User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Preventing psoriasis relapse after ustekinumab withdrawal using abatacept: A failed attempt
Key clinical point: Abatacept-mediated CD28-CD80/CD86 blockade was inept at averting psoriasis relapse following ustekinumab withdrawal in patients with moderate-to-severe plaque psoriasis.
Major finding: Between weeks 12 and 88, abatacept vs ustekinumab groups displayed similar relapse rates (91.1% vs 87.0%; P = .41) and median time to relapse from the last dose of ustekinumab (36 weeks [95% CI, 36-48] vs 32 weeks [95% CI, 28-40]).
Study details: The data come from the PAUSE trial, including 91 adult patients with moderate-to-severe plaque psoriasis who achieved Psoriasis Area Severity Index 75 at week 12 of receiving ustekinumab and who were randomly assigned to either continued ustekinumab or switch to abatacept until week 39.
Disclosures: The study was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health and Eli Lilly and Co. Some of the authors declared receiving research/institutional grants and/or personal fees from various sources, including Eli Lilly.
Source: Harris KM et al. JAMA Dermatol. 2021 Oct 13. doi: 10.1001/jamadermatol.2021.3492.
Key clinical point: Abatacept-mediated CD28-CD80/CD86 blockade was inept at averting psoriasis relapse following ustekinumab withdrawal in patients with moderate-to-severe plaque psoriasis.
Major finding: Between weeks 12 and 88, abatacept vs ustekinumab groups displayed similar relapse rates (91.1% vs 87.0%; P = .41) and median time to relapse from the last dose of ustekinumab (36 weeks [95% CI, 36-48] vs 32 weeks [95% CI, 28-40]).
Study details: The data come from the PAUSE trial, including 91 adult patients with moderate-to-severe plaque psoriasis who achieved Psoriasis Area Severity Index 75 at week 12 of receiving ustekinumab and who were randomly assigned to either continued ustekinumab or switch to abatacept until week 39.
Disclosures: The study was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health and Eli Lilly and Co. Some of the authors declared receiving research/institutional grants and/or personal fees from various sources, including Eli Lilly.
Source: Harris KM et al. JAMA Dermatol. 2021 Oct 13. doi: 10.1001/jamadermatol.2021.3492.
Key clinical point: Abatacept-mediated CD28-CD80/CD86 blockade was inept at averting psoriasis relapse following ustekinumab withdrawal in patients with moderate-to-severe plaque psoriasis.
Major finding: Between weeks 12 and 88, abatacept vs ustekinumab groups displayed similar relapse rates (91.1% vs 87.0%; P = .41) and median time to relapse from the last dose of ustekinumab (36 weeks [95% CI, 36-48] vs 32 weeks [95% CI, 28-40]).
Study details: The data come from the PAUSE trial, including 91 adult patients with moderate-to-severe plaque psoriasis who achieved Psoriasis Area Severity Index 75 at week 12 of receiving ustekinumab and who were randomly assigned to either continued ustekinumab or switch to abatacept until week 39.
Disclosures: The study was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health and Eli Lilly and Co. Some of the authors declared receiving research/institutional grants and/or personal fees from various sources, including Eli Lilly.
Source: Harris KM et al. JAMA Dermatol. 2021 Oct 13. doi: 10.1001/jamadermatol.2021.3492.
Biden seeks to return Califf as FDA chief
On Nov. 12, president Joe Biden said he will nominate Robert Califf, MD, to be commissioner of the U.S. Food and Drug Administration, the top U.S. regulator of drugs and medical devices.
Dr. Califf, a cardiologist, served as FDA chief in the Obama administration, leading the agency from Feb. 2016 to Jan. 2017.
The coming nomination ends nearly 11 months of speculation over Mr. Biden’s pick to the lead the agency during the ongoing pandemic. Janet Woodcock, MD, an FDA veteran, has been serving as acting commissioner. The White House faced a Tuesday deadline to make a nomination or see Dr. Woodcock’s tenure as acting chief expire under federal law.
The initial reaction to the idea of Dr. Califf’s return to the FDA drew mixed reactions.
The nonprofit watchdog Public Citizen issued a statement about its opposition to the potential nomination of Dr. Califf. Michael Carome, MD, director of Public Citizen’s Health Research Group, said the United States “desperately needs an FDA leader who will reverse the decades-long trend in which the agency’s relationship with the pharmaceutical and medical-device industries has grown dangerously cozier – resulting in regulatory capture of the agency by industry.”
But the idea of Dr. Califf returning to the FDA pleased Harlan Krumholz, MD, a cardiologist who has been a leader in outcomes research.
Dr. Krumholz tweeted that the Biden administration likely was testing the reaction to a possible Dr. Califf nomination before making it official. “I realize that this is being floated and not officially announced ... but the nomination of [Califf] just makes so much sense,” Dr. Krumholz tweeted. Dr. Califf’s “expertise as a researcher, policymaker, clinician are unparalleled. In a time of partisanship, he should be a slam-dunk confirmation.”
Dr. Califf’s 2016 Senate confirmation process was marked by dissent from several Democrats who questioned his ties to industry. But the chamber voted 89-4 to confirm him.
A version of this article first appeared on Medscape.com.
On Nov. 12, president Joe Biden said he will nominate Robert Califf, MD, to be commissioner of the U.S. Food and Drug Administration, the top U.S. regulator of drugs and medical devices.
Dr. Califf, a cardiologist, served as FDA chief in the Obama administration, leading the agency from Feb. 2016 to Jan. 2017.
The coming nomination ends nearly 11 months of speculation over Mr. Biden’s pick to the lead the agency during the ongoing pandemic. Janet Woodcock, MD, an FDA veteran, has been serving as acting commissioner. The White House faced a Tuesday deadline to make a nomination or see Dr. Woodcock’s tenure as acting chief expire under federal law.
The initial reaction to the idea of Dr. Califf’s return to the FDA drew mixed reactions.
The nonprofit watchdog Public Citizen issued a statement about its opposition to the potential nomination of Dr. Califf. Michael Carome, MD, director of Public Citizen’s Health Research Group, said the United States “desperately needs an FDA leader who will reverse the decades-long trend in which the agency’s relationship with the pharmaceutical and medical-device industries has grown dangerously cozier – resulting in regulatory capture of the agency by industry.”
But the idea of Dr. Califf returning to the FDA pleased Harlan Krumholz, MD, a cardiologist who has been a leader in outcomes research.
Dr. Krumholz tweeted that the Biden administration likely was testing the reaction to a possible Dr. Califf nomination before making it official. “I realize that this is being floated and not officially announced ... but the nomination of [Califf] just makes so much sense,” Dr. Krumholz tweeted. Dr. Califf’s “expertise as a researcher, policymaker, clinician are unparalleled. In a time of partisanship, he should be a slam-dunk confirmation.”
Dr. Califf’s 2016 Senate confirmation process was marked by dissent from several Democrats who questioned his ties to industry. But the chamber voted 89-4 to confirm him.
A version of this article first appeared on Medscape.com.
On Nov. 12, president Joe Biden said he will nominate Robert Califf, MD, to be commissioner of the U.S. Food and Drug Administration, the top U.S. regulator of drugs and medical devices.
Dr. Califf, a cardiologist, served as FDA chief in the Obama administration, leading the agency from Feb. 2016 to Jan. 2017.
The coming nomination ends nearly 11 months of speculation over Mr. Biden’s pick to the lead the agency during the ongoing pandemic. Janet Woodcock, MD, an FDA veteran, has been serving as acting commissioner. The White House faced a Tuesday deadline to make a nomination or see Dr. Woodcock’s tenure as acting chief expire under federal law.
The initial reaction to the idea of Dr. Califf’s return to the FDA drew mixed reactions.
The nonprofit watchdog Public Citizen issued a statement about its opposition to the potential nomination of Dr. Califf. Michael Carome, MD, director of Public Citizen’s Health Research Group, said the United States “desperately needs an FDA leader who will reverse the decades-long trend in which the agency’s relationship with the pharmaceutical and medical-device industries has grown dangerously cozier – resulting in regulatory capture of the agency by industry.”
But the idea of Dr. Califf returning to the FDA pleased Harlan Krumholz, MD, a cardiologist who has been a leader in outcomes research.
Dr. Krumholz tweeted that the Biden administration likely was testing the reaction to a possible Dr. Califf nomination before making it official. “I realize that this is being floated and not officially announced ... but the nomination of [Califf] just makes so much sense,” Dr. Krumholz tweeted. Dr. Califf’s “expertise as a researcher, policymaker, clinician are unparalleled. In a time of partisanship, he should be a slam-dunk confirmation.”
Dr. Califf’s 2016 Senate confirmation process was marked by dissent from several Democrats who questioned his ties to industry. But the chamber voted 89-4 to confirm him.
A version of this article first appeared on Medscape.com.
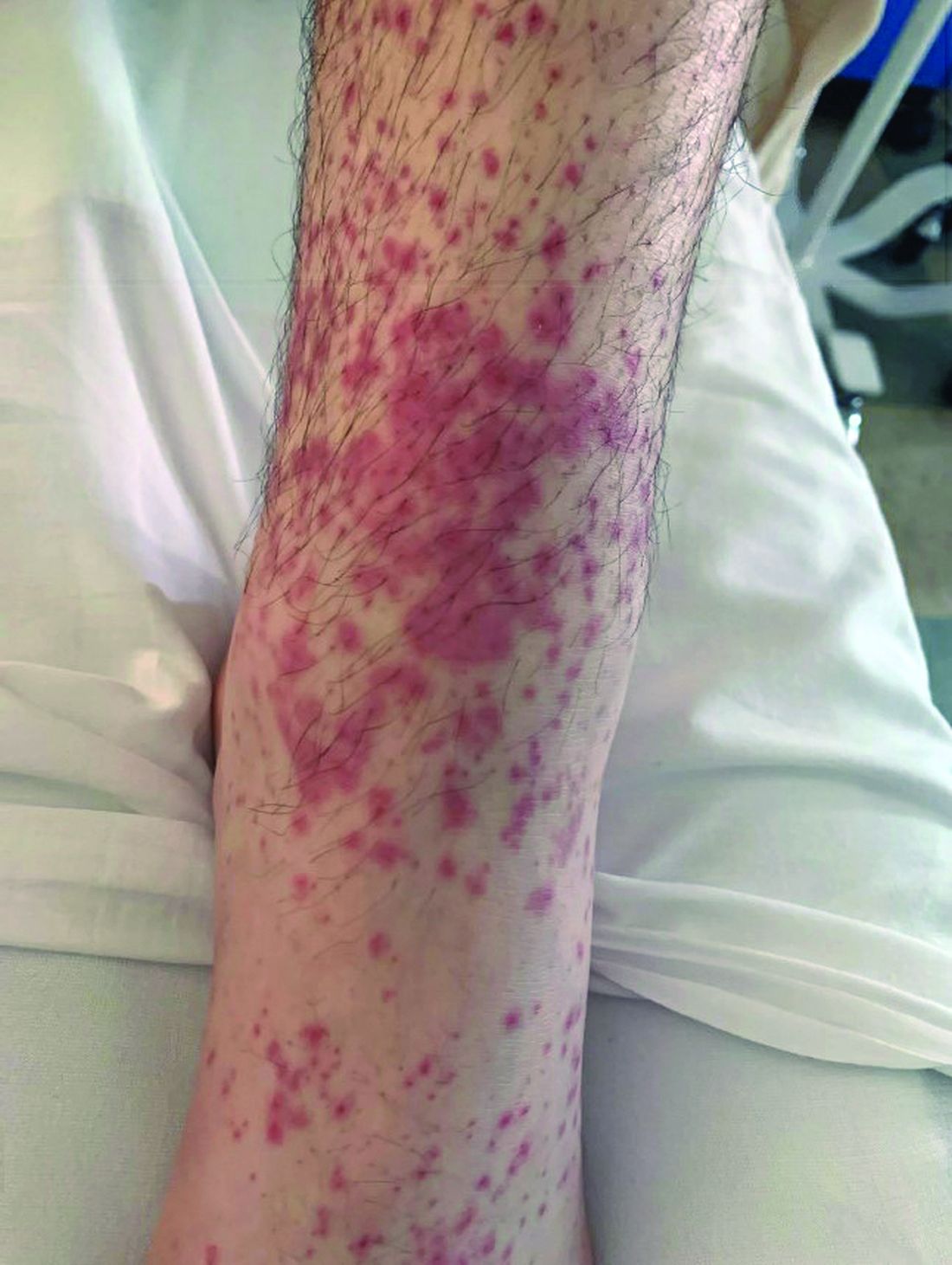
What is the diagnosis?
Numerous morphologies of skin rashes have been described in the setting of COVID-19, including pernio, livedoid rash, exanthem, and vasculitis. This classic constellation of symptoms (palpable purpura on buttocks/legs, abdominal pain, arthralgia, hematuria) is highly consistent with Henoch-Schonlein purpura (HSP). There are now multiple case reports of COVID-19–associated HSP.

