User login
CDC shortens COVID-19 quarantine time to 10 or 7 days, with conditions
Citing new evidence and an “acceptable risk” of transmission, the agency hopes reducing the 14-day quarantine will increase overall compliance and improve public health and economic constraints.
The agency also suggested people postpone travel during the upcoming winter holidays and stay home because of the pandemic.
These shorter quarantine options do not replace initial CDC guidance. “CDC continues to recommend quarantining for 14 days as the best way to reduce risk for spreading COVID-19,” said Henry Walke, MD, MPH, the CDC’s COVID-19 incident manager, during a media briefing on Wednesday.
However, “after reviewing and analyzing new research and data, CDC has identified two acceptable alternative quarantine periods.”
People can now quarantine for 10 days without a COVID-19 test if they have no symptoms. Alternatively, a quarantine can end after 7 days for someone with a negative test and no symptoms. The agency recommends a polymerase chain reaction test or an antigen assay within 48 hours before the end of a quarantine.
The agency also suggests people still monitor for symptoms for a full 14 days.
Reducing the length of quarantine “may make it easier for people to take this critical public health action, by reducing the economic hardship associated with a longer period, especially if they cannot work during that time,” Dr. Walke said. “In addition, a shorter quarantine period can lessen stress on the public health system and communities, especially when new infections are rapidly rising.”
The federal guidance leaves flexibility for local jurisdictions to make their own quarantine recommendations, as warranted, he added.
An ‘acceptable risk’ calculation
Modeling by the CDC and academic and public health partners led to the new quarantine recommendations, said John Brooks, MD, chief medical officer for the CDC’s COVID-19 response. Multiple studies “point in the same direction, which is that we can safely reduce the length of quarantine but accept there is a small residual risk that a person who is leaving quarantine early could transmit to someone else.”
The residual risk is approximately 1%, with an upper limit of 10%, when people quarantine for 10 days. A 7-day quarantine carries a residual risk of about 5% and an upper limit of 12%.
“Ten days is where the risk got into a sweet spot we like, at about 1%,” Dr. Brooks said. “That is a very acceptable risk, I think, for many people.”
Although it remains unknown what proportion of people spending 14 days in quarantine leave early, “we are hearing anecdotally from our partners in public health that many people are discontinuing quarantine ahead of time because there is pressure to go back to work, to get people back into school – and it imposes a burden on the individual,” Dr. Brooks said.
“One of our hopes is that ... if we reduce the amount of time they have to spend in quarantine, people will be more compliant,” he added.
A reporter asked why the CDC is shortening quarantines when the pandemic numbers are increasing nationwide. The timing has to do with capacity, Dr. Brooks said. “We are in situation where the number of cases is rising, the number of contacts is rising and the number of people who require quarantine is rising. That is a lot of burden, not just on the people who have to quarantine, but on public health.”
Home for the holidays
Similar to its pre-Thanksgiving advisory, the CDC also recommends people avoid travel during the upcoming winter holidays. “The best way to protect yourself and others is to postpone travel and stay home,” Dr. Walke said.
If people do decide to travel, the agency recommends COVID-19 testing 1-3 days prior to travel and again 3-5 days afterward, as well as reducing nonessential activities for a full 7 days after returning home. Furthermore, if someone does not have follow-up testing, the CDC recommends reducing nonessential activities for 10 days.
Testing does not eliminate all risk, Dr. Walke said, “but when combined with reducing nonessential activities, symptom screening and continuing with precautions like wearing masks, social distancing and hand washing, it can make travel safer.”
“We are trying to reduce the number of infections by postponing travel over the winter holiday,” Cindy Friedman, MD, chief of the CDC Travelers’ Health Branch, said during the media briefing.
“Travel volume was high during Thanksgiving,” she said, “and even if only a small percentage of those travelers were asymptomatically infected, this can translate into hundreds of thousands of additional infections moving from one community to another.”
This article first appeared on Medscape.com.
Citing new evidence and an “acceptable risk” of transmission, the agency hopes reducing the 14-day quarantine will increase overall compliance and improve public health and economic constraints.
The agency also suggested people postpone travel during the upcoming winter holidays and stay home because of the pandemic.
These shorter quarantine options do not replace initial CDC guidance. “CDC continues to recommend quarantining for 14 days as the best way to reduce risk for spreading COVID-19,” said Henry Walke, MD, MPH, the CDC’s COVID-19 incident manager, during a media briefing on Wednesday.
However, “after reviewing and analyzing new research and data, CDC has identified two acceptable alternative quarantine periods.”
People can now quarantine for 10 days without a COVID-19 test if they have no symptoms. Alternatively, a quarantine can end after 7 days for someone with a negative test and no symptoms. The agency recommends a polymerase chain reaction test or an antigen assay within 48 hours before the end of a quarantine.
The agency also suggests people still monitor for symptoms for a full 14 days.
Reducing the length of quarantine “may make it easier for people to take this critical public health action, by reducing the economic hardship associated with a longer period, especially if they cannot work during that time,” Dr. Walke said. “In addition, a shorter quarantine period can lessen stress on the public health system and communities, especially when new infections are rapidly rising.”
The federal guidance leaves flexibility for local jurisdictions to make their own quarantine recommendations, as warranted, he added.
An ‘acceptable risk’ calculation
Modeling by the CDC and academic and public health partners led to the new quarantine recommendations, said John Brooks, MD, chief medical officer for the CDC’s COVID-19 response. Multiple studies “point in the same direction, which is that we can safely reduce the length of quarantine but accept there is a small residual risk that a person who is leaving quarantine early could transmit to someone else.”
The residual risk is approximately 1%, with an upper limit of 10%, when people quarantine for 10 days. A 7-day quarantine carries a residual risk of about 5% and an upper limit of 12%.
“Ten days is where the risk got into a sweet spot we like, at about 1%,” Dr. Brooks said. “That is a very acceptable risk, I think, for many people.”
Although it remains unknown what proportion of people spending 14 days in quarantine leave early, “we are hearing anecdotally from our partners in public health that many people are discontinuing quarantine ahead of time because there is pressure to go back to work, to get people back into school – and it imposes a burden on the individual,” Dr. Brooks said.
“One of our hopes is that ... if we reduce the amount of time they have to spend in quarantine, people will be more compliant,” he added.
A reporter asked why the CDC is shortening quarantines when the pandemic numbers are increasing nationwide. The timing has to do with capacity, Dr. Brooks said. “We are in situation where the number of cases is rising, the number of contacts is rising and the number of people who require quarantine is rising. That is a lot of burden, not just on the people who have to quarantine, but on public health.”
Home for the holidays
Similar to its pre-Thanksgiving advisory, the CDC also recommends people avoid travel during the upcoming winter holidays. “The best way to protect yourself and others is to postpone travel and stay home,” Dr. Walke said.
If people do decide to travel, the agency recommends COVID-19 testing 1-3 days prior to travel and again 3-5 days afterward, as well as reducing nonessential activities for a full 7 days after returning home. Furthermore, if someone does not have follow-up testing, the CDC recommends reducing nonessential activities for 10 days.
Testing does not eliminate all risk, Dr. Walke said, “but when combined with reducing nonessential activities, symptom screening and continuing with precautions like wearing masks, social distancing and hand washing, it can make travel safer.”
“We are trying to reduce the number of infections by postponing travel over the winter holiday,” Cindy Friedman, MD, chief of the CDC Travelers’ Health Branch, said during the media briefing.
“Travel volume was high during Thanksgiving,” she said, “and even if only a small percentage of those travelers were asymptomatically infected, this can translate into hundreds of thousands of additional infections moving from one community to another.”
This article first appeared on Medscape.com.
Citing new evidence and an “acceptable risk” of transmission, the agency hopes reducing the 14-day quarantine will increase overall compliance and improve public health and economic constraints.
The agency also suggested people postpone travel during the upcoming winter holidays and stay home because of the pandemic.
These shorter quarantine options do not replace initial CDC guidance. “CDC continues to recommend quarantining for 14 days as the best way to reduce risk for spreading COVID-19,” said Henry Walke, MD, MPH, the CDC’s COVID-19 incident manager, during a media briefing on Wednesday.
However, “after reviewing and analyzing new research and data, CDC has identified two acceptable alternative quarantine periods.”
People can now quarantine for 10 days without a COVID-19 test if they have no symptoms. Alternatively, a quarantine can end after 7 days for someone with a negative test and no symptoms. The agency recommends a polymerase chain reaction test or an antigen assay within 48 hours before the end of a quarantine.
The agency also suggests people still monitor for symptoms for a full 14 days.
Reducing the length of quarantine “may make it easier for people to take this critical public health action, by reducing the economic hardship associated with a longer period, especially if they cannot work during that time,” Dr. Walke said. “In addition, a shorter quarantine period can lessen stress on the public health system and communities, especially when new infections are rapidly rising.”
The federal guidance leaves flexibility for local jurisdictions to make their own quarantine recommendations, as warranted, he added.
An ‘acceptable risk’ calculation
Modeling by the CDC and academic and public health partners led to the new quarantine recommendations, said John Brooks, MD, chief medical officer for the CDC’s COVID-19 response. Multiple studies “point in the same direction, which is that we can safely reduce the length of quarantine but accept there is a small residual risk that a person who is leaving quarantine early could transmit to someone else.”
The residual risk is approximately 1%, with an upper limit of 10%, when people quarantine for 10 days. A 7-day quarantine carries a residual risk of about 5% and an upper limit of 12%.
“Ten days is where the risk got into a sweet spot we like, at about 1%,” Dr. Brooks said. “That is a very acceptable risk, I think, for many people.”
Although it remains unknown what proportion of people spending 14 days in quarantine leave early, “we are hearing anecdotally from our partners in public health that many people are discontinuing quarantine ahead of time because there is pressure to go back to work, to get people back into school – and it imposes a burden on the individual,” Dr. Brooks said.
“One of our hopes is that ... if we reduce the amount of time they have to spend in quarantine, people will be more compliant,” he added.
A reporter asked why the CDC is shortening quarantines when the pandemic numbers are increasing nationwide. The timing has to do with capacity, Dr. Brooks said. “We are in situation where the number of cases is rising, the number of contacts is rising and the number of people who require quarantine is rising. That is a lot of burden, not just on the people who have to quarantine, but on public health.”
Home for the holidays
Similar to its pre-Thanksgiving advisory, the CDC also recommends people avoid travel during the upcoming winter holidays. “The best way to protect yourself and others is to postpone travel and stay home,” Dr. Walke said.
If people do decide to travel, the agency recommends COVID-19 testing 1-3 days prior to travel and again 3-5 days afterward, as well as reducing nonessential activities for a full 7 days after returning home. Furthermore, if someone does not have follow-up testing, the CDC recommends reducing nonessential activities for 10 days.
Testing does not eliminate all risk, Dr. Walke said, “but when combined with reducing nonessential activities, symptom screening and continuing with precautions like wearing masks, social distancing and hand washing, it can make travel safer.”
“We are trying to reduce the number of infections by postponing travel over the winter holiday,” Cindy Friedman, MD, chief of the CDC Travelers’ Health Branch, said during the media briefing.
“Travel volume was high during Thanksgiving,” she said, “and even if only a small percentage of those travelers were asymptomatically infected, this can translate into hundreds of thousands of additional infections moving from one community to another.”
This article first appeared on Medscape.com.
Real acupuncture beat sham for osteoarthritis knee pain
Electro-acupuncture resulted in significant improvement in pain and function, compared with sham acupuncture, in a randomized trial of more than 400 adults with knee OA.
The socioeconomic burden of knee OA (KOA) remains high, and will likely increase with the aging population and rising rates of obesity, wrote first author Jian-Feng Tu, MD, PhD, of Beijing University of Chinese Medicine and colleagues. “Since no disease-modifying pharmaceutical agents have been approved, current KOA treatments are mainly symptomatic,” and identifying new therapies in addition to pharmacological agents or surgery is a research priority, they added. The research on acupuncture as a treatment for KOA has increased, but remains controversial as researchers attempt to determine the number of sessions needed for effectiveness.
In a study published in Arthritis & Rheumatology, the researchers recruited 480 adults aged 45-75 years with confirmed KOA who reported knee pain for longer than 6 months. Participants were randomized to three groups: electroacupuncture (EA), manual acupuncture (MA), or sham acupuncture (SA). Each group received three treatment sessions per week. In all groups, electrodes were attached to selected acupuncture needles, but the current was turned on only in the EA treatment group.
The primary outcome was the response rate after 8 weeks of treatment, defined as patients who achieved the minimal clinically important improvement (MCII) on both the Numeric Rating Scale and the Western Ontario and McMaster Universities Osteoarthritis Index function subscale.
Overall, response rates at 8 weeks were 60.3%, 58.6%, and 47.3% for the EA, MA, and SA groups, respectively.
Between-group differences were statistically significant for EA versus SA (13%, P = .0234) but not for MA versus SA (11.3%, P = .0507) at 8 weeks; however, both EA and MA groups showed significantly higher response rates, compared with the SA group at 16 and 26 weeks. “Although a clinically meaningful response rate for KOA is not available in the literature, the difference of 11.3%, which indicates the number needed to treat of 9, is acceptable in clinical practices,” the researchers noted.
Adverse events occurred in 11.5% of the EA group, 14.2% of the MA group, and 10.8% of the SA group, and included subcutaneous hematoma, post-needling pain, and pantalgia. All adverse events related to acupuncture resolved within a week and none were serious, the researchers wrote.
The study findings were limited by several factors, including the potential burden on patients of three sessions per week, the limited study population of patients with radiologic grades of II or III only, the use of self-reports, and the lack of blinding for outcome assessors, the researchers noted.
However, the results show persistent effects in reducing pain and improving function with EA or MA, compared with SA, the researchers wrote. The findings were strengthened by “adequate dosage of acupuncture, the use of the primary outcome at an individual level, and the rigorous methodology.” The use of the MCII in the primary outcome “can provide patients and policy makers with more straightforward information to decide whether a treatment should be used.”
Optimal dosing questions remain
Current options for managing KOA are limited by factors that include low efficacy and unwanted side effects, while joint replacements increase the burden on health care systems, wrote David J. Hunter, MBBS, PhD, of the University of Sydney, and Richard E. Harris, PhD, of the University of Michigan, Ann Arbor, in an accompanying editorial. “In this context, development of new treatments or identification of efficacy of existing therapies to address the huge unmet need of pain are strongly desired.” Acupuncture continues to gain popularity in North and South America, but its efficacy for pain and KOA remain controversial.
The question of dose is challenging when assessing acupuncture because the optimal dose and how to classify it remains unknown. “In this study, the authors used three treatments a week, which is more frequent than typical studies done in the West and potentially may not be feasible in some health care settings. A recent systematic review suggests that treatment frequency matters and a dose of three sessions per week may be superior to less frequent treatment,” they emphasized. Acupuncture is generally considered to be safe, but many health systems do not reimburse for it. Patients may have large out-of-pocket expenses because of the number of visits required, which may be a barrier to further implementation in practice.
“Acupuncture is already widely practiced and readily available in many countries and health care systems,” the editorialists said. However, “more research is needed in the areas of dose-response relationships, effects of blinding the acupuncturist, feasibility of three times weekly regimens, and clarifying the mechanism of effect, particularly given the persistence of benefit.”
The study was funded by Beijing Municipal Science & Technology Commission and Beijing Municipal Administration of Hospitals. The researchers had no financial conflicts to disclose. Dr. Hunter disclosed support from a National Health and Medical Research Council Investigator Grant and providing consulting advice for Merck Serono, TLC Bio, Tissuegene, Lilly, and Pfizer.
SOURCE: Tu J-F et al. Arthritis Rheumatol. 2020 Nov 10. doi: 10.1002/art.41584.
Electro-acupuncture resulted in significant improvement in pain and function, compared with sham acupuncture, in a randomized trial of more than 400 adults with knee OA.
The socioeconomic burden of knee OA (KOA) remains high, and will likely increase with the aging population and rising rates of obesity, wrote first author Jian-Feng Tu, MD, PhD, of Beijing University of Chinese Medicine and colleagues. “Since no disease-modifying pharmaceutical agents have been approved, current KOA treatments are mainly symptomatic,” and identifying new therapies in addition to pharmacological agents or surgery is a research priority, they added. The research on acupuncture as a treatment for KOA has increased, but remains controversial as researchers attempt to determine the number of sessions needed for effectiveness.
In a study published in Arthritis & Rheumatology, the researchers recruited 480 adults aged 45-75 years with confirmed KOA who reported knee pain for longer than 6 months. Participants were randomized to three groups: electroacupuncture (EA), manual acupuncture (MA), or sham acupuncture (SA). Each group received three treatment sessions per week. In all groups, electrodes were attached to selected acupuncture needles, but the current was turned on only in the EA treatment group.
The primary outcome was the response rate after 8 weeks of treatment, defined as patients who achieved the minimal clinically important improvement (MCII) on both the Numeric Rating Scale and the Western Ontario and McMaster Universities Osteoarthritis Index function subscale.
Overall, response rates at 8 weeks were 60.3%, 58.6%, and 47.3% for the EA, MA, and SA groups, respectively.
Between-group differences were statistically significant for EA versus SA (13%, P = .0234) but not for MA versus SA (11.3%, P = .0507) at 8 weeks; however, both EA and MA groups showed significantly higher response rates, compared with the SA group at 16 and 26 weeks. “Although a clinically meaningful response rate for KOA is not available in the literature, the difference of 11.3%, which indicates the number needed to treat of 9, is acceptable in clinical practices,” the researchers noted.
Adverse events occurred in 11.5% of the EA group, 14.2% of the MA group, and 10.8% of the SA group, and included subcutaneous hematoma, post-needling pain, and pantalgia. All adverse events related to acupuncture resolved within a week and none were serious, the researchers wrote.
The study findings were limited by several factors, including the potential burden on patients of three sessions per week, the limited study population of patients with radiologic grades of II or III only, the use of self-reports, and the lack of blinding for outcome assessors, the researchers noted.
However, the results show persistent effects in reducing pain and improving function with EA or MA, compared with SA, the researchers wrote. The findings were strengthened by “adequate dosage of acupuncture, the use of the primary outcome at an individual level, and the rigorous methodology.” The use of the MCII in the primary outcome “can provide patients and policy makers with more straightforward information to decide whether a treatment should be used.”
Optimal dosing questions remain
Current options for managing KOA are limited by factors that include low efficacy and unwanted side effects, while joint replacements increase the burden on health care systems, wrote David J. Hunter, MBBS, PhD, of the University of Sydney, and Richard E. Harris, PhD, of the University of Michigan, Ann Arbor, in an accompanying editorial. “In this context, development of new treatments or identification of efficacy of existing therapies to address the huge unmet need of pain are strongly desired.” Acupuncture continues to gain popularity in North and South America, but its efficacy for pain and KOA remain controversial.
The question of dose is challenging when assessing acupuncture because the optimal dose and how to classify it remains unknown. “In this study, the authors used three treatments a week, which is more frequent than typical studies done in the West and potentially may not be feasible in some health care settings. A recent systematic review suggests that treatment frequency matters and a dose of three sessions per week may be superior to less frequent treatment,” they emphasized. Acupuncture is generally considered to be safe, but many health systems do not reimburse for it. Patients may have large out-of-pocket expenses because of the number of visits required, which may be a barrier to further implementation in practice.
“Acupuncture is already widely practiced and readily available in many countries and health care systems,” the editorialists said. However, “more research is needed in the areas of dose-response relationships, effects of blinding the acupuncturist, feasibility of three times weekly regimens, and clarifying the mechanism of effect, particularly given the persistence of benefit.”
The study was funded by Beijing Municipal Science & Technology Commission and Beijing Municipal Administration of Hospitals. The researchers had no financial conflicts to disclose. Dr. Hunter disclosed support from a National Health and Medical Research Council Investigator Grant and providing consulting advice for Merck Serono, TLC Bio, Tissuegene, Lilly, and Pfizer.
SOURCE: Tu J-F et al. Arthritis Rheumatol. 2020 Nov 10. doi: 10.1002/art.41584.
Electro-acupuncture resulted in significant improvement in pain and function, compared with sham acupuncture, in a randomized trial of more than 400 adults with knee OA.
The socioeconomic burden of knee OA (KOA) remains high, and will likely increase with the aging population and rising rates of obesity, wrote first author Jian-Feng Tu, MD, PhD, of Beijing University of Chinese Medicine and colleagues. “Since no disease-modifying pharmaceutical agents have been approved, current KOA treatments are mainly symptomatic,” and identifying new therapies in addition to pharmacological agents or surgery is a research priority, they added. The research on acupuncture as a treatment for KOA has increased, but remains controversial as researchers attempt to determine the number of sessions needed for effectiveness.
In a study published in Arthritis & Rheumatology, the researchers recruited 480 adults aged 45-75 years with confirmed KOA who reported knee pain for longer than 6 months. Participants were randomized to three groups: electroacupuncture (EA), manual acupuncture (MA), or sham acupuncture (SA). Each group received three treatment sessions per week. In all groups, electrodes were attached to selected acupuncture needles, but the current was turned on only in the EA treatment group.
The primary outcome was the response rate after 8 weeks of treatment, defined as patients who achieved the minimal clinically important improvement (MCII) on both the Numeric Rating Scale and the Western Ontario and McMaster Universities Osteoarthritis Index function subscale.
Overall, response rates at 8 weeks were 60.3%, 58.6%, and 47.3% for the EA, MA, and SA groups, respectively.
Between-group differences were statistically significant for EA versus SA (13%, P = .0234) but not for MA versus SA (11.3%, P = .0507) at 8 weeks; however, both EA and MA groups showed significantly higher response rates, compared with the SA group at 16 and 26 weeks. “Although a clinically meaningful response rate for KOA is not available in the literature, the difference of 11.3%, which indicates the number needed to treat of 9, is acceptable in clinical practices,” the researchers noted.
Adverse events occurred in 11.5% of the EA group, 14.2% of the MA group, and 10.8% of the SA group, and included subcutaneous hematoma, post-needling pain, and pantalgia. All adverse events related to acupuncture resolved within a week and none were serious, the researchers wrote.
The study findings were limited by several factors, including the potential burden on patients of three sessions per week, the limited study population of patients with radiologic grades of II or III only, the use of self-reports, and the lack of blinding for outcome assessors, the researchers noted.
However, the results show persistent effects in reducing pain and improving function with EA or MA, compared with SA, the researchers wrote. The findings were strengthened by “adequate dosage of acupuncture, the use of the primary outcome at an individual level, and the rigorous methodology.” The use of the MCII in the primary outcome “can provide patients and policy makers with more straightforward information to decide whether a treatment should be used.”
Optimal dosing questions remain
Current options for managing KOA are limited by factors that include low efficacy and unwanted side effects, while joint replacements increase the burden on health care systems, wrote David J. Hunter, MBBS, PhD, of the University of Sydney, and Richard E. Harris, PhD, of the University of Michigan, Ann Arbor, in an accompanying editorial. “In this context, development of new treatments or identification of efficacy of existing therapies to address the huge unmet need of pain are strongly desired.” Acupuncture continues to gain popularity in North and South America, but its efficacy for pain and KOA remain controversial.
The question of dose is challenging when assessing acupuncture because the optimal dose and how to classify it remains unknown. “In this study, the authors used three treatments a week, which is more frequent than typical studies done in the West and potentially may not be feasible in some health care settings. A recent systematic review suggests that treatment frequency matters and a dose of three sessions per week may be superior to less frequent treatment,” they emphasized. Acupuncture is generally considered to be safe, but many health systems do not reimburse for it. Patients may have large out-of-pocket expenses because of the number of visits required, which may be a barrier to further implementation in practice.
“Acupuncture is already widely practiced and readily available in many countries and health care systems,” the editorialists said. However, “more research is needed in the areas of dose-response relationships, effects of blinding the acupuncturist, feasibility of three times weekly regimens, and clarifying the mechanism of effect, particularly given the persistence of benefit.”
The study was funded by Beijing Municipal Science & Technology Commission and Beijing Municipal Administration of Hospitals. The researchers had no financial conflicts to disclose. Dr. Hunter disclosed support from a National Health and Medical Research Council Investigator Grant and providing consulting advice for Merck Serono, TLC Bio, Tissuegene, Lilly, and Pfizer.
SOURCE: Tu J-F et al. Arthritis Rheumatol. 2020 Nov 10. doi: 10.1002/art.41584.
FROM ARTHRITIS & RHEUMATOLOGY
Symmetric Drug-Related Intertriginous and Flexural Exanthema
To the Editor:
Symmetric drug-related intertriginous and flexural exanthema (SDRIFE) is a curious disorder that has undergone many clinical transformations since first being described by Andersen et al1 in 1984 using the term baboon syndrome. Initially described as a mercury hypersensitivity reaction resulting in an eruption resembling the red-bottomed baboon, this exanthema has expanded in definition with inciting agents, clinical features, and diagnostic criteria. Its prognosis, however, has remained stable and favorable throughout the decades. The condition is almost universally benign and self-limited.1-3 As new cases are reported in the literature and the paradigm of SDRIFE continues to shift, its prognosis also may warrant reconsideration and respect as a potentially destructive reaction.
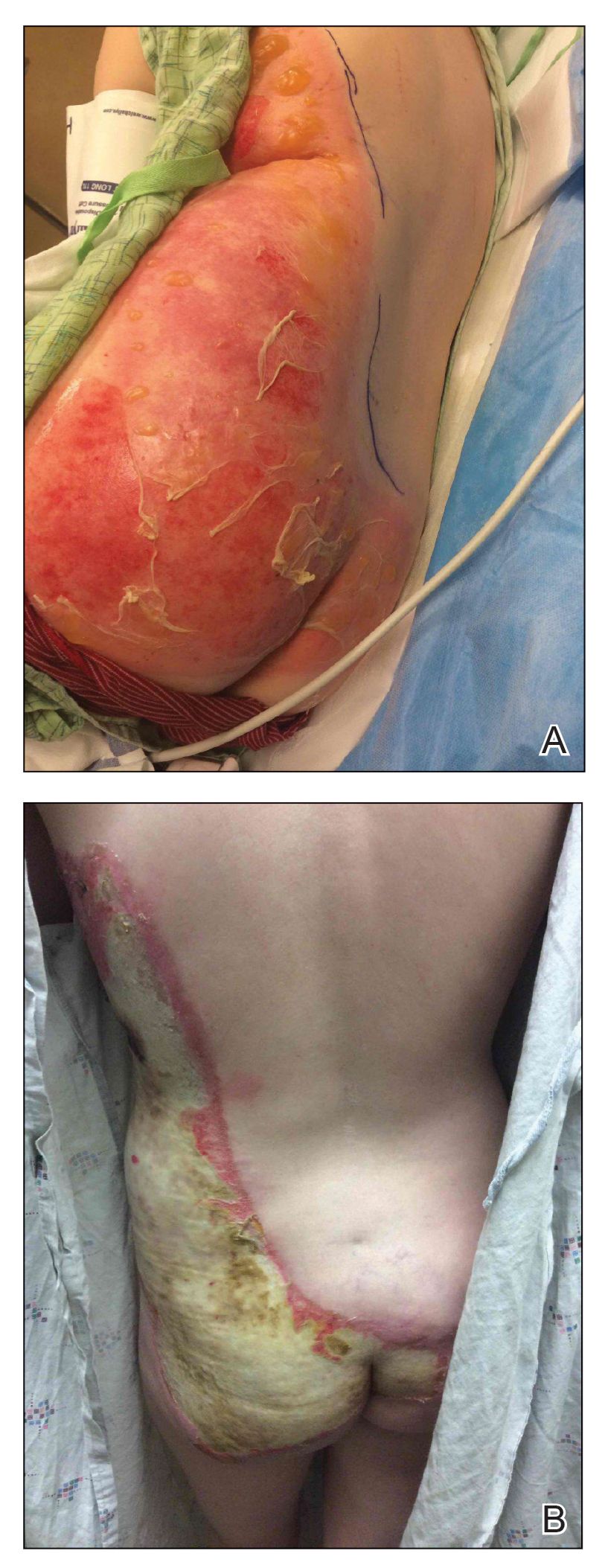
A 39-year-old woman who was otherwise healthy presented to the emergency department after developing a rapidly evolving and blistering rash on the left flank. Hours later, the rash had progressed to a sharply demarcated, confluent, erythematous plaque with central ulceration and large flaccid bullae peripherally, encompassing 18% of total body surface area and extending from the gluteal cleft to the tip of the scapula along the left flank (Figure 1) with no vaginal or mucosal involvement. The patient recently had completed a 10-day course of amoxicillin–clavulanic acid 2 days prior for a cat bite on the right dorsal wrist. Additional history confirmed the absence of prodromal fever, fatigue, or chills. Inciting trauma, including chemical and thermal burns, was denied. Potential underlying psychosocial cofounders were explored and were unrevealing.
