User login
Advocate for your LGBTQ patients
June 28, 1969, is the day that many consider to be the origin of the modern LGBTQ (Lesbian, Gay, Bisexual, Transgender, Queer/Questioning) movement.1 At that time, it was not uncommon for police officers to conduct raids on bars frequented by LGBTQ patrons, but this night was different. This night the patrons of the Stonewall Inn fought back. The subsequent violent clashes fueled the national organization of groups concentrated on the goal of advocating for LGBTQ rights. On June 28th, 1970, protests to commemorate the events at Stonewall occurred; many refer to these as the first Pride events. Since then the month of June has been seen as the unofficial Pride month for the LGBTQ community. These events began as demonstrations for equal rights and protections for LGBTQ individuals, but over time, events have grown also to become a celebration of queer lives and sexuality.2
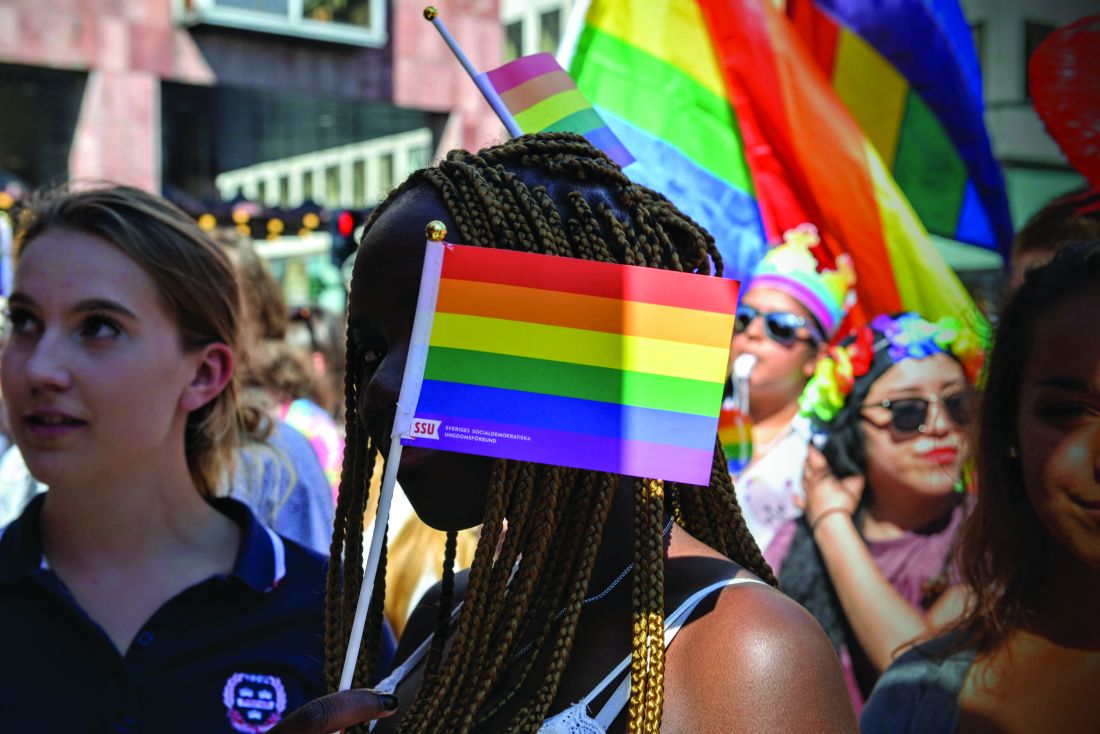
I attended my first Pride event over 10 years ago in support of a friend who had recently come out. He told me that the event was a place where he could proudly be his full self, something that he felt was not safe to do at school or work. When I participated at that event years ago, I began to understand my straight, cisgender privilege: I could walk down the street holding hands with my partner, discuss the details of a first date with colleagues at work, and wear the clothes that aligned with my gender identity without fear of being harassed or attacked. This, I realized, was not the case for everyone. Since attending that Pride event, I have had the opportunity to attend and volunteer at many local Pride events. Some have been in pouring rain, some have been in scorching heat, but all have been rejuvenating, inspiring, and fun! They have been opportunities for me to visibly show support for the local LGBTQ community and meet with other LGBTQ-serving organizations and allies.
Ways to get involved
Find out about local Pride events in your community and consider attending or volunteering. One of the contributing factors to LGBTQ health disparities is limited access to competent care. Many LGBTQ youth and adults have reported experiences of discrimination in the health care setting.3,4 When we, as health care providers, are visible at Pride events, we can have important effects on our local communities by showing them that we recognize and affirm LGBTQ identities.
Consider asking your organization or institution to provide support at local Pride events, post messages of support during Pride month, or host educational sessions about the care of LGBTQ youth.
Dr. Chelvakumar is an attending physician in the division of adolescent medicine at Nationwide Children’s Hospital and an assistant professor of clinical pediatrics at Ohio State University, both in Columbus. She said she had no relevant financial disclosures. Email her at [email protected].
Resources
Human Rights Campaign/Pride: You can learn more about the history of Pride and events in your state and community at www.hrc.org/pride.
How to contact your elected officials: You can find contact information for your local, state, and federal government representatives at www.usa.gov/elected-officials.
National LGBT Health Education Center: You can find educational resources to help optimize care of LGBT patients at www.lgbthealtheducation.org/.
U.S. Transgender Survey: You can read the report from a survey of almost 28,000 transgender respondents in the U.S. Specific information is available about experiences with health care; state level reports also available at www.ustranssurvey.org/reports/.
References
1. GLAAD Pride Month Resource Kit for Jounalists: www.glaad.org/publications/pridekit
2. Human Rights Campaign: History of LGBT Pride. www.hrc.org/blog/the-history-of-lgbt-pride-from-1970-to-now
3. The Report of the 2015 U.S. Transgender Survey (Washington, DC: National Center for Transgender Equality, 2016).
4. Healthy People 2020: Lesbian, Gay, Bisexual and Transgender Health.
June 28, 1969, is the day that many consider to be the origin of the modern LGBTQ (Lesbian, Gay, Bisexual, Transgender, Queer/Questioning) movement.1 At that time, it was not uncommon for police officers to conduct raids on bars frequented by LGBTQ patrons, but this night was different. This night the patrons of the Stonewall Inn fought back. The subsequent violent clashes fueled the national organization of groups concentrated on the goal of advocating for LGBTQ rights. On June 28th, 1970, protests to commemorate the events at Stonewall occurred; many refer to these as the first Pride events. Since then the month of June has been seen as the unofficial Pride month for the LGBTQ community. These events began as demonstrations for equal rights and protections for LGBTQ individuals, but over time, events have grown also to become a celebration of queer lives and sexuality.2
I attended my first Pride event over 10 years ago in support of a friend who had recently come out. He told me that the event was a place where he could proudly be his full self, something that he felt was not safe to do at school or work. When I participated at that event years ago, I began to understand my straight, cisgender privilege: I could walk down the street holding hands with my partner, discuss the details of a first date with colleagues at work, and wear the clothes that aligned with my gender identity without fear of being harassed or attacked. This, I realized, was not the case for everyone. Since attending that Pride event, I have had the opportunity to attend and volunteer at many local Pride events. Some have been in pouring rain, some have been in scorching heat, but all have been rejuvenating, inspiring, and fun! They have been opportunities for me to visibly show support for the local LGBTQ community and meet with other LGBTQ-serving organizations and allies.
Ways to get involved
Find out about local Pride events in your community and consider attending or volunteering. One of the contributing factors to LGBTQ health disparities is limited access to competent care. Many LGBTQ youth and adults have reported experiences of discrimination in the health care setting.3,4 When we, as health care providers, are visible at Pride events, we can have important effects on our local communities by showing them that we recognize and affirm LGBTQ identities.
Consider asking your organization or institution to provide support at local Pride events, post messages of support during Pride month, or host educational sessions about the care of LGBTQ youth.
Dr. Chelvakumar is an attending physician in the division of adolescent medicine at Nationwide Children’s Hospital and an assistant professor of clinical pediatrics at Ohio State University, both in Columbus. She said she had no relevant financial disclosures. Email her at [email protected].
Resources
Human Rights Campaign/Pride: You can learn more about the history of Pride and events in your state and community at www.hrc.org/pride.
How to contact your elected officials: You can find contact information for your local, state, and federal government representatives at www.usa.gov/elected-officials.
National LGBT Health Education Center: You can find educational resources to help optimize care of LGBT patients at www.lgbthealtheducation.org/.
U.S. Transgender Survey: You can read the report from a survey of almost 28,000 transgender respondents in the U.S. Specific information is available about experiences with health care; state level reports also available at www.ustranssurvey.org/reports/.
References
1. GLAAD Pride Month Resource Kit for Jounalists: www.glaad.org/publications/pridekit
2. Human Rights Campaign: History of LGBT Pride. www.hrc.org/blog/the-history-of-lgbt-pride-from-1970-to-now
3. The Report of the 2015 U.S. Transgender Survey (Washington, DC: National Center for Transgender Equality, 2016).
4. Healthy People 2020: Lesbian, Gay, Bisexual and Transgender Health.
June 28, 1969, is the day that many consider to be the origin of the modern LGBTQ (Lesbian, Gay, Bisexual, Transgender, Queer/Questioning) movement.1 At that time, it was not uncommon for police officers to conduct raids on bars frequented by LGBTQ patrons, but this night was different. This night the patrons of the Stonewall Inn fought back. The subsequent violent clashes fueled the national organization of groups concentrated on the goal of advocating for LGBTQ rights. On June 28th, 1970, protests to commemorate the events at Stonewall occurred; many refer to these as the first Pride events. Since then the month of June has been seen as the unofficial Pride month for the LGBTQ community. These events began as demonstrations for equal rights and protections for LGBTQ individuals, but over time, events have grown also to become a celebration of queer lives and sexuality.2
I attended my first Pride event over 10 years ago in support of a friend who had recently come out. He told me that the event was a place where he could proudly be his full self, something that he felt was not safe to do at school or work. When I participated at that event years ago, I began to understand my straight, cisgender privilege: I could walk down the street holding hands with my partner, discuss the details of a first date with colleagues at work, and wear the clothes that aligned with my gender identity without fear of being harassed or attacked. This, I realized, was not the case for everyone. Since attending that Pride event, I have had the opportunity to attend and volunteer at many local Pride events. Some have been in pouring rain, some have been in scorching heat, but all have been rejuvenating, inspiring, and fun! They have been opportunities for me to visibly show support for the local LGBTQ community and meet with other LGBTQ-serving organizations and allies.
Ways to get involved
Find out about local Pride events in your community and consider attending or volunteering. One of the contributing factors to LGBTQ health disparities is limited access to competent care. Many LGBTQ youth and adults have reported experiences of discrimination in the health care setting.3,4 When we, as health care providers, are visible at Pride events, we can have important effects on our local communities by showing them that we recognize and affirm LGBTQ identities.
Consider asking your organization or institution to provide support at local Pride events, post messages of support during Pride month, or host educational sessions about the care of LGBTQ youth.
Dr. Chelvakumar is an attending physician in the division of adolescent medicine at Nationwide Children’s Hospital and an assistant professor of clinical pediatrics at Ohio State University, both in Columbus. She said she had no relevant financial disclosures. Email her at [email protected].
Resources
Human Rights Campaign/Pride: You can learn more about the history of Pride and events in your state and community at www.hrc.org/pride.
How to contact your elected officials: You can find contact information for your local, state, and federal government representatives at www.usa.gov/elected-officials.
National LGBT Health Education Center: You can find educational resources to help optimize care of LGBT patients at www.lgbthealtheducation.org/.
U.S. Transgender Survey: You can read the report from a survey of almost 28,000 transgender respondents in the U.S. Specific information is available about experiences with health care; state level reports also available at www.ustranssurvey.org/reports/.
References
1. GLAAD Pride Month Resource Kit for Jounalists: www.glaad.org/publications/pridekit
2. Human Rights Campaign: History of LGBT Pride. www.hrc.org/blog/the-history-of-lgbt-pride-from-1970-to-now
3. The Report of the 2015 U.S. Transgender Survey (Washington, DC: National Center for Transgender Equality, 2016).
4. Healthy People 2020: Lesbian, Gay, Bisexual and Transgender Health.

Family separations could lead to irreversible health outcomes
The recent crises of the separation of stressed children from their equally stressed parents at this country’s southern border raises the specter of emotional and cognitive reactions within these children – the negative ramifications of which will manifest themselves for years to come.
Stress is ubiquitous. Children experience stress in the normal everyday frustrations that come their way: going to day care for a few hours and leaving mommy, the tripping and falling as they learn to walk, a toy breaking. These stresses help a child develop the capacity for emotional regulation and are termed “tolerable stress.” There is, however, a big difference between tolerable stress and toxic stress.

When the stress reaction in response to perceived or actual danger is beyond tolerable levels by virtue of its quality, intensity, and longevity, it saps the body’s ability to rally or handle the trauma that is being faced because of depleted neurotransmitters that normally assist the body to fight or flee from danger. Unfortunately, that’s not the end of the story: As the stressor persists, it has the capacity to produce long-term alterations in the resilience of the brain and body of the young child. This change often is irreversible.
Scientists and the lay public have begun to actively discuss the impact of adverse childhood experiences and their causal link to irreversible negative adult health outcomes. We now know without a doubt the impact of toxic versus tolerable stress on the hypothalamic-pituitary-adrenal axis of the young child. We are aware that the brain of a young child is particularly vulnerable to stress during critical and sensitive periods of development and that downstream effects of early trauma show up as disorganized behavior and cognitive underperformance.
Development plays a central role in children’s behavioral response to separation from parents. Infants develop a sense of stranger anxiety and the primacy of one central figure between ages 8 and 10 months. The baby chooses the parent over strangers for comfort and care. Roughly between ages 3 and 4, a child develops an internal representation of the parent as the primary figure in their lives so that they can tolerate short periods of being away from the primary caregiver for, let’s say, half a day. They depend on the parent for all their physical and emotional needs, which includes the need for a stable, nurturing, and predictable presence.
Familiarity of the environment, family rituals, consistency of daily routines provided by the parent help neural pathways responsible for the biologic unfolding of developmental milestones. Only recently, the science of early brain development and the role of early childhood trauma on brain biology has caught up with the longitudinal observational studies of bereaved children who lost their parents under circumstances of acute stress (the blitzkrieg, the Yom Kippur War, the Hungarian orphans) followed by the chronic stress phase of no primary caregiver for months to years. These observational studies coupled by the emerging neuroscience of early brain development and trauma are powerful informants of what is tolerable stress for children and what is not.
As a psychiatrist and expert in early child development, I am concerned about these long-term effects on migrant children. ; frantically seeking the parent/comforting familiar caregiver. Gradually as that possibility fades, with their limited ability to verbalize needs, episodic weeping can give way to disorganized behavior, despondence, and finally apathy and regression of milestones and cognitive abilities already achieved. The separation is merely the proxy face of other disasters that are probably co-occurring for the child/family and multiplies the dose of the stress: loss of siblings, loss of familiar physical elements of the landscape, loss of adequate physical sustenance, loss of routine, loss of consistency, increased vulnerability for physical illness, and the list goes on.
By age 2 and above, children are more vocal in their desire for reunification and will weep inconsolably upon separation. At this time their bodies and the psychological lens through which they view the world is changing irrevocably. Stress hormones, noxious at high levels such as cortisol/adrenocorticotropic hormone and glutamate rise to levels that no longer respond to the traditional negative feedback loop of the body and reach levels that lead to cell death in areas of the brain responsible for modulating emotions that can permanently change brain architecture.
Psychologically, the child seeks out the parent by any means (crying, flailing, tantruming). When no positive outcome occurs, this is replaced by an unwillingness to engage with the world, or the child engages but by behavior that is aggressive, disorganized, and regressed. In their own way, young children’s mistrust of the world leads to a variety of behaviors that impede normal development: rejection of others, disengagement from social connections by quietly sitting in a corner, being unwilling to eat or sleeping fitfully.
As separation from the primary caregiver continues, there is a deepening sense of hopelessness. Generally, no amount of physical props (toys, playgrounds) without the human familiar faces can rectify the feeling of abandonment and hopelessness that children in these conditions face. Assuming they are no strangers to trauma experienced through the lives of their asylum-seeking parents, this separation refreshes all the wounds of days gone by. At least in those moments of initial trauma, they had the parent as a buffer. But with this new separation, they are left to emotionally fend for themselves with no tools to manage their emotions.