HSP is the most common type of childhood systemic vasculitis. It is mediated by immunoglobulin A (IgA) immune complex deposition and has been associated with respiratory tract infections, streptococcal species, parainfluenza virus, and human parvovirus B19, medications, vaccinations, and malignancies. HSP is usually a self-limiting disease, with a course over 4-6 weeks, and can affect multiple organs, including the skin, gastrointestinal tract, joints, and the kidneys. The diagnostic criteria include palpable purpura in the presence of one or more of the following: diffuse abdominal pain, arthritis or arthralgia, any biopsy showing predominant IgA deposition, and renal involvement in the form of hematuria or proteinuria. Renal disease is variable and is the most significant indicator of long-term prognosis. This teenager was treated with oral corticosteroids because of the severe periarticular edema and responded rapidly. His subsequent urine analyses normalized.
What is on the differential?
Multisystem inflammatory syndrome in children (MIS-C) is a rare, potentially fatal, complication of COVID-19 infection that causes inflammation of multiple organs, including the heart, lungs, kidneys, brain, skin, eyes, or the gastrointestinal tract. It commonly affects children around ages 8-9 years. Initial symptoms include fever, rash, red eyes, diarrhea, and vomiting that appear 2-6 weeks post COVID-19 infection. Like HSP, MIS-C can present with edema of the extremities, worsening hand/foot pain, and hematuria; however, the absence of both fever and the pattern of system involvement seen with MIS-C and classic findings in this patient are more consistent with HSP.

Reactive infectious mucocutaneous eruption (RIME) was recently coined to encompass both infection-associated Stevens-Johnson eruptions including Mycoplasma pneumoniae-induced rash and mucositis (MIRM) and mucocutaneous eruptions caused by nonmycoplasma pathogens (including Chlamydia pneumoniae, human parainfluenza virus 2, rhinovirus, adenovirus, enterovirus, human metapneumovirus, influenza B virus, and COVID-19). It is usually seen in male children and adolescents. Prodromal symptoms include cough, fever, and malaise and they precede the prominent feature of mucositis. Our patient’s lack of mucosal involvement is not consistent with RIME.
Perniosis (chilblains) is characterized by localized edematous patches of erythema or cyanosis on exposed extremities, that may be associated with cold exposure. Lesions are usually symmetric and self-limiting, and symptoms can include numbness, tingling, pruritus, burning, or pain. Pernio-like skin lesions have been seen during the COVID-19 pandemic, though many patients have negative testing for infection by PCR and serology. Pernio may also be seen with autoimmune diseases or malignancy.
Meningococcemia is a rare disease caused by infection with gram-negative diplococci bacteria Neisseria meningitidis and spreads through saliva or respiratory secretions. Its clinical presentation can vary widely, from transient fever to fulminant disease. It is characterized by upper respiratory tract infection, fever, and petechial lesions associated with thrombocytopenia and coagulopathy.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Laborada is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Laborada have no relevant financial disclosures.
References
AlGhoozi DA, AlKhayyat HM. BMJ Case Reports CP 2021;14:e239910.
Jacobi M et al. Pediatr Infect Dis J. 2021;40(2):e93-4.
Paller A, Mancini AJ. Hurwitz clinical pediatric dermatology: A textbook of skin disorders of childhood and adolescence. 4th ed. Philadelphia (PA): Elsevier Saunders; 2011.
Radia T et al. Paediatr Respir Rev. 2021;38:51-7.
Ramien ML. Clin Exp Dermatol. 2021;46(3):420-9.
Numerous morphologies of skin rashes have been described in the setting of COVID-19, including pernio, livedoid rash, exanthem, and vasculitis. This classic constellation of symptoms (palpable purpura on buttocks/legs, abdominal pain, arthralgia, hematuria) is highly consistent with Henoch-Schonlein purpura (HSP). There are now multiple case reports of COVID-19–associated HSP.
HSP is the most common type of childhood systemic vasculitis. It is mediated by immunoglobulin A (IgA) immune complex deposition and has been associated with respiratory tract infections, streptococcal species, parainfluenza virus, and human parvovirus B19, medications, vaccinations, and malignancies. HSP is usually a self-limiting disease, with a course over 4-6 weeks, and can affect multiple organs, including the skin, gastrointestinal tract, joints, and the kidneys. The diagnostic criteria include palpable purpura in the presence of one or more of the following: diffuse abdominal pain, arthritis or arthralgia, any biopsy showing predominant IgA deposition, and renal involvement in the form of hematuria or proteinuria. Renal disease is variable and is the most significant indicator of long-term prognosis. This teenager was treated with oral corticosteroids because of the severe periarticular edema and responded rapidly. His subsequent urine analyses normalized.
What is on the differential?
Multisystem inflammatory syndrome in children (MIS-C) is a rare, potentially fatal, complication of COVID-19 infection that causes inflammation of multiple organs, including the heart, lungs, kidneys, brain, skin, eyes, or the gastrointestinal tract. It commonly affects children around ages 8-9 years. Initial symptoms include fever, rash, red eyes, diarrhea, and vomiting that appear 2-6 weeks post COVID-19 infection. Like HSP, MIS-C can present with edema of the extremities, worsening hand/foot pain, and hematuria; however, the absence of both fever and the pattern of system involvement seen with MIS-C and classic findings in this patient are more consistent with HSP.
Reactive infectious mucocutaneous eruption (RIME) was recently coined to encompass both infection-associated Stevens-Johnson eruptions including Mycoplasma pneumoniae-induced rash and mucositis (MIRM) and mucocutaneous eruptions caused by nonmycoplasma pathogens (including Chlamydia pneumoniae, human parainfluenza virus 2, rhinovirus, adenovirus, enterovirus, human metapneumovirus, influenza B virus, and COVID-19). It is usually seen in male children and adolescents. Prodromal symptoms include cough, fever, and malaise and they precede the prominent feature of mucositis. Our patient’s lack of mucosal involvement is not consistent with RIME.
Perniosis (chilblains) is characterized by localized edematous patches of erythema or cyanosis on exposed extremities, that may be associated with cold exposure. Lesions are usually symmetric and self-limiting, and symptoms can include numbness, tingling, pruritus, burning, or pain. Pernio-like skin lesions have been seen during the COVID-19 pandemic, though many patients have negative testing for infection by PCR and serology. Pernio may also be seen with autoimmune diseases or malignancy.
Meningococcemia is a rare disease caused by infection with gram-negative diplococci bacteria Neisseria meningitidis and spreads through saliva or respiratory secretions. Its clinical presentation can vary widely, from transient fever to fulminant disease. It is characterized by upper respiratory tract infection, fever, and petechial lesions associated with thrombocytopenia and coagulopathy.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Laborada is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Laborada have no relevant financial disclosures.
References
AlGhoozi DA, AlKhayyat HM. BMJ Case Reports CP 2021;14:e239910.
Jacobi M et al. Pediatr Infect Dis J. 2021;40(2):e93-4.
Paller A, Mancini AJ. Hurwitz clinical pediatric dermatology: A textbook of skin disorders of childhood and adolescence. 4th ed. Philadelphia (PA): Elsevier Saunders; 2011.
Radia T et al. Paediatr Respir Rev. 2021;38:51-7.
Ramien ML. Clin Exp Dermatol. 2021;46(3):420-9.
Numerous morphologies of skin rashes have been described in the setting of COVID-19, including pernio, livedoid rash, exanthem, and vasculitis. This classic constellation of symptoms (palpable purpura on buttocks/legs, abdominal pain, arthralgia, hematuria) is highly consistent with Henoch-Schonlein purpura (HSP). There are now multiple case reports of COVID-19–associated HSP.
HSP is the most common type of childhood systemic vasculitis. It is mediated by immunoglobulin A (IgA) immune complex deposition and has been associated with respiratory tract infections, streptococcal species, parainfluenza virus, and human parvovirus B19, medications, vaccinations, and malignancies. HSP is usually a self-limiting disease, with a course over 4-6 weeks, and can affect multiple organs, including the skin, gastrointestinal tract, joints, and the kidneys. The diagnostic criteria include palpable purpura in the presence of one or more of the following: diffuse abdominal pain, arthritis or arthralgia, any biopsy showing predominant IgA deposition, and renal involvement in the form of hematuria or proteinuria. Renal disease is variable and is the most significant indicator of long-term prognosis. This teenager was treated with oral corticosteroids because of the severe periarticular edema and responded rapidly. His subsequent urine analyses normalized.
What is on the differential?
Multisystem inflammatory syndrome in children (MIS-C) is a rare, potentially fatal, complication of COVID-19 infection that causes inflammation of multiple organs, including the heart, lungs, kidneys, brain, skin, eyes, or the gastrointestinal tract. It commonly affects children around ages 8-9 years. Initial symptoms include fever, rash, red eyes, diarrhea, and vomiting that appear 2-6 weeks post COVID-19 infection. Like HSP, MIS-C can present with edema of the extremities, worsening hand/foot pain, and hematuria; however, the absence of both fever and the pattern of system involvement seen with MIS-C and classic findings in this patient are more consistent with HSP.
Reactive infectious mucocutaneous eruption (RIME) was recently coined to encompass both infection-associated Stevens-Johnson eruptions including Mycoplasma pneumoniae-induced rash and mucositis (MIRM) and mucocutaneous eruptions caused by nonmycoplasma pathogens (including Chlamydia pneumoniae, human parainfluenza virus 2, rhinovirus, adenovirus, enterovirus, human metapneumovirus, influenza B virus, and COVID-19). It is usually seen in male children and adolescents. Prodromal symptoms include cough, fever, and malaise and they precede the prominent feature of mucositis. Our patient’s lack of mucosal involvement is not consistent with RIME.
Perniosis (chilblains) is characterized by localized edematous patches of erythema or cyanosis on exposed extremities, that may be associated with cold exposure. Lesions are usually symmetric and self-limiting, and symptoms can include numbness, tingling, pruritus, burning, or pain. Pernio-like skin lesions have been seen during the COVID-19 pandemic, though many patients have negative testing for infection by PCR and serology. Pernio may also be seen with autoimmune diseases or malignancy.
Meningococcemia is a rare disease caused by infection with gram-negative diplococci bacteria Neisseria meningitidis and spreads through saliva or respiratory secretions. Its clinical presentation can vary widely, from transient fever to fulminant disease. It is characterized by upper respiratory tract infection, fever, and petechial lesions associated with thrombocytopenia and coagulopathy.
Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Ms. Laborada is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital. Dr. Eichenfield and Ms. Laborada have no relevant financial disclosures.
References
AlGhoozi DA, AlKhayyat HM. BMJ Case Reports CP 2021;14:e239910.
Jacobi M et al. Pediatr Infect Dis J. 2021;40(2):e93-4.
Paller A, Mancini AJ. Hurwitz clinical pediatric dermatology: A textbook of skin disorders of childhood and adolescence. 4th ed. Philadelphia (PA): Elsevier Saunders; 2011.
Radia T et al. Paediatr Respir Rev. 2021;38:51-7.
Ramien ML. Clin Exp Dermatol. 2021;46(3):420-9.

Striae gravidarum: More than a ‘nuisance,’ say researchers
In the study of healthy pregnant women, “we found that SG can be associated with a host of negative reactions reflecting increased psychological and emotional distress,” reported Kaveri Karhade, MD, from the Berman Skin Institute, Los Altos, Calif., and coauthors from the University of Michigan, Ann Arbor. Dr. Karhade was with the department of dermatology at the University of Michigan at the time the study was conducted.
“We suggest that health care providers should avoid thinking of SG as merely a cosmetic ‘nuisance,’ ” they wrote in an article published in the International Journal of Women’s Dermatology. “Instead, it would be reasonable for providers to approach SG like other dermatologic concerns, and to consider asking patients whether SG cause emotional distress and whether prevention or treatment strategies should be attempted, even if not completely effective and potentially costly.”
The investigators did not evaluate treatments, but Frank Wang, MD, senior author of the study and professor of clinical dermatology at the University of Michigan Medicine, said in an interview that, “while they aren’t completely effective, some treatments can still help.” In addition, “recommending something also shows that you are listening to patients’ concerns – taking their concerns and skin lesions seriously,” he said.
Patient survey
The authors conducted a cross-sectional survey of 116 healthy pregnant women with SG. Participants were asked about the emotional and psychological effects of the lesions and how SG affects quality of life. The survey was modeled on questions from the Dermatology Life Quality Index, which asks about the impact of skin disease on embarrassment/self-consciousness, clothing choice, leisure activities, and interpersonal problems. “Content of questions was also devised from direct discussion with pregnant women attending clinic appointments or participating in other research studies on SG at our institution, and discussion with expert colleagues in obstetrics and dermatology,” the authors explained.
