free survival in both immunotherapy arms as well as a significant 2.3-month overall survival advantage with dual immunotherapy and a nonsignificant 1.6-month advantage with single agent durvalumab.
At the ELCC meeting, study presenter and lead investigator Edward Garon, MD, reported the latest data on the trial’s secondary endpoints: patient-reported outcomes. Global health status, functioning, and symptom scores were assessed using two questionnaires, the EORTC QLQ-C30 and EORTC QLQ-LC13.
Overall, Dr. Garon and colleagues reported a longer time to deterioration in all three areas – global health status, functioning, and symptoms – for patients who received immunotherapy versus chemotherapy alone, with similar results in both immunotherapy arms.
Time to deterioration in global health status, for instance, was a median of about 8 months on both immunotherapy regimens versus 5.6 months with chemotherapy alone. The positive findings held for many patient-reported treatment side effects, including dyspnea, hemoptysis, nausea/vomiting, and insomnia, but the benefits of adding immunotherapy weren’t always statistically significant.
Adding one or both checkpoint inhibitors to chemotherapy “improved efficacy while delaying deterioration in symptoms, functioning, and [health-related quality of life] versus chemotherapy alone in patients with mNSCLC,” concluded Dr. Garon, a thoracic medical oncologist at the University of California, Los Angeles. Plus, he added, “the pattern was observed across nearly all prespecified symptoms and domains of interest.”
According to study discussant Dr. Paz-Ares, “the data seem to be very consistent with all the trials asking similar questions.” The important thing here is figuring out the ideal candidates for dual inhibitor therapy, he said.
With positive efficacy and patient-reported outcomes for single and dual immunotherapy in this trial, it’s a “relatively straightforward” decision to add immunotherapy to chemotherapy for patients with mNSCLC, Massimo Di Maio, a medical oncologist at the University of Turin, Italy, said in an editorial on the ELCC’s news site.
However, that’s not always the case for every cancer type, which makes patient-reported outcomes “crucial” for determining the right treatment for each patient. Some might opt for a modest survival benefit regardless of the side effects, while others might favor a less toxic approach, even it means not living quite as long, he said.
The problem, he stressed, is that trials often release efficacy data well before patient-reported outcomes, which makes weighing the benefits and risks of a treat-ment option more difficult. The delay between efficacy and patient-reported outcome data was about 6 months in the POSEIDON trial.
“Timing is key when it comes to using [patient reported outcomes] for decision-making in oncology,” Dr. Di Maio said. “In fact, to enable a full assessment of a treatment, results should be published concurrently with the efficacy and safety data. Unfortunately, this is generally not the case.”
POSEIDON was funded by AstraZeneca, which markets durvalumab and is developing tremelimumab. Dr. Garon reported grants from the company. Dr. Paz-Ares reported honoraria and institutional research grants from AstraZeneca. Dr. Di Maio is a consultant for AstraZeneca and reported receiving honoraria and personal fees from the company.
according to an analysis presented at the annual European Lung Cancer Congress (ELCC) on March 30.
“Overall, it is very clear that chemotherapy plus immunotherapy prolongs the time to symptom deterioration and actually improves symptoms” in this patient population, said study discussant Luis Paz-Ares, MD, PhD, chair of medical oncology at the Hospital Universitario 12 de Octubre, Madrid, who was not involved in the research.
Last September, investigators reported efficacy outcomes from the phase 3 POSEIDON trial, which randomized 1,013 patients with EGFR/ALK wild-type mNSCLC to one of three first-line options: chemotherapy alone, chemotherapy plus the checkpoint inhibitor durvalumab, or chemotherapy plus two check-point inhibitors, durvalumab and tremelimumab. The analysis showed improved progression-free survival in both immunotherapy arms as well as a significant 2.3-month overall survival advantage with dual immunotherapy and a nonsignificant 1.6-month advantage with single agent durvalumab.
At the ELCC meeting, study presenter and lead investigator Edward Garon, MD, reported the latest data on the trial’s secondary endpoints: patient-reported outcomes. Global health status, functioning, and symptom scores were assessed using two questionnaires, the EORTC QLQ-C30 and EORTC QLQ-LC13.
Overall, Dr. Garon and colleagues reported a longer time to deterioration in all three areas – global health status, functioning, and symptoms – for patients who received immunotherapy versus chemotherapy alone, with similar results in both immunotherapy arms.
Time to deterioration in global health status, for instance, was a median of about 8 months on both immunotherapy regimens versus 5.6 months with chemotherapy alone. The positive findings held for many patient-reported treatment side effects, including dyspnea, hemoptysis, nausea/vomiting, and insomnia, but the benefits of adding immunotherapy weren’t always statistically significant.
Adding one or both checkpoint inhibitors to chemotherapy “improved efficacy while delaying deterioration in symptoms, functioning, and [health-related quality of life] versus chemotherapy alone in patients with mNSCLC,” concluded Dr. Garon, a thoracic medical oncologist at the University of California, Los Angeles. Plus, he added, “the pattern was observed across nearly all prespecified symptoms and domains of interest.”
According to study discussant Dr. Paz-Ares, “the data seem to be very consistent with all the trials asking similar questions.” The important thing here is figuring out the ideal candidates for dual inhibitor therapy, he said.
With positive efficacy and patient-reported outcomes for single and dual immunotherapy in this trial, it’s a “relatively straightforward” decision to add immunotherapy to chemotherapy for patients with mNSCLC, Massimo Di Maio, a medical oncologist at the University of Turin, Italy, said in an editorial on the ELCC’s news site.
However, that’s not always the case for every cancer type, which makes patient-reported outcomes “crucial” for determining the right treatment for each patient. Some might opt for a modest survival benefit regardless of the side effects, while others might favor a less toxic approach, even it means not living quite as long, he said.