We have to deal with the aftereffects of so many natural disasters that cannot be avoided: earthquakes, tornadoes, tsunamis that displace children and families with horrific outcomes. Did we really need a man-made disaster that will irrevocably change the brains and behaviors of a generation of children who just happen to be on our southern borders because of an accident of birth? If parents abandoned their children this way it would be called child abuse. Deliberate policy decisions that have such dire effects on children also take on the label of child abuse.
Although images of my years as a young parent of three boys are turning a sepia color in faded memory, in my new role as a grandmother observing my son and his wife so in love with their 2-year-old reinforces how much mutual dependency of a child and parent on each other strengthens the very fabric of life. Children are our future and are the building blocks of a just and humane society.
As of this writing, the administration has vowed to end these separations. But how will children who have been separated be reunited with their families?
The education of decision makers about the far-reaching emotional and physical consequences on normative childhood development must be a top priority for all scientists and professionals interested in kids and families: I think that means all of us.
Dr. Sood is professor of psychiatry and pediatrics, and senior professor of child mental health policy at the Virginia Treatment Center at Virginia Commonwealth University, Richmond. She also is affiliated with the department of psychiatry at VCU and Children’s Hospital of Richmond.
The recent crises of the separation of stressed children from their equally stressed parents at this country’s southern border raises the specter of emotional and cognitive reactions within these children – the negative ramifications of which will manifest themselves for years to come.
Stress is ubiquitous. Children experience stress in the normal everyday frustrations that come their way: going to day care for a few hours and leaving mommy, the tripping and falling as they learn to walk, a toy breaking. These stresses help a child develop the capacity for emotional regulation and are termed “tolerable stress.” There is, however, a big difference between tolerable stress and toxic stress.
When the stress reaction in response to perceived or actual danger is beyond tolerable levels by virtue of its quality, intensity, and longevity, it saps the body’s ability to rally or handle the trauma that is being faced because of depleted neurotransmitters that normally assist the body to fight or flee from danger. Unfortunately, that’s not the end of the story: As the stressor persists, it has the capacity to produce long-term alterations in the resilience of the brain and body of the young child. This change often is irreversible.
Scientists and the lay public have begun to actively discuss the impact of adverse childhood experiences and their causal link to irreversible negative adult health outcomes. We now know without a doubt the impact of toxic versus tolerable stress on the hypothalamic-pituitary-adrenal axis of the young child. We are aware that the brain of a young child is particularly vulnerable to stress during critical and sensitive periods of development and that downstream effects of early trauma show up as disorganized behavior and cognitive underperformance.
Development plays a central role in children’s behavioral response to separation from parents. Infants develop a sense of stranger anxiety and the primacy of one central figure between ages 8 and 10 months. The baby chooses the parent over strangers for comfort and care. Roughly between ages 3 and 4, a child develops an internal representation of the parent as the primary figure in their lives so that they can tolerate short periods of being away from the primary caregiver for, let’s say, half a day. They depend on the parent for all their physical and emotional needs, which includes the need for a stable, nurturing, and predictable presence.
Familiarity of the environment, family rituals, consistency of daily routines provided by the parent help neural pathways responsible for the biologic unfolding of developmental milestones. Only recently, the science of early brain development and the role of early childhood trauma on brain biology has caught up with the longitudinal observational studies of bereaved children who lost their parents under circumstances of acute stress (the blitzkrieg, the Yom Kippur War, the Hungarian orphans) followed by the chronic stress phase of no primary caregiver for months to years. These observational studies coupled by the emerging neuroscience of early brain development and trauma are powerful informants of what is tolerable stress for children and what is not.
As a psychiatrist and expert in early child development, I am concerned about these long-term effects on migrant children. ; frantically seeking the parent/comforting familiar caregiver. Gradually as that possibility fades, with their limited ability to verbalize needs, episodic weeping can give way to disorganized behavior, despondence, and finally apathy and regression of milestones and cognitive abilities already achieved. The separation is merely the proxy face of other disasters that are probably co-occurring for the child/family and multiplies the dose of the stress: loss of siblings, loss of familiar physical elements of the landscape, loss of adequate physical sustenance, loss of routine, loss of consistency, increased vulnerability for physical illness, and the list goes on.
By age 2 and above, children are more vocal in their desire for reunification and will weep inconsolably upon separation. At this time their bodies and the psychological lens through which they view the world is changing irrevocably. Stress hormones, noxious at high levels such as cortisol/adrenocorticotropic hormone and glutamate rise to levels that no longer respond to the traditional negative feedback loop of the body and reach levels that lead to cell death in areas of the brain responsible for modulating emotions that can permanently change brain architecture.
Psychologically, the child seeks out the parent by any means (crying, flailing, tantruming). When no positive outcome occurs, this is replaced by an unwillingness to engage with the world, or the child engages but by behavior that is aggressive, disorganized, and regressed. In their own way, young children’s mistrust of the world leads to a variety of behaviors that impede normal development: rejection of others, disengagement from social connections by quietly sitting in a corner, being unwilling to eat or sleeping fitfully.
As separation from the primary caregiver continues, there is a deepening sense of hopelessness. Generally, no amount of physical props (toys, playgrounds) without the human familiar faces can rectify the feeling of abandonment and hopelessness that children in these conditions face. Assuming they are no strangers to trauma experienced through the lives of their asylum-seeking parents, this separation refreshes all the wounds of days gone by. At least in those moments of initial trauma, they had the parent as a buffer. But with this new separation, they are left to emotionally fend for themselves with no tools to manage their emotions.
We have to deal with the aftereffects of so many natural disasters that cannot be avoided: earthquakes, tornadoes, tsunamis that displace children and families with horrific outcomes. Did we really need a man-made disaster that will irrevocably change the brains and behaviors of a generation of children who just happen to be on our southern borders because of an accident of birth? If parents abandoned their children this way it would be called child abuse. Deliberate policy decisions that have such dire effects on children also take on the label of child abuse.
Although images of my years as a young parent of three boys are turning a sepia color in faded memory, in my new role as a grandmother observing my son and his wife so in love with their 2-year-old reinforces how much mutual dependency of a child and parent on each other strengthens the very fabric of life. Children are our future and are the building blocks of a just and humane society.
As of this writing, the administration has vowed to end these separations. But how will children who have been separated be reunited with their families?
The education of decision makers about the far-reaching emotional and physical consequences on normative childhood development must be a top priority for all scientists and professionals interested in kids and families: I think that means all of us.
Dr. Sood is professor of psychiatry and pediatrics, and senior professor of child mental health policy at the Virginia Treatment Center at Virginia Commonwealth University, Richmond. She also is affiliated with the department of psychiatry at VCU and Children’s Hospital of Richmond.
The recent crises of the separation of stressed children from their equally stressed parents at this country’s southern border raises the specter of emotional and cognitive reactions within these children – the negative ramifications of which will manifest themselves for years to come.
Stress is ubiquitous. Children experience stress in the normal everyday frustrations that come their way: going to day care for a few hours and leaving mommy, the tripping and falling as they learn to walk, a toy breaking. These stresses help a child develop the capacity for emotional regulation and are termed “tolerable stress.” There is, however, a big difference between tolerable stress and toxic stress.
When the stress reaction in response to perceived or actual danger is beyond tolerable levels by virtue of its quality, intensity, and longevity, it saps the body’s ability to rally or handle the trauma that is being faced because of depleted neurotransmitters that normally assist the body to fight or flee from danger. Unfortunately, that’s not the end of the story: As the stressor persists, it has the capacity to produce long-term alterations in the resilience of the brain and body of the young child. This change often is irreversible.
Scientists and the lay public have begun to actively discuss the impact of adverse childhood experiences and their causal link to irreversible negative adult health outcomes. We now know without a doubt the impact of toxic versus tolerable stress on the hypothalamic-pituitary-adrenal axis of the young child. We are aware that the brain of a young child is particularly vulnerable to stress during critical and sensitive periods of development and that downstream effects of early trauma show up as disorganized behavior and cognitive underperformance.
Development plays a central role in children’s behavioral response to separation from parents. Infants develop a sense of stranger anxiety and the primacy of one central figure between ages 8 and 10 months. The baby chooses the parent over strangers for comfort and care. Roughly between ages 3 and 4, a child develops an internal representation of the parent as the primary figure in their lives so that they can tolerate short periods of being away from the primary caregiver for, let’s say, half a day. They depend on the parent for all their physical and emotional needs, which includes the need for a stable, nurturing, and predictable presence.
Familiarity of the environment, family rituals, consistency of daily routines provided by the parent help neural pathways responsible for the biologic unfolding of developmental milestones. Only recently, the science of early brain development and the role of early childhood trauma on brain biology has caught up with the longitudinal observational studies of bereaved children who lost their parents under circumstances of acute stress (the blitzkrieg, the Yom Kippur War, the Hungarian orphans) followed by the chronic stress phase of no primary caregiver for months to years. These observational studies coupled by the emerging neuroscience of early brain development and trauma are powerful informants of what is tolerable stress for children and what is not.
As a psychiatrist and expert in early child development, I am concerned about these long-term effects on migrant children. ; frantically seeking the parent/comforting familiar caregiver. Gradually as that possibility fades, with their limited ability to verbalize needs, episodic weeping can give way to disorganized behavior, despondence, and finally apathy and regression of milestones and cognitive abilities already achieved. The separation is merely the proxy face of other disasters that are probably co-occurring for the child/family and multiplies the dose of the stress: loss of siblings, loss of familiar physical elements of the landscape, loss of adequate physical sustenance, loss of routine, loss of consistency, increased vulnerability for physical illness, and the list goes on.
By age 2 and above, children are more vocal in their desire for reunification and will weep inconsolably upon separation. At this time their bodies and the psychological lens through which they view the world is changing irrevocably. Stress hormones, noxious at high levels such as cortisol/adrenocorticotropic hormone and glutamate rise to levels that no longer respond to the traditional negative feedback loop of the body and reach levels that lead to cell death in areas of the brain responsible for modulating emotions that can permanently change brain architecture.
Psychologically, the child seeks out the parent by any means (crying, flailing, tantruming). When no positive outcome occurs, this is replaced by an unwillingness to engage with the world, or the child engages but by behavior that is aggressive, disorganized, and regressed. In their own way, young children’s mistrust of the world leads to a variety of behaviors that impede normal development: rejection of others, disengagement from social connections by quietly sitting in a corner, being unwilling to eat or sleeping fitfully.
As separation from the primary caregiver continues, there is a deepening sense of hopelessness. Generally, no amount of physical props (toys, playgrounds) without the human familiar faces can rectify the feeling of abandonment and hopelessness that children in these conditions face. Assuming they are no strangers to trauma experienced through the lives of their asylum-seeking parents, this separation refreshes all the wounds of days gone by. At least in those moments of initial trauma, they had the parent as a buffer. But with this new separation, they are left to emotionally fend for themselves with no tools to manage their emotions.
We have to deal with the aftereffects of so many natural disasters that cannot be avoided: earthquakes, tornadoes, tsunamis that displace children and families with horrific outcomes. Did we really need a man-made disaster that will irrevocably change the brains and behaviors of a generation of children who just happen to be on our southern borders because of an accident of birth? If parents abandoned their children this way it would be called child abuse. Deliberate policy decisions that have such dire effects on children also take on the label of child abuse.
Although images of my years as a young parent of three boys are turning a sepia color in faded memory, in my new role as a grandmother observing my son and his wife so in love with their 2-year-old reinforces how much mutual dependency of a child and parent on each other strengthens the very fabric of life. Children are our future and are the building blocks of a just and humane society.
As of this writing, the administration has vowed to end these separations. But how will children who have been separated be reunited with their families?
The education of decision makers about the far-reaching emotional and physical consequences on normative childhood development must be a top priority for all scientists and professionals interested in kids and families: I think that means all of us.
Dr. Sood is professor of psychiatry and pediatrics, and senior professor of child mental health policy at the Virginia Treatment Center at Virginia Commonwealth University, Richmond. She also is affiliated with the department of psychiatry at VCU and Children’s Hospital of Richmond.
Right to Try: Mission accomplished?
On May 30, 2018, President Trump signed into law the Trickett Wendler, Frank Mongiello, Jordan McLinn, and Matthew Bellina Right to Try Act of 2017, guaranteeing patients with terminal illnesses the right to seek access to investigational drugs which have passed at least phase 1 testing. As he signed the bipartisan-backed bill into law, the president said he believes it will ultimately save “hundreds of thousands” of lives, scoring an apparent win-win-win for the president, for Congress, and for patients. A critical appraisal of the law – and of the supposition that patients will reap great benefit from it – requires that we ask two questions: Does the law degrade measures originally put in place to protect patients, and does it actually improve access to potentially life-saving drugs?
As a hematologist with about 2 decades of experience conducting clinical research involving patients with incurable cancer, I want as many patients as possible to have access to promising new therapies. Clinical trials, as the sole means of determining a candidate drug’s safety and efficacy, are conducted in a systematic fashion, with phase 1 trials focusing predominantly on safety. As any experienced researcher is aware, however, a phase 1 trial only provides a basic sense of a drug’s safety. It would be unimaginable (and unethical) to conduct subsequent phase 2 and 3 trials without including rigorous ongoing monitoring for adverse effects.

A later study evaluating all nonpediatric phase 1 trials sponsored by the National Cancer Institute’s Cancer Therapy Evaluation Program between 2001 and 2012, which included more than 8,300 patients, found the therapy-related death rate to be similarly low – approximately 1%.2
It is distinctly possible that the same drugs used in the analyzed trials would have been more toxic to patients who, for one reason or another, would not have met enrollment criteria. It is impossible to precisely quantify the degree to which risk may be increased under the Right to Try law, but even if it were tripled or quadrupled, compared with these historical expectations, the absolute risk of treatment-related death remains low. It should be noted that the incidence of nonfatal grade 3-4 adverse events in the same analyses was 10%-20% so a similar proportional increase in these events would impact a larger number of patients. Overall, it is difficult to know the degree to which Right to Try impacts the safety of fragile patients with terminal illnesses.
The answer to the second question – Is access to life-saving drugs improved? – is much easier to answer: No.
One reason is that these patients already have the right to seek access to investigational drugs through the FDA’s Expanded Access Program. An overview of the Expanded Access Program reported that approximately 11,000 patients over a period of 10 years submitted requests for access to investigational drugs and that 99% of the requests were granted.3
Proponents of the Right to Try law argue that the new law somehow simplifies the request process, but it is difficult to understand exactly how. Further, in a position paper on the topic of Right to Try policy, Lisa Kearns and Alison Bateman-House, PhD, of New York University, wrote, “In the more than 2½ years since the first [individual state right to try] law was signed, there have been no documented cases of anyone receiving access, because of a right to try law, to an experimental product that would not have been available via the FDA’s Expanded Access Program.”4
Finally, and most importantly, neither the existing Expanded Access Program nor the newly enacted Right to Try law require that pharmaceutical companies actually provide the requested drugs. Industry leaders, in testimony before Congress, have cited patient safety, costs, drug supply, and negative impact on existing clinical trial accrual as reasons pharmaceutical companies commonly deny requests for access to investigational products.
The bottom line is that the Right to Try law really lacks teeth. Despite the favorable optics for the president and Congress, it is virtually certain that Right to Try will never save hundreds of thousands of lives, and the suggestion that it will is either disingenuous or uninformed. Hopefully, though, Congress’s passage of Right to Try is indicative of a more general interest in bipartisan cooperation to improve access to affordable, high-quality health care.
Dr. Zonder is a professor in the department of oncology at the Barbara Ann Karmanos Cancer Institute and Wayne State University in Detroit. He is the leader of the KCI Myeloma and Amyloidosis Team. He is also a member of the Hematology News Editorial Advisory Board. He reported having no relevant financial disclosures.
References
1. Roberts TG Jr. et al. JAMA. 2004 Nov 3;292(17):2130-40.
2. Fukada YK et al. J Clin Oncol. 2014 May 20. doi: 10.1200/jco.2014.32.15_suppl.2552.
3. Jarow JP et al. Ther Innov Regul Sci. 2017 Mar 1;51(2):177-9.
4. Kearns L et al. Ther Innov Regul Sci. 2017 Mar 1;51(2):170-6.
On May 30, 2018, President Trump signed into law the Trickett Wendler, Frank Mongiello, Jordan McLinn, and Matthew Bellina Right to Try Act of 2017, guaranteeing patients with terminal illnesses the right to seek access to investigational drugs which have passed at least phase 1 testing. As he signed the bipartisan-backed bill into law, the president said he believes it will ultimately save “hundreds of thousands” of lives, scoring an apparent win-win-win for the president, for Congress, and for patients. A critical appraisal of the law – and of the supposition that patients will reap great benefit from it – requires that we ask two questions: Does the law degrade measures originally put in place to protect patients, and does it actually improve access to potentially life-saving drugs?
