User login
Cerliponase alfa continues to impress for CLN2 disease
BANGKOK – Biweekly cerliponase alfa continued to show durable and clinically important therapeutic benefit in children with neuronal ceroid lipofuscinosis type 2 (CLN2) disease at the 3-year mark in an ongoing international study, Marina Trivisano, MD, reported at the International Epilepsy Congress.
Cerliponase alfa, approved under the trade name Brineura by the Food and Drug Administration and European Commission, is a recombinant human tripeptidyl peptidase 1 designed as enzyme replacement therapy delivered by a surgically implanted intraventricular infusion device in children with this rare lysosomal storage disease, a form of Batten disease, she explained at the congress sponsored by the International League Against Epilepsy.
When both healthy parents carry one defective gene, each of their children has a one in four chance of inheriting this devastating disease that causes rapidly progressive dementia. CLN2 disease typically reveals itself when a child reaches about 3 years of age, with seizures, language delay, or loss of acquired language being the most common first indications.
Of 23 patients enrolled in the open-label study, 21 remained participants at 3 years of follow-up. The two dropouts weren’t caused by treatment-related adverse events, but rather by the formidable logistic challenges posed because the treatment – 300 mg of cerliponase alfa delivered by intraventricular infusion over a 4-hour period every 2 weeks – was available only at five medical centers located in Rome; London; New York; Hamburg, Germany; and Columbus, Ohio.
At 3 years of follow-up, 83% of patients met the primary study endpoint, defined as the absence of a 2-point or greater decline in the motor-language score on the 0-6 CLN2 Clinical Rating Scale. This was a success rate 12 times greater than in 42 historical controls. Indeed, at 3 years the cerliponase alfa–treated patients had an average CLN2 Clinical Rating Scale motor-language score 3.8 points better than the historical controls, reported Dr. Trivisano, a pediatric neurologist at Bambino Gesu Children’s Hospital in Rome.
Side effects included several cases of device failure, infection, and hypersensitivity reactions.
In an earlier report based upon 96 weeks of follow-up, the mean rate of decline in the motor-language score was 0.27 points per 48 weeks in treated patients, compared with 2.12 points in the historical controls (N Engl J Med. 2018 May 17;378[20]:1898-1907).
The study was funded by BioMarin Pharmaceutical, which markets Brineura. Dr. Trivisano was a subinvestigator in the trial.
SOURCE: Trivisano M et al. IEC 2019, Abstract P333.
BANGKOK – Biweekly cerliponase alfa continued to show durable and clinically important therapeutic benefit in children with neuronal ceroid lipofuscinosis type 2 (CLN2) disease at the 3-year mark in an ongoing international study, Marina Trivisano, MD, reported at the International Epilepsy Congress.
Cerliponase alfa, approved under the trade name Brineura by the Food and Drug Administration and European Commission, is a recombinant human tripeptidyl peptidase 1 designed as enzyme replacement therapy delivered by a surgically implanted intraventricular infusion device in children with this rare lysosomal storage disease, a form of Batten disease, she explained at the congress sponsored by the International League Against Epilepsy.
When both healthy parents carry one defective gene, each of their children has a one in four chance of inheriting this devastating disease that causes rapidly progressive dementia. CLN2 disease typically reveals itself when a child reaches about 3 years of age, with seizures, language delay, or loss of acquired language being the most common first indications.
Of 23 patients enrolled in the open-label study, 21 remained participants at 3 years of follow-up. The two dropouts weren’t caused by treatment-related adverse events, but rather by the formidable logistic challenges posed because the treatment – 300 mg of cerliponase alfa delivered by intraventricular infusion over a 4-hour period every 2 weeks – was available only at five medical centers located in Rome; London; New York; Hamburg, Germany; and Columbus, Ohio.
At 3 years of follow-up, 83% of patients met the primary study endpoint, defined as the absence of a 2-point or greater decline in the motor-language score on the 0-6 CLN2 Clinical Rating Scale. This was a success rate 12 times greater than in 42 historical controls. Indeed, at 3 years the cerliponase alfa–treated patients had an average CLN2 Clinical Rating Scale motor-language score 3.8 points better than the historical controls, reported Dr. Trivisano, a pediatric neurologist at Bambino Gesu Children’s Hospital in Rome.
Side effects included several cases of device failure, infection, and hypersensitivity reactions.
In an earlier report based upon 96 weeks of follow-up, the mean rate of decline in the motor-language score was 0.27 points per 48 weeks in treated patients, compared with 2.12 points in the historical controls (N Engl J Med. 2018 May 17;378[20]:1898-1907).
The study was funded by BioMarin Pharmaceutical, which markets Brineura. Dr. Trivisano was a subinvestigator in the trial.
SOURCE: Trivisano M et al. IEC 2019, Abstract P333.
BANGKOK – Biweekly cerliponase alfa continued to show durable and clinically important therapeutic benefit in children with neuronal ceroid lipofuscinosis type 2 (CLN2) disease at the 3-year mark in an ongoing international study, Marina Trivisano, MD, reported at the International Epilepsy Congress.
Cerliponase alfa, approved under the trade name Brineura by the Food and Drug Administration and European Commission, is a recombinant human tripeptidyl peptidase 1 designed as enzyme replacement therapy delivered by a surgically implanted intraventricular infusion device in children with this rare lysosomal storage disease, a form of Batten disease, she explained at the congress sponsored by the International League Against Epilepsy.
When both healthy parents carry one defective gene, each of their children has a one in four chance of inheriting this devastating disease that causes rapidly progressive dementia. CLN2 disease typically reveals itself when a child reaches about 3 years of age, with seizures, language delay, or loss of acquired language being the most common first indications.
Of 23 patients enrolled in the open-label study, 21 remained participants at 3 years of follow-up. The two dropouts weren’t caused by treatment-related adverse events, but rather by the formidable logistic challenges posed because the treatment – 300 mg of cerliponase alfa delivered by intraventricular infusion over a 4-hour period every 2 weeks – was available only at five medical centers located in Rome; London; New York; Hamburg, Germany; and Columbus, Ohio.
At 3 years of follow-up, 83% of patients met the primary study endpoint, defined as the absence of a 2-point or greater decline in the motor-language score on the 0-6 CLN2 Clinical Rating Scale. This was a success rate 12 times greater than in 42 historical controls. Indeed, at 3 years the cerliponase alfa–treated patients had an average CLN2 Clinical Rating Scale motor-language score 3.8 points better than the historical controls, reported Dr. Trivisano, a pediatric neurologist at Bambino Gesu Children’s Hospital in Rome.
Side effects included several cases of device failure, infection, and hypersensitivity reactions.
In an earlier report based upon 96 weeks of follow-up, the mean rate of decline in the motor-language score was 0.27 points per 48 weeks in treated patients, compared with 2.12 points in the historical controls (N Engl J Med. 2018 May 17;378[20]:1898-1907).
The study was funded by BioMarin Pharmaceutical, which markets Brineura. Dr. Trivisano was a subinvestigator in the trial.
SOURCE: Trivisano M et al. IEC 2019, Abstract P333.
REPORTING FROM IEC 2019
Dapagliflozin-Induced Sweet Syndrome
To the Editor:
A 75-year-old woman with a history of hypertension, gout, and adult-onset diabetes mellitus was started on dapagliflozin (5 mg) for glycemic control (hemoglobin A1c, 7.9 [reference range, 4–7]). Within 1 week of starting the medication, she developed a fine papular eruption in a photodistributed area on the neck and chest with associated malaise. The rash progressed over the next 2 weeks, evolving into edematous papules and plaques, some with vesicles involving the neck, chest, postauricular areas, and nose. Approximately 3 weeks after starting dapagliflozin, the patient also developed bilateral painful, hemorrhagic, bullous plaques (10×3 cm overall) without satellite lesions on the dorsal aspects of the hands. The borders of the bullae had rapidly expanding geographic margins and were extremely painful. The patient’s primary care physician advised to discontinue dapagliflozin, as this medication was thought to be triggering the eruption. She was administered triamcinolone (40 mg intramuscularly) and advised to take ibuprofen as needed. She had malaise and reported that she felt hot but had no known fever. No laboratory tests were ordered.
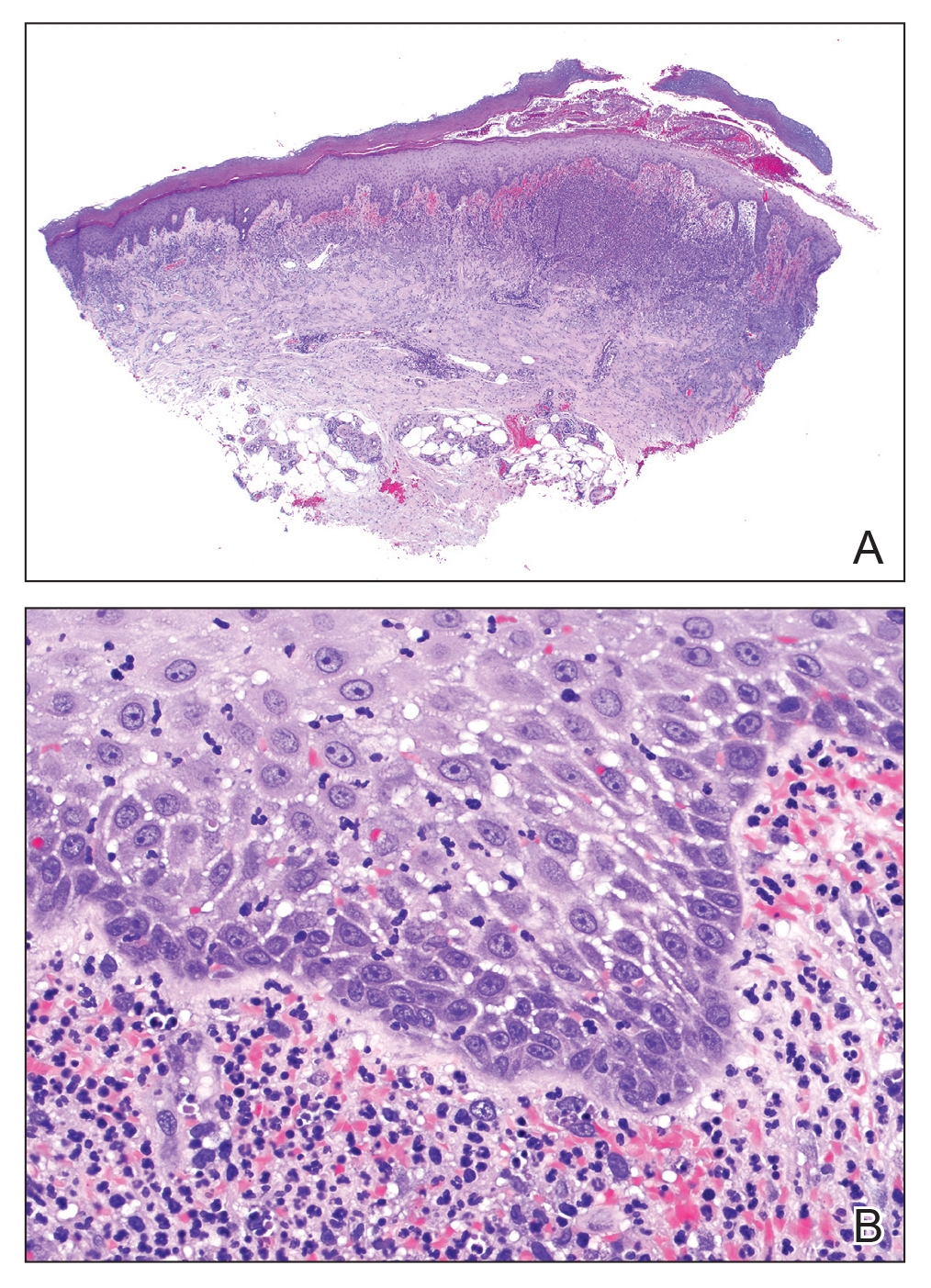
The lesions on the neck and chest began to fade within 1 week of stopping the medication and administering corticosteroids; however, the hand lesions continued to progress and began to involve the base of the digits (Figure 1). The patient was then seen by a dermatologist who biopsied the hand lesions. Histopathology was characteristic of Sweet syndrome, also known as acute febrile neutrophilic dermatosis and Gomm-Button disease. Notably, there was a dense nodular infiltrate of neutrophils, papillary dermal edema, and leukocytoclastic debris without leukocytoclastic vasculitis (Figure 2).
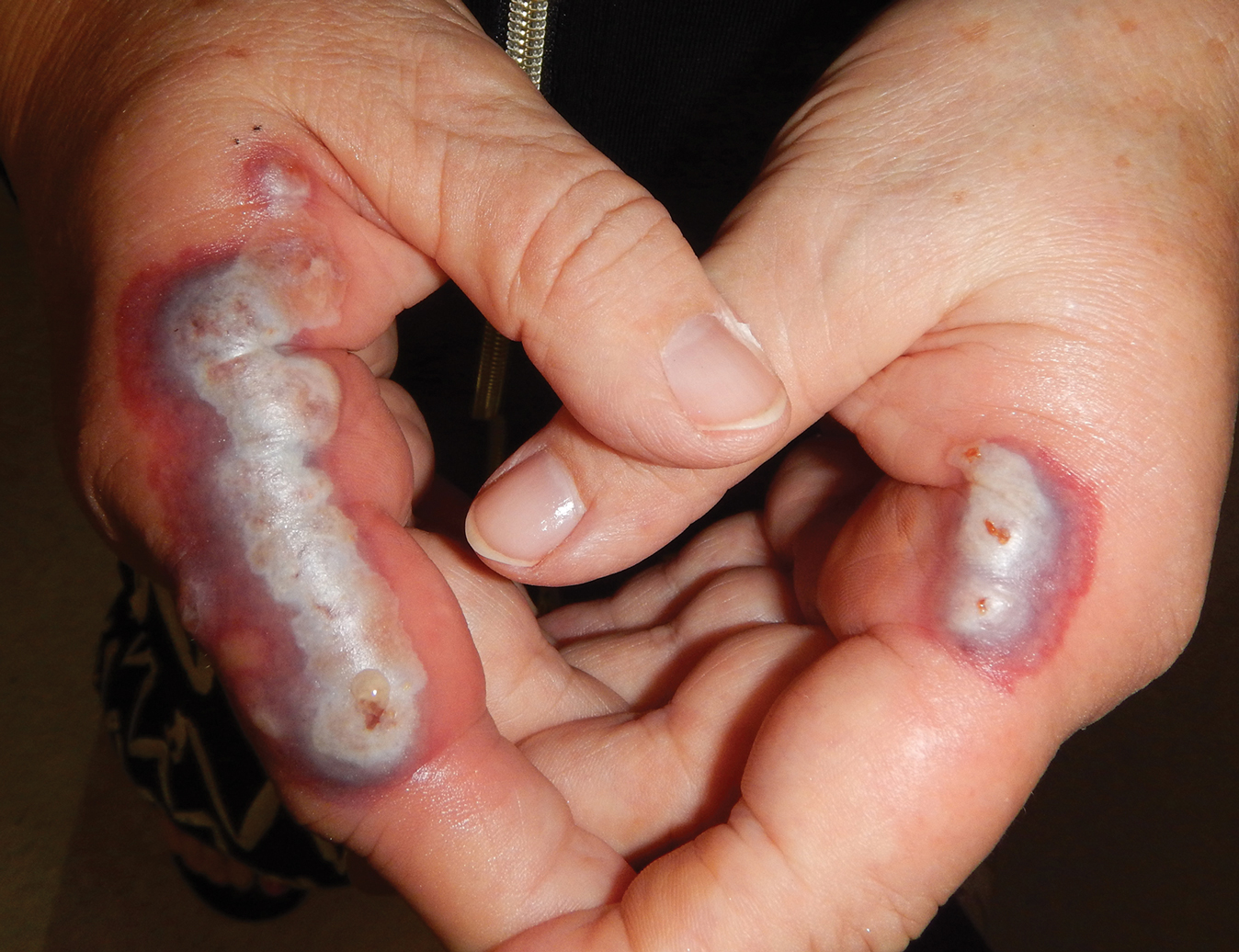
The following therapies in addition to gentle wound care were prescribed: betamethasone injectable suspension (9 mg intramuscularly), oral prednisone (60 mg daily for 5 days, tapering off over 2 months), clobetasol ointment 0.05% twice daily, and tacrolimus ointment 0.1% twice daily. The patient responded well to therapy, with complete resolution and healing of the skin lesions except for mild postinflammatory pigment alteration. The systemic steroids were slowly tapered over 2 months, and the patient remained free of symptoms or recurrences more than 3 years after discontinuation of the medication.
