User login
Laser Best Practices for Darker Skin Types

What does your patient need to know at the first visit? Does it apply to patients of all genders and ages?
Before performing laser procedures on patients with richly pigmented skin (Fitzpatrick skin types IV–VI), patients need to be informed of the higher risk for pigmentary alterations as potential complications of the procedure. Specifically, hyperpigmentation or hypopigmentation can occur postprocedure, depending on the type of device used, the treatment settings, the technique, the underlying skin disorder being treated, and the patient’s individual response to treatment. Fortunately, these pigment alterations are in most cases self-limited but can last for weeks to months depending on the severity and the nature of the dyspigmentation.
What are your go-to treatments? What are the side effects?
Notwithstanding the higher risks for pigmentary alterations, lasers can be extremely useful for the management of numerous dermatologic concerns in patients with Fitzpatrick skin types IV to VI including laser hair removal for pseudofolliculitis barbae or nonablative fractional laser resurfacing for acne scarring and pigmentary disorders. My go-to treatments include the following: long-pulsed 1064-nm Nd:YAG laser for hair removal in Fitzpatrick skin types V to VI, 808-nm diode laser with linear scanning of hair removal in Fitzpatrick skin type IV or less, 1550-nm erbium-doped nonablative fractional laser for acne scarring in Fitzpatrick skin types IV to VI, and low-power diode 1927-nm fractional laser for melasma and postinflammatory hyperpigmentation in Fitzpatrick skin types IV to VI.
All of these procedures are performed with conservative treatment settings such as low fluences and longer pulse durations for laser hair removal and low treatment densities for fractional laser procedures. Prior to laser resurfacing, I recommend hydroquinone cream 4% twice daily starting 2 weeks before the first session and for 4 weeks posttreatment. These recommendations are based on published evidence (see Suggested Readings) as well as anecdotal experience.
How do you keep patients compliant with treatment?
Emphasizing the need for broad-spectrum sunscreen and avoidance of intense sun exposure before and after laser treatments is important during the initial consultation and prior to each treatment. I warn my patients of the higher risk for hyperpigmentation if the skin is tanned or has recently had intense sun exposure.
What do you do if they refuse treatment?
If patients refuse laser treatment or recommended precautions, then I will consider nonlaser treatment options.
What resources do you recommend to patients for more information?
I recommend patients visit the Skin of Color Society website (www.skinofcolorsociety.org).
Suggested Readings
- Alexis AF. Fractional laser resurfacing of acne scarring in patients with Fitzpatrick skin types IV-VI. J Drugs Dermatol. 2011;10(12 suppl):s6-s7.
- Alexis AF. Lasers and light-based therapies in ethnic skin: treatment options and recommendations for Fitzpatrick skin types V and VI. Br J Dermatol. 2013;169(suppl 3):91-97.
- Alexis AF, Coley MK, Nijhawan RI, et al. Nonablative fractional laser resurfacing for acne scarring in patients with Fitzpatrick skin phototypes IV-VI. Dermatol Surg. 2016;42:392-402.
- Battle EF, Hobbs LM. Laser-assisted hair removal for darker skin types. Dermatol Ther. 2004;17:177-183.
- Clark CM, Silverberg JI, Alexis AF. A retrospective chart review to assess the safety of nonablative fractional laser resurfacing in Fitzpatrick skin types IV to VI. J Drugs Dermatol. 2013;12:428-431.
- Ross EV, Cooke LM, Timko AL, et al. Treatment of pseudofolliculitis barbae in skin types IV, V, and VI with a long-pulsed neodymium:yttrium aluminum garnet laser. J Am Acad Dermatol. 2002;47:263-270.
What does your patient need to know at the first visit? Does it apply to patients of all genders and ages?
Before performing laser procedures on patients with richly pigmented skin (Fitzpatrick skin types IV–VI), patients need to be informed of the higher risk for pigmentary alterations as potential complications of the procedure. Specifically, hyperpigmentation or hypopigmentation can occur postprocedure, depending on the type of device used, the treatment settings, the technique, the underlying skin disorder being treated, and the patient’s individual response to treatment. Fortunately, these pigment alterations are in most cases self-limited but can last for weeks to months depending on the severity and the nature of the dyspigmentation.
What are your go-to treatments? What are the side effects?
Notwithstanding the higher risks for pigmentary alterations, lasers can be extremely useful for the management of numerous dermatologic concerns in patients with Fitzpatrick skin types IV to VI including laser hair removal for pseudofolliculitis barbae or nonablative fractional laser resurfacing for acne scarring and pigmentary disorders. My go-to treatments include the following: long-pulsed 1064-nm Nd:YAG laser for hair removal in Fitzpatrick skin types V to VI, 808-nm diode laser with linear scanning of hair removal in Fitzpatrick skin type IV or less, 1550-nm erbium-doped nonablative fractional laser for acne scarring in Fitzpatrick skin types IV to VI, and low-power diode 1927-nm fractional laser for melasma and postinflammatory hyperpigmentation in Fitzpatrick skin types IV to VI.
All of these procedures are performed with conservative treatment settings such as low fluences and longer pulse durations for laser hair removal and low treatment densities for fractional laser procedures. Prior to laser resurfacing, I recommend hydroquinone cream 4% twice daily starting 2 weeks before the first session and for 4 weeks posttreatment. These recommendations are based on published evidence (see Suggested Readings) as well as anecdotal experience.
How do you keep patients compliant with treatment?
Emphasizing the need for broad-spectrum sunscreen and avoidance of intense sun exposure before and after laser treatments is important during the initial consultation and prior to each treatment. I warn my patients of the higher risk for hyperpigmentation if the skin is tanned or has recently had intense sun exposure.
What do you do if they refuse treatment?
If patients refuse laser treatment or recommended precautions, then I will consider nonlaser treatment options.
What resources do you recommend to patients for more information?
I recommend patients visit the Skin of Color Society website (www.skinofcolorsociety.org).
Suggested Readings
- Alexis AF. Fractional laser resurfacing of acne scarring in patients with Fitzpatrick skin types IV-VI. J Drugs Dermatol. 2011;10(12 suppl):s6-s7.
- Alexis AF. Lasers and light-based therapies in ethnic skin: treatment options and recommendations for Fitzpatrick skin types V and VI. Br J Dermatol. 2013;169(suppl 3):91-97.
- Alexis AF, Coley MK, Nijhawan RI, et al. Nonablative fractional laser resurfacing for acne scarring in patients with Fitzpatrick skin phototypes IV-VI. Dermatol Surg. 2016;42:392-402.
- Battle EF, Hobbs LM. Laser-assisted hair removal for darker skin types. Dermatol Ther. 2004;17:177-183.
- Clark CM, Silverberg JI, Alexis AF. A retrospective chart review to assess the safety of nonablative fractional laser resurfacing in Fitzpatrick skin types IV to VI. J Drugs Dermatol. 2013;12:428-431.
- Ross EV, Cooke LM, Timko AL, et al. Treatment of pseudofolliculitis barbae in skin types IV, V, and VI with a long-pulsed neodymium:yttrium aluminum garnet laser. J Am Acad Dermatol. 2002;47:263-270.
What does your patient need to know at the first visit? Does it apply to patients of all genders and ages?
Before performing laser procedures on patients with richly pigmented skin (Fitzpatrick skin types IV–VI), patients need to be informed of the higher risk for pigmentary alterations as potential complications of the procedure. Specifically, hyperpigmentation or hypopigmentation can occur postprocedure, depending on the type of device used, the treatment settings, the technique, the underlying skin disorder being treated, and the patient’s individual response to treatment. Fortunately, these pigment alterations are in most cases self-limited but can last for weeks to months depending on the severity and the nature of the dyspigmentation.
What are your go-to treatments? What are the side effects?
Notwithstanding the higher risks for pigmentary alterations, lasers can be extremely useful for the management of numerous dermatologic concerns in patients with Fitzpatrick skin types IV to VI including laser hair removal for pseudofolliculitis barbae or nonablative fractional laser resurfacing for acne scarring and pigmentary disorders. My go-to treatments include the following: long-pulsed 1064-nm Nd:YAG laser for hair removal in Fitzpatrick skin types V to VI, 808-nm diode laser with linear scanning of hair removal in Fitzpatrick skin type IV or less, 1550-nm erbium-doped nonablative fractional laser for acne scarring in Fitzpatrick skin types IV to VI, and low-power diode 1927-nm fractional laser for melasma and postinflammatory hyperpigmentation in Fitzpatrick skin types IV to VI.
All of these procedures are performed with conservative treatment settings such as low fluences and longer pulse durations for laser hair removal and low treatment densities for fractional laser procedures. Prior to laser resurfacing, I recommend hydroquinone cream 4% twice daily starting 2 weeks before the first session and for 4 weeks posttreatment. These recommendations are based on published evidence (see Suggested Readings) as well as anecdotal experience.
How do you keep patients compliant with treatment?
Emphasizing the need for broad-spectrum sunscreen and avoidance of intense sun exposure before and after laser treatments is important during the initial consultation and prior to each treatment. I warn my patients of the higher risk for hyperpigmentation if the skin is tanned or has recently had intense sun exposure.
What do you do if they refuse treatment?
If patients refuse laser treatment or recommended precautions, then I will consider nonlaser treatment options.
What resources do you recommend to patients for more information?
I recommend patients visit the Skin of Color Society website (www.skinofcolorsociety.org).
Suggested Readings
- Alexis AF. Fractional laser resurfacing of acne scarring in patients with Fitzpatrick skin types IV-VI. J Drugs Dermatol. 2011;10(12 suppl):s6-s7.
- Alexis AF. Lasers and light-based therapies in ethnic skin: treatment options and recommendations for Fitzpatrick skin types V and VI. Br J Dermatol. 2013;169(suppl 3):91-97.
- Alexis AF, Coley MK, Nijhawan RI, et al. Nonablative fractional laser resurfacing for acne scarring in patients with Fitzpatrick skin phototypes IV-VI. Dermatol Surg. 2016;42:392-402.
- Battle EF, Hobbs LM. Laser-assisted hair removal for darker skin types. Dermatol Ther. 2004;17:177-183.
- Clark CM, Silverberg JI, Alexis AF. A retrospective chart review to assess the safety of nonablative fractional laser resurfacing in Fitzpatrick skin types IV to VI. J Drugs Dermatol. 2013;12:428-431.
- Ross EV, Cooke LM, Timko AL, et al. Treatment of pseudofolliculitis barbae in skin types IV, V, and VI with a long-pulsed neodymium:yttrium aluminum garnet laser. J Am Acad Dermatol. 2002;47:263-270.

Exophytic Scalp Tumor
The Diagnosis: Primary Cutaneous Carcinosarcoma
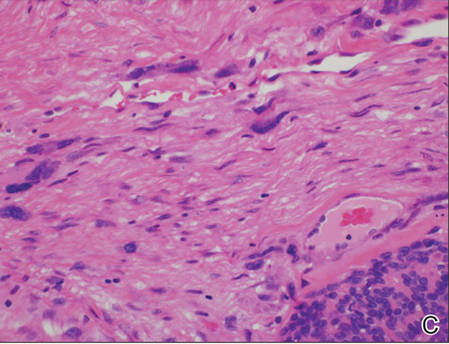
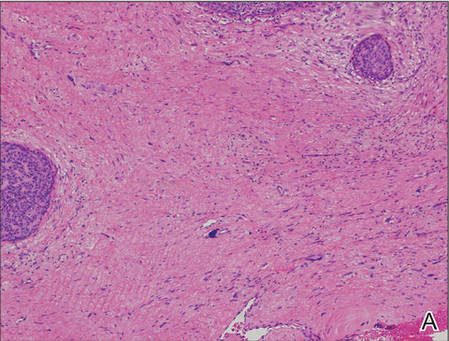
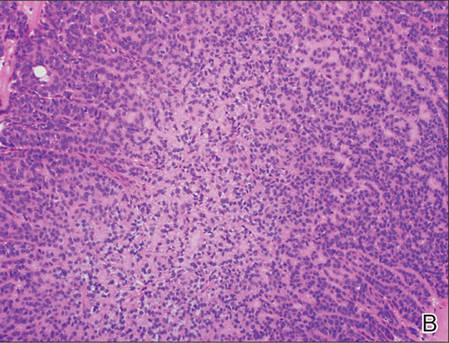
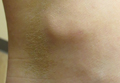
A generous shave biopsy and debulking performed on the initial visit revealed an infiltrating tumor consisting of malignant epithelial and stromal components (Figure). The basaloid and squamoid epithelial cells were keratin positive. The stromal cells demonstrated positivity for CD10 but were keratin negative. The epithelial portion of the tumor was composed mostly of basaloid islands of cells with nuclear pleomorphism, scattered mitoses, and focal sebaceous differentiation. The mesenchymal portion of the tumor displayed florid pleomorphism and polymorphism, with many large atypical cells and proliferation. A diagnosis of primary cutaneous carcinosarcoma (PCC) was rendered. Head and neck computed tomography showed tumor penetration of less than 1 cm into scalp soft tissues with no involvement of the underlying bone. There was some evidence of swelling of the supragaleal soft tissues without indication of perineural spread. An 11-mm hyperlucent lower cervical lymph node on the left side that likely represented an incidental finding was noted. Surgical excision with margin evaluation was recommended, but the patient declined. He instead received radiation therapy to the left side of the posterior scalp with a total dose of 30 Gy at 6 Gy per fraction and 1 fraction daily. The patient was found to have a well-healed scar with no evidence of recurrence at 4-week follow-up and again at 5 months after radiation therapy.
|
|
|
| A generous shave biopsy and debulking performed on the initial visit revealed an inflitrating tumor consisting on malignant epithelial and stromal components (A-C)(H&E; original magnifications ×10, ×20, and ×40, respectively). |
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm of unknown etiology that is characterized by the presence of both malignant epithelial and mesenchymal components.1 Carcinosarcomas have been reported in both the male and female reproductive tracts, urinary tract, gastrointestinal tract, lungs, breasts, larynx, thymus, and thyroid but is uncommon as a primary neoplasm of the skin.2 Epidermal PCC occurs with greater frequency in males than in females and typically presents in the eighth or ninth decades of life.3 These tumors tend to arise in sun-exposed regions, most commonly on the face and scalp.2
Morphologically, PCCs typically are exophytic growths that often feature surface ulceration and may or may not bleed upon palpation.4 Primary cutaneous carcinosarcomas may present as long-standing lesions that have undergone rapid transformation in the weeks preceding presentation.4 It is not uncommon for PCC lesions to carry the clinical diagnosis of squamous cell carcinoma, which suggests notable morphologic overlap between these entities. Histopathologically, PCC shows a basal cell carcinoma and/or a squamous cell carcinoma epithelial component intimately admixed with a sarcomatous component.5 The mesenchymal component of PCC typically resembles a superficial malignant fibrous histiocytoma characterized by pleomorphic nuclei and cytoplasm, necrosis, and an increased number of mitotic figures.2 Immunohistochemistry can be beneficial in the diagnosis of PCC. A combination of p63 and AE1/AE3 stains can be used to confirm cells of epithelial origin. Staining with vimentin, CD10, or caldesmon can help to delineate the mesenchymal component of PCC.
Epidermal PCC most commonly affects elderly individuals with a history of extensive sun exposure. It has been suggested that p53 mutations due to UV damage are key in tumor formation for both epithelial and mesenchymal elements.5 Literature supports a monoclonal origin for the epithelial and mesenchymal components of this tumor; however, there is insufficient evidence.6 Surgical excision is the primary treatment modality for epidermal PCC, but adjuvant or substitutive radiotherapy has been used in some cases.4 The prognosis of PCC is notably better than its visceral counterpart due to early diagnosis and treatment of easily visible lesions. Epidermal PCC has a 70% 5-year disease-free survival rate, while adnexal PCC tends to occur in younger patients and has a 25% 5-year disease-free survival rate.3 Due to the rarity of reported cases and limited follow-up, the long-term prognosis for PCC remains unclear.
We report an unusual case of PCC on the scalp that was successfully treated with radiation therapy alone. This modality should be considered in patients with large tumors who refuse surgery or are not good surgical candidates.
1. El Harroudi T, Ech-Charif S, Amrani M, et al. Primary carcinosarcoma of the skin. J Hand Microsurg. 2010;2:79-81.
2. Patel NK, McKee PH, Smith NP. Primary metaplastic carcinoma (carcinosarcoma) of the skin: a clinicopathologic study of four cases and review of the literature. Am J Dermatopathol. 1997;19:363-372.
3. Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
4. Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
5. Tran TA, Muller S, Chaudahri PJ, et al. Cutaneous carcinosarcoma: adnexal vs. epidermal types define high- and low-risk tumors. results of a meta-analysis. J Cutan Pathol. 2005;32:2-11.
6. Paniz Mondolfi AE, Jour G, Johnson M, et al. Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next-generation sequencing. Hum Pathol. 2013;44:2853-2860.
The Diagnosis: Primary Cutaneous Carcinosarcoma
A generous shave biopsy and debulking performed on the initial visit revealed an infiltrating tumor consisting of malignant epithelial and stromal components (Figure). The basaloid and squamoid epithelial cells were keratin positive. The stromal cells demonstrated positivity for CD10 but were keratin negative. The epithelial portion of the tumor was composed mostly of basaloid islands of cells with nuclear pleomorphism, scattered mitoses, and focal sebaceous differentiation. The mesenchymal portion of the tumor displayed florid pleomorphism and polymorphism, with many large atypical cells and proliferation. A diagnosis of primary cutaneous carcinosarcoma (PCC) was rendered. Head and neck computed tomography showed tumor penetration of less than 1 cm into scalp soft tissues with no involvement of the underlying bone. There was some evidence of swelling of the supragaleal soft tissues without indication of perineural spread. An 11-mm hyperlucent lower cervical lymph node on the left side that likely represented an incidental finding was noted. Surgical excision with margin evaluation was recommended, but the patient declined. He instead received radiation therapy to the left side of the posterior scalp with a total dose of 30 Gy at 6 Gy per fraction and 1 fraction daily. The patient was found to have a well-healed scar with no evidence of recurrence at 4-week follow-up and again at 5 months after radiation therapy.
|
|
|
|
| A generous shave biopsy and debulking performed on the initial visit revealed an inflitrating tumor consisting on malignant epithelial and stromal components (A-C)(H&E; original magnifications ×10, ×20, and ×40, respectively). |
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm of unknown etiology that is characterized by the presence of both malignant epithelial and mesenchymal components.1 Carcinosarcomas have been reported in both the male and female reproductive tracts, urinary tract, gastrointestinal tract, lungs, breasts, larynx, thymus, and thyroid but is uncommon as a primary neoplasm of the skin.2 Epidermal PCC occurs with greater frequency in males than in females and typically presents in the eighth or ninth decades of life.3 These tumors tend to arise in sun-exposed regions, most commonly on the face and scalp.2
Morphologically, PCCs typically are exophytic growths that often feature surface ulceration and may or may not bleed upon palpation.4 Primary cutaneous carcinosarcomas may present as long-standing lesions that have undergone rapid transformation in the weeks preceding presentation.4 It is not uncommon for PCC lesions to carry the clinical diagnosis of squamous cell carcinoma, which suggests notable morphologic overlap between these entities. Histopathologically, PCC shows a basal cell carcinoma and/or a squamous cell carcinoma epithelial component intimately admixed with a sarcomatous component.5 The mesenchymal component of PCC typically resembles a superficial malignant fibrous histiocytoma characterized by pleomorphic nuclei and cytoplasm, necrosis, and an increased number of mitotic figures.2 Immunohistochemistry can be beneficial in the diagnosis of PCC. A combination of p63 and AE1/AE3 stains can be used to confirm cells of epithelial origin. Staining with vimentin, CD10, or caldesmon can help to delineate the mesenchymal component of PCC.
Epidermal PCC most commonly affects elderly individuals with a history of extensive sun exposure. It has been suggested that p53 mutations due to UV damage are key in tumor formation for both epithelial and mesenchymal elements.5 Literature supports a monoclonal origin for the epithelial and mesenchymal components of this tumor; however, there is insufficient evidence.6 Surgical excision is the primary treatment modality for epidermal PCC, but adjuvant or substitutive radiotherapy has been used in some cases.4 The prognosis of PCC is notably better than its visceral counterpart due to early diagnosis and treatment of easily visible lesions. Epidermal PCC has a 70% 5-year disease-free survival rate, while adnexal PCC tends to occur in younger patients and has a 25% 5-year disease-free survival rate.3 Due to the rarity of reported cases and limited follow-up, the long-term prognosis for PCC remains unclear.
We report an unusual case of PCC on the scalp that was successfully treated with radiation therapy alone. This modality should be considered in patients with large tumors who refuse surgery or are not good surgical candidates.
The Diagnosis: Primary Cutaneous Carcinosarcoma
A generous shave biopsy and debulking performed on the initial visit revealed an infiltrating tumor consisting of malignant epithelial and stromal components (Figure). The basaloid and squamoid epithelial cells were keratin positive. The stromal cells demonstrated positivity for CD10 but were keratin negative. The epithelial portion of the tumor was composed mostly of basaloid islands of cells with nuclear pleomorphism, scattered mitoses, and focal sebaceous differentiation. The mesenchymal portion of the tumor displayed florid pleomorphism and polymorphism, with many large atypical cells and proliferation. A diagnosis of primary cutaneous carcinosarcoma (PCC) was rendered. Head and neck computed tomography showed tumor penetration of less than 1 cm into scalp soft tissues with no involvement of the underlying bone. There was some evidence of swelling of the supragaleal soft tissues without indication of perineural spread. An 11-mm hyperlucent lower cervical lymph node on the left side that likely represented an incidental finding was noted. Surgical excision with margin evaluation was recommended, but the patient declined. He instead received radiation therapy to the left side of the posterior scalp with a total dose of 30 Gy at 6 Gy per fraction and 1 fraction daily. The patient was found to have a well-healed scar with no evidence of recurrence at 4-week follow-up and again at 5 months after radiation therapy.
|
|
|
|
| A generous shave biopsy and debulking performed on the initial visit revealed an inflitrating tumor consisting on malignant epithelial and stromal components (A-C)(H&E; original magnifications ×10, ×20, and ×40, respectively). |
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm of unknown etiology that is characterized by the presence of both malignant epithelial and mesenchymal components.1 Carcinosarcomas have been reported in both the male and female reproductive tracts, urinary tract, gastrointestinal tract, lungs, breasts, larynx, thymus, and thyroid but is uncommon as a primary neoplasm of the skin.2 Epidermal PCC occurs with greater frequency in males than in females and typically presents in the eighth or ninth decades of life.3 These tumors tend to arise in sun-exposed regions, most commonly on the face and scalp.2
Morphologically, PCCs typically are exophytic growths that often feature surface ulceration and may or may not bleed upon palpation.4 Primary cutaneous carcinosarcomas may present as long-standing lesions that have undergone rapid transformation in the weeks preceding presentation.4 It is not uncommon for PCC lesions to carry the clinical diagnosis of squamous cell carcinoma, which suggests notable morphologic overlap between these entities. Histopathologically, PCC shows a basal cell carcinoma and/or a squamous cell carcinoma epithelial component intimately admixed with a sarcomatous component.5 The mesenchymal component of PCC typically resembles a superficial malignant fibrous histiocytoma characterized by pleomorphic nuclei and cytoplasm, necrosis, and an increased number of mitotic figures.2 Immunohistochemistry can be beneficial in the diagnosis of PCC. A combination of p63 and AE1/AE3 stains can be used to confirm cells of epithelial origin. Staining with vimentin, CD10, or caldesmon can help to delineate the mesenchymal component of PCC.
Epidermal PCC most commonly affects elderly individuals with a history of extensive sun exposure. It has been suggested that p53 mutations due to UV damage are key in tumor formation for both epithelial and mesenchymal elements.5 Literature supports a monoclonal origin for the epithelial and mesenchymal components of this tumor; however, there is insufficient evidence.6 Surgical excision is the primary treatment modality for epidermal PCC, but adjuvant or substitutive radiotherapy has been used in some cases.4 The prognosis of PCC is notably better than its visceral counterpart due to early diagnosis and treatment of easily visible lesions. Epidermal PCC has a 70% 5-year disease-free survival rate, while adnexal PCC tends to occur in younger patients and has a 25% 5-year disease-free survival rate.3 Due to the rarity of reported cases and limited follow-up, the long-term prognosis for PCC remains unclear.
We report an unusual case of PCC on the scalp that was successfully treated with radiation therapy alone. This modality should be considered in patients with large tumors who refuse surgery or are not good surgical candidates.
1. El Harroudi T, Ech-Charif S, Amrani M, et al. Primary carcinosarcoma of the skin. J Hand Microsurg. 2010;2:79-81.
2. Patel NK, McKee PH, Smith NP. Primary metaplastic carcinoma (carcinosarcoma) of the skin: a clinicopathologic study of four cases and review of the literature. Am J Dermatopathol. 1997;19:363-372.
3. Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
4. Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
5. Tran TA, Muller S, Chaudahri PJ, et al. Cutaneous carcinosarcoma: adnexal vs. epidermal types define high- and low-risk tumors. results of a meta-analysis. J Cutan Pathol. 2005;32:2-11.
6. Paniz Mondolfi AE, Jour G, Johnson M, et al. Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next-generation sequencing. Hum Pathol. 2013;44:2853-2860.
1. El Harroudi T, Ech-Charif S, Amrani M, et al. Primary carcinosarcoma of the skin. J Hand Microsurg. 2010;2:79-81.
2. Patel NK, McKee PH, Smith NP. Primary metaplastic carcinoma (carcinosarcoma) of the skin: a clinicopathologic study of four cases and review of the literature. Am J Dermatopathol. 1997;19:363-372.
3. Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
4. Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
5. Tran TA, Muller S, Chaudahri PJ, et al. Cutaneous carcinosarcoma: adnexal vs. epidermal types define high- and low-risk tumors. results of a meta-analysis. J Cutan Pathol. 2005;32:2-11.
6. Paniz Mondolfi AE, Jour G, Johnson M, et al. Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next-generation sequencing. Hum Pathol. 2013;44:2853-2860.
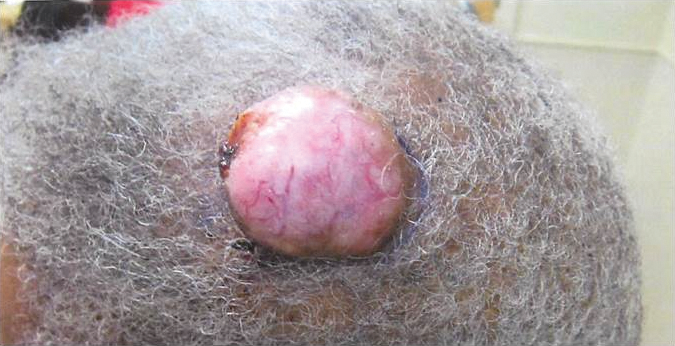
An 81-year-old man presented with a 3.5×3.0-cm pink exophytic tumor with an eroded surface and prominent vascularity on the left side of the parietal scalp. The patient reported that the tumor had been present for more than 30 years but recently had grown larger in size. He denied pain or pruritus in association with the lesion and did not report any systemic symptoms. He had received no prior treatments for the tumor.

