User login
Pruritic Eruption on the Chest
The Diagnosis: Grover Disease
Grover disease (also known as transient acantholytic dermatosis) was first described by Ralph W. Grover in 1970 as an idiopathic, acquired, monomorphous, papulovesicular eruption. Although originally characterized by solely transient acantholytic dermatosis, over time the term Grover disease has been expanded to include persistent acantholytic dermatoses. Grover disease chiefly affects white adults older than 40 years and is more prevalent in males than females. Cases generally are self-limited but correlate with age, as older adults are more likely to have prolonged eruptions.1
Grover disease typically erupts with discrete, erythematous, edematous, acneform, red-brown or flesh-colored papules, papulovesicles, or keratotic papules that primarily are seen on the trunk and anterior portion of the chest. As the rash spreads, it can erupt on the neck and thighs. The etiology of Grover disease is unknown, but many factors have been associated with the condition in a limited number of patients, including exposure to UV radiation, excessive heat or sweating, use of sulfadoxine-pyrimethamine and recombinant human IL-4, and infection with Malassezia furfur and Demodex folliculorum.1 Grover disease also has been associated with other conditions such as asteatotic eczema, allergic contact dermatitis, and atopic dermatitis.2
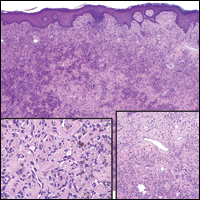
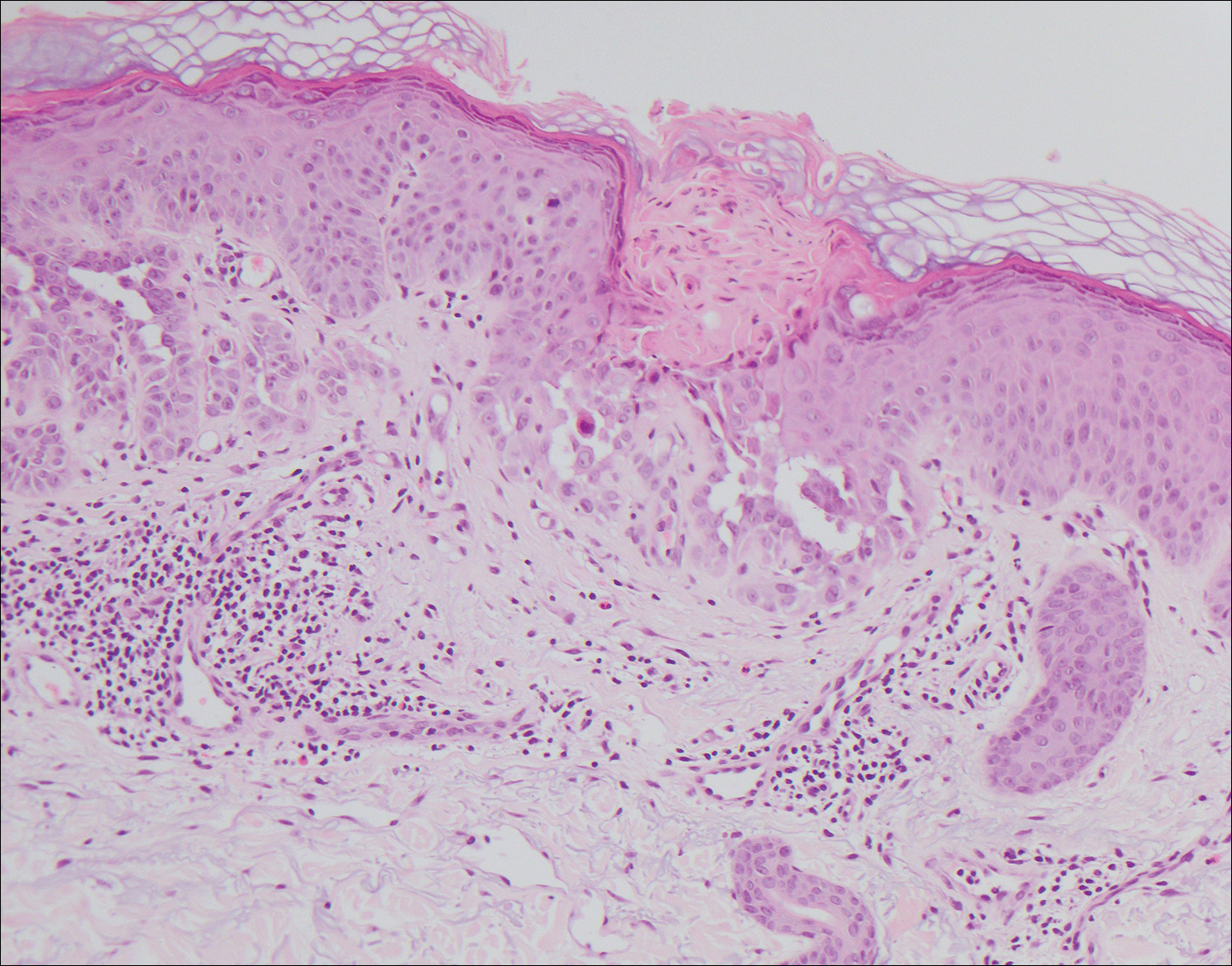
Histologically, Grover disease (Figure 1) is an acantholytic process that can exhibit dyskeratosis (corps ronds and grains). Foci often are small and multiple foci are seen on shave biopsy. There also may be spongiotic changes when associated with an eczematous element. A perivascular lymphohistiocytic infiltrate with eosinophils usually is seen.3 Basket weave keratin may be seen; however, as the lesions cause pruritus, erosions and ulcerations often are present.4
Grover disease has multiple histologic variants that may resemble Darier disease, Hailey-Hailey disease, pemphigus foliaceus, pemphigus vulgaris, and spongiotic dermatitis and can present in combination.5
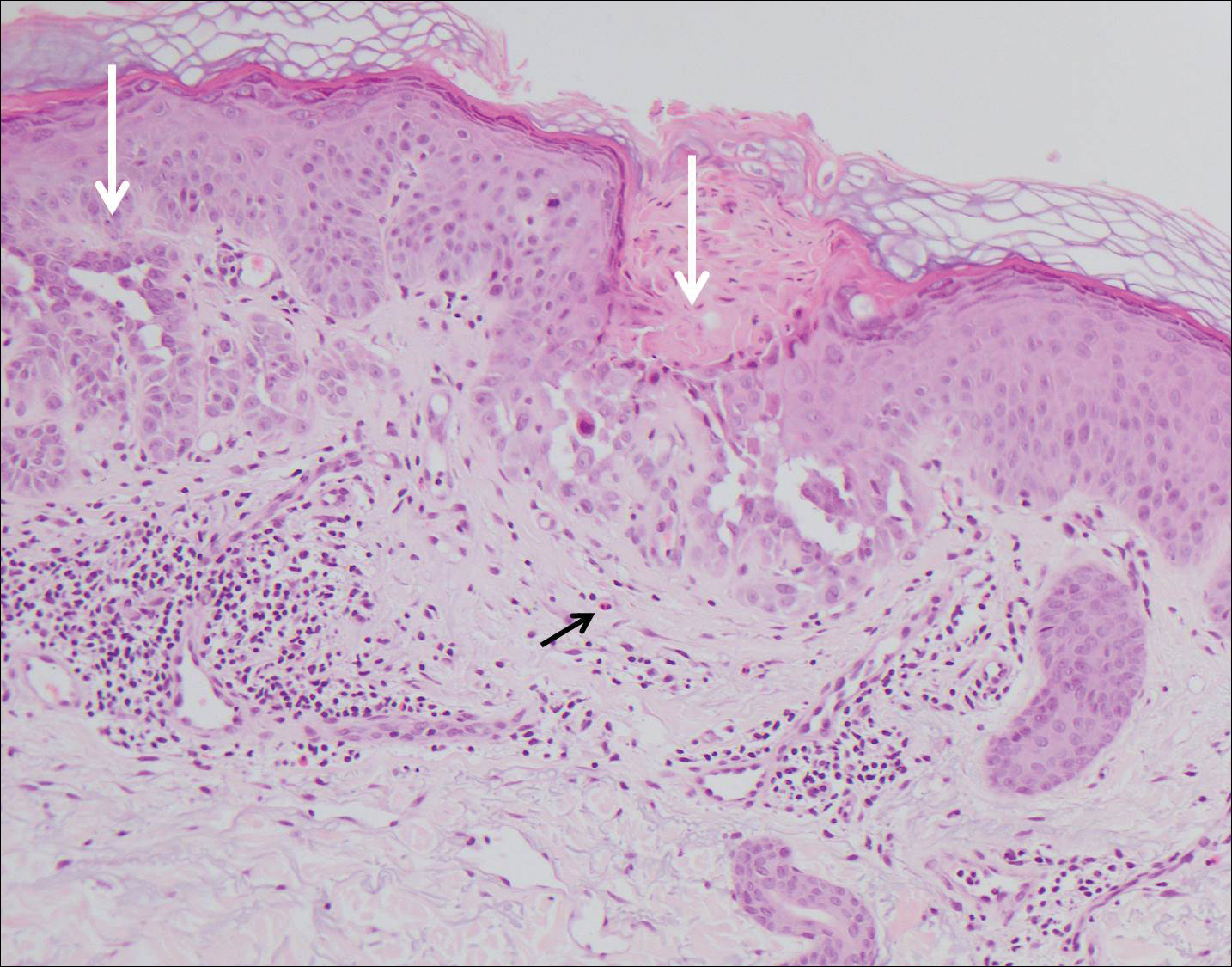
The variant of Grover disease that has a Darier-like pattern is difficult to distinguish from Darier disease, an autosomal-dominant-inherited disorder classified by small papules that emerge in seborrheic areas during childhood and adolescence. Histologically, Darier disease (Figure 2) shows broad areas of dyskeratosis and acantholysis that lead to suprabasal cleavage. Follicular extension may be present. In addition, there often is prominent vertical parakeratosis in Darier disease.6 Histologic features that favor Darier disease over the Darier-like variant of Grover disease include a broad focus of acanthotic dyskeratosis with follicular extension; the presence of a hyperkeratotic stratum corneum; and a lack of spongiosis and eosinophils, which are notably absent in Darier disease but may be present in Grover disease.4
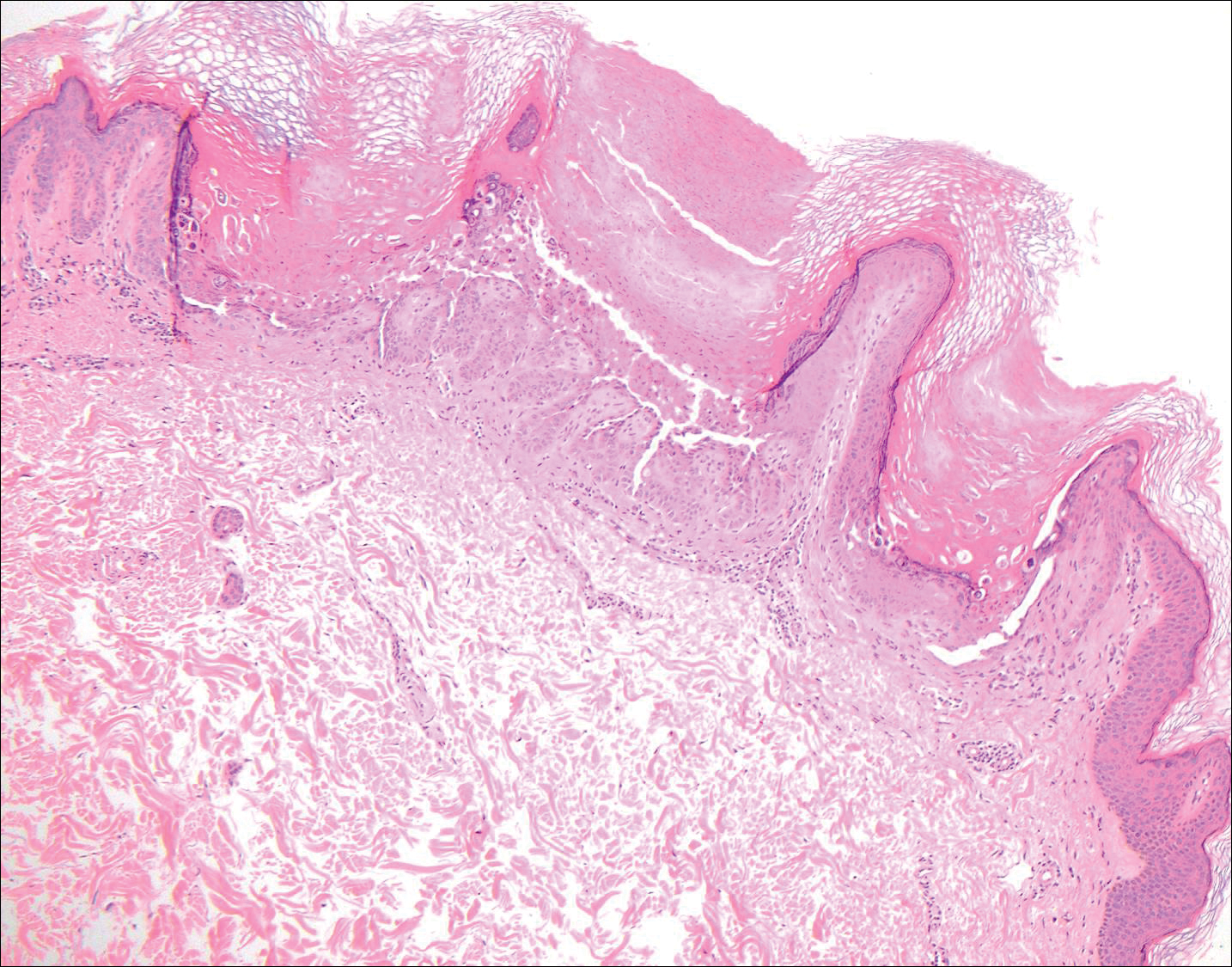
Another variant of Grover disease has a Hailey-Hailey-like pattern, which is characterized by Hailey-Hailey disease's dilapidated brick wall appearance or the diffuse suprabasal acantholysis of all epidermal layers without notable dyskeratosis.4 Hailey-Hailey disease, also known as familial benign pemphigus, is an autosomal-dominant disorder that presents with erythematous vesicular plaques in flexural areas. The plaques progress to flaccid bullae with rupture and crusting and spread peripherally.7 Pathology shows suprabasilar clefts and numerous acantholytic cells (Figure 3). Dyskeratotic keratinocytes are rare with infrequent corps ronds and rare grains. The epidermis also is less hyperplastic in Grover disease than in Hailey-Hailey disease.1
Grover disease also may present histologically with a pemphiguslike pattern, mimicking pemphigus foliaceus and pemphigus vulgaris; however, direct immunofluorescence studies are negative in Grover disease.
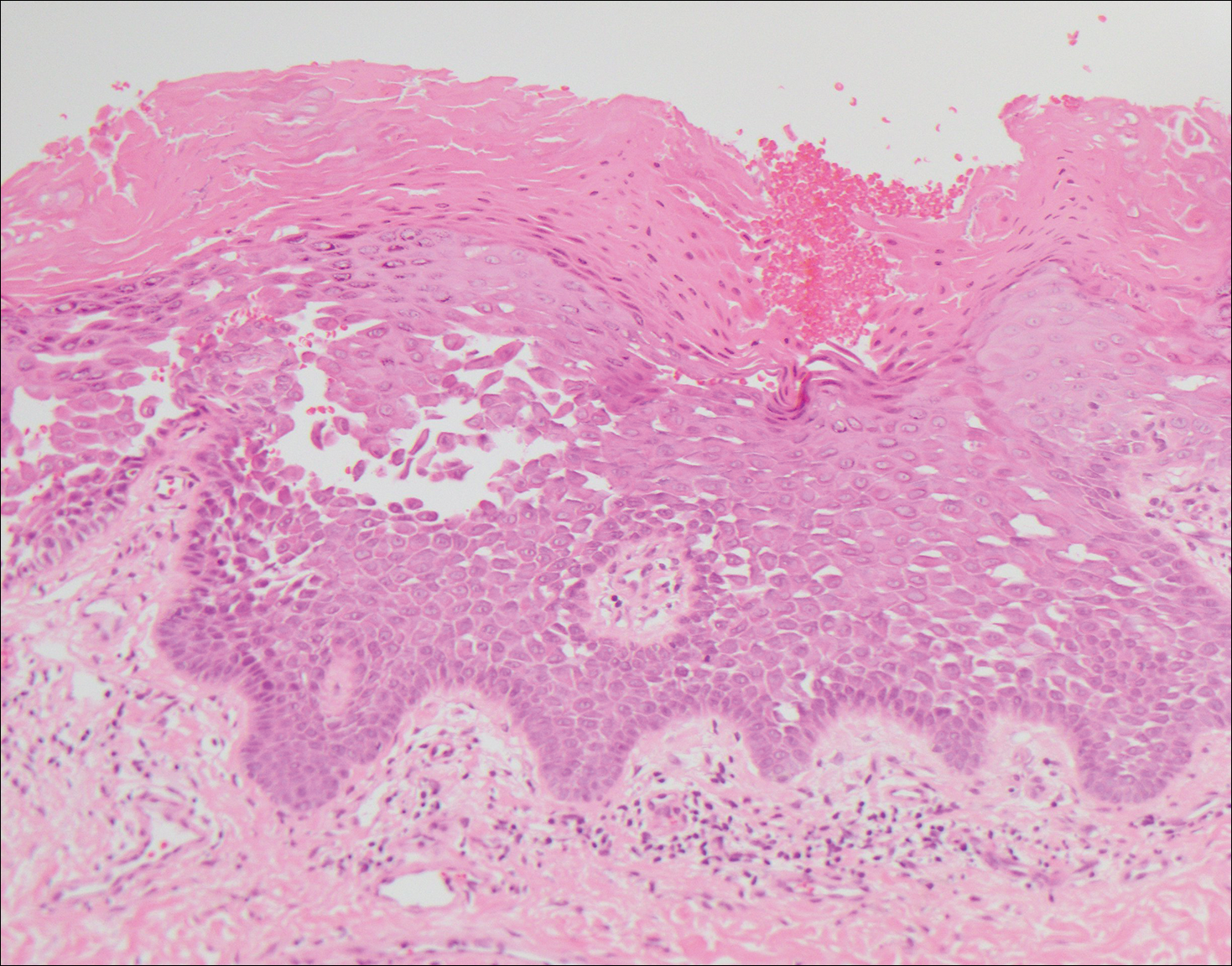
Pemphigus foliaceus is an autoimmune disorder caused by autoantibodies to desmoglein 1, which are present on the surfaces of keratinocytes, and is characterized by scaly crusts and blisters.8 Histologically, pemphigus foliaceus (Figure 4) shows a superficial epidermal blistering process. The acantholysis may be subtle and is commonly localized to the stratum granulosum, extending into the stratum corneum. Complete loss of the stratum corneum can be seen, resulting in only scattered acantholytic cells. Spongiosis also may be seen. The dermis shows a perivascular infiltrate that often contains eosinophils. Pemphigus foliaceus is confirmed by direct immunofluorescence.9
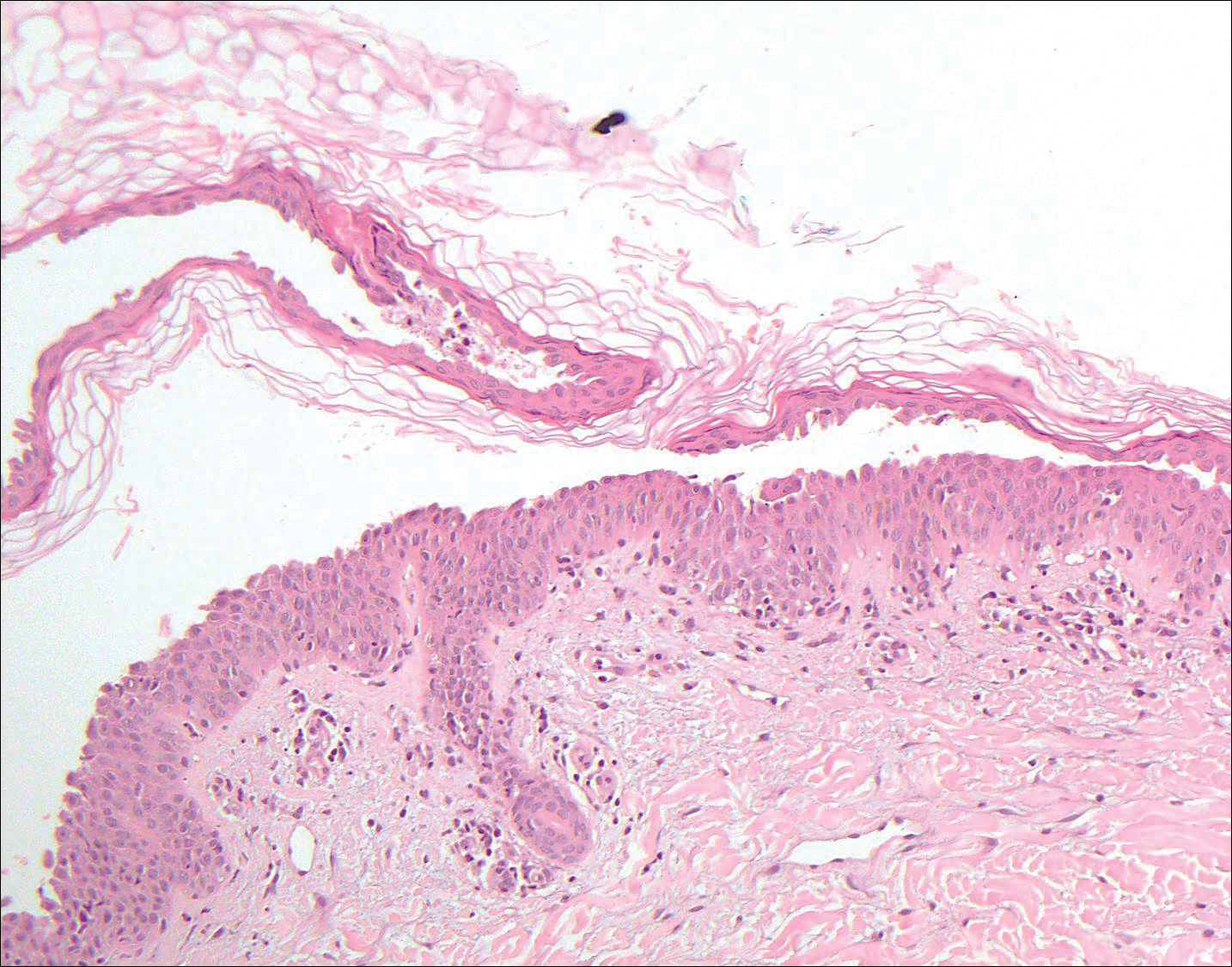
Pemphigus vulgaris is an autoimmune blistering disorder that is characterized by IgG autoantibodies to desmoglein 3, a component of desmosomes that are involved in keratinocyte-to-keratinocyte adhesion. Clinically, patients present with flaccid fragile blisters on the skin and mucous membranes that rupture easily, leading to painful erosions.10 Intraepidermal blisters are seen histologically (Figure 5) with the loss of cohesion (acantholysis) seen classically in the lower portions of the epidermis where desmoglein 3 is most prominent. When only the basal layer remains, the histology has been likened to a tombstone row.11 Extension of the blister along the adnexa is common. The underlying dermis shows a perivascular infiltrate with eosinophils. Early lesions may show only eosinophilic spongiosis. Direct immunofluorescence studies show IgG and C3 in an intercellular pattern that resembles a fish net or chicken wire.4,11
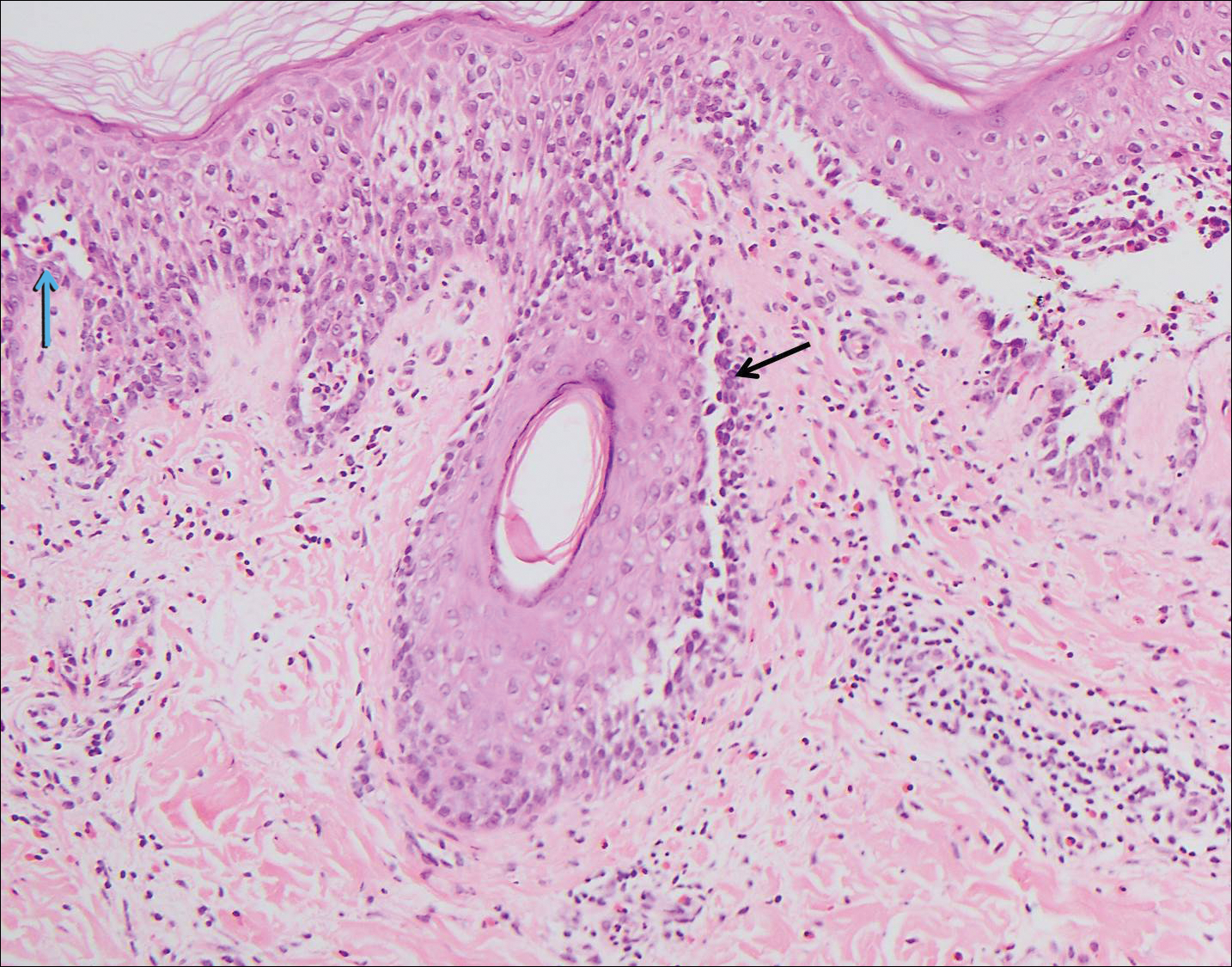
The spongioticlike pattern of Grover disease is marked by epidermal edema with separation of the keratinocytes and the revelation of their intracellular bridges,4 which manifests as vesiculation in the stratum corneum or upper layers of the epidermis.12
Grover disease is self-limited and may spontaneously resolve; however, the disease may be responsive to topical and systemic steroids. Additionally, avoidance of aggravating factors such as sunlight, heat, and sweating can improve symptoms.2
- Parsons JM. Transient acantholytic dermatosis (Grover's disease): a global perspective. J Am Acad Dermatol. 1996;35(5, pt 1):653-666; quiz 667-670.
- Quirk CJ, Heenan PJ. Grover's disease: 34 years on. Australas J Dermatol. 2004;45:83-86.
- Davis MD, Dinneen AM, Landa N, et al. Grover's disease: clinicopathologic review of 72 cases. Mayo Clin Proc. 1999;74:229-234.
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133:1490-1494.
- Chalet M, Grover R, Ackerman AB. Transient acantholytic dermatosis: a reevaluation. Arch Dermatol. 1977;133:431-435.
- Takagi A, Kamijo M, Ikeda S. Darier disease. J Dermatol. 2016;43:275-279.
- Engin B, Kutlubay Z, Celik U, et al. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33:452-455.
- de Sena Nogueira Maehara L, Huizinga J, Jonkman MF. Rituximab therapy in pemphigus foliaceus: report of 12 cases and review of recent literature [published online March 31, 2015]. Br J Dermatol. 2015;172:1420-1423.
- James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-412.
- Black M, Mignogna MD, Scully C. Number II. pemphigus vulgaris. Oral Dis. 2005;11:119-130.
- Madke B, Doshi B, Khopkar U, et al. Appearances in dermatopathology: the diagnostic and the deceptive. Indian J Dermatol Venerol Leprol. 2013;79:338-348.
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature [published online February 16, 2017]. J Cutan Pathol. 2017;44:486-489.
The Diagnosis: Grover Disease
Grover disease (also known as transient acantholytic dermatosis) was first described by Ralph W. Grover in 1970 as an idiopathic, acquired, monomorphous, papulovesicular eruption. Although originally characterized by solely transient acantholytic dermatosis, over time the term Grover disease has been expanded to include persistent acantholytic dermatoses. Grover disease chiefly affects white adults older than 40 years and is more prevalent in males than females. Cases generally are self-limited but correlate with age, as older adults are more likely to have prolonged eruptions.1
Grover disease typically erupts with discrete, erythematous, edematous, acneform, red-brown or flesh-colored papules, papulovesicles, or keratotic papules that primarily are seen on the trunk and anterior portion of the chest. As the rash spreads, it can erupt on the neck and thighs. The etiology of Grover disease is unknown, but many factors have been associated with the condition in a limited number of patients, including exposure to UV radiation, excessive heat or sweating, use of sulfadoxine-pyrimethamine and recombinant human IL-4, and infection with Malassezia furfur and Demodex folliculorum.1 Grover disease also has been associated with other conditions such as asteatotic eczema, allergic contact dermatitis, and atopic dermatitis.2
Histologically, Grover disease (Figure 1) is an acantholytic process that can exhibit dyskeratosis (corps ronds and grains). Foci often are small and multiple foci are seen on shave biopsy. There also may be spongiotic changes when associated with an eczematous element. A perivascular lymphohistiocytic infiltrate with eosinophils usually is seen.3 Basket weave keratin may be seen; however, as the lesions cause pruritus, erosions and ulcerations often are present.4
Grover disease has multiple histologic variants that may resemble Darier disease, Hailey-Hailey disease, pemphigus foliaceus, pemphigus vulgaris, and spongiotic dermatitis and can present in combination.5
The variant of Grover disease that has a Darier-like pattern is difficult to distinguish from Darier disease, an autosomal-dominant-inherited disorder classified by small papules that emerge in seborrheic areas during childhood and adolescence. Histologically, Darier disease (Figure 2) shows broad areas of dyskeratosis and acantholysis that lead to suprabasal cleavage. Follicular extension may be present. In addition, there often is prominent vertical parakeratosis in Darier disease.6 Histologic features that favor Darier disease over the Darier-like variant of Grover disease include a broad focus of acanthotic dyskeratosis with follicular extension; the presence of a hyperkeratotic stratum corneum; and a lack of spongiosis and eosinophils, which are notably absent in Darier disease but may be present in Grover disease.4
Another variant of Grover disease has a Hailey-Hailey-like pattern, which is characterized by Hailey-Hailey disease's dilapidated brick wall appearance or the diffuse suprabasal acantholysis of all epidermal layers without notable dyskeratosis.4 Hailey-Hailey disease, also known as familial benign pemphigus, is an autosomal-dominant disorder that presents with erythematous vesicular plaques in flexural areas. The plaques progress to flaccid bullae with rupture and crusting and spread peripherally.7 Pathology shows suprabasilar clefts and numerous acantholytic cells (Figure 3). Dyskeratotic keratinocytes are rare with infrequent corps ronds and rare grains. The epidermis also is less hyperplastic in Grover disease than in Hailey-Hailey disease.1
Grover disease also may present histologically with a pemphiguslike pattern, mimicking pemphigus foliaceus and pemphigus vulgaris; however, direct immunofluorescence studies are negative in Grover disease.
Pemphigus foliaceus is an autoimmune disorder caused by autoantibodies to desmoglein 1, which are present on the surfaces of keratinocytes, and is characterized by scaly crusts and blisters.8 Histologically, pemphigus foliaceus (Figure 4) shows a superficial epidermal blistering process. The acantholysis may be subtle and is commonly localized to the stratum granulosum, extending into the stratum corneum. Complete loss of the stratum corneum can be seen, resulting in only scattered acantholytic cells. Spongiosis also may be seen. The dermis shows a perivascular infiltrate that often contains eosinophils. Pemphigus foliaceus is confirmed by direct immunofluorescence.9
Pemphigus vulgaris is an autoimmune blistering disorder that is characterized by IgG autoantibodies to desmoglein 3, a component of desmosomes that are involved in keratinocyte-to-keratinocyte adhesion. Clinically, patients present with flaccid fragile blisters on the skin and mucous membranes that rupture easily, leading to painful erosions.10 Intraepidermal blisters are seen histologically (Figure 5) with the loss of cohesion (acantholysis) seen classically in the lower portions of the epidermis where desmoglein 3 is most prominent. When only the basal layer remains, the histology has been likened to a tombstone row.11 Extension of the blister along the adnexa is common. The underlying dermis shows a perivascular infiltrate with eosinophils. Early lesions may show only eosinophilic spongiosis. Direct immunofluorescence studies show IgG and C3 in an intercellular pattern that resembles a fish net or chicken wire.4,11
The spongioticlike pattern of Grover disease is marked by epidermal edema with separation of the keratinocytes and the revelation of their intracellular bridges,4 which manifests as vesiculation in the stratum corneum or upper layers of the epidermis.12
Grover disease is self-limited and may spontaneously resolve; however, the disease may be responsive to topical and systemic steroids. Additionally, avoidance of aggravating factors such as sunlight, heat, and sweating can improve symptoms.2
The Diagnosis: Grover Disease
Grover disease (also known as transient acantholytic dermatosis) was first described by Ralph W. Grover in 1970 as an idiopathic, acquired, monomorphous, papulovesicular eruption. Although originally characterized by solely transient acantholytic dermatosis, over time the term Grover disease has been expanded to include persistent acantholytic dermatoses. Grover disease chiefly affects white adults older than 40 years and is more prevalent in males than females. Cases generally are self-limited but correlate with age, as older adults are more likely to have prolonged eruptions.1
Grover disease typically erupts with discrete, erythematous, edematous, acneform, red-brown or flesh-colored papules, papulovesicles, or keratotic papules that primarily are seen on the trunk and anterior portion of the chest. As the rash spreads, it can erupt on the neck and thighs. The etiology of Grover disease is unknown, but many factors have been associated with the condition in a limited number of patients, including exposure to UV radiation, excessive heat or sweating, use of sulfadoxine-pyrimethamine and recombinant human IL-4, and infection with Malassezia furfur and Demodex folliculorum.1 Grover disease also has been associated with other conditions such as asteatotic eczema, allergic contact dermatitis, and atopic dermatitis.2
Histologically, Grover disease (Figure 1) is an acantholytic process that can exhibit dyskeratosis (corps ronds and grains). Foci often are small and multiple foci are seen on shave biopsy. There also may be spongiotic changes when associated with an eczematous element. A perivascular lymphohistiocytic infiltrate with eosinophils usually is seen.3 Basket weave keratin may be seen; however, as the lesions cause pruritus, erosions and ulcerations often are present.4
Grover disease has multiple histologic variants that may resemble Darier disease, Hailey-Hailey disease, pemphigus foliaceus, pemphigus vulgaris, and spongiotic dermatitis and can present in combination.5
The variant of Grover disease that has a Darier-like pattern is difficult to distinguish from Darier disease, an autosomal-dominant-inherited disorder classified by small papules that emerge in seborrheic areas during childhood and adolescence. Histologically, Darier disease (Figure 2) shows broad areas of dyskeratosis and acantholysis that lead to suprabasal cleavage. Follicular extension may be present. In addition, there often is prominent vertical parakeratosis in Darier disease.6 Histologic features that favor Darier disease over the Darier-like variant of Grover disease include a broad focus of acanthotic dyskeratosis with follicular extension; the presence of a hyperkeratotic stratum corneum; and a lack of spongiosis and eosinophils, which are notably absent in Darier disease but may be present in Grover disease.4
Another variant of Grover disease has a Hailey-Hailey-like pattern, which is characterized by Hailey-Hailey disease's dilapidated brick wall appearance or the diffuse suprabasal acantholysis of all epidermal layers without notable dyskeratosis.4 Hailey-Hailey disease, also known as familial benign pemphigus, is an autosomal-dominant disorder that presents with erythematous vesicular plaques in flexural areas. The plaques progress to flaccid bullae with rupture and crusting and spread peripherally.7 Pathology shows suprabasilar clefts and numerous acantholytic cells (Figure 3). Dyskeratotic keratinocytes are rare with infrequent corps ronds and rare grains. The epidermis also is less hyperplastic in Grover disease than in Hailey-Hailey disease.1
Grover disease also may present histologically with a pemphiguslike pattern, mimicking pemphigus foliaceus and pemphigus vulgaris; however, direct immunofluorescence studies are negative in Grover disease.
