User login
Practical tips help quell pseudofolliculitis barbae
LAS VEGAS – Stubble is okay, and not just because it’s trendy to sport a beard. This was a key message during a presentation on pseudofolliculitis barbae at Skin Disease Education Foundation’s annual Las Vegas dermatology seminar.
Changing up personal grooming habits is an important tactic for men who are plagued with pseudofolliculitis barbae, according to Dr. Andrew F. Alexis, chair of the department of dermatology and director of The Skin of Color Center, Mount Sinai St. Luke’s and Mount Sinai Roosevelt, New York.

This very common skin condition, affecting 45%-83% of men of African ancestry, is best managed by avoiding close shaving and preventing a sharp hair shaft tip. For those who don’t want a full beard for personal or professional reasons, using single blade razors, electric clippers, and even depilatories can help, he said.
All of these techniques prevent curly beard hairs from repenetrating or recurving before emergence – the underpinning of the pathology of pseudofolliculitis barbae. The embedded hairs eventually form a papular or pustular lesion that mimics infectious folliculitis. The inflammatory process can also prompt keloid formation in susceptible individuals.
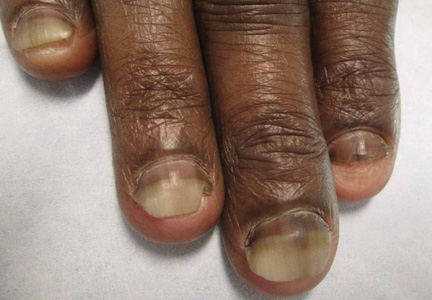
Providing treatment options is important because the condition can be disfiguring, with such long-term physical sequelae as scarring beard alopecia and postinflammatory hyperpigmentation – changes in appearance that can have a significant psychosocial impact on affected men, Dr. Alexis said.
Therapies are centered around avoiding close shaving and/or preventing a sharp hair shaft tip.
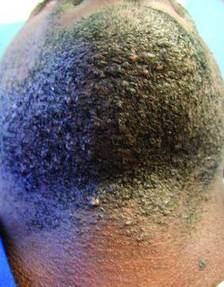
One primary treatment is to stop shaving. “Embedded hairs spontaneously release after about one centimeter of growth,” Dr. Alexis said. This process can take up to 2 months, he said, but military studies dating back to the 1970s showed that the vast majority of pseudofolliculitis barbae cases resolved when service members stopped close shaving practices.
However, many patients want a clean-shaven appearance. “We can work with them to modify their shaving practices. Historically, we have recommended single-blade razors over multiple blade razors” because they shave less closely, he said, pointing out that razor manufacturers have funded studies that challenge this finding.
“Electric clippers are a very good alternative” to razors, Dr. Alexis said. A blade setting that allows at least 0.5-1 mm stubble is desirable.
Chemical depilatories, which act by weakening keratin disulfide bonds, can be effective, since depilated hair does not have a sharp, beveled tip on regrowth and is therefore less likely to repuncture the skin. Patients should be aware, though, that these substances can cause irritant contact dermatitis, he pointed out. Newer formulations are less caustic, but also less efficacious, he said.
In terms of practical tips, shaving technique is important. “Don’t assume the patient knows. There are all sorts of varying techniques out there,” some of which can exacerbate pseudofolliculitis barbae, Dr. Alexis said.
Before shaving, men should wash with a mild cleanser, using a gentle circular technique to free any entrapped hairs, then a moisturizing shaving cream. Razors should be changed every five to seven shaves, and shaving should always be done in the direction of beard growth without pulling on the skin.
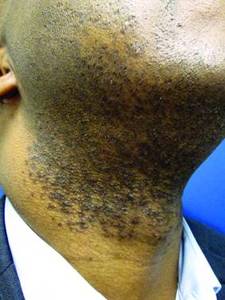
Post shave, topical benzoyl peroxide 5%/clindamycin 1% can significantly reduce papules and pustules. Topical retinoids are another effective option. A low-potency steroid can be helpful for inflammatory symptoms.
For cases that just don’t respond to conservative and medical management, laser hair removal is an option. A recent military-funded split-face study found further improvement when topical eflornithine was added to long-pulse Nd:Yag laser therapy, Dr. Alexis said.
Affected individuals may find it difficult to modify shaving practices when uniformed service regulations or office dress codes require men to be close shaven; a note from a physician can be helpful. Dr. Alexis provides patients with a form letter to show their employers, explaining that the patient has a skin disorder that is exacerbated by shaving, and that the patient should be permitted to maintain a well-groomed beard. “I end up writing a lot of these for New York police officers,” he said.
Dr. Alexis disclosed that he has received grants and research support from Allergan and Novartis, and speaker honoraria from Cipla. He has received consulting fees from Aclaris, Allergan, Amgen, Anacor, Bayer, Galderma, Johnson & Johnson, Leo, L’Oreal, Roche, Schick, Suneva, and Valeant.
SDEF and this news organization are owned by the same parent company.
On Twitter @karioakes
LAS VEGAS – Stubble is okay, and not just because it’s trendy to sport a beard. This was a key message during a presentation on pseudofolliculitis barbae at Skin Disease Education Foundation’s annual Las Vegas dermatology seminar.
Changing up personal grooming habits is an important tactic for men who are plagued with pseudofolliculitis barbae, according to Dr. Andrew F. Alexis, chair of the department of dermatology and director of The Skin of Color Center, Mount Sinai St. Luke’s and Mount Sinai Roosevelt, New York.
This very common skin condition, affecting 45%-83% of men of African ancestry, is best managed by avoiding close shaving and preventing a sharp hair shaft tip. For those who don’t want a full beard for personal or professional reasons, using single blade razors, electric clippers, and even depilatories can help, he said.
All of these techniques prevent curly beard hairs from repenetrating or recurving before emergence – the underpinning of the pathology of pseudofolliculitis barbae. The embedded hairs eventually form a papular or pustular lesion that mimics infectious folliculitis. The inflammatory process can also prompt keloid formation in susceptible individuals.
Providing treatment options is important because the condition can be disfiguring, with such long-term physical sequelae as scarring beard alopecia and postinflammatory hyperpigmentation – changes in appearance that can have a significant psychosocial impact on affected men, Dr. Alexis said.
Therapies are centered around avoiding close shaving and/or preventing a sharp hair shaft tip.
One primary treatment is to stop shaving. “Embedded hairs spontaneously release after about one centimeter of growth,” Dr. Alexis said. This process can take up to 2 months, he said, but military studies dating back to the 1970s showed that the vast majority of pseudofolliculitis barbae cases resolved when service members stopped close shaving practices.
However, many patients want a clean-shaven appearance. “We can work with them to modify their shaving practices. Historically, we have recommended single-blade razors over multiple blade razors” because they shave less closely, he said, pointing out that razor manufacturers have funded studies that challenge this finding.
“Electric clippers are a very good alternative” to razors, Dr. Alexis said. A blade setting that allows at least 0.5-1 mm stubble is desirable.
Chemical depilatories, which act by weakening keratin disulfide bonds, can be effective, since depilated hair does not have a sharp, beveled tip on regrowth and is therefore less likely to repuncture the skin. Patients should be aware, though, that these substances can cause irritant contact dermatitis, he pointed out. Newer formulations are less caustic, but also less efficacious, he said.
In terms of practical tips, shaving technique is important. “Don’t assume the patient knows. There are all sorts of varying techniques out there,” some of which can exacerbate pseudofolliculitis barbae, Dr. Alexis said.
Before shaving, men should wash with a mild cleanser, using a gentle circular technique to free any entrapped hairs, then a moisturizing shaving cream. Razors should be changed every five to seven shaves, and shaving should always be done in the direction of beard growth without pulling on the skin.
Post shave, topical benzoyl peroxide 5%/clindamycin 1% can significantly reduce papules and pustules. Topical retinoids are another effective option. A low-potency steroid can be helpful for inflammatory symptoms.
For cases that just don’t respond to conservative and medical management, laser hair removal is an option. A recent military-funded split-face study found further improvement when topical eflornithine was added to long-pulse Nd:Yag laser therapy, Dr. Alexis said.
Affected individuals may find it difficult to modify shaving practices when uniformed service regulations or office dress codes require men to be close shaven; a note from a physician can be helpful. Dr. Alexis provides patients with a form letter to show their employers, explaining that the patient has a skin disorder that is exacerbated by shaving, and that the patient should be permitted to maintain a well-groomed beard. “I end up writing a lot of these for New York police officers,” he said.
Dr. Alexis disclosed that he has received grants and research support from Allergan and Novartis, and speaker honoraria from Cipla. He has received consulting fees from Aclaris, Allergan, Amgen, Anacor, Bayer, Galderma, Johnson & Johnson, Leo, L’Oreal, Roche, Schick, Suneva, and Valeant.
SDEF and this news organization are owned by the same parent company.
On Twitter @karioakes
LAS VEGAS – Stubble is okay, and not just because it’s trendy to sport a beard. This was a key message during a presentation on pseudofolliculitis barbae at Skin Disease Education Foundation’s annual Las Vegas dermatology seminar.
Changing up personal grooming habits is an important tactic for men who are plagued with pseudofolliculitis barbae, according to Dr. Andrew F. Alexis, chair of the department of dermatology and director of The Skin of Color Center, Mount Sinai St. Luke’s and Mount Sinai Roosevelt, New York.
This very common skin condition, affecting 45%-83% of men of African ancestry, is best managed by avoiding close shaving and preventing a sharp hair shaft tip. For those who don’t want a full beard for personal or professional reasons, using single blade razors, electric clippers, and even depilatories can help, he said.
All of these techniques prevent curly beard hairs from repenetrating or recurving before emergence – the underpinning of the pathology of pseudofolliculitis barbae. The embedded hairs eventually form a papular or pustular lesion that mimics infectious folliculitis. The inflammatory process can also prompt keloid formation in susceptible individuals.
Providing treatment options is important because the condition can be disfiguring, with such long-term physical sequelae as scarring beard alopecia and postinflammatory hyperpigmentation – changes in appearance that can have a significant psychosocial impact on affected men, Dr. Alexis said.
Therapies are centered around avoiding close shaving and/or preventing a sharp hair shaft tip.
One primary treatment is to stop shaving. “Embedded hairs spontaneously release after about one centimeter of growth,” Dr. Alexis said. This process can take up to 2 months, he said, but military studies dating back to the 1970s showed that the vast majority of pseudofolliculitis barbae cases resolved when service members stopped close shaving practices.
However, many patients want a clean-shaven appearance. “We can work with them to modify their shaving practices. Historically, we have recommended single-blade razors over multiple blade razors” because they shave less closely, he said, pointing out that razor manufacturers have funded studies that challenge this finding.
“Electric clippers are a very good alternative” to razors, Dr. Alexis said. A blade setting that allows at least 0.5-1 mm stubble is desirable.
Chemical depilatories, which act by weakening keratin disulfide bonds, can be effective, since depilated hair does not have a sharp, beveled tip on regrowth and is therefore less likely to repuncture the skin. Patients should be aware, though, that these substances can cause irritant contact dermatitis, he pointed out. Newer formulations are less caustic, but also less efficacious, he said.
In terms of practical tips, shaving technique is important. “Don’t assume the patient knows. There are all sorts of varying techniques out there,” some of which can exacerbate pseudofolliculitis barbae, Dr. Alexis said.
Before shaving, men should wash with a mild cleanser, using a gentle circular technique to free any entrapped hairs, then a moisturizing shaving cream. Razors should be changed every five to seven shaves, and shaving should always be done in the direction of beard growth without pulling on the skin.
Post shave, topical benzoyl peroxide 5%/clindamycin 1% can significantly reduce papules and pustules. Topical retinoids are another effective option. A low-potency steroid can be helpful for inflammatory symptoms.
For cases that just don’t respond to conservative and medical management, laser hair removal is an option. A recent military-funded split-face study found further improvement when topical eflornithine was added to long-pulse Nd:Yag laser therapy, Dr. Alexis said.
Affected individuals may find it difficult to modify shaving practices when uniformed service regulations or office dress codes require men to be close shaven; a note from a physician can be helpful. Dr. Alexis provides patients with a form letter to show their employers, explaining that the patient has a skin disorder that is exacerbated by shaving, and that the patient should be permitted to maintain a well-groomed beard. “I end up writing a lot of these for New York police officers,” he said.
Dr. Alexis disclosed that he has received grants and research support from Allergan and Novartis, and speaker honoraria from Cipla. He has received consulting fees from Aclaris, Allergan, Amgen, Anacor, Bayer, Galderma, Johnson & Johnson, Leo, L’Oreal, Roche, Schick, Suneva, and Valeant.
SDEF and this news organization are owned by the same parent company.
On Twitter @karioakes
EXPERT ANALYSIS FROM SDEF LAS VEGAS DERMATOLOGY SEMINAR

A Novel Cream Formulation Containing Nicotinamide 4%, Arbutin 3%, Bisabolol 1%, and Retinaldehyde 0.05% for Treatment of Epidermal Melasma
Epidermal melasma is a common hyperpigmentation disorder that can be challenging to treat. The pathogenesis of melasma is not fully understood but has been associated with increased melanin and melanocyte activity.1,2 Melasma is characterized by jagged, light- to dark-brown patches on areas of the skin most often exposed to the sun—primarily the cheeks, forehead, upper lip, nose, and chin.3 Although it can affect both sexes and all races, melasma is more common in Fitzpatrick skin types II to IV and frequently is seen in Asian or Hispanic women residing in geographic locations with high levels of sun exposure (eg, tropical areas).2 Melasma presents more frequently in adult women of childbearing age, especially during pregnancy, but also can begin postmenopause. Onset may occur as early as menarche but typically is observed between the ages of 30 and 55 years.3,4 Only 10% of melasma cases are known to occur in males4 and are influenced by such factors as ethnicity, hormones, and level of sun exposure.2
Topical therapies for melasma attempt to inhibit melanocytic activation at each level of melanin formation until the deposited pigment is removed; however, results may vary greatly, as melasma often recurs due to the migration of new melanocytes from hair follicles to the skin’s surface, leading to new development of hyperpigmentation. The current standard of treatment for melasma involves the use of hydroquinone and other bleaching agents, but long-term use of these treatments has been associated with concerns regarding unstable preparations (which may lose their therapeutic properties) and adverse effects (eg, ochronosis, depigmentation).5 Cosmetic agents that recently have been evaluated for melasma treatment include nicotinamide (a form of vitamin B3), which inhibits the transfer of melanosomes from melanocytes to keratinocytes; arbutin, which inhibits melanin synthesis by inhibiting tyrosinase activity6; bisabolol, which prevents anti-inflammatory activity7; and retinaldehyde (RAL), a precursor of retinoic acid (RA) that has powerful bleaching action and low levels of cutaneous irritability.8
This prospective, single-arm, open-label study, evaluated the efficacy and safety of a novel cream formulation containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and retinaldehyde 0.05% in the treatment of epidermal melasma.
Study Product Ingredients and Background
Nicotinamide
Nicotinamide is a water-soluble amide of nicotinic acid (niacin) and one of the 2 principal forms of vitamin B3. It is a component of the coenzymes nicotinamide adenine dinucleotide and nicotinamide adenine dinucleotide phosphate. Nicotinamide essentially acts as an antioxidant, with most of its effects exerted through poly(adenosine diphosphate–ribose) polymerase inhibition. Interest has increased in the role of nicotinamide in the prevention and treatment of several skin diseases, such as acne and UV radiation–induced deleterious molecular and immunological events. Nicotinamide also has gained consideration as a potential agent in sunscreen preparations due to its possible skin-lightening effects, stimulation of DNA repair, suppression of UV photocarcinogenesis, and other antiaging effects.9
Arbutin
Arbutin is a molecule that has proven effective in treating melasma.10 Its pigment-lightening ingredients include botanicals that are structurally similar to hydroquinone. Arbutin is obtained from the leaves of the bearberry plant but also is found in lesser quantities in cranberry and blueberry leaves. A naturally occurring gluconopyranoside, arbutin reduces tyrosinase activity without affecting messenger RNA expression.11 Arbutin also inhibits melanosome maturation, is nontoxic to melanocytes, and is used in Japan in a variety of pigment-lightening preparations at 3% concentrations.12
Bisabolol
Bisabolol is a natural monocyclic sesquiterpene alcohol found in the oils of chamomile and other plants. Bisabolol often is included in cosmetics due to its favorable anti-inflammatory and depigmentation properties. Its downregulation of inducible nitric oxide synthase and cyclooxygenase-2 suggests that it may have anti-inflammatory effects.7
Retinaldehyde
Retinaldehyde is an RA precursor that forms as an intermediate metabolite in the transformation of retinol to RA in human keratinocytes. Topical RAL is well tolerated by human skin, and several of its biologic effects are identical to those of RA. Using the tails of C57BL/6 mouse models, RAL 0.05% has been found to have significantly more potent depigmenting effects than RA 0.05% (P<.001 vs P<.01, respectively) when compared to vehicle.13
Although combination therapy with RAL and arbutin could potentially cause skin irritation, the addition of bisabolol to the combination cream used in this study is believed to have conferred anti-inflammatory properties because it inhibits the release of histamine and relieves irritation.
Methods
This single-center, single-arm, prospective, open-label study evaluated the efficacy and safety of a novel cream formulation containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and RAL 0.05% in treating epidermal melasma. Clinical evaluation included assessment of Melasma Area and Severity Index (MASI) score, photographic analysis, and in vivo reflectance confocal microscopy (RCM) analysis.
The study population included women aged 18 to 50 years with Fitzpatrick skin types I through V who had clinically diagnosed epidermal melasma on the face. Eligibility requirements included confirmation of epidermal pigmentation on Wood lamp examination and RCM analysis and a MASI score of less than 10.5. A total of 35 participants were enrolled in the study (intention to treat [ITT] population). Thirty-three participants were included in the analysis of treatment effectiveness (ITTe population), as 2 were excluded due to lack of follow-up postbaseline. Four participants were prematurely withdrawn from the study—3 due to loss to follow-up and 1 due to treatment discontinuation following an adverse event (AE). The last observation carried forward method was used to input missing data from these 4 participants excluding repeated measure analysis that used the generalized estimated equation method.
At baseline, a 25-g tube of the study cream containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and RAL 0.05% was distributed to all participants for once-daily application to the entire face for 30 days. Participants were instructed to apply the product in the evening after using a gentle cleanser, which also was to be used in the morning to remove the product residue. Additionally, participants were given a sunscreen with a sun protection factor of 30 to apply daily on the entire face in the morning, after lunch, and midafternoon. During the 30-day treatment period, treatment interruption of up to 5 consecutive days or 10 nonconsecutive days in total was permitted. At day 30, participants received another 30-day supply of the study product and sunscreen to be applied according to the same regimen for an additional 30-day treatment period.
Clinical Evaluation
At baseline, demographic data and medical history was recorded for all participants and dermatologic and physical examination was performed documenting weight, height, blood pressure, heart rate, and baseline MASI score. Following Wood lamp examination, participants’ faces were photographed and catalogued using medical imaging software that allowed for measurement of the total melasma surface area (Figure 1A). The photographs also were cross-polarized for further analysis of the pigmentation (Figure 1B).

| 
| |
Figure 1. Clinical (A) and cross-polarized (B) photographs of a patient before treatment with the novel compound containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and retinaldehyde 0.05%. | ||
A questionnaire evaluating treatment satisfaction was administered to participants (ITTe population [n=33]) at baseline and days 30 and 60. Questionnaire items pertained to skin blemishes, signs of facial aging, overall appearance, texture, oiliness, brightness, and hydration. Participants were instructed to rate their satisfaction for each item on a scale of 1 to 10 (1=bad, 10=excellent). For investigator analysis, scores of 1 to 4 were classified as “dissatisfied,” scores of 5 to 6 were classified as “satisfied,” and scores of 7 to 10 were classified as “completely satisfied.” A questionnaire evaluating product appreciation was administered at day 60 to participants who completed the study (n=29). Questionnaire items asked participants to rate the study cream’s ease of application, consistency, smell, absorption, and overall satisfaction using ratings of “bad,” “regular,” “good,” “very good,” or “excellent.”
Treatment efficacy in all participants was assessed by the investigators at days 30 and 60. Investigators evaluated reductions in pigmentation and total melasma surface area using ratings of “none,” “regular,” “good,” “very good,” or “excellent.” Local tolerance also was evaluated at both time points, and AEs were recorded and analyzed with respect to their duration, intensity, frequency, and severity.
Targeted hyperpigmented skin was selected for in vivo RCM analysis. At each time point, a sequence of block images was acquired at 4 levels of skin: (1) superficial dermis, (2) suprabasal layer/ dermoepidermal junction, (3) spinous layer, and (4) superficial granular layer. Blind evaluation of these images to assess the reduction in melanin quantity was conducted by a dermatopathologist at baseline and days 30 and 60. Melanin quantity present in each layer was graded according to 4 categories (0%–25%, 25.1%–50%, 50.1%–75%, 75.1%–100%). The mean value was used for statistical evaluation.
Results
Efficacy evaluation
The primary efficacy variable was the mean reduction in MASI score from baseline to the end of treatment (day 60), which was 2.25 ± 1.87 (P<.0001). The reduction in mean MASI score was significant from baseline to day 30 (P<.0001) and from day 30 to day 60 (P<.0001). The least root-mean-square error estimates of MASI score variation at days 30 and 60 were 1.40 and 2.25, respectively.
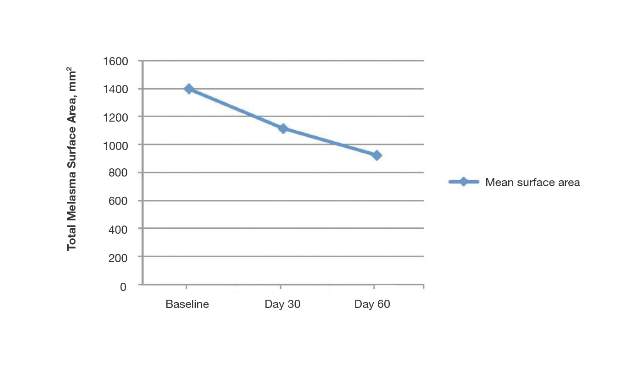
The mean total melasma surface area (as measured in analysis of clinical photographs using medical imaging software) was significantly reduced from 1398.5 mm2 at baseline to 1116.9 mm2 at day 30 (P<.0001) and 923.4 at day 60 (P<.0001). From baseline to end of treatment, the overall reduction in mean total melasma surface area was 475.1 mm2 (P<.0001)(Figure 2). Clinical and cross-polarized photographs taken at day 60 demonstrated a visible reduction in melasma surface area (Figure 3), which was confirmed using medical imaging software.

| 
| |
Figure 3. Clinical (A) and cross-polarized (B) photographs of a patient after 60 days of treatment with the novel compound containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and retinaldehyde 0.05%. | ||
In vivo RCM analyses at each time point showed reduction in pigmentation in the 4 levels of the skin that were evaluated, but the results were not statistically significant.
Participant satisfaction
There was strong statistical evidence of patient satisfaction with the treatment results at the end of the study period (P<.0001). At baseline, 75.8% (25/33) of participants were dissatisfied with the appearance of their skin as compared with 15.2% (5/33) at day 60. Additionally, 18.1% (6/33) and 6.1% (2/33) of the participants were satisfied and completely satisfied at baseline compared with 33.3% (11/33) and 51.5% (17/33) at day 60, respectively. Participant satisfaction with signs of facial aging also increased over the study period (P=.0104). At baseline, 60.6% (20/33) were dissatisfied, 12.1% (4/33) were satisfied, and 27.3% (9/33) were completely satisfied; at the end of treatment, 30.3% (10/33) were dissatisfied, 36.4% (12/33) were satisfied, and 33.3% (11/33) were completely satisfied with the improvement in signs of facial aging.
Increased patient satisfaction with facial skin texture at baseline compared to day 60 also was statistically significant (P=.0157). At baseline, 39.4% (13/33) of the participants were dissatisfied, 30.3% (10/33) were satisfied, and 30.3% (10/33) were completely satisfied with facial texture; at day 60, 15.1% (5/33) were dissatisfied, 30.3% (10/33) were satisfied, and 54.6% (18/33) were completely satisfied. Significant improvement from baseline to day 60 also was observed in participant assessment of skin oiliness (P=.0210), brightness (P=.0003), overall appearance (P<.0001), and hydration (P<.0001).
Product appreciation
At day 60, 89.7% (26/29) of the participants who completed the study rated the product’s ease of application as being at least “good,” with more than half of participants (55.2% [16/29]) rating it as “very good” or “excellent.” Overall satisfaction with the product was rated as “very good” or “excellent” by 48.3% (14/29) of the participants. Similar results were observed in participant assessments of consistency, smell, and absorption (Figure 4).

