User login
Ezetimibe-statin combo lowers liver fat in open-label trial
Ezetimibe given in combination with rosuvastatin has a beneficial effect on liver fat in people with nonalcoholic fatty liver disease (NAFLD), according results of a randomized, active-controlled trial.
The findings, which come from the investigator-initiated ESSENTIAL trial, are likely to add to the debate over whether or not the lipid-lowering combination could be of benefit beyond its effects in the blood.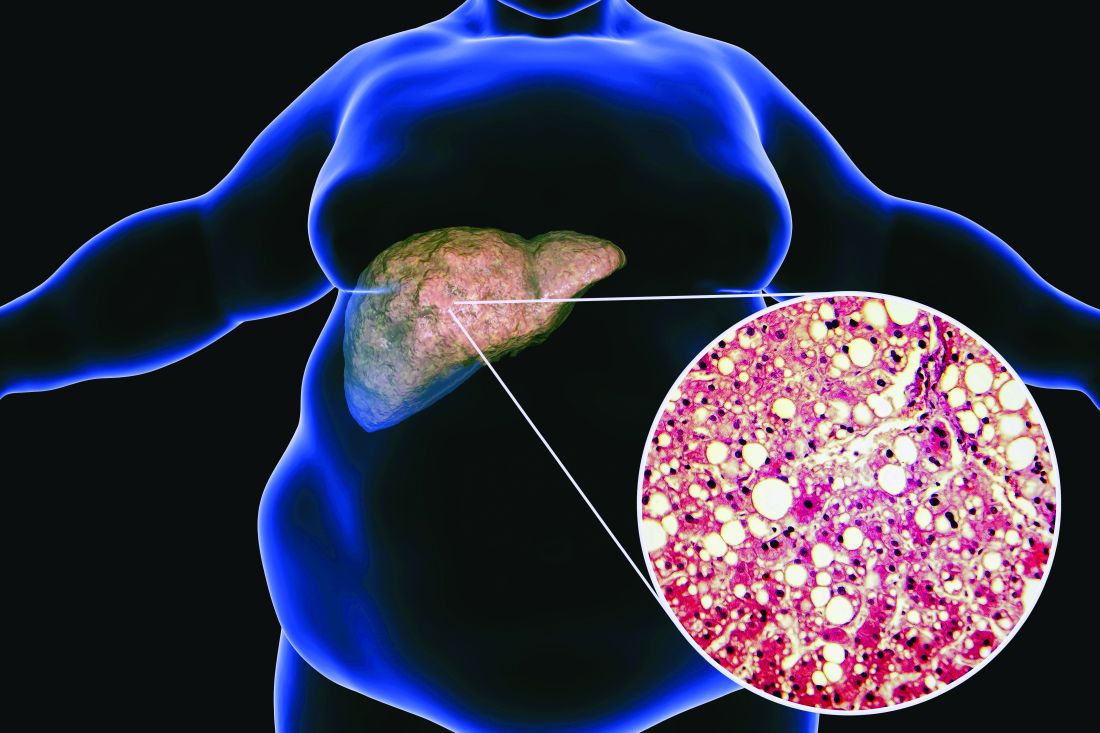
“We used magnetic resonance imaging-derived proton density fat fraction [MRI-PDFF], which is highly reliable method of assessing hepatic steatosis,” Youngjoon Kim, PhD, one of the study investigators, said at the annual meeting of the European Association for the Study of Diabetes in Barcelona.
“It enables accurate, repeatable and reproducible quantitative assessment of liver fat over the entire liver,” observed Dr. Kim, who works at Severance Hospital, part of Yonsei University in Seoul.
He reported that there was a significant 5.8% decrease in liver fat following 24 weeks’ treatment with ezetimibe and rosuvastatin comparing baseline with end of treatment MRI-PDFF values; a drop that was significant (18.2% vs. 12.3%, P < .001).
Rosuvastatin monotherapy also reduced liver fat from 15.0% at baseline to 12.4% after 24 weeks; this drop of 2.6% was also significant (P = .003).
This gave an absolute mean difference between the two study arms of 3.2% (P = .02).
Rationale for the ESSENTIAL study
Dr. Kim observed during his presentation that NAFLD is burgeoning problem around the world. Ezetimibe plus rosuvastatin was a combination treatment already used widely in clinical practice, and there had been some suggestion that ezetimibe might have an effect on liver fat.
“Although the effect of ezetimibe on hepatic steatosis is still controversial, ezetimibe has been reported to reduce visceral fat and improve insulin resistance in several studies” Dr. Kim said.
“Recently, our group reported that the use of ezetimibe affects autophagy of hepatocytes and the NLRP3 [NOD-like receptors containing pyrin domain 3] inflammasome,” he said.
Moreover, he added, “ezetimibe improved NASH [nonalcoholic steatohepatitis] in an animal model. However, the effects of ezetimibe have not been clearly shown in a human study.”
Dr. Kim also acknowledged a prior randomized control trial that had looked at the role of ezetimibe in 50 patients with NASH, but had not shown a benefit for the drug over placebo in terms of liver fat reduction.
Addressing the Hawthorne effect
“The size of the effect by that might actually be more modest due to the Hawthorne effect,” said session chair Onno Holleboom, MD, PhD, of Amsterdam UMC in the Netherlands.
“What we observe in the large clinical trials is an enormous Hawthorne effect – participating in a NAFLD trial makes people live healthier because they have health checks,” he said.
“That’s a major problem for showing efficacy for the intervention arm,” he added, but of course the open design meant that the trial only had intervention arms; “there was no placebo arm.”
A randomized, active-controlled, clinician-initiated trial
The main objective of the ESSENTIAL trial was therefore to take another look at the potential effect of ezetimibe on hepatic steatosis and doing so in the setting of statin therapy.
In all, 70 patients with NAFLD that had been confirmed via ultrasound were recruited into the prospective, single center, phase 4 trial. Participants were randomized 1:1 to received either ezetimibe 10 mg plus rosuvastatin 5 mg daily or rosuvastatin 5 mg for up to 24 weeks.
Change in liver fat was measured via MRI-PDFF, taking the average values in each of nine liver segments. Magnetic resonance elastography (MRE) was also used to measure liver fibrosis, although results did not show any differences either from baseline to end of treatment values in either group or when the two treatment groups were compared.
Dr. Kim reported that both treatment with the ezetimibe-rosuvastatin combination and rosuvastatin monotherapy reduced parameters that might be associated with a negative outcome in NAFLD, such as body mass index and waist circumference, triglycerides, and LDL cholesterol. There was also a reduction in C-reactive protein levels in the blood, and interleulin-18. There was no change in liver enzymes.
Several subgroup analyses were performed indicating that “individuals with higher BMI, type 2 diabetes, insulin resistance, and severe liver fibrosis were likely to be good responders to ezetimibe treatment,” Dr. Kim said.
“These data indicate that ezetimibe plus rosuvastatin is a safe and effective therapeutic option to treat patients with NAFLD and dyslipidemia,” he concluded.
The results of the ESSENTIAL study have been published in BMC Medicine.
The study was funded by the Yuhan Corporation. Dr. Kim had no conflicts of interest to report. Dr. Holleboom was not involved in the study and had no conflicts of interest.
Ezetimibe given in combination with rosuvastatin has a beneficial effect on liver fat in people with nonalcoholic fatty liver disease (NAFLD), according results of a randomized, active-controlled trial.
The findings, which come from the investigator-initiated ESSENTIAL trial, are likely to add to the debate over whether or not the lipid-lowering combination could be of benefit beyond its effects in the blood.
“We used magnetic resonance imaging-derived proton density fat fraction [MRI-PDFF], which is highly reliable method of assessing hepatic steatosis,” Youngjoon Kim, PhD, one of the study investigators, said at the annual meeting of the European Association for the Study of Diabetes in Barcelona.
“It enables accurate, repeatable and reproducible quantitative assessment of liver fat over the entire liver,” observed Dr. Kim, who works at Severance Hospital, part of Yonsei University in Seoul.
He reported that there was a significant 5.8% decrease in liver fat following 24 weeks’ treatment with ezetimibe and rosuvastatin comparing baseline with end of treatment MRI-PDFF values; a drop that was significant (18.2% vs. 12.3%, P < .001).
Rosuvastatin monotherapy also reduced liver fat from 15.0% at baseline to 12.4% after 24 weeks; this drop of 2.6% was also significant (P = .003).
This gave an absolute mean difference between the two study arms of 3.2% (P = .02).
Rationale for the ESSENTIAL study
Dr. Kim observed during his presentation that NAFLD is burgeoning problem around the world. Ezetimibe plus rosuvastatin was a combination treatment already used widely in clinical practice, and there had been some suggestion that ezetimibe might have an effect on liver fat.
“Although the effect of ezetimibe on hepatic steatosis is still controversial, ezetimibe has been reported to reduce visceral fat and improve insulin resistance in several studies” Dr. Kim said.
“Recently, our group reported that the use of ezetimibe affects autophagy of hepatocytes and the NLRP3 [NOD-like receptors containing pyrin domain 3] inflammasome,” he said.
Moreover, he added, “ezetimibe improved NASH [nonalcoholic steatohepatitis] in an animal model. However, the effects of ezetimibe have not been clearly shown in a human study.”
Dr. Kim also acknowledged a prior randomized control trial that had looked at the role of ezetimibe in 50 patients with NASH, but had not shown a benefit for the drug over placebo in terms of liver fat reduction.
Addressing the Hawthorne effect
“The size of the effect by that might actually be more modest due to the Hawthorne effect,” said session chair Onno Holleboom, MD, PhD, of Amsterdam UMC in the Netherlands.
“What we observe in the large clinical trials is an enormous Hawthorne effect – participating in a NAFLD trial makes people live healthier because they have health checks,” he said.
“That’s a major problem for showing efficacy for the intervention arm,” he added, but of course the open design meant that the trial only had intervention arms; “there was no placebo arm.”
A randomized, active-controlled, clinician-initiated trial
The main objective of the ESSENTIAL trial was therefore to take another look at the potential effect of ezetimibe on hepatic steatosis and doing so in the setting of statin therapy.
In all, 70 patients with NAFLD that had been confirmed via ultrasound were recruited into the prospective, single center, phase 4 trial. Participants were randomized 1:1 to received either ezetimibe 10 mg plus rosuvastatin 5 mg daily or rosuvastatin 5 mg for up to 24 weeks.
Change in liver fat was measured via MRI-PDFF, taking the average values in each of nine liver segments. Magnetic resonance elastography (MRE) was also used to measure liver fibrosis, although results did not show any differences either from baseline to end of treatment values in either group or when the two treatment groups were compared.
Dr. Kim reported that both treatment with the ezetimibe-rosuvastatin combination and rosuvastatin monotherapy reduced parameters that might be associated with a negative outcome in NAFLD, such as body mass index and waist circumference, triglycerides, and LDL cholesterol. There was also a reduction in C-reactive protein levels in the blood, and interleulin-18. There was no change in liver enzymes.
Several subgroup analyses were performed indicating that “individuals with higher BMI, type 2 diabetes, insulin resistance, and severe liver fibrosis were likely to be good responders to ezetimibe treatment,” Dr. Kim said.
“These data indicate that ezetimibe plus rosuvastatin is a safe and effective therapeutic option to treat patients with NAFLD and dyslipidemia,” he concluded.
The results of the ESSENTIAL study have been published in BMC Medicine.
The study was funded by the Yuhan Corporation. Dr. Kim had no conflicts of interest to report. Dr. Holleboom was not involved in the study and had no conflicts of interest.
Ezetimibe given in combination with rosuvastatin has a beneficial effect on liver fat in people with nonalcoholic fatty liver disease (NAFLD), according results of a randomized, active-controlled trial.
The findings, which come from the investigator-initiated ESSENTIAL trial, are likely to add to the debate over whether or not the lipid-lowering combination could be of benefit beyond its effects in the blood.
“We used magnetic resonance imaging-derived proton density fat fraction [MRI-PDFF], which is highly reliable method of assessing hepatic steatosis,” Youngjoon Kim, PhD, one of the study investigators, said at the annual meeting of the European Association for the Study of Diabetes in Barcelona.
“It enables accurate, repeatable and reproducible quantitative assessment of liver fat over the entire liver,” observed Dr. Kim, who works at Severance Hospital, part of Yonsei University in Seoul.
He reported that there was a significant 5.8% decrease in liver fat following 24 weeks’ treatment with ezetimibe and rosuvastatin comparing baseline with end of treatment MRI-PDFF values; a drop that was significant (18.2% vs. 12.3%, P < .001).
Rosuvastatin monotherapy also reduced liver fat from 15.0% at baseline to 12.4% after 24 weeks; this drop of 2.6% was also significant (P = .003).
This gave an absolute mean difference between the two study arms of 3.2% (P = .02).
Rationale for the ESSENTIAL study
Dr. Kim observed during his presentation that NAFLD is burgeoning problem around the world. Ezetimibe plus rosuvastatin was a combination treatment already used widely in clinical practice, and there had been some suggestion that ezetimibe might have an effect on liver fat.
“Although the effect of ezetimibe on hepatic steatosis is still controversial, ezetimibe has been reported to reduce visceral fat and improve insulin resistance in several studies” Dr. Kim said.
“Recently, our group reported that the use of ezetimibe affects autophagy of hepatocytes and the NLRP3 [NOD-like receptors containing pyrin domain 3] inflammasome,” he said.
Moreover, he added, “ezetimibe improved NASH [nonalcoholic steatohepatitis] in an animal model. However, the effects of ezetimibe have not been clearly shown in a human study.”
Dr. Kim also acknowledged a prior randomized control trial that had looked at the role of ezetimibe in 50 patients with NASH, but had not shown a benefit for the drug over placebo in terms of liver fat reduction.
Addressing the Hawthorne effect
“The size of the effect by that might actually be more modest due to the Hawthorne effect,” said session chair Onno Holleboom, MD, PhD, of Amsterdam UMC in the Netherlands.
“What we observe in the large clinical trials is an enormous Hawthorne effect – participating in a NAFLD trial makes people live healthier because they have health checks,” he said.
“That’s a major problem for showing efficacy for the intervention arm,” he added, but of course the open design meant that the trial only had intervention arms; “there was no placebo arm.”
A randomized, active-controlled, clinician-initiated trial
The main objective of the ESSENTIAL trial was therefore to take another look at the potential effect of ezetimibe on hepatic steatosis and doing so in the setting of statin therapy.
In all, 70 patients with NAFLD that had been confirmed via ultrasound were recruited into the prospective, single center, phase 4 trial. Participants were randomized 1:1 to received either ezetimibe 10 mg plus rosuvastatin 5 mg daily or rosuvastatin 5 mg for up to 24 weeks.
Change in liver fat was measured via MRI-PDFF, taking the average values in each of nine liver segments. Magnetic resonance elastography (MRE) was also used to measure liver fibrosis, although results did not show any differences either from baseline to end of treatment values in either group or when the two treatment groups were compared.
Dr. Kim reported that both treatment with the ezetimibe-rosuvastatin combination and rosuvastatin monotherapy reduced parameters that might be associated with a negative outcome in NAFLD, such as body mass index and waist circumference, triglycerides, and LDL cholesterol. There was also a reduction in C-reactive protein levels in the blood, and interleulin-18. There was no change in liver enzymes.
Several subgroup analyses were performed indicating that “individuals with higher BMI, type 2 diabetes, insulin resistance, and severe liver fibrosis were likely to be good responders to ezetimibe treatment,” Dr. Kim said.
“These data indicate that ezetimibe plus rosuvastatin is a safe and effective therapeutic option to treat patients with NAFLD and dyslipidemia,” he concluded.
The results of the ESSENTIAL study have been published in BMC Medicine.
The study was funded by the Yuhan Corporation. Dr. Kim had no conflicts of interest to report. Dr. Holleboom was not involved in the study and had no conflicts of interest.
FROM EASD 2022
Aspirin primary prevention benefit in those with raised Lp(a)?
Aspirin may be of specific benefit for the primary prevention of cardiovascular disease in individuals with raised Lp(a) levels, a new study has suggested.
The study analyzed data from the ASPREE (ASPirin in Reducing Events in the Elderly) trial, which randomized 19,000 individuals aged 70 years or older without a history of cardiovascular disease to aspirin (100 mg/day) or placebo. While the main results, reported previously, showed no net benefit of aspirin in the overall population, the current analysis suggests there may be a benefit in individuals with raised Lp(a) levels.
The current analysis was published online in the Journal of the American College of Cardiology.
“Our study provides evidence that aspirin may specifically benefit older individuals with genotypes for elevated plasma Lp(a) in the setting of high-risk primary prevention of cardiovascular events and that overall benefit may outweigh harm related to major bleeding,” the authors, led by Paul Lacaze, PhD, Monash University, Melbourne, conclude.
They also point out that similar observations have been previously seen in another large aspirin primary prevention study conducted in younger women, the Women’s Health Study, and the current analysis provides validation of those findings.
“Our results provide new evidence to support the potential use of aspirin to target individuals with elevated Lp(a) for the primary prevention of cardiovascular events,” the researchers say.
They acknowledge that these results would be strengthened by the use of directly measured plasma Lp(a) levels, in addition to Lp(a) genotypes.
But they add: “Nonetheless, given the lack of any currently approved therapies for targeting elevated Lp(a), our findings may have widespread clinical implications, adding evidence to the rationale that aspirin may be a viable option for reducing Lp(a)-mediated cardiovascular risk.”
Dr. Lacaze and colleagues explain that elevated plasma Lp(a) levels confer up to fourfold increased risk of cardiovascular disease, with around 20%-30% of the general population affected. Despite the high burden and prevalence of elevated plasma Lp(a), there are currently no approved pharmacologic therapies targeting this lipoprotein. Although promising candidates are in development for the secondary prevention of Lp(a)-mediated cardiovascular disease, it will be many years before these candidates are assessed for primary prevention.
For the current study, researchers analyzed data from 12,815 ASPREE participants who had undergone genotyping and compared outcomes with aspirin versus placebo in those with and without genotypes associated with elevated Lp(a) levels.
