User login
What to do when anticoagulation fails cancer patients
When a patient with cancer develops venous thromboembolism despite anticoagulation, how to help them comes down to clinical judgment, according to hematologist Neil Zakai, MD, associate professor at the University of Vermont, Burlington.
“Unfortunately,” when it comes to “anticoagulation failure, we are entering an evidence free-zone,” with no large trials to guide management and only a few guiding principles, he said during his presentation at the 2020 Update in Nonneoplastic Hematology virtual conference.
The first thing is to check if there was an inciting incident, such as medical noncompliance, an infection, or an interruption of anticoagulation. Dr. Zakai said he’s even had cancer patients develop heparin-induced thrombocytopenia when switched to enoxaparin from a direct oral anticoagulants (DOAC) for a procedure.
Once the underlying problem is addressed, patients may be able to continue with their original anticoagulant.
However, cancer progression is the main reason anticoagulation fails. “In general, it is very difficult to control cancer thrombosis if you can’t control cancer progression,” Dr. Zakai said.
In those cases, he steps up anticoagulation. Prophylactic dosing is increased to full treatment dosing, and patients on a DOAC are generally switched to a low molecular weight heparin (LMWH).
If patients are already on LMWH once daily, they will be bumped up to twice daily dosing; for instance, enoxaparin 1 mg/kg b.i.d. instead of 1.5 mg/kg q.d. Dr. Zakai said he’s gone as high at 2 or even 2.5 mg/kg to control thrombosis, without excessive bleeding.
In general, anticoagulation for thrombosis prophylaxis continues as long as the cancer is active, and certainly while patients are on hormonal treatments such as tamoxifen, which increases the risk.
Dr. Zakai stressed that both thrombosis and bleeding risk change for cancer patients over time, and treatment needs to keep up.
“I continuously assess the risk and benefit of anticoagulation. At certain times” such as during and for a few months after hospitalization, thrombosis risk increases; at other times, bleeding risk is higher. “You need to actively change your anticoagulation during those periods,” and tailor therapy based on transient risk factors. “People with cancer have peaks and troughs for their risk that we don’t take advantage of,” he said.
Dr. Zakai generally favors apixaban or enoxaparin for prophylaxis, carefully monitoring patients for bleeding and, for the DOAC, drug interactions with antiemetics, dexamethasone, and certain chemotherapy drugs.
He noted a recent trial that found a 59% reduction in venous thromboembolism risk in ambulatory cancer patients with apixaban 2.5 mg twice daily over 6 months, versus placebo, and a 6% absolute reduction, but at the cost of a twofold increase in bleeding risk, with an absolute 1.7% increase.
Dr. Zakai cautioned that patients in trials are selected for higher VTE and lower bleeding risks, so outcomes might “poorly reflect real world populations.” Dr. Zakai did not have any industry disclosures. The conference was sponsored by MedscapeLive. MedscapeLive and this news organization are owned by the same parent company.
When a patient with cancer develops venous thromboembolism despite anticoagulation, how to help them comes down to clinical judgment, according to hematologist Neil Zakai, MD, associate professor at the University of Vermont, Burlington.
“Unfortunately,” when it comes to “anticoagulation failure, we are entering an evidence free-zone,” with no large trials to guide management and only a few guiding principles, he said during his presentation at the 2020 Update in Nonneoplastic Hematology virtual conference.
The first thing is to check if there was an inciting incident, such as medical noncompliance, an infection, or an interruption of anticoagulation. Dr. Zakai said he’s even had cancer patients develop heparin-induced thrombocytopenia when switched to enoxaparin from a direct oral anticoagulants (DOAC) for a procedure.
Once the underlying problem is addressed, patients may be able to continue with their original anticoagulant.
However, cancer progression is the main reason anticoagulation fails. “In general, it is very difficult to control cancer thrombosis if you can’t control cancer progression,” Dr. Zakai said.
In those cases, he steps up anticoagulation. Prophylactic dosing is increased to full treatment dosing, and patients on a DOAC are generally switched to a low molecular weight heparin (LMWH).
If patients are already on LMWH once daily, they will be bumped up to twice daily dosing; for instance, enoxaparin 1 mg/kg b.i.d. instead of 1.5 mg/kg q.d. Dr. Zakai said he’s gone as high at 2 or even 2.5 mg/kg to control thrombosis, without excessive bleeding.
In general, anticoagulation for thrombosis prophylaxis continues as long as the cancer is active, and certainly while patients are on hormonal treatments such as tamoxifen, which increases the risk.
Dr. Zakai stressed that both thrombosis and bleeding risk change for cancer patients over time, and treatment needs to keep up.
“I continuously assess the risk and benefit of anticoagulation. At certain times” such as during and for a few months after hospitalization, thrombosis risk increases; at other times, bleeding risk is higher. “You need to actively change your anticoagulation during those periods,” and tailor therapy based on transient risk factors. “People with cancer have peaks and troughs for their risk that we don’t take advantage of,” he said.
Dr. Zakai generally favors apixaban or enoxaparin for prophylaxis, carefully monitoring patients for bleeding and, for the DOAC, drug interactions with antiemetics, dexamethasone, and certain chemotherapy drugs.
He noted a recent trial that found a 59% reduction in venous thromboembolism risk in ambulatory cancer patients with apixaban 2.5 mg twice daily over 6 months, versus placebo, and a 6% absolute reduction, but at the cost of a twofold increase in bleeding risk, with an absolute 1.7% increase.
Dr. Zakai cautioned that patients in trials are selected for higher VTE and lower bleeding risks, so outcomes might “poorly reflect real world populations.” Dr. Zakai did not have any industry disclosures. The conference was sponsored by MedscapeLive. MedscapeLive and this news organization are owned by the same parent company.
When a patient with cancer develops venous thromboembolism despite anticoagulation, how to help them comes down to clinical judgment, according to hematologist Neil Zakai, MD, associate professor at the University of Vermont, Burlington.
“Unfortunately,” when it comes to “anticoagulation failure, we are entering an evidence free-zone,” with no large trials to guide management and only a few guiding principles, he said during his presentation at the 2020 Update in Nonneoplastic Hematology virtual conference.
The first thing is to check if there was an inciting incident, such as medical noncompliance, an infection, or an interruption of anticoagulation. Dr. Zakai said he’s even had cancer patients develop heparin-induced thrombocytopenia when switched to enoxaparin from a direct oral anticoagulants (DOAC) for a procedure.
Once the underlying problem is addressed, patients may be able to continue with their original anticoagulant.
However, cancer progression is the main reason anticoagulation fails. “In general, it is very difficult to control cancer thrombosis if you can’t control cancer progression,” Dr. Zakai said.
In those cases, he steps up anticoagulation. Prophylactic dosing is increased to full treatment dosing, and patients on a DOAC are generally switched to a low molecular weight heparin (LMWH).
If patients are already on LMWH once daily, they will be bumped up to twice daily dosing; for instance, enoxaparin 1 mg/kg b.i.d. instead of 1.5 mg/kg q.d. Dr. Zakai said he’s gone as high at 2 or even 2.5 mg/kg to control thrombosis, without excessive bleeding.
In general, anticoagulation for thrombosis prophylaxis continues as long as the cancer is active, and certainly while patients are on hormonal treatments such as tamoxifen, which increases the risk.
Dr. Zakai stressed that both thrombosis and bleeding risk change for cancer patients over time, and treatment needs to keep up.
“I continuously assess the risk and benefit of anticoagulation. At certain times” such as during and for a few months after hospitalization, thrombosis risk increases; at other times, bleeding risk is higher. “You need to actively change your anticoagulation during those periods,” and tailor therapy based on transient risk factors. “People with cancer have peaks and troughs for their risk that we don’t take advantage of,” he said.
Dr. Zakai generally favors apixaban or enoxaparin for prophylaxis, carefully monitoring patients for bleeding and, for the DOAC, drug interactions with antiemetics, dexamethasone, and certain chemotherapy drugs.
He noted a recent trial that found a 59% reduction in venous thromboembolism risk in ambulatory cancer patients with apixaban 2.5 mg twice daily over 6 months, versus placebo, and a 6% absolute reduction, but at the cost of a twofold increase in bleeding risk, with an absolute 1.7% increase.
Dr. Zakai cautioned that patients in trials are selected for higher VTE and lower bleeding risks, so outcomes might “poorly reflect real world populations.” Dr. Zakai did not have any industry disclosures. The conference was sponsored by MedscapeLive. MedscapeLive and this news organization are owned by the same parent company.
FROM 2020 UNNH
Practice-changing data at this year’s ASH meeting
Instead of flying out to San Diego in California and soaking up a bit of sunshine in between listening to new research presentations, hematologists from around the world will be glued to their computer screens next weekend, tuning into the 62nd American Society of Hematology annual meeting.
Like many other conferences this year, the ASH meeting will be virtual because of the continuing COVID-19 pandemic, although the dates remain the same: Dec. 5-8.
This is the premier hematology event of the year, and the largest hematology conference in the world, with around 3,500 abstracts presented this year, commented Aaron T. Gerds, MD, chair of ASH’s Committee on Communications.
Ruxolitinib in chronic GvHD
“One of the things that people come to ASH for is to hear about practice-changing clinical trials, and this year is no exception,” said ASH secretary Robert Brodsky, MD.
In a preview webinar, he highlighted four abstracts that offer opportunities to change practice and revamp the current standards of care.
One clinical trial that is “almost certainly a practice changer,” he said, is the REACH 3 study (abstract 77) of the JAK inhibitor ruxolitinib (Jakafi, Incyte) in patients with chronic graft-versus-host disease (GvHD) after a stem cell transplant.
“This has been really hard to treat in patients undergoing allogeneic bone marrow transplants,” said Brodsky. “Steroids are the first-line treatment, but after that, nothing else has shown any improvement, and even steroids don’t work that well.”
There is currently no approved second-line therapy for chronic forms of GvHD, he emphasized. The main endpoint of the trial was overall response rate, which was doubled with ruxolitinib compared to the best available therapy (50% vs 25%).
“This is the first successful phase 3 trial for chronic GvHD,” Brodsky commented.
Transplants for older patients with MDS
Transplant offers the only curative option for myelodysplastic syndromes (MDS), but typically this option is offered to younger patients because benefits for older adults have not been well-defined, Brodsky noted.
New data from a clinical trial conducted in patients with advanced MDS aged 50-75 years (abstract 75) offers the most definitive evidence to date that allogeneic hematopoietic cell transplantation (AHCT) can significantly improve outcomes for older adults.
It’s clear that transplant is the standard of care in younger patients, Brodsky commented, and although there is a trend of offering it to older patients, some are not getting referred and instead are being offered palliative care. “The thinking is that bone marrow transplant would be too toxic in this age group,” he said. “But what is very clear here is that, in an intent-to-treat analysis, there was a significant survival advantage – 48% versus 27% at 3 years for transplantation – and it was seen across all subgroups.”
Subcutaneous daratumumab
New data on a subcutaneous formulation of daratumumab (Darzalex, Janssen), which is usually given by intravenous infusion, will be presented from the APOLLO trial (abstract 412) in patients with relapsed/refractory multiple myeloma.
Patients who received subcutaneous daratumumab combined with pomalidomide and dexamethasone had a 37% reduction in disease progression or death compared to those who received pomalidomide and dexamethasone alone.
“From previous years we’ve learned that daratumumab has had a major impact on outcomes in multiple myeloma,” said Brodsky. “The nice thing about the subcutaneous formulation is that it can be administered quickly and in an outpatient setting, which is especially important in the COVID era.”
Negative data with tranexamic acid
The fourth abstract highlighted by Brodsky is a negative study, but its findings can help guide clinical practice, he said. The a-TREAT study (abstract 2) showed that, despite being routinely used in the clinical setting, tranexamic acid does not prevent bleeding when administered prophylactically to severely thrombocytopenic patients undergoing treatment for hematologic malignancies.
“They found absolutely no difference in bleeding or need for transfusion,” said Brodsky. “What they did find was more catheter-associated blood clots in the tranexamic acid group. This is a practice changer in that it probably should not be given prophylactically to patients with thrombocytopenia.”
‘Very exciting’ news about gene therapy
Brodsky also highlighted several late-breaking abstract that will be presented at the meeting.
In particular, the first data on a gene therapy for hemophilia B (abstract LBA-6) are “very, very exciting,” he said. The HOPE-B trial showed a 96% response rate among patients with hemophilia B who were treated with etranacogene dezaparvovec, an investigational gene therapy composed of an adeno-associated virus serotype 5 (AAV5) vector containing a codon-optimized Padua variant human factor IX.
Brodsky pointed out that this was a large trial with 54 patients, but importantly, it included patients with pre-existing anti-AAV5 neutralizing antibodies. “About 40% of patients have naturally occurring antibodies to AAV5, and they have been excluded from previous trials because it was thought they wouldn’t take the vector,” said Brodsky. “But only one patient didn’t get a response.”
Following a single dose of etranacogene dezaparvovec, Factor IX activity increased into the mild to normal range without the need for prophylactic immunosuppression. Treated patients were able to discontinue prophylaxis and bleeding was controlled in most of the cohort.
“This is a big advance and we are getting very close to the point where gene therapy is going to be standard of care for some forms of hemophilia,” said Brodsky. However, he added that “we will still need to see more patients and have longer follow-up.”
He added that, with time, the technology behind gene therapy will probably become less expensive and more accessible to more patients, which will help become a standard of care.
This is also the hope for the technology behind chimeric antigen receptor T-cell (CAR-T) therapy, he added. At present, this cellular therapy is manufactured individually for each patient and is very expensive, but work on “off-the-shelf” products is underway. This topic will be explored during the presidential symposium, entitled, “Universal Donor Solutions in Hematology.”
New data on one of the currently available CAR-T cell products will be presented at the meeting. The phase 2 ZUMA-5 trial showed that axicabtagene ciloleucel (Axi-Cel) may be a viable option for some patients with high-risk non-Hodgkin lymphoma who have not responded to standard treatments (abstract 700).
At a median follow-up of almost 18 months, 92% of participants achieved an objective response, and 78% achieved a complete response to the treatment. By 12 months, 72% were still in response, and at 17.5 months, 64% were still in response.
“We were very impressed with the magnitude of the responses, and also the durability,” said senior study author Caron Jacobson, MD, of the Dana-Farber Cancer Institute, Boston, in a press release. “I was also struck early on by how favorable the safety profile was compared to what we’ve been seeing in the fast-growing lymphomas, such as large B cell lymphoma.”
Race and bloods cancers
ASH president Stephanie Lee, MD, MPH, highlighted several abstracts on disparities that will be presented at the meeting. One of these, which is to be presented during the plenary session, is an analysis of patient survival in acute myeloid leukemia (AML) (abstract 6).
It found that “self-reported race was the best indicator of survival,” noted Lee.
Overall survival at 3 years was 41% in White patients versus 32% in Black patients, a difference that was highly significant, she noted.
Part of the study also evaluated patients who were all on the same chemotherapy protocol, “so there was no effect of different treatment since they were on therapy determined by the trial,” said Lee.
Black patients were less likely to have normal cytogenetics compared with White patients (38% vs 51%; P = .01) and had a lower frequency of prognostically favorable NPM1 mutations (25% vs 38%; P = .04), but higher frequencies of spliceosome gene mutations (24% vs 12%; P = .009). Therefore, the results showed race was an independent prognosticator of poor survival in AML, aside from established molecular markers.
