User login
VHA Practice Guideline Recommendations for Diffuse Gliomas (FULL)
Over the past few decades, our understanding of the molecular underpinning of primary neoplasms of the central nervous system (CNS) has progressed substantially. Thanks in large part to this expansion in our knowledge base, the World Health Organization (WHO) has recently updated its classification of tumors of the CNS.1 One of the key elements of this update was the inclusion of molecular diagnostic criteria for the classification of infiltrating gliomas. While the previous classification system was based upon histologic subtypes of the tumor (astrocytoma, oligodendroglioma, and oligoastrocytoma), the revised classification system incorporates molecular testing to establish the genetic characteristics of the tumor to reach a final integrated diagnosis.
In this article, we present 3 cases to highlight some of these recent changes in the WHO diagnostic categories of primary CNS tumors and to illustrate the role of specific molecular tests in reaching a final integrated diagnosis. We then propose a clinical practice guideline for the Veterans Health Administration (VHA) that recommends use of molecular testing for veterans as part of the diagnostic workup of primary CNS neoplasms.
Purpose
In 2013 the VHA National Director of Pathology & Laboratory Medicine Services (P&LMS) chartered a national molecular genetics pathology workgroup (MGPW) that was charged with 4 specific tasks: (1) Provide recommendations about the effective use of molecular genetic testing for veterans; (2) Promote increased quality and availability of molecular testing within the VHA; (3) Encourage internal referral testing; and (4) Create an organizational structure and policies for molecular genetic testing and laboratory developed tests. The workgroup is currently composed of 4 subcommittees: genetic medicine, hematopathology, pharmacogenomics, and molecular oncology. The molecular oncology subcommittee is focused upon molecular genetic testing for solid tumors.
This article is intended to be the first of several publications from the molecular oncology subcommittee of the MGPW that address some of the aforementioned tasks. Similar to the recent publication from the hematopathology subcommittee of the MGPW, this article focuses on CNS neoplasms.2
Scope of Problem
The incidence of tumors of the CNS in the US population varies among age groups. It is the most common solid tumor in children aged < 14 years and represents a significant cause of mortality across all age groups.3 Of CNS tumors, diffuse gliomas comprise about 20% of the tumors and more than 70% of the primary malignant CNS tumors.3 Analysis of the VA Central Cancer Registry data from 2010 to 2014 identified 1,186 veterans (about 237 veterans per year) who were diagnosed with diffuse gliomas. (Lynch, Kulich, Colman, unpublished data, February 2018). While the majority (nearly 80%) of these cases were glioblastomas (GBMs), unfortunately a majority of these cases did not undergo molecular testing (Lynch, Kulich, Colman, unpublished data, February 2018).
Although this low rate of testing may be in part reflective of the period from which these data were gleaned (ie, prior to the WHO release of their updated the classification of tumors of the CNS), it is important to raise VA practitioners’ awareness of these recent changes to ensure that veterans receive the proper diagnosis and treatment for their disease. Thus, while the number of veterans diagnosed with diffuse gliomas within the VHA is relatively small in comparison to other malignancies, such as prostatic adenocarcinomas and lung carcinomas, the majority of diffuse gliomas do not seem to be receiving the molecular testing that would be necessary for (1) appropriate classification under the recently revised WHO recommendations; and (2) making important treatment decisions.
Case Presentations
Case 1. A veteran of the Gulf War presented with a 3-month history of possible narcoleptic events associated with a motor vehicle accident. Magnetic resonance imaging (MRI) revealed a large left frontal mass lesion with minimal surrounding edema without appreciable contrast enhancement (Figures 1A, 1B, and 1C). 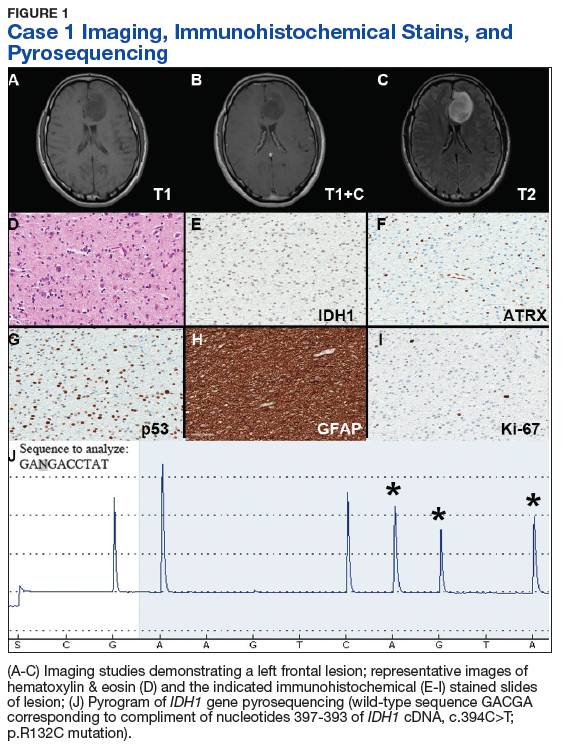
Neither mitotic figures nor endothelial proliferation were identified. Immunohistochemical stains revealed a lack of R132H mutant IDH1 protein expression, a loss of nuclear staining for ATRX protein within a substantial number of cells, and a clonal pattern of p53 protein overexpression (Figures 1E, 1F, and 1G). The lesion demonstrated diffuse glial fibrillary acidic protein (GFAP) immunoreactivity and a low proliferation index (as determined by Ki-67 staining; estimated at less than 5%) (Figures 1H and 1I).
Based upon these results, an initial morphologic diagnosis of diffuse glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. While fluorescence in situ hybridization (FISH) studies were negative for 1p/19q codeletion, pyrosequencing analysis revealed the presence of a c.394C>T (R132C) mutation of the IDH1 gene (Figure 1J). The University of Pittsburgh Medical Center’s GlioSeq targeted next-generation sequence (NGS) analysis confirmed the presence of the c.394C > T mutation in IDH1 gene.4 Based upon this additional information, a final integrated morphologic and molecular diagnosis of diffuse astrocytoma, IDH-mutant was rendered.
Case 2. A Vietnam War veteran presented with a 6-week history of new onset falls with associated left lower extremity weakness. A MRI revealed a right frontoparietal mass lesion with surrounding edema without appreciable contrast enhancement (Figures 2A, 2B, and 2C). 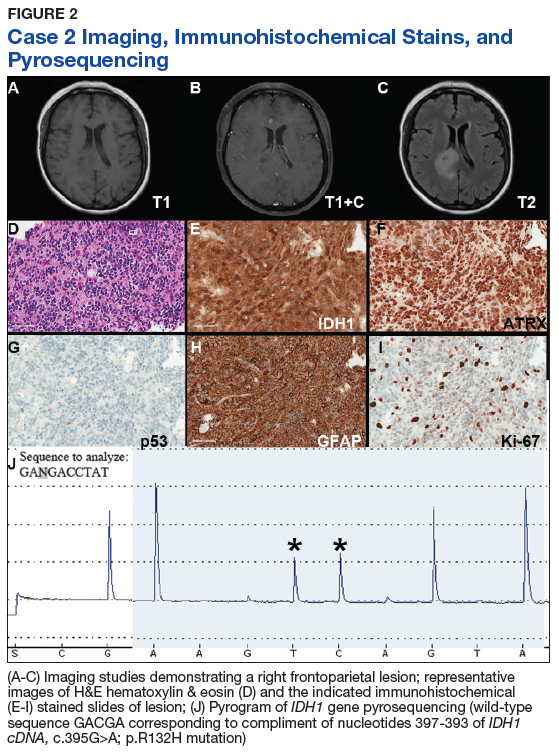
Immunohistochemical stains revealed R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, the lack of a clonal pattern of p53 protein overexpression, diffuse GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 20% (Figures 2E, 2F, 2G, 2H and 2I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were positive for 1p/19q codeletion, and pyrosequencing analysis confirmed the immunohistochemical findings of a c.395G>A (R132H) mutation of the IDH1 gene (Figure 2J). GlioSeq targeted NGS analysis confirmed the presence of the c.395G>A mutation in the IDH1 gene, a mutation in the telomerase reverse transcriptase (TERT) promoter, and possible decreased copy number of the CIC (chromosome 1p) and FUBP1 (chromosome 19q) genes.
A final integrated morphologic and molecular diagnosis of anaplastic oligodendroglioma, IDH-mutant and 1p/19q-codeleted was rendered based on the additional information. With this final diagnosis, methylation analysis of the MGMT gene promoter, which was performed for prognostic and predictive purposes, was identified in this case.5,6
Case 3. A veteran of the Vietnam War presented with a new onset seizure. A MRI revealed a focally contrast-enhancing mass with surrounding edema within the left frontal lobe (Figures 3A, 3B, and 3C). 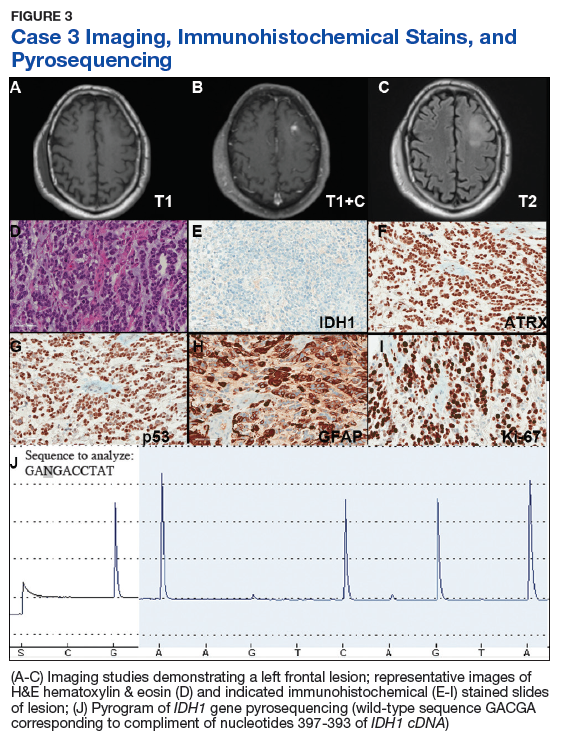
Hematoxylin and eosin (H&E) stained sections following formalin fixation and paraffin embedding demonstrated similar findings (Figure 3D), and while mitotic figures were readily identified, areas of necrosis were not identified and endothelial proliferation was not a prominent feature. Immunohistochemical stains revealed no evidence of R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, a clonal pattern of p53 protein overexpression, patchy GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 50% (Figures 3E, 3F, 3G, 3H, and 3I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and the tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were negative for EGFR gene amplification and 1p/19q codeletion, although a gain of the long arm of chromosome 1 was detected. Pyrosequencing analysis for mutations in codon 132 of the IDH1 gene revealed no mutations (Figure 3J). GlioSeq targeted NGS analysis identified mutations within the NF1, TP53, and PIK3CA genes without evidence of mutations in the IDH1, IDH2, ATRX, H3F3A, or EGFR genes or the TERT promoter. Based upon this additional information, a final integrated morphologic and molecular diagnosis of GBM, IDH wild-type was issued. The MGMT gene promoter was negative for methylation, a finding that has prognostic and predictive impact with regard to treatment with temazolamide.7-9
New Diffuse Glioma Classification
Since the issuance of the previous edition of the WHO classification of CNS tumors in 2007, several sentinel discoveries have been made that have advanced our understanding of the underlying biology of primary CNS neoplasms. Since a detailed review of these findings is beyond the scope and purpose of this manuscript and salient reviews on the topic can be found elsewhere, we will focus on the molecular findings that have been incorporated into the recently revised WHO classification.10 The importance of providing such information for proper patient management is illustrated by the recent acknowledgement by the American Academy of Neurology that molecular testing of brain tumors is a specific area in which there is a need for quality improvement.11 Therefore, it is critical that these underlying molecular abnormalities are identified to allow for proper classification and treatment of diffuse gliomas in the veteran population.
As noted previously, based on VA cancer registry data, diffuse gliomas are the most commonly encountered primary CNS cancers in the veteran population. Several of the aforementioned seminal discoveries have been incorporated into the updated classification of diffuse gliomas. While the recently updated WHO classification allows for the assignment of “not otherwise specified (NOS)” diagnostic designation, this category must be limited to cases where there is insufficient data to allow for a more precise classification due to sample limitations and not simply due to a failure of VA pathology laboratories to pursue the appropriate diagnostic testing.
Figure 4 presents the recommended diagnostic workflow for the workup of diffuse gliomas. As illustrated in the above cases, a variety of different methodologies, including immunohistochemical, FISH, loss of heterozygosity analysis, traditional and NGS may be applied when elucidating the status of molecular events at critical diagnostic branch points. 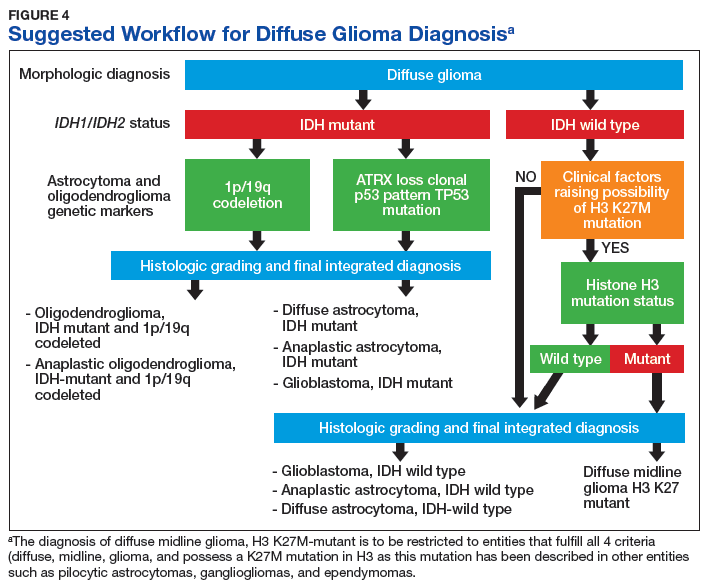
Diagnostic Uses of Molecular Testing
While the case studies in this article demonstrate the use of ancillary testing and provide a suggested strategy for properly subclassifying diffuse gliomas, inherent in this strategy is the assumption that, based upon the initial clinical and pathologic information available, one can accurately categorize the lesion as a diffuse glioma. In reality, such a distinction is not always a straightforward endeavor. It is well recognized that a proportion of low-grade, typically radiologically circumscribed, CNS neoplasms, such as pilocytic astrocytomas and glioneuronal tumors, may infiltrate the surrounding brain parenchyma. In addition, many of these low-grade CNS neoplasms also may have growth patterns that are shared with diffuse gliomas, a diagnostic challenge that often can be further hampered by the inherent limitations involved in obtaining adequate samples for diagnosis from the CNS.
Although there are limitations and caveats, molecular diagnostic testing may be invaluable in properly classifying CNS tumors in such situations. The finding of mutations in the IDH1 or IDH2 genes has been shown to be very valuable in distinguishing low-grade diffuse glioma from both nonneoplastic and low-grade circumscribed neuroepithelial neoplasms that may exhibit growth patterns that can mimic those of diffuse gliomas.15-17 Conversely, finding abnormalities in the BRAF gene in a brain neoplasm that has a low-grade morphology suggests that the lesion may represent one of these low-grade lesions such as a pleomorphic xanthoastrocytoma, pilocytic astrocytoma, or mixed neuronal-glial tumor as opposed to a diffuse glioma.18,19
Depending upon the environment in which one practices, small biopsy specimens may be prevalent, and unfortunately, it is not uncommon to obtain a biopsy that exhibits a histologic growth pattern that is discordant from what one would predict based on the clinical context and imaging findings. Molecular testing may be useful in resolving discordances in such situations. If a biopsy of a ring-enhancing lesion demonstrates a diffuse glioma that doesn’t meet WHO grade IV criteria, applying methodologies that look for genetic features commonly encountered in high-grade astrocytomas may identify genetic abnormalities that suggest a more aggressive lesion than is indicated by the histologic findings. The presence of genetic abnormalities such as homozygous deletion of the CDKN2A gene, TERT promoter mutation, loss of heterozygosity of chromosome 10q and/or phosphatase and tensin homolog (PTEN) mutations, EGFR gene amplification or the presence of the EGFR variant III are a few findings that would suggest the aforementioned sample may represent an undersampling of a higher grade diffuse astrocytoma, which would be important information to convey to the treating clinicians.20-26
Testing In the VA
The goals of the MPWG include promoting increased quality and availability of genetic testing within the VHA as well as encouraging internal referral testing. An informal survey of the chiefs of VA Pathology and Laboratory Medicine Services was conducted in November of 2017 in an attempt to identify internal VA pathology laboratories currently conducting testing that may be of use in the workup of diffuse gliomas (Table 1). 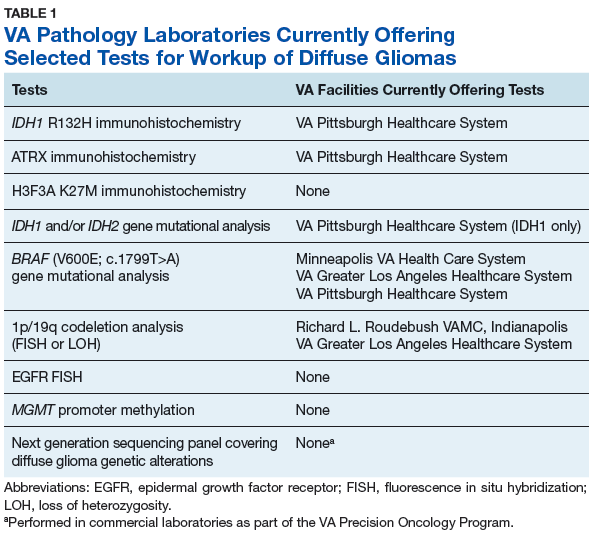
The VA currently offers NGS panels for patients with advanced-stage malignancies under the auspices of the Precision Oncology Program, whose reports provide both (1) mutational analyses for genes such as TP53, ATRX, NF1, BRAF, PTEN, TERT IDH1, and IDH2 that may be useful in the proper classifying of high-grade diffuse gliomas; and (2) information regarding clinical trials for which the veteran may be eligible for based on their glioma’s mutational profile. Interested VA providers should visit tinyurl.com/precisiononcology/ for more information about this program. Finally, although internal testing within VA laboratories is recommended to allow for the development of more cost-effective testing, testing may be performed through many nationally contracted reference laboratories.
Conclusion
In light of the recent progress made in our understanding of the molecular events of gliomagenesis, the way we diagnose diffuse gliomas within the CNS has undergone a major paradigm shift. While histology still plays a critical role in the process, we believe that additional ancillary testing is a requirement for all diffuse gliomas diagnosed within VA pathology laboratories. In the context of recently encountered cases, we have provided a recommended workflow highlighting the testing that can be performed to allow for the proper diagnosis of our veterans with diffuse gliomas (Figure 4).
Unless limited by the amount of tissue available for such tests, ancillary testing must be performed on all diffuse gliomas diagnosed within the VA system to ensure proper diagnosis and treatment of our veterans with diffuse gliomas. 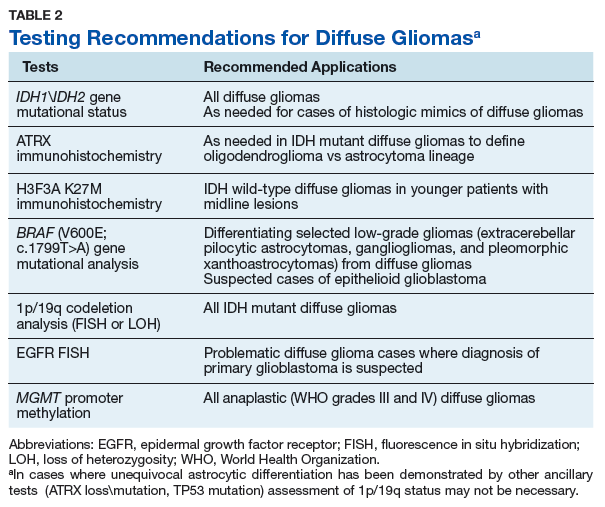

Acknowledgments
The authors thank Dr. Craig M. Horbinski (Feinberg School of Medicine, Northwestern University) and Dr. Geoffrey H. Murdoch (University of Pittsburgh) for their constructive criticism of the manuscript. We also thank the following individuals for past service as members of the molecular oncology subcommittee of the MGPW: Dr. George Ansstas (Washington University School of Medicine), Dr. Osssama Hemadeh (Bay Pines VA Health Care System), Dr. James Herman (VA Pittsburgh Healthcare System), and Dr. Ryan Phan (formerly of the VA Greater Los Angeles Healthcare System) as well as the members of the Veterans Administration pathology and laboratory medicine service molecular genetics pathology workgroup.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies.
Dr. Kulich is the Acting Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System and member of the Division of Neuropathology at University of Pittsburgh Department of Pathology, Dr. Duvvuri is an Otolaryngologist at VA Pittsburgh Healthcare System, and Dr. Passero is the Section Chief of Hematology\Oncology at VA Pittsburgh Healthcare System in Pennsylvania. Dr. Becker is an Oncologist at VA-New York Harbor Healthcare System. Dr. Dacic is a Pathologist at University of Pittsburgh Department of Pathology in Pennsylvania. Dr. Ehsan is Chief of Pathology and Laboratory Medicine Services at the South Texas Veterans Healthcare System in San Antonio. Dr. Gutkin is the former Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System. Dr. Hou is a Pathologist at St. Louis VA Medical Center in Missouri. Dr. Icardi is the VA National Director of Pathology and Laboratory Medicine Services. Dr. Lyle is a Pathologist at Bay Pine Health Care System in Florida. Dr. Lynch is an Investigator at VA Salt Lake Health Care System Informatics and Computing Infrastructure. Dr. Montgomery is an Oncologist at VA Puget Sound Health Care System, in Seattle, Washington. Dr. Przygodzki is the Director of Genomic Medicine Implementation and Associate Director of Genomic Medicine for the VA. Dr. Colman is a Neuro-Oncologist at George E. Wahlen VA Medical Center and the Director of Medical Neuro-Oncology at the Huntsman Cancer Institute, Salt Lake City, Utah.
Correspondence: Dr. Kulich ([email protected])
1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803-820.
2. Wang-Rodriguez J, Yunes A, Phan R, et al. The challenges of precision medicine and new advances in molecular diagnostic testing in hematolymphoid malignancies: impact on the VHA. Fed Pract. 2017;34(suppl 5):S38-S49.
3. Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. 2017;19(suppl 5):v1-v88.
4. Nikiforova MN, Wald AI, Melan MA, et al. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol. 2016;18(3)379-387.
5. Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90(19):1473-1479.
6. van den Bent MJ, Erdem-Eraslan L, Idbaih A, et al. MGMT-STP27 methylation status as predictive marker for response to PCV in anaplastic oligodendrogliomas and oligoastrocytomas. A report from EORTC study 26951. Clin Cancer Res. 2013;19(19):5513-5522.
7. Stupp R, Hegi ME, Mason WP, et al; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459-466.
8. Malmstrom A, Gronberg BH, Marosi C, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012;13(9):916-926.
9. van den Bent MJ, Kros JM. Predictive and prognostic markers in neuro-oncology. J Neuropathol Exp Neurol. 2007;66(12):1074-1081.
10. Chen R, Smith-Cohn M, Cohen AL, Colman H. Glioma subclassifications and their clinical significance. Neurotherapeutics. 2017;14(2):284-297.
11. Jordan JT, Sanders AE, Armstrong T, et al. Quality improvement in neurology: neuro-oncology quality measurement set. Neurology. 2018;90(14):652-658.
12. Chen L, Voronovich Z, Clark K, et al. Predicting the likelihood of an isocitrate dehydrogenase 1 or 2 mutation in diagnoses of infiltrative glioma. Neuro Oncol. 2014;16(11):1478-1483.
13. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997-1003.
14. Wick W, Platten M, Meisner C, et al; NOA-08 Study Group of Neuro-oncology Working Group (NOA) of German Cancer Society. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012;13(7):707-715.
15. Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN. Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol. 2009;68(12):1319-1325.