Dapagliflozin is a member of a new class of medications (gliflozins) used for the treatment of type 2 diabetes mellitus.3,4 The medication lowers blood glucose by inhibiting the sodium-glucose cotransport protein 2, thus lowering the blood glucose levels by increasing urinary excretion of glucose. Because many patients with type 2 diabetes mellitus are overweight, these medications are poised to gain popularity for weight loss and decreased blood pressure side effects. Three other medications in this class also have been approved by US Food and Drug Administration: empagliflozin, canagliflozin, and ertugliflozin.
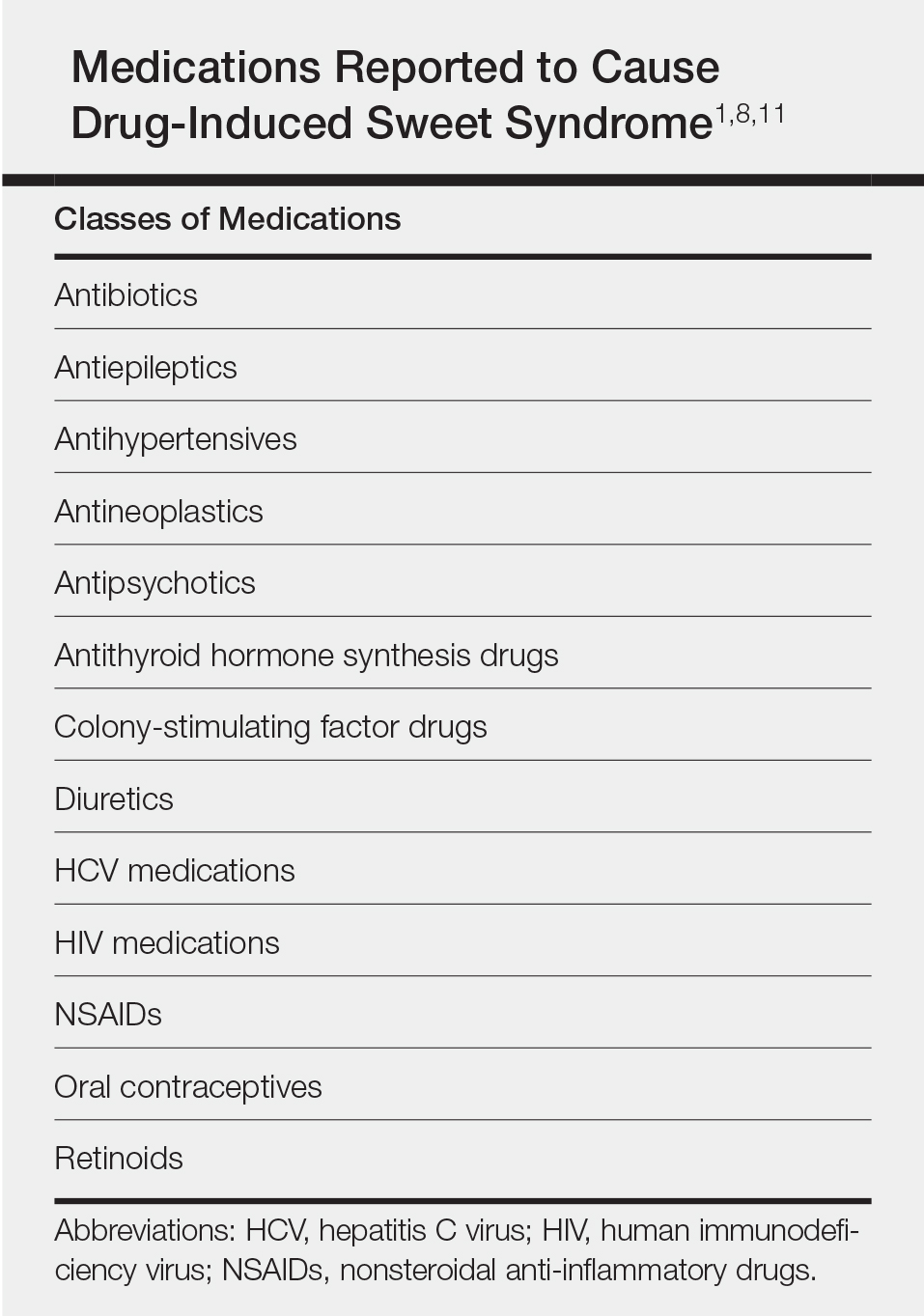
Sweet syndrome consists of 4 cardinal features that were first described in 1964: fever, leukocytosis, tender red plaques, and a dermal neutrophilic infiltrate.5 Since then, Su and Liu6 proposed guidelines consisting of major and minor criteria. In 1996, Walker and Cohen7 suggested a set of diagnostic criteria specifically for drug-induced Sweet syndrome, including painful erythematous plaques, histopathologic neutrophilic infiltrate, and fever. Additional criteria included a temporal relationship between drug ingestion and clinical presentation as well as resolution of lesions after drug withdrawal or treatment with systemic corticosteroids.7 The lesions of drug-induced Sweet syndrome often are described as painful red papules that can form plaques, may appear vesicular, and are more common in women. These lesions classically appear on the upper extremities, as well as the head, neck, trunk, and back.8 Clinically, symptoms most commonly include fever and musculoskeletal involvement, both of which were experienced by the patient who described herself as feeling feverish when the lesions first appeared and reported malaise. Our patient experienced all of these features, and although a fever was not measured in the acute stage of presentation, she reported feeling hot. Other symptoms that may occur include arthralgia, headache, and myalgia.9 Microscopically, there is a nodular infiltrate of neutrophils, papillary dermal edema, and leukocytoclastic debris. The pathogenesis of Sweet syndrome remains unclear but can be associated with malignancy, pregnancy, autoimmune disorders, and drug reactions.10 Many different classes of medications have been reported to cause drug-induced Sweet syndrome and are listed in the Table.1,8,11 The recommended treatment of Sweet syndrome is systemic corticosteroids.12
The temporal use of dapagliflozin and appearance of the hand lesions, along with the histology, favored drug-induced Sweet syndrome to dapagliflozin as the cause of the plaques. Our patient did not seek medical attention at the onset of the initial chest and neck rash but did so several weeks after the painful hand lesions that were consistent with Sweet syndrome had appeared. Discontinuation of dapagliflozin and treatment with immunosuppressive medications resulted in resolution of the skin lesions on the hands. This scenario raises the question whether or not she would have developed the inflammatory hand lesions if she had stopped the medication earlier. Because dapagliflozin is a relatively new medication and boasts the potentially beneficial side effects of reducing body weight and blood pressure in addition to glucose control, we expect additional cases may occur, especially if use of this medication notably increases. Furthermore, this reaction may be a drug-class side effect and not one specific to dapagliflozin.
- Weedon D. The vasculopathic reaction pattern. Weedon’s Skin Pathology. 3rd ed. Oxford, UK: Churchill Livingstone; 2010:218-225.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and Sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Dapagliflozin (Farxiga) for type 2 diabetes. Med Lett Drugs Ther. 2014;56:13-15.
- Aylsworth A, Dean Z, VanNorman C, et al. Dapagliflozin for the treatment of type 2 diabetes mellitus [published online June 20, 2014]. Ann Pharmacother. 2014;48:1202-1208.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Su WP, Liu HN. Diagnostic criteria for Sweet’s syndrome. Cutis. 1986;37:167-174.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34(5 pt 2):918-923.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Cohen PR, Kurzrock R. Sweet’s syndrome. a neutrophilic dermatosis classically associated with acute onset and fever. Clin Dermatol. 2000;18:265-282.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- Thompson DF, Montarella KE. Drug-induced Sweet’s syndrome. Ann Pharmacother. 2007;41:802-811.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
To the Editor:
A 75-year-old woman with a history of hypertension, gout, and adult-onset diabetes mellitus was started on dapagliflozin (5 mg) for glycemic control (hemoglobin A1c, 7.9 [reference range, 4–7]). Within 1 week of starting the medication, she developed a fine papular eruption in a photodistributed area on the neck and chest with associated malaise. The rash progressed over the next 2 weeks, evolving into edematous papules and plaques, some with vesicles involving the neck, chest, postauricular areas, and nose. Approximately 3 weeks after starting dapagliflozin, the patient also developed bilateral painful, hemorrhagic, bullous plaques (10×3 cm overall) without satellite lesions on the dorsal aspects of the hands. The borders of the bullae had rapidly expanding geographic margins and were extremely painful. The patient’s primary care physician advised to discontinue dapagliflozin, as this medication was thought to be triggering the eruption. She was administered triamcinolone (40 mg intramuscularly) and advised to take ibuprofen as needed. She had malaise and reported that she felt hot but had no known fever. No laboratory tests were ordered.
The lesions on the neck and chest began to fade within 1 week of stopping the medication and administering corticosteroids; however, the hand lesions continued to progress and began to involve the base of the digits (Figure 1). The patient was then seen by a dermatologist who biopsied the hand lesions. Histopathology was characteristic of Sweet syndrome, also known as acute febrile neutrophilic dermatosis and Gomm-Button disease. Notably, there was a dense nodular infiltrate of neutrophils, papillary dermal edema, and leukocytoclastic debris without leukocytoclastic vasculitis (Figure 2).
The following therapies in addition to gentle wound care were prescribed: betamethasone injectable suspension (9 mg intramuscularly), oral prednisone (60 mg daily for 5 days, tapering off over 2 months), clobetasol ointment 0.05% twice daily, and tacrolimus ointment 0.1% twice daily. The patient responded well to therapy, with complete resolution and healing of the skin lesions except for mild postinflammatory pigment alteration. The systemic steroids were slowly tapered over 2 months, and the patient remained free of symptoms or recurrences more than 3 years after discontinuation of the medication.
Dapagliflozin is a member of a new class of medications (gliflozins) used for the treatment of type 2 diabetes mellitus.3,4 The medication lowers blood glucose by inhibiting the sodium-glucose cotransport protein 2, thus lowering the blood glucose levels by increasing urinary excretion of glucose. Because many patients with type 2 diabetes mellitus are overweight, these medications are poised to gain popularity for weight loss and decreased blood pressure side effects. Three other medications in this class also have been approved by US Food and Drug Administration: empagliflozin, canagliflozin, and ertugliflozin.
Sweet syndrome consists of 4 cardinal features that were first described in 1964: fever, leukocytosis, tender red plaques, and a dermal neutrophilic infiltrate.5 Since then, Su and Liu6 proposed guidelines consisting of major and minor criteria. In 1996, Walker and Cohen7 suggested a set of diagnostic criteria specifically for drug-induced Sweet syndrome, including painful erythematous plaques, histopathologic neutrophilic infiltrate, and fever. Additional criteria included a temporal relationship between drug ingestion and clinical presentation as well as resolution of lesions after drug withdrawal or treatment with systemic corticosteroids.7 The lesions of drug-induced Sweet syndrome often are described as painful red papules that can form plaques, may appear vesicular, and are more common in women. These lesions classically appear on the upper extremities, as well as the head, neck, trunk, and back.8 Clinically, symptoms most commonly include fever and musculoskeletal involvement, both of which were experienced by the patient who described herself as feeling feverish when the lesions first appeared and reported malaise. Our patient experienced all of these features, and although a fever was not measured in the acute stage of presentation, she reported feeling hot. Other symptoms that may occur include arthralgia, headache, and myalgia.9 Microscopically, there is a nodular infiltrate of neutrophils, papillary dermal edema, and leukocytoclastic debris. The pathogenesis of Sweet syndrome remains unclear but can be associated with malignancy, pregnancy, autoimmune disorders, and drug reactions.10 Many different classes of medications have been reported to cause drug-induced Sweet syndrome and are listed in the Table.1,8,11 The recommended treatment of Sweet syndrome is systemic corticosteroids.12
The temporal use of dapagliflozin and appearance of the hand lesions, along with the histology, favored drug-induced Sweet syndrome to dapagliflozin as the cause of the plaques. Our patient did not seek medical attention at the onset of the initial chest and neck rash but did so several weeks after the painful hand lesions that were consistent with Sweet syndrome had appeared. Discontinuation of dapagliflozin and treatment with immunosuppressive medications resulted in resolution of the skin lesions on the hands. This scenario raises the question whether or not she would have developed the inflammatory hand lesions if she had stopped the medication earlier. Because dapagliflozin is a relatively new medication and boasts the potentially beneficial side effects of reducing body weight and blood pressure in addition to glucose control, we expect additional cases may occur, especially if use of this medication notably increases. Furthermore, this reaction may be a drug-class side effect and not one specific to dapagliflozin.
To the Editor:
A 75-year-old woman with a history of hypertension, gout, and adult-onset diabetes mellitus was started on dapagliflozin (5 mg) for glycemic control (hemoglobin A1c, 7.9 [reference range, 4–7]). Within 1 week of starting the medication, she developed a fine papular eruption in a photodistributed area on the neck and chest with associated malaise. The rash progressed over the next 2 weeks, evolving into edematous papules and plaques, some with vesicles involving the neck, chest, postauricular areas, and nose. Approximately 3 weeks after starting dapagliflozin, the patient also developed bilateral painful, hemorrhagic, bullous plaques (10×3 cm overall) without satellite lesions on the dorsal aspects of the hands. The borders of the bullae had rapidly expanding geographic margins and were extremely painful. The patient’s primary care physician advised to discontinue dapagliflozin, as this medication was thought to be triggering the eruption. She was administered triamcinolone (40 mg intramuscularly) and advised to take ibuprofen as needed. She had malaise and reported that she felt hot but had no known fever. No laboratory tests were ordered.
The lesions on the neck and chest began to fade within 1 week of stopping the medication and administering corticosteroids; however, the hand lesions continued to progress and began to involve the base of the digits (Figure 1). The patient was then seen by a dermatologist who biopsied the hand lesions. Histopathology was characteristic of Sweet syndrome, also known as acute febrile neutrophilic dermatosis and Gomm-Button disease. Notably, there was a dense nodular infiltrate of neutrophils, papillary dermal edema, and leukocytoclastic debris without leukocytoclastic vasculitis (Figure 2).
The following therapies in addition to gentle wound care were prescribed: betamethasone injectable suspension (9 mg intramuscularly), oral prednisone (60 mg daily for 5 days, tapering off over 2 months), clobetasol ointment 0.05% twice daily, and tacrolimus ointment 0.1% twice daily. The patient responded well to therapy, with complete resolution and healing of the skin lesions except for mild postinflammatory pigment alteration. The systemic steroids were slowly tapered over 2 months, and the patient remained free of symptoms or recurrences more than 3 years after discontinuation of the medication.
Dapagliflozin is a member of a new class of medications (gliflozins) used for the treatment of type 2 diabetes mellitus.3,4 The medication lowers blood glucose by inhibiting the sodium-glucose cotransport protein 2, thus lowering the blood glucose levels by increasing urinary excretion of glucose. Because many patients with type 2 diabetes mellitus are overweight, these medications are poised to gain popularity for weight loss and decreased blood pressure side effects. Three other medications in this class also have been approved by US Food and Drug Administration: empagliflozin, canagliflozin, and ertugliflozin.
