User login
Misdiagnosed Crusted Scabies in an AIDS Patient Leads to Hyperinfestation
Case Report
A recently incarcerated 34-year-old man with an 11-year history of multidrug-resistant human immunodeficiency virus/AIDS (CD4 count, 121 cells/mm3; viral load, 49,625 particles/mm3 one week prior to presentation) was admitted to the hospital for an intensely pruritic, hyperkeratotic, scaly rash involving the entire body. The rash first appeared on the feet approximately 1 year prior to admission. At that time the patient was given oral fluconazole and a steroid cream with near resolution of the rash. He was then transferred multiple times to different units with subsequent discontinuation of the medications. The rash flared and progressed to involve the knees. He was restarted on the fluconazole and steroid cream and placed in isolation by medical personnel at the prison 6 months prior to presentation. The rash continued to spread, and he was given a working diagnosis of plaque-type psoriasis by several providers after several months of nonresponse to treatment. Additional attempts at treatment at outside facilities included oral fluconazole, trimethoprim-sulfamethoxazole, and other antibiotics. He was referred to dermatology at our institution but missed the appointment and was admitted to the hospital before the appointment could be rescheduled.
On admission to the hospital, he denied similar lesions in close contacts. On review of systems he had subjective fevers and chills, decreased appetite, nausea without vomiting, dysphagia to solids, epigastric pain, and 70-lb weight loss over the last 6 months. Facial involvement of the rash impaired the ability to open the mouth, speak, and eat. He had no known drug allergies. His only medications at the time of admission were nortriptyline, trimethoprim-sulfamethoxazole, and oral combination elvitegravir-cobicistat-emtricitabine-tenofovir for hu-man immunodeficiency virus treatment.
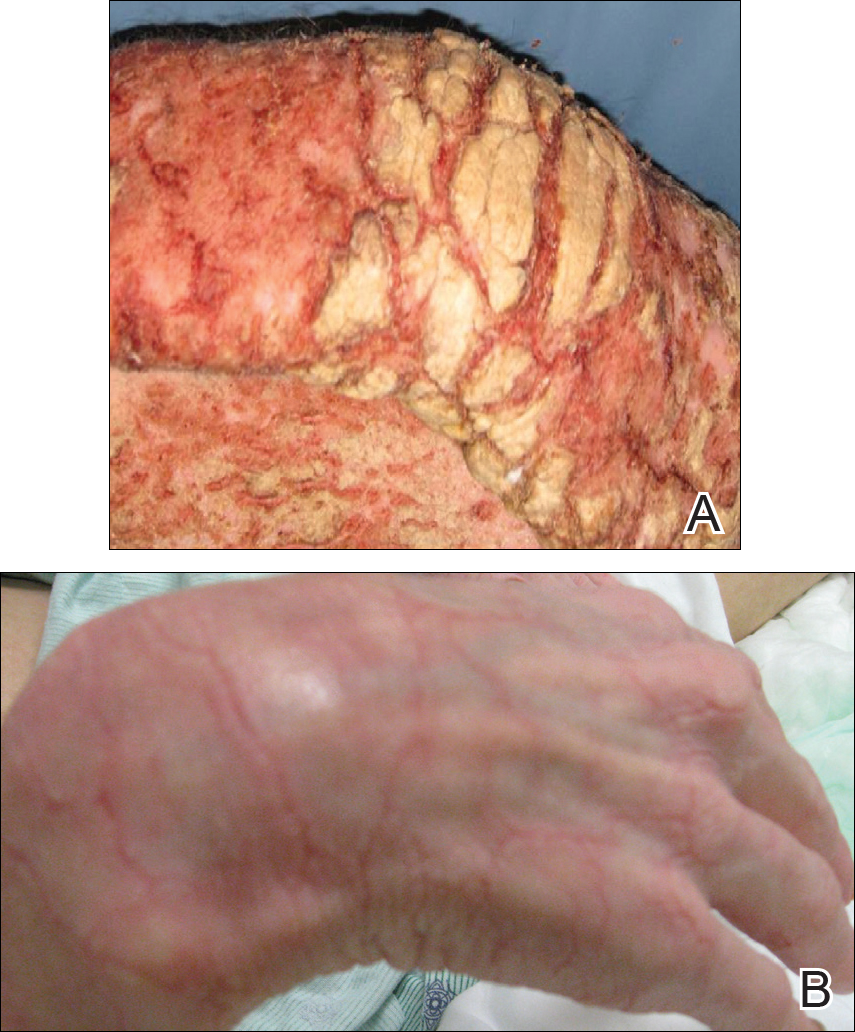
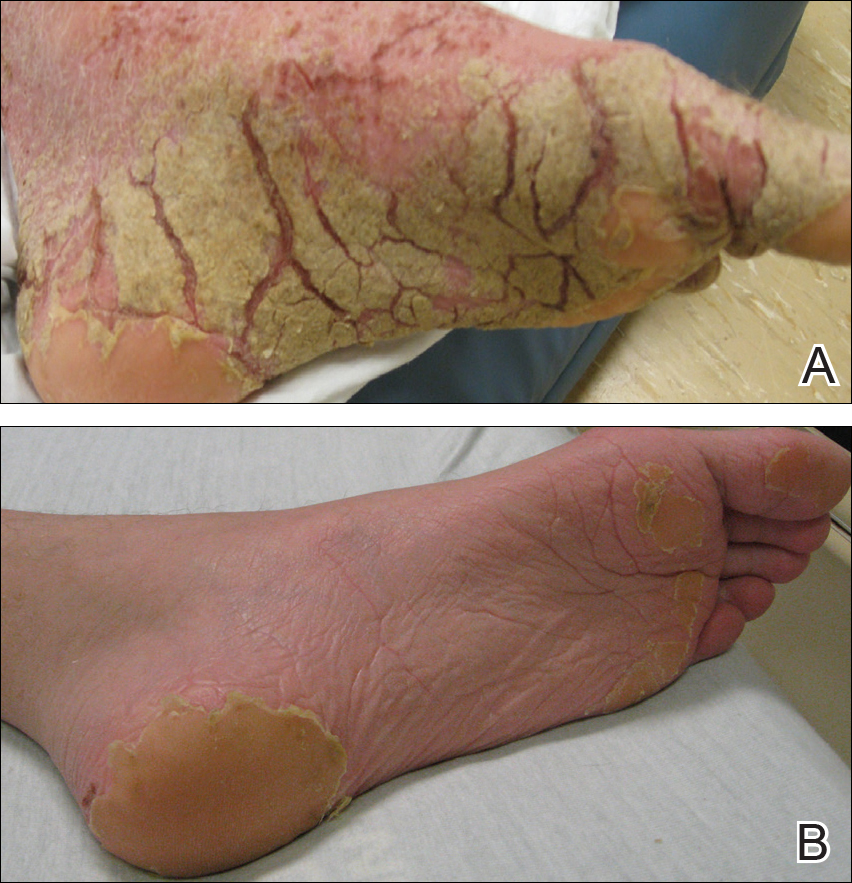
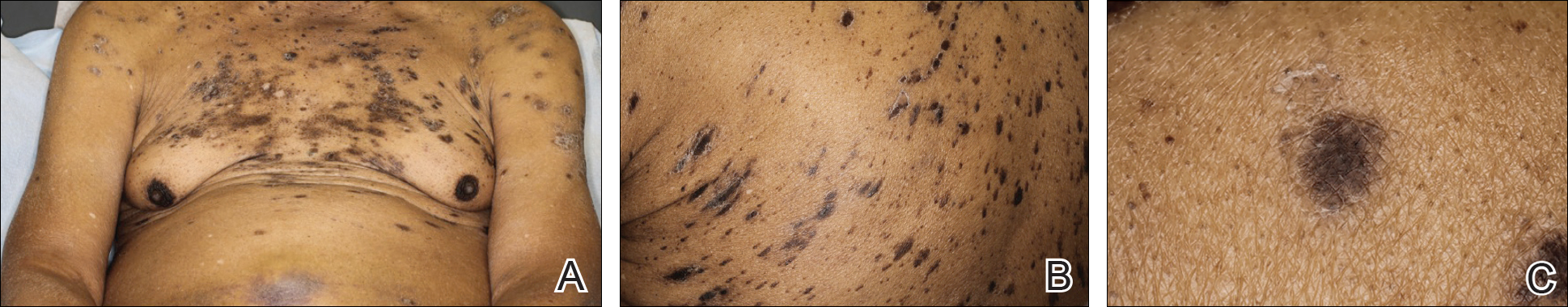
On physical examination he was cachectic, shivering, and foul smelling. He was afebrile, slightly tachycardic (112 beats per minute), and hypertensive (144/83 mm Hg) with a respiratory rate of 18 breaths per minute. His height was 1.83 m (6 ft) and weight was 48.5 kg (107 lb) with a body mass index of 14.5. Extensive erythematous, hyperkeratotic, crusted, and fissured plaques covered the entire body including the face, hands, and feet. The tongue was covered with bilateral white-colored plaques, and he had patches of alopecia, excoriations, and scales on the scalp. The elbows were fixed in a flexed position and he had decreased range of motion in the wrists and fingers due to the severe hyperkeratosis (Figure 1A). Hyperkeratosis also was prominent on the knees and feet with associated burrows (Figure 2A). He had foot drop on the left.
The differential diagnosis included a drug eruption; fungal or parasite infestation, such as crusted scabies; psoriasis; or cutaneous lymphoma. Laboratory studies were difficult to obtain, as there were limited areas suitable for vascular access. Blood work showed leukocytosis (18.9×109 cells/L [reference range, 4.8–10.8×109 cells/L) with 13.3% eosinophils (reference range, 1%–6%). This eosinophilia narrowed the likely diagnoses to a drug eruption or parasite infection.
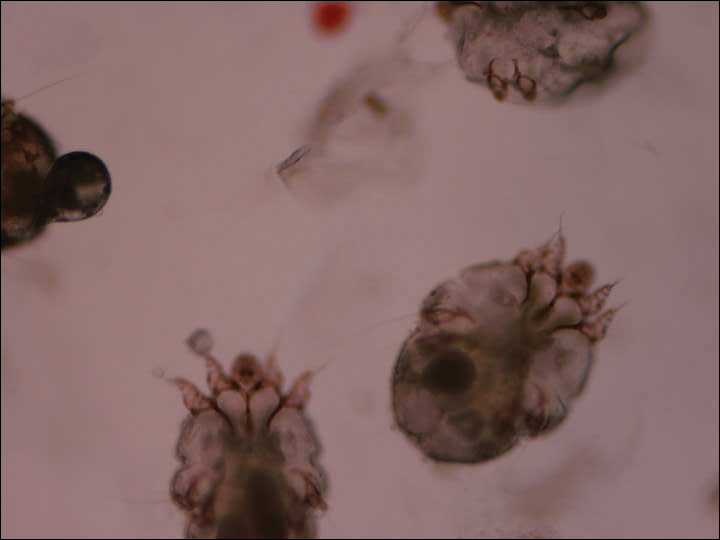
The dermatology service was consulted. A mineral oil preparation was performed and showed numerous mites and feces consistent with a diagnosis of crusted scabies (Figure 3). The patient was started on a regimen of permethrin cream 5% applied to the entire body, except the face, which was left on overnight and washed off. This regimen was repeated daily for 1 week, then twice weekly until the rash resolved after a total of 3 weeks. Due to the severity of his condition, immunocompromised status, and concern for superinfection, oral ivermectin 200 μg/kg once daily was added on days 1, 2, 8, 9, 15, 22, and 29.1
Our patient’s hospital course was further complicated by symptomatic hypoglycemia, altered mental status, and superimposed methicillin-resistant Staphylococcus aureus bacteremia, as well as Pseudomonas aeruginosa bacteremia, pneumonia, and coffee ground emesis. He was transferred to the intensive care unit but fortunately did not require intubation. His overall condition, mental status, and rash gradually improved. Three weeks after admission he only had a few residual lesions on the feet with clearing elsewhere (Figures 1B and 2B). He was discharged with a skin moisturizer and was referred for physical and occupational therapy. On follow-up clinic visits at 3 and 6 months, he had recovered well with general improvement in his condition.
Comment
Classic (noncrusted) scabies is common worldwide, with an estimated 300 million cases per year. It is caused by the mite Sarcoptes scabiei var hominis, and transmission occurs by direct skin-to-skin contact or less commonly by fomites (eg, linens, bedsheets) and therefore is common in overcrowded environments.2 Crusted scabies is a severe, highly contagious form of the disease in which the host’s immune system is overwhelmed and unable to defend against mites on the skin, resulting in hyperinfestation of the host. The mites use secretions to dissolve the epidermis and burrow through the skin, leaving feces in their tracks.3 Interestingly, the native aboriginal populations of Australia have a high incidence of crusted scabies even though they show no signs of immunosuppression. The reason remains unclear but may be due to a skewed T-cell response.4 Various mechanisms have been described for the symptoms of scabies, and it is believed that there is a hypersensitivity reaction to the mites and the feces. Increased IL-17 production by skin T cells may be responsible.5
Clinical Features
Crusted scabies is characterized by severe hyperkeratosis and plaques with desquamation and erythroderma that is worse in the acral regions and large joints, such as the elbows and the knees, as seen in our patient. Because of the deep burrows, patients are predisposed to secondary superinfections by bacteria. In our case, the patient had methicillin-resistant S aureus bacteremia, which persisted for some time despite treatment with intravenous antibiotics.
Diagnosis
Because scabies can imitate different conditions, it can be difficult to diagnose. Misdiagnosis of psoriasis in our patient led to ineffective treatment and subsequent worsening of his condition. Burrows are pathognomonic for scabies, though in severe cases, the burrows may be concealed by extreme hyperkeratosis. Diagnosis is confirmed by mineral oil preparation from the plaques showing numerous scabies mites and feces.
Treatment
It is important to control the spread of scabies, as it is highly contagious, and if the living environment is not properly cleaned, the patient can be reinfected. All clothing, bedsheets, and linens in the household must be washed in hot water and dried in a hot dryer, and nonwashable items should be placed in a closed plastic bag for 72 hours. All contacts also should be treated with 1 application of permethrin cream to the entire body including the head and neck, left on overnight, and washed off with warm water.1 The washing also helps remove some of the skin crusts. Patients should be educated that pruritus and burning may initially worsen with permethrin treatment due to the body’s reaction to the parasite.1,2 In addition, keratolytic agents such as topical urea or salicylic acid can be used as an adjuvant therapy to improve the efficacy of permethrin.
Permethrin is effective against both mites and eggs and works by inhibiting sodium channels, resulting in nerve signal conduction block and subsequent paralysis. Ivermectin is thought to act on glutamate-gated chloride channels, which are present in invertebrates but absent in vertebrates, causing hyperpolarization and paralysis of the adult mite.1,6
Conclusion
Crusted scabies is a highly contagious and intensely pruritic condition. Scabies can mimic other conditions, such as psoriasis or severe dermatitis, so it is important to keep this diagnosis in mind, especially in immunocompromised patients or populations in overcrowded areas (eg, those who are incarcerated or in nursing homes). Treatment consists of isolating the patient, starting topical permethrin and oral ivermectin (in severe cases), washing all linens, and prophylactically treating contacts. A delay in diagnosis can lead to severe debilitating disease, as seen in the extreme case of our patient. However, our patient made a full recovery with appropriate treatment and care.
- Currie BJ, McCarthy JS. Permethrin and ivermectin for scabies. N Engl J Med. 2010;362:717-725.
- World Health Organization. Water-related diseases: scabies. http://www.who.int/water_sanitation_health/diseases-risks/diseases/scabies/en/. Accessed February 23, 2017.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Roberts LJ, Huffam SE, Walton SF, et al. Crusted scabies: clinical and immunological findings in seventy-eight patients and a review of the literature. J Infect. 2005;50:375-381.
- Liu X, Walton SF, Murray HC, et al. Crusted scabies is associated with increased IL-17 secretion by skin T cells. Parasite Immunol. 2014;36:594-604.
- Geary TG. Ivermectin 20 years on: maturation of a wonder drug [published online August 26, 2005]. Trends Parasitol. 2005;21:530-532.
Case Report
A recently incarcerated 34-year-old man with an 11-year history of multidrug-resistant human immunodeficiency virus/AIDS (CD4 count, 121 cells/mm3; viral load, 49,625 particles/mm3 one week prior to presentation) was admitted to the hospital for an intensely pruritic, hyperkeratotic, scaly rash involving the entire body. The rash first appeared on the feet approximately 1 year prior to admission. At that time the patient was given oral fluconazole and a steroid cream with near resolution of the rash. He was then transferred multiple times to different units with subsequent discontinuation of the medications. The rash flared and progressed to involve the knees. He was restarted on the fluconazole and steroid cream and placed in isolation by medical personnel at the prison 6 months prior to presentation. The rash continued to spread, and he was given a working diagnosis of plaque-type psoriasis by several providers after several months of nonresponse to treatment. Additional attempts at treatment at outside facilities included oral fluconazole, trimethoprim-sulfamethoxazole, and other antibiotics. He was referred to dermatology at our institution but missed the appointment and was admitted to the hospital before the appointment could be rescheduled.
On admission to the hospital, he denied similar lesions in close contacts. On review of systems he had subjective fevers and chills, decreased appetite, nausea without vomiting, dysphagia to solids, epigastric pain, and 70-lb weight loss over the last 6 months. Facial involvement of the rash impaired the ability to open the mouth, speak, and eat. He had no known drug allergies. His only medications at the time of admission were nortriptyline, trimethoprim-sulfamethoxazole, and oral combination elvitegravir-cobicistat-emtricitabine-tenofovir for hu-man immunodeficiency virus treatment.
On physical examination he was cachectic, shivering, and foul smelling. He was afebrile, slightly tachycardic (112 beats per minute), and hypertensive (144/83 mm Hg) with a respiratory rate of 18 breaths per minute. His height was 1.83 m (6 ft) and weight was 48.5 kg (107 lb) with a body mass index of 14.5. Extensive erythematous, hyperkeratotic, crusted, and fissured plaques covered the entire body including the face, hands, and feet. The tongue was covered with bilateral white-colored plaques, and he had patches of alopecia, excoriations, and scales on the scalp. The elbows were fixed in a flexed position and he had decreased range of motion in the wrists and fingers due to the severe hyperkeratosis (Figure 1A). Hyperkeratosis also was prominent on the knees and feet with associated burrows (Figure 2A). He had foot drop on the left.
The differential diagnosis included a drug eruption; fungal or parasite infestation, such as crusted scabies; psoriasis; or cutaneous lymphoma. Laboratory studies were difficult to obtain, as there were limited areas suitable for vascular access. Blood work showed leukocytosis (18.9×109 cells/L [reference range, 4.8–10.8×109 cells/L) with 13.3% eosinophils (reference range, 1%–6%). This eosinophilia narrowed the likely diagnoses to a drug eruption or parasite infection.
The dermatology service was consulted. A mineral oil preparation was performed and showed numerous mites and feces consistent with a diagnosis of crusted scabies (Figure 3). The patient was started on a regimen of permethrin cream 5% applied to the entire body, except the face, which was left on overnight and washed off. This regimen was repeated daily for 1 week, then twice weekly until the rash resolved after a total of 3 weeks. Due to the severity of his condition, immunocompromised status, and concern for superinfection, oral ivermectin 200 μg/kg once daily was added on days 1, 2, 8, 9, 15, 22, and 29.1
Our patient’s hospital course was further complicated by symptomatic hypoglycemia, altered mental status, and superimposed methicillin-resistant Staphylococcus aureus bacteremia, as well as Pseudomonas aeruginosa bacteremia, pneumonia, and coffee ground emesis. He was transferred to the intensive care unit but fortunately did not require intubation. His overall condition, mental status, and rash gradually improved. Three weeks after admission he only had a few residual lesions on the feet with clearing elsewhere (Figures 1B and 2B). He was discharged with a skin moisturizer and was referred for physical and occupational therapy. On follow-up clinic visits at 3 and 6 months, he had recovered well with general improvement in his condition.
Comment
Classic (noncrusted) scabies is common worldwide, with an estimated 300 million cases per year. It is caused by the mite Sarcoptes scabiei var hominis, and transmission occurs by direct skin-to-skin contact or less commonly by fomites (eg, linens, bedsheets) and therefore is common in overcrowded environments.2 Crusted scabies is a severe, highly contagious form of the disease in which the host’s immune system is overwhelmed and unable to defend against mites on the skin, resulting in hyperinfestation of the host. The mites use secretions to dissolve the epidermis and burrow through the skin, leaving feces in their tracks.3 Interestingly, the native aboriginal populations of Australia have a high incidence of crusted scabies even though they show no signs of immunosuppression. The reason remains unclear but may be due to a skewed T-cell response.4 Various mechanisms have been described for the symptoms of scabies, and it is believed that there is a hypersensitivity reaction to the mites and the feces. Increased IL-17 production by skin T cells may be responsible.5
Clinical Features
Crusted scabies is characterized by severe hyperkeratosis and plaques with desquamation and erythroderma that is worse in the acral regions and large joints, such as the elbows and the knees, as seen in our patient. Because of the deep burrows, patients are predisposed to secondary superinfections by bacteria. In our case, the patient had methicillin-resistant S aureus bacteremia, which persisted for some time despite treatment with intravenous antibiotics.
Diagnosis
Because scabies can imitate different conditions, it can be difficult to diagnose. Misdiagnosis of psoriasis in our patient led to ineffective treatment and subsequent worsening of his condition. Burrows are pathognomonic for scabies, though in severe cases, the burrows may be concealed by extreme hyperkeratosis. Diagnosis is confirmed by mineral oil preparation from the plaques showing numerous scabies mites and feces.
Treatment
It is important to control the spread of scabies, as it is highly contagious, and if the living environment is not properly cleaned, the patient can be reinfected. All clothing, bedsheets, and linens in the household must be washed in hot water and dried in a hot dryer, and nonwashable items should be placed in a closed plastic bag for 72 hours. All contacts also should be treated with 1 application of permethrin cream to the entire body including the head and neck, left on overnight, and washed off with warm water.1 The washing also helps remove some of the skin crusts. Patients should be educated that pruritus and burning may initially worsen with permethrin treatment due to the body’s reaction to the parasite.1,2 In addition, keratolytic agents such as topical urea or salicylic acid can be used as an adjuvant therapy to improve the efficacy of permethrin.
Permethrin is effective against both mites and eggs and works by inhibiting sodium channels, resulting in nerve signal conduction block and subsequent paralysis. Ivermectin is thought to act on glutamate-gated chloride channels, which are present in invertebrates but absent in vertebrates, causing hyperpolarization and paralysis of the adult mite.1,6
Conclusion
Crusted scabies is a highly contagious and intensely pruritic condition. Scabies can mimic other conditions, such as psoriasis or severe dermatitis, so it is important to keep this diagnosis in mind, especially in immunocompromised patients or populations in overcrowded areas (eg, those who are incarcerated or in nursing homes). Treatment consists of isolating the patient, starting topical permethrin and oral ivermectin (in severe cases), washing all linens, and prophylactically treating contacts. A delay in diagnosis can lead to severe debilitating disease, as seen in the extreme case of our patient. However, our patient made a full recovery with appropriate treatment and care.
Case Report
A recently incarcerated 34-year-old man with an 11-year history of multidrug-resistant human immunodeficiency virus/AIDS (CD4 count, 121 cells/mm3; viral load, 49,625 particles/mm3 one week prior to presentation) was admitted to the hospital for an intensely pruritic, hyperkeratotic, scaly rash involving the entire body. The rash first appeared on the feet approximately 1 year prior to admission. At that time the patient was given oral fluconazole and a steroid cream with near resolution of the rash. He was then transferred multiple times to different units with subsequent discontinuation of the medications. The rash flared and progressed to involve the knees. He was restarted on the fluconazole and steroid cream and placed in isolation by medical personnel at the prison 6 months prior to presentation. The rash continued to spread, and he was given a working diagnosis of plaque-type psoriasis by several providers after several months of nonresponse to treatment. Additional attempts at treatment at outside facilities included oral fluconazole, trimethoprim-sulfamethoxazole, and other antibiotics. He was referred to dermatology at our institution but missed the appointment and was admitted to the hospital before the appointment could be rescheduled.
On admission to the hospital, he denied similar lesions in close contacts. On review of systems he had subjective fevers and chills, decreased appetite, nausea without vomiting, dysphagia to solids, epigastric pain, and 70-lb weight loss over the last 6 months. Facial involvement of the rash impaired the ability to open the mouth, speak, and eat. He had no known drug allergies. His only medications at the time of admission were nortriptyline, trimethoprim-sulfamethoxazole, and oral combination elvitegravir-cobicistat-emtricitabine-tenofovir for hu-man immunodeficiency virus treatment.
On physical examination he was cachectic, shivering, and foul smelling. He was afebrile, slightly tachycardic (112 beats per minute), and hypertensive (144/83 mm Hg) with a respiratory rate of 18 breaths per minute. His height was 1.83 m (6 ft) and weight was 48.5 kg (107 lb) with a body mass index of 14.5. Extensive erythematous, hyperkeratotic, crusted, and fissured plaques covered the entire body including the face, hands, and feet. The tongue was covered with bilateral white-colored plaques, and he had patches of alopecia, excoriations, and scales on the scalp. The elbows were fixed in a flexed position and he had decreased range of motion in the wrists and fingers due to the severe hyperkeratosis (Figure 1A). Hyperkeratosis also was prominent on the knees and feet with associated burrows (Figure 2A). He had foot drop on the left.
The differential diagnosis included a drug eruption; fungal or parasite infestation, such as crusted scabies; psoriasis; or cutaneous lymphoma. Laboratory studies were difficult to obtain, as there were limited areas suitable for vascular access. Blood work showed leukocytosis (18.9×109 cells/L [reference range, 4.8–10.8×109 cells/L) with 13.3% eosinophils (reference range, 1%–6%). This eosinophilia narrowed the likely diagnoses to a drug eruption or parasite infection.
The dermatology service was consulted. A mineral oil preparation was performed and showed numerous mites and feces consistent with a diagnosis of crusted scabies (Figure 3). The patient was started on a regimen of permethrin cream 5% applied to the entire body, except the face, which was left on overnight and washed off. This regimen was repeated daily for 1 week, then twice weekly until the rash resolved after a total of 3 weeks. Due to the severity of his condition, immunocompromised status, and concern for superinfection, oral ivermectin 200 μg/kg once daily was added on days 1, 2, 8, 9, 15, 22, and 29.1
Our patient’s hospital course was further complicated by symptomatic hypoglycemia, altered mental status, and superimposed methicillin-resistant Staphylococcus aureus bacteremia, as well as Pseudomonas aeruginosa bacteremia, pneumonia, and coffee ground emesis. He was transferred to the intensive care unit but fortunately did not require intubation. His overall condition, mental status, and rash gradually improved. Three weeks after admission he only had a few residual lesions on the feet with clearing elsewhere (Figures 1B and 2B). He was discharged with a skin moisturizer and was referred for physical and occupational therapy. On follow-up clinic visits at 3 and 6 months, he had recovered well with general improvement in his condition.
Comment
Classic (noncrusted) scabies is common worldwide, with an estimated 300 million cases per year. It is caused by the mite Sarcoptes scabiei var hominis, and transmission occurs by direct skin-to-skin contact or less commonly by fomites (eg, linens, bedsheets) and therefore is common in overcrowded environments.2 Crusted scabies is a severe, highly contagious form of the disease in which the host’s immune system is overwhelmed and unable to defend against mites on the skin, resulting in hyperinfestation of the host. The mites use secretions to dissolve the epidermis and burrow through the skin, leaving feces in their tracks.3 Interestingly, the native aboriginal populations of Australia have a high incidence of crusted scabies even though they show no signs of immunosuppression. The reason remains unclear but may be due to a skewed T-cell response.4 Various mechanisms have been described for the symptoms of scabies, and it is believed that there is a hypersensitivity reaction to the mites and the feces. Increased IL-17 production by skin T cells may be responsible.5
Clinical Features
Crusted scabies is characterized by severe hyperkeratosis and plaques with desquamation and erythroderma that is worse in the acral regions and large joints, such as the elbows and the knees, as seen in our patient. Because of the deep burrows, patients are predisposed to secondary superinfections by bacteria. In our case, the patient had methicillin-resistant S aureus bacteremia, which persisted for some time despite treatment with intravenous antibiotics.
Diagnosis
Because scabies can imitate different conditions, it can be difficult to diagnose. Misdiagnosis of psoriasis in our patient led to ineffective treatment and subsequent worsening of his condition. Burrows are pathognomonic for scabies, though in severe cases, the burrows may be concealed by extreme hyperkeratosis. Diagnosis is confirmed by mineral oil preparation from the plaques showing numerous scabies mites and feces.
Treatment
It is important to control the spread of scabies, as it is highly contagious, and if the living environment is not properly cleaned, the patient can be reinfected. All clothing, bedsheets, and linens in the household must be washed in hot water and dried in a hot dryer, and nonwashable items should be placed in a closed plastic bag for 72 hours. All contacts also should be treated with 1 application of permethrin cream to the entire body including the head and neck, left on overnight, and washed off with warm water.1 The washing also helps remove some of the skin crusts. Patients should be educated that pruritus and burning may initially worsen with permethrin treatment due to the body’s reaction to the parasite.1,2 In addition, keratolytic agents such as topical urea or salicylic acid can be used as an adjuvant therapy to improve the efficacy of permethrin.
Permethrin is effective against both mites and eggs and works by inhibiting sodium channels, resulting in nerve signal conduction block and subsequent paralysis. Ivermectin is thought to act on glutamate-gated chloride channels, which are present in invertebrates but absent in vertebrates, causing hyperpolarization and paralysis of the adult mite.1,6
Conclusion
Crusted scabies is a highly contagious and intensely pruritic condition. Scabies can mimic other conditions, such as psoriasis or severe dermatitis, so it is important to keep this diagnosis in mind, especially in immunocompromised patients or populations in overcrowded areas (eg, those who are incarcerated or in nursing homes). Treatment consists of isolating the patient, starting topical permethrin and oral ivermectin (in severe cases), washing all linens, and prophylactically treating contacts. A delay in diagnosis can lead to severe debilitating disease, as seen in the extreme case of our patient. However, our patient made a full recovery with appropriate treatment and care.
- Currie BJ, McCarthy JS. Permethrin and ivermectin for scabies. N Engl J Med. 2010;362:717-725.
- World Health Organization. Water-related diseases: scabies. http://www.who.int/water_sanitation_health/diseases-risks/diseases/scabies/en/. Accessed February 23, 2017.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Roberts LJ, Huffam SE, Walton SF, et al. Crusted scabies: clinical and immunological findings in seventy-eight patients and a review of the literature. J Infect. 2005;50:375-381.
- Liu X, Walton SF, Murray HC, et al. Crusted scabies is associated with increased IL-17 secretion by skin T cells. Parasite Immunol. 2014;36:594-604.
- Geary TG. Ivermectin 20 years on: maturation of a wonder drug [published online August 26, 2005]. Trends Parasitol. 2005;21:530-532.
- Currie BJ, McCarthy JS. Permethrin and ivermectin for scabies. N Engl J Med. 2010;362:717-725.
- World Health Organization. Water-related diseases: scabies. http://www.who.int/water_sanitation_health/diseases-risks/diseases/scabies/en/. Accessed February 23, 2017.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Roberts LJ, Huffam SE, Walton SF, et al. Crusted scabies: clinical and immunological findings in seventy-eight patients and a review of the literature. J Infect. 2005;50:375-381.
- Liu X, Walton SF, Murray HC, et al. Crusted scabies is associated with increased IL-17 secretion by skin T cells. Parasite Immunol. 2014;36:594-604.
- Geary TG. Ivermectin 20 years on: maturation of a wonder drug [published online August 26, 2005]. Trends Parasitol. 2005;21:530-532.
Practice Points
- Keep scabies in mind, especially in immunocompromised patients or populations in overcrowded areas.
- Treatment consists of isolating the patient, starting topical permethrin and oral ivermectin (in severe cases), washing all linens, and prophylactically treating contacts.
Imatinib Mesylate–Induced Lichenoid Drug Eruption
Imatinib mesylate is a tyrosine kinase inhibitor initially approved by the US Food and Drug Administration in 2001 for chronic myeloid leukemia (CML). The indications for imatinib have expanded since its initial approval. It is increasingly important that dermatologists recognize adverse cutaneous manifestations associated with imatinib and are aware of their management and outcomes to avoid unnecessarily discontinuing a potentially lifesaving medication.
Adverse cutaneous manifestations in response to imatinib are not infrequent, accounting for 7% to 21% of all side effects.1 The most frequent cutaneous manifestations of imatinib are dry skin, alopecia, facial edema, and photosensitivity rash, respectively.1 Other less common manifestations include exfoliative dermatitis, nail disorders, psoriasis, folliculitis, hypotrichosis, urticaria, petechiae, Stevens-Johnson syndrome, erythema multiforme, Sweet syndrome, and leukocytoclastic vasculitis.
We report a case of imatinib-induced lichenoid drug eruption (LDE), a rare cutaneous side effect of imatinib use, along with a review of the literature.
Case Report
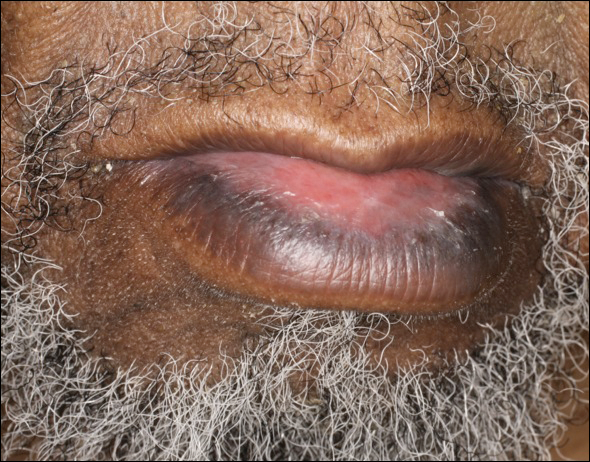
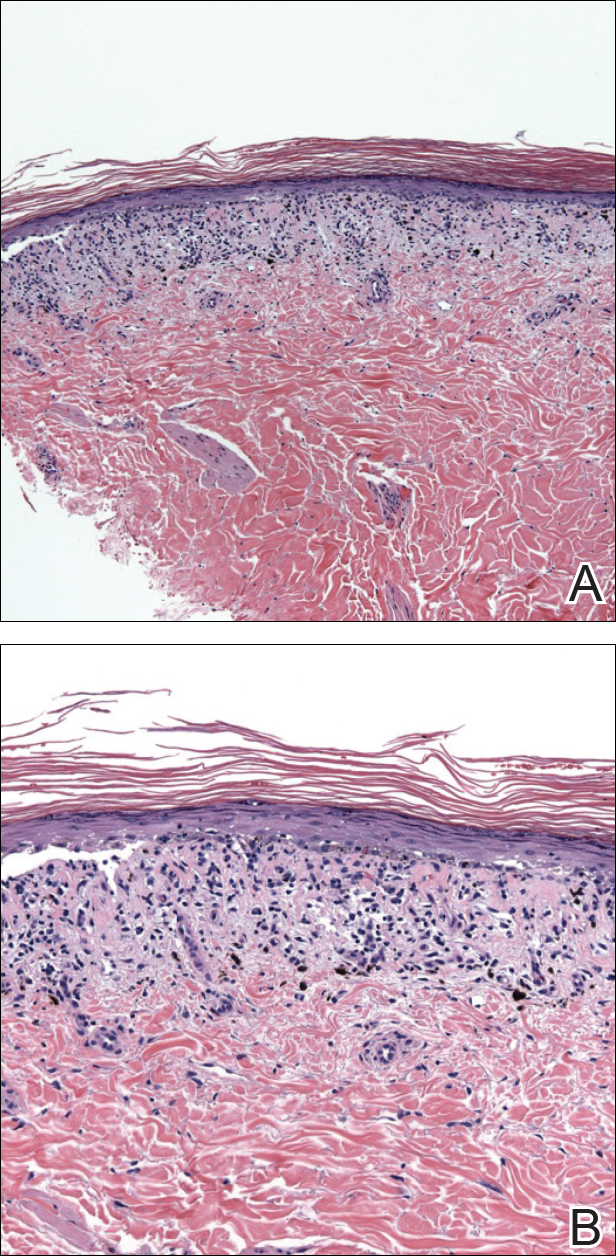
An 86-year-old man with a history of gastrointestinal stromal tumors (GISTs) and myelodysplastic syndrome presented with diffuse hyperpigmented skin lesions on the trunk, arms, legs, and lower lip of 2 weeks’ duration. He had been taking imatinib 400 mg once daily for 5 months for GIST. Although the oncologist stopped the medication 2 weeks prior, the lesions were persistent and gradually expanded to involve the trunk, arms, legs, and lower lip. He denied any pain or pruritus. Physical examination revealed multiple ill-defined, brown to violaceous, slightly scaly macules and patches on the trunk (Figures 1A and 1B), arms, and legs (Figure 1C), as well as violaceous to erythematous patches on the mucosal aspect of the lower lip (Figure 2). Two 4-mm punch biopsies were performed from the chest and back, which revealed an atrophic epidermis, lichenoid infiltration, and multiple melanophages in the upper dermis consistent with LDE (Figure 3). Direct immunofluorescence was negative. Therefore, based on the clinicopathologic correlation, the diagnosis of imatinib-induced LDE was made. He was treated with clobetasol ointment twice daily for 3 weeks with some improvement. His GIST was stable on follow-up computed tomography 3 months after presentation, and imatinib was resumed 1 month later with continued rash that was stable with topical corticosteroid treatment.
Comment
In addition to CML, imatinib has been approved for acute lymphoblastic leukemia, myelodysplastic syndromes, aggressive systemic mastocytosis, hypereosinophilic syndrome, chronic eosinophilic leukemia, dermatofibrosarcoma protuberans, and GIST. Moreover, off-label use of imatinib for various other tyrosine kinase–positive cancers and rheumatologic conditions have been documented.2,3 With the expanding use of imatinib, there will be more occasions for dermatologists to encounter cutaneous manifestations associated with its use.
According to a PubMed search of articles indexed for MEDLINE using the terms imatinib mesylate lichenoid drug, there have been few case reports of LDE associated with imatinib in the literature (eTable).4-24 Compared to classic LDE, imatinib-induced LDE has a few characteristic findings. Classic LDE frequently spares the oral mucosa and genitalia, but imatinib-induced LDE with manifestations on the oral mucosa and genitalia as well as cutaneous eruptions have been reported.4-9 In fact, the first known case of imatinib-induced LDE was an oral eruption in a patient with CML.4 In patients with oral involvement, lesions have been described as lacy reticular macules and violaceous papules, erosions, and ulcers.4,5,12 Interestingly, of those cases manifesting as concomitant oral and cutaneous LDE, the oral eruptions recurred more frequently, with 3 of 12 patients having recurrence of oral lesions after the cutaneous manifestations resolved.8,16 Genital manifestations of imatinib-induced LDE were much less common.9,11
To date, subsequent reports of imatinib-induced LDE have documented skin manifestations consistent with classic LDE occurring in a diffuse, bilateral, photodistributed pattern.10,15,16 One case presented with diffuse hyperpigmentation associated with LDE in a Japanese patient.20 The authors suggested this finding may be more prominent in patients with skin of color,20 which is consistent with the current case. Nail findings such as subungual hyperkeratosis and longitudinal ridging also have been reported.9,11
The latency period between initiation of imat-inib and onset of LDE generally ranges from 1 to 12 months, with onset most commonly occurring between 2 to 5 months or with dosage increase (eTable). Imatinib-induced LDE primarily has been documented with a 400-mg dose, with 1 case of a 600-mg dose and 1 case of an 800-mg dose, which suggests dose dependency. Furthermore, reports exist of several patients responding well to dose reduction with subsequent recurrence on dose reescalation.13,15
Historically, LDE resolves with discontinuation of the drug after a few weeks to months. When discontinuation of imatinib is unfavorable or patients report symptoms including severe pruritus or pain, treatment should be considered. Topical or oral corticosteroids can be used to treat imatinib-induced LDE, similar to lichen planus. When oral corticosteroids are contraindicated (eg, due to poor patient tolerance), oral acitretin at 25 to 35 mg once daily for 6 to 12 weeks has been reported as an alternative treatment.25
In the majority of cases of imatinib-induced LDE, it was undesirable to stop imatinib (eTable). Notably, in half the reported cases, imatinib was able to be continued and patients were treated symptomatically with either oral and/or topical steroids and/or acitretin with complete remission or tolerable recurrences. Dalmau et al9 reported 3 patients who responded poorly to topical and oral steroids and were subsequently treated with acitretin 25 mg once daily; 2 of 3 patients responded favorably to treatment and imatinib was able to be continued. In the current case imatinib initially helped, but because his rash was relatively asymptomatic, imatinib was restarted with control of rash with topical steroids. He developed some pancytopenia, which required intermittent stoppage of the imatinib.
Conclusion
We present a case of imatinib-induced cutaneous and oral LDE in a patient with GIST. Topical corticosteroids, oral acitretin, and oral steroids all may be reasonable treatment options if discontinuing imatinib is not possible in a symptomatic patient. If these therapies fail and the eruption is extensive or intolerable, dosage adjustment is another option to consider before discontinuation of imatinib.
- Scheinfeld N. Imatinib mesylate and dermatology part 2: a review of the cutaneous side effects of imatinib mesylate. J Drugs Dermatol. 2006;5:228-231.
- Kim H, Kim NH, Kang HJ, et al. Successful long-term use of imatinib mesylate in pediatric patients with sclerodermatous chronic GVHD. Pediatr Transplant. 2012;16:910-912.
- Prey S, Ezzedine K, Doussau A, et al. Imatinib mesylate in scleroderma-associated diffuse skin fibrosis: a phase II multicentre randomized double-blinded controlled trial. Br J Dermatol. 2012;167:1138-1144.
- Lim DS, Muir J. Oral lichenoid reaction to imatinib (STI 571, gleevec). Dermatology. 2002;205:169-171.
- Ena P, Chiarolini F, Siddi GM, et al. Oral lichenoid eruption secondary to imatinib (glivec). J Dermatolog Treat. 2004;15:253-255.
- Roux C, Boisseau-Garsaud AM, Saint-Cyr I, et al. Lichenoid cutaneous reaction to imatinib. Ann Dermatol Venereol. 2004;131:571-573.
- Prabhash K, Doval DC. Lichenoid eruption due to imat-inib. Indian J Dermatol Venereol Leprol. 2005;71:287-288.
- Pascual JC, Matarredona J, Miralles J, et al. Oral and cutaneous lichenoid reaction secondary to imatinib: report of two cases. Int J Dermatol. 2006;45:1471-1473.
- Dalmau J, Peramiquel L, Puig L, et al. Imatinib-associated lichenoid eruption: acitretin treatment allows maintained antineoplastic effect. Br J Dermatol. 2006;154:1213-1216.
- Chan CY, Browning J, Smith-Zagone MJ, et al. Cutaneous lichenoid dermatitis associated with imatinib mesylate. Dermatol Online J. 2007;13:29.
- Wahiduzzaman M, Pubalan M. Oral and cutaneous lichenoid reaction with nail changes secondary to imatinib: report of a case and literature review. Dermatol Online J. 2008;14:14.
- Basso FG, Boer CC, Correa ME, et al. Skin and oral lesions associated to imatinib mesylate therapy. Support Care Cancer. 2009;17:465-468.
