User login
Calciphylaxis: Diagnostic and Treatment Pearls


Weight loss medications may have a role after bariatric surgery
NASHVILLE – .

Phentermine and topiramate were each prescribed to between 10% and 12.5% of bariatric surgery patients at Boston Medical Center in recent years. That figure had been steadily increasing since 2004, when data collection began, Nawfal W. Istfan, MD, PhD, said at the meeting presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
However, the center didn’t know how patients who had received medication fared for long-term maintenance of weight loss, compared with those who had surgery alone; also, there were no clinical guidelines for prescribing weight loss medications (WLMs). “Have we done those patients any benefit by prescribing weight loss medications after gastric bypass surgery?” asked Dr. Istfan. The answer from the Boston Medical Center data is a qualified “yes;” patients with the highest rates of weight regain who were adherent to their medication did see lower rates of regain, and fewer rapid weight regain events.
Comparing patients who received prescriptions with those who did not, patients with less weight loss at nadir were more likely to receive a prescription. “This could very well be the reason they were prescribed a medication: They did not lose as much weight, and they are more likely to ask us” for WLMs, said Dr. Istfan, an endocrinologist at Boston University. However, for those who were prescribed WLMs, the slope of regain was flatter than for those who didn’t receive medication. Of the 626 patients included in the study, 91 received phentermine alone, 54 topiramate alone, and 113 were prescribed both phentermine and topiramate. Three received lorcaserin.
Those receiving medication were similar to the total bariatric surgery population in terms of age, sex, comorbidities, socioeconomic status, and preoperative body mass index, said Dr. Istfan, the senior author in the study. However, Hispanic individuals were more likely to receive WLMs, he said.
Recognizing that “the ratio of weight regain to nadir weight is more indicative of overfeeding than other parameters,” Dr. Istfan said that he and his colleagues divided patients into quartiles of regain, based on this ratio. The quartiles fell out so that those who had the least regain either lost weight or regained less than 1.4%, to make up the first quartile. The second quartile included those who regained from 1.5% to 6.26%, while the third quartile ranged up to 14.29% regain. Those who regained 14.3% or more were in the highest quartile of weight regain.
In comparing characteristics of the quartiles, there were more African Americans in the two higher quartiles (P = .017). More patients had achieved maximal weight loss in the highest quartile of regain (P less than .0001), though preoperative BMI had also been higher in this group (P = .0008).
After statistical adjustment, the investigators found that for individuals who had the highest quartile of regain, patients who were adherent to their WLMs had significantly less weight regain than those who took no medication (P = .014). However, patients considered nonadherent saw no medication effect on weight regain. The differences were small overall, with adherent patients regaining about 27% of weight relative to their nadir and those who didn’t take WLMs regaining about 30%. These significant results were seen long after bariatric surgery, at about 7 years post surgery.
In another analysis that looked just at the quartile of patients with the highest regain rate, weight regain was significantly delayed among those who were prescribed – and were adherent to – WLMs (P = .023). The analysis used a threshold weight regain rate of 1.22% per month; levels lower than that did not see a significant drug effect, and the effect was not seen for those not adherent to their WLMs.
Finally, an adjusted statistical analysis compared those taking and not taking WLMs to see whether WLMs were effective at preventing weight regain in rolling 90-day intervals throughout the study period. Again, in the highest quartile, those who were adherent to WLMs had a lower odds ratio of hitting the 1.22%/month regain rate, compared with those not taking medication (OR, 0.570; 95% confidence interval, 0.371-0.877; P = .01). The effect was not significant for the nonadherent group (OR, 0.872; 95% CI, 0.593-1.284; P = .489).
As more bariatric procedures are being done, and as more patients are living with their surgeries, physicians are seeing more weight regain, said Dr. Istfan, noting that it’s important to assess efficacy of WLMs in the postsurgical population. “Recent work showed that by 5 years after gastric bypass, half of patients had regained more than 15% of their nadir weight, and two-thirds of patients had regained more than 20% of their total maximum weight loss, said Dr. Istfan (King WC et al. JAMA.2018;320:1560).
Typically, patients will see about a 35% weight loss at their nadir, with a gradual increase in weight gain beginning about 2 years after surgery. Though it’s true that a net weight loss of 25% is still good, it can be a misleading way to look at the data, “because it does not focus on the process of weight regain itself,” said Dr. Istfan.
“Despite the maintenance of substantial weight loss, weight regain is concerning: It’s the present and future, not the past,” he said.
Regaining weight necessarily means that patients are having excess nutrient intake and a net-positive energy balance; this state can be associated with oxidative stress, inflammation, and insulin resistance – all potential contributors to the recurrence of comorbidities.
What’s to be done about weight regain, if it’s a point of concern? One option, said Dr. Istfan, is to consider more surgery. Patients might want a “re-do;” techniques that have been tried include reshaping the pouch and doing an anastomosis plication. If a gastro-gastric fistula’s developed, that can be corrected, he said.
Some factors influencing regain can be targeted by behavioral therapy. These include addressing alcohol consumption, discouraging grazing, encouraging exercise, and assessing and modifying diet quality in general.
“There is a general reluctance on the part of physicians to use weight loss medications after bariatric surgery,” said Dr. Istfan. Reasons can include concern about further nutritional compromise, especially when thinking about long-term use of appetite-suppressing medications. Importantly, there aren’t clinical guidelines for prescribing WLMs after bariatric surgery, nor is there a strong body of prospective studies in this area.
Dr. Istfan noted that the medical and surgical bariatric teams collaborate closely at Boston Medical Center to provide pre- and postoperative assessment and management.
The long observational interval and ethnic and socioeconomic diversity of the study population are strengths, said Dr. Istfan. Also, the three different multivariable models converged to similar findings.
However, the study was retrospective, with some confounding likely, and each prescriber involved in the study may have varying prescribing practices. Also, adherence was assessed by follow-up medication appointments, a measure that likely introduced some inaccuracy.
“Weight loss medications are potentially effective tools to counter weight regain after bariatric surgery; prospective studies are needed to optimize the use of weight loss medications after bariatric surgery,” said Dr. Istfan.
Dr. Istfan reported no outside sources of funding, and no conflicts of interest.
AGA provides GIs with a comprehensive, multi-disciplinary process to guide and personalize innovative obesity care for safe and effective weight management. Learn more at http://ow.ly/fFA330mWKCn.
SOURCE: Anderson W et al. Obesity Week 2018, Abstract T-OR-2016.
NASHVILLE – .
Phentermine and topiramate were each prescribed to between 10% and 12.5% of bariatric surgery patients at Boston Medical Center in recent years. That figure had been steadily increasing since 2004, when data collection began, Nawfal W. Istfan, MD, PhD, said at the meeting presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
However, the center didn’t know how patients who had received medication fared for long-term maintenance of weight loss, compared with those who had surgery alone; also, there were no clinical guidelines for prescribing weight loss medications (WLMs). “Have we done those patients any benefit by prescribing weight loss medications after gastric bypass surgery?” asked Dr. Istfan. The answer from the Boston Medical Center data is a qualified “yes;” patients with the highest rates of weight regain who were adherent to their medication did see lower rates of regain, and fewer rapid weight regain events.
Comparing patients who received prescriptions with those who did not, patients with less weight loss at nadir were more likely to receive a prescription. “This could very well be the reason they were prescribed a medication: They did not lose as much weight, and they are more likely to ask us” for WLMs, said Dr. Istfan, an endocrinologist at Boston University. However, for those who were prescribed WLMs, the slope of regain was flatter than for those who didn’t receive medication. Of the 626 patients included in the study, 91 received phentermine alone, 54 topiramate alone, and 113 were prescribed both phentermine and topiramate. Three received lorcaserin.
Those receiving medication were similar to the total bariatric surgery population in terms of age, sex, comorbidities, socioeconomic status, and preoperative body mass index, said Dr. Istfan, the senior author in the study. However, Hispanic individuals were more likely to receive WLMs, he said.
Recognizing that “the ratio of weight regain to nadir weight is more indicative of overfeeding than other parameters,” Dr. Istfan said that he and his colleagues divided patients into quartiles of regain, based on this ratio. The quartiles fell out so that those who had the least regain either lost weight or regained less than 1.4%, to make up the first quartile. The second quartile included those who regained from 1.5% to 6.26%, while the third quartile ranged up to 14.29% regain. Those who regained 14.3% or more were in the highest quartile of weight regain.
In comparing characteristics of the quartiles, there were more African Americans in the two higher quartiles (P = .017). More patients had achieved maximal weight loss in the highest quartile of regain (P less than .0001), though preoperative BMI had also been higher in this group (P = .0008).
After statistical adjustment, the investigators found that for individuals who had the highest quartile of regain, patients who were adherent to their WLMs had significantly less weight regain than those who took no medication (P = .014). However, patients considered nonadherent saw no medication effect on weight regain. The differences were small overall, with adherent patients regaining about 27% of weight relative to their nadir and those who didn’t take WLMs regaining about 30%. These significant results were seen long after bariatric surgery, at about 7 years post surgery.
In another analysis that looked just at the quartile of patients with the highest regain rate, weight regain was significantly delayed among those who were prescribed – and were adherent to – WLMs (P = .023). The analysis used a threshold weight regain rate of 1.22% per month; levels lower than that did not see a significant drug effect, and the effect was not seen for those not adherent to their WLMs.
Finally, an adjusted statistical analysis compared those taking and not taking WLMs to see whether WLMs were effective at preventing weight regain in rolling 90-day intervals throughout the study period. Again, in the highest quartile, those who were adherent to WLMs had a lower odds ratio of hitting the 1.22%/month regain rate, compared with those not taking medication (OR, 0.570; 95% confidence interval, 0.371-0.877; P = .01). The effect was not significant for the nonadherent group (OR, 0.872; 95% CI, 0.593-1.284; P = .489).
As more bariatric procedures are being done, and as more patients are living with their surgeries, physicians are seeing more weight regain, said Dr. Istfan, noting that it’s important to assess efficacy of WLMs in the postsurgical population. “Recent work showed that by 5 years after gastric bypass, half of patients had regained more than 15% of their nadir weight, and two-thirds of patients had regained more than 20% of their total maximum weight loss, said Dr. Istfan (King WC et al. JAMA.2018;320:1560).
Typically, patients will see about a 35% weight loss at their nadir, with a gradual increase in weight gain beginning about 2 years after surgery. Though it’s true that a net weight loss of 25% is still good, it can be a misleading way to look at the data, “because it does not focus on the process of weight regain itself,” said Dr. Istfan.
“Despite the maintenance of substantial weight loss, weight regain is concerning: It’s the present and future, not the past,” he said.
Regaining weight necessarily means that patients are having excess nutrient intake and a net-positive energy balance; this state can be associated with oxidative stress, inflammation, and insulin resistance – all potential contributors to the recurrence of comorbidities.
What’s to be done about weight regain, if it’s a point of concern? One option, said Dr. Istfan, is to consider more surgery. Patients might want a “re-do;” techniques that have been tried include reshaping the pouch and doing an anastomosis plication. If a gastro-gastric fistula’s developed, that can be corrected, he said.