Pemphigus foliaceus is an autoimmune disorder caused by autoantibodies to desmoglein 1, which are present on the surfaces of keratinocytes, and is characterized by scaly crusts and blisters.8 Histologically, pemphigus foliaceus (Figure 4) shows a superficial epidermal blistering process. The acantholysis may be subtle and is commonly localized to the stratum granulosum, extending into the stratum corneum. Complete loss of the stratum corneum can be seen, resulting in only scattered acantholytic cells. Spongiosis also may be seen. The dermis shows a perivascular infiltrate that often contains eosinophils. Pemphigus foliaceus is confirmed by direct immunofluorescence.9
Pemphigus vulgaris is an autoimmune blistering disorder that is characterized by IgG autoantibodies to desmoglein 3, a component of desmosomes that are involved in keratinocyte-to-keratinocyte adhesion. Clinically, patients present with flaccid fragile blisters on the skin and mucous membranes that rupture easily, leading to painful erosions.10 Intraepidermal blisters are seen histologically (Figure 5) with the loss of cohesion (acantholysis) seen classically in the lower portions of the epidermis where desmoglein 3 is most prominent. When only the basal layer remains, the histology has been likened to a tombstone row.11 Extension of the blister along the adnexa is common. The underlying dermis shows a perivascular infiltrate with eosinophils. Early lesions may show only eosinophilic spongiosis. Direct immunofluorescence studies show IgG and C3 in an intercellular pattern that resembles a fish net or chicken wire.4,11
The spongioticlike pattern of Grover disease is marked by epidermal edema with separation of the keratinocytes and the revelation of their intracellular bridges,4 which manifests as vesiculation in the stratum corneum or upper layers of the epidermis.12
Grover disease is self-limited and may spontaneously resolve; however, the disease may be responsive to topical and systemic steroids. Additionally, avoidance of aggravating factors such as sunlight, heat, and sweating can improve symptoms.2
- Parsons JM. Transient acantholytic dermatosis (Grover's disease): a global perspective. J Am Acad Dermatol. 1996;35(5, pt 1):653-666; quiz 667-670.
- Quirk CJ, Heenan PJ. Grover's disease: 34 years on. Australas J Dermatol. 2004;45:83-86.
- Davis MD, Dinneen AM, Landa N, et al. Grover's disease: clinicopathologic review of 72 cases. Mayo Clin Proc. 1999;74:229-234.
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133:1490-1494.
- Chalet M, Grover R, Ackerman AB. Transient acantholytic dermatosis: a reevaluation. Arch Dermatol. 1977;133:431-435.
- Takagi A, Kamijo M, Ikeda S. Darier disease. J Dermatol. 2016;43:275-279.
- Engin B, Kutlubay Z, Celik U, et al. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33:452-455.
- de Sena Nogueira Maehara L, Huizinga J, Jonkman MF. Rituximab therapy in pemphigus foliaceus: report of 12 cases and review of recent literature [published online March 31, 2015]. Br J Dermatol. 2015;172:1420-1423.
- James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-412.
- Black M, Mignogna MD, Scully C. Number II. pemphigus vulgaris. Oral Dis. 2005;11:119-130.
- Madke B, Doshi B, Khopkar U, et al. Appearances in dermatopathology: the diagnostic and the deceptive. Indian J Dermatol Venerol Leprol. 2013;79:338-348.
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature [published online February 16, 2017]. J Cutan Pathol. 2017;44:486-489.
- Parsons JM. Transient acantholytic dermatosis (Grover's disease): a global perspective. J Am Acad Dermatol. 1996;35(5, pt 1):653-666; quiz 667-670.
- Quirk CJ, Heenan PJ. Grover's disease: 34 years on. Australas J Dermatol. 2004;45:83-86.
- Davis MD, Dinneen AM, Landa N, et al. Grover's disease: clinicopathologic review of 72 cases. Mayo Clin Proc. 1999;74:229-234.
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133:1490-1494.
- Chalet M, Grover R, Ackerman AB. Transient acantholytic dermatosis: a reevaluation. Arch Dermatol. 1977;133:431-435.
- Takagi A, Kamijo M, Ikeda S. Darier disease. J Dermatol. 2016;43:275-279.
- Engin B, Kutlubay Z, Celik U, et al. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33:452-455.
- de Sena Nogueira Maehara L, Huizinga J, Jonkman MF. Rituximab therapy in pemphigus foliaceus: report of 12 cases and review of recent literature [published online March 31, 2015]. Br J Dermatol. 2015;172:1420-1423.
- James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-412.
- Black M, Mignogna MD, Scully C. Number II. pemphigus vulgaris. Oral Dis. 2005;11:119-130.
- Madke B, Doshi B, Khopkar U, et al. Appearances in dermatopathology: the diagnostic and the deceptive. Indian J Dermatol Venerol Leprol. 2013;79:338-348.
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature [published online February 16, 2017]. J Cutan Pathol. 2017;44:486-489.
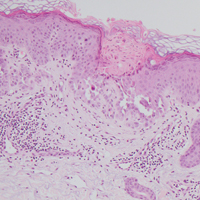
A 55-year-old man presented with small, erythematous, nonfollicular, pruritic papules on the mid chest.
Asymptomatic Cutaneous Polyarteritis Nodosa: Treatment Options and Therapeutic Guidelines
In 1931, Lindberg1 described a cutaneous variant of polyarteritis nodosa, which lacked visceral involvement and possessed a more favorable prognosis.2 Cutaneous polyarteritis nodosa (CPAN) is a localized small- to medium-vessel vasculitis restricted to the skin. Both benign and chronic courses have been described, and systemic involvement does not occur.3 Diagnostic criteria proposed by Nakamura et al3 in 2009 included cutaneous nodules, livedo reticularis, purpura, or ulcers; histopathologic fibrinoid necrotizing vasculitis of small- to medium-sized vessels; and exclusion of systemic symptoms (eg, fever, hypertension, weight loss, renal failure, cerebral hemorrhage, neuropathy, myocardial infarction, ischemic heart disease, pericarditis, pleuritis, arthralgia/myalgia). Nodules occur in 30% to 50% of cases and can remain for years if left untreated. Ulcerations occur in up to 30% of patients. Myositis, arthritis, and weakness also have been reported with this condition.4 Cutaneous polyarteritis nodosa has been associated with abnormal antibody testing with elevations of antiphospholipid cofactor antibody, lupus anticoagulant, anticardiolipin antibody, and anti-β2-glycoprotein I–dependent cardiolipin antibody, as well as elevated anti–phosphatidylserine-prothrombin complex antibody.5 These antibodies suggest increased risk for thrombosis and systemic diseases such as lupus or other autoimmune connective tissue disease. The distinction of this entity from systemic polyartertitis nodosa is key when determining treatment options and monitoring parameters.
Case Report
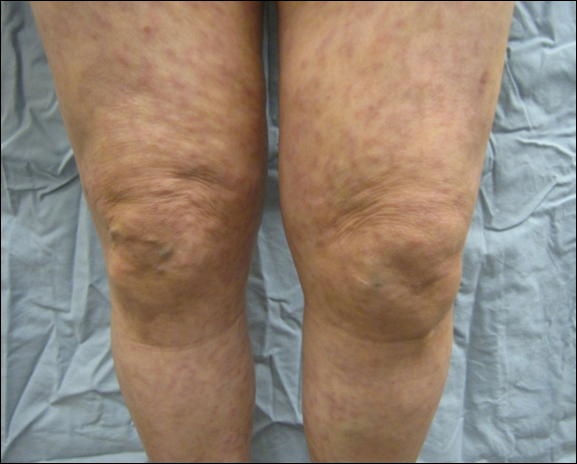
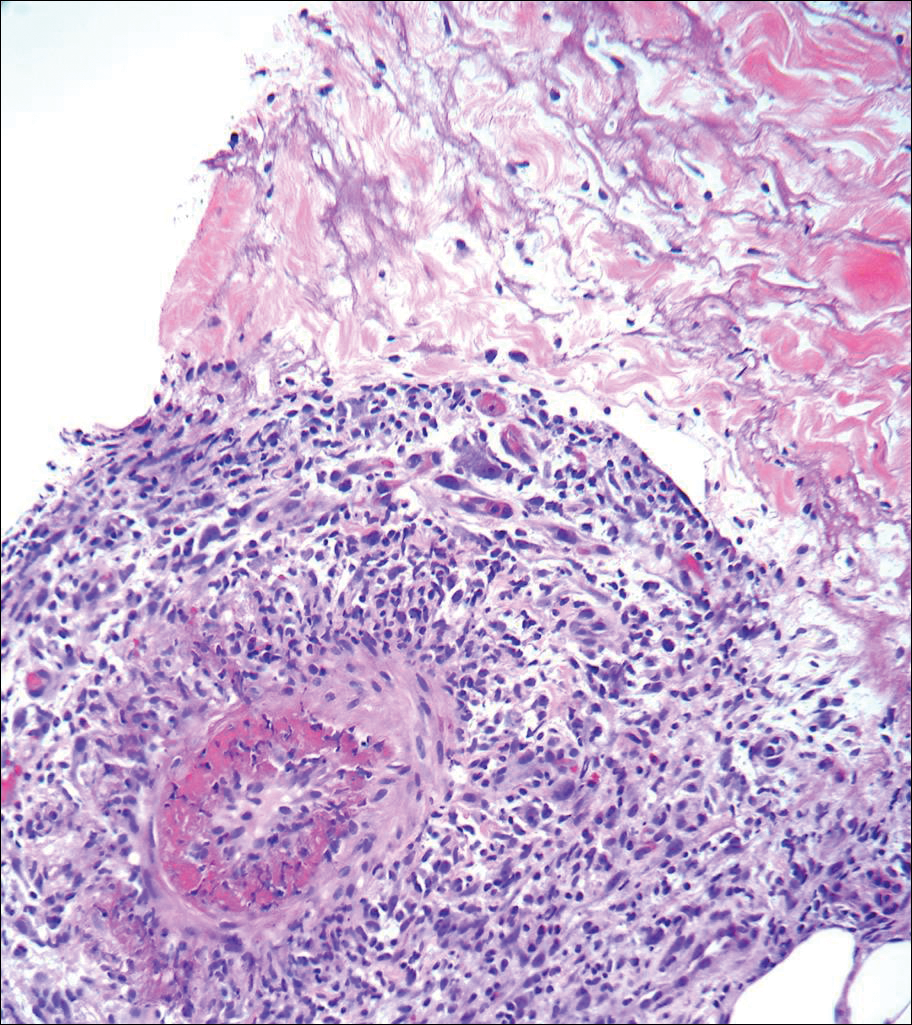
A 66-year-old woman was referred to our facility by an outside dermatologist with a mildly pruritic, blanchable, reticulated erythema on the chest and bilateral arms and legs of 3 months’ duration consistent with livedo reticularis (Figure 1). Prior systemic therapy included prednisone 10 mg 3 times daily, fexofenadine, loratadine, and hydroxyzine. When the systemic steroid was tapered, the patient developed an asymptomatic flare of her eruption. On presentation, the lesions had waxed and waned, and the patient was taking only vitamin B12 and vitamin C. Her medical history was notable for an unknown-type lymphoma of the chest wall diagnosed at 46 years of age that was treated with an unknown chemotherapeutic agent, chronic pancreatitis that resulted in a duodenectomy at 61 years of age, chronic cholecystitis, and 1 first-trimester miscarriage. Outside laboratory tests, including a comprehensive metabolic panel, complete blood cell count, urinalysis, renal function, and liver function tests were within reference range, except for the finding of mild leukocytosis (11,000/µL)(reference range, 3800–10,800/µL), which resolved after steroids were discontinued, with otherwise normal results. Punch biopsy of a specimen from the right thigh revealed medium-vessel vasculitis consistent with polyarteritis nodosa (Figure 2). Laboratory workup by our facility including hepatitis panel, perinuclear antineutrophil cytoplasmic antibody, cytoplasmic antineutrophil cytoplasmic antibody, factor V Leiden, prothrombin time/international normalized ratio, anticardiolipin antibody, and proteins C and S were all within reference range. Abnormal values included a low positive but nondiagnostic antinuclear antibody screen with negative titers, and the lupus anticoagulant titer was mildly elevated at 44 IgG binding units (reference range, <40 IgG binding units). Serum protein electrophoresis (SPEP) and urine protein electrophoresis also were performed, and SPEP was low positive for elevated κ and γ light chains. The patient was referred to oncology, and further testing revealed no underlying malignancy. The patient was monitored and no treatment was initiated; her rash completely resolved within 3 months. Laboratory monitoring at 6 months including SPEP, urine protein electrophoresis, lupus anticoagulant, and clotting studies all were within reference range.
Comment
Although the treatment of systemic polyarteritis nodosa often is necessary and typically involves high-dose corticosteroids and cyclophosphamide, the treatment of CPAN initially is less aggressive. Of the options available for treatment of CPAN, each has associated risks and side effects. Chen6 classified CPAN into 3 groups: 1 (mild), 2 (severe with no systemic involvement), and 3 (severe with progression to systemic disease)(Table). The authors performed a review of all the published treatments and their respective side effects to evaluate if treatment should be instituted for asymptomatic (group 1) disease presenting with abnormal antibody findings as demonstrated in our case.

First-line treatment of CPAN includes nonsteroidal anti-inflammatory drugs (NSAIDS) and colchicine.7 Nonsteroidal anti-inflammatory drugs are preferred; however, they also have been associated with gastrointestinal tract upset and increased risk for peptic ulcer disease with long-term use. Although colchicine often is used in conjunction with NSAIDS8 for its anti-inflammatory activity, no studies have been performed on this drug as monotherapy, and the side effect of diarrhea often limits its use.
Other therapies include dapsone, which should be monitored carefully due to the risk for dapsone hypersensitivity syndrome.8,9 Topical corticosteroids have been proven effective for mild cases of confluent erythema with remission occurring as early as 4 weeks.4 Some reports emphasize the role of streptococcal infections in CPAN, especially in children.8,10-12 Consequently it is recommended that anti–streptolysin O titers should be included in the workup for CPAN. Long-term penicillin prophylaxis and tonsillectomy have been used to prevent disease flares with limited success.8,10-12
For more severe disease, especially with neuromuscular involvement, oral methylprednisolone up to 1 mg/kg daily has been used and has proven effective in the control of acute exacerbations.7,13 However, the many adverse effects of systemic steroids limit their use long-term, and taper will often result in flare of disease.4,7 Medications used in conjunction with steroids include hydroxychloroquine, dapsone, azathioprine, cyclophosphamide, methotrexate, sulfapyridine, pentoxifylline, infliximab, etanercept, and intravenous immunoglobulin.4,9,12-17
Low-dose methotrexate has shown some improvement in skin disease with CPAN, but other case reports suggest that complete remission is not achieved with this drug.15,18 More studies are needed to assess the use of methotrexate for CPAN.
Immunomodulators have been used in multiple case reports with varying levels of success. Rogalski and Sticherling4 reported 3 cases that cleared with methylprednisolone plus azathioprine ranging from 4 weeks to 6 months; nausea limited tolerance of azathioprine in 1 case. Mycophenolate mofetil also was successfully used in 2 cases with clearance at 17 weeks and 6 months. In this series of cases, cyclosporine was ineffective for CPAN.4 Two case reports documented cutaneous clearance with cyclophosphamide in conjunction with prednisolone.9,10 No prospective trials have been performed on these medications, and immunosuppressants should only be considered in steroid-resistant cases.
The use of intravenous immunoglobulin has been reported effective in prior cases that showed resistance to more conventional trials of steroids, azathioprine, and/or cyclophosphamide.12,14 Intravenous immunoglobulin may be regarded as a treatment option for severe resistant disease. Several case reports also have documented success using tumor necrosis factor α blockers, particularly infliximab, as an adjunct to steroids and etanercept as both a steroid adjunct and monotherapy.16,17,19 More studies are necessary to evaluate these treatments.
Additionally, single case reports have outlined the use of other therapeutic agents, including tamoxifen (10 mg twice daily increased to 20 mg twice daily during episodes of breakthrough lesions),20 hyperbaric oxygen therapy (100% oxygen for 90 minutes 5 times weekly at 1.5 atm absolute followed by 2 weeks of 2 atm absolute),21 and granulocyte-macrophage colony-stimulating factor (300 µg injection in small portion to ulcer edges twice monthly for 2 months).22 All of these treatments show promise, but data are limited.
Because thrombosis is postulated to be a potential mechanism leading to CPAN, agents such as pentoxifylline, clopidogrel, and warfarin have been examined as treatment options. Pentoxifylline in combination with mycophenolate mofetil has been successful in treating a case that was resistant to other immunosuppressants.23 Clopidogrel blocks the adenosine diphosphate pathway and impairs clot retraction. Clopidogrel was reported effective in an acute flare of CPAN for clearance of skin lesions and normalization of lupus anticoagulant.24 It also was used successfully in recurrent CPAN after steroid treatments in a patient with neuromuscular symptoms. There was no recurrence in either of the patients in this case report series. Warfarin therapy at an international normalized ratio of 3.0 also has demonstrated success in halting disease progression and in facilitating the resolution of skin changes and normalization of anti–phosphatidylserine-prothrombin complex antibodies.24 Our review of the literature did not reveal evidence of a standardized length of treatment following symptom resolution or if treatment is indicated in asymptomatic disease, or as in our case, with only mild elevations of antiphospholipid antibodies.
Conclusion
Multiple treatment options exist for CPAN, but the data on their efficacies is limited and based only on anecdotal evidence, not prospective analysis. We believe that it seems reasonable to initiate treatment only for symptomatic disease or cases in which the antibody titers suggest that the patient may be at high risk for thrombosis. Mild symptoms and mild cutaneous changes would suggest the likely choice of NSAIDs, colchicine, or dapsone as treatment options versus no treatment. In patients with antibody titers, pentoxifylline, clopidogrel, or warfarin may be considered first-line therapies. With severe ulcerative lesions and neuromuscular involvement, steroids, immunosuppressants, and other investigative agents should be contemplated. In our patient, the laboratory studies were repeated and normalized on complete resolution of her livedo eruption. She remained asymptomatic and clear for 8 months without any treatment. The incidence of this presentation of CPAN is unknown and is likely underreported, as we would not expect most patients to present to their physicians for the evaluation of otherwise asymptomatic livedo reticularis. In essence, our case report suggests that it may be prudent to simply monitor patients with asymptomatic CPAN.
- Lindberg K. Ein Beitrag zur Kenntnis der Periarteritis nodosa. Acta Med Scand. 1931;76:183-225.
- Kraemer M, Linden D, Berlit P. The spectrum of differential diagnosis in neurological patients with livedo reticularis and livedo racemosa [published online August 26, 2005]. J Neurol. 2005;252:1155-1166.
- Nakamura T, Kanazawa N, Ikeda T, et al. Cutaneous polyarteritis nodosa: revisiting its definition and diagnostic criteria. Arch Dermatol Res. 2009;301:117-121.
- Rogalski C, Sticherling M. Panateritis cutanea benigna—an entity limited to the skin or cutaneous presentation of a systemic necrotizing vasculitis? report of seven cases and review of the literature. Int J Dermatol. 2007;46:817-821.
- Kawakami T, Yamazaki M, Mizoguchi M, et al. High titer of anti-phosphatidylserine-prothrombin complex antibodies in patients with cutaneous polyarteritis nodosa. Arthritis Rheum. 2007;57:1507-1513.
- Chen KR. Cutaneous polyarteritis nodosa: a clinical and histopathological study of 20 cases. J Dermatol. 1989;6:429-442.
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756.
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathologic analysis and review of the published work. J Dermatol. 2010;37:85-93.
- Flanagan N, Casey EB, Watson R, et al. Cutaneous polyartertitis nodosa with seronegative arthritis. Rheumatology (Oxford). 1999;38:1161-1162.
- Fathalla B, Miller L, Brady S, et al. Cutaneous polyarteritis nodosa in children. J Am Acad Dermatol. 2005;53:724-728.
- Misago N, Mochizuki Y, Sekiyama-Kodera H, et al. Cutaneous polyarteritis nodosa: therapy and clinical course in four cases. J Dermatol. 2001;28:719-727.
- Breda L, Franchini S, Marzetti V, et al. Intravenous immunoglobulins for cutaneous polyarteritis nodosa resistant to conventional treatment. Scand J Rheumatol. 2016;45:169-170.
- Maillard H, Szczesniak S, Martin L. Cutaneous periarteritis nodosa: diagnostic and therapeutic aspects of 9 cases. Ann Dermatol Venereol. 1999;26:125-129.
- Lobo I, Ferreira M, Silva E. Cutaneous polyarteritis nodosa treated with intravenous immunoglobulin. J Eur Acad Dermatol Venereol. 2007;22:880-882.
- Boehm I, Bauer R. Low-dose methotrexate controls a severe form of polyarteritis nodosa. Arch Dermatol. 2000;136:167-169.
- Campanilho-Marques R, Ramos F, Canhão H, et al. Remission induced by infliximab in a childhood polyarteritis nodosa refractory to conventional immunosuppression and rituximab. Joint Bone Spine. 2014;81:277-278.
- Inoue N, Shimizu M, Mizuta M, et al. Refractory cutaneous polyarteritis nodosa: successful treatment with etanercept. Pediatr Int. 2017;59:751-752.
- Schartz NE. Successful treatment in two cases of steroid dependent cutaneous polyarteritis nodosa with low-dose methotrexate. Dermatology. 2001;203:336-338.
- Valor L, Monteagudo I, de la Torre I, et al. Young male patient diagnosed with cutaneous polyarteritis nodosa successfully treated with etanercept. Mod Rheumatol. 2014;24:688-689.
- Cvancara JL, Meffert JJ, Elston DM. Estrogen sensitive cutaneous polyarteritis nodosa: response to tamoxifen. J Am Acad Dermatol. 1998;39:643-646.
- Mazokopakis E, Milkas A, Tsartsalis A, et al. Improvement of cutaneous polyarteritis nodosa with hyperbaric oxygen. Int J Dermatol. 2009;48:1017-1029.
- Tursen U, Api H, Kaya TI, et al. Rapid healing of chronic leg ulcers during perilesional injections of granulocyte-macrophage colony stimulating factor in a patient with cutaneous polyarteritis nodosa. J Eur Acad Dermatol Venereol. 2006;20:1341-1343.
- Kluger N, Guillot B, Bessis D. Ulcerative cutaneous polyarteritis nodosa treated with mycophenolate mofetil and pentoxifylline. J Dermatolog Treat. 2011;22:175-177.
- Kawakami T, Soma Y. Use of warfarin therapy at a target international normalized ratio of 3.0 for cutaneous polyarteritis nodosa. J Am Acad Dermatol. 2010;63:602-606.
In 1931, Lindberg1 described a cutaneous variant of polyarteritis nodosa, which lacked visceral involvement and possessed a more favorable prognosis.2 Cutaneous polyarteritis nodosa (CPAN) is a localized small- to medium-vessel vasculitis restricted to the skin. Both benign and chronic courses have been described, and systemic involvement does not occur.3 Diagnostic criteria proposed by Nakamura et al3 in 2009 included cutaneous nodules, livedo reticularis, purpura, or ulcers; histopathologic fibrinoid necrotizing vasculitis of small- to medium-sized vessels; and exclusion of systemic symptoms (eg, fever, hypertension, weight loss, renal failure, cerebral hemorrhage, neuropathy, myocardial infarction, ischemic heart disease, pericarditis, pleuritis, arthralgia/myalgia). Nodules occur in 30% to 50% of cases and can remain for years if left untreated. Ulcerations occur in up to 30% of patients. Myositis, arthritis, and weakness also have been reported with this condition.4 Cutaneous polyarteritis nodosa has been associated with abnormal antibody testing with elevations of antiphospholipid cofactor antibody, lupus anticoagulant, anticardiolipin antibody, and anti-β2-glycoprotein I–dependent cardiolipin antibody, as well as elevated anti–phosphatidylserine-prothrombin complex antibody.5 These antibodies suggest increased risk for thrombosis and systemic diseases such as lupus or other autoimmune connective tissue disease. The distinction of this entity from systemic polyartertitis nodosa is key when determining treatment options and monitoring parameters.
Case Report
A 66-year-old woman was referred to our facility by an outside dermatologist with a mildly pruritic, blanchable, reticulated erythema on the chest and bilateral arms and legs of 3 months’ duration consistent with livedo reticularis (Figure 1). Prior systemic therapy included prednisone 10 mg 3 times daily, fexofenadine, loratadine, and hydroxyzine. When the systemic steroid was tapered, the patient developed an asymptomatic flare of her eruption. On presentation, the lesions had waxed and waned, and the patient was taking only vitamin B12 and vitamin C. Her medical history was notable for an unknown-type lymphoma of the chest wall diagnosed at 46 years of age that was treated with an unknown chemotherapeutic agent, chronic pancreatitis that resulted in a duodenectomy at 61 years of age, chronic cholecystitis, and 1 first-trimester miscarriage. Outside laboratory tests, including a comprehensive metabolic panel, complete blood cell count, urinalysis, renal function, and liver function tests were within reference range, except for the finding of mild leukocytosis (11,000/µL)(reference range, 3800–10,800/µL), which resolved after steroids were discontinued, with otherwise normal results. Punch biopsy of a specimen from the right thigh revealed medium-vessel vasculitis consistent with polyarteritis nodosa (Figure 2). Laboratory workup by our facility including hepatitis panel, perinuclear antineutrophil cytoplasmic antibody, cytoplasmic antineutrophil cytoplasmic antibody, factor V Leiden, prothrombin time/international normalized ratio, anticardiolipin antibody, and proteins C and S were all within reference range. Abnormal values included a low positive but nondiagnostic antinuclear antibody screen with negative titers, and the lupus anticoagulant titer was mildly elevated at 44 IgG binding units (reference range, <40 IgG binding units). Serum protein electrophoresis (SPEP) and urine protein electrophoresis also were performed, and SPEP was low positive for elevated κ and γ light chains. The patient was referred to oncology, and further testing revealed no underlying malignancy. The patient was monitored and no treatment was initiated; her rash completely resolved within 3 months. Laboratory monitoring at 6 months including SPEP, urine protein electrophoresis, lupus anticoagulant, and clotting studies all were within reference range.
Comment
Although the treatment of systemic polyarteritis nodosa often is necessary and typically involves high-dose corticosteroids and cyclophosphamide, the treatment of CPAN initially is less aggressive. Of the options available for treatment of CPAN, each has associated risks and side effects. Chen6 classified CPAN into 3 groups: 1 (mild), 2 (severe with no systemic involvement), and 3 (severe with progression to systemic disease)(Table). The authors performed a review of all the published treatments and their respective side effects to evaluate if treatment should be instituted for asymptomatic (group 1) disease presenting with abnormal antibody findings as demonstrated in our case.
First-line treatment of CPAN includes nonsteroidal anti-inflammatory drugs (NSAIDS) and colchicine.7 Nonsteroidal anti-inflammatory drugs are preferred; however, they also have been associated with gastrointestinal tract upset and increased risk for peptic ulcer disease with long-term use. Although colchicine often is used in conjunction with NSAIDS8 for its anti-inflammatory activity, no studies have been performed on this drug as monotherapy, and the side effect of diarrhea often limits its use.
Other therapies include dapsone, which should be monitored carefully due to the risk for dapsone hypersensitivity syndrome.8,9 Topical corticosteroids have been proven effective for mild cases of confluent erythema with remission occurring as early as 4 weeks.4 Some reports emphasize the role of streptococcal infections in CPAN, especially in children.8,10-12 Consequently it is recommended that anti–streptolysin O titers should be included in the workup for CPAN. Long-term penicillin prophylaxis and tonsillectomy have been used to prevent disease flares with limited success.8,10-12
For more severe disease, especially with neuromuscular involvement, oral methylprednisolone up to 1 mg/kg daily has been used and has proven effective in the control of acute exacerbations.7,13 However, the many adverse effects of systemic steroids limit their use long-term, and taper will often result in flare of disease.4,7 Medications used in conjunction with steroids include hydroxychloroquine, dapsone, azathioprine, cyclophosphamide, methotrexate, sulfapyridine, pentoxifylline, infliximab, etanercept, and intravenous immunoglobulin.4,9,12-17
Low-dose methotrexate has shown some improvement in skin disease with CPAN, but other case reports suggest that complete remission is not achieved with this drug.15,18 More studies are needed to assess the use of methotrexate for CPAN.
Immunomodulators have been used in multiple case reports with varying levels of success. Rogalski and Sticherling4 reported 3 cases that cleared with methylprednisolone plus azathioprine ranging from 4 weeks to 6 months; nausea limited tolerance of azathioprine in 1 case. Mycophenolate mofetil also was successfully used in 2 cases with clearance at 17 weeks and 6 months. In this series of cases, cyclosporine was ineffective for CPAN.4 Two case reports documented cutaneous clearance with cyclophosphamide in conjunction with prednisolone.9,10 No prospective trials have been performed on these medications, and immunosuppressants should only be considered in steroid-resistant cases.
The use of intravenous immunoglobulin has been reported effective in prior cases that showed resistance to more conventional trials of steroids, azathioprine, and/or cyclophosphamide.12,14 Intravenous immunoglobulin may be regarded as a treatment option for severe resistant disease. Several case reports also have documented success using tumor necrosis factor α blockers, particularly infliximab, as an adjunct to steroids and etanercept as both a steroid adjunct and monotherapy.16,17,19 More studies are necessary to evaluate these treatments.
Additionally, single case reports have outlined the use of other therapeutic agents, including tamoxifen (10 mg twice daily increased to 20 mg twice daily during episodes of breakthrough lesions),20 hyperbaric oxygen therapy (100% oxygen for 90 minutes 5 times weekly at 1.5 atm absolute followed by 2 weeks of 2 atm absolute),21 and granulocyte-macrophage colony-stimulating factor (300 µg injection in small portion to ulcer edges twice monthly for 2 months).22 All of these treatments show promise, but data are limited.
Because thrombosis is postulated to be a potential mechanism leading to CPAN, agents such as pentoxifylline, clopidogrel, and warfarin have been examined as treatment options. Pentoxifylline in combination with mycophenolate mofetil has been successful in treating a case that was resistant to other immunosuppressants.23 Clopidogrel blocks the adenosine diphosphate pathway and impairs clot retraction. Clopidogrel was reported effective in an acute flare of CPAN for clearance of skin lesions and normalization of lupus anticoagulant.24 It also was used successfully in recurrent CPAN after steroid treatments in a patient with neuromuscular symptoms. There was no recurrence in either of the patients in this case report series. Warfarin therapy at an international normalized ratio of 3.0 also has demonstrated success in halting disease progression and in facilitating the resolution of skin changes and normalization of anti–phosphatidylserine-prothrombin complex antibodies.24 Our review of the literature did not reveal evidence of a standardized length of treatment following symptom resolution or if treatment is indicated in asymptomatic disease, or as in our case, with only mild elevations of antiphospholipid antibodies.
Conclusion
Multiple treatment options exist for CPAN, but the data on their efficacies is limited and based only on anecdotal evidence, not prospective analysis. We believe that it seems reasonable to initiate treatment only for symptomatic disease or cases in which the antibody titers suggest that the patient may be at high risk for thrombosis. Mild symptoms and mild cutaneous changes would suggest the likely choice of NSAIDs, colchicine, or dapsone as treatment options versus no treatment. In patients with antibody titers, pentoxifylline, clopidogrel, or warfarin may be considered first-line therapies. With severe ulcerative lesions and neuromuscular involvement, steroids, immunosuppressants, and other investigative agents should be contemplated. In our patient, the laboratory studies were repeated and normalized on complete resolution of her livedo eruption. She remained asymptomatic and clear for 8 months without any treatment. The incidence of this presentation of CPAN is unknown and is likely underreported, as we would not expect most patients to present to their physicians for the evaluation of otherwise asymptomatic livedo reticularis. In essence, our case report suggests that it may be prudent to simply monitor patients with asymptomatic CPAN.
In 1931, Lindberg1 described a cutaneous variant of polyarteritis nodosa, which lacked visceral involvement and possessed a more favorable prognosis.2 Cutaneous polyarteritis nodosa (CPAN) is a localized small- to medium-vessel vasculitis restricted to the skin. Both benign and chronic courses have been described, and systemic involvement does not occur.3 Diagnostic criteria proposed by Nakamura et al3 in 2009 included cutaneous nodules, livedo reticularis, purpura, or ulcers; histopathologic fibrinoid necrotizing vasculitis of small- to medium-sized vessels; and exclusion of systemic symptoms (eg, fever, hypertension, weight loss, renal failure, cerebral hemorrhage, neuropathy, myocardial infarction, ischemic heart disease, pericarditis, pleuritis, arthralgia/myalgia). Nodules occur in 30% to 50% of cases and can remain for years if left untreated. Ulcerations occur in up to 30% of patients. Myositis, arthritis, and weakness also have been reported with this condition.4 Cutaneous polyarteritis nodosa has been associated with abnormal antibody testing with elevations of antiphospholipid cofactor antibody, lupus anticoagulant, anticardiolipin antibody, and anti-β2-glycoprotein I–dependent cardiolipin antibody, as well as elevated anti–phosphatidylserine-prothrombin complex antibody.5 These antibodies suggest increased risk for thrombosis and systemic diseases such as lupus or other autoimmune connective tissue disease. The distinction of this entity from systemic polyartertitis nodosa is key when determining treatment options and monitoring parameters.
Case Report
A 66-year-old woman was referred to our facility by an outside dermatologist with a mildly pruritic, blanchable, reticulated erythema on the chest and bilateral arms and legs of 3 months’ duration consistent with livedo reticularis (Figure 1). Prior systemic therapy included prednisone 10 mg 3 times daily, fexofenadine, loratadine, and hydroxyzine. When the systemic steroid was tapered, the patient developed an asymptomatic flare of her eruption. On presentation, the lesions had waxed and waned, and the patient was taking only vitamin B12 and vitamin C. Her medical history was notable for an unknown-type lymphoma of the chest wall diagnosed at 46 years of age that was treated with an unknown chemotherapeutic agent, chronic pancreatitis that resulted in a duodenectomy at 61 years of age, chronic cholecystitis, and 1 first-trimester miscarriage. Outside laboratory tests, including a comprehensive metabolic panel, complete blood cell count, urinalysis, renal function, and liver function tests were within reference range, except for the finding of mild leukocytosis (11,000/µL)(reference range, 3800–10,800/µL), which resolved after steroids were discontinued, with otherwise normal results. Punch biopsy of a specimen from the right thigh revealed medium-vessel vasculitis consistent with polyarteritis nodosa (Figure 2). Laboratory workup by our facility including hepatitis panel, perinuclear antineutrophil cytoplasmic antibody, cytoplasmic antineutrophil cytoplasmic antibody, factor V Leiden, prothrombin time/international normalized ratio, anticardiolipin antibody, and proteins C and S were all within reference range. Abnormal values included a low positive but nondiagnostic antinuclear antibody screen with negative titers, and the lupus anticoagulant titer was mildly elevated at 44 IgG binding units (reference range, <40 IgG binding units). Serum protein electrophoresis (SPEP) and urine protein electrophoresis also were performed, and SPEP was low positive for elevated κ and γ light chains. The patient was referred to oncology, and further testing revealed no underlying malignancy. The patient was monitored and no treatment was initiated; her rash completely resolved within 3 months. Laboratory monitoring at 6 months including SPEP, urine protein electrophoresis, lupus anticoagulant, and clotting studies all were within reference range.
Comment
Although the treatment of systemic polyarteritis nodosa often is necessary and typically involves high-dose corticosteroids and cyclophosphamide, the treatment of CPAN initially is less aggressive. Of the options available for treatment of CPAN, each has associated risks and side effects. Chen6 classified CPAN into 3 groups: 1 (mild), 2 (severe with no systemic involvement), and 3 (severe with progression to systemic disease)(Table). The authors performed a review of all the published treatments and their respective side effects to evaluate if treatment should be instituted for asymptomatic (group 1) disease presenting with abnormal antibody findings as demonstrated in our case.
First-line treatment of CPAN includes nonsteroidal anti-inflammatory drugs (NSAIDS) and colchicine.7 Nonsteroidal anti-inflammatory drugs are preferred; however, they also have been associated with gastrointestinal tract upset and increased risk for peptic ulcer disease with long-term use. Although colchicine often is used in conjunction with NSAIDS8 for its anti-inflammatory activity, no studies have been performed on this drug as monotherapy, and the side effect of diarrhea often limits its use.
Other therapies include dapsone, which should be monitored carefully due to the risk for dapsone hypersensitivity syndrome.8,9 Topical corticosteroids have been proven effective for mild cases of confluent erythema with remission occurring as early as 4 weeks.4 Some reports emphasize the role of streptococcal infections in CPAN, especially in children.8,10-12 Consequently it is recommended that anti–streptolysin O titers should be included in the workup for CPAN. Long-term penicillin prophylaxis and tonsillectomy have been used to prevent disease flares with limited success.8,10-12
For more severe disease, especially with neuromuscular involvement, oral methylprednisolone up to 1 mg/kg daily has been used and has proven effective in the control of acute exacerbations.7,13 However, the many adverse effects of systemic steroids limit their use long-term, and taper will often result in flare of disease.4,7 Medications used in conjunction with steroids include hydroxychloroquine, dapsone, azathioprine, cyclophosphamide, methotrexate, sulfapyridine, pentoxifylline, infliximab, etanercept, and intravenous immunoglobulin.4,9,12-17
Low-dose methotrexate has shown some improvement in skin disease with CPAN, but other case reports suggest that complete remission is not achieved with this drug.15,18 More studies are needed to assess the use of methotrexate for CPAN.
Immunomodulators have been used in multiple case reports with varying levels of success. Rogalski and Sticherling4 reported 3 cases that cleared with methylprednisolone plus azathioprine ranging from 4 weeks to 6 months; nausea limited tolerance of azathioprine in 1 case. Mycophenolate mofetil also was successfully used in 2 cases with clearance at 17 weeks and 6 months. In this series of cases, cyclosporine was ineffective for CPAN.4 Two case reports documented cutaneous clearance with cyclophosphamide in conjunction with prednisolone.9,10 No prospective trials have been performed on these medications, and immunosuppressants should only be considered in steroid-resistant cases.
The use of intravenous immunoglobulin has been reported effective in prior cases that showed resistance to more conventional trials of steroids, azathioprine, and/or cyclophosphamide.12,14 Intravenous immunoglobulin may be regarded as a treatment option for severe resistant disease. Several case reports also have documented success using tumor necrosis factor α blockers, particularly infliximab, as an adjunct to steroids and etanercept as both a steroid adjunct and monotherapy.16,17,19 More studies are necessary to evaluate these treatments.
