User login
Adverse event reporting in second-dose varicella vaccination shows no surprises
No new or unexpected adverse events were reported in association with second-dose varicella vaccination in children age 4-18 years during 2006-2014.
During the study period of 2006-2014, 14,641 reports regarding second-dose varicella vaccinations were made to the Vaccine Adverse Event Reporting System, according to John Su, MD, PhD, and his associates. Of these reports, only 3% were serious adverse events (AE), with the most common nonserious AE being injection-site reaction, which occurred in 48% of patients age 4-6 years reporting AEs, and in 38% of patients age 7-18 years reporting AEs.
Of the 494 serious AEs reported, pyrexia was the most common in children age 4-6 years, and headache and vomiting were the most common in children age 7-18 years. A total of seven deaths from various causes were reported, though no causal relation to vaccination was seen, and significant chronic medical problems were common in these children.
“The safety data on second-dose varicella vaccination are reassuring,” the investigators noted. “Reported AEs after second-dose varicella vaccination were mild, self limiting, and similar in reported frequency to AEs after first-dose vaccination, with no new or unexpected safety concerns.”
Find the full study in Pediatrics (2017 Feb 7. doi: 1542/peds.2016-2536).
No new or unexpected adverse events were reported in association with second-dose varicella vaccination in children age 4-18 years during 2006-2014.
During the study period of 2006-2014, 14,641 reports regarding second-dose varicella vaccinations were made to the Vaccine Adverse Event Reporting System, according to John Su, MD, PhD, and his associates. Of these reports, only 3% were serious adverse events (AE), with the most common nonserious AE being injection-site reaction, which occurred in 48% of patients age 4-6 years reporting AEs, and in 38% of patients age 7-18 years reporting AEs.
Of the 494 serious AEs reported, pyrexia was the most common in children age 4-6 years, and headache and vomiting were the most common in children age 7-18 years. A total of seven deaths from various causes were reported, though no causal relation to vaccination was seen, and significant chronic medical problems were common in these children.
“The safety data on second-dose varicella vaccination are reassuring,” the investigators noted. “Reported AEs after second-dose varicella vaccination were mild, self limiting, and similar in reported frequency to AEs after first-dose vaccination, with no new or unexpected safety concerns.”
Find the full study in Pediatrics (2017 Feb 7. doi: 1542/peds.2016-2536).
No new or unexpected adverse events were reported in association with second-dose varicella vaccination in children age 4-18 years during 2006-2014.
During the study period of 2006-2014, 14,641 reports regarding second-dose varicella vaccinations were made to the Vaccine Adverse Event Reporting System, according to John Su, MD, PhD, and his associates. Of these reports, only 3% were serious adverse events (AE), with the most common nonserious AE being injection-site reaction, which occurred in 48% of patients age 4-6 years reporting AEs, and in 38% of patients age 7-18 years reporting AEs.
Of the 494 serious AEs reported, pyrexia was the most common in children age 4-6 years, and headache and vomiting were the most common in children age 7-18 years. A total of seven deaths from various causes were reported, though no causal relation to vaccination was seen, and significant chronic medical problems were common in these children.
“The safety data on second-dose varicella vaccination are reassuring,” the investigators noted. “Reported AEs after second-dose varicella vaccination were mild, self limiting, and similar in reported frequency to AEs after first-dose vaccination, with no new or unexpected safety concerns.”
Find the full study in Pediatrics (2017 Feb 7. doi: 1542/peds.2016-2536).
Hypertriglyceridemia: Identifying Secondary Causes
Screening for cardiovascular (CV) risk often includes a routine serum fasting lipid profile. However, with the focus on LDL cholesterol, triglyceride measurement is frequently overlooked. Yet this element of the lipid profile is particularly important, given its strong association with not only atherosclerotic coronary heart disease but also pancreatitis.
Hypertriglyceridemia is defined as a serum triglyceride level that exceeds 150 mg/dL. In the US, an estimated 25% of patients have hypertriglyceridemia.1 Of these, 33.1% have “borderline high” triglyceride levels (150 to 199 mg/dL), 17.8% have “high” levels (200 to 499 mg/dL), and 1.7% have “very high” levels (> 500 mg/dL).1,2
Most of the time, hypertriglyceridemia is caused (or at least exacerbated) by underlying etiology. The best way to identify and manage these secondary causes is through a systematic approach.
CONSIDER THE EVIDENCE
For mild to moderately elevated (borderline high) triglyceride levels, our reflex reaction may be to recommend a triglyceride-lowering medication, such as fenofibrate. But this may not be the best answer. Although there is increasing evidence of an independent association between elevated triglyceride levels and CV risk, it remains unclear whether targeting them specifically can reduce that risk.3
In well-designed, peer-reviewed clinical trials, statins have been shown to reduce CV risk in patients with known cardiovascular disease (CVD) and those at high risk for CVD, as well as in primary prevention. However, these trials also suggest that significant residual CV risk remains after statin therapy.4
Several trials have attempted to prove residual risk reduction following combination therapy including statins—with inconclusive results:
ACCORD: Fenofibrate showed no overall macrovascular benefit when added to a statin in patients with type 2 diabetes and a triglyceride level < 204 mg/dL.3,5
AIM-HIGH: There was a 25% reduction in triglyceride levels when niacin was added to a regimen of a statin +/- ezetimibe, with an aggressive LDL treatment target (40 to 80 mg/dL). But the study was stopped early due to the lack of expected reduction in CVD events.4,6
JELIS: A reduction in major CV events was seen with 1,800 mg/d of eicosapentaenoic acid (EPA) supplementation plus a low-dose statin, compared to statin monotherapy. However, there was minimal change in triglyceride levels, leading the researchers to hypothesize that multiple mechanisms—such as decreasing oxidative stress, platelet aggregation, plaque formation and stabilization—contributed to the outcome.4,7
Informed by the JELIS results, the much-anticipated REDUCE-IT trial is currently in progress to address the lingering question of whether combination therapy can reduce residual CV risk. In this trial, EPA omega-3 fatty acid is being added to the regimen of statin-treated patients with persistently elevated triglycerides. Results are expected in 2017 to 2018.8
Remember that a triglyceride level of 150 mg/dL is a parameter—it does not represent a therapeutic target. There is insufficient evidence that treating to this level improves CV risk beyond LDL target recommendations.7
The National Lipid Association Expert Panel’s consensus view is that non-HDL is a better primary target than triglycerides alone or LDL. Using non-HDL as a target for intervention also simplifies the management of patients with high triglycerides (200 to 499 mg/dL). The non-HDL goal is considered to be 30 mg/dL greater than the LDL target. For patients with diabetes and those with CVD, the individualized non-HDL targets are 130 mg/dL and 100 mg/dL, respectively.9
REVIEW THE MEDICATION LIST
Several commonly used medications, including ß-blockers and thiazide diuretics, can increase triglyceride levels.10 Other medications with exacerbating effects on triglycerides include corticosteroids, retrovirals, immunosuppressants, retinoids, and some antipsychotics.10 Bile acid sequestrants (eg, colesevelam) should be avoided in patients with elevated triglycerides (> 200 mg/dL).7
In women, oral estrogen (ie, menopausal hormone replacement and oral birth control) can greatly exacerbate triglyceride levels, making transdermal delivery a better option. Tamoxifen, the hormonal medication used for breast cancer prophylaxis, can also increase triglyceride levels.11
LOOK FOR UNDERLYING CONDITIONS
Among those to consider: Hypothyroidism is common and easily ruled out by a simple blood test. Nephrotic syndrome should be ruled out, particularly in patients with concomitant renal dysfunction and peripheral edema, by checking a random urine protein-to-creatinine ratio or 24-hour urine for protein. Other factors that should be explored because of their potential effect on lipid metabolism include obesity and excessive intake of sugary beverages (ie, soda, fruit juice) and alcohol.11
High triglyceride levels occurring with low HDL are characteristic of insulin resistance and concerning for metabolic syndrome and/or polycystic ovarian syndrome.3,12 Often, patients will have underlying prediabetes (fasting glucose ≥ 100 mg/dL or random glucose ≥ 140 mg/dL with an A1C > 5.7%13) or covert type 2 diabetes. Another underdiagnosed but very common condition, obstructive sleep apnea, can greatly affect insulin sensitivity and has been associated with lipid abnormalities and metabolic syndrome.14
EXAMINE YOUR PATIENT
The physical exam is an essential component of assessment for patients with high triglycerides. As discussed, elevated triglycerides and low HDL are hallmarks for insulin resistance. As triglyceride levels are affected by obesity and body fat distribution, measuring BMI and assessing for visceral adiposity are an important part of the physical exam.4
The physical exam may also yield dermatologic clues, such as skin tags or acanthosis nigricans, a dark, velvety lesion usually found on the posterior and lateral neck creases, axillae, groin, and elbows.13 In rare cases—usually those with genetic involvement from a familial lipid metabolism disorder—patients may exhibit xanthomas. These cutaneous, lipid-rich lesions can appear as flat, yellowish plaques on various parts of the body, such as the eyelids (xanthelasma) or tendons of the hands, feet, and heels. Widespread, eruptive xanthomas, which manifest as pruritic pink papules with creamy centers, are associated with severe emergent triglyceride elevation and pancreatitis.10
CONSIDER NONPHARMACOLOGIC MANAGEMENT
In mild to moderate hypertriglyceridemia, intensive lifestyle changes are considered firstline therapy. Weight loss is recommended in obese patients; a 5% to 10% reduction in body weight can lower triglycerides by 20%.15
A quick 24-hour diet recall, including beverages, is helpful for identifying key issues. The goal should be to reduce carbohydrates—in particular, simple, high glycemic index, processed foods—as well as total and saturated fats. A substantial problem in our population is the consumption of high-fructose beverages and fruit juices. Referral to a dietitian can be very helpful, not only for initial meal planning but also for continuing counseling on successful long-term weight loss and maintenance.
Exercise is also very helpful for improving lipid parameters. A daily minimum of 30 to 60 minutes of intermittent aerobic exercise or mild resistance exercise has been shown to reduce triglyceride levels.10
PRESCRIBE APPROPRIATELY
The most important indication for treatment of hypertriglyceridemia is reduction of CVD risk. However, in patients with very high triglyceride levels (> 500 mg/dL), the goal is to decrease risk for life-threatening pancreatitis.15 Lipid-lowering medications and dietary restrictions should be promptly employed.
There are medications, as discussed earlier, that specifically lower triglycerides. Fibrates offer the most robust decrease, with a 20% to 50% reduction in triglyceride levels. Fenofibrate is considered a safer option when used in combination with a statin, due to the risk for significant muscle toxicity with gemfibrozil. There is some evidence that adding a fibrate may actually increase risk for pancreatitis; since this risk is otherwise low in patients with mild to moderate triglyceride elevation, the addition of a fibrate to their regimen should be avoided.3
Statins are the drug of choice when CV risk reduction is the goal (for patients with hypertriglyceridemia < 500 mg/dL). In addition to lowering LDL, statins can reduce triglycerides by 7% to 30%, depending on the dose.15
Other triglyceride-lowering medications include omega-3 fatty acids and niacin preparations. Prescription-strength omega-3 fatty acids have been found to lower serum triglyceride levels by 50% or more; the newest preparation, icosapent ethyl, demonstrated up to 45% reduction without significant effect on LDL levels.3 (Other preparations have been shown to substantially increase LDL in many cases.) Niacin (1,500 to 2,000 mg/d) can decrease triglycerides by 15% to 25%. However, it is no longer recommended for CV risk reduction; recent data indicate it may increase stroke risk when used in combination with statins.3,10 In April 2016, the FDA revoked its approval of the co-administration of niacin and fenofibrate with statin therapy, due to a lack of CV benefit.16
Other secondline options to consider for patients with insulin resistance or diabetes are metformin and pioglitazone. These medications have been shown to improve insulin sensitivity and decrease LDL and triglycerides in patients with prediabetes. Pioglitazone has proven beneficial in the treatment of steatohepatitis.17 Insulin is an excellent rapid triglyceride-lowering agent for patients with diabetes. It is important to reinforce that reduction of glucose is a key component in reduction of triglyceride levels.3
CONCLUSION
Hypertriglyceridemia is a complex condition that requires individualized and comprehensive management strategies. Clinicians must be able to identify and address secondary causes. Treatment options should be tailored to decrease CV and pancreatitis risk, and medication recommendations should be evidenced based and carefully selected to mitigate potential adverse effects. Patients should receive education and lifestyle management support to help motivate and equip them to employ strategies to improve their health.
1. CDC. Trends in elevated triglyceride in adults: United States, 2001-2012. www.cdc.gov/nchs/data/databriefs/db198.pdf. Accessed December 27, 2016.
2. Maki KC, Bays HE, Dicklin MR. Treatment options for the management of hypertriglyceridemia: strategies based on the best-available evidence. J Clin Lipidol. 2012;6(5):413-426.
3. Rosenson RS. Approach to the patient with hypertriglyceridemia. www.uptodate.com/contents/approach-to-the-patient-with-hypertriglyceridemia. Accessed December 28, 2016.
4. Talayero BG, Sacks FM. The role of triglycerides in atherosclerosis. Curr Cardiol Rep. 2011;13(6): 544-552.
5. Ginsberg HN, Elam MB, Lovato LC, et al; ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010; 362(17):1563-1574.
6. AIM-HIGH Investigators. The role of niacin in raising high-density lipoprotein cholesterol to reduce cardiovascular events in patients with atherosclerotic cardiovascular disease and optimally treated low-density lipoprotein cholesterol. Rationale and study design. The Atherothrombosis Intervention in Metabolic syndrome with low HDL/high triglycerides: impact on Global Health outcomes (AIM-HIGH). Am Heart J. 2011;161(3):471-477.
7. Miller M, Stone NJ, Ballantyne C, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292-2333.
8. Borow KM, Nelson JR, Mason RP. Biologic plausibility, cellular effects, and molecular mechanisms of eicosapentaenoic acid (EPA) in atherosclerosis. Atherosclerosis. 2015;242(1):357-366.
9. Jacobson TA, Ito MK, Maki KC, et al. National Lipid Association recommendations for patient-centered management of dyslipidemia. J Clin Lipidol. 2015;9(2):129-169.
10. Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012; 97(9):2969-2989.
11. Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2889-2934.
12. Amini L, Sadeghi MR, Oskuie F, Maleki H. Lipid profile in women with polycystic ovary syndrome. Crescent J Med Biol Sci. 2014;1(4):147-150.
13. Mantzoros C. Insulin resistance: definition and clinical spectrum. www.uptodate.com/contents/insulin-resistance-definition-and-clinical-spectrum. Accessed December 28, 2016.
14. Lin M, Lin H, Lee P, et al. Beneficial effect of continuous positive airway pressure on lipid profiles in obstructive sleep apnea: a meta-analysis. Sleep Breath. 2015;19(3):809-817.
15. Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract. 2014;2014:943162.
16. FDA. Withdrawal of approval of indications related to the coadministration with statins in applications for niacin extended-release tablets and fenofibric acid delayed-release capsules. https://s3.amazonaws.com/public-inspection.federalregister.gov/2016-08887.pdf. Accessed December 28, 2016.
17. Mazza A, Fruci B, Garinis GA, et al. The role of metformin in the management of NAFLD. Exp Diabetes Res. 2012;2012: 716404.
Clinician Reviews in partnership with

Carrie Bowling practices at the South Georgia Medical Center Diabetes Management Center and Valdosta Specialty Clinic, Endocrinology, in Valdosta, Georgia. Joyce Ross is President of theNational Lipid Association and Past President of the Preventive Cardiovascular Nurses Association.
Ms Bowling has no disclosures relevant to the content of this article. Ms Ross is on the Speakers' Bureau for Sanofi/Regeneron, AstraZeneca, Abbott/AbbVie, Amarin, and Amgen; she is also a consultant for Amarin.
Clinician Reviews in partnership with
Carrie Bowling practices at the South Georgia Medical Center Diabetes Management Center and Valdosta Specialty Clinic, Endocrinology, in Valdosta, Georgia. Joyce Ross is President of theNational Lipid Association and Past President of the Preventive Cardiovascular Nurses Association.
