User login
At 52 weeks, complete hair regrowth rates still climbing on deuruxolitinib
BERLIN – at the annual congress of the European Academy of Dermatology and Venereology.
With response curves still climbing at follow-up to date, the results are “truly, truly remarkable,” said Brett King, MD, PhD, associate professor of dermatology, Yale University, New Haven, Conn.
Deuruxolitinib is a JAK inhibitor that has specificity for the 1 and 2 subtypes. At 24 weeks in the phase 3 THRIVE-AA1 and THRIVE-AA2 trials, presented at the American Academy of Dermatology annual meeting earlier this year, about 40% of those on the 12-mg twice-daily dose and 32% of those on the 8-mg twice-daily dose achieved a Severity of Alopecia Tool (SALT) score of ≤ 20%, signifying 80% or greater hair regrowth at 24 weeks. The placebo response was 0%.

By 52 weeks, the proportion had climbed to 62% among those on continuous deuruxolitinib whether maintained on the 8-mg or 12-mg twice daily doses. Among patients on placebo, 58.4% reached this endpoint after being switched at 24 weeks to the 12-mg twice daily dose. Of the patients on placebo switched to 8 mg twice daily, the 52-week response was 45.2%, according to Dr. King.
There were 741 patients available at 52 weeks for this on-going analysis. The mean SALT scores at entry exceeded 80%, meaning complete or near complete hair loss. The substantial proportion of patients who met the primary endpoint of SALT ≤ 20 at the end of the blinded period was encouraging, but Dr. King said that the 52-week results are important, not only showing the response was sustained, but that greater regrowth occurs over time.
“Alopecia takes time to treat,” said Dr. King, summarizing the lesson from these data. Moreover, he added that the long-term data are likely to under represent the absolute benefit even if no further growth is achieved with even longer follow-up. One reason is that missing long-term data were accounted for with a last-observation-carried-forward approach.
In other words, “this is the floor when considering response at 52 weeks,” Dr. King said. “In the real world, where adjunctive measures such as intralesional Kenalog [triamcinolone acetonide] or topical treatments are added, we are likely to do even better,” he added.
Adverse events remained low
Treatment-emergent adverse events remained low with “nothing particularly surprising,” Dr. King said. The rate of serious adverse events over 52 weeks was less than 2% on either dose of deuruxolitinib. The proportion of patients who discontinued treatment because of an adverse event was 0.7% in the 8-mg twice-daily arm and 1.1% in the 12-mg twice-daily arm.
Most approved oral JAK inhibitors carry a boxed warning based on a trial conducted with the relatively nonspecific tofacitinib. The trial enrolled older patients with rheumatoid arthritis at risk for thrombotic events, raising questions about its relevance to selective JAK inhibitors employed for other indications. There was only one thrombosis observed in the 52-week alopecia areata follow-up in a patient on deuruxolitinib. Dr. King noted that this patient, who was obese and was on the higher of the two doses, had multiple comorbidities, including systemic lupus erythematosus.
There were no major adverse cardiac events reported in long-term follow-up or cases of tuberculosis. The rate of opportunistic infections was 0.1% in the 8-mg twice-daily arm and 0.2% in the 12-mg twice-daily arm. Serious infections were observed in 0.6% and 0.4% of these two arms, respectively. There were four malignancies (0.5%) in each of the two study arms.
Of the side effects likely to be related to deuruxolitinib, acne was observed in about 10% of patients on either dose. The mechanism is unclear, but Dr. King reported this has been commonly observed with other JAK inhibitors.
Asked his opinion about the optimal starting dose of deuruxolitinib, Dr. King said, “in my mind, the efficacy of 8 mg is so impressive that I would not struggle at all in starting there,” noting that the higher dose could be considered with a slow or inadequate response.
Two JAK inhibitors are already approved
If approved for alopecia areata, deuruxolitinib will be the third JAK inhibitor available for this indication, following the recent approvals of baricitinib and ritlecitinib.
Calling JAK inhibitors “a major advance in the treatment of alopecia areata, particularly for those patients with severe, refractory disease,” Lynne Goldberg, MD, professor of dermatology at Boston University, and director of the hair clinic, Boston Medical Center, said that the proportion of patients with SALT scores ≤ 20 at 52-weeks is “huge.”
She is generally comfortable with the safety of the JAK inhibitors for alopecia areata.
“I believe that, in general, these medications are well tolerated in the alopecia areata population, particularly in otherwise healthy, young patients,” she said, indicating the benefit-to-risk ratio is particularly acceptable when disease is severe.
“This disease has tremendous emotional and functional implications, and many patients with severe or recurrent disease are willing to chance the side effects to live with a full head of hair,” she said. She added that well-informed patients can “make their own, individual assessment.”
Dr. King has financial relationships with approximately 20 pharmaceutical companies, including Concert Pharmaceuticals, which makes deuruxolitinib and provided funding for this study. Dr. Goldberg reports no financial conflicts relevant to this topic.
BERLIN – at the annual congress of the European Academy of Dermatology and Venereology.
With response curves still climbing at follow-up to date, the results are “truly, truly remarkable,” said Brett King, MD, PhD, associate professor of dermatology, Yale University, New Haven, Conn.
Deuruxolitinib is a JAK inhibitor that has specificity for the 1 and 2 subtypes. At 24 weeks in the phase 3 THRIVE-AA1 and THRIVE-AA2 trials, presented at the American Academy of Dermatology annual meeting earlier this year, about 40% of those on the 12-mg twice-daily dose and 32% of those on the 8-mg twice-daily dose achieved a Severity of Alopecia Tool (SALT) score of ≤ 20%, signifying 80% or greater hair regrowth at 24 weeks. The placebo response was 0%.
By 52 weeks, the proportion had climbed to 62% among those on continuous deuruxolitinib whether maintained on the 8-mg or 12-mg twice daily doses. Among patients on placebo, 58.4% reached this endpoint after being switched at 24 weeks to the 12-mg twice daily dose. Of the patients on placebo switched to 8 mg twice daily, the 52-week response was 45.2%, according to Dr. King.
There were 741 patients available at 52 weeks for this on-going analysis. The mean SALT scores at entry exceeded 80%, meaning complete or near complete hair loss. The substantial proportion of patients who met the primary endpoint of SALT ≤ 20 at the end of the blinded period was encouraging, but Dr. King said that the 52-week results are important, not only showing the response was sustained, but that greater regrowth occurs over time.
“Alopecia takes time to treat,” said Dr. King, summarizing the lesson from these data. Moreover, he added that the long-term data are likely to under represent the absolute benefit even if no further growth is achieved with even longer follow-up. One reason is that missing long-term data were accounted for with a last-observation-carried-forward approach.
In other words, “this is the floor when considering response at 52 weeks,” Dr. King said. “In the real world, where adjunctive measures such as intralesional Kenalog [triamcinolone acetonide] or topical treatments are added, we are likely to do even better,” he added.
Adverse events remained low
Treatment-emergent adverse events remained low with “nothing particularly surprising,” Dr. King said. The rate of serious adverse events over 52 weeks was less than 2% on either dose of deuruxolitinib. The proportion of patients who discontinued treatment because of an adverse event was 0.7% in the 8-mg twice-daily arm and 1.1% in the 12-mg twice-daily arm.
Most approved oral JAK inhibitors carry a boxed warning based on a trial conducted with the relatively nonspecific tofacitinib. The trial enrolled older patients with rheumatoid arthritis at risk for thrombotic events, raising questions about its relevance to selective JAK inhibitors employed for other indications. There was only one thrombosis observed in the 52-week alopecia areata follow-up in a patient on deuruxolitinib. Dr. King noted that this patient, who was obese and was on the higher of the two doses, had multiple comorbidities, including systemic lupus erythematosus.
There were no major adverse cardiac events reported in long-term follow-up or cases of tuberculosis. The rate of opportunistic infections was 0.1% in the 8-mg twice-daily arm and 0.2% in the 12-mg twice-daily arm. Serious infections were observed in 0.6% and 0.4% of these two arms, respectively. There were four malignancies (0.5%) in each of the two study arms.
Of the side effects likely to be related to deuruxolitinib, acne was observed in about 10% of patients on either dose. The mechanism is unclear, but Dr. King reported this has been commonly observed with other JAK inhibitors.
Asked his opinion about the optimal starting dose of deuruxolitinib, Dr. King said, “in my mind, the efficacy of 8 mg is so impressive that I would not struggle at all in starting there,” noting that the higher dose could be considered with a slow or inadequate response.
Two JAK inhibitors are already approved
If approved for alopecia areata, deuruxolitinib will be the third JAK inhibitor available for this indication, following the recent approvals of baricitinib and ritlecitinib.
Calling JAK inhibitors “a major advance in the treatment of alopecia areata, particularly for those patients with severe, refractory disease,” Lynne Goldberg, MD, professor of dermatology at Boston University, and director of the hair clinic, Boston Medical Center, said that the proportion of patients with SALT scores ≤ 20 at 52-weeks is “huge.”
She is generally comfortable with the safety of the JAK inhibitors for alopecia areata.
“I believe that, in general, these medications are well tolerated in the alopecia areata population, particularly in otherwise healthy, young patients,” she said, indicating the benefit-to-risk ratio is particularly acceptable when disease is severe.
“This disease has tremendous emotional and functional implications, and many patients with severe or recurrent disease are willing to chance the side effects to live with a full head of hair,” she said. She added that well-informed patients can “make their own, individual assessment.”
Dr. King has financial relationships with approximately 20 pharmaceutical companies, including Concert Pharmaceuticals, which makes deuruxolitinib and provided funding for this study. Dr. Goldberg reports no financial conflicts relevant to this topic.
BERLIN – at the annual congress of the European Academy of Dermatology and Venereology.
With response curves still climbing at follow-up to date, the results are “truly, truly remarkable,” said Brett King, MD, PhD, associate professor of dermatology, Yale University, New Haven, Conn.
Deuruxolitinib is a JAK inhibitor that has specificity for the 1 and 2 subtypes. At 24 weeks in the phase 3 THRIVE-AA1 and THRIVE-AA2 trials, presented at the American Academy of Dermatology annual meeting earlier this year, about 40% of those on the 12-mg twice-daily dose and 32% of those on the 8-mg twice-daily dose achieved a Severity of Alopecia Tool (SALT) score of ≤ 20%, signifying 80% or greater hair regrowth at 24 weeks. The placebo response was 0%.
By 52 weeks, the proportion had climbed to 62% among those on continuous deuruxolitinib whether maintained on the 8-mg or 12-mg twice daily doses. Among patients on placebo, 58.4% reached this endpoint after being switched at 24 weeks to the 12-mg twice daily dose. Of the patients on placebo switched to 8 mg twice daily, the 52-week response was 45.2%, according to Dr. King.
There were 741 patients available at 52 weeks for this on-going analysis. The mean SALT scores at entry exceeded 80%, meaning complete or near complete hair loss. The substantial proportion of patients who met the primary endpoint of SALT ≤ 20 at the end of the blinded period was encouraging, but Dr. King said that the 52-week results are important, not only showing the response was sustained, but that greater regrowth occurs over time.
“Alopecia takes time to treat,” said Dr. King, summarizing the lesson from these data. Moreover, he added that the long-term data are likely to under represent the absolute benefit even if no further growth is achieved with even longer follow-up. One reason is that missing long-term data were accounted for with a last-observation-carried-forward approach.
In other words, “this is the floor when considering response at 52 weeks,” Dr. King said. “In the real world, where adjunctive measures such as intralesional Kenalog [triamcinolone acetonide] or topical treatments are added, we are likely to do even better,” he added.
Adverse events remained low
Treatment-emergent adverse events remained low with “nothing particularly surprising,” Dr. King said. The rate of serious adverse events over 52 weeks was less than 2% on either dose of deuruxolitinib. The proportion of patients who discontinued treatment because of an adverse event was 0.7% in the 8-mg twice-daily arm and 1.1% in the 12-mg twice-daily arm.
Most approved oral JAK inhibitors carry a boxed warning based on a trial conducted with the relatively nonspecific tofacitinib. The trial enrolled older patients with rheumatoid arthritis at risk for thrombotic events, raising questions about its relevance to selective JAK inhibitors employed for other indications. There was only one thrombosis observed in the 52-week alopecia areata follow-up in a patient on deuruxolitinib. Dr. King noted that this patient, who was obese and was on the higher of the two doses, had multiple comorbidities, including systemic lupus erythematosus.
There were no major adverse cardiac events reported in long-term follow-up or cases of tuberculosis. The rate of opportunistic infections was 0.1% in the 8-mg twice-daily arm and 0.2% in the 12-mg twice-daily arm. Serious infections were observed in 0.6% and 0.4% of these two arms, respectively. There were four malignancies (0.5%) in each of the two study arms.
Of the side effects likely to be related to deuruxolitinib, acne was observed in about 10% of patients on either dose. The mechanism is unclear, but Dr. King reported this has been commonly observed with other JAK inhibitors.
Asked his opinion about the optimal starting dose of deuruxolitinib, Dr. King said, “in my mind, the efficacy of 8 mg is so impressive that I would not struggle at all in starting there,” noting that the higher dose could be considered with a slow or inadequate response.
Two JAK inhibitors are already approved
If approved for alopecia areata, deuruxolitinib will be the third JAK inhibitor available for this indication, following the recent approvals of baricitinib and ritlecitinib.
Calling JAK inhibitors “a major advance in the treatment of alopecia areata, particularly for those patients with severe, refractory disease,” Lynne Goldberg, MD, professor of dermatology at Boston University, and director of the hair clinic, Boston Medical Center, said that the proportion of patients with SALT scores ≤ 20 at 52-weeks is “huge.”
She is generally comfortable with the safety of the JAK inhibitors for alopecia areata.
“I believe that, in general, these medications are well tolerated in the alopecia areata population, particularly in otherwise healthy, young patients,” she said, indicating the benefit-to-risk ratio is particularly acceptable when disease is severe.
“This disease has tremendous emotional and functional implications, and many patients with severe or recurrent disease are willing to chance the side effects to live with a full head of hair,” she said. She added that well-informed patients can “make their own, individual assessment.”
Dr. King has financial relationships with approximately 20 pharmaceutical companies, including Concert Pharmaceuticals, which makes deuruxolitinib and provided funding for this study. Dr. Goldberg reports no financial conflicts relevant to this topic.
At THE EADV CONGRESS
Tapinarof effective for AD in patients as young as 2 years
BERLIN – of age, according to results of two pivotal trials presented at the at the annual congress of the European Academy of Dermatology and Venereology.
If approved for AD, one advantage of tapinarof cream relative to topical corticosteroids is potential use “without restrictions on duration, extent, or site of application,” reported Jonathan I. Silverberg, MD, PhD, MPH, director of clinical research, George Washington University, Washington.
Tapinarof cream, 1%, an aryl hydrocarbon receptor agonist, was approved in 2022 for treating plaque psoriasis in adults.

In the two phase 3 trials, ADORING 1 and ADORING 2, which were presented together at the meeting, the primary endpoint was Validated Investigator Global Assessment (vIGA) for AD of 0 (clear) or 1 (almost clear) at 8 weeks. For this endpoint and all secondary endpoints, the relative advantage of the active cream over the vehicle alone was about the same in both studies.
For example, the vIGA clear or almost clear response was met by 45.4% and 46.4% of those in the experimental arm of ADORING 1 and 2, respectively, but only 13.9% and 18.0% in the control arms (P < .0001 for both).
For the secondary endpoint of Eczema Area and Severity Index (EASI75), signifying 75% clearance of skin lesions, the response rates were 55.8% and 59.1% in the two trials, but only 22.9% and 24.1% in the respective control arms (P < .0001 for both).
The two identically designed trials randomized patients with moderate to severe AD in a 2:1 ratio to tapinarof cream or vehicle alone. There were 407 patients ages 2-81 years in ADORING I and 406 in ADORING 2. Patients were instructed to apply the active cream or vehicle once per day.
The safety data for tapinarof in these studies was generally consistent with the experience with this agent in plaque psoriasis. According to Dr. Silverberg, there was a modest increase in reports of headache early in this study, but these were transient. Follicular events were also more common on tapinarof than on its vehicle, but Dr. Silverberg said that the rate of discontinuations for adverse events, although low in both arms, was numerically lower in the active treatment arm in both trials.
“There were reports of contact dermatitis in the psoriasis studies, but we have not seen this in the AD trials,” Dr. Silverberg said.
Itch control evaluated
In a separate presentation of ADORING 1 and 2 results, Eric Simpson, MD, professor of dermatology, Oregon Health & Science University, Portland, provided detailed information about itch control, which was evaluated with the Peak Pruritus–Numerical Rating Scale (PP-NRS).

“The PP-NRS considers a person’s worst itch over the past 24 hours based on an 11-point scale,” explained Dr. Simpson, who said that patients scored itch daily with comparisons made at weeks 1, 2, 4, and 8.
Over time, pruritus scores fell in both groups, but reductions were far steeper among those in the active treatment arms.
“In ADORING 1, there were greater reductions in itch as early as day 1,” Dr. Simpson reported. Although the differences in itch were not detected until day 2 in ADORING 2, the differences were already significant and clinically meaningful in both studies by the end of the first week.
By week 8, the mean reductions in PP-NRS scores were 2.6 and 2.4 in the vehicle arms of ADORING 1 and 2, respectively. In the treatment arm, the reduction was 4.1 points in both arms (P < .0001 for both studies).
Forty-eight–week follow-up planned
More than 90% of patients in both studies have rolled over into the open-label extension ADORING 3 trial, with a planned follow-up of 48 weeks, according to Dr. Silverberg, who said that those in the placebo arm have been crossed over to tapinarof.
The response and the safety appear to be similar in adults and children, although Dr. Silverberg said that further analyses of outcomes by age are planned. He noted that there is also an ongoing study of tapinarof in children with plaque psoriasis.
In AD in particular, Dr. Silverberg said there is “an unmet need” for a topical nonsteroidal anti-inflammatory. While topical corticosteroids are a mainstay of AD therapy in children as well as adults, he noted the limitations of these drugs, including that they can only be applied for limited periods.
Tapinarof binds to the aryl hydrocarbon receptor (AhR), which regulates immune function in the skin and is expressed in many skin cell types. By inhibiting AhR, tapinarof blocks cytokine activation and has an antioxidant effect.
Adelaide A. Hebert, MD, professor and director of pediatric dermatology, McGovern Medical School at UTHealth, Houston, has participated in clinical studies of tapinarof for AD, and said she has been impressed with its efficacy and tolerability in children as well as adults. In the case of children, parents, as well as patients, “valued the rapid onset of disease control, the once-daily application regimen, and the itch control,” she said in an interview after the meeting.
If approved, Dr. Hebert said, “this novel steroid-free medication has the potential to change the management arena for pediatric and adult patients with moderate to severe atopic dermatitis.”
The recent introduction of new systemic therapies for AD, such as JAK inhibitors, has increased options for AD control, but “we still need effective and safe topical therapies, especially in children and young adults,” said Sonja Ständer, MD, head of the Interdisciplinary Center for Chronic Pruritus, University of Münster (Germany). Author of a comprehensive review article on AD in the New England Journal of Medicine 2 years ago, Dr. Ständer said results from the phase 3 topical tapinarof trials, as well as the phase 3 topical ruxolitinib trials, which were also presented as late breakers at the 2023 EADV meeting, provide “hope that an alternative to topical steroids will soon be available.”
Based on their safety and rapid control of itch in children with AD, “these will complement our current portfolio of topical therapies very well and have the potential to replace topical steroids early in therapy or to replace them altogether,” she told this news organization.
Dermavant Sciences, manufacturer of tapinarof, anticipates filing for Food and Drug Administration approval for AD in the first quarter of 2024, according to a company statement.
