User login
Common food additive makes C. difficile more virulent
, a study showed.
“Out of several carbon sources identified that supported CD2015 growth [epidemic RT027 isolate], we found the disaccharide trehalose increased the growth yield of CD2015 by approximately fivefold, compared with a non-RT027 strain,” according to James Collins, PhD, of Baylor University, Houston, and his colleagues. The increased growth of the epidemic strain of C. difficile observed by Dr. Collins and his team demonstrates that trehalose is a robust carbon source for C. difficile bacterium.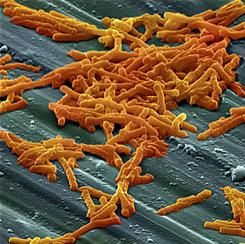
In one experiment, mice with humanized microbiota were infected with two strains of RT027, either R20291 (n = 27) or R20291-delta treA (n = 28), a phosphotrehalase enzyme (TreA) deletion mutant that cannot metabolize trehalose. Mice were then given 5 mM of trehalose ad libitum in their drinking water. Researchers observed that the mice infected with R20291-delta treA had much lower mortality rates than the R20291 group (33.3% vs.78.6%). These findings were then reinforced with a second experiment using mice with humanized microbiota, in which trehalose addition increased mortality in RT027 mice, compared with RT027-infected mice that were not given dietary trehalose.
While Dr. Collins and his team demonstrated the effect of trehalose on C. difficile in mice, they also conducted a limited analysis of ileostomy effluent from three human donors. The researchers found that in two of three samples, treA was strongly induced in CD2015, but not in another ribotype, CD2048. This demonstrates that amounts of trehalose found in food are high enough to be metabolized by certain epidemic strains of C. difficile in humans.
Prior to 2000, trehalose use was limited by a relatively high cost of production, approximately $700 per kilogram. A production innovation that utilized a novel enzymatic method that yielded trehalose from starch brought the price of trehalose to approximately $3 per kilogram, making it a commercially viable food supplement. After being considered “generally recognized as safe” by the U.S. Food and Drug Administration in 2000 and approved for use in Europe, the trehalose concentrations in food skyrocketed from around 2% to 11.25%, and trehalose became widely used in several foods, including ice cream, pasta, and ground beef.
Dr. Collins and his associates said that there is considerable evidence that the widespread use of dietary trehalose has contributed to the spread of epidemic C. difficile ribotypes. First, strains RT027 and RT078 have always had the ability to metabolize trehalose, as evidenced by outbreaks of nonepidemic C. difficile in the 1980s. But no epidemic outbreaks were reported until after 2003, several years after trehalose was approved by the FDA. Second, RT027 and RT078 are phylogenetically distant, but independently evolved the ability to metabolize low levels of trehalose. Third, increased severity of the RT027 strain, which metabolizes trehalose in mice, is consistent with increased virulence of RT078 and RT027 in human patients. Fourth, a competitive advantage is conferred to C. difficile being able to metabolize trehalose in low concentrations in a diverse intestinal setting. Finally, the levels of trehalose in ileostomy fluid from patients who eat a normal diet are high enough to be utilized by RT027 strains.
“On the basis of these observations, we propose that the widespread adoption and use of the disaccharide trehalose in the human diet has played a significant role in the emergence of these epidemic and hypervirulent strains,” Dr. Collins and his colleagues wrote in their article in Nature.
The authors of the study had no relevant financial disclosures to report.
SOURCE: Collins J et al. Nature. 2018 Jan 3. doi: 10.1038/nature25178.
, a study showed.
“Out of several carbon sources identified that supported CD2015 growth [epidemic RT027 isolate], we found the disaccharide trehalose increased the growth yield of CD2015 by approximately fivefold, compared with a non-RT027 strain,” according to James Collins, PhD, of Baylor University, Houston, and his colleagues. The increased growth of the epidemic strain of C. difficile observed by Dr. Collins and his team demonstrates that trehalose is a robust carbon source for C. difficile bacterium.
In one experiment, mice with humanized microbiota were infected with two strains of RT027, either R20291 (n = 27) or R20291-delta treA (n = 28), a phosphotrehalase enzyme (TreA) deletion mutant that cannot metabolize trehalose. Mice were then given 5 mM of trehalose ad libitum in their drinking water. Researchers observed that the mice infected with R20291-delta treA had much lower mortality rates than the R20291 group (33.3% vs.78.6%). These findings were then reinforced with a second experiment using mice with humanized microbiota, in which trehalose addition increased mortality in RT027 mice, compared with RT027-infected mice that were not given dietary trehalose.
While Dr. Collins and his team demonstrated the effect of trehalose on C. difficile in mice, they also conducted a limited analysis of ileostomy effluent from three human donors. The researchers found that in two of three samples, treA was strongly induced in CD2015, but not in another ribotype, CD2048. This demonstrates that amounts of trehalose found in food are high enough to be metabolized by certain epidemic strains of C. difficile in humans.
Prior to 2000, trehalose use was limited by a relatively high cost of production, approximately $700 per kilogram. A production innovation that utilized a novel enzymatic method that yielded trehalose from starch brought the price of trehalose to approximately $3 per kilogram, making it a commercially viable food supplement. After being considered “generally recognized as safe” by the U.S. Food and Drug Administration in 2000 and approved for use in Europe, the trehalose concentrations in food skyrocketed from around 2% to 11.25%, and trehalose became widely used in several foods, including ice cream, pasta, and ground beef.
Dr. Collins and his associates said that there is considerable evidence that the widespread use of dietary trehalose has contributed to the spread of epidemic C. difficile ribotypes. First, strains RT027 and RT078 have always had the ability to metabolize trehalose, as evidenced by outbreaks of nonepidemic C. difficile in the 1980s. But no epidemic outbreaks were reported until after 2003, several years after trehalose was approved by the FDA. Second, RT027 and RT078 are phylogenetically distant, but independently evolved the ability to metabolize low levels of trehalose. Third, increased severity of the RT027 strain, which metabolizes trehalose in mice, is consistent with increased virulence of RT078 and RT027 in human patients. Fourth, a competitive advantage is conferred to C. difficile being able to metabolize trehalose in low concentrations in a diverse intestinal setting. Finally, the levels of trehalose in ileostomy fluid from patients who eat a normal diet are high enough to be utilized by RT027 strains.
“On the basis of these observations, we propose that the widespread adoption and use of the disaccharide trehalose in the human diet has played a significant role in the emergence of these epidemic and hypervirulent strains,” Dr. Collins and his colleagues wrote in their article in Nature.
The authors of the study had no relevant financial disclosures to report.
SOURCE: Collins J et al. Nature. 2018 Jan 3. doi: 10.1038/nature25178.
, a study showed.
“Out of several carbon sources identified that supported CD2015 growth [epidemic RT027 isolate], we found the disaccharide trehalose increased the growth yield of CD2015 by approximately fivefold, compared with a non-RT027 strain,” according to James Collins, PhD, of Baylor University, Houston, and his colleagues. The increased growth of the epidemic strain of C. difficile observed by Dr. Collins and his team demonstrates that trehalose is a robust carbon source for C. difficile bacterium.
In one experiment, mice with humanized microbiota were infected with two strains of RT027, either R20291 (n = 27) or R20291-delta treA (n = 28), a phosphotrehalase enzyme (TreA) deletion mutant that cannot metabolize trehalose. Mice were then given 5 mM of trehalose ad libitum in their drinking water. Researchers observed that the mice infected with R20291-delta treA had much lower mortality rates than the R20291 group (33.3% vs.78.6%). These findings were then reinforced with a second experiment using mice with humanized microbiota, in which trehalose addition increased mortality in RT027 mice, compared with RT027-infected mice that were not given dietary trehalose.
While Dr. Collins and his team demonstrated the effect of trehalose on C. difficile in mice, they also conducted a limited analysis of ileostomy effluent from three human donors. The researchers found that in two of three samples, treA was strongly induced in CD2015, but not in another ribotype, CD2048. This demonstrates that amounts of trehalose found in food are high enough to be metabolized by certain epidemic strains of C. difficile in humans.
Prior to 2000, trehalose use was limited by a relatively high cost of production, approximately $700 per kilogram. A production innovation that utilized a novel enzymatic method that yielded trehalose from starch brought the price of trehalose to approximately $3 per kilogram, making it a commercially viable food supplement. After being considered “generally recognized as safe” by the U.S. Food and Drug Administration in 2000 and approved for use in Europe, the trehalose concentrations in food skyrocketed from around 2% to 11.25%, and trehalose became widely used in several foods, including ice cream, pasta, and ground beef.
Dr. Collins and his associates said that there is considerable evidence that the widespread use of dietary trehalose has contributed to the spread of epidemic C. difficile ribotypes. First, strains RT027 and RT078 have always had the ability to metabolize trehalose, as evidenced by outbreaks of nonepidemic C. difficile in the 1980s. But no epidemic outbreaks were reported until after 2003, several years after trehalose was approved by the FDA. Second, RT027 and RT078 are phylogenetically distant, but independently evolved the ability to metabolize low levels of trehalose. Third, increased severity of the RT027 strain, which metabolizes trehalose in mice, is consistent with increased virulence of RT078 and RT027 in human patients. Fourth, a competitive advantage is conferred to C. difficile being able to metabolize trehalose in low concentrations in a diverse intestinal setting. Finally, the levels of trehalose in ileostomy fluid from patients who eat a normal diet are high enough to be utilized by RT027 strains.
“On the basis of these observations, we propose that the widespread adoption and use of the disaccharide trehalose in the human diet has played a significant role in the emergence of these epidemic and hypervirulent strains,” Dr. Collins and his colleagues wrote in their article in Nature.
The authors of the study had no relevant financial disclosures to report.
SOURCE: Collins J et al. Nature. 2018 Jan 3. doi: 10.1038/nature25178.
FROM NATURE
Key clinical point: Metabolizing trehalose increases the virulence and mortality of C. difficile ribotype 027 (RT027).
Major finding: The ability to metabolize trehalose with the phosphotrehalase enzyme (TreA) increases mortality in RT027.
Study details: Experimental mouse models and an analysis of ileostomy effluent from three anonymous donors.
Disclosures: All authors had no financial disclosures to report.
Source: Collins J et al. Nature. 2018 Jan 3. doi: 10.1038/nature25178.
The felt pelvic anatomy model: A teaching tool for students and residents

Visit the Society of Gynecologic Surgeons online: sgsonline.org
Additional videos from SGS are available here, including these recent offerings:
Visit the Society of Gynecologic Surgeons online: sgsonline.org
Additional videos from SGS are available here, including these recent offerings:
Visit the Society of Gynecologic Surgeons online: sgsonline.org
Additional videos from SGS are available here, including these recent offerings:
This video is brought to you by![]()
Internet addiction ‘an impairing but treatable problem’
SAN DIEGO – The concept of Internet addiction is imperfect, but it has validity and describes an impairing but treatable problem, according to Diana D. Deister, MD.
“Timely diagnosis can lead to good treatment and symptom improvement,” she said at the annual meeting and scientific symposium of the American Academy of Addiction Psychiatry.
Frameworks for the diagnosis of Internet addiction (IA)/pathological Internet use (PIU) vary but are based mostly on DSM-IV substance abuse and dependency criteria, DSM-IV pathological gambling, or other models. “There are many different checklists and diagnostic instruments you can use, and common synonyms include pathological Internet use, problematic Internet use, and compulsive Internet use,” said Dr. Deister, a child and adolescent psychiatrist at Boston Children’s Hospital. “They may not all refer to the exact same problem you might encounter in clinical practice.”
The Internet Addiction Diagnostic Questionnaire, developed in 1998 by Kimberly S. Young, PsyD, is widely used in research and consists of eight yes or no questions (Cyberpsychol Behav. 1998;1[3]:237-44). Answering “yes” to five out of the eight questions gives you a diagnosis of IA. “There are no preferred symptoms that everybody has to have in order to make the diagnosis, so this is more like the current SUD diagnosis for DSM-5,” Dr. Deister explained. “And there’s no time criteria.”
Clinicians can secure a more detailed assessment of IA symptoms by using the Internet Addiction Test (IAT), which Dr. Young developed in 2013. This tool consists of 20 questions rated on a five-point Likert scale based on frequency, where 0 stands for “not applicable” and 5 stands for “always.” Questions include “How often do you find that you stay online longer than you intended?” “How often do you block out disturbing thoughts about your life with soothing thoughts of the Internet?” And “How often do you feel depressed, moody, or nervous when you are offline, which goes away once you are back online?” A score of 80-100 is considered to meet criteria for IA, a score of 50-79 is considered borderline IA, while a score of below 50 is considered not pathological. Variations of this test can be found in the medical literature, including the Internet Process Addiction Test (Behav Sci. 2015;5:341-52), which contains subscales of Internet use: surfing, online gaming, social networking, and sex.
Dr. Deister noted that imaging studies of brain pathways in IA have demonstrated that mesolimbic dopaminergic projections to the nucleus accumbens from the ventral tegmental area are involved, as well as diminished activity of the ventral medial prefrontal cortex. In a tract-based spatial statistics study, researchers used diffusor tensor imaging (DTI) to study the white matter integrity in 18 adolescents with IA, and 18 age- and gender-matched controls (PLoS ONE. 7[1]:e30253. doi:10.1371/journal.pone.0030253). Compared with controls, the IA group scored higher in the IAT, the Strengths and Difficulties Questionnaire, the Screen for Child Anxiety Related Emotional Disorders scale, and Family Assessment Device measure. DTI scans demonstrated widespread reductions of fractional anisotropy (FA; a measure of white matter health) in major white matter pathways. In addition, significantly negative correlations were found between FA values in the left genu of the corpus callosum and the Screen for Child Anxiety Related Emotional Disorders, and between FA values in the left external capsule and in the IAT.