The survey consisted of 35 questions concerning demographics, pregnancy characteristics, personal and family history of SG, specific physical concerns about SG, impact of SG on attitude toward pregnancy, willingness to prevent SG or seek treatment, severity of SG (self-evaluated), the impact of SG on specific life-quality facets, and the location of lesions.
About two-thirds of respondents were aged 25-36 years and were White; the remainder self-identified as Asian, Black, Native American, or “other.” Most women reported “average” weight gain during the current pregnancy. Almost half of participants (45%) reporting a history of SG from prior pregnancies, and 65% reported a family history of SG.
The abdomen was identified most frequently as the location of SG (75%), followed by the breasts (43%), hips (43%), thighs (36%), buttocks (19%), and other areas (6%).
For most women (75%), permanency of the lesions was their top concern. About half (51%) reported that they had attempted to prevent SG, mostly with topical creams or oils. Three-quarters (75%) expressed interest in seeking treatment for SG, but this percentage dropped significantly to 33% (P =.008) if that treatment would not be covered by insurance.
Regarding the psychological impact of SG, embarrassment/self-consciousness correlated most strongly with lesion severity, followed by general quality of life, impact on choice of attire, impact on self-image/self-esteem, feelings of anxiety/depression related to SG, alteration of social/leisure activities related to SG (all P < .0001), and creation of interpersonal problems related to SG (P = .02).
The investigators also found that an increase in the effect of SG on self-image/self-esteem was “moderately associated” with younger age (P < .001) and that increased embarrassment related to SG was “moderately associated” with weight gain during pregnancy (P < .001).
“For years, stretch marks have been a topic to avoid and something many women try to hide,” Timothy Johnson, MD, professor of obstetrics and gynecology at the University of Michigan and coauthor of the study, said in a press release from the university. “Pregnant women talk about stretch marks with me every single week at clinic, and it’s time we break the stigma and start talking about them openly with all patients. ... By doing this study, we have an opportunity to normalize stretch marks in the context of all other dermatological conditions.”
Asked to comment on the findings, Tina Alster, MD, director of the Washington Institute of Dermatologic Laser Surgery and clinical professor of dermatology at Georgetown University, Washington, said her 3 decades of clinical experience support the authors’ findings. “Most patients who have striae are very self-conscious about them and report that their presence has negatively impacted their quality of life and self-confidence,” she said in an interview. “Of course, patients who come to my office are interested in having them treated, so my patient subset is skewed.”
She said treatment strategies that she discusses with patients include topical retinol/retinoids, which she said provide “low clinical response”; microneedling, which provides “marked” clinical response; and nonablative laser treatment, which provides “good” clinical response.
Considering particular patient characteristics, including budget, Dr. Alster said, “For those on a limited budget, I would propose daily use of a topical retinol, despite the low clinical effect. Many retinol-containing products are available over the counter. Prescription-strength retinoic acid tends to be pricey, often costing as much as in-office treatments.” Medical microneedling (not the cosmetic “roller” microneedling performed by aestheticians), she added, “gives the best results for the money and produces clinical results that mirror those achieved with lasers.”
Dr. Wang agreed that even recommending less expensive and less efficacious options such as over-the-counter creams can help alleviate patients’ concerns. “It shows that you are being holistic – not just caring for medical issues around pregnancy, but that you also take the emotional/psychological concerns of pregnant individuals and new parents seriously and that you recognize the impact of skin problems on quality of life. In the end, recommending something – in other words, providing some options, like creams or other therapies, for instance – is still, in my opinion, better than not recommending anything.”
Dr. Wang is involved with a study that is currently enrolling patients and that is evaluating the formation of early SG, which includes performing skin biopsies as soon as lesions appear.
The study had no funding. The study authors and Dr. Alster disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
In the study of healthy pregnant women, “we found that SG can be associated with a host of negative reactions reflecting increased psychological and emotional distress,” reported Kaveri Karhade, MD, from the Berman Skin Institute, Los Altos, Calif., and coauthors from the University of Michigan, Ann Arbor. Dr. Karhade was with the department of dermatology at the University of Michigan at the time the study was conducted.
“We suggest that health care providers should avoid thinking of SG as merely a cosmetic ‘nuisance,’ ” they wrote in an article published in the International Journal of Women’s Dermatology. “Instead, it would be reasonable for providers to approach SG like other dermatologic concerns, and to consider asking patients whether SG cause emotional distress and whether prevention or treatment strategies should be attempted, even if not completely effective and potentially costly.”
The investigators did not evaluate treatments, but Frank Wang, MD, senior author of the study and professor of clinical dermatology at the University of Michigan Medicine, said in an interview that, “while they aren’t completely effective, some treatments can still help.” In addition, “recommending something also shows that you are listening to patients’ concerns – taking their concerns and skin lesions seriously,” he said.
Patient survey
The authors conducted a cross-sectional survey of 116 healthy pregnant women with SG. Participants were asked about the emotional and psychological effects of the lesions and how SG affects quality of life. The survey was modeled on questions from the Dermatology Life Quality Index, which asks about the impact of skin disease on embarrassment/self-consciousness, clothing choice, leisure activities, and interpersonal problems. “Content of questions was also devised from direct discussion with pregnant women attending clinic appointments or participating in other research studies on SG at our institution, and discussion with expert colleagues in obstetrics and dermatology,” the authors explained.
The survey consisted of 35 questions concerning demographics, pregnancy characteristics, personal and family history of SG, specific physical concerns about SG, impact of SG on attitude toward pregnancy, willingness to prevent SG or seek treatment, severity of SG (self-evaluated), the impact of SG on specific life-quality facets, and the location of lesions.
About two-thirds of respondents were aged 25-36 years and were White; the remainder self-identified as Asian, Black, Native American, or “other.” Most women reported “average” weight gain during the current pregnancy. Almost half of participants (45%) reporting a history of SG from prior pregnancies, and 65% reported a family history of SG.
The abdomen was identified most frequently as the location of SG (75%), followed by the breasts (43%), hips (43%), thighs (36%), buttocks (19%), and other areas (6%).
For most women (75%), permanency of the lesions was their top concern. About half (51%) reported that they had attempted to prevent SG, mostly with topical creams or oils. Three-quarters (75%) expressed interest in seeking treatment for SG, but this percentage dropped significantly to 33% (P =.008) if that treatment would not be covered by insurance.
Regarding the psychological impact of SG, embarrassment/self-consciousness correlated most strongly with lesion severity, followed by general quality of life, impact on choice of attire, impact on self-image/self-esteem, feelings of anxiety/depression related to SG, alteration of social/leisure activities related to SG (all P < .0001), and creation of interpersonal problems related to SG (P = .02).
The investigators also found that an increase in the effect of SG on self-image/self-esteem was “moderately associated” with younger age (P < .001) and that increased embarrassment related to SG was “moderately associated” with weight gain during pregnancy (P < .001).
“For years, stretch marks have been a topic to avoid and something many women try to hide,” Timothy Johnson, MD, professor of obstetrics and gynecology at the University of Michigan and coauthor of the study, said in a press release from the university. “Pregnant women talk about stretch marks with me every single week at clinic, and it’s time we break the stigma and start talking about them openly with all patients. ... By doing this study, we have an opportunity to normalize stretch marks in the context of all other dermatological conditions.”
Asked to comment on the findings, Tina Alster, MD, director of the Washington Institute of Dermatologic Laser Surgery and clinical professor of dermatology at Georgetown University, Washington, said her 3 decades of clinical experience support the authors’ findings. “Most patients who have striae are very self-conscious about them and report that their presence has negatively impacted their quality of life and self-confidence,” she said in an interview. “Of course, patients who come to my office are interested in having them treated, so my patient subset is skewed.”
She said treatment strategies that she discusses with patients include topical retinol/retinoids, which she said provide “low clinical response”; microneedling, which provides “marked” clinical response; and nonablative laser treatment, which provides “good” clinical response.
Considering particular patient characteristics, including budget, Dr. Alster said, “For those on a limited budget, I would propose daily use of a topical retinol, despite the low clinical effect. Many retinol-containing products are available over the counter. Prescription-strength retinoic acid tends to be pricey, often costing as much as in-office treatments.” Medical microneedling (not the cosmetic “roller” microneedling performed by aestheticians), she added, “gives the best results for the money and produces clinical results that mirror those achieved with lasers.”
Dr. Wang agreed that even recommending less expensive and less efficacious options such as over-the-counter creams can help alleviate patients’ concerns. “It shows that you are being holistic – not just caring for medical issues around pregnancy, but that you also take the emotional/psychological concerns of pregnant individuals and new parents seriously and that you recognize the impact of skin problems on quality of life. In the end, recommending something – in other words, providing some options, like creams or other therapies, for instance – is still, in my opinion, better than not recommending anything.”
Dr. Wang is involved with a study that is currently enrolling patients and that is evaluating the formation of early SG, which includes performing skin biopsies as soon as lesions appear.
The study had no funding. The study authors and Dr. Alster disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
In the study of healthy pregnant women, “we found that SG can be associated with a host of negative reactions reflecting increased psychological and emotional distress,” reported Kaveri Karhade, MD, from the Berman Skin Institute, Los Altos, Calif., and coauthors from the University of Michigan, Ann Arbor. Dr. Karhade was with the department of dermatology at the University of Michigan at the time the study was conducted.
“We suggest that health care providers should avoid thinking of SG as merely a cosmetic ‘nuisance,’ ” they wrote in an article published in the International Journal of Women’s Dermatology. “Instead, it would be reasonable for providers to approach SG like other dermatologic concerns, and to consider asking patients whether SG cause emotional distress and whether prevention or treatment strategies should be attempted, even if not completely effective and potentially costly.”
The investigators did not evaluate treatments, but Frank Wang, MD, senior author of the study and professor of clinical dermatology at the University of Michigan Medicine, said in an interview that, “while they aren’t completely effective, some treatments can still help.” In addition, “recommending something also shows that you are listening to patients’ concerns – taking their concerns and skin lesions seriously,” he said.
Patient survey
The authors conducted a cross-sectional survey of 116 healthy pregnant women with SG. Participants were asked about the emotional and psychological effects of the lesions and how SG affects quality of life. The survey was modeled on questions from the Dermatology Life Quality Index, which asks about the impact of skin disease on embarrassment/self-consciousness, clothing choice, leisure activities, and interpersonal problems. “Content of questions was also devised from direct discussion with pregnant women attending clinic appointments or participating in other research studies on SG at our institution, and discussion with expert colleagues in obstetrics and dermatology,” the authors explained.
The survey consisted of 35 questions concerning demographics, pregnancy characteristics, personal and family history of SG, specific physical concerns about SG, impact of SG on attitude toward pregnancy, willingness to prevent SG or seek treatment, severity of SG (self-evaluated), the impact of SG on specific life-quality facets, and the location of lesions.
About two-thirds of respondents were aged 25-36 years and were White; the remainder self-identified as Asian, Black, Native American, or “other.” Most women reported “average” weight gain during the current pregnancy. Almost half of participants (45%) reporting a history of SG from prior pregnancies, and 65% reported a family history of SG.
The abdomen was identified most frequently as the location of SG (75%), followed by the breasts (43%), hips (43%), thighs (36%), buttocks (19%), and other areas (6%).
For most women (75%), permanency of the lesions was their top concern. About half (51%) reported that they had attempted to prevent SG, mostly with topical creams or oils. Three-quarters (75%) expressed interest in seeking treatment for SG, but this percentage dropped significantly to 33% (P =.008) if that treatment would not be covered by insurance.
Regarding the psychological impact of SG, embarrassment/self-consciousness correlated most strongly with lesion severity, followed by general quality of life, impact on choice of attire, impact on self-image/self-esteem, feelings of anxiety/depression related to SG, alteration of social/leisure activities related to SG (all P < .0001), and creation of interpersonal problems related to SG (P = .02).
The investigators also found that an increase in the effect of SG on self-image/self-esteem was “moderately associated” with younger age (P < .001) and that increased embarrassment related to SG was “moderately associated” with weight gain during pregnancy (P < .001).
“For years, stretch marks have been a topic to avoid and something many women try to hide,” Timothy Johnson, MD, professor of obstetrics and gynecology at the University of Michigan and coauthor of the study, said in a press release from the university. “Pregnant women talk about stretch marks with me every single week at clinic, and it’s time we break the stigma and start talking about them openly with all patients. ... By doing this study, we have an opportunity to normalize stretch marks in the context of all other dermatological conditions.”
Asked to comment on the findings, Tina Alster, MD, director of the Washington Institute of Dermatologic Laser Surgery and clinical professor of dermatology at Georgetown University, Washington, said her 3 decades of clinical experience support the authors’ findings. “Most patients who have striae are very self-conscious about them and report that their presence has negatively impacted their quality of life and self-confidence,” she said in an interview. “Of course, patients who come to my office are interested in having them treated, so my patient subset is skewed.”