Safety evaluation
A total of 52 AEs were observed in 23 (69.7%) participants, which were recorded by participants in diary entries throughout treatment and evaluated by investigators at each time point. Among these AEs, 48 (92.3%) were considered possibly, probably, or conditionally related to treatment by the investigators based on clinical observation. The most common presumed treatment-related AE was a burning sensation on the skin, reported by 30.3% (10/33) of the participants at day 30 and 13.8% (4/29) at day 60. Of the reported AEs related to treatment, 91.7% (44/48) were of mild intensity and 93.8% (45/48) required no treatment or other action. There were no reported serious AEs related to the investigational product. Blood pressure, heart rate, and weight remained stable among all participants throughout the study.
The intensity of the AEs was described as “light” in 91.7% (44/48) of cases and “moderate” in 8.3% (4/48) of cases. The frequency of AEs was classified as “unique,” “intermittent,” or “continuous” in 45.8% (22/48), 39.6% (19/48), and14.6% (7/48) of cases, respectively. Of the 48 AEs, 3 (6.3%) occurred in 1 participant, necessitating interruption of treatment, application of the topical corticosteroid cream mometasone, and removal from the study.
Comment
Following treatment with the study cream containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and RAL 0.05%, the mean reduction in MASI score (P<.0001) and the mean reduction in total melasma surface area from baseline to end of treatment were statistically significant (P<.0001). The study product was associated with strong statistical evidence of patient satisfaction (P<.0001) regarding improvement in facial skin texture, skin oiliness, brightness, overall appearance, and hydration. Participants also responded favorably to the product and considered it safe and effective. In vivo RCM analysis demonstrated a reduction in the amount of melanin in 4 levels of the skin (superficial dermis, suprabasal layer/dermoepidermal junction, spinous layer, superficial granular layer) following treatment with the study cream; however, over the course of the 60-day treatment period, it did not reveal statistically significant reductions. This finding likely is due to the large ranges used to classify the amount of melanin present in each layer of the skin. These limitations suggest that scales used in future in vivo RCM analyses of melasma should be narrower.
Epidermal melasma is one of the most difficult dermatologic diseases to treat and control. Maintenance of clear, undamaged skin remains a treatment target for all dermatologists. This novel cream formulation containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and RAL 0.05% has proven to be an effective, safe, and tolerable treatment option for patients with epidermal melasma.
1. Grimes PE, Yamada N, Bhawan J. Light microscopic, immunohistochemical, and ultrastructural alterations in patients with melasma. Am J Dermatopathol. 2005;27:96-101.
2. Kang WH, Yoon KH, Lee ES, et al. Melasma: histopathological characteristics in 56 Korean patients. Br J Dermatol. 2002;146:228-237.
3. Cestari T, Arellano I, Hexsel D, et al. Melasma in Latin America: options the therapy and treatment algorithm. JEADV. 2009;23:760-772.
4. Miot LDB, Miot HA, Silva MG, et al. Fisiopatologia do Melasma. An Bras Dermatol. 2009;84:623-635.
5. Draelos Z. Skin lightening preparations and the hydroquinone controversy. Dermatol Ther. 2007;20:308-313.
6. Parvez S, Kang M, Chung HS, et al. Survey and mechanism of skin depigmenting and lightening agents. Phytoter Res. 2006;20:921-934.
7. Kim S, Jung E, Kim JH, et al. Inhibitory effects of (-)-α-bisabolol on LPS-induced inflammatory response in RAW264.7 macrophages. Food Chem Toxicol. 2011;49:2580-2585.
8. Ortonne JP. Retinoid therapy of pigmentary disorders. Dermatol Ther. 2006;19:280-288.
9. Namazi MR. Nicotinamide-containing sunscreens for use in Australasian countries and cancer-provoking conditions. Med Hypotheses. 2003;60:544-545.
10. Ertam I, Mutlu B, Unal I, et al. Efficiency of ellagic acid and arbutin in melasma: a randomized, prospective, open-label study. J Dermatol. 2008;35:570-574.
11. Hori I, Nihei K, Kubo I. Structural criteria for depigmenting mechanism of arbutin. Phytother Res. 2004;18:475-469.
12. Ethnic skin and pigmentation. In: Draelos ZD. Cosmetics and Dermatologic Problems and Solutions. 3rd ed. Boca Raton, FL: CRC Press; 2011:52-55.
13. Kasraee B, Tran C, Sorg O, et al. The depigmenting effect of RALGA in C57BL/6 mice. Dermatology. 2005;210(suppl 1):30-34.
Epidermal melasma is a common hyperpigmentation disorder that can be challenging to treat. The pathogenesis of melasma is not fully understood but has been associated with increased melanin and melanocyte activity.1,2 Melasma is characterized by jagged, light- to dark-brown patches on areas of the skin most often exposed to the sun—primarily the cheeks, forehead, upper lip, nose, and chin.3 Although it can affect both sexes and all races, melasma is more common in Fitzpatrick skin types II to IV and frequently is seen in Asian or Hispanic women residing in geographic locations with high levels of sun exposure (eg, tropical areas).2 Melasma presents more frequently in adult women of childbearing age, especially during pregnancy, but also can begin postmenopause. Onset may occur as early as menarche but typically is observed between the ages of 30 and 55 years.3,4 Only 10% of melasma cases are known to occur in males4 and are influenced by such factors as ethnicity, hormones, and level of sun exposure.2
Topical therapies for melasma attempt to inhibit melanocytic activation at each level of melanin formation until the deposited pigment is removed; however, results may vary greatly, as melasma often recurs due to the migration of new melanocytes from hair follicles to the skin’s surface, leading to new development of hyperpigmentation. The current standard of treatment for melasma involves the use of hydroquinone and other bleaching agents, but long-term use of these treatments has been associated with concerns regarding unstable preparations (which may lose their therapeutic properties) and adverse effects (eg, ochronosis, depigmentation).5 Cosmetic agents that recently have been evaluated for melasma treatment include nicotinamide (a form of vitamin B3), which inhibits the transfer of melanosomes from melanocytes to keratinocytes; arbutin, which inhibits melanin synthesis by inhibiting tyrosinase activity6; bisabolol, which prevents anti-inflammatory activity7; and retinaldehyde (RAL), a precursor of retinoic acid (RA) that has powerful bleaching action and low levels of cutaneous irritability.8
This prospective, single-arm, open-label study, evaluated the efficacy and safety of a novel cream formulation containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and retinaldehyde 0.05% in the treatment of epidermal melasma.
Study Product Ingredients and Background
Nicotinamide
Nicotinamide is a water-soluble amide of nicotinic acid (niacin) and one of the 2 principal forms of vitamin B3. It is a component of the coenzymes nicotinamide adenine dinucleotide and nicotinamide adenine dinucleotide phosphate. Nicotinamide essentially acts as an antioxidant, with most of its effects exerted through poly(adenosine diphosphate–ribose) polymerase inhibition. Interest has increased in the role of nicotinamide in the prevention and treatment of several skin diseases, such as acne and UV radiation–induced deleterious molecular and immunological events. Nicotinamide also has gained consideration as a potential agent in sunscreen preparations due to its possible skin-lightening effects, stimulation of DNA repair, suppression of UV photocarcinogenesis, and other antiaging effects.9
Arbutin
Arbutin is a molecule that has proven effective in treating melasma.10 Its pigment-lightening ingredients include botanicals that are structurally similar to hydroquinone. Arbutin is obtained from the leaves of the bearberry plant but also is found in lesser quantities in cranberry and blueberry leaves. A naturally occurring gluconopyranoside, arbutin reduces tyrosinase activity without affecting messenger RNA expression.11 Arbutin also inhibits melanosome maturation, is nontoxic to melanocytes, and is used in Japan in a variety of pigment-lightening preparations at 3% concentrations.12
Bisabolol
Bisabolol is a natural monocyclic sesquiterpene alcohol found in the oils of chamomile and other plants. Bisabolol often is included in cosmetics due to its favorable anti-inflammatory and depigmentation properties. Its downregulation of inducible nitric oxide synthase and cyclooxygenase-2 suggests that it may have anti-inflammatory effects.7
Retinaldehyde
Retinaldehyde is an RA precursor that forms as an intermediate metabolite in the transformation of retinol to RA in human keratinocytes. Topical RAL is well tolerated by human skin, and several of its biologic effects are identical to those of RA. Using the tails of C57BL/6 mouse models, RAL 0.05% has been found to have significantly more potent depigmenting effects than RA 0.05% (P<.001 vs P<.01, respectively) when compared to vehicle.13
Although combination therapy with RAL and arbutin could potentially cause skin irritation, the addition of bisabolol to the combination cream used in this study is believed to have conferred anti-inflammatory properties because it inhibits the release of histamine and relieves irritation.
Methods
This single-center, single-arm, prospective, open-label study evaluated the efficacy and safety of a novel cream formulation containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and RAL 0.05% in treating epidermal melasma. Clinical evaluation included assessment of Melasma Area and Severity Index (MASI) score, photographic analysis, and in vivo reflectance confocal microscopy (RCM) analysis.
The study population included women aged 18 to 50 years with Fitzpatrick skin types I through V who had clinically diagnosed epidermal melasma on the face. Eligibility requirements included confirmation of epidermal pigmentation on Wood lamp examination and RCM analysis and a MASI score of less than 10.5. A total of 35 participants were enrolled in the study (intention to treat [ITT] population). Thirty-three participants were included in the analysis of treatment effectiveness (ITTe population), as 2 were excluded due to lack of follow-up postbaseline. Four participants were prematurely withdrawn from the study—3 due to loss to follow-up and 1 due to treatment discontinuation following an adverse event (AE). The last observation carried forward method was used to input missing data from these 4 participants excluding repeated measure analysis that used the generalized estimated equation method.
At baseline, a 25-g tube of the study cream containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and RAL 0.05% was distributed to all participants for once-daily application to the entire face for 30 days. Participants were instructed to apply the product in the evening after using a gentle cleanser, which also was to be used in the morning to remove the product residue. Additionally, participants were given a sunscreen with a sun protection factor of 30 to apply daily on the entire face in the morning, after lunch, and midafternoon. During the 30-day treatment period, treatment interruption of up to 5 consecutive days or 10 nonconsecutive days in total was permitted. At day 30, participants received another 30-day supply of the study product and sunscreen to be applied according to the same regimen for an additional 30-day treatment period.
Clinical Evaluation
At baseline, demographic data and medical history was recorded for all participants and dermatologic and physical examination was performed documenting weight, height, blood pressure, heart rate, and baseline MASI score. Following Wood lamp examination, participants’ faces were photographed and catalogued using medical imaging software that allowed for measurement of the total melasma surface area (Figure 1A). The photographs also were cross-polarized for further analysis of the pigmentation (Figure 1B).
|
|
| |
Figure 1. Clinical (A) and cross-polarized (B) photographs of a patient before treatment with the novel compound containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and retinaldehyde 0.05%. | ||
A questionnaire evaluating treatment satisfaction was administered to participants (ITTe population [n=33]) at baseline and days 30 and 60. Questionnaire items pertained to skin blemishes, signs of facial aging, overall appearance, texture, oiliness, brightness, and hydration. Participants were instructed to rate their satisfaction for each item on a scale of 1 to 10 (1=bad, 10=excellent). For investigator analysis, scores of 1 to 4 were classified as “dissatisfied,” scores of 5 to 6 were classified as “satisfied,” and scores of 7 to 10 were classified as “completely satisfied.” A questionnaire evaluating product appreciation was administered at day 60 to participants who completed the study (n=29). Questionnaire items asked participants to rate the study cream’s ease of application, consistency, smell, absorption, and overall satisfaction using ratings of “bad,” “regular,” “good,” “very good,” or “excellent.”
Treatment efficacy in all participants was assessed by the investigators at days 30 and 60. Investigators evaluated reductions in pigmentation and total melasma surface area using ratings of “none,” “regular,” “good,” “very good,” or “excellent.” Local tolerance also was evaluated at both time points, and AEs were recorded and analyzed with respect to their duration, intensity, frequency, and severity.
Targeted hyperpigmented skin was selected for in vivo RCM analysis. At each time point, a sequence of block images was acquired at 4 levels of skin: (1) superficial dermis, (2) suprabasal layer/ dermoepidermal junction, (3) spinous layer, and (4) superficial granular layer. Blind evaluation of these images to assess the reduction in melanin quantity was conducted by a dermatopathologist at baseline and days 30 and 60. Melanin quantity present in each layer was graded according to 4 categories (0%–25%, 25.1%–50%, 50.1%–75%, 75.1%–100%). The mean value was used for statistical evaluation.
Results
Efficacy evaluation
The primary efficacy variable was the mean reduction in MASI score from baseline to the end of treatment (day 60), which was 2.25 ± 1.87 (P<.0001). The reduction in mean MASI score was significant from baseline to day 30 (P<.0001) and from day 30 to day 60 (P<.0001). The least root-mean-square error estimates of MASI score variation at days 30 and 60 were 1.40 and 2.25, respectively.
The mean total melasma surface area (as measured in analysis of clinical photographs using medical imaging software) was significantly reduced from 1398.5 mm2 at baseline to 1116.9 mm2 at day 30 (P<.0001) and 923.4 at day 60 (P<.0001). From baseline to end of treatment, the overall reduction in mean total melasma surface area was 475.1 mm2 (P<.0001)(Figure 2). Clinical and cross-polarized photographs taken at day 60 demonstrated a visible reduction in melasma surface area (Figure 3), which was confirmed using medical imaging software.
|
|
| |
Figure 3. Clinical (A) and cross-polarized (B) photographs of a patient after 60 days of treatment with the novel compound containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and retinaldehyde 0.05%. | ||
In vivo RCM analyses at each time point showed reduction in pigmentation in the 4 levels of the skin that were evaluated, but the results were not statistically significant.
Participant satisfaction
There was strong statistical evidence of patient satisfaction with the treatment results at the end of the study period (P<.0001). At baseline, 75.8% (25/33) of participants were dissatisfied with the appearance of their skin as compared with 15.2% (5/33) at day 60. Additionally, 18.1% (6/33) and 6.1% (2/33) of the participants were satisfied and completely satisfied at baseline compared with 33.3% (11/33) and 51.5% (17/33) at day 60, respectively. Participant satisfaction with signs of facial aging also increased over the study period (P=.0104). At baseline, 60.6% (20/33) were dissatisfied, 12.1% (4/33) were satisfied, and 27.3% (9/33) were completely satisfied; at the end of treatment, 30.3% (10/33) were dissatisfied, 36.4% (12/33) were satisfied, and 33.3% (11/33) were completely satisfied with the improvement in signs of facial aging.
Increased patient satisfaction with facial skin texture at baseline compared to day 60 also was statistically significant (P=.0157). At baseline, 39.4% (13/33) of the participants were dissatisfied, 30.3% (10/33) were satisfied, and 30.3% (10/33) were completely satisfied with facial texture; at day 60, 15.1% (5/33) were dissatisfied, 30.3% (10/33) were satisfied, and 54.6% (18/33) were completely satisfied. Significant improvement from baseline to day 60 also was observed in participant assessment of skin oiliness (P=.0210), brightness (P=.0003), overall appearance (P<.0001), and hydration (P<.0001).
Product appreciation
At day 60, 89.7% (26/29) of the participants who completed the study rated the product’s ease of application as being at least “good,” with more than half of participants (55.2% [16/29]) rating it as “very good” or “excellent.” Overall satisfaction with the product was rated as “very good” or “excellent” by 48.3% (14/29) of the participants. Similar results were observed in participant assessments of consistency, smell, and absorption (Figure 4).
Safety evaluation
A total of 52 AEs were observed in 23 (69.7%) participants, which were recorded by participants in diary entries throughout treatment and evaluated by investigators at each time point. Among these AEs, 48 (92.3%) were considered possibly, probably, or conditionally related to treatment by the investigators based on clinical observation. The most common presumed treatment-related AE was a burning sensation on the skin, reported by 30.3% (10/33) of the participants at day 30 and 13.8% (4/29) at day 60. Of the reported AEs related to treatment, 91.7% (44/48) were of mild intensity and 93.8% (45/48) required no treatment or other action. There were no reported serious AEs related to the investigational product. Blood pressure, heart rate, and weight remained stable among all participants throughout the study.
The intensity of the AEs was described as “light” in 91.7% (44/48) of cases and “moderate” in 8.3% (4/48) of cases. The frequency of AEs was classified as “unique,” “intermittent,” or “continuous” in 45.8% (22/48), 39.6% (19/48), and14.6% (7/48) of cases, respectively. Of the 48 AEs, 3 (6.3%) occurred in 1 participant, necessitating interruption of treatment, application of the topical corticosteroid cream mometasone, and removal from the study.
Comment
Following treatment with the study cream containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and RAL 0.05%, the mean reduction in MASI score (P<.0001) and the mean reduction in total melasma surface area from baseline to end of treatment were statistically significant (P<.0001). The study product was associated with strong statistical evidence of patient satisfaction (P<.0001) regarding improvement in facial skin texture, skin oiliness, brightness, overall appearance, and hydration. Participants also responded favorably to the product and considered it safe and effective. In vivo RCM analysis demonstrated a reduction in the amount of melanin in 4 levels of the skin (superficial dermis, suprabasal layer/dermoepidermal junction, spinous layer, superficial granular layer) following treatment with the study cream; however, over the course of the 60-day treatment period, it did not reveal statistically significant reductions. This finding likely is due to the large ranges used to classify the amount of melanin present in each layer of the skin. These limitations suggest that scales used in future in vivo RCM analyses of melasma should be narrower.
Epidermal melasma is one of the most difficult dermatologic diseases to treat and control. Maintenance of clear, undamaged skin remains a treatment target for all dermatologists. This novel cream formulation containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and RAL 0.05% has proven to be an effective, safe, and tolerable treatment option for patients with epidermal melasma.
Epidermal melasma is a common hyperpigmentation disorder that can be challenging to treat. The pathogenesis of melasma is not fully understood but has been associated with increased melanin and melanocyte activity.1,2 Melasma is characterized by jagged, light- to dark-brown patches on areas of the skin most often exposed to the sun—primarily the cheeks, forehead, upper lip, nose, and chin.3 Although it can affect both sexes and all races, melasma is more common in Fitzpatrick skin types II to IV and frequently is seen in Asian or Hispanic women residing in geographic locations with high levels of sun exposure (eg, tropical areas).2 Melasma presents more frequently in adult women of childbearing age, especially during pregnancy, but also can begin postmenopause. Onset may occur as early as menarche but typically is observed between the ages of 30 and 55 years.3,4 Only 10% of melasma cases are known to occur in males4 and are influenced by such factors as ethnicity, hormones, and level of sun exposure.2
Topical therapies for melasma attempt to inhibit melanocytic activation at each level of melanin formation until the deposited pigment is removed; however, results may vary greatly, as melasma often recurs due to the migration of new melanocytes from hair follicles to the skin’s surface, leading to new development of hyperpigmentation. The current standard of treatment for melasma involves the use of hydroquinone and other bleaching agents, but long-term use of these treatments has been associated with concerns regarding unstable preparations (which may lose their therapeutic properties) and adverse effects (eg, ochronosis, depigmentation).5 Cosmetic agents that recently have been evaluated for melasma treatment include nicotinamide (a form of vitamin B3), which inhibits the transfer of melanosomes from melanocytes to keratinocytes; arbutin, which inhibits melanin synthesis by inhibiting tyrosinase activity6; bisabolol, which prevents anti-inflammatory activity7; and retinaldehyde (RAL), a precursor of retinoic acid (RA) that has powerful bleaching action and low levels of cutaneous irritability.8
This prospective, single-arm, open-label study, evaluated the efficacy and safety of a novel cream formulation containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and retinaldehyde 0.05% in the treatment of epidermal melasma.
Study Product Ingredients and Background
Nicotinamide
Nicotinamide is a water-soluble amide of nicotinic acid (niacin) and one of the 2 principal forms of vitamin B3. It is a component of the coenzymes nicotinamide adenine dinucleotide and nicotinamide adenine dinucleotide phosphate. Nicotinamide essentially acts as an antioxidant, with most of its effects exerted through poly(adenosine diphosphate–ribose) polymerase inhibition. Interest has increased in the role of nicotinamide in the prevention and treatment of several skin diseases, such as acne and UV radiation–induced deleterious molecular and immunological events. Nicotinamide also has gained consideration as a potential agent in sunscreen preparations due to its possible skin-lightening effects, stimulation of DNA repair, suppression of UV photocarcinogenesis, and other antiaging effects.9
Arbutin
Arbutin is a molecule that has proven effective in treating melasma.10 Its pigment-lightening ingredients include botanicals that are structurally similar to hydroquinone. Arbutin is obtained from the leaves of the bearberry plant but also is found in lesser quantities in cranberry and blueberry leaves. A naturally occurring gluconopyranoside, arbutin reduces tyrosinase activity without affecting messenger RNA expression.11 Arbutin also inhibits melanosome maturation, is nontoxic to melanocytes, and is used in Japan in a variety of pigment-lightening preparations at 3% concentrations.12
Bisabolol
Bisabolol is a natural monocyclic sesquiterpene alcohol found in the oils of chamomile and other plants. Bisabolol often is included in cosmetics due to its favorable anti-inflammatory and depigmentation properties. Its downregulation of inducible nitric oxide synthase and cyclooxygenase-2 suggests that it may have anti-inflammatory effects.7
Retinaldehyde
Retinaldehyde is an RA precursor that forms as an intermediate metabolite in the transformation of retinol to RA in human keratinocytes. Topical RAL is well tolerated by human skin, and several of its biologic effects are identical to those of RA. Using the tails of C57BL/6 mouse models, RAL 0.05% has been found to have significantly more potent depigmenting effects than RA 0.05% (P<.001 vs P<.01, respectively) when compared to vehicle.13
Although combination therapy with RAL and arbutin could potentially cause skin irritation, the addition of bisabolol to the combination cream used in this study is believed to have conferred anti-inflammatory properties because it inhibits the release of histamine and relieves irritation.
Methods
This single-center, single-arm, prospective, open-label study evaluated the efficacy and safety of a novel cream formulation containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and RAL 0.05% in treating epidermal melasma. Clinical evaluation included assessment of Melasma Area and Severity Index (MASI) score, photographic analysis, and in vivo reflectance confocal microscopy (RCM) analysis.
The study population included women aged 18 to 50 years with Fitzpatrick skin types I through V who had clinically diagnosed epidermal melasma on the face. Eligibility requirements included confirmation of epidermal pigmentation on Wood lamp examination and RCM analysis and a MASI score of less than 10.5. A total of 35 participants were enrolled in the study (intention to treat [ITT] population). Thirty-three participants were included in the analysis of treatment effectiveness (ITTe population), as 2 were excluded due to lack of follow-up postbaseline. Four participants were prematurely withdrawn from the study—3 due to loss to follow-up and 1 due to treatment discontinuation following an adverse event (AE). The last observation carried forward method was used to input missing data from these 4 participants excluding repeated measure analysis that used the generalized estimated equation method.
At baseline, a 25-g tube of the study cream containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and RAL 0.05% was distributed to all participants for once-daily application to the entire face for 30 days. Participants were instructed to apply the product in the evening after using a gentle cleanser, which also was to be used in the morning to remove the product residue. Additionally, participants were given a sunscreen with a sun protection factor of 30 to apply daily on the entire face in the morning, after lunch, and midafternoon. During the 30-day treatment period, treatment interruption of up to 5 consecutive days or 10 nonconsecutive days in total was permitted. At day 30, participants received another 30-day supply of the study product and sunscreen to be applied according to the same regimen for an additional 30-day treatment period.
Clinical Evaluation
At baseline, demographic data and medical history was recorded for all participants and dermatologic and physical examination was performed documenting weight, height, blood pressure, heart rate, and baseline MASI score. Following Wood lamp examination, participants’ faces were photographed and catalogued using medical imaging software that allowed for measurement of the total melasma surface area (Figure 1A). The photographs also were cross-polarized for further analysis of the pigmentation (Figure 1B).
|
|
| |
Figure 1. Clinical (A) and cross-polarized (B) photographs of a patient before treatment with the novel compound containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and retinaldehyde 0.05%. | ||
A questionnaire evaluating treatment satisfaction was administered to participants (ITTe population [n=33]) at baseline and days 30 and 60. Questionnaire items pertained to skin blemishes, signs of facial aging, overall appearance, texture, oiliness, brightness, and hydration. Participants were instructed to rate their satisfaction for each item on a scale of 1 to 10 (1=bad, 10=excellent). For investigator analysis, scores of 1 to 4 were classified as “dissatisfied,” scores of 5 to 6 were classified as “satisfied,” and scores of 7 to 10 were classified as “completely satisfied.” A questionnaire evaluating product appreciation was administered at day 60 to participants who completed the study (n=29). Questionnaire items asked participants to rate the study cream’s ease of application, consistency, smell, absorption, and overall satisfaction using ratings of “bad,” “regular,” “good,” “very good,” or “excellent.”
Treatment efficacy in all participants was assessed by the investigators at days 30 and 60. Investigators evaluated reductions in pigmentation and total melasma surface area using ratings of “none,” “regular,” “good,” “very good,” or “excellent.” Local tolerance also was evaluated at both time points, and AEs were recorded and analyzed with respect to their duration, intensity, frequency, and severity.
Targeted hyperpigmented skin was selected for in vivo RCM analysis. At each time point, a sequence of block images was acquired at 4 levels of skin: (1) superficial dermis, (2) suprabasal layer/ dermoepidermal junction, (3) spinous layer, and (4) superficial granular layer. Blind evaluation of these images to assess the reduction in melanin quantity was conducted by a dermatopathologist at baseline and days 30 and 60. Melanin quantity present in each layer was graded according to 4 categories (0%–25%, 25.1%–50%, 50.1%–75%, 75.1%–100%). The mean value was used for statistical evaluation.
Results
Efficacy evaluation
The primary efficacy variable was the mean reduction in MASI score from baseline to the end of treatment (day 60), which was 2.25 ± 1.87 (P<.0001). The reduction in mean MASI score was significant from baseline to day 30 (P<.0001) and from day 30 to day 60 (P<.0001). The least root-mean-square error estimates of MASI score variation at days 30 and 60 were 1.40 and 2.25, respectively.
The mean total melasma surface area (as measured in analysis of clinical photographs using medical imaging software) was significantly reduced from 1398.5 mm2 at baseline to 1116.9 mm2 at day 30 (P<.0001) and 923.4 at day 60 (P<.0001). From baseline to end of treatment, the overall reduction in mean total melasma surface area was 475.1 mm2 (P<.0001)(Figure 2). Clinical and cross-polarized photographs taken at day 60 demonstrated a visible reduction in melasma surface area (Figure 3), which was confirmed using medical imaging software.
|
|
| |
Figure 3. Clinical (A) and cross-polarized (B) photographs of a patient after 60 days of treatment with the novel compound containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and retinaldehyde 0.05%. | ||
In vivo RCM analyses at each time point showed reduction in pigmentation in the 4 levels of the skin that were evaluated, but the results were not statistically significant.
Participant satisfaction
There was strong statistical evidence of patient satisfaction with the treatment results at the end of the study period (P<.0001). At baseline, 75.8% (25/33) of participants were dissatisfied with the appearance of their skin as compared with 15.2% (5/33) at day 60. Additionally, 18.1% (6/33) and 6.1% (2/33) of the participants were satisfied and completely satisfied at baseline compared with 33.3% (11/33) and 51.5% (17/33) at day 60, respectively. Participant satisfaction with signs of facial aging also increased over the study period (P=.0104). At baseline, 60.6% (20/33) were dissatisfied, 12.1% (4/33) were satisfied, and 27.3% (9/33) were completely satisfied; at the end of treatment, 30.3% (10/33) were dissatisfied, 36.4% (12/33) were satisfied, and 33.3% (11/33) were completely satisfied with the improvement in signs of facial aging.
Increased patient satisfaction with facial skin texture at baseline compared to day 60 also was statistically significant (P=.0157). At baseline, 39.4% (13/33) of the participants were dissatisfied, 30.3% (10/33) were satisfied, and 30.3% (10/33) were completely satisfied with facial texture; at day 60, 15.1% (5/33) were dissatisfied, 30.3% (10/33) were satisfied, and 54.6% (18/33) were completely satisfied. Significant improvement from baseline to day 60 also was observed in participant assessment of skin oiliness (P=.0210), brightness (P=.0003), overall appearance (P<.0001), and hydration (P<.0001).
Product appreciation
At day 60, 89.7% (26/29) of the participants who completed the study rated the product’s ease of application as being at least “good,” with more than half of participants (55.2% [16/29]) rating it as “very good” or “excellent.” Overall satisfaction with the product was rated as “very good” or “excellent” by 48.3% (14/29) of the participants. Similar results were observed in participant assessments of consistency, smell, and absorption (Figure 4).
Safety evaluation
A total of 52 AEs were observed in 23 (69.7%) participants, which were recorded by participants in diary entries throughout treatment and evaluated by investigators at each time point. Among these AEs, 48 (92.3%) were considered possibly, probably, or conditionally related to treatment by the investigators based on clinical observation. The most common presumed treatment-related AE was a burning sensation on the skin, reported by 30.3% (10/33) of the participants at day 30 and 13.8% (4/29) at day 60. Of the reported AEs related to treatment, 91.7% (44/48) were of mild intensity and 93.8% (45/48) required no treatment or other action. There were no reported serious AEs related to the investigational product. Blood pressure, heart rate, and weight remained stable among all participants throughout the study.
The intensity of the AEs was described as “light” in 91.7% (44/48) of cases and “moderate” in 8.3% (4/48) of cases. The frequency of AEs was classified as “unique,” “intermittent,” or “continuous” in 45.8% (22/48), 39.6% (19/48), and14.6% (7/48) of cases, respectively. Of the 48 AEs, 3 (6.3%) occurred in 1 participant, necessitating interruption of treatment, application of the topical corticosteroid cream mometasone, and removal from the study.
Comment
Following treatment with the study cream containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and RAL 0.05%, the mean reduction in MASI score (P<.0001) and the mean reduction in total melasma surface area from baseline to end of treatment were statistically significant (P<.0001). The study product was associated with strong statistical evidence of patient satisfaction (P<.0001) regarding improvement in facial skin texture, skin oiliness, brightness, overall appearance, and hydration. Participants also responded favorably to the product and considered it safe and effective. In vivo RCM analysis demonstrated a reduction in the amount of melanin in 4 levels of the skin (superficial dermis, suprabasal layer/dermoepidermal junction, spinous layer, superficial granular layer) following treatment with the study cream; however, over the course of the 60-day treatment period, it did not reveal statistically significant reductions. This finding likely is due to the large ranges used to classify the amount of melanin present in each layer of the skin. These limitations suggest that scales used in future in vivo RCM analyses of melasma should be narrower.
Epidermal melasma is one of the most difficult dermatologic diseases to treat and control. Maintenance of clear, undamaged skin remains a treatment target for all dermatologists. This novel cream formulation containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and RAL 0.05% has proven to be an effective, safe, and tolerable treatment option for patients with epidermal melasma.
1. Grimes PE, Yamada N, Bhawan J. Light microscopic, immunohistochemical, and ultrastructural alterations in patients with melasma. Am J Dermatopathol. 2005;27:96-101.
2. Kang WH, Yoon KH, Lee ES, et al. Melasma: histopathological characteristics in 56 Korean patients. Br J Dermatol. 2002;146:228-237.
3. Cestari T, Arellano I, Hexsel D, et al. Melasma in Latin America: options the therapy and treatment algorithm. JEADV. 2009;23:760-772.
4. Miot LDB, Miot HA, Silva MG, et al. Fisiopatologia do Melasma. An Bras Dermatol. 2009;84:623-635.
5. Draelos Z. Skin lightening preparations and the hydroquinone controversy. Dermatol Ther. 2007;20:308-313.
6. Parvez S, Kang M, Chung HS, et al. Survey and mechanism of skin depigmenting and lightening agents. Phytoter Res. 2006;20:921-934.
7. Kim S, Jung E, Kim JH, et al. Inhibitory effects of (-)-α-bisabolol on LPS-induced inflammatory response in RAW264.7 macrophages. Food Chem Toxicol. 2011;49:2580-2585.
8. Ortonne JP. Retinoid therapy of pigmentary disorders. Dermatol Ther. 2006;19:280-288.
9. Namazi MR. Nicotinamide-containing sunscreens for use in Australasian countries and cancer-provoking conditions. Med Hypotheses. 2003;60:544-545.
10. Ertam I, Mutlu B, Unal I, et al. Efficiency of ellagic acid and arbutin in melasma: a randomized, prospective, open-label study. J Dermatol. 2008;35:570-574.
11. Hori I, Nihei K, Kubo I. Structural criteria for depigmenting mechanism of arbutin. Phytother Res. 2004;18:475-469.
12. Ethnic skin and pigmentation. In: Draelos ZD. Cosmetics and Dermatologic Problems and Solutions. 3rd ed. Boca Raton, FL: CRC Press; 2011:52-55.
13. Kasraee B, Tran C, Sorg O, et al. The depigmenting effect of RALGA in C57BL/6 mice. Dermatology. 2005;210(suppl 1):30-34.
1. Grimes PE, Yamada N, Bhawan J. Light microscopic, immunohistochemical, and ultrastructural alterations in patients with melasma. Am J Dermatopathol. 2005;27:96-101.
2. Kang WH, Yoon KH, Lee ES, et al. Melasma: histopathological characteristics in 56 Korean patients. Br J Dermatol. 2002;146:228-237.
3. Cestari T, Arellano I, Hexsel D, et al. Melasma in Latin America: options the therapy and treatment algorithm. JEADV. 2009;23:760-772.
4. Miot LDB, Miot HA, Silva MG, et al. Fisiopatologia do Melasma. An Bras Dermatol. 2009;84:623-635.
5. Draelos Z. Skin lightening preparations and the hydroquinone controversy. Dermatol Ther. 2007;20:308-313.
6. Parvez S, Kang M, Chung HS, et al. Survey and mechanism of skin depigmenting and lightening agents. Phytoter Res. 2006;20:921-934.
7. Kim S, Jung E, Kim JH, et al. Inhibitory effects of (-)-α-bisabolol on LPS-induced inflammatory response in RAW264.7 macrophages. Food Chem Toxicol. 2011;49:2580-2585.
8. Ortonne JP. Retinoid therapy of pigmentary disorders. Dermatol Ther. 2006;19:280-288.
9. Namazi MR. Nicotinamide-containing sunscreens for use in Australasian countries and cancer-provoking conditions. Med Hypotheses. 2003;60:544-545.
10. Ertam I, Mutlu B, Unal I, et al. Efficiency of ellagic acid and arbutin in melasma: a randomized, prospective, open-label study. J Dermatol. 2008;35:570-574.
11. Hori I, Nihei K, Kubo I. Structural criteria for depigmenting mechanism of arbutin. Phytother Res. 2004;18:475-469.
12. Ethnic skin and pigmentation. In: Draelos ZD. Cosmetics and Dermatologic Problems and Solutions. 3rd ed. Boca Raton, FL: CRC Press; 2011:52-55.
13. Kasraee B, Tran C, Sorg O, et al. The depigmenting effect of RALGA in C57BL/6 mice. Dermatology. 2005;210(suppl 1):30-34.
Practice Points
- Epidermal melasma is a common hyperpigmentation disorder characterized by the appearance of abnormal melanin deposits in different layers of the skin.
- Melasma can be difficult to treat and often recurs due to the migration of new melanocytes from hair follicles to the skin’s surface.
- A novel cream formulation containing nicotinamide 4%, arbutin 3%, bisabolol 1%, and retinaldehyde 0.05% offers a safe and effective option for treatment of epidermal melasma.