As a hematologist with about 2 decades of experience conducting clinical research involving patients with incurable cancer, I want as many patients as possible to have access to promising new therapies. Clinical trials, as the sole means of determining a candidate drug’s safety and efficacy, are conducted in a systematic fashion, with phase 1 trials focusing predominantly on safety. As any experienced researcher is aware, however, a phase 1 trial only provides a basic sense of a drug’s safety. It would be unimaginable (and unethical) to conduct subsequent phase 2 and 3 trials without including rigorous ongoing monitoring for adverse effects.
A later study evaluating all nonpediatric phase 1 trials sponsored by the National Cancer Institute’s Cancer Therapy Evaluation Program between 2001 and 2012, which included more than 8,300 patients, found the therapy-related death rate to be similarly low – approximately 1%.2
It is distinctly possible that the same drugs used in the analyzed trials would have been more toxic to patients who, for one reason or another, would not have met enrollment criteria. It is impossible to precisely quantify the degree to which risk may be increased under the Right to Try law, but even if it were tripled or quadrupled, compared with these historical expectations, the absolute risk of treatment-related death remains low. It should be noted that the incidence of nonfatal grade 3-4 adverse events in the same analyses was 10%-20% so a similar proportional increase in these events would impact a larger number of patients. Overall, it is difficult to know the degree to which Right to Try impacts the safety of fragile patients with terminal illnesses.
The answer to the second question – Is access to life-saving drugs improved? – is much easier to answer: No.
One reason is that these patients already have the right to seek access to investigational drugs through the FDA’s Expanded Access Program. An overview of the Expanded Access Program reported that approximately 11,000 patients over a period of 10 years submitted requests for access to investigational drugs and that 99% of the requests were granted.3
Proponents of the Right to Try law argue that the new law somehow simplifies the request process, but it is difficult to understand exactly how. Further, in a position paper on the topic of Right to Try policy, Lisa Kearns and Alison Bateman-House, PhD, of New York University, wrote, “In the more than 2½ years since the first [individual state right to try] law was signed, there have been no documented cases of anyone receiving access, because of a right to try law, to an experimental product that would not have been available via the FDA’s Expanded Access Program.”4
Finally, and most importantly, neither the existing Expanded Access Program nor the newly enacted Right to Try law require that pharmaceutical companies actually provide the requested drugs. Industry leaders, in testimony before Congress, have cited patient safety, costs, drug supply, and negative impact on existing clinical trial accrual as reasons pharmaceutical companies commonly deny requests for access to investigational products.
The bottom line is that the Right to Try law really lacks teeth. Despite the favorable optics for the president and Congress, it is virtually certain that Right to Try will never save hundreds of thousands of lives, and the suggestion that it will is either disingenuous or uninformed. Hopefully, though, Congress’s passage of Right to Try is indicative of a more general interest in bipartisan cooperation to improve access to affordable, high-quality health care.
Dr. Zonder is a professor in the department of oncology at the Barbara Ann Karmanos Cancer Institute and Wayne State University in Detroit. He is the leader of the KCI Myeloma and Amyloidosis Team. He is also a member of the Hematology News Editorial Advisory Board. He reported having no relevant financial disclosures.
References
1. Roberts TG Jr. et al. JAMA. 2004 Nov 3;292(17):2130-40.
2. Fukada YK et al. J Clin Oncol. 2014 May 20. doi: 10.1200/jco.2014.32.15_suppl.2552.
3. Jarow JP et al. Ther Innov Regul Sci. 2017 Mar 1;51(2):177-9.
4. Kearns L et al. Ther Innov Regul Sci. 2017 Mar 1;51(2):170-6.
On May 30, 2018, President Trump signed into law the Trickett Wendler, Frank Mongiello, Jordan McLinn, and Matthew Bellina Right to Try Act of 2017, guaranteeing patients with terminal illnesses the right to seek access to investigational drugs which have passed at least phase 1 testing. As he signed the bipartisan-backed bill into law, the president said he believes it will ultimately save “hundreds of thousands” of lives, scoring an apparent win-win-win for the president, for Congress, and for patients. A critical appraisal of the law – and of the supposition that patients will reap great benefit from it – requires that we ask two questions: Does the law degrade measures originally put in place to protect patients, and does it actually improve access to potentially life-saving drugs?
As a hematologist with about 2 decades of experience conducting clinical research involving patients with incurable cancer, I want as many patients as possible to have access to promising new therapies. Clinical trials, as the sole means of determining a candidate drug’s safety and efficacy, are conducted in a systematic fashion, with phase 1 trials focusing predominantly on safety. As any experienced researcher is aware, however, a phase 1 trial only provides a basic sense of a drug’s safety. It would be unimaginable (and unethical) to conduct subsequent phase 2 and 3 trials without including rigorous ongoing monitoring for adverse effects.
A later study evaluating all nonpediatric phase 1 trials sponsored by the National Cancer Institute’s Cancer Therapy Evaluation Program between 2001 and 2012, which included more than 8,300 patients, found the therapy-related death rate to be similarly low – approximately 1%.2
It is distinctly possible that the same drugs used in the analyzed trials would have been more toxic to patients who, for one reason or another, would not have met enrollment criteria. It is impossible to precisely quantify the degree to which risk may be increased under the Right to Try law, but even if it were tripled or quadrupled, compared with these historical expectations, the absolute risk of treatment-related death remains low. It should be noted that the incidence of nonfatal grade 3-4 adverse events in the same analyses was 10%-20% so a similar proportional increase in these events would impact a larger number of patients. Overall, it is difficult to know the degree to which Right to Try impacts the safety of fragile patients with terminal illnesses.
The answer to the second question – Is access to life-saving drugs improved? – is much easier to answer: No.
One reason is that these patients already have the right to seek access to investigational drugs through the FDA’s Expanded Access Program. An overview of the Expanded Access Program reported that approximately 11,000 patients over a period of 10 years submitted requests for access to investigational drugs and that 99% of the requests were granted.3
Proponents of the Right to Try law argue that the new law somehow simplifies the request process, but it is difficult to understand exactly how. Further, in a position paper on the topic of Right to Try policy, Lisa Kearns and Alison Bateman-House, PhD, of New York University, wrote, “In the more than 2½ years since the first [individual state right to try] law was signed, there have been no documented cases of anyone receiving access, because of a right to try law, to an experimental product that would not have been available via the FDA’s Expanded Access Program.”4
Finally, and most importantly, neither the existing Expanded Access Program nor the newly enacted Right to Try law require that pharmaceutical companies actually provide the requested drugs. Industry leaders, in testimony before Congress, have cited patient safety, costs, drug supply, and negative impact on existing clinical trial accrual as reasons pharmaceutical companies commonly deny requests for access to investigational products.
The bottom line is that the Right to Try law really lacks teeth. Despite the favorable optics for the president and Congress, it is virtually certain that Right to Try will never save hundreds of thousands of lives, and the suggestion that it will is either disingenuous or uninformed. Hopefully, though, Congress’s passage of Right to Try is indicative of a more general interest in bipartisan cooperation to improve access to affordable, high-quality health care.
Dr. Zonder is a professor in the department of oncology at the Barbara Ann Karmanos Cancer Institute and Wayne State University in Detroit. He is the leader of the KCI Myeloma and Amyloidosis Team. He is also a member of the Hematology News Editorial Advisory Board. He reported having no relevant financial disclosures.
References
1. Roberts TG Jr. et al. JAMA. 2004 Nov 3;292(17):2130-40.
2. Fukada YK et al. J Clin Oncol. 2014 May 20. doi: 10.1200/jco.2014.32.15_suppl.2552.
3. Jarow JP et al. Ther Innov Regul Sci. 2017 Mar 1;51(2):177-9.
4. Kearns L et al. Ther Innov Regul Sci. 2017 Mar 1;51(2):170-6.
Q&A: Clinical implications of clonal hematopoiesis
There is growing literature around clonal hematopoiesis (CH) but many questions persist about its clinical significance. On June 14, Hematology News hosted a Twitter question-and-answer session with Aaron Viny, MD, who is on the staff of the leukemia service at Memorial Sloan Kettering Cancer Center in New York and is a member of the Hematology News editorial advisory board, to answer questions about CH and interpret the latest research. The following is an edited version of the Q&A session.
Question: Can you get us started by explaining the difference between clonal hematopoiesis (CH) and clonal cytopenia of undetermined significance?

Question: So are there differences in CH depending on the gene mutated?
Dr. Viny: Hard to say. The most common mutations found in CH are DNMT3A, TET2, PPM1D, and ASXL1, as shown in the Cell Stem Cell paper by Catherine C. Coombs, MD, and Ross Levine, MD (2017 Sep 7;21[3]:374-82.e4). So in the setting of patients with acute myeloid leukemia, there are data from several groups showing that persistence of DNMT3A, TET2, and ASXL1 have less recurrence risk, compared with the field. This, of course, does not apply to CH in the absence of a hematologic disorder.
Question: Are you aware of any screening programs for clonal hematopoiesis?
Dr. Viny: Currently at Memorial Sloan Kettering, patients who undergo MSK-IMPACT testing of their solid tumor have a germ line control from blood that is also sequenced. These samples are screened for CH mutations. In fact, there are two recent papers showing one key issue with tumor sequencing and CH: A blood sample is necessary to resolve the compartment of the CH mutation (that is, not in the solid tumor). The papers are JAMA Oncol. 2018 Jun 5. doi: 10.1001/jamaoncol.2018.2297 and Clin Cancer Res. 2018 Jun 4. doi: 10.1158/1078-0432.CCR-18-1201.
Question: Will patients with CH in screened samples be notified of the results?
Dr. Viny: Yes. The patients are being referred to the Memorial Sloan Kettering clonal hematopoiesis clinic run by Dr. Levine and Kelly Bolton, MD.
Question: Once you screen and detect CH, how should these patients be followed?
Dr. Viny: First, what are the risks these patients face? Extensive work by Siddhartha Jaiswal, MD, PhD, shows that there is an increased risk of cardiovascular disease and an increased risk for leukemia. So with regards to the latter, following with serial complete blood counts seems sufficient, with a bone marrow biopsy at the detection of any cytopenias. With regard to cardiovascular risk, I consider CH akin to an unmodifiable cardiac risk factor. Patients should be counseled to exercise, and depending on any other cardiac risk factors, interventions such as blood pressure control, lipid control, and daily aspirin use should be addressed accordingly.
Question: Is this BRCA1 all over again?
Dr. Viny: Perhaps. With BRCA1, we now have a few decades of follow-up to better understand the risks and even intervene with preventive interventions. Clonal hematopoiesis needs the follow-up and research to support clinical action.
Question: Could you please refer your readers to your favorite review on clonal hematopoiesis?
Dr. Viny: An outstanding review of the mechanisms and molecular consequences of clonal hematopoiesis is in Cell Stem Cell (2018 Feb 1;22[2]:157-70).
Question: Are there any effects of previous radiation on the development of CH?
Dr. Viny: Let’s start by saying that CH in the absence of prior cancer is probably a different entity. Radiation, tobacco use, and increased age all increase the risk for detection of a CH somatic mutation.
Question: What are the clinical implications of CH?
Dr. Viny: While I think it is still too soon to say if these are the only clinical implications, both increased risk of hematologic malignancy and increased risk of cardiovascular disease are the best studied and described to date. Here’s an excellent article on the cardiovascular risk: N Engl J Med. 2017 Jul 13;377(2):111-21. Of interest, it seems that the increased risk, while being relatively low, does not plateau over time.
Question: What do you think about TET2 mutations being among the most frequent mutations in CH, but IDH being relatively rare?
Dr. Viny: Great question. Both are affecting demethylation and epigenetic instability. While I don’t think the answer is known, perhaps the IDH mutations have a more dominant effect on hematopoietic output. The majority of the CH data uses blood; maybe the marrow tells a different story.
Question: Can we cure clonal hematopoiesis with vitamin C?
Dr. Viny: You clearly know the recent work by Iannis Aifantis, PhD, showing the effects of vitamin C on TET enzyme activity, published here: Cell. 2017 Sep 7;170(6):1079-95. For CH patients with TET2 mutations, a high-dose vitamin C regimen sounds very exciting. There is also complementary work by Sean Morrison, PhD, published in Nature (2017 Sep 28;549[7673]:476-81).
Question: Is there a limit to the sequencing depth and variant allele fraction needed to identify clonal hematopoiesis? At what point is CH not CH?
Dr. Viny: So this is as much a technical question as it is a biological question. As of now we can reliably detect variant allele fraction in CH to 0.1%, at best. What is detectable and what is clinically relevant are questions that still need to be answered.
Question: There seems to be a lot of good research going on in CH. What are the big knowledge gaps that future studies should be targeting?
Dr. Viny: First, what are the functional and molecular consequences of the varying alleles in CH? Second, are there other clinical risks to CH beyond leukemia and cardiovascular disease? And third, does inflammation cause CH or does CH cause inflammation?
Dr. Viny is with the Memorial Sloan Kettering Cancer Center, New York, where he is a clinical instructor; is on the staff of the leukemia service; and is a clinical researcher in the Ross Levine Lab. Connect with him on Twitter at @TheDoctorIsVin.
There is growing literature around clonal hematopoiesis (CH) but many questions persist about its clinical significance. On June 14, Hematology News hosted a Twitter question-and-answer session with Aaron Viny, MD, who is on the staff of the leukemia service at Memorial Sloan Kettering Cancer Center in New York and is a member of the Hematology News editorial advisory board, to answer questions about CH and interpret the latest research. The following is an edited version of the Q&A session.
Question: Can you get us started by explaining the difference between clonal hematopoiesis (CH) and clonal cytopenia of undetermined significance?
Question: So are there differences in CH depending on the gene mutated?
Dr. Viny: Hard to say. The most common mutations found in CH are DNMT3A, TET2, PPM1D, and ASXL1, as shown in the Cell Stem Cell paper by Catherine C. Coombs, MD, and Ross Levine, MD (2017 Sep 7;21[3]:374-82.e4). So in the setting of patients with acute myeloid leukemia, there are data from several groups showing that persistence of DNMT3A, TET2, and ASXL1 have less recurrence risk, compared with the field. This, of course, does not apply to CH in the absence of a hematologic disorder.
Question: Are you aware of any screening programs for clonal hematopoiesis?
Dr. Viny: Currently at Memorial Sloan Kettering, patients who undergo MSK-IMPACT testing of their solid tumor have a germ line control from blood that is also sequenced. These samples are screened for CH mutations. In fact, there are two recent papers showing one key issue with tumor sequencing and CH: A blood sample is necessary to resolve the compartment of the CH mutation (that is, not in the solid tumor). The papers are JAMA Oncol. 2018 Jun 5. doi: 10.1001/jamaoncol.2018.2297 and Clin Cancer Res. 2018 Jun 4. doi: 10.1158/1078-0432.CCR-18-1201.
Question: Will patients with CH in screened samples be notified of the results?
Dr. Viny: Yes. The patients are being referred to the Memorial Sloan Kettering clonal hematopoiesis clinic run by Dr. Levine and Kelly Bolton, MD.
Question: Once you screen and detect CH, how should these patients be followed?
Dr. Viny: First, what are the risks these patients face? Extensive work by Siddhartha Jaiswal, MD, PhD, shows that there is an increased risk of cardiovascular disease and an increased risk for leukemia. So with regards to the latter, following with serial complete blood counts seems sufficient, with a bone marrow biopsy at the detection of any cytopenias. With regard to cardiovascular risk, I consider CH akin to an unmodifiable cardiac risk factor. Patients should be counseled to exercise, and depending on any other cardiac risk factors, interventions such as blood pressure control, lipid control, and daily aspirin use should be addressed accordingly.
Question: Is this BRCA1 all over again?
Dr. Viny: Perhaps. With BRCA1, we now have a few decades of follow-up to better understand the risks and even intervene with preventive interventions. Clonal hematopoiesis needs the follow-up and research to support clinical action.
Question: Could you please refer your readers to your favorite review on clonal hematopoiesis?
Dr. Viny: An outstanding review of the mechanisms and molecular consequences of clonal hematopoiesis is in Cell Stem Cell (2018 Feb 1;22[2]:157-70).
Question: Are there any effects of previous radiation on the development of CH?
Dr. Viny: Let’s start by saying that CH in the absence of prior cancer is probably a different entity. Radiation, tobacco use, and increased age all increase the risk for detection of a CH somatic mutation.
Question: What are the clinical implications of CH?
Dr. Viny: While I think it is still too soon to say if these are the only clinical implications, both increased risk of hematologic malignancy and increased risk of cardiovascular disease are the best studied and described to date. Here’s an excellent article on the cardiovascular risk: N Engl J Med. 2017 Jul 13;377(2):111-21. Of interest, it seems that the increased risk, while being relatively low, does not plateau over time.