Prevalence of Glaucoma in Patients With Vitiligo
Vitiligo is an acquired idiopathic disease of unknown etiology. Characterized by depigmented maculae and melanocytic destruction, it usually presents in childhood or young adulthood. The incidence of vitiligo ranges from 0.5% to 2% globally and there is no racial or gender predilection.1
Patients with vitiligo may exhibit pigmentary abnormalities of the iris and retina.2 Noninflammatory depigmented lesions of the ocular fundus observed in vitiligo indicate a local loss of melanocytes.1 The fact that melanocytes are present not only in the skin and roots of the hair but also in the uvea and stria vascularis of the inner ear may explain the ophthalmologic disorders that accompany vitiligo.3 The term glaucoma refers to a large number of diseases that share a common feature: a distinctive and progressive optic neuropathy that may derive from various risks and is associated with a gradual loss of the visual field. If the disorder is not diagnosed and treated properly it could cause blindness.
Glaucoma is classified on the basis of the underlying abnormality that causes intraocular pressure (IOP) to rise. Glaucoma is first divided into open-angle and angle-closure glaucoma; glaucoma associated with developmental anomalies is then subdivided according to specific alterations.4
A PubMed search of articles indexed for MEDLINE using the terms vitiligo and glaucoma revealed only 1 study examining the incidence of glaucoma in patients with vitiligo.5 In the study reported here, we determined the presence of and possible risk factors for glaucoma in patients with vitiligo who had presented to the dermatology polyclinic.
Methods
We registered 49 patients diagnosed with vitiligo by clinical and Wood light examination and 20 age- and sex-matched healthy controls. Patients who were using topical corticosteroid treatments for vitiligo lesions located on the face were excluded from the study due to the glaucoma-inducing effects of corticosteroids. Similarly, patients who received drugs with sympathetic and parasympathetic action that can cause glaucoma were excluded.
The patients received a comprehensive ophthalmologic examination that included visual acuity testing, refraction, IOP measurement, gonioscopy, and fundus examination. All patients and controls underwent visual field tests and optic nerve head analyses using a confocal scanning laser ophthalmoscope. Glaucoma was diagnosed based on fundus examination, IOP measurement, field of vision evaluation, and optic nerve head analysis.
Informed consent was obtained from all participants. The research protocol was approved by the university hospital ethics committee.
Results
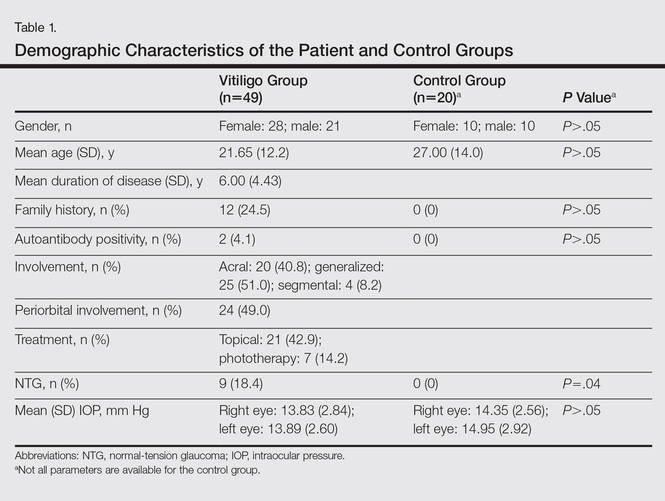
The study registered a total of 49 patients with vitiligo (28 female; 21 male) and 20 healthy controls (10 female; 10 male) with a variety of demographic and clinical characteristics (Table 1).
Mean (SD) IOP values were 13.83 (2.84) mm Hg for the right eye and 13.89 (2.60) mm Hg for the left eye in the vitiligo group. Values were 14.35 (2.56) mm Hg and 14.95 (2.92) mm Hg, respectively, in the control group. The IOP differences between the 2 groups were not statistically significant (P>.05).
Nine patients (18.4%) in the vitiligo group were found to have signs of normal-tension glaucoma (NTG). Optic nerve damage and vision loss occurs in the presence of normal IOP in NTG. There were no signs of NTG in the control group. Normal-tension glaucoma was diagnosed in the vitiligo group based on glaucomatous optic disc appearance, visual field defects, and structural analysis of the entire optic nerve head in confocal scanning laser ophthalmoscope. The NTG difference between the vitiligo and control groups was statistically significant (P=.04).
In the vitiligo group, of the 9 patients who had NTG, 6 had periorbital vitiligo lesions; the remaining 3 had none. Although patients who had periorbital lesions had a higher rate of glaucoma relative to the patients without periorbital lesions, the difference was not statistically significant (P>.05).
No statistically significant differences (P>.05) were found between patients with vitiligo with and without glaucoma in terms of age, sex, disease duration, family history of vitiligo, presence or absence of periorbital involvement, manner of involvement, percentage of the involved body areas, and IOP (Table 1).
Comment
Glaucoma is characterized by increased IOP, visual field loss, and changes in the optic nerve head. Although elevated IOP is common in ocular hypertension as well as in glaucoma, there is no glaucomatous visual field loss in ocular hypertension. In NTG, on the other hand, glaucomatous visual field loss and optic nerve head changes occur without an increase in IOP.6 Normal-tension glaucoma is a particular type of open-angle glaucoma. It is believed that NTG and high-tension glaucoma induce optic nerve head damage through different means.7 Alternative theories have been put forth to account for the glaucomatous damage to the optic nerve head that occurs in NTG, despite normal or close to normal IOP. These theories include vascular disorders (eg, ischemia, which interrupts the orthograde or retrograde axonal transport), excessive accumulation of free radicals, triggering of apoptosis, and low resistance of lamina cribrosa.8
Although there are various studies exploring ocular symptoms in patients with vitiligo,9-15 only 1 study has examined the incidence of glaucoma in this group of patients.5 Biswas et al11 examined ocular signs in 100 patients with vitiligo and found that 23% of patients had hypopigmented foci in the iris, 18% had pigmentation in the anterior chamber, 11% had chorioretinal degeneration, 9% had hypopigmentation of the retinal pigment epithelium, 5% had uveitis, and 34% were evaluated as normal. In this study, the authors concluded that there was a strong relationship between vitiligo and eye diseases.11 When Gopal et al9 compared the eye examinations of 150 vitiligo patients and 100 healthy controls, they found uveitis, iris, and retinal pigmentary abnormalities in 16% of the vitiligo patients (P<.001).
Rogosić et al5 examined the incidence of glaucoma in 42 patients with vitiligo and found primary open-angle glaucoma in 24 (57%) patients. The patients had a mean age of 56 years, mean disease duration of 13 years, and mean IOP of 18 mm Hg for the right eye and 17.5 mm Hg for the left eye. The incidence of glaucoma was significantly higher in patients with vitiligo (P<.001) and increased with disease duration.5
Similar studies, however, have failed to show a relationship between vitiligo and glaucoma. In a study that evaluated the retinal pigment epithelium and the optic nerve in patients with vitiligo, Perossini et al10 found that the fundus examination of the patients was perfectly normal.
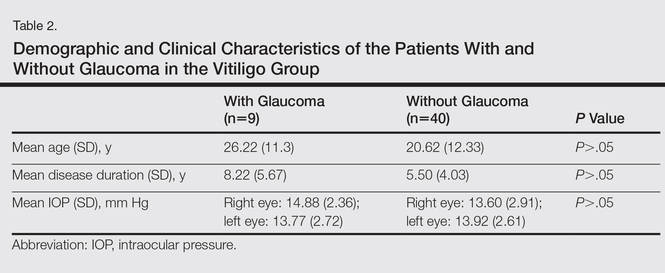
In our study, we detected NTG in 18.4% of patients with vitiligo. We did not find a significant statistical difference between patients with and without glaucoma (Table 2). Rogosić et al5 found a significant relationship between age and glaucoma incidence, but we did not find such a relationship, which we believe is because the mean age of our patients was lower than the prior study.
In vitiligo, melanocytes are destroyed through an unknown mechanism. Although the cellular and molecular mechanisms causing melanocytic destruction have not yet been determined, various hypotheses have been put forward to explain the etiopathogenesis of vitiligo. Among these, the most commonly held hypotheses are the neural, self-destruction, and autoimmune hypotheses.16
Based on the observation that stress and serious trauma could precipitate or trigger the onset of vitiligo,16 the neural hypothesis holds that neurochemical mediators released from the edges of the nerve endings exert toxic effects on melanocytes. The fact that both melanocytes and choroidal pigment cells originate from the mesenchyme and dermatomal spreading of segmental vitiligo are arguments propounded in favor of this hypothesis.17
The self-destruction hypothesis suggests that the intrinsic protective mechanisms that normally enable melanocytes to eliminate toxic intermediate products or metabolites on the melanogenesis path have been impaired in patients with vitiligo.18,19 There is evidence of increased oxidative stress over the whole epidermis of patients with vitiligo.20 Thus, free radicals affect melanin and cause membrane damage via lipid peroxidation reactions.21
The autoimmune hypothesis proposes a clinical relationship between vitiligo and several diseases believed to be autoimmune. Because the macrophage infiltration observed in vitiligo lesions is more pronounced on the perilesional skin, this hypothesis holds that macrophages may play a role in melanocyte removal.21 The Koebner phenomenon observed in vitiligo lends support to the critical role of trauma in the etiopathogenesis of the disease.
Although we could not explain the co-presence of vitiligo and glaucoma, we believe that it may result from the fact that both diseases are observed in tissues that have the same embryologic origin and etiology, perhaps vascular or neural disorders, excessive accumulation of free radicals, or the triggering of apoptosis. Dermatologists should be alert to the presence of glaucoma in patients with vitiligo because glaucoma is an eye disease that progresses slowly and may lead to vision loss.
1. Ortonne JP. Vitiligo and other disorders of hypopigmentation. In: Bolognia JB, Jorizzo JL, Rapini RP, eds. Dermatology. 1st ed. New York, NY: Mosby; 2003:947-973.
2. Ortonne JP, Bahadoran P, Fitzpatrick TB, et al. Hypomelanoses and hypermelanoses. In: Freedberg IM, Eisen AZ, Wolff K, eds. Fitzpatrick’s Dermatology in General Medicine. 6th ed. New York, NY: McGraw-Hill; 2003:836-881.
3. van den Wijngaard R, Wijngaard R, Wankowiczs-Kalinsa A, et al. Autoimmune melanocyte destruction in vitiligo. Lab Invest. 2001;81:1061-1067.
4. Shields MB, Ritch R, Krupin T. Classification of the glaucomas. In: Ritch R, Shields MB, Krupin T, eds. The Glaucomas. St Louis, MO: C.V. Mosby Co; 1996:717-725.
5. Rogosić V, Bojić L, Puizina-Ivić N, et al. Vitiligo and glaucoma–an association or a coincidence? a pilot study. Acta Dermatovenerol Croat. 2010;18:21-26.
6. Anderson DR. Normal-tension glaucoma (low-tension glaucoma). Indian J Ophthalmol. 2011;59(suppl 59):S97-S101.
7. Iwata K. Primary open angle glaucoma and low tension glaucoma–pathogenesis and mechanism of optic nerve damage [in Japanese]. Nippon Ganka Gakkai Zasshi. 1992;96:1501-1531.
8. Hitchings RA, Anderton SA. A comparative study of visual field defects seen in patients with low-tension glaucoma and chronic simple glaucoma. Br J Ophthalmol. 1983;67:818-821.
9. Gopal KV, Rama Rao GR, Kumar YH, et al. Vitiligo: a part of a systemic autoimmune process. Indian J Dermatol Venereol Leprol. 2007;73:162-165.
10. Perossini M, Turio E, Perossini T, et al. Vitiligo: ocular and electrophysiological findings. G Ital Dermatol Venereol. 2010;145:141-149.
11. Biswas G, Barbhuiya JN, Biswas MC, et al. Clinical pattern of ocular manifestations in vitiligo. J Indian Med Assoc. 2003;101:478-480.
12. Park S, Albert DM, Bolognia JL. Ocular manifestations of pigmentary disorders. Dermatol Clin. 1992;10:609-622.
13. Albert DM, Nordlund JJ, Lerner AB. Ocular abnormalities occurring with vitiligo. Ophthalmology. 1979;86:1145-1160.
14. Wagoner MD, Albert DM, Lerner AB, et al. New observations on vitiligo and ocular disease. Am J Ophthalmol. 1983;96:16-26.
15. Cowan CL Jr, Halder RM, Grimes PE, et al. Ocular disturbances in vitiligo. J Am Acad Dermatol. 1986;15:17-24.
16. Orecchia GE. Neural pathogenesis. In: Hann SK, Nordlund JJ. Vitiligo. Oxford, England: Blackwell Science Ltd; 2000:142-150.
17. Braun-Falco O, Plewig G, Wolf HH, et al. Disorders of melanin pigmentation. In: Bartels V, ed. Dermatology. Berlin, Germany: Springer; 2000:1013-1042.
18. Tüzün Y, Kotoğyan A, Aydemir EH, et al. Pigmentasyon bozuklukları. In: Baransü O. Dermatoloji. 2nd ed. Istanbul: Nobel Tıp Kitabevi; 1994:557-559.
19. Odom RB, James WD, Berger TG. Disturbances of pigmentation. In: Odom RB, James WD, Berger TG. Andrews’ Diseases of the Skin. 9th ed. Philadelphia, PA: W.B. Saunders Company; 2000:1065-1068.
20. Schallreuter KU. Biochemical theory of vitiligo: a role of pteridines in pigmentation. In: Hann SK, Nordlund JJ. Vitiligo. London, England: Blackwell Science Ltd; 2000:151-159.
21. van den Wijngaard R, Wankowicz-Kalinska A, Le Poole C, et al. Local immune response in skin of generalized vitiligo patients. destruction of melanocytes is associated with the predominent presence of CLA+T cells at the perilesional site. Lab Invest. 2000;80:1299-1309.
Vitiligo is an acquired idiopathic disease of unknown etiology. Characterized by depigmented maculae and melanocytic destruction, it usually presents in childhood or young adulthood. The incidence of vitiligo ranges from 0.5% to 2% globally and there is no racial or gender predilection.1
Patients with vitiligo may exhibit pigmentary abnormalities of the iris and retina.2 Noninflammatory depigmented lesions of the ocular fundus observed in vitiligo indicate a local loss of melanocytes.1 The fact that melanocytes are present not only in the skin and roots of the hair but also in the uvea and stria vascularis of the inner ear may explain the ophthalmologic disorders that accompany vitiligo.3 The term glaucoma refers to a large number of diseases that share a common feature: a distinctive and progressive optic neuropathy that may derive from various risks and is associated with a gradual loss of the visual field. If the disorder is not diagnosed and treated properly it could cause blindness.
Glaucoma is classified on the basis of the underlying abnormality that causes intraocular pressure (IOP) to rise. Glaucoma is first divided into open-angle and angle-closure glaucoma; glaucoma associated with developmental anomalies is then subdivided according to specific alterations.4
A PubMed search of articles indexed for MEDLINE using the terms vitiligo and glaucoma revealed only 1 study examining the incidence of glaucoma in patients with vitiligo.5 In the study reported here, we determined the presence of and possible risk factors for glaucoma in patients with vitiligo who had presented to the dermatology polyclinic.
Methods
We registered 49 patients diagnosed with vitiligo by clinical and Wood light examination and 20 age- and sex-matched healthy controls. Patients who were using topical corticosteroid treatments for vitiligo lesions located on the face were excluded from the study due to the glaucoma-inducing effects of corticosteroids. Similarly, patients who received drugs with sympathetic and parasympathetic action that can cause glaucoma were excluded.
The patients received a comprehensive ophthalmologic examination that included visual acuity testing, refraction, IOP measurement, gonioscopy, and fundus examination. All patients and controls underwent visual field tests and optic nerve head analyses using a confocal scanning laser ophthalmoscope. Glaucoma was diagnosed based on fundus examination, IOP measurement, field of vision evaluation, and optic nerve head analysis.
Informed consent was obtained from all participants. The research protocol was approved by the university hospital ethics committee.
Results
The study registered a total of 49 patients with vitiligo (28 female; 21 male) and 20 healthy controls (10 female; 10 male) with a variety of demographic and clinical characteristics (Table 1).
Mean (SD) IOP values were 13.83 (2.84) mm Hg for the right eye and 13.89 (2.60) mm Hg for the left eye in the vitiligo group. Values were 14.35 (2.56) mm Hg and 14.95 (2.92) mm Hg, respectively, in the control group. The IOP differences between the 2 groups were not statistically significant (P>.05).
Nine patients (18.4%) in the vitiligo group were found to have signs of normal-tension glaucoma (NTG). Optic nerve damage and vision loss occurs in the presence of normal IOP in NTG. There were no signs of NTG in the control group. Normal-tension glaucoma was diagnosed in the vitiligo group based on glaucomatous optic disc appearance, visual field defects, and structural analysis of the entire optic nerve head in confocal scanning laser ophthalmoscope. The NTG difference between the vitiligo and control groups was statistically significant (P=.04).
In the vitiligo group, of the 9 patients who had NTG, 6 had periorbital vitiligo lesions; the remaining 3 had none. Although patients who had periorbital lesions had a higher rate of glaucoma relative to the patients without periorbital lesions, the difference was not statistically significant (P>.05).
No statistically significant differences (P>.05) were found between patients with vitiligo with and without glaucoma in terms of age, sex, disease duration, family history of vitiligo, presence or absence of periorbital involvement, manner of involvement, percentage of the involved body areas, and IOP (Table 1).
Comment
Glaucoma is characterized by increased IOP, visual field loss, and changes in the optic nerve head. Although elevated IOP is common in ocular hypertension as well as in glaucoma, there is no glaucomatous visual field loss in ocular hypertension. In NTG, on the other hand, glaucomatous visual field loss and optic nerve head changes occur without an increase in IOP.6 Normal-tension glaucoma is a particular type of open-angle glaucoma. It is believed that NTG and high-tension glaucoma induce optic nerve head damage through different means.7 Alternative theories have been put forth to account for the glaucomatous damage to the optic nerve head that occurs in NTG, despite normal or close to normal IOP. These theories include vascular disorders (eg, ischemia, which interrupts the orthograde or retrograde axonal transport), excessive accumulation of free radicals, triggering of apoptosis, and low resistance of lamina cribrosa.8
Although there are various studies exploring ocular symptoms in patients with vitiligo,9-15 only 1 study has examined the incidence of glaucoma in this group of patients.5 Biswas et al11 examined ocular signs in 100 patients with vitiligo and found that 23% of patients had hypopigmented foci in the iris, 18% had pigmentation in the anterior chamber, 11% had chorioretinal degeneration, 9% had hypopigmentation of the retinal pigment epithelium, 5% had uveitis, and 34% were evaluated as normal. In this study, the authors concluded that there was a strong relationship between vitiligo and eye diseases.11 When Gopal et al9 compared the eye examinations of 150 vitiligo patients and 100 healthy controls, they found uveitis, iris, and retinal pigmentary abnormalities in 16% of the vitiligo patients (P<.001).
Rogosić et al5 examined the incidence of glaucoma in 42 patients with vitiligo and found primary open-angle glaucoma in 24 (57%) patients. The patients had a mean age of 56 years, mean disease duration of 13 years, and mean IOP of 18 mm Hg for the right eye and 17.5 mm Hg for the left eye. The incidence of glaucoma was significantly higher in patients with vitiligo (P<.001) and increased with disease duration.5
Similar studies, however, have failed to show a relationship between vitiligo and glaucoma. In a study that evaluated the retinal pigment epithelium and the optic nerve in patients with vitiligo, Perossini et al10 found that the fundus examination of the patients was perfectly normal.
In our study, we detected NTG in 18.4% of patients with vitiligo. We did not find a significant statistical difference between patients with and without glaucoma (Table 2). Rogosić et al5 found a significant relationship between age and glaucoma incidence, but we did not find such a relationship, which we believe is because the mean age of our patients was lower than the prior study.
In vitiligo, melanocytes are destroyed through an unknown mechanism. Although the cellular and molecular mechanisms causing melanocytic destruction have not yet been determined, various hypotheses have been put forward to explain the etiopathogenesis of vitiligo. Among these, the most commonly held hypotheses are the neural, self-destruction, and autoimmune hypotheses.16
Based on the observation that stress and serious trauma could precipitate or trigger the onset of vitiligo,16 the neural hypothesis holds that neurochemical mediators released from the edges of the nerve endings exert toxic effects on melanocytes. The fact that both melanocytes and choroidal pigment cells originate from the mesenchyme and dermatomal spreading of segmental vitiligo are arguments propounded in favor of this hypothesis.17
The self-destruction hypothesis suggests that the intrinsic protective mechanisms that normally enable melanocytes to eliminate toxic intermediate products or metabolites on the melanogenesis path have been impaired in patients with vitiligo.18,19 There is evidence of increased oxidative stress over the whole epidermis of patients with vitiligo.20 Thus, free radicals affect melanin and cause membrane damage via lipid peroxidation reactions.21
The autoimmune hypothesis proposes a clinical relationship between vitiligo and several diseases believed to be autoimmune. Because the macrophage infiltration observed in vitiligo lesions is more pronounced on the perilesional skin, this hypothesis holds that macrophages may play a role in melanocyte removal.21 The Koebner phenomenon observed in vitiligo lends support to the critical role of trauma in the etiopathogenesis of the disease.
Although we could not explain the co-presence of vitiligo and glaucoma, we believe that it may result from the fact that both diseases are observed in tissues that have the same embryologic origin and etiology, perhaps vascular or neural disorders, excessive accumulation of free radicals, or the triggering of apoptosis. Dermatologists should be alert to the presence of glaucoma in patients with vitiligo because glaucoma is an eye disease that progresses slowly and may lead to vision loss.
Vitiligo is an acquired idiopathic disease of unknown etiology. Characterized by depigmented maculae and melanocytic destruction, it usually presents in childhood or young adulthood. The incidence of vitiligo ranges from 0.5% to 2% globally and there is no racial or gender predilection.1
Patients with vitiligo may exhibit pigmentary abnormalities of the iris and retina.2 Noninflammatory depigmented lesions of the ocular fundus observed in vitiligo indicate a local loss of melanocytes.1 The fact that melanocytes are present not only in the skin and roots of the hair but also in the uvea and stria vascularis of the inner ear may explain the ophthalmologic disorders that accompany vitiligo.3 The term glaucoma refers to a large number of diseases that share a common feature: a distinctive and progressive optic neuropathy that may derive from various risks and is associated with a gradual loss of the visual field. If the disorder is not diagnosed and treated properly it could cause blindness.
Glaucoma is classified on the basis of the underlying abnormality that causes intraocular pressure (IOP) to rise. Glaucoma is first divided into open-angle and angle-closure glaucoma; glaucoma associated with developmental anomalies is then subdivided according to specific alterations.4
A PubMed search of articles indexed for MEDLINE using the terms vitiligo and glaucoma revealed only 1 study examining the incidence of glaucoma in patients with vitiligo.5 In the study reported here, we determined the presence of and possible risk factors for glaucoma in patients with vitiligo who had presented to the dermatology polyclinic.
Methods
We registered 49 patients diagnosed with vitiligo by clinical and Wood light examination and 20 age- and sex-matched healthy controls. Patients who were using topical corticosteroid treatments for vitiligo lesions located on the face were excluded from the study due to the glaucoma-inducing effects of corticosteroids. Similarly, patients who received drugs with sympathetic and parasympathetic action that can cause glaucoma were excluded.
The patients received a comprehensive ophthalmologic examination that included visual acuity testing, refraction, IOP measurement, gonioscopy, and fundus examination. All patients and controls underwent visual field tests and optic nerve head analyses using a confocal scanning laser ophthalmoscope. Glaucoma was diagnosed based on fundus examination, IOP measurement, field of vision evaluation, and optic nerve head analysis.
Informed consent was obtained from all participants. The research protocol was approved by the university hospital ethics committee.
Results
The study registered a total of 49 patients with vitiligo (28 female; 21 male) and 20 healthy controls (10 female; 10 male) with a variety of demographic and clinical characteristics (Table 1).
Mean (SD) IOP values were 13.83 (2.84) mm Hg for the right eye and 13.89 (2.60) mm Hg for the left eye in the vitiligo group. Values were 14.35 (2.56) mm Hg and 14.95 (2.92) mm Hg, respectively, in the control group. The IOP differences between the 2 groups were not statistically significant (P>.05).
Nine patients (18.4%) in the vitiligo group were found to have signs of normal-tension glaucoma (NTG). Optic nerve damage and vision loss occurs in the presence of normal IOP in NTG. There were no signs of NTG in the control group. Normal-tension glaucoma was diagnosed in the vitiligo group based on glaucomatous optic disc appearance, visual field defects, and structural analysis of the entire optic nerve head in confocal scanning laser ophthalmoscope. The NTG difference between the vitiligo and control groups was statistically significant (P=.04).
In the vitiligo group, of the 9 patients who had NTG, 6 had periorbital vitiligo lesions; the remaining 3 had none. Although patients who had periorbital lesions had a higher rate of glaucoma relative to the patients without periorbital lesions, the difference was not statistically significant (P>.05).
No statistically significant differences (P>.05) were found between patients with vitiligo with and without glaucoma in terms of age, sex, disease duration, family history of vitiligo, presence or absence of periorbital involvement, manner of involvement, percentage of the involved body areas, and IOP (Table 1).
Comment
Glaucoma is characterized by increased IOP, visual field loss, and changes in the optic nerve head. Although elevated IOP is common in ocular hypertension as well as in glaucoma, there is no glaucomatous visual field loss in ocular hypertension. In NTG, on the other hand, glaucomatous visual field loss and optic nerve head changes occur without an increase in IOP.6 Normal-tension glaucoma is a particular type of open-angle glaucoma. It is believed that NTG and high-tension glaucoma induce optic nerve head damage through different means.7 Alternative theories have been put forth to account for the glaucomatous damage to the optic nerve head that occurs in NTG, despite normal or close to normal IOP. These theories include vascular disorders (eg, ischemia, which interrupts the orthograde or retrograde axonal transport), excessive accumulation of free radicals, triggering of apoptosis, and low resistance of lamina cribrosa.8
Although there are various studies exploring ocular symptoms in patients with vitiligo,9-15 only 1 study has examined the incidence of glaucoma in this group of patients.5 Biswas et al11 examined ocular signs in 100 patients with vitiligo and found that 23% of patients had hypopigmented foci in the iris, 18% had pigmentation in the anterior chamber, 11% had chorioretinal degeneration, 9% had hypopigmentation of the retinal pigment epithelium, 5% had uveitis, and 34% were evaluated as normal. In this study, the authors concluded that there was a strong relationship between vitiligo and eye diseases.11 When Gopal et al9 compared the eye examinations of 150 vitiligo patients and 100 healthy controls, they found uveitis, iris, and retinal pigmentary abnormalities in 16% of the vitiligo patients (P<.001).
Rogosić et al5 examined the incidence of glaucoma in 42 patients with vitiligo and found primary open-angle glaucoma in 24 (57%) patients. The patients had a mean age of 56 years, mean disease duration of 13 years, and mean IOP of 18 mm Hg for the right eye and 17.5 mm Hg for the left eye. The incidence of glaucoma was significantly higher in patients with vitiligo (P<.001) and increased with disease duration.5
Similar studies, however, have failed to show a relationship between vitiligo and glaucoma. In a study that evaluated the retinal pigment epithelium and the optic nerve in patients with vitiligo, Perossini et al10 found that the fundus examination of the patients was perfectly normal.
