User login
Newborn Screening for Spinal Muscular Atrophy in the United States
Click here to read the supplement

Newborn screening for spinal muscular atrophy (SMA) is essential to achieve optimal outcomes for patients with SMA by facilitating early identification and treatment before the onset of irreversible motor neuron loss.
Click here to read the supplement
Newborn screening for spinal muscular atrophy (SMA) is essential to achieve optimal outcomes for patients with SMA by facilitating early identification and treatment before the onset of irreversible motor neuron loss.
Click here to read the supplement
Newborn screening for spinal muscular atrophy (SMA) is essential to achieve optimal outcomes for patients with SMA by facilitating early identification and treatment before the onset of irreversible motor neuron loss.
Increased AD severity linked to more frequent baths and showers, but not with duration
of showers or baths, results from a prospective observational study found.

“Patients may benefit most from counseling on showering or bathing once daily and regularly applying moisturizer after showering or bathing,” one of the study authors, Uros Rakita, MSc, told this news organization. “Recommending less than daily shower frequencies or counseling on specific shower durations may not be necessary.”
During a late-breaking abstract session at the Revolutionizing Atopic Dermatitis symposium, Mr. Rakita, a fourth-year student at Chicago Medical School, North Chicago, presented findings from a prospective, practice-based dermatology study that investigated the longitudinal relationship between different bathing practices and AD severity to help inform patient counseling about optimal bathing practices.
“AD is a chronic, inflammatory skin condition with a diverse set of environmental triggers and exacerbating factors,” Mr. Rakita said during the meeting. “Maintaining adequate skin hydration, skin hygiene, and avoiding triggers are key aspects of AD management across all disease severities. Therefore, understanding optimal shower or bath and moisturizing practices is essential.” In fact, he added, “bathing has been shown to not only hydrate the skin, but also to improve symptoms, remove allergens, and decrease [Staphylococcus] aureus colonization. However, at the same time, concern exists for the potential of inappropriate shower or bathing frequency or durations, as well as inconsistent moisturizer application to worsen disease severity and potentially compromise disease management.”
He noted that current guidelines on bathing frequency and duration among AD patients lack consensus, are limited, and are largely based on studies of pediatric populations.

Mr. Rakita, along with primary study author Jonathan I. Silverberg, MD, PhD, MPH, director of clinical research in the division of dermatology at George Washington University, Washington, and Trisha Kaundinya, a medical student at Northwestern University, Chicago, prospectively evaluated 509 adults with AD who made an average of 2.3 visits at a single dermatology clinic between 2013 and 2020. At each visit, severity of AD signs and symptoms, as well as bathing and moisturizing practices, were assessed.
AD severity was assessed using the objective component of Scoring Atopic Dermatitis (o-SCORAD), intensity of pruritus in the past 3 days (SCORAD-itch), Eczema Area and Severity Index (EASI), Patient-Oriented Eczema Measure (POEM), and Dermatology Life Quality Index (DLQI). The researchers constructed repeated measures regression models to examine associations of bathing and moisturizing practices with change in AD severity outcome measure scores over time. Multivariable models controlled for age, sex, and race.
In adjusted linear regression models, showering or bathing more than once a day versus once daily was associated with significantly higher scores for SCORAD-itch (0.74; P = .0456), o-SCORAD (4.27; P = .0171), EASI (4.20; P = .0028), POEM (2.61; P = .0021), and DLQI (2.77; P = .0004).
The researchers also found that consistent application of moisturizer after the shower or bath was associated with significantly lower scores for o-SCORAD (–7.22; P < .0001), EASI (–3.91; P = .001) and POEM (–2.68; P = .0002), compared against not applying moisturizer after a shower or bath. However, shower or bath duration of more than, compared against fewer than, 15 minutes was not associated with significantly lower scores for o-SCORAD (1.26; P = .2868), SCORAD-itch (0.17; P = .4987), EASI (0.85; P = .3454), POEM (0.24; P = .6627) or DLQI (–0.40; P = .4318).
“Interestingly, this pattern was present when the reference shower or bath durations were under 10 minutes as well as under 5 minutes,” Mr. Rakita said. “Also, shower or bath frequencies of less than daily, relative to daily frequencies, were not significantly related to longitudinal AD severity.”
Mr. Rakita acknowledged certain limitations of the study, including the fact that the researchers did not examine the potential influence of specific soap and moisturizing products, water hardness, or other bathing features such as water temperature and bath additives.
Lawrence J. Green, MD, who was asked to comment on the study, said that he was not surprised by the finding that moisturizing after bathing improved AD signs and symptoms. “On the other hand, a long-held belief that longer duration of shower/bath time worsens AD was not found to be true,” said Dr. Green, a dermatologist who practices in Rockville, Md., and is also clinical professor of dermatology at George Washington University.
“This provides useful information for practicing dermatologists who wish to provide evidenced-based education about moisturizing and bathing to their AD patients,” he said.
The study was supported by the Agency for Healthcare Research and Quality and the Dermatology Foundation. Dr. Silverberg disclosed that he is a consultant to numerous pharmaceutical companies, receives fees for non-CME/CE services from Eli Lilly, Leo Pharma, Pfizer, Regeneron, and Sanofi Genzyme, as well as contracted research fees from Galderma. Dr. Green disclosed that he is a speaker, consultant, or investigator for numerous pharmaceutical companies. There were no other disclosures.
A version of this article first appeared on Medscape.com.
of showers or baths, results from a prospective observational study found.
“Patients may benefit most from counseling on showering or bathing once daily and regularly applying moisturizer after showering or bathing,” one of the study authors, Uros Rakita, MSc, told this news organization. “Recommending less than daily shower frequencies or counseling on specific shower durations may not be necessary.”
During a late-breaking abstract session at the Revolutionizing Atopic Dermatitis symposium, Mr. Rakita, a fourth-year student at Chicago Medical School, North Chicago, presented findings from a prospective, practice-based dermatology study that investigated the longitudinal relationship between different bathing practices and AD severity to help inform patient counseling about optimal bathing practices.
“AD is a chronic, inflammatory skin condition with a diverse set of environmental triggers and exacerbating factors,” Mr. Rakita said during the meeting. “Maintaining adequate skin hydration, skin hygiene, and avoiding triggers are key aspects of AD management across all disease severities. Therefore, understanding optimal shower or bath and moisturizing practices is essential.” In fact, he added, “bathing has been shown to not only hydrate the skin, but also to improve symptoms, remove allergens, and decrease [Staphylococcus] aureus colonization. However, at the same time, concern exists for the potential of inappropriate shower or bathing frequency or durations, as well as inconsistent moisturizer application to worsen disease severity and potentially compromise disease management.”
He noted that current guidelines on bathing frequency and duration among AD patients lack consensus, are limited, and are largely based on studies of pediatric populations.
Mr. Rakita, along with primary study author Jonathan I. Silverberg, MD, PhD, MPH, director of clinical research in the division of dermatology at George Washington University, Washington, and Trisha Kaundinya, a medical student at Northwestern University, Chicago, prospectively evaluated 509 adults with AD who made an average of 2.3 visits at a single dermatology clinic between 2013 and 2020. At each visit, severity of AD signs and symptoms, as well as bathing and moisturizing practices, were assessed.
AD severity was assessed using the objective component of Scoring Atopic Dermatitis (o-SCORAD), intensity of pruritus in the past 3 days (SCORAD-itch), Eczema Area and Severity Index (EASI), Patient-Oriented Eczema Measure (POEM), and Dermatology Life Quality Index (DLQI). The researchers constructed repeated measures regression models to examine associations of bathing and moisturizing practices with change in AD severity outcome measure scores over time. Multivariable models controlled for age, sex, and race.
In adjusted linear regression models, showering or bathing more than once a day versus once daily was associated with significantly higher scores for SCORAD-itch (0.74; P = .0456), o-SCORAD (4.27; P = .0171), EASI (4.20; P = .0028), POEM (2.61; P = .0021), and DLQI (2.77; P = .0004).
The researchers also found that consistent application of moisturizer after the shower or bath was associated with significantly lower scores for o-SCORAD (–7.22; P < .0001), EASI (–3.91; P = .001) and POEM (–2.68; P = .0002), compared against not applying moisturizer after a shower or bath. However, shower or bath duration of more than, compared against fewer than, 15 minutes was not associated with significantly lower scores for o-SCORAD (1.26; P = .2868), SCORAD-itch (0.17; P = .4987), EASI (0.85; P = .3454), POEM (0.24; P = .6627) or DLQI (–0.40; P = .4318).
“Interestingly, this pattern was present when the reference shower or bath durations were under 10 minutes as well as under 5 minutes,” Mr. Rakita said. “Also, shower or bath frequencies of less than daily, relative to daily frequencies, were not significantly related to longitudinal AD severity.”
Mr. Rakita acknowledged certain limitations of the study, including the fact that the researchers did not examine the potential influence of specific soap and moisturizing products, water hardness, or other bathing features such as water temperature and bath additives.
Lawrence J. Green, MD, who was asked to comment on the study, said that he was not surprised by the finding that moisturizing after bathing improved AD signs and symptoms. “On the other hand, a long-held belief that longer duration of shower/bath time worsens AD was not found to be true,” said Dr. Green, a dermatologist who practices in Rockville, Md., and is also clinical professor of dermatology at George Washington University.
“This provides useful information for practicing dermatologists who wish to provide evidenced-based education about moisturizing and bathing to their AD patients,” he said.
The study was supported by the Agency for Healthcare Research and Quality and the Dermatology Foundation. Dr. Silverberg disclosed that he is a consultant to numerous pharmaceutical companies, receives fees for non-CME/CE services from Eli Lilly, Leo Pharma, Pfizer, Regeneron, and Sanofi Genzyme, as well as contracted research fees from Galderma. Dr. Green disclosed that he is a speaker, consultant, or investigator for numerous pharmaceutical companies. There were no other disclosures.
A version of this article first appeared on Medscape.com.
of showers or baths, results from a prospective observational study found.
“Patients may benefit most from counseling on showering or bathing once daily and regularly applying moisturizer after showering or bathing,” one of the study authors, Uros Rakita, MSc, told this news organization. “Recommending less than daily shower frequencies or counseling on specific shower durations may not be necessary.”
During a late-breaking abstract session at the Revolutionizing Atopic Dermatitis symposium, Mr. Rakita, a fourth-year student at Chicago Medical School, North Chicago, presented findings from a prospective, practice-based dermatology study that investigated the longitudinal relationship between different bathing practices and AD severity to help inform patient counseling about optimal bathing practices.
“AD is a chronic, inflammatory skin condition with a diverse set of environmental triggers and exacerbating factors,” Mr. Rakita said during the meeting. “Maintaining adequate skin hydration, skin hygiene, and avoiding triggers are key aspects of AD management across all disease severities. Therefore, understanding optimal shower or bath and moisturizing practices is essential.” In fact, he added, “bathing has been shown to not only hydrate the skin, but also to improve symptoms, remove allergens, and decrease [Staphylococcus] aureus colonization. However, at the same time, concern exists for the potential of inappropriate shower or bathing frequency or durations, as well as inconsistent moisturizer application to worsen disease severity and potentially compromise disease management.”
He noted that current guidelines on bathing frequency and duration among AD patients lack consensus, are limited, and are largely based on studies of pediatric populations.
Mr. Rakita, along with primary study author Jonathan I. Silverberg, MD, PhD, MPH, director of clinical research in the division of dermatology at George Washington University, Washington, and Trisha Kaundinya, a medical student at Northwestern University, Chicago, prospectively evaluated 509 adults with AD who made an average of 2.3 visits at a single dermatology clinic between 2013 and 2020. At each visit, severity of AD signs and symptoms, as well as bathing and moisturizing practices, were assessed.
AD severity was assessed using the objective component of Scoring Atopic Dermatitis (o-SCORAD), intensity of pruritus in the past 3 days (SCORAD-itch), Eczema Area and Severity Index (EASI), Patient-Oriented Eczema Measure (POEM), and Dermatology Life Quality Index (DLQI). The researchers constructed repeated measures regression models to examine associations of bathing and moisturizing practices with change in AD severity outcome measure scores over time. Multivariable models controlled for age, sex, and race.
In adjusted linear regression models, showering or bathing more than once a day versus once daily was associated with significantly higher scores for SCORAD-itch (0.74; P = .0456), o-SCORAD (4.27; P = .0171), EASI (4.20; P = .0028), POEM (2.61; P = .0021), and DLQI (2.77; P = .0004).
The researchers also found that consistent application of moisturizer after the shower or bath was associated with significantly lower scores for o-SCORAD (–7.22; P < .0001), EASI (–3.91; P = .001) and POEM (–2.68; P = .0002), compared against not applying moisturizer after a shower or bath. However, shower or bath duration of more than, compared against fewer than, 15 minutes was not associated with significantly lower scores for o-SCORAD (1.26; P = .2868), SCORAD-itch (0.17; P = .4987), EASI (0.85; P = .3454), POEM (0.24; P = .6627) or DLQI (–0.40; P = .4318).
“Interestingly, this pattern was present when the reference shower or bath durations were under 10 minutes as well as under 5 minutes,” Mr. Rakita said. “Also, shower or bath frequencies of less than daily, relative to daily frequencies, were not significantly related to longitudinal AD severity.”
Mr. Rakita acknowledged certain limitations of the study, including the fact that the researchers did not examine the potential influence of specific soap and moisturizing products, water hardness, or other bathing features such as water temperature and bath additives.
Lawrence J. Green, MD, who was asked to comment on the study, said that he was not surprised by the finding that moisturizing after bathing improved AD signs and symptoms. “On the other hand, a long-held belief that longer duration of shower/bath time worsens AD was not found to be true,” said Dr. Green, a dermatologist who practices in Rockville, Md., and is also clinical professor of dermatology at George Washington University.
“This provides useful information for practicing dermatologists who wish to provide evidenced-based education about moisturizing and bathing to their AD patients,” he said.
The study was supported by the Agency for Healthcare Research and Quality and the Dermatology Foundation. Dr. Silverberg disclosed that he is a consultant to numerous pharmaceutical companies, receives fees for non-CME/CE services from Eli Lilly, Leo Pharma, Pfizer, Regeneron, and Sanofi Genzyme, as well as contracted research fees from Galderma. Dr. Green disclosed that he is a speaker, consultant, or investigator for numerous pharmaceutical companies. There were no other disclosures.
A version of this article first appeared on Medscape.com.
FROM REVOLUTIONIZING AD 2021
Sleep disturbances more profound in older adults with atopic dermatitis
especially trouble staying asleep.
Those are key findings from a cross-sectional study that Jaya Manjunath, BS, and Jonathan I. Silverberg, MD, PhD, MPH, presented during a poster session at the Revolutionizing Atopic Dermatitis symposium.
“Atopic dermatitis is a chronic, pruritic skin disease associated with sleep disturbance and fatigue affecting adults of all ages,” they wrote. “When caring for geriatric patients, several factors such as sleep disturbance, polypharmacy, cognition, social support, and mobility should be considered. However, little is known about the characteristics of atopic dermatitis in the geriatric population.”
Ms. Manjunath, a student at George Washington University, Washington, and Dr. Silverberg, director of clinical research in the department of dermatology at GWU, recruited patients with AD aged 18 years and older diagnosed by Hanifin-Rajka criteria who were evaluated at an academic medical center between 2014 and 2019. They underwent full body skin exams and completed electronic questionnaires. AD severity was assessed with the Eczema Area and Severity Index (EASI), Scoring Atopic Dermatitis (SCORAD) total and itch subscores, Investigator’s Global Assessment (IGA), patient-reported Global Assessment of AD severity, and the Patient-Oriented Eczema Measure (POEM).
The researchers also assessed the frequency of sleep disturbances, including difficulty falling asleep and staying asleep, and used multivariable logistic regression models to evaluate associations of age (65 and older vs. 18-64 years) with AD severity, sleep disturbance or fatigue, controlling for total POEM score, sex, and race.
Using adjusted odds ratios, Ms. Manjunath and Dr. Silverberg found that being 65 or older was not associated with AD severity on the EASI (adjusted odds ratio, 1.47); total SCORAD (aOR, 1.10), and itch subscore (aOR, 1.00); IGA (aOR, 1.87); patient-reported Global Assessment of AD severity (aOR, 0.80), or the patient-oriented eczema measure (aOR, 0.55), associations that were not statistically significant.
However, the researchers found that older adult age was associated with an increased number of nights of sleep disturbance from AD in the past week (aOR, 2.14; P = .0142), as well as increased fatigue in the past 7 days (aOR, 1.81; P = .0313), trouble sleeping in the past 7 days (aOR, 1.98; P = .0118), and trouble staying asleep in the past 7 days (aOR, 2.26; P = .0030), but not with difficulty falling asleep in the last 7 days (aOR, 1.16; P = .5996).
“Future studies are needed to determine why geriatric AD patients have increased sleep disturbance and optimal interventions to address their sleep disturbance,” the researchers concluded.
The study was supported by the Agency for Healthcare Research and Quality, the Dermatology Foundation, and by an unrestricted grant from Galderma. Ms. Manjunath disclosed no relevant financial relationships. Dr. Silverberg reported that he is a consultant to and/or an advisory board member for several pharmaceutical companies. He is also a speaker for Regeneron and Sanofi and has received a grant from Galderma.
A version of this article first appeared on Medscape.com.
especially trouble staying asleep.
Those are key findings from a cross-sectional study that Jaya Manjunath, BS, and Jonathan I. Silverberg, MD, PhD, MPH, presented during a poster session at the Revolutionizing Atopic Dermatitis symposium.
“Atopic dermatitis is a chronic, pruritic skin disease associated with sleep disturbance and fatigue affecting adults of all ages,” they wrote. “When caring for geriatric patients, several factors such as sleep disturbance, polypharmacy, cognition, social support, and mobility should be considered. However, little is known about the characteristics of atopic dermatitis in the geriatric population.”
Ms. Manjunath, a student at George Washington University, Washington, and Dr. Silverberg, director of clinical research in the department of dermatology at GWU, recruited patients with AD aged 18 years and older diagnosed by Hanifin-Rajka criteria who were evaluated at an academic medical center between 2014 and 2019. They underwent full body skin exams and completed electronic questionnaires. AD severity was assessed with the Eczema Area and Severity Index (EASI), Scoring Atopic Dermatitis (SCORAD) total and itch subscores, Investigator’s Global Assessment (IGA), patient-reported Global Assessment of AD severity, and the Patient-Oriented Eczema Measure (POEM).