The problem, he stressed, is that trials often release efficacy data well before patient-reported outcomes, which makes weighing the benefits and risks of a treat-ment option more difficult. The delay between efficacy and patient-reported outcome data was about 6 months in the POSEIDON trial.
“Timing is key when it comes to using [patient reported outcomes] for decision-making in oncology,” Dr. Di Maio said. “In fact, to enable a full assessment of a treatment, results should be published concurrently with the efficacy and safety data. Unfortunately, this is generally not the case.”
POSEIDON was funded by AstraZeneca, which markets durvalumab and is developing tremelimumab. Dr. Garon reported grants from the company. Dr. Paz-Ares reported honoraria and institutional research grants from AstraZeneca. Dr. Di Maio is a consultant for AstraZeneca and reported receiving honoraria and personal fees from the company.
FROM ELCC 2022
Anticoagulation not routinely needed after TAVR: ADAPT-TAVR
In patients undergoing transcatheter aortic valve replacement (TAVR), the incidence of leaflet thrombosis was numerically lower in those treated with the anticoagulant edoxaban for 6 months after the procedure than in those who received dual antiplatelet therapy, although the difference was not statistically significant, in the ADAPT-TAVR study.
There was no difference in new cerebral thromboembolism or neurologic/neurocognitive function between the two groups in the study.
Also, there was no significant relation between subclinical leaflet thrombosis and increased risk for cerebral thromboembolism and neurologic dysfunction.
The ADAPT-TAVR trial was presented April 4 at the American College of Cardiology (ACC) 2022 Scientific Session by Duk-Woo Park, MD, Asan Medical Center, Seoul, South Korea. It was simultaneously published online in Circulation.

“The key messages from this study are that subclinical leaflet thrombosis has not been proven to affect clinical outcomes for patients undergoing valve replacement and that in patients in whom leaflet thrombosis causes no symptoms or complications, its presence should not dictate the type of antithrombotic therapy that patients receive following the implantation of an artificial heart valve,” Dr. Park said.
“These findings do not support the routine use of computed tomography scans to detect subclinical leaflet thrombosis,” he added.
Commenting on the study at an ACC press conference, Megan Coylewright, MD, director of the Structural Heart Program at Erlanger Health System, Chattanooga, Tennessee, said: “Oftentimes when studies are negative, we’re disappointed. In this case, I think we are pleased that the study is negative because it suggests we do not have to expose our TAVR patients to anticoagulation for benefit.”
Dr. Coylewright explained that the ADAPT-TAVR study was asking whether clots form on the valve, as defined by CT.
“We are worried about that for two reasons: could that clot cause a stroke, and could that clot cause the valve to break down over time. This study looked at the first issue. And it found that there was some clot build up on the valve, but that it wasn’t significantly different between the anticoagulant and dual antiplatelet groups. And there was no correlation with embolic events, she noted.
“It shows how fast our field moves. In the U.S. now, we are using aspirin alone at 81 mg for patients who do not have an indication for oral anticoagulation after TAVR. We are moving away from dual antiplatelet therapy because the bleeding risk is so bad,” Dr. Coylewright said.
In his presentation, Dr. Park explained that it is believed that oral anticoagulants are more effective than antiplatelet therapy at reducing subclinical leaflet thrombosis, but it is not known whether there is a causal association between subclinical leaflet thrombosis and cerebral embolism, or whether oral anticoagulation can reduce cerebral embolism related to subclinical leaflet thrombosis.
The ADAPT-TAVR was conducted to look at these issues. The open-label randomized trial was conducted in five centers in Hong Kong, South Korea, and Taiwan.
For the study, 229 patients who had undergone successful TAVR and did not have an indication for anticoagulation were randomized to edoxaban 60 mg once daily, edoxaban 30 mg once daily for patients needing a reduced dose, or dual antiplatelet therapy for 6 months.
The primary endpoint was an incidence of leaflet thrombosis on four-dimensional CT at 6 months.
Results showed a strong trend toward a lower incidence of leaflet thrombosis in the edoxaban groups than in the dual antiplatelet group (9.8% vs. 18.4%; P = .076).
There was a nonsignificant difference in the percentage of patients with new cerebral lesions identified on brain MRI between the edoxaban and dual antiplatelet groups (25.0% vs. 20.2%).
The percentage of patients with worsening of neurologic and neurocognitive function was not different among the groups.
The incidence of any or major bleeding events was not different between two therapies.
There was also no significant association of the presence or extent of leaflet thrombosis with new cerebral lesions or change of neurologic or neurocognitive function.
Dr. Park noted that the trial had several limitations, including an open-label design, use of surrogate imaging outcomes for the primary outcome, and the relatively short follow-up period, so the study was underpowered to detect any meaningful differences in clinical efficacy and safety outcomes. The results should thus be considered hypothesis-generating, highlighting the need for further research, he added.
The long-term effect of leaflet thrombosis or different antithrombotic strategies on bioprosthetic valve durability is still unknown, Dr. Park said.
He also pointed out that the findings cannot be directly extrapolated to patients with an established indication for oral anticoagulant therapy.
The ADAPT-TAVR trial was an investigator-initiated trial and was funded by the CardioVascular Research Foundation (Seoul, Korea) and Daiichi Sankyo Korea.
A version of this article first appeared on Medscape.com.
In patients undergoing transcatheter aortic valve replacement (TAVR), the incidence of leaflet thrombosis was numerically lower in those treated with the anticoagulant edoxaban for 6 months after the procedure than in those who received dual antiplatelet therapy, although the difference was not statistically significant, in the ADAPT-TAVR study.
There was no difference in new cerebral thromboembolism or neurologic/neurocognitive function between the two groups in the study.
Also, there was no significant relation between subclinical leaflet thrombosis and increased risk for cerebral thromboembolism and neurologic dysfunction.
The ADAPT-TAVR trial was presented April 4 at the American College of Cardiology (ACC) 2022 Scientific Session by Duk-Woo Park, MD, Asan Medical Center, Seoul, South Korea. It was simultaneously published online in Circulation.