Additionally, single case reports have outlined the use of other therapeutic agents, including tamoxifen (10 mg twice daily increased to 20 mg twice daily during episodes of breakthrough lesions),20 hyperbaric oxygen therapy (100% oxygen for 90 minutes 5 times weekly at 1.5 atm absolute followed by 2 weeks of 2 atm absolute),21 and granulocyte-macrophage colony-stimulating factor (300 µg injection in small portion to ulcer edges twice monthly for 2 months).22 All of these treatments show promise, but data are limited.
Because thrombosis is postulated to be a potential mechanism leading to CPAN, agents such as pentoxifylline, clopidogrel, and warfarin have been examined as treatment options. Pentoxifylline in combination with mycophenolate mofetil has been successful in treating a case that was resistant to other immunosuppressants.23 Clopidogrel blocks the adenosine diphosphate pathway and impairs clot retraction. Clopidogrel was reported effective in an acute flare of CPAN for clearance of skin lesions and normalization of lupus anticoagulant.24 It also was used successfully in recurrent CPAN after steroid treatments in a patient with neuromuscular symptoms. There was no recurrence in either of the patients in this case report series. Warfarin therapy at an international normalized ratio of 3.0 also has demonstrated success in halting disease progression and in facilitating the resolution of skin changes and normalization of anti–phosphatidylserine-prothrombin complex antibodies.24 Our review of the literature did not reveal evidence of a standardized length of treatment following symptom resolution or if treatment is indicated in asymptomatic disease, or as in our case, with only mild elevations of antiphospholipid antibodies.
Conclusion
Multiple treatment options exist for CPAN, but the data on their efficacies is limited and based only on anecdotal evidence, not prospective analysis. We believe that it seems reasonable to initiate treatment only for symptomatic disease or cases in which the antibody titers suggest that the patient may be at high risk for thrombosis. Mild symptoms and mild cutaneous changes would suggest the likely choice of NSAIDs, colchicine, or dapsone as treatment options versus no treatment. In patients with antibody titers, pentoxifylline, clopidogrel, or warfarin may be considered first-line therapies. With severe ulcerative lesions and neuromuscular involvement, steroids, immunosuppressants, and other investigative agents should be contemplated. In our patient, the laboratory studies were repeated and normalized on complete resolution of her livedo eruption. She remained asymptomatic and clear for 8 months without any treatment. The incidence of this presentation of CPAN is unknown and is likely underreported, as we would not expect most patients to present to their physicians for the evaluation of otherwise asymptomatic livedo reticularis. In essence, our case report suggests that it may be prudent to simply monitor patients with asymptomatic CPAN.
- Lindberg K. Ein Beitrag zur Kenntnis der Periarteritis nodosa. Acta Med Scand. 1931;76:183-225.
- Kraemer M, Linden D, Berlit P. The spectrum of differential diagnosis in neurological patients with livedo reticularis and livedo racemosa [published online August 26, 2005]. J Neurol. 2005;252:1155-1166.
- Nakamura T, Kanazawa N, Ikeda T, et al. Cutaneous polyarteritis nodosa: revisiting its definition and diagnostic criteria. Arch Dermatol Res. 2009;301:117-121.
- Rogalski C, Sticherling M. Panateritis cutanea benigna—an entity limited to the skin or cutaneous presentation of a systemic necrotizing vasculitis? report of seven cases and review of the literature. Int J Dermatol. 2007;46:817-821.
- Kawakami T, Yamazaki M, Mizoguchi M, et al. High titer of anti-phosphatidylserine-prothrombin complex antibodies in patients with cutaneous polyarteritis nodosa. Arthritis Rheum. 2007;57:1507-1513.
- Chen KR. Cutaneous polyarteritis nodosa: a clinical and histopathological study of 20 cases. J Dermatol. 1989;6:429-442.
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756.
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathologic analysis and review of the published work. J Dermatol. 2010;37:85-93.
- Flanagan N, Casey EB, Watson R, et al. Cutaneous polyartertitis nodosa with seronegative arthritis. Rheumatology (Oxford). 1999;38:1161-1162.
- Fathalla B, Miller L, Brady S, et al. Cutaneous polyarteritis nodosa in children. J Am Acad Dermatol. 2005;53:724-728.
- Misago N, Mochizuki Y, Sekiyama-Kodera H, et al. Cutaneous polyarteritis nodosa: therapy and clinical course in four cases. J Dermatol. 2001;28:719-727.
- Breda L, Franchini S, Marzetti V, et al. Intravenous immunoglobulins for cutaneous polyarteritis nodosa resistant to conventional treatment. Scand J Rheumatol. 2016;45:169-170.
- Maillard H, Szczesniak S, Martin L. Cutaneous periarteritis nodosa: diagnostic and therapeutic aspects of 9 cases. Ann Dermatol Venereol. 1999;26:125-129.
- Lobo I, Ferreira M, Silva E. Cutaneous polyarteritis nodosa treated with intravenous immunoglobulin. J Eur Acad Dermatol Venereol. 2007;22:880-882.
- Boehm I, Bauer R. Low-dose methotrexate controls a severe form of polyarteritis nodosa. Arch Dermatol. 2000;136:167-169.
- Campanilho-Marques R, Ramos F, Canhão H, et al. Remission induced by infliximab in a childhood polyarteritis nodosa refractory to conventional immunosuppression and rituximab. Joint Bone Spine. 2014;81:277-278.
- Inoue N, Shimizu M, Mizuta M, et al. Refractory cutaneous polyarteritis nodosa: successful treatment with etanercept. Pediatr Int. 2017;59:751-752.
- Schartz NE. Successful treatment in two cases of steroid dependent cutaneous polyarteritis nodosa with low-dose methotrexate. Dermatology. 2001;203:336-338.
- Valor L, Monteagudo I, de la Torre I, et al. Young male patient diagnosed with cutaneous polyarteritis nodosa successfully treated with etanercept. Mod Rheumatol. 2014;24:688-689.
- Cvancara JL, Meffert JJ, Elston DM. Estrogen sensitive cutaneous polyarteritis nodosa: response to tamoxifen. J Am Acad Dermatol. 1998;39:643-646.
- Mazokopakis E, Milkas A, Tsartsalis A, et al. Improvement of cutaneous polyarteritis nodosa with hyperbaric oxygen. Int J Dermatol. 2009;48:1017-1029.
- Tursen U, Api H, Kaya TI, et al. Rapid healing of chronic leg ulcers during perilesional injections of granulocyte-macrophage colony stimulating factor in a patient with cutaneous polyarteritis nodosa. J Eur Acad Dermatol Venereol. 2006;20:1341-1343.
- Kluger N, Guillot B, Bessis D. Ulcerative cutaneous polyarteritis nodosa treated with mycophenolate mofetil and pentoxifylline. J Dermatolog Treat. 2011;22:175-177.
- Kawakami T, Soma Y. Use of warfarin therapy at a target international normalized ratio of 3.0 for cutaneous polyarteritis nodosa. J Am Acad Dermatol. 2010;63:602-606.
- Lindberg K. Ein Beitrag zur Kenntnis der Periarteritis nodosa. Acta Med Scand. 1931;76:183-225.
- Kraemer M, Linden D, Berlit P. The spectrum of differential diagnosis in neurological patients with livedo reticularis and livedo racemosa [published online August 26, 2005]. J Neurol. 2005;252:1155-1166.
- Nakamura T, Kanazawa N, Ikeda T, et al. Cutaneous polyarteritis nodosa: revisiting its definition and diagnostic criteria. Arch Dermatol Res. 2009;301:117-121.
- Rogalski C, Sticherling M. Panateritis cutanea benigna—an entity limited to the skin or cutaneous presentation of a systemic necrotizing vasculitis? report of seven cases and review of the literature. Int J Dermatol. 2007;46:817-821.
- Kawakami T, Yamazaki M, Mizoguchi M, et al. High titer of anti-phosphatidylserine-prothrombin complex antibodies in patients with cutaneous polyarteritis nodosa. Arthritis Rheum. 2007;57:1507-1513.
- Chen KR. Cutaneous polyarteritis nodosa: a clinical and histopathological study of 20 cases. J Dermatol. 1989;6:429-442.
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756.
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathologic analysis and review of the published work. J Dermatol. 2010;37:85-93.
- Flanagan N, Casey EB, Watson R, et al. Cutaneous polyartertitis nodosa with seronegative arthritis. Rheumatology (Oxford). 1999;38:1161-1162.
- Fathalla B, Miller L, Brady S, et al. Cutaneous polyarteritis nodosa in children. J Am Acad Dermatol. 2005;53:724-728.
- Misago N, Mochizuki Y, Sekiyama-Kodera H, et al. Cutaneous polyarteritis nodosa: therapy and clinical course in four cases. J Dermatol. 2001;28:719-727.
- Breda L, Franchini S, Marzetti V, et al. Intravenous immunoglobulins for cutaneous polyarteritis nodosa resistant to conventional treatment. Scand J Rheumatol. 2016;45:169-170.
- Maillard H, Szczesniak S, Martin L. Cutaneous periarteritis nodosa: diagnostic and therapeutic aspects of 9 cases. Ann Dermatol Venereol. 1999;26:125-129.
- Lobo I, Ferreira M, Silva E. Cutaneous polyarteritis nodosa treated with intravenous immunoglobulin. J Eur Acad Dermatol Venereol. 2007;22:880-882.
- Boehm I, Bauer R. Low-dose methotrexate controls a severe form of polyarteritis nodosa. Arch Dermatol. 2000;136:167-169.
- Campanilho-Marques R, Ramos F, Canhão H, et al. Remission induced by infliximab in a childhood polyarteritis nodosa refractory to conventional immunosuppression and rituximab. Joint Bone Spine. 2014;81:277-278.
- Inoue N, Shimizu M, Mizuta M, et al. Refractory cutaneous polyarteritis nodosa: successful treatment with etanercept. Pediatr Int. 2017;59:751-752.
- Schartz NE. Successful treatment in two cases of steroid dependent cutaneous polyarteritis nodosa with low-dose methotrexate. Dermatology. 2001;203:336-338.
- Valor L, Monteagudo I, de la Torre I, et al. Young male patient diagnosed with cutaneous polyarteritis nodosa successfully treated with etanercept. Mod Rheumatol. 2014;24:688-689.
- Cvancara JL, Meffert JJ, Elston DM. Estrogen sensitive cutaneous polyarteritis nodosa: response to tamoxifen. J Am Acad Dermatol. 1998;39:643-646.
- Mazokopakis E, Milkas A, Tsartsalis A, et al. Improvement of cutaneous polyarteritis nodosa with hyperbaric oxygen. Int J Dermatol. 2009;48:1017-1029.
- Tursen U, Api H, Kaya TI, et al. Rapid healing of chronic leg ulcers during perilesional injections of granulocyte-macrophage colony stimulating factor in a patient with cutaneous polyarteritis nodosa. J Eur Acad Dermatol Venereol. 2006;20:1341-1343.
- Kluger N, Guillot B, Bessis D. Ulcerative cutaneous polyarteritis nodosa treated with mycophenolate mofetil and pentoxifylline. J Dermatolog Treat. 2011;22:175-177.
- Kawakami T, Soma Y. Use of warfarin therapy at a target international normalized ratio of 3.0 for cutaneous polyarteritis nodosa. J Am Acad Dermatol. 2010;63:602-606.
Practice Points
- Cutaneous polyarteritis nodosa should be in the differential of new-onset livedo reticularis.
- Workup with biopsy and specific blood work is important.
- Treatment options at this time are limited.
Necrotic Ulcer on the Thigh
The Diagnosis: Disseminated Cryptococcosis
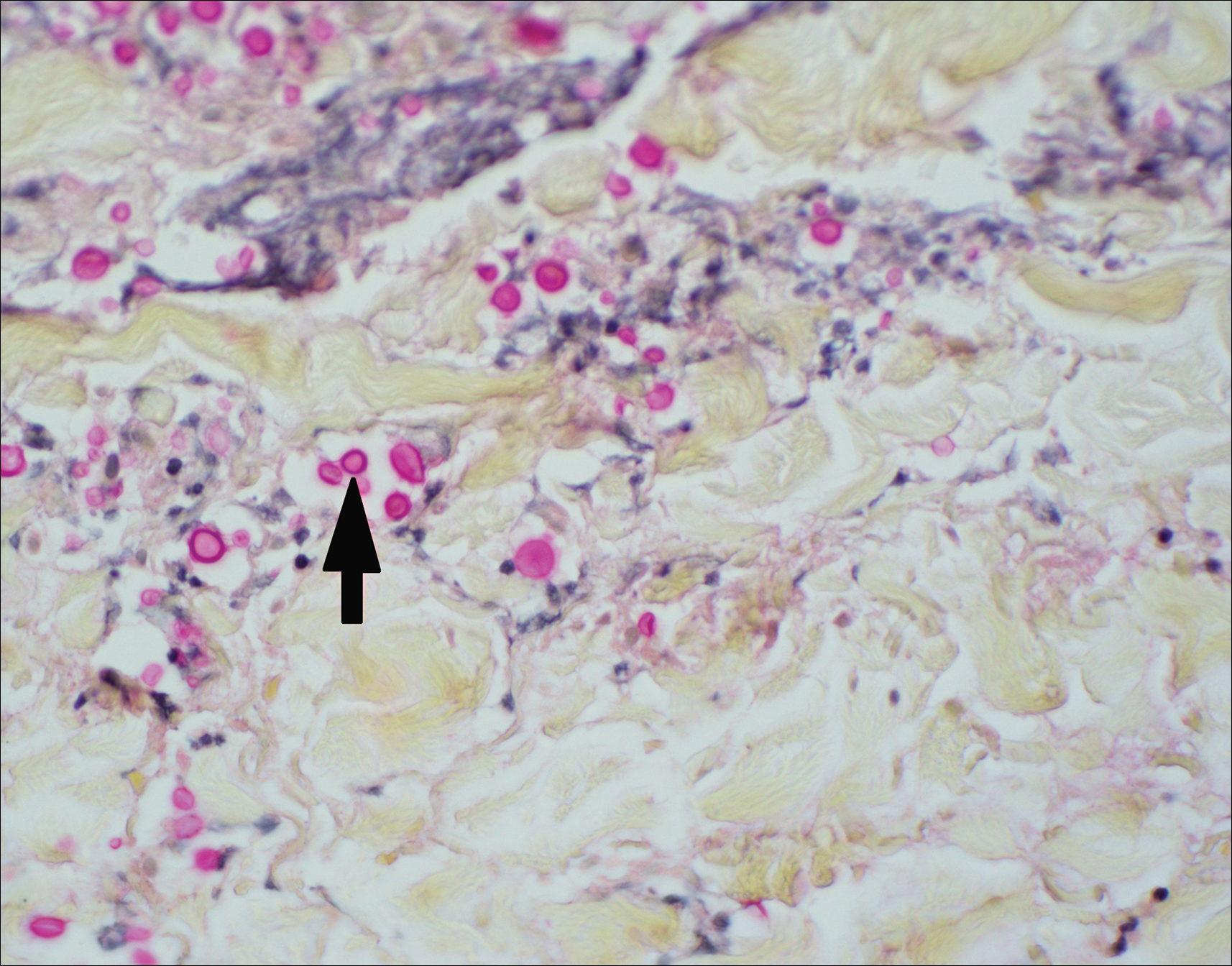
Histopathologic examination of a 3-mm punch biopsy showed a diffuse dermal neutrophilic infiltrate with necrosis and subcutaneous tissue with round yeast surrounded by a prominent halo staining bright red with mucicarmine, representing a thick mucinous capsule (Figure). Grocott-Gomori methenamine-silver and periodic acid-Schiff stains also demonstrated fungal spores morphologically. Cerebrospinal fluid culture grew Cryptococcus neoformans, and cryptococcal antigen titers were positive in both serum and cerebrospinal fluid samples (>1:4096). The patient had autolytic debridement of the ulcer after completing a 4-week induction course of intravenous liposomal amphotericin B with oral flucytosine. He was transitioned to oral fluconazole for the consolidation phase of treatment.
Cryptococcus is an opportunistic basidiomycetous yeast with worldwide distribution and 2 primary pathogenic species in humans: C neoformans and Cryptococcus gattii. It is associated with bird feces, composted food, and decayed wood.1,2 A predilection toward an immunosuppressed host is recognized in 70% to 90% of the infections caused by C neoformans; however, C gattii commonly affects individuals with apparently intact immune systems.1,3 Risk factors for infection include advanced human immunodeficiency virus infection, solid organ transplantation, chronic liver disease, autoimmune disease, hematological malignancy, and underlying genetic susceptibility.1,2
Initial exposure is through the respiratory tract with formation of latent reservoirs in the pulmonary lymph nodes with subsequent reactivation that can result in hematogenous dissemination.1,2 Cutaneous involvement was described in 108 patients (5%) in a large review of 1974 cases in France.4 Among those with cutaneous involvement, disseminated disease was diagnosed in 80 cases (74%), and 28 cases (26%) were considered primary cutaneous cryptococcosis. Primary cutaneous cryptococcosis typically presents as a single lesion, predominantly on the hand, with whitlow and more rarely with extensive cellulitis or necrotizing fasciitis.4 In disseminated cutaneous disease, there is no pathognomonic single lesion; however, it is commonly associated with multiple cutaneous lesions predominantly involving the head and neck. Plaques, abscesses, nodules, and pustular or umbilicated papules have been reported.1,5 There are few case reports that describe a single isolated necrotic ulcer with disseminated disease similar to our presented case, and more typically the necrotic ulcer is seen in transplanted patients.6 The differential diagnosis of a necrotic thigh ulcer includes pseudomonal ecthyma gangrenosum, cutaneous anthrax and aspergillosis, fusariosis, and a bite from the brown recluse spider.7 Our patient had an increased susceptibility to infection from his ongoing chemotherapy, a risk previously described in oncology patients with cell-mediated immunosuppression.8
Management for disseminated cryptococcosis is a 3-phase therapy including induction with intravenous amphotericin B and oral flucytosine for a minimum of 2 weeks, with consolidation and maintenance phases both with oral fluconazole for a length depending on underlying immunosuppression.9
- Chen SC, Meyer W, Sorrell TC. Cryptococcus gattii infections. Clin Microbiol Rev. 2014;27:980-1024.
- Williamson PR, Jarvis JN, Panackal AA, et al. Cryptococcal meningitis: epidemiology, immunology, diagnosis, and therapy [published online November 25, 2016]. Nat Rev Neurol. 2017;13:13-24.
- Speed B, Dunt D. Clinical and host differences between infections with the two varieties of Cryptococcus neoformans. Clin Infect Dis. 1995;21:28-34.
- Neuville S, Dromer F, Morin O, et al; French Cryptococcosis Study Group. Primary cutaneous cryptococcosis: a distinct clinical entity [published online January 17, 2003]. Clin Infect Dis. 2003;36:337-347.
- Murakawa GJ, Kerschmann R, Berger T. Cutaneous cryptococcus infection and AIDS: report of 12 cases and review of the literature. JAMA Dermatol. 1996;132:545-548.
- Sun HY, Alexander BD, Lortholary O, et al. Cutaneous cryptococcosis in solid organ transplant recipients. Med Mycol. 2010;48:785-791.
- Grossman ME, Fox LP, Kovarik C, et al. Cutaneous Manifestations of Infection in the Immunocompromised Host. Baltimore, MD: Williams & Wilkins; 2012.
- Korfel A, Menssen HD, Schwartz S, et al. Cryptococcosis in Hodgkin's disease: description of two cases and review of the literature. Ann Hematol. 1998;76:283-286.
- Perfect JR, Dismukes WE, Dromer F. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2010;50:291-322.
The Diagnosis: Disseminated Cryptococcosis
Histopathologic examination of a 3-mm punch biopsy showed a diffuse dermal neutrophilic infiltrate with necrosis and subcutaneous tissue with round yeast surrounded by a prominent halo staining bright red with mucicarmine, representing a thick mucinous capsule (Figure). Grocott-Gomori methenamine-silver and periodic acid-Schiff stains also demonstrated fungal spores morphologically. Cerebrospinal fluid culture grew Cryptococcus neoformans, and cryptococcal antigen titers were positive in both serum and cerebrospinal fluid samples (>1:4096). The patient had autolytic debridement of the ulcer after completing a 4-week induction course of intravenous liposomal amphotericin B with oral flucytosine. He was transitioned to oral fluconazole for the consolidation phase of treatment.
Cryptococcus is an opportunistic basidiomycetous yeast with worldwide distribution and 2 primary pathogenic species in humans: C neoformans and Cryptococcus gattii. It is associated with bird feces, composted food, and decayed wood.1,2 A predilection toward an immunosuppressed host is recognized in 70% to 90% of the infections caused by C neoformans; however, C gattii commonly affects individuals with apparently intact immune systems.1,3 Risk factors for infection include advanced human immunodeficiency virus infection, solid organ transplantation, chronic liver disease, autoimmune disease, hematological malignancy, and underlying genetic susceptibility.1,2
Initial exposure is through the respiratory tract with formation of latent reservoirs in the pulmonary lymph nodes with subsequent reactivation that can result in hematogenous dissemination.1,2 Cutaneous involvement was described in 108 patients (5%) in a large review of 1974 cases in France.4 Among those with cutaneous involvement, disseminated disease was diagnosed in 80 cases (74%), and 28 cases (26%) were considered primary cutaneous cryptococcosis. Primary cutaneous cryptococcosis typically presents as a single lesion, predominantly on the hand, with whitlow and more rarely with extensive cellulitis or necrotizing fasciitis.4 In disseminated cutaneous disease, there is no pathognomonic single lesion; however, it is commonly associated with multiple cutaneous lesions predominantly involving the head and neck. Plaques, abscesses, nodules, and pustular or umbilicated papules have been reported.1,5 There are few case reports that describe a single isolated necrotic ulcer with disseminated disease similar to our presented case, and more typically the necrotic ulcer is seen in transplanted patients.6 The differential diagnosis of a necrotic thigh ulcer includes pseudomonal ecthyma gangrenosum, cutaneous anthrax and aspergillosis, fusariosis, and a bite from the brown recluse spider.7 Our patient had an increased susceptibility to infection from his ongoing chemotherapy, a risk previously described in oncology patients with cell-mediated immunosuppression.8
Management for disseminated cryptococcosis is a 3-phase therapy including induction with intravenous amphotericin B and oral flucytosine for a minimum of 2 weeks, with consolidation and maintenance phases both with oral fluconazole for a length depending on underlying immunosuppression.9
The Diagnosis: Disseminated Cryptococcosis
Histopathologic examination of a 3-mm punch biopsy showed a diffuse dermal neutrophilic infiltrate with necrosis and subcutaneous tissue with round yeast surrounded by a prominent halo staining bright red with mucicarmine, representing a thick mucinous capsule (Figure). Grocott-Gomori methenamine-silver and periodic acid-Schiff stains also demonstrated fungal spores morphologically. Cerebrospinal fluid culture grew Cryptococcus neoformans, and cryptococcal antigen titers were positive in both serum and cerebrospinal fluid samples (>1:4096). The patient had autolytic debridement of the ulcer after completing a 4-week induction course of intravenous liposomal amphotericin B with oral flucytosine. He was transitioned to oral fluconazole for the consolidation phase of treatment.
Cryptococcus is an opportunistic basidiomycetous yeast with worldwide distribution and 2 primary pathogenic species in humans: C neoformans and Cryptococcus gattii. It is associated with bird feces, composted food, and decayed wood.1,2 A predilection toward an immunosuppressed host is recognized in 70% to 90% of the infections caused by C neoformans; however, C gattii commonly affects individuals with apparently intact immune systems.1,3 Risk factors for infection include advanced human immunodeficiency virus infection, solid organ transplantation, chronic liver disease, autoimmune disease, hematological malignancy, and underlying genetic susceptibility.1,2
Initial exposure is through the respiratory tract with formation of latent reservoirs in the pulmonary lymph nodes with subsequent reactivation that can result in hematogenous dissemination.1,2 Cutaneous involvement was described in 108 patients (5%) in a large review of 1974 cases in France.4 Among those with cutaneous involvement, disseminated disease was diagnosed in 80 cases (74%), and 28 cases (26%) were considered primary cutaneous cryptococcosis. Primary cutaneous cryptococcosis typically presents as a single lesion, predominantly on the hand, with whitlow and more rarely with extensive cellulitis or necrotizing fasciitis.4 In disseminated cutaneous disease, there is no pathognomonic single lesion; however, it is commonly associated with multiple cutaneous lesions predominantly involving the head and neck. Plaques, abscesses, nodules, and pustular or umbilicated papules have been reported.1,5 There are few case reports that describe a single isolated necrotic ulcer with disseminated disease similar to our presented case, and more typically the necrotic ulcer is seen in transplanted patients.6 The differential diagnosis of a necrotic thigh ulcer includes pseudomonal ecthyma gangrenosum, cutaneous anthrax and aspergillosis, fusariosis, and a bite from the brown recluse spider.7 Our patient had an increased susceptibility to infection from his ongoing chemotherapy, a risk previously described in oncology patients with cell-mediated immunosuppression.8
Management for disseminated cryptococcosis is a 3-phase therapy including induction with intravenous amphotericin B and oral flucytosine for a minimum of 2 weeks, with consolidation and maintenance phases both with oral fluconazole for a length depending on underlying immunosuppression.9
- Chen SC, Meyer W, Sorrell TC. Cryptococcus gattii infections. Clin Microbiol Rev. 2014;27:980-1024.
- Williamson PR, Jarvis JN, Panackal AA, et al. Cryptococcal meningitis: epidemiology, immunology, diagnosis, and therapy [published online November 25, 2016]. Nat Rev Neurol. 2017;13:13-24.
- Speed B, Dunt D. Clinical and host differences between infections with the two varieties of Cryptococcus neoformans. Clin Infect Dis. 1995;21:28-34.
- Neuville S, Dromer F, Morin O, et al; French Cryptococcosis Study Group. Primary cutaneous cryptococcosis: a distinct clinical entity [published online January 17, 2003]. Clin Infect Dis. 2003;36:337-347.
- Murakawa GJ, Kerschmann R, Berger T. Cutaneous cryptococcus infection and AIDS: report of 12 cases and review of the literature. JAMA Dermatol. 1996;132:545-548.
- Sun HY, Alexander BD, Lortholary O, et al. Cutaneous cryptococcosis in solid organ transplant recipients. Med Mycol. 2010;48:785-791.
- Grossman ME, Fox LP, Kovarik C, et al. Cutaneous Manifestations of Infection in the Immunocompromised Host. Baltimore, MD: Williams & Wilkins; 2012.
- Korfel A, Menssen HD, Schwartz S, et al. Cryptococcosis in Hodgkin's disease: description of two cases and review of the literature. Ann Hematol. 1998;76:283-286.
- Perfect JR, Dismukes WE, Dromer F. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2010;50:291-322.
- Chen SC, Meyer W, Sorrell TC. Cryptococcus gattii infections. Clin Microbiol Rev. 2014;27:980-1024.
- Williamson PR, Jarvis JN, Panackal AA, et al. Cryptococcal meningitis: epidemiology, immunology, diagnosis, and therapy [published online November 25, 2016]. Nat Rev Neurol. 2017;13:13-24.
- Speed B, Dunt D. Clinical and host differences between infections with the two varieties of Cryptococcus neoformans. Clin Infect Dis. 1995;21:28-34.
- Neuville S, Dromer F, Morin O, et al; French Cryptococcosis Study Group. Primary cutaneous cryptococcosis: a distinct clinical entity [published online January 17, 2003]. Clin Infect Dis. 2003;36:337-347.
- Murakawa GJ, Kerschmann R, Berger T. Cutaneous cryptococcus infection and AIDS: report of 12 cases and review of the literature. JAMA Dermatol. 1996;132:545-548.
- Sun HY, Alexander BD, Lortholary O, et al. Cutaneous cryptococcosis in solid organ transplant recipients. Med Mycol. 2010;48:785-791.
- Grossman ME, Fox LP, Kovarik C, et al. Cutaneous Manifestations of Infection in the Immunocompromised Host. Baltimore, MD: Williams & Wilkins; 2012.
- Korfel A, Menssen HD, Schwartz S, et al. Cryptococcosis in Hodgkin's disease: description of two cases and review of the literature. Ann Hematol. 1998;76:283-286.
- Perfect JR, Dismukes WE, Dromer F. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2010;50:291-322.
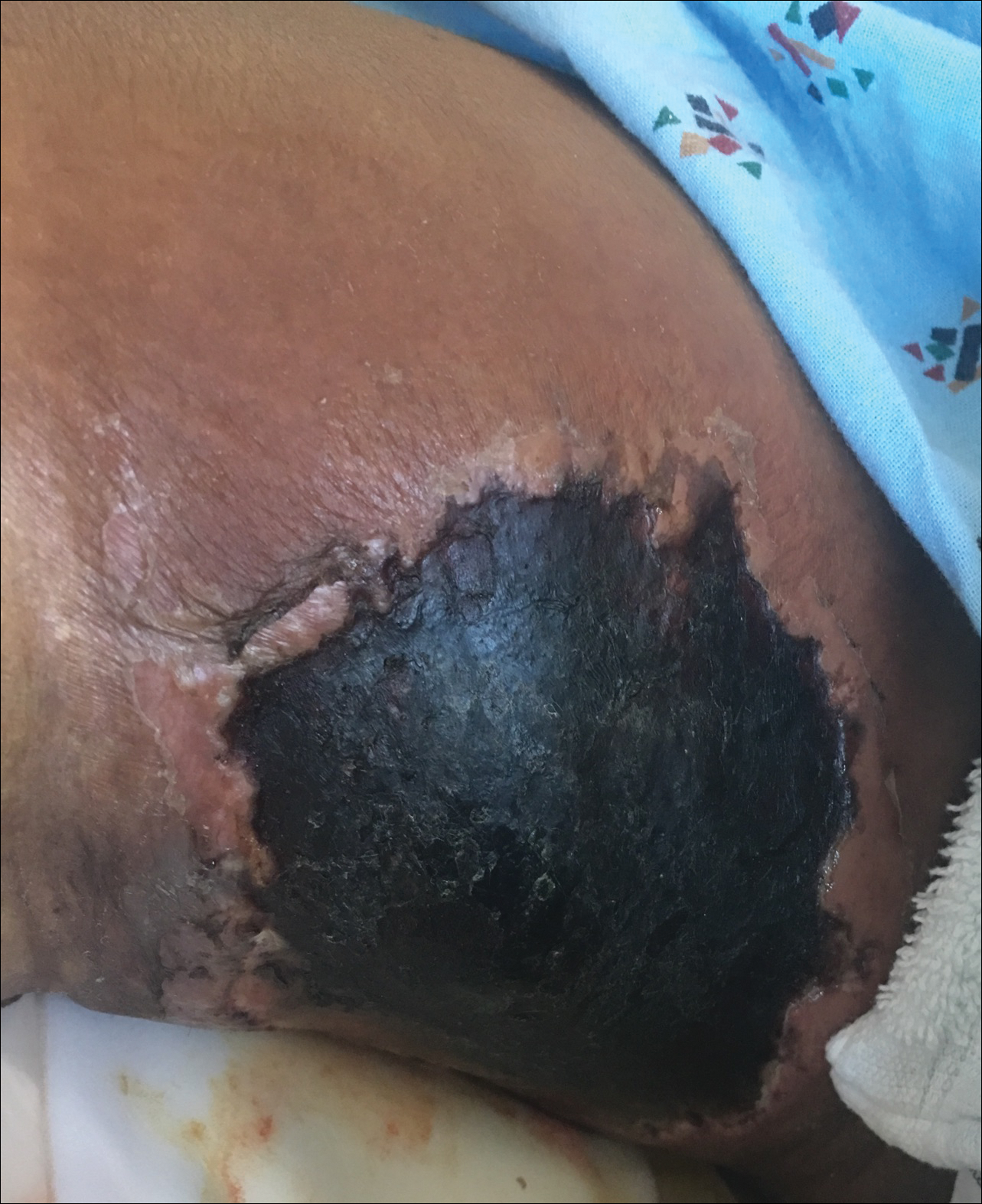
A 29-year-old man with a history of acute lymphoblastic leukemia was admitted for acute encephalopathy and a necrotic ulcer on the right thigh of 2 weeks' duration. He had received chemotherapy with pegaspargase and vincristine 6 weeks prior to admission. He reported headache with nausea and vomiting of 2 weeks' duration and had sustained a fall in the bathtub a week prior that initially resulted in a right thigh abrasion. He denied recent travel, unusual food consumption, animal exposure, exposure to sick persons, and alcohol or other drug use. On examination the patient was alert but was not oriented to person, place, or time. A 10.2 ×10-cm necrotic ulcer with surrounding mild erythema and tenderness was noted on the right inner thigh.
When the painful ‘bumps’ are calciphylaxis, what’s next?
EXPERT ANALYSIS FROM THE 2017 AAD SUMMER MEETING
NEW YORK – When patients come to the office with painful “bumps” on the legs or elsewhere, panniculitis should be in the differential. And for some patients, said Alina Bridges, DO, the panniculitis may come with the dire diagnosis of calciphylaxis.
Calciphylaxis is an underrecognized crystal deposition disease that’s associated with panniculitis, said Dr. Bridges, speaking at the American Academy of Dermatology summer meeting. When calcium accumulates in small subcutaneous vessels, an occlusive vasculopathy is created within the dermis.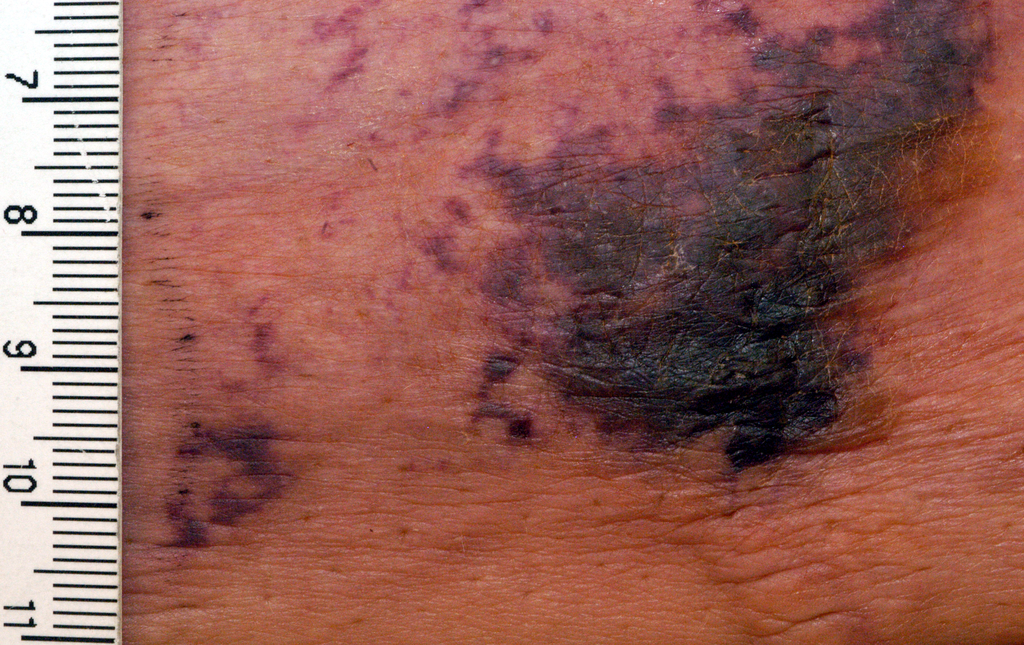
A soft-tissue radiograph of the affected area may also be helpful. Calciphylaxis shows as a fine netlike pattern of calcification, a finding that Dr. Bridges said has 90% specificity for the condition.
However, Dr. Bridges said, patients with panniculitis need a biopsy. “Careful selection of biopsy site and a deep specimen containing abundant fat obtained by incisional or excisional biopsy” is the best approach, allowing the pathologist to see the complete picture. In some cases, she said, a double-punch biopsy could also produce adequate specimens.
In addition to the calcium deposition, other pathologic findings may be lobular fat necrosis, with a pannicular vascular thrombosis. Though extravascular calcification can be seen in the panniculus, it’s not uncommon also to see intravascular calcification, said Dr. Bridges, who is the dermatopathology fellowship program director at the Mayo Clinic, Rochester, Minn.
Dr. Bridges said that the patients with calciphylaxis can present with predominant panniculitis or vasculitis, or a mixed picture; patients can also have bullae, ulcers, or livedo reticularis.
The lesions are extremely painful and become increasingly violaceous, with firm subcutaneous nodules. They are variably necrotic, and become more ulcerated over time.
Calciphylaxis is multifactorial and progressive. The prognosis is very poor for individuals with the condition, Dr. Bridges said. The median survival is 10 months, with 1-year survival rates of 46%, and just 20% of individuals with calciphylaxis surviving 2 years after diagnosis.
Gangrene is a frequent complication, and multisystem organ failure often occurs as well, she said.
Calciphylaxis most commonly occurs in individuals with chronic kidney disease and is seen in 4% of hemodialysis patients. However, it may also occur in individuals without uremia. In associations that are incompletely understood, calciphylaxis has been associated with warfarin therapy, connective tissue disorders, Crohn’s disease, liver disease, diabetes, hematologic malignancies, factor V Leiden deficiency, and protein C and S deficiency.
There’s a need for clinical suspicion of calciphylaxis when individuals with any of these conditions present with painful erythematous nodules, or with a vasculitic picture, she said.
Other, more common crystal deposition diseases can also be associated with panniculitis and can be in the differential, Dr. Bridges said. In patients with gout, sodium urate crystal deposition can occur in subcutaneous tissues.
Cutaneous oxalosis can occur as a primary disorder, when patients have metabolic errors and lack alanine-glyoxylate aminotransferase or D-glycerate dehydrogenase. Oxalosis can also be an acquired syndrome in patients with chronic renal failure who have been on long-term hemodialysis.
Although there is not a clearly effective treatment for calciphylaxis, a multitargeted, multidisciplinary approach is needed to help improve tissue health and patient quality of life. Since the primary mechanism of tissue damage is thrombotic tissue ischemia, strategies are aimed at existing clots and at preventing further clot formation.
To correct the calcium-phosphate balance, several medications have been used, including sodium thiosulfate and cinacalcet. For individuals on hemodialysis, a low-calcium dialysate may be used.
Tissue perfusion and oxygenation can be improved using tissue plasminogen activator, hyperbaric oxygen therapy, and the avoidance of warfarin if the patient requires anticoagulation.
