User login
Once-weekly insulin data published; could alter treatment
Phase 2 data for the investigational, once-weekly basal insulin analog icodec (Novo Nordisk) showing comparable efficacy and safety to once-daily insulin glargine U100 have been published in the New England Journal of Medicine.
“Insulin icodec could potentially improve acceptance and likely would facilitate management in type 2 diabetes patients needing basal insulin, and I think it will be transformational in the way we manage people with type 2 diabetes requiring insulin,” said lead author Julio Rosenstock, MD, University of Texas Southwestern Medical Center, Dallas, who also presented the data at the virtual annual meeting of the European Association for the Study of Diabetes.
Insulin icodec binds to albumin to create a circulating depot with a 196-hour (8.1 days) half-life, so the once-weekly injection is designed to cover an individual’s basal insulin requirements for a full week, with steady insulin release. Because of its concentrated formulation, its injection volume is equivalent to that of daily glargine U100.
In the 26-week, randomized, phase 2 trial involving 247 insulin-naive patients with type 2 diabetes, once-weekly icodec’s glucose-lowering and safety profiles were similar to those of once-daily insulin glargine U100. These results were previously presented by Dr. Rosenstock in June at the virtual American Diabetes Association conference, as reported by Medscape Medical News.
In addition, new data in a poster at EASD 2020 showed that switching to icodec from other basal insulins is efficacious without causing significant hypoglycemia, as reported by Harpreet Bajaj, MD, MPH, director of the Leadership Sinai Centre for Diabetes, Mount Sinai Hospital, Toronto.
Charles M. Alexander, MD, an endocrinologist and managing director of Alexander Associates, Gwynedd Valley, Pa., said in an interview that “some patients will find once-weekly basal insulin an attractive option, while other patients will be indifferent to its availability.”
Dr. Alexander also pointed out that “payers are not going to be very interested in paying for a once-weekly basal insulin when daily basal insulins have been available for many years, unless the cost is the same or less. Resource-constrained health plans will wait until the price is [similar].”
The phase 2 study: Once weekly is just as good as daily
In the phase 2, randomized, double-blind, double-dummy, parallel-group, treat-to-target trial, the patients had baseline hemoglobin A1c levels of 7.0%-9.5% despite taking metformin, with or without a dipeptidyl peptidase–4 inhibitor.
They were randomized to weekly insulin icodec plus daily placebo (n = 125) or daily insulin glargine U100 plus weekly placebo (n = 122). The primary endpoint, change in A1c from baseline to week 26, dropped 1.33 percentage points with icodec and 1.15 percentage points with glargine, down to 6.7% and 6.9%, respectively. The difference wasn’t significant (P = .08). Fasting plasma glucose levels dropped by 58 mg/dL with icodec and 54 mg/dL with glargine (P = .34).
Time in range (70-140 mg/dL or 3.9-7.8 mmol/L) as assessed by flash glucose monitoring (FreeStyle Libre Pro) was greater with Icodec, by 5.4 percentage points, corresponding to an extra 78 minutes per day in range.
Mild hypoglycemia was more common with icodec than glargine (509 vs. 211 events per 100 patient-years, but rates of moderate/clinically significant hypoglycemia (52.5 vs. 46 per 100 patient-years, respectively) and severe hypoglycemia (1.4 vs. 0 per 100 patient-years) did not differ significantly (P = .85).
And the duration of hypoglycemia wasn’t longer with icodec, compared with glargine, despite its longer duration of action, Dr. Rosenstock emphasized.
Rates of other adverse events were similar between the groups.
Use of a once-weekly basal insulin could reduce the number of annual insulin injections from 365 to just 52, the authors noted in their paper.
New data: Switching to icodec is effective, safe
The new data on switching came from a 16-week, open-label, phase 2 trial of 154 patients with type 2 diabetes with insufficient glycemic control (mean A1c 7.9%) while taking oral medication and basal insulin. They were randomized to once-weekly icodec with or without an initial loading dose, or once-daily glargine U100.
Insulin doses were titrated weekly based on blood glucose levels as measured by continuous glucose monitoring (Dexcom G6).
The primary endpoint, time in range (70-180 mg/dL or 3.9-10.0 mmol/L) during weeks 15-16 was significantly better for icodec plus loading dose, compared with glargine U100 (72.9% vs 65.0%, P = .01) and similar between icodec and glargine U100 (66.0% vs 65.0%, P = .75).
Estimated mean percentage point reductions in A1c were 0.77 for icodec plus loading dose, 0.47 for icodec without the loading dose, and 0.54 for glargine U100.
Rates of moderate to severe hypoglycemia were similar between icodec plus loading dose and glargine U100 (78.0 and 79.4 events per 100 patient-years, respectively), and lower for icodec without the loading dose (14.8/100 patient-years).
There were no unexpected safety findings.
Novo Nordisk’s phase 3 trial for icodec is set to begin in late November.
The company is also developing a coformulation of icodec with its glucagonlike peptide–1 receptor agonist semaglutide, currently in phase 1 testing. Meanwhile, Eli Lilly is also developing a once-weekly basal analog, LY3209590, currently in phase 2 trials.
Dr. Rosenstock reported receiving research support from, being on advisory boards for, and/or receiving consulting honoraria from Merck, Pfizer, Sanofi, Novo Nordisk, Eli Lilly, GlaxoSmithKline, AstraZeneca, Janssen, Genentech, Oramed, Boehringer Ingelheim, Applied Therapeutics, and Intarcia. Dr. Alexander reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Phase 2 data for the investigational, once-weekly basal insulin analog icodec (Novo Nordisk) showing comparable efficacy and safety to once-daily insulin glargine U100 have been published in the New England Journal of Medicine.
“Insulin icodec could potentially improve acceptance and likely would facilitate management in type 2 diabetes patients needing basal insulin, and I think it will be transformational in the way we manage people with type 2 diabetes requiring insulin,” said lead author Julio Rosenstock, MD, University of Texas Southwestern Medical Center, Dallas, who also presented the data at the virtual annual meeting of the European Association for the Study of Diabetes.
Insulin icodec binds to albumin to create a circulating depot with a 196-hour (8.1 days) half-life, so the once-weekly injection is designed to cover an individual’s basal insulin requirements for a full week, with steady insulin release. Because of its concentrated formulation, its injection volume is equivalent to that of daily glargine U100.
In the 26-week, randomized, phase 2 trial involving 247 insulin-naive patients with type 2 diabetes, once-weekly icodec’s glucose-lowering and safety profiles were similar to those of once-daily insulin glargine U100. These results were previously presented by Dr. Rosenstock in June at the virtual American Diabetes Association conference, as reported by Medscape Medical News.
In addition, new data in a poster at EASD 2020 showed that switching to icodec from other basal insulins is efficacious without causing significant hypoglycemia, as reported by Harpreet Bajaj, MD, MPH, director of the Leadership Sinai Centre for Diabetes, Mount Sinai Hospital, Toronto.
Charles M. Alexander, MD, an endocrinologist and managing director of Alexander Associates, Gwynedd Valley, Pa., said in an interview that “some patients will find once-weekly basal insulin an attractive option, while other patients will be indifferent to its availability.”
Dr. Alexander also pointed out that “payers are not going to be very interested in paying for a once-weekly basal insulin when daily basal insulins have been available for many years, unless the cost is the same or less. Resource-constrained health plans will wait until the price is [similar].”
The phase 2 study: Once weekly is just as good as daily
In the phase 2, randomized, double-blind, double-dummy, parallel-group, treat-to-target trial, the patients had baseline hemoglobin A1c levels of 7.0%-9.5% despite taking metformin, with or without a dipeptidyl peptidase–4 inhibitor.
They were randomized to weekly insulin icodec plus daily placebo (n = 125) or daily insulin glargine U100 plus weekly placebo (n = 122). The primary endpoint, change in A1c from baseline to week 26, dropped 1.33 percentage points with icodec and 1.15 percentage points with glargine, down to 6.7% and 6.9%, respectively. The difference wasn’t significant (P = .08). Fasting plasma glucose levels dropped by 58 mg/dL with icodec and 54 mg/dL with glargine (P = .34).
Time in range (70-140 mg/dL or 3.9-7.8 mmol/L) as assessed by flash glucose monitoring (FreeStyle Libre Pro) was greater with Icodec, by 5.4 percentage points, corresponding to an extra 78 minutes per day in range.
Mild hypoglycemia was more common with icodec than glargine (509 vs. 211 events per 100 patient-years, but rates of moderate/clinically significant hypoglycemia (52.5 vs. 46 per 100 patient-years, respectively) and severe hypoglycemia (1.4 vs. 0 per 100 patient-years) did not differ significantly (P = .85).
And the duration of hypoglycemia wasn’t longer with icodec, compared with glargine, despite its longer duration of action, Dr. Rosenstock emphasized.
Rates of other adverse events were similar between the groups.
Use of a once-weekly basal insulin could reduce the number of annual insulin injections from 365 to just 52, the authors noted in their paper.
New data: Switching to icodec is effective, safe
The new data on switching came from a 16-week, open-label, phase 2 trial of 154 patients with type 2 diabetes with insufficient glycemic control (mean A1c 7.9%) while taking oral medication and basal insulin. They were randomized to once-weekly icodec with or without an initial loading dose, or once-daily glargine U100.
Insulin doses were titrated weekly based on blood glucose levels as measured by continuous glucose monitoring (Dexcom G6).
The primary endpoint, time in range (70-180 mg/dL or 3.9-10.0 mmol/L) during weeks 15-16 was significantly better for icodec plus loading dose, compared with glargine U100 (72.9% vs 65.0%, P = .01) and similar between icodec and glargine U100 (66.0% vs 65.0%, P = .75).
Estimated mean percentage point reductions in A1c were 0.77 for icodec plus loading dose, 0.47 for icodec without the loading dose, and 0.54 for glargine U100.
Rates of moderate to severe hypoglycemia were similar between icodec plus loading dose and glargine U100 (78.0 and 79.4 events per 100 patient-years, respectively), and lower for icodec without the loading dose (14.8/100 patient-years).
There were no unexpected safety findings.
Novo Nordisk’s phase 3 trial for icodec is set to begin in late November.
The company is also developing a coformulation of icodec with its glucagonlike peptide–1 receptor agonist semaglutide, currently in phase 1 testing. Meanwhile, Eli Lilly is also developing a once-weekly basal analog, LY3209590, currently in phase 2 trials.
Dr. Rosenstock reported receiving research support from, being on advisory boards for, and/or receiving consulting honoraria from Merck, Pfizer, Sanofi, Novo Nordisk, Eli Lilly, GlaxoSmithKline, AstraZeneca, Janssen, Genentech, Oramed, Boehringer Ingelheim, Applied Therapeutics, and Intarcia. Dr. Alexander reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
Phase 2 data for the investigational, once-weekly basal insulin analog icodec (Novo Nordisk) showing comparable efficacy and safety to once-daily insulin glargine U100 have been published in the New England Journal of Medicine.
“Insulin icodec could potentially improve acceptance and likely would facilitate management in type 2 diabetes patients needing basal insulin, and I think it will be transformational in the way we manage people with type 2 diabetes requiring insulin,” said lead author Julio Rosenstock, MD, University of Texas Southwestern Medical Center, Dallas, who also presented the data at the virtual annual meeting of the European Association for the Study of Diabetes.
Insulin icodec binds to albumin to create a circulating depot with a 196-hour (8.1 days) half-life, so the once-weekly injection is designed to cover an individual’s basal insulin requirements for a full week, with steady insulin release. Because of its concentrated formulation, its injection volume is equivalent to that of daily glargine U100.
In the 26-week, randomized, phase 2 trial involving 247 insulin-naive patients with type 2 diabetes, once-weekly icodec’s glucose-lowering and safety profiles were similar to those of once-daily insulin glargine U100. These results were previously presented by Dr. Rosenstock in June at the virtual American Diabetes Association conference, as reported by Medscape Medical News.
In addition, new data in a poster at EASD 2020 showed that switching to icodec from other basal insulins is efficacious without causing significant hypoglycemia, as reported by Harpreet Bajaj, MD, MPH, director of the Leadership Sinai Centre for Diabetes, Mount Sinai Hospital, Toronto.
Charles M. Alexander, MD, an endocrinologist and managing director of Alexander Associates, Gwynedd Valley, Pa., said in an interview that “some patients will find once-weekly basal insulin an attractive option, while other patients will be indifferent to its availability.”
Dr. Alexander also pointed out that “payers are not going to be very interested in paying for a once-weekly basal insulin when daily basal insulins have been available for many years, unless the cost is the same or less. Resource-constrained health plans will wait until the price is [similar].”
The phase 2 study: Once weekly is just as good as daily
In the phase 2, randomized, double-blind, double-dummy, parallel-group, treat-to-target trial, the patients had baseline hemoglobin A1c levels of 7.0%-9.5% despite taking metformin, with or without a dipeptidyl peptidase–4 inhibitor.
They were randomized to weekly insulin icodec plus daily placebo (n = 125) or daily insulin glargine U100 plus weekly placebo (n = 122). The primary endpoint, change in A1c from baseline to week 26, dropped 1.33 percentage points with icodec and 1.15 percentage points with glargine, down to 6.7% and 6.9%, respectively. The difference wasn’t significant (P = .08). Fasting plasma glucose levels dropped by 58 mg/dL with icodec and 54 mg/dL with glargine (P = .34).
Time in range (70-140 mg/dL or 3.9-7.8 mmol/L) as assessed by flash glucose monitoring (FreeStyle Libre Pro) was greater with Icodec, by 5.4 percentage points, corresponding to an extra 78 minutes per day in range.
Mild hypoglycemia was more common with icodec than glargine (509 vs. 211 events per 100 patient-years, but rates of moderate/clinically significant hypoglycemia (52.5 vs. 46 per 100 patient-years, respectively) and severe hypoglycemia (1.4 vs. 0 per 100 patient-years) did not differ significantly (P = .85).
And the duration of hypoglycemia wasn’t longer with icodec, compared with glargine, despite its longer duration of action, Dr. Rosenstock emphasized.
Rates of other adverse events were similar between the groups.
Use of a once-weekly basal insulin could reduce the number of annual insulin injections from 365 to just 52, the authors noted in their paper.
New data: Switching to icodec is effective, safe
The new data on switching came from a 16-week, open-label, phase 2 trial of 154 patients with type 2 diabetes with insufficient glycemic control (mean A1c 7.9%) while taking oral medication and basal insulin. They were randomized to once-weekly icodec with or without an initial loading dose, or once-daily glargine U100.
Insulin doses were titrated weekly based on blood glucose levels as measured by continuous glucose monitoring (Dexcom G6).
The primary endpoint, time in range (70-180 mg/dL or 3.9-10.0 mmol/L) during weeks 15-16 was significantly better for icodec plus loading dose, compared with glargine U100 (72.9% vs 65.0%, P = .01) and similar between icodec and glargine U100 (66.0% vs 65.0%, P = .75).
Estimated mean percentage point reductions in A1c were 0.77 for icodec plus loading dose, 0.47 for icodec without the loading dose, and 0.54 for glargine U100.
Rates of moderate to severe hypoglycemia were similar between icodec plus loading dose and glargine U100 (78.0 and 79.4 events per 100 patient-years, respectively), and lower for icodec without the loading dose (14.8/100 patient-years).
There were no unexpected safety findings.
Novo Nordisk’s phase 3 trial for icodec is set to begin in late November.
The company is also developing a coformulation of icodec with its glucagonlike peptide–1 receptor agonist semaglutide, currently in phase 1 testing. Meanwhile, Eli Lilly is also developing a once-weekly basal analog, LY3209590, currently in phase 2 trials.
Dr. Rosenstock reported receiving research support from, being on advisory boards for, and/or receiving consulting honoraria from Merck, Pfizer, Sanofi, Novo Nordisk, Eli Lilly, GlaxoSmithKline, AstraZeneca, Janssen, Genentech, Oramed, Boehringer Ingelheim, Applied Therapeutics, and Intarcia. Dr. Alexander reported no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
FROM EASD 2020
Pharmacologic Management of COPD
A Discussion of the new American Thoracic Society Clinical Practice Guideline
Chronic obstructive pulmonary disease (COPD) is caused by airway and alveolar abnormalities and is the third most common cause of death worldwide. COPD results in airflow obstruction that is not fully reversible. The diagnosis of COPD should be considered in patients over 40 years who have chronic cough and/or dyspnea, particularly if they have a history of tobacco use. The diagnosis is confirmed by a diminished forced expiratory volume in 1 second (FEV1) that is not fully reversible with the use of a bronchodilator and an FEV1/forced vital capacity ratio of less than or equal to 0.7.1
Recommendation 1

Patients with COPD who report dyspnea or exercise intolerance should be treated with both a long-acting muscarinic antagonist (LAMA) and a long-acting beta agonist (LABA) (dual LAMA/LABA therapy) instead of monotherapy, the guideline says.
This recommendation represents a critical change in care and is based on strong evidence. For years practitioners have been using single bronchodilator therapy, often a LAMA as the entrance to treatment for patients with symptomatic COPD. The recommendation to begin treatment with dual bronchodilator therapy is an important one. This is the only recommendation that received a “strong” grade.
The evidence comes from the compilation of 24 randomized controlled trials that altogether included 45,441 patients. Dual therapy versus monotherapy was evaluated by examining differences in dyspnea, health-related quality of life, exacerbations (which were defined as requiring antibiotics, oral steroids, or hospitalizations), and hospitalizations independently. Marked improvements were observed for exacerbations and hospitalizations in the dual LAMA/LABA group, compared with treatment with use of a single bronchodilator. In 22,733 patients across 15 RCTs, there were 88 fewer exacerbations per 1,000 patients with a rate ratio (RR) of 0.80 (P < .002), the guideline states.
The decrease in exacerbations is a critical factor in treating patients with COPD because each exacerbation can lead to a sustained decrease in airflow and increases the risk of future exacerbations.
Recommendation 2

In COPD patients who report dyspnea or exercise intolerance, with an exacerbation in the last year, the guideline recommends triple therapy with an inhaled corticosteroid (ICS) instead of just dual LAMA/LABA therapy.
In the past many clinicians have relegated triple therapy to a “last ditch resort.” This recommendation makes it clear that triple therapy is appropriate for a broad range of patients with moderate to severe COPD.
Recommendation 3
In patients with COPD who are on triple therapy, the inhaled corticosteroid component can be withdrawn if patients have not had an exacerbation within the last year, according to the guideline.
It should be noted that the committee said that the ICS can be withdrawn, not that it necessarily needs to be withdrawn. The data showed that it would be safe to withdraw the ICS, but the data is limited in time to 1 year’s follow-up.
Recommendation 4
ATS was not able to make a recommendation for or against ICS as an additive therapy to LAMA/LABA in those without an exacerbation and elevated blood eosinophilia (defined as ≥2% blood eosinophils or >149 cell/mcL). In those with at least one exacerbation and increased blood eosinophilia, the society does recommend addition of ICS to dual LAMA/LABA therapy.
An area of ongoing discussion is at what point in disease severity, before exacerbations occur, might ICS be useful in preventing a first exacerbation. This awaits further studies and evidence.
Recommendation 5
In COPD patients with frequent and severe exacerbations who are otherwise medically optimized, the ATS advises against the use of maintenance oral corticosteroid therapy.
It has been known and accepted for years that oral steroids should be avoided if at all possible because they have little benefit and can cause significant harm. The guideline reinforces this.
The Bottom Line
Dual LAMA/LABA therapy in symptomatic patients is the standard of care. If a patient has had an exacerbation within the last year, add an ICS to the LAMA/LABA, most conveniently given in the form of triple therapy in one inhaler. Finally, even in refractory COPD, maintenance oral corticosteroids bring more harm than benefit.
Dr. Skolnik is professor of family and community medicine at the Thomas Jefferson University, Philadelphia, and associate director of the Family Medicine Residency Program at Abington (Pa.) Jefferson Health. Dr. Matthews is a second-year resident in the family medicine residency program at Abington Jefferson Health.
References
1. Wells C, Joo MJ. COPD and asthma: Diagnostic accuracy requires spirometry. J Fam Pract. 2019;68(2):76-81.
2. Nici L, Mammen MJ, Charbek E, et al. Pharmacologic management of chronic obstructive pulmonary disease. An official American Thoracic Society clinical practice guideline. Am J Respir Crit Care Med. 2020;201(9):e56-69.
A Discussion of the new American Thoracic Society Clinical Practice Guideline
A Discussion of the new American Thoracic Society Clinical Practice Guideline
Chronic obstructive pulmonary disease (COPD) is caused by airway and alveolar abnormalities and is the third most common cause of death worldwide. COPD results in airflow obstruction that is not fully reversible. The diagnosis of COPD should be considered in patients over 40 years who have chronic cough and/or dyspnea, particularly if they have a history of tobacco use. The diagnosis is confirmed by a diminished forced expiratory volume in 1 second (FEV1) that is not fully reversible with the use of a bronchodilator and an FEV1/forced vital capacity ratio of less than or equal to 0.7.1
Recommendation 1
Patients with COPD who report dyspnea or exercise intolerance should be treated with both a long-acting muscarinic antagonist (LAMA) and a long-acting beta agonist (LABA) (dual LAMA/LABA therapy) instead of monotherapy, the guideline says.
This recommendation represents a critical change in care and is based on strong evidence. For years practitioners have been using single bronchodilator therapy, often a LAMA as the entrance to treatment for patients with symptomatic COPD. The recommendation to begin treatment with dual bronchodilator therapy is an important one. This is the only recommendation that received a “strong” grade.
The evidence comes from the compilation of 24 randomized controlled trials that altogether included 45,441 patients. Dual therapy versus monotherapy was evaluated by examining differences in dyspnea, health-related quality of life, exacerbations (which were defined as requiring antibiotics, oral steroids, or hospitalizations), and hospitalizations independently. Marked improvements were observed for exacerbations and hospitalizations in the dual LAMA/LABA group, compared with treatment with use of a single bronchodilator. In 22,733 patients across 15 RCTs, there were 88 fewer exacerbations per 1,000 patients with a rate ratio (RR) of 0.80 (P < .002), the guideline states.
The decrease in exacerbations is a critical factor in treating patients with COPD because each exacerbation can lead to a sustained decrease in airflow and increases the risk of future exacerbations.
Recommendation 2
In COPD patients who report dyspnea or exercise intolerance, with an exacerbation in the last year, the guideline recommends triple therapy with an inhaled corticosteroid (ICS) instead of just dual LAMA/LABA therapy.
In the past many clinicians have relegated triple therapy to a “last ditch resort.” This recommendation makes it clear that triple therapy is appropriate for a broad range of patients with moderate to severe COPD.
Recommendation 3
In patients with COPD who are on triple therapy, the inhaled corticosteroid component can be withdrawn if patients have not had an exacerbation within the last year, according to the guideline.
It should be noted that the committee said that the ICS can be withdrawn, not that it necessarily needs to be withdrawn. The data showed that it would be safe to withdraw the ICS, but the data is limited in time to 1 year’s follow-up.
Recommendation 4
ATS was not able to make a recommendation for or against ICS as an additive therapy to LAMA/LABA in those without an exacerbation and elevated blood eosinophilia (defined as ≥2% blood eosinophils or >149 cell/mcL). In those with at least one exacerbation and increased blood eosinophilia, the society does recommend addition of ICS to dual LAMA/LABA therapy.
An area of ongoing discussion is at what point in disease severity, before exacerbations occur, might ICS be useful in preventing a first exacerbation. This awaits further studies and evidence.
Recommendation 5
In COPD patients with frequent and severe exacerbations who are otherwise medically optimized, the ATS advises against the use of maintenance oral corticosteroid therapy.
It has been known and accepted for years that oral steroids should be avoided if at all possible because they have little benefit and can cause significant harm. The guideline reinforces this.
The Bottom Line
Dual LAMA/LABA therapy in symptomatic patients is the standard of care. If a patient has had an exacerbation within the last year, add an ICS to the LAMA/LABA, most conveniently given in the form of triple therapy in one inhaler. Finally, even in refractory COPD, maintenance oral corticosteroids bring more harm than benefit.
Dr. Skolnik is professor of family and community medicine at the Thomas Jefferson University, Philadelphia, and associate director of the Family Medicine Residency Program at Abington (Pa.) Jefferson Health. Dr. Matthews is a second-year resident in the family medicine residency program at Abington Jefferson Health.
References
1. Wells C, Joo MJ. COPD and asthma: Diagnostic accuracy requires spirometry. J Fam Pract. 2019;68(2):76-81.
2. Nici L, Mammen MJ, Charbek E, et al. Pharmacologic management of chronic obstructive pulmonary disease. An official American Thoracic Society clinical practice guideline. Am J Respir Crit Care Med. 2020;201(9):e56-69.