“The key messages from this study are that subclinical leaflet thrombosis has not been proven to affect clinical outcomes for patients undergoing valve replacement and that in patients in whom leaflet thrombosis causes no symptoms or complications, its presence should not dictate the type of antithrombotic therapy that patients receive following the implantation of an artificial heart valve,” Dr. Park said.
“These findings do not support the routine use of computed tomography scans to detect subclinical leaflet thrombosis,” he added.
Commenting on the study at an ACC press conference, Megan Coylewright, MD, director of the Structural Heart Program at Erlanger Health System, Chattanooga, Tennessee, said: “Oftentimes when studies are negative, we’re disappointed. In this case, I think we are pleased that the study is negative because it suggests we do not have to expose our TAVR patients to anticoagulation for benefit.”
Dr. Coylewright explained that the ADAPT-TAVR study was asking whether clots form on the valve, as defined by CT.
“We are worried about that for two reasons: could that clot cause a stroke, and could that clot cause the valve to break down over time. This study looked at the first issue. And it found that there was some clot build up on the valve, but that it wasn’t significantly different between the anticoagulant and dual antiplatelet groups. And there was no correlation with embolic events, she noted.
“It shows how fast our field moves. In the U.S. now, we are using aspirin alone at 81 mg for patients who do not have an indication for oral anticoagulation after TAVR. We are moving away from dual antiplatelet therapy because the bleeding risk is so bad,” Dr. Coylewright said.
In his presentation, Dr. Park explained that it is believed that oral anticoagulants are more effective than antiplatelet therapy at reducing subclinical leaflet thrombosis, but it is not known whether there is a causal association between subclinical leaflet thrombosis and cerebral embolism, or whether oral anticoagulation can reduce cerebral embolism related to subclinical leaflet thrombosis.
The ADAPT-TAVR was conducted to look at these issues. The open-label randomized trial was conducted in five centers in Hong Kong, South Korea, and Taiwan.
For the study, 229 patients who had undergone successful TAVR and did not have an indication for anticoagulation were randomized to edoxaban 60 mg once daily, edoxaban 30 mg once daily for patients needing a reduced dose, or dual antiplatelet therapy for 6 months.
The primary endpoint was an incidence of leaflet thrombosis on four-dimensional CT at 6 months.
Results showed a strong trend toward a lower incidence of leaflet thrombosis in the edoxaban groups than in the dual antiplatelet group (9.8% vs. 18.4%; P = .076).
There was a nonsignificant difference in the percentage of patients with new cerebral lesions identified on brain MRI between the edoxaban and dual antiplatelet groups (25.0% vs. 20.2%).
The percentage of patients with worsening of neurologic and neurocognitive function was not different among the groups.
The incidence of any or major bleeding events was not different between two therapies.
There was also no significant association of the presence or extent of leaflet thrombosis with new cerebral lesions or change of neurologic or neurocognitive function.
Dr. Park noted that the trial had several limitations, including an open-label design, use of surrogate imaging outcomes for the primary outcome, and the relatively short follow-up period, so the study was underpowered to detect any meaningful differences in clinical efficacy and safety outcomes. The results should thus be considered hypothesis-generating, highlighting the need for further research, he added.
The long-term effect of leaflet thrombosis or different antithrombotic strategies on bioprosthetic valve durability is still unknown, Dr. Park said.
He also pointed out that the findings cannot be directly extrapolated to patients with an established indication for oral anticoagulant therapy.
The ADAPT-TAVR trial was an investigator-initiated trial and was funded by the CardioVascular Research Foundation (Seoul, Korea) and Daiichi Sankyo Korea.
A version of this article first appeared on Medscape.com.
In patients undergoing transcatheter aortic valve replacement (TAVR), the incidence of leaflet thrombosis was numerically lower in those treated with the anticoagulant edoxaban for 6 months after the procedure than in those who received dual antiplatelet therapy, although the difference was not statistically significant, in the ADAPT-TAVR study.
There was no difference in new cerebral thromboembolism or neurologic/neurocognitive function between the two groups in the study.
Also, there was no significant relation between subclinical leaflet thrombosis and increased risk for cerebral thromboembolism and neurologic dysfunction.
The ADAPT-TAVR trial was presented April 4 at the American College of Cardiology (ACC) 2022 Scientific Session by Duk-Woo Park, MD, Asan Medical Center, Seoul, South Korea. It was simultaneously published online in Circulation.
“The key messages from this study are that subclinical leaflet thrombosis has not been proven to affect clinical outcomes for patients undergoing valve replacement and that in patients in whom leaflet thrombosis causes no symptoms or complications, its presence should not dictate the type of antithrombotic therapy that patients receive following the implantation of an artificial heart valve,” Dr. Park said.
“These findings do not support the routine use of computed tomography scans to detect subclinical leaflet thrombosis,” he added.
Commenting on the study at an ACC press conference, Megan Coylewright, MD, director of the Structural Heart Program at Erlanger Health System, Chattanooga, Tennessee, said: “Oftentimes when studies are negative, we’re disappointed. In this case, I think we are pleased that the study is negative because it suggests we do not have to expose our TAVR patients to anticoagulation for benefit.”
Dr. Coylewright explained that the ADAPT-TAVR study was asking whether clots form on the valve, as defined by CT.
“We are worried about that for two reasons: could that clot cause a stroke, and could that clot cause the valve to break down over time. This study looked at the first issue. And it found that there was some clot build up on the valve, but that it wasn’t significantly different between the anticoagulant and dual antiplatelet groups. And there was no correlation with embolic events, she noted.
“It shows how fast our field moves. In the U.S. now, we are using aspirin alone at 81 mg for patients who do not have an indication for oral anticoagulation after TAVR. We are moving away from dual antiplatelet therapy because the bleeding risk is so bad,” Dr. Coylewright said.