Sweet syndrome consists of 4 cardinal features that were first described in 1964: fever, leukocytosis, tender red plaques, and a dermal neutrophilic infiltrate.5 Since then, Su and Liu6 proposed guidelines consisting of major and minor criteria. In 1996, Walker and Cohen7 suggested a set of diagnostic criteria specifically for drug-induced Sweet syndrome, including painful erythematous plaques, histopathologic neutrophilic infiltrate, and fever. Additional criteria included a temporal relationship between drug ingestion and clinical presentation as well as resolution of lesions after drug withdrawal or treatment with systemic corticosteroids.7 The lesions of drug-induced Sweet syndrome often are described as painful red papules that can form plaques, may appear vesicular, and are more common in women. These lesions classically appear on the upper extremities, as well as the head, neck, trunk, and back.8 Clinically, symptoms most commonly include fever and musculoskeletal involvement, both of which were experienced by the patient who described herself as feeling feverish when the lesions first appeared and reported malaise. Our patient experienced all of these features, and although a fever was not measured in the acute stage of presentation, she reported feeling hot. Other symptoms that may occur include arthralgia, headache, and myalgia.9 Microscopically, there is a nodular infiltrate of neutrophils, papillary dermal edema, and leukocytoclastic debris. The pathogenesis of Sweet syndrome remains unclear but can be associated with malignancy, pregnancy, autoimmune disorders, and drug reactions.10 Many different classes of medications have been reported to cause drug-induced Sweet syndrome and are listed in the Table.1,8,11 The recommended treatment of Sweet syndrome is systemic corticosteroids.12
The temporal use of dapagliflozin and appearance of the hand lesions, along with the histology, favored drug-induced Sweet syndrome to dapagliflozin as the cause of the plaques. Our patient did not seek medical attention at the onset of the initial chest and neck rash but did so several weeks after the painful hand lesions that were consistent with Sweet syndrome had appeared. Discontinuation of dapagliflozin and treatment with immunosuppressive medications resulted in resolution of the skin lesions on the hands. This scenario raises the question whether or not she would have developed the inflammatory hand lesions if she had stopped the medication earlier. Because dapagliflozin is a relatively new medication and boasts the potentially beneficial side effects of reducing body weight and blood pressure in addition to glucose control, we expect additional cases may occur, especially if use of this medication notably increases. Furthermore, this reaction may be a drug-class side effect and not one specific to dapagliflozin.
- Weedon D. The vasculopathic reaction pattern. Weedon’s Skin Pathology. 3rd ed. Oxford, UK: Churchill Livingstone; 2010:218-225.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and Sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Dapagliflozin (Farxiga) for type 2 diabetes. Med Lett Drugs Ther. 2014;56:13-15.
- Aylsworth A, Dean Z, VanNorman C, et al. Dapagliflozin for the treatment of type 2 diabetes mellitus [published online June 20, 2014]. Ann Pharmacother. 2014;48:1202-1208.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Su WP, Liu HN. Diagnostic criteria for Sweet’s syndrome. Cutis. 1986;37:167-174.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34(5 pt 2):918-923.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Cohen PR, Kurzrock R. Sweet’s syndrome. a neutrophilic dermatosis classically associated with acute onset and fever. Clin Dermatol. 2000;18:265-282.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- Thompson DF, Montarella KE. Drug-induced Sweet’s syndrome. Ann Pharmacother. 2007;41:802-811.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
- Weedon D. The vasculopathic reaction pattern. Weedon’s Skin Pathology. 3rd ed. Oxford, UK: Churchill Livingstone; 2010:218-225.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and Sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Dapagliflozin (Farxiga) for type 2 diabetes. Med Lett Drugs Ther. 2014;56:13-15.
- Aylsworth A, Dean Z, VanNorman C, et al. Dapagliflozin for the treatment of type 2 diabetes mellitus [published online June 20, 2014]. Ann Pharmacother. 2014;48:1202-1208.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Su WP, Liu HN. Diagnostic criteria for Sweet’s syndrome. Cutis. 1986;37:167-174.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34(5 pt 2):918-923.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Cohen PR, Kurzrock R. Sweet’s syndrome. a neutrophilic dermatosis classically associated with acute onset and fever. Clin Dermatol. 2000;18:265-282.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- Thompson DF, Montarella KE. Drug-induced Sweet’s syndrome. Ann Pharmacother. 2007;41:802-811.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778.
Practice Points
- Sweet syndrome consists of 4 cardinal features: fever, leukocytosis, tender red plaques, and a dermal neutrophilic infiltrate.
- In drug-induced Sweet syndrome, there is a temporal relationship between drug ingestion and clinical presentation as well as resolution of lesions after drug withdrawal or treatment with systemic corticosteroids.
- Microscopic findings of Sweet syndrome include a nodular infiltrate of neutrophils, papillary dermal edema, and leukocytoclastic debris.
- Dapagliflozin is a member of a new class of medications (gliflozins) used for treatment of type 2 diabetes mellitus, which may cause drug-induced Sweet syndrome.
Violaceous Patches on the Arm
The Diagnosis: Phacomatosis Cesioflammea
Phacomatosis pigmentovascularis (PPV) encompasses a group of diseases that have a vascular nevus coupled with a pigmented nevus.1 It is divided into 5 types: Type I is defined by the presence of a vascular malformation and epidermal nevus; type II by a vascular malformation and dermal melanosis with or without nevus anemicus; type III by a vascular malformation and nevus spilus with or without nevus anemicus; type IV by a vascular malformation, dermal melanosis, and nevus spilus with or without nevus anemicus; and type V as cutis marmorata telangiectatica congenita and dermal melanosis.1
Happle2 proposed a descriptive classification system in 2005 that eliminated type I PPV because neither linear epidermal nevus nor Becker nevus are derived from pigmentary cells. An appended "a" denotes a subtype with isolated cutaneous findings, while "b" is associated with extracutaneous manifestations. Phacomatosis cesioflammea (type IIa/b) refers to blue-hued dermal melanocytosis and nevus flammeus. Phacomatosis spilorosea (type IIIa/b) refers to nevus spilus and rose-colored nevus flammeus. Phacomatosis cesiomarmorata (type Va/b) refers to dermal melanocytosis and cutis marmorata telangiectasia congenita. The last group (type IVa/b) is unclassifiable phacomatosis pigmentovascularis.2,3
Phacomatosis pigmentovascularis can be isolated to the skin or have associated extracutaneous findings, including ocular melanocytosis, seizures, or cognitive delay due to intracerebral vascular malformations. Patients also can develop limb and soft-tissue overgrowth.4 Phacomatosis pigmentovascularis has been found to be associated with mutations in the GNA11 and GNAQ genes. The theory behind PPV is twin spotting, resulting from a somatic mutation that leads to mosaic proliferation of 2 different cell lines.5 Phacomatosis pigmentovascularis can occur in isolation or can demonstrate the phenotype of Sturge-Weber syndrome or Klippel-Trenaunay syndrome. In Sturge-Weber syndrome, capillary malformations involve the face and underlying leptomeninges and cerebral cortex. Glaucoma and epilepsy also may be present. In Klippel-Trenaunay syndrome, capillary malformations involve the extremities (usually the legs) in association with varicose veins, soft-tissue hypertrophy, and skeletal overgrowth.6-9 Tuberous sclerosis is an autosomal-dominant neurocutaneous disease in which patients develop hamartomas throughout the body, including the brain, skin, eyes, kidneys, heart, and lungs. Cutaneous manifestations include facial angiofibromas, ungual fibromas, hypomelanotic macules (ash leaf spots, confetti-like lesions), shagreen patches or connective tissue hamartomas, and fibrous plaques on the forehead. Tuberous sclerosis does not include vascular malformations.10
Our patient was diagnosed with PPV type IIb, or phacomatosis cesioflammea. He had a large port-wine stain involving the right upper arm, back (Figure, A), and chest (Figure, B) with involvement of the bilateral conjunctivae (Figure, C). Our case is unique because our patient did not have dermal melanocytosis, only ocular melanocytosis.
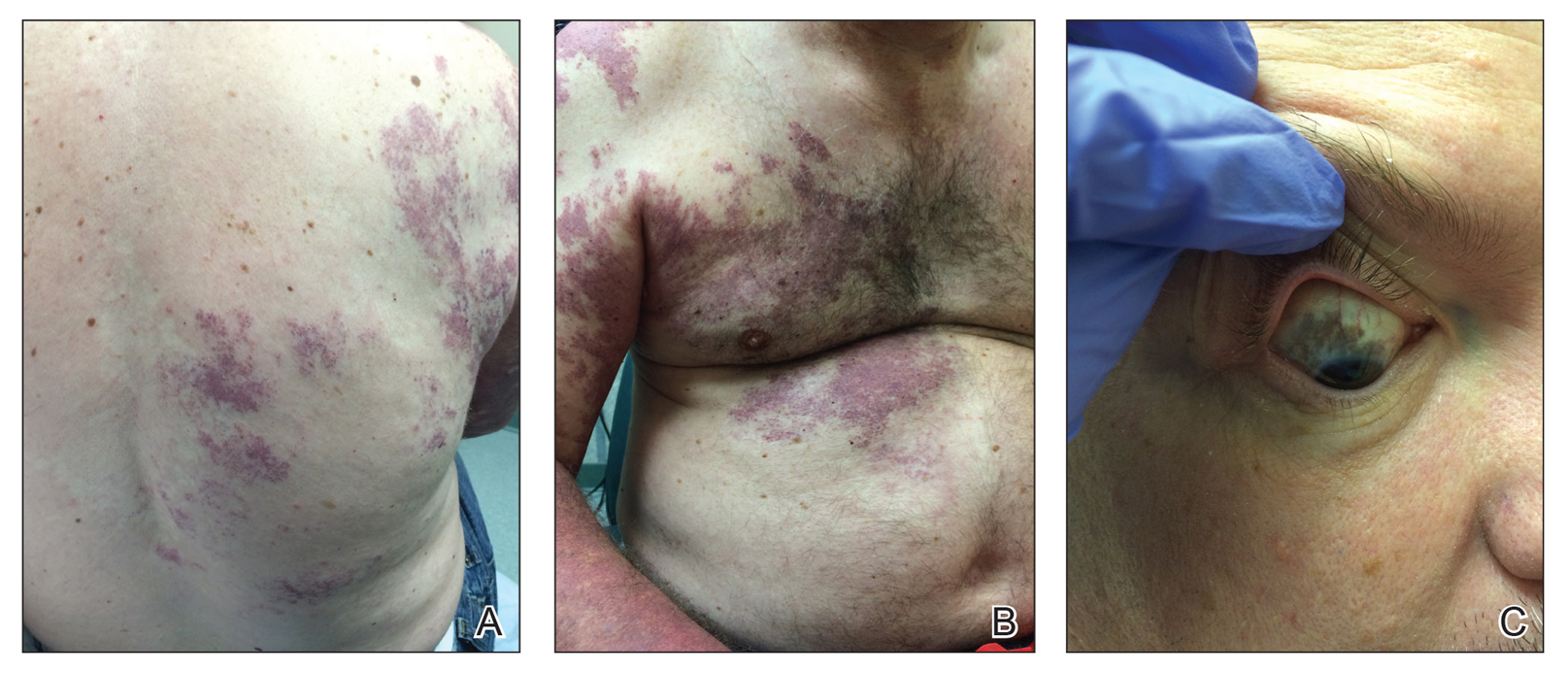
Once underlying neurologic and vascular anomalies have been ruled out, port-wine stains can be treated cosmetically. Pulsed dye laser is the gold standard therapy for capillary malformations, especially when instituted early. Follow-up with ophthalmology is advised to monitor ocular involvement. Shields et al11 suggested dilated fundoscopy for patients with port-wine stains because choroidal pigmentation often is the only ocular change seen. Ocular melanocytosis can progress to pigmented glaucoma or choroidal melanoma.
- Fernandez-Guarino M, Boixeda P, De las Heras E, et al. Phakomatosis pigmentovascularis: clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008;58:88-93.
- Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol. 2005;141:385-388.
- Villarreal DJ, Leal F. Phacomatosis pigmentovascularis of cesioflammea type. An Bras Dermatol. 2016;91(5 suppl 1):54-56.
- Thomas AC, Zeng Z, Riviere JB, et al. Mosaic activating mutations in GNA11 and GNAQ are associated with phakomatosis pigmentovascularis and extensive dermal melanocytosis. J Invest Dermatol. 2016;136:770-778.
- Krema H, Simpson R, McGowan H. Choroidal melanoma in phacomatosis pigmentovascularis cesioflammea. Can J Ophthalmol. 2013;48:E41-E42.
- Wu CY, Chen PH, Chen GS. Phacomatosis cesioflammea associated with pectus excavatum. Acta Derm Venereol. 2009;89:301-310.
- Pradhan S, Patnaik S, Padhi T, et al. Phakomatosis pigmentovascularis type IIb, Sturge-Weber syndrome and cone shaped tongue: an unusual association. Indian J Dermatol Venereol Leprol. 2015;81:614-616.
- Turk BG, Turkmen M, Tuna A, et al. Phakomatosis pigmentovascularis type IIb associated with Klippel-Trenaunay syndrome and congenital triangular alopecia. J Am Acad Dermatol. 2011;65:E46-E49.
- Sen S, Bala S, Halder C, et al. Phakomatosis pigmentovascularis presenting with Sturge-Weber syndrome and Klippel-Trenaunay syndrome. Indian J Dermatol. 2015;60:77-79.
- Schwartz RA, Fernandez G, Kotulska K, et al. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. 2007;57:189-202.
- Shields CL, Kligman BE, Suriano M, et al. Phacomatosis pigmentovascularis of cesioflammea type in 7 patients: combination of ocular pigmentation (melanocytosis or melanosis) and nevus flammeus with risk for melanoma. Arch Ophthalmol. 2011;129:746-750.
The Diagnosis: Phacomatosis Cesioflammea
Phacomatosis pigmentovascularis (PPV) encompasses a group of diseases that have a vascular nevus coupled with a pigmented nevus.1 It is divided into 5 types: Type I is defined by the presence of a vascular malformation and epidermal nevus; type II by a vascular malformation and dermal melanosis with or without nevus anemicus; type III by a vascular malformation and nevus spilus with or without nevus anemicus; type IV by a vascular malformation, dermal melanosis, and nevus spilus with or without nevus anemicus; and type V as cutis marmorata telangiectatica congenita and dermal melanosis.1
Happle2 proposed a descriptive classification system in 2005 that eliminated type I PPV because neither linear epidermal nevus nor Becker nevus are derived from pigmentary cells. An appended "a" denotes a subtype with isolated cutaneous findings, while "b" is associated with extracutaneous manifestations. Phacomatosis cesioflammea (type IIa/b) refers to blue-hued dermal melanocytosis and nevus flammeus. Phacomatosis spilorosea (type IIIa/b) refers to nevus spilus and rose-colored nevus flammeus. Phacomatosis cesiomarmorata (type Va/b) refers to dermal melanocytosis and cutis marmorata telangiectasia congenita. The last group (type IVa/b) is unclassifiable phacomatosis pigmentovascularis.2,3
Phacomatosis pigmentovascularis can be isolated to the skin or have associated extracutaneous findings, including ocular melanocytosis, seizures, or cognitive delay due to intracerebral vascular malformations. Patients also can develop limb and soft-tissue overgrowth.4 Phacomatosis pigmentovascularis has been found to be associated with mutations in the GNA11 and GNAQ genes. The theory behind PPV is twin spotting, resulting from a somatic mutation that leads to mosaic proliferation of 2 different cell lines.5 Phacomatosis pigmentovascularis can occur in isolation or can demonstrate the phenotype of Sturge-Weber syndrome or Klippel-Trenaunay syndrome. In Sturge-Weber syndrome, capillary malformations involve the face and underlying leptomeninges and cerebral cortex. Glaucoma and epilepsy also may be present. In Klippel-Trenaunay syndrome, capillary malformations involve the extremities (usually the legs) in association with varicose veins, soft-tissue hypertrophy, and skeletal overgrowth.6-9 Tuberous sclerosis is an autosomal-dominant neurocutaneous disease in which patients develop hamartomas throughout the body, including the brain, skin, eyes, kidneys, heart, and lungs. Cutaneous manifestations include facial angiofibromas, ungual fibromas, hypomelanotic macules (ash leaf spots, confetti-like lesions), shagreen patches or connective tissue hamartomas, and fibrous plaques on the forehead. Tuberous sclerosis does not include vascular malformations.10
Our patient was diagnosed with PPV type IIb, or phacomatosis cesioflammea. He had a large port-wine stain involving the right upper arm, back (Figure, A), and chest (Figure, B) with involvement of the bilateral conjunctivae (Figure, C). Our case is unique because our patient did not have dermal melanocytosis, only ocular melanocytosis.
Once underlying neurologic and vascular anomalies have been ruled out, port-wine stains can be treated cosmetically. Pulsed dye laser is the gold standard therapy for capillary malformations, especially when instituted early. Follow-up with ophthalmology is advised to monitor ocular involvement. Shields et al11 suggested dilated fundoscopy for patients with port-wine stains because choroidal pigmentation often is the only ocular change seen. Ocular melanocytosis can progress to pigmented glaucoma or choroidal melanoma.