Question: What do you think about TET2 mutations being among the most frequent mutations in CH, but IDH being relatively rare?
Dr. Viny: Great question. Both are affecting demethylation and epigenetic instability. While I don’t think the answer is known, perhaps the IDH mutations have a more dominant effect on hematopoietic output. The majority of the CH data uses blood; maybe the marrow tells a different story.
Question: Can we cure clonal hematopoiesis with vitamin C?
Dr. Viny: You clearly know the recent work by Iannis Aifantis, PhD, showing the effects of vitamin C on TET enzyme activity, published here: Cell. 2017 Sep 7;170(6):1079-95. For CH patients with TET2 mutations, a high-dose vitamin C regimen sounds very exciting. There is also complementary work by Sean Morrison, PhD, published in Nature (2017 Sep 28;549[7673]:476-81).
Question: Is there a limit to the sequencing depth and variant allele fraction needed to identify clonal hematopoiesis? At what point is CH not CH?
Dr. Viny: So this is as much a technical question as it is a biological question. As of now we can reliably detect variant allele fraction in CH to 0.1%, at best. What is detectable and what is clinically relevant are questions that still need to be answered.
Question: There seems to be a lot of good research going on in CH. What are the big knowledge gaps that future studies should be targeting?
Dr. Viny: First, what are the functional and molecular consequences of the varying alleles in CH? Second, are there other clinical risks to CH beyond leukemia and cardiovascular disease? And third, does inflammation cause CH or does CH cause inflammation?
Dr. Viny is with the Memorial Sloan Kettering Cancer Center, New York, where he is a clinical instructor; is on the staff of the leukemia service; and is a clinical researcher in the Ross Levine Lab. Connect with him on Twitter at @TheDoctorIsVin.
There is growing literature around clonal hematopoiesis (CH) but many questions persist about its clinical significance. On June 14, Hematology News hosted a Twitter question-and-answer session with Aaron Viny, MD, who is on the staff of the leukemia service at Memorial Sloan Kettering Cancer Center in New York and is a member of the Hematology News editorial advisory board, to answer questions about CH and interpret the latest research. The following is an edited version of the Q&A session.
Question: Can you get us started by explaining the difference between clonal hematopoiesis (CH) and clonal cytopenia of undetermined significance?
Question: So are there differences in CH depending on the gene mutated?
Dr. Viny: Hard to say. The most common mutations found in CH are DNMT3A, TET2, PPM1D, and ASXL1, as shown in the Cell Stem Cell paper by Catherine C. Coombs, MD, and Ross Levine, MD (2017 Sep 7;21[3]:374-82.e4). So in the setting of patients with acute myeloid leukemia, there are data from several groups showing that persistence of DNMT3A, TET2, and ASXL1 have less recurrence risk, compared with the field. This, of course, does not apply to CH in the absence of a hematologic disorder.
Question: Are you aware of any screening programs for clonal hematopoiesis?
Dr. Viny: Currently at Memorial Sloan Kettering, patients who undergo MSK-IMPACT testing of their solid tumor have a germ line control from blood that is also sequenced. These samples are screened for CH mutations. In fact, there are two recent papers showing one key issue with tumor sequencing and CH: A blood sample is necessary to resolve the compartment of the CH mutation (that is, not in the solid tumor). The papers are JAMA Oncol. 2018 Jun 5. doi: 10.1001/jamaoncol.2018.2297 and Clin Cancer Res. 2018 Jun 4. doi: 10.1158/1078-0432.CCR-18-1201.
Question: Will patients with CH in screened samples be notified of the results?
Dr. Viny: Yes. The patients are being referred to the Memorial Sloan Kettering clonal hematopoiesis clinic run by Dr. Levine and Kelly Bolton, MD.
Question: Once you screen and detect CH, how should these patients be followed?
Dr. Viny: First, what are the risks these patients face? Extensive work by Siddhartha Jaiswal, MD, PhD, shows that there is an increased risk of cardiovascular disease and an increased risk for leukemia. So with regards to the latter, following with serial complete blood counts seems sufficient, with a bone marrow biopsy at the detection of any cytopenias. With regard to cardiovascular risk, I consider CH akin to an unmodifiable cardiac risk factor. Patients should be counseled to exercise, and depending on any other cardiac risk factors, interventions such as blood pressure control, lipid control, and daily aspirin use should be addressed accordingly.
Question: Is this BRCA1 all over again?
Dr. Viny: Perhaps. With BRCA1, we now have a few decades of follow-up to better understand the risks and even intervene with preventive interventions. Clonal hematopoiesis needs the follow-up and research to support clinical action.
Question: Could you please refer your readers to your favorite review on clonal hematopoiesis?
Dr. Viny: An outstanding review of the mechanisms and molecular consequences of clonal hematopoiesis is in Cell Stem Cell (2018 Feb 1;22[2]:157-70).
Question: Are there any effects of previous radiation on the development of CH?
Dr. Viny: Let’s start by saying that CH in the absence of prior cancer is probably a different entity. Radiation, tobacco use, and increased age all increase the risk for detection of a CH somatic mutation.
Question: What are the clinical implications of CH?
Dr. Viny: While I think it is still too soon to say if these are the only clinical implications, both increased risk of hematologic malignancy and increased risk of cardiovascular disease are the best studied and described to date. Here’s an excellent article on the cardiovascular risk: N Engl J Med. 2017 Jul 13;377(2):111-21. Of interest, it seems that the increased risk, while being relatively low, does not plateau over time.
Question: What do you think about TET2 mutations being among the most frequent mutations in CH, but IDH being relatively rare?
Dr. Viny: Great question. Both are affecting demethylation and epigenetic instability. While I don’t think the answer is known, perhaps the IDH mutations have a more dominant effect on hematopoietic output. The majority of the CH data uses blood; maybe the marrow tells a different story.
Question: Can we cure clonal hematopoiesis with vitamin C?
Dr. Viny: You clearly know the recent work by Iannis Aifantis, PhD, showing the effects of vitamin C on TET enzyme activity, published here: Cell. 2017 Sep 7;170(6):1079-95. For CH patients with TET2 mutations, a high-dose vitamin C regimen sounds very exciting. There is also complementary work by Sean Morrison, PhD, published in Nature (2017 Sep 28;549[7673]:476-81).
Question: Is there a limit to the sequencing depth and variant allele fraction needed to identify clonal hematopoiesis? At what point is CH not CH?
Dr. Viny: So this is as much a technical question as it is a biological question. As of now we can reliably detect variant allele fraction in CH to 0.1%, at best. What is detectable and what is clinically relevant are questions that still need to be answered.
Question: There seems to be a lot of good research going on in CH. What are the big knowledge gaps that future studies should be targeting?
Dr. Viny: First, what are the functional and molecular consequences of the varying alleles in CH? Second, are there other clinical risks to CH beyond leukemia and cardiovascular disease? And third, does inflammation cause CH or does CH cause inflammation?
Dr. Viny is with the Memorial Sloan Kettering Cancer Center, New York, where he is a clinical instructor; is on the staff of the leukemia service; and is a clinical researcher in the Ross Levine Lab. Connect with him on Twitter at @TheDoctorIsVin.
U.S. immigration policy: What harms will persist?
The Trump policy of separating children and teenagers from their parents after crossing the U.S. border has been called un-American, immoral, cruel, and inhumane. The policy thankfully has been reversed or at least subject to delay. However, as I write today, 2,300 children and their parents are separated, not in contact, lost to each other, and with no clear plan on reunification. The ultimate outcome of immigration legislation and policy is unknown and mired in partisan politics. The policy hopefully has changed permanently, but what are the harms that will persist?
1. Many if not all of the 2,300 children taken from their parents to institutional settings will have suffered acute anxiety and despair. Following data gathered by René Spitz and John Bowlby 80 years ago, children forced to separate from their parents for long hospitalizations with limited visitation went through phases of protest, despair, and if repeated or lengthy separations, “detachment” that impaired their ability to form relationships.1
2. Many of these children have suffered traumas in their country of origin and through the journey to the U.S. border. Some of this traumatic experience was mitigated by being in the presence of their parent(s). Very likely some children have psychiatric and physical disorders that will add to the level of risk. The current trauma, forcible separation by armed guards into restrictive facilities, will compound or intensify the previous traumas without the benefit of parental support.
3. Will the harms persist? Likely this level of trauma has such a strong neurologic and psychological impact that many of the children will suffer from nightmares, depression, and persistent anxiety about trusting the safety of their setting. 2
4. The parents who are jailed, have had their children removed, and do not know where they are and aren’t able to talk to them have suffered a massive trauma. We all have lost sight of a child for a minute or two in a store or on the beach. Our anxiety is immediate, and if the separation is longer, we may remember those frightening minutes for the rest of our lives. How many immigrant parents will develop depression and posttraumatic stress disorder?
5. Guards were ordered to be the front-line implementers of the policy and must have been torn between their sworn duty and their inner knowledge that what they are doing is wrong. Hearing the children crying and calling for their parents must have elicited painful feelings of what it would have been like to have their own children taken away with no way to reach them or knowing where they were taken. Implementing this policy dehumanized them, and I believe made them feel guilty or unworthy.
6. Millions of immigrants – whether lawful, dreamers, or undocumented – must have felt fearful, powerless, and angry about this policy. Millions of their children must have been worried and lost a little bit of faith in their parents and in the United States.
7. Did U.S. citizens, many from immigrant roots, wonder if this could happen to them? How many children felt a little less secure? Was the anxiety higher for descendants of the U.S. citizens remembering the trauma of the World War II Japanese internment camps? Other descendants (like me) will remember quite vividly their mother’s story of being on the St. Louis steam ship and being turned away from the United States to face a high likelihood of death in Nazi Germany. A bit of fear will replace trust in and loyalty to the United States.

Dr. Jellinek is professor emeritus of psychiatry and pediatrics, Harvard Medical School, Boston. Email him at [email protected].
References:
1. Dev Psychol. 1992;28:759-75.
2. www.cdc.gov/violenceprevention/acestudy/index.html
The Trump policy of separating children and teenagers from their parents after crossing the U.S. border has been called un-American, immoral, cruel, and inhumane. The policy thankfully has been reversed or at least subject to delay. However, as I write today, 2,300 children and their parents are separated, not in contact, lost to each other, and with no clear plan on reunification. The ultimate outcome of immigration legislation and policy is unknown and mired in partisan politics. The policy hopefully has changed permanently, but what are the harms that will persist?
1. Many if not all of the 2,300 children taken from their parents to institutional settings will have suffered acute anxiety and despair. Following data gathered by René Spitz and John Bowlby 80 years ago, children forced to separate from their parents for long hospitalizations with limited visitation went through phases of protest, despair, and if repeated or lengthy separations, “detachment” that impaired their ability to form relationships.1
2. Many of these children have suffered traumas in their country of origin and through the journey to the U.S. border. Some of this traumatic experience was mitigated by being in the presence of their parent(s). Very likely some children have psychiatric and physical disorders that will add to the level of risk. The current trauma, forcible separation by armed guards into restrictive facilities, will compound or intensify the previous traumas without the benefit of parental support.
3. Will the harms persist? Likely this level of trauma has such a strong neurologic and psychological impact that many of the children will suffer from nightmares, depression, and persistent anxiety about trusting the safety of their setting. 2
4. The parents who are jailed, have had their children removed, and do not know where they are and aren’t able to talk to them have suffered a massive trauma. We all have lost sight of a child for a minute or two in a store or on the beach. Our anxiety is immediate, and if the separation is longer, we may remember those frightening minutes for the rest of our lives. How many immigrant parents will develop depression and posttraumatic stress disorder?
5. Guards were ordered to be the front-line implementers of the policy and must have been torn between their sworn duty and their inner knowledge that what they are doing is wrong. Hearing the children crying and calling for their parents must have elicited painful feelings of what it would have been like to have their own children taken away with no way to reach them or knowing where they were taken. Implementing this policy dehumanized them, and I believe made them feel guilty or unworthy.
6. Millions of immigrants – whether lawful, dreamers, or undocumented – must have felt fearful, powerless, and angry about this policy. Millions of their children must have been worried and lost a little bit of faith in their parents and in the United States.
7. Did U.S. citizens, many from immigrant roots, wonder if this could happen to them? How many children felt a little less secure? Was the anxiety higher for descendants of the U.S. citizens remembering the trauma of the World War II Japanese internment camps? Other descendants (like me) will remember quite vividly their mother’s story of being on the St. Louis steam ship and being turned away from the United States to face a high likelihood of death in Nazi Germany. A bit of fear will replace trust in and loyalty to the United States.
Dr. Jellinek is professor emeritus of psychiatry and pediatrics, Harvard Medical School, Boston. Email him at [email protected].
References:
1. Dev Psychol. 1992;28:759-75.
2. www.cdc.gov/violenceprevention/acestudy/index.html
The Trump policy of separating children and teenagers from their parents after crossing the U.S. border has been called un-American, immoral, cruel, and inhumane. The policy thankfully has been reversed or at least subject to delay. However, as I write today, 2,300 children and their parents are separated, not in contact, lost to each other, and with no clear plan on reunification. The ultimate outcome of immigration legislation and policy is unknown and mired in partisan politics. The policy hopefully has changed permanently, but what are the harms that will persist?
1. Many if not all of the 2,300 children taken from their parents to institutional settings will have suffered acute anxiety and despair. Following data gathered by René Spitz and John Bowlby 80 years ago, children forced to separate from their parents for long hospitalizations with limited visitation went through phases of protest, despair, and if repeated or lengthy separations, “detachment” that impaired their ability to form relationships.1
2. Many of these children have suffered traumas in their country of origin and through the journey to the U.S. border. Some of this traumatic experience was mitigated by being in the presence of their parent(s). Very likely some children have psychiatric and physical disorders that will add to the level of risk. The current trauma, forcible separation by armed guards into restrictive facilities, will compound or intensify the previous traumas without the benefit of parental support.
3. Will the harms persist? Likely this level of trauma has such a strong neurologic and psychological impact that many of the children will suffer from nightmares, depression, and persistent anxiety about trusting the safety of their setting. 2
4. The parents who are jailed, have had their children removed, and do not know where they are and aren’t able to talk to them have suffered a massive trauma. We all have lost sight of a child for a minute or two in a store or on the beach. Our anxiety is immediate, and if the separation is longer, we may remember those frightening minutes for the rest of our lives. How many immigrant parents will develop depression and posttraumatic stress disorder?
5. Guards were ordered to be the front-line implementers of the policy and must have been torn between their sworn duty and their inner knowledge that what they are doing is wrong. Hearing the children crying and calling for their parents must have elicited painful feelings of what it would have been like to have their own children taken away with no way to reach them or knowing where they were taken. Implementing this policy dehumanized them, and I believe made them feel guilty or unworthy.
6. Millions of immigrants – whether lawful, dreamers, or undocumented – must have felt fearful, powerless, and angry about this policy. Millions of their children must have been worried and lost a little bit of faith in their parents and in the United States.
7. Did U.S. citizens, many from immigrant roots, wonder if this could happen to them? How many children felt a little less secure? Was the anxiety higher for descendants of the U.S. citizens remembering the trauma of the World War II Japanese internment camps? Other descendants (like me) will remember quite vividly their mother’s story of being on the St. Louis steam ship and being turned away from the United States to face a high likelihood of death in Nazi Germany. A bit of fear will replace trust in and loyalty to the United States.
Dr. Jellinek is professor emeritus of psychiatry and pediatrics, Harvard Medical School, Boston. Email him at [email protected].
References:
1. Dev Psychol. 1992;28:759-75.
2. www.cdc.gov/violenceprevention/acestudy/index.html
Mindfulness skill can help in parenting
Behavioral parent management training (PMT), which teaches parents concrete skills to increase their attention to positive behavior and to plan for their response to undesired behavior, has abundant evidence for success for many challenging child behaviors. But sometimes parents have a hard time managing their own emotional responses in the often highly triggering situation of family conflict. Mindfulness has the potential to provide a complement to the PMT skills. Studies are beginning to explore these possibilities.
Case summary
Zoe is a bright 5-year-old who has been “strong willed” and shown intense emotional responses since early in life. The usual 2-year-old temper tantrums increased over time. She has outbursts of yelling, kicking, and hitting, especially with transitions. Her parents tried behavioral parent training, but found it frustrating. If Zoe has been yelling and hitting earlier in the day, her mother feels hurt and angry and can’t bring herself to pay warm attention when Zoe is doing better. When Zoe refuses to pick up her room, her father is flooded with thoughts about his own father hitting him for the slightest disrespect. He thinks that he is a bad, weak father, and sometimes “sees red” and ends up yelling at Zoe instead of putting into place a calm consequence.