Some factors influencing regain can be targeted by behavioral therapy. These include addressing alcohol consumption, discouraging grazing, encouraging exercise, and assessing and modifying diet quality in general.
“There is a general reluctance on the part of physicians to use weight loss medications after bariatric surgery,” said Dr. Istfan. Reasons can include concern about further nutritional compromise, especially when thinking about long-term use of appetite-suppressing medications. Importantly, there aren’t clinical guidelines for prescribing WLMs after bariatric surgery, nor is there a strong body of prospective studies in this area.
Dr. Istfan noted that the medical and surgical bariatric teams collaborate closely at Boston Medical Center to provide pre- and postoperative assessment and management.
The long observational interval and ethnic and socioeconomic diversity of the study population are strengths, said Dr. Istfan. Also, the three different multivariable models converged to similar findings.
However, the study was retrospective, with some confounding likely, and each prescriber involved in the study may have varying prescribing practices. Also, adherence was assessed by follow-up medication appointments, a measure that likely introduced some inaccuracy.
“Weight loss medications are potentially effective tools to counter weight regain after bariatric surgery; prospective studies are needed to optimize the use of weight loss medications after bariatric surgery,” said Dr. Istfan.
Dr. Istfan reported no outside sources of funding, and no conflicts of interest.
AGA provides GIs with a comprehensive, multi-disciplinary process to guide and personalize innovative obesity care for safe and effective weight management. Learn more at http://ow.ly/fFA330mWKCn.
SOURCE: Anderson W et al. Obesity Week 2018, Abstract T-OR-2016.
NASHVILLE – .
Phentermine and topiramate were each prescribed to between 10% and 12.5% of bariatric surgery patients at Boston Medical Center in recent years. That figure had been steadily increasing since 2004, when data collection began, Nawfal W. Istfan, MD, PhD, said at the meeting presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
However, the center didn’t know how patients who had received medication fared for long-term maintenance of weight loss, compared with those who had surgery alone; also, there were no clinical guidelines for prescribing weight loss medications (WLMs). “Have we done those patients any benefit by prescribing weight loss medications after gastric bypass surgery?” asked Dr. Istfan. The answer from the Boston Medical Center data is a qualified “yes;” patients with the highest rates of weight regain who were adherent to their medication did see lower rates of regain, and fewer rapid weight regain events.
Comparing patients who received prescriptions with those who did not, patients with less weight loss at nadir were more likely to receive a prescription. “This could very well be the reason they were prescribed a medication: They did not lose as much weight, and they are more likely to ask us” for WLMs, said Dr. Istfan, an endocrinologist at Boston University. However, for those who were prescribed WLMs, the slope of regain was flatter than for those who didn’t receive medication. Of the 626 patients included in the study, 91 received phentermine alone, 54 topiramate alone, and 113 were prescribed both phentermine and topiramate. Three received lorcaserin.
Those receiving medication were similar to the total bariatric surgery population in terms of age, sex, comorbidities, socioeconomic status, and preoperative body mass index, said Dr. Istfan, the senior author in the study. However, Hispanic individuals were more likely to receive WLMs, he said.
Recognizing that “the ratio of weight regain to nadir weight is more indicative of overfeeding than other parameters,” Dr. Istfan said that he and his colleagues divided patients into quartiles of regain, based on this ratio. The quartiles fell out so that those who had the least regain either lost weight or regained less than 1.4%, to make up the first quartile. The second quartile included those who regained from 1.5% to 6.26%, while the third quartile ranged up to 14.29% regain. Those who regained 14.3% or more were in the highest quartile of weight regain.
In comparing characteristics of the quartiles, there were more African Americans in the two higher quartiles (P = .017). More patients had achieved maximal weight loss in the highest quartile of regain (P less than .0001), though preoperative BMI had also been higher in this group (P = .0008).
After statistical adjustment, the investigators found that for individuals who had the highest quartile of regain, patients who were adherent to their WLMs had significantly less weight regain than those who took no medication (P = .014). However, patients considered nonadherent saw no medication effect on weight regain. The differences were small overall, with adherent patients regaining about 27% of weight relative to their nadir and those who didn’t take WLMs regaining about 30%. These significant results were seen long after bariatric surgery, at about 7 years post surgery.
In another analysis that looked just at the quartile of patients with the highest regain rate, weight regain was significantly delayed among those who were prescribed – and were adherent to – WLMs (P = .023). The analysis used a threshold weight regain rate of 1.22% per month; levels lower than that did not see a significant drug effect, and the effect was not seen for those not adherent to their WLMs.
Finally, an adjusted statistical analysis compared those taking and not taking WLMs to see whether WLMs were effective at preventing weight regain in rolling 90-day intervals throughout the study period. Again, in the highest quartile, those who were adherent to WLMs had a lower odds ratio of hitting the 1.22%/month regain rate, compared with those not taking medication (OR, 0.570; 95% confidence interval, 0.371-0.877; P = .01). The effect was not significant for the nonadherent group (OR, 0.872; 95% CI, 0.593-1.284; P = .489).
As more bariatric procedures are being done, and as more patients are living with their surgeries, physicians are seeing more weight regain, said Dr. Istfan, noting that it’s important to assess efficacy of WLMs in the postsurgical population. “Recent work showed that by 5 years after gastric bypass, half of patients had regained more than 15% of their nadir weight, and two-thirds of patients had regained more than 20% of their total maximum weight loss, said Dr. Istfan (King WC et al. JAMA.2018;320:1560).
Typically, patients will see about a 35% weight loss at their nadir, with a gradual increase in weight gain beginning about 2 years after surgery. Though it’s true that a net weight loss of 25% is still good, it can be a misleading way to look at the data, “because it does not focus on the process of weight regain itself,” said Dr. Istfan.
“Despite the maintenance of substantial weight loss, weight regain is concerning: It’s the present and future, not the past,” he said.
Regaining weight necessarily means that patients are having excess nutrient intake and a net-positive energy balance; this state can be associated with oxidative stress, inflammation, and insulin resistance – all potential contributors to the recurrence of comorbidities.
What’s to be done about weight regain, if it’s a point of concern? One option, said Dr. Istfan, is to consider more surgery. Patients might want a “re-do;” techniques that have been tried include reshaping the pouch and doing an anastomosis plication. If a gastro-gastric fistula’s developed, that can be corrected, he said.
Some factors influencing regain can be targeted by behavioral therapy. These include addressing alcohol consumption, discouraging grazing, encouraging exercise, and assessing and modifying diet quality in general.
“There is a general reluctance on the part of physicians to use weight loss medications after bariatric surgery,” said Dr. Istfan. Reasons can include concern about further nutritional compromise, especially when thinking about long-term use of appetite-suppressing medications. Importantly, there aren’t clinical guidelines for prescribing WLMs after bariatric surgery, nor is there a strong body of prospective studies in this area.
Dr. Istfan noted that the medical and surgical bariatric teams collaborate closely at Boston Medical Center to provide pre- and postoperative assessment and management.
The long observational interval and ethnic and socioeconomic diversity of the study population are strengths, said Dr. Istfan. Also, the three different multivariable models converged to similar findings.
However, the study was retrospective, with some confounding likely, and each prescriber involved in the study may have varying prescribing practices. Also, adherence was assessed by follow-up medication appointments, a measure that likely introduced some inaccuracy.
“Weight loss medications are potentially effective tools to counter weight regain after bariatric surgery; prospective studies are needed to optimize the use of weight loss medications after bariatric surgery,” said Dr. Istfan.
Dr. Istfan reported no outside sources of funding, and no conflicts of interest.
AGA provides GIs with a comprehensive, multi-disciplinary process to guide and personalize innovative obesity care for safe and effective weight management. Learn more at http://ow.ly/fFA330mWKCn.
SOURCE: Anderson W et al. Obesity Week 2018, Abstract T-OR-2016.
REPORTING FROM OBESITY WEEK 2018
Key clinical point: Weight loss medication flattened the curve of weight regain after bariatric surgery – for some patients.
Major finding: Weight loss medicine reduced regain among those who had the most weight regain (P =.014).
Study details: Retrospective single-center cohort study of 626 bariatric surgery patients.
Disclosures: The study authors reported no external sources of funding. Dr. Istfan reported no conflicts of interest.
Source: Anderson W et al. Obesity Week 2018, Abstract T-OR-2016.
Cortical Thickness Changes in Chronic Migraine
Chronic migraine (CM) patients have significantly greater cortical covariance compared to controls, according to a recent study. Cortical thickness in CM patients was predominantly accounted for by CM duration, posttraumatic stress disorder (PTSD), and poor sleep quality, while improved pain self‐efficacy buffered cortical thickness. Thirty CM cases (mean age 40 years; male‐to‐female 1:4) and 30 sex‐matched healthy controls (mean age 40 years) were enrolled. Participants completed self‐administered and standardized questionnaires assessing headache‐related clinical features and common psychological comorbidities. T1‐weighted brain images were acquired on a 3T MRI and a whole‐brain cortical thickness analysis was performed.
Researchers found:
- The whole brain cortical thickness analysis revealed no significant differences between CM patients and controls.
- However, significant associations between clinical features and cortical thickness were observed for the patients only.
- These associations included the right superior temporal sulcus (R2 = 0.72) and the right insula (R2 = 0.71) with distinct clinical variables (ie, longer history of CM, PTSD, sleep quality, pain self‐efficacy, and somatic symptoms).
Woldeamanuel YW, DeSouza DD, Sanjanwala BM, Cowan RP. Clinical features contributing to cortical thickness changes in chronic migraine–A pilot study. [Published online ahead of print November 23, 2018]. Headache. doi:10.1111/head.13452.
Chronic migraine (CM) patients have significantly greater cortical covariance compared to controls, according to a recent study. Cortical thickness in CM patients was predominantly accounted for by CM duration, posttraumatic stress disorder (PTSD), and poor sleep quality, while improved pain self‐efficacy buffered cortical thickness. Thirty CM cases (mean age 40 years; male‐to‐female 1:4) and 30 sex‐matched healthy controls (mean age 40 years) were enrolled. Participants completed self‐administered and standardized questionnaires assessing headache‐related clinical features and common psychological comorbidities. T1‐weighted brain images were acquired on a 3T MRI and a whole‐brain cortical thickness analysis was performed.
Researchers found:
- The whole brain cortical thickness analysis revealed no significant differences between CM patients and controls.
- However, significant associations between clinical features and cortical thickness were observed for the patients only.
- These associations included the right superior temporal sulcus (R2 = 0.72) and the right insula (R2 = 0.71) with distinct clinical variables (ie, longer history of CM, PTSD, sleep quality, pain self‐efficacy, and somatic symptoms).
Woldeamanuel YW, DeSouza DD, Sanjanwala BM, Cowan RP. Clinical features contributing to cortical thickness changes in chronic migraine–A pilot study. [Published online ahead of print November 23, 2018]. Headache. doi:10.1111/head.13452.
