User login
Drug crisis continues to evolve beyond opioids
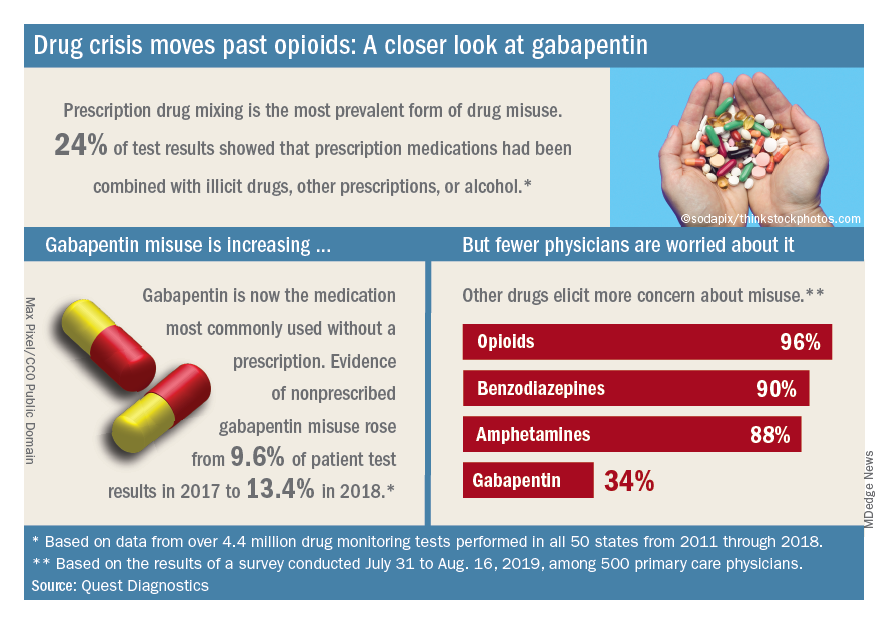
Almost three-quarters of primary care physicians believe that their patients will take their controlled medications as prescribed, but more than half of drug-monitoring lab tests show signs of misuse, according to a new report from Quest Diagnostics.
“ and may miss some of the drug misuse risks affecting their patients,” report coauthor Harvey W. Kaufman, MD, Quest’s senior medical director, said in a written statement.
Analysis of more than 4.4 million drug-monitoring tests showed that 51% involved an inconsistent result, such as detection of a nonprescribed drug or nondetection of a drug that was prescribed. The report also included a survey of 500 primary care physicians, of whom 72% said they trusted their patents to properly use opioids and other controlled substances.
“The intersection of these two data sets reveals, for the first time, the contrast between physician expectations about patient drug use and the evolution of the drug epidemic and actual patient behavior, as revealed by objective lab data, amid a national drug crisis that claimed an estimated 68,500 lives last year,” the report said.
A majority (62%) of the physicians surveyed also said that the opioid crisis will evolve into a new prescription drug crisis, and even more (72%) think that patients with chronic pain will use illicit drugs if they cannot get prescription opioids. Evidence from the drug test dataset suggests that “misuse of nonprescribed fentanyl and nonprescribed gabapentin warrant[s] a closer look,” the report said. In the survey, 78% of respondents reported prescribing gabapentin as an alternative to opioids for patients with chronic pain.
Those two drugs, along with alcohol, are the only three drug groups for which misuse increased from 2017 to 2018, and both are frequently involved in drug mixing, which is the most common form of misuse. Gabapentin went from 9.6% of all nonprescribed misuse in 2017 to 13.4% in 2018, an increase of 40%. Nonprescribed fentanyl was found in 64% of test results that were positive for heroin and 24% that were positive for cocaine, the Quest data showed.
The survey results, however, suggest that gabapentin is not on physicians’ radar, as only 34% said that they were concerned about its misuse, compared with 96% for opioids and 90% for benzodiazepines, according to the report.
“While gabapentin may not have opioids’ addictive potential, it can exaggerate euphoric effects when combined with opioids or anxiety medications. This drug mixing is dangerous,” said report coauthor Jeffrey Gudin, MD, senior medical advisor, prescription drug monitoring, for Quest Diagnostics.
The survey was conducted online among family physicians, general practitioners, and internists from July 31 to Aug. 16, 2019, by the Harris Poll on behalf of Quest and Center for Addiction. The test result data were collected in all 50 states and Washington, D.C., from 2011 to 2018, and results from drug rehabilitation clinics and addiction specialists were excluded from the analysis, so actual misuse rates are probably higher than reported.
Almost three-quarters of primary care physicians believe that their patients will take their controlled medications as prescribed, but more than half of drug-monitoring lab tests show signs of misuse, according to a new report from Quest Diagnostics.
“ and may miss some of the drug misuse risks affecting their patients,” report coauthor Harvey W. Kaufman, MD, Quest’s senior medical director, said in a written statement.
Analysis of more than 4.4 million drug-monitoring tests showed that 51% involved an inconsistent result, such as detection of a nonprescribed drug or nondetection of a drug that was prescribed. The report also included a survey of 500 primary care physicians, of whom 72% said they trusted their patents to properly use opioids and other controlled substances.
“The intersection of these two data sets reveals, for the first time, the contrast between physician expectations about patient drug use and the evolution of the drug epidemic and actual patient behavior, as revealed by objective lab data, amid a national drug crisis that claimed an estimated 68,500 lives last year,” the report said.
A majority (62%) of the physicians surveyed also said that the opioid crisis will evolve into a new prescription drug crisis, and even more (72%) think that patients with chronic pain will use illicit drugs if they cannot get prescription opioids. Evidence from the drug test dataset suggests that “misuse of nonprescribed fentanyl and nonprescribed gabapentin warrant[s] a closer look,” the report said. In the survey, 78% of respondents reported prescribing gabapentin as an alternative to opioids for patients with chronic pain.
Those two drugs, along with alcohol, are the only three drug groups for which misuse increased from 2017 to 2018, and both are frequently involved in drug mixing, which is the most common form of misuse. Gabapentin went from 9.6% of all nonprescribed misuse in 2017 to 13.4% in 2018, an increase of 40%. Nonprescribed fentanyl was found in 64% of test results that were positive for heroin and 24% that were positive for cocaine, the Quest data showed.
The survey results, however, suggest that gabapentin is not on physicians’ radar, as only 34% said that they were concerned about its misuse, compared with 96% for opioids and 90% for benzodiazepines, according to the report.
“While gabapentin may not have opioids’ addictive potential, it can exaggerate euphoric effects when combined with opioids or anxiety medications. This drug mixing is dangerous,” said report coauthor Jeffrey Gudin, MD, senior medical advisor, prescription drug monitoring, for Quest Diagnostics.
The survey was conducted online among family physicians, general practitioners, and internists from July 31 to Aug. 16, 2019, by the Harris Poll on behalf of Quest and Center for Addiction. The test result data were collected in all 50 states and Washington, D.C., from 2011 to 2018, and results from drug rehabilitation clinics and addiction specialists were excluded from the analysis, so actual misuse rates are probably higher than reported.
Almost three-quarters of primary care physicians believe that their patients will take their controlled medications as prescribed, but more than half of drug-monitoring lab tests show signs of misuse, according to a new report from Quest Diagnostics.
“ and may miss some of the drug misuse risks affecting their patients,” report coauthor Harvey W. Kaufman, MD, Quest’s senior medical director, said in a written statement.
Analysis of more than 4.4 million drug-monitoring tests showed that 51% involved an inconsistent result, such as detection of a nonprescribed drug or nondetection of a drug that was prescribed. The report also included a survey of 500 primary care physicians, of whom 72% said they trusted their patents to properly use opioids and other controlled substances.
“The intersection of these two data sets reveals, for the first time, the contrast between physician expectations about patient drug use and the evolution of the drug epidemic and actual patient behavior, as revealed by objective lab data, amid a national drug crisis that claimed an estimated 68,500 lives last year,” the report said.
A majority (62%) of the physicians surveyed also said that the opioid crisis will evolve into a new prescription drug crisis, and even more (72%) think that patients with chronic pain will use illicit drugs if they cannot get prescription opioids. Evidence from the drug test dataset suggests that “misuse of nonprescribed fentanyl and nonprescribed gabapentin warrant[s] a closer look,” the report said. In the survey, 78% of respondents reported prescribing gabapentin as an alternative to opioids for patients with chronic pain.
Those two drugs, along with alcohol, are the only three drug groups for which misuse increased from 2017 to 2018, and both are frequently involved in drug mixing, which is the most common form of misuse. Gabapentin went from 9.6% of all nonprescribed misuse in 2017 to 13.4% in 2018, an increase of 40%. Nonprescribed fentanyl was found in 64% of test results that were positive for heroin and 24% that were positive for cocaine, the Quest data showed.
The survey results, however, suggest that gabapentin is not on physicians’ radar, as only 34% said that they were concerned about its misuse, compared with 96% for opioids and 90% for benzodiazepines, according to the report.
“While gabapentin may not have opioids’ addictive potential, it can exaggerate euphoric effects when combined with opioids or anxiety medications. This drug mixing is dangerous,” said report coauthor Jeffrey Gudin, MD, senior medical advisor, prescription drug monitoring, for Quest Diagnostics.
The survey was conducted online among family physicians, general practitioners, and internists from July 31 to Aug. 16, 2019, by the Harris Poll on behalf of Quest and Center for Addiction. The test result data were collected in all 50 states and Washington, D.C., from 2011 to 2018, and results from drug rehabilitation clinics and addiction specialists were excluded from the analysis, so actual misuse rates are probably higher than reported.
Dapagliflozin approved for reducing HF hospitalization in diabetes
The Food And Drug Administration has approved the sodium-glucose cotransporter 2 (SGLT2) inhibitor dapagliflozin (Farxiga) for reducing the risk of hospitalization for heart failure in adults with type 2 diabetes and established cardiovascular disease or multiple cardiovascular risk factors, according to a statement from AstraZeneca.
The approval was based on results from the DECLARE-TIMI 58 cardiovascular outcomes trial, which evaluated dapagliflozin in more than 17,000 patients with type 2 diabetes and cardiovascular risk factors or cardiovascular disease. They showed that dapagliflozin significantly reduced the risk of the primary composite endpoint of hospitalization for heart failure by 27%, compared with placebo (2.5% vs. 3.3%; HR, 0.73; 95% confidence interval, 0.61-0.88).
The drug is an oral, once-daily SGLT2 inhibitor initially approved as a monotherapy or combination therapy for glycemic control in adults with type 2 diabetes. It has additional benefits of weight loss and reduction in blood pressure in concert with diet and exercise in the same population.
“,” Ruud Dobber, PhD, executive vice president of the company’s biopharmaceuticals business unit, said in the statement. “This is promising news for the 30 million people living with type 2 diabetes in the U.S., as heart failure is one of the earliest cardiovascular complications for them, before heart attack or stroke. [Dapagliflozin] now offers the opportunity for physicians to act sooner and reduce the risk of hospitalization for heart failure.”
In September, the agency granted dapagliflozin a Fast Track designation to reduce the risk of cardiovascular death, or the worsening of heart failure in adults with heart failure with reduced ejection fraction or preserved ejection fraction, based on the phase 3 DAPA-HF and DELIVER trials. It also gave the drug Fast Track designation to delay the progression of renal failure and prevent CV and renal death in patients with chronic kidney disease based on the phase 3 DAPA-CKD trial, the statement noted.
The Food And Drug Administration has approved the sodium-glucose cotransporter 2 (SGLT2) inhibitor dapagliflozin (Farxiga) for reducing the risk of hospitalization for heart failure in adults with type 2 diabetes and established cardiovascular disease or multiple cardiovascular risk factors, according to a statement from AstraZeneca.
The approval was based on results from the DECLARE-TIMI 58 cardiovascular outcomes trial, which evaluated dapagliflozin in more than 17,000 patients with type 2 diabetes and cardiovascular risk factors or cardiovascular disease. They showed that dapagliflozin significantly reduced the risk of the primary composite endpoint of hospitalization for heart failure by 27%, compared with placebo (2.5% vs. 3.3%; HR, 0.73; 95% confidence interval, 0.61-0.88).
The drug is an oral, once-daily SGLT2 inhibitor initially approved as a monotherapy or combination therapy for glycemic control in adults with type 2 diabetes. It has additional benefits of weight loss and reduction in blood pressure in concert with diet and exercise in the same population.