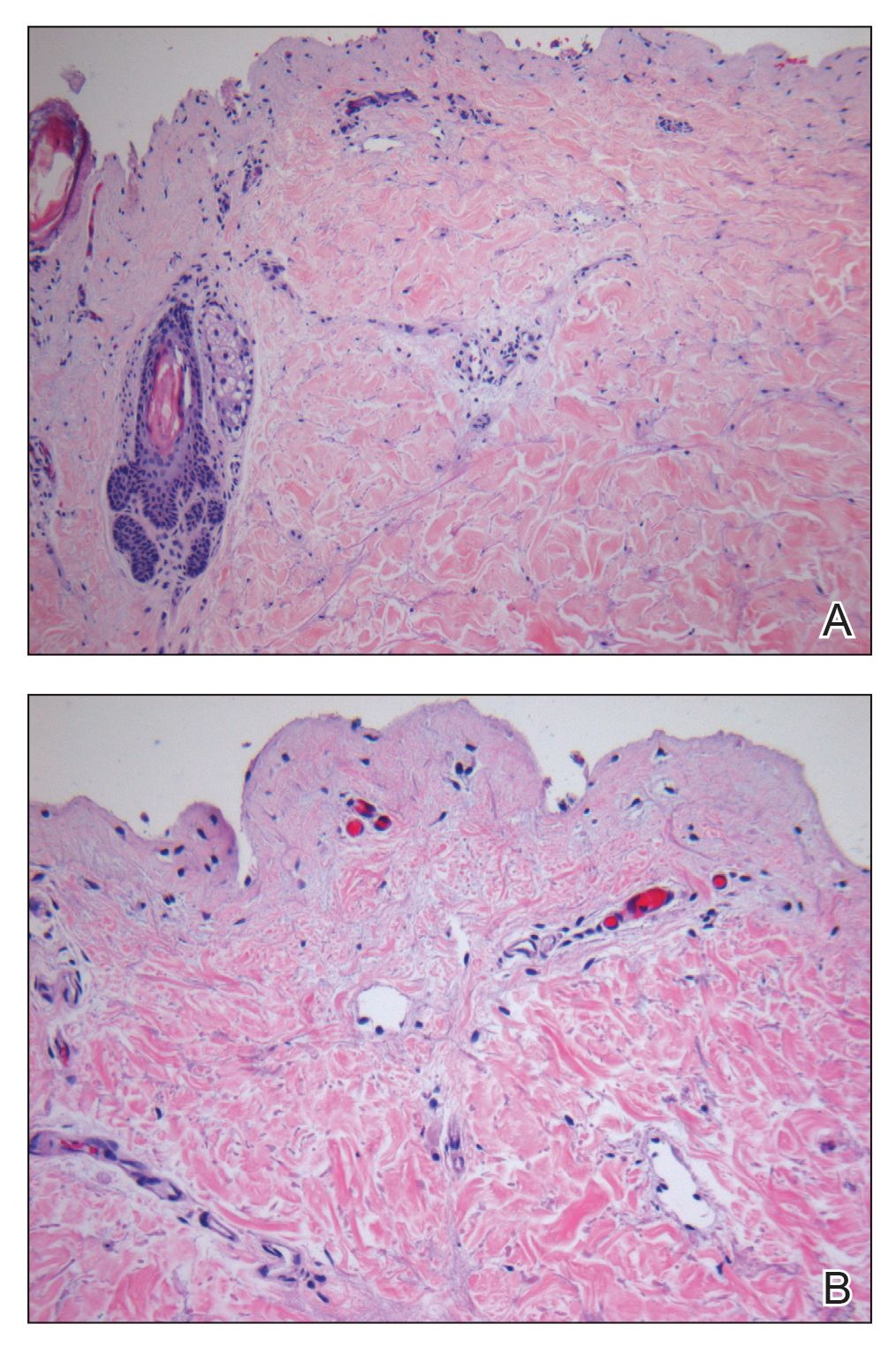
Laboratory test results, including complete blood cell count and metabolic panel as well as vital signs were unremarkable, except for slight leukocytosis at 14,000/µL (reference range 4500–11,000/µL). A punch biopsy was taken from the patient’s left upper back at the time of admission, which revealed a sparse, superficial, perivascular infiltrate of lymphocytes and rare neutrophils with largely absent epidermis and an occasional focal necrosis of adnexal epithelium (Figure 2). Immunofluorescence was negative for specific deposition of IgG, IgA, IgM, C3, or fibrinogen. Wound culture also returned negative, and the Naranjo adverse drug reaction probability scale score was calculated to be 4 out of 12, indicating possible adverse drug reaction.4
Given the extent and distribution of the rash as well as the full-thickness dermal involvement, the patient was transferred to the burn unit for subsequent care. At 8-month follow-up, she experienced severe, symptomatic, hypertrophic scarring and was awaiting intralesional triamcinolone acetonide injections. The patient subsequently was lost to follow up.
The clinical picture of SDRIFE has remained obscure over the last 30 years, likely owing to its rarity and unclear pathogenesis. Diagnostic criteria for SDRIFE were first proposed by Häusermann et al2 in 2004 and contained 5 elements: (1) occurrence after (re)exposure to systemic drugs, (2) sharply demarcated erythema of the gluteal region or V-shaped erythema of the inguinal area, (3) involvement of at least 1 other intertriginous location, (4) symmetry of affected areas, and (5) absence of systemic symptoms and signs. Based on these clinical criteria, our patients fulfilled 3 of 5 elements, with deductions for symmetry of affected areas and involvement of other intertriginous locations. Histopathologic findings in SDRIFE predominantly are nonspecific with superficial perivascular mononuclear infiltrates; however, prior reports have confirmed the potential for vacuolar changes and hydropic degeneration in the basal cell layer with subepidermal bullae formation.5,6 Similarly, although the presence of bullae are somewhat atypical in SDRIFE, it has been described.3 Taken together, we speculate that these findings may support a diagnosis of SDRIFE with atypical presentation, though an alternative diagnosis of bullous fixed drug eruption (FDE) cannot be ruled out.
Historically, SDRIFE has been associated with a benign course. The condition typically arises within a few hours to days following administration of the offending agent, most commonly amoxicillin or another β-lactam antibiotic.1 Most cases spontaneously resolve via desquamation within 1 to 2 weeks. We present an unusual case of amoxicillin-induced full-thickness epidermal necrosis resulting in symptomatic sequelae, which exhibits findings of SDRIFE, bullous FDE, or Stevens-Johnson syndrome/toxic epidermal necrolysis, suggesting the possibility for a common pathway underlying the pathogenesis of these conditions.
The diagnostic uncertainty that commonly accompanies these various toxic drug reactions may in part relate to their underlying immunopathogenesis. Although the exact mechanism by which SDRIFE results in its characteristic skin lesions has not been fully elucidated, prior work through patch testing, lymphocyte transformation assays, and immunohistochemical staining of biopsies suggests a type IV delayed hypersensitivity (DTH) reaction.7-10 Specifically, SDRIFE appears to share features of both DTH type IVa—involving CD4+ helper T cells (TH1), monocytes, and IFN-γ signaling—and DTH type IVc—involving cytotoxic CD4 and CD8 cells, granzyme B action, and FasL signaling.11,12 A similar inflammatory milieu has been implicated in numerous toxic drug eruptions, including Stevens-Johnson syndrome/toxic epidermal necrolysis and FDE.11,13 This mechanistic overlap may explain the overlap seen clinically among such conditions.
In the undifferentiated patient, categorization of the clinical syndrome proves helpful in prognostication and therapeutic approach. The complexities and commonalities intrinsic to these syndromes, however, may simultaneously preclude certain cases from neatly following the predefined rules. These atypical presentations, while diagnostically challenging, can in turn offer a unique opportunity to reexamine the current state of disease understanding to better allow for appropriate classification.
Despite its rarity, SDRIFE should be considered in the differential of undiagnosed drug eruptions, particularly as new clinical presentations emerge. Careful documentation and timely declaration of future cases will prove invaluable for diagnostic and therapeutic advancements should this once-benign condition develop a more destructive potential.
- Andersen KE, Hjorth N, Menné T. The baboon syndrome: systemically-induced allergic contact dermatitis. Contact Dermatitis. 1984;10:97-100.
- Häusermann P, Harr TH, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis. 2004;51:297-310.
- Tan SC, Tan JW. Symmetrical drug-related intertriginous and flexural exanthema. Curr Opin Allergy Clin Immunol. 2011;11:313-318.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Wolf R, Orion E, Matz H. The baboon syndrome or intertriginous drug eruption: a report of eleven cases and a second look at its pathomechanism. Dermatol Online J. 2003;9:2.
- Elmariah SB, Cheung W, Wang N, et al. Systemic drug-related intertriginous and flexural exanthema (SDRIFE). Dermatol Online J. 2009;15:3.
- Hembold P, Hegemann B, Dickert C, et al. Symptomatic psychotropic and nonpigmenting fixed drug eruption due to cimetidine (so-called baboon syndrome). Dermatology. 1998;197:402-403.
- Barbaud A, Trechot P, Granel F, et al. A baboon syndrome induced by intravenous human immunoglobulins: a report of a case and immunological analysis. Dermatology. 1999;199:258-260.
- Miyahara A, Kawashima H, Okubo Y, et al. A new proposal for a clinical-oriented subclassification of baboon syndrome and review of baboon syndrome. Asian Pac J Allergy Immunol. 2011;29:150-160.
- Goossens C, Sass U, Song M. Baboon syndrome. Dermatology. 1997;194:421-422.
- Pichler WJ. Delayed drug hypersensitivity reactions. Ann Intern Med. 2003;139:123-129.
- Ozkaya E. Current understanding of baboon syndrome. Expert Rev Dermatol. 2009;4:163-175.
- Ozakaya E. Fixed drug eruption: state of the art. J Dtsch Dermatol Ges. 2008;6:181-188.
To the Editor:
Symmetric drug-related intertriginous and flexural exanthema (SDRIFE) is a curious disorder that has undergone many clinical transformations since first being described by Andersen et al1 in 1984 using the term baboon syndrome. Initially described as a mercury hypersensitivity reaction resulting in an eruption resembling the red-bottomed baboon, this exanthema has expanded in definition with inciting agents, clinical features, and diagnostic criteria. Its prognosis, however, has remained stable and favorable throughout the decades. The condition is almost universally benign and self-limited.1-3 As new cases are reported in the literature and the paradigm of SDRIFE continues to shift, its prognosis also may warrant reconsideration and respect as a potentially destructive reaction.
A 39-year-old woman who was otherwise healthy presented to the emergency department after developing a rapidly evolving and blistering rash on the left flank. Hours later, the rash had progressed to a sharply demarcated, confluent, erythematous plaque with central ulceration and large flaccid bullae peripherally, encompassing 18% of total body surface area and extending from the gluteal cleft to the tip of the scapula along the left flank (Figure 1) with no vaginal or mucosal involvement. The patient recently had completed a 10-day course of amoxicillin–clavulanic acid 2 days prior for a cat bite on the right dorsal wrist. Additional history confirmed the absence of prodromal fever, fatigue, or chills. Inciting trauma, including chemical and thermal burns, was denied. Potential underlying psychosocial cofounders were explored and were unrevealing.
Laboratory test results, including complete blood cell count and metabolic panel as well as vital signs were unremarkable, except for slight leukocytosis at 14,000/µL (reference range 4500–11,000/µL). A punch biopsy was taken from the patient’s left upper back at the time of admission, which revealed a sparse, superficial, perivascular infiltrate of lymphocytes and rare neutrophils with largely absent epidermis and an occasional focal necrosis of adnexal epithelium (Figure 2). Immunofluorescence was negative for specific deposition of IgG, IgA, IgM, C3, or fibrinogen. Wound culture also returned negative, and the Naranjo adverse drug reaction probability scale score was calculated to be 4 out of 12, indicating possible adverse drug reaction.4
Given the extent and distribution of the rash as well as the full-thickness dermal involvement, the patient was transferred to the burn unit for subsequent care. At 8-month follow-up, she experienced severe, symptomatic, hypertrophic scarring and was awaiting intralesional triamcinolone acetonide injections. The patient subsequently was lost to follow up.
The clinical picture of SDRIFE has remained obscure over the last 30 years, likely owing to its rarity and unclear pathogenesis. Diagnostic criteria for SDRIFE were first proposed by Häusermann et al2 in 2004 and contained 5 elements: (1) occurrence after (re)exposure to systemic drugs, (2) sharply demarcated erythema of the gluteal region or V-shaped erythema of the inguinal area, (3) involvement of at least 1 other intertriginous location, (4) symmetry of affected areas, and (5) absence of systemic symptoms and signs. Based on these clinical criteria, our patients fulfilled 3 of 5 elements, with deductions for symmetry of affected areas and involvement of other intertriginous locations. Histopathologic findings in SDRIFE predominantly are nonspecific with superficial perivascular mononuclear infiltrates; however, prior reports have confirmed the potential for vacuolar changes and hydropic degeneration in the basal cell layer with subepidermal bullae formation.5,6 Similarly, although the presence of bullae are somewhat atypical in SDRIFE, it has been described.3 Taken together, we speculate that these findings may support a diagnosis of SDRIFE with atypical presentation, though an alternative diagnosis of bullous fixed drug eruption (FDE) cannot be ruled out.
Historically, SDRIFE has been associated with a benign course. The condition typically arises within a few hours to days following administration of the offending agent, most commonly amoxicillin or another β-lactam antibiotic.1 Most cases spontaneously resolve via desquamation within 1 to 2 weeks. We present an unusual case of amoxicillin-induced full-thickness epidermal necrosis resulting in symptomatic sequelae, which exhibits findings of SDRIFE, bullous FDE, or Stevens-Johnson syndrome/toxic epidermal necrolysis, suggesting the possibility for a common pathway underlying the pathogenesis of these conditions.
The diagnostic uncertainty that commonly accompanies these various toxic drug reactions may in part relate to their underlying immunopathogenesis. Although the exact mechanism by which SDRIFE results in its characteristic skin lesions has not been fully elucidated, prior work through patch testing, lymphocyte transformation assays, and immunohistochemical staining of biopsies suggests a type IV delayed hypersensitivity (DTH) reaction.7-10 Specifically, SDRIFE appears to share features of both DTH type IVa—involving CD4+ helper T cells (TH1), monocytes, and IFN-γ signaling—and DTH type IVc—involving cytotoxic CD4 and CD8 cells, granzyme B action, and FasL signaling.11,12 A similar inflammatory milieu has been implicated in numerous toxic drug eruptions, including Stevens-Johnson syndrome/toxic epidermal necrolysis and FDE.11,13 This mechanistic overlap may explain the overlap seen clinically among such conditions.
In the undifferentiated patient, categorization of the clinical syndrome proves helpful in prognostication and therapeutic approach. The complexities and commonalities intrinsic to these syndromes, however, may simultaneously preclude certain cases from neatly following the predefined rules. These atypical presentations, while diagnostically challenging, can in turn offer a unique opportunity to reexamine the current state of disease understanding to better allow for appropriate classification.
Despite its rarity, SDRIFE should be considered in the differential of undiagnosed drug eruptions, particularly as new clinical presentations emerge. Careful documentation and timely declaration of future cases will prove invaluable for diagnostic and therapeutic advancements should this once-benign condition develop a more destructive potential.
To the Editor:
Symmetric drug-related intertriginous and flexural exanthema (SDRIFE) is a curious disorder that has undergone many clinical transformations since first being described by Andersen et al1 in 1984 using the term baboon syndrome. Initially described as a mercury hypersensitivity reaction resulting in an eruption resembling the red-bottomed baboon, this exanthema has expanded in definition with inciting agents, clinical features, and diagnostic criteria. Its prognosis, however, has remained stable and favorable throughout the decades. The condition is almost universally benign and self-limited.1-3 As new cases are reported in the literature and the paradigm of SDRIFE continues to shift, its prognosis also may warrant reconsideration and respect as a potentially destructive reaction.
A 39-year-old woman who was otherwise healthy presented to the emergency department after developing a rapidly evolving and blistering rash on the left flank. Hours later, the rash had progressed to a sharply demarcated, confluent, erythematous plaque with central ulceration and large flaccid bullae peripherally, encompassing 18% of total body surface area and extending from the gluteal cleft to the tip of the scapula along the left flank (Figure 1) with no vaginal or mucosal involvement. The patient recently had completed a 10-day course of amoxicillin–clavulanic acid 2 days prior for a cat bite on the right dorsal wrist. Additional history confirmed the absence of prodromal fever, fatigue, or chills. Inciting trauma, including chemical and thermal burns, was denied. Potential underlying psychosocial cofounders were explored and were unrevealing.
Laboratory test results, including complete blood cell count and metabolic panel as well as vital signs were unremarkable, except for slight leukocytosis at 14,000/µL (reference range 4500–11,000/µL). A punch biopsy was taken from the patient’s left upper back at the time of admission, which revealed a sparse, superficial, perivascular infiltrate of lymphocytes and rare neutrophils with largely absent epidermis and an occasional focal necrosis of adnexal epithelium (Figure 2). Immunofluorescence was negative for specific deposition of IgG, IgA, IgM, C3, or fibrinogen. Wound culture also returned negative, and the Naranjo adverse drug reaction probability scale score was calculated to be 4 out of 12, indicating possible adverse drug reaction.4
Given the extent and distribution of the rash as well as the full-thickness dermal involvement, the patient was transferred to the burn unit for subsequent care. At 8-month follow-up, she experienced severe, symptomatic, hypertrophic scarring and was awaiting intralesional triamcinolone acetonide injections. The patient subsequently was lost to follow up.
The clinical picture of SDRIFE has remained obscure over the last 30 years, likely owing to its rarity and unclear pathogenesis. Diagnostic criteria for SDRIFE were first proposed by Häusermann et al2 in 2004 and contained 5 elements: (1) occurrence after (re)exposure to systemic drugs, (2) sharply demarcated erythema of the gluteal region or V-shaped erythema of the inguinal area, (3) involvement of at least 1 other intertriginous location, (4) symmetry of affected areas, and (5) absence of systemic symptoms and signs. Based on these clinical criteria, our patients fulfilled 3 of 5 elements, with deductions for symmetry of affected areas and involvement of other intertriginous locations. Histopathologic findings in SDRIFE predominantly are nonspecific with superficial perivascular mononuclear infiltrates; however, prior reports have confirmed the potential for vacuolar changes and hydropic degeneration in the basal cell layer with subepidermal bullae formation.5,6 Similarly, although the presence of bullae are somewhat atypical in SDRIFE, it has been described.3 Taken together, we speculate that these findings may support a diagnosis of SDRIFE with atypical presentation, though an alternative diagnosis of bullous fixed drug eruption (FDE) cannot be ruled out.
Historically, SDRIFE has been associated with a benign course. The condition typically arises within a few hours to days following administration of the offending agent, most commonly amoxicillin or another β-lactam antibiotic.1 Most cases spontaneously resolve via desquamation within 1 to 2 weeks. We present an unusual case of amoxicillin-induced full-thickness epidermal necrosis resulting in symptomatic sequelae, which exhibits findings of SDRIFE, bullous FDE, or Stevens-Johnson syndrome/toxic epidermal necrolysis, suggesting the possibility for a common pathway underlying the pathogenesis of these conditions.
The diagnostic uncertainty that commonly accompanies these various toxic drug reactions may in part relate to their underlying immunopathogenesis. Although the exact mechanism by which SDRIFE results in its characteristic skin lesions has not been fully elucidated, prior work through patch testing, lymphocyte transformation assays, and immunohistochemical staining of biopsies suggests a type IV delayed hypersensitivity (DTH) reaction.7-10 Specifically, SDRIFE appears to share features of both DTH type IVa—involving CD4+ helper T cells (TH1), monocytes, and IFN-γ signaling—and DTH type IVc—involving cytotoxic CD4 and CD8 cells, granzyme B action, and FasL signaling.11,12 A similar inflammatory milieu has been implicated in numerous toxic drug eruptions, including Stevens-Johnson syndrome/toxic epidermal necrolysis and FDE.11,13 This mechanistic overlap may explain the overlap seen clinically among such conditions.
In the undifferentiated patient, categorization of the clinical syndrome proves helpful in prognostication and therapeutic approach. The complexities and commonalities intrinsic to these syndromes, however, may simultaneously preclude certain cases from neatly following the predefined rules. These atypical presentations, while diagnostically challenging, can in turn offer a unique opportunity to reexamine the current state of disease understanding to better allow for appropriate classification.
Despite its rarity, SDRIFE should be considered in the differential of undiagnosed drug eruptions, particularly as new clinical presentations emerge. Careful documentation and timely declaration of future cases will prove invaluable for diagnostic and therapeutic advancements should this once-benign condition develop a more destructive potential.
- Andersen KE, Hjorth N, Menné T. The baboon syndrome: systemically-induced allergic contact dermatitis. Contact Dermatitis. 1984;10:97-100.
- Häusermann P, Harr TH, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis. 2004;51:297-310.
- Tan SC, Tan JW. Symmetrical drug-related intertriginous and flexural exanthema. Curr Opin Allergy Clin Immunol. 2011;11:313-318.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Wolf R, Orion E, Matz H. The baboon syndrome or intertriginous drug eruption: a report of eleven cases and a second look at its pathomechanism. Dermatol Online J. 2003;9:2.
- Elmariah SB, Cheung W, Wang N, et al. Systemic drug-related intertriginous and flexural exanthema (SDRIFE). Dermatol Online J. 2009;15:3.
- Hembold P, Hegemann B, Dickert C, et al. Symptomatic psychotropic and nonpigmenting fixed drug eruption due to cimetidine (so-called baboon syndrome). Dermatology. 1998;197:402-403.
- Barbaud A, Trechot P, Granel F, et al. A baboon syndrome induced by intravenous human immunoglobulins: a report of a case and immunological analysis. Dermatology. 1999;199:258-260.
- Miyahara A, Kawashima H, Okubo Y, et al. A new proposal for a clinical-oriented subclassification of baboon syndrome and review of baboon syndrome. Asian Pac J Allergy Immunol. 2011;29:150-160.
- Goossens C, Sass U, Song M. Baboon syndrome. Dermatology. 1997;194:421-422.
- Pichler WJ. Delayed drug hypersensitivity reactions. Ann Intern Med. 2003;139:123-129.
- Ozkaya E. Current understanding of baboon syndrome. Expert Rev Dermatol. 2009;4:163-175.
- Ozakaya E. Fixed drug eruption: state of the art. J Dtsch Dermatol Ges. 2008;6:181-188.
- Andersen KE, Hjorth N, Menné T. The baboon syndrome: systemically-induced allergic contact dermatitis. Contact Dermatitis. 1984;10:97-100.
- Häusermann P, Harr TH, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis. 2004;51:297-310.
- Tan SC, Tan JW. Symmetrical drug-related intertriginous and flexural exanthema. Curr Opin Allergy Clin Immunol. 2011;11:313-318.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Wolf R, Orion E, Matz H. The baboon syndrome or intertriginous drug eruption: a report of eleven cases and a second look at its pathomechanism. Dermatol Online J. 2003;9:2.
- Elmariah SB, Cheung W, Wang N, et al. Systemic drug-related intertriginous and flexural exanthema (SDRIFE). Dermatol Online J. 2009;15:3.
- Hembold P, Hegemann B, Dickert C, et al. Symptomatic psychotropic and nonpigmenting fixed drug eruption due to cimetidine (so-called baboon syndrome). Dermatology. 1998;197:402-403.
- Barbaud A, Trechot P, Granel F, et al. A baboon syndrome induced by intravenous human immunoglobulins: a report of a case and immunological analysis. Dermatology. 1999;199:258-260.
- Miyahara A, Kawashima H, Okubo Y, et al. A new proposal for a clinical-oriented subclassification of baboon syndrome and review of baboon syndrome. Asian Pac J Allergy Immunol. 2011;29:150-160.
- Goossens C, Sass U, Song M. Baboon syndrome. Dermatology. 1997;194:421-422.
- Pichler WJ. Delayed drug hypersensitivity reactions. Ann Intern Med. 2003;139:123-129.
- Ozkaya E. Current understanding of baboon syndrome. Expert Rev Dermatol. 2009;4:163-175.
- Ozakaya E. Fixed drug eruption: state of the art. J Dtsch Dermatol Ges. 2008;6:181-188.
Practice Points
- Symmetric drug-related intertriginous and flexural exanthema (SDRIFE) appears in the absence of systemic signs and symptoms such as fever, which may help differentiate it from infectious causes.
- β-Lactam antibiotics, particularly amoxicillin, are common offenders in the pathogenesis of SDRIFE, but new drug relationships frequently are being described.
- Symmetric drug-related intertriginous and flexural exanthema commonly follows a benign course but warrants respect, as it may have devastating potential.
AGA Giving Day: Our fight to eradicate disparities in digestive diseases
On Dec. 3, AGA brought together the GI community in an effort to fund health disparity research with the goal of improving care for the patients who rely on us.
The patients we serve face racial health disparities daily. It’s our responsibility to take action. With money raised through this campaign, the AGA Research Foundation will fund research projects that help understand health disparities and create strategies for overcoming them.
AGA Giving Day was the opportunity to do something about this important societal issue as it directly relates to our field. We all have a role to play in creating a just world free of health disparities in digestive diseases and free of inequities in access and effective health care delivery.
The AGA Research Foundation’s AGA Giving Day effort will help support state-of-the-art research that aligns with the realities of the current multicultural patient population and disease states to achieve health equity for all.
Learn more at gastro.org/agagivingday.
On Dec. 3, AGA brought together the GI community in an effort to fund health disparity research with the goal of improving care for the patients who rely on us.
The patients we serve face racial health disparities daily. It’s our responsibility to take action. With money raised through this campaign, the AGA Research Foundation will fund research projects that help understand health disparities and create strategies for overcoming them.
AGA Giving Day was the opportunity to do something about this important societal issue as it directly relates to our field. We all have a role to play in creating a just world free of health disparities in digestive diseases and free of inequities in access and effective health care delivery.
