User login
Bedside Swallow Examination Review
Dysphagia is a serious medical condition that can lead to aspiration pneumonia, malnutrition, and dehydration.[1] Dysphagia is the result of a variety of medical etiologies, including stroke, traumatic brain injury, progressive neurologic conditions, head and neck cancers, and general deconditioning. Prevalence estimates for dysphagia vary depending upon the etiology and patient age, but estimates as high as 38% for lifetime prevalence have been reported in those over age 65 years.[2]
To avoid adverse health outcomes, early detection of dysphagia is essential. In hospitalized patients, early detection has been associated with reduced risk of pneumonia, decreased length of hospital stay, and improved cost‐effectiveness resulting from a reduction in hospital days due to fewer cases of aspiration pneumonia.[3, 4, 5] Stroke guidelines in the United States recommend screening for dysphagia for all patients admitted with stroke.[6] Consequently, the majority of screening procedures have been designed for and tested in this population.[7, 8, 9, 10]
The videofluoroscopic swallow study (VFSS) is a commonly accepted, reference standard, instrumental evaluation technique for dysphagia, as it provides the most comprehensive information regarding anatomic and physiologic function for swallowing diagnosis and treatment. Flexible endoscopic evaluation of swallowing (FEES) is also available, as are several less commonly used techniques (scintigraphy, manometry, and ultrasound). Due to availability, patient compliance, and expertise needed, it is not possible to perform instrumental examination on every patient with suspected dysphagia. Therefore, a number of minimally invasive bedside screening procedures for dysphagia have been developed.
The value of any diagnostic screening test centers on performance characteristics, which under ideal circumstances include a positive result for all those who have dysphagia (sensitivity) and negative result for all those who do not have dysphagia (specificity). Such an ideal screening procedure would reduce unnecessary referrals and testing, thus resulting in cost savings, more effective utilization of speech‐language pathology consultation services, and less unnecessary radiation exposure. In addition, an effective screen would detect all those at risk for aspiration pneumonia in need of intervention. However, most available bedside screening tools are lacking in some or all of these desirable attributes.[11, 12] We undertook a systematic review and meta‐analysis of bedside procedures to screen for dysphagia.
METHODS
Data Sources and Searches
We conducted a comprehensive search of 7 databases, including MEDLINE, Embase, and Scopus, from each database's earliest inception through June 9, 2014 for English‐language articles and abstracts. The search strategy was designed and conducted by an experienced librarian with input from 1 researcher (J.C.O.). Controlled vocabulary supplemented with keywords was used to search for comparative studies of bedside screening tests for predicting dysphagia (see Supporting Information, Appendix 1, in the online version of this article for the full strategy).
All abstracts were screened, and potentially relevant articles were identified for full‐text review. Those references were manually inspected to identify all relevant studies.
Study Selection
A study was eligible for inclusion if it tested a diagnostic swallow study of any variety against an acceptable reference standard (VFSS or flexible endoscopic evaluation of swallowing with sensory testing [FEEST]).
Data Extraction and Quality Assessment
The primary outcome of the study was aspiration, as predicted by a bedside exam, compared to gold‐standard visualization of aspirated material entering below the vocal cords. From each study, data were abstracted based on the type of diagnostic method and reference standard study population and inclusion/exclusion characteristics, design, and prediction of aspiration. Prediction of aspiration was compared against the reference standard to yield true positives, true negatives, false positives, and false negatives. Additional potential confounding variables were abstracted using a standard form based on the Preferred Reporting Items for Systematic Reviews and Meta‐Analysis[13] (see Supporting Information, Appendix 2, in the online version of this article for the full abstraction template).
Data Synthesis and Analysis
Sensitivity and specificity for each test that identified the presence of dysphagia was calculated for each study. These were used to generate positive and negative likelihood ratios (LRs), which were plotted on a likelihood matrix, a graphic depiction of the logarithm of the +LR on the ordinate versus the logarithm of the LR on the abscissa, dividing the graphic into quadrants such that the right upper quadrant is tests that can be used for confirmation, right lower quadrant neither confirmation nor exclusion, left lower quadrant exclusion only, and left upper quadrant an ideal test with both exclusionary and confirmatory properties.[14] A good screening test would thus be on the left half of the graphic to effectively rule out dysphagia, and the ideal test with both good sensitivity and specificity would be found in the left upper quadrant. Graphics were constructed using the Stata MIDAS package (Stata Corp., College Station, TX).[15]
RESULTS
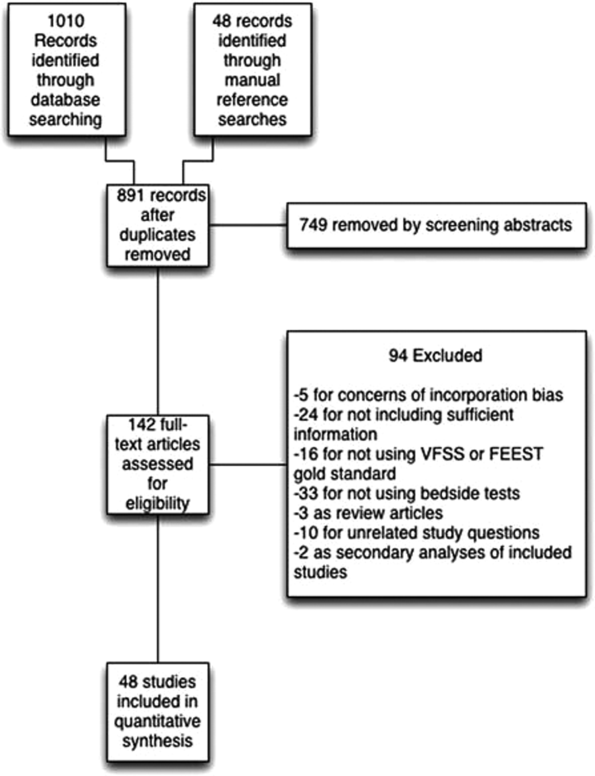
We identified 891 distinct articles. Of these, 749 were excluded based on abstract review. After reviewing the remaining 142 full‐text articles, 48 articles were determined to meet inclusion criteria, which included 10,437 observations across 7414 patients (Figure 1). We initially intended to conduct a meta‐analysis on each type, but heterogeneity in design and statistical heterogeneity in aggregate measures precluded pooling of results.
Characteristics of Included Studies
Of the 48 included studies, the majority (n=42) were prospective observational studies,[7, 8, 14, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53] whereas 2 were randomized trials,[9, 54] 2 studies were double‐blind observational,[9, 16] 1 was a case‐control design,[55] and 1 was a retrospective case series.[56] The majority of studies were exclusively inpatient,[7, 8, 9, 14, 17, 18, 19, 21, 22, 24, 25, 26, 31, 32, 33, 35, 36, 38, 41, 43, 44, 45, 46, 47, 49, 51, 52, 53, 55, 57] with 5 in mixed in and outpatient populations,[20, 27, 40, 55, 58] 2 in outpatient populations,[23, 41] and the remainder not reporting the setting from which they drew their study populations.
The indications for swallow evaluations fit broadly into 4 categories: stroke,[7, 8, 9, 14, 21, 22, 24, 25, 26, 31, 33, 34, 35, 38, 40, 41, 42, 43, 45, 48, 52, 56, 58] other neurologic disorders,[17, 18, 23, 28, 39, 47] all causes,[16, 20, 27, 29, 30, 36, 37, 44, 46, 49, 51, 52, 53, 54, 58] and postsurgical.[19, 32, 34] Most used VFSS as a reference standard,[7, 8, 9, 14, 16, 17, 18, 19, 21, 22, 23, 25, 26, 27, 28, 29, 30, 34, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 50, 51, 52, 53, 54, 56, 57, 58] with 8 using FEEST,[20, 24, 31, 32, 33, 35, 49, 55] and 1 accepting either videofluoroscopic evaluation of swallow or FEEST.[48]
Studies were placed into 1 or more of the following 4 categories: subjective bedside examination,[8, 9, 18, 19, 31, 34, 48] questionnaire‐based tools,[17, 23, 46, 53] protocolized multi‐item evaluations,[20, 21, 22, 25, 30, 33, 34, 37, 39, 44, 45, 52, 53, 57, 58] and single‐item exam maneuvers, symptoms, or signs.[7, 9, 14, 16, 24, 26, 27, 28, 29, 30, 31, 32, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 47, 48, 49, 50, 51, 56, 58, 59] The characteristics of all studies are detailed in Table 1.
| Study | Location | Design | Mean Age (SD) | Reason(s) for Dysphagia | Indx Test | Description | Reference Standard | Sample Size, No. of Patients | Sample Size, No. of Observations |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| Splaingard et al., 198844 | Milwaukee, WI, USA | Prospective observational study | NR | Multiple | Clinical bedside swallow exam | Combination of scored comprehensive physical exam, history, and observed swallow. | VFSS | 107 | 107 |
| DePippo et al., 199243 | White Plains, NY, USA | Prospective observational study | 71 (10) | Stroke | WST | Observation of swallow. | VFSS | 44 | |
| Horner et al., 199356 | Durham, NC, USA | Retrospective case series | 64* | Stroke | Clinical bedside swallow evaluation | VFSS | 38 | 114 | |
| Kidd et al., 199342 | Belfast, UK | Prospective observational study | 72 (10) | Stroke | Bedside 50‐mL swallow evaluation | Patient swallows 50 mL of water in 5‐mL aliquots, with therapist assessing for choking, coughing, or change in vocal quality after each swallow. | VFSS | 60 | 240 |
| Collins and Bakheit, 199741 | Southampton, UK | Prospective observational study | 65* | Stroke | Desaturation | Desaturation of at least 2% during videofluoroscopic study. | VFSS | 54 | 54 |
| Daniels et al., 199740 | New Orleans, LA, USA | Prospective observational study | 66 (11) | Stroke | Clinical bedside examination | 6 individual bedside assessments (dysphonia, dysphagia, cough before/after swallow, gag reflex and voice change) examined as predictors for aspiration risk. | VFSS | 59 | 354 |
| Mari et al., 199739 | Ancona, Italy | Prospective observational study | 60 (16) | Mixed neurologic diseases | Combined history and exam | Assessed symptoms of dysphagia, cough, and 3‐oz water swallow. | VFSS | 93 | 372 |
| Daniels et al., 19987 | New Orleans, LA, USA | Prospective observational study | 66 (11) | Stroke | Clinical bedside swallow evaluation | Describes sensitivity and specificity of several component physical exam maneuvers comprising the bedside exam. | VFSS | 55 | 330 |
| Smithard et al., 19988 | Ashford, UK | Prospective observational study | 79* | Stroke | Clinical bedside swallow evaluation | Not described. | VFSS | 83 | 249 |
| Addington et al., 199938 | Kansas City, MO, USA | Prospective observational study | 80* | Stroke | NR | Reflex cough. | VFSS | 40 | 40 |
| Logemann et al., 199937 | Evanston, IL, USA | Prospective observational study | 65 | Multiple | Northwestern Dysphagia Check Sheet | 28‐item screening procedure including history, observed swallow, and physical exam. | VFSS | 200 | 1400 |
| Smith et al., 20009 | Manchester, UK | Double blind observational | 69 | Stroke | Clinical bedside swallow evaluation, pulse oximetry evaluation | After eating/drinking, patient is evaluated for signs of aspiration including coughing, choking, or "wet voice." Procedure is repeated with several consistencies. Also evaluated if patient desaturates by at least 2% during evaluation. | VFSS | 53 | 53 |
| Warms et al., 200036 | Melbourne, Australia | Prospective observational study | 67 | Multiple | Wet voice | Voice was recorded and analyzed with Sony digital audio tape during videofluoroscopy. | VFSS | 23 | 708 |
| Lim et al., 200135 | Singapore, Singapore | Prospective observational study | NR | Stroke | Water swallow test, desaturation during swallow | 50‐mL swallow done in 5‐mL aliquots with assessment of phonation/choking afterward; desaturation >2% during swallow, | FEEST | 50 | 100 |
| McCullough et al., 200134 | Nashville, TN, USA | Prospective observational study | 60 (10) | Stroke | Clinical bedside swallow evaluation | 15‐item physical exam with observed swallow. | VFSS | 2040 | 60 |
| Rosen et al., 2001[74] | Newark, NJ, USA | Prospective observational study | 60 | Head and Neck cancer | Wet voice | Observation of swallow. | VFSS | 26 | 26 |
| Leder and Espinosa, 200233 | New Haven, CT, USA | Prospective observational study | 70* | Stroke | Clinical exam | Checklist evaluation of cough and voice change after swallow, volitional cough, dysphonia, dysarthria, and abnormal gag. | FEEST | 49 | 49 |
| Belafsky et al., 200332 | San Francisco, CA, USA | Prospective observational study | 65 (11) | Post‐tracheostomy patients | Modified Evans Blue Dye Test | 3 boluses of dye‐impregnated ice are given to patient. Tracheal secretions are suctioned, and evaluated for the presence of dye. | FEES | 30 | 30 |
| Chong et al., 200331 | Jalan Tan Tock Seng, Singapore | Prospective observational study | 75 (7) | Stroke | Water swallow test, desaturation during, clinical exam | Subjective exam, drinking 50 mL of water in 10‐mL aliquots, and evaluating for desaturation >2% during FEES. | FEEST | 50 | 150 |
| Tohara et al., 200330 | Tokyo, Japan | Prospective observational study | 63 (17) | Multiple | Food/water swallow tests, and a combination of the 2 | Protocolized observation of sequential food and water swallows with scored outcomes. | VFSS | 63 | 63 |
| Rosenbek et al., 200414 | Gainesville, FL, USA | Prospective observational study | 68* | Stroke | Clinical bedside swallow evaluation | Describes 5 parameters of voice quality and 15 physical examination maneuvers used. | VFSS | 60 | 1200 |
| Ryu et al., 200429 | Seoul, South Korea | Prospective observational study | 64 (14) | Multiple | Voice analysis parameters | Analysis of the/a/vowel sound with Visi‐Pitch II 3300. | VFSS | 93 | 372 |
| Shaw et al., 200428 | Sheffield, UK | Prospective observational study | 71 | Neurologic disease | Bronchial auscultation | Auscultation over the right main bronchus during trial feeding to listen for sounds of aspiration. | VFSS | 105 | 105 |
| Wu et al., 200427 | Taipei, Taiwan | Prospective observational study | 72 (11) | Multiple | 100‐mL swallow test | Patient lifts a glass of 100 mL of water and drinks as quickly as possible, and is assessed for signs of choking, coughing, or wet voice, and is timed for speed of drinking. | VFSS | 54 | 54 |
| Nishiwaki et al., 200526 | Shizuoaka, Japan | Prospective observational study | 70* | Stroke | Clinical bedside swallow evaluation | Describes sensitivity and specificity of several component physical exam maneuvers comprising the bedside exam. | VFSS | 31 | 248 |
| Wang et al., 200554 | Taipei, Taiwan | Prospective double‐blind study | 41* | Multiple | Desaturation | Desaturation of at least 2% during videofluoroscopic study. | VFSS | 60 | 60 |
| Ramsey et al., 200625 | Kent, UK | Prospective observational study | 71 (10) | Stroke | BSA | Assessment of lip seal, tongue movement, voice quality, cough, and observed 5‐mL swallow. | VFSS | 54 | 54 |
| Trapl et al., 200724 | Krems, Austria | Prospective observational study | 76 (2) | Stroke | Gugging Swallow Screen | Progressive observed swallow trials with saliva, then with 350 mL liquid, then dry bread. | FEEST | 49 | 49 |
| Suiter and Leder, 200849 | Several centers across the USA | Prospective observational study | 68.3 | Multiple | 3‐oz water swallow test | Observation of swallow. | FEEST | 3000 | 3000 |
| Wagasugi et al., 200850 | Tokyo, Japan | Prospective observational study | NR | Multiple | Cough test | Acoustic analysis of cough. | VFSS | 204 | 204 |
| Baylow et al., 200945 | New York, NY, USA | Prospective observational study | NR | Stroke | Northwestern Dysphagia Check Sheet | 28‐item screening procedure including history, observed swallow, and physical exam. | VFSS | 15 | 30 |
| Cox et al., 200923 | Leiden, the Netherlands | Prospective observational study | 68 (8) | Inclusion body myositis | Dysphagia questionnaire | Questionnaire assessing symptoms of dysphagia. | VFSS | 57 | 57 |
| Kagaya et al., 201051 | Tokyo, Japan | Prospective observational study | NR | Multiple | Simple Swallow Provocation Test | Injection of 1‐2 mL of water through nasal tube directed at the suprapharynx. | VFSS | 46 | 46 |
| Martino et al., 200957 | Toronto, Canada | Randomized trial | 69 (14) | Stroke | Toronto Bedside Swallow Screening Test | 4‐item physical assessment including Kidd water swallow test, pharyngeal sensation, tongue movement, and dysphonia (before and after water swallow). | VFSS | 59 | 59 |
| Santamato et al., 200955 | Bari, Italy | Case control | NR | Multiple | Acoustic analysis, postswallow apnea | Acoustic analysis of cough. | VFSS | 15 | 15 |
| Smith Hammond et al., 200948 | Durham, NC, USA | Prospective observational study | 67.7 (1.2) | Multiple | Cough, expiratory phase peak flow | Acoustic analysis of cough. | VFSS or FEES | 96 | 288 |
| Leigh et al., 201022 | Seoul, South Korea | Prospective observational study | NR | Stroke | Clinical bedside swallow evaluation | Not described. | VFSS | 167 | 167 |
| Pitts et al., 201047 | Gainesville, FL, USA | Prospective observational study | NR | Parkinson | Cough compression phase duration | Acoustic analysis of cough. | VFSS | 58 | 232 |
| Cohen and Manor, 201146 | Tel Aviv, Israel | Prospective observational Study | NR | Multiple | Swallow Disturbance Questionnaire | 15‐item questionnaire. | FEES | 100 | 100 |
| Edmiaston et al., 201121 | St. Louis, MO, USA | Prospective observational study | 63* | Stroke | SWALLOW‐3D Acute Stroke Dysphagia Screen | 5‐item screen including mental status; asymmetry or weakness of face, tongue, or palate; and subjective signs of aspiration when drinking 3 oz water. | VFSS | 225 | 225 |
| Mandysova et al., 201120 | Pardubice, Czech Republic | Prospective observational study | 69 (13) | Multiple | Brief Bedside Dysphagia Screening Test | 8‐item physician exam including ability to clench teeth; symmetry/strength of tongue, facial, and shoulder muscles; dysarthria; and choking, coughing, or dripping of food after taking thick liquid. | FEES | 87 | 87 |
| Steele et al., 201158 | Toronto, Canada | Double blind observational | 67 | Stroke | 4‐item bedside exam | Tongue lateralization, cough, throat clear, and voice quality. | VFSS | 400 | 40 |
| Yamamoto et al., 201117 | Kodaira, Japan | Prospective observational study | 67 (9) | Parkinson's Disease | Swallowing Disturbance Questionnaire | 15‐item questionnaire. | VFSS | 61 | 61 |
| Bhama et al., 201219 | Pittsburgh, PA, USA | Prospective observational study | 57 (14) | Post‐lung transplant | Clinical bedside swallow evaluation | Not described. | VFSS | 128 | 128 |
| Shem et al., 201218 | San Jose, CA, USA | Prospective observational study | 42 (17) | Spinal cord injuries resulting in tetraplegia | Clinical bedside swallow evaluation | After eating/drinking, patient is evaluated for signs of aspiration including coughing, choking, or "wet voice." Procedure is repeated with several consistencies. | VFSS | 26 | 26 |
| Steele et al., 201316 | Toronto, Canada | Prospective observational study | 67 (14) | Multiple | Dual‐axis accelerometry | Computed accelerometry of swallow. | VFSS | 37 | 37 |
| Edmiaston et al., 201452 | St. Louis, MO, USA | Prospective observational study | 63 (15) | Stroke | Barnes Jewish Stroke Dysphagia Screen | 5‐item screen including mental status; asymmetry or weakness of face, tongue, or palate; and subjective signs of aspiration when drinking 3 oz water. | VFSS | 225 | 225 |
| Rofes et al., 201453 | Barcelona, Spain | Prospective observational study | 74 (12) | Mixed | EAT‐10 questionnaire and variable viscosity swallow test | Symptom‐based questionnaire (EAT‐10) and repeated observations and measurements of swallow with different thickness liquids. | VFS | 134 | 134 |
Subjective Clinical Exam
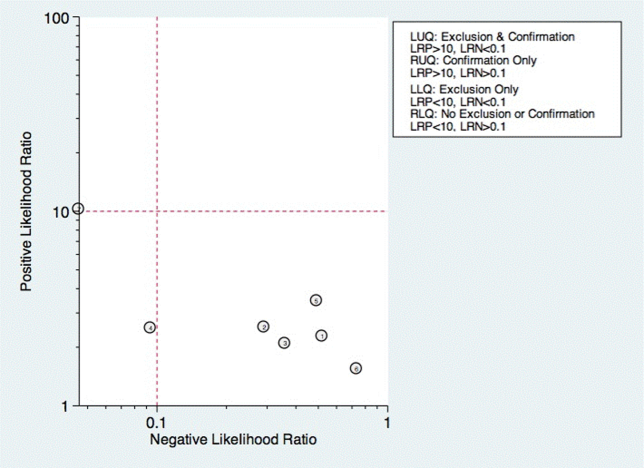
Seven studies reported the sensitivity and specificity of subjective assessments of nurses and speech‐language pathologists in observing swallowing and predicting aspiration.[8, 9, 18, 19, 31, 34, 48] The overall distribution of studies is summarized in the likelihood matrix in Figure 2. Two studies, Chong et al.[31] and Shem et al.,[18] were on the left side of the matrix, indicating a sensitive rule‐out test. However, both were small studies, and only Chong et al. reported reasonable sensitivity with incorporation bias from knowledge of a desaturation study outcome. Overall, subjective exams did not appear reliable in ruling out dysphagia.
Questionnaire‐Based Tools
Only 4 studies used questionnaire‐based tools filled out by the patient, asking about subjective assessment of dysphagia symptoms and frequency.[17, 23, 46, 53] Yamamoto et al. reported results of using the swallow dysphagia questionnaire in patients with Parkinson's disease.[17] Rofes et al. looked at the Eating Assessment Tool (EAT‐10) questionnaire among all referred patients and a small population of healthy volunteers.[53] Each was administered the questionnaire before undergoing a videofluoroscopic study. Overall, sensitivity and specificity were 77.8% and 84.6%, respectively. Cox et al. studied a different questionnaire in a group of patients with inclusion body myositis, finding 70% sensitivity and 44% specificity.[23] Cohen and Manor examined the swallow dysphagia questionnaire across several different causes of dysphagia, finding at optimum, the test is 78% specific and 73% sensitive.[46] Rofes et al. had an 86% sensitivity and 68% specificity for the EAT‐10 tool.[53]
Multi‐Item Exam Protocols
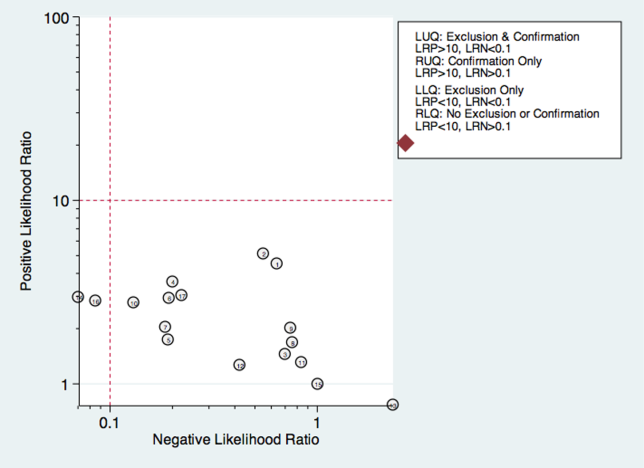
Sixteen studies reported multistep protocols for determining a patient's risk for aspiration.[9, 20, 21, 22, 25, 30, 33, 34, 37, 39, 44, 45, 52, 53, 57, 58] Each involved a combination of physical exam maneuvers and history elements, detailed in Table 1. This is shown in the likelihood matrix in Figure 3. Only 2 of these studies were in the left lower quadrant, Edmiaston et al. 201121 and 2014.[52] Both studies were restricted to stroke populations, but found reasonable sensitivity and specificity in identifying dysphagia.
Individual Exam Maneuvers
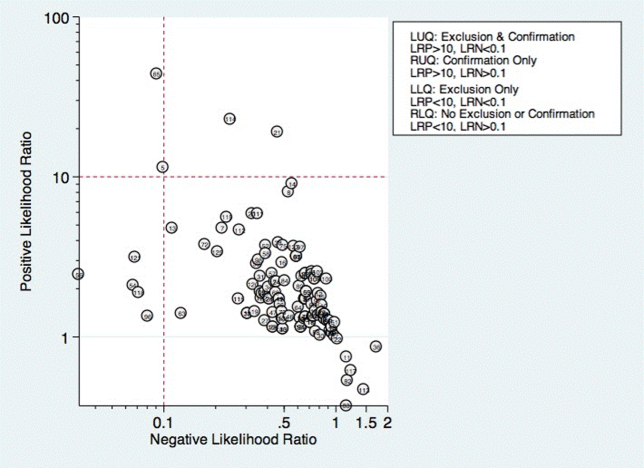
Thirty studies reported the diagnostic performance of individual exam maneuvers and signs.[7, 9, 14, 16, 24, 26, 27, 28, 29, 30, 31, 32, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 47, 48, 49, 50, 51, 54, 56, 58] Each is depicted in Figure 4 as a likelihood matrix demonstrating the +LR and LR for individual maneuvers as seen in the figure; most fall into the right lower quadrant, where they are not diagnostically useful tests. Studies in the left lower quadrant demonstrating the ability to exclude aspiration desirable in a screening test were dysphonia in McCullough et al.,[34] dual‐axis accelerometry in Steele et al.,[16] and the water swallow test in DePippo et al.[43] and Suiter and Leder.[49]
McCullough et al. found dysphonia to be the most discriminatory sign or symptom assessed, with an area under the curve (AUC) of 0.818. Dysphonia was judged by a sustained/a/and had 100% sensitivity but only 27% specificity. Wet voice within the same study was slightly less informative, with an AUC of 0.77 (sensitivity 50% and specificity 84%).[34]
Kidd et al. verified the diagnosis of stroke, and then assessed several neurologic parameters, including speech, muscle strength, and sensation. Pharyngeal sensation was assessed by touching each side of the pharyngeal wall and asking patients if they felt sensation that differed from each side. Patient report of abnormal sensation during this maneuver was 80% sensitive and 86% specific as a predictor of aspiration on VFSS.[42]
Steele et al. described the technique of dual axis accelerometry, where an accelerometer was placed at the midline of the neck over the cricoid cartilage during VFSS. The movement of the cricoid cartilage was captured for analysis in a computer algorithm to identify abnormal pharyngeal swallow behavior. Sensitivity was 100%, and specificity was 54%. Although the study was small (n=40), this novel method demonstrated good discrimination.[58]
DePippo et al. evaluated a 3‐oz water swallow in stroke patients. This protocol called for patients to drink the bolus of water without interruption, and be observed for 1 minute after for cough or wet‐hoarse voice. Presence of either sign was considered abnormal. Overall, sensitivity was 94% and specificity 30% looking for the presence of either sign.[43] Suiter and Leder used a similar protocol, with sensitivity of 97% and specificity of 49%.[49]
DISCUSSION
Our results show that most bedside swallow examinations lack the sensitivity to be used as a screening test for dysphagia across all patient populations examined. This is unfortunate as the ability to determine which patients require formal speech language pathology consultation or imaging as part of their diagnostic evaluation early in the hospital stay would lead to improved allocation of resources, cost reductions, and earlier implementation of effective therapy approaches. Furthermore, although radiation doses received during VFSS are not high when compared with other radiologic exams like computed tomography scans,[60] increasing awareness about the long‐term malignancy risks associated with medical imaging makes it desirable to reduce any test involving ionizing radiation.
There were several categories of screening procedures identified during this review process. Those classified as subjective bedside exams and protocolized multi‐item evaluations were found to have high heterogeneity in their sensitivity and specificity, though a few exam protocols did have a reasonable sensitivity and specificity.[21, 31, 52] The following individual exam maneuvers were found to demonstrate high sensitivity and an ability to exclude aspiration: a test for dysphonia through production of a sustained/a/34 and use of dual‐axis accelerometry.[16] Two other tests, the 3‐oz water swallow test[43] and testing of abnormal pharyngeal sensation,[42] were each found effective in a single study, with conflicting results from other studies.
Our results extend the findings from previous systematic reviews on this subject, most of which focused only on stroke patients.[5, 12, 61, 62] Martino and colleagues[5] conducted a review focused on screening for adults poststroke. From 13 identified articles, it was concluded that evidence to support inclusion or exclusion of screening was poor. Daniels et al. conducted a systematic review of swallowing screening tools specific to patients with acute or chronic stroke.[12] Based on 16 articles, the authors concluded that a combination of swallowing and nonswallowing features may be necessary for development of a valid screening tool. The generalizability of these reviews is limited given that all were conducted in patients poststroke, and therefore results and recommendations may not be generalizable to other patients.
Wilkinson et al.[62] conducted a recent systematic review that focused on screening techniques for inpatients 65 years or older that excluded patients with stroke or Parkinson's disease. The purpose of that review was to examine sensitivity and specificity of bedside screening tests as well as ability to accurately predict pneumonia. The authors concluded that existing evidence is not sufficient to recommend the use of bedside tests in a general older population.[62]
Specific screening tools identified by Martino and colleagues[5] to have good predictive value in detecting aspiration as a diagnostic marker of dysphagia were an abnormal test of pharyngeal sensation[42] and the 50‐mL water swallow test. Daniels et al. identified a water swallow test as an important component of a screen.[7] These results were consistent with those of this review in that the abnormal test of pharyngeal sensation[42] was identified for high levels of sensitivity. However, the 3‐oz water swallow test,[43, 49] rather than the 50‐mL water swallow test,[42] was identified in this review as the version of the water swallow test with the best predictive value in ruling out aspiration. Results of our review identified 2 additional individual items, dual‐axis accelerometry[16] and dysphonia,[34] that may be important to include in a comprehensive screening tool. In the absence of better tools, the 3 oz swallow test, properly executed, seems to be the best currently available tool validated in more than 1 study.
Several studies in the current review included an assessment of oral tongue movement that is not described thoroughly and varies between studies. Tongue movement as an individual item on a screening protocol was not found to yield high sensitivity or specificity. However, tongue movement or range of motion is only 1 aspect of oral tongue function; pressures produced by the tongue reflecting strength also may be important and warrant evaluation. Multiple studies have shown patients with dysphagia resulting from a variety of etiologies to produce lower than normal maximum isometric lingual pressures,[63, 64, 65, 66, 67, 68] or pressures produced when the tongue is pushed as hard as possible against the hard palate. Tongue strengthening protocols that result in higher maximum isometric lingual pressures have been shown to carry over to positive changes in swallow function.[69, 70, 71, 72, 73] Inclusion of tongue pressure measurement in a comprehensive screening tool may help to improve predictive capabilities.