In his presentation, Dr. Park explained that it is believed that oral anticoagulants are more effective than antiplatelet therapy at reducing subclinical leaflet thrombosis, but it is not known whether there is a causal association between subclinical leaflet thrombosis and cerebral embolism, or whether oral anticoagulation can reduce cerebral embolism related to subclinical leaflet thrombosis.
The ADAPT-TAVR was conducted to look at these issues. The open-label randomized trial was conducted in five centers in Hong Kong, South Korea, and Taiwan.
For the study, 229 patients who had undergone successful TAVR and did not have an indication for anticoagulation were randomized to edoxaban 60 mg once daily, edoxaban 30 mg once daily for patients needing a reduced dose, or dual antiplatelet therapy for 6 months.
The primary endpoint was an incidence of leaflet thrombosis on four-dimensional CT at 6 months.
Results showed a strong trend toward a lower incidence of leaflet thrombosis in the edoxaban groups than in the dual antiplatelet group (9.8% vs. 18.4%; P = .076).
There was a nonsignificant difference in the percentage of patients with new cerebral lesions identified on brain MRI between the edoxaban and dual antiplatelet groups (25.0% vs. 20.2%).
The percentage of patients with worsening of neurologic and neurocognitive function was not different among the groups.
The incidence of any or major bleeding events was not different between two therapies.
There was also no significant association of the presence or extent of leaflet thrombosis with new cerebral lesions or change of neurologic or neurocognitive function.
Dr. Park noted that the trial had several limitations, including an open-label design, use of surrogate imaging outcomes for the primary outcome, and the relatively short follow-up period, so the study was underpowered to detect any meaningful differences in clinical efficacy and safety outcomes. The results should thus be considered hypothesis-generating, highlighting the need for further research, he added.
The long-term effect of leaflet thrombosis or different antithrombotic strategies on bioprosthetic valve durability is still unknown, Dr. Park said.
He also pointed out that the findings cannot be directly extrapolated to patients with an established indication for oral anticoagulant therapy.
The ADAPT-TAVR trial was an investigator-initiated trial and was funded by the CardioVascular Research Foundation (Seoul, Korea) and Daiichi Sankyo Korea.
A version of this article first appeared on Medscape.com.
CDC recommends hep B vaccination for most adults
It also added that adults aged 60 years or older without known risk factors for hepatitis B may get vaccinated.
The agency earlier recommended the vaccination for all infants and children under the age of 19 years and for adults aged 60 years or older with known risk factors.
The CDC said it wants to expand vaccinations because, after decades of progress, the number of new hepatitis B infections is increasing among adults. Acute hepatitis B infections among adults lead to chronic hepatitis B disease in an estimated 2%-6% of cases, and can result in cirrhosis, liver cancer, and death.
Among adults aged 40-49 years, the rate of cases increased from 1.9 per 100,000 people in 2011 to 2.7 per 100,000 in 2019. Among adults aged 50-59 years, the rate increased during this period from 1.1 to 1.6 per 100,000.
Most adults aren’t vaccinated. Among adults aged 19 years or older, only 30.0% reported that they’d received at least the three recommended doses of the vaccine. The rate was 40.3% for adults aged 19-49 years, and 19.1% for adults aged 50 years or older.
Hepatitis B infection rates are particularly elevated among African Americans.
Even among adults with chronic liver disease, the vaccination rate is only 33.0%. And, among travelers to countries where the virus has been endemic since 1995, only 38.9% were vaccinated.
In a 2018 survey of internal medicine and family physicians, 68% said their patients had not told them about risk factors, making it difficult to assess whether the patients needed the vaccine according to the recommendations at the time. These risk factors include injection drug use, incarceration, and multiple sex partners, experiences the patients may not have been willing to discuss.
CDC researchers calculated that universal adult hepatitis B vaccination would cost $153,000 for every quality-adjusted life-year (QALY) gained. For adults aged 19-59 years, a QALY would cost $117,000 because infections are more prevalent in that age group.
The CDC specified that it intends its new guidelines to prompt physicians to offer the vaccine to adults aged 60 years or older rather than wait for them to request it.
The Food and Drug Administration has approved both three-dose and two-dose hepatitis B vaccines, with evidence showing similar seroprotection and adverse events.
People who have already completed their vaccination or have a history of hepatitis B infection should only receive additional vaccinations in specific cases, as detailed in the CDC’s 2018 recommendations.
A version of this article first appeared on Medscape.com.
It also added that adults aged 60 years or older without known risk factors for hepatitis B may get vaccinated.
The agency earlier recommended the vaccination for all infants and children under the age of 19 years and for adults aged 60 years or older with known risk factors.
The CDC said it wants to expand vaccinations because, after decades of progress, the number of new hepatitis B infections is increasing among adults. Acute hepatitis B infections among adults lead to chronic hepatitis B disease in an estimated 2%-6% of cases, and can result in cirrhosis, liver cancer, and death.
Among adults aged 40-49 years, the rate of cases increased from 1.9 per 100,000 people in 2011 to 2.7 per 100,000 in 2019. Among adults aged 50-59 years, the rate increased during this period from 1.1 to 1.6 per 100,000.
Most adults aren’t vaccinated. Among adults aged 19 years or older, only 30.0% reported that they’d received at least the three recommended doses of the vaccine. The rate was 40.3% for adults aged 19-49 years, and 19.1% for adults aged 50 years or older.
Hepatitis B infection rates are particularly elevated among African Americans.
Even among adults with chronic liver disease, the vaccination rate is only 33.0%. And, among travelers to countries where the virus has been endemic since 1995, only 38.9% were vaccinated.
In a 2018 survey of internal medicine and family physicians, 68% said their patients had not told them about risk factors, making it difficult to assess whether the patients needed the vaccine according to the recommendations at the time. These risk factors include injection drug use, incarceration, and multiple sex partners, experiences the patients may not have been willing to discuss.