“,” Ruud Dobber, PhD, executive vice president of the company’s biopharmaceuticals business unit, said in the statement. “This is promising news for the 30 million people living with type 2 diabetes in the U.S., as heart failure is one of the earliest cardiovascular complications for them, before heart attack or stroke. [Dapagliflozin] now offers the opportunity for physicians to act sooner and reduce the risk of hospitalization for heart failure.”
In September, the agency granted dapagliflozin a Fast Track designation to reduce the risk of cardiovascular death, or the worsening of heart failure in adults with heart failure with reduced ejection fraction or preserved ejection fraction, based on the phase 3 DAPA-HF and DELIVER trials. It also gave the drug Fast Track designation to delay the progression of renal failure and prevent CV and renal death in patients with chronic kidney disease based on the phase 3 DAPA-CKD trial, the statement noted.
The Food And Drug Administration has approved the sodium-glucose cotransporter 2 (SGLT2) inhibitor dapagliflozin (Farxiga) for reducing the risk of hospitalization for heart failure in adults with type 2 diabetes and established cardiovascular disease or multiple cardiovascular risk factors, according to a statement from AstraZeneca.
The approval was based on results from the DECLARE-TIMI 58 cardiovascular outcomes trial, which evaluated dapagliflozin in more than 17,000 patients with type 2 diabetes and cardiovascular risk factors or cardiovascular disease. They showed that dapagliflozin significantly reduced the risk of the primary composite endpoint of hospitalization for heart failure by 27%, compared with placebo (2.5% vs. 3.3%; HR, 0.73; 95% confidence interval, 0.61-0.88).
The drug is an oral, once-daily SGLT2 inhibitor initially approved as a monotherapy or combination therapy for glycemic control in adults with type 2 diabetes. It has additional benefits of weight loss and reduction in blood pressure in concert with diet and exercise in the same population.
“,” Ruud Dobber, PhD, executive vice president of the company’s biopharmaceuticals business unit, said in the statement. “This is promising news for the 30 million people living with type 2 diabetes in the U.S., as heart failure is one of the earliest cardiovascular complications for them, before heart attack or stroke. [Dapagliflozin] now offers the opportunity for physicians to act sooner and reduce the risk of hospitalization for heart failure.”
In September, the agency granted dapagliflozin a Fast Track designation to reduce the risk of cardiovascular death, or the worsening of heart failure in adults with heart failure with reduced ejection fraction or preserved ejection fraction, based on the phase 3 DAPA-HF and DELIVER trials. It also gave the drug Fast Track designation to delay the progression of renal failure and prevent CV and renal death in patients with chronic kidney disease based on the phase 3 DAPA-CKD trial, the statement noted.
Certain diabetes drugs may thwart dementia
COPENHAGEN – Selected antidiabetes medications appear to blunt the increased risk of dementia associated with type 2 diabetes, according to a Danish national case control registry study.

This benefit applies to the newer antidiabetic agents – specifically, the dipeptidyl peptidase 4 (DPP4) inhibitors, the glucagon-like peptide 1 (GLP1) analogs, and the sodium-glucose transport protein 2 (SGLT2) inhibitors – and metformin as well, Merete Osler, MD, PhD, reported at the annual congress of the European College of Neuropsychopharmacology.
In contrast, neither insulin nor the sulfonylureas showed any signal of a protective effect against development of dementia. In fact, the use of sulfonylureas was associated with a small but statistically significant 7% increased risk, added Dr. Osler, of the University of Copenhagen.
Elsewhere at the meeting, investigators tapped a Swedish national registry to demonstrate that individuals with type 1 diabetes have a sharply reduced risk of developing schizophrenia.
Type 2 diabetes medications and dementia
Dr. Osler and colleagues are among several groups of investigators who have previously shown that patients with type 2 diabetes have an increased risk of dementia.
“This has raised the question of the role of dysregulated glucose metabolism in the development of this neurodegenerative disorder, and the possible effect of antidiabetic medications,” she noted.
To further explore this issue, which links two great ongoing global epidemics, Dr. Osler and coinvestigators conducted a nested case-control study including all 176,250 patients with type 2 diabetes in the comprehensive Danish National Diabetes Register for 1995-2012. The 11,619 patients with type 2 diabetes who received a dementia diagnosis were matched with 46,476 type 2 diabetes patients without dementia. The objective was to determine associations between dementia and ever-use and cumulative dose of antidiabetes drugs, alone and in combination, in logistic regression analyses adjusted for demographics, comorbid conditions, marital status, diabetic complications, and year of dementia diagnosis.
Patients who had ever used metformin had an adjusted 6% reduction in the likelihood of dementia compared with metformin nonusers, a modest but statistically significant difference. Those on a DPP4 inhibitor had a 20% reduction in risk. The GLP1 analogs were associated with a 42% decrease in risk. So were the SGLT2 inhibitors. A dose-response relationship was evident: The higher the cumulative exposure to these agents, the lower the odds of dementia.
Combination therapy is common in type 2 diabetes, so the investigators scrutinized the impact of a variety of multidrug combinations. Combinations including a DPP4 inhibitor or GLP1 analog were also associated with significantly reduced dementia risk.
Records of glycemic control in the form of hemoglobin A1c values were available on only 1,446 type 2 diabetic dementia patients and 4,003 matched controls. An analysis that incorporated this variable showed that the observed anti-dementia effect of selected diabetes drugs was independent of glycemic control, according to Dr. Osler.
The protective effect appeared to extend to both Alzheimer’s disease and vascular dementias, although firm conclusions can’t be drawn on this score because the study was insufficiently powered to address that issue.
Dr. Osler noted that the Danish study confirms a recent Taiwanese study showing an apparent protective effect against dementia for metformin in patients with type 2 diabetes (Aging Dis. 2019 Feb 1;10(1):37-48).
“Ours is the first study on the newer diabetic drugs, so our results need to be confirmed,” she pointed out.
If confirmed, however, it would warrant exploration of these drugs more generally as potential interventions to prevent dementia. That could open a whole new chapter in the remarkable story of the SGLT2 inhibitors, a class of drugs originally developed for treatment of type 2 diabetes but which in major randomized clinical trials later proved to be so effective in the treatment of heart failure that they are now considered cardiology drugs first.
Asked if she thinks these antidiabetes agents have a general neuroprotective effect or, instead, that the observed reduced risk of dementia is a function of patients being treated better early on with modern drugs, the psychiatrist replied, “I think it might be a combination of both, especially because we find different risk estimates between the drugs.”
Dr. Osler reported having no financial conflicts of interest regarding the study, which was funded by the Danish Diabetes Foundation, the Danish Medical Association, and several other foundations.
The full study details were published online shortly before her presentation at ECNP 2019 (Eur J Endocrinol. 2019 Aug 1. pii: EJE-19-0259.R1. doi: 10.1530/EJE-19-0259).
Type 1 diabetes and schizophrenia risk
Kristina Melkersson, MD, PhD, presented a cohort study that utilized Swedish national registries to examine the relationship between type 1 diabetes and schizophrenia. The study comprised 1,745,977 individuals, of whom 10,117 had type 1 diabetes, who were followed for a median of 9.7 and maximum of 18 years from their 13th birthday. During follow-up, 1,280 individuals were diagnosed with schizophrenia and 649 others with schizoaffective disorder. The adjusted risk of schizophrenia was 70% lower in patients with type 1 diabetes. However, there was no difference in the risk of schizoaffective disorder in the type 1 diabetic versus nondiabetic subjects.
The Swedish data confirm the findings of an earlier Finnish national study showing that the risk of schizophrenia is reduced in patients with type 1 diabetes (Arch Gen Psychiatry. 2007 Aug;64(8):894-9). These findings raise the intriguing possibility that autoimmunity somehow figures into the etiology of the psychiatric disorder. Other investigators have previously reported a reduced prevalence of rheumatoid arthritis in patients with schizophrenia, noted Dr. Melkersson of the Karolinska Institute in Stockholm.
She reported having no financial conflicts regarding her study.
SOURCE: Osler M. ECNP Abstract P180. Melkersson K. Abstract 81.
COPENHAGEN – Selected antidiabetes medications appear to blunt the increased risk of dementia associated with type 2 diabetes, according to a Danish national case control registry study.
This benefit applies to the newer antidiabetic agents – specifically, the dipeptidyl peptidase 4 (DPP4) inhibitors, the glucagon-like peptide 1 (GLP1) analogs, and the sodium-glucose transport protein 2 (SGLT2) inhibitors – and metformin as well, Merete Osler, MD, PhD, reported at the annual congress of the European College of Neuropsychopharmacology.
In contrast, neither insulin nor the sulfonylureas showed any signal of a protective effect against development of dementia. In fact, the use of sulfonylureas was associated with a small but statistically significant 7% increased risk, added Dr. Osler, of the University of Copenhagen.
Elsewhere at the meeting, investigators tapped a Swedish national registry to demonstrate that individuals with type 1 diabetes have a sharply reduced risk of developing schizophrenia.
Type 2 diabetes medications and dementia
Dr. Osler and colleagues are among several groups of investigators who have previously shown that patients with type 2 diabetes have an increased risk of dementia.
“This has raised the question of the role of dysregulated glucose metabolism in the development of this neurodegenerative disorder, and the possible effect of antidiabetic medications,” she noted.
To further explore this issue, which links two great ongoing global epidemics, Dr. Osler and coinvestigators conducted a nested case-control study including all 176,250 patients with type 2 diabetes in the comprehensive Danish National Diabetes Register for 1995-2012. The 11,619 patients with type 2 diabetes who received a dementia diagnosis were matched with 46,476 type 2 diabetes patients without dementia. The objective was to determine associations between dementia and ever-use and cumulative dose of antidiabetes drugs, alone and in combination, in logistic regression analyses adjusted for demographics, comorbid conditions, marital status, diabetic complications, and year of dementia diagnosis.
Patients who had ever used metformin had an adjusted 6% reduction in the likelihood of dementia compared with metformin nonusers, a modest but statistically significant difference. Those on a DPP4 inhibitor had a 20% reduction in risk. The GLP1 analogs were associated with a 42% decrease in risk. So were the SGLT2 inhibitors. A dose-response relationship was evident: The higher the cumulative exposure to these agents, the lower the odds of dementia.
Combination therapy is common in type 2 diabetes, so the investigators scrutinized the impact of a variety of multidrug combinations. Combinations including a DPP4 inhibitor or GLP1 analog were also associated with significantly reduced dementia risk.
Records of glycemic control in the form of hemoglobin A1c values were available on only 1,446 type 2 diabetic dementia patients and 4,003 matched controls. An analysis that incorporated this variable showed that the observed anti-dementia effect of selected diabetes drugs was independent of glycemic control, according to Dr. Osler.
The protective effect appeared to extend to both Alzheimer’s disease and vascular dementias, although firm conclusions can’t be drawn on this score because the study was insufficiently powered to address that issue.
Dr. Osler noted that the Danish study confirms a recent Taiwanese study showing an apparent protective effect against dementia for metformin in patients with type 2 diabetes (Aging Dis. 2019 Feb 1;10(1):37-48).
“Ours is the first study on the newer diabetic drugs, so our results need to be confirmed,” she pointed out.
If confirmed, however, it would warrant exploration of these drugs more generally as potential interventions to prevent dementia. That could open a whole new chapter in the remarkable story of the SGLT2 inhibitors, a class of drugs originally developed for treatment of type 2 diabetes but which in major randomized clinical trials later proved to be so effective in the treatment of heart failure that they are now considered cardiology drugs first.
Asked if she thinks these antidiabetes agents have a general neuroprotective effect or, instead, that the observed reduced risk of dementia is a function of patients being treated better early on with modern drugs, the psychiatrist replied, “I think it might be a combination of both, especially because we find different risk estimates between the drugs.”
Dr. Osler reported having no financial conflicts of interest regarding the study, which was funded by the Danish Diabetes Foundation, the Danish Medical Association, and several other foundations.
The full study details were published online shortly before her presentation at ECNP 2019 (Eur J Endocrinol. 2019 Aug 1. pii: EJE-19-0259.R1. doi: 10.1530/EJE-19-0259).
Type 1 diabetes and schizophrenia risk
Kristina Melkersson, MD, PhD, presented a cohort study that utilized Swedish national registries to examine the relationship between type 1 diabetes and schizophrenia. The study comprised 1,745,977 individuals, of whom 10,117 had type 1 diabetes, who were followed for a median of 9.7 and maximum of 18 years from their 13th birthday. During follow-up, 1,280 individuals were diagnosed with schizophrenia and 649 others with schizoaffective disorder. The adjusted risk of schizophrenia was 70% lower in patients with type 1 diabetes. However, there was no difference in the risk of schizoaffective disorder in the type 1 diabetic versus nondiabetic subjects.
