User login
Disaster response, practice operations, transplant, women's health
Disaster Response and Global Health
Epigenetics and Disasters
The configuration of the DNA bordering a gene dictates under what conditions a gene is expressed. Random errors or mutations affecting the neighboring DNA or the gene itself can affect how the gene functions. Epigenetics is an emerging field of science looking at environmental and psychosocial factors that do not directly cause mutations but still affect how genes are expressed with implications for the development and inheritance of disease. These external influences are thought to affect why some segments of DNA become accessible for protein production while other segments may not.
Disasters represent stressors with potential for epigenetic impact. Women who were pregnant during the 1998 Quebec ice storm were found to have a correlation between maternal objective stress and a distinctive pattern of DNA methylation in their children 13 years later (Cao-Lei L, et al. PLoS ONE. 2014;9[9] e10765). Methylation is known to affect the activity of a DNA segment and how genes are expressed. Associations have also been found between the severity of hurricanes and the prevalence of autism in the offspring of pregnant women experiencing these disasters (Kinney DK, et al. J Autism Dev Disord. 2008;38:481).
Anthropogenic hazards may also affect the offspring of survivors as suggested by studies of civil war POWs and Dutch Hunger Winter during WW II (Costa, DL, et al. Proc Nat Acad Sci 2018;. 115:44; Heijmans BT et al. Proc Nat Acad Sci. 2008;105[44]: 17046-9).
Epigenetics represents an area for additional research as natural and man-made disasters increase.
Omesh Toolsie, MBBS
Steering Committee Fellow-in-Training
Practice Operations
Medicare Competitive Bidding Process Update
Medicare’s Competitive Bidding Program (CBP), mandated since 2003, asks providers of specific durable medical equipment (including oxygen) to submit competing proposals for services. The best offer is then awarded a 3-year contract. Recently, several reforms to CBP have been proposed. The payment structure has changed to “lead-item pricing,” where a single bid in each category is selected and payment amounts for each product are then calculated based on pricing ratios and fee schedules (CMS DMEPOS Competitive Bidding).

This is in contrast to the prior method of median pricing, which caused financial difficulty and access concerns (Council for Quality Respiratory Care. The Rationale for Reforming Medicare Home Respiratory Therapy Payment Methodology. 2018). Budget neutrality requirements should relax, and oxygen payment structures improve. These proposed changes also include improved coverage of liquid oxygen and addition of home ventilator supplies.

However, effective January 1, 2019, all CBP is suspended through CMS. During the anticipated 2-year gap, any Medicare-enrolled supplier will be able to provide items until new contracts are awarded. Pricing during the gap period is based on a current single price plus consumer price index. These changes will impact CHEST members and their patients moving forward. During the temporary gap period, some areas are seeing decreased accessibility of some DME due to demand. Once reinstated, the changes to the oxygen payment structure should improve access and reduce out-of-pocket costs. The Practice Operations NetWork will continue to provide updates on this topic as they become available.
Timothy Dempsey, MD, MPH
Steering Committee Fellow-in-Training
Megan Sisk, DO
Steering Committee Member
Transplant
Medicare Part D Plans Can Deny Coverage of Select Immunosuppressant Medications in Solid Organ Transplant Recipients
An alarming problem has emerged with some solid organ transplant recipients experiencing immunosuppressant medication claim denials by Medicare Part D plans. Affected patients are those who convert from some other insurance (ie, private insurance or state Medicaid) to Medicare after their transplant and, therefore, rely on Medicare Part D for immunosuppressant drug coverage.
Insurance companies who offer Medicare Part D plans must follow the rules described in the Medicare Prescription Drug Benefit Manual.1 Although the Manual mandates that all immunosuppressant medications are on plan formularies, Part D plans are only required to cover immunosuppressant medications when used for indications approved by the Food and Drug Administration (FDA) or for off-label indications supported by the Centers for Medicare & Medicaid Services (CMS)-approved compendia (Drugdex® and AHFS Drug Information®).

A recent study examining the extent of the problem demonstrated non-renal organ transplant recipients are frequently prescribed and maintained on at least one medication vulnerable to Medicare Part D claim denials at 1 year posttransplant (lung: 71.1%; intestine: 39.7%; pancreas: 36.8%; liver: 19.7%; heart: 18.5%).2 Lung transplant recipients are most vulnerable since no immunosuppressant is FDA-approved for use in lung transplantation, and CMS-approved compendia only support off-label use for tacrolimus and cyclosporine in this population. Therefore, mycophenolate mofetil, mycophenolic acid, azathioprine, everolimus, and sirolimus are vulnerable to denial by Medicare Part D plans when used in lung transplant recipients. Over 95% of lung transplant recipients are maintained on an anti-metabolite, with the majority (88%) maintained on mycophenolate, so this is frequently impacted.2,3 While the transplant community is aware of this issue and has begun work to correct it, it has yet to be solved.2,4 In the meantime, if transplant recipients have been denied for this off-label and off-compendia reason, and appeals of those decisions have also been denied, options for obtaining the denied immunosuppressant medication include discount programs, foundation/grant funding, and industry-sponsored assistance programs.
Jennifer K. McDermott, PharmD
NetWork Member
1. Prescription Drug Benefit Manual. Centers for Medicare & Medicaid Services. Chapter 6: Part D Drugs and Formulary Requirements. Available at: https://www.cms.gov/Medicare/Prescription-Drug-Coverage/PrescriptionDrugCovContra/Downloads/Part-D-Benefits-Manual-Chapter-6.pdf
2. Potter LM et al. Transplant recipients are vulnerable to coverage denial under Medicare Part D. Am J Transplant. 2018;18:1502.
3. Valapour M et al. OPTN/SRTR 2016 Annual Data Report: Lung. Am J Transplant. 2018;18 (Suppl 1): 363.
4. Immunusuppressant Drug Coverage Under Medicare Part D Benefit. American Society of Transplantation. Available at: www.myast.org/public-policy/key-position-statements/immunosuppressant-drug-coverage-under-medicare-part-d-benefit.
Women’s Health
Cannabis Use Affects Women Differently
As we enter an era of legalization, cannabis use is increasingly prevalent. Variances in the risks for women and men have been observed. For most age groups, men have higher rates of use or dependence on illicit drugs than women. However, women are equally likely as men to progress to a substance use disorder. Women may be more susceptible to craving and relapse , which are key phases of the addiction cycle. A study on use among adolescents concluded there was preliminary evidence of a faster transition from initiation of marijuana use to regular use in women, when compared with men (Schepis, et al. J Addict Med. 2011;5[1]:65).

Research studies suggest that marijuana impairs spatial memory in women more so than in men. Studies have suggested that teenage girls who use marijuana may have a higher risk of brain structural abnormalities associated with regular marijuana exposure than teenage boys (Tapert, et al. Addict Biol. 2009;14[4]:457).
A study published in Psychoneuroendocrinology showed that cannabinoid receptor binding site densities exhibit sex differences and can be modulated by estradiol in several limbic brain regions. These findings may account for the sex differences observed with respect to the effects of cannabinoids (Riebe, et al. Psychoneuroendocrinology. 2010;35[8]:1265).
Further research is needed to expand our understanding of the interactions between cannabinoids and sex steroids. Detoxification treatments tailored toward women and men with cannabis addiction show a promising future and necessitate further research.
Anita Rajagopal, MD
Steering Committee Member
Disaster Response and Global Health
Epigenetics and Disasters
The configuration of the DNA bordering a gene dictates under what conditions a gene is expressed. Random errors or mutations affecting the neighboring DNA or the gene itself can affect how the gene functions. Epigenetics is an emerging field of science looking at environmental and psychosocial factors that do not directly cause mutations but still affect how genes are expressed with implications for the development and inheritance of disease. These external influences are thought to affect why some segments of DNA become accessible for protein production while other segments may not.
Disasters represent stressors with potential for epigenetic impact. Women who were pregnant during the 1998 Quebec ice storm were found to have a correlation between maternal objective stress and a distinctive pattern of DNA methylation in their children 13 years later (Cao-Lei L, et al. PLoS ONE. 2014;9[9] e10765). Methylation is known to affect the activity of a DNA segment and how genes are expressed. Associations have also been found between the severity of hurricanes and the prevalence of autism in the offspring of pregnant women experiencing these disasters (Kinney DK, et al. J Autism Dev Disord. 2008;38:481).
Anthropogenic hazards may also affect the offspring of survivors as suggested by studies of civil war POWs and Dutch Hunger Winter during WW II (Costa, DL, et al. Proc Nat Acad Sci 2018;. 115:44; Heijmans BT et al. Proc Nat Acad Sci. 2008;105[44]: 17046-9).
Epigenetics represents an area for additional research as natural and man-made disasters increase.
Omesh Toolsie, MBBS
Steering Committee Fellow-in-Training
Practice Operations
Medicare Competitive Bidding Process Update
Medicare’s Competitive Bidding Program (CBP), mandated since 2003, asks providers of specific durable medical equipment (including oxygen) to submit competing proposals for services. The best offer is then awarded a 3-year contract. Recently, several reforms to CBP have been proposed. The payment structure has changed to “lead-item pricing,” where a single bid in each category is selected and payment amounts for each product are then calculated based on pricing ratios and fee schedules (CMS DMEPOS Competitive Bidding).
This is in contrast to the prior method of median pricing, which caused financial difficulty and access concerns (Council for Quality Respiratory Care. The Rationale for Reforming Medicare Home Respiratory Therapy Payment Methodology. 2018). Budget neutrality requirements should relax, and oxygen payment structures improve. These proposed changes also include improved coverage of liquid oxygen and addition of home ventilator supplies.
However, effective January 1, 2019, all CBP is suspended through CMS. During the anticipated 2-year gap, any Medicare-enrolled supplier will be able to provide items until new contracts are awarded. Pricing during the gap period is based on a current single price plus consumer price index. These changes will impact CHEST members and their patients moving forward. During the temporary gap period, some areas are seeing decreased accessibility of some DME due to demand. Once reinstated, the changes to the oxygen payment structure should improve access and reduce out-of-pocket costs. The Practice Operations NetWork will continue to provide updates on this topic as they become available.
Timothy Dempsey, MD, MPH
Steering Committee Fellow-in-Training
Megan Sisk, DO
Steering Committee Member
Transplant
Medicare Part D Plans Can Deny Coverage of Select Immunosuppressant Medications in Solid Organ Transplant Recipients
An alarming problem has emerged with some solid organ transplant recipients experiencing immunosuppressant medication claim denials by Medicare Part D plans. Affected patients are those who convert from some other insurance (ie, private insurance or state Medicaid) to Medicare after their transplant and, therefore, rely on Medicare Part D for immunosuppressant drug coverage.
Insurance companies who offer Medicare Part D plans must follow the rules described in the Medicare Prescription Drug Benefit Manual.1 Although the Manual mandates that all immunosuppressant medications are on plan formularies, Part D plans are only required to cover immunosuppressant medications when used for indications approved by the Food and Drug Administration (FDA) or for off-label indications supported by the Centers for Medicare & Medicaid Services (CMS)-approved compendia (Drugdex® and AHFS Drug Information®).
A recent study examining the extent of the problem demonstrated non-renal organ transplant recipients are frequently prescribed and maintained on at least one medication vulnerable to Medicare Part D claim denials at 1 year posttransplant (lung: 71.1%; intestine: 39.7%; pancreas: 36.8%; liver: 19.7%; heart: 18.5%).2 Lung transplant recipients are most vulnerable since no immunosuppressant is FDA-approved for use in lung transplantation, and CMS-approved compendia only support off-label use for tacrolimus and cyclosporine in this population. Therefore, mycophenolate mofetil, mycophenolic acid, azathioprine, everolimus, and sirolimus are vulnerable to denial by Medicare Part D plans when used in lung transplant recipients. Over 95% of lung transplant recipients are maintained on an anti-metabolite, with the majority (88%) maintained on mycophenolate, so this is frequently impacted.2,3 While the transplant community is aware of this issue and has begun work to correct it, it has yet to be solved.2,4 In the meantime, if transplant recipients have been denied for this off-label and off-compendia reason, and appeals of those decisions have also been denied, options for obtaining the denied immunosuppressant medication include discount programs, foundation/grant funding, and industry-sponsored assistance programs.
Jennifer K. McDermott, PharmD
NetWork Member
1. Prescription Drug Benefit Manual. Centers for Medicare & Medicaid Services. Chapter 6: Part D Drugs and Formulary Requirements. Available at: https://www.cms.gov/Medicare/Prescription-Drug-Coverage/PrescriptionDrugCovContra/Downloads/Part-D-Benefits-Manual-Chapter-6.pdf
2. Potter LM et al. Transplant recipients are vulnerable to coverage denial under Medicare Part D. Am J Transplant. 2018;18:1502.