A special scientific session on race will be held on Dec. 5, Lee noted. While other abstracts consider race from the patient side, this session will focus on the scientist’s side, she explained, and address questions such as: “What are the implications of diversity and racism? And how does that impact scientists who are from underrepresented minorities?”
COVID-19 and blood disorders
Lee also highlighted a study (abstract 215) that analyzed emerging data from the ASH Research Collaborative COVID-19 Registry for Hematology, which was developed to look at outcomes of COVID-19 infection in patients with underlying blood disorders.
An analysis of data from 250 patients at 74 sites around the world found that overall mortality was 28%. “This supports the emerging consensus that patients with hematologic malignancies experience significant morbidity and mortality from COVID-19 infection,” say the authors.
“We do need real-world data to see how SARS-CoV-2 is affecting our patients with hematologic diseases or those who don’t have a hematologic disease but who are then infected with the coronavirus and develop a hematologic problem like blood clots,” said Lee.
“More data will be coming in, but this is a good example of trying to harness real-world information to learn things until we have more controlled trials.”
‘Fireside chat’ with Fauci
COVID-19 will be on the agenda for a special session billed as a “fireside chat” with Anthony Fauci, MD, of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
“This will be kicking off our meeting on Saturday morning,” said Lee.
This article first appeared on Medscape.com.
Instead of flying out to San Diego in California and soaking up a bit of sunshine in between listening to new research presentations, hematologists from around the world will be glued to their computer screens next weekend, tuning into the 62nd American Society of Hematology annual meeting.
Like many other conferences this year, the ASH meeting will be virtual because of the continuing COVID-19 pandemic, although the dates remain the same: Dec. 5-8.
This is the premier hematology event of the year, and the largest hematology conference in the world, with around 3,500 abstracts presented this year, commented Aaron T. Gerds, MD, chair of ASH’s Committee on Communications.
Ruxolitinib in chronic GvHD
“One of the things that people come to ASH for is to hear about practice-changing clinical trials, and this year is no exception,” said ASH secretary Robert Brodsky, MD.
In a preview webinar, he highlighted four abstracts that offer opportunities to change practice and revamp the current standards of care.
One clinical trial that is “almost certainly a practice changer,” he said, is the REACH 3 study (abstract 77) of the JAK inhibitor ruxolitinib (Jakafi, Incyte) in patients with chronic graft-versus-host disease (GvHD) after a stem cell transplant.
“This has been really hard to treat in patients undergoing allogeneic bone marrow transplants,” said Brodsky. “Steroids are the first-line treatment, but after that, nothing else has shown any improvement, and even steroids don’t work that well.”
There is currently no approved second-line therapy for chronic forms of GvHD, he emphasized. The main endpoint of the trial was overall response rate, which was doubled with ruxolitinib compared to the best available therapy (50% vs 25%).
“This is the first successful phase 3 trial for chronic GvHD,” Brodsky commented.
Transplants for older patients with MDS
Transplant offers the only curative option for myelodysplastic syndromes (MDS), but typically this option is offered to younger patients because benefits for older adults have not been well-defined, Brodsky noted.
New data from a clinical trial conducted in patients with advanced MDS aged 50-75 years (abstract 75) offers the most definitive evidence to date that allogeneic hematopoietic cell transplantation (AHCT) can significantly improve outcomes for older adults.
It’s clear that transplant is the standard of care in younger patients, Brodsky commented, and although there is a trend of offering it to older patients, some are not getting referred and instead are being offered palliative care. “The thinking is that bone marrow transplant would be too toxic in this age group,” he said. “But what is very clear here is that, in an intent-to-treat analysis, there was a significant survival advantage – 48% versus 27% at 3 years for transplantation – and it was seen across all subgroups.”
Subcutaneous daratumumab
New data on a subcutaneous formulation of daratumumab (Darzalex, Janssen), which is usually given by intravenous infusion, will be presented from the APOLLO trial (abstract 412) in patients with relapsed/refractory multiple myeloma.
Patients who received subcutaneous daratumumab combined with pomalidomide and dexamethasone had a 37% reduction in disease progression or death compared to those who received pomalidomide and dexamethasone alone.
“From previous years we’ve learned that daratumumab has had a major impact on outcomes in multiple myeloma,” said Brodsky. “The nice thing about the subcutaneous formulation is that it can be administered quickly and in an outpatient setting, which is especially important in the COVID era.”
Negative data with tranexamic acid
The fourth abstract highlighted by Brodsky is a negative study, but its findings can help guide clinical practice, he said. The a-TREAT study (abstract 2) showed that, despite being routinely used in the clinical setting, tranexamic acid does not prevent bleeding when administered prophylactically to severely thrombocytopenic patients undergoing treatment for hematologic malignancies.
“They found absolutely no difference in bleeding or need for transfusion,” said Brodsky. “What they did find was more catheter-associated blood clots in the tranexamic acid group. This is a practice changer in that it probably should not be given prophylactically to patients with thrombocytopenia.”
‘Very exciting’ news about gene therapy
Brodsky also highlighted several late-breaking abstract that will be presented at the meeting.
In particular, the first data on a gene therapy for hemophilia B (abstract LBA-6) are “very, very exciting,” he said. The HOPE-B trial showed a 96% response rate among patients with hemophilia B who were treated with etranacogene dezaparvovec, an investigational gene therapy composed of an adeno-associated virus serotype 5 (AAV5) vector containing a codon-optimized Padua variant human factor IX.
Brodsky pointed out that this was a large trial with 54 patients, but importantly, it included patients with pre-existing anti-AAV5 neutralizing antibodies. “About 40% of patients have naturally occurring antibodies to AAV5, and they have been excluded from previous trials because it was thought they wouldn’t take the vector,” said Brodsky. “But only one patient didn’t get a response.”
Following a single dose of etranacogene dezaparvovec, Factor IX activity increased into the mild to normal range without the need for prophylactic immunosuppression. Treated patients were able to discontinue prophylaxis and bleeding was controlled in most of the cohort.
“This is a big advance and we are getting very close to the point where gene therapy is going to be standard of care for some forms of hemophilia,” said Brodsky. However, he added that “we will still need to see more patients and have longer follow-up.”
He added that, with time, the technology behind gene therapy will probably become less expensive and more accessible to more patients, which will help become a standard of care.
This is also the hope for the technology behind chimeric antigen receptor T-cell (CAR-T) therapy, he added. At present, this cellular therapy is manufactured individually for each patient and is very expensive, but work on “off-the-shelf” products is underway. This topic will be explored during the presidential symposium, entitled, “Universal Donor Solutions in Hematology.”
New data on one of the currently available CAR-T cell products will be presented at the meeting. The phase 2 ZUMA-5 trial showed that axicabtagene ciloleucel (Axi-Cel) may be a viable option for some patients with high-risk non-Hodgkin lymphoma who have not responded to standard treatments (abstract 700).
At a median follow-up of almost 18 months, 92% of participants achieved an objective response, and 78% achieved a complete response to the treatment. By 12 months, 72% were still in response, and at 17.5 months, 64% were still in response.
“We were very impressed with the magnitude of the responses, and also the durability,” said senior study author Caron Jacobson, MD, of the Dana-Farber Cancer Institute, Boston, in a press release. “I was also struck early on by how favorable the safety profile was compared to what we’ve been seeing in the fast-growing lymphomas, such as large B cell lymphoma.”
Race and bloods cancers
ASH president Stephanie Lee, MD, MPH, highlighted several abstracts on disparities that will be presented at the meeting. One of these, which is to be presented during the plenary session, is an analysis of patient survival in acute myeloid leukemia (AML) (abstract 6).
It found that “self-reported race was the best indicator of survival,” noted Lee.
Overall survival at 3 years was 41% in White patients versus 32% in Black patients, a difference that was highly significant, she noted.
Part of the study also evaluated patients who were all on the same chemotherapy protocol, “so there was no effect of different treatment since they were on therapy determined by the trial,” said Lee.
Black patients were less likely to have normal cytogenetics compared with White patients (38% vs 51%; P = .01) and had a lower frequency of prognostically favorable NPM1 mutations (25% vs 38%; P = .04), but higher frequencies of spliceosome gene mutations (24% vs 12%; P = .009). Therefore, the results showed race was an independent prognosticator of poor survival in AML, aside from established molecular markers.
A special scientific session on race will be held on Dec. 5, Lee noted. While other abstracts consider race from the patient side, this session will focus on the scientist’s side, she explained, and address questions such as: “What are the implications of diversity and racism? And how does that impact scientists who are from underrepresented minorities?”
COVID-19 and blood disorders
Lee also highlighted a study (abstract 215) that analyzed emerging data from the ASH Research Collaborative COVID-19 Registry for Hematology, which was developed to look at outcomes of COVID-19 infection in patients with underlying blood disorders.
An analysis of data from 250 patients at 74 sites around the world found that overall mortality was 28%. “This supports the emerging consensus that patients with hematologic malignancies experience significant morbidity and mortality from COVID-19 infection,” say the authors.
“We do need real-world data to see how SARS-CoV-2 is affecting our patients with hematologic diseases or those who don’t have a hematologic disease but who are then infected with the coronavirus and develop a hematologic problem like blood clots,” said Lee.
“More data will be coming in, but this is a good example of trying to harness real-world information to learn things until we have more controlled trials.”
‘Fireside chat’ with Fauci
COVID-19 will be on the agenda for a special session billed as a “fireside chat” with Anthony Fauci, MD, of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
“This will be kicking off our meeting on Saturday morning,” said Lee.
This article first appeared on Medscape.com.
Instead of flying out to San Diego in California and soaking up a bit of sunshine in between listening to new research presentations, hematologists from around the world will be glued to their computer screens next weekend, tuning into the 62nd American Society of Hematology annual meeting.
Like many other conferences this year, the ASH meeting will be virtual because of the continuing COVID-19 pandemic, although the dates remain the same: Dec. 5-8.
This is the premier hematology event of the year, and the largest hematology conference in the world, with around 3,500 abstracts presented this year, commented Aaron T. Gerds, MD, chair of ASH’s Committee on Communications.
Ruxolitinib in chronic GvHD
“One of the things that people come to ASH for is to hear about practice-changing clinical trials, and this year is no exception,” said ASH secretary Robert Brodsky, MD.
In a preview webinar, he highlighted four abstracts that offer opportunities to change practice and revamp the current standards of care.
One clinical trial that is “almost certainly a practice changer,” he said, is the REACH 3 study (abstract 77) of the JAK inhibitor ruxolitinib (Jakafi, Incyte) in patients with chronic graft-versus-host disease (GvHD) after a stem cell transplant.
“This has been really hard to treat in patients undergoing allogeneic bone marrow transplants,” said Brodsky. “Steroids are the first-line treatment, but after that, nothing else has shown any improvement, and even steroids don’t work that well.”
There is currently no approved second-line therapy for chronic forms of GvHD, he emphasized. The main endpoint of the trial was overall response rate, which was doubled with ruxolitinib compared to the best available therapy (50% vs 25%).
“This is the first successful phase 3 trial for chronic GvHD,” Brodsky commented.
Transplants for older patients with MDS
Transplant offers the only curative option for myelodysplastic syndromes (MDS), but typically this option is offered to younger patients because benefits for older adults have not been well-defined, Brodsky noted.
New data from a clinical trial conducted in patients with advanced MDS aged 50-75 years (abstract 75) offers the most definitive evidence to date that allogeneic hematopoietic cell transplantation (AHCT) can significantly improve outcomes for older adults.
It’s clear that transplant is the standard of care in younger patients, Brodsky commented, and although there is a trend of offering it to older patients, some are not getting referred and instead are being offered palliative care. “The thinking is that bone marrow transplant would be too toxic in this age group,” he said. “But what is very clear here is that, in an intent-to-treat analysis, there was a significant survival advantage – 48% versus 27% at 3 years for transplantation – and it was seen across all subgroups.”
Subcutaneous daratumumab
New data on a subcutaneous formulation of daratumumab (Darzalex, Janssen), which is usually given by intravenous infusion, will be presented from the APOLLO trial (abstract 412) in patients with relapsed/refractory multiple myeloma.
Patients who received subcutaneous daratumumab combined with pomalidomide and dexamethasone had a 37% reduction in disease progression or death compared to those who received pomalidomide and dexamethasone alone.
“From previous years we’ve learned that daratumumab has had a major impact on outcomes in multiple myeloma,” said Brodsky. “The nice thing about the subcutaneous formulation is that it can be administered quickly and in an outpatient setting, which is especially important in the COVID era.”
Negative data with tranexamic acid
The fourth abstract highlighted by Brodsky is a negative study, but its findings can help guide clinical practice, he said. The a-TREAT study (abstract 2) showed that, despite being routinely used in the clinical setting, tranexamic acid does not prevent bleeding when administered prophylactically to severely thrombocytopenic patients undergoing treatment for hematologic malignancies.
“They found absolutely no difference in bleeding or need for transfusion,” said Brodsky. “What they did find was more catheter-associated blood clots in the tranexamic acid group. This is a practice changer in that it probably should not be given prophylactically to patients with thrombocytopenia.”
‘Very exciting’ news about gene therapy
Brodsky also highlighted several late-breaking abstract that will be presented at the meeting.
In particular, the first data on a gene therapy for hemophilia B (abstract LBA-6) are “very, very exciting,” he said. The HOPE-B trial showed a 96% response rate among patients with hemophilia B who were treated with etranacogene dezaparvovec, an investigational gene therapy composed of an adeno-associated virus serotype 5 (AAV5) vector containing a codon-optimized Padua variant human factor IX.
Brodsky pointed out that this was a large trial with 54 patients, but importantly, it included patients with pre-existing anti-AAV5 neutralizing antibodies. “About 40% of patients have naturally occurring antibodies to AAV5, and they have been excluded from previous trials because it was thought they wouldn’t take the vector,” said Brodsky. “But only one patient didn’t get a response.”
Following a single dose of etranacogene dezaparvovec, Factor IX activity increased into the mild to normal range without the need for prophylactic immunosuppression. Treated patients were able to discontinue prophylaxis and bleeding was controlled in most of the cohort.
“This is a big advance and we are getting very close to the point where gene therapy is going to be standard of care for some forms of hemophilia,” said Brodsky. However, he added that “we will still need to see more patients and have longer follow-up.”
He added that, with time, the technology behind gene therapy will probably become less expensive and more accessible to more patients, which will help become a standard of care.
This is also the hope for the technology behind chimeric antigen receptor T-cell (CAR-T) therapy, he added. At present, this cellular therapy is manufactured individually for each patient and is very expensive, but work on “off-the-shelf” products is underway. This topic will be explored during the presidential symposium, entitled, “Universal Donor Solutions in Hematology.”
New data on one of the currently available CAR-T cell products will be presented at the meeting. The phase 2 ZUMA-5 trial showed that axicabtagene ciloleucel (Axi-Cel) may be a viable option for some patients with high-risk non-Hodgkin lymphoma who have not responded to standard treatments (abstract 700).
At a median follow-up of almost 18 months, 92% of participants achieved an objective response, and 78% achieved a complete response to the treatment. By 12 months, 72% were still in response, and at 17.5 months, 64% were still in response.
“We were very impressed with the magnitude of the responses, and also the durability,” said senior study author Caron Jacobson, MD, of the Dana-Farber Cancer Institute, Boston, in a press release. “I was also struck early on by how favorable the safety profile was compared to what we’ve been seeing in the fast-growing lymphomas, such as large B cell lymphoma.”