We believe our results have implications for practicing clinicians, and serve as a call to action for development of an easy‐to‐perform, accurate tool for dysphagia screening. Future prospective studies should focus on practical tools that can be deployed at the bedside, and correlate the results with not only gold‐standard VFSS and FEES, but with clinical outcomes such as pneumonia and aspiration events leading to prolonged length of stay.
There were several limitations to this review. High levels of heterogeneity were reported in the screening tests present in the literature, precluding meaningful meta‐analysis. In addition, the majority of studies included were in poststroke adults, which limits the generalizability of results.
In conclusion, no screening protocol has been shown to provide adequate predictive value for presence of aspiration. Several individual exam maneuvers demonstrate high sensitivity; however, the most effective combination of screening protocol components is unknown. There is a need for future research focused on the development of a comprehensive screening tool that can be applied across patient populations for accurate detection of dysphagia as well as prediction of other adverse health outcomes, including pneumonia.
Acknowledgements
The authors thank Drs. Byun‐Mo Oh and Catrionia Steele for providing additional information in response to requests for unpublished information.
Disclosures: Nasia Safdar MD, is supported by a National Institutes of Health R03 GEMSSTAR award and a VA MERIT award. The authors report no conflicts of interest.
- , , , et al. Pathophysiology, relevance and natural history of oropharyngeal dysphagia among older people. Nestle Nutr Inst Workshop Ser. 2012;72:57–66.
- , , , Dysphagia in the elderly: preliminary evidence of prevalence, risk factors, and socioemotional effects. Ann Otol Rhinol Laryngol. 2007;116(11):858–865.
- , , Formal dysphagia screening protocols prevent pneumonia. Stroke. 2006;37(3):765.
- , , Swallow management in patients on an acute stroke pathway: quality is cost effective. Arch Phys Med Rehabil. 1995;76(12):1130–1133.
- , , Screening for oropharyngeal dysphagia in stroke: insufficient evidence for guidelines. Dysphagia. 2000;15(1):19–30.
- , , , et al. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013;44(3):870–947.
- , , , , , Aspiration in patients with acute stroke. Arch Phys Med Rehabil. 1998;79(1):14–19.
- , , , et al. Can bedside assessment reliably exclude aspiration following acute stroke? Age Ageing. 1998;27(2):99–106.
- , , , The combination of bedside swallowing assessment and oxygen saturation monitoring of swallowing in acute stroke: a safe and humane screening tool. Age Ageing. 2000;29(6):495–499.
- , , , Validation of a dysphagia screening tool in acute stroke patients. Am J Crit Care. 2010;19(4):357–364.
- , Screening for dysphagia and aspiration in acute stroke: a systematic review. Dysphagia. 2001;16(1):7–18.
- , , Valid items for screening dysphagia risk in patients with stroke: a systematic review. Stroke. 2012;43(3):892–897.
- , , , , Preferred Reporting Items for Systematic Reviews and Meta‐Analyses: The PRISMA Statement. Ann Intern Med. 2009;151(4):264–269, W64.
- , , Is the information about a test important? Applying the methods of evidence‐based medicine to the clinical examination of swallowing. J Commun Disord. 2004;37(5):437–450.
- MIDAS: Stata module for meta‐analytical integration of diagnostic test accuracy studies. Statistical Software Components. S456880, Boston College Department of Economics, 2009.
- , , Noninvasive detection of thin‐liquid aspiration using dual‐axis swallowing accelerometry. Dysphagia. 2013;28(1):105–112.
- , , , , Validation of the Japanese translation of the Swallowing Disturbance Questionnaire in Parkinson's disease patients. Qual Life Res. 2012;21(7):1299–1303.
- , , , , , Diagnostic accuracy of bedside swallow evaluation versus videofluoroscopy to assess dysphagia in individuals with tetraplegia. PM R. 2012;4(4):283–289.
- , et al. 723 Aspiration after Lung Transplantation: Incidence, Risk Factors, and Accuracy of the Bedside Swallow Evaluation. The J Heart Lung Transplant. 2012;31(4 suppl 1):S247–S248.
- , , , Cerny M. Development of the brief bedside dysphagia screening test in the Czech Republic. Nurs Health Sci. 2011;13(4):388–395.
- , , SWALLOW‐3D, a simple 2‐minute bedside screening test, detects dysphagia in acute stroke patients with high sensitivity when validated against video‐fluoroscopy. Stroke. 2011;42(3):e352.
- , , , , Bedside screening and subacute reassessment of post‐stroke dysphagia: a prospective study. Int J Stroke. 2010;5:200.
- , , , , , Detecting dysphagia in inclusion body myositis. J Neurol. 2009;256(12):2009–2013.
- , , , et al. Dysphagia bedside screening for acute‐stroke patients: the Gugging Swallowing Screen. Stroke. 2007;38(11):2948–2952.
- , , Can pulse oximetry or a bedside swallowing assessment be used to detect aspiration after stroke? Stroke. 2006;37(12):2984–2988.
- , , , , , Identification of a simple screening tool for dysphagia in patients with stroke using factor analysis of multiple dysphagia variables. J Rehabil Med. 2005;37(4):247–251.
- , , , Evaluating swallowing dysfunction using a 100‐ml water swallowing test. Dysphagia. 2004;19(1):43–47.
- , , , et al. Bronchial auscultation: an effective adjunct to speech and language therapy bedside assessment when detecting dysphagia and aspiration? Dysphagia. 2004;19(4):211–218.
- , , Prediction of laryngeal aspiration using voice analysis. Am J Phys Med Rehabil. 2004;83(10):753–757.
- , , , , Three tests for predicting aspiration without videofluorography. Dysphagia. 2003;18(2):126–134.
- , , , , Bedside clinical methods useful as screening test for aspiration in elderly patients with recent and previous strokes. Ann Acad Med Singapore. 2003;32(6):790–794.
- , , , The accuracy of the modified Evan's blue dye test in predicting aspiration. Laryngoscope. 2003;113(11):1969–1972.
- , Aspiration risk after acute stroke: comparison of clinical examination and fiberoptic endoscopic evaluation of swallowing. Dysphagia. 2002;17(3):214–218.
- , , Sensitivity and specificity of clinical/bedside examination signs for detecting aspiration in adults subsequent to stroke. J Commun Disord. 2001;34(1‐2):55–72.
- , , , et al. Accuracy of bedside clinical methods compared with fiberoptic endoscopic examination of swallowing (FEES) in determining the risk of aspiration in acute stroke patients. Dysphagia. 2001;16(1):1–6.
- , “Wet Voice” as a predictor of penetration and aspiration in oropharyngeal dysphagia. Dysphagia. 2000;15(2):84–88.
- , , A screening procedure for oropharyngeal dysphagia. Dysphagia. 1999;14(1):44–51.
- , , , Assessing the laryngeal cough reflex and the risk of developing pneumonia after stroke. Arch Phys Med Rehabil. 1999;80(2):150–154.
- , , , , , Predictive value of clinical indices in detecting aspiration in patients with neurological disorders. J Neurol Neurosurg Psychiatry. 1997;63(4):456–460.
- , , , Clinical assessment of swallowing and prediction of dysphagia severity. Am J Speech Lang Pathol. 1997;6(4):17–24.
- , Does pulse oximetry reliably detect aspiration in dysphagic stroke patients? Stroke. 1997;28(9):1773–1775.
- , , , Aspiration in acute stroke: a clinical study with videofluoroscopy. Q J Med. 1993;86(12):825–829.
- , , Validation of the 3‐oz water swallow test for aspiration following stroke. Arch Neurol. 1992;49(12):1259–1261.
- , , , Aspiration in rehabilitation patients: videofluoroscopy vs bedside clinical assessment. Arch Phys Med Rehabil. 1988;69(8):637–640.
- , , , Accuracy of clinical judgment of the chin‐down posture for dysphagia during the clinical/bedside assessment as corroborated by videofluoroscopy in adults with acute stroke. Dysphagia. 2009;24(4):423–433.
- , Swallowing disturbance questionnaire for detecting dysphagia. Laryngoscope. 2011;121(7):1383–1387.
- , , , , , . Using voluntary cough to detect penetration and aspiration during oropharyngeal swallowing in patients with Parkinson disease. Chest. 2010;138(6):1426–1431.
- , , , et al. Predicting aspiration in patients with ischemic stroke: comparison of clinical signs and aerodynamic measures of voluntary cough. Chest. 2009;135(3):769–777.
- , Clinical utility of the 3‐ounce water swallow test. Dysphagia. 2008;23(3):244–250.
- , , , et al. Screening test for silent aspiration at the bedside. Dysphagia. 2008;23(4):364–370.
- , , , , , Simple swallowing provocation test has limited applicability as a screening tool for detecting aspiration, silent aspiration, or penetration. Dysphagia. 2010;25(1):6–10.
- , , , A simple bedside stroke dysphagia screen, validated against videofluoroscopy, detects dysphagia and aspiration with high sensitivity. J Stroke Cerebrovasc Dis. 2014;23 (4):712–716.
- , , , Sensitivity and specificity of the Eating Assessment Tool and the Volume‐Viscosity Swallow Test for clinical evaluation of oropharyngeal dysphagia. Neurogastroenterol Motil. 2014;26(9):1256–1265.
- , , , Pulse oximetry does not reliably detect aspiration on videofluoroscopic swallowing study. Arch Phys Med Rehabil. 2005;86(4):730–734.
- , , , et al. Acoustic analysis of swallowing sounds: a new technique for assessing dysphagia. J Rehabil Med. 2009;41(8):639–645.
- , , Aspiration in bilateral stroke patients: a validation study. Neurology. 1993;43(2):430–433.
- , , , et al. The Toronto Bedside Swallowing Screening Test (TOR‐BSST): development and validation of a dysphagia screening tool for patients with stroke. Stroke. 2009;40(2):555–561.
- , , , et al. Exploration of the utility of a brief swallow screening protocol with comparison to concurrent videofluoroscopy. Can J Speech Lang Pathol Audiol. 2011;35(3):228–242.
- , , , et al. Formal dysphagia screening protocols prevent pneumonia. Stroke. 2005;36(9):1972–1976.
- , , , et al. Radiation exposure time during MBSS: influence of swallowing impairment severity, medical diagnosis, clinician experience, and standardized protocol use. Dysphagia. 2013;28(1):77–85.
- Detection of eating difficulties after stroke: a systematic review. Int Nurs Rev. 2006;53(2):143–149.
- , , Aspiration in older patients without stroke: A systematic review of bedside diagnostic tests and predictors of pneumonia. Eur Geriatr Med. 2012;3(3):145–152.
- , , A tongue force measurement system for the assessment of oral‐phase swallowing disorders. Arch Phys Med Rehabil. 1991;72(1):38–42.
- , , Strength, Endurance, and stability of the tongue and hand in Parkinson disease. J Speech Lang Hear Res. 2000;43(1):256–267.
- , , , et al. Effects of radiotherapy with or without chemotherapy on tongue strength and swallowing in patients with oral cancer. Head Neck. 2007;29(7):632–637.
- , , , , Tongue pressure against hard palate during swallowing in post‐stroke patients. Gerodontology. 2005;22(4):227–233.
- , Tongue measures in individuals with normal and impaired swallowing. Am J Speech Lang Pathol. 2007;16(2):148–156.
- , , , et al. Tongue strength as a predictor of functional outcomes and quality of life after tongue cancer surgery. Ann Otol Rhinol Laryngol. 2013;122(6):386–397.
- , , , Effects of two types of tongue strengthening exercises in young normals. Folia Phoniatr Logop. 2003;55(4):199–205.
- , , , , , The effects of lingual exercise on swallowing in older adults. J Am Geriatr Soc. S2005;53(9):1483–1489.
- , , , et al. The effects of lingual exercise in stroke patients with dysphagia. Arch Phys Med Rehabil. 2007;88(2):150–158.
- , , , , , Pretreatment swallowing exercises improve swallow function after chemoradiation. Laryngoscope. 2008;118(1):39–43.
- , , , Effects of directional exercise on lingual strength. J Speech Lang Hear Res. 2009;52(4):1034–1047.
- , , et al. Prediction of aspiration in patients with newly diagnosed untreated advanced head and neck cancer. Archives of Otolaryngology – Head 127(8):975–979.
Dysphagia is a serious medical condition that can lead to aspiration pneumonia, malnutrition, and dehydration.[1] Dysphagia is the result of a variety of medical etiologies, including stroke, traumatic brain injury, progressive neurologic conditions, head and neck cancers, and general deconditioning. Prevalence estimates for dysphagia vary depending upon the etiology and patient age, but estimates as high as 38% for lifetime prevalence have been reported in those over age 65 years.[2]
To avoid adverse health outcomes, early detection of dysphagia is essential. In hospitalized patients, early detection has been associated with reduced risk of pneumonia, decreased length of hospital stay, and improved cost‐effectiveness resulting from a reduction in hospital days due to fewer cases of aspiration pneumonia.[3, 4, 5] Stroke guidelines in the United States recommend screening for dysphagia for all patients admitted with stroke.[6] Consequently, the majority of screening procedures have been designed for and tested in this population.[7, 8, 9, 10]
The videofluoroscopic swallow study (VFSS) is a commonly accepted, reference standard, instrumental evaluation technique for dysphagia, as it provides the most comprehensive information regarding anatomic and physiologic function for swallowing diagnosis and treatment. Flexible endoscopic evaluation of swallowing (FEES) is also available, as are several less commonly used techniques (scintigraphy, manometry, and ultrasound). Due to availability, patient compliance, and expertise needed, it is not possible to perform instrumental examination on every patient with suspected dysphagia. Therefore, a number of minimally invasive bedside screening procedures for dysphagia have been developed.
The value of any diagnostic screening test centers on performance characteristics, which under ideal circumstances include a positive result for all those who have dysphagia (sensitivity) and negative result for all those who do not have dysphagia (specificity). Such an ideal screening procedure would reduce unnecessary referrals and testing, thus resulting in cost savings, more effective utilization of speech‐language pathology consultation services, and less unnecessary radiation exposure. In addition, an effective screen would detect all those at risk for aspiration pneumonia in need of intervention. However, most available bedside screening tools are lacking in some or all of these desirable attributes.[11, 12] We undertook a systematic review and meta‐analysis of bedside procedures to screen for dysphagia.
METHODS
Data Sources and Searches
We conducted a comprehensive search of 7 databases, including MEDLINE, Embase, and Scopus, from each database's earliest inception through June 9, 2014 for English‐language articles and abstracts. The search strategy was designed and conducted by an experienced librarian with input from 1 researcher (J.C.O.). Controlled vocabulary supplemented with keywords was used to search for comparative studies of bedside screening tests for predicting dysphagia (see Supporting Information, Appendix 1, in the online version of this article for the full strategy).
All abstracts were screened, and potentially relevant articles were identified for full‐text review. Those references were manually inspected to identify all relevant studies.
Study Selection
A study was eligible for inclusion if it tested a diagnostic swallow study of any variety against an acceptable reference standard (VFSS or flexible endoscopic evaluation of swallowing with sensory testing [FEEST]).
Data Extraction and Quality Assessment
The primary outcome of the study was aspiration, as predicted by a bedside exam, compared to gold‐standard visualization of aspirated material entering below the vocal cords. From each study, data were abstracted based on the type of diagnostic method and reference standard study population and inclusion/exclusion characteristics, design, and prediction of aspiration. Prediction of aspiration was compared against the reference standard to yield true positives, true negatives, false positives, and false negatives. Additional potential confounding variables were abstracted using a standard form based on the Preferred Reporting Items for Systematic Reviews and Meta‐Analysis[13] (see Supporting Information, Appendix 2, in the online version of this article for the full abstraction template).
Data Synthesis and Analysis
Sensitivity and specificity for each test that identified the presence of dysphagia was calculated for each study. These were used to generate positive and negative likelihood ratios (LRs), which were plotted on a likelihood matrix, a graphic depiction of the logarithm of the +LR on the ordinate versus the logarithm of the LR on the abscissa, dividing the graphic into quadrants such that the right upper quadrant is tests that can be used for confirmation, right lower quadrant neither confirmation nor exclusion, left lower quadrant exclusion only, and left upper quadrant an ideal test with both exclusionary and confirmatory properties.[14] A good screening test would thus be on the left half of the graphic to effectively rule out dysphagia, and the ideal test with both good sensitivity and specificity would be found in the left upper quadrant. Graphics were constructed using the Stata MIDAS package (Stata Corp., College Station, TX).[15]
RESULTS
We identified 891 distinct articles. Of these, 749 were excluded based on abstract review. After reviewing the remaining 142 full‐text articles, 48 articles were determined to meet inclusion criteria, which included 10,437 observations across 7414 patients (Figure 1). We initially intended to conduct a meta‐analysis on each type, but heterogeneity in design and statistical heterogeneity in aggregate measures precluded pooling of results.
Characteristics of Included Studies
Of the 48 included studies, the majority (n=42) were prospective observational studies,[7, 8, 14, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53] whereas 2 were randomized trials,[9, 54] 2 studies were double‐blind observational,[9, 16] 1 was a case‐control design,[55] and 1 was a retrospective case series.[56] The majority of studies were exclusively inpatient,[7, 8, 9, 14, 17, 18, 19, 21, 22, 24, 25, 26, 31, 32, 33, 35, 36, 38, 41, 43, 44, 45, 46, 47, 49, 51, 52, 53, 55, 57] with 5 in mixed in and outpatient populations,[20, 27, 40, 55, 58] 2 in outpatient populations,[23, 41] and the remainder not reporting the setting from which they drew their study populations.
The indications for swallow evaluations fit broadly into 4 categories: stroke,[7, 8, 9, 14, 21, 22, 24, 25, 26, 31, 33, 34, 35, 38, 40, 41, 42, 43, 45, 48, 52, 56, 58] other neurologic disorders,[17, 18, 23, 28, 39, 47] all causes,[16, 20, 27, 29, 30, 36, 37, 44, 46, 49, 51, 52, 53, 54, 58] and postsurgical.[19, 32, 34] Most used VFSS as a reference standard,[7, 8, 9, 14, 16, 17, 18, 19, 21, 22, 23, 25, 26, 27, 28, 29, 30, 34, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 50, 51, 52, 53, 54, 56, 57, 58] with 8 using FEEST,[20, 24, 31, 32, 33, 35, 49, 55] and 1 accepting either videofluoroscopic evaluation of swallow or FEEST.[48]
Studies were placed into 1 or more of the following 4 categories: subjective bedside examination,[8, 9, 18, 19, 31, 34, 48] questionnaire‐based tools,[17, 23, 46, 53] protocolized multi‐item evaluations,[20, 21, 22, 25, 30, 33, 34, 37, 39, 44, 45, 52, 53, 57, 58] and single‐item exam maneuvers, symptoms, or signs.[7, 9, 14, 16, 24, 26, 27, 28, 29, 30, 31, 32, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 47, 48, 49, 50, 51, 56, 58, 59] The characteristics of all studies are detailed in Table 1.
| Study | Location | Design | Mean Age (SD) | Reason(s) for Dysphagia | Indx Test | Description | Reference Standard | Sample Size, No. of Patients | Sample Size, No. of Observations |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| Splaingard et al., 198844 | Milwaukee, WI, USA | Prospective observational study | NR | Multiple | Clinical bedside swallow exam | Combination of scored comprehensive physical exam, history, and observed swallow. | VFSS | 107 | 107 |
| DePippo et al., 199243 | White Plains, NY, USA | Prospective observational study | 71 (10) | Stroke | WST | Observation of swallow. | VFSS | 44 | |
| Horner et al., 199356 | Durham, NC, USA | Retrospective case series | 64* | Stroke | Clinical bedside swallow evaluation | VFSS | 38 | 114 | |
| Kidd et al., 199342 | Belfast, UK | Prospective observational study | 72 (10) | Stroke | Bedside 50‐mL swallow evaluation | Patient swallows 50 mL of water in 5‐mL aliquots, with therapist assessing for choking, coughing, or change in vocal quality after each swallow. | VFSS | 60 | 240 |
| Collins and Bakheit, 199741 | Southampton, UK | Prospective observational study | 65* | Stroke | Desaturation | Desaturation of at least 2% during videofluoroscopic study. | VFSS | 54 | 54 |
| Daniels et al., 199740 | New Orleans, LA, USA | Prospective observational study | 66 (11) | Stroke | Clinical bedside examination | 6 individual bedside assessments (dysphonia, dysphagia, cough before/after swallow, gag reflex and voice change) examined as predictors for aspiration risk. | VFSS | 59 | 354 |
| Mari et al., 199739 | Ancona, Italy | Prospective observational study | 60 (16) | Mixed neurologic diseases | Combined history and exam | Assessed symptoms of dysphagia, cough, and 3‐oz water swallow. | VFSS | 93 | 372 |
| Daniels et al., 19987 | New Orleans, LA, USA | Prospective observational study | 66 (11) | Stroke | Clinical bedside swallow evaluation | Describes sensitivity and specificity of several component physical exam maneuvers comprising the bedside exam. | VFSS | 55 | 330 |
| Smithard et al., 19988 | Ashford, UK | Prospective observational study | 79* | Stroke | Clinical bedside swallow evaluation | Not described. | VFSS | 83 | 249 |
| Addington et al., 199938 | Kansas City, MO, USA | Prospective observational study | 80* | Stroke | NR | Reflex cough. | VFSS | 40 | 40 |
| Logemann et al., 199937 | Evanston, IL, USA | Prospective observational study | 65 | Multiple | Northwestern Dysphagia Check Sheet | 28‐item screening procedure including history, observed swallow, and physical exam. | VFSS | 200 | 1400 |
| Smith et al., 20009 | Manchester, UK | Double blind observational | 69 | Stroke | Clinical bedside swallow evaluation, pulse oximetry evaluation | After eating/drinking, patient is evaluated for signs of aspiration including coughing, choking, or "wet voice." Procedure is repeated with several consistencies. Also evaluated if patient desaturates by at least 2% during evaluation. | VFSS | 53 | 53 |
| Warms et al., 200036 | Melbourne, Australia | Prospective observational study | 67 | Multiple | Wet voice | Voice was recorded and analyzed with Sony digital audio tape during videofluoroscopy. | VFSS | 23 | 708 |
| Lim et al., 200135 | Singapore, Singapore | Prospective observational study | NR | Stroke | Water swallow test, desaturation during swallow | 50‐mL swallow done in 5‐mL aliquots with assessment of phonation/choking afterward; desaturation >2% during swallow, | FEEST | 50 | 100 |
| McCullough et al., 200134 | Nashville, TN, USA | Prospective observational study | 60 (10) | Stroke | Clinical bedside swallow evaluation | 15‐item physical exam with observed swallow. | VFSS | 2040 | 60 |
| Rosen et al., 2001[74] | Newark, NJ, USA | Prospective observational study | 60 | Head and Neck cancer | Wet voice | Observation of swallow. | VFSS | 26 | 26 |
| Leder and Espinosa, 200233 | New Haven, CT, USA | Prospective observational study | 70* | Stroke | Clinical exam | Checklist evaluation of cough and voice change after swallow, volitional cough, dysphonia, dysarthria, and abnormal gag. | FEEST | 49 | 49 |
| Belafsky et al., 200332 | San Francisco, CA, USA | Prospective observational study | 65 (11) | Post‐tracheostomy patients | Modified Evans Blue Dye Test | 3 boluses of dye‐impregnated ice are given to patient. Tracheal secretions are suctioned, and evaluated for the presence of dye. | FEES | 30 | 30 |
| Chong et al., 200331 | Jalan Tan Tock Seng, Singapore | Prospective observational study | 75 (7) | Stroke | Water swallow test, desaturation during, clinical exam | Subjective exam, drinking 50 mL of water in 10‐mL aliquots, and evaluating for desaturation >2% during FEES. | FEEST | 50 | 150 |
| Tohara et al., 200330 | Tokyo, Japan | Prospective observational study | 63 (17) | Multiple | Food/water swallow tests, and a combination of the 2 | Protocolized observation of sequential food and water swallows with scored outcomes. | VFSS | 63 | 63 |
| Rosenbek et al., 200414 | Gainesville, FL, USA | Prospective observational study | 68* | Stroke | Clinical bedside swallow evaluation | Describes 5 parameters of voice quality and 15 physical examination maneuvers used. | VFSS | 60 | 1200 |
| Ryu et al., 200429 | Seoul, South Korea | Prospective observational study | 64 (14) | Multiple | Voice analysis parameters | Analysis of the/a/vowel sound with Visi‐Pitch II 3300. | VFSS | 93 | 372 |
| Shaw et al., 200428 | Sheffield, UK | Prospective observational study | 71 | Neurologic disease | Bronchial auscultation | Auscultation over the right main bronchus during trial feeding to listen for sounds of aspiration. | VFSS | 105 | 105 |
| Wu et al., 200427 | Taipei, Taiwan | Prospective observational study | 72 (11) | Multiple | 100‐mL swallow test | Patient lifts a glass of 100 mL of water and drinks as quickly as possible, and is assessed for signs of choking, coughing, or wet voice, and is timed for speed of drinking. | VFSS | 54 | 54 |
| Nishiwaki et al., 200526 | Shizuoaka, Japan | Prospective observational study | 70* | Stroke | Clinical bedside swallow evaluation | Describes sensitivity and specificity of several component physical exam maneuvers comprising the bedside exam. | VFSS | 31 | 248 |
| Wang et al., 200554 | Taipei, Taiwan | Prospective double‐blind study | 41* | Multiple | Desaturation | Desaturation of at least 2% during videofluoroscopic study. | VFSS | 60 | 60 |
| Ramsey et al., 200625 | Kent, UK | Prospective observational study | 71 (10) | Stroke | BSA | Assessment of lip seal, tongue movement, voice quality, cough, and observed 5‐mL swallow. | VFSS | 54 | 54 |
| Trapl et al., 200724 | Krems, Austria | Prospective observational study | 76 (2) | Stroke | Gugging Swallow Screen | Progressive observed swallow trials with saliva, then with 350 mL liquid, then dry bread. | FEEST | 49 | 49 |
| Suiter and Leder, 200849 | Several centers across the USA | Prospective observational study | 68.3 | Multiple | 3‐oz water swallow test | Observation of swallow. | FEEST | 3000 | 3000 |
| Wagasugi et al., 200850 | Tokyo, Japan | Prospective observational study | NR | Multiple | Cough test | Acoustic analysis of cough. | VFSS | 204 | 204 |
| Baylow et al., 200945 | New York, NY, USA | Prospective observational study | NR | Stroke | Northwestern Dysphagia Check Sheet | 28‐item screening procedure including history, observed swallow, and physical exam. | VFSS | 15 | 30 |
| Cox et al., 200923 | Leiden, the Netherlands | Prospective observational study | 68 (8) | Inclusion body myositis | Dysphagia questionnaire | Questionnaire assessing symptoms of dysphagia. | VFSS | 57 | 57 |
| Kagaya et al., 201051 | Tokyo, Japan | Prospective observational study | NR | Multiple | Simple Swallow Provocation Test | Injection of 1‐2 mL of water through nasal tube directed at the suprapharynx. | VFSS | 46 | 46 |
| Martino et al., 200957 | Toronto, Canada | Randomized trial | 69 (14) | Stroke | Toronto Bedside Swallow Screening Test | 4‐item physical assessment including Kidd water swallow test, pharyngeal sensation, tongue movement, and dysphonia (before and after water swallow). | VFSS | 59 | 59 |
| Santamato et al., 200955 | Bari, Italy | Case control | NR | Multiple | Acoustic analysis, postswallow apnea | Acoustic analysis of cough. | VFSS | 15 | 15 |
| Smith Hammond et al., 200948 | Durham, NC, USA | Prospective observational study | 67.7 (1.2) | Multiple | Cough, expiratory phase peak flow | Acoustic analysis of cough. | VFSS or FEES | 96 | 288 |
| Leigh et al., 201022 | Seoul, South Korea | Prospective observational study | NR | Stroke | Clinical bedside swallow evaluation | Not described. | VFSS | 167 | 167 |
| Pitts et al., 201047 | Gainesville, FL, USA | Prospective observational study | NR | Parkinson | Cough compression phase duration | Acoustic analysis of cough. | VFSS | 58 | 232 |
| Cohen and Manor, 201146 | Tel Aviv, Israel | Prospective observational Study | NR | Multiple | Swallow Disturbance Questionnaire | 15‐item questionnaire. | FEES | 100 | 100 |
| Edmiaston et al., 201121 | St. Louis, MO, USA | Prospective observational study | 63* | Stroke | SWALLOW‐3D Acute Stroke Dysphagia Screen | 5‐item screen including mental status; asymmetry or weakness of face, tongue, or palate; and subjective signs of aspiration when drinking 3 oz water. | VFSS | 225 | 225 |
| Mandysova et al., 201120 | Pardubice, Czech Republic | Prospective observational study | 69 (13) | Multiple | Brief Bedside Dysphagia Screening Test | 8‐item physician exam including ability to clench teeth; symmetry/strength of tongue, facial, and shoulder muscles; dysarthria; and choking, coughing, or dripping of food after taking thick liquid. | FEES | 87 | 87 |
| Steele et al., 201158 | Toronto, Canada | Double blind observational | 67 | Stroke | 4‐item bedside exam | Tongue lateralization, cough, throat clear, and voice quality. | VFSS | 400 | 40 |
| Yamamoto et al., 201117 | Kodaira, Japan | Prospective observational study | 67 (9) | Parkinson's Disease | Swallowing Disturbance Questionnaire | 15‐item questionnaire. | VFSS | 61 | 61 |
| Bhama et al., 201219 | Pittsburgh, PA, USA | Prospective observational study | 57 (14) | Post‐lung transplant | Clinical bedside swallow evaluation | Not described. | VFSS | 128 | 128 |
| Shem et al., 201218 | San Jose, CA, USA | Prospective observational study | 42 (17) | Spinal cord injuries resulting in tetraplegia | Clinical bedside swallow evaluation | After eating/drinking, patient is evaluated for signs of aspiration including coughing, choking, or "wet voice." Procedure is repeated with several consistencies. | VFSS | 26 | 26 |
| Steele et al., 201316 | Toronto, Canada | Prospective observational study | 67 (14) | Multiple | Dual‐axis accelerometry | Computed accelerometry of swallow. | VFSS | 37 | 37 |
| Edmiaston et al., 201452 | St. Louis, MO, USA | Prospective observational study | 63 (15) | Stroke | Barnes Jewish Stroke Dysphagia Screen | 5‐item screen including mental status; asymmetry or weakness of face, tongue, or palate; and subjective signs of aspiration when drinking 3 oz water. | VFSS | 225 | 225 |
| Rofes et al., 201453 | Barcelona, Spain | Prospective observational study | 74 (12) | Mixed | EAT‐10 questionnaire and variable viscosity swallow test | Symptom‐based questionnaire (EAT‐10) and repeated observations and measurements of swallow with different thickness liquids. | VFS | 134 | 134 |
Subjective Clinical Exam
Seven studies reported the sensitivity and specificity of subjective assessments of nurses and speech‐language pathologists in observing swallowing and predicting aspiration.[8, 9, 18, 19, 31, 34, 48] The overall distribution of studies is summarized in the likelihood matrix in Figure 2. Two studies, Chong et al.[31] and Shem et al.,[18] were on the left side of the matrix, indicating a sensitive rule‐out test. However, both were small studies, and only Chong et al. reported reasonable sensitivity with incorporation bias from knowledge of a desaturation study outcome. Overall, subjective exams did not appear reliable in ruling out dysphagia.