The Diagnosis: Phacomatosis Cesioflammea
Phacomatosis pigmentovascularis (PPV) encompasses a group of diseases that have a vascular nevus coupled with a pigmented nevus.1 It is divided into 5 types: Type I is defined by the presence of a vascular malformation and epidermal nevus; type II by a vascular malformation and dermal melanosis with or without nevus anemicus; type III by a vascular malformation and nevus spilus with or without nevus anemicus; type IV by a vascular malformation, dermal melanosis, and nevus spilus with or without nevus anemicus; and type V as cutis marmorata telangiectatica congenita and dermal melanosis.1
Happle2 proposed a descriptive classification system in 2005 that eliminated type I PPV because neither linear epidermal nevus nor Becker nevus are derived from pigmentary cells. An appended "a" denotes a subtype with isolated cutaneous findings, while "b" is associated with extracutaneous manifestations. Phacomatosis cesioflammea (type IIa/b) refers to blue-hued dermal melanocytosis and nevus flammeus. Phacomatosis spilorosea (type IIIa/b) refers to nevus spilus and rose-colored nevus flammeus. Phacomatosis cesiomarmorata (type Va/b) refers to dermal melanocytosis and cutis marmorata telangiectasia congenita. The last group (type IVa/b) is unclassifiable phacomatosis pigmentovascularis.2,3
Phacomatosis pigmentovascularis can be isolated to the skin or have associated extracutaneous findings, including ocular melanocytosis, seizures, or cognitive delay due to intracerebral vascular malformations. Patients also can develop limb and soft-tissue overgrowth.4 Phacomatosis pigmentovascularis has been found to be associated with mutations in the GNA11 and GNAQ genes. The theory behind PPV is twin spotting, resulting from a somatic mutation that leads to mosaic proliferation of 2 different cell lines.5 Phacomatosis pigmentovascularis can occur in isolation or can demonstrate the phenotype of Sturge-Weber syndrome or Klippel-Trenaunay syndrome. In Sturge-Weber syndrome, capillary malformations involve the face and underlying leptomeninges and cerebral cortex. Glaucoma and epilepsy also may be present. In Klippel-Trenaunay syndrome, capillary malformations involve the extremities (usually the legs) in association with varicose veins, soft-tissue hypertrophy, and skeletal overgrowth.6-9 Tuberous sclerosis is an autosomal-dominant neurocutaneous disease in which patients develop hamartomas throughout the body, including the brain, skin, eyes, kidneys, heart, and lungs. Cutaneous manifestations include facial angiofibromas, ungual fibromas, hypomelanotic macules (ash leaf spots, confetti-like lesions), shagreen patches or connective tissue hamartomas, and fibrous plaques on the forehead. Tuberous sclerosis does not include vascular malformations.10
Our patient was diagnosed with PPV type IIb, or phacomatosis cesioflammea. He had a large port-wine stain involving the right upper arm, back (Figure, A), and chest (Figure, B) with involvement of the bilateral conjunctivae (Figure, C). Our case is unique because our patient did not have dermal melanocytosis, only ocular melanocytosis.
Once underlying neurologic and vascular anomalies have been ruled out, port-wine stains can be treated cosmetically. Pulsed dye laser is the gold standard therapy for capillary malformations, especially when instituted early. Follow-up with ophthalmology is advised to monitor ocular involvement. Shields et al11 suggested dilated fundoscopy for patients with port-wine stains because choroidal pigmentation often is the only ocular change seen. Ocular melanocytosis can progress to pigmented glaucoma or choroidal melanoma.
- Fernandez-Guarino M, Boixeda P, De las Heras E, et al. Phakomatosis pigmentovascularis: clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008;58:88-93.
- Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol. 2005;141:385-388.
- Villarreal DJ, Leal F. Phacomatosis pigmentovascularis of cesioflammea type. An Bras Dermatol. 2016;91(5 suppl 1):54-56.
- Thomas AC, Zeng Z, Riviere JB, et al. Mosaic activating mutations in GNA11 and GNAQ are associated with phakomatosis pigmentovascularis and extensive dermal melanocytosis. J Invest Dermatol. 2016;136:770-778.
- Krema H, Simpson R, McGowan H. Choroidal melanoma in phacomatosis pigmentovascularis cesioflammea. Can J Ophthalmol. 2013;48:E41-E42.
- Wu CY, Chen PH, Chen GS. Phacomatosis cesioflammea associated with pectus excavatum. Acta Derm Venereol. 2009;89:301-310.
- Pradhan S, Patnaik S, Padhi T, et al. Phakomatosis pigmentovascularis type IIb, Sturge-Weber syndrome and cone shaped tongue: an unusual association. Indian J Dermatol Venereol Leprol. 2015;81:614-616.
- Turk BG, Turkmen M, Tuna A, et al. Phakomatosis pigmentovascularis type IIb associated with Klippel-Trenaunay syndrome and congenital triangular alopecia. J Am Acad Dermatol. 2011;65:E46-E49.
- Sen S, Bala S, Halder C, et al. Phakomatosis pigmentovascularis presenting with Sturge-Weber syndrome and Klippel-Trenaunay syndrome. Indian J Dermatol. 2015;60:77-79.
- Schwartz RA, Fernandez G, Kotulska K, et al. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. 2007;57:189-202.
- Shields CL, Kligman BE, Suriano M, et al. Phacomatosis pigmentovascularis of cesioflammea type in 7 patients: combination of ocular pigmentation (melanocytosis or melanosis) and nevus flammeus with risk for melanoma. Arch Ophthalmol. 2011;129:746-750.
- Fernandez-Guarino M, Boixeda P, De las Heras E, et al. Phakomatosis pigmentovascularis: clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008;58:88-93.
- Happle R. Phacomatosis pigmentovascularis revisited and reclassified. Arch Dermatol. 2005;141:385-388.
- Villarreal DJ, Leal F. Phacomatosis pigmentovascularis of cesioflammea type. An Bras Dermatol. 2016;91(5 suppl 1):54-56.
- Thomas AC, Zeng Z, Riviere JB, et al. Mosaic activating mutations in GNA11 and GNAQ are associated with phakomatosis pigmentovascularis and extensive dermal melanocytosis. J Invest Dermatol. 2016;136:770-778.
- Krema H, Simpson R, McGowan H. Choroidal melanoma in phacomatosis pigmentovascularis cesioflammea. Can J Ophthalmol. 2013;48:E41-E42.
- Wu CY, Chen PH, Chen GS. Phacomatosis cesioflammea associated with pectus excavatum. Acta Derm Venereol. 2009;89:301-310.
- Pradhan S, Patnaik S, Padhi T, et al. Phakomatosis pigmentovascularis type IIb, Sturge-Weber syndrome and cone shaped tongue: an unusual association. Indian J Dermatol Venereol Leprol. 2015;81:614-616.
- Turk BG, Turkmen M, Tuna A, et al. Phakomatosis pigmentovascularis type IIb associated with Klippel-Trenaunay syndrome and congenital triangular alopecia. J Am Acad Dermatol. 2011;65:E46-E49.
- Sen S, Bala S, Halder C, et al. Phakomatosis pigmentovascularis presenting with Sturge-Weber syndrome and Klippel-Trenaunay syndrome. Indian J Dermatol. 2015;60:77-79.
- Schwartz RA, Fernandez G, Kotulska K, et al. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. 2007;57:189-202.
- Shields CL, Kligman BE, Suriano M, et al. Phacomatosis pigmentovascularis of cesioflammea type in 7 patients: combination of ocular pigmentation (melanocytosis or melanosis) and nevus flammeus with risk for melanoma. Arch Ophthalmol. 2011;129:746-750.
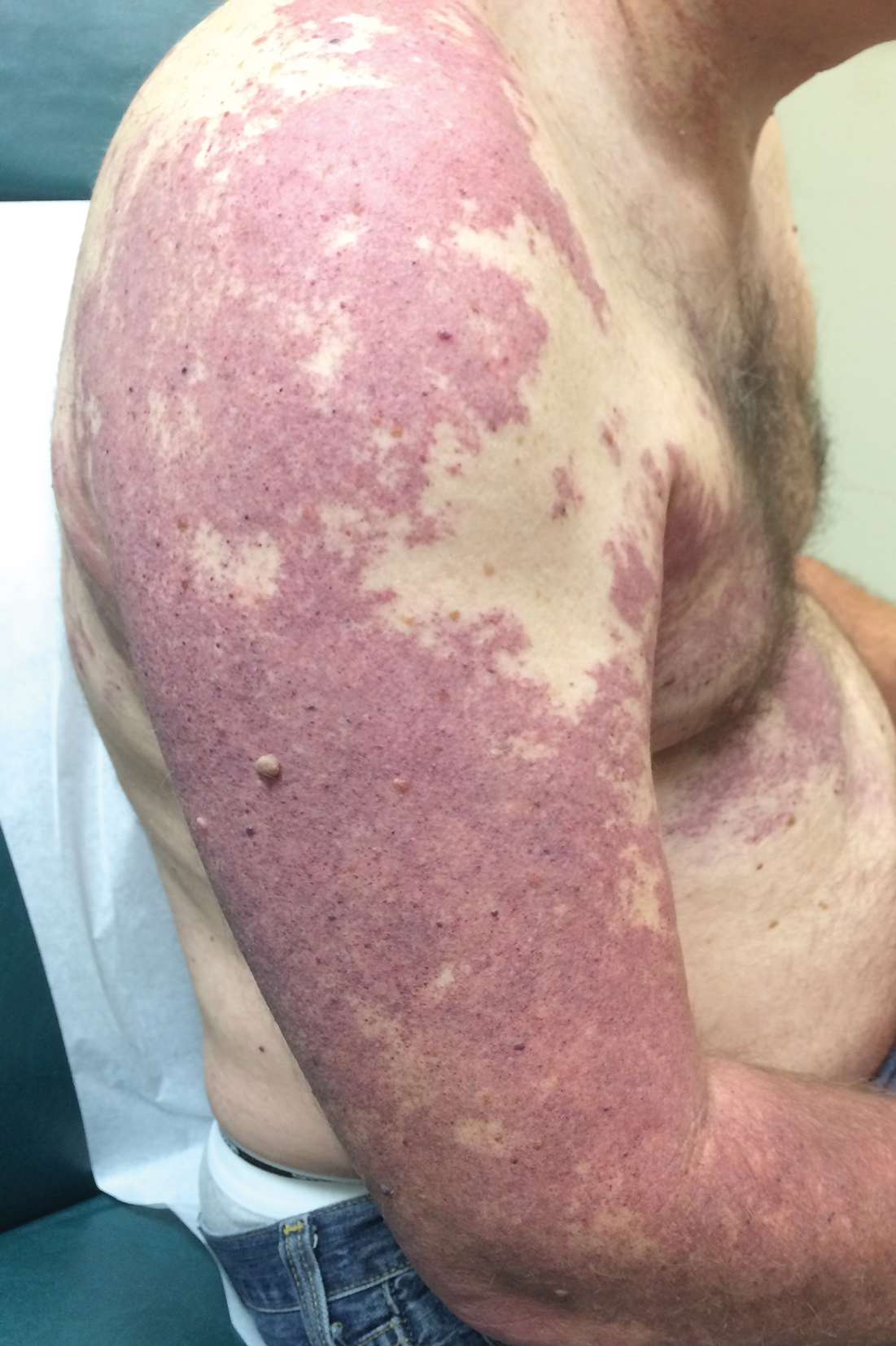
A 55-year-old man presented with red-violet patches on the right arm and chest that had been present since birth. The patches were asymptomatic and stable in size and shape. He denied any personal or family history of glaucoma or epilepsy. Physical examination demonstrated nonblanchable, violaceous to red patches on the right arm, back, and chest. No thrills or bruits were appreciable, and the right and left arms were of equal circumference and length. Further examination revealed hyperpigmented patches on the bilateral conjunctivae.
‘Pot’ is still hot for Dravet, Lennox-Gastaut
BANGKOK – Interim results of long-term, open-label extension trials of add-on prescription cannabidiol in patients with Dravet syndrome or Lennox-Gastaut syndrome show sustained, clinically meaningful seizure reductions with no new safety concerns, Anup D. Patel, MD, reported at the International Epilepsy Congress.

“Overall, this is a very promising and sustainable result that we were happy to see,” said Dr. Patel, chief of child neurology at Nationwide Children’s Hospital in Columbus, Ohio.
Epidiolex is the brand name for the plant-derived, highly purified cannabidiol (CBD) in an oil-based oral solution at 100 mg/mL. Dr. Patel has been involved in the medication’s development program since the earliest open-label compassionate use study, which was followed by rigorous phase 3, double-blind, placebo-controlled randomized trials, eventually leading to Food and Drug Administration marketing approval for the treatment of Dravet syndrome and Lennox-Gastaut syndrome in patients 2 years of age or older.
“On June 25th, 2018, history was made: for the first time in United States history, a plant-based derivative of marijuana was approved for use as a medication, and it was also the first FDA-approved treatment for Dravet syndrome,” Dr. Patel noted at the congress sponsored by the International League Against Epilepsy.
A total of 96% of the 289 children with Dravet syndrome who completed the 14-week, double-blind, controlled randomized trials enrolled in the open-label, long-term extension study, during which they were on a median of three concurrent antiepileptic drugs along with a mean modal dose of CBD at 22 mg/kg/day. Although the target maintenance dose of CBD was 20 mg/kg/day, as advised in the product labeling, physicians could reduce or increase the dose up to 30 mg/kg/day.
“In the initial compassionate-use study, our site could go up to 50 mg/kg/day,” according to Dr. Patel. “We have plenty of data showing efficacy and continued safety beyond the FDA-recommended dose.”
In the open-label extension study, the median reduction from baseline in monthly seizure frequency assessed in 12-week intervals up to a maximum of week 72 was 44%-57% for convulsive seizures and 49%-67% for total seizures. More than 80% of patients and/or caregivers reported improvement in the patient’s overall condition as assessed on the Subject/Caregiver Global Impression of Change scale.
The pattern of adverse events associated with CBD has been consistent across all of the studies. The most common side effects are diarrhea in about one-third of patients, sleepiness in one-quarter, and decreased appetite in about one-quarter. Seven percent of patients discontinued the long-term extension trial because of adverse events.
Seventy percent of patients remained in the long-term extension study at 1 year.
Twenty-six patients developed liver transaminase levels greater than three times the upper limit of normal, and of note, 23 of the 26 were on concomitant valproic acid. None met criteria for severe drug-induced liver injury, and all recovered either spontaneously or after a reduction in the dose of CBD or valproic acid. But this association between CBD, valproic acid, and increased risk of mild liver injury has been a consistent finding across the clinical trials program.
“This is a very important clinical pearl to take away,” commented Dr. Patel, who is also a pediatric neurologist at Ohio State University.
The interim results of the long-term, open-label extension study of add-on CBD in patients with Lennox-Gastaut syndrome are similar to the Dravet syndrome study. Overall, 99% of the 368 patients with Lennox-Gastaut syndrome who completed the 14-week, double-blind, randomized trials signed up for the open-label extension. During a median follow-up of 61 weeks, the median percent reduction in seizure frequency as assessed in serial 12-week windows was 48%-70% for drop seizures and 48%-63% for total seizures. Twenty-four percent of patients withdrew from the study. Eighty-eight percent of patients or caregivers reported an improvement in overall condition when assessed at weeks 24 and 48. Forty-seven patients developed elevated transaminase levels – typically within the first 2 months on CBD – and 35 of them were on concomitant valproic acid.
More on drug-drug interactions
Elsewhere at IEC 2019, Gilmour Morrison of GW Pharmaceuticals, the Cambridge, England, company that markets Epidiolex, presented the findings of a series of drug-drug interaction studies involving coadministration of their CBD with clobazam (Sympazan and Onfi), valproate, stiripentol (Diacomit), or midazolam (Versed) in adult epilepsy patients and healthy volunteers. The researchers reported a bidirectional drug-drug interaction between Epidiolex and clobazam resulting in increased levels of the active metabolites of both drugs. The mechanism is believed to involve inhibition of cytochrome P450 2C19. However, there were no interactions with midazolam or valproate, and the slight bump in stiripentol levels when given with CBD didn’t reach the level of a clinically meaningful drug-drug interaction, according to the investigators.
