User login
The new one-percenters: Children with COVID-19
according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
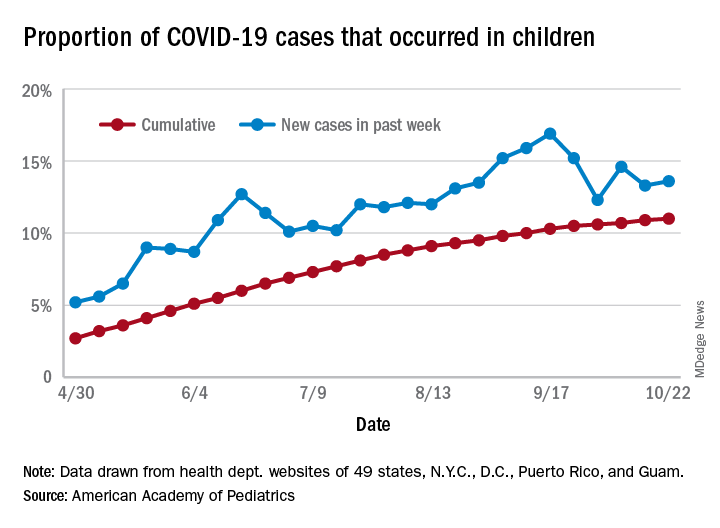
There have been 1,052 cases of COVID-19 per 100,000 children as of Oct. 22, and that works out to 1.05% of all children in the country. The cumulative number of pediatric cases is 792,188, and children now represent 11% of all COVID-19 cases, the AAP and the CHA reported Oct. 26.
There were just over 50,000 new child cases reported in the week ending Oct. 22, which was 13.6% of the national total of almost 370,000. That’s up slightly from the 13.3% the previous week but still down from the spike seen in mid-September, based on the data collected from the websites of 49 state health departments (New York does not report ages), along with the District of Columbia, New York City, Puerto Rico, and Guam.
The state-level data show that California has had more COVID-19 cases in children (92,864) than any other state, although Texas has reported ages for only 7% of its confirmed cases. Illinois is next with 46,006 cases, followed by Florida at 45,575, although Florida is using an age range of 0-14 years to define a child case, the AAP and CHA noted.
Other measures largely put small states at the extremes:
- North Dakota has the highest cumulative rate: 2,954 cases per 100,000 children.
- Vermont has the lowest cumulative rate: 190.5 per 100,000.
- Wyoming has the highest proportion of cases in children: 27.7%.
- New Jersey has the lowest proportion of child cases: 4.6%.
There were no COVID-19–related deaths in children reported the week ending Oct. 22, so the total number remains at 120, which is just 0.06% of the total for all ages, based on data from 42 states and New York City. Hospitalization figures put admissions at almost 5,600 in children, or 1.7% of all hospitalizations, although those data come from just 24 states and New York City, the AAP and CHA said.
according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
There have been 1,052 cases of COVID-19 per 100,000 children as of Oct. 22, and that works out to 1.05% of all children in the country. The cumulative number of pediatric cases is 792,188, and children now represent 11% of all COVID-19 cases, the AAP and the CHA reported Oct. 26.
There were just over 50,000 new child cases reported in the week ending Oct. 22, which was 13.6% of the national total of almost 370,000. That’s up slightly from the 13.3% the previous week but still down from the spike seen in mid-September, based on the data collected from the websites of 49 state health departments (New York does not report ages), along with the District of Columbia, New York City, Puerto Rico, and Guam.
The state-level data show that California has had more COVID-19 cases in children (92,864) than any other state, although Texas has reported ages for only 7% of its confirmed cases. Illinois is next with 46,006 cases, followed by Florida at 45,575, although Florida is using an age range of 0-14 years to define a child case, the AAP and CHA noted.
Other measures largely put small states at the extremes:
- North Dakota has the highest cumulative rate: 2,954 cases per 100,000 children.
- Vermont has the lowest cumulative rate: 190.5 per 100,000.
- Wyoming has the highest proportion of cases in children: 27.7%.
- New Jersey has the lowest proportion of child cases: 4.6%.
There were no COVID-19–related deaths in children reported the week ending Oct. 22, so the total number remains at 120, which is just 0.06% of the total for all ages, based on data from 42 states and New York City. Hospitalization figures put admissions at almost 5,600 in children, or 1.7% of all hospitalizations, although those data come from just 24 states and New York City, the AAP and CHA said.
according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
There have been 1,052 cases of COVID-19 per 100,000 children as of Oct. 22, and that works out to 1.05% of all children in the country. The cumulative number of pediatric cases is 792,188, and children now represent 11% of all COVID-19 cases, the AAP and the CHA reported Oct. 26.
There were just over 50,000 new child cases reported in the week ending Oct. 22, which was 13.6% of the national total of almost 370,000. That’s up slightly from the 13.3% the previous week but still down from the spike seen in mid-September, based on the data collected from the websites of 49 state health departments (New York does not report ages), along with the District of Columbia, New York City, Puerto Rico, and Guam.
The state-level data show that California has had more COVID-19 cases in children (92,864) than any other state, although Texas has reported ages for only 7% of its confirmed cases. Illinois is next with 46,006 cases, followed by Florida at 45,575, although Florida is using an age range of 0-14 years to define a child case, the AAP and CHA noted.
Other measures largely put small states at the extremes:
- North Dakota has the highest cumulative rate: 2,954 cases per 100,000 children.
- Vermont has the lowest cumulative rate: 190.5 per 100,000.
- Wyoming has the highest proportion of cases in children: 27.7%.
- New Jersey has the lowest proportion of child cases: 4.6%.
There were no COVID-19–related deaths in children reported the week ending Oct. 22, so the total number remains at 120, which is just 0.06% of the total for all ages, based on data from 42 states and New York City. Hospitalization figures put admissions at almost 5,600 in children, or 1.7% of all hospitalizations, although those data come from just 24 states and New York City, the AAP and CHA said.
COVID-19: Immunity from antibodies may decline rapidly
Antibody response to the SARS-CoV-2 virus wanes over time, latest research has suggested.
An ongoing study led by Imperial College London (ICL) found that the proportion of people testing positive for COVID-19 antibodies dropped by 26.5% over a 3-month period between June and September.
The findings from a non–peer reviewed preprint suggested that infection with SARS-CoV-2 confers only limited protection against reinfection.
Professor Paul Elliott, director of the REACT-2 programme at ICL, said: “Testing positive for antibodies does not mean you are immune to COVID-19.
“It remains unclear what level of immunity antibodies provide, or for how long this immunity lasts.”
Experts said that, while the findings suggested that immunity might fade over time, the severity of illness from further infections could be reduced.
Antibody prevalence declined in all adults
Results from cross-sectional studies over the 3-month period involved 365,104 adults who self-administered a lateral flow immunoassay test.
There were 17,576 positive tests over the three rounds.
Antibody prevalence, adjusted for test characteristics and weighted to the adult population of England, declined from 6.0% to 4.4%, a reduction of 26.5% over the 3 months.
The decline was seen in all age groups. However, the lowest prevalence of a positive test, and the largest fall, was seen in those aged 75 years and older.
No change was seen in positive antibody tests in health care workers over the 3 months.
The results suggested that people who did not show symptoms of COVID-19 were more likely to lose detectable antibodies sooner than those who did show symptoms.
Prof Helen Ward, one of the lead authors of the report said that, while it was clear that the proportion of people with antibodies was falling over time, “We don’t yet know whether this will leave these people at risk of reinfection with the virus that causes COVID-19, but it is essential that everyone continues to follow guidance to reduce the risk to themselves and others.”
Results ‘weaken argument for herd immunity’
Commenting on the results to the Science Media Centre, Rowland Kao, professor of veterinary epidemiology and data science at the University of Edinburgh, warned that, if the results were correct, “any strategy that relies on ‘herd immunity’ lacks credibility.”
However, he added that, “while the decline is substantial, nevertheless substantial proportions of the population do retain some immune response, over 4 months after the peak of the epidemic”.
Eleanor Riley, professor of immunology and infectious disease, also from the University of Edinburgh, said it was too early to assume that immunity to SARS-CoV-2 did not last because “the study does not look at antibody concentrations, antibody function, or other aspects of immunity such as T-cell immunity and does not look at the trajectory of antibody levels in the same individuals over time”.
However, she said the findings did not mean that a vaccine would be ineffective because vaccines contained adjuvants that could induce durable immune responses, particularly with multiple immunizations.
“What is not clear is how quickly antibody levels would rise again if a person encounters the SARS-CoV-2 virus a second time. It is possible they will still rapidly respond, and either have a milder illness, or remain protected through immune memory,” commented Dr. Alexander Edwards, associate professor in biomedical technology at the University of Reading.
Health Minister Lord Bethell said: “Regardless of the result of an antibody test, everyone must continue to comply with government guidelines including social distancing, self-isolating, and getting a test if you have symptoms, and always remember: hands, face, space.”
This article first appeared on Medscape.com.
Antibody response to the SARS-CoV-2 virus wanes over time, latest research has suggested.
An ongoing study led by Imperial College London (ICL) found that the proportion of people testing positive for COVID-19 antibodies dropped by 26.5% over a 3-month period between June and September.
The findings from a non–peer reviewed preprint suggested that infection with SARS-CoV-2 confers only limited protection against reinfection.
Professor Paul Elliott, director of the REACT-2 programme at ICL, said: “Testing positive for antibodies does not mean you are immune to COVID-19.
“It remains unclear what level of immunity antibodies provide, or for how long this immunity lasts.”
Experts said that, while the findings suggested that immunity might fade over time, the severity of illness from further infections could be reduced.
Antibody prevalence declined in all adults
Results from cross-sectional studies over the 3-month period involved 365,104 adults who self-administered a lateral flow immunoassay test.
There were 17,576 positive tests over the three rounds.
Antibody prevalence, adjusted for test characteristics and weighted to the adult population of England, declined from 6.0% to 4.4%, a reduction of 26.5% over the 3 months.
The decline was seen in all age groups. However, the lowest prevalence of a positive test, and the largest fall, was seen in those aged 75 years and older.
No change was seen in positive antibody tests in health care workers over the 3 months.
The results suggested that people who did not show symptoms of COVID-19 were more likely to lose detectable antibodies sooner than those who did show symptoms.
Prof Helen Ward, one of the lead authors of the report said that, while it was clear that the proportion of people with antibodies was falling over time, “We don’t yet know whether this will leave these people at risk of reinfection with the virus that causes COVID-19, but it is essential that everyone continues to follow guidance to reduce the risk to themselves and others.”
Results ‘weaken argument for herd immunity’
Commenting on the results to the Science Media Centre, Rowland Kao, professor of veterinary epidemiology and data science at the University of Edinburgh, warned that, if the results were correct, “any strategy that relies on ‘herd immunity’ lacks credibility.”
However, he added that, “while the decline is substantial, nevertheless substantial proportions of the population do retain some immune response, over 4 months after the peak of the epidemic”.
Eleanor Riley, professor of immunology and infectious disease, also from the University of Edinburgh, said it was too early to assume that immunity to SARS-CoV-2 did not last because “the study does not look at antibody concentrations, antibody function, or other aspects of immunity such as T-cell immunity and does not look at the trajectory of antibody levels in the same individuals over time”.
However, she said the findings did not mean that a vaccine would be ineffective because vaccines contained adjuvants that could induce durable immune responses, particularly with multiple immunizations.
“What is not clear is how quickly antibody levels would rise again if a person encounters the SARS-CoV-2 virus a second time. It is possible they will still rapidly respond, and either have a milder illness, or remain protected through immune memory,” commented Dr. Alexander Edwards, associate professor in biomedical technology at the University of Reading.
Health Minister Lord Bethell said: “Regardless of the result of an antibody test, everyone must continue to comply with government guidelines including social distancing, self-isolating, and getting a test if you have symptoms, and always remember: hands, face, space.”
This article first appeared on Medscape.com.
Antibody response to the SARS-CoV-2 virus wanes over time, latest research has suggested.
An ongoing study led by Imperial College London (ICL) found that the proportion of people testing positive for COVID-19 antibodies dropped by 26.5% over a 3-month period between June and September.
The findings from a non–peer reviewed preprint suggested that infection with SARS-CoV-2 confers only limited protection against reinfection.
Professor Paul Elliott, director of the REACT-2 programme at ICL, said: “Testing positive for antibodies does not mean you are immune to COVID-19.
“It remains unclear what level of immunity antibodies provide, or for how long this immunity lasts.”
Experts said that, while the findings suggested that immunity might fade over time, the severity of illness from further infections could be reduced.
Antibody prevalence declined in all adults
Results from cross-sectional studies over the 3-month period involved 365,104 adults who self-administered a lateral flow immunoassay test.
There were 17,576 positive tests over the three rounds.
Antibody prevalence, adjusted for test characteristics and weighted to the adult population of England, declined from 6.0% to 4.4%, a reduction of 26.5% over the 3 months.
The decline was seen in all age groups. However, the lowest prevalence of a positive test, and the largest fall, was seen in those aged 75 years and older.
