User login
Better stroke treatment moves tantalizingly within reach
Stroke is one of the most feared medical conditions, with the specter of suddenly finding oneself unable to talk, eat, walk, or live independently, according to study results.
In mid-February, results from three trials reported at the International Stroke Conference in Nashville, Tenn., changed the face of ischemic stroke treatment by proving that emergency endovascular catheterization to remove the embolus blocking cerebral blood flow produced better long-term outcomes than standard treatment with intravenous thrombolysis.
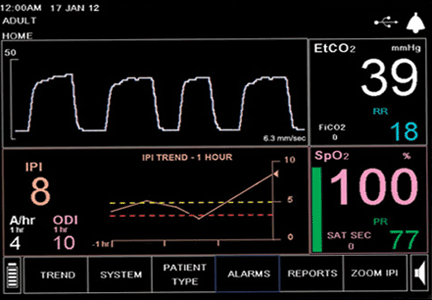
It wasn’t just that patients did better with endovascular embolectomy; it was how much they did better. In the two trials run in the United States and abroad, SWIFT PRIME and ESCAPE, the percentage of patients rated as not disabled (a modified Rankin Scale score of 0-1) when assessed after 90 days was 36% and 42% for patients treated with endovascular therapy in the two studies, compared with 17% and 19% in the two control arms. Embolectomy boosted the fraction of patients having the best stroke outcomes more than twofold, a breathtaking leap in efficacy.
Dr. Jeffrey L. Saver from UCLA, lead investigator for SWIFT PRIME, called it a “once-in-a-field” result, meaning that never again will stroke clinicians see this degree of incremental improvement by adding a new intervention.
The frustrating irony is how challenging delivery of this disease-altering treatment will be on a national scale. One problem is that it didn’t result from a single change in treatment, but from a careful mix of new diagnostic techniques with sophisticated CT imaging, new systems for expediting diagnosis, triage, transport, and treatment, in combination with new technology in the form of emboli-retrieving stents.

Stroke management specialists see a daunting series of issues to tackle as they attempt to roll out emergency endovascular interventions on a routine scale throughout much of the United States. Many more centers must open, modeled on the ones that succeeded in the trials. The centers need to be rationally positioned so they are close to patients but also give each center enough case volume to foster high interventional-skill levels. Staffing must be found for fast-moving stroke response teams that can make the diagnostics and interventions available around the clock and interpret the images to select appropriate patients. Ambulance systems have to be set up that take likely stroke patients to the centers that will best meet their treatment needs.
The stroke and public health communities will need to invest a lot of time, money, and leadership to make this happen, but it’s a clear mandate, given the promise endovascular treatment now holds to blunt the impact of one of medicine’s most feared maladies.
On Twitter @mitchelzoler
Stroke is one of the most feared medical conditions, with the specter of suddenly finding oneself unable to talk, eat, walk, or live independently, according to study results.
In mid-February, results from three trials reported at the International Stroke Conference in Nashville, Tenn., changed the face of ischemic stroke treatment by proving that emergency endovascular catheterization to remove the embolus blocking cerebral blood flow produced better long-term outcomes than standard treatment with intravenous thrombolysis.
It wasn’t just that patients did better with endovascular embolectomy; it was how much they did better. In the two trials run in the United States and abroad, SWIFT PRIME and ESCAPE, the percentage of patients rated as not disabled (a modified Rankin Scale score of 0-1) when assessed after 90 days was 36% and 42% for patients treated with endovascular therapy in the two studies, compared with 17% and 19% in the two control arms. Embolectomy boosted the fraction of patients having the best stroke outcomes more than twofold, a breathtaking leap in efficacy.
Dr. Jeffrey L. Saver from UCLA, lead investigator for SWIFT PRIME, called it a “once-in-a-field” result, meaning that never again will stroke clinicians see this degree of incremental improvement by adding a new intervention.
The frustrating irony is how challenging delivery of this disease-altering treatment will be on a national scale. One problem is that it didn’t result from a single change in treatment, but from a careful mix of new diagnostic techniques with sophisticated CT imaging, new systems for expediting diagnosis, triage, transport, and treatment, in combination with new technology in the form of emboli-retrieving stents.
Stroke management specialists see a daunting series of issues to tackle as they attempt to roll out emergency endovascular interventions on a routine scale throughout much of the United States. Many more centers must open, modeled on the ones that succeeded in the trials. The centers need to be rationally positioned so they are close to patients but also give each center enough case volume to foster high interventional-skill levels. Staffing must be found for fast-moving stroke response teams that can make the diagnostics and interventions available around the clock and interpret the images to select appropriate patients. Ambulance systems have to be set up that take likely stroke patients to the centers that will best meet their treatment needs.
The stroke and public health communities will need to invest a lot of time, money, and leadership to make this happen, but it’s a clear mandate, given the promise endovascular treatment now holds to blunt the impact of one of medicine’s most feared maladies.
On Twitter @mitchelzoler
Stroke is one of the most feared medical conditions, with the specter of suddenly finding oneself unable to talk, eat, walk, or live independently, according to study results.
In mid-February, results from three trials reported at the International Stroke Conference in Nashville, Tenn., changed the face of ischemic stroke treatment by proving that emergency endovascular catheterization to remove the embolus blocking cerebral blood flow produced better long-term outcomes than standard treatment with intravenous thrombolysis.
It wasn’t just that patients did better with endovascular embolectomy; it was how much they did better. In the two trials run in the United States and abroad, SWIFT PRIME and ESCAPE, the percentage of patients rated as not disabled (a modified Rankin Scale score of 0-1) when assessed after 90 days was 36% and 42% for patients treated with endovascular therapy in the two studies, compared with 17% and 19% in the two control arms. Embolectomy boosted the fraction of patients having the best stroke outcomes more than twofold, a breathtaking leap in efficacy.
Dr. Jeffrey L. Saver from UCLA, lead investigator for SWIFT PRIME, called it a “once-in-a-field” result, meaning that never again will stroke clinicians see this degree of incremental improvement by adding a new intervention.
The frustrating irony is how challenging delivery of this disease-altering treatment will be on a national scale. One problem is that it didn’t result from a single change in treatment, but from a careful mix of new diagnostic techniques with sophisticated CT imaging, new systems for expediting diagnosis, triage, transport, and treatment, in combination with new technology in the form of emboli-retrieving stents.
Stroke management specialists see a daunting series of issues to tackle as they attempt to roll out emergency endovascular interventions on a routine scale throughout much of the United States. Many more centers must open, modeled on the ones that succeeded in the trials. The centers need to be rationally positioned so they are close to patients but also give each center enough case volume to foster high interventional-skill levels. Staffing must be found for fast-moving stroke response teams that can make the diagnostics and interventions available around the clock and interpret the images to select appropriate patients. Ambulance systems have to be set up that take likely stroke patients to the centers that will best meet their treatment needs.
The stroke and public health communities will need to invest a lot of time, money, and leadership to make this happen, but it’s a clear mandate, given the promise endovascular treatment now holds to blunt the impact of one of medicine’s most feared maladies.
On Twitter @mitchelzoler
Diabetes therapy and cardiac risk
To the Editor: Recently, Drs. Zimmerman and Pantalone1 cited the Diabetes Control and Complications Trial (DCCT)2 and the United Kingdom Prospective Diabetes Study (UKPDS)3 as evidence that glycemic control lowers cardiac risk in type 2 diabetes. And in a related counterpoint article, Drs. Menon and Aggarwal4 also discussed the UKPDS.
These studies should not be cited in this context, since the DCCT is a study of type 1 and not type 2 diabetic patients, and the UKPDS was performed in an era when statins were not available. The UKPDS was launched in 1977 and completed in 1997, and statins were not available until 1987. Indeed, the UKPDS showed that the strongest risk factor for myocardial infarction was an elevated level of low-density lipoprotein cholesterol, followed by a low level of high-density lipoprotein cholesterol.5 It is therefore not surprising that in the initial UKPDS report the incidence of myocardial infarction was not increased in the group with a 0.9% higher hemoglobin A1c, but that in the 10-year follow-up, when statins were probably used by most patients, myocardial infarction was reduced by a significant 15% (P = .01).3,6 As would be expected in the more modern studies, ie, the Action to Control Cardiovascular Risk (ACCORD),7 the Action in Diabetes and Vascular Disease (ADVANCE),8 and the Veteran Affairs Diabetes Trial (VADT),9 cardiovascular events were not reduced with improved glycemic control.
While the UKPDS clearly demonstrated a decrease in microvascular disease due to improved glycemic control, it should not be used as evidence that improved glycemic control in type 2 diabetes decreases cardiac events.3,6
- Zimmerman RS, Pantalone KM. Diabetes management: more than just cardiovascular risk? Cleve Clin J Med 2014; 81:672–676.
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977–986.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352:837–853.
- Menon V, Aggarwal B. Why are we doing cardiovascular outcome trials in type 2 diabetes? Cleve Clin J Med 2014; 81:665–671.
- Turner RC, Millns H, Neil HA, et al. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: United Kingdom Prospective Diabetes Study (UKPDS: 23). BMJ 1998; 316:823–828.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- Duckworth W, Abraira C, Moritz T, et al; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009; 360:129–139.
To the Editor: Recently, Drs. Zimmerman and Pantalone1 cited the Diabetes Control and Complications Trial (DCCT)2 and the United Kingdom Prospective Diabetes Study (UKPDS)3 as evidence that glycemic control lowers cardiac risk in type 2 diabetes. And in a related counterpoint article, Drs. Menon and Aggarwal4 also discussed the UKPDS.
These studies should not be cited in this context, since the DCCT is a study of type 1 and not type 2 diabetic patients, and the UKPDS was performed in an era when statins were not available. The UKPDS was launched in 1977 and completed in 1997, and statins were not available until 1987. Indeed, the UKPDS showed that the strongest risk factor for myocardial infarction was an elevated level of low-density lipoprotein cholesterol, followed by a low level of high-density lipoprotein cholesterol.5 It is therefore not surprising that in the initial UKPDS report the incidence of myocardial infarction was not increased in the group with a 0.9% higher hemoglobin A1c, but that in the 10-year follow-up, when statins were probably used by most patients, myocardial infarction was reduced by a significant 15% (P = .01).3,6 As would be expected in the more modern studies, ie, the Action to Control Cardiovascular Risk (ACCORD),7 the Action in Diabetes and Vascular Disease (ADVANCE),8 and the Veteran Affairs Diabetes Trial (VADT),9 cardiovascular events were not reduced with improved glycemic control.
While the UKPDS clearly demonstrated a decrease in microvascular disease due to improved glycemic control, it should not be used as evidence that improved glycemic control in type 2 diabetes decreases cardiac events.3,6
To the Editor: Recently, Drs. Zimmerman and Pantalone1 cited the Diabetes Control and Complications Trial (DCCT)2 and the United Kingdom Prospective Diabetes Study (UKPDS)3 as evidence that glycemic control lowers cardiac risk in type 2 diabetes. And in a related counterpoint article, Drs. Menon and Aggarwal4 also discussed the UKPDS.
These studies should not be cited in this context, since the DCCT is a study of type 1 and not type 2 diabetic patients, and the UKPDS was performed in an era when statins were not available. The UKPDS was launched in 1977 and completed in 1997, and statins were not available until 1987. Indeed, the UKPDS showed that the strongest risk factor for myocardial infarction was an elevated level of low-density lipoprotein cholesterol, followed by a low level of high-density lipoprotein cholesterol.5 It is therefore not surprising that in the initial UKPDS report the incidence of myocardial infarction was not increased in the group with a 0.9% higher hemoglobin A1c, but that in the 10-year follow-up, when statins were probably used by most patients, myocardial infarction was reduced by a significant 15% (P = .01).3,6 As would be expected in the more modern studies, ie, the Action to Control Cardiovascular Risk (ACCORD),7 the Action in Diabetes and Vascular Disease (ADVANCE),8 and the Veteran Affairs Diabetes Trial (VADT),9 cardiovascular events were not reduced with improved glycemic control.
While the UKPDS clearly demonstrated a decrease in microvascular disease due to improved glycemic control, it should not be used as evidence that improved glycemic control in type 2 diabetes decreases cardiac events.3,6
- Zimmerman RS, Pantalone KM. Diabetes management: more than just cardiovascular risk? Cleve Clin J Med 2014; 81:672–676.
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977–986.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352:837–853.
- Menon V, Aggarwal B. Why are we doing cardiovascular outcome trials in type 2 diabetes? Cleve Clin J Med 2014; 81:665–671.
- Turner RC, Millns H, Neil HA, et al. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: United Kingdom Prospective Diabetes Study (UKPDS: 23). BMJ 1998; 316:823–828.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- Duckworth W, Abraira C, Moritz T, et al; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009; 360:129–139.
- Zimmerman RS, Pantalone KM. Diabetes management: more than just cardiovascular risk? Cleve Clin J Med 2014; 81:672–676.
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977–986.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352:837–853.
- Menon V, Aggarwal B. Why are we doing cardiovascular outcome trials in type 2 diabetes? Cleve Clin J Med 2014; 81:665–671.
- Turner RC, Millns H, Neil HA, et al. Risk factors for coronary artery disease in non-insulin dependent diabetes mellitus: United Kingdom Prospective Diabetes Study (UKPDS: 23). BMJ 1998; 316:823–828.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- Duckworth W, Abraira C, Moritz T, et al; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009; 360:129–139.
In reply: Diabetes therapy and cardiac risk
In Reply: We appreciate Dr. Bell’s interest in and comments regarding our recent article. Dr. Bell contends that the DCCT1 and UKPDS2 studies should not be cited since the DCCT is a study of type 1 and not type 2 diabetic patients, and the UKPDS was performed in an era when statins were not available.
While we can appreciate his point of view, we disagree with his interpretation of the available data. These studies, and their respective observational follow-up reports,3,4 provide evidence that early intervention may reduce cardiovascular risk, and that our approach to examining cardiovascular risk reduction in high-risk cardiovascular patients, as in ACCORD,5 ADVANCE,6 and VADT,7 may be short-sighted. There is an important difference between reducing long-term cardiovascular risk by treating younger and healthier patients with diabetes (type 1 or type 2) early in the disease course, before the development of complications (including cardiovascular disease), as was the case in DCCT and UKPDS, vs treating older patients with diabetes who have established cardiovascular disease or who have numerous risk factors substantially increasing their cardiovascular risk, as in ACCORD, ADVANCE, and VADT.
To his second point, that the UKPDS did not demonstrate cardiovascular risk reduction until after the 10-year follow-up when statins were probably utilized by the vast majority of patients, there would not have been a difference in cardiac events between treatment and control groups during this observational period if the statins were the cause of the reduced rate of cardiac events. The control and treatment groups would have had the same reduction in events. That was not the case. The finding of a lower risk of myocardial infarction at the completion of the follow-up period, despite ubiquitous statin use by both the treatment and control groups during this 10-year period, suggests another variable—ie, that the early differences in glycemic control achieved between the treatment and control groups during the UKPDS was responsible for the observed reduction in the risk of myocardial infarction.
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977–986.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352:837–853.
- Nathan DM, Cleary PA, Backlund JY, et al; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005; 353:2643–2653.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- Duckworth W, Abraira C, Moritz T, et al; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009; 360:129–139.
In Reply: We appreciate Dr. Bell’s interest in and comments regarding our recent article. Dr. Bell contends that the DCCT1 and UKPDS2 studies should not be cited since the DCCT is a study of type 1 and not type 2 diabetic patients, and the UKPDS was performed in an era when statins were not available.
While we can appreciate his point of view, we disagree with his interpretation of the available data. These studies, and their respective observational follow-up reports,3,4 provide evidence that early intervention may reduce cardiovascular risk, and that our approach to examining cardiovascular risk reduction in high-risk cardiovascular patients, as in ACCORD,5 ADVANCE,6 and VADT,7 may be short-sighted. There is an important difference between reducing long-term cardiovascular risk by treating younger and healthier patients with diabetes (type 1 or type 2) early in the disease course, before the development of complications (including cardiovascular disease), as was the case in DCCT and UKPDS, vs treating older patients with diabetes who have established cardiovascular disease or who have numerous risk factors substantially increasing their cardiovascular risk, as in ACCORD, ADVANCE, and VADT.
To his second point, that the UKPDS did not demonstrate cardiovascular risk reduction until after the 10-year follow-up when statins were probably utilized by the vast majority of patients, there would not have been a difference in cardiac events between treatment and control groups during this observational period if the statins were the cause of the reduced rate of cardiac events. The control and treatment groups would have had the same reduction in events. That was not the case. The finding of a lower risk of myocardial infarction at the completion of the follow-up period, despite ubiquitous statin use by both the treatment and control groups during this 10-year period, suggests another variable—ie, that the early differences in glycemic control achieved between the treatment and control groups during the UKPDS was responsible for the observed reduction in the risk of myocardial infarction.
In Reply: We appreciate Dr. Bell’s interest in and comments regarding our recent article. Dr. Bell contends that the DCCT1 and UKPDS2 studies should not be cited since the DCCT is a study of type 1 and not type 2 diabetic patients, and the UKPDS was performed in an era when statins were not available.
While we can appreciate his point of view, we disagree with his interpretation of the available data. These studies, and their respective observational follow-up reports,3,4 provide evidence that early intervention may reduce cardiovascular risk, and that our approach to examining cardiovascular risk reduction in high-risk cardiovascular patients, as in ACCORD,5 ADVANCE,6 and VADT,7 may be short-sighted. There is an important difference between reducing long-term cardiovascular risk by treating younger and healthier patients with diabetes (type 1 or type 2) early in the disease course, before the development of complications (including cardiovascular disease), as was the case in DCCT and UKPDS, vs treating older patients with diabetes who have established cardiovascular disease or who have numerous risk factors substantially increasing their cardiovascular risk, as in ACCORD, ADVANCE, and VADT.
To his second point, that the UKPDS did not demonstrate cardiovascular risk reduction until after the 10-year follow-up when statins were probably utilized by the vast majority of patients, there would not have been a difference in cardiac events between treatment and control groups during this observational period if the statins were the cause of the reduced rate of cardiac events. The control and treatment groups would have had the same reduction in events. That was not the case. The finding of a lower risk of myocardial infarction at the completion of the follow-up period, despite ubiquitous statin use by both the treatment and control groups during this 10-year period, suggests another variable—ie, that the early differences in glycemic control achieved between the treatment and control groups during the UKPDS was responsible for the observed reduction in the risk of myocardial infarction.
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977–986.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352:837–853.
- Nathan DM, Cleary PA, Backlund JY, et al; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005; 353:2643–2653.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- Duckworth W, Abraira C, Moritz T, et al; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009; 360:129–139.
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977–986.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352:837–853.
- Nathan DM, Cleary PA, Backlund JY, et al; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005; 353:2643–2653.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008; 359:1577–1589.
- Action to Control Cardiovascular Risk in Diabetes Study Group; Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358:2545–2559.
- ADVANCE Collaborative Group; Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358:2560–2572.
- Duckworth W, Abraira C, Moritz T, et al; VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009; 360:129–139.
Epithelial Ovarian Cancer: Evaluation, Staging, Surgery, and Stage I and II Disease Management
Edited by: Arthur T. Skarin, MD, FACP, FCCP
Ovarian cancer is the second most common gynecologic cancer among women in the United States. It is also the fifth leading cause of cancer mortality in women and the leading cause of death among women with gynecologic malignancies. The American Cancer Society statistics released in 2015 estimate that 21,290 new cases of ovarian cancer will occur during the year, with approximately 14,180 deaths. Globally, there were 238,719 new cases of ovarian cancer diagnosed in 2012, representing 3.6% of all cancers in women, and nearly 151,905 deaths. The highest incidence of ovarian cancer occurs in northern, central, and eastern Europe, followed by western Europe and North America, with the lowest incidence in parts of Africa and Asia. The majority of women presenting with ovarian cancer will present at an advanced stage, and the 5-year survival in this group is less than 30%.
To read the full article in PDF:
Edited by: Arthur T. Skarin, MD, FACP, FCCP
Ovarian cancer is the second most common gynecologic cancer among women in the United States. It is also the fifth leading cause of cancer mortality in women and the leading cause of death among women with gynecologic malignancies. The American Cancer Society statistics released in 2015 estimate that 21,290 new cases of ovarian cancer will occur during the year, with approximately 14,180 deaths. Globally, there were 238,719 new cases of ovarian cancer diagnosed in 2012, representing 3.6% of all cancers in women, and nearly 151,905 deaths. The highest incidence of ovarian cancer occurs in northern, central, and eastern Europe, followed by western Europe and North America, with the lowest incidence in parts of Africa and Asia. The majority of women presenting with ovarian cancer will present at an advanced stage, and the 5-year survival in this group is less than 30%.
To read the full article in PDF:
Edited by: Arthur T. Skarin, MD, FACP, FCCP
Ovarian cancer is the second most common gynecologic cancer among women in the United States. It is also the fifth leading cause of cancer mortality in women and the leading cause of death among women with gynecologic malignancies. The American Cancer Society statistics released in 2015 estimate that 21,290 new cases of ovarian cancer will occur during the year, with approximately 14,180 deaths. Globally, there were 238,719 new cases of ovarian cancer diagnosed in 2012, representing 3.6% of all cancers in women, and nearly 151,905 deaths. The highest incidence of ovarian cancer occurs in northern, central, and eastern Europe, followed by western Europe and North America, with the lowest incidence in parts of Africa and Asia. The majority of women presenting with ovarian cancer will present at an advanced stage, and the 5-year survival in this group is less than 30%.
To read the full article in PDF:
Drug seems promising for kids with severe hemophilia B
Results of a phase 3 study suggest a recombinant factor IX Fc fusion protein (rFIXFc, also known as eftrenonacog alfa and Alprolix) is a feasible treatment option for children with severe hemophilia B.
rFIXFc effectively prevented and treated bleeding episodes, patients did not develop inhibitors, and there were no serious adverse events related to treatment.
Sobi and Biogen Idec, the companies developing rFIXFc, recently announced these results from the now-complete Kids B-LONG study.
They said the successful completion of this study supports applications for pediatric indications in several regions and is an important step in seeking marketing authorization for rFIXFc in Europe.
Interim results of the Kids B-LONG study helped support the US approval of rFIXFc for use in children.
In Kids B-LONG, researchers tested rFIXFc in 30 previously treated children younger than 12 who had severe hemophilia B. Patients had at least 50 prior exposure days to factor IX therapies.
Twenty-seven patients (90%) completed the study. The median time spent on study was 49.4 weeks, and 24 participants received rFIXFc injections on at least 50 separate days.
Children who received rFIXFc prophylactically had an overall median annualized bleeding rate (ABR) of 1.97. The median ABR for spontaneous joint bleeds was 0.
Approximately 33% of patients did not experience any bleeding episodes. About 92% of bleeding episodes were controlled by 1 or 2 injections of rFIXFc.
None of the patients developed inhibitors to rFIXFc. The terminal half-life of the product was 66.5 hours for children under 6 and 70.3 hours for children ages 6 to 11.
Researchers said there were no treatment-related serious adverse events and no cases of serious allergic reactions or vascular thrombotic events. None of the patients discontinued the study due to an adverse event.
One adverse event—decreased appetite occurring in 1 patient—was considered related to rFIXFc treatment.
The pattern of treatment-emergent adverse events in this study was generally consistent with results seen in adolescents and adults in the phase 3 B-LONG study. Common adverse reactions in that study were headache and oral paresthesia.
Additional analyses of the Kids B-LONG study are ongoing, and detailed results will be presented at a future scientific meeting, according to Sobi and Biogen Idec. ![]()
Results of a phase 3 study suggest a recombinant factor IX Fc fusion protein (rFIXFc, also known as eftrenonacog alfa and Alprolix) is a feasible treatment option for children with severe hemophilia B.
rFIXFc effectively prevented and treated bleeding episodes, patients did not develop inhibitors, and there were no serious adverse events related to treatment.
Sobi and Biogen Idec, the companies developing rFIXFc, recently announced these results from the now-complete Kids B-LONG study.
They said the successful completion of this study supports applications for pediatric indications in several regions and is an important step in seeking marketing authorization for rFIXFc in Europe.
Interim results of the Kids B-LONG study helped support the US approval of rFIXFc for use in children.
In Kids B-LONG, researchers tested rFIXFc in 30 previously treated children younger than 12 who had severe hemophilia B. Patients had at least 50 prior exposure days to factor IX therapies.
Twenty-seven patients (90%) completed the study. The median time spent on study was 49.4 weeks, and 24 participants received rFIXFc injections on at least 50 separate days.
Children who received rFIXFc prophylactically had an overall median annualized bleeding rate (ABR) of 1.97. The median ABR for spontaneous joint bleeds was 0.
Approximately 33% of patients did not experience any bleeding episodes. About 92% of bleeding episodes were controlled by 1 or 2 injections of rFIXFc.
None of the patients developed inhibitors to rFIXFc. The terminal half-life of the product was 66.5 hours for children under 6 and 70.3 hours for children ages 6 to 11.
Researchers said there were no treatment-related serious adverse events and no cases of serious allergic reactions or vascular thrombotic events. None of the patients discontinued the study due to an adverse event.
One adverse event—decreased appetite occurring in 1 patient—was considered related to rFIXFc treatment.
The pattern of treatment-emergent adverse events in this study was generally consistent with results seen in adolescents and adults in the phase 3 B-LONG study. Common adverse reactions in that study were headache and oral paresthesia.
Additional analyses of the Kids B-LONG study are ongoing, and detailed results will be presented at a future scientific meeting, according to Sobi and Biogen Idec. ![]()
Results of a phase 3 study suggest a recombinant factor IX Fc fusion protein (rFIXFc, also known as eftrenonacog alfa and Alprolix) is a feasible treatment option for children with severe hemophilia B.
rFIXFc effectively prevented and treated bleeding episodes, patients did not develop inhibitors, and there were no serious adverse events related to treatment.
Sobi and Biogen Idec, the companies developing rFIXFc, recently announced these results from the now-complete Kids B-LONG study.
They said the successful completion of this study supports applications for pediatric indications in several regions and is an important step in seeking marketing authorization for rFIXFc in Europe.
Interim results of the Kids B-LONG study helped support the US approval of rFIXFc for use in children.
In Kids B-LONG, researchers tested rFIXFc in 30 previously treated children younger than 12 who had severe hemophilia B. Patients had at least 50 prior exposure days to factor IX therapies.
Twenty-seven patients (90%) completed the study. The median time spent on study was 49.4 weeks, and 24 participants received rFIXFc injections on at least 50 separate days.
Children who received rFIXFc prophylactically had an overall median annualized bleeding rate (ABR) of 1.97. The median ABR for spontaneous joint bleeds was 0.
Approximately 33% of patients did not experience any bleeding episodes. About 92% of bleeding episodes were controlled by 1 or 2 injections of rFIXFc.
None of the patients developed inhibitors to rFIXFc. The terminal half-life of the product was 66.5 hours for children under 6 and 70.3 hours for children ages 6 to 11.
Researchers said there were no treatment-related serious adverse events and no cases of serious allergic reactions or vascular thrombotic events. None of the patients discontinued the study due to an adverse event.
One adverse event—decreased appetite occurring in 1 patient—was considered related to rFIXFc treatment.
The pattern of treatment-emergent adverse events in this study was generally consistent with results seen in adolescents and adults in the phase 3 B-LONG study. Common adverse reactions in that study were headache and oral paresthesia.
Additional analyses of the Kids B-LONG study are ongoing, and detailed results will be presented at a future scientific meeting, according to Sobi and Biogen Idec. ![]()
Stress independently predicts peptic ulcers
High levels of psychological stress more than doubled the odds of peptic ulcers, and the link remained statistically significant even after controlling for factors such as Helicobacter pylori infection and cigarette smoking, according to a prospective study published in the March issue of Clinical Gastroenterology and Hepatology.
The findings contradict the widely accepted view that stress does not cause peptic ulcers, said Dr. Susan Levenstein of Aventino Medical Group in Rome and her associates. “Clinicians treating ulcer patients should investigate potential psychological stress among other risk factors,” they said.
Source: American Gastroenterological Association
Although “a vast literature links peptic ulcer to stress,” past studies suffered so many methodologic weaknesses that groups such as the U.S. National Institute of Diabetes and Digestive and Kidney Diseases rejected the evidence outright, Dr. Levenstein and her associates noted. Many studies were cross-sectional, for example, or did not control for confounders such as helicobacteriosis, they said.
To further study the effects of stress on ulcer risk, the researchers analyzed historical data from 76 patients who lacked a history of gastric and duodenal ulcers in 1982, but by 1994 had developed “distinct breach[es] in the mucosa” that were confirmed by endoscopy or contrast radiology. The researchers did not count erosions that lacked appreciable depth as ulcers, they noted (Clin. Gastroenterol. Hepatol. 2014 Aug. 8 [doi:10.1016/j.cgh.2014.07.052]).
Study subjects answered 12 questions about their stress levels, such as, “Do your hands easily shake?” “Do you often suffer from fits of dizziness?” “Do you constantly have thoughts that trouble and worry you?” and “Do you usually feel misunderstood by other people?” They answered these questions at baseline in 1982-1983, again in 1987-1988, and again in 1993-1994.
Respondents who scored in the top tertile for psychological stress had an ulcer incidence of 3.5%, compared with 1.6% for those in the lowest tertile (odds ratio, 2.2; 95% confidence interval, 1.2-3.9; P < .01), reported the investigators. And controlling for smoking, helicobacteriosis, use of nonsteroidal anti-inflammatory drugs, and low socioeconomic status only partially weakened the relationship between stress and ulcers, they said. After accounting for those risk factors, every one-point increase on the stress questionnaire still upped the odds of peptic ulcer by 12% (odds ratio, 1.12; 95% confidence interval, 1.01-1.23)they reported.
Helicobacteri pylori infection was the strongest independent predictor of ulcers (OR, 3.3; 95% CI, 2.02-5.69), while cigarette smoking came in a close second (OR, 2.91; 95% CI, 1.38-6.16), said the researchers. Notably, stress and helicobacteriosis did not seem to synergistically increase the chances of ulcers, they reported. “Stress affected H. pylori–related ulcers at least as much as those related to neither H. pylori nor nonsteroidal anti-inflammatory drugs,” they said.
Several factors might explain the stress-ulcer link, such as increased acid load, activation of the hypothalamic-pituitary-adrenal axis, shifts in blood flow, and cytokine activation that might impair gastrointestinal mucosal defenses, said the investigators. Although the baseline data in their study were more than 2 decades old, that meant that patients likely had not been treated to eradicate H. pylori and were less likely to have taken proton pump inhibitors than the current population that has over-the-counter access to PPIs, they added. They also noted that past studies found a particularly strong link between stress and bleeding or perforated ulcers, which have not declined as much as other types of ulcers. “These results support a multicausal model of peptic ulcer etiology, with intertwined biological and psychosocial components,” they concluded.
The Kirby Family Foundation funded the statistical analysis. The researchers reported no conflicts of interest.
Stress was the most frequently cited cause of ulcer disease before Helicobacter pylori was discovered. The harried executive who developed an ulcer was a widely accepted profile of an ulcer diathesis. When the role of H. pylori infection and NSAIDs became clear, the role of stress was downplayed and some articles and textbooks dismissed stress as a potential cause for ulcer disease.

|
Dr. Nimish Vakil |
Studies of New York City residents suggest a higher incidence of ulcer disease after the 9-11 attacks and studies from Japan have shown an increase in the incidence of ulcer disease after the nuclear reactor disaster. In this issue of Clinical Gastroenterology and Hepatology, Dr. Levenstein and her colleagues report the results of a study of stress and the incidence of ulcer disease in Danish subjects. In 1982-1983, a population-based study in Denmark collected sera and psychological data in over 3000 subjects and reinterviewed them in 1987-1988 and 1993-1994. An ad-hoc, unvalidated scale developed by the authors measured stress. It included a psychological scale used by the Danish military to identify recruits unsuitable for military service but also included tranquilizer use, working more than 40 hours a week, and unemployment. In multivariate analysis, they found that stress increased the risk for both gastric and duodenal ulcers, with an adjusted odds ratio of 1.19 per point increase in the stress scale for gastric ulcers (95% confidence interval, 1.03-1.37) and a odds ratio of 1.1 per point increase in the stress index for duodenal ulcers (95% CI, 0.98-1.27).
There are obvious limitations with this study: a historical cohort, an unvalidated stress scale, the inclusion of items that may not represent stress in some cultures (e.g., working more than 40 hours/week) and the lower bound of confidence intervals for risk which are very close to one. However, studies such as this tell us that we have been too quick to dismiss the role of stress in ulcer pathogenesis. With declining H. pylori prevalence and the development of safer NSAIDs, stress will undergo a renaissance in the pathogenesis of ulcer disease.
Dr. Nimish Vakil, AGAF, FASGE, FACP, is a physician specializing in gastroenterology at the Aurora Wilkinson Medical Clinic in Summit, Wisc. He is a consultant for Astra Zeneca, Ironwood, and Baxter Pharmaceuticals.
Stress was the most frequently cited cause of ulcer disease before Helicobacter pylori was discovered. The harried executive who developed an ulcer was a widely accepted profile of an ulcer diathesis. When the role of H. pylori infection and NSAIDs became clear, the role of stress was downplayed and some articles and textbooks dismissed stress as a potential cause for ulcer disease.
|
|
Dr. Nimish Vakil |
Studies of New York City residents suggest a higher incidence of ulcer disease after the 9-11 attacks and studies from Japan have shown an increase in the incidence of ulcer disease after the nuclear reactor disaster. In this issue of Clinical Gastroenterology and Hepatology, Dr. Levenstein and her colleagues report the results of a study of stress and the incidence of ulcer disease in Danish subjects. In 1982-1983, a population-based study in Denmark collected sera and psychological data in over 3000 subjects and reinterviewed them in 1987-1988 and 1993-1994. An ad-hoc, unvalidated scale developed by the authors measured stress. It included a psychological scale used by the Danish military to identify recruits unsuitable for military service but also included tranquilizer use, working more than 40 hours a week, and unemployment. In multivariate analysis, they found that stress increased the risk for both gastric and duodenal ulcers, with an adjusted odds ratio of 1.19 per point increase in the stress scale for gastric ulcers (95% confidence interval, 1.03-1.37) and a odds ratio of 1.1 per point increase in the stress index for duodenal ulcers (95% CI, 0.98-1.27).
There are obvious limitations with this study: a historical cohort, an unvalidated stress scale, the inclusion of items that may not represent stress in some cultures (e.g., working more than 40 hours/week) and the lower bound of confidence intervals for risk which are very close to one. However, studies such as this tell us that we have been too quick to dismiss the role of stress in ulcer pathogenesis. With declining H. pylori prevalence and the development of safer NSAIDs, stress will undergo a renaissance in the pathogenesis of ulcer disease.
Dr. Nimish Vakil, AGAF, FASGE, FACP, is a physician specializing in gastroenterology at the Aurora Wilkinson Medical Clinic in Summit, Wisc. He is a consultant for Astra Zeneca, Ironwood, and Baxter Pharmaceuticals.
Stress was the most frequently cited cause of ulcer disease before Helicobacter pylori was discovered. The harried executive who developed an ulcer was a widely accepted profile of an ulcer diathesis. When the role of H. pylori infection and NSAIDs became clear, the role of stress was downplayed and some articles and textbooks dismissed stress as a potential cause for ulcer disease.
|
|
Dr. Nimish Vakil |
Studies of New York City residents suggest a higher incidence of ulcer disease after the 9-11 attacks and studies from Japan have shown an increase in the incidence of ulcer disease after the nuclear reactor disaster. In this issue of Clinical Gastroenterology and Hepatology, Dr. Levenstein and her colleagues report the results of a study of stress and the incidence of ulcer disease in Danish subjects. In 1982-1983, a population-based study in Denmark collected sera and psychological data in over 3000 subjects and reinterviewed them in 1987-1988 and 1993-1994. An ad-hoc, unvalidated scale developed by the authors measured stress. It included a psychological scale used by the Danish military to identify recruits unsuitable for military service but also included tranquilizer use, working more than 40 hours a week, and unemployment. In multivariate analysis, they found that stress increased the risk for both gastric and duodenal ulcers, with an adjusted odds ratio of 1.19 per point increase in the stress scale for gastric ulcers (95% confidence interval, 1.03-1.37) and a odds ratio of 1.1 per point increase in the stress index for duodenal ulcers (95% CI, 0.98-1.27).
There are obvious limitations with this study: a historical cohort, an unvalidated stress scale, the inclusion of items that may not represent stress in some cultures (e.g., working more than 40 hours/week) and the lower bound of confidence intervals for risk which are very close to one. However, studies such as this tell us that we have been too quick to dismiss the role of stress in ulcer pathogenesis. With declining H. pylori prevalence and the development of safer NSAIDs, stress will undergo a renaissance in the pathogenesis of ulcer disease.
Dr. Nimish Vakil, AGAF, FASGE, FACP, is a physician specializing in gastroenterology at the Aurora Wilkinson Medical Clinic in Summit, Wisc. He is a consultant for Astra Zeneca, Ironwood, and Baxter Pharmaceuticals.
High levels of psychological stress more than doubled the odds of peptic ulcers, and the link remained statistically significant even after controlling for factors such as Helicobacter pylori infection and cigarette smoking, according to a prospective study published in the March issue of Clinical Gastroenterology and Hepatology.
The findings contradict the widely accepted view that stress does not cause peptic ulcers, said Dr. Susan Levenstein of Aventino Medical Group in Rome and her associates. “Clinicians treating ulcer patients should investigate potential psychological stress among other risk factors,” they said.
Source: American Gastroenterological Association
Although “a vast literature links peptic ulcer to stress,” past studies suffered so many methodologic weaknesses that groups such as the U.S. National Institute of Diabetes and Digestive and Kidney Diseases rejected the evidence outright, Dr. Levenstein and her associates noted. Many studies were cross-sectional, for example, or did not control for confounders such as helicobacteriosis, they said.
To further study the effects of stress on ulcer risk, the researchers analyzed historical data from 76 patients who lacked a history of gastric and duodenal ulcers in 1982, but by 1994 had developed “distinct breach[es] in the mucosa” that were confirmed by endoscopy or contrast radiology. The researchers did not count erosions that lacked appreciable depth as ulcers, they noted (Clin. Gastroenterol. Hepatol. 2014 Aug. 8 [doi:10.1016/j.cgh.2014.07.052]).
Study subjects answered 12 questions about their stress levels, such as, “Do your hands easily shake?” “Do you often suffer from fits of dizziness?” “Do you constantly have thoughts that trouble and worry you?” and “Do you usually feel misunderstood by other people?” They answered these questions at baseline in 1982-1983, again in 1987-1988, and again in 1993-1994.
Respondents who scored in the top tertile for psychological stress had an ulcer incidence of 3.5%, compared with 1.6% for those in the lowest tertile (odds ratio, 2.2; 95% confidence interval, 1.2-3.9; P < .01), reported the investigators. And controlling for smoking, helicobacteriosis, use of nonsteroidal anti-inflammatory drugs, and low socioeconomic status only partially weakened the relationship between stress and ulcers, they said. After accounting for those risk factors, every one-point increase on the stress questionnaire still upped the odds of peptic ulcer by 12% (odds ratio, 1.12; 95% confidence interval, 1.01-1.23)they reported.
Helicobacteri pylori infection was the strongest independent predictor of ulcers (OR, 3.3; 95% CI, 2.02-5.69), while cigarette smoking came in a close second (OR, 2.91; 95% CI, 1.38-6.16), said the researchers. Notably, stress and helicobacteriosis did not seem to synergistically increase the chances of ulcers, they reported. “Stress affected H. pylori–related ulcers at least as much as those related to neither H. pylori nor nonsteroidal anti-inflammatory drugs,” they said.
Several factors might explain the stress-ulcer link, such as increased acid load, activation of the hypothalamic-pituitary-adrenal axis, shifts in blood flow, and cytokine activation that might impair gastrointestinal mucosal defenses, said the investigators. Although the baseline data in their study were more than 2 decades old, that meant that patients likely had not been treated to eradicate H. pylori and were less likely to have taken proton pump inhibitors than the current population that has over-the-counter access to PPIs, they added. They also noted that past studies found a particularly strong link between stress and bleeding or perforated ulcers, which have not declined as much as other types of ulcers. “These results support a multicausal model of peptic ulcer etiology, with intertwined biological and psychosocial components,” they concluded.
The Kirby Family Foundation funded the statistical analysis. The researchers reported no conflicts of interest.
High levels of psychological stress more than doubled the odds of peptic ulcers, and the link remained statistically significant even after controlling for factors such as Helicobacter pylori infection and cigarette smoking, according to a prospective study published in the March issue of Clinical Gastroenterology and Hepatology.
The findings contradict the widely accepted view that stress does not cause peptic ulcers, said Dr. Susan Levenstein of Aventino Medical Group in Rome and her associates. “Clinicians treating ulcer patients should investigate potential psychological stress among other risk factors,” they said.
Source: American Gastroenterological Association
Although “a vast literature links peptic ulcer to stress,” past studies suffered so many methodologic weaknesses that groups such as the U.S. National Institute of Diabetes and Digestive and Kidney Diseases rejected the evidence outright, Dr. Levenstein and her associates noted. Many studies were cross-sectional, for example, or did not control for confounders such as helicobacteriosis, they said.
To further study the effects of stress on ulcer risk, the researchers analyzed historical data from 76 patients who lacked a history of gastric and duodenal ulcers in 1982, but by 1994 had developed “distinct breach[es] in the mucosa” that were confirmed by endoscopy or contrast radiology. The researchers did not count erosions that lacked appreciable depth as ulcers, they noted (Clin. Gastroenterol. Hepatol. 2014 Aug. 8 [doi:10.1016/j.cgh.2014.07.052]).
Study subjects answered 12 questions about their stress levels, such as, “Do your hands easily shake?” “Do you often suffer from fits of dizziness?” “Do you constantly have thoughts that trouble and worry you?” and “Do you usually feel misunderstood by other people?” They answered these questions at baseline in 1982-1983, again in 1987-1988, and again in 1993-1994.
Respondents who scored in the top tertile for psychological stress had an ulcer incidence of 3.5%, compared with 1.6% for those in the lowest tertile (odds ratio, 2.2; 95% confidence interval, 1.2-3.9; P < .01), reported the investigators. And controlling for smoking, helicobacteriosis, use of nonsteroidal anti-inflammatory drugs, and low socioeconomic status only partially weakened the relationship between stress and ulcers, they said. After accounting for those risk factors, every one-point increase on the stress questionnaire still upped the odds of peptic ulcer by 12% (odds ratio, 1.12; 95% confidence interval, 1.01-1.23)they reported.