On the horizon, Canadian researchers are investigating the possibility that since both the tetrahydrocannabinol (THC) and CBD components of marijuana have been shown to have anticonvulsant effects, adding a bit of THC to CBD will result in even better seizure control than with pure CBD in patients with Dravet syndrome. Investigators at Toronto’s Hospital for Sick Children have conducted a prospective, open-label study of a product containing CBD and THC in a 50:1 ratio as add-on therapy in 20 children with Dravet syndrome. The dose was 2-16 mg/kg/day of CBD and 0.04-0.32 mg/kg/day of THC. The cannabis plant extract used in the study was produced by Tilray, a Canadian pharmaceutical company.
Nineteen of the 20 patients completed the 20-week study. The sole noncompleter died of SUDEP (sudden unexpected death in epilepsy) deemed treatment unrelated. Patients experienced a median 71% reduction in motor seizures, compared with baseline. Sixty-three percent of patients had at least a 50% reduction in seizure frequency. Elevated liver transaminases occurred in patients on concomitant valproic acid, as did platelet abnormalities, which have not been seen in the Epidiolex studies, noted Dr. Patel, who was not involved in the Canadian study (Ann Clin Transl Neurol. 2018 Aug 1;5[9]:1077-88).
Dr. Patel reported serving as a consultant to Greenwich Biosciences, a U.S. offshoot of GW Pharmaceuticals. He receives research grants from that company as well as from the National Institutes of Health and the Pediatric Epilepsy Research Foundation.
BANGKOK – Interim results of long-term, open-label extension trials of add-on prescription cannabidiol in patients with Dravet syndrome or Lennox-Gastaut syndrome show sustained, clinically meaningful seizure reductions with no new safety concerns, Anup D. Patel, MD, reported at the International Epilepsy Congress.
“Overall, this is a very promising and sustainable result that we were happy to see,” said Dr. Patel, chief of child neurology at Nationwide Children’s Hospital in Columbus, Ohio.
Epidiolex is the brand name for the plant-derived, highly purified cannabidiol (CBD) in an oil-based oral solution at 100 mg/mL. Dr. Patel has been involved in the medication’s development program since the earliest open-label compassionate use study, which was followed by rigorous phase 3, double-blind, placebo-controlled randomized trials, eventually leading to Food and Drug Administration marketing approval for the treatment of Dravet syndrome and Lennox-Gastaut syndrome in patients 2 years of age or older.
“On June 25th, 2018, history was made: for the first time in United States history, a plant-based derivative of marijuana was approved for use as a medication, and it was also the first FDA-approved treatment for Dravet syndrome,” Dr. Patel noted at the congress sponsored by the International League Against Epilepsy.
A total of 96% of the 289 children with Dravet syndrome who completed the 14-week, double-blind, controlled randomized trials enrolled in the open-label, long-term extension study, during which they were on a median of three concurrent antiepileptic drugs along with a mean modal dose of CBD at 22 mg/kg/day. Although the target maintenance dose of CBD was 20 mg/kg/day, as advised in the product labeling, physicians could reduce or increase the dose up to 30 mg/kg/day.
“In the initial compassionate-use study, our site could go up to 50 mg/kg/day,” according to Dr. Patel. “We have plenty of data showing efficacy and continued safety beyond the FDA-recommended dose.”
In the open-label extension study, the median reduction from baseline in monthly seizure frequency assessed in 12-week intervals up to a maximum of week 72 was 44%-57% for convulsive seizures and 49%-67% for total seizures. More than 80% of patients and/or caregivers reported improvement in the patient’s overall condition as assessed on the Subject/Caregiver Global Impression of Change scale.
The pattern of adverse events associated with CBD has been consistent across all of the studies. The most common side effects are diarrhea in about one-third of patients, sleepiness in one-quarter, and decreased appetite in about one-quarter. Seven percent of patients discontinued the long-term extension trial because of adverse events.
Seventy percent of patients remained in the long-term extension study at 1 year.
Twenty-six patients developed liver transaminase levels greater than three times the upper limit of normal, and of note, 23 of the 26 were on concomitant valproic acid. None met criteria for severe drug-induced liver injury, and all recovered either spontaneously or after a reduction in the dose of CBD or valproic acid. But this association between CBD, valproic acid, and increased risk of mild liver injury has been a consistent finding across the clinical trials program.
“This is a very important clinical pearl to take away,” commented Dr. Patel, who is also a pediatric neurologist at Ohio State University.
The interim results of the long-term, open-label extension study of add-on CBD in patients with Lennox-Gastaut syndrome are similar to the Dravet syndrome study. Overall, 99% of the 368 patients with Lennox-Gastaut syndrome who completed the 14-week, double-blind, randomized trials signed up for the open-label extension. During a median follow-up of 61 weeks, the median percent reduction in seizure frequency as assessed in serial 12-week windows was 48%-70% for drop seizures and 48%-63% for total seizures. Twenty-four percent of patients withdrew from the study. Eighty-eight percent of patients or caregivers reported an improvement in overall condition when assessed at weeks 24 and 48. Forty-seven patients developed elevated transaminase levels – typically within the first 2 months on CBD – and 35 of them were on concomitant valproic acid.
More on drug-drug interactions
Elsewhere at IEC 2019, Gilmour Morrison of GW Pharmaceuticals, the Cambridge, England, company that markets Epidiolex, presented the findings of a series of drug-drug interaction studies involving coadministration of their CBD with clobazam (Sympazan and Onfi), valproate, stiripentol (Diacomit), or midazolam (Versed) in adult epilepsy patients and healthy volunteers. The researchers reported a bidirectional drug-drug interaction between Epidiolex and clobazam resulting in increased levels of the active metabolites of both drugs. The mechanism is believed to involve inhibition of cytochrome P450 2C19. However, there were no interactions with midazolam or valproate, and the slight bump in stiripentol levels when given with CBD didn’t reach the level of a clinically meaningful drug-drug interaction, according to the investigators.
On the horizon, Canadian researchers are investigating the possibility that since both the tetrahydrocannabinol (THC) and CBD components of marijuana have been shown to have anticonvulsant effects, adding a bit of THC to CBD will result in even better seizure control than with pure CBD in patients with Dravet syndrome. Investigators at Toronto’s Hospital for Sick Children have conducted a prospective, open-label study of a product containing CBD and THC in a 50:1 ratio as add-on therapy in 20 children with Dravet syndrome. The dose was 2-16 mg/kg/day of CBD and 0.04-0.32 mg/kg/day of THC. The cannabis plant extract used in the study was produced by Tilray, a Canadian pharmaceutical company.
Nineteen of the 20 patients completed the 20-week study. The sole noncompleter died of SUDEP (sudden unexpected death in epilepsy) deemed treatment unrelated. Patients experienced a median 71% reduction in motor seizures, compared with baseline. Sixty-three percent of patients had at least a 50% reduction in seizure frequency. Elevated liver transaminases occurred in patients on concomitant valproic acid, as did platelet abnormalities, which have not been seen in the Epidiolex studies, noted Dr. Patel, who was not involved in the Canadian study (Ann Clin Transl Neurol. 2018 Aug 1;5[9]:1077-88).
Dr. Patel reported serving as a consultant to Greenwich Biosciences, a U.S. offshoot of GW Pharmaceuticals. He receives research grants from that company as well as from the National Institutes of Health and the Pediatric Epilepsy Research Foundation.
BANGKOK – Interim results of long-term, open-label extension trials of add-on prescription cannabidiol in patients with Dravet syndrome or Lennox-Gastaut syndrome show sustained, clinically meaningful seizure reductions with no new safety concerns, Anup D. Patel, MD, reported at the International Epilepsy Congress.
“Overall, this is a very promising and sustainable result that we were happy to see,” said Dr. Patel, chief of child neurology at Nationwide Children’s Hospital in Columbus, Ohio.
Epidiolex is the brand name for the plant-derived, highly purified cannabidiol (CBD) in an oil-based oral solution at 100 mg/mL. Dr. Patel has been involved in the medication’s development program since the earliest open-label compassionate use study, which was followed by rigorous phase 3, double-blind, placebo-controlled randomized trials, eventually leading to Food and Drug Administration marketing approval for the treatment of Dravet syndrome and Lennox-Gastaut syndrome in patients 2 years of age or older.
“On June 25th, 2018, history was made: for the first time in United States history, a plant-based derivative of marijuana was approved for use as a medication, and it was also the first FDA-approved treatment for Dravet syndrome,” Dr. Patel noted at the congress sponsored by the International League Against Epilepsy.
A total of 96% of the 289 children with Dravet syndrome who completed the 14-week, double-blind, controlled randomized trials enrolled in the open-label, long-term extension study, during which they were on a median of three concurrent antiepileptic drugs along with a mean modal dose of CBD at 22 mg/kg/day. Although the target maintenance dose of CBD was 20 mg/kg/day, as advised in the product labeling, physicians could reduce or increase the dose up to 30 mg/kg/day.
“In the initial compassionate-use study, our site could go up to 50 mg/kg/day,” according to Dr. Patel. “We have plenty of data showing efficacy and continued safety beyond the FDA-recommended dose.”
In the open-label extension study, the median reduction from baseline in monthly seizure frequency assessed in 12-week intervals up to a maximum of week 72 was 44%-57% for convulsive seizures and 49%-67% for total seizures. More than 80% of patients and/or caregivers reported improvement in the patient’s overall condition as assessed on the Subject/Caregiver Global Impression of Change scale.
The pattern of adverse events associated with CBD has been consistent across all of the studies. The most common side effects are diarrhea in about one-third of patients, sleepiness in one-quarter, and decreased appetite in about one-quarter. Seven percent of patients discontinued the long-term extension trial because of adverse events.
Seventy percent of patients remained in the long-term extension study at 1 year.
Twenty-six patients developed liver transaminase levels greater than three times the upper limit of normal, and of note, 23 of the 26 were on concomitant valproic acid. None met criteria for severe drug-induced liver injury, and all recovered either spontaneously or after a reduction in the dose of CBD or valproic acid. But this association between CBD, valproic acid, and increased risk of mild liver injury has been a consistent finding across the clinical trials program.
“This is a very important clinical pearl to take away,” commented Dr. Patel, who is also a pediatric neurologist at Ohio State University.
The interim results of the long-term, open-label extension study of add-on CBD in patients with Lennox-Gastaut syndrome are similar to the Dravet syndrome study. Overall, 99% of the 368 patients with Lennox-Gastaut syndrome who completed the 14-week, double-blind, randomized trials signed up for the open-label extension. During a median follow-up of 61 weeks, the median percent reduction in seizure frequency as assessed in serial 12-week windows was 48%-70% for drop seizures and 48%-63% for total seizures. Twenty-four percent of patients withdrew from the study. Eighty-eight percent of patients or caregivers reported an improvement in overall condition when assessed at weeks 24 and 48. Forty-seven patients developed elevated transaminase levels – typically within the first 2 months on CBD – and 35 of them were on concomitant valproic acid.
More on drug-drug interactions
Elsewhere at IEC 2019, Gilmour Morrison of GW Pharmaceuticals, the Cambridge, England, company that markets Epidiolex, presented the findings of a series of drug-drug interaction studies involving coadministration of their CBD with clobazam (Sympazan and Onfi), valproate, stiripentol (Diacomit), or midazolam (Versed) in adult epilepsy patients and healthy volunteers. The researchers reported a bidirectional drug-drug interaction between Epidiolex and clobazam resulting in increased levels of the active metabolites of both drugs. The mechanism is believed to involve inhibition of cytochrome P450 2C19. However, there were no interactions with midazolam or valproate, and the slight bump in stiripentol levels when given with CBD didn’t reach the level of a clinically meaningful drug-drug interaction, according to the investigators.
On the horizon, Canadian researchers are investigating the possibility that since both the tetrahydrocannabinol (THC) and CBD components of marijuana have been shown to have anticonvulsant effects, adding a bit of THC to CBD will result in even better seizure control than with pure CBD in patients with Dravet syndrome. Investigators at Toronto’s Hospital for Sick Children have conducted a prospective, open-label study of a product containing CBD and THC in a 50:1 ratio as add-on therapy in 20 children with Dravet syndrome. The dose was 2-16 mg/kg/day of CBD and 0.04-0.32 mg/kg/day of THC. The cannabis plant extract used in the study was produced by Tilray, a Canadian pharmaceutical company.
Nineteen of the 20 patients completed the 20-week study. The sole noncompleter died of SUDEP (sudden unexpected death in epilepsy) deemed treatment unrelated. Patients experienced a median 71% reduction in motor seizures, compared with baseline. Sixty-three percent of patients had at least a 50% reduction in seizure frequency. Elevated liver transaminases occurred in patients on concomitant valproic acid, as did platelet abnormalities, which have not been seen in the Epidiolex studies, noted Dr. Patel, who was not involved in the Canadian study (Ann Clin Transl Neurol. 2018 Aug 1;5[9]:1077-88).
Dr. Patel reported serving as a consultant to Greenwich Biosciences, a U.S. offshoot of GW Pharmaceuticals. He receives research grants from that company as well as from the National Institutes of Health and the Pediatric Epilepsy Research Foundation.
REPORTING FROM IEC 2019
Possible role of enterovirus infection in acute flaccid myelitis cases detected
High levels of enterovirus (EV) peptides were found in the cerebrospinal fluid (CSF) and serum samples of individuals with acute flaccid myelitis (AFM) that were not present in a variety of control individuals, according to the results of a small study of patients with and without AFM published online in mBio.
In 2018, CSF samples from AFM patients were investigated by viral-capture high-throughput sequencing. These CSF and serum samples, as well as those from multiple controls, were tested for antibodies to human EVs using peptide microarrays, according to Nischay Mishra, PhD, of Columbia University, New York, and colleagues.
Although EV RNA was confirmed in CSF from only 1 adult AFM case and 1 non-AFM case, antibodies to EV peptides were present in 11 of 14 AFM patients (79%), which was a significantly higher rate than in control groups, including non-AFM patients (1 of 5, or 20%), children with Kawasaki disease (0 of 10), and adults with non-AFM CNS diseases (2 of 11, 18%), according to the authors.
In addition, 6 of 14 (43%) CSF samples and 8 of 11 (73%) serum samples from AFM patients were immunoreactive to an EV-D68–specific peptide, whereas samples from the three control groups were not immunoreactive in either CSF or sera. Previous studies have suggested that infection with EV-D68 and EV-A71 may contribute to AFM.
“There have been 570 confirmed cases since CDC began tracking AFM in August 2014. AFM outbreaks were reported to the CDC in 2014, 2016, and 2018. AFM affects the spinal cord and is characterized by the sudden onset of muscle weakness in one or more limbs. Spikes in AFM cases, primarily in children, have coincided in time and location with outbreaks of EV-D68 and a related enterovirus, EV-A71,” according to an NIH media advisory discussing the article.
In particular, as the study authors point out, a potential link to EV-D68 has also been based on the presence of viral RNA in some respiratory and stool specimens and the observation that EV-D68 infection can result in spinal cord infection.
“While other etiologies of AFM continue to be investigated, our study provides further evidence that EV infection may be a factor in AFM. In the absence of direct detection of a pathogen, antibody evidence of pathogen exposure within the CNS can be an important indicator of the underlying cause of disease,” Dr. Mishra and his colleagues added.
“These initial results may provide avenues to further explore how exposure to EV may contribute to AFM as well as the development of diagnostic tools and treatments,” the researchers concluded.
The study was funded by the National Institutes of Health. The authors reported that they had no competing financial interests.
SOURCE: Mishra N et al. mBio. 2019 Aug;10(4):e01903-19.
High levels of enterovirus (EV) peptides were found in the cerebrospinal fluid (CSF) and serum samples of individuals with acute flaccid myelitis (AFM) that were not present in a variety of control individuals, according to the results of a small study of patients with and without AFM published online in mBio.
In 2018, CSF samples from AFM patients were investigated by viral-capture high-throughput sequencing. These CSF and serum samples, as well as those from multiple controls, were tested for antibodies to human EVs using peptide microarrays, according to Nischay Mishra, PhD, of Columbia University, New York, and colleagues.