No change was seen in positive antibody tests in health care workers over the 3 months.
The results suggested that people who did not show symptoms of COVID-19 were more likely to lose detectable antibodies sooner than those who did show symptoms.
Prof Helen Ward, one of the lead authors of the report said that, while it was clear that the proportion of people with antibodies was falling over time, “We don’t yet know whether this will leave these people at risk of reinfection with the virus that causes COVID-19, but it is essential that everyone continues to follow guidance to reduce the risk to themselves and others.”
Results ‘weaken argument for herd immunity’
Commenting on the results to the Science Media Centre, Rowland Kao, professor of veterinary epidemiology and data science at the University of Edinburgh, warned that, if the results were correct, “any strategy that relies on ‘herd immunity’ lacks credibility.”
However, he added that, “while the decline is substantial, nevertheless substantial proportions of the population do retain some immune response, over 4 months after the peak of the epidemic”.
Eleanor Riley, professor of immunology and infectious disease, also from the University of Edinburgh, said it was too early to assume that immunity to SARS-CoV-2 did not last because “the study does not look at antibody concentrations, antibody function, or other aspects of immunity such as T-cell immunity and does not look at the trajectory of antibody levels in the same individuals over time”.
However, she said the findings did not mean that a vaccine would be ineffective because vaccines contained adjuvants that could induce durable immune responses, particularly with multiple immunizations.
“What is not clear is how quickly antibody levels would rise again if a person encounters the SARS-CoV-2 virus a second time. It is possible they will still rapidly respond, and either have a milder illness, or remain protected through immune memory,” commented Dr. Alexander Edwards, associate professor in biomedical technology at the University of Reading.
Health Minister Lord Bethell said: “Regardless of the result of an antibody test, everyone must continue to comply with government guidelines including social distancing, self-isolating, and getting a test if you have symptoms, and always remember: hands, face, space.”
This article first appeared on Medscape.com.
Ataluren delays disease milestones in patients with nonsense mutation DMD
(nmDMD), according to study results presented at the 2020 CNS-ICNA Conjoint Meeting, held virtually this year. Because so few patients in the study reached one of the negative pulmonary endpoints, longer follow-up will be needed to assess more conclusively the effect of ataluren on pulmonary function, said Francesco Bibbiani, MD, vice president of clinical development at PTC Therapeutics.

DMD is a rare and fatal neuromuscular disorder that causes progressive muscle weakness. Between 10% and 15% of patients with DMD have a nonsense mutation in the DMD gene. This mutation creates a premature stop codon that prevents the translation of a full-length dystrophin protein. Ataluren is designed to promote readthrough of this premature stop codon, thus enabling the production of a full-length dystrophin protein. An oral formulation of the drug has been approved in several European and South American countries.
Comparing treatment and standard of care
Study 019 was a phase 3, multicenter, open-label, long-term safety study of ataluren that enrolled international patients with nmDMD, most of whom had participated previously in a trial of ataluren. Dr. Bibbiani and colleagues conducted a post hoc analysis of Study 019 data to determine whether patients with nmDMD who received ataluren and standard of care for as long as 240 weeks had a different time to loss of ambulation and to decline of pulmonary function, compared with patients who received standard of care alone. Patients who were eligible to participate in Study 019 were male, had nmDMD, and had completed the blinded study drug treatment in a previous PTC-sponsored study. Treatment consisted of two 10-mg/kg doses and one 20-mg/kg dose of ataluren per day.
Dr. Bibbiani and colleagues used participants in the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG DNHS) as a control group. CINRG DNHS was a prospective, longitudinal study of patients with DMD who received standard of care at 20 centers worldwide from 2006 to 2016. Dr. Bibbiani and colleagues used propensity-score matching to pair participants in this study with participants in Study 019. They matched patients with respect to age at onset of first symptoms, age at initiation of corticosteroid use, duration of deflazacort use, and duration of use of other corticosteroids. These factors are established predictors of disease progression in DMD.
Patients were eligible for inclusion in the post hoc analysis if they had available data for age, loss of ambulation, and the covariates selected for matching. Of 94 Study 019 participants, 60 were eligible for propensity-score matching with participants in CINRG DNHS. Forty-five nonambulatory patients were eligible for matching in the analysis of age at the decline in pulmonary function because data for age at loss of ambulation and for the three pulmonary endpoints measured were available for them. Thus, comparable population sizes were available for each analysis.
Treatment delayed disease milestones
Kaplan–Meier analysis indicated that the median age at various disease milestones was higher among patients who received ataluren and standard of care, compared with those who received standard of care alone. The median age at loss of ambulation was 15.5 years for Study 019 participants and 13.3 years for CINRG DNHS patients. The median age at predicted forced vital capacity (FVC) of less than 60% was 18.1 years for Study 019 participants and 15.8 years for CINRG DNHS participants. The median age at predicted FVC of less than 50% was 19.1 years for Study 019 participants and 17.9 years for CINRG DNHS participants. Finally, the median age at FVC of less than 1 L was not calculable for Study 019 participants and 23.8 years for CINRG DNHS participants.
The Study 019 and CINRG DNHS study groups are sponsored by PTC Therapeutics, which developed ataluren. Dr. Bibbiani is an employee of PTC Therapeutics.
SOURCE: McDonald C, et al. CNS-ICNA 2020. Abstract PL69.
(nmDMD), according to study results presented at the 2020 CNS-ICNA Conjoint Meeting, held virtually this year. Because so few patients in the study reached one of the negative pulmonary endpoints, longer follow-up will be needed to assess more conclusively the effect of ataluren on pulmonary function, said Francesco Bibbiani, MD, vice president of clinical development at PTC Therapeutics.
DMD is a rare and fatal neuromuscular disorder that causes progressive muscle weakness. Between 10% and 15% of patients with DMD have a nonsense mutation in the DMD gene. This mutation creates a premature stop codon that prevents the translation of a full-length dystrophin protein. Ataluren is designed to promote readthrough of this premature stop codon, thus enabling the production of a full-length dystrophin protein. An oral formulation of the drug has been approved in several European and South American countries.
Comparing treatment and standard of care
Study 019 was a phase 3, multicenter, open-label, long-term safety study of ataluren that enrolled international patients with nmDMD, most of whom had participated previously in a trial of ataluren. Dr. Bibbiani and colleagues conducted a post hoc analysis of Study 019 data to determine whether patients with nmDMD who received ataluren and standard of care for as long as 240 weeks had a different time to loss of ambulation and to decline of pulmonary function, compared with patients who received standard of care alone. Patients who were eligible to participate in Study 019 were male, had nmDMD, and had completed the blinded study drug treatment in a previous PTC-sponsored study. Treatment consisted of two 10-mg/kg doses and one 20-mg/kg dose of ataluren per day.
Dr. Bibbiani and colleagues used participants in the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG DNHS) as a control group. CINRG DNHS was a prospective, longitudinal study of patients with DMD who received standard of care at 20 centers worldwide from 2006 to 2016. Dr. Bibbiani and colleagues used propensity-score matching to pair participants in this study with participants in Study 019. They matched patients with respect to age at onset of first symptoms, age at initiation of corticosteroid use, duration of deflazacort use, and duration of use of other corticosteroids. These factors are established predictors of disease progression in DMD.
Patients were eligible for inclusion in the post hoc analysis if they had available data for age, loss of ambulation, and the covariates selected for matching. Of 94 Study 019 participants, 60 were eligible for propensity-score matching with participants in CINRG DNHS. Forty-five nonambulatory patients were eligible for matching in the analysis of age at the decline in pulmonary function because data for age at loss of ambulation and for the three pulmonary endpoints measured were available for them. Thus, comparable population sizes were available for each analysis.
Treatment delayed disease milestones
Kaplan–Meier analysis indicated that the median age at various disease milestones was higher among patients who received ataluren and standard of care, compared with those who received standard of care alone. The median age at loss of ambulation was 15.5 years for Study 019 participants and 13.3 years for CINRG DNHS patients. The median age at predicted forced vital capacity (FVC) of less than 60% was 18.1 years for Study 019 participants and 15.8 years for CINRG DNHS participants. The median age at predicted FVC of less than 50% was 19.1 years for Study 019 participants and 17.9 years for CINRG DNHS participants. Finally, the median age at FVC of less than 1 L was not calculable for Study 019 participants and 23.8 years for CINRG DNHS participants.
The Study 019 and CINRG DNHS study groups are sponsored by PTC Therapeutics, which developed ataluren. Dr. Bibbiani is an employee of PTC Therapeutics.
SOURCE: McDonald C, et al. CNS-ICNA 2020. Abstract PL69.
(nmDMD), according to study results presented at the 2020 CNS-ICNA Conjoint Meeting, held virtually this year. Because so few patients in the study reached one of the negative pulmonary endpoints, longer follow-up will be needed to assess more conclusively the effect of ataluren on pulmonary function, said Francesco Bibbiani, MD, vice president of clinical development at PTC Therapeutics.
DMD is a rare and fatal neuromuscular disorder that causes progressive muscle weakness. Between 10% and 15% of patients with DMD have a nonsense mutation in the DMD gene. This mutation creates a premature stop codon that prevents the translation of a full-length dystrophin protein. Ataluren is designed to promote readthrough of this premature stop codon, thus enabling the production of a full-length dystrophin protein. An oral formulation of the drug has been approved in several European and South American countries.
Comparing treatment and standard of care
Study 019 was a phase 3, multicenter, open-label, long-term safety study of ataluren that enrolled international patients with nmDMD, most of whom had participated previously in a trial of ataluren. Dr. Bibbiani and colleagues conducted a post hoc analysis of Study 019 data to determine whether patients with nmDMD who received ataluren and standard of care for as long as 240 weeks had a different time to loss of ambulation and to decline of pulmonary function, compared with patients who received standard of care alone. Patients who were eligible to participate in Study 019 were male, had nmDMD, and had completed the blinded study drug treatment in a previous PTC-sponsored study. Treatment consisted of two 10-mg/kg doses and one 20-mg/kg dose of ataluren per day.
Dr. Bibbiani and colleagues used participants in the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG DNHS) as a control group. CINRG DNHS was a prospective, longitudinal study of patients with DMD who received standard of care at 20 centers worldwide from 2006 to 2016. Dr. Bibbiani and colleagues used propensity-score matching to pair participants in this study with participants in Study 019. They matched patients with respect to age at onset of first symptoms, age at initiation of corticosteroid use, duration of deflazacort use, and duration of use of other corticosteroids. These factors are established predictors of disease progression in DMD.
Patients were eligible for inclusion in the post hoc analysis if they had available data for age, loss of ambulation, and the covariates selected for matching. Of 94 Study 019 participants, 60 were eligible for propensity-score matching with participants in CINRG DNHS. Forty-five nonambulatory patients were eligible for matching in the analysis of age at the decline in pulmonary function because data for age at loss of ambulation and for the three pulmonary endpoints measured were available for them. Thus, comparable population sizes were available for each analysis.
Treatment delayed disease milestones
Kaplan–Meier analysis indicated that the median age at various disease milestones was higher among patients who received ataluren and standard of care, compared with those who received standard of care alone. The median age at loss of ambulation was 15.5 years for Study 019 participants and 13.3 years for CINRG DNHS patients. The median age at predicted forced vital capacity (FVC) of less than 60% was 18.1 years for Study 019 participants and 15.8 years for CINRG DNHS participants. The median age at predicted FVC of less than 50% was 19.1 years for Study 019 participants and 17.9 years for CINRG DNHS participants. Finally, the median age at FVC of less than 1 L was not calculable for Study 019 participants and 23.8 years for CINRG DNHS participants.
The Study 019 and CINRG DNHS study groups are sponsored by PTC Therapeutics, which developed ataluren. Dr. Bibbiani is an employee of PTC Therapeutics.
SOURCE: McDonald C, et al. CNS-ICNA 2020. Abstract PL69.
FROM CNS-ICNA 2020

Newer DMTs are more effective than injectable DMTs in pediatric MS
Nevertheless, all DMTs reduce children’s annualized relapse rate (ARR), according to results presented at the 2020 CNS-ICNA Conjoint Meeting, held virtually this year.
“Our study adds weight to the argument for an imminent shift in clinical practice toward the use of newer, more efficacious DMTs in the first instance,” said Omar Abdel-Mannan, MD, of Great Ormond Street Hospital in London. MRI activity continues among patients treated with DMTs, and the number of relapses is highest in the period following diagnosis. But because the effect of treatment on brain atrophy is greatest in the initial period of disease, “this time period may represent a critical therapeutic window for the use of highly effective therapies,” said Dr. Abdel-Mannan.
An examination of medical records
MS is much less prevalent among children than among adults. Compared with adults with MS, children with MS have a higher relapse rate and slower accumulation of disability. The individual response to DMTs is variable, said Dr. Abdel-Mannan. Furthermore, current standards of care for pediatric MS vary by center and are based on adult protocols.