“The reason these white matter tracts could be important is that there have been previous studies showing that you can improve the health of your white matter tracks through exercise and physical therapy,” Dr. Deister said. “So it may be that this knowledge could lead to new treatments for some of these patients.”
One study of the dose dependence of IA symptoms found that adolescents who met criteria for addictive Internet use, compared with those who met criteria for borderline-addictive Internet use, had higher rates of peer problems (adjusted odds ratio, 7.14 vs. AOR, 5.28); conduct problems (AOR, 22.31 vs. AOR, 4.77); hyperactivity (AOR, 9.49 vs. AOR, 5.58); and emotional symptoms (AOR, 19.06 vs. AOR, 2.85; Int J Adolesc Med. 2014;26[3]:369-75). Multivariate regression analysis revealed that Internet addictive behavior was independently associated with using the Internet for retrieving sexual information (AOR, 1.17) and for participating in games with monetary rewards (AOR, 1.90).
Dr. Deister said IA and aggression go hand in hand. A study of 714 middle school students in Seoul, South Korea, showed a linear association between aggression and IA (Cyberpsychol Behav Soc Netw. 2015;18[5]:260-7). A separate qualitative study of 27 university students in the United States found that the consequences of Internet use led to decreased physical activity, decreased sleep, decreased face-to-face time with other people, and poorer academic performance and concentration (PLoS ONE. 2015;10[2]:e0117372). Another study found that trait anhedonia at baseline predicted greater levels of compulsive Internet use, addiction to online activities, and greater likelihood of addiction to online/offline games (Comput Human Behav. 2016;62:475-9).
Treatment for IA takes all the usual forms, but outcome studies are limited. Cognitive-behavioral therapy for IA was first developed as a 12-week model with a familiar design of behavior modification, cognitive restructuring, harm reduction, and relapse prevention, Dr. Deister said. One study of treatment outcomes that used CBT in 128 IA patients found that 95% of patients were able to manage symptoms at the end of 12 weeks of treatment, while 78% sustained recovery 6 months after treatment (J Behav Addict. 2013;2[4]:209-15). Meanwhile, a meta-analysis of 12 studies related to IA found that medications and psychotherapy were found to be effective for improving IA status, time spent online, and depression and anxiety symptoms (Clin Psychol Rev. 2013;33:317-29). A more recent intention-to-treat analysis of IA disorders in adolescents and adults found that patients referred to treatment showed significant improvements in compulsive Internet use over time (J Behav Addict. 2017;6[4]:579-92). Differential effects were found depending on patients’ compliance, with high compliance generally resulting in significantly higher rates of change.
Dr. Deister reported having no financial disclosures.
SAN DIEGO – The concept of Internet addiction is imperfect, but it has validity and describes an impairing but treatable problem, according to Diana D. Deister, MD.
“Timely diagnosis can lead to good treatment and symptom improvement,” she said at the annual meeting and scientific symposium of the American Academy of Addiction Psychiatry.
Frameworks for the diagnosis of Internet addiction (IA)/pathological Internet use (PIU) vary but are based mostly on DSM-IV substance abuse and dependency criteria, DSM-IV pathological gambling, or other models. “There are many different checklists and diagnostic instruments you can use, and common synonyms include pathological Internet use, problematic Internet use, and compulsive Internet use,” said Dr. Deister, a child and adolescent psychiatrist at Boston Children’s Hospital. “They may not all refer to the exact same problem you might encounter in clinical practice.”
The Internet Addiction Diagnostic Questionnaire, developed in 1998 by Kimberly S. Young, PsyD, is widely used in research and consists of eight yes or no questions (Cyberpsychol Behav. 1998;1[3]:237-44). Answering “yes” to five out of the eight questions gives you a diagnosis of IA. “There are no preferred symptoms that everybody has to have in order to make the diagnosis, so this is more like the current SUD diagnosis for DSM-5,” Dr. Deister explained. “And there’s no time criteria.”
Clinicians can secure a more detailed assessment of IA symptoms by using the Internet Addiction Test (IAT), which Dr. Young developed in 2013. This tool consists of 20 questions rated on a five-point Likert scale based on frequency, where 0 stands for “not applicable” and 5 stands for “always.” Questions include “How often do you find that you stay online longer than you intended?” “How often do you block out disturbing thoughts about your life with soothing thoughts of the Internet?” And “How often do you feel depressed, moody, or nervous when you are offline, which goes away once you are back online?” A score of 80-100 is considered to meet criteria for IA, a score of 50-79 is considered borderline IA, while a score of below 50 is considered not pathological. Variations of this test can be found in the medical literature, including the Internet Process Addiction Test (Behav Sci. 2015;5:341-52), which contains subscales of Internet use: surfing, online gaming, social networking, and sex.
Dr. Deister noted that imaging studies of brain pathways in IA have demonstrated that mesolimbic dopaminergic projections to the nucleus accumbens from the ventral tegmental area are involved, as well as diminished activity of the ventral medial prefrontal cortex. In a tract-based spatial statistics study, researchers used diffusor tensor imaging (DTI) to study the white matter integrity in 18 adolescents with IA, and 18 age- and gender-matched controls (PLoS ONE. 7[1]:e30253. doi:10.1371/journal.pone.0030253). Compared with controls, the IA group scored higher in the IAT, the Strengths and Difficulties Questionnaire, the Screen for Child Anxiety Related Emotional Disorders scale, and Family Assessment Device measure. DTI scans demonstrated widespread reductions of fractional anisotropy (FA; a measure of white matter health) in major white matter pathways. In addition, significantly negative correlations were found between FA values in the left genu of the corpus callosum and the Screen for Child Anxiety Related Emotional Disorders, and between FA values in the left external capsule and in the IAT.
“The reason these white matter tracts could be important is that there have been previous studies showing that you can improve the health of your white matter tracks through exercise and physical therapy,” Dr. Deister said. “So it may be that this knowledge could lead to new treatments for some of these patients.”
One study of the dose dependence of IA symptoms found that adolescents who met criteria for addictive Internet use, compared with those who met criteria for borderline-addictive Internet use, had higher rates of peer problems (adjusted odds ratio, 7.14 vs. AOR, 5.28); conduct problems (AOR, 22.31 vs. AOR, 4.77); hyperactivity (AOR, 9.49 vs. AOR, 5.58); and emotional symptoms (AOR, 19.06 vs. AOR, 2.85; Int J Adolesc Med. 2014;26[3]:369-75). Multivariate regression analysis revealed that Internet addictive behavior was independently associated with using the Internet for retrieving sexual information (AOR, 1.17) and for participating in games with monetary rewards (AOR, 1.90).
Dr. Deister said IA and aggression go hand in hand. A study of 714 middle school students in Seoul, South Korea, showed a linear association between aggression and IA (Cyberpsychol Behav Soc Netw. 2015;18[5]:260-7). A separate qualitative study of 27 university students in the United States found that the consequences of Internet use led to decreased physical activity, decreased sleep, decreased face-to-face time with other people, and poorer academic performance and concentration (PLoS ONE. 2015;10[2]:e0117372). Another study found that trait anhedonia at baseline predicted greater levels of compulsive Internet use, addiction to online activities, and greater likelihood of addiction to online/offline games (Comput Human Behav. 2016;62:475-9).
Treatment for IA takes all the usual forms, but outcome studies are limited. Cognitive-behavioral therapy for IA was first developed as a 12-week model with a familiar design of behavior modification, cognitive restructuring, harm reduction, and relapse prevention, Dr. Deister said. One study of treatment outcomes that used CBT in 128 IA patients found that 95% of patients were able to manage symptoms at the end of 12 weeks of treatment, while 78% sustained recovery 6 months after treatment (J Behav Addict. 2013;2[4]:209-15). Meanwhile, a meta-analysis of 12 studies related to IA found that medications and psychotherapy were found to be effective for improving IA status, time spent online, and depression and anxiety symptoms (Clin Psychol Rev. 2013;33:317-29). A more recent intention-to-treat analysis of IA disorders in adolescents and adults found that patients referred to treatment showed significant improvements in compulsive Internet use over time (J Behav Addict. 2017;6[4]:579-92). Differential effects were found depending on patients’ compliance, with high compliance generally resulting in significantly higher rates of change.
Dr. Deister reported having no financial disclosures.
SAN DIEGO – The concept of Internet addiction is imperfect, but it has validity and describes an impairing but treatable problem, according to Diana D. Deister, MD.
“Timely diagnosis can lead to good treatment and symptom improvement,” she said at the annual meeting and scientific symposium of the American Academy of Addiction Psychiatry.
Frameworks for the diagnosis of Internet addiction (IA)/pathological Internet use (PIU) vary but are based mostly on DSM-IV substance abuse and dependency criteria, DSM-IV pathological gambling, or other models. “There are many different checklists and diagnostic instruments you can use, and common synonyms include pathological Internet use, problematic Internet use, and compulsive Internet use,” said Dr. Deister, a child and adolescent psychiatrist at Boston Children’s Hospital. “They may not all refer to the exact same problem you might encounter in clinical practice.”
The Internet Addiction Diagnostic Questionnaire, developed in 1998 by Kimberly S. Young, PsyD, is widely used in research and consists of eight yes or no questions (Cyberpsychol Behav. 1998;1[3]:237-44). Answering “yes” to five out of the eight questions gives you a diagnosis of IA. “There are no preferred symptoms that everybody has to have in order to make the diagnosis, so this is more like the current SUD diagnosis for DSM-5,” Dr. Deister explained. “And there’s no time criteria.”
Clinicians can secure a more detailed assessment of IA symptoms by using the Internet Addiction Test (IAT), which Dr. Young developed in 2013. This tool consists of 20 questions rated on a five-point Likert scale based on frequency, where 0 stands for “not applicable” and 5 stands for “always.” Questions include “How often do you find that you stay online longer than you intended?” “How often do you block out disturbing thoughts about your life with soothing thoughts of the Internet?” And “How often do you feel depressed, moody, or nervous when you are offline, which goes away once you are back online?” A score of 80-100 is considered to meet criteria for IA, a score of 50-79 is considered borderline IA, while a score of below 50 is considered not pathological. Variations of this test can be found in the medical literature, including the Internet Process Addiction Test (Behav Sci. 2015;5:341-52), which contains subscales of Internet use: surfing, online gaming, social networking, and sex.
Dr. Deister noted that imaging studies of brain pathways in IA have demonstrated that mesolimbic dopaminergic projections to the nucleus accumbens from the ventral tegmental area are involved, as well as diminished activity of the ventral medial prefrontal cortex. In a tract-based spatial statistics study, researchers used diffusor tensor imaging (DTI) to study the white matter integrity in 18 adolescents with IA, and 18 age- and gender-matched controls (PLoS ONE. 7[1]:e30253. doi:10.1371/journal.pone.0030253). Compared with controls, the IA group scored higher in the IAT, the Strengths and Difficulties Questionnaire, the Screen for Child Anxiety Related Emotional Disorders scale, and Family Assessment Device measure. DTI scans demonstrated widespread reductions of fractional anisotropy (FA; a measure of white matter health) in major white matter pathways. In addition, significantly negative correlations were found between FA values in the left genu of the corpus callosum and the Screen for Child Anxiety Related Emotional Disorders, and between FA values in the left external capsule and in the IAT.
“The reason these white matter tracts could be important is that there have been previous studies showing that you can improve the health of your white matter tracks through exercise and physical therapy,” Dr. Deister said. “So it may be that this knowledge could lead to new treatments for some of these patients.”
One study of the dose dependence of IA symptoms found that adolescents who met criteria for addictive Internet use, compared with those who met criteria for borderline-addictive Internet use, had higher rates of peer problems (adjusted odds ratio, 7.14 vs. AOR, 5.28); conduct problems (AOR, 22.31 vs. AOR, 4.77); hyperactivity (AOR, 9.49 vs. AOR, 5.58); and emotional symptoms (AOR, 19.06 vs. AOR, 2.85; Int J Adolesc Med. 2014;26[3]:369-75). Multivariate regression analysis revealed that Internet addictive behavior was independently associated with using the Internet for retrieving sexual information (AOR, 1.17) and for participating in games with monetary rewards (AOR, 1.90).
Dr. Deister said IA and aggression go hand in hand. A study of 714 middle school students in Seoul, South Korea, showed a linear association between aggression and IA (Cyberpsychol Behav Soc Netw. 2015;18[5]:260-7). A separate qualitative study of 27 university students in the United States found that the consequences of Internet use led to decreased physical activity, decreased sleep, decreased face-to-face time with other people, and poorer academic performance and concentration (PLoS ONE. 2015;10[2]:e0117372). Another study found that trait anhedonia at baseline predicted greater levels of compulsive Internet use, addiction to online activities, and greater likelihood of addiction to online/offline games (Comput Human Behav. 2016;62:475-9).