Ms Bowling has no disclosures relevant to the content of this article. Ms Ross is on the Speakers' Bureau for Sanofi/Regeneron, AstraZeneca, Abbott/AbbVie, Amarin, and Amgen; she is also a consultant for Amarin.
Clinician Reviews in partnership with
Carrie Bowling practices at the South Georgia Medical Center Diabetes Management Center and Valdosta Specialty Clinic, Endocrinology, in Valdosta, Georgia. Joyce Ross is President of theNational Lipid Association and Past President of the Preventive Cardiovascular Nurses Association.
Ms Bowling has no disclosures relevant to the content of this article. Ms Ross is on the Speakers' Bureau for Sanofi/Regeneron, AstraZeneca, Abbott/AbbVie, Amarin, and Amgen; she is also a consultant for Amarin.
Screening for cardiovascular (CV) risk often includes a routine serum fasting lipid profile. However, with the focus on LDL cholesterol, triglyceride measurement is frequently overlooked. Yet this element of the lipid profile is particularly important, given its strong association with not only atherosclerotic coronary heart disease but also pancreatitis.
Hypertriglyceridemia is defined as a serum triglyceride level that exceeds 150 mg/dL. In the US, an estimated 25% of patients have hypertriglyceridemia.1 Of these, 33.1% have “borderline high” triglyceride levels (150 to 199 mg/dL), 17.8% have “high” levels (200 to 499 mg/dL), and 1.7% have “very high” levels (> 500 mg/dL).1,2
Most of the time, hypertriglyceridemia is caused (or at least exacerbated) by underlying etiology. The best way to identify and manage these secondary causes is through a systematic approach.
CONSIDER THE EVIDENCE
For mild to moderately elevated (borderline high) triglyceride levels, our reflex reaction may be to recommend a triglyceride-lowering medication, such as fenofibrate. But this may not be the best answer. Although there is increasing evidence of an independent association between elevated triglyceride levels and CV risk, it remains unclear whether targeting them specifically can reduce that risk.3
In well-designed, peer-reviewed clinical trials, statins have been shown to reduce CV risk in patients with known cardiovascular disease (CVD) and those at high risk for CVD, as well as in primary prevention. However, these trials also suggest that significant residual CV risk remains after statin therapy.4
Several trials have attempted to prove residual risk reduction following combination therapy including statins—with inconclusive results:
ACCORD: Fenofibrate showed no overall macrovascular benefit when added to a statin in patients with type 2 diabetes and a triglyceride level < 204 mg/dL.3,5
AIM-HIGH: There was a 25% reduction in triglyceride levels when niacin was added to a regimen of a statin +/- ezetimibe, with an aggressive LDL treatment target (40 to 80 mg/dL). But the study was stopped early due to the lack of expected reduction in CVD events.4,6
JELIS: A reduction in major CV events was seen with 1,800 mg/d of eicosapentaenoic acid (EPA) supplementation plus a low-dose statin, compared to statin monotherapy. However, there was minimal change in triglyceride levels, leading the researchers to hypothesize that multiple mechanisms—such as decreasing oxidative stress, platelet aggregation, plaque formation and stabilization—contributed to the outcome.4,7
Informed by the JELIS results, the much-anticipated REDUCE-IT trial is currently in progress to address the lingering question of whether combination therapy can reduce residual CV risk. In this trial, EPA omega-3 fatty acid is being added to the regimen of statin-treated patients with persistently elevated triglycerides. Results are expected in 2017 to 2018.8
Remember that a triglyceride level of 150 mg/dL is a parameter—it does not represent a therapeutic target. There is insufficient evidence that treating to this level improves CV risk beyond LDL target recommendations.7
The National Lipid Association Expert Panel’s consensus view is that non-HDL is a better primary target than triglycerides alone or LDL. Using non-HDL as a target for intervention also simplifies the management of patients with high triglycerides (200 to 499 mg/dL). The non-HDL goal is considered to be 30 mg/dL greater than the LDL target. For patients with diabetes and those with CVD, the individualized non-HDL targets are 130 mg/dL and 100 mg/dL, respectively.9
REVIEW THE MEDICATION LIST
Several commonly used medications, including ß-blockers and thiazide diuretics, can increase triglyceride levels.10 Other medications with exacerbating effects on triglycerides include corticosteroids, retrovirals, immunosuppressants, retinoids, and some antipsychotics.10 Bile acid sequestrants (eg, colesevelam) should be avoided in patients with elevated triglycerides (> 200 mg/dL).7
In women, oral estrogen (ie, menopausal hormone replacement and oral birth control) can greatly exacerbate triglyceride levels, making transdermal delivery a better option. Tamoxifen, the hormonal medication used for breast cancer prophylaxis, can also increase triglyceride levels.11
LOOK FOR UNDERLYING CONDITIONS
Among those to consider: Hypothyroidism is common and easily ruled out by a simple blood test. Nephrotic syndrome should be ruled out, particularly in patients with concomitant renal dysfunction and peripheral edema, by checking a random urine protein-to-creatinine ratio or 24-hour urine for protein. Other factors that should be explored because of their potential effect on lipid metabolism include obesity and excessive intake of sugary beverages (ie, soda, fruit juice) and alcohol.11
High triglyceride levels occurring with low HDL are characteristic of insulin resistance and concerning for metabolic syndrome and/or polycystic ovarian syndrome.3,12 Often, patients will have underlying prediabetes (fasting glucose ≥ 100 mg/dL or random glucose ≥ 140 mg/dL with an A1C > 5.7%13) or covert type 2 diabetes. Another underdiagnosed but very common condition, obstructive sleep apnea, can greatly affect insulin sensitivity and has been associated with lipid abnormalities and metabolic syndrome.14
EXAMINE YOUR PATIENT
The physical exam is an essential component of assessment for patients with high triglycerides. As discussed, elevated triglycerides and low HDL are hallmarks for insulin resistance. As triglyceride levels are affected by obesity and body fat distribution, measuring BMI and assessing for visceral adiposity are an important part of the physical exam.4
The physical exam may also yield dermatologic clues, such as skin tags or acanthosis nigricans, a dark, velvety lesion usually found on the posterior and lateral neck creases, axillae, groin, and elbows.13 In rare cases—usually those with genetic involvement from a familial lipid metabolism disorder—patients may exhibit xanthomas. These cutaneous, lipid-rich lesions can appear as flat, yellowish plaques on various parts of the body, such as the eyelids (xanthelasma) or tendons of the hands, feet, and heels. Widespread, eruptive xanthomas, which manifest as pruritic pink papules with creamy centers, are associated with severe emergent triglyceride elevation and pancreatitis.10
CONSIDER NONPHARMACOLOGIC MANAGEMENT
In mild to moderate hypertriglyceridemia, intensive lifestyle changes are considered firstline therapy. Weight loss is recommended in obese patients; a 5% to 10% reduction in body weight can lower triglycerides by 20%.15
A quick 24-hour diet recall, including beverages, is helpful for identifying key issues. The goal should be to reduce carbohydrates—in particular, simple, high glycemic index, processed foods—as well as total and saturated fats. A substantial problem in our population is the consumption of high-fructose beverages and fruit juices. Referral to a dietitian can be very helpful, not only for initial meal planning but also for continuing counseling on successful long-term weight loss and maintenance.
Exercise is also very helpful for improving lipid parameters. A daily minimum of 30 to 60 minutes of intermittent aerobic exercise or mild resistance exercise has been shown to reduce triglyceride levels.10
PRESCRIBE APPROPRIATELY
The most important indication for treatment of hypertriglyceridemia is reduction of CVD risk. However, in patients with very high triglyceride levels (> 500 mg/dL), the goal is to decrease risk for life-threatening pancreatitis.15 Lipid-lowering medications and dietary restrictions should be promptly employed.
There are medications, as discussed earlier, that specifically lower triglycerides. Fibrates offer the most robust decrease, with a 20% to 50% reduction in triglyceride levels. Fenofibrate is considered a safer option when used in combination with a statin, due to the risk for significant muscle toxicity with gemfibrozil. There is some evidence that adding a fibrate may actually increase risk for pancreatitis; since this risk is otherwise low in patients with mild to moderate triglyceride elevation, the addition of a fibrate to their regimen should be avoided.3
Statins are the drug of choice when CV risk reduction is the goal (for patients with hypertriglyceridemia < 500 mg/dL). In addition to lowering LDL, statins can reduce triglycerides by 7% to 30%, depending on the dose.15
Other triglyceride-lowering medications include omega-3 fatty acids and niacin preparations. Prescription-strength omega-3 fatty acids have been found to lower serum triglyceride levels by 50% or more; the newest preparation, icosapent ethyl, demonstrated up to 45% reduction without significant effect on LDL levels.3 (Other preparations have been shown to substantially increase LDL in many cases.) Niacin (1,500 to 2,000 mg/d) can decrease triglycerides by 15% to 25%. However, it is no longer recommended for CV risk reduction; recent data indicate it may increase stroke risk when used in combination with statins.3,10 In April 2016, the FDA revoked its approval of the co-administration of niacin and fenofibrate with statin therapy, due to a lack of CV benefit.16
Other secondline options to consider for patients with insulin resistance or diabetes are metformin and pioglitazone. These medications have been shown to improve insulin sensitivity and decrease LDL and triglycerides in patients with prediabetes. Pioglitazone has proven beneficial in the treatment of steatohepatitis.17 Insulin is an excellent rapid triglyceride-lowering agent for patients with diabetes. It is important to reinforce that reduction of glucose is a key component in reduction of triglyceride levels.3
CONCLUSION
Hypertriglyceridemia is a complex condition that requires individualized and comprehensive management strategies. Clinicians must be able to identify and address secondary causes. Treatment options should be tailored to decrease CV and pancreatitis risk, and medication recommendations should be evidenced based and carefully selected to mitigate potential adverse effects. Patients should receive education and lifestyle management support to help motivate and equip them to employ strategies to improve their health.
Screening for cardiovascular (CV) risk often includes a routine serum fasting lipid profile. However, with the focus on LDL cholesterol, triglyceride measurement is frequently overlooked. Yet this element of the lipid profile is particularly important, given its strong association with not only atherosclerotic coronary heart disease but also pancreatitis.
Hypertriglyceridemia is defined as a serum triglyceride level that exceeds 150 mg/dL. In the US, an estimated 25% of patients have hypertriglyceridemia.1 Of these, 33.1% have “borderline high” triglyceride levels (150 to 199 mg/dL), 17.8% have “high” levels (200 to 499 mg/dL), and 1.7% have “very high” levels (> 500 mg/dL).1,2
Most of the time, hypertriglyceridemia is caused (or at least exacerbated) by underlying etiology. The best way to identify and manage these secondary causes is through a systematic approach.
CONSIDER THE EVIDENCE
For mild to moderately elevated (borderline high) triglyceride levels, our reflex reaction may be to recommend a triglyceride-lowering medication, such as fenofibrate. But this may not be the best answer. Although there is increasing evidence of an independent association between elevated triglyceride levels and CV risk, it remains unclear whether targeting them specifically can reduce that risk.3
In well-designed, peer-reviewed clinical trials, statins have been shown to reduce CV risk in patients with known cardiovascular disease (CVD) and those at high risk for CVD, as well as in primary prevention. However, these trials also suggest that significant residual CV risk remains after statin therapy.4
Several trials have attempted to prove residual risk reduction following combination therapy including statins—with inconclusive results:
ACCORD: Fenofibrate showed no overall macrovascular benefit when added to a statin in patients with type 2 diabetes and a triglyceride level < 204 mg/dL.3,5
AIM-HIGH: There was a 25% reduction in triglyceride levels when niacin was added to a regimen of a statin +/- ezetimibe, with an aggressive LDL treatment target (40 to 80 mg/dL). But the study was stopped early due to the lack of expected reduction in CVD events.4,6
JELIS: A reduction in major CV events was seen with 1,800 mg/d of eicosapentaenoic acid (EPA) supplementation plus a low-dose statin, compared to statin monotherapy. However, there was minimal change in triglyceride levels, leading the researchers to hypothesize that multiple mechanisms—such as decreasing oxidative stress, platelet aggregation, plaque formation and stabilization—contributed to the outcome.4,7
Informed by the JELIS results, the much-anticipated REDUCE-IT trial is currently in progress to address the lingering question of whether combination therapy can reduce residual CV risk. In this trial, EPA omega-3 fatty acid is being added to the regimen of statin-treated patients with persistently elevated triglycerides. Results are expected in 2017 to 2018.8
Remember that a triglyceride level of 150 mg/dL is a parameter—it does not represent a therapeutic target. There is insufficient evidence that treating to this level improves CV risk beyond LDL target recommendations.7
The National Lipid Association Expert Panel’s consensus view is that non-HDL is a better primary target than triglycerides alone or LDL. Using non-HDL as a target for intervention also simplifies the management of patients with high triglycerides (200 to 499 mg/dL). The non-HDL goal is considered to be 30 mg/dL greater than the LDL target. For patients with diabetes and those with CVD, the individualized non-HDL targets are 130 mg/dL and 100 mg/dL, respectively.9
REVIEW THE MEDICATION LIST
Several commonly used medications, including ß-blockers and thiazide diuretics, can increase triglyceride levels.10 Other medications with exacerbating effects on triglycerides include corticosteroids, retrovirals, immunosuppressants, retinoids, and some antipsychotics.10 Bile acid sequestrants (eg, colesevelam) should be avoided in patients with elevated triglycerides (> 200 mg/dL).7
In women, oral estrogen (ie, menopausal hormone replacement and oral birth control) can greatly exacerbate triglyceride levels, making transdermal delivery a better option. Tamoxifen, the hormonal medication used for breast cancer prophylaxis, can also increase triglyceride levels.11
LOOK FOR UNDERLYING CONDITIONS
Among those to consider: Hypothyroidism is common and easily ruled out by a simple blood test. Nephrotic syndrome should be ruled out, particularly in patients with concomitant renal dysfunction and peripheral edema, by checking a random urine protein-to-creatinine ratio or 24-hour urine for protein. Other factors that should be explored because of their potential effect on lipid metabolism include obesity and excessive intake of sugary beverages (ie, soda, fruit juice) and alcohol.11
High triglyceride levels occurring with low HDL are characteristic of insulin resistance and concerning for metabolic syndrome and/or polycystic ovarian syndrome.3,12 Often, patients will have underlying prediabetes (fasting glucose ≥ 100 mg/dL or random glucose ≥ 140 mg/dL with an A1C > 5.7%13) or covert type 2 diabetes. Another underdiagnosed but very common condition, obstructive sleep apnea, can greatly affect insulin sensitivity and has been associated with lipid abnormalities and metabolic syndrome.14
EXAMINE YOUR PATIENT
The physical exam is an essential component of assessment for patients with high triglycerides. As discussed, elevated triglycerides and low HDL are hallmarks for insulin resistance. As triglyceride levels are affected by obesity and body fat distribution, measuring BMI and assessing for visceral adiposity are an important part of the physical exam.4
The physical exam may also yield dermatologic clues, such as skin tags or acanthosis nigricans, a dark, velvety lesion usually found on the posterior and lateral neck creases, axillae, groin, and elbows.13 In rare cases—usually those with genetic involvement from a familial lipid metabolism disorder—patients may exhibit xanthomas. These cutaneous, lipid-rich lesions can appear as flat, yellowish plaques on various parts of the body, such as the eyelids (xanthelasma) or tendons of the hands, feet, and heels. Widespread, eruptive xanthomas, which manifest as pruritic pink papules with creamy centers, are associated with severe emergent triglyceride elevation and pancreatitis.10
CONSIDER NONPHARMACOLOGIC MANAGEMENT
In mild to moderate hypertriglyceridemia, intensive lifestyle changes are considered firstline therapy. Weight loss is recommended in obese patients; a 5% to 10% reduction in body weight can lower triglycerides by 20%.15
A quick 24-hour diet recall, including beverages, is helpful for identifying key issues. The goal should be to reduce carbohydrates—in particular, simple, high glycemic index, processed foods—as well as total and saturated fats. A substantial problem in our population is the consumption of high-fructose beverages and fruit juices. Referral to a dietitian can be very helpful, not only for initial meal planning but also for continuing counseling on successful long-term weight loss and maintenance.