3. Valapour M et al. OPTN/SRTR 2016 Annual Data Report: Lung. Am J Transplant. 2018;18 (Suppl 1): 363.
4. Immunusuppressant Drug Coverage Under Medicare Part D Benefit. American Society of Transplantation. Available at: www.myast.org/public-policy/key-position-statements/immunosuppressant-drug-coverage-under-medicare-part-d-benefit.
Women’s Health
Cannabis Use Affects Women Differently
As we enter an era of legalization, cannabis use is increasingly prevalent. Variances in the risks for women and men have been observed. For most age groups, men have higher rates of use or dependence on illicit drugs than women. However, women are equally likely as men to progress to a substance use disorder. Women may be more susceptible to craving and relapse , which are key phases of the addiction cycle. A study on use among adolescents concluded there was preliminary evidence of a faster transition from initiation of marijuana use to regular use in women, when compared with men (Schepis, et al. J Addict Med. 2011;5[1]:65).
Research studies suggest that marijuana impairs spatial memory in women more so than in men. Studies have suggested that teenage girls who use marijuana may have a higher risk of brain structural abnormalities associated with regular marijuana exposure than teenage boys (Tapert, et al. Addict Biol. 2009;14[4]:457).
A study published in Psychoneuroendocrinology showed that cannabinoid receptor binding site densities exhibit sex differences and can be modulated by estradiol in several limbic brain regions. These findings may account for the sex differences observed with respect to the effects of cannabinoids (Riebe, et al. Psychoneuroendocrinology. 2010;35[8]:1265).
Further research is needed to expand our understanding of the interactions between cannabinoids and sex steroids. Detoxification treatments tailored toward women and men with cannabis addiction show a promising future and necessitate further research.
Anita Rajagopal, MD
Steering Committee Member
Disaster Response and Global Health
Epigenetics and Disasters
The configuration of the DNA bordering a gene dictates under what conditions a gene is expressed. Random errors or mutations affecting the neighboring DNA or the gene itself can affect how the gene functions. Epigenetics is an emerging field of science looking at environmental and psychosocial factors that do not directly cause mutations but still affect how genes are expressed with implications for the development and inheritance of disease. These external influences are thought to affect why some segments of DNA become accessible for protein production while other segments may not.
Disasters represent stressors with potential for epigenetic impact. Women who were pregnant during the 1998 Quebec ice storm were found to have a correlation between maternal objective stress and a distinctive pattern of DNA methylation in their children 13 years later (Cao-Lei L, et al. PLoS ONE. 2014;9[9] e10765). Methylation is known to affect the activity of a DNA segment and how genes are expressed. Associations have also been found between the severity of hurricanes and the prevalence of autism in the offspring of pregnant women experiencing these disasters (Kinney DK, et al. J Autism Dev Disord. 2008;38:481).
Anthropogenic hazards may also affect the offspring of survivors as suggested by studies of civil war POWs and Dutch Hunger Winter during WW II (Costa, DL, et al. Proc Nat Acad Sci 2018;. 115:44; Heijmans BT et al. Proc Nat Acad Sci. 2008;105[44]: 17046-9).
Epigenetics represents an area for additional research as natural and man-made disasters increase.
Omesh Toolsie, MBBS
Steering Committee Fellow-in-Training
Practice Operations
Medicare Competitive Bidding Process Update
Medicare’s Competitive Bidding Program (CBP), mandated since 2003, asks providers of specific durable medical equipment (including oxygen) to submit competing proposals for services. The best offer is then awarded a 3-year contract. Recently, several reforms to CBP have been proposed. The payment structure has changed to “lead-item pricing,” where a single bid in each category is selected and payment amounts for each product are then calculated based on pricing ratios and fee schedules (CMS DMEPOS Competitive Bidding).
This is in contrast to the prior method of median pricing, which caused financial difficulty and access concerns (Council for Quality Respiratory Care. The Rationale for Reforming Medicare Home Respiratory Therapy Payment Methodology. 2018). Budget neutrality requirements should relax, and oxygen payment structures improve. These proposed changes also include improved coverage of liquid oxygen and addition of home ventilator supplies.
However, effective January 1, 2019, all CBP is suspended through CMS. During the anticipated 2-year gap, any Medicare-enrolled supplier will be able to provide items until new contracts are awarded. Pricing during the gap period is based on a current single price plus consumer price index. These changes will impact CHEST members and their patients moving forward. During the temporary gap period, some areas are seeing decreased accessibility of some DME due to demand. Once reinstated, the changes to the oxygen payment structure should improve access and reduce out-of-pocket costs. The Practice Operations NetWork will continue to provide updates on this topic as they become available.
Timothy Dempsey, MD, MPH
Steering Committee Fellow-in-Training
Megan Sisk, DO
Steering Committee Member
Transplant
Medicare Part D Plans Can Deny Coverage of Select Immunosuppressant Medications in Solid Organ Transplant Recipients
An alarming problem has emerged with some solid organ transplant recipients experiencing immunosuppressant medication claim denials by Medicare Part D plans. Affected patients are those who convert from some other insurance (ie, private insurance or state Medicaid) to Medicare after their transplant and, therefore, rely on Medicare Part D for immunosuppressant drug coverage.
Insurance companies who offer Medicare Part D plans must follow the rules described in the Medicare Prescription Drug Benefit Manual.1 Although the Manual mandates that all immunosuppressant medications are on plan formularies, Part D plans are only required to cover immunosuppressant medications when used for indications approved by the Food and Drug Administration (FDA) or for off-label indications supported by the Centers for Medicare & Medicaid Services (CMS)-approved compendia (Drugdex® and AHFS Drug Information®).
A recent study examining the extent of the problem demonstrated non-renal organ transplant recipients are frequently prescribed and maintained on at least one medication vulnerable to Medicare Part D claim denials at 1 year posttransplant (lung: 71.1%; intestine: 39.7%; pancreas: 36.8%; liver: 19.7%; heart: 18.5%).2 Lung transplant recipients are most vulnerable since no immunosuppressant is FDA-approved for use in lung transplantation, and CMS-approved compendia only support off-label use for tacrolimus and cyclosporine in this population. Therefore, mycophenolate mofetil, mycophenolic acid, azathioprine, everolimus, and sirolimus are vulnerable to denial by Medicare Part D plans when used in lung transplant recipients. Over 95% of lung transplant recipients are maintained on an anti-metabolite, with the majority (88%) maintained on mycophenolate, so this is frequently impacted.2,3 While the transplant community is aware of this issue and has begun work to correct it, it has yet to be solved.2,4 In the meantime, if transplant recipients have been denied for this off-label and off-compendia reason, and appeals of those decisions have also been denied, options for obtaining the denied immunosuppressant medication include discount programs, foundation/grant funding, and industry-sponsored assistance programs.
Jennifer K. McDermott, PharmD
NetWork Member
1. Prescription Drug Benefit Manual. Centers for Medicare & Medicaid Services. Chapter 6: Part D Drugs and Formulary Requirements. Available at: https://www.cms.gov/Medicare/Prescription-Drug-Coverage/PrescriptionDrugCovContra/Downloads/Part-D-Benefits-Manual-Chapter-6.pdf
2. Potter LM et al. Transplant recipients are vulnerable to coverage denial under Medicare Part D. Am J Transplant. 2018;18:1502.
3. Valapour M et al. OPTN/SRTR 2016 Annual Data Report: Lung. Am J Transplant. 2018;18 (Suppl 1): 363.
4. Immunusuppressant Drug Coverage Under Medicare Part D Benefit. American Society of Transplantation. Available at: www.myast.org/public-policy/key-position-statements/immunosuppressant-drug-coverage-under-medicare-part-d-benefit.
Women’s Health
Cannabis Use Affects Women Differently
As we enter an era of legalization, cannabis use is increasingly prevalent. Variances in the risks for women and men have been observed. For most age groups, men have higher rates of use or dependence on illicit drugs than women. However, women are equally likely as men to progress to a substance use disorder. Women may be more susceptible to craving and relapse , which are key phases of the addiction cycle. A study on use among adolescents concluded there was preliminary evidence of a faster transition from initiation of marijuana use to regular use in women, when compared with men (Schepis, et al. J Addict Med. 2011;5[1]:65).
Research studies suggest that marijuana impairs spatial memory in women more so than in men. Studies have suggested that teenage girls who use marijuana may have a higher risk of brain structural abnormalities associated with regular marijuana exposure than teenage boys (Tapert, et al. Addict Biol. 2009;14[4]:457).
A study published in Psychoneuroendocrinology showed that cannabinoid receptor binding site densities exhibit sex differences and can be modulated by estradiol in several limbic brain regions. These findings may account for the sex differences observed with respect to the effects of cannabinoids (Riebe, et al. Psychoneuroendocrinology. 2010;35[8]:1265).
Further research is needed to expand our understanding of the interactions between cannabinoids and sex steroids. Detoxification treatments tailored toward women and men with cannabis addiction show a promising future and necessitate further research.
Anita Rajagopal, MD
Steering Committee Member
CHEST Foundation’s NetWorks Challenge is just around the corner
The NetWorks Challenge is an annual fundraising competition that encourages NetWork members to contribute to the CHEST Foundation - supporting clinical research grants and community service programs and creating patient education materials - while earning travel grants for their NetWork members to the CHEST Annual Meeting 2019 in New Orleans. Because of your generosity throughout the 2018 NetWorks Challenge, the CHEST Foundation was able to send 59 early career clinicians to CHEST 2018 in San Antonio - marked growth from the 25 clinicians who received the travel grants in 2017.

As we further improve this program based on feedback from NetWorks members, a few elements of the fundraiser are changing in 2019.
Length: This year, the NetWorks Challenge will span 3 months. Contributions made between April 1 and June 30 count toward your NetWork’s fundraising total! Just be sure to list your NetWork when making your contribution on chestfoundation.org/donate. Each month has a unique theme related to CHEST, so be sure to watch our social media profiles to engage with us and each other during the drive.
Additionally, ANY contributions made to the CHEST Foundation during your membership renewal will count toward your NetWorks total amount raised - no matter when your membership is up for renewal. Contributions made in this manner after June 30 will count toward your Network’s 2020 amount raised.
Prizes: This year, every NetWork is eligible to receive travel grants to CHEST 2019 in New Orleans based on the amount raised by the NetWork. Our final winners – the NetWork with the highest amount raised, and the NetWork with the highest percentage of participation from their NetWork, will each receive two additional travel grants to CHEST 2019. Plus, the NetWork with the highest amount raised over the course of the challenge receives an additional prize – a seat in a CHEST Live Learning course of the winner’s choosing, offered at CHEST’s Innovation, Simulation, and Training Center in Glenview, Illinois.
Visit chestfoundation.org/nc for more detailed information.
The NetWorks Challenge is an annual fundraising competition that encourages NetWork members to contribute to the CHEST Foundation - supporting clinical research grants and community service programs and creating patient education materials - while earning travel grants for their NetWork members to the CHEST Annual Meeting 2019 in New Orleans. Because of your generosity throughout the 2018 NetWorks Challenge, the CHEST Foundation was able to send 59 early career clinicians to CHEST 2018 in San Antonio - marked growth from the 25 clinicians who received the travel grants in 2017.
As we further improve this program based on feedback from NetWorks members, a few elements of the fundraiser are changing in 2019.
Length: This year, the NetWorks Challenge will span 3 months. Contributions made between April 1 and June 30 count toward your NetWork’s fundraising total! Just be sure to list your NetWork when making your contribution on chestfoundation.org/donate. Each month has a unique theme related to CHEST, so be sure to watch our social media profiles to engage with us and each other during the drive.
Additionally, ANY contributions made to the CHEST Foundation during your membership renewal will count toward your NetWorks total amount raised - no matter when your membership is up for renewal. Contributions made in this manner after June 30 will count toward your Network’s 2020 amount raised.
Prizes: This year, every NetWork is eligible to receive travel grants to CHEST 2019 in New Orleans based on the amount raised by the NetWork. Our final winners – the NetWork with the highest amount raised, and the NetWork with the highest percentage of participation from their NetWork, will each receive two additional travel grants to CHEST 2019. Plus, the NetWork with the highest amount raised over the course of the challenge receives an additional prize – a seat in a CHEST Live Learning course of the winner’s choosing, offered at CHEST’s Innovation, Simulation, and Training Center in Glenview, Illinois.
Visit chestfoundation.org/nc for more detailed information.
The NetWorks Challenge is an annual fundraising competition that encourages NetWork members to contribute to the CHEST Foundation - supporting clinical research grants and community service programs and creating patient education materials - while earning travel grants for their NetWork members to the CHEST Annual Meeting 2019 in New Orleans. Because of your generosity throughout the 2018 NetWorks Challenge, the CHEST Foundation was able to send 59 early career clinicians to CHEST 2018 in San Antonio - marked growth from the 25 clinicians who received the travel grants in 2017.
As we further improve this program based on feedback from NetWorks members, a few elements of the fundraiser are changing in 2019.