16. Camelo-Piragua S, Jansen M, Ganguly A, Kim JC, Louis DN, Nutt CL. Mutant IDH1-specific immunohistochemistry distinguishes diffuse astrocytoma from astrocytosis. Acta Neuropathol. 2010;119(4):509-511.
17. Horbinski C, Kofler J, Yeaney G, et al. Isocitrate dehydrogenase 1 analysis differentiates gangliogliomas from infiltrative gliomas. Brain Pathol. 2011;21(5):564-574.
18. Berghoff AS, Preusser M. BRAF alterations in brain tumours: molecular pathology and therapeutic opportunities. Curr Opin Neurol. 2014;27(6):689-696.
19. Korshunov A, Meyer J, Capper D, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118(3):401-405.
20. Fuller CE, Schmidt RE, Roth KA, et al. Clinical utility of fluorescence in situ hybridization (FISH) in morphologically ambiguous gliomas with hybrid oligodendroglial/astrocytic features. J Neuropathol Exp Neurol. 2003;62(11):1118-1128.
21. Horbinski C. Practical molecular diagnostics in neuropathology: making a tough job a little easier. Semin Diagn Pathol. 2010;27(2):105-113.
22. Fuller GN, Bigner SH. Amplified cellular oncogenes in neoplasms of the human central nervous system. Mutat Res. 1992;276(3):299-306.
23. Brennan CW, Verhaak RG, McKenna A, et al; TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462-477.
24. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):829-848.
25. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110(15):6021-6026.
26. Nikiforova MN, Hamilton RL. Molecular diagnostics of gliomas. Arch Pathol Lab Med. 2011;135(5):558-568.
Over the past few decades, our understanding of the molecular underpinning of primary neoplasms of the central nervous system (CNS) has progressed substantially. Thanks in large part to this expansion in our knowledge base, the World Health Organization (WHO) has recently updated its classification of tumors of the CNS.1 One of the key elements of this update was the inclusion of molecular diagnostic criteria for the classification of infiltrating gliomas. While the previous classification system was based upon histologic subtypes of the tumor (astrocytoma, oligodendroglioma, and oligoastrocytoma), the revised classification system incorporates molecular testing to establish the genetic characteristics of the tumor to reach a final integrated diagnosis.
In this article, we present 3 cases to highlight some of these recent changes in the WHO diagnostic categories of primary CNS tumors and to illustrate the role of specific molecular tests in reaching a final integrated diagnosis. We then propose a clinical practice guideline for the Veterans Health Administration (VHA) that recommends use of molecular testing for veterans as part of the diagnostic workup of primary CNS neoplasms.
Purpose
In 2013 the VHA National Director of Pathology & Laboratory Medicine Services (P&LMS) chartered a national molecular genetics pathology workgroup (MGPW) that was charged with 4 specific tasks: (1) Provide recommendations about the effective use of molecular genetic testing for veterans; (2) Promote increased quality and availability of molecular testing within the VHA; (3) Encourage internal referral testing; and (4) Create an organizational structure and policies for molecular genetic testing and laboratory developed tests. The workgroup is currently composed of 4 subcommittees: genetic medicine, hematopathology, pharmacogenomics, and molecular oncology. The molecular oncology subcommittee is focused upon molecular genetic testing for solid tumors.
This article is intended to be the first of several publications from the molecular oncology subcommittee of the MGPW that address some of the aforementioned tasks. Similar to the recent publication from the hematopathology subcommittee of the MGPW, this article focuses on CNS neoplasms.2
Scope of Problem
The incidence of tumors of the CNS in the US population varies among age groups. It is the most common solid tumor in children aged < 14 years and represents a significant cause of mortality across all age groups.3 Of CNS tumors, diffuse gliomas comprise about 20% of the tumors and more than 70% of the primary malignant CNS tumors.3 Analysis of the VA Central Cancer Registry data from 2010 to 2014 identified 1,186 veterans (about 237 veterans per year) who were diagnosed with diffuse gliomas. (Lynch, Kulich, Colman, unpublished data, February 2018). While the majority (nearly 80%) of these cases were glioblastomas (GBMs), unfortunately a majority of these cases did not undergo molecular testing (Lynch, Kulich, Colman, unpublished data, February 2018).
Although this low rate of testing may be in part reflective of the period from which these data were gleaned (ie, prior to the WHO release of their updated the classification of tumors of the CNS), it is important to raise VA practitioners’ awareness of these recent changes to ensure that veterans receive the proper diagnosis and treatment for their disease. Thus, while the number of veterans diagnosed with diffuse gliomas within the VHA is relatively small in comparison to other malignancies, such as prostatic adenocarcinomas and lung carcinomas, the majority of diffuse gliomas do not seem to be receiving the molecular testing that would be necessary for (1) appropriate classification under the recently revised WHO recommendations; and (2) making important treatment decisions.
Case Presentations
Case 1. A veteran of the Gulf War presented with a 3-month history of possible narcoleptic events associated with a motor vehicle accident. Magnetic resonance imaging (MRI) revealed a large left frontal mass lesion with minimal surrounding edema without appreciable contrast enhancement (Figures 1A, 1B, and 1C).
Neither mitotic figures nor endothelial proliferation were identified. Immunohistochemical stains revealed a lack of R132H mutant IDH1 protein expression, a loss of nuclear staining for ATRX protein within a substantial number of cells, and a clonal pattern of p53 protein overexpression (Figures 1E, 1F, and 1G). The lesion demonstrated diffuse glial fibrillary acidic protein (GFAP) immunoreactivity and a low proliferation index (as determined by Ki-67 staining; estimated at less than 5%) (Figures 1H and 1I).
Based upon these results, an initial morphologic diagnosis of diffuse glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. While fluorescence in situ hybridization (FISH) studies were negative for 1p/19q codeletion, pyrosequencing analysis revealed the presence of a c.394C>T (R132C) mutation of the IDH1 gene (Figure 1J). The University of Pittsburgh Medical Center’s GlioSeq targeted next-generation sequence (NGS) analysis confirmed the presence of the c.394C > T mutation in IDH1 gene.4 Based upon this additional information, a final integrated morphologic and molecular diagnosis of diffuse astrocytoma, IDH-mutant was rendered.
Case 2. A Vietnam War veteran presented with a 6-week history of new onset falls with associated left lower extremity weakness. A MRI revealed a right frontoparietal mass lesion with surrounding edema without appreciable contrast enhancement (Figures 2A, 2B, and 2C).
Immunohistochemical stains revealed R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, the lack of a clonal pattern of p53 protein overexpression, diffuse GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 20% (Figures 2E, 2F, 2G, 2H and 2I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were positive for 1p/19q codeletion, and pyrosequencing analysis confirmed the immunohistochemical findings of a c.395G>A (R132H) mutation of the IDH1 gene (Figure 2J). GlioSeq targeted NGS analysis confirmed the presence of the c.395G>A mutation in the IDH1 gene, a mutation in the telomerase reverse transcriptase (TERT) promoter, and possible decreased copy number of the CIC (chromosome 1p) and FUBP1 (chromosome 19q) genes.
A final integrated morphologic and molecular diagnosis of anaplastic oligodendroglioma, IDH-mutant and 1p/19q-codeleted was rendered based on the additional information. With this final diagnosis, methylation analysis of the MGMT gene promoter, which was performed for prognostic and predictive purposes, was identified in this case.5,6
Case 3. A veteran of the Vietnam War presented with a new onset seizure. A MRI revealed a focally contrast-enhancing mass with surrounding edema within the left frontal lobe (Figures 3A, 3B, and 3C).
Hematoxylin and eosin (H&E) stained sections following formalin fixation and paraffin embedding demonstrated similar findings (Figure 3D), and while mitotic figures were readily identified, areas of necrosis were not identified and endothelial proliferation was not a prominent feature. Immunohistochemical stains revealed no evidence of R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, a clonal pattern of p53 protein overexpression, patchy GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 50% (Figures 3E, 3F, 3G, 3H, and 3I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and the tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were negative for EGFR gene amplification and 1p/19q codeletion, although a gain of the long arm of chromosome 1 was detected. Pyrosequencing analysis for mutations in codon 132 of the IDH1 gene revealed no mutations (Figure 3J). GlioSeq targeted NGS analysis identified mutations within the NF1, TP53, and PIK3CA genes without evidence of mutations in the IDH1, IDH2, ATRX, H3F3A, or EGFR genes or the TERT promoter. Based upon this additional information, a final integrated morphologic and molecular diagnosis of GBM, IDH wild-type was issued. The MGMT gene promoter was negative for methylation, a finding that has prognostic and predictive impact with regard to treatment with temazolamide.7-9
New Diffuse Glioma Classification
Since the issuance of the previous edition of the WHO classification of CNS tumors in 2007, several sentinel discoveries have been made that have advanced our understanding of the underlying biology of primary CNS neoplasms. Since a detailed review of these findings is beyond the scope and purpose of this manuscript and salient reviews on the topic can be found elsewhere, we will focus on the molecular findings that have been incorporated into the recently revised WHO classification.10 The importance of providing such information for proper patient management is illustrated by the recent acknowledgement by the American Academy of Neurology that molecular testing of brain tumors is a specific area in which there is a need for quality improvement.11 Therefore, it is critical that these underlying molecular abnormalities are identified to allow for proper classification and treatment of diffuse gliomas in the veteran population.
As noted previously, based on VA cancer registry data, diffuse gliomas are the most commonly encountered primary CNS cancers in the veteran population. Several of the aforementioned seminal discoveries have been incorporated into the updated classification of diffuse gliomas. While the recently updated WHO classification allows for the assignment of “not otherwise specified (NOS)” diagnostic designation, this category must be limited to cases where there is insufficient data to allow for a more precise classification due to sample limitations and not simply due to a failure of VA pathology laboratories to pursue the appropriate diagnostic testing.
Figure 4 presents the recommended diagnostic workflow for the workup of diffuse gliomas. As illustrated in the above cases, a variety of different methodologies, including immunohistochemical, FISH, loss of heterozygosity analysis, traditional and NGS may be applied when elucidating the status of molecular events at critical diagnostic branch points.
Diagnostic Uses of Molecular Testing
While the case studies in this article demonstrate the use of ancillary testing and provide a suggested strategy for properly subclassifying diffuse gliomas, inherent in this strategy is the assumption that, based upon the initial clinical and pathologic information available, one can accurately categorize the lesion as a diffuse glioma. In reality, such a distinction is not always a straightforward endeavor. It is well recognized that a proportion of low-grade, typically radiologically circumscribed, CNS neoplasms, such as pilocytic astrocytomas and glioneuronal tumors, may infiltrate the surrounding brain parenchyma. In addition, many of these low-grade CNS neoplasms also may have growth patterns that are shared with diffuse gliomas, a diagnostic challenge that often can be further hampered by the inherent limitations involved in obtaining adequate samples for diagnosis from the CNS.
Although there are limitations and caveats, molecular diagnostic testing may be invaluable in properly classifying CNS tumors in such situations. The finding of mutations in the IDH1 or IDH2 genes has been shown to be very valuable in distinguishing low-grade diffuse glioma from both nonneoplastic and low-grade circumscribed neuroepithelial neoplasms that may exhibit growth patterns that can mimic those of diffuse gliomas.15-17 Conversely, finding abnormalities in the BRAF gene in a brain neoplasm that has a low-grade morphology suggests that the lesion may represent one of these low-grade lesions such as a pleomorphic xanthoastrocytoma, pilocytic astrocytoma, or mixed neuronal-glial tumor as opposed to a diffuse glioma.18,19
Depending upon the environment in which one practices, small biopsy specimens may be prevalent, and unfortunately, it is not uncommon to obtain a biopsy that exhibits a histologic growth pattern that is discordant from what one would predict based on the clinical context and imaging findings. Molecular testing may be useful in resolving discordances in such situations. If a biopsy of a ring-enhancing lesion demonstrates a diffuse glioma that doesn’t meet WHO grade IV criteria, applying methodologies that look for genetic features commonly encountered in high-grade astrocytomas may identify genetic abnormalities that suggest a more aggressive lesion than is indicated by the histologic findings. The presence of genetic abnormalities such as homozygous deletion of the CDKN2A gene, TERT promoter mutation, loss of heterozygosity of chromosome 10q and/or phosphatase and tensin homolog (PTEN) mutations, EGFR gene amplification or the presence of the EGFR variant III are a few findings that would suggest the aforementioned sample may represent an undersampling of a higher grade diffuse astrocytoma, which would be important information to convey to the treating clinicians.20-26
Testing In the VA
The goals of the MPWG include promoting increased quality and availability of genetic testing within the VHA as well as encouraging internal referral testing. An informal survey of the chiefs of VA Pathology and Laboratory Medicine Services was conducted in November of 2017 in an attempt to identify internal VA pathology laboratories currently conducting testing that may be of use in the workup of diffuse gliomas (Table 1).
The VA currently offers NGS panels for patients with advanced-stage malignancies under the auspices of the Precision Oncology Program, whose reports provide both (1) mutational analyses for genes such as TP53, ATRX, NF1, BRAF, PTEN, TERT IDH1, and IDH2 that may be useful in the proper classifying of high-grade diffuse gliomas; and (2) information regarding clinical trials for which the veteran may be eligible for based on their glioma’s mutational profile. Interested VA providers should visit tinyurl.com/precisiononcology/ for more information about this program. Finally, although internal testing within VA laboratories is recommended to allow for the development of more cost-effective testing, testing may be performed through many nationally contracted reference laboratories.
Conclusion
In light of the recent progress made in our understanding of the molecular events of gliomagenesis, the way we diagnose diffuse gliomas within the CNS has undergone a major paradigm shift. While histology still plays a critical role in the process, we believe that additional ancillary testing is a requirement for all diffuse gliomas diagnosed within VA pathology laboratories. In the context of recently encountered cases, we have provided a recommended workflow highlighting the testing that can be performed to allow for the proper diagnosis of our veterans with diffuse gliomas (Figure 4).
Unless limited by the amount of tissue available for such tests, ancillary testing must be performed on all diffuse gliomas diagnosed within the VA system to ensure proper diagnosis and treatment of our veterans with diffuse gliomas.
Acknowledgments
The authors thank Dr. Craig M. Horbinski (Feinberg School of Medicine, Northwestern University) and Dr. Geoffrey H. Murdoch (University of Pittsburgh) for their constructive criticism of the manuscript. We also thank the following individuals for past service as members of the molecular oncology subcommittee of the MGPW: Dr. George Ansstas (Washington University School of Medicine), Dr. Osssama Hemadeh (Bay Pines VA Health Care System), Dr. James Herman (VA Pittsburgh Healthcare System), and Dr. Ryan Phan (formerly of the VA Greater Los Angeles Healthcare System) as well as the members of the Veterans Administration pathology and laboratory medicine service molecular genetics pathology workgroup.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies.
Dr. Kulich is the Acting Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System and member of the Division of Neuropathology at University of Pittsburgh Department of Pathology, Dr. Duvvuri is an Otolaryngologist at VA Pittsburgh Healthcare System, and Dr. Passero is the Section Chief of Hematology\Oncology at VA Pittsburgh Healthcare System in Pennsylvania. Dr. Becker is an Oncologist at VA-New York Harbor Healthcare System. Dr. Dacic is a Pathologist at University of Pittsburgh Department of Pathology in Pennsylvania. Dr. Ehsan is Chief of Pathology and Laboratory Medicine Services at the South Texas Veterans Healthcare System in San Antonio. Dr. Gutkin is the former Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System. Dr. Hou is a Pathologist at St. Louis VA Medical Center in Missouri. Dr. Icardi is the VA National Director of Pathology and Laboratory Medicine Services. Dr. Lyle is a Pathologist at Bay Pine Health Care System in Florida. Dr. Lynch is an Investigator at VA Salt Lake Health Care System Informatics and Computing Infrastructure. Dr. Montgomery is an Oncologist at VA Puget Sound Health Care System, in Seattle, Washington. Dr. Przygodzki is the Director of Genomic Medicine Implementation and Associate Director of Genomic Medicine for the VA. Dr. Colman is a Neuro-Oncologist at George E. Wahlen VA Medical Center and the Director of Medical Neuro-Oncology at the Huntsman Cancer Institute, Salt Lake City, Utah.
Correspondence: Dr. Kulich ([email protected])
Over the past few decades, our understanding of the molecular underpinning of primary neoplasms of the central nervous system (CNS) has progressed substantially. Thanks in large part to this expansion in our knowledge base, the World Health Organization (WHO) has recently updated its classification of tumors of the CNS.1 One of the key elements of this update was the inclusion of molecular diagnostic criteria for the classification of infiltrating gliomas. While the previous classification system was based upon histologic subtypes of the tumor (astrocytoma, oligodendroglioma, and oligoastrocytoma), the revised classification system incorporates molecular testing to establish the genetic characteristics of the tumor to reach a final integrated diagnosis.
In this article, we present 3 cases to highlight some of these recent changes in the WHO diagnostic categories of primary CNS tumors and to illustrate the role of specific molecular tests in reaching a final integrated diagnosis. We then propose a clinical practice guideline for the Veterans Health Administration (VHA) that recommends use of molecular testing for veterans as part of the diagnostic workup of primary CNS neoplasms.
Purpose
In 2013 the VHA National Director of Pathology & Laboratory Medicine Services (P&LMS) chartered a national molecular genetics pathology workgroup (MGPW) that was charged with 4 specific tasks: (1) Provide recommendations about the effective use of molecular genetic testing for veterans; (2) Promote increased quality and availability of molecular testing within the VHA; (3) Encourage internal referral testing; and (4) Create an organizational structure and policies for molecular genetic testing and laboratory developed tests. The workgroup is currently composed of 4 subcommittees: genetic medicine, hematopathology, pharmacogenomics, and molecular oncology. The molecular oncology subcommittee is focused upon molecular genetic testing for solid tumors.
This article is intended to be the first of several publications from the molecular oncology subcommittee of the MGPW that address some of the aforementioned tasks. Similar to the recent publication from the hematopathology subcommittee of the MGPW, this article focuses on CNS neoplasms.2
Scope of Problem
The incidence of tumors of the CNS in the US population varies among age groups. It is the most common solid tumor in children aged < 14 years and represents a significant cause of mortality across all age groups.3 Of CNS tumors, diffuse gliomas comprise about 20% of the tumors and more than 70% of the primary malignant CNS tumors.3 Analysis of the VA Central Cancer Registry data from 2010 to 2014 identified 1,186 veterans (about 237 veterans per year) who were diagnosed with diffuse gliomas. (Lynch, Kulich, Colman, unpublished data, February 2018). While the majority (nearly 80%) of these cases were glioblastomas (GBMs), unfortunately a majority of these cases did not undergo molecular testing (Lynch, Kulich, Colman, unpublished data, February 2018).
Although this low rate of testing may be in part reflective of the period from which these data were gleaned (ie, prior to the WHO release of their updated the classification of tumors of the CNS), it is important to raise VA practitioners’ awareness of these recent changes to ensure that veterans receive the proper diagnosis and treatment for their disease. Thus, while the number of veterans diagnosed with diffuse gliomas within the VHA is relatively small in comparison to other malignancies, such as prostatic adenocarcinomas and lung carcinomas, the majority of diffuse gliomas do not seem to be receiving the molecular testing that would be necessary for (1) appropriate classification under the recently revised WHO recommendations; and (2) making important treatment decisions.
Case Presentations
Case 1. A veteran of the Gulf War presented with a 3-month history of possible narcoleptic events associated with a motor vehicle accident. Magnetic resonance imaging (MRI) revealed a large left frontal mass lesion with minimal surrounding edema without appreciable contrast enhancement (Figures 1A, 1B, and 1C).
Neither mitotic figures nor endothelial proliferation were identified. Immunohistochemical stains revealed a lack of R132H mutant IDH1 protein expression, a loss of nuclear staining for ATRX protein within a substantial number of cells, and a clonal pattern of p53 protein overexpression (Figures 1E, 1F, and 1G). The lesion demonstrated diffuse glial fibrillary acidic protein (GFAP) immunoreactivity and a low proliferation index (as determined by Ki-67 staining; estimated at less than 5%) (Figures 1H and 1I).
Based upon these results, an initial morphologic diagnosis of diffuse glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. While fluorescence in situ hybridization (FISH) studies were negative for 1p/19q codeletion, pyrosequencing analysis revealed the presence of a c.394C>T (R132C) mutation of the IDH1 gene (Figure 1J). The University of Pittsburgh Medical Center’s GlioSeq targeted next-generation sequence (NGS) analysis confirmed the presence of the c.394C > T mutation in IDH1 gene.4 Based upon this additional information, a final integrated morphologic and molecular diagnosis of diffuse astrocytoma, IDH-mutant was rendered.
Case 2. A Vietnam War veteran presented with a 6-week history of new onset falls with associated left lower extremity weakness. A MRI revealed a right frontoparietal mass lesion with surrounding edema without appreciable contrast enhancement (Figures 2A, 2B, and 2C).
Immunohistochemical stains revealed R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, the lack of a clonal pattern of p53 protein overexpression, diffuse GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 20% (Figures 2E, 2F, 2G, 2H and 2I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were positive for 1p/19q codeletion, and pyrosequencing analysis confirmed the immunohistochemical findings of a c.395G>A (R132H) mutation of the IDH1 gene (Figure 2J). GlioSeq targeted NGS analysis confirmed the presence of the c.395G>A mutation in the IDH1 gene, a mutation in the telomerase reverse transcriptase (TERT) promoter, and possible decreased copy number of the CIC (chromosome 1p) and FUBP1 (chromosome 19q) genes.
A final integrated morphologic and molecular diagnosis of anaplastic oligodendroglioma, IDH-mutant and 1p/19q-codeleted was rendered based on the additional information. With this final diagnosis, methylation analysis of the MGMT gene promoter, which was performed for prognostic and predictive purposes, was identified in this case.5,6
Case 3. A veteran of the Vietnam War presented with a new onset seizure. A MRI revealed a focally contrast-enhancing mass with surrounding edema within the left frontal lobe (Figures 3A, 3B, and 3C).
Hematoxylin and eosin (H&E) stained sections following formalin fixation and paraffin embedding demonstrated similar findings (Figure 3D), and while mitotic figures were readily identified, areas of necrosis were not identified and endothelial proliferation was not a prominent feature. Immunohistochemical stains revealed no evidence of R132H mutant IDH1 protein expression, retention of nuclear staining for ATRX protein, a clonal pattern of p53 protein overexpression, patchy GFAP immunoreactivity, and a proliferation index (as determined by Ki-67 staining) focally approaching 50% (Figures 3E, 3F, 3G, 3H, and 3I).
Based upon these results, an initial morphologic diagnosis of diffuse (high grade) glioma was issued, and the tissue was subjected to a variety of nucleic acid-based tests. The FISH studies were negative for EGFR gene amplification and 1p/19q codeletion, although a gain of the long arm of chromosome 1 was detected. Pyrosequencing analysis for mutations in codon 132 of the IDH1 gene revealed no mutations (Figure 3J). GlioSeq targeted NGS analysis identified mutations within the NF1, TP53, and PIK3CA genes without evidence of mutations in the IDH1, IDH2, ATRX, H3F3A, or EGFR genes or the TERT promoter. Based upon this additional information, a final integrated morphologic and molecular diagnosis of GBM, IDH wild-type was issued. The MGMT gene promoter was negative for methylation, a finding that has prognostic and predictive impact with regard to treatment with temazolamide.7-9
New Diffuse Glioma Classification
Since the issuance of the previous edition of the WHO classification of CNS tumors in 2007, several sentinel discoveries have been made that have advanced our understanding of the underlying biology of primary CNS neoplasms. Since a detailed review of these findings is beyond the scope and purpose of this manuscript and salient reviews on the topic can be found elsewhere, we will focus on the molecular findings that have been incorporated into the recently revised WHO classification.10 The importance of providing such information for proper patient management is illustrated by the recent acknowledgement by the American Academy of Neurology that molecular testing of brain tumors is a specific area in which there is a need for quality improvement.11 Therefore, it is critical that these underlying molecular abnormalities are identified to allow for proper classification and treatment of diffuse gliomas in the veteran population.