The researchers also assessed the frequency of sleep disturbances, including difficulty falling asleep and staying asleep, and used multivariable logistic regression models to evaluate associations of age (65 and older vs. 18-64 years) with AD severity, sleep disturbance or fatigue, controlling for total POEM score, sex, and race.
Using adjusted odds ratios, Ms. Manjunath and Dr. Silverberg found that being 65 or older was not associated with AD severity on the EASI (adjusted odds ratio, 1.47); total SCORAD (aOR, 1.10), and itch subscore (aOR, 1.00); IGA (aOR, 1.87); patient-reported Global Assessment of AD severity (aOR, 0.80), or the patient-oriented eczema measure (aOR, 0.55), associations that were not statistically significant.
However, the researchers found that older adult age was associated with an increased number of nights of sleep disturbance from AD in the past week (aOR, 2.14; P = .0142), as well as increased fatigue in the past 7 days (aOR, 1.81; P = .0313), trouble sleeping in the past 7 days (aOR, 1.98; P = .0118), and trouble staying asleep in the past 7 days (aOR, 2.26; P = .0030), but not with difficulty falling asleep in the last 7 days (aOR, 1.16; P = .5996).
“Future studies are needed to determine why geriatric AD patients have increased sleep disturbance and optimal interventions to address their sleep disturbance,” the researchers concluded.
The study was supported by the Agency for Healthcare Research and Quality, the Dermatology Foundation, and by an unrestricted grant from Galderma. Ms. Manjunath disclosed no relevant financial relationships. Dr. Silverberg reported that he is a consultant to and/or an advisory board member for several pharmaceutical companies. He is also a speaker for Regeneron and Sanofi and has received a grant from Galderma.
A version of this article first appeared on Medscape.com.
especially trouble staying asleep.
Those are key findings from a cross-sectional study that Jaya Manjunath, BS, and Jonathan I. Silverberg, MD, PhD, MPH, presented during a poster session at the Revolutionizing Atopic Dermatitis symposium.
“Atopic dermatitis is a chronic, pruritic skin disease associated with sleep disturbance and fatigue affecting adults of all ages,” they wrote. “When caring for geriatric patients, several factors such as sleep disturbance, polypharmacy, cognition, social support, and mobility should be considered. However, little is known about the characteristics of atopic dermatitis in the geriatric population.”
Ms. Manjunath, a student at George Washington University, Washington, and Dr. Silverberg, director of clinical research in the department of dermatology at GWU, recruited patients with AD aged 18 years and older diagnosed by Hanifin-Rajka criteria who were evaluated at an academic medical center between 2014 and 2019. They underwent full body skin exams and completed electronic questionnaires. AD severity was assessed with the Eczema Area and Severity Index (EASI), Scoring Atopic Dermatitis (SCORAD) total and itch subscores, Investigator’s Global Assessment (IGA), patient-reported Global Assessment of AD severity, and the Patient-Oriented Eczema Measure (POEM).
The researchers also assessed the frequency of sleep disturbances, including difficulty falling asleep and staying asleep, and used multivariable logistic regression models to evaluate associations of age (65 and older vs. 18-64 years) with AD severity, sleep disturbance or fatigue, controlling for total POEM score, sex, and race.
Using adjusted odds ratios, Ms. Manjunath and Dr. Silverberg found that being 65 or older was not associated with AD severity on the EASI (adjusted odds ratio, 1.47); total SCORAD (aOR, 1.10), and itch subscore (aOR, 1.00); IGA (aOR, 1.87); patient-reported Global Assessment of AD severity (aOR, 0.80), or the patient-oriented eczema measure (aOR, 0.55), associations that were not statistically significant.
However, the researchers found that older adult age was associated with an increased number of nights of sleep disturbance from AD in the past week (aOR, 2.14; P = .0142), as well as increased fatigue in the past 7 days (aOR, 1.81; P = .0313), trouble sleeping in the past 7 days (aOR, 1.98; P = .0118), and trouble staying asleep in the past 7 days (aOR, 2.26; P = .0030), but not with difficulty falling asleep in the last 7 days (aOR, 1.16; P = .5996).
“Future studies are needed to determine why geriatric AD patients have increased sleep disturbance and optimal interventions to address their sleep disturbance,” the researchers concluded.
The study was supported by the Agency for Healthcare Research and Quality, the Dermatology Foundation, and by an unrestricted grant from Galderma. Ms. Manjunath disclosed no relevant financial relationships. Dr. Silverberg reported that he is a consultant to and/or an advisory board member for several pharmaceutical companies. He is also a speaker for Regeneron and Sanofi and has received a grant from Galderma.
A version of this article first appeared on Medscape.com.
FROM REVOLUTIONIZING AD 2021
A deep dive on tofacitinib’s mode of action
A new study has revealed potential cell-specific effects of the human Janus kinase (JAK) inhibitor tofacitinib, including possible targets – such as intestinal inflammation – for future research and even for increasing the drug’s effects.
The work used both mice and human cell models to explore the drug’s effect in inflammatory bowel disease (IBD). The mouse models suggested that the drug’s pharmacokinetics may be affected by intestinal inflammation. The human cell models seem to identify equilibrative nucleoside transporters as the likely route of cellular uptake of tofacitinib; this mechanism appears to be upregulated during inflammation and could present a therapeutic target to bolster the drug’s effects.
“We identify intestinal inflammation as a decisive modulator of the systemic pharmacokinetics of tofacitinib in mice, which needs to be studied and confirmed in humans. Finally, we decipher an important membrane transport mechanism that regulates cellular uptake of tofacitinib into activated immune cells, suggesting a model that explains a preferred uptake of tofacitinib into activated immune cells and a potential starting point to interfere with and channel such an uptake,” wrote the authors, led by Bernhard Texler and Andreas Zollner, both with the Christian Doppler Laboratory for Mucosal Immunology at the Johannes Kepler University in Linz, Austria, who published the results in Cellular and Molecular Gastroenterology and Hepatology.
IBD-related inflammation likely involves multiple cytokine pathways. The JAK-signal transducers and activator of transcription (JAK-STAT) pathway is downstream to more than 50 cytokines and growth factors, so disruption of their activity by JAK-STAT inhibitors like tofacitinib could counter the effects of more than one cytokine at a time.
Tofacitinib received FDA approval for the treatment of ulcerative colitis in 2018, but the details of its mechanism of action against intestinal inflammation remain poorly understood. For example, despite its efficacy against UC, the drug doesn’t work for Crohn’s disease patients. That may be because the drug affects specific cell populations involved only in UC pathogenesis.
To better understand the drug’s pharmacokinetics, the researchers examined the effects of tofacitinib in cells isolated from human peripheral blood, as well as an experimental mouse model of colitis.
The drug inhibited proliferation of both naïve and memory cytotoxic and helper T cells. At higher concentrations, it had strong effects on innate immune system cells, including monocytes, macrophages, and human intestinal epithelial organoids. It promotes the anti-inflammatory M2 phenotype among monocytes and macrophages. The drug also inhibited the pro-inflammatory M1 phenotype. The researchers observed similar effects in the mouse model of colitis.
The investigators also linked equilibrative nucleoside transporters (ENTs) with uptake of tofacitinib, specifically as a mediating role. These membrane proteins transport nucleosides, nucleobases, and therapeutic analogs like tofacitinib, which mimics the nucleotide adenosine triphosphate (ATP). Targeted inhibitors could potentially influence this process.
The researchers created three-dimensional, in vitro colonic organoids using intestinal epithelial cells from UC patients and healthy human controls. In this model, TNF-alpha can lead to production of pro-inflammatory cytokines, but tofacitinib blocked this effect. That result suggests that intestinal epithelial cells are a previously unidentified tofacitinib target.
Although a large amount of work has been done on the pharmacokinetics of therapeutic antibodies used to treat IBD, the authors point out that little is known about tofacitinib. In a mouse model, the serum concentration of the drug increased after exposure to dextran sulfate sodium (DSS), which triggers an IBD-like condition, and the spike was higher during more intense inflammation. The finding was surprising, considering that therapeutic antibodies typically get eliminated through feces during inflammation. Mice treated with DSS versus control had similar levels of tofacitinib in both urine and the feces, suggesting that inflammation may somehow inhibit the enzymes that metabolize the drug.
The researchers also noted that uptake of tofacitinib into leukocytes increased following stimulation with lipopolysaccharide. Given its structural similarity to ATP, the researchers propose that tofacitinib may enter the cells through adenosine cell membrane transporters ENT1 and ENT2, and some evidence even suggested that the pathway may be strengthened in activated immune cells.
The study received funding from: the Christian Doppler Research Association; the Austrian Federal Ministry of Science, Research, and Economy; and the National Foundation for Research, Technology, and Development. One author is receiving research support from AbbVie and Takeda under the framework of the Christian Doppler Research Society, but the remaining authors have no relevant conflicts of interest.
A new study has revealed potential cell-specific effects of the human Janus kinase (JAK) inhibitor tofacitinib, including possible targets – such as intestinal inflammation – for future research and even for increasing the drug’s effects.
The work used both mice and human cell models to explore the drug’s effect in inflammatory bowel disease (IBD). The mouse models suggested that the drug’s pharmacokinetics may be affected by intestinal inflammation. The human cell models seem to identify equilibrative nucleoside transporters as the likely route of cellular uptake of tofacitinib; this mechanism appears to be upregulated during inflammation and could present a therapeutic target to bolster the drug’s effects.
“We identify intestinal inflammation as a decisive modulator of the systemic pharmacokinetics of tofacitinib in mice, which needs to be studied and confirmed in humans. Finally, we decipher an important membrane transport mechanism that regulates cellular uptake of tofacitinib into activated immune cells, suggesting a model that explains a preferred uptake of tofacitinib into activated immune cells and a potential starting point to interfere with and channel such an uptake,” wrote the authors, led by Bernhard Texler and Andreas Zollner, both with the Christian Doppler Laboratory for Mucosal Immunology at the Johannes Kepler University in Linz, Austria, who published the results in Cellular and Molecular Gastroenterology and Hepatology.
IBD-related inflammation likely involves multiple cytokine pathways. The JAK-signal transducers and activator of transcription (JAK-STAT) pathway is downstream to more than 50 cytokines and growth factors, so disruption of their activity by JAK-STAT inhibitors like tofacitinib could counter the effects of more than one cytokine at a time.
Tofacitinib received FDA approval for the treatment of ulcerative colitis in 2018, but the details of its mechanism of action against intestinal inflammation remain poorly understood. For example, despite its efficacy against UC, the drug doesn’t work for Crohn’s disease patients. That may be because the drug affects specific cell populations involved only in UC pathogenesis.
To better understand the drug’s pharmacokinetics, the researchers examined the effects of tofacitinib in cells isolated from human peripheral blood, as well as an experimental mouse model of colitis.
The drug inhibited proliferation of both naïve and memory cytotoxic and helper T cells. At higher concentrations, it had strong effects on innate immune system cells, including monocytes, macrophages, and human intestinal epithelial organoids. It promotes the anti-inflammatory M2 phenotype among monocytes and macrophages. The drug also inhibited the pro-inflammatory M1 phenotype. The researchers observed similar effects in the mouse model of colitis.
The investigators also linked equilibrative nucleoside transporters (ENTs) with uptake of tofacitinib, specifically as a mediating role. These membrane proteins transport nucleosides, nucleobases, and therapeutic analogs like tofacitinib, which mimics the nucleotide adenosine triphosphate (ATP). Targeted inhibitors could potentially influence this process.
The researchers created three-dimensional, in vitro colonic organoids using intestinal epithelial cells from UC patients and healthy human controls. In this model, TNF-alpha can lead to production of pro-inflammatory cytokines, but tofacitinib blocked this effect. That result suggests that intestinal epithelial cells are a previously unidentified tofacitinib target.
Although a large amount of work has been done on the pharmacokinetics of therapeutic antibodies used to treat IBD, the authors point out that little is known about tofacitinib. In a mouse model, the serum concentration of the drug increased after exposure to dextran sulfate sodium (DSS), which triggers an IBD-like condition, and the spike was higher during more intense inflammation. The finding was surprising, considering that therapeutic antibodies typically get eliminated through feces during inflammation. Mice treated with DSS versus control had similar levels of tofacitinib in both urine and the feces, suggesting that inflammation may somehow inhibit the enzymes that metabolize the drug.
The researchers also noted that uptake of tofacitinib into leukocytes increased following stimulation with lipopolysaccharide. Given its structural similarity to ATP, the researchers propose that tofacitinib may enter the cells through adenosine cell membrane transporters ENT1 and ENT2, and some evidence even suggested that the pathway may be strengthened in activated immune cells.
The study received funding from: the Christian Doppler Research Association; the Austrian Federal Ministry of Science, Research, and Economy; and the National Foundation for Research, Technology, and Development. One author is receiving research support from AbbVie and Takeda under the framework of the Christian Doppler Research Society, but the remaining authors have no relevant conflicts of interest.
A new study has revealed potential cell-specific effects of the human Janus kinase (JAK) inhibitor tofacitinib, including possible targets – such as intestinal inflammation – for future research and even for increasing the drug’s effects.
The work used both mice and human cell models to explore the drug’s effect in inflammatory bowel disease (IBD). The mouse models suggested that the drug’s pharmacokinetics may be affected by intestinal inflammation. The human cell models seem to identify equilibrative nucleoside transporters as the likely route of cellular uptake of tofacitinib; this mechanism appears to be upregulated during inflammation and could present a therapeutic target to bolster the drug’s effects.
“We identify intestinal inflammation as a decisive modulator of the systemic pharmacokinetics of tofacitinib in mice, which needs to be studied and confirmed in humans. Finally, we decipher an important membrane transport mechanism that regulates cellular uptake of tofacitinib into activated immune cells, suggesting a model that explains a preferred uptake of tofacitinib into activated immune cells and a potential starting point to interfere with and channel such an uptake,” wrote the authors, led by Bernhard Texler and Andreas Zollner, both with the Christian Doppler Laboratory for Mucosal Immunology at the Johannes Kepler University in Linz, Austria, who published the results in Cellular and Molecular Gastroenterology and Hepatology.
IBD-related inflammation likely involves multiple cytokine pathways. The JAK-signal transducers and activator of transcription (JAK-STAT) pathway is downstream to more than 50 cytokines and growth factors, so disruption of their activity by JAK-STAT inhibitors like tofacitinib could counter the effects of more than one cytokine at a time.
Tofacitinib received FDA approval for the treatment of ulcerative colitis in 2018, but the details of its mechanism of action against intestinal inflammation remain poorly understood. For example, despite its efficacy against UC, the drug doesn’t work for Crohn’s disease patients. That may be because the drug affects specific cell populations involved only in UC pathogenesis.
To better understand the drug’s pharmacokinetics, the researchers examined the effects of tofacitinib in cells isolated from human peripheral blood, as well as an experimental mouse model of colitis.
The drug inhibited proliferation of both naïve and memory cytotoxic and helper T cells. At higher concentrations, it had strong effects on innate immune system cells, including monocytes, macrophages, and human intestinal epithelial organoids. It promotes the anti-inflammatory M2 phenotype among monocytes and macrophages. The drug also inhibited the pro-inflammatory M1 phenotype. The researchers observed similar effects in the mouse model of colitis.
The investigators also linked equilibrative nucleoside transporters (ENTs) with uptake of tofacitinib, specifically as a mediating role. These membrane proteins transport nucleosides, nucleobases, and therapeutic analogs like tofacitinib, which mimics the nucleotide adenosine triphosphate (ATP). Targeted inhibitors could potentially influence this process.
The researchers created three-dimensional, in vitro colonic organoids using intestinal epithelial cells from UC patients and healthy human controls. In this model, TNF-alpha can lead to production of pro-inflammatory cytokines, but tofacitinib blocked this effect. That result suggests that intestinal epithelial cells are a previously unidentified tofacitinib target.
Although a large amount of work has been done on the pharmacokinetics of therapeutic antibodies used to treat IBD, the authors point out that little is known about tofacitinib. In a mouse model, the serum concentration of the drug increased after exposure to dextran sulfate sodium (DSS), which triggers an IBD-like condition, and the spike was higher during more intense inflammation. The finding was surprising, considering that therapeutic antibodies typically get eliminated through feces during inflammation. Mice treated with DSS versus control had similar levels of tofacitinib in both urine and the feces, suggesting that inflammation may somehow inhibit the enzymes that metabolize the drug.
The researchers also noted that uptake of tofacitinib into leukocytes increased following stimulation with lipopolysaccharide. Given its structural similarity to ATP, the researchers propose that tofacitinib may enter the cells through adenosine cell membrane transporters ENT1 and ENT2, and some evidence even suggested that the pathway may be strengthened in activated immune cells.
The study received funding from: the Christian Doppler Research Association; the Austrian Federal Ministry of Science, Research, and Economy; and the National Foundation for Research, Technology, and Development. One author is receiving research support from AbbVie and Takeda under the framework of the Christian Doppler Research Society, but the remaining authors have no relevant conflicts of interest.
FROM CELLULAR AND MOLECULAR GASTROENTEROLOGY AND HEPATOLOGY
Pandemic poses short- and long-term risks to babies, especially boys
The pandemic has created a hostile environment for pregnant people and their babies.
Stress levels among expectant mothers have soared. Pregnant women with COVID are 5 times as likely as uninfected pregnant people to require intensive care and 22 times as likely to die. Infected moms are four times as likely to have a stillborn child.
Yet some of the pandemic’s greatest threats to infants’ health may not be apparent for years or even decades.
That’s because babies of COVID-infected moms are 60% more likely to be born very prematurely, which increases the danger of infant mortality and long-term disabilities such as cerebral palsy, asthma, and hearing loss, as well as a child’s risk of adult disease, including depression, anxiety, heart disease, and kidney disease.
Studies have linked fever and infection during pregnancy to developmental and psychiatric conditions such as autism, depression, and schizophrenia.
“Some of these conditions do not show up until middle childhood or early adult life, but they have their origins in fetal life,” said Evdokia Anagnostou, MD, a child neurologist at Holland Bloorview Kids Rehabilitation Hospital and a pediatrics professor at the University of Toronto.