Results showed that individuals with elevated Lp(a)-associated genotypes, defined in two different ways, showed a reduction in ischemic events with aspirin versus placebo, and this benefit was not outweighed by an increased bleeding risk.
Specifically, in the placebo group, individuals who carried the rs3798220-C allele, which is known to be associated with raised Lp(a) levels, making up 3.2% of the genotyped population in the study, had an almost twofold increased risk of major adverse cardiovascular events than those not carrying this genotype. However, the risk was attenuated in the aspirin group, with carriers of the rs3798220-C allele actually having a lower rate of cardiovascular events than noncarriers.
In addition, rs3798220-C carrier status was not significantly associated with increased risk of clinically significant bleeding events in the aspirin group.
Similar results were seen with the second way of identifying patients with a high risk of elevated Lp(a) levels using a 43-variant genetic risk score (LPA-GRS).
In the whole study population, aspirin reduced major adverse cardiovascular events by 1.7 events per 1,000 person-years and increased clinically significant bleeding events by 1.7 events per 1,000 person-years, suggesting parity between overall benefit versus harm.
However, in the rs3798220-C subgroup, aspirin reduced major adverse cardiovascular events by 11.4 events per 1,000 person-years (a more than sixfold higher magnitude of cardiovascular disease risk reduction than in the overall cohort), with a bleeding risk of 3.3 events per 1,000 person-years, the researchers report.
“Hence in rs3798220-C carriers, aspirin appeared to have a net benefit of 8.1 events per 1,000 person-years,” they state.
In the highest LPA-GRS quintile, aspirin reduced major adverse cardiovascular events by 3.3 events per 1,000 person-years (approximately twofold higher magnitude of risk reduction, compared with the overall cohort), with an increase in bleeding risk of 1.6 events per 1,000 person-years (almost identical bleeding risk to the overall cohort). This shifted the benefit versus harm balance in the highest LPA-GRS quintile to a net benefit of 1.7 events per 1,000 person-years.
Similar findings in the Women’s Health Study
Dr. Lacaze and colleagues point out that similar results have also been seen in another large aspirin primary prevention study – the Women’s Health Study (WHS).
The WHS compared aspirin 100 mg every other day with placebo in initially healthy younger women. Previously reported results showed that women carrying the rs3798220-C variant, associated with highly elevated Lp(a) levels, had a twofold higher risk of cardiovascular events than noncarrier women in the placebo group, but this risk was reduced in the aspirin group. And there was no increased risk of bleeding in women with elevated Lp(a).
“These results, in the absence of any other randomized controlled trial evidence or approved therapy for treating Lp(a)-associated risk, have been used by some physicians as justification for prescribing aspirin in patients with elevated Lp(a),” Dr. Lacaze and colleagues note.
“In the present study of the ASPREE trial population, our results were consistent with the WHS analysis, despite randomizing older individuals (both men and women),” they add.
They say this validation of the WHS result provides evidence that a very high-risk subgroup of individuals with highly elevated Lp(a) – those carrying the rs3798220-C allele – may benefit from low-dose aspirin for the primary prevention of cardiovascular events. Further, the benefits in this subgroup specifically may outweigh any bleeding risk.
But they point out that rs3798220-C carriers comprise only a small portion of all individuals with elevated Lp(a) in the general population, while the polygenic LPA-GRS explains about 60% of the variation in directly measured plasma Lp(a) levels and has the potential advantage of being able to identify a larger group of individuals at increased risk.
The researchers note, however, that it is not clear to what extent the LPA-GRS results add further evidence to suggest that individuals with elevated Lp(a), beyond rs3798220-C carriers, may be more likely to benefit from aspirin.
“If the benefit of aspirin extends beyond very high-risk rs3798220-C carriers alone, to the broader 20%-30% of individuals with elevated Lp(a), the potential utility of aspirin for the primary prevention of cardiovascular events would increase substantially,” they say.
‘Very high clinical relevance’
In an accompanying editorial, Ana Devesa, MD, Borja Ibanez, MD, PhD, and Valentin Fuster, MD, PhD, The National Center for Cardiovascular Research, Madrid, say that: “[Dr.] Lacaze et al. are to be congratulated for a study of very high clinical relevance that represents a first indication for primary prevention for patients at high cardiovascular risk.”
They explain that the pathogenic mechanism of Lp(a) is believed to be a combination of prothrombotic and proatherogenic effects, and the current findings support the hypothesis that the prothrombotic mechanism of Lp(a) is mediated by platelet aggregation.
This would explain the occurrence of thrombotic events in the presence of atherosclerosis in that elevated Lp(a) levels may induce platelet adhesion and aggregation to the activated atherosclerotic plaque, thus enhancing the atherothrombotic process. Moreover, activated platelets release several mediators that result in cell adhesion and attraction of chemokines and proinflammatory cytokines, driving an inflammatory response and mediating atherosclerosis progression, they add.
The editorialists highlight the limitations of the study already acknowledged by the authors: The analysis used genotypes rather than elevated Lp(a) levels and included only those of European ancestry, meaning the results are difficult to extrapolate to other populations.
“The next steps in clinical practice should be defined, and there are still questions to be answered,” they conclude. “Will every patient benefit from antithrombotic therapies? Should all patients who have elevated Lp(a) levels be treated with aspirin?”
The ASPREE Biobank is supported by grants from the Commonwealth Scientific and Industrial Research Organisation, Monash University, Menzies Research Institute, Australian National University, University of Melbourne, National Institutes of Health, National Health and Medical Research Council of Australia, and the Victorian Cancer Agency. Dr. Lacaze is supported by a National Heart Foundation Future Leader Fellowship.
A version of this article first appeared on Medscape.com.
Aspirin may be of specific benefit for the primary prevention of cardiovascular disease in individuals with raised Lp(a) levels, a new study has suggested.
The study analyzed data from the ASPREE (ASPirin in Reducing Events in the Elderly) trial, which randomized 19,000 individuals aged 70 years or older without a history of cardiovascular disease to aspirin (100 mg/day) or placebo. While the main results, reported previously, showed no net benefit of aspirin in the overall population, the current analysis suggests there may be a benefit in individuals with raised Lp(a) levels.
The current analysis was published online in the Journal of the American College of Cardiology.
“Our study provides evidence that aspirin may specifically benefit older individuals with genotypes for elevated plasma Lp(a) in the setting of high-risk primary prevention of cardiovascular events and that overall benefit may outweigh harm related to major bleeding,” the authors, led by Paul Lacaze, PhD, Monash University, Melbourne, conclude.
They also point out that similar observations have been previously seen in another large aspirin primary prevention study conducted in younger women, the Women’s Health Study, and the current analysis provides validation of those findings.
“Our results provide new evidence to support the potential use of aspirin to target individuals with elevated Lp(a) for the primary prevention of cardiovascular events,” the researchers say.
They acknowledge that these results would be strengthened by the use of directly measured plasma Lp(a) levels, in addition to Lp(a) genotypes.
But they add: “Nonetheless, given the lack of any currently approved therapies for targeting elevated Lp(a), our findings may have widespread clinical implications, adding evidence to the rationale that aspirin may be a viable option for reducing Lp(a)-mediated cardiovascular risk.”
Dr. Lacaze and colleagues explain that elevated plasma Lp(a) levels confer up to fourfold increased risk of cardiovascular disease, with around 20%-30% of the general population affected. Despite the high burden and prevalence of elevated plasma Lp(a), there are currently no approved pharmacologic therapies targeting this lipoprotein. Although promising candidates are in development for the secondary prevention of Lp(a)-mediated cardiovascular disease, it will be many years before these candidates are assessed for primary prevention.
For the current study, researchers analyzed data from 12,815 ASPREE participants who had undergone genotyping and compared outcomes with aspirin versus placebo in those with and without genotypes associated with elevated Lp(a) levels.
Results showed that individuals with elevated Lp(a)-associated genotypes, defined in two different ways, showed a reduction in ischemic events with aspirin versus placebo, and this benefit was not outweighed by an increased bleeding risk.
Specifically, in the placebo group, individuals who carried the rs3798220-C allele, which is known to be associated with raised Lp(a) levels, making up 3.2% of the genotyped population in the study, had an almost twofold increased risk of major adverse cardiovascular events than those not carrying this genotype. However, the risk was attenuated in the aspirin group, with carriers of the rs3798220-C allele actually having a lower rate of cardiovascular events than noncarriers.
In addition, rs3798220-C carrier status was not significantly associated with increased risk of clinically significant bleeding events in the aspirin group.
Similar results were seen with the second way of identifying patients with a high risk of elevated Lp(a) levels using a 43-variant genetic risk score (LPA-GRS).
In the whole study population, aspirin reduced major adverse cardiovascular events by 1.7 events per 1,000 person-years and increased clinically significant bleeding events by 1.7 events per 1,000 person-years, suggesting parity between overall benefit versus harm.
However, in the rs3798220-C subgroup, aspirin reduced major adverse cardiovascular events by 11.4 events per 1,000 person-years (a more than sixfold higher magnitude of cardiovascular disease risk reduction than in the overall cohort), with a bleeding risk of 3.3 events per 1,000 person-years, the researchers report.
“Hence in rs3798220-C carriers, aspirin appeared to have a net benefit of 8.1 events per 1,000 person-years,” they state.
In the highest LPA-GRS quintile, aspirin reduced major adverse cardiovascular events by 3.3 events per 1,000 person-years (approximately twofold higher magnitude of risk reduction, compared with the overall cohort), with an increase in bleeding risk of 1.6 events per 1,000 person-years (almost identical bleeding risk to the overall cohort). This shifted the benefit versus harm balance in the highest LPA-GRS quintile to a net benefit of 1.7 events per 1,000 person-years.
Similar findings in the Women’s Health Study
Dr. Lacaze and colleagues point out that similar results have also been seen in another large aspirin primary prevention study – the Women’s Health Study (WHS).
The WHS compared aspirin 100 mg every other day with placebo in initially healthy younger women. Previously reported results showed that women carrying the rs3798220-C variant, associated with highly elevated Lp(a) levels, had a twofold higher risk of cardiovascular events than noncarrier women in the placebo group, but this risk was reduced in the aspirin group. And there was no increased risk of bleeding in women with elevated Lp(a).
“These results, in the absence of any other randomized controlled trial evidence or approved therapy for treating Lp(a)-associated risk, have been used by some physicians as justification for prescribing aspirin in patients with elevated Lp(a),” Dr. Lacaze and colleagues note.
“In the present study of the ASPREE trial population, our results were consistent with the WHS analysis, despite randomizing older individuals (both men and women),” they add.
They say this validation of the WHS result provides evidence that a very high-risk subgroup of individuals with highly elevated Lp(a) – those carrying the rs3798220-C allele – may benefit from low-dose aspirin for the primary prevention of cardiovascular events. Further, the benefits in this subgroup specifically may outweigh any bleeding risk.
But they point out that rs3798220-C carriers comprise only a small portion of all individuals with elevated Lp(a) in the general population, while the polygenic LPA-GRS explains about 60% of the variation in directly measured plasma Lp(a) levels and has the potential advantage of being able to identify a larger group of individuals at increased risk.
The researchers note, however, that it is not clear to what extent the LPA-GRS results add further evidence to suggest that individuals with elevated Lp(a), beyond rs3798220-C carriers, may be more likely to benefit from aspirin.
“If the benefit of aspirin extends beyond very high-risk rs3798220-C carriers alone, to the broader 20%-30% of individuals with elevated Lp(a), the potential utility of aspirin for the primary prevention of cardiovascular events would increase substantially,” they say.
‘Very high clinical relevance’
In an accompanying editorial, Ana Devesa, MD, Borja Ibanez, MD, PhD, and Valentin Fuster, MD, PhD, The National Center for Cardiovascular Research, Madrid, say that: “[Dr.] Lacaze et al. are to be congratulated for a study of very high clinical relevance that represents a first indication for primary prevention for patients at high cardiovascular risk.”
They explain that the pathogenic mechanism of Lp(a) is believed to be a combination of prothrombotic and proatherogenic effects, and the current findings support the hypothesis that the prothrombotic mechanism of Lp(a) is mediated by platelet aggregation.
This would explain the occurrence of thrombotic events in the presence of atherosclerosis in that elevated Lp(a) levels may induce platelet adhesion and aggregation to the activated atherosclerotic plaque, thus enhancing the atherothrombotic process. Moreover, activated platelets release several mediators that result in cell adhesion and attraction of chemokines and proinflammatory cytokines, driving an inflammatory response and mediating atherosclerosis progression, they add.
The editorialists highlight the limitations of the study already acknowledged by the authors: The analysis used genotypes rather than elevated Lp(a) levels and included only those of European ancestry, meaning the results are difficult to extrapolate to other populations.
“The next steps in clinical practice should be defined, and there are still questions to be answered,” they conclude. “Will every patient benefit from antithrombotic therapies? Should all patients who have elevated Lp(a) levels be treated with aspirin?”
The ASPREE Biobank is supported by grants from the Commonwealth Scientific and Industrial Research Organisation, Monash University, Menzies Research Institute, Australian National University, University of Melbourne, National Institutes of Health, National Health and Medical Research Council of Australia, and the Victorian Cancer Agency. Dr. Lacaze is supported by a National Heart Foundation Future Leader Fellowship.
A version of this article first appeared on Medscape.com.
Aspirin may be of specific benefit for the primary prevention of cardiovascular disease in individuals with raised Lp(a) levels, a new study has suggested.
The study analyzed data from the ASPREE (ASPirin in Reducing Events in the Elderly) trial, which randomized 19,000 individuals aged 70 years or older without a history of cardiovascular disease to aspirin (100 mg/day) or placebo. While the main results, reported previously, showed no net benefit of aspirin in the overall population, the current analysis suggests there may be a benefit in individuals with raised Lp(a) levels.
The current analysis was published online in the Journal of the American College of Cardiology.
“Our study provides evidence that aspirin may specifically benefit older individuals with genotypes for elevated plasma Lp(a) in the setting of high-risk primary prevention of cardiovascular events and that overall benefit may outweigh harm related to major bleeding,” the authors, led by Paul Lacaze, PhD, Monash University, Melbourne, conclude.
They also point out that similar observations have been previously seen in another large aspirin primary prevention study conducted in younger women, the Women’s Health Study, and the current analysis provides validation of those findings.
“Our results provide new evidence to support the potential use of aspirin to target individuals with elevated Lp(a) for the primary prevention of cardiovascular events,” the researchers say.
They acknowledge that these results would be strengthened by the use of directly measured plasma Lp(a) levels, in addition to Lp(a) genotypes.
But they add: “Nonetheless, given the lack of any currently approved therapies for targeting elevated Lp(a), our findings may have widespread clinical implications, adding evidence to the rationale that aspirin may be a viable option for reducing Lp(a)-mediated cardiovascular risk.”
Dr. Lacaze and colleagues explain that elevated plasma Lp(a) levels confer up to fourfold increased risk of cardiovascular disease, with around 20%-30% of the general population affected. Despite the high burden and prevalence of elevated plasma Lp(a), there are currently no approved pharmacologic therapies targeting this lipoprotein. Although promising candidates are in development for the secondary prevention of Lp(a)-mediated cardiovascular disease, it will be many years before these candidates are assessed for primary prevention.
For the current study, researchers analyzed data from 12,815 ASPREE participants who had undergone genotyping and compared outcomes with aspirin versus placebo in those with and without genotypes associated with elevated Lp(a) levels.
Results showed that individuals with elevated Lp(a)-associated genotypes, defined in two different ways, showed a reduction in ischemic events with aspirin versus placebo, and this benefit was not outweighed by an increased bleeding risk.
Specifically, in the placebo group, individuals who carried the rs3798220-C allele, which is known to be associated with raised Lp(a) levels, making up 3.2% of the genotyped population in the study, had an almost twofold increased risk of major adverse cardiovascular events than those not carrying this genotype. However, the risk was attenuated in the aspirin group, with carriers of the rs3798220-C allele actually having a lower rate of cardiovascular events than noncarriers.
In addition, rs3798220-C carrier status was not significantly associated with increased risk of clinically significant bleeding events in the aspirin group.
Similar results were seen with the second way of identifying patients with a high risk of elevated Lp(a) levels using a 43-variant genetic risk score (LPA-GRS).
In the whole study population, aspirin reduced major adverse cardiovascular events by 1.7 events per 1,000 person-years and increased clinically significant bleeding events by 1.7 events per 1,000 person-years, suggesting parity between overall benefit versus harm.
However, in the rs3798220-C subgroup, aspirin reduced major adverse cardiovascular events by 11.4 events per 1,000 person-years (a more than sixfold higher magnitude of cardiovascular disease risk reduction than in the overall cohort), with a bleeding risk of 3.3 events per 1,000 person-years, the researchers report.
“Hence in rs3798220-C carriers, aspirin appeared to have a net benefit of 8.1 events per 1,000 person-years,” they state.
In the highest LPA-GRS quintile, aspirin reduced major adverse cardiovascular events by 3.3 events per 1,000 person-years (approximately twofold higher magnitude of risk reduction, compared with the overall cohort), with an increase in bleeding risk of 1.6 events per 1,000 person-years (almost identical bleeding risk to the overall cohort). This shifted the benefit versus harm balance in the highest LPA-GRS quintile to a net benefit of 1.7 events per 1,000 person-years.
Similar findings in the Women’s Health Study
Dr. Lacaze and colleagues point out that similar results have also been seen in another large aspirin primary prevention study – the Women’s Health Study (WHS).
The WHS compared aspirin 100 mg every other day with placebo in initially healthy younger women. Previously reported results showed that women carrying the rs3798220-C variant, associated with highly elevated Lp(a) levels, had a twofold higher risk of cardiovascular events than noncarrier women in the placebo group, but this risk was reduced in the aspirin group. And there was no increased risk of bleeding in women with elevated Lp(a).
“These results, in the absence of any other randomized controlled trial evidence or approved therapy for treating Lp(a)-associated risk, have been used by some physicians as justification for prescribing aspirin in patients with elevated Lp(a),” Dr. Lacaze and colleagues note.
“In the present study of the ASPREE trial population, our results were consistent with the WHS analysis, despite randomizing older individuals (both men and women),” they add.