Treatment for IA takes all the usual forms, but outcome studies are limited. Cognitive-behavioral therapy for IA was first developed as a 12-week model with a familiar design of behavior modification, cognitive restructuring, harm reduction, and relapse prevention, Dr. Deister said. One study of treatment outcomes that used CBT in 128 IA patients found that 95% of patients were able to manage symptoms at the end of 12 weeks of treatment, while 78% sustained recovery 6 months after treatment (J Behav Addict. 2013;2[4]:209-15). Meanwhile, a meta-analysis of 12 studies related to IA found that medications and psychotherapy were found to be effective for improving IA status, time spent online, and depression and anxiety symptoms (Clin Psychol Rev. 2013;33:317-29). A more recent intention-to-treat analysis of IA disorders in adolescents and adults found that patients referred to treatment showed significant improvements in compulsive Internet use over time (J Behav Addict. 2017;6[4]:579-92). Differential effects were found depending on patients’ compliance, with high compliance generally resulting in significantly higher rates of change.
Dr. Deister reported having no financial disclosures.
REPORTING FROM AAAP
Facial exercises improved appearance in small study of middle-aged women
The role of skin laxity and substructural fat and muscle loss in the appearance of facial aging has been recognized already, and there has been interest within the nonmedical community regarding use of facial exercise to improve appearance, Murad Alam, MD, of Northwestern University, Chicago, and his colleagues wrote in a research letter published in JAMA Dermatology.
The researchers recruited healthy women aged 40-65 years with some photodamage to the face and an interest in facial exercises. After two 90-minute, in-person training sessions with a certified instructor, the participants were asked to perform a 30-minute facial exercise session daily for 8 weeks at home, followed by sessions every other day during weeks 9-20. Sixteen patients completed the full 20-week study.
Two blinded physicians used validated assessment scales to compare photographs of the participants taken at the beginning and end of the 20-week period. Facial exercise was associated with an improved mean upper-cheek fullness score, compared with baseline (1.1 vs. 1.8, respectively; P = .003), and an improved mean lower-cheek fullness score, compared with baseline (0.9 vs. 1.6; P = .003).
In addition, blinded physicians’ estimates of the women’s ages decreased significantly: The estimates dropped from an average of 51 years at baseline to an average of 49 years after the women completed the 20 weeks of facial exercises (P = .002), Dr. Alam and his associates reported.
The study was limited by several factors, including its small sample size, the lack of a control group, and a self-selected population that may have been especially motivated to follow the exercise routine, the researchers noted.
However, the results suggest that the cause for improvements in appearance as a result of the exercises “may be exercise-actuated hypertrophy of cheek and other muscles,” they said. “Further research is warranted to isolate the causes and effects of exercise-related changes and to assess the generalizability of these findings,” Dr. Alam and his associates concluded.
The study was supported by research funds from the department of dermatology at Northwestern University. Dr. Alam disclosed serving as a consultant for Amway and Leo Pharma and has served as an investigator on studies supported in part by Allergan, Medicis Pharmaceutical, BioForm Medical, and Ulthera.
SOURCE: Alam M et al. JAMA Dermatol. 2018 Jan 3. doi: 10.1001/jamadermatol.2017.5142
The role of skin laxity and substructural fat and muscle loss in the appearance of facial aging has been recognized already, and there has been interest within the nonmedical community regarding use of facial exercise to improve appearance, Murad Alam, MD, of Northwestern University, Chicago, and his colleagues wrote in a research letter published in JAMA Dermatology.
The researchers recruited healthy women aged 40-65 years with some photodamage to the face and an interest in facial exercises. After two 90-minute, in-person training sessions with a certified instructor, the participants were asked to perform a 30-minute facial exercise session daily for 8 weeks at home, followed by sessions every other day during weeks 9-20. Sixteen patients completed the full 20-week study.
Two blinded physicians used validated assessment scales to compare photographs of the participants taken at the beginning and end of the 20-week period. Facial exercise was associated with an improved mean upper-cheek fullness score, compared with baseline (1.1 vs. 1.8, respectively; P = .003), and an improved mean lower-cheek fullness score, compared with baseline (0.9 vs. 1.6; P = .003).
In addition, blinded physicians’ estimates of the women’s ages decreased significantly: The estimates dropped from an average of 51 years at baseline to an average of 49 years after the women completed the 20 weeks of facial exercises (P = .002), Dr. Alam and his associates reported.
The study was limited by several factors, including its small sample size, the lack of a control group, and a self-selected population that may have been especially motivated to follow the exercise routine, the researchers noted.
However, the results suggest that the cause for improvements in appearance as a result of the exercises “may be exercise-actuated hypertrophy of cheek and other muscles,” they said. “Further research is warranted to isolate the causes and effects of exercise-related changes and to assess the generalizability of these findings,” Dr. Alam and his associates concluded.
The study was supported by research funds from the department of dermatology at Northwestern University. Dr. Alam disclosed serving as a consultant for Amway and Leo Pharma and has served as an investigator on studies supported in part by Allergan, Medicis Pharmaceutical, BioForm Medical, and Ulthera.
SOURCE: Alam M et al. JAMA Dermatol. 2018 Jan 3. doi: 10.1001/jamadermatol.2017.5142
The role of skin laxity and substructural fat and muscle loss in the appearance of facial aging has been recognized already, and there has been interest within the nonmedical community regarding use of facial exercise to improve appearance, Murad Alam, MD, of Northwestern University, Chicago, and his colleagues wrote in a research letter published in JAMA Dermatology.
The researchers recruited healthy women aged 40-65 years with some photodamage to the face and an interest in facial exercises. After two 90-minute, in-person training sessions with a certified instructor, the participants were asked to perform a 30-minute facial exercise session daily for 8 weeks at home, followed by sessions every other day during weeks 9-20. Sixteen patients completed the full 20-week study.
Two blinded physicians used validated assessment scales to compare photographs of the participants taken at the beginning and end of the 20-week period. Facial exercise was associated with an improved mean upper-cheek fullness score, compared with baseline (1.1 vs. 1.8, respectively; P = .003), and an improved mean lower-cheek fullness score, compared with baseline (0.9 vs. 1.6; P = .003).
In addition, blinded physicians’ estimates of the women’s ages decreased significantly: The estimates dropped from an average of 51 years at baseline to an average of 49 years after the women completed the 20 weeks of facial exercises (P = .002), Dr. Alam and his associates reported.
The study was limited by several factors, including its small sample size, the lack of a control group, and a self-selected population that may have been especially motivated to follow the exercise routine, the researchers noted.
However, the results suggest that the cause for improvements in appearance as a result of the exercises “may be exercise-actuated hypertrophy of cheek and other muscles,” they said. “Further research is warranted to isolate the causes and effects of exercise-related changes and to assess the generalizability of these findings,” Dr. Alam and his associates concluded.
The study was supported by research funds from the department of dermatology at Northwestern University. Dr. Alam disclosed serving as a consultant for Amway and Leo Pharma and has served as an investigator on studies supported in part by Allergan, Medicis Pharmaceutical, BioForm Medical, and Ulthera.
SOURCE: Alam M et al. JAMA Dermatol. 2018 Jan 3. doi: 10.1001/jamadermatol.2017.5142
FROM JAMA DERMATOLOGY
Key clinical point: A 20-week program of facial exercise significantly improved facial fullness and perceived age among women aged 40-65 years.
Major finding: Fullness of the upper and lower cheek significantly improved from baseline after the exercise program, based on a validated scale.
Data source: The data come from a study of 27 women aged 40-65 years.
Disclosures: The study was supported by research funds from the department of dermatology at Northwestern University. Dr. Alam disclosed serving as a consultant for Amway and LEO Pharma and has served as an investigator on studies supported in part by Allergan, Medicis Pharmaceutical, BioForm Medical, and Ulthera.
Source: Alam M et al. JAMA Dermatol. 2018 Jan 3. doi: 10.1001/jamadermatol.2017.5142.

Neutrophilic urticarial dermatosis is usually misdiagnosed
GENEVA – Neutrophilic urticarial dermatosis (NUD) in patients with systemic lupus erythematosus (SLE) is “almost always” initially misdiagnosed as a lupus flare and treated inappropriately, Dan Lipsker, MD, PhD, said in a plenary address at the annual congress of the European Academy of Dermatology and Venereology.
“This is a condition that is underdiagnosed and overtreated,” declared Dr. Lipsker, professor of dermatology at the University of Strasbourg (France).

NUD is not rare. Dr. Lipsker estimates it occurs in 1%-2% of patients with SLE. In a retrospective study of seven patients with NUD and SLE, he and his colleagues reported that NUD was initially misdiagnosed as a lupus flare in 4 patients, who were then treated with immunosuppressive drugs (Medicine. 2014 Dec;93[29]:e351]). “That’s quite logical because the patients had a rash, fever, and joint pain,” the dermatologist noted.
However, : It won’t alleviate the symptoms and needlessly exposes the patient to drug toxicities.
The treatment for NUD is not prednisone, mycophenolate mofetil, an antimalarial, or other drugs conventionally prescribed for SLE; it’s a drug that inhibits neutrophil migration, such as dapsone at 50-200 mg per day or colchicine at 0.5-1.0 mg per day. Typically, within just a few days after starting the appropriate therapy, the joint pain and rash of NUD are gone, according to Dr. Lipsker.
Making the diagnosis
The rash of NUD is distinctly different from a classic lupus rash. It consists of pale red macules or slightly raised nonpruritic papules. Individual lesions will disappear spontaneously within 24-48 hours.
The histopathology of NUD is characteristic of a neutrophilic dermatosis. On biopsy, an intense neutrophilic perivascular and interstitial infiltrate with leukocytoclasia is seen. There is no damage to the blood vessel walls, which readily distinguishes NUD from urticarial vasculitis.
Other neutrophilic dermatoses have also been reported with increased frequency in patients with SLE. These include Sweet syndrome, pyoderma gangrenosum, bullous SLE, amicrobial pustulosis of the folds, and palisaded neutrophilic granulomatous dermatitis. Dr. Lipsker lumps them, together with NUD, as neutrophilic cutaneous lupus erythematosus. Affected SLE patients have an exaggerated innate immune response. It is as yet unclear if these neutrophilic dermatoses have prognostic significance in the setting of SLE, he said.
Dr. Lipsker reported having no financial conflicts of interest regarding his presentation.
GENEVA – Neutrophilic urticarial dermatosis (NUD) in patients with systemic lupus erythematosus (SLE) is “almost always” initially misdiagnosed as a lupus flare and treated inappropriately, Dan Lipsker, MD, PhD, said in a plenary address at the annual congress of the European Academy of Dermatology and Venereology.
“This is a condition that is underdiagnosed and overtreated,” declared Dr. Lipsker, professor of dermatology at the University of Strasbourg (France).
NUD is not rare. Dr. Lipsker estimates it occurs in 1%-2% of patients with SLE. In a retrospective study of seven patients with NUD and SLE, he and his colleagues reported that NUD was initially misdiagnosed as a lupus flare in 4 patients, who were then treated with immunosuppressive drugs (Medicine. 2014 Dec;93[29]:e351]). “That’s quite logical because the patients had a rash, fever, and joint pain,” the dermatologist noted.
However, : It won’t alleviate the symptoms and needlessly exposes the patient to drug toxicities.
The treatment for NUD is not prednisone, mycophenolate mofetil, an antimalarial, or other drugs conventionally prescribed for SLE; it’s a drug that inhibits neutrophil migration, such as dapsone at 50-200 mg per day or colchicine at 0.5-1.0 mg per day. Typically, within just a few days after starting the appropriate therapy, the joint pain and rash of NUD are gone, according to Dr. Lipsker.
Making the diagnosis
The rash of NUD is distinctly different from a classic lupus rash. It consists of pale red macules or slightly raised nonpruritic papules. Individual lesions will disappear spontaneously within 24-48 hours.
The histopathology of NUD is characteristic of a neutrophilic dermatosis. On biopsy, an intense neutrophilic perivascular and interstitial infiltrate with leukocytoclasia is seen. There is no damage to the blood vessel walls, which readily distinguishes NUD from urticarial vasculitis.
Other neutrophilic dermatoses have also been reported with increased frequency in patients with SLE. These include Sweet syndrome, pyoderma gangrenosum, bullous SLE, amicrobial pustulosis of the folds, and palisaded neutrophilic granulomatous dermatitis. Dr. Lipsker lumps them, together with NUD, as neutrophilic cutaneous lupus erythematosus. Affected SLE patients have an exaggerated innate immune response. It is as yet unclear if these neutrophilic dermatoses have prognostic significance in the setting of SLE, he said.
Dr. Lipsker reported having no financial conflicts of interest regarding his presentation.
GENEVA – Neutrophilic urticarial dermatosis (NUD) in patients with systemic lupus erythematosus (SLE) is “almost always” initially misdiagnosed as a lupus flare and treated inappropriately, Dan Lipsker, MD, PhD, said in a plenary address at the annual congress of the European Academy of Dermatology and Venereology.
“This is a condition that is underdiagnosed and overtreated,” declared Dr. Lipsker, professor of dermatology at the University of Strasbourg (France).
NUD is not rare. Dr. Lipsker estimates it occurs in 1%-2% of patients with SLE. In a retrospective study of seven patients with NUD and SLE, he and his colleagues reported that NUD was initially misdiagnosed as a lupus flare in 4 patients, who were then treated with immunosuppressive drugs (Medicine. 2014 Dec;93[29]:e351]). “That’s quite logical because the patients had a rash, fever, and joint pain,” the dermatologist noted.
However, : It won’t alleviate the symptoms and needlessly exposes the patient to drug toxicities.