As noted previously, based on VA cancer registry data, diffuse gliomas are the most commonly encountered primary CNS cancers in the veteran population. Several of the aforementioned seminal discoveries have been incorporated into the updated classification of diffuse gliomas. While the recently updated WHO classification allows for the assignment of “not otherwise specified (NOS)” diagnostic designation, this category must be limited to cases where there is insufficient data to allow for a more precise classification due to sample limitations and not simply due to a failure of VA pathology laboratories to pursue the appropriate diagnostic testing.
Figure 4 presents the recommended diagnostic workflow for the workup of diffuse gliomas. As illustrated in the above cases, a variety of different methodologies, including immunohistochemical, FISH, loss of heterozygosity analysis, traditional and NGS may be applied when elucidating the status of molecular events at critical diagnostic branch points.
Diagnostic Uses of Molecular Testing
While the case studies in this article demonstrate the use of ancillary testing and provide a suggested strategy for properly subclassifying diffuse gliomas, inherent in this strategy is the assumption that, based upon the initial clinical and pathologic information available, one can accurately categorize the lesion as a diffuse glioma. In reality, such a distinction is not always a straightforward endeavor. It is well recognized that a proportion of low-grade, typically radiologically circumscribed, CNS neoplasms, such as pilocytic astrocytomas and glioneuronal tumors, may infiltrate the surrounding brain parenchyma. In addition, many of these low-grade CNS neoplasms also may have growth patterns that are shared with diffuse gliomas, a diagnostic challenge that often can be further hampered by the inherent limitations involved in obtaining adequate samples for diagnosis from the CNS.
Although there are limitations and caveats, molecular diagnostic testing may be invaluable in properly classifying CNS tumors in such situations. The finding of mutations in the IDH1 or IDH2 genes has been shown to be very valuable in distinguishing low-grade diffuse glioma from both nonneoplastic and low-grade circumscribed neuroepithelial neoplasms that may exhibit growth patterns that can mimic those of diffuse gliomas.15-17 Conversely, finding abnormalities in the BRAF gene in a brain neoplasm that has a low-grade morphology suggests that the lesion may represent one of these low-grade lesions such as a pleomorphic xanthoastrocytoma, pilocytic astrocytoma, or mixed neuronal-glial tumor as opposed to a diffuse glioma.18,19
Depending upon the environment in which one practices, small biopsy specimens may be prevalent, and unfortunately, it is not uncommon to obtain a biopsy that exhibits a histologic growth pattern that is discordant from what one would predict based on the clinical context and imaging findings. Molecular testing may be useful in resolving discordances in such situations. If a biopsy of a ring-enhancing lesion demonstrates a diffuse glioma that doesn’t meet WHO grade IV criteria, applying methodologies that look for genetic features commonly encountered in high-grade astrocytomas may identify genetic abnormalities that suggest a more aggressive lesion than is indicated by the histologic findings. The presence of genetic abnormalities such as homozygous deletion of the CDKN2A gene, TERT promoter mutation, loss of heterozygosity of chromosome 10q and/or phosphatase and tensin homolog (PTEN) mutations, EGFR gene amplification or the presence of the EGFR variant III are a few findings that would suggest the aforementioned sample may represent an undersampling of a higher grade diffuse astrocytoma, which would be important information to convey to the treating clinicians.20-26
Testing In the VA
The goals of the MPWG include promoting increased quality and availability of genetic testing within the VHA as well as encouraging internal referral testing. An informal survey of the chiefs of VA Pathology and Laboratory Medicine Services was conducted in November of 2017 in an attempt to identify internal VA pathology laboratories currently conducting testing that may be of use in the workup of diffuse gliomas (Table 1).
The VA currently offers NGS panels for patients with advanced-stage malignancies under the auspices of the Precision Oncology Program, whose reports provide both (1) mutational analyses for genes such as TP53, ATRX, NF1, BRAF, PTEN, TERT IDH1, and IDH2 that may be useful in the proper classifying of high-grade diffuse gliomas; and (2) information regarding clinical trials for which the veteran may be eligible for based on their glioma’s mutational profile. Interested VA providers should visit tinyurl.com/precisiononcology/ for more information about this program. Finally, although internal testing within VA laboratories is recommended to allow for the development of more cost-effective testing, testing may be performed through many nationally contracted reference laboratories.
Conclusion
In light of the recent progress made in our understanding of the molecular events of gliomagenesis, the way we diagnose diffuse gliomas within the CNS has undergone a major paradigm shift. While histology still plays a critical role in the process, we believe that additional ancillary testing is a requirement for all diffuse gliomas diagnosed within VA pathology laboratories. In the context of recently encountered cases, we have provided a recommended workflow highlighting the testing that can be performed to allow for the proper diagnosis of our veterans with diffuse gliomas (Figure 4).
Unless limited by the amount of tissue available for such tests, ancillary testing must be performed on all diffuse gliomas diagnosed within the VA system to ensure proper diagnosis and treatment of our veterans with diffuse gliomas.
Acknowledgments
The authors thank Dr. Craig M. Horbinski (Feinberg School of Medicine, Northwestern University) and Dr. Geoffrey H. Murdoch (University of Pittsburgh) for their constructive criticism of the manuscript. We also thank the following individuals for past service as members of the molecular oncology subcommittee of the MGPW: Dr. George Ansstas (Washington University School of Medicine), Dr. Osssama Hemadeh (Bay Pines VA Health Care System), Dr. James Herman (VA Pittsburgh Healthcare System), and Dr. Ryan Phan (formerly of the VA Greater Los Angeles Healthcare System) as well as the members of the Veterans Administration pathology and laboratory medicine service molecular genetics pathology workgroup.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the US Government, or any of its agencies.
Dr. Kulich is the Acting Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System and member of the Division of Neuropathology at University of Pittsburgh Department of Pathology, Dr. Duvvuri is an Otolaryngologist at VA Pittsburgh Healthcare System, and Dr. Passero is the Section Chief of Hematology\Oncology at VA Pittsburgh Healthcare System in Pennsylvania. Dr. Becker is an Oncologist at VA-New York Harbor Healthcare System. Dr. Dacic is a Pathologist at University of Pittsburgh Department of Pathology in Pennsylvania. Dr. Ehsan is Chief of Pathology and Laboratory Medicine Services at the South Texas Veterans Healthcare System in San Antonio. Dr. Gutkin is the former Chief of Pathology and Laboratory Medicine Service at VA Pittsburgh Healthcare System. Dr. Hou is a Pathologist at St. Louis VA Medical Center in Missouri. Dr. Icardi is the VA National Director of Pathology and Laboratory Medicine Services. Dr. Lyle is a Pathologist at Bay Pine Health Care System in Florida. Dr. Lynch is an Investigator at VA Salt Lake Health Care System Informatics and Computing Infrastructure. Dr. Montgomery is an Oncologist at VA Puget Sound Health Care System, in Seattle, Washington. Dr. Przygodzki is the Director of Genomic Medicine Implementation and Associate Director of Genomic Medicine for the VA. Dr. Colman is a Neuro-Oncologist at George E. Wahlen VA Medical Center and the Director of Medical Neuro-Oncology at the Huntsman Cancer Institute, Salt Lake City, Utah.
Correspondence: Dr. Kulich ([email protected])
1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803-820.
2. Wang-Rodriguez J, Yunes A, Phan R, et al. The challenges of precision medicine and new advances in molecular diagnostic testing in hematolymphoid malignancies: impact on the VHA. Fed Pract. 2017;34(suppl 5):S38-S49.
3. Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. 2017;19(suppl 5):v1-v88.
4. Nikiforova MN, Wald AI, Melan MA, et al. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol. 2016;18(3)379-387.
5. Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90(19):1473-1479.
6. van den Bent MJ, Erdem-Eraslan L, Idbaih A, et al. MGMT-STP27 methylation status as predictive marker for response to PCV in anaplastic oligodendrogliomas and oligoastrocytomas. A report from EORTC study 26951. Clin Cancer Res. 2013;19(19):5513-5522.
7. Stupp R, Hegi ME, Mason WP, et al; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459-466.
8. Malmstrom A, Gronberg BH, Marosi C, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012;13(9):916-926.
9. van den Bent MJ, Kros JM. Predictive and prognostic markers in neuro-oncology. J Neuropathol Exp Neurol. 2007;66(12):1074-1081.
10. Chen R, Smith-Cohn M, Cohen AL, Colman H. Glioma subclassifications and their clinical significance. Neurotherapeutics. 2017;14(2):284-297.
11. Jordan JT, Sanders AE, Armstrong T, et al. Quality improvement in neurology: neuro-oncology quality measurement set. Neurology. 2018;90(14):652-658.
12. Chen L, Voronovich Z, Clark K, et al. Predicting the likelihood of an isocitrate dehydrogenase 1 or 2 mutation in diagnoses of infiltrative glioma. Neuro Oncol. 2014;16(11):1478-1483.
13. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997-1003.
14. Wick W, Platten M, Meisner C, et al; NOA-08 Study Group of Neuro-oncology Working Group (NOA) of German Cancer Society. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012;13(7):707-715.
15. Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN. Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol. 2009;68(12):1319-1325.
16. Camelo-Piragua S, Jansen M, Ganguly A, Kim JC, Louis DN, Nutt CL. Mutant IDH1-specific immunohistochemistry distinguishes diffuse astrocytoma from astrocytosis. Acta Neuropathol. 2010;119(4):509-511.
17. Horbinski C, Kofler J, Yeaney G, et al. Isocitrate dehydrogenase 1 analysis differentiates gangliogliomas from infiltrative gliomas. Brain Pathol. 2011;21(5):564-574.
18. Berghoff AS, Preusser M. BRAF alterations in brain tumours: molecular pathology and therapeutic opportunities. Curr Opin Neurol. 2014;27(6):689-696.
19. Korshunov A, Meyer J, Capper D, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118(3):401-405.
20. Fuller CE, Schmidt RE, Roth KA, et al. Clinical utility of fluorescence in situ hybridization (FISH) in morphologically ambiguous gliomas with hybrid oligodendroglial/astrocytic features. J Neuropathol Exp Neurol. 2003;62(11):1118-1128.
21. Horbinski C. Practical molecular diagnostics in neuropathology: making a tough job a little easier. Semin Diagn Pathol. 2010;27(2):105-113.
22. Fuller GN, Bigner SH. Amplified cellular oncogenes in neoplasms of the human central nervous system. Mutat Res. 1992;276(3):299-306.
23. Brennan CW, Verhaak RG, McKenna A, et al; TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462-477.
24. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):829-848.
25. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110(15):6021-6026.
26. Nikiforova MN, Hamilton RL. Molecular diagnostics of gliomas. Arch Pathol Lab Med. 2011;135(5):558-568.
1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803-820.
2. Wang-Rodriguez J, Yunes A, Phan R, et al. The challenges of precision medicine and new advances in molecular diagnostic testing in hematolymphoid malignancies: impact on the VHA. Fed Pract. 2017;34(suppl 5):S38-S49.
3. Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. 2017;19(suppl 5):v1-v88.
4. Nikiforova MN, Wald AI, Melan MA, et al. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol. 2016;18(3)379-387.
5. Cairncross JG, Ueki K, Zlatescu MC, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90(19):1473-1479.
6. van den Bent MJ, Erdem-Eraslan L, Idbaih A, et al. MGMT-STP27 methylation status as predictive marker for response to PCV in anaplastic oligodendrogliomas and oligoastrocytomas. A report from EORTC study 26951. Clin Cancer Res. 2013;19(19):5513-5522.
7. Stupp R, Hegi ME, Mason WP, et al; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459-466.
8. Malmstrom A, Gronberg BH, Marosi C, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol. 2012;13(9):916-926.
9. van den Bent MJ, Kros JM. Predictive and prognostic markers in neuro-oncology. J Neuropathol Exp Neurol. 2007;66(12):1074-1081.
10. Chen R, Smith-Cohn M, Cohen AL, Colman H. Glioma subclassifications and their clinical significance. Neurotherapeutics. 2017;14(2):284-297.
11. Jordan JT, Sanders AE, Armstrong T, et al. Quality improvement in neurology: neuro-oncology quality measurement set. Neurology. 2018;90(14):652-658.
12. Chen L, Voronovich Z, Clark K, et al. Predicting the likelihood of an isocitrate dehydrogenase 1 or 2 mutation in diagnoses of infiltrative glioma. Neuro Oncol. 2014;16(11):1478-1483.
13. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997-1003.
14. Wick W, Platten M, Meisner C, et al; NOA-08 Study Group of Neuro-oncology Working Group (NOA) of German Cancer Society. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012;13(7):707-715.
15. Horbinski C, Kofler J, Kelly LM, Murdoch GH, Nikiforova MN. Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol. 2009;68(12):1319-1325.
16. Camelo-Piragua S, Jansen M, Ganguly A, Kim JC, Louis DN, Nutt CL. Mutant IDH1-specific immunohistochemistry distinguishes diffuse astrocytoma from astrocytosis. Acta Neuropathol. 2010;119(4):509-511.
17. Horbinski C, Kofler J, Yeaney G, et al. Isocitrate dehydrogenase 1 analysis differentiates gangliogliomas from infiltrative gliomas. Brain Pathol. 2011;21(5):564-574.
18. Berghoff AS, Preusser M. BRAF alterations in brain tumours: molecular pathology and therapeutic opportunities. Curr Opin Neurol. 2014;27(6):689-696.
19. Korshunov A, Meyer J, Capper D, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol. 2009;118(3):401-405.
20. Fuller CE, Schmidt RE, Roth KA, et al. Clinical utility of fluorescence in situ hybridization (FISH) in morphologically ambiguous gliomas with hybrid oligodendroglial/astrocytic features. J Neuropathol Exp Neurol. 2003;62(11):1118-1128.
21. Horbinski C. Practical molecular diagnostics in neuropathology: making a tough job a little easier. Semin Diagn Pathol. 2010;27(2):105-113.
22. Fuller GN, Bigner SH. Amplified cellular oncogenes in neoplasms of the human central nervous system. Mutat Res. 1992;276(3):299-306.
23. Brennan CW, Verhaak RG, McKenna A, et al; TCGA Research Network. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462-477.
24. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):829-848.
25. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110(15):6021-6026.
26. Nikiforova MN, Hamilton RL. Molecular diagnostics of gliomas. Arch Pathol Lab Med. 2011;135(5):558-568.
Posttraumatic headache may be associated with reduced pain thresholds
PHILADELPHIA – , according to results of a pilot study presented at the annual meeting of the American Headache Society. The findings suggest that patients with posttraumatic headache have abnormal, multimodal sensory processing, said Amaal J. Starling, MD, a neurologist at Mayo Clinic in Phoenix.

Mild traumatic brain injury (TBI) is a growing public health problem. Headache is the most common symptom after mild TBI, and often the most debilitating symptom for these patients. No Food and Drug Administration–approved treatments are available for patients with posttraumatic headache, and about three-quarters of these patients report that current treatments bring them no relief.
Identifying novel targets and developing new treatment options will require a deeper understanding of the pathophysiology of posttraumatic headache, said Dr. Starling. She and her colleagues conducted a pilot study to characterize allodynia, cutaneous heat pain thresholds, photophobia, and light-induced pain thresholds objectively in patients with posttraumatic headache, compared with healthy controls.
Participants were exposed to a bright-light stressor
The researchers enrolled 20 patients between ages 18 years and 65 years with posttraumatic headache attributed to mild TBI in their study. They matched these patients by age with 20 healthy controls. Dr. Starling and colleagues evaluated all participants prospectively using the Allodynia Symptom Checklist (ASC-12), Photosensitivity Assessment Questionnaire (PAQ), State-Trait Anxiety Inventory (STAI), and Beck Depression Inventory (BDI).
The investigators performed quantitative sensory testing to measure each participant’s cutaneous forearm heat pain threshold. Using a progressive light stimulation device, they quantified each participant’s light-induced pain threshold. Finally, Dr. Starling and colleagues obtained participants’ cutaneous heat pain thresholds immediately after, 10 minutes after, and 40 minutes after exposing them to a bright-light stressor.
The researchers found no significant differences between groups in age, gender, or race. The population’s average age was 41 years. Approximately 70% of the sample was female. Among participants with posttraumatic headache, the average time since the onset of posttraumatic headache was 46 months. The average number of headache days per month in that group was 17.2, which represented “a significantly high headache burden,” said Dr. Starling. Approximately 80% of patients with posttraumatic headache had headaches with a migraine phenotype.
Patients’ pain thresholds were lower
STAI and BDI scores were significantly higher among patients with posttraumatic headache, compared with controls. Mean PAQ score was 0.62 among patients and 0.24 among controls, representing significantly greater photophobia symptom severity among patients, said Dr. Starling.
Light-induced pain thresholds were significantly lower in patients with posttraumatic headache (median, 90.5 lux), compared with healthy controls (median, 863.5 lux), independent of depression and anxiety. Allodynia symptom severity was significantly higher in patients with posttraumatic headache (mean ASC-12 score, 5.7), compared with controls (mean ASC-12 score, 0.98).
In addition, the mean baseline cutaneous heat pain threshold was 40.8° C in patients with posttraumatic headache and 44.4° C in healthy controls. When participants were subjected to the bright-light stressor, the immediate change in heat pain threshold was significant in patients with posttraumatic headache (−1.9° C), compared with healthy controls. The difference between groups was not significant at 10 and 40 minutes after exposure to the stressor, however. The light intensity inducing moderate pain was 688 lux in patients with posttraumatic headache, compared with 6,000 lux in healthy controls.
“Our next steps are going to be replicating this [study] in a larger population, as well as determining whether any type of intervention would change these different types of sensory sensitivities and thresholds,” said Dr. Starling. She and her colleagues will use this human research model to examine whether posttraumatic headache differs from other headache disorders such as migraine and to examine potential differences between acute and persistent posttraumatic headache.
The study was funded through an intramural Mayo Clinic early career research award.
SOURCE: Starling AJ et al. AHS 2019. Abstract OR14.
PHILADELPHIA – , according to results of a pilot study presented at the annual meeting of the American Headache Society. The findings suggest that patients with posttraumatic headache have abnormal, multimodal sensory processing, said Amaal J. Starling, MD, a neurologist at Mayo Clinic in Phoenix.
Mild traumatic brain injury (TBI) is a growing public health problem. Headache is the most common symptom after mild TBI, and often the most debilitating symptom for these patients. No Food and Drug Administration–approved treatments are available for patients with posttraumatic headache, and about three-quarters of these patients report that current treatments bring them no relief.
Identifying novel targets and developing new treatment options will require a deeper understanding of the pathophysiology of posttraumatic headache, said Dr. Starling. She and her colleagues conducted a pilot study to characterize allodynia, cutaneous heat pain thresholds, photophobia, and light-induced pain thresholds objectively in patients with posttraumatic headache, compared with healthy controls.
Participants were exposed to a bright-light stressor
The researchers enrolled 20 patients between ages 18 years and 65 years with posttraumatic headache attributed to mild TBI in their study. They matched these patients by age with 20 healthy controls. Dr. Starling and colleagues evaluated all participants prospectively using the Allodynia Symptom Checklist (ASC-12), Photosensitivity Assessment Questionnaire (PAQ), State-Trait Anxiety Inventory (STAI), and Beck Depression Inventory (BDI).
The investigators performed quantitative sensory testing to measure each participant’s cutaneous forearm heat pain threshold. Using a progressive light stimulation device, they quantified each participant’s light-induced pain threshold. Finally, Dr. Starling and colleagues obtained participants’ cutaneous heat pain thresholds immediately after, 10 minutes after, and 40 minutes after exposing them to a bright-light stressor.
The researchers found no significant differences between groups in age, gender, or race. The population’s average age was 41 years. Approximately 70% of the sample was female. Among participants with posttraumatic headache, the average time since the onset of posttraumatic headache was 46 months. The average number of headache days per month in that group was 17.2, which represented “a significantly high headache burden,” said Dr. Starling. Approximately 80% of patients with posttraumatic headache had headaches with a migraine phenotype.
Patients’ pain thresholds were lower
STAI and BDI scores were significantly higher among patients with posttraumatic headache, compared with controls. Mean PAQ score was 0.62 among patients and 0.24 among controls, representing significantly greater photophobia symptom severity among patients, said Dr. Starling.
Light-induced pain thresholds were significantly lower in patients with posttraumatic headache (median, 90.5 lux), compared with healthy controls (median, 863.5 lux), independent of depression and anxiety. Allodynia symptom severity was significantly higher in patients with posttraumatic headache (mean ASC-12 score, 5.7), compared with controls (mean ASC-12 score, 0.98).
In addition, the mean baseline cutaneous heat pain threshold was 40.8° C in patients with posttraumatic headache and 44.4° C in healthy controls. When participants were subjected to the bright-light stressor, the immediate change in heat pain threshold was significant in patients with posttraumatic headache (−1.9° C), compared with healthy controls. The difference between groups was not significant at 10 and 40 minutes after exposure to the stressor, however. The light intensity inducing moderate pain was 688 lux in patients with posttraumatic headache, compared with 6,000 lux in healthy controls.
“Our next steps are going to be replicating this [study] in a larger population, as well as determining whether any type of intervention would change these different types of sensory sensitivities and thresholds,” said Dr. Starling. She and her colleagues will use this human research model to examine whether posttraumatic headache differs from other headache disorders such as migraine and to examine potential differences between acute and persistent posttraumatic headache.
The study was funded through an intramural Mayo Clinic early career research award.
SOURCE: Starling AJ et al. AHS 2019. Abstract OR14.
PHILADELPHIA – , according to results of a pilot study presented at the annual meeting of the American Headache Society. The findings suggest that patients with posttraumatic headache have abnormal, multimodal sensory processing, said Amaal J. Starling, MD, a neurologist at Mayo Clinic in Phoenix.
Mild traumatic brain injury (TBI) is a growing public health problem. Headache is the most common symptom after mild TBI, and often the most debilitating symptom for these patients. No Food and Drug Administration–approved treatments are available for patients with posttraumatic headache, and about three-quarters of these patients report that current treatments bring them no relief.
Identifying novel targets and developing new treatment options will require a deeper understanding of the pathophysiology of posttraumatic headache, said Dr. Starling. She and her colleagues conducted a pilot study to characterize allodynia, cutaneous heat pain thresholds, photophobia, and light-induced pain thresholds objectively in patients with posttraumatic headache, compared with healthy controls.
Participants were exposed to a bright-light stressor
The researchers enrolled 20 patients between ages 18 years and 65 years with posttraumatic headache attributed to mild TBI in their study. They matched these patients by age with 20 healthy controls. Dr. Starling and colleagues evaluated all participants prospectively using the Allodynia Symptom Checklist (ASC-12), Photosensitivity Assessment Questionnaire (PAQ), State-Trait Anxiety Inventory (STAI), and Beck Depression Inventory (BDI).
The investigators performed quantitative sensory testing to measure each participant’s cutaneous forearm heat pain threshold. Using a progressive light stimulation device, they quantified each participant’s light-induced pain threshold. Finally, Dr. Starling and colleagues obtained participants’ cutaneous heat pain thresholds immediately after, 10 minutes after, and 40 minutes after exposing them to a bright-light stressor.
The researchers found no significant differences between groups in age, gender, or race. The population’s average age was 41 years. Approximately 70% of the sample was female. Among participants with posttraumatic headache, the average time since the onset of posttraumatic headache was 46 months. The average number of headache days per month in that group was 17.2, which represented “a significantly high headache burden,” said Dr. Starling. Approximately 80% of patients with posttraumatic headache had headaches with a migraine phenotype.
Patients’ pain thresholds were lower
STAI and BDI scores were significantly higher among patients with posttraumatic headache, compared with controls. Mean PAQ score was 0.62 among patients and 0.24 among controls, representing significantly greater photophobia symptom severity among patients, said Dr. Starling.
Light-induced pain thresholds were significantly lower in patients with posttraumatic headache (median, 90.5 lux), compared with healthy controls (median, 863.5 lux), independent of depression and anxiety. Allodynia symptom severity was significantly higher in patients with posttraumatic headache (mean ASC-12 score, 5.7), compared with controls (mean ASC-12 score, 0.98).