Length: This year, the NetWorks Challenge will span 3 months. Contributions made between April 1 and June 30 count toward your NetWork’s fundraising total! Just be sure to list your NetWork when making your contribution on chestfoundation.org/donate. Each month has a unique theme related to CHEST, so be sure to watch our social media profiles to engage with us and each other during the drive.
Additionally, ANY contributions made to the CHEST Foundation during your membership renewal will count toward your NetWorks total amount raised - no matter when your membership is up for renewal. Contributions made in this manner after June 30 will count toward your Network’s 2020 amount raised.
Prizes: This year, every NetWork is eligible to receive travel grants to CHEST 2019 in New Orleans based on the amount raised by the NetWork. Our final winners – the NetWork with the highest amount raised, and the NetWork with the highest percentage of participation from their NetWork, will each receive two additional travel grants to CHEST 2019. Plus, the NetWork with the highest amount raised over the course of the challenge receives an additional prize – a seat in a CHEST Live Learning course of the winner’s choosing, offered at CHEST’s Innovation, Simulation, and Training Center in Glenview, Illinois.
Visit chestfoundation.org/nc for more detailed information.
Sleep Strategies
Compared with obstructive sleep apnea (OSA), the prevalence of central sleep apnea (CSA) is low in the general population. However, in adults, CSA may be highly prevalent in certain conditions, most commonly among those with left ventricular systolic dysfunction, left ventricular diastolic dysfunction, atrial fibrillation, stroke, and opioid users (Javaheri S, et al. J Am Coll Cardiol. 2017; 69:841). CSA may also be found in patients with carotid artery stenosis, cervical neck injury, and renal dysfunction. CSA can occur when OSA is treated (treatment-emergent central sleep apnea, or TECA), notably, and most frequently, with continuous positive airway pressure (CPAP) devices. Though in many individuals, this frequently resolves with continued use of the device.

In addition, unlike OSA, adequate treatment of CSA has proven difficult. Specifically, the response to CPAP, oxygen, theophylline, acetazolamide, and adaptive-servo ventilation (ASV) is highly variable, with individuals who respond well, and individuals in whom therapy fails to fully suppress the disorder.
Our interest in phrenic nerve stimulation increased after it was shown that CPAP therapy failed to improve morbidity and mortality of CSA in patients with heart failure and reduced ejection fraction (HFrEF) (CANPAP trial, Bradley et al. N Engl J Med. 2005;353(19):2025). In fact, in this trial, treatment with CPAP was associated with significantly increased mortality during the first few months of therapy. We reason that a potential mechanism was positive airway pressure that had adverse cardiovascular effects (Javaheri S. J Clin Sleep Med. 2006;2:399). This is because positive airway pressure therapy decreases venous return to the right side of the heart and increases lung volume. This could increase pulmonary vascular resistance (right ventricular afterload), which is lung volume-dependent. Therefore, the subgroup of individuals with heart failure whose right ventricular function is preload-dependent and has pulmonary hypertension is at risk for premature mortality with any PAP device.
Interestingly, investigators of the SERVE-HF trial (Cowie MR, et al. N Engl J Med. 2015;373:1095) also hypothesized that one reason for excess mortality associated with ASV use might have been due to an ASV-associated excessive rise in intrathoracic pressure, similar to the hypothesis we proposed earlier for CPAP. We expanded on this hypothesis and reasoned that based on the algorithm of the device, in some patients, it could have generated excessive minute ventilation and pressure contributing to excess mortality, either at night or daytime (Javaheri S, et al. Chest. 2016;149:900). Other deficiencies of the algorithm of the ASV device could have contributed to excess mortality as well (Javaheri S, et al. Chest. 2014;146:514). These deficiencies of the ASV device used in the SERVE-HF trial have been significantly improved in the new generation of ASV devices.
Undoubtedly, therefore, mask therapy with positive airway pressures increases intrathoracic pressure and will adversely affect cardiovascular function in some patients with heart failure. Another issue for mask therapy is adherence to the device remains poor, as demonstrated both in the CANPAP and SERVE-HF trials, confirming the need for new approaches utilizing non-mask therapies both for CSA and OSA.
Given the limitations of mask-based therapies, over the last several years, we have performed studies exploring the use of oxygen, acetazolamide, theophylline, and, most recently, phrenic nerve stimulation (PNS). In general, these therapies are devoid of increasing intrathoracic pressure and are expected to be less reliant on patients’ adherence than PAP therapy. Long-term randomized clinical trials are needed, and, most recently, the NIH approved a phase 3 trial for a randomized placebo-controlled low flow oxygen therapy for treatment of CSA in HFrEF. This is a modified trial proposed by one of us more than 20 years ago!
Regarding PNS, CSA is characterized by intermittent phrenic nerve (and intercostal nerves) deactivation. It, therefore, makes sense to have an implanted stimulator for the phrenic nerve to prevent development of central apneas during sleep. This is not a new idea. In 1948, Sarnoff and colleagues demonstrated for the first time that artificial respiration could be effectively administered to the cat, dog, monkey, and rabbit in the absence of spontaneous respiration by electrical stimulation of one (or both) phrenic nerves (Sarnoff SJ, et al. Science. 1948;108:482). In later experiments, these investigators showed that unilateral phrenic nerve stimulation is also equally effective in man as that shown in animal models.
The phrenic nerves comes in contact with veins on both the right (brachiocephalic) and the left (pericardiophrenic vein) side of the mediastinum. Like a cardiac pacemaker, an electrophysiologist places the stimulator within the vein at the point of encounter with the phrenic nerve. Only unilateral stimulation is needed for the therapy. The device is typically placed on the right side of the chest as many patients may already have a cardiac implanted electronic device such as a pacemaker. Like the hypoglossal nerve stimulation, the FDA approved this device for the treatment of OSA. The system can be programmed using an external programmer in the office.
Phrenic nerve stimulation system is initially activated 1 month after the device is placed. It is programmed to be automatically activated at night when the patient is at rest. First, a time is set on the device for when the patient typically goes to bed and awakens. This allows the therapy to activate. The device contains a position sensor and accelerometer, which determine position and activity level. Once appropriate time, position, and activity are confirmed, the device activates automatically. Therapy comes on and can increase in level over several minutes. The device senses transthoracic impedance and can use this measurement to make changes in the therapy output and activity. If the patient gets up at night, the device automatically stops and restarts when the patient is back in a sleeping position. How quickly the therapy restarts and at what energy is programmable. The device may allow from 1 to 15 minutes for the patient to get back to sleep before beginning therapy. These programming changes allow for patient acceptance and comfort with the therapy even in very sensitive patients. Importantly, no patient activation is needed, so that therapy delivery is independent of patient’s adherence over time.
In the prospective, randomized pivotal trial (Costanzo et al. Lancet. 2016;388:974), 151 eligible patients with moderate-severe central sleep apnea were implanted and randomly assigned to the treatment (n=73) or control (n=78) groups. Participants in the active arm received PNS for 6 months. All polysomnograms were centrally and blindly scored. There were significant decreases in AHI (50 to 26/per hour of sleep), CAI (32 to 6), arousal index (46 to 25), and ODI (44 to 25). Two points should be emphasized: first, changes in AHI with PNS are similar to those in CANPAP trial, and there remained a significant number of hypopneas (some of these hypopneas are at least in part related to the speed of the titration when the subject sits up and the device automatically is deactivated, only to resume therapy in supine position); second, in contrast to the CANPAP trial, there was a significant reduction in arousals. Probably for this reason, subjective daytime sleepiness, as measured by the ESS, improved. In addition, PNS improved quality of life, in contrast to lack of effect of CPAP or ASV in this domain. Regarding side effects, 138 (91%) of 151 patients had no serious-related adverse events at 12 months. Seven (9%) cases of related-serious adverse events occurred in the control group and six (8%) cases were reported in the treatment group.—3.4% needed lead repositioning, a rate which is like that of cardiac implantable devices. Seven patients died (unrelated to implant, system, or therapy), four deaths (two in treatment group and two in control group) during the 6-month randomization period when neurostimulation was delivered to only the treatment and was off in the control group, and three deaths between 6 months and 12 months of follow-up when all patients received neurostimulation. Of 73 patients in the treatment group, 27 (37%) reported nonserious therapy-related discomfort that was resolved with simple system reprogramming in 26 (36%) patients but was unresolved in one (1%) patient.
Long-term studies have shown sustained effects of PNS on CSA with improvement in both sleep metrics and QOL, as measured by the Minnesota Living with Heart Failure Questionnaire (MLWHF) and patient global assessment (PGA). Furthermore, in the subgroup of patients with concomitant heart failure with LVEF ≤ 45%, PNS was associated with both improvements in LVEF and a trend toward lower hospitalization rates (Costanzo et al. Eur J Heart Fail. 2018; doi:10.1002/ejhf.1312).
Several issues must be emphasized. One advantage of PNS is complete adherence resulting in a major reduction in apnea burden across the whole night. Second, the mechanism of action prevents any potential adverse consequences related to increased intrathoracic pressure. However, the cost of this therapy is high, similar to that of hypoglossal nerve stimulation. Large scale, long-term studies related to mortality are not yet available, and continued research should help identify those patients most likely to benefit from this therapeutic approach.
Compared with obstructive sleep apnea (OSA), the prevalence of central sleep apnea (CSA) is low in the general population. However, in adults, CSA may be highly prevalent in certain conditions, most commonly among those with left ventricular systolic dysfunction, left ventricular diastolic dysfunction, atrial fibrillation, stroke, and opioid users (Javaheri S, et al. J Am Coll Cardiol. 2017; 69:841). CSA may also be found in patients with carotid artery stenosis, cervical neck injury, and renal dysfunction. CSA can occur when OSA is treated (treatment-emergent central sleep apnea, or TECA), notably, and most frequently, with continuous positive airway pressure (CPAP) devices. Though in many individuals, this frequently resolves with continued use of the device.
In addition, unlike OSA, adequate treatment of CSA has proven difficult. Specifically, the response to CPAP, oxygen, theophylline, acetazolamide, and adaptive-servo ventilation (ASV) is highly variable, with individuals who respond well, and individuals in whom therapy fails to fully suppress the disorder.
Our interest in phrenic nerve stimulation increased after it was shown that CPAP therapy failed to improve morbidity and mortality of CSA in patients with heart failure and reduced ejection fraction (HFrEF) (CANPAP trial, Bradley et al. N Engl J Med. 2005;353(19):2025). In fact, in this trial, treatment with CPAP was associated with significantly increased mortality during the first few months of therapy. We reason that a potential mechanism was positive airway pressure that had adverse cardiovascular effects (Javaheri S. J Clin Sleep Med. 2006;2:399). This is because positive airway pressure therapy decreases venous return to the right side of the heart and increases lung volume. This could increase pulmonary vascular resistance (right ventricular afterload), which is lung volume-dependent. Therefore, the subgroup of individuals with heart failure whose right ventricular function is preload-dependent and has pulmonary hypertension is at risk for premature mortality with any PAP device.
Interestingly, investigators of the SERVE-HF trial (Cowie MR, et al. N Engl J Med. 2015;373:1095) also hypothesized that one reason for excess mortality associated with ASV use might have been due to an ASV-associated excessive rise in intrathoracic pressure, similar to the hypothesis we proposed earlier for CPAP. We expanded on this hypothesis and reasoned that based on the algorithm of the device, in some patients, it could have generated excessive minute ventilation and pressure contributing to excess mortality, either at night or daytime (Javaheri S, et al. Chest. 2016;149:900). Other deficiencies of the algorithm of the ASV device could have contributed to excess mortality as well (Javaheri S, et al. Chest. 2014;146:514). These deficiencies of the ASV device used in the SERVE-HF trial have been significantly improved in the new generation of ASV devices.
Undoubtedly, therefore, mask therapy with positive airway pressures increases intrathoracic pressure and will adversely affect cardiovascular function in some patients with heart failure. Another issue for mask therapy is adherence to the device remains poor, as demonstrated both in the CANPAP and SERVE-HF trials, confirming the need for new approaches utilizing non-mask therapies both for CSA and OSA.
Given the limitations of mask-based therapies, over the last several years, we have performed studies exploring the use of oxygen, acetazolamide, theophylline, and, most recently, phrenic nerve stimulation (PNS). In general, these therapies are devoid of increasing intrathoracic pressure and are expected to be less reliant on patients’ adherence than PAP therapy. Long-term randomized clinical trials are needed, and, most recently, the NIH approved a phase 3 trial for a randomized placebo-controlled low flow oxygen therapy for treatment of CSA in HFrEF. This is a modified trial proposed by one of us more than 20 years ago!
Regarding PNS, CSA is characterized by intermittent phrenic nerve (and intercostal nerves) deactivation. It, therefore, makes sense to have an implanted stimulator for the phrenic nerve to prevent development of central apneas during sleep. This is not a new idea. In 1948, Sarnoff and colleagues demonstrated for the first time that artificial respiration could be effectively administered to the cat, dog, monkey, and rabbit in the absence of spontaneous respiration by electrical stimulation of one (or both) phrenic nerves (Sarnoff SJ, et al. Science. 1948;108:482). In later experiments, these investigators showed that unilateral phrenic nerve stimulation is also equally effective in man as that shown in animal models.