Questionnaire‐Based Tools
Only 4 studies used questionnaire‐based tools filled out by the patient, asking about subjective assessment of dysphagia symptoms and frequency.[17, 23, 46, 53] Yamamoto et al. reported results of using the swallow dysphagia questionnaire in patients with Parkinson's disease.[17] Rofes et al. looked at the Eating Assessment Tool (EAT‐10) questionnaire among all referred patients and a small population of healthy volunteers.[53] Each was administered the questionnaire before undergoing a videofluoroscopic study. Overall, sensitivity and specificity were 77.8% and 84.6%, respectively. Cox et al. studied a different questionnaire in a group of patients with inclusion body myositis, finding 70% sensitivity and 44% specificity.[23] Cohen and Manor examined the swallow dysphagia questionnaire across several different causes of dysphagia, finding at optimum, the test is 78% specific and 73% sensitive.[46] Rofes et al. had an 86% sensitivity and 68% specificity for the EAT‐10 tool.[53]
Multi‐Item Exam Protocols
Sixteen studies reported multistep protocols for determining a patient's risk for aspiration.[9, 20, 21, 22, 25, 30, 33, 34, 37, 39, 44, 45, 52, 53, 57, 58] Each involved a combination of physical exam maneuvers and history elements, detailed in Table 1. This is shown in the likelihood matrix in Figure 3. Only 2 of these studies were in the left lower quadrant, Edmiaston et al. 201121 and 2014.[52] Both studies were restricted to stroke populations, but found reasonable sensitivity and specificity in identifying dysphagia.
Individual Exam Maneuvers
Thirty studies reported the diagnostic performance of individual exam maneuvers and signs.[7, 9, 14, 16, 24, 26, 27, 28, 29, 30, 31, 32, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 47, 48, 49, 50, 51, 54, 56, 58] Each is depicted in Figure 4 as a likelihood matrix demonstrating the +LR and LR for individual maneuvers as seen in the figure; most fall into the right lower quadrant, where they are not diagnostically useful tests. Studies in the left lower quadrant demonstrating the ability to exclude aspiration desirable in a screening test were dysphonia in McCullough et al.,[34] dual‐axis accelerometry in Steele et al.,[16] and the water swallow test in DePippo et al.[43] and Suiter and Leder.[49]
McCullough et al. found dysphonia to be the most discriminatory sign or symptom assessed, with an area under the curve (AUC) of 0.818. Dysphonia was judged by a sustained/a/and had 100% sensitivity but only 27% specificity. Wet voice within the same study was slightly less informative, with an AUC of 0.77 (sensitivity 50% and specificity 84%).[34]
Kidd et al. verified the diagnosis of stroke, and then assessed several neurologic parameters, including speech, muscle strength, and sensation. Pharyngeal sensation was assessed by touching each side of the pharyngeal wall and asking patients if they felt sensation that differed from each side. Patient report of abnormal sensation during this maneuver was 80% sensitive and 86% specific as a predictor of aspiration on VFSS.[42]
Steele et al. described the technique of dual axis accelerometry, where an accelerometer was placed at the midline of the neck over the cricoid cartilage during VFSS. The movement of the cricoid cartilage was captured for analysis in a computer algorithm to identify abnormal pharyngeal swallow behavior. Sensitivity was 100%, and specificity was 54%. Although the study was small (n=40), this novel method demonstrated good discrimination.[58]
DePippo et al. evaluated a 3‐oz water swallow in stroke patients. This protocol called for patients to drink the bolus of water without interruption, and be observed for 1 minute after for cough or wet‐hoarse voice. Presence of either sign was considered abnormal. Overall, sensitivity was 94% and specificity 30% looking for the presence of either sign.[43] Suiter and Leder used a similar protocol, with sensitivity of 97% and specificity of 49%.[49]
DISCUSSION
Our results show that most bedside swallow examinations lack the sensitivity to be used as a screening test for dysphagia across all patient populations examined. This is unfortunate as the ability to determine which patients require formal speech language pathology consultation or imaging as part of their diagnostic evaluation early in the hospital stay would lead to improved allocation of resources, cost reductions, and earlier implementation of effective therapy approaches. Furthermore, although radiation doses received during VFSS are not high when compared with other radiologic exams like computed tomography scans,[60] increasing awareness about the long‐term malignancy risks associated with medical imaging makes it desirable to reduce any test involving ionizing radiation.
There were several categories of screening procedures identified during this review process. Those classified as subjective bedside exams and protocolized multi‐item evaluations were found to have high heterogeneity in their sensitivity and specificity, though a few exam protocols did have a reasonable sensitivity and specificity.[21, 31, 52] The following individual exam maneuvers were found to demonstrate high sensitivity and an ability to exclude aspiration: a test for dysphonia through production of a sustained/a/34 and use of dual‐axis accelerometry.[16] Two other tests, the 3‐oz water swallow test[43] and testing of abnormal pharyngeal sensation,[42] were each found effective in a single study, with conflicting results from other studies.
Our results extend the findings from previous systematic reviews on this subject, most of which focused only on stroke patients.[5, 12, 61, 62] Martino and colleagues[5] conducted a review focused on screening for adults poststroke. From 13 identified articles, it was concluded that evidence to support inclusion or exclusion of screening was poor. Daniels et al. conducted a systematic review of swallowing screening tools specific to patients with acute or chronic stroke.[12] Based on 16 articles, the authors concluded that a combination of swallowing and nonswallowing features may be necessary for development of a valid screening tool. The generalizability of these reviews is limited given that all were conducted in patients poststroke, and therefore results and recommendations may not be generalizable to other patients.
Wilkinson et al.[62] conducted a recent systematic review that focused on screening techniques for inpatients 65 years or older that excluded patients with stroke or Parkinson's disease. The purpose of that review was to examine sensitivity and specificity of bedside screening tests as well as ability to accurately predict pneumonia. The authors concluded that existing evidence is not sufficient to recommend the use of bedside tests in a general older population.[62]
Specific screening tools identified by Martino and colleagues[5] to have good predictive value in detecting aspiration as a diagnostic marker of dysphagia were an abnormal test of pharyngeal sensation[42] and the 50‐mL water swallow test. Daniels et al. identified a water swallow test as an important component of a screen.[7] These results were consistent with those of this review in that the abnormal test of pharyngeal sensation[42] was identified for high levels of sensitivity. However, the 3‐oz water swallow test,[43, 49] rather than the 50‐mL water swallow test,[42] was identified in this review as the version of the water swallow test with the best predictive value in ruling out aspiration. Results of our review identified 2 additional individual items, dual‐axis accelerometry[16] and dysphonia,[34] that may be important to include in a comprehensive screening tool. In the absence of better tools, the 3 oz swallow test, properly executed, seems to be the best currently available tool validated in more than 1 study.
Several studies in the current review included an assessment of oral tongue movement that is not described thoroughly and varies between studies. Tongue movement as an individual item on a screening protocol was not found to yield high sensitivity or specificity. However, tongue movement or range of motion is only 1 aspect of oral tongue function; pressures produced by the tongue reflecting strength also may be important and warrant evaluation. Multiple studies have shown patients with dysphagia resulting from a variety of etiologies to produce lower than normal maximum isometric lingual pressures,[63, 64, 65, 66, 67, 68] or pressures produced when the tongue is pushed as hard as possible against the hard palate. Tongue strengthening protocols that result in higher maximum isometric lingual pressures have been shown to carry over to positive changes in swallow function.[69, 70, 71, 72, 73] Inclusion of tongue pressure measurement in a comprehensive screening tool may help to improve predictive capabilities.
We believe our results have implications for practicing clinicians, and serve as a call to action for development of an easy‐to‐perform, accurate tool for dysphagia screening. Future prospective studies should focus on practical tools that can be deployed at the bedside, and correlate the results with not only gold‐standard VFSS and FEES, but with clinical outcomes such as pneumonia and aspiration events leading to prolonged length of stay.
There were several limitations to this review. High levels of heterogeneity were reported in the screening tests present in the literature, precluding meaningful meta‐analysis. In addition, the majority of studies included were in poststroke adults, which limits the generalizability of results.
In conclusion, no screening protocol has been shown to provide adequate predictive value for presence of aspiration. Several individual exam maneuvers demonstrate high sensitivity; however, the most effective combination of screening protocol components is unknown. There is a need for future research focused on the development of a comprehensive screening tool that can be applied across patient populations for accurate detection of dysphagia as well as prediction of other adverse health outcomes, including pneumonia.
Acknowledgements
The authors thank Drs. Byun‐Mo Oh and Catrionia Steele for providing additional information in response to requests for unpublished information.
Disclosures: Nasia Safdar MD, is supported by a National Institutes of Health R03 GEMSSTAR award and a VA MERIT award. The authors report no conflicts of interest.
Dysphagia is a serious medical condition that can lead to aspiration pneumonia, malnutrition, and dehydration.[1] Dysphagia is the result of a variety of medical etiologies, including stroke, traumatic brain injury, progressive neurologic conditions, head and neck cancers, and general deconditioning. Prevalence estimates for dysphagia vary depending upon the etiology and patient age, but estimates as high as 38% for lifetime prevalence have been reported in those over age 65 years.[2]
To avoid adverse health outcomes, early detection of dysphagia is essential. In hospitalized patients, early detection has been associated with reduced risk of pneumonia, decreased length of hospital stay, and improved cost‐effectiveness resulting from a reduction in hospital days due to fewer cases of aspiration pneumonia.[3, 4, 5] Stroke guidelines in the United States recommend screening for dysphagia for all patients admitted with stroke.[6] Consequently, the majority of screening procedures have been designed for and tested in this population.[7, 8, 9, 10]
The videofluoroscopic swallow study (VFSS) is a commonly accepted, reference standard, instrumental evaluation technique for dysphagia, as it provides the most comprehensive information regarding anatomic and physiologic function for swallowing diagnosis and treatment. Flexible endoscopic evaluation of swallowing (FEES) is also available, as are several less commonly used techniques (scintigraphy, manometry, and ultrasound). Due to availability, patient compliance, and expertise needed, it is not possible to perform instrumental examination on every patient with suspected dysphagia. Therefore, a number of minimally invasive bedside screening procedures for dysphagia have been developed.
The value of any diagnostic screening test centers on performance characteristics, which under ideal circumstances include a positive result for all those who have dysphagia (sensitivity) and negative result for all those who do not have dysphagia (specificity). Such an ideal screening procedure would reduce unnecessary referrals and testing, thus resulting in cost savings, more effective utilization of speech‐language pathology consultation services, and less unnecessary radiation exposure. In addition, an effective screen would detect all those at risk for aspiration pneumonia in need of intervention. However, most available bedside screening tools are lacking in some or all of these desirable attributes.[11, 12] We undertook a systematic review and meta‐analysis of bedside procedures to screen for dysphagia.
METHODS
Data Sources and Searches
We conducted a comprehensive search of 7 databases, including MEDLINE, Embase, and Scopus, from each database's earliest inception through June 9, 2014 for English‐language articles and abstracts. The search strategy was designed and conducted by an experienced librarian with input from 1 researcher (J.C.O.). Controlled vocabulary supplemented with keywords was used to search for comparative studies of bedside screening tests for predicting dysphagia (see Supporting Information, Appendix 1, in the online version of this article for the full strategy).
All abstracts were screened, and potentially relevant articles were identified for full‐text review. Those references were manually inspected to identify all relevant studies.
Study Selection
A study was eligible for inclusion if it tested a diagnostic swallow study of any variety against an acceptable reference standard (VFSS or flexible endoscopic evaluation of swallowing with sensory testing [FEEST]).
Data Extraction and Quality Assessment
The primary outcome of the study was aspiration, as predicted by a bedside exam, compared to gold‐standard visualization of aspirated material entering below the vocal cords. From each study, data were abstracted based on the type of diagnostic method and reference standard study population and inclusion/exclusion characteristics, design, and prediction of aspiration. Prediction of aspiration was compared against the reference standard to yield true positives, true negatives, false positives, and false negatives. Additional potential confounding variables were abstracted using a standard form based on the Preferred Reporting Items for Systematic Reviews and Meta‐Analysis[13] (see Supporting Information, Appendix 2, in the online version of this article for the full abstraction template).
Data Synthesis and Analysis
Sensitivity and specificity for each test that identified the presence of dysphagia was calculated for each study. These were used to generate positive and negative likelihood ratios (LRs), which were plotted on a likelihood matrix, a graphic depiction of the logarithm of the +LR on the ordinate versus the logarithm of the LR on the abscissa, dividing the graphic into quadrants such that the right upper quadrant is tests that can be used for confirmation, right lower quadrant neither confirmation nor exclusion, left lower quadrant exclusion only, and left upper quadrant an ideal test with both exclusionary and confirmatory properties.[14] A good screening test would thus be on the left half of the graphic to effectively rule out dysphagia, and the ideal test with both good sensitivity and specificity would be found in the left upper quadrant. Graphics were constructed using the Stata MIDAS package (Stata Corp., College Station, TX).[15]
RESULTS
We identified 891 distinct articles. Of these, 749 were excluded based on abstract review. After reviewing the remaining 142 full‐text articles, 48 articles were determined to meet inclusion criteria, which included 10,437 observations across 7414 patients (Figure 1). We initially intended to conduct a meta‐analysis on each type, but heterogeneity in design and statistical heterogeneity in aggregate measures precluded pooling of results.
Characteristics of Included Studies
Of the 48 included studies, the majority (n=42) were prospective observational studies,[7, 8, 14, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53] whereas 2 were randomized trials,[9, 54] 2 studies were double‐blind observational,[9, 16] 1 was a case‐control design,[55] and 1 was a retrospective case series.[56] The majority of studies were exclusively inpatient,[7, 8, 9, 14, 17, 18, 19, 21, 22, 24, 25, 26, 31, 32, 33, 35, 36, 38, 41, 43, 44, 45, 46, 47, 49, 51, 52, 53, 55, 57] with 5 in mixed in and outpatient populations,[20, 27, 40, 55, 58] 2 in outpatient populations,[23, 41] and the remainder not reporting the setting from which they drew their study populations.
The indications for swallow evaluations fit broadly into 4 categories: stroke,[7, 8, 9, 14, 21, 22, 24, 25, 26, 31, 33, 34, 35, 38, 40, 41, 42, 43, 45, 48, 52, 56, 58] other neurologic disorders,[17, 18, 23, 28, 39, 47] all causes,[16, 20, 27, 29, 30, 36, 37, 44, 46, 49, 51, 52, 53, 54, 58] and postsurgical.[19, 32, 34] Most used VFSS as a reference standard,[7, 8, 9, 14, 16, 17, 18, 19, 21, 22, 23, 25, 26, 27, 28, 29, 30, 34, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 50, 51, 52, 53, 54, 56, 57, 58] with 8 using FEEST,[20, 24, 31, 32, 33, 35, 49, 55] and 1 accepting either videofluoroscopic evaluation of swallow or FEEST.[48]
Studies were placed into 1 or more of the following 4 categories: subjective bedside examination,[8, 9, 18, 19, 31, 34, 48] questionnaire‐based tools,[17, 23, 46, 53] protocolized multi‐item evaluations,[20, 21, 22, 25, 30, 33, 34, 37, 39, 44, 45, 52, 53, 57, 58] and single‐item exam maneuvers, symptoms, or signs.[7, 9, 14, 16, 24, 26, 27, 28, 29, 30, 31, 32, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 47, 48, 49, 50, 51, 56, 58, 59] The characteristics of all studies are detailed in Table 1.
| Study | Location | Design | Mean Age (SD) | Reason(s) for Dysphagia | Indx Test | Description | Reference Standard | Sample Size, No. of Patients | Sample Size, No. of Observations |
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| Splaingard et al., 198844 | Milwaukee, WI, USA | Prospective observational study | NR | Multiple | Clinical bedside swallow exam | Combination of scored comprehensive physical exam, history, and observed swallow. | VFSS | 107 | 107 |
| DePippo et al., 199243 | White Plains, NY, USA | Prospective observational study | 71 (10) | Stroke | WST | Observation of swallow. | VFSS | 44 | |
| Horner et al., 199356 | Durham, NC, USA | Retrospective case series | 64* | Stroke | Clinical bedside swallow evaluation | VFSS | 38 | 114 | |
| Kidd et al., 199342 | Belfast, UK | Prospective observational study | 72 (10) | Stroke | Bedside 50‐mL swallow evaluation | Patient swallows 50 mL of water in 5‐mL aliquots, with therapist assessing for choking, coughing, or change in vocal quality after each swallow. | VFSS | 60 | 240 |
| Collins and Bakheit, 199741 | Southampton, UK | Prospective observational study | 65* | Stroke | Desaturation | Desaturation of at least 2% during videofluoroscopic study. | VFSS | 54 | 54 |
| Daniels et al., 199740 | New Orleans, LA, USA | Prospective observational study | 66 (11) | Stroke | Clinical bedside examination | 6 individual bedside assessments (dysphonia, dysphagia, cough before/after swallow, gag reflex and voice change) examined as predictors for aspiration risk. | VFSS | 59 | 354 |
| Mari et al., 199739 | Ancona, Italy | Prospective observational study | 60 (16) | Mixed neurologic diseases | Combined history and exam | Assessed symptoms of dysphagia, cough, and 3‐oz water swallow. | VFSS | 93 | 372 |
| Daniels et al., 19987 | New Orleans, LA, USA | Prospective observational study | 66 (11) | Stroke | Clinical bedside swallow evaluation | Describes sensitivity and specificity of several component physical exam maneuvers comprising the bedside exam. | VFSS | 55 | 330 |
| Smithard et al., 19988 | Ashford, UK | Prospective observational study | 79* | Stroke | Clinical bedside swallow evaluation | Not described. | VFSS | 83 | 249 |
| Addington et al., 199938 | Kansas City, MO, USA | Prospective observational study | 80* | Stroke | NR | Reflex cough. | VFSS | 40 | 40 |
| Logemann et al., 199937 | Evanston, IL, USA | Prospective observational study | 65 | Multiple | Northwestern Dysphagia Check Sheet | 28‐item screening procedure including history, observed swallow, and physical exam. | VFSS | 200 | 1400 |
| Smith et al., 20009 | Manchester, UK | Double blind observational | 69 | Stroke | Clinical bedside swallow evaluation, pulse oximetry evaluation | After eating/drinking, patient is evaluated for signs of aspiration including coughing, choking, or "wet voice." Procedure is repeated with several consistencies. Also evaluated if patient desaturates by at least 2% during evaluation. | VFSS | 53 | 53 |
| Warms et al., 200036 | Melbourne, Australia | Prospective observational study | 67 | Multiple | Wet voice | Voice was recorded and analyzed with Sony digital audio tape during videofluoroscopy. | VFSS | 23 | 708 |
| Lim et al., 200135 | Singapore, Singapore | Prospective observational study | NR | Stroke | Water swallow test, desaturation during swallow | 50‐mL swallow done in 5‐mL aliquots with assessment of phonation/choking afterward; desaturation >2% during swallow, | FEEST | 50 | 100 |
| McCullough et al., 200134 | Nashville, TN, USA | Prospective observational study | 60 (10) | Stroke | Clinical bedside swallow evaluation | 15‐item physical exam with observed swallow. | VFSS | 2040 | 60 |
| Rosen et al., 2001[74] | Newark, NJ, USA | Prospective observational study | 60 | Head and Neck cancer | Wet voice | Observation of swallow. | VFSS | 26 | 26 |
| Leder and Espinosa, 200233 | New Haven, CT, USA | Prospective observational study | 70* | Stroke | Clinical exam | Checklist evaluation of cough and voice change after swallow, volitional cough, dysphonia, dysarthria, and abnormal gag. | FEEST | 49 | 49 |
| Belafsky et al., 200332 | San Francisco, CA, USA | Prospective observational study | 65 (11) | Post‐tracheostomy patients | Modified Evans Blue Dye Test | 3 boluses of dye‐impregnated ice are given to patient. Tracheal secretions are suctioned, and evaluated for the presence of dye. | FEES | 30 | 30 |
| Chong et al., 200331 | Jalan Tan Tock Seng, Singapore | Prospective observational study | 75 (7) | Stroke | Water swallow test, desaturation during, clinical exam | Subjective exam, drinking 50 mL of water in 10‐mL aliquots, and evaluating for desaturation >2% during FEES. | FEEST | 50 | 150 |
| Tohara et al., 200330 | Tokyo, Japan | Prospective observational study | 63 (17) | Multiple | Food/water swallow tests, and a combination of the 2 | Protocolized observation of sequential food and water swallows with scored outcomes. | VFSS | 63 | 63 |
| Rosenbek et al., 200414 | Gainesville, FL, USA | Prospective observational study | 68* | Stroke | Clinical bedside swallow evaluation | Describes 5 parameters of voice quality and 15 physical examination maneuvers used. | VFSS | 60 | 1200 |
| Ryu et al., 200429 | Seoul, South Korea | Prospective observational study | 64 (14) | Multiple | Voice analysis parameters | Analysis of the/a/vowel sound with Visi‐Pitch II 3300. | VFSS | 93 | 372 |
| Shaw et al., 200428 | Sheffield, UK | Prospective observational study | 71 | Neurologic disease | Bronchial auscultation | Auscultation over the right main bronchus during trial feeding to listen for sounds of aspiration. | VFSS | 105 | 105 |
| Wu et al., 200427 | Taipei, Taiwan | Prospective observational study | 72 (11) | Multiple | 100‐mL swallow test | Patient lifts a glass of 100 mL of water and drinks as quickly as possible, and is assessed for signs of choking, coughing, or wet voice, and is timed for speed of drinking. | VFSS | 54 | 54 |
| Nishiwaki et al., 200526 | Shizuoaka, Japan | Prospective observational study | 70* | Stroke | Clinical bedside swallow evaluation | Describes sensitivity and specificity of several component physical exam maneuvers comprising the bedside exam. | VFSS | 31 | 248 |
| Wang et al., 200554 | Taipei, Taiwan | Prospective double‐blind study | 41* | Multiple | Desaturation | Desaturation of at least 2% during videofluoroscopic study. | VFSS | 60 | 60 |
| Ramsey et al., 200625 | Kent, UK | Prospective observational study | 71 (10) | Stroke | BSA | Assessment of lip seal, tongue movement, voice quality, cough, and observed 5‐mL swallow. | VFSS | 54 | 54 |
| Trapl et al., 200724 | Krems, Austria | Prospective observational study | 76 (2) | Stroke | Gugging Swallow Screen | Progressive observed swallow trials with saliva, then with 350 mL liquid, then dry bread. | FEEST | 49 | 49 |
| Suiter and Leder, 200849 | Several centers across the USA | Prospective observational study | 68.3 | Multiple | 3‐oz water swallow test | Observation of swallow. | FEEST | 3000 | 3000 |
| Wagasugi et al., 200850 | Tokyo, Japan | Prospective observational study | NR | Multiple | Cough test | Acoustic analysis of cough. | VFSS | 204 | 204 |
| Baylow et al., 200945 | New York, NY, USA | Prospective observational study | NR | Stroke | Northwestern Dysphagia Check Sheet | 28‐item screening procedure including history, observed swallow, and physical exam. | VFSS | 15 | 30 |
| Cox et al., 200923 | Leiden, the Netherlands | Prospective observational study | 68 (8) | Inclusion body myositis | Dysphagia questionnaire | Questionnaire assessing symptoms of dysphagia. | VFSS | 57 | 57 |
| Kagaya et al., 201051 | Tokyo, Japan | Prospective observational study | NR | Multiple | Simple Swallow Provocation Test | Injection of 1‐2 mL of water through nasal tube directed at the suprapharynx. | VFSS | 46 | 46 |
| Martino et al., 200957 | Toronto, Canada | Randomized trial | 69 (14) | Stroke | Toronto Bedside Swallow Screening Test | 4‐item physical assessment including Kidd water swallow test, pharyngeal sensation, tongue movement, and dysphonia (before and after water swallow). | VFSS | 59 | 59 |
| Santamato et al., 200955 | Bari, Italy | Case control | NR | Multiple | Acoustic analysis, postswallow apnea | Acoustic analysis of cough. | VFSS | 15 | 15 |
| Smith Hammond et al., 200948 | Durham, NC, USA | Prospective observational study | 67.7 (1.2) | Multiple | Cough, expiratory phase peak flow | Acoustic analysis of cough. | VFSS or FEES | 96 | 288 |
| Leigh et al., 201022 | Seoul, South Korea | Prospective observational study | NR | Stroke | Clinical bedside swallow evaluation | Not described. | VFSS | 167 | 167 |
| Pitts et al., 201047 | Gainesville, FL, USA | Prospective observational study | NR | Parkinson | Cough compression phase duration | Acoustic analysis of cough. | VFSS | 58 | 232 |
| Cohen and Manor, 201146 | Tel Aviv, Israel | Prospective observational Study | NR | Multiple | Swallow Disturbance Questionnaire | 15‐item questionnaire. | FEES | 100 | 100 |
| Edmiaston et al., 201121 | St. Louis, MO, USA | Prospective observational study | 63* | Stroke | SWALLOW‐3D Acute Stroke Dysphagia Screen | 5‐item screen including mental status; asymmetry or weakness of face, tongue, or palate; and subjective signs of aspiration when drinking 3 oz water. | VFSS | 225 | 225 |
| Mandysova et al., 201120 | Pardubice, Czech Republic | Prospective observational study | 69 (13) | Multiple | Brief Bedside Dysphagia Screening Test | 8‐item physician exam including ability to clench teeth; symmetry/strength of tongue, facial, and shoulder muscles; dysarthria; and choking, coughing, or dripping of food after taking thick liquid. | FEES | 87 | 87 |
| Steele et al., 201158 | Toronto, Canada | Double blind observational | 67 | Stroke | 4‐item bedside exam | Tongue lateralization, cough, throat clear, and voice quality. | VFSS | 400 | 40 |
| Yamamoto et al., 201117 | Kodaira, Japan | Prospective observational study | 67 (9) | Parkinson's Disease | Swallowing Disturbance Questionnaire | 15‐item questionnaire. | VFSS | 61 | 61 |
| Bhama et al., 201219 | Pittsburgh, PA, USA | Prospective observational study | 57 (14) | Post‐lung transplant | Clinical bedside swallow evaluation | Not described. | VFSS | 128 | 128 |
| Shem et al., 201218 | San Jose, CA, USA | Prospective observational study | 42 (17) | Spinal cord injuries resulting in tetraplegia | Clinical bedside swallow evaluation | After eating/drinking, patient is evaluated for signs of aspiration including coughing, choking, or "wet voice." Procedure is repeated with several consistencies. | VFSS | 26 | 26 |
| Steele et al., 201316 | Toronto, Canada | Prospective observational study | 67 (14) | Multiple | Dual‐axis accelerometry | Computed accelerometry of swallow. | VFSS | 37 | 37 |
| Edmiaston et al., 201452 | St. Louis, MO, USA | Prospective observational study | 63 (15) | Stroke | Barnes Jewish Stroke Dysphagia Screen | 5‐item screen including mental status; asymmetry or weakness of face, tongue, or palate; and subjective signs of aspiration when drinking 3 oz water. | VFSS | 225 | 225 |
| Rofes et al., 201453 | Barcelona, Spain | Prospective observational study | 74 (12) | Mixed | EAT‐10 questionnaire and variable viscosity swallow test | Symptom‐based questionnaire (EAT‐10) and repeated observations and measurements of swallow with different thickness liquids. | VFS | 134 | 134 |
Subjective Clinical Exam
Seven studies reported the sensitivity and specificity of subjective assessments of nurses and speech‐language pathologists in observing swallowing and predicting aspiration.[8, 9, 18, 19, 31, 34, 48] The overall distribution of studies is summarized in the likelihood matrix in Figure 2. Two studies, Chong et al.[31] and Shem et al.,[18] were on the left side of the matrix, indicating a sensitive rule‐out test. However, both were small studies, and only Chong et al. reported reasonable sensitivity with incorporation bias from knowledge of a desaturation study outcome. Overall, subjective exams did not appear reliable in ruling out dysphagia.
Questionnaire‐Based Tools
Only 4 studies used questionnaire‐based tools filled out by the patient, asking about subjective assessment of dysphagia symptoms and frequency.[17, 23, 46, 53] Yamamoto et al. reported results of using the swallow dysphagia questionnaire in patients with Parkinson's disease.[17] Rofes et al. looked at the Eating Assessment Tool (EAT‐10) questionnaire among all referred patients and a small population of healthy volunteers.[53] Each was administered the questionnaire before undergoing a videofluoroscopic study. Overall, sensitivity and specificity were 77.8% and 84.6%, respectively. Cox et al. studied a different questionnaire in a group of patients with inclusion body myositis, finding 70% sensitivity and 44% specificity.[23] Cohen and Manor examined the swallow dysphagia questionnaire across several different causes of dysphagia, finding at optimum, the test is 78% specific and 73% sensitive.[46] Rofes et al. had an 86% sensitivity and 68% specificity for the EAT‐10 tool.[53]
Multi‐Item Exam Protocols
Sixteen studies reported multistep protocols for determining a patient's risk for aspiration.[9, 20, 21, 22, 25, 30, 33, 34, 37, 39, 44, 45, 52, 53, 57, 58] Each involved a combination of physical exam maneuvers and history elements, detailed in Table 1. This is shown in the likelihood matrix in Figure 3. Only 2 of these studies were in the left lower quadrant, Edmiaston et al. 201121 and 2014.[52] Both studies were restricted to stroke populations, but found reasonable sensitivity and specificity in identifying dysphagia.