They say this validation of the WHS result provides evidence that a very high-risk subgroup of individuals with highly elevated Lp(a) – those carrying the rs3798220-C allele – may benefit from low-dose aspirin for the primary prevention of cardiovascular events. Further, the benefits in this subgroup specifically may outweigh any bleeding risk.
But they point out that rs3798220-C carriers comprise only a small portion of all individuals with elevated Lp(a) in the general population, while the polygenic LPA-GRS explains about 60% of the variation in directly measured plasma Lp(a) levels and has the potential advantage of being able to identify a larger group of individuals at increased risk.
The researchers note, however, that it is not clear to what extent the LPA-GRS results add further evidence to suggest that individuals with elevated Lp(a), beyond rs3798220-C carriers, may be more likely to benefit from aspirin.
“If the benefit of aspirin extends beyond very high-risk rs3798220-C carriers alone, to the broader 20%-30% of individuals with elevated Lp(a), the potential utility of aspirin for the primary prevention of cardiovascular events would increase substantially,” they say.
‘Very high clinical relevance’
In an accompanying editorial, Ana Devesa, MD, Borja Ibanez, MD, PhD, and Valentin Fuster, MD, PhD, The National Center for Cardiovascular Research, Madrid, say that: “[Dr.] Lacaze et al. are to be congratulated for a study of very high clinical relevance that represents a first indication for primary prevention for patients at high cardiovascular risk.”
They explain that the pathogenic mechanism of Lp(a) is believed to be a combination of prothrombotic and proatherogenic effects, and the current findings support the hypothesis that the prothrombotic mechanism of Lp(a) is mediated by platelet aggregation.
This would explain the occurrence of thrombotic events in the presence of atherosclerosis in that elevated Lp(a) levels may induce platelet adhesion and aggregation to the activated atherosclerotic plaque, thus enhancing the atherothrombotic process. Moreover, activated platelets release several mediators that result in cell adhesion and attraction of chemokines and proinflammatory cytokines, driving an inflammatory response and mediating atherosclerosis progression, they add.
The editorialists highlight the limitations of the study already acknowledged by the authors: The analysis used genotypes rather than elevated Lp(a) levels and included only those of European ancestry, meaning the results are difficult to extrapolate to other populations.
“The next steps in clinical practice should be defined, and there are still questions to be answered,” they conclude. “Will every patient benefit from antithrombotic therapies? Should all patients who have elevated Lp(a) levels be treated with aspirin?”
The ASPREE Biobank is supported by grants from the Commonwealth Scientific and Industrial Research Organisation, Monash University, Menzies Research Institute, Australian National University, University of Melbourne, National Institutes of Health, National Health and Medical Research Council of Australia, and the Victorian Cancer Agency. Dr. Lacaze is supported by a National Heart Foundation Future Leader Fellowship.
A version of this article first appeared on Medscape.com.
Positive top-line phase 3 data for lecanemab in early Alzheimer’s
compared with placebo and decreased amyloid levels in the brain of adults enrolled in a phase 3 trial.
The Clarity AD trial included 1,795 adults with early AD and confirmed amyloid pathology in the brain. Treatment consisted of lecanemab 10 mg/kg biweekly or matching placebo.
Treatment with lecanemab met the primary endpoint, reducing clinical decline on the global cognitive and functional scale, the Clinical Dementia Rating–Sum of Boxes (CDR-SB), at 18 months by 27%, compared with placebo, with a treatment difference in the score change of –0.45 (P = .00005), the companies reported.
Starting as early as 6 months, across all time points, treatment with lecanemab yielded highly statistically significant changes in CDR-SB from baseline, compared with placebo (all P < .01).
The study also met all key secondary endpoints with highly statistically significant results, compared with placebo (P < .01).
Key secondary endpoints, in comparison with placebo, were change from baseline at 18 months in amyloid levels in the brain measured by amyloid PET, the AD Assessment Scale–cognitive subscale14 (ADAS-cog14), the AD Composite Score (ADCOMS), and the AD Cooperative Study–Activities of Daily Living Scale for Mild Cognitive Impairment (ADCS MCI-ADL).
Imaging abnormalities within expectations
Overall, rates of amyloid-related imaging abnormalities (ARIA) related to lecanemab were “within expectations,” the companies said.
The incidence of ARIA related to edema (ARIA-E) was 12.5% in the lecanemab group and 1.7% in the placebo group.
The incidence of symptomatic ARIA-E was 2.8% and 0.0%, respectively, and the rate of cerebral hemorrhage (ARIA-H) was 17.0% and 8.7%. The total incidence of ARIA (ARIA-E and/or ARIA-H) was 21.3% in the lecanemab group and 9.3% in the placebo group.
Full results of the Clarity AD trial will be presented in November at the Clinical Trials on Alzheimer’s Congress.
Incremental benefit
Responding to the findings, the Alzheimer’s Association said in a statement that it “enthusiastically welcomes” the positive findings. It noted that these are “the most encouraging results in clinical trials treating the underlying causes of Alzheimer’s to date.
“For people in the earliest stages of Alzheimer’s, this treatment has the potential to change the course of the disease in a clinically meaningful way. These results indicate lecanemab may give people more time at or near their full abilities to participate in daily life, remain independent and make future health care decisions,” the Alzheimer’s Association added.
Also weighing in, Howard Fillit, MD, cofounder and chief science officer at the Alzheimer’s Drug Discovery Foundation, said in a release that “the combination of the biomarker change – reduced amyloid – plus slowing of cognitive decline in this study is encouraging news for the 57 million patients around the world living with Alzheimer’s.
“However, amyloid-clearing drugs will provide an incremental benefit at best, and there is still a pressing need for the next generation of drugs focused on other targets based on our knowledge of the biology of aging,” Dr. Fillit cautioned.
“We are optimistic about the future as many of these drugs are in development, with 75% of drugs in the pipeline now targeting nonamyloid pathways of neurodegeneration,” he added.
In July 2022, the Food and Drug Administration accepted Eisai’s biologics license application for lecanemab under the accelerated approval pathway and granted priority review. Lecanemab has a prescription Drugs User Fee Act action date of Jan. 6, 2023.
A version of this article first appeared on Medscape.com.
compared with placebo and decreased amyloid levels in the brain of adults enrolled in a phase 3 trial.
The Clarity AD trial included 1,795 adults with early AD and confirmed amyloid pathology in the brain. Treatment consisted of lecanemab 10 mg/kg biweekly or matching placebo.
Treatment with lecanemab met the primary endpoint, reducing clinical decline on the global cognitive and functional scale, the Clinical Dementia Rating–Sum of Boxes (CDR-SB), at 18 months by 27%, compared with placebo, with a treatment difference in the score change of –0.45 (P = .00005), the companies reported.
Starting as early as 6 months, across all time points, treatment with lecanemab yielded highly statistically significant changes in CDR-SB from baseline, compared with placebo (all P < .01).
The study also met all key secondary endpoints with highly statistically significant results, compared with placebo (P < .01).
Key secondary endpoints, in comparison with placebo, were change from baseline at 18 months in amyloid levels in the brain measured by amyloid PET, the AD Assessment Scale–cognitive subscale14 (ADAS-cog14), the AD Composite Score (ADCOMS), and the AD Cooperative Study–Activities of Daily Living Scale for Mild Cognitive Impairment (ADCS MCI-ADL).
Imaging abnormalities within expectations
Overall, rates of amyloid-related imaging abnormalities (ARIA) related to lecanemab were “within expectations,” the companies said.
The incidence of ARIA related to edema (ARIA-E) was 12.5% in the lecanemab group and 1.7% in the placebo group.
The incidence of symptomatic ARIA-E was 2.8% and 0.0%, respectively, and the rate of cerebral hemorrhage (ARIA-H) was 17.0% and 8.7%. The total incidence of ARIA (ARIA-E and/or ARIA-H) was 21.3% in the lecanemab group and 9.3% in the placebo group.
Full results of the Clarity AD trial will be presented in November at the Clinical Trials on Alzheimer’s Congress.
Incremental benefit
Responding to the findings, the Alzheimer’s Association said in a statement that it “enthusiastically welcomes” the positive findings. It noted that these are “the most encouraging results in clinical trials treating the underlying causes of Alzheimer’s to date.
“For people in the earliest stages of Alzheimer’s, this treatment has the potential to change the course of the disease in a clinically meaningful way. These results indicate lecanemab may give people more time at or near their full abilities to participate in daily life, remain independent and make future health care decisions,” the Alzheimer’s Association added.
Also weighing in, Howard Fillit, MD, cofounder and chief science officer at the Alzheimer’s Drug Discovery Foundation, said in a release that “the combination of the biomarker change – reduced amyloid – plus slowing of cognitive decline in this study is encouraging news for the 57 million patients around the world living with Alzheimer’s.
“However, amyloid-clearing drugs will provide an incremental benefit at best, and there is still a pressing need for the next generation of drugs focused on other targets based on our knowledge of the biology of aging,” Dr. Fillit cautioned.
“We are optimistic about the future as many of these drugs are in development, with 75% of drugs in the pipeline now targeting nonamyloid pathways of neurodegeneration,” he added.
In July 2022, the Food and Drug Administration accepted Eisai’s biologics license application for lecanemab under the accelerated approval pathway and granted priority review. Lecanemab has a prescription Drugs User Fee Act action date of Jan. 6, 2023.
A version of this article first appeared on Medscape.com.
compared with placebo and decreased amyloid levels in the brain of adults enrolled in a phase 3 trial.
The Clarity AD trial included 1,795 adults with early AD and confirmed amyloid pathology in the brain. Treatment consisted of lecanemab 10 mg/kg biweekly or matching placebo.
Treatment with lecanemab met the primary endpoint, reducing clinical decline on the global cognitive and functional scale, the Clinical Dementia Rating–Sum of Boxes (CDR-SB), at 18 months by 27%, compared with placebo, with a treatment difference in the score change of –0.45 (P = .00005), the companies reported.
Starting as early as 6 months, across all time points, treatment with lecanemab yielded highly statistically significant changes in CDR-SB from baseline, compared with placebo (all P < .01).
The study also met all key secondary endpoints with highly statistically significant results, compared with placebo (P < .01).
Key secondary endpoints, in comparison with placebo, were change from baseline at 18 months in amyloid levels in the brain measured by amyloid PET, the AD Assessment Scale–cognitive subscale14 (ADAS-cog14), the AD Composite Score (ADCOMS), and the AD Cooperative Study–Activities of Daily Living Scale for Mild Cognitive Impairment (ADCS MCI-ADL).
Imaging abnormalities within expectations
Overall, rates of amyloid-related imaging abnormalities (ARIA) related to lecanemab were “within expectations,” the companies said.
The incidence of ARIA related to edema (ARIA-E) was 12.5% in the lecanemab group and 1.7% in the placebo group.
The incidence of symptomatic ARIA-E was 2.8% and 0.0%, respectively, and the rate of cerebral hemorrhage (ARIA-H) was 17.0% and 8.7%. The total incidence of ARIA (ARIA-E and/or ARIA-H) was 21.3% in the lecanemab group and 9.3% in the placebo group.
Full results of the Clarity AD trial will be presented in November at the Clinical Trials on Alzheimer’s Congress.
Incremental benefit
Responding to the findings, the Alzheimer’s Association said in a statement that it “enthusiastically welcomes” the positive findings. It noted that these are “the most encouraging results in clinical trials treating the underlying causes of Alzheimer’s to date.
“For people in the earliest stages of Alzheimer’s, this treatment has the potential to change the course of the disease in a clinically meaningful way. These results indicate lecanemab may give people more time at or near their full abilities to participate in daily life, remain independent and make future health care decisions,” the Alzheimer’s Association added.
Also weighing in, Howard Fillit, MD, cofounder and chief science officer at the Alzheimer’s Drug Discovery Foundation, said in a release that “the combination of the biomarker change – reduced amyloid – plus slowing of cognitive decline in this study is encouraging news for the 57 million patients around the world living with Alzheimer’s.
“However, amyloid-clearing drugs will provide an incremental benefit at best, and there is still a pressing need for the next generation of drugs focused on other targets based on our knowledge of the biology of aging,” Dr. Fillit cautioned.
“We are optimistic about the future as many of these drugs are in development, with 75% of drugs in the pipeline now targeting nonamyloid pathways of neurodegeneration,” he added.
In July 2022, the Food and Drug Administration accepted Eisai’s biologics license application for lecanemab under the accelerated approval pathway and granted priority review. Lecanemab has a prescription Drugs User Fee Act action date of Jan. 6, 2023.
A version of this article first appeared on Medscape.com.
Heart failure drug a new treatment option for alcoholism?
(AUD), new research suggests.
Researchers at the National Institute on Drug Abuse, the National Institute on Alcohol Abuse and Alcoholism, and Yale University, New Haven, Conn., investigated the impact of spironolactone on AUD.
Initially, they studied rodents and found that spironolactone reduced binge drinking in mice and reduced self-administration of alcohol in rats without adversely affecting food or water intake or causing motor or coordination problems.
They also analyzed electronic health records of patients drawn from the United States Veterans Affairs health care system to explore potential changes in alcohol use after spironolactone treatment was initiated for other conditions and found a significant link between spironolactone treatment and reduction in self-reported alcohol consumption, with the largest effects observed among those who reported hazardous/heavy episodic alcohol use prior to starting spironolactone treatment.
“Combining findings across three species and different types of research studies, and then seeing similarities in these data, gives us confidence that we are onto something potentially important scientifically and clinically,” senior coauthor Lorenzo Leggio, MD, PhD, senior investigator in the Clinical Psychoneuroendocrinology and Neuropsychopharmacology Section, a joint NIDA and NIAAA laboratory, said in a news release.
The study was published online in Molecular Psychiatry.
There is a “critical need to increase the armamentarium of pharmacotherapies to treat individuals with AUD,” the authors note, adding that neuroendocrine systems involved in alcohol craving and drinking “offer promising pharmacologic targets in this regard.”
“Both our team and others have observed that patients with AUD often present with changes in peripheral hormones, including aldosterone, which plays a key role in regulating blood pressure and electrolytes,” Dr. Leggio said in an interview.
Spironolactone is a nonselective mineralocorticoid receptor (MT) antagonist. In studies in animal models, investigators said they found “an inverse correlation between alcohol drinking and the expression of the MR in the amygdala, a key brain region in the development and maintenance of AUD and addiction in general.”
Taken together, this led them to hypothesize that blocking the MR, which is the mechanism of action of spironolactone, “could be a novel pharmacotherapeutic approach for AUD,” he said.
Previous research by the same group of researchers suggested spironolactone “may be a potential new medication to treat patients with AUD.” The present study expanded on those findings and consisted of a three-part investigation.
In the current study, the investigators tested different dosages of spironolactone on binge-like alcohol consumption in male and female mice and assessed food and water intake, blood alcohol levels, motor coordination, and spontaneous locomotion.
They then tested the effects of different dosages of spironolactone injections on operant alcohol self-administration in alcohol-dependent and nondependent male and female rats, also testing blood alcohol levels and motor coordination.
Finally, they analyzed health records of veterans to examine the association between at least 60 continuous days of spironolactone treatment and self-reported alcohol consumption (measured by the Alcohol Use Disorders Identification Test-Consumption [AUDIT-C]).
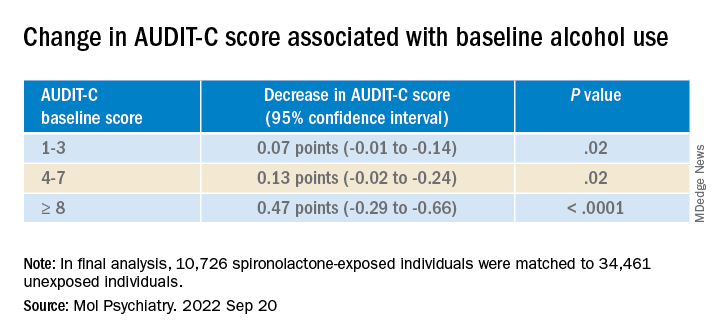
Each of the spironolactone-exposed patients was matched using propensity scores with up to five unexposed patients who had reported alcohol consumption in the 2 years prior to the index date.
The final analysis included a matched cohort of 10,726 spironolactone-exposed individuals who were matched to 34,461 unexposed individuals.
New targets
Spironolactone reduced alcohol intake in mice drinking a sweetened alcohol solution; a 2-way ANOVA revealed a main effect of dose (F 4,52 = 9.09; P < .0001) and sex, with female mice drinking more alcohol, compared to male mice (F 1,13 = 6.05; P = .02).
Post hoc comparisons showed that spironolactone at doses of 50, 100, and 200 mg/kg significantly reduced alcohol intake (P values = .007, .002, and .0001, respectively).
In mice drinking an unsweetened alcohol solution, the 2-way repeated measures ANOVA similarly found a main effect of dose (F 4,52 = 5.77; P = .0006), but not of sex (F 1,13 = 1.41; P = .25).
Spironolactone had no effect on the mice’s intake of a sweet solution without alcohol and had no impact on the consumption of food and water or on locomotion and coordination.
In rats, a 2-way ANOVA revealed a significant spironolactone effect of dose (F 3,66 = 43.95; P < .001), with a post hoc test indicating that spironolactone at 25, 50, and 75 mg/kg reduced alcohol self-administration in alcohol-dependent and nondependent rats (all P values = .0001).
In humans, among the exposed individuals in the matched cohort, 25%, 57%, and 18% received daily doses of spironolactone of less than 25 mg/day, 25-49 mg/day, and 50 mg/day or higher, respectively, with a median follow-up time of 542 (interquartile range, 337-730) days.
The AUDIT-C scores decreased during the study period in both treatment groups, with a larger decrease in average AUDIT-C scores among the exposed vs. unexposed individuals.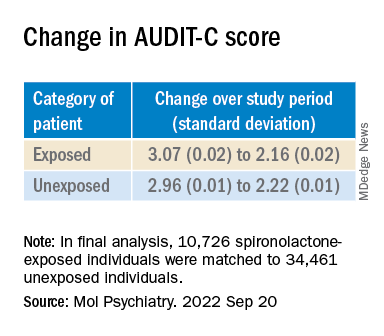
“These are very exciting times because, thanks to the progress in the addiction biomedical research field, we are increasing our understanding of the mechanisms how some people develop AUD; hence we can use this knowledge to identify new targets.” The current study “is an example of these ongoing efforts,” said Dr. Leggio.