What Is Your Diagnosis? Idiopathic Guttate Hypomelanosis
The Diagnosis: Idiopathic Guttate Hypomelanosis
A biopsy of the largest lesion from the left leg superior to the lateral malleolus was performed. Histopathologic examination revealed solar elastosis, diminished number of focal melanocytes and pigment within keratinocytes compared to uninvolved skin, and presence of hyperkeratosis with flattening of rete ridges. The clinical presentation along with histopathologic analysis confirmed a diagnosis of idiopathic guttate hypomelanosis (IGH). The lesions were treated with short-exposure cryotherapy, which resulted in partial repigmentation after several treatments.
Idiopathic guttate hypomelanosis is a common but underreported condition in elderly patients that usually presents with small, discrete, asymptomatic, hypopigmented macules. The frequency of IGH increases with age.1 Frequency of the condition is much lower in patients aged 21 to 30 years and does not exceed 7%. Lesions of IGH have a predilection for sun-exposed areas such as the arms and legs but rarely can be seen on the face and trunk. Facial lesions of IGH are more frequently reported in women.1 The size of lesions can be up to 1.5 cm in diameter. The condition generally is self-limited, but some patients may express aesthetic concerns. Rare cases of IGH in children have been associated with prolonged sun exposure.2
The etiology of IGH is unknown but an association with sun exposure has been noted. Patients with IGH frequently show other signs of photoaging, such as numerous seborrheic keratoses, solar lentigines, xeroses, freckles, and actinic keratoses.1 Short-term exposure to UVB radiation and psoralen plus UVA therapy has been shown to cause IGH in patients with chronic diseases such as mycosis fungoides.3-5 One small study that examined renal transplant recipients determined an association between HLA-DQ3 antigens and IGH, whereas HLA-DR8 antigens were not identified in any patients with IGH, indicating it may have some advantage in preventing the development of IGH.6 Shin et al1 reported that IGH was prevalent among patients who regularly traumatized their skin by scrubbing.
Clinically, IGH should be differentiated from other conditions characterized by hypopigmentation, such as pityriasis alba, pityriasis versicolor, postinflammatory hypopigmentation, progressive macular hypomelanosis, and vitiligo. Aside from clinical examination, histopathologic studies are helpful in making a definitive diagnosis. The differential diagnosis of IGH is presented in the Table.

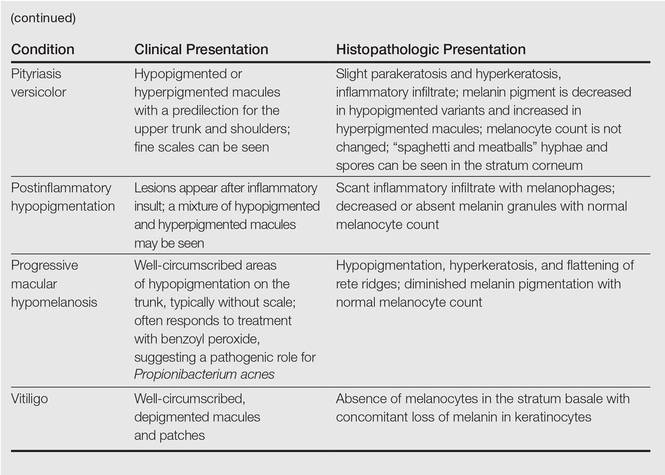
Histopathology of IGH lesions usually reveals slight atrophy of the epidermis with flattening of rete ridges and concomitant hyperkeratosis. A thickened stratum granulosum also has been noted in lesions of IGH.2 The diminished number of melanocytes and melanin pigment granules along with hyperkeratosis both appear to contribute to the hypopigmentation noted in IGH.7 Ultrastructural studies of lesions of IGH can confirm melanocytic degeneration and a decreased number of melanosomes in melanocytes and keratinocytes.2,8
There is no uniformly effective treatment of IGH. Topical application of tacrolimus and tretinoin have shown efficacy in repigmenting IGH lesions.8,9 Short-exposure cryotherapy with a duration of 3 to 5 seconds, localized chemical peels, and/or local dermabrasion can be helpful.10-12 CO2 lasers also have demonstrated promising results.13
- Shin MK, Jeong KH, Oh IH, et al. Clinical features of idiopathic guttate hypomelanosis in 646 subjects and association with other aspects of photoaging. Int J Dermatol. 2011;50:798-805.
- Kim SK, Kim EH, Kang HY, et al. Comprehensive understanding of idiopathic guttate hypomelanosis: clinical and histopathological correlation. Int J Dermatol. 2010;49:162-166.
- Friedland R, David M, Feinmesser M, et al. Idiopathic guttate hypomelanosis-like lesions in patients with mycosis fungoides: a new adverse effect of phototherapy. J Eur Acad Dermatol Venereol. 2010;24:1026-1030.
- Kaya TI, Yazici AC, Tursen U, et al. Idiopathic guttate hypomelanosis: idiopathic or ultraviolet induced? Photodermatol Photoimmunol Photomed. 2005;21:270-271.
- Loquai C, Metze D, Nashan D, et al. Confetti-like lesions with hyperkeratosis: a novel ultraviolet-induced hypomelanotic disorder? Br J Dermatol. 2005;153:190-193.
- Arrunategui A, Trujillo RA, Marulanda MP, et al. HLA-DQ3 is associated with idiopathic guttate hypomelanosis, whereas HLA-DR8 is not, in a group of renal transplant patients. Int J Dermatol. 2002;41:744-747.
- Wallace ML, Grichnik JM, Prieto VG, et al. Numbers and differentiation status of melanocytes in idiopathic guttate hypomelanosis. J Cutan Pathol. 1998;25:375-379.
- Ortonne JP, Perrot H. Idiopathic guttate hypomelanosis. ultrastructural study. Arch Dermatol. 1980;116:664-668.
- Rerknimitr P, Disphanurat W, Achariyakul M. Topical tacrolimus significantly promotes repigmentation in idiopathic guttate hypomelanosis: a double-blind, randomized, placebo-controlled study. J Eur Acad Dermatol Venereol. 2013;27:460-464.
- Pagnoni A, Kligman AM, Sadiq I, et al. Hypopigmented macules of photodamaged skin and their treatment with topical tretinoin. Acta Derm Venereol. 1999;79:305-310.
- Kumarasinghe SP. 3-5 second cryotherapy is effective in idiopathic guttate hypomelanosis. J Dermatol. 2004;31:457-459.
- Hexsel DM. Treatment of idiopathic guttate hypomelanosis by localized superficial dermabrasion. Dermatol Surg. 1999;25:917-918.
- Shin J, Kim M, Park SH, et al. The effect of fractional carbon dioxide lasers on idiopathic guttate hypomelanosis: a preliminary study. J Eur Acad Dermatol Venereol. 2013;27:e243-e246.
The Diagnosis: Idiopathic Guttate Hypomelanosis
A biopsy of the largest lesion from the left leg superior to the lateral malleolus was performed. Histopathologic examination revealed solar elastosis, diminished number of focal melanocytes and pigment within keratinocytes compared to uninvolved skin, and presence of hyperkeratosis with flattening of rete ridges. The clinical presentation along with histopathologic analysis confirmed a diagnosis of idiopathic guttate hypomelanosis (IGH). The lesions were treated with short-exposure cryotherapy, which resulted in partial repigmentation after several treatments.
Idiopathic guttate hypomelanosis is a common but underreported condition in elderly patients that usually presents with small, discrete, asymptomatic, hypopigmented macules. The frequency of IGH increases with age.1 Frequency of the condition is much lower in patients aged 21 to 30 years and does not exceed 7%. Lesions of IGH have a predilection for sun-exposed areas such as the arms and legs but rarely can be seen on the face and trunk. Facial lesions of IGH are more frequently reported in women.1 The size of lesions can be up to 1.5 cm in diameter. The condition generally is self-limited, but some patients may express aesthetic concerns. Rare cases of IGH in children have been associated with prolonged sun exposure.2
The etiology of IGH is unknown but an association with sun exposure has been noted. Patients with IGH frequently show other signs of photoaging, such as numerous seborrheic keratoses, solar lentigines, xeroses, freckles, and actinic keratoses.1 Short-term exposure to UVB radiation and psoralen plus UVA therapy has been shown to cause IGH in patients with chronic diseases such as mycosis fungoides.3-5 One small study that examined renal transplant recipients determined an association between HLA-DQ3 antigens and IGH, whereas HLA-DR8 antigens were not identified in any patients with IGH, indicating it may have some advantage in preventing the development of IGH.6 Shin et al1 reported that IGH was prevalent among patients who regularly traumatized their skin by scrubbing.
Clinically, IGH should be differentiated from other conditions characterized by hypopigmentation, such as pityriasis alba, pityriasis versicolor, postinflammatory hypopigmentation, progressive macular hypomelanosis, and vitiligo. Aside from clinical examination, histopathologic studies are helpful in making a definitive diagnosis. The differential diagnosis of IGH is presented in the Table.
Histopathology of IGH lesions usually reveals slight atrophy of the epidermis with flattening of rete ridges and concomitant hyperkeratosis. A thickened stratum granulosum also has been noted in lesions of IGH.2 The diminished number of melanocytes and melanin pigment granules along with hyperkeratosis both appear to contribute to the hypopigmentation noted in IGH.7 Ultrastructural studies of lesions of IGH can confirm melanocytic degeneration and a decreased number of melanosomes in melanocytes and keratinocytes.2,8
There is no uniformly effective treatment of IGH. Topical application of tacrolimus and tretinoin have shown efficacy in repigmenting IGH lesions.8,9 Short-exposure cryotherapy with a duration of 3 to 5 seconds, localized chemical peels, and/or local dermabrasion can be helpful.10-12 CO2 lasers also have demonstrated promising results.13
The Diagnosis: Idiopathic Guttate Hypomelanosis
A biopsy of the largest lesion from the left leg superior to the lateral malleolus was performed. Histopathologic examination revealed solar elastosis, diminished number of focal melanocytes and pigment within keratinocytes compared to uninvolved skin, and presence of hyperkeratosis with flattening of rete ridges. The clinical presentation along with histopathologic analysis confirmed a diagnosis of idiopathic guttate hypomelanosis (IGH). The lesions were treated with short-exposure cryotherapy, which resulted in partial repigmentation after several treatments.
Idiopathic guttate hypomelanosis is a common but underreported condition in elderly patients that usually presents with small, discrete, asymptomatic, hypopigmented macules. The frequency of IGH increases with age.1 Frequency of the condition is much lower in patients aged 21 to 30 years and does not exceed 7%. Lesions of IGH have a predilection for sun-exposed areas such as the arms and legs but rarely can be seen on the face and trunk. Facial lesions of IGH are more frequently reported in women.1 The size of lesions can be up to 1.5 cm in diameter. The condition generally is self-limited, but some patients may express aesthetic concerns. Rare cases of IGH in children have been associated with prolonged sun exposure.2
The etiology of IGH is unknown but an association with sun exposure has been noted. Patients with IGH frequently show other signs of photoaging, such as numerous seborrheic keratoses, solar lentigines, xeroses, freckles, and actinic keratoses.1 Short-term exposure to UVB radiation and psoralen plus UVA therapy has been shown to cause IGH in patients with chronic diseases such as mycosis fungoides.3-5 One small study that examined renal transplant recipients determined an association between HLA-DQ3 antigens and IGH, whereas HLA-DR8 antigens were not identified in any patients with IGH, indicating it may have some advantage in preventing the development of IGH.6 Shin et al1 reported that IGH was prevalent among patients who regularly traumatized their skin by scrubbing.
Clinically, IGH should be differentiated from other conditions characterized by hypopigmentation, such as pityriasis alba, pityriasis versicolor, postinflammatory hypopigmentation, progressive macular hypomelanosis, and vitiligo. Aside from clinical examination, histopathologic studies are helpful in making a definitive diagnosis. The differential diagnosis of IGH is presented in the Table.
Histopathology of IGH lesions usually reveals slight atrophy of the epidermis with flattening of rete ridges and concomitant hyperkeratosis. A thickened stratum granulosum also has been noted in lesions of IGH.2 The diminished number of melanocytes and melanin pigment granules along with hyperkeratosis both appear to contribute to the hypopigmentation noted in IGH.7 Ultrastructural studies of lesions of IGH can confirm melanocytic degeneration and a decreased number of melanosomes in melanocytes and keratinocytes.2,8
There is no uniformly effective treatment of IGH. Topical application of tacrolimus and tretinoin have shown efficacy in repigmenting IGH lesions.8,9 Short-exposure cryotherapy with a duration of 3 to 5 seconds, localized chemical peels, and/or local dermabrasion can be helpful.10-12 CO2 lasers also have demonstrated promising results.13
- Shin MK, Jeong KH, Oh IH, et al. Clinical features of idiopathic guttate hypomelanosis in 646 subjects and association with other aspects of photoaging. Int J Dermatol. 2011;50:798-805.
- Kim SK, Kim EH, Kang HY, et al. Comprehensive understanding of idiopathic guttate hypomelanosis: clinical and histopathological correlation. Int J Dermatol. 2010;49:162-166.
- Friedland R, David M, Feinmesser M, et al. Idiopathic guttate hypomelanosis-like lesions in patients with mycosis fungoides: a new adverse effect of phototherapy. J Eur Acad Dermatol Venereol. 2010;24:1026-1030.
- Kaya TI, Yazici AC, Tursen U, et al. Idiopathic guttate hypomelanosis: idiopathic or ultraviolet induced? Photodermatol Photoimmunol Photomed. 2005;21:270-271.
- Loquai C, Metze D, Nashan D, et al. Confetti-like lesions with hyperkeratosis: a novel ultraviolet-induced hypomelanotic disorder? Br J Dermatol. 2005;153:190-193.
- Arrunategui A, Trujillo RA, Marulanda MP, et al. HLA-DQ3 is associated with idiopathic guttate hypomelanosis, whereas HLA-DR8 is not, in a group of renal transplant patients. Int J Dermatol. 2002;41:744-747.
- Wallace ML, Grichnik JM, Prieto VG, et al. Numbers and differentiation status of melanocytes in idiopathic guttate hypomelanosis. J Cutan Pathol. 1998;25:375-379.
- Ortonne JP, Perrot H. Idiopathic guttate hypomelanosis. ultrastructural study. Arch Dermatol. 1980;116:664-668.
- Rerknimitr P, Disphanurat W, Achariyakul M. Topical tacrolimus significantly promotes repigmentation in idiopathic guttate hypomelanosis: a double-blind, randomized, placebo-controlled study. J Eur Acad Dermatol Venereol. 2013;27:460-464.
- Pagnoni A, Kligman AM, Sadiq I, et al. Hypopigmented macules of photodamaged skin and their treatment with topical tretinoin. Acta Derm Venereol. 1999;79:305-310.
- Kumarasinghe SP. 3-5 second cryotherapy is effective in idiopathic guttate hypomelanosis. J Dermatol. 2004;31:457-459.
- Hexsel DM. Treatment of idiopathic guttate hypomelanosis by localized superficial dermabrasion. Dermatol Surg. 1999;25:917-918.
- Shin J, Kim M, Park SH, et al. The effect of fractional carbon dioxide lasers on idiopathic guttate hypomelanosis: a preliminary study. J Eur Acad Dermatol Venereol. 2013;27:e243-e246.
- Shin MK, Jeong KH, Oh IH, et al. Clinical features of idiopathic guttate hypomelanosis in 646 subjects and association with other aspects of photoaging. Int J Dermatol. 2011;50:798-805.
- Kim SK, Kim EH, Kang HY, et al. Comprehensive understanding of idiopathic guttate hypomelanosis: clinical and histopathological correlation. Int J Dermatol. 2010;49:162-166.
- Friedland R, David M, Feinmesser M, et al. Idiopathic guttate hypomelanosis-like lesions in patients with mycosis fungoides: a new adverse effect of phototherapy. J Eur Acad Dermatol Venereol. 2010;24:1026-1030.
- Kaya TI, Yazici AC, Tursen U, et al. Idiopathic guttate hypomelanosis: idiopathic or ultraviolet induced? Photodermatol Photoimmunol Photomed. 2005;21:270-271.
- Loquai C, Metze D, Nashan D, et al. Confetti-like lesions with hyperkeratosis: a novel ultraviolet-induced hypomelanotic disorder? Br J Dermatol. 2005;153:190-193.
- Arrunategui A, Trujillo RA, Marulanda MP, et al. HLA-DQ3 is associated with idiopathic guttate hypomelanosis, whereas HLA-DR8 is not, in a group of renal transplant patients. Int J Dermatol. 2002;41:744-747.
- Wallace ML, Grichnik JM, Prieto VG, et al. Numbers and differentiation status of melanocytes in idiopathic guttate hypomelanosis. J Cutan Pathol. 1998;25:375-379.
- Ortonne JP, Perrot H. Idiopathic guttate hypomelanosis. ultrastructural study. Arch Dermatol. 1980;116:664-668.
- Rerknimitr P, Disphanurat W, Achariyakul M. Topical tacrolimus significantly promotes repigmentation in idiopathic guttate hypomelanosis: a double-blind, randomized, placebo-controlled study. J Eur Acad Dermatol Venereol. 2013;27:460-464.
- Pagnoni A, Kligman AM, Sadiq I, et al. Hypopigmented macules of photodamaged skin and their treatment with topical tretinoin. Acta Derm Venereol. 1999;79:305-310.
- Kumarasinghe SP. 3-5 second cryotherapy is effective in idiopathic guttate hypomelanosis. J Dermatol. 2004;31:457-459.
- Hexsel DM. Treatment of idiopathic guttate hypomelanosis by localized superficial dermabrasion. Dermatol Surg. 1999;25:917-918.
- Shin J, Kim M, Park SH, et al. The effect of fractional carbon dioxide lasers on idiopathic guttate hypomelanosis: a preliminary study. J Eur Acad Dermatol Venereol. 2013;27:e243-e246.
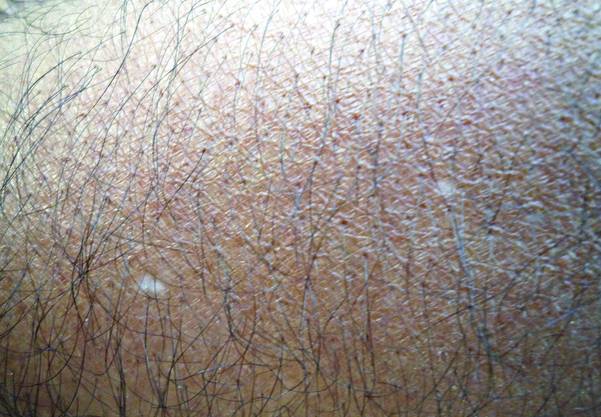
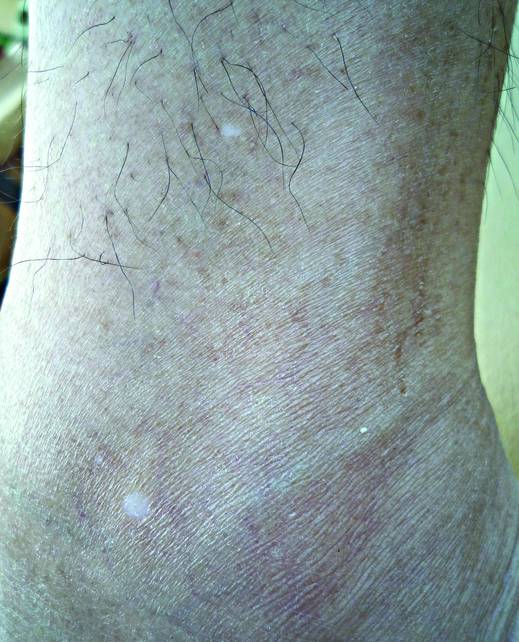
A 58-year-old man presented with disseminated, hypopigmented, asymptomatic lesions on the right arm (top) and left leg (bottom) that had been present for approximately 6 years. The patient reported that the lesions had become more visible and greater in number within the last year. Multiple circular hypopigmented macules of various sizes ranging from 1 to 3 mm in diameter were identified. No scaling was seen. Physical examination was otherwise unremarkable.
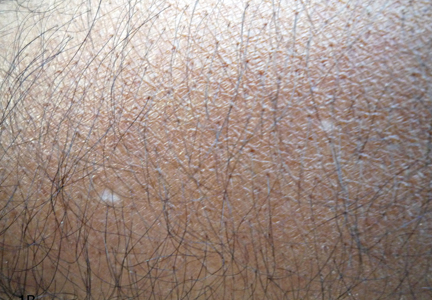
Top 10 treatments for vitiligo
PARK CITY, UTAH – At the annual meeting of the Pacific Dermatologic Association, Dr. Sancy A. Leachman offered a top 10 list of new agents and technologies for the treatment of vitiligo.
No. 10: Ultraviolet A1 (UVA1) phototherapy
Dr. Harvey Lui at the University of British Columbia in Vancouver is leading a phase II trial to evaluate the potential for UVA1 to induce repigmentation within vitiligo patches and to assess the side effect profile of the treatment. “I think it might work,” said Dr. Leachman, professor and chair of dermatology at Oregon Health & Science University (OHSU), Portland.

No. 9: Ginkgo biloba
The use of ginko biloba 40-60 mg 2-3 times per day, 10 minutes before a meal, was mentioned in a Cochrane Review of vitiligo treatments published on Feb. 24, 2015. “I think I’m going to give this a try in people who have failed other treatments and see if I can get some response,” Dr. Leachman said.
No. 8: Red light
Dr. Lui is leading a randomized phase II trial of low-intensity and high-intensity red light versus no treatment for vitiligo patches. Treatments will be given twice weekly for 10 weeks, with follow-up assessments at 4, 8, and 12 weeks post treatment.
No. 7: Micrografting
A novel suction blister device known as the CelluTome epidermal harvesting system uses heat and slight vacuum pressure to harvest healthy epidermal skin tissue without damaging the donor site. Dr. Leachman characterized the technology as “semiautomating the process of suction graft transplantation.”
No. 6: The ReCell device
Manufactured by Avita Medical, this investigational autologous cell harvesting device is used after CO2 abrasion and enables clinicians to create regenerative epithelial suspension with a small sample of the patient’s skin. A phase IV trial in the Netherlands is underway to assess the efficacy and safety of autologous epidermal cell suspension grafting with the ReCell device after CO2 laser abrasion, compared with CO2 laser abrasion alone and no treatment, in patients with piebaldism and stable vitiligo.
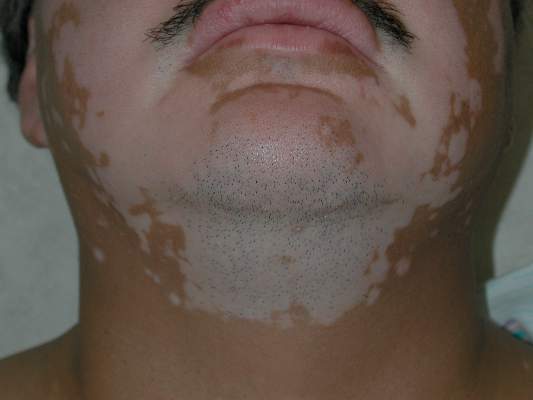
No. 5: Topical Photocil
In a pilot study sponsored by Applied Biology, researchers are enrolling patients with vitiligo to assess the safety and efficacy of Photocil. The primary outcome measure is the Vitiligo Area Severity Index (VASI). “When this cream is activated by sunlight, it degrades into narrow-band and UVB light, so you can put a topical cream on that will administer narrow-band UVB only in that spot,” said Dr. Leachman, who is also director of OHSU’s Knight Melanoma Research Program. “That’s amazing to me.”
No. 4: Afamelanotide
This is an analogue of a melanocyte-stimulating hormone. A randomized study conducted at two academic medical centers found that the combination of afamelanotide implant and narrow-band UVB phototherapy resulted in statistically superior and faster repigmentation, compared with narrow-band UVB monotherapy (JAMA Dermatol. 2015 Jan;151(1):42-50).
No. 3: Abatacept (Orencia)
This is a soluble fusion protein consisting of human cytotoxic T-lymphocyte–associated antigen 4 (CTLA4), which prevents T-cell activation. A phase I trial is underway at Brigham and Women’s Hospital in Boston to determine if weekly self-injections of the agent lead to clinical improvements of vitiligo lesions. The primary outcome measure is change in repigmentation with abatacept therapy based on the VASI score.
No. 2. Simvastatin
The notion of its use is based on STAT1 inhibition reducing interferon-gamma–dependent activation of CD8-positive T cells, according to Dr. Leachman. The concept has been successful in a mouse model, and a study in humans was recently completed by Dr. John Harris at the University of Massachusetts, Worcester. “What we have is the ability to apply an existing drug (Simvastatin) to the process and see if it works,” she said. “Wouldn’t it be cool if we could give a statin and improve vitiligo?”
No 1: Tofacitinib
This is a Janus kinase inhibitor commonly used for rheumatoid arthritis. According to Dr. Leachman, Janus kinase inhibition prevents STAT activation, “which prevents [interferon]-gamma production, which reduces activation of CD8-positive T cells via CXCL10 binding to CXCR3,” she said. A case report demonstrating its efficacy in a 53-year-old patient was recently published in JAMA Dermatology by Dr. Brett A. King and Dr. Brittany Craiglow, dermatologists at Yale School of Medicine, New Haven, Conn. “I’m hopeful that this [agent] will be made into a topical cream because these drugs do have substantial side effects,” Dr. Leachman said.
Dr. Leachman disclosed that she is a member of the medical and scientific advisory board for Myriad Genetics Laboratory. She has also participated in an advisory board meeting for Castle Biosciences and has participated in the DecisionDx registry.
PARK CITY, UTAH – At the annual meeting of the Pacific Dermatologic Association, Dr. Sancy A. Leachman offered a top 10 list of new agents and technologies for the treatment of vitiligo.
No. 10: Ultraviolet A1 (UVA1) phototherapy
Dr. Harvey Lui at the University of British Columbia in Vancouver is leading a phase II trial to evaluate the potential for UVA1 to induce repigmentation within vitiligo patches and to assess the side effect profile of the treatment. “I think it might work,” said Dr. Leachman, professor and chair of dermatology at Oregon Health & Science University (OHSU), Portland.
No. 9: Ginkgo biloba
The use of ginko biloba 40-60 mg 2-3 times per day, 10 minutes before a meal, was mentioned in a Cochrane Review of vitiligo treatments published on Feb. 24, 2015. “I think I’m going to give this a try in people who have failed other treatments and see if I can get some response,” Dr. Leachman said.
No. 8: Red light
Dr. Lui is leading a randomized phase II trial of low-intensity and high-intensity red light versus no treatment for vitiligo patches. Treatments will be given twice weekly for 10 weeks, with follow-up assessments at 4, 8, and 12 weeks post treatment.
No. 7: Micrografting
A novel suction blister device known as the CelluTome epidermal harvesting system uses heat and slight vacuum pressure to harvest healthy epidermal skin tissue without damaging the donor site. Dr. Leachman characterized the technology as “semiautomating the process of suction graft transplantation.”
No. 6: The ReCell device
Manufactured by Avita Medical, this investigational autologous cell harvesting device is used after CO2 abrasion and enables clinicians to create regenerative epithelial suspension with a small sample of the patient’s skin. A phase IV trial in the Netherlands is underway to assess the efficacy and safety of autologous epidermal cell suspension grafting with the ReCell device after CO2 laser abrasion, compared with CO2 laser abrasion alone and no treatment, in patients with piebaldism and stable vitiligo.
No. 5: Topical Photocil
In a pilot study sponsored by Applied Biology, researchers are enrolling patients with vitiligo to assess the safety and efficacy of Photocil. The primary outcome measure is the Vitiligo Area Severity Index (VASI). “When this cream is activated by sunlight, it degrades into narrow-band and UVB light, so you can put a topical cream on that will administer narrow-band UVB only in that spot,” said Dr. Leachman, who is also director of OHSU’s Knight Melanoma Research Program. “That’s amazing to me.”
No. 4: Afamelanotide
This is an analogue of a melanocyte-stimulating hormone. A randomized study conducted at two academic medical centers found that the combination of afamelanotide implant and narrow-band UVB phototherapy resulted in statistically superior and faster repigmentation, compared with narrow-band UVB monotherapy (JAMA Dermatol. 2015 Jan;151(1):42-50).
No. 3: Abatacept (Orencia)
This is a soluble fusion protein consisting of human cytotoxic T-lymphocyte–associated antigen 4 (CTLA4), which prevents T-cell activation. A phase I trial is underway at Brigham and Women’s Hospital in Boston to determine if weekly self-injections of the agent lead to clinical improvements of vitiligo lesions. The primary outcome measure is change in repigmentation with abatacept therapy based on the VASI score.
No. 2. Simvastatin
The notion of its use is based on STAT1 inhibition reducing interferon-gamma–dependent activation of CD8-positive T cells, according to Dr. Leachman. The concept has been successful in a mouse model, and a study in humans was recently completed by Dr. John Harris at the University of Massachusetts, Worcester. “What we have is the ability to apply an existing drug (Simvastatin) to the process and see if it works,” she said. “Wouldn’t it be cool if we could give a statin and improve vitiligo?”
No 1: Tofacitinib
This is a Janus kinase inhibitor commonly used for rheumatoid arthritis. According to Dr. Leachman, Janus kinase inhibition prevents STAT activation, “which prevents [interferon]-gamma production, which reduces activation of CD8-positive T cells via CXCL10 binding to CXCR3,” she said. A case report demonstrating its efficacy in a 53-year-old patient was recently published in JAMA Dermatology by Dr. Brett A. King and Dr. Brittany Craiglow, dermatologists at Yale School of Medicine, New Haven, Conn. “I’m hopeful that this [agent] will be made into a topical cream because these drugs do have substantial side effects,” Dr. Leachman said.
Dr. Leachman disclosed that she is a member of the medical and scientific advisory board for Myriad Genetics Laboratory. She has also participated in an advisory board meeting for Castle Biosciences and has participated in the DecisionDx registry.
PARK CITY, UTAH – At the annual meeting of the Pacific Dermatologic Association, Dr. Sancy A. Leachman offered a top 10 list of new agents and technologies for the treatment of vitiligo.
No. 10: Ultraviolet A1 (UVA1) phototherapy
Dr. Harvey Lui at the University of British Columbia in Vancouver is leading a phase II trial to evaluate the potential for UVA1 to induce repigmentation within vitiligo patches and to assess the side effect profile of the treatment. “I think it might work,” said Dr. Leachman, professor and chair of dermatology at Oregon Health & Science University (OHSU), Portland.
No. 9: Ginkgo biloba
The use of ginko biloba 40-60 mg 2-3 times per day, 10 minutes before a meal, was mentioned in a Cochrane Review of vitiligo treatments published on Feb. 24, 2015. “I think I’m going to give this a try in people who have failed other treatments and see if I can get some response,” Dr. Leachman said.
No. 8: Red light
Dr. Lui is leading a randomized phase II trial of low-intensity and high-intensity red light versus no treatment for vitiligo patches. Treatments will be given twice weekly for 10 weeks, with follow-up assessments at 4, 8, and 12 weeks post treatment.
No. 7: Micrografting
A novel suction blister device known as the CelluTome epidermal harvesting system uses heat and slight vacuum pressure to harvest healthy epidermal skin tissue without damaging the donor site. Dr. Leachman characterized the technology as “semiautomating the process of suction graft transplantation.”
No. 6: The ReCell device
Manufactured by Avita Medical, this investigational autologous cell harvesting device is used after CO2 abrasion and enables clinicians to create regenerative epithelial suspension with a small sample of the patient’s skin. A phase IV trial in the Netherlands is underway to assess the efficacy and safety of autologous epidermal cell suspension grafting with the ReCell device after CO2 laser abrasion, compared with CO2 laser abrasion alone and no treatment, in patients with piebaldism and stable vitiligo.
No. 5: Topical Photocil
In a pilot study sponsored by Applied Biology, researchers are enrolling patients with vitiligo to assess the safety and efficacy of Photocil. The primary outcome measure is the Vitiligo Area Severity Index (VASI). “When this cream is activated by sunlight, it degrades into narrow-band and UVB light, so you can put a topical cream on that will administer narrow-band UVB only in that spot,” said Dr. Leachman, who is also director of OHSU’s Knight Melanoma Research Program. “That’s amazing to me.”
No. 4: Afamelanotide
This is an analogue of a melanocyte-stimulating hormone. A randomized study conducted at two academic medical centers found that the combination of afamelanotide implant and narrow-band UVB phototherapy resulted in statistically superior and faster repigmentation, compared with narrow-band UVB monotherapy (JAMA Dermatol. 2015 Jan;151(1):42-50).
No. 3: Abatacept (Orencia)
This is a soluble fusion protein consisting of human cytotoxic T-lymphocyte–associated antigen 4 (CTLA4), which prevents T-cell activation. A phase I trial is underway at Brigham and Women’s Hospital in Boston to determine if weekly self-injections of the agent lead to clinical improvements of vitiligo lesions. The primary outcome measure is change in repigmentation with abatacept therapy based on the VASI score.
No. 2. Simvastatin
The notion of its use is based on STAT1 inhibition reducing interferon-gamma–dependent activation of CD8-positive T cells, according to Dr. Leachman. The concept has been successful in a mouse model, and a study in humans was recently completed by Dr. John Harris at the University of Massachusetts, Worcester. “What we have is the ability to apply an existing drug (Simvastatin) to the process and see if it works,” she said. “Wouldn’t it be cool if we could give a statin and improve vitiligo?”
No 1: Tofacitinib
This is a Janus kinase inhibitor commonly used for rheumatoid arthritis. According to Dr. Leachman, Janus kinase inhibition prevents STAT activation, “which prevents [interferon]-gamma production, which reduces activation of CD8-positive T cells via CXCL10 binding to CXCR3,” she said. A case report demonstrating its efficacy in a 53-year-old patient was recently published in JAMA Dermatology by Dr. Brett A. King and Dr. Brittany Craiglow, dermatologists at Yale School of Medicine, New Haven, Conn. “I’m hopeful that this [agent] will be made into a topical cream because these drugs do have substantial side effects,” Dr. Leachman said.
Dr. Leachman disclosed that she is a member of the medical and scientific advisory board for Myriad Genetics Laboratory. She has also participated in an advisory board meeting for Castle Biosciences and has participated in the DecisionDx registry.
EXPERT ANALYSIS AT PDA 2015