Although EV RNA was confirmed in CSF from only 1 adult AFM case and 1 non-AFM case, antibodies to EV peptides were present in 11 of 14 AFM patients (79%), which was a significantly higher rate than in control groups, including non-AFM patients (1 of 5, or 20%), children with Kawasaki disease (0 of 10), and adults with non-AFM CNS diseases (2 of 11, 18%), according to the authors.
In addition, 6 of 14 (43%) CSF samples and 8 of 11 (73%) serum samples from AFM patients were immunoreactive to an EV-D68–specific peptide, whereas samples from the three control groups were not immunoreactive in either CSF or sera. Previous studies have suggested that infection with EV-D68 and EV-A71 may contribute to AFM.
“There have been 570 confirmed cases since CDC began tracking AFM in August 2014. AFM outbreaks were reported to the CDC in 2014, 2016, and 2018. AFM affects the spinal cord and is characterized by the sudden onset of muscle weakness in one or more limbs. Spikes in AFM cases, primarily in children, have coincided in time and location with outbreaks of EV-D68 and a related enterovirus, EV-A71,” according to an NIH media advisory discussing the article.
In particular, as the study authors point out, a potential link to EV-D68 has also been based on the presence of viral RNA in some respiratory and stool specimens and the observation that EV-D68 infection can result in spinal cord infection.
“While other etiologies of AFM continue to be investigated, our study provides further evidence that EV infection may be a factor in AFM. In the absence of direct detection of a pathogen, antibody evidence of pathogen exposure within the CNS can be an important indicator of the underlying cause of disease,” Dr. Mishra and his colleagues added.
“These initial results may provide avenues to further explore how exposure to EV may contribute to AFM as well as the development of diagnostic tools and treatments,” the researchers concluded.
The study was funded by the National Institutes of Health. The authors reported that they had no competing financial interests.
SOURCE: Mishra N et al. mBio. 2019 Aug;10(4):e01903-19.
High levels of enterovirus (EV) peptides were found in the cerebrospinal fluid (CSF) and serum samples of individuals with acute flaccid myelitis (AFM) that were not present in a variety of control individuals, according to the results of a small study of patients with and without AFM published online in mBio.
In 2018, CSF samples from AFM patients were investigated by viral-capture high-throughput sequencing. These CSF and serum samples, as well as those from multiple controls, were tested for antibodies to human EVs using peptide microarrays, according to Nischay Mishra, PhD, of Columbia University, New York, and colleagues.
Although EV RNA was confirmed in CSF from only 1 adult AFM case and 1 non-AFM case, antibodies to EV peptides were present in 11 of 14 AFM patients (79%), which was a significantly higher rate than in control groups, including non-AFM patients (1 of 5, or 20%), children with Kawasaki disease (0 of 10), and adults with non-AFM CNS diseases (2 of 11, 18%), according to the authors.
In addition, 6 of 14 (43%) CSF samples and 8 of 11 (73%) serum samples from AFM patients were immunoreactive to an EV-D68–specific peptide, whereas samples from the three control groups were not immunoreactive in either CSF or sera. Previous studies have suggested that infection with EV-D68 and EV-A71 may contribute to AFM.
“There have been 570 confirmed cases since CDC began tracking AFM in August 2014. AFM outbreaks were reported to the CDC in 2014, 2016, and 2018. AFM affects the spinal cord and is characterized by the sudden onset of muscle weakness in one or more limbs. Spikes in AFM cases, primarily in children, have coincided in time and location with outbreaks of EV-D68 and a related enterovirus, EV-A71,” according to an NIH media advisory discussing the article.
In particular, as the study authors point out, a potential link to EV-D68 has also been based on the presence of viral RNA in some respiratory and stool specimens and the observation that EV-D68 infection can result in spinal cord infection.
“While other etiologies of AFM continue to be investigated, our study provides further evidence that EV infection may be a factor in AFM. In the absence of direct detection of a pathogen, antibody evidence of pathogen exposure within the CNS can be an important indicator of the underlying cause of disease,” Dr. Mishra and his colleagues added.
“These initial results may provide avenues to further explore how exposure to EV may contribute to AFM as well as the development of diagnostic tools and treatments,” the researchers concluded.
The study was funded by the National Institutes of Health. The authors reported that they had no competing financial interests.
SOURCE: Mishra N et al. mBio. 2019 Aug;10(4):e01903-19.
FROM MBIO
Key clinical point:
Major finding: EV peptide antibodies were present in 11 of 14 AFM patients (79%), significantly higher than in controls.
Study details: A peptide microarray analysis was performed on CSF and sera from 14 AFM patients, as well as three control groups of 5 pediatric and adult patients with a non-AFM CNS diseases, 10 children with Kawasaki disease, and 10 adult patients with non-AFM CNS diseases.
Disclosures: The study was funded by the National Institutes of Health. The authors reported that they had no conflicts.
Source: Mishra N et al. mBio. 2019 Aug;10(4):e01903-19.
Favorable Ebola results lead to drug trial termination, new focus
An investigational agent known as REGN-EB3 has met an early stopping criterion in the protocol of an Ebola therapeutics trial, according to a National Institutes of Health media advisory.
Preliminary results in 499 study participants showed that individuals receiving either of two treatments, REGN-EB3 or mAb114, had a greater chance of survival, compared with participants in the other two study arms.
The randomized, controlled Pamoja Tulinde Maisha (PALM) study, which began Nov. 20, 2018, was designed to evaluate four investigational agents (ZMapp, remdesivir, mAb114, and REGN-EB3) for the treatment of patients with Ebola virus disease in the Democratic Republic of the Congo (DRC) as part of the emergency response to an ongoing outbreak in the North Kivu and Ituri provinces.
As of Aug. 9, 2019, the trial had enrolled 681 patients at four Ebola treatment centers in live outbreak regions of the DRC, with the goal of enrolling 725 patients in total.
The trial investigators and study cosponsors accepted the recommendation for early termination, and staff at the trial sites in the DRC were promptly informed, according to the media advisory. Additional patient randomizations in the now-revised trial will be limited to treatment either with REGN-EB3 or mAb114. Patients randomized to the ZMapp or remdesivir arms in the last 10 days of the original trial will be given the option, at the discretion of their treating physician, to receive either of the two more effective treatments, according to the NIH.
“While the final analysis of the data can occur only after all the data are generated and collected (likely late September/early October 2019), the DSMB [Data and Safety Monitoring Board] and the study leadership felt the preliminary analysis of the existing data was compelling enough to recommend and implement these changes in the trial immediately. The complete results will be submitted for publication in the peer-reviewed medical literature as soon as possible,” the NIH stated.
The study is cosponsored and funded by the NIH, carried out by an international research consortium coordinated by the World Health Organization, and supported by four pharmaceutical companies (MappBio, Gilead, Regeneron, and Ridgeback Biotherapeutics).
An investigational agent known as REGN-EB3 has met an early stopping criterion in the protocol of an Ebola therapeutics trial, according to a National Institutes of Health media advisory.
Preliminary results in 499 study participants showed that individuals receiving either of two treatments, REGN-EB3 or mAb114, had a greater chance of survival, compared with participants in the other two study arms.
The randomized, controlled Pamoja Tulinde Maisha (PALM) study, which began Nov. 20, 2018, was designed to evaluate four investigational agents (ZMapp, remdesivir, mAb114, and REGN-EB3) for the treatment of patients with Ebola virus disease in the Democratic Republic of the Congo (DRC) as part of the emergency response to an ongoing outbreak in the North Kivu and Ituri provinces.
As of Aug. 9, 2019, the trial had enrolled 681 patients at four Ebola treatment centers in live outbreak regions of the DRC, with the goal of enrolling 725 patients in total.
The trial investigators and study cosponsors accepted the recommendation for early termination, and staff at the trial sites in the DRC were promptly informed, according to the media advisory. Additional patient randomizations in the now-revised trial will be limited to treatment either with REGN-EB3 or mAb114. Patients randomized to the ZMapp or remdesivir arms in the last 10 days of the original trial will be given the option, at the discretion of their treating physician, to receive either of the two more effective treatments, according to the NIH.
“While the final analysis of the data can occur only after all the data are generated and collected (likely late September/early October 2019), the DSMB [Data and Safety Monitoring Board] and the study leadership felt the preliminary analysis of the existing data was compelling enough to recommend and implement these changes in the trial immediately. The complete results will be submitted for publication in the peer-reviewed medical literature as soon as possible,” the NIH stated.
The study is cosponsored and funded by the NIH, carried out by an international research consortium coordinated by the World Health Organization, and supported by four pharmaceutical companies (MappBio, Gilead, Regeneron, and Ridgeback Biotherapeutics).
An investigational agent known as REGN-EB3 has met an early stopping criterion in the protocol of an Ebola therapeutics trial, according to a National Institutes of Health media advisory.
Preliminary results in 499 study participants showed that individuals receiving either of two treatments, REGN-EB3 or mAb114, had a greater chance of survival, compared with participants in the other two study arms.
The randomized, controlled Pamoja Tulinde Maisha (PALM) study, which began Nov. 20, 2018, was designed to evaluate four investigational agents (ZMapp, remdesivir, mAb114, and REGN-EB3) for the treatment of patients with Ebola virus disease in the Democratic Republic of the Congo (DRC) as part of the emergency response to an ongoing outbreak in the North Kivu and Ituri provinces.
As of Aug. 9, 2019, the trial had enrolled 681 patients at four Ebola treatment centers in live outbreak regions of the DRC, with the goal of enrolling 725 patients in total.
The trial investigators and study cosponsors accepted the recommendation for early termination, and staff at the trial sites in the DRC were promptly informed, according to the media advisory. Additional patient randomizations in the now-revised trial will be limited to treatment either with REGN-EB3 or mAb114. Patients randomized to the ZMapp or remdesivir arms in the last 10 days of the original trial will be given the option, at the discretion of their treating physician, to receive either of the two more effective treatments, according to the NIH.
“While the final analysis of the data can occur only after all the data are generated and collected (likely late September/early October 2019), the DSMB [Data and Safety Monitoring Board] and the study leadership felt the preliminary analysis of the existing data was compelling enough to recommend and implement these changes in the trial immediately. The complete results will be submitted for publication in the peer-reviewed medical literature as soon as possible,” the NIH stated.
The study is cosponsored and funded by the NIH, carried out by an international research consortium coordinated by the World Health Organization, and supported by four pharmaceutical companies (MappBio, Gilead, Regeneron, and Ridgeback Biotherapeutics).
Treatment Facility: An Important Prognostic Factor for Dedifferentiated Liposarcoma Survival (FULL)
Approximately 17% to 25% of all softtissue sarcomas (STS) are liposarcomas, making liposarcoma the most common type of STS.1 The 2013 World Health Organization (WHO) classification separates liposarcoma into 4 histologic subtypes: atypical lipomatous tumor/well-differentiated (ALT/ WDLPS), dedifferentiated (DDLPS), myxoid, and pleomorphic.2 Each subtype has unique histology, morphology, and natural history. WDLPS and DDLPS are the most common histologic subtypes, comprising approximately 50% of all sarcomas that arise in the retroperitoneum.3 DDLPS represents 18% of all liposarcomas, making it the second most common subtype of liposarcoma.4
In 1979, DDLPS was first characterized.5 Most (90%) cases of DDLPS present de novo, whereas the other 10% transform from preexisting low-grade WDLPS.2 DDLPSs are formed by an amplification of 12q14-15 involving the MDM2 gene.4 These malignancies most commonly present in the retroperitoneum as a large painless mass, consisting of both fatty and nonfatty components.2 Primary site has been previously reported as a major prognostic factor for DDLPSs, with retroperitoneal DDLPSs demonstrating the worst prognosis.6 DDLPSs have a high risk of local recurrence, with some reports estimating recurrence rates approaching 40%.2 Overall mortality at 5 years for DDLPS is estimated to be between 30% and 40%.4
Previous literature has determined that median income, race, health insurance, and facility type are related to survival outcomes for patients with DDLPS.7-9 When comparing the most common types of cancers, residents of poorer US counties consistently had a higher risk of mortality than residents in affluent US counties, and all racial minorities showed worse survival outcomes when compared with white patients.7 Differences in survival outcomes have been reported in patients attending different treatment facilities for other cancers including pancreatic cancers, glioblastomas, and oral cancers, with multiple studies concluding that academic and research programs are associated with the longest survival outcomes.10-12 For many cancers, insurance status has been shown to be a significant prognostic factor, with private insurance typically resulting in the best prognosis.8,9
The goal of this retrospective study was to assess the prognostic effects of socioeconomic variables on the overall survival (OS) probabilities in a large cohort of DDLPS patients in order to inform clinicians about a potentially at-risk population.
Method
The National Cancer Database (NCDB) was created by the Commission on Cancer (CoC) of the American College of Surgeons and the American Cancer Society. The NCDB is the largest cancer database in the US and includes data on almost 70% of US patients with cancer. CoC-accredited cancer programs add data on patients with cancer to the NCDB. The authors accessed the NCDB data through the use of the NCDB Participant Use File program.
Patients’ data from 2004 through 2015 were abstracted. Only patients with the International Classification of Diseases for Oncology histology code 8858, corresponding to DDLPS, were analyzed. Patients with other comorbid malignant tumors were excluded to accurately capture the true survival rates for DDLPS. Variables analyzed included age, sex, race, insurance status, treatment facility type, median household income by zip code, and percentage of adults in the patient’s zip code with no high school (HS) education.
Median survival, 5- and 10-year OS probabilities, and Kaplan-Meier survival curves were calculated for multiple variables, specifically race, insurance status, treatment facility type, median family income, and percentage of adults without a HS degree. Both 5- and 10-year OS probabilities were determined by race with the patients separated into white, African American, Asian, American Indian/Alaska Native (AI/AN), and Asian Indian or Pakistani groups. Our study categorized Chinese, Japanese, Filipino, Hmong, Korean, Vietnamese, Thai, Guamanian, Asian not otherwise specified, and other Asian ethnicity patients together into one collective Asian group. Insurance status was classified into Medicare, Medicaid, other government insurance, and private insurance groups. Other government insurance consisted of US Department of Veterans Affairs, Indian Health Service, Public Health Service, and other government health care programs. Further analysis could not be performed into the distribution of the other government insurance variable.
Facility types were divided into 4 groups: community, comprehensive community, academic/ research, and integrated network cancer treatment facilities. Median income quartiles and the percentage of adults with no high school degree were estimated by comparison of the patient’s zip code with US Census Bureau data. Median household income was separated into 4 groups, including lowest level of household income (< $38,000), low level of household income ($38,000 to $47,999), moderate level of household income ($48,000 to $62,999), and highest level of household income (≥ $63,000). The percentages of adults with no high school degree were divided into 4 groups: lowest level of HS education (≥ 21% ), low level of HS education (13.0% to 20.9%), moderate level of HS education (7.0% to 12.9%), and highest level of HS education (≤ 7%). The 5- and 10-year survival probabilities were calculated using the number of months between the date of diagnosis and the date of death or last known contact.
Continuous variables are presented as median and interquartile range (IQR) whereas categorical variables are presented as frequencies and proportion. IBM SPSS version 25.0 was used to produce Kaplan-Meier survival curves and descriptive statistics. This study used Kaplan- Meier survival tables and log-rank tests to analyze both the 5- and 10-year OS rates for the 5 variables listed above. This study also used a multivariable Cox regression model that accommodated the correlative nature of outcomes within facilities to study the association of the treatment facility type and other socioeconomic factors, while controlling for age, race (which was collapsed into 3 categories), sex, primary site, tumor stage, and treatment approaches. The proportional hazards assumption was individually checked for all pertinent variables. Any patient records that were missing data were excluded from the multivariable Cox regression model, which was analyzed with SAS version 9.4 (Cary, NC). P < 0.05 was used to indicate statistical significance for all analyses.
Results
Table 1 provides descriptive analysis for demographic characteristics of the 3573 patients including age, sex, and race. The median age at diagnosis was 64 years. There were 1073 more men (65%) than women (35%) in this analysis. Whites were the predominant racial category, comprising 87.7% of the patient population, followed by African Americans (6.5%) and Asians (2.5%).