In addition, the mean baseline cutaneous heat pain threshold was 40.8° C in patients with posttraumatic headache and 44.4° C in healthy controls. When participants were subjected to the bright-light stressor, the immediate change in heat pain threshold was significant in patients with posttraumatic headache (−1.9° C), compared with healthy controls. The difference between groups was not significant at 10 and 40 minutes after exposure to the stressor, however. The light intensity inducing moderate pain was 688 lux in patients with posttraumatic headache, compared with 6,000 lux in healthy controls.
“Our next steps are going to be replicating this [study] in a larger population, as well as determining whether any type of intervention would change these different types of sensory sensitivities and thresholds,” said Dr. Starling. She and her colleagues will use this human research model to examine whether posttraumatic headache differs from other headache disorders such as migraine and to examine potential differences between acute and persistent posttraumatic headache.
The study was funded through an intramural Mayo Clinic early career research award.
SOURCE: Starling AJ et al. AHS 2019. Abstract OR14.
REPORTING FROM AHS 2019
Gastroparesis
To the Editor: We read with great pleasure the article by Sharayah et al about acute gastroparesis in a patient with diabetic ketoacidosis.1 However, in the case description, the authors reached a diagnosis of gastroparesis secondary to diabetic ketoacidosis without aptly ruling out some of its most common causes such as hypokalemia and other electrolyte imbalances seen in diabetic patients (in the setting of recurrent vomiting).
The authors also did not include the patient’s duration of diabetes or hemoglobin A1c level, both of which are linked with gastroparesis in diabetic patients.2 Pertinent biochemical information that can help readers formulate a rational approach and journey to making a diagnosis appears elusive in their article.
- Sharayah AM, Hajjaj N, Osman R, Livornese D. Gastroparesis in a patient with diabetic ketoacidosis. Cleve Clin J Med 2019; 86(4):238–239. doi:10.3949/ccjm.86a.18116
- Bharucha AE, Kudva Y, Basu A, et al. Relationship between glycemic control and gastric emptying in poorly controlled type 2 diabetes. Clin Gastroenterol Hepatol 2015; 13(3):466–476.e461. doi:10.1016/j.cgh.2014.06.034
To the Editor: We read with great pleasure the article by Sharayah et al about acute gastroparesis in a patient with diabetic ketoacidosis.1 However, in the case description, the authors reached a diagnosis of gastroparesis secondary to diabetic ketoacidosis without aptly ruling out some of its most common causes such as hypokalemia and other electrolyte imbalances seen in diabetic patients (in the setting of recurrent vomiting).
The authors also did not include the patient’s duration of diabetes or hemoglobin A1c level, both of which are linked with gastroparesis in diabetic patients.2 Pertinent biochemical information that can help readers formulate a rational approach and journey to making a diagnosis appears elusive in their article.
To the Editor: We read with great pleasure the article by Sharayah et al about acute gastroparesis in a patient with diabetic ketoacidosis.1 However, in the case description, the authors reached a diagnosis of gastroparesis secondary to diabetic ketoacidosis without aptly ruling out some of its most common causes such as hypokalemia and other electrolyte imbalances seen in diabetic patients (in the setting of recurrent vomiting).
The authors also did not include the patient’s duration of diabetes or hemoglobin A1c level, both of which are linked with gastroparesis in diabetic patients.2 Pertinent biochemical information that can help readers formulate a rational approach and journey to making a diagnosis appears elusive in their article.
- Sharayah AM, Hajjaj N, Osman R, Livornese D. Gastroparesis in a patient with diabetic ketoacidosis. Cleve Clin J Med 2019; 86(4):238–239. doi:10.3949/ccjm.86a.18116
- Bharucha AE, Kudva Y, Basu A, et al. Relationship between glycemic control and gastric emptying in poorly controlled type 2 diabetes. Clin Gastroenterol Hepatol 2015; 13(3):466–476.e461. doi:10.1016/j.cgh.2014.06.034
- Sharayah AM, Hajjaj N, Osman R, Livornese D. Gastroparesis in a patient with diabetic ketoacidosis. Cleve Clin J Med 2019; 86(4):238–239. doi:10.3949/ccjm.86a.18116
- Bharucha AE, Kudva Y, Basu A, et al. Relationship between glycemic control and gastric emptying in poorly controlled type 2 diabetes. Clin Gastroenterol Hepatol 2015; 13(3):466–476.e461. doi:10.1016/j.cgh.2014.06.034
In reply: Gastroparesis
In Reply: We thank the readers for their letter. Our patient’s laboratory values at the time of presentation were as follows:
- Corrected sodium 142 mmol/L
- Potassium 5.5 mmol/L
- Phosphorus 6.6 mmol/L.
The rest of the electrolyte levels were within normal limits.
These reported electrolyte levels were unlikely to cause such gastroparesis. The patient’s hemoglobin A1c was 8.7% at the time of presentation, with no previous values available. However, since abdominal computed tomography done 1 year before this presentation did not show stomach dilation and the patient was asymptomatic, his gastroparesis was presumed to be acute.
In Reply: We thank the readers for their letter. Our patient’s laboratory values at the time of presentation were as follows:
- Corrected sodium 142 mmol/L
- Potassium 5.5 mmol/L
- Phosphorus 6.6 mmol/L.
The rest of the electrolyte levels were within normal limits.
These reported electrolyte levels were unlikely to cause such gastroparesis. The patient’s hemoglobin A1c was 8.7% at the time of presentation, with no previous values available. However, since abdominal computed tomography done 1 year before this presentation did not show stomach dilation and the patient was asymptomatic, his gastroparesis was presumed to be acute.
In Reply: We thank the readers for their letter. Our patient’s laboratory values at the time of presentation were as follows:
- Corrected sodium 142 mmol/L
- Potassium 5.5 mmol/L
- Phosphorus 6.6 mmol/L.
The rest of the electrolyte levels were within normal limits.
These reported electrolyte levels were unlikely to cause such gastroparesis. The patient’s hemoglobin A1c was 8.7% at the time of presentation, with no previous values available. However, since abdominal computed tomography done 1 year before this presentation did not show stomach dilation and the patient was asymptomatic, his gastroparesis was presumed to be acute.
Seizure-like episodes, but is it really epilepsy?
CASE Increasingly frequent paroxysmal episodes
Ms. N, age 12, comes to the hospital for evaluation of paroxysmal episodes of pain, weakness, and muscle spasms. A neurologist who evaluated her as an outpatient had recommended a routine electroencephalogram (EEG); after those results were inconclusive, Ms. N’s mother brought her to the hospital for a 24-hour video EEG.
Ms. N has a history of asthma. She has no history of seizures or headache, but her mother has an unspecified seizure disorder that has been stable with antiepileptic medication for many years. Ms. N has no other family history of autoimmune or neurologic disorders.
Ms. N’s episodes began 6 months ago and have progressively increased in frequency from 5 to 12 episodes a day. She says that before she has an episode, she “ feels tingling in her fingers and mouth, and butterflies in her belly,” and then her “whole body clenches up.” She denies experiencing tongue biting, facial or extremity weakness, incontinence, or loss of consciousness during these episodes.
Shortly before her hospitalization, Ms. N had won a scholarship to attend an overnight art camp. Because her episodes were becoming more frequent and their etiology remained unclear, Ms. N and her mother decided it would be unsafe for her to attend, and that she should go to the hospital for evaluation instead.
EVALUATION Tough questions reveal answers
The pediatric team evaluates Ms. N. Her physical exam, laboratory values, and imaging are all within normal limits. Her neurologic exam demonstrates full strength, tone, and sensation in all extremities. All cranial nerves and reflexes are intact. No dysmorphic features or gait abnormalities are noted. All laboratory and imaging tests are normal, including complete blood cell count, electrolytes, calcium, magnesium, phosphorus, glucose, creatine kinase, liver enzymes, urine drug screen, human chorionic gonadotropin (hCG) urine test, and h
After the initial workup, the pediatric team consults the child and adolescent psychiatry team for a complete assessment of Ms. N due to concerns that a psychological component is contributing to her episodes. According to the psychosocial history obtained from Ms. N and her mother, Ms. N had experienced disrupted attachment, trauma, and loss. At age 5, Ms. N was temporarily removed from her mother’s custody after a fight between her mother and brother. At age 9, Ms. N’s stepfather, her primary father figure, died of a brain tumor.
Ms. N also has significant trauma stemming from her relationship with her biological father. Ms. N’s mother reports that her daughter was conceived during nonconsensual sexual intercourse. Ms. N did not have much contact with her biological father until 6 months ago, when he started picking her up at school and taking her to his home for several hours without permission or supervision. Afterwards, Ms. N confided to her mother and a teacher that her father sexually assaulted her during those visits.
Continue to: Ms. N and her mother...
Ms. N and her mother reported the assault to the police and were awaiting legal action.
During the interview with the psychiatry team, Ms. N denies that any thoughts or actions trigger the episodes and reports that she cannot control when they happen. Because she cannot anticipate the episodes, she says she is afraid to leave her house. She does not know why the episodes are happening and feels frustrated that they are getting worse. Ms. N says, “I have been feeling down lately,” but she denies hopelessness, worthlessness, suicidal ideation, homicidal ideation, delusions, or hallucinations.
In the hospital, when the psychiatry team asks Ms. N about her visits with her father, she says that they are “too painful to talk about,” and fears that discussing them will trigger an episode. However, her mother suggests that her daughter’s sexual trauma, as well as ongoing frustrations with the legal system, are influencing her mood; she has had low energy, poor appetite, and is spending more time in bed. Her mother also reports that Ms. N “avoids going out in the sun and spending time with her friends outside. She doesn’t seem to enjoy shopping and art like she used to.” Ms. N told her mother that she was having nightmares about the trauma and “could not stop thinking about some of the bad stuff that happened during the day.”
Ten minutes into the interview, while being questioned about her father, Ms. N experiences a spastic episode. She curls up in bed on her left side, clenches her entire body, and shuts her eyes. Her mother quickly runs to her bedside and counts the seconds until the end of the episode. After 25 seconds, Ms. N awakes with full recollection of the episode. On review of the video EEG during the episode, no ictal patterns are seen.
[polldaddy:10375873]
The authors’ observations
Paroxysmal episodes of weakness, numbness, and muscle spasms in a young female are suggestive of either epilepsy or nonepileptic seizure (NES).1,2 The negative EEG and physical features are inconsistent with epileptiform seizure, and Ms. N’s history and evaluation are suggestive of NES. Nonepileptic seizures are a type of a conversion disorder, or functional neurologic symptom disorder, in which a patient experiences weakness, abnormal movements, or seizure-like episodes that are inconsistent with organic neurologic disease.3 When a diagnosis of conversion disorder is suspected, a clinician must always consider other pathology that can explain the symptoms, such as migraine, vasovagal syncope, or intracranial mass. If a patient has focal neurologic deficits, head imaging should be pursued. Additionally, the clinician must screen for malingering and factitious disorder before establishing a definitive diagnosis. However, conversion disorder is not a diagnosis of exclusion. For example, a negative EEG does not rule out epilepsy, and patients can have both epilepsy and concomitant NES.
Continue to: Although NES is a common...
Although NES is a common type of conversion disorder, it is often difficult to diagnose, manage, and treat. Patients often receive antiepileptic medications but continue to have worsening events that are refractory to treatment. Various clinical features can suggest NES instead of epilepsy. Forced eye closure on video recording is a specific finding suggestive of NES, yet this feature is not sufficient to make the diagnosis.4 A video EEG must be performed to assess for epilepsy. The diagnosis of NES does not exclude the possibility that a patient has epilepsy, as NES can occur in up to 40% of patients with epilepsy.5 A video EEG without ictal patterns before, during, and after an observed episode is diagnostic of NES.6
[polldaddy:10375874]
The authors’ observations
Conversion disorders such as NES are a presentation of neurologic symptoms that cannot be readily accounted for by other conditions and are often associated with antecedent trauma. Multiple factors in Ms. N’s history increase her risk of NES, including loss of multiple loved ones, ongoing legal involvement, and alleged sexual abuse by her father.
Victims of sexual abuse are more likely than the general population to demonstrate symptoms of conversion disorder, especially NES.7,8 The onset of paroxysmal episodes after incestuous abuse in a teenage girl is characteristic of NES. Compared with patients with complex partial epilepsy (CPE), patients with NES are 3 times more likely to report sexual trauma.9,10 Children who report sexual abuse that precedes NES are more likely to have been victimized by a first-degree relative than patients with CPE who report sexual abuse.11 Risk factors for victims developing NES may be related to the severity of adversity, stress sensitivity, and decreased hippocampal volume.12,13
Ms. N endorsed many psychiatric symptoms that accompany her paroxysmal episodes; this is similar to findings in other patients with NES.14 One study found that depression is 3 times more prevalent and PTSD is 8 times more prevalent in patients with NES.12 During the evaluation, Ms. N’s mother said her daughter had low energy, poor appetite, lethargy, and anhedonia for the preceding 5 months, which is consistent with adjustment disorder.3 Her flashbacks, nightmares, difficulty sleeping, and agoraphobia, along with her trouble engaging with the people and activities that used to bring her joy, are symptoms of PTSD. Nonepileptic seizure is often associated with PTSD and can be viewed as an expression of a dissociated subtype.15
In a literature review, Durrant et al16 isolated prognostic indicators for NES (Table16). This study found that 70% of children and 40% of adults achieve remission from NES. Ms. N’s case has multiple concerning features, such as her comorbid psychiatric conditions, ongoing involvement in a legal case, and sexual trauma; this last factor is associated with the most severe symptoms and worse outcomes.16,17 Despite this somber reality, Ms. N has the support of her mother and is relatively young, which play a vital role in recovery.

Continue to: TREATMENT A strategy for minimizing the episodes
TREATMENT A strategy for minimizing the episodes
Ms. N’s medical workup remains unremarkable throughout the rest of her hospital stay. The psychiatry and pediatric teams discuss their assessments and agree that NES is the most likely diagnosis. The psychiatry team counsels Ms. N and her mother on the diagnosis and etiology of NES.
[polldaddy:10375876]
The authors’ observations
Cognitive-behavioral therapy is currently the treatment of choice for reducing seizure frequency in patients with NES.18,19 The use of CBT was suggested due to the theory that NES represents a dissociative response to trauma. Therapy focuses on changing a patient’s beliefs and perceptions associated with attacks.5 A randomized study of 66 patients with NES compared the use of CBT plus standard medical care with standard medical care alone.18 The standard medical care consisted of supportive treatment, an explanation of NES from a neuropsychiatrist, and supervised withdrawal of antiepileptic drugs. The CBT treatment group was offered weekly hour-long sessions for 12 weeks, accompanied by CBT homework and journaling the frequency and nature of seizure episodes (the CBT techniques are outlined in the Figure18). After 4 months, the CBT treatment group had fewer seizures, and after a 6-month follow-up, they were more likely to be seizure-free. However, in this study, CBT treatment did not improve mood or employment status.

A later investigation looked at using selective serotonin reuptake inhibitors to treat NES in adults.19 This study divided participants into 4 treatment groups: CBT with informed psychotherapy (CBT-ip), CBT-ip plus sertraline, sertraline alone, and treatment as usual. Sertraline was titrated up to a dose of 200 mg/d as tolerated. After 16 weeks of sertraline alone, seizure frequency did not decrease. Although both CBT groups showed a reduction in symptoms of up to 60%, the CBT-ip group reported fewer psychiatric symptoms with better social interactions, quality of life, and global functioning compared with patients treated with CBT-ip plus sertraline. The authors suggested that this may be due to the somatic adverse effects associated with sertraline. This study suggests that CBT without medication is the treatment of choice.
In addition to CBT, studies of psychodynamic psychotherapy for NES have had promising findings.20 Psychodynamic psychotherapy focuses on addressing conscious and unconscious anger, loss, feelings of isolation, and trauma. Through improving emotional processing, insight, coping skills and self-regulation, patients often benefit from an improvement in seizures, psychosocial functioning and health care utilization.
Metin et al21 found that group therapy alongside a family-centered approach elicited a strong and durable reduction in seizures in patients with NES. At enrollment, investigators distributed information on NES to patients and families. Psychoeducation and psychoanalysis with behavior modification techniques were provided in 90-minute weekly group sessions over 3 months. Participants also underwent monthly individualized sessions for standard psychiatric care for 9 months. During the group sessions, operant conditioning techniques were used to prevent secondary gain from seizure-like activity. Families met 4 times for 1 hour each to discuss seizures, receive psychoeducation on a subconscious etiology of NES, and learn behavior modification techniques. All 9 participants who completed group and individual therapy reported a significant and sustained reduction in seizure frequency by at least 50% at 12-month follow-up. Patients also demonstrated improvements in mood, anxiety, and quality of life.
Continue to: A meta-analysis...
A meta-analysis by Carlson and Perry22 that included 13 studies and 228 participants, examined different treatment modalities and their effectiveness for NES. They found that patients who received psychological intervention had a 47% remission rate and 82% improvement in seizure frequency compared with only 14% to 23% of those who did not receive therapy. They postulated that therapy for this illness must be flexible to properly address the socially, psychologically, and functionally heterogenous patient population. Although there are few randomized controlled trials for NES to determine the best evidence-based intervention, there is now consensus that NES has a favorable prognosis when barriers to psychological care are eliminated.
OUTCOME Referral for CBT
The treatment team advises Ms. N to engage in outpatient therapy after discharge from the hospital. Ms. N and her mother agree to the treatment plan, and leave the hospital with a referral for CBT the next day.
Bottom Line
Nonepileptic seizure (NES) is a type of conversion disorder characterized by seizure-like episodes without ictal qualities. Risk factors for NES include concomitant epilepsy, psychiatric disorders, unstable psychosocial situations, and antecedent trauma. Patients with a history of incestuous sexual abuse are most at risk for developing NES. A normal EEG that fully captures a seizure-like episode is diagnostic of NES. Cognitive-behavioral therapy can minimize seizure frequency and intensity.
Related Resources
- Marsh P, Benbadis S, Fernandez F. Psychogenic nonepileptic seizures: ways to win over skeptical patients. Current Psychiatry. 2008;7(1):21-24, 32-35.
- LaFrance WC Jr. Eye-opening behaviors help diagnose nonepileptic seizures. Current Psychiatry. 2006;5(11):121-122, 124, 130.
- LaFrance WC Jr, Kanner AM, Barry JJ. Treating patients with psychological nonepileptic seizures. In: Ettinger AB, Kanner AM, eds. Psychiatric issues in epilepsy: a practical guide to diagnosis and treatment. 2nd ed. Philadelphia, PA: Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2007:461-488.
Drug Brand Name
Sertraline • Zoloft
1. Lesser R. Psychogenic seizures. Neurology. 1996;46(6):1499-1507.
2. Stone J, LaFrance W, Brown R, et al. Conversion disorder: current problems and potential solutions for DSM-5. J Psychosom Res. 2011;71(6):369-376.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Syed T, Arozullah A, Suciu G, et al. Do observer and self-reports of ictal eye closure predict psychogenic nonepileptic seizures? Epilepsia. 2008;49(5):898-904.
5. Vega-Zelaya L, Alvarez M, Ezquiaga E, et al. Psychogenic non-epileptic seizures in a surgical epilepsy unit: experience and a comprehensive review. Epilepsy Topics. 2014. doi: 10.5772/57439.
6. LaFrance W, Baker G, Duncan R, et al. Minimum requirements for the diagnosis of psychogenic nonepileptic seizures: a staged approach. Epilepsia. 2013;54(11):2005-2018.
7. Roeloes K, Pasman J. Stress, childhood trauma, and cognitive functions in functional neurologic disorders. In: Hallett M, Stone J, Carson A, eds. Handbook of clinical neurology: functional neurologic disorders. 3rd ed. New York, NY: Elsevier; 2017:139-155.
8. Paras M, Murad M, Chen L, et al. Sexual abuse and lifetime diagnosis of somatic disorders. JAMA. 2009;302(5):550.
9. Fiszman A, Alves-Leon SV, Nunes RG, et al. Traumatic events and posttraumatic stress disorder in patients with psychogenic nonepileptic seizures: a critical review. Epilepsy Behav. 2004;5(6):818-825.
10. Sharpe D, Faye C. Non-epileptic seizures and child sexual abuse: a critical review of the literature. Clin Psychol Rev. 2006;26(8):1020-1040.
11. Alper K, Devinsky O, Perrine K, et al. Nonepileptic seizures and childhood sexual and physical abuse. Neurology. 1993;43(10):1950-1953.
12. Plioplys S, Doss J, Siddarth P et al. A multisite controlled study of risk factors in pediatric psychogenic nonepileptic seizures. Epilepsia. 2014;55(11):1739-1747.
13. Andersen S, Tomada A, Vincow E, et al. Preliminary evidence for sensitive periods in the effect of childhood sexual abuse on regional brain development. J Neuropsychiatry Clin Neurosci. 2008;20(3):292-301.
14. Sar V. Childhood trauma, dissociation, and psychiatric comorbidity in patients with conversion disorder. Am J Psychiatry. 2004;161(12):2271-2276.
15. Rosenberg HJ, Rosenberg SD, Williamson PD, et al. A comparative study of trauma and posttraumatic stress disorder prevalence in epilepsy patients and psychogenic nonepileptic seizure patients. Epilepsia. 2000;41(4):447-452.
16. Durrant J, Rickards H, Cavanna A. Prognosis and outcome predictors in psychogenic nonepileptic seizures. Epilepsy Res Treat. 2011;2011:1-7.
17. Selkirk M, Duncan R, Oto M, et al. Clinical differences between patients with nonepileptic seizures who report antecedent sexual abuse and those who do not. Epilepsia. 2008;49(8):1446-1450.
18. Goldstein L, Chalder T, Chigwedere C, et al. Cognitive-behavioral therapy for psychogenic nonepileptic seizures: a pilot RCT. Neurology. 2010;74(24):1986-1994.
19. LaFrance W, Baird G, Barry J, et al. Multicenter pilot treatment trial for psychogenic nonepileptic seizures. JAMA Psychiatry. 2014;71(9):997.
20. Howlett S, Reuber M. An augmented model of brief psychodynamic interpersonal therapy for patients with nonepileptic seizures. Psychotherapy (Chic). 2009;46(1):125-138.
21. Metin SZ, Ozmen M, Metin B, et al. Treatment with group psychotherapy for chronic psychogenic nonepileptic seizures. Epilepsy Behav. 2013;28(1):91-94.
22. Carlson P, Perry KN. Psychological interventions for psychogenic non-epileptic seizures: a meta-analysis. Seizure. 2017;45:142-150.
CASE Increasingly frequent paroxysmal episodes
Ms. N, age 12, comes to the hospital for evaluation of paroxysmal episodes of pain, weakness, and muscle spasms. A neurologist who evaluated her as an outpatient had recommended a routine electroencephalogram (EEG); after those results were inconclusive, Ms. N’s mother brought her to the hospital for a 24-hour video EEG.
Ms. N has a history of asthma. She has no history of seizures or headache, but her mother has an unspecified seizure disorder that has been stable with antiepileptic medication for many years. Ms. N has no other family history of autoimmune or neurologic disorders.
Ms. N’s episodes began 6 months ago and have progressively increased in frequency from 5 to 12 episodes a day. She says that before she has an episode, she “ feels tingling in her fingers and mouth, and butterflies in her belly,” and then her “whole body clenches up.” She denies experiencing tongue biting, facial or extremity weakness, incontinence, or loss of consciousness during these episodes.
Shortly before her hospitalization, Ms. N had won a scholarship to attend an overnight art camp. Because her episodes were becoming more frequent and their etiology remained unclear, Ms. N and her mother decided it would be unsafe for her to attend, and that she should go to the hospital for evaluation instead.
EVALUATION Tough questions reveal answers
The pediatric team evaluates Ms. N. Her physical exam, laboratory values, and imaging are all within normal limits. Her neurologic exam demonstrates full strength, tone, and sensation in all extremities. All cranial nerves and reflexes are intact. No dysmorphic features or gait abnormalities are noted. All laboratory and imaging tests are normal, including complete blood cell count, electrolytes, calcium, magnesium, phosphorus, glucose, creatine kinase, liver enzymes, urine drug screen, human chorionic gonadotropin (hCG) urine test, and h
After the initial workup, the pediatric team consults the child and adolescent psychiatry team for a complete assessment of Ms. N due to concerns that a psychological component is contributing to her episodes. According to the psychosocial history obtained from Ms. N and her mother, Ms. N had experienced disrupted attachment, trauma, and loss. At age 5, Ms. N was temporarily removed from her mother’s custody after a fight between her mother and brother. At age 9, Ms. N’s stepfather, her primary father figure, died of a brain tumor.