Chronic obstructive pulmonary disease (COPD) is caused by airway and alveolar abnormalities and is the third most common cause of death worldwide. COPD results in airflow obstruction that is not fully reversible. The diagnosis of COPD should be considered in patients over 40 years who have chronic cough and/or dyspnea, particularly if they have a history of tobacco use. The diagnosis is confirmed by a diminished forced expiratory volume in 1 second (FEV1) that is not fully reversible with the use of a bronchodilator and an FEV1/forced vital capacity ratio of less than or equal to 0.7.1
Recommendation 1
Patients with COPD who report dyspnea or exercise intolerance should be treated with both a long-acting muscarinic antagonist (LAMA) and a long-acting beta agonist (LABA) (dual LAMA/LABA therapy) instead of monotherapy, the guideline says.
This recommendation represents a critical change in care and is based on strong evidence. For years practitioners have been using single bronchodilator therapy, often a LAMA as the entrance to treatment for patients with symptomatic COPD. The recommendation to begin treatment with dual bronchodilator therapy is an important one. This is the only recommendation that received a “strong” grade.
The evidence comes from the compilation of 24 randomized controlled trials that altogether included 45,441 patients. Dual therapy versus monotherapy was evaluated by examining differences in dyspnea, health-related quality of life, exacerbations (which were defined as requiring antibiotics, oral steroids, or hospitalizations), and hospitalizations independently. Marked improvements were observed for exacerbations and hospitalizations in the dual LAMA/LABA group, compared with treatment with use of a single bronchodilator. In 22,733 patients across 15 RCTs, there were 88 fewer exacerbations per 1,000 patients with a rate ratio (RR) of 0.80 (P < .002), the guideline states.
The decrease in exacerbations is a critical factor in treating patients with COPD because each exacerbation can lead to a sustained decrease in airflow and increases the risk of future exacerbations.
Recommendation 2
In COPD patients who report dyspnea or exercise intolerance, with an exacerbation in the last year, the guideline recommends triple therapy with an inhaled corticosteroid (ICS) instead of just dual LAMA/LABA therapy.
In the past many clinicians have relegated triple therapy to a “last ditch resort.” This recommendation makes it clear that triple therapy is appropriate for a broad range of patients with moderate to severe COPD.
Recommendation 3
In patients with COPD who are on triple therapy, the inhaled corticosteroid component can be withdrawn if patients have not had an exacerbation within the last year, according to the guideline.
It should be noted that the committee said that the ICS can be withdrawn, not that it necessarily needs to be withdrawn. The data showed that it would be safe to withdraw the ICS, but the data is limited in time to 1 year’s follow-up.
Recommendation 4
ATS was not able to make a recommendation for or against ICS as an additive therapy to LAMA/LABA in those without an exacerbation and elevated blood eosinophilia (defined as ≥2% blood eosinophils or >149 cell/mcL). In those with at least one exacerbation and increased blood eosinophilia, the society does recommend addition of ICS to dual LAMA/LABA therapy.
An area of ongoing discussion is at what point in disease severity, before exacerbations occur, might ICS be useful in preventing a first exacerbation. This awaits further studies and evidence.
Recommendation 5
In COPD patients with frequent and severe exacerbations who are otherwise medically optimized, the ATS advises against the use of maintenance oral corticosteroid therapy.
It has been known and accepted for years that oral steroids should be avoided if at all possible because they have little benefit and can cause significant harm. The guideline reinforces this.
The Bottom Line
Dual LAMA/LABA therapy in symptomatic patients is the standard of care. If a patient has had an exacerbation within the last year, add an ICS to the LAMA/LABA, most conveniently given in the form of triple therapy in one inhaler. Finally, even in refractory COPD, maintenance oral corticosteroids bring more harm than benefit.
Dr. Skolnik is professor of family and community medicine at the Thomas Jefferson University, Philadelphia, and associate director of the Family Medicine Residency Program at Abington (Pa.) Jefferson Health. Dr. Matthews is a second-year resident in the family medicine residency program at Abington Jefferson Health.
References
1. Wells C, Joo MJ. COPD and asthma: Diagnostic accuracy requires spirometry. J Fam Pract. 2019;68(2):76-81.
2. Nici L, Mammen MJ, Charbek E, et al. Pharmacologic management of chronic obstructive pulmonary disease. An official American Thoracic Society clinical practice guideline. Am J Respir Crit Care Med. 2020;201(9):e56-69.
If PPIs are onboard, atezolizumab may not work for bladder cancer
according to a post hoc analysis of 1,360 subjects from two atezolizumab trials.
Proton pump inhibitor (PPI) use was associated with worse overall and progression-free survival among patients on atezolizumab, but there was no such association in a matched cohort receiving chemotherapy alone. In short, concomitant “PPI users had no atezolizumab benefit,” wrote the investigators led by Ashley Hopkins, PhD, a research fellow at Flinders University in Adelaide, Australia.
This is the first time that PPI use has been shown to be an independent prognostic factor for worse survival in this setting with atezolizumab use – but not with chemotherapy, wrote the authors of the study, published online in Clinical Cancer Research.
“PPIs are overused, or inappropriately used, in patients with cancer by up to 50%, seemingly from a perspective that they will cause no harm. The findings from this study suggest that noncritical PPI use needs to be approached very cautiously, particularly when an immune checkpoint inhibitor is being used to treat urothelial cancer,” Hopkins said in a press release.
Although about one third of cancer patients use PPIs, there has been growing evidence that the changes they induce in the gut microbiome impact immune checkpoint inhibitor (ICI) effectiveness. A similar study of pooled trial data recently found that PPIs, as well as antibiotics, were associated with worse survival in advanced non–small cell lung cancer treated with atezolizumab, while no such tie was found with chemotherapy (Ann Oncol. 2020;31:525-31. doi: 10.1016/j.annonc.2020.01.006).
The mechanism is uncertain. PPIs have been associated with T-cell tolerance, pharmacokinetic changes, and decreased gut microbiota diversity. High diversity, the investigators noted, has been associated with stronger ICI responses in melanoma. Antibiotics have been associated with similar gut dysbiosis.
“It is increasingly evident that altered gut microbiota impacts homeostasis, immune response, cancer prognosis, and ICI efficacy. The hypothetical basis of [our] research is that PPIs are associated with marked changes to the gut microbiota, driven by both altered stomach acidity and direct compound effects, and these changes may impact immunotherapy,” Hopkins said in an email to Medscape.
The associations with urothelial cancer hadn’t been investigated before, so Hopkins and his team pooled patient-level data from the single-arm IMvigor210 trial of atezolizumab for urothelial cancer and the randomized IMvigor211 trial, which pitted atezolizumab against chemotherapy for the indication.
The investigators compared the outcomes of the 471 subjects who were on a PPI from 30 days before to 30 days after starting atezolizumab with the outcomes of 889 subjects who were not on a PPI. Findings were adjusted for tumor histology and the number of prior treatments and metastases sites, as well as age, body mass index, performance status, and other potential confounders.
PPI use was associated with markedly worse overall survival (hazard ratio, 1.52; 95% confidence interval, 1.27-1.83; P < .001) and progression-free survival (HR, 1.38; 95% CI, 1.18-1.62; P < .001) in patients on atezolizumab but not chemotherapy. PPI use was also associated with worse objective response to the ICI (HR, 0.51; 95% CI, 0.32-0.82; P = .006).
In the randomized trial, atezolizumab seemed to offer no overall survival benefit versus chemotherapy when PPIs were onboard (HR, 1.04; 95% CI, 0.81-1.34), but atezolizumab offered a substantial benefit when PPIs were not in use (HR, 0.69; 95% CI, 0.56-0.84). Findings were consistent when limited to the PD-L1 IC2/3 population.
It seems that PPIs negate “the magnitude of atezolizumab efficacy,” the investigators wrote.
Concomitant antibiotics made the effect of PPIs on overall survival with atezolizumab even worse (antibiotics plus PPI: HR 2.51; 95% CI, 1.12-5.59; versus no antibiotics with PPI: HR, 1.44; 95% CI, 1.19-1.74).
The investigators cautioned that, although “the conducted analyses have been adjusted, there is the potential that PPI use constitutes a surrogate marker for an unfit or immunodeficient patient.” They called for further investigation with other ICIs, cancer types, and chemotherapy regimens.
The dose and compliance with PPI therapy were unknown, but the team noted that over 90% of the PPI subjects were on PPIs for long-term reasons, most commonly gastric protection and gastroesophageal reflux disease (GERD). Omeprazole, pantoprazole, and esomeprazole were the most frequently used.
There were no significant associations between PPI use and the first occurrence of atezolizumab-induced adverse events.
The study was funded by the National Breast Cancer Foundation (Australia) and the Cancer Council South Australia. Hopkins has disclosed no relevant financial relationships. Multiple study authors have financial ties to industry, including makers of ICIs. The full list can be found with the original article.
This article first appeared on Medscape.com.
according to a post hoc analysis of 1,360 subjects from two atezolizumab trials.
Proton pump inhibitor (PPI) use was associated with worse overall and progression-free survival among patients on atezolizumab, but there was no such association in a matched cohort receiving chemotherapy alone. In short, concomitant “PPI users had no atezolizumab benefit,” wrote the investigators led by Ashley Hopkins, PhD, a research fellow at Flinders University in Adelaide, Australia.
This is the first time that PPI use has been shown to be an independent prognostic factor for worse survival in this setting with atezolizumab use – but not with chemotherapy, wrote the authors of the study, published online in Clinical Cancer Research.
“PPIs are overused, or inappropriately used, in patients with cancer by up to 50%, seemingly from a perspective that they will cause no harm. The findings from this study suggest that noncritical PPI use needs to be approached very cautiously, particularly when an immune checkpoint inhibitor is being used to treat urothelial cancer,” Hopkins said in a press release.
Although about one third of cancer patients use PPIs, there has been growing evidence that the changes they induce in the gut microbiome impact immune checkpoint inhibitor (ICI) effectiveness. A similar study of pooled trial data recently found that PPIs, as well as antibiotics, were associated with worse survival in advanced non–small cell lung cancer treated with atezolizumab, while no such tie was found with chemotherapy (Ann Oncol. 2020;31:525-31. doi: 10.1016/j.annonc.2020.01.006).
The mechanism is uncertain. PPIs have been associated with T-cell tolerance, pharmacokinetic changes, and decreased gut microbiota diversity. High diversity, the investigators noted, has been associated with stronger ICI responses in melanoma. Antibiotics have been associated with similar gut dysbiosis.
“It is increasingly evident that altered gut microbiota impacts homeostasis, immune response, cancer prognosis, and ICI efficacy. The hypothetical basis of [our] research is that PPIs are associated with marked changes to the gut microbiota, driven by both altered stomach acidity and direct compound effects, and these changes may impact immunotherapy,” Hopkins said in an email to Medscape.
The associations with urothelial cancer hadn’t been investigated before, so Hopkins and his team pooled patient-level data from the single-arm IMvigor210 trial of atezolizumab for urothelial cancer and the randomized IMvigor211 trial, which pitted atezolizumab against chemotherapy for the indication.
The investigators compared the outcomes of the 471 subjects who were on a PPI from 30 days before to 30 days after starting atezolizumab with the outcomes of 889 subjects who were not on a PPI. Findings were adjusted for tumor histology and the number of prior treatments and metastases sites, as well as age, body mass index, performance status, and other potential confounders.
PPI use was associated with markedly worse overall survival (hazard ratio, 1.52; 95% confidence interval, 1.27-1.83; P < .001) and progression-free survival (HR, 1.38; 95% CI, 1.18-1.62; P < .001) in patients on atezolizumab but not chemotherapy. PPI use was also associated with worse objective response to the ICI (HR, 0.51; 95% CI, 0.32-0.82; P = .006).
In the randomized trial, atezolizumab seemed to offer no overall survival benefit versus chemotherapy when PPIs were onboard (HR, 1.04; 95% CI, 0.81-1.34), but atezolizumab offered a substantial benefit when PPIs were not in use (HR, 0.69; 95% CI, 0.56-0.84). Findings were consistent when limited to the PD-L1 IC2/3 population.
It seems that PPIs negate “the magnitude of atezolizumab efficacy,” the investigators wrote.
Concomitant antibiotics made the effect of PPIs on overall survival with atezolizumab even worse (antibiotics plus PPI: HR 2.51; 95% CI, 1.12-5.59; versus no antibiotics with PPI: HR, 1.44; 95% CI, 1.19-1.74).
The investigators cautioned that, although “the conducted analyses have been adjusted, there is the potential that PPI use constitutes a surrogate marker for an unfit or immunodeficient patient.” They called for further investigation with other ICIs, cancer types, and chemotherapy regimens.
The dose and compliance with PPI therapy were unknown, but the team noted that over 90% of the PPI subjects were on PPIs for long-term reasons, most commonly gastric protection and gastroesophageal reflux disease (GERD). Omeprazole, pantoprazole, and esomeprazole were the most frequently used.
There were no significant associations between PPI use and the first occurrence of atezolizumab-induced adverse events.
The study was funded by the National Breast Cancer Foundation (Australia) and the Cancer Council South Australia. Hopkins has disclosed no relevant financial relationships. Multiple study authors have financial ties to industry, including makers of ICIs. The full list can be found with the original article.
This article first appeared on Medscape.com.
according to a post hoc analysis of 1,360 subjects from two atezolizumab trials.
Proton pump inhibitor (PPI) use was associated with worse overall and progression-free survival among patients on atezolizumab, but there was no such association in a matched cohort receiving chemotherapy alone. In short, concomitant “PPI users had no atezolizumab benefit,” wrote the investigators led by Ashley Hopkins, PhD, a research fellow at Flinders University in Adelaide, Australia.
This is the first time that PPI use has been shown to be an independent prognostic factor for worse survival in this setting with atezolizumab use – but not with chemotherapy, wrote the authors of the study, published online in Clinical Cancer Research.
“PPIs are overused, or inappropriately used, in patients with cancer by up to 50%, seemingly from a perspective that they will cause no harm. The findings from this study suggest that noncritical PPI use needs to be approached very cautiously, particularly when an immune checkpoint inhibitor is being used to treat urothelial cancer,” Hopkins said in a press release.
Although about one third of cancer patients use PPIs, there has been growing evidence that the changes they induce in the gut microbiome impact immune checkpoint inhibitor (ICI) effectiveness. A similar study of pooled trial data recently found that PPIs, as well as antibiotics, were associated with worse survival in advanced non–small cell lung cancer treated with atezolizumab, while no such tie was found with chemotherapy (Ann Oncol. 2020;31:525-31. doi: 10.1016/j.annonc.2020.01.006).
The mechanism is uncertain. PPIs have been associated with T-cell tolerance, pharmacokinetic changes, and decreased gut microbiota diversity. High diversity, the investigators noted, has been associated with stronger ICI responses in melanoma. Antibiotics have been associated with similar gut dysbiosis.
“It is increasingly evident that altered gut microbiota impacts homeostasis, immune response, cancer prognosis, and ICI efficacy. The hypothetical basis of [our] research is that PPIs are associated with marked changes to the gut microbiota, driven by both altered stomach acidity and direct compound effects, and these changes may impact immunotherapy,” Hopkins said in an email to Medscape.
The associations with urothelial cancer hadn’t been investigated before, so Hopkins and his team pooled patient-level data from the single-arm IMvigor210 trial of atezolizumab for urothelial cancer and the randomized IMvigor211 trial, which pitted atezolizumab against chemotherapy for the indication.
The investigators compared the outcomes of the 471 subjects who were on a PPI from 30 days before to 30 days after starting atezolizumab with the outcomes of 889 subjects who were not on a PPI. Findings were adjusted for tumor histology and the number of prior treatments and metastases sites, as well as age, body mass index, performance status, and other potential confounders.
PPI use was associated with markedly worse overall survival (hazard ratio, 1.52; 95% confidence interval, 1.27-1.83; P < .001) and progression-free survival (HR, 1.38; 95% CI, 1.18-1.62; P < .001) in patients on atezolizumab but not chemotherapy. PPI use was also associated with worse objective response to the ICI (HR, 0.51; 95% CI, 0.32-0.82; P = .006).
In the randomized trial, atezolizumab seemed to offer no overall survival benefit versus chemotherapy when PPIs were onboard (HR, 1.04; 95% CI, 0.81-1.34), but atezolizumab offered a substantial benefit when PPIs were not in use (HR, 0.69; 95% CI, 0.56-0.84). Findings were consistent when limited to the PD-L1 IC2/3 population.
It seems that PPIs negate “the magnitude of atezolizumab efficacy,” the investigators wrote.
Concomitant antibiotics made the effect of PPIs on overall survival with atezolizumab even worse (antibiotics plus PPI: HR 2.51; 95% CI, 1.12-5.59; versus no antibiotics with PPI: HR, 1.44; 95% CI, 1.19-1.74).
The investigators cautioned that, although “the conducted analyses have been adjusted, there is the potential that PPI use constitutes a surrogate marker for an unfit or immunodeficient patient.” They called for further investigation with other ICIs, cancer types, and chemotherapy regimens.
The dose and compliance with PPI therapy were unknown, but the team noted that over 90% of the PPI subjects were on PPIs for long-term reasons, most commonly gastric protection and gastroesophageal reflux disease (GERD). Omeprazole, pantoprazole, and esomeprazole were the most frequently used.
There were no significant associations between PPI use and the first occurrence of atezolizumab-induced adverse events.
The study was funded by the National Breast Cancer Foundation (Australia) and the Cancer Council South Australia. Hopkins has disclosed no relevant financial relationships. Multiple study authors have financial ties to industry, including makers of ICIs. The full list can be found with the original article.
This article first appeared on Medscape.com.
More U.S. states cap insulin cost, but activists will ‘fight harder’
Twelve U.S. states have now passed laws aimed at making insulin more affordable – and more than 30 are considering such legislation – but they all have gaps that still put the cost of this basic and essential medication out of reach for many with diabetes.
The laws only apply to health insurance through state-regulated plans, and not to the majority of health plans that cover most Americans: Medicare, Medicaid, the Veterans Affairs health system, or self-funded employer-sponsored plans.
Overall, Hannah Crabtree, an activist who writes the blog Data for Insulin, estimates state laws that limit copays, deductibles, or other out-of-pocket costs for insulin cover an average of 27% of people with diabetes across the United States.
And while diabetes activists have applauded state actions, most want more help for the under- and uninsured.
“Our chapter will be fighting harder next legislative session for the uninsured,” said Mindie Hooley, the leader of the Utah #insulin4all chapter, which successfully lobbied legislators to pass a bill signed by the state’s governor on March 30.
“With so many losing their jobs because of the pandemic, there’s no better time than now to fight for these patients who don’t have insurance,” Ms. Hooley said in an interview.
The American Diabetes Association has also been lobbying for state caps as one of many avenues for making insulin more affordable, said Stephen Habbe, the ADA’s director for state government affairs.
One in four insulin users report rationing the medication, Mr. Habbe said.
The state laws “can really provide important relief in terms of affordability for their insulin costs, which we know can be critical in terms of preserving their life and helping to prevent complications that can potentially be disabling or even deadly,” he said in an interview.
Activists with T1 International, which created the #insulin4all campaign, are working nationwide to convince state legislators to back measures that limit out-of-pocket costs for insulin, or for other diabetes medications and supplies.
Colorado, Connecticut, Delaware, Illinois, Maine, New Hampshire, New Mexico, New York, Utah, Virginia, Washington, and West Virginia have enacted such limits, with caps ranging from $25 to $100.
Insulin makers unfazed, blame insurers, PBMs for high prices
The three insulin manufacturers in the United States – Eli Lilly, Novo Nordisk, and Sanofi– have not overtly fought against the laws, although in July, the Pharmaceutical Research and Manufacturers of America did sue to block a related Minnesota law that provides a free emergency supply of insulin.
And the nonprofit news organization FairWarning reported in August that a lobbyist from Eli Lilly had attempted to push a Tennessee legislator to keep the uninsured from being eligible for any out-of-pocket limits.
The insulin makers have also not lowered prices in response to the mounting number of state laws.
They see no need, said Tara O’Neill Hayes, director of human welfare policy at the American Action Forum, a center right–leaning Washington, D.C., think tank.
“You’re going to do what you can get away with,” Ms. O’Neill Hayes said in an interview. “To the extent that they can keep their prices high and people are still buying, they have limited incentives to lower those costs.”
The insulin market is dysfunctional, she added. “The increasing cost of insulin seems primarily to be the result of a lack of competition in the market and convoluted drug pricing and insurance practices,” Ms. O’Neill Hayes and colleagues wrote in a report in April on federal and state attempts to address insulin affordability.
Novo Nordisk, however, maintains that drugmakers are not solely to blame.
“Everyone in the health care system has a role to play in affordability,” said Ken Inchausti, Novo Nordisk’s senior director for corporate communications. State legislation “attempts to address a systemic issue in [U.S.] health care: How benefit design can make medicines unaffordable for many, especially for those in high-deductible health plans,” he said in an interview.
“Efforts to place copay caps on insurance plans covering insulin can certainly help lower out-of-pocket costs,” said Mr. Inchausti.
Sanofi spokesperson Jon Florio said the company supports actions that increase affordable access to insulin. However, “while we support capped copays, we feel this should not be limited to just one class of medicines,” he said. Mr. Florio also noted that Sanofi provides out-of-pocket caps to anyone with commercial insurance and that anyone without insurance can buy one or multiple Sanofi insulins for a fixed price of $99 per month, up to 10 boxes of pens and/or 10-mL vials.
And Sanofi will take part in the Centers for Medicare & Medicaid Services’ new insulin demonstration program. Starting in 2021, CMS will cap insulin copays at $35 for people in Part D plans that participate.
Eli Lilly spokesperson Brad Jacklin said the company “believes in the common goal of ensuring affordable access to insulin and other life-saving medicines because nobody should have to forgo or ration because of cost.”
Lilly supports efforts “that more directly affect patients’ cost-sharing based on their health care coverage,” he said. Insurers and pharmacy benefit managers (PBMs) should pass savings on to patients, Mr. Jacklin urged. Lilly caps some insulins at $35 for the uninsured or commercially insured. The company will also participate in the CMS program.
Meanwhile, a PhRMA-sponsored website www.letstalkaboutcost.org said that, because they do not share savings, insurers and PBMs are responsible for high insulin costs.
Manufacturer assistance programs for patients with diabetes and other chronic diseases, on the other hand, can save individuals $300-$500 a year, PhRMA said in August.
PBMs point back at insulin manufacturers
PBMs, however, point back at drug companies. “PBMs have been able to moderate insulin costs for most consumers with insurance,” said J.C. Scott, president and CEO of the Pharmaceutical Care Management Association, the PBM trade group, in a statement.
The rising cost of insulin is caused by a lack of competition and overuse of patent extensions, PCMA maintains.
Health insurers, which, in tandem with PBMs, give insulins formulary preference based on a discounted price, are most likely to feel the impact of laws limiting out-of-pocket costs.
If they have to make up the shortfall from a patient’s reduced payment for a prescription, they will likely raise premiums, said Ms. O’Neill Hayes.
And if patients pay the same price for insulin – regardless of who makes it – drugmakers won’t have much incentive to offer discounts or rebates for formulary placement, she said. Again, that would likely lead to higher premiums.
David Allen, a spokesperson for America’s Health Insurance Plans, said in an interview that AHIP believes lack of competition has driven up insulin prices.
“High prices for insulin correspond with high health insurance costs for insulin,” he said. When CMS starts requiring drugmakers to discount their insulins for Medicare that will allow “health plans to use those savings to reduce out-of-pocket [costs] for seniors.”
He did not respond to a question as to why health insurers were not already passing savings on to commercially insured patients, especially in states with out-of-pocket limits.
Mr. Allen did say that AHIP’s plans “stand ready to work with state policymakers to remove barriers to lower insulin prices for Americans.”
Utah savings hopefully saving lives already
In Utah, legislators tuned out the blame game, and instead were keen to listen to patients, who had many stories about how the high cost of insulin had hurt them, said Ms. Hooley.
She noted an estimated 50,000 Utahans rely on insulin to stay alive.
Ms. Hooley and her chapter convinced legislators to pass a bill that gives insurers the option to cap patient copays at $30 per month, or to put insulin on its lowest formulary tier and waive any patient deductible. That aspect of the law does not go into effect until January 2021, but insurers are already starting to move insulin to the lowest formulary tier.
That has helped some people immediately. One state resident said her most recent insulin prescription cost $7 – instead of the usual $200.
The uninsured are not left totally high and dry either. Starting June 1, anyone in the state could buy through a state bulk-purchasing program, which guaranteed a 60% discount.