In our study, we detected NTG in 18.4% of patients with vitiligo. We did not find a significant statistical difference between patients with and without glaucoma (Table 2). Rogosić et al5 found a significant relationship between age and glaucoma incidence, but we did not find such a relationship, which we believe is because the mean age of our patients was lower than the prior study.
In vitiligo, melanocytes are destroyed through an unknown mechanism. Although the cellular and molecular mechanisms causing melanocytic destruction have not yet been determined, various hypotheses have been put forward to explain the etiopathogenesis of vitiligo. Among these, the most commonly held hypotheses are the neural, self-destruction, and autoimmune hypotheses.16
Based on the observation that stress and serious trauma could precipitate or trigger the onset of vitiligo,16 the neural hypothesis holds that neurochemical mediators released from the edges of the nerve endings exert toxic effects on melanocytes. The fact that both melanocytes and choroidal pigment cells originate from the mesenchyme and dermatomal spreading of segmental vitiligo are arguments propounded in favor of this hypothesis.17
The self-destruction hypothesis suggests that the intrinsic protective mechanisms that normally enable melanocytes to eliminate toxic intermediate products or metabolites on the melanogenesis path have been impaired in patients with vitiligo.18,19 There is evidence of increased oxidative stress over the whole epidermis of patients with vitiligo.20 Thus, free radicals affect melanin and cause membrane damage via lipid peroxidation reactions.21
The autoimmune hypothesis proposes a clinical relationship between vitiligo and several diseases believed to be autoimmune. Because the macrophage infiltration observed in vitiligo lesions is more pronounced on the perilesional skin, this hypothesis holds that macrophages may play a role in melanocyte removal.21 The Koebner phenomenon observed in vitiligo lends support to the critical role of trauma in the etiopathogenesis of the disease.
Although we could not explain the co-presence of vitiligo and glaucoma, we believe that it may result from the fact that both diseases are observed in tissues that have the same embryologic origin and etiology, perhaps vascular or neural disorders, excessive accumulation of free radicals, or the triggering of apoptosis. Dermatologists should be alert to the presence of glaucoma in patients with vitiligo because glaucoma is an eye disease that progresses slowly and may lead to vision loss.
1. Ortonne JP. Vitiligo and other disorders of hypopigmentation. In: Bolognia JB, Jorizzo JL, Rapini RP, eds. Dermatology. 1st ed. New York, NY: Mosby; 2003:947-973.
2. Ortonne JP, Bahadoran P, Fitzpatrick TB, et al. Hypomelanoses and hypermelanoses. In: Freedberg IM, Eisen AZ, Wolff K, eds. Fitzpatrick’s Dermatology in General Medicine. 6th ed. New York, NY: McGraw-Hill; 2003:836-881.
3. van den Wijngaard R, Wijngaard R, Wankowiczs-Kalinsa A, et al. Autoimmune melanocyte destruction in vitiligo. Lab Invest. 2001;81:1061-1067.
4. Shields MB, Ritch R, Krupin T. Classification of the glaucomas. In: Ritch R, Shields MB, Krupin T, eds. The Glaucomas. St Louis, MO: C.V. Mosby Co; 1996:717-725.
5. Rogosić V, Bojić L, Puizina-Ivić N, et al. Vitiligo and glaucoma–an association or a coincidence? a pilot study. Acta Dermatovenerol Croat. 2010;18:21-26.
6. Anderson DR. Normal-tension glaucoma (low-tension glaucoma). Indian J Ophthalmol. 2011;59(suppl 59):S97-S101.
7. Iwata K. Primary open angle glaucoma and low tension glaucoma–pathogenesis and mechanism of optic nerve damage [in Japanese]. Nippon Ganka Gakkai Zasshi. 1992;96:1501-1531.
8. Hitchings RA, Anderton SA. A comparative study of visual field defects seen in patients with low-tension glaucoma and chronic simple glaucoma. Br J Ophthalmol. 1983;67:818-821.
9. Gopal KV, Rama Rao GR, Kumar YH, et al. Vitiligo: a part of a systemic autoimmune process. Indian J Dermatol Venereol Leprol. 2007;73:162-165.
10. Perossini M, Turio E, Perossini T, et al. Vitiligo: ocular and electrophysiological findings. G Ital Dermatol Venereol. 2010;145:141-149.
11. Biswas G, Barbhuiya JN, Biswas MC, et al. Clinical pattern of ocular manifestations in vitiligo. J Indian Med Assoc. 2003;101:478-480.
12. Park S, Albert DM, Bolognia JL. Ocular manifestations of pigmentary disorders. Dermatol Clin. 1992;10:609-622.
13. Albert DM, Nordlund JJ, Lerner AB. Ocular abnormalities occurring with vitiligo. Ophthalmology. 1979;86:1145-1160.
14. Wagoner MD, Albert DM, Lerner AB, et al. New observations on vitiligo and ocular disease. Am J Ophthalmol. 1983;96:16-26.
15. Cowan CL Jr, Halder RM, Grimes PE, et al. Ocular disturbances in vitiligo. J Am Acad Dermatol. 1986;15:17-24.
16. Orecchia GE. Neural pathogenesis. In: Hann SK, Nordlund JJ. Vitiligo. Oxford, England: Blackwell Science Ltd; 2000:142-150.
17. Braun-Falco O, Plewig G, Wolf HH, et al. Disorders of melanin pigmentation. In: Bartels V, ed. Dermatology. Berlin, Germany: Springer; 2000:1013-1042.
18. Tüzün Y, Kotoğyan A, Aydemir EH, et al. Pigmentasyon bozuklukları. In: Baransü O. Dermatoloji. 2nd ed. Istanbul: Nobel Tıp Kitabevi; 1994:557-559.
19. Odom RB, James WD, Berger TG. Disturbances of pigmentation. In: Odom RB, James WD, Berger TG. Andrews’ Diseases of the Skin. 9th ed. Philadelphia, PA: W.B. Saunders Company; 2000:1065-1068.
20. Schallreuter KU. Biochemical theory of vitiligo: a role of pteridines in pigmentation. In: Hann SK, Nordlund JJ. Vitiligo. London, England: Blackwell Science Ltd; 2000:151-159.
21. van den Wijngaard R, Wankowicz-Kalinska A, Le Poole C, et al. Local immune response in skin of generalized vitiligo patients. destruction of melanocytes is associated with the predominent presence of CLA+T cells at the perilesional site. Lab Invest. 2000;80:1299-1309.
1. Ortonne JP. Vitiligo and other disorders of hypopigmentation. In: Bolognia JB, Jorizzo JL, Rapini RP, eds. Dermatology. 1st ed. New York, NY: Mosby; 2003:947-973.
2. Ortonne JP, Bahadoran P, Fitzpatrick TB, et al. Hypomelanoses and hypermelanoses. In: Freedberg IM, Eisen AZ, Wolff K, eds. Fitzpatrick’s Dermatology in General Medicine. 6th ed. New York, NY: McGraw-Hill; 2003:836-881.
3. van den Wijngaard R, Wijngaard R, Wankowiczs-Kalinsa A, et al. Autoimmune melanocyte destruction in vitiligo. Lab Invest. 2001;81:1061-1067.
4. Shields MB, Ritch R, Krupin T. Classification of the glaucomas. In: Ritch R, Shields MB, Krupin T, eds. The Glaucomas. St Louis, MO: C.V. Mosby Co; 1996:717-725.
5. Rogosić V, Bojić L, Puizina-Ivić N, et al. Vitiligo and glaucoma–an association or a coincidence? a pilot study. Acta Dermatovenerol Croat. 2010;18:21-26.
6. Anderson DR. Normal-tension glaucoma (low-tension glaucoma). Indian J Ophthalmol. 2011;59(suppl 59):S97-S101.
7. Iwata K. Primary open angle glaucoma and low tension glaucoma–pathogenesis and mechanism of optic nerve damage [in Japanese]. Nippon Ganka Gakkai Zasshi. 1992;96:1501-1531.
8. Hitchings RA, Anderton SA. A comparative study of visual field defects seen in patients with low-tension glaucoma and chronic simple glaucoma. Br J Ophthalmol. 1983;67:818-821.
9. Gopal KV, Rama Rao GR, Kumar YH, et al. Vitiligo: a part of a systemic autoimmune process. Indian J Dermatol Venereol Leprol. 2007;73:162-165.
10. Perossini M, Turio E, Perossini T, et al. Vitiligo: ocular and electrophysiological findings. G Ital Dermatol Venereol. 2010;145:141-149.
11. Biswas G, Barbhuiya JN, Biswas MC, et al. Clinical pattern of ocular manifestations in vitiligo. J Indian Med Assoc. 2003;101:478-480.
12. Park S, Albert DM, Bolognia JL. Ocular manifestations of pigmentary disorders. Dermatol Clin. 1992;10:609-622.
13. Albert DM, Nordlund JJ, Lerner AB. Ocular abnormalities occurring with vitiligo. Ophthalmology. 1979;86:1145-1160.
14. Wagoner MD, Albert DM, Lerner AB, et al. New observations on vitiligo and ocular disease. Am J Ophthalmol. 1983;96:16-26.
15. Cowan CL Jr, Halder RM, Grimes PE, et al. Ocular disturbances in vitiligo. J Am Acad Dermatol. 1986;15:17-24.
16. Orecchia GE. Neural pathogenesis. In: Hann SK, Nordlund JJ. Vitiligo. Oxford, England: Blackwell Science Ltd; 2000:142-150.
17. Braun-Falco O, Plewig G, Wolf HH, et al. Disorders of melanin pigmentation. In: Bartels V, ed. Dermatology. Berlin, Germany: Springer; 2000:1013-1042.
18. Tüzün Y, Kotoğyan A, Aydemir EH, et al. Pigmentasyon bozuklukları. In: Baransü O. Dermatoloji. 2nd ed. Istanbul: Nobel Tıp Kitabevi; 1994:557-559.
19. Odom RB, James WD, Berger TG. Disturbances of pigmentation. In: Odom RB, James WD, Berger TG. Andrews’ Diseases of the Skin. 9th ed. Philadelphia, PA: W.B. Saunders Company; 2000:1065-1068.
20. Schallreuter KU. Biochemical theory of vitiligo: a role of pteridines in pigmentation. In: Hann SK, Nordlund JJ. Vitiligo. London, England: Blackwell Science Ltd; 2000:151-159.
21. van den Wijngaard R, Wankowicz-Kalinska A, Le Poole C, et al. Local immune response in skin of generalized vitiligo patients. destruction of melanocytes is associated with the predominent presence of CLA+T cells at the perilesional site. Lab Invest. 2000;80:1299-1309.
Practice Points
- Patients with vitiligo may exhibit pigmentary abnormalities of the iris and retina.
- Normal-tension glaucoma may develop in patients with vitiligo.
- Glaucoma progresses slowly and may lead to vision loss; as a result, dermatologists should be alert to the presence of glaucoma in vitiligo patients.

Clinical Pearl: Increasing Utility of Isopropyl Alcohol for Cutaneous Dyschromia
Practice Gap
Conditions with dyschromia including terra firma-forme dermatosis (TFFD), confluent and reticulate papillomatosis (CARP), and acanthosis nigricans are difficult to distinguish from one another.
Diagnostic Tools
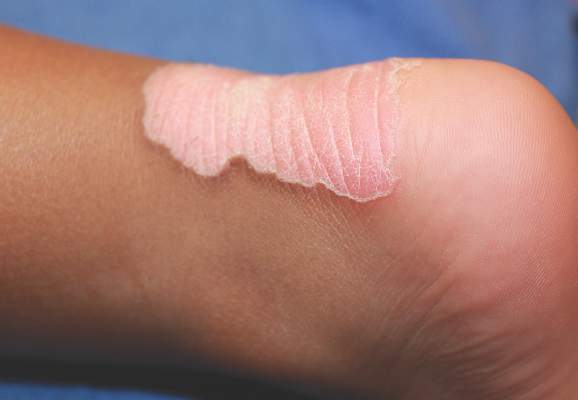
Since its development in 1920, dermatologists have utilized isopropyl alcohol in ways that exceed conventional antimicrobial purposes. If TFFP, CARP, and acanthosis nigricans are suspected, the first step in any algorithmic approach should be to rub the skin with an alcohol pad using firm continuous pressure in an attempt to remove pigmentation. Complete resolution of dyspigmentation strongly supports a diagnosis of TFFD1 and can be curative (Figure). Alcohol can similarly lighten CARP but to a lesser degree than TFFD.2 In contrast, acanthosis nigricans will display minimal to no improvement with isopropyl alcohol.
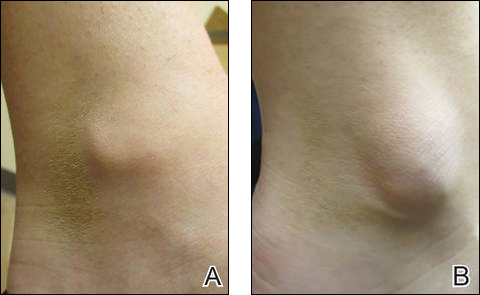
Practice Implications
Isopropyl alcohol has few side effects and each swab costs less than a dime. It is extremely cost effective compared to biopsy and subsequent pathology and laboratory costs. Patients appreciate a noninvasive initial approach, and it is rewarding to treat a cosmetically disturbing condition with ease.
Swabbing the skin with alcohol pads reflects light and improves visualization of veins that should be avoided during surgery. Alcohol-based gel inhibits bacterial colonization, reduces dermatoscope-related nosocomial infection, and enhances dermoscopic resolution.3 Alcohol swabs quickly remove gentian violet, which aids in porokeratosis diagnosis; the pathognomonic cornoid lamella of porokeratosis retains gentian violet.4 A solution of 70% isopropyl alcohol preserves myiasis larvae better than formalin, which causes larval tissue hardening. Alcohol also can be squeezed into the central punctum in myiasis as a form of treatment.5 In conclusion, alcohol represents a convenient, inexpensive, and helpful tool in the dermatologist’s armamentarium that should not be forgotten.
- Browning J, Rosen T. Terra firma-forme dermatosis revisited. Dermatol Online J. 2005;11:15.
- Berk DR. Confluent and reticulated papillomatosis response to 70% alcohol swabbing. Arch Dermatol. 2011;147:247-248.
- Kelly SC, Purcell SM. Prevention of nosocomial infection during dermoscopy? Dermatol Surg. 2006;32:552-555.
- Thomas CJ, Elston DM. Medical pearl: Gentian violet to highlight the cornoid lamella in disseminated superficial actinic porokeratosis. J Am Acad Dermatol. 2005;52(3, pt 1):513-514.
- Meinking TL, Burkhart CN, Burkhart CG. Changing paradigms in parasitic infections: common dermatological helminthic infections and cutaneous myiasis. Clin Dermatol. 2003;21:407-416.
Practice Gap
Conditions with dyschromia including terra firma-forme dermatosis (TFFD), confluent and reticulate papillomatosis (CARP), and acanthosis nigricans are difficult to distinguish from one another.
Diagnostic Tools
Since its development in 1920, dermatologists have utilized isopropyl alcohol in ways that exceed conventional antimicrobial purposes. If TFFP, CARP, and acanthosis nigricans are suspected, the first step in any algorithmic approach should be to rub the skin with an alcohol pad using firm continuous pressure in an attempt to remove pigmentation. Complete resolution of dyspigmentation strongly supports a diagnosis of TFFD1 and can be curative (Figure). Alcohol can similarly lighten CARP but to a lesser degree than TFFD.2 In contrast, acanthosis nigricans will display minimal to no improvement with isopropyl alcohol.
Practice Implications
Isopropyl alcohol has few side effects and each swab costs less than a dime. It is extremely cost effective compared to biopsy and subsequent pathology and laboratory costs. Patients appreciate a noninvasive initial approach, and it is rewarding to treat a cosmetically disturbing condition with ease.
Swabbing the skin with alcohol pads reflects light and improves visualization of veins that should be avoided during surgery. Alcohol-based gel inhibits bacterial colonization, reduces dermatoscope-related nosocomial infection, and enhances dermoscopic resolution.3 Alcohol swabs quickly remove gentian violet, which aids in porokeratosis diagnosis; the pathognomonic cornoid lamella of porokeratosis retains gentian violet.4 A solution of 70% isopropyl alcohol preserves myiasis larvae better than formalin, which causes larval tissue hardening. Alcohol also can be squeezed into the central punctum in myiasis as a form of treatment.5 In conclusion, alcohol represents a convenient, inexpensive, and helpful tool in the dermatologist’s armamentarium that should not be forgotten.
Practice Gap
Conditions with dyschromia including terra firma-forme dermatosis (TFFD), confluent and reticulate papillomatosis (CARP), and acanthosis nigricans are difficult to distinguish from one another.
Diagnostic Tools
Since its development in 1920, dermatologists have utilized isopropyl alcohol in ways that exceed conventional antimicrobial purposes. If TFFP, CARP, and acanthosis nigricans are suspected, the first step in any algorithmic approach should be to rub the skin with an alcohol pad using firm continuous pressure in an attempt to remove pigmentation. Complete resolution of dyspigmentation strongly supports a diagnosis of TFFD1 and can be curative (Figure). Alcohol can similarly lighten CARP but to a lesser degree than TFFD.2 In contrast, acanthosis nigricans will display minimal to no improvement with isopropyl alcohol.
Practice Implications
Isopropyl alcohol has few side effects and each swab costs less than a dime. It is extremely cost effective compared to biopsy and subsequent pathology and laboratory costs. Patients appreciate a noninvasive initial approach, and it is rewarding to treat a cosmetically disturbing condition with ease.
Swabbing the skin with alcohol pads reflects light and improves visualization of veins that should be avoided during surgery. Alcohol-based gel inhibits bacterial colonization, reduces dermatoscope-related nosocomial infection, and enhances dermoscopic resolution.3 Alcohol swabs quickly remove gentian violet, which aids in porokeratosis diagnosis; the pathognomonic cornoid lamella of porokeratosis retains gentian violet.4 A solution of 70% isopropyl alcohol preserves myiasis larvae better than formalin, which causes larval tissue hardening. Alcohol also can be squeezed into the central punctum in myiasis as a form of treatment.5 In conclusion, alcohol represents a convenient, inexpensive, and helpful tool in the dermatologist’s armamentarium that should not be forgotten.
- Browning J, Rosen T. Terra firma-forme dermatosis revisited. Dermatol Online J. 2005;11:15.
- Berk DR. Confluent and reticulated papillomatosis response to 70% alcohol swabbing. Arch Dermatol. 2011;147:247-248.
- Kelly SC, Purcell SM. Prevention of nosocomial infection during dermoscopy? Dermatol Surg. 2006;32:552-555.
- Thomas CJ, Elston DM. Medical pearl: Gentian violet to highlight the cornoid lamella in disseminated superficial actinic porokeratosis. J Am Acad Dermatol. 2005;52(3, pt 1):513-514.
- Meinking TL, Burkhart CN, Burkhart CG. Changing paradigms in parasitic infections: common dermatological helminthic infections and cutaneous myiasis. Clin Dermatol. 2003;21:407-416.
- Browning J, Rosen T. Terra firma-forme dermatosis revisited. Dermatol Online J. 2005;11:15.
- Berk DR. Confluent and reticulated papillomatosis response to 70% alcohol swabbing. Arch Dermatol. 2011;147:247-248.
- Kelly SC, Purcell SM. Prevention of nosocomial infection during dermoscopy? Dermatol Surg. 2006;32:552-555.
- Thomas CJ, Elston DM. Medical pearl: Gentian violet to highlight the cornoid lamella in disseminated superficial actinic porokeratosis. J Am Acad Dermatol. 2005;52(3, pt 1):513-514.
- Meinking TL, Burkhart CN, Burkhart CG. Changing paradigms in parasitic infections: common dermatological helminthic infections and cutaneous myiasis. Clin Dermatol. 2003;21:407-416.
Diagnosing Porokeratosis of Mibelli Every Time: A Novel Biopsy Technique to Maximize Histopathologic Confirmation
Porokeratosis of Mibelli (PM) is a lesion characterized by a surrounding cornoid lamella with variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) in the center of the lesion that typically presents in infancy to early childhood.1 We report a case of PM in which a prior biopsy from the center of the lesion demonstrated papulosquamous dermatitis. We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM.
Case Report
A 3-year-old girl presented with an erythematous, hypopigmented, scaling plaque on the posterior aspect of the left ankle surrounded by a hard rim. The plaque was first noted at 12 months of age and had slowly enlarged as the patient grew. Six months prior, a biopsy from the center of the lesion performed at another facility demonstrated a papulosquamous dermatitis.
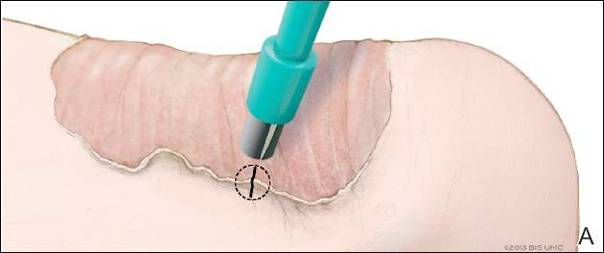
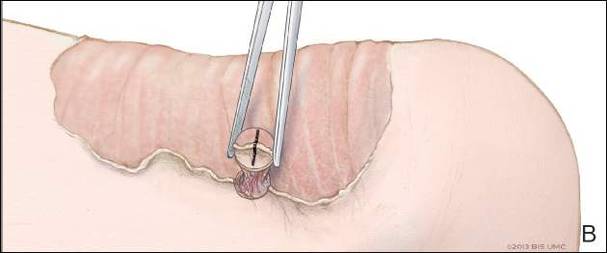
Physical examination revealed a lesion that was 4.2-cm long, 2.2-cm wide at the superior pole, and 3.5-cm wide at the inferior pole (Figure 1). A line was drawn with a skin marker perpendicular to the rim of the lesion (Figure 2A) and a 6-mm punch biopsy was performed, centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). The tissue was then bisected at the bedside along the skin marker line with a #15 blade (Figure 2C) and submitted in formalin for histologic processing. Histologic examination revealed an invagination of the epidermis producing a tier of parakeratotic cells with its apex pointed away from the center of the lesion. Dyskeratotic cells were noted at the base of the parakeratosis (Figure 3). Verrucous hyperplasia was present in the central portion of the specimen adjacent to the cornoid lamella. Based on these histopathologic findings, the correct diagnosis of PM was made.
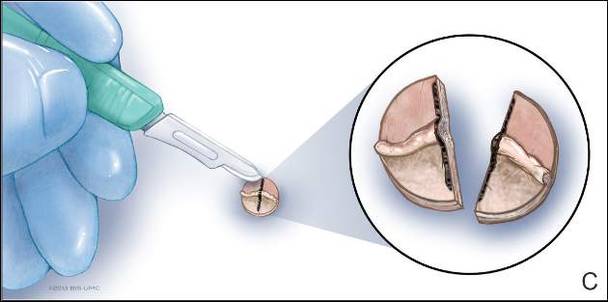
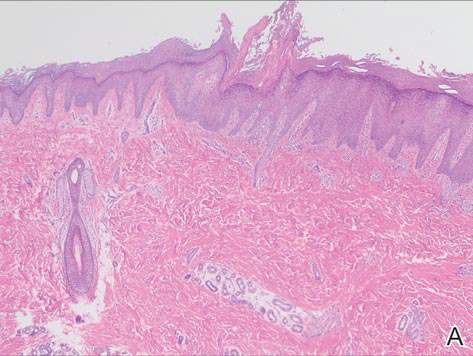
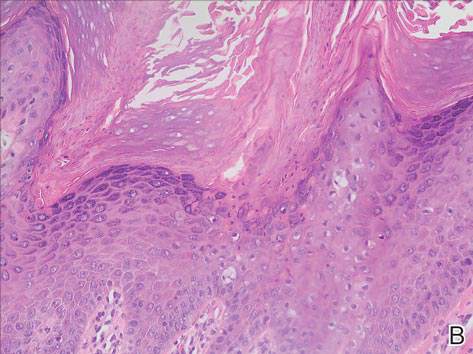
Comment
Porokeratosis of Mibelli is a rare condition that typically presents in infancy to early childhood.1 It may appear as small keratotic papules or larger plaques that reach several centimeters in diameter.2 There is a 7.5% risk for malignant transformation (eg, basal cell carcinoma, squamous cell carcinoma, Bowen disease).3 Variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) typically are present in the center of the lesion. In our case, a biopsy from the center of the plaque demonstrated verrucous hyperplasia. The incorrect diagnosis of PM as psoriasis also has been reported.4
We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM. First, draw a line perpendicular to the rim of the lesion to mark the biopsy site (Figure 2A). Second, perform a punch biopsy centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). Third, section the biopsied tissue with a #15 blade along the perpendicular line at the bedside (Figure 2C). The surgical pathology requisition should mention that the specimen has been transected and the cut edges should be placed down in the cassette, ensuring that the cornoid lamella will be present in cross-section on the slides.
If the punch biopsy specimen is not bisected, it can be difficult to orient it in the pathology laboratory, especially if the cornoid lamellae are not prominent. Furthermore, the technician processing the tissue may not be aware of the importance of sectioning the specimen perpendicular to the cornoid lamella. Following this procedure, diagnosis can be confirmed in virtually every case of PM.
- Richard G, Irvine A, Traupe H, et al. Ichthyosis and disorders of other conification. In: Schachner L, Hansen R, Krafchik B, et al, eds. Pediatric Dermatology. Philadelphia, PA: Elsevier Health Sciences; 2011:640-643.
- Pierson D, Bandel C, Ehrig, et al. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo J, Rapini R, et al, eds. Dermatology. 1st ed. Vol 2. Edinburgh, Scotland: Elsevier; 2003:1707-1709.
- Cort DF, Abdel-Aziz AH. Epithelioma arising in porokeratosis of Mibelli. Br J Plast Surg. 1972;25:318-328.
- De Simone C, Paradisi A, Massi G, et al. Giant verrucous porokeratosis of Mibelli mimicking psoriasis in a patient with psoriasis. J Am Acad Dermatol. 2007;57:665-668.
Porokeratosis of Mibelli (PM) is a lesion characterized by a surrounding cornoid lamella with variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) in the center of the lesion that typically presents in infancy to early childhood.1 We report a case of PM in which a prior biopsy from the center of the lesion demonstrated papulosquamous dermatitis. We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM.
Case Report
A 3-year-old girl presented with an erythematous, hypopigmented, scaling plaque on the posterior aspect of the left ankle surrounded by a hard rim. The plaque was first noted at 12 months of age and had slowly enlarged as the patient grew. Six months prior, a biopsy from the center of the lesion performed at another facility demonstrated a papulosquamous dermatitis.
Physical examination revealed a lesion that was 4.2-cm long, 2.2-cm wide at the superior pole, and 3.5-cm wide at the inferior pole (Figure 1). A line was drawn with a skin marker perpendicular to the rim of the lesion (Figure 2A) and a 6-mm punch biopsy was performed, centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). The tissue was then bisected at the bedside along the skin marker line with a #15 blade (Figure 2C) and submitted in formalin for histologic processing. Histologic examination revealed an invagination of the epidermis producing a tier of parakeratotic cells with its apex pointed away from the center of the lesion. Dyskeratotic cells were noted at the base of the parakeratosis (Figure 3). Verrucous hyperplasia was present in the central portion of the specimen adjacent to the cornoid lamella. Based on these histopathologic findings, the correct diagnosis of PM was made.