Ms. N also has significant trauma stemming from her relationship with her biological father. Ms. N’s mother reports that her daughter was conceived during nonconsensual sexual intercourse. Ms. N did not have much contact with her biological father until 6 months ago, when he started picking her up at school and taking her to his home for several hours without permission or supervision. Afterwards, Ms. N confided to her mother and a teacher that her father sexually assaulted her during those visits.
Continue to: Ms. N and her mother...
Ms. N and her mother reported the assault to the police and were awaiting legal action.
During the interview with the psychiatry team, Ms. N denies that any thoughts or actions trigger the episodes and reports that she cannot control when they happen. Because she cannot anticipate the episodes, she says she is afraid to leave her house. She does not know why the episodes are happening and feels frustrated that they are getting worse. Ms. N says, “I have been feeling down lately,” but she denies hopelessness, worthlessness, suicidal ideation, homicidal ideation, delusions, or hallucinations.
In the hospital, when the psychiatry team asks Ms. N about her visits with her father, she says that they are “too painful to talk about,” and fears that discussing them will trigger an episode. However, her mother suggests that her daughter’s sexual trauma, as well as ongoing frustrations with the legal system, are influencing her mood; she has had low energy, poor appetite, and is spending more time in bed. Her mother also reports that Ms. N “avoids going out in the sun and spending time with her friends outside. She doesn’t seem to enjoy shopping and art like she used to.” Ms. N told her mother that she was having nightmares about the trauma and “could not stop thinking about some of the bad stuff that happened during the day.”
Ten minutes into the interview, while being questioned about her father, Ms. N experiences a spastic episode. She curls up in bed on her left side, clenches her entire body, and shuts her eyes. Her mother quickly runs to her bedside and counts the seconds until the end of the episode. After 25 seconds, Ms. N awakes with full recollection of the episode. On review of the video EEG during the episode, no ictal patterns are seen.
[polldaddy:10375873]
The authors’ observations
Paroxysmal episodes of weakness, numbness, and muscle spasms in a young female are suggestive of either epilepsy or nonepileptic seizure (NES).1,2 The negative EEG and physical features are inconsistent with epileptiform seizure, and Ms. N’s history and evaluation are suggestive of NES. Nonepileptic seizures are a type of a conversion disorder, or functional neurologic symptom disorder, in which a patient experiences weakness, abnormal movements, or seizure-like episodes that are inconsistent with organic neurologic disease.3 When a diagnosis of conversion disorder is suspected, a clinician must always consider other pathology that can explain the symptoms, such as migraine, vasovagal syncope, or intracranial mass. If a patient has focal neurologic deficits, head imaging should be pursued. Additionally, the clinician must screen for malingering and factitious disorder before establishing a definitive diagnosis. However, conversion disorder is not a diagnosis of exclusion. For example, a negative EEG does not rule out epilepsy, and patients can have both epilepsy and concomitant NES.
Continue to: Although NES is a common...
Although NES is a common type of conversion disorder, it is often difficult to diagnose, manage, and treat. Patients often receive antiepileptic medications but continue to have worsening events that are refractory to treatment. Various clinical features can suggest NES instead of epilepsy. Forced eye closure on video recording is a specific finding suggestive of NES, yet this feature is not sufficient to make the diagnosis.4 A video EEG must be performed to assess for epilepsy. The diagnosis of NES does not exclude the possibility that a patient has epilepsy, as NES can occur in up to 40% of patients with epilepsy.5 A video EEG without ictal patterns before, during, and after an observed episode is diagnostic of NES.6
[polldaddy:10375874]
The authors’ observations
Conversion disorders such as NES are a presentation of neurologic symptoms that cannot be readily accounted for by other conditions and are often associated with antecedent trauma. Multiple factors in Ms. N’s history increase her risk of NES, including loss of multiple loved ones, ongoing legal involvement, and alleged sexual abuse by her father.
Victims of sexual abuse are more likely than the general population to demonstrate symptoms of conversion disorder, especially NES.7,8 The onset of paroxysmal episodes after incestuous abuse in a teenage girl is characteristic of NES. Compared with patients with complex partial epilepsy (CPE), patients with NES are 3 times more likely to report sexual trauma.9,10 Children who report sexual abuse that precedes NES are more likely to have been victimized by a first-degree relative than patients with CPE who report sexual abuse.11 Risk factors for victims developing NES may be related to the severity of adversity, stress sensitivity, and decreased hippocampal volume.12,13
Ms. N endorsed many psychiatric symptoms that accompany her paroxysmal episodes; this is similar to findings in other patients with NES.14 One study found that depression is 3 times more prevalent and PTSD is 8 times more prevalent in patients with NES.12 During the evaluation, Ms. N’s mother said her daughter had low energy, poor appetite, lethargy, and anhedonia for the preceding 5 months, which is consistent with adjustment disorder.3 Her flashbacks, nightmares, difficulty sleeping, and agoraphobia, along with her trouble engaging with the people and activities that used to bring her joy, are symptoms of PTSD. Nonepileptic seizure is often associated with PTSD and can be viewed as an expression of a dissociated subtype.15
In a literature review, Durrant et al16 isolated prognostic indicators for NES (Table16). This study found that 70% of children and 40% of adults achieve remission from NES. Ms. N’s case has multiple concerning features, such as her comorbid psychiatric conditions, ongoing involvement in a legal case, and sexual trauma; this last factor is associated with the most severe symptoms and worse outcomes.16,17 Despite this somber reality, Ms. N has the support of her mother and is relatively young, which play a vital role in recovery.
Continue to: TREATMENT A strategy for minimizing the episodes
TREATMENT A strategy for minimizing the episodes
Ms. N’s medical workup remains unremarkable throughout the rest of her hospital stay. The psychiatry and pediatric teams discuss their assessments and agree that NES is the most likely diagnosis. The psychiatry team counsels Ms. N and her mother on the diagnosis and etiology of NES.
[polldaddy:10375876]
The authors’ observations
Cognitive-behavioral therapy is currently the treatment of choice for reducing seizure frequency in patients with NES.18,19 The use of CBT was suggested due to the theory that NES represents a dissociative response to trauma. Therapy focuses on changing a patient’s beliefs and perceptions associated with attacks.5 A randomized study of 66 patients with NES compared the use of CBT plus standard medical care with standard medical care alone.18 The standard medical care consisted of supportive treatment, an explanation of NES from a neuropsychiatrist, and supervised withdrawal of antiepileptic drugs. The CBT treatment group was offered weekly hour-long sessions for 12 weeks, accompanied by CBT homework and journaling the frequency and nature of seizure episodes (the CBT techniques are outlined in the Figure18). After 4 months, the CBT treatment group had fewer seizures, and after a 6-month follow-up, they were more likely to be seizure-free. However, in this study, CBT treatment did not improve mood or employment status.
A later investigation looked at using selective serotonin reuptake inhibitors to treat NES in adults.19 This study divided participants into 4 treatment groups: CBT with informed psychotherapy (CBT-ip), CBT-ip plus sertraline, sertraline alone, and treatment as usual. Sertraline was titrated up to a dose of 200 mg/d as tolerated. After 16 weeks of sertraline alone, seizure frequency did not decrease. Although both CBT groups showed a reduction in symptoms of up to 60%, the CBT-ip group reported fewer psychiatric symptoms with better social interactions, quality of life, and global functioning compared with patients treated with CBT-ip plus sertraline. The authors suggested that this may be due to the somatic adverse effects associated with sertraline. This study suggests that CBT without medication is the treatment of choice.
In addition to CBT, studies of psychodynamic psychotherapy for NES have had promising findings.20 Psychodynamic psychotherapy focuses on addressing conscious and unconscious anger, loss, feelings of isolation, and trauma. Through improving emotional processing, insight, coping skills and self-regulation, patients often benefit from an improvement in seizures, psychosocial functioning and health care utilization.
Metin et al21 found that group therapy alongside a family-centered approach elicited a strong and durable reduction in seizures in patients with NES. At enrollment, investigators distributed information on NES to patients and families. Psychoeducation and psychoanalysis with behavior modification techniques were provided in 90-minute weekly group sessions over 3 months. Participants also underwent monthly individualized sessions for standard psychiatric care for 9 months. During the group sessions, operant conditioning techniques were used to prevent secondary gain from seizure-like activity. Families met 4 times for 1 hour each to discuss seizures, receive psychoeducation on a subconscious etiology of NES, and learn behavior modification techniques. All 9 participants who completed group and individual therapy reported a significant and sustained reduction in seizure frequency by at least 50% at 12-month follow-up. Patients also demonstrated improvements in mood, anxiety, and quality of life.
Continue to: A meta-analysis...
A meta-analysis by Carlson and Perry22 that included 13 studies and 228 participants, examined different treatment modalities and their effectiveness for NES. They found that patients who received psychological intervention had a 47% remission rate and 82% improvement in seizure frequency compared with only 14% to 23% of those who did not receive therapy. They postulated that therapy for this illness must be flexible to properly address the socially, psychologically, and functionally heterogenous patient population. Although there are few randomized controlled trials for NES to determine the best evidence-based intervention, there is now consensus that NES has a favorable prognosis when barriers to psychological care are eliminated.
OUTCOME Referral for CBT
The treatment team advises Ms. N to engage in outpatient therapy after discharge from the hospital. Ms. N and her mother agree to the treatment plan, and leave the hospital with a referral for CBT the next day.
Bottom Line
Nonepileptic seizure (NES) is a type of conversion disorder characterized by seizure-like episodes without ictal qualities. Risk factors for NES include concomitant epilepsy, psychiatric disorders, unstable psychosocial situations, and antecedent trauma. Patients with a history of incestuous sexual abuse are most at risk for developing NES. A normal EEG that fully captures a seizure-like episode is diagnostic of NES. Cognitive-behavioral therapy can minimize seizure frequency and intensity.
Related Resources
- Marsh P, Benbadis S, Fernandez F. Psychogenic nonepileptic seizures: ways to win over skeptical patients. Current Psychiatry. 2008;7(1):21-24, 32-35.
- LaFrance WC Jr. Eye-opening behaviors help diagnose nonepileptic seizures. Current Psychiatry. 2006;5(11):121-122, 124, 130.
- LaFrance WC Jr, Kanner AM, Barry JJ. Treating patients with psychological nonepileptic seizures. In: Ettinger AB, Kanner AM, eds. Psychiatric issues in epilepsy: a practical guide to diagnosis and treatment. 2nd ed. Philadelphia, PA: Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2007:461-488.
Drug Brand Name
Sertraline • Zoloft
CASE Increasingly frequent paroxysmal episodes
Ms. N, age 12, comes to the hospital for evaluation of paroxysmal episodes of pain, weakness, and muscle spasms. A neurologist who evaluated her as an outpatient had recommended a routine electroencephalogram (EEG); after those results were inconclusive, Ms. N’s mother brought her to the hospital for a 24-hour video EEG.
Ms. N has a history of asthma. She has no history of seizures or headache, but her mother has an unspecified seizure disorder that has been stable with antiepileptic medication for many years. Ms. N has no other family history of autoimmune or neurologic disorders.
Ms. N’s episodes began 6 months ago and have progressively increased in frequency from 5 to 12 episodes a day. She says that before she has an episode, she “ feels tingling in her fingers and mouth, and butterflies in her belly,” and then her “whole body clenches up.” She denies experiencing tongue biting, facial or extremity weakness, incontinence, or loss of consciousness during these episodes.
Shortly before her hospitalization, Ms. N had won a scholarship to attend an overnight art camp. Because her episodes were becoming more frequent and their etiology remained unclear, Ms. N and her mother decided it would be unsafe for her to attend, and that she should go to the hospital for evaluation instead.
EVALUATION Tough questions reveal answers
The pediatric team evaluates Ms. N. Her physical exam, laboratory values, and imaging are all within normal limits. Her neurologic exam demonstrates full strength, tone, and sensation in all extremities. All cranial nerves and reflexes are intact. No dysmorphic features or gait abnormalities are noted. All laboratory and imaging tests are normal, including complete blood cell count, electrolytes, calcium, magnesium, phosphorus, glucose, creatine kinase, liver enzymes, urine drug screen, human chorionic gonadotropin (hCG) urine test, and h
After the initial workup, the pediatric team consults the child and adolescent psychiatry team for a complete assessment of Ms. N due to concerns that a psychological component is contributing to her episodes. According to the psychosocial history obtained from Ms. N and her mother, Ms. N had experienced disrupted attachment, trauma, and loss. At age 5, Ms. N was temporarily removed from her mother’s custody after a fight between her mother and brother. At age 9, Ms. N’s stepfather, her primary father figure, died of a brain tumor.
Ms. N also has significant trauma stemming from her relationship with her biological father. Ms. N’s mother reports that her daughter was conceived during nonconsensual sexual intercourse. Ms. N did not have much contact with her biological father until 6 months ago, when he started picking her up at school and taking her to his home for several hours without permission or supervision. Afterwards, Ms. N confided to her mother and a teacher that her father sexually assaulted her during those visits.
Continue to: Ms. N and her mother...
Ms. N and her mother reported the assault to the police and were awaiting legal action.
During the interview with the psychiatry team, Ms. N denies that any thoughts or actions trigger the episodes and reports that she cannot control when they happen. Because she cannot anticipate the episodes, she says she is afraid to leave her house. She does not know why the episodes are happening and feels frustrated that they are getting worse. Ms. N says, “I have been feeling down lately,” but she denies hopelessness, worthlessness, suicidal ideation, homicidal ideation, delusions, or hallucinations.
In the hospital, when the psychiatry team asks Ms. N about her visits with her father, she says that they are “too painful to talk about,” and fears that discussing them will trigger an episode. However, her mother suggests that her daughter’s sexual trauma, as well as ongoing frustrations with the legal system, are influencing her mood; she has had low energy, poor appetite, and is spending more time in bed. Her mother also reports that Ms. N “avoids going out in the sun and spending time with her friends outside. She doesn’t seem to enjoy shopping and art like she used to.” Ms. N told her mother that she was having nightmares about the trauma and “could not stop thinking about some of the bad stuff that happened during the day.”
Ten minutes into the interview, while being questioned about her father, Ms. N experiences a spastic episode. She curls up in bed on her left side, clenches her entire body, and shuts her eyes. Her mother quickly runs to her bedside and counts the seconds until the end of the episode. After 25 seconds, Ms. N awakes with full recollection of the episode. On review of the video EEG during the episode, no ictal patterns are seen.
[polldaddy:10375873]
The authors’ observations
Paroxysmal episodes of weakness, numbness, and muscle spasms in a young female are suggestive of either epilepsy or nonepileptic seizure (NES).1,2 The negative EEG and physical features are inconsistent with epileptiform seizure, and Ms. N’s history and evaluation are suggestive of NES. Nonepileptic seizures are a type of a conversion disorder, or functional neurologic symptom disorder, in which a patient experiences weakness, abnormal movements, or seizure-like episodes that are inconsistent with organic neurologic disease.3 When a diagnosis of conversion disorder is suspected, a clinician must always consider other pathology that can explain the symptoms, such as migraine, vasovagal syncope, or intracranial mass. If a patient has focal neurologic deficits, head imaging should be pursued. Additionally, the clinician must screen for malingering and factitious disorder before establishing a definitive diagnosis. However, conversion disorder is not a diagnosis of exclusion. For example, a negative EEG does not rule out epilepsy, and patients can have both epilepsy and concomitant NES.
Continue to: Although NES is a common...
Although NES is a common type of conversion disorder, it is often difficult to diagnose, manage, and treat. Patients often receive antiepileptic medications but continue to have worsening events that are refractory to treatment. Various clinical features can suggest NES instead of epilepsy. Forced eye closure on video recording is a specific finding suggestive of NES, yet this feature is not sufficient to make the diagnosis.4 A video EEG must be performed to assess for epilepsy. The diagnosis of NES does not exclude the possibility that a patient has epilepsy, as NES can occur in up to 40% of patients with epilepsy.5 A video EEG without ictal patterns before, during, and after an observed episode is diagnostic of NES.6
[polldaddy:10375874]
The authors’ observations
Conversion disorders such as NES are a presentation of neurologic symptoms that cannot be readily accounted for by other conditions and are often associated with antecedent trauma. Multiple factors in Ms. N’s history increase her risk of NES, including loss of multiple loved ones, ongoing legal involvement, and alleged sexual abuse by her father.
Victims of sexual abuse are more likely than the general population to demonstrate symptoms of conversion disorder, especially NES.7,8 The onset of paroxysmal episodes after incestuous abuse in a teenage girl is characteristic of NES. Compared with patients with complex partial epilepsy (CPE), patients with NES are 3 times more likely to report sexual trauma.9,10 Children who report sexual abuse that precedes NES are more likely to have been victimized by a first-degree relative than patients with CPE who report sexual abuse.11 Risk factors for victims developing NES may be related to the severity of adversity, stress sensitivity, and decreased hippocampal volume.12,13
Ms. N endorsed many psychiatric symptoms that accompany her paroxysmal episodes; this is similar to findings in other patients with NES.14 One study found that depression is 3 times more prevalent and PTSD is 8 times more prevalent in patients with NES.12 During the evaluation, Ms. N’s mother said her daughter had low energy, poor appetite, lethargy, and anhedonia for the preceding 5 months, which is consistent with adjustment disorder.3 Her flashbacks, nightmares, difficulty sleeping, and agoraphobia, along with her trouble engaging with the people and activities that used to bring her joy, are symptoms of PTSD. Nonepileptic seizure is often associated with PTSD and can be viewed as an expression of a dissociated subtype.15
In a literature review, Durrant et al16 isolated prognostic indicators for NES (Table16). This study found that 70% of children and 40% of adults achieve remission from NES. Ms. N’s case has multiple concerning features, such as her comorbid psychiatric conditions, ongoing involvement in a legal case, and sexual trauma; this last factor is associated with the most severe symptoms and worse outcomes.16,17 Despite this somber reality, Ms. N has the support of her mother and is relatively young, which play a vital role in recovery.
Continue to: TREATMENT A strategy for minimizing the episodes
TREATMENT A strategy for minimizing the episodes
Ms. N’s medical workup remains unremarkable throughout the rest of her hospital stay. The psychiatry and pediatric teams discuss their assessments and agree that NES is the most likely diagnosis. The psychiatry team counsels Ms. N and her mother on the diagnosis and etiology of NES.
[polldaddy:10375876]
The authors’ observations
Cognitive-behavioral therapy is currently the treatment of choice for reducing seizure frequency in patients with NES.18,19 The use of CBT was suggested due to the theory that NES represents a dissociative response to trauma. Therapy focuses on changing a patient’s beliefs and perceptions associated with attacks.5 A randomized study of 66 patients with NES compared the use of CBT plus standard medical care with standard medical care alone.18 The standard medical care consisted of supportive treatment, an explanation of NES from a neuropsychiatrist, and supervised withdrawal of antiepileptic drugs. The CBT treatment group was offered weekly hour-long sessions for 12 weeks, accompanied by CBT homework and journaling the frequency and nature of seizure episodes (the CBT techniques are outlined in the Figure18). After 4 months, the CBT treatment group had fewer seizures, and after a 6-month follow-up, they were more likely to be seizure-free. However, in this study, CBT treatment did not improve mood or employment status.
A later investigation looked at using selective serotonin reuptake inhibitors to treat NES in adults.19 This study divided participants into 4 treatment groups: CBT with informed psychotherapy (CBT-ip), CBT-ip plus sertraline, sertraline alone, and treatment as usual. Sertraline was titrated up to a dose of 200 mg/d as tolerated. After 16 weeks of sertraline alone, seizure frequency did not decrease. Although both CBT groups showed a reduction in symptoms of up to 60%, the CBT-ip group reported fewer psychiatric symptoms with better social interactions, quality of life, and global functioning compared with patients treated with CBT-ip plus sertraline. The authors suggested that this may be due to the somatic adverse effects associated with sertraline. This study suggests that CBT without medication is the treatment of choice.
In addition to CBT, studies of psychodynamic psychotherapy for NES have had promising findings.20 Psychodynamic psychotherapy focuses on addressing conscious and unconscious anger, loss, feelings of isolation, and trauma. Through improving emotional processing, insight, coping skills and self-regulation, patients often benefit from an improvement in seizures, psychosocial functioning and health care utilization.
Metin et al21 found that group therapy alongside a family-centered approach elicited a strong and durable reduction in seizures in patients with NES. At enrollment, investigators distributed information on NES to patients and families. Psychoeducation and psychoanalysis with behavior modification techniques were provided in 90-minute weekly group sessions over 3 months. Participants also underwent monthly individualized sessions for standard psychiatric care for 9 months. During the group sessions, operant conditioning techniques were used to prevent secondary gain from seizure-like activity. Families met 4 times for 1 hour each to discuss seizures, receive psychoeducation on a subconscious etiology of NES, and learn behavior modification techniques. All 9 participants who completed group and individual therapy reported a significant and sustained reduction in seizure frequency by at least 50% at 12-month follow-up. Patients also demonstrated improvements in mood, anxiety, and quality of life.
Continue to: A meta-analysis...
A meta-analysis by Carlson and Perry22 that included 13 studies and 228 participants, examined different treatment modalities and their effectiveness for NES. They found that patients who received psychological intervention had a 47% remission rate and 82% improvement in seizure frequency compared with only 14% to 23% of those who did not receive therapy. They postulated that therapy for this illness must be flexible to properly address the socially, psychologically, and functionally heterogenous patient population. Although there are few randomized controlled trials for NES to determine the best evidence-based intervention, there is now consensus that NES has a favorable prognosis when barriers to psychological care are eliminated.
OUTCOME Referral for CBT
The treatment team advises Ms. N to engage in outpatient therapy after discharge from the hospital. Ms. N and her mother agree to the treatment plan, and leave the hospital with a referral for CBT the next day.
Bottom Line
Nonepileptic seizure (NES) is a type of conversion disorder characterized by seizure-like episodes without ictal qualities. Risk factors for NES include concomitant epilepsy, psychiatric disorders, unstable psychosocial situations, and antecedent trauma. Patients with a history of incestuous sexual abuse are most at risk for developing NES. A normal EEG that fully captures a seizure-like episode is diagnostic of NES. Cognitive-behavioral therapy can minimize seizure frequency and intensity.
Related Resources
- Marsh P, Benbadis S, Fernandez F. Psychogenic nonepileptic seizures: ways to win over skeptical patients. Current Psychiatry. 2008;7(1):21-24, 32-35.
- LaFrance WC Jr. Eye-opening behaviors help diagnose nonepileptic seizures. Current Psychiatry. 2006;5(11):121-122, 124, 130.
- LaFrance WC Jr, Kanner AM, Barry JJ. Treating patients with psychological nonepileptic seizures. In: Ettinger AB, Kanner AM, eds. Psychiatric issues in epilepsy: a practical guide to diagnosis and treatment. 2nd ed. Philadelphia, PA: Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2007:461-488.
Drug Brand Name
Sertraline • Zoloft
1. Lesser R. Psychogenic seizures. Neurology. 1996;46(6):1499-1507.
2. Stone J, LaFrance W, Brown R, et al. Conversion disorder: current problems and potential solutions for DSM-5. J Psychosom Res. 2011;71(6):369-376.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Syed T, Arozullah A, Suciu G, et al. Do observer and self-reports of ictal eye closure predict psychogenic nonepileptic seizures? Epilepsia. 2008;49(5):898-904.
5. Vega-Zelaya L, Alvarez M, Ezquiaga E, et al. Psychogenic non-epileptic seizures in a surgical epilepsy unit: experience and a comprehensive review. Epilepsy Topics. 2014. doi: 10.5772/57439.
6. LaFrance W, Baker G, Duncan R, et al. Minimum requirements for the diagnosis of psychogenic nonepileptic seizures: a staged approach. Epilepsia. 2013;54(11):2005-2018.