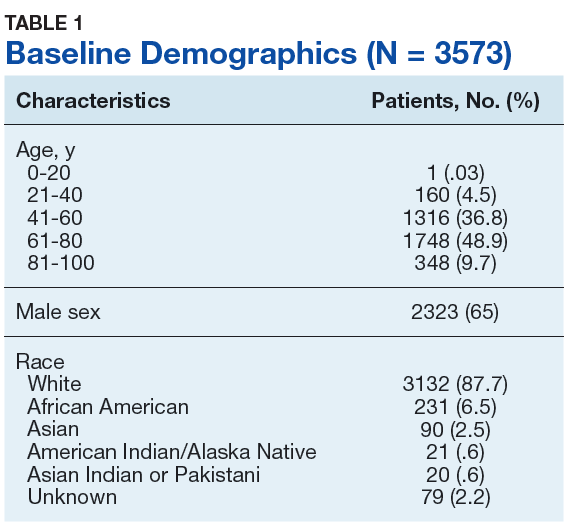
Socioeconomic Variables
The largest proportion of the patient population (45.5%) had private insurance (Table 2). Medicare came in a close second covering almost 42.2% of the population, followed by Medicaid (5.0%), uninsured (2.8%), and other government insurance (1.5%). About half (53.7%) of the patients were treated at academic or research facilities, while the fewest number of patients (5.2%) underwent treatment at community cancer facilities. The largest percentage (36.6%) of patients lived in zip codes with the highest level of median household income, while 26.0% and 22.3% had moderate and low levels of income, respectively. About 14% of patients lived within an area of the lowest level of income. Similarly, almost 15% of patients lived in an area of lowest level of HS education. The greatest percentage of the patient population (34.5%) lived in a zip code with moderate level of HS education. Surgery was the most common treatment modality with 90.8% of the cohort undergoing surgery, while 35.4% and 16.5% were treated with radiation and chemotherapy, respectively (some patients received more than one type of treatment modality).
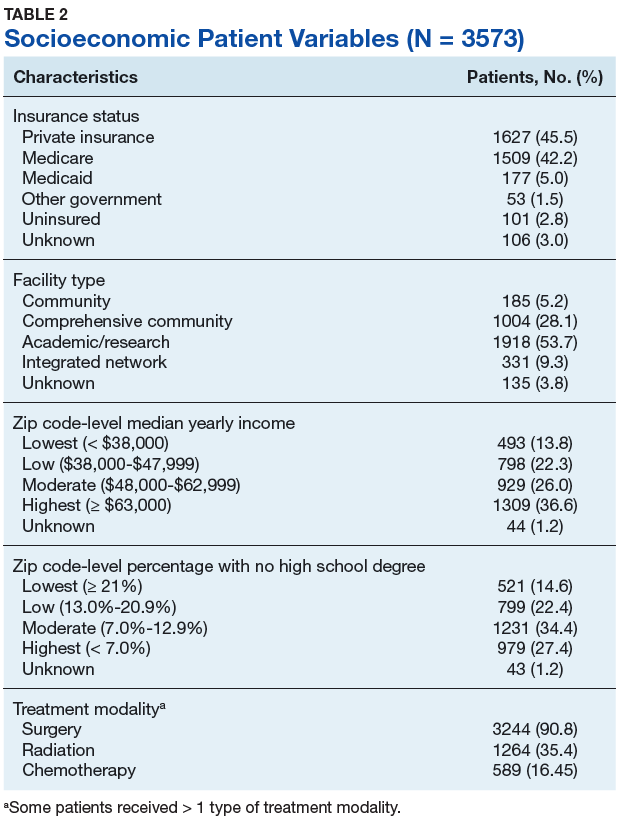
Survival Data
Survival data were available for 3112 patients. Kaplan-Meier survival curves were used to analyze OS according to insurance status, racial background, treatment facility type, median family income, and percentage of adults with no high school education. Overall 5- and 10- year OS probabilities were 51.5% and 34.8%, respectively, while the median OS (SD) was 63.57 (2.8) months (Table 3).
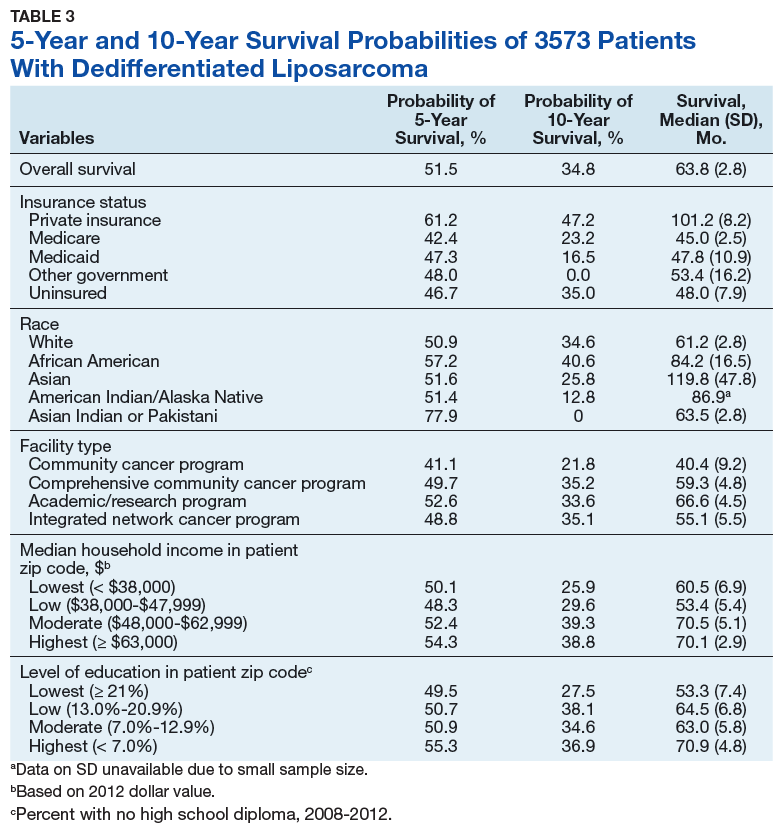
Private insurance showed significantly higher 5- and 10-year OS probabilities and median OS: 5-year OS was 61.2%, 10-year OS was 47.2%, and median survival (SD) was 101.2 (8.2) months compared with that of all other insurance groups (Medicare, Medicaid, other government insurance, and uninsured) (Figure 1). These other insurance types were fairly similar in their 5-year and median OS, but surprisingly, patients with no insurance had the second longest 10-year OS. The difference between the 5-year OS probabilities of private insurance compared with an average of the other insurances was 15.1%, which had almost doubled to 28.5% at 10 years, with a median OS difference of almost 5 years (56 months; data not shown).
Using the Kaplan-Meier survival curve, Asian Indians had the longest 5-year OS probability of 77.9% and African Americans had the longest 10-year OS probability of 40.6%. However, Asians as a group demonstrated the longest median (SD) OS outcome with 119.8 (47.8) months (Figure 2).
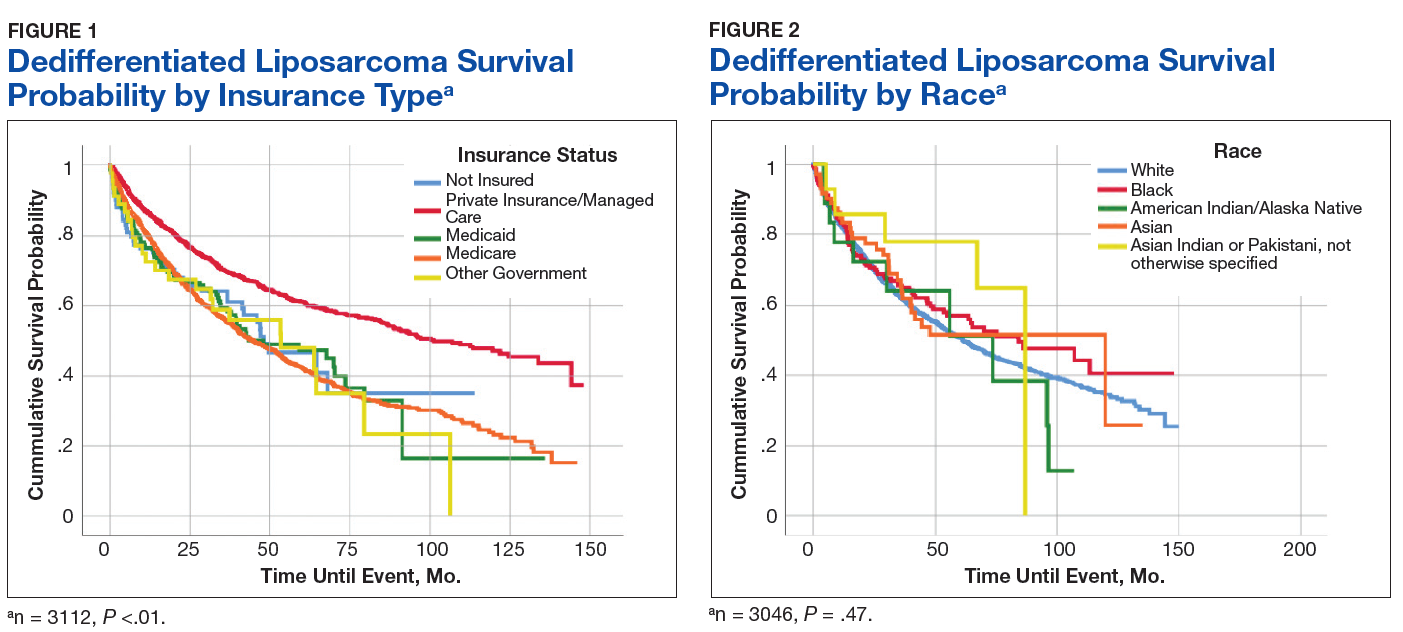
Overall, academic/research programs had the longest median OS and 5-year OS probability (SD) of 66.6 (4.5) months and 52.6%, respectively (Figure 3). Comprehensive community cancer programs and integrated network cancer programs had nearly identical 10-year OS rates (35.2% vs 35.1%, respectively). Community cancer programs had the worst 5- and 10-year OS probabilities (41.1% and 21.8%, respectively).
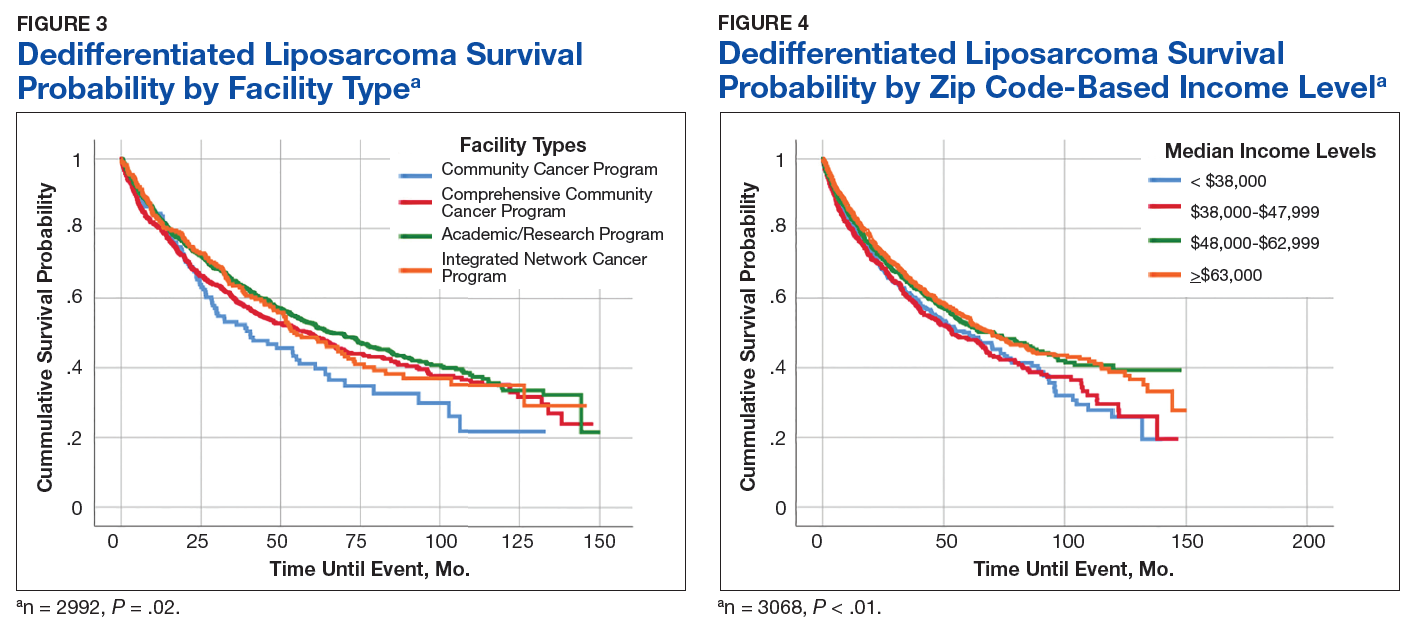
The top 2 income quartiles combined to demonstrate the longest median, 5-year, and 10-year OS probabilities and were very similar. Patients living in a zip code with the highest income level had the longest 5-year OS rates of 54.3%, while patients living in zip codes with a moderate income level had the longest 10-year OS at 39.3% and the longest median OS of about 71 months. Patients with the lowest level of median household income had the worst 5-year OS rates (48.3%) and a median (SD) OS of 53.4 (5.4) months (Figure 4).
A Kaplan-Meier curve for percentage of adults without a HS degree is displayed in Figure 5. Zip codes with the highest level of education had the longest 5-year OS rates and median (SD) OS of 55.3% and 70.9 (4.8) months, respectively. The longest 10-year OS outcomes at 38.1% were found in patients who lived in areas of low-education levels. The worst 5- and 10- year OS outcomes and median OS were found in the least educated zip codes.
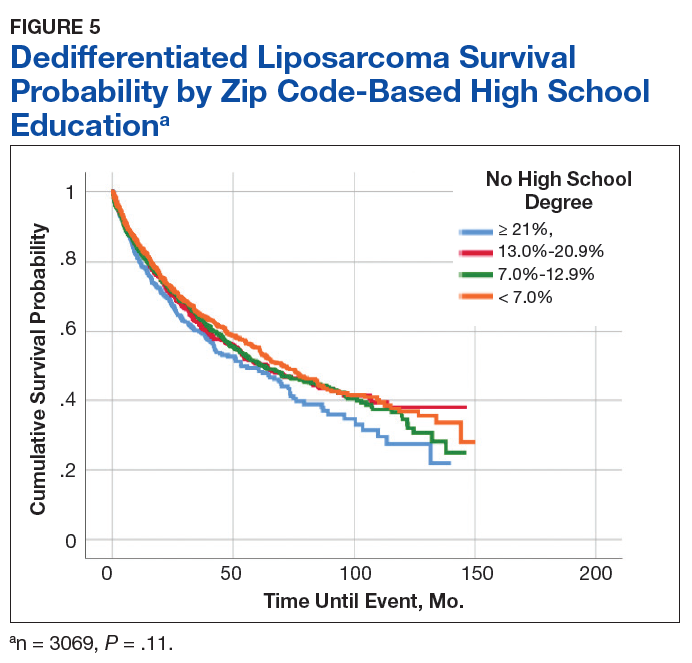
Results from the Cox regression model of OS are displayed in Table 4. Race and ethnicity, zip code-level median household income, and zip code-level education were not associated with OS. Patients with no insurance had an increased risk of death (hazard ratio [HR], 1.84; 95% CI, 1.17-2.88; P < .01) when compared with patients with private insurance. Patients with other government insurance also had an increased risk of death (HR, 2.12; 95% CI, 1.27-3.54; P < .01) when compared with patients with private insurance while controlling for all other variables. Patients with Medicare had a decreased risk of death when compared with patients with other government insurance and no insurance (HR, 0.53; 95% CI, 0.31-0.92; P = .02 and HR, 0.62; 95% CI, 0.38-0.99; P = .05, respectively). Patients treated at academic centers had better OS when compared with patients treated at comprehensive treatment centers (HR, 0.77; 95% CI, 0.65-0.92;P < .01) and community treatment centers (HR, 0.62; 95% CI, 0.44-0.86; P < .01).