Helicobacteri pylori infection was the strongest independent predictor of ulcers (OR, 3.3; 95% CI, 2.02-5.69), while cigarette smoking came in a close second (OR, 2.91; 95% CI, 1.38-6.16), said the researchers. Notably, stress and helicobacteriosis did not seem to synergistically increase the chances of ulcers, they reported. “Stress affected H. pylori–related ulcers at least as much as those related to neither H. pylori nor nonsteroidal anti-inflammatory drugs,” they said.
Several factors might explain the stress-ulcer link, such as increased acid load, activation of the hypothalamic-pituitary-adrenal axis, shifts in blood flow, and cytokine activation that might impair gastrointestinal mucosal defenses, said the investigators. Although the baseline data in their study were more than 2 decades old, that meant that patients likely had not been treated to eradicate H. pylori and were less likely to have taken proton pump inhibitors than the current population that has over-the-counter access to PPIs, they added. They also noted that past studies found a particularly strong link between stress and bleeding or perforated ulcers, which have not declined as much as other types of ulcers. “These results support a multicausal model of peptic ulcer etiology, with intertwined biological and psychosocial components,” they concluded.
The Kirby Family Foundation funded the statistical analysis. The researchers reported no conflicts of interest.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Key clinical point: High stress levels independently predicted peptic ulcers.
Major finding: After adjustment for other risk factors, every one-point increase on a 12-item stress questionnaire increased the odds of peptic ulcers by 12% (OR, 1.12; 95% CI, 1.01-1.23).
Data source: Prospective, population-based study of 76 patients with peptic ulcers.
Disclosures: The Kirby Family Foundation funded the statistical analysis. The researchers reported no conflicts of interest.

Cancer-Related Anemia
Anemia occurs in more than half of patients with cancer and is associated with worse performance status, quality of life, and survival. Anemia is often attributed to the effects of chemotherapy; however, a 2004 European Cancer Anemia Survey reported that 39% of patients with cancer were anemic prior to starting chemotherapy and the incidence of anemia may be as high as 90% in patients on chemotherapy. The pathogenesis of cancer-related anemia is multifactorial; it can be a direct result of cancer invading the bone marrow, or result from the effects of radiation, chemotherapy-induced anemia, chronic renal disease, and cancer-related inflammation leading to functional iron deficiency anemia.
To read the full article in PDF:
Anemia occurs in more than half of patients with cancer and is associated with worse performance status, quality of life, and survival. Anemia is often attributed to the effects of chemotherapy; however, a 2004 European Cancer Anemia Survey reported that 39% of patients with cancer were anemic prior to starting chemotherapy and the incidence of anemia may be as high as 90% in patients on chemotherapy. The pathogenesis of cancer-related anemia is multifactorial; it can be a direct result of cancer invading the bone marrow, or result from the effects of radiation, chemotherapy-induced anemia, chronic renal disease, and cancer-related inflammation leading to functional iron deficiency anemia.
To read the full article in PDF:
Anemia occurs in more than half of patients with cancer and is associated with worse performance status, quality of life, and survival. Anemia is often attributed to the effects of chemotherapy; however, a 2004 European Cancer Anemia Survey reported that 39% of patients with cancer were anemic prior to starting chemotherapy and the incidence of anemia may be as high as 90% in patients on chemotherapy. The pathogenesis of cancer-related anemia is multifactorial; it can be a direct result of cancer invading the bone marrow, or result from the effects of radiation, chemotherapy-induced anemia, chronic renal disease, and cancer-related inflammation leading to functional iron deficiency anemia.
To read the full article in PDF:
Managing aneurysmal subarachnoid hemorrhage: It takes a team
Aneurysmal subarachnoid hemorrhage is a devastating condition, with an estimated death rate of 30% during the initial episode.1,2 Approximately the same number of patients survive but leave the hospital with disabling neurologic deficits.3
However, better outcomes can be achieved by systems that are able to work as a team on the collective goal of quick intervention to secure the ruptured aneurysm, followed by the implementation of measures to minimize secondary brain injury. Although the search for new diagnostic, prognostic, and therapeutic modalities continues, it is clear that there exists no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of small advances that will ultimately maximize the patient’s chances of survival and neurologic recovery.
This review focuses on the management of aneurysmal subarachnoid hemorrhage and its systemic and neurologic complications.
ANEURYSM IS THE MOST COMMON CAUSE OF SUBARACHNOID BLEEDING
Aneurysmal subarachnoid hemorrhage, ie, rupture of an intracranial aneurysm, flooding the subarachnoid space with blood, affects about 24,000 Americans each year.1,2 A ruptured aneurysm is the most common cause of subarachnoid hemorrhage, accounting for about 85% of cases. Less common causes include idiopathic benign perimesencephalic hemorrhage, arteriovenous malformation, dural arteriovenous fistula, and hemorrhagic mycotic aneurysm. These have their own natural history, pathophysiology, and specific treatment, and will not be addressed in this article.
Risk factors for aneurysmal subarachnoid hemorrhage include having a first-degree relative who had the disease, hypertension, smoking, and consuming more than 150 g of alcohol per week.4
CLINICAL PRESENTATION AND DIAGNOSIS
The key symptom of aneurysmal subarachnoid hemorrhage is the abrupt onset of severe headache that peaks in intensity over 1 hour,5 often described as “the worst headache of my life.” Headache is accompanied by brief loss of consciousness in 53% of cases (conversely, nearly half of patients maintain normal mental status), by nausea or vomiting in 77%, and by meningismus (neck pain or stiffness) in 35%.6
These clinical manifestations and risk factors have been incorporated into a decision rule:
Obtain brain imaging if the patient has acute headache reaching maximal intensity within 1 hour, associated with any of the following factors:
- Age 40 or older
- Neck pain or stiffness
- Witnessed loss of consciousness
- Onset during exertion
- “Thunderclap” headache (ie, instantly peaking pain)
- Limited neck flexion on examination.5
This decision rule has nearly 100% sensitivity for aneurysmal subarachnoid hemorrhage in clinical practice.5 All patients require brain imaging if they have a severe headache plus either abnormal neurologic findings (eg, a focal neurologic deficit) or a history of cerebral aneurysm.
Emergency physicians should have a low threshold for ordering noncontrast computed tomography (CT) of the head in patients with even mild symptoms suggesting aneurysmal subarachnoid hemorrhage. Failure to order CT is the most common diagnostic error in this situation.6 CT performed within 6 hours of headache onset is nearly 100% sensitive for this condition,7 but the sensitivity falls to 93% after the first 24 hours and to less than 60% after 5 days.8 In patients who have symptoms highly suggestive of aneurysmal subarachnoid hemorrhage but a normal CT, lumbar puncture is the next diagnostic step.
There are two alternatives to CT followed by lumbar puncture: ie, noncontrast CT followed by CT angiography,9,10 and magnetic resonance imaging followed by magnetic resonance angiography. In patients with suspicious clinical symptoms but negative CT results, CT followed by CT angiography can rule out aneurysmal subarachnoid hemorrhage with a 99% probability.9,10 However, CT followed by lumbar puncture remains the standard of care and carries a class I recommendation in the American Heart Association guidelines for ruling out subarachnoid hemorrhage.5
GRADING THE SEVERITY OF SUBARACHNOID HEMORRHAGE
Age, the thickness of the blood layer in the subarachnoid space, intraventricular hemorrhage and the findings of the neurologic examination at presentation are predictors of long-term outcomes in aneurysmal subarachnoid hemorrhage (Figure 1).
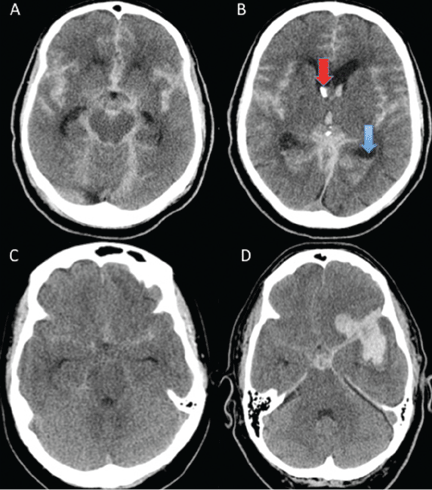
Different grading systems used in clinical practice are based on the findings on the initial neurologic examination and on the initial noncontrast CT (ie, the thickness of the blood, and whether intraventricular hemorrhage is present). Among the most widely used are those developed by Hunt and Hess12 and by the World Federation of Neurological Surgeons11 (WFNS), and the CT grading scales (Fisher13 or its modified version14) (Tables 1 and 2). With either the Hunt and Hess scale or the WFNS scale, the higher the score, the worse the patient’s probable outcome. Scores on both Fisher scales correlate with the risk of angiographic vasospasm. The higher the grade, the higher the risk of angiographic vasospasm.


The VASOGRADE score—a combination of the WFNS score and the modified Fisher scale—stratifies patients at risk of delayed cerebral ischemia, allowing for a tailored monitoring strategy.15 There are three variations:
- VASOGRADE green—Modified Fisher 1 or 2 and WFNS 1 or 2
- VASOGRADE yellow—Modified Fisher 3 or 4 and WFNS 1, 2, or 3
- VASOGRADE red—WFNS 4 or 5.
After the initial bleeding event, patients with aneurysmal subarachnoid hemorrhage are at high risk of delayed systemic and neurologic complications, with poor functional outcomes. Delayed cerebral ischemia holds the greatest risk of an unfavorable outcome and ultimately can lead to cerebral infarction, disability, and death.6,7
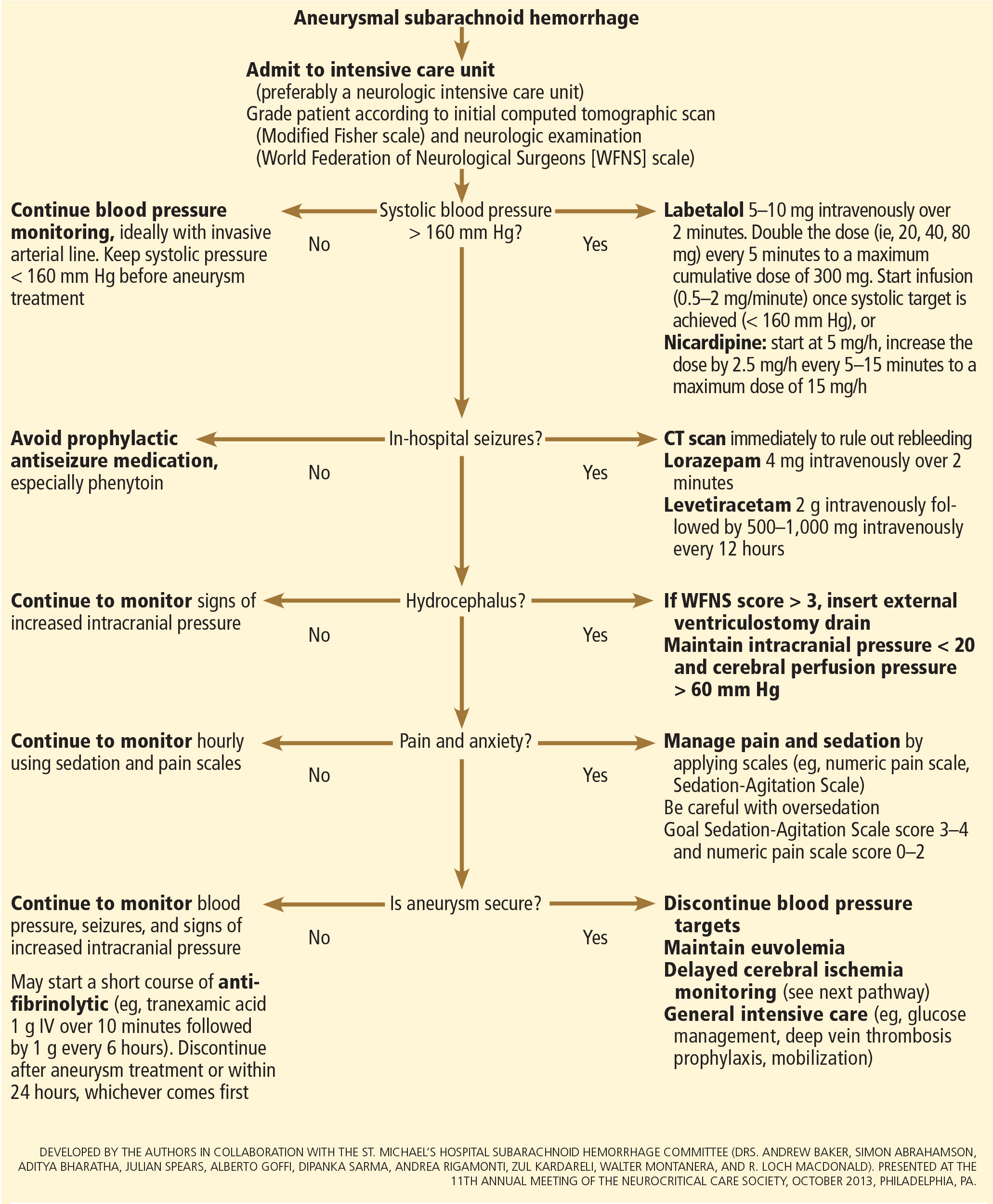
INITIAL MANAGEMENT
After aneurysmal subarachnoid hemorrhage is diagnosed, the initial management (Figure 2) includes appropriate medical prevention of rebleeding (which includes supportive care, blood pressure management, and, perhaps, the early use of a short course of an antifibrinolytic drug) and early transfer to a high-volume center for securing the aneurysm. The reported incidence of rebleeding varies from 5% to 22% in the first 72 hours. “Ultra-early” rebleeding (within 24 hours of hemorrhage) has been reported, with an incidence as high as 15% and a fatality rate around 70%. Patients with poor-grade aneurysmal subarachnoid hemorrhage, larger aneurysms, and “sentinel bleeds” are at higher risk of rebleeding.16
Outcomes are much better when patients are managed in a high-volume center, with a specialized neurointensive care unit17 and access to an interdisciplinary team.18 Regardless of the initial grade, patients with aneurysmal subarachnoid hemorrhage should be quickly transferred to a high-volume center, defined as one treating at least 35 cases per year, and the benefit is greater in centers treating more than 60 cases per year.19 The higher the caseload in any given hospital, the better the clinical outcomes in this population.20
Treating cerebral aneurysm: Clipping or coiling
Early aneurysm repair is generally considered the standard of care and the best strategy to reduce the risk of rebleeding. Further, early treatment may be associated with a lower risk of delayed cerebral ischemia21 and better outcomes.22
Three randomized clinical trials have compared surgical clipping and endovascular repair (placement of small metal coils within the aneurysm to promote clotting).
The International Subarachnoid Aneurysm Trial23 showed a reduction of 23% in relative risk and of 7% in absolute risk in patients who underwent endovascular treatment compared with surgery. The survival benefit persisted at a mean of 9 years (range 6–14 years), but with a higher annual rate of aneurysm recurrence in the coiling group (2.9% vs 0.9%).24 Of note, this trial included only patients with aneurysms deemed suitable for both coiling and clipping, so that the exclusion rate was high. Most of the patients presented with good-grade (WFNS score 1–3), small aneurysms (< 5 mm) in the anterior circulation.
A single-center Finnish study25 found no differences in rates of recovery, disability, and death at 1 year, comparing surgery and endovascular treatment. Additionally, survival rates at a mean follow-up of 39 months were similar, with no late recurrences or aneurysmal bleeding.
Lastly, the Barrow Ruptured Aneurysm Trial26,27 found that patients assigned to endovascular treatment had better 1-year neurologic outcomes, defined as a modified Rankin score of 2 or less. Importantly, 37.7% of patients originally assigned to endovascular treatment crossed over to surgical treatment. The authors then performed intention-to-treat and as-treated analyses. Either way, patients treated by endovascular means had better neurologic outcomes at 1 year. However, no difference in the relative risk reduction in worse outcome was found on 3-year follow-up, and patients treated surgically had higher rates of aneurysm obliteration and required less aneurysm retreatment, both of which were statistically significant.
The question that remains is not whether to clip or whether to coil, but whom to clip and whom to coil.28 That question must be answered on a patient-to-patient basis and requires the expertise of an interventional neuroradiologist and a vascular neurosurgeon—one of the reasons these patients are best cared for in high-volume centers providing such expertise.
MEDICAL PREVENTION OF REBLEEDING
Blood pressure management
There are no systematic data on the optimal blood pressure before securing an aneurysm. Early studies of hemodynamic augmentation in cases of ruptured untreated aneurysm reported rebleeding when the systolic blood pressure was allowed to rise above 160 mm Hg.29,30 A recent study evaluating hypertensive intracerebral hemorrhage revealed better functional outcomes with intensive lowering of blood pressure (defined as systolic blood pressure < 140 mm Hg) but no significant reduction in the combined rate of death or severe disability.31 It is difficult to know if these results can be extrapolated to patients with aneurysmal subarachnoid hemorrhage. Current guidelines3,32 say that before the aneurysm is treated, the systolic pressure should be lower than 160 mm Hg.
There is no specific drug of choice, but a short-acting, titratable medication is preferable. Nicardipine is a very good option, and labetalol might be an appropriate alternative.33 Once the aneurysm is secured, all antihypertensive drugs should be held. Hypertension should not be treated unless the patient has clinical signs of a hypertensive crisis, such as flash pulmonary edema, myocardial infarction, or hypertensive encephalopathy.
Antifibrinolytic therapy
The role of antifibrinolytic therapy in aneurysmal subarachnoid hemorrhage is controversial and has been studied in 10 clinical trials. In a Swedish study,34 early use of tranexamic acid (1 g intravenously over 10 minutes followed by 1 g every 6 hours for a maximum of 24 hours) reduced the rebleeding rate substantially, from 10.8% to 2.4%, with an 80% reduction in the mortality rate from ultra-early rebleeding. However, a recent Cochrane review that included this study found no overall benefit.35
An ongoing multicenter randomized trial in the Netherlands will, we hope, answer this question in the near future.36 At present, some centers would consider a short course of tranexamic acid before aneurysm treatment.
DIAGNOSIS AND TREATMENT OF COMPLICATIONS
Medical complications are extremely common after aneurysmal subarachnoid hemorrhage. Between 75% and 100% of patients develop some type of systemic or further neurologic derangement, which in turn has a negative impact on the long-term outcome.37,38 In the first 72 hours, rebleeding is the most feared complication, and as mentioned previously, appropriate blood pressure management and early securing of the aneurysm minimize its risk.
NEUROLOGIC COMPLICATIONS
Hydrocephalus
Hydrocephalus is the most common early neurologic complication after aneurysmal subarachnoid hemorrhage, with an overall incidence of 50%.39 Many patients with poor-grade aneurysmal subarachnoid hemorrhage and patients whose condition deteriorates due to worsening of hydrocephalus require the insertion of an external ventricular drain (Figure 1).
Up to 30% of patients who have a poor-grade aneurysmal subarachnoid hemorrhage improve neurologically with cerebrospinal fluid drainage.40 An external ventricular drain can be safely placed, even before aneurysm treatment, and placement does not appear to increase the risk of rebleeding.39,41 After placement, rapid weaning from the drain (clamping within 24 hours of insertion) is safe, decreases length of stay in the intensive care unit and hospital, and may be more cost-effective than gradual weaning over 96 hours.42
Increased intracranial pressure
Intracranial hypertension is another potential early complication, and is frequently due to the development of hydrocephalus, cerebral edema, or rebleeding. The treatment of increased intracranial pressure does not differ from the approach used in managing severe traumatic brain injury, which includes elevating the head of the bed, sedation, analgesia, normoventilation, and cerebrospinal fluid drainage.
Hypertonic saline has been tested in several studies that were very small but nevertheless consistently showed control of intracranial pressure levels and improvement in cerebral blood flow measured by xenon CT.43–47 Two of these studies even showed better outcomes at discharge.43,44 However, the small number of patients prevents any meaningful conclusion regarding the use of hypertonic saline and functional outcomes.
Barbiturates, hypothermia, and decompressive craniectomy could be tried in refractory cases.48 Seule et al49 evaluated the role of therapeutic hypothermia with or without barbiturate coma in 100 patients with refractory intracranial hypertension. Only 13 patients received hypothermia by itself. At 1 year, 32 patients had achieved a good functional outcome (Glasgow Outcome Scale score 4 or 5). The remaining patients were severely disabled or had died. Of interest, the median duration of hypothermia was 7 days, and 93% of patients developed some medical complication such as electrolyte disorders (77%), pneumonia (52%), thrombocytopenia (47%), or septic shock syndrome (40%). Six patients died as a consequence of one of these complications.
Decompressive craniectomy can be life-saving in patients with refractory intracranial hypertension. However, most of these patients will die or remain severely disabled or comatose.50
Seizure prophylaxis is controversial
Seizures can occur at the onset of intracranial hemorrhage, perioperatively, or later (ie, after the first week). The incidence varied considerably in different reports, ranging from 4% to 26%.51 Seizures occurring perioperatively, ie, after hospital admission, are less frequent and are usually the manifestation of aneurysm rebleeding.24
Seizure prophylaxis remains controversial, especially because the use of phenytoin is associated with increased incidence of cerebral vasospasm, infarction, and worse cognitive outcomes after aneurysmal subarachnoid hemorrhage.52,53 Therefore, routine prophylactic use of phenytoin is not recommended in these patients,3 although the effect of other antiepileptic drugs is less studied and less clear. Patients may be considered for this therapy if they have multiple risk factors for seizures, such as intraparenchymal hematoma, advanced age (> 65), middle cerebral artery aneurysm, craniotomy for aneurysm clipping, and a short course (≤ 72 hours) of an antiepileptic drug other than phenytoin, especially while the aneurysm is unsecured.3
Levetiracetam may be an alternative to phenytoin, having better pharmacodynamic and kinetic profiles, minimal protein binding, and absence of hepatic metabolism, resulting in a very low risk of drug interaction and better tolerability.54,55 Because of these advantages, levetiracetam has become the drug of choice in several centers treating aneurysmal subarachnoid hemorrhage in the United States.
Addressing this question, a survey was sent to 25 high-volume aneurysmal subarachnoid hemorrhage academic centers in the United States. All 25 institutions answered the survey, and interestingly, levetiracetam was the first-line agent for 16 (94%) of the 17 responders that used prophylaxis, while only 1 used phenytoin as the agent of choice.56
A retrospective cohort study by Murphy-Human et al57 showed that a short course of levetiracetam (≤ 72 hours) was associated with higher rates of in-hospital seizures compared with an extended course of phenytoin (eg, entire hospital stay). However, the study did not address functional outcomes.57
Continuous electroencephalographic monitoring may be considered in comatose patients, in patients requiring controlled ventilation and sedation, or in patients with unexplained alteration in consciousness. In one series of patients with aneurysmal subarachnoid hemorrhage who received continuous monitoring, the incidence of nonconvulsive status epilepticus was 19%, with an associated mortality rate of 100%.58
Continuous quantitative electroencephalography is useful to monitor and to detect angiographic vasospasm and delayed cerebral ischemia. Relative alpha variability and the alpha-delta ratio decrease with ischemia, and this effect can precede angiographic vasospasm by 3 days.59,60
Delayed cerebral ischemia
Delayed cerebral ischemia is defined as the occurrence of focal neurologic impairment, or a decrease of at least 2 points on the Glasgow Coma Scale that lasts for at least 1 hour, is not apparent immediately after aneurysm occlusion, and not attributable to other causes (eg, hyponatremia, fever).61
Classically, neurologic deficits that occurred within 2 weeks of aneurysm rupture were ascribed to reduced cerebral blood flow caused by delayed large-vessel vasospasm causing cerebral ischemia.62 However, perfusion abnormalities have also been observed with either mild or no demonstrable vasospasm.63 Almost 70% of patients who survive the initial hemorrhage develop some degree of angiographic vasospasm. However, only 30% of those patients will experience symptoms.
In addition to vasospasm of large cerebral arteries, impaired autoregulation and early brain injury within the first 72 hours following subarachnoid hemorrhage may play important roles in the development of delayed cerebral ischemia.64 Therefore, the modern concept of delayed cerebral ischemia monitoring should focus on cerebral perfusion rather than vessel diameter measurements. This underscores the importance of comprehensive, standardized monitoring techniques that provide information not only on microvasculature, but also at the level of the microcirculation, with information on perfusion, oxygen utilization and extraction, and autoregulation.
Although transcranial Doppler has been the most commonly applied tool to monitor for angiographic vasospasm, it has a low sensitivity and negative predictive value.37 It is nevertheless a useful technique to monitor good-grade aneurysmal subarachnoid hemorrhage patients (WFNS score 1–3) combined with frequent neurologic examinations (Figure 3).
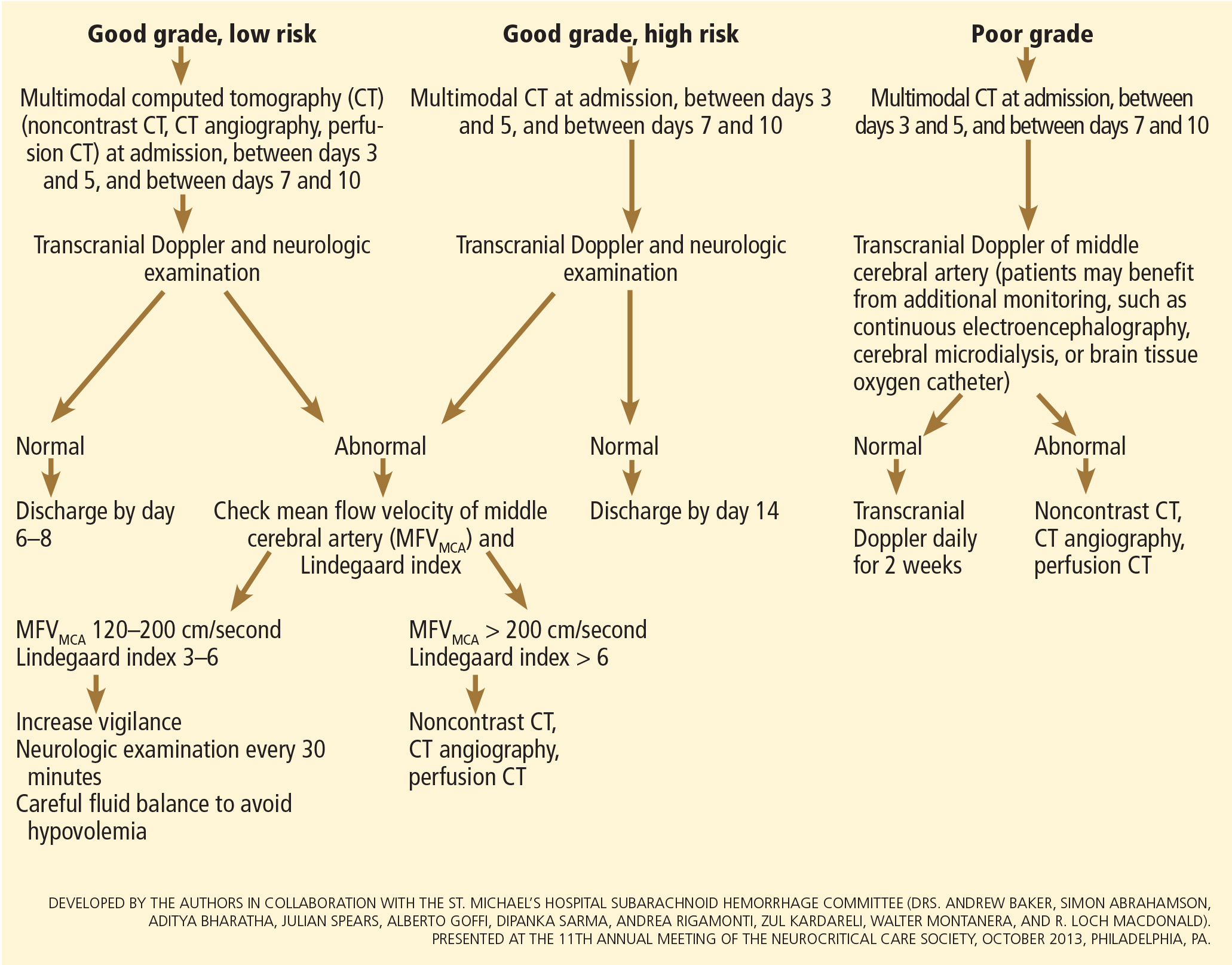
CT angiography is a good noninvasive alternative to digital subtraction angiography. However, it tends to overestimate the degree of vasoconstriction and does not provide information about perfusion and autoregulation.65 Nevertheless, CT angiography combined with a CT perfusion scan can add information about autoregulation and cerebral perfusion and has been shown to be more sensitive for the diagnosis of angiographic vasospasm than transcranial Doppler and digital subtraction angiography (Figure 4).
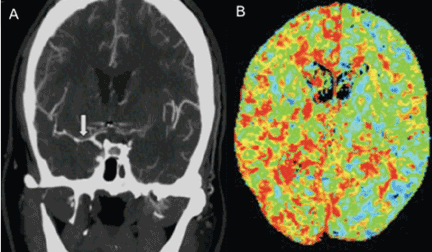
Patients with a poor clinical condition (WFNS score 4 or 5) or receiving continuous sedation constitute a challenge in monitoring for delayed neurologic deterioration. Neurologic examination is not sensitive enough in this setting to detect subtle changes. In these specific and challenging circumstances, multimodality neuromonitoring may be useful in the early detection of delayed cerebral ischemia and may help guide therapy.67
Several noninvasive and invasive techniques have been studied to monitor patients at risk of delayed cerebral ischemia after subarachnoid hemorrhage.66 These include continuous electroencephalography, brain tissue oxygenation monitoring (Ptio2), cerebral microdialysis, thermal diffusion flowmetry, and near-infrared spectroscopy. Of these techniques, Ptio2, cerebral microdialysis, and continuous electroencephalography (see discussion of seizure prophylaxis above) have been more extensively studied. However, most of the studies were observational and very small, limiting any recommendations for using these techniques in routine clinical practice.68
Ptio2 is measured by inserting an intraparenchymal oxygen-sensitive microelectrode, and microdialysis requires a microcatheter with a semipermeable membrane that allows small soluble substances to cross it into the dialysate. These substances, which include markers of ischemia (ie, glucose, lactate, and pyruvate), excitotoxins (ie, glutamate and aspartate), and membrane cell damage products (ie, glycerol), can be measured. Low Ptio2 values (< 15 mm Hg) and abnormal mycrodialysate findings (eg, glucose < 0.8 mmol/L, lactate-to-pyruvate ratio > 40) have both been associated with cerebral ischemic events and poor outcome.68
Preventing delayed cerebral ischemia
Oral nimodipine 60 mg every 4 hours for 21 days, started on admission, carries a class I, level of evidence A recommendation in the management of aneurysmal subarachnoid hemorrhage.3,32,69 It improves clinical outcome despite having no effect on the risk of angiographic vasospasm. The mechanism of improved outcome is unclear, but the effect may be a neuroprotective phenomenon limiting the extension of delayed cerebral ischemia.70
If hypotension occurs, the dose can be lowered to 30 mg every 2 hours. Whether to discontinue nimodipine in this situation is controversial. Of note, the clinical benefits of nimodipine have not been replicated with other calcium channel blockers (eg, nicardipine).71
Prophylactic hyperdynamic fluid therapy, known as “triple-H” (hypervolemia, hemodilution, and hypertension) was for years the mainstay of treatment in preventing delayed cerebral ischemia due to vasospasm. However, the clinical data supporting this intervention have been called into question, as analysis of two trials found that hypervolemia did not improve outcomes or reduce the incidence of delayed cerebral ischemia, and in fact increased the rate of complications.72,73 Based on these findings, current guidelines recommend maintaining euvolemia rather than prophylactic hypervolemia in patients with aneurysmal subarachnoid hemorrhage.3,32,69
TREATING DELAYED CEREBRAL ISCHEMIA
Hemodynamic augmentation
In patients with neurologic deterioration due to delayed cerebral ischemia, hemodynamic augmentation is the cornerstone of treatment. This is done according to a protocol, started early, involving specific physiologic goals, clinical improvement, and escalation to invasive therapies in a timely fashion in patients at high risk of further neurologic insult (Figure 5).
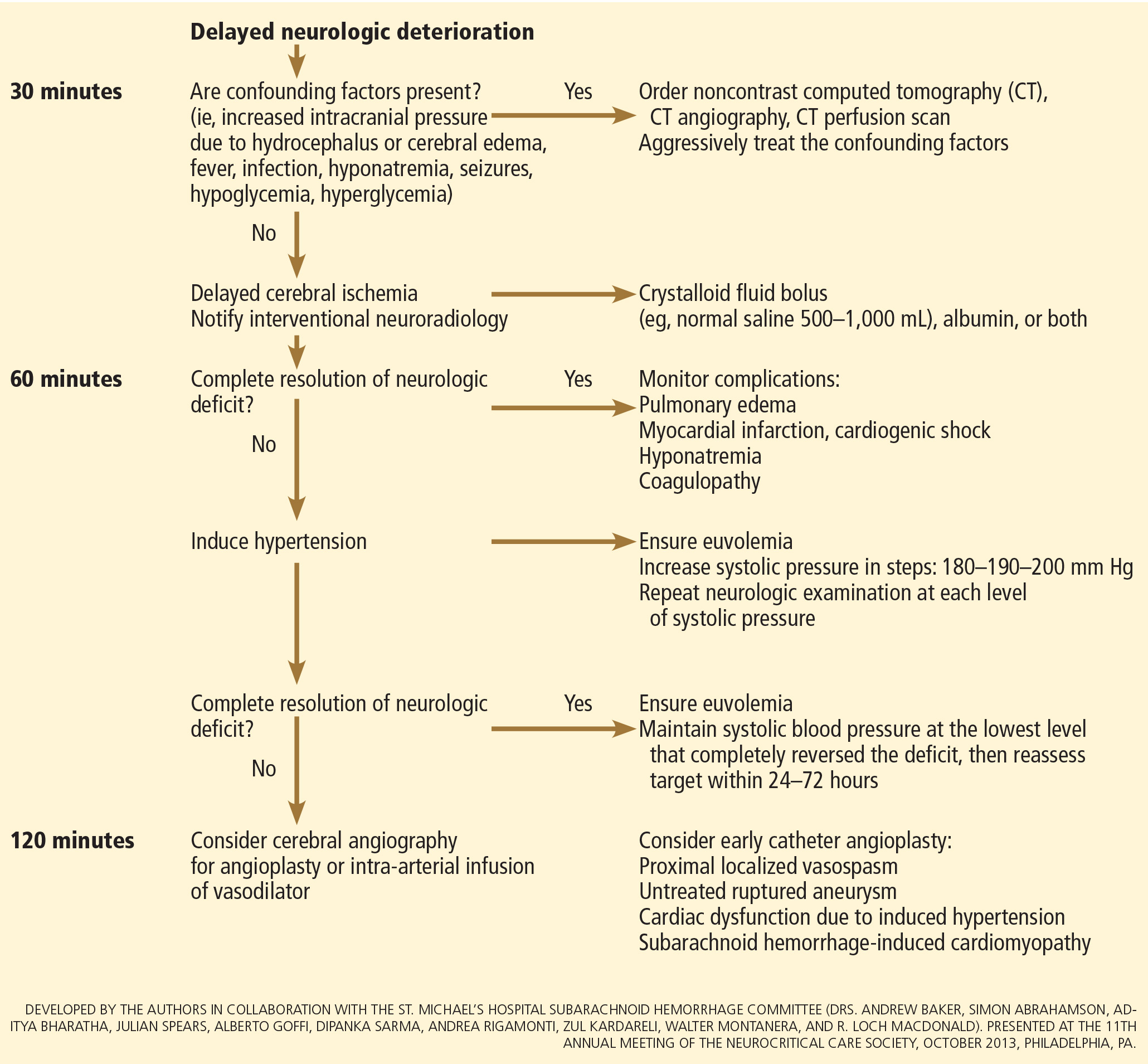
The physiologic goal is to increase the delivery of oxygen and glucose to the ischemic brain. Hypertension seems to be the most effective component of hemodynamic augmentation regardless of volume status, increasing cerebral blood flow and brain tissue oxygenation, with reversal of delayed cerebral ischemic symptoms in up to two-thirds of treated patients.74,75 However, this information comes from very small studies, with no randomized trials of induced hypertension available.
The effect of a normal saline fluid bolus in patients suspected of having delayed cerebral ischemia has been shown to increase cerebral blood flow in areas of cerebral ischemia.74 If volume augmentation fails to improve the neurologic status, the next step is to artificially induce hypertension using vasopressors. The blood pressure target should be based on clinical improvement. A stepwise approach is reasonable in this situation, and the lowest level of blood pressure at which there is a complete reversal of the new focal neurologic deficit should be maintained.3,29
Inotropic agents such as dobutamine or milrinone can be considered as alternatives in patients who have new neurologic deficits that are refractory to fluid boluses and vasopressors, or in a setting of subarachnoid hemorrhage-induced cardiomyopathy.76,77
Once the neurologic deficit is reversed by hemodynamic augmentation, the blood pressure should be maintained for 48 to 72 hours at the level that reversed the deficit completely, carefully reassessed thereafter, and the patient weaned slowly. Unruptured unsecured aneurysms should not prevent blood pressure augmentation in a setting of delayed cerebral ischemia if the culprit aneurysm is treated.3,32 If the ruptured aneurysm has not been secured, careful blood pressure augmentation can be attempted, keeping in mind that hypertension (> 160/95 mm Hg) is a risk factor for fatal aneurysm rupture.
Endovascular management of delayed cerebral ischemia
When medical augmentation fails to completely reverse the neurologic deficits, endovascular treatment can be considered. Although patients treated early in the course of delayed cerebral ischemia have better neurologic recovery, prophylactic endovascular treatment in asymptomatic patients, even if angiographic signs of spasm are present, does not improve clinical outcomes and carries the risk of fatal arterial rupture.78
SYSTEMIC COMPLICATIONS
Hyponatremia and hypovolemia
Aneurysmal subarachnoid hemorrhage is commonly associated with abnormalities of fluid balance and electrolyte derangements. Hyponatremia (serum sodium < 135 mmol/L) occurs in 30% to 50% of patients, while the rate of hypovolemia (decreased circulating blood volume) ranges from 17% to 30%.79 Both can negatively affect long-term outcomes.80,81
Decreased circulating blood volume is a well-described contributor to delayed cerebral ischemia and cerebral infarction after aneurysmal subarachnoid hemorrhage.80–82 Clinical variables such as heart rate, blood pressure, fluid balance, and serum sodium concentration are usually the cornerstones of intravascular volume status assessment. However, these variables correlate poorly with measured circulating blood volumes in those with aneurysmal subarachnoid hemorrhage.83,84
The mechanisms responsible for the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage are not completely understood. Several factors have been described and may contribute to the increased natriuresis and, hence, to a reduction in circulating blood volume: increased circulating natriuretic peptide concentrations,85–87 sympathetic nervous system hyperactivation,88 and hyperreninemic hypo-
aldosteronism syndrome.89,90
Lastly, the cerebral salt wasting syndrome, described in the 1950s,91 was thought to be a key mechanism in the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage. In contrast to the syndrome of inappropriate antidiuretic hormone, which is characterized by hyponatremia with a normal or slightly elevated intravascular volume, the characteristic feature of cerebral salt wasting syndrome is the development of hyponatremia in a setting of intravascular volume depletion.92 In critically ill neurologic and neurosurgical patients, this differential diagnosis is very difficult, especially in those with aneurysmal subarachnoid hemorrhage in whom the clinical assessment of fluid status is not reliable. These two syndromes might coexist and contribute to the development of hyponatremia after aneurysmal subarachnoid hemorrhage.92,93
Hoff et al83,84 prospectively compared the clinical assessment of fluid status by critical and intermediate care nurses and direct measurements of blood volume using pulse dye densitometry. The clinical assessment failed to accurately assess patients’ volume status. Using the same technique to measure circulating blood volume, this group showed that calculation of fluid balance does not provide adequate assessment of fluid status.83,84
Hemodynamic monitoring tools can help guide fluid replacement in this population. Mutoh et al94 randomized 160 patients within 24 hours of hemorrhage to receive early goal-directed fluid therapy (ie, preload volume and cardiac output monitored by transpulmonary thermodilution) vs standard therapy (ie, fluid balance or central venous pressure). Overall, no difference was found in the rates of delayed cerebral ischemia (33% vs 42%; P = .33) or favorable outcome (67% vs 57%; P = .22). However, in the subgroup of poor-grade patients (WFNS score 4 or 5), early goal-directed therapy was associated with a lower rate of delayed cerebral ischemia (5% vs 14%; P = .036) and with better functional outcomes at 3 months (52% vs 36%; P = .026).
Fluid restriction to treat hyponatremia in aneurysmal subarachnoid hemorrhage is no longer recommended because of the increased risk of cerebral infarction due to hypovolemic hypoperfusion.82
Prophylactic use of mineralocorticoids (eg, fludrocortisone, hydrocortisone) has been shown to limit natriuresis, hyponatremia, and the amount of fluid required to maintain euvolemia.95,96 Higher rates of hypokalemia and hyperglycemia, which can be easily treated, are the most common complications associated with this approach. Additionally, hypertonic saline (eg, 3% saline) can be used to correct hyponatremia in a setting of aneurysmal subarachnoid hemorrhage.79
Cardiac complications
Cardiac complications after subarachnoid hemorrhage are most likely related to sympathetic hyperactivity and catecholamine-induced myocyte dysfunction. The pathophysiology is complex, but cardiac complications have a significant negative impact on long-term outcome in these patients.97
Electrocardiographic changes and positive cardiac enzymes associated with aneurysmal subarachnoid hemorrhage have been extensively reported. More recently, data from studies of two-dimensional echocardiography have shown that subarachnoid hemorrhage can also be associated with significant wall-motion abnormalities and even overt cardiogenic shock.98–100
There is no specific curative therapy; the treatment is mainly supportive. Vasopressors and inotropes may be used for hemodynamic augmentation.
Pulmonary complications
Pulmonary complications occur in 20% to 30% of all aneurysmal subarachnoid hemorrhage patients and are associated with a higher risk of delayed cerebral ischemia and death. Common pulmonary complications in this population are mild acute respiratory distress syndrome (27%), hospital-acquired pneumonia (9%), cardiogenic pulmonary edema (8%), aspiration pneumonia (6%), neurogenic pulmonary edema (2%), and pulmonary embolism (1%).101–103
SUPPORTIVE CARE
Hyperthermia, hyperglycemia, and liberal use of transfusions have all been associated with longer stays in the intensive care unit and hospital, poorer neurologic outcomes, and higher mortality rates in patients with acute brain injury.104 Noninfectious fever is the most common systemic complication after subarachnoid hemorrhage.