“It is important to note that [these results] are important but preliminary.” At this juncture, “it would be too premature to think about prescribing spironolactone to treat AUD,” he added.
Exciting findings
Commenting on the study, Joyce Besheer, PhD, professor, department of psychiatry and Bowles Center for Alcohol Studies, University of North Carolina at Chapel Hill, called the study an “elegant demonstration of translational science.”
“While clinical trials will be needed to determine whether this medication is effective at reducing drinking in patients with AUD, these findings are exciting as they suggest that spironolactone may be a promising compound and new treatment options for AUD are much needed,” said Dr. Besheer, who was not involved with the current study.
Dr. Leggio agreed. “We now need prospective, placebo-controlled studies to assess the potential safety and efficacy of spironolactone in people with AUD,” he said.
This work was supported by the National Institutes of Health and the NIAAA. Dr. Leggio, study coauthors, and Dr. Besheer declare no relevant financial relationships.
A version of this article first appeared on Medscape.com.
(AUD), new research suggests.
Researchers at the National Institute on Drug Abuse, the National Institute on Alcohol Abuse and Alcoholism, and Yale University, New Haven, Conn., investigated the impact of spironolactone on AUD.
Initially, they studied rodents and found that spironolactone reduced binge drinking in mice and reduced self-administration of alcohol in rats without adversely affecting food or water intake or causing motor or coordination problems.
They also analyzed electronic health records of patients drawn from the United States Veterans Affairs health care system to explore potential changes in alcohol use after spironolactone treatment was initiated for other conditions and found a significant link between spironolactone treatment and reduction in self-reported alcohol consumption, with the largest effects observed among those who reported hazardous/heavy episodic alcohol use prior to starting spironolactone treatment.
“Combining findings across three species and different types of research studies, and then seeing similarities in these data, gives us confidence that we are onto something potentially important scientifically and clinically,” senior coauthor Lorenzo Leggio, MD, PhD, senior investigator in the Clinical Psychoneuroendocrinology and Neuropsychopharmacology Section, a joint NIDA and NIAAA laboratory, said in a news release.
The study was published online in Molecular Psychiatry.
There is a “critical need to increase the armamentarium of pharmacotherapies to treat individuals with AUD,” the authors note, adding that neuroendocrine systems involved in alcohol craving and drinking “offer promising pharmacologic targets in this regard.”
“Both our team and others have observed that patients with AUD often present with changes in peripheral hormones, including aldosterone, which plays a key role in regulating blood pressure and electrolytes,” Dr. Leggio said in an interview.
Spironolactone is a nonselective mineralocorticoid receptor (MT) antagonist. In studies in animal models, investigators said they found “an inverse correlation between alcohol drinking and the expression of the MR in the amygdala, a key brain region in the development and maintenance of AUD and addiction in general.”
Taken together, this led them to hypothesize that blocking the MR, which is the mechanism of action of spironolactone, “could be a novel pharmacotherapeutic approach for AUD,” he said.
Previous research by the same group of researchers suggested spironolactone “may be a potential new medication to treat patients with AUD.” The present study expanded on those findings and consisted of a three-part investigation.
In the current study, the investigators tested different dosages of spironolactone on binge-like alcohol consumption in male and female mice and assessed food and water intake, blood alcohol levels, motor coordination, and spontaneous locomotion.
They then tested the effects of different dosages of spironolactone injections on operant alcohol self-administration in alcohol-dependent and nondependent male and female rats, also testing blood alcohol levels and motor coordination.
Finally, they analyzed health records of veterans to examine the association between at least 60 continuous days of spironolactone treatment and self-reported alcohol consumption (measured by the Alcohol Use Disorders Identification Test-Consumption [AUDIT-C]).
Each of the spironolactone-exposed patients was matched using propensity scores with up to five unexposed patients who had reported alcohol consumption in the 2 years prior to the index date.
The final analysis included a matched cohort of 10,726 spironolactone-exposed individuals who were matched to 34,461 unexposed individuals.
New targets
Spironolactone reduced alcohol intake in mice drinking a sweetened alcohol solution; a 2-way ANOVA revealed a main effect of dose (F 4,52 = 9.09; P < .0001) and sex, with female mice drinking more alcohol, compared to male mice (F 1,13 = 6.05; P = .02).
Post hoc comparisons showed that spironolactone at doses of 50, 100, and 200 mg/kg significantly reduced alcohol intake (P values = .007, .002, and .0001, respectively).
In mice drinking an unsweetened alcohol solution, the 2-way repeated measures ANOVA similarly found a main effect of dose (F 4,52 = 5.77; P = .0006), but not of sex (F 1,13 = 1.41; P = .25).
Spironolactone had no effect on the mice’s intake of a sweet solution without alcohol and had no impact on the consumption of food and water or on locomotion and coordination.
In rats, a 2-way ANOVA revealed a significant spironolactone effect of dose (F 3,66 = 43.95; P < .001), with a post hoc test indicating that spironolactone at 25, 50, and 75 mg/kg reduced alcohol self-administration in alcohol-dependent and nondependent rats (all P values = .0001).
In humans, among the exposed individuals in the matched cohort, 25%, 57%, and 18% received daily doses of spironolactone of less than 25 mg/day, 25-49 mg/day, and 50 mg/day or higher, respectively, with a median follow-up time of 542 (interquartile range, 337-730) days.
The AUDIT-C scores decreased during the study period in both treatment groups, with a larger decrease in average AUDIT-C scores among the exposed vs. unexposed individuals.
“These are very exciting times because, thanks to the progress in the addiction biomedical research field, we are increasing our understanding of the mechanisms how some people develop AUD; hence we can use this knowledge to identify new targets.” The current study “is an example of these ongoing efforts,” said Dr. Leggio.
“It is important to note that [these results] are important but preliminary.” At this juncture, “it would be too premature to think about prescribing spironolactone to treat AUD,” he added.
Exciting findings
Commenting on the study, Joyce Besheer, PhD, professor, department of psychiatry and Bowles Center for Alcohol Studies, University of North Carolina at Chapel Hill, called the study an “elegant demonstration of translational science.”
“While clinical trials will be needed to determine whether this medication is effective at reducing drinking in patients with AUD, these findings are exciting as they suggest that spironolactone may be a promising compound and new treatment options for AUD are much needed,” said Dr. Besheer, who was not involved with the current study.
Dr. Leggio agreed. “We now need prospective, placebo-controlled studies to assess the potential safety and efficacy of spironolactone in people with AUD,” he said.
This work was supported by the National Institutes of Health and the NIAAA. Dr. Leggio, study coauthors, and Dr. Besheer declare no relevant financial relationships.
A version of this article first appeared on Medscape.com.
(AUD), new research suggests.
Researchers at the National Institute on Drug Abuse, the National Institute on Alcohol Abuse and Alcoholism, and Yale University, New Haven, Conn., investigated the impact of spironolactone on AUD.
Initially, they studied rodents and found that spironolactone reduced binge drinking in mice and reduced self-administration of alcohol in rats without adversely affecting food or water intake or causing motor or coordination problems.
They also analyzed electronic health records of patients drawn from the United States Veterans Affairs health care system to explore potential changes in alcohol use after spironolactone treatment was initiated for other conditions and found a significant link between spironolactone treatment and reduction in self-reported alcohol consumption, with the largest effects observed among those who reported hazardous/heavy episodic alcohol use prior to starting spironolactone treatment.
“Combining findings across three species and different types of research studies, and then seeing similarities in these data, gives us confidence that we are onto something potentially important scientifically and clinically,” senior coauthor Lorenzo Leggio, MD, PhD, senior investigator in the Clinical Psychoneuroendocrinology and Neuropsychopharmacology Section, a joint NIDA and NIAAA laboratory, said in a news release.
The study was published online in Molecular Psychiatry.
There is a “critical need to increase the armamentarium of pharmacotherapies to treat individuals with AUD,” the authors note, adding that neuroendocrine systems involved in alcohol craving and drinking “offer promising pharmacologic targets in this regard.”
“Both our team and others have observed that patients with AUD often present with changes in peripheral hormones, including aldosterone, which plays a key role in regulating blood pressure and electrolytes,” Dr. Leggio said in an interview.
Spironolactone is a nonselective mineralocorticoid receptor (MT) antagonist. In studies in animal models, investigators said they found “an inverse correlation between alcohol drinking and the expression of the MR in the amygdala, a key brain region in the development and maintenance of AUD and addiction in general.”
Taken together, this led them to hypothesize that blocking the MR, which is the mechanism of action of spironolactone, “could be a novel pharmacotherapeutic approach for AUD,” he said.
Previous research by the same group of researchers suggested spironolactone “may be a potential new medication to treat patients with AUD.” The present study expanded on those findings and consisted of a three-part investigation.
In the current study, the investigators tested different dosages of spironolactone on binge-like alcohol consumption in male and female mice and assessed food and water intake, blood alcohol levels, motor coordination, and spontaneous locomotion.
They then tested the effects of different dosages of spironolactone injections on operant alcohol self-administration in alcohol-dependent and nondependent male and female rats, also testing blood alcohol levels and motor coordination.
Finally, they analyzed health records of veterans to examine the association between at least 60 continuous days of spironolactone treatment and self-reported alcohol consumption (measured by the Alcohol Use Disorders Identification Test-Consumption [AUDIT-C]).
Each of the spironolactone-exposed patients was matched using propensity scores with up to five unexposed patients who had reported alcohol consumption in the 2 years prior to the index date.
The final analysis included a matched cohort of 10,726 spironolactone-exposed individuals who were matched to 34,461 unexposed individuals.
New targets
Spironolactone reduced alcohol intake in mice drinking a sweetened alcohol solution; a 2-way ANOVA revealed a main effect of dose (F 4,52 = 9.09; P < .0001) and sex, with female mice drinking more alcohol, compared to male mice (F 1,13 = 6.05; P = .02).
Post hoc comparisons showed that spironolactone at doses of 50, 100, and 200 mg/kg significantly reduced alcohol intake (P values = .007, .002, and .0001, respectively).
In mice drinking an unsweetened alcohol solution, the 2-way repeated measures ANOVA similarly found a main effect of dose (F 4,52 = 5.77; P = .0006), but not of sex (F 1,13 = 1.41; P = .25).
Spironolactone had no effect on the mice’s intake of a sweet solution without alcohol and had no impact on the consumption of food and water or on locomotion and coordination.
In rats, a 2-way ANOVA revealed a significant spironolactone effect of dose (F 3,66 = 43.95; P < .001), with a post hoc test indicating that spironolactone at 25, 50, and 75 mg/kg reduced alcohol self-administration in alcohol-dependent and nondependent rats (all P values = .0001).
In humans, among the exposed individuals in the matched cohort, 25%, 57%, and 18% received daily doses of spironolactone of less than 25 mg/day, 25-49 mg/day, and 50 mg/day or higher, respectively, with a median follow-up time of 542 (interquartile range, 337-730) days.
The AUDIT-C scores decreased during the study period in both treatment groups, with a larger decrease in average AUDIT-C scores among the exposed vs. unexposed individuals.
“These are very exciting times because, thanks to the progress in the addiction biomedical research field, we are increasing our understanding of the mechanisms how some people develop AUD; hence we can use this knowledge to identify new targets.” The current study “is an example of these ongoing efforts,” said Dr. Leggio.
“It is important to note that [these results] are important but preliminary.” At this juncture, “it would be too premature to think about prescribing spironolactone to treat AUD,” he added.
Exciting findings
Commenting on the study, Joyce Besheer, PhD, professor, department of psychiatry and Bowles Center for Alcohol Studies, University of North Carolina at Chapel Hill, called the study an “elegant demonstration of translational science.”
“While clinical trials will be needed to determine whether this medication is effective at reducing drinking in patients with AUD, these findings are exciting as they suggest that spironolactone may be a promising compound and new treatment options for AUD are much needed,” said Dr. Besheer, who was not involved with the current study.
Dr. Leggio agreed. “We now need prospective, placebo-controlled studies to assess the potential safety and efficacy of spironolactone in people with AUD,” he said.
This work was supported by the National Institutes of Health and the NIAAA. Dr. Leggio, study coauthors, and Dr. Besheer declare no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM MOLECULAR PSYCHIATRY
Apremilast may have some cardiometabolic benefits in patients with psoriasis
The trial, led by Joel M. Gelfand, MD, MSCE, professor of dermatology and epidemiology and vice chair of clinical research in dermatology at the University of Pennsylvania, Philadelphia, found that apremilast (Otezla) has a neutral effect on aortic vascular inflammation in patients with moderate to severe psoriasis.
It also had variable, but generally favorable, associations with 68 cardiometabolic biomarkers tested and associations with reductions in both visceral and subcutaneous fat. Findings of the study were published online in JAMA Dermatology.
Fat reductions maintained at 1-year mark
The researchers found a 5%-6% reduction in subcutaneous and visceral fat at week 16 of the study that was maintained at the 1-year mark. “The fact that it was rock stable a year later is pretty encouraging,” Dr. Gelfand told this news organization.

As for effects on vascular inflammation, Dr. Gelfand said, “The good news is we didn’t find any adverse effects on aortic vascular inflammation, but we didn’t find any beneficial effects either. That was a little disappointing.
“The most surprising thing was really the effects on visceral adiposity,” he added. “I’m not aware of any other drug having demonstrated that effect.”
Michael S. Garshick, MD, a cardiologist with NYU Langone Health in New York, who was not involved with the trial, told this news organization that despite seemingly good epidemiologic evidence in observational studies that by treating psoriasis surrogates of cardiovascular risk can be reduced, this trial, like others before it, failed to reduce aortic vascular inflammation.

The trial does help answer the question of whether apremilast can induce weight loss, he said, something that earlier trials suggested. “This trial confirms that, which is exciting,” he said. The reduction in both visceral and subcutaneous fat “deserves a lot further study.”
Several questions remain, Dr. Garshick said. Both he and Dr. Gelfand pointed to the need for large, placebo-controlled trials. “We still don’t know which medications may be preferrable in psoriasis to reduce [cardiovascular] risk if any at all,” Dr. Garshick said.
Seventy patients enrolled
In total, 70 patients with moderate to severe psoriasis were enrolled, 60 completed week 16, and 39 completed week 52 of the single-arm, open-label trial conducted between April 2017 and August 2021 at seven dermatology sites in the United States.
Participants took 30 mg of apremilast, an oral phosphodiesterase-4 (PDE-4) inhibitor approved for treating psoriasis and psoriatic arthritis, twice daily. Participants’ average age was 47.5 years; most were male (77.1%) and White (82.9%); almost 6% were Black. Average body mass index was 30 kg/m2. Patients could not have received biologics within 90 days of study baseline (or 180 days for ustekinumab [Stelara]).
There was no change in aortic vascular inflammation at week 16 (target to background ratio, −0.02; 95% confidence interval [CI], −0.08 to 0.05; P = .61) or week 52 (target to background ratio, −0.07; 95% CI, −0.15 to 0.01; P = .09) compared with baseline.
“At week 16, there were reductions in levels of interleukin-1b, fetuin A, valine, leucine, and isoleucine,” the authors wrote, adding that at week 52, compared with baseline, “there were reductions in levels of ferritin, cholesterol efflux capacity, beta-hydroxybutyrate, acetone, and ketone bodies, and an increase in levels of apolipoprotein A-1.”
This study highlights the importance of screening, Dr. Garshick said.
He and Dr. Gelfand said people with psoriatic disease tend to be vastly underscreened for cardiovascular risk factors.
Dr. Gelfand said, “If we did what we knew worked – meaning we screened them for diabetes, we screen their cholesterol, we check their blood pressure, and we adequately treated those traditional cardiovascular risk factors, we probably could narrow the gap quite a bit” in terms of the lower life expectancy people face when they have more significant psoriasis.
Celgene was the initial funding sponsor; sponsorship was then transferred to Amgen. The authors designed, executed, analyzed, and reported the study. Celgene provided nonbinding input into study design, and Amgen provided nonbinding input into the reporting of results. Dr. Gelfand reported numerous disclosures with various pharmaceutical companies and organizations. Dr. Garshick reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The trial, led by Joel M. Gelfand, MD, MSCE, professor of dermatology and epidemiology and vice chair of clinical research in dermatology at the University of Pennsylvania, Philadelphia, found that apremilast (Otezla) has a neutral effect on aortic vascular inflammation in patients with moderate to severe psoriasis.
It also had variable, but generally favorable, associations with 68 cardiometabolic biomarkers tested and associations with reductions in both visceral and subcutaneous fat. Findings of the study were published online in JAMA Dermatology.
Fat reductions maintained at 1-year mark
The researchers found a 5%-6% reduction in subcutaneous and visceral fat at week 16 of the study that was maintained at the 1-year mark. “The fact that it was rock stable a year later is pretty encouraging,” Dr. Gelfand told this news organization.
As for effects on vascular inflammation, Dr. Gelfand said, “The good news is we didn’t find any adverse effects on aortic vascular inflammation, but we didn’t find any beneficial effects either. That was a little disappointing.
“The most surprising thing was really the effects on visceral adiposity,” he added. “I’m not aware of any other drug having demonstrated that effect.”
Michael S. Garshick, MD, a cardiologist with NYU Langone Health in New York, who was not involved with the trial, told this news organization that despite seemingly good epidemiologic evidence in observational studies that by treating psoriasis surrogates of cardiovascular risk can be reduced, this trial, like others before it, failed to reduce aortic vascular inflammation.
The trial does help answer the question of whether apremilast can induce weight loss, he said, something that earlier trials suggested. “This trial confirms that, which is exciting,” he said. The reduction in both visceral and subcutaneous fat “deserves a lot further study.”
Several questions remain, Dr. Garshick said. Both he and Dr. Gelfand pointed to the need for large, placebo-controlled trials. “We still don’t know which medications may be preferrable in psoriasis to reduce [cardiovascular] risk if any at all,” Dr. Garshick said.