Individual Exam Maneuvers
Thirty studies reported the diagnostic performance of individual exam maneuvers and signs.[7, 9, 14, 16, 24, 26, 27, 28, 29, 30, 31, 32, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 47, 48, 49, 50, 51, 54, 56, 58] Each is depicted in Figure 4 as a likelihood matrix demonstrating the +LR and LR for individual maneuvers as seen in the figure; most fall into the right lower quadrant, where they are not diagnostically useful tests. Studies in the left lower quadrant demonstrating the ability to exclude aspiration desirable in a screening test were dysphonia in McCullough et al.,[34] dual‐axis accelerometry in Steele et al.,[16] and the water swallow test in DePippo et al.[43] and Suiter and Leder.[49]
McCullough et al. found dysphonia to be the most discriminatory sign or symptom assessed, with an area under the curve (AUC) of 0.818. Dysphonia was judged by a sustained/a/and had 100% sensitivity but only 27% specificity. Wet voice within the same study was slightly less informative, with an AUC of 0.77 (sensitivity 50% and specificity 84%).[34]
Kidd et al. verified the diagnosis of stroke, and then assessed several neurologic parameters, including speech, muscle strength, and sensation. Pharyngeal sensation was assessed by touching each side of the pharyngeal wall and asking patients if they felt sensation that differed from each side. Patient report of abnormal sensation during this maneuver was 80% sensitive and 86% specific as a predictor of aspiration on VFSS.[42]
Steele et al. described the technique of dual axis accelerometry, where an accelerometer was placed at the midline of the neck over the cricoid cartilage during VFSS. The movement of the cricoid cartilage was captured for analysis in a computer algorithm to identify abnormal pharyngeal swallow behavior. Sensitivity was 100%, and specificity was 54%. Although the study was small (n=40), this novel method demonstrated good discrimination.[58]
DePippo et al. evaluated a 3‐oz water swallow in stroke patients. This protocol called for patients to drink the bolus of water without interruption, and be observed for 1 minute after for cough or wet‐hoarse voice. Presence of either sign was considered abnormal. Overall, sensitivity was 94% and specificity 30% looking for the presence of either sign.[43] Suiter and Leder used a similar protocol, with sensitivity of 97% and specificity of 49%.[49]
DISCUSSION
Our results show that most bedside swallow examinations lack the sensitivity to be used as a screening test for dysphagia across all patient populations examined. This is unfortunate as the ability to determine which patients require formal speech language pathology consultation or imaging as part of their diagnostic evaluation early in the hospital stay would lead to improved allocation of resources, cost reductions, and earlier implementation of effective therapy approaches. Furthermore, although radiation doses received during VFSS are not high when compared with other radiologic exams like computed tomography scans,[60] increasing awareness about the long‐term malignancy risks associated with medical imaging makes it desirable to reduce any test involving ionizing radiation.
There were several categories of screening procedures identified during this review process. Those classified as subjective bedside exams and protocolized multi‐item evaluations were found to have high heterogeneity in their sensitivity and specificity, though a few exam protocols did have a reasonable sensitivity and specificity.[21, 31, 52] The following individual exam maneuvers were found to demonstrate high sensitivity and an ability to exclude aspiration: a test for dysphonia through production of a sustained/a/34 and use of dual‐axis accelerometry.[16] Two other tests, the 3‐oz water swallow test[43] and testing of abnormal pharyngeal sensation,[42] were each found effective in a single study, with conflicting results from other studies.
Our results extend the findings from previous systematic reviews on this subject, most of which focused only on stroke patients.[5, 12, 61, 62] Martino and colleagues[5] conducted a review focused on screening for adults poststroke. From 13 identified articles, it was concluded that evidence to support inclusion or exclusion of screening was poor. Daniels et al. conducted a systematic review of swallowing screening tools specific to patients with acute or chronic stroke.[12] Based on 16 articles, the authors concluded that a combination of swallowing and nonswallowing features may be necessary for development of a valid screening tool. The generalizability of these reviews is limited given that all were conducted in patients poststroke, and therefore results and recommendations may not be generalizable to other patients.
Wilkinson et al.[62] conducted a recent systematic review that focused on screening techniques for inpatients 65 years or older that excluded patients with stroke or Parkinson's disease. The purpose of that review was to examine sensitivity and specificity of bedside screening tests as well as ability to accurately predict pneumonia. The authors concluded that existing evidence is not sufficient to recommend the use of bedside tests in a general older population.[62]
Specific screening tools identified by Martino and colleagues[5] to have good predictive value in detecting aspiration as a diagnostic marker of dysphagia were an abnormal test of pharyngeal sensation[42] and the 50‐mL water swallow test. Daniels et al. identified a water swallow test as an important component of a screen.[7] These results were consistent with those of this review in that the abnormal test of pharyngeal sensation[42] was identified for high levels of sensitivity. However, the 3‐oz water swallow test,[43, 49] rather than the 50‐mL water swallow test,[42] was identified in this review as the version of the water swallow test with the best predictive value in ruling out aspiration. Results of our review identified 2 additional individual items, dual‐axis accelerometry[16] and dysphonia,[34] that may be important to include in a comprehensive screening tool. In the absence of better tools, the 3 oz swallow test, properly executed, seems to be the best currently available tool validated in more than 1 study.
Several studies in the current review included an assessment of oral tongue movement that is not described thoroughly and varies between studies. Tongue movement as an individual item on a screening protocol was not found to yield high sensitivity or specificity. However, tongue movement or range of motion is only 1 aspect of oral tongue function; pressures produced by the tongue reflecting strength also may be important and warrant evaluation. Multiple studies have shown patients with dysphagia resulting from a variety of etiologies to produce lower than normal maximum isometric lingual pressures,[63, 64, 65, 66, 67, 68] or pressures produced when the tongue is pushed as hard as possible against the hard palate. Tongue strengthening protocols that result in higher maximum isometric lingual pressures have been shown to carry over to positive changes in swallow function.[69, 70, 71, 72, 73] Inclusion of tongue pressure measurement in a comprehensive screening tool may help to improve predictive capabilities.
We believe our results have implications for practicing clinicians, and serve as a call to action for development of an easy‐to‐perform, accurate tool for dysphagia screening. Future prospective studies should focus on practical tools that can be deployed at the bedside, and correlate the results with not only gold‐standard VFSS and FEES, but with clinical outcomes such as pneumonia and aspiration events leading to prolonged length of stay.
There were several limitations to this review. High levels of heterogeneity were reported in the screening tests present in the literature, precluding meaningful meta‐analysis. In addition, the majority of studies included were in poststroke adults, which limits the generalizability of results.
In conclusion, no screening protocol has been shown to provide adequate predictive value for presence of aspiration. Several individual exam maneuvers demonstrate high sensitivity; however, the most effective combination of screening protocol components is unknown. There is a need for future research focused on the development of a comprehensive screening tool that can be applied across patient populations for accurate detection of dysphagia as well as prediction of other adverse health outcomes, including pneumonia.
Acknowledgements
The authors thank Drs. Byun‐Mo Oh and Catrionia Steele for providing additional information in response to requests for unpublished information.
Disclosures: Nasia Safdar MD, is supported by a National Institutes of Health R03 GEMSSTAR award and a VA MERIT award. The authors report no conflicts of interest.
- , , , et al. Pathophysiology, relevance and natural history of oropharyngeal dysphagia among older people. Nestle Nutr Inst Workshop Ser. 2012;72:57–66.
- , , , Dysphagia in the elderly: preliminary evidence of prevalence, risk factors, and socioemotional effects. Ann Otol Rhinol Laryngol. 2007;116(11):858–865.
- , , Formal dysphagia screening protocols prevent pneumonia. Stroke. 2006;37(3):765.
- , , Swallow management in patients on an acute stroke pathway: quality is cost effective. Arch Phys Med Rehabil. 1995;76(12):1130–1133.
- , , Screening for oropharyngeal dysphagia in stroke: insufficient evidence for guidelines. Dysphagia. 2000;15(1):19–30.
- , , , et al. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013;44(3):870–947.
- , , , , , Aspiration in patients with acute stroke. Arch Phys Med Rehabil. 1998;79(1):14–19.
- , , , et al. Can bedside assessment reliably exclude aspiration following acute stroke? Age Ageing. 1998;27(2):99–106.
- , , , The combination of bedside swallowing assessment and oxygen saturation monitoring of swallowing in acute stroke: a safe and humane screening tool. Age Ageing. 2000;29(6):495–499.
- , , , Validation of a dysphagia screening tool in acute stroke patients. Am J Crit Care. 2010;19(4):357–364.
- , Screening for dysphagia and aspiration in acute stroke: a systematic review. Dysphagia. 2001;16(1):7–18.
- , , Valid items for screening dysphagia risk in patients with stroke: a systematic review. Stroke. 2012;43(3):892–897.
- , , , , Preferred Reporting Items for Systematic Reviews and Meta‐Analyses: The PRISMA Statement. Ann Intern Med. 2009;151(4):264–269, W64.
- , , Is the information about a test important? Applying the methods of evidence‐based medicine to the clinical examination of swallowing. J Commun Disord. 2004;37(5):437–450.
- MIDAS: Stata module for meta‐analytical integration of diagnostic test accuracy studies. Statistical Software Components. S456880, Boston College Department of Economics, 2009.
- , , Noninvasive detection of thin‐liquid aspiration using dual‐axis swallowing accelerometry. Dysphagia. 2013;28(1):105–112.
- , , , , Validation of the Japanese translation of the Swallowing Disturbance Questionnaire in Parkinson's disease patients. Qual Life Res. 2012;21(7):1299–1303.
- , , , , , Diagnostic accuracy of bedside swallow evaluation versus videofluoroscopy to assess dysphagia in individuals with tetraplegia. PM R. 2012;4(4):283–289.
- , et al. 723 Aspiration after Lung Transplantation: Incidence, Risk Factors, and Accuracy of the Bedside Swallow Evaluation. The J Heart Lung Transplant. 2012;31(4 suppl 1):S247–S248.
- , , , Cerny M. Development of the brief bedside dysphagia screening test in the Czech Republic. Nurs Health Sci. 2011;13(4):388–395.
- , , SWALLOW‐3D, a simple 2‐minute bedside screening test, detects dysphagia in acute stroke patients with high sensitivity when validated against video‐fluoroscopy. Stroke. 2011;42(3):e352.
- , , , , Bedside screening and subacute reassessment of post‐stroke dysphagia: a prospective study. Int J Stroke. 2010;5:200.
- , , , , , Detecting dysphagia in inclusion body myositis. J Neurol. 2009;256(12):2009–2013.
- , , , et al. Dysphagia bedside screening for acute‐stroke patients: the Gugging Swallowing Screen. Stroke. 2007;38(11):2948–2952.
- , , Can pulse oximetry or a bedside swallowing assessment be used to detect aspiration after stroke? Stroke. 2006;37(12):2984–2988.
- , , , , , Identification of a simple screening tool for dysphagia in patients with stroke using factor analysis of multiple dysphagia variables. J Rehabil Med. 2005;37(4):247–251.
- , , , Evaluating swallowing dysfunction using a 100‐ml water swallowing test. Dysphagia. 2004;19(1):43–47.
- , , , et al. Bronchial auscultation: an effective adjunct to speech and language therapy bedside assessment when detecting dysphagia and aspiration? Dysphagia. 2004;19(4):211–218.
- , , Prediction of laryngeal aspiration using voice analysis. Am J Phys Med Rehabil. 2004;83(10):753–757.
- , , , , Three tests for predicting aspiration without videofluorography. Dysphagia. 2003;18(2):126–134.
- , , , , Bedside clinical methods useful as screening test for aspiration in elderly patients with recent and previous strokes. Ann Acad Med Singapore. 2003;32(6):790–794.
- , , , The accuracy of the modified Evan's blue dye test in predicting aspiration. Laryngoscope. 2003;113(11):1969–1972.
- , Aspiration risk after acute stroke: comparison of clinical examination and fiberoptic endoscopic evaluation of swallowing. Dysphagia. 2002;17(3):214–218.
- , , Sensitivity and specificity of clinical/bedside examination signs for detecting aspiration in adults subsequent to stroke. J Commun Disord. 2001;34(1‐2):55–72.
- , , , et al. Accuracy of bedside clinical methods compared with fiberoptic endoscopic examination of swallowing (FEES) in determining the risk of aspiration in acute stroke patients. Dysphagia. 2001;16(1):1–6.
- , “Wet Voice” as a predictor of penetration and aspiration in oropharyngeal dysphagia. Dysphagia. 2000;15(2):84–88.
- , , A screening procedure for oropharyngeal dysphagia. Dysphagia. 1999;14(1):44–51.
- , , , Assessing the laryngeal cough reflex and the risk of developing pneumonia after stroke. Arch Phys Med Rehabil. 1999;80(2):150–154.
- , , , , , Predictive value of clinical indices in detecting aspiration in patients with neurological disorders. J Neurol Neurosurg Psychiatry. 1997;63(4):456–460.
- , , , Clinical assessment of swallowing and prediction of dysphagia severity. Am J Speech Lang Pathol. 1997;6(4):17–24.
- , Does pulse oximetry reliably detect aspiration in dysphagic stroke patients? Stroke. 1997;28(9):1773–1775.
- , , , Aspiration in acute stroke: a clinical study with videofluoroscopy. Q J Med. 1993;86(12):825–829.
- , , Validation of the 3‐oz water swallow test for aspiration following stroke. Arch Neurol. 1992;49(12):1259–1261.
- , , , Aspiration in rehabilitation patients: videofluoroscopy vs bedside clinical assessment. Arch Phys Med Rehabil. 1988;69(8):637–640.
- , , , Accuracy of clinical judgment of the chin‐down posture for dysphagia during the clinical/bedside assessment as corroborated by videofluoroscopy in adults with acute stroke. Dysphagia. 2009;24(4):423–433.
- , Swallowing disturbance questionnaire for detecting dysphagia. Laryngoscope. 2011;121(7):1383–1387.
- , , , , , . Using voluntary cough to detect penetration and aspiration during oropharyngeal swallowing in patients with Parkinson disease. Chest. 2010;138(6):1426–1431.
- , , , et al. Predicting aspiration in patients with ischemic stroke: comparison of clinical signs and aerodynamic measures of voluntary cough. Chest. 2009;135(3):769–777.
- , Clinical utility of the 3‐ounce water swallow test. Dysphagia. 2008;23(3):244–250.
- , , , et al. Screening test for silent aspiration at the bedside. Dysphagia. 2008;23(4):364–370.
- , , , , , Simple swallowing provocation test has limited applicability as a screening tool for detecting aspiration, silent aspiration, or penetration. Dysphagia. 2010;25(1):6–10.
- , , , A simple bedside stroke dysphagia screen, validated against videofluoroscopy, detects dysphagia and aspiration with high sensitivity. J Stroke Cerebrovasc Dis. 2014;23 (4):712–716.
- , , , Sensitivity and specificity of the Eating Assessment Tool and the Volume‐Viscosity Swallow Test for clinical evaluation of oropharyngeal dysphagia. Neurogastroenterol Motil. 2014;26(9):1256–1265.
- , , , Pulse oximetry does not reliably detect aspiration on videofluoroscopic swallowing study. Arch Phys Med Rehabil. 2005;86(4):730–734.
- , , , et al. Acoustic analysis of swallowing sounds: a new technique for assessing dysphagia. J Rehabil Med. 2009;41(8):639–645.
- , , Aspiration in bilateral stroke patients: a validation study. Neurology. 1993;43(2):430–433.
- , , , et al. The Toronto Bedside Swallowing Screening Test (TOR‐BSST): development and validation of a dysphagia screening tool for patients with stroke. Stroke. 2009;40(2):555–561.
- , , , et al. Exploration of the utility of a brief swallow screening protocol with comparison to concurrent videofluoroscopy. Can J Speech Lang Pathol Audiol. 2011;35(3):228–242.
- , , , et al. Formal dysphagia screening protocols prevent pneumonia. Stroke. 2005;36(9):1972–1976.
- , , , et al. Radiation exposure time during MBSS: influence of swallowing impairment severity, medical diagnosis, clinician experience, and standardized protocol use. Dysphagia. 2013;28(1):77–85.
- Detection of eating difficulties after stroke: a systematic review. Int Nurs Rev. 2006;53(2):143–149.
- , , Aspiration in older patients without stroke: A systematic review of bedside diagnostic tests and predictors of pneumonia. Eur Geriatr Med. 2012;3(3):145–152.
- , , A tongue force measurement system for the assessment of oral‐phase swallowing disorders. Arch Phys Med Rehabil. 1991;72(1):38–42.
- , , Strength, Endurance, and stability of the tongue and hand in Parkinson disease. J Speech Lang Hear Res. 2000;43(1):256–267.
- , , , et al. Effects of radiotherapy with or without chemotherapy on tongue strength and swallowing in patients with oral cancer. Head Neck. 2007;29(7):632–637.
- , , , , Tongue pressure against hard palate during swallowing in post‐stroke patients. Gerodontology. 2005;22(4):227–233.
- , Tongue measures in individuals with normal and impaired swallowing. Am J Speech Lang Pathol. 2007;16(2):148–156.
- , , , et al. Tongue strength as a predictor of functional outcomes and quality of life after tongue cancer surgery. Ann Otol Rhinol Laryngol. 2013;122(6):386–397.
- , , , Effects of two types of tongue strengthening exercises in young normals. Folia Phoniatr Logop. 2003;55(4):199–205.
- , , , , , The effects of lingual exercise on swallowing in older adults. J Am Geriatr Soc. S2005;53(9):1483–1489.
- , , , et al. The effects of lingual exercise in stroke patients with dysphagia. Arch Phys Med Rehabil. 2007;88(2):150–158.
- , , , , , Pretreatment swallowing exercises improve swallow function after chemoradiation. Laryngoscope. 2008;118(1):39–43.
- , , , Effects of directional exercise on lingual strength. J Speech Lang Hear Res. 2009;52(4):1034–1047.
- , , et al. Prediction of aspiration in patients with newly diagnosed untreated advanced head and neck cancer. Archives of Otolaryngology – Head 127(8):975–979.
- , , , et al. Pathophysiology, relevance and natural history of oropharyngeal dysphagia among older people. Nestle Nutr Inst Workshop Ser. 2012;72:57–66.
- , , , Dysphagia in the elderly: preliminary evidence of prevalence, risk factors, and socioemotional effects. Ann Otol Rhinol Laryngol. 2007;116(11):858–865.
- , , Formal dysphagia screening protocols prevent pneumonia. Stroke. 2006;37(3):765.
- , , Swallow management in patients on an acute stroke pathway: quality is cost effective. Arch Phys Med Rehabil. 1995;76(12):1130–1133.
- , , Screening for oropharyngeal dysphagia in stroke: insufficient evidence for guidelines. Dysphagia. 2000;15(1):19–30.
- , , , et al. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013;44(3):870–947.
- , , , , , Aspiration in patients with acute stroke. Arch Phys Med Rehabil. 1998;79(1):14–19.
- , , , et al. Can bedside assessment reliably exclude aspiration following acute stroke? Age Ageing. 1998;27(2):99–106.
- , , , The combination of bedside swallowing assessment and oxygen saturation monitoring of swallowing in acute stroke: a safe and humane screening tool. Age Ageing. 2000;29(6):495–499.
- , , , Validation of a dysphagia screening tool in acute stroke patients. Am J Crit Care. 2010;19(4):357–364.
- , Screening for dysphagia and aspiration in acute stroke: a systematic review. Dysphagia. 2001;16(1):7–18.
- , , Valid items for screening dysphagia risk in patients with stroke: a systematic review. Stroke. 2012;43(3):892–897.
- , , , , Preferred Reporting Items for Systematic Reviews and Meta‐Analyses: The PRISMA Statement. Ann Intern Med. 2009;151(4):264–269, W64.
- , , Is the information about a test important? Applying the methods of evidence‐based medicine to the clinical examination of swallowing. J Commun Disord. 2004;37(5):437–450.
- MIDAS: Stata module for meta‐analytical integration of diagnostic test accuracy studies. Statistical Software Components. S456880, Boston College Department of Economics, 2009.
- , , Noninvasive detection of thin‐liquid aspiration using dual‐axis swallowing accelerometry. Dysphagia. 2013;28(1):105–112.
- , , , , Validation of the Japanese translation of the Swallowing Disturbance Questionnaire in Parkinson's disease patients. Qual Life Res. 2012;21(7):1299–1303.
- , , , , , Diagnostic accuracy of bedside swallow evaluation versus videofluoroscopy to assess dysphagia in individuals with tetraplegia. PM R. 2012;4(4):283–289.
- , et al. 723 Aspiration after Lung Transplantation: Incidence, Risk Factors, and Accuracy of the Bedside Swallow Evaluation. The J Heart Lung Transplant. 2012;31(4 suppl 1):S247–S248.
- , , , Cerny M. Development of the brief bedside dysphagia screening test in the Czech Republic. Nurs Health Sci. 2011;13(4):388–395.
- , , SWALLOW‐3D, a simple 2‐minute bedside screening test, detects dysphagia in acute stroke patients with high sensitivity when validated against video‐fluoroscopy. Stroke. 2011;42(3):e352.
- , , , , Bedside screening and subacute reassessment of post‐stroke dysphagia: a prospective study. Int J Stroke. 2010;5:200.
- , , , , , Detecting dysphagia in inclusion body myositis. J Neurol. 2009;256(12):2009–2013.
- , , , et al. Dysphagia bedside screening for acute‐stroke patients: the Gugging Swallowing Screen. Stroke. 2007;38(11):2948–2952.
- , , Can pulse oximetry or a bedside swallowing assessment be used to detect aspiration after stroke? Stroke. 2006;37(12):2984–2988.
- , , , , , Identification of a simple screening tool for dysphagia in patients with stroke using factor analysis of multiple dysphagia variables. J Rehabil Med. 2005;37(4):247–251.
- , , , Evaluating swallowing dysfunction using a 100‐ml water swallowing test. Dysphagia. 2004;19(1):43–47.
- , , , et al. Bronchial auscultation: an effective adjunct to speech and language therapy bedside assessment when detecting dysphagia and aspiration? Dysphagia. 2004;19(4):211–218.
- , , Prediction of laryngeal aspiration using voice analysis. Am J Phys Med Rehabil. 2004;83(10):753–757.
- , , , , Three tests for predicting aspiration without videofluorography. Dysphagia. 2003;18(2):126–134.
- , , , , Bedside clinical methods useful as screening test for aspiration in elderly patients with recent and previous strokes. Ann Acad Med Singapore. 2003;32(6):790–794.
- , , , The accuracy of the modified Evan's blue dye test in predicting aspiration. Laryngoscope. 2003;113(11):1969–1972.
- , Aspiration risk after acute stroke: comparison of clinical examination and fiberoptic endoscopic evaluation of swallowing. Dysphagia. 2002;17(3):214–218.
- , , Sensitivity and specificity of clinical/bedside examination signs for detecting aspiration in adults subsequent to stroke. J Commun Disord. 2001;34(1‐2):55–72.
- , , , et al. Accuracy of bedside clinical methods compared with fiberoptic endoscopic examination of swallowing (FEES) in determining the risk of aspiration in acute stroke patients. Dysphagia. 2001;16(1):1–6.
- , “Wet Voice” as a predictor of penetration and aspiration in oropharyngeal dysphagia. Dysphagia. 2000;15(2):84–88.
- , , A screening procedure for oropharyngeal dysphagia. Dysphagia. 1999;14(1):44–51.
- , , , Assessing the laryngeal cough reflex and the risk of developing pneumonia after stroke. Arch Phys Med Rehabil. 1999;80(2):150–154.
- , , , , , Predictive value of clinical indices in detecting aspiration in patients with neurological disorders. J Neurol Neurosurg Psychiatry. 1997;63(4):456–460.
- , , , Clinical assessment of swallowing and prediction of dysphagia severity. Am J Speech Lang Pathol. 1997;6(4):17–24.
- , Does pulse oximetry reliably detect aspiration in dysphagic stroke patients? Stroke. 1997;28(9):1773–1775.
- , , , Aspiration in acute stroke: a clinical study with videofluoroscopy. Q J Med. 1993;86(12):825–829.
- , , Validation of the 3‐oz water swallow test for aspiration following stroke. Arch Neurol. 1992;49(12):1259–1261.
- , , , Aspiration in rehabilitation patients: videofluoroscopy vs bedside clinical assessment. Arch Phys Med Rehabil. 1988;69(8):637–640.
- , , , Accuracy of clinical judgment of the chin‐down posture for dysphagia during the clinical/bedside assessment as corroborated by videofluoroscopy in adults with acute stroke. Dysphagia. 2009;24(4):423–433.
- , Swallowing disturbance questionnaire for detecting dysphagia. Laryngoscope. 2011;121(7):1383–1387.
- , , , , , . Using voluntary cough to detect penetration and aspiration during oropharyngeal swallowing in patients with Parkinson disease. Chest. 2010;138(6):1426–1431.
- , , , et al. Predicting aspiration in patients with ischemic stroke: comparison of clinical signs and aerodynamic measures of voluntary cough. Chest. 2009;135(3):769–777.
- , Clinical utility of the 3‐ounce water swallow test. Dysphagia. 2008;23(3):244–250.
- , , , et al. Screening test for silent aspiration at the bedside. Dysphagia. 2008;23(4):364–370.
- , , , , , Simple swallowing provocation test has limited applicability as a screening tool for detecting aspiration, silent aspiration, or penetration. Dysphagia. 2010;25(1):6–10.
- , , , A simple bedside stroke dysphagia screen, validated against videofluoroscopy, detects dysphagia and aspiration with high sensitivity. J Stroke Cerebrovasc Dis. 2014;23 (4):712–716.
- , , , Sensitivity and specificity of the Eating Assessment Tool and the Volume‐Viscosity Swallow Test for clinical evaluation of oropharyngeal dysphagia. Neurogastroenterol Motil. 2014;26(9):1256–1265.
- , , , Pulse oximetry does not reliably detect aspiration on videofluoroscopic swallowing study. Arch Phys Med Rehabil. 2005;86(4):730–734.
- , , , et al. Acoustic analysis of swallowing sounds: a new technique for assessing dysphagia. J Rehabil Med. 2009;41(8):639–645.
- , , Aspiration in bilateral stroke patients: a validation study. Neurology. 1993;43(2):430–433.
- , , , et al. The Toronto Bedside Swallowing Screening Test (TOR‐BSST): development and validation of a dysphagia screening tool for patients with stroke. Stroke. 2009;40(2):555–561.
- , , , et al. Exploration of the utility of a brief swallow screening protocol with comparison to concurrent videofluoroscopy. Can J Speech Lang Pathol Audiol. 2011;35(3):228–242.
- , , , et al. Formal dysphagia screening protocols prevent pneumonia. Stroke. 2005;36(9):1972–1976.
- , , , et al. Radiation exposure time during MBSS: influence of swallowing impairment severity, medical diagnosis, clinician experience, and standardized protocol use. Dysphagia. 2013;28(1):77–85.
- Detection of eating difficulties after stroke: a systematic review. Int Nurs Rev. 2006;53(2):143–149.
- , , Aspiration in older patients without stroke: A systematic review of bedside diagnostic tests and predictors of pneumonia. Eur Geriatr Med. 2012;3(3):145–152.
- , , A tongue force measurement system for the assessment of oral‐phase swallowing disorders. Arch Phys Med Rehabil. 1991;72(1):38–42.
- , , Strength, Endurance, and stability of the tongue and hand in Parkinson disease. J Speech Lang Hear Res. 2000;43(1):256–267.
- , , , et al. Effects of radiotherapy with or without chemotherapy on tongue strength and swallowing in patients with oral cancer. Head Neck. 2007;29(7):632–637.
- , , , , Tongue pressure against hard palate during swallowing in post‐stroke patients. Gerodontology. 2005;22(4):227–233.
- , Tongue measures in individuals with normal and impaired swallowing. Am J Speech Lang Pathol. 2007;16(2):148–156.
- , , , et al. Tongue strength as a predictor of functional outcomes and quality of life after tongue cancer surgery. Ann Otol Rhinol Laryngol. 2013;122(6):386–397.
- , , , Effects of two types of tongue strengthening exercises in young normals. Folia Phoniatr Logop. 2003;55(4):199–205.
- , , , , , The effects of lingual exercise on swallowing in older adults. J Am Geriatr Soc. S2005;53(9):1483–1489.
- , , , et al. The effects of lingual exercise in stroke patients with dysphagia. Arch Phys Med Rehabil. 2007;88(2):150–158.
- , , , , , Pretreatment swallowing exercises improve swallow function after chemoradiation. Laryngoscope. 2008;118(1):39–43.
- , , , Effects of directional exercise on lingual strength. J Speech Lang Hear Res. 2009;52(4):1034–1047.
- , , et al. Prediction of aspiration in patients with newly diagnosed untreated advanced head and neck cancer. Archives of Otolaryngology – Head 127(8):975–979.
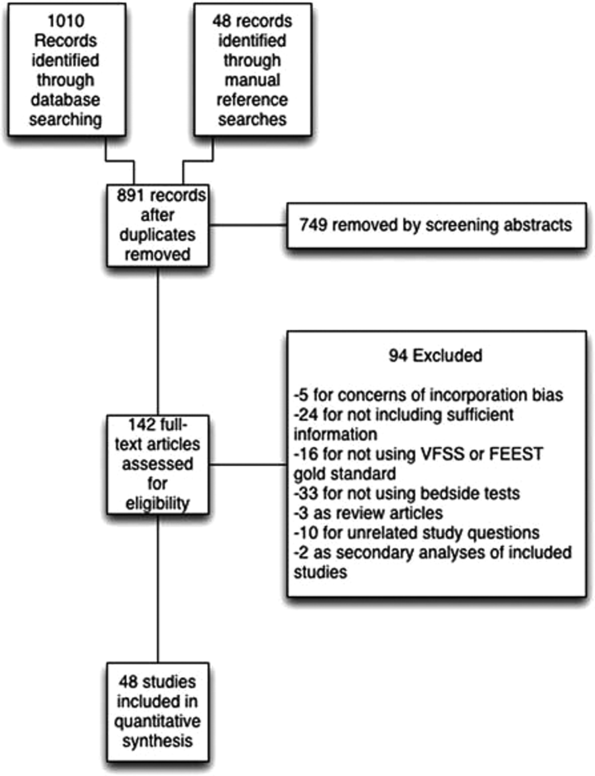
Biopsies don’t promote cancer spread, group finds
A study of more than 2000 patients refutes the idea that biopsies cause cancer to spread.
In a study published in Gut, researchers showed that patients who received a biopsy had better overall survival and similar cancer-free survival rates as patients who did not have a biopsy.
The team studied pancreatic cancer but said their findings likely apply to other cancers because the diagnostic technique used in this study—fine needle aspiration—is commonly used across tumor types.
“This study shows that physicians and patients should feel reassured that a biopsy is very safe,” said study author Michael Wallace, MD, of the Mayo Clinic in Jacksonville, Florida.
“We do millions of biopsies of cancer a year in the US, but one or two case studies have led to this common myth that biopsies spread cancer.”
This is the second study Dr Wallace and his team have conducted to examine the risk of biopsy.
In a 2013 study published in Endoscopy, the researchers examined outcomes in 256 pancreatic cancer patients treated at the Mayo Clinic in Jacksonville. The team found no difference in cancer recurrence between 208 patients who had ultrasound-guided fine needle aspiration (EUS-FNA) and the 48 patients who did not have a biopsy.
In the current study, the researchers examined 11 years (1998-2009) of Medicare data on patients with non-metastatic pancreatic cancer who underwent surgery. The team examined overall survival and pancreatic cancer-specific survival in 498 patients who had EUS-FNA and in 1536 patients who did not have a biopsy.