The phrenic nerves comes in contact with veins on both the right (brachiocephalic) and the left (pericardiophrenic vein) side of the mediastinum. Like a cardiac pacemaker, an electrophysiologist places the stimulator within the vein at the point of encounter with the phrenic nerve. Only unilateral stimulation is needed for the therapy. The device is typically placed on the right side of the chest as many patients may already have a cardiac implanted electronic device such as a pacemaker. Like the hypoglossal nerve stimulation, the FDA approved this device for the treatment of OSA. The system can be programmed using an external programmer in the office.
Phrenic nerve stimulation system is initially activated 1 month after the device is placed. It is programmed to be automatically activated at night when the patient is at rest. First, a time is set on the device for when the patient typically goes to bed and awakens. This allows the therapy to activate. The device contains a position sensor and accelerometer, which determine position and activity level. Once appropriate time, position, and activity are confirmed, the device activates automatically. Therapy comes on and can increase in level over several minutes. The device senses transthoracic impedance and can use this measurement to make changes in the therapy output and activity. If the patient gets up at night, the device automatically stops and restarts when the patient is back in a sleeping position. How quickly the therapy restarts and at what energy is programmable. The device may allow from 1 to 15 minutes for the patient to get back to sleep before beginning therapy. These programming changes allow for patient acceptance and comfort with the therapy even in very sensitive patients. Importantly, no patient activation is needed, so that therapy delivery is independent of patient’s adherence over time.
In the prospective, randomized pivotal trial (Costanzo et al. Lancet. 2016;388:974), 151 eligible patients with moderate-severe central sleep apnea were implanted and randomly assigned to the treatment (n=73) or control (n=78) groups. Participants in the active arm received PNS for 6 months. All polysomnograms were centrally and blindly scored. There were significant decreases in AHI (50 to 26/per hour of sleep), CAI (32 to 6), arousal index (46 to 25), and ODI (44 to 25). Two points should be emphasized: first, changes in AHI with PNS are similar to those in CANPAP trial, and there remained a significant number of hypopneas (some of these hypopneas are at least in part related to the speed of the titration when the subject sits up and the device automatically is deactivated, only to resume therapy in supine position); second, in contrast to the CANPAP trial, there was a significant reduction in arousals. Probably for this reason, subjective daytime sleepiness, as measured by the ESS, improved. In addition, PNS improved quality of life, in contrast to lack of effect of CPAP or ASV in this domain. Regarding side effects, 138 (91%) of 151 patients had no serious-related adverse events at 12 months. Seven (9%) cases of related-serious adverse events occurred in the control group and six (8%) cases were reported in the treatment group.—3.4% needed lead repositioning, a rate which is like that of cardiac implantable devices. Seven patients died (unrelated to implant, system, or therapy), four deaths (two in treatment group and two in control group) during the 6-month randomization period when neurostimulation was delivered to only the treatment and was off in the control group, and three deaths between 6 months and 12 months of follow-up when all patients received neurostimulation. Of 73 patients in the treatment group, 27 (37%) reported nonserious therapy-related discomfort that was resolved with simple system reprogramming in 26 (36%) patients but was unresolved in one (1%) patient.
Long-term studies have shown sustained effects of PNS on CSA with improvement in both sleep metrics and QOL, as measured by the Minnesota Living with Heart Failure Questionnaire (MLWHF) and patient global assessment (PGA). Furthermore, in the subgroup of patients with concomitant heart failure with LVEF ≤ 45%, PNS was associated with both improvements in LVEF and a trend toward lower hospitalization rates (Costanzo et al. Eur J Heart Fail. 2018; doi:10.1002/ejhf.1312).
Several issues must be emphasized. One advantage of PNS is complete adherence resulting in a major reduction in apnea burden across the whole night. Second, the mechanism of action prevents any potential adverse consequences related to increased intrathoracic pressure. However, the cost of this therapy is high, similar to that of hypoglossal nerve stimulation. Large scale, long-term studies related to mortality are not yet available, and continued research should help identify those patients most likely to benefit from this therapeutic approach.
Compared with obstructive sleep apnea (OSA), the prevalence of central sleep apnea (CSA) is low in the general population. However, in adults, CSA may be highly prevalent in certain conditions, most commonly among those with left ventricular systolic dysfunction, left ventricular diastolic dysfunction, atrial fibrillation, stroke, and opioid users (Javaheri S, et al. J Am Coll Cardiol. 2017; 69:841). CSA may also be found in patients with carotid artery stenosis, cervical neck injury, and renal dysfunction. CSA can occur when OSA is treated (treatment-emergent central sleep apnea, or TECA), notably, and most frequently, with continuous positive airway pressure (CPAP) devices. Though in many individuals, this frequently resolves with continued use of the device.
In addition, unlike OSA, adequate treatment of CSA has proven difficult. Specifically, the response to CPAP, oxygen, theophylline, acetazolamide, and adaptive-servo ventilation (ASV) is highly variable, with individuals who respond well, and individuals in whom therapy fails to fully suppress the disorder.
Our interest in phrenic nerve stimulation increased after it was shown that CPAP therapy failed to improve morbidity and mortality of CSA in patients with heart failure and reduced ejection fraction (HFrEF) (CANPAP trial, Bradley et al. N Engl J Med. 2005;353(19):2025). In fact, in this trial, treatment with CPAP was associated with significantly increased mortality during the first few months of therapy. We reason that a potential mechanism was positive airway pressure that had adverse cardiovascular effects (Javaheri S. J Clin Sleep Med. 2006;2:399). This is because positive airway pressure therapy decreases venous return to the right side of the heart and increases lung volume. This could increase pulmonary vascular resistance (right ventricular afterload), which is lung volume-dependent. Therefore, the subgroup of individuals with heart failure whose right ventricular function is preload-dependent and has pulmonary hypertension is at risk for premature mortality with any PAP device.
Interestingly, investigators of the SERVE-HF trial (Cowie MR, et al. N Engl J Med. 2015;373:1095) also hypothesized that one reason for excess mortality associated with ASV use might have been due to an ASV-associated excessive rise in intrathoracic pressure, similar to the hypothesis we proposed earlier for CPAP. We expanded on this hypothesis and reasoned that based on the algorithm of the device, in some patients, it could have generated excessive minute ventilation and pressure contributing to excess mortality, either at night or daytime (Javaheri S, et al. Chest. 2016;149:900). Other deficiencies of the algorithm of the ASV device could have contributed to excess mortality as well (Javaheri S, et al. Chest. 2014;146:514). These deficiencies of the ASV device used in the SERVE-HF trial have been significantly improved in the new generation of ASV devices.
Undoubtedly, therefore, mask therapy with positive airway pressures increases intrathoracic pressure and will adversely affect cardiovascular function in some patients with heart failure. Another issue for mask therapy is adherence to the device remains poor, as demonstrated both in the CANPAP and SERVE-HF trials, confirming the need for new approaches utilizing non-mask therapies both for CSA and OSA.
Given the limitations of mask-based therapies, over the last several years, we have performed studies exploring the use of oxygen, acetazolamide, theophylline, and, most recently, phrenic nerve stimulation (PNS). In general, these therapies are devoid of increasing intrathoracic pressure and are expected to be less reliant on patients’ adherence than PAP therapy. Long-term randomized clinical trials are needed, and, most recently, the NIH approved a phase 3 trial for a randomized placebo-controlled low flow oxygen therapy for treatment of CSA in HFrEF. This is a modified trial proposed by one of us more than 20 years ago!
Regarding PNS, CSA is characterized by intermittent phrenic nerve (and intercostal nerves) deactivation. It, therefore, makes sense to have an implanted stimulator for the phrenic nerve to prevent development of central apneas during sleep. This is not a new idea. In 1948, Sarnoff and colleagues demonstrated for the first time that artificial respiration could be effectively administered to the cat, dog, monkey, and rabbit in the absence of spontaneous respiration by electrical stimulation of one (or both) phrenic nerves (Sarnoff SJ, et al. Science. 1948;108:482). In later experiments, these investigators showed that unilateral phrenic nerve stimulation is also equally effective in man as that shown in animal models.
The phrenic nerves comes in contact with veins on both the right (brachiocephalic) and the left (pericardiophrenic vein) side of the mediastinum. Like a cardiac pacemaker, an electrophysiologist places the stimulator within the vein at the point of encounter with the phrenic nerve. Only unilateral stimulation is needed for the therapy. The device is typically placed on the right side of the chest as many patients may already have a cardiac implanted electronic device such as a pacemaker. Like the hypoglossal nerve stimulation, the FDA approved this device for the treatment of OSA. The system can be programmed using an external programmer in the office.
Phrenic nerve stimulation system is initially activated 1 month after the device is placed. It is programmed to be automatically activated at night when the patient is at rest. First, a time is set on the device for when the patient typically goes to bed and awakens. This allows the therapy to activate. The device contains a position sensor and accelerometer, which determine position and activity level. Once appropriate time, position, and activity are confirmed, the device activates automatically. Therapy comes on and can increase in level over several minutes. The device senses transthoracic impedance and can use this measurement to make changes in the therapy output and activity. If the patient gets up at night, the device automatically stops and restarts when the patient is back in a sleeping position. How quickly the therapy restarts and at what energy is programmable. The device may allow from 1 to 15 minutes for the patient to get back to sleep before beginning therapy. These programming changes allow for patient acceptance and comfort with the therapy even in very sensitive patients. Importantly, no patient activation is needed, so that therapy delivery is independent of patient’s adherence over time.
In the prospective, randomized pivotal trial (Costanzo et al. Lancet. 2016;388:974), 151 eligible patients with moderate-severe central sleep apnea were implanted and randomly assigned to the treatment (n=73) or control (n=78) groups. Participants in the active arm received PNS for 6 months. All polysomnograms were centrally and blindly scored. There were significant decreases in AHI (50 to 26/per hour of sleep), CAI (32 to 6), arousal index (46 to 25), and ODI (44 to 25). Two points should be emphasized: first, changes in AHI with PNS are similar to those in CANPAP trial, and there remained a significant number of hypopneas (some of these hypopneas are at least in part related to the speed of the titration when the subject sits up and the device automatically is deactivated, only to resume therapy in supine position); second, in contrast to the CANPAP trial, there was a significant reduction in arousals. Probably for this reason, subjective daytime sleepiness, as measured by the ESS, improved. In addition, PNS improved quality of life, in contrast to lack of effect of CPAP or ASV in this domain. Regarding side effects, 138 (91%) of 151 patients had no serious-related adverse events at 12 months. Seven (9%) cases of related-serious adverse events occurred in the control group and six (8%) cases were reported in the treatment group.—3.4% needed lead repositioning, a rate which is like that of cardiac implantable devices. Seven patients died (unrelated to implant, system, or therapy), four deaths (two in treatment group and two in control group) during the 6-month randomization period when neurostimulation was delivered to only the treatment and was off in the control group, and three deaths between 6 months and 12 months of follow-up when all patients received neurostimulation. Of 73 patients in the treatment group, 27 (37%) reported nonserious therapy-related discomfort that was resolved with simple system reprogramming in 26 (36%) patients but was unresolved in one (1%) patient.
Long-term studies have shown sustained effects of PNS on CSA with improvement in both sleep metrics and QOL, as measured by the Minnesota Living with Heart Failure Questionnaire (MLWHF) and patient global assessment (PGA). Furthermore, in the subgroup of patients with concomitant heart failure with LVEF ≤ 45%, PNS was associated with both improvements in LVEF and a trend toward lower hospitalization rates (Costanzo et al. Eur J Heart Fail. 2018; doi:10.1002/ejhf.1312).
Several issues must be emphasized. One advantage of PNS is complete adherence resulting in a major reduction in apnea burden across the whole night. Second, the mechanism of action prevents any potential adverse consequences related to increased intrathoracic pressure. However, the cost of this therapy is high, similar to that of hypoglossal nerve stimulation. Large scale, long-term studies related to mortality are not yet available, and continued research should help identify those patients most likely to benefit from this therapeutic approach.
Blood-based signature helps predict status of early AD indicator
A recently developed blood-based signature can help predict the status of an early Alzheimer’s disease risk indicator with high accuracy, investigators are reporting.
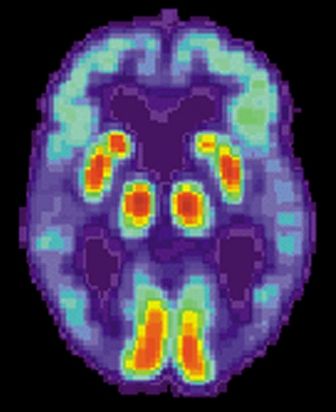
By analyzing as few as four proteins, the machine learning-derived test can predict the status of cerebrospinal fluid (CSF) amyloid beta1-42 (Abeta1-42), according to Noel G. Faux, PHD, of IBM Australia and the University of Melbourne, and co-investigators.