She said treatment strategies that she discusses with patients include topical retinol/retinoids, which she said provide “low clinical response”; microneedling, which provides “marked” clinical response; and nonablative laser treatment, which provides “good” clinical response.
Considering particular patient characteristics, including budget, Dr. Alster said, “For those on a limited budget, I would propose daily use of a topical retinol, despite the low clinical effect. Many retinol-containing products are available over the counter. Prescription-strength retinoic acid tends to be pricey, often costing as much as in-office treatments.” Medical microneedling (not the cosmetic “roller” microneedling performed by aestheticians), she added, “gives the best results for the money and produces clinical results that mirror those achieved with lasers.”
Dr. Wang agreed that even recommending less expensive and less efficacious options such as over-the-counter creams can help alleviate patients’ concerns. “It shows that you are being holistic – not just caring for medical issues around pregnancy, but that you also take the emotional/psychological concerns of pregnant individuals and new parents seriously and that you recognize the impact of skin problems on quality of life. In the end, recommending something – in other words, providing some options, like creams or other therapies, for instance – is still, in my opinion, better than not recommending anything.”
Dr. Wang is involved with a study that is currently enrolling patients and that is evaluating the formation of early SG, which includes performing skin biopsies as soon as lesions appear.
The study had no funding. The study authors and Dr. Alster disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Pandemic stresses harder on physician moms than physician dads: Study
COVID-19 has been difficult for parents trying to balance careers, home life, and keeping their loved ones safe. A new study indicates that, not only are physicians not immune to these stressors, but the long-term effects could be devastating for health care overall.

In a study published Nov. 11, 2021, in JAMA Network Open , researchers found that stresses to work/life balance and family life caused by the pandemic have differed among men and women physicians.
Physicians and other health care workers have been at the front lines of the COVID-19 pandemic, and their work lives have been the focus of a lot of attention in the media and by researchers. Their family lives, not so much. But physicians have families, and the pandemic has upended almost everything about their lives, particularly where work life and home life intersect. School and day care closures, working from home, working extra hours, or working less – all of these changes have consequences on family life and the mental health of parents who are also physicians.
Findings from a Medscape survey published in early 2021 indicate that more female physicians than male physicians were either “conflicted” or “very conflicted” as parents because of work demands (42% vs. 23%) nearly 6 months into the pandemic.
In the current study, researchers from the University of Michigan, Harvard University, and the Medical University of South Carolina teamed up to investigate gender differences in how work/family factors affected the mental health of early-career physician parents in the United States during the first year of the COVID-19 pandemic. The results suggest that the pandemic has increased gender disparity and added disproportionately to the burden of female physicians.
Managing the household falls mostly on moms
Participants were physicians enrolled in the Intern Health Study, a longitudinal study that regularly surveys medical interns in the United States to assess stress and mood. When researchers compared survey results from before the onset of the pandemic (2018) with later results (2020), they found a striking gender difference in how the pandemic has changed family and work duties for physicians.
The authors of the study pointed out that previous research had found that female physicians take on a greater share of household and childcare duties than male physicians. The current study found that their share had increased with the pandemic. Physician moms are now 30 times more likely to be in charge of these tasks than physician dads.
In families in which both parents were physicians, none of the men said they took the primary role in managing the extra demands caused by the pandemic. In addition, women were twice as likely as men to work primarily from home and to work reduced hours.
The extra stress seems to be taking a toll on women physicians. In the 2020 survey, physician mothers had higher scores for anxiety and depression symptoms, compared with men. Notably, the 2018 survey did not show a significant difference in depression scores between men and women. Nor were there significant differences in depression and anxiety scores between women and men who were not parents or in reports of work/family conflict before and after the pandemic.
In general, the results indicate that the pandemic has only widened the gender gap between women and men physicians when it comes to managing family life and dealing with the stresses of maintaining a suitable work-life balance.
‘Long-term repercussions’ for gender equity in medicine
Although these are serious problems for women physicians and their families, the effects go beyond the home and beyond individuals. Even before the pandemic, women in medicine struggled for parity in career advancement and opportunities as well as in pay, and this new setback could make those challenges even greater.
“Even short-term adjustments can have serious long-term repercussions as they may lead to lower earnings and negatively impact opportunities for promotion, further exacerbating gender inequalities in compensation and advancement,” the study’s authors wrote.
The potential damage extends to the entire profession and the health care system itself. The profession is already struggling to retain young female physicians, and this situation is likely to make that problem worse and have long-term consequences. Citing data showing that female physicians spend more time with patients and that their patients may have better outcomes, the authors wrote that the consequences of losing more early-career female physicians “could be devastating to the U.S. health care system, particularly in the context of a global pandemic and an impending physician shortage.”
The sample size was small (276 U.S. physicians), and the study relied on self-reported data. The findings suggest that more research on this topic is needed, especially research that includes other demographic factors, such as sexual orientation and ethnicity. The authors recommend that institutional and public policymakers take into account the effects of the pandemic on physician mothers to ensure that recent gains in gender equity for women physicians do not fall victim to COVID-19.
A version of this article first appeared on Medscape.com.
COVID-19 has been difficult for parents trying to balance careers, home life, and keeping their loved ones safe. A new study indicates that, not only are physicians not immune to these stressors, but the long-term effects could be devastating for health care overall.
In a study published Nov. 11, 2021, in JAMA Network Open , researchers found that stresses to work/life balance and family life caused by the pandemic have differed among men and women physicians.
Physicians and other health care workers have been at the front lines of the COVID-19 pandemic, and their work lives have been the focus of a lot of attention in the media and by researchers. Their family lives, not so much. But physicians have families, and the pandemic has upended almost everything about their lives, particularly where work life and home life intersect. School and day care closures, working from home, working extra hours, or working less – all of these changes have consequences on family life and the mental health of parents who are also physicians.
Findings from a Medscape survey published in early 2021 indicate that more female physicians than male physicians were either “conflicted” or “very conflicted” as parents because of work demands (42% vs. 23%) nearly 6 months into the pandemic.
In the current study, researchers from the University of Michigan, Harvard University, and the Medical University of South Carolina teamed up to investigate gender differences in how work/family factors affected the mental health of early-career physician parents in the United States during the first year of the COVID-19 pandemic. The results suggest that the pandemic has increased gender disparity and added disproportionately to the burden of female physicians.
Managing the household falls mostly on moms
Participants were physicians enrolled in the Intern Health Study, a longitudinal study that regularly surveys medical interns in the United States to assess stress and mood. When researchers compared survey results from before the onset of the pandemic (2018) with later results (2020), they found a striking gender difference in how the pandemic has changed family and work duties for physicians.
The authors of the study pointed out that previous research had found that female physicians take on a greater share of household and childcare duties than male physicians. The current study found that their share had increased with the pandemic. Physician moms are now 30 times more likely to be in charge of these tasks than physician dads.
In families in which both parents were physicians, none of the men said they took the primary role in managing the extra demands caused by the pandemic. In addition, women were twice as likely as men to work primarily from home and to work reduced hours.
The extra stress seems to be taking a toll on women physicians. In the 2020 survey, physician mothers had higher scores for anxiety and depression symptoms, compared with men. Notably, the 2018 survey did not show a significant difference in depression scores between men and women. Nor were there significant differences in depression and anxiety scores between women and men who were not parents or in reports of work/family conflict before and after the pandemic.
In general, the results indicate that the pandemic has only widened the gender gap between women and men physicians when it comes to managing family life and dealing with the stresses of maintaining a suitable work-life balance.
‘Long-term repercussions’ for gender equity in medicine
Although these are serious problems for women physicians and their families, the effects go beyond the home and beyond individuals. Even before the pandemic, women in medicine struggled for parity in career advancement and opportunities as well as in pay, and this new setback could make those challenges even greater.
“Even short-term adjustments can have serious long-term repercussions as they may lead to lower earnings and negatively impact opportunities for promotion, further exacerbating gender inequalities in compensation and advancement,” the study’s authors wrote.
The potential damage extends to the entire profession and the health care system itself. The profession is already struggling to retain young female physicians, and this situation is likely to make that problem worse and have long-term consequences. Citing data showing that female physicians spend more time with patients and that their patients may have better outcomes, the authors wrote that the consequences of losing more early-career female physicians “could be devastating to the U.S. health care system, particularly in the context of a global pandemic and an impending physician shortage.”
The sample size was small (276 U.S. physicians), and the study relied on self-reported data. The findings suggest that more research on this topic is needed, especially research that includes other demographic factors, such as sexual orientation and ethnicity. The authors recommend that institutional and public policymakers take into account the effects of the pandemic on physician mothers to ensure that recent gains in gender equity for women physicians do not fall victim to COVID-19.
A version of this article first appeared on Medscape.com.
COVID-19 has been difficult for parents trying to balance careers, home life, and keeping their loved ones safe. A new study indicates that, not only are physicians not immune to these stressors, but the long-term effects could be devastating for health care overall.
In a study published Nov. 11, 2021, in JAMA Network Open , researchers found that stresses to work/life balance and family life caused by the pandemic have differed among men and women physicians.
Physicians and other health care workers have been at the front lines of the COVID-19 pandemic, and their work lives have been the focus of a lot of attention in the media and by researchers. Their family lives, not so much. But physicians have families, and the pandemic has upended almost everything about their lives, particularly where work life and home life intersect. School and day care closures, working from home, working extra hours, or working less – all of these changes have consequences on family life and the mental health of parents who are also physicians.
Findings from a Medscape survey published in early 2021 indicate that more female physicians than male physicians were either “conflicted” or “very conflicted” as parents because of work demands (42% vs. 23%) nearly 6 months into the pandemic.
In the current study, researchers from the University of Michigan, Harvard University, and the Medical University of South Carolina teamed up to investigate gender differences in how work/family factors affected the mental health of early-career physician parents in the United States during the first year of the COVID-19 pandemic. The results suggest that the pandemic has increased gender disparity and added disproportionately to the burden of female physicians.
Managing the household falls mostly on moms
Participants were physicians enrolled in the Intern Health Study, a longitudinal study that regularly surveys medical interns in the United States to assess stress and mood. When researchers compared survey results from before the onset of the pandemic (2018) with later results (2020), they found a striking gender difference in how the pandemic has changed family and work duties for physicians.
The authors of the study pointed out that previous research had found that female physicians take on a greater share of household and childcare duties than male physicians. The current study found that their share had increased with the pandemic. Physician moms are now 30 times more likely to be in charge of these tasks than physician dads.
In families in which both parents were physicians, none of the men said they took the primary role in managing the extra demands caused by the pandemic. In addition, women were twice as likely as men to work primarily from home and to work reduced hours.
The extra stress seems to be taking a toll on women physicians. In the 2020 survey, physician mothers had higher scores for anxiety and depression symptoms, compared with men. Notably, the 2018 survey did not show a significant difference in depression scores between men and women. Nor were there significant differences in depression and anxiety scores between women and men who were not parents or in reports of work/family conflict before and after the pandemic.
In general, the results indicate that the pandemic has only widened the gender gap between women and men physicians when it comes to managing family life and dealing with the stresses of maintaining a suitable work-life balance.
‘Long-term repercussions’ for gender equity in medicine
Although these are serious problems for women physicians and their families, the effects go beyond the home and beyond individuals. Even before the pandemic, women in medicine struggled for parity in career advancement and opportunities as well as in pay, and this new setback could make those challenges even greater.
“Even short-term adjustments can have serious long-term repercussions as they may lead to lower earnings and negatively impact opportunities for promotion, further exacerbating gender inequalities in compensation and advancement,” the study’s authors wrote.
The potential damage extends to the entire profession and the health care system itself. The profession is already struggling to retain young female physicians, and this situation is likely to make that problem worse and have long-term consequences. Citing data showing that female physicians spend more time with patients and that their patients may have better outcomes, the authors wrote that the consequences of losing more early-career female physicians “could be devastating to the U.S. health care system, particularly in the context of a global pandemic and an impending physician shortage.”
The sample size was small (276 U.S. physicians), and the study relied on self-reported data. The findings suggest that more research on this topic is needed, especially research that includes other demographic factors, such as sexual orientation and ethnicity. The authors recommend that institutional and public policymakers take into account the effects of the pandemic on physician mothers to ensure that recent gains in gender equity for women physicians do not fall victim to COVID-19.
A version of this article first appeared on Medscape.com.
COVID-19 vaccine mandates are working, public health experts say
Some organizations have reported vaccination rates that jumped from less than 50% to more than 90%, according to ABC News. Workplace mandates have especially encouraged employees who were on the fence to get a shot.
“In general, vaccine mandates work,” James Colgrove, a public health professor at Columbia University’s Mailman School of Public Health, told ABC News.
For decades, the United States has monitored the effectiveness of vaccine mandates in schools, he noted, which have successfully required shots against measles, mumps, and other illnesses that used to be widespread. Certain employees, such as hospital workers, must take vaccines for their jobs, he said, and those requirements have also been effective over the years.
