User login
Accuracy and Sources of Images From Direct Google Image Searches for Common Dermatology Terms
To the Editor:
Prior studies have assessed the quality of text-based dermatology information on the Internet using traditional search engine queries.1 However, little is understood about the sources, accuracy, and quality of online dermatology images derived from direct image searches. Previous work has shown that direct search engine image queries were largely accurate for 3 pediatric dermatology diagnosis searches: atopic dermatitis, lichen striatus, and subcutaneous fat necrosis.2 We assessed images obtained for common dermatologic conditions from a Google image search (GIS) compared to a traditional text-based Google web search (GWS).
Image results for 32 unique dermatologic search terms were analyzed (Table 1). These search terms were selected using the results of a prior study that identified the most common dermatologic diagnoses that led users to the 2 most popular dermatology-specific websites worldwide: the American Academy of Dermatology (www.aad.org) and DermNet New Zealand (www.dermnetnz.org).3 The Alexa directory (www.alexa.com), a large publicly available Internet analytics resource, was used to determine the most common dermatology search terms that led a user to either www.dermnetnz.org or www.aad.org. In addition, searches for the 3 most common types of skin cancer—melanoma, squamous cell carcinoma, and basal cell carcinoma—were included. Each term was entered into a GIS and a GWS. The first 10 results, which represent 92% of the websites ultimately visited by users,4 were analyzed. The source, diagnostic accuracy, and Fitzpatrick skin type of the images was determined. Website sources were organized into 11 categories. All data collection occurred within a 1-week period in August 2015.
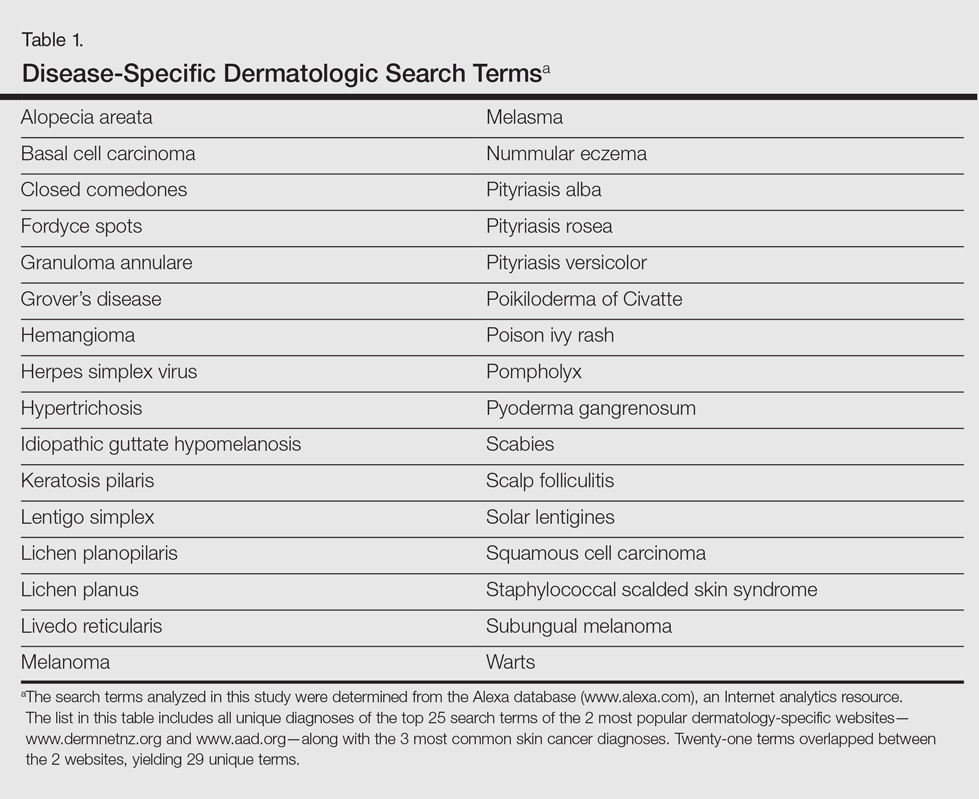
A total of 320 images were analyzed. In the GIS, private websites (36%), dermatology association websites (28%), and general health information websites (10%) were the 3 most common sources. In the GWS, health information websites (35%), private websites (21%), and dermatology association websites (20%) accounted for the most common sources (Table 2). The majority of images were of Fitzpatrick skin types I and II (89%) and nearly all images were diagnostically accurate (98%). There was no statistically significant difference in accuracy of diagnosis between physician-associated websites (100% accuracy) versus nonphysician-associated sites (98% accuracy, P=.25).
Our results showed high diagnostic accuracy among the top GIS results for common dermatology search terms. Diagnostic accuracy did not vary between websites that were physician associated versus those that were not. Our results are comparable to the reported accuracy of online dermatologic health information.1 In GIS results, the majority of images were provided by private websites, whereas the top websites in GWS results were health information websites.
Only 1% of images were of Fitzpatrick skin types VI and VII. Presentation of skin diseases is remarkably different based on the patient’s skin type.5 The shortage of readily accessible images of skin of color is in line with the lack of familiarity physicians and trainees have with dermatologic conditions in ethnic skin.6
Based on the results from this analysis, providers and patients searching for dermatologic conditions via a direct GIS should be cognizant of several considerations. Although our results showed that GIS was accurate, the searcher should note that image-based searches are not accompanied by relevant text that can help confirm relevancy and accuracy. Image searches depend on textual tags added by the source website. Websites that represent dermatological associations and academic centers can add an additional layer of confidence for users. Patients and clinicians also should be aware that the consideration of a patient’s Fitzpatrick skin type is critical when assessing the relevancy of a GIS result. In conclusion, search results via GIS queries are accurate for the dermatological diagnoses tested but may be lacking in skin of color variations, suggesting a potential unmet need based on our growing ethnic skin population.
- Jensen JD, Dunnick CA, Arbuckle HA, et al. Dermatology information on the Internet: an appraisal by dermatologists and dermatology residents. J Am Acad Dermatol. 2010;63:1101-1103.
- Cutrone M, Grimalt R. Dermatological image search engines on the Internet: do they work? J Eur Acad Dermatol Venereol. 2007;21:175-177.
- Xu S, Nault A, Bhatia A. Search and engagement analysis of association websites representing dermatologists—implications and opportunities for web visibility and patient education: website rankings of dermatology associations. Pract Dermatol. In press.
- comScore releases July 2015 U.S. desktop search engine rankings [press release]. Reston, VA: comScore, Inc; August 14, 2015. http://www.comscore.com/Insights/Market-Rankings/comScore-Releases-July-2015-U.S.-Desktop-Search-Engine-Rankings. Accessed October 18, 2016.
- Kundu RV, Patterson S. Dermatologic conditions in skin of color: part I. special considerations for common skin disorders. Am Fam Physician. 2013;87:850-856.
- Nijhawan RI, Jacob SE, Woolery-Lloyd H. Skin of color education in dermatology residency programs: does residency training reflect the changing demographics of the United States? J Am Acad Dermatol. 2008;59:615-618.
To the Editor:
Prior studies have assessed the quality of text-based dermatology information on the Internet using traditional search engine queries.1 However, little is understood about the sources, accuracy, and quality of online dermatology images derived from direct image searches. Previous work has shown that direct search engine image queries were largely accurate for 3 pediatric dermatology diagnosis searches: atopic dermatitis, lichen striatus, and subcutaneous fat necrosis.2 We assessed images obtained for common dermatologic conditions from a Google image search (GIS) compared to a traditional text-based Google web search (GWS).
Image results for 32 unique dermatologic search terms were analyzed (Table 1). These search terms were selected using the results of a prior study that identified the most common dermatologic diagnoses that led users to the 2 most popular dermatology-specific websites worldwide: the American Academy of Dermatology (www.aad.org) and DermNet New Zealand (www.dermnetnz.org).3 The Alexa directory (www.alexa.com), a large publicly available Internet analytics resource, was used to determine the most common dermatology search terms that led a user to either www.dermnetnz.org or www.aad.org. In addition, searches for the 3 most common types of skin cancer—melanoma, squamous cell carcinoma, and basal cell carcinoma—were included. Each term was entered into a GIS and a GWS. The first 10 results, which represent 92% of the websites ultimately visited by users,4 were analyzed. The source, diagnostic accuracy, and Fitzpatrick skin type of the images was determined. Website sources were organized into 11 categories. All data collection occurred within a 1-week period in August 2015.
A total of 320 images were analyzed. In the GIS, private websites (36%), dermatology association websites (28%), and general health information websites (10%) were the 3 most common sources. In the GWS, health information websites (35%), private websites (21%), and dermatology association websites (20%) accounted for the most common sources (Table 2). The majority of images were of Fitzpatrick skin types I and II (89%) and nearly all images were diagnostically accurate (98%). There was no statistically significant difference in accuracy of diagnosis between physician-associated websites (100% accuracy) versus nonphysician-associated sites (98% accuracy, P=.25).
Our results showed high diagnostic accuracy among the top GIS results for common dermatology search terms. Diagnostic accuracy did not vary between websites that were physician associated versus those that were not. Our results are comparable to the reported accuracy of online dermatologic health information.1 In GIS results, the majority of images were provided by private websites, whereas the top websites in GWS results were health information websites.
Only 1% of images were of Fitzpatrick skin types VI and VII. Presentation of skin diseases is remarkably different based on the patient’s skin type.5 The shortage of readily accessible images of skin of color is in line with the lack of familiarity physicians and trainees have with dermatologic conditions in ethnic skin.6
Based on the results from this analysis, providers and patients searching for dermatologic conditions via a direct GIS should be cognizant of several considerations. Although our results showed that GIS was accurate, the searcher should note that image-based searches are not accompanied by relevant text that can help confirm relevancy and accuracy. Image searches depend on textual tags added by the source website. Websites that represent dermatological associations and academic centers can add an additional layer of confidence for users. Patients and clinicians also should be aware that the consideration of a patient’s Fitzpatrick skin type is critical when assessing the relevancy of a GIS result. In conclusion, search results via GIS queries are accurate for the dermatological diagnoses tested but may be lacking in skin of color variations, suggesting a potential unmet need based on our growing ethnic skin population.
To the Editor:
Prior studies have assessed the quality of text-based dermatology information on the Internet using traditional search engine queries.1 However, little is understood about the sources, accuracy, and quality of online dermatology images derived from direct image searches. Previous work has shown that direct search engine image queries were largely accurate for 3 pediatric dermatology diagnosis searches: atopic dermatitis, lichen striatus, and subcutaneous fat necrosis.2 We assessed images obtained for common dermatologic conditions from a Google image search (GIS) compared to a traditional text-based Google web search (GWS).
Image results for 32 unique dermatologic search terms were analyzed (Table 1). These search terms were selected using the results of a prior study that identified the most common dermatologic diagnoses that led users to the 2 most popular dermatology-specific websites worldwide: the American Academy of Dermatology (www.aad.org) and DermNet New Zealand (www.dermnetnz.org).3 The Alexa directory (www.alexa.com), a large publicly available Internet analytics resource, was used to determine the most common dermatology search terms that led a user to either www.dermnetnz.org or www.aad.org. In addition, searches for the 3 most common types of skin cancer—melanoma, squamous cell carcinoma, and basal cell carcinoma—were included. Each term was entered into a GIS and a GWS. The first 10 results, which represent 92% of the websites ultimately visited by users,4 were analyzed. The source, diagnostic accuracy, and Fitzpatrick skin type of the images was determined. Website sources were organized into 11 categories. All data collection occurred within a 1-week period in August 2015.
A total of 320 images were analyzed. In the GIS, private websites (36%), dermatology association websites (28%), and general health information websites (10%) were the 3 most common sources. In the GWS, health information websites (35%), private websites (21%), and dermatology association websites (20%) accounted for the most common sources (Table 2). The majority of images were of Fitzpatrick skin types I and II (89%) and nearly all images were diagnostically accurate (98%). There was no statistically significant difference in accuracy of diagnosis between physician-associated websites (100% accuracy) versus nonphysician-associated sites (98% accuracy, P=.25).
Our results showed high diagnostic accuracy among the top GIS results for common dermatology search terms. Diagnostic accuracy did not vary between websites that were physician associated versus those that were not. Our results are comparable to the reported accuracy of online dermatologic health information.1 In GIS results, the majority of images were provided by private websites, whereas the top websites in GWS results were health information websites.
Only 1% of images were of Fitzpatrick skin types VI and VII. Presentation of skin diseases is remarkably different based on the patient’s skin type.5 The shortage of readily accessible images of skin of color is in line with the lack of familiarity physicians and trainees have with dermatologic conditions in ethnic skin.6
Based on the results from this analysis, providers and patients searching for dermatologic conditions via a direct GIS should be cognizant of several considerations. Although our results showed that GIS was accurate, the searcher should note that image-based searches are not accompanied by relevant text that can help confirm relevancy and accuracy. Image searches depend on textual tags added by the source website. Websites that represent dermatological associations and academic centers can add an additional layer of confidence for users. Patients and clinicians also should be aware that the consideration of a patient’s Fitzpatrick skin type is critical when assessing the relevancy of a GIS result. In conclusion, search results via GIS queries are accurate for the dermatological diagnoses tested but may be lacking in skin of color variations, suggesting a potential unmet need based on our growing ethnic skin population.
- Jensen JD, Dunnick CA, Arbuckle HA, et al. Dermatology information on the Internet: an appraisal by dermatologists and dermatology residents. J Am Acad Dermatol. 2010;63:1101-1103.
- Cutrone M, Grimalt R. Dermatological image search engines on the Internet: do they work? J Eur Acad Dermatol Venereol. 2007;21:175-177.
- Xu S, Nault A, Bhatia A. Search and engagement analysis of association websites representing dermatologists—implications and opportunities for web visibility and patient education: website rankings of dermatology associations. Pract Dermatol. In press.
- comScore releases July 2015 U.S. desktop search engine rankings [press release]. Reston, VA: comScore, Inc; August 14, 2015. http://www.comscore.com/Insights/Market-Rankings/comScore-Releases-July-2015-U.S.-Desktop-Search-Engine-Rankings. Accessed October 18, 2016.
- Kundu RV, Patterson S. Dermatologic conditions in skin of color: part I. special considerations for common skin disorders. Am Fam Physician. 2013;87:850-856.
- Nijhawan RI, Jacob SE, Woolery-Lloyd H. Skin of color education in dermatology residency programs: does residency training reflect the changing demographics of the United States? J Am Acad Dermatol. 2008;59:615-618.
- Jensen JD, Dunnick CA, Arbuckle HA, et al. Dermatology information on the Internet: an appraisal by dermatologists and dermatology residents. J Am Acad Dermatol. 2010;63:1101-1103.
- Cutrone M, Grimalt R. Dermatological image search engines on the Internet: do they work? J Eur Acad Dermatol Venereol. 2007;21:175-177.
- Xu S, Nault A, Bhatia A. Search and engagement analysis of association websites representing dermatologists—implications and opportunities for web visibility and patient education: website rankings of dermatology associations. Pract Dermatol. In press.
- comScore releases July 2015 U.S. desktop search engine rankings [press release]. Reston, VA: comScore, Inc; August 14, 2015. http://www.comscore.com/Insights/Market-Rankings/comScore-Releases-July-2015-U.S.-Desktop-Search-Engine-Rankings. Accessed October 18, 2016.
- Kundu RV, Patterson S. Dermatologic conditions in skin of color: part I. special considerations for common skin disorders. Am Fam Physician. 2013;87:850-856.
- Nijhawan RI, Jacob SE, Woolery-Lloyd H. Skin of color education in dermatology residency programs: does residency training reflect the changing demographics of the United States? J Am Acad Dermatol. 2008;59:615-618.
Practice Points
- Direct Google image searches largely deliver accurate results for common dermatological diagnoses.
- Greater effort should be made to include more publicly available images for dermatological diseases in darker skin types.
Novel De Novo Heterozygous Frameshift Mutation of the ADAR1 Gene in Heavy Dyschromatosis Symmetrica Hereditaria
To the Editor:
Dyschromatosis symmetrica hereditaria (DSH)(Online Mendelian Inheritance in Man 127400), also called reticulate acropigmentation of Dohi, is a pigmentary genodermatosis characterized by a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Linkage analysis has revealed that the DSH gene locus resides on chromosome 1q11-q21,1 and the adenosine deaminase RNA specific gene, ADAR1 (also called DSRAD), in this region has been identified as being responsible for the development of DSH.2 We report a sporadic case of severe DSH with the ADAR1 gene detected in a mutation analysis.
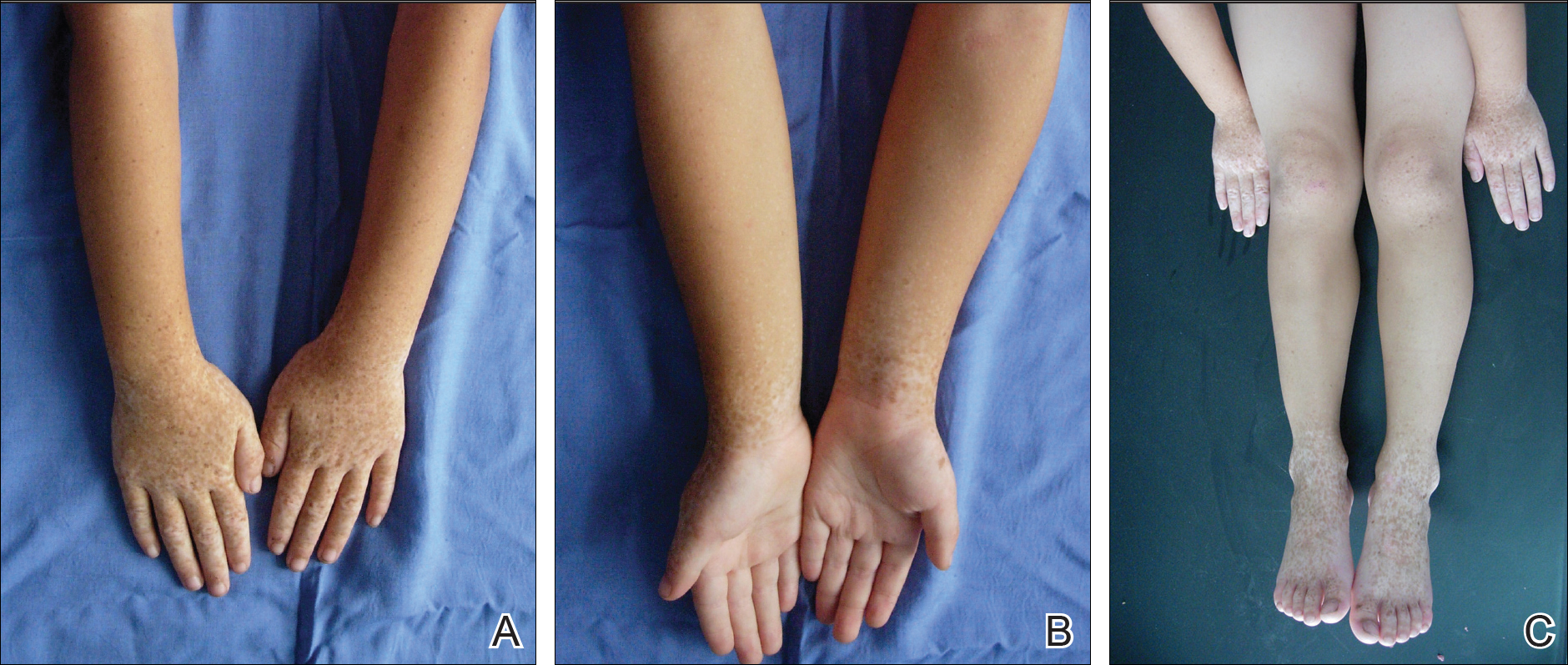
A 6-year-old girl presented with a mixture of hyperpigmented and hypopigmented macules on the dorsal aspects of the hands and feet and the curved side of the wrists, heels, and knees, as well as scattered frecklelike and depigmented spots on the face, ears, neck, arms, and upper back (Figure 1). Her parents noted that hyperpigmented and hypopigmented macules on the dorsal aspects of the hands developed at 5 months of age. Exacerbation after exposure to sunlight resulted in the eruption becoming remarkable in summer and fainter in winter. The skin lesions gradually became more progressive. Physical examination revealed that the patient generally was healthy.
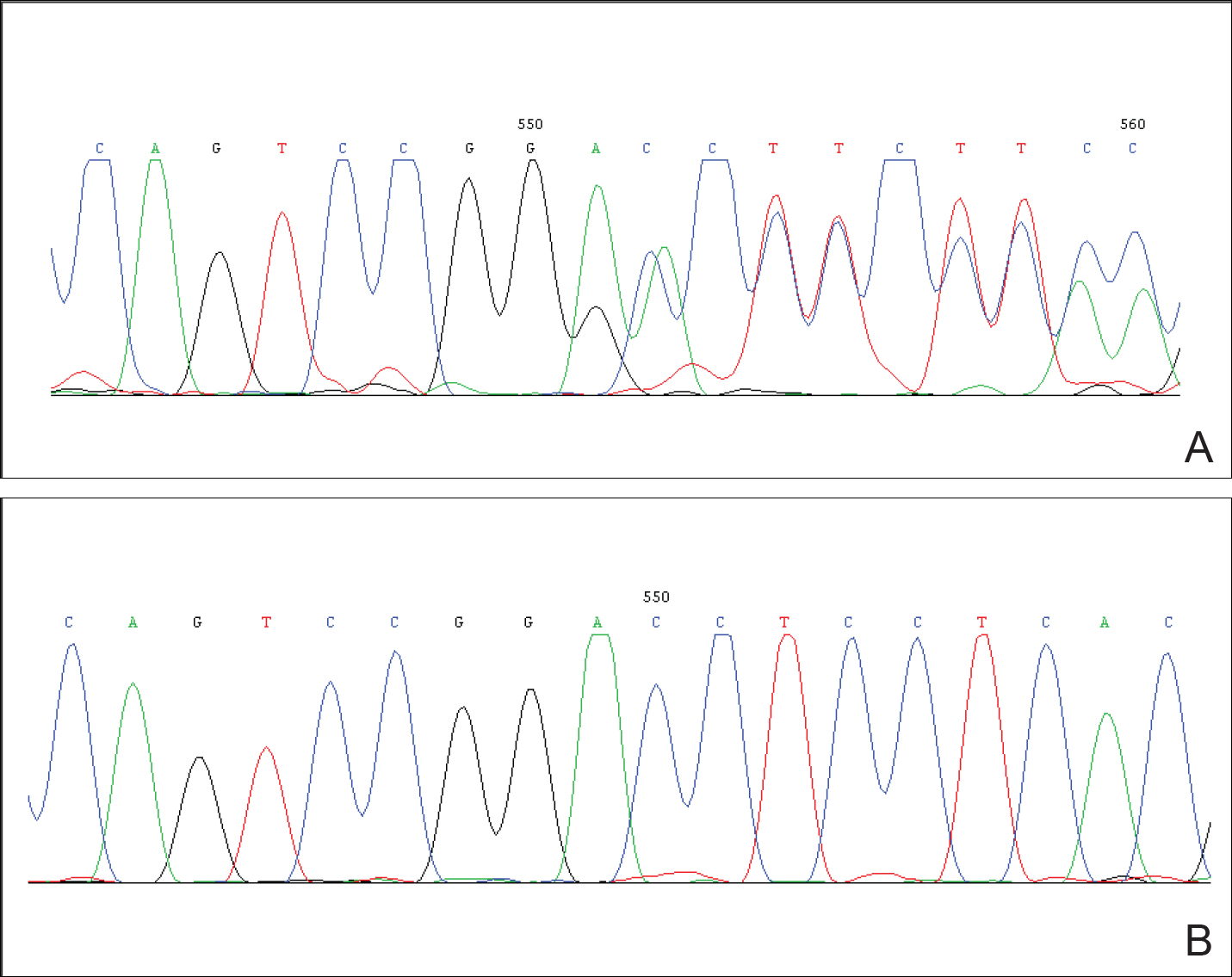
After obtaining informed consent, we performed a mutation analysis of the ADAR1 gene in our patient and her parents. We used a kit to extract genomic DNA from peripheral blood, which was then used to amplify the exons of the ADAR1 gene with intronic flanking sequences by polymerase chain reaction with the primer.3 After amplification, polymerase chain reaction products were purified. We sequenced the ADAR1 gene. Sequence comparisons and analysis found that the patient (proband) carried a heterozygous insertional mutation c.2253insG in exon 6 of the ADAR1 gene. This mutation was not detected in the proband’s healthy parents and 100 normal individuals (Figure 2).