While shifts in Abeta1-42 may signal the presence of disease long before significant cognitive decline is clinically apparent, collection of CSF is highly invasive and expensive, Faux and investigators said in their report.
By contrast, blood biomarkers could prove to be a useful alternative not only to invasive lumbar punctures, they said, but also to the positron emission tomography (PET) evaluation of Abeta1-42, which is expensive and limited in some regions.
“In conjunction with biomarkers for neocortical amyloid burden, the CSF Abeta1-42biomarkers presented in this work may help yield a cheap, non-invasive tool for both improving clinical trials targeting amyloid and population screening,” Dr. Faux and co-authors said in Scientific Reports.
Dr. Faux and colleagues used a Random Forest approach to build models for CSF Abeta1-42 using blood biomarkers and other variables.
They found that a model incorporating age, APOEe4 carrier status, and a number of plasma protein levels predicted Abeta1-42 normal/abnormalstatus with an AUC, sensitivity and specificity of 0.84, 0.78 and 0.73 respectively.
In a model they said was more suitable for clinical application, they narrowed down the variables to 4 plasma analytes and APOEe4 carrier status, which had an AUC, sensitivity, and specificity of 0.81, 0.81 and 0.64 respectively.
They validated the models on a cohort of individuals in the Alzheimer’s Disease Neuroimaging Initiative (ADNI), a large, longitudinal, multicenter study.
Patients with mild cognitive impairment with predicted abnormal CSF Abeta1-42 levels indeed did transition to a diagnosis of Alzheimer’s disease more quickly than those with predicted normal levels, according to investigators.
That helps provide “strong evidence” that the blood-based model is generalizable, robust, and could help stratify patients based on risk of progressing to Alzheimer’s disease, they said in their report.
Dr. Faux and colleagues declared no conflicts of interest related to the research.
SOURCE: Goudey B, et al. Sci Rep. 2019 Mar 10. doi: 10.1101/190207v3.
A recently developed blood-based signature can help predict the status of an early Alzheimer’s disease risk indicator with high accuracy, investigators are reporting.
By analyzing as few as four proteins, the machine learning-derived test can predict the status of cerebrospinal fluid (CSF) amyloid beta1-42 (Abeta1-42), according to Noel G. Faux, PHD, of IBM Australia and the University of Melbourne, and co-investigators.
While shifts in Abeta1-42 may signal the presence of disease long before significant cognitive decline is clinically apparent, collection of CSF is highly invasive and expensive, Faux and investigators said in their report.
By contrast, blood biomarkers could prove to be a useful alternative not only to invasive lumbar punctures, they said, but also to the positron emission tomography (PET) evaluation of Abeta1-42, which is expensive and limited in some regions.
“In conjunction with biomarkers for neocortical amyloid burden, the CSF Abeta1-42biomarkers presented in this work may help yield a cheap, non-invasive tool for both improving clinical trials targeting amyloid and population screening,” Dr. Faux and co-authors said in Scientific Reports.
Dr. Faux and colleagues used a Random Forest approach to build models for CSF Abeta1-42 using blood biomarkers and other variables.
They found that a model incorporating age, APOEe4 carrier status, and a number of plasma protein levels predicted Abeta1-42 normal/abnormalstatus with an AUC, sensitivity and specificity of 0.84, 0.78 and 0.73 respectively.
In a model they said was more suitable for clinical application, they narrowed down the variables to 4 plasma analytes and APOEe4 carrier status, which had an AUC, sensitivity, and specificity of 0.81, 0.81 and 0.64 respectively.
They validated the models on a cohort of individuals in the Alzheimer’s Disease Neuroimaging Initiative (ADNI), a large, longitudinal, multicenter study.
Patients with mild cognitive impairment with predicted abnormal CSF Abeta1-42 levels indeed did transition to a diagnosis of Alzheimer’s disease more quickly than those with predicted normal levels, according to investigators.
That helps provide “strong evidence” that the blood-based model is generalizable, robust, and could help stratify patients based on risk of progressing to Alzheimer’s disease, they said in their report.
Dr. Faux and colleagues declared no conflicts of interest related to the research.
SOURCE: Goudey B, et al. Sci Rep. 2019 Mar 10. doi: 10.1101/190207v3.
A recently developed blood-based signature can help predict the status of an early Alzheimer’s disease risk indicator with high accuracy, investigators are reporting.
By analyzing as few as four proteins, the machine learning-derived test can predict the status of cerebrospinal fluid (CSF) amyloid beta1-42 (Abeta1-42), according to Noel G. Faux, PHD, of IBM Australia and the University of Melbourne, and co-investigators.
While shifts in Abeta1-42 may signal the presence of disease long before significant cognitive decline is clinically apparent, collection of CSF is highly invasive and expensive, Faux and investigators said in their report.
By contrast, blood biomarkers could prove to be a useful alternative not only to invasive lumbar punctures, they said, but also to the positron emission tomography (PET) evaluation of Abeta1-42, which is expensive and limited in some regions.
“In conjunction with biomarkers for neocortical amyloid burden, the CSF Abeta1-42biomarkers presented in this work may help yield a cheap, non-invasive tool for both improving clinical trials targeting amyloid and population screening,” Dr. Faux and co-authors said in Scientific Reports.
Dr. Faux and colleagues used a Random Forest approach to build models for CSF Abeta1-42 using blood biomarkers and other variables.
They found that a model incorporating age, APOEe4 carrier status, and a number of plasma protein levels predicted Abeta1-42 normal/abnormalstatus with an AUC, sensitivity and specificity of 0.84, 0.78 and 0.73 respectively.
In a model they said was more suitable for clinical application, they narrowed down the variables to 4 plasma analytes and APOEe4 carrier status, which had an AUC, sensitivity, and specificity of 0.81, 0.81 and 0.64 respectively.
They validated the models on a cohort of individuals in the Alzheimer’s Disease Neuroimaging Initiative (ADNI), a large, longitudinal, multicenter study.
Patients with mild cognitive impairment with predicted abnormal CSF Abeta1-42 levels indeed did transition to a diagnosis of Alzheimer’s disease more quickly than those with predicted normal levels, according to investigators.
That helps provide “strong evidence” that the blood-based model is generalizable, robust, and could help stratify patients based on risk of progressing to Alzheimer’s disease, they said in their report.
Dr. Faux and colleagues declared no conflicts of interest related to the research.
SOURCE: Goudey B, et al. Sci Rep. 2019 Mar 10. doi: 10.1101/190207v3.
FROM SCIENTIFIC REPORTS
Key clinical point: A blood-based signature can help predict the status of an early Alzheimer’s disease risk indicator.
Major finding:
Study details: Machine learning analysis of blood biomarkers and other variables in a validation cohort of 198 individuals.
Disclosures: The study authors declared no conflicts of interest.
Source: Goudey B, et al. Sci Rep. 2019 Mar 10. doi: 10.1101/190207v3.
Evaluations for possible MS often turn up one of its many mimics
DALLAS – Of 95 patients referred to two multiple sclerosis (MS) centers for a possible diagnosis of MS, 74% did not have MS, according to a study presented at ACTRIMS Forum 2019. A majority had clinical syndromes or imaging findings that are atypical for MS, which “underscores the importance of familiarity with typical MS clinical and imaging findings in avoiding misdiagnosis,” said Marwa Kaisey, MD, and her research colleagues. Dr. Kaisey is a neurologist at Cedars-Sinai Medical Center in Los Angeles.

Physicians often refer patients to academic MS centers to determine whether patients have MS or one of its many mimics. To study the characteristics and final diagnoses of patients referred to MS centers for evaluation of possible MS, the investigators reviewed electronic medical records and MRI from all new patient evaluations at the Cedars-Sinai Medical Center and University of California, Los Angeles MS clinics between July 2016 and June 2017. The researchers excluded patients referred with a previously established diagnosis of MS.
There were 366 new patients evaluated, including 236 patients with previously established MS diagnoses and 35 patients whose evaluations were not related to MS. Of the 95 patients referred for a question of MS diagnosis, 60% had clinical syndromes that were atypical for MS, 22% had normal neurologic exams, and a third had pain or sensory changes that were not localizable to the CNS.
Sixty-seven percent had MRI that was atypical for MS, and nearly half of the patients without MS had nonspecific MRI changes. “Often, these MRI changes alone prompted referral for an MS evaluation,” Dr. Kaisey and colleagues reported. “This suggests that novel, specific imaging tools may increase diagnostic confidence in the clinical setting.”
In all, the referred patients received 28 diagnoses other than MS, most commonly migraine (10 patients), anxiety or conversion disorder (9), postinfectious or idiopathic transverse myelitis (8), compression myelopathy or spondylopathy (8), and peripheral neuropathy or radiculopathy (7).
The researchers did not have any relevant disclosures.
SOURCE: Kaisey M et al. ACTRIMS Forum 2019, Abstract 90.
DALLAS – Of 95 patients referred to two multiple sclerosis (MS) centers for a possible diagnosis of MS, 74% did not have MS, according to a study presented at ACTRIMS Forum 2019. A majority had clinical syndromes or imaging findings that are atypical for MS, which “underscores the importance of familiarity with typical MS clinical and imaging findings in avoiding misdiagnosis,” said Marwa Kaisey, MD, and her research colleagues. Dr. Kaisey is a neurologist at Cedars-Sinai Medical Center in Los Angeles.
Physicians often refer patients to academic MS centers to determine whether patients have MS or one of its many mimics. To study the characteristics and final diagnoses of patients referred to MS centers for evaluation of possible MS, the investigators reviewed electronic medical records and MRI from all new patient evaluations at the Cedars-Sinai Medical Center and University of California, Los Angeles MS clinics between July 2016 and June 2017. The researchers excluded patients referred with a previously established diagnosis of MS.
There were 366 new patients evaluated, including 236 patients with previously established MS diagnoses and 35 patients whose evaluations were not related to MS. Of the 95 patients referred for a question of MS diagnosis, 60% had clinical syndromes that were atypical for MS, 22% had normal neurologic exams, and a third had pain or sensory changes that were not localizable to the CNS.
Sixty-seven percent had MRI that was atypical for MS, and nearly half of the patients without MS had nonspecific MRI changes. “Often, these MRI changes alone prompted referral for an MS evaluation,” Dr. Kaisey and colleagues reported. “This suggests that novel, specific imaging tools may increase diagnostic confidence in the clinical setting.”
In all, the referred patients received 28 diagnoses other than MS, most commonly migraine (10 patients), anxiety or conversion disorder (9), postinfectious or idiopathic transverse myelitis (8), compression myelopathy or spondylopathy (8), and peripheral neuropathy or radiculopathy (7).
The researchers did not have any relevant disclosures.
SOURCE: Kaisey M et al. ACTRIMS Forum 2019, Abstract 90.
DALLAS – Of 95 patients referred to two multiple sclerosis (MS) centers for a possible diagnosis of MS, 74% did not have MS, according to a study presented at ACTRIMS Forum 2019. A majority had clinical syndromes or imaging findings that are atypical for MS, which “underscores the importance of familiarity with typical MS clinical and imaging findings in avoiding misdiagnosis,” said Marwa Kaisey, MD, and her research colleagues. Dr. Kaisey is a neurologist at Cedars-Sinai Medical Center in Los Angeles.
Physicians often refer patients to academic MS centers to determine whether patients have MS or one of its many mimics. To study the characteristics and final diagnoses of patients referred to MS centers for evaluation of possible MS, the investigators reviewed electronic medical records and MRI from all new patient evaluations at the Cedars-Sinai Medical Center and University of California, Los Angeles MS clinics between July 2016 and June 2017. The researchers excluded patients referred with a previously established diagnosis of MS.
There were 366 new patients evaluated, including 236 patients with previously established MS diagnoses and 35 patients whose evaluations were not related to MS. Of the 95 patients referred for a question of MS diagnosis, 60% had clinical syndromes that were atypical for MS, 22% had normal neurologic exams, and a third had pain or sensory changes that were not localizable to the CNS.
Sixty-seven percent had MRI that was atypical for MS, and nearly half of the patients without MS had nonspecific MRI changes. “Often, these MRI changes alone prompted referral for an MS evaluation,” Dr. Kaisey and colleagues reported. “This suggests that novel, specific imaging tools may increase diagnostic confidence in the clinical setting.”
In all, the referred patients received 28 diagnoses other than MS, most commonly migraine (10 patients), anxiety or conversion disorder (9), postinfectious or idiopathic transverse myelitis (8), compression myelopathy or spondylopathy (8), and peripheral neuropathy or radiculopathy (7).
The researchers did not have any relevant disclosures.
SOURCE: Kaisey M et al. ACTRIMS Forum 2019, Abstract 90.