Dr. Abdel-Mannan and colleagues conducted a retrospective study to evaluate the real-world effectiveness of the newer oral and infusion DMTs, compared with the older injectable DMTs, in children with relapsing-remitting MS. They examined data from seven tertiary pediatric neurology centers in the United Kingdom and identified patients under age 18 years with relapsing-remitting MS who were treated with DMTs between 2012 and 2018. The investigators reviewed clinical and paraclinical data retrospectively using electronic medical records. They compared patients’ ARR, new radiological activity, and Expanded Disability Status Scale score pretreatment and on treatment.
The researchers included 103 patients in their analysis. The population’s median age was 14 years. The ratio of girls to boys was approximately 3:1. Whites and other races/ethnicities accounted for approximately equal groups of patients. About one-third of patients presented with a clinically isolated syndrome (CIS) in the form of transverse myelitis or optic neuritis. Two-thirds presented with other CIS phenotypes. Almost all children had an abnormal MRI at onset.
Most patients initiated injectable DMTs
Of the 103 patients, 89 started treatment with an injectable (e.g., glatiramer or interferon) or an older DMT. Fourteen patients began treatment with a newer DMT (e.g., dimethyl fumarate, fingolimod, natalizumab, and alemtuzumab). Three of the 89 patients on an injectable DMT switched to another injectable DMT, and two of these patients later escalated to a newer DMT. Thirty-five of the 89 patients who initiated an injectable DMT were escalated immediately to a newer DMT. One of these patients later switched to another newer DMT. Two of the 14 patients who started on a newer DMT as their first drug switched to another newer DMT.
The investigators observed a reduction in ARR for all DMTs used during the study period. Nevertheless, a significant number of patients receiving injectable DMTs continued to relapse on treatment. Almost all patients receiving newer DMTs, however, had a reduction in relapses. When Dr. Abdel-Mannan and colleagues performed Kaplan–Meier survival analysis, they found that patients receiving newer DMTs had a longer time to first relapse and a longer time to switch treatment over 2 years, compared with patients receiving injectable DMTs. In addition, patients receiving newer DMTs had a longer time to develop new radiological activity, compared with patients receiving injectables. The analysis also indicated that the proportion of patients with new radiological activity was higher than the proportion who had clinical relapses and an Expanded Disability Status Scale score increase of more than 1 point over 2 years.
In all, 55 of the children receiving injectable DMTs and 18 of the patients receiving newer DMTs had side effects. The most commonly reported side effects were flulike symptoms and injection-site reactions. Five patients discontinued or switched their DMTs because of side effects. “Reassuringly, no pediatric-specific side effects were reported,” said Dr. Abdel-Mannan. The newer DMTs had similar short-term safety, tolerability, and side-effect profiles in these children as in adult patients.
The study was conducted on behalf of the UK Childhood Inflammatory Demyelination Network. Dr. Abdel-Mannan had no relevant disclosures.
SOURCE: Abdel-Mannan O et al. CNS-ICNA 2020, Abstract PL10.
Nevertheless, all DMTs reduce children’s annualized relapse rate (ARR), according to results presented at the 2020 CNS-ICNA Conjoint Meeting, held virtually this year.
“Our study adds weight to the argument for an imminent shift in clinical practice toward the use of newer, more efficacious DMTs in the first instance,” said Omar Abdel-Mannan, MD, of Great Ormond Street Hospital in London. MRI activity continues among patients treated with DMTs, and the number of relapses is highest in the period following diagnosis. But because the effect of treatment on brain atrophy is greatest in the initial period of disease, “this time period may represent a critical therapeutic window for the use of highly effective therapies,” said Dr. Abdel-Mannan.
An examination of medical records
MS is much less prevalent among children than among adults. Compared with adults with MS, children with MS have a higher relapse rate and slower accumulation of disability. The individual response to DMTs is variable, said Dr. Abdel-Mannan. Furthermore, current standards of care for pediatric MS vary by center and are based on adult protocols.
Dr. Abdel-Mannan and colleagues conducted a retrospective study to evaluate the real-world effectiveness of the newer oral and infusion DMTs, compared with the older injectable DMTs, in children with relapsing-remitting MS. They examined data from seven tertiary pediatric neurology centers in the United Kingdom and identified patients under age 18 years with relapsing-remitting MS who were treated with DMTs between 2012 and 2018. The investigators reviewed clinical and paraclinical data retrospectively using electronic medical records. They compared patients’ ARR, new radiological activity, and Expanded Disability Status Scale score pretreatment and on treatment.
The researchers included 103 patients in their analysis. The population’s median age was 14 years. The ratio of girls to boys was approximately 3:1. Whites and other races/ethnicities accounted for approximately equal groups of patients. About one-third of patients presented with a clinically isolated syndrome (CIS) in the form of transverse myelitis or optic neuritis. Two-thirds presented with other CIS phenotypes. Almost all children had an abnormal MRI at onset.
Most patients initiated injectable DMTs
Of the 103 patients, 89 started treatment with an injectable (e.g., glatiramer or interferon) or an older DMT. Fourteen patients began treatment with a newer DMT (e.g., dimethyl fumarate, fingolimod, natalizumab, and alemtuzumab). Three of the 89 patients on an injectable DMT switched to another injectable DMT, and two of these patients later escalated to a newer DMT. Thirty-five of the 89 patients who initiated an injectable DMT were escalated immediately to a newer DMT. One of these patients later switched to another newer DMT. Two of the 14 patients who started on a newer DMT as their first drug switched to another newer DMT.
The investigators observed a reduction in ARR for all DMTs used during the study period. Nevertheless, a significant number of patients receiving injectable DMTs continued to relapse on treatment. Almost all patients receiving newer DMTs, however, had a reduction in relapses. When Dr. Abdel-Mannan and colleagues performed Kaplan–Meier survival analysis, they found that patients receiving newer DMTs had a longer time to first relapse and a longer time to switch treatment over 2 years, compared with patients receiving injectable DMTs. In addition, patients receiving newer DMTs had a longer time to develop new radiological activity, compared with patients receiving injectables. The analysis also indicated that the proportion of patients with new radiological activity was higher than the proportion who had clinical relapses and an Expanded Disability Status Scale score increase of more than 1 point over 2 years.
In all, 55 of the children receiving injectable DMTs and 18 of the patients receiving newer DMTs had side effects. The most commonly reported side effects were flulike symptoms and injection-site reactions. Five patients discontinued or switched their DMTs because of side effects. “Reassuringly, no pediatric-specific side effects were reported,” said Dr. Abdel-Mannan. The newer DMTs had similar short-term safety, tolerability, and side-effect profiles in these children as in adult patients.
The study was conducted on behalf of the UK Childhood Inflammatory Demyelination Network. Dr. Abdel-Mannan had no relevant disclosures.
SOURCE: Abdel-Mannan O et al. CNS-ICNA 2020, Abstract PL10.
Nevertheless, all DMTs reduce children’s annualized relapse rate (ARR), according to results presented at the 2020 CNS-ICNA Conjoint Meeting, held virtually this year.
“Our study adds weight to the argument for an imminent shift in clinical practice toward the use of newer, more efficacious DMTs in the first instance,” said Omar Abdel-Mannan, MD, of Great Ormond Street Hospital in London. MRI activity continues among patients treated with DMTs, and the number of relapses is highest in the period following diagnosis. But because the effect of treatment on brain atrophy is greatest in the initial period of disease, “this time period may represent a critical therapeutic window for the use of highly effective therapies,” said Dr. Abdel-Mannan.
An examination of medical records
MS is much less prevalent among children than among adults. Compared with adults with MS, children with MS have a higher relapse rate and slower accumulation of disability. The individual response to DMTs is variable, said Dr. Abdel-Mannan. Furthermore, current standards of care for pediatric MS vary by center and are based on adult protocols.
Dr. Abdel-Mannan and colleagues conducted a retrospective study to evaluate the real-world effectiveness of the newer oral and infusion DMTs, compared with the older injectable DMTs, in children with relapsing-remitting MS. They examined data from seven tertiary pediatric neurology centers in the United Kingdom and identified patients under age 18 years with relapsing-remitting MS who were treated with DMTs between 2012 and 2018. The investigators reviewed clinical and paraclinical data retrospectively using electronic medical records. They compared patients’ ARR, new radiological activity, and Expanded Disability Status Scale score pretreatment and on treatment.
The researchers included 103 patients in their analysis. The population’s median age was 14 years. The ratio of girls to boys was approximately 3:1. Whites and other races/ethnicities accounted for approximately equal groups of patients. About one-third of patients presented with a clinically isolated syndrome (CIS) in the form of transverse myelitis or optic neuritis. Two-thirds presented with other CIS phenotypes. Almost all children had an abnormal MRI at onset.
Most patients initiated injectable DMTs
Of the 103 patients, 89 started treatment with an injectable (e.g., glatiramer or interferon) or an older DMT. Fourteen patients began treatment with a newer DMT (e.g., dimethyl fumarate, fingolimod, natalizumab, and alemtuzumab). Three of the 89 patients on an injectable DMT switched to another injectable DMT, and two of these patients later escalated to a newer DMT. Thirty-five of the 89 patients who initiated an injectable DMT were escalated immediately to a newer DMT. One of these patients later switched to another newer DMT. Two of the 14 patients who started on a newer DMT as their first drug switched to another newer DMT.
The investigators observed a reduction in ARR for all DMTs used during the study period. Nevertheless, a significant number of patients receiving injectable DMTs continued to relapse on treatment. Almost all patients receiving newer DMTs, however, had a reduction in relapses. When Dr. Abdel-Mannan and colleagues performed Kaplan–Meier survival analysis, they found that patients receiving newer DMTs had a longer time to first relapse and a longer time to switch treatment over 2 years, compared with patients receiving injectable DMTs. In addition, patients receiving newer DMTs had a longer time to develop new radiological activity, compared with patients receiving injectables. The analysis also indicated that the proportion of patients with new radiological activity was higher than the proportion who had clinical relapses and an Expanded Disability Status Scale score increase of more than 1 point over 2 years.
In all, 55 of the children receiving injectable DMTs and 18 of the patients receiving newer DMTs had side effects. The most commonly reported side effects were flulike symptoms and injection-site reactions. Five patients discontinued or switched their DMTs because of side effects. “Reassuringly, no pediatric-specific side effects were reported,” said Dr. Abdel-Mannan. The newer DMTs had similar short-term safety, tolerability, and side-effect profiles in these children as in adult patients.
The study was conducted on behalf of the UK Childhood Inflammatory Demyelination Network. Dr. Abdel-Mannan had no relevant disclosures.
SOURCE: Abdel-Mannan O et al. CNS-ICNA 2020, Abstract PL10.
FROM CNS-ICNA 2020
Study supports halting antiseizure medications after neonatal seizures
Maintaining antiseizure medication in infants who have had acute symptomatic neonatal seizures has been standard practice, but a prospective, observational, comparative effectiveness study calls that practice into question, providing evidence that discontinuing therapy at discharge poses no harm to children and has no effect on the development of epilepsies.

“,” said Hannah C. Glass, MDCM, MAS, of the University of California, San Francisco, Benioff Children’s Hospital, co-principal investigator, who presented results of the study at the 2020 CNS-ICNA Conjoint Meeting, held virtually this year. Renee Shellhaas, MD, MS, clinical associate professor of pediatrics at C.S. Mott Children’s Hospital, University of Michigan, was the other co-principal investigator.
“Although other, smaller studies have suggested it is safe to discontinue antiseizure medication after resolution of acute symptomatic seizures, the practice of early discontinuation has been very variable and depends largely on individual provider preference,” Dr. Glass said in an interview. “In our study, two-thirds of newborns with acute symptomatic seizures were maintained on antiseizure medication at the time of hospital discharge. Thus, a change to early medication discontinuation represents a major shift.”
The study evaluated 270 infants at nine centers enrolled in the Neonatal Seizure Registry and born from July 2015 through March 2018. Inclusion criteria were acute symptomatic seizures that occurred at up to 44 weeks postmenstrual age. In this cohort, 36% of patients had antiseizure medication discontinued after a median of 6 days; the remainder stayed on antiseizure medication after discharge at a median of 4 months.

The patients were followed for 2 years. The primary outcome was functional development measured by the Warner Initial Development Evaluation of Adaptive and Functional Skills (WIDEA-FS) assessment. The secondary outcome was epilepsy defined by International League Against Epilepsy (ILAE) criteria. Follow-up consisted of phone calls and chart reviews at 12, 18, and 24 months.
“The primary outcome, functional development, was not significantly different between those children who were maintained on antiseizure medication as compared with those who were discontinued,” Dr. Glass said.
After propensity adjustment, the discontinued ASM group had an estimated WIDEA-FS score 4 points higher on average, she said. “The confidence intervals met our a priori noninferiority limit, indicating no harm to neurodevelopment for discontinuing antiseizure medication before discharge home from the neonatal seizure admission,” Dr. Glass noted.
The study also found that 13% of all participants developed epilepsy at a median of 8 months. “There was no significant difference in the frequency or timing of epilepsy between the two groups,” she said.