Seventy patients enrolled
In total, 70 patients with moderate to severe psoriasis were enrolled, 60 completed week 16, and 39 completed week 52 of the single-arm, open-label trial conducted between April 2017 and August 2021 at seven dermatology sites in the United States.
Participants took 30 mg of apremilast, an oral phosphodiesterase-4 (PDE-4) inhibitor approved for treating psoriasis and psoriatic arthritis, twice daily. Participants’ average age was 47.5 years; most were male (77.1%) and White (82.9%); almost 6% were Black. Average body mass index was 30 kg/m2. Patients could not have received biologics within 90 days of study baseline (or 180 days for ustekinumab [Stelara]).
There was no change in aortic vascular inflammation at week 16 (target to background ratio, −0.02; 95% confidence interval [CI], −0.08 to 0.05; P = .61) or week 52 (target to background ratio, −0.07; 95% CI, −0.15 to 0.01; P = .09) compared with baseline.
“At week 16, there were reductions in levels of interleukin-1b, fetuin A, valine, leucine, and isoleucine,” the authors wrote, adding that at week 52, compared with baseline, “there were reductions in levels of ferritin, cholesterol efflux capacity, beta-hydroxybutyrate, acetone, and ketone bodies, and an increase in levels of apolipoprotein A-1.”
This study highlights the importance of screening, Dr. Garshick said.
He and Dr. Gelfand said people with psoriatic disease tend to be vastly underscreened for cardiovascular risk factors.
Dr. Gelfand said, “If we did what we knew worked – meaning we screened them for diabetes, we screen their cholesterol, we check their blood pressure, and we adequately treated those traditional cardiovascular risk factors, we probably could narrow the gap quite a bit” in terms of the lower life expectancy people face when they have more significant psoriasis.
Celgene was the initial funding sponsor; sponsorship was then transferred to Amgen. The authors designed, executed, analyzed, and reported the study. Celgene provided nonbinding input into study design, and Amgen provided nonbinding input into the reporting of results. Dr. Gelfand reported numerous disclosures with various pharmaceutical companies and organizations. Dr. Garshick reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The trial, led by Joel M. Gelfand, MD, MSCE, professor of dermatology and epidemiology and vice chair of clinical research in dermatology at the University of Pennsylvania, Philadelphia, found that apremilast (Otezla) has a neutral effect on aortic vascular inflammation in patients with moderate to severe psoriasis.
It also had variable, but generally favorable, associations with 68 cardiometabolic biomarkers tested and associations with reductions in both visceral and subcutaneous fat. Findings of the study were published online in JAMA Dermatology.
Fat reductions maintained at 1-year mark
The researchers found a 5%-6% reduction in subcutaneous and visceral fat at week 16 of the study that was maintained at the 1-year mark. “The fact that it was rock stable a year later is pretty encouraging,” Dr. Gelfand told this news organization.
As for effects on vascular inflammation, Dr. Gelfand said, “The good news is we didn’t find any adverse effects on aortic vascular inflammation, but we didn’t find any beneficial effects either. That was a little disappointing.
“The most surprising thing was really the effects on visceral adiposity,” he added. “I’m not aware of any other drug having demonstrated that effect.”
Michael S. Garshick, MD, a cardiologist with NYU Langone Health in New York, who was not involved with the trial, told this news organization that despite seemingly good epidemiologic evidence in observational studies that by treating psoriasis surrogates of cardiovascular risk can be reduced, this trial, like others before it, failed to reduce aortic vascular inflammation.
The trial does help answer the question of whether apremilast can induce weight loss, he said, something that earlier trials suggested. “This trial confirms that, which is exciting,” he said. The reduction in both visceral and subcutaneous fat “deserves a lot further study.”
Several questions remain, Dr. Garshick said. Both he and Dr. Gelfand pointed to the need for large, placebo-controlled trials. “We still don’t know which medications may be preferrable in psoriasis to reduce [cardiovascular] risk if any at all,” Dr. Garshick said.
Seventy patients enrolled
In total, 70 patients with moderate to severe psoriasis were enrolled, 60 completed week 16, and 39 completed week 52 of the single-arm, open-label trial conducted between April 2017 and August 2021 at seven dermatology sites in the United States.
Participants took 30 mg of apremilast, an oral phosphodiesterase-4 (PDE-4) inhibitor approved for treating psoriasis and psoriatic arthritis, twice daily. Participants’ average age was 47.5 years; most were male (77.1%) and White (82.9%); almost 6% were Black. Average body mass index was 30 kg/m2. Patients could not have received biologics within 90 days of study baseline (or 180 days for ustekinumab [Stelara]).
There was no change in aortic vascular inflammation at week 16 (target to background ratio, −0.02; 95% confidence interval [CI], −0.08 to 0.05; P = .61) or week 52 (target to background ratio, −0.07; 95% CI, −0.15 to 0.01; P = .09) compared with baseline.
“At week 16, there were reductions in levels of interleukin-1b, fetuin A, valine, leucine, and isoleucine,” the authors wrote, adding that at week 52, compared with baseline, “there were reductions in levels of ferritin, cholesterol efflux capacity, beta-hydroxybutyrate, acetone, and ketone bodies, and an increase in levels of apolipoprotein A-1.”
This study highlights the importance of screening, Dr. Garshick said.
He and Dr. Gelfand said people with psoriatic disease tend to be vastly underscreened for cardiovascular risk factors.
Dr. Gelfand said, “If we did what we knew worked – meaning we screened them for diabetes, we screen their cholesterol, we check their blood pressure, and we adequately treated those traditional cardiovascular risk factors, we probably could narrow the gap quite a bit” in terms of the lower life expectancy people face when they have more significant psoriasis.
Celgene was the initial funding sponsor; sponsorship was then transferred to Amgen. The authors designed, executed, analyzed, and reported the study. Celgene provided nonbinding input into study design, and Amgen provided nonbinding input into the reporting of results. Dr. Gelfand reported numerous disclosures with various pharmaceutical companies and organizations. Dr. Garshick reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JAMA DERMATOLOGY
Add PCSK9 inhibitor to high-intensity statin at primary PCI, proposes sham-controlled EPIC-STEMI
It’s best to have patients on aggressive lipid-lowering therapy before discharge after an acute ST-segment elevation myocardial infarction (STEMI), so why not start it right away – even in the cath lab – using some of the most potent LDL cholesterol–lowering agents available?
That was a main idea behind the randomized, sham-controlled EPIC-STEMI trial, in which STEMI patients were started on a PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitor immediately before direct percutaneous coronary intervention (PCI) and on top of high-intensity statins.
Those in the trial getting the active agent showed a 22% drop in LDL cholesterol levels by 6 weeks, compared with the control group given a sham injection along with high-intensity statins. They were also more likely to meet LDL cholesterol goals specified in some guidelines, including reduction by at least 50%. And those outcomes were achieved regardless of baseline LDL cholesterol levels or prior statin use.
Adoption of the trial’s early, aggressive LDL cholesterolreduction strategy in practice “has the potential to substantially reduce morbidity and mortality” in such cases “by further reducing LDL beyond statins in a much greater number of high-risk patients than are currently being treated with these agents,” suggested principal investigator Shamir R. Mehta, MD, MSc, when presenting the findings at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
Adherence to secondary prevention measures in patients with acute coronary syndromes (ACS) is much better if they are started before hospital discharge, explained Dr. Mehta, senior scientist with Population Health Research Institute and professor of medicine at McMaster University, Hamilton, Ont. But “as soon as the patient has left the hospital, it is much more difficult to get these therapies on board.”
Routine adoption of such aggressive in-hospital, lipid-lowering therapy for the vast population with ACS would likely mean far fewer deaths and cardiovascular events “across a broader patient population.”
EPIC-STEMI is among the first studies to explore the strategy. “I think that’s the point of the trial that we wanted to make, that we don’t yet have data on this. We’re treading very carefully with PCSK9 inhibitors, and it’s just inching forward in populations. And I think we need a bold trial to see whether or not this changes things.”
The PCSK9 inhibitor alirocumab (Praluent) was used in EPIC-STEMI, which was published in EuroIntervention, with Dr. Mehta as lead author, the same day as his presentation. The drug and its sham injection were given on top of either atorvastatin 40-80 mg or rosuvastatin 40 mg.
Early initiation of statins in patients with acute STEMI has become standard, but there’s good evidence from intracoronary imaging studies suggesting that the addition of PCSK9 inhibitors might promote further stabilization of plaques that could potentially cause recurrent ischemic events.
Treatment with the injectable drugs plus statins led to significant coronary lesion regression in the GLAGOV trial of patients with stable coronary disease. And initiation of PCSK9 inhibitors with high-intensity statins soon after PCI for ACS improved atheroma shrinkage in non–infarct-related arteries over 1 year in the recent, placebo-controlled PACMAN-AMI trial.
Dr. Mehta pointed out that LDL reductions on PCSK9 inhibition, compared with the sham control, weren’t necessarily as impressive as might be expected from the major trials of long-term therapy with the drugs.
“You need longer [therapy] in order to see a difference in LDL levels when you use a PCSK9 inhibitor acutely. This is shown also on measures of infarct size.” There was no difference between treatment groups in infarct size as measured by levels of the MB fraction of creatine kinase, he reported.
“What this is telling us is that the acute use of a PCSK9 inhibitor did not modify the size or the severity of the baseline STEMI event.”
And EPIC-STEMI was too small and never intended to assess clinical outcomes; it was more about feasibility and what degree of LDL cholesterol lowering might be expected.
The trial was needed, Dr. Mehta said, because the PCSK9 inhibitors haven’t been extensively adopted into clinical practice and are not getting to the patients who could most benefit. One of the reasons for that is quite clear to him. “We are missing the high-risk patients because we are not treating them acutely,” Dr. Mehta said in an interview.
The strategy “has not yet been evaluated, and there have been barriers,” he observed. “Cost has been a barrier. Access to the drug has been a barrier. But in terms of the science, in terms of reducing cardiovascular events, this is a strategy that has to be tested.”

The aggressive, early LDL cholesterol reduction strategy should be evaluated for its effect on long-term outcomes, “especially knowing that in the first 30 days to 6 months post STEMI there’s a tremendous uptick in ischemic events, including recurrent myocardial infarction,” Roxana Mehran, MD, said at a media briefing on EPIC-STEMI held before Dr. Mehta’s formal presentation.
The “fantastic reduction acutely” with a PCSK9 inhibitor on top of statins, “hopefully reducing inflammation” similarly to what’s been observed in past trials, “absolutely warrants” a STEMI clinical outcomes trial, said Dr. Mehran, Icahn School of Medicine at Mount Sinai, New York, who isn’t connected with EPIC-STEMI.
If better post-discharge medication adherence is one of the acute strategy’s goals, it will be important to consider the potential influence of prescribing a periodically injected drug, proposed Eric A. Cohen, MD, Sunnybrook Health Sciences Center, Toronto, at the press conference.
“Keep in mind that STEMI patients typically come to the hospital on zero medications and leave 2 days later on five medications,” Dr. Cohen observed. “I’m curious whether having one of those as a sub-Q injection every 2 weeks, and reducing the pill burden, will help or deter adherence to therapy. I think it’s worth studying.”
The trial originally included 97 patients undergoing PCI for STEMI who were randomly assigned to receive the PCSK9 inhibitor or a sham injection on top of high-intensity statins, without regard to LDL cholesterol levels. Randomization took place after diagnostic angiography but before PCI.
The analysis, however, subsequently excluded 29 patients who could not continue with the study, “mainly because of hospital research clinic closure due to the COVID-19 pandemic,” the published report states.
That left 68 patients who had received at least one dose of PCSK9 inhibitor, alirocumab 150 mg subcutaneously, or the sham injection, and had at least one blood draw for LDL cholesterol response which, Dr. Mehta said, still left adequate statistical power for the LDL cholesterol–based primary endpoint.
By 6 weeks, LDL cholesterol levels had fallen 72.9% in the active-therapy group and by 48.1% in the control group (P < .001). Also, 92.1% and 56.7% of patients, respectively (P = .002), had achieved levels below the 1.4 mmol/L (54 mg/dL) goal in the European guidelines, Dr. Mehta reported.
Levels fell more than 50% compared with baseline in 89.5% of alirocumab patients and 60% (P = .007) of controls, respectively.
There was no significant difference in rates of attaining LDL cholesterol levels below the 70 mg/dL (1.8 mmol/L) threshold specified in U.S. guidelines for very high-risk patients: 94.7% of alirocumab patients and 83.4% of controls (P = .26).
Nor did the groups differ significantly in natriuretic peptide levels, which reflect ventricular remodeling; or in 6-week change in the inflammatory biomarker high-sensitivity C-reactive protein.
An open-label, randomized trial scheduled to launch before the end of 2022 will explore similarly early initiation of a PCSK9 inhibitor, compared with standard lipid management, in an estimated 4,000 patients hospitalized with STEMI or non-STEMI.
The EVOLVE MI trial is looking at the monoclonal antibody evolocumab (Repatha) for its effect on the primary endpoint of myocardial infarction, ischemic stroke, arterial revascularization, or death from any cause over an expected 3-4 years.
EPIC-STEMI was supported in part by Sanofi. Dr. Mehta reported an unrestricted grant from Sanofi to Hamilton Health Sciences for the present study and consulting fees from Amgen, Sanofi, and Novartis. Dr. Cohen disclosed receiving grant support from and holding research contracts with Abbott Vascular; and receiving fees for consulting, honoraria, or serving on a speaker’s bureau for Abbott Vascular, Medtronic, and Baylis. Dr. Mehran disclosed receiving grants or research support from numerous pharmaceutical companies; receiving consultant fee or honoraria or serving on a speaker’s bureau for Novartis, Abbott Vascular, Janssen, Medtronic, Medscape/WebMD, and Cine-Med Research; and holding equity, stock, or stock options with Control Rad, Applied Therapeutics, and Elixir Medical.
A version of this article first appeared on Medscape.com.
It’s best to have patients on aggressive lipid-lowering therapy before discharge after an acute ST-segment elevation myocardial infarction (STEMI), so why not start it right away – even in the cath lab – using some of the most potent LDL cholesterol–lowering agents available?
That was a main idea behind the randomized, sham-controlled EPIC-STEMI trial, in which STEMI patients were started on a PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitor immediately before direct percutaneous coronary intervention (PCI) and on top of high-intensity statins.
Those in the trial getting the active agent showed a 22% drop in LDL cholesterol levels by 6 weeks, compared with the control group given a sham injection along with high-intensity statins. They were also more likely to meet LDL cholesterol goals specified in some guidelines, including reduction by at least 50%. And those outcomes were achieved regardless of baseline LDL cholesterol levels or prior statin use.
Adoption of the trial’s early, aggressive LDL cholesterolreduction strategy in practice “has the potential to substantially reduce morbidity and mortality” in such cases “by further reducing LDL beyond statins in a much greater number of high-risk patients than are currently being treated with these agents,” suggested principal investigator Shamir R. Mehta, MD, MSc, when presenting the findings at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
Adherence to secondary prevention measures in patients with acute coronary syndromes (ACS) is much better if they are started before hospital discharge, explained Dr. Mehta, senior scientist with Population Health Research Institute and professor of medicine at McMaster University, Hamilton, Ont. But “as soon as the patient has left the hospital, it is much more difficult to get these therapies on board.”
Routine adoption of such aggressive in-hospital, lipid-lowering therapy for the vast population with ACS would likely mean far fewer deaths and cardiovascular events “across a broader patient population.”
EPIC-STEMI is among the first studies to explore the strategy. “I think that’s the point of the trial that we wanted to make, that we don’t yet have data on this. We’re treading very carefully with PCSK9 inhibitors, and it’s just inching forward in populations. And I think we need a bold trial to see whether or not this changes things.”
The PCSK9 inhibitor alirocumab (Praluent) was used in EPIC-STEMI, which was published in EuroIntervention, with Dr. Mehta as lead author, the same day as his presentation. The drug and its sham injection were given on top of either atorvastatin 40-80 mg or rosuvastatin 40 mg.
Early initiation of statins in patients with acute STEMI has become standard, but there’s good evidence from intracoronary imaging studies suggesting that the addition of PCSK9 inhibitors might promote further stabilization of plaques that could potentially cause recurrent ischemic events.
Treatment with the injectable drugs plus statins led to significant coronary lesion regression in the GLAGOV trial of patients with stable coronary disease. And initiation of PCSK9 inhibitors with high-intensity statins soon after PCI for ACS improved atheroma shrinkage in non–infarct-related arteries over 1 year in the recent, placebo-controlled PACMAN-AMI trial.
Dr. Mehta pointed out that LDL reductions on PCSK9 inhibition, compared with the sham control, weren’t necessarily as impressive as might be expected from the major trials of long-term therapy with the drugs.
“You need longer [therapy] in order to see a difference in LDL levels when you use a PCSK9 inhibitor acutely. This is shown also on measures of infarct size.” There was no difference between treatment groups in infarct size as measured by levels of the MB fraction of creatine kinase, he reported.
“What this is telling us is that the acute use of a PCSK9 inhibitor did not modify the size or the severity of the baseline STEMI event.”
And EPIC-STEMI was too small and never intended to assess clinical outcomes; it was more about feasibility and what degree of LDL cholesterol lowering might be expected.
The trial was needed, Dr. Mehta said, because the PCSK9 inhibitors haven’t been extensively adopted into clinical practice and are not getting to the patients who could most benefit. One of the reasons for that is quite clear to him. “We are missing the high-risk patients because we are not treating them acutely,” Dr. Mehta said in an interview.
The strategy “has not yet been evaluated, and there have been barriers,” he observed. “Cost has been a barrier. Access to the drug has been a barrier. But in terms of the science, in terms of reducing cardiovascular events, this is a strategy that has to be tested.”
The aggressive, early LDL cholesterol reduction strategy should be evaluated for its effect on long-term outcomes, “especially knowing that in the first 30 days to 6 months post STEMI there’s a tremendous uptick in ischemic events, including recurrent myocardial infarction,” Roxana Mehran, MD, said at a media briefing on EPIC-STEMI held before Dr. Mehta’s formal presentation.