Discussion
Mindfulness is defined by Jon Kabat-Zinn as “paying attention in a particular way – on purpose, in the present moment, and nonjudgmentally.” A central feature of mindfulness is strengthening the ability to focus our attention. We learn to pay attention to aspects of the present moment, be that breathing, the sensations in our body, or the experiences of our senses. Often the first skill in behavioral training methods is getting parents to pay attention to their children by participating in child-led play or spending attentive time with older children. This means attending to what the child is doing or talking about rather than jumping in and taking over with suggestions, instructions, or judgments. This meshes very well with this central aspect of mindfulness.
As we practice paying attention, we observe that the mind naturally jumps around from what we mean to be attending to, to a host of distractions, worries, plans, memories, thoughts, and emotions. Mindfulness encourages practitioners to notice these thoughts, to avoid criticizing or judging oneself for becoming involved with these, but instead gently lead the mind back to what you had intended to focus on. This observation of the mind’s activity gives the mindfulness practitioner a bit of space from the thought or emotion itself. We are encouraged to name the thought or emotional processes we notice: “I am worrying, I am planning, I am remembering.”
In the heat of a difficult moment with the child, parents often are flooded with intense emotions (such as anger, fear, anxiety, panic, despair) and thoughts (such as “If my child keeps acting this way he is going to go to jail when he grows up,” “I am a terrible parent,” “Why is my child doing this to me?” or “He is just like his father”). These emotions and thoughts can drive intense, impulsive responses from the parents. As they practice mindfulness, they can gain the ability to observe themselves having these thoughts; observe harsh judgments of themselves or their children or their partners; have some space from them; and realize they may change in a few minutes or realize they may be painful but don’t necessarily have to spur impulsive action. In that moment, parents can give themselves time and space to think through possible actions, and then choose one.

From a behavioral parenting standpoint, we know that parents and humans often react intensely to negative behaviors and inadvertently make them worse with intense emotional reactivity. We want parents to have a plan about how they will respond, to remain calm in the moment, and then put the plan in place. Mindfulness may enhance parents’ ability to notice their own responses and have the space to remember what the plan was and then put it into place. It also can give them space to consider what the child might be experiencing and respond in light of this awareness. This ability does require a significant amount of mindfulness practice.
The combination of mindfulness and parenting is just beginning to be studied in research trials using a range of study designs. Some of these programs have looked at the effect of mindfulness courses, especially mindfulness-based stress reduction without any specific parenting content or indices of parent stress and child behavior. Others have looked at programs which add mindfulness to standard behavioral parenting programs, and still others are specific mindfulness/parenting programs. So far, many of these studies are quasi-experimental in nature. A recent systematic review by Townshend et al. found seven randomized controlled trials of low to moderate quality with some suggestion of ability to decrease parental stress and ADHD symptoms (JBI Database System Rev Implement Rep. 2016 Mar;14[3]:139-80). There is a clear need for randomized controlled trials with larger sample sizes.
While we may not have specific, highly evidence-based mindful parenting programs available, individuals with experience in yoga, meditation, mindfulness, dialectical behavioral therapy, and acceptance and commitment therapy can be encouraged to bring these skills to bear as parents.
Zoe’s parents had pursued outside mindfulness programs. Mindfulness concepts were brought into a standard parenting program. Her parents were encouraged to engage in child-led play with Zoe in a mindful way, fully attending to her actions and experience. Zoe’s parents also were encouraged to observe their own emotional reactions and thoughts in stressful moments and to take a breathing space before taking action.
Dr. Hall is assistant professor of psychiatry and pediatrics at the University of Vermont, Burlington. She said she had no relevant financial disclosures. Email her at [email protected].
Resources
“Mindful Parenting” (New York: Norton & Co., 2015).
“Integrating mindfulness with parent training: Effects of the mindfulness-enhanced strengthening families program” (Dev Psychol. 2015;51[1]:26-35).
“Everyday blessings: The inner work of mindful parenting,” (New York: Hyperion, 1997).
Behavioral parent management training (PMT), which teaches parents concrete skills to increase their attention to positive behavior and to plan for their response to undesired behavior, has abundant evidence for success for many challenging child behaviors. But sometimes parents have a hard time managing their own emotional responses in the often highly triggering situation of family conflict. Mindfulness has the potential to provide a complement to the PMT skills. Studies are beginning to explore these possibilities.
Case summary
Zoe is a bright 5-year-old who has been “strong willed” and shown intense emotional responses since early in life. The usual 2-year-old temper tantrums increased over time. She has outbursts of yelling, kicking, and hitting, especially with transitions. Her parents tried behavioral parent training, but found it frustrating. If Zoe has been yelling and hitting earlier in the day, her mother feels hurt and angry and can’t bring herself to pay warm attention when Zoe is doing better. When Zoe refuses to pick up her room, her father is flooded with thoughts about his own father hitting him for the slightest disrespect. He thinks that he is a bad, weak father, and sometimes “sees red” and ends up yelling at Zoe instead of putting into place a calm consequence.
Discussion
Mindfulness is defined by Jon Kabat-Zinn as “paying attention in a particular way – on purpose, in the present moment, and nonjudgmentally.” A central feature of mindfulness is strengthening the ability to focus our attention. We learn to pay attention to aspects of the present moment, be that breathing, the sensations in our body, or the experiences of our senses. Often the first skill in behavioral training methods is getting parents to pay attention to their children by participating in child-led play or spending attentive time with older children. This means attending to what the child is doing or talking about rather than jumping in and taking over with suggestions, instructions, or judgments. This meshes very well with this central aspect of mindfulness.
As we practice paying attention, we observe that the mind naturally jumps around from what we mean to be attending to, to a host of distractions, worries, plans, memories, thoughts, and emotions. Mindfulness encourages practitioners to notice these thoughts, to avoid criticizing or judging oneself for becoming involved with these, but instead gently lead the mind back to what you had intended to focus on. This observation of the mind’s activity gives the mindfulness practitioner a bit of space from the thought or emotion itself. We are encouraged to name the thought or emotional processes we notice: “I am worrying, I am planning, I am remembering.”
In the heat of a difficult moment with the child, parents often are flooded with intense emotions (such as anger, fear, anxiety, panic, despair) and thoughts (such as “If my child keeps acting this way he is going to go to jail when he grows up,” “I am a terrible parent,” “Why is my child doing this to me?” or “He is just like his father”). These emotions and thoughts can drive intense, impulsive responses from the parents. As they practice mindfulness, they can gain the ability to observe themselves having these thoughts; observe harsh judgments of themselves or their children or their partners; have some space from them; and realize they may change in a few minutes or realize they may be painful but don’t necessarily have to spur impulsive action. In that moment, parents can give themselves time and space to think through possible actions, and then choose one.
From a behavioral parenting standpoint, we know that parents and humans often react intensely to negative behaviors and inadvertently make them worse with intense emotional reactivity. We want parents to have a plan about how they will respond, to remain calm in the moment, and then put the plan in place. Mindfulness may enhance parents’ ability to notice their own responses and have the space to remember what the plan was and then put it into place. It also can give them space to consider what the child might be experiencing and respond in light of this awareness. This ability does require a significant amount of mindfulness practice.
The combination of mindfulness and parenting is just beginning to be studied in research trials using a range of study designs. Some of these programs have looked at the effect of mindfulness courses, especially mindfulness-based stress reduction without any specific parenting content or indices of parent stress and child behavior. Others have looked at programs which add mindfulness to standard behavioral parenting programs, and still others are specific mindfulness/parenting programs. So far, many of these studies are quasi-experimental in nature. A recent systematic review by Townshend et al. found seven randomized controlled trials of low to moderate quality with some suggestion of ability to decrease parental stress and ADHD symptoms (JBI Database System Rev Implement Rep. 2016 Mar;14[3]:139-80). There is a clear need for randomized controlled trials with larger sample sizes.
While we may not have specific, highly evidence-based mindful parenting programs available, individuals with experience in yoga, meditation, mindfulness, dialectical behavioral therapy, and acceptance and commitment therapy can be encouraged to bring these skills to bear as parents.
Zoe’s parents had pursued outside mindfulness programs. Mindfulness concepts were brought into a standard parenting program. Her parents were encouraged to engage in child-led play with Zoe in a mindful way, fully attending to her actions and experience. Zoe’s parents also were encouraged to observe their own emotional reactions and thoughts in stressful moments and to take a breathing space before taking action.
Dr. Hall is assistant professor of psychiatry and pediatrics at the University of Vermont, Burlington. She said she had no relevant financial disclosures. Email her at [email protected].
Resources
“Mindful Parenting” (New York: Norton & Co., 2015).
“Integrating mindfulness with parent training: Effects of the mindfulness-enhanced strengthening families program” (Dev Psychol. 2015;51[1]:26-35).
“Everyday blessings: The inner work of mindful parenting,” (New York: Hyperion, 1997).
Behavioral parent management training (PMT), which teaches parents concrete skills to increase their attention to positive behavior and to plan for their response to undesired behavior, has abundant evidence for success for many challenging child behaviors. But sometimes parents have a hard time managing their own emotional responses in the often highly triggering situation of family conflict. Mindfulness has the potential to provide a complement to the PMT skills. Studies are beginning to explore these possibilities.
Case summary
Zoe is a bright 5-year-old who has been “strong willed” and shown intense emotional responses since early in life. The usual 2-year-old temper tantrums increased over time. She has outbursts of yelling, kicking, and hitting, especially with transitions. Her parents tried behavioral parent training, but found it frustrating. If Zoe has been yelling and hitting earlier in the day, her mother feels hurt and angry and can’t bring herself to pay warm attention when Zoe is doing better. When Zoe refuses to pick up her room, her father is flooded with thoughts about his own father hitting him for the slightest disrespect. He thinks that he is a bad, weak father, and sometimes “sees red” and ends up yelling at Zoe instead of putting into place a calm consequence.
Discussion
Mindfulness is defined by Jon Kabat-Zinn as “paying attention in a particular way – on purpose, in the present moment, and nonjudgmentally.” A central feature of mindfulness is strengthening the ability to focus our attention. We learn to pay attention to aspects of the present moment, be that breathing, the sensations in our body, or the experiences of our senses. Often the first skill in behavioral training methods is getting parents to pay attention to their children by participating in child-led play or spending attentive time with older children. This means attending to what the child is doing or talking about rather than jumping in and taking over with suggestions, instructions, or judgments. This meshes very well with this central aspect of mindfulness.
As we practice paying attention, we observe that the mind naturally jumps around from what we mean to be attending to, to a host of distractions, worries, plans, memories, thoughts, and emotions. Mindfulness encourages practitioners to notice these thoughts, to avoid criticizing or judging oneself for becoming involved with these, but instead gently lead the mind back to what you had intended to focus on. This observation of the mind’s activity gives the mindfulness practitioner a bit of space from the thought or emotion itself. We are encouraged to name the thought or emotional processes we notice: “I am worrying, I am planning, I am remembering.”
In the heat of a difficult moment with the child, parents often are flooded with intense emotions (such as anger, fear, anxiety, panic, despair) and thoughts (such as “If my child keeps acting this way he is going to go to jail when he grows up,” “I am a terrible parent,” “Why is my child doing this to me?” or “He is just like his father”). These emotions and thoughts can drive intense, impulsive responses from the parents. As they practice mindfulness, they can gain the ability to observe themselves having these thoughts; observe harsh judgments of themselves or their children or their partners; have some space from them; and realize they may change in a few minutes or realize they may be painful but don’t necessarily have to spur impulsive action. In that moment, parents can give themselves time and space to think through possible actions, and then choose one.
From a behavioral parenting standpoint, we know that parents and humans often react intensely to negative behaviors and inadvertently make them worse with intense emotional reactivity. We want parents to have a plan about how they will respond, to remain calm in the moment, and then put the plan in place. Mindfulness may enhance parents’ ability to notice their own responses and have the space to remember what the plan was and then put it into place. It also can give them space to consider what the child might be experiencing and respond in light of this awareness. This ability does require a significant amount of mindfulness practice.
The combination of mindfulness and parenting is just beginning to be studied in research trials using a range of study designs. Some of these programs have looked at the effect of mindfulness courses, especially mindfulness-based stress reduction without any specific parenting content or indices of parent stress and child behavior. Others have looked at programs which add mindfulness to standard behavioral parenting programs, and still others are specific mindfulness/parenting programs. So far, many of these studies are quasi-experimental in nature. A recent systematic review by Townshend et al. found seven randomized controlled trials of low to moderate quality with some suggestion of ability to decrease parental stress and ADHD symptoms (JBI Database System Rev Implement Rep. 2016 Mar;14[3]:139-80). There is a clear need for randomized controlled trials with larger sample sizes.
While we may not have specific, highly evidence-based mindful parenting programs available, individuals with experience in yoga, meditation, mindfulness, dialectical behavioral therapy, and acceptance and commitment therapy can be encouraged to bring these skills to bear as parents.
Zoe’s parents had pursued outside mindfulness programs. Mindfulness concepts were brought into a standard parenting program. Her parents were encouraged to engage in child-led play with Zoe in a mindful way, fully attending to her actions and experience. Zoe’s parents also were encouraged to observe their own emotional reactions and thoughts in stressful moments and to take a breathing space before taking action.
Dr. Hall is assistant professor of psychiatry and pediatrics at the University of Vermont, Burlington. She said she had no relevant financial disclosures. Email her at [email protected].
Resources
“Mindful Parenting” (New York: Norton & Co., 2015).
“Integrating mindfulness with parent training: Effects of the mindfulness-enhanced strengthening families program” (Dev Psychol. 2015;51[1]:26-35).
“Everyday blessings: The inner work of mindful parenting,” (New York: Hyperion, 1997).
Will a cocaine epidemic follow the opioid crisis?
Ongoing efforts to understand the impact of cocaine on the brain and on behavior have gained considerable momentum over the past few decades. It was only 30 years ago when cocaine was widely considered both safe and nonaddicting – “the champagne” of drugs.1 Progress has been steady since the cocaine-dopamine depletion theory was proposed, and ultimately supported by functional and PET imaging.2,3
Additional discoveries promise further insights into the neuroscience of addiction, pleasure, and mood. While cocaine use, abuse, and dependence might seem relatively quiescent, compared with the scourge of opiate-related deaths and addiction, it remains a public health concern – and now is the second-leading cause of drug deaths. Cocaine cultivation, smuggling, use, and the number of first-time users are all escalating.4
These developments suggest that cocaine problems might get much worse and beg an important question: Will new research give us insight into better solutions?
What the neuroscience shows
As discussed, we already know quite a bit about the neuroscience behind cocaine addiction. The positron emission tomography studies conducted by Nora D. Volkow, MD, and her associates have shown long-lasting changes in abstinent cocaine addicts. Specifically, their findings clearly demonstrated that cocaine changes the brain and depletes dopamine-rich areas. Furthermore, dopamine recovery is negligible after months of abstinence.5
However, large gaps in our understanding remain. The realm of epigenetic study and protein expression behind abuse will be key in bridging our understanding of phenotype to genotype. A recent article by Eric J. Nestler, MD, PhD, and his research team, published in the Journal of Biological Psychiatry and titled “Cocaine self-administration alters transcriptome-wide responses in the brain’s reward circuitry,” offers exciting new insights (Biol Psychiatry. 2018 Apr. doi: 10.1016/jbiopsych.2018.04.009).
The study, led by Deena M. Walker, PhD, offers perhaps the most complete illumination to date of the genetic and epigenetic changes seen in the brain after cocaine self-administration and application.
Dr. Walker and her associates used a mouse model and sorted them into one of several groups. One group examined self-administered acute cocaine exposure only, with the mice immediately harvested thereafter. Two longer-term groups included one that had cocaine exposure with prolonged (30-day) withdrawal followed by context re-exposure (context re-exposure defined as being placed back in the special chamber and lighting they first received cocaine in) and another that had a cocaine exposure with prolonged withdrawal followed by both context and cocaine re-exposure.
The researchers also ran a parallel set of control groups substituting saline for cocaine with otherwise identical durations of observation and context re-exposure. Reward-related brain regions were harvested from each subject and examined with RNA-sequencing analysis to investigate the full genomic/transcriptomic profile of each. Pairwise comparison of the various experimental groups against the control groups (for example, the theoretical baseline of genetic expression in a non–cocaine-exposed brain) uncovered telling patterns, which the investigators aptly described as a comprehensive picture of transcriptome-wide change cocaine causes within the reward circuit.