Comment
Porokeratosis of Mibelli is a rare condition that typically presents in infancy to early childhood.1 It may appear as small keratotic papules or larger plaques that reach several centimeters in diameter.2 There is a 7.5% risk for malignant transformation (eg, basal cell carcinoma, squamous cell carcinoma, Bowen disease).3 Variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) typically are present in the center of the lesion. In our case, a biopsy from the center of the plaque demonstrated verrucous hyperplasia. The incorrect diagnosis of PM as psoriasis also has been reported.4
We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM. First, draw a line perpendicular to the rim of the lesion to mark the biopsy site (Figure 2A). Second, perform a punch biopsy centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). Third, section the biopsied tissue with a #15 blade along the perpendicular line at the bedside (Figure 2C). The surgical pathology requisition should mention that the specimen has been transected and the cut edges should be placed down in the cassette, ensuring that the cornoid lamella will be present in cross-section on the slides.
If the punch biopsy specimen is not bisected, it can be difficult to orient it in the pathology laboratory, especially if the cornoid lamellae are not prominent. Furthermore, the technician processing the tissue may not be aware of the importance of sectioning the specimen perpendicular to the cornoid lamella. Following this procedure, diagnosis can be confirmed in virtually every case of PM.
Porokeratosis of Mibelli (PM) is a lesion characterized by a surrounding cornoid lamella with variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) in the center of the lesion that typically presents in infancy to early childhood.1 We report a case of PM in which a prior biopsy from the center of the lesion demonstrated papulosquamous dermatitis. We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM.
Case Report
A 3-year-old girl presented with an erythematous, hypopigmented, scaling plaque on the posterior aspect of the left ankle surrounded by a hard rim. The plaque was first noted at 12 months of age and had slowly enlarged as the patient grew. Six months prior, a biopsy from the center of the lesion performed at another facility demonstrated a papulosquamous dermatitis.
Physical examination revealed a lesion that was 4.2-cm long, 2.2-cm wide at the superior pole, and 3.5-cm wide at the inferior pole (Figure 1). A line was drawn with a skin marker perpendicular to the rim of the lesion (Figure 2A) and a 6-mm punch biopsy was performed, centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). The tissue was then bisected at the bedside along the skin marker line with a #15 blade (Figure 2C) and submitted in formalin for histologic processing. Histologic examination revealed an invagination of the epidermis producing a tier of parakeratotic cells with its apex pointed away from the center of the lesion. Dyskeratotic cells were noted at the base of the parakeratosis (Figure 3). Verrucous hyperplasia was present in the central portion of the specimen adjacent to the cornoid lamella. Based on these histopathologic findings, the correct diagnosis of PM was made.
Comment
Porokeratosis of Mibelli is a rare condition that typically presents in infancy to early childhood.1 It may appear as small keratotic papules or larger plaques that reach several centimeters in diameter.2 There is a 7.5% risk for malignant transformation (eg, basal cell carcinoma, squamous cell carcinoma, Bowen disease).3 Variable nonspecific findings (eg, atrophy, acanthosis, verrucous hyperplasia) typically are present in the center of the lesion. In our case, a biopsy from the center of the plaque demonstrated verrucous hyperplasia. The incorrect diagnosis of PM as psoriasis also has been reported.4
We propose a 3-step technique to ensure proper orientation of a punch biopsy in cases of suspected PM. First, draw a line perpendicular to the rim of the lesion to mark the biopsy site (Figure 2A). Second, perform a punch biopsy centered at the intersection of the drawn line and the cornoid lamella (Figure 2B). Third, section the biopsied tissue with a #15 blade along the perpendicular line at the bedside (Figure 2C). The surgical pathology requisition should mention that the specimen has been transected and the cut edges should be placed down in the cassette, ensuring that the cornoid lamella will be present in cross-section on the slides.
If the punch biopsy specimen is not bisected, it can be difficult to orient it in the pathology laboratory, especially if the cornoid lamellae are not prominent. Furthermore, the technician processing the tissue may not be aware of the importance of sectioning the specimen perpendicular to the cornoid lamella. Following this procedure, diagnosis can be confirmed in virtually every case of PM.
- Richard G, Irvine A, Traupe H, et al. Ichthyosis and disorders of other conification. In: Schachner L, Hansen R, Krafchik B, et al, eds. Pediatric Dermatology. Philadelphia, PA: Elsevier Health Sciences; 2011:640-643.
- Pierson D, Bandel C, Ehrig, et al. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo J, Rapini R, et al, eds. Dermatology. 1st ed. Vol 2. Edinburgh, Scotland: Elsevier; 2003:1707-1709.
- Cort DF, Abdel-Aziz AH. Epithelioma arising in porokeratosis of Mibelli. Br J Plast Surg. 1972;25:318-328.
- De Simone C, Paradisi A, Massi G, et al. Giant verrucous porokeratosis of Mibelli mimicking psoriasis in a patient with psoriasis. J Am Acad Dermatol. 2007;57:665-668.
- Richard G, Irvine A, Traupe H, et al. Ichthyosis and disorders of other conification. In: Schachner L, Hansen R, Krafchik B, et al, eds. Pediatric Dermatology. Philadelphia, PA: Elsevier Health Sciences; 2011:640-643.
- Pierson D, Bandel C, Ehrig, et al. Benign epidermal tumors and proliferations. In: Bolognia J, Jorizzo J, Rapini R, et al, eds. Dermatology. 1st ed. Vol 2. Edinburgh, Scotland: Elsevier; 2003:1707-1709.
- Cort DF, Abdel-Aziz AH. Epithelioma arising in porokeratosis of Mibelli. Br J Plast Surg. 1972;25:318-328.
- De Simone C, Paradisi A, Massi G, et al. Giant verrucous porokeratosis of Mibelli mimicking psoriasis in a patient with psoriasis. J Am Acad Dermatol. 2007;57:665-668.
Practice Points
- A biopsy from the center of a plaque of porokeratosis will produce nonspecific findings.
- Bisecting the punch specimen at the bedside along a line drawn perpendicular to the cornoid lamella guarantees proper orientation of the specimen.
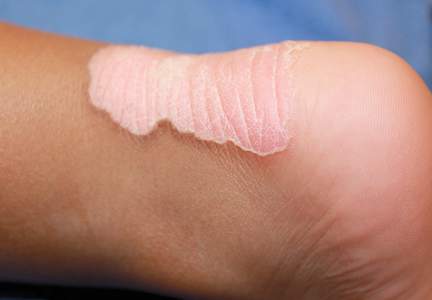
MRSA incidence decreased in children as clindamycin resistance increased
The incidence of methicillin-resistant Staphylococcus aureus (MRSA) infections has decreased in children in recent years, but resistance to clindamycin has increased over the same period, a study showed.
“The epidemic of skin and soft tissue infections and invasive MRSA led to modifications of antimicrobial prescribing practices for suspected S. aureus infections,” reported Dr. Deena E. Sutter of the San Antonio Military Medical Center in Fort Sam Houston, Tex., and her associates. “Over the study period, erythromycin susceptibility among methicillin-susceptible S. aureus (MSSA) remained stable, suggesting that declining clindamycin susceptibility is a result of an increase in inducible resistance.”
The steady decline in clindamycin susceptibility “may lead to some concern about the continued reliance on clindamycin for the empirical treatment of presumptive S. aureus infections, although it is probably premature to abandon this effective antibiotic choice,” they wrote (Pediatrics. 2016 Mar. 1. doi: 10.1542/peds.2015-3099). “It is crucial that clinicians remain knowledgeable about local susceptibility rates as it would be prudent to consider [alternative] antimicrobial agents for empirical use when the local clindamycin susceptibility rate drops below 85%.”
The researchers retrospectively analyzed lab results from 39,209 patients under age 18 who were treated for S. aureus infections at one of the 266 U.S. facilities of the Military Health System from 2005 to 2014. The data included 41,745 S. aureus isolates, classified as MRSA if found resistant to cefoxitin, methicillin, or oxacillin and as methicillin susceptible (MSSA) if susceptible to those antimicrobials. The isolates had also been tested for susceptibility to ciprofloxacin, clindamycin, erythromycin, gentamicin, oxacillin, penicillin, rifampin, tetracycline, and trimethoprim/sulfamethoxazole (TMP/SMX).
During that decade, overall S. aureus susceptibility to clindamycin, ciprofloxacin, and TMP/SMX decreased – although susceptibility to TMP/SMX in 2014 stayed high at 98% – while overall susceptibility to erythromycin, gentamicin, and oxacillin increased. Specifically, 59% of S. aureus isolates were susceptible to oxacillin in 2005, which dropped briefly to 54% in 2007 before climbing to the 2014 rate of 68%.
Meanwhile, overall susceptibility to clindamycin dropped from 91% in 2005 to 86% in 2014, and MSSA susceptibility to clindamycin dropped from 91% in 2005 to 84% in 2014. “Erythromycin susceptibility remained stable among MSSA isolates throughout the study period at 63.5%, whereas MRSA susceptibility to erythromycin increased from 12.1% to 20.5%,” Dr. Sutter and her associates reported. “Ciprofloxacin susceptibility significantly decreased overall, although an initial decrease of 10.6% over the first 7 years of the study was subsequently followed by an increase of 6% between 2011 and 2014.”
Most of the isolates came from patients with skin and soft tissue infections, which were less likely to be susceptible to oxacillin than were other infections. Infections in children aged 1-5 years also were less likely to be susceptible to oxacillin, compared with infections in children of other age groups.
If the local clindamycin susceptibility rate falls below 85%, “beta-lactams, TMP/SMX, or tetracyclines may be used for less severe infections with intravenous vancomycin employed in severe cases,” the investigators said. “If overall MRSA rates continue to decline and clindamycin resistance among MSSA continues to increase, we may see a return to antistaphylococcal beta-lactam antimicrobial agents such as oxacillin or first-generation cephalosporins as preferred empirical therapy for presumed S. aureus infections.”
The research did not use external funding, and the authors reported no relevant financial disclosures.
Staphylococcus aureus is one of the most common organisms isolated from children with health care–associated infections, regardless of whether these infections had their onset in the community or were acquired in the hospital. Thus, the initial empiric treatment of a skin or soft tissue infection or invasive infection in a child almost always includes an antibiotic effective against S. aureus.
However, over the years, clindamycin susceptibility among S. aureus isolates has declined, likely related to the increased use of this agent for empiric as well as definitive treatment of community-acquired (CA) MRSA infections, encouraging the transmission of the genes associated with clindamycin resistance.
What are the implications of the findings from the report by Sutter et al. with respect to the selection of empiric antibiotics for children with suspected S. aureus infections? Currently, considering the still substantial MRSA resistance rates that exceed the 10%-15% level suggested by many experts as the threshold above which agents effective against CA-MRSA isolates should be administered for empiric treatment, changes in the selection of empiric antibiotics are not warranted. If rates of MRSA among S. aureus isolates from otherwise normal children are documented to drop below the 10%-15% threshold in different communities, a modification of current recommendations should be considered. It would also be important to understand why methicillin resistance is declining among S. aureus isolates from CA infections; this information may provide clues for preventing CA-MRSA infections with the use of vaccines or other means. The epidemiology of S. aureus infections in children has been changing over the past 2 decades, which is why it is critical to keep a very close eye on this common pathogen.
These comments were excerpted from an accompanying commentary by Dr. Sheldon L. Kaplan of the infectious disease service at Texas Children’s Hospital in Houston (Pediatrics. 2016 Mar 1. doi: 10.1542/peds.2016-0101). Dr. Kaplan has received research funds from Pfizer, Forest Laboratories, and Cubist.
Staphylococcus aureus is one of the most common organisms isolated from children with health care–associated infections, regardless of whether these infections had their onset in the community or were acquired in the hospital. Thus, the initial empiric treatment of a skin or soft tissue infection or invasive infection in a child almost always includes an antibiotic effective against S. aureus.
However, over the years, clindamycin susceptibility among S. aureus isolates has declined, likely related to the increased use of this agent for empiric as well as definitive treatment of community-acquired (CA) MRSA infections, encouraging the transmission of the genes associated with clindamycin resistance.
What are the implications of the findings from the report by Sutter et al. with respect to the selection of empiric antibiotics for children with suspected S. aureus infections? Currently, considering the still substantial MRSA resistance rates that exceed the 10%-15% level suggested by many experts as the threshold above which agents effective against CA-MRSA isolates should be administered for empiric treatment, changes in the selection of empiric antibiotics are not warranted. If rates of MRSA among S. aureus isolates from otherwise normal children are documented to drop below the 10%-15% threshold in different communities, a modification of current recommendations should be considered. It would also be important to understand why methicillin resistance is declining among S. aureus isolates from CA infections; this information may provide clues for preventing CA-MRSA infections with the use of vaccines or other means. The epidemiology of S. aureus infections in children has been changing over the past 2 decades, which is why it is critical to keep a very close eye on this common pathogen.
These comments were excerpted from an accompanying commentary by Dr. Sheldon L. Kaplan of the infectious disease service at Texas Children’s Hospital in Houston (Pediatrics. 2016 Mar 1. doi: 10.1542/peds.2016-0101). Dr. Kaplan has received research funds from Pfizer, Forest Laboratories, and Cubist.
Staphylococcus aureus is one of the most common organisms isolated from children with health care–associated infections, regardless of whether these infections had their onset in the community or were acquired in the hospital. Thus, the initial empiric treatment of a skin or soft tissue infection or invasive infection in a child almost always includes an antibiotic effective against S. aureus.
However, over the years, clindamycin susceptibility among S. aureus isolates has declined, likely related to the increased use of this agent for empiric as well as definitive treatment of community-acquired (CA) MRSA infections, encouraging the transmission of the genes associated with clindamycin resistance.
What are the implications of the findings from the report by Sutter et al. with respect to the selection of empiric antibiotics for children with suspected S. aureus infections? Currently, considering the still substantial MRSA resistance rates that exceed the 10%-15% level suggested by many experts as the threshold above which agents effective against CA-MRSA isolates should be administered for empiric treatment, changes in the selection of empiric antibiotics are not warranted. If rates of MRSA among S. aureus isolates from otherwise normal children are documented to drop below the 10%-15% threshold in different communities, a modification of current recommendations should be considered. It would also be important to understand why methicillin resistance is declining among S. aureus isolates from CA infections; this information may provide clues for preventing CA-MRSA infections with the use of vaccines or other means. The epidemiology of S. aureus infections in children has been changing over the past 2 decades, which is why it is critical to keep a very close eye on this common pathogen.
These comments were excerpted from an accompanying commentary by Dr. Sheldon L. Kaplan of the infectious disease service at Texas Children’s Hospital in Houston (Pediatrics. 2016 Mar 1. doi: 10.1542/peds.2016-0101). Dr. Kaplan has received research funds from Pfizer, Forest Laboratories, and Cubist.
The incidence of methicillin-resistant Staphylococcus aureus (MRSA) infections has decreased in children in recent years, but resistance to clindamycin has increased over the same period, a study showed.
“The epidemic of skin and soft tissue infections and invasive MRSA led to modifications of antimicrobial prescribing practices for suspected S. aureus infections,” reported Dr. Deena E. Sutter of the San Antonio Military Medical Center in Fort Sam Houston, Tex., and her associates. “Over the study period, erythromycin susceptibility among methicillin-susceptible S. aureus (MSSA) remained stable, suggesting that declining clindamycin susceptibility is a result of an increase in inducible resistance.”
The steady decline in clindamycin susceptibility “may lead to some concern about the continued reliance on clindamycin for the empirical treatment of presumptive S. aureus infections, although it is probably premature to abandon this effective antibiotic choice,” they wrote (Pediatrics. 2016 Mar. 1. doi: 10.1542/peds.2015-3099). “It is crucial that clinicians remain knowledgeable about local susceptibility rates as it would be prudent to consider [alternative] antimicrobial agents for empirical use when the local clindamycin susceptibility rate drops below 85%.”
The researchers retrospectively analyzed lab results from 39,209 patients under age 18 who were treated for S. aureus infections at one of the 266 U.S. facilities of the Military Health System from 2005 to 2014. The data included 41,745 S. aureus isolates, classified as MRSA if found resistant to cefoxitin, methicillin, or oxacillin and as methicillin susceptible (MSSA) if susceptible to those antimicrobials. The isolates had also been tested for susceptibility to ciprofloxacin, clindamycin, erythromycin, gentamicin, oxacillin, penicillin, rifampin, tetracycline, and trimethoprim/sulfamethoxazole (TMP/SMX).
During that decade, overall S. aureus susceptibility to clindamycin, ciprofloxacin, and TMP/SMX decreased – although susceptibility to TMP/SMX in 2014 stayed high at 98% – while overall susceptibility to erythromycin, gentamicin, and oxacillin increased. Specifically, 59% of S. aureus isolates were susceptible to oxacillin in 2005, which dropped briefly to 54% in 2007 before climbing to the 2014 rate of 68%.
Meanwhile, overall susceptibility to clindamycin dropped from 91% in 2005 to 86% in 2014, and MSSA susceptibility to clindamycin dropped from 91% in 2005 to 84% in 2014. “Erythromycin susceptibility remained stable among MSSA isolates throughout the study period at 63.5%, whereas MRSA susceptibility to erythromycin increased from 12.1% to 20.5%,” Dr. Sutter and her associates reported. “Ciprofloxacin susceptibility significantly decreased overall, although an initial decrease of 10.6% over the first 7 years of the study was subsequently followed by an increase of 6% between 2011 and 2014.”
Most of the isolates came from patients with skin and soft tissue infections, which were less likely to be susceptible to oxacillin than were other infections. Infections in children aged 1-5 years also were less likely to be susceptible to oxacillin, compared with infections in children of other age groups.
If the local clindamycin susceptibility rate falls below 85%, “beta-lactams, TMP/SMX, or tetracyclines may be used for less severe infections with intravenous vancomycin employed in severe cases,” the investigators said. “If overall MRSA rates continue to decline and clindamycin resistance among MSSA continues to increase, we may see a return to antistaphylococcal beta-lactam antimicrobial agents such as oxacillin or first-generation cephalosporins as preferred empirical therapy for presumed S. aureus infections.”
The research did not use external funding, and the authors reported no relevant financial disclosures.
The incidence of methicillin-resistant Staphylococcus aureus (MRSA) infections has decreased in children in recent years, but resistance to clindamycin has increased over the same period, a study showed.
“The epidemic of skin and soft tissue infections and invasive MRSA led to modifications of antimicrobial prescribing practices for suspected S. aureus infections,” reported Dr. Deena E. Sutter of the San Antonio Military Medical Center in Fort Sam Houston, Tex., and her associates. “Over the study period, erythromycin susceptibility among methicillin-susceptible S. aureus (MSSA) remained stable, suggesting that declining clindamycin susceptibility is a result of an increase in inducible resistance.”
The steady decline in clindamycin susceptibility “may lead to some concern about the continued reliance on clindamycin for the empirical treatment of presumptive S. aureus infections, although it is probably premature to abandon this effective antibiotic choice,” they wrote (Pediatrics. 2016 Mar. 1. doi: 10.1542/peds.2015-3099). “It is crucial that clinicians remain knowledgeable about local susceptibility rates as it would be prudent to consider [alternative] antimicrobial agents for empirical use when the local clindamycin susceptibility rate drops below 85%.”
The researchers retrospectively analyzed lab results from 39,209 patients under age 18 who were treated for S. aureus infections at one of the 266 U.S. facilities of the Military Health System from 2005 to 2014. The data included 41,745 S. aureus isolates, classified as MRSA if found resistant to cefoxitin, methicillin, or oxacillin and as methicillin susceptible (MSSA) if susceptible to those antimicrobials. The isolates had also been tested for susceptibility to ciprofloxacin, clindamycin, erythromycin, gentamicin, oxacillin, penicillin, rifampin, tetracycline, and trimethoprim/sulfamethoxazole (TMP/SMX).
During that decade, overall S. aureus susceptibility to clindamycin, ciprofloxacin, and TMP/SMX decreased – although susceptibility to TMP/SMX in 2014 stayed high at 98% – while overall susceptibility to erythromycin, gentamicin, and oxacillin increased. Specifically, 59% of S. aureus isolates were susceptible to oxacillin in 2005, which dropped briefly to 54% in 2007 before climbing to the 2014 rate of 68%.
Meanwhile, overall susceptibility to clindamycin dropped from 91% in 2005 to 86% in 2014, and MSSA susceptibility to clindamycin dropped from 91% in 2005 to 84% in 2014. “Erythromycin susceptibility remained stable among MSSA isolates throughout the study period at 63.5%, whereas MRSA susceptibility to erythromycin increased from 12.1% to 20.5%,” Dr. Sutter and her associates reported. “Ciprofloxacin susceptibility significantly decreased overall, although an initial decrease of 10.6% over the first 7 years of the study was subsequently followed by an increase of 6% between 2011 and 2014.”
Most of the isolates came from patients with skin and soft tissue infections, which were less likely to be susceptible to oxacillin than were other infections. Infections in children aged 1-5 years also were less likely to be susceptible to oxacillin, compared with infections in children of other age groups.
If the local clindamycin susceptibility rate falls below 85%, “beta-lactams, TMP/SMX, or tetracyclines may be used for less severe infections with intravenous vancomycin employed in severe cases,” the investigators said. “If overall MRSA rates continue to decline and clindamycin resistance among MSSA continues to increase, we may see a return to antistaphylococcal beta-lactam antimicrobial agents such as oxacillin or first-generation cephalosporins as preferred empirical therapy for presumed S. aureus infections.”
The research did not use external funding, and the authors reported no relevant financial disclosures.
FROM PEDIATRICS
Key clinical point: The incidence of methicillin-resistant Staphylococcus aureus infections has decreased in children in recent years while resistance to clindamycin has increased.
Major finding: MRSA susceptibility to oxacillin increased to 68.4% in 2014, and susceptibility dropped to 86% for clindamycin.
Data source: A retrospective analysis of 41,745 S. aureus isolates from 39,209 patients under age 18 years in the U.S. Military Health System between 2005 and 2014.
Disclosures: The research did not use external funding, and the authors reported no relevant financial disclosures.
Piebaldism in Children
Case Report
A 14-year-old adolescent girl presented with multiple asymptomatic light-colored patches on the forehead, bilateral arms, and legs that had been present since birth. The patient reported that the size of the patches had increased in proportion to her overall growth and that “brown spots” had gradually started to form within and around the patches. She noted that her father and paternal grandfather also had similar clinical findings. A review of systems was negative for hearing impairment, ocular abnormalities, and recurrent infections.
Physical examination revealed an otherwise healthy adolescent girl with Fitzpatrick skin type I and homogeneous blue eyes. Large symmetric depigmented patches were noted on the extensor surfaces of the mid legs and mid forearms (Figure). Macules of baseline pigment and hyperpigmentation were irregularly scattered within and at the periphery of the patches. A triangular hypopigmented patch at the hairline on the mid frontal scalp hairline was accompanied by depigmentation of terminal hairs in this region.
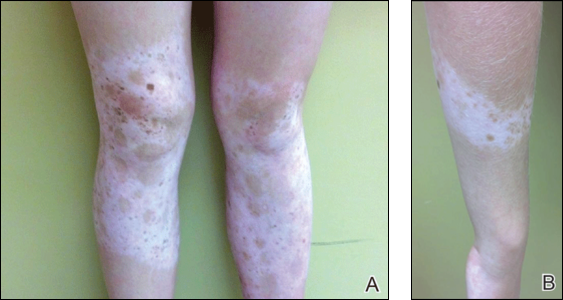
A clinical diagnosis of piebaldism was made and was discussed at length with the patient. Due to the benign nature of the condition and patient preference, no therapeutic intervention was pursued. It was recommended that she apply sunscreen daily for protection of the depigmented areas.
Comment
Piebaldism is a rare hereditary disorder of melanocyte development characterized clinically by the presence of congenital poliosis and leukoderma.1 The exact prevalence of piebaldism is unknown, but it has been estimated that less than 1 in 20,000 children are born with this condition.2 Poliosis circumscripta, traditionally known as white forelock, may be the only manifestation in 80% to 90% of cases and is present at birth.3 The white forelock typically appears in a triangular shape and the underlying skin of the scalp also is amelanotic. The eyebrows and eyelashes also may be involved.3
Characteristically, lesions of leukoderma are well-circumscribed, irregular, white patches that are often accompanied by hyperpigmented macules noted on both depigmented and unaffected adjacent skin.1 The lesions are classically distributed on the central forehead and anterior trunk, with extension to the flanks, anterior mid arms, and mid legs. Sparing of the dorsal midline, hands, feet, and periorificial area is characteristic.1
Depigmented patches typically are nonprogressive and persist into adulthood. Additional hyperpigmented macules may develop at or within the margins of the white patches. Partial or complete repigmentation may occur spontaneously or after trauma in some patients.2 Some children may develop café au lait lesions and may be misdiagnosed as concurrently having neurofibromatosis type I and piebaldism. If neurofibromatosis type I is suspected, patients should be thoroughly evaluated for other diagnostic criteria of this syndrome, as there may be cases of coexistence and overlap with piebaldism.4
Piebaldism is an autosomal-dominant inherited disorder and most commonly develops as a consequence of a mutation in the c-kit proto-oncogene (located on chromosome arm 14q12), which affects melanoblast migration, proliferation, differentiation, and survival.2 In piebaldism, the site of mutation within the gene correlates with the severity of the phenotype.5 Melanocytes are histologically and ultrastructurally absent or considerably reduced in depigmented patches but are normal in number in the hyperpigmented areas.2
Rare cases of piebaldism have been reported in association with other diseases, including congenital megacolon, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease (transient acantholytic dermatosis), and glycogen-storage disease type 1a.1,6 Poliosis alone may be the initial presentation of certain genetic syndromes, including Waardenburg syndrome (WS) and tuberous sclerosis; it also may be acquired in the setting of several conditions, including vitiligo, Vogt-Koyanagi-Harada syndrome, Alezzandrini syndrome, alopecia areata, and sarcoidosis.3
Notably, the diagnosis of piebaldism should alert the clinician to the possibility of WS, an autosomal-dominant disease characterized by a congenital white forelock, leukoderma in a piebaldlike distribution, lateral displacement of the medial canthi, a hypertrophic nasal root, heterochromia iridis, and progressive sensorineural hearing loss.7 Four clinical subtypes of WS have been described, with various gene mutations implicated: type 1 is the classic form, type 2 lacks dystopia canthorum and has a stonger association with deafness, type 3 is associated with limb abnormalities, and type 4 is associated with congenital megacolon. A case of WS type 1 has been described in association with facial nerve palsy and lingua plicata, 2 main features of Melkerson-Rosenthal syndrome.8 Depigmentation in WS is caused by the absence of melanocytes in the affected areas as well as failed migration of melanocytes to the ears and eyes.3 Waardenburg syndrome may be distinguished from piebaldism by characteristic facial features of the disease and should prompt a thorough ocular and auditory examination in affected patients.9
Although not a diagnostic criterion, poliosis rarely has been reported as one of the earliest associated findings of tuberous sclerosis.3,10 Major cutaneous features of this disease include facial angiofibromas, hypomelanotic macules, shagreen patches (connective tissue nevi), periungual fibromas, molluscum pendulum, and café au lait macules.