Dyschromatosis symmetrica hereditaria is acquired by autosomal-dominant inheritance and is mainly reported in Asians, especially in Japan and China. Oyama et al4 reviewed 185 cases of DSH in Japan and found the onset of this disease usually was during infancy or childhood; 73% of patients developed the skin lesions before 6 years of age. Suzuki et al5 reported 10 unrelated Japanese patients and found the onset of disease ranged from 1 year of age to childhood. Zhang et al1,6 investigated 78 Chinese patients with DSH including 8 multigenerational families and 2 sporadic patients and found the age of disease onset ranged from 6 months to 15 years of age. The age of onset in our patient (5 months) was younger than these prior reports.
Patients with DSH have a characteristic appearance including a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Few patients have similar lesions on the knees and elbows. Many patients have frecklelike macules on the face and arms.1-6 One patient has been described with scattered depigmented spots on the face and chest.1 Our patient had a characteristic appearance as well as some special manifestations including skin lesions on the curved side of the wrist, ears, neck, and upper back.
The human ADAR1 gene spans 30 kilobase and contains 15 exons. It encodes RNA-specific adenosine deaminase composed of 1226 amino acid residues. This enzyme is important for various functions such as site-specific RNA editing and nuclear translation. This enzyme has 2 Z-alpha domains, 3 double-stranded RNA–binding domains, and the putative deaminase domain corresponding to exon 2, exons 2 to 7, and exons 9 to 14 of ADAR1, respectively.6
Mutation analysis of the ADAR1 gene in this case showed heterozygous insertion mutation c.2253insG in exon 6 of the ADAR1 gene, which changed the reading frame, and 475 amino acid residues in C-terminus are replaced by 90 amino acid residues (TSSRAQVRLPSKSWGSLVPSRLRTQQEA RQAGSSRCGSPCLDWGEREGRTHGFHRG NPSDRGQSQKNYAPPLKVPRSTAKT DTPSHWQHLP). This mutation was not detected in the proband’s healthy parents and the 100 control individuals, which indicated that it was a de novo mutation and the pathogenic mutation of DSH rather than a common polymorphism.
In conclusion, we report a novel mutation of the ADAR1 gene with a heavy clinical phenotype in DSH. This study expands the spectrum of clinical manifestations and demonstrates the ADAR1 mutation in DSH.
Acknowledgments
We are most grateful to the patient and her family for taking part in our study.
- Zhang XJ, Gao M, Li M, et al. Identification of a locus for dyschromatosis symmetrica hereditaria at chromosome 1q11-1q21. J Invest Dermatol. 2003;120:776-780.
- Miyamura Y, Suzuki T, Kono M, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria [published online August 11, 2003]. Am J Hum Genet. 2003;73:693-699.
- Li M, Li C, Hua H, et al. Identification of two novel mutations in Chinese patients with dyschromatosis symmetrica hereditaria [published online October 8, 2005]. Arch Dermatol Res. 2005;297:196-200.
- Oyama M, Shimizu H, Ohata Y, et al. Dyschromatosis symmetrica hereditaria (reticulate acropigmentation of Dohi): report of a Japanese family with the condition and a literature review of 185 cases. Br J Dermatol. 1999;140:491-496.
- Suzuki N, Suzuki T, Inagaki K, et al. Ten novel mutations of the ADAR1 gene in Japanese patients with dyschromatosis symmetrica hereditaria [published online August 17, 2006]. J Invest Dermatol. 2007;127:309-311.
- Zhang XJ, He PP, Li M, et al. Seven novel mutations of the ADAR gene in Chinese families and sporadic patients with dyschromatosis symmetrica hereditaria (DSH). Hum Mutat. 2004;23:629-630.
To the Editor:
Dyschromatosis symmetrica hereditaria (DSH)(Online Mendelian Inheritance in Man 127400), also called reticulate acropigmentation of Dohi, is a pigmentary genodermatosis characterized by a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Linkage analysis has revealed that the DSH gene locus resides on chromosome 1q11-q21,1 and the adenosine deaminase RNA specific gene, ADAR1 (also called DSRAD), in this region has been identified as being responsible for the development of DSH.2 We report a sporadic case of severe DSH with the ADAR1 gene detected in a mutation analysis.
A 6-year-old girl presented with a mixture of hyperpigmented and hypopigmented macules on the dorsal aspects of the hands and feet and the curved side of the wrists, heels, and knees, as well as scattered frecklelike and depigmented spots on the face, ears, neck, arms, and upper back (Figure 1). Her parents noted that hyperpigmented and hypopigmented macules on the dorsal aspects of the hands developed at 5 months of age. Exacerbation after exposure to sunlight resulted in the eruption becoming remarkable in summer and fainter in winter. The skin lesions gradually became more progressive. Physical examination revealed that the patient generally was healthy.
After obtaining informed consent, we performed a mutation analysis of the ADAR1 gene in our patient and her parents. We used a kit to extract genomic DNA from peripheral blood, which was then used to amplify the exons of the ADAR1 gene with intronic flanking sequences by polymerase chain reaction with the primer.3 After amplification, polymerase chain reaction products were purified. We sequenced the ADAR1 gene. Sequence comparisons and analysis found that the patient (proband) carried a heterozygous insertional mutation c.2253insG in exon 6 of the ADAR1 gene. This mutation was not detected in the proband’s healthy parents and 100 normal individuals (Figure 2).
Dyschromatosis symmetrica hereditaria is acquired by autosomal-dominant inheritance and is mainly reported in Asians, especially in Japan and China. Oyama et al4 reviewed 185 cases of DSH in Japan and found the onset of this disease usually was during infancy or childhood; 73% of patients developed the skin lesions before 6 years of age. Suzuki et al5 reported 10 unrelated Japanese patients and found the onset of disease ranged from 1 year of age to childhood. Zhang et al1,6 investigated 78 Chinese patients with DSH including 8 multigenerational families and 2 sporadic patients and found the age of disease onset ranged from 6 months to 15 years of age. The age of onset in our patient (5 months) was younger than these prior reports.
Patients with DSH have a characteristic appearance including a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Few patients have similar lesions on the knees and elbows. Many patients have frecklelike macules on the face and arms.1-6 One patient has been described with scattered depigmented spots on the face and chest.1 Our patient had a characteristic appearance as well as some special manifestations including skin lesions on the curved side of the wrist, ears, neck, and upper back.
The human ADAR1 gene spans 30 kilobase and contains 15 exons. It encodes RNA-specific adenosine deaminase composed of 1226 amino acid residues. This enzyme is important for various functions such as site-specific RNA editing and nuclear translation. This enzyme has 2 Z-alpha domains, 3 double-stranded RNA–binding domains, and the putative deaminase domain corresponding to exon 2, exons 2 to 7, and exons 9 to 14 of ADAR1, respectively.6
Mutation analysis of the ADAR1 gene in this case showed heterozygous insertion mutation c.2253insG in exon 6 of the ADAR1 gene, which changed the reading frame, and 475 amino acid residues in C-terminus are replaced by 90 amino acid residues (TSSRAQVRLPSKSWGSLVPSRLRTQQEA RQAGSSRCGSPCLDWGEREGRTHGFHRG NPSDRGQSQKNYAPPLKVPRSTAKT DTPSHWQHLP). This mutation was not detected in the proband’s healthy parents and the 100 control individuals, which indicated that it was a de novo mutation and the pathogenic mutation of DSH rather than a common polymorphism.
In conclusion, we report a novel mutation of the ADAR1 gene with a heavy clinical phenotype in DSH. This study expands the spectrum of clinical manifestations and demonstrates the ADAR1 mutation in DSH.
Acknowledgments
We are most grateful to the patient and her family for taking part in our study.
To the Editor:
Dyschromatosis symmetrica hereditaria (DSH)(Online Mendelian Inheritance in Man 127400), also called reticulate acropigmentation of Dohi, is a pigmentary genodermatosis characterized by a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Linkage analysis has revealed that the DSH gene locus resides on chromosome 1q11-q21,1 and the adenosine deaminase RNA specific gene, ADAR1 (also called DSRAD), in this region has been identified as being responsible for the development of DSH.2 We report a sporadic case of severe DSH with the ADAR1 gene detected in a mutation analysis.
A 6-year-old girl presented with a mixture of hyperpigmented and hypopigmented macules on the dorsal aspects of the hands and feet and the curved side of the wrists, heels, and knees, as well as scattered frecklelike and depigmented spots on the face, ears, neck, arms, and upper back (Figure 1). Her parents noted that hyperpigmented and hypopigmented macules on the dorsal aspects of the hands developed at 5 months of age. Exacerbation after exposure to sunlight resulted in the eruption becoming remarkable in summer and fainter in winter. The skin lesions gradually became more progressive. Physical examination revealed that the patient generally was healthy.
After obtaining informed consent, we performed a mutation analysis of the ADAR1 gene in our patient and her parents. We used a kit to extract genomic DNA from peripheral blood, which was then used to amplify the exons of the ADAR1 gene with intronic flanking sequences by polymerase chain reaction with the primer.3 After amplification, polymerase chain reaction products were purified. We sequenced the ADAR1 gene. Sequence comparisons and analysis found that the patient (proband) carried a heterozygous insertional mutation c.2253insG in exon 6 of the ADAR1 gene. This mutation was not detected in the proband’s healthy parents and 100 normal individuals (Figure 2).
Dyschromatosis symmetrica hereditaria is acquired by autosomal-dominant inheritance and is mainly reported in Asians, especially in Japan and China. Oyama et al4 reviewed 185 cases of DSH in Japan and found the onset of this disease usually was during infancy or childhood; 73% of patients developed the skin lesions before 6 years of age. Suzuki et al5 reported 10 unrelated Japanese patients and found the onset of disease ranged from 1 year of age to childhood. Zhang et al1,6 investigated 78 Chinese patients with DSH including 8 multigenerational families and 2 sporadic patients and found the age of disease onset ranged from 6 months to 15 years of age. The age of onset in our patient (5 months) was younger than these prior reports.
Patients with DSH have a characteristic appearance including a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet. Few patients have similar lesions on the knees and elbows. Many patients have frecklelike macules on the face and arms.1-6 One patient has been described with scattered depigmented spots on the face and chest.1 Our patient had a characteristic appearance as well as some special manifestations including skin lesions on the curved side of the wrist, ears, neck, and upper back.
The human ADAR1 gene spans 30 kilobase and contains 15 exons. It encodes RNA-specific adenosine deaminase composed of 1226 amino acid residues. This enzyme is important for various functions such as site-specific RNA editing and nuclear translation. This enzyme has 2 Z-alpha domains, 3 double-stranded RNA–binding domains, and the putative deaminase domain corresponding to exon 2, exons 2 to 7, and exons 9 to 14 of ADAR1, respectively.6
Mutation analysis of the ADAR1 gene in this case showed heterozygous insertion mutation c.2253insG in exon 6 of the ADAR1 gene, which changed the reading frame, and 475 amino acid residues in C-terminus are replaced by 90 amino acid residues (TSSRAQVRLPSKSWGSLVPSRLRTQQEA RQAGSSRCGSPCLDWGEREGRTHGFHRG NPSDRGQSQKNYAPPLKVPRSTAKT DTPSHWQHLP). This mutation was not detected in the proband’s healthy parents and the 100 control individuals, which indicated that it was a de novo mutation and the pathogenic mutation of DSH rather than a common polymorphism.
In conclusion, we report a novel mutation of the ADAR1 gene with a heavy clinical phenotype in DSH. This study expands the spectrum of clinical manifestations and demonstrates the ADAR1 mutation in DSH.
Acknowledgments
We are most grateful to the patient and her family for taking part in our study.
- Zhang XJ, Gao M, Li M, et al. Identification of a locus for dyschromatosis symmetrica hereditaria at chromosome 1q11-1q21. J Invest Dermatol. 2003;120:776-780.
- Miyamura Y, Suzuki T, Kono M, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria [published online August 11, 2003]. Am J Hum Genet. 2003;73:693-699.
- Li M, Li C, Hua H, et al. Identification of two novel mutations in Chinese patients with dyschromatosis symmetrica hereditaria [published online October 8, 2005]. Arch Dermatol Res. 2005;297:196-200.
- Oyama M, Shimizu H, Ohata Y, et al. Dyschromatosis symmetrica hereditaria (reticulate acropigmentation of Dohi): report of a Japanese family with the condition and a literature review of 185 cases. Br J Dermatol. 1999;140:491-496.
- Suzuki N, Suzuki T, Inagaki K, et al. Ten novel mutations of the ADAR1 gene in Japanese patients with dyschromatosis symmetrica hereditaria [published online August 17, 2006]. J Invest Dermatol. 2007;127:309-311.
- Zhang XJ, He PP, Li M, et al. Seven novel mutations of the ADAR gene in Chinese families and sporadic patients with dyschromatosis symmetrica hereditaria (DSH). Hum Mutat. 2004;23:629-630.
- Zhang XJ, Gao M, Li M, et al. Identification of a locus for dyschromatosis symmetrica hereditaria at chromosome 1q11-1q21. J Invest Dermatol. 2003;120:776-780.
- Miyamura Y, Suzuki T, Kono M, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria [published online August 11, 2003]. Am J Hum Genet. 2003;73:693-699.
- Li M, Li C, Hua H, et al. Identification of two novel mutations in Chinese patients with dyschromatosis symmetrica hereditaria [published online October 8, 2005]. Arch Dermatol Res. 2005;297:196-200.
- Oyama M, Shimizu H, Ohata Y, et al. Dyschromatosis symmetrica hereditaria (reticulate acropigmentation of Dohi): report of a Japanese family with the condition and a literature review of 185 cases. Br J Dermatol. 1999;140:491-496.
- Suzuki N, Suzuki T, Inagaki K, et al. Ten novel mutations of the ADAR1 gene in Japanese patients with dyschromatosis symmetrica hereditaria [published online August 17, 2006]. J Invest Dermatol. 2007;127:309-311.
- Zhang XJ, He PP, Li M, et al. Seven novel mutations of the ADAR gene in Chinese families and sporadic patients with dyschromatosis symmetrica hereditaria (DSH). Hum Mutat. 2004;23:629-630.
Practice Points
- The adenosine deaminase RNA specific gene, ADAR1, has been identified as being responsible for the development of dyschromatosis symmetrica hereditaria (DSH).
- The characteristic appearance of DSH is a mixture of hyperpigmented and hypopigmented macules of various sizes on the dorsal aspects of the hands and feet.
Desmoplastic Hairless Hypopigmented Nevus
To the Editor:
We report 2 cases of desmoplastic hairless hypopigmented nevi (DHHN), which are giant congenital melanocytic nevi (GCMN) that show sclerosis with progressive loss of pigment and hair. These changes in GCMN could be considered signs of regression.
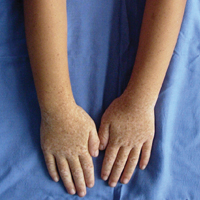
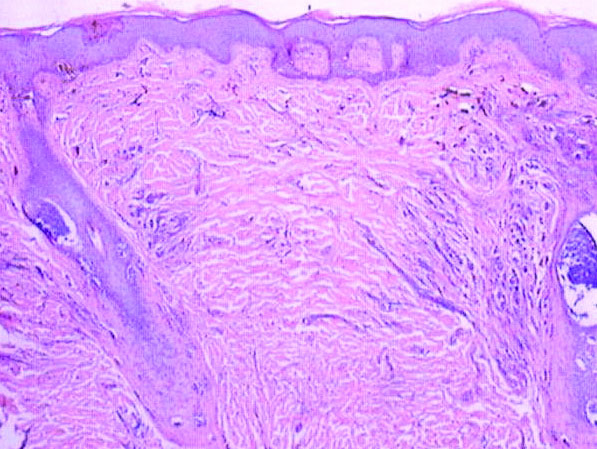
A 6-year-old boy presented in the dermatology department with an asymptomatic skin lesion on the right buttock since birth. The parents claimed that the lesion was darkly pigmented at birth and gradually increased in size, with progressive reduction in color in the last 2 years. Physical examination revealed a 10×6-cm, well-defined, raised plaque on the upper medial side of the right buttock (Figure 1). The plaque was firm with a shiny smooth surface and was devoid of hair. The surface was flesh colored with scattered pigmented spots. A punch biopsy of the lesion showed increased melanin content in the basal cell layer. The upper dermis showed small nests of epithelioid nevus cells, most of them containing melanin pigment (Figure 2). In the lower two-thirds of the dermis, nevus cells were both epithelioid and spindle shaped and were arranged in between thick sclerotic collagen bundles with an increased number of fibroblasts. There was a marked reduction in the number of hair follicles. Immunohistochemical staining results were S-100 positive and CD34 negative.
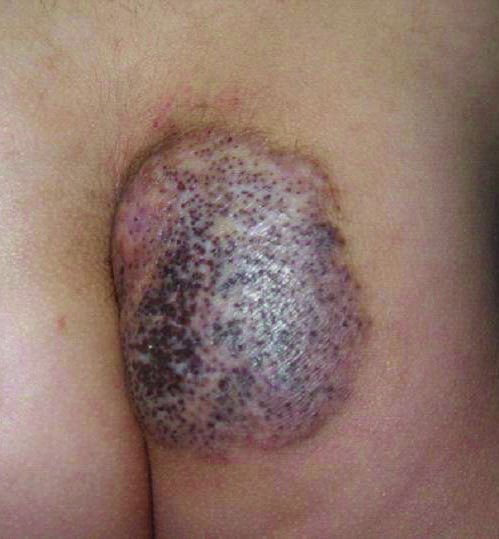
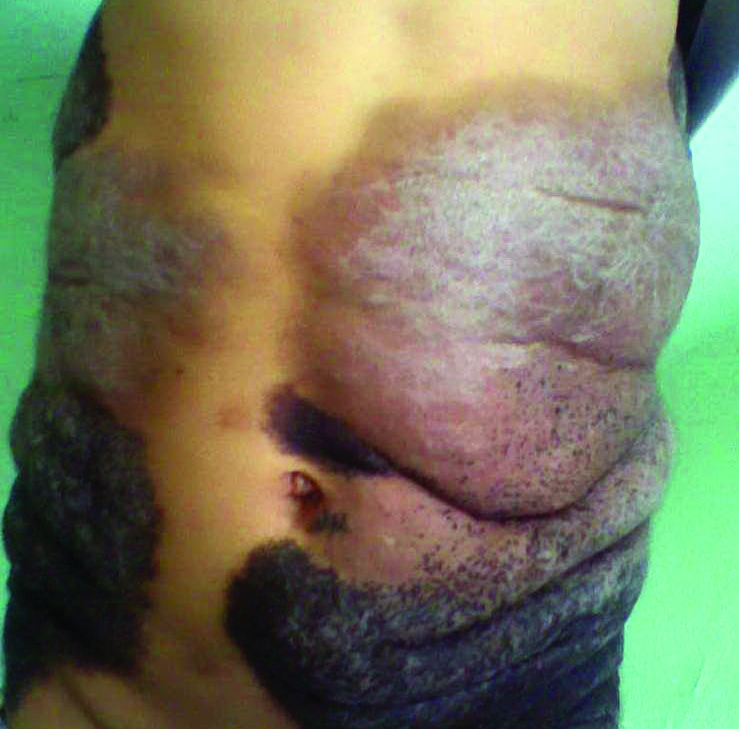
A 5-year-old boy presented in the dermatology department with a large hairy GCMN covering most of the trunk since birth. In the last 1.5 years the parents noted gradual fading of color, decreased hair density, and increased induration of the nevus. Physical examination revealed a large plaque covering the anterior aspect of the trunk (Figure 3) and the back extending down to the buttocks. The lesion formed large skin folds that were more pronounced on the back. The nevus was darkly pigmented with large areas of lighter color that were indurated, devoid of hair, and showed small spots of dark pigmentation. A punch biopsy from the lesion showed small nests of nevus cells in the upper part of the reticular dermis. In the lower part of the dermis, nevus cells were arranged in single units in between thick collagen bundles.
In 2003, Ruiz-Maldonado et al1 described 4 cases of GCMN that showed progressive loss of pigmentation, sclerosis, and hair loss. They proposed the term desmoplastic hairless hypopigmented nevus for their cases and considered it as a variant of GCMN.1 Prior to these reported cases, 2 similar cases were described. The first was a report by Hogan et al2 in 1988 of a 7-month-old girl with a GCMN involving the occipital area and the upper back that became indurated and ulcerated with progressive involution that led to complete disappearance of the nevus. The second was a report by Pattee et al3 in 2001 of a newborn with a GCMN located on the trunk with progressive sclerodermiform reaction. After surgical excision of the nevus, the sclerotic margin disappeared.3
Following the report by Ruiz-Maldonaldo et al,1 5 more cases of DHHN were described.4-8 All cases of DHHN share the same clinical and histopathological features. The clinical features include a GCMN present since birth with progressive sclerosis over time and loss of both pigmentation and hair. Histologically, DHHN shows the typical changes of a congenital melanocytic nevus with decreased numbers of nevus cells, thick sclerotic collagen bundles of the reticular dermis, increased number of fibroblasts, and decreased number of hair follicles. The progressive reduction in the number of nevus cells in melanocytic nevi is considered a sign of regression. Spontaneous regression was rarely described in GCMN, and all the reported cases of regression were associated with desmoplasia.4 Desmoplasia is thought to be induced by either melanocytes that function as adaptive fibroblasts or by fibroblasts themselves, as fibroblasts can show multifunctional differentiation capabilities.9 The direct correlation between the increased induration of DHHN and pigment depletion supports the former hypothesis. The absence of inflammatory cells within the sections of DHHN lesions is against the possibility of an immune-mediated reaction as a cause for the clinical and histological changes seen in this rare form of GCMN. The progressive hair loss in DHHN may be explained by the progressive fibrotic changes in the reticular dermis that affect the blood supply to follicles, leading to atrophy or even absence of the follicles. The progressive reduction in the number of nevus cells in DHHN reduces the potential for malignant transformation and hence following a watchful waiting strategy is a reasonable way to manage these nevi.