The treatment for NUD is not prednisone, mycophenolate mofetil, an antimalarial, or other drugs conventionally prescribed for SLE; it’s a drug that inhibits neutrophil migration, such as dapsone at 50-200 mg per day or colchicine at 0.5-1.0 mg per day. Typically, within just a few days after starting the appropriate therapy, the joint pain and rash of NUD are gone, according to Dr. Lipsker.
Making the diagnosis
The rash of NUD is distinctly different from a classic lupus rash. It consists of pale red macules or slightly raised nonpruritic papules. Individual lesions will disappear spontaneously within 24-48 hours.
The histopathology of NUD is characteristic of a neutrophilic dermatosis. On biopsy, an intense neutrophilic perivascular and interstitial infiltrate with leukocytoclasia is seen. There is no damage to the blood vessel walls, which readily distinguishes NUD from urticarial vasculitis.
Other neutrophilic dermatoses have also been reported with increased frequency in patients with SLE. These include Sweet syndrome, pyoderma gangrenosum, bullous SLE, amicrobial pustulosis of the folds, and palisaded neutrophilic granulomatous dermatitis. Dr. Lipsker lumps them, together with NUD, as neutrophilic cutaneous lupus erythematosus. Affected SLE patients have an exaggerated innate immune response. It is as yet unclear if these neutrophilic dermatoses have prognostic significance in the setting of SLE, he said.
Dr. Lipsker reported having no financial conflicts of interest regarding his presentation.
EXPERT ANALYSIS FROM THE EADV CONGRESS
In Brazil, few patients get second- and third-line treatment for metastatic RCC
, a retrospective study showed.
Of 3,990 patients with metastatic renal cell carcinoma (mRCC), 79% received an appropriate first-line treatment – mainly a vascular endothelial growth factor agent. But only 20% went on to get a second-line agent, and just 5% received a third-line agent, Paulo G. Bergerot, MD, and his colleagues reported in the Journal of Global Oncology.
Patients in private institutions were significantly more likely to receive appropriate first- and second-line treatment than those in public institutions, although the numbers receiving third-line agents were similarly low, reported Dr. Bergerot of the Federal University of São Paulo and his coauthors.
The study highlights sharp discrepancies between treatment in Brazil and more developed countries, the team noted.
“Previous reports from the International Metastatic Renal Cell Carcinoma Database Consortium suggest that approximately 48% of patients who receive first-line therapy proceed to second-line therapy. In addition, among patients who received first-line therapy in this experience, approximately 21% received third-line therapy,” the investigators wrote.
The reasons behind the differences aren’t entirely clear, but cost and clinicians’ knowledge of emerging study data could be major factors, they suggested.
“In particular, we suspect limited availability and cost of second-line treatments to be a barrier, although our data set did not have the capability of confirming this. Another barrier to receipt of second-line therapy might be educational gaps among practitioners. Emerging data from phase 3 studies supporting the use of agents in the refractory setting may not be widely broadcast. The discordance in receipt of therapies in private and public settings is perhaps the greatest indication that financial and social barriers likely affect treatment paradigms in Brazil,” the authors wrote.
Slow dissemination of clinical knowledge may also be reflected in another of the team’s findings: 240 patients received “nontraditional” first-line cytotoxic treatments, which lacked regulatory approval and had little supporting evidence for treating mRCC, the investigators reported.
Dr. Bergerot had no relevant financial disclosures, although several of his coauthors reported financial relationships with various pharmaceutical companies.
SOURCE: Bergerot et al. J Glob Oncol. 2017 Dec 27. doi: 10.1200/JGO.17.00113.
, a retrospective study showed.
Of 3,990 patients with metastatic renal cell carcinoma (mRCC), 79% received an appropriate first-line treatment – mainly a vascular endothelial growth factor agent. But only 20% went on to get a second-line agent, and just 5% received a third-line agent, Paulo G. Bergerot, MD, and his colleagues reported in the Journal of Global Oncology.
Patients in private institutions were significantly more likely to receive appropriate first- and second-line treatment than those in public institutions, although the numbers receiving third-line agents were similarly low, reported Dr. Bergerot of the Federal University of São Paulo and his coauthors.
The study highlights sharp discrepancies between treatment in Brazil and more developed countries, the team noted.
“Previous reports from the International Metastatic Renal Cell Carcinoma Database Consortium suggest that approximately 48% of patients who receive first-line therapy proceed to second-line therapy. In addition, among patients who received first-line therapy in this experience, approximately 21% received third-line therapy,” the investigators wrote.
The reasons behind the differences aren’t entirely clear, but cost and clinicians’ knowledge of emerging study data could be major factors, they suggested.
“In particular, we suspect limited availability and cost of second-line treatments to be a barrier, although our data set did not have the capability of confirming this. Another barrier to receipt of second-line therapy might be educational gaps among practitioners. Emerging data from phase 3 studies supporting the use of agents in the refractory setting may not be widely broadcast. The discordance in receipt of therapies in private and public settings is perhaps the greatest indication that financial and social barriers likely affect treatment paradigms in Brazil,” the authors wrote.
Slow dissemination of clinical knowledge may also be reflected in another of the team’s findings: 240 patients received “nontraditional” first-line cytotoxic treatments, which lacked regulatory approval and had little supporting evidence for treating mRCC, the investigators reported.
Dr. Bergerot had no relevant financial disclosures, although several of his coauthors reported financial relationships with various pharmaceutical companies.
SOURCE: Bergerot et al. J Glob Oncol. 2017 Dec 27. doi: 10.1200/JGO.17.00113.
, a retrospective study showed.
Of 3,990 patients with metastatic renal cell carcinoma (mRCC), 79% received an appropriate first-line treatment – mainly a vascular endothelial growth factor agent. But only 20% went on to get a second-line agent, and just 5% received a third-line agent, Paulo G. Bergerot, MD, and his colleagues reported in the Journal of Global Oncology.
Patients in private institutions were significantly more likely to receive appropriate first- and second-line treatment than those in public institutions, although the numbers receiving third-line agents were similarly low, reported Dr. Bergerot of the Federal University of São Paulo and his coauthors.
The study highlights sharp discrepancies between treatment in Brazil and more developed countries, the team noted.
“Previous reports from the International Metastatic Renal Cell Carcinoma Database Consortium suggest that approximately 48% of patients who receive first-line therapy proceed to second-line therapy. In addition, among patients who received first-line therapy in this experience, approximately 21% received third-line therapy,” the investigators wrote.
The reasons behind the differences aren’t entirely clear, but cost and clinicians’ knowledge of emerging study data could be major factors, they suggested.
“In particular, we suspect limited availability and cost of second-line treatments to be a barrier, although our data set did not have the capability of confirming this. Another barrier to receipt of second-line therapy might be educational gaps among practitioners. Emerging data from phase 3 studies supporting the use of agents in the refractory setting may not be widely broadcast. The discordance in receipt of therapies in private and public settings is perhaps the greatest indication that financial and social barriers likely affect treatment paradigms in Brazil,” the authors wrote.
Slow dissemination of clinical knowledge may also be reflected in another of the team’s findings: 240 patients received “nontraditional” first-line cytotoxic treatments, which lacked regulatory approval and had little supporting evidence for treating mRCC, the investigators reported.
Dr. Bergerot had no relevant financial disclosures, although several of his coauthors reported financial relationships with various pharmaceutical companies.
SOURCE: Bergerot et al. J Glob Oncol. 2017 Dec 27. doi: 10.1200/JGO.17.00113.
FROM THE JOURNAL OF GLOBAL ONCOLOGY
Key clinical point: Few Brazilians with mRCC receive anything after their first-line treatment.
Major finding: First-line agents were used in 79% of the cohort, but only 20% got second-line treatments and just 5%, third-line treatment.
Study details: A retrospective database study involving 3,990 patients with mRCC.
Disclosures: Dr. Bergerot had no relevant financial disclosures, although several of his coauthors disclosed financial relationships with pharmaceutical companies.
Source: Bergerot et al. J Glob Oncol. 2017 Dec 27. doi: 10.1200/JGO.17.00113.
2017 update to McDonald criteria loosens MS diagnosis somewhat
Updates to the McDonald criteria for diagnosing multiple sclerosis (MS), first introduced in 2010, should allow initiation of therapy earlier in the time point of disease. The changes expand some of the criteria that can be used for diagnosis of disease.
The McDonald criteria should be applied primarily to patients with a typical clinically isolated syndrome – that is, in whom the probability of MS is high. They are best applied to patients 11 years or older, according to first author Alan J. Thompson, MD, and his colleagues on the International Panel on Diagnosis of Multiple Sclerosis. Their update to the 2010 criteria was published online Dec. 21, 2017, in The Lancet Neurology.
The changes include:
• Cerebrospinal fluid-specific oligoclonal bands can now be used to diagnose MS in patients with a typical clinically isolated syndrome in whom MRI or clinical signs point to dissemination in space (DIS), and if there is no other, better explanation for clinical signs.
• Symptomatic or asymptomatic MRI lesions can be used in the determination of DIS or dissemination in time (DIT). An exception is MRI lesions in the optic nerve in patients with optic neuritis, due to insufficient evidence. Specifically, the panel notes that DIS “can be demonstrated by one or more T2-hyperintense lesions [symptomatic or asymptomatic] that are characteristic of multiple sclerosis in two or more of four areas of the CNS: periventricular, cortical or juxtacortical, and infratentorial brain regions, and the spinal cord.” DIT is defined by “the simultaneous presence of gadolinium-enhancing and non-enhancing lesions [symptomatic or asymptomatic] at any time or by a new T2-hyperintense or gadolinium-enhancing lesion on follow-up MRI, with reference to a baseline scan, irrespective of the timing of the baseline MRI.”
• In patients experiencing brainstem or spinal cord clinically isolated syndrome, symptomatic lesions are sufficient to determine DIS or DIT.
• Cortical or juxtacortical lesions can be used in determining DIS.
• When MS is diagnosed, physicians should determine disease course (relapsing-remitting, primary progressive, or secondary progressive), whether the disease is active or not, and whether it is progressive, using the clinical history over the previous year.
The update was driven by a range of factors, including ongoing developments in imaging, the performance of the 2010 guidelines in diverse populations, and potential confusion between MS and other conditions with similar imaging characteristics, such as neuromyelitis optica spectrum disorders, which should always be considered because symptoms can overlap with MS. These conditions demand different treatment protocols. In addition, in 2016, the European Magnetic Resonance Imaging in Multiple Sclerosis (MAGNIMS) network suggested changes to the MRI criteria for diagnosing multiple sclerosis.
Updates to the guidelines are hardly finished. The panel called for future examination of optic nerve involvement, diverse populations, advanced imaging techniques, and biomarkers.
Some of the panel members reported financial ties to the pharmaceutical industry.
SOURCE: Thompson A et al., Lancet Neurol. 2017 Dec 21. doi: 10.1016/S1474-4422(17)30470-2
Earlier diagnosis and treatment initiation is increasingly important to prevent long-term disability. In recent years, MRI has become a more powerful diagnostic tool. These guidelines present useful improvements, but challenges remain. Due to a lack of data, it is not clear how well the McDonald criteria perform in nonwhite and non-Western populations, as well as in patients with atypical presentation of a clinically isolated syndrome. The guidelines also offer little help in the treatment of asymptomatic patients with radiologically isolated syndromes who eventually develop dissemination in time or space as determined by MRI findings.
Careful application of the new criteria should reduce misdiagnosis between MS and migraine, fibromyalgia, psychiatric disorders, and neuromyelitis optica spectrum disorders, but vigilance is required and clinicians should keep an open mind regarding alternative diagnoses.
Stephen Hauser, MD, and Riley Bove, MD, are with the University of California, San Francisco. Dr. Hauser serves on the board of trustees for Neurona Therapeutics, and scientific advisory boards for Symbiotix, Annexon, Bionure, and Molecular Stethoscope, and he has received travel reimbursement and writing support from F. Hoffmann La Roche. Dr. Bove has received fees from Genzyme-Sanofi, Roche-Genentech, and Novartis. Their comments are derived from an editorial accompanying the 2017 McDonald criteria (Lancet Neurol. 2017 Dec 21. doi: 10.1016/S1474-4422[17]30461-1)
Earlier diagnosis and treatment initiation is increasingly important to prevent long-term disability. In recent years, MRI has become a more powerful diagnostic tool. These guidelines present useful improvements, but challenges remain. Due to a lack of data, it is not clear how well the McDonald criteria perform in nonwhite and non-Western populations, as well as in patients with atypical presentation of a clinically isolated syndrome. The guidelines also offer little help in the treatment of asymptomatic patients with radiologically isolated syndromes who eventually develop dissemination in time or space as determined by MRI findings.
Careful application of the new criteria should reduce misdiagnosis between MS and migraine, fibromyalgia, psychiatric disorders, and neuromyelitis optica spectrum disorders, but vigilance is required and clinicians should keep an open mind regarding alternative diagnoses.
Stephen Hauser, MD, and Riley Bove, MD, are with the University of California, San Francisco. Dr. Hauser serves on the board of trustees for Neurona Therapeutics, and scientific advisory boards for Symbiotix, Annexon, Bionure, and Molecular Stethoscope, and he has received travel reimbursement and writing support from F. Hoffmann La Roche. Dr. Bove has received fees from Genzyme-Sanofi, Roche-Genentech, and Novartis. Their comments are derived from an editorial accompanying the 2017 McDonald criteria (Lancet Neurol. 2017 Dec 21. doi: 10.1016/S1474-4422[17]30461-1)
Earlier diagnosis and treatment initiation is increasingly important to prevent long-term disability. In recent years, MRI has become a more powerful diagnostic tool. These guidelines present useful improvements, but challenges remain. Due to a lack of data, it is not clear how well the McDonald criteria perform in nonwhite and non-Western populations, as well as in patients with atypical presentation of a clinically isolated syndrome. The guidelines also offer little help in the treatment of asymptomatic patients with radiologically isolated syndromes who eventually develop dissemination in time or space as determined by MRI findings.