Vitiligo also may be considered in the differential diagnosis of piebaldism and can be distinguished by the presence of depigmented patches in a typical acral and periorificial distribution, lack of congential presentation, and relatively progressive course. Vitiligo is characterized by an acquired loss of epidermal melanocytes, leading to depigmented macules and patches.1,3
Vitiligo, poliosis, and alopecia areata usually are late clinical manifestations of Vogt-Koyanagi-Harada syndrome, a rare condition characterized by an autoimmune response to melanocyte-associated antigens. This condition initially presents with neurologic and ocular manifestations including headache, muscle weakness, tinnitus, uveitis, and choroiditis prior to dermatologic manifestations.11
Alezzandrini syndrome, a rare and closely related disorder, is distinctly characterized by whitening of scalp hair, eyebrows, and eyelashes, along with unilateral depigmentation of facial skin. This presentation is associated with ipsilateral visual changes and hearing abnormalities.12
The absence of abnormal ocular, auditory, and neurologic examinations, along with lack of characteristic cutaneous features indicating any of the aforementioned disorders, highly suggests a diagnosis of piebaldism.
Piebaldism is considered a relatively benign disorder but can be highly socially disabling, which presents a therapeutic challenge in affected children. Depigmented skin in piebaldism generally is considered unresponsive to medical or light therapy.1 Topical treatments with makeup or artificial pigmenting agents (eg, dihydroxyacetone [an ingredient used in sunless tanning products]) are useful but temporary. Sunscreen should be used judiciously to avoid sunburn and reduce carcinogenic potential.13
Several surgical techniques have been reported for treatment of leukoderma but with variable success. Of those reported, micropunch transplantation (minigrafting) using epidermal donor sites of 1 to 1.25 mm is a relatively inexpensive and effective method but is limited by scarring at the donor site.14 Autologous cultured epidermal cellular grafting with a controlled number of melanocytes is reported to achieve greater than 75% repigmentation. It requires fewer donor sites and, therefore, results in less scarring.15 Additionally, use of the erbium-doped:YAG laser aids in deepithelialization of the recipient site, allowing for treatment of large piebald lesions during a single operation.16 Despite these advances, additional studies are needed to improve quality of life in those affected.
- Janjua SA, Khachemoune A, Guldbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
- Agarwal S, Ojha A. Piebaldism: a brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
- Sleiman R, Kurban M, Succaria F, et al. Poliosis circumscripta: overview and underlying causes. J Am Acad Dermatol. 2013;69:625-633.
- Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-355.
- López V, Jordá E. Piebaldism in a 2-year-old girl. Dermatol Online J. 2011;17:13.
- Ghoshal B, Sarkar N, Bhattacharjee M, et al. Glycogen storage disease 1a with piebaldism. Indian Pediatr. 2012;49:235-236.
- Salvatore S, Carnevale C, Infussi R, et al. Waardenburg syndrome: a review of literature and case reports. Clin Ter. 2012;163:e85-e94.
- Dourmishev AL, Dourmishev LA, Schwartz RA, et al. Waardenburg syndrome. Int J Dermatol. 1999;38:656-663.
- Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges. 2010;8:187-201.
- McWilliam RC, Stephenson JB. Depigmented hair. the earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53:961-963.
- Chan EW, Sanjay S, Chang BC. Headache, red eyes, blurred vision and hearing loss. diagnosis: Vogt-Koyanagi-Harada syndrome. CMAJ. 2010;182:1205-1209.
- Andrade A, Pithon M. Alezzandrini syndrome: report of a sixth clinical case. Dermatology (Basel). 2011;222:8-9.
- Suga Y, Ikejima A, Matsuba S, et al. Medical pearl: DHA application for camouflaging segmental vitiligo and piebald lesions. J Am Acad Dermatol. 2002;47:436-438.
- Neves DR, Régis Júnior JR, Oliveira PJ, et al. Melanocyte transplant in piebaldism: case report. An Bras Dermatol. 2010;85:384-388.
- Van geel N, Wallaeys E, Goh BK, et al. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186-1193.
- Guerra L, Primavera G, Raskovic D, et al. Permanent repigmentation of piebaldism by erbium:YAG laser and autologous cultured epidermis. Br J Dermatol. 2004;150:715-721.
Case Report
A 14-year-old adolescent girl presented with multiple asymptomatic light-colored patches on the forehead, bilateral arms, and legs that had been present since birth. The patient reported that the size of the patches had increased in proportion to her overall growth and that “brown spots” had gradually started to form within and around the patches. She noted that her father and paternal grandfather also had similar clinical findings. A review of systems was negative for hearing impairment, ocular abnormalities, and recurrent infections.
Physical examination revealed an otherwise healthy adolescent girl with Fitzpatrick skin type I and homogeneous blue eyes. Large symmetric depigmented patches were noted on the extensor surfaces of the mid legs and mid forearms (Figure). Macules of baseline pigment and hyperpigmentation were irregularly scattered within and at the periphery of the patches. A triangular hypopigmented patch at the hairline on the mid frontal scalp hairline was accompanied by depigmentation of terminal hairs in this region.
A clinical diagnosis of piebaldism was made and was discussed at length with the patient. Due to the benign nature of the condition and patient preference, no therapeutic intervention was pursued. It was recommended that she apply sunscreen daily for protection of the depigmented areas.
Comment
Piebaldism is a rare hereditary disorder of melanocyte development characterized clinically by the presence of congenital poliosis and leukoderma.1 The exact prevalence of piebaldism is unknown, but it has been estimated that less than 1 in 20,000 children are born with this condition.2 Poliosis circumscripta, traditionally known as white forelock, may be the only manifestation in 80% to 90% of cases and is present at birth.3 The white forelock typically appears in a triangular shape and the underlying skin of the scalp also is amelanotic. The eyebrows and eyelashes also may be involved.3
Characteristically, lesions of leukoderma are well-circumscribed, irregular, white patches that are often accompanied by hyperpigmented macules noted on both depigmented and unaffected adjacent skin.1 The lesions are classically distributed on the central forehead and anterior trunk, with extension to the flanks, anterior mid arms, and mid legs. Sparing of the dorsal midline, hands, feet, and periorificial area is characteristic.1
Depigmented patches typically are nonprogressive and persist into adulthood. Additional hyperpigmented macules may develop at or within the margins of the white patches. Partial or complete repigmentation may occur spontaneously or after trauma in some patients.2 Some children may develop café au lait lesions and may be misdiagnosed as concurrently having neurofibromatosis type I and piebaldism. If neurofibromatosis type I is suspected, patients should be thoroughly evaluated for other diagnostic criteria of this syndrome, as there may be cases of coexistence and overlap with piebaldism.4
Piebaldism is an autosomal-dominant inherited disorder and most commonly develops as a consequence of a mutation in the c-kit proto-oncogene (located on chromosome arm 14q12), which affects melanoblast migration, proliferation, differentiation, and survival.2 In piebaldism, the site of mutation within the gene correlates with the severity of the phenotype.5 Melanocytes are histologically and ultrastructurally absent or considerably reduced in depigmented patches but are normal in number in the hyperpigmented areas.2
Rare cases of piebaldism have been reported in association with other diseases, including congenital megacolon, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease (transient acantholytic dermatosis), and glycogen-storage disease type 1a.1,6 Poliosis alone may be the initial presentation of certain genetic syndromes, including Waardenburg syndrome (WS) and tuberous sclerosis; it also may be acquired in the setting of several conditions, including vitiligo, Vogt-Koyanagi-Harada syndrome, Alezzandrini syndrome, alopecia areata, and sarcoidosis.3
Notably, the diagnosis of piebaldism should alert the clinician to the possibility of WS, an autosomal-dominant disease characterized by a congenital white forelock, leukoderma in a piebaldlike distribution, lateral displacement of the medial canthi, a hypertrophic nasal root, heterochromia iridis, and progressive sensorineural hearing loss.7 Four clinical subtypes of WS have been described, with various gene mutations implicated: type 1 is the classic form, type 2 lacks dystopia canthorum and has a stonger association with deafness, type 3 is associated with limb abnormalities, and type 4 is associated with congenital megacolon. A case of WS type 1 has been described in association with facial nerve palsy and lingua plicata, 2 main features of Melkerson-Rosenthal syndrome.8 Depigmentation in WS is caused by the absence of melanocytes in the affected areas as well as failed migration of melanocytes to the ears and eyes.3 Waardenburg syndrome may be distinguished from piebaldism by characteristic facial features of the disease and should prompt a thorough ocular and auditory examination in affected patients.9
Although not a diagnostic criterion, poliosis rarely has been reported as one of the earliest associated findings of tuberous sclerosis.3,10 Major cutaneous features of this disease include facial angiofibromas, hypomelanotic macules, shagreen patches (connective tissue nevi), periungual fibromas, molluscum pendulum, and café au lait macules.
Vitiligo also may be considered in the differential diagnosis of piebaldism and can be distinguished by the presence of depigmented patches in a typical acral and periorificial distribution, lack of congential presentation, and relatively progressive course. Vitiligo is characterized by an acquired loss of epidermal melanocytes, leading to depigmented macules and patches.1,3
Vitiligo, poliosis, and alopecia areata usually are late clinical manifestations of Vogt-Koyanagi-Harada syndrome, a rare condition characterized by an autoimmune response to melanocyte-associated antigens. This condition initially presents with neurologic and ocular manifestations including headache, muscle weakness, tinnitus, uveitis, and choroiditis prior to dermatologic manifestations.11
Alezzandrini syndrome, a rare and closely related disorder, is distinctly characterized by whitening of scalp hair, eyebrows, and eyelashes, along with unilateral depigmentation of facial skin. This presentation is associated with ipsilateral visual changes and hearing abnormalities.12
The absence of abnormal ocular, auditory, and neurologic examinations, along with lack of characteristic cutaneous features indicating any of the aforementioned disorders, highly suggests a diagnosis of piebaldism.
Piebaldism is considered a relatively benign disorder but can be highly socially disabling, which presents a therapeutic challenge in affected children. Depigmented skin in piebaldism generally is considered unresponsive to medical or light therapy.1 Topical treatments with makeup or artificial pigmenting agents (eg, dihydroxyacetone [an ingredient used in sunless tanning products]) are useful but temporary. Sunscreen should be used judiciously to avoid sunburn and reduce carcinogenic potential.13
Several surgical techniques have been reported for treatment of leukoderma but with variable success. Of those reported, micropunch transplantation (minigrafting) using epidermal donor sites of 1 to 1.25 mm is a relatively inexpensive and effective method but is limited by scarring at the donor site.14 Autologous cultured epidermal cellular grafting with a controlled number of melanocytes is reported to achieve greater than 75% repigmentation. It requires fewer donor sites and, therefore, results in less scarring.15 Additionally, use of the erbium-doped:YAG laser aids in deepithelialization of the recipient site, allowing for treatment of large piebald lesions during a single operation.16 Despite these advances, additional studies are needed to improve quality of life in those affected.
Case Report
A 14-year-old adolescent girl presented with multiple asymptomatic light-colored patches on the forehead, bilateral arms, and legs that had been present since birth. The patient reported that the size of the patches had increased in proportion to her overall growth and that “brown spots” had gradually started to form within and around the patches. She noted that her father and paternal grandfather also had similar clinical findings. A review of systems was negative for hearing impairment, ocular abnormalities, and recurrent infections.
Physical examination revealed an otherwise healthy adolescent girl with Fitzpatrick skin type I and homogeneous blue eyes. Large symmetric depigmented patches were noted on the extensor surfaces of the mid legs and mid forearms (Figure). Macules of baseline pigment and hyperpigmentation were irregularly scattered within and at the periphery of the patches. A triangular hypopigmented patch at the hairline on the mid frontal scalp hairline was accompanied by depigmentation of terminal hairs in this region.
A clinical diagnosis of piebaldism was made and was discussed at length with the patient. Due to the benign nature of the condition and patient preference, no therapeutic intervention was pursued. It was recommended that she apply sunscreen daily for protection of the depigmented areas.
Comment
Piebaldism is a rare hereditary disorder of melanocyte development characterized clinically by the presence of congenital poliosis and leukoderma.1 The exact prevalence of piebaldism is unknown, but it has been estimated that less than 1 in 20,000 children are born with this condition.2 Poliosis circumscripta, traditionally known as white forelock, may be the only manifestation in 80% to 90% of cases and is present at birth.3 The white forelock typically appears in a triangular shape and the underlying skin of the scalp also is amelanotic. The eyebrows and eyelashes also may be involved.3
Characteristically, lesions of leukoderma are well-circumscribed, irregular, white patches that are often accompanied by hyperpigmented macules noted on both depigmented and unaffected adjacent skin.1 The lesions are classically distributed on the central forehead and anterior trunk, with extension to the flanks, anterior mid arms, and mid legs. Sparing of the dorsal midline, hands, feet, and periorificial area is characteristic.1
Depigmented patches typically are nonprogressive and persist into adulthood. Additional hyperpigmented macules may develop at or within the margins of the white patches. Partial or complete repigmentation may occur spontaneously or after trauma in some patients.2 Some children may develop café au lait lesions and may be misdiagnosed as concurrently having neurofibromatosis type I and piebaldism. If neurofibromatosis type I is suspected, patients should be thoroughly evaluated for other diagnostic criteria of this syndrome, as there may be cases of coexistence and overlap with piebaldism.4
Piebaldism is an autosomal-dominant inherited disorder and most commonly develops as a consequence of a mutation in the c-kit proto-oncogene (located on chromosome arm 14q12), which affects melanoblast migration, proliferation, differentiation, and survival.2 In piebaldism, the site of mutation within the gene correlates with the severity of the phenotype.5 Melanocytes are histologically and ultrastructurally absent or considerably reduced in depigmented patches but are normal in number in the hyperpigmented areas.2
Rare cases of piebaldism have been reported in association with other diseases, including congenital megacolon, congenital dyserythropoietic anemia type II, Diamond-Blackfan anemia, Grover disease (transient acantholytic dermatosis), and glycogen-storage disease type 1a.1,6 Poliosis alone may be the initial presentation of certain genetic syndromes, including Waardenburg syndrome (WS) and tuberous sclerosis; it also may be acquired in the setting of several conditions, including vitiligo, Vogt-Koyanagi-Harada syndrome, Alezzandrini syndrome, alopecia areata, and sarcoidosis.3
Notably, the diagnosis of piebaldism should alert the clinician to the possibility of WS, an autosomal-dominant disease characterized by a congenital white forelock, leukoderma in a piebaldlike distribution, lateral displacement of the medial canthi, a hypertrophic nasal root, heterochromia iridis, and progressive sensorineural hearing loss.7 Four clinical subtypes of WS have been described, with various gene mutations implicated: type 1 is the classic form, type 2 lacks dystopia canthorum and has a stonger association with deafness, type 3 is associated with limb abnormalities, and type 4 is associated with congenital megacolon. A case of WS type 1 has been described in association with facial nerve palsy and lingua plicata, 2 main features of Melkerson-Rosenthal syndrome.8 Depigmentation in WS is caused by the absence of melanocytes in the affected areas as well as failed migration of melanocytes to the ears and eyes.3 Waardenburg syndrome may be distinguished from piebaldism by characteristic facial features of the disease and should prompt a thorough ocular and auditory examination in affected patients.9
Although not a diagnostic criterion, poliosis rarely has been reported as one of the earliest associated findings of tuberous sclerosis.3,10 Major cutaneous features of this disease include facial angiofibromas, hypomelanotic macules, shagreen patches (connective tissue nevi), periungual fibromas, molluscum pendulum, and café au lait macules.
Vitiligo also may be considered in the differential diagnosis of piebaldism and can be distinguished by the presence of depigmented patches in a typical acral and periorificial distribution, lack of congential presentation, and relatively progressive course. Vitiligo is characterized by an acquired loss of epidermal melanocytes, leading to depigmented macules and patches.1,3
Vitiligo, poliosis, and alopecia areata usually are late clinical manifestations of Vogt-Koyanagi-Harada syndrome, a rare condition characterized by an autoimmune response to melanocyte-associated antigens. This condition initially presents with neurologic and ocular manifestations including headache, muscle weakness, tinnitus, uveitis, and choroiditis prior to dermatologic manifestations.11
Alezzandrini syndrome, a rare and closely related disorder, is distinctly characterized by whitening of scalp hair, eyebrows, and eyelashes, along with unilateral depigmentation of facial skin. This presentation is associated with ipsilateral visual changes and hearing abnormalities.12
The absence of abnormal ocular, auditory, and neurologic examinations, along with lack of characteristic cutaneous features indicating any of the aforementioned disorders, highly suggests a diagnosis of piebaldism.
Piebaldism is considered a relatively benign disorder but can be highly socially disabling, which presents a therapeutic challenge in affected children. Depigmented skin in piebaldism generally is considered unresponsive to medical or light therapy.1 Topical treatments with makeup or artificial pigmenting agents (eg, dihydroxyacetone [an ingredient used in sunless tanning products]) are useful but temporary. Sunscreen should be used judiciously to avoid sunburn and reduce carcinogenic potential.13
Several surgical techniques have been reported for treatment of leukoderma but with variable success. Of those reported, micropunch transplantation (minigrafting) using epidermal donor sites of 1 to 1.25 mm is a relatively inexpensive and effective method but is limited by scarring at the donor site.14 Autologous cultured epidermal cellular grafting with a controlled number of melanocytes is reported to achieve greater than 75% repigmentation. It requires fewer donor sites and, therefore, results in less scarring.15 Additionally, use of the erbium-doped:YAG laser aids in deepithelialization of the recipient site, allowing for treatment of large piebald lesions during a single operation.16 Despite these advances, additional studies are needed to improve quality of life in those affected.
- Janjua SA, Khachemoune A, Guldbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
- Agarwal S, Ojha A. Piebaldism: a brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
- Sleiman R, Kurban M, Succaria F, et al. Poliosis circumscripta: overview and underlying causes. J Am Acad Dermatol. 2013;69:625-633.
- Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-355.
- López V, Jordá E. Piebaldism in a 2-year-old girl. Dermatol Online J. 2011;17:13.
- Ghoshal B, Sarkar N, Bhattacharjee M, et al. Glycogen storage disease 1a with piebaldism. Indian Pediatr. 2012;49:235-236.
- Salvatore S, Carnevale C, Infussi R, et al. Waardenburg syndrome: a review of literature and case reports. Clin Ter. 2012;163:e85-e94.
- Dourmishev AL, Dourmishev LA, Schwartz RA, et al. Waardenburg syndrome. Int J Dermatol. 1999;38:656-663.
- Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges. 2010;8:187-201.
- McWilliam RC, Stephenson JB. Depigmented hair. the earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53:961-963.
- Chan EW, Sanjay S, Chang BC. Headache, red eyes, blurred vision and hearing loss. diagnosis: Vogt-Koyanagi-Harada syndrome. CMAJ. 2010;182:1205-1209.
- Andrade A, Pithon M. Alezzandrini syndrome: report of a sixth clinical case. Dermatology (Basel). 2011;222:8-9.
- Suga Y, Ikejima A, Matsuba S, et al. Medical pearl: DHA application for camouflaging segmental vitiligo and piebald lesions. J Am Acad Dermatol. 2002;47:436-438.
- Neves DR, Régis Júnior JR, Oliveira PJ, et al. Melanocyte transplant in piebaldism: case report. An Bras Dermatol. 2010;85:384-388.
- Van geel N, Wallaeys E, Goh BK, et al. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186-1193.
- Guerra L, Primavera G, Raskovic D, et al. Permanent repigmentation of piebaldism by erbium:YAG laser and autologous cultured epidermis. Br J Dermatol. 2004;150:715-721.
- Janjua SA, Khachemoune A, Guldbakke KK. Piebaldism: a case report and a concise review of the literature. Cutis. 2007;80:411-414.
- Agarwal S, Ojha A. Piebaldism: a brief report and review of the literature. Indian Dermatol Online J. 2012;3:144-147.
- Sleiman R, Kurban M, Succaria F, et al. Poliosis circumscripta: overview and underlying causes. J Am Acad Dermatol. 2013;69:625-633.
- Oiso N, Fukai K, Kawada A, et al. Piebaldism. J Dermatol. 2013;40:330-355.
- López V, Jordá E. Piebaldism in a 2-year-old girl. Dermatol Online J. 2011;17:13.
- Ghoshal B, Sarkar N, Bhattacharjee M, et al. Glycogen storage disease 1a with piebaldism. Indian Pediatr. 2012;49:235-236.
- Salvatore S, Carnevale C, Infussi R, et al. Waardenburg syndrome: a review of literature and case reports. Clin Ter. 2012;163:e85-e94.
- Dourmishev AL, Dourmishev LA, Schwartz RA, et al. Waardenburg syndrome. Int J Dermatol. 1999;38:656-663.
- Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges. 2010;8:187-201.
- McWilliam RC, Stephenson JB. Depigmented hair. the earliest sign of tuberous sclerosis. Arch Dis Child. 1978;53:961-963.
- Chan EW, Sanjay S, Chang BC. Headache, red eyes, blurred vision and hearing loss. diagnosis: Vogt-Koyanagi-Harada syndrome. CMAJ. 2010;182:1205-1209.
- Andrade A, Pithon M. Alezzandrini syndrome: report of a sixth clinical case. Dermatology (Basel). 2011;222:8-9.
- Suga Y, Ikejima A, Matsuba S, et al. Medical pearl: DHA application for camouflaging segmental vitiligo and piebald lesions. J Am Acad Dermatol. 2002;47:436-438.
- Neves DR, Régis Júnior JR, Oliveira PJ, et al. Melanocyte transplant in piebaldism: case report. An Bras Dermatol. 2010;85:384-388.
- Van geel N, Wallaeys E, Goh BK, et al. Long-term results of noncultured epidermal cellular grafting in vitiligo, halo naevi, piebaldism and naevus depigmentosus. Br J Dermatol. 2010;163:1186-1193.
- Guerra L, Primavera G, Raskovic D, et al. Permanent repigmentation of piebaldism by erbium:YAG laser and autologous cultured epidermis. Br J Dermatol. 2004;150:715-721.
Practice Points
- Poliosis circumscripta (or white forelock) is commonly the only manifestation of piebaldism in children.
- Affected areas of leukoderma in piebaldism are classically distributed on the central forehead, anterior trunk, and mid extremities.
- The presence of congenital leukoderma should prompt a thorough skin examination and review of the patient’s medical history for evidence of ocular, auditory, and/or neurologic abnormalities.
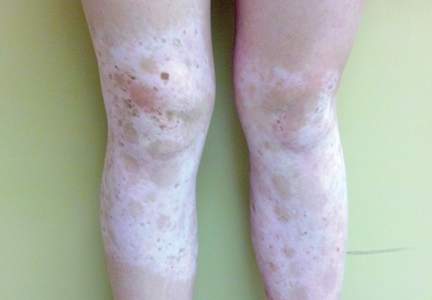
A Case of Bloom Syndrome With Uncommon Clinical Manifestations Confirmed on Genetic Testing
Bloom syndrome, also called congenital telangiectatic erythema and stunted growth, was first described by David Bloom in 1954.1 It is a rare autosomal-recessive disorder (Online Mendelian Inheritance in Man 210900) characterized by specific clinical manifestations including photosensitivity, telangiectatic facial erythema, proportionate growth deficiency, hypogonadism, immunodeficiency, and a tendency to develop various malignancies.2 Linkage analysis revealed that the Bloom syndrome gene locus resides on chromosome arm 15q26.1,3 and the BLM gene in this region has been identified as being responsible for the development of Bloom syndrome.4,5 We report the case of a 12-year-old Chinese girl with Bloom syndrome and detected BLM gene. The evaluation was approved by the Institutional Ethical Review Boards of Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China).
Case Report
We evaluated a Bloom syndrome family, which consisted of the patient and her parents. The patient was a 12-year-old Chinese girl who was apparently healthy until 3 months of age when her parents noticed an erythematous eruption with blisters on the face. Exacerbation after exposure to sunlight is usual, which results in the eruption becoming prominent in summer and fainter in winter.2 Gradually, the patient’s skin lesions became more progressive, extending to the forehead, nose, and ears, with oozing, crusting, atrophy, and telangiectases developing on the face despite treatment. In the last 3 years, no blisters were present on the patient’s face because of her efforts to avoid sun exposure. She had no history of recurrent infections.
On physical examination, the patient was generally healthy with normal intelligence and short stature. She weighed 26 kg and was approximately 122-cm tall. Telangiectatic erythema and slight scaling were noted on the face, which simulated lupus erythematosus (Figures 1A and 1B). She had additional abnormalities including alopecia areata (Figure 1C), eyebrow hair loss, flat nose, reticular pigmentation on the forehead and trunk, and finger swelling. The distal phalanges on all 10 fingers became short and sharpened and the fingernails became wider than they were long (Figure 1D). Laboratory investigations, including a complete blood cell count, liver and kidney function tests, stool examination, serum complement, and albumin and globulin levels, were within reference range.

After informed consent was obtained, a mutation analysis of the BLM gene was performed in the patient and her parents. We used a genomic DNA purification kit to extract genomic DNA from peripheral blood according to the manufacturer’s protocol. Genomic DNA was used to amplify the exons of the BLM gene with intron flanking sequences by polymerase chain reaction with the primer described elsewhere.6 After the amplification, the polymerase chain reaction products were purified and the BLM gene was sequenced. Sequence comparisons and analysis were performed using Phred/Phrap/Consed version 12.0.
The patient was found to carry changes in 2 heterozygous nucleotide sites, including c.2603C>T in exon 13 and c.3961G>A in exon 21 of the BLM gene. The patient’s father was found to carry c.2603C>T and her mother carried c.3961G>A (Figure 2).
Comment
Patients with Bloom syndrome have a characteristic clinical appearance that typically includes photosensitivity, telangiectatic facial erythema, and growth deficiency. Telangiectatic erythema of the face develops during infancy or early childhood as red macules or plaques and may simulate lupus erythematosus. The lesions are described as a butterfly rash affecting the bridge of the nose and cheeks but also may involve the margins of the eyelids, forehead, ears, and sometimes the dorsa of the hands and forearms. Moderate and proportionate growth deficiencies develop both in utero and postnatally. Patients with Bloom syndrome characteristically have narrow, slender, distinct facial features with micrognathism and a relatively prominent nose. They usually may have mild microcephaly, meaning the head is longer and narrower than normal.2,7-10
German and Takebe11 reported 14 Japanese patients with Bloom syndrome. The phenotype differs somewhat from most cases recognized elsewhere in that dolichocephaly was a less constant feature, the facial skin was less prominent, and life-threatening infections were less common. Our patient had typical telangiectatic facial erythema without microcephaly, dolichocephaly, or any infections. She also had some uncommon manifestations such as alopecia areata, eyebrow hair loss, flat nose, reticular pigmentation, and short sharpened distal phalanges with fingernails that were wider than they were long. Although she had no recurrent infections and laboratory tests were within reference range, the alopecia areata and eyebrow hair loss may be associated with an abnormal immune response. The reasons for the short sharpened distal phalanges and the fingernail findings are unclear. The presence of reticular pigmentation also is unclear but may be associated with photosensitivity. Since the BLM gene was discovered to be the disease-causing gene of Bloom syndrome in 1995,4,5 approximately 70 mutations were reported. The BLM gene encodes for the Bloom syndrome protein, a DNA helicase of the highly conserved RecQ subfamily of helicases, a group of nuclear proteins important in the maintenance of genomic stability.12
Mutation analysis of the BLM gene in our patient showed changes in 2 heterozygous nucleotide sites, including c.2603C>T in exon 13 and c.3961G>A in exon 21 of the BLM gene, which altered proline residue with leucine residue at 868 and valine residue with isoleucine residue at 1321, respectively. According to GenBank,13,14 c.2603C>T and c.3961G>A are single nucleotide polymorphisms of the BLM gene. The genotypic distribution of International HapMap Project15 showed that C=602/602 and T=0/602 on c.2603 in 301 unrelated Chinese patients and G=585/602 and A=17/602 on c.3961 in 301 unrelated Chinese patients. Because of the low prevalence of genotypes c.2603T and c.3961A in China, the relationship between clinical features and c.2603C>T and c.3961G>A of the BLM gene in our patient requires further study.