Dr. Silverberg and Dr. Simpson reported financial relationships with multiple pharmaceutical companies, including Dermavant, which provided funding for the ADORING trials. Dr. Hebert has financial relationship with more than 15 pharmaceutical companies, including Dermavent and other companies that have or are developing therapies for AD. Dr. Ständer reported financial relationships with Beiersdorf, Eli Lilly, Galderma, Kiniksa, Pfizer, and Sanofi.
BERLIN – of age, according to results of two pivotal trials presented at the at the annual congress of the European Academy of Dermatology and Venereology.
If approved for AD, one advantage of tapinarof cream relative to topical corticosteroids is potential use “without restrictions on duration, extent, or site of application,” reported Jonathan I. Silverberg, MD, PhD, MPH, director of clinical research, George Washington University, Washington.
Tapinarof cream, 1%, an aryl hydrocarbon receptor agonist, was approved in 2022 for treating plaque psoriasis in adults.
In the two phase 3 trials, ADORING 1 and ADORING 2, which were presented together at the meeting, the primary endpoint was Validated Investigator Global Assessment (vIGA) for AD of 0 (clear) or 1 (almost clear) at 8 weeks. For this endpoint and all secondary endpoints, the relative advantage of the active cream over the vehicle alone was about the same in both studies.
For example, the vIGA clear or almost clear response was met by 45.4% and 46.4% of those in the experimental arm of ADORING 1 and 2, respectively, but only 13.9% and 18.0% in the control arms (P < .0001 for both).
For the secondary endpoint of Eczema Area and Severity Index (EASI75), signifying 75% clearance of skin lesions, the response rates were 55.8% and 59.1% in the two trials, but only 22.9% and 24.1% in the respective control arms (P < .0001 for both).
The two identically designed trials randomized patients with moderate to severe AD in a 2:1 ratio to tapinarof cream or vehicle alone. There were 407 patients ages 2-81 years in ADORING I and 406 in ADORING 2. Patients were instructed to apply the active cream or vehicle once per day.
The safety data for tapinarof in these studies was generally consistent with the experience with this agent in plaque psoriasis. According to Dr. Silverberg, there was a modest increase in reports of headache early in this study, but these were transient. Follicular events were also more common on tapinarof than on its vehicle, but Dr. Silverberg said that the rate of discontinuations for adverse events, although low in both arms, was numerically lower in the active treatment arm in both trials.
“There were reports of contact dermatitis in the psoriasis studies, but we have not seen this in the AD trials,” Dr. Silverberg said.
Itch control evaluated
In a separate presentation of ADORING 1 and 2 results, Eric Simpson, MD, professor of dermatology, Oregon Health & Science University, Portland, provided detailed information about itch control, which was evaluated with the Peak Pruritus–Numerical Rating Scale (PP-NRS).
“The PP-NRS considers a person’s worst itch over the past 24 hours based on an 11-point scale,” explained Dr. Simpson, who said that patients scored itch daily with comparisons made at weeks 1, 2, 4, and 8.
Over time, pruritus scores fell in both groups, but reductions were far steeper among those in the active treatment arms.
“In ADORING 1, there were greater reductions in itch as early as day 1,” Dr. Simpson reported. Although the differences in itch were not detected until day 2 in ADORING 2, the differences were already significant and clinically meaningful in both studies by the end of the first week.
By week 8, the mean reductions in PP-NRS scores were 2.6 and 2.4 in the vehicle arms of ADORING 1 and 2, respectively. In the treatment arm, the reduction was 4.1 points in both arms (P < .0001 for both studies).
Forty-eight–week follow-up planned
More than 90% of patients in both studies have rolled over into the open-label extension ADORING 3 trial, with a planned follow-up of 48 weeks, according to Dr. Silverberg, who said that those in the placebo arm have been crossed over to tapinarof.
The response and the safety appear to be similar in adults and children, although Dr. Silverberg said that further analyses of outcomes by age are planned. He noted that there is also an ongoing study of tapinarof in children with plaque psoriasis.
In AD in particular, Dr. Silverberg said there is “an unmet need” for a topical nonsteroidal anti-inflammatory. While topical corticosteroids are a mainstay of AD therapy in children as well as adults, he noted the limitations of these drugs, including that they can only be applied for limited periods.
Tapinarof binds to the aryl hydrocarbon receptor (AhR), which regulates immune function in the skin and is expressed in many skin cell types. By inhibiting AhR, tapinarof blocks cytokine activation and has an antioxidant effect.
Adelaide A. Hebert, MD, professor and director of pediatric dermatology, McGovern Medical School at UTHealth, Houston, has participated in clinical studies of tapinarof for AD, and said she has been impressed with its efficacy and tolerability in children as well as adults. In the case of children, parents, as well as patients, “valued the rapid onset of disease control, the once-daily application regimen, and the itch control,” she said in an interview after the meeting.
If approved, Dr. Hebert said, “this novel steroid-free medication has the potential to change the management arena for pediatric and adult patients with moderate to severe atopic dermatitis.”
The recent introduction of new systemic therapies for AD, such as JAK inhibitors, has increased options for AD control, but “we still need effective and safe topical therapies, especially in children and young adults,” said Sonja Ständer, MD, head of the Interdisciplinary Center for Chronic Pruritus, University of Münster (Germany). Author of a comprehensive review article on AD in the New England Journal of Medicine 2 years ago, Dr. Ständer said results from the phase 3 topical tapinarof trials, as well as the phase 3 topical ruxolitinib trials, which were also presented as late breakers at the 2023 EADV meeting, provide “hope that an alternative to topical steroids will soon be available.”
Based on their safety and rapid control of itch in children with AD, “these will complement our current portfolio of topical therapies very well and have the potential to replace topical steroids early in therapy or to replace them altogether,” she told this news organization.
Dermavant Sciences, manufacturer of tapinarof, anticipates filing for Food and Drug Administration approval for AD in the first quarter of 2024, according to a company statement.
Dr. Silverberg and Dr. Simpson reported financial relationships with multiple pharmaceutical companies, including Dermavant, which provided funding for the ADORING trials. Dr. Hebert has financial relationship with more than 15 pharmaceutical companies, including Dermavent and other companies that have or are developing therapies for AD. Dr. Ständer reported financial relationships with Beiersdorf, Eli Lilly, Galderma, Kiniksa, Pfizer, and Sanofi.
BERLIN – of age, according to results of two pivotal trials presented at the at the annual congress of the European Academy of Dermatology and Venereology.
If approved for AD, one advantage of tapinarof cream relative to topical corticosteroids is potential use “without restrictions on duration, extent, or site of application,” reported Jonathan I. Silverberg, MD, PhD, MPH, director of clinical research, George Washington University, Washington.
Tapinarof cream, 1%, an aryl hydrocarbon receptor agonist, was approved in 2022 for treating plaque psoriasis in adults.
In the two phase 3 trials, ADORING 1 and ADORING 2, which were presented together at the meeting, the primary endpoint was Validated Investigator Global Assessment (vIGA) for AD of 0 (clear) or 1 (almost clear) at 8 weeks. For this endpoint and all secondary endpoints, the relative advantage of the active cream over the vehicle alone was about the same in both studies.
For example, the vIGA clear or almost clear response was met by 45.4% and 46.4% of those in the experimental arm of ADORING 1 and 2, respectively, but only 13.9% and 18.0% in the control arms (P < .0001 for both).
For the secondary endpoint of Eczema Area and Severity Index (EASI75), signifying 75% clearance of skin lesions, the response rates were 55.8% and 59.1% in the two trials, but only 22.9% and 24.1% in the respective control arms (P < .0001 for both).
The two identically designed trials randomized patients with moderate to severe AD in a 2:1 ratio to tapinarof cream or vehicle alone. There were 407 patients ages 2-81 years in ADORING I and 406 in ADORING 2. Patients were instructed to apply the active cream or vehicle once per day.
The safety data for tapinarof in these studies was generally consistent with the experience with this agent in plaque psoriasis. According to Dr. Silverberg, there was a modest increase in reports of headache early in this study, but these were transient. Follicular events were also more common on tapinarof than on its vehicle, but Dr. Silverberg said that the rate of discontinuations for adverse events, although low in both arms, was numerically lower in the active treatment arm in both trials.
“There were reports of contact dermatitis in the psoriasis studies, but we have not seen this in the AD trials,” Dr. Silverberg said.
Itch control evaluated
In a separate presentation of ADORING 1 and 2 results, Eric Simpson, MD, professor of dermatology, Oregon Health & Science University, Portland, provided detailed information about itch control, which was evaluated with the Peak Pruritus–Numerical Rating Scale (PP-NRS).
“The PP-NRS considers a person’s worst itch over the past 24 hours based on an 11-point scale,” explained Dr. Simpson, who said that patients scored itch daily with comparisons made at weeks 1, 2, 4, and 8.
Over time, pruritus scores fell in both groups, but reductions were far steeper among those in the active treatment arms.
“In ADORING 1, there were greater reductions in itch as early as day 1,” Dr. Simpson reported. Although the differences in itch were not detected until day 2 in ADORING 2, the differences were already significant and clinically meaningful in both studies by the end of the first week.
By week 8, the mean reductions in PP-NRS scores were 2.6 and 2.4 in the vehicle arms of ADORING 1 and 2, respectively. In the treatment arm, the reduction was 4.1 points in both arms (P < .0001 for both studies).
Forty-eight–week follow-up planned
More than 90% of patients in both studies have rolled over into the open-label extension ADORING 3 trial, with a planned follow-up of 48 weeks, according to Dr. Silverberg, who said that those in the placebo arm have been crossed over to tapinarof.
The response and the safety appear to be similar in adults and children, although Dr. Silverberg said that further analyses of outcomes by age are planned. He noted that there is also an ongoing study of tapinarof in children with plaque psoriasis.
In AD in particular, Dr. Silverberg said there is “an unmet need” for a topical nonsteroidal anti-inflammatory. While topical corticosteroids are a mainstay of AD therapy in children as well as adults, he noted the limitations of these drugs, including that they can only be applied for limited periods.
Tapinarof binds to the aryl hydrocarbon receptor (AhR), which regulates immune function in the skin and is expressed in many skin cell types. By inhibiting AhR, tapinarof blocks cytokine activation and has an antioxidant effect.
Adelaide A. Hebert, MD, professor and director of pediatric dermatology, McGovern Medical School at UTHealth, Houston, has participated in clinical studies of tapinarof for AD, and said she has been impressed with its efficacy and tolerability in children as well as adults. In the case of children, parents, as well as patients, “valued the rapid onset of disease control, the once-daily application regimen, and the itch control,” she said in an interview after the meeting.
If approved, Dr. Hebert said, “this novel steroid-free medication has the potential to change the management arena for pediatric and adult patients with moderate to severe atopic dermatitis.”
The recent introduction of new systemic therapies for AD, such as JAK inhibitors, has increased options for AD control, but “we still need effective and safe topical therapies, especially in children and young adults,” said Sonja Ständer, MD, head of the Interdisciplinary Center for Chronic Pruritus, University of Münster (Germany). Author of a comprehensive review article on AD in the New England Journal of Medicine 2 years ago, Dr. Ständer said results from the phase 3 topical tapinarof trials, as well as the phase 3 topical ruxolitinib trials, which were also presented as late breakers at the 2023 EADV meeting, provide “hope that an alternative to topical steroids will soon be available.”
Based on their safety and rapid control of itch in children with AD, “these will complement our current portfolio of topical therapies very well and have the potential to replace topical steroids early in therapy or to replace them altogether,” she told this news organization.
Dermavant Sciences, manufacturer of tapinarof, anticipates filing for Food and Drug Administration approval for AD in the first quarter of 2024, according to a company statement.
Dr. Silverberg and Dr. Simpson reported financial relationships with multiple pharmaceutical companies, including Dermavant, which provided funding for the ADORING trials. Dr. Hebert has financial relationship with more than 15 pharmaceutical companies, including Dermavent and other companies that have or are developing therapies for AD. Dr. Ständer reported financial relationships with Beiersdorf, Eli Lilly, Galderma, Kiniksa, Pfizer, and Sanofi.
AT THE EADV CONGRESS
Hidradenitis suppurativa: Two anti-IL17A/F therapies yield positive results
BERLIN – In separate trials conducted in patients with hidradenitis suppurativa (HS), two biologics that inhibit the activity of interleukin-17A (IL-17A) and IL-17F were associated with highly encouraging rates of control.
One of the trials evaluated a nanobody inhibitor, sonelokimab, a molecule with a substantially smaller size than traditional monoclonal antibodies (40 kilodaltons vs. 150 kilodaltons). After 24 weeks of treatment, the most effective of the two study doses almost doubled the proportion of patients with complete resolution of draining tunnels (41.1% vs. 23.8%; P < .05) relative to placebo.
“I think the size of sonelokimab is important,” Brian Kirby, MD, a consultant dermatologist at St. Vincent’s Hospital, Dublin, said at the annual congress of the European Academy of Dermatology and Venereology. “We think the smaller size results in better penetration of inflamed tissue,” he added, noting that penetration of abscesses, fistulae, and tunnels has been recognized in the past as a potential weakness of the larger monoclonal antibodies.

The other set of anti-17-A/F set of data were generated by a pooled 48-week maintenance from the BE HEARD I and II trials with bimekizumab. The 16-week data from these two trials were presented at the annual meeting of the American Academy of Dermatology earlier this year.
IL-17A/F trials
Both the
In the sonelokimab trial, called MIRA, 234 adults with HS were randomized in a 2:2:2:1 ratio to one of the two experimental arms, placebo, or a reference arm with the tumor necrosis factor (TNF) inhibitor adalimumab. Nearly 64% had Hurley stage II HS.
The primary endpoint was a 75% or greater reduction in total abscesses and nodules with no increase in draining tunnel count (HiSCR75) from baseline. Dr. Kirby said that this is more rigorous than the HiSCR50 endpoint more commonly used in HS clinical trials. Treatments were administered every 2 weeks for the first 8 weeks of a planned follow-up of 24 weeks and then every 4 weeks thereafter.
At 16 weeks, according to the data Dr. Kirby presented, both doses of sonelokimab were more active than placebo, but Dr. Kirby reported that the lower dose performed better for most objective endpoints.
For example, the HiSCR75 was reached by 43.3% of those randomized to the 120-mg dose (P < .001 vs. placebo), 34.8% of those randomized to the 240-mg dose (P <.01), and 14.7% of those randomized to placebo.
For HiSCR50, response rates were 65.7%, 53.0%, and 27.9%, for the 120-mg, 240-mg, and placebo arms, respectively. Again, both the lower dose (P < .001) and the higher dose (P < .01) were significantly superior to placebo.
On the International Hidradenitis Suppurativa Severity Score System (IHS4), which counts nodules and abscesses, score reductions were 19.3, 14.5, and 7.9 for the lower dose, higher dose, and placebo, respectively, with a greater statistical advantage for the lower relative to the higher dose over placebo (P <.001 vs. P <.01).
However, patient-focused outcomes were not necessarily greater for the lower dose. For the patient-completed measure, the Numerical Rating Scale 50% reduction in skin pain (NRS50), the proportion of patients responding at 12 weeks was numerically greater for the 240-mg dose (41.3%) than with the 120-mg dose (32.0%), although both reached the same statistical advantage (P < .001) over the 4.3% who reached this level of response on placebo.
For the Dermatology Life Quality Index (DLQI) and the Patient Global Impression of Severity (PGI-S), improvements from baseline were similar for the lower and higher dose, although there was a modest numerical and statistical advantage for the higher dose over placebo (P < .001 vs. P <.01).
The HiSCR50 (57.6%) and HiSCR75 (36.4%) responses were both lower for those randomized to the TNF inhibitor adalimumab relative to sonelokimab, but the smaller number of patients in this arm prohibited a statistical comparison.
Although oral candidiasis was more common among patients receiving either dose of sonelokimab than placebo, these were of mild to moderate severity. Dr. Kirby said that there were no unexpected safety issues, and sonelokimab was generally well tolerated.
The results are encouraging, but Dr. Kirby acknowledged that data are now needed to confirm that resolution of tunnels and fistulae is greater with a nanobody inhibitor of IL-17A/F than other targeted therapies. Even if this is validated, he said studies are needed to prove that the small relative molecule size is the reason behind the benefits.
Forty-eight–week bimekizumab data
From the pooled BE HEARD I and BE HEARD II maintenance data, the major message is that the robust responses observed at 16 weeks versus placebo were maintained at 48 weeks. More than 75% of patients retained a HiSCR50 response and more than 55% achieved a HiSCR75 response at the 48-week follow-up. The durable response was also reflected in other measures, according to Christos C. Zouboulis, MD, PhD, director of the department of dermatology, Brandenburg Medical School, Neuruppin, Germany.
“Improvements in disease severity were seen over time,” Dr. Zouboulis reported. “The majority of patients with severe HS at baseline shifted to mild to moderate disease according to the IHS4 classification.”
To the degree that both sonelokimab and bimekizumab target IL-17A/F, these data are mutually reinforcing. Dr. Kirby said that there is a sizable body of data implicating IL-17A/F in driving HS, and the activity of inhibitors in support the clinical value of IL-17A/F suppression.
On Oct. 18, shortly after the EADV meeting concluded, the Food and Drug Administration approved bimekizumab for treating moderate to severe plaque psoriasis, the first approved indication in the United States. In the European Union, it was approved for psoriasis in 2021, and for psoriatic arthritis and ankylosing spondylitis in June 2023.
Dr. Kirby has financial relationships with more than 10 pharmaceutical companies, including MoonLake, which is developing sonelokimab and sponsored the MIRA trial. Dr. Christos, president of the European HS Foundation, has financial relationships with multiple pharmaceutical companies, including UCB, which makes bimekizumab and provided funding for the BE HEARD I and II trials.
BERLIN – In separate trials conducted in patients with hidradenitis suppurativa (HS), two biologics that inhibit the activity of interleukin-17A (IL-17A) and IL-17F were associated with highly encouraging rates of control.
One of the trials evaluated a nanobody inhibitor, sonelokimab, a molecule with a substantially smaller size than traditional monoclonal antibodies (40 kilodaltons vs. 150 kilodaltons). After 24 weeks of treatment, the most effective of the two study doses almost doubled the proportion of patients with complete resolution of draining tunnels (41.1% vs. 23.8%; P < .05) relative to placebo.
“I think the size of sonelokimab is important,” Brian Kirby, MD, a consultant dermatologist at St. Vincent’s Hospital, Dublin, said at the annual congress of the European Academy of Dermatology and Venereology. “We think the smaller size results in better penetration of inflamed tissue,” he added, noting that penetration of abscesses, fistulae, and tunnels has been recognized in the past as a potential weakness of the larger monoclonal antibodies.
The other set of anti-17-A/F set of data were generated by a pooled 48-week maintenance from the BE HEARD I and II trials with bimekizumab. The 16-week data from these two trials were presented at the annual meeting of the American Academy of Dermatology earlier this year.
IL-17A/F trials
Both the
In the sonelokimab trial, called MIRA, 234 adults with HS were randomized in a 2:2:2:1 ratio to one of the two experimental arms, placebo, or a reference arm with the tumor necrosis factor (TNF) inhibitor adalimumab. Nearly 64% had Hurley stage II HS.
The primary endpoint was a 75% or greater reduction in total abscesses and nodules with no increase in draining tunnel count (HiSCR75) from baseline. Dr. Kirby said that this is more rigorous than the HiSCR50 endpoint more commonly used in HS clinical trials. Treatments were administered every 2 weeks for the first 8 weeks of a planned follow-up of 24 weeks and then every 4 weeks thereafter.
At 16 weeks, according to the data Dr. Kirby presented, both doses of sonelokimab were more active than placebo, but Dr. Kirby reported that the lower dose performed better for most objective endpoints.
For example, the HiSCR75 was reached by 43.3% of those randomized to the 120-mg dose (P < .001 vs. placebo), 34.8% of those randomized to the 240-mg dose (P <.01), and 14.7% of those randomized to placebo.
For HiSCR50, response rates were 65.7%, 53.0%, and 27.9%, for the 120-mg, 240-mg, and placebo arms, respectively. Again, both the lower dose (P < .001) and the higher dose (P < .01) were significantly superior to placebo.