Exercise is also very helpful for improving lipid parameters. A daily minimum of 30 to 60 minutes of intermittent aerobic exercise or mild resistance exercise has been shown to reduce triglyceride levels.10
PRESCRIBE APPROPRIATELY
The most important indication for treatment of hypertriglyceridemia is reduction of CVD risk. However, in patients with very high triglyceride levels (> 500 mg/dL), the goal is to decrease risk for life-threatening pancreatitis.15 Lipid-lowering medications and dietary restrictions should be promptly employed.
There are medications, as discussed earlier, that specifically lower triglycerides. Fibrates offer the most robust decrease, with a 20% to 50% reduction in triglyceride levels. Fenofibrate is considered a safer option when used in combination with a statin, due to the risk for significant muscle toxicity with gemfibrozil. There is some evidence that adding a fibrate may actually increase risk for pancreatitis; since this risk is otherwise low in patients with mild to moderate triglyceride elevation, the addition of a fibrate to their regimen should be avoided.3
Statins are the drug of choice when CV risk reduction is the goal (for patients with hypertriglyceridemia < 500 mg/dL). In addition to lowering LDL, statins can reduce triglycerides by 7% to 30%, depending on the dose.15
Other triglyceride-lowering medications include omega-3 fatty acids and niacin preparations. Prescription-strength omega-3 fatty acids have been found to lower serum triglyceride levels by 50% or more; the newest preparation, icosapent ethyl, demonstrated up to 45% reduction without significant effect on LDL levels.3 (Other preparations have been shown to substantially increase LDL in many cases.) Niacin (1,500 to 2,000 mg/d) can decrease triglycerides by 15% to 25%. However, it is no longer recommended for CV risk reduction; recent data indicate it may increase stroke risk when used in combination with statins.3,10 In April 2016, the FDA revoked its approval of the co-administration of niacin and fenofibrate with statin therapy, due to a lack of CV benefit.16
Other secondline options to consider for patients with insulin resistance or diabetes are metformin and pioglitazone. These medications have been shown to improve insulin sensitivity and decrease LDL and triglycerides in patients with prediabetes. Pioglitazone has proven beneficial in the treatment of steatohepatitis.17 Insulin is an excellent rapid triglyceride-lowering agent for patients with diabetes. It is important to reinforce that reduction of glucose is a key component in reduction of triglyceride levels.3
CONCLUSION
Hypertriglyceridemia is a complex condition that requires individualized and comprehensive management strategies. Clinicians must be able to identify and address secondary causes. Treatment options should be tailored to decrease CV and pancreatitis risk, and medication recommendations should be evidenced based and carefully selected to mitigate potential adverse effects. Patients should receive education and lifestyle management support to help motivate and equip them to employ strategies to improve their health.
1. CDC. Trends in elevated triglyceride in adults: United States, 2001-2012. www.cdc.gov/nchs/data/databriefs/db198.pdf. Accessed December 27, 2016.
2. Maki KC, Bays HE, Dicklin MR. Treatment options for the management of hypertriglyceridemia: strategies based on the best-available evidence. J Clin Lipidol. 2012;6(5):413-426.
3. Rosenson RS. Approach to the patient with hypertriglyceridemia. www.uptodate.com/contents/approach-to-the-patient-with-hypertriglyceridemia. Accessed December 28, 2016.
4. Talayero BG, Sacks FM. The role of triglycerides in atherosclerosis. Curr Cardiol Rep. 2011;13(6): 544-552.
5. Ginsberg HN, Elam MB, Lovato LC, et al; ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010; 362(17):1563-1574.
6. AIM-HIGH Investigators. The role of niacin in raising high-density lipoprotein cholesterol to reduce cardiovascular events in patients with atherosclerotic cardiovascular disease and optimally treated low-density lipoprotein cholesterol. Rationale and study design. The Atherothrombosis Intervention in Metabolic syndrome with low HDL/high triglycerides: impact on Global Health outcomes (AIM-HIGH). Am Heart J. 2011;161(3):471-477.
7. Miller M, Stone NJ, Ballantyne C, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292-2333.
8. Borow KM, Nelson JR, Mason RP. Biologic plausibility, cellular effects, and molecular mechanisms of eicosapentaenoic acid (EPA) in atherosclerosis. Atherosclerosis. 2015;242(1):357-366.
9. Jacobson TA, Ito MK, Maki KC, et al. National Lipid Association recommendations for patient-centered management of dyslipidemia. J Clin Lipidol. 2015;9(2):129-169.
10. Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012; 97(9):2969-2989.
11. Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2889-2934.
12. Amini L, Sadeghi MR, Oskuie F, Maleki H. Lipid profile in women with polycystic ovary syndrome. Crescent J Med Biol Sci. 2014;1(4):147-150.
13. Mantzoros C. Insulin resistance: definition and clinical spectrum. www.uptodate.com/contents/insulin-resistance-definition-and-clinical-spectrum. Accessed December 28, 2016.
14. Lin M, Lin H, Lee P, et al. Beneficial effect of continuous positive airway pressure on lipid profiles in obstructive sleep apnea: a meta-analysis. Sleep Breath. 2015;19(3):809-817.
15. Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract. 2014;2014:943162.
16. FDA. Withdrawal of approval of indications related to the coadministration with statins in applications for niacin extended-release tablets and fenofibric acid delayed-release capsules. https://s3.amazonaws.com/public-inspection.federalregister.gov/2016-08887.pdf. Accessed December 28, 2016.
17. Mazza A, Fruci B, Garinis GA, et al. The role of metformin in the management of NAFLD. Exp Diabetes Res. 2012;2012: 716404.
1. CDC. Trends in elevated triglyceride in adults: United States, 2001-2012. www.cdc.gov/nchs/data/databriefs/db198.pdf. Accessed December 27, 2016.
2. Maki KC, Bays HE, Dicklin MR. Treatment options for the management of hypertriglyceridemia: strategies based on the best-available evidence. J Clin Lipidol. 2012;6(5):413-426.
3. Rosenson RS. Approach to the patient with hypertriglyceridemia. www.uptodate.com/contents/approach-to-the-patient-with-hypertriglyceridemia. Accessed December 28, 2016.
4. Talayero BG, Sacks FM. The role of triglycerides in atherosclerosis. Curr Cardiol Rep. 2011;13(6): 544-552.
5. Ginsberg HN, Elam MB, Lovato LC, et al; ACCORD Study Group. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010; 362(17):1563-1574.
6. AIM-HIGH Investigators. The role of niacin in raising high-density lipoprotein cholesterol to reduce cardiovascular events in patients with atherosclerotic cardiovascular disease and optimally treated low-density lipoprotein cholesterol. Rationale and study design. The Atherothrombosis Intervention in Metabolic syndrome with low HDL/high triglycerides: impact on Global Health outcomes (AIM-HIGH). Am Heart J. 2011;161(3):471-477.
7. Miller M, Stone NJ, Ballantyne C, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292-2333.
8. Borow KM, Nelson JR, Mason RP. Biologic plausibility, cellular effects, and molecular mechanisms of eicosapentaenoic acid (EPA) in atherosclerosis. Atherosclerosis. 2015;242(1):357-366.
9. Jacobson TA, Ito MK, Maki KC, et al. National Lipid Association recommendations for patient-centered management of dyslipidemia. J Clin Lipidol. 2015;9(2):129-169.
10. Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012; 97(9):2969-2989.
11. Stone NJ, Robinson JG, Lichtenstein AH, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 pt B):2889-2934.
12. Amini L, Sadeghi MR, Oskuie F, Maleki H. Lipid profile in women with polycystic ovary syndrome. Crescent J Med Biol Sci. 2014;1(4):147-150.
13. Mantzoros C. Insulin resistance: definition and clinical spectrum. www.uptodate.com/contents/insulin-resistance-definition-and-clinical-spectrum. Accessed December 28, 2016.
14. Lin M, Lin H, Lee P, et al. Beneficial effect of continuous positive airway pressure on lipid profiles in obstructive sleep apnea: a meta-analysis. Sleep Breath. 2015;19(3):809-817.
15. Kaur J. A comprehensive review on metabolic syndrome. Cardiol Res Pract. 2014;2014:943162.
16. FDA. Withdrawal of approval of indications related to the coadministration with statins in applications for niacin extended-release tablets and fenofibric acid delayed-release capsules. https://s3.amazonaws.com/public-inspection.federalregister.gov/2016-08887.pdf. Accessed December 28, 2016.
17. Mazza A, Fruci B, Garinis GA, et al. The role of metformin in the management of NAFLD. Exp Diabetes Res. 2012;2012: 716404.
Scholar grants help future hospitalists explore career pathways
Editor’s note: Each month, SHM puts the spotlight on some of our most active members who are making substantial contributions to hospital medicine. Log on to www.hospitalmedicine.org/getinvolved for more information on how you can lend your expertise to help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Ernie L. Esquivel, MD, FACP, FHM, the clerkship director, medicine, and assistant professor of clinical medicine in the Division of General Internal Medicine at the Weill Cornell Medical College in New York City. Dr. Esquivel is involved with SHM’s Physicians in Training Committee, and has spearheaded the creation of the Student Hospitalist Scholar Grant program.
What inspired you to become a hospitalist?
I became a hospitalist serendipitously. At a critical juncture in my life about 8 years ago (when a change in career direction became necessary), I chanced upon a locum tenens position in a small community hospital in Lansdale, Pa., as a hospitalist. Having been a primary care track resident who subsequently chose to specialize in nephrology, I rediscovered my generalist inclinations during this job. I fell in love with the fast pace of the hospitalist’s work, the complexity of delivering care and the diversity of diseases, and of personal life stories on the general medicine wards and the ICU.

Subsequently, I worked as an intensivist in Philadelphia for a year before joining the Academic Hospital Medicine Division at Weill Cornell in New York City. At Cornell, I have managed to cultivate my passion for medical education, especially for working with and mentoring students and residents, while continuing to care for patients on the general medicine wards. As the medicine clerkship director, I have had the privilege of creating an innovative curriculum that I hope prepares medical students for the challenges in, and the richness of, encounters in the practice of inpatient medicine.
How and why did you become a member of SHM and the Physicians in Training Committee (PIT)?
I joined SHM 6 years ago as I started to explore my career options more deeply. In 2010, I attended the Academic Hospitalist Academy, and that really offered me a closer look at the different ways in which SHM could help me advance. I went to my first SHM annual meeting 5 years ago; it motivated me to become involved in committee work. Because of my interest in medical education, I volunteered for the PIT Committee and it has given me the opportunity to work closely with other hospitalists around the country, and develop programming specifically targeted toward future hospitalists.
What is the PIT Committee working on?
The committee has continued to find ways for increased engagement of residents and students in SHM. Dr. Brian Kwan, an academic hospitalist at UC San Diego, and I have been developing a travel grant program for resident trainees and hospital medicine fellows to attend the annual meeting. By offering them a stipend to defray the costs of travel if their quality improvement innovation or research project is accepted, we hope that the annual meeting can become a venue for them to highlight their work, while becoming exposed to the many activities and opportunities offered by our society. In addition, it could be a way for them to network with other future hospitalists and established future mentors.
What prompted you to lead the creation of the Student Hospitalist Scholar Grant summer program?
Before I became a hospitalist, I spent about 7 years in research, studying renal genetics. I have always been fascinated by science and asked how I can help to advance our knowledge. As a hospitalist, it became clear to me early on that there are many questions that one can pose about the clinical work we do, the way we practice medicine, or ways to innovate education, and that there are many academic hospitalists who engage in advancing the field. I spearheaded this program because I would like students to see the field of hospital medicine as one in which they can develop a future career in academic medicine, not only by caring for patients, but also by involving themselves in research questions or QI projects.
Do you have any specific advice for students and residents interested in hospital medicine? In what ways can early-career hospitalists utilize SHM resources to leverage their careers?
The decision to pursue a career as a hospitalist will open up many more questions in the future, because there are so many opportunities available. I would suggest that trainees ask in which ways they see themselves growing in the future – clinical research, medical education, QI/patient safety, operations, and hospital leadership are the main avenues. When I interview future faculty, I always pose the same question to each of them: “Every year you are allocated X amount of money that you can use for CME, etc. How are you going to use this money to improve your skills in any particular area?” The ability of candidates to answer this question reflects for me their preparedness to develop themselves as career hospitalists and their willingness to contribute to their group or division in an innovative manner.
The reality is that as one gets older, most will find it difficult to sustain a 26-week/year schedule. So find ways for your energies, in whichever area, to be noticed and developed toward a position of leadership in the hospital or medical school.
As you take care of patients in the hospital or consider your education and training, identify ways in which things can be done better. Invariably, someone in the Society of Hospital Medicine is interested in the same issue(s). Explore your ideas, share them at the meeting, talk to people, go to the SHM website and identify what resources are already available.
If SHM will be your future academic home, volunteer to engage in activities at the chapter or national levels. Our society is really dedicated to identifying ways to welcome you into our exciting and continually evolving field.
Felicia Steele is SHM’s communications coordinator.
Editor’s note: Each month, SHM puts the spotlight on some of our most active members who are making substantial contributions to hospital medicine. Log on to www.hospitalmedicine.org/getinvolved for more information on how you can lend your expertise to help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Ernie L. Esquivel, MD, FACP, FHM, the clerkship director, medicine, and assistant professor of clinical medicine in the Division of General Internal Medicine at the Weill Cornell Medical College in New York City. Dr. Esquivel is involved with SHM’s Physicians in Training Committee, and has spearheaded the creation of the Student Hospitalist Scholar Grant program.
What inspired you to become a hospitalist?
I became a hospitalist serendipitously. At a critical juncture in my life about 8 years ago (when a change in career direction became necessary), I chanced upon a locum tenens position in a small community hospital in Lansdale, Pa., as a hospitalist. Having been a primary care track resident who subsequently chose to specialize in nephrology, I rediscovered my generalist inclinations during this job. I fell in love with the fast pace of the hospitalist’s work, the complexity of delivering care and the diversity of diseases, and of personal life stories on the general medicine wards and the ICU.
Subsequently, I worked as an intensivist in Philadelphia for a year before joining the Academic Hospital Medicine Division at Weill Cornell in New York City. At Cornell, I have managed to cultivate my passion for medical education, especially for working with and mentoring students and residents, while continuing to care for patients on the general medicine wards. As the medicine clerkship director, I have had the privilege of creating an innovative curriculum that I hope prepares medical students for the challenges in, and the richness of, encounters in the practice of inpatient medicine.
How and why did you become a member of SHM and the Physicians in Training Committee (PIT)?
I joined SHM 6 years ago as I started to explore my career options more deeply. In 2010, I attended the Academic Hospitalist Academy, and that really offered me a closer look at the different ways in which SHM could help me advance. I went to my first SHM annual meeting 5 years ago; it motivated me to become involved in committee work. Because of my interest in medical education, I volunteered for the PIT Committee and it has given me the opportunity to work closely with other hospitalists around the country, and develop programming specifically targeted toward future hospitalists.
What is the PIT Committee working on?
The committee has continued to find ways for increased engagement of residents and students in SHM. Dr. Brian Kwan, an academic hospitalist at UC San Diego, and I have been developing a travel grant program for resident trainees and hospital medicine fellows to attend the annual meeting. By offering them a stipend to defray the costs of travel if their quality improvement innovation or research project is accepted, we hope that the annual meeting can become a venue for them to highlight their work, while becoming exposed to the many activities and opportunities offered by our society. In addition, it could be a way for them to network with other future hospitalists and established future mentors.
What prompted you to lead the creation of the Student Hospitalist Scholar Grant summer program?
Before I became a hospitalist, I spent about 7 years in research, studying renal genetics. I have always been fascinated by science and asked how I can help to advance our knowledge. As a hospitalist, it became clear to me early on that there are many questions that one can pose about the clinical work we do, the way we practice medicine, or ways to innovate education, and that there are many academic hospitalists who engage in advancing the field. I spearheaded this program because I would like students to see the field of hospital medicine as one in which they can develop a future career in academic medicine, not only by caring for patients, but also by involving themselves in research questions or QI projects.