The novel and creative approach used by Dr. Walker and her associates allowed them to uncover a wealth of clinically significant findings. Much could be said about their spotlighting of specific gene/protein targets for potential future pharmacological therapies toward cocaine treatment – an area that is indeed in sore need of invention. While we have highly efficacious medications for overdose and chronic treatment of opiate abuse, the landscape of treatment options for cocaine is far bleaker and shrouded in theory. With that in mind, perhaps the most salient take-home point is the evidence that cocaine, even after one exposure/withdrawal event, causes a dramatic rewiring in the very way genes are expressed across the reward circuit. The researchers found large shifts in the patterns of genetic transcription, unique and specific to discrete regions of the examined brain tissue, such as the ventral tegmental area, ventral hippocampus, and basolateral amygdala.
More interestingly, similar patterns of these genetic alternations were observed based on the exact history of the cocaine exposure. Dr. Walker and her associates concluded that the withdrawal phase and context re-exposure appear to be crucial components in the re-sculpting of the transcriptomic profile of the reward circuity.
The brain is unprepared by evolution for the reinforcing and reorganizing effects of cocaine. Clinicians, too, have learned that cocaine is addicting and can quickly replace drives such as food, water, sex, and survival. These new data from Dr. Nestler’s team reinforce the importance of prevention. In addition, they are reminders to physicians that cocaine causes changes in brain and behavior that are persistent and not necessarily reversible. Patterns of transcriptomic change are alarming enough and have only recent begun to be fleshed out, but patterns of global substance use trends suggest that we need to begin cocaine prevention activities.
Is another cocaine epidemic inevitable?
The late David F. Musto, MD, who was revered as both expert medical historian and physician at Yale University, New Haven, Conn., offered perhaps the most poignant observation in this regard: He argued that almost every opiate epidemic seems to transition into a psychostimulant epidemic.6 Experts have been looking at cocaine and methamphetamine as a way to try to understand the current opioid epidemic. Indeed, the Centers for Disease Control and Prevention’s most recent report on emerging trends in cocaine use shows numerous, concerning upticks in several realms germane to a possible emerging epidemic. One of the more upstream concerns is a gigantic spike in the shear production of coca leaves and cocaine thought to be occurring in Colombia (the principal source of cocaine in the United States). Current U.S. government estimates based on seizure rates from 2016 indicate that Colombia is producing about 910 metric tons of export quality cocaine. That represents a large increase from the 670-ton estimate the year before and the 325-ton estimate the year before that.
Similarly, a sharp rise in cocaine-related deaths, an approximate 52% increase, has been charted from 2015 to 2016. This finding is likely related to the growing presence of adulterants, such as fentanyl and carfentanil, found in seized cocaine samples. However, a rise in first-time cocaine users in the past year, which, according to the National Survey on Drug Use and Health, is up by about 12% (1.1 million people) in the 2015-2016 period, shows that the danger of cocaine-related deaths might not lie solely in adulteration but also increases in use. These signals might herald a grim return of cocaine to the center stage of public health, a development that would be an encore of the crack cocaine epidemic experienced throughout the 1980s and early 1990s.

All the above findings support cocaine as an agent of swift and massive change to our reward systems that might be poised to again surge across the United States at epidemic levels. Given this insight into just how extensively it rewires brains and the unfortunate truth that direct pharmacotherapy treatments remain mostly theoretical, it is evident that the best course of action is simply to keep cocaine from ever reaching the brain in the first place. Prevention does work, and these findings underline the importance of that message. Direct psychoeducation, awareness programs, and deterrence are the best defense we can offer to our patients at this time. In addition to these tried and true techniques, fascinating new models of prevention for cocaine abuse also are in development: vaccines. Synthesized by binding cocaine to inert proteins, these vaccines are designed to prevent addiction by training the immune system to bind cocaine and thus prevent it from crossing the blood brain barrier.7 Currently approved for clinical study in humans, these might offer a game-changing new method in the prevention of substance abuse.

In summary, continued research has enriched us with a deeper appreciation of just how profoundly cocaine, even after a single exposure, rewires the brain. Some people might have a cavalier attitude about drugs and even use terms such as experimentation to describe teen use, but cocaine is not cannabis. Not only initial cocaine self-administration, but also withdrawal and context of use (a bathroom, a bar table, a countertop) all serve to debase the natural transcriptome balance of the brain’s reward system. Our knowledge of what exactly contributes to the path of the cocaine addiction has grown, but options for how to treat cocaine overdose and addiction remain slim. This is particularly concerning, as history and data indicate a likelihood that a cocaine epidemic might come on the heels of the opiate epidemic. Now more than ever we need to emphasize the importance of preventing cocaine use – and continue to develop new interventions.
Dr. Wenzinger is a clinical fellow, PGY-4, in the department of child and adolescent psychiatry at St. Louis Children’s Hospital. Dr. Gold is the 17th Distinguished Alumni Professor at the University of Florida, Gainesville, and professor of psychiatry (adjunct) at Washington University in St. Louis. He also serves as chairman of the scientific advisory boards for RiverMend Health.
References
1. Yale J Biol Med. 1988 Mar-Apr;61(2):149-55.
2. Neurosci Biobehav Rev. 1985 Fall;9(3):469-77.
3. Am J Psychiatry. 1990;147(6):719-24.
4. DEA Museum. “A New Look at Old and Not So Old Drugs: A 2018 Update on Cocaine.” Drug Enforcement Administration. Retrieved from https://deamuseum.org/lecture-series/new-look-old-not-old-drugs-2018-update-cocaine.
5. J Addict Dis. 1996;15(4):55-71.
6. The American Disease: Origins of Narcotic Control (New York: Oxford University Press, 1999).
7. Br J Clin Pharmacol. 2014 Feb;77(2):368-74.
Ongoing efforts to understand the impact of cocaine on the brain and on behavior have gained considerable momentum over the past few decades. It was only 30 years ago when cocaine was widely considered both safe and nonaddicting – “the champagne” of drugs.1 Progress has been steady since the cocaine-dopamine depletion theory was proposed, and ultimately supported by functional and PET imaging.2,3
Additional discoveries promise further insights into the neuroscience of addiction, pleasure, and mood. While cocaine use, abuse, and dependence might seem relatively quiescent, compared with the scourge of opiate-related deaths and addiction, it remains a public health concern – and now is the second-leading cause of drug deaths. Cocaine cultivation, smuggling, use, and the number of first-time users are all escalating.4
These developments suggest that cocaine problems might get much worse and beg an important question: Will new research give us insight into better solutions?
What the neuroscience shows
As discussed, we already know quite a bit about the neuroscience behind cocaine addiction. The positron emission tomography studies conducted by Nora D. Volkow, MD, and her associates have shown long-lasting changes in abstinent cocaine addicts. Specifically, their findings clearly demonstrated that cocaine changes the brain and depletes dopamine-rich areas. Furthermore, dopamine recovery is negligible after months of abstinence.5
However, large gaps in our understanding remain. The realm of epigenetic study and protein expression behind abuse will be key in bridging our understanding of phenotype to genotype. A recent article by Eric J. Nestler, MD, PhD, and his research team, published in the Journal of Biological Psychiatry and titled “Cocaine self-administration alters transcriptome-wide responses in the brain’s reward circuitry,” offers exciting new insights (Biol Psychiatry. 2018 Apr. doi: 10.1016/jbiopsych.2018.04.009).
The study, led by Deena M. Walker, PhD, offers perhaps the most complete illumination to date of the genetic and epigenetic changes seen in the brain after cocaine self-administration and application.
Dr. Walker and her associates used a mouse model and sorted them into one of several groups. One group examined self-administered acute cocaine exposure only, with the mice immediately harvested thereafter. Two longer-term groups included one that had cocaine exposure with prolonged (30-day) withdrawal followed by context re-exposure (context re-exposure defined as being placed back in the special chamber and lighting they first received cocaine in) and another that had a cocaine exposure with prolonged withdrawal followed by both context and cocaine re-exposure.
The researchers also ran a parallel set of control groups substituting saline for cocaine with otherwise identical durations of observation and context re-exposure. Reward-related brain regions were harvested from each subject and examined with RNA-sequencing analysis to investigate the full genomic/transcriptomic profile of each. Pairwise comparison of the various experimental groups against the control groups (for example, the theoretical baseline of genetic expression in a non–cocaine-exposed brain) uncovered telling patterns, which the investigators aptly described as a comprehensive picture of transcriptome-wide change cocaine causes within the reward circuit.
The novel and creative approach used by Dr. Walker and her associates allowed them to uncover a wealth of clinically significant findings. Much could be said about their spotlighting of specific gene/protein targets for potential future pharmacological therapies toward cocaine treatment – an area that is indeed in sore need of invention. While we have highly efficacious medications for overdose and chronic treatment of opiate abuse, the landscape of treatment options for cocaine is far bleaker and shrouded in theory. With that in mind, perhaps the most salient take-home point is the evidence that cocaine, even after one exposure/withdrawal event, causes a dramatic rewiring in the very way genes are expressed across the reward circuit. The researchers found large shifts in the patterns of genetic transcription, unique and specific to discrete regions of the examined brain tissue, such as the ventral tegmental area, ventral hippocampus, and basolateral amygdala.
More interestingly, similar patterns of these genetic alternations were observed based on the exact history of the cocaine exposure. Dr. Walker and her associates concluded that the withdrawal phase and context re-exposure appear to be crucial components in the re-sculpting of the transcriptomic profile of the reward circuity.
The brain is unprepared by evolution for the reinforcing and reorganizing effects of cocaine. Clinicians, too, have learned that cocaine is addicting and can quickly replace drives such as food, water, sex, and survival. These new data from Dr. Nestler’s team reinforce the importance of prevention. In addition, they are reminders to physicians that cocaine causes changes in brain and behavior that are persistent and not necessarily reversible. Patterns of transcriptomic change are alarming enough and have only recent begun to be fleshed out, but patterns of global substance use trends suggest that we need to begin cocaine prevention activities.
Is another cocaine epidemic inevitable?
The late David F. Musto, MD, who was revered as both expert medical historian and physician at Yale University, New Haven, Conn., offered perhaps the most poignant observation in this regard: He argued that almost every opiate epidemic seems to transition into a psychostimulant epidemic.6 Experts have been looking at cocaine and methamphetamine as a way to try to understand the current opioid epidemic. Indeed, the Centers for Disease Control and Prevention’s most recent report on emerging trends in cocaine use shows numerous, concerning upticks in several realms germane to a possible emerging epidemic. One of the more upstream concerns is a gigantic spike in the shear production of coca leaves and cocaine thought to be occurring in Colombia (the principal source of cocaine in the United States). Current U.S. government estimates based on seizure rates from 2016 indicate that Colombia is producing about 910 metric tons of export quality cocaine. That represents a large increase from the 670-ton estimate the year before and the 325-ton estimate the year before that.
Similarly, a sharp rise in cocaine-related deaths, an approximate 52% increase, has been charted from 2015 to 2016. This finding is likely related to the growing presence of adulterants, such as fentanyl and carfentanil, found in seized cocaine samples. However, a rise in first-time cocaine users in the past year, which, according to the National Survey on Drug Use and Health, is up by about 12% (1.1 million people) in the 2015-2016 period, shows that the danger of cocaine-related deaths might not lie solely in adulteration but also increases in use. These signals might herald a grim return of cocaine to the center stage of public health, a development that would be an encore of the crack cocaine epidemic experienced throughout the 1980s and early 1990s.
All the above findings support cocaine as an agent of swift and massive change to our reward systems that might be poised to again surge across the United States at epidemic levels. Given this insight into just how extensively it rewires brains and the unfortunate truth that direct pharmacotherapy treatments remain mostly theoretical, it is evident that the best course of action is simply to keep cocaine from ever reaching the brain in the first place. Prevention does work, and these findings underline the importance of that message. Direct psychoeducation, awareness programs, and deterrence are the best defense we can offer to our patients at this time. In addition to these tried and true techniques, fascinating new models of prevention for cocaine abuse also are in development: vaccines. Synthesized by binding cocaine to inert proteins, these vaccines are designed to prevent addiction by training the immune system to bind cocaine and thus prevent it from crossing the blood brain barrier.7 Currently approved for clinical study in humans, these might offer a game-changing new method in the prevention of substance abuse.
In summary, continued research has enriched us with a deeper appreciation of just how profoundly cocaine, even after a single exposure, rewires the brain. Some people might have a cavalier attitude about drugs and even use terms such as experimentation to describe teen use, but cocaine is not cannabis. Not only initial cocaine self-administration, but also withdrawal and context of use (a bathroom, a bar table, a countertop) all serve to debase the natural transcriptome balance of the brain’s reward system. Our knowledge of what exactly contributes to the path of the cocaine addiction has grown, but options for how to treat cocaine overdose and addiction remain slim. This is particularly concerning, as history and data indicate a likelihood that a cocaine epidemic might come on the heels of the opiate epidemic. Now more than ever we need to emphasize the importance of preventing cocaine use – and continue to develop new interventions.
Dr. Wenzinger is a clinical fellow, PGY-4, in the department of child and adolescent psychiatry at St. Louis Children’s Hospital. Dr. Gold is the 17th Distinguished Alumni Professor at the University of Florida, Gainesville, and professor of psychiatry (adjunct) at Washington University in St. Louis. He also serves as chairman of the scientific advisory boards for RiverMend Health.
References
1. Yale J Biol Med. 1988 Mar-Apr;61(2):149-55.
2. Neurosci Biobehav Rev. 1985 Fall;9(3):469-77.
3. Am J Psychiatry. 1990;147(6):719-24.
4. DEA Museum. “A New Look at Old and Not So Old Drugs: A 2018 Update on Cocaine.” Drug Enforcement Administration. Retrieved from https://deamuseum.org/lecture-series/new-look-old-not-old-drugs-2018-update-cocaine.
5. J Addict Dis. 1996;15(4):55-71.
6. The American Disease: Origins of Narcotic Control (New York: Oxford University Press, 1999).
7. Br J Clin Pharmacol. 2014 Feb;77(2):368-74.
Ongoing efforts to understand the impact of cocaine on the brain and on behavior have gained considerable momentum over the past few decades. It was only 30 years ago when cocaine was widely considered both safe and nonaddicting – “the champagne” of drugs.1 Progress has been steady since the cocaine-dopamine depletion theory was proposed, and ultimately supported by functional and PET imaging.2,3
Additional discoveries promise further insights into the neuroscience of addiction, pleasure, and mood. While cocaine use, abuse, and dependence might seem relatively quiescent, compared with the scourge of opiate-related deaths and addiction, it remains a public health concern – and now is the second-leading cause of drug deaths. Cocaine cultivation, smuggling, use, and the number of first-time users are all escalating.4
These developments suggest that cocaine problems might get much worse and beg an important question: Will new research give us insight into better solutions?
What the neuroscience shows
As discussed, we already know quite a bit about the neuroscience behind cocaine addiction. The positron emission tomography studies conducted by Nora D. Volkow, MD, and her associates have shown long-lasting changes in abstinent cocaine addicts. Specifically, their findings clearly demonstrated that cocaine changes the brain and depletes dopamine-rich areas. Furthermore, dopamine recovery is negligible after months of abstinence.5
However, large gaps in our understanding remain. The realm of epigenetic study and protein expression behind abuse will be key in bridging our understanding of phenotype to genotype. A recent article by Eric J. Nestler, MD, PhD, and his research team, published in the Journal of Biological Psychiatry and titled “Cocaine self-administration alters transcriptome-wide responses in the brain’s reward circuitry,” offers exciting new insights (Biol Psychiatry. 2018 Apr. doi: 10.1016/jbiopsych.2018.04.009).
The study, led by Deena M. Walker, PhD, offers perhaps the most complete illumination to date of the genetic and epigenetic changes seen in the brain after cocaine self-administration and application.
Dr. Walker and her associates used a mouse model and sorted them into one of several groups. One group examined self-administered acute cocaine exposure only, with the mice immediately harvested thereafter. Two longer-term groups included one that had cocaine exposure with prolonged (30-day) withdrawal followed by context re-exposure (context re-exposure defined as being placed back in the special chamber and lighting they first received cocaine in) and another that had a cocaine exposure with prolonged withdrawal followed by both context and cocaine re-exposure.
The researchers also ran a parallel set of control groups substituting saline for cocaine with otherwise identical durations of observation and context re-exposure. Reward-related brain regions were harvested from each subject and examined with RNA-sequencing analysis to investigate the full genomic/transcriptomic profile of each. Pairwise comparison of the various experimental groups against the control groups (for example, the theoretical baseline of genetic expression in a non–cocaine-exposed brain) uncovered telling patterns, which the investigators aptly described as a comprehensive picture of transcriptome-wide change cocaine causes within the reward circuit.