We present 2 patients with DHHN, which is a rare form of GCMN that shows signs of regression. The cause of these changes is still unclear.
- Ruiz-Maldonado R, Orozco-Covarrubias L, Ridaura-Sanz C, et al. Desmoplastic hairless hypopigmented naevus: a variant of giant congenital melanocytic naevus. Br J Dermatol. 2003;148:1253-1257.
- Hogan DJ, Murphy F, Bremner RM. Spontaneous resolution of a giant congenital melanocytic nevus. Pediatr Dermatol. 1988;5:170-172.
- Pattee SF, Hansen RC, Bangert JL, et al. Giant congenital nevus with progressive sclerodermoid reaction in a newborn. Pediatr Dermatol. 2001;18:321-324.
- Boente MC, Asial RA. Desmoplastic hairless hypopigmented nevus (DHHN). a distinct variant of giant melanocytic nevus. Eur J Dermatol. 2005;15:451-453.
- Bushby SA, Rajan NJ, Shehade SA. Spontaneous resolution of a giant melanocytic naevus involving a desmoplastic process. Br J Dermatol. 2005;153(suppl 1):13-19.
- Martin JM, Jorda E, Monteagudo C, et al. Desmoplastic giant congenital nevus with progressive depigmentation. J Am Acad Dermatol. 2007;56(suppl 2):S10-S14.
- Hermandez-Martin A, Torrelo A, Echevarria C, et al. Ulcerated sclerotic giant congenital melanocytic naevus: case report and review of the literature. Clin Exp Dermatol. 2007;32:529-532.
- Werner B, Carvalho VO, Nacif SB, et al. Desmoplastic hypopigmented hairless nevus: a variant with progressive depigmentation, induration and overgrowth [published online May 16, 2011]. Pediatr Dermatol. 2012;29:336-340.
- Fearns C, Dowdle EB. The desmoplastic response: induction of collagen synthesis by melanoma cells in vitro. Int J Cancer. 1992;50:621-627.
To the Editor:
We report 2 cases of desmoplastic hairless hypopigmented nevi (DHHN), which are giant congenital melanocytic nevi (GCMN) that show sclerosis with progressive loss of pigment and hair. These changes in GCMN could be considered signs of regression.
A 6-year-old boy presented in the dermatology department with an asymptomatic skin lesion on the right buttock since birth. The parents claimed that the lesion was darkly pigmented at birth and gradually increased in size, with progressive reduction in color in the last 2 years. Physical examination revealed a 10×6-cm, well-defined, raised plaque on the upper medial side of the right buttock (Figure 1). The plaque was firm with a shiny smooth surface and was devoid of hair. The surface was flesh colored with scattered pigmented spots. A punch biopsy of the lesion showed increased melanin content in the basal cell layer. The upper dermis showed small nests of epithelioid nevus cells, most of them containing melanin pigment (Figure 2). In the lower two-thirds of the dermis, nevus cells were both epithelioid and spindle shaped and were arranged in between thick sclerotic collagen bundles with an increased number of fibroblasts. There was a marked reduction in the number of hair follicles. Immunohistochemical staining results were S-100 positive and CD34 negative.
A 5-year-old boy presented in the dermatology department with a large hairy GCMN covering most of the trunk since birth. In the last 1.5 years the parents noted gradual fading of color, decreased hair density, and increased induration of the nevus. Physical examination revealed a large plaque covering the anterior aspect of the trunk (Figure 3) and the back extending down to the buttocks. The lesion formed large skin folds that were more pronounced on the back. The nevus was darkly pigmented with large areas of lighter color that were indurated, devoid of hair, and showed small spots of dark pigmentation. A punch biopsy from the lesion showed small nests of nevus cells in the upper part of the reticular dermis. In the lower part of the dermis, nevus cells were arranged in single units in between thick collagen bundles.
In 2003, Ruiz-Maldonado et al1 described 4 cases of GCMN that showed progressive loss of pigmentation, sclerosis, and hair loss. They proposed the term desmoplastic hairless hypopigmented nevus for their cases and considered it as a variant of GCMN.1 Prior to these reported cases, 2 similar cases were described. The first was a report by Hogan et al2 in 1988 of a 7-month-old girl with a GCMN involving the occipital area and the upper back that became indurated and ulcerated with progressive involution that led to complete disappearance of the nevus. The second was a report by Pattee et al3 in 2001 of a newborn with a GCMN located on the trunk with progressive sclerodermiform reaction. After surgical excision of the nevus, the sclerotic margin disappeared.3
Following the report by Ruiz-Maldonaldo et al,1 5 more cases of DHHN were described.4-8 All cases of DHHN share the same clinical and histopathological features. The clinical features include a GCMN present since birth with progressive sclerosis over time and loss of both pigmentation and hair. Histologically, DHHN shows the typical changes of a congenital melanocytic nevus with decreased numbers of nevus cells, thick sclerotic collagen bundles of the reticular dermis, increased number of fibroblasts, and decreased number of hair follicles. The progressive reduction in the number of nevus cells in melanocytic nevi is considered a sign of regression. Spontaneous regression was rarely described in GCMN, and all the reported cases of regression were associated with desmoplasia.4 Desmoplasia is thought to be induced by either melanocytes that function as adaptive fibroblasts or by fibroblasts themselves, as fibroblasts can show multifunctional differentiation capabilities.9 The direct correlation between the increased induration of DHHN and pigment depletion supports the former hypothesis. The absence of inflammatory cells within the sections of DHHN lesions is against the possibility of an immune-mediated reaction as a cause for the clinical and histological changes seen in this rare form of GCMN. The progressive hair loss in DHHN may be explained by the progressive fibrotic changes in the reticular dermis that affect the blood supply to follicles, leading to atrophy or even absence of the follicles. The progressive reduction in the number of nevus cells in DHHN reduces the potential for malignant transformation and hence following a watchful waiting strategy is a reasonable way to manage these nevi.
We present 2 patients with DHHN, which is a rare form of GCMN that shows signs of regression. The cause of these changes is still unclear.
To the Editor:
We report 2 cases of desmoplastic hairless hypopigmented nevi (DHHN), which are giant congenital melanocytic nevi (GCMN) that show sclerosis with progressive loss of pigment and hair. These changes in GCMN could be considered signs of regression.
A 6-year-old boy presented in the dermatology department with an asymptomatic skin lesion on the right buttock since birth. The parents claimed that the lesion was darkly pigmented at birth and gradually increased in size, with progressive reduction in color in the last 2 years. Physical examination revealed a 10×6-cm, well-defined, raised plaque on the upper medial side of the right buttock (Figure 1). The plaque was firm with a shiny smooth surface and was devoid of hair. The surface was flesh colored with scattered pigmented spots. A punch biopsy of the lesion showed increased melanin content in the basal cell layer. The upper dermis showed small nests of epithelioid nevus cells, most of them containing melanin pigment (Figure 2). In the lower two-thirds of the dermis, nevus cells were both epithelioid and spindle shaped and were arranged in between thick sclerotic collagen bundles with an increased number of fibroblasts. There was a marked reduction in the number of hair follicles. Immunohistochemical staining results were S-100 positive and CD34 negative.
A 5-year-old boy presented in the dermatology department with a large hairy GCMN covering most of the trunk since birth. In the last 1.5 years the parents noted gradual fading of color, decreased hair density, and increased induration of the nevus. Physical examination revealed a large plaque covering the anterior aspect of the trunk (Figure 3) and the back extending down to the buttocks. The lesion formed large skin folds that were more pronounced on the back. The nevus was darkly pigmented with large areas of lighter color that were indurated, devoid of hair, and showed small spots of dark pigmentation. A punch biopsy from the lesion showed small nests of nevus cells in the upper part of the reticular dermis. In the lower part of the dermis, nevus cells were arranged in single units in between thick collagen bundles.
In 2003, Ruiz-Maldonado et al1 described 4 cases of GCMN that showed progressive loss of pigmentation, sclerosis, and hair loss. They proposed the term desmoplastic hairless hypopigmented nevus for their cases and considered it as a variant of GCMN.1 Prior to these reported cases, 2 similar cases were described. The first was a report by Hogan et al2 in 1988 of a 7-month-old girl with a GCMN involving the occipital area and the upper back that became indurated and ulcerated with progressive involution that led to complete disappearance of the nevus. The second was a report by Pattee et al3 in 2001 of a newborn with a GCMN located on the trunk with progressive sclerodermiform reaction. After surgical excision of the nevus, the sclerotic margin disappeared.3
Following the report by Ruiz-Maldonaldo et al,1 5 more cases of DHHN were described.4-8 All cases of DHHN share the same clinical and histopathological features. The clinical features include a GCMN present since birth with progressive sclerosis over time and loss of both pigmentation and hair. Histologically, DHHN shows the typical changes of a congenital melanocytic nevus with decreased numbers of nevus cells, thick sclerotic collagen bundles of the reticular dermis, increased number of fibroblasts, and decreased number of hair follicles. The progressive reduction in the number of nevus cells in melanocytic nevi is considered a sign of regression. Spontaneous regression was rarely described in GCMN, and all the reported cases of regression were associated with desmoplasia.4 Desmoplasia is thought to be induced by either melanocytes that function as adaptive fibroblasts or by fibroblasts themselves, as fibroblasts can show multifunctional differentiation capabilities.9 The direct correlation between the increased induration of DHHN and pigment depletion supports the former hypothesis. The absence of inflammatory cells within the sections of DHHN lesions is against the possibility of an immune-mediated reaction as a cause for the clinical and histological changes seen in this rare form of GCMN. The progressive hair loss in DHHN may be explained by the progressive fibrotic changes in the reticular dermis that affect the blood supply to follicles, leading to atrophy or even absence of the follicles. The progressive reduction in the number of nevus cells in DHHN reduces the potential for malignant transformation and hence following a watchful waiting strategy is a reasonable way to manage these nevi.
We present 2 patients with DHHN, which is a rare form of GCMN that shows signs of regression. The cause of these changes is still unclear.
- Ruiz-Maldonado R, Orozco-Covarrubias L, Ridaura-Sanz C, et al. Desmoplastic hairless hypopigmented naevus: a variant of giant congenital melanocytic naevus. Br J Dermatol. 2003;148:1253-1257.
- Hogan DJ, Murphy F, Bremner RM. Spontaneous resolution of a giant congenital melanocytic nevus. Pediatr Dermatol. 1988;5:170-172.
- Pattee SF, Hansen RC, Bangert JL, et al. Giant congenital nevus with progressive sclerodermoid reaction in a newborn. Pediatr Dermatol. 2001;18:321-324.
- Boente MC, Asial RA. Desmoplastic hairless hypopigmented nevus (DHHN). a distinct variant of giant melanocytic nevus. Eur J Dermatol. 2005;15:451-453.
- Bushby SA, Rajan NJ, Shehade SA. Spontaneous resolution of a giant melanocytic naevus involving a desmoplastic process. Br J Dermatol. 2005;153(suppl 1):13-19.
- Martin JM, Jorda E, Monteagudo C, et al. Desmoplastic giant congenital nevus with progressive depigmentation. J Am Acad Dermatol. 2007;56(suppl 2):S10-S14.
- Hermandez-Martin A, Torrelo A, Echevarria C, et al. Ulcerated sclerotic giant congenital melanocytic naevus: case report and review of the literature. Clin Exp Dermatol. 2007;32:529-532.
- Werner B, Carvalho VO, Nacif SB, et al. Desmoplastic hypopigmented hairless nevus: a variant with progressive depigmentation, induration and overgrowth [published online May 16, 2011]. Pediatr Dermatol. 2012;29:336-340.
- Fearns C, Dowdle EB. The desmoplastic response: induction of collagen synthesis by melanoma cells in vitro. Int J Cancer. 1992;50:621-627.
- Ruiz-Maldonado R, Orozco-Covarrubias L, Ridaura-Sanz C, et al. Desmoplastic hairless hypopigmented naevus: a variant of giant congenital melanocytic naevus. Br J Dermatol. 2003;148:1253-1257.
- Hogan DJ, Murphy F, Bremner RM. Spontaneous resolution of a giant congenital melanocytic nevus. Pediatr Dermatol. 1988;5:170-172.
- Pattee SF, Hansen RC, Bangert JL, et al. Giant congenital nevus with progressive sclerodermoid reaction in a newborn. Pediatr Dermatol. 2001;18:321-324.
- Boente MC, Asial RA. Desmoplastic hairless hypopigmented nevus (DHHN). a distinct variant of giant melanocytic nevus. Eur J Dermatol. 2005;15:451-453.
- Bushby SA, Rajan NJ, Shehade SA. Spontaneous resolution of a giant melanocytic naevus involving a desmoplastic process. Br J Dermatol. 2005;153(suppl 1):13-19.
- Martin JM, Jorda E, Monteagudo C, et al. Desmoplastic giant congenital nevus with progressive depigmentation. J Am Acad Dermatol. 2007;56(suppl 2):S10-S14.
- Hermandez-Martin A, Torrelo A, Echevarria C, et al. Ulcerated sclerotic giant congenital melanocytic naevus: case report and review of the literature. Clin Exp Dermatol. 2007;32:529-532.
- Werner B, Carvalho VO, Nacif SB, et al. Desmoplastic hypopigmented hairless nevus: a variant with progressive depigmentation, induration and overgrowth [published online May 16, 2011]. Pediatr Dermatol. 2012;29:336-340.
- Fearns C, Dowdle EB. The desmoplastic response: induction of collagen synthesis by melanoma cells in vitro. Int J Cancer. 1992;50:621-627.
Nevus Spilus: Is the Presence of Hair Associated With an Increased Risk for Melanoma?
The term nevus spilus (NS), also known as speckled lentiginous nevus, was first used in the 19th century to describe lesions with background café au lait–like lentiginous melanocytic hyperplasia speckled with small, 1- to 3-mm, darker foci. The dark spots reflect lentigines; junctional, compound, and intradermal nevus cell nests; and more rarely Spitz and blue nevi. Both macular and papular subtypes have been described.1 This birthmark is quite common, occurring in 1.3% to 2.3% of the adult population worldwide.2 Hypertrichosis has been described in NS.3-9 Two subsequent cases of malignant melanoma in hairy NS suggested that lesions may be particularly prone to malignant degeneration.4,8 We report an additional case of hairy NS that was not associated with melanoma and consider whether dermatologists should warn their patients about this association.
Case Report
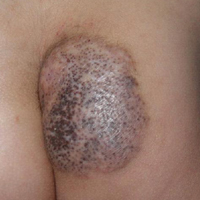
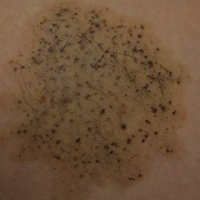
A 26-year-old woman presented with a stable 7×8-cm, tan-brown, macular, pigmented birthmark studded with darker 1- to 2-mm, irregular, brown-black and blue, confettilike macules on the left proximal lateral thigh that had been present since birth (Figure 1). Dark terminal hairs were present, arising from both the darker and lighter pigmented areas but not the surrounding normal skin.
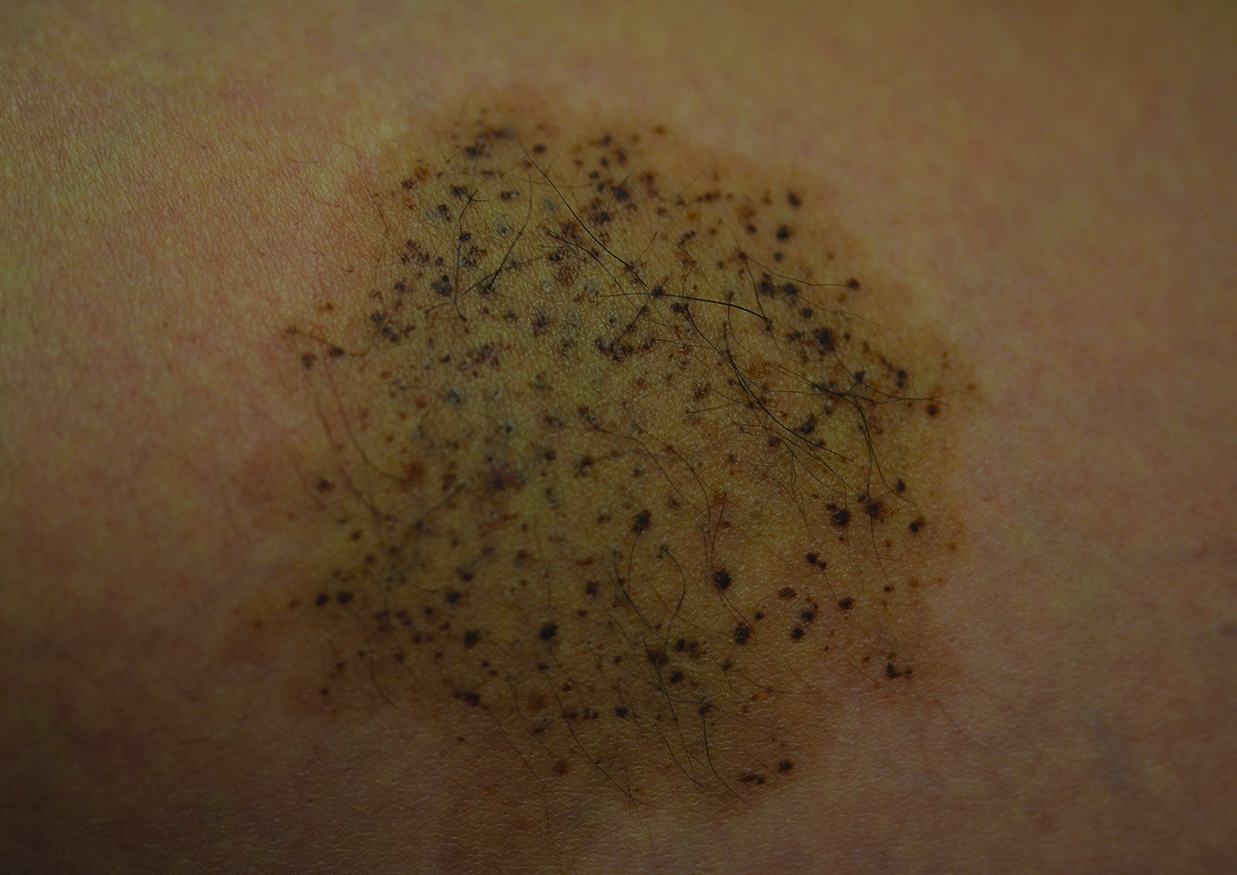
A 4-mm punch biopsy from one of the dark blue macules demonstrated uniform lentiginous melanocytic hyperplasia and nevus cell nests adjacent to the sweat glands extending into the mid dermis (Figure 2). No clinical evidence of malignant degeneration was present.
Comment
The risk for melanoma is increased in classic nonspeckled congenital nevi and the risk correlates with the size of the lesion and most probably the number of nevus cells in the lesion that increase the risk for a random mutation.8,10,11 It is likely that NS with or without hair presages a small increased risk for melanoma,6,9,12 which is not surprising because NS is a subtype of congenital melanocytic nevus (CMN), a condition that is present at birth and results from a proliferation of melanocytes.6 Nevus spilus, however, appears to have a notably lower risk for malignant degeneration than other classic CMN of the same size. The following support for this hypothesis is offered: First, CMN have nevus cells broadly filling the dermis that extend more deeply into the dermis than NS (Figure 2A).10 In our estimation, CMN have at least 100 times the number of nevus cells per square centimeter compared to NS. The potential for malignant degeneration of any one melanocyte is greater when more are present. Second, although some NS lesions evolve, classic CMN are universally more proliferative than NS.10,13 The involved skin in CMN thickens over time with increased numbers of melanocytes and marked overgrowth of adjacent tissue. Melanocytes in a proliferative phase may be more likely to undergo malignant degeneration.10
A PubMed search of articles indexed for MEDLINE using the search term nevus spilus and melanoma yielded 2 cases4,8 of melanoma arising among 15 cases of hairy NS in the literature, which led to the suggestion that the presence of hair could be associated with an increased risk for malignant degeneration in NS (Table). This apparent high incidence of melanoma most likely reflects referral/publication bias rather than a statistically significant association. In fact, the clinical lesion most clinically similar to hairy NS is Becker nevus, with tan macules demonstrating lentiginous melanocytic hyperplasia associated with numerous coarse terminal hairs. There is no indication that Becker nevi have a considerable premalignant potential, though one case of melanoma arising in a Becker nevus has been reported.9 There is no evidence to suggest that classic CMN with hypertrichosis has a greater premalignant potential than similar lesions without hypertrichosis.

We noticed the presence of hair in our patient’s lesion only after reports in the literature caused us to look for this phenomenon.9 This occurrence may actually be quite common. We do not recommend prophylactic excision of NS and believe the risk for malignant degeneration is low in NS with or without hair, though larger NS (>4 cm), especially giant, zosteriform, or segmental lesions, may have a greater risk.1,6,9,10 It is prudent for physicians to carefully examine NS and sample suspicious foci, especially when patients describe a lesion as changing.
- Vidaurri-de la Cruz H, Happle R. Two distinct types of speckled lentiginous nevi characterized by macular versus papular speckles. Dermatology. 2006;212:53-58.
- Ly L, Christie M, Swain S, et al. Melanoma(s) arising in large segmental speckled lentiginous nevi: a case series. J Am Acad Dermatol. 2011;64:1190-1193.
- Prose NS, Heilman E, Felman YM, et al. Multiple benign juvenile melanoma. J Am Acad Dermatol. 1983;9:236-242.
- Grinspan D, Casala A, Abulafia J, et al. Melanoma on dysplastic nevus spilus. Int J Dermatol. 1997;36:499-502 .
- Langenbach N, Pfau A, Landthaler M, et al. Naevi spili, café-au-lait spots and melanocytic naevi aggregated alongside Blaschko’s lines, with a review of segmental melanocytic lesions. Acta Derm Venereol. 1998;78:378-380.
- Schaffer JV, Orlow SJ, Lazova R, et al. Speckled lentiginous nevus: within the spectrum of congenital melanocytic nevi. Arch Dermatol. 2001;137:172-178.
- Saraswat A, Dogra S, Bansali A, et al. Phakomatosis pigmentokeratotica associated with hypophosphataemic vitamin D–resistant rickets: improvement in phosphate homeostasis after partial laser ablation. Br J Dermatol. 2003;148:1074-1076.