On the International Hidradenitis Suppurativa Severity Score System (IHS4), which counts nodules and abscesses, score reductions were 19.3, 14.5, and 7.9 for the lower dose, higher dose, and placebo, respectively, with a greater statistical advantage for the lower relative to the higher dose over placebo (P <.001 vs. P <.01).
However, patient-focused outcomes were not necessarily greater for the lower dose. For the patient-completed measure, the Numerical Rating Scale 50% reduction in skin pain (NRS50), the proportion of patients responding at 12 weeks was numerically greater for the 240-mg dose (41.3%) than with the 120-mg dose (32.0%), although both reached the same statistical advantage (P < .001) over the 4.3% who reached this level of response on placebo.
For the Dermatology Life Quality Index (DLQI) and the Patient Global Impression of Severity (PGI-S), improvements from baseline were similar for the lower and higher dose, although there was a modest numerical and statistical advantage for the higher dose over placebo (P < .001 vs. P <.01).
The HiSCR50 (57.6%) and HiSCR75 (36.4%) responses were both lower for those randomized to the TNF inhibitor adalimumab relative to sonelokimab, but the smaller number of patients in this arm prohibited a statistical comparison.
Although oral candidiasis was more common among patients receiving either dose of sonelokimab than placebo, these were of mild to moderate severity. Dr. Kirby said that there were no unexpected safety issues, and sonelokimab was generally well tolerated.
The results are encouraging, but Dr. Kirby acknowledged that data are now needed to confirm that resolution of tunnels and fistulae is greater with a nanobody inhibitor of IL-17A/F than other targeted therapies. Even if this is validated, he said studies are needed to prove that the small relative molecule size is the reason behind the benefits.
Forty-eight–week bimekizumab data
From the pooled BE HEARD I and BE HEARD II maintenance data, the major message is that the robust responses observed at 16 weeks versus placebo were maintained at 48 weeks. More than 75% of patients retained a HiSCR50 response and more than 55% achieved a HiSCR75 response at the 48-week follow-up. The durable response was also reflected in other measures, according to Christos C. Zouboulis, MD, PhD, director of the department of dermatology, Brandenburg Medical School, Neuruppin, Germany.
“Improvements in disease severity were seen over time,” Dr. Zouboulis reported. “The majority of patients with severe HS at baseline shifted to mild to moderate disease according to the IHS4 classification.”
To the degree that both sonelokimab and bimekizumab target IL-17A/F, these data are mutually reinforcing. Dr. Kirby said that there is a sizable body of data implicating IL-17A/F in driving HS, and the activity of inhibitors in support the clinical value of IL-17A/F suppression.
On Oct. 18, shortly after the EADV meeting concluded, the Food and Drug Administration approved bimekizumab for treating moderate to severe plaque psoriasis, the first approved indication in the United States. In the European Union, it was approved for psoriasis in 2021, and for psoriatic arthritis and ankylosing spondylitis in June 2023.
Dr. Kirby has financial relationships with more than 10 pharmaceutical companies, including MoonLake, which is developing sonelokimab and sponsored the MIRA trial. Dr. Christos, president of the European HS Foundation, has financial relationships with multiple pharmaceutical companies, including UCB, which makes bimekizumab and provided funding for the BE HEARD I and II trials.
BERLIN – In separate trials conducted in patients with hidradenitis suppurativa (HS), two biologics that inhibit the activity of interleukin-17A (IL-17A) and IL-17F were associated with highly encouraging rates of control.
One of the trials evaluated a nanobody inhibitor, sonelokimab, a molecule with a substantially smaller size than traditional monoclonal antibodies (40 kilodaltons vs. 150 kilodaltons). After 24 weeks of treatment, the most effective of the two study doses almost doubled the proportion of patients with complete resolution of draining tunnels (41.1% vs. 23.8%; P < .05) relative to placebo.
“I think the size of sonelokimab is important,” Brian Kirby, MD, a consultant dermatologist at St. Vincent’s Hospital, Dublin, said at the annual congress of the European Academy of Dermatology and Venereology. “We think the smaller size results in better penetration of inflamed tissue,” he added, noting that penetration of abscesses, fistulae, and tunnels has been recognized in the past as a potential weakness of the larger monoclonal antibodies.
The other set of anti-17-A/F set of data were generated by a pooled 48-week maintenance from the BE HEARD I and II trials with bimekizumab. The 16-week data from these two trials were presented at the annual meeting of the American Academy of Dermatology earlier this year.
IL-17A/F trials
Both the
In the sonelokimab trial, called MIRA, 234 adults with HS were randomized in a 2:2:2:1 ratio to one of the two experimental arms, placebo, or a reference arm with the tumor necrosis factor (TNF) inhibitor adalimumab. Nearly 64% had Hurley stage II HS.
The primary endpoint was a 75% or greater reduction in total abscesses and nodules with no increase in draining tunnel count (HiSCR75) from baseline. Dr. Kirby said that this is more rigorous than the HiSCR50 endpoint more commonly used in HS clinical trials. Treatments were administered every 2 weeks for the first 8 weeks of a planned follow-up of 24 weeks and then every 4 weeks thereafter.
At 16 weeks, according to the data Dr. Kirby presented, both doses of sonelokimab were more active than placebo, but Dr. Kirby reported that the lower dose performed better for most objective endpoints.
For example, the HiSCR75 was reached by 43.3% of those randomized to the 120-mg dose (P < .001 vs. placebo), 34.8% of those randomized to the 240-mg dose (P <.01), and 14.7% of those randomized to placebo.
For HiSCR50, response rates were 65.7%, 53.0%, and 27.9%, for the 120-mg, 240-mg, and placebo arms, respectively. Again, both the lower dose (P < .001) and the higher dose (P < .01) were significantly superior to placebo.
On the International Hidradenitis Suppurativa Severity Score System (IHS4), which counts nodules and abscesses, score reductions were 19.3, 14.5, and 7.9 for the lower dose, higher dose, and placebo, respectively, with a greater statistical advantage for the lower relative to the higher dose over placebo (P <.001 vs. P <.01).
However, patient-focused outcomes were not necessarily greater for the lower dose. For the patient-completed measure, the Numerical Rating Scale 50% reduction in skin pain (NRS50), the proportion of patients responding at 12 weeks was numerically greater for the 240-mg dose (41.3%) than with the 120-mg dose (32.0%), although both reached the same statistical advantage (P < .001) over the 4.3% who reached this level of response on placebo.
For the Dermatology Life Quality Index (DLQI) and the Patient Global Impression of Severity (PGI-S), improvements from baseline were similar for the lower and higher dose, although there was a modest numerical and statistical advantage for the higher dose over placebo (P < .001 vs. P <.01).
The HiSCR50 (57.6%) and HiSCR75 (36.4%) responses were both lower for those randomized to the TNF inhibitor adalimumab relative to sonelokimab, but the smaller number of patients in this arm prohibited a statistical comparison.
Although oral candidiasis was more common among patients receiving either dose of sonelokimab than placebo, these were of mild to moderate severity. Dr. Kirby said that there were no unexpected safety issues, and sonelokimab was generally well tolerated.
The results are encouraging, but Dr. Kirby acknowledged that data are now needed to confirm that resolution of tunnels and fistulae is greater with a nanobody inhibitor of IL-17A/F than other targeted therapies. Even if this is validated, he said studies are needed to prove that the small relative molecule size is the reason behind the benefits.
Forty-eight–week bimekizumab data
From the pooled BE HEARD I and BE HEARD II maintenance data, the major message is that the robust responses observed at 16 weeks versus placebo were maintained at 48 weeks. More than 75% of patients retained a HiSCR50 response and more than 55% achieved a HiSCR75 response at the 48-week follow-up. The durable response was also reflected in other measures, according to Christos C. Zouboulis, MD, PhD, director of the department of dermatology, Brandenburg Medical School, Neuruppin, Germany.
“Improvements in disease severity were seen over time,” Dr. Zouboulis reported. “The majority of patients with severe HS at baseline shifted to mild to moderate disease according to the IHS4 classification.”
To the degree that both sonelokimab and bimekizumab target IL-17A/F, these data are mutually reinforcing. Dr. Kirby said that there is a sizable body of data implicating IL-17A/F in driving HS, and the activity of inhibitors in support the clinical value of IL-17A/F suppression.
On Oct. 18, shortly after the EADV meeting concluded, the Food and Drug Administration approved bimekizumab for treating moderate to severe plaque psoriasis, the first approved indication in the United States. In the European Union, it was approved for psoriasis in 2021, and for psoriatic arthritis and ankylosing spondylitis in June 2023.
Dr. Kirby has financial relationships with more than 10 pharmaceutical companies, including MoonLake, which is developing sonelokimab and sponsored the MIRA trial. Dr. Christos, president of the European HS Foundation, has financial relationships with multiple pharmaceutical companies, including UCB, which makes bimekizumab and provided funding for the BE HEARD I and II trials.
AT THE EADV CONGRESS
Topical ivermectin study sheds light on dysbiosis in rosacea
, according to a report presented at the recent European Academy of Dermatology and Venereology (EADV) 2023 Congress.
“This is the first hint that the host’s cutaneous microbiome plays a secondary role in the immunopathogenesis of rosacea,” said Bernard Homey, MD, director of the department of dermatology at University Hospital Düsseldorf in Germany.
“In rosacea, we are well aware of trigger factors such as stress, UV light, heat, cold, food, and alcohol,” he said. “We are also well aware that there is an increase in Demodex mites in the pilosebaceous unit.”
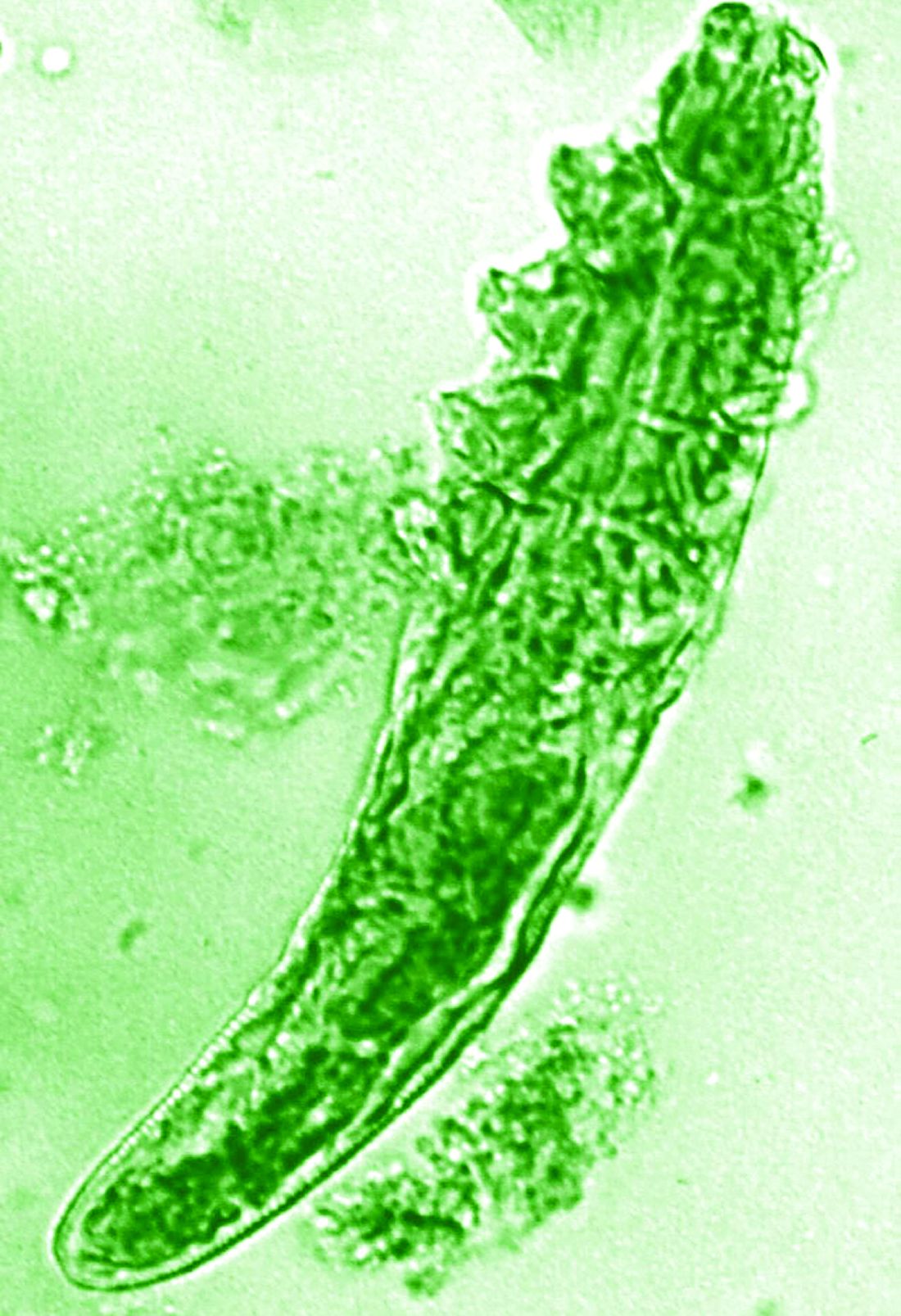
Research over the past decade has also started to look at the potential role of the skin microbiome in the disease process, but answers have remained “largely elusive,” Dr. Homey said.
Ivermectin helps, but how?
Ivermectin 1% cream (Soolantra) has been approved by the U.S. Food and Drug Administration since 2014 for the treatment of the inflammatory lesions that are characteristic of rosacea, but its mechanism of action is not clear.
Dr. Homey presented the results of a study of 61 patients designed to look at how ivermectin might be working in the treatment of people with rosacea and investigate if there was any relation to the skin microbiome and transcriptome of patients.
The trial included 41 individuals with papulopustular rosacea and 20 individuals who did not have rosacea. For all patients, surface skin biopsies were performed twice 30 days apart using cyanoacrylate glue; patients with rosacea were treated with topical ivermectin 1% between biopsies. Skin samples obtained at day 0 and day 30 were examined under the microscope, and Demodex counts (mites/cm2) of skin and RNA sequencing of the cutaneous microbiome were undertaken.
The mean age of the patients with rosacea was 54.9 years, and the mean Demodex counts before and after treatment were a respective 7.2 cm2 and 0.9 cm2.
Using the Investigator’s General Assessment to assess the severity of rosacea, Homey reported that 43.9% of patients with rosacea had a decrease in scores at day 30, indicating improvement.
In addition, topical ivermectin resulted in a marked or total decrease in Demodex mite density for 87.5% of patients (n = 24) who were identified as having the mites.
Skin microbiome changes seen
As a form of quality control, skin microbiome changes among the patients were compared with control patients using 16S rRNA sequencing.
“The taxa we find within the cutaneous niche of inflammatory lesions of rosacea patients are significantly different from healthy volunteers,” Dr. Homey said.
Cutibacterium species are predominant in healthy control persons but are not present when there is inflammation in patients with rosacea. Instead, staphylococcus species “take over the niche, similar to atopic dermatitis,” he noted.
Looking at how treatment with ivermectin influences the organisms, the decrease in C. acnes seen in patients with rosacea persisted despite treatment, and the abundance of Staphylococcus epidermidis, S. hominis, and S. capitis increased further. This suggests a possible protective or homeostatic role of C. acnes but a pathogenic role for staphylococci, explained Dr. Homey.
“Surprisingly, although inflammatory lesions decrease, patients get better, the cutaneous microbiome does not revert to homeostatic conditions during topical ivermectin treatment,” he observed.
There is, of course, variability among individuals.
Dr. Homey also reported that Snodgrassella alvi – a microorganism believed to reside in the gut of Demodex folliculorum mites – was found in the skin microbiome of patients with rosacea before but not after ivermectin treatment. This may mean that this microorganism could be partially triggering inflammation in rosacea patients.
Looking at the transcriptome of patients, Dr. Homey said that there was downregulation of distinct genes that might make for more favorable conditions for Demodex mites.
Moreover, insufficient upregulation of interleukin-17 pathways might be working together with barrier defects in the skin and metabolic changes to “pave the way” for colonization by S. epidermidis.
Pulling it together
Dr. Homey and associates conclude in their abstract that the findings “support that rosacea lesions are associated with dysbiosis.”
Although treatment with ivermectin did not normalize the skin’s microbiome, it was associated with a decrease in Demodex mite density and the reduction of microbes associated with Demodex.
Margarida Gonçalo, MD, PhD, professor of dermatology at the University of Coimbra in Portugal, who cochaired the late-breaking news session where the data were presented, asked whether healthy and affected skin in patients with rosacea had been compared, rather than comparing the skin of rosacea lesions with healthy control samples.
“No, we did not this, as this is methodologically a little bit more difficult,” Dr. Homey responded.
Also cochairing the session was Michel Gilliet, MD, chair of the department of dermatology at the University Hospital CHUV in Lausanne, Switzerland. He commented that these “data suggest that there’s an intimate link between Demodex and the skin microbiota and dysbiosis in in rosacea.”
Dr. Gilliet added: “You have a whole dysbiosis going on in rosacea, which is probably only dependent on these bacteria.”
It would be “very interesting,” as a “proof-of-concept” study, to look at whether depleting Demodex would also delete S. alvi, he suggested.
The study was funded by Galderma. Dr. Homey has acted as a consultant, speaker or investigator for many pharmaceutical companies including Galderma.
A version of this article first appeared on Medscape.com.
, according to a report presented at the recent European Academy of Dermatology and Venereology (EADV) 2023 Congress.
“This is the first hint that the host’s cutaneous microbiome plays a secondary role in the immunopathogenesis of rosacea,” said Bernard Homey, MD, director of the department of dermatology at University Hospital Düsseldorf in Germany.
“In rosacea, we are well aware of trigger factors such as stress, UV light, heat, cold, food, and alcohol,” he said. “We are also well aware that there is an increase in Demodex mites in the pilosebaceous unit.”
Research over the past decade has also started to look at the potential role of the skin microbiome in the disease process, but answers have remained “largely elusive,” Dr. Homey said.
Ivermectin helps, but how?
Ivermectin 1% cream (Soolantra) has been approved by the U.S. Food and Drug Administration since 2014 for the treatment of the inflammatory lesions that are characteristic of rosacea, but its mechanism of action is not clear.
Dr. Homey presented the results of a study of 61 patients designed to look at how ivermectin might be working in the treatment of people with rosacea and investigate if there was any relation to the skin microbiome and transcriptome of patients.
The trial included 41 individuals with papulopustular rosacea and 20 individuals who did not have rosacea. For all patients, surface skin biopsies were performed twice 30 days apart using cyanoacrylate glue; patients with rosacea were treated with topical ivermectin 1% between biopsies. Skin samples obtained at day 0 and day 30 were examined under the microscope, and Demodex counts (mites/cm2) of skin and RNA sequencing of the cutaneous microbiome were undertaken.
The mean age of the patients with rosacea was 54.9 years, and the mean Demodex counts before and after treatment were a respective 7.2 cm2 and 0.9 cm2.
Using the Investigator’s General Assessment to assess the severity of rosacea, Homey reported that 43.9% of patients with rosacea had a decrease in scores at day 30, indicating improvement.
In addition, topical ivermectin resulted in a marked or total decrease in Demodex mite density for 87.5% of patients (n = 24) who were identified as having the mites.
Skin microbiome changes seen
As a form of quality control, skin microbiome changes among the patients were compared with control patients using 16S rRNA sequencing.
“The taxa we find within the cutaneous niche of inflammatory lesions of rosacea patients are significantly different from healthy volunteers,” Dr. Homey said.
Cutibacterium species are predominant in healthy control persons but are not present when there is inflammation in patients with rosacea. Instead, staphylococcus species “take over the niche, similar to atopic dermatitis,” he noted.