The novel and creative approach used by Dr. Walker and her associates allowed them to uncover a wealth of clinically significant findings. Much could be said about their spotlighting of specific gene/protein targets for potential future pharmacological therapies toward cocaine treatment – an area that is indeed in sore need of invention. While we have highly efficacious medications for overdose and chronic treatment of opiate abuse, the landscape of treatment options for cocaine is far bleaker and shrouded in theory. With that in mind, perhaps the most salient take-home point is the evidence that cocaine, even after one exposure/withdrawal event, causes a dramatic rewiring in the very way genes are expressed across the reward circuit. The researchers found large shifts in the patterns of genetic transcription, unique and specific to discrete regions of the examined brain tissue, such as the ventral tegmental area, ventral hippocampus, and basolateral amygdala.
More interestingly, similar patterns of these genetic alternations were observed based on the exact history of the cocaine exposure. Dr. Walker and her associates concluded that the withdrawal phase and context re-exposure appear to be crucial components in the re-sculpting of the transcriptomic profile of the reward circuity.
The brain is unprepared by evolution for the reinforcing and reorganizing effects of cocaine. Clinicians, too, have learned that cocaine is addicting and can quickly replace drives such as food, water, sex, and survival. These new data from Dr. Nestler’s team reinforce the importance of prevention. In addition, they are reminders to physicians that cocaine causes changes in brain and behavior that are persistent and not necessarily reversible. Patterns of transcriptomic change are alarming enough and have only recent begun to be fleshed out, but patterns of global substance use trends suggest that we need to begin cocaine prevention activities.
Is another cocaine epidemic inevitable?
The late David F. Musto, MD, who was revered as both expert medical historian and physician at Yale University, New Haven, Conn., offered perhaps the most poignant observation in this regard: He argued that almost every opiate epidemic seems to transition into a psychostimulant epidemic.6 Experts have been looking at cocaine and methamphetamine as a way to try to understand the current opioid epidemic. Indeed, the Centers for Disease Control and Prevention’s most recent report on emerging trends in cocaine use shows numerous, concerning upticks in several realms germane to a possible emerging epidemic. One of the more upstream concerns is a gigantic spike in the shear production of coca leaves and cocaine thought to be occurring in Colombia (the principal source of cocaine in the United States). Current U.S. government estimates based on seizure rates from 2016 indicate that Colombia is producing about 910 metric tons of export quality cocaine. That represents a large increase from the 670-ton estimate the year before and the 325-ton estimate the year before that.
Similarly, a sharp rise in cocaine-related deaths, an approximate 52% increase, has been charted from 2015 to 2016. This finding is likely related to the growing presence of adulterants, such as fentanyl and carfentanil, found in seized cocaine samples. However, a rise in first-time cocaine users in the past year, which, according to the National Survey on Drug Use and Health, is up by about 12% (1.1 million people) in the 2015-2016 period, shows that the danger of cocaine-related deaths might not lie solely in adulteration but also increases in use. These signals might herald a grim return of cocaine to the center stage of public health, a development that would be an encore of the crack cocaine epidemic experienced throughout the 1980s and early 1990s.
All the above findings support cocaine as an agent of swift and massive change to our reward systems that might be poised to again surge across the United States at epidemic levels. Given this insight into just how extensively it rewires brains and the unfortunate truth that direct pharmacotherapy treatments remain mostly theoretical, it is evident that the best course of action is simply to keep cocaine from ever reaching the brain in the first place. Prevention does work, and these findings underline the importance of that message. Direct psychoeducation, awareness programs, and deterrence are the best defense we can offer to our patients at this time. In addition to these tried and true techniques, fascinating new models of prevention for cocaine abuse also are in development: vaccines. Synthesized by binding cocaine to inert proteins, these vaccines are designed to prevent addiction by training the immune system to bind cocaine and thus prevent it from crossing the blood brain barrier.7 Currently approved for clinical study in humans, these might offer a game-changing new method in the prevention of substance abuse.
In summary, continued research has enriched us with a deeper appreciation of just how profoundly cocaine, even after a single exposure, rewires the brain. Some people might have a cavalier attitude about drugs and even use terms such as experimentation to describe teen use, but cocaine is not cannabis. Not only initial cocaine self-administration, but also withdrawal and context of use (a bathroom, a bar table, a countertop) all serve to debase the natural transcriptome balance of the brain’s reward system. Our knowledge of what exactly contributes to the path of the cocaine addiction has grown, but options for how to treat cocaine overdose and addiction remain slim. This is particularly concerning, as history and data indicate a likelihood that a cocaine epidemic might come on the heels of the opiate epidemic. Now more than ever we need to emphasize the importance of preventing cocaine use – and continue to develop new interventions.
Dr. Wenzinger is a clinical fellow, PGY-4, in the department of child and adolescent psychiatry at St. Louis Children’s Hospital. Dr. Gold is the 17th Distinguished Alumni Professor at the University of Florida, Gainesville, and professor of psychiatry (adjunct) at Washington University in St. Louis. He also serves as chairman of the scientific advisory boards for RiverMend Health.
References
1. Yale J Biol Med. 1988 Mar-Apr;61(2):149-55.
2. Neurosci Biobehav Rev. 1985 Fall;9(3):469-77.
3. Am J Psychiatry. 1990;147(6):719-24.
4. DEA Museum. “A New Look at Old and Not So Old Drugs: A 2018 Update on Cocaine.” Drug Enforcement Administration. Retrieved from https://deamuseum.org/lecture-series/new-look-old-not-old-drugs-2018-update-cocaine.
5. J Addict Dis. 1996;15(4):55-71.
6. The American Disease: Origins of Narcotic Control (New York: Oxford University Press, 1999).
7. Br J Clin Pharmacol. 2014 Feb;77(2):368-74.
Are we using the right metrics to measure cesarean rates?
St. Joseph Hospital in Orange, California, like most institutions performing deliveries in 2016, started releasing metrics internally before subsequently releasing them to the public. Data for the first 9 months of 2016 were released. As I am often an outlier, I was gratified to see that I ranked 1st in the vaginal birth after cesarean delivery (VBAC) rate at 36.8% and 4th at 15.9% for my cesarean delivery (CD) rate in the low-risk nulliparous term singleton vertex (NTSV) population.
I have been an avid proponent of VBAC since 1984 when one of the fathers of modern obstetric care, Edward J. Quilligan, MD, presented the benefits and safety of VBAC at our institution.
Experiences that may alter a reported rate
I list here a few circumstances of a CD on maternal request:
- A primagravida with a 10-cm nonphysiologic, nonmalignant ovarian cyst at term elects a primary CD with ovarian cystectomy.
- A woman who is concerned about pelvic organ prolapse and urinary incontinence later in life requests a CD. After all, normal babies do not weigh 5 and 6 lb anymore.
- An elderly primagravida with an in vitro fertilization pregnancy requests a CD.
Should these experiences adversely affect a physician’s statistics? Personally, I don’t think so. Is the morbidity and mortality from a CD really all that much higher than a normal spontaneous vaginal delivery (NSVD)? Granted, the cost is more. But are we really helping all our patients by insisting on a NSVD? Thousands of people have medically indicated and elective surgery in the United States each day.
Of course, these data points depend on the denominator (the number of deliveries attributed to each ObGyn). Those with a contradictory opinion will say that this evens out over time. I dispute that claim. This might be closer to being true for the ObGyn with the highest number, say, 134 in the NTSV denominator versus someone with a low number, such as 4. For VBAC, the denominator range at our institution was 1 to 115 cases.
Rethinking my position
Two recent cases have caused me to rethink my position on using VBAC and CD rates to evaluate ObGyns.
Uterine rupture
A 31-year-old G3P1 woman at 39 6/7 weeks’ gestation was admitted in early labor for a VBAC. She had undergone a CD with her first baby because of fetal intolerance to labor. Her prenatal course was complicated by white-coat hypertension, but I monitored her blood pressure at home and it had been normal. She took aspirin 81 mg during the pregnancy. The fetus was not reactive to a nonstress test on the day of admission.
That evening, amniotomy results showed clear fluid. I placed an intrauterine pressure catheter. The patient’s labor progressed well during the night, she received an epidural anesthetic, and labor was augmented with intravenous oxytocin. She progressed to complete dilation. I was notified of severe, prolonged, variable fetal heart-rate decelerations.
The Laborist who evaluated the patient recommended an emergency CD. I came immediately to Labor and Delivery and performed a CD with delivery of a 7 lb 4 oz infant whose Apgars were 2, 5, and 8 at 1, 5, and 10 minutes, respectively. Arterial cord blood gas tests revealed: pH, 6.94; pCO2, 95 mm Hg; pO2, 19.9 mm Hg; HCO3, 19.9 mmol/L; and base excess (BE), –14.4 mmol/L. Venous cord blood gas tests revealed: pH, 7.25; pCO2, 45 mm Hg; pO2, 35 mm Hg; HCO3, 19.2 mmol/L; BE, −8.0 mmol/L. The cord blood gases revealed that the baby was becoming compromised, but was delivered in time to avoid complications.
After advocating and performing many successful VBACs for 33 years, this was my first uterine rupture.
The uterus had ruptured in the lower segment from the mid-portion extending inferolaterally on the right side and was hemorrhaging. I successfully repaired the rupture. Maternal quantitative blood loss was 1,020 mL.
The baby initially was apneic and was limp. He required continuous positive airway pressure (CPAP) and positive pressure ventilation in the operating room. The baby was transferred to the neonatal intensive care unit (NICU), recovered well, and was discharged home with the mother on the 4th day of life.
Commentary: Why should this necessary, emergency CD count against me on my core measure rate? Although I have advocated for VBACs for 33 years, perhaps they aren’t so safe. After this experience, I do not ever want to have to deal with a ruptured uterus, a compromised baby, and maternal hemorrhage again.
Read Dr. Kanofsky’s solution to using this metric.
Depressed baby
A 24-year-old G1P0 woman at 39 weeks’ gestation was admitted for induction of labor because of mild pregnancy-induced hypertension. Her prenatal course was complicated by Class A1 gestational diabetes mellitus, which was untreated due to compliance issues, Group B streptococcus, and cholelithiasis. Clinically, I suspected she was going to have a large (9 lb) baby. An ultrasound to estimate fetal weight at 37 2/7 weeks’ gestation showed the fetus at 3.937 kg. I was concerned, but, because the mother was 5 ft 5 in tall and weighed 282 lbs, I thought it was reasonable for her to attempt a NSVD.
Induction and labor progressed normally. Her labor curve decelerated at an anterior lip, but subsequently stage 2 progressed normally and lasted 2 hrs. Her temperature was elevated in stage 2 to 100.00F. The fetal heart rate tracings were reassuring.
Immediately after delivery of the fetal vertex, a turtleneck sign was seen and shoulder dystocia occurred. A Wood’s maneuver was performed in both directions, the nurse applied suprapubic pressure, and the infant was delivered. A loose nuchal cord x2 was reduced. The infant was apneic and had no tone. She was taken to the warmer, given oxygen, suctioned, and stimulated until the NICU team arrived. Her Apgar scores were 2, 5, and 9 at 1, 5, and 10 minutes, respectively. The birthweight was 9 lb 0 oz.
A depressed baby of this magnitude was certainly not expected from the FHR tracing or the shoulder dystocia. Venous cord gas evaluation revealed pH, 7.16; pCO2, 57 mm Hg; pO2, 17 mm Hg; HCO3, 20.2 mmol/L; and BE, –19.1 mmol/L.
The baby recovered quickly in the labor and delivery recovery room, went to the NICU on CPAP, subsequently transitioned to room air, and was discharged on the 4th day of life with her mother.
Commentary: Did I do the best I could for this mother and baby? In hindsight, I should have performed a CD because of my concerns for a large fetus. The “retrospectoscope” always makes cases more clear! Note that, if I had performed an elective CD for fetal macrosomia, it would have counted against me on this metric. Prior to labor, if I thought an elective CD was the right approach to this patient, and was providing the best care I could for this mother and fetus, why should it count against me?
Is there a solution?
With my newfound concerns, it is my opinion that VBAC and CD/NTSV rates may not be the correct things to use as quality metric measures without some additional qualifying information.
Better metrics of quality and safety that might be more helpful to measure include:
- Prophylactic oxytocin after delivery of the baby’s anterior shoulder
- Since “6 is the new 4,” in order to increase the NTSV rate, we could measure1:
- patients admitted before active labor
- patients receiving an epidural before active labor.
- Since NTSV is a goal, measure the number of patients in an advanced stage of labor whose labor pattern has become dysfunctional, no interventions are taken, and who subsequently deliver by primary CD.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Committee on Obstetric Practice, American College of Obstetricians and Gynecologists. Committee Opinion No. 687: Approaches to limit intervention during labor and birth. Obstet Gynecol. 2017;129(2):e20–e28.

Dr. Kanofsky is in private ObGyn practice in Orange, California. He is a member of the Perinatal Safety Work Group and the Perinatal Collaborative Committee at St. Joseph Hospital, Orange, and is Chairman, Division of Gynecology, Department of Surgery, at CHOC Children’s Hospital of Orange County.
The author reports that he is a consultant to the St. Joseph Hospital Clinical Information Systems Project.
Dr. Kanofsky is in private ObGyn practice in Orange, California. He is a member of the Perinatal Safety Work Group and the Perinatal Collaborative Committee at St. Joseph Hospital, Orange, and is Chairman, Division of Gynecology, Department of Surgery, at CHOC Children’s Hospital of Orange County.
The author reports that he is a consultant to the St. Joseph Hospital Clinical Information Systems Project.
Dr. Kanofsky is in private ObGyn practice in Orange, California. He is a member of the Perinatal Safety Work Group and the Perinatal Collaborative Committee at St. Joseph Hospital, Orange, and is Chairman, Division of Gynecology, Department of Surgery, at CHOC Children’s Hospital of Orange County.
The author reports that he is a consultant to the St. Joseph Hospital Clinical Information Systems Project.
St. Joseph Hospital in Orange, California, like most institutions performing deliveries in 2016, started releasing metrics internally before subsequently releasing them to the public. Data for the first 9 months of 2016 were released. As I am often an outlier, I was gratified to see that I ranked 1st in the vaginal birth after cesarean delivery (VBAC) rate at 36.8% and 4th at 15.9% for my cesarean delivery (CD) rate in the low-risk nulliparous term singleton vertex (NTSV) population.
I have been an avid proponent of VBAC since 1984 when one of the fathers of modern obstetric care, Edward J. Quilligan, MD, presented the benefits and safety of VBAC at our institution.
Experiences that may alter a reported rate
I list here a few circumstances of a CD on maternal request:
- A primagravida with a 10-cm nonphysiologic, nonmalignant ovarian cyst at term elects a primary CD with ovarian cystectomy.
- A woman who is concerned about pelvic organ prolapse and urinary incontinence later in life requests a CD. After all, normal babies do not weigh 5 and 6 lb anymore.
- An elderly primagravida with an in vitro fertilization pregnancy requests a CD.
Should these experiences adversely affect a physician’s statistics? Personally, I don’t think so. Is the morbidity and mortality from a CD really all that much higher than a normal spontaneous vaginal delivery (NSVD)? Granted, the cost is more. But are we really helping all our patients by insisting on a NSVD? Thousands of people have medically indicated and elective surgery in the United States each day.
Of course, these data points depend on the denominator (the number of deliveries attributed to each ObGyn). Those with a contradictory opinion will say that this evens out over time. I dispute that claim. This might be closer to being true for the ObGyn with the highest number, say, 134 in the NTSV denominator versus someone with a low number, such as 4. For VBAC, the denominator range at our institution was 1 to 115 cases.
Rethinking my position
Two recent cases have caused me to rethink my position on using VBAC and CD rates to evaluate ObGyns.
Uterine rupture
A 31-year-old G3P1 woman at 39 6/7 weeks’ gestation was admitted in early labor for a VBAC. She had undergone a CD with her first baby because of fetal intolerance to labor. Her prenatal course was complicated by white-coat hypertension, but I monitored her blood pressure at home and it had been normal. She took aspirin 81 mg during the pregnancy. The fetus was not reactive to a nonstress test on the day of admission.
That evening, amniotomy results showed clear fluid. I placed an intrauterine pressure catheter. The patient’s labor progressed well during the night, she received an epidural anesthetic, and labor was augmented with intravenous oxytocin. She progressed to complete dilation. I was notified of severe, prolonged, variable fetal heart-rate decelerations.
The Laborist who evaluated the patient recommended an emergency CD. I came immediately to Labor and Delivery and performed a CD with delivery of a 7 lb 4 oz infant whose Apgars were 2, 5, and 8 at 1, 5, and 10 minutes, respectively. Arterial cord blood gas tests revealed: pH, 6.94; pCO2, 95 mm Hg; pO2, 19.9 mm Hg; HCO3, 19.9 mmol/L; and base excess (BE), –14.4 mmol/L. Venous cord blood gas tests revealed: pH, 7.25; pCO2, 45 mm Hg; pO2, 35 mm Hg; HCO3, 19.2 mmol/L; BE, −8.0 mmol/L. The cord blood gases revealed that the baby was becoming compromised, but was delivered in time to avoid complications.