Antipyretic drugs such as acetaminophen and ibuprofen are not very effective in reducing fever in the subarachnoid hemorrhage population, but should still be used as first-line therapy. The use of surface and intravascular devices can be considered when fevers do not respond to nonsteroidal anti-inflammatory drugs.
Although no prospective randomized trial has addressed the impact of induced normothermia on long-term outcome and mortality in aneurysmal subarachnoid hemorrhage patients, fever control has been shown to reduce cerebral metabolic distress, irrespective of intracranial pressure.105 Maintenance of normothermia (< 37.5°C) seems reasonable, especially in aneurysmal subarachnoid hemorrhage patients at risk of or with active delayed cerebral ischemia.106
Current guidelines3,32,69 strongly recommend avoiding hypoglycemia, defined as a serum glucose level less than 80 mg/dL, but suggest keeping the blood sugar level below 180 or 200 mg/dL.
At the moment, there is no clear threshold for transfusion in patients with aneurysmal subarachnoid hemorrhage. Current guidelines suggest keeping hemoglobin levels between 8 and 10 g/dL.3
Preventing venous thromboembolism
The incidence of venous thromboembolism after aneurysmal subarachnoid hemorrhage varies widely, from 1.5% to 18%.107 Active surveillance with venous Doppler ultrasonography has found asymptomatic deep vein thrombosis in up to 3.4% of poor-grade aneurysmal subarachnoid hemorrhage patients receiving pharmacologic thromboprophylaxis.108
In a retrospective study of 170 patients, our group showed that giving drugs to prevent venous thromboembolism (unfractionated heparin 5,000 IU subcutaneously every 12 hours or dalteparin 5,000 IU subcutaneously daily), starting within 24 hours of aneurysm treatment, could be safe.109 Fifty-eight percent of these patients had an external ventricular drain in place. One patient developed a major cerebral hemorrhagic complication and died while on unfractionated heparin; however, the patient was also on dual antiplatelet therapy with aspirin and clopidogrel.109
Current guidelines suggest that intermittent compression devices be applied in all patients before aneurysm treatment. Pharmacologic thromboprophylaxis with a heparinoid can be started 12 to 24 hours after aneurysm treatment.3,109
A TEAM APPROACH
Patients with subarachnoid hemorrhage need integrated care from different medical and nursing specialties. The best outcomes are achieved by systems that can focus as a team on the collective goal of quick intervention to secure the aneurysm, followed by measures to minimize secondary brain injury.
The modern concept of cerebral monitoring in a setting of subarachnoid hemorrhage should focus on brain perfusion rather than vascular diameter. Although the search continues for new diagnostic, prognostic, and therapeutic tools, there is no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of many small advances that will ultimately lead to better outcomes.
ACKNOWLEDGMENT
This work was supported by research funding provided by the Bitove Foundation, which has been supportive of our clinical and research work for several years.
- van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain 2001; 124:249–278.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 2014; 129:e28–e292.
- Diringer MN, Bleck TP, Claude Hemphill J 3rd, et al; Neurocritical Care Society. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit Care 2011; 15:211–240.
- Feigin VL, Rinkel GJ, Lawes CM, et al. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke 2005; 36:2773–2780.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Clinical decision rules to rule out subarachnoid hemorrhage for acute headache. JAMA 2013; 310:1248–1255.
- Kowalski RG, Claassen J, Kreiter KT, et al. Initial misdiagnosis and outcome after subarachnoid hemorrhage. JAMA 2004; 291:866–869.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- van Gijn J, van Dongen KJ. The time course of aneurysmal haemorrhage on computed tomograms. Neuroradiology 1982; 23:153–156.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Agid R, Andersson T, Almqvist H, et al. Negative CT angiography findings in patients with spontaneous subarachnoid hemorrhage: when is digital subtraction angiography still needed? AJNR Am J Neuroradiol 2010; 31:696–705.
- Teasdale GM, Drake CG, Hunt W, et al. A universal subarachnoid hemorrhage scale: report of a committee of the World Federation of Neurosurgical Societies. J Neurol Neurosurg Psychiatry 1988; 51:1457.
- Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 1968; 28:14–20.
- Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980; 6:1–9.
- Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery 2006; 59:21–27.
- de Oliveira Manoel AL, Turkel-Parrella D, Kouzmina E, et al. The VASOGRADE—a simple, reliable grading scale for aneurysmal subarachnoid hemorrhage. Neurology 2014; 82(suppl 10): P5.123.
- Naidech AM, Janjua N, Kreiter KT, et al. Predictors and impact of aneurysm rebleeding after subarachnoid hemorrhage. Arch Neurol 2005; 62:410–416.
- Rincon F, Mayer SA. Neurocritical care: a distinct discipline? Curr Opin Crit Care 2007; 13:115–121.
- Rabinstein AA, Lanzino G, Wijdicks EF. Multidisciplinary management and emerging therapeutic strategies in aneurysmal subarachnoid haemorrhage. Lancet Neurol 2010; 9:504–519.
- Vespa P, Diringer MN; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. High-volume centers. Neurocrit Care 2011; 15:369–372.
- McNeill L, English SW, Borg N, Matta BF, Menon DK. Effects of institutional caseload of subarachnoid hemorrhage on mortality: a secondary analysis of administrative data. Stroke 2013; 44:647–652.
- Dorhout Mees SM, Molyneux AJ, Kerr RS, Algra A, Rinkel GJ. Timing of aneurysm treatment after subarachnoid hemorrhage: relationship with delayed cerebral ischemia and poor outcome. Stroke 2012; 43:2126–2129.
- Laidlaw JD, Siu KH. Ultra-early surgery for aneurysmal subarachnoid hemorrhage: outcomes for a consecutive series of 391 patients not selected by grade or age. J Neurosurg 2002; 97:250–259.
- Molyneux A, Kerr R, Stratton I, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised trial. Lancet 2002; 360:1267–1274.
- Molyneux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid aneurysm trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Koivisto T, Vanninen R, Hurskainen H, Saari T, Hernesniemi J, Vapalahti M. Outcomes of early endovascular versus surgical treatment of ruptured cerebral aneurysms. A prospective randomized study. Stroke 2000; 31:2369–2377.
- McDougall CG, Spetzler RF, Zabramski JM, et al. The Barrow Ruptured Aneurysm Trial. J Neurosurg 2012; 116:135–144.
- Spetzler RF, McDougall CG, Albuquerque FC, et al. The Barrow Ruptured Aneurysm Trial: 3-year results. J Neurosurg 2013; 119:146–157.
- Connolly ES Jr, Meyers PM. Cerebral aneurysms: to clip or to coil? That is no longer the question. Nat Rev Neurol 2009; 5:412–413.
- Kassell NF, Peerless SJ, Durward QJ, Beck DW, Drake CG, Adams HP. Treatment of ischemic deficits from vasospasm with intravascular volume expansion and induced arterial hypertension. Neurosurgery 1982; 11:337–343.
- Otsubo H, Takemae T, Inoue T, Kobayashi S, Sugita K. Normovolaemic induced hypertension therapy for cerebral vasospasm after subarachnoid haemorrhage. Acta Neurochir (Wien) 1990; 103:18–26.
- Anderson CS, Heeley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Ortega-Gutierrez S, Thomas J, Reccius A, et al. Effectiveness and safety of nicardipine and labetalol infusion for blood pressure management in patients with intracerebral and subarachnoid hemorrhage. Neurocrit Care 2013; 18:13–19.
- Hillman J, Fridriksson S, Nilsson O, Yu Z, Saveland H, Jakobsson KE. Immediate administration of tranexamic acid and reduced incidence of early rebleeding after aneurysmal subarachnoid hemorrhage: a prospective randomized study. J Neurosurg 2002; 97:771–778.
- Baharoglu MI, Germans MR, Rinkel GJ, et al. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2013; 8:CD001245.
- Germans MR, Post R, Coert BA, Rinkel GJ, Vandertop WP, Verbaan D. Ultra-early tranexamic acid after subarachnoid hemorrhage (ULTRA): study protocol for a randomized controlled trial. Trials 2013; 14:143.
- Sloan MA, Alexandrov AV, Tegeler CH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004; 62:1468–1481.
- Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med 2006; 34:617–623;
- Hellingman CA, van den Bergh WM, Beijer IS, et al. Risk of rebleeding after treatment of acute hydrocephalus in patients with aneurysmal subarachnoid hemorrhage. Stroke 2007; 38:96–99.
- Ransom ER, Mocco J, Komotar RJ, et al. External ventricular drainage response in poor grade aneurysmal subarachnoid hemorrhage: effect on preoperative grading and prognosis. Neurocrit Care 2007; 6:174–180.
- McIver JI, Friedman JA, Wijdicks EF, et al. Preoperative ventriculostomy and rebleeding after aneurysmal subarachnoid hemorrhage. J Neurosurg 2002; 97:1042–1044.
- Klopfenstein JD, Kim LJ, Feiz-Erfan I, et al. Comparison of rapid and gradual weaning from external ventricular drainage in patients with aneurysmal subarachnoid hemorrhage: a prospective randomized trial. J Neurosurg 2004; 100:225–229.
- Tseng MY, Al-Rawi PG, Czosnyka M, et al. Enhancement of cerebral blood flow using systemic hypertonic saline therapy improves outcome in patients with poor-grade spontaneous subarachnoid hemorrhage. J Neurosurg 2007; 107:274–282.
- Al-Rawi PG, Tseng MY, Richards HK, et al. Hypertonic saline in patients with poor-grade subarachnoid hemorrhage improves cerebral blood flow, brain tissue oxygen, and pH. Stroke 2010; 41:122–128.
- Tseng MY, Al-Rawi PG, Pickard JD, Rasulo FA, Kirkpatrick PJ. Effect of hypertonic saline on cerebral blood flow in poor-grade patients with subarachnoid hemorrhage. Stroke 2003; 34:1389–1396.
- Bentsen G, Breivik H, Lundar T, Stubhaug A. Hypertonic saline (7.2%) in 6% hydroxyethyl starch reduces intracranial pressure and improves hemodynamics in a placebo-controlled study involving stable patients with subarachnoid hemorrhage. Crit Care Med 2006; 34:2912–2917.
- Suarez JI, Qureshi AI, Parekh PD, et al. Administration of hypertonic (3%) sodium chloride/acetate in hyponatremic patients with symptomatic vasospasm following subarachnoid hemorrhage. J Neurosurg Anesthesiol 1999; 11:178–184.
- Stevens RD, Huff JS, Duckworth J, Papangelou A, Weingart SD, Smith WS. Emergency neurological life support: intracranial hypertension and herniation. Neurocrit Care 2012;17(suppl 1):S60–S65.
- Seule MA, Muroi C, Mink S, Yonekawa Y, Keller E. Therapeutic hypothermia in patients with aneurysmal subarachnoid hemorrhage, refractory intracranial hypertension, or cerebral vasospasm. Neurosurgery 2009; 64:86–93.
- Otani N, Takasato Y, Masaoka H, et al. Surgical outcome following decompressive craniectomy for poor-grade aneurysmal subarachnoid hemorrhage in patients with associated massive intracerebral or Sylvian hematomas. Cerebrovasc Dis 2008; 26:612–617.
- Lanzino G, D’Urso PI, Suarez J; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Seizures and anticonvulsants after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2011; 15:247–256.
- Naidech AM, Kreiter KT, Janjua N, et al. Phenytoin exposure is associated with functional and cognitive disability after subarachnoid hemorrhage. Stroke 2005; 36:583–587.
- Rosengart AJ, Huo JD, Tolentino J, et al. Outcome in patients with subarachnoid hemorrhage treated with antiepileptic drugs. J Neurosurg 2007; 107:253–260.
- Shah D, Husain AM. Utility of levetiracetam in patients with subarachnoid hemorrhage. Seizure 2009; 18:676–679.
- Patsalos PN. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol Ther 2000; 85:77–85.
- Dewan MC, Mocco J. Current practice regarding seizure prophylaxis in aneurysmal subarachnoid hemorrhage across academic centers. J Neurointerv Surg 2014 Jan 28. doi: 10.1136/neurintsurg-2013-011075 [Epub ahead of print]
- Murphy-Human T, Welch E, Zipfel G, Diringer MN, Dhar R. Comparison of short-duration levetiracetam with extended-course phenytoin for seizure prophylaxis after subarachnoid hemorrhage. World Neurosurg 2011; 75:269–274.
- Dennis LJ, Claassen J, Hirsch LJ, Emerson RG, Connolly ES, Mayer SA. Nonconvulsive status epilepticus after subarachnoid hemorrhage. Neurosurgery 2002; 51:1136–1144.
- Vespa PM, Nuwer MR, Juhász C, et al. Early detection of vasospasm after acute subarachnoid hemorrhage using continuous EEG ICU monitoring. Electroencephalogr Clin Neurophysiol 1997; 103:607–615.
- Claassen J, Hirsch LJ, Kreiter KT, et al. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin Neurophysiol 2004; 115:2699–2710.
- Vergouwen MD, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke 2010; 41:2391–2395.
- Kelly PJ, Gorten RJ, Grossman RG, Eisenberg HM. Cerebral perfusion, vascular spasm, and outcome in patients with ruptured intracranial aneurysms. J Neurosurg 1977; 47:44–49.
- Aralasmak A, Akyuz M, Ozkaynak C, Sindel T, Tuncer R. CT angiography and perfusion imaging in patients with subarachnoid hemorrhage: correlation of vasospasm to perfusion abnormality. Neuroradiology 2009; 51:85–93.
- Sabri M, Lass E, Macdonald RL. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res Treat 2013 Feb 28. doi: 10.1155/2013/394036 [Epub 2013 ahead of print]
- Yoon DY, Choi CS, Kim KH, Cho BM. Multidetector-row CT angiography of cerebral vasospasm after aneurysmal subarachnoid hemorrhage: comparison of volume-rendered images and digital subtraction angiography. AJNR Am J Neuroradiol 2006; 27:370–377.
- Wintermark M1, Ko NU, Smith WS, Liu S, Higashida RT, Dillon WP. Vasospasm after subarachnoid hemorrhage: utility of perfusion CT and CT angiography on diagnosis and management. AJNR Am J Neuroradiol 2006; 27:26–34.
- Helbok R, Madineni RC, Schmidt MJ, et al. Intracerebral monitoring of silent infarcts after subarachnoid hemorrhage. Neurocrit Care 2011; 14:162–167.
- Hänggi D; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Monitoring and detection of vasospasm II: EEG and invasive monitoring. Neurocrit Care 2011; 15:318–323.
- Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G; European Stroke Organization. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis 2013; 35:93–112.
- Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989; 298:636–642.
- Dorhout Mees SM, Rinkel GJ, Feigin VL, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2007; 3:CD000277.
- Lennihan L, Mayer SA, Fink ME, et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage: a randomized controlled trial. Stroke 2000; 31:383–391.
- Egge A, Waterloo K, Sjøholm H, Solberg T, Ingebrigtsen T, Romner B. Prophylactic hyperdynamic postoperative fluid therapy after aneurysmal subarachnoid hemorrhage: a clinical, prospective, randomized, controlled study. Neurosurgery 2001; 49:593–606.
- Jost SC, Diringer MN, Zazulia AR, et al. Effect of normal saline bolus on cerebral blood flow in regions with low baseline flow in patients with vasospasm following subarachnoid hemorrhage. J Neurosurg 2005; 103:25–30.
- Muizelaar JP, Becker DP. Induced hypertension for the treatment of cerebral ischemia after subarachnoid hemorrhage. Direct effect on cerebral blood flow. Surg Neurol 1986; 25:317–325.
- Levy ML, Rabb CH, Zelman V, Giannotta SL. Cardiac performance enhancement from dobutamine in patients refractory to hypervolemic therapy for cerebral vasospasm. J Neurosurg 1993; 79:494–499.
- Lannes M, Teitelbaum J, del Pilar Cortés M, Cardoso M, Angle M. Milrinone and homeostasis to treat cerebral vasospasm associated with subarachnoid hemorrhage: the Montreal Neurological Hospital protocol. Neurocrit Care 2012; 16:354–362.
- Zwienenberg-Lee M, Hartman J, Rudisill N, et al; Balloon Prophylaxis for Aneurysmal Vasospasm (BPAV) Study Group. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage: results of a phase II multicenter, randomized, clinical trial. Stroke 2008; 39:1759–1765.
- Rabinstein AA, Bruder N. Management of hyponatremia and volume contraction. Neurocrit Care 2011; 15:354–360.
- Wijdicks EF, Vermeulen M, Hijdra A, van Gijn J. Hyponatremia and cerebral infarction in patients with ruptured intracranial aneurysms: is fluid restriction harmful? Ann Neurol 1985; 17:137–140.
- Hasan D, Wijdicks EF, Vermeulen M. Hyponatremia is associated with cerebral ischemia in patients with aneurysmal subarachnoid hemorrhage. Ann Neurol 1990; 27:106–108.
- Wijdicks EF, Vermeulen M, ten Haaf JA, Hijdra A, Bakker WH, van Gijn J. Volume depletion and natriuresis in patients with a ruptured intracranial aneurysm. Ann Neurol 1985; 18:211–216.
- Hoff RG, Rinkel GJ, Verweij BH, Algra A, Kalkman CJ. Nurses’ prediction of volume status after aneurysmal subarachnoid haemorrhage: a prospective cohort study. Crit Care 2008; 12:R153.
- Hoff RG, van Dijk GW, Algra A, Kalkman CJ, Rinkel GJ. Fluid balance and blood volume measurement after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2008; 8:391–397.
- Berendes E, Walter M, Cullen P, et al. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet 1997; 349:245–249.
- Espiner EA, Leikis R, Ferch RD, et al. The neuro-cardio-endocrine response to acute subarachnoid haemorrhage. Clin Endocrinol (Oxf) 2002; 56:629–635.
- Isotani E, Suzuki R, Tomita K, et al. Alterations in plasma concentrations of natriuretic peptides and antidiuretic hormone after subarachnoid hemorrhage. Stroke 1994; 25:2198–2203.
- Benedict CR, Loach AB. Sympathetic nervous system activity in patients with subarachnoid hemorrhage. Stroke 1978; 9:237–244.
- Findling JW, Waters VO, Raff H. The dissociation of renin and aldosterone during critical illness. J Clin Endocrinol Metab 1987; 64:592–595.
- Solomon RA, Post KD, McMurtry JG 3rd. Depression of circulating blood volume in patients after subarachnoid hemorrhage: implications for the management of symptomatic vasospasm. Neurosurgery 1984; 15:354–361.
- Peters JP, Welt LG, Sims EA, Orloff J, Needham J. A salt-wasting syndrome associated with cerebral disease. Trans Assoc Am Physicians 1950; 63:57–64.
- Brimioulle S, Orellana-Jimenez C, Aminian A, Vincent JL. Hyponatremia in neurological patients: cerebral salt wasting versus inappropriate antidiuretic hormone secretion. Intensive Care Med 2008; 34:125–131.
- Singh S, Bohn D, Carlotti AP, Cusimano M, Rutka JT, Halperin ML. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med 2002; 30:2575–2579.
- Mutoh T, Kazumata K, Terasaka S, Taki Y, Suzuki A, Ishikawa T. Early intensive versus minimally invasive approach to postoperative hemodynamic management after subarachnoid hemorrhage. Stroke 2014; 45:1280–1284.
- Hasan D, Lindsay KW, Wijdicks EF, et al. Effect of fludrocortisone acetate in patients with subarachnoid hemorrhage. Stroke 1989; 20:1156–1161.
- Moro N, Katayama Y, Kojima J, Mori T, Kawamata T. Prophylactic management of excessive natriuresis with hydrocortisone for efficient hypervolemic therapy after subarachnoid hemorrhage. Stroke 2003; 34:2807–2811.
- Kilbourn KJ, Levy S, Staff I, Kureshi I, McCullough L. Clinical characteristics and outcomes of neurogenic stress cadiomyopathy in aneurysmal subarachnoid hemorrhage. Clin Neurol Neurosurg 2013; 115:909–914.
- Mayer SA, LiMandri G, Sherman D, et al. Electrocardiographic markers of abnormal left ventricular wall motion in acute subarachnoid hemorrhage. J Neurosurg 1995; 83:889–896.
- Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg 2003; 98:741–746.
- Banki N, Kopelnik A, Tung P, et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J Neurosurg 2006; 105:15–20.
- Kahn JM, Caldwell EC, Deem S, Newell DW, Heckbert SR, Rubenfeld GD. Acute lung injury in patients with subarachnoid hemorrhage: incidence, risk factors, and outcome. Crit Care Med 2006; 34:196–202.
- Kitamura Y, Nomura M, Shima H, et al. Acute lung injury associated with systemic inflammatory response syndrome following subarachnoid hemorrhage: a survey by the Shonan Neurosurgical Association. Neurol Med Chir (Tokyo) 2010; 50:456–460.
- Friedman JA, Pichelmann MA, Piepgras DG, et al. Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neurosurgery 2003; 52:1025–1032.
- Oh HS, Jeong HS, Seo WS. Non-infectious hyperthermia in acute brain injury patients: relationships to mortality, blood pressure, intracranial pressure and cerebral perfusion pressure. Int J Nurs Pract 2012; 18:295–302.
- Oddo M, Frangos S, Milby A, et al. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke 2009; 40:1913–1916.
- Badjatia N, Fernandez L, Schmidt JM, et al. Impact of induced normothermia on outcome after subarachnoid hemorrhage: a case-control study. Neurosurgery 2010; 66:696-701.
- Serrone JC1, Wash EM, Hartings JA, Andaluz N, Zuccarello M. Venous thromboembolism in subarachnoid hemorrhage. World Neurosurg 2013; 80:859–863.
- Mack WJ, Ducruet AF, Hickman ZL, et al. Doppler ultrasonography screening of poor-grade subarachnoid hemorrhage patients increases the diagnosis of deep venous thrombosis. Neurol Res 2008; 30:889–892.
- de Oliveira Manoel AL, Turkel-Parrella D, Germans M, et al. Safety of early pharmacological thromboprophylaxis after subarachnoid hemorrhage. Can J Neurol Sci 2014; 41:554–561.
Aneurysmal subarachnoid hemorrhage is a devastating condition, with an estimated death rate of 30% during the initial episode.1,2 Approximately the same number of patients survive but leave the hospital with disabling neurologic deficits.3
However, better outcomes can be achieved by systems that are able to work as a team on the collective goal of quick intervention to secure the ruptured aneurysm, followed by the implementation of measures to minimize secondary brain injury. Although the search for new diagnostic, prognostic, and therapeutic modalities continues, it is clear that there exists no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of small advances that will ultimately maximize the patient’s chances of survival and neurologic recovery.
This review focuses on the management of aneurysmal subarachnoid hemorrhage and its systemic and neurologic complications.
ANEURYSM IS THE MOST COMMON CAUSE OF SUBARACHNOID BLEEDING
Aneurysmal subarachnoid hemorrhage, ie, rupture of an intracranial aneurysm, flooding the subarachnoid space with blood, affects about 24,000 Americans each year.1,2 A ruptured aneurysm is the most common cause of subarachnoid hemorrhage, accounting for about 85% of cases. Less common causes include idiopathic benign perimesencephalic hemorrhage, arteriovenous malformation, dural arteriovenous fistula, and hemorrhagic mycotic aneurysm. These have their own natural history, pathophysiology, and specific treatment, and will not be addressed in this article.
Risk factors for aneurysmal subarachnoid hemorrhage include having a first-degree relative who had the disease, hypertension, smoking, and consuming more than 150 g of alcohol per week.4
CLINICAL PRESENTATION AND DIAGNOSIS
The key symptom of aneurysmal subarachnoid hemorrhage is the abrupt onset of severe headache that peaks in intensity over 1 hour,5 often described as “the worst headache of my life.” Headache is accompanied by brief loss of consciousness in 53% of cases (conversely, nearly half of patients maintain normal mental status), by nausea or vomiting in 77%, and by meningismus (neck pain or stiffness) in 35%.6
These clinical manifestations and risk factors have been incorporated into a decision rule:
Obtain brain imaging if the patient has acute headache reaching maximal intensity within 1 hour, associated with any of the following factors:
- Age 40 or older
- Neck pain or stiffness
- Witnessed loss of consciousness
- Onset during exertion
- “Thunderclap” headache (ie, instantly peaking pain)
- Limited neck flexion on examination.5
This decision rule has nearly 100% sensitivity for aneurysmal subarachnoid hemorrhage in clinical practice.5 All patients require brain imaging if they have a severe headache plus either abnormal neurologic findings (eg, a focal neurologic deficit) or a history of cerebral aneurysm.
Emergency physicians should have a low threshold for ordering noncontrast computed tomography (CT) of the head in patients with even mild symptoms suggesting aneurysmal subarachnoid hemorrhage. Failure to order CT is the most common diagnostic error in this situation.6 CT performed within 6 hours of headache onset is nearly 100% sensitive for this condition,7 but the sensitivity falls to 93% after the first 24 hours and to less than 60% after 5 days.8 In patients who have symptoms highly suggestive of aneurysmal subarachnoid hemorrhage but a normal CT, lumbar puncture is the next diagnostic step.
There are two alternatives to CT followed by lumbar puncture: ie, noncontrast CT followed by CT angiography,9,10 and magnetic resonance imaging followed by magnetic resonance angiography. In patients with suspicious clinical symptoms but negative CT results, CT followed by CT angiography can rule out aneurysmal subarachnoid hemorrhage with a 99% probability.9,10 However, CT followed by lumbar puncture remains the standard of care and carries a class I recommendation in the American Heart Association guidelines for ruling out subarachnoid hemorrhage.5
GRADING THE SEVERITY OF SUBARACHNOID HEMORRHAGE
Age, the thickness of the blood layer in the subarachnoid space, intraventricular hemorrhage and the findings of the neurologic examination at presentation are predictors of long-term outcomes in aneurysmal subarachnoid hemorrhage (Figure 1).
Different grading systems used in clinical practice are based on the findings on the initial neurologic examination and on the initial noncontrast CT (ie, the thickness of the blood, and whether intraventricular hemorrhage is present). Among the most widely used are those developed by Hunt and Hess12 and by the World Federation of Neurological Surgeons11 (WFNS), and the CT grading scales (Fisher13 or its modified version14) (Tables 1 and 2). With either the Hunt and Hess scale or the WFNS scale, the higher the score, the worse the patient’s probable outcome. Scores on both Fisher scales correlate with the risk of angiographic vasospasm. The higher the grade, the higher the risk of angiographic vasospasm.
The VASOGRADE score—a combination of the WFNS score and the modified Fisher scale—stratifies patients at risk of delayed cerebral ischemia, allowing for a tailored monitoring strategy.15 There are three variations:
- VASOGRADE green—Modified Fisher 1 or 2 and WFNS 1 or 2
- VASOGRADE yellow—Modified Fisher 3 or 4 and WFNS 1, 2, or 3
- VASOGRADE red—WFNS 4 or 5.
After the initial bleeding event, patients with aneurysmal subarachnoid hemorrhage are at high risk of delayed systemic and neurologic complications, with poor functional outcomes. Delayed cerebral ischemia holds the greatest risk of an unfavorable outcome and ultimately can lead to cerebral infarction, disability, and death.6,7
INITIAL MANAGEMENT
After aneurysmal subarachnoid hemorrhage is diagnosed, the initial management (Figure 2) includes appropriate medical prevention of rebleeding (which includes supportive care, blood pressure management, and, perhaps, the early use of a short course of an antifibrinolytic drug) and early transfer to a high-volume center for securing the aneurysm. The reported incidence of rebleeding varies from 5% to 22% in the first 72 hours. “Ultra-early” rebleeding (within 24 hours of hemorrhage) has been reported, with an incidence as high as 15% and a fatality rate around 70%. Patients with poor-grade aneurysmal subarachnoid hemorrhage, larger aneurysms, and “sentinel bleeds” are at higher risk of rebleeding.16
Outcomes are much better when patients are managed in a high-volume center, with a specialized neurointensive care unit17 and access to an interdisciplinary team.18 Regardless of the initial grade, patients with aneurysmal subarachnoid hemorrhage should be quickly transferred to a high-volume center, defined as one treating at least 35 cases per year, and the benefit is greater in centers treating more than 60 cases per year.19 The higher the caseload in any given hospital, the better the clinical outcomes in this population.20
Treating cerebral aneurysm: Clipping or coiling
Early aneurysm repair is generally considered the standard of care and the best strategy to reduce the risk of rebleeding. Further, early treatment may be associated with a lower risk of delayed cerebral ischemia21 and better outcomes.22
Three randomized clinical trials have compared surgical clipping and endovascular repair (placement of small metal coils within the aneurysm to promote clotting).
The International Subarachnoid Aneurysm Trial23 showed a reduction of 23% in relative risk and of 7% in absolute risk in patients who underwent endovascular treatment compared with surgery. The survival benefit persisted at a mean of 9 years (range 6–14 years), but with a higher annual rate of aneurysm recurrence in the coiling group (2.9% vs 0.9%).24 Of note, this trial included only patients with aneurysms deemed suitable for both coiling and clipping, so that the exclusion rate was high. Most of the patients presented with good-grade (WFNS score 1–3), small aneurysms (< 5 mm) in the anterior circulation.
A single-center Finnish study25 found no differences in rates of recovery, disability, and death at 1 year, comparing surgery and endovascular treatment. Additionally, survival rates at a mean follow-up of 39 months were similar, with no late recurrences or aneurysmal bleeding.
Lastly, the Barrow Ruptured Aneurysm Trial26,27 found that patients assigned to endovascular treatment had better 1-year neurologic outcomes, defined as a modified Rankin score of 2 or less. Importantly, 37.7% of patients originally assigned to endovascular treatment crossed over to surgical treatment. The authors then performed intention-to-treat and as-treated analyses. Either way, patients treated by endovascular means had better neurologic outcomes at 1 year. However, no difference in the relative risk reduction in worse outcome was found on 3-year follow-up, and patients treated surgically had higher rates of aneurysm obliteration and required less aneurysm retreatment, both of which were statistically significant.
The question that remains is not whether to clip or whether to coil, but whom to clip and whom to coil.28 That question must be answered on a patient-to-patient basis and requires the expertise of an interventional neuroradiologist and a vascular neurosurgeon—one of the reasons these patients are best cared for in high-volume centers providing such expertise.
MEDICAL PREVENTION OF REBLEEDING
Blood pressure management
There are no systematic data on the optimal blood pressure before securing an aneurysm. Early studies of hemodynamic augmentation in cases of ruptured untreated aneurysm reported rebleeding when the systolic blood pressure was allowed to rise above 160 mm Hg.29,30 A recent study evaluating hypertensive intracerebral hemorrhage revealed better functional outcomes with intensive lowering of blood pressure (defined as systolic blood pressure < 140 mm Hg) but no significant reduction in the combined rate of death or severe disability.31 It is difficult to know if these results can be extrapolated to patients with aneurysmal subarachnoid hemorrhage. Current guidelines3,32 say that before the aneurysm is treated, the systolic pressure should be lower than 160 mm Hg.
There is no specific drug of choice, but a short-acting, titratable medication is preferable. Nicardipine is a very good option, and labetalol might be an appropriate alternative.33 Once the aneurysm is secured, all antihypertensive drugs should be held. Hypertension should not be treated unless the patient has clinical signs of a hypertensive crisis, such as flash pulmonary edema, myocardial infarction, or hypertensive encephalopathy.
Antifibrinolytic therapy
The role of antifibrinolytic therapy in aneurysmal subarachnoid hemorrhage is controversial and has been studied in 10 clinical trials. In a Swedish study,34 early use of tranexamic acid (1 g intravenously over 10 minutes followed by 1 g every 6 hours for a maximum of 24 hours) reduced the rebleeding rate substantially, from 10.8% to 2.4%, with an 80% reduction in the mortality rate from ultra-early rebleeding. However, a recent Cochrane review that included this study found no overall benefit.35
An ongoing multicenter randomized trial in the Netherlands will, we hope, answer this question in the near future.36 At present, some centers would consider a short course of tranexamic acid before aneurysm treatment.
DIAGNOSIS AND TREATMENT OF COMPLICATIONS
Medical complications are extremely common after aneurysmal subarachnoid hemorrhage. Between 75% and 100% of patients develop some type of systemic or further neurologic derangement, which in turn has a negative impact on the long-term outcome.37,38 In the first 72 hours, rebleeding is the most feared complication, and as mentioned previously, appropriate blood pressure management and early securing of the aneurysm minimize its risk.
NEUROLOGIC COMPLICATIONS
Hydrocephalus
Hydrocephalus is the most common early neurologic complication after aneurysmal subarachnoid hemorrhage, with an overall incidence of 50%.39 Many patients with poor-grade aneurysmal subarachnoid hemorrhage and patients whose condition deteriorates due to worsening of hydrocephalus require the insertion of an external ventricular drain (Figure 1).
Up to 30% of patients who have a poor-grade aneurysmal subarachnoid hemorrhage improve neurologically with cerebrospinal fluid drainage.40 An external ventricular drain can be safely placed, even before aneurysm treatment, and placement does not appear to increase the risk of rebleeding.39,41 After placement, rapid weaning from the drain (clamping within 24 hours of insertion) is safe, decreases length of stay in the intensive care unit and hospital, and may be more cost-effective than gradual weaning over 96 hours.42
Increased intracranial pressure
Intracranial hypertension is another potential early complication, and is frequently due to the development of hydrocephalus, cerebral edema, or rebleeding. The treatment of increased intracranial pressure does not differ from the approach used in managing severe traumatic brain injury, which includes elevating the head of the bed, sedation, analgesia, normoventilation, and cerebrospinal fluid drainage.
Hypertonic saline has been tested in several studies that were very small but nevertheless consistently showed control of intracranial pressure levels and improvement in cerebral blood flow measured by xenon CT.43–47 Two of these studies even showed better outcomes at discharge.43,44 However, the small number of patients prevents any meaningful conclusion regarding the use of hypertonic saline and functional outcomes.
Barbiturates, hypothermia, and decompressive craniectomy could be tried in refractory cases.48 Seule et al49 evaluated the role of therapeutic hypothermia with or without barbiturate coma in 100 patients with refractory intracranial hypertension. Only 13 patients received hypothermia by itself. At 1 year, 32 patients had achieved a good functional outcome (Glasgow Outcome Scale score 4 or 5). The remaining patients were severely disabled or had died. Of interest, the median duration of hypothermia was 7 days, and 93% of patients developed some medical complication such as electrolyte disorders (77%), pneumonia (52%), thrombocytopenia (47%), or septic shock syndrome (40%). Six patients died as a consequence of one of these complications.
Decompressive craniectomy can be life-saving in patients with refractory intracranial hypertension. However, most of these patients will die or remain severely disabled or comatose.50
Seizure prophylaxis is controversial
Seizures can occur at the onset of intracranial hemorrhage, perioperatively, or later (ie, after the first week). The incidence varied considerably in different reports, ranging from 4% to 26%.51 Seizures occurring perioperatively, ie, after hospital admission, are less frequent and are usually the manifestation of aneurysm rebleeding.24
Seizure prophylaxis remains controversial, especially because the use of phenytoin is associated with increased incidence of cerebral vasospasm, infarction, and worse cognitive outcomes after aneurysmal subarachnoid hemorrhage.52,53 Therefore, routine prophylactic use of phenytoin is not recommended in these patients,3 although the effect of other antiepileptic drugs is less studied and less clear. Patients may be considered for this therapy if they have multiple risk factors for seizures, such as intraparenchymal hematoma, advanced age (> 65), middle cerebral artery aneurysm, craniotomy for aneurysm clipping, and a short course (≤ 72 hours) of an antiepileptic drug other than phenytoin, especially while the aneurysm is unsecured.3
Levetiracetam may be an alternative to phenytoin, having better pharmacodynamic and kinetic profiles, minimal protein binding, and absence of hepatic metabolism, resulting in a very low risk of drug interaction and better tolerability.54,55 Because of these advantages, levetiracetam has become the drug of choice in several centers treating aneurysmal subarachnoid hemorrhage in the United States.
Addressing this question, a survey was sent to 25 high-volume aneurysmal subarachnoid hemorrhage academic centers in the United States. All 25 institutions answered the survey, and interestingly, levetiracetam was the first-line agent for 16 (94%) of the 17 responders that used prophylaxis, while only 1 used phenytoin as the agent of choice.56
A retrospective cohort study by Murphy-Human et al57 showed that a short course of levetiracetam (≤ 72 hours) was associated with higher rates of in-hospital seizures compared with an extended course of phenytoin (eg, entire hospital stay). However, the study did not address functional outcomes.57
Continuous electroencephalographic monitoring may be considered in comatose patients, in patients requiring controlled ventilation and sedation, or in patients with unexplained alteration in consciousness. In one series of patients with aneurysmal subarachnoid hemorrhage who received continuous monitoring, the incidence of nonconvulsive status epilepticus was 19%, with an associated mortality rate of 100%.58
Continuous quantitative electroencephalography is useful to monitor and to detect angiographic vasospasm and delayed cerebral ischemia. Relative alpha variability and the alpha-delta ratio decrease with ischemia, and this effect can precede angiographic vasospasm by 3 days.59,60
Delayed cerebral ischemia
Delayed cerebral ischemia is defined as the occurrence of focal neurologic impairment, or a decrease of at least 2 points on the Glasgow Coma Scale that lasts for at least 1 hour, is not apparent immediately after aneurysm occlusion, and not attributable to other causes (eg, hyponatremia, fever).61
Classically, neurologic deficits that occurred within 2 weeks of aneurysm rupture were ascribed to reduced cerebral blood flow caused by delayed large-vessel vasospasm causing cerebral ischemia.62 However, perfusion abnormalities have also been observed with either mild or no demonstrable vasospasm.63 Almost 70% of patients who survive the initial hemorrhage develop some degree of angiographic vasospasm. However, only 30% of those patients will experience symptoms.
In addition to vasospasm of large cerebral arteries, impaired autoregulation and early brain injury within the first 72 hours following subarachnoid hemorrhage may play important roles in the development of delayed cerebral ischemia.64 Therefore, the modern concept of delayed cerebral ischemia monitoring should focus on cerebral perfusion rather than vessel diameter measurements. This underscores the importance of comprehensive, standardized monitoring techniques that provide information not only on microvasculature, but also at the level of the microcirculation, with information on perfusion, oxygen utilization and extraction, and autoregulation.
Although transcranial Doppler has been the most commonly applied tool to monitor for angiographic vasospasm, it has a low sensitivity and negative predictive value.37 It is nevertheless a useful technique to monitor good-grade aneurysmal subarachnoid hemorrhage patients (WFNS score 1–3) combined with frequent neurologic examinations (Figure 3).
CT angiography is a good noninvasive alternative to digital subtraction angiography. However, it tends to overestimate the degree of vasoconstriction and does not provide information about perfusion and autoregulation.65 Nevertheless, CT angiography combined with a CT perfusion scan can add information about autoregulation and cerebral perfusion and has been shown to be more sensitive for the diagnosis of angiographic vasospasm than transcranial Doppler and digital subtraction angiography (Figure 4).
Patients with a poor clinical condition (WFNS score 4 or 5) or receiving continuous sedation constitute a challenge in monitoring for delayed neurologic deterioration. Neurologic examination is not sensitive enough in this setting to detect subtle changes. In these specific and challenging circumstances, multimodality neuromonitoring may be useful in the early detection of delayed cerebral ischemia and may help guide therapy.67
Several noninvasive and invasive techniques have been studied to monitor patients at risk of delayed cerebral ischemia after subarachnoid hemorrhage.66 These include continuous electroencephalography, brain tissue oxygenation monitoring (Ptio2), cerebral microdialysis, thermal diffusion flowmetry, and near-infrared spectroscopy. Of these techniques, Ptio2, cerebral microdialysis, and continuous electroencephalography (see discussion of seizure prophylaxis above) have been more extensively studied. However, most of the studies were observational and very small, limiting any recommendations for using these techniques in routine clinical practice.68
Ptio2 is measured by inserting an intraparenchymal oxygen-sensitive microelectrode, and microdialysis requires a microcatheter with a semipermeable membrane that allows small soluble substances to cross it into the dialysate. These substances, which include markers of ischemia (ie, glucose, lactate, and pyruvate), excitotoxins (ie, glutamate and aspartate), and membrane cell damage products (ie, glycerol), can be measured. Low Ptio2 values (< 15 mm Hg) and abnormal mycrodialysate findings (eg, glucose < 0.8 mmol/L, lactate-to-pyruvate ratio > 40) have both been associated with cerebral ischemic events and poor outcome.68
Preventing delayed cerebral ischemia
Oral nimodipine 60 mg every 4 hours for 21 days, started on admission, carries a class I, level of evidence A recommendation in the management of aneurysmal subarachnoid hemorrhage.3,32,69 It improves clinical outcome despite having no effect on the risk of angiographic vasospasm. The mechanism of improved outcome is unclear, but the effect may be a neuroprotective phenomenon limiting the extension of delayed cerebral ischemia.70
If hypotension occurs, the dose can be lowered to 30 mg every 2 hours. Whether to discontinue nimodipine in this situation is controversial. Of note, the clinical benefits of nimodipine have not been replicated with other calcium channel blockers (eg, nicardipine).71
Prophylactic hyperdynamic fluid therapy, known as “triple-H” (hypervolemia, hemodilution, and hypertension) was for years the mainstay of treatment in preventing delayed cerebral ischemia due to vasospasm. However, the clinical data supporting this intervention have been called into question, as analysis of two trials found that hypervolemia did not improve outcomes or reduce the incidence of delayed cerebral ischemia, and in fact increased the rate of complications.72,73 Based on these findings, current guidelines recommend maintaining euvolemia rather than prophylactic hypervolemia in patients with aneurysmal subarachnoid hemorrhage.3,32,69
TREATING DELAYED CEREBRAL ISCHEMIA
Hemodynamic augmentation
In patients with neurologic deterioration due to delayed cerebral ischemia, hemodynamic augmentation is the cornerstone of treatment. This is done according to a protocol, started early, involving specific physiologic goals, clinical improvement, and escalation to invasive therapies in a timely fashion in patients at high risk of further neurologic insult (Figure 5).