CDC researchers calculated that universal adult hepatitis B vaccination would cost $153,000 for every quality-adjusted life-year (QALY) gained. For adults aged 19-59 years, a QALY would cost $117,000 because infections are more prevalent in that age group.
The CDC specified that it intends its new guidelines to prompt physicians to offer the vaccine to adults aged 60 years or older rather than wait for them to request it.
The Food and Drug Administration has approved both three-dose and two-dose hepatitis B vaccines, with evidence showing similar seroprotection and adverse events.
People who have already completed their vaccination or have a history of hepatitis B infection should only receive additional vaccinations in specific cases, as detailed in the CDC’s 2018 recommendations.
A version of this article first appeared on Medscape.com.
It also added that adults aged 60 years or older without known risk factors for hepatitis B may get vaccinated.
The agency earlier recommended the vaccination for all infants and children under the age of 19 years and for adults aged 60 years or older with known risk factors.
The CDC said it wants to expand vaccinations because, after decades of progress, the number of new hepatitis B infections is increasing among adults. Acute hepatitis B infections among adults lead to chronic hepatitis B disease in an estimated 2%-6% of cases, and can result in cirrhosis, liver cancer, and death.
Among adults aged 40-49 years, the rate of cases increased from 1.9 per 100,000 people in 2011 to 2.7 per 100,000 in 2019. Among adults aged 50-59 years, the rate increased during this period from 1.1 to 1.6 per 100,000.
Most adults aren’t vaccinated. Among adults aged 19 years or older, only 30.0% reported that they’d received at least the three recommended doses of the vaccine. The rate was 40.3% for adults aged 19-49 years, and 19.1% for adults aged 50 years or older.
Hepatitis B infection rates are particularly elevated among African Americans.
Even among adults with chronic liver disease, the vaccination rate is only 33.0%. And, among travelers to countries where the virus has been endemic since 1995, only 38.9% were vaccinated.
In a 2018 survey of internal medicine and family physicians, 68% said their patients had not told them about risk factors, making it difficult to assess whether the patients needed the vaccine according to the recommendations at the time. These risk factors include injection drug use, incarceration, and multiple sex partners, experiences the patients may not have been willing to discuss.
CDC researchers calculated that universal adult hepatitis B vaccination would cost $153,000 for every quality-adjusted life-year (QALY) gained. For adults aged 19-59 years, a QALY would cost $117,000 because infections are more prevalent in that age group.
The CDC specified that it intends its new guidelines to prompt physicians to offer the vaccine to adults aged 60 years or older rather than wait for them to request it.
The Food and Drug Administration has approved both three-dose and two-dose hepatitis B vaccines, with evidence showing similar seroprotection and adverse events.
People who have already completed their vaccination or have a history of hepatitis B infection should only receive additional vaccinations in specific cases, as detailed in the CDC’s 2018 recommendations.
A version of this article first appeared on Medscape.com.
FROM THE MMWR
New HF guidelines feature ‘quad’ therapy, tweaked terminology
The new heart failure (HF) guidelines released by three North American societies had a lot of catching up to do given the significant, even paradigm-shifting, additions to available treatment options in the last few years.
The landscape now includes both new and repurposed drug therapies that benefit almost without regard to ejection fraction (EF), and evidence-based urgency to engage patients early on with at least four core medication classes, so-called quadruple therapy.

The guideline document offers a roadmap for navigating those key issues and many others and uses some creative tactics. They include the introduction of generalist-friendly labels for the traditional but obscurely named four stages of HF severity that, it is hoped, will have wider reach and expand the use of effective therapies.
It introduces additional disease-staging terminology that characterizes the syndrome as a continuum:
- “At risk for HF” for stage A, applied to asymptomatic patients with risk factors such as diabetes or hypertension but no known cardiac changes.
- “Pre-HF” for stage B, which adds cardiac structural changes or elevated natriuretic peptides, still in the absence of symptoms.
- “Symptomatic HF” for stage C, that is, structural disease with current or previous symptoms.
- “Advanced HF” for stage D, characterized by severe debilitating symptoms or repeated hospitalizations even with guideline-directed medical therapy (GDMT).
The new terms should be “easier for primary care physicians as well as nonspecialists” to remember and use effectively “and easier to translate to the patients,” compared with the solely alphabetical staging labels appearing in the guidelines for more than 15 years, Biykem Bozkurt, MD, PhD, Baylor College of Medicine, Houston, said in an interview.
An emphasis on “at risk for HF” and “pre-HF” in the new document may help efforts to expand primary prevention of HF and management of preclinical HF. The guideline, Dr. Bozkurt said, includes specific treatment recommendations for those early stages.
The document also updates and sometimes introduces “recommendations for advanced heart failure, acute heart failure, and comorbidities – specifically for atrial fibrillation, iron deficiency, sleep apnea, coronary artery disease, and valvular heart disease,” Dr. Bozkurt observed, as well as for cardiomyopathy and HF related to pregnancy and cancer chemotherapy. “So, it’s a very comprehensive guideline.”
Dr. Bozkurt is vice chair of the guideline writing committee and helped introduce the guideline at the annual scientific sessions of the American College of Cardiology. The document, developed by the ACC, the American Heart Association, and the Heart Failure Society of America, was published April 1, 2022, in the societies’ flagship journals, Journal of the American College of Cardiology, Circulation, and the Journal of Cardiac Failure, respectively. It replaces the 2013 guideline from the ACC and AHA and the ACC/AHA/HFSA–focused update from 2017.

“We really need to treat early, and then we need to treat appropriately,” Douglas L. Mann, MD, Washington University in St. Louis, said in an interview. Dr. Mann, who was not involved in development of the new guideline, said he is “enthusiastic” about the new staging terminology.
“I think it makes it easier to convey the message that these people do need medicines, will benefit from medicines, and in some cases heart failure can be preventable,” he said. “I’m in favor of anything that simplifies it and makes it more readily interpretable by busy doctors who aren’t specialists.”