Race and bloods cancers
ASH president Stephanie Lee, MD, MPH, highlighted several abstracts on disparities that will be presented at the meeting. One of these, which is to be presented during the plenary session, is an analysis of patient survival in acute myeloid leukemia (AML) (abstract 6).
It found that “self-reported race was the best indicator of survival,” noted Lee.
Overall survival at 3 years was 41% in White patients versus 32% in Black patients, a difference that was highly significant, she noted.
Part of the study also evaluated patients who were all on the same chemotherapy protocol, “so there was no effect of different treatment since they were on therapy determined by the trial,” said Lee.
Black patients were less likely to have normal cytogenetics compared with White patients (38% vs 51%; P = .01) and had a lower frequency of prognostically favorable NPM1 mutations (25% vs 38%; P = .04), but higher frequencies of spliceosome gene mutations (24% vs 12%; P = .009). Therefore, the results showed race was an independent prognosticator of poor survival in AML, aside from established molecular markers.
A special scientific session on race will be held on Dec. 5, Lee noted. While other abstracts consider race from the patient side, this session will focus on the scientist’s side, she explained, and address questions such as: “What are the implications of diversity and racism? And how does that impact scientists who are from underrepresented minorities?”
COVID-19 and blood disorders
Lee also highlighted a study (abstract 215) that analyzed emerging data from the ASH Research Collaborative COVID-19 Registry for Hematology, which was developed to look at outcomes of COVID-19 infection in patients with underlying blood disorders.
An analysis of data from 250 patients at 74 sites around the world found that overall mortality was 28%. “This supports the emerging consensus that patients with hematologic malignancies experience significant morbidity and mortality from COVID-19 infection,” say the authors.
“We do need real-world data to see how SARS-CoV-2 is affecting our patients with hematologic diseases or those who don’t have a hematologic disease but who are then infected with the coronavirus and develop a hematologic problem like blood clots,” said Lee.
“More data will be coming in, but this is a good example of trying to harness real-world information to learn things until we have more controlled trials.”
‘Fireside chat’ with Fauci
COVID-19 will be on the agenda for a special session billed as a “fireside chat” with Anthony Fauci, MD, of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
“This will be kicking off our meeting on Saturday morning,” said Lee.
This article first appeared on Medscape.com.
International expert group agrees on redefining psoriasis severity
It’s high time to say farewell to the traditional categorization of psoriasis severity into mild, moderate, or severe disease, according to the International Psoriasis Council.

The mild/moderate/severe categorization is vague and defined differently by different organizations and in different countries. It often underestimates disease severity because it ignores psoriasis involvement in particularly tough-to-treat special areas, including the scalp, palms, soles, face, nails, and genitalia, Bruce E. Strober, MD, PhD, asserted at MedscapeLive’s annual Las Vegas Dermatology Seminar, held virtually this year. He chaired an IPC project in which prominent psoriasis experts in 32 countries employed a Delphi consensus approach aimed at achieving agreement on a more practical recategorization of psoriasis severity for use in both daily clinical practice and enrolling appropriate participants in clinical trials. What emerged was a simplified dichotomous categorization system.
“What we came up with is a very sensible approach to defining whether patients should get either topical or systemic therapy. In fact, there are only two groups of patients in psoriasis: those who should get topicals alone, and those who should get systemic therapy. It’s topicals or systemics,” explained Dr. Strober, a dermatologist at Yale University, New Haven, Conn., who also works in private practice in Cromwell, Conn.
Under the IPC classification, psoriasis patients are candidates for systemic therapy if they meet at least one of three criteria: body surface area of involvement greater than 10%, disease involving the previously mentioned special areas, or failure of topical therapy.
“This approach is about practically treating patients who are in need,” Dr. Strober said. “If patients meet just one of these three criteria they can move on to our current toolbox of systemic therapies, be they older systemic treatments, apremilast, phototherapy, or 1 of the 11 biologics currently approved for the treatment of psoriasis. The key point is that for patients with moderate to severe psoriasis – or should I say, systemic therapy–appropriate psoriasis – treatment should be based on individual patient characteristics. We don’t work on a stepwise approach. If a patient walks in with more than 10% body surface area involved, don’t make them fail topicals; you can go right to systemics.”
European dermatologists often use the Psoriasis Area and Severity Index (PASI) score to characterize disease severity and monitor response to therapy. In contrast, American dermatologists generally find PASI too complex and time-consuming for use in clinical practice, relying instead on the amount of body surface area involved with psoriasis. Neither of these measures incorporates disease involvement in special areas, which when present ought to automatically place a patient in the systemic therapy–appropriate category, according to Dr. Strober.
“I find this [IPC recategorization] a very practical approach. I hope you write this down and use this in your own practice,” Dr. Strober said.
The full IPC report was published in the Journal of the American Academy of Dermatology.
The IPC psoriasis severity reclassification project was unfunded. Dr. Strober reported receiving institutional research funding from and serving as a paid consultant to more than two dozen pharmaceutical companies.
MedscapeLive and this news organization are owned by the same parent company.
It’s high time to say farewell to the traditional categorization of psoriasis severity into mild, moderate, or severe disease, according to the International Psoriasis Council.
The mild/moderate/severe categorization is vague and defined differently by different organizations and in different countries. It often underestimates disease severity because it ignores psoriasis involvement in particularly tough-to-treat special areas, including the scalp, palms, soles, face, nails, and genitalia, Bruce E. Strober, MD, PhD, asserted at MedscapeLive’s annual Las Vegas Dermatology Seminar, held virtually this year. He chaired an IPC project in which prominent psoriasis experts in 32 countries employed a Delphi consensus approach aimed at achieving agreement on a more practical recategorization of psoriasis severity for use in both daily clinical practice and enrolling appropriate participants in clinical trials. What emerged was a simplified dichotomous categorization system.
“What we came up with is a very sensible approach to defining whether patients should get either topical or systemic therapy. In fact, there are only two groups of patients in psoriasis: those who should get topicals alone, and those who should get systemic therapy. It’s topicals or systemics,” explained Dr. Strober, a dermatologist at Yale University, New Haven, Conn., who also works in private practice in Cromwell, Conn.
Under the IPC classification, psoriasis patients are candidates for systemic therapy if they meet at least one of three criteria: body surface area of involvement greater than 10%, disease involving the previously mentioned special areas, or failure of topical therapy.
“This approach is about practically treating patients who are in need,” Dr. Strober said. “If patients meet just one of these three criteria they can move on to our current toolbox of systemic therapies, be they older systemic treatments, apremilast, phototherapy, or 1 of the 11 biologics currently approved for the treatment of psoriasis. The key point is that for patients with moderate to severe psoriasis – or should I say, systemic therapy–appropriate psoriasis – treatment should be based on individual patient characteristics. We don’t work on a stepwise approach. If a patient walks in with more than 10% body surface area involved, don’t make them fail topicals; you can go right to systemics.”
European dermatologists often use the Psoriasis Area and Severity Index (PASI) score to characterize disease severity and monitor response to therapy. In contrast, American dermatologists generally find PASI too complex and time-consuming for use in clinical practice, relying instead on the amount of body surface area involved with psoriasis. Neither of these measures incorporates disease involvement in special areas, which when present ought to automatically place a patient in the systemic therapy–appropriate category, according to Dr. Strober.
“I find this [IPC recategorization] a very practical approach. I hope you write this down and use this in your own practice,” Dr. Strober said.
The full IPC report was published in the Journal of the American Academy of Dermatology.
The IPC psoriasis severity reclassification project was unfunded. Dr. Strober reported receiving institutional research funding from and serving as a paid consultant to more than two dozen pharmaceutical companies.
MedscapeLive and this news organization are owned by the same parent company.
It’s high time to say farewell to the traditional categorization of psoriasis severity into mild, moderate, or severe disease, according to the International Psoriasis Council.
The mild/moderate/severe categorization is vague and defined differently by different organizations and in different countries. It often underestimates disease severity because it ignores psoriasis involvement in particularly tough-to-treat special areas, including the scalp, palms, soles, face, nails, and genitalia, Bruce E. Strober, MD, PhD, asserted at MedscapeLive’s annual Las Vegas Dermatology Seminar, held virtually this year. He chaired an IPC project in which prominent psoriasis experts in 32 countries employed a Delphi consensus approach aimed at achieving agreement on a more practical recategorization of psoriasis severity for use in both daily clinical practice and enrolling appropriate participants in clinical trials. What emerged was a simplified dichotomous categorization system.
“What we came up with is a very sensible approach to defining whether patients should get either topical or systemic therapy. In fact, there are only two groups of patients in psoriasis: those who should get topicals alone, and those who should get systemic therapy. It’s topicals or systemics,” explained Dr. Strober, a dermatologist at Yale University, New Haven, Conn., who also works in private practice in Cromwell, Conn.
Under the IPC classification, psoriasis patients are candidates for systemic therapy if they meet at least one of three criteria: body surface area of involvement greater than 10%, disease involving the previously mentioned special areas, or failure of topical therapy.
“This approach is about practically treating patients who are in need,” Dr. Strober said. “If patients meet just one of these three criteria they can move on to our current toolbox of systemic therapies, be they older systemic treatments, apremilast, phototherapy, or 1 of the 11 biologics currently approved for the treatment of psoriasis. The key point is that for patients with moderate to severe psoriasis – or should I say, systemic therapy–appropriate psoriasis – treatment should be based on individual patient characteristics. We don’t work on a stepwise approach. If a patient walks in with more than 10% body surface area involved, don’t make them fail topicals; you can go right to systemics.”
European dermatologists often use the Psoriasis Area and Severity Index (PASI) score to characterize disease severity and monitor response to therapy. In contrast, American dermatologists generally find PASI too complex and time-consuming for use in clinical practice, relying instead on the amount of body surface area involved with psoriasis. Neither of these measures incorporates disease involvement in special areas, which when present ought to automatically place a patient in the systemic therapy–appropriate category, according to Dr. Strober.
“I find this [IPC recategorization] a very practical approach. I hope you write this down and use this in your own practice,” Dr. Strober said.
The full IPC report was published in the Journal of the American Academy of Dermatology.
The IPC psoriasis severity reclassification project was unfunded. Dr. Strober reported receiving institutional research funding from and serving as a paid consultant to more than two dozen pharmaceutical companies.
MedscapeLive and this news organization are owned by the same parent company.
FROM MEDSCAPELIVE LAS VEGAS DERMATOLOGY SEMINAR
Histologic remission fails to be related to UC relapse
Relapse in ulcerative colitis patients with endoscopic remission was unaffected by histologic remission status, based on data from a retrospective study of 269 adults.
Data from previous studies suggest that histologic remission may be the strongest predictor of prognosis of disease course, wrote Neeraj Narula, MD, of McMaster University, Hamilton, Ont., and colleagues.
“However, it is unclear if UC patients who have achieved endoscopic healing have additional benefit in clinical outcomes if they have achieved histologic remission as well compared to those with ongoing histology activity,” they said.
In a study published in Alimentary Pharmacology and Therapeutics, the researchers identified 269 adults with ulcerative colitis who had endoscopic remission. Of these, 53 had normal histology, 138 had histologically inactive colitis, and 78 had histologically active colitis.
Overall, clinical relapse occurred in 64 patients, including 12 with normal histology (22.6%), 32 with inactive colitis (23.2%), and 29 with active colitis (25.6%).
No significant difference occurred in the time to relapse in patients with inactive vs. active colitis (adjusted hazard ratio 1.17, P = .67) or in patients with normal histology vs. inactive histology (AHR 0.67, P = .39). The median time to relapse was 2.92 years, 3.0 years, and 4.0 years in the normal, inactive, and active groups, respectively. Factors associated with a shorter time to relapse included older age at colonoscopy, use of 5-aminosalicylic acid, and disease extent in cases of pancolitis and left-sided colitis.
The study findings were limited by several factors including the possibility of bias in histologic scoring, lack of objective measures of disease activity, and the lack of uniformity is histologic assessment, the researchers noted. However, the results were strengthened by the large size compared with previous studies and by the adjustments for known confounding factors, they said.
“While clinical and endoscopic remission [is the target] of therapy for patients with UC, our study does not support targeting histologic remission in patients who have already achieved endoscopic remission,” they concluded.
More research may support clinical applications
“I was rather surprised by the findings, as a majority of studies have shown that histologic healing more accurately predicts clinical relapse than endoscopic remission in UC,” Atsushi Sakuraba, MD, of the University of Chicago, said in an interview.
“Although of a good sample size, this was a retrospective study, so no firm conclusion can be made,” said Dr. Sakuraba. “Using histologic healing as a therapeutic goal is still an evolving field, and it is too early to draw a conclusion as to whether (or not) to introduce histologic healing in clinical decision making,” he emphasized.
Going forward, prospective studies are needed that match for confounders such as postendoscopy medication use, age, and disease extent, Dr. Sakuraba said.
The study received no outside funding. Lead author Dr. Narula disclosed honoraria from Janssen, AbbVie, Takeda, Pfizer, Merck, and Ferring. Dr. Sakuraba had no financial conflicts to disclose.
SOURCE: Narula N et al. Aliment Pharmacol Ther. 2020 Nov 1. doi: 10.1111/apt.16147.
Relapse in ulcerative colitis patients with endoscopic remission was unaffected by histologic remission status, based on data from a retrospective study of 269 adults.
Data from previous studies suggest that histologic remission may be the strongest predictor of prognosis of disease course, wrote Neeraj Narula, MD, of McMaster University, Hamilton, Ont., and colleagues.
“However, it is unclear if UC patients who have achieved endoscopic healing have additional benefit in clinical outcomes if they have achieved histologic remission as well compared to those with ongoing histology activity,” they said.
In a study published in Alimentary Pharmacology and Therapeutics, the researchers identified 269 adults with ulcerative colitis who had endoscopic remission. Of these, 53 had normal histology, 138 had histologically inactive colitis, and 78 had histologically active colitis.
Overall, clinical relapse occurred in 64 patients, including 12 with normal histology (22.6%), 32 with inactive colitis (23.2%), and 29 with active colitis (25.6%).
No significant difference occurred in the time to relapse in patients with inactive vs. active colitis (adjusted hazard ratio 1.17, P = .67) or in patients with normal histology vs. inactive histology (AHR 0.67, P = .39). The median time to relapse was 2.92 years, 3.0 years, and 4.0 years in the normal, inactive, and active groups, respectively. Factors associated with a shorter time to relapse included older age at colonoscopy, use of 5-aminosalicylic acid, and disease extent in cases of pancolitis and left-sided colitis.
The study findings were limited by several factors including the possibility of bias in histologic scoring, lack of objective measures of disease activity, and the lack of uniformity is histologic assessment, the researchers noted. However, the results were strengthened by the large size compared with previous studies and by the adjustments for known confounding factors, they said.
“While clinical and endoscopic remission [is the target] of therapy for patients with UC, our study does not support targeting histologic remission in patients who have already achieved endoscopic remission,” they concluded.
More research may support clinical applications
“I was rather surprised by the findings, as a majority of studies have shown that histologic healing more accurately predicts clinical relapse than endoscopic remission in UC,” Atsushi Sakuraba, MD, of the University of Chicago, said in an interview.
“Although of a good sample size, this was a retrospective study, so no firm conclusion can be made,” said Dr. Sakuraba. “Using histologic healing as a therapeutic goal is still an evolving field, and it is too early to draw a conclusion as to whether (or not) to introduce histologic healing in clinical decision making,” he emphasized.
Going forward, prospective studies are needed that match for confounders such as postendoscopy medication use, age, and disease extent, Dr. Sakuraba said.