In conclusion, we report a patient with Bloom syndrome with uncommon clinical manifestations. Our findings indicate that c.2603C>T and c.3961G>A of the BLM gene may be the pathogenic nature for Bloom syndrome in China.
Acknowledgments
The authors would like to thank the patient and her family for their participation in the study. The authors also thank Li Qi, BA, Beijing, China, for his contribution to the review of the data in the literature.
- Bloom D. Congenital telangiectatic erythema resembling lupus erythematosus in dwarfs; probably a syndrome entity. AMA Am J Dis Child. 1954;88:754-758.
- German J. Bloom’s syndrome, I: genetical and clinical observations in the first twenty-seven patients. Am J Hum Genet. 1969;21:196-227.
- German J, Roe AM, Leppert MF, et al. Bloom syndrome: an analysis of consanguineous families assigns the locus mutated to chromosome band 15q26.1. Proc Natl Acad Sci U S A. 1994;91:6669-6673.
- Passarge E. A DNA helicase in full Bloom. Nat Genet. 1995;11:356-357.
- Ellis NA, Groden J, Ye TZ, et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655-666.
- German J, Sanz MM, Ciocci S, et al. Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Hum Mutat. 2007;28:743-753.
- Landau JW, Sasaki MS, Newcomer VD, et al. Bloom’s syndrome: the syndrome of telangiectatic erythema and growth retardation. Arch Dermatol. 1966;94:687-694.
- Gretzula JC, Hevia O, Weber PJ. Bloom’s syndrome. J Am Acad Dermatol. 1987;17:479-488.
- Passarge E. Bloom’s syndrome: the German experience. Ann Genet. 1991;34:179-197.
- German J. Bloom’s syndrome. Dermatol Clin. 1995;13:7-18.
- German J, Takebe H. Bloom’s syndrome, XIV: the disorder in Japan. Clin Genet. 1989;35:93-110.
- Bennett RJ, Keck JL. Structure and function of RecQ DNA helicases. Crit Rev Biochem Mol Biol. 2004;39:79-97.
- Reference SNP (refSNP) Cluster Report: rs2227935. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=2227935. Accessed February 3, 2016.
- Reference SNP (refSNP) Cluster Report: rs7167216. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=7167216. Accessed February 3, 2016.
- Homo sapiens:GRCh37.p13 (GCF_000001405.25)Chr 1 (NC_000001.10):1 - 249.3M. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/variationtools/1000genomes/?=%EF%BC%86=. Accessed February 3, 2016.
Bloom syndrome, also called congenital telangiectatic erythema and stunted growth, was first described by David Bloom in 1954.1 It is a rare autosomal-recessive disorder (Online Mendelian Inheritance in Man 210900) characterized by specific clinical manifestations including photosensitivity, telangiectatic facial erythema, proportionate growth deficiency, hypogonadism, immunodeficiency, and a tendency to develop various malignancies.2 Linkage analysis revealed that the Bloom syndrome gene locus resides on chromosome arm 15q26.1,3 and the BLM gene in this region has been identified as being responsible for the development of Bloom syndrome.4,5 We report the case of a 12-year-old Chinese girl with Bloom syndrome and detected BLM gene. The evaluation was approved by the Institutional Ethical Review Boards of Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China).
Case Report
We evaluated a Bloom syndrome family, which consisted of the patient and her parents. The patient was a 12-year-old Chinese girl who was apparently healthy until 3 months of age when her parents noticed an erythematous eruption with blisters on the face. Exacerbation after exposure to sunlight is usual, which results in the eruption becoming prominent in summer and fainter in winter.2 Gradually, the patient’s skin lesions became more progressive, extending to the forehead, nose, and ears, with oozing, crusting, atrophy, and telangiectases developing on the face despite treatment. In the last 3 years, no blisters were present on the patient’s face because of her efforts to avoid sun exposure. She had no history of recurrent infections.
On physical examination, the patient was generally healthy with normal intelligence and short stature. She weighed 26 kg and was approximately 122-cm tall. Telangiectatic erythema and slight scaling were noted on the face, which simulated lupus erythematosus (Figures 1A and 1B). She had additional abnormalities including alopecia areata (Figure 1C), eyebrow hair loss, flat nose, reticular pigmentation on the forehead and trunk, and finger swelling. The distal phalanges on all 10 fingers became short and sharpened and the fingernails became wider than they were long (Figure 1D). Laboratory investigations, including a complete blood cell count, liver and kidney function tests, stool examination, serum complement, and albumin and globulin levels, were within reference range.
After informed consent was obtained, a mutation analysis of the BLM gene was performed in the patient and her parents. We used a genomic DNA purification kit to extract genomic DNA from peripheral blood according to the manufacturer’s protocol. Genomic DNA was used to amplify the exons of the BLM gene with intron flanking sequences by polymerase chain reaction with the primer described elsewhere.6 After the amplification, the polymerase chain reaction products were purified and the BLM gene was sequenced. Sequence comparisons and analysis were performed using Phred/Phrap/Consed version 12.0.
The patient was found to carry changes in 2 heterozygous nucleotide sites, including c.2603C>T in exon 13 and c.3961G>A in exon 21 of the BLM gene. The patient’s father was found to carry c.2603C>T and her mother carried c.3961G>A (Figure 2).
Comment
Patients with Bloom syndrome have a characteristic clinical appearance that typically includes photosensitivity, telangiectatic facial erythema, and growth deficiency. Telangiectatic erythema of the face develops during infancy or early childhood as red macules or plaques and may simulate lupus erythematosus. The lesions are described as a butterfly rash affecting the bridge of the nose and cheeks but also may involve the margins of the eyelids, forehead, ears, and sometimes the dorsa of the hands and forearms. Moderate and proportionate growth deficiencies develop both in utero and postnatally. Patients with Bloom syndrome characteristically have narrow, slender, distinct facial features with micrognathism and a relatively prominent nose. They usually may have mild microcephaly, meaning the head is longer and narrower than normal.2,7-10
German and Takebe11 reported 14 Japanese patients with Bloom syndrome. The phenotype differs somewhat from most cases recognized elsewhere in that dolichocephaly was a less constant feature, the facial skin was less prominent, and life-threatening infections were less common. Our patient had typical telangiectatic facial erythema without microcephaly, dolichocephaly, or any infections. She also had some uncommon manifestations such as alopecia areata, eyebrow hair loss, flat nose, reticular pigmentation, and short sharpened distal phalanges with fingernails that were wider than they were long. Although she had no recurrent infections and laboratory tests were within reference range, the alopecia areata and eyebrow hair loss may be associated with an abnormal immune response. The reasons for the short sharpened distal phalanges and the fingernail findings are unclear. The presence of reticular pigmentation also is unclear but may be associated with photosensitivity. Since the BLM gene was discovered to be the disease-causing gene of Bloom syndrome in 1995,4,5 approximately 70 mutations were reported. The BLM gene encodes for the Bloom syndrome protein, a DNA helicase of the highly conserved RecQ subfamily of helicases, a group of nuclear proteins important in the maintenance of genomic stability.12
Mutation analysis of the BLM gene in our patient showed changes in 2 heterozygous nucleotide sites, including c.2603C>T in exon 13 and c.3961G>A in exon 21 of the BLM gene, which altered proline residue with leucine residue at 868 and valine residue with isoleucine residue at 1321, respectively. According to GenBank,13,14 c.2603C>T and c.3961G>A are single nucleotide polymorphisms of the BLM gene. The genotypic distribution of International HapMap Project15 showed that C=602/602 and T=0/602 on c.2603 in 301 unrelated Chinese patients and G=585/602 and A=17/602 on c.3961 in 301 unrelated Chinese patients. Because of the low prevalence of genotypes c.2603T and c.3961A in China, the relationship between clinical features and c.2603C>T and c.3961G>A of the BLM gene in our patient requires further study.
In conclusion, we report a patient with Bloom syndrome with uncommon clinical manifestations. Our findings indicate that c.2603C>T and c.3961G>A of the BLM gene may be the pathogenic nature for Bloom syndrome in China.
Acknowledgments
The authors would like to thank the patient and her family for their participation in the study. The authors also thank Li Qi, BA, Beijing, China, for his contribution to the review of the data in the literature.
Bloom syndrome, also called congenital telangiectatic erythema and stunted growth, was first described by David Bloom in 1954.1 It is a rare autosomal-recessive disorder (Online Mendelian Inheritance in Man 210900) characterized by specific clinical manifestations including photosensitivity, telangiectatic facial erythema, proportionate growth deficiency, hypogonadism, immunodeficiency, and a tendency to develop various malignancies.2 Linkage analysis revealed that the Bloom syndrome gene locus resides on chromosome arm 15q26.1,3 and the BLM gene in this region has been identified as being responsible for the development of Bloom syndrome.4,5 We report the case of a 12-year-old Chinese girl with Bloom syndrome and detected BLM gene. The evaluation was approved by the Institutional Ethical Review Boards of Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China).
Case Report
We evaluated a Bloom syndrome family, which consisted of the patient and her parents. The patient was a 12-year-old Chinese girl who was apparently healthy until 3 months of age when her parents noticed an erythematous eruption with blisters on the face. Exacerbation after exposure to sunlight is usual, which results in the eruption becoming prominent in summer and fainter in winter.2 Gradually, the patient’s skin lesions became more progressive, extending to the forehead, nose, and ears, with oozing, crusting, atrophy, and telangiectases developing on the face despite treatment. In the last 3 years, no blisters were present on the patient’s face because of her efforts to avoid sun exposure. She had no history of recurrent infections.
On physical examination, the patient was generally healthy with normal intelligence and short stature. She weighed 26 kg and was approximately 122-cm tall. Telangiectatic erythema and slight scaling were noted on the face, which simulated lupus erythematosus (Figures 1A and 1B). She had additional abnormalities including alopecia areata (Figure 1C), eyebrow hair loss, flat nose, reticular pigmentation on the forehead and trunk, and finger swelling. The distal phalanges on all 10 fingers became short and sharpened and the fingernails became wider than they were long (Figure 1D). Laboratory investigations, including a complete blood cell count, liver and kidney function tests, stool examination, serum complement, and albumin and globulin levels, were within reference range.
After informed consent was obtained, a mutation analysis of the BLM gene was performed in the patient and her parents. We used a genomic DNA purification kit to extract genomic DNA from peripheral blood according to the manufacturer’s protocol. Genomic DNA was used to amplify the exons of the BLM gene with intron flanking sequences by polymerase chain reaction with the primer described elsewhere.6 After the amplification, the polymerase chain reaction products were purified and the BLM gene was sequenced. Sequence comparisons and analysis were performed using Phred/Phrap/Consed version 12.0.
The patient was found to carry changes in 2 heterozygous nucleotide sites, including c.2603C>T in exon 13 and c.3961G>A in exon 21 of the BLM gene. The patient’s father was found to carry c.2603C>T and her mother carried c.3961G>A (Figure 2).
Comment
Patients with Bloom syndrome have a characteristic clinical appearance that typically includes photosensitivity, telangiectatic facial erythema, and growth deficiency. Telangiectatic erythema of the face develops during infancy or early childhood as red macules or plaques and may simulate lupus erythematosus. The lesions are described as a butterfly rash affecting the bridge of the nose and cheeks but also may involve the margins of the eyelids, forehead, ears, and sometimes the dorsa of the hands and forearms. Moderate and proportionate growth deficiencies develop both in utero and postnatally. Patients with Bloom syndrome characteristically have narrow, slender, distinct facial features with micrognathism and a relatively prominent nose. They usually may have mild microcephaly, meaning the head is longer and narrower than normal.2,7-10
German and Takebe11 reported 14 Japanese patients with Bloom syndrome. The phenotype differs somewhat from most cases recognized elsewhere in that dolichocephaly was a less constant feature, the facial skin was less prominent, and life-threatening infections were less common. Our patient had typical telangiectatic facial erythema without microcephaly, dolichocephaly, or any infections. She also had some uncommon manifestations such as alopecia areata, eyebrow hair loss, flat nose, reticular pigmentation, and short sharpened distal phalanges with fingernails that were wider than they were long. Although she had no recurrent infections and laboratory tests were within reference range, the alopecia areata and eyebrow hair loss may be associated with an abnormal immune response. The reasons for the short sharpened distal phalanges and the fingernail findings are unclear. The presence of reticular pigmentation also is unclear but may be associated with photosensitivity. Since the BLM gene was discovered to be the disease-causing gene of Bloom syndrome in 1995,4,5 approximately 70 mutations were reported. The BLM gene encodes for the Bloom syndrome protein, a DNA helicase of the highly conserved RecQ subfamily of helicases, a group of nuclear proteins important in the maintenance of genomic stability.12
Mutation analysis of the BLM gene in our patient showed changes in 2 heterozygous nucleotide sites, including c.2603C>T in exon 13 and c.3961G>A in exon 21 of the BLM gene, which altered proline residue with leucine residue at 868 and valine residue with isoleucine residue at 1321, respectively. According to GenBank,13,14 c.2603C>T and c.3961G>A are single nucleotide polymorphisms of the BLM gene. The genotypic distribution of International HapMap Project15 showed that C=602/602 and T=0/602 on c.2603 in 301 unrelated Chinese patients and G=585/602 and A=17/602 on c.3961 in 301 unrelated Chinese patients. Because of the low prevalence of genotypes c.2603T and c.3961A in China, the relationship between clinical features and c.2603C>T and c.3961G>A of the BLM gene in our patient requires further study.
In conclusion, we report a patient with Bloom syndrome with uncommon clinical manifestations. Our findings indicate that c.2603C>T and c.3961G>A of the BLM gene may be the pathogenic nature for Bloom syndrome in China.
Acknowledgments
The authors would like to thank the patient and her family for their participation in the study. The authors also thank Li Qi, BA, Beijing, China, for his contribution to the review of the data in the literature.
- Bloom D. Congenital telangiectatic erythema resembling lupus erythematosus in dwarfs; probably a syndrome entity. AMA Am J Dis Child. 1954;88:754-758.
- German J. Bloom’s syndrome, I: genetical and clinical observations in the first twenty-seven patients. Am J Hum Genet. 1969;21:196-227.
- German J, Roe AM, Leppert MF, et al. Bloom syndrome: an analysis of consanguineous families assigns the locus mutated to chromosome band 15q26.1. Proc Natl Acad Sci U S A. 1994;91:6669-6673.
- Passarge E. A DNA helicase in full Bloom. Nat Genet. 1995;11:356-357.
- Ellis NA, Groden J, Ye TZ, et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655-666.
- German J, Sanz MM, Ciocci S, et al. Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Hum Mutat. 2007;28:743-753.
- Landau JW, Sasaki MS, Newcomer VD, et al. Bloom’s syndrome: the syndrome of telangiectatic erythema and growth retardation. Arch Dermatol. 1966;94:687-694.
- Gretzula JC, Hevia O, Weber PJ. Bloom’s syndrome. J Am Acad Dermatol. 1987;17:479-488.
- Passarge E. Bloom’s syndrome: the German experience. Ann Genet. 1991;34:179-197.
- German J. Bloom’s syndrome. Dermatol Clin. 1995;13:7-18.
- German J, Takebe H. Bloom’s syndrome, XIV: the disorder in Japan. Clin Genet. 1989;35:93-110.
- Bennett RJ, Keck JL. Structure and function of RecQ DNA helicases. Crit Rev Biochem Mol Biol. 2004;39:79-97.
- Reference SNP (refSNP) Cluster Report: rs2227935. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=2227935. Accessed February 3, 2016.
- Reference SNP (refSNP) Cluster Report: rs7167216. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=7167216. Accessed February 3, 2016.
- Homo sapiens:GRCh37.p13 (GCF_000001405.25)Chr 1 (NC_000001.10):1 - 249.3M. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/variationtools/1000genomes/?=%EF%BC%86=. Accessed February 3, 2016.
- Bloom D. Congenital telangiectatic erythema resembling lupus erythematosus in dwarfs; probably a syndrome entity. AMA Am J Dis Child. 1954;88:754-758.
- German J. Bloom’s syndrome, I: genetical and clinical observations in the first twenty-seven patients. Am J Hum Genet. 1969;21:196-227.
- German J, Roe AM, Leppert MF, et al. Bloom syndrome: an analysis of consanguineous families assigns the locus mutated to chromosome band 15q26.1. Proc Natl Acad Sci U S A. 1994;91:6669-6673.
- Passarge E. A DNA helicase in full Bloom. Nat Genet. 1995;11:356-357.
- Ellis NA, Groden J, Ye TZ, et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655-666.
- German J, Sanz MM, Ciocci S, et al. Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Hum Mutat. 2007;28:743-753.
- Landau JW, Sasaki MS, Newcomer VD, et al. Bloom’s syndrome: the syndrome of telangiectatic erythema and growth retardation. Arch Dermatol. 1966;94:687-694.
- Gretzula JC, Hevia O, Weber PJ. Bloom’s syndrome. J Am Acad Dermatol. 1987;17:479-488.
- Passarge E. Bloom’s syndrome: the German experience. Ann Genet. 1991;34:179-197.
- German J. Bloom’s syndrome. Dermatol Clin. 1995;13:7-18.
- German J, Takebe H. Bloom’s syndrome, XIV: the disorder in Japan. Clin Genet. 1989;35:93-110.
- Bennett RJ, Keck JL. Structure and function of RecQ DNA helicases. Crit Rev Biochem Mol Biol. 2004;39:79-97.
- Reference SNP (refSNP) Cluster Report: rs2227935. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=2227935. Accessed February 3, 2016.
- Reference SNP (refSNP) Cluster Report: rs7167216. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=7167216. Accessed February 3, 2016.
- Homo sapiens:GRCh37.p13 (GCF_000001405.25)Chr 1 (NC_000001.10):1 - 249.3M. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/variationtools/1000genomes/?=%EF%BC%86=. Accessed February 3, 2016.
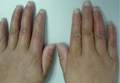
Postinflammatory Hyperpigmentation in Patients With Skin of Color
Postinflammatory hyperpigmentation (PIH) develops as darkly pigmented macules that occur after an inflammatory process of the skin such as acne, folliculitis, eczema, or shaving irritation. Patients with Fitzpatrick skin types III to VI usually are most commonly affected, and for many, the remnant pigmentation can be an even greater concern than the original inflammatory process.1,2 Reported treatments of PIH include tretinoin, hydroquinone, azelaic acid, and chemical peels. The ideal combination of therapy has yet to be delineated.
Tretinoin (Vitamin A Derivative)
Bulengo-Ransby et al3 performed one of the first clinical trials testing tretinoin cream 0.1% for PIH in patients with Fitzpatrick skin types IV to VI . The study included 54 patients (24 applied tretinoin and 30 applied a vehicle) with moderate to severe PIH on the face and arms. The patients were divided into therapy and placebo groups and were evaluated for 40 weeks. Changes were evaluated through colorimetry, light microscopy, histology, and photography, with significant clinical improvement in the tretinoin-treated group (P<.001).3 A double-blind, randomized study of 45 photoaged Chinese and Japanese patients using tretinoin cream 0.1% also was conducted for treatment of photoaging-associated hyperpigmented lesions of the face and hands. Assessment was done with clinical, colorimetric, and histological evaluation, with an overall statistical improvement noted in hyperpigmentation.4 Both of the above studies showed mild irritation (ie, retinoid dermatitis) with application of tretinoin, which creates a compliance issue in patients who are recommended to continue therapy with higher-strength tretinoin. This side-effect profile can be circumvented through gradual elevation in the strength of tretinoin.5
Combination Therapies
Combination therapies with tretinoin also have been used to improve PIH. Callender et al6 conducted a study evaluating the efficacy of clindamycin phosphate 1.2%–tretinoin 0.025% gel for the treatment of PIH secondary to mild to moderate acne in patients with Fitzpatrick skin types IV to VI. Thirty patients participated in the randomized, double-blinded, placebo-controlled study, with 15 patients in the clindamycin-tretinoin gel group and 15 in the placebo control group. Based on objective assessment using a chromameter and evaluator global acne severity scale score, clinical efficacy was demonstrated for treating acne and PIH as well as preventing further PIH.6
Hydroquinone Formulation (Tyrosine Inhibitor)
Hydroquinone bleaching cream has been the standard therapy for hyperpigmentation. It works by blocking the conversion of dihydroxyphenylalanine to melanin by inhibiting tyrosinase.7 Topical steroids directly inhibit the synthesis of melanin, and when combined with hydroquinone and tretinoin, they can be effective for short periods of time and may decrease the irritation of application.7,8 The most widely accepted formula consists of a topical steroid (triamcinolone cream 0.1%) in combination with hydroquinone 4% and tretinoin cream 0.05%.8 In a similar 12-week open-label study of 25 patients with darker skin types, Grimes9 used an alternative combination formula of hydroquinone 4% and retinol 0.15%. Overall improvement and tolerance was demonstrated through the use of colorimetry measurement. A combination of hydroquinone 4%, tretinoin 0.05%, and fluocinolone acetonide 0.01% also has been used effectively for the treatment of melasma.10 This formulation has been used more anecdotally for the treatment of PIH and has yet to have a randomized-controlled trial. The concern with repeated long-term use of hydroquinone remains. Permanent leukoderma, exogenous ochronosis, and hyperpigmentation of the surrounding normal skin (halo effect) can occur.
Azelaic Acid (Tyrosinase Inhibitor)
Azelaic acid is a dicarboxylic acid isolated from pityriasis versicolor that acts similar to a tyrosine inhibitor and has an antiproliferative effect toward abnormal melanocytes. Lowe et al11 conducted a randomized, double-blind, vehicle-controlled trial in patients with Fitzpatrick skin types IV through VI with facial hyperpigmentation using azelaic acid cream 20%. Over the course of 24 weeks, patients noted a decrease in overall pigment using both an investigator subjective scale and chromometer analysis.11
Kojic Acid (Tyrosinase Inhibitor)
Kojic acid is a tyrosinase inhibitor found in fungal metabolite species such as Acetobacter, Aspergillus, and Penicillium. It is commonly combined with other skin lightening agents such as hydroquinone or vitamin C to further enhance its efficacy. A randomized, 12-week, split-face study of Chinese women with melasma compared treatment with a glycolic acid 10%–hydroquinone 2% gel versus the combination plus kojic acid 2%. The results showed that 60% (24/40) of patients improved with the use of kojic acid as compared to those using the medication without kojic acid.12 Anecdotal data suggest kojic acid may be effective for PIH13; however, no studies specifically for PIH have been conducted.
Chemical Peels
Chemical peels have been used for a number of years, though their benefits in patients with skin of color is still being elucidated. The ideal chemical peels for Fitzpatrick skin types IV through VI are superficial to medium-depth peeling agents and techniques.14 Glycolic acid is a naturally occurring α-hydroxy acid that causes an increase in collagen synthesis, stimulates epidermolysis, and disperses basal layer melanin. Neutralization of glycolic acid peels can be done with the use of water, sodium bicarbonate, or sodium hydroxide to avoid unnecessary epidermal damage. Multiple clinical trials have been conducted to determine the response of glycolic acid peels in clearing PIH in patients with skin of color. Kessler et al15 compared glycolic acid 30% to salicylic acid 30% in 20 patients with mild to moderate acne and associated PIH. Chemical peels were performed every 2 weeks for 12 weeks. The study showed that salicylic acid was better tolerated than glycolic acid and both were equally effective after the second application (P<.05) for PIH.15 Finally, another study conducted for PIH in patients with Fitzpatrick skin types III and IV utilized glycolic acid peels with 20%, 35%, and 70% concentrations. The results showed overall improvement of PIH and acne from the use of all concentrations of glycolic peels, though faster efficacy was noted at higher concentrations.16
Other self-neutralizing peeling agents include salicylic acid and Jessner solution. Salicylic acid is a β-hydroxy acid that works through keratolysis and disrupting intercellular linkages. Jessner solution is a combination of resorcinol 14%, lactic acid 14%, and salicylic acid 14% in an alcohol base. Salicylic acid is well-tolerated in patients with Fitzpatrick skin types I through VI and has been helpful in treating acne, rosacea, melasma, hyperpigmentation, texturally rough skin, and mild photoaging. Jessner peeling solution has been used for a number of years and works as a keratolytic agent causing intercellular and intracellular edema, and due to its self-neutralizing agent, it is fairly superficial.17 Overall, superficial peeling agents should be used on patients with darker skin types to avoid the risk for worsening dyspigmentation, keloid formation, or deep scarring.18
Conclusion
These treatments are only some of the topical and chemical modalities for PIH in patients with skin of color. The patient history, evaluation, skin type, and underlying medical problems should be considered prior to using any topical or peeling agent. Lastly, photoprotection should be heavily emphasized with both sun protective gear and use of broad-spectrum sunscreens with a high sun protection factor, as UV radiation can cause darkening of PIH areas regardless of skin type and can reverse the progress made by a given therapy.18
- Savory SA, Agim NG, Mao R, et al. Reliability assessment and validation of the postacne hyperpigmentation index (PAHPI), a new instrument to measure postinflammatory hyperpigmentation from acne vulgaris. J Am Acad Dermatol. 2014;70:108-114.
- Halder RM. The role of retinoids in the management of cutaneous conditions in blacks. J Am Acad Dermatol. 1998;39(2, pt 3):S98-S103.
- Bulengo-Ransby SM, Griffiths CE, Kimbrough-Green CK, et al. Topical tretinoin (retinoid acid) therapy for hyperpigmented lesions caused by inflammation of the skin in black patients. N Engl J Med. 1993;328:1438-1443.
- Griffiths CE, Goldfarb MT, Finkel LJ, et al. Topical tretinoin (retinoic acid) treatment of hyperpigmented lesions associated with photoaging in Chinese and Japanese patients: a vehicle-controlled trial. J Am Acad Dermatol. 1994;30:76-84.
- Callendar VD. Acne in ethnic skin: special considerations for therapy. Dermatol Ther. 2004;17:184-195.
- Callender VD, Young CM, Kindred C, et al. Efficacy and safety of clindamycin phosphate 1.2% and tretinoin 0.025% gel for the treatment of acne and acne-induced post-inflammatory hyperpigmentation in patients with skin of color. J Clin Aesthet Dermatol. 2012;5:25-32.
- Badreshia-Bansal S, Draelos ZD. Insight into skin lightening cosmeceuticals for women of color. J Drugs Dermatol. 2007;6:32-39.
- Kligman AM, Willis I. A new formula for depigmenting human skin. Arch Dermatol. 1975;111:40-48.
- Grimes PE. A microsponge formulation of hydroquinone 4% and retinol 0.15% in the treatment of melasma and postinflammatory hyperpigmentation. Cutis. 2004;74:326-328.
- Chan R, Park KC, Lee MH, et al. A randomized controlled trial of the efficacy and safety of a fixed triple combination (fluocinolone acetonide 0.01%, hydroquinone 4%, tretinoin 0.05%) compared with hydroquinone 4% cream in Asian patients with moderate to severe melasma. Br J Dermatol. 2008;159:697-703.