Looking at how treatment with ivermectin influences the organisms, the decrease in C. acnes seen in patients with rosacea persisted despite treatment, and the abundance of Staphylococcus epidermidis, S. hominis, and S. capitis increased further. This suggests a possible protective or homeostatic role of C. acnes but a pathogenic role for staphylococci, explained Dr. Homey.
“Surprisingly, although inflammatory lesions decrease, patients get better, the cutaneous microbiome does not revert to homeostatic conditions during topical ivermectin treatment,” he observed.
There is, of course, variability among individuals.
Dr. Homey also reported that Snodgrassella alvi – a microorganism believed to reside in the gut of Demodex folliculorum mites – was found in the skin microbiome of patients with rosacea before but not after ivermectin treatment. This may mean that this microorganism could be partially triggering inflammation in rosacea patients.
Looking at the transcriptome of patients, Dr. Homey said that there was downregulation of distinct genes that might make for more favorable conditions for Demodex mites.
Moreover, insufficient upregulation of interleukin-17 pathways might be working together with barrier defects in the skin and metabolic changes to “pave the way” for colonization by S. epidermidis.
Pulling it together
Dr. Homey and associates conclude in their abstract that the findings “support that rosacea lesions are associated with dysbiosis.”
Although treatment with ivermectin did not normalize the skin’s microbiome, it was associated with a decrease in Demodex mite density and the reduction of microbes associated with Demodex.
Margarida Gonçalo, MD, PhD, professor of dermatology at the University of Coimbra in Portugal, who cochaired the late-breaking news session where the data were presented, asked whether healthy and affected skin in patients with rosacea had been compared, rather than comparing the skin of rosacea lesions with healthy control samples.
“No, we did not this, as this is methodologically a little bit more difficult,” Dr. Homey responded.
Also cochairing the session was Michel Gilliet, MD, chair of the department of dermatology at the University Hospital CHUV in Lausanne, Switzerland. He commented that these “data suggest that there’s an intimate link between Demodex and the skin microbiota and dysbiosis in in rosacea.”
Dr. Gilliet added: “You have a whole dysbiosis going on in rosacea, which is probably only dependent on these bacteria.”
It would be “very interesting,” as a “proof-of-concept” study, to look at whether depleting Demodex would also delete S. alvi, he suggested.
The study was funded by Galderma. Dr. Homey has acted as a consultant, speaker or investigator for many pharmaceutical companies including Galderma.
A version of this article first appeared on Medscape.com.
, according to a report presented at the recent European Academy of Dermatology and Venereology (EADV) 2023 Congress.
“This is the first hint that the host’s cutaneous microbiome plays a secondary role in the immunopathogenesis of rosacea,” said Bernard Homey, MD, director of the department of dermatology at University Hospital Düsseldorf in Germany.
“In rosacea, we are well aware of trigger factors such as stress, UV light, heat, cold, food, and alcohol,” he said. “We are also well aware that there is an increase in Demodex mites in the pilosebaceous unit.”
Research over the past decade has also started to look at the potential role of the skin microbiome in the disease process, but answers have remained “largely elusive,” Dr. Homey said.
Ivermectin helps, but how?
Ivermectin 1% cream (Soolantra) has been approved by the U.S. Food and Drug Administration since 2014 for the treatment of the inflammatory lesions that are characteristic of rosacea, but its mechanism of action is not clear.
Dr. Homey presented the results of a study of 61 patients designed to look at how ivermectin might be working in the treatment of people with rosacea and investigate if there was any relation to the skin microbiome and transcriptome of patients.
The trial included 41 individuals with papulopustular rosacea and 20 individuals who did not have rosacea. For all patients, surface skin biopsies were performed twice 30 days apart using cyanoacrylate glue; patients with rosacea were treated with topical ivermectin 1% between biopsies. Skin samples obtained at day 0 and day 30 were examined under the microscope, and Demodex counts (mites/cm2) of skin and RNA sequencing of the cutaneous microbiome were undertaken.
The mean age of the patients with rosacea was 54.9 years, and the mean Demodex counts before and after treatment were a respective 7.2 cm2 and 0.9 cm2.
Using the Investigator’s General Assessment to assess the severity of rosacea, Homey reported that 43.9% of patients with rosacea had a decrease in scores at day 30, indicating improvement.
In addition, topical ivermectin resulted in a marked or total decrease in Demodex mite density for 87.5% of patients (n = 24) who were identified as having the mites.
Skin microbiome changes seen
As a form of quality control, skin microbiome changes among the patients were compared with control patients using 16S rRNA sequencing.
“The taxa we find within the cutaneous niche of inflammatory lesions of rosacea patients are significantly different from healthy volunteers,” Dr. Homey said.
Cutibacterium species are predominant in healthy control persons but are not present when there is inflammation in patients with rosacea. Instead, staphylococcus species “take over the niche, similar to atopic dermatitis,” he noted.
Looking at how treatment with ivermectin influences the organisms, the decrease in C. acnes seen in patients with rosacea persisted despite treatment, and the abundance of Staphylococcus epidermidis, S. hominis, and S. capitis increased further. This suggests a possible protective or homeostatic role of C. acnes but a pathogenic role for staphylococci, explained Dr. Homey.
“Surprisingly, although inflammatory lesions decrease, patients get better, the cutaneous microbiome does not revert to homeostatic conditions during topical ivermectin treatment,” he observed.
There is, of course, variability among individuals.
Dr. Homey also reported that Snodgrassella alvi – a microorganism believed to reside in the gut of Demodex folliculorum mites – was found in the skin microbiome of patients with rosacea before but not after ivermectin treatment. This may mean that this microorganism could be partially triggering inflammation in rosacea patients.
Looking at the transcriptome of patients, Dr. Homey said that there was downregulation of distinct genes that might make for more favorable conditions for Demodex mites.
Moreover, insufficient upregulation of interleukin-17 pathways might be working together with barrier defects in the skin and metabolic changes to “pave the way” for colonization by S. epidermidis.
Pulling it together
Dr. Homey and associates conclude in their abstract that the findings “support that rosacea lesions are associated with dysbiosis.”
Although treatment with ivermectin did not normalize the skin’s microbiome, it was associated with a decrease in Demodex mite density and the reduction of microbes associated with Demodex.
Margarida Gonçalo, MD, PhD, professor of dermatology at the University of Coimbra in Portugal, who cochaired the late-breaking news session where the data were presented, asked whether healthy and affected skin in patients with rosacea had been compared, rather than comparing the skin of rosacea lesions with healthy control samples.
“No, we did not this, as this is methodologically a little bit more difficult,” Dr. Homey responded.
Also cochairing the session was Michel Gilliet, MD, chair of the department of dermatology at the University Hospital CHUV in Lausanne, Switzerland. He commented that these “data suggest that there’s an intimate link between Demodex and the skin microbiota and dysbiosis in in rosacea.”
Dr. Gilliet added: “You have a whole dysbiosis going on in rosacea, which is probably only dependent on these bacteria.”
It would be “very interesting,” as a “proof-of-concept” study, to look at whether depleting Demodex would also delete S. alvi, he suggested.
The study was funded by Galderma. Dr. Homey has acted as a consultant, speaker or investigator for many pharmaceutical companies including Galderma.
A version of this article first appeared on Medscape.com.
FROM EADV 2023
Body dysmorphic disorder diagnosis guidelines completed in Europe
BERLIN – were outlined in a late-breaker presentation at the annual Congress of the European Academy of Dermatology and Venereology.
The development of guidelines for BDD, a disorder familiar to many clinical dermatologists, is intended as a practical tool, according to Maria-Angeliki Gkini, MD, who has appointments at both Bart’s Health NHS Trust in London and the 401 General Army Hospital in Athens.

“BDD is a relatively common disorder in which the patients are preoccupied with a perceived defect or defects,” Dr. Gkini explained. “This affects them so intensely that it affects their mental health and their quality of life.”
In the DSM-5, published by the American Psychiatric Association, BDD is specifically defined as a preoccupation with “one or more perceived defects or flaws in physical appearance that are not observable or appear slight to others.” But Dr. Gkini said that BDD can also develop as a comorbidity of dermatological disorders that are visible.
These patients are challenging because they are difficult to please, added Dr. Gkini, who said they commonly become involved in doctor shopping, leaving negative reviews on social media for the clinicians they have cycled through. The problem is that the defects they seek to resolve typically stem from distorted perceptions.
BDD is related to obsessive-compulsive disorder by the frequency with which patients pursue repetitive behaviors related to their preoccupation, such as intensive grooming, frequent trips to the mirror, or difficulty in focusing on topics other than their own appearance.
The process to develop the soon-to-be-published guidelines began with a literature search. Of the approximately 3,200 articles identified on BDD, only 10 involved randomized controlled trials. Moreover, even the quality of these trials was considered “low to very low” by the experts who reviewed them, Dr. Gkini said.
One explanation is that psychodermatology has only recently started to attract more research interest, and better studies are now underway, she noted.
However, because of the dearth of high quality evidence now available, the guideline development relied on a Delphi method to reach consensus based on expert opinion in discussion of the available data.
Consensus reached by 17 experts
Specifically, 17 experts, all of whom were members of the European Society for Dermatology and Psychiatry proceeded to systematically address a series of clinical questions and recommendations. Consensus was defined as at least 75% of the participants strongly agreeing or agreeing. Several rounds of discussion were often required.
Among the conclusions, the guidelines support uniform screening for BDD in all patients prior to cosmetic procedures. In identifying depression, anxiety, and distorted perceptions, simple tools, such as the Patient Health Questionnaire might be adequate for an initial evaluation, but Dr. Gkini also recommended routinely inquiring about suicidal ideation, which has been reported in up to 80% of individuals with BDD.
Other instruments for screening that can be considered include DSM-5 criteria for BDD and the Body Dysmorphic Disorder Questionnaire–Dermatology Version, which might be particularly useful and appropriate for dermatologists.
One of the reasons to screen for BDD is that these patients often convince themselves that some specific procedure is needed to resolve the source of their obsession. The goal of screening is to verify that it is the dermatologic concern, not an underlying psychiatric disorder that is driving their search for relief. The risk of dermatologic interventions is not only that expectations are not met, but the patient’s perception of a failed intervention “sometimes makes these worse,” Dr. Gkini explained.
Collaboration with psychiatrists recommended
The guidelines include suggestions for treatment of BDD. Of these, SSRIs are recommended at high relative doses, according to Dr. Gkini. Consistent with the consensus recommendation of collaborating with mental health specialists, she said that the recommendations acknowledge evidence of greater benefits when SSRIs are combined with psychotherapy.
Katharine A. Phillips, MD, professor of psychiatry at Weill Cornell Medicine, New York, has been conducting BDD research for several years and has written numerous books and articles about this topic, including a review in the journal Focus. She cautioned that, because of a normal concern for appearance, BDD is easily missed by dermatologists.
“For BDD to be diagnosed, the preoccupation with a nonexistent or slight defect in appearance must cause clinically significant distress or impairment in functioning,” she said in an interview. “This is necessary to differentiate BDD from more normal and common appearance concerns that do not qualify for the diagnosis”
She specified that patients should be considered for cognitive-behavioral therapy rather than psychotherapy, a generic term that covers many forms of treatment. She said that most other types of psychotherapy “are probably not effective” for BDD.
Dr. Phillips highly endorsed the development of BDD guidelines for dermatologists because of the frequency with which physicians in this specialty encounter BDD – and believes that more attention to this diagnosis is needed.
“I recommend that dermatologists who have a patient with BDD collaborate with a psychiatrist in delivering care with an SSRI,” she said. “High doses of these medications are often needed to effectively treat BDD.”
Dr. Gkini reported financial relationships with AbbVie, Almirall, Celgene, Eli Lilly, Janssen, LEO, Novartis, Sanofi, and Regenlab. Dr. Phillips reported no relevant financial relationships.
BERLIN – were outlined in a late-breaker presentation at the annual Congress of the European Academy of Dermatology and Venereology.
The development of guidelines for BDD, a disorder familiar to many clinical dermatologists, is intended as a practical tool, according to Maria-Angeliki Gkini, MD, who has appointments at both Bart’s Health NHS Trust in London and the 401 General Army Hospital in Athens.
“BDD is a relatively common disorder in which the patients are preoccupied with a perceived defect or defects,” Dr. Gkini explained. “This affects them so intensely that it affects their mental health and their quality of life.”
In the DSM-5, published by the American Psychiatric Association, BDD is specifically defined as a preoccupation with “one or more perceived defects or flaws in physical appearance that are not observable or appear slight to others.” But Dr. Gkini said that BDD can also develop as a comorbidity of dermatological disorders that are visible.
These patients are challenging because they are difficult to please, added Dr. Gkini, who said they commonly become involved in doctor shopping, leaving negative reviews on social media for the clinicians they have cycled through. The problem is that the defects they seek to resolve typically stem from distorted perceptions.
BDD is related to obsessive-compulsive disorder by the frequency with which patients pursue repetitive behaviors related to their preoccupation, such as intensive grooming, frequent trips to the mirror, or difficulty in focusing on topics other than their own appearance.
The process to develop the soon-to-be-published guidelines began with a literature search. Of the approximately 3,200 articles identified on BDD, only 10 involved randomized controlled trials. Moreover, even the quality of these trials was considered “low to very low” by the experts who reviewed them, Dr. Gkini said.
One explanation is that psychodermatology has only recently started to attract more research interest, and better studies are now underway, she noted.
However, because of the dearth of high quality evidence now available, the guideline development relied on a Delphi method to reach consensus based on expert opinion in discussion of the available data.
Consensus reached by 17 experts
Specifically, 17 experts, all of whom were members of the European Society for Dermatology and Psychiatry proceeded to systematically address a series of clinical questions and recommendations. Consensus was defined as at least 75% of the participants strongly agreeing or agreeing. Several rounds of discussion were often required.
Among the conclusions, the guidelines support uniform screening for BDD in all patients prior to cosmetic procedures. In identifying depression, anxiety, and distorted perceptions, simple tools, such as the Patient Health Questionnaire might be adequate for an initial evaluation, but Dr. Gkini also recommended routinely inquiring about suicidal ideation, which has been reported in up to 80% of individuals with BDD.
Other instruments for screening that can be considered include DSM-5 criteria for BDD and the Body Dysmorphic Disorder Questionnaire–Dermatology Version, which might be particularly useful and appropriate for dermatologists.
One of the reasons to screen for BDD is that these patients often convince themselves that some specific procedure is needed to resolve the source of their obsession. The goal of screening is to verify that it is the dermatologic concern, not an underlying psychiatric disorder that is driving their search for relief. The risk of dermatologic interventions is not only that expectations are not met, but the patient’s perception of a failed intervention “sometimes makes these worse,” Dr. Gkini explained.
Collaboration with psychiatrists recommended
The guidelines include suggestions for treatment of BDD. Of these, SSRIs are recommended at high relative doses, according to Dr. Gkini. Consistent with the consensus recommendation of collaborating with mental health specialists, she said that the recommendations acknowledge evidence of greater benefits when SSRIs are combined with psychotherapy.
Katharine A. Phillips, MD, professor of psychiatry at Weill Cornell Medicine, New York, has been conducting BDD research for several years and has written numerous books and articles about this topic, including a review in the journal Focus. She cautioned that, because of a normal concern for appearance, BDD is easily missed by dermatologists.
“For BDD to be diagnosed, the preoccupation with a nonexistent or slight defect in appearance must cause clinically significant distress or impairment in functioning,” she said in an interview. “This is necessary to differentiate BDD from more normal and common appearance concerns that do not qualify for the diagnosis”
She specified that patients should be considered for cognitive-behavioral therapy rather than psychotherapy, a generic term that covers many forms of treatment. She said that most other types of psychotherapy “are probably not effective” for BDD.
Dr. Phillips highly endorsed the development of BDD guidelines for dermatologists because of the frequency with which physicians in this specialty encounter BDD – and believes that more attention to this diagnosis is needed.
“I recommend that dermatologists who have a patient with BDD collaborate with a psychiatrist in delivering care with an SSRI,” she said. “High doses of these medications are often needed to effectively treat BDD.”
Dr. Gkini reported financial relationships with AbbVie, Almirall, Celgene, Eli Lilly, Janssen, LEO, Novartis, Sanofi, and Regenlab. Dr. Phillips reported no relevant financial relationships.
BERLIN – were outlined in a late-breaker presentation at the annual Congress of the European Academy of Dermatology and Venereology.
The development of guidelines for BDD, a disorder familiar to many clinical dermatologists, is intended as a practical tool, according to Maria-Angeliki Gkini, MD, who has appointments at both Bart’s Health NHS Trust in London and the 401 General Army Hospital in Athens.
“BDD is a relatively common disorder in which the patients are preoccupied with a perceived defect or defects,” Dr. Gkini explained. “This affects them so intensely that it affects their mental health and their quality of life.”
In the DSM-5, published by the American Psychiatric Association, BDD is specifically defined as a preoccupation with “one or more perceived defects or flaws in physical appearance that are not observable or appear slight to others.” But Dr. Gkini said that BDD can also develop as a comorbidity of dermatological disorders that are visible.
These patients are challenging because they are difficult to please, added Dr. Gkini, who said they commonly become involved in doctor shopping, leaving negative reviews on social media for the clinicians they have cycled through. The problem is that the defects they seek to resolve typically stem from distorted perceptions.
BDD is related to obsessive-compulsive disorder by the frequency with which patients pursue repetitive behaviors related to their preoccupation, such as intensive grooming, frequent trips to the mirror, or difficulty in focusing on topics other than their own appearance.
The process to develop the soon-to-be-published guidelines began with a literature search. Of the approximately 3,200 articles identified on BDD, only 10 involved randomized controlled trials. Moreover, even the quality of these trials was considered “low to very low” by the experts who reviewed them, Dr. Gkini said.
One explanation is that psychodermatology has only recently started to attract more research interest, and better studies are now underway, she noted.
However, because of the dearth of high quality evidence now available, the guideline development relied on a Delphi method to reach consensus based on expert opinion in discussion of the available data.
Consensus reached by 17 experts
Specifically, 17 experts, all of whom were members of the European Society for Dermatology and Psychiatry proceeded to systematically address a series of clinical questions and recommendations. Consensus was defined as at least 75% of the participants strongly agreeing or agreeing. Several rounds of discussion were often required.
Among the conclusions, the guidelines support uniform screening for BDD in all patients prior to cosmetic procedures. In identifying depression, anxiety, and distorted perceptions, simple tools, such as the Patient Health Questionnaire might be adequate for an initial evaluation, but Dr. Gkini also recommended routinely inquiring about suicidal ideation, which has been reported in up to 80% of individuals with BDD.
Other instruments for screening that can be considered include DSM-5 criteria for BDD and the Body Dysmorphic Disorder Questionnaire–Dermatology Version, which might be particularly useful and appropriate for dermatologists.
One of the reasons to screen for BDD is that these patients often convince themselves that some specific procedure is needed to resolve the source of their obsession. The goal of screening is to verify that it is the dermatologic concern, not an underlying psychiatric disorder that is driving their search for relief. The risk of dermatologic interventions is not only that expectations are not met, but the patient’s perception of a failed intervention “sometimes makes these worse,” Dr. Gkini explained.
Collaboration with psychiatrists recommended
The guidelines include suggestions for treatment of BDD. Of these, SSRIs are recommended at high relative doses, according to Dr. Gkini. Consistent with the consensus recommendation of collaborating with mental health specialists, she said that the recommendations acknowledge evidence of greater benefits when SSRIs are combined with psychotherapy.
Katharine A. Phillips, MD, professor of psychiatry at Weill Cornell Medicine, New York, has been conducting BDD research for several years and has written numerous books and articles about this topic, including a review in the journal Focus. She cautioned that, because of a normal concern for appearance, BDD is easily missed by dermatologists.