The study received no outside funding. Lead author Dr. Narula disclosed honoraria from Janssen, AbbVie, Takeda, Pfizer, Merck, and Ferring. Dr. Sakuraba had no financial conflicts to disclose.
SOURCE: Narula N et al. Aliment Pharmacol Ther. 2020 Nov 1. doi: 10.1111/apt.16147.
Relapse in ulcerative colitis patients with endoscopic remission was unaffected by histologic remission status, based on data from a retrospective study of 269 adults.
Data from previous studies suggest that histologic remission may be the strongest predictor of prognosis of disease course, wrote Neeraj Narula, MD, of McMaster University, Hamilton, Ont., and colleagues.
“However, it is unclear if UC patients who have achieved endoscopic healing have additional benefit in clinical outcomes if they have achieved histologic remission as well compared to those with ongoing histology activity,” they said.
In a study published in Alimentary Pharmacology and Therapeutics, the researchers identified 269 adults with ulcerative colitis who had endoscopic remission. Of these, 53 had normal histology, 138 had histologically inactive colitis, and 78 had histologically active colitis.
Overall, clinical relapse occurred in 64 patients, including 12 with normal histology (22.6%), 32 with inactive colitis (23.2%), and 29 with active colitis (25.6%).
No significant difference occurred in the time to relapse in patients with inactive vs. active colitis (adjusted hazard ratio 1.17, P = .67) or in patients with normal histology vs. inactive histology (AHR 0.67, P = .39). The median time to relapse was 2.92 years, 3.0 years, and 4.0 years in the normal, inactive, and active groups, respectively. Factors associated with a shorter time to relapse included older age at colonoscopy, use of 5-aminosalicylic acid, and disease extent in cases of pancolitis and left-sided colitis.
The study findings were limited by several factors including the possibility of bias in histologic scoring, lack of objective measures of disease activity, and the lack of uniformity is histologic assessment, the researchers noted. However, the results were strengthened by the large size compared with previous studies and by the adjustments for known confounding factors, they said.
“While clinical and endoscopic remission [is the target] of therapy for patients with UC, our study does not support targeting histologic remission in patients who have already achieved endoscopic remission,” they concluded.
More research may support clinical applications
“I was rather surprised by the findings, as a majority of studies have shown that histologic healing more accurately predicts clinical relapse than endoscopic remission in UC,” Atsushi Sakuraba, MD, of the University of Chicago, said in an interview.
“Although of a good sample size, this was a retrospective study, so no firm conclusion can be made,” said Dr. Sakuraba. “Using histologic healing as a therapeutic goal is still an evolving field, and it is too early to draw a conclusion as to whether (or not) to introduce histologic healing in clinical decision making,” he emphasized.
Going forward, prospective studies are needed that match for confounders such as postendoscopy medication use, age, and disease extent, Dr. Sakuraba said.
The study received no outside funding. Lead author Dr. Narula disclosed honoraria from Janssen, AbbVie, Takeda, Pfizer, Merck, and Ferring. Dr. Sakuraba had no financial conflicts to disclose.
SOURCE: Narula N et al. Aliment Pharmacol Ther. 2020 Nov 1. doi: 10.1111/apt.16147.
FROM ALIMENTARY PHARMACOLOGY AND THERAPEUTICS
CMS launches hospital-at-home program to free up hospital capacity
As an increasing number of health systems implement “hospital-at-home” (HaH) programs to increase their traditional hospital capacity, the Centers for Medicare & Medicaid Services has given the movement a boost by changing its regulations to allow acute care to be provided in a patient’s home under certain conditions.
The CMS announced Nov. 25 that it was launching its Acute Hospital Care at Home program “to increase the capacity of the American health care system” during the COVID-19 pandemic.
At the same time, the agency announced it was giving more flexibility to ambulatory surgery centers (ASCs) to provide hospital-level care.
The CMS said its new HaH program is an expansion of the Hospitals Without Walls initiative that was unveiled last March. Hospitals Without Walls is a set of “temporary new rules” that provide flexibility for hospitals to provide acute care outside of inpatient settings. Under those rules, hospitals are able to transfer patients to outside facilities, such as ASCs, inpatient rehabilitation hospitals, hotels, and dormitories, while still receiving Medicare hospital payments.
Under CMS’ new Acute Hospital Care at Home, which is not described as temporary, patients can be transferred from emergency departments or inpatient wards to hospital-level care at home. The CMS said the HaH program is designed for people with conditions such as the acute phases of asthma, heart failure, pneumonia, and chronic obstructive pulmonary disease. Altogether, the agency said, more than 60 acute conditions can be treated safely at home.
However, the agency didn’t say that facilities can’t admit COVID-19 patients to the hospital at home. Rami Karjian, MBA, cofounder and CEO of Medically Home, a firm that supplies health systems with technical services and software for HaH programs, said in an interview that several Medically Home clients plan to treat both COVID-19 and non-COVID-19 patients at home when they begin to participate in the CMS program in the near future.
The CMS said it consulted extensively with academic and private industry leaders in building its HaH program. Before rolling out the initiative, the agency noted, it conducted successful pilot programs in leading hospitals and health systems. The results of some of these pilots have been reported in academic journals.
Participating hospitals will be required to have specified screening protocols in place before beginning acute care at home, the CMS announced. An in-person physician evaluation will be required before starting care at home. A nurse will evaluate each patient once daily in person or remotely, and either nurses or paramedics will visit the patient in person twice a day.
In contrast, Medicare regulations require nursing staff to be available around the clock in traditional hospitals. So the CMS has to grant waivers to hospitals for HaH programs.
While not going into detail on the telemonitoring capabilities that will be required in the acute hospital care at home, the release said, “Today’s announcement builds upon the critical work by CMS to expand telehealth coverage to keep beneficiaries safe and prevent the spread of COVID-19.”
More flexibility for ASCs
The agency is also giving ASCs the flexibility to provide 24-hour nursing services only when one or more patients are receiving care on site. This flexibility will be available to any of the 5,700 ASCs that wish to participate, and will be immediately effective for the 85 ASCs currently participating in the Hospital Without Walls initiative, the CMS said.
The new ASC regulations, the CMS said, are aimed at allowing communities “to maintain surgical capacity and other life-saving non-COVID-19 [care], like cancer surgeries.” Patients who need such procedures will be able to receive them in ASCs without being exposed to known COVID-19 cases.
Similarly, the CMS said patients and families not diagnosed with COVID-19 may prefer to receive acute care at home if local hospitals are full of COVID-19 patients. In addition, the CMS said it anticipates patients may value the ability to be treated at home without the visitation restrictions of hospitals.
Early HaH participants
Six health systems with extensive experience in providing acute hospital care at home have been approved for the new HaH waivers from Medicare rules. They include Brigham and Women’s Hospital (Massachusetts); Huntsman Cancer Institute (Utah); Massachusetts General Hospital (Massachusetts); Mount Sinai Health System (New York City); Presbyterian Healthcare Services (New Mexico); and UnityPoint Health (Iowa).
The CMS said that it’s in discussions with other health care systems and expects new applications to be submitted soon.
To support these efforts, the CMS has launched an online portal to streamline the waiver request process. The agency said it will closely monitor the program to safeguard beneficiaries and will require participating hospitals to report quality and safety data on a regular basis.
Support from hospitals
The first health systems participating in the CMS HaH appear to be supportive of the program, with some hospital leaders submitting comments to the CMS about their view of the initiative.
“The CMS has taken an extraordinary step today, facilitating the rapid expansion of Hospitalization at Home, an innovative care model with proven results,” said Kenneth L. Davis, MD, president and CEO of the Mount Sinai Health System in New York City. “This important and timely move will enable hospitals across the country to use effective tools to safely care for patients during this pandemic.”
David Levine, MD, assistant professor of medicine and medical director of strategy and innovation for Brigham Health Home Hospital in Boston, was similarly laudatory: “Our research at Brigham Health Home has shown that we can deliver hospital-level care in our patients’ homes with lower readmission rates, more physical mobility, and a positive patient experience,” he said. “During these challenging times, a focus on the home is critical. We are so encouraged that CMS is taking this important step, which will allow hospitals across the country to increase their capacity while delivering the care all patients deserve.”
Scaling up quickly
If other hospitals and health systems recognize the value of HaH, how long might it take them to develop and implement these programs in the midst of a pandemic?
Atrium Health, a large health system in the Southeast, ramped up a hospital-at-home initiative last spring for its 10 hospitals in the Charlotte, N.C., area, in just 2 weeks. However, it had been working on the project for some time before the pandemic struck. Focusing mostly on COVID-19 patients, the initiative reduced the COVID-19 patient load by 20%-25% in Atrium’s hospitals.
Medically Home, the HaH infrastructure company, said in a news release that it “enables health systems to establish new hospital-at-home services in as little as 30 days.” Medically Home has partnered in this venture with Huron Consulting Group, which has about 200 HaH-trained consultants, and Cardinal Health, a large global medical supplies distributor.
Mr. Karjian said in an interview that he expects private insurers to follow CMS’ example, as they often do. “We think this decision will cause not only CMS but private insurers to cover hospital at home after the pandemic, if it becomes the standard of care, because patients have better outcomes when treated at home,” he said.
Asked for his view on why the CMS specified that patients could be admitted to an HaH only from emergency departments or inpatient settings, Mr. Karjian said that the CMS wants to make sure that patients have access to brick-and-mortar hospital care if that’s what they need. Also, he noted, this model is new to most hospitals, so the CMS wants to make sure it starts “with all the safety guardrails” in place.
Overall, Mr. Karjian said, “This is an exciting development for patients across the country. What CMS has done is terrific in terms of letting patients get the care they want, where they want it, and get the benefit of better outcomes while the nation is going through this capacity crunch for hospital beds.”
A version of this article originally appeared on Medscape.com.
As an increasing number of health systems implement “hospital-at-home” (HaH) programs to increase their traditional hospital capacity, the Centers for Medicare & Medicaid Services has given the movement a boost by changing its regulations to allow acute care to be provided in a patient’s home under certain conditions.
The CMS announced Nov. 25 that it was launching its Acute Hospital Care at Home program “to increase the capacity of the American health care system” during the COVID-19 pandemic.
At the same time, the agency announced it was giving more flexibility to ambulatory surgery centers (ASCs) to provide hospital-level care.
The CMS said its new HaH program is an expansion of the Hospitals Without Walls initiative that was unveiled last March. Hospitals Without Walls is a set of “temporary new rules” that provide flexibility for hospitals to provide acute care outside of inpatient settings. Under those rules, hospitals are able to transfer patients to outside facilities, such as ASCs, inpatient rehabilitation hospitals, hotels, and dormitories, while still receiving Medicare hospital payments.
Under CMS’ new Acute Hospital Care at Home, which is not described as temporary, patients can be transferred from emergency departments or inpatient wards to hospital-level care at home. The CMS said the HaH program is designed for people with conditions such as the acute phases of asthma, heart failure, pneumonia, and chronic obstructive pulmonary disease. Altogether, the agency said, more than 60 acute conditions can be treated safely at home.
However, the agency didn’t say that facilities can’t admit COVID-19 patients to the hospital at home. Rami Karjian, MBA, cofounder and CEO of Medically Home, a firm that supplies health systems with technical services and software for HaH programs, said in an interview that several Medically Home clients plan to treat both COVID-19 and non-COVID-19 patients at home when they begin to participate in the CMS program in the near future.
The CMS said it consulted extensively with academic and private industry leaders in building its HaH program. Before rolling out the initiative, the agency noted, it conducted successful pilot programs in leading hospitals and health systems. The results of some of these pilots have been reported in academic journals.
Participating hospitals will be required to have specified screening protocols in place before beginning acute care at home, the CMS announced. An in-person physician evaluation will be required before starting care at home. A nurse will evaluate each patient once daily in person or remotely, and either nurses or paramedics will visit the patient in person twice a day.
In contrast, Medicare regulations require nursing staff to be available around the clock in traditional hospitals. So the CMS has to grant waivers to hospitals for HaH programs.
While not going into detail on the telemonitoring capabilities that will be required in the acute hospital care at home, the release said, “Today’s announcement builds upon the critical work by CMS to expand telehealth coverage to keep beneficiaries safe and prevent the spread of COVID-19.”
More flexibility for ASCs
The agency is also giving ASCs the flexibility to provide 24-hour nursing services only when one or more patients are receiving care on site. This flexibility will be available to any of the 5,700 ASCs that wish to participate, and will be immediately effective for the 85 ASCs currently participating in the Hospital Without Walls initiative, the CMS said.
The new ASC regulations, the CMS said, are aimed at allowing communities “to maintain surgical capacity and other life-saving non-COVID-19 [care], like cancer surgeries.” Patients who need such procedures will be able to receive them in ASCs without being exposed to known COVID-19 cases.
Similarly, the CMS said patients and families not diagnosed with COVID-19 may prefer to receive acute care at home if local hospitals are full of COVID-19 patients. In addition, the CMS said it anticipates patients may value the ability to be treated at home without the visitation restrictions of hospitals.
Early HaH participants
Six health systems with extensive experience in providing acute hospital care at home have been approved for the new HaH waivers from Medicare rules. They include Brigham and Women’s Hospital (Massachusetts); Huntsman Cancer Institute (Utah); Massachusetts General Hospital (Massachusetts); Mount Sinai Health System (New York City); Presbyterian Healthcare Services (New Mexico); and UnityPoint Health (Iowa).
The CMS said that it’s in discussions with other health care systems and expects new applications to be submitted soon.
To support these efforts, the CMS has launched an online portal to streamline the waiver request process. The agency said it will closely monitor the program to safeguard beneficiaries and will require participating hospitals to report quality and safety data on a regular basis.
Support from hospitals
The first health systems participating in the CMS HaH appear to be supportive of the program, with some hospital leaders submitting comments to the CMS about their view of the initiative.
“The CMS has taken an extraordinary step today, facilitating the rapid expansion of Hospitalization at Home, an innovative care model with proven results,” said Kenneth L. Davis, MD, president and CEO of the Mount Sinai Health System in New York City. “This important and timely move will enable hospitals across the country to use effective tools to safely care for patients during this pandemic.”
David Levine, MD, assistant professor of medicine and medical director of strategy and innovation for Brigham Health Home Hospital in Boston, was similarly laudatory: “Our research at Brigham Health Home has shown that we can deliver hospital-level care in our patients’ homes with lower readmission rates, more physical mobility, and a positive patient experience,” he said. “During these challenging times, a focus on the home is critical. We are so encouraged that CMS is taking this important step, which will allow hospitals across the country to increase their capacity while delivering the care all patients deserve.”
Scaling up quickly
If other hospitals and health systems recognize the value of HaH, how long might it take them to develop and implement these programs in the midst of a pandemic?
Atrium Health, a large health system in the Southeast, ramped up a hospital-at-home initiative last spring for its 10 hospitals in the Charlotte, N.C., area, in just 2 weeks. However, it had been working on the project for some time before the pandemic struck. Focusing mostly on COVID-19 patients, the initiative reduced the COVID-19 patient load by 20%-25% in Atrium’s hospitals.
Medically Home, the HaH infrastructure company, said in a news release that it “enables health systems to establish new hospital-at-home services in as little as 30 days.” Medically Home has partnered in this venture with Huron Consulting Group, which has about 200 HaH-trained consultants, and Cardinal Health, a large global medical supplies distributor.
Mr. Karjian said in an interview that he expects private insurers to follow CMS’ example, as they often do. “We think this decision will cause not only CMS but private insurers to cover hospital at home after the pandemic, if it becomes the standard of care, because patients have better outcomes when treated at home,” he said.