“The more normalized it becomes, the more people [know] someone else who is vaccinated, the more people will comply,” he said. “With any vaccine, the longer it’s been around, the more people get with it.”
With the widespread and contagious nature of COVID-19, workplaces have been forced to consider vaccine mandates to protect their employees and prevent worker shortages, Dr. Colgrove said.
Some companies began to issue vaccine rules this summer as the Delta variant caused a jump in cases, hospitalizations, and deaths. Major companies, including Google, Tyson Foods, United Airlines, and the Walt Disney Company, required in-person employees to get a shot. So far, the results from those mandates have been strong, ABC News reported.
For instance, Tyson announced a mandate in August, when less than half of its 140,000 employees were vaccinated. When the deadline came at the end of October, more than 60,000 additional employees had been vaccinated, and the vaccination rate was 96%.
“Has this made a difference in the health and safety of our team members? Absolutely. We’ve seen a significant decline in the number of active cases companywide,” Donnie King, CEO and president of Tyson Foods, said in a statement.
United Airlines has also shared that 99.7% of its 67,000 employees are vaccinated. Within 48 hours of announcing its mandate, the number of unvaccinated staffers fell from 593 to 320 people, ABC News reported.
Vaccine mandates appear to be working in the public sector as well. State health department officials in Washington told ABC News that the percentage of public employees who were vaccinated jumped from 49% in September to 96% by the vaccine mandate deadline in October.
Vaccination rates have also increased in New York City, where some employees in the fire, police, and sanitation departments protested the mandate. By the deadline, vaccination rates shifted from less than 75% to 82% in the fire department, 86% in the police department, and 91% of EMS personnel, ABC News reported.
Overall, vaccine mandates tend to reach groups who aren’t completely against the vaccine, medical experts told the news outlet. A small percentage of the population truly opposes the shot, and in most cases, unvaccinated people are on the fence or haven’t seen good enough messaging for it.
“When you look at vaccine resistance, the people who are the most opposed often make a very large amount of noise that is at odds with the actual numbers who are against vaccination,” Dr. Colgrove said.
A version of this article first appeared on WebMD.com.
Some organizations have reported vaccination rates that jumped from less than 50% to more than 90%, according to ABC News. Workplace mandates have especially encouraged employees who were on the fence to get a shot.
“In general, vaccine mandates work,” James Colgrove, a public health professor at Columbia University’s Mailman School of Public Health, told ABC News.
For decades, the United States has monitored the effectiveness of vaccine mandates in schools, he noted, which have successfully required shots against measles, mumps, and other illnesses that used to be widespread. Certain employees, such as hospital workers, must take vaccines for their jobs, he said, and those requirements have also been effective over the years.
“The more normalized it becomes, the more people [know] someone else who is vaccinated, the more people will comply,” he said. “With any vaccine, the longer it’s been around, the more people get with it.”
With the widespread and contagious nature of COVID-19, workplaces have been forced to consider vaccine mandates to protect their employees and prevent worker shortages, Dr. Colgrove said.
Some companies began to issue vaccine rules this summer as the Delta variant caused a jump in cases, hospitalizations, and deaths. Major companies, including Google, Tyson Foods, United Airlines, and the Walt Disney Company, required in-person employees to get a shot. So far, the results from those mandates have been strong, ABC News reported.
For instance, Tyson announced a mandate in August, when less than half of its 140,000 employees were vaccinated. When the deadline came at the end of October, more than 60,000 additional employees had been vaccinated, and the vaccination rate was 96%.
“Has this made a difference in the health and safety of our team members? Absolutely. We’ve seen a significant decline in the number of active cases companywide,” Donnie King, CEO and president of Tyson Foods, said in a statement.
United Airlines has also shared that 99.7% of its 67,000 employees are vaccinated. Within 48 hours of announcing its mandate, the number of unvaccinated staffers fell from 593 to 320 people, ABC News reported.
Vaccine mandates appear to be working in the public sector as well. State health department officials in Washington told ABC News that the percentage of public employees who were vaccinated jumped from 49% in September to 96% by the vaccine mandate deadline in October.
Vaccination rates have also increased in New York City, where some employees in the fire, police, and sanitation departments protested the mandate. By the deadline, vaccination rates shifted from less than 75% to 82% in the fire department, 86% in the police department, and 91% of EMS personnel, ABC News reported.
Overall, vaccine mandates tend to reach groups who aren’t completely against the vaccine, medical experts told the news outlet. A small percentage of the population truly opposes the shot, and in most cases, unvaccinated people are on the fence or haven’t seen good enough messaging for it.
“When you look at vaccine resistance, the people who are the most opposed often make a very large amount of noise that is at odds with the actual numbers who are against vaccination,” Dr. Colgrove said.
A version of this article first appeared on WebMD.com.
Some organizations have reported vaccination rates that jumped from less than 50% to more than 90%, according to ABC News. Workplace mandates have especially encouraged employees who were on the fence to get a shot.
“In general, vaccine mandates work,” James Colgrove, a public health professor at Columbia University’s Mailman School of Public Health, told ABC News.
For decades, the United States has monitored the effectiveness of vaccine mandates in schools, he noted, which have successfully required shots against measles, mumps, and other illnesses that used to be widespread. Certain employees, such as hospital workers, must take vaccines for their jobs, he said, and those requirements have also been effective over the years.
“The more normalized it becomes, the more people [know] someone else who is vaccinated, the more people will comply,” he said. “With any vaccine, the longer it’s been around, the more people get with it.”
With the widespread and contagious nature of COVID-19, workplaces have been forced to consider vaccine mandates to protect their employees and prevent worker shortages, Dr. Colgrove said.
Some companies began to issue vaccine rules this summer as the Delta variant caused a jump in cases, hospitalizations, and deaths. Major companies, including Google, Tyson Foods, United Airlines, and the Walt Disney Company, required in-person employees to get a shot. So far, the results from those mandates have been strong, ABC News reported.
For instance, Tyson announced a mandate in August, when less than half of its 140,000 employees were vaccinated. When the deadline came at the end of October, more than 60,000 additional employees had been vaccinated, and the vaccination rate was 96%.
“Has this made a difference in the health and safety of our team members? Absolutely. We’ve seen a significant decline in the number of active cases companywide,” Donnie King, CEO and president of Tyson Foods, said in a statement.
United Airlines has also shared that 99.7% of its 67,000 employees are vaccinated. Within 48 hours of announcing its mandate, the number of unvaccinated staffers fell from 593 to 320 people, ABC News reported.
Vaccine mandates appear to be working in the public sector as well. State health department officials in Washington told ABC News that the percentage of public employees who were vaccinated jumped from 49% in September to 96% by the vaccine mandate deadline in October.
Vaccination rates have also increased in New York City, where some employees in the fire, police, and sanitation departments protested the mandate. By the deadline, vaccination rates shifted from less than 75% to 82% in the fire department, 86% in the police department, and 91% of EMS personnel, ABC News reported.
Overall, vaccine mandates tend to reach groups who aren’t completely against the vaccine, medical experts told the news outlet. A small percentage of the population truly opposes the shot, and in most cases, unvaccinated people are on the fence or haven’t seen good enough messaging for it.
“When you look at vaccine resistance, the people who are the most opposed often make a very large amount of noise that is at odds with the actual numbers who are against vaccination,” Dr. Colgrove said.
A version of this article first appeared on WebMD.com.
Contact allergens in medical devices: A cause for concern?
Despite the clinical value of medical devices, there is a potential for these products to cause adverse skin reactions in some patients. highlighting the possibility of a high prevalence of contact allergens in these devices.
“We found it important to publish these findings, because up until now no clear figures have been reported regarding this particular clinical problem,” said study author Olivier Aerts, MD, a researcher in the contact allergy unit at the University Hospital Antwerp, Belgium, in an interview with this news organization.
For the study, Dr. Aerts and colleagues conducted a retrospective analysis of medical device users with suspected allergic contact dermatitis. All patients had been patch tested at a tertiary European clinic between 2018 and 2020.
The cohort included patients who experienced suspected contact allergy from medical adhesives (n = 57), gloves (n = 38), topical and surface medical devices (n = 38), glucose sensors and insulin pumps (n = 74), and prostheses (n = 75). Other medical products associated with contact allergy in another 44 patients included surgical glues, face masks, compression stockings, condoms, and suture materials.
Overall, 326 patients had been patch-tested during the 30-month study period. Approximately 25.8% of all patients – including 299 adults and 27 children – were referred for contact allergy associated with medical devices.
Acrylates were the most frequently encountered contact allergens and were found in diabetes devices and medical adhesives. Potential skin sensitizers included colophonium-related substances, D-limonene, isothiazolinone derivatives, salicylates, and sulphites, all of which were identified across most products.
According to the investigators, many of the labels for the medical devices made no mention of the potential skin sensitizers, except in the cases of some topical and surface disinfectants. And many topical products are often marketed as medical devices rather than cosmetics, further complicating labeling issues, according to Dr. Aerts.
“What should be done to help any patient suffering from allergic contact due to medical devices is that these devices should be labeled with all their components, or at the very least with the potential skin sensitizers these may contain,” Dr. Aerts explained. He added that manufacturers should “establish more cooperation with physicians/dermatologists who evaluate such patients,” a cooperation that often exists with cosmetic companies.
Dr. Aerts noted that while it’s important for patch testers and dermatologists to be aware of the prevalence of allergic contact dermatitis in medical device users, companies producing these devices should also be aware of these potential issues. “Additionally, legislators/regulators should perhaps focus some more on the cutaneous side effects these products may provoke,” he said, “as this awareness may hopefully also serve as a stimulant to perform more clinical allergy research in this field.”
Leonard Bielory, MD, an allergist at Robert Wood Johnson University Hospital in Rahway, New Jersey, told this news organization that the findings are “alarming” and should heighten clinicians’ awareness of the possibility of allergic contact dermatitis among medical device users.
Dr. Bielory, who wasn’t involved in the research, noted that the findings from this study may not be entirely generalizable to the U.S., given the study was performed in Europe. “In contrast to other countries, the U.S. is very conscientious about allergic responses to items being used in hospitals,” he added, “or such that the issue here is that many of these things would be an adverse reaction, which you have to report.” He suggested that further research in this field is needed to determine the prevalence of possible skin sensitizers in products specifically developed and marketed in the U.S.
The study had no specific funding. Dr. Aerts and Dr. Bielory have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Despite the clinical value of medical devices, there is a potential for these products to cause adverse skin reactions in some patients. highlighting the possibility of a high prevalence of contact allergens in these devices.
“We found it important to publish these findings, because up until now no clear figures have been reported regarding this particular clinical problem,” said study author Olivier Aerts, MD, a researcher in the contact allergy unit at the University Hospital Antwerp, Belgium, in an interview with this news organization.
For the study, Dr. Aerts and colleagues conducted a retrospective analysis of medical device users with suspected allergic contact dermatitis. All patients had been patch tested at a tertiary European clinic between 2018 and 2020.
The cohort included patients who experienced suspected contact allergy from medical adhesives (n = 57), gloves (n = 38), topical and surface medical devices (n = 38), glucose sensors and insulin pumps (n = 74), and prostheses (n = 75). Other medical products associated with contact allergy in another 44 patients included surgical glues, face masks, compression stockings, condoms, and suture materials.
Overall, 326 patients had been patch-tested during the 30-month study period. Approximately 25.8% of all patients – including 299 adults and 27 children – were referred for contact allergy associated with medical devices.
Acrylates were the most frequently encountered contact allergens and were found in diabetes devices and medical adhesives. Potential skin sensitizers included colophonium-related substances, D-limonene, isothiazolinone derivatives, salicylates, and sulphites, all of which were identified across most products.
According to the investigators, many of the labels for the medical devices made no mention of the potential skin sensitizers, except in the cases of some topical and surface disinfectants. And many topical products are often marketed as medical devices rather than cosmetics, further complicating labeling issues, according to Dr. Aerts.
“What should be done to help any patient suffering from allergic contact due to medical devices is that these devices should be labeled with all their components, or at the very least with the potential skin sensitizers these may contain,” Dr. Aerts explained. He added that manufacturers should “establish more cooperation with physicians/dermatologists who evaluate such patients,” a cooperation that often exists with cosmetic companies.
Dr. Aerts noted that while it’s important for patch testers and dermatologists to be aware of the prevalence of allergic contact dermatitis in medical device users, companies producing these devices should also be aware of these potential issues. “Additionally, legislators/regulators should perhaps focus some more on the cutaneous side effects these products may provoke,” he said, “as this awareness may hopefully also serve as a stimulant to perform more clinical allergy research in this field.”
Leonard Bielory, MD, an allergist at Robert Wood Johnson University Hospital in Rahway, New Jersey, told this news organization that the findings are “alarming” and should heighten clinicians’ awareness of the possibility of allergic contact dermatitis among medical device users.