To address wounds directly, debridement can begin with whirlpool time for patients. Surgical debridement may be required, and maggots can also help clean up wound beds.
Palliative care for patients should always include optimizing pain control and improving quality of life for patients with this serious and often life-limiting condition, Dr. Bridges said.
She reported no relevant conflicts of interest.
[email protected]
On Twitter @karioakes
EXPERT ANALYSIS FROM THE 2017 AAD SUMMER MEETING
NEW YORK – When patients come to the office with painful “bumps” on the legs or elsewhere, panniculitis should be in the differential. And for some patients, said Alina Bridges, DO, the panniculitis may come with the dire diagnosis of calciphylaxis.
Calciphylaxis is an underrecognized crystal deposition disease that’s associated with panniculitis, said Dr. Bridges, speaking at the American Academy of Dermatology summer meeting. When calcium accumulates in small subcutaneous vessels, an occlusive vasculopathy is created within the dermis.
A soft-tissue radiograph of the affected area may also be helpful. Calciphylaxis shows as a fine netlike pattern of calcification, a finding that Dr. Bridges said has 90% specificity for the condition.
However, Dr. Bridges said, patients with panniculitis need a biopsy. “Careful selection of biopsy site and a deep specimen containing abundant fat obtained by incisional or excisional biopsy” is the best approach, allowing the pathologist to see the complete picture. In some cases, she said, a double-punch biopsy could also produce adequate specimens.
In addition to the calcium deposition, other pathologic findings may be lobular fat necrosis, with a pannicular vascular thrombosis. Though extravascular calcification can be seen in the panniculus, it’s not uncommon also to see intravascular calcification, said Dr. Bridges, who is the dermatopathology fellowship program director at the Mayo Clinic, Rochester, Minn.
Dr. Bridges said that the patients with calciphylaxis can present with predominant panniculitis or vasculitis, or a mixed picture; patients can also have bullae, ulcers, or livedo reticularis.
The lesions are extremely painful and become increasingly violaceous, with firm subcutaneous nodules. They are variably necrotic, and become more ulcerated over time.
Calciphylaxis is multifactorial and progressive. The prognosis is very poor for individuals with the condition, Dr. Bridges said. The median survival is 10 months, with 1-year survival rates of 46%, and just 20% of individuals with calciphylaxis surviving 2 years after diagnosis.
Gangrene is a frequent complication, and multisystem organ failure often occurs as well, she said.
Calciphylaxis most commonly occurs in individuals with chronic kidney disease and is seen in 4% of hemodialysis patients. However, it may also occur in individuals without uremia. In associations that are incompletely understood, calciphylaxis has been associated with warfarin therapy, connective tissue disorders, Crohn’s disease, liver disease, diabetes, hematologic malignancies, factor V Leiden deficiency, and protein C and S deficiency.
There’s a need for clinical suspicion of calciphylaxis when individuals with any of these conditions present with painful erythematous nodules, or with a vasculitic picture, she said.
Other, more common crystal deposition diseases can also be associated with panniculitis and can be in the differential, Dr. Bridges said. In patients with gout, sodium urate crystal deposition can occur in subcutaneous tissues.
Cutaneous oxalosis can occur as a primary disorder, when patients have metabolic errors and lack alanine-glyoxylate aminotransferase or D-glycerate dehydrogenase. Oxalosis can also be an acquired syndrome in patients with chronic renal failure who have been on long-term hemodialysis.
Although there is not a clearly effective treatment for calciphylaxis, a multitargeted, multidisciplinary approach is needed to help improve tissue health and patient quality of life. Since the primary mechanism of tissue damage is thrombotic tissue ischemia, strategies are aimed at existing clots and at preventing further clot formation.
To correct the calcium-phosphate balance, several medications have been used, including sodium thiosulfate and cinacalcet. For individuals on hemodialysis, a low-calcium dialysate may be used.
Tissue perfusion and oxygenation can be improved using tissue plasminogen activator, hyperbaric oxygen therapy, and the avoidance of warfarin if the patient requires anticoagulation.
To address wounds directly, debridement can begin with whirlpool time for patients. Surgical debridement may be required, and maggots can also help clean up wound beds.
Palliative care for patients should always include optimizing pain control and improving quality of life for patients with this serious and often life-limiting condition, Dr. Bridges said.
She reported no relevant conflicts of interest.
[email protected]
On Twitter @karioakes
EXPERT ANALYSIS FROM THE 2017 AAD SUMMER MEETING
NEW YORK – When patients come to the office with painful “bumps” on the legs or elsewhere, panniculitis should be in the differential. And for some patients, said Alina Bridges, DO, the panniculitis may come with the dire diagnosis of calciphylaxis.
Calciphylaxis is an underrecognized crystal deposition disease that’s associated with panniculitis, said Dr. Bridges, speaking at the American Academy of Dermatology summer meeting. When calcium accumulates in small subcutaneous vessels, an occlusive vasculopathy is created within the dermis.
A soft-tissue radiograph of the affected area may also be helpful. Calciphylaxis shows as a fine netlike pattern of calcification, a finding that Dr. Bridges said has 90% specificity for the condition.
However, Dr. Bridges said, patients with panniculitis need a biopsy. “Careful selection of biopsy site and a deep specimen containing abundant fat obtained by incisional or excisional biopsy” is the best approach, allowing the pathologist to see the complete picture. In some cases, she said, a double-punch biopsy could also produce adequate specimens.
In addition to the calcium deposition, other pathologic findings may be lobular fat necrosis, with a pannicular vascular thrombosis. Though extravascular calcification can be seen in the panniculus, it’s not uncommon also to see intravascular calcification, said Dr. Bridges, who is the dermatopathology fellowship program director at the Mayo Clinic, Rochester, Minn.
Dr. Bridges said that the patients with calciphylaxis can present with predominant panniculitis or vasculitis, or a mixed picture; patients can also have bullae, ulcers, or livedo reticularis.
The lesions are extremely painful and become increasingly violaceous, with firm subcutaneous nodules. They are variably necrotic, and become more ulcerated over time.
Calciphylaxis is multifactorial and progressive. The prognosis is very poor for individuals with the condition, Dr. Bridges said. The median survival is 10 months, with 1-year survival rates of 46%, and just 20% of individuals with calciphylaxis surviving 2 years after diagnosis.
Gangrene is a frequent complication, and multisystem organ failure often occurs as well, she said.
Calciphylaxis most commonly occurs in individuals with chronic kidney disease and is seen in 4% of hemodialysis patients. However, it may also occur in individuals without uremia. In associations that are incompletely understood, calciphylaxis has been associated with warfarin therapy, connective tissue disorders, Crohn’s disease, liver disease, diabetes, hematologic malignancies, factor V Leiden deficiency, and protein C and S deficiency.
There’s a need for clinical suspicion of calciphylaxis when individuals with any of these conditions present with painful erythematous nodules, or with a vasculitic picture, she said.
Other, more common crystal deposition diseases can also be associated with panniculitis and can be in the differential, Dr. Bridges said. In patients with gout, sodium urate crystal deposition can occur in subcutaneous tissues.
Cutaneous oxalosis can occur as a primary disorder, when patients have metabolic errors and lack alanine-glyoxylate aminotransferase or D-glycerate dehydrogenase. Oxalosis can also be an acquired syndrome in patients with chronic renal failure who have been on long-term hemodialysis.
Although there is not a clearly effective treatment for calciphylaxis, a multitargeted, multidisciplinary approach is needed to help improve tissue health and patient quality of life. Since the primary mechanism of tissue damage is thrombotic tissue ischemia, strategies are aimed at existing clots and at preventing further clot formation.
To correct the calcium-phosphate balance, several medications have been used, including sodium thiosulfate and cinacalcet. For individuals on hemodialysis, a low-calcium dialysate may be used.
Tissue perfusion and oxygenation can be improved using tissue plasminogen activator, hyperbaric oxygen therapy, and the avoidance of warfarin if the patient requires anticoagulation.
To address wounds directly, debridement can begin with whirlpool time for patients. Surgical debridement may be required, and maggots can also help clean up wound beds.
Palliative care for patients should always include optimizing pain control and improving quality of life for patients with this serious and often life-limiting condition, Dr. Bridges said.
She reported no relevant conflicts of interest.
[email protected]
On Twitter @karioakes
Co-occurrence of Steatocystoma Multiplex, Eruptive Vellus Hair Cysts, and Trichofolliculomas
An association between steatocystoma multiplex (SCM) and eruptive vellus hair cysts (EVHCs) has been recognized. They are related conditions representing nevoid malformations of the pilosebaceous junctions1-10 that have similar clinical features but distinctive histologic features. Both conditions most commonly involve the anterior aspect of the chest. Six cases of a rare facial variant of SCM have been reported,11-16 3 involving lesions limited to the forehead.13-15 Two patients with a rare facial variant of EVHC also have been reported.17 The development of separate lesions of SCM and EVHC on the trunk can uncommonly occur.5,6,10 One case of SCM and EVHC on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. The simultaneous occurrence of multiple trichoblastomas, trichoepitheliomas, and SCM on the face and trunk has been reported in 1 case.18 Milia, SCM, and EVHC on the face and trunk have been reported in 1 family.4 A report of facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus also has occurred in 1 patient.19 Here, we report the simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead.
Case Report
A 37-year-old man had an increasing number of flesh-colored to yellow papules on the forehead that had been present since puberty. Although the lesions were asymptomatic, some had recently become tender, which led him to seek medical care. There was no history of trauma, burns, irradiation, or application of topical agents to the area or use of eyeglasses or goggles. The patient’s father had similar lesions limited to the forehead, which developed during adolescence.
On evaluation at our clinic, skin examination revealed 16 discrete, 0.3- to 1-cm, flesh-colored, yellow to blue, mobile, smooth papules, as well as flesh-colored papules with a central black punctum, on the forehead (Figure 1). Similar lesions were not present on the rest of the face; around the ears; or on the scalp, neck, chest, back, abdomen, genitalia, buttocks, palms, soles, axillae, arms, or legs. There were no nail abnormalities.
Multiple 3-, 4-, and 6-mm punch and excisional biopsies were performed to remove all 16 lesions on the forehead. Histologic examination revealed a collapsed cystic structure in the mid dermis in 10 lesions. The cysts were lined with a squamous epithelium without a granular layer but with an eosinophilic corrugated lining, and the cyst cavity contained scant homogeneous eosinophilic secretion. Mature sebaceous glands were adjacent to the outer portion of the cyst wall. These histologic findings were consistent with SCM (Figure 2).
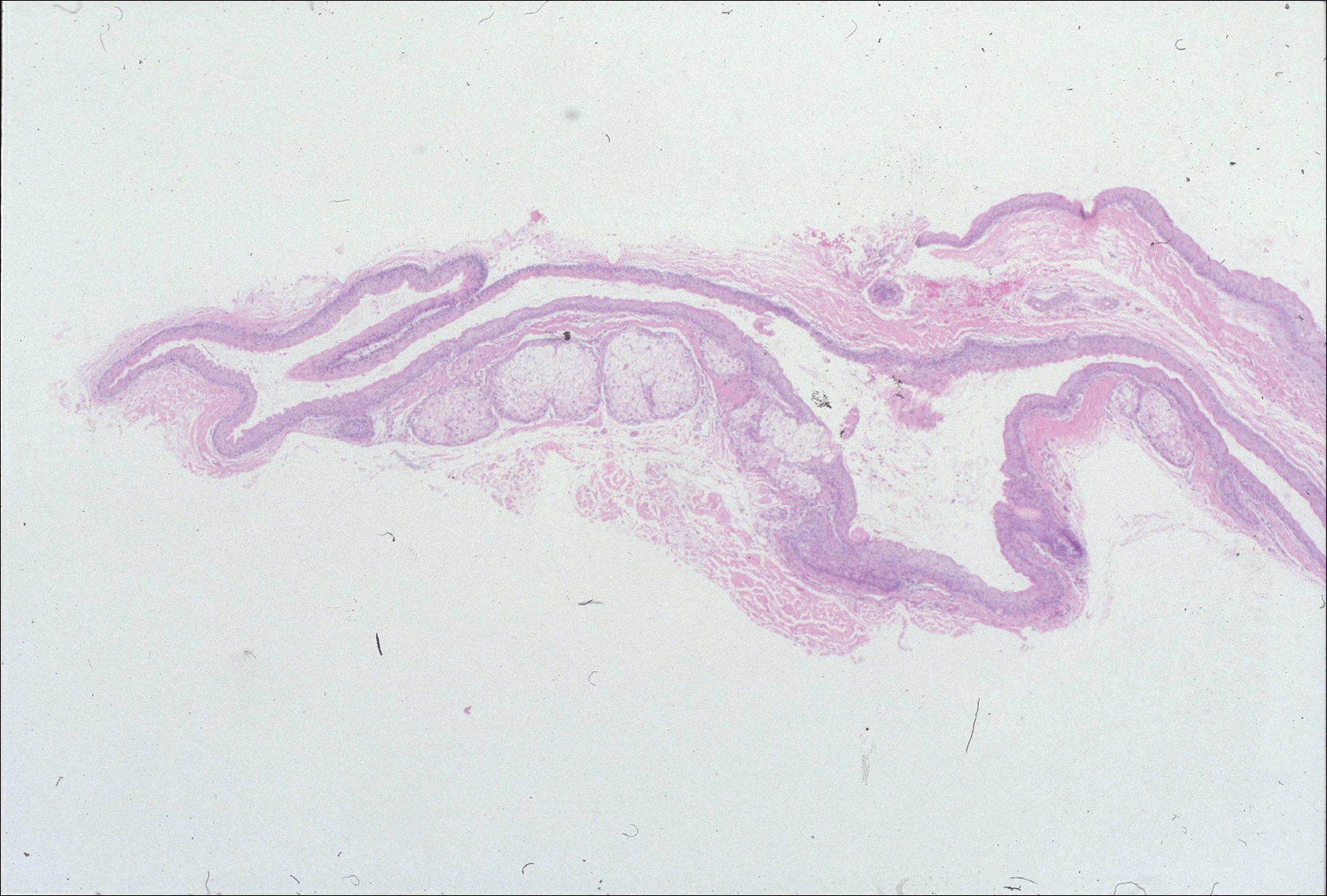
In 3 lesions, histologic examination revealed a cystic structure lined by a few layers of stratified squamous epithelium in the mid dermis. The cyst cavity contained numerous small vellus hairs and laminated keratin. These histologic findings were consistent with EVHC (Figure 3).
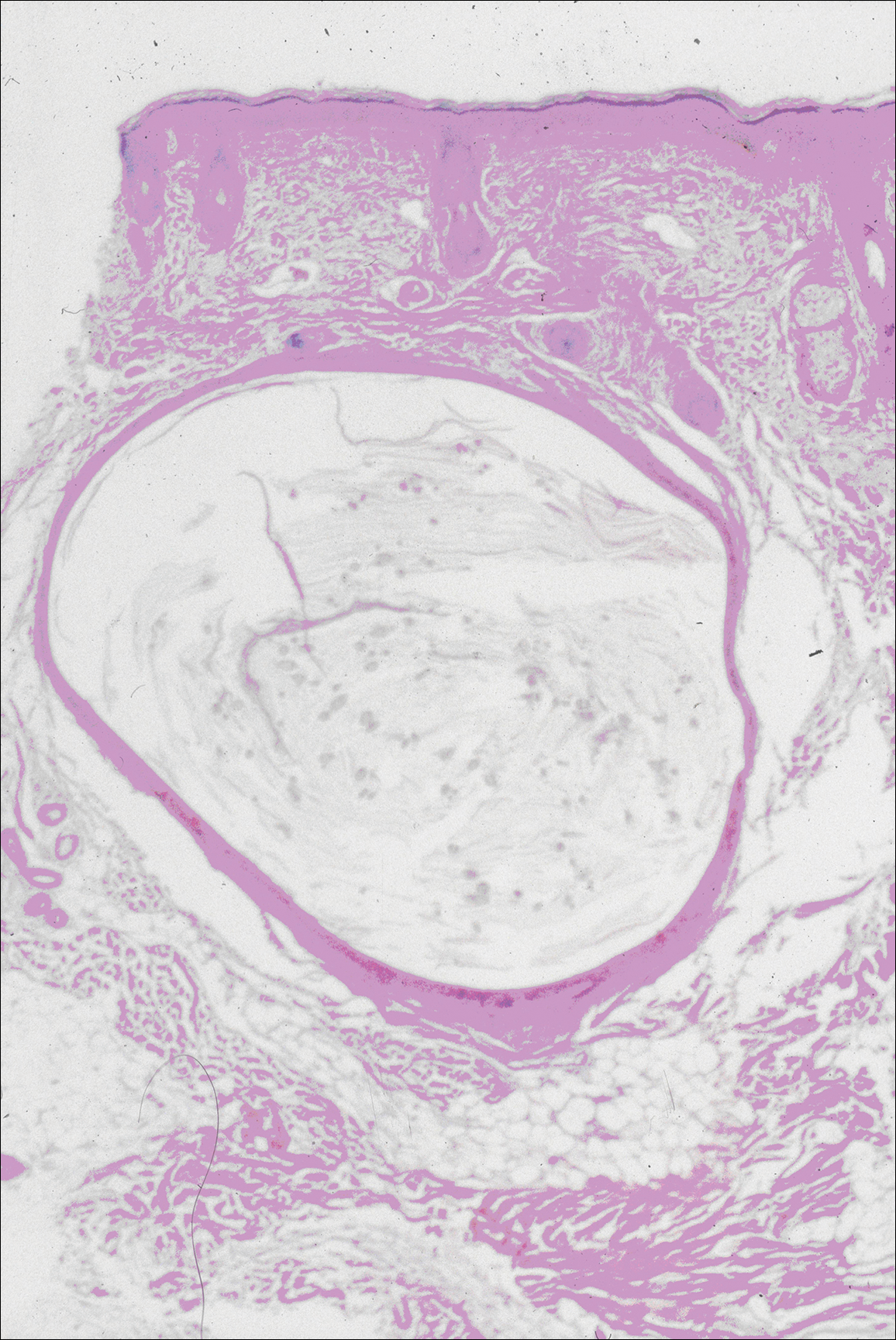
In the other 3 lesions, histologic examination revealed a dilated central cystic cavity filled with laminated keratin in the mid dermis. Multiple small follicles arose from the cysts and showed differentiation toward germinative epithelium. The surrounding stroma was fibrotic and contained a patchy lymphocytic infiltrate. These histologic findings were consistent with trichofolliculomas (Figure 4).
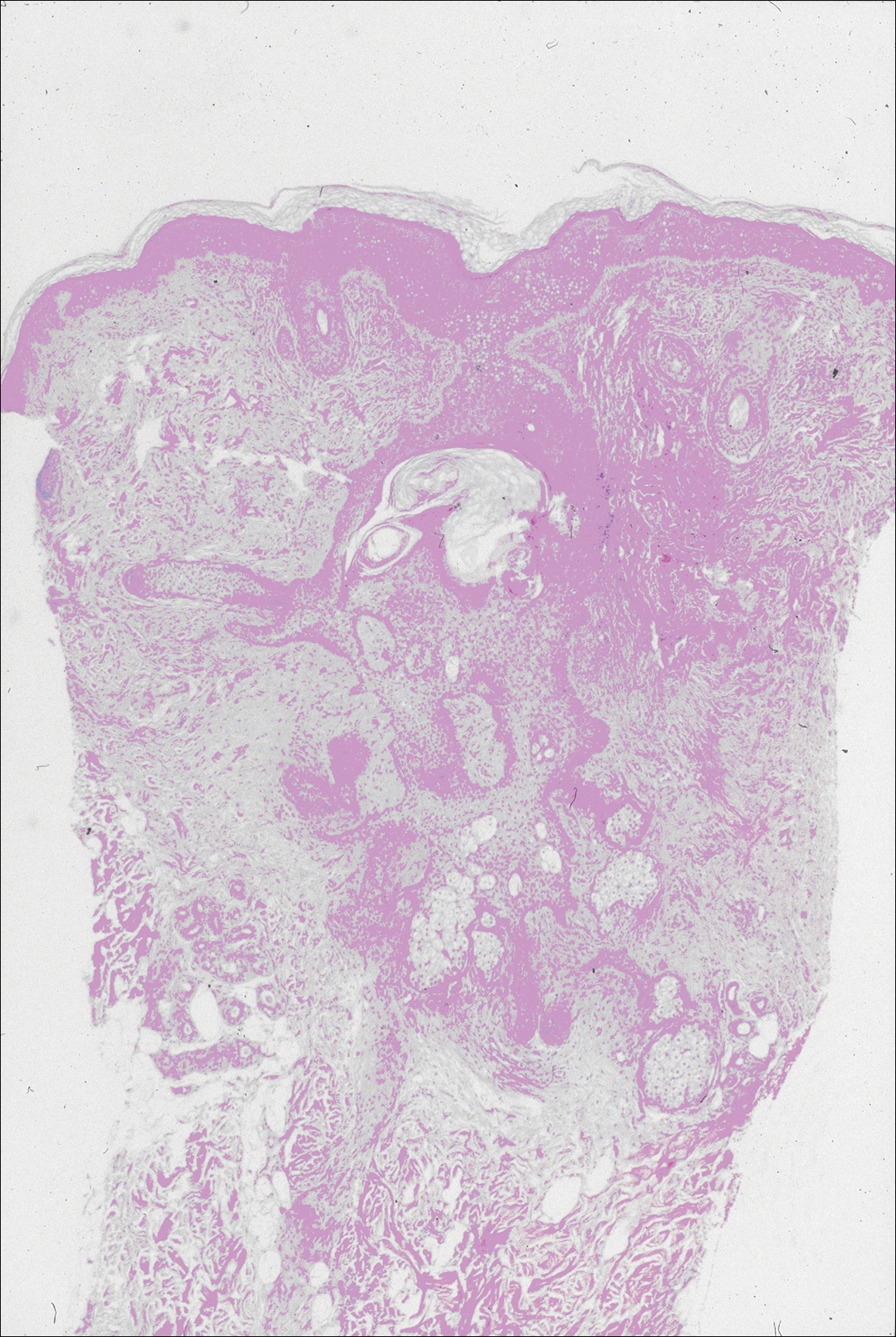
Comment
Characteristics of SCM
Steatocystoma multiplex is an uncommon condition characterized by the formation of asymptomatic, 0.2- to 2-cm, yellow to flesh-colored, soft, mobile papules or nodules on the trunk, extremities, axillae, genitalia, and/or chest. The lesions contain a clear or opaque, oily, milky or yellow, odorless fluid and most commonly are located on the anterior aspect of the chest. The face is not a commonly involved site in this condition. Six cases of a rare facial variant of SCM have been reported,11-16 with lesions limited to the forehead in 3 cases.13-15
In 1937, Mount20 credited Bozellini for describing the first case, though 3 cases reported in the late 1800s probably were SCM.21 In 1899, Pringle22 coined the term steatocystoma multiplex for this condition. It can be sporadic or have an autosomal-dominant inheritance pattern. Steatocystoma multiplex can occur at any age, though lesions develop most frequently in adolescence or young adulthood. There is no sex predilection.
Steatocystoma multiplex with pachyonychia congenita has been reported in a familial case.23 Other findings reported in patients with SCM include ichthyosis, koilonychia, acrokeratosis verruciformis of Hopf and hypertrophic lichen planus, hidradenitis suppurativa, hypotrichosis, multiple keratoacanthomas, and rheumatoid arthritis.12,24-26
Steatocystoma multiplex is a cyst lined by stratified squamous epithelium without a granular layer but with a thick eosinophilic cuticle. Mature sebaceous lobules are closely associated with the cyst wall. Steatocystoma multiplex arises from the sebaceous duct because the lining of the lumen is composed of undulating eosinophilic cuticle.
Characteristics of EVHCs
Eruptive vellus hair cysts, which were first described by Esterly et al,27 can occur at any age but develop most frequently in adolescents or young adults. Sometimes the lesions are congenital or appear in childhood. There is no sex predilection. They can be sporadic or have an autosomal-dominant inheritance pattern.
Eruptive vellus hair cysts are asymptomatic, 1- to 2-mm, smooth, crusted, or umbilicated papules on the chest or arms and legs. Eruptive vellus hair cysts most commonly involve the anterior aspect of the chest. The lesions are flesh-colored to yellow, though they have a slate gray color in darker-skinned individuals. A rare facial variant has been reported in 2 patients of Asian descent.17
Eruptive vellus hair cysts are small cystic structures lined by a stratified squamous epithelium with a granular layer. The cyst cavity contains numerous small vellus hair shafts and laminated keratin. Eruptive vellus hair cysts originate from the infundibulum or less frequently the isthmus or infundibular-isthmic junction of the hair follicle.
Characteristics of Trichofolliculomas
Trichofolliculomas are solitary, 3- to 5-mm, flesh-colored papules that occur on the face. They are highly differentiated, benign, neoplastic proliferations of an actively trichogenic epithelium, with structural components reflecting all portions of the pilosebaceous unit. Trichofolliculomas consist of a central dilated primary follicle contiguous with the surface epidermis embedded in a fibrous stroma. Multiple small secondary follicles with varying degrees of follicular differentiation arise from the primary follicle.
Co-occurrence of Lesions
An association between SCM and EVHC has been recognized.5-10 Steatocystoma multiplex and EVHC have similar clinical features but distinctive histologic features. They also have a similar age of onset, location/appearance of lesions, and mode of inheritance. Steatocystoma multiplex and EVHC can be distinguished by immunohistochemical techniques: SCM shows expression of keratin 10 and keratin 17, whereas EVHCs express only keratin 17.28
Steatocystoma multiplex and EVHC have only rarely been reported to occur together on the trunk. One case of SCM and EVHC occurring on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. Milia, SCM, and EVHC on the face and trunk have been reported in 1 family,4 and facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus was reported in 1 patient.19 Although trichofolliculomas have not been reported to occur with SCM or EVHC, 2 related follicular neoplasms—trichoepitheliomas and trichoblastomas—have been reported to occur in association with SCM on the face and chest and around the ears in 1 case.18
Differential Diagnosis
The clinical differential diagnosis includes multiple epidermoid cysts, dermoid cysts, Gardner syndrome, sebaceous adenomas, Muir-Torre syndrome, syringomas, milia, leiomyomas, lipomas, acneiform folliculitis, multiple familial and nonfamilial trichoepitheliomas, cylindromas, and angiofibromas.3,29
Conclusion
Our patient represents a rare case of simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead. The patient had multiple neoplasms involving differentiation toward various regions of the pilosebaceous unit. This case gives further support to the hypothesis that these benign follicular neoplasms are closely related but are distinct conditions within the spectrum of the same disease process. They represent nevoid malformations of the pilosebaceous unit that can be sporadic or inherited in an autosomal-dominant pattern. Pure types of these lesions may represent one end of the spectrum, but in some patients, there are overlapping features or hybrids of each condition. Several biopsies from patients with multiple lesions should be performed to establish an accurate diagnosis.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Ogawa Y, Nogita T, Kawashima M. The coexistence of eruptive vellus hair cysts and steatocystoma multiplex. J Dermatol. 1992;19:570-571.
- Sanchez Yus E, Requena L. Eruptive vellus hair cyst and steatocystoma multiplex. Am J Dermatopathol. 1990;12:536-537.
- Patrizi A, Neri I, Guerrini V, et al. Persistent milia, steatocystoma multiplex and eruptive vellus hair cysts: variable expression of multiple pilosebaceous cysts within an affected family. Dermatology. 1998;196:392-396.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5, pt 2):876-878.
- Kiene P, Hauschild A, Christophers E. Eruptive vellus hair cysts and steatocystoma multiplex: variants of one entity? Br J Dermatol. 1996;134:365-367.
- Hurlimann AF, Panizzon RG, Burg G. Eruptive vellus hair cyst and steatocystoma multiplex: hybrid cysts. Dermatology. 1996;192:64-66.
- Sexton M, Murdock DK. Eruptive vellus hair cysts: a follicular cyst of the sebaceous duct (sometimes). Am J Dermatopathol. 1989;11:364-368.
- Sanchez-Yus E, Aguilar-Martinez A, Cristobal-Gil MC, et al. Eruptive vellus hair cyst and steatocystoma multiplex: two related conditions? J Cutan Pathol. 1988;15:40-42.
- Ahn SK, Chung J, Lee WS, et al. Hybrid cysts showing alternate combination of eruptive vellus hair cyst, steatocystoma multiplex, and epidermoid cyst, and an association among the three conditions. Am J Dermatopathol. 1996;18:645-649.
- Ahn SK, Hwang SM, Lee SH, et al. Steatocystoma multiplex localized only in the face. Int J Dermatol. 1997;36:372-373.
- Cole LA. Steatocystoma multiplex. Arch Dermatol. 1976;112:1437-1439.
- Hansen KK, Troy JL, Fairley JA. Multiple papules of the scalp and forehead. steatocystoma multiplex (facial papular variant). Arch Dermatol. 1995;131:835-838.
- Nishimura M, Kohda H, Urabe A. Steatocystoma multiplex: a facial popular variant. Arch Dermatol. 1986;122:205-207.
- Requena L, Martin L, Renedo G, et al. A facial variant of steatocystoma multiplex. Cutis. 1993;51:449-452.
- Holmes R, Black MM. Steatocystoma multiplex with unusually prominent cysts on the face. Br J Dermatol. 1980;102:711-713.
- Kumakiri M, Takashima I, Iju M, et al. Eruptive vellus hair cysts: a facial variant. J Am Acad Dermatol. 1982;7:461-467.
- Gianotti R, Cavicchini S, Alessi E. Simultaneous occurrence of multiple trichoblastomas and steatocystoma multiplex. Am J Dermatopathol. 1997;19:294-298.
- Sardana K, Sharma RC, Jain A, et al. Facial steatocystoma multiplex associated with pilar cyst and bilateral preauricular sinus. J Dermatol. 2002;29:157-159.
- Mount LB. Steatocystoma multiplex. Arch Dermatol Syphilol. 1937;36:31-39.
- Dubreuilh W, Auche B. Kystes grassieux sudoripares. Arch Clin de Bordeaux. 1896;5:387-391.
- Pringle JJ. A case of peculiar multiple sebaceous cysts (steatocystoma multiplex). Br J Dermatol. 1899;11:381-88.
- Vineyard WR, Scott RA. Steatocystoma multiplex with pachyonychia congenital: eight cases in four generations. Arch Dermatol. 1961;84:824-827.
- Contreras MA, Costello MJ. Steatocystoma multiplex with embryonal hair formation: case presentation and consideration of pathogenesis. AMA Arch Derm. 1957;76:720-725.
- Sohn D, Chin TC, Fellner MJ. Multiple keratoacanthomas associated with steatocystoma multiplex and rheumatoid arthritis: a case report. Arch Dermatol. 1980;116:913-915.
- Verbov J. Acrokeratosis verruciformis of Hopf with steatocystoma multiplex and hypertrophic lichen planus. Br J Dermatol. 1972;86:91-94.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Tomkova H, Fujimoto W, Arata J. Expression of keratins (K10 and K17) in steatocystoma multiplex, eruptive vellus hair cysts, and epidermoid and trichilemmal cysts. Am J Dermatopathol. 1997;19:250-253.
- Feinstein A, Trau H, Movshovitz M, et al. Steatocystoma multiplex. Cutis. 1983;31:425-427.
An association between steatocystoma multiplex (SCM) and eruptive vellus hair cysts (EVHCs) has been recognized. They are related conditions representing nevoid malformations of the pilosebaceous junctions1-10 that have similar clinical features but distinctive histologic features. Both conditions most commonly involve the anterior aspect of the chest. Six cases of a rare facial variant of SCM have been reported,11-16 3 involving lesions limited to the forehead.13-15 Two patients with a rare facial variant of EVHC also have been reported.17 The development of separate lesions of SCM and EVHC on the trunk can uncommonly occur.5,6,10 One case of SCM and EVHC on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. The simultaneous occurrence of multiple trichoblastomas, trichoepitheliomas, and SCM on the face and trunk has been reported in 1 case.18 Milia, SCM, and EVHC on the face and trunk have been reported in 1 family.4 A report of facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus also has occurred in 1 patient.19 Here, we report the simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead.
Case Report
A 37-year-old man had an increasing number of flesh-colored to yellow papules on the forehead that had been present since puberty. Although the lesions were asymptomatic, some had recently become tender, which led him to seek medical care. There was no history of trauma, burns, irradiation, or application of topical agents to the area or use of eyeglasses or goggles. The patient’s father had similar lesions limited to the forehead, which developed during adolescence.
On evaluation at our clinic, skin examination revealed 16 discrete, 0.3- to 1-cm, flesh-colored, yellow to blue, mobile, smooth papules, as well as flesh-colored papules with a central black punctum, on the forehead (Figure 1). Similar lesions were not present on the rest of the face; around the ears; or on the scalp, neck, chest, back, abdomen, genitalia, buttocks, palms, soles, axillae, arms, or legs. There were no nail abnormalities.
Multiple 3-, 4-, and 6-mm punch and excisional biopsies were performed to remove all 16 lesions on the forehead. Histologic examination revealed a collapsed cystic structure in the mid dermis in 10 lesions. The cysts were lined with a squamous epithelium without a granular layer but with an eosinophilic corrugated lining, and the cyst cavity contained scant homogeneous eosinophilic secretion. Mature sebaceous glands were adjacent to the outer portion of the cyst wall. These histologic findings were consistent with SCM (Figure 2).
In 3 lesions, histologic examination revealed a cystic structure lined by a few layers of stratified squamous epithelium in the mid dermis. The cyst cavity contained numerous small vellus hairs and laminated keratin. These histologic findings were consistent with EVHC (Figure 3).
In the other 3 lesions, histologic examination revealed a dilated central cystic cavity filled with laminated keratin in the mid dermis. Multiple small follicles arose from the cysts and showed differentiation toward germinative epithelium. The surrounding stroma was fibrotic and contained a patchy lymphocytic infiltrate. These histologic findings were consistent with trichofolliculomas (Figure 4).
Comment
Characteristics of SCM
Steatocystoma multiplex is an uncommon condition characterized by the formation of asymptomatic, 0.2- to 2-cm, yellow to flesh-colored, soft, mobile papules or nodules on the trunk, extremities, axillae, genitalia, and/or chest. The lesions contain a clear or opaque, oily, milky or yellow, odorless fluid and most commonly are located on the anterior aspect of the chest. The face is not a commonly involved site in this condition. Six cases of a rare facial variant of SCM have been reported,11-16 with lesions limited to the forehead in 3 cases.13-15
In 1937, Mount20 credited Bozellini for describing the first case, though 3 cases reported in the late 1800s probably were SCM.21 In 1899, Pringle22 coined the term steatocystoma multiplex for this condition. It can be sporadic or have an autosomal-dominant inheritance pattern. Steatocystoma multiplex can occur at any age, though lesions develop most frequently in adolescence or young adulthood. There is no sex predilection.
Steatocystoma multiplex with pachyonychia congenita has been reported in a familial case.23 Other findings reported in patients with SCM include ichthyosis, koilonychia, acrokeratosis verruciformis of Hopf and hypertrophic lichen planus, hidradenitis suppurativa, hypotrichosis, multiple keratoacanthomas, and rheumatoid arthritis.12,24-26
Steatocystoma multiplex is a cyst lined by stratified squamous epithelium without a granular layer but with a thick eosinophilic cuticle. Mature sebaceous lobules are closely associated with the cyst wall. Steatocystoma multiplex arises from the sebaceous duct because the lining of the lumen is composed of undulating eosinophilic cuticle.
Characteristics of EVHCs
Eruptive vellus hair cysts, which were first described by Esterly et al,27 can occur at any age but develop most frequently in adolescents or young adults. Sometimes the lesions are congenital or appear in childhood. There is no sex predilection. They can be sporadic or have an autosomal-dominant inheritance pattern.
Eruptive vellus hair cysts are asymptomatic, 1- to 2-mm, smooth, crusted, or umbilicated papules on the chest or arms and legs. Eruptive vellus hair cysts most commonly involve the anterior aspect of the chest. The lesions are flesh-colored to yellow, though they have a slate gray color in darker-skinned individuals. A rare facial variant has been reported in 2 patients of Asian descent.17
Eruptive vellus hair cysts are small cystic structures lined by a stratified squamous epithelium with a granular layer. The cyst cavity contains numerous small vellus hair shafts and laminated keratin. Eruptive vellus hair cysts originate from the infundibulum or less frequently the isthmus or infundibular-isthmic junction of the hair follicle.
Characteristics of Trichofolliculomas
Trichofolliculomas are solitary, 3- to 5-mm, flesh-colored papules that occur on the face. They are highly differentiated, benign, neoplastic proliferations of an actively trichogenic epithelium, with structural components reflecting all portions of the pilosebaceous unit. Trichofolliculomas consist of a central dilated primary follicle contiguous with the surface epidermis embedded in a fibrous stroma. Multiple small secondary follicles with varying degrees of follicular differentiation arise from the primary follicle.
Co-occurrence of Lesions
An association between SCM and EVHC has been recognized.5-10 Steatocystoma multiplex and EVHC have similar clinical features but distinctive histologic features. They also have a similar age of onset, location/appearance of lesions, and mode of inheritance. Steatocystoma multiplex and EVHC can be distinguished by immunohistochemical techniques: SCM shows expression of keratin 10 and keratin 17, whereas EVHCs express only keratin 17.28
Steatocystoma multiplex and EVHC have only rarely been reported to occur together on the trunk. One case of SCM and EVHC occurring on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. Milia, SCM, and EVHC on the face and trunk have been reported in 1 family,4 and facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus was reported in 1 patient.19 Although trichofolliculomas have not been reported to occur with SCM or EVHC, 2 related follicular neoplasms—trichoepitheliomas and trichoblastomas—have been reported to occur in association with SCM on the face and chest and around the ears in 1 case.18
Differential Diagnosis
The clinical differential diagnosis includes multiple epidermoid cysts, dermoid cysts, Gardner syndrome, sebaceous adenomas, Muir-Torre syndrome, syringomas, milia, leiomyomas, lipomas, acneiform folliculitis, multiple familial and nonfamilial trichoepitheliomas, cylindromas, and angiofibromas.3,29
Conclusion
Our patient represents a rare case of simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead. The patient had multiple neoplasms involving differentiation toward various regions of the pilosebaceous unit. This case gives further support to the hypothesis that these benign follicular neoplasms are closely related but are distinct conditions within the spectrum of the same disease process. They represent nevoid malformations of the pilosebaceous unit that can be sporadic or inherited in an autosomal-dominant pattern. Pure types of these lesions may represent one end of the spectrum, but in some patients, there are overlapping features or hybrids of each condition. Several biopsies from patients with multiple lesions should be performed to establish an accurate diagnosis.