Do you have any specific advice for students and residents interested in hospital medicine? In what ways can early-career hospitalists utilize SHM resources to leverage their careers?
The decision to pursue a career as a hospitalist will open up many more questions in the future, because there are so many opportunities available. I would suggest that trainees ask in which ways they see themselves growing in the future – clinical research, medical education, QI/patient safety, operations, and hospital leadership are the main avenues. When I interview future faculty, I always pose the same question to each of them: “Every year you are allocated X amount of money that you can use for CME, etc. How are you going to use this money to improve your skills in any particular area?” The ability of candidates to answer this question reflects for me their preparedness to develop themselves as career hospitalists and their willingness to contribute to their group or division in an innovative manner.
The reality is that as one gets older, most will find it difficult to sustain a 26-week/year schedule. So find ways for your energies, in whichever area, to be noticed and developed toward a position of leadership in the hospital or medical school.
As you take care of patients in the hospital or consider your education and training, identify ways in which things can be done better. Invariably, someone in the Society of Hospital Medicine is interested in the same issue(s). Explore your ideas, share them at the meeting, talk to people, go to the SHM website and identify what resources are already available.
If SHM will be your future academic home, volunteer to engage in activities at the chapter or national levels. Our society is really dedicated to identifying ways to welcome you into our exciting and continually evolving field.
Felicia Steele is SHM’s communications coordinator.
Editor’s note: Each month, SHM puts the spotlight on some of our most active members who are making substantial contributions to hospital medicine. Log on to www.hospitalmedicine.org/getinvolved for more information on how you can lend your expertise to help SHM improve the care of hospitalized patients.
This month, The Hospitalist spotlights Ernie L. Esquivel, MD, FACP, FHM, the clerkship director, medicine, and assistant professor of clinical medicine in the Division of General Internal Medicine at the Weill Cornell Medical College in New York City. Dr. Esquivel is involved with SHM’s Physicians in Training Committee, and has spearheaded the creation of the Student Hospitalist Scholar Grant program.
What inspired you to become a hospitalist?
I became a hospitalist serendipitously. At a critical juncture in my life about 8 years ago (when a change in career direction became necessary), I chanced upon a locum tenens position in a small community hospital in Lansdale, Pa., as a hospitalist. Having been a primary care track resident who subsequently chose to specialize in nephrology, I rediscovered my generalist inclinations during this job. I fell in love with the fast pace of the hospitalist’s work, the complexity of delivering care and the diversity of diseases, and of personal life stories on the general medicine wards and the ICU.
Subsequently, I worked as an intensivist in Philadelphia for a year before joining the Academic Hospital Medicine Division at Weill Cornell in New York City. At Cornell, I have managed to cultivate my passion for medical education, especially for working with and mentoring students and residents, while continuing to care for patients on the general medicine wards. As the medicine clerkship director, I have had the privilege of creating an innovative curriculum that I hope prepares medical students for the challenges in, and the richness of, encounters in the practice of inpatient medicine.
How and why did you become a member of SHM and the Physicians in Training Committee (PIT)?
I joined SHM 6 years ago as I started to explore my career options more deeply. In 2010, I attended the Academic Hospitalist Academy, and that really offered me a closer look at the different ways in which SHM could help me advance. I went to my first SHM annual meeting 5 years ago; it motivated me to become involved in committee work. Because of my interest in medical education, I volunteered for the PIT Committee and it has given me the opportunity to work closely with other hospitalists around the country, and develop programming specifically targeted toward future hospitalists.
What is the PIT Committee working on?
The committee has continued to find ways for increased engagement of residents and students in SHM. Dr. Brian Kwan, an academic hospitalist at UC San Diego, and I have been developing a travel grant program for resident trainees and hospital medicine fellows to attend the annual meeting. By offering them a stipend to defray the costs of travel if their quality improvement innovation or research project is accepted, we hope that the annual meeting can become a venue for them to highlight their work, while becoming exposed to the many activities and opportunities offered by our society. In addition, it could be a way for them to network with other future hospitalists and established future mentors.
What prompted you to lead the creation of the Student Hospitalist Scholar Grant summer program?
Before I became a hospitalist, I spent about 7 years in research, studying renal genetics. I have always been fascinated by science and asked how I can help to advance our knowledge. As a hospitalist, it became clear to me early on that there are many questions that one can pose about the clinical work we do, the way we practice medicine, or ways to innovate education, and that there are many academic hospitalists who engage in advancing the field. I spearheaded this program because I would like students to see the field of hospital medicine as one in which they can develop a future career in academic medicine, not only by caring for patients, but also by involving themselves in research questions or QI projects.
Do you have any specific advice for students and residents interested in hospital medicine? In what ways can early-career hospitalists utilize SHM resources to leverage their careers?
The decision to pursue a career as a hospitalist will open up many more questions in the future, because there are so many opportunities available. I would suggest that trainees ask in which ways they see themselves growing in the future – clinical research, medical education, QI/patient safety, operations, and hospital leadership are the main avenues. When I interview future faculty, I always pose the same question to each of them: “Every year you are allocated X amount of money that you can use for CME, etc. How are you going to use this money to improve your skills in any particular area?” The ability of candidates to answer this question reflects for me their preparedness to develop themselves as career hospitalists and their willingness to contribute to their group or division in an innovative manner.
The reality is that as one gets older, most will find it difficult to sustain a 26-week/year schedule. So find ways for your energies, in whichever area, to be noticed and developed toward a position of leadership in the hospital or medical school.
As you take care of patients in the hospital or consider your education and training, identify ways in which things can be done better. Invariably, someone in the Society of Hospital Medicine is interested in the same issue(s). Explore your ideas, share them at the meeting, talk to people, go to the SHM website and identify what resources are already available.
If SHM will be your future academic home, volunteer to engage in activities at the chapter or national levels. Our society is really dedicated to identifying ways to welcome you into our exciting and continually evolving field.
Felicia Steele is SHM’s communications coordinator.
Postdischarge antibiotics for complicated pneumonia
Clinical question: Are oral antibiotics as effective as intravenous (IV) antibiotics in the treatment of complicated pneumonia after discharge to home?
Background: Pneumonia is the most common illness among hospitalized children and adolescents (excluding neonates). Among children admitted with community acquired pneumonia, 15% may develop a complicated pneumonia (one with a pleural effusion or empyema). Treatment for these complicated pneumonias may include a variety of invasive procedures, such as video-assisted thorascopic surgery or chest tube placement.
Typically, a long course of antibiotics is prescribed on discharge, which may be oral or parenterally administered via a peripherally inserted central catheter (PICC). Previous studies have shown that oral antibiotics are equivalent to parenteral antibiotics for outpatient treatment of osteomyelitis. However, little evidence exists comparing the effectiveness of the two routes in treating complicated pneumonia.

The rate of PICC complications in complicated pneumonia also has not been well studied.
Study design: Retrospective cohort study.
Setting: Thirty-eight children’s hospitals affiliated with the Children’s Hospital Association.
Synopsis: Over 4 years, 7,820 encounters were identified with 2,123 patients ultimately being included in the cohort. Inclusion criteria were age 2 months to 18 years, and discharge diagnoses of pneumonia and pleural effusion. The authors excluded patients with chronic medical conditions, length of stay (LOS) less than 4 and more than 14 days, patients transferred to or from other institutions, and patients receiving no antibiotics on hospital day 1. The final criteria attempted to avoid inclusion of patients with nosocomial pneumonia. After application of these criteria, individual patient records were reviewed.
Patients were categorized as PICC or oral antibiotics based upon antibiotic route at their initial discharge. Treatment failure was defined as an ED revisit or rehospitalization that led to a change in antibiotic, lengthening of antibiotic course, or pleural drainage. Records were searched for evidence of PICC complications, adverse drug reactions, and other illness-related revisits. Patients in the PICC arm and oral arm were matched by age, race, insurance, LOS, positive vs. negative blood culture, ICU admission, and timing and type of pleural drainage.
Fifty-seven patients had treatment failure (2.7%). In matched analysis, there was no difference in treatment failure between PICC and oral routes (PICC treatment failure OR, 1.26 95% CI, 0.54-2.94). PICC complications were found in 7.1% of patients. Patients with PICC had significantly higher rates of adverse drug reactions (OR, 19.1 95% CI, 4.2-87.3) and illness-related revisits (OR 3.27 95% CI, 1.65-6.48), and all revisits (OR, 4.71 95% CI, 2.97-7.46).
PICC use varied markedly across geographic regions and institutions, with rates varying from less than 10% of cases to approximately 70%. Of geographic regions, the Mid-Atlantic used PICCs least often while the East North Central used them the most.
Bottom line: Treatment failure with both oral and PICC treatment of complicated pneumonia occur at the same rate, and are uncommon. Patients with PICCs had an increased rate of complications, including adverse drug reactions and revisits.
Citation: Shah SS, Srivastava R, Wu S, et al. Intravenous versus oral antibiotics for postdischarge treatment of complicated pneumonia. Pediatrics. 2016;138(6):e20161692. doi: 10.1542/peds.2016-1692.
Dr. Stubblefield is a pediatric hospitalist at Nemours/Alfred I. duPont Hospital for Children in Wilmington, Del., and clinical assistant professor of pediatrics at Sidney Kimmel Medical College at Thomas Jefferson University in Philadelphia.
Clinical question: Are oral antibiotics as effective as intravenous (IV) antibiotics in the treatment of complicated pneumonia after discharge to home?
Background: Pneumonia is the most common illness among hospitalized children and adolescents (excluding neonates). Among children admitted with community acquired pneumonia, 15% may develop a complicated pneumonia (one with a pleural effusion or empyema). Treatment for these complicated pneumonias may include a variety of invasive procedures, such as video-assisted thorascopic surgery or chest tube placement.
Typically, a long course of antibiotics is prescribed on discharge, which may be oral or parenterally administered via a peripherally inserted central catheter (PICC). Previous studies have shown that oral antibiotics are equivalent to parenteral antibiotics for outpatient treatment of osteomyelitis. However, little evidence exists comparing the effectiveness of the two routes in treating complicated pneumonia.
The rate of PICC complications in complicated pneumonia also has not been well studied.
Study design: Retrospective cohort study.
Setting: Thirty-eight children’s hospitals affiliated with the Children’s Hospital Association.
Synopsis: Over 4 years, 7,820 encounters were identified with 2,123 patients ultimately being included in the cohort. Inclusion criteria were age 2 months to 18 years, and discharge diagnoses of pneumonia and pleural effusion. The authors excluded patients with chronic medical conditions, length of stay (LOS) less than 4 and more than 14 days, patients transferred to or from other institutions, and patients receiving no antibiotics on hospital day 1. The final criteria attempted to avoid inclusion of patients with nosocomial pneumonia. After application of these criteria, individual patient records were reviewed.
Patients were categorized as PICC or oral antibiotics based upon antibiotic route at their initial discharge. Treatment failure was defined as an ED revisit or rehospitalization that led to a change in antibiotic, lengthening of antibiotic course, or pleural drainage. Records were searched for evidence of PICC complications, adverse drug reactions, and other illness-related revisits. Patients in the PICC arm and oral arm were matched by age, race, insurance, LOS, positive vs. negative blood culture, ICU admission, and timing and type of pleural drainage.
Fifty-seven patients had treatment failure (2.7%). In matched analysis, there was no difference in treatment failure between PICC and oral routes (PICC treatment failure OR, 1.26 95% CI, 0.54-2.94). PICC complications were found in 7.1% of patients. Patients with PICC had significantly higher rates of adverse drug reactions (OR, 19.1 95% CI, 4.2-87.3) and illness-related revisits (OR 3.27 95% CI, 1.65-6.48), and all revisits (OR, 4.71 95% CI, 2.97-7.46).
PICC use varied markedly across geographic regions and institutions, with rates varying from less than 10% of cases to approximately 70%. Of geographic regions, the Mid-Atlantic used PICCs least often while the East North Central used them the most.
Bottom line: Treatment failure with both oral and PICC treatment of complicated pneumonia occur at the same rate, and are uncommon. Patients with PICCs had an increased rate of complications, including adverse drug reactions and revisits.
Citation: Shah SS, Srivastava R, Wu S, et al. Intravenous versus oral antibiotics for postdischarge treatment of complicated pneumonia. Pediatrics. 2016;138(6):e20161692. doi: 10.1542/peds.2016-1692.
Dr. Stubblefield is a pediatric hospitalist at Nemours/Alfred I. duPont Hospital for Children in Wilmington, Del., and clinical assistant professor of pediatrics at Sidney Kimmel Medical College at Thomas Jefferson University in Philadelphia.
Clinical question: Are oral antibiotics as effective as intravenous (IV) antibiotics in the treatment of complicated pneumonia after discharge to home?
Background: Pneumonia is the most common illness among hospitalized children and adolescents (excluding neonates). Among children admitted with community acquired pneumonia, 15% may develop a complicated pneumonia (one with a pleural effusion or empyema). Treatment for these complicated pneumonias may include a variety of invasive procedures, such as video-assisted thorascopic surgery or chest tube placement.
Typically, a long course of antibiotics is prescribed on discharge, which may be oral or parenterally administered via a peripherally inserted central catheter (PICC). Previous studies have shown that oral antibiotics are equivalent to parenteral antibiotics for outpatient treatment of osteomyelitis. However, little evidence exists comparing the effectiveness of the two routes in treating complicated pneumonia.
The rate of PICC complications in complicated pneumonia also has not been well studied.
Study design: Retrospective cohort study.
Setting: Thirty-eight children’s hospitals affiliated with the Children’s Hospital Association.
Synopsis: Over 4 years, 7,820 encounters were identified with 2,123 patients ultimately being included in the cohort. Inclusion criteria were age 2 months to 18 years, and discharge diagnoses of pneumonia and pleural effusion. The authors excluded patients with chronic medical conditions, length of stay (LOS) less than 4 and more than 14 days, patients transferred to or from other institutions, and patients receiving no antibiotics on hospital day 1. The final criteria attempted to avoid inclusion of patients with nosocomial pneumonia. After application of these criteria, individual patient records were reviewed.
Patients were categorized as PICC or oral antibiotics based upon antibiotic route at their initial discharge. Treatment failure was defined as an ED revisit or rehospitalization that led to a change in antibiotic, lengthening of antibiotic course, or pleural drainage. Records were searched for evidence of PICC complications, adverse drug reactions, and other illness-related revisits. Patients in the PICC arm and oral arm were matched by age, race, insurance, LOS, positive vs. negative blood culture, ICU admission, and timing and type of pleural drainage.
Fifty-seven patients had treatment failure (2.7%). In matched analysis, there was no difference in treatment failure between PICC and oral routes (PICC treatment failure OR, 1.26 95% CI, 0.54-2.94). PICC complications were found in 7.1% of patients. Patients with PICC had significantly higher rates of adverse drug reactions (OR, 19.1 95% CI, 4.2-87.3) and illness-related revisits (OR 3.27 95% CI, 1.65-6.48), and all revisits (OR, 4.71 95% CI, 2.97-7.46).
PICC use varied markedly across geographic regions and institutions, with rates varying from less than 10% of cases to approximately 70%. Of geographic regions, the Mid-Atlantic used PICCs least often while the East North Central used them the most.
Bottom line: Treatment failure with both oral and PICC treatment of complicated pneumonia occur at the same rate, and are uncommon. Patients with PICCs had an increased rate of complications, including adverse drug reactions and revisits.
Citation: Shah SS, Srivastava R, Wu S, et al. Intravenous versus oral antibiotics for postdischarge treatment of complicated pneumonia. Pediatrics. 2016;138(6):e20161692. doi: 10.1542/peds.2016-1692.
Dr. Stubblefield is a pediatric hospitalist at Nemours/Alfred I. duPont Hospital for Children in Wilmington, Del., and clinical assistant professor of pediatrics at Sidney Kimmel Medical College at Thomas Jefferson University in Philadelphia.