The “fantastic reduction acutely” with a PCSK9 inhibitor on top of statins, “hopefully reducing inflammation” similarly to what’s been observed in past trials, “absolutely warrants” a STEMI clinical outcomes trial, said Dr. Mehran, Icahn School of Medicine at Mount Sinai, New York, who isn’t connected with EPIC-STEMI.
If better post-discharge medication adherence is one of the acute strategy’s goals, it will be important to consider the potential influence of prescribing a periodically injected drug, proposed Eric A. Cohen, MD, Sunnybrook Health Sciences Center, Toronto, at the press conference.
“Keep in mind that STEMI patients typically come to the hospital on zero medications and leave 2 days later on five medications,” Dr. Cohen observed. “I’m curious whether having one of those as a sub-Q injection every 2 weeks, and reducing the pill burden, will help or deter adherence to therapy. I think it’s worth studying.”
The trial originally included 97 patients undergoing PCI for STEMI who were randomly assigned to receive the PCSK9 inhibitor or a sham injection on top of high-intensity statins, without regard to LDL cholesterol levels. Randomization took place after diagnostic angiography but before PCI.
The analysis, however, subsequently excluded 29 patients who could not continue with the study, “mainly because of hospital research clinic closure due to the COVID-19 pandemic,” the published report states.
That left 68 patients who had received at least one dose of PCSK9 inhibitor, alirocumab 150 mg subcutaneously, or the sham injection, and had at least one blood draw for LDL cholesterol response which, Dr. Mehta said, still left adequate statistical power for the LDL cholesterol–based primary endpoint.
By 6 weeks, LDL cholesterol levels had fallen 72.9% in the active-therapy group and by 48.1% in the control group (P < .001). Also, 92.1% and 56.7% of patients, respectively (P = .002), had achieved levels below the 1.4 mmol/L (54 mg/dL) goal in the European guidelines, Dr. Mehta reported.
Levels fell more than 50% compared with baseline in 89.5% of alirocumab patients and 60% (P = .007) of controls, respectively.
There was no significant difference in rates of attaining LDL cholesterol levels below the 70 mg/dL (1.8 mmol/L) threshold specified in U.S. guidelines for very high-risk patients: 94.7% of alirocumab patients and 83.4% of controls (P = .26).
Nor did the groups differ significantly in natriuretic peptide levels, which reflect ventricular remodeling; or in 6-week change in the inflammatory biomarker high-sensitivity C-reactive protein.
An open-label, randomized trial scheduled to launch before the end of 2022 will explore similarly early initiation of a PCSK9 inhibitor, compared with standard lipid management, in an estimated 4,000 patients hospitalized with STEMI or non-STEMI.
The EVOLVE MI trial is looking at the monoclonal antibody evolocumab (Repatha) for its effect on the primary endpoint of myocardial infarction, ischemic stroke, arterial revascularization, or death from any cause over an expected 3-4 years.
EPIC-STEMI was supported in part by Sanofi. Dr. Mehta reported an unrestricted grant from Sanofi to Hamilton Health Sciences for the present study and consulting fees from Amgen, Sanofi, and Novartis. Dr. Cohen disclosed receiving grant support from and holding research contracts with Abbott Vascular; and receiving fees for consulting, honoraria, or serving on a speaker’s bureau for Abbott Vascular, Medtronic, and Baylis. Dr. Mehran disclosed receiving grants or research support from numerous pharmaceutical companies; receiving consultant fee or honoraria or serving on a speaker’s bureau for Novartis, Abbott Vascular, Janssen, Medtronic, Medscape/WebMD, and Cine-Med Research; and holding equity, stock, or stock options with Control Rad, Applied Therapeutics, and Elixir Medical.
A version of this article first appeared on Medscape.com.
It’s best to have patients on aggressive lipid-lowering therapy before discharge after an acute ST-segment elevation myocardial infarction (STEMI), so why not start it right away – even in the cath lab – using some of the most potent LDL cholesterol–lowering agents available?
That was a main idea behind the randomized, sham-controlled EPIC-STEMI trial, in which STEMI patients were started on a PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitor immediately before direct percutaneous coronary intervention (PCI) and on top of high-intensity statins.
Those in the trial getting the active agent showed a 22% drop in LDL cholesterol levels by 6 weeks, compared with the control group given a sham injection along with high-intensity statins. They were also more likely to meet LDL cholesterol goals specified in some guidelines, including reduction by at least 50%. And those outcomes were achieved regardless of baseline LDL cholesterol levels or prior statin use.
Adoption of the trial’s early, aggressive LDL cholesterolreduction strategy in practice “has the potential to substantially reduce morbidity and mortality” in such cases “by further reducing LDL beyond statins in a much greater number of high-risk patients than are currently being treated with these agents,” suggested principal investigator Shamir R. Mehta, MD, MSc, when presenting the findings at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
Adherence to secondary prevention measures in patients with acute coronary syndromes (ACS) is much better if they are started before hospital discharge, explained Dr. Mehta, senior scientist with Population Health Research Institute and professor of medicine at McMaster University, Hamilton, Ont. But “as soon as the patient has left the hospital, it is much more difficult to get these therapies on board.”
Routine adoption of such aggressive in-hospital, lipid-lowering therapy for the vast population with ACS would likely mean far fewer deaths and cardiovascular events “across a broader patient population.”
EPIC-STEMI is among the first studies to explore the strategy. “I think that’s the point of the trial that we wanted to make, that we don’t yet have data on this. We’re treading very carefully with PCSK9 inhibitors, and it’s just inching forward in populations. And I think we need a bold trial to see whether or not this changes things.”
The PCSK9 inhibitor alirocumab (Praluent) was used in EPIC-STEMI, which was published in EuroIntervention, with Dr. Mehta as lead author, the same day as his presentation. The drug and its sham injection were given on top of either atorvastatin 40-80 mg or rosuvastatin 40 mg.
Early initiation of statins in patients with acute STEMI has become standard, but there’s good evidence from intracoronary imaging studies suggesting that the addition of PCSK9 inhibitors might promote further stabilization of plaques that could potentially cause recurrent ischemic events.
Treatment with the injectable drugs plus statins led to significant coronary lesion regression in the GLAGOV trial of patients with stable coronary disease. And initiation of PCSK9 inhibitors with high-intensity statins soon after PCI for ACS improved atheroma shrinkage in non–infarct-related arteries over 1 year in the recent, placebo-controlled PACMAN-AMI trial.
Dr. Mehta pointed out that LDL reductions on PCSK9 inhibition, compared with the sham control, weren’t necessarily as impressive as might be expected from the major trials of long-term therapy with the drugs.
“You need longer [therapy] in order to see a difference in LDL levels when you use a PCSK9 inhibitor acutely. This is shown also on measures of infarct size.” There was no difference between treatment groups in infarct size as measured by levels of the MB fraction of creatine kinase, he reported.
“What this is telling us is that the acute use of a PCSK9 inhibitor did not modify the size or the severity of the baseline STEMI event.”
And EPIC-STEMI was too small and never intended to assess clinical outcomes; it was more about feasibility and what degree of LDL cholesterol lowering might be expected.
The trial was needed, Dr. Mehta said, because the PCSK9 inhibitors haven’t been extensively adopted into clinical practice and are not getting to the patients who could most benefit. One of the reasons for that is quite clear to him. “We are missing the high-risk patients because we are not treating them acutely,” Dr. Mehta said in an interview.
The strategy “has not yet been evaluated, and there have been barriers,” he observed. “Cost has been a barrier. Access to the drug has been a barrier. But in terms of the science, in terms of reducing cardiovascular events, this is a strategy that has to be tested.”
The aggressive, early LDL cholesterol reduction strategy should be evaluated for its effect on long-term outcomes, “especially knowing that in the first 30 days to 6 months post STEMI there’s a tremendous uptick in ischemic events, including recurrent myocardial infarction,” Roxana Mehran, MD, said at a media briefing on EPIC-STEMI held before Dr. Mehta’s formal presentation.
The “fantastic reduction acutely” with a PCSK9 inhibitor on top of statins, “hopefully reducing inflammation” similarly to what’s been observed in past trials, “absolutely warrants” a STEMI clinical outcomes trial, said Dr. Mehran, Icahn School of Medicine at Mount Sinai, New York, who isn’t connected with EPIC-STEMI.
If better post-discharge medication adherence is one of the acute strategy’s goals, it will be important to consider the potential influence of prescribing a periodically injected drug, proposed Eric A. Cohen, MD, Sunnybrook Health Sciences Center, Toronto, at the press conference.
“Keep in mind that STEMI patients typically come to the hospital on zero medications and leave 2 days later on five medications,” Dr. Cohen observed. “I’m curious whether having one of those as a sub-Q injection every 2 weeks, and reducing the pill burden, will help or deter adherence to therapy. I think it’s worth studying.”
The trial originally included 97 patients undergoing PCI for STEMI who were randomly assigned to receive the PCSK9 inhibitor or a sham injection on top of high-intensity statins, without regard to LDL cholesterol levels. Randomization took place after diagnostic angiography but before PCI.
The analysis, however, subsequently excluded 29 patients who could not continue with the study, “mainly because of hospital research clinic closure due to the COVID-19 pandemic,” the published report states.
That left 68 patients who had received at least one dose of PCSK9 inhibitor, alirocumab 150 mg subcutaneously, or the sham injection, and had at least one blood draw for LDL cholesterol response which, Dr. Mehta said, still left adequate statistical power for the LDL cholesterol–based primary endpoint.
By 6 weeks, LDL cholesterol levels had fallen 72.9% in the active-therapy group and by 48.1% in the control group (P < .001). Also, 92.1% and 56.7% of patients, respectively (P = .002), had achieved levels below the 1.4 mmol/L (54 mg/dL) goal in the European guidelines, Dr. Mehta reported.
Levels fell more than 50% compared with baseline in 89.5% of alirocumab patients and 60% (P = .007) of controls, respectively.
There was no significant difference in rates of attaining LDL cholesterol levels below the 70 mg/dL (1.8 mmol/L) threshold specified in U.S. guidelines for very high-risk patients: 94.7% of alirocumab patients and 83.4% of controls (P = .26).
Nor did the groups differ significantly in natriuretic peptide levels, which reflect ventricular remodeling; or in 6-week change in the inflammatory biomarker high-sensitivity C-reactive protein.
An open-label, randomized trial scheduled to launch before the end of 2022 will explore similarly early initiation of a PCSK9 inhibitor, compared with standard lipid management, in an estimated 4,000 patients hospitalized with STEMI or non-STEMI.
The EVOLVE MI trial is looking at the monoclonal antibody evolocumab (Repatha) for its effect on the primary endpoint of myocardial infarction, ischemic stroke, arterial revascularization, or death from any cause over an expected 3-4 years.
EPIC-STEMI was supported in part by Sanofi. Dr. Mehta reported an unrestricted grant from Sanofi to Hamilton Health Sciences for the present study and consulting fees from Amgen, Sanofi, and Novartis. Dr. Cohen disclosed receiving grant support from and holding research contracts with Abbott Vascular; and receiving fees for consulting, honoraria, or serving on a speaker’s bureau for Abbott Vascular, Medtronic, and Baylis. Dr. Mehran disclosed receiving grants or research support from numerous pharmaceutical companies; receiving consultant fee or honoraria or serving on a speaker’s bureau for Novartis, Abbott Vascular, Janssen, Medtronic, Medscape/WebMD, and Cine-Med Research; and holding equity, stock, or stock options with Control Rad, Applied Therapeutics, and Elixir Medical.
A version of this article first appeared on Medscape.com.
FROM TCT 2022

Limiting antibiotic overprescription in pandemics: New guidelines
A statement by the Society for Healthcare Epidemiology of America, published online in Infection Control & Hospital Epidemiology, offers health care providers guidelines on how to prevent inappropriate antibiotic use in future pandemics and to avoid some of the negative scenarios that have been seen with COVID-19.
According to the U.S. Centers of Disease Control and Prevention,
The culprit might be the widespread antibiotic overprescription during the current pandemic. A 2022 meta-analysis revealed that in high-income countries, 58% of patients with COVID-19 were given antibiotics, whereas in lower- and middle-income countries, 89% of patients were put on such drugs. Some hospitals in Europe and the United States reported similarly elevated numbers, sometimes approaching 100%.
“We’ve lost control,” Natasha Pettit, PharmD, pharmacy director at University of Chicago Medicine, told this news organization. Dr. Pettit was not involved in the SHEA study. “Even if CDC didn’t come out with that data, I can tell you right now more of my time is spent trying to figure out how to manage these multi-drug–resistant infections, and we are running out of options for these patients,”
“Dealing with uncertainty, exhaustion, [and] critical illness in often young, otherwise healthy patients meant doctors wanted to do something for their patients,” said Tamar Barlam, MD, an infectious diseases expert at the Boston Medical Center who led the development of the SHEA white paper, in an interview.
That something often was a prescription for antibiotics, even without a clear indication that they were actually needed. A British study revealed that in times of pandemic uncertainty, clinicians often reached for antibiotics “just in case” and referred to conservative prescribing as “bravery.”
Studies have shown, however, that bacterial co-infections in COVID-19 are rare. A 2020 meta-analysis of 24 studies concluded that only 3.5% of patients had a bacterial co-infection on presentation, and 14.3% had a secondary infection. Similar patterns had previously been observed in other viral outbreaks. Research on MERS-CoV, for example, documented only 1% of patients with a bacterial co-infection on admission. During the 2009 H1N1 influenza pandemic, that number was 12% of non–ICU hospitalized patients.
Yet, according to Dr. Pettit, even when such data became available, it didn’t necessarily change prescribing patterns. “Information was coming at us so quickly, I think the providers didn’t have a moment to see the data, to understand what it meant for their prescribing. Having external guidance earlier on would have been hugely helpful,” she told this news organization.
That’s where the newly published SHEA statement comes in: It outlines recommendations on when to prescribe antibiotics during a respiratory viral pandemic, what tests to order, and when to de-escalate or discontinue the treatment. These recommendations include, for instance, advice to not trust inflammatory markers as reliable indicators of bacterial or fungal infection and to not use procalcitonin routinely to aid in the decision to initiate antibiotics.
According to Dr. Barlam, one of the crucial lessons here is that if clinicians see patients with symptoms that are consistent with the current pandemic, they should trust their own impressions and avoid reaching for antimicrobials “just in case.”
Another important lesson is that antibiotic stewardship programs have a huge role to play during pandemics. They should not only monitor prescribing but also compile new information on bacterial co-infections as it gets released and make sure it reaches the clinicians in a clear form.
Evidence suggests that such programs and guidelines do work to limit unnecessary antibiotic use. In one medical center in Chicago, for example, before recommendations on when to initiate and discontinue antimicrobials were released, over 74% of COVID-19 patients received antibiotics. After guidelines were put in place, the use of such drugs fell to 42%.
Dr. Pettit believes, however, that it’s important not to leave each medical center to its own devices. “Hindsight is always twenty-twenty,” she said, “but I think it would be great that, if we start hearing about a pathogen that might lead to another pandemic, we should have a mechanism in place to call together an expert body to get guidance for how antimicrobial stewardship programs should get involved.”
One of the authors of the SHEA statement, Susan Seo, reports an investigator-initiated Merck grant on cost-effectiveness of letermovir in hematopoietic stem cell transplant patients. Another author, Graeme Forrest, reports a clinical study grant from Regeneron for inpatient monoclonals against SARS-CoV-2. All other authors report no conflicts of interest. The study was independently supported.
A version of this article first appeared on Medscape.com.
A statement by the Society for Healthcare Epidemiology of America, published online in Infection Control & Hospital Epidemiology, offers health care providers guidelines on how to prevent inappropriate antibiotic use in future pandemics and to avoid some of the negative scenarios that have been seen with COVID-19.
According to the U.S. Centers of Disease Control and Prevention,
The culprit might be the widespread antibiotic overprescription during the current pandemic. A 2022 meta-analysis revealed that in high-income countries, 58% of patients with COVID-19 were given antibiotics, whereas in lower- and middle-income countries, 89% of patients were put on such drugs. Some hospitals in Europe and the United States reported similarly elevated numbers, sometimes approaching 100%.
“We’ve lost control,” Natasha Pettit, PharmD, pharmacy director at University of Chicago Medicine, told this news organization. Dr. Pettit was not involved in the SHEA study. “Even if CDC didn’t come out with that data, I can tell you right now more of my time is spent trying to figure out how to manage these multi-drug–resistant infections, and we are running out of options for these patients,”
“Dealing with uncertainty, exhaustion, [and] critical illness in often young, otherwise healthy patients meant doctors wanted to do something for their patients,” said Tamar Barlam, MD, an infectious diseases expert at the Boston Medical Center who led the development of the SHEA white paper, in an interview.
That something often was a prescription for antibiotics, even without a clear indication that they were actually needed. A British study revealed that in times of pandemic uncertainty, clinicians often reached for antibiotics “just in case” and referred to conservative prescribing as “bravery.”
Studies have shown, however, that bacterial co-infections in COVID-19 are rare. A 2020 meta-analysis of 24 studies concluded that only 3.5% of patients had a bacterial co-infection on presentation, and 14.3% had a secondary infection. Similar patterns had previously been observed in other viral outbreaks. Research on MERS-CoV, for example, documented only 1% of patients with a bacterial co-infection on admission. During the 2009 H1N1 influenza pandemic, that number was 12% of non–ICU hospitalized patients.
Yet, according to Dr. Pettit, even when such data became available, it didn’t necessarily change prescribing patterns. “Information was coming at us so quickly, I think the providers didn’t have a moment to see the data, to understand what it meant for their prescribing. Having external guidance earlier on would have been hugely helpful,” she told this news organization.
That’s where the newly published SHEA statement comes in: It outlines recommendations on when to prescribe antibiotics during a respiratory viral pandemic, what tests to order, and when to de-escalate or discontinue the treatment. These recommendations include, for instance, advice to not trust inflammatory markers as reliable indicators of bacterial or fungal infection and to not use procalcitonin routinely to aid in the decision to initiate antibiotics.
According to Dr. Barlam, one of the crucial lessons here is that if clinicians see patients with symptoms that are consistent with the current pandemic, they should trust their own impressions and avoid reaching for antimicrobials “just in case.”