“For BDD to be diagnosed, the preoccupation with a nonexistent or slight defect in appearance must cause clinically significant distress or impairment in functioning,” she said in an interview. “This is necessary to differentiate BDD from more normal and common appearance concerns that do not qualify for the diagnosis”
She specified that patients should be considered for cognitive-behavioral therapy rather than psychotherapy, a generic term that covers many forms of treatment. She said that most other types of psychotherapy “are probably not effective” for BDD.
Dr. Phillips highly endorsed the development of BDD guidelines for dermatologists because of the frequency with which physicians in this specialty encounter BDD – and believes that more attention to this diagnosis is needed.
“I recommend that dermatologists who have a patient with BDD collaborate with a psychiatrist in delivering care with an SSRI,” she said. “High doses of these medications are often needed to effectively treat BDD.”
Dr. Gkini reported financial relationships with AbbVie, Almirall, Celgene, Eli Lilly, Janssen, LEO, Novartis, Sanofi, and Regenlab. Dr. Phillips reported no relevant financial relationships.
AT THE EADV CONGRESS
More phase 3 data support use of nemolizumab for prurigo nodularis
reported at the annual Congress of the European Academy of Dermatology and Venereology.
In the OLYMPIA 1 study, clinically significant improvements in both itch and skin lesions were seen after 16 weeks of treatment with nemolizumab compared with placebo (P < .0001).
Indeed, among the 286 patients who participated in the trial (190 on nemolizumab and 96 on placebo), 58.4% of those treated with nemolizumab and 16.7% of those who received placebo had an improvement of 4 points or more in the weekly average peak pruritus numeric rating scale (PP-NRS) at week 16 (P < .0001).
Skin lesions were assessed using an investigators general assessment (IGA) score, where IGA success was defined as a score of 0/1 indicating clear or almost clear skin or where there had been at least a 2-point change from baseline values. Over a quarter (26.3%) of nemolizumab-treated patients met these criteria versus 7.3% for those on placebo (P = .0001).
“These results confirm the results of the OLYMPIA 2 study, the other phase 3 study, and now I hope you understand why we are so excited,” lead investigator Sonja Ständer, MD, of the Center for Chronic Pruritus at University Hospital Münster, Germany, said at the meeting, where she presented the data.
The OLYMPIA 2 study included 274 patients and the results showed a weekly average PP-NRS score improvement of 56.3% vs. 20.9% for placebo and IGA success in 37.7% and 11% of patients, respectively, at 16 weeks.
First-in-class therapy
“We know how difficult it is to treat patients; they are refractory to treatment, frustrated, and this really impacts them regarding their quality of life,” said Dr. Ständer. New options are needed to help patients, and nemolizumab, a first-in-class interleukin-31 (IL-31) receptor alpha antagonist, is one treatment that may answer this call.
Prurigo nodularis is a chronic neuroimmune skin condition characterized by severe itch and multiple nodular skin lesions, Dr. Ständer explained. She added that there is evidence that IL-31 has a key role to play in the development of itch, and in differentiation of keratinocytes, type 2 and type 17 immune responses, and fibrosis associated with the condition.
The OLYMPIA 1 and 2 trials are part of a large developmental program that includes two ongoing trials. One is assessing the durability of response over 24 weeks in 40 patients and the other is a long-term extension trial involving 450 patients from the OLYMPIA 1 and 2 trials.
Inclusion criteria and additional results
For inclusion in the study, adults with prurigo nodularis for at least 6 months had to have 20 or more nodules on the body with a bilateral distribution, an IGA score of 3 or more, and an average PP-NRS of 7 or higher. The latter “was really a high bar for them to qualify for the trial,” said Dr. Ständer.
After an initial 4-week screening period, patients were randomly assigned to 24 weeks of treatment with nemolizumab or placebo given as a subcutaneous injection every 4 weeks. An 8-week “off-treatment” period followed.
The nemolizumab dose was based on the patient’s body weight, with patients weighing less than 90 kg (198 pounds) getting a loading dose of 60 mg followed by further doses of 30 mg; while patients weighing 90 kg or more receiving 50 mg of nemolizumab.
Dr. Ständer reported that nemolizumab met all of the trials’ secondary endpoints; this included at least a 4-point improvement in sleep disturbance. She noted that changes in itch and subsequent sleep disturbance occurred early, at 4 weeks of treatment – after just one injection of nemolizumab.
The response rates seen in the moderate to severe prurigo nodularis population studies are quite unique when compared with conventional therapies, Dr. Ständer maintained. “We’ve never seen something like this before.”
No safety concerns
No significant difference in tolerability was seen between the nemolizumab and placebo groups, Dr. Ständer observed. Any adverse event occurred in 71.7% and 65.3% of patients, respectively, and serious adverse events in 8.6% and 10.5%.
There was a similar rate of adverse events leading to discontinuation, respectively (4.8% vs. 4.2%).
Headache was seen more frequently among those on nemolizumab than those on placebo (7.0% vs. 2.1%), and there was a higher number of eczema cases among those on nemolizumab (5.3% vs. 1.1%). The latter is somewhat paradoxical because nemolizumab is also being studied as a treatment for atopic dermatitis, with good results seen in phase 3 trials. Asked about this finding after her presentation, Dr. Ständer said “we are following up on that to know exactly what is going on; this is a side effect of nemolizumab that is seen also with other biologics.”
JAK inhibitor trial for PN, CPUO
Nemolizumab is not the only promising new approach to treating prurigo nodularis. During a separate late-breaking news session at the meeting, Shawn Kwatra, MD, director of the Johns Hopkins Itch Center in Baltimore, presented “dramatic” data from a “proof-of-concept” phase 2 study with the Janus kinase (JAK) inhibitor abrocitinib (Cibinqo), which is approved for atopic dermatitis in the United States and Europe.

The investigator-initiated trial took a different approach from most other trials, Dr. Kwatra said. The starting point was to look at studying multiple rather than single dermatologic diseases that were perhaps being left a little by the wayside but may share some common ground. Those two diseases were prurigo nodularis and chronic pruritus of unknown origin (CPUO).
“They’re actually very analogous conditions in the way we treat, so I thought those would be a good pair,” Dr. Kwatra said, noting that there were several studies that made him think that JAK inhibition “would be an interesting concept to try.”
On that basis, 10 women with prurigo nodularis (mean age, 58 years) and two women and eight men with CPUO (mean age, 70 years) were recruited and all were treated with abrocitinib at a once-daily oral dose of 200 mg for 12 weeks.
“They all had really intense itch,” before treatment, Dr. Kwatra said. The mean baseline PP-NRS was 9.2 and 8.2 in the prurigo nodularis and CPUO groups, respectively. By the end of treatment, however, “the improvement in itch was pretty dramatic,” especially for prurigo nodularis, he said.
At 12 weeks, the PP-NRS score had fallen to 2.0 in the prurigo nodularis group, equating to a significant 78% change from baseline (P < .001). And, in the CPUO group, the 12-week PP-NRS score was 3.8, nearly a 54% drop from baseline (P = .01).
Sleep disturbance was improved for both conditions, and in the patients with prurigo nodularis, there were improvements in skin lesions. Looking at the patients who responded to treatment, Dr. Kwatra noted that “if you responded, you respond fast, and you respond almost entirely.”
Additional findings from cutaneous transcriptome analysis showed that JAK inhibition with abrocitinib was modulating Th1-, Th2-, Th17-, and Th22-mediated pathways in both groups of patients.
The overall frequency of adverse events was low, and no serious adverse events occurred.
Commenting on the potential use of abrocitinib in managing patients with PN and CPUO, Tiago dos Reis Matos, MD, PhD, MSc, Amsterdam University Medical Centers, told this news organization that JAK1 inhibitors “are showing promising results in treating several diseases.”
Dr. Matos, who was not involved in the study, added that JAK inhibition was “of special interest in prurigo nodularis and chronic pruritus, since these are some of the most difficult diseases to treat with limited therapeutic options.”
Dr. Kwatra observed: “Obviously, we need further development. But we also have clues here about how to design phase 3 trials.”
Galderma funded the OLYMPIA 1 and 2 studies. Dr. Ständer was an investigator for the trial and reported serving as a consultant, speaker, or investigator for multiple pharmaceutical companies, including Galderma.
Johns Hopkins University supported the abrocitinib study with funding from Pfizer. Dr. Kwatra is an advisory board member or consultant to several pharmaceutical companies and is an investigator for Galderma, Incyte, Pfizer, and Sanofi.
A version of this article first appeared on Medscape.com.
reported at the annual Congress of the European Academy of Dermatology and Venereology.
In the OLYMPIA 1 study, clinically significant improvements in both itch and skin lesions were seen after 16 weeks of treatment with nemolizumab compared with placebo (P < .0001).
Indeed, among the 286 patients who participated in the trial (190 on nemolizumab and 96 on placebo), 58.4% of those treated with nemolizumab and 16.7% of those who received placebo had an improvement of 4 points or more in the weekly average peak pruritus numeric rating scale (PP-NRS) at week 16 (P < .0001).
Skin lesions were assessed using an investigators general assessment (IGA) score, where IGA success was defined as a score of 0/1 indicating clear or almost clear skin or where there had been at least a 2-point change from baseline values. Over a quarter (26.3%) of nemolizumab-treated patients met these criteria versus 7.3% for those on placebo (P = .0001).
“These results confirm the results of the OLYMPIA 2 study, the other phase 3 study, and now I hope you understand why we are so excited,” lead investigator Sonja Ständer, MD, of the Center for Chronic Pruritus at University Hospital Münster, Germany, said at the meeting, where she presented the data.
The OLYMPIA 2 study included 274 patients and the results showed a weekly average PP-NRS score improvement of 56.3% vs. 20.9% for placebo and IGA success in 37.7% and 11% of patients, respectively, at 16 weeks.
First-in-class therapy
“We know how difficult it is to treat patients; they are refractory to treatment, frustrated, and this really impacts them regarding their quality of life,” said Dr. Ständer. New options are needed to help patients, and nemolizumab, a first-in-class interleukin-31 (IL-31) receptor alpha antagonist, is one treatment that may answer this call.
Prurigo nodularis is a chronic neuroimmune skin condition characterized by severe itch and multiple nodular skin lesions, Dr. Ständer explained. She added that there is evidence that IL-31 has a key role to play in the development of itch, and in differentiation of keratinocytes, type 2 and type 17 immune responses, and fibrosis associated with the condition.
The OLYMPIA 1 and 2 trials are part of a large developmental program that includes two ongoing trials. One is assessing the durability of response over 24 weeks in 40 patients and the other is a long-term extension trial involving 450 patients from the OLYMPIA 1 and 2 trials.
Inclusion criteria and additional results
For inclusion in the study, adults with prurigo nodularis for at least 6 months had to have 20 or more nodules on the body with a bilateral distribution, an IGA score of 3 or more, and an average PP-NRS of 7 or higher. The latter “was really a high bar for them to qualify for the trial,” said Dr. Ständer.
After an initial 4-week screening period, patients were randomly assigned to 24 weeks of treatment with nemolizumab or placebo given as a subcutaneous injection every 4 weeks. An 8-week “off-treatment” period followed.
The nemolizumab dose was based on the patient’s body weight, with patients weighing less than 90 kg (198 pounds) getting a loading dose of 60 mg followed by further doses of 30 mg; while patients weighing 90 kg or more receiving 50 mg of nemolizumab.
Dr. Ständer reported that nemolizumab met all of the trials’ secondary endpoints; this included at least a 4-point improvement in sleep disturbance. She noted that changes in itch and subsequent sleep disturbance occurred early, at 4 weeks of treatment – after just one injection of nemolizumab.
The response rates seen in the moderate to severe prurigo nodularis population studies are quite unique when compared with conventional therapies, Dr. Ständer maintained. “We’ve never seen something like this before.”
No safety concerns
No significant difference in tolerability was seen between the nemolizumab and placebo groups, Dr. Ständer observed. Any adverse event occurred in 71.7% and 65.3% of patients, respectively, and serious adverse events in 8.6% and 10.5%.
There was a similar rate of adverse events leading to discontinuation, respectively (4.8% vs. 4.2%).
Headache was seen more frequently among those on nemolizumab than those on placebo (7.0% vs. 2.1%), and there was a higher number of eczema cases among those on nemolizumab (5.3% vs. 1.1%). The latter is somewhat paradoxical because nemolizumab is also being studied as a treatment for atopic dermatitis, with good results seen in phase 3 trials. Asked about this finding after her presentation, Dr. Ständer said “we are following up on that to know exactly what is going on; this is a side effect of nemolizumab that is seen also with other biologics.”
JAK inhibitor trial for PN, CPUO
Nemolizumab is not the only promising new approach to treating prurigo nodularis. During a separate late-breaking news session at the meeting, Shawn Kwatra, MD, director of the Johns Hopkins Itch Center in Baltimore, presented “dramatic” data from a “proof-of-concept” phase 2 study with the Janus kinase (JAK) inhibitor abrocitinib (Cibinqo), which is approved for atopic dermatitis in the United States and Europe.
The investigator-initiated trial took a different approach from most other trials, Dr. Kwatra said. The starting point was to look at studying multiple rather than single dermatologic diseases that were perhaps being left a little by the wayside but may share some common ground. Those two diseases were prurigo nodularis and chronic pruritus of unknown origin (CPUO).
“They’re actually very analogous conditions in the way we treat, so I thought those would be a good pair,” Dr. Kwatra said, noting that there were several studies that made him think that JAK inhibition “would be an interesting concept to try.”
On that basis, 10 women with prurigo nodularis (mean age, 58 years) and two women and eight men with CPUO (mean age, 70 years) were recruited and all were treated with abrocitinib at a once-daily oral dose of 200 mg for 12 weeks.
“They all had really intense itch,” before treatment, Dr. Kwatra said. The mean baseline PP-NRS was 9.2 and 8.2 in the prurigo nodularis and CPUO groups, respectively. By the end of treatment, however, “the improvement in itch was pretty dramatic,” especially for prurigo nodularis, he said.
At 12 weeks, the PP-NRS score had fallen to 2.0 in the prurigo nodularis group, equating to a significant 78% change from baseline (P < .001). And, in the CPUO group, the 12-week PP-NRS score was 3.8, nearly a 54% drop from baseline (P = .01).
Sleep disturbance was improved for both conditions, and in the patients with prurigo nodularis, there were improvements in skin lesions. Looking at the patients who responded to treatment, Dr. Kwatra noted that “if you responded, you respond fast, and you respond almost entirely.”
Additional findings from cutaneous transcriptome analysis showed that JAK inhibition with abrocitinib was modulating Th1-, Th2-, Th17-, and Th22-mediated pathways in both groups of patients.
The overall frequency of adverse events was low, and no serious adverse events occurred.
Commenting on the potential use of abrocitinib in managing patients with PN and CPUO, Tiago dos Reis Matos, MD, PhD, MSc, Amsterdam University Medical Centers, told this news organization that JAK1 inhibitors “are showing promising results in treating several diseases.”
Dr. Matos, who was not involved in the study, added that JAK inhibition was “of special interest in prurigo nodularis and chronic pruritus, since these are some of the most difficult diseases to treat with limited therapeutic options.”
Dr. Kwatra observed: “Obviously, we need further development. But we also have clues here about how to design phase 3 trials.”
Galderma funded the OLYMPIA 1 and 2 studies. Dr. Ständer was an investigator for the trial and reported serving as a consultant, speaker, or investigator for multiple pharmaceutical companies, including Galderma.
Johns Hopkins University supported the abrocitinib study with funding from Pfizer. Dr. Kwatra is an advisory board member or consultant to several pharmaceutical companies and is an investigator for Galderma, Incyte, Pfizer, and Sanofi.
A version of this article first appeared on Medscape.com.
reported at the annual Congress of the European Academy of Dermatology and Venereology.
In the OLYMPIA 1 study, clinically significant improvements in both itch and skin lesions were seen after 16 weeks of treatment with nemolizumab compared with placebo (P < .0001).
Indeed, among the 286 patients who participated in the trial (190 on nemolizumab and 96 on placebo), 58.4% of those treated with nemolizumab and 16.7% of those who received placebo had an improvement of 4 points or more in the weekly average peak pruritus numeric rating scale (PP-NRS) at week 16 (P < .0001).
Skin lesions were assessed using an investigators general assessment (IGA) score, where IGA success was defined as a score of 0/1 indicating clear or almost clear skin or where there had been at least a 2-point change from baseline values. Over a quarter (26.3%) of nemolizumab-treated patients met these criteria versus 7.3% for those on placebo (P = .0001).
“These results confirm the results of the OLYMPIA 2 study, the other phase 3 study, and now I hope you understand why we are so excited,” lead investigator Sonja Ständer, MD, of the Center for Chronic Pruritus at University Hospital Münster, Germany, said at the meeting, where she presented the data.
The OLYMPIA 2 study included 274 patients and the results showed a weekly average PP-NRS score improvement of 56.3% vs. 20.9% for placebo and IGA success in 37.7% and 11% of patients, respectively, at 16 weeks.
First-in-class therapy
“We know how difficult it is to treat patients; they are refractory to treatment, frustrated, and this really impacts them regarding their quality of life,” said Dr. Ständer. New options are needed to help patients, and nemolizumab, a first-in-class interleukin-31 (IL-31) receptor alpha antagonist, is one treatment that may answer this call.
Prurigo nodularis is a chronic neuroimmune skin condition characterized by severe itch and multiple nodular skin lesions, Dr. Ständer explained. She added that there is evidence that IL-31 has a key role to play in the development of itch, and in differentiation of keratinocytes, type 2 and type 17 immune responses, and fibrosis associated with the condition.
The OLYMPIA 1 and 2 trials are part of a large developmental program that includes two ongoing trials. One is assessing the durability of response over 24 weeks in 40 patients and the other is a long-term extension trial involving 450 patients from the OLYMPIA 1 and 2 trials.
Inclusion criteria and additional results
For inclusion in the study, adults with prurigo nodularis for at least 6 months had to have 20 or more nodules on the body with a bilateral distribution, an IGA score of 3 or more, and an average PP-NRS of 7 or higher. The latter “was really a high bar for them to qualify for the trial,” said Dr. Ständer.
After an initial 4-week screening period, patients were randomly assigned to 24 weeks of treatment with nemolizumab or placebo given as a subcutaneous injection every 4 weeks. An 8-week “off-treatment” period followed.
The nemolizumab dose was based on the patient’s body weight, with patients weighing less than 90 kg (198 pounds) getting a loading dose of 60 mg followed by further doses of 30 mg; while patients weighing 90 kg or more receiving 50 mg of nemolizumab.
Dr. Ständer reported that nemolizumab met all of the trials’ secondary endpoints; this included at least a 4-point improvement in sleep disturbance. She noted that changes in itch and subsequent sleep disturbance occurred early, at 4 weeks of treatment – after just one injection of nemolizumab.
The response rates seen in the moderate to severe prurigo nodularis population studies are quite unique when compared with conventional therapies, Dr. Ständer maintained. “We’ve never seen something like this before.”
No safety concerns
No significant difference in tolerability was seen between the nemolizumab and placebo groups, Dr. Ständer observed. Any adverse event occurred in 71.7% and 65.3% of patients, respectively, and serious adverse events in 8.6% and 10.5%.
There was a similar rate of adverse events leading to discontinuation, respectively (4.8% vs. 4.2%).
Headache was seen more frequently among those on nemolizumab than those on placebo (7.0% vs. 2.1%), and there was a higher number of eczema cases among those on nemolizumab (5.3% vs. 1.1%). The latter is somewhat paradoxical because nemolizumab is also being studied as a treatment for atopic dermatitis, with good results seen in phase 3 trials. Asked about this finding after her presentation, Dr. Ständer said “we are following up on that to know exactly what is going on; this is a side effect of nemolizumab that is seen also with other biologics.”
JAK inhibitor trial for PN, CPUO
Nemolizumab is not the only promising new approach to treating prurigo nodularis. During a separate late-breaking news session at the meeting, Shawn Kwatra, MD, director of the Johns Hopkins Itch Center in Baltimore, presented “dramatic” data from a “proof-of-concept” phase 2 study with the Janus kinase (JAK) inhibitor abrocitinib (Cibinqo), which is approved for atopic dermatitis in the United States and Europe.