- Kawakami T, Kawanabe T, Soma Y. Cutaneous lichenoid eruption caused by imatinib mesylate in a Japanese patient with chronic myeloid leukaemia. Acta Derm Venereol. 2009;89:325-326.
- Sendagorta E, Herranz P, Feito M, et al. Lichenoid drug eruption related to imatinib: report of a new case and review of the literature. Clin Exp Dermatol. 2009;34:E315-E316.
- Kuraishi N, Nagai Y, Hasegawa M, et al. Lichenoid drug eruption with palmoplantar hyperkeratosis due to imatinib mesylate: a case report and a review of the literature. Acta Derm Venereol. 2010;90:73-76.
- Brazzelli V, Muzio F, Manna G, et al. Photo-induced dermatitis and oral lichenoid reaction in a chronic myeloid leukemia patient treated with imatinib mesylate. Photodermatol Photoimmunol Photomed. 2012;28:2-5.
- Ghosh SK. Generalized lichenoid drug eruption associated with imatinib mesylate therapy. Indian J Dermatol. 2013;58:388-392.
- Lee J, Chung J, Jung M, et al. Lichenoid drug eruption after low-dose imatinib mesylate therapy. Ann Dermatol. 2013;25:500-502.
- Machaczka M, Gossart M. Multiple skin lesions caused by imatinib mesylate treatment of chronic myeloid leukemia. Pol Arch Med Wewn. 2013;123:251-252.
- Kagimoto Y, Mizuashi M, Kikuchi K, et al. Lichenoid drug eruption with hyperpigmentation caused by imatinib mesylate [published online June 20, 2013]. Int J Dermatol. 2014;53:E161-E162.
- Arshdeep, De D, Malhotra P, et al. Imatinib mesylate-induced severe lichenoid rash. Indian J Dermatol Venereol Leprol. 2014;80:93-95.
- Lau YM, Lam YK, Leung KH, et al. Trachyonychia in a patient with chronic myeloid leukaemia after imatinib mesylate. Hong Kong Med J. 2014;20:464.e2.
- Bhatia A, Kanish B, Chaudhary P. Lichenoid drug eruption due to imatinib mesylate. Int J Appl Basic Med Res. 2015;5:68-69.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Laurberg G, Geiger JM, Hjorth N, et al. Treatment of lichen planus with acitretin. a double-blind, placebo-controlled study in 65 patients. J Am Acad Dermatol. 1991;24:434-437.
Imatinib mesylate is a tyrosine kinase inhibitor initially approved by the US Food and Drug Administration in 2001 for chronic myeloid leukemia (CML). The indications for imatinib have expanded since its initial approval. It is increasingly important that dermatologists recognize adverse cutaneous manifestations associated with imatinib and are aware of their management and outcomes to avoid unnecessarily discontinuing a potentially lifesaving medication.
Adverse cutaneous manifestations in response to imatinib are not infrequent, accounting for 7% to 21% of all side effects.1 The most frequent cutaneous manifestations of imatinib are dry skin, alopecia, facial edema, and photosensitivity rash, respectively.1 Other less common manifestations include exfoliative dermatitis, nail disorders, psoriasis, folliculitis, hypotrichosis, urticaria, petechiae, Stevens-Johnson syndrome, erythema multiforme, Sweet syndrome, and leukocytoclastic vasculitis.
We report a case of imatinib-induced lichenoid drug eruption (LDE), a rare cutaneous side effect of imatinib use, along with a review of the literature.
Case Report
An 86-year-old man with a history of gastrointestinal stromal tumors (GISTs) and myelodysplastic syndrome presented with diffuse hyperpigmented skin lesions on the trunk, arms, legs, and lower lip of 2 weeks’ duration. He had been taking imatinib 400 mg once daily for 5 months for GIST. Although the oncologist stopped the medication 2 weeks prior, the lesions were persistent and gradually expanded to involve the trunk, arms, legs, and lower lip. He denied any pain or pruritus. Physical examination revealed multiple ill-defined, brown to violaceous, slightly scaly macules and patches on the trunk (Figures 1A and 1B), arms, and legs (Figure 1C), as well as violaceous to erythematous patches on the mucosal aspect of the lower lip (Figure 2). Two 4-mm punch biopsies were performed from the chest and back, which revealed an atrophic epidermis, lichenoid infiltration, and multiple melanophages in the upper dermis consistent with LDE (Figure 3). Direct immunofluorescence was negative. Therefore, based on the clinicopathologic correlation, the diagnosis of imatinib-induced LDE was made. He was treated with clobetasol ointment twice daily for 3 weeks with some improvement. His GIST was stable on follow-up computed tomography 3 months after presentation, and imatinib was resumed 1 month later with continued rash that was stable with topical corticosteroid treatment.
Comment
In addition to CML, imatinib has been approved for acute lymphoblastic leukemia, myelodysplastic syndromes, aggressive systemic mastocytosis, hypereosinophilic syndrome, chronic eosinophilic leukemia, dermatofibrosarcoma protuberans, and GIST. Moreover, off-label use of imatinib for various other tyrosine kinase–positive cancers and rheumatologic conditions have been documented.2,3 With the expanding use of imatinib, there will be more occasions for dermatologists to encounter cutaneous manifestations associated with its use.
According to a PubMed search of articles indexed for MEDLINE using the terms imatinib mesylate lichenoid drug, there have been few case reports of LDE associated with imatinib in the literature (eTable).4-24 Compared to classic LDE, imatinib-induced LDE has a few characteristic findings. Classic LDE frequently spares the oral mucosa and genitalia, but imatinib-induced LDE with manifestations on the oral mucosa and genitalia as well as cutaneous eruptions have been reported.4-9 In fact, the first known case of imatinib-induced LDE was an oral eruption in a patient with CML.4 In patients with oral involvement, lesions have been described as lacy reticular macules and violaceous papules, erosions, and ulcers.4,5,12 Interestingly, of those cases manifesting as concomitant oral and cutaneous LDE, the oral eruptions recurred more frequently, with 3 of 12 patients having recurrence of oral lesions after the cutaneous manifestations resolved.8,16 Genital manifestations of imatinib-induced LDE were much less common.9,11
To date, subsequent reports of imatinib-induced LDE have documented skin manifestations consistent with classic LDE occurring in a diffuse, bilateral, photodistributed pattern.10,15,16 One case presented with diffuse hyperpigmentation associated with LDE in a Japanese patient.20 The authors suggested this finding may be more prominent in patients with skin of color,20 which is consistent with the current case. Nail findings such as subungual hyperkeratosis and longitudinal ridging also have been reported.9,11
The latency period between initiation of imat-inib and onset of LDE generally ranges from 1 to 12 months, with onset most commonly occurring between 2 to 5 months or with dosage increase (eTable). Imatinib-induced LDE primarily has been documented with a 400-mg dose, with 1 case of a 600-mg dose and 1 case of an 800-mg dose, which suggests dose dependency. Furthermore, reports exist of several patients responding well to dose reduction with subsequent recurrence on dose reescalation.13,15
Historically, LDE resolves with discontinuation of the drug after a few weeks to months. When discontinuation of imatinib is unfavorable or patients report symptoms including severe pruritus or pain, treatment should be considered. Topical or oral corticosteroids can be used to treat imatinib-induced LDE, similar to lichen planus. When oral corticosteroids are contraindicated (eg, due to poor patient tolerance), oral acitretin at 25 to 35 mg once daily for 6 to 12 weeks has been reported as an alternative treatment.25
In the majority of cases of imatinib-induced LDE, it was undesirable to stop imatinib (eTable). Notably, in half the reported cases, imatinib was able to be continued and patients were treated symptomatically with either oral and/or topical steroids and/or acitretin with complete remission or tolerable recurrences. Dalmau et al9 reported 3 patients who responded poorly to topical and oral steroids and were subsequently treated with acitretin 25 mg once daily; 2 of 3 patients responded favorably to treatment and imatinib was able to be continued. In the current case imatinib initially helped, but because his rash was relatively asymptomatic, imatinib was restarted with control of rash with topical steroids. He developed some pancytopenia, which required intermittent stoppage of the imatinib.
Conclusion
We present a case of imatinib-induced cutaneous and oral LDE in a patient with GIST. Topical corticosteroids, oral acitretin, and oral steroids all may be reasonable treatment options if discontinuing imatinib is not possible in a symptomatic patient. If these therapies fail and the eruption is extensive or intolerable, dosage adjustment is another option to consider before discontinuation of imatinib.
Imatinib mesylate is a tyrosine kinase inhibitor initially approved by the US Food and Drug Administration in 2001 for chronic myeloid leukemia (CML). The indications for imatinib have expanded since its initial approval. It is increasingly important that dermatologists recognize adverse cutaneous manifestations associated with imatinib and are aware of their management and outcomes to avoid unnecessarily discontinuing a potentially lifesaving medication.
Adverse cutaneous manifestations in response to imatinib are not infrequent, accounting for 7% to 21% of all side effects.1 The most frequent cutaneous manifestations of imatinib are dry skin, alopecia, facial edema, and photosensitivity rash, respectively.1 Other less common manifestations include exfoliative dermatitis, nail disorders, psoriasis, folliculitis, hypotrichosis, urticaria, petechiae, Stevens-Johnson syndrome, erythema multiforme, Sweet syndrome, and leukocytoclastic vasculitis.
We report a case of imatinib-induced lichenoid drug eruption (LDE), a rare cutaneous side effect of imatinib use, along with a review of the literature.
Case Report
An 86-year-old man with a history of gastrointestinal stromal tumors (GISTs) and myelodysplastic syndrome presented with diffuse hyperpigmented skin lesions on the trunk, arms, legs, and lower lip of 2 weeks’ duration. He had been taking imatinib 400 mg once daily for 5 months for GIST. Although the oncologist stopped the medication 2 weeks prior, the lesions were persistent and gradually expanded to involve the trunk, arms, legs, and lower lip. He denied any pain or pruritus. Physical examination revealed multiple ill-defined, brown to violaceous, slightly scaly macules and patches on the trunk (Figures 1A and 1B), arms, and legs (Figure 1C), as well as violaceous to erythematous patches on the mucosal aspect of the lower lip (Figure 2). Two 4-mm punch biopsies were performed from the chest and back, which revealed an atrophic epidermis, lichenoid infiltration, and multiple melanophages in the upper dermis consistent with LDE (Figure 3). Direct immunofluorescence was negative. Therefore, based on the clinicopathologic correlation, the diagnosis of imatinib-induced LDE was made. He was treated with clobetasol ointment twice daily for 3 weeks with some improvement. His GIST was stable on follow-up computed tomography 3 months after presentation, and imatinib was resumed 1 month later with continued rash that was stable with topical corticosteroid treatment.
Comment
In addition to CML, imatinib has been approved for acute lymphoblastic leukemia, myelodysplastic syndromes, aggressive systemic mastocytosis, hypereosinophilic syndrome, chronic eosinophilic leukemia, dermatofibrosarcoma protuberans, and GIST. Moreover, off-label use of imatinib for various other tyrosine kinase–positive cancers and rheumatologic conditions have been documented.2,3 With the expanding use of imatinib, there will be more occasions for dermatologists to encounter cutaneous manifestations associated with its use.
According to a PubMed search of articles indexed for MEDLINE using the terms imatinib mesylate lichenoid drug, there have been few case reports of LDE associated with imatinib in the literature (eTable).4-24 Compared to classic LDE, imatinib-induced LDE has a few characteristic findings. Classic LDE frequently spares the oral mucosa and genitalia, but imatinib-induced LDE with manifestations on the oral mucosa and genitalia as well as cutaneous eruptions have been reported.4-9 In fact, the first known case of imatinib-induced LDE was an oral eruption in a patient with CML.4 In patients with oral involvement, lesions have been described as lacy reticular macules and violaceous papules, erosions, and ulcers.4,5,12 Interestingly, of those cases manifesting as concomitant oral and cutaneous LDE, the oral eruptions recurred more frequently, with 3 of 12 patients having recurrence of oral lesions after the cutaneous manifestations resolved.8,16 Genital manifestations of imatinib-induced LDE were much less common.9,11
To date, subsequent reports of imatinib-induced LDE have documented skin manifestations consistent with classic LDE occurring in a diffuse, bilateral, photodistributed pattern.10,15,16 One case presented with diffuse hyperpigmentation associated with LDE in a Japanese patient.20 The authors suggested this finding may be more prominent in patients with skin of color,20 which is consistent with the current case. Nail findings such as subungual hyperkeratosis and longitudinal ridging also have been reported.9,11
The latency period between initiation of imat-inib and onset of LDE generally ranges from 1 to 12 months, with onset most commonly occurring between 2 to 5 months or with dosage increase (eTable). Imatinib-induced LDE primarily has been documented with a 400-mg dose, with 1 case of a 600-mg dose and 1 case of an 800-mg dose, which suggests dose dependency. Furthermore, reports exist of several patients responding well to dose reduction with subsequent recurrence on dose reescalation.13,15
Historically, LDE resolves with discontinuation of the drug after a few weeks to months. When discontinuation of imatinib is unfavorable or patients report symptoms including severe pruritus or pain, treatment should be considered. Topical or oral corticosteroids can be used to treat imatinib-induced LDE, similar to lichen planus. When oral corticosteroids are contraindicated (eg, due to poor patient tolerance), oral acitretin at 25 to 35 mg once daily for 6 to 12 weeks has been reported as an alternative treatment.25
In the majority of cases of imatinib-induced LDE, it was undesirable to stop imatinib (eTable). Notably, in half the reported cases, imatinib was able to be continued and patients were treated symptomatically with either oral and/or topical steroids and/or acitretin with complete remission or tolerable recurrences. Dalmau et al9 reported 3 patients who responded poorly to topical and oral steroids and were subsequently treated with acitretin 25 mg once daily; 2 of 3 patients responded favorably to treatment and imatinib was able to be continued. In the current case imatinib initially helped, but because his rash was relatively asymptomatic, imatinib was restarted with control of rash with topical steroids. He developed some pancytopenia, which required intermittent stoppage of the imatinib.
Conclusion
We present a case of imatinib-induced cutaneous and oral LDE in a patient with GIST. Topical corticosteroids, oral acitretin, and oral steroids all may be reasonable treatment options if discontinuing imatinib is not possible in a symptomatic patient. If these therapies fail and the eruption is extensive or intolerable, dosage adjustment is another option to consider before discontinuation of imatinib.
- Scheinfeld N. Imatinib mesylate and dermatology part 2: a review of the cutaneous side effects of imatinib mesylate. J Drugs Dermatol. 2006;5:228-231.
- Kim H, Kim NH, Kang HJ, et al. Successful long-term use of imatinib mesylate in pediatric patients with sclerodermatous chronic GVHD. Pediatr Transplant. 2012;16:910-912.
- Prey S, Ezzedine K, Doussau A, et al. Imatinib mesylate in scleroderma-associated diffuse skin fibrosis: a phase II multicentre randomized double-blinded controlled trial. Br J Dermatol. 2012;167:1138-1144.
- Lim DS, Muir J. Oral lichenoid reaction to imatinib (STI 571, gleevec). Dermatology. 2002;205:169-171.
- Ena P, Chiarolini F, Siddi GM, et al. Oral lichenoid eruption secondary to imatinib (glivec). J Dermatolog Treat. 2004;15:253-255.
- Roux C, Boisseau-Garsaud AM, Saint-Cyr I, et al. Lichenoid cutaneous reaction to imatinib. Ann Dermatol Venereol. 2004;131:571-573.
- Prabhash K, Doval DC. Lichenoid eruption due to imat-inib. Indian J Dermatol Venereol Leprol. 2005;71:287-288.
- Pascual JC, Matarredona J, Miralles J, et al. Oral and cutaneous lichenoid reaction secondary to imatinib: report of two cases. Int J Dermatol. 2006;45:1471-1473.
- Dalmau J, Peramiquel L, Puig L, et al. Imatinib-associated lichenoid eruption: acitretin treatment allows maintained antineoplastic effect. Br J Dermatol. 2006;154:1213-1216.
- Chan CY, Browning J, Smith-Zagone MJ, et al. Cutaneous lichenoid dermatitis associated with imatinib mesylate. Dermatol Online J. 2007;13:29.
- Wahiduzzaman M, Pubalan M. Oral and cutaneous lichenoid reaction with nail changes secondary to imatinib: report of a case and literature review. Dermatol Online J. 2008;14:14.
- Basso FG, Boer CC, Correa ME, et al. Skin and oral lesions associated to imatinib mesylate therapy. Support Care Cancer. 2009;17:465-468.
- Kawakami T, Kawanabe T, Soma Y. Cutaneous lichenoid eruption caused by imatinib mesylate in a Japanese patient with chronic myeloid leukaemia. Acta Derm Venereol. 2009;89:325-326.
- Sendagorta E, Herranz P, Feito M, et al. Lichenoid drug eruption related to imatinib: report of a new case and review of the literature. Clin Exp Dermatol. 2009;34:E315-E316.
- Kuraishi N, Nagai Y, Hasegawa M, et al. Lichenoid drug eruption with palmoplantar hyperkeratosis due to imatinib mesylate: a case report and a review of the literature. Acta Derm Venereol. 2010;90:73-76.
- Brazzelli V, Muzio F, Manna G, et al. Photo-induced dermatitis and oral lichenoid reaction in a chronic myeloid leukemia patient treated with imatinib mesylate. Photodermatol Photoimmunol Photomed. 2012;28:2-5.
- Ghosh SK. Generalized lichenoid drug eruption associated with imatinib mesylate therapy. Indian J Dermatol. 2013;58:388-392.
- Lee J, Chung J, Jung M, et al. Lichenoid drug eruption after low-dose imatinib mesylate therapy. Ann Dermatol. 2013;25:500-502.
- Machaczka M, Gossart M. Multiple skin lesions caused by imatinib mesylate treatment of chronic myeloid leukemia. Pol Arch Med Wewn. 2013;123:251-252.
- Kagimoto Y, Mizuashi M, Kikuchi K, et al. Lichenoid drug eruption with hyperpigmentation caused by imatinib mesylate [published online June 20, 2013]. Int J Dermatol. 2014;53:E161-E162.
- Arshdeep, De D, Malhotra P, et al. Imatinib mesylate-induced severe lichenoid rash. Indian J Dermatol Venereol Leprol. 2014;80:93-95.
- Lau YM, Lam YK, Leung KH, et al. Trachyonychia in a patient with chronic myeloid leukaemia after imatinib mesylate. Hong Kong Med J. 2014;20:464.e2.
- Bhatia A, Kanish B, Chaudhary P. Lichenoid drug eruption due to imatinib mesylate. Int J Appl Basic Med Res. 2015;5:68-69.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Laurberg G, Geiger JM, Hjorth N, et al. Treatment of lichen planus with acitretin. a double-blind, placebo-controlled study in 65 patients. J Am Acad Dermatol. 1991;24:434-437.
- Scheinfeld N. Imatinib mesylate and dermatology part 2: a review of the cutaneous side effects of imatinib mesylate. J Drugs Dermatol. 2006;5:228-231.
- Kim H, Kim NH, Kang HJ, et al. Successful long-term use of imatinib mesylate in pediatric patients with sclerodermatous chronic GVHD. Pediatr Transplant. 2012;16:910-912.
- Prey S, Ezzedine K, Doussau A, et al. Imatinib mesylate in scleroderma-associated diffuse skin fibrosis: a phase II multicentre randomized double-blinded controlled trial. Br J Dermatol. 2012;167:1138-1144.
- Lim DS, Muir J. Oral lichenoid reaction to imatinib (STI 571, gleevec). Dermatology. 2002;205:169-171.
- Ena P, Chiarolini F, Siddi GM, et al. Oral lichenoid eruption secondary to imatinib (glivec). J Dermatolog Treat. 2004;15:253-255.
- Roux C, Boisseau-Garsaud AM, Saint-Cyr I, et al. Lichenoid cutaneous reaction to imatinib. Ann Dermatol Venereol. 2004;131:571-573.
- Prabhash K, Doval DC. Lichenoid eruption due to imat-inib. Indian J Dermatol Venereol Leprol. 2005;71:287-288.
- Pascual JC, Matarredona J, Miralles J, et al. Oral and cutaneous lichenoid reaction secondary to imatinib: report of two cases. Int J Dermatol. 2006;45:1471-1473.
- Dalmau J, Peramiquel L, Puig L, et al. Imatinib-associated lichenoid eruption: acitretin treatment allows maintained antineoplastic effect. Br J Dermatol. 2006;154:1213-1216.
- Chan CY, Browning J, Smith-Zagone MJ, et al. Cutaneous lichenoid dermatitis associated with imatinib mesylate. Dermatol Online J. 2007;13:29.
- Wahiduzzaman M, Pubalan M. Oral and cutaneous lichenoid reaction with nail changes secondary to imatinib: report of a case and literature review. Dermatol Online J. 2008;14:14.
- Basso FG, Boer CC, Correa ME, et al. Skin and oral lesions associated to imatinib mesylate therapy. Support Care Cancer. 2009;17:465-468.
- Kawakami T, Kawanabe T, Soma Y. Cutaneous lichenoid eruption caused by imatinib mesylate in a Japanese patient with chronic myeloid leukaemia. Acta Derm Venereol. 2009;89:325-326.
- Sendagorta E, Herranz P, Feito M, et al. Lichenoid drug eruption related to imatinib: report of a new case and review of the literature. Clin Exp Dermatol. 2009;34:E315-E316.
- Kuraishi N, Nagai Y, Hasegawa M, et al. Lichenoid drug eruption with palmoplantar hyperkeratosis due to imatinib mesylate: a case report and a review of the literature. Acta Derm Venereol. 2010;90:73-76.
- Brazzelli V, Muzio F, Manna G, et al. Photo-induced dermatitis and oral lichenoid reaction in a chronic myeloid leukemia patient treated with imatinib mesylate. Photodermatol Photoimmunol Photomed. 2012;28:2-5.
- Ghosh SK. Generalized lichenoid drug eruption associated with imatinib mesylate therapy. Indian J Dermatol. 2013;58:388-392.
- Lee J, Chung J, Jung M, et al. Lichenoid drug eruption after low-dose imatinib mesylate therapy. Ann Dermatol. 2013;25:500-502.
- Machaczka M, Gossart M. Multiple skin lesions caused by imatinib mesylate treatment of chronic myeloid leukemia. Pol Arch Med Wewn. 2013;123:251-252.
- Kagimoto Y, Mizuashi M, Kikuchi K, et al. Lichenoid drug eruption with hyperpigmentation caused by imatinib mesylate [published online June 20, 2013]. Int J Dermatol. 2014;53:E161-E162.
- Arshdeep, De D, Malhotra P, et al. Imatinib mesylate-induced severe lichenoid rash. Indian J Dermatol Venereol Leprol. 2014;80:93-95.
- Lau YM, Lam YK, Leung KH, et al. Trachyonychia in a patient with chronic myeloid leukaemia after imatinib mesylate. Hong Kong Med J. 2014;20:464.e2.
- Bhatia A, Kanish B, Chaudhary P. Lichenoid drug eruption due to imatinib mesylate. Int J Appl Basic Med Res. 2015;5:68-69.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Laurberg G, Geiger JM, Hjorth N, et al. Treatment of lichen planus with acitretin. a double-blind, placebo-controlled study in 65 patients. J Am Acad Dermatol. 1991;24:434-437.
Practice Points
- Imatinib mesylate can cause cutaneous adverse reactions including dry skin, alopecia, facial edema, photosensitivity rash, and lichenoid drug eruption (LDE).
- Topical corticosteroids, oral acitretin, and oral steroids may be reasonable treatment options for imatinib-induced LDE if discontinuing imatinib is not possible in a symptomatic patient.
Weakness and pain in arms and legs • dark urine • history of vertebral osteomyelitis • Dx?
THE CASE
A 76-year-old Caucasian woman presented to the emergency department with a 7-day history of weakness and pain in her arms and legs. She had a history of Candida albicans vertebral osteomyelitis that had been treated for 3 months with fluconazole; non-Hodgkin lymphoma that had been in remission for 6 months; diabetes mellitus; hyperlipidemia; and hypothyroidism. The woman had dark urine, but denied chills, fever, respiratory symptoms, bowel or bladder leakage, falls/trauma, or grapefruit juice intake.
Her current medications included oral fluconazole 400 mg/d, simvastatin 20 mg/d, levothyroxine 88 mcg/d, pregabalin 75 mg/d, metformin 1000 mg twice daily, 6 units of subcutaneous insulin glargine at bedtime, and 2 units of insulin lispro with each meal. During the examination, we noted marked proximal muscle weakness, significant tenderness in all extremities, and diminished deep tendon reflexes. The patient had no saddle anesthesia, impaired rectal tone, or sensory abnormalities.
THE DIAGNOSIS
Magnetic resonance imaging of the patient’s spine confirmed multilevel discitis and osteomyelitis (T7-T9, L5-S1) with no cord compression. Laboratory data included a creatinine level of 1.42 mg/dL (the patient’s baseline was 0.8 mg/dL); a creatine kinase (CK) level of 8876 U/L (normal range, 0-220 U/L); a thyroid-stimulating hormone (TSH) level of 9.35 mIU/L (normal range, 0.4-5.5 mIU/L); and an erythrocyte sedimentation rate of 27 mm/hr (normal range, 0-31 mm/hr).
The patient received aggressive fluid hydration, orally and intravenously. On Day 2, the patient’s serum myoglobin level was 14,301 ng/mL (normal range, 30-90 ng/mL) and her aldolase level was 87.6 U/L (normal range, 1.5-8.5 U/L).
Zeroing in on the cause. There were no signs of drug abuse or use of other non-statin culprit medications that could have caused the patient’s rhabdomyolysis. She also did not describe any triggers of rhabdomyolysis, such as trauma, viral infection, metabolic disturbances, or temperature dysregulation. We believed the most likely cause of our patient’s signs and symptoms was statin-induced rhabdomyolysis, likely due to an interaction between simvastatin and fluconazole. We considered hypothyroidism-induced rhabdomyolysis, but thought it was unlikely because the patient had a mildly increased TSH level on admission, and one would expect to see levels higher than 100 mIU/L.1-3
We also considered viral myositis in the differential, but it was an unlikely culprit because the patient lacked any history of fever or respiratory or gastrointestinal symptoms. And while paraneoplastic polymyositis could have caused the patient’s weakness, the marked muscle pain and acute kidney injury were far more suggestive of rhabdomyolysis.
DISCUSSION
Rhabdomyolysis is a serious complication of statin treatment. Both higher statin doses and pharmacokinetic factors can raise statin levels, leading to this serious muscle-related syndrome.4,5 Co-administration of statins with drugs that are strong inhibitors of cytochrome P450 (CYP) 3A4 (the main cytochrome P450 isoform that metabolizes most statins) can increase statin levels several fold.6,7 The trigger for our patient’s statin-induced rhabdomyolysis was fluconazole, a known moderate inhibitor of CYP3A4, which is comparatively weaker than certain potent azoles like itraconazole or ketoconazole.7-10 Doses of fluconazole generally ≥200 mg/d are needed to produce clinical interactions with CYP3A4 substrates.7 There are only 3 reported cases of fluconazole-simvastatin–induced rhabdomyolysis (TABLE 1).11-13
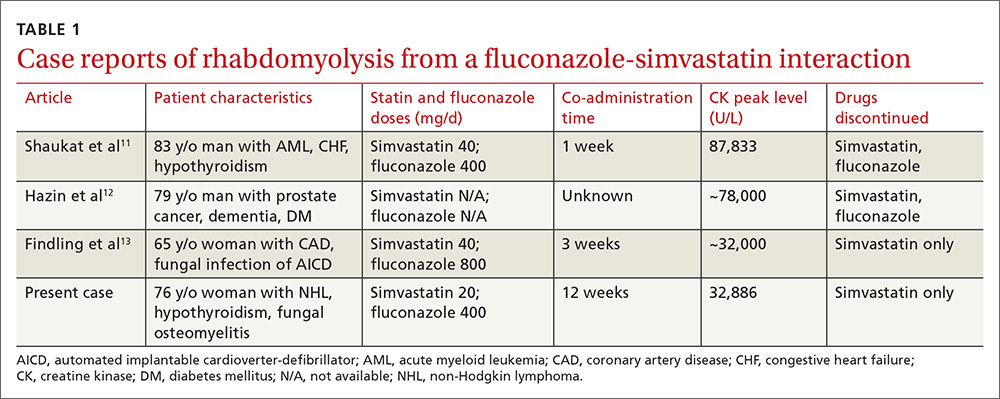
The Food and Drug Administration advises against simvastatin co-prescription with itraconazole and ketoconazole, but doesn’t mention fluconazole in its Drug Safety communication on simvastatin.14
Lexicomp places the simvastatin-fluconazole drug interaction into category C, which means that the agents can interact in a clinically significant manner (and a monitoring plan should be implemented), but that the benefits of concomitant use usually outweigh the risks.15
How our patient’s case differs from previous cases
Several features distinguish our patient’s scenario from previous cases. First, unlike other cases in which both drugs were stopped, only simvastatin was discontinued in our patient. Simvastatin and fluconazole have a half-life of 3 hours6 and 32 hours,7 respectively, suggesting that when simvastatin has fully cleared, fluconazole’s concentration will not even have halved. Thus, fluconazole was safely continued to treat the patient’s osteomyelitis.
Second, compared to previous case reports, our patient was taking a lower dose of simvastatin (20 mg). A 20-mg dose can make the drug interaction easier to miss; pharmacists are more likely to inform the physician of a potential drug interaction when the dose of a statin is ≥40 mg compared to when it is <40 mg (odds ratio=1.89; 95% confidence interval, 0.98-3.63).16
Researchers involved in the British randomized trial SEARCH (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine) sought to evaluate any added benefit to a higher dose of simvastatin in post-myocardial infarction patients. Among approximately 12,000 patients in the trial, there were 7 cases of rhabdomyolysis for the 80-mg simvastatin group and none for the 20-mg group.5 Another large case-control study showed that a 40-mg simvastatin dose was 5 times more likely to cause rhabdomyolysis than a 20-mg dose.17 Yet, based on our patient’s case, even 20 mg/d simvastatin should not decrease physician suspicion for rhabdomyolysis if patients are also taking a CYP3A4 inhibitor.
Third, the simvastatin-fluconazole co-administration time in our patient was 12 weeks, which is longer than previously reported (TABLE 111-13). Azole inhibition of CYP450 occurs relatively rapidly, but that does not mean that rhabdomyolysis will always occur immediately. For example, in cases of statin monotherapy, rhabdomyolysis secondary to statin biochemical toxicity can occur up to 1050 (mean=348) days after the drug’s initiation.18
Avoiding a drug-drug interaction in your patient
Physicians can use pharmacokinetic profiles to choose among different statins and azoles to help avoid a drug interaction (TABLE 26,7,10,19). Pravastatin’s serum concentration, for example, is not influenced by CYP3A4 inhibitors such as itraconazole11 because pravastatin is metabolized by sulfation6 and not by the CYP450 system. Rosuvastatin and pitavastatin are minimally metabolized by the CYP450 system.19,20

Among approximately 2700 statin-treated outpatients,4 the prevalence of potentially harmful statin interactions with other drugs (including CYP3A4 inhibitors), was significantly higher among patients treated with simvastatin or atorvastatin (CYP3A4-metabolized statins), than among patients treated with fluvastatin (CYP2C9-metabolized statin) or pravastatin (metabolized by sulfation). Apart from drug-drug interactions, other risk factors for statin-induced rhabdomyolysis include use of lipophilic statins, advanced age, and female gender.21
We discontinued our patient’s simvastatin on the day she was admitted to the hospital, but continued with the fluconazole throughout her hospitalization. Her CK level continued to rise, peaked on hospital Day 3 at 32,886 U/L, and then progressively decreased. The patient’s weakness and pain improved and her acute kidney injury resolved with hydration. She was discharged on hospital Day 7 on oral fluconazole, but no statin, and her muscle symptoms have since resolved.
THE TAKEAWAY
When hyperlipidemic patients have to take an azole for an extended period (eg, cancer prophylaxis or chronic osteomyelitis) and the azole is a strong CYP450 inhibitor (eg, itraconazole), switching to a statin that is not primarily metabolized by the CYP450 system (eg, pravastatin, pitavastatin) is wise. If the azole is a moderate CYP450 inhibitor (eg, fluconazole), we suggest that therapy should be closely monitored. In the case of short-term azole treatment (eg, such as for oral candidiasis), the statin should be stopped or the dose reduced by at least 50% (eg, from 40 or 20 mg to 10 mg).6
Prescriber knowledge is sometimes a limiting factor in identifying clinically significant interactions.22 This is especially pertinent in a case like this one, where a lower statin dose may result in a lower chance of the pharmacist alerting the prescribing physician16 and when an azole is used that is a comparatively weaker CYP450 inhibitor than other azoles such as itraconazole. Even in the era of electronic medical records, approximately 90% of drug interaction alerts are overridden by physicians, and alert fatigue is pronounced.23
The intricacies and pharmacokinetic principles of this case should contribute to greater provider familiarity with even low-dose simvastatin-fluconazole interactions and help prevent iatrogenic complications such as rhabdomyolysis.
1. Kisakol G, Tunc R, Kaya A. Rhabdomyolysis in a patient with hypothyroidism. Endocr J. 2003;50:221-223.
2. Scott KR, Simmons Z, Boyer PJ. Hypothyroid myopathy with a strikingly elevated serum creatine kinase level. Muscle Nerve. 2002;26:141-144.
3. Barahona MJ, Mauri A, Sucunza N, et al. Hypothyroidism as a cause of rhabdomyolysis. Endocr J. 2002;49:621-623.
4. Rätz Bravo AE, Tchambaz L, Krähenbühl-Melcher A, et al. Prevalence of potentially severe drug-drug interactions in ambulatory patients with dyslipidaemia receiving HMG-CoA reductase inhibitor therapy. Drug Saf. 2005;28:263-275.
5. Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group, Armitage J, Bowman L, Wallendszus K, et al. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet. 2010;376:1658-1669.
6. Chong PH, Seeger JD, Franklin C. Clinically relevant differences between the statins: implications for therapeutic selection. Am J Med. 2001;111:390-400.
7. Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of the antifungal agents on oxidative drug metabolism: clinical relevance. Clin Pharmacokinet. 2000;38:111-180.
8. Malhotra B, Dickins M, Alvey C, et al. Effects of the moderate CYP3A4 inhibitor, fluconazole, on the pharmacokinetics of fesoterodine in healthy subjects. Br J Clin Pharmacol. 2011;72:263-269.
9. US Food and Drug Administration. Drug development and drug interactions: Table of substrates, inhibitors and inducers. Available at: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm. Accessed February 9, 2017.
10. Niwa T, Shiraga T, Takagi A. Effect of antifungal drugs on cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 activities in human liver microsomes. Biol Pharm Bull. 2005;28:1805-1808.
11. Shaukat A, Benekli M, Vladutiu GD, et al. Simvastatin-fluconazole causing rhabdomyolysis. Ann Pharmacother. 2003;37:1032-1035.
12. Hazin R, Abuzetun JY, Suker M, et al. Rhabdomyolysis induced by simvastatin-fluconazole combination. J Natl Med Assoc. 2008;100:444-446.
13. Findling O, Meier N, Sellner J, et al. Clinical reasoning: rhabdomyolysis after combined treatment with simvastatin and fluconazole. Neurology. 2008;71:e34-e37.
14. US Food and Drug Administration. FDA Drug Safety Communication: New restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. June 8, 2011. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm256581.htm. Accessed February 1, 2017.
15. Wolters Kluwer. Lexicomp online. Available at: http://www.wolterskluwercdi.com/lexicomp-online/. Accessed February 9, 2017.
16. Molden E, Skovlund E, Braathen P. Risk management of simvastatin or atorvastatin interactions with CYP3A4 inhibitors. Drug Saf. 2008;31:587-596.
17. Parkin L, Paul C, Herbison GP. Simvastatin dose and risk of rhabdomyolysis: nested case-control study based on national health and drug dispensing data. Int J Cardiol. 2014;174:83-89.
18. Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292:2585-2590.
19. Saito Y. Pitavastatin: an overview. Atheroscler Suppl. 2011;12:271-276.
20. Olsson AG, McTaggart F, Raza A. Rosuvastatin: a highly effective new HMG-CoA reductase inhibitor. Cardiovasc Drug Rev. 2002;20:303-328.
21. Magni P, Macchi C, Morlotti B, et al. Risk identification and possible countermeasures for muscle adverse effects during statin therapy. Eur J Intern Med. 2015;26:82-88.
22. Ko Y, Malone DC, Skrepnek GH, et al. Prescribers’ knowledge of and sources of information for potential drug-drug interactions: a postal survey of US prescribers. Drug Saf. 2008;31:525-536.
23. Phansalkar S, van der Sijs H, Tucker AD, et al. Drug-drug interactions that should be non-interruptive in order to reduce alert fatigue in electronic health records. J Am Med Inform Assoc. 2013;20:489-493.
THE CASE
A 76-year-old Caucasian woman presented to the emergency department with a 7-day history of weakness and pain in her arms and legs. She had a history of Candida albicans vertebral osteomyelitis that had been treated for 3 months with fluconazole; non-Hodgkin lymphoma that had been in remission for 6 months; diabetes mellitus; hyperlipidemia; and hypothyroidism. The woman had dark urine, but denied chills, fever, respiratory symptoms, bowel or bladder leakage, falls/trauma, or grapefruit juice intake.
Her current medications included oral fluconazole 400 mg/d, simvastatin 20 mg/d, levothyroxine 88 mcg/d, pregabalin 75 mg/d, metformin 1000 mg twice daily, 6 units of subcutaneous insulin glargine at bedtime, and 2 units of insulin lispro with each meal. During the examination, we noted marked proximal muscle weakness, significant tenderness in all extremities, and diminished deep tendon reflexes. The patient had no saddle anesthesia, impaired rectal tone, or sensory abnormalities.
THE DIAGNOSIS
Magnetic resonance imaging of the patient’s spine confirmed multilevel discitis and osteomyelitis (T7-T9, L5-S1) with no cord compression. Laboratory data included a creatinine level of 1.42 mg/dL (the patient’s baseline was 0.8 mg/dL); a creatine kinase (CK) level of 8876 U/L (normal range, 0-220 U/L); a thyroid-stimulating hormone (TSH) level of 9.35 mIU/L (normal range, 0.4-5.5 mIU/L); and an erythrocyte sedimentation rate of 27 mm/hr (normal range, 0-31 mm/hr).
The patient received aggressive fluid hydration, orally and intravenously. On Day 2, the patient’s serum myoglobin level was 14,301 ng/mL (normal range, 30-90 ng/mL) and her aldolase level was 87.6 U/L (normal range, 1.5-8.5 U/L).
Zeroing in on the cause. There were no signs of drug abuse or use of other non-statin culprit medications that could have caused the patient’s rhabdomyolysis. She also did not describe any triggers of rhabdomyolysis, such as trauma, viral infection, metabolic disturbances, or temperature dysregulation. We believed the most likely cause of our patient’s signs and symptoms was statin-induced rhabdomyolysis, likely due to an interaction between simvastatin and fluconazole. We considered hypothyroidism-induced rhabdomyolysis, but thought it was unlikely because the patient had a mildly increased TSH level on admission, and one would expect to see levels higher than 100 mIU/L.1-3
We also considered viral myositis in the differential, but it was an unlikely culprit because the patient lacked any history of fever or respiratory or gastrointestinal symptoms. And while paraneoplastic polymyositis could have caused the patient’s weakness, the marked muscle pain and acute kidney injury were far more suggestive of rhabdomyolysis.
DISCUSSION
Rhabdomyolysis is a serious complication of statin treatment. Both higher statin doses and pharmacokinetic factors can raise statin levels, leading to this serious muscle-related syndrome.4,5 Co-administration of statins with drugs that are strong inhibitors of cytochrome P450 (CYP) 3A4 (the main cytochrome P450 isoform that metabolizes most statins) can increase statin levels several fold.6,7 The trigger for our patient’s statin-induced rhabdomyolysis was fluconazole, a known moderate inhibitor of CYP3A4, which is comparatively weaker than certain potent azoles like itraconazole or ketoconazole.7-10 Doses of fluconazole generally ≥200 mg/d are needed to produce clinical interactions with CYP3A4 substrates.7 There are only 3 reported cases of fluconazole-simvastatin–induced rhabdomyolysis (TABLE 1).11-13
The Food and Drug Administration advises against simvastatin co-prescription with itraconazole and ketoconazole, but doesn’t mention fluconazole in its Drug Safety communication on simvastatin.14
Lexicomp places the simvastatin-fluconazole drug interaction into category C, which means that the agents can interact in a clinically significant manner (and a monitoring plan should be implemented), but that the benefits of concomitant use usually outweigh the risks.15
How our patient’s case differs from previous cases
Several features distinguish our patient’s scenario from previous cases. First, unlike other cases in which both drugs were stopped, only simvastatin was discontinued in our patient. Simvastatin and fluconazole have a half-life of 3 hours6 and 32 hours,7 respectively, suggesting that when simvastatin has fully cleared, fluconazole’s concentration will not even have halved. Thus, fluconazole was safely continued to treat the patient’s osteomyelitis.
Second, compared to previous case reports, our patient was taking a lower dose of simvastatin (20 mg). A 20-mg dose can make the drug interaction easier to miss; pharmacists are more likely to inform the physician of a potential drug interaction when the dose of a statin is ≥40 mg compared to when it is <40 mg (odds ratio=1.89; 95% confidence interval, 0.98-3.63).16
Researchers involved in the British randomized trial SEARCH (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine) sought to evaluate any added benefit to a higher dose of simvastatin in post-myocardial infarction patients. Among approximately 12,000 patients in the trial, there were 7 cases of rhabdomyolysis for the 80-mg simvastatin group and none for the 20-mg group.5 Another large case-control study showed that a 40-mg simvastatin dose was 5 times more likely to cause rhabdomyolysis than a 20-mg dose.17 Yet, based on our patient’s case, even 20 mg/d simvastatin should not decrease physician suspicion for rhabdomyolysis if patients are also taking a CYP3A4 inhibitor.
Third, the simvastatin-fluconazole co-administration time in our patient was 12 weeks, which is longer than previously reported (TABLE 111-13). Azole inhibition of CYP450 occurs relatively rapidly, but that does not mean that rhabdomyolysis will always occur immediately. For example, in cases of statin monotherapy, rhabdomyolysis secondary to statin biochemical toxicity can occur up to 1050 (mean=348) days after the drug’s initiation.18
Avoiding a drug-drug interaction in your patient
Physicians can use pharmacokinetic profiles to choose among different statins and azoles to help avoid a drug interaction (TABLE 26,7,10,19). Pravastatin’s serum concentration, for example, is not influenced by CYP3A4 inhibitors such as itraconazole11 because pravastatin is metabolized by sulfation6 and not by the CYP450 system. Rosuvastatin and pitavastatin are minimally metabolized by the CYP450 system.19,20
Among approximately 2700 statin-treated outpatients,4 the prevalence of potentially harmful statin interactions with other drugs (including CYP3A4 inhibitors), was significantly higher among patients treated with simvastatin or atorvastatin (CYP3A4-metabolized statins), than among patients treated with fluvastatin (CYP2C9-metabolized statin) or pravastatin (metabolized by sulfation). Apart from drug-drug interactions, other risk factors for statin-induced rhabdomyolysis include use of lipophilic statins, advanced age, and female gender.21
We discontinued our patient’s simvastatin on the day she was admitted to the hospital, but continued with the fluconazole throughout her hospitalization. Her CK level continued to rise, peaked on hospital Day 3 at 32,886 U/L, and then progressively decreased. The patient’s weakness and pain improved and her acute kidney injury resolved with hydration. She was discharged on hospital Day 7 on oral fluconazole, but no statin, and her muscle symptoms have since resolved.
