User login
CDC chief lays out attack plan for COVID variants
earlier this week.
As part of JAMA’s Q&A series with JAMA editor in chief Howard Bauchner, MD, Dr. Walensky referenced the blueprint she coathored with Anthony Fauci, MD, the nation’s top infectious disease expert, and Henry T. Walke, MD, MPH, of the CDC, which was published on Feb. 17 in JAMA.
In the viewpoint article, they explain that the Department of Health & Human Services has established the SARS-CoV-2 Interagency Group to improve coordination among the CDC, the National Institutes of Health, the Food and Drug Administration, the Biomedical Advanced Research and Development Authority, the Department of Agriculture, and the Department of Defense.
Dr. Walensky said the first objective is to reinforce vigilance regarding public health mitigation strategies to decrease the amount of virus that’s circulating.
As part of that strategy, she said, the CDC strongly urges against nonessential travel.
In addition, public health leaders are working on a surveillance system to better understand the SARS-CoV-2 variants. That will take ramping up genome sequencing of the SARS-CoV-2 virus and ensuring that sampling is geographically representative.
She said the CDC is partnering with state health labs to obtain about 750 samples every week and is teaming up with commercial labs and academic centers to obtain an interim target of 6,000 samples per week.
She acknowledged the United States “is not where we need to be” with sequencing but has come a long way since January. At that time, they were sequencing 250 samples every week; they are currently sequencing thousands each week.
Data analysis is another concern: “We need to be able to understand at the basic science level what the information means,” Dr. Walensky said.
Researchers aren’t sure how the variants might affect use of convalescent plasma or monoclonal antibody treatments. It is expected that 5% of persons who are vaccinated against COVID-19 will nevertheless contract the disease. Sequencing will help answer whether such persons who have been vaccinated and who subsequently contract the virus are among those 5% or whether have been infected by a variant that evades the vaccine.
Accelerating vaccine administration globally and in the United States is essential, Dr. Walensky said.
As of Feb. 17, 56 million doses had been administered in the United States.
Top three threats
She updated the numbers on the three biggest variant threats.
Regarding B.1.1.7, which originated in the United Kingdom, she said: “So far, we’ve had over 1,200 cases in 41 states.” She noted that the variant is likely to be about 50% more transmissible and 30% to 50% more virulent.
“So far, it looks like that strain doesn’t have any real decrease in susceptibility to our vaccines,” she said.
The strain from South Africa (B.1.351) has been found in 19 cases in the United States.
The P.1. variant, which originated in Brazil, has been identified in two cases in two states.
Outlook for March and April
Dr. Bauchner asked Dr. Walensky what she envisions for March and April. He noted that public optimism is high in light of the continued reductions in COVID-19 case numbers, hospitalizations, and deaths, as well as the fact that warmer weather is coming and that more vaccinations are on the horizon.
“While I really am hopeful for what could happen in March and April,” Dr. Walensky said, “I really do know that this could go bad so fast. We saw it in November. We saw it in December.”
CDC models have projected that, by March, the more transmissible B.1.1.7 strain is likely to be the dominant strain, she reiterated.
“I worry that it will be spring, and we will all have had enough,” Dr. Walensky said. She noted that some states are already relaxing mask mandates.
“Around that time, life will look and feel a little better, and the motivation for those who might be vaccine hesitant may be diminished,” she said.
Dr. Bauchner also asked her to weigh in on whether a third vaccine, from Johnson & Johnson (J&J), may soon gain FDA emergency-use authorization – and whether its lower expected efficacy rate may result in a tiered system of vaccinations, with higher-risk populations receiving the more efficacious vaccines.
Dr. Walensky said more data are needed before that question can be answered.
“It may very well be that the data point us to the best populations in which to use this vaccine,” she said.
In phase 3 data, the J&J vaccine was shown to be 72% effective in the United States for moderate to severe disease.
Dr. Walensky said it’s important to remember that the projected efficacy for that vaccine is higher than that for the flu shot as well as many other vaccines currently in use for other diseases.
She said it also has several advantages. The vaccine has less-stringent storage requirements, requires just one dose, and protects against hospitalization and death, although it’s less efficacious in protecting against contracting the disease.
“I think many people would opt to get that one if they could get it sooner,” she said.
A version of this article first appeared on Medscape.com.
earlier this week.
As part of JAMA’s Q&A series with JAMA editor in chief Howard Bauchner, MD, Dr. Walensky referenced the blueprint she coathored with Anthony Fauci, MD, the nation’s top infectious disease expert, and Henry T. Walke, MD, MPH, of the CDC, which was published on Feb. 17 in JAMA.
In the viewpoint article, they explain that the Department of Health & Human Services has established the SARS-CoV-2 Interagency Group to improve coordination among the CDC, the National Institutes of Health, the Food and Drug Administration, the Biomedical Advanced Research and Development Authority, the Department of Agriculture, and the Department of Defense.
Dr. Walensky said the first objective is to reinforce vigilance regarding public health mitigation strategies to decrease the amount of virus that’s circulating.
As part of that strategy, she said, the CDC strongly urges against nonessential travel.
In addition, public health leaders are working on a surveillance system to better understand the SARS-CoV-2 variants. That will take ramping up genome sequencing of the SARS-CoV-2 virus and ensuring that sampling is geographically representative.
She said the CDC is partnering with state health labs to obtain about 750 samples every week and is teaming up with commercial labs and academic centers to obtain an interim target of 6,000 samples per week.
She acknowledged the United States “is not where we need to be” with sequencing but has come a long way since January. At that time, they were sequencing 250 samples every week; they are currently sequencing thousands each week.
Data analysis is another concern: “We need to be able to understand at the basic science level what the information means,” Dr. Walensky said.
Researchers aren’t sure how the variants might affect use of convalescent plasma or monoclonal antibody treatments. It is expected that 5% of persons who are vaccinated against COVID-19 will nevertheless contract the disease. Sequencing will help answer whether such persons who have been vaccinated and who subsequently contract the virus are among those 5% or whether have been infected by a variant that evades the vaccine.
Accelerating vaccine administration globally and in the United States is essential, Dr. Walensky said.
As of Feb. 17, 56 million doses had been administered in the United States.
Top three threats
She updated the numbers on the three biggest variant threats.
Regarding B.1.1.7, which originated in the United Kingdom, she said: “So far, we’ve had over 1,200 cases in 41 states.” She noted that the variant is likely to be about 50% more transmissible and 30% to 50% more virulent.
“So far, it looks like that strain doesn’t have any real decrease in susceptibility to our vaccines,” she said.
The strain from South Africa (B.1.351) has been found in 19 cases in the United States.
The P.1. variant, which originated in Brazil, has been identified in two cases in two states.
Outlook for March and April
Dr. Bauchner asked Dr. Walensky what she envisions for March and April. He noted that public optimism is high in light of the continued reductions in COVID-19 case numbers, hospitalizations, and deaths, as well as the fact that warmer weather is coming and that more vaccinations are on the horizon.
“While I really am hopeful for what could happen in March and April,” Dr. Walensky said, “I really do know that this could go bad so fast. We saw it in November. We saw it in December.”
CDC models have projected that, by March, the more transmissible B.1.1.7 strain is likely to be the dominant strain, she reiterated.
“I worry that it will be spring, and we will all have had enough,” Dr. Walensky said. She noted that some states are already relaxing mask mandates.
“Around that time, life will look and feel a little better, and the motivation for those who might be vaccine hesitant may be diminished,” she said.
Dr. Bauchner also asked her to weigh in on whether a third vaccine, from Johnson & Johnson (J&J), may soon gain FDA emergency-use authorization – and whether its lower expected efficacy rate may result in a tiered system of vaccinations, with higher-risk populations receiving the more efficacious vaccines.
Dr. Walensky said more data are needed before that question can be answered.
“It may very well be that the data point us to the best populations in which to use this vaccine,” she said.
In phase 3 data, the J&J vaccine was shown to be 72% effective in the United States for moderate to severe disease.
Dr. Walensky said it’s important to remember that the projected efficacy for that vaccine is higher than that for the flu shot as well as many other vaccines currently in use for other diseases.
She said it also has several advantages. The vaccine has less-stringent storage requirements, requires just one dose, and protects against hospitalization and death, although it’s less efficacious in protecting against contracting the disease.
“I think many people would opt to get that one if they could get it sooner,” she said.
A version of this article first appeared on Medscape.com.
earlier this week.
As part of JAMA’s Q&A series with JAMA editor in chief Howard Bauchner, MD, Dr. Walensky referenced the blueprint she coathored with Anthony Fauci, MD, the nation’s top infectious disease expert, and Henry T. Walke, MD, MPH, of the CDC, which was published on Feb. 17 in JAMA.
In the viewpoint article, they explain that the Department of Health & Human Services has established the SARS-CoV-2 Interagency Group to improve coordination among the CDC, the National Institutes of Health, the Food and Drug Administration, the Biomedical Advanced Research and Development Authority, the Department of Agriculture, and the Department of Defense.
Dr. Walensky said the first objective is to reinforce vigilance regarding public health mitigation strategies to decrease the amount of virus that’s circulating.
As part of that strategy, she said, the CDC strongly urges against nonessential travel.
In addition, public health leaders are working on a surveillance system to better understand the SARS-CoV-2 variants. That will take ramping up genome sequencing of the SARS-CoV-2 virus and ensuring that sampling is geographically representative.
She said the CDC is partnering with state health labs to obtain about 750 samples every week and is teaming up with commercial labs and academic centers to obtain an interim target of 6,000 samples per week.
She acknowledged the United States “is not where we need to be” with sequencing but has come a long way since January. At that time, they were sequencing 250 samples every week; they are currently sequencing thousands each week.
Data analysis is another concern: “We need to be able to understand at the basic science level what the information means,” Dr. Walensky said.
Researchers aren’t sure how the variants might affect use of convalescent plasma or monoclonal antibody treatments. It is expected that 5% of persons who are vaccinated against COVID-19 will nevertheless contract the disease. Sequencing will help answer whether such persons who have been vaccinated and who subsequently contract the virus are among those 5% or whether have been infected by a variant that evades the vaccine.
Accelerating vaccine administration globally and in the United States is essential, Dr. Walensky said.
As of Feb. 17, 56 million doses had been administered in the United States.
Top three threats
She updated the numbers on the three biggest variant threats.
Regarding B.1.1.7, which originated in the United Kingdom, she said: “So far, we’ve had over 1,200 cases in 41 states.” She noted that the variant is likely to be about 50% more transmissible and 30% to 50% more virulent.
“So far, it looks like that strain doesn’t have any real decrease in susceptibility to our vaccines,” she said.
The strain from South Africa (B.1.351) has been found in 19 cases in the United States.
The P.1. variant, which originated in Brazil, has been identified in two cases in two states.
Outlook for March and April
Dr. Bauchner asked Dr. Walensky what she envisions for March and April. He noted that public optimism is high in light of the continued reductions in COVID-19 case numbers, hospitalizations, and deaths, as well as the fact that warmer weather is coming and that more vaccinations are on the horizon.
“While I really am hopeful for what could happen in March and April,” Dr. Walensky said, “I really do know that this could go bad so fast. We saw it in November. We saw it in December.”
CDC models have projected that, by March, the more transmissible B.1.1.7 strain is likely to be the dominant strain, she reiterated.
“I worry that it will be spring, and we will all have had enough,” Dr. Walensky said. She noted that some states are already relaxing mask mandates.
“Around that time, life will look and feel a little better, and the motivation for those who might be vaccine hesitant may be diminished,” she said.
Dr. Bauchner also asked her to weigh in on whether a third vaccine, from Johnson & Johnson (J&J), may soon gain FDA emergency-use authorization – and whether its lower expected efficacy rate may result in a tiered system of vaccinations, with higher-risk populations receiving the more efficacious vaccines.
Dr. Walensky said more data are needed before that question can be answered.
“It may very well be that the data point us to the best populations in which to use this vaccine,” she said.
In phase 3 data, the J&J vaccine was shown to be 72% effective in the United States for moderate to severe disease.
Dr. Walensky said it’s important to remember that the projected efficacy for that vaccine is higher than that for the flu shot as well as many other vaccines currently in use for other diseases.
She said it also has several advantages. The vaccine has less-stringent storage requirements, requires just one dose, and protects against hospitalization and death, although it’s less efficacious in protecting against contracting the disease.
“I think many people would opt to get that one if they could get it sooner,” she said.
A version of this article first appeared on Medscape.com.
Super Bowl ad for diabetes device prompts debate
A commercial for the continuous glucose monitor (CGM) Dexcom G6 shown during the Super Bowl has provoked strong reactions in the diabetes community, both positive and negative.
The 30-second ad, which aired between the first two quarters of the American football game yesterday, features singer-songwriter-actor Nick Jonas, who has type 1 diabetes. During the ad, Mr. Jonas asks – with so much technology available today, including drones that deliver packages and self-driving cars – why are people with diabetes still pricking their fingers to test their blood sugar?
Mr. Jonas goes on to demonstrate the Dexcom G6 smartphone glucose app as it displays three different glucose levels including two trending upward, explaining: “It shows your glucose right in your phone, and where it’s heading, without fingersticks. Finally, technology that makes it easier to manage our diabetes.”
Diabetes type or insulin treatment are not mentioned in the ad, despite the fact that most insurance plans typically only cover CGMs for people with type 1 diabetes and sometimes for those with type 2 diabetes who take multiple daily insulin doses (given the risk for hypoglycemia).
Ad prompts mixed reaction on social media
Reactions rolled in on Twitter after the ad debuted Feb. 2, and then again after it aired during the game.
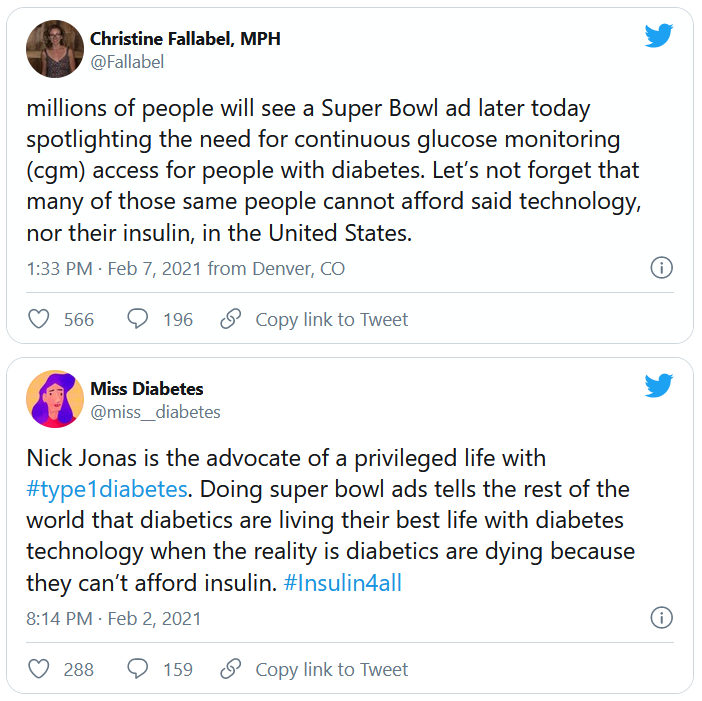
Some people who have type 1 diabetes themselves or have children with the disease who use the product were thrilled.
“Thanks to @NickJonas for his advocacy on T1. My 11-year old has been on the Dexcom for 3 weeks. For a newly diagnosed kid, it removes a lot of anxiety (and for his parents, too!) Plus, he is thrilled his meter has a Super Bowl commercial!” tweeted @KatisJewell.
Another positive tweet, from @rturnerroy, read: “@nickjonas Thank you for bringing representation to #type1diabetes. And hey #Dexcom, you’re the best.”
But many others were critical, both of Jonas and Dexcom. @hb_herrick tweeted: “Diabetes awareness is fantastic. Dexcom being able to afford Nick Jonas for a #SuperBowl commercial is not. This is a health care product. Make it more affordable for those who need it.”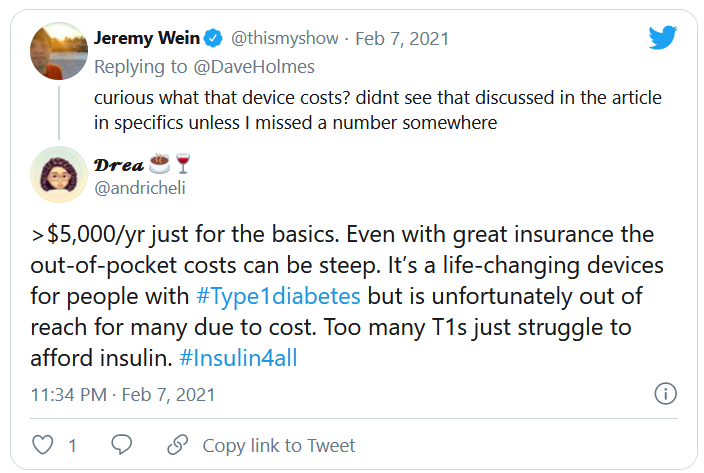
Another Twitter user, @universeofdust, tweeted: “Feeling ambivalent about the #Dexcom ad tbh. I love the awareness & representation. But also not a big fan of dexcom spending $5.5 mill+ to make the CGM seem like this ~cool & trendy~ thing when many type 1s can’t afford their insulin, let alone a CGM.”
And @andricheli wrote: “Only people lucky enough to have excellent insurance and be able to afford the out-of-pocket costs have access. Many others do not.”