During a mean follow-up time of 21 months, 285 patients (57%) in the EUS-FNA group and 1167 patients (76%) in the non-EUS-FNA group died. Pancreatic cancer was identified as the cause of death for 251 patients (50%) in the EUS-FNA group and 980 patients (64%) in the non-EUS-FNA group.
The median overall survival in the EUS-FNA group was 22 months, compared to 15 months in the non-EUS-FNA group. Multivariate analysis showed that receipt of EUS-FNA had a borderline significant association with improved overall survival (hazard ratio=0.84, P=0.03).
The median cancer-specific survival was 24 months in the EUS-FNA group and 18 months in the non-EUS-FNA group. Multivariate analysis revealed no significant difference between the two groups (hazard ratio=0.87, P=0.12).
“[Biopsies provide] very valuable information that allow us to tailor treatment,” Dr Wallace noted. “In some cases, we can offer chemotherapy and radiation before surgery for a better outcome, and, in other cases, we can avoid surgery and other therapy altogether.” ![]()
A study of more than 2000 patients refutes the idea that biopsies cause cancer to spread.
In a study published in Gut, researchers showed that patients who received a biopsy had better overall survival and similar cancer-free survival rates as patients who did not have a biopsy.
The team studied pancreatic cancer but said their findings likely apply to other cancers because the diagnostic technique used in this study—fine needle aspiration—is commonly used across tumor types.
“This study shows that physicians and patients should feel reassured that a biopsy is very safe,” said study author Michael Wallace, MD, of the Mayo Clinic in Jacksonville, Florida.
“We do millions of biopsies of cancer a year in the US, but one or two case studies have led to this common myth that biopsies spread cancer.”
This is the second study Dr Wallace and his team have conducted to examine the risk of biopsy.
In a 2013 study published in Endoscopy, the researchers examined outcomes in 256 pancreatic cancer patients treated at the Mayo Clinic in Jacksonville. The team found no difference in cancer recurrence between 208 patients who had ultrasound-guided fine needle aspiration (EUS-FNA) and the 48 patients who did not have a biopsy.
In the current study, the researchers examined 11 years (1998-2009) of Medicare data on patients with non-metastatic pancreatic cancer who underwent surgery. The team examined overall survival and pancreatic cancer-specific survival in 498 patients who had EUS-FNA and in 1536 patients who did not have a biopsy.
During a mean follow-up time of 21 months, 285 patients (57%) in the EUS-FNA group and 1167 patients (76%) in the non-EUS-FNA group died. Pancreatic cancer was identified as the cause of death for 251 patients (50%) in the EUS-FNA group and 980 patients (64%) in the non-EUS-FNA group.
The median overall survival in the EUS-FNA group was 22 months, compared to 15 months in the non-EUS-FNA group. Multivariate analysis showed that receipt of EUS-FNA had a borderline significant association with improved overall survival (hazard ratio=0.84, P=0.03).
The median cancer-specific survival was 24 months in the EUS-FNA group and 18 months in the non-EUS-FNA group. Multivariate analysis revealed no significant difference between the two groups (hazard ratio=0.87, P=0.12).
“[Biopsies provide] very valuable information that allow us to tailor treatment,” Dr Wallace noted. “In some cases, we can offer chemotherapy and radiation before surgery for a better outcome, and, in other cases, we can avoid surgery and other therapy altogether.” ![]()
A study of more than 2000 patients refutes the idea that biopsies cause cancer to spread.
In a study published in Gut, researchers showed that patients who received a biopsy had better overall survival and similar cancer-free survival rates as patients who did not have a biopsy.
The team studied pancreatic cancer but said their findings likely apply to other cancers because the diagnostic technique used in this study—fine needle aspiration—is commonly used across tumor types.
“This study shows that physicians and patients should feel reassured that a biopsy is very safe,” said study author Michael Wallace, MD, of the Mayo Clinic in Jacksonville, Florida.
“We do millions of biopsies of cancer a year in the US, but one or two case studies have led to this common myth that biopsies spread cancer.”
This is the second study Dr Wallace and his team have conducted to examine the risk of biopsy.
In a 2013 study published in Endoscopy, the researchers examined outcomes in 256 pancreatic cancer patients treated at the Mayo Clinic in Jacksonville. The team found no difference in cancer recurrence between 208 patients who had ultrasound-guided fine needle aspiration (EUS-FNA) and the 48 patients who did not have a biopsy.
In the current study, the researchers examined 11 years (1998-2009) of Medicare data on patients with non-metastatic pancreatic cancer who underwent surgery. The team examined overall survival and pancreatic cancer-specific survival in 498 patients who had EUS-FNA and in 1536 patients who did not have a biopsy.
During a mean follow-up time of 21 months, 285 patients (57%) in the EUS-FNA group and 1167 patients (76%) in the non-EUS-FNA group died. Pancreatic cancer was identified as the cause of death for 251 patients (50%) in the EUS-FNA group and 980 patients (64%) in the non-EUS-FNA group.
The median overall survival in the EUS-FNA group was 22 months, compared to 15 months in the non-EUS-FNA group. Multivariate analysis showed that receipt of EUS-FNA had a borderline significant association with improved overall survival (hazard ratio=0.84, P=0.03).
The median cancer-specific survival was 24 months in the EUS-FNA group and 18 months in the non-EUS-FNA group. Multivariate analysis revealed no significant difference between the two groups (hazard ratio=0.87, P=0.12).
“[Biopsies provide] very valuable information that allow us to tailor treatment,” Dr Wallace noted. “In some cases, we can offer chemotherapy and radiation before surgery for a better outcome, and, in other cases, we can avoid surgery and other therapy altogether.” ![]()
Drug reverses anticoagulation activity of rivaroxaban

Credit: Kevin MacKenzie
An intravenous (IV) bolus of the factor Xa inhibitor antidote andexanet alfa can significantly and immediately reverse the steady-state anticoagulation activity of rivaroxaban in healthy subjects, according to initial results of the phase 3 ANNEXA-R study.
Portola Pharmaceuticals, the company developing andexanet alfa, recently announced these results from the first part of the study.
The company expects to present the full data set on March 16 at the American College of Cardiology’s 64th Annual Scientific Session & Expo in San Diego.
The second part of the ANNEXA-R study, in which researchers are evaluating a bolus plus a continuous infusion of andexanet alfa to sustain reversal, is ongoing.
Portola is developing andexanet alfa as a universal antidote for patients treated with oral and injectable factor Xa inhibitors who are experiencing a major bleeding episode or who require emergency surgery.
Andexanet alfa acts as a factor Xa decoy that targets and sequesters both oral and injectable factor Xa inhibitors in the blood. Once bound, the factor Xa inhibitors are unable to bind to and inhibit native factor Xa, thus allowing for the restoration of normal hemostatic processes.
ANNEXA-R details
This randomized, double-blind, placebo-controlled study is an evaluation of andexanet alfa in reversing rivaroxaban-induced anticoagulation in healthy volunteers ages 50 to 75 years.
In the first part of the study, 41 subjects received rivaroxaban at 20 mg once daily for 4 days. Then, they were randomized in a 2:1 ratio to receive either andexanet alfa administered as an 800 mg IV bolus (n=27) or to placebo (n=14).
Results showed that andexanet alfa significantly and immediately reversed the anticoagulation activity of rivaroxaban. Furthermore, andexanet alfa appeared to be well tolerated.
For the second part of the ANNEXA-R study, researchers plan to enroll about 40 healthy volunteers and give them rivaroxaban at 20 mg once daily for 4 days.
Then, subjects will be randomized to receive either placebo or andexanet alfa administered as an 800 mg IV bolus, followed by a continuous infusion of 8 mg/min for 120 minutes. Data from this part of the study are expected in mid-2015.
Andexanet alfa development
Andexanet alfa is the only compound being studied as a reversal agent for factor Xa inhibitors that directly and specifically corrects anti-factor Xa activity.
Portola is evaluating andexanet alfa in randomized, placebo-controlled phase 3 ANNEXA registration studies using pharmacodynamic endpoints agreed to with the US Food and Drug Administration (FDA), such as anti-factor Xa inhibitor units, to demonstrate efficacy.
Researchers recently reported statistically significant results from the first part of the phase 3 ANNEXA-A study, in which researchers evaluated andexanet alfa administered as a single IV bolus dose with the direct factor Xa inhibitor apixaban.
The second part of the study is ongoing. It’s an evaluation of an IV bolus plus a continuous infusion of andexanet alfa to sustain the reversal of anticoagulation activity.
“The statistically significant phase 3 ANNEXA-R study data, together with results presented previously with apixaban, provide compelling evidence that this ground-breaking agent could serve as a universal antidote for factor Xa inhibitor anticoagulants,” said John T. Curnutte, MD, PhD, executive vice president, research and development for Portola.
“Andexanet alfa is unique among the other reversal agents in development in that it has been the only agent to immediately and significantly reverse all of the key pharmacodynamic measurements of coagulation that have been agreed to with the FDA for accelerated approval. These include anti-factor Xa levels, thrombin generation, and unbound anticoagulant (free-fraction). This has been demonstrated with all of the factor Xa inhibitors studied to date—apixaban, rivaroxaban, edoxaban, and enoxaparin.” ![]()
Credit: Kevin MacKenzie
An intravenous (IV) bolus of the factor Xa inhibitor antidote andexanet alfa can significantly and immediately reverse the steady-state anticoagulation activity of rivaroxaban in healthy subjects, according to initial results of the phase 3 ANNEXA-R study.
Portola Pharmaceuticals, the company developing andexanet alfa, recently announced these results from the first part of the study.
The company expects to present the full data set on March 16 at the American College of Cardiology’s 64th Annual Scientific Session & Expo in San Diego.
The second part of the ANNEXA-R study, in which researchers are evaluating a bolus plus a continuous infusion of andexanet alfa to sustain reversal, is ongoing.
Portola is developing andexanet alfa as a universal antidote for patients treated with oral and injectable factor Xa inhibitors who are experiencing a major bleeding episode or who require emergency surgery.
Andexanet alfa acts as a factor Xa decoy that targets and sequesters both oral and injectable factor Xa inhibitors in the blood. Once bound, the factor Xa inhibitors are unable to bind to and inhibit native factor Xa, thus allowing for the restoration of normal hemostatic processes.
ANNEXA-R details
This randomized, double-blind, placebo-controlled study is an evaluation of andexanet alfa in reversing rivaroxaban-induced anticoagulation in healthy volunteers ages 50 to 75 years.
In the first part of the study, 41 subjects received rivaroxaban at 20 mg once daily for 4 days. Then, they were randomized in a 2:1 ratio to receive either andexanet alfa administered as an 800 mg IV bolus (n=27) or to placebo (n=14).
Results showed that andexanet alfa significantly and immediately reversed the anticoagulation activity of rivaroxaban. Furthermore, andexanet alfa appeared to be well tolerated.
For the second part of the ANNEXA-R study, researchers plan to enroll about 40 healthy volunteers and give them rivaroxaban at 20 mg once daily for 4 days.
Then, subjects will be randomized to receive either placebo or andexanet alfa administered as an 800 mg IV bolus, followed by a continuous infusion of 8 mg/min for 120 minutes. Data from this part of the study are expected in mid-2015.
Andexanet alfa development
Andexanet alfa is the only compound being studied as a reversal agent for factor Xa inhibitors that directly and specifically corrects anti-factor Xa activity.
Portola is evaluating andexanet alfa in randomized, placebo-controlled phase 3 ANNEXA registration studies using pharmacodynamic endpoints agreed to with the US Food and Drug Administration (FDA), such as anti-factor Xa inhibitor units, to demonstrate efficacy.
Researchers recently reported statistically significant results from the first part of the phase 3 ANNEXA-A study, in which researchers evaluated andexanet alfa administered as a single IV bolus dose with the direct factor Xa inhibitor apixaban.
The second part of the study is ongoing. It’s an evaluation of an IV bolus plus a continuous infusion of andexanet alfa to sustain the reversal of anticoagulation activity.
“The statistically significant phase 3 ANNEXA-R study data, together with results presented previously with apixaban, provide compelling evidence that this ground-breaking agent could serve as a universal antidote for factor Xa inhibitor anticoagulants,” said John T. Curnutte, MD, PhD, executive vice president, research and development for Portola.
“Andexanet alfa is unique among the other reversal agents in development in that it has been the only agent to immediately and significantly reverse all of the key pharmacodynamic measurements of coagulation that have been agreed to with the FDA for accelerated approval. These include anti-factor Xa levels, thrombin generation, and unbound anticoagulant (free-fraction). This has been demonstrated with all of the factor Xa inhibitors studied to date—apixaban, rivaroxaban, edoxaban, and enoxaparin.” ![]()
Credit: Kevin MacKenzie
An intravenous (IV) bolus of the factor Xa inhibitor antidote andexanet alfa can significantly and immediately reverse the steady-state anticoagulation activity of rivaroxaban in healthy subjects, according to initial results of the phase 3 ANNEXA-R study.
Portola Pharmaceuticals, the company developing andexanet alfa, recently announced these results from the first part of the study.
The company expects to present the full data set on March 16 at the American College of Cardiology’s 64th Annual Scientific Session & Expo in San Diego.
The second part of the ANNEXA-R study, in which researchers are evaluating a bolus plus a continuous infusion of andexanet alfa to sustain reversal, is ongoing.
Portola is developing andexanet alfa as a universal antidote for patients treated with oral and injectable factor Xa inhibitors who are experiencing a major bleeding episode or who require emergency surgery.
Andexanet alfa acts as a factor Xa decoy that targets and sequesters both oral and injectable factor Xa inhibitors in the blood. Once bound, the factor Xa inhibitors are unable to bind to and inhibit native factor Xa, thus allowing for the restoration of normal hemostatic processes.
ANNEXA-R details
This randomized, double-blind, placebo-controlled study is an evaluation of andexanet alfa in reversing rivaroxaban-induced anticoagulation in healthy volunteers ages 50 to 75 years.
In the first part of the study, 41 subjects received rivaroxaban at 20 mg once daily for 4 days. Then, they were randomized in a 2:1 ratio to receive either andexanet alfa administered as an 800 mg IV bolus (n=27) or to placebo (n=14).
Results showed that andexanet alfa significantly and immediately reversed the anticoagulation activity of rivaroxaban. Furthermore, andexanet alfa appeared to be well tolerated.
For the second part of the ANNEXA-R study, researchers plan to enroll about 40 healthy volunteers and give them rivaroxaban at 20 mg once daily for 4 days.
Then, subjects will be randomized to receive either placebo or andexanet alfa administered as an 800 mg IV bolus, followed by a continuous infusion of 8 mg/min for 120 minutes. Data from this part of the study are expected in mid-2015.
Andexanet alfa development
Andexanet alfa is the only compound being studied as a reversal agent for factor Xa inhibitors that directly and specifically corrects anti-factor Xa activity.
Portola is evaluating andexanet alfa in randomized, placebo-controlled phase 3 ANNEXA registration studies using pharmacodynamic endpoints agreed to with the US Food and Drug Administration (FDA), such as anti-factor Xa inhibitor units, to demonstrate efficacy.
Researchers recently reported statistically significant results from the first part of the phase 3 ANNEXA-A study, in which researchers evaluated andexanet alfa administered as a single IV bolus dose with the direct factor Xa inhibitor apixaban.
The second part of the study is ongoing. It’s an evaluation of an IV bolus plus a continuous infusion of andexanet alfa to sustain the reversal of anticoagulation activity.
“The statistically significant phase 3 ANNEXA-R study data, together with results presented previously with apixaban, provide compelling evidence that this ground-breaking agent could serve as a universal antidote for factor Xa inhibitor anticoagulants,” said John T. Curnutte, MD, PhD, executive vice president, research and development for Portola.
“Andexanet alfa is unique among the other reversal agents in development in that it has been the only agent to immediately and significantly reverse all of the key pharmacodynamic measurements of coagulation that have been agreed to with the FDA for accelerated approval. These include anti-factor Xa levels, thrombin generation, and unbound anticoagulant (free-fraction). This has been demonstrated with all of the factor Xa inhibitors studied to date—apixaban, rivaroxaban, edoxaban, and enoxaparin.” ![]()
Localized Argyria With Pseudo-ochronosis
Localized cutaneous argyria often presents as asymptomatic black or blue-gray pigmented macules in areas of the skin exposed to silver-containing compounds.1 Silver may enter the skin by traumatic implantation or absorption via eccrine sweat glands.2 Our patient witnessed a gun fight several years ago while on a mission trip and sustained multiple shrapnel wounds.
As in our patient, hyperpigmentation may appear years following initial exposure. Over time, incident light reduces colorless silver salts and compounds to black elemental silver.3 It also has been suggested that metallic silver granules stimulate tyrosine kinase activity, leading to locally increased melanin production.4 Together, these processes result in the clinical appearance of a blue-black macule. Despite its long-standing association with silver, this appearance also has been noted with deposition of other metals.5 Histologically, metal deposits can be seen as black granules surrounding eccrine glands, blood vessels, and elastic fibers on higher magnification.6 Granules also may be found in sebaceous glands and arrector pili muscle fibers. These findings do not distinguish from generalized argyria due to increased serum silver levels; however, some cases of localized cutaneous argyria have demonstrated spheroid black globules with surrounding collagen necrosis,1 which have not been reported with generalized disease. Localized cutaneous argyria also may be associated with ocher pigmentation of thickened collagen fibers, resembling changes typically found in alkaptonuria, an inherited deficiency of homogentisic acid oxidase (an enzyme involved in tyrosine metabolism).7 The resulting buildup of metabolic intermediates leads to ochronosis, a deposition of ocher-pigmented intermediates in connective tissue throughout the body. In the skin, ocher pigmentation occurs in elastic fibers of the reticular dermis.1 Grossly, these changes result in a blue-gray discoloration of the skin due to a light-scattering phenomenon known as the Tyndall effect. Exogenous ochronosis also can occur, most commonly from the topical application of hydroquinone or other skin-lightening compounds.1,5 Ocher pigmentation occurring in the setting of localized cutaneous argyria is referred to as pseudo-ochronosis, a finding first described by Robinson-Bostom et al.1 The etiology of this condition is poorly understood, but Robinson-Bostom et al1 noted the appearance of dark metal granules surrounding collagen bundles and hypothesized that metal aggregates surrounding collagen bundles in pseudo-ochronosis cause a homogenized appearance under light microscopy. Yellow-brown, swollen, homogenized collagen bundles can be visualized in the reticular dermis with surrounding deposition of metal granules (Figures 1 and 2).1 Typical patterns of granule deposition in localized argyria also are present.

A blue nevus is a collection of proliferating dermal melanocytes. Many histologic subtypes exist and there may be extensive variability in the extent of sclerosis, cellular architecture, and tissue cellularity between each variant.8 Blue nevi commonly present as blue-black hyperpigmentation in the dermis and subcutaneous tissue.9 Histologically, they are characterized by slender, bipolar, dendritic melanocytes in a sclerotic stroma (Figure 3).8 Melanocytes are highly pigmented and contain small monomorphic nuclei. Lesions are relatively homogenous and typically are restricted to the dermis with epidermal sparing.9 Dark granules and ocher fibers are absent.
Long-term use of hydroxychloroquine or other antimalarials may cause a macular pattern of blue-gray hyperpigmentation.10 Biopsy specimens typically reveal coarse, yellow-brown pigment granules primarily affecting the superficial dermis (Figure 4). Granules are found both extracellularly and within macrophages. Fontana-Masson silver staining may identify melanin, as hydroxychloroquine-melanin binding may contribute to patterns of hyperpigmentation.10 Hemosiderin often is present in cases of hydroxychloroquine pigmentation. Preceding ecchymosis appears to favor the deposition of hydroxychloroquine in the skin.11 The absence of dark metal granules helps distinguish hydroxychloroquine pigmentation from argyria.
Regressed melanomas may appear clinically as gray macules. These lesions arise in cases of malignant melanoma that spontaneously regress without treatment. Spontaneous regression occurs in 10% to 35% of cases depending on tumor subtype.12 Lesions can have a variable appearance based on the degree of regression. Partial regression is demonstrated by mixed melanosis and fibrosis in the dermis (Figure 5).13,14 Melanin is housed within melanophages present in a variably expanded papillary dermis. Tumors in early stages of regression can be surrounded by an inflammatory infiltrate, which becomes diminished at later stages. However, a few exceptional cases have been noted with extensive inflammatory infiltrate and no residual tumor.14 Completely regressed lesions typically appear as a band of dermal melanophages in the absence of inflammation or melanocytic atypia.15 The finding of regressed melanoma should prompt further investigation including sentinel lymph node biopsy, as it may be associated with metastasis.
Tattooing occurs following traumatic penetration of the skin with impregnation of pigmented foreign material into deep dermal layers.16 Histologic examination usually reveals clumps of fine particulate material in the dermis (Figure 6). The color of the pigment depends on the agent used. For example, graphite appears as black particles that may be confused with localized cutaneous argyria. Distinction can be made using elemental identification techniques such as energy-dispersive X-ray spectroscopy.1 The intensity of the pigment in granules found in tattoos or localized cutaneous argyria will fail to diminish with the application of melanin bleach.6
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol. 2002;46:222-227.
- Tajirian AL, Campbell RM, Robinson-Bostom L. Localized argyria after exposure to aerosolized solder. Cutis. 2006;78:305-308.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Shimizu I, Dill SW, McBean J, et al. Metal-induced granule deposition with pseudo-ochronosis. J Am Acad Dermatol. 2010;63:357-359.
- Rackoff EMJ, Benbenisty KM, Maize JC, et al. Localized cutaneous argyria from an acupuncture needle clini-cally concerning for metastatic melanoma. Cutis. 2007;80:423-426.
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:5-6.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Granter SR, McKee PH, Calonje E, et al. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a clinicopathologic study of 10 cases on the spectrum of so-called ‘malignant blue nevus.’ Am J Surg Pathol. 2001;25:316.
- Puri PK, Lountzis NI, Tyler W, et al. Hydroxychloroquine-induced hyperpigmentation: the staining pattern. J Cutan Pathol. 2008;35:1134-1137.
- Jallouli M, Francès C, Piette JC, et al. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus: a case-control study. JAMA Dermatol. 2013;149:935-940.
- Blessing K, McLaren KM. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology. 1992;20:315-322.
- LeBoit PE. Melanosis and its meanings. Am J Dermatopathol. 2002;24:369-372.
- Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
- Yang CH, Yeh JT, Shen SC, et al. Regressed subungual melanoma simulating cellular blue nevus: managed with sentinel lymph node biopsy. Dermatol Surg. 2006;32:577-581.
- Apfelberg DB, Manchester GH. Decorative and traumatic tattoo biophysics and removal. Clin Plast Surg. 1987;14:243-251.
Localized cutaneous argyria often presents as asymptomatic black or blue-gray pigmented macules in areas of the skin exposed to silver-containing compounds.1 Silver may enter the skin by traumatic implantation or absorption via eccrine sweat glands.2 Our patient witnessed a gun fight several years ago while on a mission trip and sustained multiple shrapnel wounds.
As in our patient, hyperpigmentation may appear years following initial exposure. Over time, incident light reduces colorless silver salts and compounds to black elemental silver.3 It also has been suggested that metallic silver granules stimulate tyrosine kinase activity, leading to locally increased melanin production.4 Together, these processes result in the clinical appearance of a blue-black macule. Despite its long-standing association with silver, this appearance also has been noted with deposition of other metals.5 Histologically, metal deposits can be seen as black granules surrounding eccrine glands, blood vessels, and elastic fibers on higher magnification.6 Granules also may be found in sebaceous glands and arrector pili muscle fibers. These findings do not distinguish from generalized argyria due to increased serum silver levels; however, some cases of localized cutaneous argyria have demonstrated spheroid black globules with surrounding collagen necrosis,1 which have not been reported with generalized disease. Localized cutaneous argyria also may be associated with ocher pigmentation of thickened collagen fibers, resembling changes typically found in alkaptonuria, an inherited deficiency of homogentisic acid oxidase (an enzyme involved in tyrosine metabolism).7 The resulting buildup of metabolic intermediates leads to ochronosis, a deposition of ocher-pigmented intermediates in connective tissue throughout the body. In the skin, ocher pigmentation occurs in elastic fibers of the reticular dermis.1 Grossly, these changes result in a blue-gray discoloration of the skin due to a light-scattering phenomenon known as the Tyndall effect. Exogenous ochronosis also can occur, most commonly from the topical application of hydroquinone or other skin-lightening compounds.1,5 Ocher pigmentation occurring in the setting of localized cutaneous argyria is referred to as pseudo-ochronosis, a finding first described by Robinson-Bostom et al.1 The etiology of this condition is poorly understood, but Robinson-Bostom et al1 noted the appearance of dark metal granules surrounding collagen bundles and hypothesized that metal aggregates surrounding collagen bundles in pseudo-ochronosis cause a homogenized appearance under light microscopy. Yellow-brown, swollen, homogenized collagen bundles can be visualized in the reticular dermis with surrounding deposition of metal granules (Figures 1 and 2).1 Typical patterns of granule deposition in localized argyria also are present.
A blue nevus is a collection of proliferating dermal melanocytes. Many histologic subtypes exist and there may be extensive variability in the extent of sclerosis, cellular architecture, and tissue cellularity between each variant.8 Blue nevi commonly present as blue-black hyperpigmentation in the dermis and subcutaneous tissue.9 Histologically, they are characterized by slender, bipolar, dendritic melanocytes in a sclerotic stroma (Figure 3).8 Melanocytes are highly pigmented and contain small monomorphic nuclei. Lesions are relatively homogenous and typically are restricted to the dermis with epidermal sparing.9 Dark granules and ocher fibers are absent.
Long-term use of hydroxychloroquine or other antimalarials may cause a macular pattern of blue-gray hyperpigmentation.10 Biopsy specimens typically reveal coarse, yellow-brown pigment granules primarily affecting the superficial dermis (Figure 4). Granules are found both extracellularly and within macrophages. Fontana-Masson silver staining may identify melanin, as hydroxychloroquine-melanin binding may contribute to patterns of hyperpigmentation.10 Hemosiderin often is present in cases of hydroxychloroquine pigmentation. Preceding ecchymosis appears to favor the deposition of hydroxychloroquine in the skin.11 The absence of dark metal granules helps distinguish hydroxychloroquine pigmentation from argyria.
Regressed melanomas may appear clinically as gray macules. These lesions arise in cases of malignant melanoma that spontaneously regress without treatment. Spontaneous regression occurs in 10% to 35% of cases depending on tumor subtype.12 Lesions can have a variable appearance based on the degree of regression. Partial regression is demonstrated by mixed melanosis and fibrosis in the dermis (Figure 5).13,14 Melanin is housed within melanophages present in a variably expanded papillary dermis. Tumors in early stages of regression can be surrounded by an inflammatory infiltrate, which becomes diminished at later stages. However, a few exceptional cases have been noted with extensive inflammatory infiltrate and no residual tumor.14 Completely regressed lesions typically appear as a band of dermal melanophages in the absence of inflammation or melanocytic atypia.15 The finding of regressed melanoma should prompt further investigation including sentinel lymph node biopsy, as it may be associated with metastasis.
Tattooing occurs following traumatic penetration of the skin with impregnation of pigmented foreign material into deep dermal layers.16 Histologic examination usually reveals clumps of fine particulate material in the dermis (Figure 6). The color of the pigment depends on the agent used. For example, graphite appears as black particles that may be confused with localized cutaneous argyria. Distinction can be made using elemental identification techniques such as energy-dispersive X-ray spectroscopy.1 The intensity of the pigment in granules found in tattoos or localized cutaneous argyria will fail to diminish with the application of melanin bleach.6
Localized cutaneous argyria often presents as asymptomatic black or blue-gray pigmented macules in areas of the skin exposed to silver-containing compounds.1 Silver may enter the skin by traumatic implantation or absorption via eccrine sweat glands.2 Our patient witnessed a gun fight several years ago while on a mission trip and sustained multiple shrapnel wounds.
As in our patient, hyperpigmentation may appear years following initial exposure. Over time, incident light reduces colorless silver salts and compounds to black elemental silver.3 It also has been suggested that metallic silver granules stimulate tyrosine kinase activity, leading to locally increased melanin production.4 Together, these processes result in the clinical appearance of a blue-black macule. Despite its long-standing association with silver, this appearance also has been noted with deposition of other metals.5 Histologically, metal deposits can be seen as black granules surrounding eccrine glands, blood vessels, and elastic fibers on higher magnification.6 Granules also may be found in sebaceous glands and arrector pili muscle fibers. These findings do not distinguish from generalized argyria due to increased serum silver levels; however, some cases of localized cutaneous argyria have demonstrated spheroid black globules with surrounding collagen necrosis,1 which have not been reported with generalized disease. Localized cutaneous argyria also may be associated with ocher pigmentation of thickened collagen fibers, resembling changes typically found in alkaptonuria, an inherited deficiency of homogentisic acid oxidase (an enzyme involved in tyrosine metabolism).7 The resulting buildup of metabolic intermediates leads to ochronosis, a deposition of ocher-pigmented intermediates in connective tissue throughout the body. In the skin, ocher pigmentation occurs in elastic fibers of the reticular dermis.1 Grossly, these changes result in a blue-gray discoloration of the skin due to a light-scattering phenomenon known as the Tyndall effect. Exogenous ochronosis also can occur, most commonly from the topical application of hydroquinone or other skin-lightening compounds.1,5 Ocher pigmentation occurring in the setting of localized cutaneous argyria is referred to as pseudo-ochronosis, a finding first described by Robinson-Bostom et al.1 The etiology of this condition is poorly understood, but Robinson-Bostom et al1 noted the appearance of dark metal granules surrounding collagen bundles and hypothesized that metal aggregates surrounding collagen bundles in pseudo-ochronosis cause a homogenized appearance under light microscopy. Yellow-brown, swollen, homogenized collagen bundles can be visualized in the reticular dermis with surrounding deposition of metal granules (Figures 1 and 2).1 Typical patterns of granule deposition in localized argyria also are present.
A blue nevus is a collection of proliferating dermal melanocytes. Many histologic subtypes exist and there may be extensive variability in the extent of sclerosis, cellular architecture, and tissue cellularity between each variant.8 Blue nevi commonly present as blue-black hyperpigmentation in the dermis and subcutaneous tissue.9 Histologically, they are characterized by slender, bipolar, dendritic melanocytes in a sclerotic stroma (Figure 3).8 Melanocytes are highly pigmented and contain small monomorphic nuclei. Lesions are relatively homogenous and typically are restricted to the dermis with epidermal sparing.9 Dark granules and ocher fibers are absent.
Long-term use of hydroxychloroquine or other antimalarials may cause a macular pattern of blue-gray hyperpigmentation.10 Biopsy specimens typically reveal coarse, yellow-brown pigment granules primarily affecting the superficial dermis (Figure 4). Granules are found both extracellularly and within macrophages. Fontana-Masson silver staining may identify melanin, as hydroxychloroquine-melanin binding may contribute to patterns of hyperpigmentation.10 Hemosiderin often is present in cases of hydroxychloroquine pigmentation. Preceding ecchymosis appears to favor the deposition of hydroxychloroquine in the skin.11 The absence of dark metal granules helps distinguish hydroxychloroquine pigmentation from argyria.