THE TAKEAWAY
When hyperlipidemic patients have to take an azole for an extended period (eg, cancer prophylaxis or chronic osteomyelitis) and the azole is a strong CYP450 inhibitor (eg, itraconazole), switching to a statin that is not primarily metabolized by the CYP450 system (eg, pravastatin, pitavastatin) is wise. If the azole is a moderate CYP450 inhibitor (eg, fluconazole), we suggest that therapy should be closely monitored. In the case of short-term azole treatment (eg, such as for oral candidiasis), the statin should be stopped or the dose reduced by at least 50% (eg, from 40 or 20 mg to 10 mg).6
Prescriber knowledge is sometimes a limiting factor in identifying clinically significant interactions.22 This is especially pertinent in a case like this one, where a lower statin dose may result in a lower chance of the pharmacist alerting the prescribing physician16 and when an azole is used that is a comparatively weaker CYP450 inhibitor than other azoles such as itraconazole. Even in the era of electronic medical records, approximately 90% of drug interaction alerts are overridden by physicians, and alert fatigue is pronounced.23
The intricacies and pharmacokinetic principles of this case should contribute to greater provider familiarity with even low-dose simvastatin-fluconazole interactions and help prevent iatrogenic complications such as rhabdomyolysis.
THE CASE
A 76-year-old Caucasian woman presented to the emergency department with a 7-day history of weakness and pain in her arms and legs. She had a history of Candida albicans vertebral osteomyelitis that had been treated for 3 months with fluconazole; non-Hodgkin lymphoma that had been in remission for 6 months; diabetes mellitus; hyperlipidemia; and hypothyroidism. The woman had dark urine, but denied chills, fever, respiratory symptoms, bowel or bladder leakage, falls/trauma, or grapefruit juice intake.
Her current medications included oral fluconazole 400 mg/d, simvastatin 20 mg/d, levothyroxine 88 mcg/d, pregabalin 75 mg/d, metformin 1000 mg twice daily, 6 units of subcutaneous insulin glargine at bedtime, and 2 units of insulin lispro with each meal. During the examination, we noted marked proximal muscle weakness, significant tenderness in all extremities, and diminished deep tendon reflexes. The patient had no saddle anesthesia, impaired rectal tone, or sensory abnormalities.
THE DIAGNOSIS
Magnetic resonance imaging of the patient’s spine confirmed multilevel discitis and osteomyelitis (T7-T9, L5-S1) with no cord compression. Laboratory data included a creatinine level of 1.42 mg/dL (the patient’s baseline was 0.8 mg/dL); a creatine kinase (CK) level of 8876 U/L (normal range, 0-220 U/L); a thyroid-stimulating hormone (TSH) level of 9.35 mIU/L (normal range, 0.4-5.5 mIU/L); and an erythrocyte sedimentation rate of 27 mm/hr (normal range, 0-31 mm/hr).
The patient received aggressive fluid hydration, orally and intravenously. On Day 2, the patient’s serum myoglobin level was 14,301 ng/mL (normal range, 30-90 ng/mL) and her aldolase level was 87.6 U/L (normal range, 1.5-8.5 U/L).
Zeroing in on the cause. There were no signs of drug abuse or use of other non-statin culprit medications that could have caused the patient’s rhabdomyolysis. She also did not describe any triggers of rhabdomyolysis, such as trauma, viral infection, metabolic disturbances, or temperature dysregulation. We believed the most likely cause of our patient’s signs and symptoms was statin-induced rhabdomyolysis, likely due to an interaction between simvastatin and fluconazole. We considered hypothyroidism-induced rhabdomyolysis, but thought it was unlikely because the patient had a mildly increased TSH level on admission, and one would expect to see levels higher than 100 mIU/L.1-3
We also considered viral myositis in the differential, but it was an unlikely culprit because the patient lacked any history of fever or respiratory or gastrointestinal symptoms. And while paraneoplastic polymyositis could have caused the patient’s weakness, the marked muscle pain and acute kidney injury were far more suggestive of rhabdomyolysis.
DISCUSSION
Rhabdomyolysis is a serious complication of statin treatment. Both higher statin doses and pharmacokinetic factors can raise statin levels, leading to this serious muscle-related syndrome.4,5 Co-administration of statins with drugs that are strong inhibitors of cytochrome P450 (CYP) 3A4 (the main cytochrome P450 isoform that metabolizes most statins) can increase statin levels several fold.6,7 The trigger for our patient’s statin-induced rhabdomyolysis was fluconazole, a known moderate inhibitor of CYP3A4, which is comparatively weaker than certain potent azoles like itraconazole or ketoconazole.7-10 Doses of fluconazole generally ≥200 mg/d are needed to produce clinical interactions with CYP3A4 substrates.7 There are only 3 reported cases of fluconazole-simvastatin–induced rhabdomyolysis (TABLE 1).11-13
The Food and Drug Administration advises against simvastatin co-prescription with itraconazole and ketoconazole, but doesn’t mention fluconazole in its Drug Safety communication on simvastatin.14
Lexicomp places the simvastatin-fluconazole drug interaction into category C, which means that the agents can interact in a clinically significant manner (and a monitoring plan should be implemented), but that the benefits of concomitant use usually outweigh the risks.15
How our patient’s case differs from previous cases
Several features distinguish our patient’s scenario from previous cases. First, unlike other cases in which both drugs were stopped, only simvastatin was discontinued in our patient. Simvastatin and fluconazole have a half-life of 3 hours6 and 32 hours,7 respectively, suggesting that when simvastatin has fully cleared, fluconazole’s concentration will not even have halved. Thus, fluconazole was safely continued to treat the patient’s osteomyelitis.
Second, compared to previous case reports, our patient was taking a lower dose of simvastatin (20 mg). A 20-mg dose can make the drug interaction easier to miss; pharmacists are more likely to inform the physician of a potential drug interaction when the dose of a statin is ≥40 mg compared to when it is <40 mg (odds ratio=1.89; 95% confidence interval, 0.98-3.63).16
Researchers involved in the British randomized trial SEARCH (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine) sought to evaluate any added benefit to a higher dose of simvastatin in post-myocardial infarction patients. Among approximately 12,000 patients in the trial, there were 7 cases of rhabdomyolysis for the 80-mg simvastatin group and none for the 20-mg group.5 Another large case-control study showed that a 40-mg simvastatin dose was 5 times more likely to cause rhabdomyolysis than a 20-mg dose.17 Yet, based on our patient’s case, even 20 mg/d simvastatin should not decrease physician suspicion for rhabdomyolysis if patients are also taking a CYP3A4 inhibitor.
Third, the simvastatin-fluconazole co-administration time in our patient was 12 weeks, which is longer than previously reported (TABLE 111-13). Azole inhibition of CYP450 occurs relatively rapidly, but that does not mean that rhabdomyolysis will always occur immediately. For example, in cases of statin monotherapy, rhabdomyolysis secondary to statin biochemical toxicity can occur up to 1050 (mean=348) days after the drug’s initiation.18
Avoiding a drug-drug interaction in your patient
Physicians can use pharmacokinetic profiles to choose among different statins and azoles to help avoid a drug interaction (TABLE 26,7,10,19). Pravastatin’s serum concentration, for example, is not influenced by CYP3A4 inhibitors such as itraconazole11 because pravastatin is metabolized by sulfation6 and not by the CYP450 system. Rosuvastatin and pitavastatin are minimally metabolized by the CYP450 system.19,20
Among approximately 2700 statin-treated outpatients,4 the prevalence of potentially harmful statin interactions with other drugs (including CYP3A4 inhibitors), was significantly higher among patients treated with simvastatin or atorvastatin (CYP3A4-metabolized statins), than among patients treated with fluvastatin (CYP2C9-metabolized statin) or pravastatin (metabolized by sulfation). Apart from drug-drug interactions, other risk factors for statin-induced rhabdomyolysis include use of lipophilic statins, advanced age, and female gender.21
We discontinued our patient’s simvastatin on the day she was admitted to the hospital, but continued with the fluconazole throughout her hospitalization. Her CK level continued to rise, peaked on hospital Day 3 at 32,886 U/L, and then progressively decreased. The patient’s weakness and pain improved and her acute kidney injury resolved with hydration. She was discharged on hospital Day 7 on oral fluconazole, but no statin, and her muscle symptoms have since resolved.
THE TAKEAWAY
When hyperlipidemic patients have to take an azole for an extended period (eg, cancer prophylaxis or chronic osteomyelitis) and the azole is a strong CYP450 inhibitor (eg, itraconazole), switching to a statin that is not primarily metabolized by the CYP450 system (eg, pravastatin, pitavastatin) is wise. If the azole is a moderate CYP450 inhibitor (eg, fluconazole), we suggest that therapy should be closely monitored. In the case of short-term azole treatment (eg, such as for oral candidiasis), the statin should be stopped or the dose reduced by at least 50% (eg, from 40 or 20 mg to 10 mg).6
Prescriber knowledge is sometimes a limiting factor in identifying clinically significant interactions.22 This is especially pertinent in a case like this one, where a lower statin dose may result in a lower chance of the pharmacist alerting the prescribing physician16 and when an azole is used that is a comparatively weaker CYP450 inhibitor than other azoles such as itraconazole. Even in the era of electronic medical records, approximately 90% of drug interaction alerts are overridden by physicians, and alert fatigue is pronounced.23
The intricacies and pharmacokinetic principles of this case should contribute to greater provider familiarity with even low-dose simvastatin-fluconazole interactions and help prevent iatrogenic complications such as rhabdomyolysis.
1. Kisakol G, Tunc R, Kaya A. Rhabdomyolysis in a patient with hypothyroidism. Endocr J. 2003;50:221-223.
2. Scott KR, Simmons Z, Boyer PJ. Hypothyroid myopathy with a strikingly elevated serum creatine kinase level. Muscle Nerve. 2002;26:141-144.
3. Barahona MJ, Mauri A, Sucunza N, et al. Hypothyroidism as a cause of rhabdomyolysis. Endocr J. 2002;49:621-623.
4. Rätz Bravo AE, Tchambaz L, Krähenbühl-Melcher A, et al. Prevalence of potentially severe drug-drug interactions in ambulatory patients with dyslipidaemia receiving HMG-CoA reductase inhibitor therapy. Drug Saf. 2005;28:263-275.
5. Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group, Armitage J, Bowman L, Wallendszus K, et al. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet. 2010;376:1658-1669.
6. Chong PH, Seeger JD, Franklin C. Clinically relevant differences between the statins: implications for therapeutic selection. Am J Med. 2001;111:390-400.
7. Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of the antifungal agents on oxidative drug metabolism: clinical relevance. Clin Pharmacokinet. 2000;38:111-180.
8. Malhotra B, Dickins M, Alvey C, et al. Effects of the moderate CYP3A4 inhibitor, fluconazole, on the pharmacokinetics of fesoterodine in healthy subjects. Br J Clin Pharmacol. 2011;72:263-269.
9. US Food and Drug Administration. Drug development and drug interactions: Table of substrates, inhibitors and inducers. Available at: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm. Accessed February 9, 2017.
10. Niwa T, Shiraga T, Takagi A. Effect of antifungal drugs on cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 activities in human liver microsomes. Biol Pharm Bull. 2005;28:1805-1808.
11. Shaukat A, Benekli M, Vladutiu GD, et al. Simvastatin-fluconazole causing rhabdomyolysis. Ann Pharmacother. 2003;37:1032-1035.
12. Hazin R, Abuzetun JY, Suker M, et al. Rhabdomyolysis induced by simvastatin-fluconazole combination. J Natl Med Assoc. 2008;100:444-446.
13. Findling O, Meier N, Sellner J, et al. Clinical reasoning: rhabdomyolysis after combined treatment with simvastatin and fluconazole. Neurology. 2008;71:e34-e37.
14. US Food and Drug Administration. FDA Drug Safety Communication: New restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. June 8, 2011. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm256581.htm. Accessed February 1, 2017.
15. Wolters Kluwer. Lexicomp online. Available at: http://www.wolterskluwercdi.com/lexicomp-online/. Accessed February 9, 2017.
16. Molden E, Skovlund E, Braathen P. Risk management of simvastatin or atorvastatin interactions with CYP3A4 inhibitors. Drug Saf. 2008;31:587-596.
17. Parkin L, Paul C, Herbison GP. Simvastatin dose and risk of rhabdomyolysis: nested case-control study based on national health and drug dispensing data. Int J Cardiol. 2014;174:83-89.
18. Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292:2585-2590.
19. Saito Y. Pitavastatin: an overview. Atheroscler Suppl. 2011;12:271-276.
20. Olsson AG, McTaggart F, Raza A. Rosuvastatin: a highly effective new HMG-CoA reductase inhibitor. Cardiovasc Drug Rev. 2002;20:303-328.
21. Magni P, Macchi C, Morlotti B, et al. Risk identification and possible countermeasures for muscle adverse effects during statin therapy. Eur J Intern Med. 2015;26:82-88.
22. Ko Y, Malone DC, Skrepnek GH, et al. Prescribers’ knowledge of and sources of information for potential drug-drug interactions: a postal survey of US prescribers. Drug Saf. 2008;31:525-536.
23. Phansalkar S, van der Sijs H, Tucker AD, et al. Drug-drug interactions that should be non-interruptive in order to reduce alert fatigue in electronic health records. J Am Med Inform Assoc. 2013;20:489-493.
1. Kisakol G, Tunc R, Kaya A. Rhabdomyolysis in a patient with hypothyroidism. Endocr J. 2003;50:221-223.
2. Scott KR, Simmons Z, Boyer PJ. Hypothyroid myopathy with a strikingly elevated serum creatine kinase level. Muscle Nerve. 2002;26:141-144.
3. Barahona MJ, Mauri A, Sucunza N, et al. Hypothyroidism as a cause of rhabdomyolysis. Endocr J. 2002;49:621-623.
4. Rätz Bravo AE, Tchambaz L, Krähenbühl-Melcher A, et al. Prevalence of potentially severe drug-drug interactions in ambulatory patients with dyslipidaemia receiving HMG-CoA reductase inhibitor therapy. Drug Saf. 2005;28:263-275.
5. Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group, Armitage J, Bowman L, Wallendszus K, et al. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet. 2010;376:1658-1669.
6. Chong PH, Seeger JD, Franklin C. Clinically relevant differences between the statins: implications for therapeutic selection. Am J Med. 2001;111:390-400.
7. Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of the antifungal agents on oxidative drug metabolism: clinical relevance. Clin Pharmacokinet. 2000;38:111-180.
8. Malhotra B, Dickins M, Alvey C, et al. Effects of the moderate CYP3A4 inhibitor, fluconazole, on the pharmacokinetics of fesoterodine in healthy subjects. Br J Clin Pharmacol. 2011;72:263-269.
9. US Food and Drug Administration. Drug development and drug interactions: Table of substrates, inhibitors and inducers. Available at: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm. Accessed February 9, 2017.
10. Niwa T, Shiraga T, Takagi A. Effect of antifungal drugs on cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 activities in human liver microsomes. Biol Pharm Bull. 2005;28:1805-1808.
11. Shaukat A, Benekli M, Vladutiu GD, et al. Simvastatin-fluconazole causing rhabdomyolysis. Ann Pharmacother. 2003;37:1032-1035.
12. Hazin R, Abuzetun JY, Suker M, et al. Rhabdomyolysis induced by simvastatin-fluconazole combination. J Natl Med Assoc. 2008;100:444-446.
13. Findling O, Meier N, Sellner J, et al. Clinical reasoning: rhabdomyolysis after combined treatment with simvastatin and fluconazole. Neurology. 2008;71:e34-e37.
14. US Food and Drug Administration. FDA Drug Safety Communication: New restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. June 8, 2011. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm256581.htm. Accessed February 1, 2017.
15. Wolters Kluwer. Lexicomp online. Available at: http://www.wolterskluwercdi.com/lexicomp-online/. Accessed February 9, 2017.
16. Molden E, Skovlund E, Braathen P. Risk management of simvastatin or atorvastatin interactions with CYP3A4 inhibitors. Drug Saf. 2008;31:587-596.
17. Parkin L, Paul C, Herbison GP. Simvastatin dose and risk of rhabdomyolysis: nested case-control study based on national health and drug dispensing data. Int J Cardiol. 2014;174:83-89.
18. Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292:2585-2590.
19. Saito Y. Pitavastatin: an overview. Atheroscler Suppl. 2011;12:271-276.
20. Olsson AG, McTaggart F, Raza A. Rosuvastatin: a highly effective new HMG-CoA reductase inhibitor. Cardiovasc Drug Rev. 2002;20:303-328.
21. Magni P, Macchi C, Morlotti B, et al. Risk identification and possible countermeasures for muscle adverse effects during statin therapy. Eur J Intern Med. 2015;26:82-88.
22. Ko Y, Malone DC, Skrepnek GH, et al. Prescribers’ knowledge of and sources of information for potential drug-drug interactions: a postal survey of US prescribers. Drug Saf. 2008;31:525-536.
23. Phansalkar S, van der Sijs H, Tucker AD, et al. Drug-drug interactions that should be non-interruptive in order to reduce alert fatigue in electronic health records. J Am Med Inform Assoc. 2013;20:489-493.
Allergic Reaction to Phenylephrine
Phenylephrine, a sympathomimetic drug, is commonly used in eye exams to dilate the pupil of the eye and to differentiate scleritis from episcleritis. Common adverse effects (AEs) of phenylephrine include subjective burning, stinging with lacrimation, rebound hyperemia, and liberation of iris pigment into the anterior chamber. Less common, systemic AEs include tachycardia and elevation of systemic blood pressure. Although instances of allergic reactions are rare, phenylephrine has been reported to cause contact dermatitis, blepharoconjunctivitis, and as in this case, keratoconjunctivitis.
Case Report
An 83-year-old white male presented for a red eye evaluation 2 days after having undergone a comprehensive eye exam with dilation at the Malcom Randall VAMC clinic in Gainesville, Florida. The patient reported onset of blurred vision, which he described as looking through a fog. He further compared the feeling to pins sticking in his eyes. The patient noted he had experienced similar symptoms on a few other occasions following eye exams. At the most recent eye exam, proparacaine and fluorescein had been used for tonometry, and phenylephrine 2.5% and tropicamide 0.5% had been used for pupillary dilation.
The patient’s best-corrected visual acuity was counting fingers at 2 feet in the right eye (OD) and left eye (OS). The best-corrected visual acuity 2 days prior had been 20/20 OD and OS. Pupils and extraocular motilities were unremarkable. Intraocular pressures were not obtained due to concern for a possible adverse reaction to proparacaine.
Slit-lamp evaluation revealed the lids to be lax, erythematous, and edematous in both eyes (Figure 1).

The initial diagnosis was acute chemical conjunctivitis most likely due to an AE to proparacaine. The plan was to start the patient on antibiotic eye drops qid OU, prednisolone qid OU, and artificial tears every hour OU. The patient was scheduled to return to clinic 4 days later for an anterior segment follow-up.
At the follow-up visit, the patient reported significant visual improvement. His best-corrected visual acuity was 20/40-2 without improvement on pinhole OD and 20/50-2 with improvement to 20/30+ on pinhole OS. Slit-lamp evaluation revealed 1+ bulbar conjunctival injection OU, intact corneal epithelium OU, and no cells or flare in the anterior chambers OU. Due to improving punctate epitheliopathy, the frequency of the antibiotic drops, the prednisolone, and the artificial tears was reduced to bid. After 3 days, he was instructed to discontinue them. The patient was scheduled to return in 2 weeks for an anterior segment follow-up.
At the next follow-up visit, the patient reported that his vision had returned to normal, and he had no further ocular AEs. His best-corrected visual acuity was 20/20-2 OD and 20/20 OS. Slit-lamp evaluation revealed mild blepharitis OU, trace bulbar conjunctival injection OU, and complete resolution of the keratitis OU. The assessment was acute allergic conjunctivitis thought to be secondary to an AE to proparacaine OU, yet the need to rule out hypersensitivity to tropicamide and/or phenylephrine remained. The plan was to educate the patient of the possibility of allergic reaction on future visits and to recommend continued use of artificial tears as needed.
Through a careful and extensive chart review of all past visits, it was suspected that phenylephrine might be to blame rather than proparacaine. At the subsequent visit, the patient agreed to undergo testing to determine the culprit via instillation of proparacaine in one eye and tropicamide in the other. The patient had no reaction to either drop (checked 45 minutes after instillation and the following day). By process of elimination, phenylephrine was determined to be the offending agent.
Discussion
Following a thorough review of the patient’s chart, it was found that on other occasions he had presented with suspected allergic reactions following routine eye examinations. The patient reported he had experienced a reaction in 2007 but could not recall what drops were instilled in his eyes at the time. In addition, there was no documentation in his medical record of the subsequent reaction following that visit. Another reaction occurred in July 2010 with instillation of tropicamide 1%, phenylephrine 2.5%, and Fluress (fluorescein sodium and benoxinate hydrochloride ophthalmic solution USP). In October 2013, when tropicamide 0.5%, proparacaine, and fluorescein strips were instilled, there was no reaction. The next reaction occurred in October 2014, when tropicamide 0.5%, phenylephrine 2.5%, proparacaine, and fluorescein strips were instilled.
This careful review of past exam notes revealed that phenylephrine and Fluress were the only drops that had not been instilled at the October 2013 visit when no AE was reported. However, Fluress was an unlikely culprit since it was not instilled in October 2014, and the patient still experienced an AE. Therefore, the agent most likely responsible for the allergic reaction in the patient, as confirmed by a review of the past notes and by the aforementioned pharmacologic test, was deemed to be phenylephrine (Table).
Adverse reactions to topical ocular medications and specifically to diagnostic eye drops have long been recognized. Mathias, Camarasa, Barber, Ducombs, and Monsálvezhave reported on variations of conjunctivitis and periorbital erythema with positive patch testing to phenylephrine.1-5 Geyer and colleagues reported on a study of 21 patients who had blepharoconjunctivitis after instillation of phenylephrine.6 In this case study patient, severe keratoconjunctivitis was the clinical manifestation observed.
Villarreal and colleagues studied 31 patients who had a previous reaction to mydriatic drops. The study found that phenylephrine was the drug that most frequently caused an AE (93.5%).7 One patient reacted to the preservative thimerosal, and 1 patient reacted to benoxiprocaine. Tropicamide was demonstrated to be very well tolerated as none of the patients tested positive on either the patch test or the pharmacologic test.
Tropicamide is a nonselective muscarinic antagonist commonly used for mydriasis due to its fast onset and short duration.8 Adverse reactions to tropicamide are rare. Three studies reported on patients who had a positive patch test to tropicamide.9-11 However, the reaction was not provoked by direct instillation of tropicamide into the eye.
Common in-office topical anesthetics, proparacaine, tetracaine, benoxinate, and lidocaine also can cause AEs. Corneal toxicity is a well-known complication with topical anesthetic abuse, whereas allergic reactions are considered rare. The most common symptoms include stingingand discomfort upon instillation. Common signs include punctate corneal epithelial erosionsresulting indirectly from a decrease in reflex tearing, infrequent blinking, and increased tear evaporation.12 Topical anesthetics also inhibit the migration of corneal epithelial cells and cause direct damage to the cells that are present, leading to impaired healing and epithelial defects.13
Manifestations of allergic reaction to topical anesthetics can include conjunctival hyperemia and edema, edematous eyelids, and lacrimation. One published case described a 60-year-old woman who developed eczematous dermatitis of the eyelids after ophthalmic anesthetic drops were instilled prior to laser surgery. Patch testing showed a positive response to benzocaine 5%, proparacaine, and tetracaine 0.5%.14
Preservatives, in general, can cause an allergic reaction. Benzalkonium chloride’s (BAK) cytotoxic sequelae include possible trabecular cell death in glaucoma patients, disruption of tear film stability (even at low concentrations), and immune-allergenic properties. One article reported BAK as one of the 30 most frequent allergens causing allergic periorbital dermatitis.15 Benzalkonium chloride is used in most brands of phenylephrine. However, preservatives in this patient’s case were ruled out as instigating agents since both phenylephrine and tropicamide contain the same preservative, BAK 0.01%, yet this patient did not develop a reaction to tropicamide when used without phenylephrine. Expired medications also were not considered to be a factor as none of the medications used on the patient were indeed expired (the Malcom Randall VAMC clinic maintains a strict policy of discarding medications 28 days after being opened).
Although uncommon, phenylephrine sometimes has been found to cause a type 4 hypersensitivity reaction, also known as cell-mediated or delayed-type hypersensitivity.16 First, helper T cells secrete cytokines. Activation of cytokines recruits and activates cytotoxic T cells, monocytes, and macrophages, leading to inflammation of the surrounding tissue. Examples of cell-mediated hypersensitivity include reactions to the tuberculin skin test and to poison ivy.
Type 1 hypersensitivity reactions, also known as immediate or anaphylactic hypersensitivity reactions, are not triggered by phenylephrine. In this type of reaction, IgE binds to the mast cell on initial exposure to an allergen. On second exposure, the allergen binds to the IgE, causing the mast cell to release mediators of inflammation, triggering physiologic responses. Examples of this type of hypersensitivity include those seen with penicillin, bee stings, hay fever, bronchial asthma, and food allergies, for example, to shellfish.
A toxic reaction’s mechanism differs from that of a type 4 hypersensitivity reaction. Toxic reactions occur due to direct cytotoxicity of a drug caused by a low or high pH and either hyper- or hypo-osmolarity. Toxicity can lead to corneal and conjunctival cell necrosis or induce apoptosis, stimulating inflammatory reactions. Clinically, toxic reactions will present with follicles, whereas allergic reactions will present with papillae.
The definitive diagnostic methods used to determine the allergic agent causing ocular or periocular AEs are patch testing and conjunctival challenge.7 Mathias, Camarasa, Barber, Ducombs,and Monsálvezused patch testing to confirm phenylephrine as the allergic agent in their series of cases. Patch testing entails the application of a small amount of an allergic agent that is taped onto the skin. The allergic agent is confirmed if the patient has a dermal reaction, wherein the area patched will become erythematous. When patch testing is negative or inconclusive, a conjunctival challenge is performed by instillation of the suspected allergic agent into the eye with subsequent observation to determine whether a reaction occurs. The sequelae found in Villarreal’s study included itching, lacrimation, edema, erythema, and sometimes blepharitis.7
A direct conjunctival challenge with the suspected culprit was not pursued in this patient’s case due to the known severity of the potential resulting reaction. The authors instead chose an indirect method of determining the implicating agent and used the process of elimination to whittle down the most likely suspect. A challenge with the medications suspected not to be likely offenders was undertaken. This spared the patient a likely repeat of the AE he had just recovered from.
Management
Allergic reactions can resolve without medical intervention. The first step is to remove the allergen. For delayed hypersensitivity reactions, treatments may include topical decongestants, cool compresses, and corticosteroids.8 The treatment for immediate hypersensitivity reaction differs from that of delayed hypersensitivity reaction in that antihistamines are used.17,18
This patient reported receiving no treatment for his ocular symptoms following eye examinations in the past, yet he experienced complete resolution after each AE. In this case, both a steroid and a prophylactic antibiotic to facilitate a more rapid improvement were used.
Conclusion
Although uncommon, cases of allergic reaction to phenylephrine can occur. The incidence of phenylephrine allergy is 0.6%.6 The case study patient presented with a severe keratoconjunctivitis following routine eye examination with an accompanying history of adverse ocular signs and symptoms following multiple past exams.
It is important for all eye care clinicians to realize that AEs to diagnostic eye drops are possible and can occur following the most routine of visits. Such reactions can be caused by dilating agents, anesthetics, or preservatives, and these may be allergic or toxic. Clinicians should take special care to identify the instigating agent, and if possible, to avoid using such agents on patients during future exams. Clinicians also should understand how best to manage iatrogenic AEs when they encounter them in order to restore a patient’s visual function as quickly as possible.
1. Mathias CG, Maibach HI, Irvine A, Adler W. Allergic contact dermatitis to echothiophate iodide and phenylephrine. Arch Ophthalmol. 1979;97(2):286-287.
2. Camarasa JG. Contact dermatitis to phenylephrine. Contact Dermatitis. 1984;10(3):182.
3. Barber K. Allergic contact eczema to phenylephrine. Contact Dermatitis. 1983;9(4):274-277.
4. Ducombs G, de Casamayor J, Verin P, Maleville J. Allergic contact dermatitis to phenylephrine. Contact Dermatitis. 1986;15(2):107-108.
5. Monsálvez V, Fuertes L, García-Cano I, Vanaclocha F, Ortez de Frutos J. Blepharoconjunctivitis due to phenylephrine [in Spanish]. Actas Dermosifiliogr. 2010;101(5):466-467.
6. Geyer O, Yust I, Lazar M. Allergic blepharoconjunctivitis due to phenylephrine. J Ocul Pharmacol. 1988;4(2):123-126.
7. Villarreal O. Reliability of diagnostic tests for contact allergy to mydriatic eyedrops. Contact Dermatitis. 1998;38(3):150-154.
8. Frazier M, Jaanus SD. Cycloplegics. In: Bartlett JD, Jaanus SD. Clinical Ocular Pharmacology. 5th ed. St. Louis, MO: Butterworth-Heinemann; 2009:125-138.
9. Decraene T, Goossens A. Contact allergy to atropine and other mydriatic agents in eye drops. Contact Dermatitis. 2001;45(5):309-310.
10. Boukhman MP, Maibach HI. Allergic contact dermatitis from tropicamide ophthalmic solution. Contact Dermatitis. 1999;41(1):47-48.
11. Yoshikawa K, Kawahara S. Contact allergy to atropine and other mydriatic agents. Contact Dermatitis. 1985;12(1):56-57.
12. Mcgee HT, Fraunfelder FW. Toxicities of topical ophthalmic anesthetics. Expert Opin Drug Saf. 2007;6(6):637-640.
13. Dass BA, Soong HK, Lee B. Effects of proparacaine of actin cytoskeleton of corneal epithelium. J Ocul Pharmacol. 1988;4(3):187-194.
14. Dannaker CJ, Maibach HI, Austin E. Allergic contact dermatitis to proparacaine with subsequent cross-sensitization to tetracaine from ophthalmic preparations. Am J Contact Dermat. 2001;12(3):177-179.
15. Hong J, Bielory L. Allergy to ophthalmic preservatives. Curr Opin Allergy Clin Immunol. 2009;9(5):447-453.
16. Gonzalo-Garijo MA, Pérez-Calderón R, de Argila D, Rodríguez-Nevado I. Erythrodermia to pseudoephedrine in a patient with contact allergy to phenylephrine. Allergol Immunopathol (Madr). 2002;30(4):239-242.
17. Platts-Mills TAE. Immediate hypersensitivity (Type I). In: Male D, Brostoff J, Roth DB, Roitt I. Immunology. 7th ed. Canada: Elsevier Limited; 2006:423-446.
18. Britton W. Type IV hypersensitivity. In: Male D, Brostoff J, Roth DB, Roitt I. Immunology. 7th ed. Canada: Elsevier Limited; 2006:477-491.
Phenylephrine, a sympathomimetic drug, is commonly used in eye exams to dilate the pupil of the eye and to differentiate scleritis from episcleritis. Common adverse effects (AEs) of phenylephrine include subjective burning, stinging with lacrimation, rebound hyperemia, and liberation of iris pigment into the anterior chamber. Less common, systemic AEs include tachycardia and elevation of systemic blood pressure. Although instances of allergic reactions are rare, phenylephrine has been reported to cause contact dermatitis, blepharoconjunctivitis, and as in this case, keratoconjunctivitis.
Case Report
An 83-year-old white male presented for a red eye evaluation 2 days after having undergone a comprehensive eye exam with dilation at the Malcom Randall VAMC clinic in Gainesville, Florida. The patient reported onset of blurred vision, which he described as looking through a fog. He further compared the feeling to pins sticking in his eyes. The patient noted he had experienced similar symptoms on a few other occasions following eye exams. At the most recent eye exam, proparacaine and fluorescein had been used for tonometry, and phenylephrine 2.5% and tropicamide 0.5% had been used for pupillary dilation.
The patient’s best-corrected visual acuity was counting fingers at 2 feet in the right eye (OD) and left eye (OS). The best-corrected visual acuity 2 days prior had been 20/20 OD and OS. Pupils and extraocular motilities were unremarkable. Intraocular pressures were not obtained due to concern for a possible adverse reaction to proparacaine.
Slit-lamp evaluation revealed the lids to be lax, erythematous, and edematous in both eyes (Figure 1).
The initial diagnosis was acute chemical conjunctivitis most likely due to an AE to proparacaine. The plan was to start the patient on antibiotic eye drops qid OU, prednisolone qid OU, and artificial tears every hour OU. The patient was scheduled to return to clinic 4 days later for an anterior segment follow-up.
At the follow-up visit, the patient reported significant visual improvement. His best-corrected visual acuity was 20/40-2 without improvement on pinhole OD and 20/50-2 with improvement to 20/30+ on pinhole OS. Slit-lamp evaluation revealed 1+ bulbar conjunctival injection OU, intact corneal epithelium OU, and no cells or flare in the anterior chambers OU. Due to improving punctate epitheliopathy, the frequency of the antibiotic drops, the prednisolone, and the artificial tears was reduced to bid. After 3 days, he was instructed to discontinue them. The patient was scheduled to return in 2 weeks for an anterior segment follow-up.
At the next follow-up visit, the patient reported that his vision had returned to normal, and he had no further ocular AEs. His best-corrected visual acuity was 20/20-2 OD and 20/20 OS. Slit-lamp evaluation revealed mild blepharitis OU, trace bulbar conjunctival injection OU, and complete resolution of the keratitis OU. The assessment was acute allergic conjunctivitis thought to be secondary to an AE to proparacaine OU, yet the need to rule out hypersensitivity to tropicamide and/or phenylephrine remained. The plan was to educate the patient of the possibility of allergic reaction on future visits and to recommend continued use of artificial tears as needed.
Through a careful and extensive chart review of all past visits, it was suspected that phenylephrine might be to blame rather than proparacaine. At the subsequent visit, the patient agreed to undergo testing to determine the culprit via instillation of proparacaine in one eye and tropicamide in the other. The patient had no reaction to either drop (checked 45 minutes after instillation and the following day). By process of elimination, phenylephrine was determined to be the offending agent.
Discussion
Following a thorough review of the patient’s chart, it was found that on other occasions he had presented with suspected allergic reactions following routine eye examinations. The patient reported he had experienced a reaction in 2007 but could not recall what drops were instilled in his eyes at the time. In addition, there was no documentation in his medical record of the subsequent reaction following that visit. Another reaction occurred in July 2010 with instillation of tropicamide 1%, phenylephrine 2.5%, and Fluress (fluorescein sodium and benoxinate hydrochloride ophthalmic solution USP). In October 2013, when tropicamide 0.5%, proparacaine, and fluorescein strips were instilled, there was no reaction. The next reaction occurred in October 2014, when tropicamide 0.5%, phenylephrine 2.5%, proparacaine, and fluorescein strips were instilled.
This careful review of past exam notes revealed that phenylephrine and Fluress were the only drops that had not been instilled at the October 2013 visit when no AE was reported. However, Fluress was an unlikely culprit since it was not instilled in October 2014, and the patient still experienced an AE. Therefore, the agent most likely responsible for the allergic reaction in the patient, as confirmed by a review of the past notes and by the aforementioned pharmacologic test, was deemed to be phenylephrine (Table).
Adverse reactions to topical ocular medications and specifically to diagnostic eye drops have long been recognized. Mathias, Camarasa, Barber, Ducombs, and Monsálvezhave reported on variations of conjunctivitis and periorbital erythema with positive patch testing to phenylephrine.1-5 Geyer and colleagues reported on a study of 21 patients who had blepharoconjunctivitis after instillation of phenylephrine.6 In this case study patient, severe keratoconjunctivitis was the clinical manifestation observed.
Villarreal and colleagues studied 31 patients who had a previous reaction to mydriatic drops. The study found that phenylephrine was the drug that most frequently caused an AE (93.5%).7 One patient reacted to the preservative thimerosal, and 1 patient reacted to benoxiprocaine. Tropicamide was demonstrated to be very well tolerated as none of the patients tested positive on either the patch test or the pharmacologic test.
Tropicamide is a nonselective muscarinic antagonist commonly used for mydriasis due to its fast onset and short duration.8 Adverse reactions to tropicamide are rare. Three studies reported on patients who had a positive patch test to tropicamide.9-11 However, the reaction was not provoked by direct instillation of tropicamide into the eye.
Common in-office topical anesthetics, proparacaine, tetracaine, benoxinate, and lidocaine also can cause AEs. Corneal toxicity is a well-known complication with topical anesthetic abuse, whereas allergic reactions are considered rare. The most common symptoms include stingingand discomfort upon instillation. Common signs include punctate corneal epithelial erosionsresulting indirectly from a decrease in reflex tearing, infrequent blinking, and increased tear evaporation.12 Topical anesthetics also inhibit the migration of corneal epithelial cells and cause direct damage to the cells that are present, leading to impaired healing and epithelial defects.13
Manifestations of allergic reaction to topical anesthetics can include conjunctival hyperemia and edema, edematous eyelids, and lacrimation. One published case described a 60-year-old woman who developed eczematous dermatitis of the eyelids after ophthalmic anesthetic drops were instilled prior to laser surgery. Patch testing showed a positive response to benzocaine 5%, proparacaine, and tetracaine 0.5%.14
Preservatives, in general, can cause an allergic reaction. Benzalkonium chloride’s (BAK) cytotoxic sequelae include possible trabecular cell death in glaucoma patients, disruption of tear film stability (even at low concentrations), and immune-allergenic properties. One article reported BAK as one of the 30 most frequent allergens causing allergic periorbital dermatitis.15 Benzalkonium chloride is used in most brands of phenylephrine. However, preservatives in this patient’s case were ruled out as instigating agents since both phenylephrine and tropicamide contain the same preservative, BAK 0.01%, yet this patient did not develop a reaction to tropicamide when used without phenylephrine. Expired medications also were not considered to be a factor as none of the medications used on the patient were indeed expired (the Malcom Randall VAMC clinic maintains a strict policy of discarding medications 28 days after being opened).
Although uncommon, phenylephrine sometimes has been found to cause a type 4 hypersensitivity reaction, also known as cell-mediated or delayed-type hypersensitivity.16 First, helper T cells secrete cytokines. Activation of cytokines recruits and activates cytotoxic T cells, monocytes, and macrophages, leading to inflammation of the surrounding tissue. Examples of cell-mediated hypersensitivity include reactions to the tuberculin skin test and to poison ivy.
Type 1 hypersensitivity reactions, also known as immediate or anaphylactic hypersensitivity reactions, are not triggered by phenylephrine. In this type of reaction, IgE binds to the mast cell on initial exposure to an allergen. On second exposure, the allergen binds to the IgE, causing the mast cell to release mediators of inflammation, triggering physiologic responses. Examples of this type of hypersensitivity include those seen with penicillin, bee stings, hay fever, bronchial asthma, and food allergies, for example, to shellfish.
A toxic reaction’s mechanism differs from that of a type 4 hypersensitivity reaction. Toxic reactions occur due to direct cytotoxicity of a drug caused by a low or high pH and either hyper- or hypo-osmolarity. Toxicity can lead to corneal and conjunctival cell necrosis or induce apoptosis, stimulating inflammatory reactions. Clinically, toxic reactions will present with follicles, whereas allergic reactions will present with papillae.
The definitive diagnostic methods used to determine the allergic agent causing ocular or periocular AEs are patch testing and conjunctival challenge.7 Mathias, Camarasa, Barber, Ducombs,and Monsálvezused patch testing to confirm phenylephrine as the allergic agent in their series of cases. Patch testing entails the application of a small amount of an allergic agent that is taped onto the skin. The allergic agent is confirmed if the patient has a dermal reaction, wherein the area patched will become erythematous. When patch testing is negative or inconclusive, a conjunctival challenge is performed by instillation of the suspected allergic agent into the eye with subsequent observation to determine whether a reaction occurs. The sequelae found in Villarreal’s study included itching, lacrimation, edema, erythema, and sometimes blepharitis.7
A direct conjunctival challenge with the suspected culprit was not pursued in this patient’s case due to the known severity of the potential resulting reaction. The authors instead chose an indirect method of determining the implicating agent and used the process of elimination to whittle down the most likely suspect. A challenge with the medications suspected not to be likely offenders was undertaken. This spared the patient a likely repeat of the AE he had just recovered from.
Management
Allergic reactions can resolve without medical intervention. The first step is to remove the allergen. For delayed hypersensitivity reactions, treatments may include topical decongestants, cool compresses, and corticosteroids.8 The treatment for immediate hypersensitivity reaction differs from that of delayed hypersensitivity reaction in that antihistamines are used.17,18
This patient reported receiving no treatment for his ocular symptoms following eye examinations in the past, yet he experienced complete resolution after each AE. In this case, both a steroid and a prophylactic antibiotic to facilitate a more rapid improvement were used.
Conclusion
Although uncommon, cases of allergic reaction to phenylephrine can occur. The incidence of phenylephrine allergy is 0.6%.6 The case study patient presented with a severe keratoconjunctivitis following routine eye examination with an accompanying history of adverse ocular signs and symptoms following multiple past exams.
It is important for all eye care clinicians to realize that AEs to diagnostic eye drops are possible and can occur following the most routine of visits. Such reactions can be caused by dilating agents, anesthetics, or preservatives, and these may be allergic or toxic. Clinicians should take special care to identify the instigating agent, and if possible, to avoid using such agents on patients during future exams. Clinicians also should understand how best to manage iatrogenic AEs when they encounter them in order to restore a patient’s visual function as quickly as possible.
Phenylephrine, a sympathomimetic drug, is commonly used in eye exams to dilate the pupil of the eye and to differentiate scleritis from episcleritis. Common adverse effects (AEs) of phenylephrine include subjective burning, stinging with lacrimation, rebound hyperemia, and liberation of iris pigment into the anterior chamber. Less common, systemic AEs include tachycardia and elevation of systemic blood pressure. Although instances of allergic reactions are rare, phenylephrine has been reported to cause contact dermatitis, blepharoconjunctivitis, and as in this case, keratoconjunctivitis.
Case Report
An 83-year-old white male presented for a red eye evaluation 2 days after having undergone a comprehensive eye exam with dilation at the Malcom Randall VAMC clinic in Gainesville, Florida. The patient reported onset of blurred vision, which he described as looking through a fog. He further compared the feeling to pins sticking in his eyes. The patient noted he had experienced similar symptoms on a few other occasions following eye exams. At the most recent eye exam, proparacaine and fluorescein had been used for tonometry, and phenylephrine 2.5% and tropicamide 0.5% had been used for pupillary dilation.
The patient’s best-corrected visual acuity was counting fingers at 2 feet in the right eye (OD) and left eye (OS). The best-corrected visual acuity 2 days prior had been 20/20 OD and OS. Pupils and extraocular motilities were unremarkable. Intraocular pressures were not obtained due to concern for a possible adverse reaction to proparacaine.
Slit-lamp evaluation revealed the lids to be lax, erythematous, and edematous in both eyes (Figure 1).