And in another tweet the same user said, “The #Dexcom is an amazing device. It’s literally lifesaving and life extending. But it’s also very expensive and not available to everyone. Maybe instead of spending $5 mil on a Super Bowl ad, @dexcom should spend that on getting Dex into the handle of people who need it.”
Others, including @1hitwonderdate, criticized Mr. Jonas directly, asking him: “As someone who has struggled with diabetes and is trying to support themselves along with millions of others, why not use this platform to help those who can’t afford their supplies or are rationing them?!”
Dexcom and Jonas’ organization respond
This news organization reached out to both Dexcom and to Beyond Type 1, a nonprofit organization cofounded by Mr. Jonas, for comment. Both emailed responses.
Regarding the intended audience for the ad, Dexcom acknowledged that it hoped to reach a much wider group than just people with type 1 diabetes or even just insulin users.
“We believe our CGM technology has the ability to empower any person with diabetes and significantly improve their treatment and quality of life, whether they are using insulin or not,” the company said, adding that the ad was also aimed at “loved ones, caregivers, and even health care professionals who need to know about this technology.”
According to Dexcom, the G6 is covered by 99% of commercial insurance in the United States, in addition to Medicare, and by Medicaid in more than 40 states. Over 70% of Dexcom patients with pharmacy coverage in the United States pay under $60 per month for CGM, and a third pay $0 out-of-pocket.
“That said, we know there’s more to be done to improve access, and we are working with several partners to broaden access to Dexcom CGM, especially for people with type 2 diabetes not on mealtime insulin,” the company noted.
Beyond Type 1 responded to the criticisms about Mr. Jonas personally, noting that the celebrity is, in fact, heavily involved in advocacy.
“Nick was involved in the launch of GetInsulin.org this past October,” they said. “GetInsulin.org is a tool created by Beyond Type 1 to connect people with diabetes in the United States to the insulin access and affordability options that match their unique circumstances. ... Beyond Type 1 will continue driving awareness of short-term solutions related to insulin access and affordability while fighting for systemic change.”
The organization “is also advocating for systemic payment policies that will make devices less expensive and avoid the same pitfalls (and rising prices) as the drug pricing system in the U.S.”
Mr. Jonas himself appears aware of the concerns.
Is 2021’s most expensive Super Bowl ad justified?
Meanwhile, in a piece in Esquire, Dave Holmes, who has type 1 diabetes, weighs up the pros and cons of the ad.
He writes: “While Jonas makes it look fun and easy to use a Dexcom G6 – a program to just get with like you would a drone or LED eyelashes – the process of acquiring one is complicated and often very expensive, even for people with good insurance. Which makes the year’s most expensive ad buy, for a product that only a small percentage of the U.S. population needs, confusing to me and others.”
Mr. Holmes also spoke with Craig Stubing, founder of the Beta Cell Foundation, a nonprofit that aims to educate and empower those with type 1 diabetes.
“Spending all this money on an ad, when people’s lives are at stake. I don’t know if offensive is the right word, but it seems out of touch with the reality that their patients are facing,” Mr. Stubing told Mr. Holmes.
A version of this article first appeared on Medscape.com.
A commercial for the continuous glucose monitor (CGM) Dexcom G6 shown during the Super Bowl has provoked strong reactions in the diabetes community, both positive and negative.
The 30-second ad, which aired between the first two quarters of the American football game yesterday, features singer-songwriter-actor Nick Jonas, who has type 1 diabetes. During the ad, Mr. Jonas asks – with so much technology available today, including drones that deliver packages and self-driving cars – why are people with diabetes still pricking their fingers to test their blood sugar?
Mr. Jonas goes on to demonstrate the Dexcom G6 smartphone glucose app as it displays three different glucose levels including two trending upward, explaining: “It shows your glucose right in your phone, and where it’s heading, without fingersticks. Finally, technology that makes it easier to manage our diabetes.”
Diabetes type or insulin treatment are not mentioned in the ad, despite the fact that most insurance plans typically only cover CGMs for people with type 1 diabetes and sometimes for those with type 2 diabetes who take multiple daily insulin doses (given the risk for hypoglycemia).
Ad prompts mixed reaction on social media
Reactions rolled in on Twitter after the ad debuted Feb. 2, and then again after it aired during the game.
Some people who have type 1 diabetes themselves or have children with the disease who use the product were thrilled.
“Thanks to @NickJonas for his advocacy on T1. My 11-year old has been on the Dexcom for 3 weeks. For a newly diagnosed kid, it removes a lot of anxiety (and for his parents, too!) Plus, he is thrilled his meter has a Super Bowl commercial!” tweeted @KatisJewell.
Another positive tweet, from @rturnerroy, read: “@nickjonas Thank you for bringing representation to #type1diabetes. And hey #Dexcom, you’re the best.”
But many others were critical, both of Jonas and Dexcom. @hb_herrick tweeted: “Diabetes awareness is fantastic. Dexcom being able to afford Nick Jonas for a #SuperBowl commercial is not. This is a health care product. Make it more affordable for those who need it.”
Another Twitter user, @universeofdust, tweeted: “Feeling ambivalent about the #Dexcom ad tbh. I love the awareness & representation. But also not a big fan of dexcom spending $5.5 mill+ to make the CGM seem like this ~cool & trendy~ thing when many type 1s can’t afford their insulin, let alone a CGM.”
And @andricheli wrote: “Only people lucky enough to have excellent insurance and be able to afford the out-of-pocket costs have access. Many others do not.”
And in another tweet the same user said, “The #Dexcom is an amazing device. It’s literally lifesaving and life extending. But it’s also very expensive and not available to everyone. Maybe instead of spending $5 mil on a Super Bowl ad, @dexcom should spend that on getting Dex into the handle of people who need it.”
Others, including @1hitwonderdate, criticized Mr. Jonas directly, asking him: “As someone who has struggled with diabetes and is trying to support themselves along with millions of others, why not use this platform to help those who can’t afford their supplies or are rationing them?!”
Dexcom and Jonas’ organization respond
This news organization reached out to both Dexcom and to Beyond Type 1, a nonprofit organization cofounded by Mr. Jonas, for comment. Both emailed responses.
Regarding the intended audience for the ad, Dexcom acknowledged that it hoped to reach a much wider group than just people with type 1 diabetes or even just insulin users.
“We believe our CGM technology has the ability to empower any person with diabetes and significantly improve their treatment and quality of life, whether they are using insulin or not,” the company said, adding that the ad was also aimed at “loved ones, caregivers, and even health care professionals who need to know about this technology.”
According to Dexcom, the G6 is covered by 99% of commercial insurance in the United States, in addition to Medicare, and by Medicaid in more than 40 states. Over 70% of Dexcom patients with pharmacy coverage in the United States pay under $60 per month for CGM, and a third pay $0 out-of-pocket.
“That said, we know there’s more to be done to improve access, and we are working with several partners to broaden access to Dexcom CGM, especially for people with type 2 diabetes not on mealtime insulin,” the company noted.
Beyond Type 1 responded to the criticisms about Mr. Jonas personally, noting that the celebrity is, in fact, heavily involved in advocacy.
“Nick was involved in the launch of GetInsulin.org this past October,” they said. “GetInsulin.org is a tool created by Beyond Type 1 to connect people with diabetes in the United States to the insulin access and affordability options that match their unique circumstances. ... Beyond Type 1 will continue driving awareness of short-term solutions related to insulin access and affordability while fighting for systemic change.”
The organization “is also advocating for systemic payment policies that will make devices less expensive and avoid the same pitfalls (and rising prices) as the drug pricing system in the U.S.”
Mr. Jonas himself appears aware of the concerns.
Is 2021’s most expensive Super Bowl ad justified?
Meanwhile, in a piece in Esquire, Dave Holmes, who has type 1 diabetes, weighs up the pros and cons of the ad.
He writes: “While Jonas makes it look fun and easy to use a Dexcom G6 – a program to just get with like you would a drone or LED eyelashes – the process of acquiring one is complicated and often very expensive, even for people with good insurance. Which makes the year’s most expensive ad buy, for a product that only a small percentage of the U.S. population needs, confusing to me and others.”
Mr. Holmes also spoke with Craig Stubing, founder of the Beta Cell Foundation, a nonprofit that aims to educate and empower those with type 1 diabetes.
“Spending all this money on an ad, when people’s lives are at stake. I don’t know if offensive is the right word, but it seems out of touch with the reality that their patients are facing,” Mr. Stubing told Mr. Holmes.
A version of this article first appeared on Medscape.com.
A commercial for the continuous glucose monitor (CGM) Dexcom G6 shown during the Super Bowl has provoked strong reactions in the diabetes community, both positive and negative.
The 30-second ad, which aired between the first two quarters of the American football game yesterday, features singer-songwriter-actor Nick Jonas, who has type 1 diabetes. During the ad, Mr. Jonas asks – with so much technology available today, including drones that deliver packages and self-driving cars – why are people with diabetes still pricking their fingers to test their blood sugar?
Mr. Jonas goes on to demonstrate the Dexcom G6 smartphone glucose app as it displays three different glucose levels including two trending upward, explaining: “It shows your glucose right in your phone, and where it’s heading, without fingersticks. Finally, technology that makes it easier to manage our diabetes.”
Diabetes type or insulin treatment are not mentioned in the ad, despite the fact that most insurance plans typically only cover CGMs for people with type 1 diabetes and sometimes for those with type 2 diabetes who take multiple daily insulin doses (given the risk for hypoglycemia).
Ad prompts mixed reaction on social media
Reactions rolled in on Twitter after the ad debuted Feb. 2, and then again after it aired during the game.
Some people who have type 1 diabetes themselves or have children with the disease who use the product were thrilled.
“Thanks to @NickJonas for his advocacy on T1. My 11-year old has been on the Dexcom for 3 weeks. For a newly diagnosed kid, it removes a lot of anxiety (and for his parents, too!) Plus, he is thrilled his meter has a Super Bowl commercial!” tweeted @KatisJewell.
Another positive tweet, from @rturnerroy, read: “@nickjonas Thank you for bringing representation to #type1diabetes. And hey #Dexcom, you’re the best.”
But many others were critical, both of Jonas and Dexcom. @hb_herrick tweeted: “Diabetes awareness is fantastic. Dexcom being able to afford Nick Jonas for a #SuperBowl commercial is not. This is a health care product. Make it more affordable for those who need it.”
Another Twitter user, @universeofdust, tweeted: “Feeling ambivalent about the #Dexcom ad tbh. I love the awareness & representation. But also not a big fan of dexcom spending $5.5 mill+ to make the CGM seem like this ~cool & trendy~ thing when many type 1s can’t afford their insulin, let alone a CGM.”
And @andricheli wrote: “Only people lucky enough to have excellent insurance and be able to afford the out-of-pocket costs have access. Many others do not.”
And in another tweet the same user said, “The #Dexcom is an amazing device. It’s literally lifesaving and life extending. But it’s also very expensive and not available to everyone. Maybe instead of spending $5 mil on a Super Bowl ad, @dexcom should spend that on getting Dex into the handle of people who need it.”
Others, including @1hitwonderdate, criticized Mr. Jonas directly, asking him: “As someone who has struggled with diabetes and is trying to support themselves along with millions of others, why not use this platform to help those who can’t afford their supplies or are rationing them?!”
Dexcom and Jonas’ organization respond
This news organization reached out to both Dexcom and to Beyond Type 1, a nonprofit organization cofounded by Mr. Jonas, for comment. Both emailed responses.
Regarding the intended audience for the ad, Dexcom acknowledged that it hoped to reach a much wider group than just people with type 1 diabetes or even just insulin users.
“We believe our CGM technology has the ability to empower any person with diabetes and significantly improve their treatment and quality of life, whether they are using insulin or not,” the company said, adding that the ad was also aimed at “loved ones, caregivers, and even health care professionals who need to know about this technology.”
According to Dexcom, the G6 is covered by 99% of commercial insurance in the United States, in addition to Medicare, and by Medicaid in more than 40 states. Over 70% of Dexcom patients with pharmacy coverage in the United States pay under $60 per month for CGM, and a third pay $0 out-of-pocket.
“That said, we know there’s more to be done to improve access, and we are working with several partners to broaden access to Dexcom CGM, especially for people with type 2 diabetes not on mealtime insulin,” the company noted.
Beyond Type 1 responded to the criticisms about Mr. Jonas personally, noting that the celebrity is, in fact, heavily involved in advocacy.
“Nick was involved in the launch of GetInsulin.org this past October,” they said. “GetInsulin.org is a tool created by Beyond Type 1 to connect people with diabetes in the United States to the insulin access and affordability options that match their unique circumstances. ... Beyond Type 1 will continue driving awareness of short-term solutions related to insulin access and affordability while fighting for systemic change.”
The organization “is also advocating for systemic payment policies that will make devices less expensive and avoid the same pitfalls (and rising prices) as the drug pricing system in the U.S.”
Mr. Jonas himself appears aware of the concerns.
Is 2021’s most expensive Super Bowl ad justified?
Meanwhile, in a piece in Esquire, Dave Holmes, who has type 1 diabetes, weighs up the pros and cons of the ad.
He writes: “While Jonas makes it look fun and easy to use a Dexcom G6 – a program to just get with like you would a drone or LED eyelashes – the process of acquiring one is complicated and often very expensive, even for people with good insurance. Which makes the year’s most expensive ad buy, for a product that only a small percentage of the U.S. population needs, confusing to me and others.”
Mr. Holmes also spoke with Craig Stubing, founder of the Beta Cell Foundation, a nonprofit that aims to educate and empower those with type 1 diabetes.
“Spending all this money on an ad, when people’s lives are at stake. I don’t know if offensive is the right word, but it seems out of touch with the reality that their patients are facing,” Mr. Stubing told Mr. Holmes.
A version of this article first appeared on Medscape.com.
Burnout rates in ICU staff fueled by shortages, overtime
Health care professionals working in critical care settings have been overburdened because of the plethora of COVID-19 cases, which has led to symptoms of burnout in both physicians and nurses, findings from a new study show.
“Overburdening ICU professionals during an extended period of time leads to burnout,” said lead study author Niek Kok, MSc, of IQ healthcare, Radboud University Medical Center, Radboud Institute for Health Sciences, Nijmegen, the Netherlands. “All ICU professionals are at the risk of this, and in our study, the incidence of physicians experiencing burnout was significantly higher than that of nurses in June 2020.”
This burnout can be explained by conditions caused by the pandemic, he noted, such as the scarcity of staff and resources and having to work with colleagues who were not qualified to work in critical care but who were there out of necessity.
Mr. Kok presented the findings of the study at the Critical Care Congress sponsored by the Society of Critical Care Medicine.
Burnout highest among critical care physicians
The ICU can be a stressful environment for both patients and health care personnel, and burnout is not uncommon among ICU clinicians. However, COVID-19 has amplified the degree of burnout being experienced by clinicians working in this setting. Critical care physicians now top the list of physicians experiencing burnout, at 51%, up from 44% last year, according to the Medscape report ‘Death by 1000 Thousand Cuts’: Physician Burnout and Suicide Report 2021.
The Medscape Nurse Career Satisfaction Report 2020, while not restricted to those working in critical care, also reported higher rates of burnout, compared with the prepandemic period. The percentage of nurses reporting being “very burned out” prior to the pandemic was 4%. Six months into the pandemic, that percentage soared to 18%.
In this study, Mr. Kok and colleagues examined the prevalence and incidence of burnout symptoms and moral distress in health care professionals working in the ICU, both before and during the COVID-19 pandemic.
“When the COVID-19 pandemic surfaced in the Netherlands, the health care professionals in our hospitals were motivated to do everything they could to provide the best care possible,” said Mr. Kok. “Many of the ICU professionals immediately realized that they would have to work longer hours.”
However, the health care professionals that he spoke with did have mixed feelings. Some were afraid of being infected with the virus, while others said that “it was very interesting times for them and that gave them extra motivation to do the work.
“Some physicians [and] the WHO warned that COVID-19 is not going to weathered by a heroic sprint – it is an arduous marathon that is going to go hand in hand with burnout symptoms,” Mr. Kok added. “It will eat away at our qualified ICU staff.”
Before and after data on burnout
It was widely believed that the COVID-19 pandemic would increase burnout symptoms, as had been demonstrated in studies of previous pandemics. However, Mr. Kok emphasized that there are no before and after measurements that transcend cross-sectional designs.
“The claim [has been] that it increases burnout – but there are no assessments of how it progresses in ICU professionals through time,” he said. “So what we really need is a comparison [of] before and after the pandemic.”
It is quite difficult to obtain this type of information because disruptive events like the COVID-19 pandemic cannot be predicted, he said. Thus, it is challenging to get a baseline measurement. But Mr. Kok pointed out that the study has both “before and after” measurements.
“By coincidence really, we had baseline data to measure the impact of the COVID-19 pandemic and had information that was collected before the pandemic,” he said.
In January 2020, a study began looking at the effects of ethics meetings on moral distress in ICU professionals. Data had been collected on moral distress and burnout on ICU professionals in December 2019. The first COVID-19 cases appeared in the Netherlands in February 2020.
A follow-up study was then conducted in May and June 2020, several months into the pandemic.
The longitudinal open cohort study included all ICU personnel who were working in five units within a single university medical center, plus another adult ICU that was based in a separate teaching hospital.
A total of 352 health care professionals responded to a baseline survey in October through December 2019, and then 233 responded to a follow-up survey sent in May and June 2020. The authors measured burnout symptoms and moral distress with the Maslach Burnout Inventory and the Moral Distress Scale, respectively.