WCD: Dapsone gel effective for acne in women of color
VANCOUVER – Dapsone gel 5% proved effective and well tolerated for facial acne in women with skin of color in a multicenter pilot study.
The study was conducted because even though dapsone gel 5% (Aczone) is approved for the treatment of acne on the strength of two pivotal randomized, double-blind clinical trials totaling more than 3,000 patients, scant data exist on the topical agent’s performance in women with skin of color, Dr. Andrew F. Alexis explained at the World Congress of Dermatology.

He presented an open-label, seven-center, 12-week, single-arm study involving 68 women of color – three-quarters of whom were black – who treated their facial acne with dapsone gel 5% twice daily as monotherapy.
Participants averaged a mean baseline score of 2.6 on the 0-4 Global Acne Assessment Score (GAAS), with a mean total of 50 inflammatory and noninflammatory acne lesions on the face.
The primary endpoint was change in GAAS at 12 weeks, although patients also were formally assessed at 2 and 6 weeks. The average reduction in GAAS was 8.8% at 2 weeks, 20% at 6 weeks, and 39% at 12 weeks. At week 12, 43% of the women were categorized as responders, meaning they had a GAAS of 0 (meaning no acne lesions) or 1 (indicating mild disease), reported Dr. Alexis of Mt. Sinai Hospital in New York.
Total lesion counts dropped steadily throughout the 12-week trial: by 16% from baseline to week 2, 30% at week 6, and 52% at week 12. Inflammatory lesions responded best, with a 65% reduction in number at week 12.
Patient-reported outcomes on the validated, 17-item Acne Symptom and Impact Scale were favorable: Reductions of roughly 50% were documented over 12 weeks on the scale’s two domains, acne signs and quality of life impact.
No clinically meaningful treatment-related adverse events were reported in the study, although a handful of women reported trace levels of redness, burning, dryness, and/or oiliness.
Acne is more common among African American than white women. In a large epidemiologic study of adolescent and adult women, the prevalence of acne vulgaris was 37% in African Americans, compared with 24% in whites (J. Eur. Acad. Dermatol. Venereol. 2011;25:1054-60).
Dr. Alexis’ study was sponsored by Allergan. He reported serving as a consultant to and receiving research grants from the company.
VANCOUVER – Dapsone gel 5% proved effective and well tolerated for facial acne in women with skin of color in a multicenter pilot study.
The study was conducted because even though dapsone gel 5% (Aczone) is approved for the treatment of acne on the strength of two pivotal randomized, double-blind clinical trials totaling more than 3,000 patients, scant data exist on the topical agent’s performance in women with skin of color, Dr. Andrew F. Alexis explained at the World Congress of Dermatology.
He presented an open-label, seven-center, 12-week, single-arm study involving 68 women of color – three-quarters of whom were black – who treated their facial acne with dapsone gel 5% twice daily as monotherapy.
Participants averaged a mean baseline score of 2.6 on the 0-4 Global Acne Assessment Score (GAAS), with a mean total of 50 inflammatory and noninflammatory acne lesions on the face.
The primary endpoint was change in GAAS at 12 weeks, although patients also were formally assessed at 2 and 6 weeks. The average reduction in GAAS was 8.8% at 2 weeks, 20% at 6 weeks, and 39% at 12 weeks. At week 12, 43% of the women were categorized as responders, meaning they had a GAAS of 0 (meaning no acne lesions) or 1 (indicating mild disease), reported Dr. Alexis of Mt. Sinai Hospital in New York.
Total lesion counts dropped steadily throughout the 12-week trial: by 16% from baseline to week 2, 30% at week 6, and 52% at week 12. Inflammatory lesions responded best, with a 65% reduction in number at week 12.
Patient-reported outcomes on the validated, 17-item Acne Symptom and Impact Scale were favorable: Reductions of roughly 50% were documented over 12 weeks on the scale’s two domains, acne signs and quality of life impact.
No clinically meaningful treatment-related adverse events were reported in the study, although a handful of women reported trace levels of redness, burning, dryness, and/or oiliness.
Acne is more common among African American than white women. In a large epidemiologic study of adolescent and adult women, the prevalence of acne vulgaris was 37% in African Americans, compared with 24% in whites (J. Eur. Acad. Dermatol. Venereol. 2011;25:1054-60).
Dr. Alexis’ study was sponsored by Allergan. He reported serving as a consultant to and receiving research grants from the company.
VANCOUVER – Dapsone gel 5% proved effective and well tolerated for facial acne in women with skin of color in a multicenter pilot study.
The study was conducted because even though dapsone gel 5% (Aczone) is approved for the treatment of acne on the strength of two pivotal randomized, double-blind clinical trials totaling more than 3,000 patients, scant data exist on the topical agent’s performance in women with skin of color, Dr. Andrew F. Alexis explained at the World Congress of Dermatology.
He presented an open-label, seven-center, 12-week, single-arm study involving 68 women of color – three-quarters of whom were black – who treated their facial acne with dapsone gel 5% twice daily as monotherapy.
Participants averaged a mean baseline score of 2.6 on the 0-4 Global Acne Assessment Score (GAAS), with a mean total of 50 inflammatory and noninflammatory acne lesions on the face.
The primary endpoint was change in GAAS at 12 weeks, although patients also were formally assessed at 2 and 6 weeks. The average reduction in GAAS was 8.8% at 2 weeks, 20% at 6 weeks, and 39% at 12 weeks. At week 12, 43% of the women were categorized as responders, meaning they had a GAAS of 0 (meaning no acne lesions) or 1 (indicating mild disease), reported Dr. Alexis of Mt. Sinai Hospital in New York.
Total lesion counts dropped steadily throughout the 12-week trial: by 16% from baseline to week 2, 30% at week 6, and 52% at week 12. Inflammatory lesions responded best, with a 65% reduction in number at week 12.
Patient-reported outcomes on the validated, 17-item Acne Symptom and Impact Scale were favorable: Reductions of roughly 50% were documented over 12 weeks on the scale’s two domains, acne signs and quality of life impact.
No clinically meaningful treatment-related adverse events were reported in the study, although a handful of women reported trace levels of redness, burning, dryness, and/or oiliness.
Acne is more common among African American than white women. In a large epidemiologic study of adolescent and adult women, the prevalence of acne vulgaris was 37% in African Americans, compared with 24% in whites (J. Eur. Acad. Dermatol. Venereol. 2011;25:1054-60).
Dr. Alexis’ study was sponsored by Allergan. He reported serving as a consultant to and receiving research grants from the company.
AT WCD 2015
Key clinical point: Dapsone gel 5% is effective and well tolerated for treatment of facial acne in women with skin of color.
Major finding: Women of color experienced a mean 39% reduction in Global Acne Assessment Scores after 12 weeks of self-treatment of facial acne using dapsone gel 5% twice daily as monotherapy.
Data source: This was a 68-patient, open-label, seven-site, single-arm, 12-week study.
Disclosures: The study was sponsored by Allergan. Dr. Andrew F. Alexis reported serving as a consultant to and receiving research grants from the company.

Progressive Cribriform and Zosteriform Hyperpigmentation
To the Editor:
Progressive cribriform and zosteriform hyperpigmentation (PCZH) was first described by Rower et al1 in 1978. The diagnostic criteria included the following: (1) uniformly tan cribriform macular pigmentation in a zosteriform distribution; (2) a histologic pattern that consisted of a mild increase in melanin pigment in the basal cell layer and complete absence of nevus cells; (3) no history of rash, injury, or inflammation to suggest postinflammatory hyperpigmentation; (4) onset occurring well after birth with gradual extension; and (5) lack of other associated cutaneous or internal abnormalities.1
Many pigmentary disorders occurring along the Blaschko lines are included in differential diagnosis of PCZH such as incontinentia pigmenti (IP), progressive zosteriform macular pigmented lesion (PZMPL), and linear and whorled nevoid hypermelanosis (LWNH). However, PCZH is considered to be the localized variant (the late onset) of LWNH.2 We report a case of PCZH, a segmented and delayed form of LWNH.
A 25-year-old woman presented with asymptomatic progressive multiple brownish macular eruptions arranged in a zosteriform pattern on the left arm and thigh of 3 months’ duration. There was no history of injury or any prior cutaneous changes. There was no personal or family history of similar eruptions and she was otherwise in good health. She was not taking any medications. Physical examination showed linear, uniformly tanned, cribriform hyperpigmentation along the Blaschko lines on the left arm and thigh (Figure 1). Routine laboratory tests, including complete blood cell count with differential, were normal. Assuming a diagnosis of PCZH or PZMPL, we performed a punch biopsy on the left upper arm. The histopathologic findings showed increased pigmentation of the basal layer. There were a few dermal melanophages and no nevus cells present (Figure 2A). Fontana-Masson stain showed an increase in melanin in the basal layer (Figure 2B). On the basis of these clinical and histological findings, a diagnosis of PCZH was made. She was observed without treatment for 6 months showing no change.
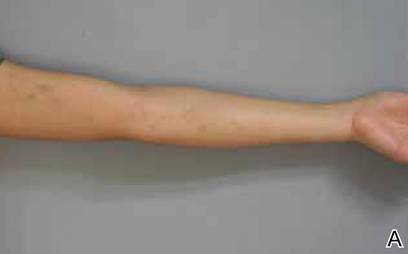
|
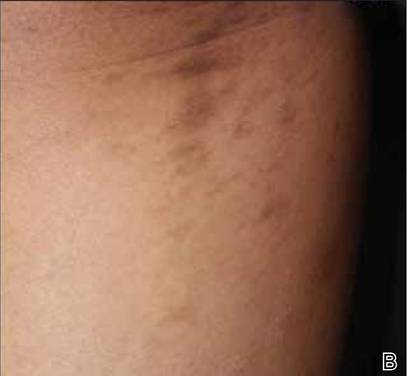
|
Progressive cribriform and zosteriform hyperpigmentation is a disorder of pigmentation along the Blaschko lines. The trunk is the most common site of involvement.3 In the differential diagnosis, other pigmentary disorders along the Blaschko lines must be excluded, including the pigmentary stage of IP, PZMPL, and LWNH. In IP, characteristic inflammatory vesicular and verrucous stages usually precede the whorled pigmentation.4 In approximately 80% of cases, IP is associated with various congenital abnormalities, particularly of the central nervous system, eyes, and teeth.5 Progressive zosteriform macular pigmented lesion is a chronic pigmentary dermatosis similar to PCZH but is characteristically accompanied by pruritus as a prodromal symptom. It is usually preceded by multiple pruritic macular pigmentation in part of the dermatome for a period of time. Then the size and number of the pigmented macules abruptly increases and coalesces into patches.6 Linear and whorled nevoid hypermelanosis was first described by Kalter et al7 in 1988. It is characterized by swirls and whorls of hyperpigmented macules without preceding bullae or verrucae along Blaschko lines, usually occurring within the first 2 years of life. The lesions are stable in some patients but can spread in others, stabilizing by 2 to 3 years of age.7-10 It has been referred to as zosteriform lentiginous nevus, zebralike hyperpigmentation, and reticulate hyperpigmentation distributed in a zosteriform fashion.2,9
Linear and whorled nevoid hypermelanosis can be distinguished from PCZH by a diffuse or localized pattern and an association of congenital anomalies.3 However, neurologic and skeletal anomalies also can be observed in PCZH.11 Additionally, not all LWNH cases show a diffuse type.2 Therefore, LWNH has been used to encompass a wide spectrum of clinical entities, ranging from the congenital or perinatal form described by Kalter et al7 to the segmented and delayed form described by Rower et al1 for which there is a tendency to use the term progressive cribriform and zosteriform hyperpigmentation.2,10,11 There are no clinical and histologic differences between PCZH and LWNH, other than a later onset.2 Although some authors reported that PCZH and LWNH have increased hyperpigmentation of the basal layer and prominent melanocytes without incontinence of pigment on histopathology,2,7,8 other reports have demonstrated that both could show pigment incontinence,3,10,12-14 such as in our case.
Figure 2. Histopathologic findings showed increased pigmentation of the basal layer with a few dermal melanophages. No nevus cells were present (A)(H&E, original magnification ×100). Fontana-Masson stain showed an increase in melanin in the basal layer (B)(original magnification ×100). |
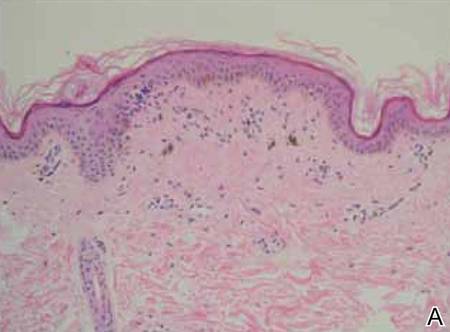
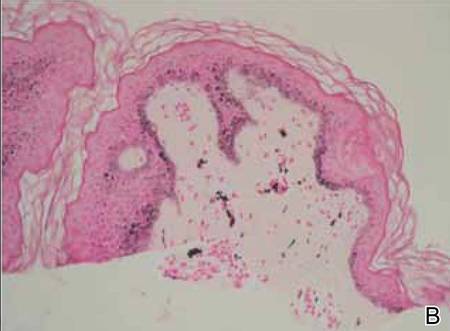
Progressive cribriform and zosteriform hyperpigmentation is considered to be the localized variant as well as the late onset of LWNH.2 We report a case of PCZH, a segmented and delayed form of LWNH without systemic abnormalities.
1. Rower JM, Carr RD, Lowney ED. Progressive cribriform and zosteriform hyperpigmentation. Arch Dermatol. 1978;114:98-99.
2. Di Lernia V. Linear and whorled hypermelanosis. Pediatr Dermatol. 2007;24:205-210.
3. Cho E, Cho SH, Lee JD. Progressive cribriform and zosteriform hyperpigmentation: a clinicopathologic study. Int J Dermatol. 2012;51:399-405.
4. Hong SP, Ahn SY, Lee WS. Linear and whorled nevoid hypermelanosis: unique clinical presentations and their possible association with chromosomal abnormality inv(9). Arch Dermatol. 2008;144:415-416.
5. Carney RG. Incontinentia pigmenti: a world statistical analysis. Arch Dermatol. 1976;112:535-542.
6. Hong JW, Lee KY, Jeon SY, et al. Progressive zosteriform macular pigmented lesion. Korean J Dermatol. 2011;49:621-624
7. Kalter DC, Griffiths WA, Atherton AJ. Linear and whorled nevoid hypermelanosis. J Am Acad Dermatol. 1988;19:1037-1044.
8. Ertam I, Turk BG, Urkmez A, et al. Linear and whorled nevoid hypermelanosis: dermatoscopic features. J Am Acad Dermatol. 2009;60:328-331.
9. Mehta V, Vasanth V, Balachandran C, et al. Linear and whorled nevoid hypermelanosis. Int J Dermatol. 2011;50:491-492.
10. Choi JC, Yang JH, Lee UH, et al. Progressive cribriform and zosteriform hyperpigmentation—the late onset linear and whorled nevoid hypermelanosis. J Eur Acad Dermatol Venereol. 2005;19:638-639.
11. Schepis C, Alberti A, Siragusa M, et al. Progressive cribriform and zosteriform hyperpigmentation: the late onset feature of linear and whorled nevoid hypermelanosis associated with congenital neurological, skeletal and cutaneous anomalies. Dermatology. 1999;199:72-73.
12. Kovarik CL, Spielvogel RL, Kantor GR. Pigmentary disorders of the skin. In: Elder DE, Elenitsas R, Murphy GF, et al, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:690.
13. Kim SJ, Kim MB, Oh CK, et al. Three cases of progressive cribriform and zosteriform hyperpigmentation. Korean J Dermatol. 2002;40:181-186.
14. Cho SH, Ha JH, Choi HC, et al. A case of atypical progressive cribriform and zosteriform hyperpigmentation. Korean J Dermatol. 2003;41:792-795.
To the Editor:
Progressive cribriform and zosteriform hyperpigmentation (PCZH) was first described by Rower et al1 in 1978. The diagnostic criteria included the following: (1) uniformly tan cribriform macular pigmentation in a zosteriform distribution; (2) a histologic pattern that consisted of a mild increase in melanin pigment in the basal cell layer and complete absence of nevus cells; (3) no history of rash, injury, or inflammation to suggest postinflammatory hyperpigmentation; (4) onset occurring well after birth with gradual extension; and (5) lack of other associated cutaneous or internal abnormalities.1
Many pigmentary disorders occurring along the Blaschko lines are included in differential diagnosis of PCZH such as incontinentia pigmenti (IP), progressive zosteriform macular pigmented lesion (PZMPL), and linear and whorled nevoid hypermelanosis (LWNH). However, PCZH is considered to be the localized variant (the late onset) of LWNH.2 We report a case of PCZH, a segmented and delayed form of LWNH.
A 25-year-old woman presented with asymptomatic progressive multiple brownish macular eruptions arranged in a zosteriform pattern on the left arm and thigh of 3 months’ duration. There was no history of injury or any prior cutaneous changes. There was no personal or family history of similar eruptions and she was otherwise in good health. She was not taking any medications. Physical examination showed linear, uniformly tanned, cribriform hyperpigmentation along the Blaschko lines on the left arm and thigh (Figure 1). Routine laboratory tests, including complete blood cell count with differential, were normal. Assuming a diagnosis of PCZH or PZMPL, we performed a punch biopsy on the left upper arm. The histopathologic findings showed increased pigmentation of the basal layer. There were a few dermal melanophages and no nevus cells present (Figure 2A). Fontana-Masson stain showed an increase in melanin in the basal layer (Figure 2B). On the basis of these clinical and histological findings, a diagnosis of PCZH was made. She was observed without treatment for 6 months showing no change.
|
|
|
|
Progressive cribriform and zosteriform hyperpigmentation is a disorder of pigmentation along the Blaschko lines. The trunk is the most common site of involvement.3 In the differential diagnosis, other pigmentary disorders along the Blaschko lines must be excluded, including the pigmentary stage of IP, PZMPL, and LWNH. In IP, characteristic inflammatory vesicular and verrucous stages usually precede the whorled pigmentation.4 In approximately 80% of cases, IP is associated with various congenital abnormalities, particularly of the central nervous system, eyes, and teeth.5 Progressive zosteriform macular pigmented lesion is a chronic pigmentary dermatosis similar to PCZH but is characteristically accompanied by pruritus as a prodromal symptom. It is usually preceded by multiple pruritic macular pigmentation in part of the dermatome for a period of time. Then the size and number of the pigmented macules abruptly increases and coalesces into patches.6 Linear and whorled nevoid hypermelanosis was first described by Kalter et al7 in 1988. It is characterized by swirls and whorls of hyperpigmented macules without preceding bullae or verrucae along Blaschko lines, usually occurring within the first 2 years of life. The lesions are stable in some patients but can spread in others, stabilizing by 2 to 3 years of age.7-10 It has been referred to as zosteriform lentiginous nevus, zebralike hyperpigmentation, and reticulate hyperpigmentation distributed in a zosteriform fashion.2,9
Linear and whorled nevoid hypermelanosis can be distinguished from PCZH by a diffuse or localized pattern and an association of congenital anomalies.3 However, neurologic and skeletal anomalies also can be observed in PCZH.11 Additionally, not all LWNH cases show a diffuse type.2 Therefore, LWNH has been used to encompass a wide spectrum of clinical entities, ranging from the congenital or perinatal form described by Kalter et al7 to the segmented and delayed form described by Rower et al1 for which there is a tendency to use the term progressive cribriform and zosteriform hyperpigmentation.2,10,11 There are no clinical and histologic differences between PCZH and LWNH, other than a later onset.2 Although some authors reported that PCZH and LWNH have increased hyperpigmentation of the basal layer and prominent melanocytes without incontinence of pigment on histopathology,2,7,8 other reports have demonstrated that both could show pigment incontinence,3,10,12-14 such as in our case.
Figure 2. Histopathologic findings showed increased pigmentation of the basal layer with a few dermal melanophages. No nevus cells were present (A)(H&E, original magnification ×100). Fontana-Masson stain showed an increase in melanin in the basal layer (B)(original magnification ×100). |
Progressive cribriform and zosteriform hyperpigmentation is considered to be the localized variant as well as the late onset of LWNH.2 We report a case of PCZH, a segmented and delayed form of LWNH without systemic abnormalities.
To the Editor:
Progressive cribriform and zosteriform hyperpigmentation (PCZH) was first described by Rower et al1 in 1978. The diagnostic criteria included the following: (1) uniformly tan cribriform macular pigmentation in a zosteriform distribution; (2) a histologic pattern that consisted of a mild increase in melanin pigment in the basal cell layer and complete absence of nevus cells; (3) no history of rash, injury, or inflammation to suggest postinflammatory hyperpigmentation; (4) onset occurring well after birth with gradual extension; and (5) lack of other associated cutaneous or internal abnormalities.1
Many pigmentary disorders occurring along the Blaschko lines are included in differential diagnosis of PCZH such as incontinentia pigmenti (IP), progressive zosteriform macular pigmented lesion (PZMPL), and linear and whorled nevoid hypermelanosis (LWNH). However, PCZH is considered to be the localized variant (the late onset) of LWNH.2 We report a case of PCZH, a segmented and delayed form of LWNH.
A 25-year-old woman presented with asymptomatic progressive multiple brownish macular eruptions arranged in a zosteriform pattern on the left arm and thigh of 3 months’ duration. There was no history of injury or any prior cutaneous changes. There was no personal or family history of similar eruptions and she was otherwise in good health. She was not taking any medications. Physical examination showed linear, uniformly tanned, cribriform hyperpigmentation along the Blaschko lines on the left arm and thigh (Figure 1). Routine laboratory tests, including complete blood cell count with differential, were normal. Assuming a diagnosis of PCZH or PZMPL, we performed a punch biopsy on the left upper arm. The histopathologic findings showed increased pigmentation of the basal layer. There were a few dermal melanophages and no nevus cells present (Figure 2A). Fontana-Masson stain showed an increase in melanin in the basal layer (Figure 2B). On the basis of these clinical and histological findings, a diagnosis of PCZH was made. She was observed without treatment for 6 months showing no change.
|
|
|
|
Progressive cribriform and zosteriform hyperpigmentation is a disorder of pigmentation along the Blaschko lines. The trunk is the most common site of involvement.3 In the differential diagnosis, other pigmentary disorders along the Blaschko lines must be excluded, including the pigmentary stage of IP, PZMPL, and LWNH. In IP, characteristic inflammatory vesicular and verrucous stages usually precede the whorled pigmentation.4 In approximately 80% of cases, IP is associated with various congenital abnormalities, particularly of the central nervous system, eyes, and teeth.5 Progressive zosteriform macular pigmented lesion is a chronic pigmentary dermatosis similar to PCZH but is characteristically accompanied by pruritus as a prodromal symptom. It is usually preceded by multiple pruritic macular pigmentation in part of the dermatome for a period of time. Then the size and number of the pigmented macules abruptly increases and coalesces into patches.6 Linear and whorled nevoid hypermelanosis was first described by Kalter et al7 in 1988. It is characterized by swirls and whorls of hyperpigmented macules without preceding bullae or verrucae along Blaschko lines, usually occurring within the first 2 years of life. The lesions are stable in some patients but can spread in others, stabilizing by 2 to 3 years of age.7-10 It has been referred to as zosteriform lentiginous nevus, zebralike hyperpigmentation, and reticulate hyperpigmentation distributed in a zosteriform fashion.2,9
Linear and whorled nevoid hypermelanosis can be distinguished from PCZH by a diffuse or localized pattern and an association of congenital anomalies.3 However, neurologic and skeletal anomalies also can be observed in PCZH.11 Additionally, not all LWNH cases show a diffuse type.2 Therefore, LWNH has been used to encompass a wide spectrum of clinical entities, ranging from the congenital or perinatal form described by Kalter et al7 to the segmented and delayed form described by Rower et al1 for which there is a tendency to use the term progressive cribriform and zosteriform hyperpigmentation.2,10,11 There are no clinical and histologic differences between PCZH and LWNH, other than a later onset.2 Although some authors reported that PCZH and LWNH have increased hyperpigmentation of the basal layer and prominent melanocytes without incontinence of pigment on histopathology,2,7,8 other reports have demonstrated that both could show pigment incontinence,3,10,12-14 such as in our case.
Figure 2. Histopathologic findings showed increased pigmentation of the basal layer with a few dermal melanophages. No nevus cells were present (A)(H&E, original magnification ×100). Fontana-Masson stain showed an increase in melanin in the basal layer (B)(original magnification ×100). |
Progressive cribriform and zosteriform hyperpigmentation is considered to be the localized variant as well as the late onset of LWNH.2 We report a case of PCZH, a segmented and delayed form of LWNH without systemic abnormalities.
1. Rower JM, Carr RD, Lowney ED. Progressive cribriform and zosteriform hyperpigmentation. Arch Dermatol. 1978;114:98-99.
2. Di Lernia V. Linear and whorled hypermelanosis. Pediatr Dermatol. 2007;24:205-210.
3. Cho E, Cho SH, Lee JD. Progressive cribriform and zosteriform hyperpigmentation: a clinicopathologic study. Int J Dermatol. 2012;51:399-405.
4. Hong SP, Ahn SY, Lee WS. Linear and whorled nevoid hypermelanosis: unique clinical presentations and their possible association with chromosomal abnormality inv(9). Arch Dermatol. 2008;144:415-416.
5. Carney RG. Incontinentia pigmenti: a world statistical analysis. Arch Dermatol. 1976;112:535-542.
6. Hong JW, Lee KY, Jeon SY, et al. Progressive zosteriform macular pigmented lesion. Korean J Dermatol. 2011;49:621-624
7. Kalter DC, Griffiths WA, Atherton AJ. Linear and whorled nevoid hypermelanosis. J Am Acad Dermatol. 1988;19:1037-1044.
8. Ertam I, Turk BG, Urkmez A, et al. Linear and whorled nevoid hypermelanosis: dermatoscopic features. J Am Acad Dermatol. 2009;60:328-331.
9. Mehta V, Vasanth V, Balachandran C, et al. Linear and whorled nevoid hypermelanosis. Int J Dermatol. 2011;50:491-492.
10. Choi JC, Yang JH, Lee UH, et al. Progressive cribriform and zosteriform hyperpigmentation—the late onset linear and whorled nevoid hypermelanosis. J Eur Acad Dermatol Venereol. 2005;19:638-639.
11. Schepis C, Alberti A, Siragusa M, et al. Progressive cribriform and zosteriform hyperpigmentation: the late onset feature of linear and whorled nevoid hypermelanosis associated with congenital neurological, skeletal and cutaneous anomalies. Dermatology. 1999;199:72-73.
12. Kovarik CL, Spielvogel RL, Kantor GR. Pigmentary disorders of the skin. In: Elder DE, Elenitsas R, Murphy GF, et al, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:690.
13. Kim SJ, Kim MB, Oh CK, et al. Three cases of progressive cribriform and zosteriform hyperpigmentation. Korean J Dermatol. 2002;40:181-186.
14. Cho SH, Ha JH, Choi HC, et al. A case of atypical progressive cribriform and zosteriform hyperpigmentation. Korean J Dermatol. 2003;41:792-795.
1. Rower JM, Carr RD, Lowney ED. Progressive cribriform and zosteriform hyperpigmentation. Arch Dermatol. 1978;114:98-99.
2. Di Lernia V. Linear and whorled hypermelanosis. Pediatr Dermatol. 2007;24:205-210.
3. Cho E, Cho SH, Lee JD. Progressive cribriform and zosteriform hyperpigmentation: a clinicopathologic study. Int J Dermatol. 2012;51:399-405.
4. Hong SP, Ahn SY, Lee WS. Linear and whorled nevoid hypermelanosis: unique clinical presentations and their possible association with chromosomal abnormality inv(9). Arch Dermatol. 2008;144:415-416.
5. Carney RG. Incontinentia pigmenti: a world statistical analysis. Arch Dermatol. 1976;112:535-542.
6. Hong JW, Lee KY, Jeon SY, et al. Progressive zosteriform macular pigmented lesion. Korean J Dermatol. 2011;49:621-624
7. Kalter DC, Griffiths WA, Atherton AJ. Linear and whorled nevoid hypermelanosis. J Am Acad Dermatol. 1988;19:1037-1044.
8. Ertam I, Turk BG, Urkmez A, et al. Linear and whorled nevoid hypermelanosis: dermatoscopic features. J Am Acad Dermatol. 2009;60:328-331.
9. Mehta V, Vasanth V, Balachandran C, et al. Linear and whorled nevoid hypermelanosis. Int J Dermatol. 2011;50:491-492.
10. Choi JC, Yang JH, Lee UH, et al. Progressive cribriform and zosteriform hyperpigmentation—the late onset linear and whorled nevoid hypermelanosis. J Eur Acad Dermatol Venereol. 2005;19:638-639.
11. Schepis C, Alberti A, Siragusa M, et al. Progressive cribriform and zosteriform hyperpigmentation: the late onset feature of linear and whorled nevoid hypermelanosis associated with congenital neurological, skeletal and cutaneous anomalies. Dermatology. 1999;199:72-73.
12. Kovarik CL, Spielvogel RL, Kantor GR. Pigmentary disorders of the skin. In: Elder DE, Elenitsas R, Murphy GF, et al, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:690.
13. Kim SJ, Kim MB, Oh CK, et al. Three cases of progressive cribriform and zosteriform hyperpigmentation. Korean J Dermatol. 2002;40:181-186.
14. Cho SH, Ha JH, Choi HC, et al. A case of atypical progressive cribriform and zosteriform hyperpigmentation. Korean J Dermatol. 2003;41:792-795.
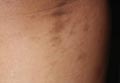
Vitiligo Disease Triggers: Psychological Stressors Preceding the Onset of Disease
Vitiligo is the loss of skin pigmentation caused by autoimmune destruction of melanocytes. Multiple pathogenic factors for vitiligo have been described, including CD8+ T lymphocyte/T helper 1 infiltrates in lesional skin1,2 with increased expression of IFN-γ3 and tumor necrosis factor α,3-6 decreased transforming growth factor β,7 and circulating autoantibodies against tyrosine hydroxylase.8 Additionally, several studies have found a high prevalence of antecedent psychological stressors in vitiligo patients, suggesting that specific stressors may trigger and/or exacerbate vitiligo.9-12
The relationship between antecedent psychological stressors and vitiligo extent has not been well studied. Potential mechanisms for stress-triggered vitiligo include increased catecholamines13 and neuropeptides,14 which have been found in vitiligo patients. However, the complex relationship between stressors and subsequent vitiligo is not well defined. We hypothesized that persistent stressors are associated with increased vitiligo extent.
Vitiligo is classically considered to be a silent pigmentary disorder with few or no symptoms. Prior studies have demonstrated that one-third of vitiligo patients report skin symptoms (eg, pruritus, burning), which may be specifically associated with early-onset disease.15-17 Further, we observed that some vitiligo patients report abdominal cramping associated with their disease. Few studies have described the burden of skin symptoms and other associated symptoms in vitiligo or their determinants.
We conducted a prospective questionnaire-based study of 1541 adult vitiligo patients to identify psychological factors that may precede vitiligo onset. We hypothesized that some types of stressors that occur within 2 years prior to disease onset would have specific associations with vitiligo and/or somatic symptoms.
Methods
Study Population and Questionnaire Distribution
This prospective questionnaire-based study was approved by the institutional review board at St. Luke’s-Roosevelt Hospital Center (now Mount Sinai St. Luke’s-Roosevelt) (New York, New York) for adults (>18 years; male or female) with vitiligo. The survey was validated in paper format at St. Luke’s-Roosevelt Hospital Center and distributed online to members of nonprofit support groups for vitiligo vulgaris, as previously described.15
Questionnaire
The a priori aim of this questionnaire was to identify psychological factors that may precede vitiligo onset. The questionnaire consisted of 77 items (55 closed questions and 22 open questions) pertaining to participant demographics/vitiligo phenotype and psychological stressors preceding vitiligo onset. The questions related to this study and response rates are listed in eTable 1. Responses were verified by screening for noninteger or implausible values (eg, <0 or >100 years of age).
Sample Size
The primary outcome used for sample size calculation was the potential association between vitiligo and the presence of antecedent psychological stressors. Using a 2-tailed test, we determined that a sample size of 1264 participants would have 90% power at α=.05 and a baseline proportion of 0.01 (1% presumed prevalence of vitiligo) to detect an odds ratio (OR) of 2.5 or higher.18
Data and Statistical Analysis
Closed question responses were analyzed using descriptive statistics. Open-ended question responses were analyzed using content analysis. Related comments were coded and grouped, with similarities and differences noted. All data processing and statistics were done with SAS version 9.2. Age at diagnosis (years) and number of anatomic sites affected were divided into tertiles for statistical analysis due to wide skewing.
Logistic regression models were constructed with numbers of reported deaths or stressors per participant within the 2 years prior to vitiligo onset as independent variables (0, 1, or ≥2), and symptoms associated with vitiligo as dependent variables. Adjusted ORs were calculated from multivariate models that included sex, current age (continuous), and comorbid autoimmune disease (binary) as covariates. Linear interaction terms were tested and were included in final models if statistically significant (P<.05).
Ordinal logistic regression was used to analyze the relationship between stressors (and other independent variables) and number of anatomic sites affected with vitiligo (tertiles). Ordinal logistic regression models were constructed to examine the impact of psychological stressors on pruritus secondary to vitiligo (not relevant combined with not at all, a little, a lot, very much) as the dependent variable. The proportional odds assumption was met in both models, as judged by score testing (P>.05). Binary logistic regression was used to analyze laterality, body surface area (BSA) greater than 25%, and involvement of the face and/or body with vitiligo lesions (binary).
Binary logistic regression models were constructed with impact of psychological stressors preceding vitiligo onset on comorbid abdominal cramping and specific etiologies as the dependent variables. There were 20 candidate stressors occurring within the 2 years prior to vitiligo onset. Selection methods for predictors were used to identify significant covariates within the context of the other covariates included in the final models. The results of forward, backward, and stepwise approaches were similar, and the stepwise selection output was presented.
Missing values were encountered because some participants did not respond to all the questionnaire items. A complete case analysis was performed (ie, missing values were ignored throughout the study). Data imputation was considered by multiple imputations; however, there were few or no differences between the estimates from the 2 approaches. Therefore, final models did not involve data imputation.
The statistical significance for all estimates was considered to be P<.05. However, a P value near .05 should be interpreted with caution given the multiple dependent tests performed in this study with increased risk for falsely rejecting the null hypothesis.
Results
Survey Population Characteristics
One thousand seven hundred participants started the survey; 1632 completed the survey (96.0% completion rate) and 1553 had been diagnosed with vitiligo by a physician. Twelve participants were excluded because they were younger than 18 years, leaving 1541 evaluable participants. Five hundred thirty-eight participants (34.9%) had comorbid autoimmune disorders. Demographics and disease phenotypes of the study participants are listed in Table 1.