With the new staging terminology and in other ways, the guideline seems to appreciate cardiomyopathy as a journey from preclinical to advanced symptomatic stages – the preclinical “at-risk” stage tightening focus on primary prevention – and updated thinking on classification of HF by EF.
For example, there is new consideration of “HF with improved ejection fraction” (HFimpEF), which suggests the patient may be evolving from HF with reduced EF (HFrEF) to HF with EF that is preserved or mildly reduced, or vice versa.
With HFimpEF, which identifies patients previously with an EF of 40% or lower that improves to beyond 40% at follow-up testing, patients should continue on the medications they had been previously taking for HFrEF, Dr. Bozkurt said.
Patients at risk for HF, in stage A by the older terminology, are characterized by one or more significant HF risk factors, such as hypertension, diabetes, or coronary disease, as they have been in prior guidelines. But the new document, Dr. Bozkurt observed, adds genetic cardiomyopathies and exposure to cardiotoxic agents to the list.
Perhaps surprisingly, the guideline also includes elevated natriuretic peptides as an indicator of “at risk for HF,” with implications for screening. The evidence suggests that, “for patients who are at risk for heart failure, natriuretic peptide-based screening, followed by team-based care, can prevent development of left ventricular dysfunction in heart failure,” Dr. Bozkurt said.
Persons at risk for HF realistically encompass a huge swath of the population given the world prevalence of high blood pressure, obesity, and diabetes. Management of stage A, therefore, focuses on established tenets of primary cardiovascular prevention, such as weight and BP control, exercise, and healthy dietary choices.
They may well be eligible for treatment with sodium-glucose transporter 2 (SGLT2) inhibitors, which have been “game changers,” Dr. Mann said. “Now you can give them to diabetics and it’s going to prevent heart failure and [cardiovascular] events. We didn’t have a drug like that before, so I think that places a lot of emphasis on aggressive treatment of diabetes.”
For patients with symptomatic HF, the document touts multidisciplinary care and early initiation of drugs from each of four drug classes. Such quadruple therapy includes an SGLT2 inhibitor along with a beta-blocker, a mineralocorticoid receptor antagonist (MRA), and a renin-angiotensin system (RAS) inhibitor: the “core foundational therapies” for patients with HFrEF, Dr. Bozkurt observed.
Of note, she said, the angiotensin receptor–neprilysin inhibitor sacubitril/valsartan (Entresto, Novartis) is the preferred RAS inhibitor. But “if the ARNI cannot be used, then use ACE inhibitors.” If the patient is intolerant of ACE inhibitors because of cough or angioedema, then the choice should be an angiotensin-receptor blocker.
“We have very effective therapies offering survival and morbidity benefits as well as improvements in quality of life and reverse remodeling,” Dr. Bozkurt observed. “The most important message is that optimization of therapies, including all of these medication classes, saves lives.”
The guideline also includes, for the first time, a series of “value statements” on cost-effectiveness of different therapies that assign a “high-value” rating to MRAs, hydralazine, and isosorbide dinitrate in otherwise optimally treated self-identified African Americans, and device therapy in appropriately selected patients. The statements hold SGLT2 inhibitors in chronic symptomatic HF and cardiac transplantation in advanced GDMT-resistant HF to be of “intermediate” value.
The value statements, Dr. Bozkurt noted, “are included throughout the document when there is evidence; when there is a high-quality cost-effectiveness study published.”
Dr. Bozkurt disclosed receiving honoraria or consulting fees from Amgen, AstraZeneca, Baxter International, Bristol-Myers Squibb, Sanofi-Aventis, scPharmaceuticals, and Vifor Pharma; serving on a data safety monitoring board for LivaNova USA; and holding other relationships with Abbott Laboratories and Relypsa. Dr. Mann disclosed receiving honoraria or consulting fees from MyoKardia, Novartis, and Novo Nordisk.
A version of this article first appeared on Medscape.com.
The new heart failure (HF) guidelines released by three North American societies had a lot of catching up to do given the significant, even paradigm-shifting, additions to available treatment options in the last few years.
The landscape now includes both new and repurposed drug therapies that benefit almost without regard to ejection fraction (EF), and evidence-based urgency to engage patients early on with at least four core medication classes, so-called quadruple therapy.
The guideline document offers a roadmap for navigating those key issues and many others and uses some creative tactics. They include the introduction of generalist-friendly labels for the traditional but obscurely named four stages of HF severity that, it is hoped, will have wider reach and expand the use of effective therapies.
It introduces additional disease-staging terminology that characterizes the syndrome as a continuum:
- “At risk for HF” for stage A, applied to asymptomatic patients with risk factors such as diabetes or hypertension but no known cardiac changes.
- “Pre-HF” for stage B, which adds cardiac structural changes or elevated natriuretic peptides, still in the absence of symptoms.
- “Symptomatic HF” for stage C, that is, structural disease with current or previous symptoms.
- “Advanced HF” for stage D, characterized by severe debilitating symptoms or repeated hospitalizations even with guideline-directed medical therapy (GDMT).
The new terms should be “easier for primary care physicians as well as nonspecialists” to remember and use effectively “and easier to translate to the patients,” compared with the solely alphabetical staging labels appearing in the guidelines for more than 15 years, Biykem Bozkurt, MD, PhD, Baylor College of Medicine, Houston, said in an interview.
An emphasis on “at risk for HF” and “pre-HF” in the new document may help efforts to expand primary prevention of HF and management of preclinical HF. The guideline, Dr. Bozkurt said, includes specific treatment recommendations for those early stages.
The document also updates and sometimes introduces “recommendations for advanced heart failure, acute heart failure, and comorbidities – specifically for atrial fibrillation, iron deficiency, sleep apnea, coronary artery disease, and valvular heart disease,” Dr. Bozkurt observed, as well as for cardiomyopathy and HF related to pregnancy and cancer chemotherapy. “So, it’s a very comprehensive guideline.”