Findings
The overall prevalence of burnout symptoms was 23.0% prior to the pandemic, and that jumped to 36.1% at post-peak time. Higher rates of burnout were reported by nurses (38.0%) than physicians (28.6%).
However, the incidence rate of new burnout cases was higher among physicians, compared with nurses (26.7% vs 21.9%). Not surprisingly, a higher prevalence of burnout symptoms was observed in the post-peak period for all clinicians (odds ratio, 1.83; 95% confidence interval, 1.32-2.53), and was higher for nurses (odds ratio, 1.77; 95% confidence interval, 1.03-3.04), for those working overtime (OR, 2.11; 95% CI, 1.48-3.02), and for personnel who directly engaged in patient care (OR, 1.87; 95% CI, 1.35-2.60).
Physicians in general were much more likely to develop burnout symptoms related to the pandemic, compared with nurses (OR, 3.56; 95% CI, 1.06-12.21).
When looking at findings on moral distress, Kok pointed out that it often arises in situations when the health care professional knows the right thing to do but is prevented from doing so. “Morally distressful situations all rose from December to June,” said Mr. Kok. “Scarcity was the most distressing. The other was where colleagues were perceived to be less skilled, and this had to do with the recruitment of people from outside of the ICU to provide care.”
Moral distress from scarcity and unskilled colleagues were both significantly related to burnout, he noted.
In the final model, working in a COVID-19 unit, stress from scarcity of resources and people, stress from unskilled colleagues, and stress from unsafe conditions were all related to burnout. “The stress of physicians was significantly higher,” said Kok. “Even though nurses had higher baseline burnout, it became less pronounced in June 2020. This indicates that burnout was significantly higher in physicians.”
Thus, Mr. Kok and colleagues concluded that overburdening ICU professionals during an extended period of time leads to burnout, and all ICU workers are at risk.
Burnout rates higher in physicians
Weighing in on the study, Greg S. Martin, MD, FCCP, professor of medicine in the division of pulmonary, allergy, critical care and sleep medicine, Emory University, Atlanta, noted that the differences observed between physicians and nurses may have to do with the fact that “nurses have been smoldering all along and experiencing higher rates of burnout.
“They may have adapted better to the pandemic conditions, since they are more used to working overtime and short staffed, and spending far more time at the bedside,” he said. “Because of the volume of patients, physicians may be spending more hours doing patient care and are experiencing more burnout.”

For physicians, this may be a more significant change in the workload, as well as the complexity of the situation because of the pandemic. “Many things layer into it, such as [the fact] that there are no families present to give patients support, the complexity of care of these patients, and things like lack of PPE,” Dr. Martin said.
The study did not differentiate among physician groups, so it is unclear if the affected physicians were residents, fellows, or more senior staff. “Residents are often quite busy already, and don’t usually have the capacity to add more to their schedules, and maybe attendings were having to spend more time doing patient care,” Dr. Martin said. “In the United States, at least some personnel were restricted from working with COVID-19 patients. Medical students were removed in many places as well as nonessential staff, so that may have also added to their burnout.”
The study was conducted in the Netherlands, so there may be differences in the work environment, responsibilities of nurses vs. physicians, staffing, and so on. “But it still shows that burnout is very real among doctors and nurses working in the ICU in pandemic conditions,” he said.
Health care professionals working in critical care settings have been overburdened because of the plethora of COVID-19 cases, which has led to symptoms of burnout in both physicians and nurses, findings from a new study show.
“Overburdening ICU professionals during an extended period of time leads to burnout,” said lead study author Niek Kok, MSc, of IQ healthcare, Radboud University Medical Center, Radboud Institute for Health Sciences, Nijmegen, the Netherlands. “All ICU professionals are at the risk of this, and in our study, the incidence of physicians experiencing burnout was significantly higher than that of nurses in June 2020.”
This burnout can be explained by conditions caused by the pandemic, he noted, such as the scarcity of staff and resources and having to work with colleagues who were not qualified to work in critical care but who were there out of necessity.
Mr. Kok presented the findings of the study at the Critical Care Congress sponsored by the Society of Critical Care Medicine.
Burnout highest among critical care physicians
The ICU can be a stressful environment for both patients and health care personnel, and burnout is not uncommon among ICU clinicians. However, COVID-19 has amplified the degree of burnout being experienced by clinicians working in this setting. Critical care physicians now top the list of physicians experiencing burnout, at 51%, up from 44% last year, according to the Medscape report ‘Death by 1000 Thousand Cuts’: Physician Burnout and Suicide Report 2021.
The Medscape Nurse Career Satisfaction Report 2020, while not restricted to those working in critical care, also reported higher rates of burnout, compared with the prepandemic period. The percentage of nurses reporting being “very burned out” prior to the pandemic was 4%. Six months into the pandemic, that percentage soared to 18%.
In this study, Mr. Kok and colleagues examined the prevalence and incidence of burnout symptoms and moral distress in health care professionals working in the ICU, both before and during the COVID-19 pandemic.
“When the COVID-19 pandemic surfaced in the Netherlands, the health care professionals in our hospitals were motivated to do everything they could to provide the best care possible,” said Mr. Kok. “Many of the ICU professionals immediately realized that they would have to work longer hours.”
However, the health care professionals that he spoke with did have mixed feelings. Some were afraid of being infected with the virus, while others said that “it was very interesting times for them and that gave them extra motivation to do the work.
“Some physicians [and] the WHO warned that COVID-19 is not going to weathered by a heroic sprint – it is an arduous marathon that is going to go hand in hand with burnout symptoms,” Mr. Kok added. “It will eat away at our qualified ICU staff.”
Before and after data on burnout
It was widely believed that the COVID-19 pandemic would increase burnout symptoms, as had been demonstrated in studies of previous pandemics. However, Mr. Kok emphasized that there are no before and after measurements that transcend cross-sectional designs.
“The claim [has been] that it increases burnout – but there are no assessments of how it progresses in ICU professionals through time,” he said. “So what we really need is a comparison [of] before and after the pandemic.”
It is quite difficult to obtain this type of information because disruptive events like the COVID-19 pandemic cannot be predicted, he said. Thus, it is challenging to get a baseline measurement. But Mr. Kok pointed out that the study has both “before and after” measurements.
“By coincidence really, we had baseline data to measure the impact of the COVID-19 pandemic and had information that was collected before the pandemic,” he said.
In January 2020, a study began looking at the effects of ethics meetings on moral distress in ICU professionals. Data had been collected on moral distress and burnout on ICU professionals in December 2019. The first COVID-19 cases appeared in the Netherlands in February 2020.
A follow-up study was then conducted in May and June 2020, several months into the pandemic.
The longitudinal open cohort study included all ICU personnel who were working in five units within a single university medical center, plus another adult ICU that was based in a separate teaching hospital.
A total of 352 health care professionals responded to a baseline survey in October through December 2019, and then 233 responded to a follow-up survey sent in May and June 2020. The authors measured burnout symptoms and moral distress with the Maslach Burnout Inventory and the Moral Distress Scale, respectively.
Findings
The overall prevalence of burnout symptoms was 23.0% prior to the pandemic, and that jumped to 36.1% at post-peak time. Higher rates of burnout were reported by nurses (38.0%) than physicians (28.6%).
However, the incidence rate of new burnout cases was higher among physicians, compared with nurses (26.7% vs 21.9%). Not surprisingly, a higher prevalence of burnout symptoms was observed in the post-peak period for all clinicians (odds ratio, 1.83; 95% confidence interval, 1.32-2.53), and was higher for nurses (odds ratio, 1.77; 95% confidence interval, 1.03-3.04), for those working overtime (OR, 2.11; 95% CI, 1.48-3.02), and for personnel who directly engaged in patient care (OR, 1.87; 95% CI, 1.35-2.60).
Physicians in general were much more likely to develop burnout symptoms related to the pandemic, compared with nurses (OR, 3.56; 95% CI, 1.06-12.21).
When looking at findings on moral distress, Kok pointed out that it often arises in situations when the health care professional knows the right thing to do but is prevented from doing so. “Morally distressful situations all rose from December to June,” said Mr. Kok. “Scarcity was the most distressing. The other was where colleagues were perceived to be less skilled, and this had to do with the recruitment of people from outside of the ICU to provide care.”
Moral distress from scarcity and unskilled colleagues were both significantly related to burnout, he noted.
In the final model, working in a COVID-19 unit, stress from scarcity of resources and people, stress from unskilled colleagues, and stress from unsafe conditions were all related to burnout. “The stress of physicians was significantly higher,” said Kok. “Even though nurses had higher baseline burnout, it became less pronounced in June 2020. This indicates that burnout was significantly higher in physicians.”
Thus, Mr. Kok and colleagues concluded that overburdening ICU professionals during an extended period of time leads to burnout, and all ICU workers are at risk.
Burnout rates higher in physicians
Weighing in on the study, Greg S. Martin, MD, FCCP, professor of medicine in the division of pulmonary, allergy, critical care and sleep medicine, Emory University, Atlanta, noted that the differences observed between physicians and nurses may have to do with the fact that “nurses have been smoldering all along and experiencing higher rates of burnout.
“They may have adapted better to the pandemic conditions, since they are more used to working overtime and short staffed, and spending far more time at the bedside,” he said. “Because of the volume of patients, physicians may be spending more hours doing patient care and are experiencing more burnout.”
For physicians, this may be a more significant change in the workload, as well as the complexity of the situation because of the pandemic. “Many things layer into it, such as [the fact] that there are no families present to give patients support, the complexity of care of these patients, and things like lack of PPE,” Dr. Martin said.
The study did not differentiate among physician groups, so it is unclear if the affected physicians were residents, fellows, or more senior staff. “Residents are often quite busy already, and don’t usually have the capacity to add more to their schedules, and maybe attendings were having to spend more time doing patient care,” Dr. Martin said. “In the United States, at least some personnel were restricted from working with COVID-19 patients. Medical students were removed in many places as well as nonessential staff, so that may have also added to their burnout.”
The study was conducted in the Netherlands, so there may be differences in the work environment, responsibilities of nurses vs. physicians, staffing, and so on. “But it still shows that burnout is very real among doctors and nurses working in the ICU in pandemic conditions,” he said.
Health care professionals working in critical care settings have been overburdened because of the plethora of COVID-19 cases, which has led to symptoms of burnout in both physicians and nurses, findings from a new study show.
“Overburdening ICU professionals during an extended period of time leads to burnout,” said lead study author Niek Kok, MSc, of IQ healthcare, Radboud University Medical Center, Radboud Institute for Health Sciences, Nijmegen, the Netherlands. “All ICU professionals are at the risk of this, and in our study, the incidence of physicians experiencing burnout was significantly higher than that of nurses in June 2020.”
This burnout can be explained by conditions caused by the pandemic, he noted, such as the scarcity of staff and resources and having to work with colleagues who were not qualified to work in critical care but who were there out of necessity.
Mr. Kok presented the findings of the study at the Critical Care Congress sponsored by the Society of Critical Care Medicine.
Burnout highest among critical care physicians
The ICU can be a stressful environment for both patients and health care personnel, and burnout is not uncommon among ICU clinicians. However, COVID-19 has amplified the degree of burnout being experienced by clinicians working in this setting. Critical care physicians now top the list of physicians experiencing burnout, at 51%, up from 44% last year, according to the Medscape report ‘Death by 1000 Thousand Cuts’: Physician Burnout and Suicide Report 2021.
The Medscape Nurse Career Satisfaction Report 2020, while not restricted to those working in critical care, also reported higher rates of burnout, compared with the prepandemic period. The percentage of nurses reporting being “very burned out” prior to the pandemic was 4%. Six months into the pandemic, that percentage soared to 18%.
In this study, Mr. Kok and colleagues examined the prevalence and incidence of burnout symptoms and moral distress in health care professionals working in the ICU, both before and during the COVID-19 pandemic.
“When the COVID-19 pandemic surfaced in the Netherlands, the health care professionals in our hospitals were motivated to do everything they could to provide the best care possible,” said Mr. Kok. “Many of the ICU professionals immediately realized that they would have to work longer hours.”
However, the health care professionals that he spoke with did have mixed feelings. Some were afraid of being infected with the virus, while others said that “it was very interesting times for them and that gave them extra motivation to do the work.
“Some physicians [and] the WHO warned that COVID-19 is not going to weathered by a heroic sprint – it is an arduous marathon that is going to go hand in hand with burnout symptoms,” Mr. Kok added. “It will eat away at our qualified ICU staff.”
Before and after data on burnout
It was widely believed that the COVID-19 pandemic would increase burnout symptoms, as had been demonstrated in studies of previous pandemics. However, Mr. Kok emphasized that there are no before and after measurements that transcend cross-sectional designs.
“The claim [has been] that it increases burnout – but there are no assessments of how it progresses in ICU professionals through time,” he said. “So what we really need is a comparison [of] before and after the pandemic.”
It is quite difficult to obtain this type of information because disruptive events like the COVID-19 pandemic cannot be predicted, he said. Thus, it is challenging to get a baseline measurement. But Mr. Kok pointed out that the study has both “before and after” measurements.
“By coincidence really, we had baseline data to measure the impact of the COVID-19 pandemic and had information that was collected before the pandemic,” he said.
In January 2020, a study began looking at the effects of ethics meetings on moral distress in ICU professionals. Data had been collected on moral distress and burnout on ICU professionals in December 2019. The first COVID-19 cases appeared in the Netherlands in February 2020.
A follow-up study was then conducted in May and June 2020, several months into the pandemic.
The longitudinal open cohort study included all ICU personnel who were working in five units within a single university medical center, plus another adult ICU that was based in a separate teaching hospital.
A total of 352 health care professionals responded to a baseline survey in October through December 2019, and then 233 responded to a follow-up survey sent in May and June 2020. The authors measured burnout symptoms and moral distress with the Maslach Burnout Inventory and the Moral Distress Scale, respectively.
Findings
The overall prevalence of burnout symptoms was 23.0% prior to the pandemic, and that jumped to 36.1% at post-peak time. Higher rates of burnout were reported by nurses (38.0%) than physicians (28.6%).
However, the incidence rate of new burnout cases was higher among physicians, compared with nurses (26.7% vs 21.9%). Not surprisingly, a higher prevalence of burnout symptoms was observed in the post-peak period for all clinicians (odds ratio, 1.83; 95% confidence interval, 1.32-2.53), and was higher for nurses (odds ratio, 1.77; 95% confidence interval, 1.03-3.04), for those working overtime (OR, 2.11; 95% CI, 1.48-3.02), and for personnel who directly engaged in patient care (OR, 1.87; 95% CI, 1.35-2.60).
Physicians in general were much more likely to develop burnout symptoms related to the pandemic, compared with nurses (OR, 3.56; 95% CI, 1.06-12.21).
When looking at findings on moral distress, Kok pointed out that it often arises in situations when the health care professional knows the right thing to do but is prevented from doing so. “Morally distressful situations all rose from December to June,” said Mr. Kok. “Scarcity was the most distressing. The other was where colleagues were perceived to be less skilled, and this had to do with the recruitment of people from outside of the ICU to provide care.”
Moral distress from scarcity and unskilled colleagues were both significantly related to burnout, he noted.
In the final model, working in a COVID-19 unit, stress from scarcity of resources and people, stress from unskilled colleagues, and stress from unsafe conditions were all related to burnout. “The stress of physicians was significantly higher,” said Kok. “Even though nurses had higher baseline burnout, it became less pronounced in June 2020. This indicates that burnout was significantly higher in physicians.”
Thus, Mr. Kok and colleagues concluded that overburdening ICU professionals during an extended period of time leads to burnout, and all ICU workers are at risk.
Burnout rates higher in physicians
Weighing in on the study, Greg S. Martin, MD, FCCP, professor of medicine in the division of pulmonary, allergy, critical care and sleep medicine, Emory University, Atlanta, noted that the differences observed between physicians and nurses may have to do with the fact that “nurses have been smoldering all along and experiencing higher rates of burnout.
“They may have adapted better to the pandemic conditions, since they are more used to working overtime and short staffed, and spending far more time at the bedside,” he said. “Because of the volume of patients, physicians may be spending more hours doing patient care and are experiencing more burnout.”
For physicians, this may be a more significant change in the workload, as well as the complexity of the situation because of the pandemic. “Many things layer into it, such as [the fact] that there are no families present to give patients support, the complexity of care of these patients, and things like lack of PPE,” Dr. Martin said.
The study did not differentiate among physician groups, so it is unclear if the affected physicians were residents, fellows, or more senior staff. “Residents are often quite busy already, and don’t usually have the capacity to add more to their schedules, and maybe attendings were having to spend more time doing patient care,” Dr. Martin said. “In the United States, at least some personnel were restricted from working with COVID-19 patients. Medical students were removed in many places as well as nonessential staff, so that may have also added to their burnout.”
The study was conducted in the Netherlands, so there may be differences in the work environment, responsibilities of nurses vs. physicians, staffing, and so on. “But it still shows that burnout is very real among doctors and nurses working in the ICU in pandemic conditions,” he said.
FROM CCC50
Mask mandates reduced COVID-19 hospitalizations
States that implemented mask mandates in 2020 saw a decline in the growth of COVID-19 hospitalizations between March and October 2020, according to a new study published Feb. 5 in the CDC’s Morbidity and Mortality Weekly Report.
Hospitalization growth rates declined by 5.5 percentage points for adults between ages 18-64 about 3 weeks after the mandates were implemented, compared with climbing growth rates in the 4 weeks before mandates.
CDC Director Rochelle Walensky said she was pleased to see the results, but that it’s “too early” to tell whether President Joe Biden’s recent mask orders have had an effect on cases and hospitalizations in 2021.