The initial diagnosis was acute chemical conjunctivitis most likely due to an AE to proparacaine. The plan was to start the patient on antibiotic eye drops qid OU, prednisolone qid OU, and artificial tears every hour OU. The patient was scheduled to return to clinic 4 days later for an anterior segment follow-up.
At the follow-up visit, the patient reported significant visual improvement. His best-corrected visual acuity was 20/40-2 without improvement on pinhole OD and 20/50-2 with improvement to 20/30+ on pinhole OS. Slit-lamp evaluation revealed 1+ bulbar conjunctival injection OU, intact corneal epithelium OU, and no cells or flare in the anterior chambers OU. Due to improving punctate epitheliopathy, the frequency of the antibiotic drops, the prednisolone, and the artificial tears was reduced to bid. After 3 days, he was instructed to discontinue them. The patient was scheduled to return in 2 weeks for an anterior segment follow-up.
At the next follow-up visit, the patient reported that his vision had returned to normal, and he had no further ocular AEs. His best-corrected visual acuity was 20/20-2 OD and 20/20 OS. Slit-lamp evaluation revealed mild blepharitis OU, trace bulbar conjunctival injection OU, and complete resolution of the keratitis OU. The assessment was acute allergic conjunctivitis thought to be secondary to an AE to proparacaine OU, yet the need to rule out hypersensitivity to tropicamide and/or phenylephrine remained. The plan was to educate the patient of the possibility of allergic reaction on future visits and to recommend continued use of artificial tears as needed.
Through a careful and extensive chart review of all past visits, it was suspected that phenylephrine might be to blame rather than proparacaine. At the subsequent visit, the patient agreed to undergo testing to determine the culprit via instillation of proparacaine in one eye and tropicamide in the other. The patient had no reaction to either drop (checked 45 minutes after instillation and the following day). By process of elimination, phenylephrine was determined to be the offending agent.
Discussion
Following a thorough review of the patient’s chart, it was found that on other occasions he had presented with suspected allergic reactions following routine eye examinations. The patient reported he had experienced a reaction in 2007 but could not recall what drops were instilled in his eyes at the time. In addition, there was no documentation in his medical record of the subsequent reaction following that visit. Another reaction occurred in July 2010 with instillation of tropicamide 1%, phenylephrine 2.5%, and Fluress (fluorescein sodium and benoxinate hydrochloride ophthalmic solution USP). In October 2013, when tropicamide 0.5%, proparacaine, and fluorescein strips were instilled, there was no reaction. The next reaction occurred in October 2014, when tropicamide 0.5%, phenylephrine 2.5%, proparacaine, and fluorescein strips were instilled.
This careful review of past exam notes revealed that phenylephrine and Fluress were the only drops that had not been instilled at the October 2013 visit when no AE was reported. However, Fluress was an unlikely culprit since it was not instilled in October 2014, and the patient still experienced an AE. Therefore, the agent most likely responsible for the allergic reaction in the patient, as confirmed by a review of the past notes and by the aforementioned pharmacologic test, was deemed to be phenylephrine (Table).
Adverse reactions to topical ocular medications and specifically to diagnostic eye drops have long been recognized. Mathias, Camarasa, Barber, Ducombs, and Monsálvezhave reported on variations of conjunctivitis and periorbital erythema with positive patch testing to phenylephrine.1-5 Geyer and colleagues reported on a study of 21 patients who had blepharoconjunctivitis after instillation of phenylephrine.6 In this case study patient, severe keratoconjunctivitis was the clinical manifestation observed.
Villarreal and colleagues studied 31 patients who had a previous reaction to mydriatic drops. The study found that phenylephrine was the drug that most frequently caused an AE (93.5%).7 One patient reacted to the preservative thimerosal, and 1 patient reacted to benoxiprocaine. Tropicamide was demonstrated to be very well tolerated as none of the patients tested positive on either the patch test or the pharmacologic test.
Tropicamide is a nonselective muscarinic antagonist commonly used for mydriasis due to its fast onset and short duration.8 Adverse reactions to tropicamide are rare. Three studies reported on patients who had a positive patch test to tropicamide.9-11 However, the reaction was not provoked by direct instillation of tropicamide into the eye.
Common in-office topical anesthetics, proparacaine, tetracaine, benoxinate, and lidocaine also can cause AEs. Corneal toxicity is a well-known complication with topical anesthetic abuse, whereas allergic reactions are considered rare. The most common symptoms include stingingand discomfort upon instillation. Common signs include punctate corneal epithelial erosionsresulting indirectly from a decrease in reflex tearing, infrequent blinking, and increased tear evaporation.12 Topical anesthetics also inhibit the migration of corneal epithelial cells and cause direct damage to the cells that are present, leading to impaired healing and epithelial defects.13
Manifestations of allergic reaction to topical anesthetics can include conjunctival hyperemia and edema, edematous eyelids, and lacrimation. One published case described a 60-year-old woman who developed eczematous dermatitis of the eyelids after ophthalmic anesthetic drops were instilled prior to laser surgery. Patch testing showed a positive response to benzocaine 5%, proparacaine, and tetracaine 0.5%.14
Preservatives, in general, can cause an allergic reaction. Benzalkonium chloride’s (BAK) cytotoxic sequelae include possible trabecular cell death in glaucoma patients, disruption of tear film stability (even at low concentrations), and immune-allergenic properties. One article reported BAK as one of the 30 most frequent allergens causing allergic periorbital dermatitis.15 Benzalkonium chloride is used in most brands of phenylephrine. However, preservatives in this patient’s case were ruled out as instigating agents since both phenylephrine and tropicamide contain the same preservative, BAK 0.01%, yet this patient did not develop a reaction to tropicamide when used without phenylephrine. Expired medications also were not considered to be a factor as none of the medications used on the patient were indeed expired (the Malcom Randall VAMC clinic maintains a strict policy of discarding medications 28 days after being opened).
Although uncommon, phenylephrine sometimes has been found to cause a type 4 hypersensitivity reaction, also known as cell-mediated or delayed-type hypersensitivity.16 First, helper T cells secrete cytokines. Activation of cytokines recruits and activates cytotoxic T cells, monocytes, and macrophages, leading to inflammation of the surrounding tissue. Examples of cell-mediated hypersensitivity include reactions to the tuberculin skin test and to poison ivy.
Type 1 hypersensitivity reactions, also known as immediate or anaphylactic hypersensitivity reactions, are not triggered by phenylephrine. In this type of reaction, IgE binds to the mast cell on initial exposure to an allergen. On second exposure, the allergen binds to the IgE, causing the mast cell to release mediators of inflammation, triggering physiologic responses. Examples of this type of hypersensitivity include those seen with penicillin, bee stings, hay fever, bronchial asthma, and food allergies, for example, to shellfish.
A toxic reaction’s mechanism differs from that of a type 4 hypersensitivity reaction. Toxic reactions occur due to direct cytotoxicity of a drug caused by a low or high pH and either hyper- or hypo-osmolarity. Toxicity can lead to corneal and conjunctival cell necrosis or induce apoptosis, stimulating inflammatory reactions. Clinically, toxic reactions will present with follicles, whereas allergic reactions will present with papillae.
The definitive diagnostic methods used to determine the allergic agent causing ocular or periocular AEs are patch testing and conjunctival challenge.7 Mathias, Camarasa, Barber, Ducombs,and Monsálvezused patch testing to confirm phenylephrine as the allergic agent in their series of cases. Patch testing entails the application of a small amount of an allergic agent that is taped onto the skin. The allergic agent is confirmed if the patient has a dermal reaction, wherein the area patched will become erythematous. When patch testing is negative or inconclusive, a conjunctival challenge is performed by instillation of the suspected allergic agent into the eye with subsequent observation to determine whether a reaction occurs. The sequelae found in Villarreal’s study included itching, lacrimation, edema, erythema, and sometimes blepharitis.7
A direct conjunctival challenge with the suspected culprit was not pursued in this patient’s case due to the known severity of the potential resulting reaction. The authors instead chose an indirect method of determining the implicating agent and used the process of elimination to whittle down the most likely suspect. A challenge with the medications suspected not to be likely offenders was undertaken. This spared the patient a likely repeat of the AE he had just recovered from.
Management
Allergic reactions can resolve without medical intervention. The first step is to remove the allergen. For delayed hypersensitivity reactions, treatments may include topical decongestants, cool compresses, and corticosteroids.8 The treatment for immediate hypersensitivity reaction differs from that of delayed hypersensitivity reaction in that antihistamines are used.17,18
This patient reported receiving no treatment for his ocular symptoms following eye examinations in the past, yet he experienced complete resolution after each AE. In this case, both a steroid and a prophylactic antibiotic to facilitate a more rapid improvement were used.
Conclusion
Although uncommon, cases of allergic reaction to phenylephrine can occur. The incidence of phenylephrine allergy is 0.6%.6 The case study patient presented with a severe keratoconjunctivitis following routine eye examination with an accompanying history of adverse ocular signs and symptoms following multiple past exams.
It is important for all eye care clinicians to realize that AEs to diagnostic eye drops are possible and can occur following the most routine of visits. Such reactions can be caused by dilating agents, anesthetics, or preservatives, and these may be allergic or toxic. Clinicians should take special care to identify the instigating agent, and if possible, to avoid using such agents on patients during future exams. Clinicians also should understand how best to manage iatrogenic AEs when they encounter them in order to restore a patient’s visual function as quickly as possible.
1. Mathias CG, Maibach HI, Irvine A, Adler W. Allergic contact dermatitis to echothiophate iodide and phenylephrine. Arch Ophthalmol. 1979;97(2):286-287.
2. Camarasa JG. Contact dermatitis to phenylephrine. Contact Dermatitis. 1984;10(3):182.
3. Barber K. Allergic contact eczema to phenylephrine. Contact Dermatitis. 1983;9(4):274-277.
4. Ducombs G, de Casamayor J, Verin P, Maleville J. Allergic contact dermatitis to phenylephrine. Contact Dermatitis. 1986;15(2):107-108.
5. Monsálvez V, Fuertes L, García-Cano I, Vanaclocha F, Ortez de Frutos J. Blepharoconjunctivitis due to phenylephrine [in Spanish]. Actas Dermosifiliogr. 2010;101(5):466-467.
6. Geyer O, Yust I, Lazar M. Allergic blepharoconjunctivitis due to phenylephrine. J Ocul Pharmacol. 1988;4(2):123-126.
7. Villarreal O. Reliability of diagnostic tests for contact allergy to mydriatic eyedrops. Contact Dermatitis. 1998;38(3):150-154.
8. Frazier M, Jaanus SD. Cycloplegics. In: Bartlett JD, Jaanus SD. Clinical Ocular Pharmacology. 5th ed. St. Louis, MO: Butterworth-Heinemann; 2009:125-138.
9. Decraene T, Goossens A. Contact allergy to atropine and other mydriatic agents in eye drops. Contact Dermatitis. 2001;45(5):309-310.
10. Boukhman MP, Maibach HI. Allergic contact dermatitis from tropicamide ophthalmic solution. Contact Dermatitis. 1999;41(1):47-48.
11. Yoshikawa K, Kawahara S. Contact allergy to atropine and other mydriatic agents. Contact Dermatitis. 1985;12(1):56-57.
12. Mcgee HT, Fraunfelder FW. Toxicities of topical ophthalmic anesthetics. Expert Opin Drug Saf. 2007;6(6):637-640.
13. Dass BA, Soong HK, Lee B. Effects of proparacaine of actin cytoskeleton of corneal epithelium. J Ocul Pharmacol. 1988;4(3):187-194.
14. Dannaker CJ, Maibach HI, Austin E. Allergic contact dermatitis to proparacaine with subsequent cross-sensitization to tetracaine from ophthalmic preparations. Am J Contact Dermat. 2001;12(3):177-179.
15. Hong J, Bielory L. Allergy to ophthalmic preservatives. Curr Opin Allergy Clin Immunol. 2009;9(5):447-453.
16. Gonzalo-Garijo MA, Pérez-Calderón R, de Argila D, Rodríguez-Nevado I. Erythrodermia to pseudoephedrine in a patient with contact allergy to phenylephrine. Allergol Immunopathol (Madr). 2002;30(4):239-242.
17. Platts-Mills TAE. Immediate hypersensitivity (Type I). In: Male D, Brostoff J, Roth DB, Roitt I. Immunology. 7th ed. Canada: Elsevier Limited; 2006:423-446.
18. Britton W. Type IV hypersensitivity. In: Male D, Brostoff J, Roth DB, Roitt I. Immunology. 7th ed. Canada: Elsevier Limited; 2006:477-491.
1. Mathias CG, Maibach HI, Irvine A, Adler W. Allergic contact dermatitis to echothiophate iodide and phenylephrine. Arch Ophthalmol. 1979;97(2):286-287.
2. Camarasa JG. Contact dermatitis to phenylephrine. Contact Dermatitis. 1984;10(3):182.
3. Barber K. Allergic contact eczema to phenylephrine. Contact Dermatitis. 1983;9(4):274-277.
4. Ducombs G, de Casamayor J, Verin P, Maleville J. Allergic contact dermatitis to phenylephrine. Contact Dermatitis. 1986;15(2):107-108.
5. Monsálvez V, Fuertes L, García-Cano I, Vanaclocha F, Ortez de Frutos J. Blepharoconjunctivitis due to phenylephrine [in Spanish]. Actas Dermosifiliogr. 2010;101(5):466-467.
6. Geyer O, Yust I, Lazar M. Allergic blepharoconjunctivitis due to phenylephrine. J Ocul Pharmacol. 1988;4(2):123-126.
7. Villarreal O. Reliability of diagnostic tests for contact allergy to mydriatic eyedrops. Contact Dermatitis. 1998;38(3):150-154.
8. Frazier M, Jaanus SD. Cycloplegics. In: Bartlett JD, Jaanus SD. Clinical Ocular Pharmacology. 5th ed. St. Louis, MO: Butterworth-Heinemann; 2009:125-138.
9. Decraene T, Goossens A. Contact allergy to atropine and other mydriatic agents in eye drops. Contact Dermatitis. 2001;45(5):309-310.
10. Boukhman MP, Maibach HI. Allergic contact dermatitis from tropicamide ophthalmic solution. Contact Dermatitis. 1999;41(1):47-48.
11. Yoshikawa K, Kawahara S. Contact allergy to atropine and other mydriatic agents. Contact Dermatitis. 1985;12(1):56-57.
12. Mcgee HT, Fraunfelder FW. Toxicities of topical ophthalmic anesthetics. Expert Opin Drug Saf. 2007;6(6):637-640.
13. Dass BA, Soong HK, Lee B. Effects of proparacaine of actin cytoskeleton of corneal epithelium. J Ocul Pharmacol. 1988;4(3):187-194.
14. Dannaker CJ, Maibach HI, Austin E. Allergic contact dermatitis to proparacaine with subsequent cross-sensitization to tetracaine from ophthalmic preparations. Am J Contact Dermat. 2001;12(3):177-179.
15. Hong J, Bielory L. Allergy to ophthalmic preservatives. Curr Opin Allergy Clin Immunol. 2009;9(5):447-453.
16. Gonzalo-Garijo MA, Pérez-Calderón R, de Argila D, Rodríguez-Nevado I. Erythrodermia to pseudoephedrine in a patient with contact allergy to phenylephrine. Allergol Immunopathol (Madr). 2002;30(4):239-242.
17. Platts-Mills TAE. Immediate hypersensitivity (Type I). In: Male D, Brostoff J, Roth DB, Roitt I. Immunology. 7th ed. Canada: Elsevier Limited; 2006:423-446.
18. Britton W. Type IV hypersensitivity. In: Male D, Brostoff J, Roth DB, Roitt I. Immunology. 7th ed. Canada: Elsevier Limited; 2006:477-491.
Late-Onset Bexarotene-Induced CD4 Lymphopenia in a Cutaneous T-cell Lymphoma Patient
Infections, autoimmune disease, bone marrow failure, medications, and total-body irradiation may induce CD4 lymphopenia, defined as a CD4 T-cell count below 300 cells/mL or less than 20% of total lymphocytes.1 Human immunodeficiency virus (HIV) is the most common cause of CD4 lymphopenia, with sepsis (bacterial and fungal) and postoperative states the most common causes in hospital settings.2 No underlying factors are found in 0.02% of CD4 lymphopenia cases, which are considered to be idiopathic.3,4 We report a patient with cutaneous T-cell lymphoma (CTCL) who developed profound CD4 lymphopenia in the setting of long-term bexarotene therapy.
Case Report
A 63-year-old man with hypertension presented to our dermatology clinic with pruritic scaly plaques on the scalp of 4 months’ duration that had progressed to full-body exfoliative erythroderma (Figure 1). He had diffuse palmoplantar keratoderma and lymphadenopathy. His only long-term medications were terazosin for benign prostatic hyperplasia and atenolol for hypertension; he reported no new medications. Laboratory evaluation revealed normal liver and kidney function. A complete blood cell count (CBC) revealed a white blood cell (WBC) count within reference range (8000/µL [reference range, 4500–11,000/µL]) but with increased eosinophils (12.9% [reference range, 2.7%]) and monocytes (11.8% [reference range, 4%]) and reduced lymphocytes (16.8% [reference range, 34%]). Flow cytometry showed a CD4:CD8 ratio of 1.18 to 1 (reference range, 0.8–4.2)(absolute CD4+ cells, 764/µL [reference range, 297–1551/µL]; absolute CD8+ cells, 654/µL [reference range, 100–1047/µL]). Skin biopsy revealed subacute spongiotic dermatitis with numerous eosinophils, exocytosis including folliculotropism, and rare atypical lymphocytes (Figure 2). Molecular studies showed T-cell receptor γ gene rearrangement. The patient did not have any other underlying conditions that would predispose him to lymphopenia. Based on these findings, a diagnosis of CTCL stage IIIA was made and agreed on by experts at the University of California, San Diego Dermatology Grand Rounds.

The patient was subsequently started on acitretin, topical corticosteroids, and hydroxyzine. However, the erythroderma progressed and he developed fever, chills, and malaise, and he was hospitalized 2 months later for intensive therapy and to rule out infection. He improved on daily wet wraps, topical steroids, oral antibiotics, and initiation of narrowband UVB therapy. He was discharged 1 week later. Acitretin was switched to bexarotene 3 months later due to peeling and cracking of the palmoplantar skin. The initial dose was 225 mg once daily, which was steadily increased over the next 4 months to a therapeutic dose of 600 mg once daily, which was much lower than the maximum dose of 400 mg/m2 daily (calculated at 750 mg/d in our patient). The patient achieved clinical remission 1 year after initiation of bexarotene in conjunction with narrowband UVB therapy. Serum eosinophils also normalized. Because there were no intolerable side effects, this dose was continued for 2 more years before it was slowly tapered to 375 mg once daily over a 1-year period. The new dose was maintained thereafter. Secondary hypertriglyceridemia and hypothyroidism, known side effects of bexarotene, developed 1 and 5 months after initiating therapy, respectively, and were treated with levothyroxine and fenofibrate. Blood counts were checked every 3 months and remained within reference range. Within the first few months of therapy, lymphocytes did trend down to 16.8%, but segmented neutrophils were normal at 59.4%. For the next 5 years the total WBC count and differential remained within reference range. T-cell subsets and flow cytometry data were not measured. No new medications were started during this period, and none of his existing medications had lymphopenia as a known side effect.
Five years after the initial diagnosis, the patient was still on bexarotene and was suspected to have pneumonia that was treated by his primary care provider with cefuroxime and azithromycin for 2 weeks with no improvement. He was then admitted to the hospital with shortness of breath, productive cough, night sweats, and dyspnea of 1 month’s duration. There was no associated weight loss or fever. Notably, the skin was clear. He was further treated for community-acquired pneumonia, first with vancomycin and ceftazidime, then with ciprofloxacin and sulfamethoxazole-trimethoprim, with no improvement. A CBC with differential was obtained on the patient’s first admission and revealed a WBC count of 3600/µL with decreased lymphocytes (8.6%), no eosinophilia, and anemia (hemoglobin, 10.5 g/dL [reference range, 33–37 g/dL]). T-cell subset studies revealed a CD4:CD8 ratio of 0.06 to 1 (absolute CD4+ cells, 6/µL; absolute CD8+ cells, 107/µL). The patient also had an elevated lactate dehydrogenase level of 1015 U/L (reference range, 100–200 U/L) and a normal comprehensive metabolic panel. A comprehensive workup, including urine and blood cultures, serum Cryptococcus and coccidioidomycosis IgG/IgM, histoplasmosis urine antigen, legionella, HIV, purified protein derivative (tuberculin), and aspergillosis galactomannan antigen panel, was negative. Blood tests for HIV and human T-lymphotropic virus also were negative. Bronchoscopy with cytology and sputum cultures for fungi, acid-fast bacteria, and viruses identified Pneumocystis jiroveci in the bronchial wash. Pneumocystis pneumonia was treated with intravenous clindamycin, primaquine, and leucovorin. The patient’s WBC count continued to drop over the next 2 weeks to a nadir of 1.7% with few lymphocytes noted on the differential. At that point, the bexarotene was stopped and was considered causative in inducing CD4 lymphopenia, resulting in opportunistic infection. The patient steadily improved and was discharged on sulfamethoxazole-trimethoprim prophyla
His CD4 count slowly improved over the next 18 months; however, his skin disease recurred and progressed to exfoliative erythroderma with marked scarring alopecia (Figure 3), facial swelling, extreme pruritus, and notable eosinophilia. Repeat computed tomography was negative for extracutaneous involvement. A repeat skin biopsy showed recurrent mycosis fungoides similar to the original biopsy (Figure 4). Topical steroids and narrowband UVB therapy were restarted. A bone marrow biopsy revealed no definitive lymphoma, but the peripheral blood showed occasional CD8+ “flower cells” and no CD4+ Sézary cells. Two repeat molecular studies failed to show the T-cell receptor gene rearrangement. Localized electron beam radiation therapy, lenalidomide, and clobetasol were tried without benefit. The patient was hospitalized 3 months later and was started on wet wraps as well as weekly infusions of the histone deacetylase inhibitor romidepsin (14 mg/m2 over a 4-hour period) on days 1, 8, and 15 of a 28-day cycle with rapid improvement. He experienced transient slight neutropenia with the first several treatments that quickly resolved. His skin was clear while on a regimen of triamcinolone, wet wraps, and intravenous romidepsin. He demonstrated visible improvement after 3 weekly infusions of romedepsin (Figure 5). His skin disease cleared after 9 infusions of romidepsin, and he currently remains in remission; however, he developed presumed bronchopneumonia after approximately 3 to 4 infusions. He then presented with severe headaches after his ninth infusion and was found to have cryptococcal meningitis. Romedepsin was stopped and he was treated with systemic antifungal therapy. His CTCL never recurred despite not restarting romidepsin.
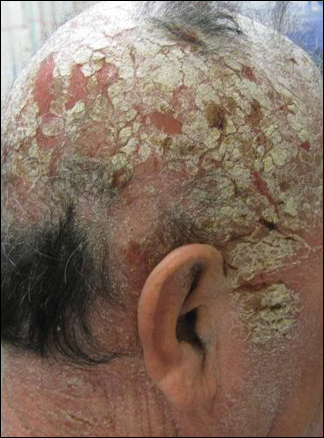
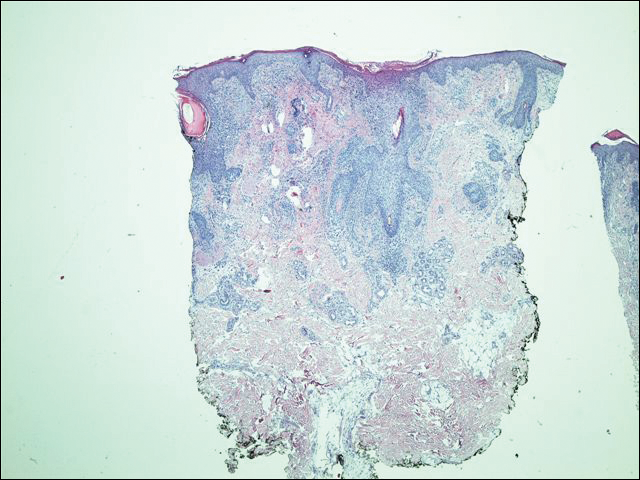
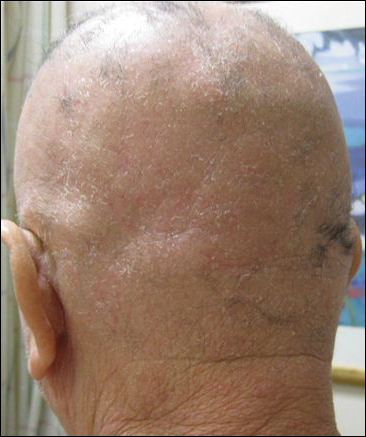
Comment
The retinoids are chemically related to vitamin A. They regulate epithelial cell growth and are beneficial in inflammatory skin disorders and in patients with increased cell turnover as well as in skin cancer and precancer prevention/treatment.5 The first- and second-generation retinoids, isotretinoin and acitretin, respectively, cause anemia or leukopenia in less than 10% of patients; adverse effects are noted more commonly in doses greater than 1 mg/kg daily.6-8
Bexarotene is a third-generation retinoid drug that is more selective for retinoid X receptors. It was approved in 1991 for treatment of advanced CTCL (stages IIB–IVB) in adult patients who have failed at least 1 prior systemic therapy. Bexarotene is noted to promote cell cycle arrest and apoptosis in CTCL cell lines.9 However, one study suggested that for bexarotene, inhibition of proliferation is more important than causing apoptosis in CTCL cells, and this effect is achieved through triggering the p53/p73-dependent cell cycle inhibition pathway.10 Studies in patients with Sézary syndrome have shown that bexarotene changes the chemokine receptor expression in circulating malignant T cells, making them less likely to traffic to the skin (lower chemokine receptor type 4 expression),11 which may explain why some CTCL cases have shown improvement of skin disease on bexarotene despite progression of extradermal disease.12
Common side effects of bexarotene include hyperlipidemia and central hypothyroidism.13 In addition, dose-related myelosuppression with isolated leukopenia, particularly neutropenia, also has been reported (18% of patients at a dosage of 300 mg/m2/d and 43% of patients with a dosage greater than 300 mg/m2/d). Leukopenia generally occurs within the first 4 to 8 weeks of treatment, is relatively mild (WBC, 1000–2999/µL), and generally is reversible.13-15 One review of 66 mycosis fungoides patients treated with bexarotene described a patient who developed leukopenia 15 months after initiating bexarotene therapy.14 The manufacturer recommends that treatment with bexarotene be continued as long as the patient is receiving benefit from the treatment. One trial of 70 mycosis fungoides patient treated with bexarotene reported response rates of 48% on bexarotene monotherapy (n=54) and 69% on bexarotene plus an additional agent (n=16).15 The authors noted higher response rates in patients on 2 lipid-lowering agents. They concluded that bexarotene was a safe and effective agent for treatment of cutaneous T-cell lymphoma and recommended continued treatment with a lowered dose of bexarotene in those achieving complete responses for a period of 2 years. Although the recommended initial dose is 300 mg/m2/d, bexarotene can be increased to 400 mg/m2/d after 8 weeks if no response to treatment is appreciated.16 Our patient was on a maximum bexarotene dose of 600 mg once daily (280 mg/m2/d) for the first 2 years, and a maintenance dose of 300 mg once daily for the next 3 years. He was not on any medicines known to induce leukopenia and he was not given any known cytochrome P450 3A4 inhibitors that could increase the toxicity of bexarotene.
The patient’s CBC was checked routinely every 2 to 3 months after he was started on bexarotene. For 5 years, the CBC and differential remained within reference range; however, his CD4 counts were not followed during those 5 years. We attribute his CD4 lymphopenia and subsequent pneumocystis pneumonia to bexarotene. After our patient’s CD4 lymphopenia was discovered, he developed a precipitous drop in his WBC and lymphocyte counts while hospitalized that worsened over a 2-week period. At this point, the bexarotene was discontinued and his WBC count slowly recovered. We believe that one of the initial antibiotics prescribed by the patient’s primary care physician at initial onset of pneumonia symptoms as an outpatient could have acted synergistically with bexarotene to worsen lymphopenia. Specifically, ceftazidime, vancomycin, and ciprofloxacin have all been reported to cause leukopenia; however, it was neutropenia in these cases, not lymphopenia.17,18 Notwithstanding, the opportunistic pneumonia and therefore CD4 lymphopenia was present prior to any antibiotic use.
The CD4 lymphopenia was unlikely due to underlying infection(s) because an extensive workup was negative, except for the pneumocystis, which likely resulted from the lymphopenia. The CD4 lymphopenia also could be idiopathic, as it has been reported in 3 patients with mycosis fungoides.19 All 3 patients were erythrodermic at presentation and were noted to have numerous CD4+ lymphocytes in the cutaneous lesions but few circulating CD4+ T lymphocytes in the blood. The authors attributed the CD4 lymphopenia to cutaneous sequestration of CD4+ T lymphocytes.19 These cases contrast with our patient who was in clinical remission at the time of CD4 lymphopenia, which improved and normalized following discontinuation of bexarotene.
Conclusion
This case emphasizes the importance of monitoring for leukopenia, specifically CD4 lymphopenia, in patients on long-term bexarotene therapy. Routine CBC as well as T-cell subset counts should be performed during treatment. Rotation off bexarotene after several years of therapy should be considered, even in patients with continuous benefit from this systemic therapy.
- Smith DK, Neal JJ, Holmberg SD. Unexplained opportunistic infections and CD4+ T-lymphocytopenia without HIV infection. an investigation of cases in the United States. The Centers for Disease Control Idiopathic CD4+ T-lymphocytopenia Task Force. N Engl J Med. 1993;328:373-379.
- Castelino DJ, McNair P, Kay TW. Lymphocytopenia in a hospital population: what does it signify? Aust N Z J Med. 1997;27:170-174.
- Zonios DI, Falloon J, Bennett JE, et al. Idiopathic CD4+ lymphocytopenia: natural history and prognostic factors. Blood. 2008;112:287-294.
- Duncan RA, von Reyn CF, Alliegro GM, et al. Idiopathic CD4+ T-lymphocytopenia: four patients with opportunistic infections and no evidence of HIV infection. N Engl J Med. 1993;328:393-398.
- Bruno NP, Beacham BE, Burnett JW. Adverse effects of isotretinoin therapy. Cutis. 1984;33:484-486, 489.
- Strauss JS, Rapini RP, Shalita AR, et al. Isotretinoin therapy for acne: results of a multicenter dose-response study. J Am Acad Dermatol. 1984;10:490-496.
- Windhorst DB, Nigra T. General clinical toxicology of oral retinoids. J Am Acad Dermatol.1982;6:675-682.
- Glinnick SE. Leucopenia from accutane: in ten percent? Schoch Let. 1985;35:9.
- Wilcox RA. Cutaneous T-cell lymphoma: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol. 2011;86:928-948.
- Nieto-Rementería N, Pérez-Yarza G, Boyano MD, et al. Bexarotene activates the p53/p73 pathway in human cutaneous T-cell lymphoma. Br J Dermatol. 2009;160:519-526.
- Richardson SK, Newton SB, Bach TL, et al. Bexarotene blunts malignant T-cell chemotaxis in Sézary syndrome: reduction of chemokine receptor 4-positive lymphocytes and decreased chemotaxis to thymus and activation-regulated chemokine. Am J Hematol. 2007;82:792-797.
- Bouwhuis SA, Davis MD, el-Azhary RA, et al. Bexarotene treatment of late-stage mycosis fungoides and Sézary syndrome: development of extracutaneous lymphoma in 6 patients. J Am Acad Dermatol. 2005;52:991-996.
- Targretin [package insert]. Bridgewater, NJ: Valeant Pharmaceuticals International, Inc; 2015.
- , , , et al. Bexarotene therapy for mycosis fungoides and Sézary syndrome. Br J Dermatol. 2009;160:1299-1307.
- , , et al. Optimizing bexarotene therapy for cutaneous T-cell lymphoma. J Am Acad Dermatol. 2002;47:672-684.
- Scarisbrick JJ, Morris S, Azurdia R, et al. U.K. consensus statement on safe clinical prescribing of bexarotene for patients with cutaneous T-cell lymphoma. Br J Dermatol. 2013;168:192-200.
- Black E, Lau TT, Ensom MH. Vancomycin-induced neutropenia: is it dose-or duration related? Ann Pharmacother. 2011;45:629-638.
- Choo PW, Gantz NM. Reversible leukopenia related to ciprofloxacin therapy. South Med J. 1990;83:597-598.
- Stevens SR, Griffiths TW, Cooper KD. Idiopathic CD4+ T lymphocytopenia in a patient with mycosis fungoides. J Am Acad Dermatol. 1995;32:1063-1064.
Infections, autoimmune disease, bone marrow failure, medications, and total-body irradiation may induce CD4 lymphopenia, defined as a CD4 T-cell count below 300 cells/mL or less than 20% of total lymphocytes.1 Human immunodeficiency virus (HIV) is the most common cause of CD4 lymphopenia, with sepsis (bacterial and fungal) and postoperative states the most common causes in hospital settings.2 No underlying factors are found in 0.02% of CD4 lymphopenia cases, which are considered to be idiopathic.3,4 We report a patient with cutaneous T-cell lymphoma (CTCL) who developed profound CD4 lymphopenia in the setting of long-term bexarotene therapy.
Case Report
A 63-year-old man with hypertension presented to our dermatology clinic with pruritic scaly plaques on the scalp of 4 months’ duration that had progressed to full-body exfoliative erythroderma (Figure 1). He had diffuse palmoplantar keratoderma and lymphadenopathy. His only long-term medications were terazosin for benign prostatic hyperplasia and atenolol for hypertension; he reported no new medications. Laboratory evaluation revealed normal liver and kidney function. A complete blood cell count (CBC) revealed a white blood cell (WBC) count within reference range (8000/µL [reference range, 4500–11,000/µL]) but with increased eosinophils (12.9% [reference range, 2.7%]) and monocytes (11.8% [reference range, 4%]) and reduced lymphocytes (16.8% [reference range, 34%]). Flow cytometry showed a CD4:CD8 ratio of 1.18 to 1 (reference range, 0.8–4.2)(absolute CD4+ cells, 764/µL [reference range, 297–1551/µL]; absolute CD8+ cells, 654/µL [reference range, 100–1047/µL]). Skin biopsy revealed subacute spongiotic dermatitis with numerous eosinophils, exocytosis including folliculotropism, and rare atypical lymphocytes (Figure 2). Molecular studies showed T-cell receptor γ gene rearrangement. The patient did not have any other underlying conditions that would predispose him to lymphopenia. Based on these findings, a diagnosis of CTCL stage IIIA was made and agreed on by experts at the University of California, San Diego Dermatology Grand Rounds.
The patient was subsequently started on acitretin, topical corticosteroids, and hydroxyzine. However, the erythroderma progressed and he developed fever, chills, and malaise, and he was hospitalized 2 months later for intensive therapy and to rule out infection. He improved on daily wet wraps, topical steroids, oral antibiotics, and initiation of narrowband UVB therapy. He was discharged 1 week later. Acitretin was switched to bexarotene 3 months later due to peeling and cracking of the palmoplantar skin. The initial dose was 225 mg once daily, which was steadily increased over the next 4 months to a therapeutic dose of 600 mg once daily, which was much lower than the maximum dose of 400 mg/m2 daily (calculated at 750 mg/d in our patient). The patient achieved clinical remission 1 year after initiation of bexarotene in conjunction with narrowband UVB therapy. Serum eosinophils also normalized. Because there were no intolerable side effects, this dose was continued for 2 more years before it was slowly tapered to 375 mg once daily over a 1-year period. The new dose was maintained thereafter. Secondary hypertriglyceridemia and hypothyroidism, known side effects of bexarotene, developed 1 and 5 months after initiating therapy, respectively, and were treated with levothyroxine and fenofibrate. Blood counts were checked every 3 months and remained within reference range. Within the first few months of therapy, lymphocytes did trend down to 16.8%, but segmented neutrophils were normal at 59.4%. For the next 5 years the total WBC count and differential remained within reference range. T-cell subsets and flow cytometry data were not measured. No new medications were started during this period, and none of his existing medications had lymphopenia as a known side effect.
Five years after the initial diagnosis, the patient was still on bexarotene and was suspected to have pneumonia that was treated by his primary care provider with cefuroxime and azithromycin for 2 weeks with no improvement. He was then admitted to the hospital with shortness of breath, productive cough, night sweats, and dyspnea of 1 month’s duration. There was no associated weight loss or fever. Notably, the skin was clear. He was further treated for community-acquired pneumonia, first with vancomycin and ceftazidime, then with ciprofloxacin and sulfamethoxazole-trimethoprim, with no improvement. A CBC with differential was obtained on the patient’s first admission and revealed a WBC count of 3600/µL with decreased lymphocytes (8.6%), no eosinophilia, and anemia (hemoglobin, 10.5 g/dL [reference range, 33–37 g/dL]). T-cell subset studies revealed a CD4:CD8 ratio of 0.06 to 1 (absolute CD4+ cells, 6/µL; absolute CD8+ cells, 107/µL). The patient also had an elevated lactate dehydrogenase level of 1015 U/L (reference range, 100–200 U/L) and a normal comprehensive metabolic panel. A comprehensive workup, including urine and blood cultures, serum Cryptococcus and coccidioidomycosis IgG/IgM, histoplasmosis urine antigen, legionella, HIV, purified protein derivative (tuberculin), and aspergillosis galactomannan antigen panel, was negative. Blood tests for HIV and human T-lymphotropic virus also were negative. Bronchoscopy with cytology and sputum cultures for fungi, acid-fast bacteria, and viruses identified Pneumocystis jiroveci in the bronchial wash. Pneumocystis pneumonia was treated with intravenous clindamycin, primaquine, and leucovorin. The patient’s WBC count continued to drop over the next 2 weeks to a nadir of 1.7% with few lymphocytes noted on the differential. At that point, the bexarotene was stopped and was considered causative in inducing CD4 lymphopenia, resulting in opportunistic infection. The patient steadily improved and was discharged on sulfamethoxazole-trimethoprim prophyla
His CD4 count slowly improved over the next 18 months; however, his skin disease recurred and progressed to exfoliative erythroderma with marked scarring alopecia (Figure 3), facial swelling, extreme pruritus, and notable eosinophilia. Repeat computed tomography was negative for extracutaneous involvement. A repeat skin biopsy showed recurrent mycosis fungoides similar to the original biopsy (Figure 4). Topical steroids and narrowband UVB therapy were restarted. A bone marrow biopsy revealed no definitive lymphoma, but the peripheral blood showed occasional CD8+ “flower cells” and no CD4+ Sézary cells. Two repeat molecular studies failed to show the T-cell receptor gene rearrangement. Localized electron beam radiation therapy, lenalidomide, and clobetasol were tried without benefit. The patient was hospitalized 3 months later and was started on wet wraps as well as weekly infusions of the histone deacetylase inhibitor romidepsin (14 mg/m2 over a 4-hour period) on days 1, 8, and 15 of a 28-day cycle with rapid improvement. He experienced transient slight neutropenia with the first several treatments that quickly resolved. His skin was clear while on a regimen of triamcinolone, wet wraps, and intravenous romidepsin. He demonstrated visible improvement after 3 weekly infusions of romedepsin (Figure 5). His skin disease cleared after 9 infusions of romidepsin, and he currently remains in remission; however, he developed presumed bronchopneumonia after approximately 3 to 4 infusions. He then presented with severe headaches after his ninth infusion and was found to have cryptococcal meningitis. Romedepsin was stopped and he was treated with systemic antifungal therapy. His CTCL never recurred despite not restarting romidepsin.
Comment
The retinoids are chemically related to vitamin A. They regulate epithelial cell growth and are beneficial in inflammatory skin disorders and in patients with increased cell turnover as well as in skin cancer and precancer prevention/treatment.5 The first- and second-generation retinoids, isotretinoin and acitretin, respectively, cause anemia or leukopenia in less than 10% of patients; adverse effects are noted more commonly in doses greater than 1 mg/kg daily.6-8
Bexarotene is a third-generation retinoid drug that is more selective for retinoid X receptors. It was approved in 1991 for treatment of advanced CTCL (stages IIB–IVB) in adult patients who have failed at least 1 prior systemic therapy. Bexarotene is noted to promote cell cycle arrest and apoptosis in CTCL cell lines.9 However, one study suggested that for bexarotene, inhibition of proliferation is more important than causing apoptosis in CTCL cells, and this effect is achieved through triggering the p53/p73-dependent cell cycle inhibition pathway.10 Studies in patients with Sézary syndrome have shown that bexarotene changes the chemokine receptor expression in circulating malignant T cells, making them less likely to traffic to the skin (lower chemokine receptor type 4 expression),11 which may explain why some CTCL cases have shown improvement of skin disease on bexarotene despite progression of extradermal disease.12
Common side effects of bexarotene include hyperlipidemia and central hypothyroidism.13 In addition, dose-related myelosuppression with isolated leukopenia, particularly neutropenia, also has been reported (18% of patients at a dosage of 300 mg/m2/d and 43% of patients with a dosage greater than 300 mg/m2/d). Leukopenia generally occurs within the first 4 to 8 weeks of treatment, is relatively mild (WBC, 1000–2999/µL), and generally is reversible.13-15 One review of 66 mycosis fungoides patients treated with bexarotene described a patient who developed leukopenia 15 months after initiating bexarotene therapy.14 The manufacturer recommends that treatment with bexarotene be continued as long as the patient is receiving benefit from the treatment. One trial of 70 mycosis fungoides patient treated with bexarotene reported response rates of 48% on bexarotene monotherapy (n=54) and 69% on bexarotene plus an additional agent (n=16).15 The authors noted higher response rates in patients on 2 lipid-lowering agents. They concluded that bexarotene was a safe and effective agent for treatment of cutaneous T-cell lymphoma and recommended continued treatment with a lowered dose of bexarotene in those achieving complete responses for a period of 2 years. Although the recommended initial dose is 300 mg/m2/d, bexarotene can be increased to 400 mg/m2/d after 8 weeks if no response to treatment is appreciated.16 Our patient was on a maximum bexarotene dose of 600 mg once daily (280 mg/m2/d) for the first 2 years, and a maintenance dose of 300 mg once daily for the next 3 years. He was not on any medicines known to induce leukopenia and he was not given any known cytochrome P450 3A4 inhibitors that could increase the toxicity of bexarotene.
The patient’s CBC was checked routinely every 2 to 3 months after he was started on bexarotene. For 5 years, the CBC and differential remained within reference range; however, his CD4 counts were not followed during those 5 years. We attribute his CD4 lymphopenia and subsequent pneumocystis pneumonia to bexarotene. After our patient’s CD4 lymphopenia was discovered, he developed a precipitous drop in his WBC and lymphocyte counts while hospitalized that worsened over a 2-week period. At this point, the bexarotene was discontinued and his WBC count slowly recovered. We believe that one of the initial antibiotics prescribed by the patient’s primary care physician at initial onset of pneumonia symptoms as an outpatient could have acted synergistically with bexarotene to worsen lymphopenia. Specifically, ceftazidime, vancomycin, and ciprofloxacin have all been reported to cause leukopenia; however, it was neutropenia in these cases, not lymphopenia.17,18 Notwithstanding, the opportunistic pneumonia and therefore CD4 lymphopenia was present prior to any antibiotic use.