The physiologic goal is to increase the delivery of oxygen and glucose to the ischemic brain. Hypertension seems to be the most effective component of hemodynamic augmentation regardless of volume status, increasing cerebral blood flow and brain tissue oxygenation, with reversal of delayed cerebral ischemic symptoms in up to two-thirds of treated patients.74,75 However, this information comes from very small studies, with no randomized trials of induced hypertension available.
The effect of a normal saline fluid bolus in patients suspected of having delayed cerebral ischemia has been shown to increase cerebral blood flow in areas of cerebral ischemia.74 If volume augmentation fails to improve the neurologic status, the next step is to artificially induce hypertension using vasopressors. The blood pressure target should be based on clinical improvement. A stepwise approach is reasonable in this situation, and the lowest level of blood pressure at which there is a complete reversal of the new focal neurologic deficit should be maintained.3,29
Inotropic agents such as dobutamine or milrinone can be considered as alternatives in patients who have new neurologic deficits that are refractory to fluid boluses and vasopressors, or in a setting of subarachnoid hemorrhage-induced cardiomyopathy.76,77
Once the neurologic deficit is reversed by hemodynamic augmentation, the blood pressure should be maintained for 48 to 72 hours at the level that reversed the deficit completely, carefully reassessed thereafter, and the patient weaned slowly. Unruptured unsecured aneurysms should not prevent blood pressure augmentation in a setting of delayed cerebral ischemia if the culprit aneurysm is treated.3,32 If the ruptured aneurysm has not been secured, careful blood pressure augmentation can be attempted, keeping in mind that hypertension (> 160/95 mm Hg) is a risk factor for fatal aneurysm rupture.
Endovascular management of delayed cerebral ischemia
When medical augmentation fails to completely reverse the neurologic deficits, endovascular treatment can be considered. Although patients treated early in the course of delayed cerebral ischemia have better neurologic recovery, prophylactic endovascular treatment in asymptomatic patients, even if angiographic signs of spasm are present, does not improve clinical outcomes and carries the risk of fatal arterial rupture.78
SYSTEMIC COMPLICATIONS
Hyponatremia and hypovolemia
Aneurysmal subarachnoid hemorrhage is commonly associated with abnormalities of fluid balance and electrolyte derangements. Hyponatremia (serum sodium < 135 mmol/L) occurs in 30% to 50% of patients, while the rate of hypovolemia (decreased circulating blood volume) ranges from 17% to 30%.79 Both can negatively affect long-term outcomes.80,81
Decreased circulating blood volume is a well-described contributor to delayed cerebral ischemia and cerebral infarction after aneurysmal subarachnoid hemorrhage.80–82 Clinical variables such as heart rate, blood pressure, fluid balance, and serum sodium concentration are usually the cornerstones of intravascular volume status assessment. However, these variables correlate poorly with measured circulating blood volumes in those with aneurysmal subarachnoid hemorrhage.83,84
The mechanisms responsible for the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage are not completely understood. Several factors have been described and may contribute to the increased natriuresis and, hence, to a reduction in circulating blood volume: increased circulating natriuretic peptide concentrations,85–87 sympathetic nervous system hyperactivation,88 and hyperreninemic hypo-
aldosteronism syndrome.89,90
Lastly, the cerebral salt wasting syndrome, described in the 1950s,91 was thought to be a key mechanism in the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage. In contrast to the syndrome of inappropriate antidiuretic hormone, which is characterized by hyponatremia with a normal or slightly elevated intravascular volume, the characteristic feature of cerebral salt wasting syndrome is the development of hyponatremia in a setting of intravascular volume depletion.92 In critically ill neurologic and neurosurgical patients, this differential diagnosis is very difficult, especially in those with aneurysmal subarachnoid hemorrhage in whom the clinical assessment of fluid status is not reliable. These two syndromes might coexist and contribute to the development of hyponatremia after aneurysmal subarachnoid hemorrhage.92,93
Hoff et al83,84 prospectively compared the clinical assessment of fluid status by critical and intermediate care nurses and direct measurements of blood volume using pulse dye densitometry. The clinical assessment failed to accurately assess patients’ volume status. Using the same technique to measure circulating blood volume, this group showed that calculation of fluid balance does not provide adequate assessment of fluid status.83,84
Hemodynamic monitoring tools can help guide fluid replacement in this population. Mutoh et al94 randomized 160 patients within 24 hours of hemorrhage to receive early goal-directed fluid therapy (ie, preload volume and cardiac output monitored by transpulmonary thermodilution) vs standard therapy (ie, fluid balance or central venous pressure). Overall, no difference was found in the rates of delayed cerebral ischemia (33% vs 42%; P = .33) or favorable outcome (67% vs 57%; P = .22). However, in the subgroup of poor-grade patients (WFNS score 4 or 5), early goal-directed therapy was associated with a lower rate of delayed cerebral ischemia (5% vs 14%; P = .036) and with better functional outcomes at 3 months (52% vs 36%; P = .026).
Fluid restriction to treat hyponatremia in aneurysmal subarachnoid hemorrhage is no longer recommended because of the increased risk of cerebral infarction due to hypovolemic hypoperfusion.82
Prophylactic use of mineralocorticoids (eg, fludrocortisone, hydrocortisone) has been shown to limit natriuresis, hyponatremia, and the amount of fluid required to maintain euvolemia.95,96 Higher rates of hypokalemia and hyperglycemia, which can be easily treated, are the most common complications associated with this approach. Additionally, hypertonic saline (eg, 3% saline) can be used to correct hyponatremia in a setting of aneurysmal subarachnoid hemorrhage.79
Cardiac complications
Cardiac complications after subarachnoid hemorrhage are most likely related to sympathetic hyperactivity and catecholamine-induced myocyte dysfunction. The pathophysiology is complex, but cardiac complications have a significant negative impact on long-term outcome in these patients.97
Electrocardiographic changes and positive cardiac enzymes associated with aneurysmal subarachnoid hemorrhage have been extensively reported. More recently, data from studies of two-dimensional echocardiography have shown that subarachnoid hemorrhage can also be associated with significant wall-motion abnormalities and even overt cardiogenic shock.98–100
There is no specific curative therapy; the treatment is mainly supportive. Vasopressors and inotropes may be used for hemodynamic augmentation.
Pulmonary complications
Pulmonary complications occur in 20% to 30% of all aneurysmal subarachnoid hemorrhage patients and are associated with a higher risk of delayed cerebral ischemia and death. Common pulmonary complications in this population are mild acute respiratory distress syndrome (27%), hospital-acquired pneumonia (9%), cardiogenic pulmonary edema (8%), aspiration pneumonia (6%), neurogenic pulmonary edema (2%), and pulmonary embolism (1%).101–103
SUPPORTIVE CARE
Hyperthermia, hyperglycemia, and liberal use of transfusions have all been associated with longer stays in the intensive care unit and hospital, poorer neurologic outcomes, and higher mortality rates in patients with acute brain injury.104 Noninfectious fever is the most common systemic complication after subarachnoid hemorrhage.
Antipyretic drugs such as acetaminophen and ibuprofen are not very effective in reducing fever in the subarachnoid hemorrhage population, but should still be used as first-line therapy. The use of surface and intravascular devices can be considered when fevers do not respond to nonsteroidal anti-inflammatory drugs.
Although no prospective randomized trial has addressed the impact of induced normothermia on long-term outcome and mortality in aneurysmal subarachnoid hemorrhage patients, fever control has been shown to reduce cerebral metabolic distress, irrespective of intracranial pressure.105 Maintenance of normothermia (< 37.5°C) seems reasonable, especially in aneurysmal subarachnoid hemorrhage patients at risk of or with active delayed cerebral ischemia.106
Current guidelines3,32,69 strongly recommend avoiding hypoglycemia, defined as a serum glucose level less than 80 mg/dL, but suggest keeping the blood sugar level below 180 or 200 mg/dL.
At the moment, there is no clear threshold for transfusion in patients with aneurysmal subarachnoid hemorrhage. Current guidelines suggest keeping hemoglobin levels between 8 and 10 g/dL.3
Preventing venous thromboembolism
The incidence of venous thromboembolism after aneurysmal subarachnoid hemorrhage varies widely, from 1.5% to 18%.107 Active surveillance with venous Doppler ultrasonography has found asymptomatic deep vein thrombosis in up to 3.4% of poor-grade aneurysmal subarachnoid hemorrhage patients receiving pharmacologic thromboprophylaxis.108
In a retrospective study of 170 patients, our group showed that giving drugs to prevent venous thromboembolism (unfractionated heparin 5,000 IU subcutaneously every 12 hours or dalteparin 5,000 IU subcutaneously daily), starting within 24 hours of aneurysm treatment, could be safe.109 Fifty-eight percent of these patients had an external ventricular drain in place. One patient developed a major cerebral hemorrhagic complication and died while on unfractionated heparin; however, the patient was also on dual antiplatelet therapy with aspirin and clopidogrel.109
Current guidelines suggest that intermittent compression devices be applied in all patients before aneurysm treatment. Pharmacologic thromboprophylaxis with a heparinoid can be started 12 to 24 hours after aneurysm treatment.3,109
A TEAM APPROACH
Patients with subarachnoid hemorrhage need integrated care from different medical and nursing specialties. The best outcomes are achieved by systems that can focus as a team on the collective goal of quick intervention to secure the aneurysm, followed by measures to minimize secondary brain injury.
The modern concept of cerebral monitoring in a setting of subarachnoid hemorrhage should focus on brain perfusion rather than vascular diameter. Although the search continues for new diagnostic, prognostic, and therapeutic tools, there is no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of many small advances that will ultimately lead to better outcomes.
ACKNOWLEDGMENT
This work was supported by research funding provided by the Bitove Foundation, which has been supportive of our clinical and research work for several years.
Aneurysmal subarachnoid hemorrhage is a devastating condition, with an estimated death rate of 30% during the initial episode.1,2 Approximately the same number of patients survive but leave the hospital with disabling neurologic deficits.3
However, better outcomes can be achieved by systems that are able to work as a team on the collective goal of quick intervention to secure the ruptured aneurysm, followed by the implementation of measures to minimize secondary brain injury. Although the search for new diagnostic, prognostic, and therapeutic modalities continues, it is clear that there exists no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of small advances that will ultimately maximize the patient’s chances of survival and neurologic recovery.
This review focuses on the management of aneurysmal subarachnoid hemorrhage and its systemic and neurologic complications.
ANEURYSM IS THE MOST COMMON CAUSE OF SUBARACHNOID BLEEDING
Aneurysmal subarachnoid hemorrhage, ie, rupture of an intracranial aneurysm, flooding the subarachnoid space with blood, affects about 24,000 Americans each year.1,2 A ruptured aneurysm is the most common cause of subarachnoid hemorrhage, accounting for about 85% of cases. Less common causes include idiopathic benign perimesencephalic hemorrhage, arteriovenous malformation, dural arteriovenous fistula, and hemorrhagic mycotic aneurysm. These have their own natural history, pathophysiology, and specific treatment, and will not be addressed in this article.
Risk factors for aneurysmal subarachnoid hemorrhage include having a first-degree relative who had the disease, hypertension, smoking, and consuming more than 150 g of alcohol per week.4
CLINICAL PRESENTATION AND DIAGNOSIS
The key symptom of aneurysmal subarachnoid hemorrhage is the abrupt onset of severe headache that peaks in intensity over 1 hour,5 often described as “the worst headache of my life.” Headache is accompanied by brief loss of consciousness in 53% of cases (conversely, nearly half of patients maintain normal mental status), by nausea or vomiting in 77%, and by meningismus (neck pain or stiffness) in 35%.6
These clinical manifestations and risk factors have been incorporated into a decision rule:
Obtain brain imaging if the patient has acute headache reaching maximal intensity within 1 hour, associated with any of the following factors:
- Age 40 or older
- Neck pain or stiffness
- Witnessed loss of consciousness
- Onset during exertion
- “Thunderclap” headache (ie, instantly peaking pain)
- Limited neck flexion on examination.5
This decision rule has nearly 100% sensitivity for aneurysmal subarachnoid hemorrhage in clinical practice.5 All patients require brain imaging if they have a severe headache plus either abnormal neurologic findings (eg, a focal neurologic deficit) or a history of cerebral aneurysm.
Emergency physicians should have a low threshold for ordering noncontrast computed tomography (CT) of the head in patients with even mild symptoms suggesting aneurysmal subarachnoid hemorrhage. Failure to order CT is the most common diagnostic error in this situation.6 CT performed within 6 hours of headache onset is nearly 100% sensitive for this condition,7 but the sensitivity falls to 93% after the first 24 hours and to less than 60% after 5 days.8 In patients who have symptoms highly suggestive of aneurysmal subarachnoid hemorrhage but a normal CT, lumbar puncture is the next diagnostic step.
There are two alternatives to CT followed by lumbar puncture: ie, noncontrast CT followed by CT angiography,9,10 and magnetic resonance imaging followed by magnetic resonance angiography. In patients with suspicious clinical symptoms but negative CT results, CT followed by CT angiography can rule out aneurysmal subarachnoid hemorrhage with a 99% probability.9,10 However, CT followed by lumbar puncture remains the standard of care and carries a class I recommendation in the American Heart Association guidelines for ruling out subarachnoid hemorrhage.5
GRADING THE SEVERITY OF SUBARACHNOID HEMORRHAGE
Age, the thickness of the blood layer in the subarachnoid space, intraventricular hemorrhage and the findings of the neurologic examination at presentation are predictors of long-term outcomes in aneurysmal subarachnoid hemorrhage (Figure 1).
Different grading systems used in clinical practice are based on the findings on the initial neurologic examination and on the initial noncontrast CT (ie, the thickness of the blood, and whether intraventricular hemorrhage is present). Among the most widely used are those developed by Hunt and Hess12 and by the World Federation of Neurological Surgeons11 (WFNS), and the CT grading scales (Fisher13 or its modified version14) (Tables 1 and 2). With either the Hunt and Hess scale or the WFNS scale, the higher the score, the worse the patient’s probable outcome. Scores on both Fisher scales correlate with the risk of angiographic vasospasm. The higher the grade, the higher the risk of angiographic vasospasm.
The VASOGRADE score—a combination of the WFNS score and the modified Fisher scale—stratifies patients at risk of delayed cerebral ischemia, allowing for a tailored monitoring strategy.15 There are three variations:
- VASOGRADE green—Modified Fisher 1 or 2 and WFNS 1 or 2
- VASOGRADE yellow—Modified Fisher 3 or 4 and WFNS 1, 2, or 3
- VASOGRADE red—WFNS 4 or 5.
After the initial bleeding event, patients with aneurysmal subarachnoid hemorrhage are at high risk of delayed systemic and neurologic complications, with poor functional outcomes. Delayed cerebral ischemia holds the greatest risk of an unfavorable outcome and ultimately can lead to cerebral infarction, disability, and death.6,7
INITIAL MANAGEMENT
After aneurysmal subarachnoid hemorrhage is diagnosed, the initial management (Figure 2) includes appropriate medical prevention of rebleeding (which includes supportive care, blood pressure management, and, perhaps, the early use of a short course of an antifibrinolytic drug) and early transfer to a high-volume center for securing the aneurysm. The reported incidence of rebleeding varies from 5% to 22% in the first 72 hours. “Ultra-early” rebleeding (within 24 hours of hemorrhage) has been reported, with an incidence as high as 15% and a fatality rate around 70%. Patients with poor-grade aneurysmal subarachnoid hemorrhage, larger aneurysms, and “sentinel bleeds” are at higher risk of rebleeding.16
Outcomes are much better when patients are managed in a high-volume center, with a specialized neurointensive care unit17 and access to an interdisciplinary team.18 Regardless of the initial grade, patients with aneurysmal subarachnoid hemorrhage should be quickly transferred to a high-volume center, defined as one treating at least 35 cases per year, and the benefit is greater in centers treating more than 60 cases per year.19 The higher the caseload in any given hospital, the better the clinical outcomes in this population.20
Treating cerebral aneurysm: Clipping or coiling
Early aneurysm repair is generally considered the standard of care and the best strategy to reduce the risk of rebleeding. Further, early treatment may be associated with a lower risk of delayed cerebral ischemia21 and better outcomes.22
Three randomized clinical trials have compared surgical clipping and endovascular repair (placement of small metal coils within the aneurysm to promote clotting).
The International Subarachnoid Aneurysm Trial23 showed a reduction of 23% in relative risk and of 7% in absolute risk in patients who underwent endovascular treatment compared with surgery. The survival benefit persisted at a mean of 9 years (range 6–14 years), but with a higher annual rate of aneurysm recurrence in the coiling group (2.9% vs 0.9%).24 Of note, this trial included only patients with aneurysms deemed suitable for both coiling and clipping, so that the exclusion rate was high. Most of the patients presented with good-grade (WFNS score 1–3), small aneurysms (< 5 mm) in the anterior circulation.
A single-center Finnish study25 found no differences in rates of recovery, disability, and death at 1 year, comparing surgery and endovascular treatment. Additionally, survival rates at a mean follow-up of 39 months were similar, with no late recurrences or aneurysmal bleeding.
Lastly, the Barrow Ruptured Aneurysm Trial26,27 found that patients assigned to endovascular treatment had better 1-year neurologic outcomes, defined as a modified Rankin score of 2 or less. Importantly, 37.7% of patients originally assigned to endovascular treatment crossed over to surgical treatment. The authors then performed intention-to-treat and as-treated analyses. Either way, patients treated by endovascular means had better neurologic outcomes at 1 year. However, no difference in the relative risk reduction in worse outcome was found on 3-year follow-up, and patients treated surgically had higher rates of aneurysm obliteration and required less aneurysm retreatment, both of which were statistically significant.
The question that remains is not whether to clip or whether to coil, but whom to clip and whom to coil.28 That question must be answered on a patient-to-patient basis and requires the expertise of an interventional neuroradiologist and a vascular neurosurgeon—one of the reasons these patients are best cared for in high-volume centers providing such expertise.
MEDICAL PREVENTION OF REBLEEDING
Blood pressure management
There are no systematic data on the optimal blood pressure before securing an aneurysm. Early studies of hemodynamic augmentation in cases of ruptured untreated aneurysm reported rebleeding when the systolic blood pressure was allowed to rise above 160 mm Hg.29,30 A recent study evaluating hypertensive intracerebral hemorrhage revealed better functional outcomes with intensive lowering of blood pressure (defined as systolic blood pressure < 140 mm Hg) but no significant reduction in the combined rate of death or severe disability.31 It is difficult to know if these results can be extrapolated to patients with aneurysmal subarachnoid hemorrhage. Current guidelines3,32 say that before the aneurysm is treated, the systolic pressure should be lower than 160 mm Hg.
There is no specific drug of choice, but a short-acting, titratable medication is preferable. Nicardipine is a very good option, and labetalol might be an appropriate alternative.33 Once the aneurysm is secured, all antihypertensive drugs should be held. Hypertension should not be treated unless the patient has clinical signs of a hypertensive crisis, such as flash pulmonary edema, myocardial infarction, or hypertensive encephalopathy.
Antifibrinolytic therapy
The role of antifibrinolytic therapy in aneurysmal subarachnoid hemorrhage is controversial and has been studied in 10 clinical trials. In a Swedish study,34 early use of tranexamic acid (1 g intravenously over 10 minutes followed by 1 g every 6 hours for a maximum of 24 hours) reduced the rebleeding rate substantially, from 10.8% to 2.4%, with an 80% reduction in the mortality rate from ultra-early rebleeding. However, a recent Cochrane review that included this study found no overall benefit.35
An ongoing multicenter randomized trial in the Netherlands will, we hope, answer this question in the near future.36 At present, some centers would consider a short course of tranexamic acid before aneurysm treatment.
DIAGNOSIS AND TREATMENT OF COMPLICATIONS
Medical complications are extremely common after aneurysmal subarachnoid hemorrhage. Between 75% and 100% of patients develop some type of systemic or further neurologic derangement, which in turn has a negative impact on the long-term outcome.37,38 In the first 72 hours, rebleeding is the most feared complication, and as mentioned previously, appropriate blood pressure management and early securing of the aneurysm minimize its risk.
NEUROLOGIC COMPLICATIONS
Hydrocephalus
Hydrocephalus is the most common early neurologic complication after aneurysmal subarachnoid hemorrhage, with an overall incidence of 50%.39 Many patients with poor-grade aneurysmal subarachnoid hemorrhage and patients whose condition deteriorates due to worsening of hydrocephalus require the insertion of an external ventricular drain (Figure 1).
Up to 30% of patients who have a poor-grade aneurysmal subarachnoid hemorrhage improve neurologically with cerebrospinal fluid drainage.40 An external ventricular drain can be safely placed, even before aneurysm treatment, and placement does not appear to increase the risk of rebleeding.39,41 After placement, rapid weaning from the drain (clamping within 24 hours of insertion) is safe, decreases length of stay in the intensive care unit and hospital, and may be more cost-effective than gradual weaning over 96 hours.42
Increased intracranial pressure
Intracranial hypertension is another potential early complication, and is frequently due to the development of hydrocephalus, cerebral edema, or rebleeding. The treatment of increased intracranial pressure does not differ from the approach used in managing severe traumatic brain injury, which includes elevating the head of the bed, sedation, analgesia, normoventilation, and cerebrospinal fluid drainage.
Hypertonic saline has been tested in several studies that were very small but nevertheless consistently showed control of intracranial pressure levels and improvement in cerebral blood flow measured by xenon CT.43–47 Two of these studies even showed better outcomes at discharge.43,44 However, the small number of patients prevents any meaningful conclusion regarding the use of hypertonic saline and functional outcomes.
Barbiturates, hypothermia, and decompressive craniectomy could be tried in refractory cases.48 Seule et al49 evaluated the role of therapeutic hypothermia with or without barbiturate coma in 100 patients with refractory intracranial hypertension. Only 13 patients received hypothermia by itself. At 1 year, 32 patients had achieved a good functional outcome (Glasgow Outcome Scale score 4 or 5). The remaining patients were severely disabled or had died. Of interest, the median duration of hypothermia was 7 days, and 93% of patients developed some medical complication such as electrolyte disorders (77%), pneumonia (52%), thrombocytopenia (47%), or septic shock syndrome (40%). Six patients died as a consequence of one of these complications.
Decompressive craniectomy can be life-saving in patients with refractory intracranial hypertension. However, most of these patients will die or remain severely disabled or comatose.50
Seizure prophylaxis is controversial
Seizures can occur at the onset of intracranial hemorrhage, perioperatively, or later (ie, after the first week). The incidence varied considerably in different reports, ranging from 4% to 26%.51 Seizures occurring perioperatively, ie, after hospital admission, are less frequent and are usually the manifestation of aneurysm rebleeding.24
Seizure prophylaxis remains controversial, especially because the use of phenytoin is associated with increased incidence of cerebral vasospasm, infarction, and worse cognitive outcomes after aneurysmal subarachnoid hemorrhage.52,53 Therefore, routine prophylactic use of phenytoin is not recommended in these patients,3 although the effect of other antiepileptic drugs is less studied and less clear. Patients may be considered for this therapy if they have multiple risk factors for seizures, such as intraparenchymal hematoma, advanced age (> 65), middle cerebral artery aneurysm, craniotomy for aneurysm clipping, and a short course (≤ 72 hours) of an antiepileptic drug other than phenytoin, especially while the aneurysm is unsecured.3
Levetiracetam may be an alternative to phenytoin, having better pharmacodynamic and kinetic profiles, minimal protein binding, and absence of hepatic metabolism, resulting in a very low risk of drug interaction and better tolerability.54,55 Because of these advantages, levetiracetam has become the drug of choice in several centers treating aneurysmal subarachnoid hemorrhage in the United States.
Addressing this question, a survey was sent to 25 high-volume aneurysmal subarachnoid hemorrhage academic centers in the United States. All 25 institutions answered the survey, and interestingly, levetiracetam was the first-line agent for 16 (94%) of the 17 responders that used prophylaxis, while only 1 used phenytoin as the agent of choice.56
A retrospective cohort study by Murphy-Human et al57 showed that a short course of levetiracetam (≤ 72 hours) was associated with higher rates of in-hospital seizures compared with an extended course of phenytoin (eg, entire hospital stay). However, the study did not address functional outcomes.57
Continuous electroencephalographic monitoring may be considered in comatose patients, in patients requiring controlled ventilation and sedation, or in patients with unexplained alteration in consciousness. In one series of patients with aneurysmal subarachnoid hemorrhage who received continuous monitoring, the incidence of nonconvulsive status epilepticus was 19%, with an associated mortality rate of 100%.58
Continuous quantitative electroencephalography is useful to monitor and to detect angiographic vasospasm and delayed cerebral ischemia. Relative alpha variability and the alpha-delta ratio decrease with ischemia, and this effect can precede angiographic vasospasm by 3 days.59,60
Delayed cerebral ischemia
Delayed cerebral ischemia is defined as the occurrence of focal neurologic impairment, or a decrease of at least 2 points on the Glasgow Coma Scale that lasts for at least 1 hour, is not apparent immediately after aneurysm occlusion, and not attributable to other causes (eg, hyponatremia, fever).61
Classically, neurologic deficits that occurred within 2 weeks of aneurysm rupture were ascribed to reduced cerebral blood flow caused by delayed large-vessel vasospasm causing cerebral ischemia.62 However, perfusion abnormalities have also been observed with either mild or no demonstrable vasospasm.63 Almost 70% of patients who survive the initial hemorrhage develop some degree of angiographic vasospasm. However, only 30% of those patients will experience symptoms.
In addition to vasospasm of large cerebral arteries, impaired autoregulation and early brain injury within the first 72 hours following subarachnoid hemorrhage may play important roles in the development of delayed cerebral ischemia.64 Therefore, the modern concept of delayed cerebral ischemia monitoring should focus on cerebral perfusion rather than vessel diameter measurements. This underscores the importance of comprehensive, standardized monitoring techniques that provide information not only on microvasculature, but also at the level of the microcirculation, with information on perfusion, oxygen utilization and extraction, and autoregulation.
Although transcranial Doppler has been the most commonly applied tool to monitor for angiographic vasospasm, it has a low sensitivity and negative predictive value.37 It is nevertheless a useful technique to monitor good-grade aneurysmal subarachnoid hemorrhage patients (WFNS score 1–3) combined with frequent neurologic examinations (Figure 3).
CT angiography is a good noninvasive alternative to digital subtraction angiography. However, it tends to overestimate the degree of vasoconstriction and does not provide information about perfusion and autoregulation.65 Nevertheless, CT angiography combined with a CT perfusion scan can add information about autoregulation and cerebral perfusion and has been shown to be more sensitive for the diagnosis of angiographic vasospasm than transcranial Doppler and digital subtraction angiography (Figure 4).
Patients with a poor clinical condition (WFNS score 4 or 5) or receiving continuous sedation constitute a challenge in monitoring for delayed neurologic deterioration. Neurologic examination is not sensitive enough in this setting to detect subtle changes. In these specific and challenging circumstances, multimodality neuromonitoring may be useful in the early detection of delayed cerebral ischemia and may help guide therapy.67
Several noninvasive and invasive techniques have been studied to monitor patients at risk of delayed cerebral ischemia after subarachnoid hemorrhage.66 These include continuous electroencephalography, brain tissue oxygenation monitoring (Ptio2), cerebral microdialysis, thermal diffusion flowmetry, and near-infrared spectroscopy. Of these techniques, Ptio2, cerebral microdialysis, and continuous electroencephalography (see discussion of seizure prophylaxis above) have been more extensively studied. However, most of the studies were observational and very small, limiting any recommendations for using these techniques in routine clinical practice.68
Ptio2 is measured by inserting an intraparenchymal oxygen-sensitive microelectrode, and microdialysis requires a microcatheter with a semipermeable membrane that allows small soluble substances to cross it into the dialysate. These substances, which include markers of ischemia (ie, glucose, lactate, and pyruvate), excitotoxins (ie, glutamate and aspartate), and membrane cell damage products (ie, glycerol), can be measured. Low Ptio2 values (< 15 mm Hg) and abnormal mycrodialysate findings (eg, glucose < 0.8 mmol/L, lactate-to-pyruvate ratio > 40) have both been associated with cerebral ischemic events and poor outcome.68
Preventing delayed cerebral ischemia
Oral nimodipine 60 mg every 4 hours for 21 days, started on admission, carries a class I, level of evidence A recommendation in the management of aneurysmal subarachnoid hemorrhage.3,32,69 It improves clinical outcome despite having no effect on the risk of angiographic vasospasm. The mechanism of improved outcome is unclear, but the effect may be a neuroprotective phenomenon limiting the extension of delayed cerebral ischemia.70
If hypotension occurs, the dose can be lowered to 30 mg every 2 hours. Whether to discontinue nimodipine in this situation is controversial. Of note, the clinical benefits of nimodipine have not been replicated with other calcium channel blockers (eg, nicardipine).71
Prophylactic hyperdynamic fluid therapy, known as “triple-H” (hypervolemia, hemodilution, and hypertension) was for years the mainstay of treatment in preventing delayed cerebral ischemia due to vasospasm. However, the clinical data supporting this intervention have been called into question, as analysis of two trials found that hypervolemia did not improve outcomes or reduce the incidence of delayed cerebral ischemia, and in fact increased the rate of complications.72,73 Based on these findings, current guidelines recommend maintaining euvolemia rather than prophylactic hypervolemia in patients with aneurysmal subarachnoid hemorrhage.3,32,69
TREATING DELAYED CEREBRAL ISCHEMIA
Hemodynamic augmentation
In patients with neurologic deterioration due to delayed cerebral ischemia, hemodynamic augmentation is the cornerstone of treatment. This is done according to a protocol, started early, involving specific physiologic goals, clinical improvement, and escalation to invasive therapies in a timely fashion in patients at high risk of further neurologic insult (Figure 5).
The physiologic goal is to increase the delivery of oxygen and glucose to the ischemic brain. Hypertension seems to be the most effective component of hemodynamic augmentation regardless of volume status, increasing cerebral blood flow and brain tissue oxygenation, with reversal of delayed cerebral ischemic symptoms in up to two-thirds of treated patients.74,75 However, this information comes from very small studies, with no randomized trials of induced hypertension available.
The effect of a normal saline fluid bolus in patients suspected of having delayed cerebral ischemia has been shown to increase cerebral blood flow in areas of cerebral ischemia.74 If volume augmentation fails to improve the neurologic status, the next step is to artificially induce hypertension using vasopressors. The blood pressure target should be based on clinical improvement. A stepwise approach is reasonable in this situation, and the lowest level of blood pressure at which there is a complete reversal of the new focal neurologic deficit should be maintained.3,29
Inotropic agents such as dobutamine or milrinone can be considered as alternatives in patients who have new neurologic deficits that are refractory to fluid boluses and vasopressors, or in a setting of subarachnoid hemorrhage-induced cardiomyopathy.76,77
Once the neurologic deficit is reversed by hemodynamic augmentation, the blood pressure should be maintained for 48 to 72 hours at the level that reversed the deficit completely, carefully reassessed thereafter, and the patient weaned slowly. Unruptured unsecured aneurysms should not prevent blood pressure augmentation in a setting of delayed cerebral ischemia if the culprit aneurysm is treated.3,32 If the ruptured aneurysm has not been secured, careful blood pressure augmentation can be attempted, keeping in mind that hypertension (> 160/95 mm Hg) is a risk factor for fatal aneurysm rupture.
Endovascular management of delayed cerebral ischemia
When medical augmentation fails to completely reverse the neurologic deficits, endovascular treatment can be considered. Although patients treated early in the course of delayed cerebral ischemia have better neurologic recovery, prophylactic endovascular treatment in asymptomatic patients, even if angiographic signs of spasm are present, does not improve clinical outcomes and carries the risk of fatal arterial rupture.78
SYSTEMIC COMPLICATIONS
Hyponatremia and hypovolemia
Aneurysmal subarachnoid hemorrhage is commonly associated with abnormalities of fluid balance and electrolyte derangements. Hyponatremia (serum sodium < 135 mmol/L) occurs in 30% to 50% of patients, while the rate of hypovolemia (decreased circulating blood volume) ranges from 17% to 30%.79 Both can negatively affect long-term outcomes.80,81
Decreased circulating blood volume is a well-described contributor to delayed cerebral ischemia and cerebral infarction after aneurysmal subarachnoid hemorrhage.80–82 Clinical variables such as heart rate, blood pressure, fluid balance, and serum sodium concentration are usually the cornerstones of intravascular volume status assessment. However, these variables correlate poorly with measured circulating blood volumes in those with aneurysmal subarachnoid hemorrhage.83,84
The mechanisms responsible for the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage are not completely understood. Several factors have been described and may contribute to the increased natriuresis and, hence, to a reduction in circulating blood volume: increased circulating natriuretic peptide concentrations,85–87 sympathetic nervous system hyperactivation,88 and hyperreninemic hypo-
aldosteronism syndrome.89,90
Lastly, the cerebral salt wasting syndrome, described in the 1950s,91 was thought to be a key mechanism in the development of hyponatremia and hypovolemia after aneurysmal subarachnoid hemorrhage. In contrast to the syndrome of inappropriate antidiuretic hormone, which is characterized by hyponatremia with a normal or slightly elevated intravascular volume, the characteristic feature of cerebral salt wasting syndrome is the development of hyponatremia in a setting of intravascular volume depletion.92 In critically ill neurologic and neurosurgical patients, this differential diagnosis is very difficult, especially in those with aneurysmal subarachnoid hemorrhage in whom the clinical assessment of fluid status is not reliable. These two syndromes might coexist and contribute to the development of hyponatremia after aneurysmal subarachnoid hemorrhage.92,93
Hoff et al83,84 prospectively compared the clinical assessment of fluid status by critical and intermediate care nurses and direct measurements of blood volume using pulse dye densitometry. The clinical assessment failed to accurately assess patients’ volume status. Using the same technique to measure circulating blood volume, this group showed that calculation of fluid balance does not provide adequate assessment of fluid status.83,84
Hemodynamic monitoring tools can help guide fluid replacement in this population. Mutoh et al94 randomized 160 patients within 24 hours of hemorrhage to receive early goal-directed fluid therapy (ie, preload volume and cardiac output monitored by transpulmonary thermodilution) vs standard therapy (ie, fluid balance or central venous pressure). Overall, no difference was found in the rates of delayed cerebral ischemia (33% vs 42%; P = .33) or favorable outcome (67% vs 57%; P = .22). However, in the subgroup of poor-grade patients (WFNS score 4 or 5), early goal-directed therapy was associated with a lower rate of delayed cerebral ischemia (5% vs 14%; P = .036) and with better functional outcomes at 3 months (52% vs 36%; P = .026).
Fluid restriction to treat hyponatremia in aneurysmal subarachnoid hemorrhage is no longer recommended because of the increased risk of cerebral infarction due to hypovolemic hypoperfusion.82
Prophylactic use of mineralocorticoids (eg, fludrocortisone, hydrocortisone) has been shown to limit natriuresis, hyponatremia, and the amount of fluid required to maintain euvolemia.95,96 Higher rates of hypokalemia and hyperglycemia, which can be easily treated, are the most common complications associated with this approach. Additionally, hypertonic saline (eg, 3% saline) can be used to correct hyponatremia in a setting of aneurysmal subarachnoid hemorrhage.79
Cardiac complications
Cardiac complications after subarachnoid hemorrhage are most likely related to sympathetic hyperactivity and catecholamine-induced myocyte dysfunction. The pathophysiology is complex, but cardiac complications have a significant negative impact on long-term outcome in these patients.97
Electrocardiographic changes and positive cardiac enzymes associated with aneurysmal subarachnoid hemorrhage have been extensively reported. More recently, data from studies of two-dimensional echocardiography have shown that subarachnoid hemorrhage can also be associated with significant wall-motion abnormalities and even overt cardiogenic shock.98–100
There is no specific curative therapy; the treatment is mainly supportive. Vasopressors and inotropes may be used for hemodynamic augmentation.
Pulmonary complications
Pulmonary complications occur in 20% to 30% of all aneurysmal subarachnoid hemorrhage patients and are associated with a higher risk of delayed cerebral ischemia and death. Common pulmonary complications in this population are mild acute respiratory distress syndrome (27%), hospital-acquired pneumonia (9%), cardiogenic pulmonary edema (8%), aspiration pneumonia (6%), neurogenic pulmonary edema (2%), and pulmonary embolism (1%).101–103
SUPPORTIVE CARE
Hyperthermia, hyperglycemia, and liberal use of transfusions have all been associated with longer stays in the intensive care unit and hospital, poorer neurologic outcomes, and higher mortality rates in patients with acute brain injury.104 Noninfectious fever is the most common systemic complication after subarachnoid hemorrhage.
Antipyretic drugs such as acetaminophen and ibuprofen are not very effective in reducing fever in the subarachnoid hemorrhage population, but should still be used as first-line therapy. The use of surface and intravascular devices can be considered when fevers do not respond to nonsteroidal anti-inflammatory drugs.
Although no prospective randomized trial has addressed the impact of induced normothermia on long-term outcome and mortality in aneurysmal subarachnoid hemorrhage patients, fever control has been shown to reduce cerebral metabolic distress, irrespective of intracranial pressure.105 Maintenance of normothermia (< 37.5°C) seems reasonable, especially in aneurysmal subarachnoid hemorrhage patients at risk of or with active delayed cerebral ischemia.106
Current guidelines3,32,69 strongly recommend avoiding hypoglycemia, defined as a serum glucose level less than 80 mg/dL, but suggest keeping the blood sugar level below 180 or 200 mg/dL.
At the moment, there is no clear threshold for transfusion in patients with aneurysmal subarachnoid hemorrhage. Current guidelines suggest keeping hemoglobin levels between 8 and 10 g/dL.3
Preventing venous thromboembolism
The incidence of venous thromboembolism after aneurysmal subarachnoid hemorrhage varies widely, from 1.5% to 18%.107 Active surveillance with venous Doppler ultrasonography has found asymptomatic deep vein thrombosis in up to 3.4% of poor-grade aneurysmal subarachnoid hemorrhage patients receiving pharmacologic thromboprophylaxis.108
In a retrospective study of 170 patients, our group showed that giving drugs to prevent venous thromboembolism (unfractionated heparin 5,000 IU subcutaneously every 12 hours or dalteparin 5,000 IU subcutaneously daily), starting within 24 hours of aneurysm treatment, could be safe.109 Fifty-eight percent of these patients had an external ventricular drain in place. One patient developed a major cerebral hemorrhagic complication and died while on unfractionated heparin; however, the patient was also on dual antiplatelet therapy with aspirin and clopidogrel.109
Current guidelines suggest that intermittent compression devices be applied in all patients before aneurysm treatment. Pharmacologic thromboprophylaxis with a heparinoid can be started 12 to 24 hours after aneurysm treatment.3,109
A TEAM APPROACH
Patients with subarachnoid hemorrhage need integrated care from different medical and nursing specialties. The best outcomes are achieved by systems that can focus as a team on the collective goal of quick intervention to secure the aneurysm, followed by measures to minimize secondary brain injury.
The modern concept of cerebral monitoring in a setting of subarachnoid hemorrhage should focus on brain perfusion rather than vascular diameter. Although the search continues for new diagnostic, prognostic, and therapeutic tools, there is no “silver bullet” that will help all patients. Instead, it is the systematic integration and application of many small advances that will ultimately lead to better outcomes.
ACKNOWLEDGMENT
This work was supported by research funding provided by the Bitove Foundation, which has been supportive of our clinical and research work for several years.
- van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain 2001; 124:249–278.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 2014; 129:e28–e292.
- Diringer MN, Bleck TP, Claude Hemphill J 3rd, et al; Neurocritical Care Society. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit Care 2011; 15:211–240.
- Feigin VL, Rinkel GJ, Lawes CM, et al. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke 2005; 36:2773–2780.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Clinical decision rules to rule out subarachnoid hemorrhage for acute headache. JAMA 2013; 310:1248–1255.
- Kowalski RG, Claassen J, Kreiter KT, et al. Initial misdiagnosis and outcome after subarachnoid hemorrhage. JAMA 2004; 291:866–869.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- van Gijn J, van Dongen KJ. The time course of aneurysmal haemorrhage on computed tomograms. Neuroradiology 1982; 23:153–156.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Agid R, Andersson T, Almqvist H, et al. Negative CT angiography findings in patients with spontaneous subarachnoid hemorrhage: when is digital subtraction angiography still needed? AJNR Am J Neuroradiol 2010; 31:696–705.
- Teasdale GM, Drake CG, Hunt W, et al. A universal subarachnoid hemorrhage scale: report of a committee of the World Federation of Neurosurgical Societies. J Neurol Neurosurg Psychiatry 1988; 51:1457.
- Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 1968; 28:14–20.
- Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980; 6:1–9.
- Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery 2006; 59:21–27.
- de Oliveira Manoel AL, Turkel-Parrella D, Kouzmina E, et al. The VASOGRADE—a simple, reliable grading scale for aneurysmal subarachnoid hemorrhage. Neurology 2014; 82(suppl 10): P5.123.
- Naidech AM, Janjua N, Kreiter KT, et al. Predictors and impact of aneurysm rebleeding after subarachnoid hemorrhage. Arch Neurol 2005; 62:410–416.
- Rincon F, Mayer SA. Neurocritical care: a distinct discipline? Curr Opin Crit Care 2007; 13:115–121.
- Rabinstein AA, Lanzino G, Wijdicks EF. Multidisciplinary management and emerging therapeutic strategies in aneurysmal subarachnoid haemorrhage. Lancet Neurol 2010; 9:504–519.
- Vespa P, Diringer MN; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. High-volume centers. Neurocrit Care 2011; 15:369–372.
- McNeill L, English SW, Borg N, Matta BF, Menon DK. Effects of institutional caseload of subarachnoid hemorrhage on mortality: a secondary analysis of administrative data. Stroke 2013; 44:647–652.
- Dorhout Mees SM, Molyneux AJ, Kerr RS, Algra A, Rinkel GJ. Timing of aneurysm treatment after subarachnoid hemorrhage: relationship with delayed cerebral ischemia and poor outcome. Stroke 2012; 43:2126–2129.
- Laidlaw JD, Siu KH. Ultra-early surgery for aneurysmal subarachnoid hemorrhage: outcomes for a consecutive series of 391 patients not selected by grade or age. J Neurosurg 2002; 97:250–259.
- Molyneux A, Kerr R, Stratton I, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised trial. Lancet 2002; 360:1267–1274.
- Molyneux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid aneurysm trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Koivisto T, Vanninen R, Hurskainen H, Saari T, Hernesniemi J, Vapalahti M. Outcomes of early endovascular versus surgical treatment of ruptured cerebral aneurysms. A prospective randomized study. Stroke 2000; 31:2369–2377.