Stressors Preceding Vitiligo Onset
Eight hundred twenty-one participants (56.6%) experienced at least one death or stressor within 2 years prior to vitiligo onset (Table 2), including death of a loved one (16.6%) and stressful life events (51.0%) within the 2 years prior to the onset of vitiligo, especially work/financial problems (10.8%), end of a long-term relationship (10.2%), and family problems (not otherwise specified)(7.8%). Two hundred (13.5%) participants reported experiencing 1 death and 46 (3.1%) reported multiple deaths. Five hundred participants (33.6%) reported experiencing 1 stressor and 259 (17.4%) reported multiple stressors.
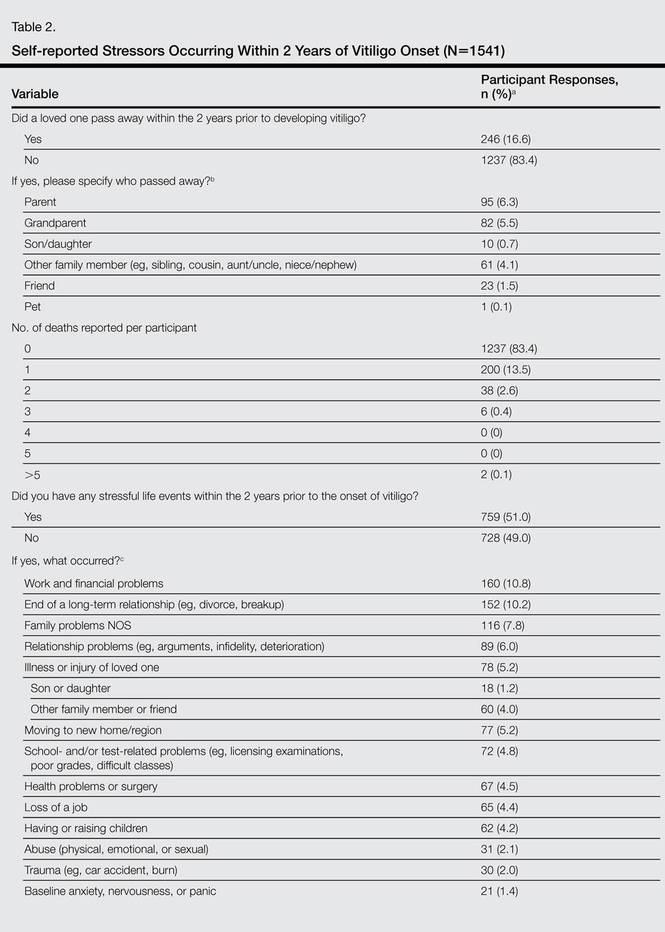
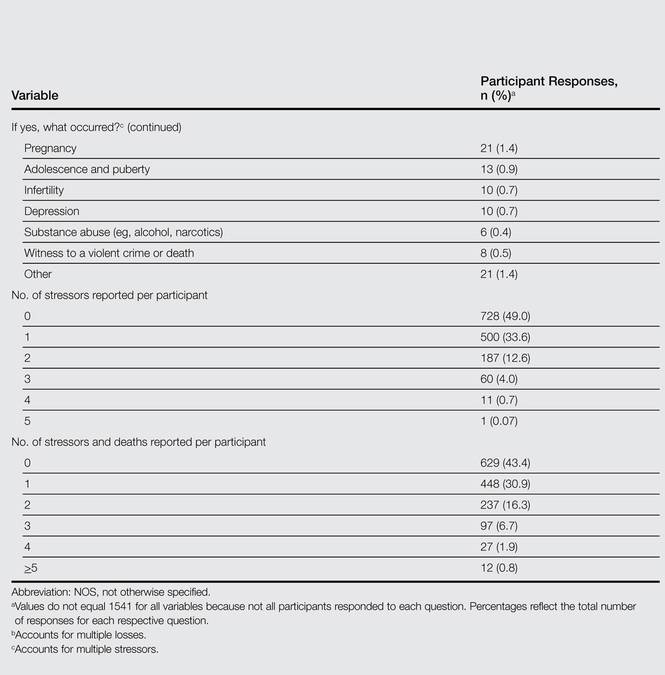
Stressors Not Associated With Vitiligo Extent
The number of deaths or stressors reported per participant within the 2 years prior to vitiligo onset were not associated with BSA, laterality, or distribution of lesions (Table 3 and eTable 2–eTable 4).
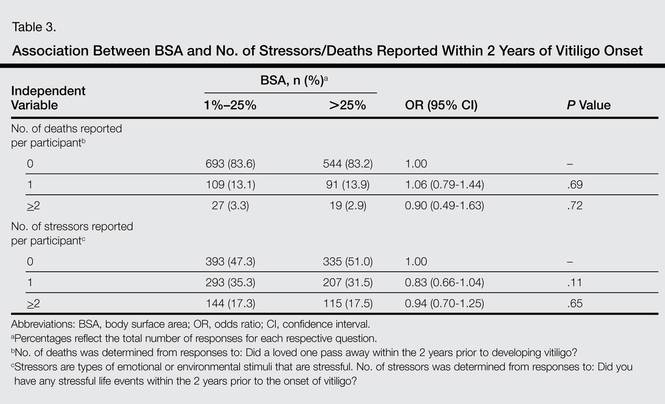
Symptoms Associated With Vitiligo
Five hundred twenty-two participants (34.5%) reported intermittent abdominal cramping, including premenstrual and/or menstrual cramping in women (9.7%), food-related abdominal cramping (4.4%), inflammatory bowel syndrome (IBS)(2.6%), anxiety-related abdominal cramping (1.5%), autoimmune gastrointestinal disorders (1.2%), and “other” etiologies (20.4%). Five hundred ten participants reported itching and/or burning associated with vitiligo lesions (35.1%).
Intermittent abdominal cramping overall was associated with a BSA greater than 75% (OR, 1.65; 95% confidence interval (CI), 1.17-2.32; P=.004). However, specific etiologies of abdominal cramping were not significantly associated with BSA (P≥.11). In contrast, itching and/or burning from vitiligo lesions was associated with a BSA greater than 25% (OR, 1.53; 95% CI, 1.23-1.90; P<.0001).
Association Between Number of Stressors and Symptoms in Vitiligo
A history of multiple stressors (≥2) within the 2 years prior to vitiligo onset was associated with intermittent abdominal cramping overall (OR, 1.84; 95% CI, 1.38-2.47; P<.0001), including premenstrual and/or menstrual cramping in women (OR, 1.84; 95% CI, 1.15-2.95; P=.01), IBS (OR, 3.29; 95% CI, 1.34-8.05; P=.01), and autoimmune gastrointestinal disorders (OR, 4.02; 95% CI, 1.27-12.80; P=.02)(eTable 5). These associations remained significant in multivariate models that included age, sex, and BSA as covariates. However, a history of 1 stressor or death or multiple deaths in the 2 years prior to vitiligo onset was not associated with any etiology of abdominal cramping.
Experiencing 1 (OR, 1.43; 95% CI, 1.12-1.82; P=.005) or multiple stressors (OR, 1.51; 95% CI, 1.12-2.04; P=.007) also was associated with itching and/or burning secondary to vitiligo. This association remained significant in a multivariate model that included age, sex, and BSA as covariates. However, a history of 1 or multiple deaths in the 2 years prior to vitiligo onset was not associated with itching and/or burning.
Association Between Specific Stressors and Vitiligo Symptoms
Perimenstrual (premenstrual and/or menstrual) cramping in women was associated with family problems (not otherwise specified) within the 2 years prior to vitiligo onset (Table 4). Food-related abdominal cramping was associated with school- and/or test-related stressors. Diagnosis of IBS was associated with health problems or surgery and being a victim of abuse within the 2 years prior to onset of vitiligo. Autoimmune gastrointestinal disorders were associated with moving to a new home/region, health problems or surgery, and witness to a violent crime or death. Finally, itching and/or burning of vitiligo lesions was associated with work and financial problems.
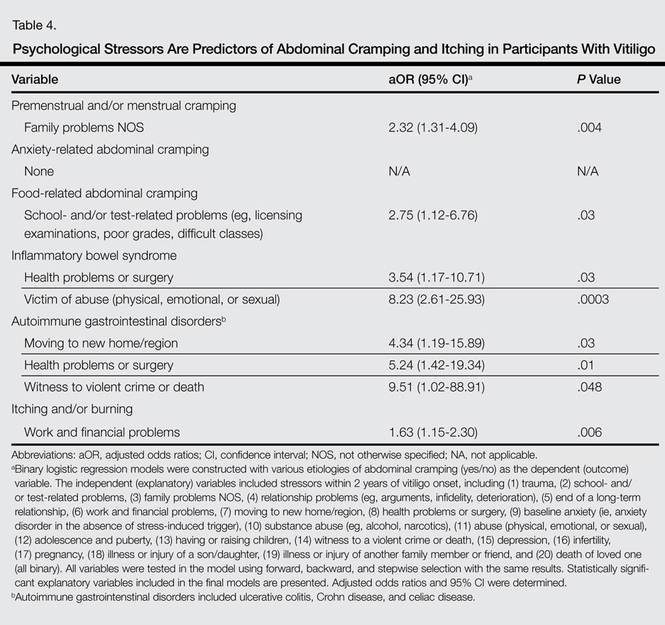
Comment
The present study found a high frequency of stressful life events and deaths of loved ones occurring within the 2 years preceding vitiligo onset. A history of multiple stressors but not deaths of loved ones was associated with more frequent symptoms in vitiligo patients, including itching and/or burning and intermittent abdominal pain. Specific stressors were associated with intermittent abdominal cramping, which occurred in approximately one-third of vitiligo patients. Abdominal cramping was related to menses in women, anxiety, foods, IBS, autoimmune gastrointestinal disorders, and other etiologies of abdominal cramping, which underscores the complex relationship between stressors, vitiligo, and inflammation. It is possible that stress-related immune abnormalities occur in vitiligo, which may influence the development of other autoimmune disorders. Alternatively, abdominal symptoms may precede and perhaps contribute to psychological stressors and impaired quality of life in vitiligo patients; however, the cross-sectional nature of the study did not allow us to elucidate this temporal relationship.
The present study found that 56.6% of participants experienced 1 or more deaths (17%) and/or stressful life events (51%) within the 2 years prior to vitiligo onset. These results are consistent with prior smaller studies that demonstrated a high frequency of stressful events preceding vitiligo onset. A case-controlled study found stressful events in 12 of 21 (57%) Romanian children with vitiligo, which was higher than controls.19 Another questionnaire-based, case-controlled study compared a heterogeneous group of 32 adolescent and adult Romanian patients with vitiligo and found higher odds of a stressful event in women preceding vitiligo diagnosis compared to controls.10 A retrospective analysis of 65 Croatian patients with vitiligo also reported that 56.9% (37/65) had some associated psychological factors.9 Another retrospective study of 31 adults with vitiligo found increased occurrence of 3 or more uncontrollable events, decreased perceived social support, and increased anxiety in vitiligo patients versus 116 other dermatologic disease controls.12 A questionnaire-based study found increased bereavements, changes in sleeping and eating habits, and personal injuries/illnesses in 73 British adults with vitiligo compared to 73 other age- and sex-matched dermatologic disease controls.11 All of these studies were limited by a small sample size, and the patient populations were localized to a regional dermatology referral center. The present study provided a larger analysis of stressful life events preceding vitiligo onset and included a diverse patient population.
The present study found that stressful life events and deaths of a loved one are not associated with vitiligo extent and distribution. This finding suggests that stressful life events may act as vitiligo triggers in genetically predisposed individuals, but ultimately the disease course and prognosis are driven by other factors, such as increased systemic inflammation or other immunologic abnormalities. Indeed, Silverberg and Silverberg20 and other investigators21,22 reported relative deficiencies of 25-hydroxyvitamin D,23 vitamins B6 and B12, and folic acid,20 as well as elevated serum homocysteine levels in vitiligo patients. Increased serum homocysteine levels were associated with increased BSA of vitiligo lesions.20 Elevated serum homocysteine levels also have been associated with increased inflammation in coronary artery disease,24 psoriasis,25,26 and in vitro.27 These laboratory anomalies likely reflect an underlying predisposition toward vitiligo, which might be triggered by stress responses or secondarily altered immune responses.
The present study had several strengths, including being prospective with a large sample size. The patient population included a large sample of men and women with representation of various adult ages and vitiligo extent. However, this study also had potential limitations. Measures of vitiligo extent were self-reported and were not clinically assessed. To address this limitation, we validated the questionnaire before posting it online.15 Invitation to participate in the survey was distributed by vitiligo support groups, which may have resulted in a selection bias toward participants with greater disease severity or with a poorer quality of life associated with vitiligo. Invitation to participate in this study was sent to members of vitiligo support groups, which allowed for recruitment of a large number of vitiligo patients despite a relatively low prevalence of disease in the general population. However, there are several challenges using this approach for nonvitiligo controls. Using participants with another dermatological disease as a control group may yield spurious results. Ideally, a large randomized sample of healthy participants with minimization of bias should be used for controls, which is an ambitious undertaking that was beyond the scope of this pilot study and will be the subject of future studies. Finally, this analysis found associations between stressors that occurred in the 2 years prior to vitiligo onset with symptomatic disease. We chose a broad interval for stressors because early vitiligo lesions may go unnoticed, making recognition of stressors occurring within days or weeks of onset infeasible. Further, we considered that chronic and prolonged stressors are more likely to have harmful consequences than acute stressors. Thus, stressors occurring within a more narrow interval (eg, 2 months) may not have the same association with vitiligo. Future studies are warranted to precisely identify the type and timing of psychological stressors preceding vitiligo onset.
Conclusion
In conclusion, there is a high prevalence of stressful life events preceding vitiligo, which may play an important role as disease triggers as well as predict the presence of intermittent abdominal cramping and itching or burning of skin. These associations indicate that screening of vitiligo patients for psychological stressors, abdominal cramping, and itching and/or burning of skin should be included in the routine assessment of vitiligo patients.
Appendix
1. Goronzy J, Weyand CM, Waase I. T cell subpopulations in inflammatory bowel disease: evidence for a defective induction of T8+ suppressor/cytotoxic T lymphocytes. Clin Exp Immunol. 1985;61:593-600.
2. Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003;16:90-100.
3. Grimes PE, Morris R, Avaniss-Aghajani E, et al. Topical tacrolimus therapy for vitiligo: therapeutic responses and skin messenger RNA expression of proinflammatory cytokines. J Am Acad Dermatol. 2004;51:52-61.
4. Birol A, Kisa U, Kurtipek GS, et al. Increased tumor necrosis factor alpha (TNF-alpha) and interleukin 1 alpha (IL1-alpha) levels in the lesional skin of patients with nonsegmental vitiligo. Int J Dermatol. 2006;45:992-993.
5. Moretti S, Spallanzani A, Amato L, et al. New insights into the pathogenesis of vitiligo: imbalance of epidermal cytokines at sites of lesions. Pigment Cell Res. 2002;15:87-92.
6. Zailaie MZ. Decreased proinflammatory cytokine production by peripheral blood mononuclear cells from vitiligo patients following aspirin treatment. Saudi Med J. 2005;26:799-805.
7. Basak PY, Adiloglu AK, Ceyhan AM, et al. The role of helper and regulatory T cells in the pathogenesis of vitiligo. J Am Acad Dermatol. 2009;60:256-260.
8. Kemp EH, Emhemad S, Akhtar S, et al. Autoantibodies against tyrosine hydroxylase in patients with non-segmental (generalised) vitiligo. Exp Dermatol. 2011;20:35-40.
9. Barisic´-Drusko V, Rucevic I. Trigger factors in childhood psoriasis and vitiligo. Coll Antropol. 2004;28:277-285.
10. Manolache L, Benea V. Stress in patients with alopecia areata and vitiligo. J Eur Acad Dermatol Venereol. 2007;21:921-928.
11. Papadopoulos L, Bor R, Legg C, et al. Impact of life events on the onset of vitiligo in adults: preliminary evidence for a psychological dimension in aetiology. Clin Exp Dermatol. 1998;23:243-248.
12. Picardi A, Pasquini P, Cattaruzza MS, et al. Stressful life events, social support, attachment security and alexithymia in vitiligo. a case-control study. Psychother Psychosom. 2003;72:150-158.
13. Salzer BA, Schallreuter KU. Investigation of the personality structure in patients with vitiligo and a possible association with impaired catecholamine metabolism. Dermatology. 1995;190:109-115.
14. Al’Abadie MS, Senior HJ, Bleehen SS, et al. Neuropeptide and neuronal marker studies in vitiligo. Br J Dermatol. 1994;131:160-165.
15. Silverberg JI, Silverberg NB. Association between vitiligo extent and distribution and quality-of-life impairment. JAMA Dermatol. 2013;149:159-164.
16. Silverberg JI, Silverberg NB. Quality of life impairments in children and adolescents with vitiligo. Pediatr Dermatol. 2014;31:309-318.
17. Kanwar AJ, Mahajan R, Parsad D. Effect of age at onset on disease characteristics in vitiligo. J Cutan Med Surg. 2013;17:253-258.
18. Hsieh FY, Bloch DA, Larsen MD. A simple method of sample size calculation for linear and logistic regression. Stat Med. 1998;17:1623-1634.
19. Manolache L, Petrescu-Seceleanu D, Benea V. Correlation of stressful events with onset of vitiligo in children. J Eur Acad Dermatol Venereol. 2009;23:187-188.
20. Silverberg JI, Silverberg NB. Serum homocysteine as a biomarker of vitiligo vulgaris severity: a pilot study. J Am Acad Dermatol. 2011;64:445-447.
21. Shaker OG, El-Tahlawi SM. Is there a relationship between homocysteine and vitiligo? a pilot study. Br J Dermatol. 2008;159:720-724.
22. Balci DD, Yonden Z, Yenin JZ, et al. Serum homocysteine, folic acid and vitamin B12 levels in vitiligo. Eur J Dermatol. 2009;19:382-383.
23. Silverberg JI, Silverberg AI, Malka E, et al. A pilot study assessing the role of 25 hydroxy vitamin D levels in patients with vitiligo vulgaris. J Am Acad Dermatol. 2010;62:937-941.
24. Jonasson T, Ohlin AK, Gottsater A, et al. Plasma homocysteine and markers for oxidative stress and inflammation in patients with coronary artery disease—a prospective randomized study of vitamin supplementation. Clin Chem Lab Med. 2005;43:628-634.
25. Cakmak SK, Gul U, Kilic C, et al. Homocysteine, vitamin B12 and folic acid levels in psoriasis patients. J Eur Acad Dermatol Venereol. 2009;23:300-303.
26. Malerba M, Gisondi P, Radaeli A, et al. Plasma homocysteine and folate levels in patients with chronic plaque psoriasis. Br J Dermatol. 2006;155:1165-1169.
27. Shastry S, James LR. Homocysteine-induced macrophage inflammatory protein-2 production by glomerular mesangial cells is mediated by PI3 Kinase and p38 MAPK. J Inflamm (Lond). 2009;6:27.
Vitiligo is the loss of skin pigmentation caused by autoimmune destruction of melanocytes. Multiple pathogenic factors for vitiligo have been described, including CD8+ T lymphocyte/T helper 1 infiltrates in lesional skin1,2 with increased expression of IFN-γ3 and tumor necrosis factor α,3-6 decreased transforming growth factor β,7 and circulating autoantibodies against tyrosine hydroxylase.8 Additionally, several studies have found a high prevalence of antecedent psychological stressors in vitiligo patients, suggesting that specific stressors may trigger and/or exacerbate vitiligo.9-12
The relationship between antecedent psychological stressors and vitiligo extent has not been well studied. Potential mechanisms for stress-triggered vitiligo include increased catecholamines13 and neuropeptides,14 which have been found in vitiligo patients. However, the complex relationship between stressors and subsequent vitiligo is not well defined. We hypothesized that persistent stressors are associated with increased vitiligo extent.
Vitiligo is classically considered to be a silent pigmentary disorder with few or no symptoms. Prior studies have demonstrated that one-third of vitiligo patients report skin symptoms (eg, pruritus, burning), which may be specifically associated with early-onset disease.15-17 Further, we observed that some vitiligo patients report abdominal cramping associated with their disease. Few studies have described the burden of skin symptoms and other associated symptoms in vitiligo or their determinants.
We conducted a prospective questionnaire-based study of 1541 adult vitiligo patients to identify psychological factors that may precede vitiligo onset. We hypothesized that some types of stressors that occur within 2 years prior to disease onset would have specific associations with vitiligo and/or somatic symptoms.
Methods
Study Population and Questionnaire Distribution
This prospective questionnaire-based study was approved by the institutional review board at St. Luke’s-Roosevelt Hospital Center (now Mount Sinai St. Luke’s-Roosevelt) (New York, New York) for adults (>18 years; male or female) with vitiligo. The survey was validated in paper format at St. Luke’s-Roosevelt Hospital Center and distributed online to members of nonprofit support groups for vitiligo vulgaris, as previously described.15
Questionnaire
The a priori aim of this questionnaire was to identify psychological factors that may precede vitiligo onset. The questionnaire consisted of 77 items (55 closed questions and 22 open questions) pertaining to participant demographics/vitiligo phenotype and psychological stressors preceding vitiligo onset. The questions related to this study and response rates are listed in eTable 1. Responses were verified by screening for noninteger or implausible values (eg, <0 or >100 years of age).
Sample Size
The primary outcome used for sample size calculation was the potential association between vitiligo and the presence of antecedent psychological stressors. Using a 2-tailed test, we determined that a sample size of 1264 participants would have 90% power at α=.05 and a baseline proportion of 0.01 (1% presumed prevalence of vitiligo) to detect an odds ratio (OR) of 2.5 or higher.18
Data and Statistical Analysis
Closed question responses were analyzed using descriptive statistics. Open-ended question responses were analyzed using content analysis. Related comments were coded and grouped, with similarities and differences noted. All data processing and statistics were done with SAS version 9.2. Age at diagnosis (years) and number of anatomic sites affected were divided into tertiles for statistical analysis due to wide skewing.
Logistic regression models were constructed with numbers of reported deaths or stressors per participant within the 2 years prior to vitiligo onset as independent variables (0, 1, or ≥2), and symptoms associated with vitiligo as dependent variables. Adjusted ORs were calculated from multivariate models that included sex, current age (continuous), and comorbid autoimmune disease (binary) as covariates. Linear interaction terms were tested and were included in final models if statistically significant (P<.05).
Ordinal logistic regression was used to analyze the relationship between stressors (and other independent variables) and number of anatomic sites affected with vitiligo (tertiles). Ordinal logistic regression models were constructed to examine the impact of psychological stressors on pruritus secondary to vitiligo (not relevant combined with not at all, a little, a lot, very much) as the dependent variable. The proportional odds assumption was met in both models, as judged by score testing (P>.05). Binary logistic regression was used to analyze laterality, body surface area (BSA) greater than 25%, and involvement of the face and/or body with vitiligo lesions (binary).
Binary logistic regression models were constructed with impact of psychological stressors preceding vitiligo onset on comorbid abdominal cramping and specific etiologies as the dependent variables. There were 20 candidate stressors occurring within the 2 years prior to vitiligo onset. Selection methods for predictors were used to identify significant covariates within the context of the other covariates included in the final models. The results of forward, backward, and stepwise approaches were similar, and the stepwise selection output was presented.
Missing values were encountered because some participants did not respond to all the questionnaire items. A complete case analysis was performed (ie, missing values were ignored throughout the study). Data imputation was considered by multiple imputations; however, there were few or no differences between the estimates from the 2 approaches. Therefore, final models did not involve data imputation.
The statistical significance for all estimates was considered to be P<.05. However, a P value near .05 should be interpreted with caution given the multiple dependent tests performed in this study with increased risk for falsely rejecting the null hypothesis.
Results
Survey Population Characteristics
One thousand seven hundred participants started the survey; 1632 completed the survey (96.0% completion rate) and 1553 had been diagnosed with vitiligo by a physician. Twelve participants were excluded because they were younger than 18 years, leaving 1541 evaluable participants. Five hundred thirty-eight participants (34.9%) had comorbid autoimmune disorders. Demographics and disease phenotypes of the study participants are listed in Table 1.
Stressors Preceding Vitiligo Onset
Eight hundred twenty-one participants (56.6%) experienced at least one death or stressor within 2 years prior to vitiligo onset (Table 2), including death of a loved one (16.6%) and stressful life events (51.0%) within the 2 years prior to the onset of vitiligo, especially work/financial problems (10.8%), end of a long-term relationship (10.2%), and family problems (not otherwise specified)(7.8%). Two hundred (13.5%) participants reported experiencing 1 death and 46 (3.1%) reported multiple deaths. Five hundred participants (33.6%) reported experiencing 1 stressor and 259 (17.4%) reported multiple stressors.
Stressors Not Associated With Vitiligo Extent
The number of deaths or stressors reported per participant within the 2 years prior to vitiligo onset were not associated with BSA, laterality, or distribution of lesions (Table 3 and eTable 2–eTable 4).
Symptoms Associated With Vitiligo
Five hundred twenty-two participants (34.5%) reported intermittent abdominal cramping, including premenstrual and/or menstrual cramping in women (9.7%), food-related abdominal cramping (4.4%), inflammatory bowel syndrome (IBS)(2.6%), anxiety-related abdominal cramping (1.5%), autoimmune gastrointestinal disorders (1.2%), and “other” etiologies (20.4%). Five hundred ten participants reported itching and/or burning associated with vitiligo lesions (35.1%).
Intermittent abdominal cramping overall was associated with a BSA greater than 75% (OR, 1.65; 95% confidence interval (CI), 1.17-2.32; P=.004). However, specific etiologies of abdominal cramping were not significantly associated with BSA (P≥.11). In contrast, itching and/or burning from vitiligo lesions was associated with a BSA greater than 25% (OR, 1.53; 95% CI, 1.23-1.90; P<.0001).
Association Between Number of Stressors and Symptoms in Vitiligo
A history of multiple stressors (≥2) within the 2 years prior to vitiligo onset was associated with intermittent abdominal cramping overall (OR, 1.84; 95% CI, 1.38-2.47; P<.0001), including premenstrual and/or menstrual cramping in women (OR, 1.84; 95% CI, 1.15-2.95; P=.01), IBS (OR, 3.29; 95% CI, 1.34-8.05; P=.01), and autoimmune gastrointestinal disorders (OR, 4.02; 95% CI, 1.27-12.80; P=.02)(eTable 5). These associations remained significant in multivariate models that included age, sex, and BSA as covariates. However, a history of 1 stressor or death or multiple deaths in the 2 years prior to vitiligo onset was not associated with any etiology of abdominal cramping.
Experiencing 1 (OR, 1.43; 95% CI, 1.12-1.82; P=.005) or multiple stressors (OR, 1.51; 95% CI, 1.12-2.04; P=.007) also was associated with itching and/or burning secondary to vitiligo. This association remained significant in a multivariate model that included age, sex, and BSA as covariates. However, a history of 1 or multiple deaths in the 2 years prior to vitiligo onset was not associated with itching and/or burning.
Association Between Specific Stressors and Vitiligo Symptoms
Perimenstrual (premenstrual and/or menstrual) cramping in women was associated with family problems (not otherwise specified) within the 2 years prior to vitiligo onset (Table 4). Food-related abdominal cramping was associated with school- and/or test-related stressors. Diagnosis of IBS was associated with health problems or surgery and being a victim of abuse within the 2 years prior to onset of vitiligo. Autoimmune gastrointestinal disorders were associated with moving to a new home/region, health problems or surgery, and witness to a violent crime or death. Finally, itching and/or burning of vitiligo lesions was associated with work and financial problems.
Comment
The present study found a high frequency of stressful life events and deaths of loved ones occurring within the 2 years preceding vitiligo onset. A history of multiple stressors but not deaths of loved ones was associated with more frequent symptoms in vitiligo patients, including itching and/or burning and intermittent abdominal pain. Specific stressors were associated with intermittent abdominal cramping, which occurred in approximately one-third of vitiligo patients. Abdominal cramping was related to menses in women, anxiety, foods, IBS, autoimmune gastrointestinal disorders, and other etiologies of abdominal cramping, which underscores the complex relationship between stressors, vitiligo, and inflammation. It is possible that stress-related immune abnormalities occur in vitiligo, which may influence the development of other autoimmune disorders. Alternatively, abdominal symptoms may precede and perhaps contribute to psychological stressors and impaired quality of life in vitiligo patients; however, the cross-sectional nature of the study did not allow us to elucidate this temporal relationship.
The present study found that 56.6% of participants experienced 1 or more deaths (17%) and/or stressful life events (51%) within the 2 years prior to vitiligo onset. These results are consistent with prior smaller studies that demonstrated a high frequency of stressful events preceding vitiligo onset. A case-controlled study found stressful events in 12 of 21 (57%) Romanian children with vitiligo, which was higher than controls.19 Another questionnaire-based, case-controlled study compared a heterogeneous group of 32 adolescent and adult Romanian patients with vitiligo and found higher odds of a stressful event in women preceding vitiligo diagnosis compared to controls.10 A retrospective analysis of 65 Croatian patients with vitiligo also reported that 56.9% (37/65) had some associated psychological factors.9 Another retrospective study of 31 adults with vitiligo found increased occurrence of 3 or more uncontrollable events, decreased perceived social support, and increased anxiety in vitiligo patients versus 116 other dermatologic disease controls.12 A questionnaire-based study found increased bereavements, changes in sleeping and eating habits, and personal injuries/illnesses in 73 British adults with vitiligo compared to 73 other age- and sex-matched dermatologic disease controls.11 All of these studies were limited by a small sample size, and the patient populations were localized to a regional dermatology referral center. The present study provided a larger analysis of stressful life events preceding vitiligo onset and included a diverse patient population.
The present study found that stressful life events and deaths of a loved one are not associated with vitiligo extent and distribution. This finding suggests that stressful life events may act as vitiligo triggers in genetically predisposed individuals, but ultimately the disease course and prognosis are driven by other factors, such as increased systemic inflammation or other immunologic abnormalities. Indeed, Silverberg and Silverberg20 and other investigators21,22 reported relative deficiencies of 25-hydroxyvitamin D,23 vitamins B6 and B12, and folic acid,20 as well as elevated serum homocysteine levels in vitiligo patients. Increased serum homocysteine levels were associated with increased BSA of vitiligo lesions.20 Elevated serum homocysteine levels also have been associated with increased inflammation in coronary artery disease,24 psoriasis,25,26 and in vitro.27 These laboratory anomalies likely reflect an underlying predisposition toward vitiligo, which might be triggered by stress responses or secondarily altered immune responses.
The present study had several strengths, including being prospective with a large sample size. The patient population included a large sample of men and women with representation of various adult ages and vitiligo extent. However, this study also had potential limitations. Measures of vitiligo extent were self-reported and were not clinically assessed. To address this limitation, we validated the questionnaire before posting it online.15 Invitation to participate in the survey was distributed by vitiligo support groups, which may have resulted in a selection bias toward participants with greater disease severity or with a poorer quality of life associated with vitiligo. Invitation to participate in this study was sent to members of vitiligo support groups, which allowed for recruitment of a large number of vitiligo patients despite a relatively low prevalence of disease in the general population. However, there are several challenges using this approach for nonvitiligo controls. Using participants with another dermatological disease as a control group may yield spurious results. Ideally, a large randomized sample of healthy participants with minimization of bias should be used for controls, which is an ambitious undertaking that was beyond the scope of this pilot study and will be the subject of future studies. Finally, this analysis found associations between stressors that occurred in the 2 years prior to vitiligo onset with symptomatic disease. We chose a broad interval for stressors because early vitiligo lesions may go unnoticed, making recognition of stressors occurring within days or weeks of onset infeasible. Further, we considered that chronic and prolonged stressors are more likely to have harmful consequences than acute stressors. Thus, stressors occurring within a more narrow interval (eg, 2 months) may not have the same association with vitiligo. Future studies are warranted to precisely identify the type and timing of psychological stressors preceding vitiligo onset.
Conclusion
In conclusion, there is a high prevalence of stressful life events preceding vitiligo, which may play an important role as disease triggers as well as predict the presence of intermittent abdominal cramping and itching or burning of skin. These associations indicate that screening of vitiligo patients for psychological stressors, abdominal cramping, and itching and/or burning of skin should be included in the routine assessment of vitiligo patients.
Appendix
Vitiligo is the loss of skin pigmentation caused by autoimmune destruction of melanocytes. Multiple pathogenic factors for vitiligo have been described, including CD8+ T lymphocyte/T helper 1 infiltrates in lesional skin1,2 with increased expression of IFN-γ3 and tumor necrosis factor α,3-6 decreased transforming growth factor β,7 and circulating autoantibodies against tyrosine hydroxylase.8 Additionally, several studies have found a high prevalence of antecedent psychological stressors in vitiligo patients, suggesting that specific stressors may trigger and/or exacerbate vitiligo.9-12
The relationship between antecedent psychological stressors and vitiligo extent has not been well studied. Potential mechanisms for stress-triggered vitiligo include increased catecholamines13 and neuropeptides,14 which have been found in vitiligo patients. However, the complex relationship between stressors and subsequent vitiligo is not well defined. We hypothesized that persistent stressors are associated with increased vitiligo extent.
Vitiligo is classically considered to be a silent pigmentary disorder with few or no symptoms. Prior studies have demonstrated that one-third of vitiligo patients report skin symptoms (eg, pruritus, burning), which may be specifically associated with early-onset disease.15-17 Further, we observed that some vitiligo patients report abdominal cramping associated with their disease. Few studies have described the burden of skin symptoms and other associated symptoms in vitiligo or their determinants.
We conducted a prospective questionnaire-based study of 1541 adult vitiligo patients to identify psychological factors that may precede vitiligo onset. We hypothesized that some types of stressors that occur within 2 years prior to disease onset would have specific associations with vitiligo and/or somatic symptoms.
Methods
Study Population and Questionnaire Distribution
This prospective questionnaire-based study was approved by the institutional review board at St. Luke’s-Roosevelt Hospital Center (now Mount Sinai St. Luke’s-Roosevelt) (New York, New York) for adults (>18 years; male or female) with vitiligo. The survey was validated in paper format at St. Luke’s-Roosevelt Hospital Center and distributed online to members of nonprofit support groups for vitiligo vulgaris, as previously described.15
Questionnaire
The a priori aim of this questionnaire was to identify psychological factors that may precede vitiligo onset. The questionnaire consisted of 77 items (55 closed questions and 22 open questions) pertaining to participant demographics/vitiligo phenotype and psychological stressors preceding vitiligo onset. The questions related to this study and response rates are listed in eTable 1. Responses were verified by screening for noninteger or implausible values (eg, <0 or >100 years of age).
Sample Size
The primary outcome used for sample size calculation was the potential association between vitiligo and the presence of antecedent psychological stressors. Using a 2-tailed test, we determined that a sample size of 1264 participants would have 90% power at α=.05 and a baseline proportion of 0.01 (1% presumed prevalence of vitiligo) to detect an odds ratio (OR) of 2.5 or higher.18
Data and Statistical Analysis
Closed question responses were analyzed using descriptive statistics. Open-ended question responses were analyzed using content analysis. Related comments were coded and grouped, with similarities and differences noted. All data processing and statistics were done with SAS version 9.2. Age at diagnosis (years) and number of anatomic sites affected were divided into tertiles for statistical analysis due to wide skewing.
Logistic regression models were constructed with numbers of reported deaths or stressors per participant within the 2 years prior to vitiligo onset as independent variables (0, 1, or ≥2), and symptoms associated with vitiligo as dependent variables. Adjusted ORs were calculated from multivariate models that included sex, current age (continuous), and comorbid autoimmune disease (binary) as covariates. Linear interaction terms were tested and were included in final models if statistically significant (P<.05).
Ordinal logistic regression was used to analyze the relationship between stressors (and other independent variables) and number of anatomic sites affected with vitiligo (tertiles). Ordinal logistic regression models were constructed to examine the impact of psychological stressors on pruritus secondary to vitiligo (not relevant combined with not at all, a little, a lot, very much) as the dependent variable. The proportional odds assumption was met in both models, as judged by score testing (P>.05). Binary logistic regression was used to analyze laterality, body surface area (BSA) greater than 25%, and involvement of the face and/or body with vitiligo lesions (binary).
Binary logistic regression models were constructed with impact of psychological stressors preceding vitiligo onset on comorbid abdominal cramping and specific etiologies as the dependent variables. There were 20 candidate stressors occurring within the 2 years prior to vitiligo onset. Selection methods for predictors were used to identify significant covariates within the context of the other covariates included in the final models. The results of forward, backward, and stepwise approaches were similar, and the stepwise selection output was presented.
Missing values were encountered because some participants did not respond to all the questionnaire items. A complete case analysis was performed (ie, missing values were ignored throughout the study). Data imputation was considered by multiple imputations; however, there were few or no differences between the estimates from the 2 approaches. Therefore, final models did not involve data imputation.
The statistical significance for all estimates was considered to be P<.05. However, a P value near .05 should be interpreted with caution given the multiple dependent tests performed in this study with increased risk for falsely rejecting the null hypothesis.
Results
Survey Population Characteristics
One thousand seven hundred participants started the survey; 1632 completed the survey (96.0% completion rate) and 1553 had been diagnosed with vitiligo by a physician. Twelve participants were excluded because they were younger than 18 years, leaving 1541 evaluable participants. Five hundred thirty-eight participants (34.9%) had comorbid autoimmune disorders. Demographics and disease phenotypes of the study participants are listed in Table 1.
Stressors Preceding Vitiligo Onset
Eight hundred twenty-one participants (56.6%) experienced at least one death or stressor within 2 years prior to vitiligo onset (Table 2), including death of a loved one (16.6%) and stressful life events (51.0%) within the 2 years prior to the onset of vitiligo, especially work/financial problems (10.8%), end of a long-term relationship (10.2%), and family problems (not otherwise specified)(7.8%). Two hundred (13.5%) participants reported experiencing 1 death and 46 (3.1%) reported multiple deaths. Five hundred participants (33.6%) reported experiencing 1 stressor and 259 (17.4%) reported multiple stressors.
Stressors Not Associated With Vitiligo Extent
The number of deaths or stressors reported per participant within the 2 years prior to vitiligo onset were not associated with BSA, laterality, or distribution of lesions (Table 3 and eTable 2–eTable 4).
Symptoms Associated With Vitiligo
Five hundred twenty-two participants (34.5%) reported intermittent abdominal cramping, including premenstrual and/or menstrual cramping in women (9.7%), food-related abdominal cramping (4.4%), inflammatory bowel syndrome (IBS)(2.6%), anxiety-related abdominal cramping (1.5%), autoimmune gastrointestinal disorders (1.2%), and “other” etiologies (20.4%). Five hundred ten participants reported itching and/or burning associated with vitiligo lesions (35.1%).
Intermittent abdominal cramping overall was associated with a BSA greater than 75% (OR, 1.65; 95% confidence interval (CI), 1.17-2.32; P=.004). However, specific etiologies of abdominal cramping were not significantly associated with BSA (P≥.11). In contrast, itching and/or burning from vitiligo lesions was associated with a BSA greater than 25% (OR, 1.53; 95% CI, 1.23-1.90; P<.0001).
Association Between Number of Stressors and Symptoms in Vitiligo
A history of multiple stressors (≥2) within the 2 years prior to vitiligo onset was associated with intermittent abdominal cramping overall (OR, 1.84; 95% CI, 1.38-2.47; P<.0001), including premenstrual and/or menstrual cramping in women (OR, 1.84; 95% CI, 1.15-2.95; P=.01), IBS (OR, 3.29; 95% CI, 1.34-8.05; P=.01), and autoimmune gastrointestinal disorders (OR, 4.02; 95% CI, 1.27-12.80; P=.02)(eTable 5). These associations remained significant in multivariate models that included age, sex, and BSA as covariates. However, a history of 1 stressor or death or multiple deaths in the 2 years prior to vitiligo onset was not associated with any etiology of abdominal cramping.
Experiencing 1 (OR, 1.43; 95% CI, 1.12-1.82; P=.005) or multiple stressors (OR, 1.51; 95% CI, 1.12-2.04; P=.007) also was associated with itching and/or burning secondary to vitiligo. This association remained significant in a multivariate model that included age, sex, and BSA as covariates. However, a history of 1 or multiple deaths in the 2 years prior to vitiligo onset was not associated with itching and/or burning.
Association Between Specific Stressors and Vitiligo Symptoms
Perimenstrual (premenstrual and/or menstrual) cramping in women was associated with family problems (not otherwise specified) within the 2 years prior to vitiligo onset (Table 4). Food-related abdominal cramping was associated with school- and/or test-related stressors. Diagnosis of IBS was associated with health problems or surgery and being a victim of abuse within the 2 years prior to onset of vitiligo. Autoimmune gastrointestinal disorders were associated with moving to a new home/region, health problems or surgery, and witness to a violent crime or death. Finally, itching and/or burning of vitiligo lesions was associated with work and financial problems.
Comment
The present study found a high frequency of stressful life events and deaths of loved ones occurring within the 2 years preceding vitiligo onset. A history of multiple stressors but not deaths of loved ones was associated with more frequent symptoms in vitiligo patients, including itching and/or burning and intermittent abdominal pain. Specific stressors were associated with intermittent abdominal cramping, which occurred in approximately one-third of vitiligo patients. Abdominal cramping was related to menses in women, anxiety, foods, IBS, autoimmune gastrointestinal disorders, and other etiologies of abdominal cramping, which underscores the complex relationship between stressors, vitiligo, and inflammation. It is possible that stress-related immune abnormalities occur in vitiligo, which may influence the development of other autoimmune disorders. Alternatively, abdominal symptoms may precede and perhaps contribute to psychological stressors and impaired quality of life in vitiligo patients; however, the cross-sectional nature of the study did not allow us to elucidate this temporal relationship.
The present study found that 56.6% of participants experienced 1 or more deaths (17%) and/or stressful life events (51%) within the 2 years prior to vitiligo onset. These results are consistent with prior smaller studies that demonstrated a high frequency of stressful events preceding vitiligo onset. A case-controlled study found stressful events in 12 of 21 (57%) Romanian children with vitiligo, which was higher than controls.19 Another questionnaire-based, case-controlled study compared a heterogeneous group of 32 adolescent and adult Romanian patients with vitiligo and found higher odds of a stressful event in women preceding vitiligo diagnosis compared to controls.10 A retrospective analysis of 65 Croatian patients with vitiligo also reported that 56.9% (37/65) had some associated psychological factors.9 Another retrospective study of 31 adults with vitiligo found increased occurrence of 3 or more uncontrollable events, decreased perceived social support, and increased anxiety in vitiligo patients versus 116 other dermatologic disease controls.12 A questionnaire-based study found increased bereavements, changes in sleeping and eating habits, and personal injuries/illnesses in 73 British adults with vitiligo compared to 73 other age- and sex-matched dermatologic disease controls.11 All of these studies were limited by a small sample size, and the patient populations were localized to a regional dermatology referral center. The present study provided a larger analysis of stressful life events preceding vitiligo onset and included a diverse patient population.
The present study found that stressful life events and deaths of a loved one are not associated with vitiligo extent and distribution. This finding suggests that stressful life events may act as vitiligo triggers in genetically predisposed individuals, but ultimately the disease course and prognosis are driven by other factors, such as increased systemic inflammation or other immunologic abnormalities. Indeed, Silverberg and Silverberg20 and other investigators21,22 reported relative deficiencies of 25-hydroxyvitamin D,23 vitamins B6 and B12, and folic acid,20 as well as elevated serum homocysteine levels in vitiligo patients. Increased serum homocysteine levels were associated with increased BSA of vitiligo lesions.20 Elevated serum homocysteine levels also have been associated with increased inflammation in coronary artery disease,24 psoriasis,25,26 and in vitro.27 These laboratory anomalies likely reflect an underlying predisposition toward vitiligo, which might be triggered by stress responses or secondarily altered immune responses.
The present study had several strengths, including being prospective with a large sample size. The patient population included a large sample of men and women with representation of various adult ages and vitiligo extent. However, this study also had potential limitations. Measures of vitiligo extent were self-reported and were not clinically assessed. To address this limitation, we validated the questionnaire before posting it online.15 Invitation to participate in the survey was distributed by vitiligo support groups, which may have resulted in a selection bias toward participants with greater disease severity or with a poorer quality of life associated with vitiligo. Invitation to participate in this study was sent to members of vitiligo support groups, which allowed for recruitment of a large number of vitiligo patients despite a relatively low prevalence of disease in the general population. However, there are several challenges using this approach for nonvitiligo controls. Using participants with another dermatological disease as a control group may yield spurious results. Ideally, a large randomized sample of healthy participants with minimization of bias should be used for controls, which is an ambitious undertaking that was beyond the scope of this pilot study and will be the subject of future studies. Finally, this analysis found associations between stressors that occurred in the 2 years prior to vitiligo onset with symptomatic disease. We chose a broad interval for stressors because early vitiligo lesions may go unnoticed, making recognition of stressors occurring within days or weeks of onset infeasible. Further, we considered that chronic and prolonged stressors are more likely to have harmful consequences than acute stressors. Thus, stressors occurring within a more narrow interval (eg, 2 months) may not have the same association with vitiligo. Future studies are warranted to precisely identify the type and timing of psychological stressors preceding vitiligo onset.
Conclusion
In conclusion, there is a high prevalence of stressful life events preceding vitiligo, which may play an important role as disease triggers as well as predict the presence of intermittent abdominal cramping and itching or burning of skin. These associations indicate that screening of vitiligo patients for psychological stressors, abdominal cramping, and itching and/or burning of skin should be included in the routine assessment of vitiligo patients.
Appendix
1. Goronzy J, Weyand CM, Waase I. T cell subpopulations in inflammatory bowel disease: evidence for a defective induction of T8+ suppressor/cytotoxic T lymphocytes. Clin Exp Immunol. 1985;61:593-600.
2. Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003;16:90-100.
3. Grimes PE, Morris R, Avaniss-Aghajani E, et al. Topical tacrolimus therapy for vitiligo: therapeutic responses and skin messenger RNA expression of proinflammatory cytokines. J Am Acad Dermatol. 2004;51:52-61.
4. Birol A, Kisa U, Kurtipek GS, et al. Increased tumor necrosis factor alpha (TNF-alpha) and interleukin 1 alpha (IL1-alpha) levels in the lesional skin of patients with nonsegmental vitiligo. Int J Dermatol. 2006;45:992-993.
5. Moretti S, Spallanzani A, Amato L, et al. New insights into the pathogenesis of vitiligo: imbalance of epidermal cytokines at sites of lesions. Pigment Cell Res. 2002;15:87-92.
6. Zailaie MZ. Decreased proinflammatory cytokine production by peripheral blood mononuclear cells from vitiligo patients following aspirin treatment. Saudi Med J. 2005;26:799-805.
7. Basak PY, Adiloglu AK, Ceyhan AM, et al. The role of helper and regulatory T cells in the pathogenesis of vitiligo. J Am Acad Dermatol. 2009;60:256-260.
8. Kemp EH, Emhemad S, Akhtar S, et al. Autoantibodies against tyrosine hydroxylase in patients with non-segmental (generalised) vitiligo. Exp Dermatol. 2011;20:35-40.
9. Barisic´-Drusko V, Rucevic I. Trigger factors in childhood psoriasis and vitiligo. Coll Antropol. 2004;28:277-285.
10. Manolache L, Benea V. Stress in patients with alopecia areata and vitiligo. J Eur Acad Dermatol Venereol. 2007;21:921-928.
11. Papadopoulos L, Bor R, Legg C, et al. Impact of life events on the onset of vitiligo in adults: preliminary evidence for a psychological dimension in aetiology. Clin Exp Dermatol. 1998;23:243-248.
12. Picardi A, Pasquini P, Cattaruzza MS, et al. Stressful life events, social support, attachment security and alexithymia in vitiligo. a case-control study. Psychother Psychosom. 2003;72:150-158.
13. Salzer BA, Schallreuter KU. Investigation of the personality structure in patients with vitiligo and a possible association with impaired catecholamine metabolism. Dermatology. 1995;190:109-115.
14. Al’Abadie MS, Senior HJ, Bleehen SS, et al. Neuropeptide and neuronal marker studies in vitiligo. Br J Dermatol. 1994;131:160-165.
15. Silverberg JI, Silverberg NB. Association between vitiligo extent and distribution and quality-of-life impairment. JAMA Dermatol. 2013;149:159-164.
16. Silverberg JI, Silverberg NB. Quality of life impairments in children and adolescents with vitiligo. Pediatr Dermatol. 2014;31:309-318.
17. Kanwar AJ, Mahajan R, Parsad D. Effect of age at onset on disease characteristics in vitiligo. J Cutan Med Surg. 2013;17:253-258.
18. Hsieh FY, Bloch DA, Larsen MD. A simple method of sample size calculation for linear and logistic regression. Stat Med. 1998;17:1623-1634.
19. Manolache L, Petrescu-Seceleanu D, Benea V. Correlation of stressful events with onset of vitiligo in children. J Eur Acad Dermatol Venereol. 2009;23:187-188.
20. Silverberg JI, Silverberg NB. Serum homocysteine as a biomarker of vitiligo vulgaris severity: a pilot study. J Am Acad Dermatol. 2011;64:445-447.
21. Shaker OG, El-Tahlawi SM. Is there a relationship between homocysteine and vitiligo? a pilot study. Br J Dermatol. 2008;159:720-724.
22. Balci DD, Yonden Z, Yenin JZ, et al. Serum homocysteine, folic acid and vitamin B12 levels in vitiligo. Eur J Dermatol. 2009;19:382-383.
23. Silverberg JI, Silverberg AI, Malka E, et al. A pilot study assessing the role of 25 hydroxy vitamin D levels in patients with vitiligo vulgaris. J Am Acad Dermatol. 2010;62:937-941.
24. Jonasson T, Ohlin AK, Gottsater A, et al. Plasma homocysteine and markers for oxidative stress and inflammation in patients with coronary artery disease—a prospective randomized study of vitamin supplementation. Clin Chem Lab Med. 2005;43:628-634.
25. Cakmak SK, Gul U, Kilic C, et al. Homocysteine, vitamin B12 and folic acid levels in psoriasis patients. J Eur Acad Dermatol Venereol. 2009;23:300-303.
26. Malerba M, Gisondi P, Radaeli A, et al. Plasma homocysteine and folate levels in patients with chronic plaque psoriasis. Br J Dermatol. 2006;155:1165-1169.
27. Shastry S, James LR. Homocysteine-induced macrophage inflammatory protein-2 production by glomerular mesangial cells is mediated by PI3 Kinase and p38 MAPK. J Inflamm (Lond). 2009;6:27.
1. Goronzy J, Weyand CM, Waase I. T cell subpopulations in inflammatory bowel disease: evidence for a defective induction of T8+ suppressor/cytotoxic T lymphocytes. Clin Exp Immunol. 1985;61:593-600.
2. Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003;16:90-100.
3. Grimes PE, Morris R, Avaniss-Aghajani E, et al. Topical tacrolimus therapy for vitiligo: therapeutic responses and skin messenger RNA expression of proinflammatory cytokines. J Am Acad Dermatol. 2004;51:52-61.
4. Birol A, Kisa U, Kurtipek GS, et al. Increased tumor necrosis factor alpha (TNF-alpha) and interleukin 1 alpha (IL1-alpha) levels in the lesional skin of patients with nonsegmental vitiligo. Int J Dermatol. 2006;45:992-993.
5. Moretti S, Spallanzani A, Amato L, et al. New insights into the pathogenesis of vitiligo: imbalance of epidermal cytokines at sites of lesions. Pigment Cell Res. 2002;15:87-92.
6. Zailaie MZ. Decreased proinflammatory cytokine production by peripheral blood mononuclear cells from vitiligo patients following aspirin treatment. Saudi Med J. 2005;26:799-805.
7. Basak PY, Adiloglu AK, Ceyhan AM, et al. The role of helper and regulatory T cells in the pathogenesis of vitiligo. J Am Acad Dermatol. 2009;60:256-260.
8. Kemp EH, Emhemad S, Akhtar S, et al. Autoantibodies against tyrosine hydroxylase in patients with non-segmental (generalised) vitiligo. Exp Dermatol. 2011;20:35-40.
9. Barisic´-Drusko V, Rucevic I. Trigger factors in childhood psoriasis and vitiligo. Coll Antropol. 2004;28:277-285.
10. Manolache L, Benea V. Stress in patients with alopecia areata and vitiligo. J Eur Acad Dermatol Venereol. 2007;21:921-928.
11. Papadopoulos L, Bor R, Legg C, et al. Impact of life events on the onset of vitiligo in adults: preliminary evidence for a psychological dimension in aetiology. Clin Exp Dermatol. 1998;23:243-248.
12. Picardi A, Pasquini P, Cattaruzza MS, et al. Stressful life events, social support, attachment security and alexithymia in vitiligo. a case-control study. Psychother Psychosom. 2003;72:150-158.
13. Salzer BA, Schallreuter KU. Investigation of the personality structure in patients with vitiligo and a possible association with impaired catecholamine metabolism. Dermatology. 1995;190:109-115.
14. Al’Abadie MS, Senior HJ, Bleehen SS, et al. Neuropeptide and neuronal marker studies in vitiligo. Br J Dermatol. 1994;131:160-165.
15. Silverberg JI, Silverberg NB. Association between vitiligo extent and distribution and quality-of-life impairment. JAMA Dermatol. 2013;149:159-164.
16. Silverberg JI, Silverberg NB. Quality of life impairments in children and adolescents with vitiligo. Pediatr Dermatol. 2014;31:309-318.
17. Kanwar AJ, Mahajan R, Parsad D. Effect of age at onset on disease characteristics in vitiligo. J Cutan Med Surg. 2013;17:253-258.
18. Hsieh FY, Bloch DA, Larsen MD. A simple method of sample size calculation for linear and logistic regression. Stat Med. 1998;17:1623-1634.
19. Manolache L, Petrescu-Seceleanu D, Benea V. Correlation of stressful events with onset of vitiligo in children. J Eur Acad Dermatol Venereol. 2009;23:187-188.
20. Silverberg JI, Silverberg NB. Serum homocysteine as a biomarker of vitiligo vulgaris severity: a pilot study. J Am Acad Dermatol. 2011;64:445-447.
21. Shaker OG, El-Tahlawi SM. Is there a relationship between homocysteine and vitiligo? a pilot study. Br J Dermatol. 2008;159:720-724.
22. Balci DD, Yonden Z, Yenin JZ, et al. Serum homocysteine, folic acid and vitamin B12 levels in vitiligo. Eur J Dermatol. 2009;19:382-383.
23. Silverberg JI, Silverberg AI, Malka E, et al. A pilot study assessing the role of 25 hydroxy vitamin D levels in patients with vitiligo vulgaris. J Am Acad Dermatol. 2010;62:937-941.
24. Jonasson T, Ohlin AK, Gottsater A, et al. Plasma homocysteine and markers for oxidative stress and inflammation in patients with coronary artery disease—a prospective randomized study of vitamin supplementation. Clin Chem Lab Med. 2005;43:628-634.
25. Cakmak SK, Gul U, Kilic C, et al. Homocysteine, vitamin B12 and folic acid levels in psoriasis patients. J Eur Acad Dermatol Venereol. 2009;23:300-303.
26. Malerba M, Gisondi P, Radaeli A, et al. Plasma homocysteine and folate levels in patients with chronic plaque psoriasis. Br J Dermatol. 2006;155:1165-1169.
27. Shastry S, James LR. Homocysteine-induced macrophage inflammatory protein-2 production by glomerular mesangial cells is mediated by PI3 Kinase and p38 MAPK. J Inflamm (Lond). 2009;6:27.
Practice Points
- Psychological stressors (eg, loss of a loved one) that occurred within 2 years prior to vitiligo onset should be considered as potential disease triggers.
- Psychological stressors have been associated with symptoms of abdominal cramping and itching/burning in vitiligo patients but not disease extent or distribution.
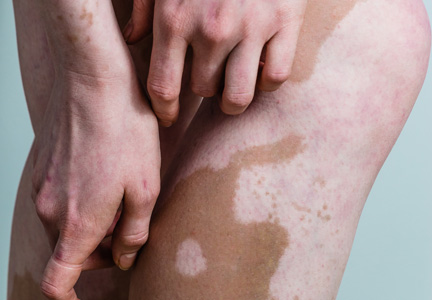
VIDEO: Sun protection urged for Asian, Hispanic women
SAN FRANCISCO – Among Asian and Hispanic patients, women are more likely than men are to get nonmelanoma skin cancer, according to a review of 4,029 cases at the University of California, San Diego.
That’s a surprise, because the reverse is true in whites, and skin cancer is generally thought to be more common in men.
About 96% of the cases were in white patients, and two-thirds of those were in men. Among Hispanic and Asian patients, about two-thirds of the cases were in women.
The reason for the gender reversal is unclear, but the study has a clear message, according to study investigator Dr. Arisa Ortiz, director of laser and cosmetic dermatology at the university. She shared that message in an interview at the American Academy of Dermatology annual meeting.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN FRANCISCO – Among Asian and Hispanic patients, women are more likely than men are to get nonmelanoma skin cancer, according to a review of 4,029 cases at the University of California, San Diego.
That’s a surprise, because the reverse is true in whites, and skin cancer is generally thought to be more common in men.
About 96% of the cases were in white patients, and two-thirds of those were in men. Among Hispanic and Asian patients, about two-thirds of the cases were in women.
The reason for the gender reversal is unclear, but the study has a clear message, according to study investigator Dr. Arisa Ortiz, director of laser and cosmetic dermatology at the university. She shared that message in an interview at the American Academy of Dermatology annual meeting.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
SAN FRANCISCO – Among Asian and Hispanic patients, women are more likely than men are to get nonmelanoma skin cancer, according to a review of 4,029 cases at the University of California, San Diego.
That’s a surprise, because the reverse is true in whites, and skin cancer is generally thought to be more common in men.
About 96% of the cases were in white patients, and two-thirds of those were in men. Among Hispanic and Asian patients, about two-thirds of the cases were in women.
The reason for the gender reversal is unclear, but the study has a clear message, according to study investigator Dr. Arisa Ortiz, director of laser and cosmetic dermatology at the university. She shared that message in an interview at the American Academy of Dermatology annual meeting.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT AAD 2015