The CD4 lymphopenia was unlikely due to underlying infection(s) because an extensive workup was negative, except for the pneumocystis, which likely resulted from the lymphopenia. The CD4 lymphopenia also could be idiopathic, as it has been reported in 3 patients with mycosis fungoides.19 All 3 patients were erythrodermic at presentation and were noted to have numerous CD4+ lymphocytes in the cutaneous lesions but few circulating CD4+ T lymphocytes in the blood. The authors attributed the CD4 lymphopenia to cutaneous sequestration of CD4+ T lymphocytes.19 These cases contrast with our patient who was in clinical remission at the time of CD4 lymphopenia, which improved and normalized following discontinuation of bexarotene.
Conclusion
This case emphasizes the importance of monitoring for leukopenia, specifically CD4 lymphopenia, in patients on long-term bexarotene therapy. Routine CBC as well as T-cell subset counts should be performed during treatment. Rotation off bexarotene after several years of therapy should be considered, even in patients with continuous benefit from this systemic therapy.
Infections, autoimmune disease, bone marrow failure, medications, and total-body irradiation may induce CD4 lymphopenia, defined as a CD4 T-cell count below 300 cells/mL or less than 20% of total lymphocytes.1 Human immunodeficiency virus (HIV) is the most common cause of CD4 lymphopenia, with sepsis (bacterial and fungal) and postoperative states the most common causes in hospital settings.2 No underlying factors are found in 0.02% of CD4 lymphopenia cases, which are considered to be idiopathic.3,4 We report a patient with cutaneous T-cell lymphoma (CTCL) who developed profound CD4 lymphopenia in the setting of long-term bexarotene therapy.
Case Report
A 63-year-old man with hypertension presented to our dermatology clinic with pruritic scaly plaques on the scalp of 4 months’ duration that had progressed to full-body exfoliative erythroderma (Figure 1). He had diffuse palmoplantar keratoderma and lymphadenopathy. His only long-term medications were terazosin for benign prostatic hyperplasia and atenolol for hypertension; he reported no new medications. Laboratory evaluation revealed normal liver and kidney function. A complete blood cell count (CBC) revealed a white blood cell (WBC) count within reference range (8000/µL [reference range, 4500–11,000/µL]) but with increased eosinophils (12.9% [reference range, 2.7%]) and monocytes (11.8% [reference range, 4%]) and reduced lymphocytes (16.8% [reference range, 34%]). Flow cytometry showed a CD4:CD8 ratio of 1.18 to 1 (reference range, 0.8–4.2)(absolute CD4+ cells, 764/µL [reference range, 297–1551/µL]; absolute CD8+ cells, 654/µL [reference range, 100–1047/µL]). Skin biopsy revealed subacute spongiotic dermatitis with numerous eosinophils, exocytosis including folliculotropism, and rare atypical lymphocytes (Figure 2). Molecular studies showed T-cell receptor γ gene rearrangement. The patient did not have any other underlying conditions that would predispose him to lymphopenia. Based on these findings, a diagnosis of CTCL stage IIIA was made and agreed on by experts at the University of California, San Diego Dermatology Grand Rounds.
The patient was subsequently started on acitretin, topical corticosteroids, and hydroxyzine. However, the erythroderma progressed and he developed fever, chills, and malaise, and he was hospitalized 2 months later for intensive therapy and to rule out infection. He improved on daily wet wraps, topical steroids, oral antibiotics, and initiation of narrowband UVB therapy. He was discharged 1 week later. Acitretin was switched to bexarotene 3 months later due to peeling and cracking of the palmoplantar skin. The initial dose was 225 mg once daily, which was steadily increased over the next 4 months to a therapeutic dose of 600 mg once daily, which was much lower than the maximum dose of 400 mg/m2 daily (calculated at 750 mg/d in our patient). The patient achieved clinical remission 1 year after initiation of bexarotene in conjunction with narrowband UVB therapy. Serum eosinophils also normalized. Because there were no intolerable side effects, this dose was continued for 2 more years before it was slowly tapered to 375 mg once daily over a 1-year period. The new dose was maintained thereafter. Secondary hypertriglyceridemia and hypothyroidism, known side effects of bexarotene, developed 1 and 5 months after initiating therapy, respectively, and were treated with levothyroxine and fenofibrate. Blood counts were checked every 3 months and remained within reference range. Within the first few months of therapy, lymphocytes did trend down to 16.8%, but segmented neutrophils were normal at 59.4%. For the next 5 years the total WBC count and differential remained within reference range. T-cell subsets and flow cytometry data were not measured. No new medications were started during this period, and none of his existing medications had lymphopenia as a known side effect.
Five years after the initial diagnosis, the patient was still on bexarotene and was suspected to have pneumonia that was treated by his primary care provider with cefuroxime and azithromycin for 2 weeks with no improvement. He was then admitted to the hospital with shortness of breath, productive cough, night sweats, and dyspnea of 1 month’s duration. There was no associated weight loss or fever. Notably, the skin was clear. He was further treated for community-acquired pneumonia, first with vancomycin and ceftazidime, then with ciprofloxacin and sulfamethoxazole-trimethoprim, with no improvement. A CBC with differential was obtained on the patient’s first admission and revealed a WBC count of 3600/µL with decreased lymphocytes (8.6%), no eosinophilia, and anemia (hemoglobin, 10.5 g/dL [reference range, 33–37 g/dL]). T-cell subset studies revealed a CD4:CD8 ratio of 0.06 to 1 (absolute CD4+ cells, 6/µL; absolute CD8+ cells, 107/µL). The patient also had an elevated lactate dehydrogenase level of 1015 U/L (reference range, 100–200 U/L) and a normal comprehensive metabolic panel. A comprehensive workup, including urine and blood cultures, serum Cryptococcus and coccidioidomycosis IgG/IgM, histoplasmosis urine antigen, legionella, HIV, purified protein derivative (tuberculin), and aspergillosis galactomannan antigen panel, was negative. Blood tests for HIV and human T-lymphotropic virus also were negative. Bronchoscopy with cytology and sputum cultures for fungi, acid-fast bacteria, and viruses identified Pneumocystis jiroveci in the bronchial wash. Pneumocystis pneumonia was treated with intravenous clindamycin, primaquine, and leucovorin. The patient’s WBC count continued to drop over the next 2 weeks to a nadir of 1.7% with few lymphocytes noted on the differential. At that point, the bexarotene was stopped and was considered causative in inducing CD4 lymphopenia, resulting in opportunistic infection. The patient steadily improved and was discharged on sulfamethoxazole-trimethoprim prophyla
His CD4 count slowly improved over the next 18 months; however, his skin disease recurred and progressed to exfoliative erythroderma with marked scarring alopecia (Figure 3), facial swelling, extreme pruritus, and notable eosinophilia. Repeat computed tomography was negative for extracutaneous involvement. A repeat skin biopsy showed recurrent mycosis fungoides similar to the original biopsy (Figure 4). Topical steroids and narrowband UVB therapy were restarted. A bone marrow biopsy revealed no definitive lymphoma, but the peripheral blood showed occasional CD8+ “flower cells” and no CD4+ Sézary cells. Two repeat molecular studies failed to show the T-cell receptor gene rearrangement. Localized electron beam radiation therapy, lenalidomide, and clobetasol were tried without benefit. The patient was hospitalized 3 months later and was started on wet wraps as well as weekly infusions of the histone deacetylase inhibitor romidepsin (14 mg/m2 over a 4-hour period) on days 1, 8, and 15 of a 28-day cycle with rapid improvement. He experienced transient slight neutropenia with the first several treatments that quickly resolved. His skin was clear while on a regimen of triamcinolone, wet wraps, and intravenous romidepsin. He demonstrated visible improvement after 3 weekly infusions of romedepsin (Figure 5). His skin disease cleared after 9 infusions of romidepsin, and he currently remains in remission; however, he developed presumed bronchopneumonia after approximately 3 to 4 infusions. He then presented with severe headaches after his ninth infusion and was found to have cryptococcal meningitis. Romedepsin was stopped and he was treated with systemic antifungal therapy. His CTCL never recurred despite not restarting romidepsin.
Comment
The retinoids are chemically related to vitamin A. They regulate epithelial cell growth and are beneficial in inflammatory skin disorders and in patients with increased cell turnover as well as in skin cancer and precancer prevention/treatment.5 The first- and second-generation retinoids, isotretinoin and acitretin, respectively, cause anemia or leukopenia in less than 10% of patients; adverse effects are noted more commonly in doses greater than 1 mg/kg daily.6-8
Bexarotene is a third-generation retinoid drug that is more selective for retinoid X receptors. It was approved in 1991 for treatment of advanced CTCL (stages IIB–IVB) in adult patients who have failed at least 1 prior systemic therapy. Bexarotene is noted to promote cell cycle arrest and apoptosis in CTCL cell lines.9 However, one study suggested that for bexarotene, inhibition of proliferation is more important than causing apoptosis in CTCL cells, and this effect is achieved through triggering the p53/p73-dependent cell cycle inhibition pathway.10 Studies in patients with Sézary syndrome have shown that bexarotene changes the chemokine receptor expression in circulating malignant T cells, making them less likely to traffic to the skin (lower chemokine receptor type 4 expression),11 which may explain why some CTCL cases have shown improvement of skin disease on bexarotene despite progression of extradermal disease.12
Common side effects of bexarotene include hyperlipidemia and central hypothyroidism.13 In addition, dose-related myelosuppression with isolated leukopenia, particularly neutropenia, also has been reported (18% of patients at a dosage of 300 mg/m2/d and 43% of patients with a dosage greater than 300 mg/m2/d). Leukopenia generally occurs within the first 4 to 8 weeks of treatment, is relatively mild (WBC, 1000–2999/µL), and generally is reversible.13-15 One review of 66 mycosis fungoides patients treated with bexarotene described a patient who developed leukopenia 15 months after initiating bexarotene therapy.14 The manufacturer recommends that treatment with bexarotene be continued as long as the patient is receiving benefit from the treatment. One trial of 70 mycosis fungoides patient treated with bexarotene reported response rates of 48% on bexarotene monotherapy (n=54) and 69% on bexarotene plus an additional agent (n=16).15 The authors noted higher response rates in patients on 2 lipid-lowering agents. They concluded that bexarotene was a safe and effective agent for treatment of cutaneous T-cell lymphoma and recommended continued treatment with a lowered dose of bexarotene in those achieving complete responses for a period of 2 years. Although the recommended initial dose is 300 mg/m2/d, bexarotene can be increased to 400 mg/m2/d after 8 weeks if no response to treatment is appreciated.16 Our patient was on a maximum bexarotene dose of 600 mg once daily (280 mg/m2/d) for the first 2 years, and a maintenance dose of 300 mg once daily for the next 3 years. He was not on any medicines known to induce leukopenia and he was not given any known cytochrome P450 3A4 inhibitors that could increase the toxicity of bexarotene.
The patient’s CBC was checked routinely every 2 to 3 months after he was started on bexarotene. For 5 years, the CBC and differential remained within reference range; however, his CD4 counts were not followed during those 5 years. We attribute his CD4 lymphopenia and subsequent pneumocystis pneumonia to bexarotene. After our patient’s CD4 lymphopenia was discovered, he developed a precipitous drop in his WBC and lymphocyte counts while hospitalized that worsened over a 2-week period. At this point, the bexarotene was discontinued and his WBC count slowly recovered. We believe that one of the initial antibiotics prescribed by the patient’s primary care physician at initial onset of pneumonia symptoms as an outpatient could have acted synergistically with bexarotene to worsen lymphopenia. Specifically, ceftazidime, vancomycin, and ciprofloxacin have all been reported to cause leukopenia; however, it was neutropenia in these cases, not lymphopenia.17,18 Notwithstanding, the opportunistic pneumonia and therefore CD4 lymphopenia was present prior to any antibiotic use.
The CD4 lymphopenia was unlikely due to underlying infection(s) because an extensive workup was negative, except for the pneumocystis, which likely resulted from the lymphopenia. The CD4 lymphopenia also could be idiopathic, as it has been reported in 3 patients with mycosis fungoides.19 All 3 patients were erythrodermic at presentation and were noted to have numerous CD4+ lymphocytes in the cutaneous lesions but few circulating CD4+ T lymphocytes in the blood. The authors attributed the CD4 lymphopenia to cutaneous sequestration of CD4+ T lymphocytes.19 These cases contrast with our patient who was in clinical remission at the time of CD4 lymphopenia, which improved and normalized following discontinuation of bexarotene.
Conclusion
This case emphasizes the importance of monitoring for leukopenia, specifically CD4 lymphopenia, in patients on long-term bexarotene therapy. Routine CBC as well as T-cell subset counts should be performed during treatment. Rotation off bexarotene after several years of therapy should be considered, even in patients with continuous benefit from this systemic therapy.
- Smith DK, Neal JJ, Holmberg SD. Unexplained opportunistic infections and CD4+ T-lymphocytopenia without HIV infection. an investigation of cases in the United States. The Centers for Disease Control Idiopathic CD4+ T-lymphocytopenia Task Force. N Engl J Med. 1993;328:373-379.
- Castelino DJ, McNair P, Kay TW. Lymphocytopenia in a hospital population: what does it signify? Aust N Z J Med. 1997;27:170-174.
- Zonios DI, Falloon J, Bennett JE, et al. Idiopathic CD4+ lymphocytopenia: natural history and prognostic factors. Blood. 2008;112:287-294.
- Duncan RA, von Reyn CF, Alliegro GM, et al. Idiopathic CD4+ T-lymphocytopenia: four patients with opportunistic infections and no evidence of HIV infection. N Engl J Med. 1993;328:393-398.
- Bruno NP, Beacham BE, Burnett JW. Adverse effects of isotretinoin therapy. Cutis. 1984;33:484-486, 489.
- Strauss JS, Rapini RP, Shalita AR, et al. Isotretinoin therapy for acne: results of a multicenter dose-response study. J Am Acad Dermatol. 1984;10:490-496.
- Windhorst DB, Nigra T. General clinical toxicology of oral retinoids. J Am Acad Dermatol.1982;6:675-682.
- Glinnick SE. Leucopenia from accutane: in ten percent? Schoch Let. 1985;35:9.
- Wilcox RA. Cutaneous T-cell lymphoma: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol. 2011;86:928-948.
- Nieto-Rementería N, Pérez-Yarza G, Boyano MD, et al. Bexarotene activates the p53/p73 pathway in human cutaneous T-cell lymphoma. Br J Dermatol. 2009;160:519-526.
- Richardson SK, Newton SB, Bach TL, et al. Bexarotene blunts malignant T-cell chemotaxis in Sézary syndrome: reduction of chemokine receptor 4-positive lymphocytes and decreased chemotaxis to thymus and activation-regulated chemokine. Am J Hematol. 2007;82:792-797.
- Bouwhuis SA, Davis MD, el-Azhary RA, et al. Bexarotene treatment of late-stage mycosis fungoides and Sézary syndrome: development of extracutaneous lymphoma in 6 patients. J Am Acad Dermatol. 2005;52:991-996.
- Targretin [package insert]. Bridgewater, NJ: Valeant Pharmaceuticals International, Inc; 2015.
- , , , et al. Bexarotene therapy for mycosis fungoides and Sézary syndrome. Br J Dermatol. 2009;160:1299-1307.
- , , et al. Optimizing bexarotene therapy for cutaneous T-cell lymphoma. J Am Acad Dermatol. 2002;47:672-684.
- Scarisbrick JJ, Morris S, Azurdia R, et al. U.K. consensus statement on safe clinical prescribing of bexarotene for patients with cutaneous T-cell lymphoma. Br J Dermatol. 2013;168:192-200.
- Black E, Lau TT, Ensom MH. Vancomycin-induced neutropenia: is it dose-or duration related? Ann Pharmacother. 2011;45:629-638.
- Choo PW, Gantz NM. Reversible leukopenia related to ciprofloxacin therapy. South Med J. 1990;83:597-598.
- Stevens SR, Griffiths TW, Cooper KD. Idiopathic CD4+ T lymphocytopenia in a patient with mycosis fungoides. J Am Acad Dermatol. 1995;32:1063-1064.
- Smith DK, Neal JJ, Holmberg SD. Unexplained opportunistic infections and CD4+ T-lymphocytopenia without HIV infection. an investigation of cases in the United States. The Centers for Disease Control Idiopathic CD4+ T-lymphocytopenia Task Force. N Engl J Med. 1993;328:373-379.
- Castelino DJ, McNair P, Kay TW. Lymphocytopenia in a hospital population: what does it signify? Aust N Z J Med. 1997;27:170-174.
- Zonios DI, Falloon J, Bennett JE, et al. Idiopathic CD4+ lymphocytopenia: natural history and prognostic factors. Blood. 2008;112:287-294.
- Duncan RA, von Reyn CF, Alliegro GM, et al. Idiopathic CD4+ T-lymphocytopenia: four patients with opportunistic infections and no evidence of HIV infection. N Engl J Med. 1993;328:393-398.
- Bruno NP, Beacham BE, Burnett JW. Adverse effects of isotretinoin therapy. Cutis. 1984;33:484-486, 489.
- Strauss JS, Rapini RP, Shalita AR, et al. Isotretinoin therapy for acne: results of a multicenter dose-response study. J Am Acad Dermatol. 1984;10:490-496.
- Windhorst DB, Nigra T. General clinical toxicology of oral retinoids. J Am Acad Dermatol.1982;6:675-682.
- Glinnick SE. Leucopenia from accutane: in ten percent? Schoch Let. 1985;35:9.
- Wilcox RA. Cutaneous T-cell lymphoma: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol. 2011;86:928-948.
- Nieto-Rementería N, Pérez-Yarza G, Boyano MD, et al. Bexarotene activates the p53/p73 pathway in human cutaneous T-cell lymphoma. Br J Dermatol. 2009;160:519-526.
- Richardson SK, Newton SB, Bach TL, et al. Bexarotene blunts malignant T-cell chemotaxis in Sézary syndrome: reduction of chemokine receptor 4-positive lymphocytes and decreased chemotaxis to thymus and activation-regulated chemokine. Am J Hematol. 2007;82:792-797.
- Bouwhuis SA, Davis MD, el-Azhary RA, et al. Bexarotene treatment of late-stage mycosis fungoides and Sézary syndrome: development of extracutaneous lymphoma in 6 patients. J Am Acad Dermatol. 2005;52:991-996.
- Targretin [package insert]. Bridgewater, NJ: Valeant Pharmaceuticals International, Inc; 2015.
- , , , et al. Bexarotene therapy for mycosis fungoides and Sézary syndrome. Br J Dermatol. 2009;160:1299-1307.
- , , et al. Optimizing bexarotene therapy for cutaneous T-cell lymphoma. J Am Acad Dermatol. 2002;47:672-684.
- Scarisbrick JJ, Morris S, Azurdia R, et al. U.K. consensus statement on safe clinical prescribing of bexarotene for patients with cutaneous T-cell lymphoma. Br J Dermatol. 2013;168:192-200.
- Black E, Lau TT, Ensom MH. Vancomycin-induced neutropenia: is it dose-or duration related? Ann Pharmacother. 2011;45:629-638.
- Choo PW, Gantz NM. Reversible leukopenia related to ciprofloxacin therapy. South Med J. 1990;83:597-598.
- Stevens SR, Griffiths TW, Cooper KD. Idiopathic CD4+ T lymphocytopenia in a patient with mycosis fungoides. J Am Acad Dermatol. 1995;32:1063-1064.
Practice Points
- Most adverse effects of bexarotene (eg, hypothyroidism, hyperlipidemia, leukopenia) occur within the first several months of therapy.
- Delayed-onset leukopenia, including CD4 lymphopenia, may occur several years after initiating bexarotene therapy, resulting in opportunistic infections.
- Long-term periodic monitoring of T lymphocyte counts at least twice yearly in addition to standard quarterly complete blood cell count with differential are recommended.
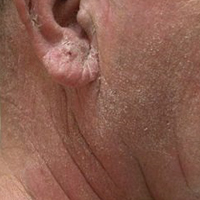
Lupus Erythematosus Tumidus of the Scalp Masquerading as Alopecia Areata
Lupus erythematosus tumidus (LET) is a relatively rare condition but may simply be underdiagnosed in the literature. It presents as urticarialike papules and plaques in sun-exposed areas, characterized by induration and erythema. Lesions occur on the face, neck, upper extremities, and trunk and heal without scarring.1,2 Rarely, lesions can show fine scaling and associated pruritus, but most often the lesions are asymptomatic.3
Case Report
A 45-year-old woman presented with 2 asymptomatic self-described bald spots on the top of the head of 2 months’ duration. The patient denied prior treatment of the lesions and noted one patch was resolving. She reported no involvement of the eyebrows, eyelashes, and axillary and pubic hair. A review of systems was negative. The patient denied personal or family history of lupus, thyroid disease, or vitiligo.
Clinical examination revealed a 1.1-cm round patch of nonscarring alopecia on the right vertex scalp and a 0.9-cm round patch of nonscarring alopecia with moderate hair regrowth on the left vertex scalp. There was no erythema, scaling, or induration. The rest of the scalp was normal in appearance and the eyebrows and eyelashes were uninvolved. The patient was diagnosed with alopecia areata and was treated with 10 mg/mL of intralesional triamcinolone once monthly for 4 months.
The patient initially showed improvement with moderate hair regrowth. After 4 months of treatment, she developed 3 new 1- to 1.5-cm erythematous alopecic patches on the vertex scalp and had worsening in the initial patches (Figure 1). Given the resistance to standard therapy and the onset of multiple new areas with evidence of inflammatory involvement, a punch biopsy was performed. Histopathologic examination revealed a fairly unremarkable epidermis and a dense dermal inflammatory infiltrate that was present both in the superficial and deep dermis (Figure 2). The inflammatory cells, which appeared to be predominantly comprised of lymphocytes, had a predilection for the vasculature but also were observed within the interstitial dermis. Additionally, mucin appeared to be slightly increased in the deep dermis. The lymphocytic phenotype was confirmed by immunohistochemical studies for CD20 and CD3. The most likely possibilities for this reaction pattern were LET, Jessner lymphocytic infiltrate of the skin (JLIS), gyrate erythema, and lymphoma; however, the immunohistochemical studies effectively ruled out lymphoma. Additionally, there was pronounced dermal mucin noted in the specimen. The patient was diagnosed with LET of the scalp based on the constellation of findings.
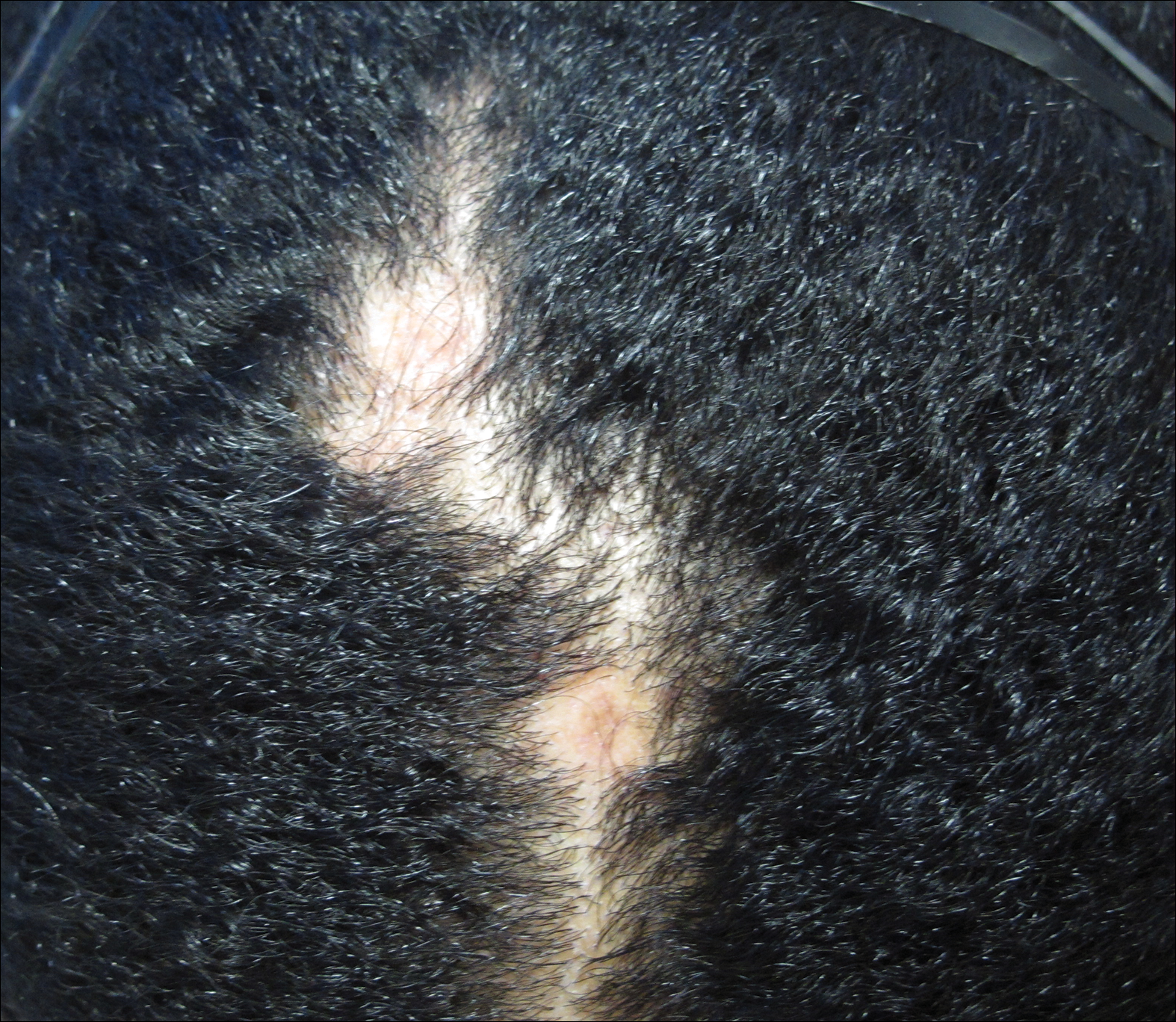
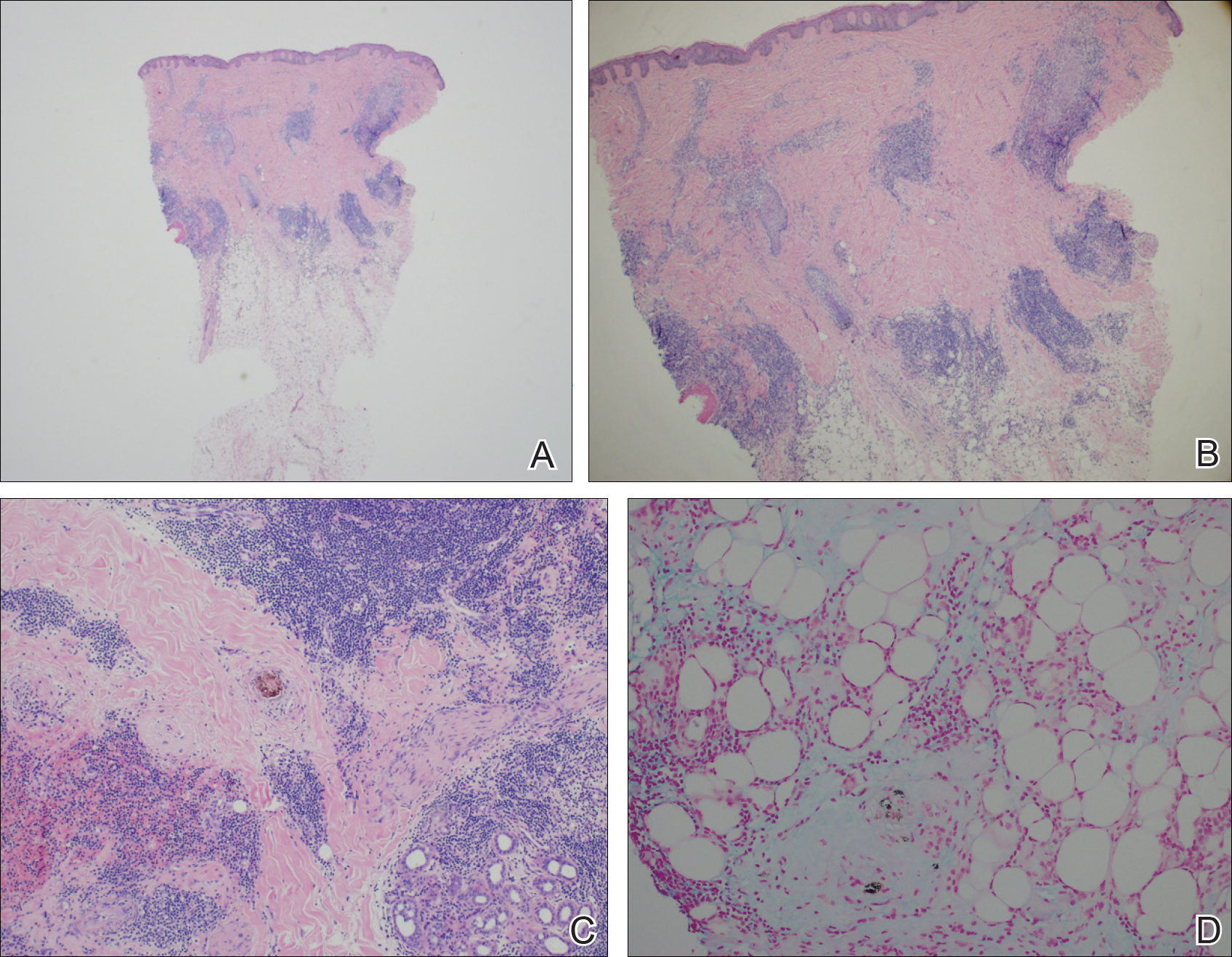
Comment
The classification of LET as a single unique entity or disease process sui generis has been in flux in the last decade. Its similarities to JLIS and other forms of chronic cutaneous lupus erythematosus (CCLE) have brought debate.4-6 In 1930, Gougerot and Burnier7 documented the first case of LET in the literature, describing smooth, infiltrated, erythematous lesions with no desquamation or other superficial changes seen in 5 patients.
In 2000, interest in LET and other forms of CCLE was increasing, and reports in the literature paralleled. That year, Kuhn et al4 reported 40 cases of LET, characterizing the clinical and histological features of each case to demonstrate that LET should be separate from other forms of CCLE. Until then, it is likely that many lesions that should have been classified as LET were instead classified as various forms of CCLE. The investigators maintained that LET also should be distinct from JLIS because it is associated with UV exposure.4 Kuhn et al8 reviewed phototesting in 60 patients with LET in 2001 and confirmed this subset was the most photosensitive type of lupus erythematosus.
In general, the histopathologic and immunohistochemical studies in LET and JLIS can be quite similar. Relatively distinguishing histopathologic findings in JLIS include no evidence of epidermal atrophy, basal vacuolar change, or follicular plugging, as well as negative immunofluorescence studies. Both entities show a predominantly T-cell population with a smaller component of B cells and thus a distinction cannot be made based on relative proportions of T and B cells in lesions.2
In 2003, Alexiades-Armenakas et al6 determined immunohistochemical criteria for LET, finding a predominance of T cells and more CD4 lymphocytes than CD8 lymphocytes with a mean ratio of roughly 3 to 1. Their study results maintained LET should be classified as a form of CCLE due to the chronicity of the lesions, the serologic profile with negative anti–double-stranded DNA, anticentromere, anti-Smith, anti-Ro/Sjögren syndrome antigen A, anti-La/Sjögren syndrome antigen B, and anti-nuclear ribonucleoprotein antibodies and the rare association with systemic disease.6 This conclusion was further solidified by a review published that same year citing unique histopathological features when compared to subacute cutaneous LE and discoid lupus erythematosus.5
This case illustrates the importance of histologic evaluation in determining the correct diagnosis in a patient with alopecia areata recalcitrant to treatment. Including LET in the differential of alopecic patches on the scalp could prove beneficial for patients, as LET responds well to antimalarial drugs and photoprotection.9 This patient had a normal antinuclear antibody panel and no signs or symptoms of systemic lupus. It was recommended that she avoid sun exposure and begin treatment with hydroxychloroquine but she declined. At a follow-up visit 6 months later she reported the lesions had improved, but a permanent wig had been sewn over the area, so it could not be examined.
- Lee L, Werth V. Rheumatologic disease. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. 3rd ed. Mosby Elsevier; 2008:615-629.
- Weedon D. The lichenoid reaction pattern. In: Weedon D. Skin Pathology. 2nd ed. Edinburgh, Scotland: Churchill Livingstone; 2002:35-70.
- Dekle CL, Mannes KD, Davis LS, et al. Lupus tumidus. J Am Acad Dermatol. 1999;41:250-253.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus—a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Kuhn A, Sonntag M, Ruzicka T, et al. Histopathologic findings in lupus erythematosus tumidus: review of 80 patients. J Am Acad Dermatol. 2003;48:901-908.
- Alexiades-Armenakas MR, Baldassano M, Bince B, et al. Tumid lupus erythematosus: criteria for classification with immunohistochemical analysis. Arthritis Rheum. 2003;49:494-500.
- Gougerot H, Burnier R. Lupuse rythe mateux “tumidus.” Bull Soc Fr Dermatol Syph. 1930;37:1291-1292.
- Kuhn A, Sonntag M, Richter-Hintz D, et al. Phototesting in lupus erythematosus tumidus—review of 60 patients. Photochem Photobiol. 2001;73:532-536.
- Cozzani E, Christana K, Rongioletti F, et al. Lupus erythematosus tumidus: clinical, histopathological and serological aspects and therapy response of 21 patients. Eur J Dermatol. 2010;20:797-801.
Lupus erythematosus tumidus (LET) is a relatively rare condition but may simply be underdiagnosed in the literature. It presents as urticarialike papules and plaques in sun-exposed areas, characterized by induration and erythema. Lesions occur on the face, neck, upper extremities, and trunk and heal without scarring.1,2 Rarely, lesions can show fine scaling and associated pruritus, but most often the lesions are asymptomatic.3
Case Report
A 45-year-old woman presented with 2 asymptomatic self-described bald spots on the top of the head of 2 months’ duration. The patient denied prior treatment of the lesions and noted one patch was resolving. She reported no involvement of the eyebrows, eyelashes, and axillary and pubic hair. A review of systems was negative. The patient denied personal or family history of lupus, thyroid disease, or vitiligo.
Clinical examination revealed a 1.1-cm round patch of nonscarring alopecia on the right vertex scalp and a 0.9-cm round patch of nonscarring alopecia with moderate hair regrowth on the left vertex scalp. There was no erythema, scaling, or induration. The rest of the scalp was normal in appearance and the eyebrows and eyelashes were uninvolved. The patient was diagnosed with alopecia areata and was treated with 10 mg/mL of intralesional triamcinolone once monthly for 4 months.
The patient initially showed improvement with moderate hair regrowth. After 4 months of treatment, she developed 3 new 1- to 1.5-cm erythematous alopecic patches on the vertex scalp and had worsening in the initial patches (Figure 1). Given the resistance to standard therapy and the onset of multiple new areas with evidence of inflammatory involvement, a punch biopsy was performed. Histopathologic examination revealed a fairly unremarkable epidermis and a dense dermal inflammatory infiltrate that was present both in the superficial and deep dermis (Figure 2). The inflammatory cells, which appeared to be predominantly comprised of lymphocytes, had a predilection for the vasculature but also were observed within the interstitial dermis. Additionally, mucin appeared to be slightly increased in the deep dermis. The lymphocytic phenotype was confirmed by immunohistochemical studies for CD20 and CD3. The most likely possibilities for this reaction pattern were LET, Jessner lymphocytic infiltrate of the skin (JLIS), gyrate erythema, and lymphoma; however, the immunohistochemical studies effectively ruled out lymphoma. Additionally, there was pronounced dermal mucin noted in the specimen. The patient was diagnosed with LET of the scalp based on the constellation of findings.
Comment
The classification of LET as a single unique entity or disease process sui generis has been in flux in the last decade. Its similarities to JLIS and other forms of chronic cutaneous lupus erythematosus (CCLE) have brought debate.4-6 In 1930, Gougerot and Burnier7 documented the first case of LET in the literature, describing smooth, infiltrated, erythematous lesions with no desquamation or other superficial changes seen in 5 patients.
In 2000, interest in LET and other forms of CCLE was increasing, and reports in the literature paralleled. That year, Kuhn et al4 reported 40 cases of LET, characterizing the clinical and histological features of each case to demonstrate that LET should be separate from other forms of CCLE. Until then, it is likely that many lesions that should have been classified as LET were instead classified as various forms of CCLE. The investigators maintained that LET also should be distinct from JLIS because it is associated with UV exposure.4 Kuhn et al8 reviewed phototesting in 60 patients with LET in 2001 and confirmed this subset was the most photosensitive type of lupus erythematosus.
In general, the histopathologic and immunohistochemical studies in LET and JLIS can be quite similar. Relatively distinguishing histopathologic findings in JLIS include no evidence of epidermal atrophy, basal vacuolar change, or follicular plugging, as well as negative immunofluorescence studies. Both entities show a predominantly T-cell population with a smaller component of B cells and thus a distinction cannot be made based on relative proportions of T and B cells in lesions.2
In 2003, Alexiades-Armenakas et al6 determined immunohistochemical criteria for LET, finding a predominance of T cells and more CD4 lymphocytes than CD8 lymphocytes with a mean ratio of roughly 3 to 1. Their study results maintained LET should be classified as a form of CCLE due to the chronicity of the lesions, the serologic profile with negative anti–double-stranded DNA, anticentromere, anti-Smith, anti-Ro/Sjögren syndrome antigen A, anti-La/Sjögren syndrome antigen B, and anti-nuclear ribonucleoprotein antibodies and the rare association with systemic disease.6 This conclusion was further solidified by a review published that same year citing unique histopathological features when compared to subacute cutaneous LE and discoid lupus erythematosus.5
This case illustrates the importance of histologic evaluation in determining the correct diagnosis in a patient with alopecia areata recalcitrant to treatment. Including LET in the differential of alopecic patches on the scalp could prove beneficial for patients, as LET responds well to antimalarial drugs and photoprotection.9 This patient had a normal antinuclear antibody panel and no signs or symptoms of systemic lupus. It was recommended that she avoid sun exposure and begin treatment with hydroxychloroquine but she declined. At a follow-up visit 6 months later she reported the lesions had improved, but a permanent wig had been sewn over the area, so it could not be examined.
Lupus erythematosus tumidus (LET) is a relatively rare condition but may simply be underdiagnosed in the literature. It presents as urticarialike papules and plaques in sun-exposed areas, characterized by induration and erythema. Lesions occur on the face, neck, upper extremities, and trunk and heal without scarring.1,2 Rarely, lesions can show fine scaling and associated pruritus, but most often the lesions are asymptomatic.3
Case Report
A 45-year-old woman presented with 2 asymptomatic self-described bald spots on the top of the head of 2 months’ duration. The patient denied prior treatment of the lesions and noted one patch was resolving. She reported no involvement of the eyebrows, eyelashes, and axillary and pubic hair. A review of systems was negative. The patient denied personal or family history of lupus, thyroid disease, or vitiligo.
Clinical examination revealed a 1.1-cm round patch of nonscarring alopecia on the right vertex scalp and a 0.9-cm round patch of nonscarring alopecia with moderate hair regrowth on the left vertex scalp. There was no erythema, scaling, or induration. The rest of the scalp was normal in appearance and the eyebrows and eyelashes were uninvolved. The patient was diagnosed with alopecia areata and was treated with 10 mg/mL of intralesional triamcinolone once monthly for 4 months.
The patient initially showed improvement with moderate hair regrowth. After 4 months of treatment, she developed 3 new 1- to 1.5-cm erythematous alopecic patches on the vertex scalp and had worsening in the initial patches (Figure 1). Given the resistance to standard therapy and the onset of multiple new areas with evidence of inflammatory involvement, a punch biopsy was performed. Histopathologic examination revealed a fairly unremarkable epidermis and a dense dermal inflammatory infiltrate that was present both in the superficial and deep dermis (Figure 2). The inflammatory cells, which appeared to be predominantly comprised of lymphocytes, had a predilection for the vasculature but also were observed within the interstitial dermis. Additionally, mucin appeared to be slightly increased in the deep dermis. The lymphocytic phenotype was confirmed by immunohistochemical studies for CD20 and CD3. The most likely possibilities for this reaction pattern were LET, Jessner lymphocytic infiltrate of the skin (JLIS), gyrate erythema, and lymphoma; however, the immunohistochemical studies effectively ruled out lymphoma. Additionally, there was pronounced dermal mucin noted in the specimen. The patient was diagnosed with LET of the scalp based on the constellation of findings.
Comment
The classification of LET as a single unique entity or disease process sui generis has been in flux in the last decade. Its similarities to JLIS and other forms of chronic cutaneous lupus erythematosus (CCLE) have brought debate.4-6 In 1930, Gougerot and Burnier7 documented the first case of LET in the literature, describing smooth, infiltrated, erythematous lesions with no desquamation or other superficial changes seen in 5 patients.
In 2000, interest in LET and other forms of CCLE was increasing, and reports in the literature paralleled. That year, Kuhn et al4 reported 40 cases of LET, characterizing the clinical and histological features of each case to demonstrate that LET should be separate from other forms of CCLE. Until then, it is likely that many lesions that should have been classified as LET were instead classified as various forms of CCLE. The investigators maintained that LET also should be distinct from JLIS because it is associated with UV exposure.4 Kuhn et al8 reviewed phototesting in 60 patients with LET in 2001 and confirmed this subset was the most photosensitive type of lupus erythematosus.
In general, the histopathologic and immunohistochemical studies in LET and JLIS can be quite similar. Relatively distinguishing histopathologic findings in JLIS include no evidence of epidermal atrophy, basal vacuolar change, or follicular plugging, as well as negative immunofluorescence studies. Both entities show a predominantly T-cell population with a smaller component of B cells and thus a distinction cannot be made based on relative proportions of T and B cells in lesions.2
In 2003, Alexiades-Armenakas et al6 determined immunohistochemical criteria for LET, finding a predominance of T cells and more CD4 lymphocytes than CD8 lymphocytes with a mean ratio of roughly 3 to 1. Their study results maintained LET should be classified as a form of CCLE due to the chronicity of the lesions, the serologic profile with negative anti–double-stranded DNA, anticentromere, anti-Smith, anti-Ro/Sjögren syndrome antigen A, anti-La/Sjögren syndrome antigen B, and anti-nuclear ribonucleoprotein antibodies and the rare association with systemic disease.6 This conclusion was further solidified by a review published that same year citing unique histopathological features when compared to subacute cutaneous LE and discoid lupus erythematosus.5
This case illustrates the importance of histologic evaluation in determining the correct diagnosis in a patient with alopecia areata recalcitrant to treatment. Including LET in the differential of alopecic patches on the scalp could prove beneficial for patients, as LET responds well to antimalarial drugs and photoprotection.9 This patient had a normal antinuclear antibody panel and no signs or symptoms of systemic lupus. It was recommended that she avoid sun exposure and begin treatment with hydroxychloroquine but she declined. At a follow-up visit 6 months later she reported the lesions had improved, but a permanent wig had been sewn over the area, so it could not be examined.
- Lee L, Werth V. Rheumatologic disease. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. 3rd ed. Mosby Elsevier; 2008:615-629.
- Weedon D. The lichenoid reaction pattern. In: Weedon D. Skin Pathology. 2nd ed. Edinburgh, Scotland: Churchill Livingstone; 2002:35-70.