Careful application of the new criteria should reduce misdiagnosis between MS and migraine, fibromyalgia, psychiatric disorders, and neuromyelitis optica spectrum disorders, but vigilance is required and clinicians should keep an open mind regarding alternative diagnoses.
Stephen Hauser, MD, and Riley Bove, MD, are with the University of California, San Francisco. Dr. Hauser serves on the board of trustees for Neurona Therapeutics, and scientific advisory boards for Symbiotix, Annexon, Bionure, and Molecular Stethoscope, and he has received travel reimbursement and writing support from F. Hoffmann La Roche. Dr. Bove has received fees from Genzyme-Sanofi, Roche-Genentech, and Novartis. Their comments are derived from an editorial accompanying the 2017 McDonald criteria (Lancet Neurol. 2017 Dec 21. doi: 10.1016/S1474-4422[17]30461-1)
Updates to the McDonald criteria for diagnosing multiple sclerosis (MS), first introduced in 2010, should allow initiation of therapy earlier in the time point of disease. The changes expand some of the criteria that can be used for diagnosis of disease.
The McDonald criteria should be applied primarily to patients with a typical clinically isolated syndrome – that is, in whom the probability of MS is high. They are best applied to patients 11 years or older, according to first author Alan J. Thompson, MD, and his colleagues on the International Panel on Diagnosis of Multiple Sclerosis. Their update to the 2010 criteria was published online Dec. 21, 2017, in The Lancet Neurology.
The changes include:
• Cerebrospinal fluid-specific oligoclonal bands can now be used to diagnose MS in patients with a typical clinically isolated syndrome in whom MRI or clinical signs point to dissemination in space (DIS), and if there is no other, better explanation for clinical signs.
• Symptomatic or asymptomatic MRI lesions can be used in the determination of DIS or dissemination in time (DIT). An exception is MRI lesions in the optic nerve in patients with optic neuritis, due to insufficient evidence. Specifically, the panel notes that DIS “can be demonstrated by one or more T2-hyperintense lesions [symptomatic or asymptomatic] that are characteristic of multiple sclerosis in two or more of four areas of the CNS: periventricular, cortical or juxtacortical, and infratentorial brain regions, and the spinal cord.” DIT is defined by “the simultaneous presence of gadolinium-enhancing and non-enhancing lesions [symptomatic or asymptomatic] at any time or by a new T2-hyperintense or gadolinium-enhancing lesion on follow-up MRI, with reference to a baseline scan, irrespective of the timing of the baseline MRI.”
• In patients experiencing brainstem or spinal cord clinically isolated syndrome, symptomatic lesions are sufficient to determine DIS or DIT.
• Cortical or juxtacortical lesions can be used in determining DIS.
• When MS is diagnosed, physicians should determine disease course (relapsing-remitting, primary progressive, or secondary progressive), whether the disease is active or not, and whether it is progressive, using the clinical history over the previous year.
The update was driven by a range of factors, including ongoing developments in imaging, the performance of the 2010 guidelines in diverse populations, and potential confusion between MS and other conditions with similar imaging characteristics, such as neuromyelitis optica spectrum disorders, which should always be considered because symptoms can overlap with MS. These conditions demand different treatment protocols. In addition, in 2016, the European Magnetic Resonance Imaging in Multiple Sclerosis (MAGNIMS) network suggested changes to the MRI criteria for diagnosing multiple sclerosis.
Updates to the guidelines are hardly finished. The panel called for future examination of optic nerve involvement, diverse populations, advanced imaging techniques, and biomarkers.
Some of the panel members reported financial ties to the pharmaceutical industry.
SOURCE: Thompson A et al., Lancet Neurol. 2017 Dec 21. doi: 10.1016/S1474-4422(17)30470-2
Updates to the McDonald criteria for diagnosing multiple sclerosis (MS), first introduced in 2010, should allow initiation of therapy earlier in the time point of disease. The changes expand some of the criteria that can be used for diagnosis of disease.
The McDonald criteria should be applied primarily to patients with a typical clinically isolated syndrome – that is, in whom the probability of MS is high. They are best applied to patients 11 years or older, according to first author Alan J. Thompson, MD, and his colleagues on the International Panel on Diagnosis of Multiple Sclerosis. Their update to the 2010 criteria was published online Dec. 21, 2017, in The Lancet Neurology.
The changes include:
• Cerebrospinal fluid-specific oligoclonal bands can now be used to diagnose MS in patients with a typical clinically isolated syndrome in whom MRI or clinical signs point to dissemination in space (DIS), and if there is no other, better explanation for clinical signs.
• Symptomatic or asymptomatic MRI lesions can be used in the determination of DIS or dissemination in time (DIT). An exception is MRI lesions in the optic nerve in patients with optic neuritis, due to insufficient evidence. Specifically, the panel notes that DIS “can be demonstrated by one or more T2-hyperintense lesions [symptomatic or asymptomatic] that are characteristic of multiple sclerosis in two or more of four areas of the CNS: periventricular, cortical or juxtacortical, and infratentorial brain regions, and the spinal cord.” DIT is defined by “the simultaneous presence of gadolinium-enhancing and non-enhancing lesions [symptomatic or asymptomatic] at any time or by a new T2-hyperintense or gadolinium-enhancing lesion on follow-up MRI, with reference to a baseline scan, irrespective of the timing of the baseline MRI.”
• In patients experiencing brainstem or spinal cord clinically isolated syndrome, symptomatic lesions are sufficient to determine DIS or DIT.
• Cortical or juxtacortical lesions can be used in determining DIS.
• When MS is diagnosed, physicians should determine disease course (relapsing-remitting, primary progressive, or secondary progressive), whether the disease is active or not, and whether it is progressive, using the clinical history over the previous year.
The update was driven by a range of factors, including ongoing developments in imaging, the performance of the 2010 guidelines in diverse populations, and potential confusion between MS and other conditions with similar imaging characteristics, such as neuromyelitis optica spectrum disorders, which should always be considered because symptoms can overlap with MS. These conditions demand different treatment protocols. In addition, in 2016, the European Magnetic Resonance Imaging in Multiple Sclerosis (MAGNIMS) network suggested changes to the MRI criteria for diagnosing multiple sclerosis.
Updates to the guidelines are hardly finished. The panel called for future examination of optic nerve involvement, diverse populations, advanced imaging techniques, and biomarkers.
Some of the panel members reported financial ties to the pharmaceutical industry.
SOURCE: Thompson A et al., Lancet Neurol. 2017 Dec 21. doi: 10.1016/S1474-4422(17)30470-2
FROM THE LANCET NEUROLOGY
Biologic mesh safe, effective for prevention of parastomal hernia recurrence
at 18.9%.
Theadore Hufford, MD, of the University of Illinois at Chicago and his colleagues conducted a study to look at the effects of placement and type of mesh on postop morbidity and recurrence of parastomal hernia (PSH). The study was a retrospective analysis of 58 patients who had undergone local PSH repair with biological mesh between July 2006 and July 2015 at a single medical center.
All procedures were conducted by three board-certified surgeons at a tertiary medical center, and decisions such as the mesh type, placement and incision type were determined by the attending surgeon’s operative preferences.
In the study group, mesh placement (overlay, underlay, or sandwich technique) was found to have a statistically significant effect on recurrence. Of the patients who received an underlay of biologic mesh, 33% had a recurrence, compared with 25% of those who had overlays. The sandwich technique (a combination of overlay and underlay) was found to have the lowest rate of recurrence at 6.7%. The type of mesh (human origin, bovine, or porcine) and type of stoma (colostomy vs. ileostomy) had no statistically significant effect on the rate of recurrence.
Total recurrences in the study patients was 18.9%, a figure consistent with the current literature on parastomal hernia repair, the investigators wrote.
A key factor in recurrence was type of incision. Keyhole incisions had a much lower rate of recurrence than did circular incisions (32% vs. 9.1%; P = .042).
In the study group, “one patient was readmitted for mesh infection within 30 days of the repair and required mesh removal. Even with the biologic mesh in place there was an overlying skin infection that warranted reoperation that resulted in the stoma being moved to a new site altogether,” the investigators wrote.
The limitations of this study include the retrospective nature of the research and the difficulty in diagnosing PSH, which is often asymptomatic, the investigators mentioned. In addition, the techniques for local PSH repair with biologic mesh are not fully standardized. Mesh type and location decisions are often made on a case-by-case basis which limits the applicability of the study data for general PSH repairs.
Dr. Hufford and his associates wrote, “Our results suggest local parastomal hernia repair with biological mesh is both a safe and effective method, especially when used with the sandwich technique for mesh placement, for definitive treatment of parastomal hernias with very low morbidity, and acceptable recurrence rate.”
The investigators reported no disclosures.
at 18.9%.
Theadore Hufford, MD, of the University of Illinois at Chicago and his colleagues conducted a study to look at the effects of placement and type of mesh on postop morbidity and recurrence of parastomal hernia (PSH). The study was a retrospective analysis of 58 patients who had undergone local PSH repair with biological mesh between July 2006 and July 2015 at a single medical center.
All procedures were conducted by three board-certified surgeons at a tertiary medical center, and decisions such as the mesh type, placement and incision type were determined by the attending surgeon’s operative preferences.
In the study group, mesh placement (overlay, underlay, or sandwich technique) was found to have a statistically significant effect on recurrence. Of the patients who received an underlay of biologic mesh, 33% had a recurrence, compared with 25% of those who had overlays. The sandwich technique (a combination of overlay and underlay) was found to have the lowest rate of recurrence at 6.7%. The type of mesh (human origin, bovine, or porcine) and type of stoma (colostomy vs. ileostomy) had no statistically significant effect on the rate of recurrence.
Total recurrences in the study patients was 18.9%, a figure consistent with the current literature on parastomal hernia repair, the investigators wrote.
A key factor in recurrence was type of incision. Keyhole incisions had a much lower rate of recurrence than did circular incisions (32% vs. 9.1%; P = .042).
In the study group, “one patient was readmitted for mesh infection within 30 days of the repair and required mesh removal. Even with the biologic mesh in place there was an overlying skin infection that warranted reoperation that resulted in the stoma being moved to a new site altogether,” the investigators wrote.
The limitations of this study include the retrospective nature of the research and the difficulty in diagnosing PSH, which is often asymptomatic, the investigators mentioned. In addition, the techniques for local PSH repair with biologic mesh are not fully standardized. Mesh type and location decisions are often made on a case-by-case basis which limits the applicability of the study data for general PSH repairs.
Dr. Hufford and his associates wrote, “Our results suggest local parastomal hernia repair with biological mesh is both a safe and effective method, especially when used with the sandwich technique for mesh placement, for definitive treatment of parastomal hernias with very low morbidity, and acceptable recurrence rate.”
The investigators reported no disclosures.
at 18.9%.
Theadore Hufford, MD, of the University of Illinois at Chicago and his colleagues conducted a study to look at the effects of placement and type of mesh on postop morbidity and recurrence of parastomal hernia (PSH). The study was a retrospective analysis of 58 patients who had undergone local PSH repair with biological mesh between July 2006 and July 2015 at a single medical center.
All procedures were conducted by three board-certified surgeons at a tertiary medical center, and decisions such as the mesh type, placement and incision type were determined by the attending surgeon’s operative preferences.
In the study group, mesh placement (overlay, underlay, or sandwich technique) was found to have a statistically significant effect on recurrence. Of the patients who received an underlay of biologic mesh, 33% had a recurrence, compared with 25% of those who had overlays. The sandwich technique (a combination of overlay and underlay) was found to have the lowest rate of recurrence at 6.7%. The type of mesh (human origin, bovine, or porcine) and type of stoma (colostomy vs. ileostomy) had no statistically significant effect on the rate of recurrence.
Total recurrences in the study patients was 18.9%, a figure consistent with the current literature on parastomal hernia repair, the investigators wrote.
A key factor in recurrence was type of incision. Keyhole incisions had a much lower rate of recurrence than did circular incisions (32% vs. 9.1%; P = .042).
In the study group, “one patient was readmitted for mesh infection within 30 days of the repair and required mesh removal. Even with the biologic mesh in place there was an overlying skin infection that warranted reoperation that resulted in the stoma being moved to a new site altogether,” the investigators wrote.
The limitations of this study include the retrospective nature of the research and the difficulty in diagnosing PSH, which is often asymptomatic, the investigators mentioned. In addition, the techniques for local PSH repair with biologic mesh are not fully standardized. Mesh type and location decisions are often made on a case-by-case basis which limits the applicability of the study data for general PSH repairs.
Dr. Hufford and his associates wrote, “Our results suggest local parastomal hernia repair with biological mesh is both a safe and effective method, especially when used with the sandwich technique for mesh placement, for definitive treatment of parastomal hernias with very low morbidity, and acceptable recurrence rate.”
The investigators reported no disclosures.
FROM THE AMERICAN JOURNAL OF SURGERY
Key clinical point: In a comparison of the biologic meshes and placement techniques, the sandwich technique was most successful in preventing recurrence.