Asked for his view on why the CMS specified that patients could be admitted to an HaH only from emergency departments or inpatient settings, Mr. Karjian said that the CMS wants to make sure that patients have access to brick-and-mortar hospital care if that’s what they need. Also, he noted, this model is new to most hospitals, so the CMS wants to make sure it starts “with all the safety guardrails” in place.
Overall, Mr. Karjian said, “This is an exciting development for patients across the country. What CMS has done is terrific in terms of letting patients get the care they want, where they want it, and get the benefit of better outcomes while the nation is going through this capacity crunch for hospital beds.”
A version of this article originally appeared on Medscape.com.
As an increasing number of health systems implement “hospital-at-home” (HaH) programs to increase their traditional hospital capacity, the Centers for Medicare & Medicaid Services has given the movement a boost by changing its regulations to allow acute care to be provided in a patient’s home under certain conditions.
The CMS announced Nov. 25 that it was launching its Acute Hospital Care at Home program “to increase the capacity of the American health care system” during the COVID-19 pandemic.
At the same time, the agency announced it was giving more flexibility to ambulatory surgery centers (ASCs) to provide hospital-level care.
The CMS said its new HaH program is an expansion of the Hospitals Without Walls initiative that was unveiled last March. Hospitals Without Walls is a set of “temporary new rules” that provide flexibility for hospitals to provide acute care outside of inpatient settings. Under those rules, hospitals are able to transfer patients to outside facilities, such as ASCs, inpatient rehabilitation hospitals, hotels, and dormitories, while still receiving Medicare hospital payments.
Under CMS’ new Acute Hospital Care at Home, which is not described as temporary, patients can be transferred from emergency departments or inpatient wards to hospital-level care at home. The CMS said the HaH program is designed for people with conditions such as the acute phases of asthma, heart failure, pneumonia, and chronic obstructive pulmonary disease. Altogether, the agency said, more than 60 acute conditions can be treated safely at home.
However, the agency didn’t say that facilities can’t admit COVID-19 patients to the hospital at home. Rami Karjian, MBA, cofounder and CEO of Medically Home, a firm that supplies health systems with technical services and software for HaH programs, said in an interview that several Medically Home clients plan to treat both COVID-19 and non-COVID-19 patients at home when they begin to participate in the CMS program in the near future.
The CMS said it consulted extensively with academic and private industry leaders in building its HaH program. Before rolling out the initiative, the agency noted, it conducted successful pilot programs in leading hospitals and health systems. The results of some of these pilots have been reported in academic journals.
Participating hospitals will be required to have specified screening protocols in place before beginning acute care at home, the CMS announced. An in-person physician evaluation will be required before starting care at home. A nurse will evaluate each patient once daily in person or remotely, and either nurses or paramedics will visit the patient in person twice a day.
In contrast, Medicare regulations require nursing staff to be available around the clock in traditional hospitals. So the CMS has to grant waivers to hospitals for HaH programs.
While not going into detail on the telemonitoring capabilities that will be required in the acute hospital care at home, the release said, “Today’s announcement builds upon the critical work by CMS to expand telehealth coverage to keep beneficiaries safe and prevent the spread of COVID-19.”
More flexibility for ASCs
The agency is also giving ASCs the flexibility to provide 24-hour nursing services only when one or more patients are receiving care on site. This flexibility will be available to any of the 5,700 ASCs that wish to participate, and will be immediately effective for the 85 ASCs currently participating in the Hospital Without Walls initiative, the CMS said.
The new ASC regulations, the CMS said, are aimed at allowing communities “to maintain surgical capacity and other life-saving non-COVID-19 [care], like cancer surgeries.” Patients who need such procedures will be able to receive them in ASCs without being exposed to known COVID-19 cases.
Similarly, the CMS said patients and families not diagnosed with COVID-19 may prefer to receive acute care at home if local hospitals are full of COVID-19 patients. In addition, the CMS said it anticipates patients may value the ability to be treated at home without the visitation restrictions of hospitals.
Early HaH participants
Six health systems with extensive experience in providing acute hospital care at home have been approved for the new HaH waivers from Medicare rules. They include Brigham and Women’s Hospital (Massachusetts); Huntsman Cancer Institute (Utah); Massachusetts General Hospital (Massachusetts); Mount Sinai Health System (New York City); Presbyterian Healthcare Services (New Mexico); and UnityPoint Health (Iowa).
The CMS said that it’s in discussions with other health care systems and expects new applications to be submitted soon.
To support these efforts, the CMS has launched an online portal to streamline the waiver request process. The agency said it will closely monitor the program to safeguard beneficiaries and will require participating hospitals to report quality and safety data on a regular basis.
Support from hospitals
The first health systems participating in the CMS HaH appear to be supportive of the program, with some hospital leaders submitting comments to the CMS about their view of the initiative.
“The CMS has taken an extraordinary step today, facilitating the rapid expansion of Hospitalization at Home, an innovative care model with proven results,” said Kenneth L. Davis, MD, president and CEO of the Mount Sinai Health System in New York City. “This important and timely move will enable hospitals across the country to use effective tools to safely care for patients during this pandemic.”
David Levine, MD, assistant professor of medicine and medical director of strategy and innovation for Brigham Health Home Hospital in Boston, was similarly laudatory: “Our research at Brigham Health Home has shown that we can deliver hospital-level care in our patients’ homes with lower readmission rates, more physical mobility, and a positive patient experience,” he said. “During these challenging times, a focus on the home is critical. We are so encouraged that CMS is taking this important step, which will allow hospitals across the country to increase their capacity while delivering the care all patients deserve.”
Scaling up quickly
If other hospitals and health systems recognize the value of HaH, how long might it take them to develop and implement these programs in the midst of a pandemic?
Atrium Health, a large health system in the Southeast, ramped up a hospital-at-home initiative last spring for its 10 hospitals in the Charlotte, N.C., area, in just 2 weeks. However, it had been working on the project for some time before the pandemic struck. Focusing mostly on COVID-19 patients, the initiative reduced the COVID-19 patient load by 20%-25% in Atrium’s hospitals.
Medically Home, the HaH infrastructure company, said in a news release that it “enables health systems to establish new hospital-at-home services in as little as 30 days.” Medically Home has partnered in this venture with Huron Consulting Group, which has about 200 HaH-trained consultants, and Cardinal Health, a large global medical supplies distributor.
Mr. Karjian said in an interview that he expects private insurers to follow CMS’ example, as they often do. “We think this decision will cause not only CMS but private insurers to cover hospital at home after the pandemic, if it becomes the standard of care, because patients have better outcomes when treated at home,” he said.
Asked for his view on why the CMS specified that patients could be admitted to an HaH only from emergency departments or inpatient settings, Mr. Karjian said that the CMS wants to make sure that patients have access to brick-and-mortar hospital care if that’s what they need. Also, he noted, this model is new to most hospitals, so the CMS wants to make sure it starts “with all the safety guardrails” in place.
Overall, Mr. Karjian said, “This is an exciting development for patients across the country. What CMS has done is terrific in terms of letting patients get the care they want, where they want it, and get the benefit of better outcomes while the nation is going through this capacity crunch for hospital beds.”
A version of this article originally appeared on Medscape.com.
Are more female physicians leaving medicine as pandemic surges?
For mid-career oncologist Tanya Wildes, MD, the pandemic was the last straw. In late September, she tweeted: “I have done the academically unfathomable: I am resigning my faculty position without another job lined up.”
She wasn’t burned out, she insisted. She loved her patients and her research. But she was also “100% confident” in her decision and “also 100% sad. This did not have to happen,” she lamented, asking not to disclose her workplace for fear of retribution.
Being a woman in medicine “is a hard life to start with,” Dr. Wildes said in an interview. “We all have that tenuous balance going on and the pandemic made everything just a little bit harder.”
She describes her prepandemic work-life balance as a “Jenga tower, with everything only just in place.” But she realized that the balance had tipped, when after a difficult clinic she felt emotionally wrung out. Her 11-year-old son had asked her to help him fly his model airplane. “I told him, ‘Honey, I can’t do it because if it crashes or gets stuck in a tree ... you’re going to be devastated and I have nothing left for you.’ “
This was a eureka moment, as “I realized, this is not who I want to be,” she said, holding back tears. “Seventy years from now my son is going to tell his grandchildren about the pandemic and I don’t want his memory of his mom to be that she couldn’t be there for him because she was too spent.”
When Dr. Wildes shared her story on Twitter, other female oncologists and physicians responded that they too have felt they’re under increased pressure this year, with the extra stress of the pandemic leading others to quit as well.
The trend of doctors leaving medicine has been noticeable. A July survey from the Physicians Foundation found that roughly 16,000 medical practices had already closed during the pandemic, with another 8,000 predicted to close within the next year.
“Similar patterns” were evident in another analysis by the Larry A. Green Center and the Primary Care Collaborative, as reported in The New York Times. In that survey, nearly one-fifth of primary care clinicians said “someone in their practice plans to retire early or has already retired because of COVID-19,” and 15% say “someone has left or plans to leave the practice.” About half said their mental exhaustion was at an all-time high, the survey found.
“COVID-19 is a burden, and that added burden has tipped people over the edge of many things,” said Monica Bertagnolli, MD, chief of the division of surgical oncology at Brigham and Women’s Hospital, Boston, and former president of the American Society of Clinical Oncology.
“It has illustrated that we do have a lot of people who are working kind of on the edge of not being able to handle everything,” she said.
While many in medicine are struggling, the pandemic seems to be pushing more women to leave, highlighting longtime gender disparities and increased caregiving burdens. And their absence may be felt for years to come.
Firm numbers are hard to come by, said Julie Silver, MD, associate professor, associate chair, and director of cancer rehabilitation in the department of physical medicine and rehabilitation at Harvard Medical School, Boston, and an expert in gender equity in medicine. But she sees some troubling trends.
“There are many indications that women are leaving medicine in disproportionately high numbers,” Dr. Silver said in an interview. “A lack of fair pay and promotion opportunities that were present before COVID-19 are now combined with a host of pandemic-related challenges.”
A survey of 1,809 women conducted in mid-April with the Physician Moms Facebook Group and accepted for online publication by the American Journal of Psychiatry found that 41% scored over the cutoff points for moderate or severe anxiety, with 46% meeting these criteria among front-line workers.
“It’s really important for society to recognize the extraordinary impact this pandemic is having on physician mothers, as there will be profound ripple effects on the ability of this key segment of the health care workforce to serve others if we do not address this problem urgently,” co-senior author Reshma Jagsi, MD, DPhil, a radiation oncologist at the University of Michigan, Ann Arbor, said in an interview.
Women weighed in on Twitter, in response to Dr. Silver’s tweet to #WomenInMedicine: “If you are thinking of leaving #medicine & need a reason to stay: we value you & need you.”
In reply, Emmy Betz, MD, MPH, associate professor of emergency medicine at the University of Colorado at Denver, Aurora, said via Twitter, “I’ve had lots of conversations with women considering leaving medicine.”
“I have thought about leaving many times. I love what I do, but medicine can be an unkind world at times,” responded Valerie Fitzhugh MD, associate professor and pathologist at New Jersey Medical School, Newark.
“Too late. Left at the end of July and it was the best decision ever,” wrote Michelle Gordon, DO, who was previously a board-certified general surgeon at Northern Westchester Surgical Associates in Putnam Valley, N.Y.
Prepandemic disparities accentuated
The pandemic “has merely accentuated – or made more apparent – some of the longstanding issues and struggles of women in oncology, women in medicine, women in academia,” said Sarah Holstein, MD, PhD, another mid-career oncologist and associate professor at the University of Nebraska Medical Center, Omaha.
“There are disparities in first-author/last-author publications, disparities in being asked to give speaking engagements, disparities in leadership,” Dr. Holstein said in an interview. “And then ... put on top of that the various surges with the pandemic where you are being asked to do clinical responsibilities you don’t normally do, perhaps some things you haven’t done since your training 10 or 20 years ago.”
This is backed up with data: There is already a “robust” body of prepandemic literature demonstrating pay gaps for female physicians and scientists, noted Dr. Silver, who founded the Her Time Is Now campaign for gender equity in medicine and runs a women’s leadership course at Harvard.
In addition, female physicians are more likely to be involved in “nonpromotable” work, group projects and educator roles that are often underappreciated and undercompensated, she said.
Writing recently in a blog post for the BMJ, Dr. Silver and colleagues predict that as a result of the pandemic, female physicians will “face disadvantages from unconscious bias in decisions about whose pay should be cut, whose operating schedules should take priority when resources are limited, and whose contributions merit retention ... The ground that women lose now will likely have a profound effect for many years to come, perhaps putting them at a disadvantage for the rest of their careers.”
There is already evidence of reduced publishing by female scientists during the pandemic, something that “could undermine the careers of an entire generation of women scholars,” noted Caitlyn Collins, PhD, assistant professor of sociology at Washington University in St. Louis.
“Science needs to address the culture of overwork,” Dr. Collins said in an interview. “Parents and other caregivers deserve support. The stress and ‘overwhelm’ they feel is not inevitable. A more fair, just, and humane approach to combining work and family is possible – what we need is the political will to pass better policies and a massive shift in our cultural understandings about how work should fit into family life, not the other way around.”
Lack of support for “vulnerable scientists,” particularly “junior scientists who are parents, women, or minorities” could lead to “severe attrition in cancer research in the coming years,” Cullen Taniguchi, MD, PhD, a radiation oncologist and associate professor at the University of Texas MD Anderson Cancer Center, Houston, and colleagues, warned in a recent letter to the journal Cancer Cell.
“The biggest worries of attrition will come from young faculty who started just before or after the pandemic,” Dr. Taniguchi said in an interview. “The first year in an academic setting is incredibly challenging but also important for establishing research efforts and building networks of colleagues to collaborate with. While completely necessary, the restrictions put in place during the pandemic made doing these things even more difficult.”
Another stressor: Caregiving at home
Another reason female physicians may be marginalized during the pandemic is that they are more often the primary caregivers at home.
“Anyone who is a caregiver, be it to kids, parents, or spouses, can relate to the challenges brought [on] by the pandemic,” said Ishwaria Subbiah, MD, a palliative care physician and medical oncologist at MD Anderson.
“Most of us work toward meeting our responsibilities by engaging a network of support, whether it’s home care workers, center-based or at-home child care, schools, or activities outside of school. The pandemic led to a level of disruption that brought most (if not all) of those responsibilities onto the caregivers themselves,” she said in an interview.
As the mother of an adult son with severe epilepsy, Dr. Bertagnolli has certainly experienced the challenges of parenting during the pandemic. “Our son is now 24 but he is handicapped, and lives with us. The care issues we have to deal with as professionals have been enormously magnified by COVID,” she said.
But she cautions against making gender distinctions when it comes to caregiving. “Has it fallen on the women? Well, this kind of stuff generally falls on the women, but I am certain it has fallen on an awful lot of men as well, because I think the world is changing that way, so it’s fallen on all of us.”
There is no question that female oncologists are bearing the brunt, both at work and at home, contended Dr. Taniguchi. “Absolutely. I have seen this first-hand,” he said.
“If it was difficult for women, underrepresented minorities, and junior faculty to find a voice in the room prepandemic, I think it can be harder in the times of virtual meetings when it is difficult to engage audiences,” he said.