REPORTING FROM ACTRIMS FORUM 2019
Low Risk TAVR trial shows 3% mortality at 1 year
WASHINGTON –Anticipating two pivotal trials scheduled for presentation at the 2019 annual meeting of the American College of Cardiology (ACC), an investigator-led study of transaortic valve replacement (TAVR) for aortic stenosis presented as a latebreaker at 2019 CRT meeting produced excellent results.

Not least impressive, “our mortality rates are the lowest ever reported in any TAVR study at one year,” said Ronald Waksman, MD, Associate Director, Division of Cardiology, Medstar Heart Institute, Washington, DC.
In a population of patients with a median age of 71.1 years, all-cause mortality was just 3% at one year while the rate of deaths due to cardiovascular causes was only 1%, according to results of the 200-patient Low Risk TAVR study (LRT 1.0, NCT02628899) that Dr. Waksman presented.
In addition, there were low rates at one year for stroke (2.1%, none of which was deemed disability), myocardial infarction (1%), new onset atrial fibrillation (6.2%), and pacemaker placement (7.3%). The rate of rehospitalization for any cause was 20.4% but only 3.1% were considered related to TAVR. Rehospitalization for any cardiovascular cause at one year occurred in 6.8%.
Although leaflet thickening was observed at one year with imaging in 14%, this has not had any identifiable clinical consequences so far, and hemodynamics have remained stable, according to Dr. Waksman, who presented the interim 30-day outcomes at the 2018 CRT meeting.
These findings are raising expectations for two phase 3 TAVR trials in low-risk patients that are being presented as latebreakers at the 2019 ACC annual meeting. Both are large randomized trials comparing TAVR to surgical aortic valve replacement (SAVR) in low risk patients. Each trial is testing a single type of value and is funded by the valve manufacturers.
In the PARTNER-3 trial, patients randomized to TAVR received the Sapien 3 valve (Edwards Lifesciences). In the other latebreaking trial, patients randomized to TAVR received an Evolut valve (Medtronic Cardiovascular). Both are comparing TAVR to SAVR with a composite primary outcome that includes mortality and stroke measured at 30 days and one year.
In contrast to these trials, LRT 1.0 was conducted with no funding from a third party, according to Dr. Waksman. The eleven centers participated in the study at their own cost. Also, the choice of TAVR device was left to the discretion of the interventional cardiologist. Finally, most of the participating centers, although experienced in TAVR, did not have a high-volume case load. In general, with the exception of Dr. Waksman’s center, most performed 100 to 150 TAVRs per year.
“We were struck by the excellence of the performance of these sites,” said Dr. Waksman, noting that a comparison of outcomes at his center relative to the lower volume centers showed no significant differences in outcome.
This real-world experience raises the bar for the pivotal phase 3 trials, which, if positive, are expected to lead the FDA to grant an indication for TAVR in low-risk patients, according to Dr. Waksman. He announced that an LRT 2.0 trial, which will again include centers performing TAVRs at moderate volumes, is now enrolling.
SOURCE: 2019 Cardiovascular Research Technologies (CRT) Meeting.
WASHINGTON –Anticipating two pivotal trials scheduled for presentation at the 2019 annual meeting of the American College of Cardiology (ACC), an investigator-led study of transaortic valve replacement (TAVR) for aortic stenosis presented as a latebreaker at 2019 CRT meeting produced excellent results.
Not least impressive, “our mortality rates are the lowest ever reported in any TAVR study at one year,” said Ronald Waksman, MD, Associate Director, Division of Cardiology, Medstar Heart Institute, Washington, DC.
In a population of patients with a median age of 71.1 years, all-cause mortality was just 3% at one year while the rate of deaths due to cardiovascular causes was only 1%, according to results of the 200-patient Low Risk TAVR study (LRT 1.0, NCT02628899) that Dr. Waksman presented.
In addition, there were low rates at one year for stroke (2.1%, none of which was deemed disability), myocardial infarction (1%), new onset atrial fibrillation (6.2%), and pacemaker placement (7.3%). The rate of rehospitalization for any cause was 20.4% but only 3.1% were considered related to TAVR. Rehospitalization for any cardiovascular cause at one year occurred in 6.8%.
Although leaflet thickening was observed at one year with imaging in 14%, this has not had any identifiable clinical consequences so far, and hemodynamics have remained stable, according to Dr. Waksman, who presented the interim 30-day outcomes at the 2018 CRT meeting.
These findings are raising expectations for two phase 3 TAVR trials in low-risk patients that are being presented as latebreakers at the 2019 ACC annual meeting. Both are large randomized trials comparing TAVR to surgical aortic valve replacement (SAVR) in low risk patients. Each trial is testing a single type of value and is funded by the valve manufacturers.
In the PARTNER-3 trial, patients randomized to TAVR received the Sapien 3 valve (Edwards Lifesciences). In the other latebreaking trial, patients randomized to TAVR received an Evolut valve (Medtronic Cardiovascular). Both are comparing TAVR to SAVR with a composite primary outcome that includes mortality and stroke measured at 30 days and one year.
In contrast to these trials, LRT 1.0 was conducted with no funding from a third party, according to Dr. Waksman. The eleven centers participated in the study at their own cost. Also, the choice of TAVR device was left to the discretion of the interventional cardiologist. Finally, most of the participating centers, although experienced in TAVR, did not have a high-volume case load. In general, with the exception of Dr. Waksman’s center, most performed 100 to 150 TAVRs per year.
“We were struck by the excellence of the performance of these sites,” said Dr. Waksman, noting that a comparison of outcomes at his center relative to the lower volume centers showed no significant differences in outcome.
This real-world experience raises the bar for the pivotal phase 3 trials, which, if positive, are expected to lead the FDA to grant an indication for TAVR in low-risk patients, according to Dr. Waksman. He announced that an LRT 2.0 trial, which will again include centers performing TAVRs at moderate volumes, is now enrolling.
SOURCE: 2019 Cardiovascular Research Technologies (CRT) Meeting.
WASHINGTON –Anticipating two pivotal trials scheduled for presentation at the 2019 annual meeting of the American College of Cardiology (ACC), an investigator-led study of transaortic valve replacement (TAVR) for aortic stenosis presented as a latebreaker at 2019 CRT meeting produced excellent results.
Not least impressive, “our mortality rates are the lowest ever reported in any TAVR study at one year,” said Ronald Waksman, MD, Associate Director, Division of Cardiology, Medstar Heart Institute, Washington, DC.
In a population of patients with a median age of 71.1 years, all-cause mortality was just 3% at one year while the rate of deaths due to cardiovascular causes was only 1%, according to results of the 200-patient Low Risk TAVR study (LRT 1.0, NCT02628899) that Dr. Waksman presented.
In addition, there were low rates at one year for stroke (2.1%, none of which was deemed disability), myocardial infarction (1%), new onset atrial fibrillation (6.2%), and pacemaker placement (7.3%). The rate of rehospitalization for any cause was 20.4% but only 3.1% were considered related to TAVR. Rehospitalization for any cardiovascular cause at one year occurred in 6.8%.
Although leaflet thickening was observed at one year with imaging in 14%, this has not had any identifiable clinical consequences so far, and hemodynamics have remained stable, according to Dr. Waksman, who presented the interim 30-day outcomes at the 2018 CRT meeting.
These findings are raising expectations for two phase 3 TAVR trials in low-risk patients that are being presented as latebreakers at the 2019 ACC annual meeting. Both are large randomized trials comparing TAVR to surgical aortic valve replacement (SAVR) in low risk patients. Each trial is testing a single type of value and is funded by the valve manufacturers.
In the PARTNER-3 trial, patients randomized to TAVR received the Sapien 3 valve (Edwards Lifesciences). In the other latebreaking trial, patients randomized to TAVR received an Evolut valve (Medtronic Cardiovascular). Both are comparing TAVR to SAVR with a composite primary outcome that includes mortality and stroke measured at 30 days and one year.
In contrast to these trials, LRT 1.0 was conducted with no funding from a third party, according to Dr. Waksman. The eleven centers participated in the study at their own cost. Also, the choice of TAVR device was left to the discretion of the interventional cardiologist. Finally, most of the participating centers, although experienced in TAVR, did not have a high-volume case load. In general, with the exception of Dr. Waksman’s center, most performed 100 to 150 TAVRs per year.
“We were struck by the excellence of the performance of these sites,” said Dr. Waksman, noting that a comparison of outcomes at his center relative to the lower volume centers showed no significant differences in outcome.
This real-world experience raises the bar for the pivotal phase 3 trials, which, if positive, are expected to lead the FDA to grant an indication for TAVR in low-risk patients, according to Dr. Waksman. He announced that an LRT 2.0 trial, which will again include centers performing TAVRs at moderate volumes, is now enrolling.
SOURCE: 2019 Cardiovascular Research Technologies (CRT) Meeting.
REPORTING FROM CRT 2019
In utero infections raise risk for autism
Children whose mothers experienced any type of infection during pregnancy were nearly 80 times more likely to be diagnosed with autism than those whose mothers did not have infections, based on data from more than one million children in Sweden.

Although previous studies have shown associations between specific infections in utero and specific conditions, such as schizophrenia, “Whether maternal infection and inflammation can alter fetal neurodevelopment to a degree that imparts risk for a broad spectrum of psychopathologic conditions across the child’s lifetime is unknown,” wrote Benjamin J. S. al-Haddad, MD, formerly of Seattle Children’s Hospital, Washington, currently with Doctors without Borders,Katiola, Côte d’Ivoire, and his colleagues.
In a study published In JAMA Psychiatry, the researchers followed 1,791,520 children (48.6% girls) born between Jan. 1, 1973, and Dec. 31, 2014, for up to 41 years using population-based registry data.
Overall, researchers found a 79% increased risk of an autism diagnosis (hazard ratio 1.79) and a 24% increased risk of a depression diagnosis (HR 1.24) for individuals exposed to any maternal infection in utero compared with those not exposed.
Similar increases in risk appeared when the data were broken down by type of infection. Hazard ratios for an autism diagnosis were 1.81 for exposure to a severe maternal infection and 1.89 for a maternal urinary tract infection; hazard ratios for depression were 1.24 and 1.30, respectively, for severe maternal infection and maternal urinary tract infection.
No increased risk in bipolar disorder, or other psychoses including schizophrenia were observed.
The findings were limited by several factors including the inclusion only of infections diagnosed in a hospital setting, and thus may not be generalizable to infections diagnosed in an outpatient setting, the researchers noted. However, the results “amplify the urgency to better understand the role of maternal infection during pregnancy on fetal brain development and suggest that prevention of infection (such as by influenza vaccination) or anti-inflammatory therapies may be important strategies for the primary prevention of some portion of autism and depression,” they said.
The researchers had no conflicts to disclose. The study was funded by grants from several organizations including the National Institutes of Health.
SOURCE: al-Haddad BJS et al. JAMA Psychiatry. doi:10.1001/jamapsychiatry.2019.0029.
Children whose mothers experienced any type of infection during pregnancy were nearly 80 times more likely to be diagnosed with autism than those whose mothers did not have infections, based on data from more than one million children in Sweden.
Although previous studies have shown associations between specific infections in utero and specific conditions, such as schizophrenia, “Whether maternal infection and inflammation can alter fetal neurodevelopment to a degree that imparts risk for a broad spectrum of psychopathologic conditions across the child’s lifetime is unknown,” wrote Benjamin J. S. al-Haddad, MD, formerly of Seattle Children’s Hospital, Washington, currently with Doctors without Borders,Katiola, Côte d’Ivoire, and his colleagues.
In a study published In JAMA Psychiatry, the researchers followed 1,791,520 children (48.6% girls) born between Jan. 1, 1973, and Dec. 31, 2014, for up to 41 years using population-based registry data.
Overall, researchers found a 79% increased risk of an autism diagnosis (hazard ratio 1.79) and a 24% increased risk of a depression diagnosis (HR 1.24) for individuals exposed to any maternal infection in utero compared with those not exposed.
Similar increases in risk appeared when the data were broken down by type of infection. Hazard ratios for an autism diagnosis were 1.81 for exposure to a severe maternal infection and 1.89 for a maternal urinary tract infection; hazard ratios for depression were 1.24 and 1.30, respectively, for severe maternal infection and maternal urinary tract infection.
No increased risk in bipolar disorder, or other psychoses including schizophrenia were observed.
The findings were limited by several factors including the inclusion only of infections diagnosed in a hospital setting, and thus may not be generalizable to infections diagnosed in an outpatient setting, the researchers noted. However, the results “amplify the urgency to better understand the role of maternal infection during pregnancy on fetal brain development and suggest that prevention of infection (such as by influenza vaccination) or anti-inflammatory therapies may be important strategies for the primary prevention of some portion of autism and depression,” they said.
The researchers had no conflicts to disclose. The study was funded by grants from several organizations including the National Institutes of Health.