Regressed melanomas may appear clinically as gray macules. These lesions arise in cases of malignant melanoma that spontaneously regress without treatment. Spontaneous regression occurs in 10% to 35% of cases depending on tumor subtype.12 Lesions can have a variable appearance based on the degree of regression. Partial regression is demonstrated by mixed melanosis and fibrosis in the dermis (Figure 5).13,14 Melanin is housed within melanophages present in a variably expanded papillary dermis. Tumors in early stages of regression can be surrounded by an inflammatory infiltrate, which becomes diminished at later stages. However, a few exceptional cases have been noted with extensive inflammatory infiltrate and no residual tumor.14 Completely regressed lesions typically appear as a band of dermal melanophages in the absence of inflammation or melanocytic atypia.15 The finding of regressed melanoma should prompt further investigation including sentinel lymph node biopsy, as it may be associated with metastasis.
Tattooing occurs following traumatic penetration of the skin with impregnation of pigmented foreign material into deep dermal layers.16 Histologic examination usually reveals clumps of fine particulate material in the dermis (Figure 6). The color of the pigment depends on the agent used. For example, graphite appears as black particles that may be confused with localized cutaneous argyria. Distinction can be made using elemental identification techniques such as energy-dispersive X-ray spectroscopy.1 The intensity of the pigment in granules found in tattoos or localized cutaneous argyria will fail to diminish with the application of melanin bleach.6
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol. 2002;46:222-227.
- Tajirian AL, Campbell RM, Robinson-Bostom L. Localized argyria after exposure to aerosolized solder. Cutis. 2006;78:305-308.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Shimizu I, Dill SW, McBean J, et al. Metal-induced granule deposition with pseudo-ochronosis. J Am Acad Dermatol. 2010;63:357-359.
- Rackoff EMJ, Benbenisty KM, Maize JC, et al. Localized cutaneous argyria from an acupuncture needle clini-cally concerning for metastatic melanoma. Cutis. 2007;80:423-426.
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:5-6.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Granter SR, McKee PH, Calonje E, et al. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a clinicopathologic study of 10 cases on the spectrum of so-called ‘malignant blue nevus.’ Am J Surg Pathol. 2001;25:316.
- Puri PK, Lountzis NI, Tyler W, et al. Hydroxychloroquine-induced hyperpigmentation: the staining pattern. J Cutan Pathol. 2008;35:1134-1137.
- Jallouli M, Francès C, Piette JC, et al. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus: a case-control study. JAMA Dermatol. 2013;149:935-940.
- Blessing K, McLaren KM. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology. 1992;20:315-322.
- LeBoit PE. Melanosis and its meanings. Am J Dermatopathol. 2002;24:369-372.
- Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
- Yang CH, Yeh JT, Shen SC, et al. Regressed subungual melanoma simulating cellular blue nevus: managed with sentinel lymph node biopsy. Dermatol Surg. 2006;32:577-581.
- Apfelberg DB, Manchester GH. Decorative and traumatic tattoo biophysics and removal. Clin Plast Surg. 1987;14:243-251.
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol. 2002;46:222-227.
- Tajirian AL, Campbell RM, Robinson-Bostom L. Localized argyria after exposure to aerosolized solder. Cutis. 2006;78:305-308.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Shimizu I, Dill SW, McBean J, et al. Metal-induced granule deposition with pseudo-ochronosis. J Am Acad Dermatol. 2010;63:357-359.
- Rackoff EMJ, Benbenisty KM, Maize JC, et al. Localized cutaneous argyria from an acupuncture needle clini-cally concerning for metastatic melanoma. Cutis. 2007;80:423-426.
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:5-6.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Granter SR, McKee PH, Calonje E, et al. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a clinicopathologic study of 10 cases on the spectrum of so-called ‘malignant blue nevus.’ Am J Surg Pathol. 2001;25:316.
- Puri PK, Lountzis NI, Tyler W, et al. Hydroxychloroquine-induced hyperpigmentation: the staining pattern. J Cutan Pathol. 2008;35:1134-1137.
- Jallouli M, Francès C, Piette JC, et al. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus: a case-control study. JAMA Dermatol. 2013;149:935-940.
- Blessing K, McLaren KM. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology. 1992;20:315-322.
- LeBoit PE. Melanosis and its meanings. Am J Dermatopathol. 2002;24:369-372.
- Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
- Yang CH, Yeh JT, Shen SC, et al. Regressed subungual melanoma simulating cellular blue nevus: managed with sentinel lymph node biopsy. Dermatol Surg. 2006;32:577-581.
- Apfelberg DB, Manchester GH. Decorative and traumatic tattoo biophysics and removal. Clin Plast Surg. 1987;14:243-251.
Edoxaban approved for atrial fib, DVT, and PE indications
Edoxaban, a selective factor Xa-inhibitor, has been approved by the Food and Drug Administration for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, with a statement in the boxed warning that it should not be used in patients with normal renal function.
The warning reflects the results of a subgroup analysis in the pivotal trial, which found that the 60-mg dose was superior to warfarin in terms of reducing the stroke risk in mildly renally impaired patients, but was worse in patients with normal renal function. This was the main focus of a meeting of the FDA’s Cardiovascular and Renal Drugs Advisory Panel meeting in October, in which the panel voted 9-1 to recommend approval of edoxaban for this indication, but had mixed opinions on whether approval should be limited to patients with mild to moderate renal impairment.
The approved prescribing information recommends that a patient’s creatinine clearance should be checked before edoxaban is prescribed. “Patients with creatinine clearance greater than 95 mL/min have an increased risk of stroke, compared to similar patients given warfarin,” and should be treated with another anticoagulant, the FDA said in the Jan. 9 statement announcing the approval. The recommended dose for those with a creatinine clearance between 50 mL/min and 95 mL/min is 60 mg once a day; for those with a creatinine clearance of 15-50 mL/min, the recommended dose is 30 mg once a day, according to the prescribing information.
Edoxaban, the fourth novel oral anticoagulant drug approved by the FDA, will be marketed as Savaysa by Daiichi Sankyo. It was also approved to treat deep vein thrombosis and pulmonary embolism following 5-10 days of initial therapy with a parenteral anticoagulant. The recommended dose for this indication is 60 mg once a day. For patients with a creatinine clearance of 15-50 mL/min, or who weigh up to 60 kg (about 132 pounds), or who are taking “certain P-glycoprotein inhibitors,” the 30-mg/day dose is recommended.
Approval for the nonvalvular AF indication was based on ENGAGE AF-TIMI 48 (Effective Anticoagulation with Factor Xa Next Generation in Atrial Fibrillation) study, comparing once-daily edoxaban (60 mg and 30 mg) to warfarin in 21,015 patients with nonvalvular AF, at a moderate to high risk of thromboembolic events (N. Engl. J. Med. 2013;369:2093-104). Over a median of almost 3 years, both doses were noninferior to warfarin in the primary efficacy endpoint, the occurrence of first stroke or of a systemic embolic event. Overall, major bleeding events were significantly lower among those on the 60-mg and 30-mg doses, compared with those on warfarin. However, the rate of ischemic stroke was higher relative to warfarin in patients with a creatinine clearance over 95 mL/min.
About half of the edoxaban dose is eliminated by the kidneys, and patients with a creatinine clearance above 95 mL/min have lower plasma edoxaban levels, according to a statement in the clinical trials section of the prescribing information, which adds: “Given the clear relationship of dose and blood levels to effectiveness in the ENGAGE AF-TIMI 48 study, it could be anticipated that patients with better renal function would show a smaller effect of Savaysa, compared to warfarin than would patients with mildly impaired renal function, and this was in fact observed.”
Approval of the DVT and PE indication was based on the Hokusai-VTE study of about 8,200 people comparing edoxaban to warfarin, which found that the edoxaban 60 mg once a day was noninferior to warfarin in the rate of symptomatic venous thromboembolism (3.2% vs. 3.5% in those on warfarin). The rate of major or clinically relevant nonmajor bleeding events was 8.5% among those on edoxaban vs. 10.3% in those on warfarin (N. Engl. J. Med. 2013;369:1406-15).
Bleeding and anemia were the most common adverse events among patients with nonvalvular atrial fibrillation in clinical trials, and “as with other FDA-approved anticlotting drugs, bleeding, including life-threatening bleeding, is the most serious risk with Savaysa,” the FDA statement said. Among those treated for DVT and PE, the most common adverse events were bleeding, rash, abnormal liver function tests, and anemia.
Savaysa is the fourth novel oral anticoagulant to be cleared by the FDA, after dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis).
Serious adverse events associated with edoxaban should be reported to the FDA’s MedWatch program or at 800-332-1088.
A precedent does not come readily to mind where one restricts the use of a drug to patients with mild to moderately impaired renal function to optimize the benefit-risk balance. Typically, one avoids the drug or reduces the dose in such subgroups. For this reason, I think the drug is likely going to be a “nonstarter” for many clinicians.
During the FDA Advisory Committee panel meeting on edoxaban, the emphasis was on excluding patients with a creatinine clearance greater than 80 mL/min (representing about 37% of patients in the pivotal trial). The FDA approval used a cut-off of 95 mL/min (representing about 22% of patients in the pivotal trial). Even though the latter increases the eligible pool of patients for edoxaban, the major challenge is going to be clinical acceptability and marketability, especially given no unique advantage of this drug over other approved novel anticoagulants.
The CrCl cut off of 95mL/min applies to nonvalvular atrial fibrillation indication, and not to the venous thromboembolism (DVT/PE) indication. However, the latter indication requires 5-10 days of parenteral anticoagulant therapy, which puts it at a disadvantage compared with rivaroxaban or apixaban.
It is interesting to note that active pathological bleeding qualifies as a contraindication, but use in patients with normal renal function does not. There is a twofold increased risk of overall stroke or systemic embolism (primary endpoint) and ischemic stroke in patients with normal renal function (CrCl > 95 mL/min) along with an attenuation of the hemorrhagic risk advantage in this subgroup. Even though adjudicated major bleeding is still favorable for edoxaban in this subgroup, in my opinion the ischemic stroke risk outweighs the major bleeding advantage.
Sanjay Kaul, M.D., is professor of medicine at the University of California, Los Angeles. He was a member of the FDA’s Cardiovascular and Renal Drugs Advisory Committee that reviewed edoxaban at a meeting on Oct. 30, 2014.
A precedent does not come readily to mind where one restricts the use of a drug to patients with mild to moderately impaired renal function to optimize the benefit-risk balance. Typically, one avoids the drug or reduces the dose in such subgroups. For this reason, I think the drug is likely going to be a “nonstarter” for many clinicians.
During the FDA Advisory Committee panel meeting on edoxaban, the emphasis was on excluding patients with a creatinine clearance greater than 80 mL/min (representing about 37% of patients in the pivotal trial). The FDA approval used a cut-off of 95 mL/min (representing about 22% of patients in the pivotal trial). Even though the latter increases the eligible pool of patients for edoxaban, the major challenge is going to be clinical acceptability and marketability, especially given no unique advantage of this drug over other approved novel anticoagulants.
The CrCl cut off of 95mL/min applies to nonvalvular atrial fibrillation indication, and not to the venous thromboembolism (DVT/PE) indication. However, the latter indication requires 5-10 days of parenteral anticoagulant therapy, which puts it at a disadvantage compared with rivaroxaban or apixaban.
It is interesting to note that active pathological bleeding qualifies as a contraindication, but use in patients with normal renal function does not. There is a twofold increased risk of overall stroke or systemic embolism (primary endpoint) and ischemic stroke in patients with normal renal function (CrCl > 95 mL/min) along with an attenuation of the hemorrhagic risk advantage in this subgroup. Even though adjudicated major bleeding is still favorable for edoxaban in this subgroup, in my opinion the ischemic stroke risk outweighs the major bleeding advantage.
Sanjay Kaul, M.D., is professor of medicine at the University of California, Los Angeles. He was a member of the FDA’s Cardiovascular and Renal Drugs Advisory Committee that reviewed edoxaban at a meeting on Oct. 30, 2014.
A precedent does not come readily to mind where one restricts the use of a drug to patients with mild to moderately impaired renal function to optimize the benefit-risk balance. Typically, one avoids the drug or reduces the dose in such subgroups. For this reason, I think the drug is likely going to be a “nonstarter” for many clinicians.
During the FDA Advisory Committee panel meeting on edoxaban, the emphasis was on excluding patients with a creatinine clearance greater than 80 mL/min (representing about 37% of patients in the pivotal trial). The FDA approval used a cut-off of 95 mL/min (representing about 22% of patients in the pivotal trial). Even though the latter increases the eligible pool of patients for edoxaban, the major challenge is going to be clinical acceptability and marketability, especially given no unique advantage of this drug over other approved novel anticoagulants.
The CrCl cut off of 95mL/min applies to nonvalvular atrial fibrillation indication, and not to the venous thromboembolism (DVT/PE) indication. However, the latter indication requires 5-10 days of parenteral anticoagulant therapy, which puts it at a disadvantage compared with rivaroxaban or apixaban.
It is interesting to note that active pathological bleeding qualifies as a contraindication, but use in patients with normal renal function does not. There is a twofold increased risk of overall stroke or systemic embolism (primary endpoint) and ischemic stroke in patients with normal renal function (CrCl > 95 mL/min) along with an attenuation of the hemorrhagic risk advantage in this subgroup. Even though adjudicated major bleeding is still favorable for edoxaban in this subgroup, in my opinion the ischemic stroke risk outweighs the major bleeding advantage.
Sanjay Kaul, M.D., is professor of medicine at the University of California, Los Angeles. He was a member of the FDA’s Cardiovascular and Renal Drugs Advisory Committee that reviewed edoxaban at a meeting on Oct. 30, 2014.
Edoxaban, a selective factor Xa-inhibitor, has been approved by the Food and Drug Administration for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, with a statement in the boxed warning that it should not be used in patients with normal renal function.
The warning reflects the results of a subgroup analysis in the pivotal trial, which found that the 60-mg dose was superior to warfarin in terms of reducing the stroke risk in mildly renally impaired patients, but was worse in patients with normal renal function. This was the main focus of a meeting of the FDA’s Cardiovascular and Renal Drugs Advisory Panel meeting in October, in which the panel voted 9-1 to recommend approval of edoxaban for this indication, but had mixed opinions on whether approval should be limited to patients with mild to moderate renal impairment.
The approved prescribing information recommends that a patient’s creatinine clearance should be checked before edoxaban is prescribed. “Patients with creatinine clearance greater than 95 mL/min have an increased risk of stroke, compared to similar patients given warfarin,” and should be treated with another anticoagulant, the FDA said in the Jan. 9 statement announcing the approval. The recommended dose for those with a creatinine clearance between 50 mL/min and 95 mL/min is 60 mg once a day; for those with a creatinine clearance of 15-50 mL/min, the recommended dose is 30 mg once a day, according to the prescribing information.
Edoxaban, the fourth novel oral anticoagulant drug approved by the FDA, will be marketed as Savaysa by Daiichi Sankyo. It was also approved to treat deep vein thrombosis and pulmonary embolism following 5-10 days of initial therapy with a parenteral anticoagulant. The recommended dose for this indication is 60 mg once a day. For patients with a creatinine clearance of 15-50 mL/min, or who weigh up to 60 kg (about 132 pounds), or who are taking “certain P-glycoprotein inhibitors,” the 30-mg/day dose is recommended.
Approval for the nonvalvular AF indication was based on ENGAGE AF-TIMI 48 (Effective Anticoagulation with Factor Xa Next Generation in Atrial Fibrillation) study, comparing once-daily edoxaban (60 mg and 30 mg) to warfarin in 21,015 patients with nonvalvular AF, at a moderate to high risk of thromboembolic events (N. Engl. J. Med. 2013;369:2093-104). Over a median of almost 3 years, both doses were noninferior to warfarin in the primary efficacy endpoint, the occurrence of first stroke or of a systemic embolic event. Overall, major bleeding events were significantly lower among those on the 60-mg and 30-mg doses, compared with those on warfarin. However, the rate of ischemic stroke was higher relative to warfarin in patients with a creatinine clearance over 95 mL/min.
About half of the edoxaban dose is eliminated by the kidneys, and patients with a creatinine clearance above 95 mL/min have lower plasma edoxaban levels, according to a statement in the clinical trials section of the prescribing information, which adds: “Given the clear relationship of dose and blood levels to effectiveness in the ENGAGE AF-TIMI 48 study, it could be anticipated that patients with better renal function would show a smaller effect of Savaysa, compared to warfarin than would patients with mildly impaired renal function, and this was in fact observed.”
Approval of the DVT and PE indication was based on the Hokusai-VTE study of about 8,200 people comparing edoxaban to warfarin, which found that the edoxaban 60 mg once a day was noninferior to warfarin in the rate of symptomatic venous thromboembolism (3.2% vs. 3.5% in those on warfarin). The rate of major or clinically relevant nonmajor bleeding events was 8.5% among those on edoxaban vs. 10.3% in those on warfarin (N. Engl. J. Med. 2013;369:1406-15).
Bleeding and anemia were the most common adverse events among patients with nonvalvular atrial fibrillation in clinical trials, and “as with other FDA-approved anticlotting drugs, bleeding, including life-threatening bleeding, is the most serious risk with Savaysa,” the FDA statement said. Among those treated for DVT and PE, the most common adverse events were bleeding, rash, abnormal liver function tests, and anemia.
Savaysa is the fourth novel oral anticoagulant to be cleared by the FDA, after dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis).
Serious adverse events associated with edoxaban should be reported to the FDA’s MedWatch program or at 800-332-1088.
Edoxaban, a selective factor Xa-inhibitor, has been approved by the Food and Drug Administration for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, with a statement in the boxed warning that it should not be used in patients with normal renal function.
The warning reflects the results of a subgroup analysis in the pivotal trial, which found that the 60-mg dose was superior to warfarin in terms of reducing the stroke risk in mildly renally impaired patients, but was worse in patients with normal renal function. This was the main focus of a meeting of the FDA’s Cardiovascular and Renal Drugs Advisory Panel meeting in October, in which the panel voted 9-1 to recommend approval of edoxaban for this indication, but had mixed opinions on whether approval should be limited to patients with mild to moderate renal impairment.
The approved prescribing information recommends that a patient’s creatinine clearance should be checked before edoxaban is prescribed. “Patients with creatinine clearance greater than 95 mL/min have an increased risk of stroke, compared to similar patients given warfarin,” and should be treated with another anticoagulant, the FDA said in the Jan. 9 statement announcing the approval. The recommended dose for those with a creatinine clearance between 50 mL/min and 95 mL/min is 60 mg once a day; for those with a creatinine clearance of 15-50 mL/min, the recommended dose is 30 mg once a day, according to the prescribing information.
Edoxaban, the fourth novel oral anticoagulant drug approved by the FDA, will be marketed as Savaysa by Daiichi Sankyo. It was also approved to treat deep vein thrombosis and pulmonary embolism following 5-10 days of initial therapy with a parenteral anticoagulant. The recommended dose for this indication is 60 mg once a day. For patients with a creatinine clearance of 15-50 mL/min, or who weigh up to 60 kg (about 132 pounds), or who are taking “certain P-glycoprotein inhibitors,” the 30-mg/day dose is recommended.
Approval for the nonvalvular AF indication was based on ENGAGE AF-TIMI 48 (Effective Anticoagulation with Factor Xa Next Generation in Atrial Fibrillation) study, comparing once-daily edoxaban (60 mg and 30 mg) to warfarin in 21,015 patients with nonvalvular AF, at a moderate to high risk of thromboembolic events (N. Engl. J. Med. 2013;369:2093-104). Over a median of almost 3 years, both doses were noninferior to warfarin in the primary efficacy endpoint, the occurrence of first stroke or of a systemic embolic event. Overall, major bleeding events were significantly lower among those on the 60-mg and 30-mg doses, compared with those on warfarin. However, the rate of ischemic stroke was higher relative to warfarin in patients with a creatinine clearance over 95 mL/min.
About half of the edoxaban dose is eliminated by the kidneys, and patients with a creatinine clearance above 95 mL/min have lower plasma edoxaban levels, according to a statement in the clinical trials section of the prescribing information, which adds: “Given the clear relationship of dose and blood levels to effectiveness in the ENGAGE AF-TIMI 48 study, it could be anticipated that patients with better renal function would show a smaller effect of Savaysa, compared to warfarin than would patients with mildly impaired renal function, and this was in fact observed.”
Approval of the DVT and PE indication was based on the Hokusai-VTE study of about 8,200 people comparing edoxaban to warfarin, which found that the edoxaban 60 mg once a day was noninferior to warfarin in the rate of symptomatic venous thromboembolism (3.2% vs. 3.5% in those on warfarin). The rate of major or clinically relevant nonmajor bleeding events was 8.5% among those on edoxaban vs. 10.3% in those on warfarin (N. Engl. J. Med. 2013;369:1406-15).
Bleeding and anemia were the most common adverse events among patients with nonvalvular atrial fibrillation in clinical trials, and “as with other FDA-approved anticlotting drugs, bleeding, including life-threatening bleeding, is the most serious risk with Savaysa,” the FDA statement said. Among those treated for DVT and PE, the most common adverse events were bleeding, rash, abnormal liver function tests, and anemia.
Savaysa is the fourth novel oral anticoagulant to be cleared by the FDA, after dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis).
Serious adverse events associated with edoxaban should be reported to the FDA’s MedWatch program or at 800-332-1088.
Taking a look at neurologist burnout
There’s a lot in the news these days about doctor burnout. More specifically, neurologist burnout.
In a 2012 survey study, about 53% of neurologists reported burnout, which was third among all specialties surveyed, behind emergency medicine physicians and general internists. Neurologists also reported the fourth lowest job satisfaction with work-life balance, with about 41% satisfied that work leaves enough time for personal or family life. Neurology was the only one out of five specialties with the highest rates of burnout that was also among the five specialties with the lowest work-life balance.

Granted, the term “burnout” can mean a lot, but these days seems to refer to the fall of the American physician: Overworked, with rising costs, and falling reimbursements, sandwiched between patients who want to be cured immediately and those who want to sue us, and even on a good day facing a litany of terrible diseases.
Heck, I’d be burned out, too. Maybe I am.
Some say this is from the worries of solo practice, since we’re usually more pressed for time and money. I disagree, as I’ve seen it on both sides.
Recently, I saw my own internist. Six months ago she closed her own solo practice to join a large, hospital-owned group. She looked exhausted, worse than I’d ever seen her. She told me that she now gets a secure paycheck, but her stress level is worse. The hospital sets her schedule, tells her how much time she can spend with each patient, gives her quotas she has to meet, and has supplied an electronic health record (EHR) system that’s less than user friendly. (Personally, all of the ones I’ve tried are terrible.) When she goes home, she told me that now after dinner she still has to log on and do 2-3 more hours of charting just to catch up.
The grass is always greener. In her, I see a doctor who doesn’t have to watch each penny and worry about whether she’ll get a paycheck next week. In me, she looks at someone who’s free to pick their vacation days and isn’t chained to a quota system and a burdensome EHR.
Who’s right? I suppose it depends on what your life preferences are. Are we both burned out? We probably are, but in different ways.
But why the high rate of burnout for neurologists? Likely because of the issues I mentioned above. For myself, I’ve seen my salary drop 50% since its highest point in 2005. We’re faced with rising costs (like many other businesses). Unlike other professions, however, we don’t have much control over our reimbursement. Peculiar to medicine is the simple fact that what we charge has no bearing on what we get paid. Those rates are set by factors over which we have no control. Worse, they’re often set by politicians and insurance executives, who see us as the enemy.
There’s also the way reimbursements are set-up: they still favor docs who do a lot of procedures. While neurologists have a few, most of our job is thinking. And that’s not compensated nearly as well as jabbing needles and scalpels in people.
Then you get beyond financial issues. Many of us go through the day feeling like we have a target on our backs, in fear of patients becoming plaintiffs. What else? The nature of our field is such that we deal with diseases that are often challenging to diagnose and sometimes difficult, if not impossible, to treat. Yet, we still have to put on our best show and attitude for those afflicted. Part of why they come to us is to have questions answered and be given any glimmer of hope we can find.
In spite of this, the majority of us go on. Even burned out, we came here to help others. It’s part of what makes us tick and drives us to look in the mirror and head to the office. I wouldn’t trade what I do for anything. But I wish I could do it in a less adversarial world where I’m forced to choose between freedom and a (even temporary) sense of security.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
There’s a lot in the news these days about doctor burnout. More specifically, neurologist burnout.
In a 2012 survey study, about 53% of neurologists reported burnout, which was third among all specialties surveyed, behind emergency medicine physicians and general internists. Neurologists also reported the fourth lowest job satisfaction with work-life balance, with about 41% satisfied that work leaves enough time for personal or family life. Neurology was the only one out of five specialties with the highest rates of burnout that was also among the five specialties with the lowest work-life balance.
Granted, the term “burnout” can mean a lot, but these days seems to refer to the fall of the American physician: Overworked, with rising costs, and falling reimbursements, sandwiched between patients who want to be cured immediately and those who want to sue us, and even on a good day facing a litany of terrible diseases.
Heck, I’d be burned out, too. Maybe I am.
Some say this is from the worries of solo practice, since we’re usually more pressed for time and money. I disagree, as I’ve seen it on both sides.
Recently, I saw my own internist. Six months ago she closed her own solo practice to join a large, hospital-owned group. She looked exhausted, worse than I’d ever seen her. She told me that she now gets a secure paycheck, but her stress level is worse. The hospital sets her schedule, tells her how much time she can spend with each patient, gives her quotas she has to meet, and has supplied an electronic health record (EHR) system that’s less than user friendly. (Personally, all of the ones I’ve tried are terrible.) When she goes home, she told me that now after dinner she still has to log on and do 2-3 more hours of charting just to catch up.
The grass is always greener. In her, I see a doctor who doesn’t have to watch each penny and worry about whether she’ll get a paycheck next week. In me, she looks at someone who’s free to pick their vacation days and isn’t chained to a quota system and a burdensome EHR.
Who’s right? I suppose it depends on what your life preferences are. Are we both burned out? We probably are, but in different ways.
But why the high rate of burnout for neurologists? Likely because of the issues I mentioned above. For myself, I’ve seen my salary drop 50% since its highest point in 2005. We’re faced with rising costs (like many other businesses). Unlike other professions, however, we don’t have much control over our reimbursement. Peculiar to medicine is the simple fact that what we charge has no bearing on what we get paid. Those rates are set by factors over which we have no control. Worse, they’re often set by politicians and insurance executives, who see us as the enemy.
There’s also the way reimbursements are set-up: they still favor docs who do a lot of procedures. While neurologists have a few, most of our job is thinking. And that’s not compensated nearly as well as jabbing needles and scalpels in people.
Then you get beyond financial issues. Many of us go through the day feeling like we have a target on our backs, in fear of patients becoming plaintiffs. What else? The nature of our field is such that we deal with diseases that are often challenging to diagnose and sometimes difficult, if not impossible, to treat. Yet, we still have to put on our best show and attitude for those afflicted. Part of why they come to us is to have questions answered and be given any glimmer of hope we can find.
In spite of this, the majority of us go on. Even burned out, we came here to help others. It’s part of what makes us tick and drives us to look in the mirror and head to the office. I wouldn’t trade what I do for anything. But I wish I could do it in a less adversarial world where I’m forced to choose between freedom and a (even temporary) sense of security.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
There’s a lot in the news these days about doctor burnout. More specifically, neurologist burnout.
In a 2012 survey study, about 53% of neurologists reported burnout, which was third among all specialties surveyed, behind emergency medicine physicians and general internists. Neurologists also reported the fourth lowest job satisfaction with work-life balance, with about 41% satisfied that work leaves enough time for personal or family life. Neurology was the only one out of five specialties with the highest rates of burnout that was also among the five specialties with the lowest work-life balance.
Granted, the term “burnout” can mean a lot, but these days seems to refer to the fall of the American physician: Overworked, with rising costs, and falling reimbursements, sandwiched between patients who want to be cured immediately and those who want to sue us, and even on a good day facing a litany of terrible diseases.
Heck, I’d be burned out, too. Maybe I am.
Some say this is from the worries of solo practice, since we’re usually more pressed for time and money. I disagree, as I’ve seen it on both sides.
Recently, I saw my own internist. Six months ago she closed her own solo practice to join a large, hospital-owned group. She looked exhausted, worse than I’d ever seen her. She told me that she now gets a secure paycheck, but her stress level is worse. The hospital sets her schedule, tells her how much time she can spend with each patient, gives her quotas she has to meet, and has supplied an electronic health record (EHR) system that’s less than user friendly. (Personally, all of the ones I’ve tried are terrible.) When she goes home, she told me that now after dinner she still has to log on and do 2-3 more hours of charting just to catch up.
The grass is always greener. In her, I see a doctor who doesn’t have to watch each penny and worry about whether she’ll get a paycheck next week. In me, she looks at someone who’s free to pick their vacation days and isn’t chained to a quota system and a burdensome EHR.
Who’s right? I suppose it depends on what your life preferences are. Are we both burned out? We probably are, but in different ways.
But why the high rate of burnout for neurologists? Likely because of the issues I mentioned above. For myself, I’ve seen my salary drop 50% since its highest point in 2005. We’re faced with rising costs (like many other businesses). Unlike other professions, however, we don’t have much control over our reimbursement. Peculiar to medicine is the simple fact that what we charge has no bearing on what we get paid. Those rates are set by factors over which we have no control. Worse, they’re often set by politicians and insurance executives, who see us as the enemy.
There’s also the way reimbursements are set-up: they still favor docs who do a lot of procedures. While neurologists have a few, most of our job is thinking. And that’s not compensated nearly as well as jabbing needles and scalpels in people.
Then you get beyond financial issues. Many of us go through the day feeling like we have a target on our backs, in fear of patients becoming plaintiffs. What else? The nature of our field is such that we deal with diseases that are often challenging to diagnose and sometimes difficult, if not impossible, to treat. Yet, we still have to put on our best show and attitude for those afflicted. Part of why they come to us is to have questions answered and be given any glimmer of hope we can find.
In spite of this, the majority of us go on. Even burned out, we came here to help others. It’s part of what makes us tick and drives us to look in the mirror and head to the office. I wouldn’t trade what I do for anything. But I wish I could do it in a less adversarial world where I’m forced to choose between freedom and a (even temporary) sense of security.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.

Cutaneous Side Effects of Chemotherapy in Pediatric Oncology Patients
Pediatric oncology patients can present with various skin lesions related to both their primary disease and immunosuppressive treatments. In the majority of cases, cutaneous findings are associated with the use of chemotherapeutic agents. The toxic effects of chemotherapeutic agents, which generally are associated with treatment of solid organ malignancies (eg, liver, kidneys), can be detected by oncologists using clinical signs and laboratory tests.1-3 However, it also is important for dermatologists to recognize and evaluate cutaneous side effects associated with chemotherapeutic agents. Reports in the literature of cutaneous side effects of chemotherapy in pediatric patients generally are limited to case studies. This study aimed to evaluate the characteristics of cutaneous side effects of chemotherapy in pediatric oncology patients.