Ms. Hooley said she’d recently heard about a patient who usually spent $300 per prescription but was able to buy insulin for $100 through the program.
“Although $100 is still too much, it is nice knowing the Utah Insulin Savings Program is saving lives,” Ms. Hooley concluded.
A version of this article originally appeared on Medscape.com.
Twelve U.S. states have now passed laws aimed at making insulin more affordable – and more than 30 are considering such legislation – but they all have gaps that still put the cost of this basic and essential medication out of reach for many with diabetes.
The laws only apply to health insurance through state-regulated plans, and not to the majority of health plans that cover most Americans: Medicare, Medicaid, the Veterans Affairs health system, or self-funded employer-sponsored plans.
Overall, Hannah Crabtree, an activist who writes the blog Data for Insulin, estimates state laws that limit copays, deductibles, or other out-of-pocket costs for insulin cover an average of 27% of people with diabetes across the United States.
And while diabetes activists have applauded state actions, most want more help for the under- and uninsured.
“Our chapter will be fighting harder next legislative session for the uninsured,” said Mindie Hooley, the leader of the Utah #insulin4all chapter, which successfully lobbied legislators to pass a bill signed by the state’s governor on March 30.
“With so many losing their jobs because of the pandemic, there’s no better time than now to fight for these patients who don’t have insurance,” Ms. Hooley said in an interview.
The American Diabetes Association has also been lobbying for state caps as one of many avenues for making insulin more affordable, said Stephen Habbe, the ADA’s director for state government affairs.
One in four insulin users report rationing the medication, Mr. Habbe said.
The state laws “can really provide important relief in terms of affordability for their insulin costs, which we know can be critical in terms of preserving their life and helping to prevent complications that can potentially be disabling or even deadly,” he said in an interview.
Activists with T1 International, which created the #insulin4all campaign, are working nationwide to convince state legislators to back measures that limit out-of-pocket costs for insulin, or for other diabetes medications and supplies.
Colorado, Connecticut, Delaware, Illinois, Maine, New Hampshire, New Mexico, New York, Utah, Virginia, Washington, and West Virginia have enacted such limits, with caps ranging from $25 to $100.
Insulin makers unfazed, blame insurers, PBMs for high prices
The three insulin manufacturers in the United States – Eli Lilly, Novo Nordisk, and Sanofi– have not overtly fought against the laws, although in July, the Pharmaceutical Research and Manufacturers of America did sue to block a related Minnesota law that provides a free emergency supply of insulin.
And the nonprofit news organization FairWarning reported in August that a lobbyist from Eli Lilly had attempted to push a Tennessee legislator to keep the uninsured from being eligible for any out-of-pocket limits.
The insulin makers have also not lowered prices in response to the mounting number of state laws.
They see no need, said Tara O’Neill Hayes, director of human welfare policy at the American Action Forum, a center right–leaning Washington, D.C., think tank.
“You’re going to do what you can get away with,” Ms. O’Neill Hayes said in an interview. “To the extent that they can keep their prices high and people are still buying, they have limited incentives to lower those costs.”
The insulin market is dysfunctional, she added. “The increasing cost of insulin seems primarily to be the result of a lack of competition in the market and convoluted drug pricing and insurance practices,” Ms. O’Neill Hayes and colleagues wrote in a report in April on federal and state attempts to address insulin affordability.
Novo Nordisk, however, maintains that drugmakers are not solely to blame.
“Everyone in the health care system has a role to play in affordability,” said Ken Inchausti, Novo Nordisk’s senior director for corporate communications. State legislation “attempts to address a systemic issue in [U.S.] health care: How benefit design can make medicines unaffordable for many, especially for those in high-deductible health plans,” he said in an interview.
“Efforts to place copay caps on insurance plans covering insulin can certainly help lower out-of-pocket costs,” said Mr. Inchausti.
Sanofi spokesperson Jon Florio said the company supports actions that increase affordable access to insulin. However, “while we support capped copays, we feel this should not be limited to just one class of medicines,” he said. Mr. Florio also noted that Sanofi provides out-of-pocket caps to anyone with commercial insurance and that anyone without insurance can buy one or multiple Sanofi insulins for a fixed price of $99 per month, up to 10 boxes of pens and/or 10-mL vials.
And Sanofi will take part in the Centers for Medicare & Medicaid Services’ new insulin demonstration program. Starting in 2021, CMS will cap insulin copays at $35 for people in Part D plans that participate.
Eli Lilly spokesperson Brad Jacklin said the company “believes in the common goal of ensuring affordable access to insulin and other life-saving medicines because nobody should have to forgo or ration because of cost.”
Lilly supports efforts “that more directly affect patients’ cost-sharing based on their health care coverage,” he said. Insurers and pharmacy benefit managers (PBMs) should pass savings on to patients, Mr. Jacklin urged. Lilly caps some insulins at $35 for the uninsured or commercially insured. The company will also participate in the CMS program.
Meanwhile, a PhRMA-sponsored website www.letstalkaboutcost.org said that, because they do not share savings, insurers and PBMs are responsible for high insulin costs.
Manufacturer assistance programs for patients with diabetes and other chronic diseases, on the other hand, can save individuals $300-$500 a year, PhRMA said in August.
PBMs point back at insulin manufacturers
PBMs, however, point back at drug companies. “PBMs have been able to moderate insulin costs for most consumers with insurance,” said J.C. Scott, president and CEO of the Pharmaceutical Care Management Association, the PBM trade group, in a statement.
The rising cost of insulin is caused by a lack of competition and overuse of patent extensions, PCMA maintains.
Health insurers, which, in tandem with PBMs, give insulins formulary preference based on a discounted price, are most likely to feel the impact of laws limiting out-of-pocket costs.
If they have to make up the shortfall from a patient’s reduced payment for a prescription, they will likely raise premiums, said Ms. O’Neill Hayes.
And if patients pay the same price for insulin – regardless of who makes it – drugmakers won’t have much incentive to offer discounts or rebates for formulary placement, she said. Again, that would likely lead to higher premiums.
David Allen, a spokesperson for America’s Health Insurance Plans, said in an interview that AHIP believes lack of competition has driven up insulin prices.
“High prices for insulin correspond with high health insurance costs for insulin,” he said. When CMS starts requiring drugmakers to discount their insulins for Medicare that will allow “health plans to use those savings to reduce out-of-pocket [costs] for seniors.”
He did not respond to a question as to why health insurers were not already passing savings on to commercially insured patients, especially in states with out-of-pocket limits.
Mr. Allen did say that AHIP’s plans “stand ready to work with state policymakers to remove barriers to lower insulin prices for Americans.”
Utah savings hopefully saving lives already
In Utah, legislators tuned out the blame game, and instead were keen to listen to patients, who had many stories about how the high cost of insulin had hurt them, said Ms. Hooley.
She noted an estimated 50,000 Utahans rely on insulin to stay alive.
Ms. Hooley and her chapter convinced legislators to pass a bill that gives insurers the option to cap patient copays at $30 per month, or to put insulin on its lowest formulary tier and waive any patient deductible. That aspect of the law does not go into effect until January 2021, but insurers are already starting to move insulin to the lowest formulary tier.
That has helped some people immediately. One state resident said her most recent insulin prescription cost $7 – instead of the usual $200.
The uninsured are not left totally high and dry either. Starting June 1, anyone in the state could buy through a state bulk-purchasing program, which guaranteed a 60% discount.
Ms. Hooley said she’d recently heard about a patient who usually spent $300 per prescription but was able to buy insulin for $100 through the program.
“Although $100 is still too much, it is nice knowing the Utah Insulin Savings Program is saving lives,” Ms. Hooley concluded.
A version of this article originally appeared on Medscape.com.
Twelve U.S. states have now passed laws aimed at making insulin more affordable – and more than 30 are considering such legislation – but they all have gaps that still put the cost of this basic and essential medication out of reach for many with diabetes.
The laws only apply to health insurance through state-regulated plans, and not to the majority of health plans that cover most Americans: Medicare, Medicaid, the Veterans Affairs health system, or self-funded employer-sponsored plans.
Overall, Hannah Crabtree, an activist who writes the blog Data for Insulin, estimates state laws that limit copays, deductibles, or other out-of-pocket costs for insulin cover an average of 27% of people with diabetes across the United States.
And while diabetes activists have applauded state actions, most want more help for the under- and uninsured.
“Our chapter will be fighting harder next legislative session for the uninsured,” said Mindie Hooley, the leader of the Utah #insulin4all chapter, which successfully lobbied legislators to pass a bill signed by the state’s governor on March 30.
“With so many losing their jobs because of the pandemic, there’s no better time than now to fight for these patients who don’t have insurance,” Ms. Hooley said in an interview.
The American Diabetes Association has also been lobbying for state caps as one of many avenues for making insulin more affordable, said Stephen Habbe, the ADA’s director for state government affairs.
One in four insulin users report rationing the medication, Mr. Habbe said.
The state laws “can really provide important relief in terms of affordability for their insulin costs, which we know can be critical in terms of preserving their life and helping to prevent complications that can potentially be disabling or even deadly,” he said in an interview.
Activists with T1 International, which created the #insulin4all campaign, are working nationwide to convince state legislators to back measures that limit out-of-pocket costs for insulin, or for other diabetes medications and supplies.
Colorado, Connecticut, Delaware, Illinois, Maine, New Hampshire, New Mexico, New York, Utah, Virginia, Washington, and West Virginia have enacted such limits, with caps ranging from $25 to $100.
Insulin makers unfazed, blame insurers, PBMs for high prices
The three insulin manufacturers in the United States – Eli Lilly, Novo Nordisk, and Sanofi– have not overtly fought against the laws, although in July, the Pharmaceutical Research and Manufacturers of America did sue to block a related Minnesota law that provides a free emergency supply of insulin.
And the nonprofit news organization FairWarning reported in August that a lobbyist from Eli Lilly had attempted to push a Tennessee legislator to keep the uninsured from being eligible for any out-of-pocket limits.
The insulin makers have also not lowered prices in response to the mounting number of state laws.
They see no need, said Tara O’Neill Hayes, director of human welfare policy at the American Action Forum, a center right–leaning Washington, D.C., think tank.
“You’re going to do what you can get away with,” Ms. O’Neill Hayes said in an interview. “To the extent that they can keep their prices high and people are still buying, they have limited incentives to lower those costs.”
The insulin market is dysfunctional, she added. “The increasing cost of insulin seems primarily to be the result of a lack of competition in the market and convoluted drug pricing and insurance practices,” Ms. O’Neill Hayes and colleagues wrote in a report in April on federal and state attempts to address insulin affordability.
Novo Nordisk, however, maintains that drugmakers are not solely to blame.
“Everyone in the health care system has a role to play in affordability,” said Ken Inchausti, Novo Nordisk’s senior director for corporate communications. State legislation “attempts to address a systemic issue in [U.S.] health care: How benefit design can make medicines unaffordable for many, especially for those in high-deductible health plans,” he said in an interview.
“Efforts to place copay caps on insurance plans covering insulin can certainly help lower out-of-pocket costs,” said Mr. Inchausti.
Sanofi spokesperson Jon Florio said the company supports actions that increase affordable access to insulin. However, “while we support capped copays, we feel this should not be limited to just one class of medicines,” he said. Mr. Florio also noted that Sanofi provides out-of-pocket caps to anyone with commercial insurance and that anyone without insurance can buy one or multiple Sanofi insulins for a fixed price of $99 per month, up to 10 boxes of pens and/or 10-mL vials.
And Sanofi will take part in the Centers for Medicare & Medicaid Services’ new insulin demonstration program. Starting in 2021, CMS will cap insulin copays at $35 for people in Part D plans that participate.
Eli Lilly spokesperson Brad Jacklin said the company “believes in the common goal of ensuring affordable access to insulin and other life-saving medicines because nobody should have to forgo or ration because of cost.”
Lilly supports efforts “that more directly affect patients’ cost-sharing based on their health care coverage,” he said. Insurers and pharmacy benefit managers (PBMs) should pass savings on to patients, Mr. Jacklin urged. Lilly caps some insulins at $35 for the uninsured or commercially insured. The company will also participate in the CMS program.
Meanwhile, a PhRMA-sponsored website www.letstalkaboutcost.org said that, because they do not share savings, insurers and PBMs are responsible for high insulin costs.
Manufacturer assistance programs for patients with diabetes and other chronic diseases, on the other hand, can save individuals $300-$500 a year, PhRMA said in August.
PBMs point back at insulin manufacturers
PBMs, however, point back at drug companies. “PBMs have been able to moderate insulin costs for most consumers with insurance,” said J.C. Scott, president and CEO of the Pharmaceutical Care Management Association, the PBM trade group, in a statement.
The rising cost of insulin is caused by a lack of competition and overuse of patent extensions, PCMA maintains.
Health insurers, which, in tandem with PBMs, give insulins formulary preference based on a discounted price, are most likely to feel the impact of laws limiting out-of-pocket costs.
If they have to make up the shortfall from a patient’s reduced payment for a prescription, they will likely raise premiums, said Ms. O’Neill Hayes.
And if patients pay the same price for insulin – regardless of who makes it – drugmakers won’t have much incentive to offer discounts or rebates for formulary placement, she said. Again, that would likely lead to higher premiums.
David Allen, a spokesperson for America’s Health Insurance Plans, said in an interview that AHIP believes lack of competition has driven up insulin prices.
“High prices for insulin correspond with high health insurance costs for insulin,” he said. When CMS starts requiring drugmakers to discount their insulins for Medicare that will allow “health plans to use those savings to reduce out-of-pocket [costs] for seniors.”
He did not respond to a question as to why health insurers were not already passing savings on to commercially insured patients, especially in states with out-of-pocket limits.
Mr. Allen did say that AHIP’s plans “stand ready to work with state policymakers to remove barriers to lower insulin prices for Americans.”
Utah savings hopefully saving lives already
In Utah, legislators tuned out the blame game, and instead were keen to listen to patients, who had many stories about how the high cost of insulin had hurt them, said Ms. Hooley.
She noted an estimated 50,000 Utahans rely on insulin to stay alive.
Ms. Hooley and her chapter convinced legislators to pass a bill that gives insurers the option to cap patient copays at $30 per month, or to put insulin on its lowest formulary tier and waive any patient deductible. That aspect of the law does not go into effect until January 2021, but insurers are already starting to move insulin to the lowest formulary tier.
That has helped some people immediately. One state resident said her most recent insulin prescription cost $7 – instead of the usual $200.
The uninsured are not left totally high and dry either. Starting June 1, anyone in the state could buy through a state bulk-purchasing program, which guaranteed a 60% discount.
Ms. Hooley said she’d recently heard about a patient who usually spent $300 per prescription but was able to buy insulin for $100 through the program.
“Although $100 is still too much, it is nice knowing the Utah Insulin Savings Program is saving lives,” Ms. Hooley concluded.
A version of this article originally appeared on Medscape.com.
Drug Overdose and Suicide Among Veteran Enrollees in the VHA: Comparison Among Local, Regional, and National Data
Suicide is the 10th leading cause of death in the US. In 2017, there were 47,173 deaths by suicide (14 deaths per 100,000 people), representing a 33% increase from 1999.1 In 2017 veterans accounted for 13.5% of all suicide deaths among US adults, although veterans comprised only 7.9% of the adult population; the age- and sex-adjusted suicide rate was 1.5 times higher for veterans than that of nonveteran adults.2,3
Among veteran users of Veterans Health Administration (VHA) services, mental health and substance use disorders, chronic medical conditions, and chronic pain are associated with an increased risk for suicide.3 About one-half of VHA veterans have been diagnosed with chronic pain.4 A chronic pain diagnosis (eg, back pain, migraine, and psychogenic pain) increased the risk of death by suicide even after adjusting for comorbid psychiatric diagnoses, according to a study on pain and suicide among US veterans.5
One-quarter of veterans received an opioid prescription during VHA outpatient care in 2012.4 Increased prescribing of opioid medications has been associated with opioid overdose and suicides.6-10 Opioids are the most common drugs found in suicide by overdose.11 The rate of opioid-related suicide deaths is 13 times higher among individuals with opioid use disorder (OUD) than it is for those without OUD.12 The rate of OUD diagnosis among VHA users was 7 times higher than that for non-VHA users.13
In the US the age-adjusted rate of drug overdose deaths increased from 6 per 100,000 persons in 1999 to 22 per 100,000 in 2017.14 Drug overdoses accounted for 52,404 US deaths in 2015; 33,091 (63.1%) were from opioids.15 In 2017, there were 70,237 drug overdose deaths; 67.8% involved opioids (ie, 5 per 100,000 population represent prescription opioids).16
The VHA is committed to reducing opioid use and veteran suicide prevention. In 2013 the VHA launched the Opioid Safety Initiative employing 4 strategies: education, pain management, risk management, and addiction treatment.17 To address the opioid epidemic, the North Florida/South Georgia Veteran Health System (NF/SGVHS) developed and implemented a multispecialty Opioid Risk Reduction Program that is fully integrated with mental health and addiction services. The purpose of the NF/SGVHS one-stop pain addiction clinic is to provide a treatment program for chronic pain and addiction. The program includes elements of a whole health approach to pain care, including battlefield and traditional acupuncture. The focus went beyond replacing pharmacologic treatments with a complementary integrative health approach to helping veterans regain control of their lives through empowerment, skill building, shared goal setting, and reinforcing self-management.
The self-management programs include a pain school for patient education, a pain psychology program, and a yoga program, all stressing self-management offered onsite and via telehealth. Special effort was directed to identify patients with OUD and opioid dependence. Many of these patients were transitioned to buprenorphine, a potent analgesic that suppresses opioid cravings and withdrawal symptoms associated with stopping opioids. The clinic was structured so that patients could be seen often for follow-up and support. In addition, open lines of communication and referral were set up between this clinic, the interventional pain clinic, and the physical medicine and rehabilitation service. A detailed description of this program has been published elsewhere.18
The number of veterans receiving opioid prescription across the VHA system decreased by 172,000 prescriptions quarterly between 2012 and 2016.19 Fewer veterans were prescribed high doses of opioids or concomitant interacting medicines and more veterans were receiving nonopioid therapies.19 The prescription reduction across the VHA has varied. For example, from 2012 to 2017 the NF/SGVHS reported an 87% reduction of opioid prescriptions (≥ 100 mg morphine equivalents/d), compared with the VHA national average reduction of 49%.18
Vigorous opioid reduction is controversial. In a systematic review on opioid reduction, Frank and colleagues reported some beneficial effects of opioid reduction, such as increased health-related quality of life.20 However, another study suggested a risk of increased pain with opioid tapering.21 The literature findings on the association between prescription opioid use and suicide are mixed. The VHA Office of Mental Health and Suicide Prevention literature review reported that veterans were at increased risk of committing suicide within the first 6 months of discontinuing opioid therapy.22 Another study reported that veterans who discontinued long-term opioid treatment had an increased risk for suicidal ideation.23 However, higher doses of opioids were associated with an increased risk for suicide among individuals with chronic pain.10 The link between opioid tapering and the risk of suicide or overdose is uncertain.
Bohnert and Ilgen suggested that discontinuing prescription opioids leads to suicide without examining the risk factors that influenced discontinuation is ill-informed.7 Strong evidence about the association or relationship among opioid use, overdose, and suicide is needed. To increase our understanding of that association, Bohnert and Ilgen argued for multifaceted interventions that simultaneously address the shared causes and risk factors for OUD,7 such as the multispecialty Opioid Risk Reduction Program at NF/SGVHS.
Because of the reported association between robust integrated mental health and addiction, primary care pain clinic intervention, and the higher rate of opioid tapering in NF/SGVHS,18 this study aims to describe the pattern of overdose diagnosis (opioid overdose and nonopioid overdose) and pattern of suicide rates among veterans enrolled in NF/SGVHS, Veterans Integrated Service Network (VISN) 8, and the entire VA health care system during 2012 to 2016.The study reviewed and compared overdose diagnosis and suicide rates among veterans across NF/SGVHS and 2 other levels of the VA health care system to determine whether there were variances in the pattern of overdose/suicide rates and to explore these differences.
Methods
In this retrospective study, aggregate data were obtained from several sources. First, the drug overdose data were extracted from the VA Support Service Center (VSSC) medical diagnosis cube. We reviewed the literature for opioid codes reported in the literature and compared these reported opioid International Classification of Diseases, Ninth Revision (ICD-9) and International Classification of Diseases, 10th Revision (ICD-10) codes with the local facility patient-level comprehensive overdose diagnosis codes. Based on the comparison, we found 98 ICD-9 and ICD-10 overdose diagnosis codes and ran the modified codes against the VSSC national database. Overdose data were aggregated by facility and fiscal year, and the overdose rates (per 1,000) were calculated for unique veteran users at the 3 levels (NF/SGVHS, VISN 8, and VA national) as the denominator.
Each of the 18 VISNs comprise multiple VAMCs and clinics within a geographic region. VISN 8 encompasses most of Florida and portions of southern Georgia and the Caribbean (Puerto Rico, US Virgin Islands), including NF/SGVHS.
In this study, drug overdose refers to the overdose or poisoning from all drugs (ie, opioids, cocaine, amphetamines, sedatives, etc) and defined as any unintentional (accidental), deliberate, or intent undetermined drug poisoning.24 The suicide data for this study were drawn from the VA Suicide Prevention Program at 3 different levels: NF/SGVHS, VISN 8, and VHA national. Suicide is death caused by an intentional act of injuring oneself with the intent to die.25
This descriptive study compared the rate of annual drug overdoses (per 1,000 enrollees) between NF/SGVHS, VISN 8, and VHA national from 2012 to 2016. It also compared the annual rate of suicide per 100,000 enrollees across these 3 levels of the VHA. The overdose and suicide rates and numbers are mutually exclusive, meaning the VISN 8 data do not include the NF/SGVHS information, and the national data excluded data from VISN 8 and NF/SGVHS. This approach helped improve the quality of multiple level comparisons for different levels of the VHA system.
Results
Figure 1 shows the pattern of overdose diagnosis by rates (per 1,000) across the study period (2012 to 2016) and compares patterns at 3 levels of VHA (NF/SGVHS, VISN 8, and VHA national). The average annual rate of overdose diagnoses for NF/SGVHS during the study was slightly higher (16.8 per 1,000) than that of VISN 8 (16 per 1,000) and VHA national (15.3 per 1,000), but by the end of the study period the NF/SGVHS rate (18.6 per 1,000) nearly matched the national rate (18.2 per 1,000) and was lower than the VISN 8 rate (20.4 per 1,000). Additionally, NF/SGVHS had less variability (SD, 1.34) in yearly average overdose rates compared with VISN 8 (SD, 2.96), and VHA national (SD, 1.69).
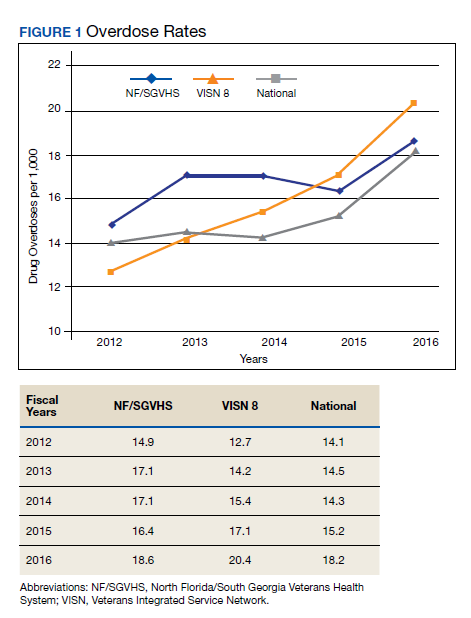
From 2013 to 2014 the overdose diagnosis rate for NF/SGVHS remained the same (17.1 per 1,000). A similar pattern was observed for the VHA national data, whereas the VISN 8 data showed a steady increase during the same period. In 2015, the NF/SGVHS had 0.7 per 1,000 decrease in overdose diagnosis rate, whereas VISN 8 and VHA national data showed 1.7 per 1,000 and 0.9 per 1,000 increases, respectively. During the last year of the study (2016), there was a dramatic increase in overdose diagnosis for all the health care systems, ranging from 2.2 per 1,000 for NF/SGVHS to 3.3 per 1,000 for VISN 8.
Figure 2 shows the annual rates (per 100,000 individuals) of suicide for NF/SGVHS, VISN 8, and VHA national. The suicide pattern for VISN 8 shows a cyclical acceleration and deceleration trend across the study period. From 2012 to 2014, the VHA national data show a steady increase of about 1 per 100,000 from year to year. On the contrary, NF/SGVHS shows a low suicide rate from year to year within the same period with a rate of 10 per 100,000 in 2013 compared with the previous year. Although the NF/SGVHS suicide rate increased in 2016 (10.4 per 100,000), it remained lower than that of VISN 8 (10.7 per 100,00) and VHA national (38.2 per 100,000).