The AGA Research Foundation’s AGA Giving Day effort will help support state-of-the-art research that aligns with the realities of the current multicultural patient population and disease states to achieve health equity for all.
Learn more at gastro.org/agagivingday.
On Dec. 3, AGA brought together the GI community in an effort to fund health disparity research with the goal of improving care for the patients who rely on us.
The patients we serve face racial health disparities daily. It’s our responsibility to take action. With money raised through this campaign, the AGA Research Foundation will fund research projects that help understand health disparities and create strategies for overcoming them.
AGA Giving Day was the opportunity to do something about this important societal issue as it directly relates to our field. We all have a role to play in creating a just world free of health disparities in digestive diseases and free of inequities in access and effective health care delivery.
The AGA Research Foundation’s AGA Giving Day effort will help support state-of-the-art research that aligns with the realities of the current multicultural patient population and disease states to achieve health equity for all.
Learn more at gastro.org/agagivingday.
Challenges in the Management of Peptic Ulcer Disease
From the University of Alabama at Birmingham, Birmingham, AL.
Abstract
Objective: To review current challenges in the management of peptic ulcer disease.
Methods: Review of the literature.
Results: Peptic ulcer disease affects 5% to 10% of the population worldwide, with recent decreases in lifetime prevalence in high-income countries. Helicobacter pylori infection and nonsteroidal anti-inflammatory drug (NSAID) use are the most important drivers of peptic ulcer disease. Current management strategies for peptic ulcer disease focus on ulcer healing; management of complications such as bleeding, perforation, and obstruction; and prevention of ulcer recurrence. Proton pump inhibitors (PPIs) are the cornerstone of medical therapy for peptic ulcers, and complement testing for and treatment of H. pylori infection as well as elimination of NSAID use. Although advances have been made in the medical and endoscopic treatment of peptic ulcer disease and the management of ulcer complications, such as bleeding and obstruction, challenges remain.
Conclusion: Peptic ulcer disease is a common health problem globally, with persistent challenges related to refractory ulcers, antiplatelet and anticoagulant use, and continued bleeding in the face of endoscopic therapy. These challenges should be met with PPI therapy of adequate frequency and duration, vigilant attention to and treatment of ulcer etiology, evidence-based handling of antiplatelet and anticoagulant medications, and utilization of novel endoscopic tools to obtain improved clinical outcomes.
Keywords: H. pylori; nonsteroidal anti-inflammatory drugs; NSAIDs; proton pump inhibitor; PPI; bleeding; perforation; obstruction; refractory ulcer; salvage endoscopic therapy; transcatheter angiographic embolization.
A peptic ulcer is a fibrin-covered break in the mucosa of the digestive tract extending to the submucosa that is caused by acid injury (Figure 1). Most peptic ulcers occur in the stomach or proximal duodenum, though they may also occur in the esophagus or, less frequently, in a Meckel’s diverticulum.1,2 The estimated worldwide prevalence of peptic ulcer disease is 5% to 10%, with an annual incidence of 0.1% to 0.3%1; both rates are declining.3 The annual incidence of peptic ulcer disease requiring medical or surgical treatment is also declining, and currently is estimated to be 0.1% to 0.2%.4 The lifetime prevalence of peptic ulcers has been decreasing in high-income countries since the mid-20th century due to both the widespread use of medications that suppress gastric acid secretion and the declining prevalence of Helicobacter pylori infection.1,3

Peptic ulcer disease in most individuals results from H. pylori infection, chronic use of nonsteroidal anti-inflammatory drugs (NSAIDs), including aspirin, or both. A combination of H. pylori factors and host factors lead to mucosal disruption in infected individuals who develop peptic ulcers. H. pylori–specific factors include the expression of virulence factors such as CagA and VacA, which interact with the host inflammatory response to cause mucosal injury. The mucosal inflammatory response is at least partially determined by polymorphisms in the host’s cytokine genes.1,4 NSAIDs inhibit the production of cyclooxygenase-1-derived prostaglandins, with subsequent decreases in epithelial mucous formation, bicarbonate secretion, cell proliferation, and mucosal blood flow, all of which are key elements in the maintenance of mucosal integrity.1,5 Less common causes of peptic ulcers include gastrinoma, adenocarcinoma, idiopathic ulcers, use of sympathomimetic drugs (eg, cocaine or methamphetamine), certain anticancer agents, and bariatric surgery.4,6
This article provides an overview of current management principles for peptic ulcer disease and discusses current challenges in peptic ulcer management, including proton pump inhibitor (PPI) therapy, refractory ulcers, handling of antiplatelet and anticoagulants during and after peptic ulcer bleeding, and ulcer bleeding that continues despite salvage endoscopic therapy.
Methods
We searched MEDLINE using the term peptic ulcer disease in combination with the terms current challenges, epidemiology, bleeding, anticoagulant, antiplatelet, PPI potency, etiology, treatment, management, and refractory. We selected publications from the past 35 years that we judged to be relevant.
Current Management
The goals of peptic ulcer disease management are ulcer healing and prevention of recurrence. The primary interventions used in the management of peptic ulcer disease are medical therapy and implementation of measures that address the underlying etiology of the disease.
Medical Therapy
Introduced in the late 1980s, PPIs are the cornerstone of medical therapy for peptic ulcer disease.6 These agents irreversibly inhibit the H+/K+-ATPase pump in the gastric mucosa and thereby inhibit gastric acid secretion, promoting ulcer healing. PPIs improve rates of ulcer healing compared to H2-receptor antagonists.4,7
Underlying Causes
The underlying cause of peptic ulcer disease should be addressed, in addition to initiating medical therapy. A detailed history of NSAID use should be obtained, and patients with peptic ulcers caused by NSAIDs should be counseled to avoid them, if possible. Patients with peptic ulcer disease who require long-term use of NSAIDs should be placed on long-term PPI therapy.6 Any patient with peptic ulcer disease, regardless of any history of H. pylori infection or treatment, should be tested for infection. Tests that identify active infection, such as urea breath test, stool antigen assay, or mucosal biopsy–based testing, are preferred to IgG antibody testing, although the latter is acceptable in the context of peptic ulcer disease with a high pretest probability of infection.8 Any evidence of active infection warrants appropriate treatment to allow ulcer healing and prevent recurrence.1H. pylori infection is most often treated with clarithromycin triple therapy or bismuth quadruple therapy for 14 days, with regimens selected based on the presence or absence of penicillin allergy, prior antibiotic exposure, and local clarithromycin resistance rates, when known.4,8
Managing Complications
An additional aspect of care in peptic ulcer disease is managing the complications of bleeding, perforation, and gastric outlet obstruction. Acute upper gastrointestinal bleeding (GIB) is the most common complication of peptic ulcer disease, which accounts for 40% to 60% of nonvariceal acute upper GIB.1,6 The first step in the management of acute GIB from a peptic ulcer is fluid resuscitation to ensure hemodynamic stability. If there is associated anemia with a hemoglobin level < 8 g/dL, blood transfusion should be undertaken to target a hemoglobin level > 8 g/dL. In patients with peptic ulcer disease–related acute upper GIB and comorbid cardiovascular disease, the transfusion threshold is higher, with the specific cutoff depending on clinical status, type and severity of cardiovascular disease, and degree of bleeding. Endoscopic management should generally be undertaken within 24 hours of presentation and should not be delayed in patients taking anticoagulants.9 Combination endoscopic treatment with through-the-scope clips plus thermocoagulation or sclerosant injection is recommended for acutely bleeding peptic ulcers with high-risk stigmata.
Pharmacologic management of patients with bleeding peptic ulcers with high-risk stigmata includes PPI therapy, with an 80 mg intravenous (IV) loading dose followed by continuous infusion of 8 mg/hr for 72 hours to reduce rebleeding and mortality. Following completion of IV therapy, oral PPI therapy should be continued twice daily for 14 days, followed by once-daily dosing thereafter.9Patients with peptic ulcer perforation present with sudden-onset epigastric abdominal pain and have tenderness to palpation, guarding, and rigidity on examination, often along with tachycardia and hypotension.1,4 Computed tomography (CT) of the abdomen is 98% sensitive for identifying and localizing a perforation. Most perforations occur in the duodenum or antrum.
Management of a peptic ulcer perforation requires consultation with a surgeon to determine whether a nonoperative approach may be employed (eg, a stable patient with a contained perforation), or if surgery is indicated. The surgical approach to peptic ulcer perforation has been impacted by the clinical success of gastric acid suppression with PPIs and H. pylori eradication, but a range of surgical approaches are still used to repair perforations, from omental patch repair with peritoneal drain placement, to more extensive surgeries such as wedge resection or partial gastrectomy.4 Perforation carries a high mortality risk, up to 20% to 30%, and is the leading cause of death in patients with peptic ulcer disease.1,4
Gastric outlet obstruction, a rare complication of peptic ulcer disease, results from recurrent ulcer formation and scarring. Obstruction often presents with hypovolemia and metabolic alkalosis from prolonged vomiting. CT imaging with oral contrast is often the first diagnostic test employed to demonstrate obstruction. Upper endoscopy should be performed to evaluate the appearance and degree of obstruction as well as to obtain biopsies to evaluate for a malignant etiology of the ulcer disease. Endoscopic balloon dilation has become the cornerstone of initial therapy for obstruction from peptic ulcer disease, especially in the case of ulcers due to reversible causes. Surgery is now typically reserved for cases of refractory obstruction, after repeated endoscopic balloon dilation has failed to remove the obstruction. However, because nearly all patients with gastric outlet obstruction present with malnutrition, nutritional deficiencies should be addressed prior to the patient undergoing surgical intervention. Surgical options include pyloroplasty, antrectomy, and gastrojejunostomy.4
Current Challenges
Rapid Metabolism of PPIs
High-dose PPI therapy is a key component of therapy for peptic ulcer healing. PPIs are metabolized by the cytochrome P450 system, which is comprised of multiple isoenzymes. CYP2C19, an isoenzyme involved in PPI metabolism, has 21 polymorphisms, which have variable effects leading to ultra-rapid, extensive, intermediate, or poor metabolism of PPIs.10 With rapid metabolism of PPIs, standard dosing can result in inadequate suppression of acid secretion. Despite this knowledge, routine testing of CYP2C19 phenotype is not recommended due to the cost of testing. Instead, inadequate ulcer healing should prompt consideration of increased PPI dosing to 80 mg orally twice daily, which may be sufficient to overcome rapid PPI metabolism.11
Relative Potency of PPIs
In addition to variation in PPI metabolism, the relative potency of various PPIs has been questioned. A review of all available clinical studies of the effects of PPIs on mean 24-hour intragastric pH reported a quantitative difference in the potency of 5 PPIs, with omeprazole as the reference standard. Potencies ranged from 0.23 omeprazole equivalents for pantoprazole to 1.82 omeprazole equivalents for rabeprazole.12 An additional study of data from 56 randomized clinical trials confirmed that PPIs vary in potency, which was measured as time that gastric pH is less than 4. A linear increase in intragastric pH time less than 4 was observed from 9 to 64 mg omeprazole equivalents; higher doses yielded no additional benefit. An increase in PPI dosing from once daily to twice daily also increased the duration of intragastric pH time less than 4 from 15 to 21 hours.13 Earlier modeling of the relationship between duodenal ulcer healing and antisecretory therapy showed a strong correlation of ulcer healing with the duration of acid suppression, length of therapy, and the degree of acid suppression. Additional benefit was not observed after intragastric pH rose above 3.14 Thus, as the frequency and duration of acid suppression therapy are more important than PPI potency, PPIs can be used interchangeably.13,14
Addressing Underlying Causes
Continued NSAID Use. Refractory peptic ulcers are defined as those that do not heal despite adherence to 8 to 12 weeks of standard acid-suppression therapy. A cause of refractory peptic ulcer disease that must be considered is continued NSAID use.1,15 In a study of patients with refractory peptic ulcers, 27% of patients continued NSAID use, as determined by eventual disclosure by the patients or platelet cyclooxygenase activity assay, despite extensive counseling to avoid NSAIDs at the time of the diagnosis of their refractory ulcer and at subsequent visits.16 Pain may make NSAID cessation difficult for some patients, while others do not realize that over-the-counter preparations they take contain NSAIDs.15
Another group of patients with continued NSAID exposure are those who require long-term NSAID therapy for control of arthritis or the management of cardiovascular conditions. If NSAID therapy cannot be discontinued, the risk of NSAID-related gastrointestinal injury can be assessed based on the presence of multiple risk factors, including age > 65 years, high-dose NSAID therapy, a history of peptic ulcer, and concurrent use of aspirin, corticosteroids, or anticoagulants. Individuals with 3 or more of the preceding risk factors or a history of a peptic ulcer with a complication, especially if recent, are considered to be at high risk of developing an NSAID-related ulcer and possible subsequent complications.17 In these individuals, NSAID therapy should be continued with agents that have the lowest risk for gastrointestinal toxicity and at the lowest possible dose. A meta-analysis comparing nonselective NSAIDs to placebo demonstrated naproxen to have the highest risk of gastrointestinal complications, including GIB, perforation, and obstruction (adjusted rate ratio, 4.2), while diclofenac demonstrated the lowest risk (adjusted rate ratio, 1.89). High-dose NSAID therapy demonstrated a 2-fold increase in risk of peptic ulcer formation as compared to low-dose therapy.18
In addition to selecting the NSAID with the least gastrointestinal toxicity at the lowest possible dose, additional strategies to prevent peptic ulcer disease and its complications in chronic NSAID users include co-administration of a PPI and substitution of a COX-2 selective NSAID for nonselective NSAIDs.1,9 Prior double-blind, placebo-controlled, randomized, multicenter trials with patients requiring daily NSAIDs demonstrated an up to 15% absolute reduction in the risk of developing peptic ulcers over 6 months while taking esomeprazole.19
Persistent Infection. Persistent H. pylori infection, due either to initial false-negative testing or ongoing infection despite first-line therapy, is another cause of refractory peptic ulcer disease.1,15 Because antibiotics and PPIs can reduce the number of H. pylori bacteria, use of these medications concurrent with H. pylori testing can lead to false-negative results with several testing modalities. When suspicion for H. pylori is high, 2 or more diagnostic tests may be needed to effectively rule out infection.15
When H. pylori is detected, successful eradication is becoming more difficult due to an increasing prevalence of antibiotic resistance, leading to persistent infection in many cases and maintained risk of peptic ulcer disease, despite appropriate first-line therapy.8 Options for salvage therapy for persistent H. pylori, as well as information on the role and best timing of susceptibility testing, are beyond the scope of this review, but are reviewed by Lanas and Chan1 and in the American College of Gastroenterology guideline on the treatment of H. pylori infection.8
Other Causes. In a meta-analysis of rigorously designed studies from North America, 20% of patients experienced ulcer recurrence at 6 months, despite successful H. pylori eradication and no NSAID use.20 In addition, as H. pylori prevalence is decreasing, idiopathic ulcers are increasingly being diagnosed, and such ulcers may be associated with high rates of GIB and mortality.1 In this subset of patients with non-H. pylori, non-NSAID ulcers, increased effort is required to further evaluate the differential diagnosis for rarer causes of upper GI tract ulcer disease (Table). Certain malignancies, including adenocarcinoma and lymphoma, can cause ulcer formation and should be considered in refractory cases. Repeat biopsy at follow-up endoscopy for persistent ulcers should always be obtained to further evaluate for malignancy.1,15 Infectious diseases other than H. pylori infection, such as tuberculosis, syphilis, cytomegalovirus, and herpes simplex virus, are also reported as etiologies of refractory ulcers, and require specific antimicrobial treatment over and above PPI monotherapy. Special attention in biopsy sampling and sample processing is often required when infectious etiologies are being considered, as specific histologic stains and cultures may be needed for identification.15

Systemic conditions, including sarcoidosis,21 Behçet disease,22 and polyarteritis nodosa,15,23 can also cause refractory ulcers. Approximately 15% of patients with Crohn disease have gastroduodenal involvement, which may include ulcers of variable sizes.1,15,24 The increased gastric acid production seen in Zollinger-Ellison syndrome commonly presents as refractory peptic ulcers in the duodenum beyond the bulb that do not heal with standard doses of PPIs.1,15 More rare causes of acid hypersecretion leading to refractory ulcers include idiopathic gastric acid hypersecretion and retained gastric antrum syndrome after partial gastrectomy with Billroth II anastomosis.15 Smoking is a known risk factor for impaired tissue healing throughout the body, and can contribute to impaired healing of peptic ulcers through decreased prostaglandin synthesis25 and reduced gastric mucosal blood flow.26 Smoking should always be addressed in patients with refractory peptic ulcers, and cessation should be strongly encouraged. Other less common causes of refractory upper GI tract ulcers include radiation therapy, crack cocaine use, and mesenteric ischemia.15
Managing Antiplatelet and Anticoagulant Medications
Use of antiplatelets and anticoagulants, alone or in combination, increases the risk of peptic ulcer bleeding. In patients who continue to take aspirin after a peptic ulcer bleed, recurrent bleeding occurs in up to 300 cases per 1000 person-years. The rate of GIB associated with aspirin use ranges from 1.1% to 2.5%, depending on the dose. Prior peptic ulcer disease, age greater than 70 years, and concurrent NSAID, steroid, anticoagulant, or dual antiplatelet therapy (DAPT) use increase the risk of bleeding while on aspirin. The rate of GIB while taking a thienopyridine alone is slightly less than that when taking aspirin, ranging from 0.5% to 1.6%. Studies to date have yielded mixed estimates of the effect of DAPT on the risk of GIB. Estimates of the risk of GIB with DAPT range from an odds ratio for serious GIB of 7.4 to an absolute risk increase of only 1.3% when compared to clopidogrel alone.27
Many patients are also on warfarin or a direct oral anticoagulant (DOAC). In a study from the United Kingdom, the adjusted rate ratio of GIB with warfarin alone was 1.94, and this increased to 6.48 when warfarin was used with aspirin.28 The use of warfarin and DAPT, often called triple therapy, further increases the risk of GIB, with a hazard ratio of 5.0 compared to DAPT alone, and 5.38 when compared to warfarin alone. DOACs are increasingly prescribed for the treatment and prevention of thromboembolism, and by 2014 were prescribed as often as warfarin for stroke prevention in atrial fibrillation in the United States. A meta-analysis showed the risk of major GIB did not differ between DOACs and warfarin or low-molecular-weight heparin, but among DOACs factor Xa inhibitors showed a reduced risk of GIB compared with dabigatran, a direct thrombin inhibitor.29
The use of antiplatelets and anticoagulants in the context of peptic ulcer bleeding is a current management challenge. Data to guide decision-making in patients on antiplatelet and/or anticoagulant therapy who experience peptic ulcer bleeding are scarce. Decision-making in this group of patients requires balancing the severity and risk of bleeding with the risk of thromboembolism.1,27 In patients on antiplatelet therapy for primary prophylaxis of atherothrombosis who develop bleeding from a peptic ulcer, the antiplatelet should generally be held and the indication for the medication reassessed. In patients on antiplatelet therapy for secondary prevention, the agent may be immediately resumed after endoscopy if bleeding is found to be due to an ulcer with low-risk stigmata. With bleeding resulting from an ulcer with high-risk stigmata, antiplatelet agents employed for secondary prevention may be held initially, with consideration given to early reintroduction, as early as day 3 after endoscopy.1 In patients at high risk for atherothrombotic events, including those on aspirin for secondary prophylaxis, withholding aspirin leads to a 3-fold increase in the risk of a major adverse cardiac event, with events occurring as early as 5 days after aspirin cessation in some cases.27 A randomized controlled trial of continuing low-dose aspirin versus withholding it for 8 weeks in patients on aspirin for secondary prophylaxis of cardiovascular events who experienced peptic ulcer bleeding that required endoscopic therapy demonstrated lower all-cause mortality (1.3% vs 12.9%), including death from cardiovascular or cerebrovascular events, among those who continued aspirin therapy, with a small increased risk of recurrent ulcer bleeding (10.3% vs 5.4%).30 Thus, it is recommended that antiplatelet therapy, when held, be resumed as early as possible when the risk of a cardiovascular or cerebrovascular event is considered to be higher than the risk of bleeding.27
When patients are on DAPT for a history of drug-eluting stent placement, withholding both antiplatelet medications should be avoided, even for a brief period of time, given the risk of in-stent thrombosis. When DAPT is employed for other reasons, it should be continued, if indicated, after bleeding that is found to be due to peptic ulcers with low-risk stigmata. If bleeding is due to a peptic ulcer with high-risk stigmata at endoscopy, then aspirin monotherapy should be continued and consultation should be obtained with a cardiologist to determine optimal timing to resume the second antiplatelet agent.1 In patients on anticoagulants, anticoagulation should be resumed once hemostasis is achieved when the risk of withholding anticoagulation is thought to be greater than the risk of rebleeding. For example, anticoagulation should be resumed early in a patient with a mechanical heart valve to prevent thrombosis.1,27 Following upper GIB from peptic ulcer disease, patients who will require long-term aspirin, DAPT, or anticoagulation with either warfarin or DOACs should be maintained on long-term PPI therapy to reduce the risk of recurrent bleeding.9,27
Failure of Endoscopic Therapy to Control Peptic Ulcer Bleeding
Bleeding recurs in as many as 10% to 20% of patients after initial endoscopic control of peptic ulcer bleeding.4,31 In this context, repeat upper endoscopy for hemostasis is preferred to surgery, as it leads to less morbidity while providing long-term control of bleeding in more than 70% of cases.31,32 Two potential endoscopic rescue therapies that may be employed are over-the-scope clips (OTSCs) and hemostatic powder.32,33
While through-the-scope (TTS) hemostatic clips are often used during endoscopy to control active peptic ulcer bleeding, their use may be limited in large or fibrotic ulcers due to the smaller size of the clips and method of application. OTSCs have several advantages over TTS clips; notably, their larger size allows the endoscopist to achieve deeper mucosal or submucosal clip attachment via suction of the targeted tissue into the endoscopic cap (Figure 2). In a systematic review of OTSCs, successful hemostasis was achieved in 84% of 761 lesions, including 75% of lesions due to peptic ulcer disease.34 Some have argued that OTSCs may be preferred as first-line therapy over epinephrine with TTS clips for hemostasis in bleeding from high-risk peptic ulcers (ie, those with visualized arterial bleeding or a visible vessel) given observed decreases in rebleeding events.35

Despite the advantages of OTSCs, endoscopists should be mindful of the potential complications of OTSC use, including luminal obstruction, particularly in the duodenum, and perforation, which occurs in 0.3% to 2% of cases. Additionally, retrieval of misplaced OTSCs presents a significant challenge. Careful decision-making with consideration of the location, size, and depth of lesions is required when deciding on OTSC placement.34,36
A newer endoscopic tool developed for refractory bleeding from peptic ulcers and other causes is hemostatic powder. Hemostatic powders accelerate the coagulation cascade, leading to shortened coagulation times and enhanced clot formation.37 A recent meta-analysis showed that immediate hemostasis could be achieved in 95% of cases of bleeding, including in 96% of cases of bleeding from peptic ulcer disease.38 The primary limitation of hemostatic powders is the temporary nature of hemostasis, which requires the underlying etiology of bleeding to be addressed in order to provide long-term hemostasis. In the above meta-analysis, rebleeding occurred in 17% of cases after 30 days.38
Hypotension and ulcer diameter ≥ 2 cm are independent predictors of failure of endoscopic salvage therapy.31 When severe bleeding is not controlled with initial endoscopic therapy or bleeding recurs despite salvage endoscopic therapy, transcatheter angiographic embolization (TAE) is the treatment of choice.4 Systematic reviews and meta-analyses of studies that compared TAE to surgery have shown that the rate of rebleeding may be higher with TAE, but with less morbidity and either decreased or equivalent rates of mortality, with no increased need for additional interventions.4,32 In a case series examining 5 years of experience at a single medical center in China, massive GIB from duodenal ulcers was successfully treated with TAE in 27 of 29 cases (93% clinical success rate), with no mucosal ischemic necrosis observed.39
If repeated endoscopic therapy has not led to hemostasis of a bleeding peptic ulcer and TAE is not available, then surgery is the next best option. Bleeding gastric ulcers may be excised, wedge resected, or oversewn after an anterior gastrostomy. Bleeding duodenal ulcers may require use of a Kocher maneuver and linear incision of the anterior duodenum followed by ligation of the gastroduodenal artery. Fortunately, such surgical management is rarely necessary given the availability of TAE at most centers.4
Conclusion
Peptic ulcer disease is a common health problem globally, with persistent challenges related to refractory ulcers, antiplatelet and anticoagulant use, and continued bleeding in the face of endoscopic therapy. These challenges should be met with adequate frequency and duration of PPI therapy, vigilant attention to and treatment of ulcer etiology, evidence-based handling of antiplatelet and anticoagulant medications, and utilization of novel endoscopic tools to obtain improved clinical outcomes.
Acknowledgment: We thank Dr. Nipun Reddy from our institution for providing the endoscopic images used in this article.
Corresponding author: Adam L. Edwards, MD, MS; [email protected].
Financial disclosures: None.
1. Lanas A, Chan FKL. Peptic ulcer disease. Lancet. 2017;390:613-624.
2. Malfertheiner P, Chan FK, McColl KE. Peptic ulcer disease. Lancet. 2009;374:1449-1461.
3. Roberts-Thomson IC. Rise and fall of peptic ulceration: A disease of civilization? J Gastroenterol Hepatol. 2018;33:1321-1326.
4. Kempenich JW, Sirinek KR. Acid peptic disease. Surg Clin North Am. 2018;98:933-944.
5. Cryer B, Feldman M. Effects of very low dose daily, long-term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury in healthy humans. Gastroenterology. 1999;117:17-25.
6. Kavitt RT, Lipowska AM, Anyane-Yeboa A, Gralnek IM. Diagnosis and treatment of peptic ulcer disease. Am J Med. 2019;132:447-456.
7. Walan A, Bader JP, Classen M, et al. Effect of omeprazole and ranitidine on ulcer healing and relapse rates in patients with benign gastric ulcer. New Engl J Med. 1989;320:69-75.
8. Chey WD, Leontiadis GI, Howden CW, Moss SF. ACG Clinical Guideline: Treatment of Helicobacter pylori Infection. Am J Gastroenterol. 2017;112:212-239.
9. Barkun AN, Almadi M, Kuipers EJ, et al. Management of nonvariceal upper gastrointestinal bleeding: Guideline recommendations from the International Consensus Group. Ann Intern Med. 2019;171:805-822.
10. Arevalo Galvis A, Trespalacios Rangel AA, Otero Regino W. Personalized therapy for Helicobacter pylori: CYP2C19 genotype effect on first-line triple therapy. Helicobacter. 2019;24:e12574.
11. Furuta T, Ohashi K, Kamata T, et al. Effect of genetic differences in omeprazole metabolism on cure rates for Helicobacter pylori infection and peptic ulcer. Ann Intern Med. 1998;129:1027-1030.
12. Kirchheiner J, Glatt S, Fuhr U, et al. Relative potency of proton-pump inhibitors-comparison of effects on intragastric pH. Eur J Clin Pharmacol. 2009;65:19-31.
13. Graham DY, Tansel A. Interchangeable use of proton pump inhibitors based on relative potency. Clin Gastroenterol Hepatol. 2018;16:800-808.e7.
14. Burget DW, Chiverton SG, Hunt RH. Is there an optimal degree of acid suppression for healing of duodenal ulcers? A model of the relationship between ulcer healing and acid suppression. Gastroenterology. 1990;99:345-351.