- McDougall CG, Spetzler RF, Zabramski JM, et al. The Barrow Ruptured Aneurysm Trial. J Neurosurg 2012; 116:135–144.
- Spetzler RF, McDougall CG, Albuquerque FC, et al. The Barrow Ruptured Aneurysm Trial: 3-year results. J Neurosurg 2013; 119:146–157.
- Connolly ES Jr, Meyers PM. Cerebral aneurysms: to clip or to coil? That is no longer the question. Nat Rev Neurol 2009; 5:412–413.
- Kassell NF, Peerless SJ, Durward QJ, Beck DW, Drake CG, Adams HP. Treatment of ischemic deficits from vasospasm with intravascular volume expansion and induced arterial hypertension. Neurosurgery 1982; 11:337–343.
- Otsubo H, Takemae T, Inoue T, Kobayashi S, Sugita K. Normovolaemic induced hypertension therapy for cerebral vasospasm after subarachnoid haemorrhage. Acta Neurochir (Wien) 1990; 103:18–26.
- Anderson CS, Heeley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Ortega-Gutierrez S, Thomas J, Reccius A, et al. Effectiveness and safety of nicardipine and labetalol infusion for blood pressure management in patients with intracerebral and subarachnoid hemorrhage. Neurocrit Care 2013; 18:13–19.
- Hillman J, Fridriksson S, Nilsson O, Yu Z, Saveland H, Jakobsson KE. Immediate administration of tranexamic acid and reduced incidence of early rebleeding after aneurysmal subarachnoid hemorrhage: a prospective randomized study. J Neurosurg 2002; 97:771–778.
- Baharoglu MI, Germans MR, Rinkel GJ, et al. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2013; 8:CD001245.
- Germans MR, Post R, Coert BA, Rinkel GJ, Vandertop WP, Verbaan D. Ultra-early tranexamic acid after subarachnoid hemorrhage (ULTRA): study protocol for a randomized controlled trial. Trials 2013; 14:143.
- Sloan MA, Alexandrov AV, Tegeler CH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004; 62:1468–1481.
- Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med 2006; 34:617–623;
- Hellingman CA, van den Bergh WM, Beijer IS, et al. Risk of rebleeding after treatment of acute hydrocephalus in patients with aneurysmal subarachnoid hemorrhage. Stroke 2007; 38:96–99.
- Ransom ER, Mocco J, Komotar RJ, et al. External ventricular drainage response in poor grade aneurysmal subarachnoid hemorrhage: effect on preoperative grading and prognosis. Neurocrit Care 2007; 6:174–180.
- McIver JI, Friedman JA, Wijdicks EF, et al. Preoperative ventriculostomy and rebleeding after aneurysmal subarachnoid hemorrhage. J Neurosurg 2002; 97:1042–1044.
- Klopfenstein JD, Kim LJ, Feiz-Erfan I, et al. Comparison of rapid and gradual weaning from external ventricular drainage in patients with aneurysmal subarachnoid hemorrhage: a prospective randomized trial. J Neurosurg 2004; 100:225–229.
- Tseng MY, Al-Rawi PG, Czosnyka M, et al. Enhancement of cerebral blood flow using systemic hypertonic saline therapy improves outcome in patients with poor-grade spontaneous subarachnoid hemorrhage. J Neurosurg 2007; 107:274–282.
- Al-Rawi PG, Tseng MY, Richards HK, et al. Hypertonic saline in patients with poor-grade subarachnoid hemorrhage improves cerebral blood flow, brain tissue oxygen, and pH. Stroke 2010; 41:122–128.
- Tseng MY, Al-Rawi PG, Pickard JD, Rasulo FA, Kirkpatrick PJ. Effect of hypertonic saline on cerebral blood flow in poor-grade patients with subarachnoid hemorrhage. Stroke 2003; 34:1389–1396.
- Bentsen G, Breivik H, Lundar T, Stubhaug A. Hypertonic saline (7.2%) in 6% hydroxyethyl starch reduces intracranial pressure and improves hemodynamics in a placebo-controlled study involving stable patients with subarachnoid hemorrhage. Crit Care Med 2006; 34:2912–2917.
- Suarez JI, Qureshi AI, Parekh PD, et al. Administration of hypertonic (3%) sodium chloride/acetate in hyponatremic patients with symptomatic vasospasm following subarachnoid hemorrhage. J Neurosurg Anesthesiol 1999; 11:178–184.
- Stevens RD, Huff JS, Duckworth J, Papangelou A, Weingart SD, Smith WS. Emergency neurological life support: intracranial hypertension and herniation. Neurocrit Care 2012;17(suppl 1):S60–S65.
- Seule MA, Muroi C, Mink S, Yonekawa Y, Keller E. Therapeutic hypothermia in patients with aneurysmal subarachnoid hemorrhage, refractory intracranial hypertension, or cerebral vasospasm. Neurosurgery 2009; 64:86–93.
- Otani N, Takasato Y, Masaoka H, et al. Surgical outcome following decompressive craniectomy for poor-grade aneurysmal subarachnoid hemorrhage in patients with associated massive intracerebral or Sylvian hematomas. Cerebrovasc Dis 2008; 26:612–617.
- Lanzino G, D’Urso PI, Suarez J; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Seizures and anticonvulsants after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2011; 15:247–256.
- Naidech AM, Kreiter KT, Janjua N, et al. Phenytoin exposure is associated with functional and cognitive disability after subarachnoid hemorrhage. Stroke 2005; 36:583–587.
- Rosengart AJ, Huo JD, Tolentino J, et al. Outcome in patients with subarachnoid hemorrhage treated with antiepileptic drugs. J Neurosurg 2007; 107:253–260.
- Shah D, Husain AM. Utility of levetiracetam in patients with subarachnoid hemorrhage. Seizure 2009; 18:676–679.
- Patsalos PN. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol Ther 2000; 85:77–85.
- Dewan MC, Mocco J. Current practice regarding seizure prophylaxis in aneurysmal subarachnoid hemorrhage across academic centers. J Neurointerv Surg 2014 Jan 28. doi: 10.1136/neurintsurg-2013-011075 [Epub ahead of print]
- Murphy-Human T, Welch E, Zipfel G, Diringer MN, Dhar R. Comparison of short-duration levetiracetam with extended-course phenytoin for seizure prophylaxis after subarachnoid hemorrhage. World Neurosurg 2011; 75:269–274.
- Dennis LJ, Claassen J, Hirsch LJ, Emerson RG, Connolly ES, Mayer SA. Nonconvulsive status epilepticus after subarachnoid hemorrhage. Neurosurgery 2002; 51:1136–1144.
- Vespa PM, Nuwer MR, Juhász C, et al. Early detection of vasospasm after acute subarachnoid hemorrhage using continuous EEG ICU monitoring. Electroencephalogr Clin Neurophysiol 1997; 103:607–615.
- Claassen J, Hirsch LJ, Kreiter KT, et al. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin Neurophysiol 2004; 115:2699–2710.
- Vergouwen MD, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke 2010; 41:2391–2395.
- Kelly PJ, Gorten RJ, Grossman RG, Eisenberg HM. Cerebral perfusion, vascular spasm, and outcome in patients with ruptured intracranial aneurysms. J Neurosurg 1977; 47:44–49.
- Aralasmak A, Akyuz M, Ozkaynak C, Sindel T, Tuncer R. CT angiography and perfusion imaging in patients with subarachnoid hemorrhage: correlation of vasospasm to perfusion abnormality. Neuroradiology 2009; 51:85–93.
- Sabri M, Lass E, Macdonald RL. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res Treat 2013 Feb 28. doi: 10.1155/2013/394036 [Epub 2013 ahead of print]
- Yoon DY, Choi CS, Kim KH, Cho BM. Multidetector-row CT angiography of cerebral vasospasm after aneurysmal subarachnoid hemorrhage: comparison of volume-rendered images and digital subtraction angiography. AJNR Am J Neuroradiol 2006; 27:370–377.
- Wintermark M1, Ko NU, Smith WS, Liu S, Higashida RT, Dillon WP. Vasospasm after subarachnoid hemorrhage: utility of perfusion CT and CT angiography on diagnosis and management. AJNR Am J Neuroradiol 2006; 27:26–34.
- Helbok R, Madineni RC, Schmidt MJ, et al. Intracerebral monitoring of silent infarcts after subarachnoid hemorrhage. Neurocrit Care 2011; 14:162–167.
- Hänggi D; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Monitoring and detection of vasospasm II: EEG and invasive monitoring. Neurocrit Care 2011; 15:318–323.
- Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G; European Stroke Organization. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis 2013; 35:93–112.
- Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989; 298:636–642.
- Dorhout Mees SM, Rinkel GJ, Feigin VL, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2007; 3:CD000277.
- Lennihan L, Mayer SA, Fink ME, et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage: a randomized controlled trial. Stroke 2000; 31:383–391.
- Egge A, Waterloo K, Sjøholm H, Solberg T, Ingebrigtsen T, Romner B. Prophylactic hyperdynamic postoperative fluid therapy after aneurysmal subarachnoid hemorrhage: a clinical, prospective, randomized, controlled study. Neurosurgery 2001; 49:593–606.
- Jost SC, Diringer MN, Zazulia AR, et al. Effect of normal saline bolus on cerebral blood flow in regions with low baseline flow in patients with vasospasm following subarachnoid hemorrhage. J Neurosurg 2005; 103:25–30.
- Muizelaar JP, Becker DP. Induced hypertension for the treatment of cerebral ischemia after subarachnoid hemorrhage. Direct effect on cerebral blood flow. Surg Neurol 1986; 25:317–325.
- Levy ML, Rabb CH, Zelman V, Giannotta SL. Cardiac performance enhancement from dobutamine in patients refractory to hypervolemic therapy for cerebral vasospasm. J Neurosurg 1993; 79:494–499.
- Lannes M, Teitelbaum J, del Pilar Cortés M, Cardoso M, Angle M. Milrinone and homeostasis to treat cerebral vasospasm associated with subarachnoid hemorrhage: the Montreal Neurological Hospital protocol. Neurocrit Care 2012; 16:354–362.
- Zwienenberg-Lee M, Hartman J, Rudisill N, et al; Balloon Prophylaxis for Aneurysmal Vasospasm (BPAV) Study Group. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage: results of a phase II multicenter, randomized, clinical trial. Stroke 2008; 39:1759–1765.
- Rabinstein AA, Bruder N. Management of hyponatremia and volume contraction. Neurocrit Care 2011; 15:354–360.
- Wijdicks EF, Vermeulen M, Hijdra A, van Gijn J. Hyponatremia and cerebral infarction in patients with ruptured intracranial aneurysms: is fluid restriction harmful? Ann Neurol 1985; 17:137–140.
- Hasan D, Wijdicks EF, Vermeulen M. Hyponatremia is associated with cerebral ischemia in patients with aneurysmal subarachnoid hemorrhage. Ann Neurol 1990; 27:106–108.
- Wijdicks EF, Vermeulen M, ten Haaf JA, Hijdra A, Bakker WH, van Gijn J. Volume depletion and natriuresis in patients with a ruptured intracranial aneurysm. Ann Neurol 1985; 18:211–216.
- Hoff RG, Rinkel GJ, Verweij BH, Algra A, Kalkman CJ. Nurses’ prediction of volume status after aneurysmal subarachnoid haemorrhage: a prospective cohort study. Crit Care 2008; 12:R153.
- Hoff RG, van Dijk GW, Algra A, Kalkman CJ, Rinkel GJ. Fluid balance and blood volume measurement after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2008; 8:391–397.
- Berendes E, Walter M, Cullen P, et al. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet 1997; 349:245–249.
- Espiner EA, Leikis R, Ferch RD, et al. The neuro-cardio-endocrine response to acute subarachnoid haemorrhage. Clin Endocrinol (Oxf) 2002; 56:629–635.
- Isotani E, Suzuki R, Tomita K, et al. Alterations in plasma concentrations of natriuretic peptides and antidiuretic hormone after subarachnoid hemorrhage. Stroke 1994; 25:2198–2203.
- Benedict CR, Loach AB. Sympathetic nervous system activity in patients with subarachnoid hemorrhage. Stroke 1978; 9:237–244.
- Findling JW, Waters VO, Raff H. The dissociation of renin and aldosterone during critical illness. J Clin Endocrinol Metab 1987; 64:592–595.
- Solomon RA, Post KD, McMurtry JG 3rd. Depression of circulating blood volume in patients after subarachnoid hemorrhage: implications for the management of symptomatic vasospasm. Neurosurgery 1984; 15:354–361.
- Peters JP, Welt LG, Sims EA, Orloff J, Needham J. A salt-wasting syndrome associated with cerebral disease. Trans Assoc Am Physicians 1950; 63:57–64.
- Brimioulle S, Orellana-Jimenez C, Aminian A, Vincent JL. Hyponatremia in neurological patients: cerebral salt wasting versus inappropriate antidiuretic hormone secretion. Intensive Care Med 2008; 34:125–131.
- Singh S, Bohn D, Carlotti AP, Cusimano M, Rutka JT, Halperin ML. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med 2002; 30:2575–2579.
- Mutoh T, Kazumata K, Terasaka S, Taki Y, Suzuki A, Ishikawa T. Early intensive versus minimally invasive approach to postoperative hemodynamic management after subarachnoid hemorrhage. Stroke 2014; 45:1280–1284.
- Hasan D, Lindsay KW, Wijdicks EF, et al. Effect of fludrocortisone acetate in patients with subarachnoid hemorrhage. Stroke 1989; 20:1156–1161.
- Moro N, Katayama Y, Kojima J, Mori T, Kawamata T. Prophylactic management of excessive natriuresis with hydrocortisone for efficient hypervolemic therapy after subarachnoid hemorrhage. Stroke 2003; 34:2807–2811.
- Kilbourn KJ, Levy S, Staff I, Kureshi I, McCullough L. Clinical characteristics and outcomes of neurogenic stress cadiomyopathy in aneurysmal subarachnoid hemorrhage. Clin Neurol Neurosurg 2013; 115:909–914.
- Mayer SA, LiMandri G, Sherman D, et al. Electrocardiographic markers of abnormal left ventricular wall motion in acute subarachnoid hemorrhage. J Neurosurg 1995; 83:889–896.
- Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg 2003; 98:741–746.
- Banki N, Kopelnik A, Tung P, et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J Neurosurg 2006; 105:15–20.
- Kahn JM, Caldwell EC, Deem S, Newell DW, Heckbert SR, Rubenfeld GD. Acute lung injury in patients with subarachnoid hemorrhage: incidence, risk factors, and outcome. Crit Care Med 2006; 34:196–202.
- Kitamura Y, Nomura M, Shima H, et al. Acute lung injury associated with systemic inflammatory response syndrome following subarachnoid hemorrhage: a survey by the Shonan Neurosurgical Association. Neurol Med Chir (Tokyo) 2010; 50:456–460.
- Friedman JA, Pichelmann MA, Piepgras DG, et al. Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neurosurgery 2003; 52:1025–1032.
- Oh HS, Jeong HS, Seo WS. Non-infectious hyperthermia in acute brain injury patients: relationships to mortality, blood pressure, intracranial pressure and cerebral perfusion pressure. Int J Nurs Pract 2012; 18:295–302.
- Oddo M, Frangos S, Milby A, et al. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke 2009; 40:1913–1916.
- Badjatia N, Fernandez L, Schmidt JM, et al. Impact of induced normothermia on outcome after subarachnoid hemorrhage: a case-control study. Neurosurgery 2010; 66:696-701.
- Serrone JC1, Wash EM, Hartings JA, Andaluz N, Zuccarello M. Venous thromboembolism in subarachnoid hemorrhage. World Neurosurg 2013; 80:859–863.
- Mack WJ, Ducruet AF, Hickman ZL, et al. Doppler ultrasonography screening of poor-grade subarachnoid hemorrhage patients increases the diagnosis of deep venous thrombosis. Neurol Res 2008; 30:889–892.
- de Oliveira Manoel AL, Turkel-Parrella D, Germans M, et al. Safety of early pharmacological thromboprophylaxis after subarachnoid hemorrhage. Can J Neurol Sci 2014; 41:554–561.
- van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain 2001; 124:249–278.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 2014; 129:e28–e292.
- Diringer MN, Bleck TP, Claude Hemphill J 3rd, et al; Neurocritical Care Society. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit Care 2011; 15:211–240.
- Feigin VL, Rinkel GJ, Lawes CM, et al. Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke 2005; 36:2773–2780.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Clinical decision rules to rule out subarachnoid hemorrhage for acute headache. JAMA 2013; 310:1248–1255.
- Kowalski RG, Claassen J, Kreiter KT, et al. Initial misdiagnosis and outcome after subarachnoid hemorrhage. JAMA 2004; 291:866–869.
- Perry JJ, Stiell IG, Sivilotti ML, et al. Sensitivity of computed tomography performed within six hours of onset of headache for diagnosis of subarachnoid haemorrhage: prospective cohort study. BMJ 2011; 343:d4277.
- van Gijn J, van Dongen KJ. The time course of aneurysmal haemorrhage on computed tomograms. Neuroradiology 1982; 23:153–156.
- McCormack RF, Hutson A. Can computed tomography angiography of the brain replace lumbar puncture in the evaluation of acute-onset headache after a negative noncontrast cranial computed tomography scan? Acad Emerg Med 2010; 17:444–451.
- Agid R, Andersson T, Almqvist H, et al. Negative CT angiography findings in patients with spontaneous subarachnoid hemorrhage: when is digital subtraction angiography still needed? AJNR Am J Neuroradiol 2010; 31:696–705.
- Teasdale GM, Drake CG, Hunt W, et al. A universal subarachnoid hemorrhage scale: report of a committee of the World Federation of Neurosurgical Societies. J Neurol Neurosurg Psychiatry 1988; 51:1457.
- Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg 1968; 28:14–20.
- Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery 1980; 6:1–9.
- Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified Fisher scale. Neurosurgery 2006; 59:21–27.
- de Oliveira Manoel AL, Turkel-Parrella D, Kouzmina E, et al. The VASOGRADE—a simple, reliable grading scale for aneurysmal subarachnoid hemorrhage. Neurology 2014; 82(suppl 10): P5.123.
- Naidech AM, Janjua N, Kreiter KT, et al. Predictors and impact of aneurysm rebleeding after subarachnoid hemorrhage. Arch Neurol 2005; 62:410–416.
- Rincon F, Mayer SA. Neurocritical care: a distinct discipline? Curr Opin Crit Care 2007; 13:115–121.
- Rabinstein AA, Lanzino G, Wijdicks EF. Multidisciplinary management and emerging therapeutic strategies in aneurysmal subarachnoid haemorrhage. Lancet Neurol 2010; 9:504–519.
- Vespa P, Diringer MN; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. High-volume centers. Neurocrit Care 2011; 15:369–372.
- McNeill L, English SW, Borg N, Matta BF, Menon DK. Effects of institutional caseload of subarachnoid hemorrhage on mortality: a secondary analysis of administrative data. Stroke 2013; 44:647–652.
- Dorhout Mees SM, Molyneux AJ, Kerr RS, Algra A, Rinkel GJ. Timing of aneurysm treatment after subarachnoid hemorrhage: relationship with delayed cerebral ischemia and poor outcome. Stroke 2012; 43:2126–2129.
- Laidlaw JD, Siu KH. Ultra-early surgery for aneurysmal subarachnoid hemorrhage: outcomes for a consecutive series of 391 patients not selected by grade or age. J Neurosurg 2002; 97:250–259.
- Molyneux A, Kerr R, Stratton I, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised trial. Lancet 2002; 360:1267–1274.
- Molyneux AJ, Kerr RS, Yu LM, et al; International Subarachnoid Aneurysm Trial (ISAT) Collaborative Group. International subarachnoid aneurysm trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2,143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet 2005; 366:809–817.
- Koivisto T, Vanninen R, Hurskainen H, Saari T, Hernesniemi J, Vapalahti M. Outcomes of early endovascular versus surgical treatment of ruptured cerebral aneurysms. A prospective randomized study. Stroke 2000; 31:2369–2377.
- McDougall CG, Spetzler RF, Zabramski JM, et al. The Barrow Ruptured Aneurysm Trial. J Neurosurg 2012; 116:135–144.
- Spetzler RF, McDougall CG, Albuquerque FC, et al. The Barrow Ruptured Aneurysm Trial: 3-year results. J Neurosurg 2013; 119:146–157.
- Connolly ES Jr, Meyers PM. Cerebral aneurysms: to clip or to coil? That is no longer the question. Nat Rev Neurol 2009; 5:412–413.
- Kassell NF, Peerless SJ, Durward QJ, Beck DW, Drake CG, Adams HP. Treatment of ischemic deficits from vasospasm with intravascular volume expansion and induced arterial hypertension. Neurosurgery 1982; 11:337–343.
- Otsubo H, Takemae T, Inoue T, Kobayashi S, Sugita K. Normovolaemic induced hypertension therapy for cerebral vasospasm after subarachnoid haemorrhage. Acta Neurochir (Wien) 1990; 103:18–26.
- Anderson CS, Heeley E, Huang Y, et al; INTERACT2 Investigators. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage. N Engl J Med 2013; 368:2355–2365.
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2012; 43:1711–1737.
- Ortega-Gutierrez S, Thomas J, Reccius A, et al. Effectiveness and safety of nicardipine and labetalol infusion for blood pressure management in patients with intracerebral and subarachnoid hemorrhage. Neurocrit Care 2013; 18:13–19.
- Hillman J, Fridriksson S, Nilsson O, Yu Z, Saveland H, Jakobsson KE. Immediate administration of tranexamic acid and reduced incidence of early rebleeding after aneurysmal subarachnoid hemorrhage: a prospective randomized study. J Neurosurg 2002; 97:771–778.
- Baharoglu MI, Germans MR, Rinkel GJ, et al. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2013; 8:CD001245.
- Germans MR, Post R, Coert BA, Rinkel GJ, Vandertop WP, Verbaan D. Ultra-early tranexamic acid after subarachnoid hemorrhage (ULTRA): study protocol for a randomized controlled trial. Trials 2013; 14:143.
- Sloan MA, Alexandrov AV, Tegeler CH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004; 62:1468–1481.
- Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med 2006; 34:617–623;
- Hellingman CA, van den Bergh WM, Beijer IS, et al. Risk of rebleeding after treatment of acute hydrocephalus in patients with aneurysmal subarachnoid hemorrhage. Stroke 2007; 38:96–99.
- Ransom ER, Mocco J, Komotar RJ, et al. External ventricular drainage response in poor grade aneurysmal subarachnoid hemorrhage: effect on preoperative grading and prognosis. Neurocrit Care 2007; 6:174–180.
- McIver JI, Friedman JA, Wijdicks EF, et al. Preoperative ventriculostomy and rebleeding after aneurysmal subarachnoid hemorrhage. J Neurosurg 2002; 97:1042–1044.
- Klopfenstein JD, Kim LJ, Feiz-Erfan I, et al. Comparison of rapid and gradual weaning from external ventricular drainage in patients with aneurysmal subarachnoid hemorrhage: a prospective randomized trial. J Neurosurg 2004; 100:225–229.
- Tseng MY, Al-Rawi PG, Czosnyka M, et al. Enhancement of cerebral blood flow using systemic hypertonic saline therapy improves outcome in patients with poor-grade spontaneous subarachnoid hemorrhage. J Neurosurg 2007; 107:274–282.
- Al-Rawi PG, Tseng MY, Richards HK, et al. Hypertonic saline in patients with poor-grade subarachnoid hemorrhage improves cerebral blood flow, brain tissue oxygen, and pH. Stroke 2010; 41:122–128.
- Tseng MY, Al-Rawi PG, Pickard JD, Rasulo FA, Kirkpatrick PJ. Effect of hypertonic saline on cerebral blood flow in poor-grade patients with subarachnoid hemorrhage. Stroke 2003; 34:1389–1396.
- Bentsen G, Breivik H, Lundar T, Stubhaug A. Hypertonic saline (7.2%) in 6% hydroxyethyl starch reduces intracranial pressure and improves hemodynamics in a placebo-controlled study involving stable patients with subarachnoid hemorrhage. Crit Care Med 2006; 34:2912–2917.
- Suarez JI, Qureshi AI, Parekh PD, et al. Administration of hypertonic (3%) sodium chloride/acetate in hyponatremic patients with symptomatic vasospasm following subarachnoid hemorrhage. J Neurosurg Anesthesiol 1999; 11:178–184.
- Stevens RD, Huff JS, Duckworth J, Papangelou A, Weingart SD, Smith WS. Emergency neurological life support: intracranial hypertension and herniation. Neurocrit Care 2012;17(suppl 1):S60–S65.
- Seule MA, Muroi C, Mink S, Yonekawa Y, Keller E. Therapeutic hypothermia in patients with aneurysmal subarachnoid hemorrhage, refractory intracranial hypertension, or cerebral vasospasm. Neurosurgery 2009; 64:86–93.
- Otani N, Takasato Y, Masaoka H, et al. Surgical outcome following decompressive craniectomy for poor-grade aneurysmal subarachnoid hemorrhage in patients with associated massive intracerebral or Sylvian hematomas. Cerebrovasc Dis 2008; 26:612–617.
- Lanzino G, D’Urso PI, Suarez J; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Seizures and anticonvulsants after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2011; 15:247–256.
- Naidech AM, Kreiter KT, Janjua N, et al. Phenytoin exposure is associated with functional and cognitive disability after subarachnoid hemorrhage. Stroke 2005; 36:583–587.
- Rosengart AJ, Huo JD, Tolentino J, et al. Outcome in patients with subarachnoid hemorrhage treated with antiepileptic drugs. J Neurosurg 2007; 107:253–260.
- Shah D, Husain AM. Utility of levetiracetam in patients with subarachnoid hemorrhage. Seizure 2009; 18:676–679.
- Patsalos PN. Pharmacokinetic profile of levetiracetam: toward ideal characteristics. Pharmacol Ther 2000; 85:77–85.
- Dewan MC, Mocco J. Current practice regarding seizure prophylaxis in aneurysmal subarachnoid hemorrhage across academic centers. J Neurointerv Surg 2014 Jan 28. doi: 10.1136/neurintsurg-2013-011075 [Epub ahead of print]
- Murphy-Human T, Welch E, Zipfel G, Diringer MN, Dhar R. Comparison of short-duration levetiracetam with extended-course phenytoin for seizure prophylaxis after subarachnoid hemorrhage. World Neurosurg 2011; 75:269–274.
- Dennis LJ, Claassen J, Hirsch LJ, Emerson RG, Connolly ES, Mayer SA. Nonconvulsive status epilepticus after subarachnoid hemorrhage. Neurosurgery 2002; 51:1136–1144.
- Vespa PM, Nuwer MR, Juhász C, et al. Early detection of vasospasm after acute subarachnoid hemorrhage using continuous EEG ICU monitoring. Electroencephalogr Clin Neurophysiol 1997; 103:607–615.
- Claassen J, Hirsch LJ, Kreiter KT, et al. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin Neurophysiol 2004; 115:2699–2710.
- Vergouwen MD, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke 2010; 41:2391–2395.
- Kelly PJ, Gorten RJ, Grossman RG, Eisenberg HM. Cerebral perfusion, vascular spasm, and outcome in patients with ruptured intracranial aneurysms. J Neurosurg 1977; 47:44–49.
- Aralasmak A, Akyuz M, Ozkaynak C, Sindel T, Tuncer R. CT angiography and perfusion imaging in patients with subarachnoid hemorrhage: correlation of vasospasm to perfusion abnormality. Neuroradiology 2009; 51:85–93.
- Sabri M, Lass E, Macdonald RL. Early brain injury: a common mechanism in subarachnoid hemorrhage and global cerebral ischemia. Stroke Res Treat 2013 Feb 28. doi: 10.1155/2013/394036 [Epub 2013 ahead of print]
- Yoon DY, Choi CS, Kim KH, Cho BM. Multidetector-row CT angiography of cerebral vasospasm after aneurysmal subarachnoid hemorrhage: comparison of volume-rendered images and digital subtraction angiography. AJNR Am J Neuroradiol 2006; 27:370–377.
- Wintermark M1, Ko NU, Smith WS, Liu S, Higashida RT, Dillon WP. Vasospasm after subarachnoid hemorrhage: utility of perfusion CT and CT angiography on diagnosis and management. AJNR Am J Neuroradiol 2006; 27:26–34.
- Helbok R, Madineni RC, Schmidt MJ, et al. Intracerebral monitoring of silent infarcts after subarachnoid hemorrhage. Neurocrit Care 2011; 14:162–167.
- Hänggi D; Participants in the International Multi-Disciplinary Consensus Conference on the Critical Care Management of Subarachnoid Hemorrhage. Monitoring and detection of vasospasm II: EEG and invasive monitoring. Neurocrit Care 2011; 15:318–323.
- Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G; European Stroke Organization. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis 2013; 35:93–112.
- Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989; 298:636–642.
- Dorhout Mees SM, Rinkel GJ, Feigin VL, et al. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2007; 3:CD000277.
- Lennihan L, Mayer SA, Fink ME, et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage: a randomized controlled trial. Stroke 2000; 31:383–391.
- Egge A, Waterloo K, Sjøholm H, Solberg T, Ingebrigtsen T, Romner B. Prophylactic hyperdynamic postoperative fluid therapy after aneurysmal subarachnoid hemorrhage: a clinical, prospective, randomized, controlled study. Neurosurgery 2001; 49:593–606.
- Jost SC, Diringer MN, Zazulia AR, et al. Effect of normal saline bolus on cerebral blood flow in regions with low baseline flow in patients with vasospasm following subarachnoid hemorrhage. J Neurosurg 2005; 103:25–30.
- Muizelaar JP, Becker DP. Induced hypertension for the treatment of cerebral ischemia after subarachnoid hemorrhage. Direct effect on cerebral blood flow. Surg Neurol 1986; 25:317–325.
- Levy ML, Rabb CH, Zelman V, Giannotta SL. Cardiac performance enhancement from dobutamine in patients refractory to hypervolemic therapy for cerebral vasospasm. J Neurosurg 1993; 79:494–499.
- Lannes M, Teitelbaum J, del Pilar Cortés M, Cardoso M, Angle M. Milrinone and homeostasis to treat cerebral vasospasm associated with subarachnoid hemorrhage: the Montreal Neurological Hospital protocol. Neurocrit Care 2012; 16:354–362.
- Zwienenberg-Lee M, Hartman J, Rudisill N, et al; Balloon Prophylaxis for Aneurysmal Vasospasm (BPAV) Study Group. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage: results of a phase II multicenter, randomized, clinical trial. Stroke 2008; 39:1759–1765.
- Rabinstein AA, Bruder N. Management of hyponatremia and volume contraction. Neurocrit Care 2011; 15:354–360.
- Wijdicks EF, Vermeulen M, Hijdra A, van Gijn J. Hyponatremia and cerebral infarction in patients with ruptured intracranial aneurysms: is fluid restriction harmful? Ann Neurol 1985; 17:137–140.
- Hasan D, Wijdicks EF, Vermeulen M. Hyponatremia is associated with cerebral ischemia in patients with aneurysmal subarachnoid hemorrhage. Ann Neurol 1990; 27:106–108.
- Wijdicks EF, Vermeulen M, ten Haaf JA, Hijdra A, Bakker WH, van Gijn J. Volume depletion and natriuresis in patients with a ruptured intracranial aneurysm. Ann Neurol 1985; 18:211–216.
- Hoff RG, Rinkel GJ, Verweij BH, Algra A, Kalkman CJ. Nurses’ prediction of volume status after aneurysmal subarachnoid haemorrhage: a prospective cohort study. Crit Care 2008; 12:R153.
- Hoff RG, van Dijk GW, Algra A, Kalkman CJ, Rinkel GJ. Fluid balance and blood volume measurement after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2008; 8:391–397.
- Berendes E, Walter M, Cullen P, et al. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet 1997; 349:245–249.
- Espiner EA, Leikis R, Ferch RD, et al. The neuro-cardio-endocrine response to acute subarachnoid haemorrhage. Clin Endocrinol (Oxf) 2002; 56:629–635.
- Isotani E, Suzuki R, Tomita K, et al. Alterations in plasma concentrations of natriuretic peptides and antidiuretic hormone after subarachnoid hemorrhage. Stroke 1994; 25:2198–2203.
- Benedict CR, Loach AB. Sympathetic nervous system activity in patients with subarachnoid hemorrhage. Stroke 1978; 9:237–244.
- Findling JW, Waters VO, Raff H. The dissociation of renin and aldosterone during critical illness. J Clin Endocrinol Metab 1987; 64:592–595.
- Solomon RA, Post KD, McMurtry JG 3rd. Depression of circulating blood volume in patients after subarachnoid hemorrhage: implications for the management of symptomatic vasospasm. Neurosurgery 1984; 15:354–361.
- Peters JP, Welt LG, Sims EA, Orloff J, Needham J. A salt-wasting syndrome associated with cerebral disease. Trans Assoc Am Physicians 1950; 63:57–64.
- Brimioulle S, Orellana-Jimenez C, Aminian A, Vincent JL. Hyponatremia in neurological patients: cerebral salt wasting versus inappropriate antidiuretic hormone secretion. Intensive Care Med 2008; 34:125–131.
- Singh S, Bohn D, Carlotti AP, Cusimano M, Rutka JT, Halperin ML. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med 2002; 30:2575–2579.
- Mutoh T, Kazumata K, Terasaka S, Taki Y, Suzuki A, Ishikawa T. Early intensive versus minimally invasive approach to postoperative hemodynamic management after subarachnoid hemorrhage. Stroke 2014; 45:1280–1284.
- Hasan D, Lindsay KW, Wijdicks EF, et al. Effect of fludrocortisone acetate in patients with subarachnoid hemorrhage. Stroke 1989; 20:1156–1161.
- Moro N, Katayama Y, Kojima J, Mori T, Kawamata T. Prophylactic management of excessive natriuresis with hydrocortisone for efficient hypervolemic therapy after subarachnoid hemorrhage. Stroke 2003; 34:2807–2811.
- Kilbourn KJ, Levy S, Staff I, Kureshi I, McCullough L. Clinical characteristics and outcomes of neurogenic stress cadiomyopathy in aneurysmal subarachnoid hemorrhage. Clin Neurol Neurosurg 2013; 115:909–914.
- Mayer SA, LiMandri G, Sherman D, et al. Electrocardiographic markers of abnormal left ventricular wall motion in acute subarachnoid hemorrhage. J Neurosurg 1995; 83:889–896.
- Deibert E, Barzilai B, Braverman AC, et al. Clinical significance of elevated troponin I levels in patients with nontraumatic subarachnoid hemorrhage. J Neurosurg 2003; 98:741–746.
- Banki N, Kopelnik A, Tung P, et al. Prospective analysis of prevalence, distribution, and rate of recovery of left ventricular systolic dysfunction in patients with subarachnoid hemorrhage. J Neurosurg 2006; 105:15–20.
- Kahn JM, Caldwell EC, Deem S, Newell DW, Heckbert SR, Rubenfeld GD. Acute lung injury in patients with subarachnoid hemorrhage: incidence, risk factors, and outcome. Crit Care Med 2006; 34:196–202.
- Kitamura Y, Nomura M, Shima H, et al. Acute lung injury associated with systemic inflammatory response syndrome following subarachnoid hemorrhage: a survey by the Shonan Neurosurgical Association. Neurol Med Chir (Tokyo) 2010; 50:456–460.
- Friedman JA, Pichelmann MA, Piepgras DG, et al. Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neurosurgery 2003; 52:1025–1032.
- Oh HS, Jeong HS, Seo WS. Non-infectious hyperthermia in acute brain injury patients: relationships to mortality, blood pressure, intracranial pressure and cerebral perfusion pressure. Int J Nurs Pract 2012; 18:295–302.
- Oddo M, Frangos S, Milby A, et al. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke 2009; 40:1913–1916.
- Badjatia N, Fernandez L, Schmidt JM, et al. Impact of induced normothermia on outcome after subarachnoid hemorrhage: a case-control study. Neurosurgery 2010; 66:696-701.
- Serrone JC1, Wash EM, Hartings JA, Andaluz N, Zuccarello M. Venous thromboembolism in subarachnoid hemorrhage. World Neurosurg 2013; 80:859–863.
- Mack WJ, Ducruet AF, Hickman ZL, et al. Doppler ultrasonography screening of poor-grade subarachnoid hemorrhage patients increases the diagnosis of deep venous thrombosis. Neurol Res 2008; 30:889–892.
- de Oliveira Manoel AL, Turkel-Parrella D, Germans M, et al. Safety of early pharmacological thromboprophylaxis after subarachnoid hemorrhage. Can J Neurol Sci 2014; 41:554–561.
KEY POINTS
- The key symptom is the abrupt onset of severe headache, commonly described as “the worst headache of my life.
- Computed tomography without contrast should be done promptly when this condition is suspected.
- Outcomes are improved when patients are managed in a high-volume center with a specialized neurointensive care unit and access to an interdisciplinary team.
- Early aneurysm repair by surgical clipping or endovascular coiling is considered the standard of care and is the best strategy to reduce the risk of rebleeding.
- Medical and neurologic complications are extremely common and include hydrocephalus, increased intracranial pressure, seizures, delayed cerebral ischemia, hyponatremia, hypovolemia, and cardiac and pulmonary abnormalities.
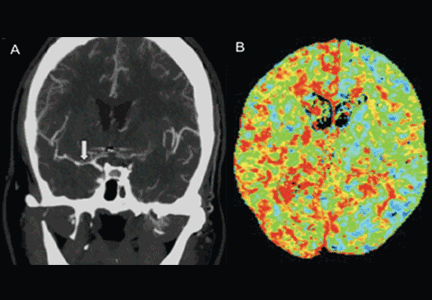
Left atrial appendage closure: An emerging option in atrial fibrillation when oral anticoagulants are not tolerated
Can patients with atrial fibrillation undergo a percutaneous procedure to reduce their risk of stroke, thereby eliminating the need for lifelong treatment with an oral anticoagulant drug? The data are preliminary, but this is an emerging option that physicians should be aware of.
We review here the current evidence and techniques aimed at isolating the left atrial appendage to prevent stroke, and we emphasize the need for continued systematic comparisons between oral anticoagulation and percutaneous treatment options.
NOVEL TREATMENTS ARE NEEDED
Atrial fibrillation is the most common cardiac arrhythmia,1 affecting an estimated 1% to 2% of people worldwide. In 2001, an estimated 2.3 million persons in the United States had atrial fibrillation, and that number is expected to more than double by 2050.2
Atrial fibrillation independently increases the risk of stroke by a factor of 4 to 5.3 The American Heart Association ranks stroke as the fourth most common cause of death and the leading cause of disability in the United States.4 Atrial fibrillation accounts for 15% of strokes in people of all ages and 30% in those over age 80.5 Untreated, 2% to 5% of patients with atrial fibrillation suffer a stroke in any given year.6 Most of these strokes are cardioembolic, with thrombi originating in the left atrial appendage.7 Furthermore, it has been estimated8,9 that patients with atrial fibrillation who have already had a stroke and cannot tolerate oral anticoagulants have an annual risk of stroke close to 12% and a relative risk of approximately 3.0 compared with those with atrial fibrillation and prior stroke who can tolerate anticoagulation.
Oral anticoagulation effectively prevents thromboembolic events associated with atrial fibrillation,10 but several factors limit its efficacy and applicability. The risk of bleeding complications, the need for frequent monitoring, and challenges with compliance create a large population of patients who would benefit from alternative approaches. Consequently, physicians have looked for other ways to prevent stroke—especially surgical and transcatheter procedures—that are not associated with an ongoing risk of hemorrhage and a lifelong need to take an anticoagulant.
THE LEFT ATRIAL APPENDAGE: A SITE OF CLOT FORMATION
The left atrial appendage is the most common site of thrombus formation, particularly in patients with nonvalvular atrial fibrillation. Nearly 90% of thrombi discovered in the left atrium form in the appendage.7 A study of 233 patients not on long-term anticoagulation revealed that after 48 hours of atrial fibrillation, 15% had a left atrial thrombus, and all but one of the thrombi were in the appendage.11
Believed to function as a decompression chamber during left ventricular systole, the left atrial appendage is embryologically derived from the left wall of the primary atrium. It is in close proximity to the free wall of the left ventricle, and therefore its flow can vary with left ventricular function. Relative stasis due to its location and extensive trabeculations, especially in times of poor forward flow, make it a high-risk site for clot formation.12
ANTICOAGULATION: EFFECTIVE BUT IMPERFECT
In deciding whether a patient with atrial fibrillation should be prescribed anticoagulation therapy, the physician must balance the risk of stroke against the risk of bleeding. Several tools for assessing these two risks have been developed. Of note, some of the risk factors for stroke are the same as some of the risk factors for bleeding.
Calculating the risk of stroke
CHADS2 and CHA2DS2-VASc are the two most commonly used tools for assessing the risk of stroke, but only the newer CHA2DS2-VASc has received a class I recommendation (the highest) from the European Society of Cardiology (ESC).13
CHADS2 risk factors are Congestive heart failure (1 point), Hypertension (1 point), Age 75 or older (1 point), Diabetes (1 point), and Stroke or transient ischemic attack (2 points). Risk of stroke is considered low with a score of 0, intermediate with a score of 1, and high with a score of 2 or more.
CHA2DS2-VASc risk factors are Congestive heart failure or left ventricular ejection fraction ≤ 40% (1 point), Hypertension (1 point), Age ≥ 75 (2 points), Age 65–74 (1 point), Diabetes mellitus (1 point), Stroke, transient ischemic attack, or thromboembolism (2 points), Vascular disease (1 point), and female Sex (1 point). Low risk is defined as a score of 0 for a man or, for a woman with no other risk factors, a score of 1. A score of 1 for a man indicates moderate risk, and a score of 2 or more is high risk.
Calculating the risk of bleeding
Tools for assessing bleeding risk include ATRIA2 and HAS-BLED,14 the latter carrying a class I recommendation from the ESC.13
HAS-BLED risk factors are Hypertension (1 point), Abnormal renal or liver function (1 point each), Stroke (1 point), Bleeding (1 point), Labile international normalized ratio (INR) (1 point), Elderly (age > 65) (1 point), and Drug or alcohol use (1 point each). The risk of bleeding is considered high with a score of 3 or higher.
Disadvantages of oral anticoagulation
Oral anticoagulation is the standard treatment for preventing stroke in patients with atrial fibrillation, and the vitamin K antagonist warfarin remains the foundation.