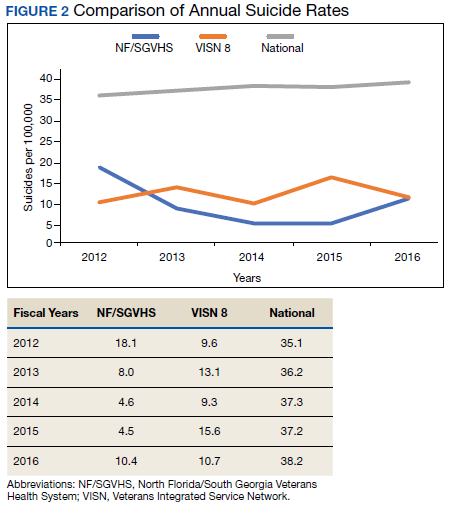
This study shows that NF/SGVHS had the lowest average annual rate of suicide (9.1 per 100,000) during the study period, which was 4 times lower than that of VHA national and 2.6 times lower than VISN 8.
Discussion
This study described and compared the distribution pattern of overdose (nonopioid and opioid) and suicide rates at different levels of the VHA system. Although VHA implemented systemwide opioid tapering in 2013, little is known about the association between opioid tapering and overdose and suicide. We believe a retrospective examination regarding overdose and suicide among VHA users at 3 different levels of the system from 2012 to 2016 could contribute to the discussion regarding the potential risks and benefits of discontinuing opioids.
First, the average annual rate of overdose diagnosis for NF/SGVHS during the study period was slightly higher (16.8 per 1,000) compared with those of VISN 8 (16.0 per 1,000) and VHA national (15.3 per 1,000) with a general pattern of increase and minimum variations in the rates observed during the study period among the 3 levels of the system. These increased overdose patterns are consistent with other reports in the literature.14 By the end of the study period, the NF/SGVHS rate (18.6 per 1,000) nearly matched the national rate (18.2 per 1,000) and was lower than VISN 8 (20.4 per 1,000). During the last year of the study period (2016), there was a dramatic increase in overdose diagnosis for all health care systems ranging from 2.2 per 1,000 for NF/SGVHS to 3.3 per 1,000 for VISN 8, which might be because of the VHA systemwide change of diagnosis code from ICD-9 to ICD-10, which includes more detailed diagnosis codes.
Second, our results showed that NF/SGVHS had the lowest average annual suicide rate (9.1 per 100,000) during the study period, which is one-fourth the VHA national rate and 2.6 per 100,000 lower than the VISN 8 rate. According to Bohnert and Ilgen,programs that improve the quality of pain care, expand access to psychotherapy, and increase access to medication-assisted treatment for OUDs could reduce suicide by drug overdose.7 We suggest that the low suicide rate at NF/SGVHS and the difference in the suicide rates between the NF/SGVHS and VISN 8 and VHA national data might be associated with the practice-based biopsychosocial interventions implemented at NF/SGVHS.
Our data showed a rise in the incidence of suicide at the NF/SGVHS in 2016. We are not aware of a local change in conditions, policy, and practice that would account for this increase. Suicide is variable, and data are likely to show spikes and valleys. Based on the available data, although the incidence of suicides at the NF/SGVHS in 2016 was higher, it remained below the VISN 8 and national VHA rate. This study seems to support the practice of tapering or stopping opioids within the context of a multidisciplinary approach that offers frequent follow-up, nonopioid options, and treatment of opioid addiction/dependence.
Limitations
The research findings of this study are limited by the retrospective and descriptive nature of its design. However, the findings might provide important information for understanding variations of overdose and suicide among VHA enrollees. Studies that use more robust methodologies are warranted to clinically investigate the impact of a multispecialty opioid risk reduction program targeting chronic pain and addiction management and identify best practices of opioid reduction and any unintended consequences that might arise from opioid tapering.26 Further, we did not have access to the VA national overdose and suicide data after 2016. Similar to most retrospective data studies, ours might be limited by availability of national overdose and suicide data after 2016. It is important for future studies to cross-validate our study findings.
Conclusions
The NF/SGVHS developed and implemented a biopsychosocial model of pain treatment that includes multicomponent primary care integrated with mental health and addiction services as well as the interventional pain and physical medicine and rehabilitation services. The presence of this program, during a period when the facility was tapering opioids is likely to account for at least part of the relative reduction in suicide.
1. American Foundation for Suicide Prevention. Suicide statistics. https://afsp.org/about-suicide/suicide-statistics. Updated 2019. Accessed September 2, 2020.
2. Shane L 3rd. New veteran suicide numbers raise concerns among experts hoping for positive news. https://www.militarytimes.com/news/pentagon-congress/2019/10/09/new-veteran-suicide-numbers-raise-concerns-among-experts-hoping-for-positive-news. Published October 9, 2019. Accessed July 23, 2020.
3. Veterans Health Administration, Office of Mental Health and Suicide Prevention. Veteran suicide data report, 2005–2017. https://www.mentalhealth.va.gov/docs/data-sheets/2019/2019_National_Veteran_Suicide_Prevention_Annual_Report_508.pdf. Published September 2019. Accessed July 20, 2020.
4. Gallagher RM. Advancing the pain agenda in the veteran population. Anesthesiol Clin. 2016;34(2):357-378. doi:10.1016/j.anclin.2016.01.003
5. Ilgen MA, Kleinberg F, Ignacio RV, et al. Noncancer pain conditions and risk of suicide. JAMA Psychiatry. 2013;70(7):692-697. doi:10.1001/jamapsychiatry.2013.908
6. Frenk SM, Porter KS, Paulozzi LJ. Prescription opioid analgesic use among adults: United States, 1999-2012. National Center for Health Statistics data brief. https://www.cdc.gov/nchs/products/databriefs/db189.htm. Published February 25, 2015. Accessed July 20, 2020.
7. Bohnert ASB, Ilgen MA. Understanding links among opioid use, overdose, and suicide. N Engl J Med. 2019;380(14):71-79. doi:10.1056/NEJMc1901540
8. Dunn KM, Saunders KW, Rutter CM, et al. Opioid prescriptions for chronic pain and overdose: a cohort study. Ann Intern Med. 2010;152(2):85-92. doi:10.7326/0003-4819-152-2-201001190-00006
9. Gomes T, Mamdani MM, Dhalla IA, Paterson JM, Juurlink DN. Opioid dose and drug-related mortality in patients with nonmalignant pain. Arch Intern Med. 2011;171(7):686-691. doi:10.1001/archinternmed.2011.117
10. Ilgen MA, Bohnert AS, Ganoczy D, Bair MJ, McCarthy JF, Blow FC. Opioid dose and risk of suicide. Pain. 2016;157(5):1079-1084. doi:10.1097/j.pain.0000000000000484
11. Sinyor M, Howlett A, Cheung AH, Schaffer A. Substances used in completed suicide by overdose in Toronto: an observational study of coroner’s data. Can J Psychiatry. 2012;57(3):184-191. doi:10.1177/070674371205700308
12. Wilcox HC, Conner KR, Caine ED. Association of alcohol and drug use disorders and completed suicide: an empirical review of cohort studies. Drug Alcohol Depend. 2004;76(suppl):S11-S19 doi:10.1016/j.drugalcdep.2004.08.003.
13. Baser OL, Mardekian XJ, Schaaf D, Wang L, Joshi AV. Prevalence of diagnosed opioid abuse and its economic burden in the Veterans Health Administration. Pain Pract. 2014;14(5):437-445. doi:10.1111/papr.12097
14. Hedegaard H, Warner M, Miniño AM. Drug overdose deaths in the united states, 1999-2015. National Center for Health Statistics data brief. https://www.cdc.gov/nchs/data/databriefs/db273.pdf. Published February 2017. Accessed July 20, 2020.
15. Rudd RA, Seth P, David F, Scholl L. Increases in drug and opioid-involved overdose deaths—United States, 2010-2015. MMWR Morb Mortal Wkly Rep. 2016;65(50-51):1445-1452. doi:10.15585/mmwr.mm655051e1
16. Scholl L, Seth P, Kariisa M, Wilson N, Baldwin G. Drug and opioid-involved overdose deaths—United States, 2013-2017. MMWR Morb Mortal Wkly Rep. 2019,67(5152):1419-1427. doi:10.15585/mmwr.mm675152e1
17. US Department of Veterans Affairs and Department of Defense. VA/DOD clinical practice guideline for opioid therapy for chronic pain version 3.0. https://www.healthquality.va.gov/guidelines/pain/cot. Updated March 1, 2018. Accessed July 20, 2020.
18. Vaughn IA, Beyth RJ, Ayers ML, et al. Multispecialty opioid risk reduction program targeting chronic pain and addiction management in veterans. Fed Pract. 2019;36(9):406-411.
19. Gellad WF, Good CB, Shulkin DJ. Addressing the opioid epidemic in the United States: lessons from the Department of Veterans Affairs. JAMA Intern Med. 2017;177(5):611-612. doi:10.1001/jamainternmed.2017.0147
20. Frank JW, Lovejoy TI, Becker WC, et al. Patient outcomes in dose reduction or discontinuation of long-term opioid therapy: a systematic review. Ann Intern Med. 2017;167(3):181-191. doi:10.7326/M17-0598
21. Berna C, Kulich RJ, Rathmell JP. Tapering long-term opioid therapy in chronic noncancer pain: evidence and recommendations for everyday practice. Mayo Clin Proc. 2015;90(6):828-842. doi:10.1016/j.mayocp.2015.04.003
22. Veterans Health Administration, Office of Mental Health and Suicide Prevention. Opioid use and suicide risk. https://www.mentalhealth.va.gov/suicide_prevention/docs/Literature_Review_Opioid_Use_and_Suicide_Risk_508_FINAL_04-26-2019.pdf. Published April 26, 2019. Accessed July 20, 2020.
23. Demidenko MI, Dobscha SK, Morasco BJ, Meath THA, Ilgen MA, Lovejoy TI. Suicidal ideation and suicidal self-directed violence following clinician-initiated prescription opioid discontinuation among long-term opioid users. Gen Hosp Psychiatry. 2017;47:29-35. doi:10.1016/j.genhosppsych.2017.04.011
24. National Institute on Drug Abuse. Intentional versus unintentional overdose deaths. https://www.drugabuse.gov/related-topics/treatment/intentional-vs-unintentional-overdose-deaths. Updated February 13, 2017. Accessed July 20, 2020.
25. Centers for Disease Control and Prevention. Preventing suicide. https://www.cdc.gov/violenceprevention/pdf/suicide-factsheet.pdf. Published 2018. Accessed July 20, 2020.
26. Webster LR. Pain and suicide: the other side of the opioid story. Pain Med. 2014;15(3):345-346. doi:10.1111/pme.12398
Suicide is the 10th leading cause of death in the US. In 2017, there were 47,173 deaths by suicide (14 deaths per 100,000 people), representing a 33% increase from 1999.1 In 2017 veterans accounted for 13.5% of all suicide deaths among US adults, although veterans comprised only 7.9% of the adult population; the age- and sex-adjusted suicide rate was 1.5 times higher for veterans than that of nonveteran adults.2,3
Among veteran users of Veterans Health Administration (VHA) services, mental health and substance use disorders, chronic medical conditions, and chronic pain are associated with an increased risk for suicide.3 About one-half of VHA veterans have been diagnosed with chronic pain.4 A chronic pain diagnosis (eg, back pain, migraine, and psychogenic pain) increased the risk of death by suicide even after adjusting for comorbid psychiatric diagnoses, according to a study on pain and suicide among US veterans.5
One-quarter of veterans received an opioid prescription during VHA outpatient care in 2012.4 Increased prescribing of opioid medications has been associated with opioid overdose and suicides.6-10 Opioids are the most common drugs found in suicide by overdose.11 The rate of opioid-related suicide deaths is 13 times higher among individuals with opioid use disorder (OUD) than it is for those without OUD.12 The rate of OUD diagnosis among VHA users was 7 times higher than that for non-VHA users.13
In the US the age-adjusted rate of drug overdose deaths increased from 6 per 100,000 persons in 1999 to 22 per 100,000 in 2017.14 Drug overdoses accounted for 52,404 US deaths in 2015; 33,091 (63.1%) were from opioids.15 In 2017, there were 70,237 drug overdose deaths; 67.8% involved opioids (ie, 5 per 100,000 population represent prescription opioids).16
The VHA is committed to reducing opioid use and veteran suicide prevention. In 2013 the VHA launched the Opioid Safety Initiative employing 4 strategies: education, pain management, risk management, and addiction treatment.17 To address the opioid epidemic, the North Florida/South Georgia Veteran Health System (NF/SGVHS) developed and implemented a multispecialty Opioid Risk Reduction Program that is fully integrated with mental health and addiction services. The purpose of the NF/SGVHS one-stop pain addiction clinic is to provide a treatment program for chronic pain and addiction. The program includes elements of a whole health approach to pain care, including battlefield and traditional acupuncture. The focus went beyond replacing pharmacologic treatments with a complementary integrative health approach to helping veterans regain control of their lives through empowerment, skill building, shared goal setting, and reinforcing self-management.
The self-management programs include a pain school for patient education, a pain psychology program, and a yoga program, all stressing self-management offered onsite and via telehealth. Special effort was directed to identify patients with OUD and opioid dependence. Many of these patients were transitioned to buprenorphine, a potent analgesic that suppresses opioid cravings and withdrawal symptoms associated with stopping opioids. The clinic was structured so that patients could be seen often for follow-up and support. In addition, open lines of communication and referral were set up between this clinic, the interventional pain clinic, and the physical medicine and rehabilitation service. A detailed description of this program has been published elsewhere.18
The number of veterans receiving opioid prescription across the VHA system decreased by 172,000 prescriptions quarterly between 2012 and 2016.19 Fewer veterans were prescribed high doses of opioids or concomitant interacting medicines and more veterans were receiving nonopioid therapies.19 The prescription reduction across the VHA has varied. For example, from 2012 to 2017 the NF/SGVHS reported an 87% reduction of opioid prescriptions (≥ 100 mg morphine equivalents/d), compared with the VHA national average reduction of 49%.18
Vigorous opioid reduction is controversial. In a systematic review on opioid reduction, Frank and colleagues reported some beneficial effects of opioid reduction, such as increased health-related quality of life.20 However, another study suggested a risk of increased pain with opioid tapering.21 The literature findings on the association between prescription opioid use and suicide are mixed. The VHA Office of Mental Health and Suicide Prevention literature review reported that veterans were at increased risk of committing suicide within the first 6 months of discontinuing opioid therapy.22 Another study reported that veterans who discontinued long-term opioid treatment had an increased risk for suicidal ideation.23 However, higher doses of opioids were associated with an increased risk for suicide among individuals with chronic pain.10 The link between opioid tapering and the risk of suicide or overdose is uncertain.
Bohnert and Ilgen suggested that discontinuing prescription opioids leads to suicide without examining the risk factors that influenced discontinuation is ill-informed.7 Strong evidence about the association or relationship among opioid use, overdose, and suicide is needed. To increase our understanding of that association, Bohnert and Ilgen argued for multifaceted interventions that simultaneously address the shared causes and risk factors for OUD,7 such as the multispecialty Opioid Risk Reduction Program at NF/SGVHS.
Because of the reported association between robust integrated mental health and addiction, primary care pain clinic intervention, and the higher rate of opioid tapering in NF/SGVHS,18 this study aims to describe the pattern of overdose diagnosis (opioid overdose and nonopioid overdose) and pattern of suicide rates among veterans enrolled in NF/SGVHS, Veterans Integrated Service Network (VISN) 8, and the entire VA health care system during 2012 to 2016.The study reviewed and compared overdose diagnosis and suicide rates among veterans across NF/SGVHS and 2 other levels of the VA health care system to determine whether there were variances in the pattern of overdose/suicide rates and to explore these differences.
Methods
In this retrospective study, aggregate data were obtained from several sources. First, the drug overdose data were extracted from the VA Support Service Center (VSSC) medical diagnosis cube. We reviewed the literature for opioid codes reported in the literature and compared these reported opioid International Classification of Diseases, Ninth Revision (ICD-9) and International Classification of Diseases, 10th Revision (ICD-10) codes with the local facility patient-level comprehensive overdose diagnosis codes. Based on the comparison, we found 98 ICD-9 and ICD-10 overdose diagnosis codes and ran the modified codes against the VSSC national database. Overdose data were aggregated by facility and fiscal year, and the overdose rates (per 1,000) were calculated for unique veteran users at the 3 levels (NF/SGVHS, VISN 8, and VA national) as the denominator.
Each of the 18 VISNs comprise multiple VAMCs and clinics within a geographic region. VISN 8 encompasses most of Florida and portions of southern Georgia and the Caribbean (Puerto Rico, US Virgin Islands), including NF/SGVHS.
In this study, drug overdose refers to the overdose or poisoning from all drugs (ie, opioids, cocaine, amphetamines, sedatives, etc) and defined as any unintentional (accidental), deliberate, or intent undetermined drug poisoning.24 The suicide data for this study were drawn from the VA Suicide Prevention Program at 3 different levels: NF/SGVHS, VISN 8, and VHA national. Suicide is death caused by an intentional act of injuring oneself with the intent to die.25
This descriptive study compared the rate of annual drug overdoses (per 1,000 enrollees) between NF/SGVHS, VISN 8, and VHA national from 2012 to 2016. It also compared the annual rate of suicide per 100,000 enrollees across these 3 levels of the VHA. The overdose and suicide rates and numbers are mutually exclusive, meaning the VISN 8 data do not include the NF/SGVHS information, and the national data excluded data from VISN 8 and NF/SGVHS. This approach helped improve the quality of multiple level comparisons for different levels of the VHA system.
Results
Figure 1 shows the pattern of overdose diagnosis by rates (per 1,000) across the study period (2012 to 2016) and compares patterns at 3 levels of VHA (NF/SGVHS, VISN 8, and VHA national). The average annual rate of overdose diagnoses for NF/SGVHS during the study was slightly higher (16.8 per 1,000) than that of VISN 8 (16 per 1,000) and VHA national (15.3 per 1,000), but by the end of the study period the NF/SGVHS rate (18.6 per 1,000) nearly matched the national rate (18.2 per 1,000) and was lower than the VISN 8 rate (20.4 per 1,000). Additionally, NF/SGVHS had less variability (SD, 1.34) in yearly average overdose rates compared with VISN 8 (SD, 2.96), and VHA national (SD, 1.69).
From 2013 to 2014 the overdose diagnosis rate for NF/SGVHS remained the same (17.1 per 1,000). A similar pattern was observed for the VHA national data, whereas the VISN 8 data showed a steady increase during the same period. In 2015, the NF/SGVHS had 0.7 per 1,000 decrease in overdose diagnosis rate, whereas VISN 8 and VHA national data showed 1.7 per 1,000 and 0.9 per 1,000 increases, respectively. During the last year of the study (2016), there was a dramatic increase in overdose diagnosis for all the health care systems, ranging from 2.2 per 1,000 for NF/SGVHS to 3.3 per 1,000 for VISN 8.
Figure 2 shows the annual rates (per 100,000 individuals) of suicide for NF/SGVHS, VISN 8, and VHA national. The suicide pattern for VISN 8 shows a cyclical acceleration and deceleration trend across the study period. From 2012 to 2014, the VHA national data show a steady increase of about 1 per 100,000 from year to year. On the contrary, NF/SGVHS shows a low suicide rate from year to year within the same period with a rate of 10 per 100,000 in 2013 compared with the previous year. Although the NF/SGVHS suicide rate increased in 2016 (10.4 per 100,000), it remained lower than that of VISN 8 (10.7 per 100,00) and VHA national (38.2 per 100,000).
This study shows that NF/SGVHS had the lowest average annual rate of suicide (9.1 per 100,000) during the study period, which was 4 times lower than that of VHA national and 2.6 times lower than VISN 8.
Discussion
This study described and compared the distribution pattern of overdose (nonopioid and opioid) and suicide rates at different levels of the VHA system. Although VHA implemented systemwide opioid tapering in 2013, little is known about the association between opioid tapering and overdose and suicide. We believe a retrospective examination regarding overdose and suicide among VHA users at 3 different levels of the system from 2012 to 2016 could contribute to the discussion regarding the potential risks and benefits of discontinuing opioids.
First, the average annual rate of overdose diagnosis for NF/SGVHS during the study period was slightly higher (16.8 per 1,000) compared with those of VISN 8 (16.0 per 1,000) and VHA national (15.3 per 1,000) with a general pattern of increase and minimum variations in the rates observed during the study period among the 3 levels of the system. These increased overdose patterns are consistent with other reports in the literature.14 By the end of the study period, the NF/SGVHS rate (18.6 per 1,000) nearly matched the national rate (18.2 per 1,000) and was lower than VISN 8 (20.4 per 1,000). During the last year of the study period (2016), there was a dramatic increase in overdose diagnosis for all health care systems ranging from 2.2 per 1,000 for NF/SGVHS to 3.3 per 1,000 for VISN 8, which might be because of the VHA systemwide change of diagnosis code from ICD-9 to ICD-10, which includes more detailed diagnosis codes.
Second, our results showed that NF/SGVHS had the lowest average annual suicide rate (9.1 per 100,000) during the study period, which is one-fourth the VHA national rate and 2.6 per 100,000 lower than the VISN 8 rate. According to Bohnert and Ilgen,programs that improve the quality of pain care, expand access to psychotherapy, and increase access to medication-assisted treatment for OUDs could reduce suicide by drug overdose.7 We suggest that the low suicide rate at NF/SGVHS and the difference in the suicide rates between the NF/SGVHS and VISN 8 and VHA national data might be associated with the practice-based biopsychosocial interventions implemented at NF/SGVHS.
Our data showed a rise in the incidence of suicide at the NF/SGVHS in 2016. We are not aware of a local change in conditions, policy, and practice that would account for this increase. Suicide is variable, and data are likely to show spikes and valleys. Based on the available data, although the incidence of suicides at the NF/SGVHS in 2016 was higher, it remained below the VISN 8 and national VHA rate. This study seems to support the practice of tapering or stopping opioids within the context of a multidisciplinary approach that offers frequent follow-up, nonopioid options, and treatment of opioid addiction/dependence.
Limitations
The research findings of this study are limited by the retrospective and descriptive nature of its design. However, the findings might provide important information for understanding variations of overdose and suicide among VHA enrollees. Studies that use more robust methodologies are warranted to clinically investigate the impact of a multispecialty opioid risk reduction program targeting chronic pain and addiction management and identify best practices of opioid reduction and any unintended consequences that might arise from opioid tapering.26 Further, we did not have access to the VA national overdose and suicide data after 2016. Similar to most retrospective data studies, ours might be limited by availability of national overdose and suicide data after 2016. It is important for future studies to cross-validate our study findings.
Conclusions
The NF/SGVHS developed and implemented a biopsychosocial model of pain treatment that includes multicomponent primary care integrated with mental health and addiction services as well as the interventional pain and physical medicine and rehabilitation services. The presence of this program, during a period when the facility was tapering opioids is likely to account for at least part of the relative reduction in suicide.
Suicide is the 10th leading cause of death in the US. In 2017, there were 47,173 deaths by suicide (14 deaths per 100,000 people), representing a 33% increase from 1999.1 In 2017 veterans accounted for 13.5% of all suicide deaths among US adults, although veterans comprised only 7.9% of the adult population; the age- and sex-adjusted suicide rate was 1.5 times higher for veterans than that of nonveteran adults.2,3
Among veteran users of Veterans Health Administration (VHA) services, mental health and substance use disorders, chronic medical conditions, and chronic pain are associated with an increased risk for suicide.3 About one-half of VHA veterans have been diagnosed with chronic pain.4 A chronic pain diagnosis (eg, back pain, migraine, and psychogenic pain) increased the risk of death by suicide even after adjusting for comorbid psychiatric diagnoses, according to a study on pain and suicide among US veterans.5
One-quarter of veterans received an opioid prescription during VHA outpatient care in 2012.4 Increased prescribing of opioid medications has been associated with opioid overdose and suicides.6-10 Opioids are the most common drugs found in suicide by overdose.11 The rate of opioid-related suicide deaths is 13 times higher among individuals with opioid use disorder (OUD) than it is for those without OUD.12 The rate of OUD diagnosis among VHA users was 7 times higher than that for non-VHA users.13
In the US the age-adjusted rate of drug overdose deaths increased from 6 per 100,000 persons in 1999 to 22 per 100,000 in 2017.14 Drug overdoses accounted for 52,404 US deaths in 2015; 33,091 (63.1%) were from opioids.15 In 2017, there were 70,237 drug overdose deaths; 67.8% involved opioids (ie, 5 per 100,000 population represent prescription opioids).16
The VHA is committed to reducing opioid use and veteran suicide prevention. In 2013 the VHA launched the Opioid Safety Initiative employing 4 strategies: education, pain management, risk management, and addiction treatment.17 To address the opioid epidemic, the North Florida/South Georgia Veteran Health System (NF/SGVHS) developed and implemented a multispecialty Opioid Risk Reduction Program that is fully integrated with mental health and addiction services. The purpose of the NF/SGVHS one-stop pain addiction clinic is to provide a treatment program for chronic pain and addiction. The program includes elements of a whole health approach to pain care, including battlefield and traditional acupuncture. The focus went beyond replacing pharmacologic treatments with a complementary integrative health approach to helping veterans regain control of their lives through empowerment, skill building, shared goal setting, and reinforcing self-management.