Localized Argyria With Pseudo-ochronosis
Localized cutaneous argyria often presents as asymptomatic black or blue-gray pigmented macules in areas of the skin exposed to silver-containing compounds.1 Silver may enter the skin by traumatic implantation or absorption via eccrine sweat glands.2 Our patient witnessed a gun fight several years ago while on a mission trip and sustained multiple shrapnel wounds.
As in our patient, hyperpigmentation may appear years following initial exposure. Over time, incident light reduces colorless silver salts and compounds to black elemental silver.3 It also has been suggested that metallic silver granules stimulate tyrosine kinase activity, leading to locally increased melanin production.4 Together, these processes result in the clinical appearance of a blue-black macule. Despite its long-standing association with silver, this appearance also has been noted with deposition of other metals.5 Histologically, metal deposits can be seen as black granules surrounding eccrine glands, blood vessels, and elastic fibers on higher magnification.6 Granules also may be found in sebaceous glands and arrector pili muscle fibers. These findings do not distinguish from generalized argyria due to increased serum silver levels; however, some cases of localized cutaneous argyria have demonstrated spheroid black globules with surrounding collagen necrosis,1 which have not been reported with generalized disease. Localized cutaneous argyria also may be associated with ocher pigmentation of thickened collagen fibers, resembling changes typically found in alkaptonuria, an inherited deficiency of homogentisic acid oxidase (an enzyme involved in tyrosine metabolism).7 The resulting buildup of metabolic intermediates leads to ochronosis, a deposition of ocher-pigmented intermediates in connective tissue throughout the body. In the skin, ocher pigmentation occurs in elastic fibers of the reticular dermis.1 Grossly, these changes result in a blue-gray discoloration of the skin due to a light-scattering phenomenon known as the Tyndall effect. Exogenous ochronosis also can occur, most commonly from the topical application of hydroquinone or other skin-lightening compounds.1,5 Ocher pigmentation occurring in the setting of localized cutaneous argyria is referred to as pseudo-ochronosis, a finding first described by Robinson-Bostom et al.1 The etiology of this condition is poorly understood, but Robinson-Bostom et al1 noted the appearance of dark metal granules surrounding collagen bundles and hypothesized that metal aggregates surrounding collagen bundles in pseudo-ochronosis cause a homogenized appearance under light microscopy. Yellow-brown, swollen, homogenized collagen bundles can be visualized in the reticular dermis with surrounding deposition of metal granules (Figures 1 and 2).1 Typical patterns of granule deposition in localized argyria also are present.
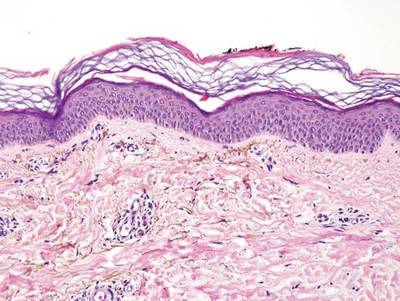