The Swedish data confirm the findings of an earlier Finnish national study showing that the risk of schizophrenia is reduced in patients with type 1 diabetes (Arch Gen Psychiatry. 2007 Aug;64(8):894-9). These findings raise the intriguing possibility that autoimmunity somehow figures into the etiology of the psychiatric disorder. Other investigators have previously reported a reduced prevalence of rheumatoid arthritis in patients with schizophrenia, noted Dr. Melkersson of the Karolinska Institute in Stockholm.
She reported having no financial conflicts regarding her study.
SOURCE: Osler M. ECNP Abstract P180. Melkersson K. Abstract 81.
COPENHAGEN – Selected antidiabetes medications appear to blunt the increased risk of dementia associated with type 2 diabetes, according to a Danish national case control registry study.
This benefit applies to the newer antidiabetic agents – specifically, the dipeptidyl peptidase 4 (DPP4) inhibitors, the glucagon-like peptide 1 (GLP1) analogs, and the sodium-glucose transport protein 2 (SGLT2) inhibitors – and metformin as well, Merete Osler, MD, PhD, reported at the annual congress of the European College of Neuropsychopharmacology.
In contrast, neither insulin nor the sulfonylureas showed any signal of a protective effect against development of dementia. In fact, the use of sulfonylureas was associated with a small but statistically significant 7% increased risk, added Dr. Osler, of the University of Copenhagen.
Elsewhere at the meeting, investigators tapped a Swedish national registry to demonstrate that individuals with type 1 diabetes have a sharply reduced risk of developing schizophrenia.
Type 2 diabetes medications and dementia
Dr. Osler and colleagues are among several groups of investigators who have previously shown that patients with type 2 diabetes have an increased risk of dementia.
“This has raised the question of the role of dysregulated glucose metabolism in the development of this neurodegenerative disorder, and the possible effect of antidiabetic medications,” she noted.
To further explore this issue, which links two great ongoing global epidemics, Dr. Osler and coinvestigators conducted a nested case-control study including all 176,250 patients with type 2 diabetes in the comprehensive Danish National Diabetes Register for 1995-2012. The 11,619 patients with type 2 diabetes who received a dementia diagnosis were matched with 46,476 type 2 diabetes patients without dementia. The objective was to determine associations between dementia and ever-use and cumulative dose of antidiabetes drugs, alone and in combination, in logistic regression analyses adjusted for demographics, comorbid conditions, marital status, diabetic complications, and year of dementia diagnosis.
Patients who had ever used metformin had an adjusted 6% reduction in the likelihood of dementia compared with metformin nonusers, a modest but statistically significant difference. Those on a DPP4 inhibitor had a 20% reduction in risk. The GLP1 analogs were associated with a 42% decrease in risk. So were the SGLT2 inhibitors. A dose-response relationship was evident: The higher the cumulative exposure to these agents, the lower the odds of dementia.
Combination therapy is common in type 2 diabetes, so the investigators scrutinized the impact of a variety of multidrug combinations. Combinations including a DPP4 inhibitor or GLP1 analog were also associated with significantly reduced dementia risk.
Records of glycemic control in the form of hemoglobin A1c values were available on only 1,446 type 2 diabetic dementia patients and 4,003 matched controls. An analysis that incorporated this variable showed that the observed anti-dementia effect of selected diabetes drugs was independent of glycemic control, according to Dr. Osler.
The protective effect appeared to extend to both Alzheimer’s disease and vascular dementias, although firm conclusions can’t be drawn on this score because the study was insufficiently powered to address that issue.
Dr. Osler noted that the Danish study confirms a recent Taiwanese study showing an apparent protective effect against dementia for metformin in patients with type 2 diabetes (Aging Dis. 2019 Feb 1;10(1):37-48).
“Ours is the first study on the newer diabetic drugs, so our results need to be confirmed,” she pointed out.
If confirmed, however, it would warrant exploration of these drugs more generally as potential interventions to prevent dementia. That could open a whole new chapter in the remarkable story of the SGLT2 inhibitors, a class of drugs originally developed for treatment of type 2 diabetes but which in major randomized clinical trials later proved to be so effective in the treatment of heart failure that they are now considered cardiology drugs first.
Asked if she thinks these antidiabetes agents have a general neuroprotective effect or, instead, that the observed reduced risk of dementia is a function of patients being treated better early on with modern drugs, the psychiatrist replied, “I think it might be a combination of both, especially because we find different risk estimates between the drugs.”
Dr. Osler reported having no financial conflicts of interest regarding the study, which was funded by the Danish Diabetes Foundation, the Danish Medical Association, and several other foundations.
The full study details were published online shortly before her presentation at ECNP 2019 (Eur J Endocrinol. 2019 Aug 1. pii: EJE-19-0259.R1. doi: 10.1530/EJE-19-0259).
Type 1 diabetes and schizophrenia risk
Kristina Melkersson, MD, PhD, presented a cohort study that utilized Swedish national registries to examine the relationship between type 1 diabetes and schizophrenia. The study comprised 1,745,977 individuals, of whom 10,117 had type 1 diabetes, who were followed for a median of 9.7 and maximum of 18 years from their 13th birthday. During follow-up, 1,280 individuals were diagnosed with schizophrenia and 649 others with schizoaffective disorder. The adjusted risk of schizophrenia was 70% lower in patients with type 1 diabetes. However, there was no difference in the risk of schizoaffective disorder in the type 1 diabetic versus nondiabetic subjects.
The Swedish data confirm the findings of an earlier Finnish national study showing that the risk of schizophrenia is reduced in patients with type 1 diabetes (Arch Gen Psychiatry. 2007 Aug;64(8):894-9). These findings raise the intriguing possibility that autoimmunity somehow figures into the etiology of the psychiatric disorder. Other investigators have previously reported a reduced prevalence of rheumatoid arthritis in patients with schizophrenia, noted Dr. Melkersson of the Karolinska Institute in Stockholm.
She reported having no financial conflicts regarding her study.
SOURCE: Osler M. ECNP Abstract P180. Melkersson K. Abstract 81.
REPORTING FROM ECNP 2019
Primary periodic paralysis attacks reduced with long-term dichlorphenamide
AUSTIN, TEX. – Dichlorphenamide continues to reduce attacks from primary periodic paralysis (PPP) through 1 year with mild or moderate paresthesia and cognition-related adverse events, according to new research.
“These adverse events rarely resulted in discontinuation from the study and were sometimes managed by dichlorphenamide dose reductions,” concluded Nicholas E. Johnson, MD, of Virginia Commonwealth University, Richmond, and colleagues. “Reduction in dose was frequently associated with resolution of these events, suggesting a potential intervention to hasten resolution.” Dr. Johnson presented the findings in an abstract at the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine.
Dichlorphenamide (Keveyis) was approved by the Food and Drug Administration in 2015 for treating primary hyperkalemic and hypokalemic periodic paralysis and similar variants. The original hyperkalemic/hypokalemic PPP trial was a phase 3 randomized, double-blind, placebo-controlled trial that lasted 9 weeks and assessed the efficacy of dichlorphenamide in reducing PPP attacks and its adverse events. In the dichlorphenamide group, 47% experienced paresthesia, compared with 14% in the placebo group, and 19% experienced cognitive disorder, compared with 7% in the placebo.
In a 52-week open-label extension, participants who had been receiving the placebo switched to receiving 50 mg of dichlorphenamide twice daily. The intervention group continued with the dose they had been receiving when the 9-week double-blind phase ended. (During the initial intervention, they took either 50 mg twice daily or the dose they had at baseline for those taking it before the study began.)
The researchers then tracked rates of attacks and their severity over the next year – through week 61 after baseline – to compare these endpoints both within the intervention groups and between them.
Among the 63 predominantly white (84.1%) male (61.9%) adults who began the trial, 36 received dichlorphenamide and 27 received placebo. Just over two-thirds (68.3%) had hypokalemic PPP. Among the 47 patients (74.6%) who completed the open-label extension phase, 26 had been in the original dichlorphenamide group and 21 had been in the placebo group.
The median weekly attack rate in the dichlorphenamide group dropped from 1.75 at baseline to 0.06 at week 61 (median decrease 1.00, 93.8%; P less than .0001). In the placebo group that switched over to dichlorphenamide at week 9, the median weekly attack rate dropped from 3.00 at baseline to 0.25 at week 61 (median decrease 0.63, 75%; P = .01).
The median attack rate weighted for severity in the dichlorphenamide group dropped from 2.25 at baseline to 0.06 at week 61 (median decrease 2.25, 97.1%; P less than .0001). In the placebo group, it dropped from 5.88 to 0.50 (median decrease 1.69, 80.8%; P = .01).
No significant difference in median weekly attack rates and severity-weighted attack rates was found between the intervention groups through week 61.
Across all patients during the extension, 39.7% patients experienced at least one paresthesia adverse event, none of which were determined to be severe and resulting in one discontinuation.
A quarter of the participants (25.4%) experienced at least one cognition-related adverse event, and four patients (6.3%) discontinued because of these side effects. Most (14.3%) were mild with 7.9% reporting moderate and 3.2% reporting severe effects.
Dr. Johnson has received research support from or consulted with a variety of pharmaceutical companies including Strongbridge Biopharma, the manufacturer of the drug. Other authors consulted for several pharmaceutical companies, and one author is an employee of Strongbridge Biopharma.
SOURCE: Johnson NE et al. AANEM 2019. Abstract 102. Long-term efficacy and adverse event characterization of dichlorphenamide for the treatment of primary periodic paralysis.
AUSTIN, TEX. – Dichlorphenamide continues to reduce attacks from primary periodic paralysis (PPP) through 1 year with mild or moderate paresthesia and cognition-related adverse events, according to new research.
“These adverse events rarely resulted in discontinuation from the study and were sometimes managed by dichlorphenamide dose reductions,” concluded Nicholas E. Johnson, MD, of Virginia Commonwealth University, Richmond, and colleagues. “Reduction in dose was frequently associated with resolution of these events, suggesting a potential intervention to hasten resolution.” Dr. Johnson presented the findings in an abstract at the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine.
Dichlorphenamide (Keveyis) was approved by the Food and Drug Administration in 2015 for treating primary hyperkalemic and hypokalemic periodic paralysis and similar variants. The original hyperkalemic/hypokalemic PPP trial was a phase 3 randomized, double-blind, placebo-controlled trial that lasted 9 weeks and assessed the efficacy of dichlorphenamide in reducing PPP attacks and its adverse events. In the dichlorphenamide group, 47% experienced paresthesia, compared with 14% in the placebo group, and 19% experienced cognitive disorder, compared with 7% in the placebo.
In a 52-week open-label extension, participants who had been receiving the placebo switched to receiving 50 mg of dichlorphenamide twice daily. The intervention group continued with the dose they had been receiving when the 9-week double-blind phase ended. (During the initial intervention, they took either 50 mg twice daily or the dose they had at baseline for those taking it before the study began.)
The researchers then tracked rates of attacks and their severity over the next year – through week 61 after baseline – to compare these endpoints both within the intervention groups and between them.
Among the 63 predominantly white (84.1%) male (61.9%) adults who began the trial, 36 received dichlorphenamide and 27 received placebo. Just over two-thirds (68.3%) had hypokalemic PPP. Among the 47 patients (74.6%) who completed the open-label extension phase, 26 had been in the original dichlorphenamide group and 21 had been in the placebo group.
The median weekly attack rate in the dichlorphenamide group dropped from 1.75 at baseline to 0.06 at week 61 (median decrease 1.00, 93.8%; P less than .0001). In the placebo group that switched over to dichlorphenamide at week 9, the median weekly attack rate dropped from 3.00 at baseline to 0.25 at week 61 (median decrease 0.63, 75%; P = .01).
The median attack rate weighted for severity in the dichlorphenamide group dropped from 2.25 at baseline to 0.06 at week 61 (median decrease 2.25, 97.1%; P less than .0001). In the placebo group, it dropped from 5.88 to 0.50 (median decrease 1.69, 80.8%; P = .01).
No significant difference in median weekly attack rates and severity-weighted attack rates was found between the intervention groups through week 61.
Across all patients during the extension, 39.7% patients experienced at least one paresthesia adverse event, none of which were determined to be severe and resulting in one discontinuation.
A quarter of the participants (25.4%) experienced at least one cognition-related adverse event, and four patients (6.3%) discontinued because of these side effects. Most (14.3%) were mild with 7.9% reporting moderate and 3.2% reporting severe effects.