7. Roeloes K, Pasman J. Stress, childhood trauma, and cognitive functions in functional neurologic disorders. In: Hallett M, Stone J, Carson A, eds. Handbook of clinical neurology: functional neurologic disorders. 3rd ed. New York, NY: Elsevier; 2017:139-155.
8. Paras M, Murad M, Chen L, et al. Sexual abuse and lifetime diagnosis of somatic disorders. JAMA. 2009;302(5):550.
9. Fiszman A, Alves-Leon SV, Nunes RG, et al. Traumatic events and posttraumatic stress disorder in patients with psychogenic nonepileptic seizures: a critical review. Epilepsy Behav. 2004;5(6):818-825.
10. Sharpe D, Faye C. Non-epileptic seizures and child sexual abuse: a critical review of the literature. Clin Psychol Rev. 2006;26(8):1020-1040.
11. Alper K, Devinsky O, Perrine K, et al. Nonepileptic seizures and childhood sexual and physical abuse. Neurology. 1993;43(10):1950-1953.
12. Plioplys S, Doss J, Siddarth P et al. A multisite controlled study of risk factors in pediatric psychogenic nonepileptic seizures. Epilepsia. 2014;55(11):1739-1747.
13. Andersen S, Tomada A, Vincow E, et al. Preliminary evidence for sensitive periods in the effect of childhood sexual abuse on regional brain development. J Neuropsychiatry Clin Neurosci. 2008;20(3):292-301.
14. Sar V. Childhood trauma, dissociation, and psychiatric comorbidity in patients with conversion disorder. Am J Psychiatry. 2004;161(12):2271-2276.
15. Rosenberg HJ, Rosenberg SD, Williamson PD, et al. A comparative study of trauma and posttraumatic stress disorder prevalence in epilepsy patients and psychogenic nonepileptic seizure patients. Epilepsia. 2000;41(4):447-452.
16. Durrant J, Rickards H, Cavanna A. Prognosis and outcome predictors in psychogenic nonepileptic seizures. Epilepsy Res Treat. 2011;2011:1-7.
17. Selkirk M, Duncan R, Oto M, et al. Clinical differences between patients with nonepileptic seizures who report antecedent sexual abuse and those who do not. Epilepsia. 2008;49(8):1446-1450.
18. Goldstein L, Chalder T, Chigwedere C, et al. Cognitive-behavioral therapy for psychogenic nonepileptic seizures: a pilot RCT. Neurology. 2010;74(24):1986-1994.
19. LaFrance W, Baird G, Barry J, et al. Multicenter pilot treatment trial for psychogenic nonepileptic seizures. JAMA Psychiatry. 2014;71(9):997.
20. Howlett S, Reuber M. An augmented model of brief psychodynamic interpersonal therapy for patients with nonepileptic seizures. Psychotherapy (Chic). 2009;46(1):125-138.
21. Metin SZ, Ozmen M, Metin B, et al. Treatment with group psychotherapy for chronic psychogenic nonepileptic seizures. Epilepsy Behav. 2013;28(1):91-94.
22. Carlson P, Perry KN. Psychological interventions for psychogenic non-epileptic seizures: a meta-analysis. Seizure. 2017;45:142-150.
1. Lesser R. Psychogenic seizures. Neurology. 1996;46(6):1499-1507.
2. Stone J, LaFrance W, Brown R, et al. Conversion disorder: current problems and potential solutions for DSM-5. J Psychosom Res. 2011;71(6):369-376.
3. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
4. Syed T, Arozullah A, Suciu G, et al. Do observer and self-reports of ictal eye closure predict psychogenic nonepileptic seizures? Epilepsia. 2008;49(5):898-904.
5. Vega-Zelaya L, Alvarez M, Ezquiaga E, et al. Psychogenic non-epileptic seizures in a surgical epilepsy unit: experience and a comprehensive review. Epilepsy Topics. 2014. doi: 10.5772/57439.
6. LaFrance W, Baker G, Duncan R, et al. Minimum requirements for the diagnosis of psychogenic nonepileptic seizures: a staged approach. Epilepsia. 2013;54(11):2005-2018.
7. Roeloes K, Pasman J. Stress, childhood trauma, and cognitive functions in functional neurologic disorders. In: Hallett M, Stone J, Carson A, eds. Handbook of clinical neurology: functional neurologic disorders. 3rd ed. New York, NY: Elsevier; 2017:139-155.
8. Paras M, Murad M, Chen L, et al. Sexual abuse and lifetime diagnosis of somatic disorders. JAMA. 2009;302(5):550.
9. Fiszman A, Alves-Leon SV, Nunes RG, et al. Traumatic events and posttraumatic stress disorder in patients with psychogenic nonepileptic seizures: a critical review. Epilepsy Behav. 2004;5(6):818-825.
10. Sharpe D, Faye C. Non-epileptic seizures and child sexual abuse: a critical review of the literature. Clin Psychol Rev. 2006;26(8):1020-1040.
11. Alper K, Devinsky O, Perrine K, et al. Nonepileptic seizures and childhood sexual and physical abuse. Neurology. 1993;43(10):1950-1953.
12. Plioplys S, Doss J, Siddarth P et al. A multisite controlled study of risk factors in pediatric psychogenic nonepileptic seizures. Epilepsia. 2014;55(11):1739-1747.
13. Andersen S, Tomada A, Vincow E, et al. Preliminary evidence for sensitive periods in the effect of childhood sexual abuse on regional brain development. J Neuropsychiatry Clin Neurosci. 2008;20(3):292-301.
14. Sar V. Childhood trauma, dissociation, and psychiatric comorbidity in patients with conversion disorder. Am J Psychiatry. 2004;161(12):2271-2276.
15. Rosenberg HJ, Rosenberg SD, Williamson PD, et al. A comparative study of trauma and posttraumatic stress disorder prevalence in epilepsy patients and psychogenic nonepileptic seizure patients. Epilepsia. 2000;41(4):447-452.
16. Durrant J, Rickards H, Cavanna A. Prognosis and outcome predictors in psychogenic nonepileptic seizures. Epilepsy Res Treat. 2011;2011:1-7.
17. Selkirk M, Duncan R, Oto M, et al. Clinical differences between patients with nonepileptic seizures who report antecedent sexual abuse and those who do not. Epilepsia. 2008;49(8):1446-1450.
18. Goldstein L, Chalder T, Chigwedere C, et al. Cognitive-behavioral therapy for psychogenic nonepileptic seizures: a pilot RCT. Neurology. 2010;74(24):1986-1994.
19. LaFrance W, Baird G, Barry J, et al. Multicenter pilot treatment trial for psychogenic nonepileptic seizures. JAMA Psychiatry. 2014;71(9):997.
20. Howlett S, Reuber M. An augmented model of brief psychodynamic interpersonal therapy for patients with nonepileptic seizures. Psychotherapy (Chic). 2009;46(1):125-138.
21. Metin SZ, Ozmen M, Metin B, et al. Treatment with group psychotherapy for chronic psychogenic nonepileptic seizures. Epilepsy Behav. 2013;28(1):91-94.
22. Carlson P, Perry KN. Psychological interventions for psychogenic non-epileptic seizures: a meta-analysis. Seizure. 2017;45:142-150.

A solution for reducing referrals (and malpractice suits)
I agree with Dr. Hickner’s editorial “To refer—or not?” (J Fam Pract. 2019;68:8) that family physicians could manage about 30% of the patients they refer to specialists. Still, it’s worth noting that many referrals are motivated by the threat of unmerited malpractice suits. Until the medical liability system becomes less adversarial and unmerited suits are eliminated, all primary care doctors—not just family physicians—will continue to send patients to specialists—even when these physicians are themselves capable of treating such patients.
What might help mitigate malpractice suits? There could be benefit from oversight of health courts, which would be presided over by judges with special training in medical malpractice. Being nonadversarial, health courts would cut down on legal wrangling, settle suits, and get awards to patients quicker. They would also cut down on attorney and court fees, which account for almost half of the total amount spent on litigation. These courts wouldn’t completely eliminate unnecessary referrals to specialists, but they could help make a difference.
Edward Volpintesta, MD
Bethel, Conn
I agree with Dr. Hickner’s editorial “To refer—or not?” (J Fam Pract. 2019;68:8) that family physicians could manage about 30% of the patients they refer to specialists. Still, it’s worth noting that many referrals are motivated by the threat of unmerited malpractice suits. Until the medical liability system becomes less adversarial and unmerited suits are eliminated, all primary care doctors—not just family physicians—will continue to send patients to specialists—even when these physicians are themselves capable of treating such patients.
What might help mitigate malpractice suits? There could be benefit from oversight of health courts, which would be presided over by judges with special training in medical malpractice. Being nonadversarial, health courts would cut down on legal wrangling, settle suits, and get awards to patients quicker. They would also cut down on attorney and court fees, which account for almost half of the total amount spent on litigation. These courts wouldn’t completely eliminate unnecessary referrals to specialists, but they could help make a difference.
Edward Volpintesta, MD
Bethel, Conn
I agree with Dr. Hickner’s editorial “To refer—or not?” (J Fam Pract. 2019;68:8) that family physicians could manage about 30% of the patients they refer to specialists. Still, it’s worth noting that many referrals are motivated by the threat of unmerited malpractice suits. Until the medical liability system becomes less adversarial and unmerited suits are eliminated, all primary care doctors—not just family physicians—will continue to send patients to specialists—even when these physicians are themselves capable of treating such patients.
What might help mitigate malpractice suits? There could be benefit from oversight of health courts, which would be presided over by judges with special training in medical malpractice. Being nonadversarial, health courts would cut down on legal wrangling, settle suits, and get awards to patients quicker. They would also cut down on attorney and court fees, which account for almost half of the total amount spent on litigation. These courts wouldn’t completely eliminate unnecessary referrals to specialists, but they could help make a difference.
Edward Volpintesta, MD
Bethel, Conn
Acute hearing loss, tinnitus, and fullness in the left ear • Weber test lateralized to the right ear • Positive Rinne test and normal tympanometry • Dx?
THE CASE
A healthy 48-year-old man presented to our otolaryngology clinic with a 2-hour history of hearing loss, tinnitus, and fullness in the left ear. He denied any vertigo, nausea, vomiting, otalgia, or otorrhea. He had noticed signs of a possible upper respiratory infection, including a sore throat and headache, the day before his symptoms started. His medical history was unremarkable. He denied any history of otologic surgery, trauma, or vision problems, and he was not taking any medications.
The patient was afebrile on physical examination with a heart rate of 48 beats/min and blood pressure of 117/68 mm Hg. A Weber test performed using a 512-Hz tuning fork lateralized to the right ear. A Rinne test showed air conduction was louder than bone conduction in the affected left ear—a normal finding. Tympanometry and otoscopic examination showed the bilateral tympanic membranes were normal.
THE DIAGNOSIS
Pure tone audiometry showed severe sensorineural hearing loss in the left ear and a poor speech discrimination score. The Weber test confirmed the hearing loss was sensorineural and not conductive, ruling out a middle ear effusion. Additionally, the normal tympanogram made conductive hearing loss from a middle ear effusion or tympanic membrane perforation unlikely. The positive Rinne test was consistent with a diagnosis of idiopathic sudden sensorineural hearing loss (SSNHL).
DISCUSSION
SSNHL is defined by hearing loss of more than 30 dB in at least 3 consecutive frequencies with acute onset of less than 72 hours.1,2 The most common symptoms include acute hearing loss, tinnitus, and fullness in the affected ear.1 The majority of cases of SSNHL are unilateral. The typical age of onset is in the fourth and fifth decades, occurring with equal distribution in both sexes.
Etiology.
Diagnosis. The initial evaluation should include an otoscopic examination, tuning fork tests, and pure tone audiometry.1-3 Weber and Rinne tests are essential when evaluating patients for unilateral hearing loss and determining the type of loss (ie, sensorineural vs conductive). The Weber test (ideally using a 512-Hz tuning fork) can detect either conductive or sensorineural hearing loss. In a normal Weber test, the patient should hear the vibration of the tuning fork equally in both ears. The tuning fork will be heard in both ears in conductive hearing loss but will only be heard in the unaffected hear if sensorineural hearing loss is present. So, for instance, if a patient has a perforation in the right tympanic membrane causing conductive hearing loss in the right hear, the tuning fork would be heard in both ears. If the patient has sensorineural hearing loss in the right ear, the tuning fork would only be heard in the left ear.
The Rinne test compares the perception of sound waves transmitted by air conduction vs bone conduction and serves as a rapid screen for conductive hearing loss.
Continue to: Magnetic resonance imaging...
Magnetic resonance imaging (MRI) of the brain and brainstem with gadolinium contrast can reveal vascular events (thrombotic or hemorrhagic), demyelinating disorders, or retrocochlear lesions such as vestibular schwannoma and is indicated in all cases of suspected SSNHL.4,5
Treatment and management. The current standard of care for treatment of idiopathic SSNHL is systemic steroids.1,2 Although the gold standard currently is oral prednisolone or methylprednisolone (1 mg/kg/d for 10 to 14 days with a taper,1,2 the evidence for this regimen stems from a single placebo-controlled trial (N = 67) that demonstrated greater improvement in the steroid group compared with the placebo group (61% vs 32%).6 A Cochrane review and other systematic analyses have not demonstrated clear efficacy of corticosteroid treatment for the management of idiopathic SSNHL.7,8
Because of the potential systemic adverse effects associated with oral corticosteroids, intratympanic (IT) corticosteroids have been advocated as an alternative treatment option. A prospective, randomized, noninferiority trial comparing the efficacy of oral vs IT corticosteroids for idiopathic SSNHL found IT corticosteroids to be noninferior to systemic treatment.9 IT treatment also has been advocated as a rescue therapy for patients who do not respond to systemic treatment.10
A combination of oral and IT corticosteroids was investigated in a retrospective study analyzing multiple treatment modalities.10 Researchers first compared 122 patients receiving one of 3 treatments: (1) IT corticosteroids, (2) oral corticosteroids, and (3) combination treatment (IT + oral corticosteroids). There was no difference in hearing recovery among any of the treatments. Fifty-eight patients who were refractory to initial treatment were then included in a second analysis in which they were divided into those who received additional IT corticosteroids (salvage treatment) vs no treatment (control). There was no difference in hearing recovery between the 2 groups. The authors concluded that IT corticosteroids were as effective as oral treatment and that salvage IT treatment did not add any benefit.10
The American Academy of Otolaryngology-Head and Neck Surgery (AAO-HNS) recently published guidelines on the diagnosis and management of SSNHL.11 The guidelines state that IT steroids should be considered in patients who cannot tolerate oral steroids, such as patients with diabetes. It is important to note, however, that the high cost of IT treatment (~$2000 for dexamethasone or methylprednisolone vs < $10 for oral prednisolone) is an issue that needs to be considered as health care costs continue to rise.
Continue to: Antivirals
Antivirals. Because an underlying viral etiology has been speculated as a potential cause of idiopathic SSNHL, antiviral agents such as valacyclovir or famciclovir also are potential treatment agents.12 Antiviral medications have minimal adverse effects and are relatively inexpensive, but the benefits have not yet been proven in randomized controlled trials,and they currently are not endorsed by the AAO-HNS in their guidelines for the management of SSNHL.11
Spontaneous recovery occurs in up to 40% of patients with idiopathic SSNHL. As many as 65% of those who experience recovery do so within 2 weeks of the onset of symptoms, regardless of treatment.1,2 Treatment beyond 2 weeks after onset of symptoms is unlikely to be of any benefit, although some otolaryngologists will treat for up to 6 weeks after the onset of hearing loss.
A substantial number of patients with SSNHL may not recover. Management of these patients begins with referral to an appropriate specialist to initiate counseling and lifestyle changes. Depending on the degree of hearing loss, audiologic rehabilitation may include use of a traditional or bone-anchored hearing aid or a frequency-modulation system.1,2,11 Tinnitus retraining therapy might be of benefit for patients with persistent tinnitus.11
Our patient. After a discussion of his treatment options, our patient decided on a combination of oral prednisolone (60 mg once daily for 9 days followed by a taper for 5 days) and intratympanic dexamethasone injections (1 mL [10 mg/mL] once weekly for 3 weeks).
The rationale for this approach was the minimal adverse effects associated with short-term (ie, days to 1–2 weeks) use of high-dose (ie, > 30 mg/d) corticosteroids. Although steroid therapy has been associated with adverse effects such as aseptic necrosis of the hip, these complications usually arise after longer periods (ie, months to years) of high-dose steroid therapy with a mean cumulative dose much higher than what was used in our patient.13
Continue to: Our patient...
Our patient noticed slight improvement within 48 hours of the initial onset of symptoms that continued for the next several weeks until full recovery was attained. An MRI performed 5 days after the onset of symptoms was negative for retrocochlear pathology.
THE TAKEAWAY
SSNHL is a medical emergency that requires prompt recognition and diagnosis. The steps in evaluating sudden hearing loss include: (1) appropriate history and physical examination (eg, otoscopic examination, tuning fork tests), (2) urgent audiometry to confirm hearing loss, (3) immediate referral to an otolaryngologist for further testing (eg, tympanometry, blood tests, MRI), and (4) initiation of treatment.
If a specific etiology is identified (eg, vestibular schwannoma), the patient should be referred to a specialist for appropriate treatment. If there is no identifiable cause (idiopathic SSNHL), the patient should be treated with oral and/or intratympanic steroids. Patients who do not recover following treatment should be offered audiologic rehabilitation.
CORRESPONDENCE
Sergio Huerta, MD, UT Southwestern Medical Center, 4500 S Lancaster Road #112L, Dallas, TX 75216; [email protected]
1. Schreiber BE, Agrup C, Haskard DO, et al. Sudden sensorineural hearing loss. Lancet. 2010;375:1203-1211.
2. Rauch SD. Clinical practice. Idiopathic sudden sensorineural hearing loss. N Engl J Med. 2008;359:833-840.
3. Paul BC, Roland JT Jr. An abnormal audiogram. JAMA. 2015;313:85-86.
4. Aarnisalo AA, Suoranta H, Ylikoski J. Magnetic resonance imaging findings in the auditory pathway of patients with sudden deafness. Otol Neurotol. 2004;25:245-249.
5. Cadoni G, Cianfoni A, Agostino S, et al. Magnetic resonance imaging findings in sudden sensorineural hearing loss. J Otolaryngol. 2006;35:310-316.
6. Wilson WR, Byl FM, Laird N. The efficacy of steroids in the treatment of idiopathic sudden hearing loss. A double-blind clinical study. Arch Otolaryngol. 1980;106:772-776.
7. Wei BPC, Stathopoulos D, O’Leary S. Steroids for idiopathic sudden sensorineural hearing loss. Cochrane Database Syst Rev. 2013. doi:10.1002/14651858.CD003998.pub3.
8. Conlin AE, Parnes LS. Treatment of sudden sensorineural hearing loss: II. a meta-analysis. Arch Otolaryngol Head Neck Surg. 2007;133:582-586.
9. Rauch SD, Halpin CF, Antonelli PJ, et al. Oral vs intratympanic corticosteroid therapy for idiopathic sudden sensorineural hearing loss: a randomized trial. JAMA. 2011;305:2071-2079.
10. Lee KH, Ryu SH, Lee HM, et al. Is intratympanic dexamethasone injection effective for the treatment of idiopathic sudden sensorineural hearing loss? J Audiol Otol. 2015;19:154-158.
11. Stachler RJ, Chandrasekhar SS, Archer SM, et al. Clinical practice guideline: sudden hearing loss. Otolaryngol Head Neck Surg. 2012;146(3 suppl):S1-S35.
12. Westerlaken BO, Stokroos RJ, Dhooge IJ, et al. Treatment of idiopathic sudden sensorineural hearing loss with antiviral therapy: a prospective, randomized, double-blind clinical trial. Ann Otol Rhinol Laryngol. 2003;112:993-1000.
13. Nowak DA, Yeung J. Steroid-induced osteonecrosis in dermatology: a review [published online March 30, 2015]. J Cutan Med Surg. 2015;19:358-360.
THE CASE
A healthy 48-year-old man presented to our otolaryngology clinic with a 2-hour history of hearing loss, tinnitus, and fullness in the left ear. He denied any vertigo, nausea, vomiting, otalgia, or otorrhea. He had noticed signs of a possible upper respiratory infection, including a sore throat and headache, the day before his symptoms started. His medical history was unremarkable. He denied any history of otologic surgery, trauma, or vision problems, and he was not taking any medications.
The patient was afebrile on physical examination with a heart rate of 48 beats/min and blood pressure of 117/68 mm Hg. A Weber test performed using a 512-Hz tuning fork lateralized to the right ear. A Rinne test showed air conduction was louder than bone conduction in the affected left ear—a normal finding. Tympanometry and otoscopic examination showed the bilateral tympanic membranes were normal.
THE DIAGNOSIS
Pure tone audiometry showed severe sensorineural hearing loss in the left ear and a poor speech discrimination score. The Weber test confirmed the hearing loss was sensorineural and not conductive, ruling out a middle ear effusion. Additionally, the normal tympanogram made conductive hearing loss from a middle ear effusion or tympanic membrane perforation unlikely. The positive Rinne test was consistent with a diagnosis of idiopathic sudden sensorineural hearing loss (SSNHL).
DISCUSSION
SSNHL is defined by hearing loss of more than 30 dB in at least 3 consecutive frequencies with acute onset of less than 72 hours.1,2 The most common symptoms include acute hearing loss, tinnitus, and fullness in the affected ear.1 The majority of cases of SSNHL are unilateral. The typical age of onset is in the fourth and fifth decades, occurring with equal distribution in both sexes.
Etiology.
Diagnosis. The initial evaluation should include an otoscopic examination, tuning fork tests, and pure tone audiometry.1-3 Weber and Rinne tests are essential when evaluating patients for unilateral hearing loss and determining the type of loss (ie, sensorineural vs conductive). The Weber test (ideally using a 512-Hz tuning fork) can detect either conductive or sensorineural hearing loss. In a normal Weber test, the patient should hear the vibration of the tuning fork equally in both ears. The tuning fork will be heard in both ears in conductive hearing loss but will only be heard in the unaffected hear if sensorineural hearing loss is present. So, for instance, if a patient has a perforation in the right tympanic membrane causing conductive hearing loss in the right hear, the tuning fork would be heard in both ears. If the patient has sensorineural hearing loss in the right ear, the tuning fork would only be heard in the left ear.
The Rinne test compares the perception of sound waves transmitted by air conduction vs bone conduction and serves as a rapid screen for conductive hearing loss.
Continue to: Magnetic resonance imaging...
Magnetic resonance imaging (MRI) of the brain and brainstem with gadolinium contrast can reveal vascular events (thrombotic or hemorrhagic), demyelinating disorders, or retrocochlear lesions such as vestibular schwannoma and is indicated in all cases of suspected SSNHL.4,5
Treatment and management. The current standard of care for treatment of idiopathic SSNHL is systemic steroids.1,2 Although the gold standard currently is oral prednisolone or methylprednisolone (1 mg/kg/d for 10 to 14 days with a taper,1,2 the evidence for this regimen stems from a single placebo-controlled trial (N = 67) that demonstrated greater improvement in the steroid group compared with the placebo group (61% vs 32%).6 A Cochrane review and other systematic analyses have not demonstrated clear efficacy of corticosteroid treatment for the management of idiopathic SSNHL.7,8
Because of the potential systemic adverse effects associated with oral corticosteroids, intratympanic (IT) corticosteroids have been advocated as an alternative treatment option. A prospective, randomized, noninferiority trial comparing the efficacy of oral vs IT corticosteroids for idiopathic SSNHL found IT corticosteroids to be noninferior to systemic treatment.9 IT treatment also has been advocated as a rescue therapy for patients who do not respond to systemic treatment.10
A combination of oral and IT corticosteroids was investigated in a retrospective study analyzing multiple treatment modalities.10 Researchers first compared 122 patients receiving one of 3 treatments: (1) IT corticosteroids, (2) oral corticosteroids, and (3) combination treatment (IT + oral corticosteroids). There was no difference in hearing recovery among any of the treatments. Fifty-eight patients who were refractory to initial treatment were then included in a second analysis in which they were divided into those who received additional IT corticosteroids (salvage treatment) vs no treatment (control). There was no difference in hearing recovery between the 2 groups. The authors concluded that IT corticosteroids were as effective as oral treatment and that salvage IT treatment did not add any benefit.10
The American Academy of Otolaryngology-Head and Neck Surgery (AAO-HNS) recently published guidelines on the diagnosis and management of SSNHL.11 The guidelines state that IT steroids should be considered in patients who cannot tolerate oral steroids, such as patients with diabetes. It is important to note, however, that the high cost of IT treatment (~$2000 for dexamethasone or methylprednisolone vs < $10 for oral prednisolone) is an issue that needs to be considered as health care costs continue to rise.