The investigator-initiated trial took a different approach from most other trials, Dr. Kwatra said. The starting point was to look at studying multiple rather than single dermatologic diseases that were perhaps being left a little by the wayside but may share some common ground. Those two diseases were prurigo nodularis and chronic pruritus of unknown origin (CPUO).
“They’re actually very analogous conditions in the way we treat, so I thought those would be a good pair,” Dr. Kwatra said, noting that there were several studies that made him think that JAK inhibition “would be an interesting concept to try.”
On that basis, 10 women with prurigo nodularis (mean age, 58 years) and two women and eight men with CPUO (mean age, 70 years) were recruited and all were treated with abrocitinib at a once-daily oral dose of 200 mg for 12 weeks.
“They all had really intense itch,” before treatment, Dr. Kwatra said. The mean baseline PP-NRS was 9.2 and 8.2 in the prurigo nodularis and CPUO groups, respectively. By the end of treatment, however, “the improvement in itch was pretty dramatic,” especially for prurigo nodularis, he said.
At 12 weeks, the PP-NRS score had fallen to 2.0 in the prurigo nodularis group, equating to a significant 78% change from baseline (P < .001). And, in the CPUO group, the 12-week PP-NRS score was 3.8, nearly a 54% drop from baseline (P = .01).
Sleep disturbance was improved for both conditions, and in the patients with prurigo nodularis, there were improvements in skin lesions. Looking at the patients who responded to treatment, Dr. Kwatra noted that “if you responded, you respond fast, and you respond almost entirely.”
Additional findings from cutaneous transcriptome analysis showed that JAK inhibition with abrocitinib was modulating Th1-, Th2-, Th17-, and Th22-mediated pathways in both groups of patients.
The overall frequency of adverse events was low, and no serious adverse events occurred.
Commenting on the potential use of abrocitinib in managing patients with PN and CPUO, Tiago dos Reis Matos, MD, PhD, MSc, Amsterdam University Medical Centers, told this news organization that JAK1 inhibitors “are showing promising results in treating several diseases.”
Dr. Matos, who was not involved in the study, added that JAK inhibition was “of special interest in prurigo nodularis and chronic pruritus, since these are some of the most difficult diseases to treat with limited therapeutic options.”
Dr. Kwatra observed: “Obviously, we need further development. But we also have clues here about how to design phase 3 trials.”
Galderma funded the OLYMPIA 1 and 2 studies. Dr. Ständer was an investigator for the trial and reported serving as a consultant, speaker, or investigator for multiple pharmaceutical companies, including Galderma.
Johns Hopkins University supported the abrocitinib study with funding from Pfizer. Dr. Kwatra is an advisory board member or consultant to several pharmaceutical companies and is an investigator for Galderma, Incyte, Pfizer, and Sanofi.
A version of this article first appeared on Medscape.com.
FROM THE EADV CONGRESS
Birch bark–derived treatment reduces daily dressings in patients with epidermolysis bullosa
Additional when compared with a control gel.
In a final, post hoc analysis to come from the trial, 15 of 45 (33%) patients treated with Oleogel-S10 versus 5 of 48 (10.4%) treated with the control gel were reported as no longer needing daily dressing changes at 45 days of follow-up.
Moreover, the effect was sustained, with similar percentages of patients no longer requiring daily dressing changes at 60 days (34% vs. 13%, respectively) and 90 days (36% vs. 11%) of follow-up.
The mean reduction in daily dressing changes was 1.36 for Oleogel-S10 and 0.41 for the control gel (P = .005).
“Patients who, in the beginning, had daily dressing changes had almost three fewer dressing changes every 2 weeks if they were treated with Oleogel-S10,” Dimitra Kiritsi, MD, PhD, of the department of dermatology at the University of Freiburg (Germany), reported at the annual congress of the European Academy of Dermatology and Venereology. By comparison, patients in the control group had just one fewer daily dressing change in 2 weeks.
“You might say okay, but what does this mean in terms of time?” added Dr. Kiritsi. Using historical data on the time required for whole body care (Orphanet J Rare Dis. 2020 Jan 3. doi: 10.1186/s13023-019-1279-y), it was estimated that treatment with Oleogel-S10 was associated with an overall time-saving per week of 11 hours (6.6 hours for the patient and 4.4 hours for the caregiver) and use of the control gel was associated with an overall time-saving of 4 hours (2.4 hours for the patient and 1.6 hours for the caregiver).
“This is, for our patients, important,” said Dr. Kiritsi, as “it is time that they can spend doing something nice with the family” instead, avoiding the pain and distress associated with frequent dressing changes.
Approved in Europe, not in the United States
Oleogel-S10, classified as an herbal product, contains triterpenes derived from birch bark extract, which have been formulated with sunflower oil to form a gel.
Despite being approved for use in Europe, Oleogel-S10 has not yet been approved to treat EB in the United States. The FDA did not approve Amryt Pharma’s new drug application in February 2022. The application had included data from the EASE trial.
EASE included 223 patients with dystrophic or junctional EB, including 156 children, at 58 sites in 28 countries. As such, this makes it the largest treatment study in this rare genetic disease to date.
The trial had consisted of an initial 90-day, double-blind treatment period, during which time 109 patients had used Oleogel-S10 and 114 had used a control gel. This was followed by a 24-month open-label phase, during which time all remaining patients (n = 205) had used Oleogel-S10 on top of their standard of care.
Dr. Kiritsi summarized the main results of the EASE trial as follows.
- Complete healing of target wounds (primary endpoint) in 41.3% of patients treated with Oleogel-S10 and 28.9% of patients treated with the control gel (P = .013).
- Improved total body wound burden measured by both Epidermolysis Bullosa Disease Activity and Scarring Index and Body Surface Area Percentage scores.
- Reduced frequency of dressing changes (1 less per 2 weeks for Oleogel-S10 versus 0 less per 2 weeks for control gel).
- Improved pain among participants aged 4 years and older while their dressings were being changed.
- Reduced rates of wound infection (0.9% Oleogel-S10 vs. 4.4% control gel).
- Similar rates of treatment-emergent adverse events (24.8% vs. 22.8%, respectively), which were mostly deemed to be mild or moderate.
The EASE study – an important trial for EB
EASE is an important trial for EB, the study’s principal investigator Dédée Murrell, MD, DSc, University of New South Wales, Sydney, has pointed out previously.
“This was the first EB study to meet its primary endpoint and demonstrated a statistically significant acceleration of target wound healing by day 45,” Dr. Murrell said in a press release issued by Amryt Pharma to coincide with the online publication of the trial results.
“In addition, the favorable trends we see with key secondary endpoints such as reduced wound burden, pain, and frequency of dressing changes are considered as being very meaningful for patients,” Dr. Murrell said.
The EASE study was funded by Amryt Research Limited. Dr. Kiritsi reported receiving honoraria or consultation fees from Amryt, RHEACELL GmbH, and Fibrx Derm. She also acknowledged grant or research support from DEBRA International, EB Research Partnership, Fritz-Thyssen Foundation, German Research Foundation, and RHEACELL. Dr. Murrell has ties to Amryt and Amicus and is a co-owner of the patent for topical sirolimus for EB simplex.
A version of this article appeared on Medscape.com.
Additional when compared with a control gel.
In a final, post hoc analysis to come from the trial, 15 of 45 (33%) patients treated with Oleogel-S10 versus 5 of 48 (10.4%) treated with the control gel were reported as no longer needing daily dressing changes at 45 days of follow-up.
Moreover, the effect was sustained, with similar percentages of patients no longer requiring daily dressing changes at 60 days (34% vs. 13%, respectively) and 90 days (36% vs. 11%) of follow-up.
The mean reduction in daily dressing changes was 1.36 for Oleogel-S10 and 0.41 for the control gel (P = .005).
“Patients who, in the beginning, had daily dressing changes had almost three fewer dressing changes every 2 weeks if they were treated with Oleogel-S10,” Dimitra Kiritsi, MD, PhD, of the department of dermatology at the University of Freiburg (Germany), reported at the annual congress of the European Academy of Dermatology and Venereology. By comparison, patients in the control group had just one fewer daily dressing change in 2 weeks.
“You might say okay, but what does this mean in terms of time?” added Dr. Kiritsi. Using historical data on the time required for whole body care (Orphanet J Rare Dis. 2020 Jan 3. doi: 10.1186/s13023-019-1279-y), it was estimated that treatment with Oleogel-S10 was associated with an overall time-saving per week of 11 hours (6.6 hours for the patient and 4.4 hours for the caregiver) and use of the control gel was associated with an overall time-saving of 4 hours (2.4 hours for the patient and 1.6 hours for the caregiver).
“This is, for our patients, important,” said Dr. Kiritsi, as “it is time that they can spend doing something nice with the family” instead, avoiding the pain and distress associated with frequent dressing changes.
Approved in Europe, not in the United States
Oleogel-S10, classified as an herbal product, contains triterpenes derived from birch bark extract, which have been formulated with sunflower oil to form a gel.
Despite being approved for use in Europe, Oleogel-S10 has not yet been approved to treat EB in the United States. The FDA did not approve Amryt Pharma’s new drug application in February 2022. The application had included data from the EASE trial.
EASE included 223 patients with dystrophic or junctional EB, including 156 children, at 58 sites in 28 countries. As such, this makes it the largest treatment study in this rare genetic disease to date.
The trial had consisted of an initial 90-day, double-blind treatment period, during which time 109 patients had used Oleogel-S10 and 114 had used a control gel. This was followed by a 24-month open-label phase, during which time all remaining patients (n = 205) had used Oleogel-S10 on top of their standard of care.
Dr. Kiritsi summarized the main results of the EASE trial as follows.
- Complete healing of target wounds (primary endpoint) in 41.3% of patients treated with Oleogel-S10 and 28.9% of patients treated with the control gel (P = .013).
- Improved total body wound burden measured by both Epidermolysis Bullosa Disease Activity and Scarring Index and Body Surface Area Percentage scores.
- Reduced frequency of dressing changes (1 less per 2 weeks for Oleogel-S10 versus 0 less per 2 weeks for control gel).
- Improved pain among participants aged 4 years and older while their dressings were being changed.
- Reduced rates of wound infection (0.9% Oleogel-S10 vs. 4.4% control gel).
- Similar rates of treatment-emergent adverse events (24.8% vs. 22.8%, respectively), which were mostly deemed to be mild or moderate.
The EASE study – an important trial for EB
EASE is an important trial for EB, the study’s principal investigator Dédée Murrell, MD, DSc, University of New South Wales, Sydney, has pointed out previously.
“This was the first EB study to meet its primary endpoint and demonstrated a statistically significant acceleration of target wound healing by day 45,” Dr. Murrell said in a press release issued by Amryt Pharma to coincide with the online publication of the trial results.
“In addition, the favorable trends we see with key secondary endpoints such as reduced wound burden, pain, and frequency of dressing changes are considered as being very meaningful for patients,” Dr. Murrell said.
The EASE study was funded by Amryt Research Limited. Dr. Kiritsi reported receiving honoraria or consultation fees from Amryt, RHEACELL GmbH, and Fibrx Derm. She also acknowledged grant or research support from DEBRA International, EB Research Partnership, Fritz-Thyssen Foundation, German Research Foundation, and RHEACELL. Dr. Murrell has ties to Amryt and Amicus and is a co-owner of the patent for topical sirolimus for EB simplex.
A version of this article appeared on Medscape.com.
Additional when compared with a control gel.
In a final, post hoc analysis to come from the trial, 15 of 45 (33%) patients treated with Oleogel-S10 versus 5 of 48 (10.4%) treated with the control gel were reported as no longer needing daily dressing changes at 45 days of follow-up.
Moreover, the effect was sustained, with similar percentages of patients no longer requiring daily dressing changes at 60 days (34% vs. 13%, respectively) and 90 days (36% vs. 11%) of follow-up.
The mean reduction in daily dressing changes was 1.36 for Oleogel-S10 and 0.41 for the control gel (P = .005).
“Patients who, in the beginning, had daily dressing changes had almost three fewer dressing changes every 2 weeks if they were treated with Oleogel-S10,” Dimitra Kiritsi, MD, PhD, of the department of dermatology at the University of Freiburg (Germany), reported at the annual congress of the European Academy of Dermatology and Venereology. By comparison, patients in the control group had just one fewer daily dressing change in 2 weeks.
“You might say okay, but what does this mean in terms of time?” added Dr. Kiritsi. Using historical data on the time required for whole body care (Orphanet J Rare Dis. 2020 Jan 3. doi: 10.1186/s13023-019-1279-y), it was estimated that treatment with Oleogel-S10 was associated with an overall time-saving per week of 11 hours (6.6 hours for the patient and 4.4 hours for the caregiver) and use of the control gel was associated with an overall time-saving of 4 hours (2.4 hours for the patient and 1.6 hours for the caregiver).
“This is, for our patients, important,” said Dr. Kiritsi, as “it is time that they can spend doing something nice with the family” instead, avoiding the pain and distress associated with frequent dressing changes.
Approved in Europe, not in the United States
Oleogel-S10, classified as an herbal product, contains triterpenes derived from birch bark extract, which have been formulated with sunflower oil to form a gel.
Despite being approved for use in Europe, Oleogel-S10 has not yet been approved to treat EB in the United States. The FDA did not approve Amryt Pharma’s new drug application in February 2022. The application had included data from the EASE trial.
EASE included 223 patients with dystrophic or junctional EB, including 156 children, at 58 sites in 28 countries. As such, this makes it the largest treatment study in this rare genetic disease to date.
The trial had consisted of an initial 90-day, double-blind treatment period, during which time 109 patients had used Oleogel-S10 and 114 had used a control gel. This was followed by a 24-month open-label phase, during which time all remaining patients (n = 205) had used Oleogel-S10 on top of their standard of care.
Dr. Kiritsi summarized the main results of the EASE trial as follows.
- Complete healing of target wounds (primary endpoint) in 41.3% of patients treated with Oleogel-S10 and 28.9% of patients treated with the control gel (P = .013).
- Improved total body wound burden measured by both Epidermolysis Bullosa Disease Activity and Scarring Index and Body Surface Area Percentage scores.
- Reduced frequency of dressing changes (1 less per 2 weeks for Oleogel-S10 versus 0 less per 2 weeks for control gel).
- Improved pain among participants aged 4 years and older while their dressings were being changed.
- Reduced rates of wound infection (0.9% Oleogel-S10 vs. 4.4% control gel).
- Similar rates of treatment-emergent adverse events (24.8% vs. 22.8%, respectively), which were mostly deemed to be mild or moderate.
The EASE study – an important trial for EB
EASE is an important trial for EB, the study’s principal investigator Dédée Murrell, MD, DSc, University of New South Wales, Sydney, has pointed out previously.
“This was the first EB study to meet its primary endpoint and demonstrated a statistically significant acceleration of target wound healing by day 45,” Dr. Murrell said in a press release issued by Amryt Pharma to coincide with the online publication of the trial results.
“In addition, the favorable trends we see with key secondary endpoints such as reduced wound burden, pain, and frequency of dressing changes are considered as being very meaningful for patients,” Dr. Murrell said.
The EASE study was funded by Amryt Research Limited. Dr. Kiritsi reported receiving honoraria or consultation fees from Amryt, RHEACELL GmbH, and Fibrx Derm. She also acknowledged grant or research support from DEBRA International, EB Research Partnership, Fritz-Thyssen Foundation, German Research Foundation, and RHEACELL. Dr. Murrell has ties to Amryt and Amicus and is a co-owner of the patent for topical sirolimus for EB simplex.
A version of this article appeared on Medscape.com.
FROM THE EADV CONGRESS
AI flagged skin cancer with near-perfect accuracy, in UK study
. AI detected more than 99% of all skin cancers.
The researchers tested the AI by integrating it into a clinical diagnosis process – anticipating a future in which AI helps doctors catch skin cancer faster and triage patients.
Skin cancer is the most common cancer in the United States; one in five 5 Americans develop skin cancer by age 70. With melanoma, the deadliest skin cancer, the 5-year survival rate is better than 99% if caught early, though only about three-quarters of melanomas are caught at this stage.
Amid rising skin cancer rates come concerns that the number of dermatologists in the workforce isn’t keeping pace. That may be why the average wait time for a dermatology appointment is trending up – in 2022, it reached 34.5 days.
The study, which was presented at the European Academy of Dermatology and Venereology Congress recently and has not yet been published, involved 6,900 patients in the United Kingdom with suspected skin cancer. The patients had been referred by their primary care physicians. The researchers took images of the suspicious areas and uploaded them to the AI software. The AI’s assessment was then shared with a dermatologist.
“Note that the diagnosis issued by the AI was not hidden from the dermatologist doing the second assessment,” said lead researcher Kashini Andrew, MBBS, a dermatologist and specialist registrar at University Hospitals Birmingham NHS Foundation Trust.
Dr. Andrew acknowledged that this may have influenced the dermatologist’s opinion. But that’s the vision of how doctors could use this tool.
The AI caught 59 of 59 melanomas and 189 of 190 total skin cancers (99.5%). (The one case that the AI missed was caught by the dermatologist.) It also flagged 541 of 585 precancerous lesions (92.5%). This represented a big improvement from a 2021 version of the model, which detected 86% of melanomas, 84% of all skin cancers, and 54% of precancerous lesions.
Over the 10-month period of the study, the system saved more than 1,000 face-to-face consultations, freeing dermatologists’ time to catch more cancers and serve more patients.
Limitations
The patients in the study were from “one hospital in a single region of the UK,” and the sample was not large enough to allow broad statements to be made about the use of AI in dermatology, Dr. Andrew said.
But it can open the conversation. Roxana Daneshjou, MD, PhD, a dermatologist at Stanford (Calif.) University who has studied the pros and cons of AI in medicine, had some concerns. For one thing, doctors can gather more in-depth information during an in-person exam than AI can glean from a photo, Dr. Daneshjou noted. They can examine skin texture, gather patient history, and take photos with special lighting and magnification.

And the AI needs to get better at ruling out malignancy, Dr. Daneshjou said. In this study, the AI identified 75% of benign lesions, a decline from the earlier version. The researchers noted in the abstract that this is a potential trade-off for increased sensitivity.
“[Unnecessary] biopsies can clog up the health care system, cost money, and cause stress and scarring,” said Dr. Daneshjou. “You don’t want to increase the burden of that.”
Still, if AI software such as the kind used in the study proves just as accurate in larger, more diverse sample sizes, then it could be a powerful tool for triage, Dr. Daneshjou said. “If AI gets particularly good at finding malignancy and also ruling it out, that would be a win.”
A version of this article appeared on Medscape.com.
. AI detected more than 99% of all skin cancers.
The researchers tested the AI by integrating it into a clinical diagnosis process – anticipating a future in which AI helps doctors catch skin cancer faster and triage patients.
Skin cancer is the most common cancer in the United States; one in five 5 Americans develop skin cancer by age 70. With melanoma, the deadliest skin cancer, the 5-year survival rate is better than 99% if caught early, though only about three-quarters of melanomas are caught at this stage.
Amid rising skin cancer rates come concerns that the number of dermatologists in the workforce isn’t keeping pace. That may be why the average wait time for a dermatology appointment is trending up – in 2022, it reached 34.5 days.
The study, which was presented at the European Academy of Dermatology and Venereology Congress recently and has not yet been published, involved 6,900 patients in the United Kingdom with suspected skin cancer. The patients had been referred by their primary care physicians. The researchers took images of the suspicious areas and uploaded them to the AI software. The AI’s assessment was then shared with a dermatologist.
“Note that the diagnosis issued by the AI was not hidden from the dermatologist doing the second assessment,” said lead researcher Kashini Andrew, MBBS, a dermatologist and specialist registrar at University Hospitals Birmingham NHS Foundation Trust.
Dr. Andrew acknowledged that this may have influenced the dermatologist’s opinion. But that’s the vision of how doctors could use this tool.