- Zeren-Bilgin
i , Gür S, Aydın O, et al. Melanoma arising in a hairy nevus spilus. Int J Dermatol. 2006;45:1362-1364. - Singh S, Jain N, Khanna N, et al. Hairy nevus spilus: a case series. Pediatr Dermatol. 2013;30:100-104.
- Price HN, Schaffer JV. Congenital melanocytic nevi—when to worry and how to treat: facts and controversies. Clin Dermatol. 2010;28:293-302.
- Alikhan Ali, Ibrahimi OA, Eisen DB. Congenital melanocytic nevi: where are we now? J Am Acad Dermatol. 2012;67:495.e1-495.e17.
- Haenssle HA, Kaune KM, Buhl T, et al. Melanoma arising in segmental nevus spilus: detection by sequential digital dermatoscopy. J Am Acad Dermatol. 2009;61:337-341.
- Cohen LM. Nevus spilus: congenital or acquired? Arch Dermatol. 2001;137:215-216.
The term nevus spilus (NS), also known as speckled lentiginous nevus, was first used in the 19th century to describe lesions with background café au lait–like lentiginous melanocytic hyperplasia speckled with small, 1- to 3-mm, darker foci. The dark spots reflect lentigines; junctional, compound, and intradermal nevus cell nests; and more rarely Spitz and blue nevi. Both macular and papular subtypes have been described.1 This birthmark is quite common, occurring in 1.3% to 2.3% of the adult population worldwide.2 Hypertrichosis has been described in NS.3-9 Two subsequent cases of malignant melanoma in hairy NS suggested that lesions may be particularly prone to malignant degeneration.4,8 We report an additional case of hairy NS that was not associated with melanoma and consider whether dermatologists should warn their patients about this association.
Case Report
A 26-year-old woman presented with a stable 7×8-cm, tan-brown, macular, pigmented birthmark studded with darker 1- to 2-mm, irregular, brown-black and blue, confettilike macules on the left proximal lateral thigh that had been present since birth (Figure 1). Dark terminal hairs were present, arising from both the darker and lighter pigmented areas but not the surrounding normal skin.
A 4-mm punch biopsy from one of the dark blue macules demonstrated uniform lentiginous melanocytic hyperplasia and nevus cell nests adjacent to the sweat glands extending into the mid dermis (Figure 2). No clinical evidence of malignant degeneration was present.
Comment
The risk for melanoma is increased in classic nonspeckled congenital nevi and the risk correlates with the size of the lesion and most probably the number of nevus cells in the lesion that increase the risk for a random mutation.8,10,11 It is likely that NS with or without hair presages a small increased risk for melanoma,6,9,12 which is not surprising because NS is a subtype of congenital melanocytic nevus (CMN), a condition that is present at birth and results from a proliferation of melanocytes.6 Nevus spilus, however, appears to have a notably lower risk for malignant degeneration than other classic CMN of the same size. The following support for this hypothesis is offered: First, CMN have nevus cells broadly filling the dermis that extend more deeply into the dermis than NS (Figure 2A).10 In our estimation, CMN have at least 100 times the number of nevus cells per square centimeter compared to NS. The potential for malignant degeneration of any one melanocyte is greater when more are present. Second, although some NS lesions evolve, classic CMN are universally more proliferative than NS.10,13 The involved skin in CMN thickens over time with increased numbers of melanocytes and marked overgrowth of adjacent tissue. Melanocytes in a proliferative phase may be more likely to undergo malignant degeneration.10
A PubMed search of articles indexed for MEDLINE using the search term nevus spilus and melanoma yielded 2 cases4,8 of melanoma arising among 15 cases of hairy NS in the literature, which led to the suggestion that the presence of hair could be associated with an increased risk for malignant degeneration in NS (Table). This apparent high incidence of melanoma most likely reflects referral/publication bias rather than a statistically significant association. In fact, the clinical lesion most clinically similar to hairy NS is Becker nevus, with tan macules demonstrating lentiginous melanocytic hyperplasia associated with numerous coarse terminal hairs. There is no indication that Becker nevi have a considerable premalignant potential, though one case of melanoma arising in a Becker nevus has been reported.9 There is no evidence to suggest that classic CMN with hypertrichosis has a greater premalignant potential than similar lesions without hypertrichosis.
We noticed the presence of hair in our patient’s lesion only after reports in the literature caused us to look for this phenomenon.9 This occurrence may actually be quite common. We do not recommend prophylactic excision of NS and believe the risk for malignant degeneration is low in NS with or without hair, though larger NS (>4 cm), especially giant, zosteriform, or segmental lesions, may have a greater risk.1,6,9,10 It is prudent for physicians to carefully examine NS and sample suspicious foci, especially when patients describe a lesion as changing.
The term nevus spilus (NS), also known as speckled lentiginous nevus, was first used in the 19th century to describe lesions with background café au lait–like lentiginous melanocytic hyperplasia speckled with small, 1- to 3-mm, darker foci. The dark spots reflect lentigines; junctional, compound, and intradermal nevus cell nests; and more rarely Spitz and blue nevi. Both macular and papular subtypes have been described.1 This birthmark is quite common, occurring in 1.3% to 2.3% of the adult population worldwide.2 Hypertrichosis has been described in NS.3-9 Two subsequent cases of malignant melanoma in hairy NS suggested that lesions may be particularly prone to malignant degeneration.4,8 We report an additional case of hairy NS that was not associated with melanoma and consider whether dermatologists should warn their patients about this association.
Case Report
A 26-year-old woman presented with a stable 7×8-cm, tan-brown, macular, pigmented birthmark studded with darker 1- to 2-mm, irregular, brown-black and blue, confettilike macules on the left proximal lateral thigh that had been present since birth (Figure 1). Dark terminal hairs were present, arising from both the darker and lighter pigmented areas but not the surrounding normal skin.
A 4-mm punch biopsy from one of the dark blue macules demonstrated uniform lentiginous melanocytic hyperplasia and nevus cell nests adjacent to the sweat glands extending into the mid dermis (Figure 2). No clinical evidence of malignant degeneration was present.
Comment
The risk for melanoma is increased in classic nonspeckled congenital nevi and the risk correlates with the size of the lesion and most probably the number of nevus cells in the lesion that increase the risk for a random mutation.8,10,11 It is likely that NS with or without hair presages a small increased risk for melanoma,6,9,12 which is not surprising because NS is a subtype of congenital melanocytic nevus (CMN), a condition that is present at birth and results from a proliferation of melanocytes.6 Nevus spilus, however, appears to have a notably lower risk for malignant degeneration than other classic CMN of the same size. The following support for this hypothesis is offered: First, CMN have nevus cells broadly filling the dermis that extend more deeply into the dermis than NS (Figure 2A).10 In our estimation, CMN have at least 100 times the number of nevus cells per square centimeter compared to NS. The potential for malignant degeneration of any one melanocyte is greater when more are present. Second, although some NS lesions evolve, classic CMN are universally more proliferative than NS.10,13 The involved skin in CMN thickens over time with increased numbers of melanocytes and marked overgrowth of adjacent tissue. Melanocytes in a proliferative phase may be more likely to undergo malignant degeneration.10
A PubMed search of articles indexed for MEDLINE using the search term nevus spilus and melanoma yielded 2 cases4,8 of melanoma arising among 15 cases of hairy NS in the literature, which led to the suggestion that the presence of hair could be associated with an increased risk for malignant degeneration in NS (Table). This apparent high incidence of melanoma most likely reflects referral/publication bias rather than a statistically significant association. In fact, the clinical lesion most clinically similar to hairy NS is Becker nevus, with tan macules demonstrating lentiginous melanocytic hyperplasia associated with numerous coarse terminal hairs. There is no indication that Becker nevi have a considerable premalignant potential, though one case of melanoma arising in a Becker nevus has been reported.9 There is no evidence to suggest that classic CMN with hypertrichosis has a greater premalignant potential than similar lesions without hypertrichosis.
We noticed the presence of hair in our patient’s lesion only after reports in the literature caused us to look for this phenomenon.9 This occurrence may actually be quite common. We do not recommend prophylactic excision of NS and believe the risk for malignant degeneration is low in NS with or without hair, though larger NS (>4 cm), especially giant, zosteriform, or segmental lesions, may have a greater risk.1,6,9,10 It is prudent for physicians to carefully examine NS and sample suspicious foci, especially when patients describe a lesion as changing.
- Vidaurri-de la Cruz H, Happle R. Two distinct types of speckled lentiginous nevi characterized by macular versus papular speckles. Dermatology. 2006;212:53-58.
- Ly L, Christie M, Swain S, et al. Melanoma(s) arising in large segmental speckled lentiginous nevi: a case series. J Am Acad Dermatol. 2011;64:1190-1193.
- Prose NS, Heilman E, Felman YM, et al. Multiple benign juvenile melanoma. J Am Acad Dermatol. 1983;9:236-242.
- Grinspan D, Casala A, Abulafia J, et al. Melanoma on dysplastic nevus spilus. Int J Dermatol. 1997;36:499-502 .
- Langenbach N, Pfau A, Landthaler M, et al. Naevi spili, café-au-lait spots and melanocytic naevi aggregated alongside Blaschko’s lines, with a review of segmental melanocytic lesions. Acta Derm Venereol. 1998;78:378-380.
- Schaffer JV, Orlow SJ, Lazova R, et al. Speckled lentiginous nevus: within the spectrum of congenital melanocytic nevi. Arch Dermatol. 2001;137:172-178.
- Saraswat A, Dogra S, Bansali A, et al. Phakomatosis pigmentokeratotica associated with hypophosphataemic vitamin D–resistant rickets: improvement in phosphate homeostasis after partial laser ablation. Br J Dermatol. 2003;148:1074-1076.
- Zeren-Bilgin
i , Gür S, Aydın O, et al. Melanoma arising in a hairy nevus spilus. Int J Dermatol. 2006;45:1362-1364. - Singh S, Jain N, Khanna N, et al. Hairy nevus spilus: a case series. Pediatr Dermatol. 2013;30:100-104.
- Price HN, Schaffer JV. Congenital melanocytic nevi—when to worry and how to treat: facts and controversies. Clin Dermatol. 2010;28:293-302.
- Alikhan Ali, Ibrahimi OA, Eisen DB. Congenital melanocytic nevi: where are we now? J Am Acad Dermatol. 2012;67:495.e1-495.e17.
- Haenssle HA, Kaune KM, Buhl T, et al. Melanoma arising in segmental nevus spilus: detection by sequential digital dermatoscopy. J Am Acad Dermatol. 2009;61:337-341.
- Cohen LM. Nevus spilus: congenital or acquired? Arch Dermatol. 2001;137:215-216.
- Vidaurri-de la Cruz H, Happle R. Two distinct types of speckled lentiginous nevi characterized by macular versus papular speckles. Dermatology. 2006;212:53-58.
- Ly L, Christie M, Swain S, et al. Melanoma(s) arising in large segmental speckled lentiginous nevi: a case series. J Am Acad Dermatol. 2011;64:1190-1193.
- Prose NS, Heilman E, Felman YM, et al. Multiple benign juvenile melanoma. J Am Acad Dermatol. 1983;9:236-242.
- Grinspan D, Casala A, Abulafia J, et al. Melanoma on dysplastic nevus spilus. Int J Dermatol. 1997;36:499-502 .
- Langenbach N, Pfau A, Landthaler M, et al. Naevi spili, café-au-lait spots and melanocytic naevi aggregated alongside Blaschko’s lines, with a review of segmental melanocytic lesions. Acta Derm Venereol. 1998;78:378-380.
- Schaffer JV, Orlow SJ, Lazova R, et al. Speckled lentiginous nevus: within the spectrum of congenital melanocytic nevi. Arch Dermatol. 2001;137:172-178.
- Saraswat A, Dogra S, Bansali A, et al. Phakomatosis pigmentokeratotica associated with hypophosphataemic vitamin D–resistant rickets: improvement in phosphate homeostasis after partial laser ablation. Br J Dermatol. 2003;148:1074-1076.
- Zeren-Bilgin
i , Gür S, Aydın O, et al. Melanoma arising in a hairy nevus spilus. Int J Dermatol. 2006;45:1362-1364. - Singh S, Jain N, Khanna N, et al. Hairy nevus spilus: a case series. Pediatr Dermatol. 2013;30:100-104.
- Price HN, Schaffer JV. Congenital melanocytic nevi—when to worry and how to treat: facts and controversies. Clin Dermatol. 2010;28:293-302.
- Alikhan Ali, Ibrahimi OA, Eisen DB. Congenital melanocytic nevi: where are we now? J Am Acad Dermatol. 2012;67:495.e1-495.e17.
- Haenssle HA, Kaune KM, Buhl T, et al. Melanoma arising in segmental nevus spilus: detection by sequential digital dermatoscopy. J Am Acad Dermatol. 2009;61:337-341.
- Cohen LM. Nevus spilus: congenital or acquired? Arch Dermatol. 2001;137:215-216.
Practice Points
- Nevus spilus (NS) appears as a café au lait macule studded with darker brown “moles.”
- Although melanoma has been described in NS, it is rare.
- There is no evidence that hairy NS are predisposed to melanoma.
Painful Ulcerations Above the Malleoli
The Diagnosis: Livedoid Vasculopathy
Livedoid vasculopathy (LV) is a rare cutaneous disorder that most commonly affects the lower legs. It has an estimated incidence of 1 case per 100,000 per year and predominantly affects women.1 The disease pathogenesis is not fully understood but is thought to involve thrombosis and occlusion of dermal vessels resulting in tissue hypoxia.2 Both inherited and acquired thrombophilic conditions frequently are seen in patients with LV.3,4 Livedoid vasculopathy also has been described as idiopathic5 and is associated with immune complex deposition.6 However, the number of cases of idiopathic LV may be overestimated; as technological advancements to detect coagulation abnormalities improve, it is hypothesized that this entity will be identified less often.2,4
Livedoid vasculopathy has been described in the literature using the term PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities).7 The triad of livedo racemosa, recurrent painful ulcerations, and residual healing with atrophie blanche characterizes the clinical manifestations of LV; however, all 3 characteristics do not need to appear simultaneously for a diagnosis to be made. The condition has a chronic course with spontaneous remissions and exacerbations. Episodic ulcerations occur, especially in the summertime, and heal slowly, leaving behind atrophic, porcelain white, stellate-shaped scars called atrophie blanche. Livedo racemosa also may be seen in Sneddon syndrome; however, these patients experience neurologic symptoms secondary to cerebrovascular occlusion. In contrast to livedo racemosa, acquired livedo reticularis represents a physiologic hypoperfusion pattern that occurs in response to cold exposure.8 A localized sharp pain, known as angina cutis, typically precedes the clinical symptom of painful ulcerations.9 Atrophie blanche once was thought to be specific to LV but has been seen in other diseases such as systemic lupus erythematosus and chronic venous insufficiency.2
The diagnosis of LV is based on identification of characteristic clinical features and skin biopsy. In almost all biopsy specimens, histopathology reveals fibrinoid occlusion of vessels in the superficial and mid dermis.4 Other findings may include epidermal necrosis and vessel wall hyalinization and infarction2 (Figure). Because LV is commonly misdiagnosed as vasculitis, the absence of hallmark features of vasculitis such as neutrophilic infiltrate of blood vessel walls and fibrinoid necrosis suggest the diagnosis. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities should be performed.
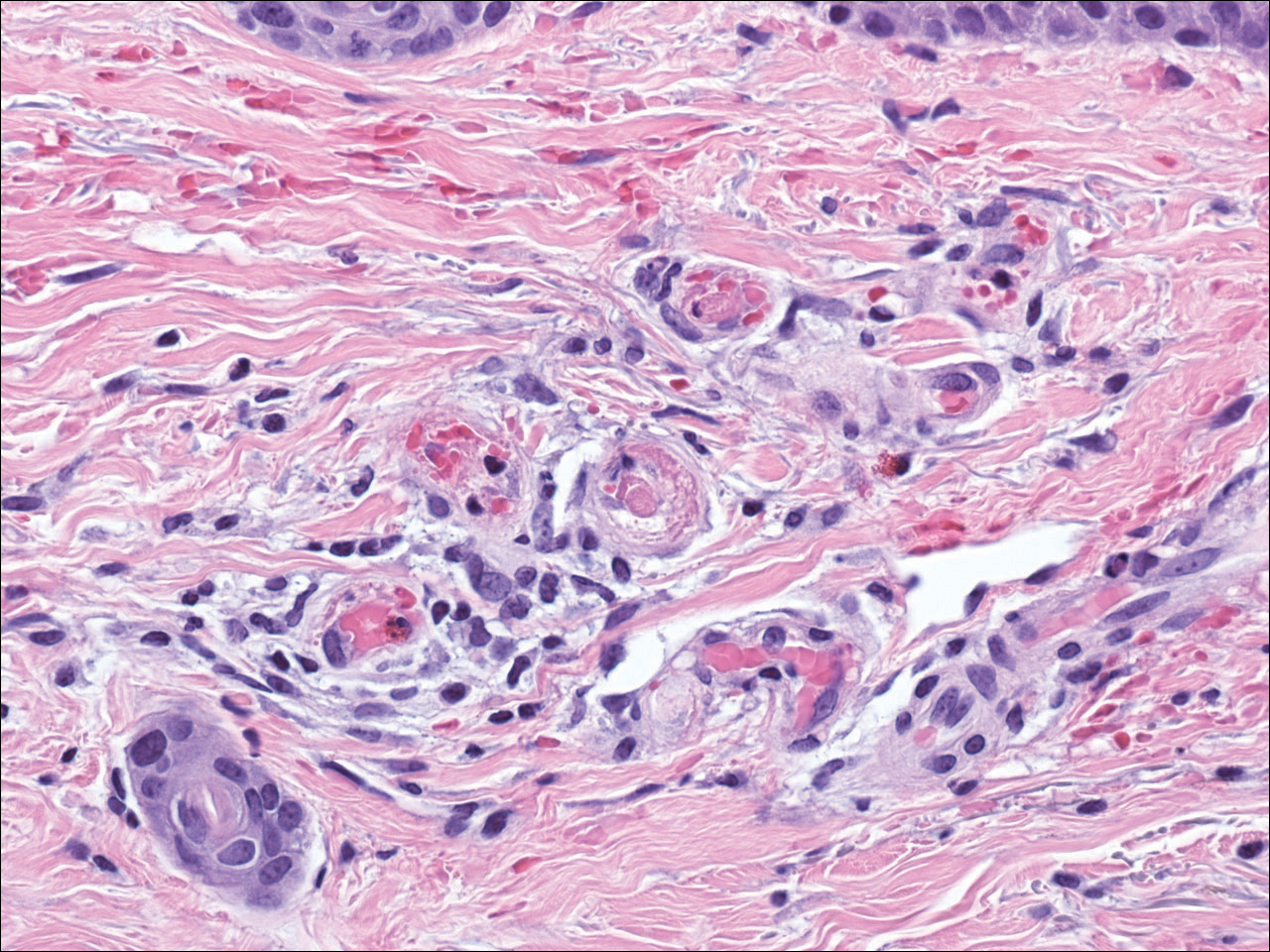
Treatment of LV is difficult, as there is currently no consensus on optimal therapy. The mainstay of therapy is to reduce pain, prevent infection, and reduce ulceration and development of atrophie blanche. Underlying causes should be identified and appropriately treated. Because the primary pathogenesis of LV is considered to be a hypercoagulable state, first-line treatment often includes therapies to enhance blood flow and prevent thrombosis such as smoking cessation, antiplatelet therapy, and pentoxifylline. Vasodilating agents, anti-inflammatory agents, anticoagulation, and fibrinolytic therapy also have been used with varying degrees of success.7
- Fritsch P, Zelger B. Livedo vasculitis [in German]. Hautarzt. 1995;46:215-224; quiz 222-223.
- Kerk N, Goerge T. Livedoid vasculopathy—a thrombotic disease. Vasa. 2013;42:317-322.
- Stevanovic DV. Atrophie blanche. a sign of dermal blood occlusion. Arch Dermatol. 1974;109:858-862.
- Hairston BR, Davis MD, Pittelkow MR, et al. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol. 2006;142:1413-1418.
- Shornick JK, Nicholes BK, Bergstresser PR, et al. Idiopathic atrophie blanche. J Am Acad Dermatol. 1983;8:792-798.
- Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with ulcerations. Circulation. 1956;13:196-216.
- Callen JP. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol. 2006;142:1481-1482.
- Copeman PW. Livedo reticularis. signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975;93:519-529.
- Goerge T. Livedoid vasculopathy. pathogenesis, diagnosis and treatment of cutaneous infarction [in German]. Hautarzt. 2011;62:627-634; quiz 635.
The Diagnosis: Livedoid Vasculopathy
Livedoid vasculopathy (LV) is a rare cutaneous disorder that most commonly affects the lower legs. It has an estimated incidence of 1 case per 100,000 per year and predominantly affects women.1 The disease pathogenesis is not fully understood but is thought to involve thrombosis and occlusion of dermal vessels resulting in tissue hypoxia.2 Both inherited and acquired thrombophilic conditions frequently are seen in patients with LV.3,4 Livedoid vasculopathy also has been described as idiopathic5 and is associated with immune complex deposition.6 However, the number of cases of idiopathic LV may be overestimated; as technological advancements to detect coagulation abnormalities improve, it is hypothesized that this entity will be identified less often.2,4
Livedoid vasculopathy has been described in the literature using the term PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities).7 The triad of livedo racemosa, recurrent painful ulcerations, and residual healing with atrophie blanche characterizes the clinical manifestations of LV; however, all 3 characteristics do not need to appear simultaneously for a diagnosis to be made. The condition has a chronic course with spontaneous remissions and exacerbations. Episodic ulcerations occur, especially in the summertime, and heal slowly, leaving behind atrophic, porcelain white, stellate-shaped scars called atrophie blanche. Livedo racemosa also may be seen in Sneddon syndrome; however, these patients experience neurologic symptoms secondary to cerebrovascular occlusion. In contrast to livedo racemosa, acquired livedo reticularis represents a physiologic hypoperfusion pattern that occurs in response to cold exposure.8 A localized sharp pain, known as angina cutis, typically precedes the clinical symptom of painful ulcerations.9 Atrophie blanche once was thought to be specific to LV but has been seen in other diseases such as systemic lupus erythematosus and chronic venous insufficiency.2
The diagnosis of LV is based on identification of characteristic clinical features and skin biopsy. In almost all biopsy specimens, histopathology reveals fibrinoid occlusion of vessels in the superficial and mid dermis.4 Other findings may include epidermal necrosis and vessel wall hyalinization and infarction2 (Figure). Because LV is commonly misdiagnosed as vasculitis, the absence of hallmark features of vasculitis such as neutrophilic infiltrate of blood vessel walls and fibrinoid necrosis suggest the diagnosis. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities should be performed.