Continue to: Antivirals
Antivirals. Because an underlying viral etiology has been speculated as a potential cause of idiopathic SSNHL, antiviral agents such as valacyclovir or famciclovir also are potential treatment agents.12 Antiviral medications have minimal adverse effects and are relatively inexpensive, but the benefits have not yet been proven in randomized controlled trials,and they currently are not endorsed by the AAO-HNS in their guidelines for the management of SSNHL.11
Spontaneous recovery occurs in up to 40% of patients with idiopathic SSNHL. As many as 65% of those who experience recovery do so within 2 weeks of the onset of symptoms, regardless of treatment.1,2 Treatment beyond 2 weeks after onset of symptoms is unlikely to be of any benefit, although some otolaryngologists will treat for up to 6 weeks after the onset of hearing loss.
A substantial number of patients with SSNHL may not recover. Management of these patients begins with referral to an appropriate specialist to initiate counseling and lifestyle changes. Depending on the degree of hearing loss, audiologic rehabilitation may include use of a traditional or bone-anchored hearing aid or a frequency-modulation system.1,2,11 Tinnitus retraining therapy might be of benefit for patients with persistent tinnitus.11
Our patient. After a discussion of his treatment options, our patient decided on a combination of oral prednisolone (60 mg once daily for 9 days followed by a taper for 5 days) and intratympanic dexamethasone injections (1 mL [10 mg/mL] once weekly for 3 weeks).
The rationale for this approach was the minimal adverse effects associated with short-term (ie, days to 1–2 weeks) use of high-dose (ie, > 30 mg/d) corticosteroids. Although steroid therapy has been associated with adverse effects such as aseptic necrosis of the hip, these complications usually arise after longer periods (ie, months to years) of high-dose steroid therapy with a mean cumulative dose much higher than what was used in our patient.13
Continue to: Our patient...
Our patient noticed slight improvement within 48 hours of the initial onset of symptoms that continued for the next several weeks until full recovery was attained. An MRI performed 5 days after the onset of symptoms was negative for retrocochlear pathology.
THE TAKEAWAY
SSNHL is a medical emergency that requires prompt recognition and diagnosis. The steps in evaluating sudden hearing loss include: (1) appropriate history and physical examination (eg, otoscopic examination, tuning fork tests), (2) urgent audiometry to confirm hearing loss, (3) immediate referral to an otolaryngologist for further testing (eg, tympanometry, blood tests, MRI), and (4) initiation of treatment.
If a specific etiology is identified (eg, vestibular schwannoma), the patient should be referred to a specialist for appropriate treatment. If there is no identifiable cause (idiopathic SSNHL), the patient should be treated with oral and/or intratympanic steroids. Patients who do not recover following treatment should be offered audiologic rehabilitation.
CORRESPONDENCE
Sergio Huerta, MD, UT Southwestern Medical Center, 4500 S Lancaster Road #112L, Dallas, TX 75216; [email protected]
THE CASE
A healthy 48-year-old man presented to our otolaryngology clinic with a 2-hour history of hearing loss, tinnitus, and fullness in the left ear. He denied any vertigo, nausea, vomiting, otalgia, or otorrhea. He had noticed signs of a possible upper respiratory infection, including a sore throat and headache, the day before his symptoms started. His medical history was unremarkable. He denied any history of otologic surgery, trauma, or vision problems, and he was not taking any medications.
The patient was afebrile on physical examination with a heart rate of 48 beats/min and blood pressure of 117/68 mm Hg. A Weber test performed using a 512-Hz tuning fork lateralized to the right ear. A Rinne test showed air conduction was louder than bone conduction in the affected left ear—a normal finding. Tympanometry and otoscopic examination showed the bilateral tympanic membranes were normal.
THE DIAGNOSIS
Pure tone audiometry showed severe sensorineural hearing loss in the left ear and a poor speech discrimination score. The Weber test confirmed the hearing loss was sensorineural and not conductive, ruling out a middle ear effusion. Additionally, the normal tympanogram made conductive hearing loss from a middle ear effusion or tympanic membrane perforation unlikely. The positive Rinne test was consistent with a diagnosis of idiopathic sudden sensorineural hearing loss (SSNHL).
DISCUSSION
SSNHL is defined by hearing loss of more than 30 dB in at least 3 consecutive frequencies with acute onset of less than 72 hours.1,2 The most common symptoms include acute hearing loss, tinnitus, and fullness in the affected ear.1 The majority of cases of SSNHL are unilateral. The typical age of onset is in the fourth and fifth decades, occurring with equal distribution in both sexes.
Etiology.
Diagnosis. The initial evaluation should include an otoscopic examination, tuning fork tests, and pure tone audiometry.1-3 Weber and Rinne tests are essential when evaluating patients for unilateral hearing loss and determining the type of loss (ie, sensorineural vs conductive). The Weber test (ideally using a 512-Hz tuning fork) can detect either conductive or sensorineural hearing loss. In a normal Weber test, the patient should hear the vibration of the tuning fork equally in both ears. The tuning fork will be heard in both ears in conductive hearing loss but will only be heard in the unaffected hear if sensorineural hearing loss is present. So, for instance, if a patient has a perforation in the right tympanic membrane causing conductive hearing loss in the right hear, the tuning fork would be heard in both ears. If the patient has sensorineural hearing loss in the right ear, the tuning fork would only be heard in the left ear.
The Rinne test compares the perception of sound waves transmitted by air conduction vs bone conduction and serves as a rapid screen for conductive hearing loss.
Continue to: Magnetic resonance imaging...
Magnetic resonance imaging (MRI) of the brain and brainstem with gadolinium contrast can reveal vascular events (thrombotic or hemorrhagic), demyelinating disorders, or retrocochlear lesions such as vestibular schwannoma and is indicated in all cases of suspected SSNHL.4,5
Treatment and management. The current standard of care for treatment of idiopathic SSNHL is systemic steroids.1,2 Although the gold standard currently is oral prednisolone or methylprednisolone (1 mg/kg/d for 10 to 14 days with a taper,1,2 the evidence for this regimen stems from a single placebo-controlled trial (N = 67) that demonstrated greater improvement in the steroid group compared with the placebo group (61% vs 32%).6 A Cochrane review and other systematic analyses have not demonstrated clear efficacy of corticosteroid treatment for the management of idiopathic SSNHL.7,8
Because of the potential systemic adverse effects associated with oral corticosteroids, intratympanic (IT) corticosteroids have been advocated as an alternative treatment option. A prospective, randomized, noninferiority trial comparing the efficacy of oral vs IT corticosteroids for idiopathic SSNHL found IT corticosteroids to be noninferior to systemic treatment.9 IT treatment also has been advocated as a rescue therapy for patients who do not respond to systemic treatment.10
A combination of oral and IT corticosteroids was investigated in a retrospective study analyzing multiple treatment modalities.10 Researchers first compared 122 patients receiving one of 3 treatments: (1) IT corticosteroids, (2) oral corticosteroids, and (3) combination treatment (IT + oral corticosteroids). There was no difference in hearing recovery among any of the treatments. Fifty-eight patients who were refractory to initial treatment were then included in a second analysis in which they were divided into those who received additional IT corticosteroids (salvage treatment) vs no treatment (control). There was no difference in hearing recovery between the 2 groups. The authors concluded that IT corticosteroids were as effective as oral treatment and that salvage IT treatment did not add any benefit.10
The American Academy of Otolaryngology-Head and Neck Surgery (AAO-HNS) recently published guidelines on the diagnosis and management of SSNHL.11 The guidelines state that IT steroids should be considered in patients who cannot tolerate oral steroids, such as patients with diabetes. It is important to note, however, that the high cost of IT treatment (~$2000 for dexamethasone or methylprednisolone vs < $10 for oral prednisolone) is an issue that needs to be considered as health care costs continue to rise.
Continue to: Antivirals
Antivirals. Because an underlying viral etiology has been speculated as a potential cause of idiopathic SSNHL, antiviral agents such as valacyclovir or famciclovir also are potential treatment agents.12 Antiviral medications have minimal adverse effects and are relatively inexpensive, but the benefits have not yet been proven in randomized controlled trials,and they currently are not endorsed by the AAO-HNS in their guidelines for the management of SSNHL.11
Spontaneous recovery occurs in up to 40% of patients with idiopathic SSNHL. As many as 65% of those who experience recovery do so within 2 weeks of the onset of symptoms, regardless of treatment.1,2 Treatment beyond 2 weeks after onset of symptoms is unlikely to be of any benefit, although some otolaryngologists will treat for up to 6 weeks after the onset of hearing loss.
A substantial number of patients with SSNHL may not recover. Management of these patients begins with referral to an appropriate specialist to initiate counseling and lifestyle changes. Depending on the degree of hearing loss, audiologic rehabilitation may include use of a traditional or bone-anchored hearing aid or a frequency-modulation system.1,2,11 Tinnitus retraining therapy might be of benefit for patients with persistent tinnitus.11
Our patient. After a discussion of his treatment options, our patient decided on a combination of oral prednisolone (60 mg once daily for 9 days followed by a taper for 5 days) and intratympanic dexamethasone injections (1 mL [10 mg/mL] once weekly for 3 weeks).
The rationale for this approach was the minimal adverse effects associated with short-term (ie, days to 1–2 weeks) use of high-dose (ie, > 30 mg/d) corticosteroids. Although steroid therapy has been associated with adverse effects such as aseptic necrosis of the hip, these complications usually arise after longer periods (ie, months to years) of high-dose steroid therapy with a mean cumulative dose much higher than what was used in our patient.13
Continue to: Our patient...
Our patient noticed slight improvement within 48 hours of the initial onset of symptoms that continued for the next several weeks until full recovery was attained. An MRI performed 5 days after the onset of symptoms was negative for retrocochlear pathology.
THE TAKEAWAY
SSNHL is a medical emergency that requires prompt recognition and diagnosis. The steps in evaluating sudden hearing loss include: (1) appropriate history and physical examination (eg, otoscopic examination, tuning fork tests), (2) urgent audiometry to confirm hearing loss, (3) immediate referral to an otolaryngologist for further testing (eg, tympanometry, blood tests, MRI), and (4) initiation of treatment.
If a specific etiology is identified (eg, vestibular schwannoma), the patient should be referred to a specialist for appropriate treatment. If there is no identifiable cause (idiopathic SSNHL), the patient should be treated with oral and/or intratympanic steroids. Patients who do not recover following treatment should be offered audiologic rehabilitation.
CORRESPONDENCE
Sergio Huerta, MD, UT Southwestern Medical Center, 4500 S Lancaster Road #112L, Dallas, TX 75216; [email protected]
1. Schreiber BE, Agrup C, Haskard DO, et al. Sudden sensorineural hearing loss. Lancet. 2010;375:1203-1211.
2. Rauch SD. Clinical practice. Idiopathic sudden sensorineural hearing loss. N Engl J Med. 2008;359:833-840.
3. Paul BC, Roland JT Jr. An abnormal audiogram. JAMA. 2015;313:85-86.
4. Aarnisalo AA, Suoranta H, Ylikoski J. Magnetic resonance imaging findings in the auditory pathway of patients with sudden deafness. Otol Neurotol. 2004;25:245-249.
5. Cadoni G, Cianfoni A, Agostino S, et al. Magnetic resonance imaging findings in sudden sensorineural hearing loss. J Otolaryngol. 2006;35:310-316.
6. Wilson WR, Byl FM, Laird N. The efficacy of steroids in the treatment of idiopathic sudden hearing loss. A double-blind clinical study. Arch Otolaryngol. 1980;106:772-776.
7. Wei BPC, Stathopoulos D, O’Leary S. Steroids for idiopathic sudden sensorineural hearing loss. Cochrane Database Syst Rev. 2013. doi:10.1002/14651858.CD003998.pub3.
8. Conlin AE, Parnes LS. Treatment of sudden sensorineural hearing loss: II. a meta-analysis. Arch Otolaryngol Head Neck Surg. 2007;133:582-586.
9. Rauch SD, Halpin CF, Antonelli PJ, et al. Oral vs intratympanic corticosteroid therapy for idiopathic sudden sensorineural hearing loss: a randomized trial. JAMA. 2011;305:2071-2079.
10. Lee KH, Ryu SH, Lee HM, et al. Is intratympanic dexamethasone injection effective for the treatment of idiopathic sudden sensorineural hearing loss? J Audiol Otol. 2015;19:154-158.
11. Stachler RJ, Chandrasekhar SS, Archer SM, et al. Clinical practice guideline: sudden hearing loss. Otolaryngol Head Neck Surg. 2012;146(3 suppl):S1-S35.
12. Westerlaken BO, Stokroos RJ, Dhooge IJ, et al. Treatment of idiopathic sudden sensorineural hearing loss with antiviral therapy: a prospective, randomized, double-blind clinical trial. Ann Otol Rhinol Laryngol. 2003;112:993-1000.
13. Nowak DA, Yeung J. Steroid-induced osteonecrosis in dermatology: a review [published online March 30, 2015]. J Cutan Med Surg. 2015;19:358-360.
1. Schreiber BE, Agrup C, Haskard DO, et al. Sudden sensorineural hearing loss. Lancet. 2010;375:1203-1211.
2. Rauch SD. Clinical practice. Idiopathic sudden sensorineural hearing loss. N Engl J Med. 2008;359:833-840.
3. Paul BC, Roland JT Jr. An abnormal audiogram. JAMA. 2015;313:85-86.
4. Aarnisalo AA, Suoranta H, Ylikoski J. Magnetic resonance imaging findings in the auditory pathway of patients with sudden deafness. Otol Neurotol. 2004;25:245-249.
5. Cadoni G, Cianfoni A, Agostino S, et al. Magnetic resonance imaging findings in sudden sensorineural hearing loss. J Otolaryngol. 2006;35:310-316.
6. Wilson WR, Byl FM, Laird N. The efficacy of steroids in the treatment of idiopathic sudden hearing loss. A double-blind clinical study. Arch Otolaryngol. 1980;106:772-776.
7. Wei BPC, Stathopoulos D, O’Leary S. Steroids for idiopathic sudden sensorineural hearing loss. Cochrane Database Syst Rev. 2013. doi:10.1002/14651858.CD003998.pub3.
8. Conlin AE, Parnes LS. Treatment of sudden sensorineural hearing loss: II. a meta-analysis. Arch Otolaryngol Head Neck Surg. 2007;133:582-586.
9. Rauch SD, Halpin CF, Antonelli PJ, et al. Oral vs intratympanic corticosteroid therapy for idiopathic sudden sensorineural hearing loss: a randomized trial. JAMA. 2011;305:2071-2079.
10. Lee KH, Ryu SH, Lee HM, et al. Is intratympanic dexamethasone injection effective for the treatment of idiopathic sudden sensorineural hearing loss? J Audiol Otol. 2015;19:154-158.
11. Stachler RJ, Chandrasekhar SS, Archer SM, et al. Clinical practice guideline: sudden hearing loss. Otolaryngol Head Neck Surg. 2012;146(3 suppl):S1-S35.
12. Westerlaken BO, Stokroos RJ, Dhooge IJ, et al. Treatment of idiopathic sudden sensorineural hearing loss with antiviral therapy: a prospective, randomized, double-blind clinical trial. Ann Otol Rhinol Laryngol. 2003;112:993-1000.
13. Nowak DA, Yeung J. Steroid-induced osteonecrosis in dermatology: a review [published online March 30, 2015]. J Cutan Med Surg. 2015;19:358-360.

Hemoglobin levels are associated with long-term dementia risk
This U-shaped association “may relate to differences in white matter integrity and cerebral perfusion,” the researchers wrote in Neurology.

“With around 10% of people over age 65 having anemia in the Americas and Europe and up to 45% in African and southeast Asian countries, these results could have important implications for the burden of dementia,” said study author M. Arfan Ikram, MD, PhD, in a news release. Dr. Ikram is a professor of epidemiology at Erasmus Medical Center in Rotterdam, the Netherlands.
Prior studies have found that low hemoglobin levels are associated with adverse health outcomes, such as coronary heart disease, stroke, and mortality, but data about the relationship between hemoglobin levels and dementia risk have been limited.
A population-based cohort study
To examine the long-term association of hemoglobin levels and anemia with risk of dementia, Dr. Ikram and coauthors analyzed data from the Rotterdam Study, an ongoing population-based cohort study in the Netherlands that started in 1990. Their analysis included data from 12,305 participants without dementia who had serum hemoglobin measured at baseline (mean age, 64.6 years; 57.7% women).
During a mean follow-up of 12.1 years, 1,520 participants developed dementia, 1,194 of whom had Alzheimer’s disease.
“Both low and high hemoglobin levels were associated with increased dementia risk,” the authors wrote. Compared with participants in the middle quintile of hemoglobin levels (8.57-8.99 mmol/L), participants in the lowest quintile (less than 8.11 mmol/L) had a hazard ratio of dementia of 1.29, and participants in the highest quintile (greater than 9.40 mmol/L) had an HR of 1.20.
About 6% of the participants had anemia – that is, a hemoglobin level of less than 8.1 mmol/L for men and less than 7.5 mmol/L for women. Anemia was associated with a 34% increased risk of dementia and a 41% increased risk of Alzheimer’s disease.
Of the 745 people with anemia, 128 developed dementia, compared with 1,392 of the 11,560 people who did not have anemia (17% vs. 12%).
A U-shaped association
The researchers also examined hemoglobin in relation to vascular brain disease, structural connectivity, and global cerebral perfusion among 5,267 participants without dementia who had brain MRI. White matter hyperintensity volume and hemoglobin had a U-shaped association, similar to that for dementia and hemoglobin. In addition, hemoglobin inversely correlated to cerebral perfusion.
The results remained consistent after adjustment for factors such as smoking, high blood pressure, high cholesterol, and alcohol use.
A limitation of the study is that the participants lived in the Netherlands and were primarily of European descent, so the results may not apply to other populations, the authors wrote.
Dr. Ikram noted that the study does not prove that low or high hemoglobin levels cause dementia. “More research is needed to determine whether hemoglobin levels play a direct role in this increased risk or whether these associations can be explained by underlying issues or other vascular or metabolic changes.”
The study was supported by the Netherlands Cardiovascular Research Initiative; Erasmus Medical Centre; Erasmus University Rotterdam; Netherlands Organization for Scientific Research; Netherlands Organization for Health Research and Development; Research Institute for Diseases in the Elderly; Netherlands Genomic Initiative; Dutch Ministry of Education, Culture, and Science; Dutch Ministry of Health, Welfare, and Sports; European Commission; Municipality of Rotterdam; Netherlands Consortium for Healthy Aging; and Dutch Heart Foundation. The authors reported no relevant disclosures.
SOURCE: Ikram MA et al. Neurology. 2019 Jul 31. doi: 10.1212/WNL.0000000000008003.
This U-shaped association “may relate to differences in white matter integrity and cerebral perfusion,” the researchers wrote in Neurology.
“With around 10% of people over age 65 having anemia in the Americas and Europe and up to 45% in African and southeast Asian countries, these results could have important implications for the burden of dementia,” said study author M. Arfan Ikram, MD, PhD, in a news release. Dr. Ikram is a professor of epidemiology at Erasmus Medical Center in Rotterdam, the Netherlands.
Prior studies have found that low hemoglobin levels are associated with adverse health outcomes, such as coronary heart disease, stroke, and mortality, but data about the relationship between hemoglobin levels and dementia risk have been limited.
A population-based cohort study
To examine the long-term association of hemoglobin levels and anemia with risk of dementia, Dr. Ikram and coauthors analyzed data from the Rotterdam Study, an ongoing population-based cohort study in the Netherlands that started in 1990. Their analysis included data from 12,305 participants without dementia who had serum hemoglobin measured at baseline (mean age, 64.6 years; 57.7% women).
During a mean follow-up of 12.1 years, 1,520 participants developed dementia, 1,194 of whom had Alzheimer’s disease.
“Both low and high hemoglobin levels were associated with increased dementia risk,” the authors wrote. Compared with participants in the middle quintile of hemoglobin levels (8.57-8.99 mmol/L), participants in the lowest quintile (less than 8.11 mmol/L) had a hazard ratio of dementia of 1.29, and participants in the highest quintile (greater than 9.40 mmol/L) had an HR of 1.20.
About 6% of the participants had anemia – that is, a hemoglobin level of less than 8.1 mmol/L for men and less than 7.5 mmol/L for women. Anemia was associated with a 34% increased risk of dementia and a 41% increased risk of Alzheimer’s disease.
Of the 745 people with anemia, 128 developed dementia, compared with 1,392 of the 11,560 people who did not have anemia (17% vs. 12%).
A U-shaped association
The researchers also examined hemoglobin in relation to vascular brain disease, structural connectivity, and global cerebral perfusion among 5,267 participants without dementia who had brain MRI. White matter hyperintensity volume and hemoglobin had a U-shaped association, similar to that for dementia and hemoglobin. In addition, hemoglobin inversely correlated to cerebral perfusion.
The results remained consistent after adjustment for factors such as smoking, high blood pressure, high cholesterol, and alcohol use.
A limitation of the study is that the participants lived in the Netherlands and were primarily of European descent, so the results may not apply to other populations, the authors wrote.
Dr. Ikram noted that the study does not prove that low or high hemoglobin levels cause dementia. “More research is needed to determine whether hemoglobin levels play a direct role in this increased risk or whether these associations can be explained by underlying issues or other vascular or metabolic changes.”
The study was supported by the Netherlands Cardiovascular Research Initiative; Erasmus Medical Centre; Erasmus University Rotterdam; Netherlands Organization for Scientific Research; Netherlands Organization for Health Research and Development; Research Institute for Diseases in the Elderly; Netherlands Genomic Initiative; Dutch Ministry of Education, Culture, and Science; Dutch Ministry of Health, Welfare, and Sports; European Commission; Municipality of Rotterdam; Netherlands Consortium for Healthy Aging; and Dutch Heart Foundation. The authors reported no relevant disclosures.
SOURCE: Ikram MA et al. Neurology. 2019 Jul 31. doi: 10.1212/WNL.0000000000008003.
This U-shaped association “may relate to differences in white matter integrity and cerebral perfusion,” the researchers wrote in Neurology.
“With around 10% of people over age 65 having anemia in the Americas and Europe and up to 45% in African and southeast Asian countries, these results could have important implications for the burden of dementia,” said study author M. Arfan Ikram, MD, PhD, in a news release. Dr. Ikram is a professor of epidemiology at Erasmus Medical Center in Rotterdam, the Netherlands.
Prior studies have found that low hemoglobin levels are associated with adverse health outcomes, such as coronary heart disease, stroke, and mortality, but data about the relationship between hemoglobin levels and dementia risk have been limited.
A population-based cohort study
To examine the long-term association of hemoglobin levels and anemia with risk of dementia, Dr. Ikram and coauthors analyzed data from the Rotterdam Study, an ongoing population-based cohort study in the Netherlands that started in 1990. Their analysis included data from 12,305 participants without dementia who had serum hemoglobin measured at baseline (mean age, 64.6 years; 57.7% women).
During a mean follow-up of 12.1 years, 1,520 participants developed dementia, 1,194 of whom had Alzheimer’s disease.
“Both low and high hemoglobin levels were associated with increased dementia risk,” the authors wrote. Compared with participants in the middle quintile of hemoglobin levels (8.57-8.99 mmol/L), participants in the lowest quintile (less than 8.11 mmol/L) had a hazard ratio of dementia of 1.29, and participants in the highest quintile (greater than 9.40 mmol/L) had an HR of 1.20.