- Dekle CL, Mannes KD, Davis LS, et al. Lupus tumidus. J Am Acad Dermatol. 1999;41:250-253.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus—a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Kuhn A, Sonntag M, Ruzicka T, et al. Histopathologic findings in lupus erythematosus tumidus: review of 80 patients. J Am Acad Dermatol. 2003;48:901-908.
- Alexiades-Armenakas MR, Baldassano M, Bince B, et al. Tumid lupus erythematosus: criteria for classification with immunohistochemical analysis. Arthritis Rheum. 2003;49:494-500.
- Gougerot H, Burnier R. Lupuse rythe mateux “tumidus.” Bull Soc Fr Dermatol Syph. 1930;37:1291-1292.
- Kuhn A, Sonntag M, Richter-Hintz D, et al. Phototesting in lupus erythematosus tumidus—review of 60 patients. Photochem Photobiol. 2001;73:532-536.
- Cozzani E, Christana K, Rongioletti F, et al. Lupus erythematosus tumidus: clinical, histopathological and serological aspects and therapy response of 21 patients. Eur J Dermatol. 2010;20:797-801.
- Lee L, Werth V. Rheumatologic disease. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. 3rd ed. Mosby Elsevier; 2008:615-629.
- Weedon D. The lichenoid reaction pattern. In: Weedon D. Skin Pathology. 2nd ed. Edinburgh, Scotland: Churchill Livingstone; 2002:35-70.
- Dekle CL, Mannes KD, Davis LS, et al. Lupus tumidus. J Am Acad Dermatol. 1999;41:250-253.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus—a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Kuhn A, Sonntag M, Ruzicka T, et al. Histopathologic findings in lupus erythematosus tumidus: review of 80 patients. J Am Acad Dermatol. 2003;48:901-908.
- Alexiades-Armenakas MR, Baldassano M, Bince B, et al. Tumid lupus erythematosus: criteria for classification with immunohistochemical analysis. Arthritis Rheum. 2003;49:494-500.
- Gougerot H, Burnier R. Lupuse rythe mateux “tumidus.” Bull Soc Fr Dermatol Syph. 1930;37:1291-1292.
- Kuhn A, Sonntag M, Richter-Hintz D, et al. Phototesting in lupus erythematosus tumidus—review of 60 patients. Photochem Photobiol. 2001;73:532-536.
- Cozzani E, Christana K, Rongioletti F, et al. Lupus erythematosus tumidus: clinical, histopathological and serological aspects and therapy response of 21 patients. Eur J Dermatol. 2010;20:797-801.
Practice Points
- Lupus erythematosus tumidus (LET) of the scalp can mimic alopecia areata on clinical presentation.
- A unique variant of chronic cutaneous lupus erythematosus, LET presents in sun-exposed areas without any corresponding systemic signs.
- Lupus erythematosus tumidus may respond well to antimalarial drugs.
Resolution of Psoriatic Lesions on the Gingiva and Hard Palate Following Administration of Adalimumab for Cutaneous Psoriasis
Psoriasis is a chronic, relapsing, inflammatory systemic disorder of the skin with an incidence of 2% to 3% and is estimated to affect 125 million individuals worldwide.1 Environmental triggers of disease modulation may include cutaneous microbiota, smoking, alcohol use, drugs (ie, beta-blockers, lithium, antimalarials), stress, and trauma.2 Comorbidities associated with cutaneous lesions include psoriatic arthritis, Crohn disease, type 2 diabetes mellitus, metabolic syndrome, stroke, and cardiovascular disease.3 In some studies, patients with psoriasis also had a 24% to 27% increased propensity for periodontal bone loss versus 10% of controls.4,5
Oral psoriasis is rare and case reports have been preferentially published in dental journals, usually with regard to glossal lesions, leaving gingival and palatal psoriatic involvement infrequently reported in the dermatologic literature.6,7 In fact, oral assessments involving 535 psoriatic patients from a dermatology center only yielded cases of geographic and fissured tongue.8 Another study at a psoriasis clinic found 3.8% (21/547) of patients with geographic tongue, 3.1% (17/547) with buccal mucosal plaques, and only 0.4% (2/547) with palatal lesions.9 To extend the knowledge of oral psoriasis, we provide the clinical and histopathologic findings of a patient with synchronous oral and cutaneous psoriatic lesions that responded well to the administration of adalimumab for management of recurrent cutaneous disease.
Case Report
A 51-year-old man presented to the attending periodontist for comprehensive treatment of multiple quadrants of gingival recession. His medical history was remarkable for psoriasis; Prinzmetal angina, which led to myocardial infarction; and diverticulitis. The cutaneous psoriasis began approximately 18 years prior to the current presentation and was initially managed with various topical therapeutics. At an 11-year follow-up, the patient was experiencing poor lesional control as well as severe pruritus and was prescribed etanercept by a dermatologist. His inconsistent compliance with frequency and dosing failed to achieve satisfactory disease suppression and etanercept was discontinued after approximately 2.5 years. Two years later the patient was switched to adalimumab by a dermatologist, and around this time he had developed psoriatic arthritis of the hands and knees and pitting of the nail plates. The patient elected to discontinue adalimumab usage after 3 years due to successful management of the skin lesions, cost considerations, and his perception that the psoriasis could “remain in remission.” After a 6-month lapse, the patient resumed adalimumab due to cutaneous lesional recurrence (Figure 1A).
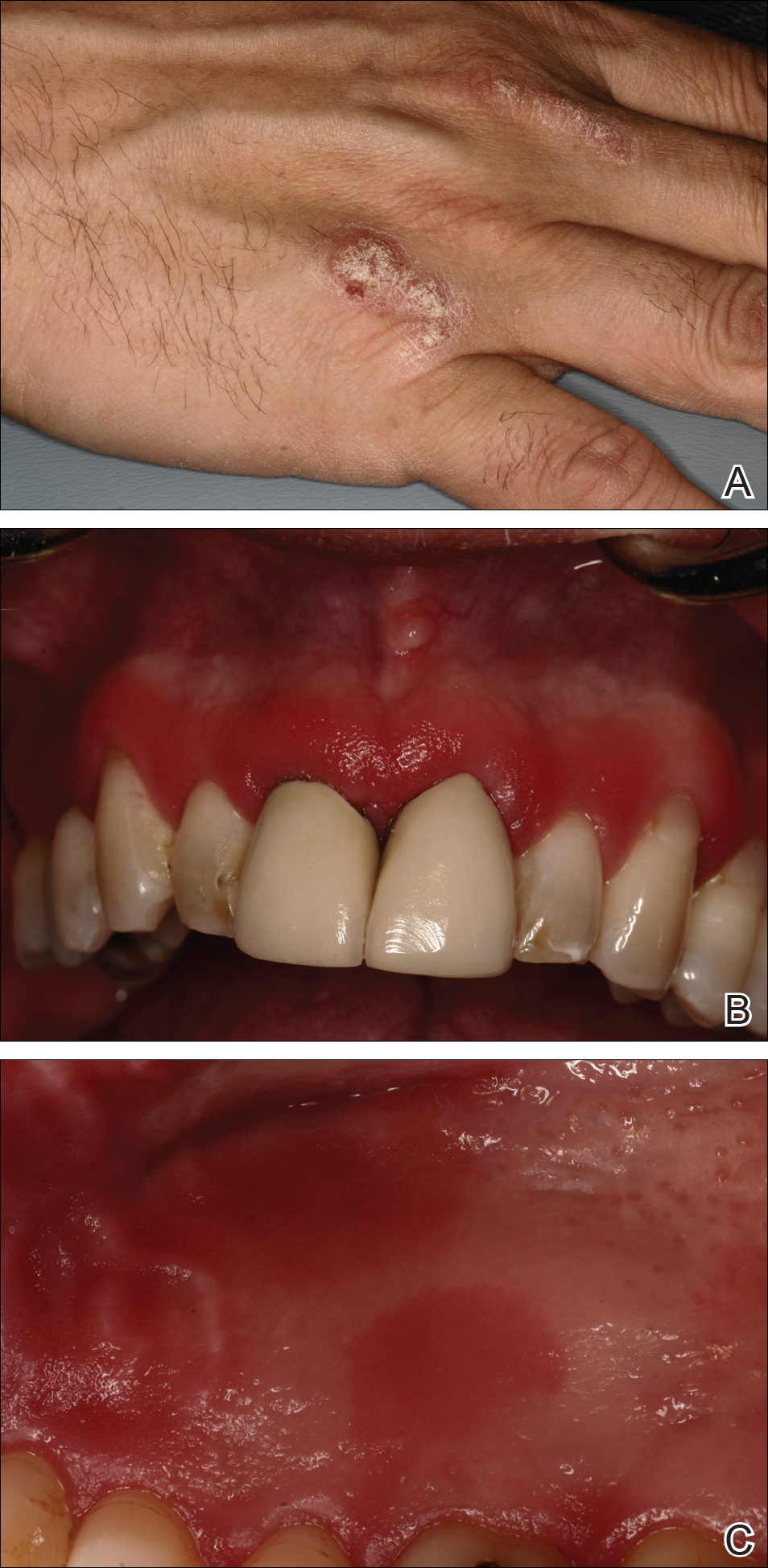
At the current presentation, an oral examination performed 2 days after the reinstitution of adalim-umab revealed generalized severe gingivitis with an atypical inflammatory response that extended from just beyond the mucogingival junction to the marginal gingiva. The gingiva also appeared edematous with a conspicuously granular surface (Figure 1B). The hard palate displayed multiple red macules of varying sizes (Figure 1C). A maxillary gingival biopsy demonstrated hyperkeratosis, parakeratosis, spongiosis, acanthosis, elongation of the rete ridges, numerous collections of neutrophils (Munro microabscesses), and abundant lymphocytes in the subjacent connective tissue (Figure 2). Periodic acid–Schiff staining was negative for fungal hyphae. These features were consistent with oral mucosal psoriasis.
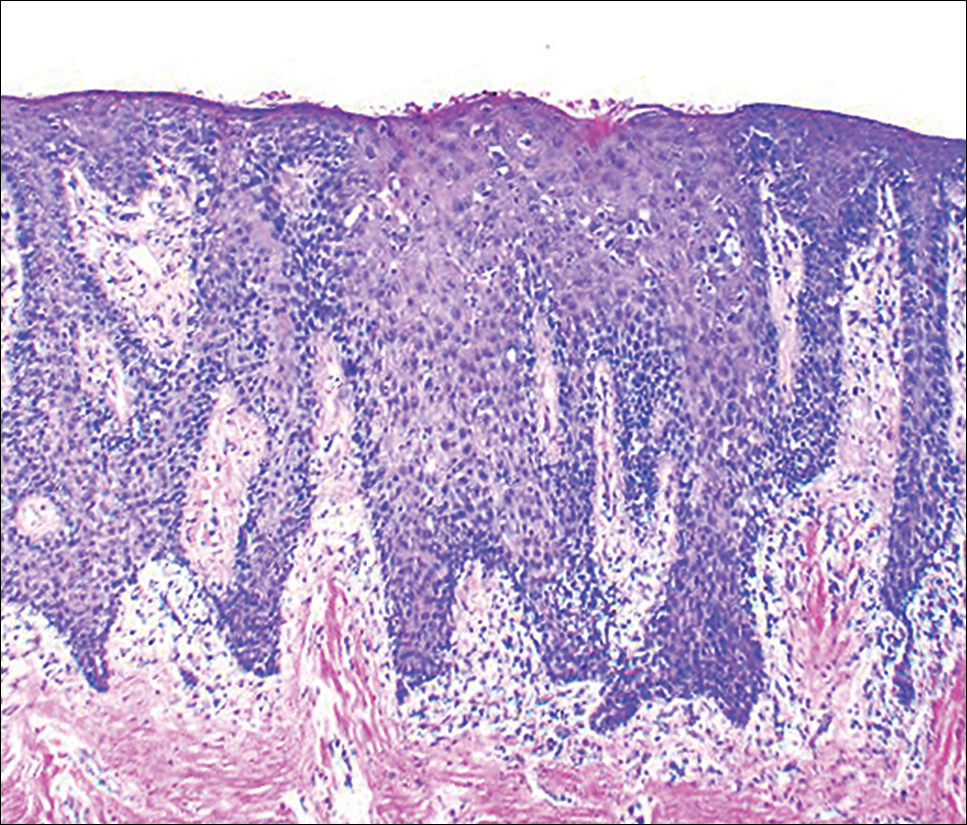
At a 2-month follow-up, the biopsy site had healed without incident and without loss of the gingival architecture. There was an almost-complete resolution of the gingival erythema (Figure 3A) and the patient has since noticed a lack of bleeding using floss. Additionally, the red macules on the palate were no longer present (Figure 3B). The cutaneous plaques were greatly reduced in size and the patient experienced a proportionate decline in pruritus. Based on the uneventful surgical biopsy procedure, the patient was advised to undergo gingival grafting and has not returned for periodontal care.
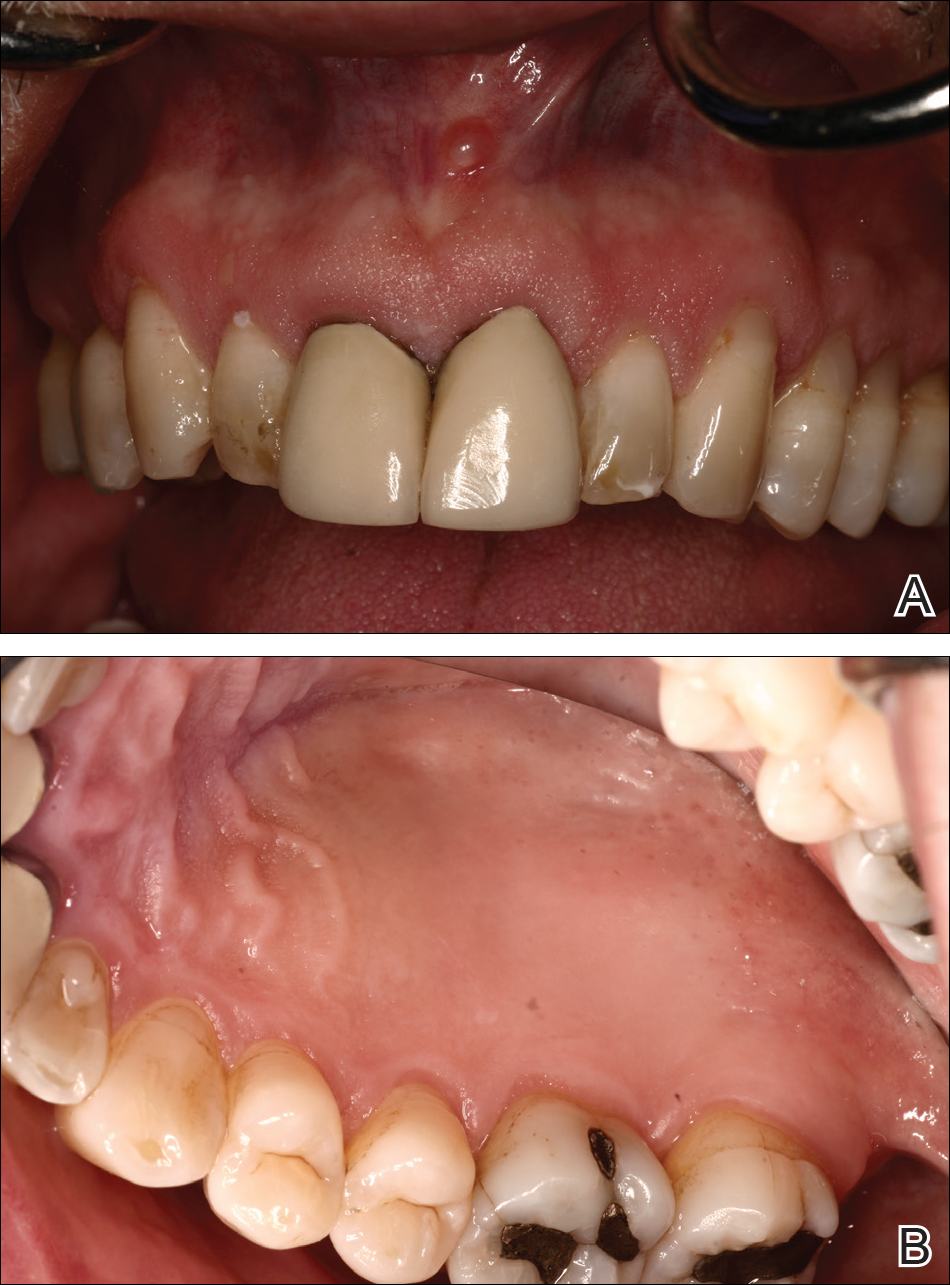
Comment
Psoriasis of the oral cavity is rare and typically occurs on the tongue and less frequently on the hard palate, lip, buccal mucosa, and gingiva.2,7 The lesions are almost always concordant with cutaneous psoriasis, and only sporadic examples exclusive to the oral mucosa have been recognized.7,10 Gingival psoriasis usually is described as intensely erythematous and occasionally laced with white scaly streaks involving the marginal gingiva that extend toward the mucogingival junction. In general, the erythematous presentation of gingival psoriasis may not be commensurate with the degree of inflammation induced by dental plaque-based periodontal disease. Doben11 documented gingival psoriasis as appearing “deeply stippled and grainy” and commented that the tissue was “friable” and incapable of maintaining a “clean incision line” during periodontal surgery. In our patient, the gingiva also had exhibited a granular surface. Patients with oral psoriasis often report soreness or a burning sensation of the gingiva, which may easily bleed on manipulation or brushing the teeth, whereas other patients are asymptomatic,12 as in our case. Psoriasis of the hard palate usually presents as multiple painless red macules. Unlike cutaneous psoriasis, oral lesions rarely evoke pruritus.10 Histopathologically, oral psoriasis bears a striking resemblance to its cutaneous counterpart. The epithelium has a pronounced parakeratinized surface with elongated rete ridges and aggregations of Munro microabscesses. The connective tissue often is composed of dilated capillaries that closely approximate the epithelium as well as infiltrations of lymphocytes. Specimens suspected for oral psoriasis should routinely be stained with periodic acid–Schiff to rule out candidiasis coinfection. The microscopic findings of our patient were congruent with prior reports of oral psoriasis.7,10-12 Some clinicians have questioned if psoriasis can actually occur in the oral cavity, but most authorities in the field have recognized its true existence, as evidenced by various shared HLA antigens, specifically HLA-Cw.13
Another group of oral lesions collectively referred to as psoriasiform mucositis, notably geographic tongue (benign migratory glossitis, erythema migrans) and its extraglossal variant geographic stomatitis,14,15 have histopathologic features and HLAs similar to those seen in cutaneous psoriasis.13 Interestingly, geographic tongue has been found in 3.8% to 9.1% of cohorts with cutaneous psoriasis,8,9 but in the extant population, the vast majority of patients with oral psoriasiform mucositis do not have cutaneous psoriasis. Other differential diagnoses for gingival psoriasis are lichen planus, human immunodeficiency virus–associated periodontitis, desquamative gingivitis, plasma cell gingivitis, erythematous candidiasis, mucous membrane pemphigoid, pemphigus vulgaris, leukemia, systemic lupus erythematosus, granulomatosis with polyangiitis, orofacial granulomatosis, localized juvenile spongiotic gingivitis hyperplasia, and primary gingivostomatitis.
Management of gingival psoriasis focuses on strategies to reduce inflammation and discomfort and measures to achieve meticulous oral plaque control. Judicious efforts should be exercised to avoid oral soft-tissue injury when performing periodontal scaling, although it has not been established whether gingival psoriasis is associated with the Köbner phenomenon, as seen with cutaneous lesions. Adjunctive measures employed for symptomatic patients have involved the use of corticosteroids (eg, lesional injection, oral rinse, systemic) and oral rinses with retinoic acid, chlorhexidine gluconate, and warm saline.7,10,16 Prolonged utilization of corticosteroids, however, may necessitate supplemental administration of antifungal agents.
This case report represents a rare documentation of a successful outcome of gingival and palatal psoriasis subsequent to the reinstitution of adalimumab solely for treatment of recurrent cutaneous disease. There likely is a pharmacologic basis for the amelioration of oral psoriasis in our patient. Adalimumab is a bivalent IgG monoclonal antibody that binds to activated dermal dendritic cell receptors of tumor necrosis factor α, thereby attenuating a cytokine-derived inflammatory response and apoptosis.17 In fact, patients with rheumatoid arthritis showed notable reductions in both gingival inflammation and bleeding following a 3-month regimen of adalimumab.18
Conclusion
Practitioners should be aware of the phenotypic overlap of cutaneous and oral psoriasis, particularly involving the gingiva and palate. It is recommended that psoriasis patients routinely receive a dental prophylaxis and engage in oral hygiene efforts to reduce the presence of oral microbiota. Furthermore, it is emphasized that psoriatic patients who maintain an atypical erythematous presentation on the oral mucosa undergo a biopsy for identification of the lesions and correlation with disease dissemination. Prospective studies are needed to characterize the clinical courses of oral psoriasis, ascertain their correlative behavior with cutaneous flares, and determine if lesional improvement can be achieved with the use of biologic agents or other therapeutic modalities.
- Gupta R, Debbaneh MG, Liao W. Genetic epidemiology of psoriasis. Curr Dermatol Rep. 2014;3:61-78.
- Younai FS, Phelan JA. Oral mucositis with features of psoriasis: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:61-67.
- Xu T, Zhang YH. Association of psoriasis with stroke and myocardial infarction: meta-analysis of cohort studies. Br J Dermatol. 2012;167:1345-1350.
- Lazaridou E, Tsikrikoni A, Fotiadou C, et al. Association of chronic plaque psoriasis and severe periodontitis: a hospital based case-control study. J Eur Acad Dermatol Venereol. 2013;27:967-972.
- Skudutyte-Rysstad R, Slevolden EM, Hansen BF, et al. Association between moderate to severe psoriasis and periodontitis in a Scandinavian population. BMC Oral Health. 2014;14:139.
- Zunt SL, Tomich CE. Erythema migrans—a psoriasiform lesion of the oral mucosa. J Dermatol Surg Oncol. 1989;15:1067-1070.
- Reis V, Artico G, Seo J, et al. Psoriasiform mucositis on the gingival and palatal mucosae treated with retinoic-acid mouthwash. Int J Dermatol. 2013;52:113-115.
- Germi L, De Giorgi V, Bergamo F, et al. Psoriasis and oral lesions: multicentric study of oral mucosa diseases Italian group (GIPMO). Dermatol Online J. 2012;18:11.
- Kaur I, Handa S, Kumar B. Oral lesions in psoriasis. Int J Dermatol. 1997;36:78-79.
- Brayshaw HA, Orban B. Psoriasis gingivae. J Periodontol. 1953;24:156-160.
- Doben DI. Psoriasis of the attached gingiva. J Periodontol. 1976;47:38-40.
- Mattsson U, Warfvinge G, Jontell M. Oral psoriasis—a diagnostic dilemma: a report of two cases and a review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;120:e183-e189.
- Dermatologic diseases. In: Neville BW, Damm DD, Allen CM, et al, eds. Oral and Maxillofacial Pathology. 3rd ed. St. Louis, MO: Saunders/Elsevier; 2009:792-794.
- Brooks JK, Balciunas BA. Geographic stomatitis: review of the literature and report of five cases. J Am Dent Assoc. 1987;115:421-424.
- Brooks JK, Nikitakis NG. Multiple mucosal lesions. erythema migrans. Gen Dent. 2007;55:160, 163.
- Ulmansky M, Michelle R, Azaz B. Oral psoriasis: report of six new cases. J Oral Pathol Med. 1995;24:42-45.
- Lis K, Kuzawinska O, Bałkowiec-Iskra E. Tumor necrosis factor inhibitors—state of knowledge. Arch Med Sci. 2014;10:1175-1185.
- Kobayashi T, Yokoyama T, Ito S, et al. Periodontal and serum protein profiles in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitor adalimumab. J Periodontol. 2014;85:1480-1488.
Psoriasis is a chronic, relapsing, inflammatory systemic disorder of the skin with an incidence of 2% to 3% and is estimated to affect 125 million individuals worldwide.1 Environmental triggers of disease modulation may include cutaneous microbiota, smoking, alcohol use, drugs (ie, beta-blockers, lithium, antimalarials), stress, and trauma.2 Comorbidities associated with cutaneous lesions include psoriatic arthritis, Crohn disease, type 2 diabetes mellitus, metabolic syndrome, stroke, and cardiovascular disease.3 In some studies, patients with psoriasis also had a 24% to 27% increased propensity for periodontal bone loss versus 10% of controls.4,5
Oral psoriasis is rare and case reports have been preferentially published in dental journals, usually with regard to glossal lesions, leaving gingival and palatal psoriatic involvement infrequently reported in the dermatologic literature.6,7 In fact, oral assessments involving 535 psoriatic patients from a dermatology center only yielded cases of geographic and fissured tongue.8 Another study at a psoriasis clinic found 3.8% (21/547) of patients with geographic tongue, 3.1% (17/547) with buccal mucosal plaques, and only 0.4% (2/547) with palatal lesions.9 To extend the knowledge of oral psoriasis, we provide the clinical and histopathologic findings of a patient with synchronous oral and cutaneous psoriatic lesions that responded well to the administration of adalimumab for management of recurrent cutaneous disease.
Case Report
A 51-year-old man presented to the attending periodontist for comprehensive treatment of multiple quadrants of gingival recession. His medical history was remarkable for psoriasis; Prinzmetal angina, which led to myocardial infarction; and diverticulitis. The cutaneous psoriasis began approximately 18 years prior to the current presentation and was initially managed with various topical therapeutics. At an 11-year follow-up, the patient was experiencing poor lesional control as well as severe pruritus and was prescribed etanercept by a dermatologist. His inconsistent compliance with frequency and dosing failed to achieve satisfactory disease suppression and etanercept was discontinued after approximately 2.5 years. Two years later the patient was switched to adalimumab by a dermatologist, and around this time he had developed psoriatic arthritis of the hands and knees and pitting of the nail plates. The patient elected to discontinue adalimumab usage after 3 years due to successful management of the skin lesions, cost considerations, and his perception that the psoriasis could “remain in remission.” After a 6-month lapse, the patient resumed adalimumab due to cutaneous lesional recurrence (Figure 1A).
At the current presentation, an oral examination performed 2 days after the reinstitution of adalim-umab revealed generalized severe gingivitis with an atypical inflammatory response that extended from just beyond the mucogingival junction to the marginal gingiva. The gingiva also appeared edematous with a conspicuously granular surface (Figure 1B). The hard palate displayed multiple red macules of varying sizes (Figure 1C). A maxillary gingival biopsy demonstrated hyperkeratosis, parakeratosis, spongiosis, acanthosis, elongation of the rete ridges, numerous collections of neutrophils (Munro microabscesses), and abundant lymphocytes in the subjacent connective tissue (Figure 2). Periodic acid–Schiff staining was negative for fungal hyphae. These features were consistent with oral mucosal psoriasis.
At a 2-month follow-up, the biopsy site had healed without incident and without loss of the gingival architecture. There was an almost-complete resolution of the gingival erythema (Figure 3A) and the patient has since noticed a lack of bleeding using floss. Additionally, the red macules on the palate were no longer present (Figure 3B). The cutaneous plaques were greatly reduced in size and the patient experienced a proportionate decline in pruritus. Based on the uneventful surgical biopsy procedure, the patient was advised to undergo gingival grafting and has not returned for periodontal care.
Comment
Psoriasis of the oral cavity is rare and typically occurs on the tongue and less frequently on the hard palate, lip, buccal mucosa, and gingiva.2,7 The lesions are almost always concordant with cutaneous psoriasis, and only sporadic examples exclusive to the oral mucosa have been recognized.7,10 Gingival psoriasis usually is described as intensely erythematous and occasionally laced with white scaly streaks involving the marginal gingiva that extend toward the mucogingival junction. In general, the erythematous presentation of gingival psoriasis may not be commensurate with the degree of inflammation induced by dental plaque-based periodontal disease. Doben11 documented gingival psoriasis as appearing “deeply stippled and grainy” and commented that the tissue was “friable” and incapable of maintaining a “clean incision line” during periodontal surgery. In our patient, the gingiva also had exhibited a granular surface. Patients with oral psoriasis often report soreness or a burning sensation of the gingiva, which may easily bleed on manipulation or brushing the teeth, whereas other patients are asymptomatic,12 as in our case. Psoriasis of the hard palate usually presents as multiple painless red macules. Unlike cutaneous psoriasis, oral lesions rarely evoke pruritus.10 Histopathologically, oral psoriasis bears a striking resemblance to its cutaneous counterpart. The epithelium has a pronounced parakeratinized surface with elongated rete ridges and aggregations of Munro microabscesses. The connective tissue often is composed of dilated capillaries that closely approximate the epithelium as well as infiltrations of lymphocytes. Specimens suspected for oral psoriasis should routinely be stained with periodic acid–Schiff to rule out candidiasis coinfection. The microscopic findings of our patient were congruent with prior reports of oral psoriasis.7,10-12 Some clinicians have questioned if psoriasis can actually occur in the oral cavity, but most authorities in the field have recognized its true existence, as evidenced by various shared HLA antigens, specifically HLA-Cw.13
Another group of oral lesions collectively referred to as psoriasiform mucositis, notably geographic tongue (benign migratory glossitis, erythema migrans) and its extraglossal variant geographic stomatitis,14,15 have histopathologic features and HLAs similar to those seen in cutaneous psoriasis.13 Interestingly, geographic tongue has been found in 3.8% to 9.1% of cohorts with cutaneous psoriasis,8,9 but in the extant population, the vast majority of patients with oral psoriasiform mucositis do not have cutaneous psoriasis. Other differential diagnoses for gingival psoriasis are lichen planus, human immunodeficiency virus–associated periodontitis, desquamative gingivitis, plasma cell gingivitis, erythematous candidiasis, mucous membrane pemphigoid, pemphigus vulgaris, leukemia, systemic lupus erythematosus, granulomatosis with polyangiitis, orofacial granulomatosis, localized juvenile spongiotic gingivitis hyperplasia, and primary gingivostomatitis.
Management of gingival psoriasis focuses on strategies to reduce inflammation and discomfort and measures to achieve meticulous oral plaque control. Judicious efforts should be exercised to avoid oral soft-tissue injury when performing periodontal scaling, although it has not been established whether gingival psoriasis is associated with the Köbner phenomenon, as seen with cutaneous lesions. Adjunctive measures employed for symptomatic patients have involved the use of corticosteroids (eg, lesional injection, oral rinse, systemic) and oral rinses with retinoic acid, chlorhexidine gluconate, and warm saline.7,10,16 Prolonged utilization of corticosteroids, however, may necessitate supplemental administration of antifungal agents.
This case report represents a rare documentation of a successful outcome of gingival and palatal psoriasis subsequent to the reinstitution of adalimumab solely for treatment of recurrent cutaneous disease. There likely is a pharmacologic basis for the amelioration of oral psoriasis in our patient. Adalimumab is a bivalent IgG monoclonal antibody that binds to activated dermal dendritic cell receptors of tumor necrosis factor α, thereby attenuating a cytokine-derived inflammatory response and apoptosis.17 In fact, patients with rheumatoid arthritis showed notable reductions in both gingival inflammation and bleeding following a 3-month regimen of adalimumab.18
Conclusion
Practitioners should be aware of the phenotypic overlap of cutaneous and oral psoriasis, particularly involving the gingiva and palate. It is recommended that psoriasis patients routinely receive a dental prophylaxis and engage in oral hygiene efforts to reduce the presence of oral microbiota. Furthermore, it is emphasized that psoriatic patients who maintain an atypical erythematous presentation on the oral mucosa undergo a biopsy for identification of the lesions and correlation with disease dissemination. Prospective studies are needed to characterize the clinical courses of oral psoriasis, ascertain their correlative behavior with cutaneous flares, and determine if lesional improvement can be achieved with the use of biologic agents or other therapeutic modalities.
Psoriasis is a chronic, relapsing, inflammatory systemic disorder of the skin with an incidence of 2% to 3% and is estimated to affect 125 million individuals worldwide.1 Environmental triggers of disease modulation may include cutaneous microbiota, smoking, alcohol use, drugs (ie, beta-blockers, lithium, antimalarials), stress, and trauma.2 Comorbidities associated with cutaneous lesions include psoriatic arthritis, Crohn disease, type 2 diabetes mellitus, metabolic syndrome, stroke, and cardiovascular disease.3 In some studies, patients with psoriasis also had a 24% to 27% increased propensity for periodontal bone loss versus 10% of controls.4,5
Oral psoriasis is rare and case reports have been preferentially published in dental journals, usually with regard to glossal lesions, leaving gingival and palatal psoriatic involvement infrequently reported in the dermatologic literature.6,7 In fact, oral assessments involving 535 psoriatic patients from a dermatology center only yielded cases of geographic and fissured tongue.8 Another study at a psoriasis clinic found 3.8% (21/547) of patients with geographic tongue, 3.1% (17/547) with buccal mucosal plaques, and only 0.4% (2/547) with palatal lesions.9 To extend the knowledge of oral psoriasis, we provide the clinical and histopathologic findings of a patient with synchronous oral and cutaneous psoriatic lesions that responded well to the administration of adalimumab for management of recurrent cutaneous disease.
Case Report
A 51-year-old man presented to the attending periodontist for comprehensive treatment of multiple quadrants of gingival recession. His medical history was remarkable for psoriasis; Prinzmetal angina, which led to myocardial infarction; and diverticulitis. The cutaneous psoriasis began approximately 18 years prior to the current presentation and was initially managed with various topical therapeutics. At an 11-year follow-up, the patient was experiencing poor lesional control as well as severe pruritus and was prescribed etanercept by a dermatologist. His inconsistent compliance with frequency and dosing failed to achieve satisfactory disease suppression and etanercept was discontinued after approximately 2.5 years. Two years later the patient was switched to adalimumab by a dermatologist, and around this time he had developed psoriatic arthritis of the hands and knees and pitting of the nail plates. The patient elected to discontinue adalimumab usage after 3 years due to successful management of the skin lesions, cost considerations, and his perception that the psoriasis could “remain in remission.” After a 6-month lapse, the patient resumed adalimumab due to cutaneous lesional recurrence (Figure 1A).
At the current presentation, an oral examination performed 2 days after the reinstitution of adalim-umab revealed generalized severe gingivitis with an atypical inflammatory response that extended from just beyond the mucogingival junction to the marginal gingiva. The gingiva also appeared edematous with a conspicuously granular surface (Figure 1B). The hard palate displayed multiple red macules of varying sizes (Figure 1C). A maxillary gingival biopsy demonstrated hyperkeratosis, parakeratosis, spongiosis, acanthosis, elongation of the rete ridges, numerous collections of neutrophils (Munro microabscesses), and abundant lymphocytes in the subjacent connective tissue (Figure 2). Periodic acid–Schiff staining was negative for fungal hyphae. These features were consistent with oral mucosal psoriasis.
At a 2-month follow-up, the biopsy site had healed without incident and without loss of the gingival architecture. There was an almost-complete resolution of the gingival erythema (Figure 3A) and the patient has since noticed a lack of bleeding using floss. Additionally, the red macules on the palate were no longer present (Figure 3B). The cutaneous plaques were greatly reduced in size and the patient experienced a proportionate decline in pruritus. Based on the uneventful surgical biopsy procedure, the patient was advised to undergo gingival grafting and has not returned for periodontal care.
Comment
Psoriasis of the oral cavity is rare and typically occurs on the tongue and less frequently on the hard palate, lip, buccal mucosa, and gingiva.2,7 The lesions are almost always concordant with cutaneous psoriasis, and only sporadic examples exclusive to the oral mucosa have been recognized.7,10 Gingival psoriasis usually is described as intensely erythematous and occasionally laced with white scaly streaks involving the marginal gingiva that extend toward the mucogingival junction. In general, the erythematous presentation of gingival psoriasis may not be commensurate with the degree of inflammation induced by dental plaque-based periodontal disease. Doben11 documented gingival psoriasis as appearing “deeply stippled and grainy” and commented that the tissue was “friable” and incapable of maintaining a “clean incision line” during periodontal surgery. In our patient, the gingiva also had exhibited a granular surface. Patients with oral psoriasis often report soreness or a burning sensation of the gingiva, which may easily bleed on manipulation or brushing the teeth, whereas other patients are asymptomatic,12 as in our case. Psoriasis of the hard palate usually presents as multiple painless red macules. Unlike cutaneous psoriasis, oral lesions rarely evoke pruritus.10 Histopathologically, oral psoriasis bears a striking resemblance to its cutaneous counterpart. The epithelium has a pronounced parakeratinized surface with elongated rete ridges and aggregations of Munro microabscesses. The connective tissue often is composed of dilated capillaries that closely approximate the epithelium as well as infiltrations of lymphocytes. Specimens suspected for oral psoriasis should routinely be stained with periodic acid–Schiff to rule out candidiasis coinfection. The microscopic findings of our patient were congruent with prior reports of oral psoriasis.7,10-12 Some clinicians have questioned if psoriasis can actually occur in the oral cavity, but most authorities in the field have recognized its true existence, as evidenced by various shared HLA antigens, specifically HLA-Cw.13
Another group of oral lesions collectively referred to as psoriasiform mucositis, notably geographic tongue (benign migratory glossitis, erythema migrans) and its extraglossal variant geographic stomatitis,14,15 have histopathologic features and HLAs similar to those seen in cutaneous psoriasis.13 Interestingly, geographic tongue has been found in 3.8% to 9.1% of cohorts with cutaneous psoriasis,8,9 but in the extant population, the vast majority of patients with oral psoriasiform mucositis do not have cutaneous psoriasis. Other differential diagnoses for gingival psoriasis are lichen planus, human immunodeficiency virus–associated periodontitis, desquamative gingivitis, plasma cell gingivitis, erythematous candidiasis, mucous membrane pemphigoid, pemphigus vulgaris, leukemia, systemic lupus erythematosus, granulomatosis with polyangiitis, orofacial granulomatosis, localized juvenile spongiotic gingivitis hyperplasia, and primary gingivostomatitis.
Management of gingival psoriasis focuses on strategies to reduce inflammation and discomfort and measures to achieve meticulous oral plaque control. Judicious efforts should be exercised to avoid oral soft-tissue injury when performing periodontal scaling, although it has not been established whether gingival psoriasis is associated with the Köbner phenomenon, as seen with cutaneous lesions. Adjunctive measures employed for symptomatic patients have involved the use of corticosteroids (eg, lesional injection, oral rinse, systemic) and oral rinses with retinoic acid, chlorhexidine gluconate, and warm saline.7,10,16 Prolonged utilization of corticosteroids, however, may necessitate supplemental administration of antifungal agents.
This case report represents a rare documentation of a successful outcome of gingival and palatal psoriasis subsequent to the reinstitution of adalimumab solely for treatment of recurrent cutaneous disease. There likely is a pharmacologic basis for the amelioration of oral psoriasis in our patient. Adalimumab is a bivalent IgG monoclonal antibody that binds to activated dermal dendritic cell receptors of tumor necrosis factor α, thereby attenuating a cytokine-derived inflammatory response and apoptosis.17 In fact, patients with rheumatoid arthritis showed notable reductions in both gingival inflammation and bleeding following a 3-month regimen of adalimumab.18
Conclusion
Practitioners should be aware of the phenotypic overlap of cutaneous and oral psoriasis, particularly involving the gingiva and palate. It is recommended that psoriasis patients routinely receive a dental prophylaxis and engage in oral hygiene efforts to reduce the presence of oral microbiota. Furthermore, it is emphasized that psoriatic patients who maintain an atypical erythematous presentation on the oral mucosa undergo a biopsy for identification of the lesions and correlation with disease dissemination. Prospective studies are needed to characterize the clinical courses of oral psoriasis, ascertain their correlative behavior with cutaneous flares, and determine if lesional improvement can be achieved with the use of biologic agents or other therapeutic modalities.
- Gupta R, Debbaneh MG, Liao W. Genetic epidemiology of psoriasis. Curr Dermatol Rep. 2014;3:61-78.
- Younai FS, Phelan JA. Oral mucositis with features of psoriasis: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:61-67.
- Xu T, Zhang YH. Association of psoriasis with stroke and myocardial infarction: meta-analysis of cohort studies. Br J Dermatol. 2012;167:1345-1350.
- Lazaridou E, Tsikrikoni A, Fotiadou C, et al. Association of chronic plaque psoriasis and severe periodontitis: a hospital based case-control study. J Eur Acad Dermatol Venereol. 2013;27:967-972.
- Skudutyte-Rysstad R, Slevolden EM, Hansen BF, et al. Association between moderate to severe psoriasis and periodontitis in a Scandinavian population. BMC Oral Health. 2014;14:139.
- Zunt SL, Tomich CE. Erythema migrans—a psoriasiform lesion of the oral mucosa. J Dermatol Surg Oncol. 1989;15:1067-1070.
- Reis V, Artico G, Seo J, et al. Psoriasiform mucositis on the gingival and palatal mucosae treated with retinoic-acid mouthwash. Int J Dermatol. 2013;52:113-115.
- Germi L, De Giorgi V, Bergamo F, et al. Psoriasis and oral lesions: multicentric study of oral mucosa diseases Italian group (GIPMO). Dermatol Online J. 2012;18:11.
- Kaur I, Handa S, Kumar B. Oral lesions in psoriasis. Int J Dermatol. 1997;36:78-79.
- Brayshaw HA, Orban B. Psoriasis gingivae. J Periodontol. 1953;24:156-160.
- Doben DI. Psoriasis of the attached gingiva. J Periodontol. 1976;47:38-40.
- Mattsson U, Warfvinge G, Jontell M. Oral psoriasis—a diagnostic dilemma: a report of two cases and a review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;120:e183-e189.
- Dermatologic diseases. In: Neville BW, Damm DD, Allen CM, et al, eds. Oral and Maxillofacial Pathology. 3rd ed. St. Louis, MO: Saunders/Elsevier; 2009:792-794.
- Brooks JK, Balciunas BA. Geographic stomatitis: review of the literature and report of five cases. J Am Dent Assoc. 1987;115:421-424.
- Brooks JK, Nikitakis NG. Multiple mucosal lesions. erythema migrans. Gen Dent. 2007;55:160, 163.
- Ulmansky M, Michelle R, Azaz B. Oral psoriasis: report of six new cases. J Oral Pathol Med. 1995;24:42-45.
- Lis K, Kuzawinska O, Bałkowiec-Iskra E. Tumor necrosis factor inhibitors—state of knowledge. Arch Med Sci. 2014;10:1175-1185.
- Kobayashi T, Yokoyama T, Ito S, et al. Periodontal and serum protein profiles in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitor adalimumab. J Periodontol. 2014;85:1480-1488.
- Gupta R, Debbaneh MG, Liao W. Genetic epidemiology of psoriasis. Curr Dermatol Rep. 2014;3:61-78.
- Younai FS, Phelan JA. Oral mucositis with features of psoriasis: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:61-67.
- Xu T, Zhang YH. Association of psoriasis with stroke and myocardial infarction: meta-analysis of cohort studies. Br J Dermatol. 2012;167:1345-1350.
- Lazaridou E, Tsikrikoni A, Fotiadou C, et al. Association of chronic plaque psoriasis and severe periodontitis: a hospital based case-control study. J Eur Acad Dermatol Venereol. 2013;27:967-972.