For fetuses exposed to COVID, the greatest danger is usually not the coronavirus itself, but the mother’s immune system.
Both severe COVID infections and the strain of the pandemic can expose fetuses to harmful inflammation, which can occur when a mother’s immune system is fighting a virus or when stress hormones send nonstop alarm signals.
Prenatal inflammation “changes the way the brain develops and, depending on the timing of the infection, it can change the way the heart or kidneys develop,” Dr. Anagnostou said.
Although health officials have strongly recommended COVID vaccines for pregnant people, only 35% are fully vaccinated.
At least 150,000 pregnant women have been diagnosed with COVID; more than 25,000 of them have been hospitalized, and 249 have died, according to the Centers for Disease Control and Prevention.
Although most babies will be fine, even a small increase in the percentage of children with special medical or educational needs could have a large effect on the population, given the huge number of COVID infections, Dr. Anagnostou said.
“If someone has a baby who is doing well, that is what they should focus on,” Dr. Anagnostou said. “But from a public health point of view, we need to follow women who experienced severe COVID and their babies to understand the impact.”
Learning from history
Researchers in the United States and other countries are already studying “the COVID generation” to see whether these children have more health issues than those conceived or born before 2020.
Previous crises have shown that the challenges fetuses face in the womb – such as maternal infections, hunger, stress, and hormone-disrupting chemicals – can leave a lasting imprint on their health, as well as that of their children and grandchildren, said Frederick Kaskel, MD, director of pediatric nephrology at the Children’s Hospital at Montefiore, New York.
People whose mothers were pregnant during surges in the 1918 influenza pandemic, for example, had poorer health throughout their lives, compared with Americans born at other times, said John McCarthy, who is a medical student at Albert Einstein College of Medicine, New York, and cowrote a recent review in JAMA Pediatrics with Dr. Kaskel.
Researchers don’t know exactly which moms were infected with pandemic flu, Mr. McCarthy said. But women who were pregnant during major surges – when infection was widespread – had children with higher rates of heart disease or diabetes. These children were also less successful in school, less economically productive, and more likely to live with a disability.
Because organ systems develop during different periods of pregnancy, fetuses exposed during the first trimester may face different risks than those exposed toward the end of pregnancy, Mr. McCarthy said. For example, people born in the fall of 1918 were 50% more likely than others to develop kidney disease; that may reflect an exposure to the pandemic in the third trimester, while the kidneys were still developing.
Nearly 2 years into the COVID pandemic, researchers have begun to publish preliminary observations of infants exposed to COVID infections and stress before birth.
Although Dr. Anagnostou noted that it’s too early to reach definitive conclusions, “there is evidence that babies born to moms with severe COVID infections have changes to their immune system,” she said. “It’s enough to make us worry a little bit.”
Damaging a fetal security system
The good news about the coronavirus is that it seldom crosses the placenta, the organ tasked with protecting a developing fetus from infections and providing it with oxygen. So moms with COVID rarely give the virus to their children before birth.
That’s important, because some viruses that directly infect the fetus – such as Zika – can cause devastating birth defects, said Karin Nielsen-Saines, MD, a specialist in pediatric infectious diseases at University of California, Los Angeles.
But studies also suggest that inflammation from a mother’s COVID infection can injure the placenta, said Jeffery Goldstein, MD, an assistant professor of pathology at Northwestern University, Chicago. In a study published in American Journal of Clinical Pathology , Dr. Goldstein and his coauthors found that placentas from COVID-infected moms had more abnormal blood vessels than placentas from patients without COVID, making it harder for them to deliver sufficient oxygen to the fetus.
Placental damage can also lead to preeclampsia, a serious complication of pregnancy that can cause a mother’s blood pressure to spike.
Preeclampsia occurs when blood vessels in the placenta don’t develop or function properly, forcing the mother’s heart to work harder to get blood to the fetus, which may not receive enough oxygen and nutrients. Preeclampsia also predisposes women to heart attacks and strokes later in life.
Rewiring the immune system
In some cases, COVID also appears to rewire a baby’s immune response, Dr. Nielsen-Saines said.
In an October study in the journal Cell Reports Medicine, Dr. Nielsen-Saines and her coauthors found that infants born to people with severe COVID infections had a different mix of immune cells and proteins than other babies. None of the newborns tested positive for the coronavirus.
The immune changes are concerning, Dr. Nielsen-Saines said, because this pattern of immune cells and proteins has previously been found in infants with respiratory problems and in some cases poor neurodevelopment.
Notably, all the babies in her study appear healthy, said Dr. Nielsen-Saines, who plans to follow them for 3 years to see whether these early signals translate into developmental delays, such as problems talking, walking, or interacting with others.
“How big of a difference does any of this make in the baby?” asked Dr. Anagnostou. “We won’t know for a few years. All we can do is try to be as prepared as possible.”
Increasing the risk for boys
Boys could face higher risks from COVID, even before birth.
Males are generally more vulnerable than females as fetuses and newborns; they’re more likely to be born prematurely and to die as infants. Preterm boys also have a higher risk of disability and death.
But coronavirus infection poses special dangers, said Sabra Klein, PhD, a professor of molecular microbiology and immunology at the Johns Hopkins Bloomberg School of Public Health, Baltimore.
That’s because boys are disproportionately affected by conditions linked to maternal infections. Boys are four times as likely as girls to be diagnosed with autism or attention-deficit/hyperactivity disorder, for example, while men are 75% more likely than women to develop schizophrenia.
Scientists don’t fully understand why boys appear more fragile in the womb, although testosterone – which can dampen immune response – may play a role, said Kristina Adams Waldorf, MD, a professor of obstetrics and gynecology at the University of Washington.
Men generally mount weaker immune responses than women and more often develop severe COVID infections. Recent research suggests boys with COVID are more likely than girls to become seriously ill or develop a rare inflammatory condition called multisystem inflammatory syndrome.
New research on COVID could help illuminate this vulnerability.
In a study published in October, researchers found that the sex of a fetus influences the way its placenta responds to COVID, as well as how its mother’s immune system responds.
Pregnant people infected with COVID made fewer antibodies against the coronavirus if they were carrying male fetuses than if they were carrying females. Mothers also transferred fewer antibodies to boys than to girls, said Andrea Edlow, MD, senior author of the study and a maternal-fetal medicine specialist at Massachusetts General Hospital, Boston.
When examining the placentas of male fetuses after delivery, researchers found changes that could leave boys less protected against damaging inflammation.
The sex of a fetus can influence its mother’s response to other illnesses, as well.
For example, research shows that pregnant women with asthma have worse symptoms if they’re carrying a female. Women carrying males are slightly more likely to develop gestational diabetes.
Dr. Edlow said her findings raise questions about the “cross talk” between mother and baby. “The mom’s immune system is sensing there is a male fetus,” Dr. Edlow said. “And the fetus is actively communicating with the mom’s immune system.”
Boosting toxic stress
Rates of depression and stress among pregnant women have increased dramatically during the pandemic.
That’s concerning because chronic stress can lead to inflammation, affecting the babies of both infected and uninfected women, Dr. Anagnostou said.
Studies consistently show that infants born to mothers who experience significant stress during pregnancy have higher rates of short- and long-term health damage – including heart defects and obesity – than babies born to women with less stress.
“We know that inflammation directly influences the way a baby’s brain develops,” said Elinor Sullivan, PhD, an associate professor in psychiatry at Oregon Health & Science University, Portland.
Lockdowns, travel restrictions and physical distancing left many pregnant women without the support of family and friends. The stress of losing a loved one, a job, or a home further heightens the risks to moms and babies, said Dr. Sullivan, who is following children born during the pandemic for 5 years.
In research that has not yet been published, Dr. Sullivan found that babies of women who were pregnant during the pandemic showed more sadness and negative emotions in the first year of life, compared with infants of women who were pregnant before the pandemic.
The findings show the importance of helping and protecting pregnant people before and after delivery, said Dr. Sullivan, who conducted a separate study that found women who received more social support were less depressed.
Italian researchers are also studying the effect of maternal stress on infants’ behavior, as well as the way their genes are regulated.
Although stress-related inflammation doesn’t alter the structure of a baby’s genes, it can influence whether they’re turned on and off, said Livio Provenzi, PhD, a psychologist at the C. Mondino National Institute of Neurology Foundation in Pavia, Italy.
In Dr. Provenzi’s study of 163 mother-baby pairs he found differences in how genes that regulate the stress response were activated. Genes that help people respond to stress were more likely to be turned off in babies whose moms reported the most stress during pregnancy. The same moms also reported that their babies cried more and were fussier when they were 3 months old.
Researchers usually prefer to make in-person observations of babies as they interact with their mothers, Dr. Provenzi said. But because of the pandemic, Dr. Provenzi asked mothers to fill out questionnaires about infant behavior. He plans to observe mothers and babies in person when the children are 12 months old.
While vaccinating pregnant people is the best way to protect them and their fetuses from the virus, Dr. Anagnostou said, society needs to do more to preserve expectant mothers’ mental health.
“We can’t escape the fact that we’ve lived through 2 years of a pandemic,” Dr. Anagnostou said. “But we can think about opportunities for reducing the risk.”
KHN (Kaiser Health News) is a national newsroom that produces in-depth journalism about health issues. Together with Policy Analysis and Polling, KHN is one of the three major operating programs at KFF (Kaiser Family Foundation). KFF is an endowed nonprofit organization providing information on health issues to the nation.
The pandemic has created a hostile environment for pregnant people and their babies.
Stress levels among expectant mothers have soared. Pregnant women with COVID are 5 times as likely as uninfected pregnant people to require intensive care and 22 times as likely to die. Infected moms are four times as likely to have a stillborn child.
Yet some of the pandemic’s greatest threats to infants’ health may not be apparent for years or even decades.
That’s because babies of COVID-infected moms are 60% more likely to be born very prematurely, which increases the danger of infant mortality and long-term disabilities such as cerebral palsy, asthma, and hearing loss, as well as a child’s risk of adult disease, including depression, anxiety, heart disease, and kidney disease.
Studies have linked fever and infection during pregnancy to developmental and psychiatric conditions such as autism, depression, and schizophrenia.
“Some of these conditions do not show up until middle childhood or early adult life, but they have their origins in fetal life,” said Evdokia Anagnostou, MD, a child neurologist at Holland Bloorview Kids Rehabilitation Hospital and a pediatrics professor at the University of Toronto.
For fetuses exposed to COVID, the greatest danger is usually not the coronavirus itself, but the mother’s immune system.
Both severe COVID infections and the strain of the pandemic can expose fetuses to harmful inflammation, which can occur when a mother’s immune system is fighting a virus or when stress hormones send nonstop alarm signals.
Prenatal inflammation “changes the way the brain develops and, depending on the timing of the infection, it can change the way the heart or kidneys develop,” Dr. Anagnostou said.
Although health officials have strongly recommended COVID vaccines for pregnant people, only 35% are fully vaccinated.
At least 150,000 pregnant women have been diagnosed with COVID; more than 25,000 of them have been hospitalized, and 249 have died, according to the Centers for Disease Control and Prevention.
Although most babies will be fine, even a small increase in the percentage of children with special medical or educational needs could have a large effect on the population, given the huge number of COVID infections, Dr. Anagnostou said.
“If someone has a baby who is doing well, that is what they should focus on,” Dr. Anagnostou said. “But from a public health point of view, we need to follow women who experienced severe COVID and their babies to understand the impact.”
Learning from history
Researchers in the United States and other countries are already studying “the COVID generation” to see whether these children have more health issues than those conceived or born before 2020.
Previous crises have shown that the challenges fetuses face in the womb – such as maternal infections, hunger, stress, and hormone-disrupting chemicals – can leave a lasting imprint on their health, as well as that of their children and grandchildren, said Frederick Kaskel, MD, director of pediatric nephrology at the Children’s Hospital at Montefiore, New York.
People whose mothers were pregnant during surges in the 1918 influenza pandemic, for example, had poorer health throughout their lives, compared with Americans born at other times, said John McCarthy, who is a medical student at Albert Einstein College of Medicine, New York, and cowrote a recent review in JAMA Pediatrics with Dr. Kaskel.
Researchers don’t know exactly which moms were infected with pandemic flu, Mr. McCarthy said. But women who were pregnant during major surges – when infection was widespread – had children with higher rates of heart disease or diabetes. These children were also less successful in school, less economically productive, and more likely to live with a disability.
Because organ systems develop during different periods of pregnancy, fetuses exposed during the first trimester may face different risks than those exposed toward the end of pregnancy, Mr. McCarthy said. For example, people born in the fall of 1918 were 50% more likely than others to develop kidney disease; that may reflect an exposure to the pandemic in the third trimester, while the kidneys were still developing.
Nearly 2 years into the COVID pandemic, researchers have begun to publish preliminary observations of infants exposed to COVID infections and stress before birth.
Although Dr. Anagnostou noted that it’s too early to reach definitive conclusions, “there is evidence that babies born to moms with severe COVID infections have changes to their immune system,” she said. “It’s enough to make us worry a little bit.”
Damaging a fetal security system
The good news about the coronavirus is that it seldom crosses the placenta, the organ tasked with protecting a developing fetus from infections and providing it with oxygen. So moms with COVID rarely give the virus to their children before birth.
That’s important, because some viruses that directly infect the fetus – such as Zika – can cause devastating birth defects, said Karin Nielsen-Saines, MD, a specialist in pediatric infectious diseases at University of California, Los Angeles.
But studies also suggest that inflammation from a mother’s COVID infection can injure the placenta, said Jeffery Goldstein, MD, an assistant professor of pathology at Northwestern University, Chicago. In a study published in American Journal of Clinical Pathology , Dr. Goldstein and his coauthors found that placentas from COVID-infected moms had more abnormal blood vessels than placentas from patients without COVID, making it harder for them to deliver sufficient oxygen to the fetus.
Placental damage can also lead to preeclampsia, a serious complication of pregnancy that can cause a mother’s blood pressure to spike.
Preeclampsia occurs when blood vessels in the placenta don’t develop or function properly, forcing the mother’s heart to work harder to get blood to the fetus, which may not receive enough oxygen and nutrients. Preeclampsia also predisposes women to heart attacks and strokes later in life.
Rewiring the immune system
In some cases, COVID also appears to rewire a baby’s immune response, Dr. Nielsen-Saines said.
In an October study in the journal Cell Reports Medicine, Dr. Nielsen-Saines and her coauthors found that infants born to people with severe COVID infections had a different mix of immune cells and proteins than other babies. None of the newborns tested positive for the coronavirus.
The immune changes are concerning, Dr. Nielsen-Saines said, because this pattern of immune cells and proteins has previously been found in infants with respiratory problems and in some cases poor neurodevelopment.
Notably, all the babies in her study appear healthy, said Dr. Nielsen-Saines, who plans to follow them for 3 years to see whether these early signals translate into developmental delays, such as problems talking, walking, or interacting with others.
“How big of a difference does any of this make in the baby?” asked Dr. Anagnostou. “We won’t know for a few years. All we can do is try to be as prepared as possible.”
Increasing the risk for boys
Boys could face higher risks from COVID, even before birth.
Males are generally more vulnerable than females as fetuses and newborns; they’re more likely to be born prematurely and to die as infants. Preterm boys also have a higher risk of disability and death.
But coronavirus infection poses special dangers, said Sabra Klein, PhD, a professor of molecular microbiology and immunology at the Johns Hopkins Bloomberg School of Public Health, Baltimore.
That’s because boys are disproportionately affected by conditions linked to maternal infections. Boys are four times as likely as girls to be diagnosed with autism or attention-deficit/hyperactivity disorder, for example, while men are 75% more likely than women to develop schizophrenia.
Scientists don’t fully understand why boys appear more fragile in the womb, although testosterone – which can dampen immune response – may play a role, said Kristina Adams Waldorf, MD, a professor of obstetrics and gynecology at the University of Washington.
Men generally mount weaker immune responses than women and more often develop severe COVID infections. Recent research suggests boys with COVID are more likely than girls to become seriously ill or develop a rare inflammatory condition called multisystem inflammatory syndrome.
New research on COVID could help illuminate this vulnerability.
In a study published in October, researchers found that the sex of a fetus influences the way its placenta responds to COVID, as well as how its mother’s immune system responds.
Pregnant people infected with COVID made fewer antibodies against the coronavirus if they were carrying male fetuses than if they were carrying females. Mothers also transferred fewer antibodies to boys than to girls, said Andrea Edlow, MD, senior author of the study and a maternal-fetal medicine specialist at Massachusetts General Hospital, Boston.
When examining the placentas of male fetuses after delivery, researchers found changes that could leave boys less protected against damaging inflammation.
The sex of a fetus can influence its mother’s response to other illnesses, as well.
For example, research shows that pregnant women with asthma have worse symptoms if they’re carrying a female. Women carrying males are slightly more likely to develop gestational diabetes.
Dr. Edlow said her findings raise questions about the “cross talk” between mother and baby. “The mom’s immune system is sensing there is a male fetus,” Dr. Edlow said. “And the fetus is actively communicating with the mom’s immune system.”
Boosting toxic stress
Rates of depression and stress among pregnant women have increased dramatically during the pandemic.
That’s concerning because chronic stress can lead to inflammation, affecting the babies of both infected and uninfected women, Dr. Anagnostou said.
Studies consistently show that infants born to mothers who experience significant stress during pregnancy have higher rates of short- and long-term health damage – including heart defects and obesity – than babies born to women with less stress.
“We know that inflammation directly influences the way a baby’s brain develops,” said Elinor Sullivan, PhD, an associate professor in psychiatry at Oregon Health & Science University, Portland.
Lockdowns, travel restrictions and physical distancing left many pregnant women without the support of family and friends. The stress of losing a loved one, a job, or a home further heightens the risks to moms and babies, said Dr. Sullivan, who is following children born during the pandemic for 5 years.
In research that has not yet been published, Dr. Sullivan found that babies of women who were pregnant during the pandemic showed more sadness and negative emotions in the first year of life, compared with infants of women who were pregnant before the pandemic.
The findings show the importance of helping and protecting pregnant people before and after delivery, said Dr. Sullivan, who conducted a separate study that found women who received more social support were less depressed.
Italian researchers are also studying the effect of maternal stress on infants’ behavior, as well as the way their genes are regulated.