Major finding: Rate of recurrence of parastomal hernia was 33% with underlay surgical mesh placement, 25% with overlay placement, and 6.7% with sandwich placement.
Study details: A retrospective analysis of 58 patients who had undergone local parastomal hernia repair with biological mesh at a single medical center.
Disclosures: The investigators reported no disclosures.
Source: Hufford T et al. Am J Surg. 2018 Jan. 215(1):88-90.
Phoenix Children’s Hospital integrates care from ground up
About 4 years ago, officials at Phoenix Children’s Hospital stopped and took a look around. The adult health care landscape was zooming toward value-based models and integrating care so that previously separate components were now working together. The health of whole populations mattered more than ever.
But their hospital, they found, accounted for just 9% of the “care touches” – interactions between a patient and a doctor – of their half-million pediatric population. They were not working closely with primary care doctors and independent specialists. Patients would come to the hospital, get treated, and then – poof, they were gone. The hospital came to a realization that there was a better way to provide care.

They began a process that led to what appears to be a “first-of-its-kind” model, an integrated care network created from the ground up by a hospital venturing out into the community and actively recruiting private primary care doctors and specialists. Now, more than 1,000 physicians from more than 100 practices in the Phoenix metro area are part of the network, joined with the hospital through contracts laden with incentives for meeting care and wellness goals. When good care is being given, the network gets paid, and everyone benefits financially.
“It’s amazing the difference we’re able to provide when we start linking together what used to be very disparate systems,” Mr. Johnson said.
Here are some features of the network:
Every group and practice, including the hospital, is now sharing their data. When a child shows up at the ER, the ER doctor can quickly see things like who the primary care doctor is, allergies, medications, and care history.
- Targets such as asthma control, providing basic wellness exams, and following patients appropriately, are tied to financial rewards.
- Children with complex or special health care needs, and patients who are high utilizers, have a care coordinator assigned to look more closely at their cases.
- A corporate entity created by the hospital and its independent community physician partners has a doctor-heavy board of directors composed of community primary care physicians, specialists, hospital-employed physicians, and hospital administrators.
Some of the care improvements have been dramatic, Mr. Johnson said. One teenager had made 55 ER visits and 21 inpatient visits over 9 months, but the pattern went unnoticed. With new tools – reports drawn from hospital records, insurance records, and records from other doctor visits – the problem became apparent. A care coordinator found that the mother didn’t quite understand how to administer the boy’s medication, prompting repeated medical crises and hundred of thousands of dollars in unnecessary costs. The teenager has since re-enrolled in school and has had no more hospital admissions, Johnson said.
He said that, at first, many community doctors had a “real skepticism” of being too closely tied to a hospital financially, but now doctors are getting in touch with the network about joining.
“There’s a leap of faith that has to happen in the initial stages,” he said. “When you get the insurance companies at the table to really work with you to build the right incentives around truly impactful and quality care, you can really start to move the needle. When you see – with data – that what you’re doing is having success, and they see the additional money coming from the incentives, that really helps.”
Amy Knight, MHA, chief operating officer of the Washington-based Children’s Hospital Association, said that, while other children’s hospitals have migrated toward more integrated care, they either haven’t needed to recruit community physicians as they have in Phoenix, because they already employed many primary care physicians, or market conditions have been such that they haven’t expanded as quickly.
“Phoenix saw a huge opportunity and was very smart about how they approached their own market,” she said. “They are definitely on the front end, the cutting edge of doing that.”
Since its network has expanded, Phoenix Children’s has hosted visitors who hope to draw lessons from their experience, she said.
“I think what most people go away with is: ‘Very interesting, very cool – not sure it would work in our market,’” she said. Still, lessons on thinking about risk and building a governance structure are widely applicable, she said.
“The house may look a little bit different, but some of the things you’re doing inside it may actually all look the same,” Ms. Knight said.
She expects a continued move toward more integrated care networks, despite the on-again, off-again political talk about repealing and replacing the Affordable Care Act.
“There’s probably some people stepping back in hesitancy, but I don’t think that the political discourse right now will necessarily change the trajectory that we’re on.”
About 4 years ago, officials at Phoenix Children’s Hospital stopped and took a look around. The adult health care landscape was zooming toward value-based models and integrating care so that previously separate components were now working together. The health of whole populations mattered more than ever.
But their hospital, they found, accounted for just 9% of the “care touches” – interactions between a patient and a doctor – of their half-million pediatric population. They were not working closely with primary care doctors and independent specialists. Patients would come to the hospital, get treated, and then – poof, they were gone. The hospital came to a realization that there was a better way to provide care.
They began a process that led to what appears to be a “first-of-its-kind” model, an integrated care network created from the ground up by a hospital venturing out into the community and actively recruiting private primary care doctors and specialists. Now, more than 1,000 physicians from more than 100 practices in the Phoenix metro area are part of the network, joined with the hospital through contracts laden with incentives for meeting care and wellness goals. When good care is being given, the network gets paid, and everyone benefits financially.
“It’s amazing the difference we’re able to provide when we start linking together what used to be very disparate systems,” Mr. Johnson said.
Here are some features of the network:
Every group and practice, including the hospital, is now sharing their data. When a child shows up at the ER, the ER doctor can quickly see things like who the primary care doctor is, allergies, medications, and care history.
- Targets such as asthma control, providing basic wellness exams, and following patients appropriately, are tied to financial rewards.
- Children with complex or special health care needs, and patients who are high utilizers, have a care coordinator assigned to look more closely at their cases.
- A corporate entity created by the hospital and its independent community physician partners has a doctor-heavy board of directors composed of community primary care physicians, specialists, hospital-employed physicians, and hospital administrators.
Some of the care improvements have been dramatic, Mr. Johnson said. One teenager had made 55 ER visits and 21 inpatient visits over 9 months, but the pattern went unnoticed. With new tools – reports drawn from hospital records, insurance records, and records from other doctor visits – the problem became apparent. A care coordinator found that the mother didn’t quite understand how to administer the boy’s medication, prompting repeated medical crises and hundred of thousands of dollars in unnecessary costs. The teenager has since re-enrolled in school and has had no more hospital admissions, Johnson said.
He said that, at first, many community doctors had a “real skepticism” of being too closely tied to a hospital financially, but now doctors are getting in touch with the network about joining.
“There’s a leap of faith that has to happen in the initial stages,” he said. “When you get the insurance companies at the table to really work with you to build the right incentives around truly impactful and quality care, you can really start to move the needle. When you see – with data – that what you’re doing is having success, and they see the additional money coming from the incentives, that really helps.”
Amy Knight, MHA, chief operating officer of the Washington-based Children’s Hospital Association, said that, while other children’s hospitals have migrated toward more integrated care, they either haven’t needed to recruit community physicians as they have in Phoenix, because they already employed many primary care physicians, or market conditions have been such that they haven’t expanded as quickly.
“Phoenix saw a huge opportunity and was very smart about how they approached their own market,” she said. “They are definitely on the front end, the cutting edge of doing that.”
Since its network has expanded, Phoenix Children’s has hosted visitors who hope to draw lessons from their experience, she said.
“I think what most people go away with is: ‘Very interesting, very cool – not sure it would work in our market,’” she said. Still, lessons on thinking about risk and building a governance structure are widely applicable, she said.
“The house may look a little bit different, but some of the things you’re doing inside it may actually all look the same,” Ms. Knight said.
She expects a continued move toward more integrated care networks, despite the on-again, off-again political talk about repealing and replacing the Affordable Care Act.
“There’s probably some people stepping back in hesitancy, but I don’t think that the political discourse right now will necessarily change the trajectory that we’re on.”
About 4 years ago, officials at Phoenix Children’s Hospital stopped and took a look around. The adult health care landscape was zooming toward value-based models and integrating care so that previously separate components were now working together. The health of whole populations mattered more than ever.
But their hospital, they found, accounted for just 9% of the “care touches” – interactions between a patient and a doctor – of their half-million pediatric population. They were not working closely with primary care doctors and independent specialists. Patients would come to the hospital, get treated, and then – poof, they were gone. The hospital came to a realization that there was a better way to provide care.
They began a process that led to what appears to be a “first-of-its-kind” model, an integrated care network created from the ground up by a hospital venturing out into the community and actively recruiting private primary care doctors and specialists. Now, more than 1,000 physicians from more than 100 practices in the Phoenix metro area are part of the network, joined with the hospital through contracts laden with incentives for meeting care and wellness goals. When good care is being given, the network gets paid, and everyone benefits financially.
“It’s amazing the difference we’re able to provide when we start linking together what used to be very disparate systems,” Mr. Johnson said.
Here are some features of the network:
Every group and practice, including the hospital, is now sharing their data. When a child shows up at the ER, the ER doctor can quickly see things like who the primary care doctor is, allergies, medications, and care history.
- Targets such as asthma control, providing basic wellness exams, and following patients appropriately, are tied to financial rewards.
- Children with complex or special health care needs, and patients who are high utilizers, have a care coordinator assigned to look more closely at their cases.
- A corporate entity created by the hospital and its independent community physician partners has a doctor-heavy board of directors composed of community primary care physicians, specialists, hospital-employed physicians, and hospital administrators.
Some of the care improvements have been dramatic, Mr. Johnson said. One teenager had made 55 ER visits and 21 inpatient visits over 9 months, but the pattern went unnoticed. With new tools – reports drawn from hospital records, insurance records, and records from other doctor visits – the problem became apparent. A care coordinator found that the mother didn’t quite understand how to administer the boy’s medication, prompting repeated medical crises and hundred of thousands of dollars in unnecessary costs. The teenager has since re-enrolled in school and has had no more hospital admissions, Johnson said.
He said that, at first, many community doctors had a “real skepticism” of being too closely tied to a hospital financially, but now doctors are getting in touch with the network about joining.
“There’s a leap of faith that has to happen in the initial stages,” he said. “When you get the insurance companies at the table to really work with you to build the right incentives around truly impactful and quality care, you can really start to move the needle. When you see – with data – that what you’re doing is having success, and they see the additional money coming from the incentives, that really helps.”
Amy Knight, MHA, chief operating officer of the Washington-based Children’s Hospital Association, said that, while other children’s hospitals have migrated toward more integrated care, they either haven’t needed to recruit community physicians as they have in Phoenix, because they already employed many primary care physicians, or market conditions have been such that they haven’t expanded as quickly.
“Phoenix saw a huge opportunity and was very smart about how they approached their own market,” she said. “They are definitely on the front end, the cutting edge of doing that.”
Since its network has expanded, Phoenix Children’s has hosted visitors who hope to draw lessons from their experience, she said.
“I think what most people go away with is: ‘Very interesting, very cool – not sure it would work in our market,’” she said. Still, lessons on thinking about risk and building a governance structure are widely applicable, she said.
“The house may look a little bit different, but some of the things you’re doing inside it may actually all look the same,” Ms. Knight said.
She expects a continued move toward more integrated care networks, despite the on-again, off-again political talk about repealing and replacing the Affordable Care Act.
“There’s probably some people stepping back in hesitancy, but I don’t think that the political discourse right now will necessarily change the trajectory that we’re on.”
Sexual harassment
Question: A medical assistant alleged that Dr. Y sexually harassed her by sending anonymous gifts and messages such as, “you’re gorgeous,” and “I love your figure.” It was a repeat of Dr. Y’s previous behavior pattern directed at a different worker, who had lodged a complaint with the human resources department. The medical assistant now files a sexual harassment action under Title VII of the federal Civil Rights Act of 1964 against the health care institution, alleging a hostile work environment.
Which of the following is false?
A. Sexual harassment is a form of sexual misconduct regulated by state medical boards.
B. Mere words, without physical action, may suffice to be deemed sexual harassment.
C. A hostile environment arises when offensive conduct is so severe and pervasive as to amount to job discrimination.
D. Sexual harassment is a civil rights violation unique to the workplace.
E. Liability may attach to the supervisor, institution, or the harasser.
Answer: D. This hypothetical is modified from an actual Connecticut case that was recently decided in favor of the plaintiff.1 In that case, which involved a dentist, the federal Second Circuit unanimously rejected the University of Connecticut Health Center’s appeal against a jury’s verdict holding it responsible for its employee’s sexual harassment of a coworker, who was awarded $125,000. It ruled that the health center should have known of its employee’s harassing behavior.
Sexual harassment, a current hot topic, is pervasive, affecting a diversity of individuals in the fields of media, sports, politics, judiciary, education, entertainment, and others. The medical profession is no exception, and studies indicate that sexual harassment affects patients and physicians alike, occurring in hospitals, private offices, and academic centers.
In a large questionnaire study involving 4,501 female physicians, the authors found a prevalence rate of 47.7%. Harassment was more common while in medical school or during internship, residency, or fellowship than in practice.2 Patients may be the harassers. In 599 of the 1,064 licensed female family physicians in Ontario, more than 75% reported sexual harassment by patients at some time during their careers, either in their own offices by their own patients, or in settings such as emergency departments and clinics, where unknown patients presented an even higher risk.3
When physicians sexually harass fellow workers such as nurses, they distract their victims from providing attentive and competent care. In a review of the subject, researchers cited a study of 188 critical care nurses in hospitals, where nearly half (46%) reported experiencing sexual harassment that included “offensive sexual remarks, unwanted physical contact, unwanted nonverbal attention, requests for unwanted dates, sexual propositions, and physical assault.”4 To this list must now be added misconduct via the use of social media. In the study, physicians (82%), coworkers (20%), and immediate supervisors (7%) accounted for most of the incidents.