An association between steatocystoma multiplex (SCM) and eruptive vellus hair cysts (EVHCs) has been recognized. They are related conditions representing nevoid malformations of the pilosebaceous junctions1-10 that have similar clinical features but distinctive histologic features. Both conditions most commonly involve the anterior aspect of the chest. Six cases of a rare facial variant of SCM have been reported,11-16 3 involving lesions limited to the forehead.13-15 Two patients with a rare facial variant of EVHC also have been reported.17 The development of separate lesions of SCM and EVHC on the trunk can uncommonly occur.5,6,10 One case of SCM and EVHC on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. The simultaneous occurrence of multiple trichoblastomas, trichoepitheliomas, and SCM on the face and trunk has been reported in 1 case.18 Milia, SCM, and EVHC on the face and trunk have been reported in 1 family.4 A report of facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus also has occurred in 1 patient.19 Here, we report the simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead.
Case Report
A 37-year-old man had an increasing number of flesh-colored to yellow papules on the forehead that had been present since puberty. Although the lesions were asymptomatic, some had recently become tender, which led him to seek medical care. There was no history of trauma, burns, irradiation, or application of topical agents to the area or use of eyeglasses or goggles. The patient’s father had similar lesions limited to the forehead, which developed during adolescence.
On evaluation at our clinic, skin examination revealed 16 discrete, 0.3- to 1-cm, flesh-colored, yellow to blue, mobile, smooth papules, as well as flesh-colored papules with a central black punctum, on the forehead (Figure 1). Similar lesions were not present on the rest of the face; around the ears; or on the scalp, neck, chest, back, abdomen, genitalia, buttocks, palms, soles, axillae, arms, or legs. There were no nail abnormalities.
Multiple 3-, 4-, and 6-mm punch and excisional biopsies were performed to remove all 16 lesions on the forehead. Histologic examination revealed a collapsed cystic structure in the mid dermis in 10 lesions. The cysts were lined with a squamous epithelium without a granular layer but with an eosinophilic corrugated lining, and the cyst cavity contained scant homogeneous eosinophilic secretion. Mature sebaceous glands were adjacent to the outer portion of the cyst wall. These histologic findings were consistent with SCM (Figure 2).
In 3 lesions, histologic examination revealed a cystic structure lined by a few layers of stratified squamous epithelium in the mid dermis. The cyst cavity contained numerous small vellus hairs and laminated keratin. These histologic findings were consistent with EVHC (Figure 3).
In the other 3 lesions, histologic examination revealed a dilated central cystic cavity filled with laminated keratin in the mid dermis. Multiple small follicles arose from the cysts and showed differentiation toward germinative epithelium. The surrounding stroma was fibrotic and contained a patchy lymphocytic infiltrate. These histologic findings were consistent with trichofolliculomas (Figure 4).
Comment
Characteristics of SCM
Steatocystoma multiplex is an uncommon condition characterized by the formation of asymptomatic, 0.2- to 2-cm, yellow to flesh-colored, soft, mobile papules or nodules on the trunk, extremities, axillae, genitalia, and/or chest. The lesions contain a clear or opaque, oily, milky or yellow, odorless fluid and most commonly are located on the anterior aspect of the chest. The face is not a commonly involved site in this condition. Six cases of a rare facial variant of SCM have been reported,11-16 with lesions limited to the forehead in 3 cases.13-15
In 1937, Mount20 credited Bozellini for describing the first case, though 3 cases reported in the late 1800s probably were SCM.21 In 1899, Pringle22 coined the term steatocystoma multiplex for this condition. It can be sporadic or have an autosomal-dominant inheritance pattern. Steatocystoma multiplex can occur at any age, though lesions develop most frequently in adolescence or young adulthood. There is no sex predilection.
Steatocystoma multiplex with pachyonychia congenita has been reported in a familial case.23 Other findings reported in patients with SCM include ichthyosis, koilonychia, acrokeratosis verruciformis of Hopf and hypertrophic lichen planus, hidradenitis suppurativa, hypotrichosis, multiple keratoacanthomas, and rheumatoid arthritis.12,24-26
Steatocystoma multiplex is a cyst lined by stratified squamous epithelium without a granular layer but with a thick eosinophilic cuticle. Mature sebaceous lobules are closely associated with the cyst wall. Steatocystoma multiplex arises from the sebaceous duct because the lining of the lumen is composed of undulating eosinophilic cuticle.
Characteristics of EVHCs
Eruptive vellus hair cysts, which were first described by Esterly et al,27 can occur at any age but develop most frequently in adolescents or young adults. Sometimes the lesions are congenital or appear in childhood. There is no sex predilection. They can be sporadic or have an autosomal-dominant inheritance pattern.
Eruptive vellus hair cysts are asymptomatic, 1- to 2-mm, smooth, crusted, or umbilicated papules on the chest or arms and legs. Eruptive vellus hair cysts most commonly involve the anterior aspect of the chest. The lesions are flesh-colored to yellow, though they have a slate gray color in darker-skinned individuals. A rare facial variant has been reported in 2 patients of Asian descent.17
Eruptive vellus hair cysts are small cystic structures lined by a stratified squamous epithelium with a granular layer. The cyst cavity contains numerous small vellus hair shafts and laminated keratin. Eruptive vellus hair cysts originate from the infundibulum or less frequently the isthmus or infundibular-isthmic junction of the hair follicle.
Characteristics of Trichofolliculomas
Trichofolliculomas are solitary, 3- to 5-mm, flesh-colored papules that occur on the face. They are highly differentiated, benign, neoplastic proliferations of an actively trichogenic epithelium, with structural components reflecting all portions of the pilosebaceous unit. Trichofolliculomas consist of a central dilated primary follicle contiguous with the surface epidermis embedded in a fibrous stroma. Multiple small secondary follicles with varying degrees of follicular differentiation arise from the primary follicle.
Co-occurrence of Lesions
An association between SCM and EVHC has been recognized.5-10 Steatocystoma multiplex and EVHC have similar clinical features but distinctive histologic features. They also have a similar age of onset, location/appearance of lesions, and mode of inheritance. Steatocystoma multiplex and EVHC can be distinguished by immunohistochemical techniques: SCM shows expression of keratin 10 and keratin 17, whereas EVHCs express only keratin 17.28
Steatocystoma multiplex and EVHC have only rarely been reported to occur together on the trunk. One case of SCM and EVHC occurring on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. Milia, SCM, and EVHC on the face and trunk have been reported in 1 family,4 and facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus was reported in 1 patient.19 Although trichofolliculomas have not been reported to occur with SCM or EVHC, 2 related follicular neoplasms—trichoepitheliomas and trichoblastomas—have been reported to occur in association with SCM on the face and chest and around the ears in 1 case.18
Differential Diagnosis
The clinical differential diagnosis includes multiple epidermoid cysts, dermoid cysts, Gardner syndrome, sebaceous adenomas, Muir-Torre syndrome, syringomas, milia, leiomyomas, lipomas, acneiform folliculitis, multiple familial and nonfamilial trichoepitheliomas, cylindromas, and angiofibromas.3,29
Conclusion
Our patient represents a rare case of simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead. The patient had multiple neoplasms involving differentiation toward various regions of the pilosebaceous unit. This case gives further support to the hypothesis that these benign follicular neoplasms are closely related but are distinct conditions within the spectrum of the same disease process. They represent nevoid malformations of the pilosebaceous unit that can be sporadic or inherited in an autosomal-dominant pattern. Pure types of these lesions may represent one end of the spectrum, but in some patients, there are overlapping features or hybrids of each condition. Several biopsies from patients with multiple lesions should be performed to establish an accurate diagnosis.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Ogawa Y, Nogita T, Kawashima M. The coexistence of eruptive vellus hair cysts and steatocystoma multiplex. J Dermatol. 1992;19:570-571.
- Sanchez Yus E, Requena L. Eruptive vellus hair cyst and steatocystoma multiplex. Am J Dermatopathol. 1990;12:536-537.
- Patrizi A, Neri I, Guerrini V, et al. Persistent milia, steatocystoma multiplex and eruptive vellus hair cysts: variable expression of multiple pilosebaceous cysts within an affected family. Dermatology. 1998;196:392-396.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5, pt 2):876-878.
- Kiene P, Hauschild A, Christophers E. Eruptive vellus hair cysts and steatocystoma multiplex: variants of one entity? Br J Dermatol. 1996;134:365-367.
- Hurlimann AF, Panizzon RG, Burg G. Eruptive vellus hair cyst and steatocystoma multiplex: hybrid cysts. Dermatology. 1996;192:64-66.
- Sexton M, Murdock DK. Eruptive vellus hair cysts: a follicular cyst of the sebaceous duct (sometimes). Am J Dermatopathol. 1989;11:364-368.
- Sanchez-Yus E, Aguilar-Martinez A, Cristobal-Gil MC, et al. Eruptive vellus hair cyst and steatocystoma multiplex: two related conditions? J Cutan Pathol. 1988;15:40-42.
- Ahn SK, Chung J, Lee WS, et al. Hybrid cysts showing alternate combination of eruptive vellus hair cyst, steatocystoma multiplex, and epidermoid cyst, and an association among the three conditions. Am J Dermatopathol. 1996;18:645-649.
- Ahn SK, Hwang SM, Lee SH, et al. Steatocystoma multiplex localized only in the face. Int J Dermatol. 1997;36:372-373.
- Cole LA. Steatocystoma multiplex. Arch Dermatol. 1976;112:1437-1439.
- Hansen KK, Troy JL, Fairley JA. Multiple papules of the scalp and forehead. steatocystoma multiplex (facial papular variant). Arch Dermatol. 1995;131:835-838.
- Nishimura M, Kohda H, Urabe A. Steatocystoma multiplex: a facial popular variant. Arch Dermatol. 1986;122:205-207.
- Requena L, Martin L, Renedo G, et al. A facial variant of steatocystoma multiplex. Cutis. 1993;51:449-452.
- Holmes R, Black MM. Steatocystoma multiplex with unusually prominent cysts on the face. Br J Dermatol. 1980;102:711-713.
- Kumakiri M, Takashima I, Iju M, et al. Eruptive vellus hair cysts: a facial variant. J Am Acad Dermatol. 1982;7:461-467.
- Gianotti R, Cavicchini S, Alessi E. Simultaneous occurrence of multiple trichoblastomas and steatocystoma multiplex. Am J Dermatopathol. 1997;19:294-298.
- Sardana K, Sharma RC, Jain A, et al. Facial steatocystoma multiplex associated with pilar cyst and bilateral preauricular sinus. J Dermatol. 2002;29:157-159.
- Mount LB. Steatocystoma multiplex. Arch Dermatol Syphilol. 1937;36:31-39.
- Dubreuilh W, Auche B. Kystes grassieux sudoripares. Arch Clin de Bordeaux. 1896;5:387-391.
- Pringle JJ. A case of peculiar multiple sebaceous cysts (steatocystoma multiplex). Br J Dermatol. 1899;11:381-88.
- Vineyard WR, Scott RA. Steatocystoma multiplex with pachyonychia congenital: eight cases in four generations. Arch Dermatol. 1961;84:824-827.
- Contreras MA, Costello MJ. Steatocystoma multiplex with embryonal hair formation: case presentation and consideration of pathogenesis. AMA Arch Derm. 1957;76:720-725.
- Sohn D, Chin TC, Fellner MJ. Multiple keratoacanthomas associated with steatocystoma multiplex and rheumatoid arthritis: a case report. Arch Dermatol. 1980;116:913-915.
- Verbov J. Acrokeratosis verruciformis of Hopf with steatocystoma multiplex and hypertrophic lichen planus. Br J Dermatol. 1972;86:91-94.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Tomkova H, Fujimoto W, Arata J. Expression of keratins (K10 and K17) in steatocystoma multiplex, eruptive vellus hair cysts, and epidermoid and trichilemmal cysts. Am J Dermatopathol. 1997;19:250-253.
- Feinstein A, Trau H, Movshovitz M, et al. Steatocystoma multiplex. Cutis. 1983;31:425-427.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Ogawa Y, Nogita T, Kawashima M. The coexistence of eruptive vellus hair cysts and steatocystoma multiplex. J Dermatol. 1992;19:570-571.
- Sanchez Yus E, Requena L. Eruptive vellus hair cyst and steatocystoma multiplex. Am J Dermatopathol. 1990;12:536-537.
- Patrizi A, Neri I, Guerrini V, et al. Persistent milia, steatocystoma multiplex and eruptive vellus hair cysts: variable expression of multiple pilosebaceous cysts within an affected family. Dermatology. 1998;196:392-396.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5, pt 2):876-878.
- Kiene P, Hauschild A, Christophers E. Eruptive vellus hair cysts and steatocystoma multiplex: variants of one entity? Br J Dermatol. 1996;134:365-367.
- Hurlimann AF, Panizzon RG, Burg G. Eruptive vellus hair cyst and steatocystoma multiplex: hybrid cysts. Dermatology. 1996;192:64-66.
- Sexton M, Murdock DK. Eruptive vellus hair cysts: a follicular cyst of the sebaceous duct (sometimes). Am J Dermatopathol. 1989;11:364-368.
- Sanchez-Yus E, Aguilar-Martinez A, Cristobal-Gil MC, et al. Eruptive vellus hair cyst and steatocystoma multiplex: two related conditions? J Cutan Pathol. 1988;15:40-42.
- Ahn SK, Chung J, Lee WS, et al. Hybrid cysts showing alternate combination of eruptive vellus hair cyst, steatocystoma multiplex, and epidermoid cyst, and an association among the three conditions. Am J Dermatopathol. 1996;18:645-649.
- Ahn SK, Hwang SM, Lee SH, et al. Steatocystoma multiplex localized only in the face. Int J Dermatol. 1997;36:372-373.
- Cole LA. Steatocystoma multiplex. Arch Dermatol. 1976;112:1437-1439.
- Hansen KK, Troy JL, Fairley JA. Multiple papules of the scalp and forehead. steatocystoma multiplex (facial papular variant). Arch Dermatol. 1995;131:835-838.
- Nishimura M, Kohda H, Urabe A. Steatocystoma multiplex: a facial popular variant. Arch Dermatol. 1986;122:205-207.
- Requena L, Martin L, Renedo G, et al. A facial variant of steatocystoma multiplex. Cutis. 1993;51:449-452.
- Holmes R, Black MM. Steatocystoma multiplex with unusually prominent cysts on the face. Br J Dermatol. 1980;102:711-713.
- Kumakiri M, Takashima I, Iju M, et al. Eruptive vellus hair cysts: a facial variant. J Am Acad Dermatol. 1982;7:461-467.
- Gianotti R, Cavicchini S, Alessi E. Simultaneous occurrence of multiple trichoblastomas and steatocystoma multiplex. Am J Dermatopathol. 1997;19:294-298.
- Sardana K, Sharma RC, Jain A, et al. Facial steatocystoma multiplex associated with pilar cyst and bilateral preauricular sinus. J Dermatol. 2002;29:157-159.
- Mount LB. Steatocystoma multiplex. Arch Dermatol Syphilol. 1937;36:31-39.
- Dubreuilh W, Auche B. Kystes grassieux sudoripares. Arch Clin de Bordeaux. 1896;5:387-391.
- Pringle JJ. A case of peculiar multiple sebaceous cysts (steatocystoma multiplex). Br J Dermatol. 1899;11:381-88.
- Vineyard WR, Scott RA. Steatocystoma multiplex with pachyonychia congenital: eight cases in four generations. Arch Dermatol. 1961;84:824-827.
- Contreras MA, Costello MJ. Steatocystoma multiplex with embryonal hair formation: case presentation and consideration of pathogenesis. AMA Arch Derm. 1957;76:720-725.
- Sohn D, Chin TC, Fellner MJ. Multiple keratoacanthomas associated with steatocystoma multiplex and rheumatoid arthritis: a case report. Arch Dermatol. 1980;116:913-915.
- Verbov J. Acrokeratosis verruciformis of Hopf with steatocystoma multiplex and hypertrophic lichen planus. Br J Dermatol. 1972;86:91-94.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Tomkova H, Fujimoto W, Arata J. Expression of keratins (K10 and K17) in steatocystoma multiplex, eruptive vellus hair cysts, and epidermoid and trichilemmal cysts. Am J Dermatopathol. 1997;19:250-253.
- Feinstein A, Trau H, Movshovitz M, et al. Steatocystoma multiplex. Cutis. 1983;31:425-427.
Practice Points
- Steatocystoma multiplex (SCM) and eruptive vellus hair cysts (EVHCs) have similar clinical features but distinctive histologic features.
- Milia, pilar cyst, trichoepitheliomas, and trichoblastomas simultaneously developing in association with SCM or EVHC on the face are rare.
- This case supports the hypothesis that these benign follicular neoplasms are related but distinct nevoid malformations of the pilosebaceous unit within the same disease spectrum.
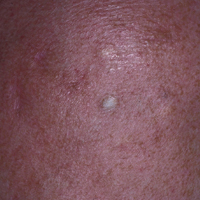
Rowell Syndrome: Targeting a True Definition
Case Report
A 37-year-old woman was admitted to the intensive care unit secondary to the acute development of an erythematous rash with tissue sloughing that involved acral sites and mucosal surfaces. Her medical history was notable for anti-Ro/Sjögren syndrome antigen A (SS-A)–positive lupus erythematosus (LE) with a morphologic semblance to subacute cutaneous LE (SCLE). Prior treatment had included oral corticosteroids. In addition, she reported a concurrent history of acral and mucosal lesions that appeared to flare with her lupus. The nature of these lesions was not clear to the patient or her physicians. Before this particular episode, her primary care physician had attempted to wean her off of the corticosteroids. As she dropped below 20 mg of prednisone daily, new lesions developed. The patient stated that her social situation was poor and that these lesions did seem to develop more frequently during times of physical and emotional stress. She recounted her first episode developing during her second pregnancy. Oral prednisone and over-the-counter calcium with vitamin D were her only reported medications. She denied the use of any other medications, including nonsteroidal anti-inflammatory drugs, acetaminophen, and recent antibiotic therapy.
Dermatology was called in for consultation, and physical examination revealed areas of epidermal sloughing on the hands and feet. Complete clinical exposure of the underlying dermis was noted with remarkable tenderness. These lesions were noted to be in various stages of healing (Figure 1). Figure 2 displays a lesion in early development. The mucosal surfaces of the lips and eyes demonstrated hemorrhagic crusting, and some tissue sloughing was noted on the ears. A widespread erythematous exanthema with fine scaling was noted on the face, neck, chest, back, abdomen, arms, and legs (Figure 3).
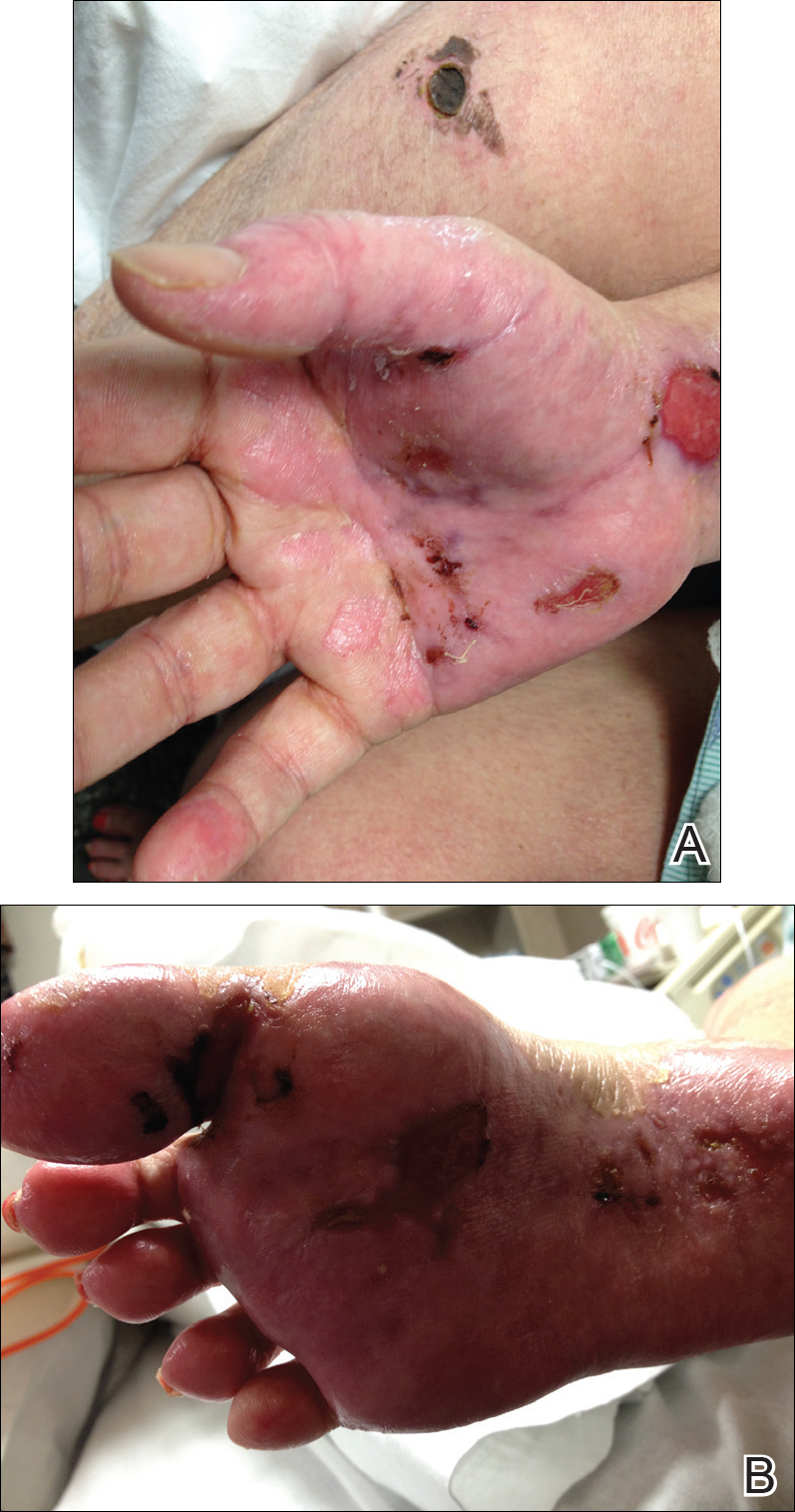
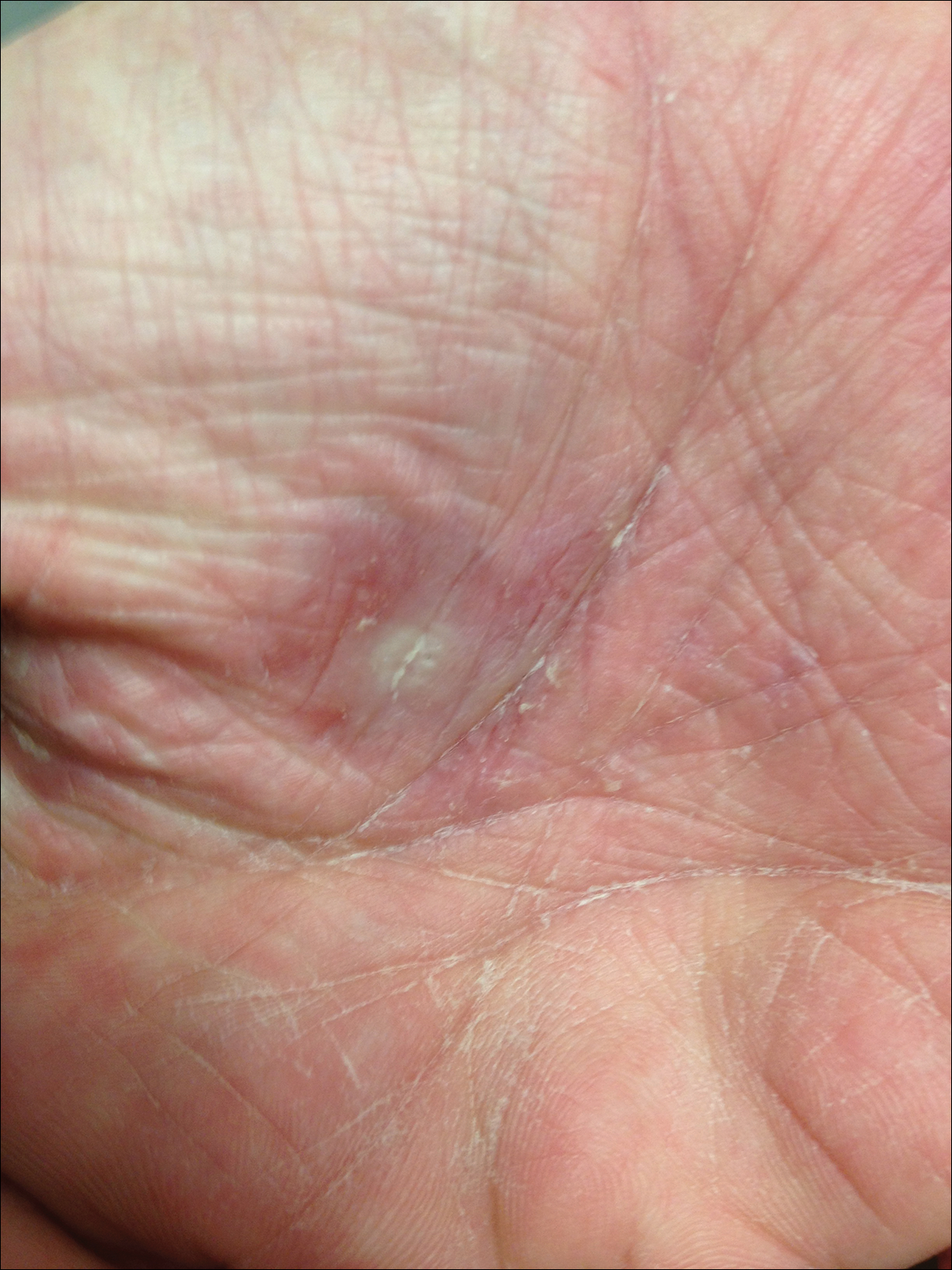
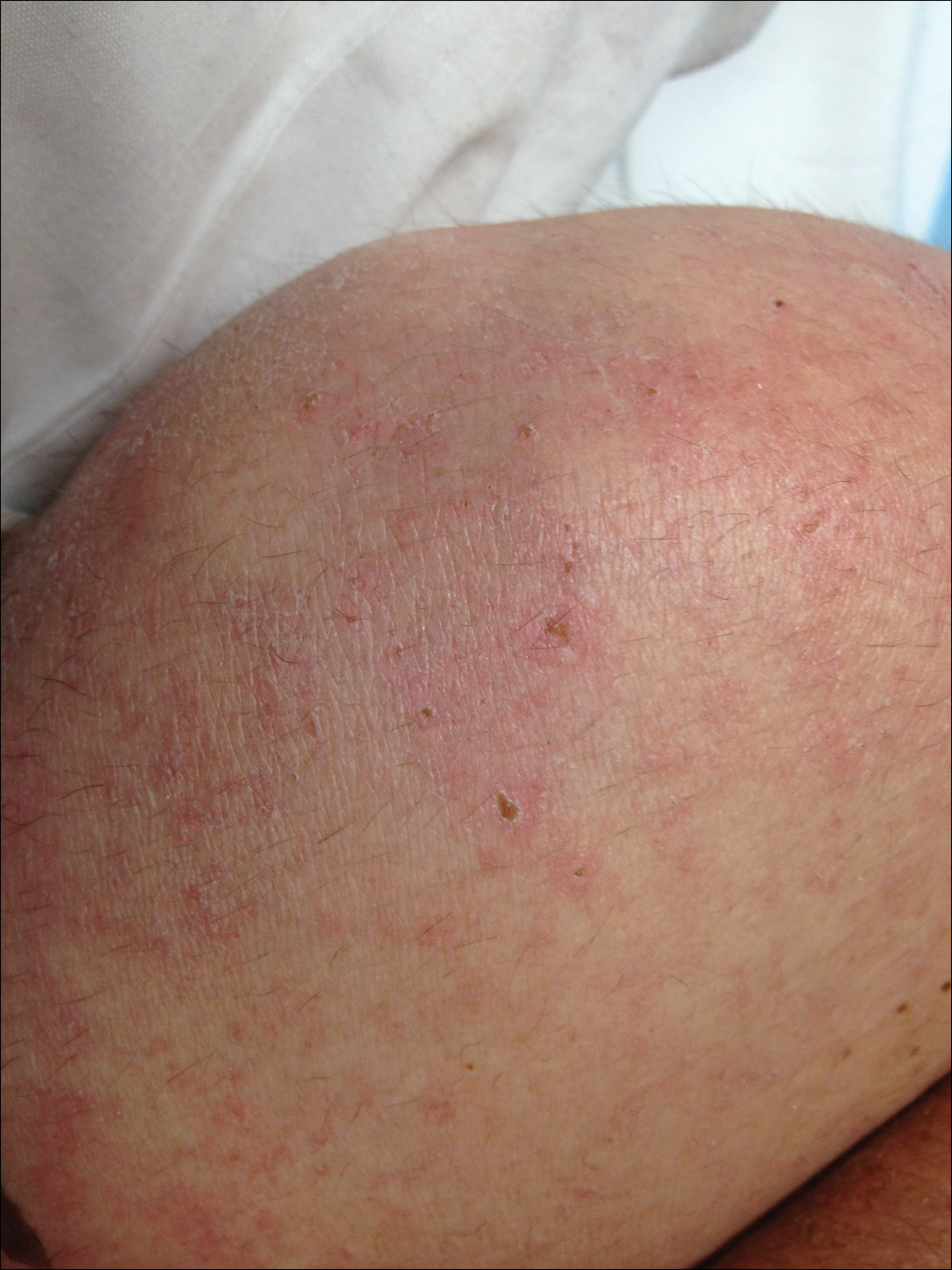
Laboratory evaluation revealed positive antinuclear antibodies (ANAs), anti-Ro/SS-A antibodies, anti-La/Sjögren syndrome antigen B (SS-B) antibodies, and anti–double-stranded DNA. The hemoglobin level was 9.4 g/dL (reference range, 12–15 g/dL) and hematocrit was 28.8% (reference range, 36%–47%). The mean corpuscular hemoglobin level was 32 pg/cell (reference range, 27–31 pg/cell), and the mean corpuscular hemoglobin concentration was 32.5 g/dL (reference range, 30–35 g/dL). Rheumatoid factor (RF) and herpes simplex virus types 1 and 2 IgM were all found to be negative.
A deep shave biopsy obtained from the patient’s right knee revealed an atrophic interface dermatitis associated with a lymphocytic eccrine hidradenitis accompanied by abundant mesenchymal mucin deposition (Figure 4). Direct immunofluorescence (DIF) from the same area demonstrated IgG and IgM along the dermoepidermal junction with some granular deposition. Frozen sections performed on acral lesions demonstrated epidermal necrosis (Figure 5). Direct immunofluorescence of acral lesions was negative. In light of these findings, a diagnosis of Rowell syndrome (RS) was suspected to be the most likely explanation for the presentation.
Intravenous corticosteroids and antibiotics were administered, and over a 2-week hospitalization, the lesions on the feet and hands slowly reepithelialized. Physical therapy was required to aid in ambulation. The patient was discharged on a tapering course of oral prednisone and hydroxychloroquine. After 6 months of therapy with hydroxychloroquine 200 mg twice daily, the patient continued to experience recurrent bouts of acral lesions, and pulse doses of oral prednisone were required. The lesions currently are controlled with azathioprine 50 mg twice daily and prednisone 10 mg by mouth daily.

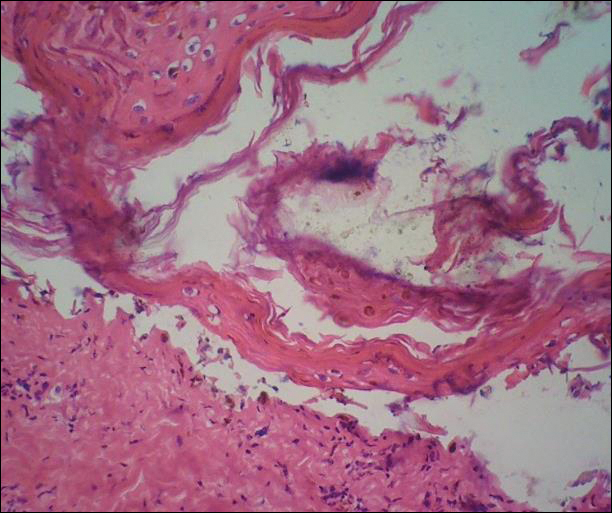
Comment
The 4 prototypical patients identified by Rowell et al1 in 1963 in the first account of the eponymous syndrome were all females with discoid lupus erythematosus (DLE) and perniosis. In addition, they all displayed positive RF and saline extract of human tissue antibodies (analogous to anti-Ro/SS-A and anti-La/SS-B).2 Since then, at least 132 patients with clinical symptoms suspicious of RS have been identified with variations on these original criteria.3 The reported permutations of the lupus component of the disease include cutaneous LE (CLE), bullous systemic LE, necrotic lesions associated with antiphospholipid syndrome, annular/polycyclic SCLE, systemic LE (SLE) without CLE, SLE with lupus nephritis, SLE with pericarditis, SLE with systemic vasculitis, Sjögren syndrome, rheumatoid arthritis, and necrotizing lymphadenitis.2 In addition, variations of the erythema multiforme (EM)–like lesions found in reported cases include changes to their gross appearance (flat vs raised), location (acral or mucosal involvement), and resemblance to other conditions (Stevens-Johnson syndrome or toxic epidermal necrolysis).2,3 From this information alone, it is clear that, as further cases have been chronicled, defining exact criteria for the disease has been challenging.
The essential question concerning the existence of RS hinges on the strength of its distinctiveness: Is it a unique disorder or merely another variant of lupus? Antiga et al2 concluded that it should be characterized as a variant of SCLE. Lee at al4 agreed, stating that “[i]n view of the lack of specific features that distinguish RS from LE, Kuhn et al5 suggested that [RS] is probably not a distinct entity and is now widely considered to be a variant of SCLE.” One of the primary contributors to this conclusion is that the laboratory findings of reported patients with SCLE have more closely mirrored the original cases from Rowell et al’s1 report than those of typical LE. Patients with SCLE have demonstrated positive ANA antibodies in 60% to 80% of cases, positive anti-Ro/SS-A antibodies in 40% to 100% of cases, positive anti-La/SS-B antibodies in 12% to 42% of cases, positive anti–double-stranded DNA in 1.2% to 10% of cases, and positive RF antibodies in 33% of cases.2 An argument could certainly be made to ascribe our patient’s condition to an SCLE variant, as 4 of 5 preceding laboratory findings were found to be positive; however, the majority of reported cases of SCLE have been linked to drugs (ie, hydrochlorothiazide, angiotensin-converting enzyme inhibitors, calcium channel blockers, terbinafine),2 which has not commonly been the attributable etiology of other cases of RS, including the 4 cases reported by Rowell et al.1
In a review of the literature on RS since 2010 in addition to their report of 132 new cases, Torchia et al3 outlined a set of diagnostic standards for the condition consisting of major and minor criteria. According to the authors, if all 4 major and 1 minor criteria are met, the patient meets the standards for true RS. The major criteria include the following: (1) presence of chronic CLE [DLE and/or chilblain]; (2) presence of EM-like lesions [typical or atypical targets]; (3) at least 1 positivity among speckled ANA, anti-Ro/SS-A, and anti-La/SS-B antibodies; and (4) negative DIF on lesional EM-like targetoid lesions. The minor criteria include the following: (1) absence of infectious or pharmacologic triggers; (2) absence of typical EM location (acral and mucosal); and (3) presence of at least 1 additional American College of Rheumatology criterion for diagnosis of SLE8 besides discoid rash and positive ANA antibodies and excluding photosensitivity, malar rash, and oral ulcers. Using these criteria, the patient in our case met the standards for diagnosis of RS.
One area of disagreement that has been encountered in the literature is the exact histologic determination of true RS, specifically related to the microscopic findings of the EM-like lesions. Two cases presented by Modi et al6 were interpreted under the stipulation that true RS must contain histologic LE and histologic EM. Because the EM-appearing lesions revealed LE histology, the cases were concluded to be variants of LE. These cases are similar to our case in that the EM-like lesions in our patient demonstrated LE pathology. Torchia et al,3 as demonstrated in the above criteria, seemed to be less concerned about the histology of the EM-like lesions, only requiring them to show negative DIF.
Conclusion
In the search for answers concerning RS, many unanswered questions remain: Where should the line be drawn in the inclusion of so many variations of both the LE and EM components of the condition? Also, should these elements even be approached as distinct components in the first place? Viewing the majority of RS cases as simply simultaneous LE and EM, Shteyngarts et al7 concluded that “the concomitant occurrence of EM with LE did not change the course, therapy, or prognosis of either disease. SLE and DLE can coexist with EM, but the coexistence does not impart any unusual characteristic to either illness. Rowell’s syndrome is not reproducible, and the immunologic disturbances in such patients are probably coincidental.”
If the condition is a genuine pathological individuality, should we not view the seemingly separate LE and EM as the product of a single underlying biochemical process? These questions and others in the search for a true definition of the disease should continue to be debated. It is clear that further investigation is warranted in the understanding of the underlying mechanism of the pathology.
- Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions: a syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;88:176-180.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577-585.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417-421.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme). Dermatol Online J. 2009;15:1.
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1140.
- Modi GM, Shen A, Mazloom A, et al. Lupus erythematosus masquerading as erythema multiforme: does Rowell syndrome really exist? Dermatol Online J. 2009;15:5.