Although stress-related inflammation doesn’t alter the structure of a baby’s genes, it can influence whether they’re turned on and off, said Livio Provenzi, PhD, a psychologist at the C. Mondino National Institute of Neurology Foundation in Pavia, Italy.
In Dr. Provenzi’s study of 163 mother-baby pairs he found differences in how genes that regulate the stress response were activated. Genes that help people respond to stress were more likely to be turned off in babies whose moms reported the most stress during pregnancy. The same moms also reported that their babies cried more and were fussier when they were 3 months old.
Researchers usually prefer to make in-person observations of babies as they interact with their mothers, Dr. Provenzi said. But because of the pandemic, Dr. Provenzi asked mothers to fill out questionnaires about infant behavior. He plans to observe mothers and babies in person when the children are 12 months old.
While vaccinating pregnant people is the best way to protect them and their fetuses from the virus, Dr. Anagnostou said, society needs to do more to preserve expectant mothers’ mental health.
“We can’t escape the fact that we’ve lived through 2 years of a pandemic,” Dr. Anagnostou said. “But we can think about opportunities for reducing the risk.”
KHN (Kaiser Health News) is a national newsroom that produces in-depth journalism about health issues. Together with Policy Analysis and Polling, KHN is one of the three major operating programs at KFF (Kaiser Family Foundation). KFF is an endowed nonprofit organization providing information on health issues to the nation.
The pandemic has created a hostile environment for pregnant people and their babies.
Stress levels among expectant mothers have soared. Pregnant women with COVID are 5 times as likely as uninfected pregnant people to require intensive care and 22 times as likely to die. Infected moms are four times as likely to have a stillborn child.
Yet some of the pandemic’s greatest threats to infants’ health may not be apparent for years or even decades.
That’s because babies of COVID-infected moms are 60% more likely to be born very prematurely, which increases the danger of infant mortality and long-term disabilities such as cerebral palsy, asthma, and hearing loss, as well as a child’s risk of adult disease, including depression, anxiety, heart disease, and kidney disease.
Studies have linked fever and infection during pregnancy to developmental and psychiatric conditions such as autism, depression, and schizophrenia.
“Some of these conditions do not show up until middle childhood or early adult life, but they have their origins in fetal life,” said Evdokia Anagnostou, MD, a child neurologist at Holland Bloorview Kids Rehabilitation Hospital and a pediatrics professor at the University of Toronto.
For fetuses exposed to COVID, the greatest danger is usually not the coronavirus itself, but the mother’s immune system.
Both severe COVID infections and the strain of the pandemic can expose fetuses to harmful inflammation, which can occur when a mother’s immune system is fighting a virus or when stress hormones send nonstop alarm signals.
Prenatal inflammation “changes the way the brain develops and, depending on the timing of the infection, it can change the way the heart or kidneys develop,” Dr. Anagnostou said.
Although health officials have strongly recommended COVID vaccines for pregnant people, only 35% are fully vaccinated.
At least 150,000 pregnant women have been diagnosed with COVID; more than 25,000 of them have been hospitalized, and 249 have died, according to the Centers for Disease Control and Prevention.
Although most babies will be fine, even a small increase in the percentage of children with special medical or educational needs could have a large effect on the population, given the huge number of COVID infections, Dr. Anagnostou said.
“If someone has a baby who is doing well, that is what they should focus on,” Dr. Anagnostou said. “But from a public health point of view, we need to follow women who experienced severe COVID and their babies to understand the impact.”
Learning from history
Researchers in the United States and other countries are already studying “the COVID generation” to see whether these children have more health issues than those conceived or born before 2020.
Previous crises have shown that the challenges fetuses face in the womb – such as maternal infections, hunger, stress, and hormone-disrupting chemicals – can leave a lasting imprint on their health, as well as that of their children and grandchildren, said Frederick Kaskel, MD, director of pediatric nephrology at the Children’s Hospital at Montefiore, New York.
People whose mothers were pregnant during surges in the 1918 influenza pandemic, for example, had poorer health throughout their lives, compared with Americans born at other times, said John McCarthy, who is a medical student at Albert Einstein College of Medicine, New York, and cowrote a recent review in JAMA Pediatrics with Dr. Kaskel.
Researchers don’t know exactly which moms were infected with pandemic flu, Mr. McCarthy said. But women who were pregnant during major surges – when infection was widespread – had children with higher rates of heart disease or diabetes. These children were also less successful in school, less economically productive, and more likely to live with a disability.
Because organ systems develop during different periods of pregnancy, fetuses exposed during the first trimester may face different risks than those exposed toward the end of pregnancy, Mr. McCarthy said. For example, people born in the fall of 1918 were 50% more likely than others to develop kidney disease; that may reflect an exposure to the pandemic in the third trimester, while the kidneys were still developing.
Nearly 2 years into the COVID pandemic, researchers have begun to publish preliminary observations of infants exposed to COVID infections and stress before birth.
Although Dr. Anagnostou noted that it’s too early to reach definitive conclusions, “there is evidence that babies born to moms with severe COVID infections have changes to their immune system,” she said. “It’s enough to make us worry a little bit.”
Damaging a fetal security system
The good news about the coronavirus is that it seldom crosses the placenta, the organ tasked with protecting a developing fetus from infections and providing it with oxygen. So moms with COVID rarely give the virus to their children before birth.
That’s important, because some viruses that directly infect the fetus – such as Zika – can cause devastating birth defects, said Karin Nielsen-Saines, MD, a specialist in pediatric infectious diseases at University of California, Los Angeles.
But studies also suggest that inflammation from a mother’s COVID infection can injure the placenta, said Jeffery Goldstein, MD, an assistant professor of pathology at Northwestern University, Chicago. In a study published in American Journal of Clinical Pathology , Dr. Goldstein and his coauthors found that placentas from COVID-infected moms had more abnormal blood vessels than placentas from patients without COVID, making it harder for them to deliver sufficient oxygen to the fetus.
Placental damage can also lead to preeclampsia, a serious complication of pregnancy that can cause a mother’s blood pressure to spike.
Preeclampsia occurs when blood vessels in the placenta don’t develop or function properly, forcing the mother’s heart to work harder to get blood to the fetus, which may not receive enough oxygen and nutrients. Preeclampsia also predisposes women to heart attacks and strokes later in life.
Rewiring the immune system
In some cases, COVID also appears to rewire a baby’s immune response, Dr. Nielsen-Saines said.
In an October study in the journal Cell Reports Medicine, Dr. Nielsen-Saines and her coauthors found that infants born to people with severe COVID infections had a different mix of immune cells and proteins than other babies. None of the newborns tested positive for the coronavirus.
The immune changes are concerning, Dr. Nielsen-Saines said, because this pattern of immune cells and proteins has previously been found in infants with respiratory problems and in some cases poor neurodevelopment.
Notably, all the babies in her study appear healthy, said Dr. Nielsen-Saines, who plans to follow them for 3 years to see whether these early signals translate into developmental delays, such as problems talking, walking, or interacting with others.
“How big of a difference does any of this make in the baby?” asked Dr. Anagnostou. “We won’t know for a few years. All we can do is try to be as prepared as possible.”
Increasing the risk for boys
Boys could face higher risks from COVID, even before birth.
Males are generally more vulnerable than females as fetuses and newborns; they’re more likely to be born prematurely and to die as infants. Preterm boys also have a higher risk of disability and death.
But coronavirus infection poses special dangers, said Sabra Klein, PhD, a professor of molecular microbiology and immunology at the Johns Hopkins Bloomberg School of Public Health, Baltimore.
That’s because boys are disproportionately affected by conditions linked to maternal infections. Boys are four times as likely as girls to be diagnosed with autism or attention-deficit/hyperactivity disorder, for example, while men are 75% more likely than women to develop schizophrenia.
Scientists don’t fully understand why boys appear more fragile in the womb, although testosterone – which can dampen immune response – may play a role, said Kristina Adams Waldorf, MD, a professor of obstetrics and gynecology at the University of Washington.
Men generally mount weaker immune responses than women and more often develop severe COVID infections. Recent research suggests boys with COVID are more likely than girls to become seriously ill or develop a rare inflammatory condition called multisystem inflammatory syndrome.
New research on COVID could help illuminate this vulnerability.
In a study published in October, researchers found that the sex of a fetus influences the way its placenta responds to COVID, as well as how its mother’s immune system responds.
Pregnant people infected with COVID made fewer antibodies against the coronavirus if they were carrying male fetuses than if they were carrying females. Mothers also transferred fewer antibodies to boys than to girls, said Andrea Edlow, MD, senior author of the study and a maternal-fetal medicine specialist at Massachusetts General Hospital, Boston.
When examining the placentas of male fetuses after delivery, researchers found changes that could leave boys less protected against damaging inflammation.
The sex of a fetus can influence its mother’s response to other illnesses, as well.
For example, research shows that pregnant women with asthma have worse symptoms if they’re carrying a female. Women carrying males are slightly more likely to develop gestational diabetes.
Dr. Edlow said her findings raise questions about the “cross talk” between mother and baby. “The mom’s immune system is sensing there is a male fetus,” Dr. Edlow said. “And the fetus is actively communicating with the mom’s immune system.”
Boosting toxic stress
Rates of depression and stress among pregnant women have increased dramatically during the pandemic.
That’s concerning because chronic stress can lead to inflammation, affecting the babies of both infected and uninfected women, Dr. Anagnostou said.
Studies consistently show that infants born to mothers who experience significant stress during pregnancy have higher rates of short- and long-term health damage – including heart defects and obesity – than babies born to women with less stress.
“We know that inflammation directly influences the way a baby’s brain develops,” said Elinor Sullivan, PhD, an associate professor in psychiatry at Oregon Health & Science University, Portland.
Lockdowns, travel restrictions and physical distancing left many pregnant women without the support of family and friends. The stress of losing a loved one, a job, or a home further heightens the risks to moms and babies, said Dr. Sullivan, who is following children born during the pandemic for 5 years.
In research that has not yet been published, Dr. Sullivan found that babies of women who were pregnant during the pandemic showed more sadness and negative emotions in the first year of life, compared with infants of women who were pregnant before the pandemic.
The findings show the importance of helping and protecting pregnant people before and after delivery, said Dr. Sullivan, who conducted a separate study that found women who received more social support were less depressed.
Italian researchers are also studying the effect of maternal stress on infants’ behavior, as well as the way their genes are regulated.
Although stress-related inflammation doesn’t alter the structure of a baby’s genes, it can influence whether they’re turned on and off, said Livio Provenzi, PhD, a psychologist at the C. Mondino National Institute of Neurology Foundation in Pavia, Italy.
In Dr. Provenzi’s study of 163 mother-baby pairs he found differences in how genes that regulate the stress response were activated. Genes that help people respond to stress were more likely to be turned off in babies whose moms reported the most stress during pregnancy. The same moms also reported that their babies cried more and were fussier when they were 3 months old.
Researchers usually prefer to make in-person observations of babies as they interact with their mothers, Dr. Provenzi said. But because of the pandemic, Dr. Provenzi asked mothers to fill out questionnaires about infant behavior. He plans to observe mothers and babies in person when the children are 12 months old.
While vaccinating pregnant people is the best way to protect them and their fetuses from the virus, Dr. Anagnostou said, society needs to do more to preserve expectant mothers’ mental health.
“We can’t escape the fact that we’ve lived through 2 years of a pandemic,” Dr. Anagnostou said. “But we can think about opportunities for reducing the risk.”
KHN (Kaiser Health News) is a national newsroom that produces in-depth journalism about health issues. Together with Policy Analysis and Polling, KHN is one of the three major operating programs at KFF (Kaiser Family Foundation). KFF is an endowed nonprofit organization providing information on health issues to the nation.
Transcranial magnetic stimulation shows promise for alcohol addiction
Deep, repetitive transcranial magnetic stimulation (TMS) is safe and effective in decreasing symptoms of alcohol addiction and brain reactivity, new research suggests.

In a randomized, double-blind, sham-controlled trial, participants who received TMS targeting the medial prefrontal cortex (mPFC) and anterior cingulate cortex (ACC) for 3 weeks showed significantly reduced heavy drinking days, compared with a group who received a sham treatment.
and showed less functional connectivity on MRI in areas of the brain that can trigger craving and relapse.
Clinicians should “keep their eyes open” in the wake of this phase 2 trial, cocorresponding author Markus Heilig, MD, PhD, professor of psychiatry and director at the Center for Social and Affective Neuroscience, department of biomedical and clinical sciences, Linköping (Sweden) University, said in an interview.
“If and when this replicates in the equivalent of a phase 3 study, we will actually have a completely novel treatment available for this difficult to treat and very impactful disease,” Dr. Heilig said.
The findings were published online Dec. 5, 2021, in Biological Psychiatry.
Proof of concept
In the proof-of-concept trial, researchers enrolled and randomly assigned 51 treatment-seeking adults with moderate to severe alcohol dependence to receive active or sham treatment. Before treatment, participants completed “craving induction,” which included holding and smelling but not consuming an alcoholic beverage.
Dr. Heilig noted that, before stimulating the brain, “you want to make it as malleable as possible;” and brain networks tend to be more malleable when they are active.
During the 3-week treatment phase, active or sham stimulations were delivered in five 30-minute sessions per week. During the sessions, all participants wore a helmet with a deep TMS coil produced by BrainsWay.
In the active-stimulation group, each session included 100 trains of 30 pulses at 10 Hz (3 seconds) with 15-second intervals, for a total of 3,000 pulses. The sham stimulation produced the same acoustic artifact and generated skin sensations mimicking those of the active stimulation, but it did not involve a magnetic field.
Participants, operators, and raters were blinded to the type of coil used.
Five participants relapsed during the first 3 weeks of treatment and were excluded from the analysis. The mean age of those completing treatment (n = 23 in each group) was 43 years, and two-thirds were men.
The gender makeup of the study reflects “what a real treatment-seeking group looks like,” Dr. Heilig said.
During the 12-week follow-up phase, five additional participants dropped out.
‘Pretty robust’ treatment effect
The primary outcome was reduction in percentage of heavy drinking days (pHDD), defined as consuming at least five drinks of 12 grams of alcohol per day for men and at least four such drinks for women.
Initially, pHDD dropped in both groups, which is something generally seen in alcohol studies, said Dr. Heilig. “The moment people decide to participate in a study, everybody drops their consumption, [which] biases a study like this against picking up an effect.”
However, heavy drinking days increased during follow-up in the sham group but remained low in the active-treatment group. The mean pHDD was significantly lower in the active versus sham groups (2.9% vs. 10.6%, P = .037).
“So despite the bias, a treatment effect does emerge,” and was “pretty robust,” Dr. Heilig said.
This was supported by a significant group difference in weekly alcohol consumption and a trend-level difference in percentage of alcohol-positive urine samples.
A secondary outcome was change in alcohol craving, assessed with the Penn Alcohol Craving Scale; PACS scores decreased in both groups during treatment but was more steeply reduced in the active group. During follow-up, craving levels increased to a lesser extent in the active group.
MRI scans showed reduced connectivity from the mPFC to the subgenual ACC, an area involved in negative emotions that can trigger craving and relapse, said Dr. Heilig. There was also reduced connectivity between the dorsal ACC and caudate, a circuit involved in the reward system.
In treatment trials, researchers look for a biomarker of target engagement. However, “to date, there has been no study using TMS that has actually demonstrated the intervention had a measurable effect on brain activity. So to me, this is a biomarker; it did something to the brain,” Dr. Heilig said.
Delving deeper into the brain
The results underline the importance of stimulating deeper parts of the brain, cocorresponding author Abraham Zangen, PhD, head of the Brain Stimulation and Behavior Lab and chair of the psychobiology brain program, Ben Gurion University, Be’er Sheva, Israel, said in an interview.
Early TMS studies, which involved superficial brain stimulation, reduced cigarette consumption but was not associated with quitting, Dr. Zangen said. “It was only when we targeted deeper parts of the prefrontal cortex that we were able to induce smoking cessation.”
It was this research that led to approval by the Food and Drug Administration of deep TMS for smoking cessation.
This same deep-brain approach was used in the current study. “So the emphasis on the technology that allows penetration into deeper parts of the brain and targeting the relevant pathological circuitry of addiction is a key complement of the success of this study,” Dr. Zangen said.
Results also showed no serious adverse events. Only a few participants reported transient headaches, which all resolved spontaneously; and frequency did not differ between groups.
Dr. Heilig now hopes to carry out a multisite phase 3 study of the intervention and would suggest it involve 4 (instead of 3) weeks of initial treatment and then a weekly booster session. “There are biological reasons to believe that might be more efficient, although we don’t have the data,” he said.
On the other hand, he noted, the longer the trial, the more difficult it might be to recruit patients.
Clinically significant?
Commenting on the study, Derek Blevins, MD, assistant professor of clinical psychiatry at Columbia University and research psychiatrist, division on substance use disorders, New York State Psychiatric Institute, both in New York, called the research “really exciting.”
To date, most TMS studies have been relatively small and looked at a target such as craving. Although these studies did show some effect, the clinical significance of that effect was unclear, said Dr. Blevins, who was not involved with the current research.
“I think this new study actually demonstrated a clinically significant effect of a noninvasive treatment for a disease that’s very difficult to treat,” he said.
A potential limitation of the study, however, was it required abstinence, Dr. Blevins noted. It would be “really helpful” to understand how TMS might aid individuals such as those who relapsed during the study, “because they’re the more treatment-refractory individuals we see in clinical practice.”
If a multicenter trial is launched, Dr. Blevins said he would also like it to include an ethnically and racially diverse population.
The study was supported by grants from the European Union’s Horizon 2020 research and innovation program and the Swedish Research Council. Dr. Heilig reported having received consulting fees, research support, or other compensation from Indivior, Camurus, BrainsWay, Aelis Farma, and Janssen Pharmaceuticals. Dr. Zangen is an inventor of deep TMS coils and has financial interest in BrainsWay, which produces and markets these coils. Dr. Blevins reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Deep, repetitive transcranial magnetic stimulation (TMS) is safe and effective in decreasing symptoms of alcohol addiction and brain reactivity, new research suggests.
In a randomized, double-blind, sham-controlled trial, participants who received TMS targeting the medial prefrontal cortex (mPFC) and anterior cingulate cortex (ACC) for 3 weeks showed significantly reduced heavy drinking days, compared with a group who received a sham treatment.
and showed less functional connectivity on MRI in areas of the brain that can trigger craving and relapse.