The AI caught 59 of 59 melanomas and 189 of 190 total skin cancers (99.5%). (The one case that the AI missed was caught by the dermatologist.) It also flagged 541 of 585 precancerous lesions (92.5%). This represented a big improvement from a 2021 version of the model, which detected 86% of melanomas, 84% of all skin cancers, and 54% of precancerous lesions.
Over the 10-month period of the study, the system saved more than 1,000 face-to-face consultations, freeing dermatologists’ time to catch more cancers and serve more patients.
Limitations
The patients in the study were from “one hospital in a single region of the UK,” and the sample was not large enough to allow broad statements to be made about the use of AI in dermatology, Dr. Andrew said.
But it can open the conversation. Roxana Daneshjou, MD, PhD, a dermatologist at Stanford (Calif.) University who has studied the pros and cons of AI in medicine, had some concerns. For one thing, doctors can gather more in-depth information during an in-person exam than AI can glean from a photo, Dr. Daneshjou noted. They can examine skin texture, gather patient history, and take photos with special lighting and magnification.
And the AI needs to get better at ruling out malignancy, Dr. Daneshjou said. In this study, the AI identified 75% of benign lesions, a decline from the earlier version. The researchers noted in the abstract that this is a potential trade-off for increased sensitivity.
“[Unnecessary] biopsies can clog up the health care system, cost money, and cause stress and scarring,” said Dr. Daneshjou. “You don’t want to increase the burden of that.”
Still, if AI software such as the kind used in the study proves just as accurate in larger, more diverse sample sizes, then it could be a powerful tool for triage, Dr. Daneshjou said. “If AI gets particularly good at finding malignancy and also ruling it out, that would be a win.”
A version of this article appeared on Medscape.com.
. AI detected more than 99% of all skin cancers.
The researchers tested the AI by integrating it into a clinical diagnosis process – anticipating a future in which AI helps doctors catch skin cancer faster and triage patients.
Skin cancer is the most common cancer in the United States; one in five 5 Americans develop skin cancer by age 70. With melanoma, the deadliest skin cancer, the 5-year survival rate is better than 99% if caught early, though only about three-quarters of melanomas are caught at this stage.
Amid rising skin cancer rates come concerns that the number of dermatologists in the workforce isn’t keeping pace. That may be why the average wait time for a dermatology appointment is trending up – in 2022, it reached 34.5 days.
The study, which was presented at the European Academy of Dermatology and Venereology Congress recently and has not yet been published, involved 6,900 patients in the United Kingdom with suspected skin cancer. The patients had been referred by their primary care physicians. The researchers took images of the suspicious areas and uploaded them to the AI software. The AI’s assessment was then shared with a dermatologist.
“Note that the diagnosis issued by the AI was not hidden from the dermatologist doing the second assessment,” said lead researcher Kashini Andrew, MBBS, a dermatologist and specialist registrar at University Hospitals Birmingham NHS Foundation Trust.
Dr. Andrew acknowledged that this may have influenced the dermatologist’s opinion. But that’s the vision of how doctors could use this tool.
The AI caught 59 of 59 melanomas and 189 of 190 total skin cancers (99.5%). (The one case that the AI missed was caught by the dermatologist.) It also flagged 541 of 585 precancerous lesions (92.5%). This represented a big improvement from a 2021 version of the model, which detected 86% of melanomas, 84% of all skin cancers, and 54% of precancerous lesions.
Over the 10-month period of the study, the system saved more than 1,000 face-to-face consultations, freeing dermatologists’ time to catch more cancers and serve more patients.
Limitations
The patients in the study were from “one hospital in a single region of the UK,” and the sample was not large enough to allow broad statements to be made about the use of AI in dermatology, Dr. Andrew said.
But it can open the conversation. Roxana Daneshjou, MD, PhD, a dermatologist at Stanford (Calif.) University who has studied the pros and cons of AI in medicine, had some concerns. For one thing, doctors can gather more in-depth information during an in-person exam than AI can glean from a photo, Dr. Daneshjou noted. They can examine skin texture, gather patient history, and take photos with special lighting and magnification.
And the AI needs to get better at ruling out malignancy, Dr. Daneshjou said. In this study, the AI identified 75% of benign lesions, a decline from the earlier version. The researchers noted in the abstract that this is a potential trade-off for increased sensitivity.
“[Unnecessary] biopsies can clog up the health care system, cost money, and cause stress and scarring,” said Dr. Daneshjou. “You don’t want to increase the burden of that.”
Still, if AI software such as the kind used in the study proves just as accurate in larger, more diverse sample sizes, then it could be a powerful tool for triage, Dr. Daneshjou said. “If AI gets particularly good at finding malignancy and also ruling it out, that would be a win.”
A version of this article appeared on Medscape.com.
FROM THE EADV CONGRESS
Phase 3 trial supports topical JAK inhibitor for AD in young children
BERLIN – as previously shown in adolescents and adults for whom it already has an approved indication.
In this study – TRUE-AD3 – systemic exposure to ruxolitinib, which is selective for JAK1 and 2, was followed closely, and the low mean plasma concentrations “suggest systemic JAK inhibition is highly unlikely,” Lawrence F. Eichenfield, MD, professor of dermatology and pediatrics at the University of California, San Diego, said at the annual congress of the European Academy of Dermatology and Venereology.

For example, at a plasma concentration no greater than 27 nM in both younger and older patients at 4 weeks and again at 8 weeks, the systemic exposure was about a tenth of that (281 nM) previously associated with myelosuppression, he reported.
Given the boxed warning for oral JAK inhibitors, which was based largely on a 2022 study in adults with rheumatoid arthritis that associated tofacitinib, a nonspecific JAK inhibitor, with an increased risk of thrombotic events in adults already at risk for these events, safety was a focus of this phase 3 trial. The boxed warning is also in the labeling for topical ruxolitinib, 1.5% (Opzelura), approved for treating to mild to moderate atopic dermatitis in patients 12 years of age and older.
Dr. Eichenfield said there were no significant safety signals in the younger pediatric population. “There were no treatment-emergent adverse events suggestive of systemic JAK inhibition,” he said. This not only included the absence of serious infections, cardiac events, thromboses, or malignancies, but there was no signal of hematologic abnormalities, such as change in hemoglobin or neutrophil count.
Application site reactions
Rather, in the study of children ages 2-11, the only adverse events associated with topical ruxolitinib not observed in the control arm, which received the vehicle alone, were application site reactions, such as pain, erythema, and irritation. None of these occurred in more than 3% of those randomized to ruxolitinib regardless of dose.
Overall, in the trial, which randomized 329 patients ages from 2 to under 12 years with mild to moderate AD to ruxolitinib 1.5% cream, ruxolitinib 0.75% cream, or vehicle in a 2:2:1 fashion, there were just two (0.8%) discontinuations in the ruxolitinib groups (one in each dosing arm). There were none in the vehicle arm.
The safety supports an expansion of the AD indication for topical ruxolitinib in young children, because the rates of response were very similar to that seen in adolescents and adults in the previously published TRUE AD-1 and TRUE AD-2 trials, he said.
For the primary endpoint of Investigator’s Global Assessment (IGA) score of 0 (clear) or 1 (almost clear) with at least a 2 grade improvement in IGA score from baseline, the response rates were 56.5%, 36.6%, and 10.8% for ruxolitinib 1.5%, ruxolitinib 0.75%, and vehicle respectively, at 8 weeks (P < .0001 for both doses relative to vehicle).
For the secondary efficacy endpoint of 75% or greater clearance on the Eczema Area and Severity Index, the rates were 67.2%, 51.5%, and 15.4%, for ruxolitinib 1.5%, ruxolitinib 0.75%, and vehicle respectively. Again, the advantage of both doses of ruxolitinib relative to vehicle was highly statistically significant (P < .0001).
Control of itch, evaluated with the Numerical Rating Scale was only evaluated in children 6-2 because of concern of the reliability of reporting in younger children. Control was defined as at least a 4-point improvement from baseline. It was achieved by 43.4%, 37.5%, and 29.7% by week 8 in the arms receiving the higher dose of ruxolitinib, the lower dose, and vehicle, respectively. The median time to achieving itch control was 11 days, 13 days, and 23 days, respectively. For all of these endpoints, the separation of the curves was readily apparent within the first 2 weeks.
The efficacy and tolerability of ruxolitinib appeared to be similar in younger children (ages 2-6) relative to older children.
Extension study in children near completion
Most of the patients who participated in TRUE AD-3 have been rolled over to the open-label extension trial, which is nearing completion. Those originally randomized to vehicle have been rerandomized to the lower or higher dose of ruxolitinib.
While this trial was focused on ruxolitinib as monotherapy, Thrasyvoulos Tzellos, MD, head of the department of dermatology, Nordland Hospital Trust, Bødo, Norway, questioned whether this is will be how it will be used in clinical practice. With the increasing array of therapies for AD, the “concept of combination therapy becomes more and more relevant,” he said after Dr. Eichenfield’s presentation.
Questioning whether an effective nonsteroidal anti-inflammatory agent like ruxolitinib should be considered a first-line treatment in mild disease or an adjunctive treatment for AD of any severity, he suggested that it might be best considered within a combination.
Dr. Eichenfield agreed. “Once we get the drug approved in a controlled trial, I think we then figure out how to use it in clinical practice.” Based on his own use of ruxolitinib in adults, he noted that he has not seen this drug replace other therapies so much as provide another option for control.
“We have an increasing armamentarium of drugs to use for involvement in different areas of the body in order to get more long-term control of disease,” he said. As an effective topical nonsteroidal drug, he believes its addition to clinical care in younger children, if approved, will be meaningful.
Dr. Eichenfield disclosed financial relationships with more multiple pharmaceutical companies, including Incyte, the manufacturer of ruxolitinib cream that provided funding for the True-AD trials. Dr. Tzellos reported financial relationships with AbbVie and UCB.
BERLIN – as previously shown in adolescents and adults for whom it already has an approved indication.
In this study – TRUE-AD3 – systemic exposure to ruxolitinib, which is selective for JAK1 and 2, was followed closely, and the low mean plasma concentrations “suggest systemic JAK inhibition is highly unlikely,” Lawrence F. Eichenfield, MD, professor of dermatology and pediatrics at the University of California, San Diego, said at the annual congress of the European Academy of Dermatology and Venereology.
For example, at a plasma concentration no greater than 27 nM in both younger and older patients at 4 weeks and again at 8 weeks, the systemic exposure was about a tenth of that (281 nM) previously associated with myelosuppression, he reported.
Given the boxed warning for oral JAK inhibitors, which was based largely on a 2022 study in adults with rheumatoid arthritis that associated tofacitinib, a nonspecific JAK inhibitor, with an increased risk of thrombotic events in adults already at risk for these events, safety was a focus of this phase 3 trial. The boxed warning is also in the labeling for topical ruxolitinib, 1.5% (Opzelura), approved for treating to mild to moderate atopic dermatitis in patients 12 years of age and older.
Dr. Eichenfield said there were no significant safety signals in the younger pediatric population. “There were no treatment-emergent adverse events suggestive of systemic JAK inhibition,” he said. This not only included the absence of serious infections, cardiac events, thromboses, or malignancies, but there was no signal of hematologic abnormalities, such as change in hemoglobin or neutrophil count.
Application site reactions
Rather, in the study of children ages 2-11, the only adverse events associated with topical ruxolitinib not observed in the control arm, which received the vehicle alone, were application site reactions, such as pain, erythema, and irritation. None of these occurred in more than 3% of those randomized to ruxolitinib regardless of dose.
Overall, in the trial, which randomized 329 patients ages from 2 to under 12 years with mild to moderate AD to ruxolitinib 1.5% cream, ruxolitinib 0.75% cream, or vehicle in a 2:2:1 fashion, there were just two (0.8%) discontinuations in the ruxolitinib groups (one in each dosing arm). There were none in the vehicle arm.
The safety supports an expansion of the AD indication for topical ruxolitinib in young children, because the rates of response were very similar to that seen in adolescents and adults in the previously published TRUE AD-1 and TRUE AD-2 trials, he said.
For the primary endpoint of Investigator’s Global Assessment (IGA) score of 0 (clear) or 1 (almost clear) with at least a 2 grade improvement in IGA score from baseline, the response rates were 56.5%, 36.6%, and 10.8% for ruxolitinib 1.5%, ruxolitinib 0.75%, and vehicle respectively, at 8 weeks (P < .0001 for both doses relative to vehicle).
For the secondary efficacy endpoint of 75% or greater clearance on the Eczema Area and Severity Index, the rates were 67.2%, 51.5%, and 15.4%, for ruxolitinib 1.5%, ruxolitinib 0.75%, and vehicle respectively. Again, the advantage of both doses of ruxolitinib relative to vehicle was highly statistically significant (P < .0001).
Control of itch, evaluated with the Numerical Rating Scale was only evaluated in children 6-2 because of concern of the reliability of reporting in younger children. Control was defined as at least a 4-point improvement from baseline. It was achieved by 43.4%, 37.5%, and 29.7% by week 8 in the arms receiving the higher dose of ruxolitinib, the lower dose, and vehicle, respectively. The median time to achieving itch control was 11 days, 13 days, and 23 days, respectively. For all of these endpoints, the separation of the curves was readily apparent within the first 2 weeks.
The efficacy and tolerability of ruxolitinib appeared to be similar in younger children (ages 2-6) relative to older children.
Extension study in children near completion
Most of the patients who participated in TRUE AD-3 have been rolled over to the open-label extension trial, which is nearing completion. Those originally randomized to vehicle have been rerandomized to the lower or higher dose of ruxolitinib.
While this trial was focused on ruxolitinib as monotherapy, Thrasyvoulos Tzellos, MD, head of the department of dermatology, Nordland Hospital Trust, Bødo, Norway, questioned whether this is will be how it will be used in clinical practice. With the increasing array of therapies for AD, the “concept of combination therapy becomes more and more relevant,” he said after Dr. Eichenfield’s presentation.
Questioning whether an effective nonsteroidal anti-inflammatory agent like ruxolitinib should be considered a first-line treatment in mild disease or an adjunctive treatment for AD of any severity, he suggested that it might be best considered within a combination.
Dr. Eichenfield agreed. “Once we get the drug approved in a controlled trial, I think we then figure out how to use it in clinical practice.” Based on his own use of ruxolitinib in adults, he noted that he has not seen this drug replace other therapies so much as provide another option for control.
“We have an increasing armamentarium of drugs to use for involvement in different areas of the body in order to get more long-term control of disease,” he said. As an effective topical nonsteroidal drug, he believes its addition to clinical care in younger children, if approved, will be meaningful.
Dr. Eichenfield disclosed financial relationships with more multiple pharmaceutical companies, including Incyte, the manufacturer of ruxolitinib cream that provided funding for the True-AD trials. Dr. Tzellos reported financial relationships with AbbVie and UCB.
BERLIN – as previously shown in adolescents and adults for whom it already has an approved indication.
In this study – TRUE-AD3 – systemic exposure to ruxolitinib, which is selective for JAK1 and 2, was followed closely, and the low mean plasma concentrations “suggest systemic JAK inhibition is highly unlikely,” Lawrence F. Eichenfield, MD, professor of dermatology and pediatrics at the University of California, San Diego, said at the annual congress of the European Academy of Dermatology and Venereology.
For example, at a plasma concentration no greater than 27 nM in both younger and older patients at 4 weeks and again at 8 weeks, the systemic exposure was about a tenth of that (281 nM) previously associated with myelosuppression, he reported.
Given the boxed warning for oral JAK inhibitors, which was based largely on a 2022 study in adults with rheumatoid arthritis that associated tofacitinib, a nonspecific JAK inhibitor, with an increased risk of thrombotic events in adults already at risk for these events, safety was a focus of this phase 3 trial. The boxed warning is also in the labeling for topical ruxolitinib, 1.5% (Opzelura), approved for treating to mild to moderate atopic dermatitis in patients 12 years of age and older.
Dr. Eichenfield said there were no significant safety signals in the younger pediatric population. “There were no treatment-emergent adverse events suggestive of systemic JAK inhibition,” he said. This not only included the absence of serious infections, cardiac events, thromboses, or malignancies, but there was no signal of hematologic abnormalities, such as change in hemoglobin or neutrophil count.
Application site reactions
Rather, in the study of children ages 2-11, the only adverse events associated with topical ruxolitinib not observed in the control arm, which received the vehicle alone, were application site reactions, such as pain, erythema, and irritation. None of these occurred in more than 3% of those randomized to ruxolitinib regardless of dose.
Overall, in the trial, which randomized 329 patients ages from 2 to under 12 years with mild to moderate AD to ruxolitinib 1.5% cream, ruxolitinib 0.75% cream, or vehicle in a 2:2:1 fashion, there were just two (0.8%) discontinuations in the ruxolitinib groups (one in each dosing arm). There were none in the vehicle arm.
The safety supports an expansion of the AD indication for topical ruxolitinib in young children, because the rates of response were very similar to that seen in adolescents and adults in the previously published TRUE AD-1 and TRUE AD-2 trials, he said.
For the primary endpoint of Investigator’s Global Assessment (IGA) score of 0 (clear) or 1 (almost clear) with at least a 2 grade improvement in IGA score from baseline, the response rates were 56.5%, 36.6%, and 10.8% for ruxolitinib 1.5%, ruxolitinib 0.75%, and vehicle respectively, at 8 weeks (P < .0001 for both doses relative to vehicle).
For the secondary efficacy endpoint of 75% or greater clearance on the Eczema Area and Severity Index, the rates were 67.2%, 51.5%, and 15.4%, for ruxolitinib 1.5%, ruxolitinib 0.75%, and vehicle respectively. Again, the advantage of both doses of ruxolitinib relative to vehicle was highly statistically significant (P < .0001).
Control of itch, evaluated with the Numerical Rating Scale was only evaluated in children 6-2 because of concern of the reliability of reporting in younger children. Control was defined as at least a 4-point improvement from baseline. It was achieved by 43.4%, 37.5%, and 29.7% by week 8 in the arms receiving the higher dose of ruxolitinib, the lower dose, and vehicle, respectively. The median time to achieving itch control was 11 days, 13 days, and 23 days, respectively. For all of these endpoints, the separation of the curves was readily apparent within the first 2 weeks.
The efficacy and tolerability of ruxolitinib appeared to be similar in younger children (ages 2-6) relative to older children.
Extension study in children near completion
Most of the patients who participated in TRUE AD-3 have been rolled over to the open-label extension trial, which is nearing completion. Those originally randomized to vehicle have been rerandomized to the lower or higher dose of ruxolitinib.
While this trial was focused on ruxolitinib as monotherapy, Thrasyvoulos Tzellos, MD, head of the department of dermatology, Nordland Hospital Trust, Bødo, Norway, questioned whether this is will be how it will be used in clinical practice. With the increasing array of therapies for AD, the “concept of combination therapy becomes more and more relevant,” he said after Dr. Eichenfield’s presentation.
Questioning whether an effective nonsteroidal anti-inflammatory agent like ruxolitinib should be considered a first-line treatment in mild disease or an adjunctive treatment for AD of any severity, he suggested that it might be best considered within a combination.
Dr. Eichenfield agreed. “Once we get the drug approved in a controlled trial, I think we then figure out how to use it in clinical practice.” Based on his own use of ruxolitinib in adults, he noted that he has not seen this drug replace other therapies so much as provide another option for control.
“We have an increasing armamentarium of drugs to use for involvement in different areas of the body in order to get more long-term control of disease,” he said. As an effective topical nonsteroidal drug, he believes its addition to clinical care in younger children, if approved, will be meaningful.
Dr. Eichenfield disclosed financial relationships with more multiple pharmaceutical companies, including Incyte, the manufacturer of ruxolitinib cream that provided funding for the True-AD trials. Dr. Tzellos reported financial relationships with AbbVie and UCB.