Dr. Johnson has received research support from or consulted with a variety of pharmaceutical companies including Strongbridge Biopharma, the manufacturer of the drug. Other authors consulted for several pharmaceutical companies, and one author is an employee of Strongbridge Biopharma.
SOURCE: Johnson NE et al. AANEM 2019. Abstract 102. Long-term efficacy and adverse event characterization of dichlorphenamide for the treatment of primary periodic paralysis.
AUSTIN, TEX. – Dichlorphenamide continues to reduce attacks from primary periodic paralysis (PPP) through 1 year with mild or moderate paresthesia and cognition-related adverse events, according to new research.
“These adverse events rarely resulted in discontinuation from the study and were sometimes managed by dichlorphenamide dose reductions,” concluded Nicholas E. Johnson, MD, of Virginia Commonwealth University, Richmond, and colleagues. “Reduction in dose was frequently associated with resolution of these events, suggesting a potential intervention to hasten resolution.” Dr. Johnson presented the findings in an abstract at the annual meeting of the American Association for Neuromuscular and Electrodiagnostic Medicine.
Dichlorphenamide (Keveyis) was approved by the Food and Drug Administration in 2015 for treating primary hyperkalemic and hypokalemic periodic paralysis and similar variants. The original hyperkalemic/hypokalemic PPP trial was a phase 3 randomized, double-blind, placebo-controlled trial that lasted 9 weeks and assessed the efficacy of dichlorphenamide in reducing PPP attacks and its adverse events. In the dichlorphenamide group, 47% experienced paresthesia, compared with 14% in the placebo group, and 19% experienced cognitive disorder, compared with 7% in the placebo.
In a 52-week open-label extension, participants who had been receiving the placebo switched to receiving 50 mg of dichlorphenamide twice daily. The intervention group continued with the dose they had been receiving when the 9-week double-blind phase ended. (During the initial intervention, they took either 50 mg twice daily or the dose they had at baseline for those taking it before the study began.)
The researchers then tracked rates of attacks and their severity over the next year – through week 61 after baseline – to compare these endpoints both within the intervention groups and between them.
Among the 63 predominantly white (84.1%) male (61.9%) adults who began the trial, 36 received dichlorphenamide and 27 received placebo. Just over two-thirds (68.3%) had hypokalemic PPP. Among the 47 patients (74.6%) who completed the open-label extension phase, 26 had been in the original dichlorphenamide group and 21 had been in the placebo group.
The median weekly attack rate in the dichlorphenamide group dropped from 1.75 at baseline to 0.06 at week 61 (median decrease 1.00, 93.8%; P less than .0001). In the placebo group that switched over to dichlorphenamide at week 9, the median weekly attack rate dropped from 3.00 at baseline to 0.25 at week 61 (median decrease 0.63, 75%; P = .01).
The median attack rate weighted for severity in the dichlorphenamide group dropped from 2.25 at baseline to 0.06 at week 61 (median decrease 2.25, 97.1%; P less than .0001). In the placebo group, it dropped from 5.88 to 0.50 (median decrease 1.69, 80.8%; P = .01).
No significant difference in median weekly attack rates and severity-weighted attack rates was found between the intervention groups through week 61.
Across all patients during the extension, 39.7% patients experienced at least one paresthesia adverse event, none of which were determined to be severe and resulting in one discontinuation.
A quarter of the participants (25.4%) experienced at least one cognition-related adverse event, and four patients (6.3%) discontinued because of these side effects. Most (14.3%) were mild with 7.9% reporting moderate and 3.2% reporting severe effects.
Dr. Johnson has received research support from or consulted with a variety of pharmaceutical companies including Strongbridge Biopharma, the manufacturer of the drug. Other authors consulted for several pharmaceutical companies, and one author is an employee of Strongbridge Biopharma.
SOURCE: Johnson NE et al. AANEM 2019. Abstract 102. Long-term efficacy and adverse event characterization of dichlorphenamide for the treatment of primary periodic paralysis.
REPORTING FROM AANEM
FDA gives nod to earlier use of Nplate in adults with ITP
Romiplostim (Nplate) earned a new indication from the Food and Drug Administration, allowing for earlier usage in adult patients with immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.

The new indication is for newly diagnosed and persistent adult patients.
Romiplostim is already approved for the treatment of pediatric patients aged 1 year and older who have had ITP for at least 6 months and had an insufficient response to other treatments, as well as for adults patients with chronic ITP who had insufficient response to other therapies.
The latest approval is based on positive results from a single-arm, phase 2 trial in adults with ITP who had an insufficient response to first-line treatment. The study met its primary endpoint – platelet response (50 x 109/L or greater). The median number of months with platelet response was 11 months during a 12-month treatment period. The median time to first platelet response was just over 2 weeks.
Adverse events of at least 5% incidence in patients taking romiplostim with an ITP duration up to 12 months included bronchitis, sinusitis, vomiting, arthralgia, myalgia, headache, dizziness, diarrhea, upper respiratory tract infection, cough, nausea, and oropharyngeal pain. There was a 2% incidence of thrombocytosis among adults with an ITP duration up to 12 months.
Romiplostim (Nplate) earned a new indication from the Food and Drug Administration, allowing for earlier usage in adult patients with immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
The new indication is for newly diagnosed and persistent adult patients.
Romiplostim is already approved for the treatment of pediatric patients aged 1 year and older who have had ITP for at least 6 months and had an insufficient response to other treatments, as well as for adults patients with chronic ITP who had insufficient response to other therapies.
The latest approval is based on positive results from a single-arm, phase 2 trial in adults with ITP who had an insufficient response to first-line treatment. The study met its primary endpoint – platelet response (50 x 109/L or greater). The median number of months with platelet response was 11 months during a 12-month treatment period. The median time to first platelet response was just over 2 weeks.
Adverse events of at least 5% incidence in patients taking romiplostim with an ITP duration up to 12 months included bronchitis, sinusitis, vomiting, arthralgia, myalgia, headache, dizziness, diarrhea, upper respiratory tract infection, cough, nausea, and oropharyngeal pain. There was a 2% incidence of thrombocytosis among adults with an ITP duration up to 12 months.
Romiplostim (Nplate) earned a new indication from the Food and Drug Administration, allowing for earlier usage in adult patients with immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
The new indication is for newly diagnosed and persistent adult patients.
Romiplostim is already approved for the treatment of pediatric patients aged 1 year and older who have had ITP for at least 6 months and had an insufficient response to other treatments, as well as for adults patients with chronic ITP who had insufficient response to other therapies.
The latest approval is based on positive results from a single-arm, phase 2 trial in adults with ITP who had an insufficient response to first-line treatment. The study met its primary endpoint – platelet response (50 x 109/L or greater). The median number of months with platelet response was 11 months during a 12-month treatment period. The median time to first platelet response was just over 2 weeks.
Adverse events of at least 5% incidence in patients taking romiplostim with an ITP duration up to 12 months included bronchitis, sinusitis, vomiting, arthralgia, myalgia, headache, dizziness, diarrhea, upper respiratory tract infection, cough, nausea, and oropharyngeal pain. There was a 2% incidence of thrombocytosis among adults with an ITP duration up to 12 months.
Surgery better than medical therapy in some GERD patients
Surgery may be more effective than medical therapy, according to results from a randomized trial in 78 patients with reflux-related heartburn refractory to proton pump inhibitors (PPIs).
Stuart J. Spechler, MD, from Baylor University Medical Center, Dallas, and coauthors wrote in the New England Journal of Medicine that, for these patients, there were no medical treatment options that had been shown to have long-term benefit, so PPIs were often continued despite not offering adequate symptom relief. Other medical options such as baclofen and neuromodulators often have unacceptable side effects, and studies of their efficacy were few and of short duration.
In this study, patients were randomized either to laparoscopic Nissen fundoplication, treatment with omeprazole plus baclofen with desipramine depending on symptoms, or a control treatment of omeprazole plus placebo.
At 1 year, researchers saw a significantly higher rate of treatment success – defined as 50% or greater improvement in gastroesophageal reflux disease health-related quality of life score – in the surgery group (67%), compared with the medical-treatment group (28%) and control-medical group (12%).
This translated to an unadjusted 138% greater chance of treatment success with surgery, compared with active medical treatment, and a greater than 400% increase for surgery, compared with the control medical treatment.
Researchers also did a prespecified subgroup analysis among people with reflex hypersensitivity or abnormal acid reflux, and found the incidence of success with surgery was 71% and 62%, respectively.
They described this finding as “noteworthy,” given that reflux hypersensitivity was considered a functional disorder that would not be expected to improve with a procedure that didn’t alter abnormal esophageal pain perception.
However, they acknowledged that, as the study did not include a sham-surgery group, they couldn’t determine how much the placebo effect might have contributed to the treatment success of surgery.
They also stressed that the randomized group was a highly selected group of patients, and that the systematic work-up including esophageal multichannel intraluminal impedance pH monitoring could identify a subgroup that might have a better response to surgery than to medical treatment.
Four patients in the surgery group experienced a total of five serious adverse events, including one patients who had a herniated fundoplication treated with repeat surgery; four patients in the active-medical group experienced four serious adverse events; and three patients in the control-medical group experienced five serious adverse events.
The authors noted that 366 patients with PPI-refractory heartburn were originally enrolled in the study, then treated with 20 mg of omeprazole twice daily for 2 weeks with strict instructions to take 20 minutes before breakfast and dinner. Of these patients, 42 had their symptoms relieved by the omeprazole treatment and so were excluded from the randomization.
The “strict instructions” on how to take omeprazole were important, because PPIs only bind to gastric proton pumps that are actively secreting acid, the authors wrote. They also commented that the relative potencies of individual PPIs can vary, so patients not on omeprazole before the study may have responded better to this than other PPIs.
Before randomizations, patients also underwent endoscopy, esophageal biopsy, esophageal manometry, and multichannel intraluminal impedance pH monitoring. This excluded another 23 patients who were found to have non–gastroesophageal reflux disease, including eosinophilic esophagitis, other endoscopic or histologic abnormalities, and manometric abnormalities.
“This trial highlights the critical importance of systematic evaluation, similar to that recommended by Gyawali and Fass for managing the care of patients with PPI-refractory heartburn,” they wrote. “Many patients would not complete this rigorous evaluation, and among those who did, the cause of heartburn in most of them was not [gastroesophageal reflux disease].”
The study was funded by the Department of Veterans Affairs Cooperative Studies Program. Four authors declared consultancies with and/or grants from the pharmaceutical sector.
SOURCE: Spechler SJ et al. N Engl J Med. 2019 Oct 16. doi: 10.1056/NEJMoa1811424.
Around 40% of troublesome heartburn fails to respond to proton pump inhibitor therapy, which may reflect a diverse range of underlying causes of the condition. Therefore we cannot treat it as a single disease process that will respond to higher and higher doses of acid suppression.
The results of a study of surgical intervention in a carefully selected group of patients are striking in showing surgery’s superiority to medical treatment, but it is important to note that 79% of patients enrolled in the study did not meet the criteria for surgery. Therefore these findings cannot be generalized to all patients with refractory heartburn, and each case should only be considered for surgery after extended trials of medical therapy.
Nicholas J. Talley, MD, PhD, is from the faculty of health and medicine at the University of Newcastle (Australia) and Hunter Medical Research Institute, also in Newcastle. These comments are adapted from an accompanying editorial (N Engl J Med. 2019 Oct 17. doi: 10.1056/NEJMe1911623). Dr. Talley declared a range of consultancies, grants, personal fees, and patents unrelated to the study.
Around 40% of troublesome heartburn fails to respond to proton pump inhibitor therapy, which may reflect a diverse range of underlying causes of the condition. Therefore we cannot treat it as a single disease process that will respond to higher and higher doses of acid suppression.
The results of a study of surgical intervention in a carefully selected group of patients are striking in showing surgery’s superiority to medical treatment, but it is important to note that 79% of patients enrolled in the study did not meet the criteria for surgery. Therefore these findings cannot be generalized to all patients with refractory heartburn, and each case should only be considered for surgery after extended trials of medical therapy.
Nicholas J. Talley, MD, PhD, is from the faculty of health and medicine at the University of Newcastle (Australia) and Hunter Medical Research Institute, also in Newcastle. These comments are adapted from an accompanying editorial (N Engl J Med. 2019 Oct 17. doi: 10.1056/NEJMe1911623). Dr. Talley declared a range of consultancies, grants, personal fees, and patents unrelated to the study.