“We conclude there is no clear rationale for antiseizure medication maintenance,” Dr. Glass said. “There is no benefit to neurodevelopment, it prolongs the exposure to potentially harmful antiseizure medications, it does not significantly delay the onset of epilepsy, and the earliest-onset epilepsies occur in spite of antiseizure medication.”
The Patient-Centered Outcomes Research Institute (PCORI) and Pediatric Epilepsy Research Foundation funded the study. Dr. Glass has no other financial relationships to disclose.
SOURCE: Glass HC et al. CNS-ICNA 2020. Presentation PL58.
Maintaining antiseizure medication in infants who have had acute symptomatic neonatal seizures has been standard practice, but a prospective, observational, comparative effectiveness study calls that practice into question, providing evidence that discontinuing therapy at discharge poses no harm to children and has no effect on the development of epilepsies.
“,” said Hannah C. Glass, MDCM, MAS, of the University of California, San Francisco, Benioff Children’s Hospital, co-principal investigator, who presented results of the study at the 2020 CNS-ICNA Conjoint Meeting, held virtually this year. Renee Shellhaas, MD, MS, clinical associate professor of pediatrics at C.S. Mott Children’s Hospital, University of Michigan, was the other co-principal investigator.
“Although other, smaller studies have suggested it is safe to discontinue antiseizure medication after resolution of acute symptomatic seizures, the practice of early discontinuation has been very variable and depends largely on individual provider preference,” Dr. Glass said in an interview. “In our study, two-thirds of newborns with acute symptomatic seizures were maintained on antiseizure medication at the time of hospital discharge. Thus, a change to early medication discontinuation represents a major shift.”
The study evaluated 270 infants at nine centers enrolled in the Neonatal Seizure Registry and born from July 2015 through March 2018. Inclusion criteria were acute symptomatic seizures that occurred at up to 44 weeks postmenstrual age. In this cohort, 36% of patients had antiseizure medication discontinued after a median of 6 days; the remainder stayed on antiseizure medication after discharge at a median of 4 months.
The patients were followed for 2 years. The primary outcome was functional development measured by the Warner Initial Development Evaluation of Adaptive and Functional Skills (WIDEA-FS) assessment. The secondary outcome was epilepsy defined by International League Against Epilepsy (ILAE) criteria. Follow-up consisted of phone calls and chart reviews at 12, 18, and 24 months.
“The primary outcome, functional development, was not significantly different between those children who were maintained on antiseizure medication as compared with those who were discontinued,” Dr. Glass said.
After propensity adjustment, the discontinued ASM group had an estimated WIDEA-FS score 4 points higher on average, she said. “The confidence intervals met our a priori noninferiority limit, indicating no harm to neurodevelopment for discontinuing antiseizure medication before discharge home from the neonatal seizure admission,” Dr. Glass noted.
The study also found that 13% of all participants developed epilepsy at a median of 8 months. “There was no significant difference in the frequency or timing of epilepsy between the two groups,” she said.
“We conclude there is no clear rationale for antiseizure medication maintenance,” Dr. Glass said. “There is no benefit to neurodevelopment, it prolongs the exposure to potentially harmful antiseizure medications, it does not significantly delay the onset of epilepsy, and the earliest-onset epilepsies occur in spite of antiseizure medication.”
The Patient-Centered Outcomes Research Institute (PCORI) and Pediatric Epilepsy Research Foundation funded the study. Dr. Glass has no other financial relationships to disclose.
SOURCE: Glass HC et al. CNS-ICNA 2020. Presentation PL58.
Maintaining antiseizure medication in infants who have had acute symptomatic neonatal seizures has been standard practice, but a prospective, observational, comparative effectiveness study calls that practice into question, providing evidence that discontinuing therapy at discharge poses no harm to children and has no effect on the development of epilepsies.
“,” said Hannah C. Glass, MDCM, MAS, of the University of California, San Francisco, Benioff Children’s Hospital, co-principal investigator, who presented results of the study at the 2020 CNS-ICNA Conjoint Meeting, held virtually this year. Renee Shellhaas, MD, MS, clinical associate professor of pediatrics at C.S. Mott Children’s Hospital, University of Michigan, was the other co-principal investigator.
“Although other, smaller studies have suggested it is safe to discontinue antiseizure medication after resolution of acute symptomatic seizures, the practice of early discontinuation has been very variable and depends largely on individual provider preference,” Dr. Glass said in an interview. “In our study, two-thirds of newborns with acute symptomatic seizures were maintained on antiseizure medication at the time of hospital discharge. Thus, a change to early medication discontinuation represents a major shift.”
The study evaluated 270 infants at nine centers enrolled in the Neonatal Seizure Registry and born from July 2015 through March 2018. Inclusion criteria were acute symptomatic seizures that occurred at up to 44 weeks postmenstrual age. In this cohort, 36% of patients had antiseizure medication discontinued after a median of 6 days; the remainder stayed on antiseizure medication after discharge at a median of 4 months.
The patients were followed for 2 years. The primary outcome was functional development measured by the Warner Initial Development Evaluation of Adaptive and Functional Skills (WIDEA-FS) assessment. The secondary outcome was epilepsy defined by International League Against Epilepsy (ILAE) criteria. Follow-up consisted of phone calls and chart reviews at 12, 18, and 24 months.
“The primary outcome, functional development, was not significantly different between those children who were maintained on antiseizure medication as compared with those who were discontinued,” Dr. Glass said.
After propensity adjustment, the discontinued ASM group had an estimated WIDEA-FS score 4 points higher on average, she said. “The confidence intervals met our a priori noninferiority limit, indicating no harm to neurodevelopment for discontinuing antiseizure medication before discharge home from the neonatal seizure admission,” Dr. Glass noted.
The study also found that 13% of all participants developed epilepsy at a median of 8 months. “There was no significant difference in the frequency or timing of epilepsy between the two groups,” she said.
“We conclude there is no clear rationale for antiseizure medication maintenance,” Dr. Glass said. “There is no benefit to neurodevelopment, it prolongs the exposure to potentially harmful antiseizure medications, it does not significantly delay the onset of epilepsy, and the earliest-onset epilepsies occur in spite of antiseizure medication.”
The Patient-Centered Outcomes Research Institute (PCORI) and Pediatric Epilepsy Research Foundation funded the study. Dr. Glass has no other financial relationships to disclose.
SOURCE: Glass HC et al. CNS-ICNA 2020. Presentation PL58.
FROM CNS-ICNA 2020
Valvular disease and COVID-19 are a deadly mix; don’t delay intervention
Danny Dvir, MD, has a message for physicians who have patients with severe valvular heart disease who are deferring valve replacement or repair until after the COVID-19 pandemic: Urge them not to wait.

Data from the Multicenter International Valve Disease Registry vividly demonstrate that clinical outcomes are poor in patients with uncorrected valve disease who become hospitalized with COVID-19. Indeed, the mortality rate within 30 days after hospital admission in 136 such patients enrolled in the registry from centers in Europe, North America, and Israel was 42%, Dr. Dvir reported at the Transcatheter Cardiovascular Research Therapeutics virtual annual meeting.
“That’s dramatically higher than for an age-matched population infected with COVID-19 without valvular heart disease, which is 10%-15%,” he noted at the meeting sponsored by the Cardiovascular Research Foundation.
The bright spot was that, in the small subgroup of 15 registry participants who underwent transcatheter or, much less frequently, surgical treatment of their failing valve while COVID-19 infected, 30-day mortality was far lower. In fact, it was comparable with the background rate in hospitalized COVID-19 patients without valve disease, according to Dr. Dvir, an interventional cardiologist at Shaare Zedek Medical Center, Hebrew University, Jerusalem.
He personally did several of the transcatheter aortic valve replacements.
“It’s doable. I truly believe that when you get a severe aortic stenosis patient who’s infected with the coronavirus, they get very unstable, but we can treat them. We can treat them even during the infection,” Dr. Dvir said.
The majority of patients in the registry had severe aortic stenosis. In the 42 such patients aged 80 years or more who didn’t undergo transcatheter aortic valve replacement (TAVR) or surgical valve replacement, 30-day mortality was 60%. In contrast, only one of the six patients in this advanced-age category who underwent valve replacement while infected died. Similarly, 30-day mortality was 24% among those younger than age 80 who valve remained untreated, but it dropped to 11% in those who received a prosthetic valve.
“We try our best to protect our patients through social distancing, but we have a treatment that can potentially reduce their mortality risk if they get infected later on. So I say to my patients: ‘Don’t wait at home. Do not wait! If you get infected when you have severe aortic stenosis, the clinical outcome is bad.’ But it seems reasonable that if they get infected when they’ve already been treated for their aortic stenosis or mitral regurgitation, they will do better.”
Dr. Dvir noted that, although the case numbers in the registry series were small and subject to potential bias, the data suggest this treatment approach may be lifesaving.

Session comoderator Timothy D. Henry, MD, commented that this registry study contains a great take-home point: “This is really consistent with what see in a lot of the other areas of COVID, that what we know to be best clinical care, we should do it, with or without the COVID.”
He asked Dr. Dvir about any special measures he takes while doing TAVR in this extreme setting. In the United States, for example, interventionalists are increasingly using transesophageal echocardiography to guide their procedures using conscious sedation, without intubation, noted Dr. Henry, medical director of the Carl and Edyth Lindner Center for Research at the Christ Hospital, Cincinnati.
“We try to minimize the procedure time; that’s one of the important things,” Dr. Dvir replied. “And you need to be protected during the procedure in a very cautious and meticulous way. You need many fans in the room because you sweat a lot.”
Discussant Renu Virmani, MD, president of the CVPath Institute in Gaithersburg, Md., commented: “The main thing I get from this presentation is the need for patients to be educated that if you’ve got valve disease, you’re better off getting it treated before you’ve got COVID. Obviously, try to prevent getting COVID – that’s the best thing you can do – but you can’t always control that.”
Discussant Mamas Mamas, MD, professor of cardiology at Keele University, Staffordshire, England, said deferred treatment of severe valvular heart disease during the pandemic has created a looming public health crisis in the United Kingdom.
“We’ve analyzed the U.K. management of aortic stenosis, and what we’ve found is that during the COVID pandemic there have been 2,500 fewer cases of aortic stenosis that have been treated. We’ve got 2,500 patients on the waiting list, and we’ve got to work out how we’re going to treat them. We estimate with simulations that about 300 of them are going to die before we can get them treated for their aortic stenosis,” according to Dr. Mamas.
Dr. Henry commented that deferral of valve procedures is “really challenging” for a couple of reasons: Not only are patients scared to come into the hospital because they fear getting COVID, but they don’t want to be hospitalized during the pandemic because their family can’t visit them there.
“These patients are mostly over 80 years old. No one wants to come in the hospital when the family won’t be around, especially when you’re 90 years old,” the interventional cardiologist said.
Dr. Dvir reported serving as a consultant to Medtronic, Edwards Lifesciences, Abbott, and Jena.
Danny Dvir, MD, has a message for physicians who have patients with severe valvular heart disease who are deferring valve replacement or repair until after the COVID-19 pandemic: Urge them not to wait.
Data from the Multicenter International Valve Disease Registry vividly demonstrate that clinical outcomes are poor in patients with uncorrected valve disease who become hospitalized with COVID-19. Indeed, the mortality rate within 30 days after hospital admission in 136 such patients enrolled in the registry from centers in Europe, North America, and Israel was 42%, Dr. Dvir reported at the Transcatheter Cardiovascular Research Therapeutics virtual annual meeting.
“That’s dramatically higher than for an age-matched population infected with COVID-19 without valvular heart disease, which is 10%-15%,” he noted at the meeting sponsored by the Cardiovascular Research Foundation.
The bright spot was that, in the small subgroup of 15 registry participants who underwent transcatheter or, much less frequently, surgical treatment of their failing valve while COVID-19 infected, 30-day mortality was far lower. In fact, it was comparable with the background rate in hospitalized COVID-19 patients without valve disease, according to Dr. Dvir, an interventional cardiologist at Shaare Zedek Medical Center, Hebrew University, Jerusalem.
He personally did several of the transcatheter aortic valve replacements.
“It’s doable. I truly believe that when you get a severe aortic stenosis patient who’s infected with the coronavirus, they get very unstable, but we can treat them. We can treat them even during the infection,” Dr. Dvir said.
The majority of patients in the registry had severe aortic stenosis. In the 42 such patients aged 80 years or more who didn’t undergo transcatheter aortic valve replacement (TAVR) or surgical valve replacement, 30-day mortality was 60%. In contrast, only one of the six patients in this advanced-age category who underwent valve replacement while infected died. Similarly, 30-day mortality was 24% among those younger than age 80 who valve remained untreated, but it dropped to 11% in those who received a prosthetic valve.
“We try our best to protect our patients through social distancing, but we have a treatment that can potentially reduce their mortality risk if they get infected later on. So I say to my patients: ‘Don’t wait at home. Do not wait! If you get infected when you have severe aortic stenosis, the clinical outcome is bad.’ But it seems reasonable that if they get infected when they’ve already been treated for their aortic stenosis or mitral regurgitation, they will do better.”
Dr. Dvir noted that, although the case numbers in the registry series were small and subject to potential bias, the data suggest this treatment approach may be lifesaving.