AT THE EADV CONGRESS
Recombinant IL-2 shows potential in atopic dermatitis
BERLIN – A novel regulatory T cell–stimulating therapy appears to significantly improve atopic dermatitis in patients with moderate to severe disease and may even benefit quality of life, suggest results from a phase 1b trial.
The research was presented at the annual congress of the European Academy of Dermatology and Venereology.
, after which responders were observed out to 48 weeks. The higher dosage was associated with significant improvements in Eczema Area and Severity Index (EASI) and Body Surface Area (BSA) scores, which were maintained over the course of the study, as well as trends for improved patient-reported outcomes.
“This is the first study to demonstrate the therapeutic potential of rezpegaldesleukin,” said presenter Jonathan Silverberg, MD, PhD, MPH, professor of dermatology and director of clinical research at George Washington University, Washington. He added, “These may be some of the most compelling data to date for the field, proving that, at a high level, if you causally increase regulator T cells, you will take down inflammation and improve a disease state.
“For me, this is proof of concept for so many things, and it gets me very excited.”
Dr. Silverberg noted that with the response maintained out to 48 weeks, despite stopping therapy at week 12, the “hope” with the approach of inducing regulator T cells “is that we could induce tolerance and that we could have some potential for disease modification.”
He continued, “Maybe I daren’t use the word ‘cure,’ but can we at least get to something that is truly remitted, where they can stop the drug and maintain that response?”
Dr. Silverberg said rezpegaldesleukin is now being evaluated in a phase 2b study for moderate to severe atopic dermatitis, and a phase 2b trial for alopecia areata is in development.
Tiago dos Reis Matos, MD, PhD, MSc, Amsterdam University Medical Centers, who was not involved in the study, told this news organization that “recombinant human interleukin-2 is an original therapy.”
Instead of blocking or inhibiting inflammation, it stimulates the patient’s immune system to “restore a healthy balance.”
He explained that it “stimulates regulatory T cells, which can be seen as the Peace Corps of the immune system, responsible for maintaining the equilibrium and avoiding uncontrolled inflammation.”
At the meeting, Dr. Silverberg told the audience that although they are the “beneficiaries of riches of new advances” in atopic dermatitis, “still, many observational studies have shown that the majority of patients do not achieve adequate control by the end of their induction periods and clinical trials, in the real world,” with currently available treatments.
Moreover, “there are challenges that come up with any of the different therapies,” he said, with adverse effects an important issue. For example, biologic therapies are associated with conjunctivitis, facial erythema, and arthralgia, and there are boxed warnings for Janus kinase inhibitors.
Dr. Silverberg continued, “Even patients with a favorable response can experience a loss of disease control when they come off therapy.” Consequently, “new strategies are certainly welcome that could potentially induce both deep and potentially therapy-free remission.”
To those ends, he explained that regulatory T cells play a central role in immune homeostasis but have not been “therapeutically relevant until very recently,” when it was posited that increasing their function can “induce that homeostasis, to normalize the inflammatory cascades” seen in a range of conditions, including atopic dermatitis.
Rezpegaldesleukin has high selectivity for regulatory T cells, without causing activation of effector T cells, and has been shown to increase cell numbers in a dose-dependent manner that is sustained for up to 30 days.
The current study involved patients aged 18-70 years with moderate to severe atopic dermatitis and a history of inadequate responses or intolerance to topical medications, and an EASI score ≥ 16.
Participants were randomly assigned to receive subcutaneous rezpegaldesleukin 12 mcg/kg or 24 mcg/kg or placebo every 2 weeks for 12 weeks. They then discontinued treatment and were followed up until week 19, when responders, defined as having a reduction in EASI score ≥ 50%, continued follow-up out to week 48.
Seventeen patients were randomized to higher-dose rezpegaldesleukin, whereas 16 received the lower dose and 10 were assigned to placebo. Dr. Silverberg said that the three groups were “fairly well balanced,” with “fairly good representation” across age, race, and ethnicity groups.
The mean baseline EASI score was between 21.9 and 23.7, and the Validated Investigator Global Assessment for Atopic Dermatitis (vIGA-AD) suggested that there was an even split between moderate and severe atopic dermatitis, although the higher-dose rezpegaldesleukin group had more patients with moderate disease.
By week 12, rezpegaldesleukin was associated with significantly greater improvements in EASI scores vs. placebo. Patients on the higher dose had a mean 83% improvement over baseline vs. 65% with the lower dose and 47% with placebo (P = .002 for the higher dose vs. placebo).
Crucially, these differences were maintained up to week 48 in patients, particularly in the higher-dose group.
There was also a nonsignificant increase in the proportion of patients who achieved a reduction in EASI scores ≥ 75% over baseline with the active drug: 41% at week 12 with higher-dose rezpegaldesleukin, 25% with the lower dose, and 20% with placebo. Again, the benefit was maintained up to week 48.
The mean improvement in BSA score from baseline with rezpegaldesleukin was significantly greater than that seen with placebo, at 72% with the higher dose, 55% with the lower dose, and 36% with placebo (P = .0158 for the higher dose vs. placebo).
Although improvements in vIGA-AD scores over baseline with rezpegaldesleukin were not substantial at week 12, by week 48 there was a marked difference between higher-dose rezpegaldesleukin and placebo, with 40.0% of patients responding to the drug vs. 0% in the latter group.
A similar pattern was seen for the Itch Numeric Rating Scale, in which 55.6% of patients treated with higher-dose rezpegaldesleukin responding by week 48, compared with 0% of those who received placebo.
Greater improvements in the Dermatology Life Quality Index (DLQI) and Patient Oriented Eczema Measure (POEM) over baseline with higher-dose rezpegaldesleukin vs. plain placebo were also noted, despite a strong response in the latter group.
Dr. Silverberg reported that all treatment-emergent adverse effects in the two rezpegaldesleukin treatment arms were mild to moderate, with no severe or serious events observed.
The most common adverse events were mild to moderate injection-site reactions, seen in 75.0% of the lower-dose rezpegaldesleukin group and 58.8% the of higher-dose group. There were no cases of conjunctivitis.
The study was sponsored by Eli Lilly and Company in collaboration with Nektar Therapeutics.
Dr. Silverberg declares relationships with AbbVie, Alamar, Aldena, Amgen, AOBiome, Arcutis, Arena, Asana, ASLAN, BioMX, Biosion, Bodewell, Boehringer-Ingelheim, Bristol-Myers Squibb, Cara, Castle Biosciences, Celgene, Connect Biopharma, CorEvitas, Dermavant, DermTech, Eli Lilly, Galderma, GlaxoSmithKline, Incyte, Kiniksa, LEO Pharma, Nektar, Novartis, Optum, Pfizer, RAPT, Recludix, Regeneron, Sanofi-Genzyme, Shaperon, Target RWE, Union, and UpToDate.
A version of this article appeared on Medscape.com.
BERLIN – A novel regulatory T cell–stimulating therapy appears to significantly improve atopic dermatitis in patients with moderate to severe disease and may even benefit quality of life, suggest results from a phase 1b trial.
The research was presented at the annual congress of the European Academy of Dermatology and Venereology.
, after which responders were observed out to 48 weeks. The higher dosage was associated with significant improvements in Eczema Area and Severity Index (EASI) and Body Surface Area (BSA) scores, which were maintained over the course of the study, as well as trends for improved patient-reported outcomes.
“This is the first study to demonstrate the therapeutic potential of rezpegaldesleukin,” said presenter Jonathan Silverberg, MD, PhD, MPH, professor of dermatology and director of clinical research at George Washington University, Washington. He added, “These may be some of the most compelling data to date for the field, proving that, at a high level, if you causally increase regulator T cells, you will take down inflammation and improve a disease state.
“For me, this is proof of concept for so many things, and it gets me very excited.”
Dr. Silverberg noted that with the response maintained out to 48 weeks, despite stopping therapy at week 12, the “hope” with the approach of inducing regulator T cells “is that we could induce tolerance and that we could have some potential for disease modification.”
He continued, “Maybe I daren’t use the word ‘cure,’ but can we at least get to something that is truly remitted, where they can stop the drug and maintain that response?”
Dr. Silverberg said rezpegaldesleukin is now being evaluated in a phase 2b study for moderate to severe atopic dermatitis, and a phase 2b trial for alopecia areata is in development.
Tiago dos Reis Matos, MD, PhD, MSc, Amsterdam University Medical Centers, who was not involved in the study, told this news organization that “recombinant human interleukin-2 is an original therapy.”
Instead of blocking or inhibiting inflammation, it stimulates the patient’s immune system to “restore a healthy balance.”
He explained that it “stimulates regulatory T cells, which can be seen as the Peace Corps of the immune system, responsible for maintaining the equilibrium and avoiding uncontrolled inflammation.”
At the meeting, Dr. Silverberg told the audience that although they are the “beneficiaries of riches of new advances” in atopic dermatitis, “still, many observational studies have shown that the majority of patients do not achieve adequate control by the end of their induction periods and clinical trials, in the real world,” with currently available treatments.
Moreover, “there are challenges that come up with any of the different therapies,” he said, with adverse effects an important issue. For example, biologic therapies are associated with conjunctivitis, facial erythema, and arthralgia, and there are boxed warnings for Janus kinase inhibitors.
Dr. Silverberg continued, “Even patients with a favorable response can experience a loss of disease control when they come off therapy.” Consequently, “new strategies are certainly welcome that could potentially induce both deep and potentially therapy-free remission.”
To those ends, he explained that regulatory T cells play a central role in immune homeostasis but have not been “therapeutically relevant until very recently,” when it was posited that increasing their function can “induce that homeostasis, to normalize the inflammatory cascades” seen in a range of conditions, including atopic dermatitis.
Rezpegaldesleukin has high selectivity for regulatory T cells, without causing activation of effector T cells, and has been shown to increase cell numbers in a dose-dependent manner that is sustained for up to 30 days.
The current study involved patients aged 18-70 years with moderate to severe atopic dermatitis and a history of inadequate responses or intolerance to topical medications, and an EASI score ≥ 16.
Participants were randomly assigned to receive subcutaneous rezpegaldesleukin 12 mcg/kg or 24 mcg/kg or placebo every 2 weeks for 12 weeks. They then discontinued treatment and were followed up until week 19, when responders, defined as having a reduction in EASI score ≥ 50%, continued follow-up out to week 48.
Seventeen patients were randomized to higher-dose rezpegaldesleukin, whereas 16 received the lower dose and 10 were assigned to placebo. Dr. Silverberg said that the three groups were “fairly well balanced,” with “fairly good representation” across age, race, and ethnicity groups.
The mean baseline EASI score was between 21.9 and 23.7, and the Validated Investigator Global Assessment for Atopic Dermatitis (vIGA-AD) suggested that there was an even split between moderate and severe atopic dermatitis, although the higher-dose rezpegaldesleukin group had more patients with moderate disease.
By week 12, rezpegaldesleukin was associated with significantly greater improvements in EASI scores vs. placebo. Patients on the higher dose had a mean 83% improvement over baseline vs. 65% with the lower dose and 47% with placebo (P = .002 for the higher dose vs. placebo).
Crucially, these differences were maintained up to week 48 in patients, particularly in the higher-dose group.
There was also a nonsignificant increase in the proportion of patients who achieved a reduction in EASI scores ≥ 75% over baseline with the active drug: 41% at week 12 with higher-dose rezpegaldesleukin, 25% with the lower dose, and 20% with placebo. Again, the benefit was maintained up to week 48.
The mean improvement in BSA score from baseline with rezpegaldesleukin was significantly greater than that seen with placebo, at 72% with the higher dose, 55% with the lower dose, and 36% with placebo (P = .0158 for the higher dose vs. placebo).
Although improvements in vIGA-AD scores over baseline with rezpegaldesleukin were not substantial at week 12, by week 48 there was a marked difference between higher-dose rezpegaldesleukin and placebo, with 40.0% of patients responding to the drug vs. 0% in the latter group.
A similar pattern was seen for the Itch Numeric Rating Scale, in which 55.6% of patients treated with higher-dose rezpegaldesleukin responding by week 48, compared with 0% of those who received placebo.
Greater improvements in the Dermatology Life Quality Index (DLQI) and Patient Oriented Eczema Measure (POEM) over baseline with higher-dose rezpegaldesleukin vs. plain placebo were also noted, despite a strong response in the latter group.
Dr. Silverberg reported that all treatment-emergent adverse effects in the two rezpegaldesleukin treatment arms were mild to moderate, with no severe or serious events observed.
The most common adverse events were mild to moderate injection-site reactions, seen in 75.0% of the lower-dose rezpegaldesleukin group and 58.8% the of higher-dose group. There were no cases of conjunctivitis.
The study was sponsored by Eli Lilly and Company in collaboration with Nektar Therapeutics.
Dr. Silverberg declares relationships with AbbVie, Alamar, Aldena, Amgen, AOBiome, Arcutis, Arena, Asana, ASLAN, BioMX, Biosion, Bodewell, Boehringer-Ingelheim, Bristol-Myers Squibb, Cara, Castle Biosciences, Celgene, Connect Biopharma, CorEvitas, Dermavant, DermTech, Eli Lilly, Galderma, GlaxoSmithKline, Incyte, Kiniksa, LEO Pharma, Nektar, Novartis, Optum, Pfizer, RAPT, Recludix, Regeneron, Sanofi-Genzyme, Shaperon, Target RWE, Union, and UpToDate.
A version of this article appeared on Medscape.com.
BERLIN – A novel regulatory T cell–stimulating therapy appears to significantly improve atopic dermatitis in patients with moderate to severe disease and may even benefit quality of life, suggest results from a phase 1b trial.
The research was presented at the annual congress of the European Academy of Dermatology and Venereology.
, after which responders were observed out to 48 weeks. The higher dosage was associated with significant improvements in Eczema Area and Severity Index (EASI) and Body Surface Area (BSA) scores, which were maintained over the course of the study, as well as trends for improved patient-reported outcomes.
“This is the first study to demonstrate the therapeutic potential of rezpegaldesleukin,” said presenter Jonathan Silverberg, MD, PhD, MPH, professor of dermatology and director of clinical research at George Washington University, Washington. He added, “These may be some of the most compelling data to date for the field, proving that, at a high level, if you causally increase regulator T cells, you will take down inflammation and improve a disease state.
“For me, this is proof of concept for so many things, and it gets me very excited.”
Dr. Silverberg noted that with the response maintained out to 48 weeks, despite stopping therapy at week 12, the “hope” with the approach of inducing regulator T cells “is that we could induce tolerance and that we could have some potential for disease modification.”
He continued, “Maybe I daren’t use the word ‘cure,’ but can we at least get to something that is truly remitted, where they can stop the drug and maintain that response?”
Dr. Silverberg said rezpegaldesleukin is now being evaluated in a phase 2b study for moderate to severe atopic dermatitis, and a phase 2b trial for alopecia areata is in development.
Tiago dos Reis Matos, MD, PhD, MSc, Amsterdam University Medical Centers, who was not involved in the study, told this news organization that “recombinant human interleukin-2 is an original therapy.”
Instead of blocking or inhibiting inflammation, it stimulates the patient’s immune system to “restore a healthy balance.”
He explained that it “stimulates regulatory T cells, which can be seen as the Peace Corps of the immune system, responsible for maintaining the equilibrium and avoiding uncontrolled inflammation.”
At the meeting, Dr. Silverberg told the audience that although they are the “beneficiaries of riches of new advances” in atopic dermatitis, “still, many observational studies have shown that the majority of patients do not achieve adequate control by the end of their induction periods and clinical trials, in the real world,” with currently available treatments.
Moreover, “there are challenges that come up with any of the different therapies,” he said, with adverse effects an important issue. For example, biologic therapies are associated with conjunctivitis, facial erythema, and arthralgia, and there are boxed warnings for Janus kinase inhibitors.
Dr. Silverberg continued, “Even patients with a favorable response can experience a loss of disease control when they come off therapy.” Consequently, “new strategies are certainly welcome that could potentially induce both deep and potentially therapy-free remission.”
To those ends, he explained that regulatory T cells play a central role in immune homeostasis but have not been “therapeutically relevant until very recently,” when it was posited that increasing their function can “induce that homeostasis, to normalize the inflammatory cascades” seen in a range of conditions, including atopic dermatitis.
Rezpegaldesleukin has high selectivity for regulatory T cells, without causing activation of effector T cells, and has been shown to increase cell numbers in a dose-dependent manner that is sustained for up to 30 days.
The current study involved patients aged 18-70 years with moderate to severe atopic dermatitis and a history of inadequate responses or intolerance to topical medications, and an EASI score ≥ 16.
Participants were randomly assigned to receive subcutaneous rezpegaldesleukin 12 mcg/kg or 24 mcg/kg or placebo every 2 weeks for 12 weeks. They then discontinued treatment and were followed up until week 19, when responders, defined as having a reduction in EASI score ≥ 50%, continued follow-up out to week 48.
Seventeen patients were randomized to higher-dose rezpegaldesleukin, whereas 16 received the lower dose and 10 were assigned to placebo. Dr. Silverberg said that the three groups were “fairly well balanced,” with “fairly good representation” across age, race, and ethnicity groups.
The mean baseline EASI score was between 21.9 and 23.7, and the Validated Investigator Global Assessment for Atopic Dermatitis (vIGA-AD) suggested that there was an even split between moderate and severe atopic dermatitis, although the higher-dose rezpegaldesleukin group had more patients with moderate disease.
By week 12, rezpegaldesleukin was associated with significantly greater improvements in EASI scores vs. placebo. Patients on the higher dose had a mean 83% improvement over baseline vs. 65% with the lower dose and 47% with placebo (P = .002 for the higher dose vs. placebo).
Crucially, these differences were maintained up to week 48 in patients, particularly in the higher-dose group.
There was also a nonsignificant increase in the proportion of patients who achieved a reduction in EASI scores ≥ 75% over baseline with the active drug: 41% at week 12 with higher-dose rezpegaldesleukin, 25% with the lower dose, and 20% with placebo. Again, the benefit was maintained up to week 48.
The mean improvement in BSA score from baseline with rezpegaldesleukin was significantly greater than that seen with placebo, at 72% with the higher dose, 55% with the lower dose, and 36% with placebo (P = .0158 for the higher dose vs. placebo).
Although improvements in vIGA-AD scores over baseline with rezpegaldesleukin were not substantial at week 12, by week 48 there was a marked difference between higher-dose rezpegaldesleukin and placebo, with 40.0% of patients responding to the drug vs. 0% in the latter group.
A similar pattern was seen for the Itch Numeric Rating Scale, in which 55.6% of patients treated with higher-dose rezpegaldesleukin responding by week 48, compared with 0% of those who received placebo.
Greater improvements in the Dermatology Life Quality Index (DLQI) and Patient Oriented Eczema Measure (POEM) over baseline with higher-dose rezpegaldesleukin vs. plain placebo were also noted, despite a strong response in the latter group.
Dr. Silverberg reported that all treatment-emergent adverse effects in the two rezpegaldesleukin treatment arms were mild to moderate, with no severe or serious events observed.
The most common adverse events were mild to moderate injection-site reactions, seen in 75.0% of the lower-dose rezpegaldesleukin group and 58.8% the of higher-dose group. There were no cases of conjunctivitis.
The study was sponsored by Eli Lilly and Company in collaboration with Nektar Therapeutics.
Dr. Silverberg declares relationships with AbbVie, Alamar, Aldena, Amgen, AOBiome, Arcutis, Arena, Asana, ASLAN, BioMX, Biosion, Bodewell, Boehringer-Ingelheim, Bristol-Myers Squibb, Cara, Castle Biosciences, Celgene, Connect Biopharma, CorEvitas, Dermavant, DermTech, Eli Lilly, Galderma, GlaxoSmithKline, Incyte, Kiniksa, LEO Pharma, Nektar, Novartis, Optum, Pfizer, RAPT, Recludix, Regeneron, Sanofi-Genzyme, Shaperon, Target RWE, Union, and UpToDate.
A version of this article appeared on Medscape.com.
AT THE EADV CONGRESS