About 6% of the participants had anemia – that is, a hemoglobin level of less than 8.1 mmol/L for men and less than 7.5 mmol/L for women. Anemia was associated with a 34% increased risk of dementia and a 41% increased risk of Alzheimer’s disease.
Of the 745 people with anemia, 128 developed dementia, compared with 1,392 of the 11,560 people who did not have anemia (17% vs. 12%).
A U-shaped association
The researchers also examined hemoglobin in relation to vascular brain disease, structural connectivity, and global cerebral perfusion among 5,267 participants without dementia who had brain MRI. White matter hyperintensity volume and hemoglobin had a U-shaped association, similar to that for dementia and hemoglobin. In addition, hemoglobin inversely correlated to cerebral perfusion.
The results remained consistent after adjustment for factors such as smoking, high blood pressure, high cholesterol, and alcohol use.
A limitation of the study is that the participants lived in the Netherlands and were primarily of European descent, so the results may not apply to other populations, the authors wrote.
Dr. Ikram noted that the study does not prove that low or high hemoglobin levels cause dementia. “More research is needed to determine whether hemoglobin levels play a direct role in this increased risk or whether these associations can be explained by underlying issues or other vascular or metabolic changes.”
The study was supported by the Netherlands Cardiovascular Research Initiative; Erasmus Medical Centre; Erasmus University Rotterdam; Netherlands Organization for Scientific Research; Netherlands Organization for Health Research and Development; Research Institute for Diseases in the Elderly; Netherlands Genomic Initiative; Dutch Ministry of Education, Culture, and Science; Dutch Ministry of Health, Welfare, and Sports; European Commission; Municipality of Rotterdam; Netherlands Consortium for Healthy Aging; and Dutch Heart Foundation. The authors reported no relevant disclosures.
SOURCE: Ikram MA et al. Neurology. 2019 Jul 31. doi: 10.1212/WNL.0000000000008003.
FROM NEUROLOGY
Key clinical point: Adults with low levels of hemoglobin and adults with high levels of hemoglobin may have an increased risk of dementia.
Major finding: Compared with participants in the middle quintile of hemoglobin levels (8.57-8.99 mmol/L), participants in the lowest quintile (less than 8.11 mmol/L) had a hazard ratio of dementia of 1.29, and participants in the highest quintile (greater than 9.40 mmol/L) had an HR of 1.20.
Study details: An analysis of data from 12,305 participants in the Rotterdam Study, a population-based cohort study in the Netherlands, who were followed up for an average of 12 years.
Disclosures: The study was supported by the Netherlands Cardiovascular Research Initiative; Erasmus Medical Centre; Erasmus University Rotterdam; Netherlands Organization for Scientific Research; Netherlands Organization for Health Research and Development; Research Institute for Diseases in the Elderly; Netherlands Genomic Initiative; Dutch Ministry of Education, Culture, and Science; Dutch Ministry of Health, Welfare, and Sports; European Commission; Municipality of Rotterdam; Netherlands Consortium for Healthy Aging; and Dutch Heart Foundation. The authors reported no relevant disclosures.
Source: Ikram MA et al. Neurology. 2019 Jul 31. doi: 10.1212/WNL.0000000000008003.
NIH launches 5-year, $10 million study on acute flaccid myelitis
Researchers at the University of Alabama at Birmingham will lead a 5-year, federally-funded study of acute flaccid myelitis (AFM) – a rare pediatric neurologic disease.

The National Institute of Allergy and Infectious Diseases (NIAID) awarded the $10 million grant to primary investigator David Kimberlin, MD, a UAB professor of pediatrics. Carlos Pardo-Villamizar, MD, professor of neurology and pathology at Johns Hopkins University, Baltimore, is the co-principal investigator.
The university will organize and implement the international, multisite study. Its primary goal is to examine the incidence and distribution of AFM, and its pathogenesis and progression. Enrollment is expected to commence next fall. Investigators will enroll children with symptoms of AFM and follow them for 1 year. Household contacts of the subjects will serve as comparators.
In addition to collecting data about risk factors and disease progression, the researchers will collect clinical specimens, including blood and cerebrospinal fluid. More details about the design and study sites will be released then, according to a press statement issued by NIAID.
AFM targets spinal nerves and often develops after a mild respiratory illness. The disease mounted a global epidemic comeback in 2014, primarily affecting children; it has occurred concurrently with enterovirus outbreaks.
“Growing epidemiological evidence suggests that enterovirus-D68 [EV-D68] could play a role,” the statement noted. “Most people who become infected with EV-D68 are asymptomatic or experience mild, cold-like symptoms. Researchers and physicians are working to understand if there is a connection between these viral outbreaks and AFM, and if so, why some children but not others experience this sudden muscle weakness and paralysis.”
The study will draw on the expertise of the AFM Task Force, established last fall. The group comprises physicians, scientists, and public health experts from diverse disciplines and institutions who will assist in the ongoing investigation.
The AFM natural history study is funded under contract HHSN272201600018C.
Researchers at the University of Alabama at Birmingham will lead a 5-year, federally-funded study of acute flaccid myelitis (AFM) – a rare pediatric neurologic disease.
The National Institute of Allergy and Infectious Diseases (NIAID) awarded the $10 million grant to primary investigator David Kimberlin, MD, a UAB professor of pediatrics. Carlos Pardo-Villamizar, MD, professor of neurology and pathology at Johns Hopkins University, Baltimore, is the co-principal investigator.
The university will organize and implement the international, multisite study. Its primary goal is to examine the incidence and distribution of AFM, and its pathogenesis and progression. Enrollment is expected to commence next fall. Investigators will enroll children with symptoms of AFM and follow them for 1 year. Household contacts of the subjects will serve as comparators.
In addition to collecting data about risk factors and disease progression, the researchers will collect clinical specimens, including blood and cerebrospinal fluid. More details about the design and study sites will be released then, according to a press statement issued by NIAID.
AFM targets spinal nerves and often develops after a mild respiratory illness. The disease mounted a global epidemic comeback in 2014, primarily affecting children; it has occurred concurrently with enterovirus outbreaks.
“Growing epidemiological evidence suggests that enterovirus-D68 [EV-D68] could play a role,” the statement noted. “Most people who become infected with EV-D68 are asymptomatic or experience mild, cold-like symptoms. Researchers and physicians are working to understand if there is a connection between these viral outbreaks and AFM, and if so, why some children but not others experience this sudden muscle weakness and paralysis.”
The study will draw on the expertise of the AFM Task Force, established last fall. The group comprises physicians, scientists, and public health experts from diverse disciplines and institutions who will assist in the ongoing investigation.
The AFM natural history study is funded under contract HHSN272201600018C.
Researchers at the University of Alabama at Birmingham will lead a 5-year, federally-funded study of acute flaccid myelitis (AFM) – a rare pediatric neurologic disease.
The National Institute of Allergy and Infectious Diseases (NIAID) awarded the $10 million grant to primary investigator David Kimberlin, MD, a UAB professor of pediatrics. Carlos Pardo-Villamizar, MD, professor of neurology and pathology at Johns Hopkins University, Baltimore, is the co-principal investigator.
The university will organize and implement the international, multisite study. Its primary goal is to examine the incidence and distribution of AFM, and its pathogenesis and progression. Enrollment is expected to commence next fall. Investigators will enroll children with symptoms of AFM and follow them for 1 year. Household contacts of the subjects will serve as comparators.
In addition to collecting data about risk factors and disease progression, the researchers will collect clinical specimens, including blood and cerebrospinal fluid. More details about the design and study sites will be released then, according to a press statement issued by NIAID.
AFM targets spinal nerves and often develops after a mild respiratory illness. The disease mounted a global epidemic comeback in 2014, primarily affecting children; it has occurred concurrently with enterovirus outbreaks.
“Growing epidemiological evidence suggests that enterovirus-D68 [EV-D68] could play a role,” the statement noted. “Most people who become infected with EV-D68 are asymptomatic or experience mild, cold-like symptoms. Researchers and physicians are working to understand if there is a connection between these viral outbreaks and AFM, and if so, why some children but not others experience this sudden muscle weakness and paralysis.”
The study will draw on the expertise of the AFM Task Force, established last fall. The group comprises physicians, scientists, and public health experts from diverse disciplines and institutions who will assist in the ongoing investigation.
The AFM natural history study is funded under contract HHSN272201600018C.
Summary: American Medical Society for Sports Medicine position statement on concussion in sport
An estimated 1-1.8 million sport-related concussions (SRC) occur per year in patients younger than 18 years of age. Concussion is defined as “a traumatically induced transient disturbance of brain function.” More than 50% of concussions among high school youth are not related to organized sports and between 2% and 15% of athletes in organized sports will sustain a concussion during a season of play.

which will be described in this article. The guidelines include recommendations for imaging, treatment, and decision making regarding when as well as whether to return to play. Here is a brief summary of those recommendations.
Preseason: Preseason evaluation includes a preparticipation physical evaluation and discussion of concussion history as well as risk factors associated with prolonged concussion recovery. Neurocognitive tests are available for baseline evaluation. While these may assist with diagnosis and return-to-play decisions, there can be considerable variation in an individual’s baseline score as well as the possibility of changes in that baseline over time. Because of this potential for variability, these tests are not required or accepted as the standard of care.

Sideline assessment: Familiarity with the athlete is the best way to detect subtle changes in personality or performance. Looking at symptoms is still the most sensitive way to diagnose a concussion. Loss of consciousness, seizure, tonic posturing, lack of motor coordination, confusion, amnesia, difficulty with balance, or any cognitive difficulty should prompt removal from play for possible concussion. Once a potential injury is identified, how the athlete responds to the elements of orientation, memory, concentration, speech pattern, and balance should be evaluated. If an athlete has a probable or definite concussion, the athlete needs to be removed from play and cannot return to same-day play, and a more detailed evaluation needs to be done.

Office assessment: It is not unusual for symptoms and testing to normalize by the time an office visit occurs. If this is the case, the visit should focus on recommendations for safe return to school and sport. A standard office evaluation should include taking a history with details of the mechanism of injury and preexisting conditions – such as depression and prior concussion – that can affect concussion recovery. The history should focus on detecting symptoms that typically cause impairment from concussion: headache, ocular-vestibular issues leading to problems with balance, and cognitive issues with difficulty concentrating and remembering, as well as fatigue and mood issues such as anxiety, irritability, and depression. The physical exam should include assessment of ocular and vestibular function, gait, and balance in addition to a neurological exam.
Imaging: Head CT or MRI are rarely indicated. Intracranial bleeds are rare in the context of SRC but can occur. If there is concern for a bleed, then CT scan is the imaging test of choice. MRI may have value for evaluation for atypical or prolonged recovery.
Recovery time: The large majority (80%-90%) of concussed older adolescents and adults return to preinjury levels of function within 2 weeks; in younger athletes, clinical recovery may take up to 4 weeks. The best predictor of recovery from SRC is the number and severity of symptoms.
Treatment: For decades, cognitive and physical rest has been the standard of treatment. However, this is no longer the “gold standard” as it has been shown that strict rest (“cocoon therapy”) after SRC slows recovery and leads to an increased chance of prolonged symptoms. Current consensus guidelines support 24-48 hours of symptom-limited rest, both cognitive and physical, followed by a gradual increase in activity, staying below symptom-exacerbation thresholds. Activity, along with good sleep hygiene, appears to be helpful in facilitating recovery from SRC. In athletes with persistent post concussive symptoms that continue beyond the expected recovery time frame, activities of daily living, school, and exercise that do not significantly exacerbate symptoms are recommended.
Return to learning/play: A concussion can cause temporary deficits in attention, cognitive processing, short-term memory, and executive functioning. School personnel should be informed of the injury and assist in employing an individualized return to learn plan, including academic accommodations. Ultimately, return to sports activities should follow a successful return to the classroom. Return to play involves a stepwise increase in physical demands/activity without symptoms before a student is allowed to participate in full contact play.
Concussion-related risks: Continuing to participate in sports before resolution of concussion can worsen and prolong symptoms of SRC. Returning too early after concussion, before full recovery, increases the risk of recurrent SRC. During the initial post-injury period, returning to sports too early increases the risk for a rare but devastating possibility of second impact syndrome that can be a life-threatening repeat head injury. Studies of long-term mental health diagnoses are conflicting and inconsistent. Chronic traumatic encephalopathy has been described in athletes with a long history of concussions and repetitive sub-symptom head impacts. The degree of exposure needed appears to be variable and dependent on the individual.
Disqualification from play: Because each athlete is individually assessed after SRC, there are no evidence-based studies indicating how many concussions are “safe” for an athlete to have in a lifetime. The decision to stop playing sports is both serious and difficult for most athletes and requires shared decision making between clinician, the athlete, and the athlete’s parents. Factors to consider when determining if disqualification from play is warranted include:
- The total number of concussions experienced by a patient.
- Whether a patient has sustained subsequent concussions with progressively less forceful blows to the head.
- If a patient has sustained multiple concussions,whether the time to complete a full recovery after each concussion event increased.
The bottom line: “Cocoon therapy” is no longer recommended. Consensus guidelines endorse 24-48 hours of symptom-limited cognitive and physical rest followed by a gradual increase in activity, including noncontact physical activity that does not provoke symptoms.
Dr. Belogorodsky is a second-year resident and Dr. Fidler is an associate director in the Family Medicine Residency Program at Abington (Pa.) Jefferson Health. Dr. Skolnik is professor of family and community medicine at Jefferson Medical College, Philadelphia, and an associate director of the family medicine residency program at Abington Jefferson Health.
Reference
Harmon KG et al. American Medical Society for Sports Medicine position statement on concussion in sport. Br J Sports Med. 2019;53:213-25.
An estimated 1-1.8 million sport-related concussions (SRC) occur per year in patients younger than 18 years of age. Concussion is defined as “a traumatically induced transient disturbance of brain function.” More than 50% of concussions among high school youth are not related to organized sports and between 2% and 15% of athletes in organized sports will sustain a concussion during a season of play.
which will be described in this article. The guidelines include recommendations for imaging, treatment, and decision making regarding when as well as whether to return to play. Here is a brief summary of those recommendations.
Preseason: Preseason evaluation includes a preparticipation physical evaluation and discussion of concussion history as well as risk factors associated with prolonged concussion recovery. Neurocognitive tests are available for baseline evaluation. While these may assist with diagnosis and return-to-play decisions, there can be considerable variation in an individual’s baseline score as well as the possibility of changes in that baseline over time. Because of this potential for variability, these tests are not required or accepted as the standard of care.
Sideline assessment: Familiarity with the athlete is the best way to detect subtle changes in personality or performance. Looking at symptoms is still the most sensitive way to diagnose a concussion. Loss of consciousness, seizure, tonic posturing, lack of motor coordination, confusion, amnesia, difficulty with balance, or any cognitive difficulty should prompt removal from play for possible concussion. Once a potential injury is identified, how the athlete responds to the elements of orientation, memory, concentration, speech pattern, and balance should be evaluated. If an athlete has a probable or definite concussion, the athlete needs to be removed from play and cannot return to same-day play, and a more detailed evaluation needs to be done.
Office assessment: It is not unusual for symptoms and testing to normalize by the time an office visit occurs. If this is the case, the visit should focus on recommendations for safe return to school and sport. A standard office evaluation should include taking a history with details of the mechanism of injury and preexisting conditions – such as depression and prior concussion – that can affect concussion recovery. The history should focus on detecting symptoms that typically cause impairment from concussion: headache, ocular-vestibular issues leading to problems with balance, and cognitive issues with difficulty concentrating and remembering, as well as fatigue and mood issues such as anxiety, irritability, and depression. The physical exam should include assessment of ocular and vestibular function, gait, and balance in addition to a neurological exam.
Imaging: Head CT or MRI are rarely indicated. Intracranial bleeds are rare in the context of SRC but can occur. If there is concern for a bleed, then CT scan is the imaging test of choice. MRI may have value for evaluation for atypical or prolonged recovery.
Recovery time: The large majority (80%-90%) of concussed older adolescents and adults return to preinjury levels of function within 2 weeks; in younger athletes, clinical recovery may take up to 4 weeks. The best predictor of recovery from SRC is the number and severity of symptoms.
Treatment: For decades, cognitive and physical rest has been the standard of treatment. However, this is no longer the “gold standard” as it has been shown that strict rest (“cocoon therapy”) after SRC slows recovery and leads to an increased chance of prolonged symptoms. Current consensus guidelines support 24-48 hours of symptom-limited rest, both cognitive and physical, followed by a gradual increase in activity, staying below symptom-exacerbation thresholds. Activity, along with good sleep hygiene, appears to be helpful in facilitating recovery from SRC. In athletes with persistent post concussive symptoms that continue beyond the expected recovery time frame, activities of daily living, school, and exercise that do not significantly exacerbate symptoms are recommended.
Return to learning/play: A concussion can cause temporary deficits in attention, cognitive processing, short-term memory, and executive functioning. School personnel should be informed of the injury and assist in employing an individualized return to learn plan, including academic accommodations. Ultimately, return to sports activities should follow a successful return to the classroom. Return to play involves a stepwise increase in physical demands/activity without symptoms before a student is allowed to participate in full contact play.
Concussion-related risks: Continuing to participate in sports before resolution of concussion can worsen and prolong symptoms of SRC. Returning too early after concussion, before full recovery, increases the risk of recurrent SRC. During the initial post-injury period, returning to sports too early increases the risk for a rare but devastating possibility of second impact syndrome that can be a life-threatening repeat head injury. Studies of long-term mental health diagnoses are conflicting and inconsistent. Chronic traumatic encephalopathy has been described in athletes with a long history of concussions and repetitive sub-symptom head impacts. The degree of exposure needed appears to be variable and dependent on the individual.
Disqualification from play: Because each athlete is individually assessed after SRC, there are no evidence-based studies indicating how many concussions are “safe” for an athlete to have in a lifetime. The decision to stop playing sports is both serious and difficult for most athletes and requires shared decision making between clinician, the athlete, and the athlete’s parents. Factors to consider when determining if disqualification from play is warranted include:
- The total number of concussions experienced by a patient.
- Whether a patient has sustained subsequent concussions with progressively less forceful blows to the head.
- If a patient has sustained multiple concussions,whether the time to complete a full recovery after each concussion event increased.
The bottom line: “Cocoon therapy” is no longer recommended. Consensus guidelines endorse 24-48 hours of symptom-limited cognitive and physical rest followed by a gradual increase in activity, including noncontact physical activity that does not provoke symptoms.
Dr. Belogorodsky is a second-year resident and Dr. Fidler is an associate director in the Family Medicine Residency Program at Abington (Pa.) Jefferson Health. Dr. Skolnik is professor of family and community medicine at Jefferson Medical College, Philadelphia, and an associate director of the family medicine residency program at Abington Jefferson Health.
Reference
Harmon KG et al. American Medical Society for Sports Medicine position statement on concussion in sport. Br J Sports Med. 2019;53:213-25.
An estimated 1-1.8 million sport-related concussions (SRC) occur per year in patients younger than 18 years of age. Concussion is defined as “a traumatically induced transient disturbance of brain function.” More than 50% of concussions among high school youth are not related to organized sports and between 2% and 15% of athletes in organized sports will sustain a concussion during a season of play.
which will be described in this article. The guidelines include recommendations for imaging, treatment, and decision making regarding when as well as whether to return to play. Here is a brief summary of those recommendations.
Preseason: Preseason evaluation includes a preparticipation physical evaluation and discussion of concussion history as well as risk factors associated with prolonged concussion recovery. Neurocognitive tests are available for baseline evaluation. While these may assist with diagnosis and return-to-play decisions, there can be considerable variation in an individual’s baseline score as well as the possibility of changes in that baseline over time. Because of this potential for variability, these tests are not required or accepted as the standard of care.
Sideline assessment: Familiarity with the athlete is the best way to detect subtle changes in personality or performance. Looking at symptoms is still the most sensitive way to diagnose a concussion. Loss of consciousness, seizure, tonic posturing, lack of motor coordination, confusion, amnesia, difficulty with balance, or any cognitive difficulty should prompt removal from play for possible concussion. Once a potential injury is identified, how the athlete responds to the elements of orientation, memory, concentration, speech pattern, and balance should be evaluated. If an athlete has a probable or definite concussion, the athlete needs to be removed from play and cannot return to same-day play, and a more detailed evaluation needs to be done.
Office assessment: It is not unusual for symptoms and testing to normalize by the time an office visit occurs. If this is the case, the visit should focus on recommendations for safe return to school and sport. A standard office evaluation should include taking a history with details of the mechanism of injury and preexisting conditions – such as depression and prior concussion – that can affect concussion recovery. The history should focus on detecting symptoms that typically cause impairment from concussion: headache, ocular-vestibular issues leading to problems with balance, and cognitive issues with difficulty concentrating and remembering, as well as fatigue and mood issues such as anxiety, irritability, and depression. The physical exam should include assessment of ocular and vestibular function, gait, and balance in addition to a neurological exam.
Imaging: Head CT or MRI are rarely indicated. Intracranial bleeds are rare in the context of SRC but can occur. If there is concern for a bleed, then CT scan is the imaging test of choice. MRI may have value for evaluation for atypical or prolonged recovery.
Recovery time: The large majority (80%-90%) of concussed older adolescents and adults return to preinjury levels of function within 2 weeks; in younger athletes, clinical recovery may take up to 4 weeks. The best predictor of recovery from SRC is the number and severity of symptoms.
Treatment: For decades, cognitive and physical rest has been the standard of treatment. However, this is no longer the “gold standard” as it has been shown that strict rest (“cocoon therapy”) after SRC slows recovery and leads to an increased chance of prolonged symptoms. Current consensus guidelines support 24-48 hours of symptom-limited rest, both cognitive and physical, followed by a gradual increase in activity, staying below symptom-exacerbation thresholds. Activity, along with good sleep hygiene, appears to be helpful in facilitating recovery from SRC. In athletes with persistent post concussive symptoms that continue beyond the expected recovery time frame, activities of daily living, school, and exercise that do not significantly exacerbate symptoms are recommended.
Return to learning/play: A concussion can cause temporary deficits in attention, cognitive processing, short-term memory, and executive functioning. School personnel should be informed of the injury and assist in employing an individualized return to learn plan, including academic accommodations. Ultimately, return to sports activities should follow a successful return to the classroom. Return to play involves a stepwise increase in physical demands/activity without symptoms before a student is allowed to participate in full contact play.
Concussion-related risks: Continuing to participate in sports before resolution of concussion can worsen and prolong symptoms of SRC. Returning too early after concussion, before full recovery, increases the risk of recurrent SRC. During the initial post-injury period, returning to sports too early increases the risk for a rare but devastating possibility of second impact syndrome that can be a life-threatening repeat head injury. Studies of long-term mental health diagnoses are conflicting and inconsistent. Chronic traumatic encephalopathy has been described in athletes with a long history of concussions and repetitive sub-symptom head impacts. The degree of exposure needed appears to be variable and dependent on the individual.
Disqualification from play: Because each athlete is individually assessed after SRC, there are no evidence-based studies indicating how many concussions are “safe” for an athlete to have in a lifetime. The decision to stop playing sports is both serious and difficult for most athletes and requires shared decision making between clinician, the athlete, and the athlete’s parents. Factors to consider when determining if disqualification from play is warranted include:
- The total number of concussions experienced by a patient.
- Whether a patient has sustained subsequent concussions with progressively less forceful blows to the head.
- If a patient has sustained multiple concussions,whether the time to complete a full recovery after each concussion event increased.
The bottom line: “Cocoon therapy” is no longer recommended. Consensus guidelines endorse 24-48 hours of symptom-limited cognitive and physical rest followed by a gradual increase in activity, including noncontact physical activity that does not provoke symptoms.
Dr. Belogorodsky is a second-year resident and Dr. Fidler is an associate director in the Family Medicine Residency Program at Abington (Pa.) Jefferson Health. Dr. Skolnik is professor of family and community medicine at Jefferson Medical College, Philadelphia, and an associate director of the family medicine residency program at Abington Jefferson Health.
Reference
Harmon KG et al. American Medical Society for Sports Medicine position statement on concussion in sport. Br J Sports Med. 2019;53:213-25.