Around 40% of troublesome heartburn fails to respond to proton pump inhibitor therapy, which may reflect a diverse range of underlying causes of the condition. Therefore we cannot treat it as a single disease process that will respond to higher and higher doses of acid suppression.
The results of a study of surgical intervention in a carefully selected group of patients are striking in showing surgery’s superiority to medical treatment, but it is important to note that 79% of patients enrolled in the study did not meet the criteria for surgery. Therefore these findings cannot be generalized to all patients with refractory heartburn, and each case should only be considered for surgery after extended trials of medical therapy.
Nicholas J. Talley, MD, PhD, is from the faculty of health and medicine at the University of Newcastle (Australia) and Hunter Medical Research Institute, also in Newcastle. These comments are adapted from an accompanying editorial (N Engl J Med. 2019 Oct 17. doi: 10.1056/NEJMe1911623). Dr. Talley declared a range of consultancies, grants, personal fees, and patents unrelated to the study.
Surgery may be more effective than medical therapy, according to results from a randomized trial in 78 patients with reflux-related heartburn refractory to proton pump inhibitors (PPIs).
Stuart J. Spechler, MD, from Baylor University Medical Center, Dallas, and coauthors wrote in the New England Journal of Medicine that, for these patients, there were no medical treatment options that had been shown to have long-term benefit, so PPIs were often continued despite not offering adequate symptom relief. Other medical options such as baclofen and neuromodulators often have unacceptable side effects, and studies of their efficacy were few and of short duration.
In this study, patients were randomized either to laparoscopic Nissen fundoplication, treatment with omeprazole plus baclofen with desipramine depending on symptoms, or a control treatment of omeprazole plus placebo.
At 1 year, researchers saw a significantly higher rate of treatment success – defined as 50% or greater improvement in gastroesophageal reflux disease health-related quality of life score – in the surgery group (67%), compared with the medical-treatment group (28%) and control-medical group (12%).
This translated to an unadjusted 138% greater chance of treatment success with surgery, compared with active medical treatment, and a greater than 400% increase for surgery, compared with the control medical treatment.
Researchers also did a prespecified subgroup analysis among people with reflex hypersensitivity or abnormal acid reflux, and found the incidence of success with surgery was 71% and 62%, respectively.
They described this finding as “noteworthy,” given that reflux hypersensitivity was considered a functional disorder that would not be expected to improve with a procedure that didn’t alter abnormal esophageal pain perception.
However, they acknowledged that, as the study did not include a sham-surgery group, they couldn’t determine how much the placebo effect might have contributed to the treatment success of surgery.
They also stressed that the randomized group was a highly selected group of patients, and that the systematic work-up including esophageal multichannel intraluminal impedance pH monitoring could identify a subgroup that might have a better response to surgery than to medical treatment.
Four patients in the surgery group experienced a total of five serious adverse events, including one patients who had a herniated fundoplication treated with repeat surgery; four patients in the active-medical group experienced four serious adverse events; and three patients in the control-medical group experienced five serious adverse events.
The authors noted that 366 patients with PPI-refractory heartburn were originally enrolled in the study, then treated with 20 mg of omeprazole twice daily for 2 weeks with strict instructions to take 20 minutes before breakfast and dinner. Of these patients, 42 had their symptoms relieved by the omeprazole treatment and so were excluded from the randomization.
The “strict instructions” on how to take omeprazole were important, because PPIs only bind to gastric proton pumps that are actively secreting acid, the authors wrote. They also commented that the relative potencies of individual PPIs can vary, so patients not on omeprazole before the study may have responded better to this than other PPIs.
Before randomizations, patients also underwent endoscopy, esophageal biopsy, esophageal manometry, and multichannel intraluminal impedance pH monitoring. This excluded another 23 patients who were found to have non–gastroesophageal reflux disease, including eosinophilic esophagitis, other endoscopic or histologic abnormalities, and manometric abnormalities.
“This trial highlights the critical importance of systematic evaluation, similar to that recommended by Gyawali and Fass for managing the care of patients with PPI-refractory heartburn,” they wrote. “Many patients would not complete this rigorous evaluation, and among those who did, the cause of heartburn in most of them was not [gastroesophageal reflux disease].”
The study was funded by the Department of Veterans Affairs Cooperative Studies Program. Four authors declared consultancies with and/or grants from the pharmaceutical sector.
SOURCE: Spechler SJ et al. N Engl J Med. 2019 Oct 16. doi: 10.1056/NEJMoa1811424.
Surgery may be more effective than medical therapy, according to results from a randomized trial in 78 patients with reflux-related heartburn refractory to proton pump inhibitors (PPIs).
Stuart J. Spechler, MD, from Baylor University Medical Center, Dallas, and coauthors wrote in the New England Journal of Medicine that, for these patients, there were no medical treatment options that had been shown to have long-term benefit, so PPIs were often continued despite not offering adequate symptom relief. Other medical options such as baclofen and neuromodulators often have unacceptable side effects, and studies of their efficacy were few and of short duration.
In this study, patients were randomized either to laparoscopic Nissen fundoplication, treatment with omeprazole plus baclofen with desipramine depending on symptoms, or a control treatment of omeprazole plus placebo.
At 1 year, researchers saw a significantly higher rate of treatment success – defined as 50% or greater improvement in gastroesophageal reflux disease health-related quality of life score – in the surgery group (67%), compared with the medical-treatment group (28%) and control-medical group (12%).
This translated to an unadjusted 138% greater chance of treatment success with surgery, compared with active medical treatment, and a greater than 400% increase for surgery, compared with the control medical treatment.
Researchers also did a prespecified subgroup analysis among people with reflex hypersensitivity or abnormal acid reflux, and found the incidence of success with surgery was 71% and 62%, respectively.
They described this finding as “noteworthy,” given that reflux hypersensitivity was considered a functional disorder that would not be expected to improve with a procedure that didn’t alter abnormal esophageal pain perception.
However, they acknowledged that, as the study did not include a sham-surgery group, they couldn’t determine how much the placebo effect might have contributed to the treatment success of surgery.
They also stressed that the randomized group was a highly selected group of patients, and that the systematic work-up including esophageal multichannel intraluminal impedance pH monitoring could identify a subgroup that might have a better response to surgery than to medical treatment.
Four patients in the surgery group experienced a total of five serious adverse events, including one patients who had a herniated fundoplication treated with repeat surgery; four patients in the active-medical group experienced four serious adverse events; and three patients in the control-medical group experienced five serious adverse events.
The authors noted that 366 patients with PPI-refractory heartburn were originally enrolled in the study, then treated with 20 mg of omeprazole twice daily for 2 weeks with strict instructions to take 20 minutes before breakfast and dinner. Of these patients, 42 had their symptoms relieved by the omeprazole treatment and so were excluded from the randomization.
The “strict instructions” on how to take omeprazole were important, because PPIs only bind to gastric proton pumps that are actively secreting acid, the authors wrote. They also commented that the relative potencies of individual PPIs can vary, so patients not on omeprazole before the study may have responded better to this than other PPIs.
Before randomizations, patients also underwent endoscopy, esophageal biopsy, esophageal manometry, and multichannel intraluminal impedance pH monitoring. This excluded another 23 patients who were found to have non–gastroesophageal reflux disease, including eosinophilic esophagitis, other endoscopic or histologic abnormalities, and manometric abnormalities.
“This trial highlights the critical importance of systematic evaluation, similar to that recommended by Gyawali and Fass for managing the care of patients with PPI-refractory heartburn,” they wrote. “Many patients would not complete this rigorous evaluation, and among those who did, the cause of heartburn in most of them was not [gastroesophageal reflux disease].”
The study was funded by the Department of Veterans Affairs Cooperative Studies Program. Four authors declared consultancies with and/or grants from the pharmaceutical sector.
SOURCE: Spechler SJ et al. N Engl J Med. 2019 Oct 16. doi: 10.1056/NEJMoa1811424.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Surgery may be more effective than medical therapy in some patients with treatment-refractory gastroesophageal reflux disease.
Major finding: Patients with treatment-refractory gastroesophageal reflux disease treated with surgery were 2.38 times more likely to respond to surgery than medical treatment.
Study details: A randomized, controlled trial in 78 patients with gastroesophageal reflux disease.
Disclosures: The study was funded by the Department of Veterans Affairs Cooperative Studies Program. Four authors declared consultancies with and/or grants from the pharmaceutical sector.
Source: Spechler SJ et al. N Engl J Med. 2019 Oct 16. doi: 10.1056/NEJMoa1811424.
California bans “Pay for Delay,” promotes black maternal health, PrEP access

AB 824, the Pay for Delay bill, bans pharmaceutical companies from keeping cheaper generic drugs off the market. The bill prohibits agreements between brand name and generic drug manufacturers to delay the release of generic drugs, defining them as presumptively anticompetitive. A Federal Trade Commission study found that “these anticompetitive deals cost consumers and taxpayers $3.5 billion in higher drug costs every year,” according to a statement from the governor’s office.
The second bill, SB 464, is intended to improve black maternal health care. The bill is designed to reduce preventable maternal mortality among black women by requiring all perinatal health care providers to undergo implicit bias training to curb the effects of bias on maternal health and by improving data collection at the California Department of Public Health to better understand pregnancy-related deaths. “We know that black women have been dying at alarming rates during and after giving birth. The disproportionate effect of the maternal mortality rate on this community is a public health crisis and a major health equity issue. We must do everything in our power to take implicit bias out of the medical system – it is literally a matter of life and death,” said Gov. Newsom.
The third bill, SB 159, aims to facilitate the use of pre-exposure prophylaxis and postexposure prophylaxis against HIV infection. The bill allows pharmacists in the state to dispense PrEP and PEP without a physician’s prescription and prohibits insurance companies from requiring prior authorization for patients to obtain PrEP coverage. “All Californians deserve access to PrEP and PEP, two treatments that have transformed our fight against HIV and AIDS,” Gov. Newsom said in a statement.
AB 824, the Pay for Delay bill, bans pharmaceutical companies from keeping cheaper generic drugs off the market. The bill prohibits agreements between brand name and generic drug manufacturers to delay the release of generic drugs, defining them as presumptively anticompetitive. A Federal Trade Commission study found that “these anticompetitive deals cost consumers and taxpayers $3.5 billion in higher drug costs every year,” according to a statement from the governor’s office.
The second bill, SB 464, is intended to improve black maternal health care. The bill is designed to reduce preventable maternal mortality among black women by requiring all perinatal health care providers to undergo implicit bias training to curb the effects of bias on maternal health and by improving data collection at the California Department of Public Health to better understand pregnancy-related deaths. “We know that black women have been dying at alarming rates during and after giving birth. The disproportionate effect of the maternal mortality rate on this community is a public health crisis and a major health equity issue. We must do everything in our power to take implicit bias out of the medical system – it is literally a matter of life and death,” said Gov. Newsom.
The third bill, SB 159, aims to facilitate the use of pre-exposure prophylaxis and postexposure prophylaxis against HIV infection. The bill allows pharmacists in the state to dispense PrEP and PEP without a physician’s prescription and prohibits insurance companies from requiring prior authorization for patients to obtain PrEP coverage. “All Californians deserve access to PrEP and PEP, two treatments that have transformed our fight against HIV and AIDS,” Gov. Newsom said in a statement.
AB 824, the Pay for Delay bill, bans pharmaceutical companies from keeping cheaper generic drugs off the market. The bill prohibits agreements between brand name and generic drug manufacturers to delay the release of generic drugs, defining them as presumptively anticompetitive. A Federal Trade Commission study found that “these anticompetitive deals cost consumers and taxpayers $3.5 billion in higher drug costs every year,” according to a statement from the governor’s office.
The second bill, SB 464, is intended to improve black maternal health care. The bill is designed to reduce preventable maternal mortality among black women by requiring all perinatal health care providers to undergo implicit bias training to curb the effects of bias on maternal health and by improving data collection at the California Department of Public Health to better understand pregnancy-related deaths. “We know that black women have been dying at alarming rates during and after giving birth. The disproportionate effect of the maternal mortality rate on this community is a public health crisis and a major health equity issue. We must do everything in our power to take implicit bias out of the medical system – it is literally a matter of life and death,” said Gov. Newsom.
The third bill, SB 159, aims to facilitate the use of pre-exposure prophylaxis and postexposure prophylaxis against HIV infection. The bill allows pharmacists in the state to dispense PrEP and PEP without a physician’s prescription and prohibits insurance companies from requiring prior authorization for patients to obtain PrEP coverage. “All Californians deserve access to PrEP and PEP, two treatments that have transformed our fight against HIV and AIDS,” Gov. Newsom said in a statement.