Guideline: CRC patients should receive RAS mutation testing
Current evidence supports testing colorectal cancers for genetic mutations in the epidermal growth factor receptor (EGFR) signaling pathway because the results provide clinically actionable information, according to a new clinical practice guideline published in the Journal of Clinical Oncology.
In contrast, the evidence is inadequate at this time to recommend either for or against several other mutational analyses, such as PIK3CA, PTEN, or BRAF V600 testing. This guideline is intended “to help establish standard molecular biomarker testing, guide targeted therapies, and advance personalized care for colorectal cancer patients,” said Antonia R. Sepulveda, MD, PhD, and her associates on the guideline committee.
The guideline is meant for use by oncologists and other clinicians, pathologists, laboratory employees, molecular diagnostics professionals, scientists, government agencies, nonprofit organizations, patients, and patient advocates. It includes 21 recommendations and was developed jointly by the American Society for Clinical Pathology, the College of American Pathologists, the Association for Molecular Pathology, and the American Society of Clinical Oncology. The groups based these recommendations on a comprehensive review of 123 randomized controlled studies, comparative studies, existing practice guidelines, consensus documents, and meta-analyses published in the medical literature or presented at meetings since 2008, said Dr. Sepulveda, professor of pathology and cell biology at Columbia University, New York, and her associates (J Clin Oncol. 2017 Feb 6. doi: 10.1200/JCO.2016.71.9807.
Among the strongest recommendations based on the highest-quality evidence:
• Patients with colorectal carcinoma who are being considered for anti-EGFR therapy must undergo RAS mutational testing, and KRAS and NRAS mutations are reliable predictors that this therapy will not be beneficial.
• Analysis of BRAF and MMR mutations have clear prognostic value, and MMR status is emerging as predictive in patients with advanced disease being considered for anti-PD-1/PD-L1 treatment.
• Laboratories must use validated molecular testing methods and follow accepted standards for molecular diagnostic tests. They should include in their reports a “results and interpretation” section readily understandable to oncologists and pathologists.
• Laboratories must incorporate colorectal carcinoma molecular biomarker testing methods into their overall quality improvement programs and must participate in formal proficiency testing programs or an appropriate alternative.
Several recommendations address the importance of timeliness in performing genetic testing on colorectal cancers. Professionals and staff must expedite sending specimens to specialty laboratories, performing all biomarker tests, and reporting all test results to clinicians. It is “suggested” that molecular diagnostics laboratories aim for a benchmark of 90% of reports available within 10 working days from date of receipt of the specimen.
Current evidence supports testing colorectal cancers for genetic mutations in the epidermal growth factor receptor (EGFR) signaling pathway because the results provide clinically actionable information, according to a new clinical practice guideline published in the Journal of Clinical Oncology.
In contrast, the evidence is inadequate at this time to recommend either for or against several other mutational analyses, such as PIK3CA, PTEN, or BRAF V600 testing. This guideline is intended “to help establish standard molecular biomarker testing, guide targeted therapies, and advance personalized care for colorectal cancer patients,” said Antonia R. Sepulveda, MD, PhD, and her associates on the guideline committee.
The guideline is meant for use by oncologists and other clinicians, pathologists, laboratory employees, molecular diagnostics professionals, scientists, government agencies, nonprofit organizations, patients, and patient advocates. It includes 21 recommendations and was developed jointly by the American Society for Clinical Pathology, the College of American Pathologists, the Association for Molecular Pathology, and the American Society of Clinical Oncology. The groups based these recommendations on a comprehensive review of 123 randomized controlled studies, comparative studies, existing practice guidelines, consensus documents, and meta-analyses published in the medical literature or presented at meetings since 2008, said Dr. Sepulveda, professor of pathology and cell biology at Columbia University, New York, and her associates (J Clin Oncol. 2017 Feb 6. doi: 10.1200/JCO.2016.71.9807.
Among the strongest recommendations based on the highest-quality evidence:
• Patients with colorectal carcinoma who are being considered for anti-EGFR therapy must undergo RAS mutational testing, and KRAS and NRAS mutations are reliable predictors that this therapy will not be beneficial.
• Analysis of BRAF and MMR mutations have clear prognostic value, and MMR status is emerging as predictive in patients with advanced disease being considered for anti-PD-1/PD-L1 treatment.
• Laboratories must use validated molecular testing methods and follow accepted standards for molecular diagnostic tests. They should include in their reports a “results and interpretation” section readily understandable to oncologists and pathologists.
• Laboratories must incorporate colorectal carcinoma molecular biomarker testing methods into their overall quality improvement programs and must participate in formal proficiency testing programs or an appropriate alternative.
Several recommendations address the importance of timeliness in performing genetic testing on colorectal cancers. Professionals and staff must expedite sending specimens to specialty laboratories, performing all biomarker tests, and reporting all test results to clinicians. It is “suggested” that molecular diagnostics laboratories aim for a benchmark of 90% of reports available within 10 working days from date of receipt of the specimen.
Current evidence supports testing colorectal cancers for genetic mutations in the epidermal growth factor receptor (EGFR) signaling pathway because the results provide clinically actionable information, according to a new clinical practice guideline published in the Journal of Clinical Oncology.
In contrast, the evidence is inadequate at this time to recommend either for or against several other mutational analyses, such as PIK3CA, PTEN, or BRAF V600 testing. This guideline is intended “to help establish standard molecular biomarker testing, guide targeted therapies, and advance personalized care for colorectal cancer patients,” said Antonia R. Sepulveda, MD, PhD, and her associates on the guideline committee.
The guideline is meant for use by oncologists and other clinicians, pathologists, laboratory employees, molecular diagnostics professionals, scientists, government agencies, nonprofit organizations, patients, and patient advocates. It includes 21 recommendations and was developed jointly by the American Society for Clinical Pathology, the College of American Pathologists, the Association for Molecular Pathology, and the American Society of Clinical Oncology. The groups based these recommendations on a comprehensive review of 123 randomized controlled studies, comparative studies, existing practice guidelines, consensus documents, and meta-analyses published in the medical literature or presented at meetings since 2008, said Dr. Sepulveda, professor of pathology and cell biology at Columbia University, New York, and her associates (J Clin Oncol. 2017 Feb 6. doi: 10.1200/JCO.2016.71.9807.
Among the strongest recommendations based on the highest-quality evidence:
• Patients with colorectal carcinoma who are being considered for anti-EGFR therapy must undergo RAS mutational testing, and KRAS and NRAS mutations are reliable predictors that this therapy will not be beneficial.
• Analysis of BRAF and MMR mutations have clear prognostic value, and MMR status is emerging as predictive in patients with advanced disease being considered for anti-PD-1/PD-L1 treatment.
• Laboratories must use validated molecular testing methods and follow accepted standards for molecular diagnostic tests. They should include in their reports a “results and interpretation” section readily understandable to oncologists and pathologists.
• Laboratories must incorporate colorectal carcinoma molecular biomarker testing methods into their overall quality improvement programs and must participate in formal proficiency testing programs or an appropriate alternative.
Several recommendations address the importance of timeliness in performing genetic testing on colorectal cancers. Professionals and staff must expedite sending specimens to specialty laboratories, performing all biomarker tests, and reporting all test results to clinicians. It is “suggested” that molecular diagnostics laboratories aim for a benchmark of 90% of reports available within 10 working days from date of receipt of the specimen.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: The current evidence supports testing colorectal cancers for mutations of genes in the EGFR signaling pathway.
Key numerical finding: Molecular diagnostics laboratories should aim for a benchmark of 90% of reports available to pathologists and clinicians within 10 working days from the date of receipt of the colorectal cancer specimen.
Data source: A review of 123 reports in the literature and a compilation of 21 recommendations regarding molecular testing of colorectal cancers.
Disclosures: No industry funds were used in the development of this guideline.
Aspirin reduces recurrent preeclampsia in real-world study
LAS VEGAS – Using low-dose aspirin during pregnancy significantly reduced the risk of recurrent preeclampsia, according to results of a new study.
“The net benefit of aspirin is substantial,” Mary Catherine Tolcher, MD, the study’s lead author said. “The number needed to treat to prevent one case of recurrent preeclampsia is six... The cost of daily low-dose aspirin for the duration of one pregnancy is about $4.00. Comparatively, the cost to prevent one case of eclampsia is approximately $21,000.”
Dr. Tolcher, a postdoctorate fellow in ob.gyn. at Baylor College of Medicine, Houston, and her colleagues found a total of 417 at-risk women in the institution’s labor and delivery database during the August 2011–June 2016 study period; 284 were identified before the guidelines were implemented in 2014, while 133 women were identified as high-risk after guideline implementation.
While nearly one-third (32.4%) of women with a history of preeclampsia had a recurrence in the before group, the recurrence rate fell to 16.5% in the after group, who had been instructed to take low-dose aspirin in accordance with the guidelines. When the investigators calculated the fully adjusted odds ratio for recurrent preeclampsia, they found a reduction of about 30% in recurrent preeclampsia [aOR, 0.71; 95% confidence interval, 0.52-0.95].
“This decrease is greater than the approximately 10 to 15 percent reduction that has been previously reported in clinical trials, and different from the meta-analysis that prompted the national recommendations,” Dr. Tolcher said at the annual Pregnancy Meeting sponsored by the Society for Maternal-Fetal Medicine.
She and her colleagues hypothesize that the greater effect size may be attributable to limiting the study population to the higher-risk group of women with a history of preeclampsia. Alternatively, she said, aspirin’s pharmacodynamics can differ by race, so racial differences between study populations may also be at play.
One of the causative mechanisms for preeclampsia is thought to be impaired trophoblastic remodeling that impedes development of the low-resistance vascular system of the maternal-fetal unit. “The resulting pathophysiology is multisystemic and includes vascular and endothelial dysfunction, placental ischemia, and an inflammatory and stress response,” Dr. Tolcher said. Overproduction of thromboxanes, she said, plays a role in this process. Aspirin’s inhibition of cyclooxygenase-2 (COX-2) enzymes and thromboxanes is thought to mitigate the vasoconstriction and endothelial dysfunction seen in preeclampsia, she said.
A secondary outcome measure in the study was use of magnesium sulfate, and after guideline implementation “there was a trend toward reduced use of magnesium sulfate, which we reserved for preeclampsia with severe features,” Dr. Tolcher said.
There was no difference in the incidence of preterm deliveries, another secondary outcome measure, after the aspirin guidelines were implemented.
There were some differences in characteristics between the before and after groups: the ratio of Hispanic women with preeclampsia was significantly lower in the after group (P less than .0001). The distribution of method of payment shifted, with more private pay patients, fewer Children’s Health Insurance Program (CHIP) patients, and fewer “other” payment method patients in the after group. Though Dr. Tolcher reported that type 1 diabetes was seen more often in the after group, the rates of any kind of pre-pregnancy diabetes were similar between the groups (6.33% before; 4.26% after).
Potentially confounding variables were controlled by means of logistic regression analysis. Women with multiple gestations were excluded, and only the first pregnancy after a previous episode of preeclampsia was studied.
Otherwise, demographics and other patient characteristics – including rates of chronic hypertension, were similar between the before and after groups. About a quarter of the patients in each group had a history of preterm delivery, and the median age at delivery for both was 38 years.
Within the total group that had recurrent preeclampsia, “maternal age greater than 35, type 2 diabetes, chronic hypertension, and a history of preterm preeclampsia were significantly associated with recurrent preeclampsia,” Dr. Tolcher said.
Study limitations included the retrospective nature, the inclusion of only women who had a prior history of preeclampsia, and the investigators’ inability to determine whether patients were adherent to recommendations for aspirin use. However, the study represents actual clinical use, Dr. Tolcher said, and addresses a real need. “Preeclampsia is responsible for 75,000 maternal deaths annually, and accounts for 15% of the preterm births in the U.S.,” she said.
Dr. Tolcher reported having no relevant financial disclosures.
[email protected]
On Twitter @karioakes
LAS VEGAS – Using low-dose aspirin during pregnancy significantly reduced the risk of recurrent preeclampsia, according to results of a new study.
“The net benefit of aspirin is substantial,” Mary Catherine Tolcher, MD, the study’s lead author said. “The number needed to treat to prevent one case of recurrent preeclampsia is six... The cost of daily low-dose aspirin for the duration of one pregnancy is about $4.00. Comparatively, the cost to prevent one case of eclampsia is approximately $21,000.”
Dr. Tolcher, a postdoctorate fellow in ob.gyn. at Baylor College of Medicine, Houston, and her colleagues found a total of 417 at-risk women in the institution’s labor and delivery database during the August 2011–June 2016 study period; 284 were identified before the guidelines were implemented in 2014, while 133 women were identified as high-risk after guideline implementation.
While nearly one-third (32.4%) of women with a history of preeclampsia had a recurrence in the before group, the recurrence rate fell to 16.5% in the after group, who had been instructed to take low-dose aspirin in accordance with the guidelines. When the investigators calculated the fully adjusted odds ratio for recurrent preeclampsia, they found a reduction of about 30% in recurrent preeclampsia [aOR, 0.71; 95% confidence interval, 0.52-0.95].
“This decrease is greater than the approximately 10 to 15 percent reduction that has been previously reported in clinical trials, and different from the meta-analysis that prompted the national recommendations,” Dr. Tolcher said at the annual Pregnancy Meeting sponsored by the Society for Maternal-Fetal Medicine.
She and her colleagues hypothesize that the greater effect size may be attributable to limiting the study population to the higher-risk group of women with a history of preeclampsia. Alternatively, she said, aspirin’s pharmacodynamics can differ by race, so racial differences between study populations may also be at play.
One of the causative mechanisms for preeclampsia is thought to be impaired trophoblastic remodeling that impedes development of the low-resistance vascular system of the maternal-fetal unit. “The resulting pathophysiology is multisystemic and includes vascular and endothelial dysfunction, placental ischemia, and an inflammatory and stress response,” Dr. Tolcher said. Overproduction of thromboxanes, she said, plays a role in this process. Aspirin’s inhibition of cyclooxygenase-2 (COX-2) enzymes and thromboxanes is thought to mitigate the vasoconstriction and endothelial dysfunction seen in preeclampsia, she said.
A secondary outcome measure in the study was use of magnesium sulfate, and after guideline implementation “there was a trend toward reduced use of magnesium sulfate, which we reserved for preeclampsia with severe features,” Dr. Tolcher said.
There was no difference in the incidence of preterm deliveries, another secondary outcome measure, after the aspirin guidelines were implemented.
There were some differences in characteristics between the before and after groups: the ratio of Hispanic women with preeclampsia was significantly lower in the after group (P less than .0001). The distribution of method of payment shifted, with more private pay patients, fewer Children’s Health Insurance Program (CHIP) patients, and fewer “other” payment method patients in the after group. Though Dr. Tolcher reported that type 1 diabetes was seen more often in the after group, the rates of any kind of pre-pregnancy diabetes were similar between the groups (6.33% before; 4.26% after).
Potentially confounding variables were controlled by means of logistic regression analysis. Women with multiple gestations were excluded, and only the first pregnancy after a previous episode of preeclampsia was studied.
Otherwise, demographics and other patient characteristics – including rates of chronic hypertension, were similar between the before and after groups. About a quarter of the patients in each group had a history of preterm delivery, and the median age at delivery for both was 38 years.
Within the total group that had recurrent preeclampsia, “maternal age greater than 35, type 2 diabetes, chronic hypertension, and a history of preterm preeclampsia were significantly associated with recurrent preeclampsia,” Dr. Tolcher said.
Study limitations included the retrospective nature, the inclusion of only women who had a prior history of preeclampsia, and the investigators’ inability to determine whether patients were adherent to recommendations for aspirin use. However, the study represents actual clinical use, Dr. Tolcher said, and addresses a real need. “Preeclampsia is responsible for 75,000 maternal deaths annually, and accounts for 15% of the preterm births in the U.S.,” she said.
Dr. Tolcher reported having no relevant financial disclosures.
[email protected]
On Twitter @karioakes
LAS VEGAS – Using low-dose aspirin during pregnancy significantly reduced the risk of recurrent preeclampsia, according to results of a new study.