A blue nevus is a collection of proliferating dermal melanocytes. Many histologic subtypes exist and there may be extensive variability in the extent of sclerosis, cellular architecture, and tissue cellularity between each variant.8 Blue nevi commonly present as blue-black hyperpigmentation in the dermis and subcutaneous tissue.9 Histologically, they are characterized by slender, bipolar, dendritic melanocytes in a sclerotic stroma (Figure 3).8 Melanocytes are highly pigmented and contain small monomorphic nuclei. Lesions are relatively homogenous and typically are restricted to the dermis with epidermal sparing.9 Dark granules and ocher fibers are absent.
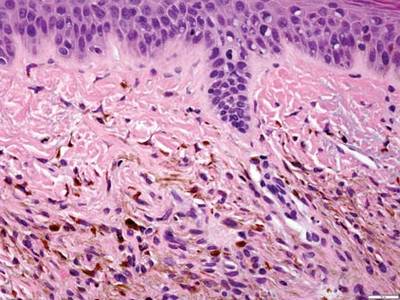
Long-term use of hydroxychloroquine or other antimalarials may cause a macular pattern of blue-gray hyperpigmentation.10 Biopsy specimens typically reveal coarse, yellow-brown pigment granules primarily affecting the superficial dermis (Figure 4). Granules are found both extracellularly and within macrophages. Fontana-Masson silver staining may identify melanin, as hydroxychloroquine-melanin binding may contribute to patterns of hyperpigmentation.10 Hemosiderin often is present in cases of hydroxychloroquine pigmentation. Preceding ecchymosis appears to favor the deposition of hydroxychloroquine in the skin.11 The absence of dark metal granules helps distinguish hydroxychloroquine pigmentation from argyria.
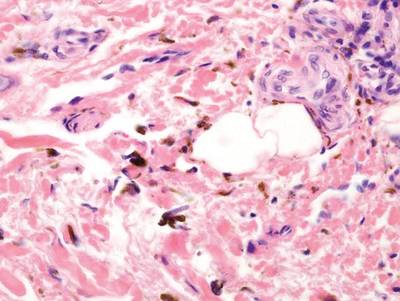
Regressed melanomas may appear clinically as gray macules. These lesions arise in cases of malignant melanoma that spontaneously regress without treatment. Spontaneous regression occurs in 10% to 35% of cases depending on tumor subtype.12 Lesions can have a variable appearance based on the degree of regression. Partial regression is demonstrated by mixed melanosis and fibrosis in the dermis (Figure 5).13,14 Melanin is housed within melanophages present in a variably expanded papillary dermis. Tumors in early stages of regression can be surrounded by an inflammatory infiltrate, which becomes diminished at later stages. However, a few exceptional cases have been noted with extensive inflammatory infiltrate and no residual tumor.14 Completely regressed lesions typically appear as a band of dermal melanophages in the absence of inflammation or melanocytic atypia.15 The finding of regressed melanoma should prompt further investigation including sentinel lymph node biopsy, as it may be associated with metastasis.
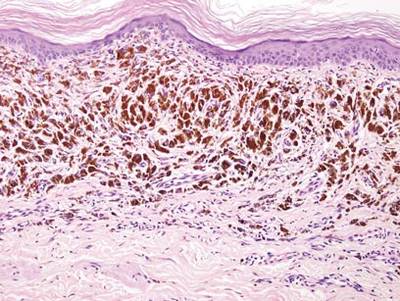
Tattooing occurs following traumatic penetration of the skin with impregnation of pigmented foreign material into deep dermal layers.16 Histologic examination usually reveals clumps of fine particulate material in the dermis (Figure 6). The color of the pigment depends on the agent used. For example, graphite appears as black particles that may be confused with localized cutaneous argyria. Distinction can be made using elemental identification techniques such as energy-dispersive X-ray spectroscopy.1 The intensity of the pigment in granules found in tattoos or localized cutaneous argyria will fail to diminish with the application of melanin bleach.6
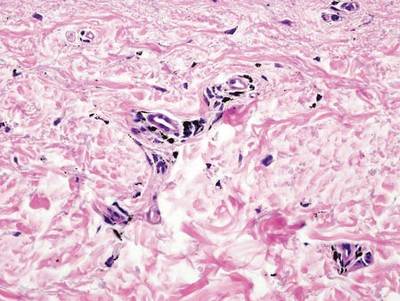
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol. 2002;46:222-227.
- Tajirian AL, Campbell RM, Robinson-Bostom L. Localized argyria after exposure to aerosolized solder. Cutis. 2006;78:305-308.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Shimizu I, Dill SW, McBean J, et al. Metal-induced granule deposition with pseudo-ochronosis. J Am Acad Dermatol. 2010;63:357-359.
- Rackoff EMJ, Benbenisty KM, Maize JC, et al. Localized cutaneous argyria from an acupuncture needle clini-cally concerning for metastatic melanoma. Cutis. 2007;80:423-426.
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:5-6.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Granter SR, McKee PH, Calonje E, et al. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a clinicopathologic study of 10 cases on the spectrum of so-called ‘malignant blue nevus.’ Am J Surg Pathol. 2001;25:316.
- Puri PK, Lountzis NI, Tyler W, et al. Hydroxychloroquine-induced hyperpigmentation: the staining pattern. J Cutan Pathol. 2008;35:1134-1137.
- Jallouli M, Francès C, Piette JC, et al. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus: a case-control study. JAMA Dermatol. 2013;149:935-940.
- Blessing K, McLaren KM. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology. 1992;20:315-322.
- LeBoit PE. Melanosis and its meanings. Am J Dermatopathol. 2002;24:369-372.
- Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
- Yang CH, Yeh JT, Shen SC, et al. Regressed subungual melanoma simulating cellular blue nevus: managed with sentinel lymph node biopsy. Dermatol Surg. 2006;32:577-581.
- Apfelberg DB, Manchester GH. Decorative and traumatic tattoo biophysics and removal. Clin Plast Surg. 1987;14:243-251.
Localized cutaneous argyria often presents as asymptomatic black or blue-gray pigmented macules in areas of the skin exposed to silver-containing compounds.1 Silver may enter the skin by traumatic implantation or absorption via eccrine sweat glands.2 Our patient witnessed a gun fight several years ago while on a mission trip and sustained multiple shrapnel wounds.
As in our patient, hyperpigmentation may appear years following initial exposure. Over time, incident light reduces colorless silver salts and compounds to black elemental silver.3 It also has been suggested that metallic silver granules stimulate tyrosine kinase activity, leading to locally increased melanin production.4 Together, these processes result in the clinical appearance of a blue-black macule. Despite its long-standing association with silver, this appearance also has been noted with deposition of other metals.5 Histologically, metal deposits can be seen as black granules surrounding eccrine glands, blood vessels, and elastic fibers on higher magnification.6 Granules also may be found in sebaceous glands and arrector pili muscle fibers. These findings do not distinguish from generalized argyria due to increased serum silver levels; however, some cases of localized cutaneous argyria have demonstrated spheroid black globules with surrounding collagen necrosis,1 which have not been reported with generalized disease. Localized cutaneous argyria also may be associated with ocher pigmentation of thickened collagen fibers, resembling changes typically found in alkaptonuria, an inherited deficiency of homogentisic acid oxidase (an enzyme involved in tyrosine metabolism).7 The resulting buildup of metabolic intermediates leads to ochronosis, a deposition of ocher-pigmented intermediates in connective tissue throughout the body. In the skin, ocher pigmentation occurs in elastic fibers of the reticular dermis.1 Grossly, these changes result in a blue-gray discoloration of the skin due to a light-scattering phenomenon known as the Tyndall effect. Exogenous ochronosis also can occur, most commonly from the topical application of hydroquinone or other skin-lightening compounds.1,5 Ocher pigmentation occurring in the setting of localized cutaneous argyria is referred to as pseudo-ochronosis, a finding first described by Robinson-Bostom et al.1 The etiology of this condition is poorly understood, but Robinson-Bostom et al1 noted the appearance of dark metal granules surrounding collagen bundles and hypothesized that metal aggregates surrounding collagen bundles in pseudo-ochronosis cause a homogenized appearance under light microscopy. Yellow-brown, swollen, homogenized collagen bundles can be visualized in the reticular dermis with surrounding deposition of metal granules (Figures 1 and 2).1 Typical patterns of granule deposition in localized argyria also are present.
A blue nevus is a collection of proliferating dermal melanocytes. Many histologic subtypes exist and there may be extensive variability in the extent of sclerosis, cellular architecture, and tissue cellularity between each variant.8 Blue nevi commonly present as blue-black hyperpigmentation in the dermis and subcutaneous tissue.9 Histologically, they are characterized by slender, bipolar, dendritic melanocytes in a sclerotic stroma (Figure 3).8 Melanocytes are highly pigmented and contain small monomorphic nuclei. Lesions are relatively homogenous and typically are restricted to the dermis with epidermal sparing.9 Dark granules and ocher fibers are absent.
Long-term use of hydroxychloroquine or other antimalarials may cause a macular pattern of blue-gray hyperpigmentation.10 Biopsy specimens typically reveal coarse, yellow-brown pigment granules primarily affecting the superficial dermis (Figure 4). Granules are found both extracellularly and within macrophages. Fontana-Masson silver staining may identify melanin, as hydroxychloroquine-melanin binding may contribute to patterns of hyperpigmentation.10 Hemosiderin often is present in cases of hydroxychloroquine pigmentation. Preceding ecchymosis appears to favor the deposition of hydroxychloroquine in the skin.11 The absence of dark metal granules helps distinguish hydroxychloroquine pigmentation from argyria.
Regressed melanomas may appear clinically as gray macules. These lesions arise in cases of malignant melanoma that spontaneously regress without treatment. Spontaneous regression occurs in 10% to 35% of cases depending on tumor subtype.12 Lesions can have a variable appearance based on the degree of regression. Partial regression is demonstrated by mixed melanosis and fibrosis in the dermis (Figure 5).13,14 Melanin is housed within melanophages present in a variably expanded papillary dermis. Tumors in early stages of regression can be surrounded by an inflammatory infiltrate, which becomes diminished at later stages. However, a few exceptional cases have been noted with extensive inflammatory infiltrate and no residual tumor.14 Completely regressed lesions typically appear as a band of dermal melanophages in the absence of inflammation or melanocytic atypia.15 The finding of regressed melanoma should prompt further investigation including sentinel lymph node biopsy, as it may be associated with metastasis.
Tattooing occurs following traumatic penetration of the skin with impregnation of pigmented foreign material into deep dermal layers.16 Histologic examination usually reveals clumps of fine particulate material in the dermis (Figure 6). The color of the pigment depends on the agent used. For example, graphite appears as black particles that may be confused with localized cutaneous argyria. Distinction can be made using elemental identification techniques such as energy-dispersive X-ray spectroscopy.1 The intensity of the pigment in granules found in tattoos or localized cutaneous argyria will fail to diminish with the application of melanin bleach.6
Localized cutaneous argyria often presents as asymptomatic black or blue-gray pigmented macules in areas of the skin exposed to silver-containing compounds.1 Silver may enter the skin by traumatic implantation or absorption via eccrine sweat glands.2 Our patient witnessed a gun fight several years ago while on a mission trip and sustained multiple shrapnel wounds.
As in our patient, hyperpigmentation may appear years following initial exposure. Over time, incident light reduces colorless silver salts and compounds to black elemental silver.3 It also has been suggested that metallic silver granules stimulate tyrosine kinase activity, leading to locally increased melanin production.4 Together, these processes result in the clinical appearance of a blue-black macule. Despite its long-standing association with silver, this appearance also has been noted with deposition of other metals.5 Histologically, metal deposits can be seen as black granules surrounding eccrine glands, blood vessels, and elastic fibers on higher magnification.6 Granules also may be found in sebaceous glands and arrector pili muscle fibers. These findings do not distinguish from generalized argyria due to increased serum silver levels; however, some cases of localized cutaneous argyria have demonstrated spheroid black globules with surrounding collagen necrosis,1 which have not been reported with generalized disease. Localized cutaneous argyria also may be associated with ocher pigmentation of thickened collagen fibers, resembling changes typically found in alkaptonuria, an inherited deficiency of homogentisic acid oxidase (an enzyme involved in tyrosine metabolism).7 The resulting buildup of metabolic intermediates leads to ochronosis, a deposition of ocher-pigmented intermediates in connective tissue throughout the body. In the skin, ocher pigmentation occurs in elastic fibers of the reticular dermis.1 Grossly, these changes result in a blue-gray discoloration of the skin due to a light-scattering phenomenon known as the Tyndall effect. Exogenous ochronosis also can occur, most commonly from the topical application of hydroquinone or other skin-lightening compounds.1,5 Ocher pigmentation occurring in the setting of localized cutaneous argyria is referred to as pseudo-ochronosis, a finding first described by Robinson-Bostom et al.1 The etiology of this condition is poorly understood, but Robinson-Bostom et al1 noted the appearance of dark metal granules surrounding collagen bundles and hypothesized that metal aggregates surrounding collagen bundles in pseudo-ochronosis cause a homogenized appearance under light microscopy. Yellow-brown, swollen, homogenized collagen bundles can be visualized in the reticular dermis with surrounding deposition of metal granules (Figures 1 and 2).1 Typical patterns of granule deposition in localized argyria also are present.
A blue nevus is a collection of proliferating dermal melanocytes. Many histologic subtypes exist and there may be extensive variability in the extent of sclerosis, cellular architecture, and tissue cellularity between each variant.8 Blue nevi commonly present as blue-black hyperpigmentation in the dermis and subcutaneous tissue.9 Histologically, they are characterized by slender, bipolar, dendritic melanocytes in a sclerotic stroma (Figure 3).8 Melanocytes are highly pigmented and contain small monomorphic nuclei. Lesions are relatively homogenous and typically are restricted to the dermis with epidermal sparing.9 Dark granules and ocher fibers are absent.
Long-term use of hydroxychloroquine or other antimalarials may cause a macular pattern of blue-gray hyperpigmentation.10 Biopsy specimens typically reveal coarse, yellow-brown pigment granules primarily affecting the superficial dermis (Figure 4). Granules are found both extracellularly and within macrophages. Fontana-Masson silver staining may identify melanin, as hydroxychloroquine-melanin binding may contribute to patterns of hyperpigmentation.10 Hemosiderin often is present in cases of hydroxychloroquine pigmentation. Preceding ecchymosis appears to favor the deposition of hydroxychloroquine in the skin.11 The absence of dark metal granules helps distinguish hydroxychloroquine pigmentation from argyria.
Regressed melanomas may appear clinically as gray macules. These lesions arise in cases of malignant melanoma that spontaneously regress without treatment. Spontaneous regression occurs in 10% to 35% of cases depending on tumor subtype.12 Lesions can have a variable appearance based on the degree of regression. Partial regression is demonstrated by mixed melanosis and fibrosis in the dermis (Figure 5).13,14 Melanin is housed within melanophages present in a variably expanded papillary dermis. Tumors in early stages of regression can be surrounded by an inflammatory infiltrate, which becomes diminished at later stages. However, a few exceptional cases have been noted with extensive inflammatory infiltrate and no residual tumor.14 Completely regressed lesions typically appear as a band of dermal melanophages in the absence of inflammation or melanocytic atypia.15 The finding of regressed melanoma should prompt further investigation including sentinel lymph node biopsy, as it may be associated with metastasis.
Tattooing occurs following traumatic penetration of the skin with impregnation of pigmented foreign material into deep dermal layers.16 Histologic examination usually reveals clumps of fine particulate material in the dermis (Figure 6). The color of the pigment depends on the agent used. For example, graphite appears as black particles that may be confused with localized cutaneous argyria. Distinction can be made using elemental identification techniques such as energy-dispersive X-ray spectroscopy.1 The intensity of the pigment in granules found in tattoos or localized cutaneous argyria will fail to diminish with the application of melanin bleach.6
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol. 2002;46:222-227.
- Tajirian AL, Campbell RM, Robinson-Bostom L. Localized argyria after exposure to aerosolized solder. Cutis. 2006;78:305-308.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Shimizu I, Dill SW, McBean J, et al. Metal-induced granule deposition with pseudo-ochronosis. J Am Acad Dermatol. 2010;63:357-359.
- Rackoff EMJ, Benbenisty KM, Maize JC, et al. Localized cutaneous argyria from an acupuncture needle clini-cally concerning for metastatic melanoma. Cutis. 2007;80:423-426.
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:5-6.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Granter SR, McKee PH, Calonje E, et al. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a clinicopathologic study of 10 cases on the spectrum of so-called ‘malignant blue nevus.’ Am J Surg Pathol. 2001;25:316.
- Puri PK, Lountzis NI, Tyler W, et al. Hydroxychloroquine-induced hyperpigmentation: the staining pattern. J Cutan Pathol. 2008;35:1134-1137.
- Jallouli M, Francès C, Piette JC, et al. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus: a case-control study. JAMA Dermatol. 2013;149:935-940.
- Blessing K, McLaren KM. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology. 1992;20:315-322.
- LeBoit PE. Melanosis and its meanings. Am J Dermatopathol. 2002;24:369-372.
- Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
- Yang CH, Yeh JT, Shen SC, et al. Regressed subungual melanoma simulating cellular blue nevus: managed with sentinel lymph node biopsy. Dermatol Surg. 2006;32:577-581.
- Apfelberg DB, Manchester GH. Decorative and traumatic tattoo biophysics and removal. Clin Plast Surg. 1987;14:243-251.
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol. 2002;46:222-227.
- Tajirian AL, Campbell RM, Robinson-Bostom L. Localized argyria after exposure to aerosolized solder. Cutis. 2006;78:305-308.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Shimizu I, Dill SW, McBean J, et al. Metal-induced granule deposition with pseudo-ochronosis. J Am Acad Dermatol. 2010;63:357-359.
- Rackoff EMJ, Benbenisty KM, Maize JC, et al. Localized cutaneous argyria from an acupuncture needle clini-cally concerning for metastatic melanoma. Cutis. 2007;80:423-426.
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:5-6.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Granter SR, McKee PH, Calonje E, et al. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a clinicopathologic study of 10 cases on the spectrum of so-called ‘malignant blue nevus.’ Am J Surg Pathol. 2001;25:316.
- Puri PK, Lountzis NI, Tyler W, et al. Hydroxychloroquine-induced hyperpigmentation: the staining pattern. J Cutan Pathol. 2008;35:1134-1137.
- Jallouli M, Francès C, Piette JC, et al. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus: a case-control study. JAMA Dermatol. 2013;149:935-940.
- Blessing K, McLaren KM. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology. 1992;20:315-322.
- LeBoit PE. Melanosis and its meanings. Am J Dermatopathol. 2002;24:369-372.
- Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
- Yang CH, Yeh JT, Shen SC, et al. Regressed subungual melanoma simulating cellular blue nevus: managed with sentinel lymph node biopsy. Dermatol Surg. 2006;32:577-581.
- Apfelberg DB, Manchester GH. Decorative and traumatic tattoo biophysics and removal. Clin Plast Surg. 1987;14:243-251.
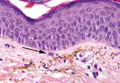
Paclitaxel-Associated Melanonychia
To the Editor:
Taxane-based chemotherapy including paclitaxel and docetaxel is commonly used to treat solid tumor malignancies including lung, breast, ovarian, and bladder cancers.1 Taxanes work by interrupting normal microtubule function by inducing tubulin polymerization and inhibiting microtubule depolymerization, thereby leading to cell cycle arrest at the gap 2 (premitotic) and mitotic phase and the blockade of cell division.2
Cutaneous side effects have been reported with taxane-based therapies, including alopecia, skin rash and erythema, and desquamation of the hands and feet (hand-foot syndrome).3 Nail changes also have been reported to occur in 0% to 44% of treated patients,4 with one study reporting an incidence as high as 50.5%.5 Nail abnormalities that have been described primarily include onycholysis, and less frequently Beau lines, subungual hemorrhagic bullae, subungual hyperkeratosis, splinter hemorrhages, acute paronychia, and pigmentary changes such as nail bed dyschromia. Among the taxanes, nail abnormalities are more commonly seen with docetaxel; few reports address paclitaxel-induced nail changes.4 Onycholysis, diffuse fingernail orange discoloration, Beau lines, subungual distal hyperkeratosis, and brown discoloration of 3 fingernail beds sparing the lunula have been reported with paclitaxel.6-9 We report a unique case of paclitaxel-associated melanonychia.
A 54-year-old black woman with a history of multiple myeloma and breast cancer who was being treated with paclitaxel for breast cancer presented with nail changes including nail darkening since initiating paclitaxel. She was diagnosed with multiple myeloma in 2010 and received bortezomib, dexamethasone, and an autologous stem cell transplant in August 2011. She never achieved complete remission but had been on lenalidomide with stable disease. She underwent a lumpectomy in December 2012, which revealed intraductal carcinoma with ductal carcinoma in situ that was estrogen receptor and progesterone receptor negative and ERBB2 (formerly HER2) positive. She was started on weekly paclitaxel (80 mg/m2) to complete 12 cycles and trastuzumab (6 mg/kg) every 3 weeks. While on paclitaxel, she developed grade 2 neuropathy of the hands, leading to subsequent dose reduction at week 9. She denied any other changes to her medications. On clinical examination she had diffuse and well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis involving all 20 nails (Figure, A and B). A nail clipping of the right hallux nail was sent for analysis. Pathology results showed evidence of scattered clusters of brown melanin pigment in the nail plate. Periodic acid–Schiff staining revealed numerous yeasts at the nail base but no infiltrating hyphae. Iron stain was negative for hemosiderin. The right index finger was injected with triamcinolone acetonide to treat the onycholysis. Four months after completing the paclitaxel, she began to notice lightening of the nails and improvement of the onycholysis in all nails (Figure, C and D).