Neglecting to look seriously into complaints or to monitor and remedy the situation may create a hostile environment and trigger liability.
An example is the recent well-publicized case of Olympics team physician Dr. Larry Nassar, who was also a faculty member at Michigan State University. Olympic gold medalist McKayla Maroney named both the university and the U.S. Olympic Committee as codefendants in a lawsuit alleging that the institutions failed to properly investigate the team doctor’s criminal sexual conduct.
In Anania v. Daubenspeck Chiropractic, two employees of a chiropractor alleged that his patients sexually harassed them, but he did not remedy the situation.5 The trial court initially dismissed their lawsuit, holding that Ohio law did not recognize a cause of action for sexual harassment by a nonemployee patient, and that liability for sexual harassment can only exist in the context of respondeat superior (employer-employee) liability.
However, the court of appeals held that so long as the chiropractor knew or should have known of the harassment, and failed to take corrective action, he could be liable for allowing a hostile environment to exist.
Negligent supervision is another favorite plaintiff’s cause of action. In Doe v. Borromeo, a patient sought to hold the hospital liable for sexual assault by a physician during a medical exam.6 The lower state court had summarily dismissed the case, which was based on vicarious liability, but the state court of appeals reversed, finding the patient’s complaint against the hospital included a negligent supervision claim.
The appeals court reasoned that this was distinguishable from one based upon vicarious liability, so long as the supervising entity had a duty to protect the victim – and such a duty can only be established if the supervising entity knew or should have known of the existence of the harasser’s propensities, if any, to commit criminal and tortious acts.
Sexual harassment is a form of sex discrimination under Title VII of the Civil Rights Act of 1964, which is enforced by the Equal Employment Opportunity Commission. The commission’s website explains the law in clear and simple language:
“It is unlawful to harass a person (an applicant or employee) because of that person’s sex. Harassment can include ‘sexual harassment’ or unwelcome sexual advances, requests for sexual favors, and other verbal or physical harassment of a sexual nature. Harassment does not have to be of a sexual nature, however, and can include offensive remarks about a person’s sex.
“For example, it is illegal to harass a woman by making offensive comments about women in general. Both victim and the harasser can be either a woman or a man, and the victim and harasser can be the same sex.
“Although the law doesn’t prohibit simple teasing, offhand comments, or isolated incidents that are not very serious, harassment is illegal when it is so frequent or severe that it creates a hostile or offensive work environment or when it results in an adverse employment decision (such as the victim being fired or demoted). The harasser can be the victim’s supervisor, a supervisor in another area, a coworker, or someone who is not an employee of the employer, such as a client or customer.”7
For sexual harassment to occur, the aggrieved party must either show a “hostile environment” or “quid pro quo” situation.
In a hostile environment case, the harassment is serious and persistent, creating unacceptable and offensive work conditions. The plaintiff has to show that the employer knew or should have known of the situation but failed to remedy it.
The “quid pro quo” type of case requires a showing that a person in authority conditioned some aspect of the employee’s employment, such as promotion or retention, upon a sexual favor or relationship.
The U.S. Supreme Court has both clarified and muddied the law’s position on these two previously distinct types of sexual harassment.
In the landmark case of Burlington Industries v. Ellerth, the plaintiff, who was a salesperson, alleged that a supervisor made advances to her and threatened to deny her certain job benefits if she did not cooperate.8 The threats were never carried out, and she was in fact promoted; but her lawsuit alleged that the harassment caused her resignation and amounted to a “constructive” discharge.
Likewise, in Faragher v. City of Boca Raton, the plaintiff, employed as a lifeguard, alleged that her work environment was riddled with crude remarks and obscenities.9 One of the two supervisors reportedly once said to Faragher, “Date me or clean toilets for a year.” Another lifeguard had previously lodged similar complaints. The plaintiff ultimately resigned and brought suit.
The U.S. Supreme Court characterized both of these as “hostile environment” rather than “quid pro quo” cases, because the plaintiffs did not suffer any direct adverse job action. In its decisions, the court defined the scope of liability and affirmative defenses, holding that employers can be subject to vicarious liability when supervisors create actionable hostile work environments.
In other cases, the Supreme Court has ruled for the use of “the reasonable person in the plaintiff’s position” standard in judging the severity of sexual harassment. The court has also held that the genders of the harasser and the harassed employee are not material in determining whether sexual harassment has occurred.
A physician can be accused of harassing an employee, a nurse, an assistant, a fellow worker, a third party, or a patient. Focusing on misconduct within the doctor-patient relationship, the Federation of State Medical Boards adopted in May 2006 a policy entitled “Addressing Sexual Boundaries: Guidelines for State Medical Boards.”10
Although it did not use the term sexual harassment, the policy emphasized that physician sexual misconduct may include behavior that is verbal or physical, and may include expressions of thoughts and feelings or gestures that are sexual. It used the term “sexual impropriety” to denote behavior, gestures, or expressions that are seductive, sexually suggestive, disrespectful of patient privacy, or sexually demeaning to a patient. Together with “sexual violation,” a term the FSMB used when referring to physical sexual contact, they form the basis for disciplinary action by a state medical board.
Caveat: When performing a physical exam, physicians should always use good judgment and sensitivity, relying on the presence of a medical assistant to ensure patient comfort and to alleviate possible embarrassment or anxiety.
Under the federal EEOC rules, the employer rather than the harasser is the defendant. But there are other legal recourses, including tort and criminal actions, that directly target the harasser. Successful plaintiffs may be awarded lost wages, as well as damages for emotional distress, medical expenses, and punitive damages. They may also recover attorney fees.
In one case, a psychiatric nurse was awarded $1.2 million (later reduced to $850,000); in another, a nurse successfully sued a physician’s medical practice and received $150,000 in damages.4 And in an unusual case, a plaintiff was awarded only $1 in damages, but her counsel was paid $41,598 in fees.11 For the practicing doctor, medical board sanction, notoriety, and loss of professional standing and privileges constitute additional costs.
The medical profession is as susceptible as any other – perhaps more so – to allegations of sexual harassment. The magic words for actionable sexual harassment are severe, pervasive, and unwelcome. Although laws in the workplace generally do not prohibit simple teasing, offhand comments, or minor isolated incidents, the line separating these behaviors from bona fide sexual harassment is thin.
Erring on the side of strict and sober professional propriety seems prudent, given the current climate of zero tolerance.

References
1. MacCluskey v. University of Connecticut Health Center, United States Court of Appeals, Second Circuit, No. 17-0807-cv, Dec. 19, 2017.
2. Arch Intern Med. 1998 Feb 23;158(4):352-8.
3. N Engl J Med. 1993 Dec 23;329(26):1936-9.
4. J Nurs Care Qual. 2004 Jul-Sep;19(3):234-41.
5. Anania v. Daubenspeck Chiropractic, 718 N.E. 2d 480 (Ohio 1998).
6. Doe v. Borromeo, Nos. 305162, 305163 (Mich. Ct. App. Sept. 20, 2012).
7. Available at https://www.eeoc.gov/laws/types/sexual_harassment.cfm.
8. Burlington Industries, Inc. v. Ellerth, 524 US 742 (1998).
9. Faragher v. City of Boca Raton, 524 U.S. 775 (1998).
10. Federation of State Medical Boards, “Addressing Sexual Boundaries: Guidelines for State Medical Boards.”
11. J Healthc Risk Manag. 1999 Summer;19(3):14-25.
Question: A medical assistant alleged that Dr. Y sexually harassed her by sending anonymous gifts and messages such as, “you’re gorgeous,” and “I love your figure.” It was a repeat of Dr. Y’s previous behavior pattern directed at a different worker, who had lodged a complaint with the human resources department. The medical assistant now files a sexual harassment action under Title VII of the federal Civil Rights Act of 1964 against the health care institution, alleging a hostile work environment.
Which of the following is false?
A. Sexual harassment is a form of sexual misconduct regulated by state medical boards.
B. Mere words, without physical action, may suffice to be deemed sexual harassment.
C. A hostile environment arises when offensive conduct is so severe and pervasive as to amount to job discrimination.
D. Sexual harassment is a civil rights violation unique to the workplace.
E. Liability may attach to the supervisor, institution, or the harasser.
Answer: D. This hypothetical is modified from an actual Connecticut case that was recently decided in favor of the plaintiff.1 In that case, which involved a dentist, the federal Second Circuit unanimously rejected the University of Connecticut Health Center’s appeal against a jury’s verdict holding it responsible for its employee’s sexual harassment of a coworker, who was awarded $125,000. It ruled that the health center should have known of its employee’s harassing behavior.
Sexual harassment, a current hot topic, is pervasive, affecting a diversity of individuals in the fields of media, sports, politics, judiciary, education, entertainment, and others. The medical profession is no exception, and studies indicate that sexual harassment affects patients and physicians alike, occurring in hospitals, private offices, and academic centers.
In a large questionnaire study involving 4,501 female physicians, the authors found a prevalence rate of 47.7%. Harassment was more common while in medical school or during internship, residency, or fellowship than in practice.2 Patients may be the harassers. In 599 of the 1,064 licensed female family physicians in Ontario, more than 75% reported sexual harassment by patients at some time during their careers, either in their own offices by their own patients, or in settings such as emergency departments and clinics, where unknown patients presented an even higher risk.3
When physicians sexually harass fellow workers such as nurses, they distract their victims from providing attentive and competent care. In a review of the subject, researchers cited a study of 188 critical care nurses in hospitals, where nearly half (46%) reported experiencing sexual harassment that included “offensive sexual remarks, unwanted physical contact, unwanted nonverbal attention, requests for unwanted dates, sexual propositions, and physical assault.”4 To this list must now be added misconduct via the use of social media. In the study, physicians (82%), coworkers (20%), and immediate supervisors (7%) accounted for most of the incidents.
Neglecting to look seriously into complaints or to monitor and remedy the situation may create a hostile environment and trigger liability.
An example is the recent well-publicized case of Olympics team physician Dr. Larry Nassar, who was also a faculty member at Michigan State University. Olympic gold medalist McKayla Maroney named both the university and the U.S. Olympic Committee as codefendants in a lawsuit alleging that the institutions failed to properly investigate the team doctor’s criminal sexual conduct.
In Anania v. Daubenspeck Chiropractic, two employees of a chiropractor alleged that his patients sexually harassed them, but he did not remedy the situation.5 The trial court initially dismissed their lawsuit, holding that Ohio law did not recognize a cause of action for sexual harassment by a nonemployee patient, and that liability for sexual harassment can only exist in the context of respondeat superior (employer-employee) liability.
However, the court of appeals held that so long as the chiropractor knew or should have known of the harassment, and failed to take corrective action, he could be liable for allowing a hostile environment to exist.
Negligent supervision is another favorite plaintiff’s cause of action. In Doe v. Borromeo, a patient sought to hold the hospital liable for sexual assault by a physician during a medical exam.6 The lower state court had summarily dismissed the case, which was based on vicarious liability, but the state court of appeals reversed, finding the patient’s complaint against the hospital included a negligent supervision claim.
The appeals court reasoned that this was distinguishable from one based upon vicarious liability, so long as the supervising entity had a duty to protect the victim – and such a duty can only be established if the supervising entity knew or should have known of the existence of the harasser’s propensities, if any, to commit criminal and tortious acts.
Sexual harassment is a form of sex discrimination under Title VII of the Civil Rights Act of 1964, which is enforced by the Equal Employment Opportunity Commission. The commission’s website explains the law in clear and simple language:
“It is unlawful to harass a person (an applicant or employee) because of that person’s sex. Harassment can include ‘sexual harassment’ or unwelcome sexual advances, requests for sexual favors, and other verbal or physical harassment of a sexual nature. Harassment does not have to be of a sexual nature, however, and can include offensive remarks about a person’s sex.
“For example, it is illegal to harass a woman by making offensive comments about women in general. Both victim and the harasser can be either a woman or a man, and the victim and harasser can be the same sex.
“Although the law doesn’t prohibit simple teasing, offhand comments, or isolated incidents that are not very serious, harassment is illegal when it is so frequent or severe that it creates a hostile or offensive work environment or when it results in an adverse employment decision (such as the victim being fired or demoted). The harasser can be the victim’s supervisor, a supervisor in another area, a coworker, or someone who is not an employee of the employer, such as a client or customer.”7
For sexual harassment to occur, the aggrieved party must either show a “hostile environment” or “quid pro quo” situation.
In a hostile environment case, the harassment is serious and persistent, creating unacceptable and offensive work conditions. The plaintiff has to show that the employer knew or should have known of the situation but failed to remedy it.
The “quid pro quo” type of case requires a showing that a person in authority conditioned some aspect of the employee’s employment, such as promotion or retention, upon a sexual favor or relationship.
The U.S. Supreme Court has both clarified and muddied the law’s position on these two previously distinct types of sexual harassment.
In the landmark case of Burlington Industries v. Ellerth, the plaintiff, who was a salesperson, alleged that a supervisor made advances to her and threatened to deny her certain job benefits if she did not cooperate.8 The threats were never carried out, and she was in fact promoted; but her lawsuit alleged that the harassment caused her resignation and amounted to a “constructive” discharge.
Likewise, in Faragher v. City of Boca Raton, the plaintiff, employed as a lifeguard, alleged that her work environment was riddled with crude remarks and obscenities.9 One of the two supervisors reportedly once said to Faragher, “Date me or clean toilets for a year.” Another lifeguard had previously lodged similar complaints. The plaintiff ultimately resigned and brought suit.