“The net benefit of aspirin is substantial,” Mary Catherine Tolcher, MD, the study’s lead author said. “The number needed to treat to prevent one case of recurrent preeclampsia is six... The cost of daily low-dose aspirin for the duration of one pregnancy is about $4.00. Comparatively, the cost to prevent one case of eclampsia is approximately $21,000.”
Dr. Tolcher, a postdoctorate fellow in ob.gyn. at Baylor College of Medicine, Houston, and her colleagues found a total of 417 at-risk women in the institution’s labor and delivery database during the August 2011–June 2016 study period; 284 were identified before the guidelines were implemented in 2014, while 133 women were identified as high-risk after guideline implementation.
While nearly one-third (32.4%) of women with a history of preeclampsia had a recurrence in the before group, the recurrence rate fell to 16.5% in the after group, who had been instructed to take low-dose aspirin in accordance with the guidelines. When the investigators calculated the fully adjusted odds ratio for recurrent preeclampsia, they found a reduction of about 30% in recurrent preeclampsia [aOR, 0.71; 95% confidence interval, 0.52-0.95].
“This decrease is greater than the approximately 10 to 15 percent reduction that has been previously reported in clinical trials, and different from the meta-analysis that prompted the national recommendations,” Dr. Tolcher said at the annual Pregnancy Meeting sponsored by the Society for Maternal-Fetal Medicine.
She and her colleagues hypothesize that the greater effect size may be attributable to limiting the study population to the higher-risk group of women with a history of preeclampsia. Alternatively, she said, aspirin’s pharmacodynamics can differ by race, so racial differences between study populations may also be at play.
One of the causative mechanisms for preeclampsia is thought to be impaired trophoblastic remodeling that impedes development of the low-resistance vascular system of the maternal-fetal unit. “The resulting pathophysiology is multisystemic and includes vascular and endothelial dysfunction, placental ischemia, and an inflammatory and stress response,” Dr. Tolcher said. Overproduction of thromboxanes, she said, plays a role in this process. Aspirin’s inhibition of cyclooxygenase-2 (COX-2) enzymes and thromboxanes is thought to mitigate the vasoconstriction and endothelial dysfunction seen in preeclampsia, she said.
A secondary outcome measure in the study was use of magnesium sulfate, and after guideline implementation “there was a trend toward reduced use of magnesium sulfate, which we reserved for preeclampsia with severe features,” Dr. Tolcher said.
There was no difference in the incidence of preterm deliveries, another secondary outcome measure, after the aspirin guidelines were implemented.
There were some differences in characteristics between the before and after groups: the ratio of Hispanic women with preeclampsia was significantly lower in the after group (P less than .0001). The distribution of method of payment shifted, with more private pay patients, fewer Children’s Health Insurance Program (CHIP) patients, and fewer “other” payment method patients in the after group. Though Dr. Tolcher reported that type 1 diabetes was seen more often in the after group, the rates of any kind of pre-pregnancy diabetes were similar between the groups (6.33% before; 4.26% after).
Potentially confounding variables were controlled by means of logistic regression analysis. Women with multiple gestations were excluded, and only the first pregnancy after a previous episode of preeclampsia was studied.
Otherwise, demographics and other patient characteristics – including rates of chronic hypertension, were similar between the before and after groups. About a quarter of the patients in each group had a history of preterm delivery, and the median age at delivery for both was 38 years.
Within the total group that had recurrent preeclampsia, “maternal age greater than 35, type 2 diabetes, chronic hypertension, and a history of preterm preeclampsia were significantly associated with recurrent preeclampsia,” Dr. Tolcher said.
Study limitations included the retrospective nature, the inclusion of only women who had a prior history of preeclampsia, and the investigators’ inability to determine whether patients were adherent to recommendations for aspirin use. However, the study represents actual clinical use, Dr. Tolcher said, and addresses a real need. “Preeclampsia is responsible for 75,000 maternal deaths annually, and accounts for 15% of the preterm births in the U.S.,” she said.
Dr. Tolcher reported having no relevant financial disclosures.
[email protected]
On Twitter @karioakes
AT THE PREGNANCY MEETING
Key clinical point:
Major finding: The adjusted odds ratio for recurrent preeclampsia was 0.71 after implementing U.S. Preventive Services Task Force (USPSTF) guidelines for aspirin administration.
Data source: Retrospective cohort study of 17,256 deliveries at a single academic medical center before and after implementation of USPSTF aspirin guidelines, with 417 cases of recurrent preeclampsia identified.
Disclosures: Dr. Tolcher reported having no relevant financial disclosures.
Children with psychotic illness aren’t treated soon enough
SAN FRANCISCO – Early detection and treatment of psychotic illness is critical in children, according to Devanand Manoli, MD, PhD, of the University of California, San Francisco.
“After the conversion to psychotic illness, one of the most important prognostic factors is the duration of untreated psychosis.” A longer duration is associated with a greater symptom burden and lower functioning, which have “significant prognostic implications,” but sometimes treatment doesn’t come for a year or more. “There are many patients out there not receiving treatment,” the pediatric psychiatrist said at a psychopharmacology update held by the American Academy of Child and Adolescent Psychiatry.

The impact of early treatment raises the question of what to do with those who seem to be at risk, but don’t meet criteria for formal diagnosis.
It’s tough to be sure who exactly is at risk. Signs and symptoms can be nonspecific in adolescents, including disorganized communication, suspiciousness, verbal memory deficiencies, and decline in social functioning.
It’s even a tougher call in younger children. Most who exhibit psychotic or psychoticlike symptoms do not have a true psychotic disorder. Loose associations and illogical thinking are fairly common in early childhood, and preschool children can have transient tactile, visual, and other hallucinations that are benign. If there does seem to be a problem, metabolic issues are the most likely culprit before the age of 6 or so.
Again, comprehensive early intervention – including reducing environmental stressors – helps with functioning, symptoms, and other issues even before a formal diagnosis. However, antipsychotics do not decrease the rate of conversion to psychotic illness in truly high-risk children; their use is for symptom management, Dr. Manoli said.
It’s important adolescents avoid cannabis; it’s become clear in recent years that marijuana increases the risk of conversion to schizophrenia and the risk of relapse. “It’s a very important thing to emphasize. Cannabis use is something to counsel against,” he said.
Atypicals seem to be about equally effective, so their selection mostly comes down to side effects and cost. An exception might be clozapine. “It’s a medication we are very resistant to prescribe” because of the need for aggressive neutrophil monitoring and other issues, “but in patients with acute psychotic symptoms, it can be very effective” in both treatment-naïve and treatment-resistant children and help with aggression, he said, noting also that it’s important to remember children are more sensitive than adults to akathisia and other extrapyramidal symptoms with antipsychotics.
In response to audience questions, Dr. Manoli said there isn’t really a need to monitor for prolactin elevations with atypical antipsychotics unless there are symptoms, such as inappropriate breast tissue or lactation. In those cases, augmentation with aripiprazole can normalize levels.
He also noted that he and his colleagues do order baseline and follow-up ECGs when prescribing ziprasidone; prolonged QTc interval is a concern, regardless of family cardiac history.
Dr. Manoli reported research funding from the Brain & Behavior Research Foundation, the One Mind Institute, the National Institutes of Health, and other sources.
SAN FRANCISCO – Early detection and treatment of psychotic illness is critical in children, according to Devanand Manoli, MD, PhD, of the University of California, San Francisco.
“After the conversion to psychotic illness, one of the most important prognostic factors is the duration of untreated psychosis.” A longer duration is associated with a greater symptom burden and lower functioning, which have “significant prognostic implications,” but sometimes treatment doesn’t come for a year or more. “There are many patients out there not receiving treatment,” the pediatric psychiatrist said at a psychopharmacology update held by the American Academy of Child and Adolescent Psychiatry.
The impact of early treatment raises the question of what to do with those who seem to be at risk, but don’t meet criteria for formal diagnosis.
It’s tough to be sure who exactly is at risk. Signs and symptoms can be nonspecific in adolescents, including disorganized communication, suspiciousness, verbal memory deficiencies, and decline in social functioning.
It’s even a tougher call in younger children. Most who exhibit psychotic or psychoticlike symptoms do not have a true psychotic disorder. Loose associations and illogical thinking are fairly common in early childhood, and preschool children can have transient tactile, visual, and other hallucinations that are benign. If there does seem to be a problem, metabolic issues are the most likely culprit before the age of 6 or so.
Again, comprehensive early intervention – including reducing environmental stressors – helps with functioning, symptoms, and other issues even before a formal diagnosis. However, antipsychotics do not decrease the rate of conversion to psychotic illness in truly high-risk children; their use is for symptom management, Dr. Manoli said.
It’s important adolescents avoid cannabis; it’s become clear in recent years that marijuana increases the risk of conversion to schizophrenia and the risk of relapse. “It’s a very important thing to emphasize. Cannabis use is something to counsel against,” he said.
Atypicals seem to be about equally effective, so their selection mostly comes down to side effects and cost. An exception might be clozapine. “It’s a medication we are very resistant to prescribe” because of the need for aggressive neutrophil monitoring and other issues, “but in patients with acute psychotic symptoms, it can be very effective” in both treatment-naïve and treatment-resistant children and help with aggression, he said, noting also that it’s important to remember children are more sensitive than adults to akathisia and other extrapyramidal symptoms with antipsychotics.
In response to audience questions, Dr. Manoli said there isn’t really a need to monitor for prolactin elevations with atypical antipsychotics unless there are symptoms, such as inappropriate breast tissue or lactation. In those cases, augmentation with aripiprazole can normalize levels.
He also noted that he and his colleagues do order baseline and follow-up ECGs when prescribing ziprasidone; prolonged QTc interval is a concern, regardless of family cardiac history.
Dr. Manoli reported research funding from the Brain & Behavior Research Foundation, the One Mind Institute, the National Institutes of Health, and other sources.
SAN FRANCISCO – Early detection and treatment of psychotic illness is critical in children, according to Devanand Manoli, MD, PhD, of the University of California, San Francisco.
“After the conversion to psychotic illness, one of the most important prognostic factors is the duration of untreated psychosis.” A longer duration is associated with a greater symptom burden and lower functioning, which have “significant prognostic implications,” but sometimes treatment doesn’t come for a year or more. “There are many patients out there not receiving treatment,” the pediatric psychiatrist said at a psychopharmacology update held by the American Academy of Child and Adolescent Psychiatry.
The impact of early treatment raises the question of what to do with those who seem to be at risk, but don’t meet criteria for formal diagnosis.
It’s tough to be sure who exactly is at risk. Signs and symptoms can be nonspecific in adolescents, including disorganized communication, suspiciousness, verbal memory deficiencies, and decline in social functioning.
It’s even a tougher call in younger children. Most who exhibit psychotic or psychoticlike symptoms do not have a true psychotic disorder. Loose associations and illogical thinking are fairly common in early childhood, and preschool children can have transient tactile, visual, and other hallucinations that are benign. If there does seem to be a problem, metabolic issues are the most likely culprit before the age of 6 or so.
Again, comprehensive early intervention – including reducing environmental stressors – helps with functioning, symptoms, and other issues even before a formal diagnosis. However, antipsychotics do not decrease the rate of conversion to psychotic illness in truly high-risk children; their use is for symptom management, Dr. Manoli said.
It’s important adolescents avoid cannabis; it’s become clear in recent years that marijuana increases the risk of conversion to schizophrenia and the risk of relapse. “It’s a very important thing to emphasize. Cannabis use is something to counsel against,” he said.
Atypicals seem to be about equally effective, so their selection mostly comes down to side effects and cost. An exception might be clozapine. “It’s a medication we are very resistant to prescribe” because of the need for aggressive neutrophil monitoring and other issues, “but in patients with acute psychotic symptoms, it can be very effective” in both treatment-naïve and treatment-resistant children and help with aggression, he said, noting also that it’s important to remember children are more sensitive than adults to akathisia and other extrapyramidal symptoms with antipsychotics.
In response to audience questions, Dr. Manoli said there isn’t really a need to monitor for prolactin elevations with atypical antipsychotics unless there are symptoms, such as inappropriate breast tissue or lactation. In those cases, augmentation with aripiprazole can normalize levels.
He also noted that he and his colleagues do order baseline and follow-up ECGs when prescribing ziprasidone; prolonged QTc interval is a concern, regardless of family cardiac history.
Dr. Manoli reported research funding from the Brain & Behavior Research Foundation, the One Mind Institute, the National Institutes of Health, and other sources.
EXPERT ANALYSIS FROM THE PSYCHOPHARMACOLOGY UPDATE INSTITUTE

Teen vaccines: Where we are now, and how can we go further?
Adolescents remain undervaccinated for several diseases, and clinicians need to lead the charge to change this reality, according to new clinical reports issued by the American Academy of Pediatrics Committee on Infectious Diseases.
Teens are lagging behind the Healthy People 2020 vaccination goals for the Tdap, quadrivalent meningococcal conjugate (menACWY), and human papillomavirus (HPV) vaccines, lead authors Henry H. Bernstein, DO, and Joseph A. Bocchini Jr., MD, wrote in the Feb. 6 issue of Pediatrics.

“Although HPV vaccination rates are slowly improving, they continue to lag far behind Tdap and menACWY rates for both boys and girls,” the authors wrote. Only 63% of girls and 50% of boys have gotten at least one HPV vaccine dose; just 42% of girls and 28% of boys finished the entire three-dose series in 2015.
A recent study found that parents declined the HPV vaccine 56% of the time. The three most common reasons were the belief that their child had a low risk of acquiring HPV; fear that the risk of adverse events was too great and belief that the vaccine wasn’t well researched and hasn’t been on the market long enough.
But teens also are falling behind with less controversial vaccines, the report noted. Fewer than half of teens got a flu shot in the 2015-2016 season. Rates for the MenACWY and Tdap are better, but not optimal (81% and 86%, respectively).
Barriers to optimal immunization
The clinical report puts clinicians on the first line of responsibility and makes no apologies for that.
“One of the greatest challenges is health care provider recommendation, which often lacks consistency and urgency,” the authors said. “Many health care providers do not universally recommend vaccines to eligible populations and do not offer concomitant vaccination with indicated vaccines during a single patient encounter.”
In fact, they noted, a recent physician survey found that only about 60% of pediatricians and family doctors strongly recommend the HPV vaccine for 11- and 12-year-old girls. Several parent surveys have determined that lack of provider recommendation is a leading reason for undervaccination among adolescents.