| 
| |
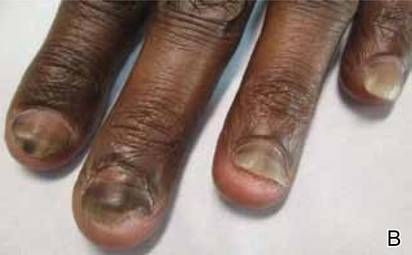
| 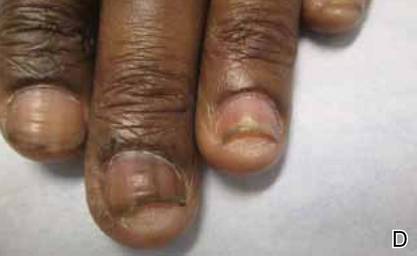
|
Initial appearance of diffuse, well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis in the nails on the right hand (A) and left hand (B). Four months after completing paclitaxel, the patient began to notice lightening of the nails and improvement of the onycholysis in the nails on the right hand (C) and left hand (D). |
The highly proliferating cells that comprise the nail matrix epithelium mature, differentiate, and keratinize to form the nail plate and are susceptible to the antimitotic effects of systemic chemotherapy. As a result, systemic chemotherapies may lead to abnormal nail plate production and keratinization of the nail plate, causing the clinical manifestations of Beau lines, onychomadesis, and leukonychia.10
Melanonychia is the development of melanin pigmentation of the nail plate and is typically caused by matrix melanin deposition through the activation of nail matrix melanocytes. There are 3 patterns of melanonychia: longitudinal, transverse, and diffuse. A single nail plate can involve more than one pattern of melanonychia and several nails may be affected. Longitudinal melanonychia typically develops from the activation of a group of melanocytes in the nail matrix, while diffuse pigmentation arises from diffuse melanocyte activation.11 Longitudinal melanonychia is common in darker-pigmented individuals12 and can be associated with systemic diseases.10 Transverse melanonychia has been reported in association with medications including many chemotherapy agents, and each band of transverse melanonychia may correspond to a cycle of therapy.11 Drug-induced melanonychia can affect several nails and tends to resolve after completion of therapy. Melanonychia has previously been described with vincristine, doxorubicin, hydroxyurea, cyclophosphamide, 5-fluorouracil, bleomycin, dacarbazine, methotrexate, and electron beam therapy.11 Nail pigmentation changes have been reported with docetaxel; a patient developed blue discoloration on the right and left thumb lunulae that improved 3 months after discontinuation of docetaxel therapy.13 While on docetaxel, another patient developed acral erythema, onycholysis, and longitudinal melanonychia in photoexposed areas, which was thought to be secondary to possible photosensitization.14 Possible explanations for paclitaxel-induced melanonychia include a direct toxic effect on the nail bed or nail matrix, focal stimulation of nail matrix melanocytes, or photosensitization. Drug-induced melanonychia commonly appears 3 to 8 weeks after drug intake and typically resolves 6 to 8 weeks after drug discontinuation.15
Predictors of taxane-related nail changes have been studied.5 Taxane-induced nail toxicity was more prevalent in patients who were female, had a history of diabetes mellitus, had received capecitabine with docetaxel, and had a diagnosis of breast or gynecological cancer. The nail changes increased with greater number of taxane cycles administered, body mass index, and severity of treatment-related neuropathy.5 Although nail changes often are temporary and typically resolve with drug withdrawal, they may persist in some patients.16 Possible measures have been proposed to prevent taxane-induced nail toxicity including frozen gloves,17 nail cutting, and avoiding potential fingernail irritants.18
It is possible that the nails of our darker-skinned patient may have been affected by some degree of melanonychia prior to starting the therapy, which cannot be ruled out. However, according to the patient, she only noticed the change after starting paclitaxel, raising the possibility of either new, worsening, or more diffuse involvement following initiation of paclitaxel therapy. Additionally, she was receiving weekly administration of paclitaxel and experienced severe neuropathy, both predictors of nail toxicity.5 No reports of melanonychia from lenalidomide have been reported in the literature indexed for MEDLINE. Although these nail changes are not life threatening, clinicians should be aware of these side effects, as they are cosmetically distressing to many patients and can impact quality of life.19
1. Crown J, O’Leary M. The taxanes: an update. Lancet. 2000;356:507-508.
2. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by Taxol. Nature. 1979;277:665-667.
3. Heidary N, Naik H, Burgin S. Chemotherapeutic agents and the skin: an update. J Am Acad Dermatol. 2008;58:545-570.
4. Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;14:333-337.
5. Can G, Aydiner A, Cavdar I. Taxane-induced nail changes: predictors and efficacy of the use of frozen gloves and socks in the prevention of nail toxicity. Eur J Oncol Nurs. 2012;16:270-275.
6. Lüftner D, Flath B, Akrivakis C, et al. Dose-intensified weekly paclitaxel induces multiple nail disorders. Ann Oncol. 1998;9:1139-1141.
7. Hussain S, Anderson DN, Salvatti ME, et al. Onycholysis as a complication of systemic chemotherapy. report of five cases associated with prolonged weekly paclitaxel therapy and review of the literature. Cancer. 2000;88:2367-2371.
8. Almagro M, Del Pozo J, Garcia-Silva J, et al. Nail alterations secondary to paclitaxel therapy. Eur J Dermatol. 2000;10:146-147.
9. Flory SM, Solimando DA Jr, Webster GF, et al. Onycholysis associated with weekly administration of paclitaxel. Ann Pharmacother. 1999;33:584-586.
10. Hinds G, Thomas VD. Malignancy and cancer treatment-related hair and nail changes. Dermatol Clin. 2008;26:59-68.
11. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Practice. 2009;15:143-55.
12. Buka R, Friedman KA, Phelps RG, et al. Childhood longitudinal melanonychia: case reports and review of the literature. Mt Sinai J Med. 2001;68:331-335.
13. Halvorson CR, Erickson CL, Gaspari AA. A rare manifestation of nail changes with docetaxel therapy. Skinmed. 2010;8:179-180.
14. Ferreira O, Baudrier T, Mota A, et al. Docetaxel-induced acral erythema and nail changes distributed to photoexposed areas. Cutan Ocul Toxicol. 2010;29:296-299.
15. Piraccini BM, Iorizzo M. Drug reactions affecting the nail unit: diagnosis and management. Dermatol Clin. 2007;25:215-221.
16. Piraccini BM, Tosti A. Drug-induced nail disorders: incidence, management and prognosis. Drug Saf. 1999;21:187-201.
17. Scotté F, Tourani JM, Banu E, et al. Multicenter study of a frozen glove to prevent docetaxel-induced onycholysis and cutaneous toxicity of the hand. J Clin Oncol. 2005;23:4424-4429.
18. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Pract. 2009;15:143-155.
19. Hackbarth M, Haas N, Fotopoulou C, et al. Chemotherapy-induced dermatological toxicity: frequencies and impact on quality of life in women’s cancers. results of a prospective study. Support Care Cancer. 2008;16:267-273.
To the Editor:
Taxane-based chemotherapy including paclitaxel and docetaxel is commonly used to treat solid tumor malignancies including lung, breast, ovarian, and bladder cancers.1 Taxanes work by interrupting normal microtubule function by inducing tubulin polymerization and inhibiting microtubule depolymerization, thereby leading to cell cycle arrest at the gap 2 (premitotic) and mitotic phase and the blockade of cell division.2
Cutaneous side effects have been reported with taxane-based therapies, including alopecia, skin rash and erythema, and desquamation of the hands and feet (hand-foot syndrome).3 Nail changes also have been reported to occur in 0% to 44% of treated patients,4 with one study reporting an incidence as high as 50.5%.5 Nail abnormalities that have been described primarily include onycholysis, and less frequently Beau lines, subungual hemorrhagic bullae, subungual hyperkeratosis, splinter hemorrhages, acute paronychia, and pigmentary changes such as nail bed dyschromia. Among the taxanes, nail abnormalities are more commonly seen with docetaxel; few reports address paclitaxel-induced nail changes.4 Onycholysis, diffuse fingernail orange discoloration, Beau lines, subungual distal hyperkeratosis, and brown discoloration of 3 fingernail beds sparing the lunula have been reported with paclitaxel.6-9 We report a unique case of paclitaxel-associated melanonychia.
A 54-year-old black woman with a history of multiple myeloma and breast cancer who was being treated with paclitaxel for breast cancer presented with nail changes including nail darkening since initiating paclitaxel. She was diagnosed with multiple myeloma in 2010 and received bortezomib, dexamethasone, and an autologous stem cell transplant in August 2011. She never achieved complete remission but had been on lenalidomide with stable disease. She underwent a lumpectomy in December 2012, which revealed intraductal carcinoma with ductal carcinoma in situ that was estrogen receptor and progesterone receptor negative and ERBB2 (formerly HER2) positive. She was started on weekly paclitaxel (80 mg/m2) to complete 12 cycles and trastuzumab (6 mg/kg) every 3 weeks. While on paclitaxel, she developed grade 2 neuropathy of the hands, leading to subsequent dose reduction at week 9. She denied any other changes to her medications. On clinical examination she had diffuse and well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis involving all 20 nails (Figure, A and B). A nail clipping of the right hallux nail was sent for analysis. Pathology results showed evidence of scattered clusters of brown melanin pigment in the nail plate. Periodic acid–Schiff staining revealed numerous yeasts at the nail base but no infiltrating hyphae. Iron stain was negative for hemosiderin. The right index finger was injected with triamcinolone acetonide to treat the onycholysis. Four months after completing the paclitaxel, she began to notice lightening of the nails and improvement of the onycholysis in all nails (Figure, C and D).
|
|
| |
|
|
|
Initial appearance of diffuse, well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis in the nails on the right hand (A) and left hand (B). Four months after completing paclitaxel, the patient began to notice lightening of the nails and improvement of the onycholysis in the nails on the right hand (C) and left hand (D). |
The highly proliferating cells that comprise the nail matrix epithelium mature, differentiate, and keratinize to form the nail plate and are susceptible to the antimitotic effects of systemic chemotherapy. As a result, systemic chemotherapies may lead to abnormal nail plate production and keratinization of the nail plate, causing the clinical manifestations of Beau lines, onychomadesis, and leukonychia.10
Melanonychia is the development of melanin pigmentation of the nail plate and is typically caused by matrix melanin deposition through the activation of nail matrix melanocytes. There are 3 patterns of melanonychia: longitudinal, transverse, and diffuse. A single nail plate can involve more than one pattern of melanonychia and several nails may be affected. Longitudinal melanonychia typically develops from the activation of a group of melanocytes in the nail matrix, while diffuse pigmentation arises from diffuse melanocyte activation.11 Longitudinal melanonychia is common in darker-pigmented individuals12 and can be associated with systemic diseases.10 Transverse melanonychia has been reported in association with medications including many chemotherapy agents, and each band of transverse melanonychia may correspond to a cycle of therapy.11 Drug-induced melanonychia can affect several nails and tends to resolve after completion of therapy. Melanonychia has previously been described with vincristine, doxorubicin, hydroxyurea, cyclophosphamide, 5-fluorouracil, bleomycin, dacarbazine, methotrexate, and electron beam therapy.11 Nail pigmentation changes have been reported with docetaxel; a patient developed blue discoloration on the right and left thumb lunulae that improved 3 months after discontinuation of docetaxel therapy.13 While on docetaxel, another patient developed acral erythema, onycholysis, and longitudinal melanonychia in photoexposed areas, which was thought to be secondary to possible photosensitization.14 Possible explanations for paclitaxel-induced melanonychia include a direct toxic effect on the nail bed or nail matrix, focal stimulation of nail matrix melanocytes, or photosensitization. Drug-induced melanonychia commonly appears 3 to 8 weeks after drug intake and typically resolves 6 to 8 weeks after drug discontinuation.15
Predictors of taxane-related nail changes have been studied.5 Taxane-induced nail toxicity was more prevalent in patients who were female, had a history of diabetes mellitus, had received capecitabine with docetaxel, and had a diagnosis of breast or gynecological cancer. The nail changes increased with greater number of taxane cycles administered, body mass index, and severity of treatment-related neuropathy.5 Although nail changes often are temporary and typically resolve with drug withdrawal, they may persist in some patients.16 Possible measures have been proposed to prevent taxane-induced nail toxicity including frozen gloves,17 nail cutting, and avoiding potential fingernail irritants.18
It is possible that the nails of our darker-skinned patient may have been affected by some degree of melanonychia prior to starting the therapy, which cannot be ruled out. However, according to the patient, she only noticed the change after starting paclitaxel, raising the possibility of either new, worsening, or more diffuse involvement following initiation of paclitaxel therapy. Additionally, she was receiving weekly administration of paclitaxel and experienced severe neuropathy, both predictors of nail toxicity.5 No reports of melanonychia from lenalidomide have been reported in the literature indexed for MEDLINE. Although these nail changes are not life threatening, clinicians should be aware of these side effects, as they are cosmetically distressing to many patients and can impact quality of life.19
To the Editor:
Taxane-based chemotherapy including paclitaxel and docetaxel is commonly used to treat solid tumor malignancies including lung, breast, ovarian, and bladder cancers.1 Taxanes work by interrupting normal microtubule function by inducing tubulin polymerization and inhibiting microtubule depolymerization, thereby leading to cell cycle arrest at the gap 2 (premitotic) and mitotic phase and the blockade of cell division.2
Cutaneous side effects have been reported with taxane-based therapies, including alopecia, skin rash and erythema, and desquamation of the hands and feet (hand-foot syndrome).3 Nail changes also have been reported to occur in 0% to 44% of treated patients,4 with one study reporting an incidence as high as 50.5%.5 Nail abnormalities that have been described primarily include onycholysis, and less frequently Beau lines, subungual hemorrhagic bullae, subungual hyperkeratosis, splinter hemorrhages, acute paronychia, and pigmentary changes such as nail bed dyschromia. Among the taxanes, nail abnormalities are more commonly seen with docetaxel; few reports address paclitaxel-induced nail changes.4 Onycholysis, diffuse fingernail orange discoloration, Beau lines, subungual distal hyperkeratosis, and brown discoloration of 3 fingernail beds sparing the lunula have been reported with paclitaxel.6-9 We report a unique case of paclitaxel-associated melanonychia.
A 54-year-old black woman with a history of multiple myeloma and breast cancer who was being treated with paclitaxel for breast cancer presented with nail changes including nail darkening since initiating paclitaxel. She was diagnosed with multiple myeloma in 2010 and received bortezomib, dexamethasone, and an autologous stem cell transplant in August 2011. She never achieved complete remission but had been on lenalidomide with stable disease. She underwent a lumpectomy in December 2012, which revealed intraductal carcinoma with ductal carcinoma in situ that was estrogen receptor and progesterone receptor negative and ERBB2 (formerly HER2) positive. She was started on weekly paclitaxel (80 mg/m2) to complete 12 cycles and trastuzumab (6 mg/kg) every 3 weeks. While on paclitaxel, she developed grade 2 neuropathy of the hands, leading to subsequent dose reduction at week 9. She denied any other changes to her medications. On clinical examination she had diffuse and well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis involving all 20 nails (Figure, A and B). A nail clipping of the right hallux nail was sent for analysis. Pathology results showed evidence of scattered clusters of brown melanin pigment in the nail plate. Periodic acid–Schiff staining revealed numerous yeasts at the nail base but no infiltrating hyphae. Iron stain was negative for hemosiderin. The right index finger was injected with triamcinolone acetonide to treat the onycholysis. Four months after completing the paclitaxel, she began to notice lightening of the nails and improvement of the onycholysis in all nails (Figure, C and D).
|
|
| |
|
|
|
Initial appearance of diffuse, well-demarcated, brown-black, longitudinal and transverse bands beginning at the proximal nail plate and progressing distally, with onycholysis in the nails on the right hand (A) and left hand (B). Four months after completing paclitaxel, the patient began to notice lightening of the nails and improvement of the onycholysis in the nails on the right hand (C) and left hand (D). |
The highly proliferating cells that comprise the nail matrix epithelium mature, differentiate, and keratinize to form the nail plate and are susceptible to the antimitotic effects of systemic chemotherapy. As a result, systemic chemotherapies may lead to abnormal nail plate production and keratinization of the nail plate, causing the clinical manifestations of Beau lines, onychomadesis, and leukonychia.10
Melanonychia is the development of melanin pigmentation of the nail plate and is typically caused by matrix melanin deposition through the activation of nail matrix melanocytes. There are 3 patterns of melanonychia: longitudinal, transverse, and diffuse. A single nail plate can involve more than one pattern of melanonychia and several nails may be affected. Longitudinal melanonychia typically develops from the activation of a group of melanocytes in the nail matrix, while diffuse pigmentation arises from diffuse melanocyte activation.11 Longitudinal melanonychia is common in darker-pigmented individuals12 and can be associated with systemic diseases.10 Transverse melanonychia has been reported in association with medications including many chemotherapy agents, and each band of transverse melanonychia may correspond to a cycle of therapy.11 Drug-induced melanonychia can affect several nails and tends to resolve after completion of therapy. Melanonychia has previously been described with vincristine, doxorubicin, hydroxyurea, cyclophosphamide, 5-fluorouracil, bleomycin, dacarbazine, methotrexate, and electron beam therapy.11 Nail pigmentation changes have been reported with docetaxel; a patient developed blue discoloration on the right and left thumb lunulae that improved 3 months after discontinuation of docetaxel therapy.13 While on docetaxel, another patient developed acral erythema, onycholysis, and longitudinal melanonychia in photoexposed areas, which was thought to be secondary to possible photosensitization.14 Possible explanations for paclitaxel-induced melanonychia include a direct toxic effect on the nail bed or nail matrix, focal stimulation of nail matrix melanocytes, or photosensitization. Drug-induced melanonychia commonly appears 3 to 8 weeks after drug intake and typically resolves 6 to 8 weeks after drug discontinuation.15
Predictors of taxane-related nail changes have been studied.5 Taxane-induced nail toxicity was more prevalent in patients who were female, had a history of diabetes mellitus, had received capecitabine with docetaxel, and had a diagnosis of breast or gynecological cancer. The nail changes increased with greater number of taxane cycles administered, body mass index, and severity of treatment-related neuropathy.5 Although nail changes often are temporary and typically resolve with drug withdrawal, they may persist in some patients.16 Possible measures have been proposed to prevent taxane-induced nail toxicity including frozen gloves,17 nail cutting, and avoiding potential fingernail irritants.18
It is possible that the nails of our darker-skinned patient may have been affected by some degree of melanonychia prior to starting the therapy, which cannot be ruled out. However, according to the patient, she only noticed the change after starting paclitaxel, raising the possibility of either new, worsening, or more diffuse involvement following initiation of paclitaxel therapy. Additionally, she was receiving weekly administration of paclitaxel and experienced severe neuropathy, both predictors of nail toxicity.5 No reports of melanonychia from lenalidomide have been reported in the literature indexed for MEDLINE. Although these nail changes are not life threatening, clinicians should be aware of these side effects, as they are cosmetically distressing to many patients and can impact quality of life.19
1. Crown J, O’Leary M. The taxanes: an update. Lancet. 2000;356:507-508.
2. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by Taxol. Nature. 1979;277:665-667.
3. Heidary N, Naik H, Burgin S. Chemotherapeutic agents and the skin: an update. J Am Acad Dermatol. 2008;58:545-570.
4. Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;14:333-337.
5. Can G, Aydiner A, Cavdar I. Taxane-induced nail changes: predictors and efficacy of the use of frozen gloves and socks in the prevention of nail toxicity. Eur J Oncol Nurs. 2012;16:270-275.
6. Lüftner D, Flath B, Akrivakis C, et al. Dose-intensified weekly paclitaxel induces multiple nail disorders. Ann Oncol. 1998;9:1139-1141.
7. Hussain S, Anderson DN, Salvatti ME, et al. Onycholysis as a complication of systemic chemotherapy. report of five cases associated with prolonged weekly paclitaxel therapy and review of the literature. Cancer. 2000;88:2367-2371.
8. Almagro M, Del Pozo J, Garcia-Silva J, et al. Nail alterations secondary to paclitaxel therapy. Eur J Dermatol. 2000;10:146-147.
9. Flory SM, Solimando DA Jr, Webster GF, et al. Onycholysis associated with weekly administration of paclitaxel. Ann Pharmacother. 1999;33:584-586.
10. Hinds G, Thomas VD. Malignancy and cancer treatment-related hair and nail changes. Dermatol Clin. 2008;26:59-68.
11. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Practice. 2009;15:143-55.
12. Buka R, Friedman KA, Phelps RG, et al. Childhood longitudinal melanonychia: case reports and review of the literature. Mt Sinai J Med. 2001;68:331-335.
13. Halvorson CR, Erickson CL, Gaspari AA. A rare manifestation of nail changes with docetaxel therapy. Skinmed. 2010;8:179-180.
14. Ferreira O, Baudrier T, Mota A, et al. Docetaxel-induced acral erythema and nail changes distributed to photoexposed areas. Cutan Ocul Toxicol. 2010;29:296-299.
15. Piraccini BM, Iorizzo M. Drug reactions affecting the nail unit: diagnosis and management. Dermatol Clin. 2007;25:215-221.
16. Piraccini BM, Tosti A. Drug-induced nail disorders: incidence, management and prognosis. Drug Saf. 1999;21:187-201.
17. Scotté F, Tourani JM, Banu E, et al. Multicenter study of a frozen glove to prevent docetaxel-induced onycholysis and cutaneous toxicity of the hand. J Clin Oncol. 2005;23:4424-4429.
18. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Pract. 2009;15:143-155.
19. Hackbarth M, Haas N, Fotopoulou C, et al. Chemotherapy-induced dermatological toxicity: frequencies and impact on quality of life in women’s cancers. results of a prospective study. Support Care Cancer. 2008;16:267-273.
1. Crown J, O’Leary M. The taxanes: an update. Lancet. 2000;356:507-508.
2. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro by Taxol. Nature. 1979;277:665-667.
3. Heidary N, Naik H, Burgin S. Chemotherapeutic agents and the skin: an update. J Am Acad Dermatol. 2008;58:545-570.
4. Minisini AM, Tosti A, Sobrero AF, et al. Taxane-induced nail changes: incidence, clinical presentation and outcome. Ann Oncol. 2003;14:333-337.
5. Can G, Aydiner A, Cavdar I. Taxane-induced nail changes: predictors and efficacy of the use of frozen gloves and socks in the prevention of nail toxicity. Eur J Oncol Nurs. 2012;16:270-275.
6. Lüftner D, Flath B, Akrivakis C, et al. Dose-intensified weekly paclitaxel induces multiple nail disorders. Ann Oncol. 1998;9:1139-1141.
7. Hussain S, Anderson DN, Salvatti ME, et al. Onycholysis as a complication of systemic chemotherapy. report of five cases associated with prolonged weekly paclitaxel therapy and review of the literature. Cancer. 2000;88:2367-2371.
8. Almagro M, Del Pozo J, Garcia-Silva J, et al. Nail alterations secondary to paclitaxel therapy. Eur J Dermatol. 2000;10:146-147.
9. Flory SM, Solimando DA Jr, Webster GF, et al. Onycholysis associated with weekly administration of paclitaxel. Ann Pharmacother. 1999;33:584-586.
10. Hinds G, Thomas VD. Malignancy and cancer treatment-related hair and nail changes. Dermatol Clin. 2008;26:59-68.
11. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Practice. 2009;15:143-55.
12. Buka R, Friedman KA, Phelps RG, et al. Childhood longitudinal melanonychia: case reports and review of the literature. Mt Sinai J Med. 2001;68:331-335.
13. Halvorson CR, Erickson CL, Gaspari AA. A rare manifestation of nail changes with docetaxel therapy. Skinmed. 2010;8:179-180.
14. Ferreira O, Baudrier T, Mota A, et al. Docetaxel-induced acral erythema and nail changes distributed to photoexposed areas. Cutan Ocul Toxicol. 2010;29:296-299.
15. Piraccini BM, Iorizzo M. Drug reactions affecting the nail unit: diagnosis and management. Dermatol Clin. 2007;25:215-221.
16. Piraccini BM, Tosti A. Drug-induced nail disorders: incidence, management and prognosis. Drug Saf. 1999;21:187-201.
17. Scotté F, Tourani JM, Banu E, et al. Multicenter study of a frozen glove to prevent docetaxel-induced onycholysis and cutaneous toxicity of the hand. J Clin Oncol. 2005;23:4424-4429.
18. Gilbar P, Hain A, Peereboom VM. Nail toxicity induced by cancer chemotherapy. J Oncol Pharm Pract. 2009;15:143-155.
19. Hackbarth M, Haas N, Fotopoulou C, et al. Chemotherapy-induced dermatological toxicity: frequencies and impact on quality of life in women’s cancers. results of a prospective study. Support Care Cancer. 2008;16:267-273.