Treatment of LV is difficult, as there is currently no consensus on optimal therapy. The mainstay of therapy is to reduce pain, prevent infection, and reduce ulceration and development of atrophie blanche. Underlying causes should be identified and appropriately treated. Because the primary pathogenesis of LV is considered to be a hypercoagulable state, first-line treatment often includes therapies to enhance blood flow and prevent thrombosis such as smoking cessation, antiplatelet therapy, and pentoxifylline. Vasodilating agents, anti-inflammatory agents, anticoagulation, and fibrinolytic therapy also have been used with varying degrees of success.7
The Diagnosis: Livedoid Vasculopathy
Livedoid vasculopathy (LV) is a rare cutaneous disorder that most commonly affects the lower legs. It has an estimated incidence of 1 case per 100,000 per year and predominantly affects women.1 The disease pathogenesis is not fully understood but is thought to involve thrombosis and occlusion of dermal vessels resulting in tissue hypoxia.2 Both inherited and acquired thrombophilic conditions frequently are seen in patients with LV.3,4 Livedoid vasculopathy also has been described as idiopathic5 and is associated with immune complex deposition.6 However, the number of cases of idiopathic LV may be overestimated; as technological advancements to detect coagulation abnormalities improve, it is hypothesized that this entity will be identified less often.2,4
Livedoid vasculopathy has been described in the literature using the term PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities).7 The triad of livedo racemosa, recurrent painful ulcerations, and residual healing with atrophie blanche characterizes the clinical manifestations of LV; however, all 3 characteristics do not need to appear simultaneously for a diagnosis to be made. The condition has a chronic course with spontaneous remissions and exacerbations. Episodic ulcerations occur, especially in the summertime, and heal slowly, leaving behind atrophic, porcelain white, stellate-shaped scars called atrophie blanche. Livedo racemosa also may be seen in Sneddon syndrome; however, these patients experience neurologic symptoms secondary to cerebrovascular occlusion. In contrast to livedo racemosa, acquired livedo reticularis represents a physiologic hypoperfusion pattern that occurs in response to cold exposure.8 A localized sharp pain, known as angina cutis, typically precedes the clinical symptom of painful ulcerations.9 Atrophie blanche once was thought to be specific to LV but has been seen in other diseases such as systemic lupus erythematosus and chronic venous insufficiency.2
The diagnosis of LV is based on identification of characteristic clinical features and skin biopsy. In almost all biopsy specimens, histopathology reveals fibrinoid occlusion of vessels in the superficial and mid dermis.4 Other findings may include epidermal necrosis and vessel wall hyalinization and infarction2 (Figure). Because LV is commonly misdiagnosed as vasculitis, the absence of hallmark features of vasculitis such as neutrophilic infiltrate of blood vessel walls and fibrinoid necrosis suggest the diagnosis. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities should be performed.
Treatment of LV is difficult, as there is currently no consensus on optimal therapy. The mainstay of therapy is to reduce pain, prevent infection, and reduce ulceration and development of atrophie blanche. Underlying causes should be identified and appropriately treated. Because the primary pathogenesis of LV is considered to be a hypercoagulable state, first-line treatment often includes therapies to enhance blood flow and prevent thrombosis such as smoking cessation, antiplatelet therapy, and pentoxifylline. Vasodilating agents, anti-inflammatory agents, anticoagulation, and fibrinolytic therapy also have been used with varying degrees of success.7
- Fritsch P, Zelger B. Livedo vasculitis [in German]. Hautarzt. 1995;46:215-224; quiz 222-223.
- Kerk N, Goerge T. Livedoid vasculopathy—a thrombotic disease. Vasa. 2013;42:317-322.
- Stevanovic DV. Atrophie blanche. a sign of dermal blood occlusion. Arch Dermatol. 1974;109:858-862.
- Hairston BR, Davis MD, Pittelkow MR, et al. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol. 2006;142:1413-1418.
- Shornick JK, Nicholes BK, Bergstresser PR, et al. Idiopathic atrophie blanche. J Am Acad Dermatol. 1983;8:792-798.
- Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with ulcerations. Circulation. 1956;13:196-216.
- Callen JP. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol. 2006;142:1481-1482.
- Copeman PW. Livedo reticularis. signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975;93:519-529.
- Goerge T. Livedoid vasculopathy. pathogenesis, diagnosis and treatment of cutaneous infarction [in German]. Hautarzt. 2011;62:627-634; quiz 635.
- Fritsch P, Zelger B. Livedo vasculitis [in German]. Hautarzt. 1995;46:215-224; quiz 222-223.
- Kerk N, Goerge T. Livedoid vasculopathy—a thrombotic disease. Vasa. 2013;42:317-322.
- Stevanovic DV. Atrophie blanche. a sign of dermal blood occlusion. Arch Dermatol. 1974;109:858-862.
- Hairston BR, Davis MD, Pittelkow MR, et al. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol. 2006;142:1413-1418.
- Shornick JK, Nicholes BK, Bergstresser PR, et al. Idiopathic atrophie blanche. J Am Acad Dermatol. 1983;8:792-798.
- Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with ulcerations. Circulation. 1956;13:196-216.
- Callen JP. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol. 2006;142:1481-1482.
- Copeman PW. Livedo reticularis. signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975;93:519-529.
- Goerge T. Livedoid vasculopathy. pathogenesis, diagnosis and treatment of cutaneous infarction [in German]. Hautarzt. 2011;62:627-634; quiz 635.
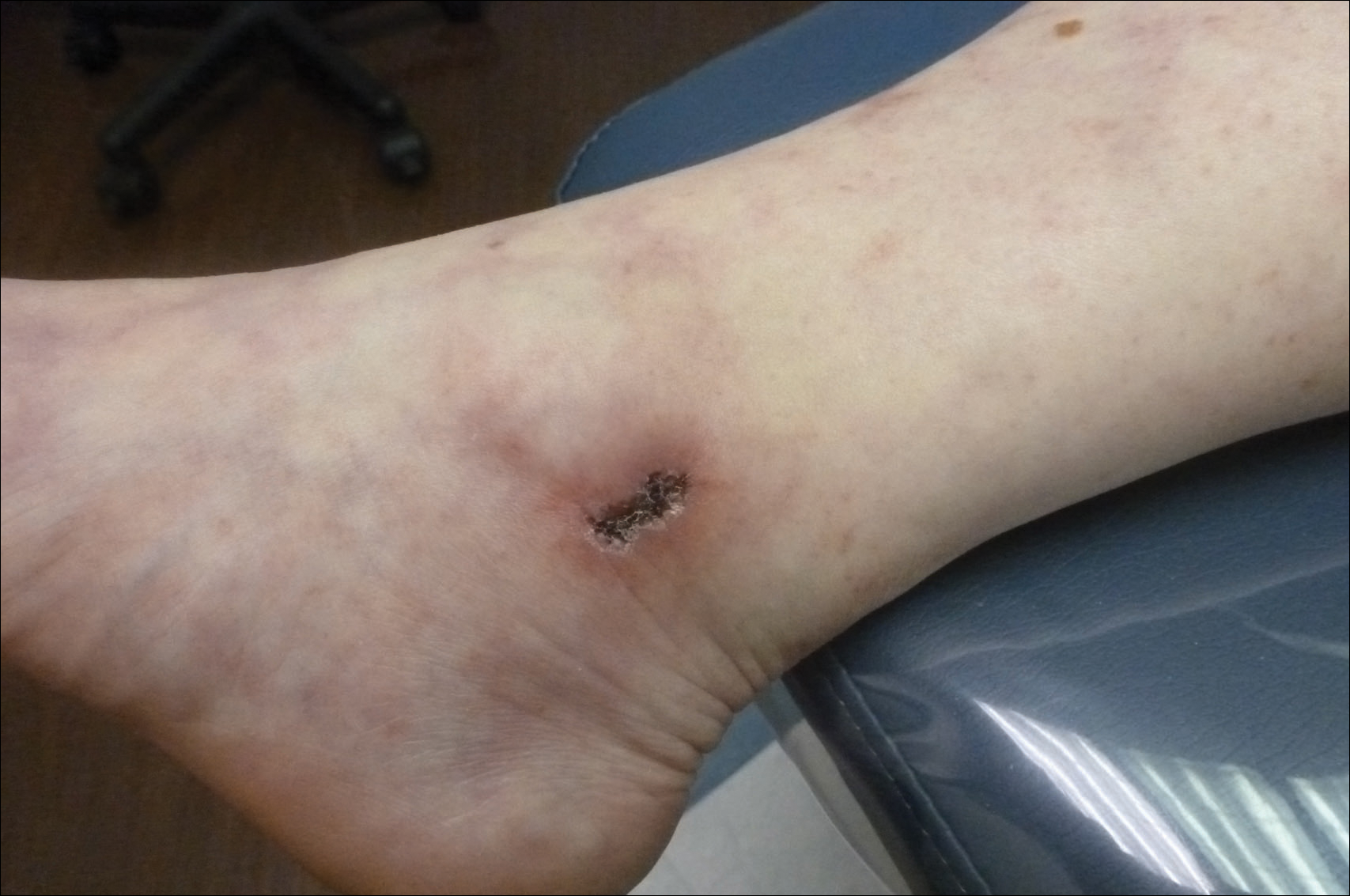
A 58-year-old woman presented in the summertime with skin discoloration of the bilateral lower legs and painful ulcerations above the medial and lateral malleoli of 15 years’ duration. She denied any recent trauma to the area or change in skin lesion appearance with cold exposure. Extensive laboratory evaluation for inherited and acquired coagulation abnormalities was negative. A punch biopsy specimen obtained from the left anterior lower leg revealed vascular thrombi with extravasated erythrocytes and a sparse perivascular inflammatory cell infiltrate.
Subungual Onycholemmal Cyst of the Toenail Mimicking Subungual Melanoma
Case Report
A 23-year-old woman presented with a horizontal split along the midline of the right great toenail associated with some tenderness of 2 to 3 months’ duration. Approximately 5 years prior, she noticed a bluish-colored area under the nail that had been steadily increasing in size. She denied a history of trauma, drainage, or bleeding. There was no history of other nail abnormalities. Her medications and personal, family, and social history were noncontributory.
Physical examination of the right great toenail revealed a horizontal split of the nail plate with a bluish hue visible under the nail plate (Figure 1A). The remaining toenails and fingernails were normal. A punch biopsy of the nail bed was performed with a presumptive clinical diagnosis of subungual melanoma versus melanocytic nevus versus cyst (Figure 1B). Nail plate avulsion revealed a blackened nail bed dotted with areas of bluish color and a red friable nodule present focally. Upon further inspection, extension was apparent into the distal matrix.
Histopathologic examination revealed a cystic structure with an epithelial lining mostly reminiscent of an isthmus catagen cyst admixed with the presence of both an intermittent focal granular layer and an eosinophilic cuticle surrounding pink laminated keratin, most consistent with a diagnosis of subungual onycholemmal cyst (SOC)(Figure 2). A reexcision was performed with removal of half of the nail bed, including a portion of the distal matrix extending inferiorly to the bone. Variably sized, epithelium-lined, keratin-filled cystic structures emanated from the nail bed epithelium. There were foci of hemorrhage and granulation tissue secondary to cyst rupture (Figure 3). The defect healed by secondary intention. No clinical evidence of recurrence was seen at 6-month follow-up.
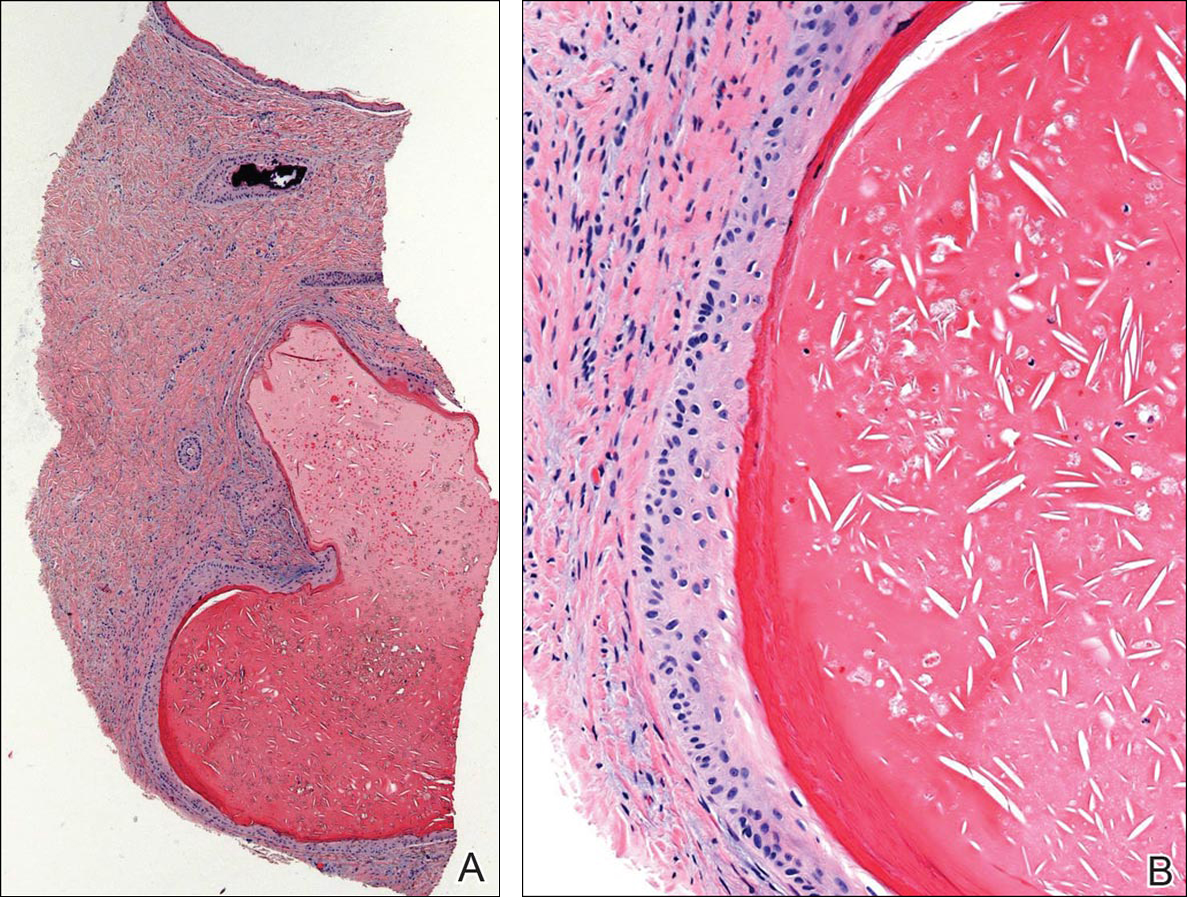

Subungual onycholemmal cysts, also known as subungual epidermoid cysts or subungual epidermoid inclusions, are rare and distinctive nail abnormalities occurring in the dermis of the nail bed. We present a case of an SOC in a toenail mimicking subungual malignant melanoma.
Originally described by Samman1 in 1959, SOCs were attributed to trauma to the nail with resultant implantation of the epidermis into the deeper tissue. Lewin2,3 examined 90 postmortem fingernail and nail bed samples and found 8 subungual epidermoid cysts associated with clubbing of the fingernails. He postulated that the early pathogenesis of clubbing involved dermal fibroblast proliferation in the nail bed, leading to sequestration of nail bed epithelium into the dermis with resultant cyst formation. Microscopic subungual cysts also were identified in normal-appearing nails without evidence of trauma, thought to have arisen from the tips of the nail bed rete ridges by a process of bulbous proliferation rather than sequestration. These findings in normal nails suggest that SOCs may represent a more common entity than previously recognized.
It is imperative to recognize the presence of nail unit tumors early because of the risk for permanent nail plate dystrophy and the possibility of a malignant tumor.4,5 Subungual onycholemmal cysts may present with a wide spectrum of clinical findings including marked subungual hyperkeratosis, onychodystrophy, ridging, nail bed pigmentation, clubbing, thickening, or less often a normal-appearing nail. Based on reported cases, several trends are evident. Although nail dystrophy is most often asymptomatic, pain is not uncommon.5,6 It most commonly involves single digits, predominantly thumbs and great toenails.7,8 This predilection suggests that trauma or other local factors may be involved in its pathogenesis. Of note, trauma to the nail may occur years before the development of the lesions or it may not be recalled at all.
Diagnosis requires a degree of clinical suspicion and a nail bed biopsy with partial or total nail plate avulsion to visualize the pathologic portion of the nail bed. Because surgical intervention may lead to the implantation of epithelium, recurrences after nail biopsy or excision may occur.
In contrast to epidermal inclusion cysts arising in the skin, most SOCs do not have a granular layer.9 Hair and nails represent analogous differentiation products of the ectoderm. The nail matrix is homologous to portions of the hair matrix, while the nail bed epithelium is comparable to the outer root sheath of the hair follicle.7 Subungual onycholemmal cysts originate from the nail bed epithelium, which keratinizes in the absence of a granular layer, similar to the follicular isthmus outer root sheath. Thus, SOCs are comparable to the outer root sheath–derived isthmus-catagen cysts because of their abrupt central keratinization.8
Subungual onycholemmal cysts also must be distinguished from slowly growing malignant tumors of the nail bed epithelium, referred to as onycholemmal carcinomas by Alessi et al.10 This entity characteristically presents in elderly patients as a slowly growing, circumscribed, subungual discoloration that may ulcerate, destroying the nail apparatus and penetrating the phalangeal bone. On histopathology, it is characterized by small cysts filled with eosinophilic keratin devoid of a granular layer and lined by atypical squamous epithelium accompanied by solid nests and strands of atypical keratinocytes within the dermis.11 When a cystic component and clear cells predominate, the designation of malignant proliferating onycholemmal cyst has been applied. Its infiltrative growth pattern with destruction of the underlying bone makes it an important entity to exclude when considering the differential diagnosis of tumors of the nail bed.
Subungual melanomas comprise only 1% to 3% of malignant melanomas and 85% are initially misdiagnosed due to their rarity and nonspecific variable presentation. Aside from clinical evidence of Hutchinson sign in the early stages in almost all cases, accurate diagnosis of subungual melanoma and differentiation from SOCs relies on histopathology. A biopsy is necessary to make the diagnosis, but even microscopic findings may be nonspecific during the early stages.
Conclusion
We report a case of a 23-year-old woman with horizontal ridging and tenderness of the right great toenail associated with pigmentation of 5 years’ duration due to an SOC. The etiology of these subungual cysts, with or without nail abnormalities, still remains unclear. Its predilection for the thumbs and great toenails suggests that trauma or other local factors may be involved in its pathogenesis. Because of the rarity of this entity, there are no guidelines for surgical treatment. Subungual onycholemmal cysts may be an underrecognized and more common entity that must be considered when discussing tumors of the nail unit.
- Samman PD. The human toe nail. its genesis and blood supply. Br J Dermatol. 1959;71:296-302.
- Lewin K. The normal fingernail. Br J Dermatol. 1965;77:421-430.
- Lewin K. Subungual epidermoid inclusions. Br J Dermatol. 1969;81:671-675.
- Dominguez-Cherit J, Chanussot-Deprez C, Maria-Sarti H, et al. Nail unit tumors: a study of 234 patients in the dermatology department of the “Dr. Manuel Gea González” General Hospital in Mexico City. Dermatol Surg. 2008;34:1363-1371.
- Sáez-de-Ocariz MM, Domínguez-Cherit J, García-Corona C. Subungual epidermoid cysts. Int J Dermatol. 2001;40:524-526.
- Molly DO, Herbert K. Subungual epidermoid cyst. J Hand Surg Br. 2006;31:345.
- Telang GH, Jellinek N. Multiple calcified subungual epidermoid inclusions. J Am Acad Dermatol. 2007;56:336-339.
- Fanti PA, Tosti A. Subungual epidermoid inclusions: report of 8 cases. Dermatologica. 1989;178:209-212.
- Takiyoshi N, Nakano H, Matsuzaki T, et al. An eclipse in the subungual space: a diagnostic sign for a subungual epidermal cyst? Br J Dermatol. 2009;161:962-963.
- Alessi E, Coggi A, Gianotti R, et al. Onycholemmal carcinoma. Am J Dermatopathol. 2004;26:397-402.
- Inaoki M, Makino E, Adachi M, et al. Onycholemmal carcinoma. J Cutan Pathol. 2006;33:577-580.
Case Report
A 23-year-old woman presented with a horizontal split along the midline of the right great toenail associated with some tenderness of 2 to 3 months’ duration. Approximately 5 years prior, she noticed a bluish-colored area under the nail that had been steadily increasing in size. She denied a history of trauma, drainage, or bleeding. There was no history of other nail abnormalities. Her medications and personal, family, and social history were noncontributory.
Physical examination of the right great toenail revealed a horizontal split of the nail plate with a bluish hue visible under the nail plate (Figure 1A). The remaining toenails and fingernails were normal. A punch biopsy of the nail bed was performed with a presumptive clinical diagnosis of subungual melanoma versus melanocytic nevus versus cyst (Figure 1B). Nail plate avulsion revealed a blackened nail bed dotted with areas of bluish color and a red friable nodule present focally. Upon further inspection, extension was apparent into the distal matrix.
Histopathologic examination revealed a cystic structure with an epithelial lining mostly reminiscent of an isthmus catagen cyst admixed with the presence of both an intermittent focal granular layer and an eosinophilic cuticle surrounding pink laminated keratin, most consistent with a diagnosis of subungual onycholemmal cyst (SOC)(Figure 2). A reexcision was performed with removal of half of the nail bed, including a portion of the distal matrix extending inferiorly to the bone. Variably sized, epithelium-lined, keratin-filled cystic structures emanated from the nail bed epithelium. There were foci of hemorrhage and granulation tissue secondary to cyst rupture (Figure 3). The defect healed by secondary intention. No clinical evidence of recurrence was seen at 6-month follow-up.
Subungual onycholemmal cysts, also known as subungual epidermoid cysts or subungual epidermoid inclusions, are rare and distinctive nail abnormalities occurring in the dermis of the nail bed. We present a case of an SOC in a toenail mimicking subungual malignant melanoma.
Originally described by Samman1 in 1959, SOCs were attributed to trauma to the nail with resultant implantation of the epidermis into the deeper tissue. Lewin2,3 examined 90 postmortem fingernail and nail bed samples and found 8 subungual epidermoid cysts associated with clubbing of the fingernails. He postulated that the early pathogenesis of clubbing involved dermal fibroblast proliferation in the nail bed, leading to sequestration of nail bed epithelium into the dermis with resultant cyst formation. Microscopic subungual cysts also were identified in normal-appearing nails without evidence of trauma, thought to have arisen from the tips of the nail bed rete ridges by a process of bulbous proliferation rather than sequestration. These findings in normal nails suggest that SOCs may represent a more common entity than previously recognized.
It is imperative to recognize the presence of nail unit tumors early because of the risk for permanent nail plate dystrophy and the possibility of a malignant tumor.4,5 Subungual onycholemmal cysts may present with a wide spectrum of clinical findings including marked subungual hyperkeratosis, onychodystrophy, ridging, nail bed pigmentation, clubbing, thickening, or less often a normal-appearing nail. Based on reported cases, several trends are evident. Although nail dystrophy is most often asymptomatic, pain is not uncommon.5,6 It most commonly involves single digits, predominantly thumbs and great toenails.7,8 This predilection suggests that trauma or other local factors may be involved in its pathogenesis. Of note, trauma to the nail may occur years before the development of the lesions or it may not be recalled at all.