15. Kim HU. Diagnostic and treatment approaches for refractory peptic ulcers. Clin Endosc. 2015;48:285-290.
16. Lanas AI, Remacha B, Esteva F, Sainz R. Risk factors associated with refractory peptic ulcers. Gastroenterology. 1995;109:124-133.
17. Lanza FL, Chan FK, Quigley EM. Guidelines for prevention of NSAID-related ulcer complications. Am J Gastroenterol. 2009;104:728-738.
18. Richy F, Bruyere O, Ethgen O, et al. Time dependent risk of gastrointestinal complications induced by non-steroidal anti-inflammatory drug use: a consensus statement using a meta-analytic approach. Ann Rheum Dis. 2004;63:759-766.
19. Scheiman JM, Yeomans ND, Talley NJ, et al. Prevention of ulcers by esomeprazole in at-risk patients using non-selective NSAIDs and COX-2 inhibitors. Am J Gastroenterol. 2006;101:701-710.
20. Laine L, Hopkins RJ, Girardi LS. Has the impact of Helicobacter pylori therapy on ulcer recurrence in the United States been overstated? A meta-analysis of rigorously designed trials. Am J Gastroenterol. 1998;93:1409-1415.
21. Akiyama T, Endo H, Inamori M, et al. Symptomatic gastric sarcoidosis with multiple antral ulcers. Endoscopy. 2009;41 Suppl 2:E159.
22. Sonoda A, Ogawa R, Mizukami K, et al. Marked improvement in gastric involvement in Behcet’s disease with adalimumab treatment. Turk J Gastroenterol. 2017;28:405-407.
23. Saikia N, Talukdar R, Mazumder S, et al. Polyarteritis nodosa presenting as massive upper gastrointestinal hemorrhage. Gastrointest Endosc. 2006;63:868-870.
24. Annunziata ML, Caviglia R, Papparella LG, Cicala M. Upper gastrointestinal involvement of Crohn’s disease: a prospective study on the role of upper endoscopy in the diagnostic work-up. Dig Dis Sci. 2012;57:1618-1623.
25. Quimby GF, Bonnice CA, Burstein SH, Eastwood GL. Active smoking depresses prostaglandin synthesis in human gastric mucosa. Ann Intern Med. 1986;104:616-619.
26. Iwao T, Toyonaga A, Ikegami M, et al. Gastric mucosal blood flow after smoking in healthy human beings assessed by laser Doppler flowmetry. Gastrointest Endosc. 1993;39:400-403.
27. Almadi MA, Barkun A, Brophy J. Antiplatelet and anticoagulant therapy in patients with gastrointestinal bleeding: an 86-year-old woman with peptic ulcer disease. JAMA. 2011;306:2367-2374.
28. Delaney JA, Opatrny L, Brophy JM, Suissa S. Drug drug interactions between antithrombotic medications and the risk of gastrointestinal bleeding. CMAJ. 2007;177:347-351.
29. Burr N, Lummis K, Sood R, et al. Risk of gastrointestinal bleeding with direct oral anticoagulants: a systematic review and network meta-analysis. Lancet Gastroenterol Hepatol. 2017;2:85-93.
30. Sung JJ, Lau JY, Ching JY, et al. Continuation of low-dose aspirin therapy in peptic ulcer bleeding: a randomized trial. Ann Intern Med. 2010;152:1-9.
31. Lau JY, Sung JJ, Lam YH, et al. Endoscopic retreatment compared with surgery in patients with recurrent bleeding after initial endoscopic control of bleeding ulcers. N Engl J Med. 1999;340:751-756.
32. Gralnek IM, Dumonceau JM, Kuipers EJ, et al. Diagnosis and management of nonvariceal upper gastrointestinal hemorrhage: European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy. 2015;47:a1-46.
33. Skinner M, Gutierrez JP, Neumann H, et al. Over-the-scope clip placement is effective rescue therapy for severe acute upper gastrointestinal bleeding. Endosc Int Open. 2014;2:E37-40.
34. Zhong C, Tan S, Ren Y, et al. Clinical outcomes of over-the-scope-clip system for the treatment of acute upper non-variceal gastrointestinal bleeding: a systematic review and meta-analysis. BMC Gastroenterol. 2019;19:225.
35. Mangiafico S, Pigo F, Bertani H, et al. Over-the-scope clip vs epinephrine with clip for first-line hemostasis in non-variceal upper gastrointestinal bleeding: a propensity score match analysis. Endosc Int Open. 2020;8:E50-e8.
36. Wedi E, Gonzalez S, Menke D, et al. One hundred and one over-the-scope-clip applications for severe gastrointestinal bleeding, leaks and fistulas. World J Gastroenterol. 2016;22:1844-1853.
37. Holster IL, van Beusekom HM, Kuipers EJ, et al. Effects of a hemostatic powder hemospray on coagulation and clot formation. Endoscopy. 2015;47:638-645.
38. Facciorusso A, Straus Takahashi M, et al. Efficacy of hemostatic powders in upper gastrointestinal bleeding: A systematic review and meta-analysis. Dig Liver Dis. 2019;51:1633-1640.
39. Wang YL, Cheng YS, et al. Emergency transcatheter arterial embolization for patients with acute massive duodenal ulcer hemorrhage. World J Gastroenterol. 2012;18:4765-4770.
From the University of Alabama at Birmingham, Birmingham, AL.
Abstract
Objective: To review current challenges in the management of peptic ulcer disease.
Methods: Review of the literature.
Results: Peptic ulcer disease affects 5% to 10% of the population worldwide, with recent decreases in lifetime prevalence in high-income countries. Helicobacter pylori infection and nonsteroidal anti-inflammatory drug (NSAID) use are the most important drivers of peptic ulcer disease. Current management strategies for peptic ulcer disease focus on ulcer healing; management of complications such as bleeding, perforation, and obstruction; and prevention of ulcer recurrence. Proton pump inhibitors (PPIs) are the cornerstone of medical therapy for peptic ulcers, and complement testing for and treatment of H. pylori infection as well as elimination of NSAID use. Although advances have been made in the medical and endoscopic treatment of peptic ulcer disease and the management of ulcer complications, such as bleeding and obstruction, challenges remain.
Conclusion: Peptic ulcer disease is a common health problem globally, with persistent challenges related to refractory ulcers, antiplatelet and anticoagulant use, and continued bleeding in the face of endoscopic therapy. These challenges should be met with PPI therapy of adequate frequency and duration, vigilant attention to and treatment of ulcer etiology, evidence-based handling of antiplatelet and anticoagulant medications, and utilization of novel endoscopic tools to obtain improved clinical outcomes.
Keywords: H. pylori; nonsteroidal anti-inflammatory drugs; NSAIDs; proton pump inhibitor; PPI; bleeding; perforation; obstruction; refractory ulcer; salvage endoscopic therapy; transcatheter angiographic embolization.
A peptic ulcer is a fibrin-covered break in the mucosa of the digestive tract extending to the submucosa that is caused by acid injury (Figure 1). Most peptic ulcers occur in the stomach or proximal duodenum, though they may also occur in the esophagus or, less frequently, in a Meckel’s diverticulum.1,2 The estimated worldwide prevalence of peptic ulcer disease is 5% to 10%, with an annual incidence of 0.1% to 0.3%1; both rates are declining.3 The annual incidence of peptic ulcer disease requiring medical or surgical treatment is also declining, and currently is estimated to be 0.1% to 0.2%.4 The lifetime prevalence of peptic ulcers has been decreasing in high-income countries since the mid-20th century due to both the widespread use of medications that suppress gastric acid secretion and the declining prevalence of Helicobacter pylori infection.1,3
Peptic ulcer disease in most individuals results from H. pylori infection, chronic use of nonsteroidal anti-inflammatory drugs (NSAIDs), including aspirin, or both. A combination of H. pylori factors and host factors lead to mucosal disruption in infected individuals who develop peptic ulcers. H. pylori–specific factors include the expression of virulence factors such as CagA and VacA, which interact with the host inflammatory response to cause mucosal injury. The mucosal inflammatory response is at least partially determined by polymorphisms in the host’s cytokine genes.1,4 NSAIDs inhibit the production of cyclooxygenase-1-derived prostaglandins, with subsequent decreases in epithelial mucous formation, bicarbonate secretion, cell proliferation, and mucosal blood flow, all of which are key elements in the maintenance of mucosal integrity.1,5 Less common causes of peptic ulcers include gastrinoma, adenocarcinoma, idiopathic ulcers, use of sympathomimetic drugs (eg, cocaine or methamphetamine), certain anticancer agents, and bariatric surgery.4,6
This article provides an overview of current management principles for peptic ulcer disease and discusses current challenges in peptic ulcer management, including proton pump inhibitor (PPI) therapy, refractory ulcers, handling of antiplatelet and anticoagulants during and after peptic ulcer bleeding, and ulcer bleeding that continues despite salvage endoscopic therapy.
Methods
We searched MEDLINE using the term peptic ulcer disease in combination with the terms current challenges, epidemiology, bleeding, anticoagulant, antiplatelet, PPI potency, etiology, treatment, management, and refractory. We selected publications from the past 35 years that we judged to be relevant.
Current Management
The goals of peptic ulcer disease management are ulcer healing and prevention of recurrence. The primary interventions used in the management of peptic ulcer disease are medical therapy and implementation of measures that address the underlying etiology of the disease.
Medical Therapy
Introduced in the late 1980s, PPIs are the cornerstone of medical therapy for peptic ulcer disease.6 These agents irreversibly inhibit the H+/K+-ATPase pump in the gastric mucosa and thereby inhibit gastric acid secretion, promoting ulcer healing. PPIs improve rates of ulcer healing compared to H2-receptor antagonists.4,7
Underlying Causes
The underlying cause of peptic ulcer disease should be addressed, in addition to initiating medical therapy. A detailed history of NSAID use should be obtained, and patients with peptic ulcers caused by NSAIDs should be counseled to avoid them, if possible. Patients with peptic ulcer disease who require long-term use of NSAIDs should be placed on long-term PPI therapy.6 Any patient with peptic ulcer disease, regardless of any history of H. pylori infection or treatment, should be tested for infection. Tests that identify active infection, such as urea breath test, stool antigen assay, or mucosal biopsy–based testing, are preferred to IgG antibody testing, although the latter is acceptable in the context of peptic ulcer disease with a high pretest probability of infection.8 Any evidence of active infection warrants appropriate treatment to allow ulcer healing and prevent recurrence.1H. pylori infection is most often treated with clarithromycin triple therapy or bismuth quadruple therapy for 14 days, with regimens selected based on the presence or absence of penicillin allergy, prior antibiotic exposure, and local clarithromycin resistance rates, when known.4,8
Managing Complications
An additional aspect of care in peptic ulcer disease is managing the complications of bleeding, perforation, and gastric outlet obstruction. Acute upper gastrointestinal bleeding (GIB) is the most common complication of peptic ulcer disease, which accounts for 40% to 60% of nonvariceal acute upper GIB.1,6 The first step in the management of acute GIB from a peptic ulcer is fluid resuscitation to ensure hemodynamic stability. If there is associated anemia with a hemoglobin level < 8 g/dL, blood transfusion should be undertaken to target a hemoglobin level > 8 g/dL. In patients with peptic ulcer disease–related acute upper GIB and comorbid cardiovascular disease, the transfusion threshold is higher, with the specific cutoff depending on clinical status, type and severity of cardiovascular disease, and degree of bleeding. Endoscopic management should generally be undertaken within 24 hours of presentation and should not be delayed in patients taking anticoagulants.9 Combination endoscopic treatment with through-the-scope clips plus thermocoagulation or sclerosant injection is recommended for acutely bleeding peptic ulcers with high-risk stigmata.
Pharmacologic management of patients with bleeding peptic ulcers with high-risk stigmata includes PPI therapy, with an 80 mg intravenous (IV) loading dose followed by continuous infusion of 8 mg/hr for 72 hours to reduce rebleeding and mortality. Following completion of IV therapy, oral PPI therapy should be continued twice daily for 14 days, followed by once-daily dosing thereafter.9Patients with peptic ulcer perforation present with sudden-onset epigastric abdominal pain and have tenderness to palpation, guarding, and rigidity on examination, often along with tachycardia and hypotension.1,4 Computed tomography (CT) of the abdomen is 98% sensitive for identifying and localizing a perforation. Most perforations occur in the duodenum or antrum.
Management of a peptic ulcer perforation requires consultation with a surgeon to determine whether a nonoperative approach may be employed (eg, a stable patient with a contained perforation), or if surgery is indicated. The surgical approach to peptic ulcer perforation has been impacted by the clinical success of gastric acid suppression with PPIs and H. pylori eradication, but a range of surgical approaches are still used to repair perforations, from omental patch repair with peritoneal drain placement, to more extensive surgeries such as wedge resection or partial gastrectomy.4 Perforation carries a high mortality risk, up to 20% to 30%, and is the leading cause of death in patients with peptic ulcer disease.1,4
Gastric outlet obstruction, a rare complication of peptic ulcer disease, results from recurrent ulcer formation and scarring. Obstruction often presents with hypovolemia and metabolic alkalosis from prolonged vomiting. CT imaging with oral contrast is often the first diagnostic test employed to demonstrate obstruction. Upper endoscopy should be performed to evaluate the appearance and degree of obstruction as well as to obtain biopsies to evaluate for a malignant etiology of the ulcer disease. Endoscopic balloon dilation has become the cornerstone of initial therapy for obstruction from peptic ulcer disease, especially in the case of ulcers due to reversible causes. Surgery is now typically reserved for cases of refractory obstruction, after repeated endoscopic balloon dilation has failed to remove the obstruction. However, because nearly all patients with gastric outlet obstruction present with malnutrition, nutritional deficiencies should be addressed prior to the patient undergoing surgical intervention. Surgical options include pyloroplasty, antrectomy, and gastrojejunostomy.4
Current Challenges
Rapid Metabolism of PPIs
High-dose PPI therapy is a key component of therapy for peptic ulcer healing. PPIs are metabolized by the cytochrome P450 system, which is comprised of multiple isoenzymes. CYP2C19, an isoenzyme involved in PPI metabolism, has 21 polymorphisms, which have variable effects leading to ultra-rapid, extensive, intermediate, or poor metabolism of PPIs.10 With rapid metabolism of PPIs, standard dosing can result in inadequate suppression of acid secretion. Despite this knowledge, routine testing of CYP2C19 phenotype is not recommended due to the cost of testing. Instead, inadequate ulcer healing should prompt consideration of increased PPI dosing to 80 mg orally twice daily, which may be sufficient to overcome rapid PPI metabolism.11
Relative Potency of PPIs
In addition to variation in PPI metabolism, the relative potency of various PPIs has been questioned. A review of all available clinical studies of the effects of PPIs on mean 24-hour intragastric pH reported a quantitative difference in the potency of 5 PPIs, with omeprazole as the reference standard. Potencies ranged from 0.23 omeprazole equivalents for pantoprazole to 1.82 omeprazole equivalents for rabeprazole.12 An additional study of data from 56 randomized clinical trials confirmed that PPIs vary in potency, which was measured as time that gastric pH is less than 4. A linear increase in intragastric pH time less than 4 was observed from 9 to 64 mg omeprazole equivalents; higher doses yielded no additional benefit. An increase in PPI dosing from once daily to twice daily also increased the duration of intragastric pH time less than 4 from 15 to 21 hours.13 Earlier modeling of the relationship between duodenal ulcer healing and antisecretory therapy showed a strong correlation of ulcer healing with the duration of acid suppression, length of therapy, and the degree of acid suppression. Additional benefit was not observed after intragastric pH rose above 3.14 Thus, as the frequency and duration of acid suppression therapy are more important than PPI potency, PPIs can be used interchangeably.13,14
Addressing Underlying Causes
Continued NSAID Use. Refractory peptic ulcers are defined as those that do not heal despite adherence to 8 to 12 weeks of standard acid-suppression therapy. A cause of refractory peptic ulcer disease that must be considered is continued NSAID use.1,15 In a study of patients with refractory peptic ulcers, 27% of patients continued NSAID use, as determined by eventual disclosure by the patients or platelet cyclooxygenase activity assay, despite extensive counseling to avoid NSAIDs at the time of the diagnosis of their refractory ulcer and at subsequent visits.16 Pain may make NSAID cessation difficult for some patients, while others do not realize that over-the-counter preparations they take contain NSAIDs.15
Another group of patients with continued NSAID exposure are those who require long-term NSAID therapy for control of arthritis or the management of cardiovascular conditions. If NSAID therapy cannot be discontinued, the risk of NSAID-related gastrointestinal injury can be assessed based on the presence of multiple risk factors, including age > 65 years, high-dose NSAID therapy, a history of peptic ulcer, and concurrent use of aspirin, corticosteroids, or anticoagulants. Individuals with 3 or more of the preceding risk factors or a history of a peptic ulcer with a complication, especially if recent, are considered to be at high risk of developing an NSAID-related ulcer and possible subsequent complications.17 In these individuals, NSAID therapy should be continued with agents that have the lowest risk for gastrointestinal toxicity and at the lowest possible dose. A meta-analysis comparing nonselective NSAIDs to placebo demonstrated naproxen to have the highest risk of gastrointestinal complications, including GIB, perforation, and obstruction (adjusted rate ratio, 4.2), while diclofenac demonstrated the lowest risk (adjusted rate ratio, 1.89). High-dose NSAID therapy demonstrated a 2-fold increase in risk of peptic ulcer formation as compared to low-dose therapy.18
In addition to selecting the NSAID with the least gastrointestinal toxicity at the lowest possible dose, additional strategies to prevent peptic ulcer disease and its complications in chronic NSAID users include co-administration of a PPI and substitution of a COX-2 selective NSAID for nonselective NSAIDs.1,9 Prior double-blind, placebo-controlled, randomized, multicenter trials with patients requiring daily NSAIDs demonstrated an up to 15% absolute reduction in the risk of developing peptic ulcers over 6 months while taking esomeprazole.19
Persistent Infection. Persistent H. pylori infection, due either to initial false-negative testing or ongoing infection despite first-line therapy, is another cause of refractory peptic ulcer disease.1,15 Because antibiotics and PPIs can reduce the number of H. pylori bacteria, use of these medications concurrent with H. pylori testing can lead to false-negative results with several testing modalities. When suspicion for H. pylori is high, 2 or more diagnostic tests may be needed to effectively rule out infection.15
When H. pylori is detected, successful eradication is becoming more difficult due to an increasing prevalence of antibiotic resistance, leading to persistent infection in many cases and maintained risk of peptic ulcer disease, despite appropriate first-line therapy.8 Options for salvage therapy for persistent H. pylori, as well as information on the role and best timing of susceptibility testing, are beyond the scope of this review, but are reviewed by Lanas and Chan1 and in the American College of Gastroenterology guideline on the treatment of H. pylori infection.8
Other Causes. In a meta-analysis of rigorously designed studies from North America, 20% of patients experienced ulcer recurrence at 6 months, despite successful H. pylori eradication and no NSAID use.20 In addition, as H. pylori prevalence is decreasing, idiopathic ulcers are increasingly being diagnosed, and such ulcers may be associated with high rates of GIB and mortality.1 In this subset of patients with non-H. pylori, non-NSAID ulcers, increased effort is required to further evaluate the differential diagnosis for rarer causes of upper GI tract ulcer disease (Table). Certain malignancies, including adenocarcinoma and lymphoma, can cause ulcer formation and should be considered in refractory cases. Repeat biopsy at follow-up endoscopy for persistent ulcers should always be obtained to further evaluate for malignancy.1,15 Infectious diseases other than H. pylori infection, such as tuberculosis, syphilis, cytomegalovirus, and herpes simplex virus, are also reported as etiologies of refractory ulcers, and require specific antimicrobial treatment over and above PPI monotherapy. Special attention in biopsy sampling and sample processing is often required when infectious etiologies are being considered, as specific histologic stains and cultures may be needed for identification.15
Systemic conditions, including sarcoidosis,21 Behçet disease,22 and polyarteritis nodosa,15,23 can also cause refractory ulcers. Approximately 15% of patients with Crohn disease have gastroduodenal involvement, which may include ulcers of variable sizes.1,15,24 The increased gastric acid production seen in Zollinger-Ellison syndrome commonly presents as refractory peptic ulcers in the duodenum beyond the bulb that do not heal with standard doses of PPIs.1,15 More rare causes of acid hypersecretion leading to refractory ulcers include idiopathic gastric acid hypersecretion and retained gastric antrum syndrome after partial gastrectomy with Billroth II anastomosis.15 Smoking is a known risk factor for impaired tissue healing throughout the body, and can contribute to impaired healing of peptic ulcers through decreased prostaglandin synthesis25 and reduced gastric mucosal blood flow.26 Smoking should always be addressed in patients with refractory peptic ulcers, and cessation should be strongly encouraged. Other less common causes of refractory upper GI tract ulcers include radiation therapy, crack cocaine use, and mesenteric ischemia.15
Managing Antiplatelet and Anticoagulant Medications
Use of antiplatelets and anticoagulants, alone or in combination, increases the risk of peptic ulcer bleeding. In patients who continue to take aspirin after a peptic ulcer bleed, recurrent bleeding occurs in up to 300 cases per 1000 person-years. The rate of GIB associated with aspirin use ranges from 1.1% to 2.5%, depending on the dose. Prior peptic ulcer disease, age greater than 70 years, and concurrent NSAID, steroid, anticoagulant, or dual antiplatelet therapy (DAPT) use increase the risk of bleeding while on aspirin. The rate of GIB while taking a thienopyridine alone is slightly less than that when taking aspirin, ranging from 0.5% to 1.6%. Studies to date have yielded mixed estimates of the effect of DAPT on the risk of GIB. Estimates of the risk of GIB with DAPT range from an odds ratio for serious GIB of 7.4 to an absolute risk increase of only 1.3% when compared to clopidogrel alone.27
Many patients are also on warfarin or a direct oral anticoagulant (DOAC). In a study from the United Kingdom, the adjusted rate ratio of GIB with warfarin alone was 1.94, and this increased to 6.48 when warfarin was used with aspirin.28 The use of warfarin and DAPT, often called triple therapy, further increases the risk of GIB, with a hazard ratio of 5.0 compared to DAPT alone, and 5.38 when compared to warfarin alone. DOACs are increasingly prescribed for the treatment and prevention of thromboembolism, and by 2014 were prescribed as often as warfarin for stroke prevention in atrial fibrillation in the United States. A meta-analysis showed the risk of major GIB did not differ between DOACs and warfarin or low-molecular-weight heparin, but among DOACs factor Xa inhibitors showed a reduced risk of GIB compared with dabigatran, a direct thrombin inhibitor.29
The use of antiplatelets and anticoagulants in the context of peptic ulcer bleeding is a current management challenge. Data to guide decision-making in patients on antiplatelet and/or anticoagulant therapy who experience peptic ulcer bleeding are scarce. Decision-making in this group of patients requires balancing the severity and risk of bleeding with the risk of thromboembolism.1,27 In patients on antiplatelet therapy for primary prophylaxis of atherothrombosis who develop bleeding from a peptic ulcer, the antiplatelet should generally be held and the indication for the medication reassessed. In patients on antiplatelet therapy for secondary prevention, the agent may be immediately resumed after endoscopy if bleeding is found to be due to an ulcer with low-risk stigmata. With bleeding resulting from an ulcer with high-risk stigmata, antiplatelet agents employed for secondary prevention may be held initially, with consideration given to early reintroduction, as early as day 3 after endoscopy.1 In patients at high risk for atherothrombotic events, including those on aspirin for secondary prophylaxis, withholding aspirin leads to a 3-fold increase in the risk of a major adverse cardiac event, with events occurring as early as 5 days after aspirin cessation in some cases.27 A randomized controlled trial of continuing low-dose aspirin versus withholding it for 8 weeks in patients on aspirin for secondary prophylaxis of cardiovascular events who experienced peptic ulcer bleeding that required endoscopic therapy demonstrated lower all-cause mortality (1.3% vs 12.9%), including death from cardiovascular or cerebrovascular events, among those who continued aspirin therapy, with a small increased risk of recurrent ulcer bleeding (10.3% vs 5.4%).30 Thus, it is recommended that antiplatelet therapy, when held, be resumed as early as possible when the risk of a cardiovascular or cerebrovascular event is considered to be higher than the risk of bleeding.27
When patients are on DAPT for a history of drug-eluting stent placement, withholding both antiplatelet medications should be avoided, even for a brief period of time, given the risk of in-stent thrombosis. When DAPT is employed for other reasons, it should be continued, if indicated, after bleeding that is found to be due to peptic ulcers with low-risk stigmata. If bleeding is due to a peptic ulcer with high-risk stigmata at endoscopy, then aspirin monotherapy should be continued and consultation should be obtained with a cardiologist to determine optimal timing to resume the second antiplatelet agent.1 In patients on anticoagulants, anticoagulation should be resumed once hemostasis is achieved when the risk of withholding anticoagulation is thought to be greater than the risk of rebleeding. For example, anticoagulation should be resumed early in a patient with a mechanical heart valve to prevent thrombosis.1,27 Following upper GIB from peptic ulcer disease, patients who will require long-term aspirin, DAPT, or anticoagulation with either warfarin or DOACs should be maintained on long-term PPI therapy to reduce the risk of recurrent bleeding.9,27
Failure of Endoscopic Therapy to Control Peptic Ulcer Bleeding
Bleeding recurs in as many as 10% to 20% of patients after initial endoscopic control of peptic ulcer bleeding.4,31 In this context, repeat upper endoscopy for hemostasis is preferred to surgery, as it leads to less morbidity while providing long-term control of bleeding in more than 70% of cases.31,32 Two potential endoscopic rescue therapies that may be employed are over-the-scope clips (OTSCs) and hemostatic powder.32,33
While through-the-scope (TTS) hemostatic clips are often used during endoscopy to control active peptic ulcer bleeding, their use may be limited in large or fibrotic ulcers due to the smaller size of the clips and method of application. OTSCs have several advantages over TTS clips; notably, their larger size allows the endoscopist to achieve deeper mucosal or submucosal clip attachment via suction of the targeted tissue into the endoscopic cap (Figure 2). In a systematic review of OTSCs, successful hemostasis was achieved in 84% of 761 lesions, including 75% of lesions due to peptic ulcer disease.34 Some have argued that OTSCs may be preferred as first-line therapy over epinephrine with TTS clips for hemostasis in bleeding from high-risk peptic ulcers (ie, those with visualized arterial bleeding or a visible vessel) given observed decreases in rebleeding events.35
Despite the advantages of OTSCs, endoscopists should be mindful of the potential complications of OTSC use, including luminal obstruction, particularly in the duodenum, and perforation, which occurs in 0.3% to 2% of cases. Additionally, retrieval of misplaced OTSCs presents a significant challenge. Careful decision-making with consideration of the location, size, and depth of lesions is required when deciding on OTSC placement.34,36
A newer endoscopic tool developed for refractory bleeding from peptic ulcers and other causes is hemostatic powder. Hemostatic powders accelerate the coagulation cascade, leading to shortened coagulation times and enhanced clot formation.37 A recent meta-analysis showed that immediate hemostasis could be achieved in 95% of cases of bleeding, including in 96% of cases of bleeding from peptic ulcer disease.38 The primary limitation of hemostatic powders is the temporary nature of hemostasis, which requires the underlying etiology of bleeding to be addressed in order to provide long-term hemostasis. In the above meta-analysis, rebleeding occurred in 17% of cases after 30 days.38
Hypotension and ulcer diameter ≥ 2 cm are independent predictors of failure of endoscopic salvage therapy.31 When severe bleeding is not controlled with initial endoscopic therapy or bleeding recurs despite salvage endoscopic therapy, transcatheter angiographic embolization (TAE) is the treatment of choice.4 Systematic reviews and meta-analyses of studies that compared TAE to surgery have shown that the rate of rebleeding may be higher with TAE, but with less morbidity and either decreased or equivalent rates of mortality, with no increased need for additional interventions.4,32 In a case series examining 5 years of experience at a single medical center in China, massive GIB from duodenal ulcers was successfully treated with TAE in 27 of 29 cases (93% clinical success rate), with no mucosal ischemic necrosis observed.39
If repeated endoscopic therapy has not led to hemostasis of a bleeding peptic ulcer and TAE is not available, then surgery is the next best option. Bleeding gastric ulcers may be excised, wedge resected, or oversewn after an anterior gastrostomy. Bleeding duodenal ulcers may require use of a Kocher maneuver and linear incision of the anterior duodenum followed by ligation of the gastroduodenal artery. Fortunately, such surgical management is rarely necessary given the availability of TAE at most centers.4
Conclusion
Peptic ulcer disease is a common health problem globally, with persistent challenges related to refractory ulcers, antiplatelet and anticoagulant use, and continued bleeding in the face of endoscopic therapy. These challenges should be met with adequate frequency and duration of PPI therapy, vigilant attention to and treatment of ulcer etiology, evidence-based handling of antiplatelet and anticoagulant medications, and utilization of novel endoscopic tools to obtain improved clinical outcomes.