Dr. Bielory, who wasn’t involved in the research, noted that the findings from this study may not be entirely generalizable to the U.S., given the study was performed in Europe. “In contrast to other countries, the U.S. is very conscientious about allergic responses to items being used in hospitals,” he added, “or such that the issue here is that many of these things would be an adverse reaction, which you have to report.” He suggested that further research in this field is needed to determine the prevalence of possible skin sensitizers in products specifically developed and marketed in the U.S.
The study had no specific funding. Dr. Aerts and Dr. Bielory have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Despite the clinical value of medical devices, there is a potential for these products to cause adverse skin reactions in some patients. highlighting the possibility of a high prevalence of contact allergens in these devices.
“We found it important to publish these findings, because up until now no clear figures have been reported regarding this particular clinical problem,” said study author Olivier Aerts, MD, a researcher in the contact allergy unit at the University Hospital Antwerp, Belgium, in an interview with this news organization.
For the study, Dr. Aerts and colleagues conducted a retrospective analysis of medical device users with suspected allergic contact dermatitis. All patients had been patch tested at a tertiary European clinic between 2018 and 2020.
The cohort included patients who experienced suspected contact allergy from medical adhesives (n = 57), gloves (n = 38), topical and surface medical devices (n = 38), glucose sensors and insulin pumps (n = 74), and prostheses (n = 75). Other medical products associated with contact allergy in another 44 patients included surgical glues, face masks, compression stockings, condoms, and suture materials.
Overall, 326 patients had been patch-tested during the 30-month study period. Approximately 25.8% of all patients – including 299 adults and 27 children – were referred for contact allergy associated with medical devices.
Acrylates were the most frequently encountered contact allergens and were found in diabetes devices and medical adhesives. Potential skin sensitizers included colophonium-related substances, D-limonene, isothiazolinone derivatives, salicylates, and sulphites, all of which were identified across most products.
According to the investigators, many of the labels for the medical devices made no mention of the potential skin sensitizers, except in the cases of some topical and surface disinfectants. And many topical products are often marketed as medical devices rather than cosmetics, further complicating labeling issues, according to Dr. Aerts.
“What should be done to help any patient suffering from allergic contact due to medical devices is that these devices should be labeled with all their components, or at the very least with the potential skin sensitizers these may contain,” Dr. Aerts explained. He added that manufacturers should “establish more cooperation with physicians/dermatologists who evaluate such patients,” a cooperation that often exists with cosmetic companies.
Dr. Aerts noted that while it’s important for patch testers and dermatologists to be aware of the prevalence of allergic contact dermatitis in medical device users, companies producing these devices should also be aware of these potential issues. “Additionally, legislators/regulators should perhaps focus some more on the cutaneous side effects these products may provoke,” he said, “as this awareness may hopefully also serve as a stimulant to perform more clinical allergy research in this field.”
Leonard Bielory, MD, an allergist at Robert Wood Johnson University Hospital in Rahway, New Jersey, told this news organization that the findings are “alarming” and should heighten clinicians’ awareness of the possibility of allergic contact dermatitis among medical device users.
Dr. Bielory, who wasn’t involved in the research, noted that the findings from this study may not be entirely generalizable to the U.S., given the study was performed in Europe. “In contrast to other countries, the U.S. is very conscientious about allergic responses to items being used in hospitals,” he added, “or such that the issue here is that many of these things would be an adverse reaction, which you have to report.” He suggested that further research in this field is needed to determine the prevalence of possible skin sensitizers in products specifically developed and marketed in the U.S.
The study had no specific funding. Dr. Aerts and Dr. Bielory have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Cannabinoids being studied for a variety of dermatologic conditions
.

“When you walk into places like CVS or Walgreens, you see lots of displays for CBD creams and oils,” Todd S. Anhalt, MD, said during the annual meeting of the Pacific Dermatologic Association. “The problem is, we don’t know what’s in them or who made them or how good they are. That’s going to be a problem for a while.”
According to Dr. Anhalt, clinical professor emeritus of dermatology at Stanford (Calif.) University, there are about 140 active cannabinoid compounds in cannabis, but the most important ones are THC and cannabidiol (CBD). There are three types of cannabinoids, based on where the cannabidiol is produced: endocannabinoids, which are produced in the human body; phytocannabinoids, which are derived from plants such as marijuana and hemp; and synthetic cannabinoids, which are derived in labs.
Dr. Anhalt described the endocannabinoid system as a conserved network of molecular signaling made of several components: signaling molecules (endocannabinoids), endocannabinoid receptors (CB-1 and CB-2), enzymes, and transporters. There is also overlap between cannabinoids and terpenes, which are responsible for flavor and aroma in plants and marijuana and can enhance the effects of CBD.
“For the most part, CB-1 receptors are in the central nervous system and CB-2 [receptors] are mostly in the periphery,” including the skin and digestive system, said Dr. Anhalt, who practices at the California Skin Institute in Los Altos, Calif. “This is interesting because one of the main conditions I recommend cannabidiol for is in patients with peripheral neuropathy, despite the fact they may be on all sorts of medications such as Neurontin and Lyrica or tricyclic antidepressants. Sometimes they don’t get much relief from those. I have had many patients tell me that they have had reduction of pain and increased functionality using the CBD creams.” CB-2 receptors, he noted, are located in keratinocytes, sensory receptors, sweat glands, fibroblasts, Langerhans cells, melanocytes, and sebaceous glands.
Recent research shows that the endocannabinoid system is involved in modulation of the CNS and in immune function, particularly skin homeostasis and barrier function. “We know that barrier function can be affected by the generation of oxidative species,” he said. “The stress that it causes can decrease barrier function and lead to cytokine release and itch. CBDs have been shown to enter cells, target and upregulate genes with decreased oxidation and inflammation, and protect membrane integrity in skin cells. Therefore, this might be helpful in atopic dermatitis.” Other potential uses in dermatology include wound healing, acne, hair growth modulation, skin and hair pigmentation, skin infections, psoriasis, and cutaneous malignancies, as well as neuropathic pain.
Evidence is strongest for neuropathic pain, he said, which is mediated by CB-1 receptors peripherally, followed by itch and atopic dermatitis. The authors of a 2017 systematic review concluded that “low-strength” evidence exists to suggest that cannabis alleviates neuropathic pain, with insufficient evidence for other types of pain.
Topical CBD comes in various forms: oils (usually hemp oil), creams, and lotions, Dr. Anhalt said. “I advise patients to apply it 2-4 times per day depending on how anxious or uncomfortable they are. It takes my patients 10 days to 2 weeks before they notice anything at all.”
For atopic dermatitis, it could be useful “not to use it instead of a moisturizer, but as a moisturizer,” Dr. Anhalt advised. “You can have a patient get big jars of CBD creams and lotions. They may have to try a few before they find one that they really like, but you can replace all of the other moisturizers that you’re using right now in patients who have a lot of itch.”
As for CBD’s effect on peripheral neuropathy, the medical literature is lacking, but some studies show low to moderate evidence of efficacy. For example, a Cochrane Review found that a 30% or greater pain reduction was achieved by 39% of patients who used cannabis-based treatments, vs. 33% of those on placebo.
“I would not suggest CBD as a first-line drug unless it’s very mild peripheral neuropathy, but for patients who are on gabapentin who are better but not better enough, this is an excellent adjunct,” Dr. Anhalt said. “It’s worth trying. It’s not too expensive and it’s really safe.”
The application of topical CBD to treat cutaneous malignancies has not yet shown evidence of significant efficacy, while using CBDs for acne holds promise. “The endogenous cannabinoid system is involved in the production of lipids,” he said. “Cannabinoids have an antilipogenic activity, so they decrease sebum production. CBD could help patients with mild acne who are reluctant to use other types of medications. For this and other potential dermatologic applications, lots more studies need to be done.”
Dr. Anhalt reported having no financial disclosures.
.
“When you walk into places like CVS or Walgreens, you see lots of displays for CBD creams and oils,” Todd S. Anhalt, MD, said during the annual meeting of the Pacific Dermatologic Association. “The problem is, we don’t know what’s in them or who made them or how good they are. That’s going to be a problem for a while.”
According to Dr. Anhalt, clinical professor emeritus of dermatology at Stanford (Calif.) University, there are about 140 active cannabinoid compounds in cannabis, but the most important ones are THC and cannabidiol (CBD). There are three types of cannabinoids, based on where the cannabidiol is produced: endocannabinoids, which are produced in the human body; phytocannabinoids, which are derived from plants such as marijuana and hemp; and synthetic cannabinoids, which are derived in labs.
Dr. Anhalt described the endocannabinoid system as a conserved network of molecular signaling made of several components: signaling molecules (endocannabinoids), endocannabinoid receptors (CB-1 and CB-2), enzymes, and transporters. There is also overlap between cannabinoids and terpenes, which are responsible for flavor and aroma in plants and marijuana and can enhance the effects of CBD.
“For the most part, CB-1 receptors are in the central nervous system and CB-2 [receptors] are mostly in the periphery,” including the skin and digestive system, said Dr. Anhalt, who practices at the California Skin Institute in Los Altos, Calif. “This is interesting because one of the main conditions I recommend cannabidiol for is in patients with peripheral neuropathy, despite the fact they may be on all sorts of medications such as Neurontin and Lyrica or tricyclic antidepressants. Sometimes they don’t get much relief from those. I have had many patients tell me that they have had reduction of pain and increased functionality using the CBD creams.” CB-2 receptors, he noted, are located in keratinocytes, sensory receptors, sweat glands, fibroblasts, Langerhans cells, melanocytes, and sebaceous glands.
Recent research shows that the endocannabinoid system is involved in modulation of the CNS and in immune function, particularly skin homeostasis and barrier function. “We know that barrier function can be affected by the generation of oxidative species,” he said. “The stress that it causes can decrease barrier function and lead to cytokine release and itch. CBDs have been shown to enter cells, target and upregulate genes with decreased oxidation and inflammation, and protect membrane integrity in skin cells. Therefore, this might be helpful in atopic dermatitis.” Other potential uses in dermatology include wound healing, acne, hair growth modulation, skin and hair pigmentation, skin infections, psoriasis, and cutaneous malignancies, as well as neuropathic pain.
Evidence is strongest for neuropathic pain, he said, which is mediated by CB-1 receptors peripherally, followed by itch and atopic dermatitis. The authors of a 2017 systematic review concluded that “low-strength” evidence exists to suggest that cannabis alleviates neuropathic pain, with insufficient evidence for other types of pain.
Topical CBD comes in various forms: oils (usually hemp oil), creams, and lotions, Dr. Anhalt said. “I advise patients to apply it 2-4 times per day depending on how anxious or uncomfortable they are. It takes my patients 10 days to 2 weeks before they notice anything at all.”
For atopic dermatitis, it could be useful “not to use it instead of a moisturizer, but as a moisturizer,” Dr. Anhalt advised. “You can have a patient get big jars of CBD creams and lotions. They may have to try a few before they find one that they really like, but you can replace all of the other moisturizers that you’re using right now in patients who have a lot of itch.”
As for CBD’s effect on peripheral neuropathy, the medical literature is lacking, but some studies show low to moderate evidence of efficacy. For example, a Cochrane Review found that a 30% or greater pain reduction was achieved by 39% of patients who used cannabis-based treatments, vs. 33% of those on placebo.
“I would not suggest CBD as a first-line drug unless it’s very mild peripheral neuropathy, but for patients who are on gabapentin who are better but not better enough, this is an excellent adjunct,” Dr. Anhalt said. “It’s worth trying. It’s not too expensive and it’s really safe.”
The application of topical CBD to treat cutaneous malignancies has not yet shown evidence of significant efficacy, while using CBDs for acne holds promise. “The endogenous cannabinoid system is involved in the production of lipids,” he said. “Cannabinoids have an antilipogenic activity, so they decrease sebum production. CBD could help patients with mild acne who are reluctant to use other types of medications. For this and other potential dermatologic applications, lots more studies need to be done.”
Dr. Anhalt reported having no financial disclosures.
.
“When you walk into places like CVS or Walgreens, you see lots of displays for CBD creams and oils,” Todd S. Anhalt, MD, said during the annual meeting of the Pacific Dermatologic Association. “The problem is, we don’t know what’s in them or who made them or how good they are. That’s going to be a problem for a while.”
According to Dr. Anhalt, clinical professor emeritus of dermatology at Stanford (Calif.) University, there are about 140 active cannabinoid compounds in cannabis, but the most important ones are THC and cannabidiol (CBD). There are three types of cannabinoids, based on where the cannabidiol is produced: endocannabinoids, which are produced in the human body; phytocannabinoids, which are derived from plants such as marijuana and hemp; and synthetic cannabinoids, which are derived in labs.