Dr. Holstein said she is lucky to be well-supported at her institution, with both a female chief of hematology/oncology and a female chair of internal medicine, but still, she worries about the long-term consequences of the pandemic on the gender landscape of medicine.
“If you’re having to put aside research projects because you have extra responsibilities – again because women just tend to have a lot of other things going on – that might not be a big deal for 3 months, 6 months, but this is going to be a year or 2 years before ‘normal’ comes back,” she says. “One to two years of underpublishing or not getting the grants could be career killers for women in academic oncology.”
Cancer COVID-19 combo
As Dr. Wildes completed her final weeks of seeing cancer patients, she received an outpouring of support, which she says convinced her of the shared experience of all doctors, and especially female doctors, during the pandemic. But even more specifically, she feels that she has tapped into the unique burden shouldered by oncologists during this time.
“It’s intimidating being an oncologist; we are literally giving people poison for a living. Then throw into it a pandemic where early in March we had so little data. I was helping my patients make decisions about their cancer care based on a case series of four patients in China. The burden of those conversations is something I never want to have to live through again,” she said.
“Oncology is a particularly intense subspecialty within medicine,” agreed Dr. Subbiah. “The people we care for have received a life-altering and potentially life-limiting diagnosis. Coupled with that, the COVID-19 pandemic has brought an unprecedented cloud of uncertainty ... Whether the patients can see it overtly or not, oncologists carry the weight of this worry with them for not just one but all of their patients.”
Dr. Wildes said she plans to return to academic medicine and clinical care “in time,” but for now, the gap that she and others like her leave is troubling to those who have stayed on.
“We need these women in medicine,” said Dr. Holstein. “We have data suggesting that women take more time with their patients than men, that patient outcomes are better if they have a female physician. But also for the generations coming up, we need the mid-career and senior women to be in place to mentor and guide and make sure we continue to increase women in leadership.”
A version of this article originally appeared on Medscape.com.
For mid-career oncologist Tanya Wildes, MD, the pandemic was the last straw. In late September, she tweeted: “I have done the academically unfathomable: I am resigning my faculty position without another job lined up.”
She wasn’t burned out, she insisted. She loved her patients and her research. But she was also “100% confident” in her decision and “also 100% sad. This did not have to happen,” she lamented, asking not to disclose her workplace for fear of retribution.
Being a woman in medicine “is a hard life to start with,” Dr. Wildes said in an interview. “We all have that tenuous balance going on and the pandemic made everything just a little bit harder.”
She describes her prepandemic work-life balance as a “Jenga tower, with everything only just in place.” But she realized that the balance had tipped, when after a difficult clinic she felt emotionally wrung out. Her 11-year-old son had asked her to help him fly his model airplane. “I told him, ‘Honey, I can’t do it because if it crashes or gets stuck in a tree ... you’re going to be devastated and I have nothing left for you.’ “
This was a eureka moment, as “I realized, this is not who I want to be,” she said, holding back tears. “Seventy years from now my son is going to tell his grandchildren about the pandemic and I don’t want his memory of his mom to be that she couldn’t be there for him because she was too spent.”
When Dr. Wildes shared her story on Twitter, other female oncologists and physicians responded that they too have felt they’re under increased pressure this year, with the extra stress of the pandemic leading others to quit as well.
The trend of doctors leaving medicine has been noticeable. A July survey from the Physicians Foundation found that roughly 16,000 medical practices had already closed during the pandemic, with another 8,000 predicted to close within the next year.
“Similar patterns” were evident in another analysis by the Larry A. Green Center and the Primary Care Collaborative, as reported in The New York Times. In that survey, nearly one-fifth of primary care clinicians said “someone in their practice plans to retire early or has already retired because of COVID-19,” and 15% say “someone has left or plans to leave the practice.” About half said their mental exhaustion was at an all-time high, the survey found.
“COVID-19 is a burden, and that added burden has tipped people over the edge of many things,” said Monica Bertagnolli, MD, chief of the division of surgical oncology at Brigham and Women’s Hospital, Boston, and former president of the American Society of Clinical Oncology.
“It has illustrated that we do have a lot of people who are working kind of on the edge of not being able to handle everything,” she said.
While many in medicine are struggling, the pandemic seems to be pushing more women to leave, highlighting longtime gender disparities and increased caregiving burdens. And their absence may be felt for years to come.
Firm numbers are hard to come by, said Julie Silver, MD, associate professor, associate chair, and director of cancer rehabilitation in the department of physical medicine and rehabilitation at Harvard Medical School, Boston, and an expert in gender equity in medicine. But she sees some troubling trends.
“There are many indications that women are leaving medicine in disproportionately high numbers,” Dr. Silver said in an interview. “A lack of fair pay and promotion opportunities that were present before COVID-19 are now combined with a host of pandemic-related challenges.”
A survey of 1,809 women conducted in mid-April with the Physician Moms Facebook Group and accepted for online publication by the American Journal of Psychiatry found that 41% scored over the cutoff points for moderate or severe anxiety, with 46% meeting these criteria among front-line workers.
“It’s really important for society to recognize the extraordinary impact this pandemic is having on physician mothers, as there will be profound ripple effects on the ability of this key segment of the health care workforce to serve others if we do not address this problem urgently,” co-senior author Reshma Jagsi, MD, DPhil, a radiation oncologist at the University of Michigan, Ann Arbor, said in an interview.
Women weighed in on Twitter, in response to Dr. Silver’s tweet to #WomenInMedicine: “If you are thinking of leaving #medicine & need a reason to stay: we value you & need you.”
In reply, Emmy Betz, MD, MPH, associate professor of emergency medicine at the University of Colorado at Denver, Aurora, said via Twitter, “I’ve had lots of conversations with women considering leaving medicine.”
“I have thought about leaving many times. I love what I do, but medicine can be an unkind world at times,” responded Valerie Fitzhugh MD, associate professor and pathologist at New Jersey Medical School, Newark.
“Too late. Left at the end of July and it was the best decision ever,” wrote Michelle Gordon, DO, who was previously a board-certified general surgeon at Northern Westchester Surgical Associates in Putnam Valley, N.Y.
Prepandemic disparities accentuated
The pandemic “has merely accentuated – or made more apparent – some of the longstanding issues and struggles of women in oncology, women in medicine, women in academia,” said Sarah Holstein, MD, PhD, another mid-career oncologist and associate professor at the University of Nebraska Medical Center, Omaha.
“There are disparities in first-author/last-author publications, disparities in being asked to give speaking engagements, disparities in leadership,” Dr. Holstein said in an interview. “And then ... put on top of that the various surges with the pandemic where you are being asked to do clinical responsibilities you don’t normally do, perhaps some things you haven’t done since your training 10 or 20 years ago.”
This is backed up with data: There is already a “robust” body of prepandemic literature demonstrating pay gaps for female physicians and scientists, noted Dr. Silver, who founded the Her Time Is Now campaign for gender equity in medicine and runs a women’s leadership course at Harvard.
In addition, female physicians are more likely to be involved in “nonpromotable” work, group projects and educator roles that are often underappreciated and undercompensated, she said.
Writing recently in a blog post for the BMJ, Dr. Silver and colleagues predict that as a result of the pandemic, female physicians will “face disadvantages from unconscious bias in decisions about whose pay should be cut, whose operating schedules should take priority when resources are limited, and whose contributions merit retention ... The ground that women lose now will likely have a profound effect for many years to come, perhaps putting them at a disadvantage for the rest of their careers.”
There is already evidence of reduced publishing by female scientists during the pandemic, something that “could undermine the careers of an entire generation of women scholars,” noted Caitlyn Collins, PhD, assistant professor of sociology at Washington University in St. Louis.
“Science needs to address the culture of overwork,” Dr. Collins said in an interview. “Parents and other caregivers deserve support. The stress and ‘overwhelm’ they feel is not inevitable. A more fair, just, and humane approach to combining work and family is possible – what we need is the political will to pass better policies and a massive shift in our cultural understandings about how work should fit into family life, not the other way around.”
Lack of support for “vulnerable scientists,” particularly “junior scientists who are parents, women, or minorities” could lead to “severe attrition in cancer research in the coming years,” Cullen Taniguchi, MD, PhD, a radiation oncologist and associate professor at the University of Texas MD Anderson Cancer Center, Houston, and colleagues, warned in a recent letter to the journal Cancer Cell.
“The biggest worries of attrition will come from young faculty who started just before or after the pandemic,” Dr. Taniguchi said in an interview. “The first year in an academic setting is incredibly challenging but also important for establishing research efforts and building networks of colleagues to collaborate with. While completely necessary, the restrictions put in place during the pandemic made doing these things even more difficult.”
Another stressor: Caregiving at home
Another reason female physicians may be marginalized during the pandemic is that they are more often the primary caregivers at home.
“Anyone who is a caregiver, be it to kids, parents, or spouses, can relate to the challenges brought [on] by the pandemic,” said Ishwaria Subbiah, MD, a palliative care physician and medical oncologist at MD Anderson.
“Most of us work toward meeting our responsibilities by engaging a network of support, whether it’s home care workers, center-based or at-home child care, schools, or activities outside of school. The pandemic led to a level of disruption that brought most (if not all) of those responsibilities onto the caregivers themselves,” she said in an interview.
As the mother of an adult son with severe epilepsy, Dr. Bertagnolli has certainly experienced the challenges of parenting during the pandemic. “Our son is now 24 but he is handicapped, and lives with us. The care issues we have to deal with as professionals have been enormously magnified by COVID,” she said.
But she cautions against making gender distinctions when it comes to caregiving. “Has it fallen on the women? Well, this kind of stuff generally falls on the women, but I am certain it has fallen on an awful lot of men as well, because I think the world is changing that way, so it’s fallen on all of us.”
There is no question that female oncologists are bearing the brunt, both at work and at home, contended Dr. Taniguchi. “Absolutely. I have seen this first-hand,” he said.
“If it was difficult for women, underrepresented minorities, and junior faculty to find a voice in the room prepandemic, I think it can be harder in the times of virtual meetings when it is difficult to engage audiences,” he said.
Dr. Holstein said she is lucky to be well-supported at her institution, with both a female chief of hematology/oncology and a female chair of internal medicine, but still, she worries about the long-term consequences of the pandemic on the gender landscape of medicine.
“If you’re having to put aside research projects because you have extra responsibilities – again because women just tend to have a lot of other things going on – that might not be a big deal for 3 months, 6 months, but this is going to be a year or 2 years before ‘normal’ comes back,” she says. “One to two years of underpublishing or not getting the grants could be career killers for women in academic oncology.”
Cancer COVID-19 combo
As Dr. Wildes completed her final weeks of seeing cancer patients, she received an outpouring of support, which she says convinced her of the shared experience of all doctors, and especially female doctors, during the pandemic. But even more specifically, she feels that she has tapped into the unique burden shouldered by oncologists during this time.
“It’s intimidating being an oncologist; we are literally giving people poison for a living. Then throw into it a pandemic where early in March we had so little data. I was helping my patients make decisions about their cancer care based on a case series of four patients in China. The burden of those conversations is something I never want to have to live through again,” she said.
“Oncology is a particularly intense subspecialty within medicine,” agreed Dr. Subbiah. “The people we care for have received a life-altering and potentially life-limiting diagnosis. Coupled with that, the COVID-19 pandemic has brought an unprecedented cloud of uncertainty ... Whether the patients can see it overtly or not, oncologists carry the weight of this worry with them for not just one but all of their patients.”
Dr. Wildes said she plans to return to academic medicine and clinical care “in time,” but for now, the gap that she and others like her leave is troubling to those who have stayed on.
“We need these women in medicine,” said Dr. Holstein. “We have data suggesting that women take more time with their patients than men, that patient outcomes are better if they have a female physician. But also for the generations coming up, we need the mid-career and senior women to be in place to mentor and guide and make sure we continue to increase women in leadership.”
A version of this article originally appeared on Medscape.com.
For mid-career oncologist Tanya Wildes, MD, the pandemic was the last straw. In late September, she tweeted: “I have done the academically unfathomable: I am resigning my faculty position without another job lined up.”
She wasn’t burned out, she insisted. She loved her patients and her research. But she was also “100% confident” in her decision and “also 100% sad. This did not have to happen,” she lamented, asking not to disclose her workplace for fear of retribution.
Being a woman in medicine “is a hard life to start with,” Dr. Wildes said in an interview. “We all have that tenuous balance going on and the pandemic made everything just a little bit harder.”
She describes her prepandemic work-life balance as a “Jenga tower, with everything only just in place.” But she realized that the balance had tipped, when after a difficult clinic she felt emotionally wrung out. Her 11-year-old son had asked her to help him fly his model airplane. “I told him, ‘Honey, I can’t do it because if it crashes or gets stuck in a tree ... you’re going to be devastated and I have nothing left for you.’ “
This was a eureka moment, as “I realized, this is not who I want to be,” she said, holding back tears. “Seventy years from now my son is going to tell his grandchildren about the pandemic and I don’t want his memory of his mom to be that she couldn’t be there for him because she was too spent.”
When Dr. Wildes shared her story on Twitter, other female oncologists and physicians responded that they too have felt they’re under increased pressure this year, with the extra stress of the pandemic leading others to quit as well.
The trend of doctors leaving medicine has been noticeable. A July survey from the Physicians Foundation found that roughly 16,000 medical practices had already closed during the pandemic, with another 8,000 predicted to close within the next year.
“Similar patterns” were evident in another analysis by the Larry A. Green Center and the Primary Care Collaborative, as reported in The New York Times. In that survey, nearly one-fifth of primary care clinicians said “someone in their practice plans to retire early or has already retired because of COVID-19,” and 15% say “someone has left or plans to leave the practice.” About half said their mental exhaustion was at an all-time high, the survey found.
“COVID-19 is a burden, and that added burden has tipped people over the edge of many things,” said Monica Bertagnolli, MD, chief of the division of surgical oncology at Brigham and Women’s Hospital, Boston, and former president of the American Society of Clinical Oncology.
“It has illustrated that we do have a lot of people who are working kind of on the edge of not being able to handle everything,” she said.
While many in medicine are struggling, the pandemic seems to be pushing more women to leave, highlighting longtime gender disparities and increased caregiving burdens. And their absence may be felt for years to come.
Firm numbers are hard to come by, said Julie Silver, MD, associate professor, associate chair, and director of cancer rehabilitation in the department of physical medicine and rehabilitation at Harvard Medical School, Boston, and an expert in gender equity in medicine. But she sees some troubling trends.
“There are many indications that women are leaving medicine in disproportionately high numbers,” Dr. Silver said in an interview. “A lack of fair pay and promotion opportunities that were present before COVID-19 are now combined with a host of pandemic-related challenges.”
A survey of 1,809 women conducted in mid-April with the Physician Moms Facebook Group and accepted for online publication by the American Journal of Psychiatry found that 41% scored over the cutoff points for moderate or severe anxiety, with 46% meeting these criteria among front-line workers.
“It’s really important for society to recognize the extraordinary impact this pandemic is having on physician mothers, as there will be profound ripple effects on the ability of this key segment of the health care workforce to serve others if we do not address this problem urgently,” co-senior author Reshma Jagsi, MD, DPhil, a radiation oncologist at the University of Michigan, Ann Arbor, said in an interview.
Women weighed in on Twitter, in response to Dr. Silver’s tweet to #WomenInMedicine: “If you are thinking of leaving #medicine & need a reason to stay: we value you & need you.”
In reply, Emmy Betz, MD, MPH, associate professor of emergency medicine at the University of Colorado at Denver, Aurora, said via Twitter, “I’ve had lots of conversations with women considering leaving medicine.”