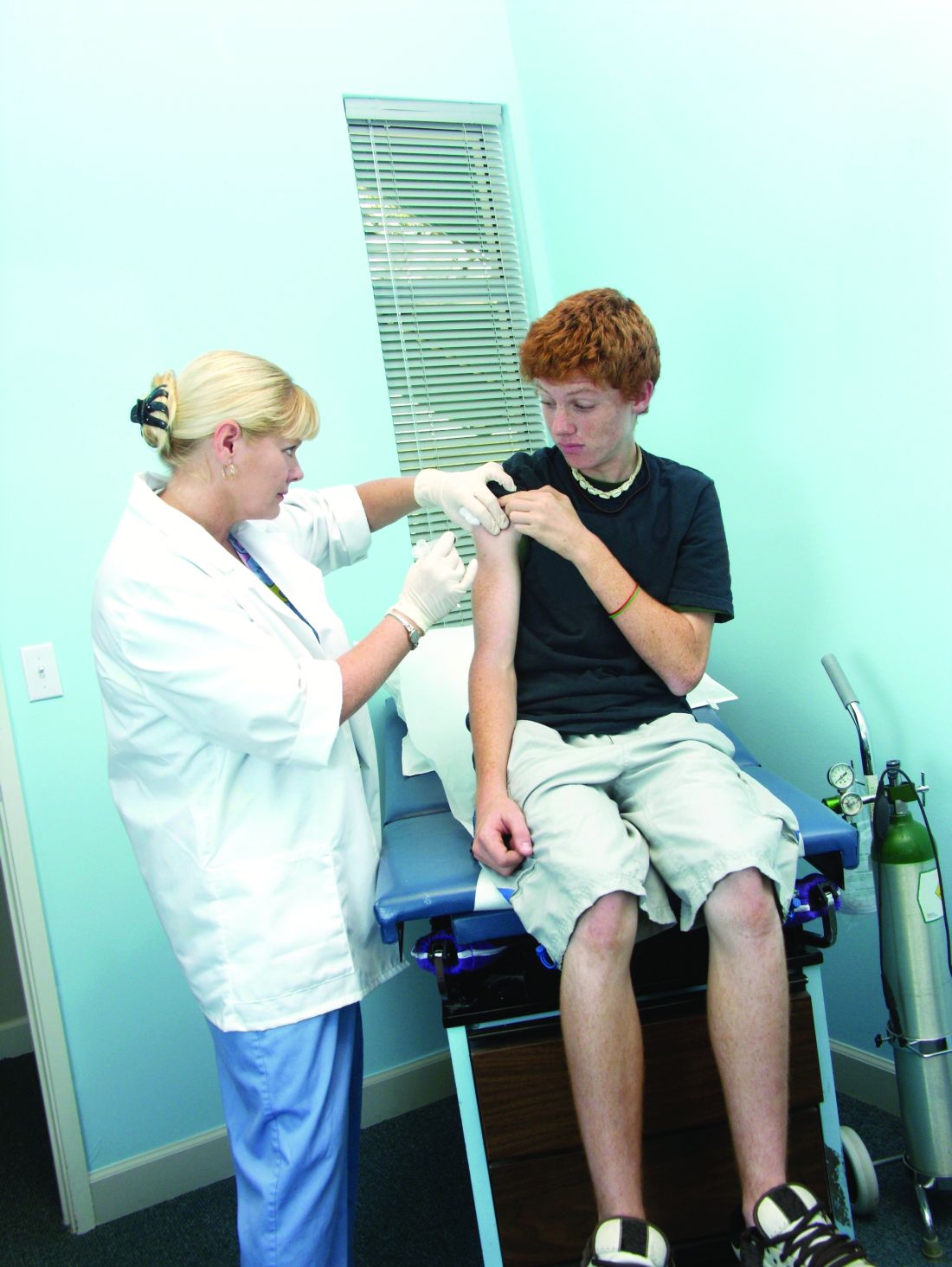
A companion clinical report, also authored by Dr. Bernstein and Dr. Bocchini, discusses practical ways to confront these barriers (Pediatrics. 2017 Feb 6. doi: 10.1542/peds.2016-4187). It opens with a strong call to physicians to lead the charge in increasing immunization rates.
“Up to 65% of parents have reported not receiving a recommendation … for immunizations. The major reason for nonreceipt [of Tdap and menACWY] was lack of a health care provider recommendation,” they said.
Overcoming parental vaccine hesitancy is a thorny struggle, the authors acknowledge. But they offer some helpful suggestions, including:
• Don’t offer immunizations as “optional.” This opens the door to vaccine dismissal. Instead, “strongly endorse all universally recommended vaccines as important for adolescents’ health.”
• Ask open-ended questions to get at the root of parental hesitancy. Give fact-based, medically sound information. Stress that doctors are parent partners in raising healthy children and protecting them from disease.
• Stress the big-picture benefits of vaccines. Telling parents that the HPV vaccine prevents cancer lifelong is a powerful message. “Up-to-date information on current events and disease outbreaks is a tool to bring into the conversation about vaccines as well.”
• Review the vaccine timeline. Do everything possible to ensure parents follow up and complete each series. “Follow-up immunization visits should be scheduled before the family leaves the care setting.”
• Don’t give up when a parent refuses a vaccine. “Although it may be challenging, it’s important that health care providers offer the vaccine at the next most appropriate time. Perseverance is critical for vaccine uptake and immunization rates.”
Dr. Bernstein and Dr. Bocchini also suggest that vaccine uptake could be boosted by a mental re-set of the office visit. “Missed opportunities for adolescent immunizations,” such as sick calls, are a prime example. “The majority of vaccines are administered during well-child visit [sports or camp physicals]. … However, acute care visits or sick visits in the patient-centered medical home are also an opportunity to deliver vaccines or to discuss upcoming vaccines,” they said.
Education and communication are essential to improving vaccine uptake, the report concludes. “Appropriate techniques for approaching adolescent patients and their parents in the office to encourage immunizations are important skills for healthcare providers. The key to increasing immunization rates and decreasing vaccine-preventable disease … is to focus on educating adolescents and strengthening health care providers’ recommendations by using all clinical opportunities to assess immunization status and provide needed vaccinations.”
Neither Dr Bernstein nor Dr. Bocchini had any financial disclosures.
Need help talking about teen vaccines? Check out these resources
The American Academy of Pediatrics offered the following resources designed to help clinicians communicate effectively when talking to patients and parents about vaccines:
• Talking to parents about the HPV vaccine. This website hosted by the Centers for Disease Control and Prevention includes a link to the latest vaccination recommendations, as well as a tip sheet with HPV vaccine talking points, a fact sheet on the vaccine’s safety data, and a video with four clinical vignettes that model effective communication.
• The HPV Champion Toolkit. This contains numerous resources to help clinicians learn to educate other health care professionals, discuss HPV vaccination with parents, and make practice changes to increase HPV vaccine uptake.
• You Are the Key to HPV Prevention. This CDC video presents up-to-date information on HPV infection and disease, the HPV vaccine, and ways to successfully communicate with patients and their parents about HPV vaccination.
• Adolescentvaccination.org. The National Foundation for Infectious Diseases maintains a website is entirely devoted to adolescent vaccination issues. It contains resources for clinicians, and parents.
• Top Strategies for Increasing Vaccine Coverage. This is a report by the American Academy of Pediatrics.
• You Call the Shots. This is an interactive, Web-based immunization training course created by the CDC.
• Suggestions to Improve Your Immunization Services. This is a checklist of suggestions, by the Immunization Action.
[email protected]
On Twitter @Alz_Gal
Adolescents remain undervaccinated for several diseases, and clinicians need to lead the charge to change this reality, according to new clinical reports issued by the American Academy of Pediatrics Committee on Infectious Diseases.
Teens are lagging behind the Healthy People 2020 vaccination goals for the Tdap, quadrivalent meningococcal conjugate (menACWY), and human papillomavirus (HPV) vaccines, lead authors Henry H. Bernstein, DO, and Joseph A. Bocchini Jr., MD, wrote in the Feb. 6 issue of Pediatrics.
“Although HPV vaccination rates are slowly improving, they continue to lag far behind Tdap and menACWY rates for both boys and girls,” the authors wrote. Only 63% of girls and 50% of boys have gotten at least one HPV vaccine dose; just 42% of girls and 28% of boys finished the entire three-dose series in 2015.
A recent study found that parents declined the HPV vaccine 56% of the time. The three most common reasons were the belief that their child had a low risk of acquiring HPV; fear that the risk of adverse events was too great and belief that the vaccine wasn’t well researched and hasn’t been on the market long enough.
But teens also are falling behind with less controversial vaccines, the report noted. Fewer than half of teens got a flu shot in the 2015-2016 season. Rates for the MenACWY and Tdap are better, but not optimal (81% and 86%, respectively).
Barriers to optimal immunization
The clinical report puts clinicians on the first line of responsibility and makes no apologies for that.
“One of the greatest challenges is health care provider recommendation, which often lacks consistency and urgency,” the authors said. “Many health care providers do not universally recommend vaccines to eligible populations and do not offer concomitant vaccination with indicated vaccines during a single patient encounter.”
In fact, they noted, a recent physician survey found that only about 60% of pediatricians and family doctors strongly recommend the HPV vaccine for 11- and 12-year-old girls. Several parent surveys have determined that lack of provider recommendation is a leading reason for undervaccination among adolescents.
A companion clinical report, also authored by Dr. Bernstein and Dr. Bocchini, discusses practical ways to confront these barriers (Pediatrics. 2017 Feb 6. doi: 10.1542/peds.2016-4187). It opens with a strong call to physicians to lead the charge in increasing immunization rates.
“Up to 65% of parents have reported not receiving a recommendation … for immunizations. The major reason for nonreceipt [of Tdap and menACWY] was lack of a health care provider recommendation,” they said.
Overcoming parental vaccine hesitancy is a thorny struggle, the authors acknowledge. But they offer some helpful suggestions, including:
• Don’t offer immunizations as “optional.” This opens the door to vaccine dismissal. Instead, “strongly endorse all universally recommended vaccines as important for adolescents’ health.”
• Ask open-ended questions to get at the root of parental hesitancy. Give fact-based, medically sound information. Stress that doctors are parent partners in raising healthy children and protecting them from disease.
• Stress the big-picture benefits of vaccines. Telling parents that the HPV vaccine prevents cancer lifelong is a powerful message. “Up-to-date information on current events and disease outbreaks is a tool to bring into the conversation about vaccines as well.”
• Review the vaccine timeline. Do everything possible to ensure parents follow up and complete each series. “Follow-up immunization visits should be scheduled before the family leaves the care setting.”
• Don’t give up when a parent refuses a vaccine. “Although it may be challenging, it’s important that health care providers offer the vaccine at the next most appropriate time. Perseverance is critical for vaccine uptake and immunization rates.”
Dr. Bernstein and Dr. Bocchini also suggest that vaccine uptake could be boosted by a mental re-set of the office visit. “Missed opportunities for adolescent immunizations,” such as sick calls, are a prime example. “The majority of vaccines are administered during well-child visit [sports or camp physicals]. … However, acute care visits or sick visits in the patient-centered medical home are also an opportunity to deliver vaccines or to discuss upcoming vaccines,” they said.
Education and communication are essential to improving vaccine uptake, the report concludes. “Appropriate techniques for approaching adolescent patients and their parents in the office to encourage immunizations are important skills for healthcare providers. The key to increasing immunization rates and decreasing vaccine-preventable disease … is to focus on educating adolescents and strengthening health care providers’ recommendations by using all clinical opportunities to assess immunization status and provide needed vaccinations.”
Neither Dr Bernstein nor Dr. Bocchini had any financial disclosures.
Need help talking about teen vaccines? Check out these resources
The American Academy of Pediatrics offered the following resources designed to help clinicians communicate effectively when talking to patients and parents about vaccines:
• Talking to parents about the HPV vaccine. This website hosted by the Centers for Disease Control and Prevention includes a link to the latest vaccination recommendations, as well as a tip sheet with HPV vaccine talking points, a fact sheet on the vaccine’s safety data, and a video with four clinical vignettes that model effective communication.
• The HPV Champion Toolkit. This contains numerous resources to help clinicians learn to educate other health care professionals, discuss HPV vaccination with parents, and make practice changes to increase HPV vaccine uptake.
• You Are the Key to HPV Prevention. This CDC video presents up-to-date information on HPV infection and disease, the HPV vaccine, and ways to successfully communicate with patients and their parents about HPV vaccination.
• Adolescentvaccination.org. The National Foundation for Infectious Diseases maintains a website is entirely devoted to adolescent vaccination issues. It contains resources for clinicians, and parents.
• Top Strategies for Increasing Vaccine Coverage. This is a report by the American Academy of Pediatrics.
• You Call the Shots. This is an interactive, Web-based immunization training course created by the CDC.
• Suggestions to Improve Your Immunization Services. This is a checklist of suggestions, by the Immunization Action.
[email protected]
On Twitter @Alz_Gal
Adolescents remain undervaccinated for several diseases, and clinicians need to lead the charge to change this reality, according to new clinical reports issued by the American Academy of Pediatrics Committee on Infectious Diseases.
Teens are lagging behind the Healthy People 2020 vaccination goals for the Tdap, quadrivalent meningococcal conjugate (menACWY), and human papillomavirus (HPV) vaccines, lead authors Henry H. Bernstein, DO, and Joseph A. Bocchini Jr., MD, wrote in the Feb. 6 issue of Pediatrics.
“Although HPV vaccination rates are slowly improving, they continue to lag far behind Tdap and menACWY rates for both boys and girls,” the authors wrote. Only 63% of girls and 50% of boys have gotten at least one HPV vaccine dose; just 42% of girls and 28% of boys finished the entire three-dose series in 2015.
A recent study found that parents declined the HPV vaccine 56% of the time. The three most common reasons were the belief that their child had a low risk of acquiring HPV; fear that the risk of adverse events was too great and belief that the vaccine wasn’t well researched and hasn’t been on the market long enough.
But teens also are falling behind with less controversial vaccines, the report noted. Fewer than half of teens got a flu shot in the 2015-2016 season. Rates for the MenACWY and Tdap are better, but not optimal (81% and 86%, respectively).
Barriers to optimal immunization
The clinical report puts clinicians on the first line of responsibility and makes no apologies for that.
“One of the greatest challenges is health care provider recommendation, which often lacks consistency and urgency,” the authors said. “Many health care providers do not universally recommend vaccines to eligible populations and do not offer concomitant vaccination with indicated vaccines during a single patient encounter.”
In fact, they noted, a recent physician survey found that only about 60% of pediatricians and family doctors strongly recommend the HPV vaccine for 11- and 12-year-old girls. Several parent surveys have determined that lack of provider recommendation is a leading reason for undervaccination among adolescents.
A companion clinical report, also authored by Dr. Bernstein and Dr. Bocchini, discusses practical ways to confront these barriers (Pediatrics. 2017 Feb 6. doi: 10.1542/peds.2016-4187). It opens with a strong call to physicians to lead the charge in increasing immunization rates.
“Up to 65% of parents have reported not receiving a recommendation … for immunizations. The major reason for nonreceipt [of Tdap and menACWY] was lack of a health care provider recommendation,” they said.
Overcoming parental vaccine hesitancy is a thorny struggle, the authors acknowledge. But they offer some helpful suggestions, including:
• Don’t offer immunizations as “optional.” This opens the door to vaccine dismissal. Instead, “strongly endorse all universally recommended vaccines as important for adolescents’ health.”
• Ask open-ended questions to get at the root of parental hesitancy. Give fact-based, medically sound information. Stress that doctors are parent partners in raising healthy children and protecting them from disease.
• Stress the big-picture benefits of vaccines. Telling parents that the HPV vaccine prevents cancer lifelong is a powerful message. “Up-to-date information on current events and disease outbreaks is a tool to bring into the conversation about vaccines as well.”
• Review the vaccine timeline. Do everything possible to ensure parents follow up and complete each series. “Follow-up immunization visits should be scheduled before the family leaves the care setting.”
• Don’t give up when a parent refuses a vaccine. “Although it may be challenging, it’s important that health care providers offer the vaccine at the next most appropriate time. Perseverance is critical for vaccine uptake and immunization rates.”
Dr. Bernstein and Dr. Bocchini also suggest that vaccine uptake could be boosted by a mental re-set of the office visit. “Missed opportunities for adolescent immunizations,” such as sick calls, are a prime example. “The majority of vaccines are administered during well-child visit [sports or camp physicals]. … However, acute care visits or sick visits in the patient-centered medical home are also an opportunity to deliver vaccines or to discuss upcoming vaccines,” they said.
Education and communication are essential to improving vaccine uptake, the report concludes. “Appropriate techniques for approaching adolescent patients and their parents in the office to encourage immunizations are important skills for healthcare providers. The key to increasing immunization rates and decreasing vaccine-preventable disease … is to focus on educating adolescents and strengthening health care providers’ recommendations by using all clinical opportunities to assess immunization status and provide needed vaccinations.”
Neither Dr Bernstein nor Dr. Bocchini had any financial disclosures.
Need help talking about teen vaccines? Check out these resources
The American Academy of Pediatrics offered the following resources designed to help clinicians communicate effectively when talking to patients and parents about vaccines:
• Talking to parents about the HPV vaccine. This website hosted by the Centers for Disease Control and Prevention includes a link to the latest vaccination recommendations, as well as a tip sheet with HPV vaccine talking points, a fact sheet on the vaccine’s safety data, and a video with four clinical vignettes that model effective communication.
• The HPV Champion Toolkit. This contains numerous resources to help clinicians learn to educate other health care professionals, discuss HPV vaccination with parents, and make practice changes to increase HPV vaccine uptake.
• You Are the Key to HPV Prevention. This CDC video presents up-to-date information on HPV infection and disease, the HPV vaccine, and ways to successfully communicate with patients and their parents about HPV vaccination.