The self-management programs include a pain school for patient education, a pain psychology program, and a yoga program, all stressing self-management offered onsite and via telehealth. Special effort was directed to identify patients with OUD and opioid dependence. Many of these patients were transitioned to buprenorphine, a potent analgesic that suppresses opioid cravings and withdrawal symptoms associated with stopping opioids. The clinic was structured so that patients could be seen often for follow-up and support. In addition, open lines of communication and referral were set up between this clinic, the interventional pain clinic, and the physical medicine and rehabilitation service. A detailed description of this program has been published elsewhere.18
The number of veterans receiving opioid prescription across the VHA system decreased by 172,000 prescriptions quarterly between 2012 and 2016.19 Fewer veterans were prescribed high doses of opioids or concomitant interacting medicines and more veterans were receiving nonopioid therapies.19 The prescription reduction across the VHA has varied. For example, from 2012 to 2017 the NF/SGVHS reported an 87% reduction of opioid prescriptions (≥ 100 mg morphine equivalents/d), compared with the VHA national average reduction of 49%.18
Vigorous opioid reduction is controversial. In a systematic review on opioid reduction, Frank and colleagues reported some beneficial effects of opioid reduction, such as increased health-related quality of life.20 However, another study suggested a risk of increased pain with opioid tapering.21 The literature findings on the association between prescription opioid use and suicide are mixed. The VHA Office of Mental Health and Suicide Prevention literature review reported that veterans were at increased risk of committing suicide within the first 6 months of discontinuing opioid therapy.22 Another study reported that veterans who discontinued long-term opioid treatment had an increased risk for suicidal ideation.23 However, higher doses of opioids were associated with an increased risk for suicide among individuals with chronic pain.10 The link between opioid tapering and the risk of suicide or overdose is uncertain.
Bohnert and Ilgen suggested that discontinuing prescription opioids leads to suicide without examining the risk factors that influenced discontinuation is ill-informed.7 Strong evidence about the association or relationship among opioid use, overdose, and suicide is needed. To increase our understanding of that association, Bohnert and Ilgen argued for multifaceted interventions that simultaneously address the shared causes and risk factors for OUD,7 such as the multispecialty Opioid Risk Reduction Program at NF/SGVHS.
Because of the reported association between robust integrated mental health and addiction, primary care pain clinic intervention, and the higher rate of opioid tapering in NF/SGVHS,18 this study aims to describe the pattern of overdose diagnosis (opioid overdose and nonopioid overdose) and pattern of suicide rates among veterans enrolled in NF/SGVHS, Veterans Integrated Service Network (VISN) 8, and the entire VA health care system during 2012 to 2016.The study reviewed and compared overdose diagnosis and suicide rates among veterans across NF/SGVHS and 2 other levels of the VA health care system to determine whether there were variances in the pattern of overdose/suicide rates and to explore these differences.
Methods
In this retrospective study, aggregate data were obtained from several sources. First, the drug overdose data were extracted from the VA Support Service Center (VSSC) medical diagnosis cube. We reviewed the literature for opioid codes reported in the literature and compared these reported opioid International Classification of Diseases, Ninth Revision (ICD-9) and International Classification of Diseases, 10th Revision (ICD-10) codes with the local facility patient-level comprehensive overdose diagnosis codes. Based on the comparison, we found 98 ICD-9 and ICD-10 overdose diagnosis codes and ran the modified codes against the VSSC national database. Overdose data were aggregated by facility and fiscal year, and the overdose rates (per 1,000) were calculated for unique veteran users at the 3 levels (NF/SGVHS, VISN 8, and VA national) as the denominator.
Each of the 18 VISNs comprise multiple VAMCs and clinics within a geographic region. VISN 8 encompasses most of Florida and portions of southern Georgia and the Caribbean (Puerto Rico, US Virgin Islands), including NF/SGVHS.
In this study, drug overdose refers to the overdose or poisoning from all drugs (ie, opioids, cocaine, amphetamines, sedatives, etc) and defined as any unintentional (accidental), deliberate, or intent undetermined drug poisoning.24 The suicide data for this study were drawn from the VA Suicide Prevention Program at 3 different levels: NF/SGVHS, VISN 8, and VHA national. Suicide is death caused by an intentional act of injuring oneself with the intent to die.25
This descriptive study compared the rate of annual drug overdoses (per 1,000 enrollees) between NF/SGVHS, VISN 8, and VHA national from 2012 to 2016. It also compared the annual rate of suicide per 100,000 enrollees across these 3 levels of the VHA. The overdose and suicide rates and numbers are mutually exclusive, meaning the VISN 8 data do not include the NF/SGVHS information, and the national data excluded data from VISN 8 and NF/SGVHS. This approach helped improve the quality of multiple level comparisons for different levels of the VHA system.
Results
Figure 1 shows the pattern of overdose diagnosis by rates (per 1,000) across the study period (2012 to 2016) and compares patterns at 3 levels of VHA (NF/SGVHS, VISN 8, and VHA national). The average annual rate of overdose diagnoses for NF/SGVHS during the study was slightly higher (16.8 per 1,000) than that of VISN 8 (16 per 1,000) and VHA national (15.3 per 1,000), but by the end of the study period the NF/SGVHS rate (18.6 per 1,000) nearly matched the national rate (18.2 per 1,000) and was lower than the VISN 8 rate (20.4 per 1,000). Additionally, NF/SGVHS had less variability (SD, 1.34) in yearly average overdose rates compared with VISN 8 (SD, 2.96), and VHA national (SD, 1.69).
From 2013 to 2014 the overdose diagnosis rate for NF/SGVHS remained the same (17.1 per 1,000). A similar pattern was observed for the VHA national data, whereas the VISN 8 data showed a steady increase during the same period. In 2015, the NF/SGVHS had 0.7 per 1,000 decrease in overdose diagnosis rate, whereas VISN 8 and VHA national data showed 1.7 per 1,000 and 0.9 per 1,000 increases, respectively. During the last year of the study (2016), there was a dramatic increase in overdose diagnosis for all the health care systems, ranging from 2.2 per 1,000 for NF/SGVHS to 3.3 per 1,000 for VISN 8.
Figure 2 shows the annual rates (per 100,000 individuals) of suicide for NF/SGVHS, VISN 8, and VHA national. The suicide pattern for VISN 8 shows a cyclical acceleration and deceleration trend across the study period. From 2012 to 2014, the VHA national data show a steady increase of about 1 per 100,000 from year to year. On the contrary, NF/SGVHS shows a low suicide rate from year to year within the same period with a rate of 10 per 100,000 in 2013 compared with the previous year. Although the NF/SGVHS suicide rate increased in 2016 (10.4 per 100,000), it remained lower than that of VISN 8 (10.7 per 100,00) and VHA national (38.2 per 100,000).
This study shows that NF/SGVHS had the lowest average annual rate of suicide (9.1 per 100,000) during the study period, which was 4 times lower than that of VHA national and 2.6 times lower than VISN 8.
Discussion
This study described and compared the distribution pattern of overdose (nonopioid and opioid) and suicide rates at different levels of the VHA system. Although VHA implemented systemwide opioid tapering in 2013, little is known about the association between opioid tapering and overdose and suicide. We believe a retrospective examination regarding overdose and suicide among VHA users at 3 different levels of the system from 2012 to 2016 could contribute to the discussion regarding the potential risks and benefits of discontinuing opioids.
First, the average annual rate of overdose diagnosis for NF/SGVHS during the study period was slightly higher (16.8 per 1,000) compared with those of VISN 8 (16.0 per 1,000) and VHA national (15.3 per 1,000) with a general pattern of increase and minimum variations in the rates observed during the study period among the 3 levels of the system. These increased overdose patterns are consistent with other reports in the literature.14 By the end of the study period, the NF/SGVHS rate (18.6 per 1,000) nearly matched the national rate (18.2 per 1,000) and was lower than VISN 8 (20.4 per 1,000). During the last year of the study period (2016), there was a dramatic increase in overdose diagnosis for all health care systems ranging from 2.2 per 1,000 for NF/SGVHS to 3.3 per 1,000 for VISN 8, which might be because of the VHA systemwide change of diagnosis code from ICD-9 to ICD-10, which includes more detailed diagnosis codes.
Second, our results showed that NF/SGVHS had the lowest average annual suicide rate (9.1 per 100,000) during the study period, which is one-fourth the VHA national rate and 2.6 per 100,000 lower than the VISN 8 rate. According to Bohnert and Ilgen,programs that improve the quality of pain care, expand access to psychotherapy, and increase access to medication-assisted treatment for OUDs could reduce suicide by drug overdose.7 We suggest that the low suicide rate at NF/SGVHS and the difference in the suicide rates between the NF/SGVHS and VISN 8 and VHA national data might be associated with the practice-based biopsychosocial interventions implemented at NF/SGVHS.
Our data showed a rise in the incidence of suicide at the NF/SGVHS in 2016. We are not aware of a local change in conditions, policy, and practice that would account for this increase. Suicide is variable, and data are likely to show spikes and valleys. Based on the available data, although the incidence of suicides at the NF/SGVHS in 2016 was higher, it remained below the VISN 8 and national VHA rate. This study seems to support the practice of tapering or stopping opioids within the context of a multidisciplinary approach that offers frequent follow-up, nonopioid options, and treatment of opioid addiction/dependence.
Limitations
The research findings of this study are limited by the retrospective and descriptive nature of its design. However, the findings might provide important information for understanding variations of overdose and suicide among VHA enrollees. Studies that use more robust methodologies are warranted to clinically investigate the impact of a multispecialty opioid risk reduction program targeting chronic pain and addiction management and identify best practices of opioid reduction and any unintended consequences that might arise from opioid tapering.26 Further, we did not have access to the VA national overdose and suicide data after 2016. Similar to most retrospective data studies, ours might be limited by availability of national overdose and suicide data after 2016. It is important for future studies to cross-validate our study findings.
Conclusions
The NF/SGVHS developed and implemented a biopsychosocial model of pain treatment that includes multicomponent primary care integrated with mental health and addiction services as well as the interventional pain and physical medicine and rehabilitation services. The presence of this program, during a period when the facility was tapering opioids is likely to account for at least part of the relative reduction in suicide.
1. American Foundation for Suicide Prevention. Suicide statistics. https://afsp.org/about-suicide/suicide-statistics. Updated 2019. Accessed September 2, 2020.
2. Shane L 3rd. New veteran suicide numbers raise concerns among experts hoping for positive news. https://www.militarytimes.com/news/pentagon-congress/2019/10/09/new-veteran-suicide-numbers-raise-concerns-among-experts-hoping-for-positive-news. Published October 9, 2019. Accessed July 23, 2020.
3. Veterans Health Administration, Office of Mental Health and Suicide Prevention. Veteran suicide data report, 2005–2017. https://www.mentalhealth.va.gov/docs/data-sheets/2019/2019_National_Veteran_Suicide_Prevention_Annual_Report_508.pdf. Published September 2019. Accessed July 20, 2020.
4. Gallagher RM. Advancing the pain agenda in the veteran population. Anesthesiol Clin. 2016;34(2):357-378. doi:10.1016/j.anclin.2016.01.003
5. Ilgen MA, Kleinberg F, Ignacio RV, et al. Noncancer pain conditions and risk of suicide. JAMA Psychiatry. 2013;70(7):692-697. doi:10.1001/jamapsychiatry.2013.908
6. Frenk SM, Porter KS, Paulozzi LJ. Prescription opioid analgesic use among adults: United States, 1999-2012. National Center for Health Statistics data brief. https://www.cdc.gov/nchs/products/databriefs/db189.htm. Published February 25, 2015. Accessed July 20, 2020.
7. Bohnert ASB, Ilgen MA. Understanding links among opioid use, overdose, and suicide. N Engl J Med. 2019;380(14):71-79. doi:10.1056/NEJMc1901540
8. Dunn KM, Saunders KW, Rutter CM, et al. Opioid prescriptions for chronic pain and overdose: a cohort study. Ann Intern Med. 2010;152(2):85-92. doi:10.7326/0003-4819-152-2-201001190-00006
9. Gomes T, Mamdani MM, Dhalla IA, Paterson JM, Juurlink DN. Opioid dose and drug-related mortality in patients with nonmalignant pain. Arch Intern Med. 2011;171(7):686-691. doi:10.1001/archinternmed.2011.117
10. Ilgen MA, Bohnert AS, Ganoczy D, Bair MJ, McCarthy JF, Blow FC. Opioid dose and risk of suicide. Pain. 2016;157(5):1079-1084. doi:10.1097/j.pain.0000000000000484
11. Sinyor M, Howlett A, Cheung AH, Schaffer A. Substances used in completed suicide by overdose in Toronto: an observational study of coroner’s data. Can J Psychiatry. 2012;57(3):184-191. doi:10.1177/070674371205700308
12. Wilcox HC, Conner KR, Caine ED. Association of alcohol and drug use disorders and completed suicide: an empirical review of cohort studies. Drug Alcohol Depend. 2004;76(suppl):S11-S19 doi:10.1016/j.drugalcdep.2004.08.003.
13. Baser OL, Mardekian XJ, Schaaf D, Wang L, Joshi AV. Prevalence of diagnosed opioid abuse and its economic burden in the Veterans Health Administration. Pain Pract. 2014;14(5):437-445. doi:10.1111/papr.12097
14. Hedegaard H, Warner M, Miniño AM. Drug overdose deaths in the united states, 1999-2015. National Center for Health Statistics data brief. https://www.cdc.gov/nchs/data/databriefs/db273.pdf. Published February 2017. Accessed July 20, 2020.
15. Rudd RA, Seth P, David F, Scholl L. Increases in drug and opioid-involved overdose deaths—United States, 2010-2015. MMWR Morb Mortal Wkly Rep. 2016;65(50-51):1445-1452. doi:10.15585/mmwr.mm655051e1
16. Scholl L, Seth P, Kariisa M, Wilson N, Baldwin G. Drug and opioid-involved overdose deaths—United States, 2013-2017. MMWR Morb Mortal Wkly Rep. 2019,67(5152):1419-1427. doi:10.15585/mmwr.mm675152e1
17. US Department of Veterans Affairs and Department of Defense. VA/DOD clinical practice guideline for opioid therapy for chronic pain version 3.0. https://www.healthquality.va.gov/guidelines/pain/cot. Updated March 1, 2018. Accessed July 20, 2020.
18. Vaughn IA, Beyth RJ, Ayers ML, et al. Multispecialty opioid risk reduction program targeting chronic pain and addiction management in veterans. Fed Pract. 2019;36(9):406-411.
19. Gellad WF, Good CB, Shulkin DJ. Addressing the opioid epidemic in the United States: lessons from the Department of Veterans Affairs. JAMA Intern Med. 2017;177(5):611-612. doi:10.1001/jamainternmed.2017.0147
20. Frank JW, Lovejoy TI, Becker WC, et al. Patient outcomes in dose reduction or discontinuation of long-term opioid therapy: a systematic review. Ann Intern Med. 2017;167(3):181-191. doi:10.7326/M17-0598
21. Berna C, Kulich RJ, Rathmell JP. Tapering long-term opioid therapy in chronic noncancer pain: evidence and recommendations for everyday practice. Mayo Clin Proc. 2015;90(6):828-842. doi:10.1016/j.mayocp.2015.04.003
22. Veterans Health Administration, Office of Mental Health and Suicide Prevention. Opioid use and suicide risk. https://www.mentalhealth.va.gov/suicide_prevention/docs/Literature_Review_Opioid_Use_and_Suicide_Risk_508_FINAL_04-26-2019.pdf. Published April 26, 2019. Accessed July 20, 2020.
23. Demidenko MI, Dobscha SK, Morasco BJ, Meath THA, Ilgen MA, Lovejoy TI. Suicidal ideation and suicidal self-directed violence following clinician-initiated prescription opioid discontinuation among long-term opioid users. Gen Hosp Psychiatry. 2017;47:29-35. doi:10.1016/j.genhosppsych.2017.04.011
24. National Institute on Drug Abuse. Intentional versus unintentional overdose deaths. https://www.drugabuse.gov/related-topics/treatment/intentional-vs-unintentional-overdose-deaths. Updated February 13, 2017. Accessed July 20, 2020.
25. Centers for Disease Control and Prevention. Preventing suicide. https://www.cdc.gov/violenceprevention/pdf/suicide-factsheet.pdf. Published 2018. Accessed July 20, 2020.
26. Webster LR. Pain and suicide: the other side of the opioid story. Pain Med. 2014;15(3):345-346. doi:10.1111/pme.12398
1. American Foundation for Suicide Prevention. Suicide statistics. https://afsp.org/about-suicide/suicide-statistics. Updated 2019. Accessed September 2, 2020.
2. Shane L 3rd. New veteran suicide numbers raise concerns among experts hoping for positive news. https://www.militarytimes.com/news/pentagon-congress/2019/10/09/new-veteran-suicide-numbers-raise-concerns-among-experts-hoping-for-positive-news. Published October 9, 2019. Accessed July 23, 2020.
3. Veterans Health Administration, Office of Mental Health and Suicide Prevention. Veteran suicide data report, 2005–2017. https://www.mentalhealth.va.gov/docs/data-sheets/2019/2019_National_Veteran_Suicide_Prevention_Annual_Report_508.pdf. Published September 2019. Accessed July 20, 2020.
4. Gallagher RM. Advancing the pain agenda in the veteran population. Anesthesiol Clin. 2016;34(2):357-378. doi:10.1016/j.anclin.2016.01.003
5. Ilgen MA, Kleinberg F, Ignacio RV, et al. Noncancer pain conditions and risk of suicide. JAMA Psychiatry. 2013;70(7):692-697. doi:10.1001/jamapsychiatry.2013.908
6. Frenk SM, Porter KS, Paulozzi LJ. Prescription opioid analgesic use among adults: United States, 1999-2012. National Center for Health Statistics data brief. https://www.cdc.gov/nchs/products/databriefs/db189.htm. Published February 25, 2015. Accessed July 20, 2020.
7. Bohnert ASB, Ilgen MA. Understanding links among opioid use, overdose, and suicide. N Engl J Med. 2019;380(14):71-79. doi:10.1056/NEJMc1901540
8. Dunn KM, Saunders KW, Rutter CM, et al. Opioid prescriptions for chronic pain and overdose: a cohort study. Ann Intern Med. 2010;152(2):85-92. doi:10.7326/0003-4819-152-2-201001190-00006
9. Gomes T, Mamdani MM, Dhalla IA, Paterson JM, Juurlink DN. Opioid dose and drug-related mortality in patients with nonmalignant pain. Arch Intern Med. 2011;171(7):686-691. doi:10.1001/archinternmed.2011.117
10. Ilgen MA, Bohnert AS, Ganoczy D, Bair MJ, McCarthy JF, Blow FC. Opioid dose and risk of suicide. Pain. 2016;157(5):1079-1084. doi:10.1097/j.pain.0000000000000484
11. Sinyor M, Howlett A, Cheung AH, Schaffer A. Substances used in completed suicide by overdose in Toronto: an observational study of coroner’s data. Can J Psychiatry. 2012;57(3):184-191. doi:10.1177/070674371205700308
12. Wilcox HC, Conner KR, Caine ED. Association of alcohol and drug use disorders and completed suicide: an empirical review of cohort studies. Drug Alcohol Depend. 2004;76(suppl):S11-S19 doi:10.1016/j.drugalcdep.2004.08.003.
13. Baser OL, Mardekian XJ, Schaaf D, Wang L, Joshi AV. Prevalence of diagnosed opioid abuse and its economic burden in the Veterans Health Administration. Pain Pract. 2014;14(5):437-445. doi:10.1111/papr.12097
14. Hedegaard H, Warner M, Miniño AM. Drug overdose deaths in the united states, 1999-2015. National Center for Health Statistics data brief. https://www.cdc.gov/nchs/data/databriefs/db273.pdf. Published February 2017. Accessed July 20, 2020.
15. Rudd RA, Seth P, David F, Scholl L. Increases in drug and opioid-involved overdose deaths—United States, 2010-2015. MMWR Morb Mortal Wkly Rep. 2016;65(50-51):1445-1452. doi:10.15585/mmwr.mm655051e1
16. Scholl L, Seth P, Kariisa M, Wilson N, Baldwin G. Drug and opioid-involved overdose deaths—United States, 2013-2017. MMWR Morb Mortal Wkly Rep. 2019,67(5152):1419-1427. doi:10.15585/mmwr.mm675152e1
17. US Department of Veterans Affairs and Department of Defense. VA/DOD clinical practice guideline for opioid therapy for chronic pain version 3.0. https://www.healthquality.va.gov/guidelines/pain/cot. Updated March 1, 2018. Accessed July 20, 2020.
18. Vaughn IA, Beyth RJ, Ayers ML, et al. Multispecialty opioid risk reduction program targeting chronic pain and addiction management in veterans. Fed Pract. 2019;36(9):406-411.
19. Gellad WF, Good CB, Shulkin DJ. Addressing the opioid epidemic in the United States: lessons from the Department of Veterans Affairs. JAMA Intern Med. 2017;177(5):611-612. doi:10.1001/jamainternmed.2017.0147
20. Frank JW, Lovejoy TI, Becker WC, et al. Patient outcomes in dose reduction or discontinuation of long-term opioid therapy: a systematic review. Ann Intern Med. 2017;167(3):181-191. doi:10.7326/M17-0598
21. Berna C, Kulich RJ, Rathmell JP. Tapering long-term opioid therapy in chronic noncancer pain: evidence and recommendations for everyday practice. Mayo Clin Proc. 2015;90(6):828-842. doi:10.1016/j.mayocp.2015.04.003
22. Veterans Health Administration, Office of Mental Health and Suicide Prevention. Opioid use and suicide risk. https://www.mentalhealth.va.gov/suicide_prevention/docs/Literature_Review_Opioid_Use_and_Suicide_Risk_508_FINAL_04-26-2019.pdf. Published April 26, 2019. Accessed July 20, 2020.
23. Demidenko MI, Dobscha SK, Morasco BJ, Meath THA, Ilgen MA, Lovejoy TI. Suicidal ideation and suicidal self-directed violence following clinician-initiated prescription opioid discontinuation among long-term opioid users. Gen Hosp Psychiatry. 2017;47:29-35. doi:10.1016/j.genhosppsych.2017.04.011
24. National Institute on Drug Abuse. Intentional versus unintentional overdose deaths. https://www.drugabuse.gov/related-topics/treatment/intentional-vs-unintentional-overdose-deaths. Updated February 13, 2017. Accessed July 20, 2020.
25. Centers for Disease Control and Prevention. Preventing suicide. https://www.cdc.gov/violenceprevention/pdf/suicide-factsheet.pdf. Published 2018. Accessed July 20, 2020.
26. Webster LR. Pain and suicide: the other side of the opioid story. Pain Med. 2014;15(3):345-346. doi:10.1111/pme.12398
First randomized trial reassures on ACEIs, ARBs in COVID-19
The first randomized study to compare continuing versus stopping ACE inhibitors or angiotensin receptor blockers (ARBs) for patients with COVID-19 has shown no difference in key outcomes between the two approaches.

The BRACE CORONA trial – conducted in patients had been taking an ACE inhibitor or an ARB on a long-term basis and who were subsequently hospitalized with COVID-19 – showed no difference in the primary endpoint of number of days alive and out of hospital among those whose medication was suspended for 30 days and those who continued undergoing treatment with these agents.
“Because these data indicate that there is no clinical benefit from routinely interrupting these medications in hospitalized patients with mild to moderate COVID-19, they should generally be continued for those with an indication,” principal investigator Renato Lopes, MD, of Duke Clinical Research Institute, Durham, N.C., concluded.
The BRACE CORONA trial was presented at the European Society of Cardiology Congress 2020 on Sept. 1.
Dr. Lopes explained that there are two conflicting hypotheses about the role of ACE inhibitors and ARBs in COVID-19.