Another important lesson is that antibiotic stewardship programs have a huge role to play during pandemics. They should not only monitor prescribing but also compile new information on bacterial co-infections as it gets released and make sure it reaches the clinicians in a clear form.
Evidence suggests that such programs and guidelines do work to limit unnecessary antibiotic use. In one medical center in Chicago, for example, before recommendations on when to initiate and discontinue antimicrobials were released, over 74% of COVID-19 patients received antibiotics. After guidelines were put in place, the use of such drugs fell to 42%.
Dr. Pettit believes, however, that it’s important not to leave each medical center to its own devices. “Hindsight is always twenty-twenty,” she said, “but I think it would be great that, if we start hearing about a pathogen that might lead to another pandemic, we should have a mechanism in place to call together an expert body to get guidance for how antimicrobial stewardship programs should get involved.”
One of the authors of the SHEA statement, Susan Seo, reports an investigator-initiated Merck grant on cost-effectiveness of letermovir in hematopoietic stem cell transplant patients. Another author, Graeme Forrest, reports a clinical study grant from Regeneron for inpatient monoclonals against SARS-CoV-2. All other authors report no conflicts of interest. The study was independently supported.
A version of this article first appeared on Medscape.com.
A statement by the Society for Healthcare Epidemiology of America, published online in Infection Control & Hospital Epidemiology, offers health care providers guidelines on how to prevent inappropriate antibiotic use in future pandemics and to avoid some of the negative scenarios that have been seen with COVID-19.
According to the U.S. Centers of Disease Control and Prevention,
The culprit might be the widespread antibiotic overprescription during the current pandemic. A 2022 meta-analysis revealed that in high-income countries, 58% of patients with COVID-19 were given antibiotics, whereas in lower- and middle-income countries, 89% of patients were put on such drugs. Some hospitals in Europe and the United States reported similarly elevated numbers, sometimes approaching 100%.
“We’ve lost control,” Natasha Pettit, PharmD, pharmacy director at University of Chicago Medicine, told this news organization. Dr. Pettit was not involved in the SHEA study. “Even if CDC didn’t come out with that data, I can tell you right now more of my time is spent trying to figure out how to manage these multi-drug–resistant infections, and we are running out of options for these patients,”
“Dealing with uncertainty, exhaustion, [and] critical illness in often young, otherwise healthy patients meant doctors wanted to do something for their patients,” said Tamar Barlam, MD, an infectious diseases expert at the Boston Medical Center who led the development of the SHEA white paper, in an interview.
That something often was a prescription for antibiotics, even without a clear indication that they were actually needed. A British study revealed that in times of pandemic uncertainty, clinicians often reached for antibiotics “just in case” and referred to conservative prescribing as “bravery.”
Studies have shown, however, that bacterial co-infections in COVID-19 are rare. A 2020 meta-analysis of 24 studies concluded that only 3.5% of patients had a bacterial co-infection on presentation, and 14.3% had a secondary infection. Similar patterns had previously been observed in other viral outbreaks. Research on MERS-CoV, for example, documented only 1% of patients with a bacterial co-infection on admission. During the 2009 H1N1 influenza pandemic, that number was 12% of non–ICU hospitalized patients.
Yet, according to Dr. Pettit, even when such data became available, it didn’t necessarily change prescribing patterns. “Information was coming at us so quickly, I think the providers didn’t have a moment to see the data, to understand what it meant for their prescribing. Having external guidance earlier on would have been hugely helpful,” she told this news organization.
That’s where the newly published SHEA statement comes in: It outlines recommendations on when to prescribe antibiotics during a respiratory viral pandemic, what tests to order, and when to de-escalate or discontinue the treatment. These recommendations include, for instance, advice to not trust inflammatory markers as reliable indicators of bacterial or fungal infection and to not use procalcitonin routinely to aid in the decision to initiate antibiotics.
According to Dr. Barlam, one of the crucial lessons here is that if clinicians see patients with symptoms that are consistent with the current pandemic, they should trust their own impressions and avoid reaching for antimicrobials “just in case.”
Another important lesson is that antibiotic stewardship programs have a huge role to play during pandemics. They should not only monitor prescribing but also compile new information on bacterial co-infections as it gets released and make sure it reaches the clinicians in a clear form.
Evidence suggests that such programs and guidelines do work to limit unnecessary antibiotic use. In one medical center in Chicago, for example, before recommendations on when to initiate and discontinue antimicrobials were released, over 74% of COVID-19 patients received antibiotics. After guidelines were put in place, the use of such drugs fell to 42%.
Dr. Pettit believes, however, that it’s important not to leave each medical center to its own devices. “Hindsight is always twenty-twenty,” she said, “but I think it would be great that, if we start hearing about a pathogen that might lead to another pandemic, we should have a mechanism in place to call together an expert body to get guidance for how antimicrobial stewardship programs should get involved.”
One of the authors of the SHEA statement, Susan Seo, reports an investigator-initiated Merck grant on cost-effectiveness of letermovir in hematopoietic stem cell transplant patients. Another author, Graeme Forrest, reports a clinical study grant from Regeneron for inpatient monoclonals against SARS-CoV-2. All other authors report no conflicts of interest. The study was independently supported.
A version of this article first appeared on Medscape.com.
FROM INFECTION CONTROL & HOSPITAL EPIDEMIOLOGY
FDA okays terlipressin (Terlivaz) injection for hepatorenal syndrome
The Food and Drug Administration has approved terlipressin (Terlivaz), the first and only drug approved for patients with hepatorenal syndrome (HRS).
HRS is characterized by progressive deterioration in kidney function in people with advanced liver disease.
Terlipressin is an injectable synthetic vasopressin analogue indicated for patients with HRS who are experiencing rapid deterioration of kidney function (type 1 HRS). The condition affects an estimated 35,000 Americans annually.
The safety and efficacy of terlipressin for type 1 HRS was assessed in the phase 3 CONFIRM trial, which involved 300 patients in the United States and Canada.
Patients received an injection of terlipressin (0.85 mg) or placebo every 6 hours for a maximum of 14 days. The dose was adjusted on the basis of changes in kidney function.
Twenty-nine percent of patients in the terlipressin group experienced improvement in kidney function, vs. 16% in the placebo group.
The CONFIRM trial met its primary endpoint of verified HRS reversal, defined as renal function improvement, avoidance of dialysis, and short-term survival (P = .012).
To achieve this endpoint, patients had to have two consecutive serum creatinine (SCr) values of ≤ 1.5 mg/dL at least 2 hours apart by day 14 or be discharged from the hospital.
The most commonly observed adverse reactions that occurred in at least 4% of patients treated with terlipressin were abdominal pain (19.5%), nausea (16%), respiratory failure (15.5%), diarrhea (13%), and dyspnea (12.5%).
Results of the CONFIRM trial were published in The New England Journal of Medicine.
“Diagnosing and treating HRS can be challenging, and every minute counts when managing patients who have it,” said Steven Romano, MD, executive vice president and chief scientific officer at Mallinckrodt, which makes the drug.
“Terlivaz gives physicians the first FDA-approved option for treating HRS patients with rapid reduction in kidney function that may help them improve kidney function and lessen the associated need for renal replacement therapy, such as dialysis,” Dr. Romano said.
The company plans to launch the product in the coming weeks.
The application for terlipressin for HRS was granted priority review and fast-track status, as well as orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved terlipressin (Terlivaz), the first and only drug approved for patients with hepatorenal syndrome (HRS).
HRS is characterized by progressive deterioration in kidney function in people with advanced liver disease.
Terlipressin is an injectable synthetic vasopressin analogue indicated for patients with HRS who are experiencing rapid deterioration of kidney function (type 1 HRS). The condition affects an estimated 35,000 Americans annually.
The safety and efficacy of terlipressin for type 1 HRS was assessed in the phase 3 CONFIRM trial, which involved 300 patients in the United States and Canada.
Patients received an injection of terlipressin (0.85 mg) or placebo every 6 hours for a maximum of 14 days. The dose was adjusted on the basis of changes in kidney function.
Twenty-nine percent of patients in the terlipressin group experienced improvement in kidney function, vs. 16% in the placebo group.
The CONFIRM trial met its primary endpoint of verified HRS reversal, defined as renal function improvement, avoidance of dialysis, and short-term survival (P = .012).
To achieve this endpoint, patients had to have two consecutive serum creatinine (SCr) values of ≤ 1.5 mg/dL at least 2 hours apart by day 14 or be discharged from the hospital.
The most commonly observed adverse reactions that occurred in at least 4% of patients treated with terlipressin were abdominal pain (19.5%), nausea (16%), respiratory failure (15.5%), diarrhea (13%), and dyspnea (12.5%).
Results of the CONFIRM trial were published in The New England Journal of Medicine.
“Diagnosing and treating HRS can be challenging, and every minute counts when managing patients who have it,” said Steven Romano, MD, executive vice president and chief scientific officer at Mallinckrodt, which makes the drug.
“Terlivaz gives physicians the first FDA-approved option for treating HRS patients with rapid reduction in kidney function that may help them improve kidney function and lessen the associated need for renal replacement therapy, such as dialysis,” Dr. Romano said.
The company plans to launch the product in the coming weeks.
The application for terlipressin for HRS was granted priority review and fast-track status, as well as orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved terlipressin (Terlivaz), the first and only drug approved for patients with hepatorenal syndrome (HRS).
HRS is characterized by progressive deterioration in kidney function in people with advanced liver disease.
Terlipressin is an injectable synthetic vasopressin analogue indicated for patients with HRS who are experiencing rapid deterioration of kidney function (type 1 HRS). The condition affects an estimated 35,000 Americans annually.
The safety and efficacy of terlipressin for type 1 HRS was assessed in the phase 3 CONFIRM trial, which involved 300 patients in the United States and Canada.
Patients received an injection of terlipressin (0.85 mg) or placebo every 6 hours for a maximum of 14 days. The dose was adjusted on the basis of changes in kidney function.
Twenty-nine percent of patients in the terlipressin group experienced improvement in kidney function, vs. 16% in the placebo group.
The CONFIRM trial met its primary endpoint of verified HRS reversal, defined as renal function improvement, avoidance of dialysis, and short-term survival (P = .012).
To achieve this endpoint, patients had to have two consecutive serum creatinine (SCr) values of ≤ 1.5 mg/dL at least 2 hours apart by day 14 or be discharged from the hospital.
The most commonly observed adverse reactions that occurred in at least 4% of patients treated with terlipressin were abdominal pain (19.5%), nausea (16%), respiratory failure (15.5%), diarrhea (13%), and dyspnea (12.5%).
Results of the CONFIRM trial were published in The New England Journal of Medicine.
“Diagnosing and treating HRS can be challenging, and every minute counts when managing patients who have it,” said Steven Romano, MD, executive vice president and chief scientific officer at Mallinckrodt, which makes the drug.
“Terlivaz gives physicians the first FDA-approved option for treating HRS patients with rapid reduction in kidney function that may help them improve kidney function and lessen the associated need for renal replacement therapy, such as dialysis,” Dr. Romano said.
The company plans to launch the product in the coming weeks.
The application for terlipressin for HRS was granted priority review and fast-track status, as well as orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
A version of this article first appeared on Medscape.com.
Ketamine linked to reduced suicidal thoughts, depression, anxiety
, new research suggests.

Results from a retrospective chart review analysis, which included more than 400 participants with TRD, illustrate that ketamine is a safe and rapid treatment in a real-world patient population, lead author Patrick A. Oliver, MD, founder and medical director, MindPeace Clinics, Richmond, Va., told this news organization.
The effect was perhaps most notable for reducing suicidal ideation, he said.
“In 2 weeks, we can take somebody from being suicidal to nonsuicidal. It’s a total game changer,” Dr. Oliver added.
Every year in the United States, about 12 million individuals think about suicide, 3.2 million make a plan to kill themselves, and more than 46,000 succeed, the investigators note.
The findings were published online in the Journal of Clinical Psychiatry.
Molecule mixture
Primarily used as an anesthetic in hospitals, ketamine is also taken illegally as a recreational drug. Users may aim for an intense high or feeling of dissociation, or an out-of-body–type experience.
Ketamine is a mixture of two mirror-image molecules. An intranasal version of one of these molecules (esketamine) is approved by the U.S. Food and Drug Administration for TRD. Both esketamine and ketamine are believed to increase neurotrophic signaling that affects synaptic function.
The study included 424 patients (mean age, 41.7 years) with major depressive disorder or another mood disorder and who received at least one ketamine infusion at a specialty clinic. Most participants had failed prior medication trials.
Patients in the study were typically started on 0.5 mg/kg of ketamine, with the dose titrated to achieve symptoms of partial dissociation. The median dose administered after titration was 0.93 mg/kg over 40 minutes.
The main treatment course of at least six infusions within 21 days was completed by 70% of the patients.
At each clinic visit, all participants completed the Patient Health Questionnaire-9 (PHQ-9) and the Generalized Anxiety Disorder-7 (GAD-7).
The primary outcome was PHQ-9 total scores, for which researchers looked at seven time periods: 1 week, 2-3 weeks, 4-6 weeks, 7-12 weeks, 13-24 weeks, 25-51 weeks, and 52+ weeks.
‘Blows it out of the water’
Results showed PHQ-9 total scores declined by 50% throughout the course of treatment, with much of the improvement gained within 4-6 weeks. There was a significant difference between week 1 and all later time periods (all P values < .001) and between weeks 2 and 3 and all later periods (all P values < .001).
Other measures included treatment response, defined as at least a 50% improvement on the PHQ-9, and depression remission, defined as a PHQ-9 score of less than 5. After three infusions, 14% of the patients responded and 7% were in remission. After 10 infusions, 72% responded and 38% were in remission.
These results compare favorably to other depression treatments, said Dr. Oliver. “Truthfully, with the exception of ECT [electroconvulsive therapy], this blows it all out of the water,” he added.
Dr. Oliver noted that the success rate for repetitive transcranial magnetic stimulation is 40%-60% depending on the modality; and for selective serotonin reuptake inhibitors, the success rate “is somewhere between the mid-20s and low-30s percent range.”
Another outcome measure was the self-harm/suicidal ideation item of the PHQ-9 questionnaire, which asks about “thoughts that you would be better off dead, or of hurting yourself in some way.” About 22% of the study participants no longer reported suicidal ideation after 3 infusions, 50% by 6 infusions, and 75% by 10 infusions.
By 15 infusions, 85% no longer reported these thoughts. “Nothing else has shown that, ever,” said Dr. Oliver.
Symptoms of generalized anxiety were also substantially improved. There was about a 30% reduction in the GAD-7 score during treatment and, again, most of the response occurred by 4-6 weeks.
Study limitations
Sex, age, and other demographic characteristics did not predict response or remission, but suicide planning trended toward higher response rates (P = .083). This suggests that a more depressed subgroup can achieve greater benefit from the treatment than can less symptomatic patients, the investigators note.
A history of psychosis also trended toward better response to treatment (P = .086) but not remission.
The researchers note that study limitations include that it was retrospective, lacked a control group, and did not require patients to be hospitalized – so the study sample may have been less severely ill than in other studies.
In addition, most patients paid out of pocket for the treatment at $495 per infusion, and they self-reported their symptoms.
As well, the researchers did not assess adverse events, although nurses made follow-up calls to patients. Dr. Oliver noted the most common side effects of ketamine are nausea, vomiting, and anxiety.
Previous research has suggested that ketamine therapy is not linked to long-term side effects, such as sexual dysfunction, weight gain, lethargy, or cognitive issues, said Dr. Oliver.
The investigators point out another study limitation was lack of detailed demographic information, such as race, income, and education, which might affect its generalizability.
Concerns and questions
Pouya Movahed Rad, MD, PhD, senior consultant and researcher in psychiatry, Lund (Sweden) University, noted several concerns, including that the clinics treating the study participants with ketamine profited from it.
He also speculated about who can afford the treatment because only a few patients in the study were reimbursed through insurance.
Dr. Movahed Rad was not involved with the current research but was principal investigator for a recent study that compared intravenous ketamine to ECT.
He questioned whether the patient population in the new study really was “real world.” Well-designed randomized controlled trials have been carried out in a “naturalistic setting, [which] get closer to real-life patients,” he said.
He also noted that the median dose after clinician titration (0.93 mg/kg over 40 minutes) “may be considered very high.”
With regard to doses being titrated to achieve symptoms of partial dissociation, “there is no obvious evidence to my knowledge that patients need to develop dissociative symptoms in order to have antidepressant effect,” said Dr. Movahed Rad.
Finally, he noted that the finding that 28% of the participants were using illegal drugs “is worrying” and wondered what drugs they were taking; he also questioned why 81% of the study population needed to take antidepressants.
The study did not receive outside funding. Dr. Oliver is the founder of MindPeace Clinics, which specialize in ketamine therapeutics. Dr. Movahed Rad has reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, new research suggests.
Results from a retrospective chart review analysis, which included more than 400 participants with TRD, illustrate that ketamine is a safe and rapid treatment in a real-world patient population, lead author Patrick A. Oliver, MD, founder and medical director, MindPeace Clinics, Richmond, Va., told this news organization.
The effect was perhaps most notable for reducing suicidal ideation, he said.
“In 2 weeks, we can take somebody from being suicidal to nonsuicidal. It’s a total game changer,” Dr. Oliver added.
Every year in the United States, about 12 million individuals think about suicide, 3.2 million make a plan to kill themselves, and more than 46,000 succeed, the investigators note.
The findings were published online in the Journal of Clinical Psychiatry.
Molecule mixture
Primarily used as an anesthetic in hospitals, ketamine is also taken illegally as a recreational drug. Users may aim for an intense high or feeling of dissociation, or an out-of-body–type experience.
Ketamine is a mixture of two mirror-image molecules. An intranasal version of one of these molecules (esketamine) is approved by the U.S. Food and Drug Administration for TRD. Both esketamine and ketamine are believed to increase neurotrophic signaling that affects synaptic function.
The study included 424 patients (mean age, 41.7 years) with major depressive disorder or another mood disorder and who received at least one ketamine infusion at a specialty clinic. Most participants had failed prior medication trials.