- Lowe NJ, Rizk D, Grimes P. Azelaic acid 20% cream in the treatment of facial hyperpigmentation in darker-skinned patients. Clin Ther. 1998;20:945-959.
- Lim JT. Treatment of melasma using kojic acid in a gel containing hydroquinone and glycolic acid. Dermatol Surg. 1999;25:282-284.
- Alexis AF, Blackcloud P. Natural ingredients for darker skin types: growing options for hyperpigmentation. J Drugs Dermatol. 2013;12:123-127.
- Roberts WE. Chemical peeling in ethnic/dark skin. Dermatol Ther. 2004;17:196-205.
- Kessler E, Flanagan K, Chia C, et al. Comparison of alpha- and beta-hydroxy acid chemical peels in the treatment of mild to moderately severe facial acne vulgaris [published online December 5, 2007]. Dermatol Surg. 2008;34:45-50, discussion 51.
- Erbağci Z, Akçali C. Biweekly serial glycolic acid peels vs. long-term daily use of topical low-strength glycolic acid in the treatment of atrophic acne scars. Int J Dermatol. 2000;39:789-794.
- Jackson A. Chemical peels [published online January 31, 2014]. Facial Plast Surg. 2014;30:26-34.
- Davis EC, Callender VD. Postinflammatory hyperpigmentation: a review of the epidemiology, clinical features, and treatment options in skin of color. J Clin Aesthet Dermatol. 2010;3:20-31.
Postinflammatory hyperpigmentation (PIH) develops as darkly pigmented macules that occur after an inflammatory process of the skin such as acne, folliculitis, eczema, or shaving irritation. Patients with Fitzpatrick skin types III to VI usually are most commonly affected, and for many, the remnant pigmentation can be an even greater concern than the original inflammatory process.1,2 Reported treatments of PIH include tretinoin, hydroquinone, azelaic acid, and chemical peels. The ideal combination of therapy has yet to be delineated.
Tretinoin (Vitamin A Derivative)
Bulengo-Ransby et al3 performed one of the first clinical trials testing tretinoin cream 0.1% for PIH in patients with Fitzpatrick skin types IV to VI . The study included 54 patients (24 applied tretinoin and 30 applied a vehicle) with moderate to severe PIH on the face and arms. The patients were divided into therapy and placebo groups and were evaluated for 40 weeks. Changes were evaluated through colorimetry, light microscopy, histology, and photography, with significant clinical improvement in the tretinoin-treated group (P<.001).3 A double-blind, randomized study of 45 photoaged Chinese and Japanese patients using tretinoin cream 0.1% also was conducted for treatment of photoaging-associated hyperpigmented lesions of the face and hands. Assessment was done with clinical, colorimetric, and histological evaluation, with an overall statistical improvement noted in hyperpigmentation.4 Both of the above studies showed mild irritation (ie, retinoid dermatitis) with application of tretinoin, which creates a compliance issue in patients who are recommended to continue therapy with higher-strength tretinoin. This side-effect profile can be circumvented through gradual elevation in the strength of tretinoin.5
Combination Therapies
Combination therapies with tretinoin also have been used to improve PIH. Callender et al6 conducted a study evaluating the efficacy of clindamycin phosphate 1.2%–tretinoin 0.025% gel for the treatment of PIH secondary to mild to moderate acne in patients with Fitzpatrick skin types IV to VI. Thirty patients participated in the randomized, double-blinded, placebo-controlled study, with 15 patients in the clindamycin-tretinoin gel group and 15 in the placebo control group. Based on objective assessment using a chromameter and evaluator global acne severity scale score, clinical efficacy was demonstrated for treating acne and PIH as well as preventing further PIH.6
Hydroquinone Formulation (Tyrosine Inhibitor)
Hydroquinone bleaching cream has been the standard therapy for hyperpigmentation. It works by blocking the conversion of dihydroxyphenylalanine to melanin by inhibiting tyrosinase.7 Topical steroids directly inhibit the synthesis of melanin, and when combined with hydroquinone and tretinoin, they can be effective for short periods of time and may decrease the irritation of application.7,8 The most widely accepted formula consists of a topical steroid (triamcinolone cream 0.1%) in combination with hydroquinone 4% and tretinoin cream 0.05%.8 In a similar 12-week open-label study of 25 patients with darker skin types, Grimes9 used an alternative combination formula of hydroquinone 4% and retinol 0.15%. Overall improvement and tolerance was demonstrated through the use of colorimetry measurement. A combination of hydroquinone 4%, tretinoin 0.05%, and fluocinolone acetonide 0.01% also has been used effectively for the treatment of melasma.10 This formulation has been used more anecdotally for the treatment of PIH and has yet to have a randomized-controlled trial. The concern with repeated long-term use of hydroquinone remains. Permanent leukoderma, exogenous ochronosis, and hyperpigmentation of the surrounding normal skin (halo effect) can occur.
Azelaic Acid (Tyrosinase Inhibitor)
Azelaic acid is a dicarboxylic acid isolated from pityriasis versicolor that acts similar to a tyrosine inhibitor and has an antiproliferative effect toward abnormal melanocytes. Lowe et al11 conducted a randomized, double-blind, vehicle-controlled trial in patients with Fitzpatrick skin types IV through VI with facial hyperpigmentation using azelaic acid cream 20%. Over the course of 24 weeks, patients noted a decrease in overall pigment using both an investigator subjective scale and chromometer analysis.11
Kojic Acid (Tyrosinase Inhibitor)
Kojic acid is a tyrosinase inhibitor found in fungal metabolite species such as Acetobacter, Aspergillus, and Penicillium. It is commonly combined with other skin lightening agents such as hydroquinone or vitamin C to further enhance its efficacy. A randomized, 12-week, split-face study of Chinese women with melasma compared treatment with a glycolic acid 10%–hydroquinone 2% gel versus the combination plus kojic acid 2%. The results showed that 60% (24/40) of patients improved with the use of kojic acid as compared to those using the medication without kojic acid.12 Anecdotal data suggest kojic acid may be effective for PIH13; however, no studies specifically for PIH have been conducted.
Chemical Peels
Chemical peels have been used for a number of years, though their benefits in patients with skin of color is still being elucidated. The ideal chemical peels for Fitzpatrick skin types IV through VI are superficial to medium-depth peeling agents and techniques.14 Glycolic acid is a naturally occurring α-hydroxy acid that causes an increase in collagen synthesis, stimulates epidermolysis, and disperses basal layer melanin. Neutralization of glycolic acid peels can be done with the use of water, sodium bicarbonate, or sodium hydroxide to avoid unnecessary epidermal damage. Multiple clinical trials have been conducted to determine the response of glycolic acid peels in clearing PIH in patients with skin of color. Kessler et al15 compared glycolic acid 30% to salicylic acid 30% in 20 patients with mild to moderate acne and associated PIH. Chemical peels were performed every 2 weeks for 12 weeks. The study showed that salicylic acid was better tolerated than glycolic acid and both were equally effective after the second application (P<.05) for PIH.15 Finally, another study conducted for PIH in patients with Fitzpatrick skin types III and IV utilized glycolic acid peels with 20%, 35%, and 70% concentrations. The results showed overall improvement of PIH and acne from the use of all concentrations of glycolic peels, though faster efficacy was noted at higher concentrations.16
Other self-neutralizing peeling agents include salicylic acid and Jessner solution. Salicylic acid is a β-hydroxy acid that works through keratolysis and disrupting intercellular linkages. Jessner solution is a combination of resorcinol 14%, lactic acid 14%, and salicylic acid 14% in an alcohol base. Salicylic acid is well-tolerated in patients with Fitzpatrick skin types I through VI and has been helpful in treating acne, rosacea, melasma, hyperpigmentation, texturally rough skin, and mild photoaging. Jessner peeling solution has been used for a number of years and works as a keratolytic agent causing intercellular and intracellular edema, and due to its self-neutralizing agent, it is fairly superficial.17 Overall, superficial peeling agents should be used on patients with darker skin types to avoid the risk for worsening dyspigmentation, keloid formation, or deep scarring.18
Conclusion
These treatments are only some of the topical and chemical modalities for PIH in patients with skin of color. The patient history, evaluation, skin type, and underlying medical problems should be considered prior to using any topical or peeling agent. Lastly, photoprotection should be heavily emphasized with both sun protective gear and use of broad-spectrum sunscreens with a high sun protection factor, as UV radiation can cause darkening of PIH areas regardless of skin type and can reverse the progress made by a given therapy.18
Postinflammatory hyperpigmentation (PIH) develops as darkly pigmented macules that occur after an inflammatory process of the skin such as acne, folliculitis, eczema, or shaving irritation. Patients with Fitzpatrick skin types III to VI usually are most commonly affected, and for many, the remnant pigmentation can be an even greater concern than the original inflammatory process.1,2 Reported treatments of PIH include tretinoin, hydroquinone, azelaic acid, and chemical peels. The ideal combination of therapy has yet to be delineated.
Tretinoin (Vitamin A Derivative)
Bulengo-Ransby et al3 performed one of the first clinical trials testing tretinoin cream 0.1% for PIH in patients with Fitzpatrick skin types IV to VI . The study included 54 patients (24 applied tretinoin and 30 applied a vehicle) with moderate to severe PIH on the face and arms. The patients were divided into therapy and placebo groups and were evaluated for 40 weeks. Changes were evaluated through colorimetry, light microscopy, histology, and photography, with significant clinical improvement in the tretinoin-treated group (P<.001).3 A double-blind, randomized study of 45 photoaged Chinese and Japanese patients using tretinoin cream 0.1% also was conducted for treatment of photoaging-associated hyperpigmented lesions of the face and hands. Assessment was done with clinical, colorimetric, and histological evaluation, with an overall statistical improvement noted in hyperpigmentation.4 Both of the above studies showed mild irritation (ie, retinoid dermatitis) with application of tretinoin, which creates a compliance issue in patients who are recommended to continue therapy with higher-strength tretinoin. This side-effect profile can be circumvented through gradual elevation in the strength of tretinoin.5
Combination Therapies
Combination therapies with tretinoin also have been used to improve PIH. Callender et al6 conducted a study evaluating the efficacy of clindamycin phosphate 1.2%–tretinoin 0.025% gel for the treatment of PIH secondary to mild to moderate acne in patients with Fitzpatrick skin types IV to VI. Thirty patients participated in the randomized, double-blinded, placebo-controlled study, with 15 patients in the clindamycin-tretinoin gel group and 15 in the placebo control group. Based on objective assessment using a chromameter and evaluator global acne severity scale score, clinical efficacy was demonstrated for treating acne and PIH as well as preventing further PIH.6
Hydroquinone Formulation (Tyrosine Inhibitor)
Hydroquinone bleaching cream has been the standard therapy for hyperpigmentation. It works by blocking the conversion of dihydroxyphenylalanine to melanin by inhibiting tyrosinase.7 Topical steroids directly inhibit the synthesis of melanin, and when combined with hydroquinone and tretinoin, they can be effective for short periods of time and may decrease the irritation of application.7,8 The most widely accepted formula consists of a topical steroid (triamcinolone cream 0.1%) in combination with hydroquinone 4% and tretinoin cream 0.05%.8 In a similar 12-week open-label study of 25 patients with darker skin types, Grimes9 used an alternative combination formula of hydroquinone 4% and retinol 0.15%. Overall improvement and tolerance was demonstrated through the use of colorimetry measurement. A combination of hydroquinone 4%, tretinoin 0.05%, and fluocinolone acetonide 0.01% also has been used effectively for the treatment of melasma.10 This formulation has been used more anecdotally for the treatment of PIH and has yet to have a randomized-controlled trial. The concern with repeated long-term use of hydroquinone remains. Permanent leukoderma, exogenous ochronosis, and hyperpigmentation of the surrounding normal skin (halo effect) can occur.
Azelaic Acid (Tyrosinase Inhibitor)
Azelaic acid is a dicarboxylic acid isolated from pityriasis versicolor that acts similar to a tyrosine inhibitor and has an antiproliferative effect toward abnormal melanocytes. Lowe et al11 conducted a randomized, double-blind, vehicle-controlled trial in patients with Fitzpatrick skin types IV through VI with facial hyperpigmentation using azelaic acid cream 20%. Over the course of 24 weeks, patients noted a decrease in overall pigment using both an investigator subjective scale and chromometer analysis.11
Kojic Acid (Tyrosinase Inhibitor)
Kojic acid is a tyrosinase inhibitor found in fungal metabolite species such as Acetobacter, Aspergillus, and Penicillium. It is commonly combined with other skin lightening agents such as hydroquinone or vitamin C to further enhance its efficacy. A randomized, 12-week, split-face study of Chinese women with melasma compared treatment with a glycolic acid 10%–hydroquinone 2% gel versus the combination plus kojic acid 2%. The results showed that 60% (24/40) of patients improved with the use of kojic acid as compared to those using the medication without kojic acid.12 Anecdotal data suggest kojic acid may be effective for PIH13; however, no studies specifically for PIH have been conducted.
Chemical Peels
Chemical peels have been used for a number of years, though their benefits in patients with skin of color is still being elucidated. The ideal chemical peels for Fitzpatrick skin types IV through VI are superficial to medium-depth peeling agents and techniques.14 Glycolic acid is a naturally occurring α-hydroxy acid that causes an increase in collagen synthesis, stimulates epidermolysis, and disperses basal layer melanin. Neutralization of glycolic acid peels can be done with the use of water, sodium bicarbonate, or sodium hydroxide to avoid unnecessary epidermal damage. Multiple clinical trials have been conducted to determine the response of glycolic acid peels in clearing PIH in patients with skin of color. Kessler et al15 compared glycolic acid 30% to salicylic acid 30% in 20 patients with mild to moderate acne and associated PIH. Chemical peels were performed every 2 weeks for 12 weeks. The study showed that salicylic acid was better tolerated than glycolic acid and both were equally effective after the second application (P<.05) for PIH.15 Finally, another study conducted for PIH in patients with Fitzpatrick skin types III and IV utilized glycolic acid peels with 20%, 35%, and 70% concentrations. The results showed overall improvement of PIH and acne from the use of all concentrations of glycolic peels, though faster efficacy was noted at higher concentrations.16
Other self-neutralizing peeling agents include salicylic acid and Jessner solution. Salicylic acid is a β-hydroxy acid that works through keratolysis and disrupting intercellular linkages. Jessner solution is a combination of resorcinol 14%, lactic acid 14%, and salicylic acid 14% in an alcohol base. Salicylic acid is well-tolerated in patients with Fitzpatrick skin types I through VI and has been helpful in treating acne, rosacea, melasma, hyperpigmentation, texturally rough skin, and mild photoaging. Jessner peeling solution has been used for a number of years and works as a keratolytic agent causing intercellular and intracellular edema, and due to its self-neutralizing agent, it is fairly superficial.17 Overall, superficial peeling agents should be used on patients with darker skin types to avoid the risk for worsening dyspigmentation, keloid formation, or deep scarring.18
Conclusion
These treatments are only some of the topical and chemical modalities for PIH in patients with skin of color. The patient history, evaluation, skin type, and underlying medical problems should be considered prior to using any topical or peeling agent. Lastly, photoprotection should be heavily emphasized with both sun protective gear and use of broad-spectrum sunscreens with a high sun protection factor, as UV radiation can cause darkening of PIH areas regardless of skin type and can reverse the progress made by a given therapy.18
- Savory SA, Agim NG, Mao R, et al. Reliability assessment and validation of the postacne hyperpigmentation index (PAHPI), a new instrument to measure postinflammatory hyperpigmentation from acne vulgaris. J Am Acad Dermatol. 2014;70:108-114.
- Halder RM. The role of retinoids in the management of cutaneous conditions in blacks. J Am Acad Dermatol. 1998;39(2, pt 3):S98-S103.
- Bulengo-Ransby SM, Griffiths CE, Kimbrough-Green CK, et al. Topical tretinoin (retinoid acid) therapy for hyperpigmented lesions caused by inflammation of the skin in black patients. N Engl J Med. 1993;328:1438-1443.
- Griffiths CE, Goldfarb MT, Finkel LJ, et al. Topical tretinoin (retinoic acid) treatment of hyperpigmented lesions associated with photoaging in Chinese and Japanese patients: a vehicle-controlled trial. J Am Acad Dermatol. 1994;30:76-84.
- Callendar VD. Acne in ethnic skin: special considerations for therapy. Dermatol Ther. 2004;17:184-195.
- Callender VD, Young CM, Kindred C, et al. Efficacy and safety of clindamycin phosphate 1.2% and tretinoin 0.025% gel for the treatment of acne and acne-induced post-inflammatory hyperpigmentation in patients with skin of color. J Clin Aesthet Dermatol. 2012;5:25-32.
- Badreshia-Bansal S, Draelos ZD. Insight into skin lightening cosmeceuticals for women of color. J Drugs Dermatol. 2007;6:32-39.
- Kligman AM, Willis I. A new formula for depigmenting human skin. Arch Dermatol. 1975;111:40-48.
- Grimes PE. A microsponge formulation of hydroquinone 4% and retinol 0.15% in the treatment of melasma and postinflammatory hyperpigmentation. Cutis. 2004;74:326-328.
- Chan R, Park KC, Lee MH, et al. A randomized controlled trial of the efficacy and safety of a fixed triple combination (fluocinolone acetonide 0.01%, hydroquinone 4%, tretinoin 0.05%) compared with hydroquinone 4% cream in Asian patients with moderate to severe melasma. Br J Dermatol. 2008;159:697-703.
- Lowe NJ, Rizk D, Grimes P. Azelaic acid 20% cream in the treatment of facial hyperpigmentation in darker-skinned patients. Clin Ther. 1998;20:945-959.
- Lim JT. Treatment of melasma using kojic acid in a gel containing hydroquinone and glycolic acid. Dermatol Surg. 1999;25:282-284.
- Alexis AF, Blackcloud P. Natural ingredients for darker skin types: growing options for hyperpigmentation. J Drugs Dermatol. 2013;12:123-127.
- Roberts WE. Chemical peeling in ethnic/dark skin. Dermatol Ther. 2004;17:196-205.
- Kessler E, Flanagan K, Chia C, et al. Comparison of alpha- and beta-hydroxy acid chemical peels in the treatment of mild to moderately severe facial acne vulgaris [published online December 5, 2007]. Dermatol Surg. 2008;34:45-50, discussion 51.
- Erbağci Z, Akçali C. Biweekly serial glycolic acid peels vs. long-term daily use of topical low-strength glycolic acid in the treatment of atrophic acne scars. Int J Dermatol. 2000;39:789-794.
- Jackson A. Chemical peels [published online January 31, 2014]. Facial Plast Surg. 2014;30:26-34.
- Davis EC, Callender VD. Postinflammatory hyperpigmentation: a review of the epidemiology, clinical features, and treatment options in skin of color. J Clin Aesthet Dermatol. 2010;3:20-31.
- Savory SA, Agim NG, Mao R, et al. Reliability assessment and validation of the postacne hyperpigmentation index (PAHPI), a new instrument to measure postinflammatory hyperpigmentation from acne vulgaris. J Am Acad Dermatol. 2014;70:108-114.
- Halder RM. The role of retinoids in the management of cutaneous conditions in blacks. J Am Acad Dermatol. 1998;39(2, pt 3):S98-S103.
- Bulengo-Ransby SM, Griffiths CE, Kimbrough-Green CK, et al. Topical tretinoin (retinoid acid) therapy for hyperpigmented lesions caused by inflammation of the skin in black patients. N Engl J Med. 1993;328:1438-1443.
- Griffiths CE, Goldfarb MT, Finkel LJ, et al. Topical tretinoin (retinoic acid) treatment of hyperpigmented lesions associated with photoaging in Chinese and Japanese patients: a vehicle-controlled trial. J Am Acad Dermatol. 1994;30:76-84.
- Callendar VD. Acne in ethnic skin: special considerations for therapy. Dermatol Ther. 2004;17:184-195.
- Callender VD, Young CM, Kindred C, et al. Efficacy and safety of clindamycin phosphate 1.2% and tretinoin 0.025% gel for the treatment of acne and acne-induced post-inflammatory hyperpigmentation in patients with skin of color. J Clin Aesthet Dermatol. 2012;5:25-32.
- Badreshia-Bansal S, Draelos ZD. Insight into skin lightening cosmeceuticals for women of color. J Drugs Dermatol. 2007;6:32-39.
- Kligman AM, Willis I. A new formula for depigmenting human skin. Arch Dermatol. 1975;111:40-48.
- Grimes PE. A microsponge formulation of hydroquinone 4% and retinol 0.15% in the treatment of melasma and postinflammatory hyperpigmentation. Cutis. 2004;74:326-328.
- Chan R, Park KC, Lee MH, et al. A randomized controlled trial of the efficacy and safety of a fixed triple combination (fluocinolone acetonide 0.01%, hydroquinone 4%, tretinoin 0.05%) compared with hydroquinone 4% cream in Asian patients with moderate to severe melasma. Br J Dermatol. 2008;159:697-703.
- Lowe NJ, Rizk D, Grimes P. Azelaic acid 20% cream in the treatment of facial hyperpigmentation in darker-skinned patients. Clin Ther. 1998;20:945-959.
- Lim JT. Treatment of melasma using kojic acid in a gel containing hydroquinone and glycolic acid. Dermatol Surg. 1999;25:282-284.
- Alexis AF, Blackcloud P. Natural ingredients for darker skin types: growing options for hyperpigmentation. J Drugs Dermatol. 2013;12:123-127.
- Roberts WE. Chemical peeling in ethnic/dark skin. Dermatol Ther. 2004;17:196-205.
- Kessler E, Flanagan K, Chia C, et al. Comparison of alpha- and beta-hydroxy acid chemical peels in the treatment of mild to moderately severe facial acne vulgaris [published online December 5, 2007]. Dermatol Surg. 2008;34:45-50, discussion 51.
- Erbağci Z, Akçali C. Biweekly serial glycolic acid peels vs. long-term daily use of topical low-strength glycolic acid in the treatment of atrophic acne scars. Int J Dermatol. 2000;39:789-794.
- Jackson A. Chemical peels [published online January 31, 2014]. Facial Plast Surg. 2014;30:26-34.
- Davis EC, Callender VD. Postinflammatory hyperpigmentation: a review of the epidemiology, clinical features, and treatment options in skin of color. J Clin Aesthet Dermatol. 2010;3:20-31.

Acquired Port-wine Stain With Superimposed Eczema Following Penetrating Abdominal Trauma
Port-wine stains (PWSs) are common congenital capillary vascular malformations with an incidence of 3 per 1000 neonates.1 Rarely, acquired PWSs are seen, sometimes appearing following trauma.2-5 Port-wine stains are diagnosed clinically and present as painless, partially or entirely blanchable pink patches that respect the median (midline) plane.6 Although histopathologic examination is not necessary for diagnosis of PWS, typical findings include dilated, ectatic capillaries.7,8 Since it was first reported by Traub9 in 1939, more than 60 cases of acquired PWSs have been reported.10 A PubMed search of articles indexed for MEDLINE using the search terms acquired port-wine stain and port-wine stain and eczema yielded no cases of acquired PWS with associated eczematous changes and only 30 cases of congenital PWS with superimposed eczema.11-18 We report the case of an acquired PWS with superimposed eczema in an 18-year-old man following penetrating abdominal trauma.
Case Report
An otherwise healthy 18-year-old man presented to our dermatology office for evaluation of an eruption that had developed at the site of an abdominal stab wound he sustained 2 to 3 years prior. One year after he was stabbed, the patient developed a nonpruritic, painless red patch located 1 cm anterior to the healed wound on the left abdomen. The patch gradually grew larger to involve the entire left abdomen, extending to the left lower back. The site of the healed stab wound also became raised and pruritic, and the patient noted another pruritic plaque that formed within the larger patch. The patient reported no other skin conditions prior to the current eruption. His medical history was notable for seasonal allergies and asthma, but no childhood eczema.
Physical examination revealed a healthy, well-nourished man with Fitzpatrick skin type IV. A red, purpuric, coalescent patch with slightly arcuate borders extending from the mid abdomen to the left posterior flank was noted. The left lateral aspect of the patch blanched with pressure and respected the median plane. Within the larger patch, a 4-cm×2-cm lichenified, slightly macerated, hyperpigmented plaque was noted at the site of the stab wound (Figure 1). Based on these clinical findings, a presumptive diagnosis of an acquired PWS with superimposed eczema was made.
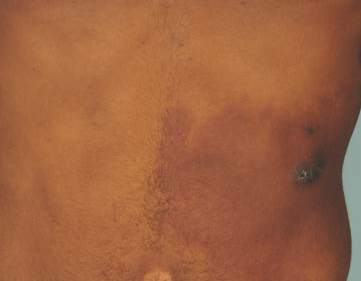
Punch biopsy specimens were taken from the large vascular patch and the smaller lichenified plaque. Histopathologic examination of the vascular patch showed an increased number of small vessels in the superficial dermis with thickened vessel walls, ectatic lumens, and no vasculopathy, consistent with a vascular malformation or a reactive vascular proliferation (Figure 2). On histopathology, the plaque showed epidermal spongiosis and hyperplasia with serum crust and a papillary dermis containing a mixed inflammatory infiltrate with occasional eosinophils, consistent with an eczematous dermatitis (Figure 3). The histologic findings confirmed the clinical diagnosis.
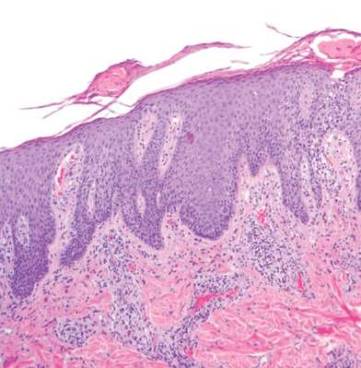
The pruritic, lichenified plaque improved with application of triamcinolone ointment 0.1% twice daily for 2 weeks. Magnetic resonance imaging to rule out an underlying arteriovenous malformation was recommended, but the patient declined.
Comment
The exact cause of PWS is unknown. There have been a multitude of genomic suspects for congenital lesions, including a somatic activating mutation (ie, a mutation acquired during fetal development) of the GNAQ (guanine nucleotide binding protein [G protein], q polypeptide) gene, which may contribute to abnormal cell proliferation including the regulation of blood vessels, and inactivating mutations in the RASA1 (RAS p21 protein activator [GTPase activating protein] 1) gene, which controls endothelial cell organization.19-22 Later mutations (ie, those occurring after the first trimester) may be more likely to result in isolated PWSs as opposed to syndromic PWSs.19 Whatever the source of genetic misinformation, it is thought that the diminished neuronal control of blood flow and the resulting alterations in dermal structure contribute to the pathogenesis of PWS and its associated histologic features.7,23
The clinical and histopathologic features of acquired PWSs are indistinguishable from those of congenital lesions, indicating that different processes may lead to the same presentation.4 Abnormal innervation and decreased supportive stroma have both been identified as contributing factors in the development of congenital and acquired PWSs.7,23-25 Rosen and Smoller23 found that diminished nerve density affects vascular tone and caliber in PWSs and had hypothesized in a prior report that decreased perivascular Schwann cells may indicate abnormal sympathetic innervation.7 Since then, PWS has been shown to lack both somatic and sensory innervation.24 Tsuji and Sawabe25 indicated that alterations to the perivascular stroma, whether congenital or as a result of trauma, decrease support for vessels, leading to ectasia.