Clinicians should “keep their eyes open” in the wake of this phase 2 trial, cocorresponding author Markus Heilig, MD, PhD, professor of psychiatry and director at the Center for Social and Affective Neuroscience, department of biomedical and clinical sciences, Linköping (Sweden) University, said in an interview.
“If and when this replicates in the equivalent of a phase 3 study, we will actually have a completely novel treatment available for this difficult to treat and very impactful disease,” Dr. Heilig said.
The findings were published online Dec. 5, 2021, in Biological Psychiatry.
Proof of concept
In the proof-of-concept trial, researchers enrolled and randomly assigned 51 treatment-seeking adults with moderate to severe alcohol dependence to receive active or sham treatment. Before treatment, participants completed “craving induction,” which included holding and smelling but not consuming an alcoholic beverage.
Dr. Heilig noted that, before stimulating the brain, “you want to make it as malleable as possible;” and brain networks tend to be more malleable when they are active.
During the 3-week treatment phase, active or sham stimulations were delivered in five 30-minute sessions per week. During the sessions, all participants wore a helmet with a deep TMS coil produced by BrainsWay.
In the active-stimulation group, each session included 100 trains of 30 pulses at 10 Hz (3 seconds) with 15-second intervals, for a total of 3,000 pulses. The sham stimulation produced the same acoustic artifact and generated skin sensations mimicking those of the active stimulation, but it did not involve a magnetic field.
Participants, operators, and raters were blinded to the type of coil used.
Five participants relapsed during the first 3 weeks of treatment and were excluded from the analysis. The mean age of those completing treatment (n = 23 in each group) was 43 years, and two-thirds were men.
The gender makeup of the study reflects “what a real treatment-seeking group looks like,” Dr. Heilig said.
During the 12-week follow-up phase, five additional participants dropped out.
‘Pretty robust’ treatment effect
The primary outcome was reduction in percentage of heavy drinking days (pHDD), defined as consuming at least five drinks of 12 grams of alcohol per day for men and at least four such drinks for women.
Initially, pHDD dropped in both groups, which is something generally seen in alcohol studies, said Dr. Heilig. “The moment people decide to participate in a study, everybody drops their consumption, [which] biases a study like this against picking up an effect.”
However, heavy drinking days increased during follow-up in the sham group but remained low in the active-treatment group. The mean pHDD was significantly lower in the active versus sham groups (2.9% vs. 10.6%, P = .037).
“So despite the bias, a treatment effect does emerge,” and was “pretty robust,” Dr. Heilig said.
This was supported by a significant group difference in weekly alcohol consumption and a trend-level difference in percentage of alcohol-positive urine samples.
A secondary outcome was change in alcohol craving, assessed with the Penn Alcohol Craving Scale; PACS scores decreased in both groups during treatment but was more steeply reduced in the active group. During follow-up, craving levels increased to a lesser extent in the active group.
MRI scans showed reduced connectivity from the mPFC to the subgenual ACC, an area involved in negative emotions that can trigger craving and relapse, said Dr. Heilig. There was also reduced connectivity between the dorsal ACC and caudate, a circuit involved in the reward system.
In treatment trials, researchers look for a biomarker of target engagement. However, “to date, there has been no study using TMS that has actually demonstrated the intervention had a measurable effect on brain activity. So to me, this is a biomarker; it did something to the brain,” Dr. Heilig said.
Delving deeper into the brain
The results underline the importance of stimulating deeper parts of the brain, cocorresponding author Abraham Zangen, PhD, head of the Brain Stimulation and Behavior Lab and chair of the psychobiology brain program, Ben Gurion University, Be’er Sheva, Israel, said in an interview.
Early TMS studies, which involved superficial brain stimulation, reduced cigarette consumption but was not associated with quitting, Dr. Zangen said. “It was only when we targeted deeper parts of the prefrontal cortex that we were able to induce smoking cessation.”
It was this research that led to approval by the Food and Drug Administration of deep TMS for smoking cessation.
This same deep-brain approach was used in the current study. “So the emphasis on the technology that allows penetration into deeper parts of the brain and targeting the relevant pathological circuitry of addiction is a key complement of the success of this study,” Dr. Zangen said.
Results also showed no serious adverse events. Only a few participants reported transient headaches, which all resolved spontaneously; and frequency did not differ between groups.
Dr. Heilig now hopes to carry out a multisite phase 3 study of the intervention and would suggest it involve 4 (instead of 3) weeks of initial treatment and then a weekly booster session. “There are biological reasons to believe that might be more efficient, although we don’t have the data,” he said.
On the other hand, he noted, the longer the trial, the more difficult it might be to recruit patients.
Clinically significant?
Commenting on the study, Derek Blevins, MD, assistant professor of clinical psychiatry at Columbia University and research psychiatrist, division on substance use disorders, New York State Psychiatric Institute, both in New York, called the research “really exciting.”
To date, most TMS studies have been relatively small and looked at a target such as craving. Although these studies did show some effect, the clinical significance of that effect was unclear, said Dr. Blevins, who was not involved with the current research.
“I think this new study actually demonstrated a clinically significant effect of a noninvasive treatment for a disease that’s very difficult to treat,” he said.
A potential limitation of the study, however, was it required abstinence, Dr. Blevins noted. It would be “really helpful” to understand how TMS might aid individuals such as those who relapsed during the study, “because they’re the more treatment-refractory individuals we see in clinical practice.”
If a multicenter trial is launched, Dr. Blevins said he would also like it to include an ethnically and racially diverse population.
The study was supported by grants from the European Union’s Horizon 2020 research and innovation program and the Swedish Research Council. Dr. Heilig reported having received consulting fees, research support, or other compensation from Indivior, Camurus, BrainsWay, Aelis Farma, and Janssen Pharmaceuticals. Dr. Zangen is an inventor of deep TMS coils and has financial interest in BrainsWay, which produces and markets these coils. Dr. Blevins reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Deep, repetitive transcranial magnetic stimulation (TMS) is safe and effective in decreasing symptoms of alcohol addiction and brain reactivity, new research suggests.
In a randomized, double-blind, sham-controlled trial, participants who received TMS targeting the medial prefrontal cortex (mPFC) and anterior cingulate cortex (ACC) for 3 weeks showed significantly reduced heavy drinking days, compared with a group who received a sham treatment.
and showed less functional connectivity on MRI in areas of the brain that can trigger craving and relapse.
Clinicians should “keep their eyes open” in the wake of this phase 2 trial, cocorresponding author Markus Heilig, MD, PhD, professor of psychiatry and director at the Center for Social and Affective Neuroscience, department of biomedical and clinical sciences, Linköping (Sweden) University, said in an interview.
“If and when this replicates in the equivalent of a phase 3 study, we will actually have a completely novel treatment available for this difficult to treat and very impactful disease,” Dr. Heilig said.
The findings were published online Dec. 5, 2021, in Biological Psychiatry.
Proof of concept
In the proof-of-concept trial, researchers enrolled and randomly assigned 51 treatment-seeking adults with moderate to severe alcohol dependence to receive active or sham treatment. Before treatment, participants completed “craving induction,” which included holding and smelling but not consuming an alcoholic beverage.
Dr. Heilig noted that, before stimulating the brain, “you want to make it as malleable as possible;” and brain networks tend to be more malleable when they are active.
During the 3-week treatment phase, active or sham stimulations were delivered in five 30-minute sessions per week. During the sessions, all participants wore a helmet with a deep TMS coil produced by BrainsWay.
In the active-stimulation group, each session included 100 trains of 30 pulses at 10 Hz (3 seconds) with 15-second intervals, for a total of 3,000 pulses. The sham stimulation produced the same acoustic artifact and generated skin sensations mimicking those of the active stimulation, but it did not involve a magnetic field.
Participants, operators, and raters were blinded to the type of coil used.
Five participants relapsed during the first 3 weeks of treatment and were excluded from the analysis. The mean age of those completing treatment (n = 23 in each group) was 43 years, and two-thirds were men.
The gender makeup of the study reflects “what a real treatment-seeking group looks like,” Dr. Heilig said.
During the 12-week follow-up phase, five additional participants dropped out.
‘Pretty robust’ treatment effect
The primary outcome was reduction in percentage of heavy drinking days (pHDD), defined as consuming at least five drinks of 12 grams of alcohol per day for men and at least four such drinks for women.
Initially, pHDD dropped in both groups, which is something generally seen in alcohol studies, said Dr. Heilig. “The moment people decide to participate in a study, everybody drops their consumption, [which] biases a study like this against picking up an effect.”
However, heavy drinking days increased during follow-up in the sham group but remained low in the active-treatment group. The mean pHDD was significantly lower in the active versus sham groups (2.9% vs. 10.6%, P = .037).
“So despite the bias, a treatment effect does emerge,” and was “pretty robust,” Dr. Heilig said.
This was supported by a significant group difference in weekly alcohol consumption and a trend-level difference in percentage of alcohol-positive urine samples.
A secondary outcome was change in alcohol craving, assessed with the Penn Alcohol Craving Scale; PACS scores decreased in both groups during treatment but was more steeply reduced in the active group. During follow-up, craving levels increased to a lesser extent in the active group.
MRI scans showed reduced connectivity from the mPFC to the subgenual ACC, an area involved in negative emotions that can trigger craving and relapse, said Dr. Heilig. There was also reduced connectivity between the dorsal ACC and caudate, a circuit involved in the reward system.
In treatment trials, researchers look for a biomarker of target engagement. However, “to date, there has been no study using TMS that has actually demonstrated the intervention had a measurable effect on brain activity. So to me, this is a biomarker; it did something to the brain,” Dr. Heilig said.
Delving deeper into the brain
The results underline the importance of stimulating deeper parts of the brain, cocorresponding author Abraham Zangen, PhD, head of the Brain Stimulation and Behavior Lab and chair of the psychobiology brain program, Ben Gurion University, Be’er Sheva, Israel, said in an interview.
Early TMS studies, which involved superficial brain stimulation, reduced cigarette consumption but was not associated with quitting, Dr. Zangen said. “It was only when we targeted deeper parts of the prefrontal cortex that we were able to induce smoking cessation.”
It was this research that led to approval by the Food and Drug Administration of deep TMS for smoking cessation.
This same deep-brain approach was used in the current study. “So the emphasis on the technology that allows penetration into deeper parts of the brain and targeting the relevant pathological circuitry of addiction is a key complement of the success of this study,” Dr. Zangen said.
Results also showed no serious adverse events. Only a few participants reported transient headaches, which all resolved spontaneously; and frequency did not differ between groups.
Dr. Heilig now hopes to carry out a multisite phase 3 study of the intervention and would suggest it involve 4 (instead of 3) weeks of initial treatment and then a weekly booster session. “There are biological reasons to believe that might be more efficient, although we don’t have the data,” he said.
On the other hand, he noted, the longer the trial, the more difficult it might be to recruit patients.
Clinically significant?
Commenting on the study, Derek Blevins, MD, assistant professor of clinical psychiatry at Columbia University and research psychiatrist, division on substance use disorders, New York State Psychiatric Institute, both in New York, called the research “really exciting.”
To date, most TMS studies have been relatively small and looked at a target such as craving. Although these studies did show some effect, the clinical significance of that effect was unclear, said Dr. Blevins, who was not involved with the current research.
“I think this new study actually demonstrated a clinically significant effect of a noninvasive treatment for a disease that’s very difficult to treat,” he said.
A potential limitation of the study, however, was it required abstinence, Dr. Blevins noted. It would be “really helpful” to understand how TMS might aid individuals such as those who relapsed during the study, “because they’re the more treatment-refractory individuals we see in clinical practice.”
If a multicenter trial is launched, Dr. Blevins said he would also like it to include an ethnically and racially diverse population.
The study was supported by grants from the European Union’s Horizon 2020 research and innovation program and the Swedish Research Council. Dr. Heilig reported having received consulting fees, research support, or other compensation from Indivior, Camurus, BrainsWay, Aelis Farma, and Janssen Pharmaceuticals. Dr. Zangen is an inventor of deep TMS coils and has financial interest in BrainsWay, which produces and markets these coils. Dr. Blevins reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM BIOLOGICAL PSYCHIATRY
Infectious disease pop quiz: Clinical challenge #7 for the ObGyn
What is the most appropriate treatment for trichomonas infection in pregnancy?
Continue to the answer...
Trichomonas infection should be treated with oral metronidazole 500 mg twice daily for 7 days. Metronidazole also can be given as a single oral 2-g dose. This treatment is not quite as effective as the multidose regimen, but it may be appropriate for patients who are not likely to be adherent with the longer course of treatment.
Resistance to metronidazole is rare; in such instances, oral tinidazole 2 g in a single dose may be effective.
- Duff P. Maternal and perinatal infections: bacterial. In: Landon MB, Galan HL, Jauniaux ERM, et al. Gabbe’s Obstetrics: Normal and Problem Pregnancies. 8th ed. Elsevier; 2021:1124-1146.
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TJ, et al. Creasy & Resnik’s Maternal-Fetal Medicine: Principles and Practice. 8th ed. Elsevier; 2019:862-919.

Dr. Edwards is a Resident in the Department of Medicine, University of Florida College of Medicine, Gainesville.
Dr. Duff is Professor of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
Dr. Edwards is a Resident in the Department of Medicine, University of Florida College of Medicine, Gainesville.
Dr. Duff is Professor of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
Dr. Edwards is a Resident in the Department of Medicine, University of Florida College of Medicine, Gainesville.
Dr. Duff is Professor of Maternal-Fetal Medicine, Department of Obstetrics and Gynecology, University of Florida College of Medicine, Gainesville.
The authors report no financial relationships relevant to this article.
What is the most appropriate treatment for trichomonas infection in pregnancy?
Continue to the answer...
Trichomonas infection should be treated with oral metronidazole 500 mg twice daily for 7 days. Metronidazole also can be given as a single oral 2-g dose. This treatment is not quite as effective as the multidose regimen, but it may be appropriate for patients who are not likely to be adherent with the longer course of treatment.
Resistance to metronidazole is rare; in such instances, oral tinidazole 2 g in a single dose may be effective.
What is the most appropriate treatment for trichomonas infection in pregnancy?
Continue to the answer...
Trichomonas infection should be treated with oral metronidazole 500 mg twice daily for 7 days. Metronidazole also can be given as a single oral 2-g dose. This treatment is not quite as effective as the multidose regimen, but it may be appropriate for patients who are not likely to be adherent with the longer course of treatment.
Resistance to metronidazole is rare; in such instances, oral tinidazole 2 g in a single dose may be effective.
- Duff P. Maternal and perinatal infections: bacterial. In: Landon MB, Galan HL, Jauniaux ERM, et al. Gabbe’s Obstetrics: Normal and Problem Pregnancies. 8th ed. Elsevier; 2021:1124-1146.
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TJ, et al. Creasy & Resnik’s Maternal-Fetal Medicine: Principles and Practice. 8th ed. Elsevier; 2019:862-919.
- Duff P. Maternal and perinatal infections: bacterial. In: Landon MB, Galan HL, Jauniaux ERM, et al. Gabbe’s Obstetrics: Normal and Problem Pregnancies. 8th ed. Elsevier; 2021:1124-1146.
- Duff P. Maternal and fetal infections. In: Resnik R, Lockwood CJ, Moore TJ, et al. Creasy & Resnik’s Maternal-Fetal Medicine: Principles and Practice. 8th ed. Elsevier; 2019:862-919.
The mess that is matching in psychiatry
The day I interviewed at Johns Hopkins in Baltimore, like every other day of residency interviews, was a very long and draining day.

I started by meeting alone with Philip Slavney, MD, the residency director, who spoke with me about the program and gave me a schedule to follow. I was to meet with residents and psychiatrists, some of whom had graduated from my medical school, and was sent to the Bayview campus a few miles away to have lunch and attend a few meetings. By the time I boarded an Amtrak train at Baltimore Penn Station, I was tired but I liked what I had seen. By the end of the interview season, I had crossed four programs off my list and had decided to rank only three.
In 1987, there were 987 residency positions in psychiatry in the United States, and 83.6% of those positions filled with a combination of U.S. and international medical graduates. Still, this was a risky move; the programs that I decided to rank would fill, but I was matching separately for an internship year in internal medicine in New York and decided that I would rather reapply in a year than risk matching at a program I didn’t want to go to.
I wasn’t quite sure where I wanted to rank Hopkins on my list, so I called Dr. Slavney and said I wanted to come back and meet more members of the department. He did not hide his surprise and was quick to tell me that no one had ever requested a second set of interviews. I mentioned specific people I wanted to meet with, and he was kind enough to accommodate my request and set up a second day of interviews for me.
Needless to say, the residency match felt very personal – at least to me – and although I felt vulnerable, I also felt empowered. Because of the low pay, patients with stigmatized illnesses, and the rampant belief that psychiatry was not “real” medicine and the patients never got better, psychiatry was not a desired specialty.
The residency application process in psychiatry (and every other specialty) has become a much different process. In 2006, the Association of American Medical Colleges called on medical schools to increase their enrollments to address the national shortage of physicians. Soon, there were more medical schools, bigger classes, and more doctors being minted, but the Balanced Budget Act of 1997 prevented a proportional increase in residency positions.
Len Marquez, senior director of government relations at the AAMC noted: “The Resident Physician Shortage Reduction Act of 2021 (S. 834), sponsored by Sen. Robert Menendez (D-N.J.), Sen. John Boozman (R-Ark.), and Majority Leader Sen. Charles Schumer (D-N.Y.), would support 2,000 additional Medicare-supported residency positions each year for 7 years, but Congress has not yet acted on the legislation. We were very pleased that last year, Congress passed the first increase in Medicare-supported graduate medical education in 25 years by including 1,000 new slots as part of the Consolidated Appropriations Act, 2021.”
In addition, the Build Back Better Act, which is currently being debated in Congress, would provide 4,000 more graduate medical education slots, including a specific requirement that 15% of them go to “psychiatry-related residencies,” he added.
Over 90% of graduates from U.S. medical schools currently match into a residency position. That statistic for international medical graduates is notably lower, with perhaps as few as 50% of all applicants matching.