“I have thought about leaving many times. I love what I do, but medicine can be an unkind world at times,” responded Valerie Fitzhugh MD, associate professor and pathologist at New Jersey Medical School, Newark.
“Too late. Left at the end of July and it was the best decision ever,” wrote Michelle Gordon, DO, who was previously a board-certified general surgeon at Northern Westchester Surgical Associates in Putnam Valley, N.Y.
Prepandemic disparities accentuated
The pandemic “has merely accentuated – or made more apparent – some of the longstanding issues and struggles of women in oncology, women in medicine, women in academia,” said Sarah Holstein, MD, PhD, another mid-career oncologist and associate professor at the University of Nebraska Medical Center, Omaha.
“There are disparities in first-author/last-author publications, disparities in being asked to give speaking engagements, disparities in leadership,” Dr. Holstein said in an interview. “And then ... put on top of that the various surges with the pandemic where you are being asked to do clinical responsibilities you don’t normally do, perhaps some things you haven’t done since your training 10 or 20 years ago.”
This is backed up with data: There is already a “robust” body of prepandemic literature demonstrating pay gaps for female physicians and scientists, noted Dr. Silver, who founded the Her Time Is Now campaign for gender equity in medicine and runs a women’s leadership course at Harvard.
In addition, female physicians are more likely to be involved in “nonpromotable” work, group projects and educator roles that are often underappreciated and undercompensated, she said.
Writing recently in a blog post for the BMJ, Dr. Silver and colleagues predict that as a result of the pandemic, female physicians will “face disadvantages from unconscious bias in decisions about whose pay should be cut, whose operating schedules should take priority when resources are limited, and whose contributions merit retention ... The ground that women lose now will likely have a profound effect for many years to come, perhaps putting them at a disadvantage for the rest of their careers.”
There is already evidence of reduced publishing by female scientists during the pandemic, something that “could undermine the careers of an entire generation of women scholars,” noted Caitlyn Collins, PhD, assistant professor of sociology at Washington University in St. Louis.
“Science needs to address the culture of overwork,” Dr. Collins said in an interview. “Parents and other caregivers deserve support. The stress and ‘overwhelm’ they feel is not inevitable. A more fair, just, and humane approach to combining work and family is possible – what we need is the political will to pass better policies and a massive shift in our cultural understandings about how work should fit into family life, not the other way around.”
Lack of support for “vulnerable scientists,” particularly “junior scientists who are parents, women, or minorities” could lead to “severe attrition in cancer research in the coming years,” Cullen Taniguchi, MD, PhD, a radiation oncologist and associate professor at the University of Texas MD Anderson Cancer Center, Houston, and colleagues, warned in a recent letter to the journal Cancer Cell.
“The biggest worries of attrition will come from young faculty who started just before or after the pandemic,” Dr. Taniguchi said in an interview. “The first year in an academic setting is incredibly challenging but also important for establishing research efforts and building networks of colleagues to collaborate with. While completely necessary, the restrictions put in place during the pandemic made doing these things even more difficult.”
Another stressor: Caregiving at home
Another reason female physicians may be marginalized during the pandemic is that they are more often the primary caregivers at home.
“Anyone who is a caregiver, be it to kids, parents, or spouses, can relate to the challenges brought [on] by the pandemic,” said Ishwaria Subbiah, MD, a palliative care physician and medical oncologist at MD Anderson.
“Most of us work toward meeting our responsibilities by engaging a network of support, whether it’s home care workers, center-based or at-home child care, schools, or activities outside of school. The pandemic led to a level of disruption that brought most (if not all) of those responsibilities onto the caregivers themselves,” she said in an interview.
As the mother of an adult son with severe epilepsy, Dr. Bertagnolli has certainly experienced the challenges of parenting during the pandemic. “Our son is now 24 but he is handicapped, and lives with us. The care issues we have to deal with as professionals have been enormously magnified by COVID,” she said.
But she cautions against making gender distinctions when it comes to caregiving. “Has it fallen on the women? Well, this kind of stuff generally falls on the women, but I am certain it has fallen on an awful lot of men as well, because I think the world is changing that way, so it’s fallen on all of us.”
There is no question that female oncologists are bearing the brunt, both at work and at home, contended Dr. Taniguchi. “Absolutely. I have seen this first-hand,” he said.
“If it was difficult for women, underrepresented minorities, and junior faculty to find a voice in the room prepandemic, I think it can be harder in the times of virtual meetings when it is difficult to engage audiences,” he said.
Dr. Holstein said she is lucky to be well-supported at her institution, with both a female chief of hematology/oncology and a female chair of internal medicine, but still, she worries about the long-term consequences of the pandemic on the gender landscape of medicine.
“If you’re having to put aside research projects because you have extra responsibilities – again because women just tend to have a lot of other things going on – that might not be a big deal for 3 months, 6 months, but this is going to be a year or 2 years before ‘normal’ comes back,” she says. “One to two years of underpublishing or not getting the grants could be career killers for women in academic oncology.”
Cancer COVID-19 combo
As Dr. Wildes completed her final weeks of seeing cancer patients, she received an outpouring of support, which she says convinced her of the shared experience of all doctors, and especially female doctors, during the pandemic. But even more specifically, she feels that she has tapped into the unique burden shouldered by oncologists during this time.
“It’s intimidating being an oncologist; we are literally giving people poison for a living. Then throw into it a pandemic where early in March we had so little data. I was helping my patients make decisions about their cancer care based on a case series of four patients in China. The burden of those conversations is something I never want to have to live through again,” she said.
“Oncology is a particularly intense subspecialty within medicine,” agreed Dr. Subbiah. “The people we care for have received a life-altering and potentially life-limiting diagnosis. Coupled with that, the COVID-19 pandemic has brought an unprecedented cloud of uncertainty ... Whether the patients can see it overtly or not, oncologists carry the weight of this worry with them for not just one but all of their patients.”
Dr. Wildes said she plans to return to academic medicine and clinical care “in time,” but for now, the gap that she and others like her leave is troubling to those who have stayed on.
“We need these women in medicine,” said Dr. Holstein. “We have data suggesting that women take more time with their patients than men, that patient outcomes are better if they have a female physician. But also for the generations coming up, we need the mid-career and senior women to be in place to mentor and guide and make sure we continue to increase women in leadership.”
A version of this article originally appeared on Medscape.com.
Inverse Distribution of Pink Macules and Patches
Punch biopsies from the right axilla (Figure) and right abdomen as well as a tangential biopsy from the left volar wrist papule showed an interstitial histiocytic infiltrate with focal palisading of histiocytes around central regions with collagen alteration and increased mucin. Grocott-Gomori methenamine-silver stain and acid-fast bacilli smear both were negative for organisms; these findings were consistent with a diagnosis of granuloma annulare (GA).

Granuloma annulare is a noninfectious granulomatous disease of unknown etiology. It most commonly appears as asymptomatic, flesh-colored, pink or violaceous annular patches or thin plaques favoring the trunk and extremities. Granuloma annulare has many documented presentations including generalized, patch, subcutaneous, and perforating forms. It can present as macules, papules, nodules, patches, or plaques. Reported associations include diabetes mellitus, hyperlipidemia, solid organ tumors, systemic infection, and thyroid disease.1 Granuloma annulare can occur in any age group but is most common between the ages of 20 and 40 years.2
Diagnosis most often is made clinically and can be confirmed by histopathology. Histologic examination most commonly shows histiocytes within the dermis that palisade around a central area of mucin deposition between degenerating collagen fibers. The histiocytes of GA stain positive with vimentin, lysozyme, and CD68. The increased mucin stains with colloidal iron and Alcian blue. Multinucleated giant cells and perivascular lymphocytic infiltrate also are commonly seen.3
Cutaneous B-cell lymphoma has a wide range of presentations but usually occurs as hyperpigmented plaques and patches with dermal atrophy. Psoriasis can present in an inverse distribution but will show epidermal changes including scale. Sarcoidosis presents as multiple erythematous plaques and papules and also can be accompanied by erythema nodosum. Tinea corporis likely would have resolved with antifungal treatment.
Many different treatments have been described as effective, including cryosurgery, topical and intralesional corticosteroids, antibiotics, immune modulators, phototherapy, and oral corticosteroids.1 We started our patient on triple-antibiotic therapy with rifampin 600 mg, minocycline 100 mg, and ofloxacin 400 mg all once monthly for 6 months, which has been shown to be efficacious in treating GA.4 The patient returned for follow-up 1 year after the initial presentation. At that time, she had faint pink patches on the waist and medial upper thighs, and the axillary lesions had cleared. In the interim, she developed more classic GA lesions—pink to violaceous smooth papules with no overlying epidermal changes—on the volar wrists and dorsal feet. These lesions were asymptomatic, and she currently is not undergoing any further treatment.
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Patterson JW, Hosler GA. The granulomatous reaction pattern. Weedon’s Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016:198-203.
- Marcus DV, Mahmoud BH, Hamzavi IH. Granuloma annulare treated with rifampin, ofloxacin, and minocycline combination therapy. Arch Dermatol. 2009;145:787-789.
Punch biopsies from the right axilla (Figure) and right abdomen as well as a tangential biopsy from the left volar wrist papule showed an interstitial histiocytic infiltrate with focal palisading of histiocytes around central regions with collagen alteration and increased mucin. Grocott-Gomori methenamine-silver stain and acid-fast bacilli smear both were negative for organisms; these findings were consistent with a diagnosis of granuloma annulare (GA).
Granuloma annulare is a noninfectious granulomatous disease of unknown etiology. It most commonly appears as asymptomatic, flesh-colored, pink or violaceous annular patches or thin plaques favoring the trunk and extremities. Granuloma annulare has many documented presentations including generalized, patch, subcutaneous, and perforating forms. It can present as macules, papules, nodules, patches, or plaques. Reported associations include diabetes mellitus, hyperlipidemia, solid organ tumors, systemic infection, and thyroid disease.1 Granuloma annulare can occur in any age group but is most common between the ages of 20 and 40 years.2
Diagnosis most often is made clinically and can be confirmed by histopathology. Histologic examination most commonly shows histiocytes within the dermis that palisade around a central area of mucin deposition between degenerating collagen fibers. The histiocytes of GA stain positive with vimentin, lysozyme, and CD68. The increased mucin stains with colloidal iron and Alcian blue. Multinucleated giant cells and perivascular lymphocytic infiltrate also are commonly seen.3
Cutaneous B-cell lymphoma has a wide range of presentations but usually occurs as hyperpigmented plaques and patches with dermal atrophy. Psoriasis can present in an inverse distribution but will show epidermal changes including scale. Sarcoidosis presents as multiple erythematous plaques and papules and also can be accompanied by erythema nodosum. Tinea corporis likely would have resolved with antifungal treatment.
Many different treatments have been described as effective, including cryosurgery, topical and intralesional corticosteroids, antibiotics, immune modulators, phototherapy, and oral corticosteroids.1 We started our patient on triple-antibiotic therapy with rifampin 600 mg, minocycline 100 mg, and ofloxacin 400 mg all once monthly for 6 months, which has been shown to be efficacious in treating GA.4 The patient returned for follow-up 1 year after the initial presentation. At that time, she had faint pink patches on the waist and medial upper thighs, and the axillary lesions had cleared. In the interim, she developed more classic GA lesions—pink to violaceous smooth papules with no overlying epidermal changes—on the volar wrists and dorsal feet. These lesions were asymptomatic, and she currently is not undergoing any further treatment.
Punch biopsies from the right axilla (Figure) and right abdomen as well as a tangential biopsy from the left volar wrist papule showed an interstitial histiocytic infiltrate with focal palisading of histiocytes around central regions with collagen alteration and increased mucin. Grocott-Gomori methenamine-silver stain and acid-fast bacilli smear both were negative for organisms; these findings were consistent with a diagnosis of granuloma annulare (GA).
Granuloma annulare is a noninfectious granulomatous disease of unknown etiology. It most commonly appears as asymptomatic, flesh-colored, pink or violaceous annular patches or thin plaques favoring the trunk and extremities. Granuloma annulare has many documented presentations including generalized, patch, subcutaneous, and perforating forms. It can present as macules, papules, nodules, patches, or plaques. Reported associations include diabetes mellitus, hyperlipidemia, solid organ tumors, systemic infection, and thyroid disease.1 Granuloma annulare can occur in any age group but is most common between the ages of 20 and 40 years.2
Diagnosis most often is made clinically and can be confirmed by histopathology. Histologic examination most commonly shows histiocytes within the dermis that palisade around a central area of mucin deposition between degenerating collagen fibers. The histiocytes of GA stain positive with vimentin, lysozyme, and CD68. The increased mucin stains with colloidal iron and Alcian blue. Multinucleated giant cells and perivascular lymphocytic infiltrate also are commonly seen.3
Cutaneous B-cell lymphoma has a wide range of presentations but usually occurs as hyperpigmented plaques and patches with dermal atrophy. Psoriasis can present in an inverse distribution but will show epidermal changes including scale. Sarcoidosis presents as multiple erythematous plaques and papules and also can be accompanied by erythema nodosum. Tinea corporis likely would have resolved with antifungal treatment.
Many different treatments have been described as effective, including cryosurgery, topical and intralesional corticosteroids, antibiotics, immune modulators, phototherapy, and oral corticosteroids.1 We started our patient on triple-antibiotic therapy with rifampin 600 mg, minocycline 100 mg, and ofloxacin 400 mg all once monthly for 6 months, which has been shown to be efficacious in treating GA.4 The patient returned for follow-up 1 year after the initial presentation. At that time, she had faint pink patches on the waist and medial upper thighs, and the axillary lesions had cleared. In the interim, she developed more classic GA lesions—pink to violaceous smooth papules with no overlying epidermal changes—on the volar wrists and dorsal feet. These lesions were asymptomatic, and she currently is not undergoing any further treatment.
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Patterson JW, Hosler GA. The granulomatous reaction pattern. Weedon’s Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016:198-203.
- Marcus DV, Mahmoud BH, Hamzavi IH. Granuloma annulare treated with rifampin, ofloxacin, and minocycline combination therapy. Arch Dermatol. 2009;145:787-789.
- Piette EW, Rosenbach M. Granuloma annulare: pathogenesis, disease associations and triggers, and therapeutic options. J Am Acad Dermatol. 2016;75:467-479.
- Piette EW, Rosenbach M. Granuloma annulare: clinical and histologic variants, epidemiology, and genetics. J Am Acad Dermatol. 2016;75:457-465.
- Patterson JW, Hosler GA. The granulomatous reaction pattern. Weedon’s Skin Pathology. 4th ed. China: Churchill Livingstone Elsevier; 2016:198-203.
- Marcus DV, Mahmoud BH, Hamzavi IH. Granuloma annulare treated with rifampin, ofloxacin, and minocycline combination therapy. Arch Dermatol. 2009;145:787-789.