Acknowledgment: We thank Dr. Nipun Reddy from our institution for providing the endoscopic images used in this article.
Corresponding author: Adam L. Edwards, MD, MS; [email protected].
Financial disclosures: None.
From the University of Alabama at Birmingham, Birmingham, AL.
Abstract
Objective: To review current challenges in the management of peptic ulcer disease.
Methods: Review of the literature.
Results: Peptic ulcer disease affects 5% to 10% of the population worldwide, with recent decreases in lifetime prevalence in high-income countries. Helicobacter pylori infection and nonsteroidal anti-inflammatory drug (NSAID) use are the most important drivers of peptic ulcer disease. Current management strategies for peptic ulcer disease focus on ulcer healing; management of complications such as bleeding, perforation, and obstruction; and prevention of ulcer recurrence. Proton pump inhibitors (PPIs) are the cornerstone of medical therapy for peptic ulcers, and complement testing for and treatment of H. pylori infection as well as elimination of NSAID use. Although advances have been made in the medical and endoscopic treatment of peptic ulcer disease and the management of ulcer complications, such as bleeding and obstruction, challenges remain.
Conclusion: Peptic ulcer disease is a common health problem globally, with persistent challenges related to refractory ulcers, antiplatelet and anticoagulant use, and continued bleeding in the face of endoscopic therapy. These challenges should be met with PPI therapy of adequate frequency and duration, vigilant attention to and treatment of ulcer etiology, evidence-based handling of antiplatelet and anticoagulant medications, and utilization of novel endoscopic tools to obtain improved clinical outcomes.
Keywords: H. pylori; nonsteroidal anti-inflammatory drugs; NSAIDs; proton pump inhibitor; PPI; bleeding; perforation; obstruction; refractory ulcer; salvage endoscopic therapy; transcatheter angiographic embolization.
A peptic ulcer is a fibrin-covered break in the mucosa of the digestive tract extending to the submucosa that is caused by acid injury (Figure 1). Most peptic ulcers occur in the stomach or proximal duodenum, though they may also occur in the esophagus or, less frequently, in a Meckel’s diverticulum.1,2 The estimated worldwide prevalence of peptic ulcer disease is 5% to 10%, with an annual incidence of 0.1% to 0.3%1; both rates are declining.3 The annual incidence of peptic ulcer disease requiring medical or surgical treatment is also declining, and currently is estimated to be 0.1% to 0.2%.4 The lifetime prevalence of peptic ulcers has been decreasing in high-income countries since the mid-20th century due to both the widespread use of medications that suppress gastric acid secretion and the declining prevalence of Helicobacter pylori infection.1,3
Peptic ulcer disease in most individuals results from H. pylori infection, chronic use of nonsteroidal anti-inflammatory drugs (NSAIDs), including aspirin, or both. A combination of H. pylori factors and host factors lead to mucosal disruption in infected individuals who develop peptic ulcers. H. pylori–specific factors include the expression of virulence factors such as CagA and VacA, which interact with the host inflammatory response to cause mucosal injury. The mucosal inflammatory response is at least partially determined by polymorphisms in the host’s cytokine genes.1,4 NSAIDs inhibit the production of cyclooxygenase-1-derived prostaglandins, with subsequent decreases in epithelial mucous formation, bicarbonate secretion, cell proliferation, and mucosal blood flow, all of which are key elements in the maintenance of mucosal integrity.1,5 Less common causes of peptic ulcers include gastrinoma, adenocarcinoma, idiopathic ulcers, use of sympathomimetic drugs (eg, cocaine or methamphetamine), certain anticancer agents, and bariatric surgery.4,6
This article provides an overview of current management principles for peptic ulcer disease and discusses current challenges in peptic ulcer management, including proton pump inhibitor (PPI) therapy, refractory ulcers, handling of antiplatelet and anticoagulants during and after peptic ulcer bleeding, and ulcer bleeding that continues despite salvage endoscopic therapy.
Methods
We searched MEDLINE using the term peptic ulcer disease in combination with the terms current challenges, epidemiology, bleeding, anticoagulant, antiplatelet, PPI potency, etiology, treatment, management, and refractory. We selected publications from the past 35 years that we judged to be relevant.
Current Management
The goals of peptic ulcer disease management are ulcer healing and prevention of recurrence. The primary interventions used in the management of peptic ulcer disease are medical therapy and implementation of measures that address the underlying etiology of the disease.
Medical Therapy
Introduced in the late 1980s, PPIs are the cornerstone of medical therapy for peptic ulcer disease.6 These agents irreversibly inhibit the H+/K+-ATPase pump in the gastric mucosa and thereby inhibit gastric acid secretion, promoting ulcer healing. PPIs improve rates of ulcer healing compared to H2-receptor antagonists.4,7
Underlying Causes
The underlying cause of peptic ulcer disease should be addressed, in addition to initiating medical therapy. A detailed history of NSAID use should be obtained, and patients with peptic ulcers caused by NSAIDs should be counseled to avoid them, if possible. Patients with peptic ulcer disease who require long-term use of NSAIDs should be placed on long-term PPI therapy.6 Any patient with peptic ulcer disease, regardless of any history of H. pylori infection or treatment, should be tested for infection. Tests that identify active infection, such as urea breath test, stool antigen assay, or mucosal biopsy–based testing, are preferred to IgG antibody testing, although the latter is acceptable in the context of peptic ulcer disease with a high pretest probability of infection.8 Any evidence of active infection warrants appropriate treatment to allow ulcer healing and prevent recurrence.1H. pylori infection is most often treated with clarithromycin triple therapy or bismuth quadruple therapy for 14 days, with regimens selected based on the presence or absence of penicillin allergy, prior antibiotic exposure, and local clarithromycin resistance rates, when known.4,8
Managing Complications
An additional aspect of care in peptic ulcer disease is managing the complications of bleeding, perforation, and gastric outlet obstruction. Acute upper gastrointestinal bleeding (GIB) is the most common complication of peptic ulcer disease, which accounts for 40% to 60% of nonvariceal acute upper GIB.1,6 The first step in the management of acute GIB from a peptic ulcer is fluid resuscitation to ensure hemodynamic stability. If there is associated anemia with a hemoglobin level < 8 g/dL, blood transfusion should be undertaken to target a hemoglobin level > 8 g/dL. In patients with peptic ulcer disease–related acute upper GIB and comorbid cardiovascular disease, the transfusion threshold is higher, with the specific cutoff depending on clinical status, type and severity of cardiovascular disease, and degree of bleeding. Endoscopic management should generally be undertaken within 24 hours of presentation and should not be delayed in patients taking anticoagulants.9 Combination endoscopic treatment with through-the-scope clips plus thermocoagulation or sclerosant injection is recommended for acutely bleeding peptic ulcers with high-risk stigmata.
Pharmacologic management of patients with bleeding peptic ulcers with high-risk stigmata includes PPI therapy, with an 80 mg intravenous (IV) loading dose followed by continuous infusion of 8 mg/hr for 72 hours to reduce rebleeding and mortality. Following completion of IV therapy, oral PPI therapy should be continued twice daily for 14 days, followed by once-daily dosing thereafter.9Patients with peptic ulcer perforation present with sudden-onset epigastric abdominal pain and have tenderness to palpation, guarding, and rigidity on examination, often along with tachycardia and hypotension.1,4 Computed tomography (CT) of the abdomen is 98% sensitive for identifying and localizing a perforation. Most perforations occur in the duodenum or antrum.
Management of a peptic ulcer perforation requires consultation with a surgeon to determine whether a nonoperative approach may be employed (eg, a stable patient with a contained perforation), or if surgery is indicated. The surgical approach to peptic ulcer perforation has been impacted by the clinical success of gastric acid suppression with PPIs and H. pylori eradication, but a range of surgical approaches are still used to repair perforations, from omental patch repair with peritoneal drain placement, to more extensive surgeries such as wedge resection or partial gastrectomy.4 Perforation carries a high mortality risk, up to 20% to 30%, and is the leading cause of death in patients with peptic ulcer disease.1,4
Gastric outlet obstruction, a rare complication of peptic ulcer disease, results from recurrent ulcer formation and scarring. Obstruction often presents with hypovolemia and metabolic alkalosis from prolonged vomiting. CT imaging with oral contrast is often the first diagnostic test employed to demonstrate obstruction. Upper endoscopy should be performed to evaluate the appearance and degree of obstruction as well as to obtain biopsies to evaluate for a malignant etiology of the ulcer disease. Endoscopic balloon dilation has become the cornerstone of initial therapy for obstruction from peptic ulcer disease, especially in the case of ulcers due to reversible causes. Surgery is now typically reserved for cases of refractory obstruction, after repeated endoscopic balloon dilation has failed to remove the obstruction. However, because nearly all patients with gastric outlet obstruction present with malnutrition, nutritional deficiencies should be addressed prior to the patient undergoing surgical intervention. Surgical options include pyloroplasty, antrectomy, and gastrojejunostomy.4
Current Challenges
Rapid Metabolism of PPIs
High-dose PPI therapy is a key component of therapy for peptic ulcer healing. PPIs are metabolized by the cytochrome P450 system, which is comprised of multiple isoenzymes. CYP2C19, an isoenzyme involved in PPI metabolism, has 21 polymorphisms, which have variable effects leading to ultra-rapid, extensive, intermediate, or poor metabolism of PPIs.10 With rapid metabolism of PPIs, standard dosing can result in inadequate suppression of acid secretion. Despite this knowledge, routine testing of CYP2C19 phenotype is not recommended due to the cost of testing. Instead, inadequate ulcer healing should prompt consideration of increased PPI dosing to 80 mg orally twice daily, which may be sufficient to overcome rapid PPI metabolism.11
Relative Potency of PPIs
In addition to variation in PPI metabolism, the relative potency of various PPIs has been questioned. A review of all available clinical studies of the effects of PPIs on mean 24-hour intragastric pH reported a quantitative difference in the potency of 5 PPIs, with omeprazole as the reference standard. Potencies ranged from 0.23 omeprazole equivalents for pantoprazole to 1.82 omeprazole equivalents for rabeprazole.12 An additional study of data from 56 randomized clinical trials confirmed that PPIs vary in potency, which was measured as time that gastric pH is less than 4. A linear increase in intragastric pH time less than 4 was observed from 9 to 64 mg omeprazole equivalents; higher doses yielded no additional benefit. An increase in PPI dosing from once daily to twice daily also increased the duration of intragastric pH time less than 4 from 15 to 21 hours.13 Earlier modeling of the relationship between duodenal ulcer healing and antisecretory therapy showed a strong correlation of ulcer healing with the duration of acid suppression, length of therapy, and the degree of acid suppression. Additional benefit was not observed after intragastric pH rose above 3.14 Thus, as the frequency and duration of acid suppression therapy are more important than PPI potency, PPIs can be used interchangeably.13,14
Addressing Underlying Causes
Continued NSAID Use. Refractory peptic ulcers are defined as those that do not heal despite adherence to 8 to 12 weeks of standard acid-suppression therapy. A cause of refractory peptic ulcer disease that must be considered is continued NSAID use.1,15 In a study of patients with refractory peptic ulcers, 27% of patients continued NSAID use, as determined by eventual disclosure by the patients or platelet cyclooxygenase activity assay, despite extensive counseling to avoid NSAIDs at the time of the diagnosis of their refractory ulcer and at subsequent visits.16 Pain may make NSAID cessation difficult for some patients, while others do not realize that over-the-counter preparations they take contain NSAIDs.15
Another group of patients with continued NSAID exposure are those who require long-term NSAID therapy for control of arthritis or the management of cardiovascular conditions. If NSAID therapy cannot be discontinued, the risk of NSAID-related gastrointestinal injury can be assessed based on the presence of multiple risk factors, including age > 65 years, high-dose NSAID therapy, a history of peptic ulcer, and concurrent use of aspirin, corticosteroids, or anticoagulants. Individuals with 3 or more of the preceding risk factors or a history of a peptic ulcer with a complication, especially if recent, are considered to be at high risk of developing an NSAID-related ulcer and possible subsequent complications.17 In these individuals, NSAID therapy should be continued with agents that have the lowest risk for gastrointestinal toxicity and at the lowest possible dose. A meta-analysis comparing nonselective NSAIDs to placebo demonstrated naproxen to have the highest risk of gastrointestinal complications, including GIB, perforation, and obstruction (adjusted rate ratio, 4.2), while diclofenac demonstrated the lowest risk (adjusted rate ratio, 1.89). High-dose NSAID therapy demonstrated a 2-fold increase in risk of peptic ulcer formation as compared to low-dose therapy.18
In addition to selecting the NSAID with the least gastrointestinal toxicity at the lowest possible dose, additional strategies to prevent peptic ulcer disease and its complications in chronic NSAID users include co-administration of a PPI and substitution of a COX-2 selective NSAID for nonselective NSAIDs.1,9 Prior double-blind, placebo-controlled, randomized, multicenter trials with patients requiring daily NSAIDs demonstrated an up to 15% absolute reduction in the risk of developing peptic ulcers over 6 months while taking esomeprazole.19
Persistent Infection. Persistent H. pylori infection, due either to initial false-negative testing or ongoing infection despite first-line therapy, is another cause of refractory peptic ulcer disease.1,15 Because antibiotics and PPIs can reduce the number of H. pylori bacteria, use of these medications concurrent with H. pylori testing can lead to false-negative results with several testing modalities. When suspicion for H. pylori is high, 2 or more diagnostic tests may be needed to effectively rule out infection.15
When H. pylori is detected, successful eradication is becoming more difficult due to an increasing prevalence of antibiotic resistance, leading to persistent infection in many cases and maintained risk of peptic ulcer disease, despite appropriate first-line therapy.8 Options for salvage therapy for persistent H. pylori, as well as information on the role and best timing of susceptibility testing, are beyond the scope of this review, but are reviewed by Lanas and Chan1 and in the American College of Gastroenterology guideline on the treatment of H. pylori infection.8
Other Causes. In a meta-analysis of rigorously designed studies from North America, 20% of patients experienced ulcer recurrence at 6 months, despite successful H. pylori eradication and no NSAID use.20 In addition, as H. pylori prevalence is decreasing, idiopathic ulcers are increasingly being diagnosed, and such ulcers may be associated with high rates of GIB and mortality.1 In this subset of patients with non-H. pylori, non-NSAID ulcers, increased effort is required to further evaluate the differential diagnosis for rarer causes of upper GI tract ulcer disease (Table). Certain malignancies, including adenocarcinoma and lymphoma, can cause ulcer formation and should be considered in refractory cases. Repeat biopsy at follow-up endoscopy for persistent ulcers should always be obtained to further evaluate for malignancy.1,15 Infectious diseases other than H. pylori infection, such as tuberculosis, syphilis, cytomegalovirus, and herpes simplex virus, are also reported as etiologies of refractory ulcers, and require specific antimicrobial treatment over and above PPI monotherapy. Special attention in biopsy sampling and sample processing is often required when infectious etiologies are being considered, as specific histologic stains and cultures may be needed for identification.15
Systemic conditions, including sarcoidosis,21 Behçet disease,22 and polyarteritis nodosa,15,23 can also cause refractory ulcers. Approximately 15% of patients with Crohn disease have gastroduodenal involvement, which may include ulcers of variable sizes.1,15,24 The increased gastric acid production seen in Zollinger-Ellison syndrome commonly presents as refractory peptic ulcers in the duodenum beyond the bulb that do not heal with standard doses of PPIs.1,15 More rare causes of acid hypersecretion leading to refractory ulcers include idiopathic gastric acid hypersecretion and retained gastric antrum syndrome after partial gastrectomy with Billroth II anastomosis.15 Smoking is a known risk factor for impaired tissue healing throughout the body, and can contribute to impaired healing of peptic ulcers through decreased prostaglandin synthesis25 and reduced gastric mucosal blood flow.26 Smoking should always be addressed in patients with refractory peptic ulcers, and cessation should be strongly encouraged. Other less common causes of refractory upper GI tract ulcers include radiation therapy, crack cocaine use, and mesenteric ischemia.15
Managing Antiplatelet and Anticoagulant Medications
Use of antiplatelets and anticoagulants, alone or in combination, increases the risk of peptic ulcer bleeding. In patients who continue to take aspirin after a peptic ulcer bleed, recurrent bleeding occurs in up to 300 cases per 1000 person-years. The rate of GIB associated with aspirin use ranges from 1.1% to 2.5%, depending on the dose. Prior peptic ulcer disease, age greater than 70 years, and concurrent NSAID, steroid, anticoagulant, or dual antiplatelet therapy (DAPT) use increase the risk of bleeding while on aspirin. The rate of GIB while taking a thienopyridine alone is slightly less than that when taking aspirin, ranging from 0.5% to 1.6%. Studies to date have yielded mixed estimates of the effect of DAPT on the risk of GIB. Estimates of the risk of GIB with DAPT range from an odds ratio for serious GIB of 7.4 to an absolute risk increase of only 1.3% when compared to clopidogrel alone.27
Many patients are also on warfarin or a direct oral anticoagulant (DOAC). In a study from the United Kingdom, the adjusted rate ratio of GIB with warfarin alone was 1.94, and this increased to 6.48 when warfarin was used with aspirin.28 The use of warfarin and DAPT, often called triple therapy, further increases the risk of GIB, with a hazard ratio of 5.0 compared to DAPT alone, and 5.38 when compared to warfarin alone. DOACs are increasingly prescribed for the treatment and prevention of thromboembolism, and by 2014 were prescribed as often as warfarin for stroke prevention in atrial fibrillation in the United States. A meta-analysis showed the risk of major GIB did not differ between DOACs and warfarin or low-molecular-weight heparin, but among DOACs factor Xa inhibitors showed a reduced risk of GIB compared with dabigatran, a direct thrombin inhibitor.29
The use of antiplatelets and anticoagulants in the context of peptic ulcer bleeding is a current management challenge. Data to guide decision-making in patients on antiplatelet and/or anticoagulant therapy who experience peptic ulcer bleeding are scarce. Decision-making in this group of patients requires balancing the severity and risk of bleeding with the risk of thromboembolism.1,27 In patients on antiplatelet therapy for primary prophylaxis of atherothrombosis who develop bleeding from a peptic ulcer, the antiplatelet should generally be held and the indication for the medication reassessed. In patients on antiplatelet therapy for secondary prevention, the agent may be immediately resumed after endoscopy if bleeding is found to be due to an ulcer with low-risk stigmata. With bleeding resulting from an ulcer with high-risk stigmata, antiplatelet agents employed for secondary prevention may be held initially, with consideration given to early reintroduction, as early as day 3 after endoscopy.1 In patients at high risk for atherothrombotic events, including those on aspirin for secondary prophylaxis, withholding aspirin leads to a 3-fold increase in the risk of a major adverse cardiac event, with events occurring as early as 5 days after aspirin cessation in some cases.27 A randomized controlled trial of continuing low-dose aspirin versus withholding it for 8 weeks in patients on aspirin for secondary prophylaxis of cardiovascular events who experienced peptic ulcer bleeding that required endoscopic therapy demonstrated lower all-cause mortality (1.3% vs 12.9%), including death from cardiovascular or cerebrovascular events, among those who continued aspirin therapy, with a small increased risk of recurrent ulcer bleeding (10.3% vs 5.4%).30 Thus, it is recommended that antiplatelet therapy, when held, be resumed as early as possible when the risk of a cardiovascular or cerebrovascular event is considered to be higher than the risk of bleeding.27
When patients are on DAPT for a history of drug-eluting stent placement, withholding both antiplatelet medications should be avoided, even for a brief period of time, given the risk of in-stent thrombosis. When DAPT is employed for other reasons, it should be continued, if indicated, after bleeding that is found to be due to peptic ulcers with low-risk stigmata. If bleeding is due to a peptic ulcer with high-risk stigmata at endoscopy, then aspirin monotherapy should be continued and consultation should be obtained with a cardiologist to determine optimal timing to resume the second antiplatelet agent.1 In patients on anticoagulants, anticoagulation should be resumed once hemostasis is achieved when the risk of withholding anticoagulation is thought to be greater than the risk of rebleeding. For example, anticoagulation should be resumed early in a patient with a mechanical heart valve to prevent thrombosis.1,27 Following upper GIB from peptic ulcer disease, patients who will require long-term aspirin, DAPT, or anticoagulation with either warfarin or DOACs should be maintained on long-term PPI therapy to reduce the risk of recurrent bleeding.9,27
Failure of Endoscopic Therapy to Control Peptic Ulcer Bleeding
Bleeding recurs in as many as 10% to 20% of patients after initial endoscopic control of peptic ulcer bleeding.4,31 In this context, repeat upper endoscopy for hemostasis is preferred to surgery, as it leads to less morbidity while providing long-term control of bleeding in more than 70% of cases.31,32 Two potential endoscopic rescue therapies that may be employed are over-the-scope clips (OTSCs) and hemostatic powder.32,33
While through-the-scope (TTS) hemostatic clips are often used during endoscopy to control active peptic ulcer bleeding, their use may be limited in large or fibrotic ulcers due to the smaller size of the clips and method of application. OTSCs have several advantages over TTS clips; notably, their larger size allows the endoscopist to achieve deeper mucosal or submucosal clip attachment via suction of the targeted tissue into the endoscopic cap (Figure 2). In a systematic review of OTSCs, successful hemostasis was achieved in 84% of 761 lesions, including 75% of lesions due to peptic ulcer disease.34 Some have argued that OTSCs may be preferred as first-line therapy over epinephrine with TTS clips for hemostasis in bleeding from high-risk peptic ulcers (ie, those with visualized arterial bleeding or a visible vessel) given observed decreases in rebleeding events.35
Despite the advantages of OTSCs, endoscopists should be mindful of the potential complications of OTSC use, including luminal obstruction, particularly in the duodenum, and perforation, which occurs in 0.3% to 2% of cases. Additionally, retrieval of misplaced OTSCs presents a significant challenge. Careful decision-making with consideration of the location, size, and depth of lesions is required when deciding on OTSC placement.34,36
A newer endoscopic tool developed for refractory bleeding from peptic ulcers and other causes is hemostatic powder. Hemostatic powders accelerate the coagulation cascade, leading to shortened coagulation times and enhanced clot formation.37 A recent meta-analysis showed that immediate hemostasis could be achieved in 95% of cases of bleeding, including in 96% of cases of bleeding from peptic ulcer disease.38 The primary limitation of hemostatic powders is the temporary nature of hemostasis, which requires the underlying etiology of bleeding to be addressed in order to provide long-term hemostasis. In the above meta-analysis, rebleeding occurred in 17% of cases after 30 days.38
Hypotension and ulcer diameter ≥ 2 cm are independent predictors of failure of endoscopic salvage therapy.31 When severe bleeding is not controlled with initial endoscopic therapy or bleeding recurs despite salvage endoscopic therapy, transcatheter angiographic embolization (TAE) is the treatment of choice.4 Systematic reviews and meta-analyses of studies that compared TAE to surgery have shown that the rate of rebleeding may be higher with TAE, but with less morbidity and either decreased or equivalent rates of mortality, with no increased need for additional interventions.4,32 In a case series examining 5 years of experience at a single medical center in China, massive GIB from duodenal ulcers was successfully treated with TAE in 27 of 29 cases (93% clinical success rate), with no mucosal ischemic necrosis observed.39
If repeated endoscopic therapy has not led to hemostasis of a bleeding peptic ulcer and TAE is not available, then surgery is the next best option. Bleeding gastric ulcers may be excised, wedge resected, or oversewn after an anterior gastrostomy. Bleeding duodenal ulcers may require use of a Kocher maneuver and linear incision of the anterior duodenum followed by ligation of the gastroduodenal artery. Fortunately, such surgical management is rarely necessary given the availability of TAE at most centers.4
Conclusion
Peptic ulcer disease is a common health problem globally, with persistent challenges related to refractory ulcers, antiplatelet and anticoagulant use, and continued bleeding in the face of endoscopic therapy. These challenges should be met with adequate frequency and duration of PPI therapy, vigilant attention to and treatment of ulcer etiology, evidence-based handling of antiplatelet and anticoagulant medications, and utilization of novel endoscopic tools to obtain improved clinical outcomes.
Acknowledgment: We thank Dr. Nipun Reddy from our institution for providing the endoscopic images used in this article.
Corresponding author: Adam L. Edwards, MD, MS; [email protected].
Financial disclosures: None.
1. Lanas A, Chan FKL. Peptic ulcer disease. Lancet. 2017;390:613-624.
2. Malfertheiner P, Chan FK, McColl KE. Peptic ulcer disease. Lancet. 2009;374:1449-1461.
3. Roberts-Thomson IC. Rise and fall of peptic ulceration: A disease of civilization? J Gastroenterol Hepatol. 2018;33:1321-1326.
4. Kempenich JW, Sirinek KR. Acid peptic disease. Surg Clin North Am. 2018;98:933-944.
5. Cryer B, Feldman M. Effects of very low dose daily, long-term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury in healthy humans. Gastroenterology. 1999;117:17-25.
6. Kavitt RT, Lipowska AM, Anyane-Yeboa A, Gralnek IM. Diagnosis and treatment of peptic ulcer disease. Am J Med. 2019;132:447-456.
7. Walan A, Bader JP, Classen M, et al. Effect of omeprazole and ranitidine on ulcer healing and relapse rates in patients with benign gastric ulcer. New Engl J Med. 1989;320:69-75.
8. Chey WD, Leontiadis GI, Howden CW, Moss SF. ACG Clinical Guideline: Treatment of Helicobacter pylori Infection. Am J Gastroenterol. 2017;112:212-239.
9. Barkun AN, Almadi M, Kuipers EJ, et al. Management of nonvariceal upper gastrointestinal bleeding: Guideline recommendations from the International Consensus Group. Ann Intern Med. 2019;171:805-822.