Chronic migraine (CM) patients have significantly greater cortical covariance compared to controls, according to a recent study. Cortical thickness in CM patients was predominantly accounted for by CM duration, posttraumatic stress disorder (PTSD), and poor sleep quality, while improved pain self‐efficacy buffered cortical thickness. Thirty CM cases (mean age 40 years; male‐to‐female 1:4) and 30 sex‐matched healthy controls (mean age 40 years) were enrolled. Participants completed self‐administered and standardized questionnaires assessing headache‐related clinical features and common psychological comorbidities. T1‐weighted brain images were acquired on a 3T MRI and a whole‐brain cortical thickness analysis was performed.
Researchers found:
- The whole brain cortical thickness analysis revealed no significant differences between CM patients and controls.
- However, significant associations between clinical features and cortical thickness were observed for the patients only.
- These associations included the right superior temporal sulcus (R2 = 0.72) and the right insula (R2 = 0.71) with distinct clinical variables (ie, longer history of CM, PTSD, sleep quality, pain self‐efficacy, and somatic symptoms).
Woldeamanuel YW, DeSouza DD, Sanjanwala BM, Cowan RP. Clinical features contributing to cortical thickness changes in chronic migraine–A pilot study. [Published online ahead of print November 23, 2018]. Headache. doi:10.1111/head.13452.
The power of the turkey sandwich
A relatively high proportion of pediatric visits to the emergency department are related to psychiatric symptoms, oftentimes with suicidal or violent ideation.1 Given that pediatric emergencies related to psychiatric symptoms are on the increase, clinicians frequently are called to assess children and adolescents with symptoms of aggression and violence. Management of these cases can be tricky.

Case presentation
Henry is a 6-year-old boy with mild developmental delays and possible anxiety who was brought to the emergency department because of concerns on the bus. For about a month, Henry, who is repeating his kindergarten year, had been struggling with getting on and off the bus and with other transitions at school. These struggles had been attributed to anxiety. He was started on sertraline and the dose was increased about 2 weeks later. Soon thereafter he complained of stomach upset with the sertraline, refused to take the medicine, and had a very hard day at school. He required one-on-one attention for unsafe behavior most of that day, and he missed most of his lunch and recess. His school support team was able to get him onto the bus at the end of the day, but he refused to get off of the bus at home. He became violent with the bus driver, kicking and biting him until the police were called. The police called EMS and he was brought into the emergency department after fighting to get on the transport stretcher. He was eventually brought into a secure exam room in the emergency department, but was unable to be fully assessed because he would only make animal noises when approached. His father already had been called, but was unable to calm him down. The emergency department physician was unable to approach Henry because he began swinging at him as soon as the physician entered the room. An emergent psychiatric consultation was called to determine what medication to give to Henry to calm him down and to assess him for possible psychosis.
Case discussion
It sounds like Henry was having a severe tantrum exacerbated by a number of factors. First of all, this is a child who struggles with transitions. That day had been loaded with transitions, eventually leading him to be in an unfamiliar environment with many unfamiliar faces. Even the familiar face of his father wasn’t enough to help because he was overly stimulated and scared. Next, he was probably hungry. We know for certain that he missed lunch, and several hours into his presentation there were no breaks to deal with his basic needs. The first approach to assessment of aggressive behavior in the emergency setting is to try to care for the basic needs of the individual to deescalate the situation. Finally, he had recently been started on sertraline, a selective serotonin reuptake inhibitor. He had been having some dyspepsia and/or nausea with the sertraline, leading to his having missed some doses. Some children and adolescents have a discontinuation syndrome, which can be more severe in younger children and with medications that have shorter half-lives.2 In Henry’s case, a missed dose or two can be enough to trigger this discontinuation response leading to more aggressive behavior.
Case follow-up
The child and adolescent psychiatrist called to the case received a history from the primary team. When he started to try to talk with the parent outside of the room, the child became upset. He was able to gather the information that Henry also had skipped breakfast. In an attempt to calm the patient down, the psychiatrist addressed Henry using a nonjudgmental, nonconfrontational, collaborative approach, incorporating play. Henry responded to this approach and allowed the psychiatrist to ask a few questions about basic needs, and admitted that he was hungry. He was offered a turkey sandwich, which was rapidly ingested. The tantrum slowly subsided. Within about 30 minutes (and with some more food), the child was able to sit on his parent’s lap and finish the interview. The decision was made to have him follow up with his primary care provider to change to an SSRI with a longer half-life, such as fluoxetine, as he did seem to be experiencing some discontinuation even after missing just a dose or two of sertraline.

When dealing with emergent, aggressive behavior, food isn’t always the best medicine, but sometimes it is. Environmental barriers to the child’s regaining control include hunger, thirst, a full bladder, constipation, or other pain. Attending to these issues first sometimes can help avoid sedating medications, which can prolong emergency visits and lead to unwelcome side effects.
Dr. Althoff is associate professor of psychiatry, psychology, and pediatrics at the University of Vermont, Burlington. He is director of the division of behavioral genetics and conducts research on the development of self-regulation in children. Email him at [email protected].
References
1. Pediatrics. 2011 May;127(5):e1356-66.
2. J Can Acad Child Adolesc Psychiatry. 2011 Feb;20(1):60-7.
A relatively high proportion of pediatric visits to the emergency department are related to psychiatric symptoms, oftentimes with suicidal or violent ideation.1 Given that pediatric emergencies related to psychiatric symptoms are on the increase, clinicians frequently are called to assess children and adolescents with symptoms of aggression and violence. Management of these cases can be tricky.
Case presentation
Henry is a 6-year-old boy with mild developmental delays and possible anxiety who was brought to the emergency department because of concerns on the bus. For about a month, Henry, who is repeating his kindergarten year, had been struggling with getting on and off the bus and with other transitions at school. These struggles had been attributed to anxiety. He was started on sertraline and the dose was increased about 2 weeks later. Soon thereafter he complained of stomach upset with the sertraline, refused to take the medicine, and had a very hard day at school. He required one-on-one attention for unsafe behavior most of that day, and he missed most of his lunch and recess. His school support team was able to get him onto the bus at the end of the day, but he refused to get off of the bus at home. He became violent with the bus driver, kicking and biting him until the police were called. The police called EMS and he was brought into the emergency department after fighting to get on the transport stretcher. He was eventually brought into a secure exam room in the emergency department, but was unable to be fully assessed because he would only make animal noises when approached. His father already had been called, but was unable to calm him down. The emergency department physician was unable to approach Henry because he began swinging at him as soon as the physician entered the room. An emergent psychiatric consultation was called to determine what medication to give to Henry to calm him down and to assess him for possible psychosis.
Case discussion
It sounds like Henry was having a severe tantrum exacerbated by a number of factors. First of all, this is a child who struggles with transitions. That day had been loaded with transitions, eventually leading him to be in an unfamiliar environment with many unfamiliar faces. Even the familiar face of his father wasn’t enough to help because he was overly stimulated and scared. Next, he was probably hungry. We know for certain that he missed lunch, and several hours into his presentation there were no breaks to deal with his basic needs. The first approach to assessment of aggressive behavior in the emergency setting is to try to care for the basic needs of the individual to deescalate the situation. Finally, he had recently been started on sertraline, a selective serotonin reuptake inhibitor. He had been having some dyspepsia and/or nausea with the sertraline, leading to his having missed some doses. Some children and adolescents have a discontinuation syndrome, which can be more severe in younger children and with medications that have shorter half-lives.2 In Henry’s case, a missed dose or two can be enough to trigger this discontinuation response leading to more aggressive behavior.
Case follow-up
The child and adolescent psychiatrist called to the case received a history from the primary team. When he started to try to talk with the parent outside of the room, the child became upset. He was able to gather the information that Henry also had skipped breakfast. In an attempt to calm the patient down, the psychiatrist addressed Henry using a nonjudgmental, nonconfrontational, collaborative approach, incorporating play. Henry responded to this approach and allowed the psychiatrist to ask a few questions about basic needs, and admitted that he was hungry. He was offered a turkey sandwich, which was rapidly ingested. The tantrum slowly subsided. Within about 30 minutes (and with some more food), the child was able to sit on his parent’s lap and finish the interview. The decision was made to have him follow up with his primary care provider to change to an SSRI with a longer half-life, such as fluoxetine, as he did seem to be experiencing some discontinuation even after missing just a dose or two of sertraline.
When dealing with emergent, aggressive behavior, food isn’t always the best medicine, but sometimes it is. Environmental barriers to the child’s regaining control include hunger, thirst, a full bladder, constipation, or other pain. Attending to these issues first sometimes can help avoid sedating medications, which can prolong emergency visits and lead to unwelcome side effects.
Dr. Althoff is associate professor of psychiatry, psychology, and pediatrics at the University of Vermont, Burlington. He is director of the division of behavioral genetics and conducts research on the development of self-regulation in children. Email him at [email protected].
References
1. Pediatrics. 2011 May;127(5):e1356-66.
2. J Can Acad Child Adolesc Psychiatry. 2011 Feb;20(1):60-7.
A relatively high proportion of pediatric visits to the emergency department are related to psychiatric symptoms, oftentimes with suicidal or violent ideation.1 Given that pediatric emergencies related to psychiatric symptoms are on the increase, clinicians frequently are called to assess children and adolescents with symptoms of aggression and violence. Management of these cases can be tricky.
Case presentation
Henry is a 6-year-old boy with mild developmental delays and possible anxiety who was brought to the emergency department because of concerns on the bus. For about a month, Henry, who is repeating his kindergarten year, had been struggling with getting on and off the bus and with other transitions at school. These struggles had been attributed to anxiety. He was started on sertraline and the dose was increased about 2 weeks later. Soon thereafter he complained of stomach upset with the sertraline, refused to take the medicine, and had a very hard day at school. He required one-on-one attention for unsafe behavior most of that day, and he missed most of his lunch and recess. His school support team was able to get him onto the bus at the end of the day, but he refused to get off of the bus at home. He became violent with the bus driver, kicking and biting him until the police were called. The police called EMS and he was brought into the emergency department after fighting to get on the transport stretcher. He was eventually brought into a secure exam room in the emergency department, but was unable to be fully assessed because he would only make animal noises when approached. His father already had been called, but was unable to calm him down. The emergency department physician was unable to approach Henry because he began swinging at him as soon as the physician entered the room. An emergent psychiatric consultation was called to determine what medication to give to Henry to calm him down and to assess him for possible psychosis.
Case discussion
It sounds like Henry was having a severe tantrum exacerbated by a number of factors. First of all, this is a child who struggles with transitions. That day had been loaded with transitions, eventually leading him to be in an unfamiliar environment with many unfamiliar faces. Even the familiar face of his father wasn’t enough to help because he was overly stimulated and scared. Next, he was probably hungry. We know for certain that he missed lunch, and several hours into his presentation there were no breaks to deal with his basic needs. The first approach to assessment of aggressive behavior in the emergency setting is to try to care for the basic needs of the individual to deescalate the situation. Finally, he had recently been started on sertraline, a selective serotonin reuptake inhibitor. He had been having some dyspepsia and/or nausea with the sertraline, leading to his having missed some doses. Some children and adolescents have a discontinuation syndrome, which can be more severe in younger children and with medications that have shorter half-lives.2 In Henry’s case, a missed dose or two can be enough to trigger this discontinuation response leading to more aggressive behavior.