- Shteyngarts AR, Warner MR, Camisa C. Lupus erythematosus associated with erythema multiforme: does Rowell’s syndrome exist? J Am Acad Dermatol. 1999;40(5 pt 1):773-777.
- Lupus diagnosis. Lupus Research Alliance website. http://lupusresearchinstitute.org/lupus-facts/lupus-diagnosis. Accessed July 11, 2017.
Case Report
A 37-year-old woman was admitted to the intensive care unit secondary to the acute development of an erythematous rash with tissue sloughing that involved acral sites and mucosal surfaces. Her medical history was notable for anti-Ro/Sjögren syndrome antigen A (SS-A)–positive lupus erythematosus (LE) with a morphologic semblance to subacute cutaneous LE (SCLE). Prior treatment had included oral corticosteroids. In addition, she reported a concurrent history of acral and mucosal lesions that appeared to flare with her lupus. The nature of these lesions was not clear to the patient or her physicians. Before this particular episode, her primary care physician had attempted to wean her off of the corticosteroids. As she dropped below 20 mg of prednisone daily, new lesions developed. The patient stated that her social situation was poor and that these lesions did seem to develop more frequently during times of physical and emotional stress. She recounted her first episode developing during her second pregnancy. Oral prednisone and over-the-counter calcium with vitamin D were her only reported medications. She denied the use of any other medications, including nonsteroidal anti-inflammatory drugs, acetaminophen, and recent antibiotic therapy.
Dermatology was called in for consultation, and physical examination revealed areas of epidermal sloughing on the hands and feet. Complete clinical exposure of the underlying dermis was noted with remarkable tenderness. These lesions were noted to be in various stages of healing (Figure 1). Figure 2 displays a lesion in early development. The mucosal surfaces of the lips and eyes demonstrated hemorrhagic crusting, and some tissue sloughing was noted on the ears. A widespread erythematous exanthema with fine scaling was noted on the face, neck, chest, back, abdomen, arms, and legs (Figure 3).
Laboratory evaluation revealed positive antinuclear antibodies (ANAs), anti-Ro/SS-A antibodies, anti-La/Sjögren syndrome antigen B (SS-B) antibodies, and anti–double-stranded DNA. The hemoglobin level was 9.4 g/dL (reference range, 12–15 g/dL) and hematocrit was 28.8% (reference range, 36%–47%). The mean corpuscular hemoglobin level was 32 pg/cell (reference range, 27–31 pg/cell), and the mean corpuscular hemoglobin concentration was 32.5 g/dL (reference range, 30–35 g/dL). Rheumatoid factor (RF) and herpes simplex virus types 1 and 2 IgM were all found to be negative.
A deep shave biopsy obtained from the patient’s right knee revealed an atrophic interface dermatitis associated with a lymphocytic eccrine hidradenitis accompanied by abundant mesenchymal mucin deposition (Figure 4). Direct immunofluorescence (DIF) from the same area demonstrated IgG and IgM along the dermoepidermal junction with some granular deposition. Frozen sections performed on acral lesions demonstrated epidermal necrosis (Figure 5). Direct immunofluorescence of acral lesions was negative. In light of these findings, a diagnosis of Rowell syndrome (RS) was suspected to be the most likely explanation for the presentation.
Intravenous corticosteroids and antibiotics were administered, and over a 2-week hospitalization, the lesions on the feet and hands slowly reepithelialized. Physical therapy was required to aid in ambulation. The patient was discharged on a tapering course of oral prednisone and hydroxychloroquine. After 6 months of therapy with hydroxychloroquine 200 mg twice daily, the patient continued to experience recurrent bouts of acral lesions, and pulse doses of oral prednisone were required. The lesions currently are controlled with azathioprine 50 mg twice daily and prednisone 10 mg by mouth daily.
Comment
The 4 prototypical patients identified by Rowell et al1 in 1963 in the first account of the eponymous syndrome were all females with discoid lupus erythematosus (DLE) and perniosis. In addition, they all displayed positive RF and saline extract of human tissue antibodies (analogous to anti-Ro/SS-A and anti-La/SS-B).2 Since then, at least 132 patients with clinical symptoms suspicious of RS have been identified with variations on these original criteria.3 The reported permutations of the lupus component of the disease include cutaneous LE (CLE), bullous systemic LE, necrotic lesions associated with antiphospholipid syndrome, annular/polycyclic SCLE, systemic LE (SLE) without CLE, SLE with lupus nephritis, SLE with pericarditis, SLE with systemic vasculitis, Sjögren syndrome, rheumatoid arthritis, and necrotizing lymphadenitis.2 In addition, variations of the erythema multiforme (EM)–like lesions found in reported cases include changes to their gross appearance (flat vs raised), location (acral or mucosal involvement), and resemblance to other conditions (Stevens-Johnson syndrome or toxic epidermal necrolysis).2,3 From this information alone, it is clear that, as further cases have been chronicled, defining exact criteria for the disease has been challenging.
The essential question concerning the existence of RS hinges on the strength of its distinctiveness: Is it a unique disorder or merely another variant of lupus? Antiga et al2 concluded that it should be characterized as a variant of SCLE. Lee at al4 agreed, stating that “[i]n view of the lack of specific features that distinguish RS from LE, Kuhn et al5 suggested that [RS] is probably not a distinct entity and is now widely considered to be a variant of SCLE.” One of the primary contributors to this conclusion is that the laboratory findings of reported patients with SCLE have more closely mirrored the original cases from Rowell et al’s1 report than those of typical LE. Patients with SCLE have demonstrated positive ANA antibodies in 60% to 80% of cases, positive anti-Ro/SS-A antibodies in 40% to 100% of cases, positive anti-La/SS-B antibodies in 12% to 42% of cases, positive anti–double-stranded DNA in 1.2% to 10% of cases, and positive RF antibodies in 33% of cases.2 An argument could certainly be made to ascribe our patient’s condition to an SCLE variant, as 4 of 5 preceding laboratory findings were found to be positive; however, the majority of reported cases of SCLE have been linked to drugs (ie, hydrochlorothiazide, angiotensin-converting enzyme inhibitors, calcium channel blockers, terbinafine),2 which has not commonly been the attributable etiology of other cases of RS, including the 4 cases reported by Rowell et al.1
In a review of the literature on RS since 2010 in addition to their report of 132 new cases, Torchia et al3 outlined a set of diagnostic standards for the condition consisting of major and minor criteria. According to the authors, if all 4 major and 1 minor criteria are met, the patient meets the standards for true RS. The major criteria include the following: (1) presence of chronic CLE [DLE and/or chilblain]; (2) presence of EM-like lesions [typical or atypical targets]; (3) at least 1 positivity among speckled ANA, anti-Ro/SS-A, and anti-La/SS-B antibodies; and (4) negative DIF on lesional EM-like targetoid lesions. The minor criteria include the following: (1) absence of infectious or pharmacologic triggers; (2) absence of typical EM location (acral and mucosal); and (3) presence of at least 1 additional American College of Rheumatology criterion for diagnosis of SLE8 besides discoid rash and positive ANA antibodies and excluding photosensitivity, malar rash, and oral ulcers. Using these criteria, the patient in our case met the standards for diagnosis of RS.
One area of disagreement that has been encountered in the literature is the exact histologic determination of true RS, specifically related to the microscopic findings of the EM-like lesions. Two cases presented by Modi et al6 were interpreted under the stipulation that true RS must contain histologic LE and histologic EM. Because the EM-appearing lesions revealed LE histology, the cases were concluded to be variants of LE. These cases are similar to our case in that the EM-like lesions in our patient demonstrated LE pathology. Torchia et al,3 as demonstrated in the above criteria, seemed to be less concerned about the histology of the EM-like lesions, only requiring them to show negative DIF.
Conclusion
In the search for answers concerning RS, many unanswered questions remain: Where should the line be drawn in the inclusion of so many variations of both the LE and EM components of the condition? Also, should these elements even be approached as distinct components in the first place? Viewing the majority of RS cases as simply simultaneous LE and EM, Shteyngarts et al7 concluded that “the concomitant occurrence of EM with LE did not change the course, therapy, or prognosis of either disease. SLE and DLE can coexist with EM, but the coexistence does not impart any unusual characteristic to either illness. Rowell’s syndrome is not reproducible, and the immunologic disturbances in such patients are probably coincidental.”
If the condition is a genuine pathological individuality, should we not view the seemingly separate LE and EM as the product of a single underlying biochemical process? These questions and others in the search for a true definition of the disease should continue to be debated. It is clear that further investigation is warranted in the understanding of the underlying mechanism of the pathology.
Case Report
A 37-year-old woman was admitted to the intensive care unit secondary to the acute development of an erythematous rash with tissue sloughing that involved acral sites and mucosal surfaces. Her medical history was notable for anti-Ro/Sjögren syndrome antigen A (SS-A)–positive lupus erythematosus (LE) with a morphologic semblance to subacute cutaneous LE (SCLE). Prior treatment had included oral corticosteroids. In addition, she reported a concurrent history of acral and mucosal lesions that appeared to flare with her lupus. The nature of these lesions was not clear to the patient or her physicians. Before this particular episode, her primary care physician had attempted to wean her off of the corticosteroids. As she dropped below 20 mg of prednisone daily, new lesions developed. The patient stated that her social situation was poor and that these lesions did seem to develop more frequently during times of physical and emotional stress. She recounted her first episode developing during her second pregnancy. Oral prednisone and over-the-counter calcium with vitamin D were her only reported medications. She denied the use of any other medications, including nonsteroidal anti-inflammatory drugs, acetaminophen, and recent antibiotic therapy.
Dermatology was called in for consultation, and physical examination revealed areas of epidermal sloughing on the hands and feet. Complete clinical exposure of the underlying dermis was noted with remarkable tenderness. These lesions were noted to be in various stages of healing (Figure 1). Figure 2 displays a lesion in early development. The mucosal surfaces of the lips and eyes demonstrated hemorrhagic crusting, and some tissue sloughing was noted on the ears. A widespread erythematous exanthema with fine scaling was noted on the face, neck, chest, back, abdomen, arms, and legs (Figure 3).
Laboratory evaluation revealed positive antinuclear antibodies (ANAs), anti-Ro/SS-A antibodies, anti-La/Sjögren syndrome antigen B (SS-B) antibodies, and anti–double-stranded DNA. The hemoglobin level was 9.4 g/dL (reference range, 12–15 g/dL) and hematocrit was 28.8% (reference range, 36%–47%). The mean corpuscular hemoglobin level was 32 pg/cell (reference range, 27–31 pg/cell), and the mean corpuscular hemoglobin concentration was 32.5 g/dL (reference range, 30–35 g/dL). Rheumatoid factor (RF) and herpes simplex virus types 1 and 2 IgM were all found to be negative.
A deep shave biopsy obtained from the patient’s right knee revealed an atrophic interface dermatitis associated with a lymphocytic eccrine hidradenitis accompanied by abundant mesenchymal mucin deposition (Figure 4). Direct immunofluorescence (DIF) from the same area demonstrated IgG and IgM along the dermoepidermal junction with some granular deposition. Frozen sections performed on acral lesions demonstrated epidermal necrosis (Figure 5). Direct immunofluorescence of acral lesions was negative. In light of these findings, a diagnosis of Rowell syndrome (RS) was suspected to be the most likely explanation for the presentation.
Intravenous corticosteroids and antibiotics were administered, and over a 2-week hospitalization, the lesions on the feet and hands slowly reepithelialized. Physical therapy was required to aid in ambulation. The patient was discharged on a tapering course of oral prednisone and hydroxychloroquine. After 6 months of therapy with hydroxychloroquine 200 mg twice daily, the patient continued to experience recurrent bouts of acral lesions, and pulse doses of oral prednisone were required. The lesions currently are controlled with azathioprine 50 mg twice daily and prednisone 10 mg by mouth daily.
Comment
The 4 prototypical patients identified by Rowell et al1 in 1963 in the first account of the eponymous syndrome were all females with discoid lupus erythematosus (DLE) and perniosis. In addition, they all displayed positive RF and saline extract of human tissue antibodies (analogous to anti-Ro/SS-A and anti-La/SS-B).2 Since then, at least 132 patients with clinical symptoms suspicious of RS have been identified with variations on these original criteria.3 The reported permutations of the lupus component of the disease include cutaneous LE (CLE), bullous systemic LE, necrotic lesions associated with antiphospholipid syndrome, annular/polycyclic SCLE, systemic LE (SLE) without CLE, SLE with lupus nephritis, SLE with pericarditis, SLE with systemic vasculitis, Sjögren syndrome, rheumatoid arthritis, and necrotizing lymphadenitis.2 In addition, variations of the erythema multiforme (EM)–like lesions found in reported cases include changes to their gross appearance (flat vs raised), location (acral or mucosal involvement), and resemblance to other conditions (Stevens-Johnson syndrome or toxic epidermal necrolysis).2,3 From this information alone, it is clear that, as further cases have been chronicled, defining exact criteria for the disease has been challenging.
The essential question concerning the existence of RS hinges on the strength of its distinctiveness: Is it a unique disorder or merely another variant of lupus? Antiga et al2 concluded that it should be characterized as a variant of SCLE. Lee at al4 agreed, stating that “[i]n view of the lack of specific features that distinguish RS from LE, Kuhn et al5 suggested that [RS] is probably not a distinct entity and is now widely considered to be a variant of SCLE.” One of the primary contributors to this conclusion is that the laboratory findings of reported patients with SCLE have more closely mirrored the original cases from Rowell et al’s1 report than those of typical LE. Patients with SCLE have demonstrated positive ANA antibodies in 60% to 80% of cases, positive anti-Ro/SS-A antibodies in 40% to 100% of cases, positive anti-La/SS-B antibodies in 12% to 42% of cases, positive anti–double-stranded DNA in 1.2% to 10% of cases, and positive RF antibodies in 33% of cases.2 An argument could certainly be made to ascribe our patient’s condition to an SCLE variant, as 4 of 5 preceding laboratory findings were found to be positive; however, the majority of reported cases of SCLE have been linked to drugs (ie, hydrochlorothiazide, angiotensin-converting enzyme inhibitors, calcium channel blockers, terbinafine),2 which has not commonly been the attributable etiology of other cases of RS, including the 4 cases reported by Rowell et al.1
In a review of the literature on RS since 2010 in addition to their report of 132 new cases, Torchia et al3 outlined a set of diagnostic standards for the condition consisting of major and minor criteria. According to the authors, if all 4 major and 1 minor criteria are met, the patient meets the standards for true RS. The major criteria include the following: (1) presence of chronic CLE [DLE and/or chilblain]; (2) presence of EM-like lesions [typical or atypical targets]; (3) at least 1 positivity among speckled ANA, anti-Ro/SS-A, and anti-La/SS-B antibodies; and (4) negative DIF on lesional EM-like targetoid lesions. The minor criteria include the following: (1) absence of infectious or pharmacologic triggers; (2) absence of typical EM location (acral and mucosal); and (3) presence of at least 1 additional American College of Rheumatology criterion for diagnosis of SLE8 besides discoid rash and positive ANA antibodies and excluding photosensitivity, malar rash, and oral ulcers. Using these criteria, the patient in our case met the standards for diagnosis of RS.
One area of disagreement that has been encountered in the literature is the exact histologic determination of true RS, specifically related to the microscopic findings of the EM-like lesions. Two cases presented by Modi et al6 were interpreted under the stipulation that true RS must contain histologic LE and histologic EM. Because the EM-appearing lesions revealed LE histology, the cases were concluded to be variants of LE. These cases are similar to our case in that the EM-like lesions in our patient demonstrated LE pathology. Torchia et al,3 as demonstrated in the above criteria, seemed to be less concerned about the histology of the EM-like lesions, only requiring them to show negative DIF.
Conclusion
In the search for answers concerning RS, many unanswered questions remain: Where should the line be drawn in the inclusion of so many variations of both the LE and EM components of the condition? Also, should these elements even be approached as distinct components in the first place? Viewing the majority of RS cases as simply simultaneous LE and EM, Shteyngarts et al7 concluded that “the concomitant occurrence of EM with LE did not change the course, therapy, or prognosis of either disease. SLE and DLE can coexist with EM, but the coexistence does not impart any unusual characteristic to either illness. Rowell’s syndrome is not reproducible, and the immunologic disturbances in such patients are probably coincidental.”
If the condition is a genuine pathological individuality, should we not view the seemingly separate LE and EM as the product of a single underlying biochemical process? These questions and others in the search for a true definition of the disease should continue to be debated. It is clear that further investigation is warranted in the understanding of the underlying mechanism of the pathology.
- Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions: a syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;88:176-180.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577-585.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417-421.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme). Dermatol Online J. 2009;15:1.
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1140.
- Modi GM, Shen A, Mazloom A, et al. Lupus erythematosus masquerading as erythema multiforme: does Rowell syndrome really exist? Dermatol Online J. 2009;15:5.
- Shteyngarts AR, Warner MR, Camisa C. Lupus erythematosus associated with erythema multiforme: does Rowell’s syndrome exist? J Am Acad Dermatol. 1999;40(5 pt 1):773-777.
- Lupus diagnosis. Lupus Research Alliance website. http://lupusresearchinstitute.org/lupus-facts/lupus-diagnosis. Accessed July 11, 2017.
- Rowell NR, Beck JS, Anderson JR. Lupus erythematosus and erythema multiforme-like lesions: a syndrome with characteristic immunological abnormalities. Arch Dermatol. 1963;88:176-180.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577-585.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol. 2012;67:417-421.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme). Dermatol Online J. 2009;15:1.
- Kuhn A, Sticherling M, Bonsmann G. Clinical manifestations of cutaneous lupus erythematosus. J Dtsch Dermatol Ges. 2007;5:1124-1140.
- Modi GM, Shen A, Mazloom A, et al. Lupus erythematosus masquerading as erythema multiforme: does Rowell syndrome really exist? Dermatol Online J. 2009;15:5.
- Shteyngarts AR, Warner MR, Camisa C. Lupus erythematosus associated with erythema multiforme: does Rowell’s syndrome exist? J Am Acad Dermatol. 1999;40(5 pt 1):773-777.
- Lupus diagnosis. Lupus Research Alliance website. http://lupusresearchinstitute.org/lupus-facts/lupus-diagnosis. Accessed July 11, 2017.
Practice Points
- Rowell syndrome (RS) is an often unrecognized unique presentation of lupus erythematosus.
- There have been a variety of historical criteria that have sought to characterize RS.
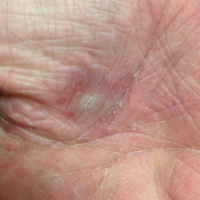
Dermatofibrosarcoma Protuberans
To the Editor:
A 41-year-old man presented with a slowly enlarging, tender, firm lesion on the left hallux of approximately 5 months' duration that initially appeared to be a blister. He reported no history of keloids or trauma to the left foot. On examination, a 3.5-cm, flesh-colored, pedunculated, firm nodule was present on the lateral aspect of the left great hallux (Figure 1). No lymphadenopathy was found. The lesion was diagnosed at that time as a keloid and treated with intralesional steroids without response. The patient was lost to follow-up, and after 5 months he presented again with pain and drainage from the lesion. Acute drainage resolved after antibiotic therapy. A shave biopsy was performed, which revealed findings consistent with a dermatofibrosarcoma protuberans (DFSP). A chest radiograph was unremarkable. Re-excision was performed with negative margins on frozen section but with positive peripheral and deep margins on permanent sections. The patient subsequently underwent amputation of the left great toe and was lost to follow-up after the initial postoperative period.
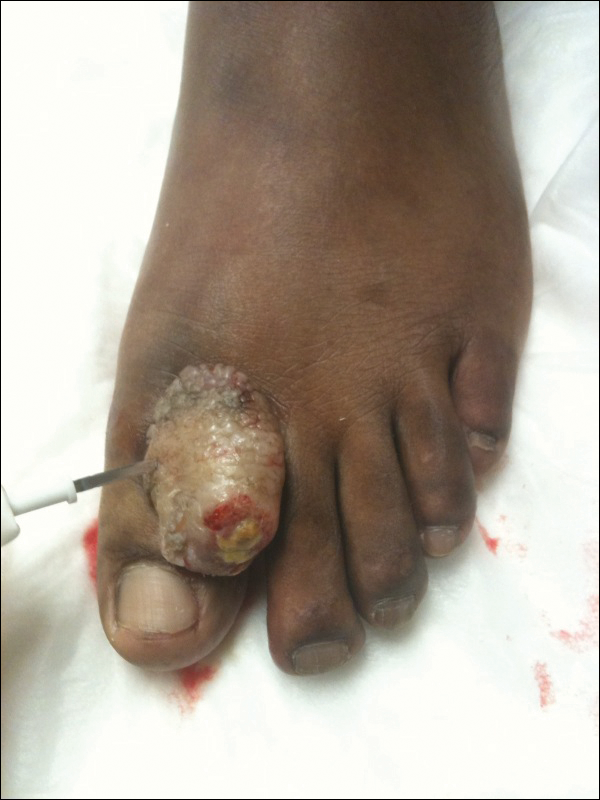
Histopathologic examination demonstrated a polypoid spindle cell tumor that filled the dermis and invaded into the subcutaneous adipose tissue (Figure 2). The spindle cells had tapered nuclei in a honeycomb arrangement with only mild nuclear pleomorphism arranged in fascicles with a herringbone formation. Areas showed a myxoid stroma with abundant mucin (Figure 3). Immunostaining demonstrated cells strongly positive for CD34 and negative for MART (melanoma-associated antigen recognized by T cells), S-100, and smooth muscle actin immunostains.
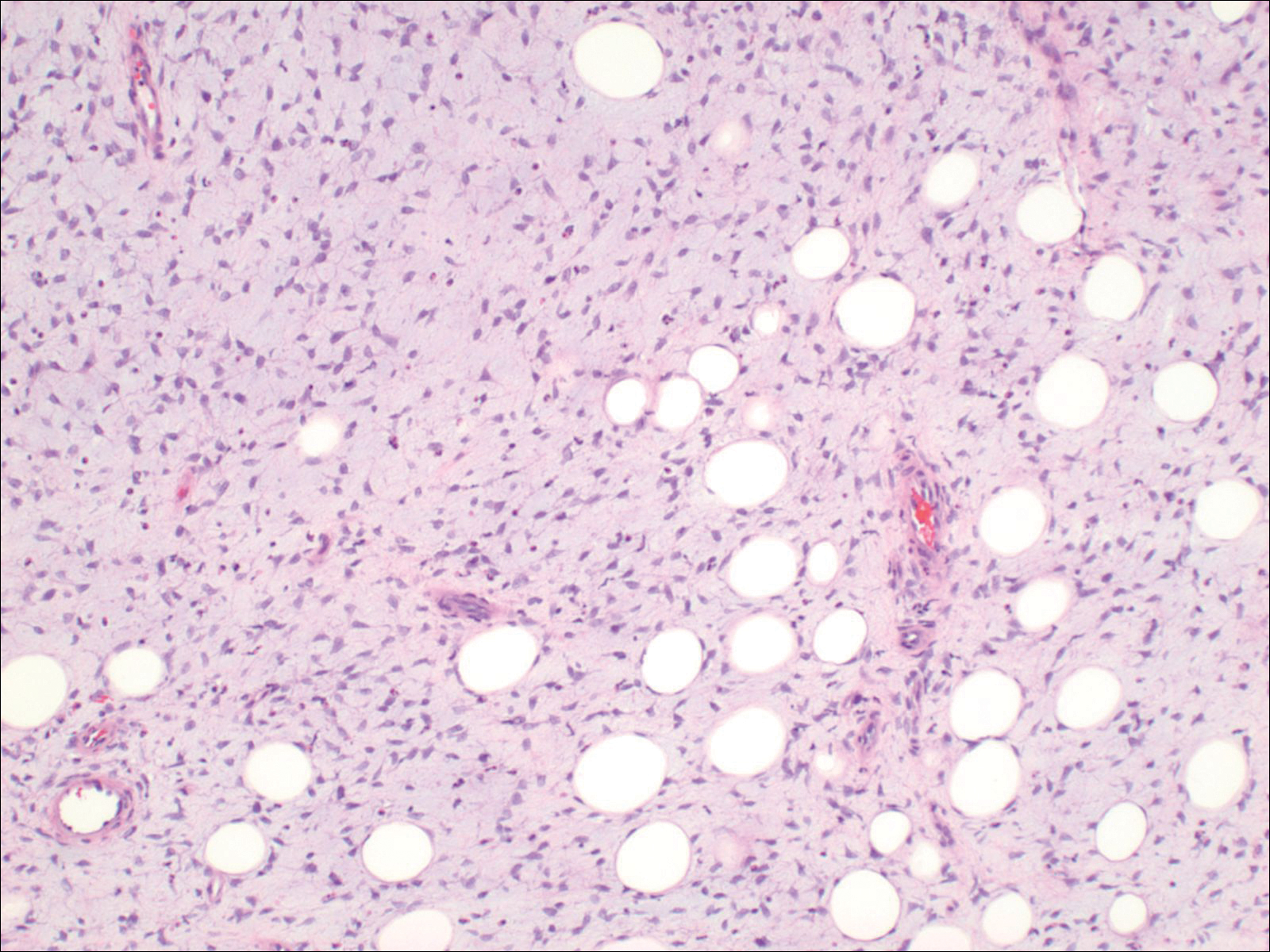
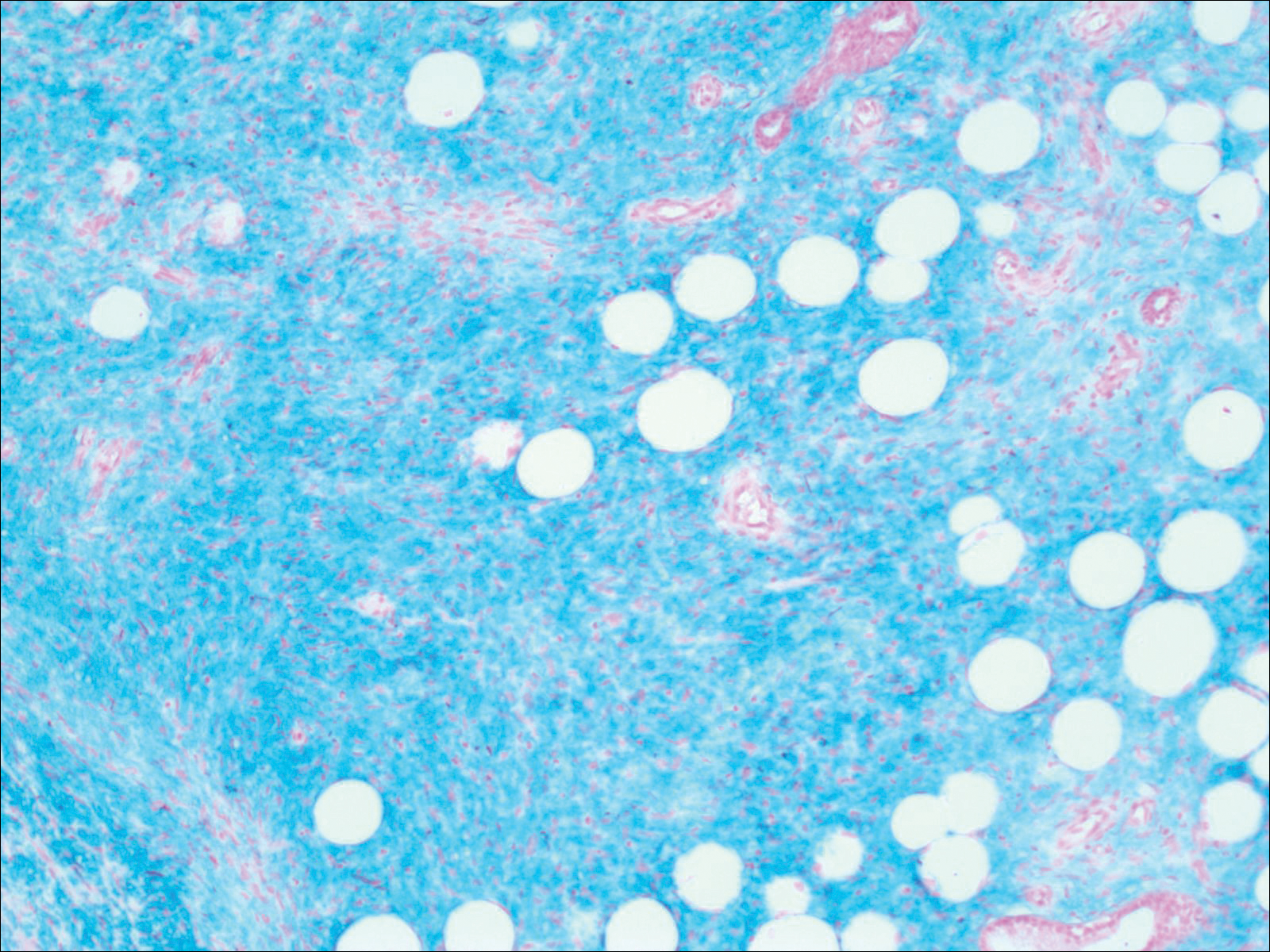
Dermatofibrosarcoma protuberans is a sarcoma that is locally aggressive and tends to recur after surgical excision, though rare cases of metastasis involving the lungs have been reported.12 Dermatofibrosarcoma protuberans usually affects young to middle-aged adults. Acral DFSP is rare in adults, with tumors most commonly occurring on the trunk (50%-60%), proximal extremities (20%-30%), or the head and neck (10%-15%).1,2 A higher rate of acral DFSP has been found in children, which may be due to the increased rate of extremity trauma. Dermatofibrosarcoma protuberans commonly presents as an asymptomatic, slowly growing, indurated plaque that may be flesh colored or hyperpigmented, followed by development of erythematous firm nodules of up to several centimeters.1,3 Dermatofibrosarcoma protuberans may be associated with a purulent exudate or ulceration, and pain may develop as the lesion grows.
Histopathologic evaluation shows an early plaque stage characterized by low cellularity, minimal nuclear atypia, and rare mitotic figures.4 In the nodular stage, the spindle cells are arranged as short fascicles in a storiform arrangement and infiltrate the subcutaneous tissue in a honeycomb pattern with hyperchromatic nuclei and mitotic figures. The nodules may develop myxomatous areas as well as less-differentiated foci with intersecting fascicles in a herringbone pattern. Anti-CD34 antibody immunostaining demonstrates strongly positive spindle cells, while DFSP is negative for stromelysin 3, factor XIIIa, and D2-40, which can help to differentiate DFSP from dermatofibroma.5 The myxoid subtype of DFSP does not differ clinically or prognostically from conventional DFSP, though its recognition can be of use in differentiating other myxoid tumors. Myxoid DFSP is nearly always positive for CD34 and negative for the neural marker S-100 protein.6
Some reports have demonstrated that Mohs micrographic surgery is superior to wide local excision in treatment of DFSP, as it results in fewer local recurrences and metastases.7,8 Because of cytogenic abnormalities such as a reciprocal chromosomal (17;22) translocation or supernumerary ring chromosome derived from t(17;22) that place the PDGFB gene under the control of COL1A1 promoter, imatinib mesylate has been tested in DFSP and resulted in dramatic responses in both adults and children.9,10 Suggested uses of imatinib include metastatic disease and locally invasive disease not suitable for surgical excision as well as a method to debulk tumors prior to resection.11
- Gloster HM Jr. Dermatofibrosarcoma protuberans. J Am Acad Dermatol. 1996;35(3, pt 1):355-374; quiz 375-376.
- Do AN, Goleno K, Geisse JK. Mohs micrographic surgery and partial amputation preserving function and aesthetics in digits: case reports of invasive melanoma and digital dermatofibrosarcoma protuberans. Dermatol Surg. 2006;32:1516-1521.
- Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans: a study of 115 cases. Cancer. 1962;15:717-725.
- Kamino H, Reddy VB, Pui J. Dermatofibrosarcoma protuberans. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. London, England: Elsevier; 2012:1961-1977.
- Cohen PR, Rapini RP, Farhood AI. Dermatofibroma and dermatofibrosarcoma protuberans: differential expression of CD34 and factor XIIIa. Am J Dermatopathol. 1994;16:573-574.
- Llombart B, Serra-Guillén C, Monteagudo C, et al. Dermatofibrosarcoma protuberans: a comprehensive review and update of diagnosis and management. Semin Diagn Pathol. 2013;30:13-28.
- Paradisi A, Abeni D, Rusciani A, et al. Dermatofibrosarcoma protuberans: wide local excision vs. Mohs micrographic surgery. Cancer Treat Rev. 2008;34:728-736.
- Foroozan M, Sei JF, Amini M, et al. Efficacy of Mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans: systematic review. Arch Dermatol. 2012;148:1055-1063.
- Patel KU, Szaebo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- McArthur GA, Demetri GD, van Oosterom A, et al. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol. 2005;23:866-873.
- Rutkowski P, Van Glabbeke M, Rankin CJ, et al; European Organisation for Research and Treatment of Cancer Soft Tissue/Bone Sarcoma Group, Southwest Oncology Group. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials [published online March 1, 2010]. J Clin Oncol. 2010;28:1772-1779.
- Mentzel T, Beham A, Katenkamp D, et al. Fibrosarcomatous ("high-grade") dermatofibrosarcoma protuberans: clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am J Surg Pathol. 1998;22:576-587.
To the Editor:
A 41-year-old man presented with a slowly enlarging, tender, firm lesion on the left hallux of approximately 5 months' duration that initially appeared to be a blister. He reported no history of keloids or trauma to the left foot. On examination, a 3.5-cm, flesh-colored, pedunculated, firm nodule was present on the lateral aspect of the left great hallux (Figure 1). No lymphadenopathy was found. The lesion was diagnosed at that time as a keloid and treated with intralesional steroids without response. The patient was lost to follow-up, and after 5 months he presented again with pain and drainage from the lesion. Acute drainage resolved after antibiotic therapy. A shave biopsy was performed, which revealed findings consistent with a dermatofibrosarcoma protuberans (DFSP). A chest radiograph was unremarkable. Re-excision was performed with negative margins on frozen section but with positive peripheral and deep margins on permanent sections. The patient subsequently underwent amputation of the left great toe and was lost to follow-up after the initial postoperative period.
Histopathologic examination demonstrated a polypoid spindle cell tumor that filled the dermis and invaded into the subcutaneous adipose tissue (Figure 2). The spindle cells had tapered nuclei in a honeycomb arrangement with only mild nuclear pleomorphism arranged in fascicles with a herringbone formation. Areas showed a myxoid stroma with abundant mucin (Figure 3). Immunostaining demonstrated cells strongly positive for CD34 and negative for MART (melanoma-associated antigen recognized by T cells), S-100, and smooth muscle actin immunostains.
Dermatofibrosarcoma protuberans is a sarcoma that is locally aggressive and tends to recur after surgical excision, though rare cases of metastasis involving the lungs have been reported.12 Dermatofibrosarcoma protuberans usually affects young to middle-aged adults. Acral DFSP is rare in adults, with tumors most commonly occurring on the trunk (50%-60%), proximal extremities (20%-30%), or the head and neck (10%-15%).1,2 A higher rate of acral DFSP has been found in children, which may be due to the increased rate of extremity trauma. Dermatofibrosarcoma protuberans commonly presents as an asymptomatic, slowly growing, indurated plaque that may be flesh colored or hyperpigmented, followed by development of erythematous firm nodules of up to several centimeters.1,3 Dermatofibrosarcoma protuberans may be associated with a purulent exudate or ulceration, and pain may develop as the lesion grows.
Histopathologic evaluation shows an early plaque stage characterized by low cellularity, minimal nuclear atypia, and rare mitotic figures.4 In the nodular stage, the spindle cells are arranged as short fascicles in a storiform arrangement and infiltrate the subcutaneous tissue in a honeycomb pattern with hyperchromatic nuclei and mitotic figures. The nodules may develop myxomatous areas as well as less-differentiated foci with intersecting fascicles in a herringbone pattern. Anti-CD34 antibody immunostaining demonstrates strongly positive spindle cells, while DFSP is negative for stromelysin 3, factor XIIIa, and D2-40, which can help to differentiate DFSP from dermatofibroma.5 The myxoid subtype of DFSP does not differ clinically or prognostically from conventional DFSP, though its recognition can be of use in differentiating other myxoid tumors. Myxoid DFSP is nearly always positive for CD34 and negative for the neural marker S-100 protein.6
Some reports have demonstrated that Mohs micrographic surgery is superior to wide local excision in treatment of DFSP, as it results in fewer local recurrences and metastases.7,8 Because of cytogenic abnormalities such as a reciprocal chromosomal (17;22) translocation or supernumerary ring chromosome derived from t(17;22) that place the PDGFB gene under the control of COL1A1 promoter, imatinib mesylate has been tested in DFSP and resulted in dramatic responses in both adults and children.9,10 Suggested uses of imatinib include metastatic disease and locally invasive disease not suitable for surgical excision as well as a method to debulk tumors prior to resection.11
To the Editor:
A 41-year-old man presented with a slowly enlarging, tender, firm lesion on the left hallux of approximately 5 months' duration that initially appeared to be a blister. He reported no history of keloids or trauma to the left foot. On examination, a 3.5-cm, flesh-colored, pedunculated, firm nodule was present on the lateral aspect of the left great hallux (Figure 1). No lymphadenopathy was found. The lesion was diagnosed at that time as a keloid and treated with intralesional steroids without response. The patient was lost to follow-up, and after 5 months he presented again with pain and drainage from the lesion. Acute drainage resolved after antibiotic therapy. A shave biopsy was performed, which revealed findings consistent with a dermatofibrosarcoma protuberans (DFSP). A chest radiograph was unremarkable. Re-excision was performed with negative margins on frozen section but with positive peripheral and deep margins on permanent sections. The patient subsequently underwent amputation of the left great toe and was lost to follow-up after the initial postoperative period.
Histopathologic examination demonstrated a polypoid spindle cell tumor that filled the dermis and invaded into the subcutaneous adipose tissue (Figure 2). The spindle cells had tapered nuclei in a honeycomb arrangement with only mild nuclear pleomorphism arranged in fascicles with a herringbone formation. Areas showed a myxoid stroma with abundant mucin (Figure 3). Immunostaining demonstrated cells strongly positive for CD34 and negative for MART (melanoma-associated antigen recognized by T cells), S-100, and smooth muscle actin immunostains.