Though highly effective, warfarin requires close monitoring and frequent dose adjustments because of its numerous food and drug interactions. Bleeding risk and the challenge of frequent monitoring rule out treatment with warfarin in 14% to 44% of patients with atrial fibrillation.15 Even in “ideal” candidates, warfarin is underused, with one study reporting that only 38% of those with clinical indications for it had been prescribed warfarin, and of those for whom it had not been prescribed, 63% were also not taking aspirin.16 Moreover, a meta-analysis suggested that the average patient treated with warfarin has his or her INR in the therapeutic range only about 55% of the time.17
Newer, target-specific oral anticoagulants such as dabigatran (a direct thrombin inhibitor) and rivaroxaban and apixaban (both factor Xa inhibitors) do not require monitoring and have fewer drug interactions. But like warfarin, they also confer a risk of serious bleeding.18–20 Most of the studies of these newer drugs have compared them with warfarin, with the preponderance of evidence showing them to be either noninferior or superior to warfarin for stroke reduction. But bleeding complication rates remain significant, apixaban having lower rates of major bleeding than dabigatran and rivaroxaban.
In a meta-analysis, Ruff et al21 concluded that the target-specific oral anticoagulants provide a favorable balance of risk and benefit. Compared with warfarin, these new drugs reduced the rate of stroke or systemic embolic events by 19%. There was also a significant reduction in rates of intracranial hemorrhage and all-cause mortality. The risk of major bleeding was similar to that with warfarin, though there was a higher risk of gastrointestinal bleeding with target-specific agents. These effects were consistent across a wide range of patients.
Given the difficulties, risks, and serious side effects of anticoagulant therapy, many patients stop taking these drugs soon after starting them, either on their own or on their physician’s recommendation. In the RE-LY trial (Dabigatran vs Warfarin in Patients With Atrial Fibrillation), 10% of patients receiving dabigatran and 17% of those receiving warfarin stopped the treatment within 1 to 2 years.22 In a similar trial of rivaroxaban vs warfarin in nonvalvular atrial fibrillation (the ROCKET-AF trial), 24% of those treated with rivaroxaban and 22% of those treated with warfarin stopped treatment during the study.19 In the ARISTOTLE trial (Apixaban vs Warfarin in Patients With Atrial Fibrillation), 25% of patients discontinued apixaban and 28% discontinued warfarin.20
The results of these trials show a clear need for treatments without high attrition rates, since patients with atrial fibrillation need protection from stroke for the rest of their life.
SURGICAL CLOSURE AS AN ADD-ON TO OTHER PROCEDURES
If the patient is undergoing cardiac surgery for another reason, the surgeon can excise, suture, staple, or clip the left atrial appendage shut at the same time. Closure is recommended as part of valve replacement.8 In a 2008 retrospective study, left atrial appendage closure was successfully performed in 40% of those undergoing the procedure during cardiac surgery, and complete closure occurred more often with excision than with suture exclusion and stapler exclusion.23 A study of patients who underwent ligation of the left atrial appendage during mitral value replacement found that 35% demonstrated incomplete closure as determined by transesophageal echocardiography.24
Newer devices have shown more success. The AtriClip (AtriCure Inc., West Chester, OH) is a self-closing, implantable clip applied epicardially by either an open surgical or a minimally invasive technique.25 Successful closure was confirmed in 60 of 61 patients at 90 days as determined by computed tomography or transesophageal echocardiography, and there were no adverse events related to implantation of the device.25 The TigerPaw system (Terumo Cardiovascular Systems, Ann Arbor, MI)26 is a fastener delivered surgically around the base of the ostium of the left atrial appendage. In an initial trial, 90 days after the procedure, transesophageal echocardiography showed no leaks in any of those who were examined (54 of 60 patients).
Amputation of the left atrial appendage is also considered part of the maze procedure for atrial fibrillation, in which the operator creates multiple small scars in the atria to prevent irregular impulses from being conducted.27
Results of these surgical approaches have been mixed, as incomplete closure or clipping actually increases the risk of left atrial thrombus formation and embolization.28 Moreover, these invasive surgical techniques are associated with significant periprocedural morbidity.29 Because of the high risk of surgical complications, cardiac specialists have sought less invasive percutaneous procedures to manage stroke risk in patients with atrial fibrillation.
PERCUTANEOUS OCCLUSION
One option for closing the left atrial appendage is a percutaneous transseptal approach in which a plug is placed in the opening connecting the appendage to the rest of the atrium.
The PLAATO device
The Percutaneous LAA Transcatheter Occlusion (PLAATO) device (Appriva Medical Inc., Sunnyvale, CA) contains an expandable nitinol-covered cage designed to be placed in the orifice of the left atrial appendage. Over time, tissue grows into the device, entirely isolating the appendage from the rest of the atrium.
In 2002, Sievert et al30 reported using this device in 15 patients. Subsequently, in a nonrandomized trial in patients with contraindications to lifelong anticoagulation, total occlusion was achieved in 108 of 111 patients, with no thrombosis or migration of the device at 6 months. The annual risk of stroke was 2.2%, a reduction in relative risk of 65% based on the CHADS2 score.31
But despite this apparent success, the PLAATO device was discontinued for unspecified commercial reasons.
Amplatzer cardiac plug
Modeled after an atrial septal occluder, the Amplatzer cardiac plug (St. Jude Medical, St. Paul, MN) consists of a lobe and a disk connected by a central waist.
In 2011, Park et al32 published their initial experience implanting this device in patients who either could not tolerate or did not desire long-term anticoagulation. They reported a 96% closure rate (137 of 143 patients), but there were serious complications in 10 patients: 3 with ischemic stroke, 2 with device embolism, and 5 with pericardial effusions.
Urena et al33 reported similar results in 52 patients with absolute contraindications to warfarin, with a 98.1% implantation rate. Patients were then maintained on either single or dual antiplatelet therapy at the discretion of the operator. At 20-month follow-up, there had been one stroke, one transient ischemic attack, and one major bleeding event. The leakage rate was 16.2% as determined by transesophageal echocardiography.
While initial results were promising, a clinical trial comparing this device and optimal medical treatment is currently on hold. Thus, there are no clear data comparing the Amplatzer device with oral anticoagulation.34
The Watchman device
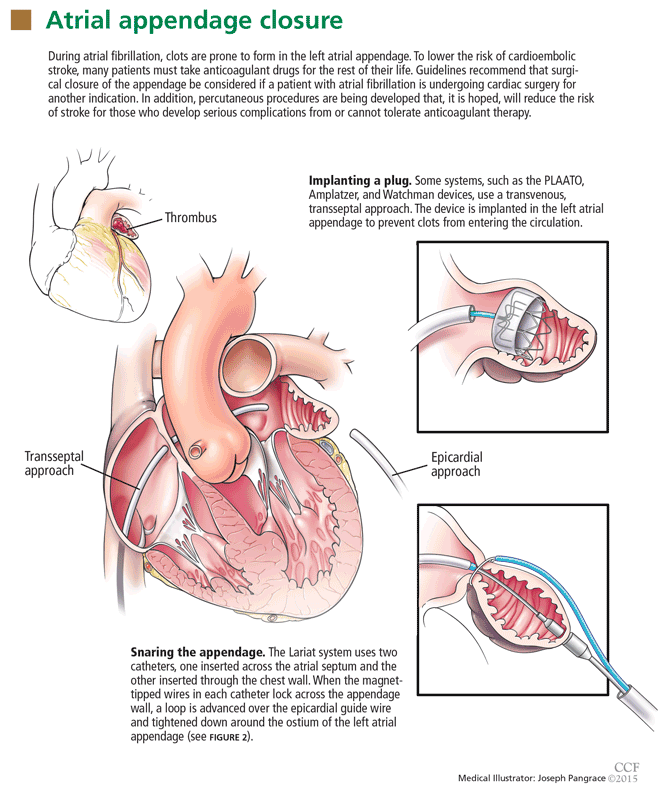
The Watchman device (Boston Scientific, Marlborough, MA), an evolution of the PLAATO device, is a self-expanding nitinol structure with fixation barbs and a polyethylene membrane to protect the atrium-facing side of the device (Figure 1).
A pilot trial reported successful implantation in 66 of 75 patients, though the device was found to migrate after placement in 5 of the first 16 patients using the original device and delivery system. The device was modified, and no further embolization of the device occurred.35
The PROTECT-AF trial (Protection in Patients With Atrial Fibrillation)36 was the first completed and published randomized controlled trial evaluating left atrial appendage closure using a device vs long-term warfarin therapy. This study randomized 707 people with nonvalvular atrial fibrillation from 59 centers worldwide to receive the Watchman device or a control treatment. The study included patients age 18 or older with nonvalvular atrial fibrillation who were able to tolerate warfarin therapy. Patients in the control group received warfarin for the duration of the study and were monitored every 2 weeks for a goal INR of 2 to 3, achieving a therapeutic INR 66% of the time. The device group was also treated with warfarin for 45 days to allow device endothelialization. Warfarin was discontinued if transesophageal echocardiography showed complete closure or significantly decreased flow around the device. Patients in the device group were then treated with aspirin and clopidogrel for 6 months, and then with aspirin indefinitely.
At 1,065 patient-years of follow-up, PROTECT-AF showed that in patients with atrial fibrillation who were candidates for warfarin therapy, percutaneous left atrial appendage closure using the Watchman device reduced the rate of hemorrhagic stroke compared with warfarin and was noninferior to warfarin in terms of all-cause mortality and stroke. A 4-year follow-up to the PROTECT-AF trial found that receiving the Watchman was better than taking warfarin in terms of risk of cardiovascular death, stroke and other systemic embolization, and all-cause mortality. The adverse event rates were 2.3% in the device group and 3.8% in the control group, a 40% relative risk reduction in the Watchman group.37
The PREVAIL trial (Prospective Randomized Evaluation of the WATCHMAN LAA Closure Device in Patients With Atrial Fibrillation vs Long-Term Warfarin Therapy) aimed to confirm the safety and efficacy of the Watchman device compared with long-term warfarin therapy.38 The event rate (defined as 7-day occurrence of death, ischemic stroke, systemic embolism, and procedure- or device-related complications requiring major cardiovascular or endovascular intervention) was 2.2%. But the PREVAIL trial was unable to show that the device was noninferior to warfarin in terms of its second primary end point of stroke, systemic embolism, and cardiovascular or unexplained death at 18 months. When performed by physicians who were new to the procedure, the procedure was successful (ie, the device was successfully implanted) in 93.2%; the rate was slightly higher (96.3%) when performed by experienced implanters.
Safety data gathered in PREVAIL in conjunction with demonstrated efficacy from PROTECT-AF suggest that the Watchman device may be a safe and effective alternative to long-term oral anticoagulation in patients with nonvalvular atrial fibrillation.
In patients with contraindications to warfarin
Most of the published data have been about the efficacy of occlusion devices compared with long-term warfarin therapy. Unfortunately, the population that has not been studied extensively is patients who have contraindications to long-term oral anticoagulation, who would benefit the most from an occlusive device.
The ASA Plavix Feasibility Study (ASAP) focused on this population, specifically those who had a CHADS2 score of 1 or higher and who were considered ineligible for warfarin, to determine whether closure using the Watchman device could be safely performed without a transition period with warfarin.39 After device implantation, trial participants were given clopidogrel for 6 months and aspirin indefinitely. The trial enrolled 150 patients and followed them for a mean of 14.4 (± 8.6) months. In that time, there were four strokes, five pericardial effusions, and six instances of device-related thrombus by transesophageal echocardiography. Three of the strokes were ischemic (1.7% per year), which is a 77% reduction from the expected rate of 7.3% based on the CHADS2 scores of the patient cohort.
These data suggest that implantation of the Watchman device may be appropriate for those who cannot tolerate warfarin even in the short term.
The Lariat system
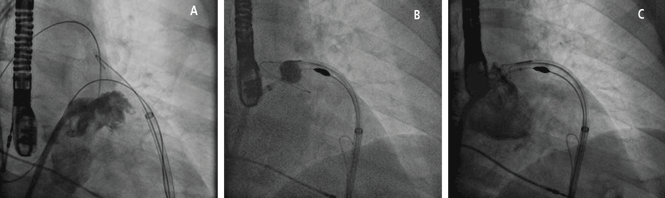
The Lariat suture delivery device (SentreHeart, Inc., Redwood City, CA) is approved by the US Food and Drug Administration (FDA) for soft-tissue closure and has been used for percutaneous left atrial appendage closure. It uses a magnet-tipped wire that is passed to the epicardial side of the left atrial appendage via pericardial access to meet a second magnet-tipped wire introduced into the appendage via transseptal access. A “lasso” is then advanced over the epicardial guide wire and tightened down around the ostium of the left atrial appendage. This tool facilitates deployment of a nonabsorbable polyester suture, which effectively ligates off the appendage from the rest of the left atrium (Figure 2).40 In theory, the Lariat’s epicardial approach could eliminate the need for short- and long-term anticoagulation, as there would be no foreign body left within the heart.
In an initial cohort of 89 patients in Poland,41 the investigators reported a 96% closure rate as determined by transesophageal echocardiography immediately after the procedure. At 1-year follow-up, there was 98% complete closure, including cases of incomplete closure detected earlier.41 Adverse events were limited, with only two cases of severe pericarditis, two strokes, and one pericardial effusion. These results were replicated in the United States in a cohort of 25 patients, with a 100% closure rate and no stroke events.42
There have been three published case reports of left atrial clot formation after successful left atrial appendage ligation using the Lariat device.43–45 These experiences further emphasize that closure does not necessarily confer instant stroke prevention, and there remains a need to investigate the need for routine imaging and possibly periprocedural anticoagulation after ligation.
More recently, Pillai et al46 published their initial experience following 71 patients with echocardiograms 3 months after left atrial appendage closure using the Lariat device. They reported leaks in 6 of the 71 patients; five of the leaks were successfully closed using the Amplatzer Septal Occluder, and one was closed with a repeat Lariat procedure.
Although the Lariat system has been used in more than 2,000 patients worldwide (SentreHeart, personal communication), there has been no published systematic comparison between it and oral anticoagulation to date.
AN EMERGING OPTION
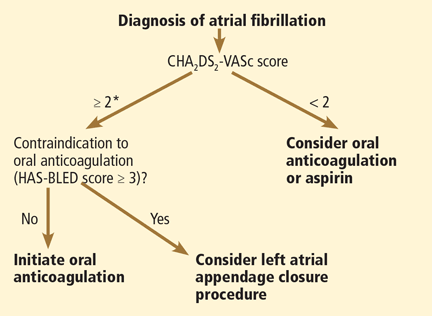
Established guidelines help determine which patients with atrial fibrillation should receive oral anticoagulant therapy. For patients who have absolute contraindications to oral anticoagulants or who are undergoing cardiac surgery, surgical ligation of the left atrial appendage is an option. But for those with contraindications to oral anticoagulation in both the short term and the long term, there is a growing body of evidence suggesting that a percutaneous intervention is at least noninferior to—and in some cases is superior to—warfarin. Figure 3 shows our recommendations for the steps to determine which patients would be most appropriate to consider for left atrial appendage closure.
Holmes et al47 propose that we may now have enough evidence to support an expedited regulatory approval process of these occlusion devices. But there are still a number of areas in which further investigation is clearly needed before left atrial appendage occlusion devices can be widely adopted.
The trials discussed above had specific inclusion and exclusion criteria, and therefore, although they support percutaneous intervention, the generalizability of their results remains in question. Indeed, the patients in PROTECT-AF36 had an average CHADS2 score of only 2.2. This study also included only patients who were able to tolerate both aspirin and clopidogrel simultaneously for a significant amount of time. Hence, one cannot assume the results would be the same in patients who have strict contraindications to warfarin or any target-specific oral anticoagulant. Concern regarding the generalizability of the conclusions from PROTECT-AF and PREVAIL has led to mixed votes (three assessments to date) from the FDA Circulatory Device Panel.48
In an encouraging review of cases, Gafoor et al49 reported safe and efficacious occlusion in octogenarians using the devices mentioned above. These patients often pose the greatest challenge in initiating long-term anticoagulation because of the many drug-drug interactions and the risk of intracranial hemorrhage secondary to falls.
Further, while occlusion devices would clearly be useful for patients in whom traditional oral anticoagulation is not an option, the newer oral anticoagulants might complicate the picture somewhat. As shown by Ruff et al,21 the risk-benefit ratio of these target-specific oral anticoagulants is quite favorable and by some measurements is superior to that of warfarin. Could there be a group of patients who cannot take warfarin but could instead do well on one of the newer anticoagulants, thus alleviating the need for percutaneous intervention? As the newer oral anticoagulants become more commonly used, the cost-benefit analysis of implanting an occlusion device could shift.
Lastly, in this era of high-value medical care, one must consider the cost-effectiveness of these novel interventions. As with any new technology, the up-front cost of implantation is certainly greater than that of warfarin therapy. If device implantation can prevent a hospitalization from a major bleed secondary to warfarin use or prevent a catastrophic stroke due to untreated atrial fibrillation, then the cost-benefit analysis may be tipped in the other direction. As these devices become more widely available and physicians have more experience implanting them, the costs will likely decrease.
As with oral anticoagulation therapy, all interventions, whether surgical or percutaneous, carry a risk of bleeding and stroke. There remains no substitute for frank and clear discussions between the physician and patient regarding the risks and benefits of each approach.
While a growing body of evidence surrounds left atrial appendage occlusion devices, many questions remain. Notably, could these devices be used in patients who can tolerate oral anticoagulants? And if so, which subgroups would benefit most? Does occlusion or ligation of the left atrial appendage affect electrical connections between it and the left atrium, thereby lowering the burden of atrial fibrillation?
We expect that continued investigation of and experience with left atrial appendage closure devices will position them one day as a viable and equal option for preventing stroke in patients with atrial fibrillation.
- Rosamond W, Flegal K, Furie K, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2008; 117:e25–146.
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001; 285:2370–2375.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991; 22:983–988.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation 2013; 127:e6–e245.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation: a major contributor to stroke in the elderly. The Framingham Study. Arch Intern Med 1987; 147:1561–1564.
- Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med 1994; 154:1449–1457.
- Blackshear JL, Odell JA. Appendage obliteration to reduce stroke in cardiac surgical patients with atrial fibrillation. Ann Thorac Surg 1996; 61:755–759.
- Bonow RO, Carabello BA, Chatterjee K, et al; 2006 Writing Committee Members; American College of Cardiology/American Heart Association Task Force. 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation 2008; 118:e523–e661.
- Odell JA, Blackshear JL, Davies E, et al. Thoracoscopic obliteration of the left atrial appendage: potential for stroke reduction? Ann Thorac Surg 1996; 61:565–569.
- Hart RG, Benavente O, McBride R, Pearce LA. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a meta-analysis. Ann Intern Med 1999; 131:492–501.
- Manning WJ, Silverman DI, Keighley CS, Oettgen P, Douglas PS. Transesophageal echocardiographically facilitated early cardioversion from atrial fibrillation using short-term anticoagulation: final results of a prospective 4.5-year study. J Am Coll Cardiol 1995; 25:1354–1361.
- Al-Saady NM, Obel OA, Camm AJ. Left atrial appendage: structure, function, and role in thromboembolism. Heart 1999; 82:547–554.
- Lip GY. Recommendations for thromboprophylaxis in the 2012 focused update of the ESC guidelines on atrial fibrillation: a commentary. J Thromb Haemost 2013; 11:615–626.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 2010; 138:1093–1100.
- Onalan O, Lashevsky I, Hamad A, Crystal E. Nonpharmacologic stroke prevention in atrial fibrillation. Expert Rev Cardiovasc Ther 2005; 3:619–633.
- Brass LM, Krumholz HM, Scinto JM, Radford M. Warfarin use among patients with atrial fibrillation. Stroke 1997; 28:2382–2389.
- Baker WL, Cios DA, Sander SD, Coleman CI. Meta-analysis to assess the quality of warfarin control in atrial fibrillation patients in the United States. J Manag Care Pharm 2009; 15:244–252.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365:981–992.
- Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet 2014; 383:955–962.
- Lip GY, Clemens A, Noack H, Ferreira J, Connolly SJ, Yusuf S. Patient outcomes using the European label for dabigatran. A post-hoc analysis from the RE-LY database. Thromb Haemost 2014; 111:933–942.
- Kanderian AS, Gillinov AM, Pettersson GB, Blackstone E, Klein AL. Success of surgical left atrial appendage closure: assessment by transesophageal echocardiography. J Am Coll Cardiol 2008; 52:924–929.
- Katz ES, Tsiamtsiouris T, Applebaum RM, Schwartzbard A, Tunick PA, Kronzon I. Surgical left atrial appendage ligation is frequently incomplete: a transesophageal echocardiograhic study. J Am Coll Cardiol 2000; 36:468–471.
- Ailawadi G, Gerdisch MW, Harvey RL, et al. Exclusion of the left atrial appendage with a novel device: early results of a multicenter trial. J Thorac Cardiovasc Surg 2011; 142:1002–1009.e1.
- Slater AD, Tatooles AJ, Coffey A, et al. Prospective clinical study of a novel left atrial appendage occlusion device. Ann Thorac Surg 2012; 93:2035-2040.
- Pinho-Gomes AC, Amorim MJ, Oliveira SM, Leite-Moreira AF. Surgical treatment of atrial fibrillation: an updated review. Eur J Cardiothorac Surg 2014; 46:167–178.
- Aryana A, Cavaco D, Arthur A, O’Neill PG, Adragão P, D’Avila A. Percutaneous endocardial occlusion of incompletely surgically ligated left atrial appendage. J Cardiovasc Electrophysiol 2013; 24:968–974.
- García-Fernández MA, Pérez-David E, Quiles J, et al. Role of left atrial appendage obliteration in stroke reduction in patients with mitral valve prosthesis: a transesophageal echocardiographic study. J Am Coll Cardiol 2003; 42:1253–1258.
- Sievert H, Lesh MD, Trepels T, et al. Percutaneous left atrial appendage transcatheter occlusion to prevent stroke in high-risk patients with atrial fibrillation: early clinical experience. Circulation 2002; 105:1887–1889.
- Ostermayer SH, Reisman M, Kramer PH, et al. Percutaneous left atrial appendage transcatheter occlusion (PLAATO system) to prevent stroke in high-risk patients with non-rheumatic atrial fibrillation: results from the international multi-center feasibility trials. J Am Coll Cardiol 2005; 46:9–14.
- Park JW, Bethencourt A, Sievert H, et al. Left atrial appendage closure with Amplatzer cardiac plug in atrial fibrillation: initial European experience. Catheter Cardiovasc Interv 2011; 77:700–706.
- Urena M, Rodés-Cabau J, Freixa X, et al. Percutaneous left atrial appendage closure with the AMPLATZER cardiac plug device in patients with nonvalvular atrial fibrillation and contraindications to anticoagulation therapy. J Am Coll Cardiol 2013; 62:96–102.
- ClinicalTrials.gov. http://clinicaltrials.gov/show/NCT01118299. Accessed January 30, 2015.
- Sick PB, Schuler G, Hauptmann KE, et al. Initial worldwide experience with the WATCHMAN left atrial appendage system for stroke prevention in atrial fibrillation. J Am Coll Cardiol 2007; 49:1490–1495.
- Holmes DR, Reddy VY, Turi ZG, et al; PROTECT AF Investigators. Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomised non-inferiority trial. Lancet 2009; 374:534–542.
- Boston Scientific. WATCHMAN™ Left Atrial Appendage Closure Device. http://www.bostonscientific.com/watchman-eu/assets/pdf/SH-158101-AA-PROTECT-AF-Reddy-HRS-2013.pdf. Accessed January 30, 2015.
- David Holmes M. Boston Scientific. March 9, 2013. Available at: http://www.bostonscientific.com/watchman-eu/assets/downloads/PREVAIL-Clinical-Results.ppt.pdf. Accessed January 30, 2015.
- Reddy VY, Möbius-Winkler S, Miller MA, et al. Left atrial appendage closure with the Watchman device in patients with a contraindication for oral anticoagulation: the ASAP study (ASA Plavix Feasibility Study With Watchman Left Atrial Appendage Closure Technology). J Am Coll Cardiol 2013; 61:2551–2556.
- Koneru JN, Badhwar N, Ellenbogen KA, Lee RJ. LAA ligation using the LARIAT suture delivery device: tips and tricks for a successful procedure. Heart Rhythm 2014; 11:911–921.
- Bartus K, Han FT, Bednarek J, et al. Percutaneous left atrial appendage suture ligation using the LARIAT device in patients with atrial fibrillation: initial clinical experience. J Am Coll Cardiol 2013; 62:108–118.
- Massumi A, Chelu MG, Nazeri A, et al. Initial experience with a novel percutaneous left atrial appendage exclusion device in patients with atrial fibrillation, increased stroke risk, and contraindications to anticoagulation. Am J Cardiol 2013; 111:869–873.
- Giedrimas E, Lin AC, Knight BP. Left atrial thrombus after appendage closure using LARIAT. Circ Arrhythm Electrophysiol 2013; 6:e52–e53.
- Briceno DF, Fernando RR, Laing ST. Left atrial appendage thrombus post LARIAT closure device. Heart Rhythm 2014; 11:1600–1601.
- Baker MS, Paul Mounsey J, Gehi AK, Chung EH. Left atrial thrombus after appendage ligation with LARIAT. Heart Rhythm 2014; 11:1489.
- Pillai AM, Kanmanthareddy A, Earnest M, et al. Initial experience with post Lariat left atrial appendage leak closure with Amplatzer septal occluder device and repeat Lariat application. Heart Rhythm 2014; 11:1877–1883.
- Holmes DR Jr, Lakkireddy DR, Whitlock RP, Waksman R, Mack MJ. Left atrial appendage occlusion: opportunities and challenges. J Am Coll Cardiol 2014; 63:291–298.
- Wood S. FDA Advisors cool on Watchman approval amid ischemic-stroke data. Medscape Multispecialty October 8, 2014. www.medscape.com/viewarticle/832993.
- Gafoor S, Franke J, Bertog S, et al. Left atrial appendage occlusion in octogenarians: short-term and 1-year follow-up. Catheter Cardiovasc Interv 2014; 83:805–810.
Can patients with atrial fibrillation undergo a percutaneous procedure to reduce their risk of stroke, thereby eliminating the need for lifelong treatment with an oral anticoagulant drug? The data are preliminary, but this is an emerging option that physicians should be aware of.
We review here the current evidence and techniques aimed at isolating the left atrial appendage to prevent stroke, and we emphasize the need for continued systematic comparisons between oral anticoagulation and percutaneous treatment options.
NOVEL TREATMENTS ARE NEEDED
Atrial fibrillation is the most common cardiac arrhythmia,1 affecting an estimated 1% to 2% of people worldwide. In 2001, an estimated 2.3 million persons in the United States had atrial fibrillation, and that number is expected to more than double by 2050.2
Atrial fibrillation independently increases the risk of stroke by a factor of 4 to 5.3 The American Heart Association ranks stroke as the fourth most common cause of death and the leading cause of disability in the United States.4 Atrial fibrillation accounts for 15% of strokes in people of all ages and 30% in those over age 80.5 Untreated, 2% to 5% of patients with atrial fibrillation suffer a stroke in any given year.6 Most of these strokes are cardioembolic, with thrombi originating in the left atrial appendage.7 Furthermore, it has been estimated8,9 that patients with atrial fibrillation who have already had a stroke and cannot tolerate oral anticoagulants have an annual risk of stroke close to 12% and a relative risk of approximately 3.0 compared with those with atrial fibrillation and prior stroke who can tolerate anticoagulation.
Oral anticoagulation effectively prevents thromboembolic events associated with atrial fibrillation,10 but several factors limit its efficacy and applicability. The risk of bleeding complications, the need for frequent monitoring, and challenges with compliance create a large population of patients who would benefit from alternative approaches. Consequently, physicians have looked for other ways to prevent stroke—especially surgical and transcatheter procedures—that are not associated with an ongoing risk of hemorrhage and a lifelong need to take an anticoagulant.
THE LEFT ATRIAL APPENDAGE: A SITE OF CLOT FORMATION
The left atrial appendage is the most common site of thrombus formation, particularly in patients with nonvalvular atrial fibrillation. Nearly 90% of thrombi discovered in the left atrium form in the appendage.7 A study of 233 patients not on long-term anticoagulation revealed that after 48 hours of atrial fibrillation, 15% had a left atrial thrombus, and all but one of the thrombi were in the appendage.11
Believed to function as a decompression chamber during left ventricular systole, the left atrial appendage is embryologically derived from the left wall of the primary atrium. It is in close proximity to the free wall of the left ventricle, and therefore its flow can vary with left ventricular function. Relative stasis due to its location and extensive trabeculations, especially in times of poor forward flow, make it a high-risk site for clot formation.12
ANTICOAGULATION: EFFECTIVE BUT IMPERFECT
In deciding whether a patient with atrial fibrillation should be prescribed anticoagulation therapy, the physician must balance the risk of stroke against the risk of bleeding. Several tools for assessing these two risks have been developed. Of note, some of the risk factors for stroke are the same as some of the risk factors for bleeding.
Calculating the risk of stroke
CHADS2 and CHA2DS2-VASc are the two most commonly used tools for assessing the risk of stroke, but only the newer CHA2DS2-VASc has received a class I recommendation (the highest) from the European Society of Cardiology (ESC).13
CHADS2 risk factors are Congestive heart failure (1 point), Hypertension (1 point), Age 75 or older (1 point), Diabetes (1 point), and Stroke or transient ischemic attack (2 points). Risk of stroke is considered low with a score of 0, intermediate with a score of 1, and high with a score of 2 or more.
CHA2DS2-VASc risk factors are Congestive heart failure or left ventricular ejection fraction ≤ 40% (1 point), Hypertension (1 point), Age ≥ 75 (2 points), Age 65–74 (1 point), Diabetes mellitus (1 point), Stroke, transient ischemic attack, or thromboembolism (2 points), Vascular disease (1 point), and female Sex (1 point). Low risk is defined as a score of 0 for a man or, for a woman with no other risk factors, a score of 1. A score of 1 for a man indicates moderate risk, and a score of 2 or more is high risk.
Calculating the risk of bleeding
Tools for assessing bleeding risk include ATRIA2 and HAS-BLED,14 the latter carrying a class I recommendation from the ESC.13
HAS-BLED risk factors are Hypertension (1 point), Abnormal renal or liver function (1 point each), Stroke (1 point), Bleeding (1 point), Labile international normalized ratio (INR) (1 point), Elderly (age > 65) (1 point), and Drug or alcohol use (1 point each). The risk of bleeding is considered high with a score of 3 or higher.
Disadvantages of oral anticoagulation
Oral anticoagulation is the standard treatment for preventing stroke in patients with atrial fibrillation, and the vitamin K antagonist warfarin remains the foundation.
Though highly effective, warfarin requires close monitoring and frequent dose adjustments because of its numerous food and drug interactions. Bleeding risk and the challenge of frequent monitoring rule out treatment with warfarin in 14% to 44% of patients with atrial fibrillation.15 Even in “ideal” candidates, warfarin is underused, with one study reporting that only 38% of those with clinical indications for it had been prescribed warfarin, and of those for whom it had not been prescribed, 63% were also not taking aspirin.16 Moreover, a meta-analysis suggested that the average patient treated with warfarin has his or her INR in the therapeutic range only about 55% of the time.17
Newer, target-specific oral anticoagulants such as dabigatran (a direct thrombin inhibitor) and rivaroxaban and apixaban (both factor Xa inhibitors) do not require monitoring and have fewer drug interactions. But like warfarin, they also confer a risk of serious bleeding.18–20 Most of the studies of these newer drugs have compared them with warfarin, with the preponderance of evidence showing them to be either noninferior or superior to warfarin for stroke reduction. But bleeding complication rates remain significant, apixaban having lower rates of major bleeding than dabigatran and rivaroxaban.
In a meta-analysis, Ruff et al21 concluded that the target-specific oral anticoagulants provide a favorable balance of risk and benefit. Compared with warfarin, these new drugs reduced the rate of stroke or systemic embolic events by 19%. There was also a significant reduction in rates of intracranial hemorrhage and all-cause mortality. The risk of major bleeding was similar to that with warfarin, though there was a higher risk of gastrointestinal bleeding with target-specific agents. These effects were consistent across a wide range of patients.
Given the difficulties, risks, and serious side effects of anticoagulant therapy, many patients stop taking these drugs soon after starting them, either on their own or on their physician’s recommendation. In the RE-LY trial (Dabigatran vs Warfarin in Patients With Atrial Fibrillation), 10% of patients receiving dabigatran and 17% of those receiving warfarin stopped the treatment within 1 to 2 years.22 In a similar trial of rivaroxaban vs warfarin in nonvalvular atrial fibrillation (the ROCKET-AF trial), 24% of those treated with rivaroxaban and 22% of those treated with warfarin stopped treatment during the study.19 In the ARISTOTLE trial (Apixaban vs Warfarin in Patients With Atrial Fibrillation), 25% of patients discontinued apixaban and 28% discontinued warfarin.20
The results of these trials show a clear need for treatments without high attrition rates, since patients with atrial fibrillation need protection from stroke for the rest of their life.
SURGICAL CLOSURE AS AN ADD-ON TO OTHER PROCEDURES
If the patient is undergoing cardiac surgery for another reason, the surgeon can excise, suture, staple, or clip the left atrial appendage shut at the same time. Closure is recommended as part of valve replacement.8 In a 2008 retrospective study, left atrial appendage closure was successfully performed in 40% of those undergoing the procedure during cardiac surgery, and complete closure occurred more often with excision than with suture exclusion and stapler exclusion.23 A study of patients who underwent ligation of the left atrial appendage during mitral value replacement found that 35% demonstrated incomplete closure as determined by transesophageal echocardiography.24
Newer devices have shown more success. The AtriClip (AtriCure Inc., West Chester, OH) is a self-closing, implantable clip applied epicardially by either an open surgical or a minimally invasive technique.25 Successful closure was confirmed in 60 of 61 patients at 90 days as determined by computed tomography or transesophageal echocardiography, and there were no adverse events related to implantation of the device.25 The TigerPaw system (Terumo Cardiovascular Systems, Ann Arbor, MI)26 is a fastener delivered surgically around the base of the ostium of the left atrial appendage. In an initial trial, 90 days after the procedure, transesophageal echocardiography showed no leaks in any of those who were examined (54 of 60 patients).
Amputation of the left atrial appendage is also considered part of the maze procedure for atrial fibrillation, in which the operator creates multiple small scars in the atria to prevent irregular impulses from being conducted.27
Results of these surgical approaches have been mixed, as incomplete closure or clipping actually increases the risk of left atrial thrombus formation and embolization.28 Moreover, these invasive surgical techniques are associated with significant periprocedural morbidity.29 Because of the high risk of surgical complications, cardiac specialists have sought less invasive percutaneous procedures to manage stroke risk in patients with atrial fibrillation.
PERCUTANEOUS OCCLUSION
One option for closing the left atrial appendage is a percutaneous transseptal approach in which a plug is placed in the opening connecting the appendage to the rest of the atrium.
The PLAATO device
The Percutaneous LAA Transcatheter Occlusion (PLAATO) device (Appriva Medical Inc., Sunnyvale, CA) contains an expandable nitinol-covered cage designed to be placed in the orifice of the left atrial appendage. Over time, tissue grows into the device, entirely isolating the appendage from the rest of the atrium.
In 2002, Sievert et al30 reported using this device in 15 patients. Subsequently, in a nonrandomized trial in patients with contraindications to lifelong anticoagulation, total occlusion was achieved in 108 of 111 patients, with no thrombosis or migration of the device at 6 months. The annual risk of stroke was 2.2%, a reduction in relative risk of 65% based on the CHADS2 score.31
But despite this apparent success, the PLAATO device was discontinued for unspecified commercial reasons.
Amplatzer cardiac plug
Modeled after an atrial septal occluder, the Amplatzer cardiac plug (St. Jude Medical, St. Paul, MN) consists of a lobe and a disk connected by a central waist.
In 2011, Park et al32 published their initial experience implanting this device in patients who either could not tolerate or did not desire long-term anticoagulation. They reported a 96% closure rate (137 of 143 patients), but there were serious complications in 10 patients: 3 with ischemic stroke, 2 with device embolism, and 5 with pericardial effusions.
Urena et al33 reported similar results in 52 patients with absolute contraindications to warfarin, with a 98.1% implantation rate. Patients were then maintained on either single or dual antiplatelet therapy at the discretion of the operator. At 20-month follow-up, there had been one stroke, one transient ischemic attack, and one major bleeding event. The leakage rate was 16.2% as determined by transesophageal echocardiography.
While initial results were promising, a clinical trial comparing this device and optimal medical treatment is currently on hold. Thus, there are no clear data comparing the Amplatzer device with oral anticoagulation.34
The Watchman device
The Watchman device (Boston Scientific, Marlborough, MA), an evolution of the PLAATO device, is a self-expanding nitinol structure with fixation barbs and a polyethylene membrane to protect the atrium-facing side of the device (Figure 1).
A pilot trial reported successful implantation in 66 of 75 patients, though the device was found to migrate after placement in 5 of the first 16 patients using the original device and delivery system. The device was modified, and no further embolization of the device occurred.35
The PROTECT-AF trial (Protection in Patients With Atrial Fibrillation)36 was the first completed and published randomized controlled trial evaluating left atrial appendage closure using a device vs long-term warfarin therapy. This study randomized 707 people with nonvalvular atrial fibrillation from 59 centers worldwide to receive the Watchman device or a control treatment. The study included patients age 18 or older with nonvalvular atrial fibrillation who were able to tolerate warfarin therapy. Patients in the control group received warfarin for the duration of the study and were monitored every 2 weeks for a goal INR of 2 to 3, achieving a therapeutic INR 66% of the time. The device group was also treated with warfarin for 45 days to allow device endothelialization. Warfarin was discontinued if transesophageal echocardiography showed complete closure or significantly decreased flow around the device. Patients in the device group were then treated with aspirin and clopidogrel for 6 months, and then with aspirin indefinitely.
At 1,065 patient-years of follow-up, PROTECT-AF showed that in patients with atrial fibrillation who were candidates for warfarin therapy, percutaneous left atrial appendage closure using the Watchman device reduced the rate of hemorrhagic stroke compared with warfarin and was noninferior to warfarin in terms of all-cause mortality and stroke. A 4-year follow-up to the PROTECT-AF trial found that receiving the Watchman was better than taking warfarin in terms of risk of cardiovascular death, stroke and other systemic embolization, and all-cause mortality. The adverse event rates were 2.3% in the device group and 3.8% in the control group, a 40% relative risk reduction in the Watchman group.37
The PREVAIL trial (Prospective Randomized Evaluation of the WATCHMAN LAA Closure Device in Patients With Atrial Fibrillation vs Long-Term Warfarin Therapy) aimed to confirm the safety and efficacy of the Watchman device compared with long-term warfarin therapy.38 The event rate (defined as 7-day occurrence of death, ischemic stroke, systemic embolism, and procedure- or device-related complications requiring major cardiovascular or endovascular intervention) was 2.2%. But the PREVAIL trial was unable to show that the device was noninferior to warfarin in terms of its second primary end point of stroke, systemic embolism, and cardiovascular or unexplained death at 18 months. When performed by physicians who were new to the procedure, the procedure was successful (ie, the device was successfully implanted) in 93.2%; the rate was slightly higher (96.3%) when performed by experienced implanters.
Safety data gathered in PREVAIL in conjunction with demonstrated efficacy from PROTECT-AF suggest that the Watchman device may be a safe and effective alternative to long-term oral anticoagulation in patients with nonvalvular atrial fibrillation.
In patients with contraindications to warfarin
Most of the published data have been about the efficacy of occlusion devices compared with long-term warfarin therapy. Unfortunately, the population that has not been studied extensively is patients who have contraindications to long-term oral anticoagulation, who would benefit the most from an occlusive device.
The ASA Plavix Feasibility Study (ASAP) focused on this population, specifically those who had a CHADS2 score of 1 or higher and who were considered ineligible for warfarin, to determine whether closure using the Watchman device could be safely performed without a transition period with warfarin.39 After device implantation, trial participants were given clopidogrel for 6 months and aspirin indefinitely. The trial enrolled 150 patients and followed them for a mean of 14.4 (± 8.6) months. In that time, there were four strokes, five pericardial effusions, and six instances of device-related thrombus by transesophageal echocardiography. Three of the strokes were ischemic (1.7% per year), which is a 77% reduction from the expected rate of 7.3% based on the CHADS2 scores of the patient cohort.
These data suggest that implantation of the Watchman device may be appropriate for those who cannot tolerate warfarin even in the short term.
The Lariat system
The Lariat suture delivery device (SentreHeart, Inc., Redwood City, CA) is approved by the US Food and Drug Administration (FDA) for soft-tissue closure and has been used for percutaneous left atrial appendage closure. It uses a magnet-tipped wire that is passed to the epicardial side of the left atrial appendage via pericardial access to meet a second magnet-tipped wire introduced into the appendage via transseptal access. A “lasso” is then advanced over the epicardial guide wire and tightened down around the ostium of the left atrial appendage. This tool facilitates deployment of a nonabsorbable polyester suture, which effectively ligates off the appendage from the rest of the left atrium (Figure 2).40 In theory, the Lariat’s epicardial approach could eliminate the need for short- and long-term anticoagulation, as there would be no foreign body left within the heart.
In an initial cohort of 89 patients in Poland,41 the investigators reported a 96% closure rate as determined by transesophageal echocardiography immediately after the procedure. At 1-year follow-up, there was 98% complete closure, including cases of incomplete closure detected earlier.41 Adverse events were limited, with only two cases of severe pericarditis, two strokes, and one pericardial effusion. These results were replicated in the United States in a cohort of 25 patients, with a 100% closure rate and no stroke events.42
There have been three published case reports of left atrial clot formation after successful left atrial appendage ligation using the Lariat device.43–45 These experiences further emphasize that closure does not necessarily confer instant stroke prevention, and there remains a need to investigate the need for routine imaging and possibly periprocedural anticoagulation after ligation.
More recently, Pillai et al46 published their initial experience following 71 patients with echocardiograms 3 months after left atrial appendage closure using the Lariat device. They reported leaks in 6 of the 71 patients; five of the leaks were successfully closed using the Amplatzer Septal Occluder, and one was closed with a repeat Lariat procedure.
Although the Lariat system has been used in more than 2,000 patients worldwide (SentreHeart, personal communication), there has been no published systematic comparison between it and oral anticoagulation to date.