Case follow-up
The child and adolescent psychiatrist called to the case received a history from the primary team. When he started to try to talk with the parent outside of the room, the child became upset. He was able to gather the information that Henry also had skipped breakfast. In an attempt to calm the patient down, the psychiatrist addressed Henry using a nonjudgmental, nonconfrontational, collaborative approach, incorporating play. Henry responded to this approach and allowed the psychiatrist to ask a few questions about basic needs, and admitted that he was hungry. He was offered a turkey sandwich, which was rapidly ingested. The tantrum slowly subsided. Within about 30 minutes (and with some more food), the child was able to sit on his parent’s lap and finish the interview. The decision was made to have him follow up with his primary care provider to change to an SSRI with a longer half-life, such as fluoxetine, as he did seem to be experiencing some discontinuation even after missing just a dose or two of sertraline.
When dealing with emergent, aggressive behavior, food isn’t always the best medicine, but sometimes it is. Environmental barriers to the child’s regaining control include hunger, thirst, a full bladder, constipation, or other pain. Attending to these issues first sometimes can help avoid sedating medications, which can prolong emergency visits and lead to unwelcome side effects.
Dr. Althoff is associate professor of psychiatry, psychology, and pediatrics at the University of Vermont, Burlington. He is director of the division of behavioral genetics and conducts research on the development of self-regulation in children. Email him at [email protected].
References
1. Pediatrics. 2011 May;127(5):e1356-66.
2. J Can Acad Child Adolesc Psychiatry. 2011 Feb;20(1):60-7.
Phase 3 data support apixaban for cancer-associated VTE
SAN DIEGO – according to the Phase 3 ADAM VTE trial.
The rates of major bleeding and clinically relevant nonmajor bleeding in patients who received apixaban were similar to those in patients who received dalteparin. However, the rate of VTE recurrence was significantly lower with apixaban than it was with dalteparin.
“[A]pixaban was associated with very low bleeding rates and venous thrombosis recurrence rates compared to dalteparin,” said Robert D. McBane II, MD, of the Mayo Clinic in Rochester, Minn., at the annual meeting of the American Society of Hematology.
The trial included 300 adults (aged 18 years and older) with active cancer and acute VTE who were randomized to receive apixaban (n = 150) or dalteparin (n = 150). The dose and schedule for oral apixaban was 10 mg twice daily for 7 days followed by 5 mg twice daily for 6 months. Dalteparin was given subcutaneously at 200 IU/kg per day for 1 month followed by 150 IU/kg daily for 6 months. Among the patients in the study, 145 patients in the apixaban arm and 142 in the dalteparin arm ultimately received their assigned treatment.
Every month, patients completed an anticoagulation satisfaction survey and bruise survey (a modification of the Duke Anticoagulation Satisfaction Scale). They also underwent lab testing (complete blood count, liver and renal function testing) and were assessed for outcomes, medication reconciliation, drug compliance, and ECOG status on a monthly basis.
Patient characteristics
Baseline characteristics were similar between the treatment arms. The mean age was 64 years in both arms, and roughly half of patients in both arms were female. Hematologic malignancies were present in 9% of patients in the apixaban arm and 11% in the dalteparin arm. Others included lung, colorectal,
pancreatic/hepatobiliary, gynecologic, breast, genitourinary, upper gastrointestinal, and brain cancers.
Of patients in the study, 65% of those in the apixaban arm and 66% in the dalteparin arm had distant metastasis, and 74% of patients in both arms were receiving chemotherapy while on study.
Patients had the following qualifying thrombotic events:
- Any pulmonary embolism (PE) – 55% of patients in the apixaban arm and 51% in the dalteparin arm
- Any deep vein thrombosis (DVT) – 48% and 47%, respectively
- PE only – 44% and 39%, respectively
- PE with DVT – 12% in both arms
- DVT only – 37% and 35%, respectively
- Lower extremity DVT – 31% and 34%, respectively
- Upper extremity DVT – 17% and 14%, respectively
- Cerebral venous thrombosis (VT) – 1% and 0%, respectively
- Splanchnic VT – 8% and 18%, respectively.
Bleeding, thrombosis, and death
The study’s primary endpoint was major bleeding, which did not occur in any of the apixaban-treated patients. However, major bleeding did occur in two (1.4%) patients in the dalteparin arm (P = .14).
A secondary endpoint was major bleeding plus clinically relevant nonmajor bleeding. This occurred in nine (6.2%) patients in the apixaban arm and nine (6.3%) in the dalteparin arm (P = .88).
The researchers also assessed VTE recurrence. One patient in the apixaban arm (0.7%) and nine in the dalteparin arm (6.3%) had VTE recurrence (P = .03).
The patient in the apixaban arm experienced cerebral VT, and the patients with recurrence in the dalteparin arm had leg (n = 4) or arm (n = 2) VTE, PE (n = 1), or splanchnic VT (n = 2).
One patient in each arm (0.7%) had arterial thrombosis.
There was no significant difference in cumulative mortality between the treatment arms (hazard ratio, 1.40; P = .3078).
Satisfaction and discontinuation
Overall, apixaban fared better than dalteparin in the monthly patient satisfaction surveys. At various time points, apixaban-treated patients were significantly less likely to be concerned about excessive bruising, find anticoagulant treatment a burden or difficult to carry out, or say anticoagulant treatment added stress to their lives, negatively impacted their quality of life, or caused them “a great deal” of worry, irritation, or frustration.
However, apixaban-treated patients were less likely than dalteparin recipients to have confidence that their drug protected them from VTE recurrence, while the apixaban recipients were more likely than the dalteparin group to report overall satisfaction with their treatment.
In addition, premature treatment discontinuation was more common in the dalteparin group than in the apixaban group – 15% and 4%, respectively (P = .0012).
“Apixaban was well tolerated with superior patient safety satisfaction, as well as significantly fewer study drug discontinuations compared to dalteparin,” Dr. McBane said. “I believe that these data support the use of apixaban for the acute treatment of cancer-associated venous thromboembolism.”
This study was funded by BMS/Pfizer Alliance. Dr. McBane declared no other conflicts of interest.
SOURCE: McBane RD et al. ASH 2018, Abstract 421.
SAN DIEGO – according to the Phase 3 ADAM VTE trial.
The rates of major bleeding and clinically relevant nonmajor bleeding in patients who received apixaban were similar to those in patients who received dalteparin. However, the rate of VTE recurrence was significantly lower with apixaban than it was with dalteparin.
“[A]pixaban was associated with very low bleeding rates and venous thrombosis recurrence rates compared to dalteparin,” said Robert D. McBane II, MD, of the Mayo Clinic in Rochester, Minn., at the annual meeting of the American Society of Hematology.
The trial included 300 adults (aged 18 years and older) with active cancer and acute VTE who were randomized to receive apixaban (n = 150) or dalteparin (n = 150). The dose and schedule for oral apixaban was 10 mg twice daily for 7 days followed by 5 mg twice daily for 6 months. Dalteparin was given subcutaneously at 200 IU/kg per day for 1 month followed by 150 IU/kg daily for 6 months. Among the patients in the study, 145 patients in the apixaban arm and 142 in the dalteparin arm ultimately received their assigned treatment.
Every month, patients completed an anticoagulation satisfaction survey and bruise survey (a modification of the Duke Anticoagulation Satisfaction Scale). They also underwent lab testing (complete blood count, liver and renal function testing) and were assessed for outcomes, medication reconciliation, drug compliance, and ECOG status on a monthly basis.
Patient characteristics
Baseline characteristics were similar between the treatment arms. The mean age was 64 years in both arms, and roughly half of patients in both arms were female. Hematologic malignancies were present in 9% of patients in the apixaban arm and 11% in the dalteparin arm. Others included lung, colorectal,
pancreatic/hepatobiliary, gynecologic, breast, genitourinary, upper gastrointestinal, and brain cancers.
Of patients in the study, 65% of those in the apixaban arm and 66% in the dalteparin arm had distant metastasis, and 74% of patients in both arms were receiving chemotherapy while on study.
Patients had the following qualifying thrombotic events:
- Any pulmonary embolism (PE) – 55% of patients in the apixaban arm and 51% in the dalteparin arm
- Any deep vein thrombosis (DVT) – 48% and 47%, respectively
- PE only – 44% and 39%, respectively
- PE with DVT – 12% in both arms
- DVT only – 37% and 35%, respectively
- Lower extremity DVT – 31% and 34%, respectively
- Upper extremity DVT – 17% and 14%, respectively
- Cerebral venous thrombosis (VT) – 1% and 0%, respectively
- Splanchnic VT – 8% and 18%, respectively.
Bleeding, thrombosis, and death
The study’s primary endpoint was major bleeding, which did not occur in any of the apixaban-treated patients. However, major bleeding did occur in two (1.4%) patients in the dalteparin arm (P = .14).
A secondary endpoint was major bleeding plus clinically relevant nonmajor bleeding. This occurred in nine (6.2%) patients in the apixaban arm and nine (6.3%) in the dalteparin arm (P = .88).
The researchers also assessed VTE recurrence. One patient in the apixaban arm (0.7%) and nine in the dalteparin arm (6.3%) had VTE recurrence (P = .03).
The patient in the apixaban arm experienced cerebral VT, and the patients with recurrence in the dalteparin arm had leg (n = 4) or arm (n = 2) VTE, PE (n = 1), or splanchnic VT (n = 2).
One patient in each arm (0.7%) had arterial thrombosis.
There was no significant difference in cumulative mortality between the treatment arms (hazard ratio, 1.40; P = .3078).
Satisfaction and discontinuation
Overall, apixaban fared better than dalteparin in the monthly patient satisfaction surveys. At various time points, apixaban-treated patients were significantly less likely to be concerned about excessive bruising, find anticoagulant treatment a burden or difficult to carry out, or say anticoagulant treatment added stress to their lives, negatively impacted their quality of life, or caused them “a great deal” of worry, irritation, or frustration.
However, apixaban-treated patients were less likely than dalteparin recipients to have confidence that their drug protected them from VTE recurrence, while the apixaban recipients were more likely than the dalteparin group to report overall satisfaction with their treatment.
In addition, premature treatment discontinuation was more common in the dalteparin group than in the apixaban group – 15% and 4%, respectively (P = .0012).
“Apixaban was well tolerated with superior patient safety satisfaction, as well as significantly fewer study drug discontinuations compared to dalteparin,” Dr. McBane said. “I believe that these data support the use of apixaban for the acute treatment of cancer-associated venous thromboembolism.”
This study was funded by BMS/Pfizer Alliance. Dr. McBane declared no other conflicts of interest.