Dermatofibrosarcoma protuberans is a sarcoma that is locally aggressive and tends to recur after surgical excision, though rare cases of metastasis involving the lungs have been reported.12 Dermatofibrosarcoma protuberans usually affects young to middle-aged adults. Acral DFSP is rare in adults, with tumors most commonly occurring on the trunk (50%-60%), proximal extremities (20%-30%), or the head and neck (10%-15%).1,2 A higher rate of acral DFSP has been found in children, which may be due to the increased rate of extremity trauma. Dermatofibrosarcoma protuberans commonly presents as an asymptomatic, slowly growing, indurated plaque that may be flesh colored or hyperpigmented, followed by development of erythematous firm nodules of up to several centimeters.1,3 Dermatofibrosarcoma protuberans may be associated with a purulent exudate or ulceration, and pain may develop as the lesion grows.
Histopathologic evaluation shows an early plaque stage characterized by low cellularity, minimal nuclear atypia, and rare mitotic figures.4 In the nodular stage, the spindle cells are arranged as short fascicles in a storiform arrangement and infiltrate the subcutaneous tissue in a honeycomb pattern with hyperchromatic nuclei and mitotic figures. The nodules may develop myxomatous areas as well as less-differentiated foci with intersecting fascicles in a herringbone pattern. Anti-CD34 antibody immunostaining demonstrates strongly positive spindle cells, while DFSP is negative for stromelysin 3, factor XIIIa, and D2-40, which can help to differentiate DFSP from dermatofibroma.5 The myxoid subtype of DFSP does not differ clinically or prognostically from conventional DFSP, though its recognition can be of use in differentiating other myxoid tumors. Myxoid DFSP is nearly always positive for CD34 and negative for the neural marker S-100 protein.6
Some reports have demonstrated that Mohs micrographic surgery is superior to wide local excision in treatment of DFSP, as it results in fewer local recurrences and metastases.7,8 Because of cytogenic abnormalities such as a reciprocal chromosomal (17;22) translocation or supernumerary ring chromosome derived from t(17;22) that place the PDGFB gene under the control of COL1A1 promoter, imatinib mesylate has been tested in DFSP and resulted in dramatic responses in both adults and children.9,10 Suggested uses of imatinib include metastatic disease and locally invasive disease not suitable for surgical excision as well as a method to debulk tumors prior to resection.11
- Gloster HM Jr. Dermatofibrosarcoma protuberans. J Am Acad Dermatol. 1996;35(3, pt 1):355-374; quiz 375-376.
- Do AN, Goleno K, Geisse JK. Mohs micrographic surgery and partial amputation preserving function and aesthetics in digits: case reports of invasive melanoma and digital dermatofibrosarcoma protuberans. Dermatol Surg. 2006;32:1516-1521.
- Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans: a study of 115 cases. Cancer. 1962;15:717-725.
- Kamino H, Reddy VB, Pui J. Dermatofibrosarcoma protuberans. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. London, England: Elsevier; 2012:1961-1977.
- Cohen PR, Rapini RP, Farhood AI. Dermatofibroma and dermatofibrosarcoma protuberans: differential expression of CD34 and factor XIIIa. Am J Dermatopathol. 1994;16:573-574.
- Llombart B, Serra-Guillén C, Monteagudo C, et al. Dermatofibrosarcoma protuberans: a comprehensive review and update of diagnosis and management. Semin Diagn Pathol. 2013;30:13-28.
- Paradisi A, Abeni D, Rusciani A, et al. Dermatofibrosarcoma protuberans: wide local excision vs. Mohs micrographic surgery. Cancer Treat Rev. 2008;34:728-736.
- Foroozan M, Sei JF, Amini M, et al. Efficacy of Mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans: systematic review. Arch Dermatol. 2012;148:1055-1063.
- Patel KU, Szaebo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- McArthur GA, Demetri GD, van Oosterom A, et al. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol. 2005;23:866-873.
- Rutkowski P, Van Glabbeke M, Rankin CJ, et al; European Organisation for Research and Treatment of Cancer Soft Tissue/Bone Sarcoma Group, Southwest Oncology Group. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials [published online March 1, 2010]. J Clin Oncol. 2010;28:1772-1779.
- Mentzel T, Beham A, Katenkamp D, et al. Fibrosarcomatous ("high-grade") dermatofibrosarcoma protuberans: clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am J Surg Pathol. 1998;22:576-587.
- Gloster HM Jr. Dermatofibrosarcoma protuberans. J Am Acad Dermatol. 1996;35(3, pt 1):355-374; quiz 375-376.
- Do AN, Goleno K, Geisse JK. Mohs micrographic surgery and partial amputation preserving function and aesthetics in digits: case reports of invasive melanoma and digital dermatofibrosarcoma protuberans. Dermatol Surg. 2006;32:1516-1521.
- Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans: a study of 115 cases. Cancer. 1962;15:717-725.
- Kamino H, Reddy VB, Pui J. Dermatofibrosarcoma protuberans. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 3rd ed. London, England: Elsevier; 2012:1961-1977.
- Cohen PR, Rapini RP, Farhood AI. Dermatofibroma and dermatofibrosarcoma protuberans: differential expression of CD34 and factor XIIIa. Am J Dermatopathol. 1994;16:573-574.
- Llombart B, Serra-Guillén C, Monteagudo C, et al. Dermatofibrosarcoma protuberans: a comprehensive review and update of diagnosis and management. Semin Diagn Pathol. 2013;30:13-28.
- Paradisi A, Abeni D, Rusciani A, et al. Dermatofibrosarcoma protuberans: wide local excision vs. Mohs micrographic surgery. Cancer Treat Rev. 2008;34:728-736.
- Foroozan M, Sei JF, Amini M, et al. Efficacy of Mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans: systematic review. Arch Dermatol. 2012;148:1055-1063.
- Patel KU, Szaebo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- McArthur GA, Demetri GD, van Oosterom A, et al. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol. 2005;23:866-873.
- Rutkowski P, Van Glabbeke M, Rankin CJ, et al; European Organisation for Research and Treatment of Cancer Soft Tissue/Bone Sarcoma Group, Southwest Oncology Group. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials [published online March 1, 2010]. J Clin Oncol. 2010;28:1772-1779.
- Mentzel T, Beham A, Katenkamp D, et al. Fibrosarcomatous ("high-grade") dermatofibrosarcoma protuberans: clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am J Surg Pathol. 1998;22:576-587.
Practice Points
- Consider dermatofibrosarcoma protuberans for a keloidlike enlarging lesion when there is no history of trauma or prior keloid formation.
- Treatments such as Mohs micrographic surgery or oral imatinib mesylate can provide lower recurrence rates in appropriate patients as stand-alone or adjuvant therapy.
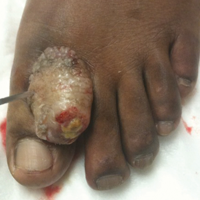
Temporal Triangular Alopecia Acquired in Adulthood
To the Editor:
Temporal triangular alopecia (TTA), a condition first described by Sabouraud1 in 1905, is a circumscribed nonscarring form of alopecia. Also referred to as congenital triangular alopecia, TTA presents as a triangular or lancet-shaped area of hair loss involving the frontotemporal hairline. Temporal triangular alopecia is characterized histologically by a normal number of miniaturized hair follicles without notable inflammation.2 Although the majority of cases arise between birth and 9 years of age,3,4 rare cases of adult-onset TTA also have been reported.5,6 Adult-onset cases can cause notable diagnostic confusion and inappropriate treatment, as reported in our patient.
A 25-year-old woman with a history of Hashimoto thyroiditis presented with hair loss affecting the right temporal scalp of 3 years' duration that was first noticed by her husband. The lesion was an asymptomatic, 6×8-cm, roughly lancet-shaped patch of alopecia located on the right temporal scalp, bordering on the frontal hairline (Figure 1). Centrally, the patch appeared almost hairless with a few retained terminal hairs. The frontal hairline was thinned but still present. There was no scaling or erythema, and fine vellus hairs and a few isolated terminal hairs covered the area. The corresponding skin on the contralateral temporal scalp showed normal hair density. The patient insisted that she had normal hair at the affected area until 22 years of age, and she denied a history of trauma or tight hairstyles. Initially diagnosed with alopecia areata by her primary care provider, the patient was treated with topical corticosteroids for 6 months without benefit. She was subsequently referred to a dermatologist who again offered a diagnosis of alopecia areata and treated the lesions with 2 intralesional corticosteroid injections without benefit. No biopsies of the affected area were performed, and the patient was given a trial of topical minoxidil.
The patient consulted a new primary care provider and was diagnosed with scarring alopecia. She was referred to our dermatology department for further treatment. An initial biopsy at the edge of the affected area was interpreted as normal, but after failing additional intralesional corticosteroid injections, she was referred to our hair clinic where another biopsy was performed in the central portion of the lesion. A 4-mm diameter punch biopsy specimen revealed a normal epidermis and dermis; however, in the lower dermis only a single terminal follicle was seen (Figure 2). Sections through the upper dermis (Figure 3) showed that the total number of hairs was normal or nearly normal with at least 22 follicles, but most were vellus and indeterminate hairs with only a single terminal hair. The dermal architecture was otherwise normal. Given the clinical and histologic findings, a diagnosis of TTA was made. Subsequent to the diagnosis, the patient did not pursue any additional treatment options and preferred to style her hair so that the area of TTA remained covered.
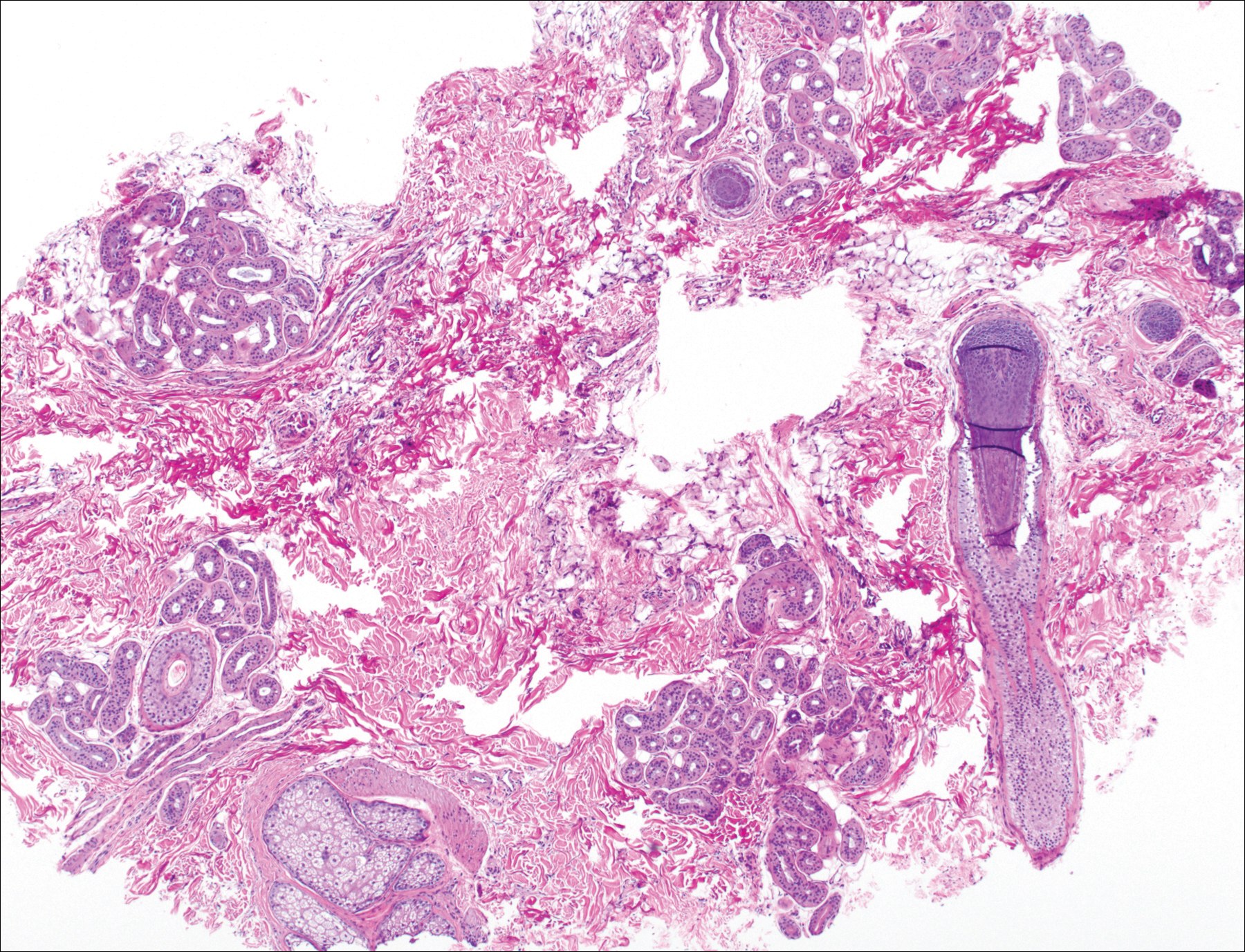
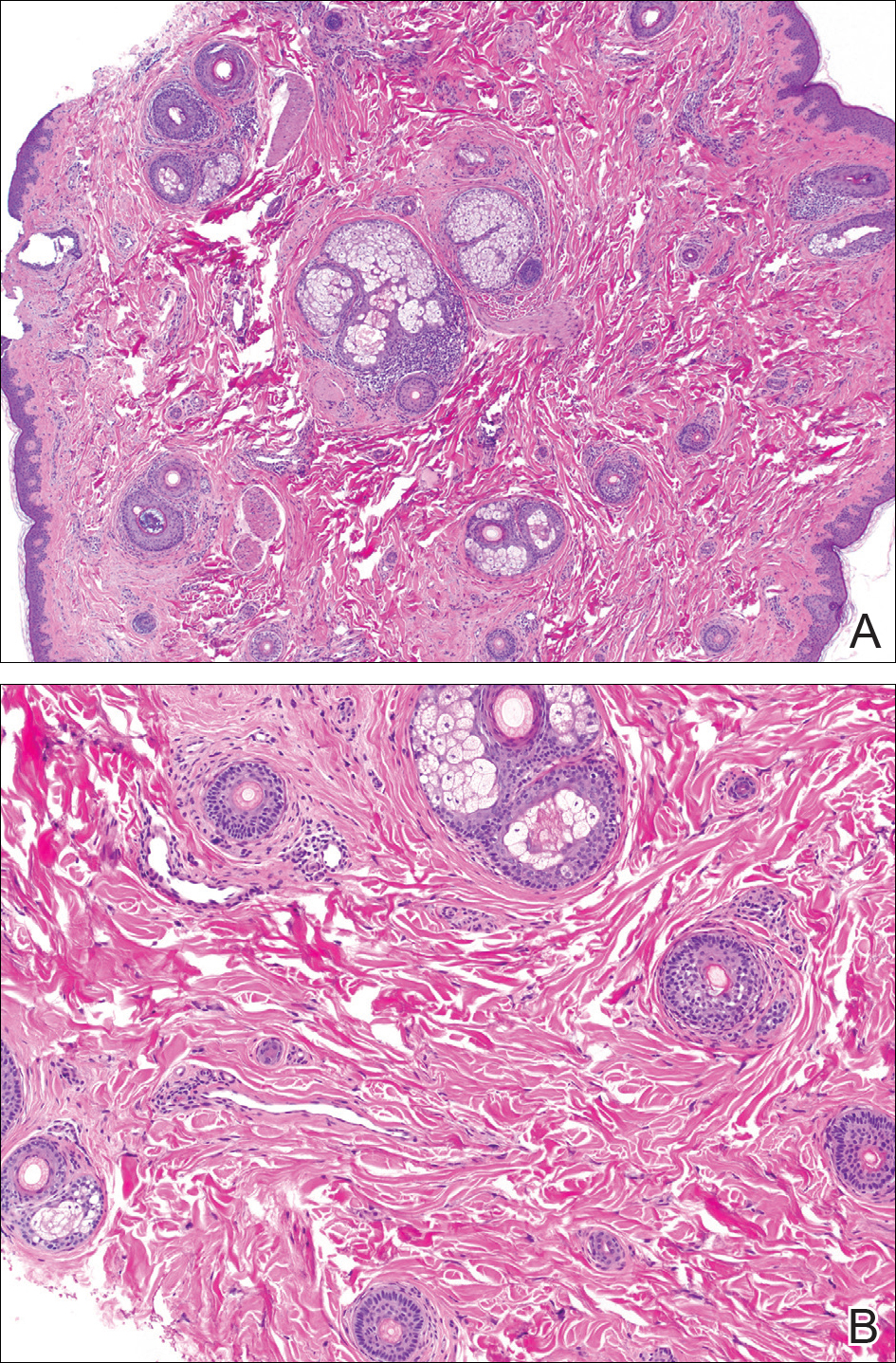
The differential diagnosis in adults presenting with a patch of localized alopecia includes alopecia areata, trichotillomania, pressure-induced alopecia, traction alopecia, lichen planopilaris, discoid lupus erythematosus, and rarely TTA. Temporal triangular alopecia is a fairly common, if underreported, nonscarring form of alopecia that mainly affects young children. A PubMed search of articles indexed for MEDLINE using the terms temporal triangular alopecia or congenital triangular alopecia or triangular alopecia documented only 76 cases of TTA including our own, with the majority of patients diagnosed before 9 years of age. Only 2 cases of adult-onset TTA have been reported,5,6 possibly leading to misdiagnosis of adult patients who present with similar areas of hair loss. As with some prior cases of TTA,5,7 our patient was misdiagnosed with alopecia areata and scarring alopecia, both treated unsuccessfully before a diagnosis of TTA was considered. Clues to the diagnosis included the location, the lack of change in size and shape, the lack of response to intralesional corticosteroids, and the presence of numerous vellus hairs on the surface. A biopsy of the visibly hairless zone was confirmatory. The normal or nearly normal number of miniaturized hairs in specimens of TTA suggest that topical minoxidil therapy (eg, 5% solution twice daily for at least 6 months) might be useful, but the authors have tried it on a few other patients with clinically typical TTA without discernible benefit. When lesions are small, excision provides a fast and permanent solution to the problem, albeit with the usual risks of minor surgery.
- Sabouraud RJA. Manuel Élémentaire de Dermatologie Topographique Régionale. Paris, France: Masson & Cie; 1905:197.
- Trakimas C, Sperling LC, Skelton HG 3rd, et al. Clinical and histologic findings in temporal triangular alopecia. J Am Acad Dermatol. 1994;31:205-209.
- Yamazaki M, Irisawa R, Tsuboi R. Temporal triangular alopecia and a review of 52 past cases. J Dermatol. 2010;37:360-362.
- Sarifakioglu E, Yilmaz AE, Gorpelioglu C, et al. Prevalence of scalp disorders and hair loss in children. Cutis. 2012;90:225-229.
- Trakimas CA, Sperling LC. Temporal triangular alopecia acquired in adulthood. J Am Acad Dermatol. 1999;40:842-844.
- Akan IM, Yildirim S, Avci G, et al. Bilateral temporal triangular alopecia acquired in adulthood. Plast Reconstr Surg. 2001;107:1616-1617.
- Gupta LK, Khare AK, Garg A, et al. Congenital triangular alopecia--a close mimicker of alopecia areata. Int J Trichology. 2011;3:40-41.
To the Editor:
Temporal triangular alopecia (TTA), a condition first described by Sabouraud1 in 1905, is a circumscribed nonscarring form of alopecia. Also referred to as congenital triangular alopecia, TTA presents as a triangular or lancet-shaped area of hair loss involving the frontotemporal hairline. Temporal triangular alopecia is characterized histologically by a normal number of miniaturized hair follicles without notable inflammation.2 Although the majority of cases arise between birth and 9 years of age,3,4 rare cases of adult-onset TTA also have been reported.5,6 Adult-onset cases can cause notable diagnostic confusion and inappropriate treatment, as reported in our patient.
A 25-year-old woman with a history of Hashimoto thyroiditis presented with hair loss affecting the right temporal scalp of 3 years' duration that was first noticed by her husband. The lesion was an asymptomatic, 6×8-cm, roughly lancet-shaped patch of alopecia located on the right temporal scalp, bordering on the frontal hairline (Figure 1). Centrally, the patch appeared almost hairless with a few retained terminal hairs. The frontal hairline was thinned but still present. There was no scaling or erythema, and fine vellus hairs and a few isolated terminal hairs covered the area. The corresponding skin on the contralateral temporal scalp showed normal hair density. The patient insisted that she had normal hair at the affected area until 22 years of age, and she denied a history of trauma or tight hairstyles. Initially diagnosed with alopecia areata by her primary care provider, the patient was treated with topical corticosteroids for 6 months without benefit. She was subsequently referred to a dermatologist who again offered a diagnosis of alopecia areata and treated the lesions with 2 intralesional corticosteroid injections without benefit. No biopsies of the affected area were performed, and the patient was given a trial of topical minoxidil.
The patient consulted a new primary care provider and was diagnosed with scarring alopecia. She was referred to our dermatology department for further treatment. An initial biopsy at the edge of the affected area was interpreted as normal, but after failing additional intralesional corticosteroid injections, she was referred to our hair clinic where another biopsy was performed in the central portion of the lesion. A 4-mm diameter punch biopsy specimen revealed a normal epidermis and dermis; however, in the lower dermis only a single terminal follicle was seen (Figure 2). Sections through the upper dermis (Figure 3) showed that the total number of hairs was normal or nearly normal with at least 22 follicles, but most were vellus and indeterminate hairs with only a single terminal hair. The dermal architecture was otherwise normal. Given the clinical and histologic findings, a diagnosis of TTA was made. Subsequent to the diagnosis, the patient did not pursue any additional treatment options and preferred to style her hair so that the area of TTA remained covered.
The differential diagnosis in adults presenting with a patch of localized alopecia includes alopecia areata, trichotillomania, pressure-induced alopecia, traction alopecia, lichen planopilaris, discoid lupus erythematosus, and rarely TTA. Temporal triangular alopecia is a fairly common, if underreported, nonscarring form of alopecia that mainly affects young children. A PubMed search of articles indexed for MEDLINE using the terms temporal triangular alopecia or congenital triangular alopecia or triangular alopecia documented only 76 cases of TTA including our own, with the majority of patients diagnosed before 9 years of age. Only 2 cases of adult-onset TTA have been reported,5,6 possibly leading to misdiagnosis of adult patients who present with similar areas of hair loss. As with some prior cases of TTA,5,7 our patient was misdiagnosed with alopecia areata and scarring alopecia, both treated unsuccessfully before a diagnosis of TTA was considered. Clues to the diagnosis included the location, the lack of change in size and shape, the lack of response to intralesional corticosteroids, and the presence of numerous vellus hairs on the surface. A biopsy of the visibly hairless zone was confirmatory. The normal or nearly normal number of miniaturized hairs in specimens of TTA suggest that topical minoxidil therapy (eg, 5% solution twice daily for at least 6 months) might be useful, but the authors have tried it on a few other patients with clinically typical TTA without discernible benefit. When lesions are small, excision provides a fast and permanent solution to the problem, albeit with the usual risks of minor surgery.
To the Editor:
Temporal triangular alopecia (TTA), a condition first described by Sabouraud1 in 1905, is a circumscribed nonscarring form of alopecia. Also referred to as congenital triangular alopecia, TTA presents as a triangular or lancet-shaped area of hair loss involving the frontotemporal hairline. Temporal triangular alopecia is characterized histologically by a normal number of miniaturized hair follicles without notable inflammation.2 Although the majority of cases arise between birth and 9 years of age,3,4 rare cases of adult-onset TTA also have been reported.5,6 Adult-onset cases can cause notable diagnostic confusion and inappropriate treatment, as reported in our patient.
A 25-year-old woman with a history of Hashimoto thyroiditis presented with hair loss affecting the right temporal scalp of 3 years' duration that was first noticed by her husband. The lesion was an asymptomatic, 6×8-cm, roughly lancet-shaped patch of alopecia located on the right temporal scalp, bordering on the frontal hairline (Figure 1). Centrally, the patch appeared almost hairless with a few retained terminal hairs. The frontal hairline was thinned but still present. There was no scaling or erythema, and fine vellus hairs and a few isolated terminal hairs covered the area. The corresponding skin on the contralateral temporal scalp showed normal hair density. The patient insisted that she had normal hair at the affected area until 22 years of age, and she denied a history of trauma or tight hairstyles. Initially diagnosed with alopecia areata by her primary care provider, the patient was treated with topical corticosteroids for 6 months without benefit. She was subsequently referred to a dermatologist who again offered a diagnosis of alopecia areata and treated the lesions with 2 intralesional corticosteroid injections without benefit. No biopsies of the affected area were performed, and the patient was given a trial of topical minoxidil.
The patient consulted a new primary care provider and was diagnosed with scarring alopecia. She was referred to our dermatology department for further treatment. An initial biopsy at the edge of the affected area was interpreted as normal, but after failing additional intralesional corticosteroid injections, she was referred to our hair clinic where another biopsy was performed in the central portion of the lesion. A 4-mm diameter punch biopsy specimen revealed a normal epidermis and dermis; however, in the lower dermis only a single terminal follicle was seen (Figure 2). Sections through the upper dermis (Figure 3) showed that the total number of hairs was normal or nearly normal with at least 22 follicles, but most were vellus and indeterminate hairs with only a single terminal hair. The dermal architecture was otherwise normal. Given the clinical and histologic findings, a diagnosis of TTA was made. Subsequent to the diagnosis, the patient did not pursue any additional treatment options and preferred to style her hair so that the area of TTA remained covered.
The differential diagnosis in adults presenting with a patch of localized alopecia includes alopecia areata, trichotillomania, pressure-induced alopecia, traction alopecia, lichen planopilaris, discoid lupus erythematosus, and rarely TTA. Temporal triangular alopecia is a fairly common, if underreported, nonscarring form of alopecia that mainly affects young children. A PubMed search of articles indexed for MEDLINE using the terms temporal triangular alopecia or congenital triangular alopecia or triangular alopecia documented only 76 cases of TTA including our own, with the majority of patients diagnosed before 9 years of age. Only 2 cases of adult-onset TTA have been reported,5,6 possibly leading to misdiagnosis of adult patients who present with similar areas of hair loss. As with some prior cases of TTA,5,7 our patient was misdiagnosed with alopecia areata and scarring alopecia, both treated unsuccessfully before a diagnosis of TTA was considered. Clues to the diagnosis included the location, the lack of change in size and shape, the lack of response to intralesional corticosteroids, and the presence of numerous vellus hairs on the surface. A biopsy of the visibly hairless zone was confirmatory. The normal or nearly normal number of miniaturized hairs in specimens of TTA suggest that topical minoxidil therapy (eg, 5% solution twice daily for at least 6 months) might be useful, but the authors have tried it on a few other patients with clinically typical TTA without discernible benefit. When lesions are small, excision provides a fast and permanent solution to the problem, albeit with the usual risks of minor surgery.
- Sabouraud RJA. Manuel Élémentaire de Dermatologie Topographique Régionale. Paris, France: Masson & Cie; 1905:197.
- Trakimas C, Sperling LC, Skelton HG 3rd, et al. Clinical and histologic findings in temporal triangular alopecia. J Am Acad Dermatol. 1994;31:205-209.
- Yamazaki M, Irisawa R, Tsuboi R. Temporal triangular alopecia and a review of 52 past cases. J Dermatol. 2010;37:360-362.
- Sarifakioglu E, Yilmaz AE, Gorpelioglu C, et al. Prevalence of scalp disorders and hair loss in children. Cutis. 2012;90:225-229.
- Trakimas CA, Sperling LC. Temporal triangular alopecia acquired in adulthood. J Am Acad Dermatol. 1999;40:842-844.
- Akan IM, Yildirim S, Avci G, et al. Bilateral temporal triangular alopecia acquired in adulthood. Plast Reconstr Surg. 2001;107:1616-1617.
- Gupta LK, Khare AK, Garg A, et al. Congenital triangular alopecia--a close mimicker of alopecia areata. Int J Trichology. 2011;3:40-41.
- Sabouraud RJA. Manuel Élémentaire de Dermatologie Topographique Régionale. Paris, France: Masson & Cie; 1905:197.
- Trakimas C, Sperling LC, Skelton HG 3rd, et al. Clinical and histologic findings in temporal triangular alopecia. J Am Acad Dermatol. 1994;31:205-209.
- Yamazaki M, Irisawa R, Tsuboi R. Temporal triangular alopecia and a review of 52 past cases. J Dermatol. 2010;37:360-362.
- Sarifakioglu E, Yilmaz AE, Gorpelioglu C, et al. Prevalence of scalp disorders and hair loss in children. Cutis. 2012;90:225-229.
- Trakimas CA, Sperling LC. Temporal triangular alopecia acquired in adulthood. J Am Acad Dermatol. 1999;40:842-844.
- Akan IM, Yildirim S, Avci G, et al. Bilateral temporal triangular alopecia acquired in adulthood. Plast Reconstr Surg. 2001;107:1616-1617.
- Gupta LK, Khare AK, Garg A, et al. Congenital triangular alopecia--a close mimicker of alopecia areata. Int J Trichology. 2011;3:40-41.
Practice Points
- Temporal triangular alopecia (TTA) in adults often is confused with alopecia areata.
- An acquired, persistent, unchanging, circumscribed hairless spot in an adult that does not respond to intralesional corticosteroids may represent TTA.
- Hair miniaturization without peribulbar inflammation is consistent with a diagnosis of TTA.
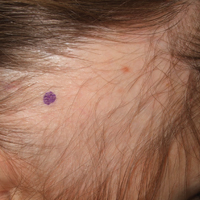
Eroded Plaque on the Lower Lip
The Diagnosis: Squamous Cell Carcinoma
The initial clinical presentation suggested a diagnosis of herpes simplex labialis. The patient reported no response to topical acyclovir, and because the plaque persisted, a biopsy was performed. Pathology demonstrated squamous cell carcinoma (SCC) that was moderately well differentiated and invasive (Figure).
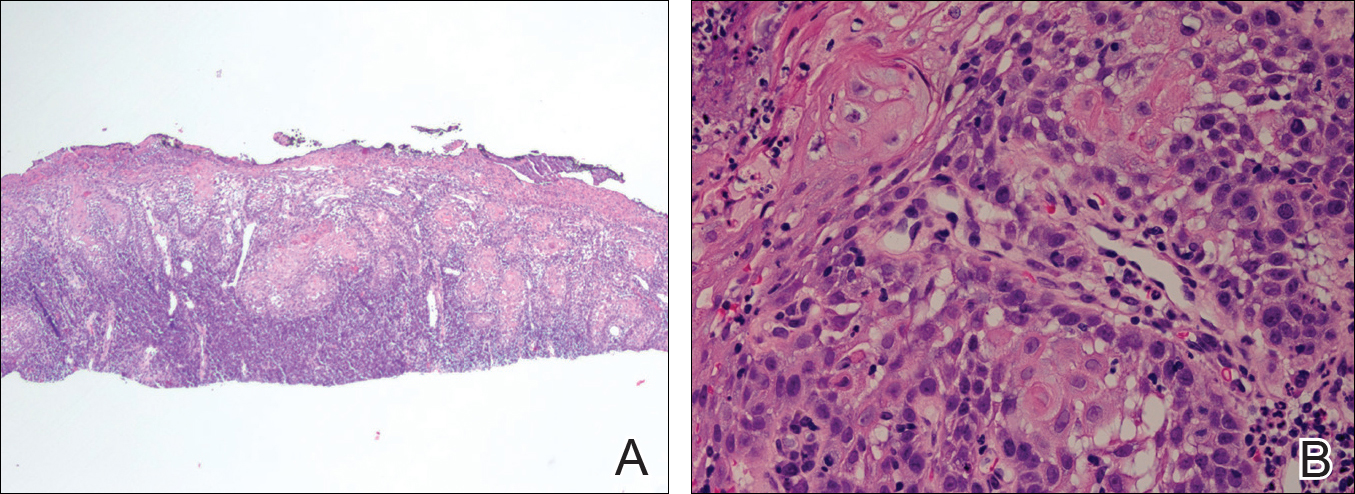
Approximately 38% of all oral SCCs in the United States occur on the lower lip and typically are solar-related cancers developing within the epidermis.1 Oral lesions initially may be asymptomatic and may not be of concern to the patient; however, it is important to recognize SCC early, as invasive lesions have the potential to metastasize. Some factors that increase the chance for the development of metastases include tumor size larger than 2 cm; location on the ear, lip, or other sites on the head and neck; and history of prior unsuccessful treatment.2 Any solitary ulcer, lump, wound, or lesion that will not heal and persists for more than 3 weeks should be regarded as cancer until proven otherwise. Although few oral SCCs are detected by clinicians at an early stage, diagnostic aids such as vital staining and molecular markers in tissues and saliva may be implemented.3 Toluidine blue is a simple, fast, and inexpensive technique that stains the nuclear material of malignant lesions, but not normal mucosa, and may be a worthwhile diagnostic adjunct to clinical inspection.4
Our patient presented with a lesion that clinically looked herpetic, though he reported no prodromal signs of tingling, burning, or pain before the occurrence of the lesion. Due to the persistence of the lesion and lack of response to treatment, a biopsy was indicated. The differential diagnoses include aphthous ulcers, which may occasionally extend on to the vermilion border of the lip and exhibit nondiagnostic histology.5 Bullous oral lichen planus is the least common variant of oral lichen planus, is unlikely to present as a solitary lesion, and is rarely seen on the lips. Histologically, the lesion demonstrated lichenoid inflammation.6 Solitary keratoacanthoma, though histologically similar to SCC, typically presents as a rapidly growing crateriform nodule without erosion or ulceration.7 The differential diagnoses are summarized in the Table.

The patient underwent wide excision with repair by mucosal advancement flap. He continues to be regularly seen in the clinic for monitoring of other skin cancers and is doing well. Clinicians encountering any wound or ulcer that does not show signs of healing should be wary of underlying malignancy and be prompted to perform a biopsy.
- Fehrenbach MJ. Extraoral and intraoral clinical assessment. In: Darby ML, Walsh MM, eds. Dental Hygiene: Theory and Practice. 4th ed. St Louis, MO: Elsevier; 2014:214-233.
- Hawrot A, Alam M, Ratner D. Squamous cell carcinoma. Curr Probl Dermatol. 2003;15:91-133.
- Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-308.
- Chhabra N, Chhabra S, Sapra N. Diagnostic modalities for squamous cell carcinoma: an extensive review of literature considering toluidine blue as a useful adjunct. J Oral Maxillofac Surg. 2015;14:188-200.
- Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 2003;9:1499-1505.
- Bricker SL. Oral lichen planus: a review. Semin Dermatol. 1994;13:87-90.
- Cabrijan L, Lipozencic´ J, Batinac T, et al. Differences between keratoacanthoma and squamous cell carcinoma using TGF-alpha. Coll Antropol. 2013;37:147-150.
- Douglas GD, Couch RB. A prospective study of chronic herpes simplex virus infection and recurrent herpes labialis in humans. J Immunol. 1970;104:289-295.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:976-983.
- van Tuyll van Serooskerken AM, van Marion AM, de Zwart-Storm E, et al. Lichen planus with bullous manifestation on the lip. Int J Dermatol. 2007;46(suppl 3):25-26.
- Messadi DV, Younai F. Apthous ulcers. Dermatol Ther. 2010;23:281-290.
- Ko CJ. Keratoacanthoma: facts and controversies. Clin Dermatol. 2010;28:254-261.
The Diagnosis: Squamous Cell Carcinoma
The initial clinical presentation suggested a diagnosis of herpes simplex labialis. The patient reported no response to topical acyclovir, and because the plaque persisted, a biopsy was performed. Pathology demonstrated squamous cell carcinoma (SCC) that was moderately well differentiated and invasive (Figure).
Approximately 38% of all oral SCCs in the United States occur on the lower lip and typically are solar-related cancers developing within the epidermis.1 Oral lesions initially may be asymptomatic and may not be of concern to the patient; however, it is important to recognize SCC early, as invasive lesions have the potential to metastasize. Some factors that increase the chance for the development of metastases include tumor size larger than 2 cm; location on the ear, lip, or other sites on the head and neck; and history of prior unsuccessful treatment.2 Any solitary ulcer, lump, wound, or lesion that will not heal and persists for more than 3 weeks should be regarded as cancer until proven otherwise. Although few oral SCCs are detected by clinicians at an early stage, diagnostic aids such as vital staining and molecular markers in tissues and saliva may be implemented.3 Toluidine blue is a simple, fast, and inexpensive technique that stains the nuclear material of malignant lesions, but not normal mucosa, and may be a worthwhile diagnostic adjunct to clinical inspection.4
Our patient presented with a lesion that clinically looked herpetic, though he reported no prodromal signs of tingling, burning, or pain before the occurrence of the lesion. Due to the persistence of the lesion and lack of response to treatment, a biopsy was indicated. The differential diagnoses include aphthous ulcers, which may occasionally extend on to the vermilion border of the lip and exhibit nondiagnostic histology.5 Bullous oral lichen planus is the least common variant of oral lichen planus, is unlikely to present as a solitary lesion, and is rarely seen on the lips. Histologically, the lesion demonstrated lichenoid inflammation.6 Solitary keratoacanthoma, though histologically similar to SCC, typically presents as a rapidly growing crateriform nodule without erosion or ulceration.7 The differential diagnoses are summarized in the Table.
The patient underwent wide excision with repair by mucosal advancement flap. He continues to be regularly seen in the clinic for monitoring of other skin cancers and is doing well. Clinicians encountering any wound or ulcer that does not show signs of healing should be wary of underlying malignancy and be prompted to perform a biopsy.
The Diagnosis: Squamous Cell Carcinoma
The initial clinical presentation suggested a diagnosis of herpes simplex labialis. The patient reported no response to topical acyclovir, and because the plaque persisted, a biopsy was performed. Pathology demonstrated squamous cell carcinoma (SCC) that was moderately well differentiated and invasive (Figure).