• Adolescentvaccination.org. The National Foundation for Infectious Diseases maintains a website is entirely devoted to adolescent vaccination issues. It contains resources for clinicians, and parents.
• Top Strategies for Increasing Vaccine Coverage. This is a report by the American Academy of Pediatrics.
• You Call the Shots. This is an interactive, Web-based immunization training course created by the CDC.
• Suggestions to Improve Your Immunization Services. This is a checklist of suggestions, by the Immunization Action.
[email protected]
On Twitter @Alz_Gal
ACIP releases updated guidance for adult vaccinations
, according to the newly issued 2017 adult Recommended Immunization Schedule released by the CDC’s Advisory Committee on Immunization Practices (ACIP).
“Changes are related to concerns regarding low effectiveness of [LAIV] (FluMist, MedImmune) against influenza A(H1N1)pdm09 in the United States during the 2013-2014 and 2015-2016 influenza seasons,” wrote the authors of the report, published in Annals of Internal Medicine and led by David K. Kim, MD, of the CDC’s Immunization Services Division in Atlanta. ![]()
Another major change involves vaccination of adults with mild or severe egg allergy. The new guidance states that those with a mild egg allergy should receive either inactivated influenza vaccine or recombinant influenza vaccine, while those with severe allergies should be given one of the same vaccinations but only in a health care setting, so a clinician can monitor any signs of reaction and treat the patient accordingly.
Vaccine doses should be administered based on the patient’s age; a patient with “severe” egg allergy is one who exhibits angioedema, respiratory distress, lightheadedness, or recurrent emesis; requires epinephrine; or requires emergency medical care of any kind after consuming egg products.
Human papillomavirus (HPV) schedules have also been noticeably altered, with the CDC now considering all men and women through the ages of 21 and 26 years, respectively, who received a two-dose series of HPV vaccinations before the age of 15 to be adequately protected. Those who took only one of those two doses still need to take another dose, while men and women who have not been vaccinated should received a three-dose series at 0, 1-2, and 6 months.
Hepatitis B recommendations were also updated, with the CDC now advising: “Adults with chronic liver disease, including, but not limited to, hepatitis C virus infection, cirrhosis, fatty liver disease, alcoholic liver disease, autoimmune hepatitis, and an alanine aminotransferase (ALT) or aspartate aminotransferase (AST) level greater than twice the upper limit of normal, should receive a HepB series.”
Meningococcal vaccination guidelines also underwent a number of small changes pertaining to adults with anatomical or functional asplenia and human immunodeficiency virus, among other risk factors.
A number of small changes to the schedule chart were implemented to help make the immunization schedule more “clean and streamlined,” according to the CDC.
“Physicians should pay careful attention to the details found in the footnotes,” the CDC said in a statement. “The footnotes clarify who needs what vaccine, when, and at what dose.”
, according to the newly issued 2017 adult Recommended Immunization Schedule released by the CDC’s Advisory Committee on Immunization Practices (ACIP).
“Changes are related to concerns regarding low effectiveness of [LAIV] (FluMist, MedImmune) against influenza A(H1N1)pdm09 in the United States during the 2013-2014 and 2015-2016 influenza seasons,” wrote the authors of the report, published in Annals of Internal Medicine and led by David K. Kim, MD, of the CDC’s Immunization Services Division in Atlanta. ![]()
Another major change involves vaccination of adults with mild or severe egg allergy. The new guidance states that those with a mild egg allergy should receive either inactivated influenza vaccine or recombinant influenza vaccine, while those with severe allergies should be given one of the same vaccinations but only in a health care setting, so a clinician can monitor any signs of reaction and treat the patient accordingly.
Vaccine doses should be administered based on the patient’s age; a patient with “severe” egg allergy is one who exhibits angioedema, respiratory distress, lightheadedness, or recurrent emesis; requires epinephrine; or requires emergency medical care of any kind after consuming egg products.
Human papillomavirus (HPV) schedules have also been noticeably altered, with the CDC now considering all men and women through the ages of 21 and 26 years, respectively, who received a two-dose series of HPV vaccinations before the age of 15 to be adequately protected. Those who took only one of those two doses still need to take another dose, while men and women who have not been vaccinated should received a three-dose series at 0, 1-2, and 6 months.
Hepatitis B recommendations were also updated, with the CDC now advising: “Adults with chronic liver disease, including, but not limited to, hepatitis C virus infection, cirrhosis, fatty liver disease, alcoholic liver disease, autoimmune hepatitis, and an alanine aminotransferase (ALT) or aspartate aminotransferase (AST) level greater than twice the upper limit of normal, should receive a HepB series.”
Meningococcal vaccination guidelines also underwent a number of small changes pertaining to adults with anatomical or functional asplenia and human immunodeficiency virus, among other risk factors.
A number of small changes to the schedule chart were implemented to help make the immunization schedule more “clean and streamlined,” according to the CDC.
“Physicians should pay careful attention to the details found in the footnotes,” the CDC said in a statement. “The footnotes clarify who needs what vaccine, when, and at what dose.”
, according to the newly issued 2017 adult Recommended Immunization Schedule released by the CDC’s Advisory Committee on Immunization Practices (ACIP).
“Changes are related to concerns regarding low effectiveness of [LAIV] (FluMist, MedImmune) against influenza A(H1N1)pdm09 in the United States during the 2013-2014 and 2015-2016 influenza seasons,” wrote the authors of the report, published in Annals of Internal Medicine and led by David K. Kim, MD, of the CDC’s Immunization Services Division in Atlanta. ![]()
Another major change involves vaccination of adults with mild or severe egg allergy. The new guidance states that those with a mild egg allergy should receive either inactivated influenza vaccine or recombinant influenza vaccine, while those with severe allergies should be given one of the same vaccinations but only in a health care setting, so a clinician can monitor any signs of reaction and treat the patient accordingly.
Vaccine doses should be administered based on the patient’s age; a patient with “severe” egg allergy is one who exhibits angioedema, respiratory distress, lightheadedness, or recurrent emesis; requires epinephrine; or requires emergency medical care of any kind after consuming egg products.
Human papillomavirus (HPV) schedules have also been noticeably altered, with the CDC now considering all men and women through the ages of 21 and 26 years, respectively, who received a two-dose series of HPV vaccinations before the age of 15 to be adequately protected. Those who took only one of those two doses still need to take another dose, while men and women who have not been vaccinated should received a three-dose series at 0, 1-2, and 6 months.
Hepatitis B recommendations were also updated, with the CDC now advising: “Adults with chronic liver disease, including, but not limited to, hepatitis C virus infection, cirrhosis, fatty liver disease, alcoholic liver disease, autoimmune hepatitis, and an alanine aminotransferase (ALT) or aspartate aminotransferase (AST) level greater than twice the upper limit of normal, should receive a HepB series.”
Meningococcal vaccination guidelines also underwent a number of small changes pertaining to adults with anatomical or functional asplenia and human immunodeficiency virus, among other risk factors.
A number of small changes to the schedule chart were implemented to help make the immunization schedule more “clean and streamlined,” according to the CDC.
“Physicians should pay careful attention to the details found in the footnotes,” the CDC said in a statement. “The footnotes clarify who needs what vaccine, when, and at what dose.”
FROM ANNALS OF INTERNAL MEDICINE
Intentional weight loss protects against endometrial cancer
Among postmenopausal women, intentional weight loss protects against endometrial cancer, even if that loss is modest (5% or more), according to a report in the Journal of Clinical Oncology.
Endometrial cancer is the malignancy most strongly associated with obesity, but to date few studies have examined the effect of intentional weight loss on risk. Researchers analyzed data from the prospective Women’s Health Initiative to assess this relationship in a large, ethnically diverse population of postmenopausal women with detailed records on potentially confounding factors such as body mass index, smoking status, physical activity level, hormone therapy use, parity, age at menarche, age at first birth, and family history.
They focused on 36,793 women who participated in the WHI and who reported whether they had maintained a stable weight, lost 5% or more of their total weight, or gained 5% or more of their total weight during the first 3 years after enrollment. During a mean of 11.4 years of follow-up, 566 of these women developed endometrial cancer, said Juhua Luo, PhD, of the department of epidemiology and biostatistics, Indiana University School of Public Health, Bloomington, and her associates.
Women who lost weight intentionally showed a significantly lower risk of endometrial cancer than did those who had stable weight (HR, 0.71). This association was most pronounced among obese women: Those who lost at least 5% of their body weight intentionally showed a dramatic reduction in endometrial cancer risk (HR, 0.44). In contrast, unintentional weight loss was not associated with a lower risk of endometrial cancer. And women who gained 10 pounds or more during the first 3 years of the study showed a significantly higher risk of endometrial cancer, the investigators said (J Clin Oncol. 2017 Feb 6. doi: 10.1200/JCO.2016.70.5822).
In a sensitivity analysis, women who intentionally lost weight and achieved a normal BMI showed the same risk of endometrial cancer as that of those who had maintained a stable BMI throughout the study.
Luo et al. showed that women who deliberately lost 5% or more of their body weight during a 3-year period reduced their long-term risk of endometrial cancer by nearly 30%, and obese women did so by 56%. At a minimum, clinicians should inform their patients about this association, especially because the link between obesity and endometrial cancer is clearly underrecognized.
But given the difficulty of losing weight, more aggressive strategies may be beneficial. Bariatric surgery and the use of progestational agents such as levonorgestrel IUDs warrant consideration.
Screening for endometrial cancer currently is very limited, so it also should be reassessed. Endometrial sampling and transvaginal ultrasound have drawbacks, including a lack of sensitivity and specificity in asymptomatic women. If more novel strategies prove reliable and cost-effective, including uterine lavage and collection of exfoliated cells from the cervix, they can be widely adapted into clinical practice.
Jason D. Wright, MD, is in the division of gynecologic oncology at Columbia University, New York, and New York Presbyterian Hospital. He reported having no relevant financial disclosures. Dr. Wright made these remarks in an editorial accompanying Dr. Luo’s report (J Clin Oncol. 2017 Feb 6. doi: 10.1200/JCO.2016.71.7991).
Luo et al. showed that women who deliberately lost 5% or more of their body weight during a 3-year period reduced their long-term risk of endometrial cancer by nearly 30%, and obese women did so by 56%. At a minimum, clinicians should inform their patients about this association, especially because the link between obesity and endometrial cancer is clearly underrecognized.
But given the difficulty of losing weight, more aggressive strategies may be beneficial. Bariatric surgery and the use of progestational agents such as levonorgestrel IUDs warrant consideration.
Screening for endometrial cancer currently is very limited, so it also should be reassessed. Endometrial sampling and transvaginal ultrasound have drawbacks, including a lack of sensitivity and specificity in asymptomatic women. If more novel strategies prove reliable and cost-effective, including uterine lavage and collection of exfoliated cells from the cervix, they can be widely adapted into clinical practice.
Jason D. Wright, MD, is in the division of gynecologic oncology at Columbia University, New York, and New York Presbyterian Hospital. He reported having no relevant financial disclosures. Dr. Wright made these remarks in an editorial accompanying Dr. Luo’s report (J Clin Oncol. 2017 Feb 6. doi: 10.1200/JCO.2016.71.7991).
Luo et al. showed that women who deliberately lost 5% or more of their body weight during a 3-year period reduced their long-term risk of endometrial cancer by nearly 30%, and obese women did so by 56%. At a minimum, clinicians should inform their patients about this association, especially because the link between obesity and endometrial cancer is clearly underrecognized.
But given the difficulty of losing weight, more aggressive strategies may be beneficial. Bariatric surgery and the use of progestational agents such as levonorgestrel IUDs warrant consideration.
Screening for endometrial cancer currently is very limited, so it also should be reassessed. Endometrial sampling and transvaginal ultrasound have drawbacks, including a lack of sensitivity and specificity in asymptomatic women. If more novel strategies prove reliable and cost-effective, including uterine lavage and collection of exfoliated cells from the cervix, they can be widely adapted into clinical practice.
Jason D. Wright, MD, is in the division of gynecologic oncology at Columbia University, New York, and New York Presbyterian Hospital. He reported having no relevant financial disclosures. Dr. Wright made these remarks in an editorial accompanying Dr. Luo’s report (J Clin Oncol. 2017 Feb 6. doi: 10.1200/JCO.2016.71.7991).
Among postmenopausal women, intentional weight loss protects against endometrial cancer, even if that loss is modest (5% or more), according to a report in the Journal of Clinical Oncology.
Endometrial cancer is the malignancy most strongly associated with obesity, but to date few studies have examined the effect of intentional weight loss on risk. Researchers analyzed data from the prospective Women’s Health Initiative to assess this relationship in a large, ethnically diverse population of postmenopausal women with detailed records on potentially confounding factors such as body mass index, smoking status, physical activity level, hormone therapy use, parity, age at menarche, age at first birth, and family history.
They focused on 36,793 women who participated in the WHI and who reported whether they had maintained a stable weight, lost 5% or more of their total weight, or gained 5% or more of their total weight during the first 3 years after enrollment. During a mean of 11.4 years of follow-up, 566 of these women developed endometrial cancer, said Juhua Luo, PhD, of the department of epidemiology and biostatistics, Indiana University School of Public Health, Bloomington, and her associates.
Women who lost weight intentionally showed a significantly lower risk of endometrial cancer than did those who had stable weight (HR, 0.71). This association was most pronounced among obese women: Those who lost at least 5% of their body weight intentionally showed a dramatic reduction in endometrial cancer risk (HR, 0.44). In contrast, unintentional weight loss was not associated with a lower risk of endometrial cancer. And women who gained 10 pounds or more during the first 3 years of the study showed a significantly higher risk of endometrial cancer, the investigators said (J Clin Oncol. 2017 Feb 6. doi: 10.1200/JCO.2016.70.5822).
In a sensitivity analysis, women who intentionally lost weight and achieved a normal BMI showed the same risk of endometrial cancer as that of those who had maintained a stable BMI throughout the study.
Among postmenopausal women, intentional weight loss protects against endometrial cancer, even if that loss is modest (5% or more), according to a report in the Journal of Clinical Oncology.
Endometrial cancer is the malignancy most strongly associated with obesity, but to date few studies have examined the effect of intentional weight loss on risk. Researchers analyzed data from the prospective Women’s Health Initiative to assess this relationship in a large, ethnically diverse population of postmenopausal women with detailed records on potentially confounding factors such as body mass index, smoking status, physical activity level, hormone therapy use, parity, age at menarche, age at first birth, and family history.
They focused on 36,793 women who participated in the WHI and who reported whether they had maintained a stable weight, lost 5% or more of their total weight, or gained 5% or more of their total weight during the first 3 years after enrollment. During a mean of 11.4 years of follow-up, 566 of these women developed endometrial cancer, said Juhua Luo, PhD, of the department of epidemiology and biostatistics, Indiana University School of Public Health, Bloomington, and her associates.
Women who lost weight intentionally showed a significantly lower risk of endometrial cancer than did those who had stable weight (HR, 0.71). This association was most pronounced among obese women: Those who lost at least 5% of their body weight intentionally showed a dramatic reduction in endometrial cancer risk (HR, 0.44). In contrast, unintentional weight loss was not associated with a lower risk of endometrial cancer. And women who gained 10 pounds or more during the first 3 years of the study showed a significantly higher risk of endometrial cancer, the investigators said (J Clin Oncol. 2017 Feb 6. doi: 10.1200/JCO.2016.70.5822).
In a sensitivity analysis, women who intentionally lost weight and achieved a normal BMI showed the same risk of endometrial cancer as that of those who had maintained a stable BMI throughout the study.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Among postmenopausal women, intentional weight loss protects against endometrial cancer, even if that loss is modest (5% or more).
Major finding: Women who intentionally lost at least 5% of their total body weight showed a significantly lower risk of endometrial cancer than did those who had stable weight (HR, 0.71), and obese women showed a dramatically reduced risk (HR, 0.44).
Data source: A secondary analysis of data from 36,793 women participating in the Women’s Health Initiative.
Disclosures: No sponsor was cited for this study. Dr. Luo reported having no relevant financial disclosures; her associates reported ties to numerous industry sources.