- Skudutyte-Rysstad R, Slevolden EM, Hansen BF, et al. Association between moderate to severe psoriasis and periodontitis in a Scandinavian population. BMC Oral Health. 2014;14:139.
- Zunt SL, Tomich CE. Erythema migrans—a psoriasiform lesion of the oral mucosa. J Dermatol Surg Oncol. 1989;15:1067-1070.
- Reis V, Artico G, Seo J, et al. Psoriasiform mucositis on the gingival and palatal mucosae treated with retinoic-acid mouthwash. Int J Dermatol. 2013;52:113-115.
- Germi L, De Giorgi V, Bergamo F, et al. Psoriasis and oral lesions: multicentric study of oral mucosa diseases Italian group (GIPMO). Dermatol Online J. 2012;18:11.
- Kaur I, Handa S, Kumar B. Oral lesions in psoriasis. Int J Dermatol. 1997;36:78-79.
- Brayshaw HA, Orban B. Psoriasis gingivae. J Periodontol. 1953;24:156-160.
- Doben DI. Psoriasis of the attached gingiva. J Periodontol. 1976;47:38-40.
- Mattsson U, Warfvinge G, Jontell M. Oral psoriasis—a diagnostic dilemma: a report of two cases and a review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;120:e183-e189.
- Dermatologic diseases. In: Neville BW, Damm DD, Allen CM, et al, eds. Oral and Maxillofacial Pathology. 3rd ed. St. Louis, MO: Saunders/Elsevier; 2009:792-794.
- Brooks JK, Balciunas BA. Geographic stomatitis: review of the literature and report of five cases. J Am Dent Assoc. 1987;115:421-424.
- Brooks JK, Nikitakis NG. Multiple mucosal lesions. erythema migrans. Gen Dent. 2007;55:160, 163.
- Ulmansky M, Michelle R, Azaz B. Oral psoriasis: report of six new cases. J Oral Pathol Med. 1995;24:42-45.
- Lis K, Kuzawinska O, Bałkowiec-Iskra E. Tumor necrosis factor inhibitors—state of knowledge. Arch Med Sci. 2014;10:1175-1185.
- Kobayashi T, Yokoyama T, Ito S, et al. Periodontal and serum protein profiles in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitor adalimumab. J Periodontol. 2014;85:1480-1488.
Practice Points
- A subset of patients with cutaneous psoriasis may be associated with oral psoriatic outbreaks.
- Oral psoriasis presents as an atypical inflammatory response, and histopathologic assessment is recommended for lesional identity.
- Use of adalimumab for management of cutaneous psoriasis may demonstrate efficacy for oral psoriasis.
Diagnosis of Severe Acute Lower Gastrointestinal Bleeding with CTA
Case
A 31-year-old white man presented to the ED with abdominal and rectal pain accompanied by multiple episodes of bloody diarrhea. He stated he had mild rectal pain the previous night but was pain-free and in his usual state of health the morning of his presentation. Approximately 2 hours before presenting to the ED, however, he began experiencing mild stomach pain, then bloody diarrhea which he described as bright red and “filling the toilet bowl with blood.” He had no history of inflammatory bowel disease or other gastrointestinal (GI) disorder, no recent travel, no complaints of nausea or vomiting, and no infectious symptoms. He described a remote history of external hemorrhoids, and review of his family history was significant for multiple paternal relatives with aortic aneurysms. He was not taking any medications and was a nonsmoker with a normal body mass index (24.3 kg/m2).
Upon arrival at the ED, the patient’s vital signs were: heart rate, 112 beats/min; and blood pressure, 139/102 mm Hg; respiratory rate and temperature were normal, as was the patient’s oxygen saturation on room air. Physical examination was notable for no subjective or objective findings of orthostatic hypotension; increased bowel sounds and diffuse mild abdominal tenderness; and no external hemorrhoids, fissures, or rectal tenderness. Laboratory evaluation was significant for hemoglobin (Hgb), 15.0 g/dL; blood urea nitrogen (BUN)-to-creatinine (Cr) ratio, 11.6; and anion gap, 17 mEq/L.
Upon initial presentation, there was some concern for an infection. However, as the patient continued to have bowel movements consisting almost entirely of frank blood and did not have any infectious signs, a vascular etiology was more strongly considered. Given the patient’s relatively stable vital signs, BUN-to-Cr ratio of less than 20, and lack of orthostatic hypotension, there was low concern for an upper GI etiology, and endoscopy was not obtained emergently. The patient instead underwent abdominal computed tomography angiography (CTA), which identified active extravasation and contrast pooling within the cecum and appendix (Figure 1).

Shortly after the patient returned from imaging, repeat laboratory studies were performed, demonstrating an Hgb drop from 15.0 g/dL to 12.3 g/dL, and surgical services was emergently consulted. The surgeon recommended that embolization first be attempted, with surgery as the option of last resort given the poor localization of the bleed on CTA and the long-term consequences of colonic resection in a young, otherwise healthy man.
Interventional radiology was consulted, and the patient was brought immediately to the angiography suite, where he was found to have “active extravasation arising from a distal descending branch off the right colic artery” (Figure 2). Coil embolization resulted in complete resolution of the hemorrhage.

Later that evening, the patient’s Hgb continued to drop, reaching nadir at 7.3 g/dL, and he continued to have severe hematochezia. His falling Hgb was thought to be indicative of the degree of hemorrhage he had sustained prior to embolization, and the clearance of such blood as the source of his ongoing hematochezia. Following transfusion of 2 U of packed red blood cells (PRBCs), the patient’s Hgb improved to 12.0 g/dL, and he did not experience any significant bleeding for the remainder of his hospital stay.
The following morning, the patient underwent an extensive colonoscopy (extending 25 cm into the terminal ileum), which was unable to detect any signs of arteriovenous malformations, angiodysplasia, or any other possible source of bleeding. After 24 hours with stable vital signs and Hgb levels, the patient was discharged home with close surgical and gastroenterological follow-up, with possible genetic testing for connective tissue diseases. The diagnosis at discharge was spontaneous mesenteric hemorrhage of unknown etiology.
Discussion
Acute lower GI bleeding has an estimated annual hospitalization rate of 36 patients per 100,000, or about half the rate for upper GI bleeding.1,2 The majority of patients (>80%) will have spontaneous resolution and can be worked up nonemergently.
Etiology and Work-Up
Assessment of the etiology of hematochezia begins with ruling out an upper GI source of the bleed; 10% to 15% of patients presenting with hematochezia without hematemesis are ultimately diagnosed with an upper GI etiology. 4,5
BUN-to-Cr Ratio. In a study of patients presenting with hematochezia but no hematemesis or renal failure, Srygley et al6 found a BUN-to-Cr ratio greater than 93% to be sensitive for an upper GI source, with a likelihood ratio of 7.5. The proposed etiology is some combination of absorption of digested blood products and prerenal azotemia due to hypovolemia.
Tachycardia and Orthostatic Hypotension. There have been discussions in the literature about other findings to rule in/out upper GI bleeding. While some studies have found statistically significant results between upper and lower GI bleeding for tachycardia and orthostatic hypotension (increased percentage of both in upper GI bleeding), there is disagreement about whether these findings are clinically significant.7-9
Nasogastric Lavage. Although nasogastric (NG) lavage is no longer the standard of care in the ED due to poor sensitivity and marked discomfort to the patient, most current gastroenterology guidelines still recommend its use; therefore, NG may be requested by the GI consultant.10-12
Diagnosis
Once an upper GI source has been ruled out, identification of the lesion is the next step. The differential diagnosis includes common sources such as diverticular disease, angiodysplasia, colitis, anorectal sources, and neoplasm.5 Less common, but associated with a high risk of mortality, is aortoenteric fistula (100% mortality without surgical intervention).5
Colonoscopy. Emergent colonoscopy can be used for both diagnosis and (potential) therapeutic intervention and is therefore the first option of choice.1,3,4,9 However, as seen in our case, some patients experience such profound hemorrhage that visualization of the colon may be difficult or impossible; patients may also be too unstable to await bowel preparation or undergo a procedure.
Computed Tomography Angiography. For patients in whom colonoscopy is contraindicated, CTA is the imaging modality of choice, and has a 91% to 92% sensitivity in identifying active bleeding (>0.35 mL/min).13-16
Computed tomography of the abdomen and pelvis with contrast alone, as opposed to CTA, is insufficient for detecting GI bleeding, as it is timed so that imaging is obtained when the contrast is in the portal venous capillary beds, rather than in the arteries or arterioles. By protocol, though, many institutions require abdominal and pelvic CTA to include both arterial phase and venous phase images, allowing for assessment of both active arterial bleeding and alternative lower GI sources of hematochezia (eg, mesenteric ischemia).
When ordering a CT study, an awareness of local practice is important in understanding the information that will be obtained from the study. Protocols for lower GI bleed that include CTA have reported accuracy and efficiency without worsening of renal function, despite the increased contrast load.17
Triphasic CT Enterography. Another CT modality to consider is triphasic CT enterography, which uses IV and oral contrast. In a preliminary trial, this modality achieved a specificity of 100% (sensitivity 42%) in detecting GI bleeding.18
Red Blood Cell Scintigraphy. An additional imaging modality that has been the subject of much debate in the GI literature is tagged RBC scintigraphy with Technetium-99m. Various studies have found bleeding-site confirmation in 24% to 97% of patients, and correct localization in 41% to 100% of patients. Given the extensive variability within the literature on selection criteria, localization, site confirmation, and other variables, as well as evidence from one prospective trial by Zink et al19 that found a significant disagreement between CTA and scintigraphy, RBC scintigraphy is not recommended as an alternative imaging modality for the rapid diagnosis of an acute lower GI bleed.
Conclusion
Severe hematochezia is a potential surgical emergency with a broad differential diagnosis. While emergent colonoscopy is an excellent first option, in patients with severe hematochezia, there may be too much blood in the colon to obtain adequate visual images; additionally, depending on practice setting, emergency colonoscopy may not be immediately available. In either case, CTA—a readily available, noninvasive, rapid, and repeatable diagnostic tool—should be considered as an alternate to colonoscopy, particularly in patients with brisk hematochezia.
If a patient with severe hematochezia presents to the ED, the emergency physician (EP) must recognize that the degree of hemorrhage may not correlate with the patient’s vital signs or initial laboratory values. For this reason, the EP must have a high index of suspicion, and consider CTA to allow for a rapid definitive diagnosis and prompt discussion between surgical, interventional radiology, and/or gastroenterology teams to improve clinical outcomes and decrease morbidity and mortality.20
1. Ghassemi K, Jensen D. Lower GI bleeding: epidemiology and management. Curr Gastroenterol Rep. 2013;15(7):333. doi:10.1007/s11894-013-0333-5.
2. Strate LL, Ayanian JZ, Kotler G, Syngal S. Risk factors for mortality in lower intestinal bleeding. Clin Gastroenterol Hepatol. 2008;6(9):1004-1010. doi:10.1016/j.cgh.2008.03.021.
3. Qayad E, Dagar G, Nanchal R. Lower gastrointestinal hemorrhage. Crit Care Clin. 2016;32(2):241-254. doi:10.1016/j.ccc.2015.12.004.
4. Strate LL. Lower GI bleeding: epidemiology and diagnosis. Gastroenterol Clin North Am. 2005;34(4):643-664.
5. Goralnick E, Meguerdichian D. Gastrointestinal bleeding. In: Marx J, Hockberger R, Walls R. (Eds.). Rosen’s Emergency Medicine, 8th Edition. Philadelphia, PA: Saunders, 2014;248-253.
6. Srygley FD, Gerando CJ, Tran T, Fisher DA. Does this patient have a severe upper gastrointestinal bleed? JAMA. 2012;307(10):1072-1079. doi:10.1001/jama.2012.253.
7. Whelen C, Chen C, Kaboli P, Siddique J, Prochaska M, Meltzer DO. Upper versus lower gastrointestinal bleeding: a direct comparison of clinical presentation, outcomes, and resource utilization. J Hosp Med. 2010;5(3):141-147. doi:10.1002/jhm.606.
8. Sittichanbunch Y, Senasu S, Thongkrau T, Keeratiksikorn C, Sawanyawisuth K. How to differentiate sites of gastrointestinal bleeding in patients with hematochezia by using clinical factors? Gastroenterol Res Pract. 2013;2013:265076. doi:10.1155/2013/265076.
9. Velayos F, Williamson A, Sousa KH, et al. Early predictors of severe lower gastrointestinal bleeding and adverse outcomes: a prospective study. Clin Gastroenterol Hepatol. 2004;2(6):485-490.
10. Palamidessi N, Sinert R, Falzon L, Zehtabchi S. Nasogastric aspiration and lavage in emergency department patients with hematochezia or melena without hematemesis. Acad Emerg Med. 2010;17(2):126-132. doi:10.1111/j.1553-2712.2009.00609.x.
11. Singer AJ, Richman PB, Kowalska A, Thode HC Jr. Comparison of patient and practitioner assessments of pain from commonly performed emergency department procedures. Ann Emerg Med. 1999;33(6):652-658.
12. Strate L, Gralnek I. ACG clinical guideline: management of patients with acute lower gastrointestinal bleeding. Am J Gastroenterol. 2016;111(4):459-474. doi:10.1038/ajg.2016.41.
13. Wu LM, Xu JR, Yin Y, Qu XH. Usefulness of CT angiography in diagnosing acute gastrointestinal bleeding: a meta-analysis. World J Gastroenterol. 2010;16(31):3957-3963.
14. Geffroy Y, Rodallec MH, Boulay-Coletta I, Julles MC, Ridereau-Zins C, Zins M. Multidetector CT angiography in acute gastrointestinal bleeding: why, when, and how. Radiographics. 2011;31(3):E35-E46.
15. Reis F, Cardia P, D’Ippolito G. Computed tomography angiography in patients with active gastrointestinal bleeding. Radiol Bras. 2015;48(6):381-390. doi:10.1590/0100-3984.2014.0014.
16. Chan V, Tse D, Dixon S, et al. Outcome following a negative CT angiogram for gastrointestinal hemorrhage. Cardiovasc Intervent Radiol. 2015;38(2):329-335. doi:10.1007/s00270-014-0928-8.
17. Jacovides T, Nadolski G, Allen S, et al. Arteriography for lower gastrointestinal hemorrhage: role of preceding abdominal computed tomographic angiogram in diagnosis and localization. JAMA Surgery. 2015;150(7):650-656. doi:10.1001/jamasurg.2015.97.
18. Hara AK, Walker FB, Silva AC, Leighton JA. Preliminary estimate of triphasic CT enterography performance in hemodynamically stable patients with suspected gastrointestinal bleeding. AJR Am J Roentgenol. 2009;193(5):1252-1260. doi:10.2214/AJR.08.1494.
19. Zink SI, Ohki SK, Stein B, et al. Noninvasive evaluation of active lower gastrointestinal bleeding: comparison between contrast-enhanced MDCT and 99mTc-labeled RBC scintigraphy. AJR Am J Roentgenol. 2008;91(4):1107-1114. doi:10.2214/AJR.07.3642.
20. Nable J, Graham A. Gastrointestinal bleeding. Emerg Med Clin N Am. 2016;34(2):309-325. doi:10.1016/j.emc.2015.12.001.
Case
A 31-year-old white man presented to the ED with abdominal and rectal pain accompanied by multiple episodes of bloody diarrhea. He stated he had mild rectal pain the previous night but was pain-free and in his usual state of health the morning of his presentation. Approximately 2 hours before presenting to the ED, however, he began experiencing mild stomach pain, then bloody diarrhea which he described as bright red and “filling the toilet bowl with blood.” He had no history of inflammatory bowel disease or other gastrointestinal (GI) disorder, no recent travel, no complaints of nausea or vomiting, and no infectious symptoms. He described a remote history of external hemorrhoids, and review of his family history was significant for multiple paternal relatives with aortic aneurysms. He was not taking any medications and was a nonsmoker with a normal body mass index (24.3 kg/m2).
Upon arrival at the ED, the patient’s vital signs were: heart rate, 112 beats/min; and blood pressure, 139/102 mm Hg; respiratory rate and temperature were normal, as was the patient’s oxygen saturation on room air. Physical examination was notable for no subjective or objective findings of orthostatic hypotension; increased bowel sounds and diffuse mild abdominal tenderness; and no external hemorrhoids, fissures, or rectal tenderness. Laboratory evaluation was significant for hemoglobin (Hgb), 15.0 g/dL; blood urea nitrogen (BUN)-to-creatinine (Cr) ratio, 11.6; and anion gap, 17 mEq/L.
Upon initial presentation, there was some concern for an infection. However, as the patient continued to have bowel movements consisting almost entirely of frank blood and did not have any infectious signs, a vascular etiology was more strongly considered. Given the patient’s relatively stable vital signs, BUN-to-Cr ratio of less than 20, and lack of orthostatic hypotension, there was low concern for an upper GI etiology, and endoscopy was not obtained emergently. The patient instead underwent abdominal computed tomography angiography (CTA), which identified active extravasation and contrast pooling within the cecum and appendix (Figure 1).
Shortly after the patient returned from imaging, repeat laboratory studies were performed, demonstrating an Hgb drop from 15.0 g/dL to 12.3 g/dL, and surgical services was emergently consulted. The surgeon recommended that embolization first be attempted, with surgery as the option of last resort given the poor localization of the bleed on CTA and the long-term consequences of colonic resection in a young, otherwise healthy man.
Interventional radiology was consulted, and the patient was brought immediately to the angiography suite, where he was found to have “active extravasation arising from a distal descending branch off the right colic artery” (Figure 2). Coil embolization resulted in complete resolution of the hemorrhage.
Later that evening, the patient’s Hgb continued to drop, reaching nadir at 7.3 g/dL, and he continued to have severe hematochezia. His falling Hgb was thought to be indicative of the degree of hemorrhage he had sustained prior to embolization, and the clearance of such blood as the source of his ongoing hematochezia. Following transfusion of 2 U of packed red blood cells (PRBCs), the patient’s Hgb improved to 12.0 g/dL, and he did not experience any significant bleeding for the remainder of his hospital stay.
The following morning, the patient underwent an extensive colonoscopy (extending 25 cm into the terminal ileum), which was unable to detect any signs of arteriovenous malformations, angiodysplasia, or any other possible source of bleeding. After 24 hours with stable vital signs and Hgb levels, the patient was discharged home with close surgical and gastroenterological follow-up, with possible genetic testing for connective tissue diseases. The diagnosis at discharge was spontaneous mesenteric hemorrhage of unknown etiology.
Discussion
Acute lower GI bleeding has an estimated annual hospitalization rate of 36 patients per 100,000, or about half the rate for upper GI bleeding.1,2 The majority of patients (>80%) will have spontaneous resolution and can be worked up nonemergently.
Etiology and Work-Up
Assessment of the etiology of hematochezia begins with ruling out an upper GI source of the bleed; 10% to 15% of patients presenting with hematochezia without hematemesis are ultimately diagnosed with an upper GI etiology. 4,5
BUN-to-Cr Ratio. In a study of patients presenting with hematochezia but no hematemesis or renal failure, Srygley et al6 found a BUN-to-Cr ratio greater than 93% to be sensitive for an upper GI source, with a likelihood ratio of 7.5. The proposed etiology is some combination of absorption of digested blood products and prerenal azotemia due to hypovolemia.
Tachycardia and Orthostatic Hypotension. There have been discussions in the literature about other findings to rule in/out upper GI bleeding. While some studies have found statistically significant results between upper and lower GI bleeding for tachycardia and orthostatic hypotension (increased percentage of both in upper GI bleeding), there is disagreement about whether these findings are clinically significant.7-9
Nasogastric Lavage. Although nasogastric (NG) lavage is no longer the standard of care in the ED due to poor sensitivity and marked discomfort to the patient, most current gastroenterology guidelines still recommend its use; therefore, NG may be requested by the GI consultant.10-12
Diagnosis
Once an upper GI source has been ruled out, identification of the lesion is the next step. The differential diagnosis includes common sources such as diverticular disease, angiodysplasia, colitis, anorectal sources, and neoplasm.5 Less common, but associated with a high risk of mortality, is aortoenteric fistula (100% mortality without surgical intervention).5
Colonoscopy. Emergent colonoscopy can be used for both diagnosis and (potential) therapeutic intervention and is therefore the first option of choice.1,3,4,9 However, as seen in our case, some patients experience such profound hemorrhage that visualization of the colon may be difficult or impossible; patients may also be too unstable to await bowel preparation or undergo a procedure.
Computed Tomography Angiography. For patients in whom colonoscopy is contraindicated, CTA is the imaging modality of choice, and has a 91% to 92% sensitivity in identifying active bleeding (>0.35 mL/min).13-16
Computed tomography of the abdomen and pelvis with contrast alone, as opposed to CTA, is insufficient for detecting GI bleeding, as it is timed so that imaging is obtained when the contrast is in the portal venous capillary beds, rather than in the arteries or arterioles. By protocol, though, many institutions require abdominal and pelvic CTA to include both arterial phase and venous phase images, allowing for assessment of both active arterial bleeding and alternative lower GI sources of hematochezia (eg, mesenteric ischemia).
When ordering a CT study, an awareness of local practice is important in understanding the information that will be obtained from the study. Protocols for lower GI bleed that include CTA have reported accuracy and efficiency without worsening of renal function, despite the increased contrast load.17
Triphasic CT Enterography. Another CT modality to consider is triphasic CT enterography, which uses IV and oral contrast. In a preliminary trial, this modality achieved a specificity of 100% (sensitivity 42%) in detecting GI bleeding.18
Red Blood Cell Scintigraphy. An additional imaging modality that has been the subject of much debate in the GI literature is tagged RBC scintigraphy with Technetium-99m. Various studies have found bleeding-site confirmation in 24% to 97% of patients, and correct localization in 41% to 100% of patients. Given the extensive variability within the literature on selection criteria, localization, site confirmation, and other variables, as well as evidence from one prospective trial by Zink et al19 that found a significant disagreement between CTA and scintigraphy, RBC scintigraphy is not recommended as an alternative imaging modality for the rapid diagnosis of an acute lower GI bleed.
Conclusion
Severe hematochezia is a potential surgical emergency with a broad differential diagnosis. While emergent colonoscopy is an excellent first option, in patients with severe hematochezia, there may be too much blood in the colon to obtain adequate visual images; additionally, depending on practice setting, emergency colonoscopy may not be immediately available. In either case, CTA—a readily available, noninvasive, rapid, and repeatable diagnostic tool—should be considered as an alternate to colonoscopy, particularly in patients with brisk hematochezia.
If a patient with severe hematochezia presents to the ED, the emergency physician (EP) must recognize that the degree of hemorrhage may not correlate with the patient’s vital signs or initial laboratory values. For this reason, the EP must have a high index of suspicion, and consider CTA to allow for a rapid definitive diagnosis and prompt discussion between surgical, interventional radiology, and/or gastroenterology teams to improve clinical outcomes and decrease morbidity and mortality.20
Case
A 31-year-old white man presented to the ED with abdominal and rectal pain accompanied by multiple episodes of bloody diarrhea. He stated he had mild rectal pain the previous night but was pain-free and in his usual state of health the morning of his presentation. Approximately 2 hours before presenting to the ED, however, he began experiencing mild stomach pain, then bloody diarrhea which he described as bright red and “filling the toilet bowl with blood.” He had no history of inflammatory bowel disease or other gastrointestinal (GI) disorder, no recent travel, no complaints of nausea or vomiting, and no infectious symptoms. He described a remote history of external hemorrhoids, and review of his family history was significant for multiple paternal relatives with aortic aneurysms. He was not taking any medications and was a nonsmoker with a normal body mass index (24.3 kg/m2).
Upon arrival at the ED, the patient’s vital signs were: heart rate, 112 beats/min; and blood pressure, 139/102 mm Hg; respiratory rate and temperature were normal, as was the patient’s oxygen saturation on room air. Physical examination was notable for no subjective or objective findings of orthostatic hypotension; increased bowel sounds and diffuse mild abdominal tenderness; and no external hemorrhoids, fissures, or rectal tenderness. Laboratory evaluation was significant for hemoglobin (Hgb), 15.0 g/dL; blood urea nitrogen (BUN)-to-creatinine (Cr) ratio, 11.6; and anion gap, 17 mEq/L.
Upon initial presentation, there was some concern for an infection. However, as the patient continued to have bowel movements consisting almost entirely of frank blood and did not have any infectious signs, a vascular etiology was more strongly considered. Given the patient’s relatively stable vital signs, BUN-to-Cr ratio of less than 20, and lack of orthostatic hypotension, there was low concern for an upper GI etiology, and endoscopy was not obtained emergently. The patient instead underwent abdominal computed tomography angiography (CTA), which identified active extravasation and contrast pooling within the cecum and appendix (Figure 1).
Shortly after the patient returned from imaging, repeat laboratory studies were performed, demonstrating an Hgb drop from 15.0 g/dL to 12.3 g/dL, and surgical services was emergently consulted. The surgeon recommended that embolization first be attempted, with surgery as the option of last resort given the poor localization of the bleed on CTA and the long-term consequences of colonic resection in a young, otherwise healthy man.
Interventional radiology was consulted, and the patient was brought immediately to the angiography suite, where he was found to have “active extravasation arising from a distal descending branch off the right colic artery” (Figure 2). Coil embolization resulted in complete resolution of the hemorrhage.
Later that evening, the patient’s Hgb continued to drop, reaching nadir at 7.3 g/dL, and he continued to have severe hematochezia. His falling Hgb was thought to be indicative of the degree of hemorrhage he had sustained prior to embolization, and the clearance of such blood as the source of his ongoing hematochezia. Following transfusion of 2 U of packed red blood cells (PRBCs), the patient’s Hgb improved to 12.0 g/dL, and he did not experience any significant bleeding for the remainder of his hospital stay.
The following morning, the patient underwent an extensive colonoscopy (extending 25 cm into the terminal ileum), which was unable to detect any signs of arteriovenous malformations, angiodysplasia, or any other possible source of bleeding. After 24 hours with stable vital signs and Hgb levels, the patient was discharged home with close surgical and gastroenterological follow-up, with possible genetic testing for connective tissue diseases. The diagnosis at discharge was spontaneous mesenteric hemorrhage of unknown etiology.
Discussion
Acute lower GI bleeding has an estimated annual hospitalization rate of 36 patients per 100,000, or about half the rate for upper GI bleeding.1,2 The majority of patients (>80%) will have spontaneous resolution and can be worked up nonemergently.
Etiology and Work-Up
Assessment of the etiology of hematochezia begins with ruling out an upper GI source of the bleed; 10% to 15% of patients presenting with hematochezia without hematemesis are ultimately diagnosed with an upper GI etiology. 4,5
BUN-to-Cr Ratio. In a study of patients presenting with hematochezia but no hematemesis or renal failure, Srygley et al6 found a BUN-to-Cr ratio greater than 93% to be sensitive for an upper GI source, with a likelihood ratio of 7.5. The proposed etiology is some combination of absorption of digested blood products and prerenal azotemia due to hypovolemia.
Tachycardia and Orthostatic Hypotension. There have been discussions in the literature about other findings to rule in/out upper GI bleeding. While some studies have found statistically significant results between upper and lower GI bleeding for tachycardia and orthostatic hypotension (increased percentage of both in upper GI bleeding), there is disagreement about whether these findings are clinically significant.7-9
Nasogastric Lavage. Although nasogastric (NG) lavage is no longer the standard of care in the ED due to poor sensitivity and marked discomfort to the patient, most current gastroenterology guidelines still recommend its use; therefore, NG may be requested by the GI consultant.10-12
Diagnosis
Once an upper GI source has been ruled out, identification of the lesion is the next step. The differential diagnosis includes common sources such as diverticular disease, angiodysplasia, colitis, anorectal sources, and neoplasm.5 Less common, but associated with a high risk of mortality, is aortoenteric fistula (100% mortality without surgical intervention).5
Colonoscopy. Emergent colonoscopy can be used for both diagnosis and (potential) therapeutic intervention and is therefore the first option of choice.1,3,4,9 However, as seen in our case, some patients experience such profound hemorrhage that visualization of the colon may be difficult or impossible; patients may also be too unstable to await bowel preparation or undergo a procedure.
Computed Tomography Angiography. For patients in whom colonoscopy is contraindicated, CTA is the imaging modality of choice, and has a 91% to 92% sensitivity in identifying active bleeding (>0.35 mL/min).13-16
Computed tomography of the abdomen and pelvis with contrast alone, as opposed to CTA, is insufficient for detecting GI bleeding, as it is timed so that imaging is obtained when the contrast is in the portal venous capillary beds, rather than in the arteries or arterioles. By protocol, though, many institutions require abdominal and pelvic CTA to include both arterial phase and venous phase images, allowing for assessment of both active arterial bleeding and alternative lower GI sources of hematochezia (eg, mesenteric ischemia).
When ordering a CT study, an awareness of local practice is important in understanding the information that will be obtained from the study. Protocols for lower GI bleed that include CTA have reported accuracy and efficiency without worsening of renal function, despite the increased contrast load.17
Triphasic CT Enterography. Another CT modality to consider is triphasic CT enterography, which uses IV and oral contrast. In a preliminary trial, this modality achieved a specificity of 100% (sensitivity 42%) in detecting GI bleeding.18
Red Blood Cell Scintigraphy. An additional imaging modality that has been the subject of much debate in the GI literature is tagged RBC scintigraphy with Technetium-99m. Various studies have found bleeding-site confirmation in 24% to 97% of patients, and correct localization in 41% to 100% of patients. Given the extensive variability within the literature on selection criteria, localization, site confirmation, and other variables, as well as evidence from one prospective trial by Zink et al19 that found a significant disagreement between CTA and scintigraphy, RBC scintigraphy is not recommended as an alternative imaging modality for the rapid diagnosis of an acute lower GI bleed.
Conclusion
Severe hematochezia is a potential surgical emergency with a broad differential diagnosis. While emergent colonoscopy is an excellent first option, in patients with severe hematochezia, there may be too much blood in the colon to obtain adequate visual images; additionally, depending on practice setting, emergency colonoscopy may not be immediately available. In either case, CTA—a readily available, noninvasive, rapid, and repeatable diagnostic tool—should be considered as an alternate to colonoscopy, particularly in patients with brisk hematochezia.
If a patient with severe hematochezia presents to the ED, the emergency physician (EP) must recognize that the degree of hemorrhage may not correlate with the patient’s vital signs or initial laboratory values. For this reason, the EP must have a high index of suspicion, and consider CTA to allow for a rapid definitive diagnosis and prompt discussion between surgical, interventional radiology, and/or gastroenterology teams to improve clinical outcomes and decrease morbidity and mortality.20
1. Ghassemi K, Jensen D. Lower GI bleeding: epidemiology and management. Curr Gastroenterol Rep. 2013;15(7):333. doi:10.1007/s11894-013-0333-5.
2. Strate LL, Ayanian JZ, Kotler G, Syngal S. Risk factors for mortality in lower intestinal bleeding. Clin Gastroenterol Hepatol. 2008;6(9):1004-1010. doi:10.1016/j.cgh.2008.03.021.
3. Qayad E, Dagar G, Nanchal R. Lower gastrointestinal hemorrhage. Crit Care Clin. 2016;32(2):241-254. doi:10.1016/j.ccc.2015.12.004.
4. Strate LL. Lower GI bleeding: epidemiology and diagnosis. Gastroenterol Clin North Am. 2005;34(4):643-664.
5. Goralnick E, Meguerdichian D. Gastrointestinal bleeding. In: Marx J, Hockberger R, Walls R. (Eds.). Rosen’s Emergency Medicine, 8th Edition. Philadelphia, PA: Saunders, 2014;248-253.
6. Srygley FD, Gerando CJ, Tran T, Fisher DA. Does this patient have a severe upper gastrointestinal bleed? JAMA. 2012;307(10):1072-1079. doi:10.1001/jama.2012.253.
7. Whelen C, Chen C, Kaboli P, Siddique J, Prochaska M, Meltzer DO. Upper versus lower gastrointestinal bleeding: a direct comparison of clinical presentation, outcomes, and resource utilization. J Hosp Med. 2010;5(3):141-147. doi:10.1002/jhm.606.
8. Sittichanbunch Y, Senasu S, Thongkrau T, Keeratiksikorn C, Sawanyawisuth K. How to differentiate sites of gastrointestinal bleeding in patients with hematochezia by using clinical factors? Gastroenterol Res Pract. 2013;2013:265076. doi:10.1155/2013/265076.
9. Velayos F, Williamson A, Sousa KH, et al. Early predictors of severe lower gastrointestinal bleeding and adverse outcomes: a prospective study. Clin Gastroenterol Hepatol. 2004;2(6):485-490.
10. Palamidessi N, Sinert R, Falzon L, Zehtabchi S. Nasogastric aspiration and lavage in emergency department patients with hematochezia or melena without hematemesis. Acad Emerg Med. 2010;17(2):126-132. doi:10.1111/j.1553-2712.2009.00609.x.
11. Singer AJ, Richman PB, Kowalska A, Thode HC Jr. Comparison of patient and practitioner assessments of pain from commonly performed emergency department procedures. Ann Emerg Med. 1999;33(6):652-658.
12. Strate L, Gralnek I. ACG clinical guideline: management of patients with acute lower gastrointestinal bleeding. Am J Gastroenterol. 2016;111(4):459-474. doi:10.1038/ajg.2016.41.
13. Wu LM, Xu JR, Yin Y, Qu XH. Usefulness of CT angiography in diagnosing acute gastrointestinal bleeding: a meta-analysis. World J Gastroenterol. 2010;16(31):3957-3963.
14. Geffroy Y, Rodallec MH, Boulay-Coletta I, Julles MC, Ridereau-Zins C, Zins M. Multidetector CT angiography in acute gastrointestinal bleeding: why, when, and how. Radiographics. 2011;31(3):E35-E46.
15. Reis F, Cardia P, D’Ippolito G. Computed tomography angiography in patients with active gastrointestinal bleeding. Radiol Bras. 2015;48(6):381-390. doi:10.1590/0100-3984.2014.0014.
16. Chan V, Tse D, Dixon S, et al. Outcome following a negative CT angiogram for gastrointestinal hemorrhage. Cardiovasc Intervent Radiol. 2015;38(2):329-335. doi:10.1007/s00270-014-0928-8.
17. Jacovides T, Nadolski G, Allen S, et al. Arteriography for lower gastrointestinal hemorrhage: role of preceding abdominal computed tomographic angiogram in diagnosis and localization. JAMA Surgery. 2015;150(7):650-656. doi:10.1001/jamasurg.2015.97.
18. Hara AK, Walker FB, Silva AC, Leighton JA. Preliminary estimate of triphasic CT enterography performance in hemodynamically stable patients with suspected gastrointestinal bleeding. AJR Am J Roentgenol. 2009;193(5):1252-1260. doi:10.2214/AJR.08.1494.
19. Zink SI, Ohki SK, Stein B, et al. Noninvasive evaluation of active lower gastrointestinal bleeding: comparison between contrast-enhanced MDCT and 99mTc-labeled RBC scintigraphy. AJR Am J Roentgenol. 2008;91(4):1107-1114. doi:10.2214/AJR.07.3642.
20. Nable J, Graham A. Gastrointestinal bleeding. Emerg Med Clin N Am. 2016;34(2):309-325. doi:10.1016/j.emc.2015.12.001.
1. Ghassemi K, Jensen D. Lower GI bleeding: epidemiology and management. Curr Gastroenterol Rep. 2013;15(7):333. doi:10.1007/s11894-013-0333-5.
2. Strate LL, Ayanian JZ, Kotler G, Syngal S. Risk factors for mortality in lower intestinal bleeding. Clin Gastroenterol Hepatol. 2008;6(9):1004-1010. doi:10.1016/j.cgh.2008.03.021.
3. Qayad E, Dagar G, Nanchal R. Lower gastrointestinal hemorrhage. Crit Care Clin. 2016;32(2):241-254. doi:10.1016/j.ccc.2015.12.004.
4. Strate LL. Lower GI bleeding: epidemiology and diagnosis. Gastroenterol Clin North Am. 2005;34(4):643-664.
5. Goralnick E, Meguerdichian D. Gastrointestinal bleeding. In: Marx J, Hockberger R, Walls R. (Eds.). Rosen’s Emergency Medicine, 8th Edition. Philadelphia, PA: Saunders, 2014;248-253.
6. Srygley FD, Gerando CJ, Tran T, Fisher DA. Does this patient have a severe upper gastrointestinal bleed? JAMA. 2012;307(10):1072-1079. doi:10.1001/jama.2012.253.
7. Whelen C, Chen C, Kaboli P, Siddique J, Prochaska M, Meltzer DO. Upper versus lower gastrointestinal bleeding: a direct comparison of clinical presentation, outcomes, and resource utilization. J Hosp Med. 2010;5(3):141-147. doi:10.1002/jhm.606.
8. Sittichanbunch Y, Senasu S, Thongkrau T, Keeratiksikorn C, Sawanyawisuth K. How to differentiate sites of gastrointestinal bleeding in patients with hematochezia by using clinical factors? Gastroenterol Res Pract. 2013;2013:265076. doi:10.1155/2013/265076.
9. Velayos F, Williamson A, Sousa KH, et al. Early predictors of severe lower gastrointestinal bleeding and adverse outcomes: a prospective study. Clin Gastroenterol Hepatol. 2004;2(6):485-490.
10. Palamidessi N, Sinert R, Falzon L, Zehtabchi S. Nasogastric aspiration and lavage in emergency department patients with hematochezia or melena without hematemesis. Acad Emerg Med. 2010;17(2):126-132. doi:10.1111/j.1553-2712.2009.00609.x.
11. Singer AJ, Richman PB, Kowalska A, Thode HC Jr. Comparison of patient and practitioner assessments of pain from commonly performed emergency department procedures. Ann Emerg Med. 1999;33(6):652-658.
12. Strate L, Gralnek I. ACG clinical guideline: management of patients with acute lower gastrointestinal bleeding. Am J Gastroenterol. 2016;111(4):459-474. doi:10.1038/ajg.2016.41.
13. Wu LM, Xu JR, Yin Y, Qu XH. Usefulness of CT angiography in diagnosing acute gastrointestinal bleeding: a meta-analysis. World J Gastroenterol. 2010;16(31):3957-3963.
14. Geffroy Y, Rodallec MH, Boulay-Coletta I, Julles MC, Ridereau-Zins C, Zins M. Multidetector CT angiography in acute gastrointestinal bleeding: why, when, and how. Radiographics. 2011;31(3):E35-E46.
15. Reis F, Cardia P, D’Ippolito G. Computed tomography angiography in patients with active gastrointestinal bleeding. Radiol Bras. 2015;48(6):381-390. doi:10.1590/0100-3984.2014.0014.
16. Chan V, Tse D, Dixon S, et al. Outcome following a negative CT angiogram for gastrointestinal hemorrhage. Cardiovasc Intervent Radiol. 2015;38(2):329-335. doi:10.1007/s00270-014-0928-8.
17. Jacovides T, Nadolski G, Allen S, et al. Arteriography for lower gastrointestinal hemorrhage: role of preceding abdominal computed tomographic angiogram in diagnosis and localization. JAMA Surgery. 2015;150(7):650-656. doi:10.1001/jamasurg.2015.97.
18. Hara AK, Walker FB, Silva AC, Leighton JA. Preliminary estimate of triphasic CT enterography performance in hemodynamically stable patients with suspected gastrointestinal bleeding. AJR Am J Roentgenol. 2009;193(5):1252-1260. doi:10.2214/AJR.08.1494.
19. Zink SI, Ohki SK, Stein B, et al. Noninvasive evaluation of active lower gastrointestinal bleeding: comparison between contrast-enhanced MDCT and 99mTc-labeled RBC scintigraphy. AJR Am J Roentgenol. 2008;91(4):1107-1114. doi:10.2214/AJR.07.3642.
20. Nable J, Graham A. Gastrointestinal bleeding. Emerg Med Clin N Am. 2016;34(2):309-325. doi:10.1016/j.emc.2015.12.001.
Idiopathic Intracranial Hypertension in a 24-Year-Old Woman
Case
A 24-year-old woman presented to the ED for evaluation of a 3-week history of worsening headache and a 5-day history of increasingly blurry vision. The patient stated that she had initially contacted her primary care physician, but instead presented to the ED because he had no open appointments until the following week and recommended that she go to the ED.
The patient described her headache as a pulsating and throbbing pain over her entire head, which only mildly improved after taking over-the-counter (OTC) ibuprofen. She further noted that her headache was somewhat worse when lying down, and reported the sensation of hearing her own pulsating heartbeat in her ears.
The patient had no personal or family history of migraines, tension headaches, aneurysms, clotting disorders, bleeding disorders, or renal disease, and stated that she had never experienced this type of headache before. She denied photophobia, phonophobia, neck stiffness, fever, vomiting, cough, numbness or weakness in her extremities, or pain anywhere else in her body.
Over the past 5 days, the patient noticed her vision had become increasingly blurry. She was not on any prescription medications, stating the only medication she used was occasional OTC ibuprofen. She had no known allergy to medications and denied smoking or recreational drug use; she admitted to occasional alcohol consumption.
The patient resided with her husband, who had no similar symptoms. Physical examination showed an obese woman (height, 5 ft 6 in; weight, 195 lb; body mass index, 32 kg/m2), lying supine in apparent discomfort. Vital signs at presentation were all normal, and oxygen saturation was normal on room air.
A bedside ocular examination showed 20/100 in both eyes while using glasses; no visual field cuts or obvious central scotoma was present. The patient was alert and oriented to time and place. The neurological examination showed intact cranial nerves, 5/5 strength in all extremities, intact sensation in all extremities, no pronator drift, negative Romberg test, and a normal gait. Fundoscopic examination revealed mildly blurred medial optic discs bilaterally. The rest of the physical examination was normal.
Discussion
Pseudotumor cerebri, more commonly referred to as idiopathic intracranial hypertension (IIH), is characterized by increased intracranial pressure (ICP) with no explanatory findings on imaging studies or in cerebrospinal fluid (CSF) analysis, and may be accompanied by symptoms of chronic headache, tinnitus, papilledema and progressive vision loss caused by optic nerve damage.1 Though historically IIH was referred to by several other names, including “benign intracranial hypertension,” the condition is not benign—when untreated, IIH can cause chronic disabling headaches and permanent vision loss.1
Clinical Course
The clinical course of IIH is unpredictable: In some patients, vision loss occurs gradually over the course of several weeks, while in others, loss occurs over a several month period. There are also patients with IIH who do not experience any alteration or loss of vision. Furthermore, some patients will experience permanent resolution of symptoms after a single lumbar puncture (LP); others have symptom recurrence after less than 24 hours; and some patients spontaneously remit on their own with no treatment whatsoever.1-4
Etiology
In the United States, IIH is a rare cause of headache, occurring in just 1 person per 100,000 annually.1 Though 90% of IIH cases occur in obese women of childbearing age, the etiology of IIH is unknown. Lumbar puncture usually alleviates the patient’s headache, but the CSF pressure typically returns to its pre-tap levels after a few hours.4,5 Neither CSF overproduction nor insufficient CSF resorption is responsible for causing IIH. One theory on the etiology of IIH proposes its cause to be due to a congenital malformation of the venous sinuses. This theory would explain why the symptoms so closely mimic those of venous sinus thrombosis, and why some IIH patients experience relief of symptoms after placement of a venous sinus stent.2
Symptoms
As noted previously, the most common symptom of IIH is headache, which patients usually describe as pressure-like and throbbing, and often involving retro-ocular pain. One feature in over half of patients is pulse-synchronous tinnitus (ie, hearing their own heartbeat in their ears). Eye pain, photophobia, blurry vision, and nausea/vomiting are all common symptoms in IIH, but these symptoms are also present in other causes of headache. The IIH headache might be relapsing and remitting, and can last from a few hours to weeks.2-4,6
Diagnosis
Imaging Studies. Noncontrast computed tomography (CT) imaging studies do not typically demonstrate any abnormal findings.1 Magnetic resonance imaging (MRI) studies show some inconsistent and subtle findings, such as flattening of the backs of the eyeballs, empty sella, or tortuous optic nerves.1
Lumbar Puncture. On LP, a very high opening pressure is a hallmark of IIH. An opening pressure <20 cm H2O is generally considered normal, 20 cm to 25 cm H2O is “equivocal,” and >25 cm H2O is abnormal.7 Patients presenting with IIH commonly have an opening pressure that exceeds 200 cm H2O.1-3 Extremely high pressures, however, are not required for the diagnosis, but some elevations in opening pressure will always be present.2,5 With the exception of a high opening pressure, the patient’s CSF analysis is normal.