One hypothesis suggests that use of these drugs could be harmful by increasing the expression of ACE2 receptors (which the SARS-CoV-2 virus uses to gain entry into cells), thus potentially enhancing viral binding and viral entry. The other suggests that ACE inhibitors and ARBs could be protective by reducing production of angiotensin II and enhancing the generation of angiotensin 1-7, which attenuates inflammation and fibrosis and therefore could attenuate lung injury.
The BRACE CORONA trial was an academic-led randomized study that tested two strategies: temporarily stopping the ACE inhibitor/ARB for 30 days or continuing these drugs for patients who had been taking these medications on a long-term basis and were hospitalized with a confirmed diagnosis of COVID-19.
The primary outcome was the number of days alive and out of hospital at 30 days. Patients who were using more than three antihypertensive drugs or sacubitril/valsartan or who were hemodynamically unstable at presentation were excluded from the study.
The trial enrolled 659 patients from 29 sites in Brazil. The mean age of patients was 56 years, 40% were women, and 52% were obese. ACE inhibitors were being taken by 15% of the trial participants; ARBs were being taken by 85%. The median duration of ACE inhibitor/ARB treatment was 5 years.
Patients were a median of 6 days from COVID-19 symptom onset. For 30% of the patients, oxygen saturation was below 94% at entry. In terms of COVID-19 symptoms, 57% were classified as mild, and 43% as moderate.
Those with severe COVID-19 symptoms who needed intubation or vasoactive drugs were excluded. Antihypertensive therapy would generally be discontinued in these patients anyway, Dr. Lopes said.
Results showed that the average number of days alive and out of hospital was 21.9 days for patients who stopped taking ACE inhibitors/ARBs and 22.9 days for patients who continued taking these medications. The average difference between groups was –1.1 days.
The average ratio of days alive and out of hospital between the suspending and continuing groups was 0.95 (95% CI, 0.90-1.01; P = .09).
The proportion of patients alive and out of hospital by the end of 30 days in the suspending ACE inhibitor/ARB group was 91.8% versus 95% in the continuing group.
A similar 30-day mortality rate was seen for patients who continued and those who suspended ACE inhibitor/ARB therapy, at 2.8% and 2.7%, respectively (hazard ratio, 0.97). The median number of days that patients were alive and out of hospital was 25 in both groups.
Dr. Lopes said that there was no difference between the two groups with regard to many other secondary outcomes. These included COVID-19 disease progression (need for intubation, ventilation, need for vasoactive drugs, or imaging results) and cardiovascular endpoints (MI, stroke, thromboembolic events, worsening heart failure, myocarditis, or hypertensive crisis).
“Our results endorse with reliable and more definitive data what most medical and cardiovascular societies are recommending – that patients do not stop ACE inhibitor or ARB medication. This has been based on observational data so far, but BRACE CORONA now provides randomized data to support this recommendation,” Dr. Lopes concluded.
Dr. Lopes noted that several subgroups had been prespecified for analysis. Factors included age, obesity, difference between ACE inhibitors/ARBs, difference in oxygen saturation at presentation, time since COVID-19 symptom onset, degree of lung involvement on CT, and symptom severity on presentation.
“We saw very consistent effects of our main findings across all these subgroups, and we plan to report more details of these in the near future,” he said.
Protective for older patients?
The discussant of the study at the ESC Hotline session, Gianfranco Parati, MD, University of Milan-Bicocca and San Luca Hospital, Milan, congratulated Lopes and his team for conducting this important trial at such a difficult time.
He pointed out that patients in the BRACE CORONA trial were quite young (average age, 56 years) and that observational data so far suggest that ACE inhibitors and ARBs have a stronger protective effect in older COVID-19 patients.
He also noted that the percentage of patients alive and out of hospital at 30 days was higher for the patients who continued on treatment in this study (95% vs. 91.8%), which suggested an advantage in maintaining the medication.
Dr. Lopes replied that one-quarter of the population in the BRACE CORONA trial was older than 65 years, which he said was a “reasonable number.”
“Subgroup analysis by age did not show a significant interaction, but the effect of continuing treatment does seem to be more favorable in older patients and also in those who were sicker and had more comorbidities,” he added.
Dr. Parati also suggested that it would have been difficult to discern differences between ACE inhibitors and ARBs in the BRACE CORONA trial, because so few patents were taking ACE inhibitors; the follow-up period of 30 days was relatively short, inasmuch as these drugs may have long-term effects; and it would have been difficult to show differences in the main outcomes used in the study – mortality and time out of hospital – in these patients with mild to moderate disease.
Franz H. Messerli, MD, and Christoph Gräni, MD, University of Bern (Switzerland), said in a joint statement: “The BRACE CORONA trial provides answers to what we know from retrospective studies: if you have already COVID, don’t stop renin-angiotensin system blocker medication.”
But they added that the study does not answer the question about the risk/benefit of ACE inhibitors or ARBs with regard to possible enhanced viral entry through the ACE2 receptor. “What about all those on these drugs who are not infected with COVID? Do they need to stop them? We simply don’t know yet,” they said.
Dr. Messerli and Dr. Gräni added that they would like to see a study that compared patients before SARS-CoV-2 infection who were without hypertension, patients with hypertension who were taking ACE inhibitors or ARBs, and patients with hypertension taking other antihypertensive drugs.
The BRACE CORONA trial was sponsored by D’Or Institute for Research and Education and the Brazilian Clinical Research Institute. Dr. Lopes has disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
The first randomized study to compare continuing versus stopping ACE inhibitors or angiotensin receptor blockers (ARBs) for patients with COVID-19 has shown no difference in key outcomes between the two approaches.
The BRACE CORONA trial – conducted in patients had been taking an ACE inhibitor or an ARB on a long-term basis and who were subsequently hospitalized with COVID-19 – showed no difference in the primary endpoint of number of days alive and out of hospital among those whose medication was suspended for 30 days and those who continued undergoing treatment with these agents.
“Because these data indicate that there is no clinical benefit from routinely interrupting these medications in hospitalized patients with mild to moderate COVID-19, they should generally be continued for those with an indication,” principal investigator Renato Lopes, MD, of Duke Clinical Research Institute, Durham, N.C., concluded.
The BRACE CORONA trial was presented at the European Society of Cardiology Congress 2020 on Sept. 1.
Dr. Lopes explained that there are two conflicting hypotheses about the role of ACE inhibitors and ARBs in COVID-19.
One hypothesis suggests that use of these drugs could be harmful by increasing the expression of ACE2 receptors (which the SARS-CoV-2 virus uses to gain entry into cells), thus potentially enhancing viral binding and viral entry. The other suggests that ACE inhibitors and ARBs could be protective by reducing production of angiotensin II and enhancing the generation of angiotensin 1-7, which attenuates inflammation and fibrosis and therefore could attenuate lung injury.
The BRACE CORONA trial was an academic-led randomized study that tested two strategies: temporarily stopping the ACE inhibitor/ARB for 30 days or continuing these drugs for patients who had been taking these medications on a long-term basis and were hospitalized with a confirmed diagnosis of COVID-19.
The primary outcome was the number of days alive and out of hospital at 30 days. Patients who were using more than three antihypertensive drugs or sacubitril/valsartan or who were hemodynamically unstable at presentation were excluded from the study.
The trial enrolled 659 patients from 29 sites in Brazil. The mean age of patients was 56 years, 40% were women, and 52% were obese. ACE inhibitors were being taken by 15% of the trial participants; ARBs were being taken by 85%. The median duration of ACE inhibitor/ARB treatment was 5 years.
Patients were a median of 6 days from COVID-19 symptom onset. For 30% of the patients, oxygen saturation was below 94% at entry. In terms of COVID-19 symptoms, 57% were classified as mild, and 43% as moderate.
Those with severe COVID-19 symptoms who needed intubation or vasoactive drugs were excluded. Antihypertensive therapy would generally be discontinued in these patients anyway, Dr. Lopes said.
Results showed that the average number of days alive and out of hospital was 21.9 days for patients who stopped taking ACE inhibitors/ARBs and 22.9 days for patients who continued taking these medications. The average difference between groups was –1.1 days.
The average ratio of days alive and out of hospital between the suspending and continuing groups was 0.95 (95% CI, 0.90-1.01; P = .09).
The proportion of patients alive and out of hospital by the end of 30 days in the suspending ACE inhibitor/ARB group was 91.8% versus 95% in the continuing group.
A similar 30-day mortality rate was seen for patients who continued and those who suspended ACE inhibitor/ARB therapy, at 2.8% and 2.7%, respectively (hazard ratio, 0.97). The median number of days that patients were alive and out of hospital was 25 in both groups.
Dr. Lopes said that there was no difference between the two groups with regard to many other secondary outcomes. These included COVID-19 disease progression (need for intubation, ventilation, need for vasoactive drugs, or imaging results) and cardiovascular endpoints (MI, stroke, thromboembolic events, worsening heart failure, myocarditis, or hypertensive crisis).
“Our results endorse with reliable and more definitive data what most medical and cardiovascular societies are recommending – that patients do not stop ACE inhibitor or ARB medication. This has been based on observational data so far, but BRACE CORONA now provides randomized data to support this recommendation,” Dr. Lopes concluded.
Dr. Lopes noted that several subgroups had been prespecified for analysis. Factors included age, obesity, difference between ACE inhibitors/ARBs, difference in oxygen saturation at presentation, time since COVID-19 symptom onset, degree of lung involvement on CT, and symptom severity on presentation.
“We saw very consistent effects of our main findings across all these subgroups, and we plan to report more details of these in the near future,” he said.
Protective for older patients?
The discussant of the study at the ESC Hotline session, Gianfranco Parati, MD, University of Milan-Bicocca and San Luca Hospital, Milan, congratulated Lopes and his team for conducting this important trial at such a difficult time.
He pointed out that patients in the BRACE CORONA trial were quite young (average age, 56 years) and that observational data so far suggest that ACE inhibitors and ARBs have a stronger protective effect in older COVID-19 patients.
He also noted that the percentage of patients alive and out of hospital at 30 days was higher for the patients who continued on treatment in this study (95% vs. 91.8%), which suggested an advantage in maintaining the medication.
Dr. Lopes replied that one-quarter of the population in the BRACE CORONA trial was older than 65 years, which he said was a “reasonable number.”
“Subgroup analysis by age did not show a significant interaction, but the effect of continuing treatment does seem to be more favorable in older patients and also in those who were sicker and had more comorbidities,” he added.
Dr. Parati also suggested that it would have been difficult to discern differences between ACE inhibitors and ARBs in the BRACE CORONA trial, because so few patents were taking ACE inhibitors; the follow-up period of 30 days was relatively short, inasmuch as these drugs may have long-term effects; and it would have been difficult to show differences in the main outcomes used in the study – mortality and time out of hospital – in these patients with mild to moderate disease.
Franz H. Messerli, MD, and Christoph Gräni, MD, University of Bern (Switzerland), said in a joint statement: “The BRACE CORONA trial provides answers to what we know from retrospective studies: if you have already COVID, don’t stop renin-angiotensin system blocker medication.”
But they added that the study does not answer the question about the risk/benefit of ACE inhibitors or ARBs with regard to possible enhanced viral entry through the ACE2 receptor. “What about all those on these drugs who are not infected with COVID? Do they need to stop them? We simply don’t know yet,” they said.
Dr. Messerli and Dr. Gräni added that they would like to see a study that compared patients before SARS-CoV-2 infection who were without hypertension, patients with hypertension who were taking ACE inhibitors or ARBs, and patients with hypertension taking other antihypertensive drugs.
The BRACE CORONA trial was sponsored by D’Or Institute for Research and Education and the Brazilian Clinical Research Institute. Dr. Lopes has disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
The first randomized study to compare continuing versus stopping ACE inhibitors or angiotensin receptor blockers (ARBs) for patients with COVID-19 has shown no difference in key outcomes between the two approaches.
The BRACE CORONA trial – conducted in patients had been taking an ACE inhibitor or an ARB on a long-term basis and who were subsequently hospitalized with COVID-19 – showed no difference in the primary endpoint of number of days alive and out of hospital among those whose medication was suspended for 30 days and those who continued undergoing treatment with these agents.
“Because these data indicate that there is no clinical benefit from routinely interrupting these medications in hospitalized patients with mild to moderate COVID-19, they should generally be continued for those with an indication,” principal investigator Renato Lopes, MD, of Duke Clinical Research Institute, Durham, N.C., concluded.
The BRACE CORONA trial was presented at the European Society of Cardiology Congress 2020 on Sept. 1.
Dr. Lopes explained that there are two conflicting hypotheses about the role of ACE inhibitors and ARBs in COVID-19.
One hypothesis suggests that use of these drugs could be harmful by increasing the expression of ACE2 receptors (which the SARS-CoV-2 virus uses to gain entry into cells), thus potentially enhancing viral binding and viral entry. The other suggests that ACE inhibitors and ARBs could be protective by reducing production of angiotensin II and enhancing the generation of angiotensin 1-7, which attenuates inflammation and fibrosis and therefore could attenuate lung injury.
The BRACE CORONA trial was an academic-led randomized study that tested two strategies: temporarily stopping the ACE inhibitor/ARB for 30 days or continuing these drugs for patients who had been taking these medications on a long-term basis and were hospitalized with a confirmed diagnosis of COVID-19.
The primary outcome was the number of days alive and out of hospital at 30 days. Patients who were using more than three antihypertensive drugs or sacubitril/valsartan or who were hemodynamically unstable at presentation were excluded from the study.
The trial enrolled 659 patients from 29 sites in Brazil. The mean age of patients was 56 years, 40% were women, and 52% were obese. ACE inhibitors were being taken by 15% of the trial participants; ARBs were being taken by 85%. The median duration of ACE inhibitor/ARB treatment was 5 years.
Patients were a median of 6 days from COVID-19 symptom onset. For 30% of the patients, oxygen saturation was below 94% at entry. In terms of COVID-19 symptoms, 57% were classified as mild, and 43% as moderate.
Those with severe COVID-19 symptoms who needed intubation or vasoactive drugs were excluded. Antihypertensive therapy would generally be discontinued in these patients anyway, Dr. Lopes said.
Results showed that the average number of days alive and out of hospital was 21.9 days for patients who stopped taking ACE inhibitors/ARBs and 22.9 days for patients who continued taking these medications. The average difference between groups was –1.1 days.
The average ratio of days alive and out of hospital between the suspending and continuing groups was 0.95 (95% CI, 0.90-1.01; P = .09).
The proportion of patients alive and out of hospital by the end of 30 days in the suspending ACE inhibitor/ARB group was 91.8% versus 95% in the continuing group.
A similar 30-day mortality rate was seen for patients who continued and those who suspended ACE inhibitor/ARB therapy, at 2.8% and 2.7%, respectively (hazard ratio, 0.97). The median number of days that patients were alive and out of hospital was 25 in both groups.
Dr. Lopes said that there was no difference between the two groups with regard to many other secondary outcomes. These included COVID-19 disease progression (need for intubation, ventilation, need for vasoactive drugs, or imaging results) and cardiovascular endpoints (MI, stroke, thromboembolic events, worsening heart failure, myocarditis, or hypertensive crisis).
“Our results endorse with reliable and more definitive data what most medical and cardiovascular societies are recommending – that patients do not stop ACE inhibitor or ARB medication. This has been based on observational data so far, but BRACE CORONA now provides randomized data to support this recommendation,” Dr. Lopes concluded.
Dr. Lopes noted that several subgroups had been prespecified for analysis. Factors included age, obesity, difference between ACE inhibitors/ARBs, difference in oxygen saturation at presentation, time since COVID-19 symptom onset, degree of lung involvement on CT, and symptom severity on presentation.
“We saw very consistent effects of our main findings across all these subgroups, and we plan to report more details of these in the near future,” he said.
Protective for older patients?
The discussant of the study at the ESC Hotline session, Gianfranco Parati, MD, University of Milan-Bicocca and San Luca Hospital, Milan, congratulated Lopes and his team for conducting this important trial at such a difficult time.
He pointed out that patients in the BRACE CORONA trial were quite young (average age, 56 years) and that observational data so far suggest that ACE inhibitors and ARBs have a stronger protective effect in older COVID-19 patients.
He also noted that the percentage of patients alive and out of hospital at 30 days was higher for the patients who continued on treatment in this study (95% vs. 91.8%), which suggested an advantage in maintaining the medication.
Dr. Lopes replied that one-quarter of the population in the BRACE CORONA trial was older than 65 years, which he said was a “reasonable number.”
“Subgroup analysis by age did not show a significant interaction, but the effect of continuing treatment does seem to be more favorable in older patients and also in those who were sicker and had more comorbidities,” he added.
Dr. Parati also suggested that it would have been difficult to discern differences between ACE inhibitors and ARBs in the BRACE CORONA trial, because so few patents were taking ACE inhibitors; the follow-up period of 30 days was relatively short, inasmuch as these drugs may have long-term effects; and it would have been difficult to show differences in the main outcomes used in the study – mortality and time out of hospital – in these patients with mild to moderate disease.
Franz H. Messerli, MD, and Christoph Gräni, MD, University of Bern (Switzerland), said in a joint statement: “The BRACE CORONA trial provides answers to what we know from retrospective studies: if you have already COVID, don’t stop renin-angiotensin system blocker medication.”
But they added that the study does not answer the question about the risk/benefit of ACE inhibitors or ARBs with regard to possible enhanced viral entry through the ACE2 receptor. “What about all those on these drugs who are not infected with COVID? Do they need to stop them? We simply don’t know yet,” they said.
Dr. Messerli and Dr. Gräni added that they would like to see a study that compared patients before SARS-CoV-2 infection who were without hypertension, patients with hypertension who were taking ACE inhibitors or ARBs, and patients with hypertension taking other antihypertensive drugs.
The BRACE CORONA trial was sponsored by D’Or Institute for Research and Education and the Brazilian Clinical Research Institute. Dr. Lopes has disclosed no relevant financial relationships.
A version of this article originally appeared on Medscape.com.
FDA expands remdesivir use for all COVID-19 hospitalized patients
An EUA of remdesivir issued in May allowed the drug to be used only for patients with severe COVID-19, specifically, COVID-19 patients with low blood oxygen levels or who need oxygen therapy or mechanical ventilation.
“Today, based on the Agency’s ongoing review of the EUA, including its review of the totality of scientific information now available, the FDA has determined that it is reasonable to believe Veklury may be effective for the treatment of suspected or laboratory-confirmed COVID-19 in all hospitalized adult and pediatric patients,” the FDA news release about the expanded EUA said. “The Agency’s review has also concluded that the known and potential benefits of Veklury outweigh the known and potential risks for these uses.”
‘Further evaluation’ needed
The EUA expansion is partially based on the results of a randomized, open-label trial that Gilead Sciences, remdesivir’s manufacturer, conducted at multiple sites.
The trial showed that a 5-day course of remdesivir was associated with statistically significant improvement among patients hospitalized with moderate COVID-19 in comparison with those receiving standard care. However, patients who were randomly assigned to a receive longer, 10-day remdesivir course had not improved significantly 11 days after treatment started, compared with those who received standard care.
Results with remdesivir in this trial and in two previously reported randomized trials varied, “raising the question of whether the discrepancies are artifacts of study design choices, including patient populations, or whether the drug is less efficacious than hoped,” wrote Erin K. McCreary, PharmD, and Derek C. Angus, MD, MPH, with the University of Pittsburgh School of Medicine, in an editorial that accompanied publication of the trials in JAMA.
Angus previously expressed concern that expanding remdesivir’s EUA could “interrupt or thwart efforts to execute the needed RCTs [randomized controlled trials].
“We think there really needs to be further evaluation of remdesivir in large-scale RCTs adequately powered to understand in which patients, at which dose, given at which point in the course of illness leads to what concrete and tangible improvement in clinical outcomes,” he told Medscape Medical News.
“At this point, remdesivir definitely holds promise, but given the cost to produce and distribute the drug, it seems crucial to know with more certainty how best to use it,” Angus said.
The EUA expansion is also partially based on results from a randomized, double-blind, placebo-controlled clinical trial that the National Institutes of Allergy and Infectious Diseases conducted. In that trial, there was a statistically significant reduction in median recovery time and higher odds of clinical improvement after 2 weeks for hospitalized patients who received remdesivir.
For hospitalized patients with mild to moderate disease, the results were consistent with the overall study results but were not statistically significant.
This article first appeared on Medscape.com.
An EUA of remdesivir issued in May allowed the drug to be used only for patients with severe COVID-19, specifically, COVID-19 patients with low blood oxygen levels or who need oxygen therapy or mechanical ventilation.
“Today, based on the Agency’s ongoing review of the EUA, including its review of the totality of scientific information now available, the FDA has determined that it is reasonable to believe Veklury may be effective for the treatment of suspected or laboratory-confirmed COVID-19 in all hospitalized adult and pediatric patients,” the FDA news release about the expanded EUA said. “The Agency’s review has also concluded that the known and potential benefits of Veklury outweigh the known and potential risks for these uses.”
‘Further evaluation’ needed
The EUA expansion is partially based on the results of a randomized, open-label trial that Gilead Sciences, remdesivir’s manufacturer, conducted at multiple sites.
The trial showed that a 5-day course of remdesivir was associated with statistically significant improvement among patients hospitalized with moderate COVID-19 in comparison with those receiving standard care. However, patients who were randomly assigned to a receive longer, 10-day remdesivir course had not improved significantly 11 days after treatment started, compared with those who received standard care.
Results with remdesivir in this trial and in two previously reported randomized trials varied, “raising the question of whether the discrepancies are artifacts of study design choices, including patient populations, or whether the drug is less efficacious than hoped,” wrote Erin K. McCreary, PharmD, and Derek C. Angus, MD, MPH, with the University of Pittsburgh School of Medicine, in an editorial that accompanied publication of the trials in JAMA.
Angus previously expressed concern that expanding remdesivir’s EUA could “interrupt or thwart efforts to execute the needed RCTs [randomized controlled trials].
“We think there really needs to be further evaluation of remdesivir in large-scale RCTs adequately powered to understand in which patients, at which dose, given at which point in the course of illness leads to what concrete and tangible improvement in clinical outcomes,” he told Medscape Medical News.
“At this point, remdesivir definitely holds promise, but given the cost to produce and distribute the drug, it seems crucial to know with more certainty how best to use it,” Angus said.
The EUA expansion is also partially based on results from a randomized, double-blind, placebo-controlled clinical trial that the National Institutes of Allergy and Infectious Diseases conducted. In that trial, there was a statistically significant reduction in median recovery time and higher odds of clinical improvement after 2 weeks for hospitalized patients who received remdesivir.
For hospitalized patients with mild to moderate disease, the results were consistent with the overall study results but were not statistically significant.
This article first appeared on Medscape.com.
An EUA of remdesivir issued in May allowed the drug to be used only for patients with severe COVID-19, specifically, COVID-19 patients with low blood oxygen levels or who need oxygen therapy or mechanical ventilation.
“Today, based on the Agency’s ongoing review of the EUA, including its review of the totality of scientific information now available, the FDA has determined that it is reasonable to believe Veklury may be effective for the treatment of suspected or laboratory-confirmed COVID-19 in all hospitalized adult and pediatric patients,” the FDA news release about the expanded EUA said. “The Agency’s review has also concluded that the known and potential benefits of Veklury outweigh the known and potential risks for these uses.”
‘Further evaluation’ needed
The EUA expansion is partially based on the results of a randomized, open-label trial that Gilead Sciences, remdesivir’s manufacturer, conducted at multiple sites.
The trial showed that a 5-day course of remdesivir was associated with statistically significant improvement among patients hospitalized with moderate COVID-19 in comparison with those receiving standard care. However, patients who were randomly assigned to a receive longer, 10-day remdesivir course had not improved significantly 11 days after treatment started, compared with those who received standard care.
Results with remdesivir in this trial and in two previously reported randomized trials varied, “raising the question of whether the discrepancies are artifacts of study design choices, including patient populations, or whether the drug is less efficacious than hoped,” wrote Erin K. McCreary, PharmD, and Derek C. Angus, MD, MPH, with the University of Pittsburgh School of Medicine, in an editorial that accompanied publication of the trials in JAMA.
Angus previously expressed concern that expanding remdesivir’s EUA could “interrupt or thwart efforts to execute the needed RCTs [randomized controlled trials].
“We think there really needs to be further evaluation of remdesivir in large-scale RCTs adequately powered to understand in which patients, at which dose, given at which point in the course of illness leads to what concrete and tangible improvement in clinical outcomes,” he told Medscape Medical News.
“At this point, remdesivir definitely holds promise, but given the cost to produce and distribute the drug, it seems crucial to know with more certainty how best to use it,” Angus said.