“We’re going to be watching the mask data very carefully,” she said during a news briefing with the White House COVID-19 Response Team on Feb. 5. “I think it’s probably still a bit too early to tell, but I’m encouraged with the decrease in case rates right now.”
In another study published Feb. 5 in the Morbidity and Mortality Weekly Report, trained observers tracked mask use at six universities with mask mandates between September and November 2020. Overall, observers reported that about 92% of people wore masks correctly indoors, which varied based on the type of mask.
About 97% of people used N95 masks correctly, compared with 92% who used cloth masks, and 79% who used bandanas, scarves, or neck gaiters. Cloth masks were most common, and bandanas and scarves were least common.
The Biden administration is considering whether to send masks directly to American households to encourage people to wear them, according to NBC News. The White House COVID-19 Response Team is debating the logistics of mailing out masks, including how many to send and what the mask material would be, the news outlet reported.
Wisconsin Gov. Tony Evers reissued a new statewide mask mandate on Feb. 4, just an hour after the Republican-controlled legislature voted to repeal his previous mandate, according to The Associated Press. Gov. Evers said his priority is to keep people safe and that wearing a mask is the easiest way to do so.
“If the legislature keeps playing politics and we don’t keep wearing masks, we’re going to see more preventable deaths,” he said. “It’s going to take even longer to get our state and our economy back on track.”
A version of this article first appeared on WebMD.com.
States that implemented mask mandates in 2020 saw a decline in the growth of COVID-19 hospitalizations between March and October 2020, according to a new study published Feb. 5 in the CDC’s Morbidity and Mortality Weekly Report.
Hospitalization growth rates declined by 5.5 percentage points for adults between ages 18-64 about 3 weeks after the mandates were implemented, compared with climbing growth rates in the 4 weeks before mandates.
CDC Director Rochelle Walensky said she was pleased to see the results, but that it’s “too early” to tell whether President Joe Biden’s recent mask orders have had an effect on cases and hospitalizations in 2021.
“We’re going to be watching the mask data very carefully,” she said during a news briefing with the White House COVID-19 Response Team on Feb. 5. “I think it’s probably still a bit too early to tell, but I’m encouraged with the decrease in case rates right now.”
In another study published Feb. 5 in the Morbidity and Mortality Weekly Report, trained observers tracked mask use at six universities with mask mandates between September and November 2020. Overall, observers reported that about 92% of people wore masks correctly indoors, which varied based on the type of mask.
About 97% of people used N95 masks correctly, compared with 92% who used cloth masks, and 79% who used bandanas, scarves, or neck gaiters. Cloth masks were most common, and bandanas and scarves were least common.
The Biden administration is considering whether to send masks directly to American households to encourage people to wear them, according to NBC News. The White House COVID-19 Response Team is debating the logistics of mailing out masks, including how many to send and what the mask material would be, the news outlet reported.
Wisconsin Gov. Tony Evers reissued a new statewide mask mandate on Feb. 4, just an hour after the Republican-controlled legislature voted to repeal his previous mandate, according to The Associated Press. Gov. Evers said his priority is to keep people safe and that wearing a mask is the easiest way to do so.
“If the legislature keeps playing politics and we don’t keep wearing masks, we’re going to see more preventable deaths,” he said. “It’s going to take even longer to get our state and our economy back on track.”
A version of this article first appeared on WebMD.com.
States that implemented mask mandates in 2020 saw a decline in the growth of COVID-19 hospitalizations between March and October 2020, according to a new study published Feb. 5 in the CDC’s Morbidity and Mortality Weekly Report.
Hospitalization growth rates declined by 5.5 percentage points for adults between ages 18-64 about 3 weeks after the mandates were implemented, compared with climbing growth rates in the 4 weeks before mandates.
CDC Director Rochelle Walensky said she was pleased to see the results, but that it’s “too early” to tell whether President Joe Biden’s recent mask orders have had an effect on cases and hospitalizations in 2021.
“We’re going to be watching the mask data very carefully,” she said during a news briefing with the White House COVID-19 Response Team on Feb. 5. “I think it’s probably still a bit too early to tell, but I’m encouraged with the decrease in case rates right now.”
In another study published Feb. 5 in the Morbidity and Mortality Weekly Report, trained observers tracked mask use at six universities with mask mandates between September and November 2020. Overall, observers reported that about 92% of people wore masks correctly indoors, which varied based on the type of mask.
About 97% of people used N95 masks correctly, compared with 92% who used cloth masks, and 79% who used bandanas, scarves, or neck gaiters. Cloth masks were most common, and bandanas and scarves were least common.
The Biden administration is considering whether to send masks directly to American households to encourage people to wear them, according to NBC News. The White House COVID-19 Response Team is debating the logistics of mailing out masks, including how many to send and what the mask material would be, the news outlet reported.
Wisconsin Gov. Tony Evers reissued a new statewide mask mandate on Feb. 4, just an hour after the Republican-controlled legislature voted to repeal his previous mandate, according to The Associated Press. Gov. Evers said his priority is to keep people safe and that wearing a mask is the easiest way to do so.
“If the legislature keeps playing politics and we don’t keep wearing masks, we’re going to see more preventable deaths,” he said. “It’s going to take even longer to get our state and our economy back on track.”
A version of this article first appeared on WebMD.com.
FDA curbs use of COVID-19 convalescent plasma, citing new data
The Food and Drug Administration has revised its emergency use authorization for COVID-19 convalescent plasma on the basis of the latest available data.
The revision states that only high-titer COVID-19 convalescent plasma can be used and only in hospitalized patients who are early in the disease course and those with impaired humoral immunity who cannot produce an adequate antibody response.
The revisions stem from new clinical trial data analyzed or reported since the original EUA was issued in August 2020. The original EUA did not have these restrictions.
“This and other changes to the EUA represent important updates to the use of convalescent plasma for the treatment of COVID-19 patients,” Peter Marks, MD, PhD, director, FDA Center for Biologics Evaluation and Research, said in a statement announcing the revisions.
“COVID-19 convalescent plasma used according to the revised EUA may have efficacy, and its known and potential benefits outweigh its known and potential risks,” the FDA said.
The agency said it revoked use of low-titer COVID-19 convalescent plasma on the basis of new data from clinical trials, including randomized, controlled trials, that have failed to demonstrate that low-titer convalescent plasma may be effective in the treatment of hospitalized patients with COVID-19.
The FDA’s updated fact sheet for health care providers on the use of COVID-19 convalescent plasma also notes that transfusion of COVID-19 convalescent plasma late in the disease course, following respiratory failure requiring intubation and mechanical ventilation, hasn’t been found to have clinical benefit.
The revised EUA also includes several additional tests that can be used to manufacture COVID-19 convalescent plasma.
“With this update, nine tests are now included in the EUA for testing plasma donations for anti-SARS-CoV-2 antibodies as a manufacturing step to determine suitability before release,” the FDA said.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has revised its emergency use authorization for COVID-19 convalescent plasma on the basis of the latest available data.
The revision states that only high-titer COVID-19 convalescent plasma can be used and only in hospitalized patients who are early in the disease course and those with impaired humoral immunity who cannot produce an adequate antibody response.
The revisions stem from new clinical trial data analyzed or reported since the original EUA was issued in August 2020. The original EUA did not have these restrictions.
“This and other changes to the EUA represent important updates to the use of convalescent plasma for the treatment of COVID-19 patients,” Peter Marks, MD, PhD, director, FDA Center for Biologics Evaluation and Research, said in a statement announcing the revisions.
“COVID-19 convalescent plasma used according to the revised EUA may have efficacy, and its known and potential benefits outweigh its known and potential risks,” the FDA said.
The agency said it revoked use of low-titer COVID-19 convalescent plasma on the basis of new data from clinical trials, including randomized, controlled trials, that have failed to demonstrate that low-titer convalescent plasma may be effective in the treatment of hospitalized patients with COVID-19.
The FDA’s updated fact sheet for health care providers on the use of COVID-19 convalescent plasma also notes that transfusion of COVID-19 convalescent plasma late in the disease course, following respiratory failure requiring intubation and mechanical ventilation, hasn’t been found to have clinical benefit.
The revised EUA also includes several additional tests that can be used to manufacture COVID-19 convalescent plasma.
“With this update, nine tests are now included in the EUA for testing plasma donations for anti-SARS-CoV-2 antibodies as a manufacturing step to determine suitability before release,” the FDA said.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has revised its emergency use authorization for COVID-19 convalescent plasma on the basis of the latest available data.
The revision states that only high-titer COVID-19 convalescent plasma can be used and only in hospitalized patients who are early in the disease course and those with impaired humoral immunity who cannot produce an adequate antibody response.
The revisions stem from new clinical trial data analyzed or reported since the original EUA was issued in August 2020. The original EUA did not have these restrictions.
“This and other changes to the EUA represent important updates to the use of convalescent plasma for the treatment of COVID-19 patients,” Peter Marks, MD, PhD, director, FDA Center for Biologics Evaluation and Research, said in a statement announcing the revisions.
“COVID-19 convalescent plasma used according to the revised EUA may have efficacy, and its known and potential benefits outweigh its known and potential risks,” the FDA said.
The agency said it revoked use of low-titer COVID-19 convalescent plasma on the basis of new data from clinical trials, including randomized, controlled trials, that have failed to demonstrate that low-titer convalescent plasma may be effective in the treatment of hospitalized patients with COVID-19.
The FDA’s updated fact sheet for health care providers on the use of COVID-19 convalescent plasma also notes that transfusion of COVID-19 convalescent plasma late in the disease course, following respiratory failure requiring intubation and mechanical ventilation, hasn’t been found to have clinical benefit.
The revised EUA also includes several additional tests that can be used to manufacture COVID-19 convalescent plasma.
“With this update, nine tests are now included in the EUA for testing plasma donations for anti-SARS-CoV-2 antibodies as a manufacturing step to determine suitability before release,” the FDA said.
A version of this article first appeared on Medscape.com.
Reimbursement for Teledermatology During the COVID-19 Public Health Emergency: Change Has Come, But Will It Stay?
The world of telemedicine—especially teledermatology—had been a sleepy underutilized afterthought for most physicians until we were faced with a global pandemic the likes of which none of us had seen in our lifetimes. And just like that, teledermatology went from an afterthought to part of the “new normal.” Although those of us already practicing telemedicine knew of potential pitfalls and concerns, this great social experiment of throwing everyone into unexplored territory led to a great deal of frustration with technology and workflows that were not optimized for dermatology visits. The process is still changing, and the technical aspects of conducting teledermatology visits will no doubt improve, but what about the bigger question of reimbursement? Without adequate payments and financial models, the long-term future of telemedicine is uncertain, so an understanding of the current and likely future landscape of telemedicine reimbursement is critical.
Waivers During the Public Health Emergency
The declaration of a public health emergency (PHE)allowed for significant flexibility by the Centers for Medicare & Medicaid Services (CMS) during the coronavirus disease 2019 (COVID-19) pandemic. Importantly, the CMS was permitted to act quickly to allow telehealth to flourish during the worst of the pandemic and throughout the declared PHE, which has been extended several times already. Currently, the PHE is set to expire on April 20, 2021, but may be extended again if the pandemic is ongoing. The most important of these waivers was probably the removal of both the originating site and geographic requirements for telehealth services.1 Prior to the COVID-19 PHE, a patient would have to travel to a doctor’s office, hospital, or skilled nursing facility to receive telehealth care (originating site requirement), and even then this was only allowed in defined rural areas of the country (geographic requirement). Both of these requirements were waived, allowing for any patient to receive telehealth services within their own homes. Concurrently, the requirement that patients must have an established relationship with the provider (ie, telehealth could not be used to provide care to new patients) also was waived.1
In the spirit of expanding access to care and providing reasonable reimbursement for medical services, other changes were made for which the CMS should be commended. In acknowledging that many Medicare/Medicaid beneficiaries may not have access to devices that permit real-time, 2-way audio/video communication, which previously were necessary to qualify for a telehealth encounter, the CMS decided to cover telephone visits and provide reimbursement at the level of an established visit.1 They also changed the billing structure to remove the place of service (POS) designation for telehealth (POS 02) and replace it with the normal physician’s office POS designation (usually POS 11), bringing back a telehealth modifier (modifier -95) in the process. The benefit of this change is solely to increase reimbursement for these services, as telehealth POS services generally are covered at lower facility rates, whereas POS 11 codes are reimbursed at the full level of a nonfacility physician’s office rate.
Finally, other waivers such as the Office of Civil Rights’ decision to waive HIPAA (Health Insurance Portability and Accountability Act) violations for telehealth platforms during the PHE allowed offices to take on telemedicine quickly without having to implement a new infrastructure.2 Numerous codes were added to the list of covered services for telehealth, but these generally are not relevant for dermatologists. The CMS also allowed physicians’ offices to waive the patient responsibility/co-pay during the COVID-19 PHE, which previously was not allowed due to concerns about the anti-kickback statute.1 These co-pay waivers were intended to remove another barrier to care for patients who were hesitant to participate in virtual visits. For the most part, the waiver of state licensing requirements is a bit less useful. As part of the CMS waiver, providers technically are allowed to see out-of-state Medicare/Medicaid beneficiaries, but state licensing laws are still in effect; thus, in the absence of a blanket state-level waiver (which some states enacted, modeled after the Uniform Emergency Volunteer Health Practitioner Act of 20063), providers still cannot see most out-of-state patients from a legal and malpractice coverage standpoint.
An important flexibility during the COVID-19 PHE is one that often is underrecognized. The CMS has been clear about the ability to provide direct supervision for advanced practice providers (APPs) and residents via telehealth during the PHE, which allows for incident-to billing for APPs at remote sites given that the supervising physician is immediately available via an interactive, 2-way, live audio/video telecommunications method. It also allows for direct supervision of APPs and residents using such technology. For dermatology, which does not have a primary care waiver, an attending must still directly supervise each patient and see the patient via a live audio/video modality but does not have to be on-site to do so. This is a very interesting concept that, if extended, could truly impact practice management for the long-term.
Response From Commercial Insurance Carriers
Tracking along with the CMS waivers and flexibilities during the PHE, most commercial carriers quickly adopted similar policies to cover telehealth services. It should be noted that for most commercial insurance carriers, the coverage was already broader than Medicare/Medicaid coverage for telehealth prior to the PHE, so in many ways it is an extension of that concept and acceptance of telemedicine as a whole. What is sometimes confusing, though, is that various policies and requirements around billing exist; for example, while most carriers emulated the POS requirements that the CMS adopted, some carriers still stuck with the telemedicine POS but paid full in-office visit rates for those codes. Some carriers adopted higher reimbursement rates for telephone visits, similar to the CMS, while others instructed providers to just bill for the established office visit codes and allowed for telephone-only visits to qualify for these billing codes. Some carriers also waived co-pays for telehealth visits for their members (whether related to COVID-19 or not). It is beyond the scope of this article to delve into the specifics, which may vary not only by carrier but by region and plan. However, it is important to stay on top of one’s insurance carriers to find out what their latest directives are for billing for telehealth.
Postpandemic Teledermatology
What about the future of teledermatology? Although many dermatologists have adopted telehealth services out of necessity during the COVID-19 PHE, the jury is still out on the long-term forecast for telemedicine in dermatology. Concerns about liability/malpractice and technology issues abound, and for many, the headaches of teledermatology—such as trying to focus on a blurry photograph of a nevus that the patient is concerned about—make it unappealing. Some of these issues will be addressed by better technology, but the reimbursement structure must continue for teledermatology to remain in widespread use.
Currently, the biggest question facing telehealth is whether the waivers for originating site and geographic requirements will be able to continue. The CMS itself does not have the statutory authority to make these changes permanent and was only allowed to act due to a waiver under section 1135 of the Social Security Act during a PHE. It would take an act of Congress to change the law to allow for this specific expansion of telehealth services. A number of federal bills, including S 2741 (Creating Opportunities Now for Necessary and Effective Care Technologies [CONNECT] for Health Act of 2019) and S 4796 (Fair Care Act of 2020) from the Senate, contain such provisions, but none have been passed at the time of writing. There does seem to be broad support of the concept of expanding telemedicine access, such as noted by New York State Governor Andrew Cuomo in his 2021 State of the State address,4 but it remains to be seen when action will come.
Some regulations, such as the HIPAA waiver and the ability to waive co-pays, are not slated to continue after the pandemic. The ability to supervise residents via telehealth (real-time audio/video) has been made permanent, but only in rural areas. Direct supervision of APPs via telehealth will continue through the end of the calendar year of the PHE or the end of 2021, whichever comes later, but it remains to be seen whether remote supervision will continue. The CMS has stated in its comments that it is looking at this issue closely and may establish certain guardrails to ensure quality of care is maintained.1 Telephone/audio-only visits also may come under further scrutiny, but research has supported the concept that patients who are more likely to gain access through audio-only modalities are older, Medicare/Medicaid (vs commercial), and Black (vs White) patients,5 so it would indeed introduce an unfair barrier to access if such coverage was rolled back.
Final Thoughts
Overall, we have made much progress in teledermatology. Once utilized by a small fraction of dermatologists, the vast majority of us turned to teledermatology to sustain our practices during the COVID-19 pandemic. Moving forward, there are 2 critical factors to consider: continued technological innovation and permanent coverage for telehealth reimbursement at in-office visit levels. With these challenges resolved, we can move forward and consider novel models that may be able to deliver dermatologic care to a broader patient population, thereby solving the critical issue of access to care for so many patients in need in our country.