Patients in the study were typically started on 0.5 mg/kg of ketamine, with the dose titrated to achieve symptoms of partial dissociation. The median dose administered after titration was 0.93 mg/kg over 40 minutes.
The main treatment course of at least six infusions within 21 days was completed by 70% of the patients.
At each clinic visit, all participants completed the Patient Health Questionnaire-9 (PHQ-9) and the Generalized Anxiety Disorder-7 (GAD-7).
The primary outcome was PHQ-9 total scores, for which researchers looked at seven time periods: 1 week, 2-3 weeks, 4-6 weeks, 7-12 weeks, 13-24 weeks, 25-51 weeks, and 52+ weeks.
‘Blows it out of the water’
Results showed PHQ-9 total scores declined by 50% throughout the course of treatment, with much of the improvement gained within 4-6 weeks. There was a significant difference between week 1 and all later time periods (all P values < .001) and between weeks 2 and 3 and all later periods (all P values < .001).
Other measures included treatment response, defined as at least a 50% improvement on the PHQ-9, and depression remission, defined as a PHQ-9 score of less than 5. After three infusions, 14% of the patients responded and 7% were in remission. After 10 infusions, 72% responded and 38% were in remission.
These results compare favorably to other depression treatments, said Dr. Oliver. “Truthfully, with the exception of ECT [electroconvulsive therapy], this blows it all out of the water,” he added.
Dr. Oliver noted that the success rate for repetitive transcranial magnetic stimulation is 40%-60% depending on the modality; and for selective serotonin reuptake inhibitors, the success rate “is somewhere between the mid-20s and low-30s percent range.”
Another outcome measure was the self-harm/suicidal ideation item of the PHQ-9 questionnaire, which asks about “thoughts that you would be better off dead, or of hurting yourself in some way.” About 22% of the study participants no longer reported suicidal ideation after 3 infusions, 50% by 6 infusions, and 75% by 10 infusions.
By 15 infusions, 85% no longer reported these thoughts. “Nothing else has shown that, ever,” said Dr. Oliver.
Symptoms of generalized anxiety were also substantially improved. There was about a 30% reduction in the GAD-7 score during treatment and, again, most of the response occurred by 4-6 weeks.
Study limitations
Sex, age, and other demographic characteristics did not predict response or remission, but suicide planning trended toward higher response rates (P = .083). This suggests that a more depressed subgroup can achieve greater benefit from the treatment than can less symptomatic patients, the investigators note.
A history of psychosis also trended toward better response to treatment (P = .086) but not remission.
The researchers note that study limitations include that it was retrospective, lacked a control group, and did not require patients to be hospitalized – so the study sample may have been less severely ill than in other studies.
In addition, most patients paid out of pocket for the treatment at $495 per infusion, and they self-reported their symptoms.
As well, the researchers did not assess adverse events, although nurses made follow-up calls to patients. Dr. Oliver noted the most common side effects of ketamine are nausea, vomiting, and anxiety.
Previous research has suggested that ketamine therapy is not linked to long-term side effects, such as sexual dysfunction, weight gain, lethargy, or cognitive issues, said Dr. Oliver.
The investigators point out another study limitation was lack of detailed demographic information, such as race, income, and education, which might affect its generalizability.
Concerns and questions
Pouya Movahed Rad, MD, PhD, senior consultant and researcher in psychiatry, Lund (Sweden) University, noted several concerns, including that the clinics treating the study participants with ketamine profited from it.
He also speculated about who can afford the treatment because only a few patients in the study were reimbursed through insurance.
Dr. Movahed Rad was not involved with the current research but was principal investigator for a recent study that compared intravenous ketamine to ECT.
He questioned whether the patient population in the new study really was “real world.” Well-designed randomized controlled trials have been carried out in a “naturalistic setting, [which] get closer to real-life patients,” he said.
He also noted that the median dose after clinician titration (0.93 mg/kg over 40 minutes) “may be considered very high.”
With regard to doses being titrated to achieve symptoms of partial dissociation, “there is no obvious evidence to my knowledge that patients need to develop dissociative symptoms in order to have antidepressant effect,” said Dr. Movahed Rad.
Finally, he noted that the finding that 28% of the participants were using illegal drugs “is worrying” and wondered what drugs they were taking; he also questioned why 81% of the study population needed to take antidepressants.
The study did not receive outside funding. Dr. Oliver is the founder of MindPeace Clinics, which specialize in ketamine therapeutics. Dr. Movahed Rad has reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, new research suggests.
Results from a retrospective chart review analysis, which included more than 400 participants with TRD, illustrate that ketamine is a safe and rapid treatment in a real-world patient population, lead author Patrick A. Oliver, MD, founder and medical director, MindPeace Clinics, Richmond, Va., told this news organization.
The effect was perhaps most notable for reducing suicidal ideation, he said.
“In 2 weeks, we can take somebody from being suicidal to nonsuicidal. It’s a total game changer,” Dr. Oliver added.
Every year in the United States, about 12 million individuals think about suicide, 3.2 million make a plan to kill themselves, and more than 46,000 succeed, the investigators note.
The findings were published online in the Journal of Clinical Psychiatry.
Molecule mixture
Primarily used as an anesthetic in hospitals, ketamine is also taken illegally as a recreational drug. Users may aim for an intense high or feeling of dissociation, or an out-of-body–type experience.
Ketamine is a mixture of two mirror-image molecules. An intranasal version of one of these molecules (esketamine) is approved by the U.S. Food and Drug Administration for TRD. Both esketamine and ketamine are believed to increase neurotrophic signaling that affects synaptic function.
The study included 424 patients (mean age, 41.7 years) with major depressive disorder or another mood disorder and who received at least one ketamine infusion at a specialty clinic. Most participants had failed prior medication trials.
Patients in the study were typically started on 0.5 mg/kg of ketamine, with the dose titrated to achieve symptoms of partial dissociation. The median dose administered after titration was 0.93 mg/kg over 40 minutes.
The main treatment course of at least six infusions within 21 days was completed by 70% of the patients.
At each clinic visit, all participants completed the Patient Health Questionnaire-9 (PHQ-9) and the Generalized Anxiety Disorder-7 (GAD-7).
The primary outcome was PHQ-9 total scores, for which researchers looked at seven time periods: 1 week, 2-3 weeks, 4-6 weeks, 7-12 weeks, 13-24 weeks, 25-51 weeks, and 52+ weeks.
‘Blows it out of the water’
Results showed PHQ-9 total scores declined by 50% throughout the course of treatment, with much of the improvement gained within 4-6 weeks. There was a significant difference between week 1 and all later time periods (all P values < .001) and between weeks 2 and 3 and all later periods (all P values < .001).
Other measures included treatment response, defined as at least a 50% improvement on the PHQ-9, and depression remission, defined as a PHQ-9 score of less than 5. After three infusions, 14% of the patients responded and 7% were in remission. After 10 infusions, 72% responded and 38% were in remission.
These results compare favorably to other depression treatments, said Dr. Oliver. “Truthfully, with the exception of ECT [electroconvulsive therapy], this blows it all out of the water,” he added.
Dr. Oliver noted that the success rate for repetitive transcranial magnetic stimulation is 40%-60% depending on the modality; and for selective serotonin reuptake inhibitors, the success rate “is somewhere between the mid-20s and low-30s percent range.”
Another outcome measure was the self-harm/suicidal ideation item of the PHQ-9 questionnaire, which asks about “thoughts that you would be better off dead, or of hurting yourself in some way.” About 22% of the study participants no longer reported suicidal ideation after 3 infusions, 50% by 6 infusions, and 75% by 10 infusions.
By 15 infusions, 85% no longer reported these thoughts. “Nothing else has shown that, ever,” said Dr. Oliver.
Symptoms of generalized anxiety were also substantially improved. There was about a 30% reduction in the GAD-7 score during treatment and, again, most of the response occurred by 4-6 weeks.
Study limitations
Sex, age, and other demographic characteristics did not predict response or remission, but suicide planning trended toward higher response rates (P = .083). This suggests that a more depressed subgroup can achieve greater benefit from the treatment than can less symptomatic patients, the investigators note.
A history of psychosis also trended toward better response to treatment (P = .086) but not remission.
The researchers note that study limitations include that it was retrospective, lacked a control group, and did not require patients to be hospitalized – so the study sample may have been less severely ill than in other studies.
In addition, most patients paid out of pocket for the treatment at $495 per infusion, and they self-reported their symptoms.
As well, the researchers did not assess adverse events, although nurses made follow-up calls to patients. Dr. Oliver noted the most common side effects of ketamine are nausea, vomiting, and anxiety.
Previous research has suggested that ketamine therapy is not linked to long-term side effects, such as sexual dysfunction, weight gain, lethargy, or cognitive issues, said Dr. Oliver.
The investigators point out another study limitation was lack of detailed demographic information, such as race, income, and education, which might affect its generalizability.
Concerns and questions
Pouya Movahed Rad, MD, PhD, senior consultant and researcher in psychiatry, Lund (Sweden) University, noted several concerns, including that the clinics treating the study participants with ketamine profited from it.
He also speculated about who can afford the treatment because only a few patients in the study were reimbursed through insurance.
Dr. Movahed Rad was not involved with the current research but was principal investigator for a recent study that compared intravenous ketamine to ECT.
He questioned whether the patient population in the new study really was “real world.” Well-designed randomized controlled trials have been carried out in a “naturalistic setting, [which] get closer to real-life patients,” he said.
He also noted that the median dose after clinician titration (0.93 mg/kg over 40 minutes) “may be considered very high.”
With regard to doses being titrated to achieve symptoms of partial dissociation, “there is no obvious evidence to my knowledge that patients need to develop dissociative symptoms in order to have antidepressant effect,” said Dr. Movahed Rad.
Finally, he noted that the finding that 28% of the participants were using illegal drugs “is worrying” and wondered what drugs they were taking; he also questioned why 81% of the study population needed to take antidepressants.
The study did not receive outside funding. Dr. Oliver is the founder of MindPeace Clinics, which specialize in ketamine therapeutics. Dr. Movahed Rad has reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JOURNAL OF CLINICAL PSYCHIATRY
Is acetaminophen really safer than NSAIDs in heart disease?
New research calls into question the assumption that acetaminophen is safer than NSAIDs for patients with known cardiovascular disease (CVD) or CVD risk factors.
The analysis found a significant correlation between the use of acetaminophen and elevated systolic blood pressure.
While acetaminophen may still be safer than NSAIDs from a bleeding risk standpoint, or in patients with known kidney disease, “the gap may not be as large as once thought,” Rahul Gupta, MD, cardiologist with Lehigh Valley Health Network, Allentown, Pa., said in an interview.
“Cautious use is recommended over the long term, especially in patients with pre-existing hypertension or cardiovascular risk factors,” Dr. Gupta said.
The study was presented at the Hypertension Scientific Sessions, San Diego, sponsored by the American Heart Association.
Acetaminophen is one of the most widely used over-the-counter medications, as it is considered a safer medication for long-term use since it lacks the anti-inflammatory effects of NSAIDs, Dr. Gupta explained.
NSAIDs have been known to raise blood pressure, but the effect of acetaminophen in this regard has not been well studied. Observational studies have shown contradictory results in terms of its effect on blood pressure, he noted.
To investigate further, Dr. Gupta and colleagues did a meta-analysis of three studies that compared the effect of acetaminophen (3-4 g/day) versus placebo on systolic and diastolic ambulatory blood pressure in patients with heart disease or hypertension. Together, the studies included 172 adults (mean age, 60 years; 73% male).
They found that patients receiving acetaminophen had significantly higher systolic blood pressure, compared with those receiving placebo (standard mean difference [SMD] = 0.35; 95% confidence interval, 0.08-0.63; P = .01).
Subgroup analysis of the effect on hypertensive patients showed significant change in systolic blood pressure as well (SMD = 0.38; 95% CI, 0.05-0.71; P = .02).
“Interestingly, there was no significant difference in the effect on diastolic blood pressure,” Dr. Gupta commented.
Reached for comment, Timothy S. Anderson, MD, clinical investigator in the Division of General Medicine at Beth Israel Deaconess Medical Center and assistant professor of medicine at the Harvard Medical School, both in Boston, said this is “an interesting and not particularly well-known issue.”
“However, most of the trials look at very high doses of acetaminophen use (for example, six to eight of the 500 mg pills each day) so we don’t really know whether the more common patterns of using one to two acetaminophen pills every once in a while is problematic,” Dr. Anderson told this news organization.
“We also don’t have data showing a direct harm from these medications with regards to strokes or heart attacks or other downstream consequences of high blood pressure. Ideally we would need a head-to-head trial comparing ibuprofen-type medications to acetaminophen-type medications,” Dr. Anderson said.
The study had no specific funding. Dr. Gupta and Dr. Anderson reported no relevant disclosures.
A version of this article first appeared on Medscape.com.
New research calls into question the assumption that acetaminophen is safer than NSAIDs for patients with known cardiovascular disease (CVD) or CVD risk factors.
The analysis found a significant correlation between the use of acetaminophen and elevated systolic blood pressure.
While acetaminophen may still be safer than NSAIDs from a bleeding risk standpoint, or in patients with known kidney disease, “the gap may not be as large as once thought,” Rahul Gupta, MD, cardiologist with Lehigh Valley Health Network, Allentown, Pa., said in an interview.
“Cautious use is recommended over the long term, especially in patients with pre-existing hypertension or cardiovascular risk factors,” Dr. Gupta said.
The study was presented at the Hypertension Scientific Sessions, San Diego, sponsored by the American Heart Association.
Acetaminophen is one of the most widely used over-the-counter medications, as it is considered a safer medication for long-term use since it lacks the anti-inflammatory effects of NSAIDs, Dr. Gupta explained.
NSAIDs have been known to raise blood pressure, but the effect of acetaminophen in this regard has not been well studied. Observational studies have shown contradictory results in terms of its effect on blood pressure, he noted.
To investigate further, Dr. Gupta and colleagues did a meta-analysis of three studies that compared the effect of acetaminophen (3-4 g/day) versus placebo on systolic and diastolic ambulatory blood pressure in patients with heart disease or hypertension. Together, the studies included 172 adults (mean age, 60 years; 73% male).
They found that patients receiving acetaminophen had significantly higher systolic blood pressure, compared with those receiving placebo (standard mean difference [SMD] = 0.35; 95% confidence interval, 0.08-0.63; P = .01).
Subgroup analysis of the effect on hypertensive patients showed significant change in systolic blood pressure as well (SMD = 0.38; 95% CI, 0.05-0.71; P = .02).
“Interestingly, there was no significant difference in the effect on diastolic blood pressure,” Dr. Gupta commented.
Reached for comment, Timothy S. Anderson, MD, clinical investigator in the Division of General Medicine at Beth Israel Deaconess Medical Center and assistant professor of medicine at the Harvard Medical School, both in Boston, said this is “an interesting and not particularly well-known issue.”
“However, most of the trials look at very high doses of acetaminophen use (for example, six to eight of the 500 mg pills each day) so we don’t really know whether the more common patterns of using one to two acetaminophen pills every once in a while is problematic,” Dr. Anderson told this news organization.
“We also don’t have data showing a direct harm from these medications with regards to strokes or heart attacks or other downstream consequences of high blood pressure. Ideally we would need a head-to-head trial comparing ibuprofen-type medications to acetaminophen-type medications,” Dr. Anderson said.
The study had no specific funding. Dr. Gupta and Dr. Anderson reported no relevant disclosures.
A version of this article first appeared on Medscape.com.
New research calls into question the assumption that acetaminophen is safer than NSAIDs for patients with known cardiovascular disease (CVD) or CVD risk factors.
The analysis found a significant correlation between the use of acetaminophen and elevated systolic blood pressure.
While acetaminophen may still be safer than NSAIDs from a bleeding risk standpoint, or in patients with known kidney disease, “the gap may not be as large as once thought,” Rahul Gupta, MD, cardiologist with Lehigh Valley Health Network, Allentown, Pa., said in an interview.
“Cautious use is recommended over the long term, especially in patients with pre-existing hypertension or cardiovascular risk factors,” Dr. Gupta said.
The study was presented at the Hypertension Scientific Sessions, San Diego, sponsored by the American Heart Association.
Acetaminophen is one of the most widely used over-the-counter medications, as it is considered a safer medication for long-term use since it lacks the anti-inflammatory effects of NSAIDs, Dr. Gupta explained.
NSAIDs have been known to raise blood pressure, but the effect of acetaminophen in this regard has not been well studied. Observational studies have shown contradictory results in terms of its effect on blood pressure, he noted.
To investigate further, Dr. Gupta and colleagues did a meta-analysis of three studies that compared the effect of acetaminophen (3-4 g/day) versus placebo on systolic and diastolic ambulatory blood pressure in patients with heart disease or hypertension. Together, the studies included 172 adults (mean age, 60 years; 73% male).
They found that patients receiving acetaminophen had significantly higher systolic blood pressure, compared with those receiving placebo (standard mean difference [SMD] = 0.35; 95% confidence interval, 0.08-0.63; P = .01).
Subgroup analysis of the effect on hypertensive patients showed significant change in systolic blood pressure as well (SMD = 0.38; 95% CI, 0.05-0.71; P = .02).
“Interestingly, there was no significant difference in the effect on diastolic blood pressure,” Dr. Gupta commented.
Reached for comment, Timothy S. Anderson, MD, clinical investigator in the Division of General Medicine at Beth Israel Deaconess Medical Center and assistant professor of medicine at the Harvard Medical School, both in Boston, said this is “an interesting and not particularly well-known issue.”
“However, most of the trials look at very high doses of acetaminophen use (for example, six to eight of the 500 mg pills each day) so we don’t really know whether the more common patterns of using one to two acetaminophen pills every once in a while is problematic,” Dr. Anderson told this news organization.
“We also don’t have data showing a direct harm from these medications with regards to strokes or heart attacks or other downstream consequences of high blood pressure. Ideally we would need a head-to-head trial comparing ibuprofen-type medications to acetaminophen-type medications,” Dr. Anderson said.
The study had no specific funding. Dr. Gupta and Dr. Anderson reported no relevant disclosures.
A version of this article first appeared on Medscape.com.
FROM HYPERTENSION 2022