The U.S. Supreme Court characterized both of these as “hostile environment” rather than “quid pro quo” cases, because the plaintiffs did not suffer any direct adverse job action. In its decisions, the court defined the scope of liability and affirmative defenses, holding that employers can be subject to vicarious liability when supervisors create actionable hostile work environments.
In other cases, the Supreme Court has ruled for the use of “the reasonable person in the plaintiff’s position” standard in judging the severity of sexual harassment. The court has also held that the genders of the harasser and the harassed employee are not material in determining whether sexual harassment has occurred.
A physician can be accused of harassing an employee, a nurse, an assistant, a fellow worker, a third party, or a patient. Focusing on misconduct within the doctor-patient relationship, the Federation of State Medical Boards adopted in May 2006 a policy entitled “Addressing Sexual Boundaries: Guidelines for State Medical Boards.”10
Although it did not use the term sexual harassment, the policy emphasized that physician sexual misconduct may include behavior that is verbal or physical, and may include expressions of thoughts and feelings or gestures that are sexual. It used the term “sexual impropriety” to denote behavior, gestures, or expressions that are seductive, sexually suggestive, disrespectful of patient privacy, or sexually demeaning to a patient. Together with “sexual violation,” a term the FSMB used when referring to physical sexual contact, they form the basis for disciplinary action by a state medical board.
Caveat: When performing a physical exam, physicians should always use good judgment and sensitivity, relying on the presence of a medical assistant to ensure patient comfort and to alleviate possible embarrassment or anxiety.
Under the federal EEOC rules, the employer rather than the harasser is the defendant. But there are other legal recourses, including tort and criminal actions, that directly target the harasser. Successful plaintiffs may be awarded lost wages, as well as damages for emotional distress, medical expenses, and punitive damages. They may also recover attorney fees.
In one case, a psychiatric nurse was awarded $1.2 million (later reduced to $850,000); in another, a nurse successfully sued a physician’s medical practice and received $150,000 in damages.4 And in an unusual case, a plaintiff was awarded only $1 in damages, but her counsel was paid $41,598 in fees.11 For the practicing doctor, medical board sanction, notoriety, and loss of professional standing and privileges constitute additional costs.
The medical profession is as susceptible as any other – perhaps more so – to allegations of sexual harassment. The magic words for actionable sexual harassment are severe, pervasive, and unwelcome. Although laws in the workplace generally do not prohibit simple teasing, offhand comments, or minor isolated incidents, the line separating these behaviors from bona fide sexual harassment is thin.
Erring on the side of strict and sober professional propriety seems prudent, given the current climate of zero tolerance.
References
1. MacCluskey v. University of Connecticut Health Center, United States Court of Appeals, Second Circuit, No. 17-0807-cv, Dec. 19, 2017.
2. Arch Intern Med. 1998 Feb 23;158(4):352-8.
3. N Engl J Med. 1993 Dec 23;329(26):1936-9.
4. J Nurs Care Qual. 2004 Jul-Sep;19(3):234-41.
5. Anania v. Daubenspeck Chiropractic, 718 N.E. 2d 480 (Ohio 1998).
6. Doe v. Borromeo, Nos. 305162, 305163 (Mich. Ct. App. Sept. 20, 2012).
7. Available at https://www.eeoc.gov/laws/types/sexual_harassment.cfm.
8. Burlington Industries, Inc. v. Ellerth, 524 US 742 (1998).
9. Faragher v. City of Boca Raton, 524 U.S. 775 (1998).
10. Federation of State Medical Boards, “Addressing Sexual Boundaries: Guidelines for State Medical Boards.”
11. J Healthc Risk Manag. 1999 Summer;19(3):14-25.
Question: A medical assistant alleged that Dr. Y sexually harassed her by sending anonymous gifts and messages such as, “you’re gorgeous,” and “I love your figure.” It was a repeat of Dr. Y’s previous behavior pattern directed at a different worker, who had lodged a complaint with the human resources department. The medical assistant now files a sexual harassment action under Title VII of the federal Civil Rights Act of 1964 against the health care institution, alleging a hostile work environment.
Which of the following is false?
A. Sexual harassment is a form of sexual misconduct regulated by state medical boards.
B. Mere words, without physical action, may suffice to be deemed sexual harassment.
C. A hostile environment arises when offensive conduct is so severe and pervasive as to amount to job discrimination.
D. Sexual harassment is a civil rights violation unique to the workplace.
E. Liability may attach to the supervisor, institution, or the harasser.
Answer: D. This hypothetical is modified from an actual Connecticut case that was recently decided in favor of the plaintiff.1 In that case, which involved a dentist, the federal Second Circuit unanimously rejected the University of Connecticut Health Center’s appeal against a jury’s verdict holding it responsible for its employee’s sexual harassment of a coworker, who was awarded $125,000. It ruled that the health center should have known of its employee’s harassing behavior.
Sexual harassment, a current hot topic, is pervasive, affecting a diversity of individuals in the fields of media, sports, politics, judiciary, education, entertainment, and others. The medical profession is no exception, and studies indicate that sexual harassment affects patients and physicians alike, occurring in hospitals, private offices, and academic centers.
In a large questionnaire study involving 4,501 female physicians, the authors found a prevalence rate of 47.7%. Harassment was more common while in medical school or during internship, residency, or fellowship than in practice.2 Patients may be the harassers. In 599 of the 1,064 licensed female family physicians in Ontario, more than 75% reported sexual harassment by patients at some time during their careers, either in their own offices by their own patients, or in settings such as emergency departments and clinics, where unknown patients presented an even higher risk.3
When physicians sexually harass fellow workers such as nurses, they distract their victims from providing attentive and competent care. In a review of the subject, researchers cited a study of 188 critical care nurses in hospitals, where nearly half (46%) reported experiencing sexual harassment that included “offensive sexual remarks, unwanted physical contact, unwanted nonverbal attention, requests for unwanted dates, sexual propositions, and physical assault.”4 To this list must now be added misconduct via the use of social media. In the study, physicians (82%), coworkers (20%), and immediate supervisors (7%) accounted for most of the incidents.
Neglecting to look seriously into complaints or to monitor and remedy the situation may create a hostile environment and trigger liability.
An example is the recent well-publicized case of Olympics team physician Dr. Larry Nassar, who was also a faculty member at Michigan State University. Olympic gold medalist McKayla Maroney named both the university and the U.S. Olympic Committee as codefendants in a lawsuit alleging that the institutions failed to properly investigate the team doctor’s criminal sexual conduct.
In Anania v. Daubenspeck Chiropractic, two employees of a chiropractor alleged that his patients sexually harassed them, but he did not remedy the situation.5 The trial court initially dismissed their lawsuit, holding that Ohio law did not recognize a cause of action for sexual harassment by a nonemployee patient, and that liability for sexual harassment can only exist in the context of respondeat superior (employer-employee) liability.
However, the court of appeals held that so long as the chiropractor knew or should have known of the harassment, and failed to take corrective action, he could be liable for allowing a hostile environment to exist.
Negligent supervision is another favorite plaintiff’s cause of action. In Doe v. Borromeo, a patient sought to hold the hospital liable for sexual assault by a physician during a medical exam.6 The lower state court had summarily dismissed the case, which was based on vicarious liability, but the state court of appeals reversed, finding the patient’s complaint against the hospital included a negligent supervision claim.
The appeals court reasoned that this was distinguishable from one based upon vicarious liability, so long as the supervising entity had a duty to protect the victim – and such a duty can only be established if the supervising entity knew or should have known of the existence of the harasser’s propensities, if any, to commit criminal and tortious acts.
Sexual harassment is a form of sex discrimination under Title VII of the Civil Rights Act of 1964, which is enforced by the Equal Employment Opportunity Commission. The commission’s website explains the law in clear and simple language:
“It is unlawful to harass a person (an applicant or employee) because of that person’s sex. Harassment can include ‘sexual harassment’ or unwelcome sexual advances, requests for sexual favors, and other verbal or physical harassment of a sexual nature. Harassment does not have to be of a sexual nature, however, and can include offensive remarks about a person’s sex.
“For example, it is illegal to harass a woman by making offensive comments about women in general. Both victim and the harasser can be either a woman or a man, and the victim and harasser can be the same sex.
“Although the law doesn’t prohibit simple teasing, offhand comments, or isolated incidents that are not very serious, harassment is illegal when it is so frequent or severe that it creates a hostile or offensive work environment or when it results in an adverse employment decision (such as the victim being fired or demoted). The harasser can be the victim’s supervisor, a supervisor in another area, a coworker, or someone who is not an employee of the employer, such as a client or customer.”7
For sexual harassment to occur, the aggrieved party must either show a “hostile environment” or “quid pro quo” situation.
In a hostile environment case, the harassment is serious and persistent, creating unacceptable and offensive work conditions. The plaintiff has to show that the employer knew or should have known of the situation but failed to remedy it.
The “quid pro quo” type of case requires a showing that a person in authority conditioned some aspect of the employee’s employment, such as promotion or retention, upon a sexual favor or relationship.
The U.S. Supreme Court has both clarified and muddied the law’s position on these two previously distinct types of sexual harassment.
In the landmark case of Burlington Industries v. Ellerth, the plaintiff, who was a salesperson, alleged that a supervisor made advances to her and threatened to deny her certain job benefits if she did not cooperate.8 The threats were never carried out, and she was in fact promoted; but her lawsuit alleged that the harassment caused her resignation and amounted to a “constructive” discharge.
Likewise, in Faragher v. City of Boca Raton, the plaintiff, employed as a lifeguard, alleged that her work environment was riddled with crude remarks and obscenities.9 One of the two supervisors reportedly once said to Faragher, “Date me or clean toilets for a year.” Another lifeguard had previously lodged similar complaints. The plaintiff ultimately resigned and brought suit.
The U.S. Supreme Court characterized both of these as “hostile environment” rather than “quid pro quo” cases, because the plaintiffs did not suffer any direct adverse job action. In its decisions, the court defined the scope of liability and affirmative defenses, holding that employers can be subject to vicarious liability when supervisors create actionable hostile work environments.
In other cases, the Supreme Court has ruled for the use of “the reasonable person in the plaintiff’s position” standard in judging the severity of sexual harassment. The court has also held that the genders of the harasser and the harassed employee are not material in determining whether sexual harassment has occurred.
A physician can be accused of harassing an employee, a nurse, an assistant, a fellow worker, a third party, or a patient. Focusing on misconduct within the doctor-patient relationship, the Federation of State Medical Boards adopted in May 2006 a policy entitled “Addressing Sexual Boundaries: Guidelines for State Medical Boards.”10
Although it did not use the term sexual harassment, the policy emphasized that physician sexual misconduct may include behavior that is verbal or physical, and may include expressions of thoughts and feelings or gestures that are sexual. It used the term “sexual impropriety” to denote behavior, gestures, or expressions that are seductive, sexually suggestive, disrespectful of patient privacy, or sexually demeaning to a patient. Together with “sexual violation,” a term the FSMB used when referring to physical sexual contact, they form the basis for disciplinary action by a state medical board.
Caveat: When performing a physical exam, physicians should always use good judgment and sensitivity, relying on the presence of a medical assistant to ensure patient comfort and to alleviate possible embarrassment or anxiety.
Under the federal EEOC rules, the employer rather than the harasser is the defendant. But there are other legal recourses, including tort and criminal actions, that directly target the harasser. Successful plaintiffs may be awarded lost wages, as well as damages for emotional distress, medical expenses, and punitive damages. They may also recover attorney fees.
In one case, a psychiatric nurse was awarded $1.2 million (later reduced to $850,000); in another, a nurse successfully sued a physician’s medical practice and received $150,000 in damages.4 And in an unusual case, a plaintiff was awarded only $1 in damages, but her counsel was paid $41,598 in fees.11 For the practicing doctor, medical board sanction, notoriety, and loss of professional standing and privileges constitute additional costs.
The medical profession is as susceptible as any other – perhaps more so – to allegations of sexual harassment. The magic words for actionable sexual harassment are severe, pervasive, and unwelcome. Although laws in the workplace generally do not prohibit simple teasing, offhand comments, or minor isolated incidents, the line separating these behaviors from bona fide sexual harassment is thin.
Erring on the side of strict and sober professional propriety seems prudent, given the current climate of zero tolerance.
References
1. MacCluskey v. University of Connecticut Health Center, United States Court of Appeals, Second Circuit, No. 17-0807-cv, Dec. 19, 2017.
2. Arch Intern Med. 1998 Feb 23;158(4):352-8.
3. N Engl J Med. 1993 Dec 23;329(26):1936-9.
4. J Nurs Care Qual. 2004 Jul-Sep;19(3):234-41.
5. Anania v. Daubenspeck Chiropractic, 718 N.E. 2d 480 (Ohio 1998).
6. Doe v. Borromeo, Nos. 305162, 305163 (Mich. Ct. App. Sept. 20, 2012).
7. Available at https://www.eeoc.gov/laws/types/sexual_harassment.cfm.
8. Burlington Industries, Inc. v. Ellerth, 524 US 742 (1998).
9. Faragher v. City of Boca Raton, 524 U.S. 775 (1998).
10. Federation of State Medical Boards, “Addressing Sexual Boundaries: Guidelines for State Medical Boards.”
11. J Healthc Risk Manag. 1999 Summer;19(3):14-25.