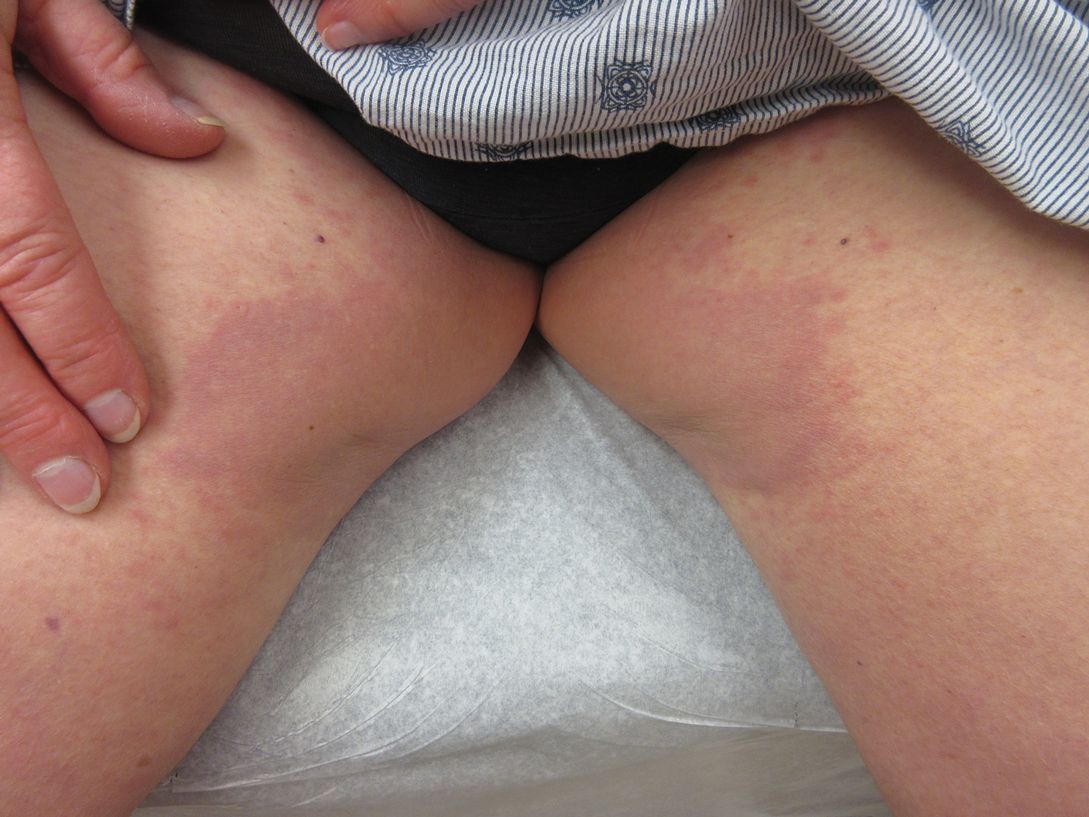
A 73-year-old woman presented for evaluation of an asymptomatic progressive rash on the left wrist, waist, groin, and inner thighs of 2 months’ duration. Her primary care provider prescribed clotrimazole and fluconazole with no improvement. Review of systems was negative. Medications included omeprazole, candesartan hydrochlorothiazide, potassium chloride, and levothyroxine. Physical examination revealed many scattered, pink to violaceous macules and patches in the axillae (sparing the vaults) and inguinal folds as well as on the waist and medial upper thighs. The lesions were without scale or other epidermal change. She also had a pink papule on the left volar wrist. A Wood lamp examination was unremarkable, and punch biopsies were performed.
A Review of ACR Convergence Abstracts on Rheumatoid Arthritis
Dr Stanley Cohen, of the University of Texas, Southwestern Medical School in Dallas, reviews key abstracts on the management of patients with rheumatoid arthritis (RA) that were presented at this year's American College of Rheumatology (ACR) annual meeting, held virtually because of COVID-19.
Dr Cohen highlights the updated ACR pharmacologic recommendations for the treatment of RA, including the need to maximize methotrexate therapy as well as the avoidance of glucocorticoids and their associated long-term toxicity.
Dr Cohen discusses two abstracts addressing the use of the recombinant zoster vaccine (RZV) in patients with RA, including research from Sweden comparing immunologic response in patients taking JAK inhibitors with that of patients not on disease-modifying therapy. He also reports on a safety study from the Cleveland Clinic which followed patients after they received RZV and measured their rate of flares over 3 months.
Additionally, he discusses recent trial data on the incidence of and risk for venous thromboembolism events in patients with RA enrolled in the upadacitinib phase 3 clinical trial program.
--
Stanley B. Cohen, MD, Clinical Professor, Department of Internal Medicine, Rheumatic Diseases Division, UT Southwestern Medical School; Director, Division of Rheumatology, Texas Health Resources, Texas Health Presbyterian Hospital Dallas, Dallas, Texas.
Stanley B. Cohen, MD, has disclosed the following relevant financial relationships:
Received research grant from: Amgen; AbbVie; Genentech; Gilead; Pfizer; Roche.
Dr Stanley Cohen, of the University of Texas, Southwestern Medical School in Dallas, reviews key abstracts on the management of patients with rheumatoid arthritis (RA) that were presented at this year's American College of Rheumatology (ACR) annual meeting, held virtually because of COVID-19.
Dr Cohen highlights the updated ACR pharmacologic recommendations for the treatment of RA, including the need to maximize methotrexate therapy as well as the avoidance of glucocorticoids and their associated long-term toxicity.
Dr Cohen discusses two abstracts addressing the use of the recombinant zoster vaccine (RZV) in patients with RA, including research from Sweden comparing immunologic response in patients taking JAK inhibitors with that of patients not on disease-modifying therapy. He also reports on a safety study from the Cleveland Clinic which followed patients after they received RZV and measured their rate of flares over 3 months.
Additionally, he discusses recent trial data on the incidence of and risk for venous thromboembolism events in patients with RA enrolled in the upadacitinib phase 3 clinical trial program.
--
Stanley B. Cohen, MD, Clinical Professor, Department of Internal Medicine, Rheumatic Diseases Division, UT Southwestern Medical School; Director, Division of Rheumatology, Texas Health Resources, Texas Health Presbyterian Hospital Dallas, Dallas, Texas.
Stanley B. Cohen, MD, has disclosed the following relevant financial relationships:
Received research grant from: Amgen; AbbVie; Genentech; Gilead; Pfizer; Roche.
Dr Stanley Cohen, of the University of Texas, Southwestern Medical School in Dallas, reviews key abstracts on the management of patients with rheumatoid arthritis (RA) that were presented at this year's American College of Rheumatology (ACR) annual meeting, held virtually because of COVID-19.
Dr Cohen highlights the updated ACR pharmacologic recommendations for the treatment of RA, including the need to maximize methotrexate therapy as well as the avoidance of glucocorticoids and their associated long-term toxicity.
Dr Cohen discusses two abstracts addressing the use of the recombinant zoster vaccine (RZV) in patients with RA, including research from Sweden comparing immunologic response in patients taking JAK inhibitors with that of patients not on disease-modifying therapy. He also reports on a safety study from the Cleveland Clinic which followed patients after they received RZV and measured their rate of flares over 3 months.
Additionally, he discusses recent trial data on the incidence of and risk for venous thromboembolism events in patients with RA enrolled in the upadacitinib phase 3 clinical trial program.
--
Stanley B. Cohen, MD, Clinical Professor, Department of Internal Medicine, Rheumatic Diseases Division, UT Southwestern Medical School; Director, Division of Rheumatology, Texas Health Resources, Texas Health Presbyterian Hospital Dallas, Dallas, Texas.
Stanley B. Cohen, MD, has disclosed the following relevant financial relationships:
Received research grant from: Amgen; AbbVie; Genentech; Gilead; Pfizer; Roche.

Patients with HF have higher risks of postop mortality, complications after ambulatory noncardiac surgery
Background: Heart failure is a known risk factor for postoperative mortality and complications. Many of the studies used to establish this association, however, have focused on major high-risk surgeries and not on outpatient surgeries. Improved medical care has increased the survival rate of patients with heart failure and an increasing number of these patients are undergoing elective surgical procedures. This has led to an increasing need to better understand the degree to which heart failure affects preoperative risk in the outpatient setting.
Study design: A retrospective cohort study.
Setting: Multiple Veteran’s Affairs Hospitals using data from the VA Surgical Quality Improvement Program (VASQIP) and the VA Corporate Data Warehouse.
Synopsis: A total of 355,121 patients who underwent outpatient surgeries were analyzed. 19,353 patients had heart failure and 334,768 did not. Patients with heart failure had a higher risk of 90-day mortality with an adjusted odds ratio of 1.95 (95% confidence interval, 1.69-2.44), and this risk progressively increased as the ejection fraction decreased. The risk of 30-day complication also increased in patients with heart failure with an adjusted OR of 1.10 (95% CI, 1.02-1.19).
Limitations of this study include the patient population, which were all veterans and mostly male. The nature of the inclusion criteria was limiting as well, in that all the patients in this study were deemed fit for surgery. There were no data available for patients who had been considered but ultimately did not undergo surgery or for patients who were considered for ambulatory surgery but ultimately underwent inpatient surgery. These limitations may have resulted in a selection bias, which limited the generalizability of the study’s findings when assessing patients for ambulatory surgery.
Bottom line: Patients with heart failure had a higher risk of 90-day postoperative mortality and 30-day postoperative complication in ambulatory noncardiac surgery. The risk of postoperative mortality increased as systolic function decreased.
Citation: Lerman BJ et al. Association between heart failure and postoperative mortality among patients undergoing ambulatory noncardiac surgery. JAMA Surg. 2019 Jul 10. doi: 10.1001/jamasurg.2019.2110.
Dr. Cheatham is a hospitalist and clinical educator at St. Louis University School of Medicine.
Background: Heart failure is a known risk factor for postoperative mortality and complications. Many of the studies used to establish this association, however, have focused on major high-risk surgeries and not on outpatient surgeries. Improved medical care has increased the survival rate of patients with heart failure and an increasing number of these patients are undergoing elective surgical procedures. This has led to an increasing need to better understand the degree to which heart failure affects preoperative risk in the outpatient setting.
Study design: A retrospective cohort study.
Setting: Multiple Veteran’s Affairs Hospitals using data from the VA Surgical Quality Improvement Program (VASQIP) and the VA Corporate Data Warehouse.
Synopsis: A total of 355,121 patients who underwent outpatient surgeries were analyzed. 19,353 patients had heart failure and 334,768 did not. Patients with heart failure had a higher risk of 90-day mortality with an adjusted odds ratio of 1.95 (95% confidence interval, 1.69-2.44), and this risk progressively increased as the ejection fraction decreased. The risk of 30-day complication also increased in patients with heart failure with an adjusted OR of 1.10 (95% CI, 1.02-1.19).
Limitations of this study include the patient population, which were all veterans and mostly male. The nature of the inclusion criteria was limiting as well, in that all the patients in this study were deemed fit for surgery. There were no data available for patients who had been considered but ultimately did not undergo surgery or for patients who were considered for ambulatory surgery but ultimately underwent inpatient surgery. These limitations may have resulted in a selection bias, which limited the generalizability of the study’s findings when assessing patients for ambulatory surgery.
Bottom line: Patients with heart failure had a higher risk of 90-day postoperative mortality and 30-day postoperative complication in ambulatory noncardiac surgery. The risk of postoperative mortality increased as systolic function decreased.
Citation: Lerman BJ et al. Association between heart failure and postoperative mortality among patients undergoing ambulatory noncardiac surgery. JAMA Surg. 2019 Jul 10. doi: 10.1001/jamasurg.2019.2110.
Dr. Cheatham is a hospitalist and clinical educator at St. Louis University School of Medicine.
Background: Heart failure is a known risk factor for postoperative mortality and complications. Many of the studies used to establish this association, however, have focused on major high-risk surgeries and not on outpatient surgeries. Improved medical care has increased the survival rate of patients with heart failure and an increasing number of these patients are undergoing elective surgical procedures. This has led to an increasing need to better understand the degree to which heart failure affects preoperative risk in the outpatient setting.
Study design: A retrospective cohort study.
Setting: Multiple Veteran’s Affairs Hospitals using data from the VA Surgical Quality Improvement Program (VASQIP) and the VA Corporate Data Warehouse.
Synopsis: A total of 355,121 patients who underwent outpatient surgeries were analyzed. 19,353 patients had heart failure and 334,768 did not. Patients with heart failure had a higher risk of 90-day mortality with an adjusted odds ratio of 1.95 (95% confidence interval, 1.69-2.44), and this risk progressively increased as the ejection fraction decreased. The risk of 30-day complication also increased in patients with heart failure with an adjusted OR of 1.10 (95% CI, 1.02-1.19).
Limitations of this study include the patient population, which were all veterans and mostly male. The nature of the inclusion criteria was limiting as well, in that all the patients in this study were deemed fit for surgery. There were no data available for patients who had been considered but ultimately did not undergo surgery or for patients who were considered for ambulatory surgery but ultimately underwent inpatient surgery. These limitations may have resulted in a selection bias, which limited the generalizability of the study’s findings when assessing patients for ambulatory surgery.
Bottom line: Patients with heart failure had a higher risk of 90-day postoperative mortality and 30-day postoperative complication in ambulatory noncardiac surgery. The risk of postoperative mortality increased as systolic function decreased.
Citation: Lerman BJ et al. Association between heart failure and postoperative mortality among patients undergoing ambulatory noncardiac surgery. JAMA Surg. 2019 Jul 10. doi: 10.1001/jamasurg.2019.2110.
Dr. Cheatham is a hospitalist and clinical educator at St. Louis University School of Medicine.
Applying Guidelines in Practice: Noninvasive Prenatal Testing

This supplement is designed to provide ObGyn clinicians with current information on the cell-free DNA screening test options available for fetal chromosomal abnormalities. These screening tests are commonly referred to as Noninvasive Prenatal Screening (NIPS). In August 2020, the American College of Obstetricians and Gynecologists (ACOG) issued a Practice Bulletin entitled “Screening for Fetal Chromosomal Abnormalities” (PB #226). This Practice Bulletin included expanded information regarding the use of NIPS in all patients regardless of maternal age or baseline risk. It also identified NIPS as the most sensitive and specific test for screening for the most common aneuploidies. The authors of this supplement provide additional information on the technology, performance, and clinical utilization of NIPS testing.
Click here to read this article
CME CREDITS: 1 CREDIT
To receive CME credit, please read the articles and go to www.omniaeducation.com/NIPT to access the post-test and evaluation.
This supplement is designed to provide ObGyn clinicians with current information on the cell-free DNA screening test options available for fetal chromosomal abnormalities. These screening tests are commonly referred to as Noninvasive Prenatal Screening (NIPS). In August 2020, the American College of Obstetricians and Gynecologists (ACOG) issued a Practice Bulletin entitled “Screening for Fetal Chromosomal Abnormalities” (PB #226). This Practice Bulletin included expanded information regarding the use of NIPS in all patients regardless of maternal age or baseline risk. It also identified NIPS as the most sensitive and specific test for screening for the most common aneuploidies. The authors of this supplement provide additional information on the technology, performance, and clinical utilization of NIPS testing.
Click here to read this article
CME CREDITS: 1 CREDIT
To receive CME credit, please read the articles and go to www.omniaeducation.com/NIPT to access the post-test and evaluation.
This supplement is designed to provide ObGyn clinicians with current information on the cell-free DNA screening test options available for fetal chromosomal abnormalities. These screening tests are commonly referred to as Noninvasive Prenatal Screening (NIPS). In August 2020, the American College of Obstetricians and Gynecologists (ACOG) issued a Practice Bulletin entitled “Screening for Fetal Chromosomal Abnormalities” (PB #226). This Practice Bulletin included expanded information regarding the use of NIPS in all patients regardless of maternal age or baseline risk. It also identified NIPS as the most sensitive and specific test for screening for the most common aneuploidies. The authors of this supplement provide additional information on the technology, performance, and clinical utilization of NIPS testing.
Click here to read this article