10. Arevalo Galvis A, Trespalacios Rangel AA, Otero Regino W. Personalized therapy for Helicobacter pylori: CYP2C19 genotype effect on first-line triple therapy. Helicobacter. 2019;24:e12574.
11. Furuta T, Ohashi K, Kamata T, et al. Effect of genetic differences in omeprazole metabolism on cure rates for Helicobacter pylori infection and peptic ulcer. Ann Intern Med. 1998;129:1027-1030.
12. Kirchheiner J, Glatt S, Fuhr U, et al. Relative potency of proton-pump inhibitors-comparison of effects on intragastric pH. Eur J Clin Pharmacol. 2009;65:19-31.
13. Graham DY, Tansel A. Interchangeable use of proton pump inhibitors based on relative potency. Clin Gastroenterol Hepatol. 2018;16:800-808.e7.
14. Burget DW, Chiverton SG, Hunt RH. Is there an optimal degree of acid suppression for healing of duodenal ulcers? A model of the relationship between ulcer healing and acid suppression. Gastroenterology. 1990;99:345-351.
15. Kim HU. Diagnostic and treatment approaches for refractory peptic ulcers. Clin Endosc. 2015;48:285-290.
16. Lanas AI, Remacha B, Esteva F, Sainz R. Risk factors associated with refractory peptic ulcers. Gastroenterology. 1995;109:124-133.
17. Lanza FL, Chan FK, Quigley EM. Guidelines for prevention of NSAID-related ulcer complications. Am J Gastroenterol. 2009;104:728-738.
18. Richy F, Bruyere O, Ethgen O, et al. Time dependent risk of gastrointestinal complications induced by non-steroidal anti-inflammatory drug use: a consensus statement using a meta-analytic approach. Ann Rheum Dis. 2004;63:759-766.
19. Scheiman JM, Yeomans ND, Talley NJ, et al. Prevention of ulcers by esomeprazole in at-risk patients using non-selective NSAIDs and COX-2 inhibitors. Am J Gastroenterol. 2006;101:701-710.
20. Laine L, Hopkins RJ, Girardi LS. Has the impact of Helicobacter pylori therapy on ulcer recurrence in the United States been overstated? A meta-analysis of rigorously designed trials. Am J Gastroenterol. 1998;93:1409-1415.
21. Akiyama T, Endo H, Inamori M, et al. Symptomatic gastric sarcoidosis with multiple antral ulcers. Endoscopy. 2009;41 Suppl 2:E159.
22. Sonoda A, Ogawa R, Mizukami K, et al. Marked improvement in gastric involvement in Behcet’s disease with adalimumab treatment. Turk J Gastroenterol. 2017;28:405-407.
23. Saikia N, Talukdar R, Mazumder S, et al. Polyarteritis nodosa presenting as massive upper gastrointestinal hemorrhage. Gastrointest Endosc. 2006;63:868-870.
24. Annunziata ML, Caviglia R, Papparella LG, Cicala M. Upper gastrointestinal involvement of Crohn’s disease: a prospective study on the role of upper endoscopy in the diagnostic work-up. Dig Dis Sci. 2012;57:1618-1623.
25. Quimby GF, Bonnice CA, Burstein SH, Eastwood GL. Active smoking depresses prostaglandin synthesis in human gastric mucosa. Ann Intern Med. 1986;104:616-619.
26. Iwao T, Toyonaga A, Ikegami M, et al. Gastric mucosal blood flow after smoking in healthy human beings assessed by laser Doppler flowmetry. Gastrointest Endosc. 1993;39:400-403.
27. Almadi MA, Barkun A, Brophy J. Antiplatelet and anticoagulant therapy in patients with gastrointestinal bleeding: an 86-year-old woman with peptic ulcer disease. JAMA. 2011;306:2367-2374.
28. Delaney JA, Opatrny L, Brophy JM, Suissa S. Drug drug interactions between antithrombotic medications and the risk of gastrointestinal bleeding. CMAJ. 2007;177:347-351.
29. Burr N, Lummis K, Sood R, et al. Risk of gastrointestinal bleeding with direct oral anticoagulants: a systematic review and network meta-analysis. Lancet Gastroenterol Hepatol. 2017;2:85-93.
30. Sung JJ, Lau JY, Ching JY, et al. Continuation of low-dose aspirin therapy in peptic ulcer bleeding: a randomized trial. Ann Intern Med. 2010;152:1-9.
31. Lau JY, Sung JJ, Lam YH, et al. Endoscopic retreatment compared with surgery in patients with recurrent bleeding after initial endoscopic control of bleeding ulcers. N Engl J Med. 1999;340:751-756.
32. Gralnek IM, Dumonceau JM, Kuipers EJ, et al. Diagnosis and management of nonvariceal upper gastrointestinal hemorrhage: European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy. 2015;47:a1-46.
33. Skinner M, Gutierrez JP, Neumann H, et al. Over-the-scope clip placement is effective rescue therapy for severe acute upper gastrointestinal bleeding. Endosc Int Open. 2014;2:E37-40.
34. Zhong C, Tan S, Ren Y, et al. Clinical outcomes of over-the-scope-clip system for the treatment of acute upper non-variceal gastrointestinal bleeding: a systematic review and meta-analysis. BMC Gastroenterol. 2019;19:225.
35. Mangiafico S, Pigo F, Bertani H, et al. Over-the-scope clip vs epinephrine with clip for first-line hemostasis in non-variceal upper gastrointestinal bleeding: a propensity score match analysis. Endosc Int Open. 2020;8:E50-e8.
36. Wedi E, Gonzalez S, Menke D, et al. One hundred and one over-the-scope-clip applications for severe gastrointestinal bleeding, leaks and fistulas. World J Gastroenterol. 2016;22:1844-1853.
37. Holster IL, van Beusekom HM, Kuipers EJ, et al. Effects of a hemostatic powder hemospray on coagulation and clot formation. Endoscopy. 2015;47:638-645.
38. Facciorusso A, Straus Takahashi M, et al. Efficacy of hemostatic powders in upper gastrointestinal bleeding: A systematic review and meta-analysis. Dig Liver Dis. 2019;51:1633-1640.
39. Wang YL, Cheng YS, et al. Emergency transcatheter arterial embolization for patients with acute massive duodenal ulcer hemorrhage. World J Gastroenterol. 2012;18:4765-4770.
1. Lanas A, Chan FKL. Peptic ulcer disease. Lancet. 2017;390:613-624.
2. Malfertheiner P, Chan FK, McColl KE. Peptic ulcer disease. Lancet. 2009;374:1449-1461.
3. Roberts-Thomson IC. Rise and fall of peptic ulceration: A disease of civilization? J Gastroenterol Hepatol. 2018;33:1321-1326.
4. Kempenich JW, Sirinek KR. Acid peptic disease. Surg Clin North Am. 2018;98:933-944.
5. Cryer B, Feldman M. Effects of very low dose daily, long-term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury in healthy humans. Gastroenterology. 1999;117:17-25.
6. Kavitt RT, Lipowska AM, Anyane-Yeboa A, Gralnek IM. Diagnosis and treatment of peptic ulcer disease. Am J Med. 2019;132:447-456.
7. Walan A, Bader JP, Classen M, et al. Effect of omeprazole and ranitidine on ulcer healing and relapse rates in patients with benign gastric ulcer. New Engl J Med. 1989;320:69-75.
8. Chey WD, Leontiadis GI, Howden CW, Moss SF. ACG Clinical Guideline: Treatment of Helicobacter pylori Infection. Am J Gastroenterol. 2017;112:212-239.
9. Barkun AN, Almadi M, Kuipers EJ, et al. Management of nonvariceal upper gastrointestinal bleeding: Guideline recommendations from the International Consensus Group. Ann Intern Med. 2019;171:805-822.
10. Arevalo Galvis A, Trespalacios Rangel AA, Otero Regino W. Personalized therapy for Helicobacter pylori: CYP2C19 genotype effect on first-line triple therapy. Helicobacter. 2019;24:e12574.
11. Furuta T, Ohashi K, Kamata T, et al. Effect of genetic differences in omeprazole metabolism on cure rates for Helicobacter pylori infection and peptic ulcer. Ann Intern Med. 1998;129:1027-1030.
12. Kirchheiner J, Glatt S, Fuhr U, et al. Relative potency of proton-pump inhibitors-comparison of effects on intragastric pH. Eur J Clin Pharmacol. 2009;65:19-31.
13. Graham DY, Tansel A. Interchangeable use of proton pump inhibitors based on relative potency. Clin Gastroenterol Hepatol. 2018;16:800-808.e7.
14. Burget DW, Chiverton SG, Hunt RH. Is there an optimal degree of acid suppression for healing of duodenal ulcers? A model of the relationship between ulcer healing and acid suppression. Gastroenterology. 1990;99:345-351.
15. Kim HU. Diagnostic and treatment approaches for refractory peptic ulcers. Clin Endosc. 2015;48:285-290.
16. Lanas AI, Remacha B, Esteva F, Sainz R. Risk factors associated with refractory peptic ulcers. Gastroenterology. 1995;109:124-133.
17. Lanza FL, Chan FK, Quigley EM. Guidelines for prevention of NSAID-related ulcer complications. Am J Gastroenterol. 2009;104:728-738.
18. Richy F, Bruyere O, Ethgen O, et al. Time dependent risk of gastrointestinal complications induced by non-steroidal anti-inflammatory drug use: a consensus statement using a meta-analytic approach. Ann Rheum Dis. 2004;63:759-766.
19. Scheiman JM, Yeomans ND, Talley NJ, et al. Prevention of ulcers by esomeprazole in at-risk patients using non-selective NSAIDs and COX-2 inhibitors. Am J Gastroenterol. 2006;101:701-710.
20. Laine L, Hopkins RJ, Girardi LS. Has the impact of Helicobacter pylori therapy on ulcer recurrence in the United States been overstated? A meta-analysis of rigorously designed trials. Am J Gastroenterol. 1998;93:1409-1415.
21. Akiyama T, Endo H, Inamori M, et al. Symptomatic gastric sarcoidosis with multiple antral ulcers. Endoscopy. 2009;41 Suppl 2:E159.
22. Sonoda A, Ogawa R, Mizukami K, et al. Marked improvement in gastric involvement in Behcet’s disease with adalimumab treatment. Turk J Gastroenterol. 2017;28:405-407.
23. Saikia N, Talukdar R, Mazumder S, et al. Polyarteritis nodosa presenting as massive upper gastrointestinal hemorrhage. Gastrointest Endosc. 2006;63:868-870.
24. Annunziata ML, Caviglia R, Papparella LG, Cicala M. Upper gastrointestinal involvement of Crohn’s disease: a prospective study on the role of upper endoscopy in the diagnostic work-up. Dig Dis Sci. 2012;57:1618-1623.
25. Quimby GF, Bonnice CA, Burstein SH, Eastwood GL. Active smoking depresses prostaglandin synthesis in human gastric mucosa. Ann Intern Med. 1986;104:616-619.
26. Iwao T, Toyonaga A, Ikegami M, et al. Gastric mucosal blood flow after smoking in healthy human beings assessed by laser Doppler flowmetry. Gastrointest Endosc. 1993;39:400-403.
27. Almadi MA, Barkun A, Brophy J. Antiplatelet and anticoagulant therapy in patients with gastrointestinal bleeding: an 86-year-old woman with peptic ulcer disease. JAMA. 2011;306:2367-2374.
28. Delaney JA, Opatrny L, Brophy JM, Suissa S. Drug drug interactions between antithrombotic medications and the risk of gastrointestinal bleeding. CMAJ. 2007;177:347-351.
29. Burr N, Lummis K, Sood R, et al. Risk of gastrointestinal bleeding with direct oral anticoagulants: a systematic review and network meta-analysis. Lancet Gastroenterol Hepatol. 2017;2:85-93.
30. Sung JJ, Lau JY, Ching JY, et al. Continuation of low-dose aspirin therapy in peptic ulcer bleeding: a randomized trial. Ann Intern Med. 2010;152:1-9.
31. Lau JY, Sung JJ, Lam YH, et al. Endoscopic retreatment compared with surgery in patients with recurrent bleeding after initial endoscopic control of bleeding ulcers. N Engl J Med. 1999;340:751-756.
32. Gralnek IM, Dumonceau JM, Kuipers EJ, et al. Diagnosis and management of nonvariceal upper gastrointestinal hemorrhage: European Society of Gastrointestinal Endoscopy (ESGE) Guideline. Endoscopy. 2015;47:a1-46.
33. Skinner M, Gutierrez JP, Neumann H, et al. Over-the-scope clip placement is effective rescue therapy for severe acute upper gastrointestinal bleeding. Endosc Int Open. 2014;2:E37-40.
34. Zhong C, Tan S, Ren Y, et al. Clinical outcomes of over-the-scope-clip system for the treatment of acute upper non-variceal gastrointestinal bleeding: a systematic review and meta-analysis. BMC Gastroenterol. 2019;19:225.
35. Mangiafico S, Pigo F, Bertani H, et al. Over-the-scope clip vs epinephrine with clip for first-line hemostasis in non-variceal upper gastrointestinal bleeding: a propensity score match analysis. Endosc Int Open. 2020;8:E50-e8.
36. Wedi E, Gonzalez S, Menke D, et al. One hundred and one over-the-scope-clip applications for severe gastrointestinal bleeding, leaks and fistulas. World J Gastroenterol. 2016;22:1844-1853.
37. Holster IL, van Beusekom HM, Kuipers EJ, et al. Effects of a hemostatic powder hemospray on coagulation and clot formation. Endoscopy. 2015;47:638-645.
38. Facciorusso A, Straus Takahashi M, et al. Efficacy of hemostatic powders in upper gastrointestinal bleeding: A systematic review and meta-analysis. Dig Liver Dis. 2019;51:1633-1640.
39. Wang YL, Cheng YS, et al. Emergency transcatheter arterial embolization for patients with acute massive duodenal ulcer hemorrhage. World J Gastroenterol. 2012;18:4765-4770.
Blood vessels in the eye may diagnose Parkinson’s disease
new research shows. Using an advanced machine-learning algorithm and fundus eye images, which depict the small blood vessels and more at the back of the eye, investigators are able to classify patients with Parkinson’s disease compared against a control group. “We discovered that micro blood vessels decreased in both size and number in patients with Parkinson’s disease,” said Maximillian Diaz, a PhD student at the University of Florida, Gainesville.
The simple eye examination may offer a way to diagnose Parkinson’s early in the disease progression.
Mr. Diaz said the test could be incorporated to a patient’s annual physical examination not only to look for Parkinson’s disease but also for other neurological diseases. Researchers at the University of Florida are also looking at whether the same technique can diagnose Alzheimer’s disease.
The beauty of this is that “the technique is simple,” he said. “What surprised us is that we can do this with fundus images, which can be taken in a clinical setting with a lens that attaches to your smartphone. It’s affordable and portable and it takes less than a minute.”
Machine learning on fundus eye images
Researchers, under the direction of Ruogu Fang, PhD, director of the J. Crayton Pruitt Department of Biomedical Engineering’s Smart Medical Informatics Learning and Evaluation Lab (SMILE) at the University of Florida, Gainesville, collected fundus eye images from 476 age- and gender-matched individuals, 238 diagnosed with Parkinson’s disease and 238 control group images. Another set of 100 images were collected from the University of Florida database using green color channels (UKB-Green and UF-UKB Green) and used to improve vessel segmentation. Of these, 28 were controls and 72 from patients with Parkinson’s disease. Researchers added 44 more control images from the U.K. Biobank to complete the second age- and gender-matched dataset.
“We used 80% of the images to develop the machine-learning network,” Mr. Diaz said. The other 20% of images, which were new to the algorithm, were used to test it, to determine true or false, Parkinson’s disease or control?
“We were able to achieve an accuracy of 85%,” Mr. Diaz said. Currently, there are no biomarkers to diagnose Parkinson’s disease. The disease is only recognizable once 80% of dopaminergic cells have already decayed. “Clinically, there’s no way to tell how long someone has had it,” Mr. Diaz said. He hopes that by doing additional research and testing earlier – with a longitudinal study of images – a pattern may be detected to better predict disease.
Eye vasculature reveals disease
“This concept [studying eye vasculature] is getting a lot of interest right now,” said Anant Madabhushi, PhD, Case Western Reserve University, Cleveland. “The eye is the proverbial window to the soul, and in this case, shows what’s happening in rest of the body.”
Dr. Madabhushi, who was not involved in the Parkinson’s research, has also been working with a team in Cleveland to look at how vessels in the eye predict response to drug therapies in diabetic macular edema, including treatment durability. “What we’ve found is the more twisted the vessels, the more constricted, and the less likely the person would respond to therapy,” he said, adding that studying the pathology of the eye makes a lot of sense. “The arrangement of vessels in the eye are likely to have implications in all kinds of diseases.”
Since Parkinson’s disease does not have any biomarkers, this technology could be very useful in diagnosis. “With specific quantitative measurements, we could have computational imaging biomarkers to predict the risk of onset of Parkinson’s, and the prognosis of disease. That’s the true utility of this approach,” he said.
Mr. Diaz disclosed no relevant financial relationships. Dr. Madabhushi has consulted for Aiforia and has had research sponsored by AstraZeneca, Bristol-Myers Squibb, and Boehringer Ingelheim.
A version of this article originally appeared on Medscape.com.
new research shows. Using an advanced machine-learning algorithm and fundus eye images, which depict the small blood vessels and more at the back of the eye, investigators are able to classify patients with Parkinson’s disease compared against a control group. “We discovered that micro blood vessels decreased in both size and number in patients with Parkinson’s disease,” said Maximillian Diaz, a PhD student at the University of Florida, Gainesville.
The simple eye examination may offer a way to diagnose Parkinson’s early in the disease progression.
Mr. Diaz said the test could be incorporated to a patient’s annual physical examination not only to look for Parkinson’s disease but also for other neurological diseases. Researchers at the University of Florida are also looking at whether the same technique can diagnose Alzheimer’s disease.
The beauty of this is that “the technique is simple,” he said. “What surprised us is that we can do this with fundus images, which can be taken in a clinical setting with a lens that attaches to your smartphone. It’s affordable and portable and it takes less than a minute.”
Machine learning on fundus eye images
Researchers, under the direction of Ruogu Fang, PhD, director of the J. Crayton Pruitt Department of Biomedical Engineering’s Smart Medical Informatics Learning and Evaluation Lab (SMILE) at the University of Florida, Gainesville, collected fundus eye images from 476 age- and gender-matched individuals, 238 diagnosed with Parkinson’s disease and 238 control group images. Another set of 100 images were collected from the University of Florida database using green color channels (UKB-Green and UF-UKB Green) and used to improve vessel segmentation. Of these, 28 were controls and 72 from patients with Parkinson’s disease. Researchers added 44 more control images from the U.K. Biobank to complete the second age- and gender-matched dataset.
“We used 80% of the images to develop the machine-learning network,” Mr. Diaz said. The other 20% of images, which were new to the algorithm, were used to test it, to determine true or false, Parkinson’s disease or control?
“We were able to achieve an accuracy of 85%,” Mr. Diaz said. Currently, there are no biomarkers to diagnose Parkinson’s disease. The disease is only recognizable once 80% of dopaminergic cells have already decayed. “Clinically, there’s no way to tell how long someone has had it,” Mr. Diaz said. He hopes that by doing additional research and testing earlier – with a longitudinal study of images – a pattern may be detected to better predict disease.
Eye vasculature reveals disease
“This concept [studying eye vasculature] is getting a lot of interest right now,” said Anant Madabhushi, PhD, Case Western Reserve University, Cleveland. “The eye is the proverbial window to the soul, and in this case, shows what’s happening in rest of the body.”
Dr. Madabhushi, who was not involved in the Parkinson’s research, has also been working with a team in Cleveland to look at how vessels in the eye predict response to drug therapies in diabetic macular edema, including treatment durability. “What we’ve found is the more twisted the vessels, the more constricted, and the less likely the person would respond to therapy,” he said, adding that studying the pathology of the eye makes a lot of sense. “The arrangement of vessels in the eye are likely to have implications in all kinds of diseases.”
Since Parkinson’s disease does not have any biomarkers, this technology could be very useful in diagnosis. “With specific quantitative measurements, we could have computational imaging biomarkers to predict the risk of onset of Parkinson’s, and the prognosis of disease. That’s the true utility of this approach,” he said.
Mr. Diaz disclosed no relevant financial relationships. Dr. Madabhushi has consulted for Aiforia and has had research sponsored by AstraZeneca, Bristol-Myers Squibb, and Boehringer Ingelheim.
A version of this article originally appeared on Medscape.com.
new research shows. Using an advanced machine-learning algorithm and fundus eye images, which depict the small blood vessels and more at the back of the eye, investigators are able to classify patients with Parkinson’s disease compared against a control group. “We discovered that micro blood vessels decreased in both size and number in patients with Parkinson’s disease,” said Maximillian Diaz, a PhD student at the University of Florida, Gainesville.
The simple eye examination may offer a way to diagnose Parkinson’s early in the disease progression.
Mr. Diaz said the test could be incorporated to a patient’s annual physical examination not only to look for Parkinson’s disease but also for other neurological diseases. Researchers at the University of Florida are also looking at whether the same technique can diagnose Alzheimer’s disease.
The beauty of this is that “the technique is simple,” he said. “What surprised us is that we can do this with fundus images, which can be taken in a clinical setting with a lens that attaches to your smartphone. It’s affordable and portable and it takes less than a minute.”
Machine learning on fundus eye images
Researchers, under the direction of Ruogu Fang, PhD, director of the J. Crayton Pruitt Department of Biomedical Engineering’s Smart Medical Informatics Learning and Evaluation Lab (SMILE) at the University of Florida, Gainesville, collected fundus eye images from 476 age- and gender-matched individuals, 238 diagnosed with Parkinson’s disease and 238 control group images. Another set of 100 images were collected from the University of Florida database using green color channels (UKB-Green and UF-UKB Green) and used to improve vessel segmentation. Of these, 28 were controls and 72 from patients with Parkinson’s disease. Researchers added 44 more control images from the U.K. Biobank to complete the second age- and gender-matched dataset.
“We used 80% of the images to develop the machine-learning network,” Mr. Diaz said. The other 20% of images, which were new to the algorithm, were used to test it, to determine true or false, Parkinson’s disease or control?
“We were able to achieve an accuracy of 85%,” Mr. Diaz said. Currently, there are no biomarkers to diagnose Parkinson’s disease. The disease is only recognizable once 80% of dopaminergic cells have already decayed. “Clinically, there’s no way to tell how long someone has had it,” Mr. Diaz said. He hopes that by doing additional research and testing earlier – with a longitudinal study of images – a pattern may be detected to better predict disease.
Eye vasculature reveals disease
“This concept [studying eye vasculature] is getting a lot of interest right now,” said Anant Madabhushi, PhD, Case Western Reserve University, Cleveland. “The eye is the proverbial window to the soul, and in this case, shows what’s happening in rest of the body.”
Dr. Madabhushi, who was not involved in the Parkinson’s research, has also been working with a team in Cleveland to look at how vessels in the eye predict response to drug therapies in diabetic macular edema, including treatment durability. “What we’ve found is the more twisted the vessels, the more constricted, and the less likely the person would respond to therapy,” he said, adding that studying the pathology of the eye makes a lot of sense. “The arrangement of vessels in the eye are likely to have implications in all kinds of diseases.”
Since Parkinson’s disease does not have any biomarkers, this technology could be very useful in diagnosis. “With specific quantitative measurements, we could have computational imaging biomarkers to predict the risk of onset of Parkinson’s, and the prognosis of disease. That’s the true utility of this approach,” he said.
Mr. Diaz disclosed no relevant financial relationships. Dr. Madabhushi has consulted for Aiforia and has had research sponsored by AstraZeneca, Bristol-Myers Squibb, and Boehringer Ingelheim.
A version of this article originally appeared on Medscape.com.
Cognitive Behavioral Therapy Plus Placebo Is Inferior to NSAID Therapy for Arthritis Pain
Study Overview
Objective. To examine whether discontinuation of nonsteroidal anti-inflammatory drug (NSAID) therapy and initiation of telephone-based cognitive behavioral therapy (CBT) is not worse than continuation of NSAIDs in the management of arthritis pain.
Design. Randomized controlled trial with noninferiority design.
Setting and participants. This study was a multicenter trial conducted across 4 Veterans Affairs health care systems in Boston, Providence, Connecticut, and North Florida/South Georgia that started September 2013 and ended September 2018. Eligibility criteria included being age 20 years or older, radiographic evidence of knee osteoarthritis, and use of an NSAID for knee pain on most days of the month for at least the past 3 months. Exclusion criteria included significant hearing impairments that may impede the conduct of the trial, current opioid prescriptions excluding tramadol, contraindications to NSAID use, recent or scheduled intra-articular injections or surgery, comorbid conditions other than knee pain that limited walking, and bilateral knee replacements or pain only in the replaced knee. Concurrent use of tramadol and other non-NSAID analgesics was permitted.
A total of 490 participants took part in the 2-week run-in period where their NSAID regimen was discontinued and they were started on a standardized dose of the NSAID meloxicam 15 mg daily. During the run-in period, 126 participants were excluded for several reasons, including worsening pain and patient withdrawal, yielding 364 participants who were randomized to continue meloxicam treatment or placebo for 4 weeks with blinding.
Intervention. Subsequent to the 4-week phase 1 placebo controlled trial, participants in the placebo group were given CBT via telephone (unblinded) for 10 weeks, and the meloxicam group continued treatment with meloxicam for phase 2. The CBT group received 10 modules over 10 weeks in 30- to 45-minute telephone contacts with a psychologist using a treatment manual modified for knee osteoarthritis. These modules consisted of 1 introductory module, 8 pain coping skills modules (eg, deep breathing and visual imagery, progressive muscle relaxation, physical activity and bodily mechanics, identifying unhealthy thoughts, balancing unhealthy thoughts, managing stress, time-based pacing, and sleep hygiene), and a final module emphasizing skill consolidation and relapse prevention. Outcomes were assessed at the end of the phase 1 and phase 2 periods.
Main outcome measures. Main study outcome measures included pain as measured with the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) at 4 weeks. Secondary outcomes included the WOMAC pain score, disability score, and global impression of change after treatment at 14 weeks. The WOMAC pain scale ranges from 0 to 20, and consists of 5 questions regarding severity of pain during walking, stair use, lying in bed at night, sitting, and standing, with 0 indicating no pain; 1, mild pain; 2, moderate pain; 3, severe pain; and 4, very severe pain for each item. The WOMAC disability scale measures self-reported difficulty in performing tasks that reflect lower-extremity physical function, including climbing stairs, rising from a chair, walking, and other activities of daily living. The global impression of change after treatment was measured on a 5-point scale (where 1 indicates much better and 5 indicates much worse). The minimum clinically important difference of the WOMAC pain scale is 2, based on prior literature. With the noninferiority design, the margin was set as a score of 1.
Main results. The placebo group consisted of 180 participants, with an average age of 58.2 years (SD, 11.8 years); 89% of them were male. The meloxicam group consisted of 184 participants, with an average age of 58.6 years (SD, 10 years); 84% of them were male. The average body mass index was 33.9 and 33.4 in each group, respectively. For the primary outcome, the placebo group had a worse pain score than the meloxicam group at 4 weeks (difference of 1.4; 95% confidence interval, 0.8- 2.0). At 14 weeks, the placebo group (with CBT) had a worse pain score than the meloxicam group (difference of 0.8; 95% CI, 0.2-1.4). There was no statistically significant difference in the disability score or global impression of change after treatment score between the 2 groups. The observed difference in pain score did not, however, exceed the minimum clinically important difference.