Session comoderator Timothy D. Henry, MD, commented that this registry study contains a great take-home point: “This is really consistent with what see in a lot of the other areas of COVID, that what we know to be best clinical care, we should do it, with or without the COVID.”
He asked Dr. Dvir about any special measures he takes while doing TAVR in this extreme setting. In the United States, for example, interventionalists are increasingly using transesophageal echocardiography to guide their procedures using conscious sedation, without intubation, noted Dr. Henry, medical director of the Carl and Edyth Lindner Center for Research at the Christ Hospital, Cincinnati.
“We try to minimize the procedure time; that’s one of the important things,” Dr. Dvir replied. “And you need to be protected during the procedure in a very cautious and meticulous way. You need many fans in the room because you sweat a lot.”
Discussant Renu Virmani, MD, president of the CVPath Institute in Gaithersburg, Md., commented: “The main thing I get from this presentation is the need for patients to be educated that if you’ve got valve disease, you’re better off getting it treated before you’ve got COVID. Obviously, try to prevent getting COVID – that’s the best thing you can do – but you can’t always control that.”
Discussant Mamas Mamas, MD, professor of cardiology at Keele University, Staffordshire, England, said deferred treatment of severe valvular heart disease during the pandemic has created a looming public health crisis in the United Kingdom.
“We’ve analyzed the U.K. management of aortic stenosis, and what we’ve found is that during the COVID pandemic there have been 2,500 fewer cases of aortic stenosis that have been treated. We’ve got 2,500 patients on the waiting list, and we’ve got to work out how we’re going to treat them. We estimate with simulations that about 300 of them are going to die before we can get them treated for their aortic stenosis,” according to Dr. Mamas.
Dr. Henry commented that deferral of valve procedures is “really challenging” for a couple of reasons: Not only are patients scared to come into the hospital because they fear getting COVID, but they don’t want to be hospitalized during the pandemic because their family can’t visit them there.
“These patients are mostly over 80 years old. No one wants to come in the hospital when the family won’t be around, especially when you’re 90 years old,” the interventional cardiologist said.
Dr. Dvir reported serving as a consultant to Medtronic, Edwards Lifesciences, Abbott, and Jena.
Danny Dvir, MD, has a message for physicians who have patients with severe valvular heart disease who are deferring valve replacement or repair until after the COVID-19 pandemic: Urge them not to wait.
Data from the Multicenter International Valve Disease Registry vividly demonstrate that clinical outcomes are poor in patients with uncorrected valve disease who become hospitalized with COVID-19. Indeed, the mortality rate within 30 days after hospital admission in 136 such patients enrolled in the registry from centers in Europe, North America, and Israel was 42%, Dr. Dvir reported at the Transcatheter Cardiovascular Research Therapeutics virtual annual meeting.
“That’s dramatically higher than for an age-matched population infected with COVID-19 without valvular heart disease, which is 10%-15%,” he noted at the meeting sponsored by the Cardiovascular Research Foundation.
The bright spot was that, in the small subgroup of 15 registry participants who underwent transcatheter or, much less frequently, surgical treatment of their failing valve while COVID-19 infected, 30-day mortality was far lower. In fact, it was comparable with the background rate in hospitalized COVID-19 patients without valve disease, according to Dr. Dvir, an interventional cardiologist at Shaare Zedek Medical Center, Hebrew University, Jerusalem.
He personally did several of the transcatheter aortic valve replacements.
“It’s doable. I truly believe that when you get a severe aortic stenosis patient who’s infected with the coronavirus, they get very unstable, but we can treat them. We can treat them even during the infection,” Dr. Dvir said.
The majority of patients in the registry had severe aortic stenosis. In the 42 such patients aged 80 years or more who didn’t undergo transcatheter aortic valve replacement (TAVR) or surgical valve replacement, 30-day mortality was 60%. In contrast, only one of the six patients in this advanced-age category who underwent valve replacement while infected died. Similarly, 30-day mortality was 24% among those younger than age 80 who valve remained untreated, but it dropped to 11% in those who received a prosthetic valve.
“We try our best to protect our patients through social distancing, but we have a treatment that can potentially reduce their mortality risk if they get infected later on. So I say to my patients: ‘Don’t wait at home. Do not wait! If you get infected when you have severe aortic stenosis, the clinical outcome is bad.’ But it seems reasonable that if they get infected when they’ve already been treated for their aortic stenosis or mitral regurgitation, they will do better.”
Dr. Dvir noted that, although the case numbers in the registry series were small and subject to potential bias, the data suggest this treatment approach may be lifesaving.
Session comoderator Timothy D. Henry, MD, commented that this registry study contains a great take-home point: “This is really consistent with what see in a lot of the other areas of COVID, that what we know to be best clinical care, we should do it, with or without the COVID.”
He asked Dr. Dvir about any special measures he takes while doing TAVR in this extreme setting. In the United States, for example, interventionalists are increasingly using transesophageal echocardiography to guide their procedures using conscious sedation, without intubation, noted Dr. Henry, medical director of the Carl and Edyth Lindner Center for Research at the Christ Hospital, Cincinnati.
“We try to minimize the procedure time; that’s one of the important things,” Dr. Dvir replied. “And you need to be protected during the procedure in a very cautious and meticulous way. You need many fans in the room because you sweat a lot.”
Discussant Renu Virmani, MD, president of the CVPath Institute in Gaithersburg, Md., commented: “The main thing I get from this presentation is the need for patients to be educated that if you’ve got valve disease, you’re better off getting it treated before you’ve got COVID. Obviously, try to prevent getting COVID – that’s the best thing you can do – but you can’t always control that.”
Discussant Mamas Mamas, MD, professor of cardiology at Keele University, Staffordshire, England, said deferred treatment of severe valvular heart disease during the pandemic has created a looming public health crisis in the United Kingdom.
“We’ve analyzed the U.K. management of aortic stenosis, and what we’ve found is that during the COVID pandemic there have been 2,500 fewer cases of aortic stenosis that have been treated. We’ve got 2,500 patients on the waiting list, and we’ve got to work out how we’re going to treat them. We estimate with simulations that about 300 of them are going to die before we can get them treated for their aortic stenosis,” according to Dr. Mamas.
Dr. Henry commented that deferral of valve procedures is “really challenging” for a couple of reasons: Not only are patients scared to come into the hospital because they fear getting COVID, but they don’t want to be hospitalized during the pandemic because their family can’t visit them there.
“These patients are mostly over 80 years old. No one wants to come in the hospital when the family won’t be around, especially when you’re 90 years old,” the interventional cardiologist said.
Dr. Dvir reported serving as a consultant to Medtronic, Edwards Lifesciences, Abbott, and Jena.
FROM TCT 2020
PPI use associated with all-cause and cause-specific mortality
Background: PPI use has previously been associated with an increased risk of acute kidney injury, Clostridium difficile infection, osteoporosis, dementia, and all-cause mortality. An estimation of the level of mortality risk, as well as cause-specific mortality risk, may better inform decisions about prescribing PPIs.

Study design: Longitudinal observational cohort.
Setting: Department of Veterans Affairs.
Synopsis: Using a cohort of veterans newly prescribed acid suppression therapy in 2002-2004, 157,625 new PPI users were compared with 56,842 new H2 receptor–blocker users. Over the following 10 years, a specific cause of death was determined using national death index data. In that period, 37.3% of patients died, with PPI use associated with 45.2 excess deaths per 1,000 patients (95% confidence interval, 28.2-61.4). There were significant associations with the following specific causes of death: circulatory system diseases (17.5 excess deaths per 1,000 patients, 95% CI, 5.5-28.8), neoplasms (12.9; 95% CI, 1.2-24.3), genitourinary system diseases including chronic kidney disease (6.3; 95% CI, 1.6-7.0), and infectious/parasitic diseases (4.2; 95% CI, 3.2-9.2).
Limitations include the observational study design and potential for confounding variables not accounted for by the researchers. There is also a question of broader applicability given the VA patient population. Nevertheless, this study adds to growing evidence regarding risks associated with PPI use. Clinicians should consider prescribing PPIs only for indications and durations where it is known to offer benefit in order minimize risk of adverse events.
Bottom line: PPI use is associated with an excess risk of death, particularly death caused by cardiovascular disease, malignancy, genitourinary diseases, and infection.
CITATION: Xie Y et al. Estimates of all-cause mortality and cause specific mortality associated with proton pump inhibitors among US veterans: Cohort study. BMJ. 2019 May 29;365:l1580.
Dr. Kruse is a hospitalist at Vanderbilt University Medical Center, Nashville, Tenn.
Background: PPI use has previously been associated with an increased risk of acute kidney injury, Clostridium difficile infection, osteoporosis, dementia, and all-cause mortality. An estimation of the level of mortality risk, as well as cause-specific mortality risk, may better inform decisions about prescribing PPIs.
Study design: Longitudinal observational cohort.
Setting: Department of Veterans Affairs.
Synopsis: Using a cohort of veterans newly prescribed acid suppression therapy in 2002-2004, 157,625 new PPI users were compared with 56,842 new H2 receptor–blocker users. Over the following 10 years, a specific cause of death was determined using national death index data. In that period, 37.3% of patients died, with PPI use associated with 45.2 excess deaths per 1,000 patients (95% confidence interval, 28.2-61.4). There were significant associations with the following specific causes of death: circulatory system diseases (17.5 excess deaths per 1,000 patients, 95% CI, 5.5-28.8), neoplasms (12.9; 95% CI, 1.2-24.3), genitourinary system diseases including chronic kidney disease (6.3; 95% CI, 1.6-7.0), and infectious/parasitic diseases (4.2; 95% CI, 3.2-9.2).
Limitations include the observational study design and potential for confounding variables not accounted for by the researchers. There is also a question of broader applicability given the VA patient population. Nevertheless, this study adds to growing evidence regarding risks associated with PPI use. Clinicians should consider prescribing PPIs only for indications and durations where it is known to offer benefit in order minimize risk of adverse events.
Bottom line: PPI use is associated with an excess risk of death, particularly death caused by cardiovascular disease, malignancy, genitourinary diseases, and infection.
CITATION: Xie Y et al. Estimates of all-cause mortality and cause specific mortality associated with proton pump inhibitors among US veterans: Cohort study. BMJ. 2019 May 29;365:l1580.
Dr. Kruse is a hospitalist at Vanderbilt University Medical Center, Nashville, Tenn.
Background: PPI use has previously been associated with an increased risk of acute kidney injury, Clostridium difficile infection, osteoporosis, dementia, and all-cause mortality. An estimation of the level of mortality risk, as well as cause-specific mortality risk, may better inform decisions about prescribing PPIs.
Study design: Longitudinal observational cohort.
Setting: Department of Veterans Affairs.
Synopsis: Using a cohort of veterans newly prescribed acid suppression therapy in 2002-2004, 157,625 new PPI users were compared with 56,842 new H2 receptor–blocker users. Over the following 10 years, a specific cause of death was determined using national death index data. In that period, 37.3% of patients died, with PPI use associated with 45.2 excess deaths per 1,000 patients (95% confidence interval, 28.2-61.4). There were significant associations with the following specific causes of death: circulatory system diseases (17.5 excess deaths per 1,000 patients, 95% CI, 5.5-28.8), neoplasms (12.9; 95% CI, 1.2-24.3), genitourinary system diseases including chronic kidney disease (6.3; 95% CI, 1.6-7.0), and infectious/parasitic diseases (4.2; 95% CI, 3.2-9.2).
Limitations include the observational study design and potential for confounding variables not accounted for by the researchers. There is also a question of broader applicability given the VA patient population. Nevertheless, this study adds to growing evidence regarding risks associated with PPI use. Clinicians should consider prescribing PPIs only for indications and durations where it is known to offer benefit in order minimize risk of adverse events.
Bottom line: PPI use is associated with an excess risk of death, particularly death caused by cardiovascular disease, malignancy, genitourinary diseases, and infection.
CITATION: Xie Y et al. Estimates of all-cause mortality and cause specific mortality associated with proton pump inhibitors among US veterans: Cohort study. BMJ. 2019 May 29;365:l1580.
Dr. Kruse is a hospitalist at Vanderbilt University Medical Center, Nashville, Tenn.
Now USPSTF also suggests start CRC screening at age 45
that is open for public comment.
“This is the only change that was made,” said task force member Michael Barry, MD, director of the Informed Medical Decisions Program in the Health Decision Sciences Center at Massachusetts General Hospital, Boston.
The recommendation is that all adults aged 45-75 years be screened for CRC.
This is an “A” recommendation for adults aged 50-75 and a “B” recommendation for adults aged 45-49. Dr. Barry explained that the reason for this difference is that the benefit is smaller for the 45- to 49-years age group. “But there’s not much difference between A and B from a practical standpoint,” he explained.
For adults aged 76-85, the benefits and harms of screening need to be weighed against the individual’s overall health and personal circumstances. This is a “C” recommendation.
Barry emphasized that the USPSTF document is not final. The draft recommendation and supporting evidence is posted on the task force website and will be available for public comments until Nov. 23.