In addition to an acquired PWS, our patient also had associated eczema within the PWS. Eczematous lesions were absent elsewhere, and he did not have a history of childhood eczema. Our review of the literature yielded 8 studies since 1996 that collectively described 30 cases of eczema within PWSs.11-18 Only 2 of these reports described adult patients with concomitant eczema and PWS and none described acquired PWS.13,18
Few studies have addressed the relationship between PWSs and eczema. It is unclear if concomitant PWS and localized eczema are collision dermatoses or if a PWS may predispose the affected skin to eczema.11-13 It has been hypothesized that the increased dermal vasculature in PWSs predisposes the skin to the development of eczema—more specifically, that ectasia may lead to increased inflammation.12,17 The concept of the “immunocompromised district” proposed by Ruocco et al26 is a unifying theory that may underlie the association noted between cases of trauma and later development of a PWS and superimposed eczematous dermatitis, such as in our case. Trauma is noted as one of a number of possible disruptive forces affecting both immunomodulation and neuromodulation within a local area of skin, leading to increased susceptibility of that district to various cutaneous diseases.26
Although our patient’s eczema responded to conservative treatment with a topical steroid, several case series have reported success with laser therapy in the treatment of PWS while preventing recurrence of associated eczematous dermatitis.12,17 Following the cessation of eczema treatment with topical steroid, which causes vasoconstriction, we suggest postponing laser therapy several weeks to allow resolution of vasoconstriction, thus providing enhanced therapeutic targeting with a vascular laser. Of particular relevance to our case, a recent study showed efficacy of the pulsed dye laser in treating PWSs in Fitzpatrick skin types IV and V.27
Conclusion
Although acquired PWS is rare, it can present later in life as an acquired lesion at a site of previous trauma.1-5 Congenital capillary malformations also can be associated with superimposed, localized eczema.11-18 We present a rarely reported case of an acquired PWS with superimposed, localized eczema. As in cases of congenital PWS with concomitant eczema, the associated eczema in our case was responsive to topical corticosteroid therapy. Additionally, pulsed dye laser has been shown to treat PWSs while preventing the recurrence of eczema, and it has been deemed effective for individuals with darker skin types.12,17, 27 Further studies are needed to explore the relationship between PWS and eczema.
- Jacobs AH, Walton RG. The incidence of birthmarks in the neonate. Pediatrics. 1976;58:218-222.
- Fegeler F. Naevus flammeus im trigeminusgebiet nach trauma im rahmen eines posttraumatisch-vegetativen syndroms. Arch Dermatol Syphilol. 1949;188:416-422.
- Kirkland CR, Mutasim DF. Acquired port-wine stain following repetitive trauma. J Am Acad Dermatol. 2011;65:462-463.
- Adams BB, Lucky AW. Acquired port-wine stains and antecedent trauma: case report and review of the literature. Arch Dermatol. 2000;136:897-899.
- Colver GB, Ryan TJ. Acquired port-wine stain. Arch Dermatol. 1986;122:1415-1416.
- Nigro J, Swerlick RA, Sepp NT, et al. Angiogenesis, vascular malformations and proliferations. In: Arndt KA, LeBoit PE, Robinson JK, Wintroub BU, eds. Cutaneous Medicine and Surgery: An Integrated Program in Dermatology. Philadelphia, PA: WB Saunders Co; 1996:1492-1521.
- Smoller BR, Rosen S. Port-wine stains. a disease of altered neural modulation of blood vessels? Arch Dermatol. 1986;122:177-179.
- Chang CJ, Yu JS, Nelson JS. Confocal microscopy study of neurovascular distribution in facial port wine stains(capillary malformation). J Formos Med Assoc. 2008;107:559-666.
- Traub EF. Naevus flammeus appearing at the age of twenty three. Arch Dermatol. 1939;39:752.
- Freysz M, Cribier B, Lipsker, D. Fegelers syndrome, acquired port-wine stain or acquired capillary malformation: three cases and a literature review [article in French]. Ann Dermatol Venereol. 2013;140:341-346.
- Tay YK, Morelli J, Weston WL. Inflammatory nuchal-occipital port-wine stains. J Am Acad Dermatol. 1996;35:811-813.
- Sidwell RU, Syed S, Harper JI. Port-wine stains and eczema. Br J Dermatol. 2001;144:1269-1270.
- Hofer T. Meyerson phenomenon within a nevus flammeus. Dermatology. 2002;205:180-183.
- Raff K, Landthaler M, Hoheleutner U. Port-wine stains with eczema. Phlebologie. 2003;32:15-17.
- Tsuboi H, Miyata T, Katsuoka K. Eczema in a port-wine stain. Clin Exp Dermatol. 2003;28:322-323.
- Rajan N, Natarahan S. Impetiginized eczema arising within a port-wine stain of the arm. J Eur Acad Dermatol Venereol. 2006;20:1009-1010.
- Fonder MA, Mamelak AJ, Kazin RA, et al. Port-wine-stain-associated dermatitis: implications for cutaneous vascular laser therapy. Pediatr Dermatol. 2007;24:376-379.
- Simon V, Wolfgan H, Katharina F. Meyerson-Phenomenon hides a nevus flammeus. J Dtsch Dermatol Ges. 2011;9:305-307.
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971-1979.
- Hershkovitz D, Bercovich D, Sprecher E, et al. RASA1 mutations may cause hereditary capillary malformations without arteriovenous malformations. Br J Dermatol. 2008;158:1035-1040.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Henkemeyer M, Rossi DJ, Holmyard DP, et al. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature. 1995;377:695-701.
- Rosen S, Smoller BR. Port-wine stains: a new hypothesis. J Am Acad Dermatol. 1987;17:164-166.
- Rydh M, Malm BM, Jernmeck J, et al. Ectatic blood vessels in port-wine stains lack innervation: possible role in pathogenesis. Plast Reconstr Surg. 1991;87:419-422.
- Tsuji T, Sawabe M. A new type of telangiectasia following trauma. J Cutan Pathol. 1988;15:22-26.
- Ruocco V, Ruocco E, Brunnetti G, et al. Opportunistic localization of skin lesions on vulnerable areas. Clin Dermatol. 2011;29:483-488.
- Thajudeheen CP, Jyothy K, Pryadarshi A. Treatment of port-wine stains with flash lamp pumped pulsed dye laser on Indian skin: a six year study. J Cutan Aesthet Surg. 2014;7:32-36.
Port-wine stains (PWSs) are common congenital capillary vascular malformations with an incidence of 3 per 1000 neonates.1 Rarely, acquired PWSs are seen, sometimes appearing following trauma.2-5 Port-wine stains are diagnosed clinically and present as painless, partially or entirely blanchable pink patches that respect the median (midline) plane.6 Although histopathologic examination is not necessary for diagnosis of PWS, typical findings include dilated, ectatic capillaries.7,8 Since it was first reported by Traub9 in 1939, more than 60 cases of acquired PWSs have been reported.10 A PubMed search of articles indexed for MEDLINE using the search terms acquired port-wine stain and port-wine stain and eczema yielded no cases of acquired PWS with associated eczematous changes and only 30 cases of congenital PWS with superimposed eczema.11-18 We report the case of an acquired PWS with superimposed eczema in an 18-year-old man following penetrating abdominal trauma.
Case Report
An otherwise healthy 18-year-old man presented to our dermatology office for evaluation of an eruption that had developed at the site of an abdominal stab wound he sustained 2 to 3 years prior. One year after he was stabbed, the patient developed a nonpruritic, painless red patch located 1 cm anterior to the healed wound on the left abdomen. The patch gradually grew larger to involve the entire left abdomen, extending to the left lower back. The site of the healed stab wound also became raised and pruritic, and the patient noted another pruritic plaque that formed within the larger patch. The patient reported no other skin conditions prior to the current eruption. His medical history was notable for seasonal allergies and asthma, but no childhood eczema.
Physical examination revealed a healthy, well-nourished man with Fitzpatrick skin type IV. A red, purpuric, coalescent patch with slightly arcuate borders extending from the mid abdomen to the left posterior flank was noted. The left lateral aspect of the patch blanched with pressure and respected the median plane. Within the larger patch, a 4-cm×2-cm lichenified, slightly macerated, hyperpigmented plaque was noted at the site of the stab wound (Figure 1). Based on these clinical findings, a presumptive diagnosis of an acquired PWS with superimposed eczema was made.
Punch biopsy specimens were taken from the large vascular patch and the smaller lichenified plaque. Histopathologic examination of the vascular patch showed an increased number of small vessels in the superficial dermis with thickened vessel walls, ectatic lumens, and no vasculopathy, consistent with a vascular malformation or a reactive vascular proliferation (Figure 2). On histopathology, the plaque showed epidermal spongiosis and hyperplasia with serum crust and a papillary dermis containing a mixed inflammatory infiltrate with occasional eosinophils, consistent with an eczematous dermatitis (Figure 3). The histologic findings confirmed the clinical diagnosis.
The pruritic, lichenified plaque improved with application of triamcinolone ointment 0.1% twice daily for 2 weeks. Magnetic resonance imaging to rule out an underlying arteriovenous malformation was recommended, but the patient declined.
Comment
The exact cause of PWS is unknown. There have been a multitude of genomic suspects for congenital lesions, including a somatic activating mutation (ie, a mutation acquired during fetal development) of the GNAQ (guanine nucleotide binding protein [G protein], q polypeptide) gene, which may contribute to abnormal cell proliferation including the regulation of blood vessels, and inactivating mutations in the RASA1 (RAS p21 protein activator [GTPase activating protein] 1) gene, which controls endothelial cell organization.19-22 Later mutations (ie, those occurring after the first trimester) may be more likely to result in isolated PWSs as opposed to syndromic PWSs.19 Whatever the source of genetic misinformation, it is thought that the diminished neuronal control of blood flow and the resulting alterations in dermal structure contribute to the pathogenesis of PWS and its associated histologic features.7,23
The clinical and histopathologic features of acquired PWSs are indistinguishable from those of congenital lesions, indicating that different processes may lead to the same presentation.4 Abnormal innervation and decreased supportive stroma have both been identified as contributing factors in the development of congenital and acquired PWSs.7,23-25 Rosen and Smoller23 found that diminished nerve density affects vascular tone and caliber in PWSs and had hypothesized in a prior report that decreased perivascular Schwann cells may indicate abnormal sympathetic innervation.7 Since then, PWS has been shown to lack both somatic and sensory innervation.24 Tsuji and Sawabe25 indicated that alterations to the perivascular stroma, whether congenital or as a result of trauma, decrease support for vessels, leading to ectasia.
In addition to an acquired PWS, our patient also had associated eczema within the PWS. Eczematous lesions were absent elsewhere, and he did not have a history of childhood eczema. Our review of the literature yielded 8 studies since 1996 that collectively described 30 cases of eczema within PWSs.11-18 Only 2 of these reports described adult patients with concomitant eczema and PWS and none described acquired PWS.13,18
Few studies have addressed the relationship between PWSs and eczema. It is unclear if concomitant PWS and localized eczema are collision dermatoses or if a PWS may predispose the affected skin to eczema.11-13 It has been hypothesized that the increased dermal vasculature in PWSs predisposes the skin to the development of eczema—more specifically, that ectasia may lead to increased inflammation.12,17 The concept of the “immunocompromised district” proposed by Ruocco et al26 is a unifying theory that may underlie the association noted between cases of trauma and later development of a PWS and superimposed eczematous dermatitis, such as in our case. Trauma is noted as one of a number of possible disruptive forces affecting both immunomodulation and neuromodulation within a local area of skin, leading to increased susceptibility of that district to various cutaneous diseases.26
Although our patient’s eczema responded to conservative treatment with a topical steroid, several case series have reported success with laser therapy in the treatment of PWS while preventing recurrence of associated eczematous dermatitis.12,17 Following the cessation of eczema treatment with topical steroid, which causes vasoconstriction, we suggest postponing laser therapy several weeks to allow resolution of vasoconstriction, thus providing enhanced therapeutic targeting with a vascular laser. Of particular relevance to our case, a recent study showed efficacy of the pulsed dye laser in treating PWSs in Fitzpatrick skin types IV and V.27
Conclusion
Although acquired PWS is rare, it can present later in life as an acquired lesion at a site of previous trauma.1-5 Congenital capillary malformations also can be associated with superimposed, localized eczema.11-18 We present a rarely reported case of an acquired PWS with superimposed, localized eczema. As in cases of congenital PWS with concomitant eczema, the associated eczema in our case was responsive to topical corticosteroid therapy. Additionally, pulsed dye laser has been shown to treat PWSs while preventing the recurrence of eczema, and it has been deemed effective for individuals with darker skin types.12,17, 27 Further studies are needed to explore the relationship between PWS and eczema.
Port-wine stains (PWSs) are common congenital capillary vascular malformations with an incidence of 3 per 1000 neonates.1 Rarely, acquired PWSs are seen, sometimes appearing following trauma.2-5 Port-wine stains are diagnosed clinically and present as painless, partially or entirely blanchable pink patches that respect the median (midline) plane.6 Although histopathologic examination is not necessary for diagnosis of PWS, typical findings include dilated, ectatic capillaries.7,8 Since it was first reported by Traub9 in 1939, more than 60 cases of acquired PWSs have been reported.10 A PubMed search of articles indexed for MEDLINE using the search terms acquired port-wine stain and port-wine stain and eczema yielded no cases of acquired PWS with associated eczematous changes and only 30 cases of congenital PWS with superimposed eczema.11-18 We report the case of an acquired PWS with superimposed eczema in an 18-year-old man following penetrating abdominal trauma.
Case Report
An otherwise healthy 18-year-old man presented to our dermatology office for evaluation of an eruption that had developed at the site of an abdominal stab wound he sustained 2 to 3 years prior. One year after he was stabbed, the patient developed a nonpruritic, painless red patch located 1 cm anterior to the healed wound on the left abdomen. The patch gradually grew larger to involve the entire left abdomen, extending to the left lower back. The site of the healed stab wound also became raised and pruritic, and the patient noted another pruritic plaque that formed within the larger patch. The patient reported no other skin conditions prior to the current eruption. His medical history was notable for seasonal allergies and asthma, but no childhood eczema.
Physical examination revealed a healthy, well-nourished man with Fitzpatrick skin type IV. A red, purpuric, coalescent patch with slightly arcuate borders extending from the mid abdomen to the left posterior flank was noted. The left lateral aspect of the patch blanched with pressure and respected the median plane. Within the larger patch, a 4-cm×2-cm lichenified, slightly macerated, hyperpigmented plaque was noted at the site of the stab wound (Figure 1). Based on these clinical findings, a presumptive diagnosis of an acquired PWS with superimposed eczema was made.
Punch biopsy specimens were taken from the large vascular patch and the smaller lichenified plaque. Histopathologic examination of the vascular patch showed an increased number of small vessels in the superficial dermis with thickened vessel walls, ectatic lumens, and no vasculopathy, consistent with a vascular malformation or a reactive vascular proliferation (Figure 2). On histopathology, the plaque showed epidermal spongiosis and hyperplasia with serum crust and a papillary dermis containing a mixed inflammatory infiltrate with occasional eosinophils, consistent with an eczematous dermatitis (Figure 3). The histologic findings confirmed the clinical diagnosis.
The pruritic, lichenified plaque improved with application of triamcinolone ointment 0.1% twice daily for 2 weeks. Magnetic resonance imaging to rule out an underlying arteriovenous malformation was recommended, but the patient declined.
Comment
The exact cause of PWS is unknown. There have been a multitude of genomic suspects for congenital lesions, including a somatic activating mutation (ie, a mutation acquired during fetal development) of the GNAQ (guanine nucleotide binding protein [G protein], q polypeptide) gene, which may contribute to abnormal cell proliferation including the regulation of blood vessels, and inactivating mutations in the RASA1 (RAS p21 protein activator [GTPase activating protein] 1) gene, which controls endothelial cell organization.19-22 Later mutations (ie, those occurring after the first trimester) may be more likely to result in isolated PWSs as opposed to syndromic PWSs.19 Whatever the source of genetic misinformation, it is thought that the diminished neuronal control of blood flow and the resulting alterations in dermal structure contribute to the pathogenesis of PWS and its associated histologic features.7,23
The clinical and histopathologic features of acquired PWSs are indistinguishable from those of congenital lesions, indicating that different processes may lead to the same presentation.4 Abnormal innervation and decreased supportive stroma have both been identified as contributing factors in the development of congenital and acquired PWSs.7,23-25 Rosen and Smoller23 found that diminished nerve density affects vascular tone and caliber in PWSs and had hypothesized in a prior report that decreased perivascular Schwann cells may indicate abnormal sympathetic innervation.7 Since then, PWS has been shown to lack both somatic and sensory innervation.24 Tsuji and Sawabe25 indicated that alterations to the perivascular stroma, whether congenital or as a result of trauma, decrease support for vessels, leading to ectasia.
In addition to an acquired PWS, our patient also had associated eczema within the PWS. Eczematous lesions were absent elsewhere, and he did not have a history of childhood eczema. Our review of the literature yielded 8 studies since 1996 that collectively described 30 cases of eczema within PWSs.11-18 Only 2 of these reports described adult patients with concomitant eczema and PWS and none described acquired PWS.13,18
Few studies have addressed the relationship between PWSs and eczema. It is unclear if concomitant PWS and localized eczema are collision dermatoses or if a PWS may predispose the affected skin to eczema.11-13 It has been hypothesized that the increased dermal vasculature in PWSs predisposes the skin to the development of eczema—more specifically, that ectasia may lead to increased inflammation.12,17 The concept of the “immunocompromised district” proposed by Ruocco et al26 is a unifying theory that may underlie the association noted between cases of trauma and later development of a PWS and superimposed eczematous dermatitis, such as in our case. Trauma is noted as one of a number of possible disruptive forces affecting both immunomodulation and neuromodulation within a local area of skin, leading to increased susceptibility of that district to various cutaneous diseases.26
Although our patient’s eczema responded to conservative treatment with a topical steroid, several case series have reported success with laser therapy in the treatment of PWS while preventing recurrence of associated eczematous dermatitis.12,17 Following the cessation of eczema treatment with topical steroid, which causes vasoconstriction, we suggest postponing laser therapy several weeks to allow resolution of vasoconstriction, thus providing enhanced therapeutic targeting with a vascular laser. Of particular relevance to our case, a recent study showed efficacy of the pulsed dye laser in treating PWSs in Fitzpatrick skin types IV and V.27
Conclusion
Although acquired PWS is rare, it can present later in life as an acquired lesion at a site of previous trauma.1-5 Congenital capillary malformations also can be associated with superimposed, localized eczema.11-18 We present a rarely reported case of an acquired PWS with superimposed, localized eczema. As in cases of congenital PWS with concomitant eczema, the associated eczema in our case was responsive to topical corticosteroid therapy. Additionally, pulsed dye laser has been shown to treat PWSs while preventing the recurrence of eczema, and it has been deemed effective for individuals with darker skin types.12,17, 27 Further studies are needed to explore the relationship between PWS and eczema.
- Jacobs AH, Walton RG. The incidence of birthmarks in the neonate. Pediatrics. 1976;58:218-222.
- Fegeler F. Naevus flammeus im trigeminusgebiet nach trauma im rahmen eines posttraumatisch-vegetativen syndroms. Arch Dermatol Syphilol. 1949;188:416-422.
- Kirkland CR, Mutasim DF. Acquired port-wine stain following repetitive trauma. J Am Acad Dermatol. 2011;65:462-463.
- Adams BB, Lucky AW. Acquired port-wine stains and antecedent trauma: case report and review of the literature. Arch Dermatol. 2000;136:897-899.
- Colver GB, Ryan TJ. Acquired port-wine stain. Arch Dermatol. 1986;122:1415-1416.
- Nigro J, Swerlick RA, Sepp NT, et al. Angiogenesis, vascular malformations and proliferations. In: Arndt KA, LeBoit PE, Robinson JK, Wintroub BU, eds. Cutaneous Medicine and Surgery: An Integrated Program in Dermatology. Philadelphia, PA: WB Saunders Co; 1996:1492-1521.
- Smoller BR, Rosen S. Port-wine stains. a disease of altered neural modulation of blood vessels? Arch Dermatol. 1986;122:177-179.
- Chang CJ, Yu JS, Nelson JS. Confocal microscopy study of neurovascular distribution in facial port wine stains(capillary malformation). J Formos Med Assoc. 2008;107:559-666.
- Traub EF. Naevus flammeus appearing at the age of twenty three. Arch Dermatol. 1939;39:752.
- Freysz M, Cribier B, Lipsker, D. Fegelers syndrome, acquired port-wine stain or acquired capillary malformation: three cases and a literature review [article in French]. Ann Dermatol Venereol. 2013;140:341-346.
- Tay YK, Morelli J, Weston WL. Inflammatory nuchal-occipital port-wine stains. J Am Acad Dermatol. 1996;35:811-813.
- Sidwell RU, Syed S, Harper JI. Port-wine stains and eczema. Br J Dermatol. 2001;144:1269-1270.
- Hofer T. Meyerson phenomenon within a nevus flammeus. Dermatology. 2002;205:180-183.
- Raff K, Landthaler M, Hoheleutner U. Port-wine stains with eczema. Phlebologie. 2003;32:15-17.
- Tsuboi H, Miyata T, Katsuoka K. Eczema in a port-wine stain. Clin Exp Dermatol. 2003;28:322-323.
- Rajan N, Natarahan S. Impetiginized eczema arising within a port-wine stain of the arm. J Eur Acad Dermatol Venereol. 2006;20:1009-1010.
- Fonder MA, Mamelak AJ, Kazin RA, et al. Port-wine-stain-associated dermatitis: implications for cutaneous vascular laser therapy. Pediatr Dermatol. 2007;24:376-379.
- Simon V, Wolfgan H, Katharina F. Meyerson-Phenomenon hides a nevus flammeus. J Dtsch Dermatol Ges. 2011;9:305-307.
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971-1979.
- Hershkovitz D, Bercovich D, Sprecher E, et al. RASA1 mutations may cause hereditary capillary malformations without arteriovenous malformations. Br J Dermatol. 2008;158:1035-1040.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Henkemeyer M, Rossi DJ, Holmyard DP, et al. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature. 1995;377:695-701.
- Rosen S, Smoller BR. Port-wine stains: a new hypothesis. J Am Acad Dermatol. 1987;17:164-166.
- Rydh M, Malm BM, Jernmeck J, et al. Ectatic blood vessels in port-wine stains lack innervation: possible role in pathogenesis. Plast Reconstr Surg. 1991;87:419-422.
- Tsuji T, Sawabe M. A new type of telangiectasia following trauma. J Cutan Pathol. 1988;15:22-26.
- Ruocco V, Ruocco E, Brunnetti G, et al. Opportunistic localization of skin lesions on vulnerable areas. Clin Dermatol. 2011;29:483-488.
- Thajudeheen CP, Jyothy K, Pryadarshi A. Treatment of port-wine stains with flash lamp pumped pulsed dye laser on Indian skin: a six year study. J Cutan Aesthet Surg. 2014;7:32-36.
- Jacobs AH, Walton RG. The incidence of birthmarks in the neonate. Pediatrics. 1976;58:218-222.
- Fegeler F. Naevus flammeus im trigeminusgebiet nach trauma im rahmen eines posttraumatisch-vegetativen syndroms. Arch Dermatol Syphilol. 1949;188:416-422.
- Kirkland CR, Mutasim DF. Acquired port-wine stain following repetitive trauma. J Am Acad Dermatol. 2011;65:462-463.
- Adams BB, Lucky AW. Acquired port-wine stains and antecedent trauma: case report and review of the literature. Arch Dermatol. 2000;136:897-899.
- Colver GB, Ryan TJ. Acquired port-wine stain. Arch Dermatol. 1986;122:1415-1416.
- Nigro J, Swerlick RA, Sepp NT, et al. Angiogenesis, vascular malformations and proliferations. In: Arndt KA, LeBoit PE, Robinson JK, Wintroub BU, eds. Cutaneous Medicine and Surgery: An Integrated Program in Dermatology. Philadelphia, PA: WB Saunders Co; 1996:1492-1521.
- Smoller BR, Rosen S. Port-wine stains. a disease of altered neural modulation of blood vessels? Arch Dermatol. 1986;122:177-179.
- Chang CJ, Yu JS, Nelson JS. Confocal microscopy study of neurovascular distribution in facial port wine stains(capillary malformation). J Formos Med Assoc. 2008;107:559-666.
- Traub EF. Naevus flammeus appearing at the age of twenty three. Arch Dermatol. 1939;39:752.
- Freysz M, Cribier B, Lipsker, D. Fegelers syndrome, acquired port-wine stain or acquired capillary malformation: three cases and a literature review [article in French]. Ann Dermatol Venereol. 2013;140:341-346.
- Tay YK, Morelli J, Weston WL. Inflammatory nuchal-occipital port-wine stains. J Am Acad Dermatol. 1996;35:811-813.
- Sidwell RU, Syed S, Harper JI. Port-wine stains and eczema. Br J Dermatol. 2001;144:1269-1270.
- Hofer T. Meyerson phenomenon within a nevus flammeus. Dermatology. 2002;205:180-183.
- Raff K, Landthaler M, Hoheleutner U. Port-wine stains with eczema. Phlebologie. 2003;32:15-17.
- Tsuboi H, Miyata T, Katsuoka K. Eczema in a port-wine stain. Clin Exp Dermatol. 2003;28:322-323.
- Rajan N, Natarahan S. Impetiginized eczema arising within a port-wine stain of the arm. J Eur Acad Dermatol Venereol. 2006;20:1009-1010.
- Fonder MA, Mamelak AJ, Kazin RA, et al. Port-wine-stain-associated dermatitis: implications for cutaneous vascular laser therapy. Pediatr Dermatol. 2007;24:376-379.
- Simon V, Wolfgan H, Katharina F. Meyerson-Phenomenon hides a nevus flammeus. J Dtsch Dermatol Ges. 2011;9:305-307.
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971-1979.
- Hershkovitz D, Bercovich D, Sprecher E, et al. RASA1 mutations may cause hereditary capillary malformations without arteriovenous malformations. Br J Dermatol. 2008;158:1035-1040.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Henkemeyer M, Rossi DJ, Holmyard DP, et al. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature. 1995;377:695-701.
- Rosen S, Smoller BR. Port-wine stains: a new hypothesis. J Am Acad Dermatol. 1987;17:164-166.
- Rydh M, Malm BM, Jernmeck J, et al. Ectatic blood vessels in port-wine stains lack innervation: possible role in pathogenesis. Plast Reconstr Surg. 1991;87:419-422.
- Tsuji T, Sawabe M. A new type of telangiectasia following trauma. J Cutan Pathol. 1988;15:22-26.
- Ruocco V, Ruocco E, Brunnetti G, et al. Opportunistic localization of skin lesions on vulnerable areas. Clin Dermatol. 2011;29:483-488.
- Thajudeheen CP, Jyothy K, Pryadarshi A. Treatment of port-wine stains with flash lamp pumped pulsed dye laser on Indian skin: a six year study. J Cutan Aesthet Surg. 2014;7:32-36.
Practice Points
- Port-wine stains (PWSs) most often are congenital lesions but can present later in life as acquired lesions with the same clinical and histologic findings.
- Magnetic resonance imaging of acquired PWSs should be considered to rule out underlying vascular anomalies (eg, deeper arteriovenous malformations).
- Pulsed dye laser therapy is safe for darker skin types and is the treatment of choice for acquired PWSs.