Materials and Methods
The study was performed through the collaboration of the departments of dermatology and venereology and pediatric oncology in the Faculty of Medicine at Ege University, Izmir, Turkey. Sixty-five pediatric oncology patients who were scheduled to undergo chemotherapy from May 2011 to May 2013 were included in the study. Clinical examination of dermatologic findings was conducted at baseline (prior to beginning chemotherapy) and at months 1, 3, and 6 of treatment. Patients were examined a total of 4 times during the study. Patients with a history of skin disease prior to diagnosis of their malignancy were excluded, as the study aimed to evaluate cutaneous side effects of chemotherapy. Patients who developed cutaneous side effects during the study period were photographed. Skin biopsy was performed to confirm clinical diagnosis. Patients were split into 5 groups according to oncological diagnoses, including hematological malignancies, solid organ tumors, bone and soft tissue tumors, central nervous system tumors, and Langerhans cell histiocytosis. Data regarding age, gender, treatments administered (ie, chemotherapeutics, antibiotics, antifungals, antivirals), and dermatologic signs were recorded. Mucocutaneous findings were classified as infectious (viral, bacterial, fungal) lesions, bullous lesions, inflammatory dermatoses (eg, diaper dermatitis, asteatotic eczema, contact dermatitis, seborrheic dermatitis), xeroderma, petechiae/ecchymoses, nail signs, alopecia, mucositis, cheilitis, oral aphthae, drug reactions confirmed by histopathology, cushingoid signs (eg, striae, acneform eruption, hypertrichosis), and cutaneous hyperpigmentation.
Statistical analysis was performed using SPSS version 15.0 and χ2 test was applied to the analysis.
Results
Of 65 patients, 62 completed the study and were included in the analysis. Three patients were excluded from the results, as 2 patients died during treatment and 1 patient withdrew from the study prior to completion. Twenty-seven (43.5%) patients were female and 35 (56.5%) were male ranging in age from 1 to 17 years (mean age, 8.14 years; median age [standard deviation], 7.25 [5.42] years). There were 31 (50%) patients in the hematological malignancies group, 11 (17.7%) in the solid organ tumors group, 10 (16.1%) in the bone and soft tissue tumors group, and 9 (14.5%) in the central nervous system tumors group; Langerhans cell histiocytosis was diagnosed in 1 (1.6%) patient. Hodgkin lymphoma made up 29.0% (n=9) of hematological malignancies. Other hematological malignancies included acute myeloblastic leukemia (n=7 [22.5%]), acute lymphoblastic leukemia (n=7 [22.5%]), T-cell lymphoma (n=5 [16.1%]), non-Hodgkin lym-phoma (n=1 [3.2%]), anaplastic giant cell lymphoma (n=1 [3.2%]), and diffuse giant cell lymphoma (n=1 [3.2%]).
In addition to chemotherapeutic agents, 7 (11.3%) patients in this study also received antibiotics and 3 (4.8%) received antivirals. The most frequently employed chemotherapeutic agents were vincristine, methotrexate, cytarabine, etoposide, and dexamethasone. Cyclophosphamide, doxorubicin, ifosfamide, asparaginase, carboplatin, procarbazine, daunorubicin, actinomycin D, vinblastine, cisplatin, bleomycin, idarubicin, 6-mercaptopurine, temozolamide, and cyclosporine also were administered. The most commonly encountered dermatological side effects were alopecia, xeroderma, inflammatory skin lesions, infectious lesions, and mucositis, respectively (Table 1). Cutaneous side effects were frequently seen at months 1 and 3 of treatment.
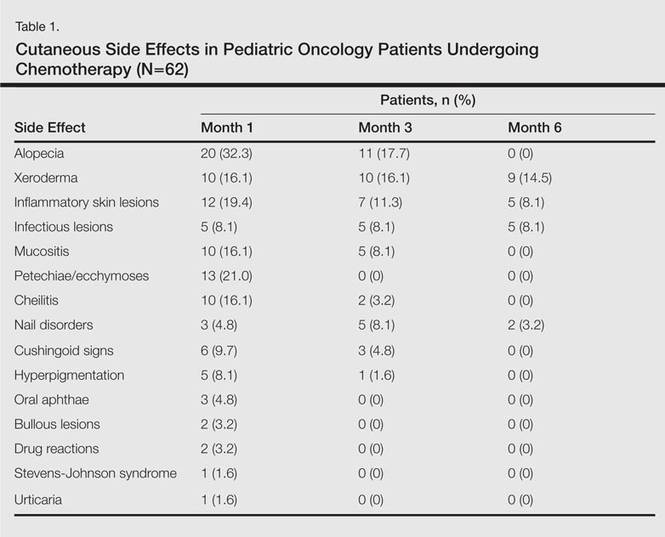
The most commonly encountered dermatologic side effect was alopecia (31/62 [50%]). Anagen effluvium (Figure 1) was detected in half of the cases, while complete scalp hair loss was noted in the rest. Alopecia was encountered more commonly in cases with central nervous system tumors (5/9 [55.6%]) and hematological malignancies (16/31 [51.6%])(Table 2).
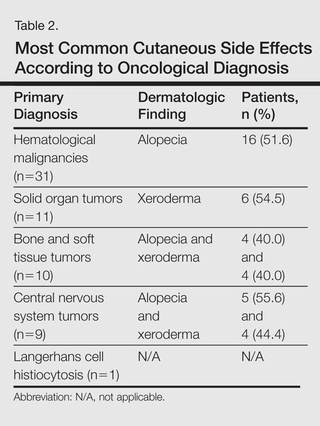
The second most commonly encountered side effect was xeroderma (29/62 [46.8%])(Figure 2). This side effect was most commonly encountered in patients with solid organ tumors (6/11 [54.5%]) and central nervous system tumors (4/9 [44.4%]), and occurred less frequently with bone and soft tissue tumors (4/10 [40.0%]).
Findings of eczema accounted for the majority of inflammatory lesions, which were the third most commonly encountered side effects. Among 24 cases of inflammatory skin lesions, 8 patients (33.3%) had diaper dermatitis, 7 (29.2%) had asteatotic eczema, 6 (25.0%) had contact dermatitis, and 3 (12.5%) had seborrheic dermatitis. Although inflammatory skin lesions were commonly encountered in patients with hematological malignancies (14/31 [45.2%]), the difference was not statistically significant.
Mucositis and oral aphthous lesions were observed in 15 (24.2%) and 3 (4.8%) patients, respectively. Nail signs were noted in 10 (16.1%) patients; 4 patients had transverse streaks on the nail plates, 3 had linear streaks, 2 had nail plate fragility, and 1 had increased pigmentation at the nail bed and periungual area. Figure 3 shows linear streaks on the nail plate. These side effects were most commonly encountered in patients with solid organ tumors (5/11 [45.5%]); however, the difference was not statistically significant when compared with the other diagnostic groups.
Dermatologic signs with infectious origins were detected in 15 (24.2%) patients; 2 patients had herpes labialis, 2 had verruca vulgaris, 3 had bacterial folliculitis, 1 had acute paronychia, 1 had soft tissue infection, 2 had tinea versicolor, and 4 had mucocutaneous candidiasis. Dermatologic side effects due to infectious causes were more commonly encountered in patients with bone and soft tissue tumors (4/11 [36.4%]), and the difference was statistically significant when compared with the other diagnostic groups (P=.04).
Petechiae and ecchymotic lesions were present in 13 (21.0%) patients. These side effects occurred mainly in the first month of chemotherapy, namely when patients were in the pancytopenic phase.
Comment
Variability among the oncological diagnosis and drugs used in treatment as well as increased numbers of chemotherapeutic agents available have led to many side effects and complications in pediatric oncology patients undergoing chemotherapy.1,2 Comprehensive studies regarding the cutaneous side effects of chemotherapeuticagents in cancer treatment have been conducted in adult patients. Side effects in pediatric patients have only been documented in case reports in the literature. In our study of pediatric oncology patients undergoing treatment with chemotherapy, the most commonly observed dermatologic side effect was alopecia, followed by xeroderma, inflammatory lesions, infectious lesions, mucositis, petechiae/ecchymoses, cheilitis, nail disorders, cushingoid signs, oral aphthae, bullous lesions, and drug reactions confirmed histopathologically (Table 1).
Because the common effects of chemotherapeutic agents used in cancer treatment are greatest in areas of rapidly dividing cells, the skin and skin appendages frequently are affected by these drugs.1-3 Cutaneous signs are frequently observed, especially in regions with increased mitotic activity such as the hair, mucosa, and nails.
Kamil et al1 reported that the incidence of alopecia was 64.3% (74/115) in a study of adult cancer patients who underwent chemotherapy. Chemotherapeutic agents that have commonly caused alopecia are vincristine, daunorubicin, doxorubicin, cyclophosphamide, etoposide, cytarabine, and carboplatin.1,2 In our study, alopecia was noted in 31 (50.0%) patients, especially with the use of vincristine (7/31 [22.6%]), daunorubicin (8/31 [25.8%]), doxorubicin (6/31 [19.4%]), and cyclophosphamide (10/31 [32.3%]).
Darkening of the skin and paleness accompanied the majority of cases of xeroderma in our study. Skin dryness was in an ichthyosiform appearance and was severe in 1 patient who was diagnosed with osteosarcoma. Asteatotic eczema and cheilitis were related to skin dryness. It has been reported that acquired paraneoplastic ichthyosis can develop in hematological malignancies, primarily in patients with Hodgkin lymphoma.4
The incidence of mucositis has been related to the doses of chemotherapeutic agents. Although it is a commonly encountered side effect, there is no standard treatment of mucositis; therefore, preventive care in patients undergoing chemotherapy is important. It has been reported that practicing good oral hygiene before the treatment period can decrease the incidence of mucositis.5-9 The lower incidence of mucositis in our study compared to the literature (55.6%)5 can be attributed to the lower doses of chemotherapy drugs administered to children due to their weights; they also had active oral mucosa care during chemotherapy.
Another common complication observed in our study was nail disorders. Transverse streaks commonly are encountered due to damage in the nail matrix. Other signs are increased linear streaks, longitudinal melanonychia, nail plate fragility, and onycholysis.10
Cancer patients acquire infections more frequently because of immunosuppression from chemotherapy and malignancy.11,12 In our study, cutaneous side effects with infectious causes were noted in 15 patients. Steroids, which are included in the majority of chemotherapeutic protocols, can cause cushingoid changes. Striae from rapid weight gain, acneform eruptions, hypertrichosis, and atrophy of the skin also have been observed among secondary changes to chemotherapy.1,11
Other skin signs observed in the study were acute urticaria in 1 patient (1.6%) following administration of intrathecal methotrexate; Stevens-Johnson syndrome related to voriconazole was noted in 1 (1.6%) patient.
Hyperpigmentation is a common side effect observed in oncology patients.13-15 It can be observed locally in the skin as well as the mucosa, teeth, hair, and nails, and it generally develops secondary to alkylating agents.16 Moreover, hyperpigmentation may develop in regions with occlusions (eg, electrocardiogram pads, adhesion sites of plasters), and commonly is associated with ifosfamide, etoposide, carboplatin, and cyclosporine. Although the development mechanism of hyperpigmentation related to chemotherapy drugs is not clearly known, it is thought to be due to direct toxicity, melanocyte stimulation, or postinflammatory changes.1,6,17 In our study, xeroderma was noted in some patients with hyperpigmentation; all of them had received cyclosporine and systemic steroid treatments. The other chemotherapeutics were defined as etoposide, cytarabine, dacarbazine, and ifosfamide.1 Our patients with hyperpigmentation were not taking these therapies.
Increased skin malignancies have been reported in adult cases with hematological malignancies.18 None of the patients in our study had a secondary skin malignancy, likely because we evaluated a pediatric population and the follow-up period (6 months) was too short for the development of a secondary malignancy.
Conclusion
A wide range of cutaneous side effects can be observed in pediatric oncology patients undergoing chemotherapy based on oncological diagnosis and treatment protocol. Although these side effects are not fatal, they may negatively affect morbidity and can lead to emotional distress. Knowing the possible cutaneous side effects of chemotherapy in pediatric patients and their causes is important for early diagnosis and minimal treatment.
- Kamil N, Kamil S, Ahmed SP, et al. Toxic effects of multiple anticancer drugs on skin. Pak J Pharm Sci. 2010;23:7-14.
- Alley E, Green R, Schuchter L. Cutaneous toxicities of cancer therapy. Curr Opin Oncol. 2002;14:212-216.
- Ozkan A, Apak H, Celkan T, et al. Toxic epidermal necrolysis after the use of high-dose cytosine arabinoside. Pediatr Dermatol. 2001;18:38-40.
- Rizos E, Milionis HJ, Pavlidis N, et al. Acquired ichthyosis: a paraneoplastic skin manifestation of Hodgkin’s disease. Lancet Oncol. 2002;3:727.
- Otmani N, Alami R, Hessissen L, et al. Determinants of severe oral mucositis in pediatric cancer patients: a prospective study. Int J Pediatr Dent. 2011;21:210-216.
- Mateus C, Robert C. New drugs in oncology and skin toxicity [in French]. Rev Med Interne. 2009;30:401-410.
- Manji A, Tomlinson D, Ethier MC, et al. Psychometric properties of the Oral Mucositis Daily Questionnaire for child self-report and importance of mucositis in children treated with chemotherapy. Support Care Cancer. 2012;20:1251-1258.
- Keefe DM. Mucositis management in patients with cancer. Support Cancer Ther. 2006;3:154-157.
- Raber-Durlacher JE, Elad S, Barasch A. Oral mucositis. Oral Oncol. 2010;46:452-456.
- Utas S, Kulluk P. A case of hydroxyurea-induced longitudinal melanonychia. Int J Dermatol. 2010;49:466-474.
- Ott H, Höger PH. Dermatologic manifestations of infections in pediatric cancer patients [in German]. Klin Padiatr. 2005;217(suppl 1):110-119.
- Ramphal R, Grant RM, Dzolganovski B, et al. Herpes simplex virus in febrile neutropenic children undergoing chemotherapy for cancer: a prospective cohort study. Pediatr Infect Dis J. 2007;26:700-704.
- Yaris N, Cakir M, Kalyoncu M, et al. Bleomycin induced hyperpigmentation with yolk sac tumor. Indian J Pediatr. 2007;74:505-506.
- Kleynberg RL, Sofi AA, Chaudhary RT. Hand-foot hyperpigmentation skin lesions associated with combination gemcitabine-carboplatin (GemCarbo) therapy. Am J Ther. 2011;18:261-263.
- Blaya M, Saba N. Chemotherapy-induced hyperpigmentation of the tongue. N Engl J Med. 2011;365:e20.
- Anandajeya WV, Corrêa ZM, Augsburger JJ. Primary acquired melanosis with atypia treated with mitomycin C. Int Ophthalmol. 2009;29:285-288.
- Torres C, Wong L, Welsh O, et al. Skin manifestations associated with chemotherapy in children with hematologic malignancies. Pediatr Dermatol. 2011;2:123-147.
- Mays SR, Cohen PR. Emerging dermatologic issues in the oncology patient. Semin Cutan Med Surg. 2006;25:179-189.
Pediatric oncology patients can present with various skin lesions related to both their primary disease and immunosuppressive treatments. In the majority of cases, cutaneous findings are associated with the use of chemotherapeutic agents. The toxic effects of chemotherapeutic agents, which generally are associated with treatment of solid organ malignancies (eg, liver, kidneys), can be detected by oncologists using clinical signs and laboratory tests.1-3 However, it also is important for dermatologists to recognize and evaluate cutaneous side effects associated with chemotherapeutic agents. Reports in the literature of cutaneous side effects of chemotherapy in pediatric patients generally are limited to case studies. This study aimed to evaluate the characteristics of cutaneous side effects of chemotherapy in pediatric oncology patients.
Materials and Methods
The study was performed through the collaboration of the departments of dermatology and venereology and pediatric oncology in the Faculty of Medicine at Ege University, Izmir, Turkey. Sixty-five pediatric oncology patients who were scheduled to undergo chemotherapy from May 2011 to May 2013 were included in the study. Clinical examination of dermatologic findings was conducted at baseline (prior to beginning chemotherapy) and at months 1, 3, and 6 of treatment. Patients were examined a total of 4 times during the study. Patients with a history of skin disease prior to diagnosis of their malignancy were excluded, as the study aimed to evaluate cutaneous side effects of chemotherapy. Patients who developed cutaneous side effects during the study period were photographed. Skin biopsy was performed to confirm clinical diagnosis. Patients were split into 5 groups according to oncological diagnoses, including hematological malignancies, solid organ tumors, bone and soft tissue tumors, central nervous system tumors, and Langerhans cell histiocytosis. Data regarding age, gender, treatments administered (ie, chemotherapeutics, antibiotics, antifungals, antivirals), and dermatologic signs were recorded. Mucocutaneous findings were classified as infectious (viral, bacterial, fungal) lesions, bullous lesions, inflammatory dermatoses (eg, diaper dermatitis, asteatotic eczema, contact dermatitis, seborrheic dermatitis), xeroderma, petechiae/ecchymoses, nail signs, alopecia, mucositis, cheilitis, oral aphthae, drug reactions confirmed by histopathology, cushingoid signs (eg, striae, acneform eruption, hypertrichosis), and cutaneous hyperpigmentation.
Statistical analysis was performed using SPSS version 15.0 and χ2 test was applied to the analysis.
Results
Of 65 patients, 62 completed the study and were included in the analysis. Three patients were excluded from the results, as 2 patients died during treatment and 1 patient withdrew from the study prior to completion. Twenty-seven (43.5%) patients were female and 35 (56.5%) were male ranging in age from 1 to 17 years (mean age, 8.14 years; median age [standard deviation], 7.25 [5.42] years). There were 31 (50%) patients in the hematological malignancies group, 11 (17.7%) in the solid organ tumors group, 10 (16.1%) in the bone and soft tissue tumors group, and 9 (14.5%) in the central nervous system tumors group; Langerhans cell histiocytosis was diagnosed in 1 (1.6%) patient. Hodgkin lymphoma made up 29.0% (n=9) of hematological malignancies. Other hematological malignancies included acute myeloblastic leukemia (n=7 [22.5%]), acute lymphoblastic leukemia (n=7 [22.5%]), T-cell lymphoma (n=5 [16.1%]), non-Hodgkin lym-phoma (n=1 [3.2%]), anaplastic giant cell lymphoma (n=1 [3.2%]), and diffuse giant cell lymphoma (n=1 [3.2%]).
In addition to chemotherapeutic agents, 7 (11.3%) patients in this study also received antibiotics and 3 (4.8%) received antivirals. The most frequently employed chemotherapeutic agents were vincristine, methotrexate, cytarabine, etoposide, and dexamethasone. Cyclophosphamide, doxorubicin, ifosfamide, asparaginase, carboplatin, procarbazine, daunorubicin, actinomycin D, vinblastine, cisplatin, bleomycin, idarubicin, 6-mercaptopurine, temozolamide, and cyclosporine also were administered. The most commonly encountered dermatological side effects were alopecia, xeroderma, inflammatory skin lesions, infectious lesions, and mucositis, respectively (Table 1). Cutaneous side effects were frequently seen at months 1 and 3 of treatment.
The most commonly encountered dermatologic side effect was alopecia (31/62 [50%]). Anagen effluvium (Figure 1) was detected in half of the cases, while complete scalp hair loss was noted in the rest. Alopecia was encountered more commonly in cases with central nervous system tumors (5/9 [55.6%]) and hematological malignancies (16/31 [51.6%])(Table 2).
The second most commonly encountered side effect was xeroderma (29/62 [46.8%])(Figure 2). This side effect was most commonly encountered in patients with solid organ tumors (6/11 [54.5%]) and central nervous system tumors (4/9 [44.4%]), and occurred less frequently with bone and soft tissue tumors (4/10 [40.0%]).
Findings of eczema accounted for the majority of inflammatory lesions, which were the third most commonly encountered side effects. Among 24 cases of inflammatory skin lesions, 8 patients (33.3%) had diaper dermatitis, 7 (29.2%) had asteatotic eczema, 6 (25.0%) had contact dermatitis, and 3 (12.5%) had seborrheic dermatitis. Although inflammatory skin lesions were commonly encountered in patients with hematological malignancies (14/31 [45.2%]), the difference was not statistically significant.
Mucositis and oral aphthous lesions were observed in 15 (24.2%) and 3 (4.8%) patients, respectively. Nail signs were noted in 10 (16.1%) patients; 4 patients had transverse streaks on the nail plates, 3 had linear streaks, 2 had nail plate fragility, and 1 had increased pigmentation at the nail bed and periungual area. Figure 3 shows linear streaks on the nail plate. These side effects were most commonly encountered in patients with solid organ tumors (5/11 [45.5%]); however, the difference was not statistically significant when compared with the other diagnostic groups.
Dermatologic signs with infectious origins were detected in 15 (24.2%) patients; 2 patients had herpes labialis, 2 had verruca vulgaris, 3 had bacterial folliculitis, 1 had acute paronychia, 1 had soft tissue infection, 2 had tinea versicolor, and 4 had mucocutaneous candidiasis. Dermatologic side effects due to infectious causes were more commonly encountered in patients with bone and soft tissue tumors (4/11 [36.4%]), and the difference was statistically significant when compared with the other diagnostic groups (P=.04).
Petechiae and ecchymotic lesions were present in 13 (21.0%) patients. These side effects occurred mainly in the first month of chemotherapy, namely when patients were in the pancytopenic phase.
Comment
Variability among the oncological diagnosis and drugs used in treatment as well as increased numbers of chemotherapeutic agents available have led to many side effects and complications in pediatric oncology patients undergoing chemotherapy.1,2 Comprehensive studies regarding the cutaneous side effects of chemotherapeuticagents in cancer treatment have been conducted in adult patients. Side effects in pediatric patients have only been documented in case reports in the literature. In our study of pediatric oncology patients undergoing treatment with chemotherapy, the most commonly observed dermatologic side effect was alopecia, followed by xeroderma, inflammatory lesions, infectious lesions, mucositis, petechiae/ecchymoses, cheilitis, nail disorders, cushingoid signs, oral aphthae, bullous lesions, and drug reactions confirmed histopathologically (Table 1).
Because the common effects of chemotherapeutic agents used in cancer treatment are greatest in areas of rapidly dividing cells, the skin and skin appendages frequently are affected by these drugs.1-3 Cutaneous signs are frequently observed, especially in regions with increased mitotic activity such as the hair, mucosa, and nails.
Kamil et al1 reported that the incidence of alopecia was 64.3% (74/115) in a study of adult cancer patients who underwent chemotherapy. Chemotherapeutic agents that have commonly caused alopecia are vincristine, daunorubicin, doxorubicin, cyclophosphamide, etoposide, cytarabine, and carboplatin.1,2 In our study, alopecia was noted in 31 (50.0%) patients, especially with the use of vincristine (7/31 [22.6%]), daunorubicin (8/31 [25.8%]), doxorubicin (6/31 [19.4%]), and cyclophosphamide (10/31 [32.3%]).
Darkening of the skin and paleness accompanied the majority of cases of xeroderma in our study. Skin dryness was in an ichthyosiform appearance and was severe in 1 patient who was diagnosed with osteosarcoma. Asteatotic eczema and cheilitis were related to skin dryness. It has been reported that acquired paraneoplastic ichthyosis can develop in hematological malignancies, primarily in patients with Hodgkin lymphoma.4
The incidence of mucositis has been related to the doses of chemotherapeutic agents. Although it is a commonly encountered side effect, there is no standard treatment of mucositis; therefore, preventive care in patients undergoing chemotherapy is important. It has been reported that practicing good oral hygiene before the treatment period can decrease the incidence of mucositis.5-9 The lower incidence of mucositis in our study compared to the literature (55.6%)5 can be attributed to the lower doses of chemotherapy drugs administered to children due to their weights; they also had active oral mucosa care during chemotherapy.
Another common complication observed in our study was nail disorders. Transverse streaks commonly are encountered due to damage in the nail matrix. Other signs are increased linear streaks, longitudinal melanonychia, nail plate fragility, and onycholysis.10
Cancer patients acquire infections more frequently because of immunosuppression from chemotherapy and malignancy.11,12 In our study, cutaneous side effects with infectious causes were noted in 15 patients. Steroids, which are included in the majority of chemotherapeutic protocols, can cause cushingoid changes. Striae from rapid weight gain, acneform eruptions, hypertrichosis, and atrophy of the skin also have been observed among secondary changes to chemotherapy.1,11
Other skin signs observed in the study were acute urticaria in 1 patient (1.6%) following administration of intrathecal methotrexate; Stevens-Johnson syndrome related to voriconazole was noted in 1 (1.6%) patient.
Hyperpigmentation is a common side effect observed in oncology patients.13-15 It can be observed locally in the skin as well as the mucosa, teeth, hair, and nails, and it generally develops secondary to alkylating agents.16 Moreover, hyperpigmentation may develop in regions with occlusions (eg, electrocardiogram pads, adhesion sites of plasters), and commonly is associated with ifosfamide, etoposide, carboplatin, and cyclosporine. Although the development mechanism of hyperpigmentation related to chemotherapy drugs is not clearly known, it is thought to be due to direct toxicity, melanocyte stimulation, or postinflammatory changes.1,6,17 In our study, xeroderma was noted in some patients with hyperpigmentation; all of them had received cyclosporine and systemic steroid treatments. The other chemotherapeutics were defined as etoposide, cytarabine, dacarbazine, and ifosfamide.1 Our patients with hyperpigmentation were not taking these therapies.
Increased skin malignancies have been reported in adult cases with hematological malignancies.18 None of the patients in our study had a secondary skin malignancy, likely because we evaluated a pediatric population and the follow-up period (6 months) was too short for the development of a secondary malignancy.
Conclusion
A wide range of cutaneous side effects can be observed in pediatric oncology patients undergoing chemotherapy based on oncological diagnosis and treatment protocol. Although these side effects are not fatal, they may negatively affect morbidity and can lead to emotional distress. Knowing the possible cutaneous side effects of chemotherapy in pediatric patients and their causes is important for early diagnosis and minimal treatment.
Pediatric oncology patients can present with various skin lesions related to both their primary disease and immunosuppressive treatments. In the majority of cases, cutaneous findings are associated with the use of chemotherapeutic agents. The toxic effects of chemotherapeutic agents, which generally are associated with treatment of solid organ malignancies (eg, liver, kidneys), can be detected by oncologists using clinical signs and laboratory tests.1-3 However, it also is important for dermatologists to recognize and evaluate cutaneous side effects associated with chemotherapeutic agents. Reports in the literature of cutaneous side effects of chemotherapy in pediatric patients generally are limited to case studies. This study aimed to evaluate the characteristics of cutaneous side effects of chemotherapy in pediatric oncology patients.
Materials and Methods
The study was performed through the collaboration of the departments of dermatology and venereology and pediatric oncology in the Faculty of Medicine at Ege University, Izmir, Turkey. Sixty-five pediatric oncology patients who were scheduled to undergo chemotherapy from May 2011 to May 2013 were included in the study. Clinical examination of dermatologic findings was conducted at baseline (prior to beginning chemotherapy) and at months 1, 3, and 6 of treatment. Patients were examined a total of 4 times during the study. Patients with a history of skin disease prior to diagnosis of their malignancy were excluded, as the study aimed to evaluate cutaneous side effects of chemotherapy. Patients who developed cutaneous side effects during the study period were photographed. Skin biopsy was performed to confirm clinical diagnosis. Patients were split into 5 groups according to oncological diagnoses, including hematological malignancies, solid organ tumors, bone and soft tissue tumors, central nervous system tumors, and Langerhans cell histiocytosis. Data regarding age, gender, treatments administered (ie, chemotherapeutics, antibiotics, antifungals, antivirals), and dermatologic signs were recorded. Mucocutaneous findings were classified as infectious (viral, bacterial, fungal) lesions, bullous lesions, inflammatory dermatoses (eg, diaper dermatitis, asteatotic eczema, contact dermatitis, seborrheic dermatitis), xeroderma, petechiae/ecchymoses, nail signs, alopecia, mucositis, cheilitis, oral aphthae, drug reactions confirmed by histopathology, cushingoid signs (eg, striae, acneform eruption, hypertrichosis), and cutaneous hyperpigmentation.
Statistical analysis was performed using SPSS version 15.0 and χ2 test was applied to the analysis.
Results
Of 65 patients, 62 completed the study and were included in the analysis. Three patients were excluded from the results, as 2 patients died during treatment and 1 patient withdrew from the study prior to completion. Twenty-seven (43.5%) patients were female and 35 (56.5%) were male ranging in age from 1 to 17 years (mean age, 8.14 years; median age [standard deviation], 7.25 [5.42] years). There were 31 (50%) patients in the hematological malignancies group, 11 (17.7%) in the solid organ tumors group, 10 (16.1%) in the bone and soft tissue tumors group, and 9 (14.5%) in the central nervous system tumors group; Langerhans cell histiocytosis was diagnosed in 1 (1.6%) patient. Hodgkin lymphoma made up 29.0% (n=9) of hematological malignancies. Other hematological malignancies included acute myeloblastic leukemia (n=7 [22.5%]), acute lymphoblastic leukemia (n=7 [22.5%]), T-cell lymphoma (n=5 [16.1%]), non-Hodgkin lym-phoma (n=1 [3.2%]), anaplastic giant cell lymphoma (n=1 [3.2%]), and diffuse giant cell lymphoma (n=1 [3.2%]).
In addition to chemotherapeutic agents, 7 (11.3%) patients in this study also received antibiotics and 3 (4.8%) received antivirals. The most frequently employed chemotherapeutic agents were vincristine, methotrexate, cytarabine, etoposide, and dexamethasone. Cyclophosphamide, doxorubicin, ifosfamide, asparaginase, carboplatin, procarbazine, daunorubicin, actinomycin D, vinblastine, cisplatin, bleomycin, idarubicin, 6-mercaptopurine, temozolamide, and cyclosporine also were administered. The most commonly encountered dermatological side effects were alopecia, xeroderma, inflammatory skin lesions, infectious lesions, and mucositis, respectively (Table 1). Cutaneous side effects were frequently seen at months 1 and 3 of treatment.
The most commonly encountered dermatologic side effect was alopecia (31/62 [50%]). Anagen effluvium (Figure 1) was detected in half of the cases, while complete scalp hair loss was noted in the rest. Alopecia was encountered more commonly in cases with central nervous system tumors (5/9 [55.6%]) and hematological malignancies (16/31 [51.6%])(Table 2).
The second most commonly encountered side effect was xeroderma (29/62 [46.8%])(Figure 2). This side effect was most commonly encountered in patients with solid organ tumors (6/11 [54.5%]) and central nervous system tumors (4/9 [44.4%]), and occurred less frequently with bone and soft tissue tumors (4/10 [40.0%]).