Mounting pressure
The move comes after mounting evidence of an increase in CRC among younger adults and mounting pressure to lower the starting age.
Two years ago, the American Cancer Society (ACS) revised its own screening guidelines and lowered the starting age to 45 years. Soon afterward, a coalition of 22 public health and patient advocacy groups joined the ACS in submitting a letter to the USPSTF asking that the task force reconsider its 2016 guidance (which recommends starting at age 50 years).
The starting age for screening is an important issue, commented Judy Yee, MD, chair of radiology at the Albert Einstein College of Medicine and the Montefiore Health System in New York and chair of the Colon Cancer Committee of the American College of Radiology.
“Right now it is very confusing to physicians and to the public,” Dr. Yee said in an interview at that time. “The USPSTF and the ACS differ as far as the age to begin screening, and insurers may not cover the cost of colorectal cancer screening before age 50.”
Dr. Barry said that the Task Force took notice of recent data showing an increase in the incidence of CRC among younger adults. “The risk now for age 45 to 49 is pretty similar to the risk for people in their early 50s. So in some ways, today’s late 40-year-olds are like yesterday’s 50-year-olds,” he commented.
The task force used simulation models that confirmed what the epidemiologic data suggested and “that we could prevent some additional colorectal cancer deaths by starting screening at age 45,” he said.
The rest of the new draft recommendation is similar to the 2016 guidelines, in which the task force says there is convincing evidence that CRC screening substantially reduces disease-related mortality. However, it does not recommend any one screening approach over another. It recommends both direct visualization, such as colonoscopy, as well as noninvasive stool-based tests. It does not recommend serum tests, urine tests, or capsule endoscopy because there is not yet enough evidence about the benefits and harms of these tests.
“The right test is the one a patient will do,” Dr. Barry commented.
Defining populations
CRC in young adults made the news in August 2020 when Chadwick Boseman, known for his role as King T’Challa in Marvel’s “Black Panther,” died of colon cancer. Diagnosed in 2016, he was only 43 years old.
“The recent passing of Chadwick Boseman is tragic, and our thoughts are with his loved ones during this difficult time,” said Dr. Barry. “As a Black man, the data show that Chadwick was at higher risk for developing colorectal cancer.”
Unfortunately, there is currently not enough evidence that screening Black men younger than 45 could help prevent tragic deaths such as Chadwick’s, he commented. “The task force is calling for more research on colorectal cancer screening in Black adults,” he added.
Limit screening to those at higher risk
In contrast to the USPSTF and ACS guidelines, which recommend screening for CRC for everyone over a certain age, a set of recommendations developed by an international panel of experts suggests screening only for individuals who are at higher risk for CRC.
As previously reported, these guidelines suggest restricting screening to adults whose cumulative cancer risk is 3% or more in the next 15 years, the point at which the balance between benefits and harms favors screening.
The authors, led by Lise Helsingen, MD, Clinical Effectiveness Research Group, University of Oslo, said “the optimal choice for each person requires shared decision-making.”
Such a risk-based approach is “increasingly regarded as the most appropriate way to discuss cancer screening.” That approach is already used in prostate and lung cancer screening, they noted.
A version of this article originally appeared on Medscape.com.
Clinicians and researchers have actively debated the pros and cons of lowering the screening age to 45 years since 2018, when the American Cancer Society released its colorectal cancer (CRC) screening guidelines. The most compelling argument in support of lowering the screening age is that recent data from Surveillance Epidemiology and End Results (SEER) show that the CRC incidence rates in 45- to 50-year-olds are similar to rates seen in 50- to 54-year-olds about 20 years ago, when the first guidelines to initiate screening at age 50 were widely established. Termed early-onset CRC (EOCRC), the underlying reasons for this increase are not completely understood, and while the absolute numbers of EOCRC cases are smaller than in older age groups, modeling studies show that screening this age group is both efficient and effective.

Over the last 20 years we have made major strides in reducing the incidence and mortality from CRC in ages 50 years and older, and now we must rise to the challenge of delivering CRC screening to this younger group in order to see similar dividends over time and curb the rising incidence curve of EOCRC. And we must do so without direct evidence to guide us as to the magnitude of the benefit of screening this younger group, the best modality to use, or tools to risk stratify who is likely to benefit from screening in this group. We must also be careful not to worsen racial and geographic disparities in CRC screening, which already exist for African Americans, Native Americans, and other minorities and rural residents. Finally, even though the goal posts are changing, our target remains to get to 80% screening rates for all age groups, and not neglect the currently underscreened 50- to 75-year-olds, who are at a much higher risk of CRC than their younger counterparts.
Aasma Shaukat, MD, MPH, is an investigator, Center for Care Delivery and Outcomes Research, section chief and staff physician, GI section, Minneapolis VA Health Care System; staff physician, Fairview University of Minnesota Medical Center, Minneapolis; and professor, University of Minnesota department of medicine, division of gastroenterology, Minneapolis. She has no conflicts of interest.
Clinicians and researchers have actively debated the pros and cons of lowering the screening age to 45 years since 2018, when the American Cancer Society released its colorectal cancer (CRC) screening guidelines. The most compelling argument in support of lowering the screening age is that recent data from Surveillance Epidemiology and End Results (SEER) show that the CRC incidence rates in 45- to 50-year-olds are similar to rates seen in 50- to 54-year-olds about 20 years ago, when the first guidelines to initiate screening at age 50 were widely established. Termed early-onset CRC (EOCRC), the underlying reasons for this increase are not completely understood, and while the absolute numbers of EOCRC cases are smaller than in older age groups, modeling studies show that screening this age group is both efficient and effective.
Over the last 20 years we have made major strides in reducing the incidence and mortality from CRC in ages 50 years and older, and now we must rise to the challenge of delivering CRC screening to this younger group in order to see similar dividends over time and curb the rising incidence curve of EOCRC. And we must do so without direct evidence to guide us as to the magnitude of the benefit of screening this younger group, the best modality to use, or tools to risk stratify who is likely to benefit from screening in this group. We must also be careful not to worsen racial and geographic disparities in CRC screening, which already exist for African Americans, Native Americans, and other minorities and rural residents. Finally, even though the goal posts are changing, our target remains to get to 80% screening rates for all age groups, and not neglect the currently underscreened 50- to 75-year-olds, who are at a much higher risk of CRC than their younger counterparts.
Aasma Shaukat, MD, MPH, is an investigator, Center for Care Delivery and Outcomes Research, section chief and staff physician, GI section, Minneapolis VA Health Care System; staff physician, Fairview University of Minnesota Medical Center, Minneapolis; and professor, University of Minnesota department of medicine, division of gastroenterology, Minneapolis. She has no conflicts of interest.
Clinicians and researchers have actively debated the pros and cons of lowering the screening age to 45 years since 2018, when the American Cancer Society released its colorectal cancer (CRC) screening guidelines. The most compelling argument in support of lowering the screening age is that recent data from Surveillance Epidemiology and End Results (SEER) show that the CRC incidence rates in 45- to 50-year-olds are similar to rates seen in 50- to 54-year-olds about 20 years ago, when the first guidelines to initiate screening at age 50 were widely established. Termed early-onset CRC (EOCRC), the underlying reasons for this increase are not completely understood, and while the absolute numbers of EOCRC cases are smaller than in older age groups, modeling studies show that screening this age group is both efficient and effective.
Over the last 20 years we have made major strides in reducing the incidence and mortality from CRC in ages 50 years and older, and now we must rise to the challenge of delivering CRC screening to this younger group in order to see similar dividends over time and curb the rising incidence curve of EOCRC. And we must do so without direct evidence to guide us as to the magnitude of the benefit of screening this younger group, the best modality to use, or tools to risk stratify who is likely to benefit from screening in this group. We must also be careful not to worsen racial and geographic disparities in CRC screening, which already exist for African Americans, Native Americans, and other minorities and rural residents. Finally, even though the goal posts are changing, our target remains to get to 80% screening rates for all age groups, and not neglect the currently underscreened 50- to 75-year-olds, who are at a much higher risk of CRC than their younger counterparts.
Aasma Shaukat, MD, MPH, is an investigator, Center for Care Delivery and Outcomes Research, section chief and staff physician, GI section, Minneapolis VA Health Care System; staff physician, Fairview University of Minnesota Medical Center, Minneapolis; and professor, University of Minnesota department of medicine, division of gastroenterology, Minneapolis. She has no conflicts of interest.
that is open for public comment.
“This is the only change that was made,” said task force member Michael Barry, MD, director of the Informed Medical Decisions Program in the Health Decision Sciences Center at Massachusetts General Hospital, Boston.
The recommendation is that all adults aged 45-75 years be screened for CRC.
This is an “A” recommendation for adults aged 50-75 and a “B” recommendation for adults aged 45-49. Dr. Barry explained that the reason for this difference is that the benefit is smaller for the 45- to 49-years age group. “But there’s not much difference between A and B from a practical standpoint,” he explained.
For adults aged 76-85, the benefits and harms of screening need to be weighed against the individual’s overall health and personal circumstances. This is a “C” recommendation.
Barry emphasized that the USPSTF document is not final. The draft recommendation and supporting evidence is posted on the task force website and will be available for public comments until Nov. 23.
Mounting pressure
The move comes after mounting evidence of an increase in CRC among younger adults and mounting pressure to lower the starting age.
Two years ago, the American Cancer Society (ACS) revised its own screening guidelines and lowered the starting age to 45 years. Soon afterward, a coalition of 22 public health and patient advocacy groups joined the ACS in submitting a letter to the USPSTF asking that the task force reconsider its 2016 guidance (which recommends starting at age 50 years).
The starting age for screening is an important issue, commented Judy Yee, MD, chair of radiology at the Albert Einstein College of Medicine and the Montefiore Health System in New York and chair of the Colon Cancer Committee of the American College of Radiology.
“Right now it is very confusing to physicians and to the public,” Dr. Yee said in an interview at that time. “The USPSTF and the ACS differ as far as the age to begin screening, and insurers may not cover the cost of colorectal cancer screening before age 50.”
Dr. Barry said that the Task Force took notice of recent data showing an increase in the incidence of CRC among younger adults. “The risk now for age 45 to 49 is pretty similar to the risk for people in their early 50s. So in some ways, today’s late 40-year-olds are like yesterday’s 50-year-olds,” he commented.
The task force used simulation models that confirmed what the epidemiologic data suggested and “that we could prevent some additional colorectal cancer deaths by starting screening at age 45,” he said.
The rest of the new draft recommendation is similar to the 2016 guidelines, in which the task force says there is convincing evidence that CRC screening substantially reduces disease-related mortality. However, it does not recommend any one screening approach over another. It recommends both direct visualization, such as colonoscopy, as well as noninvasive stool-based tests. It does not recommend serum tests, urine tests, or capsule endoscopy because there is not yet enough evidence about the benefits and harms of these tests.
“The right test is the one a patient will do,” Dr. Barry commented.
Defining populations
CRC in young adults made the news in August 2020 when Chadwick Boseman, known for his role as King T’Challa in Marvel’s “Black Panther,” died of colon cancer. Diagnosed in 2016, he was only 43 years old.
“The recent passing of Chadwick Boseman is tragic, and our thoughts are with his loved ones during this difficult time,” said Dr. Barry. “As a Black man, the data show that Chadwick was at higher risk for developing colorectal cancer.”
Unfortunately, there is currently not enough evidence that screening Black men younger than 45 could help prevent tragic deaths such as Chadwick’s, he commented. “The task force is calling for more research on colorectal cancer screening in Black adults,” he added.
Limit screening to those at higher risk
In contrast to the USPSTF and ACS guidelines, which recommend screening for CRC for everyone over a certain age, a set of recommendations developed by an international panel of experts suggests screening only for individuals who are at higher risk for CRC.
As previously reported, these guidelines suggest restricting screening to adults whose cumulative cancer risk is 3% or more in the next 15 years, the point at which the balance between benefits and harms favors screening.
The authors, led by Lise Helsingen, MD, Clinical Effectiveness Research Group, University of Oslo, said “the optimal choice for each person requires shared decision-making.”
Such a risk-based approach is “increasingly regarded as the most appropriate way to discuss cancer screening.” That approach is already used in prostate and lung cancer screening, they noted.
A version of this article originally appeared on Medscape.com.
that is open for public comment.
“This is the only change that was made,” said task force member Michael Barry, MD, director of the Informed Medical Decisions Program in the Health Decision Sciences Center at Massachusetts General Hospital, Boston.
The recommendation is that all adults aged 45-75 years be screened for CRC.
This is an “A” recommendation for adults aged 50-75 and a “B” recommendation for adults aged 45-49. Dr. Barry explained that the reason for this difference is that the benefit is smaller for the 45- to 49-years age group. “But there’s not much difference between A and B from a practical standpoint,” he explained.
For adults aged 76-85, the benefits and harms of screening need to be weighed against the individual’s overall health and personal circumstances. This is a “C” recommendation.
Barry emphasized that the USPSTF document is not final. The draft recommendation and supporting evidence is posted on the task force website and will be available for public comments until Nov. 23.