- Medicare Program; CY 2021 Payment Policies Under the Physician Fee Schedule and Other Changes to Part B Payment Policies; Medicare Shared Savings Program Requirements; Medicaid Promoting Interoperability Program Requirements for Eligible Professionals; Quality Payment Program; Coverage of Opioid Use Disorder Services Furnished by Opioid Treatment Programs; Medicare Enrollment of Opioid Treatment Programs; Electronic Prescribing for Controlled Substances for a Covered Part D Drug; Payment for Office/Outpatient Evaluation and Management Services; Hospital IQR Program; Establish New Code Categories; Medicare Diabetes Prevention Program (MDPP) Expanded Model Emergency Policy; Coding and Payment for Virtual Check-in Services Interim Final Rule Policy; Coding and Payment for Personal Protective Equipment (PPE) Interim Final Rule Policy; Regulatory Revisions in Response to the Public Health Emergency (PHE) for COVID-19; and Finalization of Certain Provisions from the March 31st, May 8th and September 2nd Interim Final Rules in Response to the PHE for COVID-19. Fed Registr. 2020;85:84472-85377. To be codified at 42 CFR §400, 410, 414, 415, 423, 424, and 425. https://www.federalregister.gov/documents/2020/12/28/2020-26815/medicare-program-cy-2021-payment-policies-under-the-physician-fee-schedule-and-other-changes-to-part
- Office for Civil Rights. Notification of enforcement discretion for telehealth remote communications during the COVID-19 nationwide public health emergency. US Department of Health and Human Services website. Reviewed January 20, 2021. Accessed January 25, 2021. https://www.hhs.gov/hipaa/for-professionals/special-topics/emergency-preparedness/notification-enforcement-discretion-telehealth/index.html
- Hoffman DA. Increasing access to care: telehealth during COVID-19 [published online June 16, 2020]. J Law Biosci. doi:10.1093/jlb/lsaa043
- Governor Cuomo announces proposal to expand access to telehealth for all as part of 2021 State of the State. New York State website. Published January 10, 2021. Accessed January 25, 021. https://www.governor.ny.gov/news/governor-cuomo-announces-proposal-expand-access-telehealth-all-part-2021-state-state#:~:text=and%20Rural%20Communities-,Governor%20Andrew%20M.,2021%20State%20of%20the%20State.&text=New%20Yorkers%20have%20adapted%20throughout,into%20our%20existing%20healthcare%20system
- Gilson SF, Umscheid CA, Laiteerapong N, et al. Growth of ambulatory virtual visit and differential use by patient sociodemographics at one urban academic medical center during the COVID-19 pandemic: retrospective analysis. JMIR Med Inform. 2020;8:E24544.
The world of telemedicine—especially teledermatology—had been a sleepy underutilized afterthought for most physicians until we were faced with a global pandemic the likes of which none of us had seen in our lifetimes. And just like that, teledermatology went from an afterthought to part of the “new normal.” Although those of us already practicing telemedicine knew of potential pitfalls and concerns, this great social experiment of throwing everyone into unexplored territory led to a great deal of frustration with technology and workflows that were not optimized for dermatology visits. The process is still changing, and the technical aspects of conducting teledermatology visits will no doubt improve, but what about the bigger question of reimbursement? Without adequate payments and financial models, the long-term future of telemedicine is uncertain, so an understanding of the current and likely future landscape of telemedicine reimbursement is critical.
Waivers During the Public Health Emergency
The declaration of a public health emergency (PHE)allowed for significant flexibility by the Centers for Medicare & Medicaid Services (CMS) during the coronavirus disease 2019 (COVID-19) pandemic. Importantly, the CMS was permitted to act quickly to allow telehealth to flourish during the worst of the pandemic and throughout the declared PHE, which has been extended several times already. Currently, the PHE is set to expire on April 20, 2021, but may be extended again if the pandemic is ongoing. The most important of these waivers was probably the removal of both the originating site and geographic requirements for telehealth services.1 Prior to the COVID-19 PHE, a patient would have to travel to a doctor’s office, hospital, or skilled nursing facility to receive telehealth care (originating site requirement), and even then this was only allowed in defined rural areas of the country (geographic requirement). Both of these requirements were waived, allowing for any patient to receive telehealth services within their own homes. Concurrently, the requirement that patients must have an established relationship with the provider (ie, telehealth could not be used to provide care to new patients) also was waived.1
In the spirit of expanding access to care and providing reasonable reimbursement for medical services, other changes were made for which the CMS should be commended. In acknowledging that many Medicare/Medicaid beneficiaries may not have access to devices that permit real-time, 2-way audio/video communication, which previously were necessary to qualify for a telehealth encounter, the CMS decided to cover telephone visits and provide reimbursement at the level of an established visit.1 They also changed the billing structure to remove the place of service (POS) designation for telehealth (POS 02) and replace it with the normal physician’s office POS designation (usually POS 11), bringing back a telehealth modifier (modifier -95) in the process. The benefit of this change is solely to increase reimbursement for these services, as telehealth POS services generally are covered at lower facility rates, whereas POS 11 codes are reimbursed at the full level of a nonfacility physician’s office rate.
Finally, other waivers such as the Office of Civil Rights’ decision to waive HIPAA (Health Insurance Portability and Accountability Act) violations for telehealth platforms during the PHE allowed offices to take on telemedicine quickly without having to implement a new infrastructure.2 Numerous codes were added to the list of covered services for telehealth, but these generally are not relevant for dermatologists. The CMS also allowed physicians’ offices to waive the patient responsibility/co-pay during the COVID-19 PHE, which previously was not allowed due to concerns about the anti-kickback statute.1 These co-pay waivers were intended to remove another barrier to care for patients who were hesitant to participate in virtual visits. For the most part, the waiver of state licensing requirements is a bit less useful. As part of the CMS waiver, providers technically are allowed to see out-of-state Medicare/Medicaid beneficiaries, but state licensing laws are still in effect; thus, in the absence of a blanket state-level waiver (which some states enacted, modeled after the Uniform Emergency Volunteer Health Practitioner Act of 20063), providers still cannot see most out-of-state patients from a legal and malpractice coverage standpoint.
An important flexibility during the COVID-19 PHE is one that often is underrecognized. The CMS has been clear about the ability to provide direct supervision for advanced practice providers (APPs) and residents via telehealth during the PHE, which allows for incident-to billing for APPs at remote sites given that the supervising physician is immediately available via an interactive, 2-way, live audio/video telecommunications method. It also allows for direct supervision of APPs and residents using such technology. For dermatology, which does not have a primary care waiver, an attending must still directly supervise each patient and see the patient via a live audio/video modality but does not have to be on-site to do so. This is a very interesting concept that, if extended, could truly impact practice management for the long-term.
Response From Commercial Insurance Carriers
Tracking along with the CMS waivers and flexibilities during the PHE, most commercial carriers quickly adopted similar policies to cover telehealth services. It should be noted that for most commercial insurance carriers, the coverage was already broader than Medicare/Medicaid coverage for telehealth prior to the PHE, so in many ways it is an extension of that concept and acceptance of telemedicine as a whole. What is sometimes confusing, though, is that various policies and requirements around billing exist; for example, while most carriers emulated the POS requirements that the CMS adopted, some carriers still stuck with the telemedicine POS but paid full in-office visit rates for those codes. Some carriers adopted higher reimbursement rates for telephone visits, similar to the CMS, while others instructed providers to just bill for the established office visit codes and allowed for telephone-only visits to qualify for these billing codes. Some carriers also waived co-pays for telehealth visits for their members (whether related to COVID-19 or not). It is beyond the scope of this article to delve into the specifics, which may vary not only by carrier but by region and plan. However, it is important to stay on top of one’s insurance carriers to find out what their latest directives are for billing for telehealth.
Postpandemic Teledermatology
What about the future of teledermatology? Although many dermatologists have adopted telehealth services out of necessity during the COVID-19 PHE, the jury is still out on the long-term forecast for telemedicine in dermatology. Concerns about liability/malpractice and technology issues abound, and for many, the headaches of teledermatology—such as trying to focus on a blurry photograph of a nevus that the patient is concerned about—make it unappealing. Some of these issues will be addressed by better technology, but the reimbursement structure must continue for teledermatology to remain in widespread use.
Currently, the biggest question facing telehealth is whether the waivers for originating site and geographic requirements will be able to continue. The CMS itself does not have the statutory authority to make these changes permanent and was only allowed to act due to a waiver under section 1135 of the Social Security Act during a PHE. It would take an act of Congress to change the law to allow for this specific expansion of telehealth services. A number of federal bills, including S 2741 (Creating Opportunities Now for Necessary and Effective Care Technologies [CONNECT] for Health Act of 2019) and S 4796 (Fair Care Act of 2020) from the Senate, contain such provisions, but none have been passed at the time of writing. There does seem to be broad support of the concept of expanding telemedicine access, such as noted by New York State Governor Andrew Cuomo in his 2021 State of the State address,4 but it remains to be seen when action will come.
Some regulations, such as the HIPAA waiver and the ability to waive co-pays, are not slated to continue after the pandemic. The ability to supervise residents via telehealth (real-time audio/video) has been made permanent, but only in rural areas. Direct supervision of APPs via telehealth will continue through the end of the calendar year of the PHE or the end of 2021, whichever comes later, but it remains to be seen whether remote supervision will continue. The CMS has stated in its comments that it is looking at this issue closely and may establish certain guardrails to ensure quality of care is maintained.1 Telephone/audio-only visits also may come under further scrutiny, but research has supported the concept that patients who are more likely to gain access through audio-only modalities are older, Medicare/Medicaid (vs commercial), and Black (vs White) patients,5 so it would indeed introduce an unfair barrier to access if such coverage was rolled back.
Final Thoughts
Overall, we have made much progress in teledermatology. Once utilized by a small fraction of dermatologists, the vast majority of us turned to teledermatology to sustain our practices during the COVID-19 pandemic. Moving forward, there are 2 critical factors to consider: continued technological innovation and permanent coverage for telehealth reimbursement at in-office visit levels. With these challenges resolved, we can move forward and consider novel models that may be able to deliver dermatologic care to a broader patient population, thereby solving the critical issue of access to care for so many patients in need in our country.
The world of telemedicine—especially teledermatology—had been a sleepy underutilized afterthought for most physicians until we were faced with a global pandemic the likes of which none of us had seen in our lifetimes. And just like that, teledermatology went from an afterthought to part of the “new normal.” Although those of us already practicing telemedicine knew of potential pitfalls and concerns, this great social experiment of throwing everyone into unexplored territory led to a great deal of frustration with technology and workflows that were not optimized for dermatology visits. The process is still changing, and the technical aspects of conducting teledermatology visits will no doubt improve, but what about the bigger question of reimbursement? Without adequate payments and financial models, the long-term future of telemedicine is uncertain, so an understanding of the current and likely future landscape of telemedicine reimbursement is critical.
Waivers During the Public Health Emergency
The declaration of a public health emergency (PHE)allowed for significant flexibility by the Centers for Medicare & Medicaid Services (CMS) during the coronavirus disease 2019 (COVID-19) pandemic. Importantly, the CMS was permitted to act quickly to allow telehealth to flourish during the worst of the pandemic and throughout the declared PHE, which has been extended several times already. Currently, the PHE is set to expire on April 20, 2021, but may be extended again if the pandemic is ongoing. The most important of these waivers was probably the removal of both the originating site and geographic requirements for telehealth services.1 Prior to the COVID-19 PHE, a patient would have to travel to a doctor’s office, hospital, or skilled nursing facility to receive telehealth care (originating site requirement), and even then this was only allowed in defined rural areas of the country (geographic requirement). Both of these requirements were waived, allowing for any patient to receive telehealth services within their own homes. Concurrently, the requirement that patients must have an established relationship with the provider (ie, telehealth could not be used to provide care to new patients) also was waived.1
In the spirit of expanding access to care and providing reasonable reimbursement for medical services, other changes were made for which the CMS should be commended. In acknowledging that many Medicare/Medicaid beneficiaries may not have access to devices that permit real-time, 2-way audio/video communication, which previously were necessary to qualify for a telehealth encounter, the CMS decided to cover telephone visits and provide reimbursement at the level of an established visit.1 They also changed the billing structure to remove the place of service (POS) designation for telehealth (POS 02) and replace it with the normal physician’s office POS designation (usually POS 11), bringing back a telehealth modifier (modifier -95) in the process. The benefit of this change is solely to increase reimbursement for these services, as telehealth POS services generally are covered at lower facility rates, whereas POS 11 codes are reimbursed at the full level of a nonfacility physician’s office rate.
Finally, other waivers such as the Office of Civil Rights’ decision to waive HIPAA (Health Insurance Portability and Accountability Act) violations for telehealth platforms during the PHE allowed offices to take on telemedicine quickly without having to implement a new infrastructure.2 Numerous codes were added to the list of covered services for telehealth, but these generally are not relevant for dermatologists. The CMS also allowed physicians’ offices to waive the patient responsibility/co-pay during the COVID-19 PHE, which previously was not allowed due to concerns about the anti-kickback statute.1 These co-pay waivers were intended to remove another barrier to care for patients who were hesitant to participate in virtual visits. For the most part, the waiver of state licensing requirements is a bit less useful. As part of the CMS waiver, providers technically are allowed to see out-of-state Medicare/Medicaid beneficiaries, but state licensing laws are still in effect; thus, in the absence of a blanket state-level waiver (which some states enacted, modeled after the Uniform Emergency Volunteer Health Practitioner Act of 20063), providers still cannot see most out-of-state patients from a legal and malpractice coverage standpoint.
An important flexibility during the COVID-19 PHE is one that often is underrecognized. The CMS has been clear about the ability to provide direct supervision for advanced practice providers (APPs) and residents via telehealth during the PHE, which allows for incident-to billing for APPs at remote sites given that the supervising physician is immediately available via an interactive, 2-way, live audio/video telecommunications method. It also allows for direct supervision of APPs and residents using such technology. For dermatology, which does not have a primary care waiver, an attending must still directly supervise each patient and see the patient via a live audio/video modality but does not have to be on-site to do so. This is a very interesting concept that, if extended, could truly impact practice management for the long-term.
Response From Commercial Insurance Carriers
Tracking along with the CMS waivers and flexibilities during the PHE, most commercial carriers quickly adopted similar policies to cover telehealth services. It should be noted that for most commercial insurance carriers, the coverage was already broader than Medicare/Medicaid coverage for telehealth prior to the PHE, so in many ways it is an extension of that concept and acceptance of telemedicine as a whole. What is sometimes confusing, though, is that various policies and requirements around billing exist; for example, while most carriers emulated the POS requirements that the CMS adopted, some carriers still stuck with the telemedicine POS but paid full in-office visit rates for those codes. Some carriers adopted higher reimbursement rates for telephone visits, similar to the CMS, while others instructed providers to just bill for the established office visit codes and allowed for telephone-only visits to qualify for these billing codes. Some carriers also waived co-pays for telehealth visits for their members (whether related to COVID-19 or not). It is beyond the scope of this article to delve into the specifics, which may vary not only by carrier but by region and plan. However, it is important to stay on top of one’s insurance carriers to find out what their latest directives are for billing for telehealth.
Postpandemic Teledermatology
What about the future of teledermatology? Although many dermatologists have adopted telehealth services out of necessity during the COVID-19 PHE, the jury is still out on the long-term forecast for telemedicine in dermatology. Concerns about liability/malpractice and technology issues abound, and for many, the headaches of teledermatology—such as trying to focus on a blurry photograph of a nevus that the patient is concerned about—make it unappealing. Some of these issues will be addressed by better technology, but the reimbursement structure must continue for teledermatology to remain in widespread use.
Currently, the biggest question facing telehealth is whether the waivers for originating site and geographic requirements will be able to continue. The CMS itself does not have the statutory authority to make these changes permanent and was only allowed to act due to a waiver under section 1135 of the Social Security Act during a PHE. It would take an act of Congress to change the law to allow for this specific expansion of telehealth services. A number of federal bills, including S 2741 (Creating Opportunities Now for Necessary and Effective Care Technologies [CONNECT] for Health Act of 2019) and S 4796 (Fair Care Act of 2020) from the Senate, contain such provisions, but none have been passed at the time of writing. There does seem to be broad support of the concept of expanding telemedicine access, such as noted by New York State Governor Andrew Cuomo in his 2021 State of the State address,4 but it remains to be seen when action will come.
Some regulations, such as the HIPAA waiver and the ability to waive co-pays, are not slated to continue after the pandemic. The ability to supervise residents via telehealth (real-time audio/video) has been made permanent, but only in rural areas. Direct supervision of APPs via telehealth will continue through the end of the calendar year of the PHE or the end of 2021, whichever comes later, but it remains to be seen whether remote supervision will continue. The CMS has stated in its comments that it is looking at this issue closely and may establish certain guardrails to ensure quality of care is maintained.1 Telephone/audio-only visits also may come under further scrutiny, but research has supported the concept that patients who are more likely to gain access through audio-only modalities are older, Medicare/Medicaid (vs commercial), and Black (vs White) patients,5 so it would indeed introduce an unfair barrier to access if such coverage was rolled back.
Final Thoughts
Overall, we have made much progress in teledermatology. Once utilized by a small fraction of dermatologists, the vast majority of us turned to teledermatology to sustain our practices during the COVID-19 pandemic. Moving forward, there are 2 critical factors to consider: continued technological innovation and permanent coverage for telehealth reimbursement at in-office visit levels. With these challenges resolved, we can move forward and consider novel models that may be able to deliver dermatologic care to a broader patient population, thereby solving the critical issue of access to care for so many patients in need in our country.