Diagnosis requires a degree of clinical suspicion and a nail bed biopsy with partial or total nail plate avulsion to visualize the pathologic portion of the nail bed. Because surgical intervention may lead to the implantation of epithelium, recurrences after nail biopsy or excision may occur.
In contrast to epidermal inclusion cysts arising in the skin, most SOCs do not have a granular layer.9 Hair and nails represent analogous differentiation products of the ectoderm. The nail matrix is homologous to portions of the hair matrix, while the nail bed epithelium is comparable to the outer root sheath of the hair follicle.7 Subungual onycholemmal cysts originate from the nail bed epithelium, which keratinizes in the absence of a granular layer, similar to the follicular isthmus outer root sheath. Thus, SOCs are comparable to the outer root sheath–derived isthmus-catagen cysts because of their abrupt central keratinization.8
Subungual onycholemmal cysts also must be distinguished from slowly growing malignant tumors of the nail bed epithelium, referred to as onycholemmal carcinomas by Alessi et al.10 This entity characteristically presents in elderly patients as a slowly growing, circumscribed, subungual discoloration that may ulcerate, destroying the nail apparatus and penetrating the phalangeal bone. On histopathology, it is characterized by small cysts filled with eosinophilic keratin devoid of a granular layer and lined by atypical squamous epithelium accompanied by solid nests and strands of atypical keratinocytes within the dermis.11 When a cystic component and clear cells predominate, the designation of malignant proliferating onycholemmal cyst has been applied. Its infiltrative growth pattern with destruction of the underlying bone makes it an important entity to exclude when considering the differential diagnosis of tumors of the nail bed.
Subungual melanomas comprise only 1% to 3% of malignant melanomas and 85% are initially misdiagnosed due to their rarity and nonspecific variable presentation. Aside from clinical evidence of Hutchinson sign in the early stages in almost all cases, accurate diagnosis of subungual melanoma and differentiation from SOCs relies on histopathology. A biopsy is necessary to make the diagnosis, but even microscopic findings may be nonspecific during the early stages.
Conclusion
We report a case of a 23-year-old woman with horizontal ridging and tenderness of the right great toenail associated with pigmentation of 5 years’ duration due to an SOC. The etiology of these subungual cysts, with or without nail abnormalities, still remains unclear. Its predilection for the thumbs and great toenails suggests that trauma or other local factors may be involved in its pathogenesis. Because of the rarity of this entity, there are no guidelines for surgical treatment. Subungual onycholemmal cysts may be an underrecognized and more common entity that must be considered when discussing tumors of the nail unit.
Case Report
A 23-year-old woman presented with a horizontal split along the midline of the right great toenail associated with some tenderness of 2 to 3 months’ duration. Approximately 5 years prior, she noticed a bluish-colored area under the nail that had been steadily increasing in size. She denied a history of trauma, drainage, or bleeding. There was no history of other nail abnormalities. Her medications and personal, family, and social history were noncontributory.
Physical examination of the right great toenail revealed a horizontal split of the nail plate with a bluish hue visible under the nail plate (Figure 1A). The remaining toenails and fingernails were normal. A punch biopsy of the nail bed was performed with a presumptive clinical diagnosis of subungual melanoma versus melanocytic nevus versus cyst (Figure 1B). Nail plate avulsion revealed a blackened nail bed dotted with areas of bluish color and a red friable nodule present focally. Upon further inspection, extension was apparent into the distal matrix.
Histopathologic examination revealed a cystic structure with an epithelial lining mostly reminiscent of an isthmus catagen cyst admixed with the presence of both an intermittent focal granular layer and an eosinophilic cuticle surrounding pink laminated keratin, most consistent with a diagnosis of subungual onycholemmal cyst (SOC)(Figure 2). A reexcision was performed with removal of half of the nail bed, including a portion of the distal matrix extending inferiorly to the bone. Variably sized, epithelium-lined, keratin-filled cystic structures emanated from the nail bed epithelium. There were foci of hemorrhage and granulation tissue secondary to cyst rupture (Figure 3). The defect healed by secondary intention. No clinical evidence of recurrence was seen at 6-month follow-up.
Subungual onycholemmal cysts, also known as subungual epidermoid cysts or subungual epidermoid inclusions, are rare and distinctive nail abnormalities occurring in the dermis of the nail bed. We present a case of an SOC in a toenail mimicking subungual malignant melanoma.
Originally described by Samman1 in 1959, SOCs were attributed to trauma to the nail with resultant implantation of the epidermis into the deeper tissue. Lewin2,3 examined 90 postmortem fingernail and nail bed samples and found 8 subungual epidermoid cysts associated with clubbing of the fingernails. He postulated that the early pathogenesis of clubbing involved dermal fibroblast proliferation in the nail bed, leading to sequestration of nail bed epithelium into the dermis with resultant cyst formation. Microscopic subungual cysts also were identified in normal-appearing nails without evidence of trauma, thought to have arisen from the tips of the nail bed rete ridges by a process of bulbous proliferation rather than sequestration. These findings in normal nails suggest that SOCs may represent a more common entity than previously recognized.
It is imperative to recognize the presence of nail unit tumors early because of the risk for permanent nail plate dystrophy and the possibility of a malignant tumor.4,5 Subungual onycholemmal cysts may present with a wide spectrum of clinical findings including marked subungual hyperkeratosis, onychodystrophy, ridging, nail bed pigmentation, clubbing, thickening, or less often a normal-appearing nail. Based on reported cases, several trends are evident. Although nail dystrophy is most often asymptomatic, pain is not uncommon.5,6 It most commonly involves single digits, predominantly thumbs and great toenails.7,8 This predilection suggests that trauma or other local factors may be involved in its pathogenesis. Of note, trauma to the nail may occur years before the development of the lesions or it may not be recalled at all.
Diagnosis requires a degree of clinical suspicion and a nail bed biopsy with partial or total nail plate avulsion to visualize the pathologic portion of the nail bed. Because surgical intervention may lead to the implantation of epithelium, recurrences after nail biopsy or excision may occur.
In contrast to epidermal inclusion cysts arising in the skin, most SOCs do not have a granular layer.9 Hair and nails represent analogous differentiation products of the ectoderm. The nail matrix is homologous to portions of the hair matrix, while the nail bed epithelium is comparable to the outer root sheath of the hair follicle.7 Subungual onycholemmal cysts originate from the nail bed epithelium, which keratinizes in the absence of a granular layer, similar to the follicular isthmus outer root sheath. Thus, SOCs are comparable to the outer root sheath–derived isthmus-catagen cysts because of their abrupt central keratinization.8
Subungual onycholemmal cysts also must be distinguished from slowly growing malignant tumors of the nail bed epithelium, referred to as onycholemmal carcinomas by Alessi et al.10 This entity characteristically presents in elderly patients as a slowly growing, circumscribed, subungual discoloration that may ulcerate, destroying the nail apparatus and penetrating the phalangeal bone. On histopathology, it is characterized by small cysts filled with eosinophilic keratin devoid of a granular layer and lined by atypical squamous epithelium accompanied by solid nests and strands of atypical keratinocytes within the dermis.11 When a cystic component and clear cells predominate, the designation of malignant proliferating onycholemmal cyst has been applied. Its infiltrative growth pattern with destruction of the underlying bone makes it an important entity to exclude when considering the differential diagnosis of tumors of the nail bed.
Subungual melanomas comprise only 1% to 3% of malignant melanomas and 85% are initially misdiagnosed due to their rarity and nonspecific variable presentation. Aside from clinical evidence of Hutchinson sign in the early stages in almost all cases, accurate diagnosis of subungual melanoma and differentiation from SOCs relies on histopathology. A biopsy is necessary to make the diagnosis, but even microscopic findings may be nonspecific during the early stages.
Conclusion
We report a case of a 23-year-old woman with horizontal ridging and tenderness of the right great toenail associated with pigmentation of 5 years’ duration due to an SOC. The etiology of these subungual cysts, with or without nail abnormalities, still remains unclear. Its predilection for the thumbs and great toenails suggests that trauma or other local factors may be involved in its pathogenesis. Because of the rarity of this entity, there are no guidelines for surgical treatment. Subungual onycholemmal cysts may be an underrecognized and more common entity that must be considered when discussing tumors of the nail unit.
- Samman PD. The human toe nail. its genesis and blood supply. Br J Dermatol. 1959;71:296-302.
- Lewin K. The normal fingernail. Br J Dermatol. 1965;77:421-430.
- Lewin K. Subungual epidermoid inclusions. Br J Dermatol. 1969;81:671-675.
- Dominguez-Cherit J, Chanussot-Deprez C, Maria-Sarti H, et al. Nail unit tumors: a study of 234 patients in the dermatology department of the “Dr. Manuel Gea González” General Hospital in Mexico City. Dermatol Surg. 2008;34:1363-1371.
- Sáez-de-Ocariz MM, Domínguez-Cherit J, García-Corona C. Subungual epidermoid cysts. Int J Dermatol. 2001;40:524-526.
- Molly DO, Herbert K. Subungual epidermoid cyst. J Hand Surg Br. 2006;31:345.
- Telang GH, Jellinek N. Multiple calcified subungual epidermoid inclusions. J Am Acad Dermatol. 2007;56:336-339.
- Fanti PA, Tosti A. Subungual epidermoid inclusions: report of 8 cases. Dermatologica. 1989;178:209-212.
- Takiyoshi N, Nakano H, Matsuzaki T, et al. An eclipse in the subungual space: a diagnostic sign for a subungual epidermal cyst? Br J Dermatol. 2009;161:962-963.
- Alessi E, Coggi A, Gianotti R, et al. Onycholemmal carcinoma. Am J Dermatopathol. 2004;26:397-402.
- Inaoki M, Makino E, Adachi M, et al. Onycholemmal carcinoma. J Cutan Pathol. 2006;33:577-580.
- Samman PD. The human toe nail. its genesis and blood supply. Br J Dermatol. 1959;71:296-302.
- Lewin K. The normal fingernail. Br J Dermatol. 1965;77:421-430.
- Lewin K. Subungual epidermoid inclusions. Br J Dermatol. 1969;81:671-675.
- Dominguez-Cherit J, Chanussot-Deprez C, Maria-Sarti H, et al. Nail unit tumors: a study of 234 patients in the dermatology department of the “Dr. Manuel Gea González” General Hospital in Mexico City. Dermatol Surg. 2008;34:1363-1371.
- Sáez-de-Ocariz MM, Domínguez-Cherit J, García-Corona C. Subungual epidermoid cysts. Int J Dermatol. 2001;40:524-526.
- Molly DO, Herbert K. Subungual epidermoid cyst. J Hand Surg Br. 2006;31:345.
- Telang GH, Jellinek N. Multiple calcified subungual epidermoid inclusions. J Am Acad Dermatol. 2007;56:336-339.
- Fanti PA, Tosti A. Subungual epidermoid inclusions: report of 8 cases. Dermatologica. 1989;178:209-212.
- Takiyoshi N, Nakano H, Matsuzaki T, et al. An eclipse in the subungual space: a diagnostic sign for a subungual epidermal cyst? Br J Dermatol. 2009;161:962-963.
- Alessi E, Coggi A, Gianotti R, et al. Onycholemmal carcinoma. Am J Dermatopathol. 2004;26:397-402.
- Inaoki M, Makino E, Adachi M, et al. Onycholemmal carcinoma. J Cutan Pathol. 2006;33:577-580.
Practice Points
- Trauma to the nail may occur years before the development of subungual onycholemmal cysts or it may not be recalled at all.
- Diagnosis requires a degree of clinical suspicion and a nail bed biopsy.
- Subungual onycholemmal cysts must be distinguished from slowly growing malignant tumors of the nail bed epithelium.
Lasers for Darker Skin Types


Linearly Curved, Blackish Macule on the Wrist
Linear Basal Cell Carcinoma
On examination, the lesion was suspected to be a nevocellular nevus, foreign body granuloma, or venous lake; however, a skin biopsy specimen from the lesion on the left wrist revealed a tumor mass of basaloid cells, peripheral palisading arrangement, and scattered pigment granules (Figure 1). Tumor cells were negative for S-100 protein staining. These findings were consistent with a diagnosis of linear basal cell carcinoma (BCC). The lesion was removed by simple excision with primary closure of the wound. The surgical margins were free of tumor cells. The lesion had not recurred at 6-month follow-up. The patient was subsequently lost to follow-up.
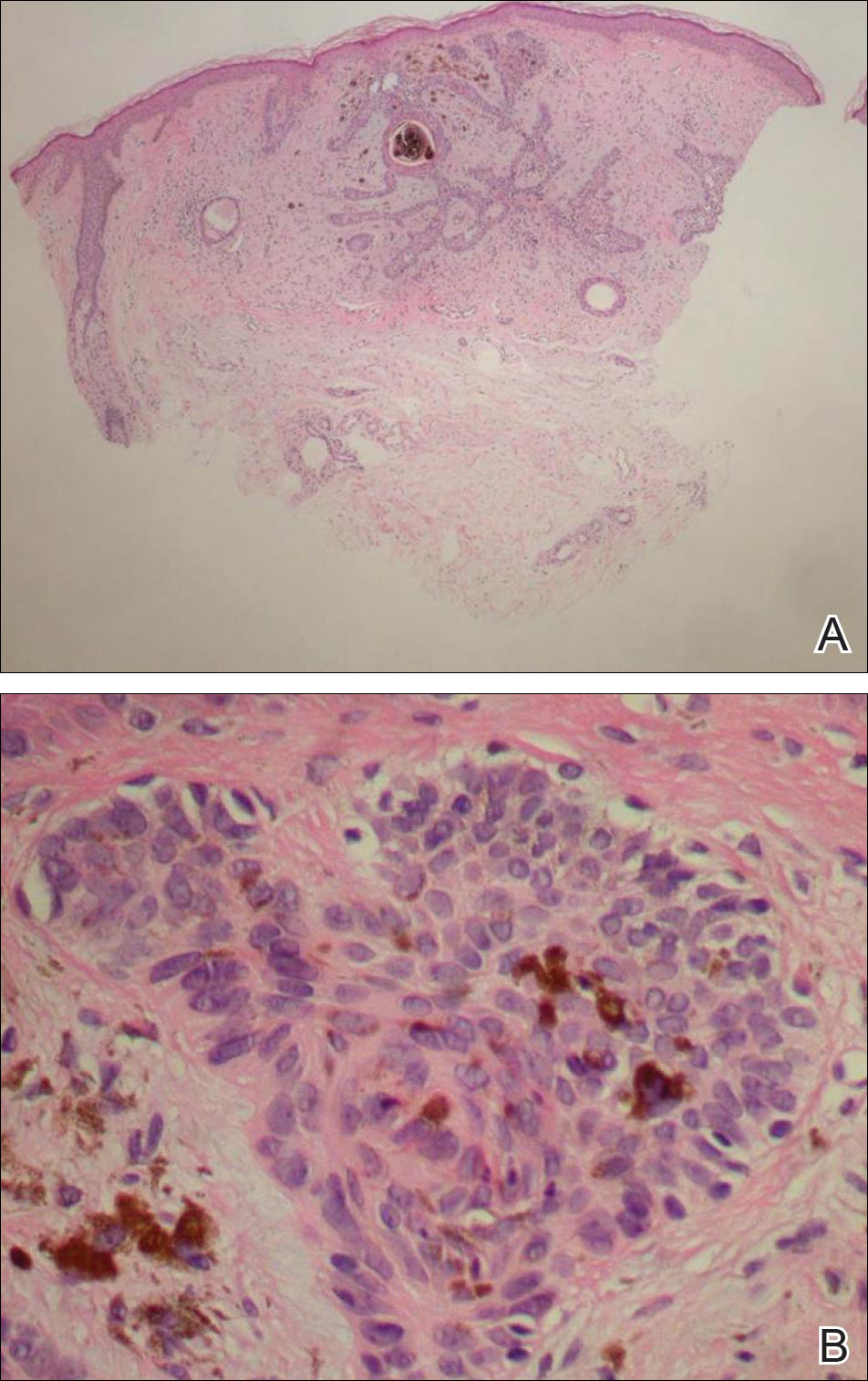
Basal cell carcinoma presents with diverse clinical features, and several morphologic and histologic variants have been reported.1 Linear BCC was described as a distinct clinical entity in 1985 by Lewis2 in a 73-year-old man with a 20-mm linear pigmented lesion on the left cheek. Linear BCC often is not recognized or categorized as such by clinicians, as some may think that linear BCC is not a distinct entity but rather is one of the diverse clinical features of BCC.3 Linear BCC is believed to have specific clinical and histologic features and can be regarded as a distinct entity.4 Mavrikakis et al5 objectively defined linear BCC as a lesion that appeared to extend preferentially in one direction, resulting in a lesion with relatively straight borders and a length much greater than the width (3:1 ratio). Our patient presented with a linearly curved lesion, which is a rare feature of BCC.
Linear BCC occurs in equal proportions in men and women aged 40 to 87 years. More than 92% of reported patients were older than 60 years.6 The most common site for linear BCC is the periocular area, with the majority of lesions occurring on the cheek or lower eyelid. The second most common site is the neck, followed by the trunk, lower face, and inguinal skin fold.3,5
The mechanism of linearity has been speculated. The majority of the reported cases of linear BCC have no history of trauma.7 However, focal trauma has been assumed to be a risk factor for the development of linear BCC, so the possibility that the Köbner phenomenon may be related to its linear pattern has been proposed.8 The Köbner phenomenon can be implicated in our case, as there was a history of surgery, which resulted in a scar.
Menzies9 described dermoscopic features of pigmented BCC and stated that the diagnosis of pigmented BCC required the presence of 1 or more of the following 6 positive features: large blue-gray ovoid nests; multiple blue-gray globules; maple leaf–like areas; spoke wheel areas; ulceration; and arborizing treelike vessels. In our case, there were multiple blue-gray globules and a streak that resembled ginseng (Figure 2).
Linear BCC is an uncommon morphological variant that requires clinical recognition. Our case was unique because of the ginsenglike streak on dermoscopy and possible association with a prior trauma.
- Sexton M, Jones DB, Maloney ME. Histologic pattern analysis of basal cell carcinoma. study of a series of 1,039 consecutive neoplasms. J Am Acad Dermatol. 1990;23(6, pt 1):1118-1126.
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1985;24:124-125.
- Mavrikakis I, Malhotra R, Selva D, et al. Linear basal cell carcinoma: a distinct clinical entity. J Plast Reconstr Aesthet Surg. 2006;59:419-423.
- Jellouli A, Triki S, Zghal M, et al. Linear basal cell carcinoma. Actas Dermosifiliogr. 2010;101:648-650.
- Mavrikakis I, Malhotra R, Barlow R, et al. Linear basal cell carcinoma: a distinct clinical entity in the periocular region [published online January 10, 2006]. Ophthalmology. 2006;113:338-342.
- Lim KK, Randle HW, Roenigk RK, et al. Linear basal cell carcinoma: report of seventeen cases and review of the presentation and treatment. Dermatol Surg. 1999;25:63-67.
- Iga N, Sakurai K, Fujii H, et al. Linear basal cell carcinoma at the external genitalia. J Dermatol. 2014;41:275-276.
- Peschen M, Lo JS, Snow SN, et al. Linear basal cell carcinoma. Cutis. 1993;51:287-289.
- Menzies SW. Dermoscopy of pigmented basal cell carcinoma. Clin Dermatol. 2002;20:268-269.
Linear Basal Cell Carcinoma
On examination, the lesion was suspected to be a nevocellular nevus, foreign body granuloma, or venous lake; however, a skin biopsy specimen from the lesion on the left wrist revealed a tumor mass of basaloid cells, peripheral palisading arrangement, and scattered pigment granules (Figure 1). Tumor cells were negative for S-100 protein staining. These findings were consistent with a diagnosis of linear basal cell carcinoma (BCC). The lesion was removed by simple excision with primary closure of the wound. The surgical margins were free of tumor cells. The lesion had not recurred at 6-month follow-up. The patient was subsequently lost to follow-up.
Basal cell carcinoma presents with diverse clinical features, and several morphologic and histologic variants have been reported.1 Linear BCC was described as a distinct clinical entity in 1985 by Lewis2 in a 73-year-old man with a 20-mm linear pigmented lesion on the left cheek. Linear BCC often is not recognized or categorized as such by clinicians, as some may think that linear BCC is not a distinct entity but rather is one of the diverse clinical features of BCC.3 Linear BCC is believed to have specific clinical and histologic features and can be regarded as a distinct entity.4 Mavrikakis et al5 objectively defined linear BCC as a lesion that appeared to extend preferentially in one direction, resulting in a lesion with relatively straight borders and a length much greater than the width (3:1 ratio). Our patient presented with a linearly curved lesion, which is a rare feature of BCC.
Linear BCC occurs in equal proportions in men and women aged 40 to 87 years. More than 92% of reported patients were older than 60 years.6 The most common site for linear BCC is the periocular area, with the majority of lesions occurring on the cheek or lower eyelid. The second most common site is the neck, followed by the trunk, lower face, and inguinal skin fold.3,5
The mechanism of linearity has been speculated. The majority of the reported cases of linear BCC have no history of trauma.7 However, focal trauma has been assumed to be a risk factor for the development of linear BCC, so the possibility that the Köbner phenomenon may be related to its linear pattern has been proposed.8 The Köbner phenomenon can be implicated in our case, as there was a history of surgery, which resulted in a scar.
Menzies9 described dermoscopic features of pigmented BCC and stated that the diagnosis of pigmented BCC required the presence of 1 or more of the following 6 positive features: large blue-gray ovoid nests; multiple blue-gray globules; maple leaf–like areas; spoke wheel areas; ulceration; and arborizing treelike vessels. In our case, there were multiple blue-gray globules and a streak that resembled ginseng (Figure 2).
Linear BCC is an uncommon morphological variant that requires clinical recognition. Our case was unique because of the ginsenglike streak on dermoscopy and possible association with a prior trauma.
Linear Basal Cell Carcinoma
On examination, the lesion was suspected to be a nevocellular nevus, foreign body granuloma, or venous lake; however, a skin biopsy specimen from the lesion on the left wrist revealed a tumor mass of basaloid cells, peripheral palisading arrangement, and scattered pigment granules (Figure 1). Tumor cells were negative for S-100 protein staining. These findings were consistent with a diagnosis of linear basal cell carcinoma (BCC). The lesion was removed by simple excision with primary closure of the wound. The surgical margins were free of tumor cells. The lesion had not recurred at 6-month follow-up. The patient was subsequently lost to follow-up.