Findings of eczema accounted for the majority of inflammatory lesions, which were the third most commonly encountered side effects. Among 24 cases of inflammatory skin lesions, 8 patients (33.3%) had diaper dermatitis, 7 (29.2%) had asteatotic eczema, 6 (25.0%) had contact dermatitis, and 3 (12.5%) had seborrheic dermatitis. Although inflammatory skin lesions were commonly encountered in patients with hematological malignancies (14/31 [45.2%]), the difference was not statistically significant.
Mucositis and oral aphthous lesions were observed in 15 (24.2%) and 3 (4.8%) patients, respectively. Nail signs were noted in 10 (16.1%) patients; 4 patients had transverse streaks on the nail plates, 3 had linear streaks, 2 had nail plate fragility, and 1 had increased pigmentation at the nail bed and periungual area. Figure 3 shows linear streaks on the nail plate. These side effects were most commonly encountered in patients with solid organ tumors (5/11 [45.5%]); however, the difference was not statistically significant when compared with the other diagnostic groups.
Dermatologic signs with infectious origins were detected in 15 (24.2%) patients; 2 patients had herpes labialis, 2 had verruca vulgaris, 3 had bacterial folliculitis, 1 had acute paronychia, 1 had soft tissue infection, 2 had tinea versicolor, and 4 had mucocutaneous candidiasis. Dermatologic side effects due to infectious causes were more commonly encountered in patients with bone and soft tissue tumors (4/11 [36.4%]), and the difference was statistically significant when compared with the other diagnostic groups (P=.04).
Petechiae and ecchymotic lesions were present in 13 (21.0%) patients. These side effects occurred mainly in the first month of chemotherapy, namely when patients were in the pancytopenic phase.
Comment
Variability among the oncological diagnosis and drugs used in treatment as well as increased numbers of chemotherapeutic agents available have led to many side effects and complications in pediatric oncology patients undergoing chemotherapy.1,2 Comprehensive studies regarding the cutaneous side effects of chemotherapeuticagents in cancer treatment have been conducted in adult patients. Side effects in pediatric patients have only been documented in case reports in the literature. In our study of pediatric oncology patients undergoing treatment with chemotherapy, the most commonly observed dermatologic side effect was alopecia, followed by xeroderma, inflammatory lesions, infectious lesions, mucositis, petechiae/ecchymoses, cheilitis, nail disorders, cushingoid signs, oral aphthae, bullous lesions, and drug reactions confirmed histopathologically (Table 1).
Because the common effects of chemotherapeutic agents used in cancer treatment are greatest in areas of rapidly dividing cells, the skin and skin appendages frequently are affected by these drugs.1-3 Cutaneous signs are frequently observed, especially in regions with increased mitotic activity such as the hair, mucosa, and nails.
Kamil et al1 reported that the incidence of alopecia was 64.3% (74/115) in a study of adult cancer patients who underwent chemotherapy. Chemotherapeutic agents that have commonly caused alopecia are vincristine, daunorubicin, doxorubicin, cyclophosphamide, etoposide, cytarabine, and carboplatin.1,2 In our study, alopecia was noted in 31 (50.0%) patients, especially with the use of vincristine (7/31 [22.6%]), daunorubicin (8/31 [25.8%]), doxorubicin (6/31 [19.4%]), and cyclophosphamide (10/31 [32.3%]).
Darkening of the skin and paleness accompanied the majority of cases of xeroderma in our study. Skin dryness was in an ichthyosiform appearance and was severe in 1 patient who was diagnosed with osteosarcoma. Asteatotic eczema and cheilitis were related to skin dryness. It has been reported that acquired paraneoplastic ichthyosis can develop in hematological malignancies, primarily in patients with Hodgkin lymphoma.4
The incidence of mucositis has been related to the doses of chemotherapeutic agents. Although it is a commonly encountered side effect, there is no standard treatment of mucositis; therefore, preventive care in patients undergoing chemotherapy is important. It has been reported that practicing good oral hygiene before the treatment period can decrease the incidence of mucositis.5-9 The lower incidence of mucositis in our study compared to the literature (55.6%)5 can be attributed to the lower doses of chemotherapy drugs administered to children due to their weights; they also had active oral mucosa care during chemotherapy.
Another common complication observed in our study was nail disorders. Transverse streaks commonly are encountered due to damage in the nail matrix. Other signs are increased linear streaks, longitudinal melanonychia, nail plate fragility, and onycholysis.10
Cancer patients acquire infections more frequently because of immunosuppression from chemotherapy and malignancy.11,12 In our study, cutaneous side effects with infectious causes were noted in 15 patients. Steroids, which are included in the majority of chemotherapeutic protocols, can cause cushingoid changes. Striae from rapid weight gain, acneform eruptions, hypertrichosis, and atrophy of the skin also have been observed among secondary changes to chemotherapy.1,11
Other skin signs observed in the study were acute urticaria in 1 patient (1.6%) following administration of intrathecal methotrexate; Stevens-Johnson syndrome related to voriconazole was noted in 1 (1.6%) patient.
Hyperpigmentation is a common side effect observed in oncology patients.13-15 It can be observed locally in the skin as well as the mucosa, teeth, hair, and nails, and it generally develops secondary to alkylating agents.16 Moreover, hyperpigmentation may develop in regions with occlusions (eg, electrocardiogram pads, adhesion sites of plasters), and commonly is associated with ifosfamide, etoposide, carboplatin, and cyclosporine. Although the development mechanism of hyperpigmentation related to chemotherapy drugs is not clearly known, it is thought to be due to direct toxicity, melanocyte stimulation, or postinflammatory changes.1,6,17 In our study, xeroderma was noted in some patients with hyperpigmentation; all of them had received cyclosporine and systemic steroid treatments. The other chemotherapeutics were defined as etoposide, cytarabine, dacarbazine, and ifosfamide.1 Our patients with hyperpigmentation were not taking these therapies.
Increased skin malignancies have been reported in adult cases with hematological malignancies.18 None of the patients in our study had a secondary skin malignancy, likely because we evaluated a pediatric population and the follow-up period (6 months) was too short for the development of a secondary malignancy.
Conclusion
A wide range of cutaneous side effects can be observed in pediatric oncology patients undergoing chemotherapy based on oncological diagnosis and treatment protocol. Although these side effects are not fatal, they may negatively affect morbidity and can lead to emotional distress. Knowing the possible cutaneous side effects of chemotherapy in pediatric patients and their causes is important for early diagnosis and minimal treatment.
- Kamil N, Kamil S, Ahmed SP, et al. Toxic effects of multiple anticancer drugs on skin. Pak J Pharm Sci. 2010;23:7-14.
- Alley E, Green R, Schuchter L. Cutaneous toxicities of cancer therapy. Curr Opin Oncol. 2002;14:212-216.
- Ozkan A, Apak H, Celkan T, et al. Toxic epidermal necrolysis after the use of high-dose cytosine arabinoside. Pediatr Dermatol. 2001;18:38-40.
- Rizos E, Milionis HJ, Pavlidis N, et al. Acquired ichthyosis: a paraneoplastic skin manifestation of Hodgkin’s disease. Lancet Oncol. 2002;3:727.
- Otmani N, Alami R, Hessissen L, et al. Determinants of severe oral mucositis in pediatric cancer patients: a prospective study. Int J Pediatr Dent. 2011;21:210-216.
- Mateus C, Robert C. New drugs in oncology and skin toxicity [in French]. Rev Med Interne. 2009;30:401-410.
- Manji A, Tomlinson D, Ethier MC, et al. Psychometric properties of the Oral Mucositis Daily Questionnaire for child self-report and importance of mucositis in children treated with chemotherapy. Support Care Cancer. 2012;20:1251-1258.
- Keefe DM. Mucositis management in patients with cancer. Support Cancer Ther. 2006;3:154-157.
- Raber-Durlacher JE, Elad S, Barasch A. Oral mucositis. Oral Oncol. 2010;46:452-456.
- Utas S, Kulluk P. A case of hydroxyurea-induced longitudinal melanonychia. Int J Dermatol. 2010;49:466-474.
- Ott H, Höger PH. Dermatologic manifestations of infections in pediatric cancer patients [in German]. Klin Padiatr. 2005;217(suppl 1):110-119.
- Ramphal R, Grant RM, Dzolganovski B, et al. Herpes simplex virus in febrile neutropenic children undergoing chemotherapy for cancer: a prospective cohort study. Pediatr Infect Dis J. 2007;26:700-704.
- Yaris N, Cakir M, Kalyoncu M, et al. Bleomycin induced hyperpigmentation with yolk sac tumor. Indian J Pediatr. 2007;74:505-506.
- Kleynberg RL, Sofi AA, Chaudhary RT. Hand-foot hyperpigmentation skin lesions associated with combination gemcitabine-carboplatin (GemCarbo) therapy. Am J Ther. 2011;18:261-263.
- Blaya M, Saba N. Chemotherapy-induced hyperpigmentation of the tongue. N Engl J Med. 2011;365:e20.
- Anandajeya WV, Corrêa ZM, Augsburger JJ. Primary acquired melanosis with atypia treated with mitomycin C. Int Ophthalmol. 2009;29:285-288.
- Torres C, Wong L, Welsh O, et al. Skin manifestations associated with chemotherapy in children with hematologic malignancies. Pediatr Dermatol. 2011;2:123-147.
- Mays SR, Cohen PR. Emerging dermatologic issues in the oncology patient. Semin Cutan Med Surg. 2006;25:179-189.
- Kamil N, Kamil S, Ahmed SP, et al. Toxic effects of multiple anticancer drugs on skin. Pak J Pharm Sci. 2010;23:7-14.
- Alley E, Green R, Schuchter L. Cutaneous toxicities of cancer therapy. Curr Opin Oncol. 2002;14:212-216.
- Ozkan A, Apak H, Celkan T, et al. Toxic epidermal necrolysis after the use of high-dose cytosine arabinoside. Pediatr Dermatol. 2001;18:38-40.
- Rizos E, Milionis HJ, Pavlidis N, et al. Acquired ichthyosis: a paraneoplastic skin manifestation of Hodgkin’s disease. Lancet Oncol. 2002;3:727.
- Otmani N, Alami R, Hessissen L, et al. Determinants of severe oral mucositis in pediatric cancer patients: a prospective study. Int J Pediatr Dent. 2011;21:210-216.
- Mateus C, Robert C. New drugs in oncology and skin toxicity [in French]. Rev Med Interne. 2009;30:401-410.
- Manji A, Tomlinson D, Ethier MC, et al. Psychometric properties of the Oral Mucositis Daily Questionnaire for child self-report and importance of mucositis in children treated with chemotherapy. Support Care Cancer. 2012;20:1251-1258.
- Keefe DM. Mucositis management in patients with cancer. Support Cancer Ther. 2006;3:154-157.
- Raber-Durlacher JE, Elad S, Barasch A. Oral mucositis. Oral Oncol. 2010;46:452-456.
- Utas S, Kulluk P. A case of hydroxyurea-induced longitudinal melanonychia. Int J Dermatol. 2010;49:466-474.
- Ott H, Höger PH. Dermatologic manifestations of infections in pediatric cancer patients [in German]. Klin Padiatr. 2005;217(suppl 1):110-119.
- Ramphal R, Grant RM, Dzolganovski B, et al. Herpes simplex virus in febrile neutropenic children undergoing chemotherapy for cancer: a prospective cohort study. Pediatr Infect Dis J. 2007;26:700-704.
- Yaris N, Cakir M, Kalyoncu M, et al. Bleomycin induced hyperpigmentation with yolk sac tumor. Indian J Pediatr. 2007;74:505-506.
- Kleynberg RL, Sofi AA, Chaudhary RT. Hand-foot hyperpigmentation skin lesions associated with combination gemcitabine-carboplatin (GemCarbo) therapy. Am J Ther. 2011;18:261-263.
- Blaya M, Saba N. Chemotherapy-induced hyperpigmentation of the tongue. N Engl J Med. 2011;365:e20.
- Anandajeya WV, Corrêa ZM, Augsburger JJ. Primary acquired melanosis with atypia treated with mitomycin C. Int Ophthalmol. 2009;29:285-288.
- Torres C, Wong L, Welsh O, et al. Skin manifestations associated with chemotherapy in children with hematologic malignancies. Pediatr Dermatol. 2011;2:123-147.
- Mays SR, Cohen PR. Emerging dermatologic issues in the oncology patient. Semin Cutan Med Surg. 2006;25:179-189.
Practice Points
- Chemotherapeutic agents can cause a variety of cutaneous side effects.
- Pediatric oncology patients should be examined regularly for cutaneous side effects of chemotherapeutics.
FDA approves oral anticoagulant for NVAF, VTE

Credit: FDA
The US Food and Drug Administration (FDA) has approved the oral, direct factor Xa inhibitor edoxaban (Savaysa) for use in two patient populations.
The anticoagulant is now approved to reduce the risk of stroke and systemic embolism in patients with non-valvular atrial fibrillation (NVAF) and to treat venous thromboembolism (VTE) in patients who have already received parenteral anticoagulation for 5 to 10 days.
The drug has been approved with a Boxed Warning.
The warning states that edoxaban is less effective in NVAF patients with a creatinine clearance greater than 95 mL/min. Patients with creatinine clearance above this limit have an increased risk of stroke if they receive edoxaban (compared to the risk with warfarin), so these patients should not receive edoxaban.
The warning also states that premature discontinuation of edoxaban increases the risk of stroke. Furthermore, spinal or epidural hematomas may occur in patients on edoxaban who are receiving anesthesia injected around the spine or undergoing spinal puncture.
Edoxaban for VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with deep vein thrombosis and 3319 with pulmonary embolism. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
Edoxaban proved superior when it came to the primary safety outcome. Clinically relevant bleeding occurred in 8.5% of edoxaban-treated patients and 10.3% of warfarin-treated patients (P=0.004 for superiority).
In the edoxaban arm, there were 2 fatal bleeds and 13 non-fatal bleeds in a critical site. With warfarin, there were 10 fatal bleeds and 25 non-fatal bleeds in a critical site.
Edoxaban in NVAF
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin for the prevention of stroke or systemic embolic events (SEE) in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or SEE was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
Annualized rates for the secondary composite endpoint of stroke, SEE, and cardiovascular death were 4.43% with warfarin, 3.85% with edoxaban at 60 mg (P=0.005), and 4.23% with edoxaban at 30 mg (P=0.32).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001).
Edoxaban is under development by Daiichi Sankyo Co., Ltd. ![]()
Credit: FDA
The US Food and Drug Administration (FDA) has approved the oral, direct factor Xa inhibitor edoxaban (Savaysa) for use in two patient populations.
The anticoagulant is now approved to reduce the risk of stroke and systemic embolism in patients with non-valvular atrial fibrillation (NVAF) and to treat venous thromboembolism (VTE) in patients who have already received parenteral anticoagulation for 5 to 10 days.
The drug has been approved with a Boxed Warning.
The warning states that edoxaban is less effective in NVAF patients with a creatinine clearance greater than 95 mL/min. Patients with creatinine clearance above this limit have an increased risk of stroke if they receive edoxaban (compared to the risk with warfarin), so these patients should not receive edoxaban.
The warning also states that premature discontinuation of edoxaban increases the risk of stroke. Furthermore, spinal or epidural hematomas may occur in patients on edoxaban who are receiving anesthesia injected around the spine or undergoing spinal puncture.
Edoxaban for VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with deep vein thrombosis and 3319 with pulmonary embolism. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
Edoxaban proved superior when it came to the primary safety outcome. Clinically relevant bleeding occurred in 8.5% of edoxaban-treated patients and 10.3% of warfarin-treated patients (P=0.004 for superiority).
In the edoxaban arm, there were 2 fatal bleeds and 13 non-fatal bleeds in a critical site. With warfarin, there were 10 fatal bleeds and 25 non-fatal bleeds in a critical site.
Edoxaban in NVAF
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin for the prevention of stroke or systemic embolic events (SEE) in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or SEE was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
Annualized rates for the secondary composite endpoint of stroke, SEE, and cardiovascular death were 4.43% with warfarin, 3.85% with edoxaban at 60 mg (P=0.005), and 4.23% with edoxaban at 30 mg (P=0.32).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001).
Edoxaban is under development by Daiichi Sankyo Co., Ltd. ![]()
Credit: FDA
The US Food and Drug Administration (FDA) has approved the oral, direct factor Xa inhibitor edoxaban (Savaysa) for use in two patient populations.
The anticoagulant is now approved to reduce the risk of stroke and systemic embolism in patients with non-valvular atrial fibrillation (NVAF) and to treat venous thromboembolism (VTE) in patients who have already received parenteral anticoagulation for 5 to 10 days.
The drug has been approved with a Boxed Warning.
The warning states that edoxaban is less effective in NVAF patients with a creatinine clearance greater than 95 mL/min. Patients with creatinine clearance above this limit have an increased risk of stroke if they receive edoxaban (compared to the risk with warfarin), so these patients should not receive edoxaban.
The warning also states that premature discontinuation of edoxaban increases the risk of stroke. Furthermore, spinal or epidural hematomas may occur in patients on edoxaban who are receiving anesthesia injected around the spine or undergoing spinal puncture.
Edoxaban for VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with deep vein thrombosis and 3319 with pulmonary embolism. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
Edoxaban proved superior when it came to the primary safety outcome. Clinically relevant bleeding occurred in 8.5% of edoxaban-treated patients and 10.3% of warfarin-treated patients (P=0.004 for superiority).
In the edoxaban arm, there were 2 fatal bleeds and 13 non-fatal bleeds in a critical site. With warfarin, there were 10 fatal bleeds and 25 non-fatal bleeds in a critical site.
Edoxaban in NVAF
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin for the prevention of stroke or systemic embolic events (SEE) in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or SEE was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
Annualized rates for the secondary composite endpoint of stroke, SEE, and cardiovascular death were 4.43% with warfarin, 3.85% with edoxaban at 60 mg (P=0.005), and 4.23% with edoxaban at 30 mg (P=0.32).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001).
Edoxaban is under development by Daiichi Sankyo Co., Ltd. ![]()
Product gets fast track designation for CTCL

mycosis fungoides
The US Food and Drug Administration (FDA) has granted fast track designation to SGX301 as a first-line treatment for cutaneous T-cell lymphoma (CTCL).
SGX301 is a photodynamic therapy utilizing safe, visible light for activation. The active ingredient in SGX301 is synthetic hypericin, a photosensitizer that is applied to skin lesions and then activated by fluorescent light 16 to 24 hours later.
Combined with photoactivation, hypericin has demonstrated significant antiproliferative effects on activated, normal human lymphoid cells and inhibited the growth of malignant T cells isolated from CTCL patients. Topical hypericin has also proven safe in a phase 1 study of healthy volunteers.
In a phase 2 trial of patients with CTCL (mycosis fungoides only) or psoriasis, topical hypericin conferred a significant improvement over placebo. Among CTCL patients, the treatment prompted a response rate of 58.3%, compared to an 8.3% response rate for placebo (P≤0.04).
Topical hypericin was also well tolerated in this trial. There were no deaths or serious adverse events related to the treatment. However, there were reports of mild to moderate burning, itching, erythema, and pruritus at the application site.
A phase 3 trial of SGX301 is set to begin in the first half of this year. In addition to its new fast track status, SGX301 also has orphan designation from the FDA.
About fast track designation
The FDA grants fast track designation to a drug that is intended to treat a serious or life-threatening condition and that demonstrates the potential to address an unmet medical need for the condition.
Fast track designation is designed to facilitate the development and expedite the review of new drugs. For instance, Soligenix, Inc., the company developing SGX301, is eligible to submit a new drug application (NDA) for SGX301 on a rolling basis, allowing the FDA to review sections of the NDA prior to receiving the complete submission.
Additionally, NDAs for fast track development programs ordinarily will be eligible for priority review, which imparts an abbreviated review time of approximately 6 months. ![]()
mycosis fungoides
The US Food and Drug Administration (FDA) has granted fast track designation to SGX301 as a first-line treatment for cutaneous T-cell lymphoma (CTCL).
SGX301 is a photodynamic therapy utilizing safe, visible light for activation. The active ingredient in SGX301 is synthetic hypericin, a photosensitizer that is applied to skin lesions and then activated by fluorescent light 16 to 24 hours later.
Combined with photoactivation, hypericin has demonstrated significant antiproliferative effects on activated, normal human lymphoid cells and inhibited the growth of malignant T cells isolated from CTCL patients. Topical hypericin has also proven safe in a phase 1 study of healthy volunteers.
In a phase 2 trial of patients with CTCL (mycosis fungoides only) or psoriasis, topical hypericin conferred a significant improvement over placebo. Among CTCL patients, the treatment prompted a response rate of 58.3%, compared to an 8.3% response rate for placebo (P≤0.04).
Topical hypericin was also well tolerated in this trial. There were no deaths or serious adverse events related to the treatment. However, there were reports of mild to moderate burning, itching, erythema, and pruritus at the application site.
A phase 3 trial of SGX301 is set to begin in the first half of this year. In addition to its new fast track status, SGX301 also has orphan designation from the FDA.
About fast track designation
The FDA grants fast track designation to a drug that is intended to treat a serious or life-threatening condition and that demonstrates the potential to address an unmet medical need for the condition.
Fast track designation is designed to facilitate the development and expedite the review of new drugs. For instance, Soligenix, Inc., the company developing SGX301, is eligible to submit a new drug application (NDA) for SGX301 on a rolling basis, allowing the FDA to review sections of the NDA prior to receiving the complete submission.
Additionally, NDAs for fast track development programs ordinarily will be eligible for priority review, which imparts an abbreviated review time of approximately 6 months. ![]()
mycosis fungoides
The US Food and Drug Administration (FDA) has granted fast track designation to SGX301 as a first-line treatment for cutaneous T-cell lymphoma (CTCL).
SGX301 is a photodynamic therapy utilizing safe, visible light for activation. The active ingredient in SGX301 is synthetic hypericin, a photosensitizer that is applied to skin lesions and then activated by fluorescent light 16 to 24 hours later.
Combined with photoactivation, hypericin has demonstrated significant antiproliferative effects on activated, normal human lymphoid cells and inhibited the growth of malignant T cells isolated from CTCL patients. Topical hypericin has also proven safe in a phase 1 study of healthy volunteers.
In a phase 2 trial of patients with CTCL (mycosis fungoides only) or psoriasis, topical hypericin conferred a significant improvement over placebo. Among CTCL patients, the treatment prompted a response rate of 58.3%, compared to an 8.3% response rate for placebo (P≤0.04).
Topical hypericin was also well tolerated in this trial. There were no deaths or serious adverse events related to the treatment. However, there were reports of mild to moderate burning, itching, erythema, and pruritus at the application site.
A phase 3 trial of SGX301 is set to begin in the first half of this year. In addition to its new fast track status, SGX301 also has orphan designation from the FDA.
About fast track designation
The FDA grants fast track designation to a drug that is intended to treat a serious or life-threatening condition and that demonstrates the potential to address an unmet medical need for the condition.
Fast track designation is designed to facilitate the development and expedite the review of new drugs. For instance, Soligenix, Inc., the company developing SGX301, is eligible to submit a new drug application (NDA) for SGX301 on a rolling basis, allowing the FDA to review sections of the NDA prior to receiving the complete submission.
Additionally, NDAs for fast track development programs ordinarily will be eligible for priority review, which imparts an abbreviated review time of approximately 6 months. ![]()
Drug granted orphan designation for MM

The US Food and Drug Administration (FDA) has granted selinexor (KPT-330) orphan drug designation to treat multiple myeloma (MM).
Selinexor already has orphan designation from the FDA to treat acute myeloid leukemia (AML) and diffuse large B-cell lymphoma (DLBCL).
The drug has also received orphan designation from the European Medicines Agency (EMA) to treat MM, AML, DLBCL, and chronic lymphocytic leukemia/small lymphocytic lymphoma, including Richter’s transformation.
“Orphan drug designation by the FDA for multiple myeloma is another significant milestone in the selinexor development program,” said Sharon Shacham, PhD, President and Chief Scientific Officer of Karyopharm Therapeutics, Inc., the company developing selinexor.
In the US, orphan designation qualifies a company for certain benefits, including an accelerated approval process, 7 years of market exclusivity following the drug’s approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
About selinexor
Selinexor (KPT-330) is a first-in-class, oral, selective inhibitor of nuclear export compound. The drug functions by inhibiting the nuclear export protein XPO1 (also called CRM1).
This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which subsequently reinitiates and amplifies their tumor suppressor function. This is thought to prompt apoptosis in cancer cells while largely sparing normal cells.
Selinexor combos in MM
In a poster presented at the 2014 ASH Annual Meeting (4773), researchers reported results observed with selinexor plus dexamethasone in preclinical models and in patients with heavily pretreated, refractory MM.
The study included 9 evaluable patients who received selinexor at 45 mg/m2 twice weekly and dexamethasone at 20 mg twice weekly. The combination prompted an overall response rate of 67%, with one stringent complete response (11%) and 5 partial responses (56%), as well as a clinical benefit rate of 89%.
The combination demonstrated a reduction in nausea grades and very little weight loss compared with selinexor alone. The most common grade 1/2 adverse events were nausea, fatigue, anorexia, and vomiting.
The combination was also associated with an increase in time on study relative to selinexor alone. Sixty-six percent of patients remained on study for at least 16 weeks, including one patient for 28 weeks and one for 43 weeks as of December 1, 2014.
During the dose-evaluation part of the study, the 60 mg/m2 selinexor dose was deemed intolerable in this heavily pretreated patient population. So 45 mg/m2 is the recommended future study dose.
In another poster presented at the 2014 ASH Annual Meeting (3443), researchers described the activity of selinexor in combination with carfilzomib. This preclinical study revealed a novel, intracellular, membrane-embedded mechanism of caspase activation.
The results suggested a model of synergy wherein the selinexor-carfilzomib combination promotes caspase activation, likely by induced proximity, cleavage of other caspases, and subsequent apoptosis as well as autophagy. ![]()
The US Food and Drug Administration (FDA) has granted selinexor (KPT-330) orphan drug designation to treat multiple myeloma (MM).
Selinexor already has orphan designation from the FDA to treat acute myeloid leukemia (AML) and diffuse large B-cell lymphoma (DLBCL).
The drug has also received orphan designation from the European Medicines Agency (EMA) to treat MM, AML, DLBCL, and chronic lymphocytic leukemia/small lymphocytic lymphoma, including Richter’s transformation.
“Orphan drug designation by the FDA for multiple myeloma is another significant milestone in the selinexor development program,” said Sharon Shacham, PhD, President and Chief Scientific Officer of Karyopharm Therapeutics, Inc., the company developing selinexor.
In the US, orphan designation qualifies a company for certain benefits, including an accelerated approval process, 7 years of market exclusivity following the drug’s approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
About selinexor
Selinexor (KPT-330) is a first-in-class, oral, selective inhibitor of nuclear export compound. The drug functions by inhibiting the nuclear export protein XPO1 (also called CRM1).
This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which subsequently reinitiates and amplifies their tumor suppressor function. This is thought to prompt apoptosis in cancer cells while largely sparing normal cells.
Selinexor combos in MM
In a poster presented at the 2014 ASH Annual Meeting (4773), researchers reported results observed with selinexor plus dexamethasone in preclinical models and in patients with heavily pretreated, refractory MM.
The study included 9 evaluable patients who received selinexor at 45 mg/m2 twice weekly and dexamethasone at 20 mg twice weekly. The combination prompted an overall response rate of 67%, with one stringent complete response (11%) and 5 partial responses (56%), as well as a clinical benefit rate of 89%.
The combination demonstrated a reduction in nausea grades and very little weight loss compared with selinexor alone. The most common grade 1/2 adverse events were nausea, fatigue, anorexia, and vomiting.
The combination was also associated with an increase in time on study relative to selinexor alone. Sixty-six percent of patients remained on study for at least 16 weeks, including one patient for 28 weeks and one for 43 weeks as of December 1, 2014.
During the dose-evaluation part of the study, the 60 mg/m2 selinexor dose was deemed intolerable in this heavily pretreated patient population. So 45 mg/m2 is the recommended future study dose.
In another poster presented at the 2014 ASH Annual Meeting (3443), researchers described the activity of selinexor in combination with carfilzomib. This preclinical study revealed a novel, intracellular, membrane-embedded mechanism of caspase activation.
The results suggested a model of synergy wherein the selinexor-carfilzomib combination promotes caspase activation, likely by induced proximity, cleavage of other caspases, and subsequent apoptosis as well as autophagy. ![]()
The US Food and Drug Administration (FDA) has granted selinexor (KPT-330) orphan drug designation to treat multiple myeloma (MM).
Selinexor already has orphan designation from the FDA to treat acute myeloid leukemia (AML) and diffuse large B-cell lymphoma (DLBCL).
The drug has also received orphan designation from the European Medicines Agency (EMA) to treat MM, AML, DLBCL, and chronic lymphocytic leukemia/small lymphocytic lymphoma, including Richter’s transformation.
“Orphan drug designation by the FDA for multiple myeloma is another significant milestone in the selinexor development program,” said Sharon Shacham, PhD, President and Chief Scientific Officer of Karyopharm Therapeutics, Inc., the company developing selinexor.
In the US, orphan designation qualifies a company for certain benefits, including an accelerated approval process, 7 years of market exclusivity following the drug’s approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
About selinexor
Selinexor (KPT-330) is a first-in-class, oral, selective inhibitor of nuclear export compound. The drug functions by inhibiting the nuclear export protein XPO1 (also called CRM1).
This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which subsequently reinitiates and amplifies their tumor suppressor function. This is thought to prompt apoptosis in cancer cells while largely sparing normal cells.
Selinexor combos in MM
In a poster presented at the 2014 ASH Annual Meeting (4773), researchers reported results observed with selinexor plus dexamethasone in preclinical models and in patients with heavily pretreated, refractory MM.
The study included 9 evaluable patients who received selinexor at 45 mg/m2 twice weekly and dexamethasone at 20 mg twice weekly. The combination prompted an overall response rate of 67%, with one stringent complete response (11%) and 5 partial responses (56%), as well as a clinical benefit rate of 89%.
The combination demonstrated a reduction in nausea grades and very little weight loss compared with selinexor alone. The most common grade 1/2 adverse events were nausea, fatigue, anorexia, and vomiting.
The combination was also associated with an increase in time on study relative to selinexor alone. Sixty-six percent of patients remained on study for at least 16 weeks, including one patient for 28 weeks and one for 43 weeks as of December 1, 2014.
During the dose-evaluation part of the study, the 60 mg/m2 selinexor dose was deemed intolerable in this heavily pretreated patient population. So 45 mg/m2 is the recommended future study dose.
In another poster presented at the 2014 ASH Annual Meeting (3443), researchers described the activity of selinexor in combination with carfilzomib. This preclinical study revealed a novel, intracellular, membrane-embedded mechanism of caspase activation.
The results suggested a model of synergy wherein the selinexor-carfilzomib combination promotes caspase activation, likely by induced proximity, cleavage of other caspases, and subsequent apoptosis as well as autophagy. ![]()