SOURCE: McBane RD et al. ASH 2018, Abstract 421.
SAN DIEGO – according to the Phase 3 ADAM VTE trial.
The rates of major bleeding and clinically relevant nonmajor bleeding in patients who received apixaban were similar to those in patients who received dalteparin. However, the rate of VTE recurrence was significantly lower with apixaban than it was with dalteparin.
“[A]pixaban was associated with very low bleeding rates and venous thrombosis recurrence rates compared to dalteparin,” said Robert D. McBane II, MD, of the Mayo Clinic in Rochester, Minn., at the annual meeting of the American Society of Hematology.
The trial included 300 adults (aged 18 years and older) with active cancer and acute VTE who were randomized to receive apixaban (n = 150) or dalteparin (n = 150). The dose and schedule for oral apixaban was 10 mg twice daily for 7 days followed by 5 mg twice daily for 6 months. Dalteparin was given subcutaneously at 200 IU/kg per day for 1 month followed by 150 IU/kg daily for 6 months. Among the patients in the study, 145 patients in the apixaban arm and 142 in the dalteparin arm ultimately received their assigned treatment.
Every month, patients completed an anticoagulation satisfaction survey and bruise survey (a modification of the Duke Anticoagulation Satisfaction Scale). They also underwent lab testing (complete blood count, liver and renal function testing) and were assessed for outcomes, medication reconciliation, drug compliance, and ECOG status on a monthly basis.
Patient characteristics
Baseline characteristics were similar between the treatment arms. The mean age was 64 years in both arms, and roughly half of patients in both arms were female. Hematologic malignancies were present in 9% of patients in the apixaban arm and 11% in the dalteparin arm. Others included lung, colorectal,
pancreatic/hepatobiliary, gynecologic, breast, genitourinary, upper gastrointestinal, and brain cancers.
Of patients in the study, 65% of those in the apixaban arm and 66% in the dalteparin arm had distant metastasis, and 74% of patients in both arms were receiving chemotherapy while on study.
Patients had the following qualifying thrombotic events:
- Any pulmonary embolism (PE) – 55% of patients in the apixaban arm and 51% in the dalteparin arm
- Any deep vein thrombosis (DVT) – 48% and 47%, respectively
- PE only – 44% and 39%, respectively
- PE with DVT – 12% in both arms
- DVT only – 37% and 35%, respectively
- Lower extremity DVT – 31% and 34%, respectively
- Upper extremity DVT – 17% and 14%, respectively
- Cerebral venous thrombosis (VT) – 1% and 0%, respectively
- Splanchnic VT – 8% and 18%, respectively.
Bleeding, thrombosis, and death
The study’s primary endpoint was major bleeding, which did not occur in any of the apixaban-treated patients. However, major bleeding did occur in two (1.4%) patients in the dalteparin arm (P = .14).
A secondary endpoint was major bleeding plus clinically relevant nonmajor bleeding. This occurred in nine (6.2%) patients in the apixaban arm and nine (6.3%) in the dalteparin arm (P = .88).
The researchers also assessed VTE recurrence. One patient in the apixaban arm (0.7%) and nine in the dalteparin arm (6.3%) had VTE recurrence (P = .03).
The patient in the apixaban arm experienced cerebral VT, and the patients with recurrence in the dalteparin arm had leg (n = 4) or arm (n = 2) VTE, PE (n = 1), or splanchnic VT (n = 2).
One patient in each arm (0.7%) had arterial thrombosis.
There was no significant difference in cumulative mortality between the treatment arms (hazard ratio, 1.40; P = .3078).
Satisfaction and discontinuation
Overall, apixaban fared better than dalteparin in the monthly patient satisfaction surveys. At various time points, apixaban-treated patients were significantly less likely to be concerned about excessive bruising, find anticoagulant treatment a burden or difficult to carry out, or say anticoagulant treatment added stress to their lives, negatively impacted their quality of life, or caused them “a great deal” of worry, irritation, or frustration.
However, apixaban-treated patients were less likely than dalteparin recipients to have confidence that their drug protected them from VTE recurrence, while the apixaban recipients were more likely than the dalteparin group to report overall satisfaction with their treatment.
In addition, premature treatment discontinuation was more common in the dalteparin group than in the apixaban group – 15% and 4%, respectively (P = .0012).
“Apixaban was well tolerated with superior patient safety satisfaction, as well as significantly fewer study drug discontinuations compared to dalteparin,” Dr. McBane said. “I believe that these data support the use of apixaban for the acute treatment of cancer-associated venous thromboembolism.”
This study was funded by BMS/Pfizer Alliance. Dr. McBane declared no other conflicts of interest.
SOURCE: McBane RD et al. ASH 2018, Abstract 421.
REPORTING FROM ASH 2018
Key clinical point: Apixaban is associated with a similar risk of major bleeding and a lower risk of VTE recurrence when compared with dalteparin in patients with cancer-associated VTE.
Major finding: There were no major bleeding events in the apixaban arm and two in the dalteparin arm (P = .14).
Study details: Phase 3 study of 300 patients.
Disclosures: This study was funded by BMS/Pfizer Alliance.
Source: McBane RD et al. ASH 2018, Abstract 421.
Open enrollment 2019: Busiest week so far at HealthCare.gov
but the weekly and cumulative totals for plans selected continued to run below last year’s levels, according to the Centers for Medicare & Medicaid Services.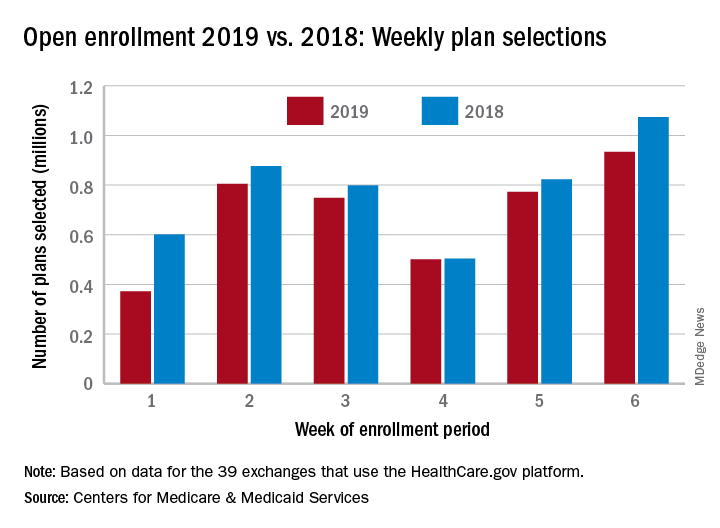
Over 934,000 plans were selected from Dec. 2 to Dec. 8, which puts the total at 4.13 million plans for the 2019 coverage year in the 39 states that use the HealthCare.gov platform, the CMS reported. Consumers renewing their coverage make up the majority of plans selected during week 6 (640,000) and cumulatively for the season (3.03 million), with new applications running at 295,000 for week 6 and 1.1 million overall.
Those numbers are down from last year, when 1.07 million plans (685,000 renewals and 389,000 new applications) were selected during week 6 of open enrollment for the 2018 coverage year, which brought the total for the season at the time to 4.68 million (3.30 million/1.38 million), CMS data show.
The deadline to enroll in a plan for 2019 is Dec. 15.
but the weekly and cumulative totals for plans selected continued to run below last year’s levels, according to the Centers for Medicare & Medicaid Services.
Over 934,000 plans were selected from Dec. 2 to Dec. 8, which puts the total at 4.13 million plans for the 2019 coverage year in the 39 states that use the HealthCare.gov platform, the CMS reported. Consumers renewing their coverage make up the majority of plans selected during week 6 (640,000) and cumulatively for the season (3.03 million), with new applications running at 295,000 for week 6 and 1.1 million overall.
Those numbers are down from last year, when 1.07 million plans (685,000 renewals and 389,000 new applications) were selected during week 6 of open enrollment for the 2018 coverage year, which brought the total for the season at the time to 4.68 million (3.30 million/1.38 million), CMS data show.
The deadline to enroll in a plan for 2019 is Dec. 15.
but the weekly and cumulative totals for plans selected continued to run below last year’s levels, according to the Centers for Medicare & Medicaid Services.
Over 934,000 plans were selected from Dec. 2 to Dec. 8, which puts the total at 4.13 million plans for the 2019 coverage year in the 39 states that use the HealthCare.gov platform, the CMS reported. Consumers renewing their coverage make up the majority of plans selected during week 6 (640,000) and cumulatively for the season (3.03 million), with new applications running at 295,000 for week 6 and 1.1 million overall.
Those numbers are down from last year, when 1.07 million plans (685,000 renewals and 389,000 new applications) were selected during week 6 of open enrollment for the 2018 coverage year, which brought the total for the season at the time to 4.68 million (3.30 million/1.38 million), CMS data show.
The deadline to enroll in a plan for 2019 is Dec. 15.
FDA issues alert over e-liquids with undeclared drugs
The Food and Drug Administration has issued an alert regarding two . E-liquid is the flavored mixture used in electronic cigarettes.

In a laboratory analysis, the FDA found that “E-Cialis HelloCig E-Liquid” contained both sildenafil and tadalafil, and that “E-Rimonabant HelloCig E-Liquid” contained sildenafil. Sildenafil and tadalafil are approved for the treatment of erectile dysfunction. Unapproved usage of these drugs in over-the-counter e-liquids is therefore illegal.
Both sildenafil and tadalafil can interact with nitrates found in some prescription drugs and can cause a dangerous lowering of blood pressure. Conditions commonly treated with nitrates include diabetes, high blood pressure, high cholesterol, or heart disease.
“FDA recently warned HelloCig of these issues and contacted the company several times to recommend they recall these products due to the risks to consumers. However, HelloCig has not responded to the agency’s recommendation. Therefore, FDA urges consumers to stop using these products and to contact their health care professional with any concerns associated with their use,” the FDA said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has issued an alert regarding two . E-liquid is the flavored mixture used in electronic cigarettes.
In a laboratory analysis, the FDA found that “E-Cialis HelloCig E-Liquid” contained both sildenafil and tadalafil, and that “E-Rimonabant HelloCig E-Liquid” contained sildenafil. Sildenafil and tadalafil are approved for the treatment of erectile dysfunction. Unapproved usage of these drugs in over-the-counter e-liquids is therefore illegal.
Both sildenafil and tadalafil can interact with nitrates found in some prescription drugs and can cause a dangerous lowering of blood pressure. Conditions commonly treated with nitrates include diabetes, high blood pressure, high cholesterol, or heart disease.
“FDA recently warned HelloCig of these issues and contacted the company several times to recommend they recall these products due to the risks to consumers. However, HelloCig has not responded to the agency’s recommendation. Therefore, FDA urges consumers to stop using these products and to contact their health care professional with any concerns associated with their use,” the FDA said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has issued an alert regarding two . E-liquid is the flavored mixture used in electronic cigarettes.