AN EMERGING OPTION
Established guidelines help determine which patients with atrial fibrillation should receive oral anticoagulant therapy. For patients who have absolute contraindications to oral anticoagulants or who are undergoing cardiac surgery, surgical ligation of the left atrial appendage is an option. But for those with contraindications to oral anticoagulation in both the short term and the long term, there is a growing body of evidence suggesting that a percutaneous intervention is at least noninferior to—and in some cases is superior to—warfarin. Figure 3 shows our recommendations for the steps to determine which patients would be most appropriate to consider for left atrial appendage closure.
Holmes et al47 propose that we may now have enough evidence to support an expedited regulatory approval process of these occlusion devices. But there are still a number of areas in which further investigation is clearly needed before left atrial appendage occlusion devices can be widely adopted.
The trials discussed above had specific inclusion and exclusion criteria, and therefore, although they support percutaneous intervention, the generalizability of their results remains in question. Indeed, the patients in PROTECT-AF36 had an average CHADS2 score of only 2.2. This study also included only patients who were able to tolerate both aspirin and clopidogrel simultaneously for a significant amount of time. Hence, one cannot assume the results would be the same in patients who have strict contraindications to warfarin or any target-specific oral anticoagulant. Concern regarding the generalizability of the conclusions from PROTECT-AF and PREVAIL has led to mixed votes (three assessments to date) from the FDA Circulatory Device Panel.48
In an encouraging review of cases, Gafoor et al49 reported safe and efficacious occlusion in octogenarians using the devices mentioned above. These patients often pose the greatest challenge in initiating long-term anticoagulation because of the many drug-drug interactions and the risk of intracranial hemorrhage secondary to falls.
Further, while occlusion devices would clearly be useful for patients in whom traditional oral anticoagulation is not an option, the newer oral anticoagulants might complicate the picture somewhat. As shown by Ruff et al,21 the risk-benefit ratio of these target-specific oral anticoagulants is quite favorable and by some measurements is superior to that of warfarin. Could there be a group of patients who cannot take warfarin but could instead do well on one of the newer anticoagulants, thus alleviating the need for percutaneous intervention? As the newer oral anticoagulants become more commonly used, the cost-benefit analysis of implanting an occlusion device could shift.
Lastly, in this era of high-value medical care, one must consider the cost-effectiveness of these novel interventions. As with any new technology, the up-front cost of implantation is certainly greater than that of warfarin therapy. If device implantation can prevent a hospitalization from a major bleed secondary to warfarin use or prevent a catastrophic stroke due to untreated atrial fibrillation, then the cost-benefit analysis may be tipped in the other direction. As these devices become more widely available and physicians have more experience implanting them, the costs will likely decrease.
As with oral anticoagulation therapy, all interventions, whether surgical or percutaneous, carry a risk of bleeding and stroke. There remains no substitute for frank and clear discussions between the physician and patient regarding the risks and benefits of each approach.
While a growing body of evidence surrounds left atrial appendage occlusion devices, many questions remain. Notably, could these devices be used in patients who can tolerate oral anticoagulants? And if so, which subgroups would benefit most? Does occlusion or ligation of the left atrial appendage affect electrical connections between it and the left atrium, thereby lowering the burden of atrial fibrillation?
We expect that continued investigation of and experience with left atrial appendage closure devices will position them one day as a viable and equal option for preventing stroke in patients with atrial fibrillation.
Can patients with atrial fibrillation undergo a percutaneous procedure to reduce their risk of stroke, thereby eliminating the need for lifelong treatment with an oral anticoagulant drug? The data are preliminary, but this is an emerging option that physicians should be aware of.
We review here the current evidence and techniques aimed at isolating the left atrial appendage to prevent stroke, and we emphasize the need for continued systematic comparisons between oral anticoagulation and percutaneous treatment options.
NOVEL TREATMENTS ARE NEEDED
Atrial fibrillation is the most common cardiac arrhythmia,1 affecting an estimated 1% to 2% of people worldwide. In 2001, an estimated 2.3 million persons in the United States had atrial fibrillation, and that number is expected to more than double by 2050.2
Atrial fibrillation independently increases the risk of stroke by a factor of 4 to 5.3 The American Heart Association ranks stroke as the fourth most common cause of death and the leading cause of disability in the United States.4 Atrial fibrillation accounts for 15% of strokes in people of all ages and 30% in those over age 80.5 Untreated, 2% to 5% of patients with atrial fibrillation suffer a stroke in any given year.6 Most of these strokes are cardioembolic, with thrombi originating in the left atrial appendage.7 Furthermore, it has been estimated8,9 that patients with atrial fibrillation who have already had a stroke and cannot tolerate oral anticoagulants have an annual risk of stroke close to 12% and a relative risk of approximately 3.0 compared with those with atrial fibrillation and prior stroke who can tolerate anticoagulation.
Oral anticoagulation effectively prevents thromboembolic events associated with atrial fibrillation,10 but several factors limit its efficacy and applicability. The risk of bleeding complications, the need for frequent monitoring, and challenges with compliance create a large population of patients who would benefit from alternative approaches. Consequently, physicians have looked for other ways to prevent stroke—especially surgical and transcatheter procedures—that are not associated with an ongoing risk of hemorrhage and a lifelong need to take an anticoagulant.
THE LEFT ATRIAL APPENDAGE: A SITE OF CLOT FORMATION
The left atrial appendage is the most common site of thrombus formation, particularly in patients with nonvalvular atrial fibrillation. Nearly 90% of thrombi discovered in the left atrium form in the appendage.7 A study of 233 patients not on long-term anticoagulation revealed that after 48 hours of atrial fibrillation, 15% had a left atrial thrombus, and all but one of the thrombi were in the appendage.11
Believed to function as a decompression chamber during left ventricular systole, the left atrial appendage is embryologically derived from the left wall of the primary atrium. It is in close proximity to the free wall of the left ventricle, and therefore its flow can vary with left ventricular function. Relative stasis due to its location and extensive trabeculations, especially in times of poor forward flow, make it a high-risk site for clot formation.12
ANTICOAGULATION: EFFECTIVE BUT IMPERFECT
In deciding whether a patient with atrial fibrillation should be prescribed anticoagulation therapy, the physician must balance the risk of stroke against the risk of bleeding. Several tools for assessing these two risks have been developed. Of note, some of the risk factors for stroke are the same as some of the risk factors for bleeding.
Calculating the risk of stroke
CHADS2 and CHA2DS2-VASc are the two most commonly used tools for assessing the risk of stroke, but only the newer CHA2DS2-VASc has received a class I recommendation (the highest) from the European Society of Cardiology (ESC).13
CHADS2 risk factors are Congestive heart failure (1 point), Hypertension (1 point), Age 75 or older (1 point), Diabetes (1 point), and Stroke or transient ischemic attack (2 points). Risk of stroke is considered low with a score of 0, intermediate with a score of 1, and high with a score of 2 or more.
CHA2DS2-VASc risk factors are Congestive heart failure or left ventricular ejection fraction ≤ 40% (1 point), Hypertension (1 point), Age ≥ 75 (2 points), Age 65–74 (1 point), Diabetes mellitus (1 point), Stroke, transient ischemic attack, or thromboembolism (2 points), Vascular disease (1 point), and female Sex (1 point). Low risk is defined as a score of 0 for a man or, for a woman with no other risk factors, a score of 1. A score of 1 for a man indicates moderate risk, and a score of 2 or more is high risk.
Calculating the risk of bleeding
Tools for assessing bleeding risk include ATRIA2 and HAS-BLED,14 the latter carrying a class I recommendation from the ESC.13
HAS-BLED risk factors are Hypertension (1 point), Abnormal renal or liver function (1 point each), Stroke (1 point), Bleeding (1 point), Labile international normalized ratio (INR) (1 point), Elderly (age > 65) (1 point), and Drug or alcohol use (1 point each). The risk of bleeding is considered high with a score of 3 or higher.
Disadvantages of oral anticoagulation
Oral anticoagulation is the standard treatment for preventing stroke in patients with atrial fibrillation, and the vitamin K antagonist warfarin remains the foundation.
Though highly effective, warfarin requires close monitoring and frequent dose adjustments because of its numerous food and drug interactions. Bleeding risk and the challenge of frequent monitoring rule out treatment with warfarin in 14% to 44% of patients with atrial fibrillation.15 Even in “ideal” candidates, warfarin is underused, with one study reporting that only 38% of those with clinical indications for it had been prescribed warfarin, and of those for whom it had not been prescribed, 63% were also not taking aspirin.16 Moreover, a meta-analysis suggested that the average patient treated with warfarin has his or her INR in the therapeutic range only about 55% of the time.17
Newer, target-specific oral anticoagulants such as dabigatran (a direct thrombin inhibitor) and rivaroxaban and apixaban (both factor Xa inhibitors) do not require monitoring and have fewer drug interactions. But like warfarin, they also confer a risk of serious bleeding.18–20 Most of the studies of these newer drugs have compared them with warfarin, with the preponderance of evidence showing them to be either noninferior or superior to warfarin for stroke reduction. But bleeding complication rates remain significant, apixaban having lower rates of major bleeding than dabigatran and rivaroxaban.
In a meta-analysis, Ruff et al21 concluded that the target-specific oral anticoagulants provide a favorable balance of risk and benefit. Compared with warfarin, these new drugs reduced the rate of stroke or systemic embolic events by 19%. There was also a significant reduction in rates of intracranial hemorrhage and all-cause mortality. The risk of major bleeding was similar to that with warfarin, though there was a higher risk of gastrointestinal bleeding with target-specific agents. These effects were consistent across a wide range of patients.
Given the difficulties, risks, and serious side effects of anticoagulant therapy, many patients stop taking these drugs soon after starting them, either on their own or on their physician’s recommendation. In the RE-LY trial (Dabigatran vs Warfarin in Patients With Atrial Fibrillation), 10% of patients receiving dabigatran and 17% of those receiving warfarin stopped the treatment within 1 to 2 years.22 In a similar trial of rivaroxaban vs warfarin in nonvalvular atrial fibrillation (the ROCKET-AF trial), 24% of those treated with rivaroxaban and 22% of those treated with warfarin stopped treatment during the study.19 In the ARISTOTLE trial (Apixaban vs Warfarin in Patients With Atrial Fibrillation), 25% of patients discontinued apixaban and 28% discontinued warfarin.20
The results of these trials show a clear need for treatments without high attrition rates, since patients with atrial fibrillation need protection from stroke for the rest of their life.
SURGICAL CLOSURE AS AN ADD-ON TO OTHER PROCEDURES
If the patient is undergoing cardiac surgery for another reason, the surgeon can excise, suture, staple, or clip the left atrial appendage shut at the same time. Closure is recommended as part of valve replacement.8 In a 2008 retrospective study, left atrial appendage closure was successfully performed in 40% of those undergoing the procedure during cardiac surgery, and complete closure occurred more often with excision than with suture exclusion and stapler exclusion.23 A study of patients who underwent ligation of the left atrial appendage during mitral value replacement found that 35% demonstrated incomplete closure as determined by transesophageal echocardiography.24
Newer devices have shown more success. The AtriClip (AtriCure Inc., West Chester, OH) is a self-closing, implantable clip applied epicardially by either an open surgical or a minimally invasive technique.25 Successful closure was confirmed in 60 of 61 patients at 90 days as determined by computed tomography or transesophageal echocardiography, and there were no adverse events related to implantation of the device.25 The TigerPaw system (Terumo Cardiovascular Systems, Ann Arbor, MI)26 is a fastener delivered surgically around the base of the ostium of the left atrial appendage. In an initial trial, 90 days after the procedure, transesophageal echocardiography showed no leaks in any of those who were examined (54 of 60 patients).
Amputation of the left atrial appendage is also considered part of the maze procedure for atrial fibrillation, in which the operator creates multiple small scars in the atria to prevent irregular impulses from being conducted.27
Results of these surgical approaches have been mixed, as incomplete closure or clipping actually increases the risk of left atrial thrombus formation and embolization.28 Moreover, these invasive surgical techniques are associated with significant periprocedural morbidity.29 Because of the high risk of surgical complications, cardiac specialists have sought less invasive percutaneous procedures to manage stroke risk in patients with atrial fibrillation.
PERCUTANEOUS OCCLUSION
One option for closing the left atrial appendage is a percutaneous transseptal approach in which a plug is placed in the opening connecting the appendage to the rest of the atrium.
The PLAATO device
The Percutaneous LAA Transcatheter Occlusion (PLAATO) device (Appriva Medical Inc., Sunnyvale, CA) contains an expandable nitinol-covered cage designed to be placed in the orifice of the left atrial appendage. Over time, tissue grows into the device, entirely isolating the appendage from the rest of the atrium.
In 2002, Sievert et al30 reported using this device in 15 patients. Subsequently, in a nonrandomized trial in patients with contraindications to lifelong anticoagulation, total occlusion was achieved in 108 of 111 patients, with no thrombosis or migration of the device at 6 months. The annual risk of stroke was 2.2%, a reduction in relative risk of 65% based on the CHADS2 score.31
But despite this apparent success, the PLAATO device was discontinued for unspecified commercial reasons.
Amplatzer cardiac plug
Modeled after an atrial septal occluder, the Amplatzer cardiac plug (St. Jude Medical, St. Paul, MN) consists of a lobe and a disk connected by a central waist.
In 2011, Park et al32 published their initial experience implanting this device in patients who either could not tolerate or did not desire long-term anticoagulation. They reported a 96% closure rate (137 of 143 patients), but there were serious complications in 10 patients: 3 with ischemic stroke, 2 with device embolism, and 5 with pericardial effusions.
Urena et al33 reported similar results in 52 patients with absolute contraindications to warfarin, with a 98.1% implantation rate. Patients were then maintained on either single or dual antiplatelet therapy at the discretion of the operator. At 20-month follow-up, there had been one stroke, one transient ischemic attack, and one major bleeding event. The leakage rate was 16.2% as determined by transesophageal echocardiography.
While initial results were promising, a clinical trial comparing this device and optimal medical treatment is currently on hold. Thus, there are no clear data comparing the Amplatzer device with oral anticoagulation.34
The Watchman device
The Watchman device (Boston Scientific, Marlborough, MA), an evolution of the PLAATO device, is a self-expanding nitinol structure with fixation barbs and a polyethylene membrane to protect the atrium-facing side of the device (Figure 1).
A pilot trial reported successful implantation in 66 of 75 patients, though the device was found to migrate after placement in 5 of the first 16 patients using the original device and delivery system. The device was modified, and no further embolization of the device occurred.35
The PROTECT-AF trial (Protection in Patients With Atrial Fibrillation)36 was the first completed and published randomized controlled trial evaluating left atrial appendage closure using a device vs long-term warfarin therapy. This study randomized 707 people with nonvalvular atrial fibrillation from 59 centers worldwide to receive the Watchman device or a control treatment. The study included patients age 18 or older with nonvalvular atrial fibrillation who were able to tolerate warfarin therapy. Patients in the control group received warfarin for the duration of the study and were monitored every 2 weeks for a goal INR of 2 to 3, achieving a therapeutic INR 66% of the time. The device group was also treated with warfarin for 45 days to allow device endothelialization. Warfarin was discontinued if transesophageal echocardiography showed complete closure or significantly decreased flow around the device. Patients in the device group were then treated with aspirin and clopidogrel for 6 months, and then with aspirin indefinitely.
At 1,065 patient-years of follow-up, PROTECT-AF showed that in patients with atrial fibrillation who were candidates for warfarin therapy, percutaneous left atrial appendage closure using the Watchman device reduced the rate of hemorrhagic stroke compared with warfarin and was noninferior to warfarin in terms of all-cause mortality and stroke. A 4-year follow-up to the PROTECT-AF trial found that receiving the Watchman was better than taking warfarin in terms of risk of cardiovascular death, stroke and other systemic embolization, and all-cause mortality. The adverse event rates were 2.3% in the device group and 3.8% in the control group, a 40% relative risk reduction in the Watchman group.37
The PREVAIL trial (Prospective Randomized Evaluation of the WATCHMAN LAA Closure Device in Patients With Atrial Fibrillation vs Long-Term Warfarin Therapy) aimed to confirm the safety and efficacy of the Watchman device compared with long-term warfarin therapy.38 The event rate (defined as 7-day occurrence of death, ischemic stroke, systemic embolism, and procedure- or device-related complications requiring major cardiovascular or endovascular intervention) was 2.2%. But the PREVAIL trial was unable to show that the device was noninferior to warfarin in terms of its second primary end point of stroke, systemic embolism, and cardiovascular or unexplained death at 18 months. When performed by physicians who were new to the procedure, the procedure was successful (ie, the device was successfully implanted) in 93.2%; the rate was slightly higher (96.3%) when performed by experienced implanters.
Safety data gathered in PREVAIL in conjunction with demonstrated efficacy from PROTECT-AF suggest that the Watchman device may be a safe and effective alternative to long-term oral anticoagulation in patients with nonvalvular atrial fibrillation.
In patients with contraindications to warfarin
Most of the published data have been about the efficacy of occlusion devices compared with long-term warfarin therapy. Unfortunately, the population that has not been studied extensively is patients who have contraindications to long-term oral anticoagulation, who would benefit the most from an occlusive device.
The ASA Plavix Feasibility Study (ASAP) focused on this population, specifically those who had a CHADS2 score of 1 or higher and who were considered ineligible for warfarin, to determine whether closure using the Watchman device could be safely performed without a transition period with warfarin.39 After device implantation, trial participants were given clopidogrel for 6 months and aspirin indefinitely. The trial enrolled 150 patients and followed them for a mean of 14.4 (± 8.6) months. In that time, there were four strokes, five pericardial effusions, and six instances of device-related thrombus by transesophageal echocardiography. Three of the strokes were ischemic (1.7% per year), which is a 77% reduction from the expected rate of 7.3% based on the CHADS2 scores of the patient cohort.
These data suggest that implantation of the Watchman device may be appropriate for those who cannot tolerate warfarin even in the short term.
The Lariat system
The Lariat suture delivery device (SentreHeart, Inc., Redwood City, CA) is approved by the US Food and Drug Administration (FDA) for soft-tissue closure and has been used for percutaneous left atrial appendage closure. It uses a magnet-tipped wire that is passed to the epicardial side of the left atrial appendage via pericardial access to meet a second magnet-tipped wire introduced into the appendage via transseptal access. A “lasso” is then advanced over the epicardial guide wire and tightened down around the ostium of the left atrial appendage. This tool facilitates deployment of a nonabsorbable polyester suture, which effectively ligates off the appendage from the rest of the left atrium (Figure 2).40 In theory, the Lariat’s epicardial approach could eliminate the need for short- and long-term anticoagulation, as there would be no foreign body left within the heart.
In an initial cohort of 89 patients in Poland,41 the investigators reported a 96% closure rate as determined by transesophageal echocardiography immediately after the procedure. At 1-year follow-up, there was 98% complete closure, including cases of incomplete closure detected earlier.41 Adverse events were limited, with only two cases of severe pericarditis, two strokes, and one pericardial effusion. These results were replicated in the United States in a cohort of 25 patients, with a 100% closure rate and no stroke events.42
There have been three published case reports of left atrial clot formation after successful left atrial appendage ligation using the Lariat device.43–45 These experiences further emphasize that closure does not necessarily confer instant stroke prevention, and there remains a need to investigate the need for routine imaging and possibly periprocedural anticoagulation after ligation.
More recently, Pillai et al46 published their initial experience following 71 patients with echocardiograms 3 months after left atrial appendage closure using the Lariat device. They reported leaks in 6 of the 71 patients; five of the leaks were successfully closed using the Amplatzer Septal Occluder, and one was closed with a repeat Lariat procedure.
Although the Lariat system has been used in more than 2,000 patients worldwide (SentreHeart, personal communication), there has been no published systematic comparison between it and oral anticoagulation to date.
AN EMERGING OPTION
Established guidelines help determine which patients with atrial fibrillation should receive oral anticoagulant therapy. For patients who have absolute contraindications to oral anticoagulants or who are undergoing cardiac surgery, surgical ligation of the left atrial appendage is an option. But for those with contraindications to oral anticoagulation in both the short term and the long term, there is a growing body of evidence suggesting that a percutaneous intervention is at least noninferior to—and in some cases is superior to—warfarin. Figure 3 shows our recommendations for the steps to determine which patients would be most appropriate to consider for left atrial appendage closure.
Holmes et al47 propose that we may now have enough evidence to support an expedited regulatory approval process of these occlusion devices. But there are still a number of areas in which further investigation is clearly needed before left atrial appendage occlusion devices can be widely adopted.
The trials discussed above had specific inclusion and exclusion criteria, and therefore, although they support percutaneous intervention, the generalizability of their results remains in question. Indeed, the patients in PROTECT-AF36 had an average CHADS2 score of only 2.2. This study also included only patients who were able to tolerate both aspirin and clopidogrel simultaneously for a significant amount of time. Hence, one cannot assume the results would be the same in patients who have strict contraindications to warfarin or any target-specific oral anticoagulant. Concern regarding the generalizability of the conclusions from PROTECT-AF and PREVAIL has led to mixed votes (three assessments to date) from the FDA Circulatory Device Panel.48
In an encouraging review of cases, Gafoor et al49 reported safe and efficacious occlusion in octogenarians using the devices mentioned above. These patients often pose the greatest challenge in initiating long-term anticoagulation because of the many drug-drug interactions and the risk of intracranial hemorrhage secondary to falls.
Further, while occlusion devices would clearly be useful for patients in whom traditional oral anticoagulation is not an option, the newer oral anticoagulants might complicate the picture somewhat. As shown by Ruff et al,21 the risk-benefit ratio of these target-specific oral anticoagulants is quite favorable and by some measurements is superior to that of warfarin. Could there be a group of patients who cannot take warfarin but could instead do well on one of the newer anticoagulants, thus alleviating the need for percutaneous intervention? As the newer oral anticoagulants become more commonly used, the cost-benefit analysis of implanting an occlusion device could shift.
Lastly, in this era of high-value medical care, one must consider the cost-effectiveness of these novel interventions. As with any new technology, the up-front cost of implantation is certainly greater than that of warfarin therapy. If device implantation can prevent a hospitalization from a major bleed secondary to warfarin use or prevent a catastrophic stroke due to untreated atrial fibrillation, then the cost-benefit analysis may be tipped in the other direction. As these devices become more widely available and physicians have more experience implanting them, the costs will likely decrease.
As with oral anticoagulation therapy, all interventions, whether surgical or percutaneous, carry a risk of bleeding and stroke. There remains no substitute for frank and clear discussions between the physician and patient regarding the risks and benefits of each approach.
While a growing body of evidence surrounds left atrial appendage occlusion devices, many questions remain. Notably, could these devices be used in patients who can tolerate oral anticoagulants? And if so, which subgroups would benefit most? Does occlusion or ligation of the left atrial appendage affect electrical connections between it and the left atrium, thereby lowering the burden of atrial fibrillation?
We expect that continued investigation of and experience with left atrial appendage closure devices will position them one day as a viable and equal option for preventing stroke in patients with atrial fibrillation.
- Rosamond W, Flegal K, Furie K, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2008; 117:e25–146.
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001; 285:2370–2375.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991; 22:983–988.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation 2013; 127:e6–e245.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation: a major contributor to stroke in the elderly. The Framingham Study. Arch Intern Med 1987; 147:1561–1564.
- Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med 1994; 154:1449–1457.
- Blackshear JL, Odell JA. Appendage obliteration to reduce stroke in cardiac surgical patients with atrial fibrillation. Ann Thorac Surg 1996; 61:755–759.
- Bonow RO, Carabello BA, Chatterjee K, et al; 2006 Writing Committee Members; American College of Cardiology/American Heart Association Task Force. 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation 2008; 118:e523–e661.
- Odell JA, Blackshear JL, Davies E, et al. Thoracoscopic obliteration of the left atrial appendage: potential for stroke reduction? Ann Thorac Surg 1996; 61:565–569.
- Hart RG, Benavente O, McBride R, Pearce LA. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a meta-analysis. Ann Intern Med 1999; 131:492–501.
- Manning WJ, Silverman DI, Keighley CS, Oettgen P, Douglas PS. Transesophageal echocardiographically facilitated early cardioversion from atrial fibrillation using short-term anticoagulation: final results of a prospective 4.5-year study. J Am Coll Cardiol 1995; 25:1354–1361.
- Al-Saady NM, Obel OA, Camm AJ. Left atrial appendage: structure, function, and role in thromboembolism. Heart 1999; 82:547–554.
- Lip GY. Recommendations for thromboprophylaxis in the 2012 focused update of the ESC guidelines on atrial fibrillation: a commentary. J Thromb Haemost 2013; 11:615–626.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 2010; 138:1093–1100.
- Onalan O, Lashevsky I, Hamad A, Crystal E. Nonpharmacologic stroke prevention in atrial fibrillation. Expert Rev Cardiovasc Ther 2005; 3:619–633.
- Brass LM, Krumholz HM, Scinto JM, Radford M. Warfarin use among patients with atrial fibrillation. Stroke 1997; 28:2382–2389.
- Baker WL, Cios DA, Sander SD, Coleman CI. Meta-analysis to assess the quality of warfarin control in atrial fibrillation patients in the United States. J Manag Care Pharm 2009; 15:244–252.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365:981–992.
- Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet 2014; 383:955–962.
- Lip GY, Clemens A, Noack H, Ferreira J, Connolly SJ, Yusuf S. Patient outcomes using the European label for dabigatran. A post-hoc analysis from the RE-LY database. Thromb Haemost 2014; 111:933–942.
- Kanderian AS, Gillinov AM, Pettersson GB, Blackstone E, Klein AL. Success of surgical left atrial appendage closure: assessment by transesophageal echocardiography. J Am Coll Cardiol 2008; 52:924–929.
- Katz ES, Tsiamtsiouris T, Applebaum RM, Schwartzbard A, Tunick PA, Kronzon I. Surgical left atrial appendage ligation is frequently incomplete: a transesophageal echocardiograhic study. J Am Coll Cardiol 2000; 36:468–471.
- Ailawadi G, Gerdisch MW, Harvey RL, et al. Exclusion of the left atrial appendage with a novel device: early results of a multicenter trial. J Thorac Cardiovasc Surg 2011; 142:1002–1009.e1.
- Slater AD, Tatooles AJ, Coffey A, et al. Prospective clinical study of a novel left atrial appendage occlusion device. Ann Thorac Surg 2012; 93:2035-2040.
- Pinho-Gomes AC, Amorim MJ, Oliveira SM, Leite-Moreira AF. Surgical treatment of atrial fibrillation: an updated review. Eur J Cardiothorac Surg 2014; 46:167–178.
- Aryana A, Cavaco D, Arthur A, O’Neill PG, Adragão P, D’Avila A. Percutaneous endocardial occlusion of incompletely surgically ligated left atrial appendage. J Cardiovasc Electrophysiol 2013; 24:968–974.
- García-Fernández MA, Pérez-David E, Quiles J, et al. Role of left atrial appendage obliteration in stroke reduction in patients with mitral valve prosthesis: a transesophageal echocardiographic study. J Am Coll Cardiol 2003; 42:1253–1258.
- Sievert H, Lesh MD, Trepels T, et al. Percutaneous left atrial appendage transcatheter occlusion to prevent stroke in high-risk patients with atrial fibrillation: early clinical experience. Circulation 2002; 105:1887–1889.
- Ostermayer SH, Reisman M, Kramer PH, et al. Percutaneous left atrial appendage transcatheter occlusion (PLAATO system) to prevent stroke in high-risk patients with non-rheumatic atrial fibrillation: results from the international multi-center feasibility trials. J Am Coll Cardiol 2005; 46:9–14.
- Park JW, Bethencourt A, Sievert H, et al. Left atrial appendage closure with Amplatzer cardiac plug in atrial fibrillation: initial European experience. Catheter Cardiovasc Interv 2011; 77:700–706.
- Urena M, Rodés-Cabau J, Freixa X, et al. Percutaneous left atrial appendage closure with the AMPLATZER cardiac plug device in patients with nonvalvular atrial fibrillation and contraindications to anticoagulation therapy. J Am Coll Cardiol 2013; 62:96–102.
- ClinicalTrials.gov. http://clinicaltrials.gov/show/NCT01118299. Accessed January 30, 2015.
- Sick PB, Schuler G, Hauptmann KE, et al. Initial worldwide experience with the WATCHMAN left atrial appendage system for stroke prevention in atrial fibrillation. J Am Coll Cardiol 2007; 49:1490–1495.
- Holmes DR, Reddy VY, Turi ZG, et al; PROTECT AF Investigators. Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomised non-inferiority trial. Lancet 2009; 374:534–542.
- Boston Scientific. WATCHMAN™ Left Atrial Appendage Closure Device. http://www.bostonscientific.com/watchman-eu/assets/pdf/SH-158101-AA-PROTECT-AF-Reddy-HRS-2013.pdf. Accessed January 30, 2015.
- David Holmes M. Boston Scientific. March 9, 2013. Available at: http://www.bostonscientific.com/watchman-eu/assets/downloads/PREVAIL-Clinical-Results.ppt.pdf. Accessed January 30, 2015.
- Reddy VY, Möbius-Winkler S, Miller MA, et al. Left atrial appendage closure with the Watchman device in patients with a contraindication for oral anticoagulation: the ASAP study (ASA Plavix Feasibility Study With Watchman Left Atrial Appendage Closure Technology). J Am Coll Cardiol 2013; 61:2551–2556.
- Koneru JN, Badhwar N, Ellenbogen KA, Lee RJ. LAA ligation using the LARIAT suture delivery device: tips and tricks for a successful procedure. Heart Rhythm 2014; 11:911–921.
- Bartus K, Han FT, Bednarek J, et al. Percutaneous left atrial appendage suture ligation using the LARIAT device in patients with atrial fibrillation: initial clinical experience. J Am Coll Cardiol 2013; 62:108–118.
- Massumi A, Chelu MG, Nazeri A, et al. Initial experience with a novel percutaneous left atrial appendage exclusion device in patients with atrial fibrillation, increased stroke risk, and contraindications to anticoagulation. Am J Cardiol 2013; 111:869–873.
- Giedrimas E, Lin AC, Knight BP. Left atrial thrombus after appendage closure using LARIAT. Circ Arrhythm Electrophysiol 2013; 6:e52–e53.
- Briceno DF, Fernando RR, Laing ST. Left atrial appendage thrombus post LARIAT closure device. Heart Rhythm 2014; 11:1600–1601.
- Baker MS, Paul Mounsey J, Gehi AK, Chung EH. Left atrial thrombus after appendage ligation with LARIAT. Heart Rhythm 2014; 11:1489.
- Pillai AM, Kanmanthareddy A, Earnest M, et al. Initial experience with post Lariat left atrial appendage leak closure with Amplatzer septal occluder device and repeat Lariat application. Heart Rhythm 2014; 11:1877–1883.
- Holmes DR Jr, Lakkireddy DR, Whitlock RP, Waksman R, Mack MJ. Left atrial appendage occlusion: opportunities and challenges. J Am Coll Cardiol 2014; 63:291–298.
- Wood S. FDA Advisors cool on Watchman approval amid ischemic-stroke data. Medscape Multispecialty October 8, 2014. www.medscape.com/viewarticle/832993.
- Gafoor S, Franke J, Bertog S, et al. Left atrial appendage occlusion in octogenarians: short-term and 1-year follow-up. Catheter Cardiovasc Interv 2014; 83:805–810.
- Rosamond W, Flegal K, Furie K, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2008; 117:e25–146.
- Go AS, Hylek EM, Phillips KA, et al. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001; 285:2370–2375.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke 1991; 22:983–988.
- Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation 2013; 127:e6–e245.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation: a major contributor to stroke in the elderly. The Framingham Study. Arch Intern Med 1987; 147:1561–1564.
- Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med 1994; 154:1449–1457.
- Blackshear JL, Odell JA. Appendage obliteration to reduce stroke in cardiac surgical patients with atrial fibrillation. Ann Thorac Surg 1996; 61:755–759.
- Bonow RO, Carabello BA, Chatterjee K, et al; 2006 Writing Committee Members; American College of Cardiology/American Heart Association Task Force. 2008 Focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease): endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. Circulation 2008; 118:e523–e661.
- Odell JA, Blackshear JL, Davies E, et al. Thoracoscopic obliteration of the left atrial appendage: potential for stroke reduction? Ann Thorac Surg 1996; 61:565–569.
- Hart RG, Benavente O, McBride R, Pearce LA. Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a meta-analysis. Ann Intern Med 1999; 131:492–501.
- Manning WJ, Silverman DI, Keighley CS, Oettgen P, Douglas PS. Transesophageal echocardiographically facilitated early cardioversion from atrial fibrillation using short-term anticoagulation: final results of a prospective 4.5-year study. J Am Coll Cardiol 1995; 25:1354–1361.
- Al-Saady NM, Obel OA, Camm AJ. Left atrial appendage: structure, function, and role in thromboembolism. Heart 1999; 82:547–554.
- Lip GY. Recommendations for thromboprophylaxis in the 2012 focused update of the ESC guidelines on atrial fibrillation: a commentary. J Thromb Haemost 2013; 11:615–626.
- Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest 2010; 138:1093–1100.
- Onalan O, Lashevsky I, Hamad A, Crystal E. Nonpharmacologic stroke prevention in atrial fibrillation. Expert Rev Cardiovasc Ther 2005; 3:619–633.
- Brass LM, Krumholz HM, Scinto JM, Radford M. Warfarin use among patients with atrial fibrillation. Stroke 1997; 28:2382–2389.
- Baker WL, Cios DA, Sander SD, Coleman CI. Meta-analysis to assess the quality of warfarin control in atrial fibrillation patients in the United States. J Manag Care Pharm 2009; 15:244–252.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365:981–992.
- Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet 2014; 383:955–962.
- Lip GY, Clemens A, Noack H, Ferreira J, Connolly SJ, Yusuf S. Patient outcomes using the European label for dabigatran. A post-hoc analysis from the RE-LY database. Thromb Haemost 2014; 111:933–942.
- Kanderian AS, Gillinov AM, Pettersson GB, Blackstone E, Klein AL. Success of surgical left atrial appendage closure: assessment by transesophageal echocardiography. J Am Coll Cardiol 2008; 52:924–929.
- Katz ES, Tsiamtsiouris T, Applebaum RM, Schwartzbard A, Tunick PA, Kronzon I. Surgical left atrial appendage ligation is frequently incomplete: a transesophageal echocardiograhic study. J Am Coll Cardiol 2000; 36:468–471.
- Ailawadi G, Gerdisch MW, Harvey RL, et al. Exclusion of the left atrial appendage with a novel device: early results of a multicenter trial. J Thorac Cardiovasc Surg 2011; 142:1002–1009.e1.
- Slater AD, Tatooles AJ, Coffey A, et al. Prospective clinical study of a novel left atrial appendage occlusion device. Ann Thorac Surg 2012; 93:2035-2040.
- Pinho-Gomes AC, Amorim MJ, Oliveira SM, Leite-Moreira AF. Surgical treatment of atrial fibrillation: an updated review. Eur J Cardiothorac Surg 2014; 46:167–178.
- Aryana A, Cavaco D, Arthur A, O’Neill PG, Adragão P, D’Avila A. Percutaneous endocardial occlusion of incompletely surgically ligated left atrial appendage. J Cardiovasc Electrophysiol 2013; 24:968–974.
- García-Fernández MA, Pérez-David E, Quiles J, et al. Role of left atrial appendage obliteration in stroke reduction in patients with mitral valve prosthesis: a transesophageal echocardiographic study. J Am Coll Cardiol 2003; 42:1253–1258.
- Sievert H, Lesh MD, Trepels T, et al. Percutaneous left atrial appendage transcatheter occlusion to prevent stroke in high-risk patients with atrial fibrillation: early clinical experience. Circulation 2002; 105:1887–1889.
- Ostermayer SH, Reisman M, Kramer PH, et al. Percutaneous left atrial appendage transcatheter occlusion (PLAATO system) to prevent stroke in high-risk patients with non-rheumatic atrial fibrillation: results from the international multi-center feasibility trials. J Am Coll Cardiol 2005; 46:9–14.
- Park JW, Bethencourt A, Sievert H, et al. Left atrial appendage closure with Amplatzer cardiac plug in atrial fibrillation: initial European experience. Catheter Cardiovasc Interv 2011; 77:700–706.
- Urena M, Rodés-Cabau J, Freixa X, et al. Percutaneous left atrial appendage closure with the AMPLATZER cardiac plug device in patients with nonvalvular atrial fibrillation and contraindications to anticoagulation therapy. J Am Coll Cardiol 2013; 62:96–102.
- ClinicalTrials.gov. http://clinicaltrials.gov/show/NCT01118299. Accessed January 30, 2015.
- Sick PB, Schuler G, Hauptmann KE, et al. Initial worldwide experience with the WATCHMAN left atrial appendage system for stroke prevention in atrial fibrillation. J Am Coll Cardiol 2007; 49:1490–1495.
- Holmes DR, Reddy VY, Turi ZG, et al; PROTECT AF Investigators. Percutaneous closure of the left atrial appendage versus warfarin therapy for prevention of stroke in patients with atrial fibrillation: a randomised non-inferiority trial. Lancet 2009; 374:534–542.
- Boston Scientific. WATCHMAN™ Left Atrial Appendage Closure Device. http://www.bostonscientific.com/watchman-eu/assets/pdf/SH-158101-AA-PROTECT-AF-Reddy-HRS-2013.pdf. Accessed January 30, 2015.
- David Holmes M. Boston Scientific. March 9, 2013. Available at: http://www.bostonscientific.com/watchman-eu/assets/downloads/PREVAIL-Clinical-Results.ppt.pdf. Accessed January 30, 2015.
- Reddy VY, Möbius-Winkler S, Miller MA, et al. Left atrial appendage closure with the Watchman device in patients with a contraindication for oral anticoagulation: the ASAP study (ASA Plavix Feasibility Study With Watchman Left Atrial Appendage Closure Technology). J Am Coll Cardiol 2013; 61:2551–2556.
- Koneru JN, Badhwar N, Ellenbogen KA, Lee RJ. LAA ligation using the LARIAT suture delivery device: tips and tricks for a successful procedure. Heart Rhythm 2014; 11:911–921.
- Bartus K, Han FT, Bednarek J, et al. Percutaneous left atrial appendage suture ligation using the LARIAT device in patients with atrial fibrillation: initial clinical experience. J Am Coll Cardiol 2013; 62:108–118.
- Massumi A, Chelu MG, Nazeri A, et al. Initial experience with a novel percutaneous left atrial appendage exclusion device in patients with atrial fibrillation, increased stroke risk, and contraindications to anticoagulation. Am J Cardiol 2013; 111:869–873.
- Giedrimas E, Lin AC, Knight BP. Left atrial thrombus after appendage closure using LARIAT. Circ Arrhythm Electrophysiol 2013; 6:e52–e53.
- Briceno DF, Fernando RR, Laing ST. Left atrial appendage thrombus post LARIAT closure device. Heart Rhythm 2014; 11:1600–1601.
- Baker MS, Paul Mounsey J, Gehi AK, Chung EH. Left atrial thrombus after appendage ligation with LARIAT. Heart Rhythm 2014; 11:1489.
- Pillai AM, Kanmanthareddy A, Earnest M, et al. Initial experience with post Lariat left atrial appendage leak closure with Amplatzer septal occluder device and repeat Lariat application. Heart Rhythm 2014; 11:1877–1883.
- Holmes DR Jr, Lakkireddy DR, Whitlock RP, Waksman R, Mack MJ. Left atrial appendage occlusion: opportunities and challenges. J Am Coll Cardiol 2014; 63:291–298.
- Wood S. FDA Advisors cool on Watchman approval amid ischemic-stroke data. Medscape Multispecialty October 8, 2014. www.medscape.com/viewarticle/832993.
- Gafoor S, Franke J, Bertog S, et al. Left atrial appendage occlusion in octogenarians: short-term and 1-year follow-up. Catheter Cardiovasc Interv 2014; 83:805–810.
KEY POINTS
- Few well-designed studies of surgical closure have been done.
- The Watchman percutaneous device was shown to be noninferior to warfarin in certain patients. Other closure devices demonstrate similar success, though trials have not compared them with warfarin.
- Occlusion of the left atrial appendage is an emerging option for general internists to be aware of when treating those with atrial fibrillation who cannot tolerate oral anticoagulation.
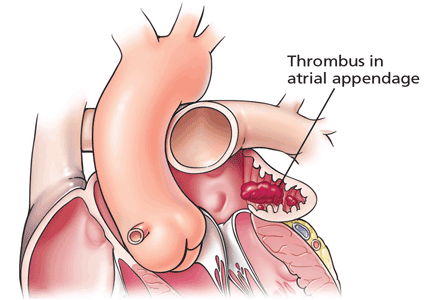
When the dissociation curve shifts to the left
A 48-year-old woman presented to the emergency department after 2 days of nonproductive cough, chest discomfort, worsening shortness of breath, and subjective fever. She had a history of systemic sclerosis. She was currently taking prednisone 20 mg daily and aspirin 81 mg daily.
Physical examination revealed tachypnea (28 breaths per minute), and bronchial breath sounds in the left lower chest posteriorly.
The initial laboratory workup revealed:
- Hemoglobin 106 g/L (reference range 115–155)
- Mean corpuscular volume 84 fL (80–100)
- White blood cell count 29.4 × 109/L (3.70–11.0), with 85% neutrophils
- Platelet count 180 × 109/L (150–350)
- Lactate dehydrogenase 312 U/L (100–220).
Chest radiography showed opacification of the lower lobe of the left lung.
She was admitted to the hospital and started treatment with intravenous azithromycin and ceftriaxone for presumed community-acquired pneumonia, based on the clinical presentation and findings on chest radiography. Because of her immunosuppression (due to chronic prednisone therapy) and her high lactate dehydrogenase level, Pneumocystis jirovecii pneumonia was suspected, and because she had a history of allergy to trimethoprim-sulfamethoxazole and pentamidine, she was started on dapsone.
During the next 24 hours, she developed worsening dyspnea, hypoxia, and cyanosis. She was placed on an air-entrainment mask, with a fraction of inspired oxygen of 0.5. Pulse oximetry showed an oxygen saturation of 85%, but arterial blood gas analysis indicated an oxyhemoglobin concentration of 95%.
THE ‘SATURATION GAP’
1. Which is most likely to have caused the discrepancy between the oxyhemoglobin concentration and the oxygen saturation by pulse oximetry in this patient?
- Methemoglobinemia
- Carbon monoxide poisoning
- Inappropriate placement of the pulse oximeter probe
- Pulmonary embolism
Methemoglobinemia is the most likely cause of the discrepancy between the oxyhemoglobin levels and the oxygen saturation by pulse oximetry, a phenomenon also known as the “saturation gap.” Other common causes are cyanide poisoning and carbon monoxide poisoning.