Approximately 38% of all oral SCCs in the United States occur on the lower lip and typically are solar-related cancers developing within the epidermis.1 Oral lesions initially may be asymptomatic and may not be of concern to the patient; however, it is important to recognize SCC early, as invasive lesions have the potential to metastasize. Some factors that increase the chance for the development of metastases include tumor size larger than 2 cm; location on the ear, lip, or other sites on the head and neck; and history of prior unsuccessful treatment.2 Any solitary ulcer, lump, wound, or lesion that will not heal and persists for more than 3 weeks should be regarded as cancer until proven otherwise. Although few oral SCCs are detected by clinicians at an early stage, diagnostic aids such as vital staining and molecular markers in tissues and saliva may be implemented.3 Toluidine blue is a simple, fast, and inexpensive technique that stains the nuclear material of malignant lesions, but not normal mucosa, and may be a worthwhile diagnostic adjunct to clinical inspection.4
Our patient presented with a lesion that clinically looked herpetic, though he reported no prodromal signs of tingling, burning, or pain before the occurrence of the lesion. Due to the persistence of the lesion and lack of response to treatment, a biopsy was indicated. The differential diagnoses include aphthous ulcers, which may occasionally extend on to the vermilion border of the lip and exhibit nondiagnostic histology.5 Bullous oral lichen planus is the least common variant of oral lichen planus, is unlikely to present as a solitary lesion, and is rarely seen on the lips. Histologically, the lesion demonstrated lichenoid inflammation.6 Solitary keratoacanthoma, though histologically similar to SCC, typically presents as a rapidly growing crateriform nodule without erosion or ulceration.7 The differential diagnoses are summarized in the Table.
The patient underwent wide excision with repair by mucosal advancement flap. He continues to be regularly seen in the clinic for monitoring of other skin cancers and is doing well. Clinicians encountering any wound or ulcer that does not show signs of healing should be wary of underlying malignancy and be prompted to perform a biopsy.
- Fehrenbach MJ. Extraoral and intraoral clinical assessment. In: Darby ML, Walsh MM, eds. Dental Hygiene: Theory and Practice. 4th ed. St Louis, MO: Elsevier; 2014:214-233.
- Hawrot A, Alam M, Ratner D. Squamous cell carcinoma. Curr Probl Dermatol. 2003;15:91-133.
- Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-308.
- Chhabra N, Chhabra S, Sapra N. Diagnostic modalities for squamous cell carcinoma: an extensive review of literature considering toluidine blue as a useful adjunct. J Oral Maxillofac Surg. 2015;14:188-200.
- Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 2003;9:1499-1505.
- Bricker SL. Oral lichen planus: a review. Semin Dermatol. 1994;13:87-90.
- Cabrijan L, Lipozencic´ J, Batinac T, et al. Differences between keratoacanthoma and squamous cell carcinoma using TGF-alpha. Coll Antropol. 2013;37:147-150.
- Douglas GD, Couch RB. A prospective study of chronic herpes simplex virus infection and recurrent herpes labialis in humans. J Immunol. 1970;104:289-295.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:976-983.
- van Tuyll van Serooskerken AM, van Marion AM, de Zwart-Storm E, et al. Lichen planus with bullous manifestation on the lip. Int J Dermatol. 2007;46(suppl 3):25-26.
- Messadi DV, Younai F. Apthous ulcers. Dermatol Ther. 2010;23:281-290.
- Ko CJ. Keratoacanthoma: facts and controversies. Clin Dermatol. 2010;28:254-261.
- Fehrenbach MJ. Extraoral and intraoral clinical assessment. In: Darby ML, Walsh MM, eds. Dental Hygiene: Theory and Practice. 4th ed. St Louis, MO: Elsevier; 2014:214-233.
- Hawrot A, Alam M, Ratner D. Squamous cell carcinoma. Curr Probl Dermatol. 2003;15:91-133.
- Scully C, Bagan J. Oral squamous cell carcinoma overview. Oral Oncol. 2009;45:301-308.
- Chhabra N, Chhabra S, Sapra N. Diagnostic modalities for squamous cell carcinoma: an extensive review of literature considering toluidine blue as a useful adjunct. J Oral Maxillofac Surg. 2015;14:188-200.
- Porter SR, Scully C, Pedersen A. Recurrent aphthous stomatitis. Crit Rev Oral Biol Med. 2003;9:1499-1505.
- Bricker SL. Oral lichen planus: a review. Semin Dermatol. 1994;13:87-90.
- Cabrijan L, Lipozencic´ J, Batinac T, et al. Differences between keratoacanthoma and squamous cell carcinoma using TGF-alpha. Coll Antropol. 2013;37:147-150.
- Douglas GD, Couch RB. A prospective study of chronic herpes simplex virus infection and recurrent herpes labialis in humans. J Immunol. 1970;104:289-295.
- Alam M, Ratner D. Cutaneous squamous-cell carcinoma. N Engl J Med. 2001;344:976-983.
- van Tuyll van Serooskerken AM, van Marion AM, de Zwart-Storm E, et al. Lichen planus with bullous manifestation on the lip. Int J Dermatol. 2007;46(suppl 3):25-26.
- Messadi DV, Younai F. Apthous ulcers. Dermatol Ther. 2010;23:281-290.
- Ko CJ. Keratoacanthoma: facts and controversies. Clin Dermatol. 2010;28:254-261.
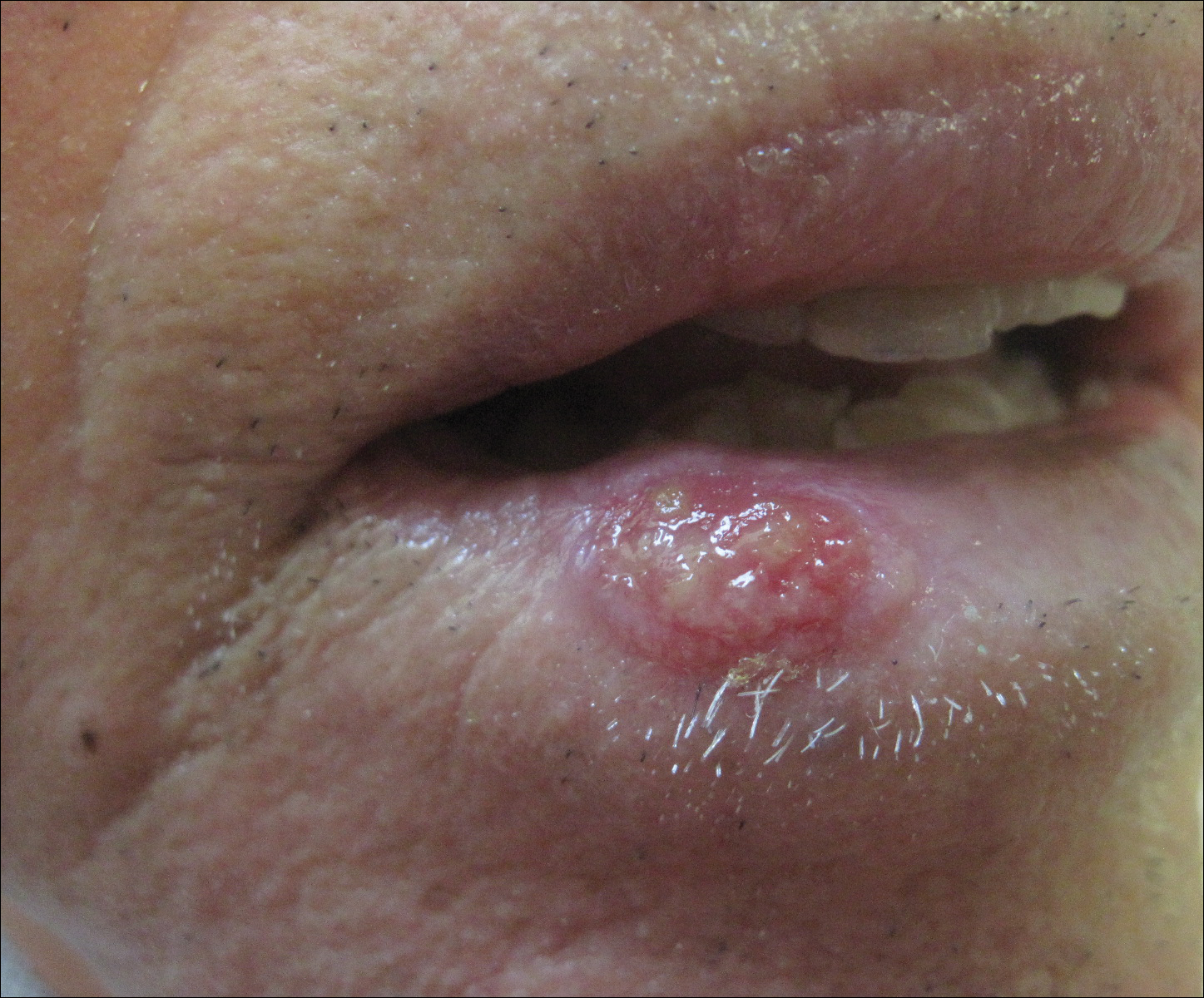
An 83-year-old man presented with a new-onset 1.2-cm eroded plaque on the vermilion border of the right lower lip that reportedly developed 2 weeks prior and was increasing in size. The plaque was moist and was composed of confluent glistening papules. Medical history was notable for the presence of both basal cell and squamous cell carcinomas.
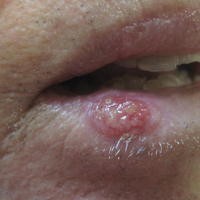
Large Hyperpigmented Nodule on the Leg
The Diagnosis: Dermatofibroma
Dermatofibroma (DF) is a commonly encountered lesion. Although usually a straightforward clinical diagnosis, histopathological diagnosis is sometimes required. Conventional histologic findings of DF are hyperkeratosis, induction of the epidermis with acanthosis, and basal layer hyperpigmentation.1,2 Within the dermis there usually is proliferation of fibroblasts, histiocytes, and blood vessels that sometimes spares the overlying papillary dermis. Nomenclature of specific variants may be assigned based on the predominant component (eg, nodular subepidermal fibrosis, histiocytoma, sclerosing hemangioma) or histologic findings (eg, fibrocollagenous, sclerotic, cellular, histiocytic, lipidized, angiomatous, aneurysmal, clear cell, monster cell, myxoid, keloidal, palisading, osteoclastic, epithelioid).3-5 Of the histologic variants, fibrocollagenous is most common, but knowledge of other variants is important for accurate diagnosis, especially to exclude malignancy.
The sclerosing hemangioma variant of DF may pre-sent a diagnostic dilemma. In addition to typical features of DF, pseudovascular spaces, abundant hemosiderin, and reactive-appearing spindled cells are histologically demonstrated. The marked sclerosis and pigment deposition may mimic a blue nevus, and the dilated pseudovascular spaces may be reminiscent of a vascular neoplasm such as angiosarcoma or Kaposi sarcoma. However, the presence of characteristic features such as peripheral collagen trapping and overlying epidermal hyperplasia provide important clues for correct diagnosis.
Angiosarcomas (Figure 1) are malignant neoplasms with vascular differentiation. Cutaneous angiosarcomas present as purple plaques or nodules on the head and/or neck in elderly individuals as well as in patients with chronic lymphedema or prior radiation exposure.6-9 They are aggressive neoplasms with high rates of recurrence and metastases. Microscopically, the tumor is composed of anastomosing vascular channels lined by atypical endothelial cells with a multilayered appearance. There is frequent red blood cell extravasation, and substantial hemosiderin deposition may be noted in long-standing lesions. Neoplastic cells are positive for vascular markers (CD34, CD31, ETS-related gene transcription factor). Notably, cases associated with radiation exposure and chronic lymphedema are positive for MYC.10

Blue nevi (Figure 2) are benign melanocytic tumors that occur most frequently in children but may pre-sent in any age group. Clinical presentation is a blue to black, slightly raised papule that may be found on any site of the body. Biopsy typically shows a wedge-shaped infiltrate of spindled melanocytes with elongated dendritic processes in a sclerotic collagenous stroma. There frequently is a striking population of heavily pigmented melanophages. The melanocytes are positive for melanoma antigen recognized by T cells (MART-1)/melan-A, S-100, and transcription factor SOX-10. In contrast to other benign nevi, human melanoma black-45 will be positive in the dermal component.
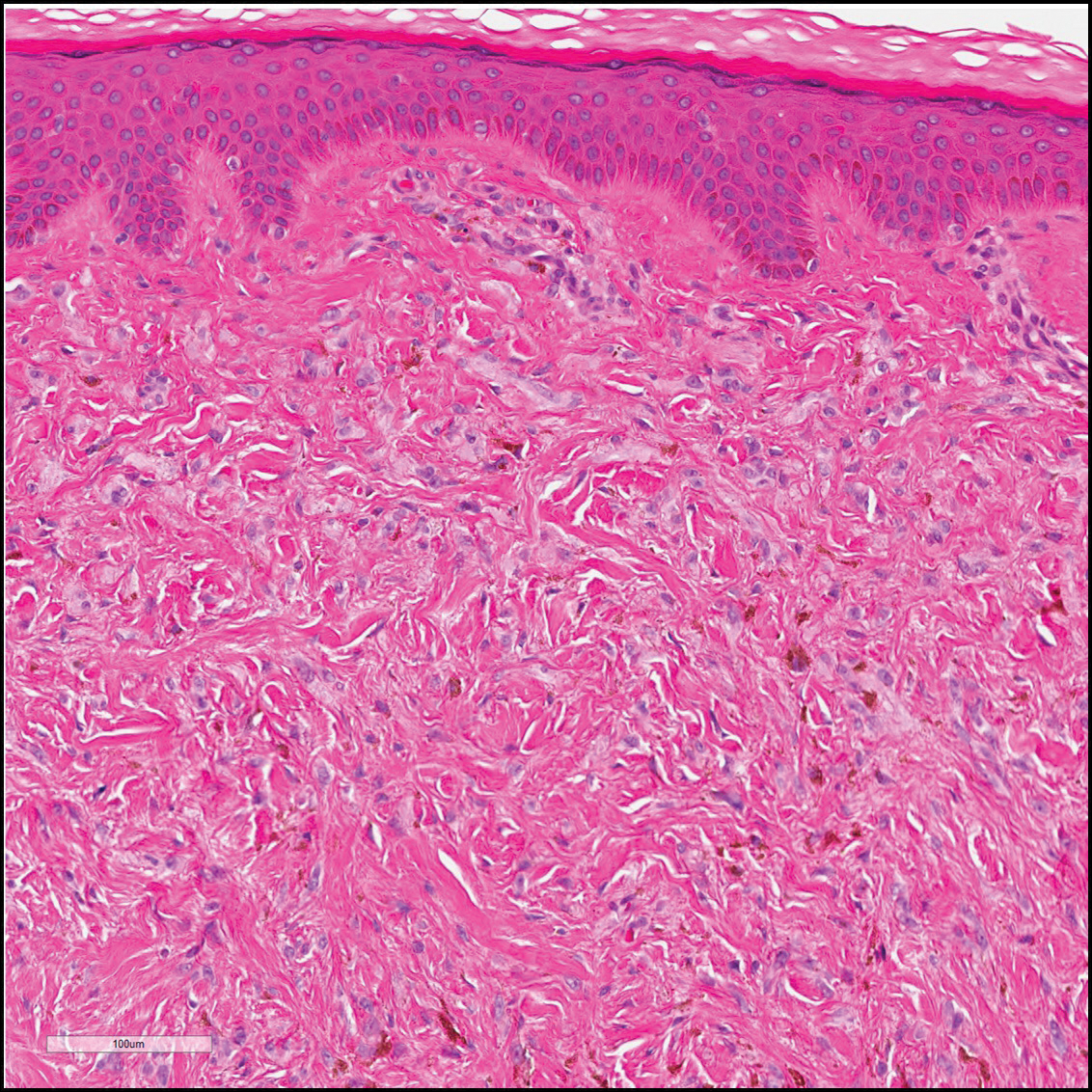
Dermatofibrosarcoma protuberans (Figure 3) is a dermal-based tumor of intermediate malignant potential with a high rate of local recurrence and potential for sarcomatous transformation. Dermatofibrosarcoma protuberans most commonly presents in young adults as firm, pink to brown plaques and can occur on any site of the body. Histologically, they show a dermal proliferation of spindled cells that infiltrate in a storiform fashion into the subcutaneous adipose tissue,11 which imparts a honeycomb or Swiss cheese pattern. The tumor characteristically demonstrates positive staining for CD34. Loss of CD34 staining, increased mitoses, nuclear atypia, and fascicular growth are features suggestive of sarcomatous transformation.11,12 Dermatofibrosarcoma protuberans is associated with chromosomal abnormalities of chromosomes 17 and 22, resulting in COL1A1 (collagen type 1 alpha 1 chain) and PDGF-β (platelet-derived growth factor subunit B) gene fusion.13
Sclerotic fibromas (also known as storiform collagenomas)(Figure 4) may represent regressed DFs and are frequently associated with prior trauma to the affected area.14,15 They usually appear as flesh-colored papules or nodules on the face and trunk. The presence of multiple sclerotic fibromas is associated with Cowden syndrome.16,17 Histologically, the lesions present as well-demarcated, nonencapsulated, dermal nodules composed of a storiform or whorled arrangement of collagen with spindled fibroblasts. The sclerotic collagen bundles often are separated by small clefts imparting a plywoodlike pattern.16
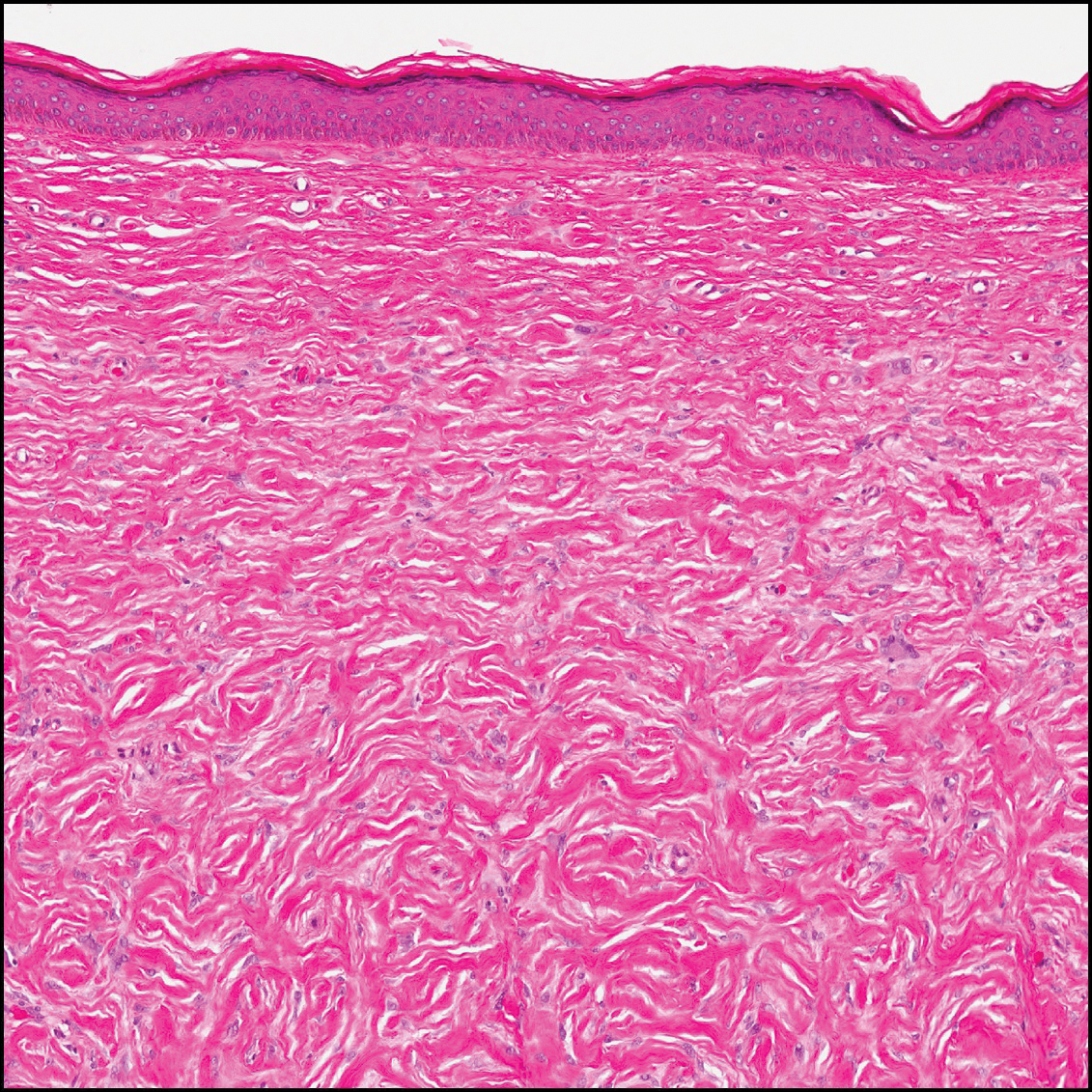
The differential diagnosis for DF expands once atypical clinical and histopathological findings are present. In this case, the nodule was much larger and darker than the usual appearance of DF (3-10 mm).2,4 Given the lesion's nodularity, the clinical dimple sign on lateral compression could not be seen. On biopsy, the predominance of blood vessels and sclerosis further complicated the diagnostic picture. In unusual cases such as this one, correlation of clinical history, histology, and immunophenotype is ever important.
- Zeidi M, North JP. Sebaceous induction in dermatofibroma: a common feature of dermatofibromas on the shoulder. J Cutan Pathol. 2015;42:400-405.
- Şenel E, Yuyucu Karabulut Y, Doğruer S¸enel S. Clinical, histopathological, dermatoscopic and digital microscopic features of dermatofibroma: a retrospective analysis of 200 lesions. J Eur Acad Dermatol Venereol. 2015;29:1958-1966.
- Vilanova JR, Flint A. The morphological variations of fibrous histiocytomas. J Cutan Pathol. 1974;1:155-164.
- Han TY, Chang HS, Lee JH, et al. A clinical and histopathological study of 122 cases of dermatofibroma (benign fibrous histiocytoma)[published online May 27, 2011]. Ann Dermatol. 2011;23:185-192.
- Alves JVP, Matos DM, Barreiros HF, et al. Variants of dermatofibroma--a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Rosai J, Sumner HW, Major MC, et al. Angiosarcoma of the skin: a clinicopathologic and fine structural study. Hum Pathol. 1976;7:83-109.
- Haustein UF. Angiosarcoma of the face and scalp. Int J Dermatol. 1991;30:851-856.
- Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema: a report of six cases in elephantiasis chirurgica. Cancer. 1948;1:64-81.
- Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12(5 pt 2):922-926.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Voth H, Landsberg J, Hinz T, et al. Management of dermatofibrosarcoma protuberans with fibrosarcomatous transformation: an evidence-based review of the literature. J Eur Acad Dermatol Venereol. 2011;25:1385-1391.
- Goldblum JR. CD34 positivity in fibrosarcomas which arise in dermatofibrosarcoma protuberans. Arch Pathol Lab Med. 1995;119:238-241.
- Patel KU, Szabo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- Sohn IB, Hwang SM, Lee SH, et al. Dermatofibroma with sclerotic areas resembling a sclerotic fibroma of the skin. J Cutan Pathol. 2002;29:44-47.
- Pujol RM, de Castro F, Schroeter AL, et al. Solitary sclerotic fibroma of the skin: a sclerotic dermatofibroma? Am J Dermatopathol. 1996;18:620-624.
- Requena L, Gutiérrez J, Sánchez Yus E. Multiple sclerotic fibromas of the skin: a cutaneous marker of Cowden's disease. J Cutan Pathol. 1992;19:346-351.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
The Diagnosis: Dermatofibroma
Dermatofibroma (DF) is a commonly encountered lesion. Although usually a straightforward clinical diagnosis, histopathological diagnosis is sometimes required. Conventional histologic findings of DF are hyperkeratosis, induction of the epidermis with acanthosis, and basal layer hyperpigmentation.1,2 Within the dermis there usually is proliferation of fibroblasts, histiocytes, and blood vessels that sometimes spares the overlying papillary dermis. Nomenclature of specific variants may be assigned based on the predominant component (eg, nodular subepidermal fibrosis, histiocytoma, sclerosing hemangioma) or histologic findings (eg, fibrocollagenous, sclerotic, cellular, histiocytic, lipidized, angiomatous, aneurysmal, clear cell, monster cell, myxoid, keloidal, palisading, osteoclastic, epithelioid).3-5 Of the histologic variants, fibrocollagenous is most common, but knowledge of other variants is important for accurate diagnosis, especially to exclude malignancy.
The sclerosing hemangioma variant of DF may pre-sent a diagnostic dilemma. In addition to typical features of DF, pseudovascular spaces, abundant hemosiderin, and reactive-appearing spindled cells are histologically demonstrated. The marked sclerosis and pigment deposition may mimic a blue nevus, and the dilated pseudovascular spaces may be reminiscent of a vascular neoplasm such as angiosarcoma or Kaposi sarcoma. However, the presence of characteristic features such as peripheral collagen trapping and overlying epidermal hyperplasia provide important clues for correct diagnosis.
Angiosarcomas (Figure 1) are malignant neoplasms with vascular differentiation. Cutaneous angiosarcomas present as purple plaques or nodules on the head and/or neck in elderly individuals as well as in patients with chronic lymphedema or prior radiation exposure.6-9 They are aggressive neoplasms with high rates of recurrence and metastases. Microscopically, the tumor is composed of anastomosing vascular channels lined by atypical endothelial cells with a multilayered appearance. There is frequent red blood cell extravasation, and substantial hemosiderin deposition may be noted in long-standing lesions. Neoplastic cells are positive for vascular markers (CD34, CD31, ETS-related gene transcription factor). Notably, cases associated with radiation exposure and chronic lymphedema are positive for MYC.10
Blue nevi (Figure 2) are benign melanocytic tumors that occur most frequently in children but may pre-sent in any age group. Clinical presentation is a blue to black, slightly raised papule that may be found on any site of the body. Biopsy typically shows a wedge-shaped infiltrate of spindled melanocytes with elongated dendritic processes in a sclerotic collagenous stroma. There frequently is a striking population of heavily pigmented melanophages. The melanocytes are positive for melanoma antigen recognized by T cells (MART-1)/melan-A, S-100, and transcription factor SOX-10. In contrast to other benign nevi, human melanoma black-45 will be positive in the dermal component.
Dermatofibrosarcoma protuberans (Figure 3) is a dermal-based tumor of intermediate malignant potential with a high rate of local recurrence and potential for sarcomatous transformation. Dermatofibrosarcoma protuberans most commonly presents in young adults as firm, pink to brown plaques and can occur on any site of the body. Histologically, they show a dermal proliferation of spindled cells that infiltrate in a storiform fashion into the subcutaneous adipose tissue,11 which imparts a honeycomb or Swiss cheese pattern. The tumor characteristically demonstrates positive staining for CD34. Loss of CD34 staining, increased mitoses, nuclear atypia, and fascicular growth are features suggestive of sarcomatous transformation.11,12 Dermatofibrosarcoma protuberans is associated with chromosomal abnormalities of chromosomes 17 and 22, resulting in COL1A1 (collagen type 1 alpha 1 chain) and PDGF-β (platelet-derived growth factor subunit B) gene fusion.13
Sclerotic fibromas (also known as storiform collagenomas)(Figure 4) may represent regressed DFs and are frequently associated with prior trauma to the affected area.14,15 They usually appear as flesh-colored papules or nodules on the face and trunk. The presence of multiple sclerotic fibromas is associated with Cowden syndrome.16,17 Histologically, the lesions present as well-demarcated, nonencapsulated, dermal nodules composed of a storiform or whorled arrangement of collagen with spindled fibroblasts. The sclerotic collagen bundles often are separated by small clefts imparting a plywoodlike pattern.16
The differential diagnosis for DF expands once atypical clinical and histopathological findings are present. In this case, the nodule was much larger and darker than the usual appearance of DF (3-10 mm).2,4 Given the lesion's nodularity, the clinical dimple sign on lateral compression could not be seen. On biopsy, the predominance of blood vessels and sclerosis further complicated the diagnostic picture. In unusual cases such as this one, correlation of clinical history, histology, and immunophenotype is ever important.
The Diagnosis: Dermatofibroma
Dermatofibroma (DF) is a commonly encountered lesion. Although usually a straightforward clinical diagnosis, histopathological diagnosis is sometimes required. Conventional histologic findings of DF are hyperkeratosis, induction of the epidermis with acanthosis, and basal layer hyperpigmentation.1,2 Within the dermis there usually is proliferation of fibroblasts, histiocytes, and blood vessels that sometimes spares the overlying papillary dermis. Nomenclature of specific variants may be assigned based on the predominant component (eg, nodular subepidermal fibrosis, histiocytoma, sclerosing hemangioma) or histologic findings (eg, fibrocollagenous, sclerotic, cellular, histiocytic, lipidized, angiomatous, aneurysmal, clear cell, monster cell, myxoid, keloidal, palisading, osteoclastic, epithelioid).3-5 Of the histologic variants, fibrocollagenous is most common, but knowledge of other variants is important for accurate diagnosis, especially to exclude malignancy.
The sclerosing hemangioma variant of DF may pre-sent a diagnostic dilemma. In addition to typical features of DF, pseudovascular spaces, abundant hemosiderin, and reactive-appearing spindled cells are histologically demonstrated. The marked sclerosis and pigment deposition may mimic a blue nevus, and the dilated pseudovascular spaces may be reminiscent of a vascular neoplasm such as angiosarcoma or Kaposi sarcoma. However, the presence of characteristic features such as peripheral collagen trapping and overlying epidermal hyperplasia provide important clues for correct diagnosis.
Angiosarcomas (Figure 1) are malignant neoplasms with vascular differentiation. Cutaneous angiosarcomas present as purple plaques or nodules on the head and/or neck in elderly individuals as well as in patients with chronic lymphedema or prior radiation exposure.6-9 They are aggressive neoplasms with high rates of recurrence and metastases. Microscopically, the tumor is composed of anastomosing vascular channels lined by atypical endothelial cells with a multilayered appearance. There is frequent red blood cell extravasation, and substantial hemosiderin deposition may be noted in long-standing lesions. Neoplastic cells are positive for vascular markers (CD34, CD31, ETS-related gene transcription factor). Notably, cases associated with radiation exposure and chronic lymphedema are positive for MYC.10
Blue nevi (Figure 2) are benign melanocytic tumors that occur most frequently in children but may pre-sent in any age group. Clinical presentation is a blue to black, slightly raised papule that may be found on any site of the body. Biopsy typically shows a wedge-shaped infiltrate of spindled melanocytes with elongated dendritic processes in a sclerotic collagenous stroma. There frequently is a striking population of heavily pigmented melanophages. The melanocytes are positive for melanoma antigen recognized by T cells (MART-1)/melan-A, S-100, and transcription factor SOX-10. In contrast to other benign nevi, human melanoma black-45 will be positive in the dermal component.
Dermatofibrosarcoma protuberans (Figure 3) is a dermal-based tumor of intermediate malignant potential with a high rate of local recurrence and potential for sarcomatous transformation. Dermatofibrosarcoma protuberans most commonly presents in young adults as firm, pink to brown plaques and can occur on any site of the body. Histologically, they show a dermal proliferation of spindled cells that infiltrate in a storiform fashion into the subcutaneous adipose tissue,11 which imparts a honeycomb or Swiss cheese pattern. The tumor characteristically demonstrates positive staining for CD34. Loss of CD34 staining, increased mitoses, nuclear atypia, and fascicular growth are features suggestive of sarcomatous transformation.11,12 Dermatofibrosarcoma protuberans is associated with chromosomal abnormalities of chromosomes 17 and 22, resulting in COL1A1 (collagen type 1 alpha 1 chain) and PDGF-β (platelet-derived growth factor subunit B) gene fusion.13
Sclerotic fibromas (also known as storiform collagenomas)(Figure 4) may represent regressed DFs and are frequently associated with prior trauma to the affected area.14,15 They usually appear as flesh-colored papules or nodules on the face and trunk. The presence of multiple sclerotic fibromas is associated with Cowden syndrome.16,17 Histologically, the lesions present as well-demarcated, nonencapsulated, dermal nodules composed of a storiform or whorled arrangement of collagen with spindled fibroblasts. The sclerotic collagen bundles often are separated by small clefts imparting a plywoodlike pattern.16
The differential diagnosis for DF expands once atypical clinical and histopathological findings are present. In this case, the nodule was much larger and darker than the usual appearance of DF (3-10 mm).2,4 Given the lesion's nodularity, the clinical dimple sign on lateral compression could not be seen. On biopsy, the predominance of blood vessels and sclerosis further complicated the diagnostic picture. In unusual cases such as this one, correlation of clinical history, histology, and immunophenotype is ever important.
- Zeidi M, North JP. Sebaceous induction in dermatofibroma: a common feature of dermatofibromas on the shoulder. J Cutan Pathol. 2015;42:400-405.
- Şenel E, Yuyucu Karabulut Y, Doğruer S¸enel S. Clinical, histopathological, dermatoscopic and digital microscopic features of dermatofibroma: a retrospective analysis of 200 lesions. J Eur Acad Dermatol Venereol. 2015;29:1958-1966.
- Vilanova JR, Flint A. The morphological variations of fibrous histiocytomas. J Cutan Pathol. 1974;1:155-164.
- Han TY, Chang HS, Lee JH, et al. A clinical and histopathological study of 122 cases of dermatofibroma (benign fibrous histiocytoma)[published online May 27, 2011]. Ann Dermatol. 2011;23:185-192.
- Alves JVP, Matos DM, Barreiros HF, et al. Variants of dermatofibroma--a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Rosai J, Sumner HW, Major MC, et al. Angiosarcoma of the skin: a clinicopathologic and fine structural study. Hum Pathol. 1976;7:83-109.
- Haustein UF. Angiosarcoma of the face and scalp. Int J Dermatol. 1991;30:851-856.
- Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema: a report of six cases in elephantiasis chirurgica. Cancer. 1948;1:64-81.
- Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12(5 pt 2):922-926.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Voth H, Landsberg J, Hinz T, et al. Management of dermatofibrosarcoma protuberans with fibrosarcomatous transformation: an evidence-based review of the literature. J Eur Acad Dermatol Venereol. 2011;25:1385-1391.
- Goldblum JR. CD34 positivity in fibrosarcomas which arise in dermatofibrosarcoma protuberans. Arch Pathol Lab Med. 1995;119:238-241.
- Patel KU, Szabo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- Sohn IB, Hwang SM, Lee SH, et al. Dermatofibroma with sclerotic areas resembling a sclerotic fibroma of the skin. J Cutan Pathol. 2002;29:44-47.
- Pujol RM, de Castro F, Schroeter AL, et al. Solitary sclerotic fibroma of the skin: a sclerotic dermatofibroma? Am J Dermatopathol. 1996;18:620-624.
- Requena L, Gutiérrez J, Sánchez Yus E. Multiple sclerotic fibromas of the skin: a cutaneous marker of Cowden's disease. J Cutan Pathol. 1992;19:346-351.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Zeidi M, North JP. Sebaceous induction in dermatofibroma: a common feature of dermatofibromas on the shoulder. J Cutan Pathol. 2015;42:400-405.
- Şenel E, Yuyucu Karabulut Y, Doğruer S¸enel S. Clinical, histopathological, dermatoscopic and digital microscopic features of dermatofibroma: a retrospective analysis of 200 lesions. J Eur Acad Dermatol Venereol. 2015;29:1958-1966.
- Vilanova JR, Flint A. The morphological variations of fibrous histiocytomas. J Cutan Pathol. 1974;1:155-164.
- Han TY, Chang HS, Lee JH, et al. A clinical and histopathological study of 122 cases of dermatofibroma (benign fibrous histiocytoma)[published online May 27, 2011]. Ann Dermatol. 2011;23:185-192.
- Alves JVP, Matos DM, Barreiros HF, et al. Variants of dermatofibroma--a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Rosai J, Sumner HW, Major MC, et al. Angiosarcoma of the skin: a clinicopathologic and fine structural study. Hum Pathol. 1976;7:83-109.
- Haustein UF. Angiosarcoma of the face and scalp. Int J Dermatol. 1991;30:851-856.
- Stewart FW, Treves N. Lymphangiosarcoma in postmastectomy lymphedema: a report of six cases in elephantiasis chirurgica. Cancer. 1948;1:64-81.
- Goette DK, Detlefs RL. Postirradiation angiosarcoma. J Am Acad Dermatol. 1985;12(5 pt 2):922-926.
- Manner J, Radlwimmer B, Hohenberger P, et al. MYC high level gene amplification is a distinctive feature of angiosarcomas after irradiation or chronic lymphedema. Am J Pathol. 2010;176:34-39.
- Voth H, Landsberg J, Hinz T, et al. Management of dermatofibrosarcoma protuberans with fibrosarcomatous transformation: an evidence-based review of the literature. J Eur Acad Dermatol Venereol. 2011;25:1385-1391.
- Goldblum JR. CD34 positivity in fibrosarcomas which arise in dermatofibrosarcoma protuberans. Arch Pathol Lab Med. 1995;119:238-241.
- Patel KU, Szabo SS, Hernandez VS, et al. Dermatofibrosarcoma protuberans COL1A1-PDGFB fusion is identified in virtually all dermatofibrosarcoma protuberans cases when investigated by newly developed multiplex reverse transcription polymerase chain reaction and fluorescence in situ hybridization assays. Hum Pathol. 2008;39:184-193.
- Sohn IB, Hwang SM, Lee SH, et al. Dermatofibroma with sclerotic areas resembling a sclerotic fibroma of the skin. J Cutan Pathol. 2002;29:44-47.
- Pujol RM, de Castro F, Schroeter AL, et al. Solitary sclerotic fibroma of the skin: a sclerotic dermatofibroma? Am J Dermatopathol. 1996;18:620-624.
- Requena L, Gutiérrez J, Sánchez Yus E. Multiple sclerotic fibromas of the skin: a cutaneous marker of Cowden's disease. J Cutan Pathol. 1992;19:346-351.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.

A 61-year-old woman presented with a 2.5-cm hyperpigmented exophytic nodule on the anterior aspect of the left shin of approximately 2 years' duration. The patient initially noticed a small lesion following a bee sting, but it subsequently grew over the ensuing 2 years. A shave biopsy was obtained.