In a laboratory analysis, the FDA found that “E-Cialis HelloCig E-Liquid” contained both sildenafil and tadalafil, and that “E-Rimonabant HelloCig E-Liquid” contained sildenafil. Sildenafil and tadalafil are approved for the treatment of erectile dysfunction. Unapproved usage of these drugs in over-the-counter e-liquids is therefore illegal.
Both sildenafil and tadalafil can interact with nitrates found in some prescription drugs and can cause a dangerous lowering of blood pressure. Conditions commonly treated with nitrates include diabetes, high blood pressure, high cholesterol, or heart disease.
“FDA recently warned HelloCig of these issues and contacted the company several times to recommend they recall these products due to the risks to consumers. However, HelloCig has not responded to the agency’s recommendation. Therefore, FDA urges consumers to stop using these products and to contact their health care professional with any concerns associated with their use,” the FDA said in the press release.
Find the full press release on the FDA website.
Severe adverse events seen in placebo arm of cancer clinical trials
A significant number of patients who receive only a placebo in a clinical trial of cancer immunotherapy still experience grade three or grade four adverse events, research suggests.
Writing in JAMA Network Open, researchers reported the outcomes of a systematic review and meta-analysis of 10 randomized, double-blind, placebo-controlled, phase 3 trials of targeted therapy or immunotherapy drugs for cancer, in which patients in the placebo group were treated only with placebo and no other anticancer drugs.
Among the 4,873 patients who were randomized to placebo, a mean of 85.1% experienced some sort of placebo adverse event. The overall incidence of grade 3-4 placebo adverse events was 18% but was as high as 25% in two trials – one in renal cell carcinoma and one in melanoma – and as low as 11% in one trial.
Hypertension was the most frequent grade 3-4 adverse event among patients on placebo, experienced by a mean of 2.8% of patients, followed by fatigue (1%) and diarrhea (0.8%).
Neither route of administration nor cancer type made a significant difference in terms of the rate of placebo adverse events. No deaths attributed to the placebo were reported, but the mean rate of discontinuation due to placebo adverse events was 3.9%, and was higher than 5% for four trials.
The median duration of placebo administration ranged from 10 to 15 months for all but one study, and the authors noted that the longer the placebo exposure, the higher the proportion of grade 3-4 adverse events.
The investigators – Matías Rodrigo Chacón, MD, and his colleagues in the research department at the Argentine Association of Clinical Oncology – said that studies with a lower incidence of grade 3-4 adverse events in the treatment arm also had a lower incidence of grade 3-4 placebo adverse events, while the higher incidences of placebo adverse events were seen in studies that also had a higher incidence of treatment-related adverse events.
They suggested that “contextual factors,” such as the information given during the informed consent process, could contribute to negative expectations of adverse events.
“To illustrate this point, in an RCT [randomized controlled trial] of aspirin as a treatment for unstable angina, a higher incidence of gastrointestinal irritation was reported in centers that specified its potential occurrence in the informed consent compared with research units that did not include that risk,” they wrote.
They also suggested that patients may experience anxiety associated with the uncertainty about whether they had received active treatment or placebo, and this could also affect their distress levels.
No conflicts of interest were declared.
SOURCE: Chacón M et al. JAMA Network Open. 2018 Dec 7. doi: 10.1001/jamanetworkopen.2018.5617.
A significant number of patients who receive only a placebo in a clinical trial of cancer immunotherapy still experience grade three or grade four adverse events, research suggests.
Writing in JAMA Network Open, researchers reported the outcomes of a systematic review and meta-analysis of 10 randomized, double-blind, placebo-controlled, phase 3 trials of targeted therapy or immunotherapy drugs for cancer, in which patients in the placebo group were treated only with placebo and no other anticancer drugs.
Among the 4,873 patients who were randomized to placebo, a mean of 85.1% experienced some sort of placebo adverse event. The overall incidence of grade 3-4 placebo adverse events was 18% but was as high as 25% in two trials – one in renal cell carcinoma and one in melanoma – and as low as 11% in one trial.
Hypertension was the most frequent grade 3-4 adverse event among patients on placebo, experienced by a mean of 2.8% of patients, followed by fatigue (1%) and diarrhea (0.8%).
Neither route of administration nor cancer type made a significant difference in terms of the rate of placebo adverse events. No deaths attributed to the placebo were reported, but the mean rate of discontinuation due to placebo adverse events was 3.9%, and was higher than 5% for four trials.
The median duration of placebo administration ranged from 10 to 15 months for all but one study, and the authors noted that the longer the placebo exposure, the higher the proportion of grade 3-4 adverse events.
The investigators – Matías Rodrigo Chacón, MD, and his colleagues in the research department at the Argentine Association of Clinical Oncology – said that studies with a lower incidence of grade 3-4 adverse events in the treatment arm also had a lower incidence of grade 3-4 placebo adverse events, while the higher incidences of placebo adverse events were seen in studies that also had a higher incidence of treatment-related adverse events.
They suggested that “contextual factors,” such as the information given during the informed consent process, could contribute to negative expectations of adverse events.
“To illustrate this point, in an RCT [randomized controlled trial] of aspirin as a treatment for unstable angina, a higher incidence of gastrointestinal irritation was reported in centers that specified its potential occurrence in the informed consent compared with research units that did not include that risk,” they wrote.
They also suggested that patients may experience anxiety associated with the uncertainty about whether they had received active treatment or placebo, and this could also affect their distress levels.
No conflicts of interest were declared.
SOURCE: Chacón M et al. JAMA Network Open. 2018 Dec 7. doi: 10.1001/jamanetworkopen.2018.5617.
A significant number of patients who receive only a placebo in a clinical trial of cancer immunotherapy still experience grade three or grade four adverse events, research suggests.
Writing in JAMA Network Open, researchers reported the outcomes of a systematic review and meta-analysis of 10 randomized, double-blind, placebo-controlled, phase 3 trials of targeted therapy or immunotherapy drugs for cancer, in which patients in the placebo group were treated only with placebo and no other anticancer drugs.
Among the 4,873 patients who were randomized to placebo, a mean of 85.1% experienced some sort of placebo adverse event. The overall incidence of grade 3-4 placebo adverse events was 18% but was as high as 25% in two trials – one in renal cell carcinoma and one in melanoma – and as low as 11% in one trial.
Hypertension was the most frequent grade 3-4 adverse event among patients on placebo, experienced by a mean of 2.8% of patients, followed by fatigue (1%) and diarrhea (0.8%).
Neither route of administration nor cancer type made a significant difference in terms of the rate of placebo adverse events. No deaths attributed to the placebo were reported, but the mean rate of discontinuation due to placebo adverse events was 3.9%, and was higher than 5% for four trials.
The median duration of placebo administration ranged from 10 to 15 months for all but one study, and the authors noted that the longer the placebo exposure, the higher the proportion of grade 3-4 adverse events.
The investigators – Matías Rodrigo Chacón, MD, and his colleagues in the research department at the Argentine Association of Clinical Oncology – said that studies with a lower incidence of grade 3-4 adverse events in the treatment arm also had a lower incidence of grade 3-4 placebo adverse events, while the higher incidences of placebo adverse events were seen in studies that also had a higher incidence of treatment-related adverse events.
They suggested that “contextual factors,” such as the information given during the informed consent process, could contribute to negative expectations of adverse events.
“To illustrate this point, in an RCT [randomized controlled trial] of aspirin as a treatment for unstable angina, a higher incidence of gastrointestinal irritation was reported in centers that specified its potential occurrence in the informed consent compared with research units that did not include that risk,” they wrote.
They also suggested that patients may experience anxiety associated with the uncertainty about whether they had received active treatment or placebo, and this could also affect their distress levels.
No conflicts of interest were declared.
SOURCE: Chacón M et al. JAMA Network Open. 2018 Dec 7. doi: 10.1001/jamanetworkopen.2018.5617.
FROM JAMA NETWORK OPEN
Key clinical point: Serious adverse events can occur in patients treated only with placebo in cancer clinical trials.
Major finding: The incidence of grade 3-4 placebo adverse events was 18% in cancer clinical trials.
Study details: Systematic review and meta-analysis of 10 randomized, placebo-controlled, double-blind trials.
Disclosures: No conflicts of interest were declared.
Source: Chacón M et al. JAMA Network Open. 2018 Dec 7. doi: 10.1001/jamanetworkopen.2018.5617.
Wasp-stung lung bugs, fat clay, Botoxed Vulcans, and ‘GOT’ mortality risk
The wonderful world of wasps

Never thought you’d be thankful for wasps, did you? Neither did we. Using mice as test subjects, scientists at MIT found that a tiny peptide in the venom was able to completely eliminate Pseudomonas aeruginosa, which causes respiratory infections and is often resistant to antibiotics. Now if you’re sick, all you have to do is go outside and get stung by a bunch of wasps!
Wait, no, that’s not right. The researchers at MIT engineered a molecule that can be used to create an antibiotic that’s safe for humans. While most insect venom is chock full of compounds that are toxic to humans, the scientists were able to transform their tiny peptide into a bacteria-defeating machine. This is a big victory in the war against antibiotic-resistant bacteria. Go, wasps!
Live long and Botox
Talk about highly illogical! Botox patients sometimes return for follow-up visits with dermatologist Kelly Stankiewicz, MD, and haven’t noticed they’ve “Spocked,” even though it may be obvious to just about everyone else.
But other Botox patients are most certainly aware that their eyebrows have arched up on the right and left sides – just like those of a certain Vulcan character on “Star Trek.” And they want to be beamed out of that uncomfortable situation pronto.
Dr. Stankiewicz, who works in Park City, Utah, explained the “Spocking” phenomenon in a presentation about facial treatments at the recent Las Vegas Dermatology Symposium. Spocking can occur in patents who get Botox treatment to eliminate the “11 line” – two vertical wrinkles – between the eyebrows, Dr. Stankiewicz said. It occurs “when the middle of the forehead doesn’t move but the outside does,” she said, causing an unsightly outwardly arched eyebrow look.
The solution to Spocking is easy, she said: “Put a tiny bit of Botox in the forehead muscle right where the eyebrow is peaking the most.”
Leonard Nimoy, the original Mr. Spock, is not available for comment, given that he died in 2015. But Dr. Stankiewicz does have a perspective on Mr. Spock’s trademark look: “It’s like the people who drew his brow knew that Botox was on the horizon.”
Add the dirt, lose the fat
Lots of things are supposed to be stronger than dirt, but it looks like obesity might not be one of them. Investigators who were trying to improve drug delivery using a type of clay – spray-dried smectite clay particles, to be exact – discovered that it has “a unique ability to ‘soak up’ fat droplets in the gut,” according to a statement from the University of South Australia, Adelaide.
“Not only were the clay materials trapping the fats within their particle structure, but they were also preventing them from being absorbed by the body, ensuring that fat simply passed through the digestive system,” researcher Tahnee J. Dening said.