Mounting pressure
The move comes after mounting evidence of an increase in CRC among younger adults and mounting pressure to lower the starting age.
Two years ago, the American Cancer Society (ACS) revised its own screening guidelines and lowered the starting age to 45 years. Soon afterward, a coalition of 22 public health and patient advocacy groups joined the ACS in submitting a letter to the USPSTF asking that the task force reconsider its 2016 guidance (which recommends starting at age 50 years).
The starting age for screening is an important issue, commented Judy Yee, MD, chair of radiology at the Albert Einstein College of Medicine and the Montefiore Health System in New York and chair of the Colon Cancer Committee of the American College of Radiology.
“Right now it is very confusing to physicians and to the public,” Dr. Yee said in an interview at that time. “The USPSTF and the ACS differ as far as the age to begin screening, and insurers may not cover the cost of colorectal cancer screening before age 50.”
Dr. Barry said that the Task Force took notice of recent data showing an increase in the incidence of CRC among younger adults. “The risk now for age 45 to 49 is pretty similar to the risk for people in their early 50s. So in some ways, today’s late 40-year-olds are like yesterday’s 50-year-olds,” he commented.
The task force used simulation models that confirmed what the epidemiologic data suggested and “that we could prevent some additional colorectal cancer deaths by starting screening at age 45,” he said.
The rest of the new draft recommendation is similar to the 2016 guidelines, in which the task force says there is convincing evidence that CRC screening substantially reduces disease-related mortality. However, it does not recommend any one screening approach over another. It recommends both direct visualization, such as colonoscopy, as well as noninvasive stool-based tests. It does not recommend serum tests, urine tests, or capsule endoscopy because there is not yet enough evidence about the benefits and harms of these tests.
“The right test is the one a patient will do,” Dr. Barry commented.
Defining populations
CRC in young adults made the news in August 2020 when Chadwick Boseman, known for his role as King T’Challa in Marvel’s “Black Panther,” died of colon cancer. Diagnosed in 2016, he was only 43 years old.
“The recent passing of Chadwick Boseman is tragic, and our thoughts are with his loved ones during this difficult time,” said Dr. Barry. “As a Black man, the data show that Chadwick was at higher risk for developing colorectal cancer.”
Unfortunately, there is currently not enough evidence that screening Black men younger than 45 could help prevent tragic deaths such as Chadwick’s, he commented. “The task force is calling for more research on colorectal cancer screening in Black adults,” he added.
Limit screening to those at higher risk
In contrast to the USPSTF and ACS guidelines, which recommend screening for CRC for everyone over a certain age, a set of recommendations developed by an international panel of experts suggests screening only for individuals who are at higher risk for CRC.
As previously reported, these guidelines suggest restricting screening to adults whose cumulative cancer risk is 3% or more in the next 15 years, the point at which the balance between benefits and harms favors screening.
The authors, led by Lise Helsingen, MD, Clinical Effectiveness Research Group, University of Oslo, said “the optimal choice for each person requires shared decision-making.”
Such a risk-based approach is “increasingly regarded as the most appropriate way to discuss cancer screening.” That approach is already used in prostate and lung cancer screening, they noted.
A version of this article originally appeared on Medscape.com.
Few women hospitalized for influenza have been vaccinated
Researchers analyzed data from 9,652 women ages 15-44 who were hospitalized with laboratory-confirmed influenza from October through April during the 2010-2019 influenza seasons. Data were pulled from the U.S. Influenza Hospitalization Surveillance Network (FluSurv-NET).
Of those women, 2,697 (28%) were pregnant. Median age was 28 and median gestational age was 32 weeks. Those studied included 36% who were non-Hispanic White; 29% non-Hispanic Black; and 20% Hispanic women.
Some 89% of the women, pregnant and nonpregnant, received antivirals while in the hospital but only 31% reported they had received the flu vaccine in the current season, despite guideline recommendations citing clear evidence that vaccination is safe for mother and baby.
Rachel Holstein, MPH, an epidemiology and information science fellow at the Centers for Disease Control and Prevention, who presented her team’s work as part of IDWeek 2020, explained that the mother’s vaccination can help protect the baby from flu infection for several months after birth, before the baby can be vaccinated.
She noted that pregnant women are at high risk for influenza-associated hospitalization.
“Changes in the immune system, heart, and lungs during pregnancy make pregnant women, and women up to 2 weeks post partum, more prone to severe illness from flu, including illness resulting in hospitalization,” she said in an interview
“Vaccination has been shown to reduce the risk of flu-associated acute respiratory infection in pregnant women by up to one-half,” she said. “A 2018 study showed that getting a flu shot reduced a pregnant woman’s risk of being hospitalized with flu by an average of 40%.»
FluSurv-NET data show hospitalizations were more common in the third trimester of pregnancy compared with the first and second, Holstein said. The most common underlying conditions among these women were asthma (23%) and obesity (10%), and 12% were current tobacco smokers. Overall, 5% of pregnant women with flu required ICU admission, 2% needed mechanical ventilation, and 6% developed pneumonia.
Vaccine uptake lowest in first two trimesters
Holstein said vaccine coverage was lowest among women in their first or second trimesters for all 9 seasons, and overall vaccination coverage increased significantly over time.
Uptake also differed by age. The data showed coverage was lower among women aged 15-34 years, compared with women 35 years and older (34% vs. 50%).
“It was as low as 15% among pregnant women aged 15-34 years in the 2011-12 season,” she added.
Jeanne Sheffield, MD, director of the division of maternal-fetal medicine at Johns Hopkins Medicine, Baltimore, said in an interview the low uptake of vaccine shown in this study is both familiar and frustrating.
She said education from health care providers has improved, but women are nonetheless frequently fearful. She pointed out the widespread phenomenon of vaccine hesitancy in the general population.
Coverage was 45.3% among adults in the 2018-2019 flu season, 8.2 percentage points higher than coverage during the 2017-18 season (37.1%) according to CDC estimates.
Added to that, she said, is further hesitancy when women believe vaccination could harm the unborn baby, despite “very good data that flu vaccine is safe in pregnancy, acceptable in pregnancy in all trimesters, and is optimal standard of care.”
Holstein added, “We know from past research that a range of factors – including negative attitudes and beliefs about vaccines, less knowledge about and access to vaccines, and a lack of trust in healthcare providers and vaccines – can contribute to lower vaccination rates.”
Healthcare providers play a key role in increasing flu vaccinations among pregnant women, she said.
“A provider recommendation, combined with an offer to administer a flu vaccine at the time of visit, remains one of the best ways to accomplish this,” Holstein said.
Holstein and Sheffield have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Researchers analyzed data from 9,652 women ages 15-44 who were hospitalized with laboratory-confirmed influenza from October through April during the 2010-2019 influenza seasons. Data were pulled from the U.S. Influenza Hospitalization Surveillance Network (FluSurv-NET).
Of those women, 2,697 (28%) were pregnant. Median age was 28 and median gestational age was 32 weeks. Those studied included 36% who were non-Hispanic White; 29% non-Hispanic Black; and 20% Hispanic women.
Some 89% of the women, pregnant and nonpregnant, received antivirals while in the hospital but only 31% reported they had received the flu vaccine in the current season, despite guideline recommendations citing clear evidence that vaccination is safe for mother and baby.
Rachel Holstein, MPH, an epidemiology and information science fellow at the Centers for Disease Control and Prevention, who presented her team’s work as part of IDWeek 2020, explained that the mother’s vaccination can help protect the baby from flu infection for several months after birth, before the baby can be vaccinated.
She noted that pregnant women are at high risk for influenza-associated hospitalization.
“Changes in the immune system, heart, and lungs during pregnancy make pregnant women, and women up to 2 weeks post partum, more prone to severe illness from flu, including illness resulting in hospitalization,” she said in an interview
“Vaccination has been shown to reduce the risk of flu-associated acute respiratory infection in pregnant women by up to one-half,” she said. “A 2018 study showed that getting a flu shot reduced a pregnant woman’s risk of being hospitalized with flu by an average of 40%.»
FluSurv-NET data show hospitalizations were more common in the third trimester of pregnancy compared with the first and second, Holstein said. The most common underlying conditions among these women were asthma (23%) and obesity (10%), and 12% were current tobacco smokers. Overall, 5% of pregnant women with flu required ICU admission, 2% needed mechanical ventilation, and 6% developed pneumonia.
Vaccine uptake lowest in first two trimesters
Holstein said vaccine coverage was lowest among women in their first or second trimesters for all 9 seasons, and overall vaccination coverage increased significantly over time.
Uptake also differed by age. The data showed coverage was lower among women aged 15-34 years, compared with women 35 years and older (34% vs. 50%).
“It was as low as 15% among pregnant women aged 15-34 years in the 2011-12 season,” she added.
Jeanne Sheffield, MD, director of the division of maternal-fetal medicine at Johns Hopkins Medicine, Baltimore, said in an interview the low uptake of vaccine shown in this study is both familiar and frustrating.
She said education from health care providers has improved, but women are nonetheless frequently fearful. She pointed out the widespread phenomenon of vaccine hesitancy in the general population.
Coverage was 45.3% among adults in the 2018-2019 flu season, 8.2 percentage points higher than coverage during the 2017-18 season (37.1%) according to CDC estimates.
Added to that, she said, is further hesitancy when women believe vaccination could harm the unborn baby, despite “very good data that flu vaccine is safe in pregnancy, acceptable in pregnancy in all trimesters, and is optimal standard of care.”
Holstein added, “We know from past research that a range of factors – including negative attitudes and beliefs about vaccines, less knowledge about and access to vaccines, and a lack of trust in healthcare providers and vaccines – can contribute to lower vaccination rates.”
Healthcare providers play a key role in increasing flu vaccinations among pregnant women, she said.
“A provider recommendation, combined with an offer to administer a flu vaccine at the time of visit, remains one of the best ways to accomplish this,” Holstein said.
Holstein and Sheffield have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Researchers analyzed data from 9,652 women ages 15-44 who were hospitalized with laboratory-confirmed influenza from October through April during the 2010-2019 influenza seasons. Data were pulled from the U.S. Influenza Hospitalization Surveillance Network (FluSurv-NET).
Of those women, 2,697 (28%) were pregnant. Median age was 28 and median gestational age was 32 weeks. Those studied included 36% who were non-Hispanic White; 29% non-Hispanic Black; and 20% Hispanic women.
Some 89% of the women, pregnant and nonpregnant, received antivirals while in the hospital but only 31% reported they had received the flu vaccine in the current season, despite guideline recommendations citing clear evidence that vaccination is safe for mother and baby.
Rachel Holstein, MPH, an epidemiology and information science fellow at the Centers for Disease Control and Prevention, who presented her team’s work as part of IDWeek 2020, explained that the mother’s vaccination can help protect the baby from flu infection for several months after birth, before the baby can be vaccinated.
She noted that pregnant women are at high risk for influenza-associated hospitalization.
“Changes in the immune system, heart, and lungs during pregnancy make pregnant women, and women up to 2 weeks post partum, more prone to severe illness from flu, including illness resulting in hospitalization,” she said in an interview
“Vaccination has been shown to reduce the risk of flu-associated acute respiratory infection in pregnant women by up to one-half,” she said. “A 2018 study showed that getting a flu shot reduced a pregnant woman’s risk of being hospitalized with flu by an average of 40%.»
FluSurv-NET data show hospitalizations were more common in the third trimester of pregnancy compared with the first and second, Holstein said. The most common underlying conditions among these women were asthma (23%) and obesity (10%), and 12% were current tobacco smokers. Overall, 5% of pregnant women with flu required ICU admission, 2% needed mechanical ventilation, and 6% developed pneumonia.
Vaccine uptake lowest in first two trimesters
Holstein said vaccine coverage was lowest among women in their first or second trimesters for all 9 seasons, and overall vaccination coverage increased significantly over time.
Uptake also differed by age. The data showed coverage was lower among women aged 15-34 years, compared with women 35 years and older (34% vs. 50%).
“It was as low as 15% among pregnant women aged 15-34 years in the 2011-12 season,” she added.
Jeanne Sheffield, MD, director of the division of maternal-fetal medicine at Johns Hopkins Medicine, Baltimore, said in an interview the low uptake of vaccine shown in this study is both familiar and frustrating.
She said education from health care providers has improved, but women are nonetheless frequently fearful. She pointed out the widespread phenomenon of vaccine hesitancy in the general population.
Coverage was 45.3% among adults in the 2018-2019 flu season, 8.2 percentage points higher than coverage during the 2017-18 season (37.1%) according to CDC estimates.
Added to that, she said, is further hesitancy when women believe vaccination could harm the unborn baby, despite “very good data that flu vaccine is safe in pregnancy, acceptable in pregnancy in all trimesters, and is optimal standard of care.”
Holstein added, “We know from past research that a range of factors – including negative attitudes and beliefs about vaccines, less knowledge about and access to vaccines, and a lack of trust in healthcare providers and vaccines – can contribute to lower vaccination rates.”
Healthcare providers play a key role in increasing flu vaccinations among pregnant women, she said.