SOURCE: al-Haddad BJS et al. JAMA Psychiatry. doi:10.1001/jamapsychiatry.2019.0029.
Children whose mothers experienced any type of infection during pregnancy were nearly 80 times more likely to be diagnosed with autism than those whose mothers did not have infections, based on data from more than one million children in Sweden.
Although previous studies have shown associations between specific infections in utero and specific conditions, such as schizophrenia, “Whether maternal infection and inflammation can alter fetal neurodevelopment to a degree that imparts risk for a broad spectrum of psychopathologic conditions across the child’s lifetime is unknown,” wrote Benjamin J. S. al-Haddad, MD, formerly of Seattle Children’s Hospital, Washington, currently with Doctors without Borders,Katiola, Côte d’Ivoire, and his colleagues.
In a study published In JAMA Psychiatry, the researchers followed 1,791,520 children (48.6% girls) born between Jan. 1, 1973, and Dec. 31, 2014, for up to 41 years using population-based registry data.
Overall, researchers found a 79% increased risk of an autism diagnosis (hazard ratio 1.79) and a 24% increased risk of a depression diagnosis (HR 1.24) for individuals exposed to any maternal infection in utero compared with those not exposed.
Similar increases in risk appeared when the data were broken down by type of infection. Hazard ratios for an autism diagnosis were 1.81 for exposure to a severe maternal infection and 1.89 for a maternal urinary tract infection; hazard ratios for depression were 1.24 and 1.30, respectively, for severe maternal infection and maternal urinary tract infection.
No increased risk in bipolar disorder, or other psychoses including schizophrenia were observed.
The findings were limited by several factors including the inclusion only of infections diagnosed in a hospital setting, and thus may not be generalizable to infections diagnosed in an outpatient setting, the researchers noted. However, the results “amplify the urgency to better understand the role of maternal infection during pregnancy on fetal brain development and suggest that prevention of infection (such as by influenza vaccination) or anti-inflammatory therapies may be important strategies for the primary prevention of some portion of autism and depression,” they said.
The researchers had no conflicts to disclose. The study was funded by grants from several organizations including the National Institutes of Health.
SOURCE: al-Haddad BJS et al. JAMA Psychiatry. doi:10.1001/jamapsychiatry.2019.0029.
FROM JAMA PSYCHIATRY
Alcohol-mediated renal denervation appears safe for BP reduction
WASHINGTON – Injection of dehydrated alcohol through the wall of the renal artery can be added to a growing list of renal denervation strategies that have been associated with sustained blood pressure reductions, according to data presented as a latebreaker at 2019 CRT meeting.

For the primary efficacy endpoint of change in systolic blood pressure at six months, the mean reduction six months after denervation was 11 mmHg as measured with 24-hour ambulatory blood pressure monitoring (ABPM), according to Horst Sievert, MD, PhD, Director of the CardioVascular Center, Frankfurt, Germany.
“Alcohol denervation was associated with efficient and safe lowering of systolic blood pressure,” reported Dr. Sievert, who said these data have prompted a new set of studies, including a phase 2 controlled trial that will evaluate the effect of renal denervation for control of blood pressure off-medication.
After consent was withdrawn from one patient, study results were available from 44 patients with treatment resistant hypertension who were enrolled in this initial study. Entry requirements included a mean systolic blood pressure greater than 150 mmHg while taking at least three antihypertensive medications from different classes.
In this study, the alcohol was delivered with a proprietary device called the Peregrine System™ infusion catheter (Ablation Solutions). This catheter is equipped with microneedles that remain retracted until the catheter is navigated into position. Once in the renal artery, the microneedles are deployed to inject alcohol into the perivascular space, which produces a neurolytic effect.
The technical success for delivery of the alcohol was achieved in 100% of the study group. There were no serious adverse events associated with treatment. Minor adverse events included those involving the access site, such as pain, as well as two dissections that resolved without treatment. One patient complained of abdominal pain on the day of the procedure, but that also resolved, according to Dr. Sievert.
Over the course of followup, patients remained on the therapies they were taking prior to the intervention. There was no change in antihypertensive therapy during the first month of followup in 84% of treated patients. Of those who did have a change in medication, all but one involved a reduction in medication prompted by improved blood pressure control. At six months, there was no change in medication for 73% of those evaluated.
Following alcohol denervation, there was a mean 7 mmHg reduction in diastolic pressure as measured with 24-hour ABBM.
Based on these data, a trials program is being launched. In addition to the phase 2 multinational off-medication trial, which is enrolling 300 patients who are being randomized to the alcohol denervation therapy or a sham control, an open-label crossover trial will be conducted to confirm the safety and tolerability of this approach.
Delivery of alcohol through the catheter device used in this study requires a renal artery diameter of at least 4 mm. This is a potential limitation for smaller individuals, but several other devices used for denervation share this requirement, according to Dr. Sievert.
The potential advantage of this approach is that “you can stay proximal,” according to Dr. Sievert, contrasting this technique with renal denervation by radiofrequency ablation. He explained that radiofrequency renal denervation requires a relatively distal approach to achieve an appropriate energy penetration for target nerve ablation. Further study is needed to determine whether more proximal delivery has any clinical advantage.
SOURCE: 2019 Cardiovascular Research Technologies (CRT) Meeting.
WASHINGTON – Injection of dehydrated alcohol through the wall of the renal artery can be added to a growing list of renal denervation strategies that have been associated with sustained blood pressure reductions, according to data presented as a latebreaker at 2019 CRT meeting.
For the primary efficacy endpoint of change in systolic blood pressure at six months, the mean reduction six months after denervation was 11 mmHg as measured with 24-hour ambulatory blood pressure monitoring (ABPM), according to Horst Sievert, MD, PhD, Director of the CardioVascular Center, Frankfurt, Germany.
“Alcohol denervation was associated with efficient and safe lowering of systolic blood pressure,” reported Dr. Sievert, who said these data have prompted a new set of studies, including a phase 2 controlled trial that will evaluate the effect of renal denervation for control of blood pressure off-medication.
After consent was withdrawn from one patient, study results were available from 44 patients with treatment resistant hypertension who were enrolled in this initial study. Entry requirements included a mean systolic blood pressure greater than 150 mmHg while taking at least three antihypertensive medications from different classes.
In this study, the alcohol was delivered with a proprietary device called the Peregrine System™ infusion catheter (Ablation Solutions). This catheter is equipped with microneedles that remain retracted until the catheter is navigated into position. Once in the renal artery, the microneedles are deployed to inject alcohol into the perivascular space, which produces a neurolytic effect.
The technical success for delivery of the alcohol was achieved in 100% of the study group. There were no serious adverse events associated with treatment. Minor adverse events included those involving the access site, such as pain, as well as two dissections that resolved without treatment. One patient complained of abdominal pain on the day of the procedure, but that also resolved, according to Dr. Sievert.
Over the course of followup, patients remained on the therapies they were taking prior to the intervention. There was no change in antihypertensive therapy during the first month of followup in 84% of treated patients. Of those who did have a change in medication, all but one involved a reduction in medication prompted by improved blood pressure control. At six months, there was no change in medication for 73% of those evaluated.
Following alcohol denervation, there was a mean 7 mmHg reduction in diastolic pressure as measured with 24-hour ABBM.
Based on these data, a trials program is being launched. In addition to the phase 2 multinational off-medication trial, which is enrolling 300 patients who are being randomized to the alcohol denervation therapy or a sham control, an open-label crossover trial will be conducted to confirm the safety and tolerability of this approach.
Delivery of alcohol through the catheter device used in this study requires a renal artery diameter of at least 4 mm. This is a potential limitation for smaller individuals, but several other devices used for denervation share this requirement, according to Dr. Sievert.
The potential advantage of this approach is that “you can stay proximal,” according to Dr. Sievert, contrasting this technique with renal denervation by radiofrequency ablation. He explained that radiofrequency renal denervation requires a relatively distal approach to achieve an appropriate energy penetration for target nerve ablation. Further study is needed to determine whether more proximal delivery has any clinical advantage.
SOURCE: 2019 Cardiovascular Research Technologies (CRT) Meeting.
WASHINGTON – Injection of dehydrated alcohol through the wall of the renal artery can be added to a growing list of renal denervation strategies that have been associated with sustained blood pressure reductions, according to data presented as a latebreaker at 2019 CRT meeting.
For the primary efficacy endpoint of change in systolic blood pressure at six months, the mean reduction six months after denervation was 11 mmHg as measured with 24-hour ambulatory blood pressure monitoring (ABPM), according to Horst Sievert, MD, PhD, Director of the CardioVascular Center, Frankfurt, Germany.
“Alcohol denervation was associated with efficient and safe lowering of systolic blood pressure,” reported Dr. Sievert, who said these data have prompted a new set of studies, including a phase 2 controlled trial that will evaluate the effect of renal denervation for control of blood pressure off-medication.
After consent was withdrawn from one patient, study results were available from 44 patients with treatment resistant hypertension who were enrolled in this initial study. Entry requirements included a mean systolic blood pressure greater than 150 mmHg while taking at least three antihypertensive medications from different classes.
In this study, the alcohol was delivered with a proprietary device called the Peregrine System™ infusion catheter (Ablation Solutions). This catheter is equipped with microneedles that remain retracted until the catheter is navigated into position. Once in the renal artery, the microneedles are deployed to inject alcohol into the perivascular space, which produces a neurolytic effect.
The technical success for delivery of the alcohol was achieved in 100% of the study group. There were no serious adverse events associated with treatment. Minor adverse events included those involving the access site, such as pain, as well as two dissections that resolved without treatment. One patient complained of abdominal pain on the day of the procedure, but that also resolved, according to Dr. Sievert.
Over the course of followup, patients remained on the therapies they were taking prior to the intervention. There was no change in antihypertensive therapy during the first month of followup in 84% of treated patients. Of those who did have a change in medication, all but one involved a reduction in medication prompted by improved blood pressure control. At six months, there was no change in medication for 73% of those evaluated.
Following alcohol denervation, there was a mean 7 mmHg reduction in diastolic pressure as measured with 24-hour ABBM.
Based on these data, a trials program is being launched. In addition to the phase 2 multinational off-medication trial, which is enrolling 300 patients who are being randomized to the alcohol denervation therapy or a sham control, an open-label crossover trial will be conducted to confirm the safety and tolerability of this approach.
Delivery of alcohol through the catheter device used in this study requires a renal artery diameter of at least 4 mm. This is a potential limitation for smaller individuals, but several other devices used for denervation share this requirement, according to Dr. Sievert.
The potential advantage of this approach is that “you can stay proximal,” according to Dr. Sievert, contrasting this technique with renal denervation by radiofrequency ablation. He explained that radiofrequency renal denervation requires a relatively distal approach to achieve an appropriate energy penetration for target nerve ablation. Further study is needed to determine whether more proximal delivery has any clinical advantage.
SOURCE: 2019 Cardiovascular Research Technologies (CRT) Meeting.
REPORTING FROM CRT 2019
Total plasma tau correlates with dementia onset, Alzheimer’s disease
The total tau level in blood plasma appears to predict both onset and progression of dementia and could be used to help refine research cohorts.

Blood samples from two large dementia research cohorts confirmed the finding: Each standard deviation in plasma tau above the median is associated with a 29% greater risk of incident all-cause dementia and a 35%increase in the risk of incident Alzheimer’s disease, Matthew P. Pase, PhD and colleagues wrote in JAMA Neurology. It also correlated positively with some neuropathological aspects of dementia: smaller hippocampus and a higher burden of neurofibrillary tangles in the medial temporal lobe, said Dr. Pase of The Florey Institute for Neuroscience and Mental Health. Victoria, Australia.
Plasma tau isn’t the highly sought Holy Grail of a simple Alzheimer’s blood test. But the finding could benefit the research world. As a study entry criteria, it could substantially decrease the number of subjects needed to validate an outcome of either all-cause dementia or Alzheimer’s disease. Abnormal tau is also a required finding for an Alzheimer’s diagnosis in revised NIA-AA Research Framework. And, the authors noted, although plasma tau wasn’t quite as accurate a predictor as CSF tau, a needle in the arm would be much more acceptable to many more patients than a lumbar puncture.
“Whereas we do not expect plasma t-tau cutoffs to enhance diagnostic certainty for any single patient, our results suggest that plasma t-tau could be associated with improved risk stratification at a population level, targeting persons for inclusion in prevention trials, thus improving the power and precision of clinical trials and potentially accelerating therapeutic pipelines and drug discovery,” the team wrote.
The study drew on stored plasma samples from subjects enrolled in the Framingham Heart Study (1,453) and the Memento study, a multicenter cohort of persons with mild cognitive impairment or subjective cognitive complaints recruited from memory clinics across France (367).
The Framingham cohort was followed for up to 10 years between baseline examination to incident event (median 6 years).
Over that time, 134 (9.2%) cases of dementia developed; most of these (105) were due possible, probable, or definite Alzheimer’s.