In the study, the smectite outperformed the weight-loss drug orlistat in rats fed a high-fat diet for 2 weeks (Pharm Res. 2019;36:21 doi: 10.1007/s11095-018-2552-9). Even better, smectite is already widely used in foods and nutraceuticals and is considered to be safe. Even even better, smectite is a really fun word to say. And with the prevalence of obesity such as it is, we’re sure that physicians will prescribe smectite just so they can say “smectite” to their patients. Smectite.
When you play the game of thrones …

… You conduct an evidence-based analysis of mortality across the Seven Kingdoms. In celebration of the approaching final season, two researchers published a study of mortality and survival in HBO’s “Game of Thrones” series. They examined data on sociodemographic factors and circumstances of death to identify predictors of mortality.
After looking at 330 characters from the show (186 who suffered death), the authors determined that being male, lowborn, and loyal had the highest correlation with mortality. Poor men who never switched their allegiance from one side to the other had the lowest chance of survival. In addition, the probability of dying in the first hour after appearing on screen was 14%. Not a good statistic if you’re trying to become famous in Westeros. And a mortality risk rivaling even that of Mr. Spock’s red-shirted shipmates down in engineering.
Nurse, sand wedge, STAT!
There really is no sport like golf. The wide green spaces, the fresh air, the thrill of launching a 300-yard drive down the fairway, the fun of double-bogeying the last three holes to shoot yet another 93; golf is a noble pursuit, indeed. And a sport everyone knows doctors love. Right?
In research published in the Christmas edition of the BMJ, a group of doctors from Massachusetts sought to find out just how accurate the stereotype was. Their answer? Doctors really do enjoy teeing it up.
Well, male doctors.
Well, old male doctors.
Using a database of about a million physicians, the researchers found that 5.5% of male physicians and 1.3% of female physicians – or 4.1% overall – maintained an official United States Golf Association handicap. Male physicians aged 61-70 years were most likely to play, and female physicians aged 31-35 years were least likely.
However, while golf is certainly a common pastime among doctors, they aren’t exactly very good at it, as the average doctor is actually slightly worse than the average nondoctor. Surgeons presented a notable exception, possessing significantly lower handicaps than their colleagues while, perhaps not coincidentally, also playing more often. We’re sure there’s a joke about overpaid surgeons in narrow specialties having too much time on their hands, and we’ll get right back to you with it after our 12 o’clock tee time.
The wonderful world of wasps
Never thought you’d be thankful for wasps, did you? Neither did we. Using mice as test subjects, scientists at MIT found that a tiny peptide in the venom was able to completely eliminate Pseudomonas aeruginosa, which causes respiratory infections and is often resistant to antibiotics. Now if you’re sick, all you have to do is go outside and get stung by a bunch of wasps!
Wait, no, that’s not right. The researchers at MIT engineered a molecule that can be used to create an antibiotic that’s safe for humans. While most insect venom is chock full of compounds that are toxic to humans, the scientists were able to transform their tiny peptide into a bacteria-defeating machine. This is a big victory in the war against antibiotic-resistant bacteria. Go, wasps!
Live long and Botox
Talk about highly illogical! Botox patients sometimes return for follow-up visits with dermatologist Kelly Stankiewicz, MD, and haven’t noticed they’ve “Spocked,” even though it may be obvious to just about everyone else.
But other Botox patients are most certainly aware that their eyebrows have arched up on the right and left sides – just like those of a certain Vulcan character on “Star Trek.” And they want to be beamed out of that uncomfortable situation pronto.
Dr. Stankiewicz, who works in Park City, Utah, explained the “Spocking” phenomenon in a presentation about facial treatments at the recent Las Vegas Dermatology Symposium. Spocking can occur in patents who get Botox treatment to eliminate the “11 line” – two vertical wrinkles – between the eyebrows, Dr. Stankiewicz said. It occurs “when the middle of the forehead doesn’t move but the outside does,” she said, causing an unsightly outwardly arched eyebrow look.
The solution to Spocking is easy, she said: “Put a tiny bit of Botox in the forehead muscle right where the eyebrow is peaking the most.”
Leonard Nimoy, the original Mr. Spock, is not available for comment, given that he died in 2015. But Dr. Stankiewicz does have a perspective on Mr. Spock’s trademark look: “It’s like the people who drew his brow knew that Botox was on the horizon.”
Add the dirt, lose the fat
Lots of things are supposed to be stronger than dirt, but it looks like obesity might not be one of them. Investigators who were trying to improve drug delivery using a type of clay – spray-dried smectite clay particles, to be exact – discovered that it has “a unique ability to ‘soak up’ fat droplets in the gut,” according to a statement from the University of South Australia, Adelaide.
“Not only were the clay materials trapping the fats within their particle structure, but they were also preventing them from being absorbed by the body, ensuring that fat simply passed through the digestive system,” researcher Tahnee J. Dening said.
In the study, the smectite outperformed the weight-loss drug orlistat in rats fed a high-fat diet for 2 weeks (Pharm Res. 2019;36:21 doi: 10.1007/s11095-018-2552-9). Even better, smectite is already widely used in foods and nutraceuticals and is considered to be safe. Even even better, smectite is a really fun word to say. And with the prevalence of obesity such as it is, we’re sure that physicians will prescribe smectite just so they can say “smectite” to their patients. Smectite.
When you play the game of thrones …
… You conduct an evidence-based analysis of mortality across the Seven Kingdoms. In celebration of the approaching final season, two researchers published a study of mortality and survival in HBO’s “Game of Thrones” series. They examined data on sociodemographic factors and circumstances of death to identify predictors of mortality.
After looking at 330 characters from the show (186 who suffered death), the authors determined that being male, lowborn, and loyal had the highest correlation with mortality. Poor men who never switched their allegiance from one side to the other had the lowest chance of survival. In addition, the probability of dying in the first hour after appearing on screen was 14%. Not a good statistic if you’re trying to become famous in Westeros. And a mortality risk rivaling even that of Mr. Spock’s red-shirted shipmates down in engineering.
Nurse, sand wedge, STAT!
There really is no sport like golf. The wide green spaces, the fresh air, the thrill of launching a 300-yard drive down the fairway, the fun of double-bogeying the last three holes to shoot yet another 93; golf is a noble pursuit, indeed. And a sport everyone knows doctors love. Right?
In research published in the Christmas edition of the BMJ, a group of doctors from Massachusetts sought to find out just how accurate the stereotype was. Their answer? Doctors really do enjoy teeing it up.
Well, male doctors.
Well, old male doctors.
Using a database of about a million physicians, the researchers found that 5.5% of male physicians and 1.3% of female physicians – or 4.1% overall – maintained an official United States Golf Association handicap. Male physicians aged 61-70 years were most likely to play, and female physicians aged 31-35 years were least likely.
However, while golf is certainly a common pastime among doctors, they aren’t exactly very good at it, as the average doctor is actually slightly worse than the average nondoctor. Surgeons presented a notable exception, possessing significantly lower handicaps than their colleagues while, perhaps not coincidentally, also playing more often. We’re sure there’s a joke about overpaid surgeons in narrow specialties having too much time on their hands, and we’ll get right back to you with it after our 12 o’clock tee time.
The wonderful world of wasps
Never thought you’d be thankful for wasps, did you? Neither did we. Using mice as test subjects, scientists at MIT found that a tiny peptide in the venom was able to completely eliminate Pseudomonas aeruginosa, which causes respiratory infections and is often resistant to antibiotics. Now if you’re sick, all you have to do is go outside and get stung by a bunch of wasps!
Wait, no, that’s not right. The researchers at MIT engineered a molecule that can be used to create an antibiotic that’s safe for humans. While most insect venom is chock full of compounds that are toxic to humans, the scientists were able to transform their tiny peptide into a bacteria-defeating machine. This is a big victory in the war against antibiotic-resistant bacteria. Go, wasps!
Live long and Botox
Talk about highly illogical! Botox patients sometimes return for follow-up visits with dermatologist Kelly Stankiewicz, MD, and haven’t noticed they’ve “Spocked,” even though it may be obvious to just about everyone else.
But other Botox patients are most certainly aware that their eyebrows have arched up on the right and left sides – just like those of a certain Vulcan character on “Star Trek.” And they want to be beamed out of that uncomfortable situation pronto.
Dr. Stankiewicz, who works in Park City, Utah, explained the “Spocking” phenomenon in a presentation about facial treatments at the recent Las Vegas Dermatology Symposium. Spocking can occur in patents who get Botox treatment to eliminate the “11 line” – two vertical wrinkles – between the eyebrows, Dr. Stankiewicz said. It occurs “when the middle of the forehead doesn’t move but the outside does,” she said, causing an unsightly outwardly arched eyebrow look.
The solution to Spocking is easy, she said: “Put a tiny bit of Botox in the forehead muscle right where the eyebrow is peaking the most.”
Leonard Nimoy, the original Mr. Spock, is not available for comment, given that he died in 2015. But Dr. Stankiewicz does have a perspective on Mr. Spock’s trademark look: “It’s like the people who drew his brow knew that Botox was on the horizon.”
Add the dirt, lose the fat
Lots of things are supposed to be stronger than dirt, but it looks like obesity might not be one of them. Investigators who were trying to improve drug delivery using a type of clay – spray-dried smectite clay particles, to be exact – discovered that it has “a unique ability to ‘soak up’ fat droplets in the gut,” according to a statement from the University of South Australia, Adelaide.
“Not only were the clay materials trapping the fats within their particle structure, but they were also preventing them from being absorbed by the body, ensuring that fat simply passed through the digestive system,” researcher Tahnee J. Dening said.
In the study, the smectite outperformed the weight-loss drug orlistat in rats fed a high-fat diet for 2 weeks (Pharm Res. 2019;36:21 doi: 10.1007/s11095-018-2552-9). Even better, smectite is already widely used in foods and nutraceuticals and is considered to be safe. Even even better, smectite is a really fun word to say. And with the prevalence of obesity such as it is, we’re sure that physicians will prescribe smectite just so they can say “smectite” to their patients. Smectite.
When you play the game of thrones …
… You conduct an evidence-based analysis of mortality across the Seven Kingdoms. In celebration of the approaching final season, two researchers published a study of mortality and survival in HBO’s “Game of Thrones” series. They examined data on sociodemographic factors and circumstances of death to identify predictors of mortality.
After looking at 330 characters from the show (186 who suffered death), the authors determined that being male, lowborn, and loyal had the highest correlation with mortality. Poor men who never switched their allegiance from one side to the other had the lowest chance of survival. In addition, the probability of dying in the first hour after appearing on screen was 14%. Not a good statistic if you’re trying to become famous in Westeros. And a mortality risk rivaling even that of Mr. Spock’s red-shirted shipmates down in engineering.
Nurse, sand wedge, STAT!
There really is no sport like golf. The wide green spaces, the fresh air, the thrill of launching a 300-yard drive down the fairway, the fun of double-bogeying the last three holes to shoot yet another 93; golf is a noble pursuit, indeed. And a sport everyone knows doctors love. Right?
In research published in the Christmas edition of the BMJ, a group of doctors from Massachusetts sought to find out just how accurate the stereotype was. Their answer? Doctors really do enjoy teeing it up.