Basal cell carcinoma presents with diverse clinical features, and several morphologic and histologic variants have been reported.1 Linear BCC was described as a distinct clinical entity in 1985 by Lewis2 in a 73-year-old man with a 20-mm linear pigmented lesion on the left cheek. Linear BCC often is not recognized or categorized as such by clinicians, as some may think that linear BCC is not a distinct entity but rather is one of the diverse clinical features of BCC.3 Linear BCC is believed to have specific clinical and histologic features and can be regarded as a distinct entity.4 Mavrikakis et al5 objectively defined linear BCC as a lesion that appeared to extend preferentially in one direction, resulting in a lesion with relatively straight borders and a length much greater than the width (3:1 ratio). Our patient presented with a linearly curved lesion, which is a rare feature of BCC.
Linear BCC occurs in equal proportions in men and women aged 40 to 87 years. More than 92% of reported patients were older than 60 years.6 The most common site for linear BCC is the periocular area, with the majority of lesions occurring on the cheek or lower eyelid. The second most common site is the neck, followed by the trunk, lower face, and inguinal skin fold.3,5
The mechanism of linearity has been speculated. The majority of the reported cases of linear BCC have no history of trauma.7 However, focal trauma has been assumed to be a risk factor for the development of linear BCC, so the possibility that the Köbner phenomenon may be related to its linear pattern has been proposed.8 The Köbner phenomenon can be implicated in our case, as there was a history of surgery, which resulted in a scar.
Menzies9 described dermoscopic features of pigmented BCC and stated that the diagnosis of pigmented BCC required the presence of 1 or more of the following 6 positive features: large blue-gray ovoid nests; multiple blue-gray globules; maple leaf–like areas; spoke wheel areas; ulceration; and arborizing treelike vessels. In our case, there were multiple blue-gray globules and a streak that resembled ginseng (Figure 2).
Linear BCC is an uncommon morphological variant that requires clinical recognition. Our case was unique because of the ginsenglike streak on dermoscopy and possible association with a prior trauma.
- Sexton M, Jones DB, Maloney ME. Histologic pattern analysis of basal cell carcinoma. study of a series of 1,039 consecutive neoplasms. J Am Acad Dermatol. 1990;23(6, pt 1):1118-1126.
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1985;24:124-125.
- Mavrikakis I, Malhotra R, Selva D, et al. Linear basal cell carcinoma: a distinct clinical entity. J Plast Reconstr Aesthet Surg. 2006;59:419-423.
- Jellouli A, Triki S, Zghal M, et al. Linear basal cell carcinoma. Actas Dermosifiliogr. 2010;101:648-650.
- Mavrikakis I, Malhotra R, Barlow R, et al. Linear basal cell carcinoma: a distinct clinical entity in the periocular region [published online January 10, 2006]. Ophthalmology. 2006;113:338-342.
- Lim KK, Randle HW, Roenigk RK, et al. Linear basal cell carcinoma: report of seventeen cases and review of the presentation and treatment. Dermatol Surg. 1999;25:63-67.
- Iga N, Sakurai K, Fujii H, et al. Linear basal cell carcinoma at the external genitalia. J Dermatol. 2014;41:275-276.
- Peschen M, Lo JS, Snow SN, et al. Linear basal cell carcinoma. Cutis. 1993;51:287-289.
- Menzies SW. Dermoscopy of pigmented basal cell carcinoma. Clin Dermatol. 2002;20:268-269.
- Sexton M, Jones DB, Maloney ME. Histologic pattern analysis of basal cell carcinoma. study of a series of 1,039 consecutive neoplasms. J Am Acad Dermatol. 1990;23(6, pt 1):1118-1126.
- Lewis JE. Linear basal cell epithelioma. Int J Dermatol. 1985;24:124-125.
- Mavrikakis I, Malhotra R, Selva D, et al. Linear basal cell carcinoma: a distinct clinical entity. J Plast Reconstr Aesthet Surg. 2006;59:419-423.
- Jellouli A, Triki S, Zghal M, et al. Linear basal cell carcinoma. Actas Dermosifiliogr. 2010;101:648-650.
- Mavrikakis I, Malhotra R, Barlow R, et al. Linear basal cell carcinoma: a distinct clinical entity in the periocular region [published online January 10, 2006]. Ophthalmology. 2006;113:338-342.
- Lim KK, Randle HW, Roenigk RK, et al. Linear basal cell carcinoma: report of seventeen cases and review of the presentation and treatment. Dermatol Surg. 1999;25:63-67.
- Iga N, Sakurai K, Fujii H, et al. Linear basal cell carcinoma at the external genitalia. J Dermatol. 2014;41:275-276.
- Peschen M, Lo JS, Snow SN, et al. Linear basal cell carcinoma. Cutis. 1993;51:287-289.
- Menzies SW. Dermoscopy of pigmented basal cell carcinoma. Clin Dermatol. 2002;20:268-269.

Spontaneous Repigmentation of Silvery Hair in an Infant With Congenital Hydrops Fetalis and Hypoproteinemia
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.


Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
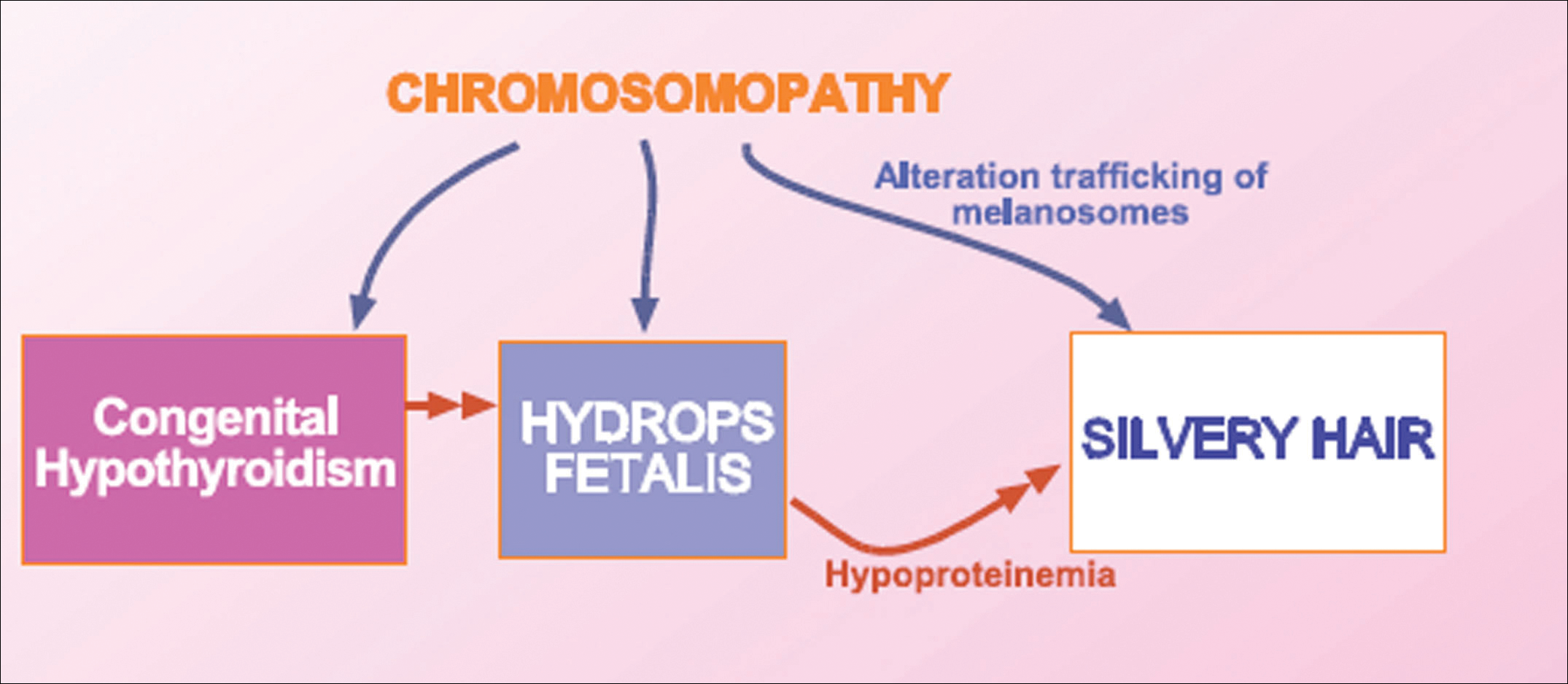
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.
Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
Silvery hair is characteristic of 3 rare autosomal-recessive disorders—Chédiak-Higashi syndrome (CHS), Elejalde syndrome (ES), and Griscelli syndrome (GS)—which are associated with mutations in various genes that encode several proteins involved in the intracellular processing and movement of melanosomes. We report the case of a 2-month-old male infant with transient silvery hair and generalized hypopigmentation of the skin and eyes who did not have any genetic mutations associated with the classic syndromes that usually are characterized by transient silvery hair.
Case Report
A 2-month-old male infant presented to the dermatology department for evaluation of silvery hair with generalized hypopigmentation of the skin and eyes (Figure 1) that had developed at 1 month of age. His parents were healthy, nonconsanguineous, and reported no family history of silvery hair. The patient was delivered by cesarean section at 35 weeks’ gestation. His medical history was remarkable for congenital hydrops fetalis with pleuropericardial effusion, ascites, soft-tissue edema, and hydrocele with no signs of any congenital infection. Both the patient and his mother were O Rh +.
Several studies were performed following delivery. A direct Coombs test was negative. Blood studies revealed hypothyroidism and hypoalbuminemia secondary to protein loss associated with fetal hydrops. Cerebral, abdominal, and renal ultrasound; echocardiogram; thoracic and abdominal computed tomography; and cerebral magnetic resonance imaging revealed no abnormalities.
Karyotype results showed 46,XY,add(2)(p23), and subsequent spectral karyotyping and fluorescence in situ hybridization tests identified a chromosomal abnormality (46,XY,add[2][p23].ish del[2][pter][2PTEL27‒], dup[4][qter][D4S2930++])(Figure 2). Parental karyotypes were normal.
After birth, the infant was admitted to the neonatal intensive care unit for 50 days and received pleural and peritoneal drainages, mechanical ventilation, vasoactive drugs, parenteral nutrition with resolution of the hypoalbuminemia, levothyroxine, and intravenous antibiotics for central venous catheter infection. No drugs known to be associated with hypopigmentation of the hair, skin, or eyes were administered.
Two weeks after discharge from the neonatal intensive care unit, the patient was referred to our department. Physical examination revealed silvery hair on the scalp, eyebrows, and eyelashes, along with generalized hypopigmentation of the skin and eyes. Abdominal, cardiovascular, respiratory, and neurologic examination revealed no abnormalities, and no hepatosplenomegaly, lymphadenopathy, nystagmus, or strabismus was noted.
Light microscopy of the hair revealed small and regular aggregates of melanin along the hair shaft, predominantly in the medulla (Figure 3). Light microscopy of a skin biopsy specimen showed normal pigmentation in the melanocytes and no giant melanosomes. The melanocyte count was within reference range. A peripheral blood smear showed no giant granules in the granulocytes. No treatment was administered and the patient was followed closely every month. When the patient returned for follow-up at 9 months of age, physical examination revealed brown hair on the head, eyebrows, and eyelashes, as well as normal pigmentation of the skin and eyes (Figure 4). Thyroid function was normal and no recurrent infections of any type were noted. At follow-up at the age of 4 years, he showed normal neurological and psychological development with brown hair, no recurrent infections, and normal thyroid function. Given that CHS, ES, and GS had been ruled out, the clinical presentation and the genetic mutation detected may indicate that this case represents a new entity characterized by transient silvery hair.
Comment
Silvery hair is a known feature of CHS, ES, and GS (Table). The characteristic hypopigmentation associated with these autosomal-recessive disorders is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes. These disorders differ from oculocutaneous albinism in that melanin synthesis is unaffected.
Chédiak-Higashi syndrome is characterized by generalized hypopigmentation of the skin and eyes, silvery hair, neurologic and immune dysfunction, lymphoproliferative disorders, and large granules in granulocytes and other cell types.1-3 A common complication of CHS is hemophagocytic lymphohistiocytosis, which is characterized by fever, jaundice, lymphadenopathy, hepatosplenomegaly, and pancytopenia.4 Pigmentary dilution of the irises also may be present, along with photophobia, strabismus, nystagmus, and impaired visual acuity. Chédiak-Higashi syndrome is the result of a genetic defect in the lysosomal trafficking regulator gene, also known as CHS1 (located on chromosome 1q42.1‒q42.2).5 Melanin in the hair shaft is distributed uniformly in multiple small aggregates. Light microscopy of the skin typically shows giant melanosomes in melanocytes and aberrant keratinocyte maturation.
Elejalde syndrome is characterized by silvery hair (eyelashes and eyebrows), neurologic defects, and normal immunologic function.6,7 The underlying molecular basis remains unknown. It appears related to or allelic to GS type 1 and thus associated with mutations in MYO5A (myosin VA); however, the gene mutation responsible has yet to be defined.8 Light microscopy of the hair shaft usually shows an irregular distribution of large melanin aggregates, primarily in the medulla.9,10 Skin biopsy generally shows irregular distribution and irregular size of melanin granules in the basal layer.11 Leukocytes usually show no abnormal cytoplasmic granules. Ocular involvement is common and may present as nystagmus, diplopia, hypopigmented retinas, and/or papilledema.
In GS, hair microscopy generally reveals large aggregates of melanin pigment distributed irregularly along the hair shaft. Granulocytes typically show no giant granules. Light microscopy of the skin usually shows increased pigment in melanocytes with sparse pigment in keratinocytes. Griscelli syndrome is classified into 3 types.12 In GS type 1, patients have silvery gray hair, light-colored skin, severe neurologic defects,13 and normal immune status. This variant is caused by a mutation in the MYO5A gene located on chromosome 15q21. In GS type 2, patients have silvery gray hair, pyogenic infections, an accelerated phase of hemophagocytic lymphohistiocytosis, and variable neurologic defects in the absence of primary neurologic disease.14,15 This variant is caused by a mutation in the RAB27A (member RAS oncogene family) gene located on chromosome 15q21. In GS type 3, patients exhibit generalized hypopigmentation of the skin and hair with no abnormalities of the nervous or immune systems. There are 2 different mutations associated with GS type 3: the first is located on chromosome 2q37.3, causing a mutation in MLPH (melanophilin), and the second is caused by an F-exon deletion in the MYO5A gene.14
Our patient had silvery hair, generalized hypopigmentation of the skin and eyes, and normal central nervous system function with no other ocular involvement and no evidence of recurrent infections of any kind. Light microscopy showed small and regular melanin pigment aggregates in the hair shaft, which differs from the irregular pigment aggregates in GS and ES.
The regular melanin pigment aggregates observed along the hair shaft were consistent with CHS, but other manifestations of this syndrome were absent: ocular, neurologic, hematologic, and immunologic abnormalities with presence of giant intracytoplasmic granules in leukocytes, and giant melanosomes in melanocytes. In our patient, the absence of these features along with the spontaneous repigmentation of the silvery hair, improvement of thyroid function, reversal of hypoalbuminemia, and the chromosomopathy detected make a diagnosis of CHS highly improbable.
We concluded that the silvery hair noted in our patient resulted from the 46,XY,add(2)(p23) chromosomal abnormality. This mutation could affect some of the genes that control the trafficking of melanosomes or could induce hypothyroidism and hypoproteinemia associated with congenital hydrops fetalis (Figure 5).
Hydrops fetalis is a potentially fatal condition characterized by severe edema (swelling) in a fetus or neonate. There are 2 types of hydrops fetalis: immune and nonimmune. Immune hydrops fetalis may develop in an Rh+ fetus with an Rh– mother, as the mother’s immune cells begin to break down the red blood cells of the fetus, resulting in anemia in the fetus with subsequent fetal heart failure, leading to an accumulation of large amounts of fluid in the tissues and organs. Nonimmune hydrops fetalis can occur secondary to diseases that interfere with the fetus’s ability to manage fluid (eg, severe anemia; congenital infections; urinary, lymphatic, heart, or thoracic defects; inborn errors of metabolism; chromosomal abnormalities). Case studies have suggested that congenital hypothyroidism could be a cause of nonimmune hydrops fetalis.16,17 Thyroid hormone deficiency reduces stimulation of adrenergic receptors in the lymphatic system and lungs, thereby decreasing lymph flow and protein efflux to the lymphatic system and decreasing clearance of liquid from the lungs. The final result is lymph vessel engorgement and subsequent leakage of lymphatic fluid to pleural spaces, causing hydrops fetalis and chylothorax.
The 46,XY,add(2)(p23) chromosomal abnormality has not been commonly associated with hypothyroidism and hydrops fetalis. The silvery hair in our patient was transient and spontaneously repigmented to brown over the course of follow-up in conjunction with improved physiologic changes. We concluded that the silvery hair in our patient was induced by his hypoproteinemic status secondary to hydrops fetalis and hypothyroidism.
Conclusion
In addition to CHS, ES, and GS, the differential diagnosis for silvery hair with abnormal skin pigmentation in children should include 46,XY,add(2)(p23) mutation, as was detected in our patient. Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
- White JG. The Chédiak-Higashi syndrome: a possible lysosomal disease. Blood. 1966;28:143-156.
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chédiak-Higashi syndrome. Mol Genet Metab. 1999;68:283-303.
- Kaplan J, De Domenico I, Ward DM. Chédiak-Higashi syndrome. Curr Opin Hematol. 2008;15:22-29.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis [published online December 7, 2006]. Eur J Pediatr. 2007;166:95-109.
- Morrone K, Wang Y, Huizing M, et al. Two novel mutations identified in an African-American child with Chédiak-Higashi syndrome [published online March 24, 2010]. Case Report Med. 2010;2010:967535.
- Ivanovich J, Mallory S, Storer T, et al. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease). Am J Med Genet. 2001;98:313-316.
- Cahali JB, Fernandez SA, Oliveira ZN, et al. Elejalde syndrome: report of a case and review of the literature. Pediatr Dermatol. 2004;21:479-482.
- Bahadoran P, Ortonne JP, Ballotti R, et al. Comment on Elejalde syndrome and relationship with Griscelli syndrome. Am J Med Genet. 2003;116:408-409.
- Duran-McKinster C, Rodriguez-Jurado R, Ridaura C, et al. Elejalde syndrome—a melanolysosomal neurocutaneous syndrome: clinical and morphological findings in 7 patients. Arch Dermatol. 1999;135:182-186.
- Happle R. Neurocutaneous diseases. In: Freedberg IM, Eisen AZ, Wolff K, et al, eds. Dermatology in General Medicine. 5th ed. New York, NY: McGraw-Hill; 1999:2131-2148.
- Sanal O, Yel L, Kucukali T, et al. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder). J Neurol. 2000;247:570-572.
- Malhotra AK, Bhaskar G, Nanda M, et al. Griscelli syndrome. J Am Acad Dermatol. 2006;55:337-340.
- Al-Idrissi E, ElGhazali G, Alzahrani M, et al. Premature birth, respiratory distress, intracerebral hemorrhage, and silvery-gray hair: differential diagnosis of the 3 types of Griscelli syndrome. J Pediatr Hematol Oncol. 2010;32:494-496.
- Ménasché G, Ho CH, Sanal O, et al. Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003;112:450-456.
- Griscelli C, Durandy A, Guy-Grand D, et al. A syndrome associating partial albinism and immunodeficiency. Am J Med. 1978;65:691-702.
- Narchi H. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;104:1416-1417.
- Kessel I, Makhoul IR, Sujov P. Congenital hypothyroidism and nonimmune hydrops fetalis: associated? Pediatrics. 1999;103:E9.
Practice Points
- Silvery hair is characteristic of 3 rare autosomal-recessive disorders: Chédiak-Higashi syndrome, Elejalde syndrome, and Griscelli syndrome.
- Hypopigmentation is the result of impaired melanosome transport leading to failed transfer of melanin to keratinocytes.
- Evaluation should include light microscopy of the hair shaft, skin biopsy, assessment of immune function, peripheral blood smear, and neurologic and eye examinations.
Management of Vitiligo Patients With Surgical Interventions
Vitiligo is a common, asymptomatic, acquired depigmentation disorder that is caused by an unknown etiology. Lesions appear as sharply demarcated, depigmented macules and patches that are scattered symmetrically or unsymmetrically over the body. The presentation can be delineated based on the segmental or nonsegmental nature of the disease. According to the revised classification/nomenclature of vitiligo,1 the disorder can be classified as nonsegmental, segmental, mixed, or unclassified. The pathogenesis of the vitiligo disease process is due to multiple modalities that contribute to melanocyte loss. Theories for melanocyte destruction include but are not limited to autoimmunity, biochemicals, epidermal cytokines, increased hydrogen peroxide and free radicals, and humoral and cellular immune alteration.2,3
Despite its long history, the most frustrating aspect of the vitiligo disease process remains its treatment due to limited efficacy, frequent application of topicals, and the need for high-potency steroids. Medical therapies usually are the first line of treatment and are most effective with few side effects for bilateral nonsegmental or evolving vitiligo.2 Some of the primary therapies with the highest efficacies appear to be calcipotriene and psoralen plus UVA, psoralen plus UVA as monotherapy, excimer laser, narrowband UVB, oral steroids, 8-methoxypsoralen, tacrolimus, and topical steroids.4 The theory is that these treatments would be successful if the patient had active melanocytes in the external root sheath that would be able to repigment a patch of vitiligo.5 Hence, it would be more difficult to treat areas such as the dorsal aspect of the fingers and toes because they lack hair-bearing areas with melanocytes.6 The alternative approach to treating vitiligo patches would be surgical intervention techniques, as they provide melanocytic cells to a previously depigmented area.3,5 The focus of this article is to evaluate the efficacy and appropriate use of some of the surgical procedures that can be used in the treatment of vitiligo patients.
Candidate Selection
First, vitiligo patients for whom first-line treatment with medical therapies has failed are candidates for surgical techniques. The second vital component is to clinically confirm the diagnosis of vitiligo as opposed to other genetic, infectious, or autoimmune causes of pigment loss. Lastly, the vitiligo patch should be stable. A stable vitiligo patch does not continue to progress and is no longer responsive to topical medications that are meant to repigment for a discernible period of time.7
Classification of Disease Stage
To classify the stage of vitiligo prior to surgical intervention, Gauthier8 created a basic grading system: grade I, with partial depletion of epidermal melanocytes in a vitiligo patch that responds to repigmentation in a follicular pattern evenly such as on the face and neck; grade II, with complete depletion of epidermal melanocytes with a usual follicular pattern of repigmentation; and grade III, indicating complete depletion of follicular melanocytes with no hope of response to medical therapy. According to Rusfianti and Wirohadidjodjo,2 the surgical techniques that have developed over the years for treatment of grade III vitiligo patients include split-thickness skin grafting, suction blister grafting, miniature punch grafting, and cultured melanocyte transplantation.
Surgical Techniques
Split-thickness skin grafting is an older procedure that entails the use of a harvesting graft site with no pigment loss and dermabrasion of the recipient area to allow interaction with the wound bed.9 With proper care and minimal movement or wrinkling of the graft site, patients can have repigmentation without skip areas.