- Medicare Program; CY 2021 Payment Policies Under the Physician Fee Schedule and Other Changes to Part B Payment Policies; Medicare Shared Savings Program Requirements; Medicaid Promoting Interoperability Program Requirements for Eligible Professionals; Quality Payment Program; Coverage of Opioid Use Disorder Services Furnished by Opioid Treatment Programs; Medicare Enrollment of Opioid Treatment Programs; Electronic Prescribing for Controlled Substances for a Covered Part D Drug; Payment for Office/Outpatient Evaluation and Management Services; Hospital IQR Program; Establish New Code Categories; Medicare Diabetes Prevention Program (MDPP) Expanded Model Emergency Policy; Coding and Payment for Virtual Check-in Services Interim Final Rule Policy; Coding and Payment for Personal Protective Equipment (PPE) Interim Final Rule Policy; Regulatory Revisions in Response to the Public Health Emergency (PHE) for COVID-19; and Finalization of Certain Provisions from the March 31st, May 8th and September 2nd Interim Final Rules in Response to the PHE for COVID-19. Fed Registr. 2020;85:84472-85377. To be codified at 42 CFR §400, 410, 414, 415, 423, 424, and 425. https://www.federalregister.gov/documents/2020/12/28/2020-26815/medicare-program-cy-2021-payment-policies-under-the-physician-fee-schedule-and-other-changes-to-part
- Office for Civil Rights. Notification of enforcement discretion for telehealth remote communications during the COVID-19 nationwide public health emergency. US Department of Health and Human Services website. Reviewed January 20, 2021. Accessed January 25, 2021. https://www.hhs.gov/hipaa/for-professionals/special-topics/emergency-preparedness/notification-enforcement-discretion-telehealth/index.html
- Hoffman DA. Increasing access to care: telehealth during COVID-19 [published online June 16, 2020]. J Law Biosci. doi:10.1093/jlb/lsaa043
- Governor Cuomo announces proposal to expand access to telehealth for all as part of 2021 State of the State. New York State website. Published January 10, 2021. Accessed January 25, 021. https://www.governor.ny.gov/news/governor-cuomo-announces-proposal-expand-access-telehealth-all-part-2021-state-state#:~:text=and%20Rural%20Communities-,Governor%20Andrew%20M.,2021%20State%20of%20the%20State.&text=New%20Yorkers%20have%20adapted%20throughout,into%20our%20existing%20healthcare%20system
- Gilson SF, Umscheid CA, Laiteerapong N, et al. Growth of ambulatory virtual visit and differential use by patient sociodemographics at one urban academic medical center during the COVID-19 pandemic: retrospective analysis. JMIR Med Inform. 2020;8:E24544.
- Medicare Program; CY 2021 Payment Policies Under the Physician Fee Schedule and Other Changes to Part B Payment Policies; Medicare Shared Savings Program Requirements; Medicaid Promoting Interoperability Program Requirements for Eligible Professionals; Quality Payment Program; Coverage of Opioid Use Disorder Services Furnished by Opioid Treatment Programs; Medicare Enrollment of Opioid Treatment Programs; Electronic Prescribing for Controlled Substances for a Covered Part D Drug; Payment for Office/Outpatient Evaluation and Management Services; Hospital IQR Program; Establish New Code Categories; Medicare Diabetes Prevention Program (MDPP) Expanded Model Emergency Policy; Coding and Payment for Virtual Check-in Services Interim Final Rule Policy; Coding and Payment for Personal Protective Equipment (PPE) Interim Final Rule Policy; Regulatory Revisions in Response to the Public Health Emergency (PHE) for COVID-19; and Finalization of Certain Provisions from the March 31st, May 8th and September 2nd Interim Final Rules in Response to the PHE for COVID-19. Fed Registr. 2020;85:84472-85377. To be codified at 42 CFR §400, 410, 414, 415, 423, 424, and 425. https://www.federalregister.gov/documents/2020/12/28/2020-26815/medicare-program-cy-2021-payment-policies-under-the-physician-fee-schedule-and-other-changes-to-part
- Office for Civil Rights. Notification of enforcement discretion for telehealth remote communications during the COVID-19 nationwide public health emergency. US Department of Health and Human Services website. Reviewed January 20, 2021. Accessed January 25, 2021. https://www.hhs.gov/hipaa/for-professionals/special-topics/emergency-preparedness/notification-enforcement-discretion-telehealth/index.html
- Hoffman DA. Increasing access to care: telehealth during COVID-19 [published online June 16, 2020]. J Law Biosci. doi:10.1093/jlb/lsaa043
- Governor Cuomo announces proposal to expand access to telehealth for all as part of 2021 State of the State. New York State website. Published January 10, 2021. Accessed January 25, 021. https://www.governor.ny.gov/news/governor-cuomo-announces-proposal-expand-access-telehealth-all-part-2021-state-state#:~:text=and%20Rural%20Communities-,Governor%20Andrew%20M.,2021%20State%20of%20the%20State.&text=New%20Yorkers%20have%20adapted%20throughout,into%20our%20existing%20healthcare%20system
- Gilson SF, Umscheid CA, Laiteerapong N, et al. Growth of ambulatory virtual visit and differential use by patient sociodemographics at one urban academic medical center during the COVID-19 pandemic: retrospective analysis. JMIR Med Inform. 2020;8:E24544.

Biden administration nixes buprenorphine waiver, docs disappointed
The Biden administration has halted a Trump administration initiative that would have allowed more physicians to prescribe buprenorphine for opioid use disorder (OUD).
Under the Trump administration’s plan, many doctors would be exempt from taking a day’s training before they could prescribe buprenorphine for OUD.
On Jan. 25, 2021, citing anonymous sources, the Washington Post reported that this action by the Biden administration was likely. At the time, there were concerns about whether the Department of Health & Human Services had the legal authority to make this policy change, the Post reported. The Substance Abuse and Mental Health Services Administration subsequently announced the derailment of the buprenorphine proposal on its website.
In SAMHSA’s view, the proposal was made “prematurely.” The SAMHSA statement did not detail the reasons for abandoning the Jan. 14 proposal. It had been scheduled to take effect upon publication in the Federal Register.
Instead of finalizing it in this way, the HHS said it would work with other federal agencies to “increase access to buprenorphine, reduce overdose rates and save lives.”
The HHS decision to scupper the proposal disappointed many physician groups. In a letter dated Jan. 27, several physician groups called on the Biden administration to proceed with the Trump proposal.
Under current federal law, physicians who wish to prescribe buprenorphine outside of opioid treatment programs must take an 8-hour course and receive a waiver from the Drug Enforcement Administration, the letter noted. It was signed by the American College of Emergency Physicians, the American Medical Association, and other organizations.
Treatment barrier
After taking the training course, it can take 60-90 days for physicians to receive the waiver. The license application can then be submitted. Physician groups argue that this so-called X-waiver requirement creates a barrier to providing medication-assisted treatment.
“Due to the stigma, some clinicians are not willing to pursue this DEA license or even engage in treatment of patients with [OUD],” the letter said.
The Trump administration’s proposal would have limited most physicians to treating no more than 30 patients with buprenorphine for OUD at any one time. This cap would not have applied to hospital-based physicians, such as those practicing emergency medicine, the HHS noted in a statement. The policy would have applied to only physicians who already have registered with the DEA.
Patrice A. Harris, MD, the immediate past president of the AMA and chair of the organization’s Opioid Task Force, was among the many physicians who supported the Trump administration proposal.
“It is estimated that more than 2 million Americans need treatment for opioid use disorder, but only a small percentage actually receive treatment,” Dr. Harris said in statement. Dr. Harris also noted that overdose deaths have reportedly accelerated during the COVID-19 pandemic.
Centers for Disease Control and Prevention data show there were more than 83,000 drug overdose deaths in the United States in the 12 months ending in June 2020. That is the highest number of overdose deaths ever recorded in a 12-month period and is an increase of more than 21%, compared with the previous year.
A ‘disappointment’
On Jan. 28, Dr. Harris said the decision to drop the plan was a disappointment.
“We encourage the current administration to quickly develop a path forward that removes the burdensome waiver requirement, thus allowing more physicians to prescribe this lifesaving medication,” she said in a statement sent to this news organization.
In a Jan. 26 statement, the American Society of Addiction Medicine urged Congress to eliminate the X-waiver and called for more education and training in the treatment of patients who struggle with opioids.
In the 116th session of Congress, which ended on Jan. 3, there was bipartisan support for proposed legislation to ease requirements for buprenorphine prescribing. A House bill had more than 90 Democratic and 21 Republican sponsors. A companion Senate bill had three Democratic and three Republican Sponsors, including Sen. Maggie Hassan (D-N.H.). On Jan. 25, Dr. Hassan tweeted that she would be seeking an explanation from the Biden administration if it halted the plan to ease the waiver restriction.
“Medication-assisted treatment can save lives, and the buprenorphine waiver requirement should be eliminated so that physicians can more easily prescribe it to those who need it,” she said.
Many clinicians and policy experts turned to Twitter to urge an easing of buprenorphine prescribing, using the hashtag “Xthexwaiver.”
Among them was the official who put forward the Jan. 14 proposal, Brett Giroir, MD. He served as assistant secretary for health during the Trump administration.
Objections
In its Jan. 25 article, the Washington Post referred to an article in Alcoholism and Drug Abuse Weekly in which a top federal official in the Trump administration objected to Dr. Giroir’s plan.
Elinore F. McCance-Katz, MD, PhD, who served as the assistant secretary of HHS for SAMHSA, had earlier proposed raising the cap for addiction experts. Alcoholism and Drug Abuse Weekly quotes Dr. McCance-Katz as saying the Trump buprenorphine proposal was “unfair to the incoming administration.”
“The Biden administration has so much work to do to get their programs and policies into place, and to do something like this at the 11th hour that could get doctors into trouble – it’s heinous,” she said in the article.
Dr. McCance-Katz had resigned before the Trump administration proposal was unveiled. On Jan. 7, she issued a public notice announcing she would resign, citing concerns about the previous day’s attack on the U.S. Capitol.
“It had been my plan to stay until the change in administration occurred, but my plans abruptly changed last evening when, on my way back from visiting an excellent residential treatment program in New York, I saw the violent takeover of the Capitol building,” she said.
On Twitter, Roland Flores, MD, an anesthesiologist and pain specialist, urged his colleagues to consider the need for more education among clinicians who treat OUD. He jousted a bit with those favoring a swift drive to “XtheXwaiver” and questioned their arguments about the burden of the current rules.
“I think ‘all this red tape’ is a little bit of an exaggeration – it’s an 8-hour online course, and an application,” Dr. Flores tweeted in one exchange. “But #XtheXwaiver is fine – it’s probably rooted in stigma. It’s unlikely to make much difference tho. The waiver wasn’t the thing keeping docs from prescribing.”
A version of this article first appeared on Medscape.com.
The Biden administration has halted a Trump administration initiative that would have allowed more physicians to prescribe buprenorphine for opioid use disorder (OUD).
Under the Trump administration’s plan, many doctors would be exempt from taking a day’s training before they could prescribe buprenorphine for OUD.
On Jan. 25, 2021, citing anonymous sources, the Washington Post reported that this action by the Biden administration was likely. At the time, there were concerns about whether the Department of Health & Human Services had the legal authority to make this policy change, the Post reported. The Substance Abuse and Mental Health Services Administration subsequently announced the derailment of the buprenorphine proposal on its website.
In SAMHSA’s view, the proposal was made “prematurely.” The SAMHSA statement did not detail the reasons for abandoning the Jan. 14 proposal. It had been scheduled to take effect upon publication in the Federal Register.
Instead of finalizing it in this way, the HHS said it would work with other federal agencies to “increase access to buprenorphine, reduce overdose rates and save lives.”
The HHS decision to scupper the proposal disappointed many physician groups. In a letter dated Jan. 27, several physician groups called on the Biden administration to proceed with the Trump proposal.
Under current federal law, physicians who wish to prescribe buprenorphine outside of opioid treatment programs must take an 8-hour course and receive a waiver from the Drug Enforcement Administration, the letter noted. It was signed by the American College of Emergency Physicians, the American Medical Association, and other organizations.
Treatment barrier
After taking the training course, it can take 60-90 days for physicians to receive the waiver. The license application can then be submitted. Physician groups argue that this so-called X-waiver requirement creates a barrier to providing medication-assisted treatment.
“Due to the stigma, some clinicians are not willing to pursue this DEA license or even engage in treatment of patients with [OUD],” the letter said.
The Trump administration’s proposal would have limited most physicians to treating no more than 30 patients with buprenorphine for OUD at any one time. This cap would not have applied to hospital-based physicians, such as those practicing emergency medicine, the HHS noted in a statement. The policy would have applied to only physicians who already have registered with the DEA.
Patrice A. Harris, MD, the immediate past president of the AMA and chair of the organization’s Opioid Task Force, was among the many physicians who supported the Trump administration proposal.
“It is estimated that more than 2 million Americans need treatment for opioid use disorder, but only a small percentage actually receive treatment,” Dr. Harris said in statement. Dr. Harris also noted that overdose deaths have reportedly accelerated during the COVID-19 pandemic.
Centers for Disease Control and Prevention data show there were more than 83,000 drug overdose deaths in the United States in the 12 months ending in June 2020. That is the highest number of overdose deaths ever recorded in a 12-month period and is an increase of more than 21%, compared with the previous year.
A ‘disappointment’
On Jan. 28, Dr. Harris said the decision to drop the plan was a disappointment.
“We encourage the current administration to quickly develop a path forward that removes the burdensome waiver requirement, thus allowing more physicians to prescribe this lifesaving medication,” she said in a statement sent to this news organization.
In a Jan. 26 statement, the American Society of Addiction Medicine urged Congress to eliminate the X-waiver and called for more education and training in the treatment of patients who struggle with opioids.
In the 116th session of Congress, which ended on Jan. 3, there was bipartisan support for proposed legislation to ease requirements for buprenorphine prescribing. A House bill had more than 90 Democratic and 21 Republican sponsors. A companion Senate bill had three Democratic and three Republican Sponsors, including Sen. Maggie Hassan (D-N.H.). On Jan. 25, Dr. Hassan tweeted that she would be seeking an explanation from the Biden administration if it halted the plan to ease the waiver restriction.
“Medication-assisted treatment can save lives, and the buprenorphine waiver requirement should be eliminated so that physicians can more easily prescribe it to those who need it,” she said.
Many clinicians and policy experts turned to Twitter to urge an easing of buprenorphine prescribing, using the hashtag “Xthexwaiver.”
Among them was the official who put forward the Jan. 14 proposal, Brett Giroir, MD. He served as assistant secretary for health during the Trump administration.
Objections
In its Jan. 25 article, the Washington Post referred to an article in Alcoholism and Drug Abuse Weekly in which a top federal official in the Trump administration objected to Dr. Giroir’s plan.
Elinore F. McCance-Katz, MD, PhD, who served as the assistant secretary of HHS for SAMHSA, had earlier proposed raising the cap for addiction experts. Alcoholism and Drug Abuse Weekly quotes Dr. McCance-Katz as saying the Trump buprenorphine proposal was “unfair to the incoming administration.”
“The Biden administration has so much work to do to get their programs and policies into place, and to do something like this at the 11th hour that could get doctors into trouble – it’s heinous,” she said in the article.
Dr. McCance-Katz had resigned before the Trump administration proposal was unveiled. On Jan. 7, she issued a public notice announcing she would resign, citing concerns about the previous day’s attack on the U.S. Capitol.
“It had been my plan to stay until the change in administration occurred, but my plans abruptly changed last evening when, on my way back from visiting an excellent residential treatment program in New York, I saw the violent takeover of the Capitol building,” she said.
On Twitter, Roland Flores, MD, an anesthesiologist and pain specialist, urged his colleagues to consider the need for more education among clinicians who treat OUD. He jousted a bit with those favoring a swift drive to “XtheXwaiver” and questioned their arguments about the burden of the current rules.
“I think ‘all this red tape’ is a little bit of an exaggeration – it’s an 8-hour online course, and an application,” Dr. Flores tweeted in one exchange. “But #XtheXwaiver is fine – it’s probably rooted in stigma. It’s unlikely to make much difference tho. The waiver wasn’t the thing keeping docs from prescribing.”
A version of this article first appeared on Medscape.com.
The Biden administration has halted a Trump administration initiative that would have allowed more physicians to prescribe buprenorphine for opioid use disorder (OUD).
Under the Trump administration’s plan, many doctors would be exempt from taking a day’s training before they could prescribe buprenorphine for OUD.
On Jan. 25, 2021, citing anonymous sources, the Washington Post reported that this action by the Biden administration was likely. At the time, there were concerns about whether the Department of Health & Human Services had the legal authority to make this policy change, the Post reported. The Substance Abuse and Mental Health Services Administration subsequently announced the derailment of the buprenorphine proposal on its website.
In SAMHSA’s view, the proposal was made “prematurely.” The SAMHSA statement did not detail the reasons for abandoning the Jan. 14 proposal. It had been scheduled to take effect upon publication in the Federal Register.
Instead of finalizing it in this way, the HHS said it would work with other federal agencies to “increase access to buprenorphine, reduce overdose rates and save lives.”
The HHS decision to scupper the proposal disappointed many physician groups. In a letter dated Jan. 27, several physician groups called on the Biden administration to proceed with the Trump proposal.
Under current federal law, physicians who wish to prescribe buprenorphine outside of opioid treatment programs must take an 8-hour course and receive a waiver from the Drug Enforcement Administration, the letter noted. It was signed by the American College of Emergency Physicians, the American Medical Association, and other organizations.