FDA approves afamelanotide for treatment of rare condition with light-induced pain
The Food and Drug Administration has approved , a rare condition that causes extremely painful reactions when skin is exposed to light, according to an FDA announcement.
This is the first treatment approved to help patients with this condition increase their exposure to light, according to the release.
Afamelanotide, administered in a subcutaneous implant, is a melanocortin-1 receptor (MC1-R) agonist, which “increases the production of eumelanin in the skin independent of exposure to sunlight or artificial light sources,” the release says.
Approval is based on a pair of parallel-group clinical trials that compared the number of hours spent in sunlight in the treatment and placebo groups. The first trial enrolled 93 patients; 48 received afamelanotide. The treated patients spent a median of 61 hours in total over 180 days in direct sunlight between 10 a.m. and 6 p.m. on days with no pain, compared with 41 hours for patients taking placebo.
The second trial assessed the total number of hours over 270 days spent outdoors between 10 a.m. and 3 p.m. on days with no pain for which “most of the day” was spent in direct sunlight. In this study, 38 patients treated with afamelanotide spent a median total of 6 hours, compared with 0.75 hours among the remaining 36 who were taking a placebo.
The most common side effects include implant site reaction, nausea, and oropharyngeal pain. The implant should be administered only by trained professionals. Because afamelanotide may cause skin darkening, it’s recommended that patients should undergo twice-yearly skin examinations. Patients are also encouraged to maintain sun protection measures to help prevent phototoxic reactions.
“Today’s approval is one example of the FDA’s ongoing commitment to encourage industry innovation of therapies to treat rare diseases, and work with drug developers to make promising new therapies available to patients as safely and efficiently as possible,” said Julie Beitz, MD, director of FDA’s Center for Drug Evaluation and Research Office of Drug Evaluation III in the FDA release.
The Food and Drug Administration has approved , a rare condition that causes extremely painful reactions when skin is exposed to light, according to an FDA announcement.
This is the first treatment approved to help patients with this condition increase their exposure to light, according to the release.
Afamelanotide, administered in a subcutaneous implant, is a melanocortin-1 receptor (MC1-R) agonist, which “increases the production of eumelanin in the skin independent of exposure to sunlight or artificial light sources,” the release says.
Approval is based on a pair of parallel-group clinical trials that compared the number of hours spent in sunlight in the treatment and placebo groups. The first trial enrolled 93 patients; 48 received afamelanotide. The treated patients spent a median of 61 hours in total over 180 days in direct sunlight between 10 a.m. and 6 p.m. on days with no pain, compared with 41 hours for patients taking placebo.
The second trial assessed the total number of hours over 270 days spent outdoors between 10 a.m. and 3 p.m. on days with no pain for which “most of the day” was spent in direct sunlight. In this study, 38 patients treated with afamelanotide spent a median total of 6 hours, compared with 0.75 hours among the remaining 36 who were taking a placebo.
The most common side effects include implant site reaction, nausea, and oropharyngeal pain. The implant should be administered only by trained professionals. Because afamelanotide may cause skin darkening, it’s recommended that patients should undergo twice-yearly skin examinations. Patients are also encouraged to maintain sun protection measures to help prevent phototoxic reactions.
“Today’s approval is one example of the FDA’s ongoing commitment to encourage industry innovation of therapies to treat rare diseases, and work with drug developers to make promising new therapies available to patients as safely and efficiently as possible,” said Julie Beitz, MD, director of FDA’s Center for Drug Evaluation and Research Office of Drug Evaluation III in the FDA release.
The Food and Drug Administration has approved , a rare condition that causes extremely painful reactions when skin is exposed to light, according to an FDA announcement.
This is the first treatment approved to help patients with this condition increase their exposure to light, according to the release.
Afamelanotide, administered in a subcutaneous implant, is a melanocortin-1 receptor (MC1-R) agonist, which “increases the production of eumelanin in the skin independent of exposure to sunlight or artificial light sources,” the release says.
Approval is based on a pair of parallel-group clinical trials that compared the number of hours spent in sunlight in the treatment and placebo groups. The first trial enrolled 93 patients; 48 received afamelanotide. The treated patients spent a median of 61 hours in total over 180 days in direct sunlight between 10 a.m. and 6 p.m. on days with no pain, compared with 41 hours for patients taking placebo.
The second trial assessed the total number of hours over 270 days spent outdoors between 10 a.m. and 3 p.m. on days with no pain for which “most of the day” was spent in direct sunlight. In this study, 38 patients treated with afamelanotide spent a median total of 6 hours, compared with 0.75 hours among the remaining 36 who were taking a placebo.
The most common side effects include implant site reaction, nausea, and oropharyngeal pain. The implant should be administered only by trained professionals. Because afamelanotide may cause skin darkening, it’s recommended that patients should undergo twice-yearly skin examinations. Patients are also encouraged to maintain sun protection measures to help prevent phototoxic reactions.
“Today’s approval is one example of the FDA’s ongoing commitment to encourage industry innovation of therapies to treat rare diseases, and work with drug developers to make promising new therapies available to patients as safely and efficiently as possible,” said Julie Beitz, MD, director of FDA’s Center for Drug Evaluation and Research Office of Drug Evaluation III in the FDA release.
Consider centralized pain in patients with rheumatic disease
Las Vegas – A fibromyalgia survey may provide important information about the degree to which patients with rheumatic disease experience centralized pain. This information may guide treatment decisions, said Daniel J. Clauw, MD, professor of anesthesiology, rheumatology, and psychiatry and director of the Chronic Pain and Fatigue Research Center at the University of Michigan in Ann Arbor.

The questionnaire that Dr. Clauw uses is a patient self-report survey for the assessment of fibromyalgia based on criteria in the 2011 modification of the American College of Rheumatology preliminary diagnostic criteria for fibromyalgia. In it, he asks patients to report where they experience pain throughout the body and symptoms such as fatigue, sleep problems, and memory problems. The survey predicts outcomes of surgery for osteoarthritis better than x-rays, MRI scans, or psychological factors do, he said.
Physicians should ask every patient with chronic pain, including patients with OA, rheumatoid arthritis, or lupus, to complete the survey, Dr. Clauw said at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education. “This score will tell you the degree to which their central nervous system is augmenting or amplifying what is going on in their body,” he said. “And the higher their score is, the more you should treat them like you would someone with fibromyalgia, even if their underlying disease might be an autoimmune disease.”
Physicians should not use a cutoff of 13 points on the fibromyalgia measure to define whether a patient has the disease, as has been done in the past, he said. The threshold is arbitrary, he said. “We should not think about fibromyalgia as ‘yes’ or ‘no.’ We should think of the degree of fibromyalgia that people have.”
A poor relationship between pain and imaging
Some patients who have severe knee OA on imaging walk without pain. Other patients have normal x-rays, but severe pain. “There is a terrible relationship between what you see on a knee x-ray or an MRI and whether someone has pain,” Dr. Clauw said. Furthermore, the poor relationship between imaging and pain is common across chronic pain conditions, he said.
This phenomenon may occur because pain manifests in different ways, similar to there being multiple ways to adjust the volume of an electric guitar, he said. How hard the strings are strummed affects the volume. But so does the amplifier setting. “In these centralized pain conditions, the problem is an amplifier problem, not a guitar problem,” he said. “The amplifier, i.e., the central nervous system, is set too high.”
Researchers have found that people who have severe OA of the knee on x-ray but do not experience pain “have a very low amplifier setting,” he said. That is, they are nontender and less sensitive to pain. Most of these patients are men. “On average, men have a much lower amplifier setting than women,” he said. “This is also why ... women have 1.5 to 2 times the rate of any type of chronic pain than men, because on average women have a higher amplifier setting. ... In OA, at any given age, men and women have the exact same percentage of radiographic OA. But if you look at the clinical condition of OA, it is always two-thirds women, one-third men.”
Opioid responsiveness
To examine whether fibromyalgia survey results correlate with outcomes after knee and hip arthroplasty, Dr. Clauw and colleagues conducted a prospective, observational cohort study that included approximately 500 people. Patients completed the questionnaire on the day of surgery.
Patients with higher levels of fibromyalgia were less responsive to opioids. “For each 1-point increase in the fibromyalgia score, people needed about one more hydrocodone tablet in the first 24-48 hours to control their pain,” he said (Anesthesiology. 2013 Dec;119[6]:1434-43). In addition, each 1-point increase in the fibromyalgia score made people about 25% less likely to have a 50% improvement in knee pain level after 6 months (Arthritis Rheumatol. 2015 May;67[5]:1386-94). The correlations were independent of psychological factors. In addition, the associations were linear. “There was nothing magical about a fibromyalgia score of 13,” Dr. Clauw said.
Dr. Clauw is a coauthor of a study to be presented at the 2019 American College of Rheumatology/Association of Rheumatology Professionals annual meeting that found pain centralization in patients with RA is associated with poor response to disease-modifying antirheumatic drugs (DMARDs).
Prior studies in patients with RA have found that the degree of fibromyalgia is a better predictor of pain and disability than erythrocyte sedimentation rate or the number of swollen joints.
Diagnosed cases are the “tip of the iceberg”
Researchers at Dr. Clauw’s institution have identified dozens of patients undergoing knee surgery who met criteria for fibromyalgia but had not received the diagnosis. “This is at the University of Michigan, which is the epicenter for fibromyalgia research. If we are not seeing fibromyalgia superimposed on OA in our patients, no one is seeing it,” he said.
Patients with diagnosed fibromyalgia are “the tip of the iceberg,” he said. “There are far greater numbers of individuals whose primary diagnosis is OA, RA, lupus, ankylosing spondylitis, cancer pain, or sickle cell disease that have the same fundamental problem as fibromyalgia patients. But you do not see it because you label them as having an autoimmune disease or osteoarthritis. And that is at your peril and at their peril. Because treating that individual as if all of their pain and other symptoms are due to a problem out on the periphery will not make that person better.”
Patients with high levels of centralized pain may be less responsive to peripherally directed therapies such as surgery or injections, Dr. Clauw said. Pharmacologic options for patients with centralized pain include gabapentinoids (e.g., pregabalin and gabapentin), serotonin-norepinephrine reuptake inhibitors (e.g., duloxetine and milnacipran), and tricyclic compounds (e.g., amitriptyline and cyclobenzaprine), he said. “Opioids are going to be quite unlikely to help these individuals,” he said. “In fact, it is likely that opioids will make this kind of pain worse.”
Dr. Clauw is a consultant for Aptinyx, Daiichi Sankyo, Eli Lilly, Intec Pharma, Pfizer, Samumed, Theravance, Tonix, and Zynerba Pharma. He has received grant or research support from Aptinyx and Pfizer and is an expert witness.
Global Academy for Medical Education and this news organization are owned by the same parent company.
Las Vegas – A fibromyalgia survey may provide important information about the degree to which patients with rheumatic disease experience centralized pain. This information may guide treatment decisions, said Daniel J. Clauw, MD, professor of anesthesiology, rheumatology, and psychiatry and director of the Chronic Pain and Fatigue Research Center at the University of Michigan in Ann Arbor.
The questionnaire that Dr. Clauw uses is a patient self-report survey for the assessment of fibromyalgia based on criteria in the 2011 modification of the American College of Rheumatology preliminary diagnostic criteria for fibromyalgia. In it, he asks patients to report where they experience pain throughout the body and symptoms such as fatigue, sleep problems, and memory problems. The survey predicts outcomes of surgery for osteoarthritis better than x-rays, MRI scans, or psychological factors do, he said.
Physicians should ask every patient with chronic pain, including patients with OA, rheumatoid arthritis, or lupus, to complete the survey, Dr. Clauw said at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education. “This score will tell you the degree to which their central nervous system is augmenting or amplifying what is going on in their body,” he said. “And the higher their score is, the more you should treat them like you would someone with fibromyalgia, even if their underlying disease might be an autoimmune disease.”
Physicians should not use a cutoff of 13 points on the fibromyalgia measure to define whether a patient has the disease, as has been done in the past, he said. The threshold is arbitrary, he said. “We should not think about fibromyalgia as ‘yes’ or ‘no.’ We should think of the degree of fibromyalgia that people have.”
A poor relationship between pain and imaging
Some patients who have severe knee OA on imaging walk without pain. Other patients have normal x-rays, but severe pain. “There is a terrible relationship between what you see on a knee x-ray or an MRI and whether someone has pain,” Dr. Clauw said. Furthermore, the poor relationship between imaging and pain is common across chronic pain conditions, he said.