Suction blister grafting is another tried and tested surgical intervention. Hasegawa et al10 conducted a study of 15 patients (13 males, 2 females; age range, 16–38 years) diagnosed with segmental vitiligo who were treated using the suction blister grafting technique with CO2 laser resurfacing. Patients were recruited 1 month prior to initiating therapy and no other treatments were used during the month or in conjunction with the surgical intervention. Suction blisters were harvested from the left thigh and transferred in saline to the recipient site, which was abraded with 1 pass of the short-pulse CO2 laser system. The recipient sites were then closed with 7-0 nylon sutures and covered tightly with tie-over dressings for at least 1 week. Within 6 months of the procedure, a treatment response of 100% was seen in 15 patients, making it an effective method for treatment-resistant vitiligo patients.10
Miniature punch grafting is another possible treatment option for resistant cases of vitiligo. Mapar et al11 conducted a study in 25 patients (21 women, 4 men; age range, 20–47 years) who had been diagnosed with stable vitiligo (ie, no progression in the last 2 years) and were treated with single hair follicle transplant versus miniature punch grafting. The theory behind the study was to use the melanocytic reservoir noted in the normal hair follicle to repigment the vitiligo patch. With follow-up of both methods of treatment, there was no statistical difference in treatment results.11 A similar study was conducted by Malakar and Lahiri12 in patients with lip leukoderma (a variant of vitiligo). One hundred eight patients (41 males, 67 females; age range, 14–62 years) who had been diagnosed with stable lip leukoderma (ie, stable vitiligo for at least 6 months) underwent treatment via autologous miniature punch grafting. Punch biopsies were performed in donor sites of the buttocks and upper thighs with 72% of patients noting complete repigmentation. Complications noted were herpes labialis–induced lip leukoderma, which ultimately led to rejection of the graft site.12 Overall, however, miniature punch grafting is a viable surgical option in stable vitiligo patients.
Cultured melanocyte transplantation, or a noncultured epidermal suspension, was first initiated in 1992.13 Silpa-Archa et al14 conducted an open, split-comparison study of 6 vitiligo patients (5 women, 1 man; age range, 20–65 years) with stable lesions. Fifty percent of patients received autologous pigmented skin cellular suspension, which was applied to vitiligo-affected skin that was treated with a fractionated CO2 laser, and 50% received dermabrasion. Composite dressing was placed overlying the site with dressing removal in 1 week. The degree of repigmentation was based on a modified vitiligo area scoring index scale of poor (0%–25%), fair (26%–50%), good (51%–75%), very good (76%–90%), or excellent (91%–100%). Overall repigmentation was very good to excellent in all 6 patients.14 Potentially, this method can far improve the surgical treatment options for future vitiligo patients.
Final Thoughts
Overall, when evaluating surgical interventions for the treatment of vitiligo, careful consideration of the patient’s disease progression, failed therapies, outcome expectations, and repigmentation is warranted prior to initiating any procedure. For appropriate candidates, a range of surgical methodologies has proven to be effective in treatment of stable vitiligo patients.
- Taïeb A, Picardo M; VETF members. The definition and assessment of vitiligo: a consensus report of the Vitiligo European Task Force. Pigment Cell Res. 2007;20:27-35. Cited by: Ezzedine K, Lim HW, Suzuki T, et al; Vitiligo Global Issue Consensus Conference Panelists. Revised classification/nomenclature of vitiligo and related issues: the Vitiligo Global Issues Consensus Conference. Pigment Cell Melanoma Res. 2012;25:E1-E13.
- Rusfianti M, Wirohadidjodjo YW. Dermatosurgical techniques for repigmentation of vitiligo. Int J Dermatol. 2006;45:411-417.
- Falabella R. Surgical therapies for vitiligo. Clin Dermatol. 1997;15:927-939.
- Whitton ME, Pinart M, Batchelor J, et al. Interventions for vitiligo. Cochrane Database Syst Rev. 2015;2:CD003263.
- Mulekar SV, Isedeh P. Surgical interventions for vitiligo: an evidence-based review. Br J Dermatol. 2013;169(suppl 3):57-66.
- Dutta AK, Mandal SB. A clinical study of 650 vitiligo cases and their classification. Indian J Dermatol. 1969;14:103-111.
- Falabella R, Arrunategui A, Barona MI, et al. The minigrafting test for vitiligo: detection of stable lesions for melanocyte transplantation. J Am Acad Dermatol. 1995;32:228-232.
- Gauthier Y. Le vitiligo. Gaz Med. 1994;101:8-12.
- Malakar S, Malakar RS. Surgical pearl: composite film and graft unit for the recipient area dressing after split-thickness skin grafting in vitiligo. J Am Acad Dermatol. 2001;44:856-858.
- Hasegawa T, Suga Y, Ikejima A, et al. Suction blister grafting with CO2 laser resurfacing of the graft recipient site for vitiligo. J Dermatol. 2007;34:490-492.
- Mapar MA, Safarpour M, Mapar M, et al. A comparative study of the mini-punch grafting and hair follicle transplantation in the treatment of refractory and stable vitiligo. J Am Acad Dermatol. 2014;70:743-747.
- Malakar S, Lahiri K. Punch grafting for lip leukoderma. Dermatology. 2004;208:125-128.
- Gauthier Y, Surleve-Bazeille JE. Autologous grafting with noncultured melanocytes: a simplified method for treatment of depigmented lesions. J Am Acad Dermatol. 1992;26(2, pt 1):191-194.
- Silpa-Archa N, Griffith JL, Williams MS, et al. Prospective comparison of recipient-site preparation with fractional carbon dioxide laser versus dermabrasion and recipient-site dressing composition in melanocyte-keratinocyte transplantation procedure in vitiligo: a preliminary study [published online January 24, 2016]. Br J Dermatol. 2016;174:895-897.
Vitiligo is a common, asymptomatic, acquired depigmentation disorder that is caused by an unknown etiology. Lesions appear as sharply demarcated, depigmented macules and patches that are scattered symmetrically or unsymmetrically over the body. The presentation can be delineated based on the segmental or nonsegmental nature of the disease. According to the revised classification/nomenclature of vitiligo,1 the disorder can be classified as nonsegmental, segmental, mixed, or unclassified. The pathogenesis of the vitiligo disease process is due to multiple modalities that contribute to melanocyte loss. Theories for melanocyte destruction include but are not limited to autoimmunity, biochemicals, epidermal cytokines, increased hydrogen peroxide and free radicals, and humoral and cellular immune alteration.2,3
Despite its long history, the most frustrating aspect of the vitiligo disease process remains its treatment due to limited efficacy, frequent application of topicals, and the need for high-potency steroids. Medical therapies usually are the first line of treatment and are most effective with few side effects for bilateral nonsegmental or evolving vitiligo.2 Some of the primary therapies with the highest efficacies appear to be calcipotriene and psoralen plus UVA, psoralen plus UVA as monotherapy, excimer laser, narrowband UVB, oral steroids, 8-methoxypsoralen, tacrolimus, and topical steroids.4 The theory is that these treatments would be successful if the patient had active melanocytes in the external root sheath that would be able to repigment a patch of vitiligo.5 Hence, it would be more difficult to treat areas such as the dorsal aspect of the fingers and toes because they lack hair-bearing areas with melanocytes.6 The alternative approach to treating vitiligo patches would be surgical intervention techniques, as they provide melanocytic cells to a previously depigmented area.3,5 The focus of this article is to evaluate the efficacy and appropriate use of some of the surgical procedures that can be used in the treatment of vitiligo patients.
Candidate Selection
First, vitiligo patients for whom first-line treatment with medical therapies has failed are candidates for surgical techniques. The second vital component is to clinically confirm the diagnosis of vitiligo as opposed to other genetic, infectious, or autoimmune causes of pigment loss. Lastly, the vitiligo patch should be stable. A stable vitiligo patch does not continue to progress and is no longer responsive to topical medications that are meant to repigment for a discernible period of time.7
Classification of Disease Stage
To classify the stage of vitiligo prior to surgical intervention, Gauthier8 created a basic grading system: grade I, with partial depletion of epidermal melanocytes in a vitiligo patch that responds to repigmentation in a follicular pattern evenly such as on the face and neck; grade II, with complete depletion of epidermal melanocytes with a usual follicular pattern of repigmentation; and grade III, indicating complete depletion of follicular melanocytes with no hope of response to medical therapy. According to Rusfianti and Wirohadidjodjo,2 the surgical techniques that have developed over the years for treatment of grade III vitiligo patients include split-thickness skin grafting, suction blister grafting, miniature punch grafting, and cultured melanocyte transplantation.
Surgical Techniques
Split-thickness skin grafting is an older procedure that entails the use of a harvesting graft site with no pigment loss and dermabrasion of the recipient area to allow interaction with the wound bed.9 With proper care and minimal movement or wrinkling of the graft site, patients can have repigmentation without skip areas.
Suction blister grafting is another tried and tested surgical intervention. Hasegawa et al10 conducted a study of 15 patients (13 males, 2 females; age range, 16–38 years) diagnosed with segmental vitiligo who were treated using the suction blister grafting technique with CO2 laser resurfacing. Patients were recruited 1 month prior to initiating therapy and no other treatments were used during the month or in conjunction with the surgical intervention. Suction blisters were harvested from the left thigh and transferred in saline to the recipient site, which was abraded with 1 pass of the short-pulse CO2 laser system. The recipient sites were then closed with 7-0 nylon sutures and covered tightly with tie-over dressings for at least 1 week. Within 6 months of the procedure, a treatment response of 100% was seen in 15 patients, making it an effective method for treatment-resistant vitiligo patients.10
Miniature punch grafting is another possible treatment option for resistant cases of vitiligo. Mapar et al11 conducted a study in 25 patients (21 women, 4 men; age range, 20–47 years) who had been diagnosed with stable vitiligo (ie, no progression in the last 2 years) and were treated with single hair follicle transplant versus miniature punch grafting. The theory behind the study was to use the melanocytic reservoir noted in the normal hair follicle to repigment the vitiligo patch. With follow-up of both methods of treatment, there was no statistical difference in treatment results.11 A similar study was conducted by Malakar and Lahiri12 in patients with lip leukoderma (a variant of vitiligo). One hundred eight patients (41 males, 67 females; age range, 14–62 years) who had been diagnosed with stable lip leukoderma (ie, stable vitiligo for at least 6 months) underwent treatment via autologous miniature punch grafting. Punch biopsies were performed in donor sites of the buttocks and upper thighs with 72% of patients noting complete repigmentation. Complications noted were herpes labialis–induced lip leukoderma, which ultimately led to rejection of the graft site.12 Overall, however, miniature punch grafting is a viable surgical option in stable vitiligo patients.
Cultured melanocyte transplantation, or a noncultured epidermal suspension, was first initiated in 1992.13 Silpa-Archa et al14 conducted an open, split-comparison study of 6 vitiligo patients (5 women, 1 man; age range, 20–65 years) with stable lesions. Fifty percent of patients received autologous pigmented skin cellular suspension, which was applied to vitiligo-affected skin that was treated with a fractionated CO2 laser, and 50% received dermabrasion. Composite dressing was placed overlying the site with dressing removal in 1 week. The degree of repigmentation was based on a modified vitiligo area scoring index scale of poor (0%–25%), fair (26%–50%), good (51%–75%), very good (76%–90%), or excellent (91%–100%). Overall repigmentation was very good to excellent in all 6 patients.14 Potentially, this method can far improve the surgical treatment options for future vitiligo patients.
Final Thoughts
Overall, when evaluating surgical interventions for the treatment of vitiligo, careful consideration of the patient’s disease progression, failed therapies, outcome expectations, and repigmentation is warranted prior to initiating any procedure. For appropriate candidates, a range of surgical methodologies has proven to be effective in treatment of stable vitiligo patients.
Vitiligo is a common, asymptomatic, acquired depigmentation disorder that is caused by an unknown etiology. Lesions appear as sharply demarcated, depigmented macules and patches that are scattered symmetrically or unsymmetrically over the body. The presentation can be delineated based on the segmental or nonsegmental nature of the disease. According to the revised classification/nomenclature of vitiligo,1 the disorder can be classified as nonsegmental, segmental, mixed, or unclassified. The pathogenesis of the vitiligo disease process is due to multiple modalities that contribute to melanocyte loss. Theories for melanocyte destruction include but are not limited to autoimmunity, biochemicals, epidermal cytokines, increased hydrogen peroxide and free radicals, and humoral and cellular immune alteration.2,3
Despite its long history, the most frustrating aspect of the vitiligo disease process remains its treatment due to limited efficacy, frequent application of topicals, and the need for high-potency steroids. Medical therapies usually are the first line of treatment and are most effective with few side effects for bilateral nonsegmental or evolving vitiligo.2 Some of the primary therapies with the highest efficacies appear to be calcipotriene and psoralen plus UVA, psoralen plus UVA as monotherapy, excimer laser, narrowband UVB, oral steroids, 8-methoxypsoralen, tacrolimus, and topical steroids.4 The theory is that these treatments would be successful if the patient had active melanocytes in the external root sheath that would be able to repigment a patch of vitiligo.5 Hence, it would be more difficult to treat areas such as the dorsal aspect of the fingers and toes because they lack hair-bearing areas with melanocytes.6 The alternative approach to treating vitiligo patches would be surgical intervention techniques, as they provide melanocytic cells to a previously depigmented area.3,5 The focus of this article is to evaluate the efficacy and appropriate use of some of the surgical procedures that can be used in the treatment of vitiligo patients.
Candidate Selection
First, vitiligo patients for whom first-line treatment with medical therapies has failed are candidates for surgical techniques. The second vital component is to clinically confirm the diagnosis of vitiligo as opposed to other genetic, infectious, or autoimmune causes of pigment loss. Lastly, the vitiligo patch should be stable. A stable vitiligo patch does not continue to progress and is no longer responsive to topical medications that are meant to repigment for a discernible period of time.7
Classification of Disease Stage
To classify the stage of vitiligo prior to surgical intervention, Gauthier8 created a basic grading system: grade I, with partial depletion of epidermal melanocytes in a vitiligo patch that responds to repigmentation in a follicular pattern evenly such as on the face and neck; grade II, with complete depletion of epidermal melanocytes with a usual follicular pattern of repigmentation; and grade III, indicating complete depletion of follicular melanocytes with no hope of response to medical therapy. According to Rusfianti and Wirohadidjodjo,2 the surgical techniques that have developed over the years for treatment of grade III vitiligo patients include split-thickness skin grafting, suction blister grafting, miniature punch grafting, and cultured melanocyte transplantation.
Surgical Techniques
Split-thickness skin grafting is an older procedure that entails the use of a harvesting graft site with no pigment loss and dermabrasion of the recipient area to allow interaction with the wound bed.9 With proper care and minimal movement or wrinkling of the graft site, patients can have repigmentation without skip areas.
Suction blister grafting is another tried and tested surgical intervention. Hasegawa et al10 conducted a study of 15 patients (13 males, 2 females; age range, 16–38 years) diagnosed with segmental vitiligo who were treated using the suction blister grafting technique with CO2 laser resurfacing. Patients were recruited 1 month prior to initiating therapy and no other treatments were used during the month or in conjunction with the surgical intervention. Suction blisters were harvested from the left thigh and transferred in saline to the recipient site, which was abraded with 1 pass of the short-pulse CO2 laser system. The recipient sites were then closed with 7-0 nylon sutures and covered tightly with tie-over dressings for at least 1 week. Within 6 months of the procedure, a treatment response of 100% was seen in 15 patients, making it an effective method for treatment-resistant vitiligo patients.10
Miniature punch grafting is another possible treatment option for resistant cases of vitiligo. Mapar et al11 conducted a study in 25 patients (21 women, 4 men; age range, 20–47 years) who had been diagnosed with stable vitiligo (ie, no progression in the last 2 years) and were treated with single hair follicle transplant versus miniature punch grafting. The theory behind the study was to use the melanocytic reservoir noted in the normal hair follicle to repigment the vitiligo patch. With follow-up of both methods of treatment, there was no statistical difference in treatment results.11 A similar study was conducted by Malakar and Lahiri12 in patients with lip leukoderma (a variant of vitiligo). One hundred eight patients (41 males, 67 females; age range, 14–62 years) who had been diagnosed with stable lip leukoderma (ie, stable vitiligo for at least 6 months) underwent treatment via autologous miniature punch grafting. Punch biopsies were performed in donor sites of the buttocks and upper thighs with 72% of patients noting complete repigmentation. Complications noted were herpes labialis–induced lip leukoderma, which ultimately led to rejection of the graft site.12 Overall, however, miniature punch grafting is a viable surgical option in stable vitiligo patients.
Cultured melanocyte transplantation, or a noncultured epidermal suspension, was first initiated in 1992.13 Silpa-Archa et al14 conducted an open, split-comparison study of 6 vitiligo patients (5 women, 1 man; age range, 20–65 years) with stable lesions. Fifty percent of patients received autologous pigmented skin cellular suspension, which was applied to vitiligo-affected skin that was treated with a fractionated CO2 laser, and 50% received dermabrasion. Composite dressing was placed overlying the site with dressing removal in 1 week. The degree of repigmentation was based on a modified vitiligo area scoring index scale of poor (0%–25%), fair (26%–50%), good (51%–75%), very good (76%–90%), or excellent (91%–100%). Overall repigmentation was very good to excellent in all 6 patients.14 Potentially, this method can far improve the surgical treatment options for future vitiligo patients.
Final Thoughts
Overall, when evaluating surgical interventions for the treatment of vitiligo, careful consideration of the patient’s disease progression, failed therapies, outcome expectations, and repigmentation is warranted prior to initiating any procedure. For appropriate candidates, a range of surgical methodologies has proven to be effective in treatment of stable vitiligo patients.
- Taïeb A, Picardo M; VETF members. The definition and assessment of vitiligo: a consensus report of the Vitiligo European Task Force. Pigment Cell Res. 2007;20:27-35. Cited by: Ezzedine K, Lim HW, Suzuki T, et al; Vitiligo Global Issue Consensus Conference Panelists. Revised classification/nomenclature of vitiligo and related issues: the Vitiligo Global Issues Consensus Conference. Pigment Cell Melanoma Res. 2012;25:E1-E13.
- Rusfianti M, Wirohadidjodjo YW. Dermatosurgical techniques for repigmentation of vitiligo. Int J Dermatol. 2006;45:411-417.
- Falabella R. Surgical therapies for vitiligo. Clin Dermatol. 1997;15:927-939.
- Whitton ME, Pinart M, Batchelor J, et al. Interventions for vitiligo. Cochrane Database Syst Rev. 2015;2:CD003263.
- Mulekar SV, Isedeh P. Surgical interventions for vitiligo: an evidence-based review. Br J Dermatol. 2013;169(suppl 3):57-66.
- Dutta AK, Mandal SB. A clinical study of 650 vitiligo cases and their classification. Indian J Dermatol. 1969;14:103-111.
- Falabella R, Arrunategui A, Barona MI, et al. The minigrafting test for vitiligo: detection of stable lesions for melanocyte transplantation. J Am Acad Dermatol. 1995;32:228-232.
- Gauthier Y. Le vitiligo. Gaz Med. 1994;101:8-12.
- Malakar S, Malakar RS. Surgical pearl: composite film and graft unit for the recipient area dressing after split-thickness skin grafting in vitiligo. J Am Acad Dermatol. 2001;44:856-858.
- Hasegawa T, Suga Y, Ikejima A, et al. Suction blister grafting with CO2 laser resurfacing of the graft recipient site for vitiligo. J Dermatol. 2007;34:490-492.
- Mapar MA, Safarpour M, Mapar M, et al. A comparative study of the mini-punch grafting and hair follicle transplantation in the treatment of refractory and stable vitiligo. J Am Acad Dermatol. 2014;70:743-747.
- Malakar S, Lahiri K. Punch grafting for lip leukoderma. Dermatology. 2004;208:125-128.
- Gauthier Y, Surleve-Bazeille JE. Autologous grafting with noncultured melanocytes: a simplified method for treatment of depigmented lesions. J Am Acad Dermatol. 1992;26(2, pt 1):191-194.
- Silpa-Archa N, Griffith JL, Williams MS, et al. Prospective comparison of recipient-site preparation with fractional carbon dioxide laser versus dermabrasion and recipient-site dressing composition in melanocyte-keratinocyte transplantation procedure in vitiligo: a preliminary study [published online January 24, 2016]. Br J Dermatol. 2016;174:895-897.
- Taïeb A, Picardo M; VETF members. The definition and assessment of vitiligo: a consensus report of the Vitiligo European Task Force. Pigment Cell Res. 2007;20:27-35. Cited by: Ezzedine K, Lim HW, Suzuki T, et al; Vitiligo Global Issue Consensus Conference Panelists. Revised classification/nomenclature of vitiligo and related issues: the Vitiligo Global Issues Consensus Conference. Pigment Cell Melanoma Res. 2012;25:E1-E13.
- Rusfianti M, Wirohadidjodjo YW. Dermatosurgical techniques for repigmentation of vitiligo. Int J Dermatol. 2006;45:411-417.
- Falabella R. Surgical therapies for vitiligo. Clin Dermatol. 1997;15:927-939.
- Whitton ME, Pinart M, Batchelor J, et al. Interventions for vitiligo. Cochrane Database Syst Rev. 2015;2:CD003263.
- Mulekar SV, Isedeh P. Surgical interventions for vitiligo: an evidence-based review. Br J Dermatol. 2013;169(suppl 3):57-66.
- Dutta AK, Mandal SB. A clinical study of 650 vitiligo cases and their classification. Indian J Dermatol. 1969;14:103-111.
- Falabella R, Arrunategui A, Barona MI, et al. The minigrafting test for vitiligo: detection of stable lesions for melanocyte transplantation. J Am Acad Dermatol. 1995;32:228-232.
- Gauthier Y. Le vitiligo. Gaz Med. 1994;101:8-12.
- Malakar S, Malakar RS. Surgical pearl: composite film and graft unit for the recipient area dressing after split-thickness skin grafting in vitiligo. J Am Acad Dermatol. 2001;44:856-858.
- Hasegawa T, Suga Y, Ikejima A, et al. Suction blister grafting with CO2 laser resurfacing of the graft recipient site for vitiligo. J Dermatol. 2007;34:490-492.
- Mapar MA, Safarpour M, Mapar M, et al. A comparative study of the mini-punch grafting and hair follicle transplantation in the treatment of refractory and stable vitiligo. J Am Acad Dermatol. 2014;70:743-747.
- Malakar S, Lahiri K. Punch grafting for lip leukoderma. Dermatology. 2004;208:125-128.
- Gauthier Y, Surleve-Bazeille JE. Autologous grafting with noncultured melanocytes: a simplified method for treatment of depigmented lesions. J Am Acad Dermatol. 1992;26(2, pt 1):191-194.
- Silpa-Archa N, Griffith JL, Williams MS, et al. Prospective comparison of recipient-site preparation with fractional carbon dioxide laser versus dermabrasion and recipient-site dressing composition in melanocyte-keratinocyte transplantation procedure in vitiligo: a preliminary study [published online January 24, 2016]. Br J Dermatol. 2016;174:895-897.