Treatment barrier
After taking the training course, it can take 60-90 days for physicians to receive the waiver. The license application can then be submitted. Physician groups argue that this so-called X-waiver requirement creates a barrier to providing medication-assisted treatment.
“Due to the stigma, some clinicians are not willing to pursue this DEA license or even engage in treatment of patients with [OUD],” the letter said.
The Trump administration’s proposal would have limited most physicians to treating no more than 30 patients with buprenorphine for OUD at any one time. This cap would not have applied to hospital-based physicians, such as those practicing emergency medicine, the HHS noted in a statement. The policy would have applied to only physicians who already have registered with the DEA.
Patrice A. Harris, MD, the immediate past president of the AMA and chair of the organization’s Opioid Task Force, was among the many physicians who supported the Trump administration proposal.
“It is estimated that more than 2 million Americans need treatment for opioid use disorder, but only a small percentage actually receive treatment,” Dr. Harris said in statement. Dr. Harris also noted that overdose deaths have reportedly accelerated during the COVID-19 pandemic.
Centers for Disease Control and Prevention data show there were more than 83,000 drug overdose deaths in the United States in the 12 months ending in June 2020. That is the highest number of overdose deaths ever recorded in a 12-month period and is an increase of more than 21%, compared with the previous year.
A ‘disappointment’
On Jan. 28, Dr. Harris said the decision to drop the plan was a disappointment.
“We encourage the current administration to quickly develop a path forward that removes the burdensome waiver requirement, thus allowing more physicians to prescribe this lifesaving medication,” she said in a statement sent to this news organization.
In a Jan. 26 statement, the American Society of Addiction Medicine urged Congress to eliminate the X-waiver and called for more education and training in the treatment of patients who struggle with opioids.
In the 116th session of Congress, which ended on Jan. 3, there was bipartisan support for proposed legislation to ease requirements for buprenorphine prescribing. A House bill had more than 90 Democratic and 21 Republican sponsors. A companion Senate bill had three Democratic and three Republican Sponsors, including Sen. Maggie Hassan (D-N.H.). On Jan. 25, Dr. Hassan tweeted that she would be seeking an explanation from the Biden administration if it halted the plan to ease the waiver restriction.
“Medication-assisted treatment can save lives, and the buprenorphine waiver requirement should be eliminated so that physicians can more easily prescribe it to those who need it,” she said.
Many clinicians and policy experts turned to Twitter to urge an easing of buprenorphine prescribing, using the hashtag “Xthexwaiver.”
Among them was the official who put forward the Jan. 14 proposal, Brett Giroir, MD. He served as assistant secretary for health during the Trump administration.
Objections
In its Jan. 25 article, the Washington Post referred to an article in Alcoholism and Drug Abuse Weekly in which a top federal official in the Trump administration objected to Dr. Giroir’s plan.
Elinore F. McCance-Katz, MD, PhD, who served as the assistant secretary of HHS for SAMHSA, had earlier proposed raising the cap for addiction experts. Alcoholism and Drug Abuse Weekly quotes Dr. McCance-Katz as saying the Trump buprenorphine proposal was “unfair to the incoming administration.”
“The Biden administration has so much work to do to get their programs and policies into place, and to do something like this at the 11th hour that could get doctors into trouble – it’s heinous,” she said in the article.
Dr. McCance-Katz had resigned before the Trump administration proposal was unveiled. On Jan. 7, she issued a public notice announcing she would resign, citing concerns about the previous day’s attack on the U.S. Capitol.
“It had been my plan to stay until the change in administration occurred, but my plans abruptly changed last evening when, on my way back from visiting an excellent residential treatment program in New York, I saw the violent takeover of the Capitol building,” she said.
On Twitter, Roland Flores, MD, an anesthesiologist and pain specialist, urged his colleagues to consider the need for more education among clinicians who treat OUD. He jousted a bit with those favoring a swift drive to “XtheXwaiver” and questioned their arguments about the burden of the current rules.
“I think ‘all this red tape’ is a little bit of an exaggeration – it’s an 8-hour online course, and an application,” Dr. Flores tweeted in one exchange. “But #XtheXwaiver is fine – it’s probably rooted in stigma. It’s unlikely to make much difference tho. The waiver wasn’t the thing keeping docs from prescribing.”
A version of this article first appeared on Medscape.com.
New NIH database will track neurologic effects of COVID-19
“We know COVID-19 can disrupt multiple body systems, but the effects of the virus and the body’s response to COVID-19 infection on the brain, spinal cord, nerves, and muscle can be particularly devastating and contribute to persistence of disability even after the virus is cleared,” said Barbara Karp, MD, program director at the National Institute of Neurological Disorders and Stroke.
“There is an urgent need to understand COVID-19–related neurological problems, which not uncommonly include headaches, fatigue, cognitive difficulties, stroke, pain, and sleep disorders as well as some very rare complications of serious infections,” said Dr. Karp.
The COVID-19 NeuroDatabank/BioBank (NeuroCOVID) is funded by the NINDS. It was created and will be maintained by researchers at NYU Langone Health in New York.
The project is led by Andrea Troxel, ScD, professor of population health, and Eva Petkova, PhD, professor of population health and child and adolescent psychiatry, both at New York University.
“We’ve built a pretty comprehensive database that will accept deidentified patient information about new neurological issues that coincide with their COVID disease or worsening of preexisting neurological problems,” said Dr. Troxel. “In addition, we have a bio repository that will accept almost any kind of biological sample, such as blood, plasma, cerebrospinal fluid, and tissue,” she said.
“Neuroimages are very difficult to store because the files are so enormous, but we’ve had some questions about that, and we’re looking into whether we can accommodate neuroimages,” Dr. Troxel noted.
Dr. Troxel said a “blast of information and invitations” has gone out in an effort to acquire data and biospecimens. “We’ve been really pleased with the amount of interest already, interest not only from large academic medical centers, as you might expect, but also from some smaller stand-alone clinics and even some individuals who have either experienced some of these neurological problems of COVID or know those who have and are really eager to try to provide information,” she added.
Researchers interested in using data and biosamples from the database may submit requests to the NeuroCOVID Steering Committee. More information is available online on the NeuroCOVID website.
A version of this article first appeared on Medscape.com.
“We know COVID-19 can disrupt multiple body systems, but the effects of the virus and the body’s response to COVID-19 infection on the brain, spinal cord, nerves, and muscle can be particularly devastating and contribute to persistence of disability even after the virus is cleared,” said Barbara Karp, MD, program director at the National Institute of Neurological Disorders and Stroke.
“There is an urgent need to understand COVID-19–related neurological problems, which not uncommonly include headaches, fatigue, cognitive difficulties, stroke, pain, and sleep disorders as well as some very rare complications of serious infections,” said Dr. Karp.
The COVID-19 NeuroDatabank/BioBank (NeuroCOVID) is funded by the NINDS. It was created and will be maintained by researchers at NYU Langone Health in New York.
The project is led by Andrea Troxel, ScD, professor of population health, and Eva Petkova, PhD, professor of population health and child and adolescent psychiatry, both at New York University.
“We’ve built a pretty comprehensive database that will accept deidentified patient information about new neurological issues that coincide with their COVID disease or worsening of preexisting neurological problems,” said Dr. Troxel. “In addition, we have a bio repository that will accept almost any kind of biological sample, such as blood, plasma, cerebrospinal fluid, and tissue,” she said.
“Neuroimages are very difficult to store because the files are so enormous, but we’ve had some questions about that, and we’re looking into whether we can accommodate neuroimages,” Dr. Troxel noted.
Dr. Troxel said a “blast of information and invitations” has gone out in an effort to acquire data and biospecimens. “We’ve been really pleased with the amount of interest already, interest not only from large academic medical centers, as you might expect, but also from some smaller stand-alone clinics and even some individuals who have either experienced some of these neurological problems of COVID or know those who have and are really eager to try to provide information,” she added.
Researchers interested in using data and biosamples from the database may submit requests to the NeuroCOVID Steering Committee. More information is available online on the NeuroCOVID website.
A version of this article first appeared on Medscape.com.
“We know COVID-19 can disrupt multiple body systems, but the effects of the virus and the body’s response to COVID-19 infection on the brain, spinal cord, nerves, and muscle can be particularly devastating and contribute to persistence of disability even after the virus is cleared,” said Barbara Karp, MD, program director at the National Institute of Neurological Disorders and Stroke.
“There is an urgent need to understand COVID-19–related neurological problems, which not uncommonly include headaches, fatigue, cognitive difficulties, stroke, pain, and sleep disorders as well as some very rare complications of serious infections,” said Dr. Karp.
The COVID-19 NeuroDatabank/BioBank (NeuroCOVID) is funded by the NINDS. It was created and will be maintained by researchers at NYU Langone Health in New York.
The project is led by Andrea Troxel, ScD, professor of population health, and Eva Petkova, PhD, professor of population health and child and adolescent psychiatry, both at New York University.
“We’ve built a pretty comprehensive database that will accept deidentified patient information about new neurological issues that coincide with their COVID disease or worsening of preexisting neurological problems,” said Dr. Troxel. “In addition, we have a bio repository that will accept almost any kind of biological sample, such as blood, plasma, cerebrospinal fluid, and tissue,” she said.
“Neuroimages are very difficult to store because the files are so enormous, but we’ve had some questions about that, and we’re looking into whether we can accommodate neuroimages,” Dr. Troxel noted.
Dr. Troxel said a “blast of information and invitations” has gone out in an effort to acquire data and biospecimens. “We’ve been really pleased with the amount of interest already, interest not only from large academic medical centers, as you might expect, but also from some smaller stand-alone clinics and even some individuals who have either experienced some of these neurological problems of COVID or know those who have and are really eager to try to provide information,” she added.
Researchers interested in using data and biosamples from the database may submit requests to the NeuroCOVID Steering Committee. More information is available online on the NeuroCOVID website.
A version of this article first appeared on Medscape.com.
COVID-19 vaccination in cancer patients: NCCN outlines priorities
Vaccination timing considerations vary based on factors such as cancer and treatment type, and reasons for delaying vaccination in the general public also apply to cancer patients (recent COVID-19 exposure, for example).
In general, however, patients with cancer should be assigned to Centers for Disease Control and Prevention priority group 1 b/c and immunized when vaccination is available to them, the guidelines state. Exceptions to this recommendation include:
- Patients undergoing hematopoietic stem cell transplant or receiving engineered cellular therapy such as chimeric antigen receptor T-cell therapy. Vaccination should be delayed for at least 3 months in these patients to maximize vaccine efficacy. Caregivers of these patients, however, should be immunized when possible.
- Patients with hematologic malignancies who are receiving intensive cytotoxic chemotherapy, such as cytarabine- or anthracycline-based regimens for acute myeloid leukemia. Vaccination in these patients should be delayed until absolute neutrophil count recovery.
- Patients undergoing major surgery. Vaccination should occur at least a few days before or after surgery.
- Patients who have experienced a severe or immediate adverse reaction to any of the ingredients in the mRNA COVID-19 vaccines.
Conversely, vaccination should occur when available in patients with hematologic malignancies and marrow failure who are expected to have limited or no recovery, patients with hematologic malignancies who are on long-term maintenance therapy, and patients with solid tumors who are receiving cytotoxic chemotherapy, targeted therapy, checkpoint inhibitors and other immunotherapy, or radiotherapy.
Caregivers, household contacts, and other close contacts who are 16 years of age and older should be vaccinated whenever they are eligible.
Unique concerns in patients with cancer
The NCCN recommendations were developed to address the unique issues and concerns with respect to patients with cancer, who have an increased risk of severe illness from SARS-CoV-2 infection. But the guidelines come with a caveat: “[t]here are limited safety and efficacy data in these patients,” the NCCN emphasized in a press statement.
“Right now, there is urgent need and limited data,” Steven Pergam, MD, co-leader of the NCCN COVID-19 Vaccination Committee, said in the statement.
“Our number one goal is helping to get the vaccine to as many people as we can,” Dr. Pergam said. “That means following existing national and regional directions for prioritizing people who are more likely to face death or severe illness from COVID-19.”
Dr. Pergam, associate professor at Fred Hutchinson Cancer Research Center in Seattle, further explained that “people receiving active cancer treatment are at greater risk for worse outcomes from COVID-19, particularly if they are older and have additional comorbidities, like immunosuppression.”
NCCN’s recommendations couldn’t have come at a better time for patients with cancer, according to Nora Disis, MD, a professor at the University of Washington in Seattle.
“The NCCN’s recommendations to prioritize COVID vaccinations for cancer patients on active treatment is an important step forward in protecting our patients from the infection,” Dr. Disis said in an interview.
“Cancer patients may be at higher risk for the complications seen with infection. In addition, cancer is a disease of older people, and a good number of our patients have the comorbidities that would predict a poorer outcome if they should become sick,” Dr. Disis added. “With the correct treatment, many patients with cancer will be long-term survivors. It is important that they be protected from infection with COVID to realize their best outcome.”
Additional vaccine considerations
The NCCN recommendations also address several other issues of importance for cancer patients, including:
- Deprioritizing other vaccines. COVID-19 vaccines should take precedence over other vaccines because data on dual vaccination are lacking. The NCCN recommends waiting 14 days after COVID-19 vaccination to deliver other vaccines.
- Vaccinating clinical trial participants. Trial leads should be consulted to prevent protocol violations or exclusions.
- Decision-making in the setting of limited vaccine availability. The NCCN noted that decisions on allocation must be made in accordance with state and local vaccine guidance but suggests prioritizing appropriate patients on active treatment, those planning to start treatment, and those who have just completed treatment. Additional risk factors for these patients, as well as other factors associated with risk for adverse COVID-19 outcomes, should also be considered. These include advanced age, comorbidities, and adverse social and demographic factors such as poverty and limited health care access.
- The need for ongoing prevention measures. Vaccines have been shown to decrease the incidence of COVID-19 and related complications, but it remains unclear whether vaccines prevent infection and subsequent transmission. This means everyone should continue following prevention recommendations, such as wearing masks and avoiding crowds.
The NCCN stressed that these recommendations are “intended to be a living document that is constantly evolving – it will be updated rapidly whenever new data comes out, as well as any potential new vaccines that may get approved in the future.” The NCCN also noted that the advisory committee will meet regularly to refine the recommendations as needed.
Dr. Pergam disclosed relationships with Chimerix Inc., Merck & Co., Global Life Technologies Inc., and Sanofi-Aventis. Dr. Disis disclosed grants from Pfizer, Bavarian Nordisk, Janssen, and Precigen. She is the founder of EpiThany and editor-in-chief of JAMA Oncology.
Vaccination timing considerations vary based on factors such as cancer and treatment type, and reasons for delaying vaccination in the general public also apply to cancer patients (recent COVID-19 exposure, for example).
In general, however, patients with cancer should be assigned to Centers for Disease Control and Prevention priority group 1 b/c and immunized when vaccination is available to them, the guidelines state. Exceptions to this recommendation include:
- Patients undergoing hematopoietic stem cell transplant or receiving engineered cellular therapy such as chimeric antigen receptor T-cell therapy. Vaccination should be delayed for at least 3 months in these patients to maximize vaccine efficacy. Caregivers of these patients, however, should be immunized when possible.
- Patients with hematologic malignancies who are receiving intensive cytotoxic chemotherapy, such as cytarabine- or anthracycline-based regimens for acute myeloid leukemia. Vaccination in these patients should be delayed until absolute neutrophil count recovery.
- Patients undergoing major surgery. Vaccination should occur at least a few days before or after surgery.
- Patients who have experienced a severe or immediate adverse reaction to any of the ingredients in the mRNA COVID-19 vaccines.
Conversely, vaccination should occur when available in patients with hematologic malignancies and marrow failure who are expected to have limited or no recovery, patients with hematologic malignancies who are on long-term maintenance therapy, and patients with solid tumors who are receiving cytotoxic chemotherapy, targeted therapy, checkpoint inhibitors and other immunotherapy, or radiotherapy.
Caregivers, household contacts, and other close contacts who are 16 years of age and older should be vaccinated whenever they are eligible.
Unique concerns in patients with cancer
The NCCN recommendations were developed to address the unique issues and concerns with respect to patients with cancer, who have an increased risk of severe illness from SARS-CoV-2 infection. But the guidelines come with a caveat: “[t]here are limited safety and efficacy data in these patients,” the NCCN emphasized in a press statement.
“Right now, there is urgent need and limited data,” Steven Pergam, MD, co-leader of the NCCN COVID-19 Vaccination Committee, said in the statement.
“Our number one goal is helping to get the vaccine to as many people as we can,” Dr. Pergam said. “That means following existing national and regional directions for prioritizing people who are more likely to face death or severe illness from COVID-19.”
Dr. Pergam, associate professor at Fred Hutchinson Cancer Research Center in Seattle, further explained that “people receiving active cancer treatment are at greater risk for worse outcomes from COVID-19, particularly if they are older and have additional comorbidities, like immunosuppression.”
NCCN’s recommendations couldn’t have come at a better time for patients with cancer, according to Nora Disis, MD, a professor at the University of Washington in Seattle.
“The NCCN’s recommendations to prioritize COVID vaccinations for cancer patients on active treatment is an important step forward in protecting our patients from the infection,” Dr. Disis said in an interview.
“Cancer patients may be at higher risk for the complications seen with infection. In addition, cancer is a disease of older people, and a good number of our patients have the comorbidities that would predict a poorer outcome if they should become sick,” Dr. Disis added. “With the correct treatment, many patients with cancer will be long-term survivors. It is important that they be protected from infection with COVID to realize their best outcome.”