Carbon monoxide poisoning, however, does not explain our patient’s cyanosis. On the contrary, carbon monoxide poisoning can actually cause the patient’s lips and mucous membranes to appear unnaturally bright pink. Also, carbon monoxide poisoning raises the blood concentration of carboxyhemoglobin (which has a high affinity for oxygen), and this usually causes pulse oximetry to read inappropriately high, whereas in our patient it read low.
Incorrect placement of the pulse oximeter probe can result in an inaccurate measurement of oxygen saturation. Visualization of the waveform on the plethysmograph or the signal quality index can be used to assess adequate placement of the pulse oximeter probe. However, inadequate probe placement does not explain our patient’s dyspnea and cyanosis.
Pulmonary embolism can lead to hypoxia as a result of ventilation-perfusion mismatch. However, pulmonary embolism leading to low oxygen saturation on pulse oximetry will also lead to concomitantly low oxyhemoglobin levels as measured by arterial blood gas analysis, and this was not seen in our patient.
BACK TO OUR PATIENT
Because there was a discrepancy between our patient’s pulse oximetry reading and oxyhemoglobin concentration by arterial blood gas measurement, her methemoglobin level was checked and was found to be 30%, thus confirming the diagnosis of methemoglobinemia.
WHAT IS METHEMOGLOBINEMIA, AND WHAT CAUSES IT?
Oxygen is normally bound to iron in its ferrous (Fe2+) form in hemoglobin to form oxyhemoglobin. Oxidative stress in the body can cause iron to change from the ferrous to the ferric (Fe3+) state, forming methemoglobin. Methemoglobin is normally present in the blood in low levels (< 1% of the total hemoglobin), and ferric iron is reduced and recycled back to the ferrous form by NADH-cytochrome b5 reductase, an enzyme present in red blood cells. This protective mechanism maintains methemoglobin levels within safe limits. But increased production can lead to accumulation of methemoglobin, resulting in dyspnea and hypoxia and the condition referred to as methemoglobinemia.1
Increased levels of methemoglobin relative to normal hemoglobin cause tissue hypoxia by several mechanisms. Methemoglobin cannot efficiently carry oxygen; instead, it binds to water or to a hydroxide ion depending on the pH of the environment.2 Therefore, the hemoglobin molecule does not carry its usual load of oxygen, and hypoxia results from the reduced delivery of oxygen to tissues. In addition, an increased concentration of methemoglobin causes a leftward shift in the oxygen-hemoglobin dissociation curve, representing an increased affinity to bound oxygen in the remaining heme groups. The tightly bound oxygen is not adequately released at the tissue level, thus causing cellular hypoxia.
Methemoglobinemia is most often caused by exposure to an oxidizing chemical or drug that increases production of methemoglobin. In rare cases, it is caused by a congenital deficiency of NADH-cytochrome b5 reductase.3
2. Which of the following drugs can cause methemoglobinemia?
- Acetaminophen
- Dapsone
- Benzocaine
- Primaquine
All four of these drugs are common culprits for causing acquired methemoglobinemia; others include chloroquine, nitroglycerin, and sulfonamides.4–6
The increased production of methemoglobin caused by these drugs overwhelms the protective effect of reducing enzymes and can lead to an accumulation of methemoglobin. However, because of variability in cellular metabolism, not every person who takes these drugs develops dangerous levels of methemoglobin.

Dapsone and benzocaine are the most commonly encountered drugs known to cause methemoglobinemia (Table 1). Dapsone is an anti-inflammatory and antimicrobial agent most commonly used for treating lepromatous leprosy and dermatitis herpetiformis. It is also often prescribed for prophylaxis and treatment of P jirovecii pneumonia in immunosuppressed individuals.7 Benzocaine is a local anesthetic and was commonly used before procedures such as oral or dental surgery, transesophageal echocardiography, and endoscopy.8–10 Even low doses of benzocaine can lead to high levels of methemoglobinemia. However, the availability of other, safer anesthetics now limits the use of benzocaine in major US centers. In addition, the topical anesthetic Emla (lidocaine plus prilocaine) has been recently reported as a cause of methemoglobinemia in infants and children.11,12
Also, potentially fatal methemoglobinemia has been reported in patients with a deficiency of G-6-phosphate dehydrogenase (G6PD) who received rasburicase, a recombinant version of urate oxidase enzyme used to prevent and treat tumor lysis syndrome.13,14
Lastly, methemoglobinemia has been reported in patients with inflammatory bowel disease treated with mesalamine.
Although this adverse reaction is rare, clinicians should be aware of it, since these agents are commonly used in everyday medical practice.15
RECOGNIZING THE DANGER SIGNS
The clinical manifestations of methemoglobinemia are directly proportional to the percentage of methemoglobin in red blood cells. Cyanosis generally becomes apparent at concentrations around 15%, at which point the patient may still have no symptoms. Anxiety, lightheadedness, tachycardia, and dizziness manifest at levels of 20% to 30%. Fatigue, confusion, dizziness, tachypnea, and worsening tachycardia occur at levels of 30% to 50%. Levels of 50% to 70% cause coma, seizures, arrhythmias, and acidosis, and levels over 70% are considered lethal.16
While these levels provide a general guideline of symptomatology in an otherwise healthy person, it is important to remember that patients with underlying conditions such as anemia, lung disease (both of which our patient had), sepsis, thalassemia, G6PD deficiency, and sickle cell disease can manifest symptoms at lower concentrations of methemoglobin.1,17
Most patients who develop clinically significant levels of methemoglobin do so within the first few hours of starting one of the culprit drugs.
DIAGNOSIS: METHEMOGLOBINEMIA AND THE SATURATION GAP
In patients with methemoglobinemia, pulse oximetry gives lower values than arterial blood gas oxygen measurements. Regular pulse oximetry works by measuring light absorbance at two distinct wavelengths (660 and 940 nm) to calculate the ratio of oxyhemoglobin to deoxyhemoglobin. Methemoglobin absorbs light at both these wavelengths, thus lowering the pulse oximetry values.1
In contrast, oxygen saturation of arterial blood gas (oxyhemoglobin) is calculated indirectly from the concentration of dissolved oxygen in the blood and does not include oxygen bound to hemoglobin. Therefore, the measured arterial oxygen saturation is often normal in patients with methemoglobinemia since it relies only on inspired oxygen content and is independent of the methemoglobin concentration.18
Oxygen supplementation can raise the level of oxyhemoglobin, which is a measure of dissolved oxygen, but the oxygen saturation as measured by pulse oximetry remains largely unchanged—ie, the saturation gap. A difference of more than 5% between the oxygen saturation by pulse oximetry and blood gas analysis is abnormal. Patients with clinically significant methemoglobinemia usually have a saturation gap greater than 10%.
Several other unique features should raise suspicion of methemoglobinemia. It should be considered in a patient presenting with cyanosis out of proportion to the oxygen saturation and in a patient with low oxygen saturation and a normal chest radiograph. Other clues include blood that is chocolate-colored on gross examination, rather than the dark red of deoxygenated blood.
Co-oximetry measures oxygen saturation using different wavelengths of light to distinguish between fractions of oxyhemoglobin, deoxyhemoglobin, and methemoglobin, but it is not widely available.
THE NEXT STEP
3. What is the next step in the management of our patient?
- Discontinue the dapsone
- Start methylene blue
- Start hyperbaric oxygen
- Give sodium thiosulfate
- Discontinue dapsone and start methylene blue
The next step in her management should be to stop the dapsone and start an infusion of methylene blue. Hyperbaric oxygen is used in treating carbon monoxide poisoning, and sodium thiosulfate is used in treating cyanide toxicity. They would not be appropriate in this patient’s care.
MANAGEMENT OF ACQUIRED METHEMOGLOBINEMIA
The first, most critical step in managing acquired methemoglobinemia is to immediately discontinue the suspected offending agent. In most patients without a concomitant condition such as anemia or lung disease and with a methemoglobin level below 20%, discontinuing the offending agent may suffice. Patients with a level of 20% or greater and patients with cardiac and pulmonary disease, who develop symptoms at lower concentrations of methemoglobin, require infusion of methylene blue.
Methylene blue is converted to its reduced form, leukomethylene blue, by NADPH-methemoglobin reductase. As it is oxidized, leukomethylene blue reduces methemoglobin to hemoglobin. A dose of 1 mg/kg intravenously is given at first. The response is usually dramatic, with a reduction in methemoglobin levels and improvement in symptoms often within 30 to 60 minutes. If levels remain high, the dose can be repeated 1 hour later.19
A caveat: methylene blue therapy should be avoided in patients with complete G6PD deficiency. Methylene blue works through the enzyme NADPH-methemoglobin reductase, and since patients with G6PD deficiency lack this enzyme, methylene blue is ineffective. In fact, since it cannot be reduced, excessive methylene blue can oxidize hemoglobin to methemoglobin, further exacerbating the condition. In patients with partial G6PD deficiency, methylene blue is still recommended as a first-line treatment, but at a lower initial dose (0.3–0.5 mg/kg). However, in patients with significant hemolysis, an exchange transfusion is the only treatment option.
CASE CONCLUDED
Since dapsone was identified as the likely cause of methemoglobinemia in our patient, it was immediately discontinued. Because she was symptomatic, 70 mg of methylene blue was given intravenously. Over the next 60 minutes, her clinical condition improved significantly. A repeat methemoglobin measurement was 3%.
She was discharged home the next day on oral antibiotics to complete treatment for community-acquired pneumonia.
TAKE-HOME POINTS
- Consider methemoglobinemia in a patient with unexplained cyanosis.
- Pulse oximetry gives lower values than arterial blood gas oxygen measurements in patients with methemoglobinemia, and pulse oximetry readings do not improve with supplemental oxygen.
- A saturation gap greater than 5% strongly suggests methemoglobinemia.
- The diagnosis of methemoglobinemia is confirmed by measuring the methemoglobin concentration.
- Most healthy patients develop symptoms at methemoglobin levels of 20%, but patients with comorbidities can develop symptoms at lower levels.
- A number of drugs can cause methemoglobinemia, even at therapeutic dosages.
- Treatment is generally indicated in patients who have symptoms or in healthy patients who have a methemoglobin level of 20% or greater.
- Identifying and promptly discontinuing the causative agent and initiating methylene blue infusion (1 mg/kg over 5 minutes) is the preferred treatment.
- Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothorac Vasc Anesth 2014; 28:1055–1059.
- Margulies DR, Manookian CM. Methemoglobinemia as a cause of respiratory failure. J Trauma 2002; 52:796–797.
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011; 104:757–761.
- Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004; 83:265–273.
- Kanji HD, Mithani S, Boucher P, Dias VC, Yarema MC. Coma, metabolic acidosis, and methemoglobinemia in a patient with acetaminophen toxicity. J Popul Ther Clin Pharmacol 2013; 20:e207–e211.
- Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013; 52:1741–1743.
- Wieringa A, Bethlehem C, Hoogendoorn M, van der Maten J, van Roon EN. Very late recovery of dapsone-induced methemoglobinemia. Clin Toxicol (Phila) 2014; 52:80–81.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011; 45:1103–1115.
- Taleb M, Ashraf Z, Valavoor S, Tinkel J. Evaluation and management of acquired methemoglobinemia associated with topical benzocaine use. Am J Cardiovasc Drugs 2013; 13:325–330.
- Chowdhary S, Bukoye B, Bhansali AM, et al. Risk of topical anesthetic-induced methemoglobinemia: a 10-year retrospective case-control study. JAMA Intern Med 2013; 173:771–776.
- Larson A, Stidham T, Banerji S, Kaufman J. Seizures and methemoglobinemia in an infant after excessive EMLA application. Pediatr Emerg Care 2013; 29:377–379.
- Schmitt C, Matulic M, Kervégant M, et al. Methaemoglobinaemia in a child treated with Emla cream: circumstances and consequences of overdose [in French]. Ann Dermatol Venereol 2012; 139:824–827.
- Bucklin MH, Groth CM. Mortality following rasburicase-induced methemoglobinemia. Ann Pharmacother 2013; 47:1353–1358.
- Cheah CY, Lew TE, Seymour JF, Burbury K. Rasburicase causing severe oxidative hemolysis and methemoglobinemia in a patient with previously unrecognized glucose-6-phosphate dehydrogenase deficiency. Acta Haematol 2013; 130:254–259.
- Druez A, Rahier JF, Hébuterne X. Methaemoglobinaemia and renal failure following mesalazine for treatment of inflammatory bowel disease. J Crohns Colitis 2014; 8:900–901.
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999; 34:646–656.
- Groeper K, Katcher K, Tobias JD. Anesthetic management of a patient with methemoglobinemia. South Med J 2003; 96:504–509.
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51:434–444.
- Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol 2013; 9:242–249.
A 48-year-old woman presented to the emergency department after 2 days of nonproductive cough, chest discomfort, worsening shortness of breath, and subjective fever. She had a history of systemic sclerosis. She was currently taking prednisone 20 mg daily and aspirin 81 mg daily.
Physical examination revealed tachypnea (28 breaths per minute), and bronchial breath sounds in the left lower chest posteriorly.
The initial laboratory workup revealed:
- Hemoglobin 106 g/L (reference range 115–155)
- Mean corpuscular volume 84 fL (80–100)
- White blood cell count 29.4 × 109/L (3.70–11.0), with 85% neutrophils
- Platelet count 180 × 109/L (150–350)
- Lactate dehydrogenase 312 U/L (100–220).
Chest radiography showed opacification of the lower lobe of the left lung.
She was admitted to the hospital and started treatment with intravenous azithromycin and ceftriaxone for presumed community-acquired pneumonia, based on the clinical presentation and findings on chest radiography. Because of her immunosuppression (due to chronic prednisone therapy) and her high lactate dehydrogenase level, Pneumocystis jirovecii pneumonia was suspected, and because she had a history of allergy to trimethoprim-sulfamethoxazole and pentamidine, she was started on dapsone.
During the next 24 hours, she developed worsening dyspnea, hypoxia, and cyanosis. She was placed on an air-entrainment mask, with a fraction of inspired oxygen of 0.5. Pulse oximetry showed an oxygen saturation of 85%, but arterial blood gas analysis indicated an oxyhemoglobin concentration of 95%.
THE ‘SATURATION GAP’
1. Which is most likely to have caused the discrepancy between the oxyhemoglobin concentration and the oxygen saturation by pulse oximetry in this patient?
- Methemoglobinemia
- Carbon monoxide poisoning
- Inappropriate placement of the pulse oximeter probe
- Pulmonary embolism
Methemoglobinemia is the most likely cause of the discrepancy between the oxyhemoglobin levels and the oxygen saturation by pulse oximetry, a phenomenon also known as the “saturation gap.” Other common causes are cyanide poisoning and carbon monoxide poisoning.
Carbon monoxide poisoning, however, does not explain our patient’s cyanosis. On the contrary, carbon monoxide poisoning can actually cause the patient’s lips and mucous membranes to appear unnaturally bright pink. Also, carbon monoxide poisoning raises the blood concentration of carboxyhemoglobin (which has a high affinity for oxygen), and this usually causes pulse oximetry to read inappropriately high, whereas in our patient it read low.
Incorrect placement of the pulse oximeter probe can result in an inaccurate measurement of oxygen saturation. Visualization of the waveform on the plethysmograph or the signal quality index can be used to assess adequate placement of the pulse oximeter probe. However, inadequate probe placement does not explain our patient’s dyspnea and cyanosis.
Pulmonary embolism can lead to hypoxia as a result of ventilation-perfusion mismatch. However, pulmonary embolism leading to low oxygen saturation on pulse oximetry will also lead to concomitantly low oxyhemoglobin levels as measured by arterial blood gas analysis, and this was not seen in our patient.
BACK TO OUR PATIENT
Because there was a discrepancy between our patient’s pulse oximetry reading and oxyhemoglobin concentration by arterial blood gas measurement, her methemoglobin level was checked and was found to be 30%, thus confirming the diagnosis of methemoglobinemia.
WHAT IS METHEMOGLOBINEMIA, AND WHAT CAUSES IT?
Oxygen is normally bound to iron in its ferrous (Fe2+) form in hemoglobin to form oxyhemoglobin. Oxidative stress in the body can cause iron to change from the ferrous to the ferric (Fe3+) state, forming methemoglobin. Methemoglobin is normally present in the blood in low levels (< 1% of the total hemoglobin), and ferric iron is reduced and recycled back to the ferrous form by NADH-cytochrome b5 reductase, an enzyme present in red blood cells. This protective mechanism maintains methemoglobin levels within safe limits. But increased production can lead to accumulation of methemoglobin, resulting in dyspnea and hypoxia and the condition referred to as methemoglobinemia.1
Increased levels of methemoglobin relative to normal hemoglobin cause tissue hypoxia by several mechanisms. Methemoglobin cannot efficiently carry oxygen; instead, it binds to water or to a hydroxide ion depending on the pH of the environment.2 Therefore, the hemoglobin molecule does not carry its usual load of oxygen, and hypoxia results from the reduced delivery of oxygen to tissues. In addition, an increased concentration of methemoglobin causes a leftward shift in the oxygen-hemoglobin dissociation curve, representing an increased affinity to bound oxygen in the remaining heme groups. The tightly bound oxygen is not adequately released at the tissue level, thus causing cellular hypoxia.
Methemoglobinemia is most often caused by exposure to an oxidizing chemical or drug that increases production of methemoglobin. In rare cases, it is caused by a congenital deficiency of NADH-cytochrome b5 reductase.3
2. Which of the following drugs can cause methemoglobinemia?
- Acetaminophen
- Dapsone
- Benzocaine
- Primaquine
All four of these drugs are common culprits for causing acquired methemoglobinemia; others include chloroquine, nitroglycerin, and sulfonamides.4–6
The increased production of methemoglobin caused by these drugs overwhelms the protective effect of reducing enzymes and can lead to an accumulation of methemoglobin. However, because of variability in cellular metabolism, not every person who takes these drugs develops dangerous levels of methemoglobin.
Dapsone and benzocaine are the most commonly encountered drugs known to cause methemoglobinemia (Table 1). Dapsone is an anti-inflammatory and antimicrobial agent most commonly used for treating lepromatous leprosy and dermatitis herpetiformis. It is also often prescribed for prophylaxis and treatment of P jirovecii pneumonia in immunosuppressed individuals.7 Benzocaine is a local anesthetic and was commonly used before procedures such as oral or dental surgery, transesophageal echocardiography, and endoscopy.8–10 Even low doses of benzocaine can lead to high levels of methemoglobinemia. However, the availability of other, safer anesthetics now limits the use of benzocaine in major US centers. In addition, the topical anesthetic Emla (lidocaine plus prilocaine) has been recently reported as a cause of methemoglobinemia in infants and children.11,12
Also, potentially fatal methemoglobinemia has been reported in patients with a deficiency of G-6-phosphate dehydrogenase (G6PD) who received rasburicase, a recombinant version of urate oxidase enzyme used to prevent and treat tumor lysis syndrome.13,14
Lastly, methemoglobinemia has been reported in patients with inflammatory bowel disease treated with mesalamine.
Although this adverse reaction is rare, clinicians should be aware of it, since these agents are commonly used in everyday medical practice.15
RECOGNIZING THE DANGER SIGNS
The clinical manifestations of methemoglobinemia are directly proportional to the percentage of methemoglobin in red blood cells. Cyanosis generally becomes apparent at concentrations around 15%, at which point the patient may still have no symptoms. Anxiety, lightheadedness, tachycardia, and dizziness manifest at levels of 20% to 30%. Fatigue, confusion, dizziness, tachypnea, and worsening tachycardia occur at levels of 30% to 50%. Levels of 50% to 70% cause coma, seizures, arrhythmias, and acidosis, and levels over 70% are considered lethal.16
While these levels provide a general guideline of symptomatology in an otherwise healthy person, it is important to remember that patients with underlying conditions such as anemia, lung disease (both of which our patient had), sepsis, thalassemia, G6PD deficiency, and sickle cell disease can manifest symptoms at lower concentrations of methemoglobin.1,17
Most patients who develop clinically significant levels of methemoglobin do so within the first few hours of starting one of the culprit drugs.
DIAGNOSIS: METHEMOGLOBINEMIA AND THE SATURATION GAP
In patients with methemoglobinemia, pulse oximetry gives lower values than arterial blood gas oxygen measurements. Regular pulse oximetry works by measuring light absorbance at two distinct wavelengths (660 and 940 nm) to calculate the ratio of oxyhemoglobin to deoxyhemoglobin. Methemoglobin absorbs light at both these wavelengths, thus lowering the pulse oximetry values.1
In contrast, oxygen saturation of arterial blood gas (oxyhemoglobin) is calculated indirectly from the concentration of dissolved oxygen in the blood and does not include oxygen bound to hemoglobin. Therefore, the measured arterial oxygen saturation is often normal in patients with methemoglobinemia since it relies only on inspired oxygen content and is independent of the methemoglobin concentration.18
Oxygen supplementation can raise the level of oxyhemoglobin, which is a measure of dissolved oxygen, but the oxygen saturation as measured by pulse oximetry remains largely unchanged—ie, the saturation gap. A difference of more than 5% between the oxygen saturation by pulse oximetry and blood gas analysis is abnormal. Patients with clinically significant methemoglobinemia usually have a saturation gap greater than 10%.
Several other unique features should raise suspicion of methemoglobinemia. It should be considered in a patient presenting with cyanosis out of proportion to the oxygen saturation and in a patient with low oxygen saturation and a normal chest radiograph. Other clues include blood that is chocolate-colored on gross examination, rather than the dark red of deoxygenated blood.
Co-oximetry measures oxygen saturation using different wavelengths of light to distinguish between fractions of oxyhemoglobin, deoxyhemoglobin, and methemoglobin, but it is not widely available.
THE NEXT STEP
3. What is the next step in the management of our patient?
- Discontinue the dapsone
- Start methylene blue
- Start hyperbaric oxygen
- Give sodium thiosulfate
- Discontinue dapsone and start methylene blue
The next step in her management should be to stop the dapsone and start an infusion of methylene blue. Hyperbaric oxygen is used in treating carbon monoxide poisoning, and sodium thiosulfate is used in treating cyanide toxicity. They would not be appropriate in this patient’s care.
MANAGEMENT OF ACQUIRED METHEMOGLOBINEMIA
The first, most critical step in managing acquired methemoglobinemia is to immediately discontinue the suspected offending agent. In most patients without a concomitant condition such as anemia or lung disease and with a methemoglobin level below 20%, discontinuing the offending agent may suffice. Patients with a level of 20% or greater and patients with cardiac and pulmonary disease, who develop symptoms at lower concentrations of methemoglobin, require infusion of methylene blue.
Methylene blue is converted to its reduced form, leukomethylene blue, by NADPH-methemoglobin reductase. As it is oxidized, leukomethylene blue reduces methemoglobin to hemoglobin. A dose of 1 mg/kg intravenously is given at first. The response is usually dramatic, with a reduction in methemoglobin levels and improvement in symptoms often within 30 to 60 minutes. If levels remain high, the dose can be repeated 1 hour later.19
A caveat: methylene blue therapy should be avoided in patients with complete G6PD deficiency. Methylene blue works through the enzyme NADPH-methemoglobin reductase, and since patients with G6PD deficiency lack this enzyme, methylene blue is ineffective. In fact, since it cannot be reduced, excessive methylene blue can oxidize hemoglobin to methemoglobin, further exacerbating the condition. In patients with partial G6PD deficiency, methylene blue is still recommended as a first-line treatment, but at a lower initial dose (0.3–0.5 mg/kg). However, in patients with significant hemolysis, an exchange transfusion is the only treatment option.
CASE CONCLUDED
Since dapsone was identified as the likely cause of methemoglobinemia in our patient, it was immediately discontinued. Because she was symptomatic, 70 mg of methylene blue was given intravenously. Over the next 60 minutes, her clinical condition improved significantly. A repeat methemoglobin measurement was 3%.
She was discharged home the next day on oral antibiotics to complete treatment for community-acquired pneumonia.
TAKE-HOME POINTS
- Consider methemoglobinemia in a patient with unexplained cyanosis.
- Pulse oximetry gives lower values than arterial blood gas oxygen measurements in patients with methemoglobinemia, and pulse oximetry readings do not improve with supplemental oxygen.
- A saturation gap greater than 5% strongly suggests methemoglobinemia.
- The diagnosis of methemoglobinemia is confirmed by measuring the methemoglobin concentration.
- Most healthy patients develop symptoms at methemoglobin levels of 20%, but patients with comorbidities can develop symptoms at lower levels.
- A number of drugs can cause methemoglobinemia, even at therapeutic dosages.
- Treatment is generally indicated in patients who have symptoms or in healthy patients who have a methemoglobin level of 20% or greater.
- Identifying and promptly discontinuing the causative agent and initiating methylene blue infusion (1 mg/kg over 5 minutes) is the preferred treatment.
A 48-year-old woman presented to the emergency department after 2 days of nonproductive cough, chest discomfort, worsening shortness of breath, and subjective fever. She had a history of systemic sclerosis. She was currently taking prednisone 20 mg daily and aspirin 81 mg daily.
Physical examination revealed tachypnea (28 breaths per minute), and bronchial breath sounds in the left lower chest posteriorly.
The initial laboratory workup revealed:
- Hemoglobin 106 g/L (reference range 115–155)
- Mean corpuscular volume 84 fL (80–100)
- White blood cell count 29.4 × 109/L (3.70–11.0), with 85% neutrophils
- Platelet count 180 × 109/L (150–350)
- Lactate dehydrogenase 312 U/L (100–220).
Chest radiography showed opacification of the lower lobe of the left lung.
She was admitted to the hospital and started treatment with intravenous azithromycin and ceftriaxone for presumed community-acquired pneumonia, based on the clinical presentation and findings on chest radiography. Because of her immunosuppression (due to chronic prednisone therapy) and her high lactate dehydrogenase level, Pneumocystis jirovecii pneumonia was suspected, and because she had a history of allergy to trimethoprim-sulfamethoxazole and pentamidine, she was started on dapsone.
During the next 24 hours, she developed worsening dyspnea, hypoxia, and cyanosis. She was placed on an air-entrainment mask, with a fraction of inspired oxygen of 0.5. Pulse oximetry showed an oxygen saturation of 85%, but arterial blood gas analysis indicated an oxyhemoglobin concentration of 95%.
THE ‘SATURATION GAP’
1. Which is most likely to have caused the discrepancy between the oxyhemoglobin concentration and the oxygen saturation by pulse oximetry in this patient?
- Methemoglobinemia
- Carbon monoxide poisoning
- Inappropriate placement of the pulse oximeter probe
- Pulmonary embolism
Methemoglobinemia is the most likely cause of the discrepancy between the oxyhemoglobin levels and the oxygen saturation by pulse oximetry, a phenomenon also known as the “saturation gap.” Other common causes are cyanide poisoning and carbon monoxide poisoning.
Carbon monoxide poisoning, however, does not explain our patient’s cyanosis. On the contrary, carbon monoxide poisoning can actually cause the patient’s lips and mucous membranes to appear unnaturally bright pink. Also, carbon monoxide poisoning raises the blood concentration of carboxyhemoglobin (which has a high affinity for oxygen), and this usually causes pulse oximetry to read inappropriately high, whereas in our patient it read low.
Incorrect placement of the pulse oximeter probe can result in an inaccurate measurement of oxygen saturation. Visualization of the waveform on the plethysmograph or the signal quality index can be used to assess adequate placement of the pulse oximeter probe. However, inadequate probe placement does not explain our patient’s dyspnea and cyanosis.
Pulmonary embolism can lead to hypoxia as a result of ventilation-perfusion mismatch. However, pulmonary embolism leading to low oxygen saturation on pulse oximetry will also lead to concomitantly low oxyhemoglobin levels as measured by arterial blood gas analysis, and this was not seen in our patient.
BACK TO OUR PATIENT
Because there was a discrepancy between our patient’s pulse oximetry reading and oxyhemoglobin concentration by arterial blood gas measurement, her methemoglobin level was checked and was found to be 30%, thus confirming the diagnosis of methemoglobinemia.
WHAT IS METHEMOGLOBINEMIA, AND WHAT CAUSES IT?
Oxygen is normally bound to iron in its ferrous (Fe2+) form in hemoglobin to form oxyhemoglobin. Oxidative stress in the body can cause iron to change from the ferrous to the ferric (Fe3+) state, forming methemoglobin. Methemoglobin is normally present in the blood in low levels (< 1% of the total hemoglobin), and ferric iron is reduced and recycled back to the ferrous form by NADH-cytochrome b5 reductase, an enzyme present in red blood cells. This protective mechanism maintains methemoglobin levels within safe limits. But increased production can lead to accumulation of methemoglobin, resulting in dyspnea and hypoxia and the condition referred to as methemoglobinemia.1
Increased levels of methemoglobin relative to normal hemoglobin cause tissue hypoxia by several mechanisms. Methemoglobin cannot efficiently carry oxygen; instead, it binds to water or to a hydroxide ion depending on the pH of the environment.2 Therefore, the hemoglobin molecule does not carry its usual load of oxygen, and hypoxia results from the reduced delivery of oxygen to tissues. In addition, an increased concentration of methemoglobin causes a leftward shift in the oxygen-hemoglobin dissociation curve, representing an increased affinity to bound oxygen in the remaining heme groups. The tightly bound oxygen is not adequately released at the tissue level, thus causing cellular hypoxia.
Methemoglobinemia is most often caused by exposure to an oxidizing chemical or drug that increases production of methemoglobin. In rare cases, it is caused by a congenital deficiency of NADH-cytochrome b5 reductase.3
2. Which of the following drugs can cause methemoglobinemia?
- Acetaminophen
- Dapsone
- Benzocaine
- Primaquine
All four of these drugs are common culprits for causing acquired methemoglobinemia; others include chloroquine, nitroglycerin, and sulfonamides.4–6
The increased production of methemoglobin caused by these drugs overwhelms the protective effect of reducing enzymes and can lead to an accumulation of methemoglobin. However, because of variability in cellular metabolism, not every person who takes these drugs develops dangerous levels of methemoglobin.
Dapsone and benzocaine are the most commonly encountered drugs known to cause methemoglobinemia (Table 1). Dapsone is an anti-inflammatory and antimicrobial agent most commonly used for treating lepromatous leprosy and dermatitis herpetiformis. It is also often prescribed for prophylaxis and treatment of P jirovecii pneumonia in immunosuppressed individuals.7 Benzocaine is a local anesthetic and was commonly used before procedures such as oral or dental surgery, transesophageal echocardiography, and endoscopy.8–10 Even low doses of benzocaine can lead to high levels of methemoglobinemia. However, the availability of other, safer anesthetics now limits the use of benzocaine in major US centers. In addition, the topical anesthetic Emla (lidocaine plus prilocaine) has been recently reported as a cause of methemoglobinemia in infants and children.11,12
Also, potentially fatal methemoglobinemia has been reported in patients with a deficiency of G-6-phosphate dehydrogenase (G6PD) who received rasburicase, a recombinant version of urate oxidase enzyme used to prevent and treat tumor lysis syndrome.13,14
Lastly, methemoglobinemia has been reported in patients with inflammatory bowel disease treated with mesalamine.
Although this adverse reaction is rare, clinicians should be aware of it, since these agents are commonly used in everyday medical practice.15
RECOGNIZING THE DANGER SIGNS
The clinical manifestations of methemoglobinemia are directly proportional to the percentage of methemoglobin in red blood cells. Cyanosis generally becomes apparent at concentrations around 15%, at which point the patient may still have no symptoms. Anxiety, lightheadedness, tachycardia, and dizziness manifest at levels of 20% to 30%. Fatigue, confusion, dizziness, tachypnea, and worsening tachycardia occur at levels of 30% to 50%. Levels of 50% to 70% cause coma, seizures, arrhythmias, and acidosis, and levels over 70% are considered lethal.16
While these levels provide a general guideline of symptomatology in an otherwise healthy person, it is important to remember that patients with underlying conditions such as anemia, lung disease (both of which our patient had), sepsis, thalassemia, G6PD deficiency, and sickle cell disease can manifest symptoms at lower concentrations of methemoglobin.1,17
Most patients who develop clinically significant levels of methemoglobin do so within the first few hours of starting one of the culprit drugs.
DIAGNOSIS: METHEMOGLOBINEMIA AND THE SATURATION GAP
In patients with methemoglobinemia, pulse oximetry gives lower values than arterial blood gas oxygen measurements. Regular pulse oximetry works by measuring light absorbance at two distinct wavelengths (660 and 940 nm) to calculate the ratio of oxyhemoglobin to deoxyhemoglobin. Methemoglobin absorbs light at both these wavelengths, thus lowering the pulse oximetry values.1
In contrast, oxygen saturation of arterial blood gas (oxyhemoglobin) is calculated indirectly from the concentration of dissolved oxygen in the blood and does not include oxygen bound to hemoglobin. Therefore, the measured arterial oxygen saturation is often normal in patients with methemoglobinemia since it relies only on inspired oxygen content and is independent of the methemoglobin concentration.18
Oxygen supplementation can raise the level of oxyhemoglobin, which is a measure of dissolved oxygen, but the oxygen saturation as measured by pulse oximetry remains largely unchanged—ie, the saturation gap. A difference of more than 5% between the oxygen saturation by pulse oximetry and blood gas analysis is abnormal. Patients with clinically significant methemoglobinemia usually have a saturation gap greater than 10%.
Several other unique features should raise suspicion of methemoglobinemia. It should be considered in a patient presenting with cyanosis out of proportion to the oxygen saturation and in a patient with low oxygen saturation and a normal chest radiograph. Other clues include blood that is chocolate-colored on gross examination, rather than the dark red of deoxygenated blood.
Co-oximetry measures oxygen saturation using different wavelengths of light to distinguish between fractions of oxyhemoglobin, deoxyhemoglobin, and methemoglobin, but it is not widely available.
THE NEXT STEP
3. What is the next step in the management of our patient?
- Discontinue the dapsone
- Start methylene blue
- Start hyperbaric oxygen
- Give sodium thiosulfate
- Discontinue dapsone and start methylene blue
The next step in her management should be to stop the dapsone and start an infusion of methylene blue. Hyperbaric oxygen is used in treating carbon monoxide poisoning, and sodium thiosulfate is used in treating cyanide toxicity. They would not be appropriate in this patient’s care.
MANAGEMENT OF ACQUIRED METHEMOGLOBINEMIA
The first, most critical step in managing acquired methemoglobinemia is to immediately discontinue the suspected offending agent. In most patients without a concomitant condition such as anemia or lung disease and with a methemoglobin level below 20%, discontinuing the offending agent may suffice. Patients with a level of 20% or greater and patients with cardiac and pulmonary disease, who develop symptoms at lower concentrations of methemoglobin, require infusion of methylene blue.
Methylene blue is converted to its reduced form, leukomethylene blue, by NADPH-methemoglobin reductase. As it is oxidized, leukomethylene blue reduces methemoglobin to hemoglobin. A dose of 1 mg/kg intravenously is given at first. The response is usually dramatic, with a reduction in methemoglobin levels and improvement in symptoms often within 30 to 60 minutes. If levels remain high, the dose can be repeated 1 hour later.19
A caveat: methylene blue therapy should be avoided in patients with complete G6PD deficiency. Methylene blue works through the enzyme NADPH-methemoglobin reductase, and since patients with G6PD deficiency lack this enzyme, methylene blue is ineffective. In fact, since it cannot be reduced, excessive methylene blue can oxidize hemoglobin to methemoglobin, further exacerbating the condition. In patients with partial G6PD deficiency, methylene blue is still recommended as a first-line treatment, but at a lower initial dose (0.3–0.5 mg/kg). However, in patients with significant hemolysis, an exchange transfusion is the only treatment option.
CASE CONCLUDED
Since dapsone was identified as the likely cause of methemoglobinemia in our patient, it was immediately discontinued. Because she was symptomatic, 70 mg of methylene blue was given intravenously. Over the next 60 minutes, her clinical condition improved significantly. A repeat methemoglobin measurement was 3%.
She was discharged home the next day on oral antibiotics to complete treatment for community-acquired pneumonia.
TAKE-HOME POINTS
- Consider methemoglobinemia in a patient with unexplained cyanosis.
- Pulse oximetry gives lower values than arterial blood gas oxygen measurements in patients with methemoglobinemia, and pulse oximetry readings do not improve with supplemental oxygen.
- A saturation gap greater than 5% strongly suggests methemoglobinemia.
- The diagnosis of methemoglobinemia is confirmed by measuring the methemoglobin concentration.
- Most healthy patients develop symptoms at methemoglobin levels of 20%, but patients with comorbidities can develop symptoms at lower levels.
- A number of drugs can cause methemoglobinemia, even at therapeutic dosages.
- Treatment is generally indicated in patients who have symptoms or in healthy patients who have a methemoglobin level of 20% or greater.
- Identifying and promptly discontinuing the causative agent and initiating methylene blue infusion (1 mg/kg over 5 minutes) is the preferred treatment.
- Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothorac Vasc Anesth 2014; 28:1055–1059.
- Margulies DR, Manookian CM. Methemoglobinemia as a cause of respiratory failure. J Trauma 2002; 52:796–797.
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011; 104:757–761.
- Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004; 83:265–273.
- Kanji HD, Mithani S, Boucher P, Dias VC, Yarema MC. Coma, metabolic acidosis, and methemoglobinemia in a patient with acetaminophen toxicity. J Popul Ther Clin Pharmacol 2013; 20:e207–e211.
- Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013; 52:1741–1743.
- Wieringa A, Bethlehem C, Hoogendoorn M, van der Maten J, van Roon EN. Very late recovery of dapsone-induced methemoglobinemia. Clin Toxicol (Phila) 2014; 52:80–81.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011; 45:1103–1115.
- Taleb M, Ashraf Z, Valavoor S, Tinkel J. Evaluation and management of acquired methemoglobinemia associated with topical benzocaine use. Am J Cardiovasc Drugs 2013; 13:325–330.
- Chowdhary S, Bukoye B, Bhansali AM, et al. Risk of topical anesthetic-induced methemoglobinemia: a 10-year retrospective case-control study. JAMA Intern Med 2013; 173:771–776.
- Larson A, Stidham T, Banerji S, Kaufman J. Seizures and methemoglobinemia in an infant after excessive EMLA application. Pediatr Emerg Care 2013; 29:377–379.
- Schmitt C, Matulic M, Kervégant M, et al. Methaemoglobinaemia in a child treated with Emla cream: circumstances and consequences of overdose [in French]. Ann Dermatol Venereol 2012; 139:824–827.
- Bucklin MH, Groth CM. Mortality following rasburicase-induced methemoglobinemia. Ann Pharmacother 2013; 47:1353–1358.
- Cheah CY, Lew TE, Seymour JF, Burbury K. Rasburicase causing severe oxidative hemolysis and methemoglobinemia in a patient with previously unrecognized glucose-6-phosphate dehydrogenase deficiency. Acta Haematol 2013; 130:254–259.
- Druez A, Rahier JF, Hébuterne X. Methaemoglobinaemia and renal failure following mesalazine for treatment of inflammatory bowel disease. J Crohns Colitis 2014; 8:900–901.
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999; 34:646–656.
- Groeper K, Katcher K, Tobias JD. Anesthetic management of a patient with methemoglobinemia. South Med J 2003; 96:504–509.
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51:434–444.
- Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol 2013; 9:242–249.
- Cortazzo JA, Lichtman AD. Methemoglobinemia: a review and recommendations for management. J Cardiothorac Vasc Anesth 2014; 28:1055–1059.
- Margulies DR, Manookian CM. Methemoglobinemia as a cause of respiratory failure. J Trauma 2002; 52:796–797.
- Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011; 104:757–761.
- Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004; 83:265–273.
- Kanji HD, Mithani S, Boucher P, Dias VC, Yarema MC. Coma, metabolic acidosis, and methemoglobinemia in a patient with acetaminophen toxicity. J Popul Ther Clin Pharmacol 2013; 20:e207–e211.
- Kawasumi H, Tanaka E, Hoshi D, Kawaguchi Y, Yamanaka H. Methemoglobinemia induced by trimethoprim-sulfamethoxazole in a patient with systemic lupus erythematosus. Intern Med 2013; 52:1741–1743.
- Wieringa A, Bethlehem C, Hoogendoorn M, van der Maten J, van Roon EN. Very late recovery of dapsone-induced methemoglobinemia. Clin Toxicol (Phila) 2014; 52:80–81.
- Barclay JA, Ziemba SE, Ibrahim RB. Dapsone-induced methemoglobinemia: a primer for clinicians. Ann Pharmacother 2011; 45:1103–1115.
- Taleb M, Ashraf Z, Valavoor S, Tinkel J. Evaluation and management of acquired methemoglobinemia associated with topical benzocaine use. Am J Cardiovasc Drugs 2013; 13:325–330.
- Chowdhary S, Bukoye B, Bhansali AM, et al. Risk of topical anesthetic-induced methemoglobinemia: a 10-year retrospective case-control study. JAMA Intern Med 2013; 173:771–776.
- Larson A, Stidham T, Banerji S, Kaufman J. Seizures and methemoglobinemia in an infant after excessive EMLA application. Pediatr Emerg Care 2013; 29:377–379.
- Schmitt C, Matulic M, Kervégant M, et al. Methaemoglobinaemia in a child treated with Emla cream: circumstances and consequences of overdose [in French]. Ann Dermatol Venereol 2012; 139:824–827.
- Bucklin MH, Groth CM. Mortality following rasburicase-induced methemoglobinemia. Ann Pharmacother 2013; 47:1353–1358.
- Cheah CY, Lew TE, Seymour JF, Burbury K. Rasburicase causing severe oxidative hemolysis and methemoglobinemia in a patient with previously unrecognized glucose-6-phosphate dehydrogenase deficiency. Acta Haematol 2013; 130:254–259.
- Druez A, Rahier JF, Hébuterne X. Methaemoglobinaemia and renal failure following mesalazine for treatment of inflammatory bowel disease. J Crohns Colitis 2014; 8:900–901.
- Wright RO, Lewander WJ, Woolf AD. Methemoglobinemia: etiology, pharmacology, and clinical management. Ann Emerg Med 1999; 34:646–656.
- Groeper K, Katcher K, Tobias JD. Anesthetic management of a patient with methemoglobinemia. South Med J 2003; 96:504–509.
- Haymond S, Cariappa R, Eby CS, Scott MG. Laboratory assessment of oxygenation in methemoglobinemia. Clin Chem 2005; 51:434–444.
- Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol 2013; 9:242–249.