Plasma tau levels rose linearly as the cohort aged. Higher plasma t-tau levels were associated with proven AD risk factors, including female sex, lower education, and higher vascular risk factors. They did not differ by apolipoprotein epsilon 4 status (APOEe4).
After adjusting for age and sex, each stand deviation unit increase in the log of tau was associated with a 29% greater risk of incident all-cause dementia, and a 35% increase in the risk of incident Alzheimer’s dementia. Subjects with tau levels above the median had a 62% increased risk of all-cause dementia and a 76% greater risk of AD. Adding APOEe4 status and vascular risk factors to the analysis didn’t alter the associations.
“Plasma t-tau level improved risk discrimination for all dementia and AD dementia beyond age and sex,” the investigators wrote. “[It] was associated with improved risk discrimination … in both APOEe4 carriers and noncarriers.”
In a hypothetical 5-year clinical trial, enrolling subjects with total plasma tau greater than the median could reduce the estimated necessary sample size by 38% for an outcome of all-cause dementia and by 50% for one of Alzheimer’s. Selecting those with both elevated plasma tau and APOEe4 carriage could reduce the required sample by 69% for all-cause dementia outcomes and by 80% for Alzheimer’s outcomes.
In the neuropathologic study, each standard deviation unit increase was associated with more neurofibrillary tangles in the medial temporal lobe, more microinfarcts, and smaller hippocampal volume. There was no association with amyloid plaque in any brain region.
Subjects in the Memento study had a meant of 4 years of follow-up. Over that time, there were 76 cases of incident dementia, 55 of which were probable Alzheimer’s.
Each standard deviation unit increase was associated with a nonsignificant 14% greater risk of all-cause dementia and a significant 54% increase in the risk of incident Alzheimer’s.
CSF was drawn on the same day as plasma in 140 of these subjects. The addition of CSF boosted the predictive value; each standard deviation increase more than doubled the risk of both Alzheimer’s (HR 2.33). Each standard deviation unit increase in CSF t-tau increased the risk by 2.14.
“Plasma t-tau was weakly correlated with CSF t-tau in our study. This finding is consistent
with previous studies showing that the associations of plasma t-tau with CSF t-tau have been weak or nonexistent,” the authors wrote. But, “Despite a weak correlation between plasma and CSF t-tau, plasma t-tau was at least as strongly associated with the development of incident AD dementia.”
“Use of plasma t-tau in this manner could be likened to the measurement of the APOEe4 allele, which is not a biomarker of AD pathology providing diagnostic certainty for AD dementia but is still routinely used to power clinical trials by selecting at-risk individuals,” they concluded.
Dr. Pase had no financial disclosures.
SOURCE: Pase, M et al JAMA Neurol 2019 doi:10.1001/jamaneurol.2018.4666
The total tau level in blood plasma appears to predict both onset and progression of dementia and could be used to help refine research cohorts.
Blood samples from two large dementia research cohorts confirmed the finding: Each standard deviation in plasma tau above the median is associated with a 29% greater risk of incident all-cause dementia and a 35%increase in the risk of incident Alzheimer’s disease, Matthew P. Pase, PhD and colleagues wrote in JAMA Neurology. It also correlated positively with some neuropathological aspects of dementia: smaller hippocampus and a higher burden of neurofibrillary tangles in the medial temporal lobe, said Dr. Pase of The Florey Institute for Neuroscience and Mental Health. Victoria, Australia.
Plasma tau isn’t the highly sought Holy Grail of a simple Alzheimer’s blood test. But the finding could benefit the research world. As a study entry criteria, it could substantially decrease the number of subjects needed to validate an outcome of either all-cause dementia or Alzheimer’s disease. Abnormal tau is also a required finding for an Alzheimer’s diagnosis in revised NIA-AA Research Framework. And, the authors noted, although plasma tau wasn’t quite as accurate a predictor as CSF tau, a needle in the arm would be much more acceptable to many more patients than a lumbar puncture.
“Whereas we do not expect plasma t-tau cutoffs to enhance diagnostic certainty for any single patient, our results suggest that plasma t-tau could be associated with improved risk stratification at a population level, targeting persons for inclusion in prevention trials, thus improving the power and precision of clinical trials and potentially accelerating therapeutic pipelines and drug discovery,” the team wrote.
The study drew on stored plasma samples from subjects enrolled in the Framingham Heart Study (1,453) and the Memento study, a multicenter cohort of persons with mild cognitive impairment or subjective cognitive complaints recruited from memory clinics across France (367).
The Framingham cohort was followed for up to 10 years between baseline examination to incident event (median 6 years).
Over that time, 134 (9.2%) cases of dementia developed; most of these (105) were due possible, probable, or definite Alzheimer’s.
Plasma tau levels rose linearly as the cohort aged. Higher plasma t-tau levels were associated with proven AD risk factors, including female sex, lower education, and higher vascular risk factors. They did not differ by apolipoprotein epsilon 4 status (APOEe4).
After adjusting for age and sex, each stand deviation unit increase in the log of tau was associated with a 29% greater risk of incident all-cause dementia, and a 35% increase in the risk of incident Alzheimer’s dementia. Subjects with tau levels above the median had a 62% increased risk of all-cause dementia and a 76% greater risk of AD. Adding APOEe4 status and vascular risk factors to the analysis didn’t alter the associations.
“Plasma t-tau level improved risk discrimination for all dementia and AD dementia beyond age and sex,” the investigators wrote. “[It] was associated with improved risk discrimination … in both APOEe4 carriers and noncarriers.”
In a hypothetical 5-year clinical trial, enrolling subjects with total plasma tau greater than the median could reduce the estimated necessary sample size by 38% for an outcome of all-cause dementia and by 50% for one of Alzheimer’s. Selecting those with both elevated plasma tau and APOEe4 carriage could reduce the required sample by 69% for all-cause dementia outcomes and by 80% for Alzheimer’s outcomes.
In the neuropathologic study, each standard deviation unit increase was associated with more neurofibrillary tangles in the medial temporal lobe, more microinfarcts, and smaller hippocampal volume. There was no association with amyloid plaque in any brain region.
Subjects in the Memento study had a meant of 4 years of follow-up. Over that time, there were 76 cases of incident dementia, 55 of which were probable Alzheimer’s.
Each standard deviation unit increase was associated with a nonsignificant 14% greater risk of all-cause dementia and a significant 54% increase in the risk of incident Alzheimer’s.
CSF was drawn on the same day as plasma in 140 of these subjects. The addition of CSF boosted the predictive value; each standard deviation increase more than doubled the risk of both Alzheimer’s (HR 2.33). Each standard deviation unit increase in CSF t-tau increased the risk by 2.14.
“Plasma t-tau was weakly correlated with CSF t-tau in our study. This finding is consistent
with previous studies showing that the associations of plasma t-tau with CSF t-tau have been weak or nonexistent,” the authors wrote. But, “Despite a weak correlation between plasma and CSF t-tau, plasma t-tau was at least as strongly associated with the development of incident AD dementia.”
“Use of plasma t-tau in this manner could be likened to the measurement of the APOEe4 allele, which is not a biomarker of AD pathology providing diagnostic certainty for AD dementia but is still routinely used to power clinical trials by selecting at-risk individuals,” they concluded.
Dr. Pase had no financial disclosures.
SOURCE: Pase, M et al JAMA Neurol 2019 doi:10.1001/jamaneurol.2018.4666
The total tau level in blood plasma appears to predict both onset and progression of dementia and could be used to help refine research cohorts.
Blood samples from two large dementia research cohorts confirmed the finding: Each standard deviation in plasma tau above the median is associated with a 29% greater risk of incident all-cause dementia and a 35%increase in the risk of incident Alzheimer’s disease, Matthew P. Pase, PhD and colleagues wrote in JAMA Neurology. It also correlated positively with some neuropathological aspects of dementia: smaller hippocampus and a higher burden of neurofibrillary tangles in the medial temporal lobe, said Dr. Pase of The Florey Institute for Neuroscience and Mental Health. Victoria, Australia.
Plasma tau isn’t the highly sought Holy Grail of a simple Alzheimer’s blood test. But the finding could benefit the research world. As a study entry criteria, it could substantially decrease the number of subjects needed to validate an outcome of either all-cause dementia or Alzheimer’s disease. Abnormal tau is also a required finding for an Alzheimer’s diagnosis in revised NIA-AA Research Framework. And, the authors noted, although plasma tau wasn’t quite as accurate a predictor as CSF tau, a needle in the arm would be much more acceptable to many more patients than a lumbar puncture.
“Whereas we do not expect plasma t-tau cutoffs to enhance diagnostic certainty for any single patient, our results suggest that plasma t-tau could be associated with improved risk stratification at a population level, targeting persons for inclusion in prevention trials, thus improving the power and precision of clinical trials and potentially accelerating therapeutic pipelines and drug discovery,” the team wrote.
The study drew on stored plasma samples from subjects enrolled in the Framingham Heart Study (1,453) and the Memento study, a multicenter cohort of persons with mild cognitive impairment or subjective cognitive complaints recruited from memory clinics across France (367).
The Framingham cohort was followed for up to 10 years between baseline examination to incident event (median 6 years).
Over that time, 134 (9.2%) cases of dementia developed; most of these (105) were due possible, probable, or definite Alzheimer’s.
Plasma tau levels rose linearly as the cohort aged. Higher plasma t-tau levels were associated with proven AD risk factors, including female sex, lower education, and higher vascular risk factors. They did not differ by apolipoprotein epsilon 4 status (APOEe4).
After adjusting for age and sex, each stand deviation unit increase in the log of tau was associated with a 29% greater risk of incident all-cause dementia, and a 35% increase in the risk of incident Alzheimer’s dementia. Subjects with tau levels above the median had a 62% increased risk of all-cause dementia and a 76% greater risk of AD. Adding APOEe4 status and vascular risk factors to the analysis didn’t alter the associations.
“Plasma t-tau level improved risk discrimination for all dementia and AD dementia beyond age and sex,” the investigators wrote. “[It] was associated with improved risk discrimination … in both APOEe4 carriers and noncarriers.”
In a hypothetical 5-year clinical trial, enrolling subjects with total plasma tau greater than the median could reduce the estimated necessary sample size by 38% for an outcome of all-cause dementia and by 50% for one of Alzheimer’s. Selecting those with both elevated plasma tau and APOEe4 carriage could reduce the required sample by 69% for all-cause dementia outcomes and by 80% for Alzheimer’s outcomes.
In the neuropathologic study, each standard deviation unit increase was associated with more neurofibrillary tangles in the medial temporal lobe, more microinfarcts, and smaller hippocampal volume. There was no association with amyloid plaque in any brain region.
Subjects in the Memento study had a meant of 4 years of follow-up. Over that time, there were 76 cases of incident dementia, 55 of which were probable Alzheimer’s.
Each standard deviation unit increase was associated with a nonsignificant 14% greater risk of all-cause dementia and a significant 54% increase in the risk of incident Alzheimer’s.
CSF was drawn on the same day as plasma in 140 of these subjects. The addition of CSF boosted the predictive value; each standard deviation increase more than doubled the risk of both Alzheimer’s (HR 2.33). Each standard deviation unit increase in CSF t-tau increased the risk by 2.14.
“Plasma t-tau was weakly correlated with CSF t-tau in our study. This finding is consistent
with previous studies showing that the associations of plasma t-tau with CSF t-tau have been weak or nonexistent,” the authors wrote. But, “Despite a weak correlation between plasma and CSF t-tau, plasma t-tau was at least as strongly associated with the development of incident AD dementia.”
“Use of plasma t-tau in this manner could be likened to the measurement of the APOEe4 allele, which is not a biomarker of AD pathology providing diagnostic certainty for AD dementia but is still routinely used to power clinical trials by selecting at-risk individuals,” they concluded.
Dr. Pase had no financial disclosures.
SOURCE: Pase, M et al JAMA Neurol 2019 doi:10.1001/jamaneurol.2018.4666
FROM JAMA NEUROLOGY
AAD 2019 meeting wrap-up
Many common dermatologic drugs can be safely used during pregnancy. The 31-GEP test predicts the likelihood of metastasis for cutaneous melanoma. And bermekimab reduces lesions and cuts pain in patients with hidradenitis suppurativa.
Amazon Alexa
Apple Podcasts
Google Podcasts
Spotify
Many common dermatologic drugs can be safely used during pregnancy. The 31-GEP test predicts the likelihood of metastasis for cutaneous melanoma. And bermekimab reduces lesions and cuts pain in patients with hidradenitis suppurativa.
Amazon Alexa
Apple Podcasts
Google Podcasts
Spotify
Many common dermatologic drugs can be safely used during pregnancy. The 31-GEP test predicts the likelihood of metastasis for cutaneous melanoma. And bermekimab reduces lesions and cuts pain in patients with hidradenitis suppurativa.
Amazon Alexa
Apple Podcasts
Google Podcasts
Spotify