Since 2014, the number of applicants to psychiatry residencies has nearly doubled. For the 2021 match, there were 2,486 applicants applying for 1,858 positions in psychiatry – so 1.34 applicants for each slot. Of the 1,117 senior medical students at U.S. schools who applied to psychiatry residencies, 129 did not match. Overall, 99.8% of residency positions in psychiatry filled.
“It used to be less competitive,” said Kaz J. Nelson, MD, the vice chair for education at the University of Minnesota’s department of psychiatry and behavioral sciences in Minneapolis, adding that interest in psychiatry has increased over the years.
“Interest has skyrocketed as the word has gotten out about how great a field it is. It helps that reimbursements are better, that there is less bias and discrimination against patients with psychiatric issues, and that psychiatric care is seen as a legitimate part of medicine. It has been exciting to watch!” Dr. Nelson said.
The numbers are only one part of the story, however.
Application submission now involves a centralized, electronic process, and it has become easier for applicants to apply to a lot of programs indiscriminately. It’s not unusual for applicants to apply to 70 or more programs. The factors that have limited applications include the cost: Electronic Residency Application Services (ERAS) charges for each application package they send to a program, and applicants traditionally pay to travel to the programs where they interview. This all changed with the 2021 cycle when in-person interviews were halted for the pandemic and interviews became virtual. While I recall applying to 7 residency programs, this year the average number of applications was 54.7 per applicant.
“It used to be that the cap on interviewing was financial,” Dr. Nelson said. “It was discriminatory and favored those who had more money to travel to interviews. There are still the ERAS fees, but COVID has been an equalizer and we are getting more applicants, and interviewing more who are not from Minnesota or the Midwest. We have been working to make our program attractive in terms of diversity, equity, inclusion, and justice. Our hospital is located a mile from where George Floyd was murdered, and it’s our responsibility to lead the effort to ensure the psychiatry workforce is diverse, and inclusive, as possible.”
Daniel E. Gih, MD, is the program director for a new psychiatry residency at the University of Nebraska, Omaha. When the program started in 2019, there were spots for four residents and the program had 588 applications. In 2020, the program grew to five positions and this year there were 553 applicants. Dr. Gih attributed the high number of applications to his program’s strong social media presence.
“Going through the applications and meeting the students are some of the most enjoyable parts of my work,” Dr. Gih said. “I feel guilty though, that I’m likely going to miss a great applicant. Each application averages 35 pages and it’s inevitable that programs have to take shortcuts. Applicants worry that they’ll be ranked by board scores. While we certainly don’t do that here, students might feel ruled out of a program if their numbers aren’t high enough. Furthermore, wealthy students can apply to more programs. The pandemic has really highlighted the inequity issues.”
Dr. Gih noted that the Zoom interview process has not been disappointing: “Two of the people we matched had never been to Omaha, and many expressed concerns about what it is like here. Of course, on Zoom you don’t catch subtle interpersonal issues, but we have been pleasantly surprised that the people we matched were consistent with what we expected. It is exciting to meet the people who will eventually replace us as psychiatrists, they will be here to deal with future challenges!” His enthusiasm was tangible.
While the program directors remain optimistic, the system is not without its stresses, as many programs receive over 1,000 applications.
“This is difficult,” Dr. Nelson said.” It’s wonderful for the programs, but for the medical students, not matching is experienced by them as being catastrophic, so they apply to a lot of programs. Getting this many applications is a challenge, yet I don’t want to interview someone if they are going to rank our program No. 80 on their list!”
Residencies have dealt with the deluge of applicants in a number of ways. Some specialties started a “signal” protocol wherein candidates and programs receive a certain number of tokens to indicate that each would rank the other highly, but psychiatry has not done this. Early on in the Zoom process, multiple applicants would be offered interviews simultaneously, and the interview would be given to the candidate who responded first. Students vented their frustrations on Twitter when they lost interview spots at their coveted programs because they hadn’t checked their email in time or had gone to take a shower.
“The American Association of Directors of Psychiatry Residency Training Programs issued guidelines saying that it is unacceptable to offer interview spots without allowing a reasonable time for the applicant to respond, and that it is not appropriate to offer multiple candidates one spot on a first-come, first-serve basis,” Dr. Nelson explained.
Her program has managed some of the application chaos by using a software program called Scutmonkey, codeveloped by David Ross, MD, PhD, the associate program director of the Yale Adult Psychiatry Residency Program.
“It lets us screen applications for candidates who specifically are interested in being here, and for those who qualify as part of the mission we are trying to fulfill.”
One fourth-year student at a mid-Atlantic medical school who is applying in psychiatry – who I’ll call Sacha to protect his anonymity – applied to 73 psychiatry programs and to date, has interviewed at 6. He describes a stressful, roller coaster experience:
“I got those six interviews right away and that was an amazing start, but then I didn’t get any more. The interviews I had went well, but it has been disappointing not to have more. Some were all-day interviews, while other programs had me meet with residents and attendings for 20 minutes each and it was all done after 2 hours.”
He has mixed opinions about not seeing the schools in person. “There are very heavy pros and cons. I’ve saved thousands of dollars in travel expenses that would have limited my applications, so logistically it’s a dream. On the other hand, I’ve interviewed in cities I have never been to, it’s hard to get a sense of the intangibles of a program, and the shorter interviews feel very impersonal.”
Sacha expressed anxieties about the process. “With so many applicants, it’s difficult for someone with a nontraditional story to get a spot and it’s easier for the programs to toss applications. it’s common enough and everyone has seen someone who has gone through this. At times, we feel powerless; we have no real agency or control. We send stuff out and then we sit in the prayer position and wait.”
I think back on my own application process with a sense of gratitude. I certainly didn’t feel powerless, and in today’s world, postinterview communications with program directors are regulated for both parties. Dr. Slavney was kind enough to humor my request, but I don’t believe this would be feasible in the current environment.
Even though it is wonderful that more doctors have figured out that careers in psychiatry are rewarding, the current situation is overwhelming for both the applicants and the programs. With over 100 applicants for every position – many of whom will have no interest in going to some of the programs they apply to – qualified candidates who go unmatched, and a roulette wheel which requires heavily indebted students to pay to apply, this is simply not sustainable in a country with a shortage of physicians – psychiatrists in particular.
We hear that mid-level practitioners are the answer to our shortages, but perhaps we need to create a system with enough residency positions to accommodate highly trained and qualified physicians in a more inviting and targeted way.
Dinah Miller, MD, is coauthor of “Committed: The Battle Over Involuntary Psychiatric Care” (Baltimore: Johns Hopkins University Press, 2016). She has a private practice and is assistant professor of psychiatry and behavioral sciences at Johns Hopkins, both in Baltimore. A version of this article first appeared on Medscape.com.
The day I interviewed at Johns Hopkins in Baltimore, like every other day of residency interviews, was a very long and draining day.
I started by meeting alone with Philip Slavney, MD, the residency director, who spoke with me about the program and gave me a schedule to follow. I was to meet with residents and psychiatrists, some of whom had graduated from my medical school, and was sent to the Bayview campus a few miles away to have lunch and attend a few meetings. By the time I boarded an Amtrak train at Baltimore Penn Station, I was tired but I liked what I had seen. By the end of the interview season, I had crossed four programs off my list and had decided to rank only three.
In 1987, there were 987 residency positions in psychiatry in the United States, and 83.6% of those positions filled with a combination of U.S. and international medical graduates. Still, this was a risky move; the programs that I decided to rank would fill, but I was matching separately for an internship year in internal medicine in New York and decided that I would rather reapply in a year than risk matching at a program I didn’t want to go to.
I wasn’t quite sure where I wanted to rank Hopkins on my list, so I called Dr. Slavney and said I wanted to come back and meet more members of the department. He did not hide his surprise and was quick to tell me that no one had ever requested a second set of interviews. I mentioned specific people I wanted to meet with, and he was kind enough to accommodate my request and set up a second day of interviews for me.
Needless to say, the residency match felt very personal – at least to me – and although I felt vulnerable, I also felt empowered. Because of the low pay, patients with stigmatized illnesses, and the rampant belief that psychiatry was not “real” medicine and the patients never got better, psychiatry was not a desired specialty.
The residency application process in psychiatry (and every other specialty) has become a much different process. In 2006, the Association of American Medical Colleges called on medical schools to increase their enrollments to address the national shortage of physicians. Soon, there were more medical schools, bigger classes, and more doctors being minted, but the Balanced Budget Act of 1997 prevented a proportional increase in residency positions.
Len Marquez, senior director of government relations at the AAMC noted: “The Resident Physician Shortage Reduction Act of 2021 (S. 834), sponsored by Sen. Robert Menendez (D-N.J.), Sen. John Boozman (R-Ark.), and Majority Leader Sen. Charles Schumer (D-N.Y.), would support 2,000 additional Medicare-supported residency positions each year for 7 years, but Congress has not yet acted on the legislation. We were very pleased that last year, Congress passed the first increase in Medicare-supported graduate medical education in 25 years by including 1,000 new slots as part of the Consolidated Appropriations Act, 2021.”
In addition, the Build Back Better Act, which is currently being debated in Congress, would provide 4,000 more graduate medical education slots, including a specific requirement that 15% of them go to “psychiatry-related residencies,” he added.
Over 90% of graduates from U.S. medical schools currently match into a residency position. That statistic for international medical graduates is notably lower, with perhaps as few as 50% of all applicants matching.
Since 2014, the number of applicants to psychiatry residencies has nearly doubled. For the 2021 match, there were 2,486 applicants applying for 1,858 positions in psychiatry – so 1.34 applicants for each slot. Of the 1,117 senior medical students at U.S. schools who applied to psychiatry residencies, 129 did not match. Overall, 99.8% of residency positions in psychiatry filled.
“It used to be less competitive,” said Kaz J. Nelson, MD, the vice chair for education at the University of Minnesota’s department of psychiatry and behavioral sciences in Minneapolis, adding that interest in psychiatry has increased over the years.
“Interest has skyrocketed as the word has gotten out about how great a field it is. It helps that reimbursements are better, that there is less bias and discrimination against patients with psychiatric issues, and that psychiatric care is seen as a legitimate part of medicine. It has been exciting to watch!” Dr. Nelson said.
The numbers are only one part of the story, however.
Application submission now involves a centralized, electronic process, and it has become easier for applicants to apply to a lot of programs indiscriminately. It’s not unusual for applicants to apply to 70 or more programs. The factors that have limited applications include the cost: Electronic Residency Application Services (ERAS) charges for each application package they send to a program, and applicants traditionally pay to travel to the programs where they interview. This all changed with the 2021 cycle when in-person interviews were halted for the pandemic and interviews became virtual. While I recall applying to 7 residency programs, this year the average number of applications was 54.7 per applicant.
“It used to be that the cap on interviewing was financial,” Dr. Nelson said. “It was discriminatory and favored those who had more money to travel to interviews. There are still the ERAS fees, but COVID has been an equalizer and we are getting more applicants, and interviewing more who are not from Minnesota or the Midwest. We have been working to make our program attractive in terms of diversity, equity, inclusion, and justice. Our hospital is located a mile from where George Floyd was murdered, and it’s our responsibility to lead the effort to ensure the psychiatry workforce is diverse, and inclusive, as possible.”
Daniel E. Gih, MD, is the program director for a new psychiatry residency at the University of Nebraska, Omaha. When the program started in 2019, there were spots for four residents and the program had 588 applications. In 2020, the program grew to five positions and this year there were 553 applicants. Dr. Gih attributed the high number of applications to his program’s strong social media presence.
“Going through the applications and meeting the students are some of the most enjoyable parts of my work,” Dr. Gih said. “I feel guilty though, that I’m likely going to miss a great applicant. Each application averages 35 pages and it’s inevitable that programs have to take shortcuts. Applicants worry that they’ll be ranked by board scores. While we certainly don’t do that here, students might feel ruled out of a program if their numbers aren’t high enough. Furthermore, wealthy students can apply to more programs. The pandemic has really highlighted the inequity issues.”
Dr. Gih noted that the Zoom interview process has not been disappointing: “Two of the people we matched had never been to Omaha, and many expressed concerns about what it is like here. Of course, on Zoom you don’t catch subtle interpersonal issues, but we have been pleasantly surprised that the people we matched were consistent with what we expected. It is exciting to meet the people who will eventually replace us as psychiatrists, they will be here to deal with future challenges!” His enthusiasm was tangible.
While the program directors remain optimistic, the system is not without its stresses, as many programs receive over 1,000 applications.
“This is difficult,” Dr. Nelson said.” It’s wonderful for the programs, but for the medical students, not matching is experienced by them as being catastrophic, so they apply to a lot of programs. Getting this many applications is a challenge, yet I don’t want to interview someone if they are going to rank our program No. 80 on their list!”
Residencies have dealt with the deluge of applicants in a number of ways. Some specialties started a “signal” protocol wherein candidates and programs receive a certain number of tokens to indicate that each would rank the other highly, but psychiatry has not done this. Early on in the Zoom process, multiple applicants would be offered interviews simultaneously, and the interview would be given to the candidate who responded first. Students vented their frustrations on Twitter when they lost interview spots at their coveted programs because they hadn’t checked their email in time or had gone to take a shower.
“The American Association of Directors of Psychiatry Residency Training Programs issued guidelines saying that it is unacceptable to offer interview spots without allowing a reasonable time for the applicant to respond, and that it is not appropriate to offer multiple candidates one spot on a first-come, first-serve basis,” Dr. Nelson explained.
Her program has managed some of the application chaos by using a software program called Scutmonkey, codeveloped by David Ross, MD, PhD, the associate program director of the Yale Adult Psychiatry Residency Program.
“It lets us screen applications for candidates who specifically are interested in being here, and for those who qualify as part of the mission we are trying to fulfill.”
One fourth-year student at a mid-Atlantic medical school who is applying in psychiatry – who I’ll call Sacha to protect his anonymity – applied to 73 psychiatry programs and to date, has interviewed at 6. He describes a stressful, roller coaster experience:
“I got those six interviews right away and that was an amazing start, but then I didn’t get any more. The interviews I had went well, but it has been disappointing not to have more. Some were all-day interviews, while other programs had me meet with residents and attendings for 20 minutes each and it was all done after 2 hours.”
He has mixed opinions about not seeing the schools in person. “There are very heavy pros and cons. I’ve saved thousands of dollars in travel expenses that would have limited my applications, so logistically it’s a dream. On the other hand, I’ve interviewed in cities I have never been to, it’s hard to get a sense of the intangibles of a program, and the shorter interviews feel very impersonal.”
Sacha expressed anxieties about the process. “With so many applicants, it’s difficult for someone with a nontraditional story to get a spot and it’s easier for the programs to toss applications. it’s common enough and everyone has seen someone who has gone through this. At times, we feel powerless; we have no real agency or control. We send stuff out and then we sit in the prayer position and wait.”
I think back on my own application process with a sense of gratitude. I certainly didn’t feel powerless, and in today’s world, postinterview communications with program directors are regulated for both parties. Dr. Slavney was kind enough to humor my request, but I don’t believe this would be feasible in the current environment.
Even though it is wonderful that more doctors have figured out that careers in psychiatry are rewarding, the current situation is overwhelming for both the applicants and the programs. With over 100 applicants for every position – many of whom will have no interest in going to some of the programs they apply to – qualified candidates who go unmatched, and a roulette wheel which requires heavily indebted students to pay to apply, this is simply not sustainable in a country with a shortage of physicians – psychiatrists in particular.
We hear that mid-level practitioners are the answer to our shortages, but perhaps we need to create a system with enough residency positions to accommodate highly trained and qualified physicians in a more inviting and targeted way.
Dinah Miller, MD, is coauthor of “Committed: The Battle Over Involuntary Psychiatric Care” (Baltimore: Johns Hopkins University Press, 2016). She has a private practice and is assistant professor of psychiatry and behavioral sciences at Johns Hopkins, both in Baltimore. A version of this article first appeared on Medscape.com.
The day I interviewed at Johns Hopkins in Baltimore, like every other day of residency interviews, was a very long and draining day.
I started by meeting alone with Philip Slavney, MD, the residency director, who spoke with me about the program and gave me a schedule to follow. I was to meet with residents and psychiatrists, some of whom had graduated from my medical school, and was sent to the Bayview campus a few miles away to have lunch and attend a few meetings. By the time I boarded an Amtrak train at Baltimore Penn Station, I was tired but I liked what I had seen. By the end of the interview season, I had crossed four programs off my list and had decided to rank only three.
In 1987, there were 987 residency positions in psychiatry in the United States, and 83.6% of those positions filled with a combination of U.S. and international medical graduates. Still, this was a risky move; the programs that I decided to rank would fill, but I was matching separately for an internship year in internal medicine in New York and decided that I would rather reapply in a year than risk matching at a program I didn’t want to go to.
I wasn’t quite sure where I wanted to rank Hopkins on my list, so I called Dr. Slavney and said I wanted to come back and meet more members of the department. He did not hide his surprise and was quick to tell me that no one had ever requested a second set of interviews. I mentioned specific people I wanted to meet with, and he was kind enough to accommodate my request and set up a second day of interviews for me.
Needless to say, the residency match felt very personal – at least to me – and although I felt vulnerable, I also felt empowered. Because of the low pay, patients with stigmatized illnesses, and the rampant belief that psychiatry was not “real” medicine and the patients never got better, psychiatry was not a desired specialty.
The residency application process in psychiatry (and every other specialty) has become a much different process. In 2006, the Association of American Medical Colleges called on medical schools to increase their enrollments to address the national shortage of physicians. Soon, there were more medical schools, bigger classes, and more doctors being minted, but the Balanced Budget Act of 1997 prevented a proportional increase in residency positions.
Len Marquez, senior director of government relations at the AAMC noted: “The Resident Physician Shortage Reduction Act of 2021 (S. 834), sponsored by Sen. Robert Menendez (D-N.J.), Sen. John Boozman (R-Ark.), and Majority Leader Sen. Charles Schumer (D-N.Y.), would support 2,000 additional Medicare-supported residency positions each year for 7 years, but Congress has not yet acted on the legislation. We were very pleased that last year, Congress passed the first increase in Medicare-supported graduate medical education in 25 years by including 1,000 new slots as part of the Consolidated Appropriations Act, 2021.”
In addition, the Build Back Better Act, which is currently being debated in Congress, would provide 4,000 more graduate medical education slots, including a specific requirement that 15% of them go to “psychiatry-related residencies,” he added.