Dr. Bozkurt is vice chair of the guideline writing committee and helped introduce the guideline at the annual scientific sessions of the American College of Cardiology. The document, developed by the ACC, the American Heart Association, and the Heart Failure Society of America, was published April 1, 2022, in the societies’ flagship journals, Journal of the American College of Cardiology, Circulation, and the Journal of Cardiac Failure, respectively. It replaces the 2013 guideline from the ACC and AHA and the ACC/AHA/HFSA–focused update from 2017.
“We really need to treat early, and then we need to treat appropriately,” Douglas L. Mann, MD, Washington University in St. Louis, said in an interview. Dr. Mann, who was not involved in development of the new guideline, said he is “enthusiastic” about the new staging terminology.
“I think it makes it easier to convey the message that these people do need medicines, will benefit from medicines, and in some cases heart failure can be preventable,” he said. “I’m in favor of anything that simplifies it and makes it more readily interpretable by busy doctors who aren’t specialists.”
With the new staging terminology and in other ways, the guideline seems to appreciate cardiomyopathy as a journey from preclinical to advanced symptomatic stages – the preclinical “at-risk” stage tightening focus on primary prevention – and updated thinking on classification of HF by EF.
For example, there is new consideration of “HF with improved ejection fraction” (HFimpEF), which suggests the patient may be evolving from HF with reduced EF (HFrEF) to HF with EF that is preserved or mildly reduced, or vice versa.
With HFimpEF, which identifies patients previously with an EF of 40% or lower that improves to beyond 40% at follow-up testing, patients should continue on the medications they had been previously taking for HFrEF, Dr. Bozkurt said.
Patients at risk for HF, in stage A by the older terminology, are characterized by one or more significant HF risk factors, such as hypertension, diabetes, or coronary disease, as they have been in prior guidelines. But the new document, Dr. Bozkurt observed, adds genetic cardiomyopathies and exposure to cardiotoxic agents to the list.
Perhaps surprisingly, the guideline also includes elevated natriuretic peptides as an indicator of “at risk for HF,” with implications for screening. The evidence suggests that, “for patients who are at risk for heart failure, natriuretic peptide-based screening, followed by team-based care, can prevent development of left ventricular dysfunction in heart failure,” Dr. Bozkurt said.
Persons at risk for HF realistically encompass a huge swath of the population given the world prevalence of high blood pressure, obesity, and diabetes. Management of stage A, therefore, focuses on established tenets of primary cardiovascular prevention, such as weight and BP control, exercise, and healthy dietary choices.
They may well be eligible for treatment with sodium-glucose transporter 2 (SGLT2) inhibitors, which have been “game changers,” Dr. Mann said. “Now you can give them to diabetics and it’s going to prevent heart failure and [cardiovascular] events. We didn’t have a drug like that before, so I think that places a lot of emphasis on aggressive treatment of diabetes.”
For patients with symptomatic HF, the document touts multidisciplinary care and early initiation of drugs from each of four drug classes. Such quadruple therapy includes an SGLT2 inhibitor along with a beta-blocker, a mineralocorticoid receptor antagonist (MRA), and a renin-angiotensin system (RAS) inhibitor: the “core foundational therapies” for patients with HFrEF, Dr. Bozkurt observed.
Of note, she said, the angiotensin receptor–neprilysin inhibitor sacubitril/valsartan (Entresto, Novartis) is the preferred RAS inhibitor. But “if the ARNI cannot be used, then use ACE inhibitors.” If the patient is intolerant of ACE inhibitors because of cough or angioedema, then the choice should be an angiotensin-receptor blocker.
“We have very effective therapies offering survival and morbidity benefits as well as improvements in quality of life and reverse remodeling,” Dr. Bozkurt observed. “The most important message is that optimization of therapies, including all of these medication classes, saves lives.”
The guideline also includes, for the first time, a series of “value statements” on cost-effectiveness of different therapies that assign a “high-value” rating to MRAs, hydralazine, and isosorbide dinitrate in otherwise optimally treated self-identified African Americans, and device therapy in appropriately selected patients. The statements hold SGLT2 inhibitors in chronic symptomatic HF and cardiac transplantation in advanced GDMT-resistant HF to be of “intermediate” value.
The value statements, Dr. Bozkurt noted, “are included throughout the document when there is evidence; when there is a high-quality cost-effectiveness study published.”
Dr. Bozkurt disclosed receiving honoraria or consulting fees from Amgen, AstraZeneca, Baxter International, Bristol-Myers Squibb, Sanofi-Aventis, scPharmaceuticals, and Vifor Pharma; serving on a data safety monitoring board for LivaNova USA; and holding other relationships with Abbott Laboratories and Relypsa. Dr. Mann disclosed receiving honoraria or consulting fees from MyoKardia, Novartis, and Novo Nordisk.
A version of this article first appeared on Medscape.com.
The new heart failure (HF) guidelines released by three North American societies had a lot of catching up to do given the significant, even paradigm-shifting, additions to available treatment options in the last few years.
The landscape now includes both new and repurposed drug therapies that benefit almost without regard to ejection fraction (EF), and evidence-based urgency to engage patients early on with at least four core medication classes, so-called quadruple therapy.
The guideline document offers a roadmap for navigating those key issues and many others and uses some creative tactics. They include the introduction of generalist-friendly labels for the traditional but obscurely named four stages of HF severity that, it is hoped, will have wider reach and expand the use of effective therapies.
It introduces additional disease-staging terminology that characterizes the syndrome as a continuum:
- “At risk for HF” for stage A, applied to asymptomatic patients with risk factors such as diabetes or hypertension but no known cardiac changes.
- “Pre-HF” for stage B, which adds cardiac structural changes or elevated natriuretic peptides, still in the absence of symptoms.
- “Symptomatic HF” for stage C, that is, structural disease with current or previous symptoms.