Dr. Anhalt described the endocannabinoid system as a conserved network of molecular signaling made of several components: signaling molecules (endocannabinoids), endocannabinoid receptors (CB-1 and CB-2), enzymes, and transporters. There is also overlap between cannabinoids and terpenes, which are responsible for flavor and aroma in plants and marijuana and can enhance the effects of CBD.
“For the most part, CB-1 receptors are in the central nervous system and CB-2 [receptors] are mostly in the periphery,” including the skin and digestive system, said Dr. Anhalt, who practices at the California Skin Institute in Los Altos, Calif. “This is interesting because one of the main conditions I recommend cannabidiol for is in patients with peripheral neuropathy, despite the fact they may be on all sorts of medications such as Neurontin and Lyrica or tricyclic antidepressants. Sometimes they don’t get much relief from those. I have had many patients tell me that they have had reduction of pain and increased functionality using the CBD creams.” CB-2 receptors, he noted, are located in keratinocytes, sensory receptors, sweat glands, fibroblasts, Langerhans cells, melanocytes, and sebaceous glands.
Recent research shows that the endocannabinoid system is involved in modulation of the CNS and in immune function, particularly skin homeostasis and barrier function. “We know that barrier function can be affected by the generation of oxidative species,” he said. “The stress that it causes can decrease barrier function and lead to cytokine release and itch. CBDs have been shown to enter cells, target and upregulate genes with decreased oxidation and inflammation, and protect membrane integrity in skin cells. Therefore, this might be helpful in atopic dermatitis.” Other potential uses in dermatology include wound healing, acne, hair growth modulation, skin and hair pigmentation, skin infections, psoriasis, and cutaneous malignancies, as well as neuropathic pain.
Evidence is strongest for neuropathic pain, he said, which is mediated by CB-1 receptors peripherally, followed by itch and atopic dermatitis. The authors of a 2017 systematic review concluded that “low-strength” evidence exists to suggest that cannabis alleviates neuropathic pain, with insufficient evidence for other types of pain.
Topical CBD comes in various forms: oils (usually hemp oil), creams, and lotions, Dr. Anhalt said. “I advise patients to apply it 2-4 times per day depending on how anxious or uncomfortable they are. It takes my patients 10 days to 2 weeks before they notice anything at all.”
For atopic dermatitis, it could be useful “not to use it instead of a moisturizer, but as a moisturizer,” Dr. Anhalt advised. “You can have a patient get big jars of CBD creams and lotions. They may have to try a few before they find one that they really like, but you can replace all of the other moisturizers that you’re using right now in patients who have a lot of itch.”
As for CBD’s effect on peripheral neuropathy, the medical literature is lacking, but some studies show low to moderate evidence of efficacy. For example, a Cochrane Review found that a 30% or greater pain reduction was achieved by 39% of patients who used cannabis-based treatments, vs. 33% of those on placebo.
“I would not suggest CBD as a first-line drug unless it’s very mild peripheral neuropathy, but for patients who are on gabapentin who are better but not better enough, this is an excellent adjunct,” Dr. Anhalt said. “It’s worth trying. It’s not too expensive and it’s really safe.”
The application of topical CBD to treat cutaneous malignancies has not yet shown evidence of significant efficacy, while using CBDs for acne holds promise. “The endogenous cannabinoid system is involved in the production of lipids,” he said. “Cannabinoids have an antilipogenic activity, so they decrease sebum production. CBD could help patients with mild acne who are reluctant to use other types of medications. For this and other potential dermatologic applications, lots more studies need to be done.”
Dr. Anhalt reported having no financial disclosures.
FROM PDA 2021
Step right up, folks, for a public dissection
The greatest autopsy on Earth?
The LOTME staff would like to apologize in advance. The following item contains historical facts.
P.T. Barnum is a rather controversial figure in American history. The greatest show on Earth was certainly popular in its day. However, Barnum got his start in 1835 by leasing a slave named Joyce Heth, an elderly Black woman who told vivid stories of caring for a young George Washington. He toured her around the country, advertising her as a 160-year-old woman who served as George Washington’s nanny. When Ms. Heth died the next year, Barnum sold tickets to the autopsy, charging the equivalent of $30 in today’s money.
When a doctor announced that Ms. Heth was actually 75-80 when she died, it caused great controversy in the press and ruined Barnum’s career. Wait, no, that’s not right. The opposite, actually. He weathered the storm, built his famous circus, and never again committed a hoax.
It’s difficult to quantify how wrong publicly dissecting a person and charging people to see said dissection is, but that was almost 200 years ago. At the very least, we can say that such terrible behavior is firmly in the distant past.
Oh wait.
David Saunders, a 98-year-old veteran of World War II and the Korean War, donated his body to science. His body, however, was purchased by DeathScience.org from a medical lab – with the buyer supposedly misleading the medical lab about its intentions, which was for use at the traveling Oddities and Curiosities Expo. Tickets went for up to $500 each to witness the public autopsy of Mr. Saunders’ body, which took place at a Marriott in Portland, Ore. It promised to be an exciting, all-day event from 9 a.m. to 4 p.m., with a break for lunch, of course. You can’t have an autopsy without a catered lunch.
Another public autopsy event was scheduled in Seattle but canceled after news of the first event broke. Oh, and for that extra little kick, Mr. Saunders died from COVID-19, meaning that all those paying customers were exposed.
P.T. Barnum is probably rolling over in his grave right now. His autopsy tickets were a bargain.
Go ahead, have that soda before math
We should all know by now that sugary drinks are bad, even artificially sweetened ones. It might not always stop us from drinking them, but we know the deal. But what if sugary drinks like soda could be helpful for girls in school?

You read that right. We said girls. A soda before class might have boys bouncing off the walls, but not girls. A recent study showed that not only was girls’ behavior unaffected by having a sugary drink, their math skills even improved.
Researchers analyzed the behavior of 4- to 6-year-old children before and after having a sugary drink. The sugar rush was actually calming for girls and helped them perform better with numerical skills, but the opposite was true for boys. “Our study is the first to provide large-scale experimental evidence on the impact of sugary drinks on preschool children. The results clearly indicate a causal impact of sugary drinks on children’s behavior and test scores,” Fritz Schiltz, PhD, said in a written statement.
This probably isn’t the green light to have as many sugary drinks as you want, but it might be interesting to see how your work is affected after a soda.
Chicken nuggets and the meat paradox
Two young children are fighting over the last chicken nugget when an adult comes in to see what’s going on.
Liam: Vegetable!
Olivia: Meat!
Liam: Chicken nuggets are vegetables!
Olivia: No, dorkface! They’re meat.
Caregiver: Good news, kids. You’re both right.
Olivia: How can we both be right?
At this point, a woman enters the room. She’s wearing a white lab coat, so she must be a scientist.
Dr. Scientist: You can’t both be right, Olivia. You are being fed a serving of the meat paradox. That’s why Liam here doesn’t know that chicken nuggets are made of chicken, which is a form of meat. Sadly, he’s not the only one.

In a recent study, scientists from Furman University in Greenville, S.C., found that 38% of 176 children aged 4-7 years thought that chicken nuggets were vegetables and more than 46% identified French fries as animal based.
Olivia: Did our caregiver lie to us, Dr. Scientist?
Dr. Scientist: Yes, Olivia. The researchers I mentioned explained that “many people experience unease while eating meat. Omnivores eat foods that entail animal suffering and death while at the same time endorsing the compassionate treatment of animals.” That’s the meat paradox.
Liam: What else did they say, Dr. Scientist?
Dr. Scientist: Over 70% of those children said that cows and pigs were not edible and 5% thought that cats and horses were. The investigators wrote “that children and youth should be viewed as agents of environmental change” in the future, but suggested that parents need to bring honesty to the table.
Caregiver: How did you get in here anyway? And how do you know their names?
Dr. Scientist: I’ve been rooting through your garbage for years. All in the name of science, of course.
Bedtimes aren’t just for children
There are multiple ways to prevent heart disease, but what if it could be as easy as switching your bedtime? A recent study in European Heart Journal–Digital Health suggests that there’s a sweet spot when it comes to sleep timing.

Through smartwatch-like devices, researchers measured the sleep-onset and wake-up times for 7 days in 88,026 participants aged 43-79 years. After 5.7 years of follow-up to see if anyone had a heart attack, stroke, or any other cardiovascular event, 3.6% developed some kind of cardiovascular disease.
Those who went to bed between 10 p.m. and 11 p.m. had a lower risk of developing heart disease. The risk was 25% higher for subjects who went to bed at midnight or later, 24% higher for bedtimes before 10 p.m., and 12% higher for bedtimes between 11 p.m. and midnight.
So, why can you go to bed before “The Tonight Show” and lower your cardiovascular risk but not before the nightly news? Well, it has something to do with your body’s natural clock.
“The optimum time to go to sleep is at a specific point in the body’s 24-hour cycle and deviations may be detrimental to health. The riskiest time was after midnight, potentially because it may reduce the likelihood of seeing morning light, which resets the body clock,” said study author Dr. David Plans of the University of Exeter, England.
Although a sleep schedule is preferred, it isn’t realistic all the time for those in certain occupations who might have to resort to other methods to keep their circadian clocks ticking optimally for their health. But if all it takes is prescribing a sleep time to reduce heart disease on a massive scale it would make a great “low-cost public health target.”
So bedtimes aren’t just for children.
The greatest autopsy on Earth?
The LOTME staff would like to apologize in advance. The following item contains historical facts.
P.T. Barnum is a rather controversial figure in American history. The greatest show on Earth was certainly popular in its day. However, Barnum got his start in 1835 by leasing a slave named Joyce Heth, an elderly Black woman who told vivid stories of caring for a young George Washington. He toured her around the country, advertising her as a 160-year-old woman who served as George Washington’s nanny. When Ms. Heth died the next year, Barnum sold tickets to the autopsy, charging the equivalent of $30 in today’s money.
When a doctor announced that Ms. Heth was actually 75-80 when she died, it caused great controversy in the press and ruined Barnum’s career. Wait, no, that’s not right. The opposite, actually. He weathered the storm, built his famous circus, and never again committed a hoax.
It’s difficult to quantify how wrong publicly dissecting a person and charging people to see said dissection is, but that was almost 200 years ago. At the very least, we can say that such terrible behavior is firmly in the distant past.
Oh wait.
David Saunders, a 98-year-old veteran of World War II and the Korean War, donated his body to science. His body, however, was purchased by DeathScience.org from a medical lab – with the buyer supposedly misleading the medical lab about its intentions, which was for use at the traveling Oddities and Curiosities Expo. Tickets went for up to $500 each to witness the public autopsy of Mr. Saunders’ body, which took place at a Marriott in Portland, Ore. It promised to be an exciting, all-day event from 9 a.m. to 4 p.m., with a break for lunch, of course. You can’t have an autopsy without a catered lunch.
Another public autopsy event was scheduled in Seattle but canceled after news of the first event broke. Oh, and for that extra little kick, Mr. Saunders died from COVID-19, meaning that all those paying customers were exposed.
P.T. Barnum is probably rolling over in his grave right now. His autopsy tickets were a bargain.
Go ahead, have that soda before math
We should all know by now that sugary drinks are bad, even artificially sweetened ones. It might not always stop us from drinking them, but we know the deal. But what if sugary drinks like soda could be helpful for girls in school?
You read that right. We said girls. A soda before class might have boys bouncing off the walls, but not girls. A recent study showed that not only was girls’ behavior unaffected by having a sugary drink, their math skills even improved.
Researchers analyzed the behavior of 4- to 6-year-old children before and after having a sugary drink. The sugar rush was actually calming for girls and helped them perform better with numerical skills, but the opposite was true for boys. “Our study is the first to provide large-scale experimental evidence on the impact of sugary drinks on preschool children. The results clearly indicate a causal impact of sugary drinks on children’s behavior and test scores,” Fritz Schiltz, PhD, said in a written statement.
This probably isn’t the green light to have as many sugary drinks as you want, but it might be interesting to see how your work is affected after a soda.
Chicken nuggets and the meat paradox
Two young children are fighting over the last chicken nugget when an adult comes in to see what’s going on.
Liam: Vegetable!
Olivia: Meat!
Liam: Chicken nuggets are vegetables!
Olivia: No, dorkface! They’re meat.
Caregiver: Good news, kids. You’re both right.
Olivia: How can we both be right?
At this point, a woman enters the room. She’s wearing a white lab coat, so she must be a scientist.
Dr. Scientist: You can’t both be right, Olivia. You are being fed a serving of the meat paradox. That’s why Liam here doesn’t know that chicken nuggets are made of chicken, which is a form of meat. Sadly, he’s not the only one.
In a recent study, scientists from Furman University in Greenville, S.C., found that 38% of 176 children aged 4-7 years thought that chicken nuggets were vegetables and more than 46% identified French fries as animal based.
Olivia: Did our caregiver lie to us, Dr. Scientist?