Over 90% of graduates from U.S. medical schools currently match into a residency position. That statistic for international medical graduates is notably lower, with perhaps as few as 50% of all applicants matching.
Since 2014, the number of applicants to psychiatry residencies has nearly doubled. For the 2021 match, there were 2,486 applicants applying for 1,858 positions in psychiatry – so 1.34 applicants for each slot. Of the 1,117 senior medical students at U.S. schools who applied to psychiatry residencies, 129 did not match. Overall, 99.8% of residency positions in psychiatry filled.
“It used to be less competitive,” said Kaz J. Nelson, MD, the vice chair for education at the University of Minnesota’s department of psychiatry and behavioral sciences in Minneapolis, adding that interest in psychiatry has increased over the years.
“Interest has skyrocketed as the word has gotten out about how great a field it is. It helps that reimbursements are better, that there is less bias and discrimination against patients with psychiatric issues, and that psychiatric care is seen as a legitimate part of medicine. It has been exciting to watch!” Dr. Nelson said.
The numbers are only one part of the story, however.
Application submission now involves a centralized, electronic process, and it has become easier for applicants to apply to a lot of programs indiscriminately. It’s not unusual for applicants to apply to 70 or more programs. The factors that have limited applications include the cost: Electronic Residency Application Services (ERAS) charges for each application package they send to a program, and applicants traditionally pay to travel to the programs where they interview. This all changed with the 2021 cycle when in-person interviews were halted for the pandemic and interviews became virtual. While I recall applying to 7 residency programs, this year the average number of applications was 54.7 per applicant.
“It used to be that the cap on interviewing was financial,” Dr. Nelson said. “It was discriminatory and favored those who had more money to travel to interviews. There are still the ERAS fees, but COVID has been an equalizer and we are getting more applicants, and interviewing more who are not from Minnesota or the Midwest. We have been working to make our program attractive in terms of diversity, equity, inclusion, and justice. Our hospital is located a mile from where George Floyd was murdered, and it’s our responsibility to lead the effort to ensure the psychiatry workforce is diverse, and inclusive, as possible.”
Daniel E. Gih, MD, is the program director for a new psychiatry residency at the University of Nebraska, Omaha. When the program started in 2019, there were spots for four residents and the program had 588 applications. In 2020, the program grew to five positions and this year there were 553 applicants. Dr. Gih attributed the high number of applications to his program’s strong social media presence.
“Going through the applications and meeting the students are some of the most enjoyable parts of my work,” Dr. Gih said. “I feel guilty though, that I’m likely going to miss a great applicant. Each application averages 35 pages and it’s inevitable that programs have to take shortcuts. Applicants worry that they’ll be ranked by board scores. While we certainly don’t do that here, students might feel ruled out of a program if their numbers aren’t high enough. Furthermore, wealthy students can apply to more programs. The pandemic has really highlighted the inequity issues.”
Dr. Gih noted that the Zoom interview process has not been disappointing: “Two of the people we matched had never been to Omaha, and many expressed concerns about what it is like here. Of course, on Zoom you don’t catch subtle interpersonal issues, but we have been pleasantly surprised that the people we matched were consistent with what we expected. It is exciting to meet the people who will eventually replace us as psychiatrists, they will be here to deal with future challenges!” His enthusiasm was tangible.
While the program directors remain optimistic, the system is not without its stresses, as many programs receive over 1,000 applications.
“This is difficult,” Dr. Nelson said.” It’s wonderful for the programs, but for the medical students, not matching is experienced by them as being catastrophic, so they apply to a lot of programs. Getting this many applications is a challenge, yet I don’t want to interview someone if they are going to rank our program No. 80 on their list!”
Residencies have dealt with the deluge of applicants in a number of ways. Some specialties started a “signal” protocol wherein candidates and programs receive a certain number of tokens to indicate that each would rank the other highly, but psychiatry has not done this. Early on in the Zoom process, multiple applicants would be offered interviews simultaneously, and the interview would be given to the candidate who responded first. Students vented their frustrations on Twitter when they lost interview spots at their coveted programs because they hadn’t checked their email in time or had gone to take a shower.
“The American Association of Directors of Psychiatry Residency Training Programs issued guidelines saying that it is unacceptable to offer interview spots without allowing a reasonable time for the applicant to respond, and that it is not appropriate to offer multiple candidates one spot on a first-come, first-serve basis,” Dr. Nelson explained.
Her program has managed some of the application chaos by using a software program called Scutmonkey, codeveloped by David Ross, MD, PhD, the associate program director of the Yale Adult Psychiatry Residency Program.
“It lets us screen applications for candidates who specifically are interested in being here, and for those who qualify as part of the mission we are trying to fulfill.”
One fourth-year student at a mid-Atlantic medical school who is applying in psychiatry – who I’ll call Sacha to protect his anonymity – applied to 73 psychiatry programs and to date, has interviewed at 6. He describes a stressful, roller coaster experience:
“I got those six interviews right away and that was an amazing start, but then I didn’t get any more. The interviews I had went well, but it has been disappointing not to have more. Some were all-day interviews, while other programs had me meet with residents and attendings for 20 minutes each and it was all done after 2 hours.”
He has mixed opinions about not seeing the schools in person. “There are very heavy pros and cons. I’ve saved thousands of dollars in travel expenses that would have limited my applications, so logistically it’s a dream. On the other hand, I’ve interviewed in cities I have never been to, it’s hard to get a sense of the intangibles of a program, and the shorter interviews feel very impersonal.”
Sacha expressed anxieties about the process. “With so many applicants, it’s difficult for someone with a nontraditional story to get a spot and it’s easier for the programs to toss applications. it’s common enough and everyone has seen someone who has gone through this. At times, we feel powerless; we have no real agency or control. We send stuff out and then we sit in the prayer position and wait.”
I think back on my own application process with a sense of gratitude. I certainly didn’t feel powerless, and in today’s world, postinterview communications with program directors are regulated for both parties. Dr. Slavney was kind enough to humor my request, but I don’t believe this would be feasible in the current environment.
Even though it is wonderful that more doctors have figured out that careers in psychiatry are rewarding, the current situation is overwhelming for both the applicants and the programs. With over 100 applicants for every position – many of whom will have no interest in going to some of the programs they apply to – qualified candidates who go unmatched, and a roulette wheel which requires heavily indebted students to pay to apply, this is simply not sustainable in a country with a shortage of physicians – psychiatrists in particular.
We hear that mid-level practitioners are the answer to our shortages, but perhaps we need to create a system with enough residency positions to accommodate highly trained and qualified physicians in a more inviting and targeted way.
Dinah Miller, MD, is coauthor of “Committed: The Battle Over Involuntary Psychiatric Care” (Baltimore: Johns Hopkins University Press, 2016). She has a private practice and is assistant professor of psychiatry and behavioral sciences at Johns Hopkins, both in Baltimore. A version of this article first appeared on Medscape.com.
Even light physical activity linked to lower dementia risk
Older adults who participate in even light physical activity (LPA) may have a lower risk of developing dementia, new research suggests.
In a retrospective analysis of more than 62,000 individuals aged 65 or older without preexisting dementia, 6% developed dementia.
Compared with inactive individuals, “insufficiently active,” “active,” and “highly active” individuals all had a 10%, 20%, and 28% lower risk for dementia, respectively. And this association was consistent regardless of age, sex, other comorbidities, or after the researchers censored for stroke.
Even the lowest amount of LPA was associated with reduced dementia risk, investigators noted.
“In older adults, an increased physical activity level, including a low amount of LPA, was associated with a reduced risk of dementia,” Minjae Yoon, MD, division of cardiology, Severance Cardiovascular Hospital, Yonsei University, Seoul, South Korea, and colleagues wrote.
“Promotion of LPA might reduce the risk of dementia in older adults,” they added.
The findings were published online in JAMA Network Open.
Reverse causation?
Physical activity has been shown previously to be associated with reduced dementia risk. Current World Health Organization guidelines recommend that adults with normal cognition should engage in PA to reduce their risk for cognitive decline.
However, some studies have not yielded this result, “suggesting that previous findings showing a lower risk of dementia in physically active people could be attributed to reverse causation,” the investigators noted. Additionally, previous research regarding exercise intensity has been “inconsistent” concerning the role of LPA in reducing dementia risk.
Many older adults with frailty and comorbidity cannot perform intense or even moderate PA, therefore “these adults would have to gain the benefits of physical activity from LPA,” the researchers noted.
To clarify the potential association between PA and new-onset dementia, they focused specifically on the “dose-response association” between PA and dementia – especially LPA.
Between 2009 and 2012, the investigators enrolled 62,286 older individuals (60.4% women; mean age, 73.2 years) with available health checkup data from the National Health Insurance Service–Senior Database of Korea. All had no history of dementia.
Leisure-time PA was assessed with self-report questionnaires that used a 7-day recall method and included three questions regarding usual frequency (in days per week):
- Vigorous PA (VPA) for at least 20 minutes
- Moderate-intensity PA (MPA) for at least 30 minutes
- LPA for at least 30 minutes
VPA was defined as “intense exercise that caused severe shortness of breath, MPA was defined as activity causing mild shortness of breath, and LPA was defined as “walking at a slow or leisurely pace.”
PA-related energy expenditure was also calculated in metabolic equivalent (MET) minutes per week by “summing the product of frequency, intensity, and duration,” the investigators noted.
Participants were stratified on the basis of their weekly total PA levels into the following groups:
- Inactive (no LPA beyond basic movements)
- Insufficiently active (less than the recommended target range of 1-499 MET-min/wk)
- Active (meeting the recommended target range of 500-999 MET-min/wk)
- Highly active (exceeding the recommended target range of at least 1,000 MET-min/wk)
Of all participants, 35% were categorized as inactive, 25% were insufficiently active, 24.4% were active, and 15.2% were highly active.
Controversy remains
During the total median follow-up of 42 months, 6% of participants had all-cause dementia. After the researchers excluded the first 2 years, incidence of dementia was 21.6 per 1000 person-years during follow-up.
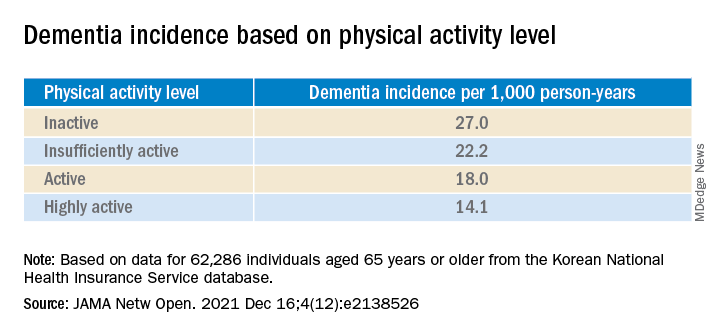
“The cumulative incidence of dementia was associated with a progressively decreasing trend with increasing physical activity” (P = .001 for trend), the investigators reported.
When using a competing-risk multivariable regression model, they found that higher levels of PA were associated with lower risk for dementia, compared with the inactive group.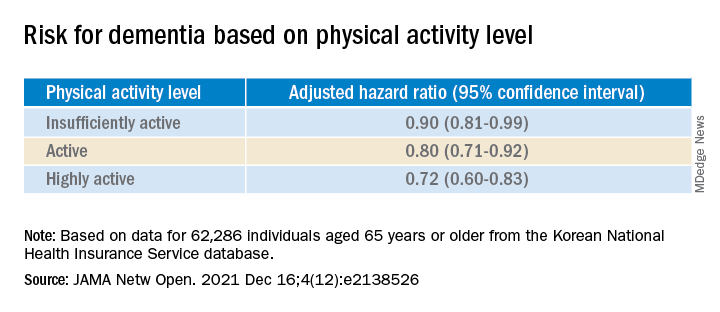
Similar findings were obtained after censoring for stroke, and were consistent for all follow-up periods. In subgroup analysis, the association between PA level and dementia risk remained consistent, regardless of age, sex, and comorbidities.
Even a low amount of LPA (1-299 MET-min/wk) was linked to reduced risk for dementia versus total sedentary behavior (adjusted HR, 0.86; 95% CI, 0.74-0.99).
The investigators noted that some “controversy” remains regarding the possibility of reverse causation and, because their study was observational in nature, “it cannot be used to establish causal relationship.”
Nevertheless, the study had important strengths, including the large number of older adults with available data, the assessment of dose-response association between PA and dementia, and the sensitivity analyses they performed, the researchers added.
Piece of important evidence
Commenting on the findings, Takashi Tarumi, PhD, senior research investigator, National Institute of Advanced Industrial Science and Technology, Ibaraki, Japan, said previous studies have suggested “an inverse association between physical activity and dementia risk, such that older adults performing a higher dose of exercise may have a greater benefit for reducing the dementia risk.”
Dr. Tarumi, an associate editor at the Journal of Alzheimer’s Disease, added the current study “significantly extends our knowledge by showing that dementia risk can also be reduced by light physical activities when they are performed for longer hours.”
This provides “another piece of important evidence” to support clinicians recommending regular physical activity for the prevention of dementia in later life, said Dr. Tarumi, who was not involved with the research.
Also commenting, Martin Underwood, MD, Warwick Medical School, Coventry, England, described the association between reduced physical inactivity and dementia as well established – and noted the current study “appears to confirm earlier observational data showing this relationship.”
The current results have “still not been able to fully exclude the possibility of reverse causation,” said Dr. Underwood, who was also not associated with the study.
However, the finding that more physically active individuals are less likely to develop dementia “only becomes of real interest if we can show that increased physical activity prevents the onset, or slows the progression, of dementia,” he noted.
“To my knowledge this has not yet been established” in randomized clinical trials, Dr. Underwood added.
The study was supported by grants from the Patient-Centered Clinical Research Coordinating Center, funded by the Ministry of Health & Welfare, Republic of Korea; and by a research grant from Yonsei University. One coauthor reported serving as a speaker for Bayer, Bristol-Myers Squibb/Pfizer, Medtronic, and Daiichi-Sankyo, and receiving research funds from Medtronic and Abbott. No other author disclosures were reported. Dr. Tarumi and Dr. Underwood have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Older adults who participate in even light physical activity (LPA) may have a lower risk of developing dementia, new research suggests.
In a retrospective analysis of more than 62,000 individuals aged 65 or older without preexisting dementia, 6% developed dementia.
Compared with inactive individuals, “insufficiently active,” “active,” and “highly active” individuals all had a 10%, 20%, and 28% lower risk for dementia, respectively. And this association was consistent regardless of age, sex, other comorbidities, or after the researchers censored for stroke.
Even the lowest amount of LPA was associated with reduced dementia risk, investigators noted.
“In older adults, an increased physical activity level, including a low amount of LPA, was associated with a reduced risk of dementia,” Minjae Yoon, MD, division of cardiology, Severance Cardiovascular Hospital, Yonsei University, Seoul, South Korea, and colleagues wrote.
“Promotion of LPA might reduce the risk of dementia in older adults,” they added.
The findings were published online in JAMA Network Open.
Reverse causation?
Physical activity has been shown previously to be associated with reduced dementia risk. Current World Health Organization guidelines recommend that adults with normal cognition should engage in PA to reduce their risk for cognitive decline.
However, some studies have not yielded this result, “suggesting that previous findings showing a lower risk of dementia in physically active people could be attributed to reverse causation,” the investigators noted. Additionally, previous research regarding exercise intensity has been “inconsistent” concerning the role of LPA in reducing dementia risk.
Many older adults with frailty and comorbidity cannot perform intense or even moderate PA, therefore “these adults would have to gain the benefits of physical activity from LPA,” the researchers noted.
To clarify the potential association between PA and new-onset dementia, they focused specifically on the “dose-response association” between PA and dementia – especially LPA.
Between 2009 and 2012, the investigators enrolled 62,286 older individuals (60.4% women; mean age, 73.2 years) with available health checkup data from the National Health Insurance Service–Senior Database of Korea. All had no history of dementia.
Leisure-time PA was assessed with self-report questionnaires that used a 7-day recall method and included three questions regarding usual frequency (in days per week):
- Vigorous PA (VPA) for at least 20 minutes
- Moderate-intensity PA (MPA) for at least 30 minutes
- LPA for at least 30 minutes
VPA was defined as “intense exercise that caused severe shortness of breath, MPA was defined as activity causing mild shortness of breath, and LPA was defined as “walking at a slow or leisurely pace.”
PA-related energy expenditure was also calculated in metabolic equivalent (MET) minutes per week by “summing the product of frequency, intensity, and duration,” the investigators noted.
Participants were stratified on the basis of their weekly total PA levels into the following groups:
- Inactive (no LPA beyond basic movements)
- Insufficiently active (less than the recommended target range of 1-499 MET-min/wk)
- Active (meeting the recommended target range of 500-999 MET-min/wk)
- Highly active (exceeding the recommended target range of at least 1,000 MET-min/wk)
Of all participants, 35% were categorized as inactive, 25% were insufficiently active, 24.4% were active, and 15.2% were highly active.
Controversy remains
During the total median follow-up of 42 months, 6% of participants had all-cause dementia. After the researchers excluded the first 2 years, incidence of dementia was 21.6 per 1000 person-years during follow-up.
“The cumulative incidence of dementia was associated with a progressively decreasing trend with increasing physical activity” (P = .001 for trend), the investigators reported.
When using a competing-risk multivariable regression model, they found that higher levels of PA were associated with lower risk for dementia, compared with the inactive group.
Similar findings were obtained after censoring for stroke, and were consistent for all follow-up periods. In subgroup analysis, the association between PA level and dementia risk remained consistent, regardless of age, sex, and comorbidities.
Even a low amount of LPA (1-299 MET-min/wk) was linked to reduced risk for dementia versus total sedentary behavior (adjusted HR, 0.86; 95% CI, 0.74-0.99).
The investigators noted that some “controversy” remains regarding the possibility of reverse causation and, because their study was observational in nature, “it cannot be used to establish causal relationship.”
Nevertheless, the study had important strengths, including the large number of older adults with available data, the assessment of dose-response association between PA and dementia, and the sensitivity analyses they performed, the researchers added.
Piece of important evidence
Commenting on the findings, Takashi Tarumi, PhD, senior research investigator, National Institute of Advanced Industrial Science and Technology, Ibaraki, Japan, said previous studies have suggested “an inverse association between physical activity and dementia risk, such that older adults performing a higher dose of exercise may have a greater benefit for reducing the dementia risk.”