Additional vaccine considerations
The NCCN recommendations also address several other issues of importance for cancer patients, including:
- Deprioritizing other vaccines. COVID-19 vaccines should take precedence over other vaccines because data on dual vaccination are lacking. The NCCN recommends waiting 14 days after COVID-19 vaccination to deliver other vaccines.
- Vaccinating clinical trial participants. Trial leads should be consulted to prevent protocol violations or exclusions.
- Decision-making in the setting of limited vaccine availability. The NCCN noted that decisions on allocation must be made in accordance with state and local vaccine guidance but suggests prioritizing appropriate patients on active treatment, those planning to start treatment, and those who have just completed treatment. Additional risk factors for these patients, as well as other factors associated with risk for adverse COVID-19 outcomes, should also be considered. These include advanced age, comorbidities, and adverse social and demographic factors such as poverty and limited health care access.
- The need for ongoing prevention measures. Vaccines have been shown to decrease the incidence of COVID-19 and related complications, but it remains unclear whether vaccines prevent infection and subsequent transmission. This means everyone should continue following prevention recommendations, such as wearing masks and avoiding crowds.
The NCCN stressed that these recommendations are “intended to be a living document that is constantly evolving – it will be updated rapidly whenever new data comes out, as well as any potential new vaccines that may get approved in the future.” The NCCN also noted that the advisory committee will meet regularly to refine the recommendations as needed.
Dr. Pergam disclosed relationships with Chimerix Inc., Merck & Co., Global Life Technologies Inc., and Sanofi-Aventis. Dr. Disis disclosed grants from Pfizer, Bavarian Nordisk, Janssen, and Precigen. She is the founder of EpiThany and editor-in-chief of JAMA Oncology.
Vaccination timing considerations vary based on factors such as cancer and treatment type, and reasons for delaying vaccination in the general public also apply to cancer patients (recent COVID-19 exposure, for example).
In general, however, patients with cancer should be assigned to Centers for Disease Control and Prevention priority group 1 b/c and immunized when vaccination is available to them, the guidelines state. Exceptions to this recommendation include:
- Patients undergoing hematopoietic stem cell transplant or receiving engineered cellular therapy such as chimeric antigen receptor T-cell therapy. Vaccination should be delayed for at least 3 months in these patients to maximize vaccine efficacy. Caregivers of these patients, however, should be immunized when possible.
- Patients with hematologic malignancies who are receiving intensive cytotoxic chemotherapy, such as cytarabine- or anthracycline-based regimens for acute myeloid leukemia. Vaccination in these patients should be delayed until absolute neutrophil count recovery.
- Patients undergoing major surgery. Vaccination should occur at least a few days before or after surgery.
- Patients who have experienced a severe or immediate adverse reaction to any of the ingredients in the mRNA COVID-19 vaccines.
Conversely, vaccination should occur when available in patients with hematologic malignancies and marrow failure who are expected to have limited or no recovery, patients with hematologic malignancies who are on long-term maintenance therapy, and patients with solid tumors who are receiving cytotoxic chemotherapy, targeted therapy, checkpoint inhibitors and other immunotherapy, or radiotherapy.
Caregivers, household contacts, and other close contacts who are 16 years of age and older should be vaccinated whenever they are eligible.
Unique concerns in patients with cancer
The NCCN recommendations were developed to address the unique issues and concerns with respect to patients with cancer, who have an increased risk of severe illness from SARS-CoV-2 infection. But the guidelines come with a caveat: “[t]here are limited safety and efficacy data in these patients,” the NCCN emphasized in a press statement.
“Right now, there is urgent need and limited data,” Steven Pergam, MD, co-leader of the NCCN COVID-19 Vaccination Committee, said in the statement.
“Our number one goal is helping to get the vaccine to as many people as we can,” Dr. Pergam said. “That means following existing national and regional directions for prioritizing people who are more likely to face death or severe illness from COVID-19.”
Dr. Pergam, associate professor at Fred Hutchinson Cancer Research Center in Seattle, further explained that “people receiving active cancer treatment are at greater risk for worse outcomes from COVID-19, particularly if they are older and have additional comorbidities, like immunosuppression.”
NCCN’s recommendations couldn’t have come at a better time for patients with cancer, according to Nora Disis, MD, a professor at the University of Washington in Seattle.
“The NCCN’s recommendations to prioritize COVID vaccinations for cancer patients on active treatment is an important step forward in protecting our patients from the infection,” Dr. Disis said in an interview.
“Cancer patients may be at higher risk for the complications seen with infection. In addition, cancer is a disease of older people, and a good number of our patients have the comorbidities that would predict a poorer outcome if they should become sick,” Dr. Disis added. “With the correct treatment, many patients with cancer will be long-term survivors. It is important that they be protected from infection with COVID to realize their best outcome.”
Additional vaccine considerations
The NCCN recommendations also address several other issues of importance for cancer patients, including:
- Deprioritizing other vaccines. COVID-19 vaccines should take precedence over other vaccines because data on dual vaccination are lacking. The NCCN recommends waiting 14 days after COVID-19 vaccination to deliver other vaccines.
- Vaccinating clinical trial participants. Trial leads should be consulted to prevent protocol violations or exclusions.
- Decision-making in the setting of limited vaccine availability. The NCCN noted that decisions on allocation must be made in accordance with state and local vaccine guidance but suggests prioritizing appropriate patients on active treatment, those planning to start treatment, and those who have just completed treatment. Additional risk factors for these patients, as well as other factors associated with risk for adverse COVID-19 outcomes, should also be considered. These include advanced age, comorbidities, and adverse social and demographic factors such as poverty and limited health care access.
- The need for ongoing prevention measures. Vaccines have been shown to decrease the incidence of COVID-19 and related complications, but it remains unclear whether vaccines prevent infection and subsequent transmission. This means everyone should continue following prevention recommendations, such as wearing masks and avoiding crowds.
The NCCN stressed that these recommendations are “intended to be a living document that is constantly evolving – it will be updated rapidly whenever new data comes out, as well as any potential new vaccines that may get approved in the future.” The NCCN also noted that the advisory committee will meet regularly to refine the recommendations as needed.
Dr. Pergam disclosed relationships with Chimerix Inc., Merck & Co., Global Life Technologies Inc., and Sanofi-Aventis. Dr. Disis disclosed grants from Pfizer, Bavarian Nordisk, Janssen, and Precigen. She is the founder of EpiThany and editor-in-chief of JAMA Oncology.
Feds look to retrofit factories to increase COVID vaccine production
The Biden administration is exploring whether factories can be retrofitted to produce more of the Pfizer/BioNTech and Moderna COVID-19 mRNA vaccines to speed up vaccination of the vast majority of Americans.
The announcement comes as the nation is on track to see 479,000-514,000 deaths by the end of February, said Rochelle Walensky, MD, the director of the Centers for Disease Control and Prevention.
Dr. Walensky, speaking to reporters Wednesday in the first briefing from the White House COVID-19 Response Team, said that 1.6 million COVID-19 shots had been administered each day over the past week and that 3.4 million Americans have been fully vaccinated with two doses.
More than 500 million doses will be needed to vaccinate every American older than 16 years, Andy Slavitt, the senior advisor to the COVID-19 response team, told reporters. Pfizer and Moderna are due to deliver an additional 200 million doses near the end of March, and President Biden is seeking to purchase another 200 million doses from the companies, said Mr. Slavitt.
But it may not be enough. Whether companies can retrofit factories to produce vaccines is “something that’s under active exploration,” Mr. Slavitt said.
“This is a national emergency,” said Jeff Zients, the White House COVID-19 response coordinator. “Everything is on the table across the whole supply chain,” he said. He noted that the administration was also buying low-dead-space syringes to help extract an additional sixth dose from every Pfizer vial.
Mr. Slavitt said the team had identified 12 areas in which Mr. Biden was authorized to use the Defense Production Act to spur the manufacture of items such as masks and COVID-19 diagnostics.
More sequencing needed
As new variants emerge, vaccine makers and the CDC are racing to stay a step ahead. “RNA viruses mutate all the time – that’s what they do, that’s their business,” said Anthony Fauci, MD, director of the National Institute of Allergy and Infectious Diseases and Mr. Biden’s chief medical adviser, in the briefing.
Three concerning variants have emerged: the B117, which is circulating widely in the United Kingdom; the B1.351 in South Africa; and the P.1 in Brazil. As of Jan. 26, no cases involving the B1.351 variant have been detected in the United States; one person with the P.1 variant was identified in Minnesota. The CDC has identified 308 cases of the U.K. variant in 26 states, said Dr. Walensky.
The United States is dismally behind in surveillance and sequencing of variants, said Zients. “We are 43rd in the world at genomic sequencing,” which he said was “totally unacceptable.”
Dr. Walensky said the CDC is working on improving data collection and sequencing, but she said more money is needed to “do the amount of sequencing and surveillance that we need in order to be able to detect these when they first start to emerge.”
Both she and Mr. Zients called on Congress to pass Mr. Biden’s proposed American Rescue package, which includes more money for sequencing.
Dr. Fauci said the National Institutes of Health was collaborating with the CDC to determine whether other newly emerging variants pose any threat – such as increased transmissibility or lethality or some other functional characteristic. Scientists will also monitor “in real-time” whether current vaccines continue to make neutralizing antibodies against these mutants.
“With the U.K. variant, what we’re seeing is a very slight, if at all, impact on vaccine-induced antibodies and very little impact on anything else,” he said. With the South African variant, there is “a multifold diminution in the in vitro neutralization by vaccine-induced antibodies,” but “it still is well within the cushion of protection” for the current vaccines.
But, he added, “we have to be concerned looking forward of what the further evolution of this might be.” The anti-COVID monoclonal antibodies – bamlanivimab and the combination of casirivimab and imdevimab – are “more seriously inhibited by this South African strain,” which is spurring development of new monoclonals.
Dr. Fauci also noted that the Johnson & Johnson/Janssen vaccine that is in development – for which phase 3 data may be released within days – was tested in South Africa and Brazil in addition to the United States. The comparative data could help researchers and clinicians make better-informed decisions about what vaccine to use if the South African variant “seeds itself in the U.S.”
A version of this article first appeared on Medscape.com.
The Biden administration is exploring whether factories can be retrofitted to produce more of the Pfizer/BioNTech and Moderna COVID-19 mRNA vaccines to speed up vaccination of the vast majority of Americans.
The announcement comes as the nation is on track to see 479,000-514,000 deaths by the end of February, said Rochelle Walensky, MD, the director of the Centers for Disease Control and Prevention.
Dr. Walensky, speaking to reporters Wednesday in the first briefing from the White House COVID-19 Response Team, said that 1.6 million COVID-19 shots had been administered each day over the past week and that 3.4 million Americans have been fully vaccinated with two doses.
More than 500 million doses will be needed to vaccinate every American older than 16 years, Andy Slavitt, the senior advisor to the COVID-19 response team, told reporters. Pfizer and Moderna are due to deliver an additional 200 million doses near the end of March, and President Biden is seeking to purchase another 200 million doses from the companies, said Mr. Slavitt.
But it may not be enough. Whether companies can retrofit factories to produce vaccines is “something that’s under active exploration,” Mr. Slavitt said.
“This is a national emergency,” said Jeff Zients, the White House COVID-19 response coordinator. “Everything is on the table across the whole supply chain,” he said. He noted that the administration was also buying low-dead-space syringes to help extract an additional sixth dose from every Pfizer vial.
Mr. Slavitt said the team had identified 12 areas in which Mr. Biden was authorized to use the Defense Production Act to spur the manufacture of items such as masks and COVID-19 diagnostics.
More sequencing needed
As new variants emerge, vaccine makers and the CDC are racing to stay a step ahead. “RNA viruses mutate all the time – that’s what they do, that’s their business,” said Anthony Fauci, MD, director of the National Institute of Allergy and Infectious Diseases and Mr. Biden’s chief medical adviser, in the briefing.
Three concerning variants have emerged: the B117, which is circulating widely in the United Kingdom; the B1.351 in South Africa; and the P.1 in Brazil. As of Jan. 26, no cases involving the B1.351 variant have been detected in the United States; one person with the P.1 variant was identified in Minnesota. The CDC has identified 308 cases of the U.K. variant in 26 states, said Dr. Walensky.
The United States is dismally behind in surveillance and sequencing of variants, said Zients. “We are 43rd in the world at genomic sequencing,” which he said was “totally unacceptable.”
Dr. Walensky said the CDC is working on improving data collection and sequencing, but she said more money is needed to “do the amount of sequencing and surveillance that we need in order to be able to detect these when they first start to emerge.”
Both she and Mr. Zients called on Congress to pass Mr. Biden’s proposed American Rescue package, which includes more money for sequencing.
Dr. Fauci said the National Institutes of Health was collaborating with the CDC to determine whether other newly emerging variants pose any threat – such as increased transmissibility or lethality or some other functional characteristic. Scientists will also monitor “in real-time” whether current vaccines continue to make neutralizing antibodies against these mutants.
“With the U.K. variant, what we’re seeing is a very slight, if at all, impact on vaccine-induced antibodies and very little impact on anything else,” he said. With the South African variant, there is “a multifold diminution in the in vitro neutralization by vaccine-induced antibodies,” but “it still is well within the cushion of protection” for the current vaccines.
But, he added, “we have to be concerned looking forward of what the further evolution of this might be.” The anti-COVID monoclonal antibodies – bamlanivimab and the combination of casirivimab and imdevimab – are “more seriously inhibited by this South African strain,” which is spurring development of new monoclonals.
Dr. Fauci also noted that the Johnson & Johnson/Janssen vaccine that is in development – for which phase 3 data may be released within days – was tested in South Africa and Brazil in addition to the United States. The comparative data could help researchers and clinicians make better-informed decisions about what vaccine to use if the South African variant “seeds itself in the U.S.”
A version of this article first appeared on Medscape.com.
The Biden administration is exploring whether factories can be retrofitted to produce more of the Pfizer/BioNTech and Moderna COVID-19 mRNA vaccines to speed up vaccination of the vast majority of Americans.
The announcement comes as the nation is on track to see 479,000-514,000 deaths by the end of February, said Rochelle Walensky, MD, the director of the Centers for Disease Control and Prevention.
Dr. Walensky, speaking to reporters Wednesday in the first briefing from the White House COVID-19 Response Team, said that 1.6 million COVID-19 shots had been administered each day over the past week and that 3.4 million Americans have been fully vaccinated with two doses.
More than 500 million doses will be needed to vaccinate every American older than 16 years, Andy Slavitt, the senior advisor to the COVID-19 response team, told reporters. Pfizer and Moderna are due to deliver an additional 200 million doses near the end of March, and President Biden is seeking to purchase another 200 million doses from the companies, said Mr. Slavitt.
But it may not be enough. Whether companies can retrofit factories to produce vaccines is “something that’s under active exploration,” Mr. Slavitt said.
“This is a national emergency,” said Jeff Zients, the White House COVID-19 response coordinator. “Everything is on the table across the whole supply chain,” he said. He noted that the administration was also buying low-dead-space syringes to help extract an additional sixth dose from every Pfizer vial.
Mr. Slavitt said the team had identified 12 areas in which Mr. Biden was authorized to use the Defense Production Act to spur the manufacture of items such as masks and COVID-19 diagnostics.
More sequencing needed
As new variants emerge, vaccine makers and the CDC are racing to stay a step ahead. “RNA viruses mutate all the time – that’s what they do, that’s their business,” said Anthony Fauci, MD, director of the National Institute of Allergy and Infectious Diseases and Mr. Biden’s chief medical adviser, in the briefing.
Three concerning variants have emerged: the B117, which is circulating widely in the United Kingdom; the B1.351 in South Africa; and the P.1 in Brazil. As of Jan. 26, no cases involving the B1.351 variant have been detected in the United States; one person with the P.1 variant was identified in Minnesota. The CDC has identified 308 cases of the U.K. variant in 26 states, said Dr. Walensky.
The United States is dismally behind in surveillance and sequencing of variants, said Zients. “We are 43rd in the world at genomic sequencing,” which he said was “totally unacceptable.”
Dr. Walensky said the CDC is working on improving data collection and sequencing, but she said more money is needed to “do the amount of sequencing and surveillance that we need in order to be able to detect these when they first start to emerge.”
Both she and Mr. Zients called on Congress to pass Mr. Biden’s proposed American Rescue package, which includes more money for sequencing.
Dr. Fauci said the National Institutes of Health was collaborating with the CDC to determine whether other newly emerging variants pose any threat – such as increased transmissibility or lethality or some other functional characteristic. Scientists will also monitor “in real-time” whether current vaccines continue to make neutralizing antibodies against these mutants.
“With the U.K. variant, what we’re seeing is a very slight, if at all, impact on vaccine-induced antibodies and very little impact on anything else,” he said. With the South African variant, there is “a multifold diminution in the in vitro neutralization by vaccine-induced antibodies,” but “it still is well within the cushion of protection” for the current vaccines.
But, he added, “we have to be concerned looking forward of what the further evolution of this might be.” The anti-COVID monoclonal antibodies – bamlanivimab and the combination of casirivimab and imdevimab – are “more seriously inhibited by this South African strain,” which is spurring development of new monoclonals.
Dr. Fauci also noted that the Johnson & Johnson/Janssen vaccine that is in development – for which phase 3 data may be released within days – was tested in South Africa and Brazil in addition to the United States. The comparative data could help researchers and clinicians make better-informed decisions about what vaccine to use if the South African variant “seeds itself in the U.S.”
A version of this article first appeared on Medscape.com.