The EUA expansion is also partially based on results from a randomized, double-blind, placebo-controlled clinical trial that the National Institutes of Allergy and Infectious Diseases conducted. In that trial, there was a statistically significant reduction in median recovery time and higher odds of clinical improvement after 2 weeks for hospitalized patients who received remdesivir.
For hospitalized patients with mild to moderate disease, the results were consistent with the overall study results but were not statistically significant.
This article first appeared on Medscape.com.
Immunotherapy should not be withheld because of sex, age, or PS
The improvement in survival in many cancer types that is seen with immune checkpoint inhibitors (ICIs), when compared to control therapies, is not affected by the patient’s sex, age, or Eastern Cooperative Oncology Group (ECOG) performance status (PS), according to a new meta-analysis.
Therefore, treatment with these immunotherapies should not be withheld on the basis of these factors, the authors concluded.
Asked whether there have been such instances of withholding ICIs, lead author Yucai Wang, MD, PhD, Mayo Clinic, Rochester, Minnesota, told Medscape Medical News: “We did this study solely based on scientific questions we had and not because we were seeing any bias at the moment in the use of ICIs.
“And we saw that the survival benefits were very similar across all of the categories [we analyzed], with a survival benefit of about 20% from immunotherapy across the board, which is clinically meaningful,” he added.
The study was published online August 7 in JAMA Network Open.
“The comparable survival advantage between patients of different sex, age, and ECOG PS may encourage more patients to receive ICI treatment regardless of cancer types, lines of therapy, agents of immunotherapy, and intervention therapies,” the authors commented.
Wang noted that there have been conflicting reports in the literature suggesting that male patients may benefit more from immunotherapy than female patients and that older patients may benefit more from the same treatment than younger patients.
However, there are also suggestions in the literature that women experience a stronger immune response than men and that, with aging, the immune system generally undergoes immunosenescence.
In addition, the PS of oncology patients has been implicated in how well patients respond to immunotherapy.
Wang noted that the findings of past studies have contradicted each other.
Findings of the Meta-Analysis
The meta-analysis included 37 randomized clinical trials that involved a total of 23,760 patients with a variety of advanced cancers. “Most of the trials were phase 3 (n = 34) and conduced for subsequent lines of therapy (n = 22),” the authors explained.
The most common cancers treated with an ICI were non–small cell lung cancer and melanoma.
Pooled overall survival (OS) hazard ratios (HRs) were calculated on the basis of sex, age (younger than 65 years and 65 years and older), and an ECOG PS of 0 and 1 or higher.
Responses were stratified on the basis of cancer type, line of therapy, the ICI used, and the immunotherapy strategy used in the ICI arm.
Most of the drugs evaluated were PD-1 and PD-L1 inhibitors. The specific drugs assessed included ipilimumab, tremelimumab, nivolumab, pembrolizumab, atezolizumab, durvalumab, and avelumab.
A total of 32 trials that involved more than 20,000 patients reported HRs for death according to the patients’ sex. Thirty-four trials that involved more than 21,000 patients reported HRs for death according to patients’ age, and 30 trials that involved more than 19,000 patients reported HRs for death according to patients’ ECOG PS.
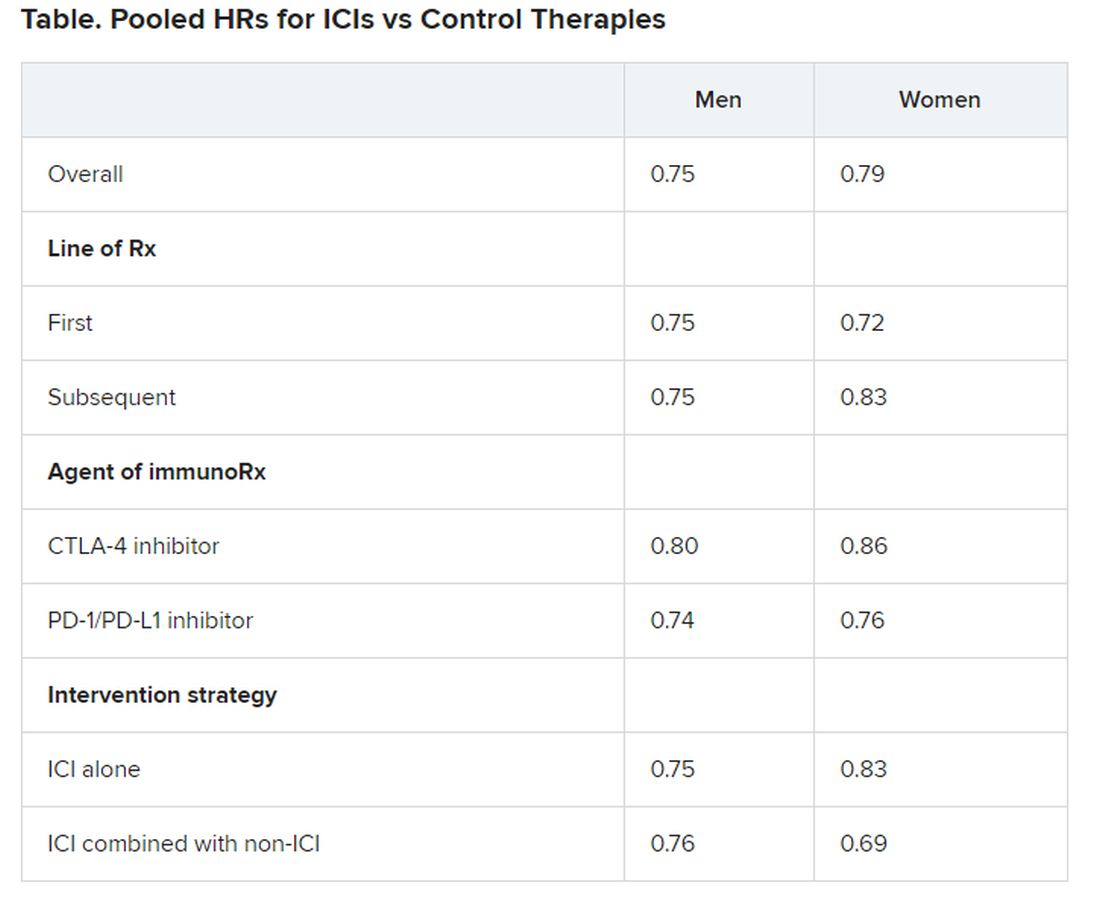
No significant differences in OS benefit were seen by cancer type, line of therapy, agent of immunotherapy, or intervention strategy, the investigators pointed out.
There were also no differences in survival benefit associated with immunotherapy vs control therapies for patients with an ECOG PS of 0 and an ECOG PS of 1 or greater. The OS benefit was 0.81 for those with an ECOG PS of 0 and 0.79 for those with an ECOG PS of 1 or greater.
Wang has disclosed no relevant financial relationships.
This article first appeared on Medscape.com .
The improvement in survival in many cancer types that is seen with immune checkpoint inhibitors (ICIs), when compared to control therapies, is not affected by the patient’s sex, age, or Eastern Cooperative Oncology Group (ECOG) performance status (PS), according to a new meta-analysis.
Therefore, treatment with these immunotherapies should not be withheld on the basis of these factors, the authors concluded.
Asked whether there have been such instances of withholding ICIs, lead author Yucai Wang, MD, PhD, Mayo Clinic, Rochester, Minnesota, told Medscape Medical News: “We did this study solely based on scientific questions we had and not because we were seeing any bias at the moment in the use of ICIs.
“And we saw that the survival benefits were very similar across all of the categories [we analyzed], with a survival benefit of about 20% from immunotherapy across the board, which is clinically meaningful,” he added.
The study was published online August 7 in JAMA Network Open.
“The comparable survival advantage between patients of different sex, age, and ECOG PS may encourage more patients to receive ICI treatment regardless of cancer types, lines of therapy, agents of immunotherapy, and intervention therapies,” the authors commented.
Wang noted that there have been conflicting reports in the literature suggesting that male patients may benefit more from immunotherapy than female patients and that older patients may benefit more from the same treatment than younger patients.
However, there are also suggestions in the literature that women experience a stronger immune response than men and that, with aging, the immune system generally undergoes immunosenescence.
In addition, the PS of oncology patients has been implicated in how well patients respond to immunotherapy.
Wang noted that the findings of past studies have contradicted each other.
Findings of the Meta-Analysis
The meta-analysis included 37 randomized clinical trials that involved a total of 23,760 patients with a variety of advanced cancers. “Most of the trials were phase 3 (n = 34) and conduced for subsequent lines of therapy (n = 22),” the authors explained.
The most common cancers treated with an ICI were non–small cell lung cancer and melanoma.
Pooled overall survival (OS) hazard ratios (HRs) were calculated on the basis of sex, age (younger than 65 years and 65 years and older), and an ECOG PS of 0 and 1 or higher.
Responses were stratified on the basis of cancer type, line of therapy, the ICI used, and the immunotherapy strategy used in the ICI arm.
Most of the drugs evaluated were PD-1 and PD-L1 inhibitors. The specific drugs assessed included ipilimumab, tremelimumab, nivolumab, pembrolizumab, atezolizumab, durvalumab, and avelumab.
A total of 32 trials that involved more than 20,000 patients reported HRs for death according to the patients’ sex. Thirty-four trials that involved more than 21,000 patients reported HRs for death according to patients’ age, and 30 trials that involved more than 19,000 patients reported HRs for death according to patients’ ECOG PS.
No significant differences in OS benefit were seen by cancer type, line of therapy, agent of immunotherapy, or intervention strategy, the investigators pointed out.
There were also no differences in survival benefit associated with immunotherapy vs control therapies for patients with an ECOG PS of 0 and an ECOG PS of 1 or greater. The OS benefit was 0.81 for those with an ECOG PS of 0 and 0.79 for those with an ECOG PS of 1 or greater.
Wang has disclosed no relevant financial relationships.
This article first appeared on Medscape.com .
The improvement in survival in many cancer types that is seen with immune checkpoint inhibitors (ICIs), when compared to control therapies, is not affected by the patient’s sex, age, or Eastern Cooperative Oncology Group (ECOG) performance status (PS), according to a new meta-analysis.
Therefore, treatment with these immunotherapies should not be withheld on the basis of these factors, the authors concluded.
Asked whether there have been such instances of withholding ICIs, lead author Yucai Wang, MD, PhD, Mayo Clinic, Rochester, Minnesota, told Medscape Medical News: “We did this study solely based on scientific questions we had and not because we were seeing any bias at the moment in the use of ICIs.
“And we saw that the survival benefits were very similar across all of the categories [we analyzed], with a survival benefit of about 20% from immunotherapy across the board, which is clinically meaningful,” he added.
The study was published online August 7 in JAMA Network Open.
“The comparable survival advantage between patients of different sex, age, and ECOG PS may encourage more patients to receive ICI treatment regardless of cancer types, lines of therapy, agents of immunotherapy, and intervention therapies,” the authors commented.
Wang noted that there have been conflicting reports in the literature suggesting that male patients may benefit more from immunotherapy than female patients and that older patients may benefit more from the same treatment than younger patients.
However, there are also suggestions in the literature that women experience a stronger immune response than men and that, with aging, the immune system generally undergoes immunosenescence.
In addition, the PS of oncology patients has been implicated in how well patients respond to immunotherapy.
Wang noted that the findings of past studies have contradicted each other.
Findings of the Meta-Analysis
The meta-analysis included 37 randomized clinical trials that involved a total of 23,760 patients with a variety of advanced cancers. “Most of the trials were phase 3 (n = 34) and conduced for subsequent lines of therapy (n = 22),” the authors explained.
The most common cancers treated with an ICI were non–small cell lung cancer and melanoma.
Pooled overall survival (OS) hazard ratios (HRs) were calculated on the basis of sex, age (younger than 65 years and 65 years and older), and an ECOG PS of 0 and 1 or higher.
Responses were stratified on the basis of cancer type, line of therapy, the ICI used, and the immunotherapy strategy used in the ICI arm.
Most of the drugs evaluated were PD-1 and PD-L1 inhibitors. The specific drugs assessed included ipilimumab, tremelimumab, nivolumab, pembrolizumab, atezolizumab, durvalumab, and avelumab.
A total of 32 trials that involved more than 20,000 patients reported HRs for death according to the patients’ sex. Thirty-four trials that involved more than 21,000 patients reported HRs for death according to patients’ age, and 30 trials that involved more than 19,000 patients reported HRs for death according to patients’ ECOG PS.
No significant differences in OS benefit were seen by cancer type, line of therapy, agent of immunotherapy, or intervention strategy, the investigators pointed out.
There were also no differences in survival benefit associated with immunotherapy vs control therapies for patients with an ECOG PS of 0 and an ECOG PS of 1 or greater. The OS benefit was 0.81 for those with an ECOG PS of 0 and 0.79 for those with an ECOG PS of 1 or greater.
Wang has disclosed no relevant financial relationships.
This article first appeared on Medscape.com .
Aspirin may accelerate cancer progression in older adults
Aspirin may accelerate the progression of advanced cancers and lead to an earlier death as a result, new data from the ASPREE study suggest.
The results showed that patients 65 years and older who started taking daily low-dose aspirin had a 19% higher chance of being diagnosed with metastatic cancer, a 22% higher chance of being diagnosed with a stage 4 tumor, and a 31% increased risk of death from stage 4 cancer, when compared with patients who took a placebo.
John J. McNeil, MBBS, PhD, of Monash University in Melbourne, Australia, and colleagues detailed these findings in the Journal of the National Cancer Institute.
“If confirmed, the clinical implications of these findings could be important for the use of aspirin in an older population,” the authors wrote.
When results of the ASPREE study were first reported in 2018, they “raised important concerns,” Ernest Hawk, MD, and Karen Colbert Maresso wrote in an editorial related to the current publication.
“Unlike ARRIVE, ASCEND, and nearly all prior primary prevention CVD [cardiovascular disease] trials of aspirin, ASPREE surprisingly demonstrated increased all-cause mortality in the aspirin group, which appeared to be driven largely by an increase in cancer-related deaths,” wrote the editorialists, who are both from the University of Texas MD Anderson Cancer Center in Houston.
Even though the ASPREE investigators have now taken a deeper dive into their data, the findings “neither explain nor alleviate the concerns raised by the initial ASPREE report,” the editorialists noted.
ASPREE design and results
ASPREE is a multicenter, double-blind trial of 19,114 older adults living in Australia (n = 16,703) or the United States (n = 2,411). Most patients were 70 years or older at baseline. However, the U.S. group also included patients 65 years and older who were racial/ethnic minorities (n = 564).
Patients were randomized to receive 100 mg of enteric-coated aspirin daily (n = 9,525) or matching placebo (n = 9,589) from March 2010 through December 2014.
At inclusion, all participants were free from cardiovascular disease, dementia, or physical disability. A previous history of cancer was not used to exclude participants, and 19.1% of patients had cancer at randomization. Most patients (89%) had not used aspirin regularly before entering the trial.
At a median follow-up of 4.7 years, there were 981 incident cancer events in the aspirin-treated group and 952 in the placebo-treated group, with an overall incident cancer rate of 10.1%.
Of the 1,933 patients with newly diagnosed cancer, 65.7% had a localized cancer, 18.8% had a new metastatic cancer, 5.8% had metastatic disease from an existing cancer, and 9.7% had a new hematologic or lymphatic cancer.
A quarter of cancer patients (n = 495) died as a result of their malignancy, with 52 dying from a cancer they already had at randomization.
Aspirin was not associated with the risk of first incident cancer diagnosis or incident localized cancer diagnosis. The hazard ratios were 1.04 for all incident cancers (95% confidence interval, 0.95-1.14) and 0.99 for incident localized cancers (95% CI, 0.89-1.11).
However, aspirin was associated with an increased risk of metastatic cancer and cancer presenting at stage 4. The HR for metastatic cancer was 1.19 (95% CI, 1.00-1.43), and the HR for newly diagnosed stage 4 cancer was 1.22 (95% CI, 1.02-1.45).
Furthermore, “an increased progression to death was observed amongst those randomized to aspirin, regardless of whether the initial cancer presentation had been localized or metastatic,” the investigators wrote.
The HRs for death were 1.35 for all cancers (95% CI, 1.13-1.61), 1.47 for localized cancers (95% CI, 1.07-2.02), and 1.30 for metastatic cancers (95% CI, 1.03-1.63).
“Deaths were particularly high among those on aspirin who were diagnosed with advanced solid cancers,” study author Andrew Chan, MD, of Massachusetts General Hospital in Boston, said in a press statement.
Indeed, HRs for death in patients with solid tumors presenting at stage 3 and 4 were a respective 2.11 (95% CI, 1.03-4.33) and 1.31 (95% CI, 1.04-1.64). This suggests a possible adverse effect of aspirin on the growth of cancers once they have already developed in older adults, Dr. Chan said.
Where does that leave aspirin for cancer prevention?
“Although these results suggest that we should be cautious about starting aspirin therapy in otherwise healthy older adults, this does not mean that individuals who are already taking aspirin – particularly if they began taking it at a younger age – should stop their aspirin regimen,” Dr. Chan said.
There are decades of data supporting the use of daily aspirin to prevent multiple cancer types, particularly colorectal cancer, in individuals under the age of 70 years. In a recent meta-analysis, for example, regular aspirin use was linked to a 27% reduced risk for colorectal cancer, a 33% reduced risk for squamous cell esophageal cancer, a 39% decreased risk for adenocarcinoma of the esophagus and gastric cardia, a 36% decreased risk for stomach cancer, a 38% decreased risk for hepatobiliary tract cancer, and a 22% decreased risk for pancreatic cancer.
While these figures are mostly based on observational and case-control studies, it “reaffirms the fact that, overall, when you look at all of the ages, that there is still a benefit of aspirin for cancer,” John Cuzick, PhD, of Queen Mary University of London (England), said in an interview.
In fact, the meta-analysis goes as far as suggesting that perhaps the dose of aspirin being used is too low, with the authors noting that there was a 35% risk reduction in colorectal cancer with a dose of 325 mg daily. That’s a new finding, Dr. Cuzick said.
He noted that the ASPREE study largely consists of patients 70 years of age or older, and the authors “draw some conclusions which we can’t ignore about potential safety.”
One of the safety concerns is the increased risk for gastrointestinal bleeding, which is why Dr. Cuzick and colleagues previously recommended caution in the use of aspirin to prevent cancer in elderly patients. The group published a study in 2015 that suggested a benefit of taking aspirin daily for 5-10 years in patients aged 50-65 years, but the risk/benefit ratio was unclear for patients 70 years and older.
The ASPREE data now add to those uncertainties and suggest “there may be some side effects that we do not understand,” Dr. Cuzick said.
“I’m still optimistic that aspirin is going to be important for cancer prevention, but probably focusing on ages 50-70,” he added. “[The ASPREE data] reinforce the caution that we have to take in terms of trying to understand what the side effects are and what’s going on at these older ages.”
Dr. Cuzick is currently leading the AsCaP Project, an international effort to better understand why aspirin might work in preventing some cancer types but not others. AsCaP is supported by Cancer Research UK and also includes Dr. Chan among the researchers attempting to find out which patients may benefit the most from aspirin and which may be at greater risk of adverse effects.
The ASPREE trial was funded by grants from the National Institute on Aging, the National Cancer Institute, the National Health and Medical Research Council of Australia, Monash University, and the Victorian Cancer Agency. Several ASPREE investigators disclosed financial relationships with Bayer Pharma. The editorialists had no conflicts of interest. Dr. Cuzick has been an advisory board member for Bayer in the past.
SOURCE: McNeil J et al. J Natl Cancer Inst. 2020 Aug 11. doi: 10.1093/jnci/djaa114.
Aspirin may accelerate the progression of advanced cancers and lead to an earlier death as a result, new data from the ASPREE study suggest.
The results showed that patients 65 years and older who started taking daily low-dose aspirin had a 19% higher chance of being diagnosed with metastatic cancer, a 22% higher chance of being diagnosed with a stage 4 tumor, and a 31% increased risk of death from stage 4 cancer, when compared with patients who took a placebo.
John J. McNeil, MBBS, PhD, of Monash University in Melbourne, Australia, and colleagues detailed these findings in the Journal of the National Cancer Institute.
“If confirmed, the clinical implications of these findings could be important for the use of aspirin in an older population,” the authors wrote.
When results of the ASPREE study were first reported in 2018, they “raised important concerns,” Ernest Hawk, MD, and Karen Colbert Maresso wrote in an editorial related to the current publication.
“Unlike ARRIVE, ASCEND, and nearly all prior primary prevention CVD [cardiovascular disease] trials of aspirin, ASPREE surprisingly demonstrated increased all-cause mortality in the aspirin group, which appeared to be driven largely by an increase in cancer-related deaths,” wrote the editorialists, who are both from the University of Texas MD Anderson Cancer Center in Houston.
Even though the ASPREE investigators have now taken a deeper dive into their data, the findings “neither explain nor alleviate the concerns raised by the initial ASPREE report,” the editorialists noted.
ASPREE design and results
ASPREE is a multicenter, double-blind trial of 19,114 older adults living in Australia (n = 16,703) or the United States (n = 2,411). Most patients were 70 years or older at baseline. However, the U.S. group also included patients 65 years and older who were racial/ethnic minorities (n = 564).
Patients were randomized to receive 100 mg of enteric-coated aspirin daily (n = 9,525) or matching placebo (n = 9,589) from March 2010 through December 2014.
At inclusion, all participants were free from cardiovascular disease, dementia, or physical disability. A previous history of cancer was not used to exclude participants, and 19.1% of patients had cancer at randomization. Most patients (89%) had not used aspirin regularly before entering the trial.
At a median follow-up of 4.7 years, there were 981 incident cancer events in the aspirin-treated group and 952 in the placebo-treated group, with an overall incident cancer rate of 10.1%.
Of the 1,933 patients with newly diagnosed cancer, 65.7% had a localized cancer, 18.8% had a new metastatic cancer, 5.8% had metastatic disease from an existing cancer, and 9.7% had a new hematologic or lymphatic cancer.
A quarter of cancer patients (n = 495) died as a result of their malignancy, with 52 dying from a cancer they already had at randomization.
Aspirin was not associated with the risk of first incident cancer diagnosis or incident localized cancer diagnosis. The hazard ratios were 1.04 for all incident cancers (95% confidence interval, 0.95-1.14) and 0.99 for incident localized cancers (95% CI, 0.89-1.11).
However, aspirin was associated with an increased risk of metastatic cancer and cancer presenting at stage 4. The HR for metastatic cancer was 1.19 (95% CI, 1.00-1.43), and the HR for newly diagnosed stage 4 cancer was 1.22 (95% CI, 1.02-1.45).
Furthermore, “an increased progression to death was observed amongst those randomized to aspirin, regardless of whether the initial cancer presentation had been localized or metastatic,” the investigators wrote.
The HRs for death were 1.35 for all cancers (95% CI, 1.13-1.61), 1.47 for localized cancers (95% CI, 1.07-2.02), and 1.30 for metastatic cancers (95% CI, 1.03-1.63).
“Deaths were particularly high among those on aspirin who were diagnosed with advanced solid cancers,” study author Andrew Chan, MD, of Massachusetts General Hospital in Boston, said in a press statement.
Indeed, HRs for death in patients with solid tumors presenting at stage 3 and 4 were a respective 2.11 (95% CI, 1.03-4.33) and 1.31 (95% CI, 1.04-1.64). This suggests a possible adverse effect of aspirin on the growth of cancers once they have already developed in older adults, Dr. Chan said.
Where does that leave aspirin for cancer prevention?
“Although these results suggest that we should be cautious about starting aspirin therapy in otherwise healthy older adults, this does not mean that individuals who are already taking aspirin – particularly if they began taking it at a younger age – should stop their aspirin regimen,” Dr. Chan said.
There are decades of data supporting the use of daily aspirin to prevent multiple cancer types, particularly colorectal cancer, in individuals under the age of 70 years. In a recent meta-analysis, for example, regular aspirin use was linked to a 27% reduced risk for colorectal cancer, a 33% reduced risk for squamous cell esophageal cancer, a 39% decreased risk for adenocarcinoma of the esophagus and gastric cardia, a 36% decreased risk for stomach cancer, a 38% decreased risk for hepatobiliary tract cancer, and a 22% decreased risk for pancreatic cancer.
While these figures are mostly based on observational and case-control studies, it “reaffirms the fact that, overall, when you look at all of the ages, that there is still a benefit of aspirin for cancer,” John Cuzick, PhD, of Queen Mary University of London (England), said in an interview.
In fact, the meta-analysis goes as far as suggesting that perhaps the dose of aspirin being used is too low, with the authors noting that there was a 35% risk reduction in colorectal cancer with a dose of 325 mg daily. That’s a new finding, Dr. Cuzick said.