Well, male doctors.
Well, old male doctors.
Using a database of about a million physicians, the researchers found that 5.5% of male physicians and 1.3% of female physicians – or 4.1% overall – maintained an official United States Golf Association handicap. Male physicians aged 61-70 years were most likely to play, and female physicians aged 31-35 years were least likely.
However, while golf is certainly a common pastime among doctors, they aren’t exactly very good at it, as the average doctor is actually slightly worse than the average nondoctor. Surgeons presented a notable exception, possessing significantly lower handicaps than their colleagues while, perhaps not coincidentally, also playing more often. We’re sure there’s a joke about overpaid surgeons in narrow specialties having too much time on their hands, and we’ll get right back to you with it after our 12 o’clock tee time.
‘Optimal’ pazopanib levels linked to lower toxicity in RCC patients
In patients with renal cell carcinoma, trough concentrations of pazopanib in a specific target range were associated with better safety and comparable efficacy, versus higher concentrations of the drug, authors of an exploratory investigation have reported.
Fewer serious toxicities were seen in patients with pazopanib concentrations in the 20- to 50-mcg/mL range, yet overall response rate was similar compared to patients with concentrations over 50 mcg/mL, according to results of the retrospective study.
The findings suggest therapeutic drug monitoring to achieve a specific trough concentration may be useful to optimize pazopanib dosing, according to investigator Tomohiro Terada, PhD, of Shiga University of Medical Science Hospital, Japan, and coinvestigators.
Therapeutic drug monitoring for sunitinib in RCC patients was recently covered by medical insurance in Japan, Dr. Terada and colleagues wrote in Clinical Genitourinary Cancer.
The strategy may be especially suited to adjusting the dose of pazopanib, which is often associated with severe toxicities and has pharmacokinetics that suggest a wide variation between individuals, they added.
The retrospective study by Dr. Terada and colleagues included 27 renal cell carcinoma patients who received pazopanib at doses of 400, 600, or 800 mg daily based on the recommendation of the treating physician, with doses reduced or discontinued based on adverse events or progression.
Trough concentrations of pazopanib 3 months after starting the treatment ranged from a low of 10.6 mcg/mL to a high of 106 mcg/mL, with a median of 49.1 mcg/mL, according to the report.
One-third of patients experienced grade 3 or greater toxicities, including anorexia, hypertension, anemia, and thrombocytopenia, among others. The median pazopanib concentration for those nine patients was 69.3 mcg/mL, compared with 41.2 mcg/mL for those not experiencing serious toxicities (P less than .05).
A concentration over 50.3 mcg/mL predicted grade 3 or greater toxicity, statistical analysis showed, with 8 out of 13 patients with concentrations above that threshold experiencing serious toxicities. By contrast, 1 of 14 patients below that threshold experienced grade 3 or greater toxicities.
No responses were observed in three patients with pazopanib concentrations below 20.5 mcg/mL, a concentration level considered to be subtherapeutic based on previous investigations.
Overall response rates were similar for patients with concentrations in the “optimal” 20.5- and 50.3-mcg/mL range and those with concentrations over 50.3 mcg/mL, according to the investigators.
Among 11 patients in that “optimal” range, partial responses were seen in 5, they reported, while for the 13 patients with high concentrations of the drug, partial responses were seen in 4 patients and a complete response was seen in 1 patient.
Trough concentrations may not predict all types of toxicities, according to the investigators, who said that the results of their small, retrospective analysis should be validated in a large, prospective study.
In particular, there was no significant relationship between trough concentration of pazopanib and development of grade 2 or greater hepatotoxicity, a common and specific side effect of the drug.
“Based on these findings, pazopanib-induced hepatotoxicity may not be related to the pharmacokinetics of pazopanib,” they wrote. “Therefore, pharmacogenomics testing is needed to predict pazopanib-induced hepatotoxicity.”
Dr. Terada and coauthors said they had no conflicts of interest to report.
SOURCE: Noda S et al. Clin Genitourin Cancer. 2018 Dec 7. doi: 10.1016/j.clgc.2018.12.001.
In patients with renal cell carcinoma, trough concentrations of pazopanib in a specific target range were associated with better safety and comparable efficacy, versus higher concentrations of the drug, authors of an exploratory investigation have reported.
Fewer serious toxicities were seen in patients with pazopanib concentrations in the 20- to 50-mcg/mL range, yet overall response rate was similar compared to patients with concentrations over 50 mcg/mL, according to results of the retrospective study.
The findings suggest therapeutic drug monitoring to achieve a specific trough concentration may be useful to optimize pazopanib dosing, according to investigator Tomohiro Terada, PhD, of Shiga University of Medical Science Hospital, Japan, and coinvestigators.
Therapeutic drug monitoring for sunitinib in RCC patients was recently covered by medical insurance in Japan, Dr. Terada and colleagues wrote in Clinical Genitourinary Cancer.
The strategy may be especially suited to adjusting the dose of pazopanib, which is often associated with severe toxicities and has pharmacokinetics that suggest a wide variation between individuals, they added.
The retrospective study by Dr. Terada and colleagues included 27 renal cell carcinoma patients who received pazopanib at doses of 400, 600, or 800 mg daily based on the recommendation of the treating physician, with doses reduced or discontinued based on adverse events or progression.
Trough concentrations of pazopanib 3 months after starting the treatment ranged from a low of 10.6 mcg/mL to a high of 106 mcg/mL, with a median of 49.1 mcg/mL, according to the report.
One-third of patients experienced grade 3 or greater toxicities, including anorexia, hypertension, anemia, and thrombocytopenia, among others. The median pazopanib concentration for those nine patients was 69.3 mcg/mL, compared with 41.2 mcg/mL for those not experiencing serious toxicities (P less than .05).
A concentration over 50.3 mcg/mL predicted grade 3 or greater toxicity, statistical analysis showed, with 8 out of 13 patients with concentrations above that threshold experiencing serious toxicities. By contrast, 1 of 14 patients below that threshold experienced grade 3 or greater toxicities.
No responses were observed in three patients with pazopanib concentrations below 20.5 mcg/mL, a concentration level considered to be subtherapeutic based on previous investigations.
Overall response rates were similar for patients with concentrations in the “optimal” 20.5- and 50.3-mcg/mL range and those with concentrations over 50.3 mcg/mL, according to the investigators.
Among 11 patients in that “optimal” range, partial responses were seen in 5, they reported, while for the 13 patients with high concentrations of the drug, partial responses were seen in 4 patients and a complete response was seen in 1 patient.
Trough concentrations may not predict all types of toxicities, according to the investigators, who said that the results of their small, retrospective analysis should be validated in a large, prospective study.
In particular, there was no significant relationship between trough concentration of pazopanib and development of grade 2 or greater hepatotoxicity, a common and specific side effect of the drug.
“Based on these findings, pazopanib-induced hepatotoxicity may not be related to the pharmacokinetics of pazopanib,” they wrote. “Therefore, pharmacogenomics testing is needed to predict pazopanib-induced hepatotoxicity.”
Dr. Terada and coauthors said they had no conflicts of interest to report.
SOURCE: Noda S et al. Clin Genitourin Cancer. 2018 Dec 7. doi: 10.1016/j.clgc.2018.12.001.
In patients with renal cell carcinoma, trough concentrations of pazopanib in a specific target range were associated with better safety and comparable efficacy, versus higher concentrations of the drug, authors of an exploratory investigation have reported.
Fewer serious toxicities were seen in patients with pazopanib concentrations in the 20- to 50-mcg/mL range, yet overall response rate was similar compared to patients with concentrations over 50 mcg/mL, according to results of the retrospective study.
The findings suggest therapeutic drug monitoring to achieve a specific trough concentration may be useful to optimize pazopanib dosing, according to investigator Tomohiro Terada, PhD, of Shiga University of Medical Science Hospital, Japan, and coinvestigators.
Therapeutic drug monitoring for sunitinib in RCC patients was recently covered by medical insurance in Japan, Dr. Terada and colleagues wrote in Clinical Genitourinary Cancer.
The strategy may be especially suited to adjusting the dose of pazopanib, which is often associated with severe toxicities and has pharmacokinetics that suggest a wide variation between individuals, they added.
The retrospective study by Dr. Terada and colleagues included 27 renal cell carcinoma patients who received pazopanib at doses of 400, 600, or 800 mg daily based on the recommendation of the treating physician, with doses reduced or discontinued based on adverse events or progression.
Trough concentrations of pazopanib 3 months after starting the treatment ranged from a low of 10.6 mcg/mL to a high of 106 mcg/mL, with a median of 49.1 mcg/mL, according to the report.
One-third of patients experienced grade 3 or greater toxicities, including anorexia, hypertension, anemia, and thrombocytopenia, among others. The median pazopanib concentration for those nine patients was 69.3 mcg/mL, compared with 41.2 mcg/mL for those not experiencing serious toxicities (P less than .05).
A concentration over 50.3 mcg/mL predicted grade 3 or greater toxicity, statistical analysis showed, with 8 out of 13 patients with concentrations above that threshold experiencing serious toxicities. By contrast, 1 of 14 patients below that threshold experienced grade 3 or greater toxicities.
No responses were observed in three patients with pazopanib concentrations below 20.5 mcg/mL, a concentration level considered to be subtherapeutic based on previous investigations.
Overall response rates were similar for patients with concentrations in the “optimal” 20.5- and 50.3-mcg/mL range and those with concentrations over 50.3 mcg/mL, according to the investigators.
Among 11 patients in that “optimal” range, partial responses were seen in 5, they reported, while for the 13 patients with high concentrations of the drug, partial responses were seen in 4 patients and a complete response was seen in 1 patient.
Trough concentrations may not predict all types of toxicities, according to the investigators, who said that the results of their small, retrospective analysis should be validated in a large, prospective study.
In particular, there was no significant relationship between trough concentration of pazopanib and development of grade 2 or greater hepatotoxicity, a common and specific side effect of the drug.
“Based on these findings, pazopanib-induced hepatotoxicity may not be related to the pharmacokinetics of pazopanib,” they wrote. “Therefore, pharmacogenomics testing is needed to predict pazopanib-induced hepatotoxicity.”
Dr. Terada and coauthors said they had no conflicts of interest to report.
SOURCE: Noda S et al. Clin Genitourin Cancer. 2018 Dec 7. doi: 10.1016/j.clgc.2018.12.001.
FROM CLINICAL GENITOURINARY CANCER
Key clinical point: Trough concentrations of pazopanib in approximately the 20- to 50-mcg/mL range were associated with less toxicity but a similar overall response rate, versus higher concentrations.
Major finding: The overall response rate was about 46% in patients with pazopanib concentrations in that range, and in patients with higher concentrations. One patient in the “optimal” range experienced a grade 3 or greater toxicity.
Study details: An exploratory analysis including 27 patients with renal cell carcinoma.
Disclosures: The authors reported no conflicts of interest.
Source: Noda S et al. Clin Genitourin Cancer. 2018 Dec 7. doi: 10.1016/j.clgc.2018.12.001.