Conclusion. Placebo treatment and CBT are inferior to NSAIDs in managing pain for patients with knee osteoarthritis. The difference in pain may not be clinically important, and there were no differences in function at 14 weeks.
Commentary
Osteoarthritis is a common chronic condition that causes pain and disability and is often treated with oral analgesics. NSAIDs, despite few high-quality trials demonstrating their efficacy, are among the most commonly used treatment for osteoarthritis pain.1 NSAID therapy, however, does have potential side effects, such as gastric reflux and renal dysfunction.2 This withdrawal trial with placebo control contributes further evidence of the effectiveness of NSAIDs on knee osteoarthritis, demonstrating that indeed NSAIDs improve pain scores to a greater degree than placebo treatment. Augmenting placebo treatment with nonpharmacologic CBT was inferior to NSAIDs in pain management. The authors pointed out that the difference in pain score may not be clinically important, and that lower-extremity function was not different between the groups, concluding that, despite the higher pain score, CBT could be a treatment option, particularly for those who may have difficulty tolerating NSAID treatment.
The study population had a number of chronic conditions in addition to having knee arthritis, and thus likely were taking multiple medications for chronic disease management. Use of multiple medications is associated with an increased risk of rug interactions and adverse effects of medications.3 Thus, this attempt to assess whether a nonpharmacologic alternative treatment is noninferior to a drug treatment is a step toward building the evidence base for deprescribing and enhancing medication safety.4 Previous studies have examined other nonpharmacologic treatments for knee arthritis, such as acupuncture,5 and it is worthwhile to consider combining nonpharmacological approaches as an alternative to oral analgesic medication use.
Applications for Clinical Practice
This study advances our understanding of the effect of NSAID use on knee osteoarthritis when compared to placebo with CBT. Although this is a negative study that failed to show that placebo combined with CBT is noninferior to NSAIDs, it did quantify the expected treatment effect of NSAIDs and showed that this effect likely is not clinically important and/or does not alter lower-extremity function. Further studies are needed to identify other nonpharmacologic approaches and test whether combinations of approaches are effective in the management of chronic pain from osteoarthritis.
–William W. Hung, MD, MPH
1. Wongrakpanich S, Wongrakpanich A, Melhado K, Rangaswami J. A comprehensive review of non-steroidal anti-inflammatory drug use in the elderly. Aging Dis. 2018;9:143-150.
2. Pilotto A, Franceschi M, Leandro G, Di Mario F. NSAID and aspirin use by the elderly in general practice: effect on gastrointestinal symptoms and therapies. Drugs Aging. 2003;20:701-710.
3. Steinman MA. Polypharmacy-time to get beyond numbers. JAMA Intern Med. 2016;176:482-483.
4. Rashid R, Chang C, Niu F, et al. Evaluation of a pharmacist-managed nonsteroidal anti-inflammatory drugs deprescribing program in an integrated health care system. J Manag Care Spec Pharm. 2020;26:918-924.
5. Sun J, Zhao Y, Zhu R, et al. Acupotomy therapy for knee osteoarthritis pain: systematic review and meta-analysis. Evid Based Complement Alternat Med. 2020;2020:2168283.
Study Overview
Objective. To examine whether discontinuation of nonsteroidal anti-inflammatory drug (NSAID) therapy and initiation of telephone-based cognitive behavioral therapy (CBT) is not worse than continuation of NSAIDs in the management of arthritis pain.
Design. Randomized controlled trial with noninferiority design.
Setting and participants. This study was a multicenter trial conducted across 4 Veterans Affairs health care systems in Boston, Providence, Connecticut, and North Florida/South Georgia that started September 2013 and ended September 2018. Eligibility criteria included being age 20 years or older, radiographic evidence of knee osteoarthritis, and use of an NSAID for knee pain on most days of the month for at least the past 3 months. Exclusion criteria included significant hearing impairments that may impede the conduct of the trial, current opioid prescriptions excluding tramadol, contraindications to NSAID use, recent or scheduled intra-articular injections or surgery, comorbid conditions other than knee pain that limited walking, and bilateral knee replacements or pain only in the replaced knee. Concurrent use of tramadol and other non-NSAID analgesics was permitted.
A total of 490 participants took part in the 2-week run-in period where their NSAID regimen was discontinued and they were started on a standardized dose of the NSAID meloxicam 15 mg daily. During the run-in period, 126 participants were excluded for several reasons, including worsening pain and patient withdrawal, yielding 364 participants who were randomized to continue meloxicam treatment or placebo for 4 weeks with blinding.
Intervention. Subsequent to the 4-week phase 1 placebo controlled trial, participants in the placebo group were given CBT via telephone (unblinded) for 10 weeks, and the meloxicam group continued treatment with meloxicam for phase 2. The CBT group received 10 modules over 10 weeks in 30- to 45-minute telephone contacts with a psychologist using a treatment manual modified for knee osteoarthritis. These modules consisted of 1 introductory module, 8 pain coping skills modules (eg, deep breathing and visual imagery, progressive muscle relaxation, physical activity and bodily mechanics, identifying unhealthy thoughts, balancing unhealthy thoughts, managing stress, time-based pacing, and sleep hygiene), and a final module emphasizing skill consolidation and relapse prevention. Outcomes were assessed at the end of the phase 1 and phase 2 periods.
Main outcome measures. Main study outcome measures included pain as measured with the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) at 4 weeks. Secondary outcomes included the WOMAC pain score, disability score, and global impression of change after treatment at 14 weeks. The WOMAC pain scale ranges from 0 to 20, and consists of 5 questions regarding severity of pain during walking, stair use, lying in bed at night, sitting, and standing, with 0 indicating no pain; 1, mild pain; 2, moderate pain; 3, severe pain; and 4, very severe pain for each item. The WOMAC disability scale measures self-reported difficulty in performing tasks that reflect lower-extremity physical function, including climbing stairs, rising from a chair, walking, and other activities of daily living. The global impression of change after treatment was measured on a 5-point scale (where 1 indicates much better and 5 indicates much worse). The minimum clinically important difference of the WOMAC pain scale is 2, based on prior literature. With the noninferiority design, the margin was set as a score of 1.
Main results. The placebo group consisted of 180 participants, with an average age of 58.2 years (SD, 11.8 years); 89% of them were male. The meloxicam group consisted of 184 participants, with an average age of 58.6 years (SD, 10 years); 84% of them were male. The average body mass index was 33.9 and 33.4 in each group, respectively. For the primary outcome, the placebo group had a worse pain score than the meloxicam group at 4 weeks (difference of 1.4; 95% confidence interval, 0.8- 2.0). At 14 weeks, the placebo group (with CBT) had a worse pain score than the meloxicam group (difference of 0.8; 95% CI, 0.2-1.4). There was no statistically significant difference in the disability score or global impression of change after treatment score between the 2 groups. The observed difference in pain score did not, however, exceed the minimum clinically important difference.
Conclusion. Placebo treatment and CBT are inferior to NSAIDs in managing pain for patients with knee osteoarthritis. The difference in pain may not be clinically important, and there were no differences in function at 14 weeks.
Commentary
Osteoarthritis is a common chronic condition that causes pain and disability and is often treated with oral analgesics. NSAIDs, despite few high-quality trials demonstrating their efficacy, are among the most commonly used treatment for osteoarthritis pain.1 NSAID therapy, however, does have potential side effects, such as gastric reflux and renal dysfunction.2 This withdrawal trial with placebo control contributes further evidence of the effectiveness of NSAIDs on knee osteoarthritis, demonstrating that indeed NSAIDs improve pain scores to a greater degree than placebo treatment. Augmenting placebo treatment with nonpharmacologic CBT was inferior to NSAIDs in pain management. The authors pointed out that the difference in pain score may not be clinically important, and that lower-extremity function was not different between the groups, concluding that, despite the higher pain score, CBT could be a treatment option, particularly for those who may have difficulty tolerating NSAID treatment.
The study population had a number of chronic conditions in addition to having knee arthritis, and thus likely were taking multiple medications for chronic disease management. Use of multiple medications is associated with an increased risk of rug interactions and adverse effects of medications.3 Thus, this attempt to assess whether a nonpharmacologic alternative treatment is noninferior to a drug treatment is a step toward building the evidence base for deprescribing and enhancing medication safety.4 Previous studies have examined other nonpharmacologic treatments for knee arthritis, such as acupuncture,5 and it is worthwhile to consider combining nonpharmacological approaches as an alternative to oral analgesic medication use.
Applications for Clinical Practice
This study advances our understanding of the effect of NSAID use on knee osteoarthritis when compared to placebo with CBT. Although this is a negative study that failed to show that placebo combined with CBT is noninferior to NSAIDs, it did quantify the expected treatment effect of NSAIDs and showed that this effect likely is not clinically important and/or does not alter lower-extremity function. Further studies are needed to identify other nonpharmacologic approaches and test whether combinations of approaches are effective in the management of chronic pain from osteoarthritis.
–William W. Hung, MD, MPH
Study Overview
Objective. To examine whether discontinuation of nonsteroidal anti-inflammatory drug (NSAID) therapy and initiation of telephone-based cognitive behavioral therapy (CBT) is not worse than continuation of NSAIDs in the management of arthritis pain.
Design. Randomized controlled trial with noninferiority design.
Setting and participants. This study was a multicenter trial conducted across 4 Veterans Affairs health care systems in Boston, Providence, Connecticut, and North Florida/South Georgia that started September 2013 and ended September 2018. Eligibility criteria included being age 20 years or older, radiographic evidence of knee osteoarthritis, and use of an NSAID for knee pain on most days of the month for at least the past 3 months. Exclusion criteria included significant hearing impairments that may impede the conduct of the trial, current opioid prescriptions excluding tramadol, contraindications to NSAID use, recent or scheduled intra-articular injections or surgery, comorbid conditions other than knee pain that limited walking, and bilateral knee replacements or pain only in the replaced knee. Concurrent use of tramadol and other non-NSAID analgesics was permitted.
A total of 490 participants took part in the 2-week run-in period where their NSAID regimen was discontinued and they were started on a standardized dose of the NSAID meloxicam 15 mg daily. During the run-in period, 126 participants were excluded for several reasons, including worsening pain and patient withdrawal, yielding 364 participants who were randomized to continue meloxicam treatment or placebo for 4 weeks with blinding.
Intervention. Subsequent to the 4-week phase 1 placebo controlled trial, participants in the placebo group were given CBT via telephone (unblinded) for 10 weeks, and the meloxicam group continued treatment with meloxicam for phase 2. The CBT group received 10 modules over 10 weeks in 30- to 45-minute telephone contacts with a psychologist using a treatment manual modified for knee osteoarthritis. These modules consisted of 1 introductory module, 8 pain coping skills modules (eg, deep breathing and visual imagery, progressive muscle relaxation, physical activity and bodily mechanics, identifying unhealthy thoughts, balancing unhealthy thoughts, managing stress, time-based pacing, and sleep hygiene), and a final module emphasizing skill consolidation and relapse prevention. Outcomes were assessed at the end of the phase 1 and phase 2 periods.
Main outcome measures. Main study outcome measures included pain as measured with the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) at 4 weeks. Secondary outcomes included the WOMAC pain score, disability score, and global impression of change after treatment at 14 weeks. The WOMAC pain scale ranges from 0 to 20, and consists of 5 questions regarding severity of pain during walking, stair use, lying in bed at night, sitting, and standing, with 0 indicating no pain; 1, mild pain; 2, moderate pain; 3, severe pain; and 4, very severe pain for each item. The WOMAC disability scale measures self-reported difficulty in performing tasks that reflect lower-extremity physical function, including climbing stairs, rising from a chair, walking, and other activities of daily living. The global impression of change after treatment was measured on a 5-point scale (where 1 indicates much better and 5 indicates much worse). The minimum clinically important difference of the WOMAC pain scale is 2, based on prior literature. With the noninferiority design, the margin was set as a score of 1.
Main results. The placebo group consisted of 180 participants, with an average age of 58.2 years (SD, 11.8 years); 89% of them were male. The meloxicam group consisted of 184 participants, with an average age of 58.6 years (SD, 10 years); 84% of them were male. The average body mass index was 33.9 and 33.4 in each group, respectively. For the primary outcome, the placebo group had a worse pain score than the meloxicam group at 4 weeks (difference of 1.4; 95% confidence interval, 0.8- 2.0). At 14 weeks, the placebo group (with CBT) had a worse pain score than the meloxicam group (difference of 0.8; 95% CI, 0.2-1.4). There was no statistically significant difference in the disability score or global impression of change after treatment score between the 2 groups. The observed difference in pain score did not, however, exceed the minimum clinically important difference.
Conclusion. Placebo treatment and CBT are inferior to NSAIDs in managing pain for patients with knee osteoarthritis. The difference in pain may not be clinically important, and there were no differences in function at 14 weeks.
Commentary
Osteoarthritis is a common chronic condition that causes pain and disability and is often treated with oral analgesics. NSAIDs, despite few high-quality trials demonstrating their efficacy, are among the most commonly used treatment for osteoarthritis pain.1 NSAID therapy, however, does have potential side effects, such as gastric reflux and renal dysfunction.2 This withdrawal trial with placebo control contributes further evidence of the effectiveness of NSAIDs on knee osteoarthritis, demonstrating that indeed NSAIDs improve pain scores to a greater degree than placebo treatment. Augmenting placebo treatment with nonpharmacologic CBT was inferior to NSAIDs in pain management. The authors pointed out that the difference in pain score may not be clinically important, and that lower-extremity function was not different between the groups, concluding that, despite the higher pain score, CBT could be a treatment option, particularly for those who may have difficulty tolerating NSAID treatment.
The study population had a number of chronic conditions in addition to having knee arthritis, and thus likely were taking multiple medications for chronic disease management. Use of multiple medications is associated with an increased risk of rug interactions and adverse effects of medications.3 Thus, this attempt to assess whether a nonpharmacologic alternative treatment is noninferior to a drug treatment is a step toward building the evidence base for deprescribing and enhancing medication safety.4 Previous studies have examined other nonpharmacologic treatments for knee arthritis, such as acupuncture,5 and it is worthwhile to consider combining nonpharmacological approaches as an alternative to oral analgesic medication use.
Applications for Clinical Practice
This study advances our understanding of the effect of NSAID use on knee osteoarthritis when compared to placebo with CBT. Although this is a negative study that failed to show that placebo combined with CBT is noninferior to NSAIDs, it did quantify the expected treatment effect of NSAIDs and showed that this effect likely is not clinically important and/or does not alter lower-extremity function. Further studies are needed to identify other nonpharmacologic approaches and test whether combinations of approaches are effective in the management of chronic pain from osteoarthritis.
–William W. Hung, MD, MPH
1. Wongrakpanich S, Wongrakpanich A, Melhado K, Rangaswami J. A comprehensive review of non-steroidal anti-inflammatory drug use in the elderly. Aging Dis. 2018;9:143-150.
2. Pilotto A, Franceschi M, Leandro G, Di Mario F. NSAID and aspirin use by the elderly in general practice: effect on gastrointestinal symptoms and therapies. Drugs Aging. 2003;20:701-710.
3. Steinman MA. Polypharmacy-time to get beyond numbers. JAMA Intern Med. 2016;176:482-483.
4. Rashid R, Chang C, Niu F, et al. Evaluation of a pharmacist-managed nonsteroidal anti-inflammatory drugs deprescribing program in an integrated health care system. J Manag Care Spec Pharm. 2020;26:918-924.
5. Sun J, Zhao Y, Zhu R, et al. Acupotomy therapy for knee osteoarthritis pain: systematic review and meta-analysis. Evid Based Complement Alternat Med. 2020;2020:2168283.
1. Wongrakpanich S, Wongrakpanich A, Melhado K, Rangaswami J. A comprehensive review of non-steroidal anti-inflammatory drug use in the elderly. Aging Dis. 2018;9:143-150.
2. Pilotto A, Franceschi M, Leandro G, Di Mario F. NSAID and aspirin use by the elderly in general practice: effect on gastrointestinal symptoms and therapies. Drugs Aging. 2003;20:701-710.
3. Steinman MA. Polypharmacy-time to get beyond numbers. JAMA Intern Med. 2016;176:482-483.
4. Rashid R, Chang C, Niu F, et al. Evaluation of a pharmacist-managed nonsteroidal anti-inflammatory drugs deprescribing program in an integrated health care system. J Manag Care Spec Pharm. 2020;26:918-924.
5. Sun J, Zhao Y, Zhu R, et al. Acupotomy therapy for knee osteoarthritis pain: systematic review and meta-analysis. Evid Based Complement Alternat Med. 2020;2020:2168283.
Noninvasive, low-cost CGM for type 2 diabetes coming in U.S. and EU
A novel lower-cost noninvasive continuous glucose monitor (CGM) combined with a digital education/guidance program is set to launch in the United States and Europe this month for use in type 2 diabetes.
With the goal of improving management, or even reversing the condition, Neumara’s SugarBEAT device is thought to be the world’s first noninvasive CGM.
Its cost is anticipated to be far lower than traditional CGM, and it’s aimed at a different patient population: those with type 2 diabetes or prediabetes who may or may not be performing fingerstick glucose monitoring, but if they are, they still aren’t using the information to guide management.
“This isn’t about handing out devices and letting patients get on about it on their own accord. This is really about supporting those individuals,” Faz Chowdhury, MD, Nemaura’s chief executive officer, said in an interview.
He pointed to studies showing improvements in glycemic control in patients with type 2 diabetes who were instructed to perform fingerstick blood glucose testing seven times a day for 3-4 days a month and given advice about how to respond to the data.
“This is well established. We’re saying we can make that process a lot more scalable and affordable and convenient for the patient. ... The behavior change side is digitized,” Dr. Chowdhury said. “We want to provide a program to help people reverse their diabetes or at least stabilize it as much as possible.”
Nicholas Argento, MD, diabetes technology director at Maryland Endocrine and Diabetes, Columbia, said in an interview: “It’s interesting. They’re taking a very different approach. I think there’s a lot of validity to what they’re looking at because we have great CGMs right now, but because of the price point it’s not accessible to a lot of people.
“I think they’re onto something that could prove to be useful to a larger group of patients,” he added.
Worn a few days per month and accurate despite being noninvasive
Instead of inserting a catheter under the skin with a needle, as do current CGMs, the device comprises a small rechargeable transmitter and adhesive patch with a sensor that sits on the top of the skin, typically the upper arm. Glucose molecules are drawn out of the interstitial fluid just below the skin and into a chamber where the transmitter measures the glucose level and transmits the data every 5 minutes via Bluetooth to a smartphone app.
Despite this noninvasive approach, the device appears to be about as accurate as traditional CGMs, with comparable mean absolute relative difference (MARD) from a gold standard glucose measure of about 11%-12% with once-daily calibration versus 10%-11% for the Abbott FreeStyle Libre.
Unlike traditional CGMs, SugarBEAT is meant to be worn for only 14 hours at a time during the day and for 2-4 days per month rather than every day.
It’s not aimed at patients with type 1 diabetes or those with type 2 diabetes who are at high risk for hypoglycemia. It requires once-daily fingerstick calibration and is not indicated to replace fingersticks for treatment decisions.
SugarBEAT received a CE Mark in Europe as a Class IIb medical device in May 2019. That version provides real-time glucose values visible to the wearer. In the United States the company submitted a premarketing approval application for the device to the Food and Drug Administration in July 2020, which awaits a decision.
However, FDA is allowing it to enter the U.S. market as a “wellness” device that won’t deliver real-time values for now but instead will generate retroactive reports available to the physician and the patient.
And last month, U.K.-based Neumara launched the BEATdiabetes site, which allows users to sign in and link to the device once it becomes available.
The site provides “scientifically validated, personalized coaching” based on a program developed at the Joslin Diabetes Clinic in Syracuse, N.Y., and will ultimately include monitoring of other cardiovascular risk factors with digital connectivity to a variety of wearables.
Fingerstick monitoring in type 2 diabetes is only so useful
“Fingerstick monitoring for type 2 diabetes is only so useful,” Dr. Argento said in an interview.
“It’s difficult to get people to monitor in a meaningful way.” If patients perform them only in the morning or at other sporadic times of the day, he said, “Then you get a one-dimensional picture ... and they don’t know what to do with the information anyway, so they stop doing it.”
In contrast, with SugarBEAT and BEATDiabetes, “I think it does address a need that fingerstick monitoring doesn’t.”
Dr. Argento did express a few caveats about the device, however. For one, it still requires one fingerstick a day for calibration. “If people don’t like needles, that might be a disincentive.”
Also, despite the apparently comparable mean absolute relative difference with that of conventional CGMs, that measure can still “hide” values that may be consistently either above or below target range.
“MARD is like A1c in that it’s useful but limited. ... It doesn’t tell you about variability or systemic bias,” he said.
Dr. Argento also said that he’d like to see data on the lag time between the interstitial fluid and blood glucose measures with this noninvasive method as compared with that of a subcutaneous catheter.
However, he acknowledged that these potentials for error would be less important for patients with type 2 diabetes who aren’t generally taking medications that increase their risk for hypoglycemia.
In all, he said, “stay tuned. I think this is part of a movement going away from point-in-time to looking at trends and wearables and data to enrich decision-making…There are still some unanswered questions I have but I think they’re onto a concept that’s useful for a broader population.”
Dr. Chowdhury is an employee of Neumara. Dr. Argento consults for Senseonics and Dexcom, and is also a speaker for Dexcom.
This article first appeared on Medscape.com.
A novel lower-cost noninvasive continuous glucose monitor (CGM) combined with a digital education/guidance program is set to launch in the United States and Europe this month for use in type 2 diabetes.
With the goal of improving management, or even reversing the condition, Neumara’s SugarBEAT device is thought to be the world’s first noninvasive CGM.
Its cost is anticipated to be far lower than traditional CGM, and it’s aimed at a different patient population: those with type 2 diabetes or prediabetes who may or may not be performing fingerstick glucose monitoring, but if they are, they still aren’t using the information to guide management.
“This isn’t about handing out devices and letting patients get on about it on their own accord. This is really about supporting those individuals,” Faz Chowdhury, MD, Nemaura’s chief executive officer, said in an interview.
He pointed to studies showing improvements in glycemic control in patients with type 2 diabetes who were instructed to perform fingerstick blood glucose testing seven times a day for 3-4 days a month and given advice about how to respond to the data.
“This is well established. We’re saying we can make that process a lot more scalable and affordable and convenient for the patient. ... The behavior change side is digitized,” Dr. Chowdhury said. “We want to provide a program to help people reverse their diabetes or at least stabilize it as much as possible.”
Nicholas Argento, MD, diabetes technology director at Maryland Endocrine and Diabetes, Columbia, said in an interview: “It’s interesting. They’re taking a very different approach. I think there’s a lot of validity to what they’re looking at because we have great CGMs right now, but because of the price point it’s not accessible to a lot of people.
“I think they’re onto something that could prove to be useful to a larger group of patients,” he added.
Worn a few days per month and accurate despite being noninvasive
Instead of inserting a catheter under the skin with a needle, as do current CGMs, the device comprises a small rechargeable transmitter and adhesive patch with a sensor that sits on the top of the skin, typically the upper arm. Glucose molecules are drawn out of the interstitial fluid just below the skin and into a chamber where the transmitter measures the glucose level and transmits the data every 5 minutes via Bluetooth to a smartphone app.
Despite this noninvasive approach, the device appears to be about as accurate as traditional CGMs, with comparable mean absolute relative difference (MARD) from a gold standard glucose measure of about 11%-12% with once-daily calibration versus 10%-11% for the Abbott FreeStyle Libre.
Unlike traditional CGMs, SugarBEAT is meant to be worn for only 14 hours at a time during the day and for 2-4 days per month rather than every day.
It’s not aimed at patients with type 1 diabetes or those with type 2 diabetes who are at high risk for hypoglycemia. It requires once-daily fingerstick calibration and is not indicated to replace fingersticks for treatment decisions.
SugarBEAT received a CE Mark in Europe as a Class IIb medical device in May 2019. That version provides real-time glucose values visible to the wearer. In the United States the company submitted a premarketing approval application for the device to the Food and Drug Administration in July 2020, which awaits a decision.
However, FDA is allowing it to enter the U.S. market as a “wellness” device that won’t deliver real-time values for now but instead will generate retroactive reports available to the physician and the patient.
And last month, U.K.-based Neumara launched the BEATdiabetes site, which allows users to sign in and link to the device once it becomes available.
The site provides “scientifically validated, personalized coaching” based on a program developed at the Joslin Diabetes Clinic in Syracuse, N.Y., and will ultimately include monitoring of other cardiovascular risk factors with digital connectivity to a variety of wearables.
Fingerstick monitoring in type 2 diabetes is only so useful
“Fingerstick monitoring for type 2 diabetes is only so useful,” Dr. Argento said in an interview.
“It’s difficult to get people to monitor in a meaningful way.” If patients perform them only in the morning or at other sporadic times of the day, he said, “Then you get a one-dimensional picture ... and they don’t know what to do with the information anyway, so they stop doing it.”
In contrast, with SugarBEAT and BEATDiabetes, “I think it does address a need that fingerstick monitoring doesn’t.”
Dr. Argento did express a few caveats about the device, however. For one, it still requires one fingerstick a day for calibration. “If people don’t like needles, that might be a disincentive.”
Also, despite the apparently comparable mean absolute relative difference with that of conventional CGMs, that measure can still “hide” values that may be consistently either above or below target range.
“MARD is like A1c in that it’s useful but limited. ... It doesn’t tell you about variability or systemic bias,” he said.
Dr. Argento also said that he’d like to see data on the lag time between the interstitial fluid and blood glucose measures with this noninvasive method as compared with that of a subcutaneous catheter.
However, he acknowledged that these potentials for error would be less important for patients with type 2 diabetes who aren’t generally taking medications that increase their risk for hypoglycemia.
In all, he said, “stay tuned. I think this is part of a movement going away from point-in-time to looking at trends and wearables and data to enrich decision-making…There are still some unanswered questions I have but I think they’re onto a concept that’s useful for a broader population.”
Dr. Chowdhury is an employee of Neumara. Dr. Argento consults for Senseonics and Dexcom, and is also a speaker for Dexcom.
This article first appeared on Medscape.com.
A novel lower-cost noninvasive continuous glucose monitor (CGM) combined with a digital education/guidance program is set to launch in the United States and Europe this month for use in type 2 diabetes.
With the goal of improving management, or even reversing the condition, Neumara’s SugarBEAT device is thought to be the world’s first noninvasive CGM.
Its cost is anticipated to be far lower than traditional CGM, and it’s aimed at a different patient population: those with type 2 diabetes or prediabetes who may or may not be performing fingerstick glucose monitoring, but if they are, they still aren’t using the information to guide management.
“This isn’t about handing out devices and letting patients get on about it on their own accord. This is really about supporting those individuals,” Faz Chowdhury, MD, Nemaura’s chief executive officer, said in an interview.
He pointed to studies showing improvements in glycemic control in patients with type 2 diabetes who were instructed to perform fingerstick blood glucose testing seven times a day for 3-4 days a month and given advice about how to respond to the data.