Dr. Scientist: Yes, Olivia. The researchers I mentioned explained that “many people experience unease while eating meat. Omnivores eat foods that entail animal suffering and death while at the same time endorsing the compassionate treatment of animals.” That’s the meat paradox.
Liam: What else did they say, Dr. Scientist?
Dr. Scientist: Over 70% of those children said that cows and pigs were not edible and 5% thought that cats and horses were. The investigators wrote “that children and youth should be viewed as agents of environmental change” in the future, but suggested that parents need to bring honesty to the table.
Caregiver: How did you get in here anyway? And how do you know their names?
Dr. Scientist: I’ve been rooting through your garbage for years. All in the name of science, of course.
Bedtimes aren’t just for children
There are multiple ways to prevent heart disease, but what if it could be as easy as switching your bedtime? A recent study in European Heart Journal–Digital Health suggests that there’s a sweet spot when it comes to sleep timing.
Through smartwatch-like devices, researchers measured the sleep-onset and wake-up times for 7 days in 88,026 participants aged 43-79 years. After 5.7 years of follow-up to see if anyone had a heart attack, stroke, or any other cardiovascular event, 3.6% developed some kind of cardiovascular disease.
Those who went to bed between 10 p.m. and 11 p.m. had a lower risk of developing heart disease. The risk was 25% higher for subjects who went to bed at midnight or later, 24% higher for bedtimes before 10 p.m., and 12% higher for bedtimes between 11 p.m. and midnight.
So, why can you go to bed before “The Tonight Show” and lower your cardiovascular risk but not before the nightly news? Well, it has something to do with your body’s natural clock.
“The optimum time to go to sleep is at a specific point in the body’s 24-hour cycle and deviations may be detrimental to health. The riskiest time was after midnight, potentially because it may reduce the likelihood of seeing morning light, which resets the body clock,” said study author Dr. David Plans of the University of Exeter, England.
Although a sleep schedule is preferred, it isn’t realistic all the time for those in certain occupations who might have to resort to other methods to keep their circadian clocks ticking optimally for their health. But if all it takes is prescribing a sleep time to reduce heart disease on a massive scale it would make a great “low-cost public health target.”
So bedtimes aren’t just for children.
The greatest autopsy on Earth?
The LOTME staff would like to apologize in advance. The following item contains historical facts.
P.T. Barnum is a rather controversial figure in American history. The greatest show on Earth was certainly popular in its day. However, Barnum got his start in 1835 by leasing a slave named Joyce Heth, an elderly Black woman who told vivid stories of caring for a young George Washington. He toured her around the country, advertising her as a 160-year-old woman who served as George Washington’s nanny. When Ms. Heth died the next year, Barnum sold tickets to the autopsy, charging the equivalent of $30 in today’s money.
When a doctor announced that Ms. Heth was actually 75-80 when she died, it caused great controversy in the press and ruined Barnum’s career. Wait, no, that’s not right. The opposite, actually. He weathered the storm, built his famous circus, and never again committed a hoax.
It’s difficult to quantify how wrong publicly dissecting a person and charging people to see said dissection is, but that was almost 200 years ago. At the very least, we can say that such terrible behavior is firmly in the distant past.
Oh wait.
David Saunders, a 98-year-old veteran of World War II and the Korean War, donated his body to science. His body, however, was purchased by DeathScience.org from a medical lab – with the buyer supposedly misleading the medical lab about its intentions, which was for use at the traveling Oddities and Curiosities Expo. Tickets went for up to $500 each to witness the public autopsy of Mr. Saunders’ body, which took place at a Marriott in Portland, Ore. It promised to be an exciting, all-day event from 9 a.m. to 4 p.m., with a break for lunch, of course. You can’t have an autopsy without a catered lunch.
Another public autopsy event was scheduled in Seattle but canceled after news of the first event broke. Oh, and for that extra little kick, Mr. Saunders died from COVID-19, meaning that all those paying customers were exposed.
P.T. Barnum is probably rolling over in his grave right now. His autopsy tickets were a bargain.
Go ahead, have that soda before math
We should all know by now that sugary drinks are bad, even artificially sweetened ones. It might not always stop us from drinking them, but we know the deal. But what if sugary drinks like soda could be helpful for girls in school?
You read that right. We said girls. A soda before class might have boys bouncing off the walls, but not girls. A recent study showed that not only was girls’ behavior unaffected by having a sugary drink, their math skills even improved.
Researchers analyzed the behavior of 4- to 6-year-old children before and after having a sugary drink. The sugar rush was actually calming for girls and helped them perform better with numerical skills, but the opposite was true for boys. “Our study is the first to provide large-scale experimental evidence on the impact of sugary drinks on preschool children. The results clearly indicate a causal impact of sugary drinks on children’s behavior and test scores,” Fritz Schiltz, PhD, said in a written statement.
This probably isn’t the green light to have as many sugary drinks as you want, but it might be interesting to see how your work is affected after a soda.
Chicken nuggets and the meat paradox
Two young children are fighting over the last chicken nugget when an adult comes in to see what’s going on.
Liam: Vegetable!
Olivia: Meat!
Liam: Chicken nuggets are vegetables!
Olivia: No, dorkface! They’re meat.
Caregiver: Good news, kids. You’re both right.
Olivia: How can we both be right?
At this point, a woman enters the room. She’s wearing a white lab coat, so she must be a scientist.
Dr. Scientist: You can’t both be right, Olivia. You are being fed a serving of the meat paradox. That’s why Liam here doesn’t know that chicken nuggets are made of chicken, which is a form of meat. Sadly, he’s not the only one.
In a recent study, scientists from Furman University in Greenville, S.C., found that 38% of 176 children aged 4-7 years thought that chicken nuggets were vegetables and more than 46% identified French fries as animal based.
Olivia: Did our caregiver lie to us, Dr. Scientist?
Dr. Scientist: Yes, Olivia. The researchers I mentioned explained that “many people experience unease while eating meat. Omnivores eat foods that entail animal suffering and death while at the same time endorsing the compassionate treatment of animals.” That’s the meat paradox.
Liam: What else did they say, Dr. Scientist?
Dr. Scientist: Over 70% of those children said that cows and pigs were not edible and 5% thought that cats and horses were. The investigators wrote “that children and youth should be viewed as agents of environmental change” in the future, but suggested that parents need to bring honesty to the table.
Caregiver: How did you get in here anyway? And how do you know their names?
Dr. Scientist: I’ve been rooting through your garbage for years. All in the name of science, of course.
Bedtimes aren’t just for children
There are multiple ways to prevent heart disease, but what if it could be as easy as switching your bedtime? A recent study in European Heart Journal–Digital Health suggests that there’s a sweet spot when it comes to sleep timing.
Through smartwatch-like devices, researchers measured the sleep-onset and wake-up times for 7 days in 88,026 participants aged 43-79 years. After 5.7 years of follow-up to see if anyone had a heart attack, stroke, or any other cardiovascular event, 3.6% developed some kind of cardiovascular disease.
Those who went to bed between 10 p.m. and 11 p.m. had a lower risk of developing heart disease. The risk was 25% higher for subjects who went to bed at midnight or later, 24% higher for bedtimes before 10 p.m., and 12% higher for bedtimes between 11 p.m. and midnight.
So, why can you go to bed before “The Tonight Show” and lower your cardiovascular risk but not before the nightly news? Well, it has something to do with your body’s natural clock.
“The optimum time to go to sleep is at a specific point in the body’s 24-hour cycle and deviations may be detrimental to health. The riskiest time was after midnight, potentially because it may reduce the likelihood of seeing morning light, which resets the body clock,” said study author Dr. David Plans of the University of Exeter, England.
Although a sleep schedule is preferred, it isn’t realistic all the time for those in certain occupations who might have to resort to other methods to keep their circadian clocks ticking optimally for their health. But if all it takes is prescribing a sleep time to reduce heart disease on a massive scale it would make a great “low-cost public health target.”
So bedtimes aren’t just for children.
Pfizer seeks EUA expansion for COVID-19 booster
Pfizer and its European partner BioNTech on Nov. 9 asked the U.S. government to expand emergency use authorization (EUA) to allow everybody over 18 to receive their COVID-19 booster shots.
If the request is approved, He announced the goal last August but backed off after some scientists said younger people may not need boosters, especially with large parts of the world unvaccinated.
Pfizer is submitting a study of booster effects on 10,000 people to make its case, according to The Associated Press.
This would be Pfizer’s second attempt. In September, a Food and Drug Administration advisory panel turned down Pfizer’s idea of booster shots for everybody over 18.
However, the committee recommended Pfizer booster shots for people 65 and over, essential workers, and people with underlying health conditions.
The FDA and the Centers for Disease Control and Prevention authorized the Pfizer booster for those other groups and later authorization was granted for the same groups with Moderna and Johnson & Johnson boosters. People who got the two-shot Pfizer or Moderna vaccines should get a booster 6 months after the second dose and people who got the one-dose J&J vaccine should get a booster 2 months later.
The pro-booster argument has strengthened because new data have come in from Israel that confirm boosters provide protection as vaccine effectiveness wanes over time, The Washington Post reported. Also, health officials are worried about a post-holiday surge and because COVID-19 case counts and deaths are not dropping in every part of the country, though they are declining overall, according to the The Post report.
The regulatory path for a booster-for-all application is unclear. The Post, citing two unnamed officials, said the FDA probably won’t send the Pfizer application to the FDA advisory committee this time because the committee has already had extensive discussions about boosters. If the FDA gives the green light, CDC Director Rochelle Walensky, MD, would have to make updated recommendations on boosters, The Post article noted.
A version of this article first appeared on WebMD.com.
Pfizer and its European partner BioNTech on Nov. 9 asked the U.S. government to expand emergency use authorization (EUA) to allow everybody over 18 to receive their COVID-19 booster shots.
If the request is approved, He announced the goal last August but backed off after some scientists said younger people may not need boosters, especially with large parts of the world unvaccinated.
Pfizer is submitting a study of booster effects on 10,000 people to make its case, according to The Associated Press.
This would be Pfizer’s second attempt. In September, a Food and Drug Administration advisory panel turned down Pfizer’s idea of booster shots for everybody over 18.
However, the committee recommended Pfizer booster shots for people 65 and over, essential workers, and people with underlying health conditions.
The FDA and the Centers for Disease Control and Prevention authorized the Pfizer booster for those other groups and later authorization was granted for the same groups with Moderna and Johnson & Johnson boosters. People who got the two-shot Pfizer or Moderna vaccines should get a booster 6 months after the second dose and people who got the one-dose J&J vaccine should get a booster 2 months later.
The pro-booster argument has strengthened because new data have come in from Israel that confirm boosters provide protection as vaccine effectiveness wanes over time, The Washington Post reported. Also, health officials are worried about a post-holiday surge and because COVID-19 case counts and deaths are not dropping in every part of the country, though they are declining overall, according to the The Post report.
The regulatory path for a booster-for-all application is unclear. The Post, citing two unnamed officials, said the FDA probably won’t send the Pfizer application to the FDA advisory committee this time because the committee has already had extensive discussions about boosters. If the FDA gives the green light, CDC Director Rochelle Walensky, MD, would have to make updated recommendations on boosters, The Post article noted.
A version of this article first appeared on WebMD.com.
Pfizer and its European partner BioNTech on Nov. 9 asked the U.S. government to expand emergency use authorization (EUA) to allow everybody over 18 to receive their COVID-19 booster shots.
If the request is approved, He announced the goal last August but backed off after some scientists said younger people may not need boosters, especially with large parts of the world unvaccinated.
Pfizer is submitting a study of booster effects on 10,000 people to make its case, according to The Associated Press.
This would be Pfizer’s second attempt. In September, a Food and Drug Administration advisory panel turned down Pfizer’s idea of booster shots for everybody over 18.
However, the committee recommended Pfizer booster shots for people 65 and over, essential workers, and people with underlying health conditions.
The FDA and the Centers for Disease Control and Prevention authorized the Pfizer booster for those other groups and later authorization was granted for the same groups with Moderna and Johnson & Johnson boosters. People who got the two-shot Pfizer or Moderna vaccines should get a booster 6 months after the second dose and people who got the one-dose J&J vaccine should get a booster 2 months later.
The pro-booster argument has strengthened because new data have come in from Israel that confirm boosters provide protection as vaccine effectiveness wanes over time, The Washington Post reported. Also, health officials are worried about a post-holiday surge and because COVID-19 case counts and deaths are not dropping in every part of the country, though they are declining overall, according to the The Post report.
The regulatory path for a booster-for-all application is unclear. The Post, citing two unnamed officials, said the FDA probably won’t send the Pfizer application to the FDA advisory committee this time because the committee has already had extensive discussions about boosters. If the FDA gives the green light, CDC Director Rochelle Walensky, MD, would have to make updated recommendations on boosters, The Post article noted.
A version of this article first appeared on WebMD.com.