Dr. Tarumi, an associate editor at the Journal of Alzheimer’s Disease, added the current study “significantly extends our knowledge by showing that dementia risk can also be reduced by light physical activities when they are performed for longer hours.”
This provides “another piece of important evidence” to support clinicians recommending regular physical activity for the prevention of dementia in later life, said Dr. Tarumi, who was not involved with the research.
Also commenting, Martin Underwood, MD, Warwick Medical School, Coventry, England, described the association between reduced physical inactivity and dementia as well established – and noted the current study “appears to confirm earlier observational data showing this relationship.”
The current results have “still not been able to fully exclude the possibility of reverse causation,” said Dr. Underwood, who was also not associated with the study.
However, the finding that more physically active individuals are less likely to develop dementia “only becomes of real interest if we can show that increased physical activity prevents the onset, or slows the progression, of dementia,” he noted.
“To my knowledge this has not yet been established” in randomized clinical trials, Dr. Underwood added.
The study was supported by grants from the Patient-Centered Clinical Research Coordinating Center, funded by the Ministry of Health & Welfare, Republic of Korea; and by a research grant from Yonsei University. One coauthor reported serving as a speaker for Bayer, Bristol-Myers Squibb/Pfizer, Medtronic, and Daiichi-Sankyo, and receiving research funds from Medtronic and Abbott. No other author disclosures were reported. Dr. Tarumi and Dr. Underwood have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Older adults who participate in even light physical activity (LPA) may have a lower risk of developing dementia, new research suggests.
In a retrospective analysis of more than 62,000 individuals aged 65 or older without preexisting dementia, 6% developed dementia.
Compared with inactive individuals, “insufficiently active,” “active,” and “highly active” individuals all had a 10%, 20%, and 28% lower risk for dementia, respectively. And this association was consistent regardless of age, sex, other comorbidities, or after the researchers censored for stroke.
Even the lowest amount of LPA was associated with reduced dementia risk, investigators noted.
“In older adults, an increased physical activity level, including a low amount of LPA, was associated with a reduced risk of dementia,” Minjae Yoon, MD, division of cardiology, Severance Cardiovascular Hospital, Yonsei University, Seoul, South Korea, and colleagues wrote.
“Promotion of LPA might reduce the risk of dementia in older adults,” they added.
The findings were published online in JAMA Network Open.
Reverse causation?
Physical activity has been shown previously to be associated with reduced dementia risk. Current World Health Organization guidelines recommend that adults with normal cognition should engage in PA to reduce their risk for cognitive decline.
However, some studies have not yielded this result, “suggesting that previous findings showing a lower risk of dementia in physically active people could be attributed to reverse causation,” the investigators noted. Additionally, previous research regarding exercise intensity has been “inconsistent” concerning the role of LPA in reducing dementia risk.
Many older adults with frailty and comorbidity cannot perform intense or even moderate PA, therefore “these adults would have to gain the benefits of physical activity from LPA,” the researchers noted.
To clarify the potential association between PA and new-onset dementia, they focused specifically on the “dose-response association” between PA and dementia – especially LPA.
Between 2009 and 2012, the investigators enrolled 62,286 older individuals (60.4% women; mean age, 73.2 years) with available health checkup data from the National Health Insurance Service–Senior Database of Korea. All had no history of dementia.
Leisure-time PA was assessed with self-report questionnaires that used a 7-day recall method and included three questions regarding usual frequency (in days per week):
- Vigorous PA (VPA) for at least 20 minutes
- Moderate-intensity PA (MPA) for at least 30 minutes
- LPA for at least 30 minutes
VPA was defined as “intense exercise that caused severe shortness of breath, MPA was defined as activity causing mild shortness of breath, and LPA was defined as “walking at a slow or leisurely pace.”
PA-related energy expenditure was also calculated in metabolic equivalent (MET) minutes per week by “summing the product of frequency, intensity, and duration,” the investigators noted.
Participants were stratified on the basis of their weekly total PA levels into the following groups:
- Inactive (no LPA beyond basic movements)
- Insufficiently active (less than the recommended target range of 1-499 MET-min/wk)
- Active (meeting the recommended target range of 500-999 MET-min/wk)
- Highly active (exceeding the recommended target range of at least 1,000 MET-min/wk)
Of all participants, 35% were categorized as inactive, 25% were insufficiently active, 24.4% were active, and 15.2% were highly active.
Controversy remains
During the total median follow-up of 42 months, 6% of participants had all-cause dementia. After the researchers excluded the first 2 years, incidence of dementia was 21.6 per 1000 person-years during follow-up.
“The cumulative incidence of dementia was associated with a progressively decreasing trend with increasing physical activity” (P = .001 for trend), the investigators reported.
When using a competing-risk multivariable regression model, they found that higher levels of PA were associated with lower risk for dementia, compared with the inactive group.
Similar findings were obtained after censoring for stroke, and were consistent for all follow-up periods. In subgroup analysis, the association between PA level and dementia risk remained consistent, regardless of age, sex, and comorbidities.
Even a low amount of LPA (1-299 MET-min/wk) was linked to reduced risk for dementia versus total sedentary behavior (adjusted HR, 0.86; 95% CI, 0.74-0.99).
The investigators noted that some “controversy” remains regarding the possibility of reverse causation and, because their study was observational in nature, “it cannot be used to establish causal relationship.”
Nevertheless, the study had important strengths, including the large number of older adults with available data, the assessment of dose-response association between PA and dementia, and the sensitivity analyses they performed, the researchers added.
Piece of important evidence
Commenting on the findings, Takashi Tarumi, PhD, senior research investigator, National Institute of Advanced Industrial Science and Technology, Ibaraki, Japan, said previous studies have suggested “an inverse association between physical activity and dementia risk, such that older adults performing a higher dose of exercise may have a greater benefit for reducing the dementia risk.”
Dr. Tarumi, an associate editor at the Journal of Alzheimer’s Disease, added the current study “significantly extends our knowledge by showing that dementia risk can also be reduced by light physical activities when they are performed for longer hours.”
This provides “another piece of important evidence” to support clinicians recommending regular physical activity for the prevention of dementia in later life, said Dr. Tarumi, who was not involved with the research.
Also commenting, Martin Underwood, MD, Warwick Medical School, Coventry, England, described the association between reduced physical inactivity and dementia as well established – and noted the current study “appears to confirm earlier observational data showing this relationship.”
The current results have “still not been able to fully exclude the possibility of reverse causation,” said Dr. Underwood, who was also not associated with the study.
However, the finding that more physically active individuals are less likely to develop dementia “only becomes of real interest if we can show that increased physical activity prevents the onset, or slows the progression, of dementia,” he noted.
“To my knowledge this has not yet been established” in randomized clinical trials, Dr. Underwood added.
The study was supported by grants from the Patient-Centered Clinical Research Coordinating Center, funded by the Ministry of Health & Welfare, Republic of Korea; and by a research grant from Yonsei University. One coauthor reported serving as a speaker for Bayer, Bristol-Myers Squibb/Pfizer, Medtronic, and Daiichi-Sankyo, and receiving research funds from Medtronic and Abbott. No other author disclosures were reported. Dr. Tarumi and Dr. Underwood have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
GI symptoms in kids with COVID may predict severe outcomes
Severe gastrointestinal involvement can be common in children who have had COVID-19, a new study shows.
Andrea Lo Veccio, MD, PhD, with the department of translational medical sciences, section of pediatrics, University of Naples (Italy) Federico II, and colleagues retrospectively analyzed data from a large cohort of children aged 18 years and younger who had been diagnosed with COVID-19 between Feb. 25, 2020, and Jan. 20, 2021, in 54 Italian institutions.
Overall, 685 Italian children (56.4% boys; average age, 7 years) were included in the study. Of these, 628 (91.7%) were diagnosed with acute SARS-CoV-2 infection and 57 (8.3%) with multisystem inflammatory syndrome in children (MIS-C).
When children had GI symptoms, the authors found a higher risk of hospitalization (odds ratio, 2.64; 95% confidence interval, 1.89-3.69) and nearly four times the risk of ICU admission (OR, 3.90; 95% CI, 1.98-7.68).
Severe GI involvement occurred in 65 children (9.5%). The authors included the following within that category: disseminated adenomesenteritis (39.6%), appendicitis (33.5%), abdominal fluid collection (21.3%), pancreatitis (6.9%), or intussusception (4.6%). Additionally, out of these 65 children, 27 (41.5%) underwent surgery.
Older children were much more likely than preschoolers to have severe GI symptoms. Children aged 5-10 years were eight times more likely than preschoolers to show severe symptoms (OR, 8.33; 95% CI, 2.62-26.5). In those older than age 10 years, severe symptoms were six times more likely (OR, 6.37; 95% CI, 2.12-19.1).
Awareness about its frequency and presentation may help practitioners to appropriately manage children at risk of severe outcomes, the authors wrote.
The findings of this study were published online Dec. 20 in JAMA Network Open.
Study highlights the GI link
Reached for comment, William Balistreri, MD, with the division of gastroenterology, hepatology, and nutrition at Cincinnati Children’s Hospital Medical Center, said that it has been known that children are more likely than adults to present with GI symptoms, and also that these symptoms are especially common in children with MIS-C.
“The symptoms most commonly cited in the literature to date include diarrhea, nausea, vomiting, or abdominal pain,” he said. “What [has not been known] is the frequency, predictive markers, and clinical course of the severe GI manifestations of COVID-19.”
The findings of this study are important to clinicians to help recognize the potential for severe GI involvement, Dr. Balistreri said, adding that “the occurrence of abdominal pain, leukopenia, and elevated inflammatory markers or MIS-C should raise suspicion and lead to early evaluation.”
Margaret E. Thew, APNP, medical director in adolescent medicine and a family nurse practitioner with Medical College of Wisconsin, Milwaukee, said that news reports typically emphasize the respiratory involvement, but this study provides a detailed analysis of the link between GI symptoms and COVID-19.
“Their data show that there may be less respiratory illness with children, regardless of whether they are generally healthy kids,” she said. “They may have more GI symptoms.
“We know that COVID-19 causes a lot of inflammation, and a large percentage of these kids had inflammation in their stomach or an inflamed bowel,” she added.
Dr. Thew said primary care doctors and urgent and emergency care clinicians will benefit from the findings of this study and should be on alert when kids come in with belly pain or vomiting.
Parents will benefit too, she said, if they are waiting for respiratory illness before they suspect COVID.
“You have to have a high suspicion this is going to be COVID positive,” she said. “You have to have that as part of your thought process.”
Though the study was done in Italy, Dr. Thew added that their experiences mimic those she’s seen locally.
Dr. Lo Vecchio reported receiving fees from Pfizer as an advisory board member outside the submitted work. A coauthor reported speaker’s fees from Angelini, Sobi, and X4 Pharma outside the submitted work. No other disclosures were reported. Dr. Balistreri and Dr. Thew reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Severe gastrointestinal involvement can be common in children who have had COVID-19, a new study shows.
Andrea Lo Veccio, MD, PhD, with the department of translational medical sciences, section of pediatrics, University of Naples (Italy) Federico II, and colleagues retrospectively analyzed data from a large cohort of children aged 18 years and younger who had been diagnosed with COVID-19 between Feb. 25, 2020, and Jan. 20, 2021, in 54 Italian institutions.
Overall, 685 Italian children (56.4% boys; average age, 7 years) were included in the study. Of these, 628 (91.7%) were diagnosed with acute SARS-CoV-2 infection and 57 (8.3%) with multisystem inflammatory syndrome in children (MIS-C).
When children had GI symptoms, the authors found a higher risk of hospitalization (odds ratio, 2.64; 95% confidence interval, 1.89-3.69) and nearly four times the risk of ICU admission (OR, 3.90; 95% CI, 1.98-7.68).
Severe GI involvement occurred in 65 children (9.5%). The authors included the following within that category: disseminated adenomesenteritis (39.6%), appendicitis (33.5%), abdominal fluid collection (21.3%), pancreatitis (6.9%), or intussusception (4.6%). Additionally, out of these 65 children, 27 (41.5%) underwent surgery.
Older children were much more likely than preschoolers to have severe GI symptoms. Children aged 5-10 years were eight times more likely than preschoolers to show severe symptoms (OR, 8.33; 95% CI, 2.62-26.5). In those older than age 10 years, severe symptoms were six times more likely (OR, 6.37; 95% CI, 2.12-19.1).
Awareness about its frequency and presentation may help practitioners to appropriately manage children at risk of severe outcomes, the authors wrote.
The findings of this study were published online Dec. 20 in JAMA Network Open.
Study highlights the GI link
Reached for comment, William Balistreri, MD, with the division of gastroenterology, hepatology, and nutrition at Cincinnati Children’s Hospital Medical Center, said that it has been known that children are more likely than adults to present with GI symptoms, and also that these symptoms are especially common in children with MIS-C.
“The symptoms most commonly cited in the literature to date include diarrhea, nausea, vomiting, or abdominal pain,” he said. “What [has not been known] is the frequency, predictive markers, and clinical course of the severe GI manifestations of COVID-19.”
The findings of this study are important to clinicians to help recognize the potential for severe GI involvement, Dr. Balistreri said, adding that “the occurrence of abdominal pain, leukopenia, and elevated inflammatory markers or MIS-C should raise suspicion and lead to early evaluation.”
Margaret E. Thew, APNP, medical director in adolescent medicine and a family nurse practitioner with Medical College of Wisconsin, Milwaukee, said that news reports typically emphasize the respiratory involvement, but this study provides a detailed analysis of the link between GI symptoms and COVID-19.
“Their data show that there may be less respiratory illness with children, regardless of whether they are generally healthy kids,” she said. “They may have more GI symptoms.
“We know that COVID-19 causes a lot of inflammation, and a large percentage of these kids had inflammation in their stomach or an inflamed bowel,” she added.
Dr. Thew said primary care doctors and urgent and emergency care clinicians will benefit from the findings of this study and should be on alert when kids come in with belly pain or vomiting.
Parents will benefit too, she said, if they are waiting for respiratory illness before they suspect COVID.
“You have to have a high suspicion this is going to be COVID positive,” she said. “You have to have that as part of your thought process.”
Though the study was done in Italy, Dr. Thew added that their experiences mimic those she’s seen locally.
Dr. Lo Vecchio reported receiving fees from Pfizer as an advisory board member outside the submitted work. A coauthor reported speaker’s fees from Angelini, Sobi, and X4 Pharma outside the submitted work. No other disclosures were reported. Dr. Balistreri and Dr. Thew reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Severe gastrointestinal involvement can be common in children who have had COVID-19, a new study shows.
Andrea Lo Veccio, MD, PhD, with the department of translational medical sciences, section of pediatrics, University of Naples (Italy) Federico II, and colleagues retrospectively analyzed data from a large cohort of children aged 18 years and younger who had been diagnosed with COVID-19 between Feb. 25, 2020, and Jan. 20, 2021, in 54 Italian institutions.
Overall, 685 Italian children (56.4% boys; average age, 7 years) were included in the study. Of these, 628 (91.7%) were diagnosed with acute SARS-CoV-2 infection and 57 (8.3%) with multisystem inflammatory syndrome in children (MIS-C).
When children had GI symptoms, the authors found a higher risk of hospitalization (odds ratio, 2.64; 95% confidence interval, 1.89-3.69) and nearly four times the risk of ICU admission (OR, 3.90; 95% CI, 1.98-7.68).
Severe GI involvement occurred in 65 children (9.5%). The authors included the following within that category: disseminated adenomesenteritis (39.6%), appendicitis (33.5%), abdominal fluid collection (21.3%), pancreatitis (6.9%), or intussusception (4.6%). Additionally, out of these 65 children, 27 (41.5%) underwent surgery.
Older children were much more likely than preschoolers to have severe GI symptoms. Children aged 5-10 years were eight times more likely than preschoolers to show severe symptoms (OR, 8.33; 95% CI, 2.62-26.5). In those older than age 10 years, severe symptoms were six times more likely (OR, 6.37; 95% CI, 2.12-19.1).
Awareness about its frequency and presentation may help practitioners to appropriately manage children at risk of severe outcomes, the authors wrote.
The findings of this study were published online Dec. 20 in JAMA Network Open.
Study highlights the GI link
Reached for comment, William Balistreri, MD, with the division of gastroenterology, hepatology, and nutrition at Cincinnati Children’s Hospital Medical Center, said that it has been known that children are more likely than adults to present with GI symptoms, and also that these symptoms are especially common in children with MIS-C.
“The symptoms most commonly cited in the literature to date include diarrhea, nausea, vomiting, or abdominal pain,” he said. “What [has not been known] is the frequency, predictive markers, and clinical course of the severe GI manifestations of COVID-19.”
The findings of this study are important to clinicians to help recognize the potential for severe GI involvement, Dr. Balistreri said, adding that “the occurrence of abdominal pain, leukopenia, and elevated inflammatory markers or MIS-C should raise suspicion and lead to early evaluation.”
Margaret E. Thew, APNP, medical director in adolescent medicine and a family nurse practitioner with Medical College of Wisconsin, Milwaukee, said that news reports typically emphasize the respiratory involvement, but this study provides a detailed analysis of the link between GI symptoms and COVID-19.
“Their data show that there may be less respiratory illness with children, regardless of whether they are generally healthy kids,” she said. “They may have more GI symptoms.
“We know that COVID-19 causes a lot of inflammation, and a large percentage of these kids had inflammation in their stomach or an inflamed bowel,” she added.
Dr. Thew said primary care doctors and urgent and emergency care clinicians will benefit from the findings of this study and should be on alert when kids come in with belly pain or vomiting.
Parents will benefit too, she said, if they are waiting for respiratory illness before they suspect COVID.
“You have to have a high suspicion this is going to be COVID positive,” she said. “You have to have that as part of your thought process.”
Though the study was done in Italy, Dr. Thew added that their experiences mimic those she’s seen locally.
Dr. Lo Vecchio reported receiving fees from Pfizer as an advisory board member outside the submitted work. A coauthor reported speaker’s fees from Angelini, Sobi, and X4 Pharma outside the submitted work. No other disclosures were reported. Dr. Balistreri and Dr. Thew reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JAMA NETWORK OPEN