“A provider recommendation, combined with an offer to administer a flu vaccine at the time of visit, remains one of the best ways to accomplish this,” Holstein said.
Holstein and Sheffield have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Acute HIV cases double in ED. Is COVID-19 responsible?
David Pitrak, MD, an infectious diseases specialist at the University of Chicago Medicine, and colleagues found that the incidence ratio of acute HIV infection (AHI) jumped to 14.4 this year, compared with the 6.8 average for the previous 4 years (IR, 2.14; 95% confidence interval, 1.01-4.54; P < .05).
At a press conference at IDWeek 2020, he said that this year, acute patients made up one quarter of all new diagnoses (9 of 35), “the highest percentage we have ever seen.
“Patients with acute infection, especially those with symptoms, have extremely high viral loads and progress more rapidly. Because of those high viral loads, there’s risk of transmission to others, so rapid linkage to care and ART [antiretroviral treatment] is really important,” he said.
After the IDWeek abstract was submitted in September, Dr. Pitrak said, three additional AHI cases were diagnosed in the ED, bringing the IR of AHI during the pandemic to 2.57 (95% CI, 1.29-5.11).
Should all EDs link HIV screening to COVID-19 testing?
The ED at UCM incorporated blood draws for HIV screening as part of COVID-19 evaluations early on during the pandemic, and they recommend that practice for EDs across the nation.
After a positive test result, the ID team was able to quickly link the HIV patients to care and initiation of antiretroviral treatment without adding staff or resources, Dr. Pitrak said in an interview.
Dr. Pitrak and colleagues reviewed data from 13 health care centers on the south and west sides of Chicago. At most of the centers, fourth- and fifth-generation antibody tests were available. The investigators found that the number of HIV screens that were conducted dropped significantly during the COVID-19 pandemic.
At the height of the pandemic, HIV screening at the sites decreased an average of 58%, the researchers found. As of the end of June, the number was decreased by 32%.
“This is a global problem,” he said. “HIV services have been severely impacted worldwide, with the greatest impact on the LGBTQ community.”
UCM performed 19,111 HIV screens (11,133 in the ED) between Jan. 1 and Aug. 17 this year. It performed 14,754 COVID polymerase chain reaction tests in the ED between March 17 and Aug. 17. All of the acute cases were identified in the ED.
Dr. Pitrak mentioned some possible causes of an increase in the number of patients with acute cases who present in the ED. People who do not suspect they have AHI may be coming to the ED because they think they have COVID-19, inasmuch as many of the symptoms overlap. One of the AHI patients actually did have a coinfection, Dr. Pitrak noted.
“There is also the possibility that this could be bad news,” Dr. Pitrak said in an interview. “It could be that there are more acute cases presenting because there are more community transmissions.”
He noted that follow-up visits have been canceled or converted to telehealth visits during the pandemic, and the number of patients who are initiating pre-exposure prophylaxis has declined significantly.
“I hope we’re not seeing an increase in new transmissions after so much work has been done to decrease transmissions over the past few years,” he said.
Partnership with emergency physicians
Critical to screening these patients is building a solid partnership between ID and ED physicians.
Coauthor Kimberly Stanford, MD, MPH, an assistant professor in emergency medicine at UCM, said, “You need a champion within the emergency department who can help make sure that the work flow is not disrupted, that however you implement your screening program, you’re not putting extra work on the staff.
“We can feel extremely confident that if I send a test and it comes back positive, I know someone is going to call that patient and make sure they get into care.”
Although the testing is performed in the ED at UCM, the follow-up, linkage to care, and initiation of treatment are conducted by the ID specialists.
Beverly E. Sha, MD, professor in the division of infectious diseases, department of internal medicine, Rush Medical College, Chicago, said in an interview that although she agrees that HIV screening programs in EDs “make absolute sense,” there are different ways to conduct such programs. Dr. Sha was not involved in Dr. Pitrak’s study.
At Rush’s ED, she says, HIV testing is linked with a complete blood count.
“If someone presents with fever, we would often be doing that test as well,” she said. “I think just globally increasing screening [in the ED] is what makes the most sense.”
Dr. Sha said they have not seen a similar surge in acute cases in the ED at Rush during the pandemic.
She noted, however, that UCM tested more than 11,000 people for HIV in the ED this year, whereas “we probably only did about 3500.
“The reason testing is so important, whether for HIV or COVID, is the more you test, the more you’re going to find,” she said, “especially in cities like Chicago.”
Dr. Pitrak received grant support from Gilead Sciences. His coauthors and Dr. Sha reported no relevant financial relationships.
This article first appeared on Medscape.com.
David Pitrak, MD, an infectious diseases specialist at the University of Chicago Medicine, and colleagues found that the incidence ratio of acute HIV infection (AHI) jumped to 14.4 this year, compared with the 6.8 average for the previous 4 years (IR, 2.14; 95% confidence interval, 1.01-4.54; P < .05).
At a press conference at IDWeek 2020, he said that this year, acute patients made up one quarter of all new diagnoses (9 of 35), “the highest percentage we have ever seen.
“Patients with acute infection, especially those with symptoms, have extremely high viral loads and progress more rapidly. Because of those high viral loads, there’s risk of transmission to others, so rapid linkage to care and ART [antiretroviral treatment] is really important,” he said.
After the IDWeek abstract was submitted in September, Dr. Pitrak said, three additional AHI cases were diagnosed in the ED, bringing the IR of AHI during the pandemic to 2.57 (95% CI, 1.29-5.11).
Should all EDs link HIV screening to COVID-19 testing?
The ED at UCM incorporated blood draws for HIV screening as part of COVID-19 evaluations early on during the pandemic, and they recommend that practice for EDs across the nation.
After a positive test result, the ID team was able to quickly link the HIV patients to care and initiation of antiretroviral treatment without adding staff or resources, Dr. Pitrak said in an interview.
Dr. Pitrak and colleagues reviewed data from 13 health care centers on the south and west sides of Chicago. At most of the centers, fourth- and fifth-generation antibody tests were available. The investigators found that the number of HIV screens that were conducted dropped significantly during the COVID-19 pandemic.
At the height of the pandemic, HIV screening at the sites decreased an average of 58%, the researchers found. As of the end of June, the number was decreased by 32%.
“This is a global problem,” he said. “HIV services have been severely impacted worldwide, with the greatest impact on the LGBTQ community.”
UCM performed 19,111 HIV screens (11,133 in the ED) between Jan. 1 and Aug. 17 this year. It performed 14,754 COVID polymerase chain reaction tests in the ED between March 17 and Aug. 17. All of the acute cases were identified in the ED.
Dr. Pitrak mentioned some possible causes of an increase in the number of patients with acute cases who present in the ED. People who do not suspect they have AHI may be coming to the ED because they think they have COVID-19, inasmuch as many of the symptoms overlap. One of the AHI patients actually did have a coinfection, Dr. Pitrak noted.
“There is also the possibility that this could be bad news,” Dr. Pitrak said in an interview. “It could be that there are more acute cases presenting because there are more community transmissions.”
He noted that follow-up visits have been canceled or converted to telehealth visits during the pandemic, and the number of patients who are initiating pre-exposure prophylaxis has declined significantly.
“I hope we’re not seeing an increase in new transmissions after so much work has been done to decrease transmissions over the past few years,” he said.
Partnership with emergency physicians
Critical to screening these patients is building a solid partnership between ID and ED physicians.
Coauthor Kimberly Stanford, MD, MPH, an assistant professor in emergency medicine at UCM, said, “You need a champion within the emergency department who can help make sure that the work flow is not disrupted, that however you implement your screening program, you’re not putting extra work on the staff.
“We can feel extremely confident that if I send a test and it comes back positive, I know someone is going to call that patient and make sure they get into care.”
Although the testing is performed in the ED at UCM, the follow-up, linkage to care, and initiation of treatment are conducted by the ID specialists.
Beverly E. Sha, MD, professor in the division of infectious diseases, department of internal medicine, Rush Medical College, Chicago, said in an interview that although she agrees that HIV screening programs in EDs “make absolute sense,” there are different ways to conduct such programs. Dr. Sha was not involved in Dr. Pitrak’s study.
At Rush’s ED, she says, HIV testing is linked with a complete blood count.
“If someone presents with fever, we would often be doing that test as well,” she said. “I think just globally increasing screening [in the ED] is what makes the most sense.”
Dr. Sha said they have not seen a similar surge in acute cases in the ED at Rush during the pandemic.
She noted, however, that UCM tested more than 11,000 people for HIV in the ED this year, whereas “we probably only did about 3500.
“The reason testing is so important, whether for HIV or COVID, is the more you test, the more you’re going to find,” she said, “especially in cities like Chicago.”
Dr. Pitrak received grant support from Gilead Sciences. His coauthors and Dr. Sha reported no relevant financial relationships.
This article first appeared on Medscape.com.
David Pitrak, MD, an infectious diseases specialist at the University of Chicago Medicine, and colleagues found that the incidence ratio of acute HIV infection (AHI) jumped to 14.4 this year, compared with the 6.8 average for the previous 4 years (IR, 2.14; 95% confidence interval, 1.01-4.54; P < .05).
At a press conference at IDWeek 2020, he said that this year, acute patients made up one quarter of all new diagnoses (9 of 35), “the highest percentage we have ever seen.
“Patients with acute infection, especially those with symptoms, have extremely high viral loads and progress more rapidly. Because of those high viral loads, there’s risk of transmission to others, so rapid linkage to care and ART [antiretroviral treatment] is really important,” he said.
After the IDWeek abstract was submitted in September, Dr. Pitrak said, three additional AHI cases were diagnosed in the ED, bringing the IR of AHI during the pandemic to 2.57 (95% CI, 1.29-5.11).
Should all EDs link HIV screening to COVID-19 testing?
The ED at UCM incorporated blood draws for HIV screening as part of COVID-19 evaluations early on during the pandemic, and they recommend that practice for EDs across the nation.
After a positive test result, the ID team was able to quickly link the HIV patients to care and initiation of antiretroviral treatment without adding staff or resources, Dr. Pitrak said in an interview.
Dr. Pitrak and colleagues reviewed data from 13 health care centers on the south and west sides of Chicago. At most of the centers, fourth- and fifth-generation antibody tests were available. The investigators found that the number of HIV screens that were conducted dropped significantly during the COVID-19 pandemic.
At the height of the pandemic, HIV screening at the sites decreased an average of 58%, the researchers found. As of the end of June, the number was decreased by 32%.
“This is a global problem,” he said. “HIV services have been severely impacted worldwide, with the greatest impact on the LGBTQ community.”
UCM performed 19,111 HIV screens (11,133 in the ED) between Jan. 1 and Aug. 17 this year. It performed 14,754 COVID polymerase chain reaction tests in the ED between March 17 and Aug. 17. All of the acute cases were identified in the ED.
Dr. Pitrak mentioned some possible causes of an increase in the number of patients with acute cases who present in the ED. People who do not suspect they have AHI may be coming to the ED because they think they have COVID-19, inasmuch as many of the symptoms overlap. One of the AHI patients actually did have a coinfection, Dr. Pitrak noted.
“There is also the possibility that this could be bad news,” Dr. Pitrak said in an interview. “It could be that there are more acute cases presenting because there are more community transmissions.”
He noted that follow-up visits have been canceled or converted to telehealth visits during the pandemic, and the number of patients who are initiating pre-exposure prophylaxis has declined significantly.
“I hope we’re not seeing an increase in new transmissions after so much work has been done to decrease transmissions over the past few years,” he said.
Partnership with emergency physicians
Critical to screening these patients is building a solid partnership between ID and ED physicians.
Coauthor Kimberly Stanford, MD, MPH, an assistant professor in emergency medicine at UCM, said, “You need a champion within the emergency department who can help make sure that the work flow is not disrupted, that however you implement your screening program, you’re not putting extra work on the staff.
“We can feel extremely confident that if I send a test and it comes back positive, I know someone is going to call that patient and make sure they get into care.”
Although the testing is performed in the ED at UCM, the follow-up, linkage to care, and initiation of treatment are conducted by the ID specialists.
Beverly E. Sha, MD, professor in the division of infectious diseases, department of internal medicine, Rush Medical College, Chicago, said in an interview that although she agrees that HIV screening programs in EDs “make absolute sense,” there are different ways to conduct such programs. Dr. Sha was not involved in Dr. Pitrak’s study.
At Rush’s ED, she says, HIV testing is linked with a complete blood count.
“If someone presents with fever, we would often be doing that test as well,” she said. “I think just globally increasing screening [in the ED] is what makes the most sense.”
Dr. Sha said they have not seen a similar surge in acute cases in the ED at Rush during the pandemic.
She noted, however, that UCM tested more than 11,000 people for HIV in the ED this year, whereas “we probably only did about 3500.
“The reason testing is so important, whether for HIV or COVID, is the more you test, the more you’re going to find,” she said, “especially in cities like Chicago.”
Dr. Pitrak received grant support from Gilead Sciences. His coauthors and Dr. Sha reported no relevant financial relationships.
This article first appeared on Medscape.com.