This phenomenon may occur because pain manifests in different ways, similar to there being multiple ways to adjust the volume of an electric guitar, he said. How hard the strings are strummed affects the volume. But so does the amplifier setting. “In these centralized pain conditions, the problem is an amplifier problem, not a guitar problem,” he said. “The amplifier, i.e., the central nervous system, is set too high.”
Researchers have found that people who have severe OA of the knee on x-ray but do not experience pain “have a very low amplifier setting,” he said. That is, they are nontender and less sensitive to pain. Most of these patients are men. “On average, men have a much lower amplifier setting than women,” he said. “This is also why ... women have 1.5 to 2 times the rate of any type of chronic pain than men, because on average women have a higher amplifier setting. ... In OA, at any given age, men and women have the exact same percentage of radiographic OA. But if you look at the clinical condition of OA, it is always two-thirds women, one-third men.”
Opioid responsiveness
To examine whether fibromyalgia survey results correlate with outcomes after knee and hip arthroplasty, Dr. Clauw and colleagues conducted a prospective, observational cohort study that included approximately 500 people. Patients completed the questionnaire on the day of surgery.
Patients with higher levels of fibromyalgia were less responsive to opioids. “For each 1-point increase in the fibromyalgia score, people needed about one more hydrocodone tablet in the first 24-48 hours to control their pain,” he said (Anesthesiology. 2013 Dec;119[6]:1434-43). In addition, each 1-point increase in the fibromyalgia score made people about 25% less likely to have a 50% improvement in knee pain level after 6 months (Arthritis Rheumatol. 2015 May;67[5]:1386-94). The correlations were independent of psychological factors. In addition, the associations were linear. “There was nothing magical about a fibromyalgia score of 13,” Dr. Clauw said.
Dr. Clauw is a coauthor of a study to be presented at the 2019 American College of Rheumatology/Association of Rheumatology Professionals annual meeting that found pain centralization in patients with RA is associated with poor response to disease-modifying antirheumatic drugs (DMARDs).
Prior studies in patients with RA have found that the degree of fibromyalgia is a better predictor of pain and disability than erythrocyte sedimentation rate or the number of swollen joints.
Diagnosed cases are the “tip of the iceberg”
Researchers at Dr. Clauw’s institution have identified dozens of patients undergoing knee surgery who met criteria for fibromyalgia but had not received the diagnosis. “This is at the University of Michigan, which is the epicenter for fibromyalgia research. If we are not seeing fibromyalgia superimposed on OA in our patients, no one is seeing it,” he said.
Patients with diagnosed fibromyalgia are “the tip of the iceberg,” he said. “There are far greater numbers of individuals whose primary diagnosis is OA, RA, lupus, ankylosing spondylitis, cancer pain, or sickle cell disease that have the same fundamental problem as fibromyalgia patients. But you do not see it because you label them as having an autoimmune disease or osteoarthritis. And that is at your peril and at their peril. Because treating that individual as if all of their pain and other symptoms are due to a problem out on the periphery will not make that person better.”
Patients with high levels of centralized pain may be less responsive to peripherally directed therapies such as surgery or injections, Dr. Clauw said. Pharmacologic options for patients with centralized pain include gabapentinoids (e.g., pregabalin and gabapentin), serotonin-norepinephrine reuptake inhibitors (e.g., duloxetine and milnacipran), and tricyclic compounds (e.g., amitriptyline and cyclobenzaprine), he said. “Opioids are going to be quite unlikely to help these individuals,” he said. “In fact, it is likely that opioids will make this kind of pain worse.”
Dr. Clauw is a consultant for Aptinyx, Daiichi Sankyo, Eli Lilly, Intec Pharma, Pfizer, Samumed, Theravance, Tonix, and Zynerba Pharma. He has received grant or research support from Aptinyx and Pfizer and is an expert witness.
Global Academy for Medical Education and this news organization are owned by the same parent company.
Las Vegas – A fibromyalgia survey may provide important information about the degree to which patients with rheumatic disease experience centralized pain. This information may guide treatment decisions, said Daniel J. Clauw, MD, professor of anesthesiology, rheumatology, and psychiatry and director of the Chronic Pain and Fatigue Research Center at the University of Michigan in Ann Arbor.
The questionnaire that Dr. Clauw uses is a patient self-report survey for the assessment of fibromyalgia based on criteria in the 2011 modification of the American College of Rheumatology preliminary diagnostic criteria for fibromyalgia. In it, he asks patients to report where they experience pain throughout the body and symptoms such as fatigue, sleep problems, and memory problems. The survey predicts outcomes of surgery for osteoarthritis better than x-rays, MRI scans, or psychological factors do, he said.
Physicians should ask every patient with chronic pain, including patients with OA, rheumatoid arthritis, or lupus, to complete the survey, Dr. Clauw said at the annual Perspectives in Rheumatic Diseases held by Global Academy for Medical Education. “This score will tell you the degree to which their central nervous system is augmenting or amplifying what is going on in their body,” he said. “And the higher their score is, the more you should treat them like you would someone with fibromyalgia, even if their underlying disease might be an autoimmune disease.”
Physicians should not use a cutoff of 13 points on the fibromyalgia measure to define whether a patient has the disease, as has been done in the past, he said. The threshold is arbitrary, he said. “We should not think about fibromyalgia as ‘yes’ or ‘no.’ We should think of the degree of fibromyalgia that people have.”
A poor relationship between pain and imaging
Some patients who have severe knee OA on imaging walk without pain. Other patients have normal x-rays, but severe pain. “There is a terrible relationship between what you see on a knee x-ray or an MRI and whether someone has pain,” Dr. Clauw said. Furthermore, the poor relationship between imaging and pain is common across chronic pain conditions, he said.
This phenomenon may occur because pain manifests in different ways, similar to there being multiple ways to adjust the volume of an electric guitar, he said. How hard the strings are strummed affects the volume. But so does the amplifier setting. “In these centralized pain conditions, the problem is an amplifier problem, not a guitar problem,” he said. “The amplifier, i.e., the central nervous system, is set too high.”
Researchers have found that people who have severe OA of the knee on x-ray but do not experience pain “have a very low amplifier setting,” he said. That is, they are nontender and less sensitive to pain. Most of these patients are men. “On average, men have a much lower amplifier setting than women,” he said. “This is also why ... women have 1.5 to 2 times the rate of any type of chronic pain than men, because on average women have a higher amplifier setting. ... In OA, at any given age, men and women have the exact same percentage of radiographic OA. But if you look at the clinical condition of OA, it is always two-thirds women, one-third men.”
Opioid responsiveness
To examine whether fibromyalgia survey results correlate with outcomes after knee and hip arthroplasty, Dr. Clauw and colleagues conducted a prospective, observational cohort study that included approximately 500 people. Patients completed the questionnaire on the day of surgery.
Patients with higher levels of fibromyalgia were less responsive to opioids. “For each 1-point increase in the fibromyalgia score, people needed about one more hydrocodone tablet in the first 24-48 hours to control their pain,” he said (Anesthesiology. 2013 Dec;119[6]:1434-43). In addition, each 1-point increase in the fibromyalgia score made people about 25% less likely to have a 50% improvement in knee pain level after 6 months (Arthritis Rheumatol. 2015 May;67[5]:1386-94). The correlations were independent of psychological factors. In addition, the associations were linear. “There was nothing magical about a fibromyalgia score of 13,” Dr. Clauw said.
Dr. Clauw is a coauthor of a study to be presented at the 2019 American College of Rheumatology/Association of Rheumatology Professionals annual meeting that found pain centralization in patients with RA is associated with poor response to disease-modifying antirheumatic drugs (DMARDs).
Prior studies in patients with RA have found that the degree of fibromyalgia is a better predictor of pain and disability than erythrocyte sedimentation rate or the number of swollen joints.
Diagnosed cases are the “tip of the iceberg”
Researchers at Dr. Clauw’s institution have identified dozens of patients undergoing knee surgery who met criteria for fibromyalgia but had not received the diagnosis. “This is at the University of Michigan, which is the epicenter for fibromyalgia research. If we are not seeing fibromyalgia superimposed on OA in our patients, no one is seeing it,” he said.
Patients with diagnosed fibromyalgia are “the tip of the iceberg,” he said. “There are far greater numbers of individuals whose primary diagnosis is OA, RA, lupus, ankylosing spondylitis, cancer pain, or sickle cell disease that have the same fundamental problem as fibromyalgia patients. But you do not see it because you label them as having an autoimmune disease or osteoarthritis. And that is at your peril and at their peril. Because treating that individual as if all of their pain and other symptoms are due to a problem out on the periphery will not make that person better.”
Patients with high levels of centralized pain may be less responsive to peripherally directed therapies such as surgery or injections, Dr. Clauw said. Pharmacologic options for patients with centralized pain include gabapentinoids (e.g., pregabalin and gabapentin), serotonin-norepinephrine reuptake inhibitors (e.g., duloxetine and milnacipran), and tricyclic compounds (e.g., amitriptyline and cyclobenzaprine), he said. “Opioids are going to be quite unlikely to help these individuals,” he said. “In fact, it is likely that opioids will make this kind of pain worse.”
Dr. Clauw is a consultant for Aptinyx, Daiichi Sankyo, Eli Lilly, Intec Pharma, Pfizer, Samumed, Theravance, Tonix, and Zynerba Pharma. He has received grant or research support from Aptinyx and Pfizer and is an expert witness.
Global Academy for Medical Education and this news organization are owned by the same parent company.
EXPERT ANALYSIS FROM PRD 2019
What is the optimal duration of maintenance in myeloma?
SAN FRANCISCO – Should patients with multiple myeloma receive maintenance therapy until progression?

Yvonne A. Efebera, MD, of The Ohio State University Comprehensive Cancer Center – Arthur G. James Cancer Hospital in Columbus, and Nina Shah, MD, of the University of California San Francisco Health, faced off on this question at the National Comprehensive Cancer Network Hematologic Malignancies Annual Congress.

Dr. Shah said maintenance therapy improves survival in myeloma patients, so it follows that treating them until progression would confer a survival advantage. While Dr. Efebera agreed that maintenance can improve survival, she said the optimal duration of that treatment is unknown.
Treat until progression
Dr. Shah cited studies suggesting that maintenance improves progression-free survival (PFS) and may prolong overall survival (OS) in multiple myeloma.
A meta-analysis of data from the IFM 2005-02, CALGB 100104, and GIMEMA RV-MM-PI-209 trials showed that lenalidomide maintenance prolonged PFS and OS. The median PFS was 52.8 months in patients who received maintenance and 23.5 months in those who received placebo or observation (hazard ratio [HR], 0.48). At a median follow-up of 79.5 months, the median OS was not reached for the maintenance group and was 86.0 months for the no-maintenance group (HR, 0.75; P = .001; J Clin Oncol. 2017 Oct 10;35[29]:3279-89).
In the Myeloma XI trial, maintenance improved PFS, but not OS, in both transplant-eligible and ineligible patients. Overall, the median PFS was 39 months in the lenalidomide maintenance arm and 20 months in the observation arm (P less than .0001). Among transplant-eligible patients, the median PFS was 57 months and 30 months, respectively (P less than .0001). Among transplant-ineligible patients, the median PFS was 26 months and 11 months, respectively (P less than .0001; Lancet Oncol. 2019 Jan;20[1]:57-73).
These data suggest maintenance can improve survival, “but the question is, how long should we have therapy,” Dr. Shah said. “No one has looked at this in a prospective manner, so we really have to look at our retrospective data.”
One study suggested a longer duration of lenalidomide maintenance improves PFS. The HR for progression or death was 0.39 for patients who received maintenance for 12-24 months, compared with those who received maintenance for less than 12 months. The HR was 0.13 for patients who received maintenance for more than 24 months, compared with less than 12 months (Leuk Lymphoma. 2019 Feb;60[2]:511-4).
Dr. Shah also cited a pooled analysis of three phase 3 trials suggesting that continuous therapy is superior to fixed-duration therapy in patients with newly diagnosed myeloma. The median PFS was 32 months with continuous therapy and 16 months with fixed-duration therapy (P less than .001). The 4-year OS was 69% and 60%, respectively (P = .003; J Clin Oncol. 2015 Oct 20;33[30]:3459-66).
These data suggest that “continuous therapy, more therapy, has a survival advantage,” Dr. Shah said.
Don’t treat until progression
Dr. Efebera also discussed data from studies showing that lenalidomide maintenance can prolong survival in multiple myeloma. However, she said, it’s unclear how long maintenance should last.