“This is well established. We’re saying we can make that process a lot more scalable and affordable and convenient for the patient. ... The behavior change side is digitized,” Dr. Chowdhury said. “We want to provide a program to help people reverse their diabetes or at least stabilize it as much as possible.”
Nicholas Argento, MD, diabetes technology director at Maryland Endocrine and Diabetes, Columbia, said in an interview: “It’s interesting. They’re taking a very different approach. I think there’s a lot of validity to what they’re looking at because we have great CGMs right now, but because of the price point it’s not accessible to a lot of people.
“I think they’re onto something that could prove to be useful to a larger group of patients,” he added.
Worn a few days per month and accurate despite being noninvasive
Instead of inserting a catheter under the skin with a needle, as do current CGMs, the device comprises a small rechargeable transmitter and adhesive patch with a sensor that sits on the top of the skin, typically the upper arm. Glucose molecules are drawn out of the interstitial fluid just below the skin and into a chamber where the transmitter measures the glucose level and transmits the data every 5 minutes via Bluetooth to a smartphone app.
Despite this noninvasive approach, the device appears to be about as accurate as traditional CGMs, with comparable mean absolute relative difference (MARD) from a gold standard glucose measure of about 11%-12% with once-daily calibration versus 10%-11% for the Abbott FreeStyle Libre.
Unlike traditional CGMs, SugarBEAT is meant to be worn for only 14 hours at a time during the day and for 2-4 days per month rather than every day.
It’s not aimed at patients with type 1 diabetes or those with type 2 diabetes who are at high risk for hypoglycemia. It requires once-daily fingerstick calibration and is not indicated to replace fingersticks for treatment decisions.
SugarBEAT received a CE Mark in Europe as a Class IIb medical device in May 2019. That version provides real-time glucose values visible to the wearer. In the United States the company submitted a premarketing approval application for the device to the Food and Drug Administration in July 2020, which awaits a decision.
However, FDA is allowing it to enter the U.S. market as a “wellness” device that won’t deliver real-time values for now but instead will generate retroactive reports available to the physician and the patient.
And last month, U.K.-based Neumara launched the BEATdiabetes site, which allows users to sign in and link to the device once it becomes available.
The site provides “scientifically validated, personalized coaching” based on a program developed at the Joslin Diabetes Clinic in Syracuse, N.Y., and will ultimately include monitoring of other cardiovascular risk factors with digital connectivity to a variety of wearables.
Fingerstick monitoring in type 2 diabetes is only so useful
“Fingerstick monitoring for type 2 diabetes is only so useful,” Dr. Argento said in an interview.
“It’s difficult to get people to monitor in a meaningful way.” If patients perform them only in the morning or at other sporadic times of the day, he said, “Then you get a one-dimensional picture ... and they don’t know what to do with the information anyway, so they stop doing it.”
In contrast, with SugarBEAT and BEATDiabetes, “I think it does address a need that fingerstick monitoring doesn’t.”
Dr. Argento did express a few caveats about the device, however. For one, it still requires one fingerstick a day for calibration. “If people don’t like needles, that might be a disincentive.”
Also, despite the apparently comparable mean absolute relative difference with that of conventional CGMs, that measure can still “hide” values that may be consistently either above or below target range.
“MARD is like A1c in that it’s useful but limited. ... It doesn’t tell you about variability or systemic bias,” he said.
Dr. Argento also said that he’d like to see data on the lag time between the interstitial fluid and blood glucose measures with this noninvasive method as compared with that of a subcutaneous catheter.
However, he acknowledged that these potentials for error would be less important for patients with type 2 diabetes who aren’t generally taking medications that increase their risk for hypoglycemia.
In all, he said, “stay tuned. I think this is part of a movement going away from point-in-time to looking at trends and wearables and data to enrich decision-making…There are still some unanswered questions I have but I think they’re onto a concept that’s useful for a broader population.”
Dr. Chowdhury is an employee of Neumara. Dr. Argento consults for Senseonics and Dexcom, and is also a speaker for Dexcom.
This article first appeared on Medscape.com.
New AHA scientific statement on menopause and CVD risk
Changes in hormones, body composition, lipids, and vascular health during the menopause transition can increase a woman’s chance of developing cardiovascular disease (CVD) after menopause, the American Heart Association said in a scientific statement.
“This statement aims to raise awareness of both healthcare providers and women about the menopause transition as a time of increasing heart disease risk,” Samar R. El Khoudary, PhD, MPH, who chaired the writing group, said in an interview.
“As such, it emphasizes the importance of monitoring women’s health during midlife and targeting this stage as a critical window for applying early intervention strategies that aim to maintain a healthy heart and reduce the risk of heart disease,” said Dr. El Khoudary, of the University of Pittsburgh.
The statement was published online Nov. 30 in Circulation.
Evolution in knowledge
During the past 20 years, knowledge of how menopause might contribute to CVD has evolved “dramatically,” Dr. El Khoudary noted. The accumulated data consistently point to the menopause transition as a time of change in heart health.
“Importantly,” she said, the latest AHA guidelines for CVD prevention in women, published in 2011, do not include data now available on the menopause transition as a time of increased CVD risk.
“As such, there is a compelling need to discuss the implications of the accumulating body of literature on this topic,” said Dr. El Khoudary.
The statement provides a contemporary synthesis of the existing data on menopause and how it relates to CVD, the leading cause of death of U.S. women.
Earlier age at natural menopause has generally been found to be a marker of greater CVD risk. Iatrogenically induced menopause (bilateral oophorectomy) during the premenopausal period is also associated with higher CVD risk, the data suggest.
Vasomotor symptoms are associated with worse levels of CVD risk factors and measures of subclinical atherosclerosis. Sleep disturbance has also been linked to greater risk for subclinical CVD and worse CV health indexes in women during midlife.
Increases in central/visceral fat and decreases in lean muscle mass are more pronounced during the menopause transition. This increased central adiposity is associated with increased risk for mortality, even among those with normal body mass index, the writing group found.
Increases in lipid levels (LDL cholesterol and apolipoprotein B), metabolic syndrome risk, and vascular remodeling at midlife are driven by the menopause transition more than aging, whereas increases in blood pressure, insulin level, and glucose level are likely more influenced by chronological aging, they reported.
Lifestyle interventions
The writing group noted that, because of the increase in overall life expectancy in the United States, a significant proportion of women will spend up to 40% of their lives after menopause.
Yet data suggest that only 7.2% of women transitioning to menopause are meeting physical activity guidelines and that fewer than 20% of those women are consistently maintaining a healthy diet.
Limited data from randomized, controlled trials suggest that a multidimensional lifestyle intervention during the menopause transition can prevent weight gain and reduce blood pressure and levels of triglycerides, blood glucose, and insulin and reduce the incidence of subclinical carotid atherosclerosis, they pointed out.
“Novel data” indicate a reversal in the associations of HDL cholesterol with CVD risk over the menopause transition, suggesting that higher HDL cholesterol levels may not consistently reflect good cardiovascular health in middle-aged women, the group noted.
There are also data suggesting that starting menopause hormone therapy when younger than 60 years or within 10 years of menopause is associated with reduced CVD risk.
The group said further research is needed into the cardiometabolic effects of menopause hormone therapy, including effects associated with form, route, and duration of administration, in women traversing menopause.
They also noted that data for the primary and secondary prevention of atherosclerotic CVD and improved survival with lipid-lowering interventions “remain elusive” for women and that further study is needed to develop evidence-based recommendations tailored specifically to women.
The research had no commercial funding. Dr. El Khoudary has disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Changes in hormones, body composition, lipids, and vascular health during the menopause transition can increase a woman’s chance of developing cardiovascular disease (CVD) after menopause, the American Heart Association said in a scientific statement.
“This statement aims to raise awareness of both healthcare providers and women about the menopause transition as a time of increasing heart disease risk,” Samar R. El Khoudary, PhD, MPH, who chaired the writing group, said in an interview.
“As such, it emphasizes the importance of monitoring women’s health during midlife and targeting this stage as a critical window for applying early intervention strategies that aim to maintain a healthy heart and reduce the risk of heart disease,” said Dr. El Khoudary, of the University of Pittsburgh.
The statement was published online Nov. 30 in Circulation.
Evolution in knowledge
During the past 20 years, knowledge of how menopause might contribute to CVD has evolved “dramatically,” Dr. El Khoudary noted. The accumulated data consistently point to the menopause transition as a time of change in heart health.
“Importantly,” she said, the latest AHA guidelines for CVD prevention in women, published in 2011, do not include data now available on the menopause transition as a time of increased CVD risk.
“As such, there is a compelling need to discuss the implications of the accumulating body of literature on this topic,” said Dr. El Khoudary.
The statement provides a contemporary synthesis of the existing data on menopause and how it relates to CVD, the leading cause of death of U.S. women.
Earlier age at natural menopause has generally been found to be a marker of greater CVD risk. Iatrogenically induced menopause (bilateral oophorectomy) during the premenopausal period is also associated with higher CVD risk, the data suggest.
Vasomotor symptoms are associated with worse levels of CVD risk factors and measures of subclinical atherosclerosis. Sleep disturbance has also been linked to greater risk for subclinical CVD and worse CV health indexes in women during midlife.
Increases in central/visceral fat and decreases in lean muscle mass are more pronounced during the menopause transition. This increased central adiposity is associated with increased risk for mortality, even among those with normal body mass index, the writing group found.
Increases in lipid levels (LDL cholesterol and apolipoprotein B), metabolic syndrome risk, and vascular remodeling at midlife are driven by the menopause transition more than aging, whereas increases in blood pressure, insulin level, and glucose level are likely more influenced by chronological aging, they reported.
Lifestyle interventions
The writing group noted that, because of the increase in overall life expectancy in the United States, a significant proportion of women will spend up to 40% of their lives after menopause.
Yet data suggest that only 7.2% of women transitioning to menopause are meeting physical activity guidelines and that fewer than 20% of those women are consistently maintaining a healthy diet.
Limited data from randomized, controlled trials suggest that a multidimensional lifestyle intervention during the menopause transition can prevent weight gain and reduce blood pressure and levels of triglycerides, blood glucose, and insulin and reduce the incidence of subclinical carotid atherosclerosis, they pointed out.
“Novel data” indicate a reversal in the associations of HDL cholesterol with CVD risk over the menopause transition, suggesting that higher HDL cholesterol levels may not consistently reflect good cardiovascular health in middle-aged women, the group noted.
There are also data suggesting that starting menopause hormone therapy when younger than 60 years or within 10 years of menopause is associated with reduced CVD risk.
The group said further research is needed into the cardiometabolic effects of menopause hormone therapy, including effects associated with form, route, and duration of administration, in women traversing menopause.
They also noted that data for the primary and secondary prevention of atherosclerotic CVD and improved survival with lipid-lowering interventions “remain elusive” for women and that further study is needed to develop evidence-based recommendations tailored specifically to women.
The research had no commercial funding. Dr. El Khoudary has disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Changes in hormones, body composition, lipids, and vascular health during the menopause transition can increase a woman’s chance of developing cardiovascular disease (CVD) after menopause, the American Heart Association said in a scientific statement.
“This statement aims to raise awareness of both healthcare providers and women about the menopause transition as a time of increasing heart disease risk,” Samar R. El Khoudary, PhD, MPH, who chaired the writing group, said in an interview.
“As such, it emphasizes the importance of monitoring women’s health during midlife and targeting this stage as a critical window for applying early intervention strategies that aim to maintain a healthy heart and reduce the risk of heart disease,” said Dr. El Khoudary, of the University of Pittsburgh.
The statement was published online Nov. 30 in Circulation.
Evolution in knowledge
During the past 20 years, knowledge of how menopause might contribute to CVD has evolved “dramatically,” Dr. El Khoudary noted. The accumulated data consistently point to the menopause transition as a time of change in heart health.
“Importantly,” she said, the latest AHA guidelines for CVD prevention in women, published in 2011, do not include data now available on the menopause transition as a time of increased CVD risk.
“As such, there is a compelling need to discuss the implications of the accumulating body of literature on this topic,” said Dr. El Khoudary.
The statement provides a contemporary synthesis of the existing data on menopause and how it relates to CVD, the leading cause of death of U.S. women.
Earlier age at natural menopause has generally been found to be a marker of greater CVD risk. Iatrogenically induced menopause (bilateral oophorectomy) during the premenopausal period is also associated with higher CVD risk, the data suggest.
Vasomotor symptoms are associated with worse levels of CVD risk factors and measures of subclinical atherosclerosis. Sleep disturbance has also been linked to greater risk for subclinical CVD and worse CV health indexes in women during midlife.
Increases in central/visceral fat and decreases in lean muscle mass are more pronounced during the menopause transition. This increased central adiposity is associated with increased risk for mortality, even among those with normal body mass index, the writing group found.
Increases in lipid levels (LDL cholesterol and apolipoprotein B), metabolic syndrome risk, and vascular remodeling at midlife are driven by the menopause transition more than aging, whereas increases in blood pressure, insulin level, and glucose level are likely more influenced by chronological aging, they reported.
Lifestyle interventions
The writing group noted that, because of the increase in overall life expectancy in the United States, a significant proportion of women will spend up to 40% of their lives after menopause.
Yet data suggest that only 7.2% of women transitioning to menopause are meeting physical activity guidelines and that fewer than 20% of those women are consistently maintaining a healthy diet.
Limited data from randomized, controlled trials suggest that a multidimensional lifestyle intervention during the menopause transition can prevent weight gain and reduce blood pressure and levels of triglycerides, blood glucose, and insulin and reduce the incidence of subclinical carotid atherosclerosis, they pointed out.
“Novel data” indicate a reversal in the associations of HDL cholesterol with CVD risk over the menopause transition, suggesting that higher HDL cholesterol levels may not consistently reflect good cardiovascular health in middle-aged women, the group noted.
There are also data suggesting that starting menopause hormone therapy when younger than 60 years or within 10 years of menopause is associated with reduced CVD risk.
The group said further research is needed into the cardiometabolic effects of menopause hormone therapy, including effects associated with form, route, and duration of administration, in women traversing menopause.
They also noted that data for the primary and secondary prevention of atherosclerotic CVD and improved survival with lipid-lowering interventions “remain elusive” for women and that further study is needed to develop evidence-based recommendations tailored specifically to women.
The research had no commercial funding. Dr. El Khoudary has disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Acute-on-chronic itch is new frontier in atopic dermatitis
Brian S. Kim, MD, said at MedscapeLive’s annual Las Vegas Dermatology Seminar, held virtually this year.
Recent years have brought enormous progress in understanding how chronic itch in patients with atopic dermatitis (AD) is mediated by type 2 cytokines, including interleukin-13, IL-4, and IL-31, as well as by Janus kinase (JAK) signaling. This has led to development of potent therapies targeting these mediators, including dupilumab (Dupixent) and the investigational agents tralokinumab, lebrikizumab, abrocitinib, upadacitinib, baricitinib, and the IL-31 inhibitor nemolizumab.
“This is now one of the most active areas in the field of dermatology,” observed Dr. Kim, a dermatologist and codirector of the Center for the Study of Itch and Sensory Disorders at Washington University in St. Louis.
He has figured prominently in this effort. He and his coinvestigators conducted translational studies in mouse models which unraveled key mechanisms by which the immune system responsible for skin inflammation in AD communicates with the nervous system to trigger the neural sensation of itch. He also led a phase 2 randomized trial in 307 patients with AD, which demonstrated that the investigational JAK1/JAK2 inhibitor ruxolitinib cream markedly improved itch within 36 hours, well before subsequent improvement in skin inflammation – and the topical JAK inhibitor did so with minimal systemic absorption.
Compared with chronic itch, much less research attention has been devoted to the phenomenon of acute itch flares superimposed upon the chronic itch of AD. These acute-on-chronic itch flares are a common feature of the disease. In a soon-to-be-published study of 159 AD patients in the placebo arm of a clinical trial, Dr. Kim and coinvestigators found that 26% exhibited a pattern of acute itch flares during the course of a single month. During the next month, 3.1% of patients under study went from an acute-on-chronic itch pattern in month 1 to a nonflare pattern, 20% went from a nonflare pattern in month 1 to acute itch flares in month 2, and 23% of the overall study population retained their pattern of acute itch flares through both months.

“This does not seem to be just a static phenotype, but rather these patients can evolve over time. And we think that this can be driven by allergen-specific IgE,” according to Dr. Kim.
Indeed, the investigators found that patients with allergen-specific IgE in their serum were roughly twice as likely to exhibit the acute-on-chronic itch flare pattern than those without allergen-specific IgE.
The classical thinking has been that IgE binds to its receptors on mast cells, causing mast cell degranulation and release of histamine and other itch-inducing molecules. Yet antihistamines have proven notoriously ineffective for the treatment of AD.
Circulating basophils capable of working their way into inflamed skin also have IgE receptors. Dr. Kim and colleagues have shown that allergen-specific IgE in mice binds to those receptors, causing the basophils to degenerate, releasing itch-promoting chemicals. They have subsequently carried over this work into the clinical arena.
“We’ve found that patients with atopic dermatitis have significantly higher expression of receptors for IgE in their basophils than in the basophils of healthy controls, indicating perhaps that the basophils in patients with atopic dermatitis are much more prone to stimulation by allergen by way of IgE. This is a new concept that we’re exploring,” Dr. Kim said.
“We haven’t really known before what IgE does in atopic dermatitis, but it turns out that it may actually play a very important role in triggering acute flares of itch,” the dermatologist explained. “What’s been surprising is that the IgE activity is not mediated so much by mast cells, which are tissue-resident; the predominant means appears to be that IgE acts on basophils. That then creates release not of histamine, but of leukotriene C4, which is a very potent pruritogen. This may be responsible for those acute itch flares.”
Asked during an audience Q&A how allergen-specific IgE–mediated basophil activation might be targeted therapeutically in order to prevent acute-on-chronic itch flares in patients with AD, Dr. Kim mentioned two possibilities. One is treatment with potent anti-IgE agents, which to date have not been adequately tested for their antipruritic prowess in AD.
“Also, there’s another molecule that seems to be relatively basophil-selective and -specific that’s just been discovered by my colleague Xinzhong Dong at Johns Hopkins University [in Baltimore] – called MRGPRX2 – that may actually be a potentially viable way to go after basophils, maybe even by depleting them if you had an antibody against that,” Dr. Kim said. He was a coinvestigator in Dr. Dong’s recent study characterizing MRGPRX2, the mast-cell-expressed Mas-related G-protein–coupled receptor activator.
Dr. Kim reported receiving research funding from Cara Therapeutics and LEO Pharma, holding a patent for the use of JAK inhibitors in chronic itch, and serving as a consultant to numerous pharmaceutical companies.
MedscapeLive and this news organization are owned by the same parent company.
Brian S. Kim, MD, said at MedscapeLive’s annual Las Vegas Dermatology Seminar, held virtually this year.
Recent years have brought enormous progress in understanding how chronic itch in patients with atopic dermatitis (AD) is mediated by type 2 cytokines, including interleukin-13, IL-4, and IL-31, as well as by Janus kinase (JAK) signaling. This has led to development of potent therapies targeting these mediators, including dupilumab (Dupixent) and the investigational agents tralokinumab, lebrikizumab, abrocitinib, upadacitinib, baricitinib, and the IL-31 inhibitor nemolizumab.
“This is now one of the most active areas in the field of dermatology,” observed Dr. Kim, a dermatologist and codirector of the Center for the Study of Itch and Sensory Disorders at Washington University in St. Louis.
He has figured prominently in this effort. He and his coinvestigators conducted translational studies in mouse models which unraveled key mechanisms by which the immune system responsible for skin inflammation in AD communicates with the nervous system to trigger the neural sensation of itch. He also led a phase 2 randomized trial in 307 patients with AD, which demonstrated that the investigational JAK1/JAK2 inhibitor ruxolitinib cream markedly improved itch within 36 hours, well before subsequent improvement in skin inflammation – and the topical JAK inhibitor did so with minimal systemic absorption.
Compared with chronic itch, much less research attention has been devoted to the phenomenon of acute itch flares superimposed upon the chronic itch of AD. These acute-on-chronic itch flares are a common feature of the disease. In a soon-to-be-published study of 159 AD patients in the placebo arm of a clinical trial, Dr. Kim and coinvestigators found that 26% exhibited a pattern of acute itch flares during the course of a single month. During the next month, 3.1% of patients under study went from an acute-on-chronic itch pattern in month 1 to a nonflare pattern, 20% went from a nonflare pattern in month 1 to acute itch flares in month 2, and 23% of the overall study population retained their pattern of acute itch flares through both months.
“This does not seem to be just a static phenotype, but rather these patients can evolve over time. And we think that this can be driven by allergen-specific IgE,” according to Dr. Kim.
Indeed, the investigators found that patients with allergen-specific IgE in their serum were roughly twice as likely to exhibit the acute-on-chronic itch flare pattern than those without allergen-specific IgE.
The classical thinking has been that IgE binds to its receptors on mast cells, causing mast cell degranulation and release of histamine and other itch-inducing molecules. Yet antihistamines have proven notoriously ineffective for the treatment of AD.
Circulating basophils capable of working their way into inflamed skin also have IgE receptors. Dr. Kim and colleagues have shown that allergen-specific IgE in mice binds to those receptors, causing the basophils to degenerate, releasing itch-promoting chemicals. They have subsequently carried over this work into the clinical arena.
“We’ve found that patients with atopic dermatitis have significantly higher expression of receptors for IgE in their basophils than in the basophils of healthy controls, indicating perhaps that the basophils in patients with atopic dermatitis are much more prone to stimulation by allergen by way of IgE. This is a new concept that we’re exploring,” Dr. Kim said.
“We haven’t really known before what IgE does in atopic dermatitis, but it turns out that it may actually play a very important role in triggering acute flares of itch,” the dermatologist explained. “What’s been surprising is that the IgE activity is not mediated so much by mast cells, which are tissue-resident; the predominant means appears to be that IgE acts on basophils. That then creates release not of histamine, but of leukotriene C4, which is a very potent pruritogen. This may be responsible for those acute itch flares.”
Asked during an audience Q&A how allergen-specific IgE–mediated basophil activation might be targeted therapeutically in order to prevent acute-on-chronic itch flares in patients with AD, Dr. Kim mentioned two possibilities. One is treatment with potent anti-IgE agents, which to date have not been adequately tested for their antipruritic prowess in AD.
“Also, there’s another molecule that seems to be relatively basophil-selective and -specific that’s just been discovered by my colleague Xinzhong Dong at Johns Hopkins University [in Baltimore] – called MRGPRX2 – that may actually be a potentially viable way to go after basophils, maybe even by depleting them if you had an antibody against that,” Dr. Kim said. He was a coinvestigator in Dr. Dong’s recent study characterizing MRGPRX2, the mast-cell-expressed Mas-related G-protein–coupled receptor activator.
Dr. Kim reported receiving research funding from Cara Therapeutics and LEO Pharma, holding a patent for the use of JAK inhibitors in chronic itch, and serving as a consultant to numerous pharmaceutical companies.
MedscapeLive and this news organization are owned by the same parent company.
Brian S. Kim, MD, said at MedscapeLive’s annual Las Vegas Dermatology Seminar, held virtually this year.
Recent years have brought enormous progress in understanding how chronic itch in patients with atopic dermatitis (AD) is mediated by type 2 cytokines, including interleukin-13, IL-4, and IL-31, as well as by Janus kinase (JAK) signaling. This has led to development of potent therapies targeting these mediators, including dupilumab (Dupixent) and the investigational agents tralokinumab, lebrikizumab, abrocitinib, upadacitinib, baricitinib, and the IL-31 inhibitor nemolizumab.
“This is now one of the most active areas in the field of dermatology,” observed Dr. Kim, a dermatologist and codirector of the Center for the Study of Itch and Sensory Disorders at Washington University in St. Louis.
He has figured prominently in this effort. He and his coinvestigators conducted translational studies in mouse models which unraveled key mechanisms by which the immune system responsible for skin inflammation in AD communicates with the nervous system to trigger the neural sensation of itch. He also led a phase 2 randomized trial in 307 patients with AD, which demonstrated that the investigational JAK1/JAK2 inhibitor ruxolitinib cream markedly improved itch within 36 hours, well before subsequent improvement in skin inflammation – and the topical JAK inhibitor did so with minimal systemic absorption.
Compared with chronic itch, much less research attention has been devoted to the phenomenon of acute itch flares superimposed upon the chronic itch of AD. These acute-on-chronic itch flares are a common feature of the disease. In a soon-to-be-published study of 159 AD patients in the placebo arm of a clinical trial, Dr. Kim and coinvestigators found that 26% exhibited a pattern of acute itch flares during the course of a single month. During the next month, 3.1% of patients under study went from an acute-on-chronic itch pattern in month 1 to a nonflare pattern, 20% went from a nonflare pattern in month 1 to acute itch flares in month 2, and 23% of the overall study population retained their pattern of acute itch flares through both months.
“This does not seem to be just a static phenotype, but rather these patients can evolve over time. And we think that this can be driven by allergen-specific IgE,” according to Dr. Kim.
Indeed, the investigators found that patients with allergen-specific IgE in their serum were roughly twice as likely to exhibit the acute-on-chronic itch flare pattern than those without allergen-specific IgE.
The classical thinking has been that IgE binds to its receptors on mast cells, causing mast cell degranulation and release of histamine and other itch-inducing molecules. Yet antihistamines have proven notoriously ineffective for the treatment of AD.
Circulating basophils capable of working their way into inflamed skin also have IgE receptors. Dr. Kim and colleagues have shown that allergen-specific IgE in mice binds to those receptors, causing the basophils to degenerate, releasing itch-promoting chemicals. They have subsequently carried over this work into the clinical arena.
“We’ve found that patients with atopic dermatitis have significantly higher expression of receptors for IgE in their basophils than in the basophils of healthy controls, indicating perhaps that the basophils in patients with atopic dermatitis are much more prone to stimulation by allergen by way of IgE. This is a new concept that we’re exploring,” Dr. Kim said.
“We haven’t really known before what IgE does in atopic dermatitis, but it turns out that it may actually play a very important role in triggering acute flares of itch,” the dermatologist explained. “What’s been surprising is that the IgE activity is not mediated so much by mast cells, which are tissue-resident; the predominant means appears to be that IgE acts on basophils. That then creates release not of histamine, but of leukotriene C4, which is a very potent pruritogen. This may be responsible for those acute itch flares.”
Asked during an audience Q&A how allergen-specific IgE–mediated basophil activation might be targeted therapeutically in order to prevent acute-on-chronic itch flares in patients with AD, Dr. Kim mentioned two possibilities. One is treatment with potent anti-IgE agents, which to date have not been adequately tested for their antipruritic prowess in AD.
“Also, there’s another molecule that seems to be relatively basophil-selective and -specific that’s just been discovered by my colleague Xinzhong Dong at Johns Hopkins University [in Baltimore] – called MRGPRX2 – that may actually be a potentially viable way to go after basophils, maybe even by depleting them if you had an antibody against that,” Dr. Kim said. He was a coinvestigator in Dr. Dong’s recent study characterizing MRGPRX2, the mast-cell-expressed Mas-related G-protein–coupled receptor activator.
Dr. Kim reported receiving research funding from Cara Therapeutics and LEO Pharma, holding a patent for the use of JAK inhibitors in chronic itch, and serving as a consultant to numerous pharmaceutical companies.
MedscapeLive and this news organization are owned by the same parent company.
FROM MEDSCAPELIVE LAS VEGAS DERMATOLOGY SEMINAR