Differential Diagnosis
Idiopathic intracranial hypertension is essentially a diagnosis of exclusion, one that is made after exclusion of all other potential causes of increased ICP (Table). Since contrast CT and MRI can identify subtle anatomical deformities and small lesions, their absence on these studies can help establish a diagnosis of IIH.

Venous Sinus Thrombosis. Venous sinus thrombosis is a rare but devastating condition that also cannot be diagnosed from a noncontrast CT but must always be considered in the differential diagnosis of IIH.8-10 Venous sinus thrombosis is characterized by a clot in one of the large venous sinuses that drain blood from the brain; the clot causes pressure to back up into the smaller cerebral vasculature, eventually inducing either a hemorrhagic stroke from a stressed vessel rupturing, or an ischemic stroke from lack of blood flow to the affected area of the brain. This condition is even more rare than IIH (0.5 cases per 100,000 population), but it can be devastating if missed, carrying a mortality rate as high as 15% in some studies.11
Risk Factors
Risk factors known to cause cerebral venous clots include genetic thrombophilias, pregnancy or recent pregnancy, oral contraceptive use, inflammatory bowel disease, severe dehydration, local infection/trauma, and substance abuse. Regardless of risk factors, the most recent guidelines of the American Heart Association/American Stroke Association recommend imaging studies of the cerebral venous sinuses for any patient presenting with new-onset symptoms suggestive of IIH (Class 1, Level of Evidence C).11 The two imaging options for evaluation of the cerebral venous sinuses are CT venography or MR venography. Since the 2013 American College of Radiology Appropriateness Criteria do not indicate a preference of one modality over the other, the choice of can be left to your radiologist.12
Patient Disposition
Patients with IIH typically do not require inpatient admission. Only about 3% of IIH patients will have a fulminant course of rapid-onset of vision loss, but even the most severe and acute cases will deteriorate over weeks, not hours or days.13 Nevertheless, close neurology follow-up is essential. If rapid and thorough outpatient neurological care is unavailable, admission is required.
Management
Not every patient with IIH experiences amelioration or resolution of symptoms following an LP; moreover, there is no clear way to differentiate patients who will experience therapeutic effects from LP from those who will not. Serial LPs as treatment for IIH have been discussed in the literature, but a ventriculoperitoneal shunt is a more practical approach in patients who do not respond to an initial LP.2,14
CSF Volume. The volume of CSF that can be removed safely may be 15 to 25 mL or more. A 1974 paper by Johnston and Paterson15 described five pseudotumor patients whose CSF was drained until their pressure had normalized; the amount removed varied from 15 to 25 mL, without adverse effects. A 1975 case series by Weisberg6 described safe removal of up to 30 mL of CSF in pseudotumor patients—the precise amount removed was determined by that which was necessary to lower the CSF pressure into the normal range. In 2007, a case report by Aly and Lawther16 of a pregnant woman with IIH describes twice weekly LP drainage of 30 mL.
There is nothing in the current literature to suggest that removing 10 to 30 mL of CSF instead of the 4 to 8 mL typically drawn in a diagnostic LP is going to pose any risk to the patient. The main complication associated with therapeutic LP is post-LP headache.5,17,18 There are currently no studies documenting outcomes after specific amounts of CSF removal.
Lifestyle Modifications: Weight Loss. No prospective, randomized controlled trials have proven weight loss to be effective in ameliorating the symptoms of IIH; however, several studies have found that rapid weight loss—whether through aggressive dieting or gastric bypass surgery—can improve symptoms dramatically within several months.19,20 One small study by Johnson et al has suggested that a 6% weight reduction is associated with marked improvement in papilledema.21Pharmacotherapy. The accepted first-line medication to alleviate symptoms of IIH is acetazolamide, and its use is supported by a recent randomized controlled trial conducted by the Neuro-Ophthalmology Research Disease Investigator Consortium (NORDIC).22 Most neurologists will administer a starting dose of acetazolamide 500 mg twice a day, and then increase the dose until symptoms are controlled or adverse effects appear (eg, fatigue, nausea/vomiting/diarrhea, electrolyte abnormalities, kidney stones) that contraindicate further dosage increases. In the NORDIC trial, patients were given up to 4 g of acetazolamide daily.22
Other medications, including loop diuretics and corticosteroids, should not be used except under the direct supervision of a neurologist.2,14
Refractory Cases
A patient who fails conservative treatment should be referred to a neurosurgeon for placement of a CSF shunt, optic nerve sheath fenestration, or placement of a venous sinus stent.23
Case Conclusion
After a noncontrast CT of the head was interpreted as completely normal, an LP was performed with the patient in the left lateral recumbent position. The opening CSF pressure exceeded 55 cm H2O (the upper limit of the manometer). The CSF was clear, and opening pressure was rechecked after each 5 mL draw. After 15 mL had been removed, the patient reported a sudden, dramatic disappearance of her headache and clearing of her vision. After 19 mL of CSF had been removed, the CSF pressure had dropped into the normal range (<20 cm H2O), and the procedure was ended.
To definitively rule out venous sinus thrombosis, a CT venogram was performed in the ED, and interpreted as normal. All other CSF results (cell count, protein, glucose, and gram stain) were normal. After complete resolution of the patient’s symptoms, she was discharged home with a prescription for acetazolamide 500 mg twice daily and instructions to follow-up with a neurologist within 48 hours. At discharge, the patient also received weight-loss counseling and was instructed to return immediately to the ED if her headache recurred or if she experienced any new neurological symptoms.
Summary
Idiopathic intracranial hypertension, also referred to as pseudotumor cerebri, is a rare but potentially vision-threatening cause of headache. Patients with signs and symptoms of IIH often initially present to the ED for evaluation and management. While the etiology of IIH is poorly understood, its clinical picture is unique: elevated ICP (sometimes markedly so) with no other significant findings on noncontrast head CT or CSF analysis. Venous sinus thrombosis, a life-threatening mimic of IIH, must always be included in the differential diagnosis.
Idiopathic intracranial hypertension is initially treated with rapid weight loss and acetazolamide. Many patients experience instant, though sometimes only transient, symptom relief from LP. No definitive studies to support any specific approach, including “therapeutic lumbar punctures.” The condition is rarely fulminant, and hospital admission is not typically required as long as urgent outpatient neurology follow-up is available.
1. Degnan AJ, Levy LM. Pseudotumor cerebri: brief review of clinical syndrome and imaging findings. AJNR Am J Neuroradiol. 2011;32(11):1986-1893. doi:10.3174/ajnr.A2404.
2. Biousse V, Bruce BB, Newman NJ. Update on the pathophysiology and management of idiopathic intracranial hypertension. J Neurol Neurosurg Psychiatry. 2012;83(5):488-494. doi:10.1136/jnnp-2011-302029. 3. Wall M, George D. Idiopathic intracranial hypertension: a prospective study of 50 patients. Brain. 1991;114(Pt 1A):155-180.
4. Wall M. Idiopathic intracranial hypertension. Neurol Clin. 2010;28(3):593-617. doi:10.1016/j.ncl.2010.03.003.
5. Friedman DI, Rausch EA. Headache diagnoses in patients with treated idiopathic intracranial hypertension. Neurology. 2002;58(10):1551-1553.
6. Weisberg LA. Benign intracranial hypertension. Medicine (Baltimore). 1975;54(3):197-207.
7. Whiteley W, Al-Shahi R, Warlow CP, Zeidler M, Lueck CJ. CSF opening pressure: reference interval and the effect of body mass index. Neurology. 2006;67(9):1690-1691.
8. Biousse V, Ameri A, Bousser MG. Isolated intracranial hypertension as the only sign of cerebral venous thrombosis. Neurology. 1999;53(7):1537-1542.
9. Leker RR, Steiner I. Features of dural sinus thrombosis simulating pseudotumor cerebri. Eur J Neurol. 1999;6(5):601-604.
10. Sylaja PN, Ahsan Moosa NV, Radhakrishnan K, Sankara Sarma P, Pradeep Kumar S. Differential diagnosis of patients with intracranial sinus venous thrombosis related isolated intracranial hypertension from those with idiopathic intracranial hypertension. J Neurol Sci. 2003;215(1-2):9-12.
11. Saposnik G, Barinagarrementeria F, Brown RD Jr, et al; American Heart Association Stroke Council and the Council on Epidemiology and Prevention. Diagnosis and management of cerebral venous thrombosis: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(4):1158-1192. doi:10.1161/STR.0b013e31820a8364.
12. American College of Radiology ACR Appropriateness Criteria: Headache. https://acsearch.acr.org/docs/69482/Narrative/. Updated 2013. Accessed January 19, 2017.
13. Thambisetty M, Lavin PJ, Newman NJ, Biousse V. Fulminant idiopathic intracranial hypertension. Neurology. 2007;68(3):229-232.
14. Mollan SP, Markey KA, Benzimra JD, et al. A practical approach to, diagnosis, assessment and management of idiopathic intracranial hypertension. Pract Neurol. 2014;14(6):380-390. doi:10.1136/practneurol-2014-000821.
15. Johnston I, Paterson A. Benign intracranial hypertension. II. CSF pressure and circulation. Brain. 1974;97(2):301-312.
16. Aly EE, Lawther BK. Anaesthetic management of uncontrolled idiopathic intracranial hypertension during labour and delivery using an intrathecal catheter. Anesthesia. 2007;62(2):178-181.
17. Panikkath R, Welker J, Johnston R, Lado-Abeal J. Intracranial hypertension and intracranial hypotension causing headache in the same patient. Proc (Bayl Univ Med Cent). 2014;27(3):217-218.
18. Nafiu OO, Monterosso D, Walton SR, Bradin S. Post dural puncture headache in a pediatric patient with idiopathic intracranial hypertension. Paediatr Anaesth. 2005;15(9):778-781. doi:10.1111/j.1460-9592.2004.01529.x.
19. Sinclair AJ, Burdon MA, Nightingale PG, et al. Low energy diet and intracranial pressure in women with idiopathic intracranial hypertension: prospective cohort study. BMJ. 2010;341:c2701. doi:10.1136/bmj.c2701.
20. Kupersmith MJ, Gamell L, Turbin R, Peck V, Spiegel P, Wall M. Effects of weight loss on the course of idiopathic intracranial hypertension in women. Neurology. 1998;50(4):1094-1098.
21. Johnson LN, Krohel GB, Madsen RW, March GA Jr. The role of weight loss and acetazolamide in the treatment of idiopathic intracranial hypertension (pseudotumor cerebri) Ophthalmology. 1998;105(12):2313-2317. doi:10.1016/S0161-6420(98)91234-9.
22. NORDIC Idiopathic Intracranial Hypertension Study Group Writing Committee; Wall M, McDermott MP, Kieburtz KD, et al. Effect of acetazolamide on visual function in patients with idiopathic intracranial hypertension and mild visual loss: the idiopathic intracranial hypertension treatment trial. JAMA. 2014;311(16):1641-1651. doi:10.1001/jama.2014.3312.
23. Fonseca PL, Rigamonti D, Miller NR, Subramanian PS. Visual outcomes of surgical intervention for pseudotumour cerebri: optic nerve sheath fenestration versus cerebrospinal fluid diversion. Br J Ophthalmol. 2014;98(10):1360-1363. doi:10.1136/bjophthalmol-2014-304953.
Case
A 24-year-old woman presented to the ED for evaluation of a 3-week history of worsening headache and a 5-day history of increasingly blurry vision. The patient stated that she had initially contacted her primary care physician, but instead presented to the ED because he had no open appointments until the following week and recommended that she go to the ED.
The patient described her headache as a pulsating and throbbing pain over her entire head, which only mildly improved after taking over-the-counter (OTC) ibuprofen. She further noted that her headache was somewhat worse when lying down, and reported the sensation of hearing her own pulsating heartbeat in her ears.
The patient had no personal or family history of migraines, tension headaches, aneurysms, clotting disorders, bleeding disorders, or renal disease, and stated that she had never experienced this type of headache before. She denied photophobia, phonophobia, neck stiffness, fever, vomiting, cough, numbness or weakness in her extremities, or pain anywhere else in her body.
Over the past 5 days, the patient noticed her vision had become increasingly blurry. She was not on any prescription medications, stating the only medication she used was occasional OTC ibuprofen. She had no known allergy to medications and denied smoking or recreational drug use; she admitted to occasional alcohol consumption.
The patient resided with her husband, who had no similar symptoms. Physical examination showed an obese woman (height, 5 ft 6 in; weight, 195 lb; body mass index, 32 kg/m2), lying supine in apparent discomfort. Vital signs at presentation were all normal, and oxygen saturation was normal on room air.
A bedside ocular examination showed 20/100 in both eyes while using glasses; no visual field cuts or obvious central scotoma was present. The patient was alert and oriented to time and place. The neurological examination showed intact cranial nerves, 5/5 strength in all extremities, intact sensation in all extremities, no pronator drift, negative Romberg test, and a normal gait. Fundoscopic examination revealed mildly blurred medial optic discs bilaterally. The rest of the physical examination was normal.
Discussion
Pseudotumor cerebri, more commonly referred to as idiopathic intracranial hypertension (IIH), is characterized by increased intracranial pressure (ICP) with no explanatory findings on imaging studies or in cerebrospinal fluid (CSF) analysis, and may be accompanied by symptoms of chronic headache, tinnitus, papilledema and progressive vision loss caused by optic nerve damage.1 Though historically IIH was referred to by several other names, including “benign intracranial hypertension,” the condition is not benign—when untreated, IIH can cause chronic disabling headaches and permanent vision loss.1
Clinical Course
The clinical course of IIH is unpredictable: In some patients, vision loss occurs gradually over the course of several weeks, while in others, loss occurs over a several month period. There are also patients with IIH who do not experience any alteration or loss of vision. Furthermore, some patients will experience permanent resolution of symptoms after a single lumbar puncture (LP); others have symptom recurrence after less than 24 hours; and some patients spontaneously remit on their own with no treatment whatsoever.1-4
Etiology
In the United States, IIH is a rare cause of headache, occurring in just 1 person per 100,000 annually.1 Though 90% of IIH cases occur in obese women of childbearing age, the etiology of IIH is unknown. Lumbar puncture usually alleviates the patient’s headache, but the CSF pressure typically returns to its pre-tap levels after a few hours.4,5 Neither CSF overproduction nor insufficient CSF resorption is responsible for causing IIH. One theory on the etiology of IIH proposes its cause to be due to a congenital malformation of the venous sinuses. This theory would explain why the symptoms so closely mimic those of venous sinus thrombosis, and why some IIH patients experience relief of symptoms after placement of a venous sinus stent.2
Symptoms
As noted previously, the most common symptom of IIH is headache, which patients usually describe as pressure-like and throbbing, and often involving retro-ocular pain. One feature in over half of patients is pulse-synchronous tinnitus (ie, hearing their own heartbeat in their ears). Eye pain, photophobia, blurry vision, and nausea/vomiting are all common symptoms in IIH, but these symptoms are also present in other causes of headache. The IIH headache might be relapsing and remitting, and can last from a few hours to weeks.2-4,6
Diagnosis
Imaging Studies. Noncontrast computed tomography (CT) imaging studies do not typically demonstrate any abnormal findings.1 Magnetic resonance imaging (MRI) studies show some inconsistent and subtle findings, such as flattening of the backs of the eyeballs, empty sella, or tortuous optic nerves.1
Lumbar Puncture. On LP, a very high opening pressure is a hallmark of IIH. An opening pressure <20 cm H2O is generally considered normal, 20 cm to 25 cm H2O is “equivocal,” and >25 cm H2O is abnormal.7 Patients presenting with IIH commonly have an opening pressure that exceeds 200 cm H2O.1-3 Extremely high pressures, however, are not required for the diagnosis, but some elevations in opening pressure will always be present.2,5 With the exception of a high opening pressure, the patient’s CSF analysis is normal.
Differential Diagnosis
Idiopathic intracranial hypertension is essentially a diagnosis of exclusion, one that is made after exclusion of all other potential causes of increased ICP (Table). Since contrast CT and MRI can identify subtle anatomical deformities and small lesions, their absence on these studies can help establish a diagnosis of IIH.
Venous Sinus Thrombosis. Venous sinus thrombosis is a rare but devastating condition that also cannot be diagnosed from a noncontrast CT but must always be considered in the differential diagnosis of IIH.8-10 Venous sinus thrombosis is characterized by a clot in one of the large venous sinuses that drain blood from the brain; the clot causes pressure to back up into the smaller cerebral vasculature, eventually inducing either a hemorrhagic stroke from a stressed vessel rupturing, or an ischemic stroke from lack of blood flow to the affected area of the brain. This condition is even more rare than IIH (0.5 cases per 100,000 population), but it can be devastating if missed, carrying a mortality rate as high as 15% in some studies.11
Risk Factors
Risk factors known to cause cerebral venous clots include genetic thrombophilias, pregnancy or recent pregnancy, oral contraceptive use, inflammatory bowel disease, severe dehydration, local infection/trauma, and substance abuse. Regardless of risk factors, the most recent guidelines of the American Heart Association/American Stroke Association recommend imaging studies of the cerebral venous sinuses for any patient presenting with new-onset symptoms suggestive of IIH (Class 1, Level of Evidence C).11 The two imaging options for evaluation of the cerebral venous sinuses are CT venography or MR venography. Since the 2013 American College of Radiology Appropriateness Criteria do not indicate a preference of one modality over the other, the choice of can be left to your radiologist.12
Patient Disposition
Patients with IIH typically do not require inpatient admission. Only about 3% of IIH patients will have a fulminant course of rapid-onset of vision loss, but even the most severe and acute cases will deteriorate over weeks, not hours or days.13 Nevertheless, close neurology follow-up is essential. If rapid and thorough outpatient neurological care is unavailable, admission is required.
Management
Not every patient with IIH experiences amelioration or resolution of symptoms following an LP; moreover, there is no clear way to differentiate patients who will experience therapeutic effects from LP from those who will not. Serial LPs as treatment for IIH have been discussed in the literature, but a ventriculoperitoneal shunt is a more practical approach in patients who do not respond to an initial LP.2,14
CSF Volume. The volume of CSF that can be removed safely may be 15 to 25 mL or more. A 1974 paper by Johnston and Paterson15 described five pseudotumor patients whose CSF was drained until their pressure had normalized; the amount removed varied from 15 to 25 mL, without adverse effects. A 1975 case series by Weisberg6 described safe removal of up to 30 mL of CSF in pseudotumor patients—the precise amount removed was determined by that which was necessary to lower the CSF pressure into the normal range. In 2007, a case report by Aly and Lawther16 of a pregnant woman with IIH describes twice weekly LP drainage of 30 mL.
There is nothing in the current literature to suggest that removing 10 to 30 mL of CSF instead of the 4 to 8 mL typically drawn in a diagnostic LP is going to pose any risk to the patient. The main complication associated with therapeutic LP is post-LP headache.5,17,18 There are currently no studies documenting outcomes after specific amounts of CSF removal.
Lifestyle Modifications: Weight Loss. No prospective, randomized controlled trials have proven weight loss to be effective in ameliorating the symptoms of IIH; however, several studies have found that rapid weight loss—whether through aggressive dieting or gastric bypass surgery—can improve symptoms dramatically within several months.19,20 One small study by Johnson et al has suggested that a 6% weight reduction is associated with marked improvement in papilledema.21Pharmacotherapy. The accepted first-line medication to alleviate symptoms of IIH is acetazolamide, and its use is supported by a recent randomized controlled trial conducted by the Neuro-Ophthalmology Research Disease Investigator Consortium (NORDIC).22 Most neurologists will administer a starting dose of acetazolamide 500 mg twice a day, and then increase the dose until symptoms are controlled or adverse effects appear (eg, fatigue, nausea/vomiting/diarrhea, electrolyte abnormalities, kidney stones) that contraindicate further dosage increases. In the NORDIC trial, patients were given up to 4 g of acetazolamide daily.22
Other medications, including loop diuretics and corticosteroids, should not be used except under the direct supervision of a neurologist.2,14
Refractory Cases
A patient who fails conservative treatment should be referred to a neurosurgeon for placement of a CSF shunt, optic nerve sheath fenestration, or placement of a venous sinus stent.23
Case Conclusion
After a noncontrast CT of the head was interpreted as completely normal, an LP was performed with the patient in the left lateral recumbent position. The opening CSF pressure exceeded 55 cm H2O (the upper limit of the manometer). The CSF was clear, and opening pressure was rechecked after each 5 mL draw. After 15 mL had been removed, the patient reported a sudden, dramatic disappearance of her headache and clearing of her vision. After 19 mL of CSF had been removed, the CSF pressure had dropped into the normal range (<20 cm H2O), and the procedure was ended.
To definitively rule out venous sinus thrombosis, a CT venogram was performed in the ED, and interpreted as normal. All other CSF results (cell count, protein, glucose, and gram stain) were normal. After complete resolution of the patient’s symptoms, she was discharged home with a prescription for acetazolamide 500 mg twice daily and instructions to follow-up with a neurologist within 48 hours. At discharge, the patient also received weight-loss counseling and was instructed to return immediately to the ED if her headache recurred or if she experienced any new neurological symptoms.
Summary
Idiopathic intracranial hypertension, also referred to as pseudotumor cerebri, is a rare but potentially vision-threatening cause of headache. Patients with signs and symptoms of IIH often initially present to the ED for evaluation and management. While the etiology of IIH is poorly understood, its clinical picture is unique: elevated ICP (sometimes markedly so) with no other significant findings on noncontrast head CT or CSF analysis. Venous sinus thrombosis, a life-threatening mimic of IIH, must always be included in the differential diagnosis.
Idiopathic intracranial hypertension is initially treated with rapid weight loss and acetazolamide. Many patients experience instant, though sometimes only transient, symptom relief from LP. No definitive studies to support any specific approach, including “therapeutic lumbar punctures.” The condition is rarely fulminant, and hospital admission is not typically required as long as urgent outpatient neurology follow-up is available.
Case
A 24-year-old woman presented to the ED for evaluation of a 3-week history of worsening headache and a 5-day history of increasingly blurry vision. The patient stated that she had initially contacted her primary care physician, but instead presented to the ED because he had no open appointments until the following week and recommended that she go to the ED.
The patient described her headache as a pulsating and throbbing pain over her entire head, which only mildly improved after taking over-the-counter (OTC) ibuprofen. She further noted that her headache was somewhat worse when lying down, and reported the sensation of hearing her own pulsating heartbeat in her ears.
The patient had no personal or family history of migraines, tension headaches, aneurysms, clotting disorders, bleeding disorders, or renal disease, and stated that she had never experienced this type of headache before. She denied photophobia, phonophobia, neck stiffness, fever, vomiting, cough, numbness or weakness in her extremities, or pain anywhere else in her body.
Over the past 5 days, the patient noticed her vision had become increasingly blurry. She was not on any prescription medications, stating the only medication she used was occasional OTC ibuprofen. She had no known allergy to medications and denied smoking or recreational drug use; she admitted to occasional alcohol consumption.
The patient resided with her husband, who had no similar symptoms. Physical examination showed an obese woman (height, 5 ft 6 in; weight, 195 lb; body mass index, 32 kg/m2), lying supine in apparent discomfort. Vital signs at presentation were all normal, and oxygen saturation was normal on room air.
A bedside ocular examination showed 20/100 in both eyes while using glasses; no visual field cuts or obvious central scotoma was present. The patient was alert and oriented to time and place. The neurological examination showed intact cranial nerves, 5/5 strength in all extremities, intact sensation in all extremities, no pronator drift, negative Romberg test, and a normal gait. Fundoscopic examination revealed mildly blurred medial optic discs bilaterally. The rest of the physical examination was normal.
Discussion
Pseudotumor cerebri, more commonly referred to as idiopathic intracranial hypertension (IIH), is characterized by increased intracranial pressure (ICP) with no explanatory findings on imaging studies or in cerebrospinal fluid (CSF) analysis, and may be accompanied by symptoms of chronic headache, tinnitus, papilledema and progressive vision loss caused by optic nerve damage.1 Though historically IIH was referred to by several other names, including “benign intracranial hypertension,” the condition is not benign—when untreated, IIH can cause chronic disabling headaches and permanent vision loss.1
Clinical Course
The clinical course of IIH is unpredictable: In some patients, vision loss occurs gradually over the course of several weeks, while in others, loss occurs over a several month period. There are also patients with IIH who do not experience any alteration or loss of vision. Furthermore, some patients will experience permanent resolution of symptoms after a single lumbar puncture (LP); others have symptom recurrence after less than 24 hours; and some patients spontaneously remit on their own with no treatment whatsoever.1-4
Etiology
In the United States, IIH is a rare cause of headache, occurring in just 1 person per 100,000 annually.1 Though 90% of IIH cases occur in obese women of childbearing age, the etiology of IIH is unknown. Lumbar puncture usually alleviates the patient’s headache, but the CSF pressure typically returns to its pre-tap levels after a few hours.4,5 Neither CSF overproduction nor insufficient CSF resorption is responsible for causing IIH. One theory on the etiology of IIH proposes its cause to be due to a congenital malformation of the venous sinuses. This theory would explain why the symptoms so closely mimic those of venous sinus thrombosis, and why some IIH patients experience relief of symptoms after placement of a venous sinus stent.2
Symptoms
As noted previously, the most common symptom of IIH is headache, which patients usually describe as pressure-like and throbbing, and often involving retro-ocular pain. One feature in over half of patients is pulse-synchronous tinnitus (ie, hearing their own heartbeat in their ears). Eye pain, photophobia, blurry vision, and nausea/vomiting are all common symptoms in IIH, but these symptoms are also present in other causes of headache. The IIH headache might be relapsing and remitting, and can last from a few hours to weeks.2-4,6
Diagnosis
Imaging Studies. Noncontrast computed tomography (CT) imaging studies do not typically demonstrate any abnormal findings.1 Magnetic resonance imaging (MRI) studies show some inconsistent and subtle findings, such as flattening of the backs of the eyeballs, empty sella, or tortuous optic nerves.1
Lumbar Puncture. On LP, a very high opening pressure is a hallmark of IIH. An opening pressure <20 cm H2O is generally considered normal, 20 cm to 25 cm H2O is “equivocal,” and >25 cm H2O is abnormal.7 Patients presenting with IIH commonly have an opening pressure that exceeds 200 cm H2O.1-3 Extremely high pressures, however, are not required for the diagnosis, but some elevations in opening pressure will always be present.2,5 With the exception of a high opening pressure, the patient’s CSF analysis is normal.
Differential Diagnosis
Idiopathic intracranial hypertension is essentially a diagnosis of exclusion, one that is made after exclusion of all other potential causes of increased ICP (Table). Since contrast CT and MRI can identify subtle anatomical deformities and small lesions, their absence on these studies can help establish a diagnosis of IIH.
Venous Sinus Thrombosis. Venous sinus thrombosis is a rare but devastating condition that also cannot be diagnosed from a noncontrast CT but must always be considered in the differential diagnosis of IIH.8-10 Venous sinus thrombosis is characterized by a clot in one of the large venous sinuses that drain blood from the brain; the clot causes pressure to back up into the smaller cerebral vasculature, eventually inducing either a hemorrhagic stroke from a stressed vessel rupturing, or an ischemic stroke from lack of blood flow to the affected area of the brain. This condition is even more rare than IIH (0.5 cases per 100,000 population), but it can be devastating if missed, carrying a mortality rate as high as 15% in some studies.11
Risk Factors
Risk factors known to cause cerebral venous clots include genetic thrombophilias, pregnancy or recent pregnancy, oral contraceptive use, inflammatory bowel disease, severe dehydration, local infection/trauma, and substance abuse. Regardless of risk factors, the most recent guidelines of the American Heart Association/American Stroke Association recommend imaging studies of the cerebral venous sinuses for any patient presenting with new-onset symptoms suggestive of IIH (Class 1, Level of Evidence C).11 The two imaging options for evaluation of the cerebral venous sinuses are CT venography or MR venography. Since the 2013 American College of Radiology Appropriateness Criteria do not indicate a preference of one modality over the other, the choice of can be left to your radiologist.12
Patient Disposition
Patients with IIH typically do not require inpatient admission. Only about 3% of IIH patients will have a fulminant course of rapid-onset of vision loss, but even the most severe and acute cases will deteriorate over weeks, not hours or days.13 Nevertheless, close neurology follow-up is essential. If rapid and thorough outpatient neurological care is unavailable, admission is required.
Management
Not every patient with IIH experiences amelioration or resolution of symptoms following an LP; moreover, there is no clear way to differentiate patients who will experience therapeutic effects from LP from those who will not. Serial LPs as treatment for IIH have been discussed in the literature, but a ventriculoperitoneal shunt is a more practical approach in patients who do not respond to an initial LP.2,14
CSF Volume. The volume of CSF that can be removed safely may be 15 to 25 mL or more. A 1974 paper by Johnston and Paterson15 described five pseudotumor patients whose CSF was drained until their pressure had normalized; the amount removed varied from 15 to 25 mL, without adverse effects. A 1975 case series by Weisberg6 described safe removal of up to 30 mL of CSF in pseudotumor patients—the precise amount removed was determined by that which was necessary to lower the CSF pressure into the normal range. In 2007, a case report by Aly and Lawther16 of a pregnant woman with IIH describes twice weekly LP drainage of 30 mL.
There is nothing in the current literature to suggest that removing 10 to 30 mL of CSF instead of the 4 to 8 mL typically drawn in a diagnostic LP is going to pose any risk to the patient. The main complication associated with therapeutic LP is post-LP headache.5,17,18 There are currently no studies documenting outcomes after specific amounts of CSF removal.
Lifestyle Modifications: Weight Loss. No prospective, randomized controlled trials have proven weight loss to be effective in ameliorating the symptoms of IIH; however, several studies have found that rapid weight loss—whether through aggressive dieting or gastric bypass surgery—can improve symptoms dramatically within several months.19,20 One small study by Johnson et al has suggested that a 6% weight reduction is associated with marked improvement in papilledema.21Pharmacotherapy. The accepted first-line medication to alleviate symptoms of IIH is acetazolamide, and its use is supported by a recent randomized controlled trial conducted by the Neuro-Ophthalmology Research Disease Investigator Consortium (NORDIC).22 Most neurologists will administer a starting dose of acetazolamide 500 mg twice a day, and then increase the dose until symptoms are controlled or adverse effects appear (eg, fatigue, nausea/vomiting/diarrhea, electrolyte abnormalities, kidney stones) that contraindicate further dosage increases. In the NORDIC trial, patients were given up to 4 g of acetazolamide daily.22
Other medications, including loop diuretics and corticosteroids, should not be used except under the direct supervision of a neurologist.2,14
Refractory Cases
A patient who fails conservative treatment should be referred to a neurosurgeon for placement of a CSF shunt, optic nerve sheath fenestration, or placement of a venous sinus stent.23
Case Conclusion
After a noncontrast CT of the head was interpreted as completely normal, an LP was performed with the patient in the left lateral recumbent position. The opening CSF pressure exceeded 55 cm H2O (the upper limit of the manometer). The CSF was clear, and opening pressure was rechecked after each 5 mL draw. After 15 mL had been removed, the patient reported a sudden, dramatic disappearance of her headache and clearing of her vision. After 19 mL of CSF had been removed, the CSF pressure had dropped into the normal range (<20 cm H2O), and the procedure was ended.
To definitively rule out venous sinus thrombosis, a CT venogram was performed in the ED, and interpreted as normal. All other CSF results (cell count, protein, glucose, and gram stain) were normal. After complete resolution of the patient’s symptoms, she was discharged home with a prescription for acetazolamide 500 mg twice daily and instructions to follow-up with a neurologist within 48 hours. At discharge, the patient also received weight-loss counseling and was instructed to return immediately to the ED if her headache recurred or if she experienced any new neurological symptoms.
Summary
Idiopathic intracranial hypertension, also referred to as pseudotumor cerebri, is a rare but potentially vision-threatening cause of headache. Patients with signs and symptoms of IIH often initially present to the ED for evaluation and management. While the etiology of IIH is poorly understood, its clinical picture is unique: elevated ICP (sometimes markedly so) with no other significant findings on noncontrast head CT or CSF analysis. Venous sinus thrombosis, a life-threatening mimic of IIH, must always be included in the differential diagnosis.
Idiopathic intracranial hypertension is initially treated with rapid weight loss and acetazolamide. Many patients experience instant, though sometimes only transient, symptom relief from LP. No definitive studies to support any specific approach, including “therapeutic lumbar punctures.” The condition is rarely fulminant, and hospital admission is not typically required as long as urgent outpatient neurology follow-up is available.
1. Degnan AJ, Levy LM. Pseudotumor cerebri: brief review of clinical syndrome and imaging findings. AJNR Am J Neuroradiol. 2011;32(11):1986-1893. doi:10.3174/ajnr.A2404.
2. Biousse V, Bruce BB, Newman NJ. Update on the pathophysiology and management of idiopathic intracranial hypertension. J Neurol Neurosurg Psychiatry. 2012;83(5):488-494. doi:10.1136/jnnp-2011-302029. 3. Wall M, George D. Idiopathic intracranial hypertension: a prospective study of 50 patients. Brain. 1991;114(Pt 1A):155-180.
4. Wall M. Idiopathic intracranial hypertension. Neurol Clin. 2010;28(3):593-617. doi:10.1016/j.ncl.2010.03.003.
5. Friedman DI, Rausch EA. Headache diagnoses in patients with treated idiopathic intracranial hypertension. Neurology. 2002;58(10):1551-1553.
6. Weisberg LA. Benign intracranial hypertension. Medicine (Baltimore). 1975;54(3):197-207.
7. Whiteley W, Al-Shahi R, Warlow CP, Zeidler M, Lueck CJ. CSF opening pressure: reference interval and the effect of body mass index. Neurology. 2006;67(9):1690-1691.
8. Biousse V, Ameri A, Bousser MG. Isolated intracranial hypertension as the only sign of cerebral venous thrombosis. Neurology. 1999;53(7):1537-1542.
9. Leker RR, Steiner I. Features of dural sinus thrombosis simulating pseudotumor cerebri. Eur J Neurol. 1999;6(5):601-604.
10. Sylaja PN, Ahsan Moosa NV, Radhakrishnan K, Sankara Sarma P, Pradeep Kumar S. Differential diagnosis of patients with intracranial sinus venous thrombosis related isolated intracranial hypertension from those with idiopathic intracranial hypertension. J Neurol Sci. 2003;215(1-2):9-12.
11. Saposnik G, Barinagarrementeria F, Brown RD Jr, et al; American Heart Association Stroke Council and the Council on Epidemiology and Prevention. Diagnosis and management of cerebral venous thrombosis: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(4):1158-1192. doi:10.1161/STR.0b013e31820a8364.
12. American College of Radiology ACR Appropriateness Criteria: Headache. https://acsearch.acr.org/docs/69482/Narrative/. Updated 2013. Accessed January 19, 2017.
13. Thambisetty M, Lavin PJ, Newman NJ, Biousse V. Fulminant idiopathic intracranial hypertension. Neurology. 2007;68(3):229-232.
14. Mollan SP, Markey KA, Benzimra JD, et al. A practical approach to, diagnosis, assessment and management of idiopathic intracranial hypertension. Pract Neurol. 2014;14(6):380-390. doi:10.1136/practneurol-2014-000821.
15. Johnston I, Paterson A. Benign intracranial hypertension. II. CSF pressure and circulation. Brain. 1974;97(2):301-312.
16. Aly EE, Lawther BK. Anaesthetic management of uncontrolled idiopathic intracranial hypertension during labour and delivery using an intrathecal catheter. Anesthesia. 2007;62(2):178-181.
17. Panikkath R, Welker J, Johnston R, Lado-Abeal J. Intracranial hypertension and intracranial hypotension causing headache in the same patient. Proc (Bayl Univ Med Cent). 2014;27(3):217-218.
18. Nafiu OO, Monterosso D, Walton SR, Bradin S. Post dural puncture headache in a pediatric patient with idiopathic intracranial hypertension. Paediatr Anaesth. 2005;15(9):778-781. doi:10.1111/j.1460-9592.2004.01529.x.
19. Sinclair AJ, Burdon MA, Nightingale PG, et al. Low energy diet and intracranial pressure in women with idiopathic intracranial hypertension: prospective cohort study. BMJ. 2010;341:c2701. doi:10.1136/bmj.c2701.
20. Kupersmith MJ, Gamell L, Turbin R, Peck V, Spiegel P, Wall M. Effects of weight loss on the course of idiopathic intracranial hypertension in women. Neurology. 1998;50(4):1094-1098.
21. Johnson LN, Krohel GB, Madsen RW, March GA Jr. The role of weight loss and acetazolamide in the treatment of idiopathic intracranial hypertension (pseudotumor cerebri) Ophthalmology. 1998;105(12):2313-2317. doi:10.1016/S0161-6420(98)91234-9.
22. NORDIC Idiopathic Intracranial Hypertension Study Group Writing Committee; Wall M, McDermott MP, Kieburtz KD, et al. Effect of acetazolamide on visual function in patients with idiopathic intracranial hypertension and mild visual loss: the idiopathic intracranial hypertension treatment trial. JAMA. 2014;311(16):1641-1651. doi:10.1001/jama.2014.3312.
23. Fonseca PL, Rigamonti D, Miller NR, Subramanian PS. Visual outcomes of surgical intervention for pseudotumour cerebri: optic nerve sheath fenestration versus cerebrospinal fluid diversion. Br J Ophthalmol. 2014;98(10):1360-1363. doi:10.1136/bjophthalmol-2014-304953.
1. Degnan AJ, Levy LM. Pseudotumor cerebri: brief review of clinical syndrome and imaging findings. AJNR Am J Neuroradiol. 2011;32(11):1986-1893. doi:10.3174/ajnr.A2404.
2. Biousse V, Bruce BB, Newman NJ. Update on the pathophysiology and management of idiopathic intracranial hypertension. J Neurol Neurosurg Psychiatry. 2012;83(5):488-494. doi:10.1136/jnnp-2011-302029. 3. Wall M, George D. Idiopathic intracranial hypertension: a prospective study of 50 patients. Brain. 1991;114(Pt 1A):155-180.
4. Wall M. Idiopathic intracranial hypertension. Neurol Clin. 2010;28(3):593-617. doi:10.1016/j.ncl.2010.03.003.
5. Friedman DI, Rausch EA. Headache diagnoses in patients with treated idiopathic intracranial hypertension. Neurology. 2002;58(10):1551-1553.
6. Weisberg LA. Benign intracranial hypertension. Medicine (Baltimore). 1975;54(3):197-207.
7. Whiteley W, Al-Shahi R, Warlow CP, Zeidler M, Lueck CJ. CSF opening pressure: reference interval and the effect of body mass index. Neurology. 2006;67(9):1690-1691.
8. Biousse V, Ameri A, Bousser MG. Isolated intracranial hypertension as the only sign of cerebral venous thrombosis. Neurology. 1999;53(7):1537-1542.
9. Leker RR, Steiner I. Features of dural sinus thrombosis simulating pseudotumor cerebri. Eur J Neurol. 1999;6(5):601-604.
10. Sylaja PN, Ahsan Moosa NV, Radhakrishnan K, Sankara Sarma P, Pradeep Kumar S. Differential diagnosis of patients with intracranial sinus venous thrombosis related isolated intracranial hypertension from those with idiopathic intracranial hypertension. J Neurol Sci. 2003;215(1-2):9-12.
11. Saposnik G, Barinagarrementeria F, Brown RD Jr, et al; American Heart Association Stroke Council and the Council on Epidemiology and Prevention. Diagnosis and management of cerebral venous thrombosis: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(4):1158-1192. doi:10.1161/STR.0b013e31820a8364.
12. American College of Radiology ACR Appropriateness Criteria: Headache. https://acsearch.acr.org/docs/69482/Narrative/. Updated 2013. Accessed January 19, 2017.
13. Thambisetty M, Lavin PJ, Newman NJ, Biousse V. Fulminant idiopathic intracranial hypertension. Neurology. 2007;68(3):229-232.
14. Mollan SP, Markey KA, Benzimra JD, et al. A practical approach to, diagnosis, assessment and management of idiopathic intracranial hypertension. Pract Neurol. 2014;14(6):380-390. doi:10.1136/practneurol-2014-000821.
15. Johnston I, Paterson A. Benign intracranial hypertension. II. CSF pressure and circulation. Brain. 1974;97(2):301-312.
16. Aly EE, Lawther BK. Anaesthetic management of uncontrolled idiopathic intracranial hypertension during labour and delivery using an intrathecal catheter. Anesthesia. 2007;62(2):178-181.
17. Panikkath R, Welker J, Johnston R, Lado-Abeal J. Intracranial hypertension and intracranial hypotension causing headache in the same patient. Proc (Bayl Univ Med Cent). 2014;27(3):217-218.
18. Nafiu OO, Monterosso D, Walton SR, Bradin S. Post dural puncture headache in a pediatric patient with idiopathic intracranial hypertension. Paediatr Anaesth. 2005;15(9):778-781. doi:10.1111/j.1460-9592.2004.01529.x.
19. Sinclair AJ, Burdon MA, Nightingale PG, et al. Low energy diet and intracranial pressure in women with idiopathic intracranial hypertension: prospective cohort study. BMJ. 2010;341:c2701. doi:10.1136/bmj.c2701.
20. Kupersmith MJ, Gamell L, Turbin R, Peck V, Spiegel P, Wall M. Effects of weight loss on the course of idiopathic intracranial hypertension in women. Neurology. 1998;50(4):1094-1098.
21. Johnson LN, Krohel GB, Madsen RW, March GA Jr. The role of weight loss and acetazolamide in the treatment of idiopathic intracranial hypertension (pseudotumor cerebri) Ophthalmology. 1998;105(12):2313-2317. doi:10.1016/S0161-6420(98)91234-9.
22. NORDIC Idiopathic Intracranial Hypertension Study Group Writing Committee; Wall M, McDermott MP, Kieburtz KD, et al. Effect of acetazolamide on visual function in patients with idiopathic intracranial hypertension and mild visual loss: the idiopathic intracranial hypertension treatment trial. JAMA. 2014;311(16):1641-1651. doi:10.1001/jama.2014.3312.
23. Fonseca PL, Rigamonti D, Miller NR, Subramanian PS. Visual outcomes of surgical intervention for pseudotumour cerebri: optic nerve sheath fenestration versus cerebrospinal fluid diversion. Br J Ophthalmol. 2014;98(10):1360-1363. doi:10.1136/bjophthalmol-2014-304953.