He noted that the ASPREE study largely consists of patients 70 years of age or older, and the authors “draw some conclusions which we can’t ignore about potential safety.”
One of the safety concerns is the increased risk for gastrointestinal bleeding, which is why Dr. Cuzick and colleagues previously recommended caution in the use of aspirin to prevent cancer in elderly patients. The group published a study in 2015 that suggested a benefit of taking aspirin daily for 5-10 years in patients aged 50-65 years, but the risk/benefit ratio was unclear for patients 70 years and older.
The ASPREE data now add to those uncertainties and suggest “there may be some side effects that we do not understand,” Dr. Cuzick said.
“I’m still optimistic that aspirin is going to be important for cancer prevention, but probably focusing on ages 50-70,” he added. “[The ASPREE data] reinforce the caution that we have to take in terms of trying to understand what the side effects are and what’s going on at these older ages.”
Dr. Cuzick is currently leading the AsCaP Project, an international effort to better understand why aspirin might work in preventing some cancer types but not others. AsCaP is supported by Cancer Research UK and also includes Dr. Chan among the researchers attempting to find out which patients may benefit the most from aspirin and which may be at greater risk of adverse effects.
The ASPREE trial was funded by grants from the National Institute on Aging, the National Cancer Institute, the National Health and Medical Research Council of Australia, Monash University, and the Victorian Cancer Agency. Several ASPREE investigators disclosed financial relationships with Bayer Pharma. The editorialists had no conflicts of interest. Dr. Cuzick has been an advisory board member for Bayer in the past.
SOURCE: McNeil J et al. J Natl Cancer Inst. 2020 Aug 11. doi: 10.1093/jnci/djaa114.
Aspirin may accelerate the progression of advanced cancers and lead to an earlier death as a result, new data from the ASPREE study suggest.
The results showed that patients 65 years and older who started taking daily low-dose aspirin had a 19% higher chance of being diagnosed with metastatic cancer, a 22% higher chance of being diagnosed with a stage 4 tumor, and a 31% increased risk of death from stage 4 cancer, when compared with patients who took a placebo.
John J. McNeil, MBBS, PhD, of Monash University in Melbourne, Australia, and colleagues detailed these findings in the Journal of the National Cancer Institute.
“If confirmed, the clinical implications of these findings could be important for the use of aspirin in an older population,” the authors wrote.
When results of the ASPREE study were first reported in 2018, they “raised important concerns,” Ernest Hawk, MD, and Karen Colbert Maresso wrote in an editorial related to the current publication.
“Unlike ARRIVE, ASCEND, and nearly all prior primary prevention CVD [cardiovascular disease] trials of aspirin, ASPREE surprisingly demonstrated increased all-cause mortality in the aspirin group, which appeared to be driven largely by an increase in cancer-related deaths,” wrote the editorialists, who are both from the University of Texas MD Anderson Cancer Center in Houston.
Even though the ASPREE investigators have now taken a deeper dive into their data, the findings “neither explain nor alleviate the concerns raised by the initial ASPREE report,” the editorialists noted.
ASPREE design and results
ASPREE is a multicenter, double-blind trial of 19,114 older adults living in Australia (n = 16,703) or the United States (n = 2,411). Most patients were 70 years or older at baseline. However, the U.S. group also included patients 65 years and older who were racial/ethnic minorities (n = 564).
Patients were randomized to receive 100 mg of enteric-coated aspirin daily (n = 9,525) or matching placebo (n = 9,589) from March 2010 through December 2014.
At inclusion, all participants were free from cardiovascular disease, dementia, or physical disability. A previous history of cancer was not used to exclude participants, and 19.1% of patients had cancer at randomization. Most patients (89%) had not used aspirin regularly before entering the trial.
At a median follow-up of 4.7 years, there were 981 incident cancer events in the aspirin-treated group and 952 in the placebo-treated group, with an overall incident cancer rate of 10.1%.
Of the 1,933 patients with newly diagnosed cancer, 65.7% had a localized cancer, 18.8% had a new metastatic cancer, 5.8% had metastatic disease from an existing cancer, and 9.7% had a new hematologic or lymphatic cancer.
A quarter of cancer patients (n = 495) died as a result of their malignancy, with 52 dying from a cancer they already had at randomization.
Aspirin was not associated with the risk of first incident cancer diagnosis or incident localized cancer diagnosis. The hazard ratios were 1.04 for all incident cancers (95% confidence interval, 0.95-1.14) and 0.99 for incident localized cancers (95% CI, 0.89-1.11).
However, aspirin was associated with an increased risk of metastatic cancer and cancer presenting at stage 4. The HR for metastatic cancer was 1.19 (95% CI, 1.00-1.43), and the HR for newly diagnosed stage 4 cancer was 1.22 (95% CI, 1.02-1.45).
Furthermore, “an increased progression to death was observed amongst those randomized to aspirin, regardless of whether the initial cancer presentation had been localized or metastatic,” the investigators wrote.
The HRs for death were 1.35 for all cancers (95% CI, 1.13-1.61), 1.47 for localized cancers (95% CI, 1.07-2.02), and 1.30 for metastatic cancers (95% CI, 1.03-1.63).
“Deaths were particularly high among those on aspirin who were diagnosed with advanced solid cancers,” study author Andrew Chan, MD, of Massachusetts General Hospital in Boston, said in a press statement.
Indeed, HRs for death in patients with solid tumors presenting at stage 3 and 4 were a respective 2.11 (95% CI, 1.03-4.33) and 1.31 (95% CI, 1.04-1.64). This suggests a possible adverse effect of aspirin on the growth of cancers once they have already developed in older adults, Dr. Chan said.
Where does that leave aspirin for cancer prevention?
“Although these results suggest that we should be cautious about starting aspirin therapy in otherwise healthy older adults, this does not mean that individuals who are already taking aspirin – particularly if they began taking it at a younger age – should stop their aspirin regimen,” Dr. Chan said.
There are decades of data supporting the use of daily aspirin to prevent multiple cancer types, particularly colorectal cancer, in individuals under the age of 70 years. In a recent meta-analysis, for example, regular aspirin use was linked to a 27% reduced risk for colorectal cancer, a 33% reduced risk for squamous cell esophageal cancer, a 39% decreased risk for adenocarcinoma of the esophagus and gastric cardia, a 36% decreased risk for stomach cancer, a 38% decreased risk for hepatobiliary tract cancer, and a 22% decreased risk for pancreatic cancer.
While these figures are mostly based on observational and case-control studies, it “reaffirms the fact that, overall, when you look at all of the ages, that there is still a benefit of aspirin for cancer,” John Cuzick, PhD, of Queen Mary University of London (England), said in an interview.
In fact, the meta-analysis goes as far as suggesting that perhaps the dose of aspirin being used is too low, with the authors noting that there was a 35% risk reduction in colorectal cancer with a dose of 325 mg daily. That’s a new finding, Dr. Cuzick said.
He noted that the ASPREE study largely consists of patients 70 years of age or older, and the authors “draw some conclusions which we can’t ignore about potential safety.”
One of the safety concerns is the increased risk for gastrointestinal bleeding, which is why Dr. Cuzick and colleagues previously recommended caution in the use of aspirin to prevent cancer in elderly patients. The group published a study in 2015 that suggested a benefit of taking aspirin daily for 5-10 years in patients aged 50-65 years, but the risk/benefit ratio was unclear for patients 70 years and older.
The ASPREE data now add to those uncertainties and suggest “there may be some side effects that we do not understand,” Dr. Cuzick said.
“I’m still optimistic that aspirin is going to be important for cancer prevention, but probably focusing on ages 50-70,” he added. “[The ASPREE data] reinforce the caution that we have to take in terms of trying to understand what the side effects are and what’s going on at these older ages.”
Dr. Cuzick is currently leading the AsCaP Project, an international effort to better understand why aspirin might work in preventing some cancer types but not others. AsCaP is supported by Cancer Research UK and also includes Dr. Chan among the researchers attempting to find out which patients may benefit the most from aspirin and which may be at greater risk of adverse effects.
The ASPREE trial was funded by grants from the National Institute on Aging, the National Cancer Institute, the National Health and Medical Research Council of Australia, Monash University, and the Victorian Cancer Agency. Several ASPREE investigators disclosed financial relationships with Bayer Pharma. The editorialists had no conflicts of interest. Dr. Cuzick has been an advisory board member for Bayer in the past.
SOURCE: McNeil J et al. J Natl Cancer Inst. 2020 Aug 11. doi: 10.1093/jnci/djaa114.
FROM JOURNAL OF THE NATIONAL CANCER INSTITUTE
Nine antihypertensive drugs associated with reduced risk of depression
The risk of depression is elevated in patients with cardiovascular diseases, but several specific antihypertensive therapies are associated with a reduced risk, and none appear to increase the risk, according to a population-based study that evaluated 10 years of data in nearly 4 million subjects.
“As the first study on individual antihypertensives and risk of depression, we found a decreased risk of depression with nine drugs,” reported a collaborative group of investigators from multiple institutions in Denmark where the study was undertaken.
In a study period spanning from 2005 to 2015, risk of a diagnosis of depression was evaluated in patients taking any of 41 antihypertensive therapies in four major categories. These were identified as angiotensin agents (ACE inhibitors or angiotensin II receptor blockers), calcium antagonists, beta-blockers, and diuretics.
Within these groups, agents associated with a reduced risk of depression were: two angiotensin agents, enalapril and ramipril; three calcium antagonists, amlodipine, verapamil, and verapamil combinations; and four beta-blockers, propranolol, atenolol, bisoprolol, and carvedilol. The remaining drugs in these classes and diuretics were not associated with a reduced risk of depression. However, no antihypertensive agent was linked to an increased risk of depression.
All people living in Denmark are assigned a unique personal identification number that permits health information to be tracked across multiple registers. In this study, information was linked for several registries, including the Danish Medical Register on Vital Statistics, the Medicinal Product Statistics, and the Danish Psychiatric Central Register.
Data from a total of 3.75 million patients exposed to antihypertensive therapy during the study period were evaluated. Roughly 1 million of them were exposed to angiotensin drugs and slightly more than a million were exposed to diuretics. For calcium antagonists or beta-blockers, the numbers were approximately 835,000 and 775,000, respectively.
After adjustment for such factors as concomitant somatic diagnoses, sex, age, and employment status, the hazard ratios for depression among drugs associated with protection identified a risk reduction of 10%-25% in most cases when those who had been given 6-10 prescriptions or more than 10 prescriptions were compared with those who received 2 or fewer.
At the level of 10 or more prescriptions, for example, the risk reductions were 17% for ramipril (HR, 0.83; 95% CI, 0.78-0.89), 8% for enalapril (HR, 0.92; 95% CI, 0.88-0.96), 18% for amlodipine (HR, 0.82; 95% CI, 0.79-0.86), 15% for verapamil (HR, 0.85; 95% CI, 0.79-0.83), 28% for propranolol (HR, 0.72; 95% CI, 0.67-0.77), 20% for atenolol (HR, 0.80; 95% CI, 0.74-0.86), 25% for bisoprolol (HR, 0.75; 95% CI, 0.67-0.84), and 16% for carvedilol (HR, 0.84; 95% CI, 0.75-0.95).
For verapamil combinations, the risk reduction was 67% (HR, 0.33; 95% CI, 0.17-0.63), but the investigators cautioned that only 130 individuals were exposed to verapamil combinations, limiting the reliability of this analysis.
Interpreting the findings
A study hypothesis, the observed protective effect against depression, was expected for angiotensin drugs and calcium-channel blockers, but not for beta-blockers, according to the investigators.
“The renin-angiotensin systems is one of the pathways known to modulate inflammation in the central nervous system and seems involved in the regulation of the stress response. Angiotensin agents may also exert anti-inflammatory effects,” the investigators explained. “Dysregulation of intracellular calcium is evident in depression, including receptor-regulated calcium signaling.”
In contrast, beta-blockers have been associated with increased risk of depression in some but not all studies, according to the investigators. They maintained that some clinicians avoid these agents in patients with a history of mood disorders.
In attempting to account for the variability within drug classes regarding protection and lack of protection against depression, the investigators speculated that differences in pharmacologic properties, such as relative lipophilicity or anti-inflammatory effect, might be important.
Despite the large amount of data, William B. White, MD, professor emeritus at the Calhoun Cardiology Center, University of Connecticut, Farmington, is not convinced.
“In observational studies, even those with very large samples sizes, bias and confounding are hard to extricate with controls and propensity-score matching,” Dr. White said. From his perspective, the protective effects of some but not all drugs within a class “give one the impression that the findings are likely random.”
A member of the editorial board of the journal in which this study appeared, Dr. White said he was not involved in the review of the manuscript. Ultimately, he believed that the results are difficult to interpret.
“For example, there is no plausible rationale for why 2 of the 16 ACE inhibitors or angiotensin II receptor blockers or 4 of the 15 beta-blockers or 3 of the 10 calcium-channel blockers would reduce depression while the others in the class would have no effect,” he said.
Despite the investigators’ conclusion that these data should drive drug choice for patients at risk of depression, “I would say the results of this analysis would not lead me to alter clinical practice,” Dr. White added.
According to the principal investigator of the study, Lars Vedel Kessing, MD, DSc, professor of psychiatry at the University of Copenhagen, many variables affect choice of antihypertensive drug. However, the depression risk is elevated in patients with cardiovascular or cerebrovascular disease and hypertension.
When risk of a mood disorder is a concern, use of one of the nine drugs associated with protection from depression should be considered, “especially in patients at increased risk of developing depression, including patients with prior depression or anxiety and patients with a family history of depression,” he and his coinvestigators concluded.
However, Dr. Kessing said in an interview that the data do not help with individual treatment choices. “We do not compare different antihypertensives against each other due to the risk of confounding by indications, so, no, it is not reasonable to consider relative risk among specific agents.”
The authors reported no potential conflicts of interest involving this topic.
SOURCE: Kessing LV et al. Hypertension. 2020 Aug 24. doi: 10.1161/HYPERTENSIONAHA.120.15605.
The risk of depression is elevated in patients with cardiovascular diseases, but several specific antihypertensive therapies are associated with a reduced risk, and none appear to increase the risk, according to a population-based study that evaluated 10 years of data in nearly 4 million subjects.
“As the first study on individual antihypertensives and risk of depression, we found a decreased risk of depression with nine drugs,” reported a collaborative group of investigators from multiple institutions in Denmark where the study was undertaken.
In a study period spanning from 2005 to 2015, risk of a diagnosis of depression was evaluated in patients taking any of 41 antihypertensive therapies in four major categories. These were identified as angiotensin agents (ACE inhibitors or angiotensin II receptor blockers), calcium antagonists, beta-blockers, and diuretics.
Within these groups, agents associated with a reduced risk of depression were: two angiotensin agents, enalapril and ramipril; three calcium antagonists, amlodipine, verapamil, and verapamil combinations; and four beta-blockers, propranolol, atenolol, bisoprolol, and carvedilol. The remaining drugs in these classes and diuretics were not associated with a reduced risk of depression. However, no antihypertensive agent was linked to an increased risk of depression.
All people living in Denmark are assigned a unique personal identification number that permits health information to be tracked across multiple registers. In this study, information was linked for several registries, including the Danish Medical Register on Vital Statistics, the Medicinal Product Statistics, and the Danish Psychiatric Central Register.
Data from a total of 3.75 million patients exposed to antihypertensive therapy during the study period were evaluated. Roughly 1 million of them were exposed to angiotensin drugs and slightly more than a million were exposed to diuretics. For calcium antagonists or beta-blockers, the numbers were approximately 835,000 and 775,000, respectively.
After adjustment for such factors as concomitant somatic diagnoses, sex, age, and employment status, the hazard ratios for depression among drugs associated with protection identified a risk reduction of 10%-25% in most cases when those who had been given 6-10 prescriptions or more than 10 prescriptions were compared with those who received 2 or fewer.
At the level of 10 or more prescriptions, for example, the risk reductions were 17% for ramipril (HR, 0.83; 95% CI, 0.78-0.89), 8% for enalapril (HR, 0.92; 95% CI, 0.88-0.96), 18% for amlodipine (HR, 0.82; 95% CI, 0.79-0.86), 15% for verapamil (HR, 0.85; 95% CI, 0.79-0.83), 28% for propranolol (HR, 0.72; 95% CI, 0.67-0.77), 20% for atenolol (HR, 0.80; 95% CI, 0.74-0.86), 25% for bisoprolol (HR, 0.75; 95% CI, 0.67-0.84), and 16% for carvedilol (HR, 0.84; 95% CI, 0.75-0.95).
For verapamil combinations, the risk reduction was 67% (HR, 0.33; 95% CI, 0.17-0.63), but the investigators cautioned that only 130 individuals were exposed to verapamil combinations, limiting the reliability of this analysis.
Interpreting the findings
A study hypothesis, the observed protective effect against depression, was expected for angiotensin drugs and calcium-channel blockers, but not for beta-blockers, according to the investigators.
“The renin-angiotensin systems is one of the pathways known to modulate inflammation in the central nervous system and seems involved in the regulation of the stress response. Angiotensin agents may also exert anti-inflammatory effects,” the investigators explained. “Dysregulation of intracellular calcium is evident in depression, including receptor-regulated calcium signaling.”
In contrast, beta-blockers have been associated with increased risk of depression in some but not all studies, according to the investigators. They maintained that some clinicians avoid these agents in patients with a history of mood disorders.
In attempting to account for the variability within drug classes regarding protection and lack of protection against depression, the investigators speculated that differences in pharmacologic properties, such as relative lipophilicity or anti-inflammatory effect, might be important.
Despite the large amount of data, William B. White, MD, professor emeritus at the Calhoun Cardiology Center, University of Connecticut, Farmington, is not convinced.
“In observational studies, even those with very large samples sizes, bias and confounding are hard to extricate with controls and propensity-score matching,” Dr. White said. From his perspective, the protective effects of some but not all drugs within a class “give one the impression that the findings are likely random.”
A member of the editorial board of the journal in which this study appeared, Dr. White said he was not involved in the review of the manuscript. Ultimately, he believed that the results are difficult to interpret.
“For example, there is no plausible rationale for why 2 of the 16 ACE inhibitors or angiotensin II receptor blockers or 4 of the 15 beta-blockers or 3 of the 10 calcium-channel blockers would reduce depression while the others in the class would have no effect,” he said.
Despite the investigators’ conclusion that these data should drive drug choice for patients at risk of depression, “I would say the results of this analysis would not lead me to alter clinical practice,” Dr. White added.
According to the principal investigator of the study, Lars Vedel Kessing, MD, DSc, professor of psychiatry at the University of Copenhagen, many variables affect choice of antihypertensive drug. However, the depression risk is elevated in patients with cardiovascular or cerebrovascular disease and hypertension.
When risk of a mood disorder is a concern, use of one of the nine drugs associated with protection from depression should be considered, “especially in patients at increased risk of developing depression, including patients with prior depression or anxiety and patients with a family history of depression,” he and his coinvestigators concluded.
However, Dr. Kessing said in an interview that the data do not help with individual treatment choices. “We do not compare different antihypertensives against each other due to the risk of confounding by indications, so, no, it is not reasonable to consider relative risk among specific agents.”
The authors reported no potential conflicts of interest involving this topic.
SOURCE: Kessing LV et al. Hypertension. 2020 Aug 24. doi: 10.1161/HYPERTENSIONAHA.120.15605.
The risk of depression is elevated in patients with cardiovascular diseases, but several specific antihypertensive therapies are associated with a reduced risk, and none appear to increase the risk, according to a population-based study that evaluated 10 years of data in nearly 4 million subjects.
“As the first study on individual antihypertensives and risk of depression, we found a decreased risk of depression with nine drugs,” reported a collaborative group of investigators from multiple institutions in Denmark where the study was undertaken.
In a study period spanning from 2005 to 2015, risk of a diagnosis of depression was evaluated in patients taking any of 41 antihypertensive therapies in four major categories. These were identified as angiotensin agents (ACE inhibitors or angiotensin II receptor blockers), calcium antagonists, beta-blockers, and diuretics.
Within these groups, agents associated with a reduced risk of depression were: two angiotensin agents, enalapril and ramipril; three calcium antagonists, amlodipine, verapamil, and verapamil combinations; and four beta-blockers, propranolol, atenolol, bisoprolol, and carvedilol. The remaining drugs in these classes and diuretics were not associated with a reduced risk of depression. However, no antihypertensive agent was linked to an increased risk of depression.
All people living in Denmark are assigned a unique personal identification number that permits health information to be tracked across multiple registers. In this study, information was linked for several registries, including the Danish Medical Register on Vital Statistics, the Medicinal Product Statistics, and the Danish Psychiatric Central Register.
Data from a total of 3.75 million patients exposed to antihypertensive therapy during the study period were evaluated. Roughly 1 million of them were exposed to angiotensin drugs and slightly more than a million were exposed to diuretics. For calcium antagonists or beta-blockers, the numbers were approximately 835,000 and 775,000, respectively.
After adjustment for such factors as concomitant somatic diagnoses, sex, age, and employment status, the hazard ratios for depression among drugs associated with protection identified a risk reduction of 10%-25% in most cases when those who had been given 6-10 prescriptions or more than 10 prescriptions were compared with those who received 2 or fewer.
At the level of 10 or more prescriptions, for example, the risk reductions were 17% for ramipril (HR, 0.83; 95% CI, 0.78-0.89), 8% for enalapril (HR, 0.92; 95% CI, 0.88-0.96), 18% for amlodipine (HR, 0.82; 95% CI, 0.79-0.86), 15% for verapamil (HR, 0.85; 95% CI, 0.79-0.83), 28% for propranolol (HR, 0.72; 95% CI, 0.67-0.77), 20% for atenolol (HR, 0.80; 95% CI, 0.74-0.86), 25% for bisoprolol (HR, 0.75; 95% CI, 0.67-0.84), and 16% for carvedilol (HR, 0.84; 95% CI, 0.75-0.95).
For verapamil combinations, the risk reduction was 67% (HR, 0.33; 95% CI, 0.17-0.63), but the investigators cautioned that only 130 individuals were exposed to verapamil combinations, limiting the reliability of this analysis.
Interpreting the findings
A study hypothesis, the observed protective effect against depression, was expected for angiotensin drugs and calcium-channel blockers, but not for beta-blockers, according to the investigators.
“The renin-angiotensin systems is one of the pathways known to modulate inflammation in the central nervous system and seems involved in the regulation of the stress response. Angiotensin agents may also exert anti-inflammatory effects,” the investigators explained. “Dysregulation of intracellular calcium is evident in depression, including receptor-regulated calcium signaling.”
In contrast, beta-blockers have been associated with increased risk of depression in some but not all studies, according to the investigators. They maintained that some clinicians avoid these agents in patients with a history of mood disorders.
In attempting to account for the variability within drug classes regarding protection and lack of protection against depression, the investigators speculated that differences in pharmacologic properties, such as relative lipophilicity or anti-inflammatory effect, might be important.
Despite the large amount of data, William B. White, MD, professor emeritus at the Calhoun Cardiology Center, University of Connecticut, Farmington, is not convinced.
“In observational studies, even those with very large samples sizes, bias and confounding are hard to extricate with controls and propensity-score matching,” Dr. White said. From his perspective, the protective effects of some but not all drugs within a class “give one the impression that the findings are likely random.”
A member of the editorial board of the journal in which this study appeared, Dr. White said he was not involved in the review of the manuscript. Ultimately, he believed that the results are difficult to interpret.
“For example, there is no plausible rationale for why 2 of the 16 ACE inhibitors or angiotensin II receptor blockers or 4 of the 15 beta-blockers or 3 of the 10 calcium-channel blockers would reduce depression while the others in the class would have no effect,” he said.
Despite the investigators’ conclusion that these data should drive drug choice for patients at risk of depression, “I would say the results of this analysis would not lead me to alter clinical practice,” Dr. White added.
According to the principal investigator of the study, Lars Vedel Kessing, MD, DSc, professor of psychiatry at the University of Copenhagen, many variables affect choice of antihypertensive drug. However, the depression risk is elevated in patients with cardiovascular or cerebrovascular disease and hypertension.
When risk of a mood disorder is a concern, use of one of the nine drugs associated with protection from depression should be considered, “especially in patients at increased risk of developing depression, including patients with prior depression or anxiety and patients with a family history of depression,” he and his coinvestigators concluded.
However, Dr. Kessing said in an interview that the data do not help with individual treatment choices. “We do not compare different antihypertensives against each other due to the risk of confounding by indications, so, no, it is not reasonable to consider relative risk among specific agents.”
The authors reported no potential conflicts of interest involving this topic.
SOURCE: Kessing LV et al. Hypertension. 2020 Aug 24. doi: 10.1161/HYPERTENSIONAHA.120.15605.
FROM HYPERTENSION