- “Advanced HF” for stage D, characterized by severe debilitating symptoms or repeated hospitalizations even with guideline-directed medical therapy (GDMT).
The new terms should be “easier for primary care physicians as well as nonspecialists” to remember and use effectively “and easier to translate to the patients,” compared with the solely alphabetical staging labels appearing in the guidelines for more than 15 years, Biykem Bozkurt, MD, PhD, Baylor College of Medicine, Houston, said in an interview.
An emphasis on “at risk for HF” and “pre-HF” in the new document may help efforts to expand primary prevention of HF and management of preclinical HF. The guideline, Dr. Bozkurt said, includes specific treatment recommendations for those early stages.
The document also updates and sometimes introduces “recommendations for advanced heart failure, acute heart failure, and comorbidities – specifically for atrial fibrillation, iron deficiency, sleep apnea, coronary artery disease, and valvular heart disease,” Dr. Bozkurt observed, as well as for cardiomyopathy and HF related to pregnancy and cancer chemotherapy. “So, it’s a very comprehensive guideline.”
Dr. Bozkurt is vice chair of the guideline writing committee and helped introduce the guideline at the annual scientific sessions of the American College of Cardiology. The document, developed by the ACC, the American Heart Association, and the Heart Failure Society of America, was published April 1, 2022, in the societies’ flagship journals, Journal of the American College of Cardiology, Circulation, and the Journal of Cardiac Failure, respectively. It replaces the 2013 guideline from the ACC and AHA and the ACC/AHA/HFSA–focused update from 2017.
“We really need to treat early, and then we need to treat appropriately,” Douglas L. Mann, MD, Washington University in St. Louis, said in an interview. Dr. Mann, who was not involved in development of the new guideline, said he is “enthusiastic” about the new staging terminology.
“I think it makes it easier to convey the message that these people do need medicines, will benefit from medicines, and in some cases heart failure can be preventable,” he said. “I’m in favor of anything that simplifies it and makes it more readily interpretable by busy doctors who aren’t specialists.”
With the new staging terminology and in other ways, the guideline seems to appreciate cardiomyopathy as a journey from preclinical to advanced symptomatic stages – the preclinical “at-risk” stage tightening focus on primary prevention – and updated thinking on classification of HF by EF.
For example, there is new consideration of “HF with improved ejection fraction” (HFimpEF), which suggests the patient may be evolving from HF with reduced EF (HFrEF) to HF with EF that is preserved or mildly reduced, or vice versa.
With HFimpEF, which identifies patients previously with an EF of 40% or lower that improves to beyond 40% at follow-up testing, patients should continue on the medications they had been previously taking for HFrEF, Dr. Bozkurt said.
Patients at risk for HF, in stage A by the older terminology, are characterized by one or more significant HF risk factors, such as hypertension, diabetes, or coronary disease, as they have been in prior guidelines. But the new document, Dr. Bozkurt observed, adds genetic cardiomyopathies and exposure to cardiotoxic agents to the list.
Perhaps surprisingly, the guideline also includes elevated natriuretic peptides as an indicator of “at risk for HF,” with implications for screening. The evidence suggests that, “for patients who are at risk for heart failure, natriuretic peptide-based screening, followed by team-based care, can prevent development of left ventricular dysfunction in heart failure,” Dr. Bozkurt said.
Persons at risk for HF realistically encompass a huge swath of the population given the world prevalence of high blood pressure, obesity, and diabetes. Management of stage A, therefore, focuses on established tenets of primary cardiovascular prevention, such as weight and BP control, exercise, and healthy dietary choices.
They may well be eligible for treatment with sodium-glucose transporter 2 (SGLT2) inhibitors, which have been “game changers,” Dr. Mann said. “Now you can give them to diabetics and it’s going to prevent heart failure and [cardiovascular] events. We didn’t have a drug like that before, so I think that places a lot of emphasis on aggressive treatment of diabetes.”
For patients with symptomatic HF, the document touts multidisciplinary care and early initiation of drugs from each of four drug classes. Such quadruple therapy includes an SGLT2 inhibitor along with a beta-blocker, a mineralocorticoid receptor antagonist (MRA), and a renin-angiotensin system (RAS) inhibitor: the “core foundational therapies” for patients with HFrEF, Dr. Bozkurt observed.
Of note, she said, the angiotensin receptor–neprilysin inhibitor sacubitril/valsartan (Entresto, Novartis) is the preferred RAS inhibitor. But “if the ARNI cannot be used, then use ACE inhibitors.” If the patient is intolerant of ACE inhibitors because of cough or angioedema, then the choice should be an angiotensin-receptor blocker.
“We have very effective therapies offering survival and morbidity benefits as well as improvements in quality of life and reverse remodeling,” Dr. Bozkurt observed. “The most important message is that optimization of therapies, including all of these medication classes, saves lives.”
The guideline also includes, for the first time, a series of “value statements” on cost-effectiveness of different therapies that assign a “high-value” rating to MRAs, hydralazine, and isosorbide dinitrate in otherwise optimally treated self-identified African Americans, and device therapy in appropriately selected patients. The statements hold SGLT2 inhibitors in chronic symptomatic HF and cardiac transplantation in advanced GDMT-resistant HF to be of “intermediate” value.
The value statements, Dr. Bozkurt noted, “are included throughout the document when there is evidence; when there is a high-quality cost-effectiveness study published.”
Dr. Bozkurt disclosed receiving honoraria or consulting fees from Amgen, AstraZeneca, Baxter International, Bristol-Myers Squibb, Sanofi-Aventis, scPharmaceuticals, and Vifor Pharma; serving on a data safety monitoring board for LivaNova USA; and holding other relationships with Abbott Laboratories and Relypsa. Dr. Mann disclosed receiving honoraria or consulting fees from MyoKardia, Novartis, and Novo Nordisk.
A version of this article first appeared on Medscape.com.
FROM ACC 2022