Different durations of maintenance have proved effective in different trials. In the CALGB 100104 trial, the median duration of maintenance was 31 months (Lancet Haematol. 2017 Sep;4[9]:e431-e442). In the meta-analysis of the CALGB, IFM, and GIMEMA trials, the median duration was 22 months. And in Myeloma XI, the median duration was 18 months.
As there is no randomized trial comparing different durations of maintenance, Dr. Efebera proposed that researchers conduct one. She said this “perfect study” would involve induction with an immunomodulatory agent, a proteasome inhibitor, dexamethasone, and perhaps an anti-CD38 therapy. Transplant-eligible patients would receive four cycles of induction before transplant. Transplant-ineligible patients would receive eight cycles of induction. Then, all patients would be randomized to lenalidomide maintenance for 3 years, 5 years, or 7-10 years.
Until a trial like this reveals the optimal duration of maintenance, we cannot conclude that treating patients until progression is better, Dr. Efebera said.
She added that maintenance has been shown to have detrimental effects, and these should be taken into consideration. For instance, neutropenia, other hematologic adverse events, and second primary malignancies have been shown to be more common among patients who receive lenalidomide maintenance (N Engl J Med. 2012; 366:1782-91).
The cost of maintenance is another factor to consider. Researchers analyzed data from the CALGB 100104 and IFM 2005-02 trials to compare the cost of lenalidomide maintenance with no maintenance. In the CALGB 100104 trial, patients who received lenalidomide maintenance had 5.72 quality-adjusted life years (QALYs), and those who received no maintenance had 4.61 QALYs. The incremental cost-utility ratio (ICUR) was more than 277,000 euros per QALY.
In the IFM2005-02 trial, patients in the lenalidomide group had 5.13 QALYs, and those who didn’t receive maintenance had 4.98 QALYs. The ICUR was more than 1.5 million euros per QALY. The researchers said the high ICURs and budgetary impact add “uncertainty about the maximum prudent duration of the treatment” (Bone Marrow Transplant. 2019 May 31. doi: 10.1038/s41409-019-0574-5).
Dr. Efebera reported relationships with Akcea Therapeutics, Janssen, and Takeda. Dr. Shah reported having no relevant financial relationships.
SAN FRANCISCO – Should patients with multiple myeloma receive maintenance therapy until progression?
Yvonne A. Efebera, MD, of The Ohio State University Comprehensive Cancer Center – Arthur G. James Cancer Hospital in Columbus, and Nina Shah, MD, of the University of California San Francisco Health, faced off on this question at the National Comprehensive Cancer Network Hematologic Malignancies Annual Congress.
Dr. Shah said maintenance therapy improves survival in myeloma patients, so it follows that treating them until progression would confer a survival advantage. While Dr. Efebera agreed that maintenance can improve survival, she said the optimal duration of that treatment is unknown.
Treat until progression
Dr. Shah cited studies suggesting that maintenance improves progression-free survival (PFS) and may prolong overall survival (OS) in multiple myeloma.
A meta-analysis of data from the IFM 2005-02, CALGB 100104, and GIMEMA RV-MM-PI-209 trials showed that lenalidomide maintenance prolonged PFS and OS. The median PFS was 52.8 months in patients who received maintenance and 23.5 months in those who received placebo or observation (hazard ratio [HR], 0.48). At a median follow-up of 79.5 months, the median OS was not reached for the maintenance group and was 86.0 months for the no-maintenance group (HR, 0.75; P = .001; J Clin Oncol. 2017 Oct 10;35[29]:3279-89).
In the Myeloma XI trial, maintenance improved PFS, but not OS, in both transplant-eligible and ineligible patients. Overall, the median PFS was 39 months in the lenalidomide maintenance arm and 20 months in the observation arm (P less than .0001). Among transplant-eligible patients, the median PFS was 57 months and 30 months, respectively (P less than .0001). Among transplant-ineligible patients, the median PFS was 26 months and 11 months, respectively (P less than .0001; Lancet Oncol. 2019 Jan;20[1]:57-73).
These data suggest maintenance can improve survival, “but the question is, how long should we have therapy,” Dr. Shah said. “No one has looked at this in a prospective manner, so we really have to look at our retrospective data.”
One study suggested a longer duration of lenalidomide maintenance improves PFS. The HR for progression or death was 0.39 for patients who received maintenance for 12-24 months, compared with those who received maintenance for less than 12 months. The HR was 0.13 for patients who received maintenance for more than 24 months, compared with less than 12 months (Leuk Lymphoma. 2019 Feb;60[2]:511-4).
Dr. Shah also cited a pooled analysis of three phase 3 trials suggesting that continuous therapy is superior to fixed-duration therapy in patients with newly diagnosed myeloma. The median PFS was 32 months with continuous therapy and 16 months with fixed-duration therapy (P less than .001). The 4-year OS was 69% and 60%, respectively (P = .003; J Clin Oncol. 2015 Oct 20;33[30]:3459-66).
These data suggest that “continuous therapy, more therapy, has a survival advantage,” Dr. Shah said.
Don’t treat until progression
Dr. Efebera also discussed data from studies showing that lenalidomide maintenance can prolong survival in multiple myeloma. However, she said, it’s unclear how long maintenance should last.
Different durations of maintenance have proved effective in different trials. In the CALGB 100104 trial, the median duration of maintenance was 31 months (Lancet Haematol. 2017 Sep;4[9]:e431-e442). In the meta-analysis of the CALGB, IFM, and GIMEMA trials, the median duration was 22 months. And in Myeloma XI, the median duration was 18 months.
As there is no randomized trial comparing different durations of maintenance, Dr. Efebera proposed that researchers conduct one. She said this “perfect study” would involve induction with an immunomodulatory agent, a proteasome inhibitor, dexamethasone, and perhaps an anti-CD38 therapy. Transplant-eligible patients would receive four cycles of induction before transplant. Transplant-ineligible patients would receive eight cycles of induction. Then, all patients would be randomized to lenalidomide maintenance for 3 years, 5 years, or 7-10 years.
Until a trial like this reveals the optimal duration of maintenance, we cannot conclude that treating patients until progression is better, Dr. Efebera said.
She added that maintenance has been shown to have detrimental effects, and these should be taken into consideration. For instance, neutropenia, other hematologic adverse events, and second primary malignancies have been shown to be more common among patients who receive lenalidomide maintenance (N Engl J Med. 2012; 366:1782-91).
The cost of maintenance is another factor to consider. Researchers analyzed data from the CALGB 100104 and IFM 2005-02 trials to compare the cost of lenalidomide maintenance with no maintenance. In the CALGB 100104 trial, patients who received lenalidomide maintenance had 5.72 quality-adjusted life years (QALYs), and those who received no maintenance had 4.61 QALYs. The incremental cost-utility ratio (ICUR) was more than 277,000 euros per QALY.
In the IFM2005-02 trial, patients in the lenalidomide group had 5.13 QALYs, and those who didn’t receive maintenance had 4.98 QALYs. The ICUR was more than 1.5 million euros per QALY. The researchers said the high ICURs and budgetary impact add “uncertainty about the maximum prudent duration of the treatment” (Bone Marrow Transplant. 2019 May 31. doi: 10.1038/s41409-019-0574-5).
Dr. Efebera reported relationships with Akcea Therapeutics, Janssen, and Takeda. Dr. Shah reported having no relevant financial relationships.
SAN FRANCISCO – Should patients with multiple myeloma receive maintenance therapy until progression?
Yvonne A. Efebera, MD, of The Ohio State University Comprehensive Cancer Center – Arthur G. James Cancer Hospital in Columbus, and Nina Shah, MD, of the University of California San Francisco Health, faced off on this question at the National Comprehensive Cancer Network Hematologic Malignancies Annual Congress.
Dr. Shah said maintenance therapy improves survival in myeloma patients, so it follows that treating them until progression would confer a survival advantage. While Dr. Efebera agreed that maintenance can improve survival, she said the optimal duration of that treatment is unknown.
Treat until progression
Dr. Shah cited studies suggesting that maintenance improves progression-free survival (PFS) and may prolong overall survival (OS) in multiple myeloma.
A meta-analysis of data from the IFM 2005-02, CALGB 100104, and GIMEMA RV-MM-PI-209 trials showed that lenalidomide maintenance prolonged PFS and OS. The median PFS was 52.8 months in patients who received maintenance and 23.5 months in those who received placebo or observation (hazard ratio [HR], 0.48). At a median follow-up of 79.5 months, the median OS was not reached for the maintenance group and was 86.0 months for the no-maintenance group (HR, 0.75; P = .001; J Clin Oncol. 2017 Oct 10;35[29]:3279-89).
In the Myeloma XI trial, maintenance improved PFS, but not OS, in both transplant-eligible and ineligible patients. Overall, the median PFS was 39 months in the lenalidomide maintenance arm and 20 months in the observation arm (P less than .0001). Among transplant-eligible patients, the median PFS was 57 months and 30 months, respectively (P less than .0001). Among transplant-ineligible patients, the median PFS was 26 months and 11 months, respectively (P less than .0001; Lancet Oncol. 2019 Jan;20[1]:57-73).
These data suggest maintenance can improve survival, “but the question is, how long should we have therapy,” Dr. Shah said. “No one has looked at this in a prospective manner, so we really have to look at our retrospective data.”
One study suggested a longer duration of lenalidomide maintenance improves PFS. The HR for progression or death was 0.39 for patients who received maintenance for 12-24 months, compared with those who received maintenance for less than 12 months. The HR was 0.13 for patients who received maintenance for more than 24 months, compared with less than 12 months (Leuk Lymphoma. 2019 Feb;60[2]:511-4).
Dr. Shah also cited a pooled analysis of three phase 3 trials suggesting that continuous therapy is superior to fixed-duration therapy in patients with newly diagnosed myeloma. The median PFS was 32 months with continuous therapy and 16 months with fixed-duration therapy (P less than .001). The 4-year OS was 69% and 60%, respectively (P = .003; J Clin Oncol. 2015 Oct 20;33[30]:3459-66).
These data suggest that “continuous therapy, more therapy, has a survival advantage,” Dr. Shah said.
Don’t treat until progression
Dr. Efebera also discussed data from studies showing that lenalidomide maintenance can prolong survival in multiple myeloma. However, she said, it’s unclear how long maintenance should last.
Different durations of maintenance have proved effective in different trials. In the CALGB 100104 trial, the median duration of maintenance was 31 months (Lancet Haematol. 2017 Sep;4[9]:e431-e442). In the meta-analysis of the CALGB, IFM, and GIMEMA trials, the median duration was 22 months. And in Myeloma XI, the median duration was 18 months.
As there is no randomized trial comparing different durations of maintenance, Dr. Efebera proposed that researchers conduct one. She said this “perfect study” would involve induction with an immunomodulatory agent, a proteasome inhibitor, dexamethasone, and perhaps an anti-CD38 therapy. Transplant-eligible patients would receive four cycles of induction before transplant. Transplant-ineligible patients would receive eight cycles of induction. Then, all patients would be randomized to lenalidomide maintenance for 3 years, 5 years, or 7-10 years.
Until a trial like this reveals the optimal duration of maintenance, we cannot conclude that treating patients until progression is better, Dr. Efebera said.
She added that maintenance has been shown to have detrimental effects, and these should be taken into consideration. For instance, neutropenia, other hematologic adverse events, and second primary malignancies have been shown to be more common among patients who receive lenalidomide maintenance (N Engl J Med. 2012; 366:1782-91).
The cost of maintenance is another factor to consider. Researchers analyzed data from the CALGB 100104 and IFM 2005-02 trials to compare the cost of lenalidomide maintenance with no maintenance. In the CALGB 100104 trial, patients who received lenalidomide maintenance had 5.72 quality-adjusted life years (QALYs), and those who received no maintenance had 4.61 QALYs. The incremental cost-utility ratio (ICUR) was more than 277,000 euros per QALY.
In the IFM2005-02 trial, patients in the lenalidomide group had 5.13 QALYs, and those who didn’t receive maintenance had 4.98 QALYs. The ICUR was more than 1.5 million euros per QALY. The researchers said the high ICURs and budgetary impact add “uncertainty about the maximum prudent duration of the treatment” (Bone Marrow Transplant. 2019 May 31. doi: 10.1038/s41409-019-0574-5).
Dr. Efebera reported relationships with Akcea Therapeutics, Janssen, and Takeda. Dr. Shah reported having no relevant financial relationships.
EXPERT ANALYSIS FROM NCCN HEMATOLOGIC MALIGNANCIES