After advocating and performing many successful VBACs for 33 years, this was my first uterine rupture.
The uterus had ruptured in the lower segment from the mid-portion extending inferolaterally on the right side and was hemorrhaging. I successfully repaired the rupture. Maternal quantitative blood loss was 1,020 mL.
The baby initially was apneic and was limp. He required continuous positive airway pressure (CPAP) and positive pressure ventilation in the operating room. The baby was transferred to the neonatal intensive care unit (NICU), recovered well, and was discharged home with the mother on the 4th day of life.
Commentary: Why should this necessary, emergency CD count against me on my core measure rate? Although I have advocated for VBACs for 33 years, perhaps they aren’t so safe. After this experience, I do not ever want to have to deal with a ruptured uterus, a compromised baby, and maternal hemorrhage again.
Read Dr. Kanofsky’s solution to using this metric.
Depressed baby
A 24-year-old G1P0 woman at 39 weeks’ gestation was admitted for induction of labor because of mild pregnancy-induced hypertension. Her prenatal course was complicated by Class A1 gestational diabetes mellitus, which was untreated due to compliance issues, Group B streptococcus, and cholelithiasis. Clinically, I suspected she was going to have a large (9 lb) baby. An ultrasound to estimate fetal weight at 37 2/7 weeks’ gestation showed the fetus at 3.937 kg. I was concerned, but, because the mother was 5 ft 5 in tall and weighed 282 lbs, I thought it was reasonable for her to attempt a NSVD.
Induction and labor progressed normally. Her labor curve decelerated at an anterior lip, but subsequently stage 2 progressed normally and lasted 2 hrs. Her temperature was elevated in stage 2 to 100.00F. The fetal heart rate tracings were reassuring.
Immediately after delivery of the fetal vertex, a turtleneck sign was seen and shoulder dystocia occurred. A Wood’s maneuver was performed in both directions, the nurse applied suprapubic pressure, and the infant was delivered. A loose nuchal cord x2 was reduced. The infant was apneic and had no tone. She was taken to the warmer, given oxygen, suctioned, and stimulated until the NICU team arrived. Her Apgar scores were 2, 5, and 9 at 1, 5, and 10 minutes, respectively. The birthweight was 9 lb 0 oz.
A depressed baby of this magnitude was certainly not expected from the FHR tracing or the shoulder dystocia. Venous cord gas evaluation revealed pH, 7.16; pCO2, 57 mm Hg; pO2, 17 mm Hg; HCO3, 20.2 mmol/L; and BE, –19.1 mmol/L.
The baby recovered quickly in the labor and delivery recovery room, went to the NICU on CPAP, subsequently transitioned to room air, and was discharged on the 4th day of life with her mother.
Commentary: Did I do the best I could for this mother and baby? In hindsight, I should have performed a CD because of my concerns for a large fetus. The “retrospectoscope” always makes cases more clear! Note that, if I had performed an elective CD for fetal macrosomia, it would have counted against me on this metric. Prior to labor, if I thought an elective CD was the right approach to this patient, and was providing the best care I could for this mother and fetus, why should it count against me?
Is there a solution?
With my newfound concerns, it is my opinion that VBAC and CD/NTSV rates may not be the correct things to use as quality metric measures without some additional qualifying information.
Better metrics of quality and safety that might be more helpful to measure include:
- Prophylactic oxytocin after delivery of the baby’s anterior shoulder
- Since “6 is the new 4,” in order to increase the NTSV rate, we could measure1:
- patients admitted before active labor
- patients receiving an epidural before active labor.
- Since NTSV is a goal, measure the number of patients in an advanced stage of labor whose labor pattern has become dysfunctional, no interventions are taken, and who subsequently deliver by primary CD.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
St. Joseph Hospital in Orange, California, like most institutions performing deliveries in 2016, started releasing metrics internally before subsequently releasing them to the public. Data for the first 9 months of 2016 were released. As I am often an outlier, I was gratified to see that I ranked 1st in the vaginal birth after cesarean delivery (VBAC) rate at 36.8% and 4th at 15.9% for my cesarean delivery (CD) rate in the low-risk nulliparous term singleton vertex (NTSV) population.
I have been an avid proponent of VBAC since 1984 when one of the fathers of modern obstetric care, Edward J. Quilligan, MD, presented the benefits and safety of VBAC at our institution.
Experiences that may alter a reported rate
I list here a few circumstances of a CD on maternal request:
- A primagravida with a 10-cm nonphysiologic, nonmalignant ovarian cyst at term elects a primary CD with ovarian cystectomy.
- A woman who is concerned about pelvic organ prolapse and urinary incontinence later in life requests a CD. After all, normal babies do not weigh 5 and 6 lb anymore.
- An elderly primagravida with an in vitro fertilization pregnancy requests a CD.
Should these experiences adversely affect a physician’s statistics? Personally, I don’t think so. Is the morbidity and mortality from a CD really all that much higher than a normal spontaneous vaginal delivery (NSVD)? Granted, the cost is more. But are we really helping all our patients by insisting on a NSVD? Thousands of people have medically indicated and elective surgery in the United States each day.
Of course, these data points depend on the denominator (the number of deliveries attributed to each ObGyn). Those with a contradictory opinion will say that this evens out over time. I dispute that claim. This might be closer to being true for the ObGyn with the highest number, say, 134 in the NTSV denominator versus someone with a low number, such as 4. For VBAC, the denominator range at our institution was 1 to 115 cases.
Rethinking my position
Two recent cases have caused me to rethink my position on using VBAC and CD rates to evaluate ObGyns.
Uterine rupture
A 31-year-old G3P1 woman at 39 6/7 weeks’ gestation was admitted in early labor for a VBAC. She had undergone a CD with her first baby because of fetal intolerance to labor. Her prenatal course was complicated by white-coat hypertension, but I monitored her blood pressure at home and it had been normal. She took aspirin 81 mg during the pregnancy. The fetus was not reactive to a nonstress test on the day of admission.
That evening, amniotomy results showed clear fluid. I placed an intrauterine pressure catheter. The patient’s labor progressed well during the night, she received an epidural anesthetic, and labor was augmented with intravenous oxytocin. She progressed to complete dilation. I was notified of severe, prolonged, variable fetal heart-rate decelerations.
The Laborist who evaluated the patient recommended an emergency CD. I came immediately to Labor and Delivery and performed a CD with delivery of a 7 lb 4 oz infant whose Apgars were 2, 5, and 8 at 1, 5, and 10 minutes, respectively. Arterial cord blood gas tests revealed: pH, 6.94; pCO2, 95 mm Hg; pO2, 19.9 mm Hg; HCO3, 19.9 mmol/L; and base excess (BE), –14.4 mmol/L. Venous cord blood gas tests revealed: pH, 7.25; pCO2, 45 mm Hg; pO2, 35 mm Hg; HCO3, 19.2 mmol/L; BE, −8.0 mmol/L. The cord blood gases revealed that the baby was becoming compromised, but was delivered in time to avoid complications.
After advocating and performing many successful VBACs for 33 years, this was my first uterine rupture.
The uterus had ruptured in the lower segment from the mid-portion extending inferolaterally on the right side and was hemorrhaging. I successfully repaired the rupture. Maternal quantitative blood loss was 1,020 mL.
The baby initially was apneic and was limp. He required continuous positive airway pressure (CPAP) and positive pressure ventilation in the operating room. The baby was transferred to the neonatal intensive care unit (NICU), recovered well, and was discharged home with the mother on the 4th day of life.
Commentary: Why should this necessary, emergency CD count against me on my core measure rate? Although I have advocated for VBACs for 33 years, perhaps they aren’t so safe. After this experience, I do not ever want to have to deal with a ruptured uterus, a compromised baby, and maternal hemorrhage again.
Read Dr. Kanofsky’s solution to using this metric.
Depressed baby
A 24-year-old G1P0 woman at 39 weeks’ gestation was admitted for induction of labor because of mild pregnancy-induced hypertension. Her prenatal course was complicated by Class A1 gestational diabetes mellitus, which was untreated due to compliance issues, Group B streptococcus, and cholelithiasis. Clinically, I suspected she was going to have a large (9 lb) baby. An ultrasound to estimate fetal weight at 37 2/7 weeks’ gestation showed the fetus at 3.937 kg. I was concerned, but, because the mother was 5 ft 5 in tall and weighed 282 lbs, I thought it was reasonable for her to attempt a NSVD.
Induction and labor progressed normally. Her labor curve decelerated at an anterior lip, but subsequently stage 2 progressed normally and lasted 2 hrs. Her temperature was elevated in stage 2 to 100.00F. The fetal heart rate tracings were reassuring.
Immediately after delivery of the fetal vertex, a turtleneck sign was seen and shoulder dystocia occurred. A Wood’s maneuver was performed in both directions, the nurse applied suprapubic pressure, and the infant was delivered. A loose nuchal cord x2 was reduced. The infant was apneic and had no tone. She was taken to the warmer, given oxygen, suctioned, and stimulated until the NICU team arrived. Her Apgar scores were 2, 5, and 9 at 1, 5, and 10 minutes, respectively. The birthweight was 9 lb 0 oz.
A depressed baby of this magnitude was certainly not expected from the FHR tracing or the shoulder dystocia. Venous cord gas evaluation revealed pH, 7.16; pCO2, 57 mm Hg; pO2, 17 mm Hg; HCO3, 20.2 mmol/L; and BE, –19.1 mmol/L.
The baby recovered quickly in the labor and delivery recovery room, went to the NICU on CPAP, subsequently transitioned to room air, and was discharged on the 4th day of life with her mother.
Commentary: Did I do the best I could for this mother and baby? In hindsight, I should have performed a CD because of my concerns for a large fetus. The “retrospectoscope” always makes cases more clear! Note that, if I had performed an elective CD for fetal macrosomia, it would have counted against me on this metric. Prior to labor, if I thought an elective CD was the right approach to this patient, and was providing the best care I could for this mother and fetus, why should it count against me?
Is there a solution?
With my newfound concerns, it is my opinion that VBAC and CD/NTSV rates may not be the correct things to use as quality metric measures without some additional qualifying information.
Better metrics of quality and safety that might be more helpful to measure include:
- Prophylactic oxytocin after delivery of the baby’s anterior shoulder
- Since “6 is the new 4,” in order to increase the NTSV rate, we could measure1:
- patients admitted before active labor
- patients receiving an epidural before active labor.
- Since NTSV is a goal, measure the number of patients in an advanced stage of labor whose labor pattern has become dysfunctional, no interventions are taken, and who subsequently deliver by primary CD.
Share your thoughts! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
- Committee on Obstetric Practice, American College of Obstetricians and Gynecologists. Committee Opinion No. 687: Approaches to limit intervention during labor and birth. Obstet Gynecol. 2017;129(2):e20–e28.
- Committee on Obstetric Practice, American College of Obstetricians and Gynecologists. Committee Opinion No. 687: Approaches to limit intervention during labor and birth. Obstet Gynecol. 2017;129(2):e20–e28.
Is the rise of suicide a medical or societal issue?
It’s hard to ignore the recent report from the Centers for Disease Control and Prevention revealing suicide rates went up by more than 30% in half of states since 1999 (“Suicide rates rising across the U.S.,” CDC Newsroom, June 7, 2018). My professional experience with suicide is limited to one former patient who died several years after aging out of my practice. His death was not a total surprise. The cornerstones of the situation he felt he couldn’t escape were well in place when he was a freshman in high school. I’m sure there was more I could have tried to do at that early stage. Although he was anxious and mildly depressed he never admitted to being suicidal.

Just as in Ms. Roberts’ family, the CDC report has elicited a fresh round of finger pointing and introspection in our country at a time when it is already struggling to find a sense of its own identity. Is it too many guns? Or too few mental health professionals? Or a broken health delivery system?
Dr. Richard A. Friedman, a psychiatrist at Weill Cornell Medical College, writes in the New York Times that “suicide is a medical problem” and that we should declare war on it “as we’ve done with other public health threats like HIV and heart disease” (“Suicide Rates Are Rising. What Should We Do About It?” June 11, 2018). Although it may be contagious with outbreaks and clusters, particularly in the wake of celebrity suicides (“The Science Behind Suicide Contagion,” by Margot Sanger-Katz, The New York Times, Aug 13, 2014), I’m not so sure that suicide is a medical problem. Certainly, it can be spread by a vector, in this case the ubiquitous news media. With more exposure, suicide has become if not the norm, at least in certain subgroups a socially acceptable management option for an unhappy life. But while there are other features of suicide that tempt us to retreat into our comfort zone of the medical model, we need to face the more realistic and unsettling explanation that the increase in the suicide rate is the symptom of a sick society.

We already are making a mistake by interpreting the apparent rise in distractible behavior as a disease that requires medication. Let’s spread a broader net as we search for answers to the alarming suicide death statistics.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
It’s hard to ignore the recent report from the Centers for Disease Control and Prevention revealing suicide rates went up by more than 30% in half of states since 1999 (“Suicide rates rising across the U.S.,” CDC Newsroom, June 7, 2018). My professional experience with suicide is limited to one former patient who died several years after aging out of my practice. His death was not a total surprise. The cornerstones of the situation he felt he couldn’t escape were well in place when he was a freshman in high school. I’m sure there was more I could have tried to do at that early stage. Although he was anxious and mildly depressed he never admitted to being suicidal.
Just as in Ms. Roberts’ family, the CDC report has elicited a fresh round of finger pointing and introspection in our country at a time when it is already struggling to find a sense of its own identity. Is it too many guns? Or too few mental health professionals? Or a broken health delivery system?
Dr. Richard A. Friedman, a psychiatrist at Weill Cornell Medical College, writes in the New York Times that “suicide is a medical problem” and that we should declare war on it “as we’ve done with other public health threats like HIV and heart disease” (“Suicide Rates Are Rising. What Should We Do About It?” June 11, 2018). Although it may be contagious with outbreaks and clusters, particularly in the wake of celebrity suicides (“The Science Behind Suicide Contagion,” by Margot Sanger-Katz, The New York Times, Aug 13, 2014), I’m not so sure that suicide is a medical problem. Certainly, it can be spread by a vector, in this case the ubiquitous news media. With more exposure, suicide has become if not the norm, at least in certain subgroups a socially acceptable management option for an unhappy life. But while there are other features of suicide that tempt us to retreat into our comfort zone of the medical model, we need to face the more realistic and unsettling explanation that the increase in the suicide rate is the symptom of a sick society.
We already are making a mistake by interpreting the apparent rise in distractible behavior as a disease that requires medication. Let’s spread a broader net as we search for answers to the alarming suicide death statistics.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].
It’s hard to ignore the recent report from the Centers for Disease Control and Prevention revealing suicide rates went up by more than 30% in half of states since 1999 (“Suicide rates rising across the U.S.,” CDC Newsroom, June 7, 2018). My professional experience with suicide is limited to one former patient who died several years after aging out of my practice. His death was not a total surprise. The cornerstones of the situation he felt he couldn’t escape were well in place when he was a freshman in high school. I’m sure there was more I could have tried to do at that early stage. Although he was anxious and mildly depressed he never admitted to being suicidal.
Just as in Ms. Roberts’ family, the CDC report has elicited a fresh round of finger pointing and introspection in our country at a time when it is already struggling to find a sense of its own identity. Is it too many guns? Or too few mental health professionals? Or a broken health delivery system?
Dr. Richard A. Friedman, a psychiatrist at Weill Cornell Medical College, writes in the New York Times that “suicide is a medical problem” and that we should declare war on it “as we’ve done with other public health threats like HIV and heart disease” (“Suicide Rates Are Rising. What Should We Do About It?” June 11, 2018). Although it may be contagious with outbreaks and clusters, particularly in the wake of celebrity suicides (“The Science Behind Suicide Contagion,” by Margot Sanger-Katz, The New York Times, Aug 13, 2014), I’m not so sure that suicide is a medical problem. Certainly, it can be spread by a vector, in this case the ubiquitous news media. With more exposure, suicide has become if not the norm, at least in certain subgroups a socially acceptable management option for an unhappy life. But while there are other features of suicide that tempt us to retreat into our comfort zone of the medical model, we need to face the more realistic and unsettling explanation that the increase in the suicide rate is the symptom of a sick society.
We already are making a mistake by interpreting the apparent rise in distractible behavior as a disease that requires medication. Let’s spread a broader net as we search for answers to the alarming suicide death statistics.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at [email protected].