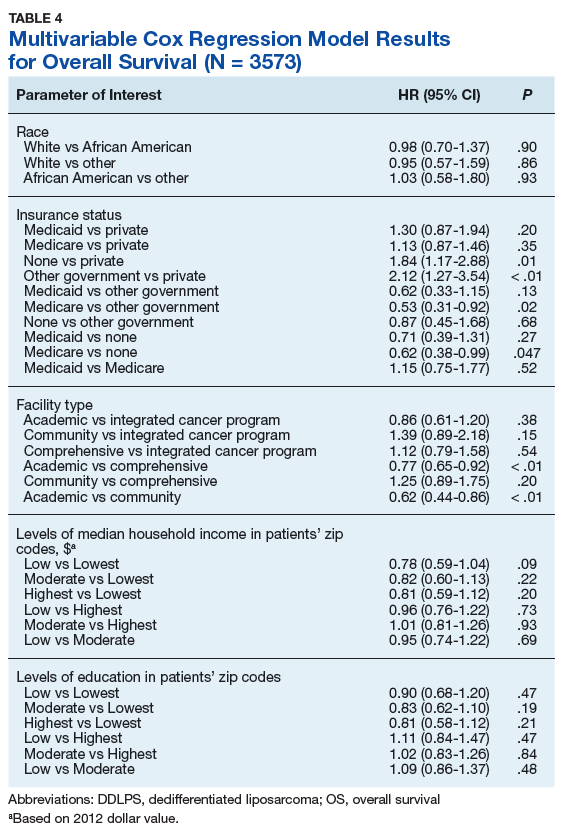
Discussion
This study is the largest study to date that specifically studies the type of treatment facilities and socioeconomic factors, including insurance status, race, income, and education, and how they affect survival of DDLPS. The overall 5- and 10-year OS probabilities for DDLPS in this study were 51.5% and 34.8%, respectively, with median OS of 63.6 months. These results were more encouraging than previous reports, which found a 5-year survival probability of 36.5% and a median OS of 45 months.13,14
The largest age grouping was aged 61 to 80 years (48.9% of the cohort), and the median age at diagnosis was 64 years. DDLPSs most typically present between the ages of 50 and 70 years.15 Our cohort was 65% male. Previous studies have indicated that DDLPSs affect the sexes equally; however, another study showed a similar male predominance (68.8%) at the MD Anderson Cancer Center in Houston, Texas.13,16
In our study, approximately 88% of patients were white, 6.5% were African American, and 2.5% were Asian, which differed from a previous study of 84 patients that had a 78.6% white, 4.8% Asian, and 1.2% African American patient population.14
Asian Indian or Pakistani patients had the best 5-year OS probability at 77.9%, followed by African American (57.2%), Asian (51.6%), AI/AN (51.4%), and white patients (50.9%). This trend had disappeared by 10 years and Asian, AI/AN, African American, and Asian Indian or Pakistani groups all demonstrated longer median OS than did white patients. In fact, Asian patients had the longest median OS at 119.8 months, which was almost double that of white patients with the lowest median OS of 61.2 months. This finding is contrary to previous studies, which reported that racial minorities typically had worse OS outcomes when compared with white patients in different types of cancer.7,17 Notably, these findings were not statistically significant in our current study in the log-rank or multivariable analyses.
Private insurance was the most common form of insurance followed in decreasing order by Medicare, Medicaid, uninsured, and other government insurance. About 42% of the cohort had Medicare, which is a federally funded US insurance program designated for patients aged ≥ 65 years and certain younger patients with disabilities.
Patients with private insurance demonstrated the longest OS, essentially twice the median OS of all other insured groups at 101 months. Medicare had the worst 5-year OS probability and median OS of all groups. A previous study of 77 patients with DDLPS reported that patients aged > 65 years had reduced OS.13 Medicare patients in this study were older, with a mean and median age at DDLPS diagnosis of 71 and 72 years, respectively, while private insurance had a mean and median age at diagnosis of 56 and 57 years, respectively. Medicare inherently covers older patients and this age difference could account for the decrease in overall survival.
Improved OS for privately insured patients was most notable compared with the uninsured or patients with other government insurance. Uninsured patients had an 83.7% increased risk of mortality when compared with patients with private insurance. When compared with patients with private insurance, patients with other government insurance had an 111.5% increased risk of mortality. Comparing patients with Medicare vs patients with no insurance or other government insurance, there was a decreased risk of mortality of 38.5% and 46.6%, respectively. This decreased OS in patients with other government insurance could be related to the choice of treatment facility, because only 31% of the patients with other government insurance went to academic or research centers when compared with the 58.4% and 50.8% of patients with private and Medicare insurance treated there (data not shown). Such centers often have access to more advanced technology and protocols that may not be available at other treatment facilities.
A little more than half of the patients in the cohort went to an academic or research center for treatment (53.7%); comprehensive community cancer programs were the second most common treatment facility at 28%. Patients treated at academic or research centers demonstrated the best outcomes with a 5-year OS of 52.6%, followed in decreasing order by comprehensive community cancer programs (49.7%), integrated network cancer programs (48.8%), and community cancer programs (41.1%). In our patient cocohort, patients treated at an academic/research center had slightly decreased 10-year OS rates compared with those patients treated at a comprehensive community cancer program, although the median OS for the academic/research centers were still the highest of all treatment facilities.
Treatment options varied significantly by facility, and the number of patients treated surgically followed a similar trend, with 92% undergoing surgery as the primary treatment at academic or research programs compared with 89% at comprehensive cancer programs and 82.7% at community cancer programs (data not shown). Another potential explaination for differing OS outcomes across facilities is the surgical margin outcome. Surgeries performed at community cancer programs or comprehensive cancer programs resulted with no residual tumor in 36% and 40% of cases, respectively, whereas cases performed at academic or research programs resulted with no residual tumor in 47% of cases (data not shown). Regardless, multivariate analysis demonstrated a marked decrease in the chance of mortality when comparing treatment received at academic facility centers with that received at comprehensive cancer centers (22.9%) and community cancer centers (38.3%) (data not shown).
A recent study demonstrated improved outcomes for patients with retroperitoneal or extremity STS treated at high-volume treatment centers.18 Patients treated at high-volume centers were found to have an 8% decreased risk of death compared with patients treated at low-volume centers. Notably, they found highvolume academic centers demonstrated the strongest improvement in survival, while highvolume community centers showed decreased survival.18 Similarly, we found that patients treated at academic/research institutions had improved 5-year OS and greater median OS than did patients treated at community cancer programs or comprehensive community cancer programs.
The top 2 income quartiles (≥ $48,000) combined to demonstrate the longest median, 5-year, and 10-year OS and were fairly similar between the quartiles. Patients living in zip codes with a median income of $38,000 to $47,999 had the worst 5-year OS and median OS. The log-rank analysis showed statistical evidence of differences in survival associated with income, but within the context of the multivariable analysis, there was no remaining evidence of a difference.
The longest 5-year OS outcomes were seen in patients living in zip codes with the highest level of education (55.3%). However, the difference in OS was not statistically significant using either the log-rank analysis or multivariate analysis.
Limitations
This study has certain inherent limitations in using a retrospective design and a large database such as the NCDB. Many different pathologists at CoC-accredited cancer programs perform the pathology that contributes to the data in the NCDB. There was no pathological review of these findings, which could potentially introduce error into the findings of this study. With the NCDB, potential selection bias is possible because patients in the database are added only from CoC-accredited cancer programs. This risk is minimized because NCDB contains data on most newly diagnosed cancer patients in the US. Further potential risks, which are unable to be controlled for, include potential interobserver error and data that may be incompletely, improperly, or inaccurately recorded from the patients’ charts. Without patient-specific information regarding income and education, it is challenging to utilize zip codes to estimate socioeconomic status and educational level. Even though a patient may live in a zip code identified with specific economic and educational characteristics, that patient may not share those characteristics. Furthermore, patients with Medicare tend to be older than patients with other forms of insurance, which limits the significance of comparisons across insurance groups. A future SEER (Surveillance, Epidemiology, and End Results) program study to confirm this study’s results and the effects of socioeconomic variables on DDLPS would be an excellent followup study.
Conclusion
This study used a large cohort of patients with DDLPS to study the effects of treatment facility, insurance status, and socioeconomic variables on survival outcomes. Although insurance status, median household income, and treatment facility were associated with differences in median OS and 5- and 10-year OS probabilities, evidence for a difference remained for only insurance status and facility type within the context of a multivariable analysis irrespective of age, race, sex, insurance status, education, and median income. Patients with private insurance and Medicaid had a decreased risk of mortality compared with other government insurance and no insurance. Patients receiving treatment at academic research programs had the highest median and 5-year OS of 66.6 months and 52.6%, respectively. Patients receiving treatment at academic centers had improved survival outcomes with a decrease in mortality of 23% and 38% compared to comprehensive or community cancer programs.
1. Dodd LG. Update on liposarcoma: a review for cytopathologists. Diagn Cytopathol. 2012;40(12):1122-1131.
2. Mangham D. World Health Organisation classification of tumours: pathology and genetics of tumours of soft tissue and bone. J Bone Joint Surg Am. 2004;86(3):466.
3. Dalal KM, Kattan MW, Antonescu CR, Brennan MF, Singer S. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg. 2006;244(3):381-391.
4. Coindre JM, Pédeutour F, Aurias A. Well-differentiated and dedifferentiated liposarcomas. Virchows Arch. 2010;456(2):167-179.
5. Evans HL. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol. 1979;3(6):507-523.
6. Henricks WH, Chu YC, Goldblum JR, Weiss SW. Dedifferentiated liposarcoma: a clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol. 1997;21(3):271-281.
7. Ward E, Jemal A, Cokkinides V, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin. 2004;54(2):78-93.
8. Halpern MT, Ward EM, Pavluck AL, Schrag NM, Bian J, Chen AY. Association of insurance status and ethnicity with cancer stage at diagnosis for 12 cancer sites: a retrospective analysis. Lancet Oncol. 2008;9(3):222-231.
9. Niu X, Roche LM, Pawlish KS, Henry KA. Cancer survival disparities by health insurance status. Cancer Med. 2013;2(3):403-411.
10. Hauser A, Dutta SW, Showalter TN, Sheehan JP, Grover S, Trifiletti DM. Impact of academic facility type and volume on post-surgical outcomes following diagnosis of glioblastoma. J Clin Neurosci. 2018;47:103-110.
11. Chu Q, Medeiros K, Zhou M, et al. Effect of facility type on outcome following pancreatectomy for pancreatic adenocarcinoma: analysis of the National Cancer Data Base [Abstract FP26-02]. HPB (Oxford). 2016;18(suppl 1):E81-E82.
12. Rubin SJ, Cohen MB, Kirke DN, Qureshi MM, Truong MT, Jalisi S. Comparison of facility type outcomes for oral cavity cancer: analysis of the National Cancer Database. Laryngoscope. 2017;127(11):2551-2557.
13. Lahat G, Anaya DA, Wang X, Tuvin D, Lev D, Pollock RE. Resectable well-differentiated versus dedifferentiated liposarcomas: two different diseases possibly requiring different treatment approaches. Ann Surg Oncol. 2008;15(6):1585-1593.
14. Livingston JA, Bugano D, Barbo A, et al. Role of chemotherapy in dedifferentiated liposarcoma of the retroperitoneum: defining the benefit and challenges of the standard. Sci Rep. 2017;7(1):11836.
15. Brennan MF, Antonescu CR, Alektiar KM, Maki RG. Management of Soft Tissue Sarcoma. 2nd ed. New York, NY: Springer; 2016.
16. Goldblum JR, Folpe AL, Weiss SW. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Philadelphia, PA: Saunders; 2014.
17. White A, Djenaba J, Rim SH, Johnson CJ, Coleman MP, Allemani C. Colon cancer survival in the United States by race and stage (2001‐2009): findings from the CONCORD‐2 study. Cancer. 2017;123 (suppl 24):5014-5036.
18. Murphy JD, Padwal J, Guss ZD, Okamoto K, Sardar R. Impact of hospital volume on patterns of care and outcomes in soft tissue sarcoma [ASCO Abstract e23550]. J Clin Oncol. 2018;36(suppl 15):e23550
Approximately 17% to 25% of all softtissue sarcomas (STS) are liposarcomas, making liposarcoma the most common type of STS.1 The 2013 World Health Organization (WHO) classification separates liposarcoma into 4 histologic subtypes: atypical lipomatous tumor/well-differentiated (ALT/ WDLPS), dedifferentiated (DDLPS), myxoid, and pleomorphic.2 Each subtype has unique histology, morphology, and natural history. WDLPS and DDLPS are the most common histologic subtypes, comprising approximately 50% of all sarcomas that arise in the retroperitoneum.3 DDLPS represents 18% of all liposarcomas, making it the second most common subtype of liposarcoma.4
In 1979, DDLPS was first characterized.5 Most (90%) cases of DDLPS present de novo, whereas the other 10% transform from preexisting low-grade WDLPS.2 DDLPSs are formed by an amplification of 12q14-15 involving the MDM2 gene.4 These malignancies most commonly present in the retroperitoneum as a large painless mass, consisting of both fatty and nonfatty components.2 Primary site has been previously reported as a major prognostic factor for DDLPSs, with retroperitoneal DDLPSs demonstrating the worst prognosis.6 DDLPSs have a high risk of local recurrence, with some reports estimating recurrence rates approaching 40%.2 Overall mortality at 5 years for DDLPS is estimated to be between 30% and 40%.4
Previous literature has determined that median income, race, health insurance, and facility type are related to survival outcomes for patients with DDLPS.7-9 When comparing the most common types of cancers, residents of poorer US counties consistently had a higher risk of mortality than residents in affluent US counties, and all racial minorities showed worse survival outcomes when compared with white patients.7 Differences in survival outcomes have been reported in patients attending different treatment facilities for other cancers including pancreatic cancers, glioblastomas, and oral cancers, with multiple studies concluding that academic and research programs are associated with the longest survival outcomes.10-12 For many cancers, insurance status has been shown to be a significant prognostic factor, with private insurance typically resulting in the best prognosis.8,9
The goal of this retrospective study was to assess the prognostic effects of socioeconomic variables on the overall survival (OS) probabilities in a large cohort of DDLPS patients in order to inform clinicians about a potentially at-risk population.
Method
The National Cancer Database (NCDB) was created by the Commission on Cancer (CoC) of the American College of Surgeons and the American Cancer Society. The NCDB is the largest cancer database in the US and includes data on almost 70% of US patients with cancer. CoC-accredited cancer programs add data on patients with cancer to the NCDB. The authors accessed the NCDB data through the use of the NCDB Participant Use File program.
Patients’ data from 2004 through 2015 were abstracted. Only patients with the International Classification of Diseases for Oncology histology code 8858, corresponding to DDLPS, were analyzed. Patients with other comorbid malignant tumors were excluded to accurately capture the true survival rates for DDLPS. Variables analyzed included age, sex, race, insurance status, treatment facility type, median household income by zip code, and percentage of adults in the patient’s zip code with no high school (HS) education.
Median survival, 5- and 10-year OS probabilities, and Kaplan-Meier survival curves were calculated for multiple variables, specifically race, insurance status, treatment facility type, median family income, and percentage of adults without a HS degree. Both 5- and 10-year OS probabilities were determined by race with the patients separated into white, African American, Asian, American Indian/Alaska Native (AI/AN), and Asian Indian or Pakistani groups. Our study categorized Chinese, Japanese, Filipino, Hmong, Korean, Vietnamese, Thai, Guamanian, Asian not otherwise specified, and other Asian ethnicity patients together into one collective Asian group. Insurance status was classified into Medicare, Medicaid, other government insurance, and private insurance groups. Other government insurance consisted of US Department of Veterans Affairs, Indian Health Service, Public Health Service, and other government health care programs. Further analysis could not be performed into the distribution of the other government insurance variable.
Facility types were divided into 4 groups: community, comprehensive community, academic/ research, and integrated network cancer treatment facilities. Median income quartiles and the percentage of adults with no high school degree were estimated by comparison of the patient’s zip code with US Census Bureau data. Median household income was separated into 4 groups, including lowest level of household income (< $38,000), low level of household income ($38,000 to $47,999), moderate level of household income ($48,000 to $62,999), and highest level of household income (≥ $63,000). The percentages of adults with no high school degree were divided into 4 groups: lowest level of HS education (≥ 21% ), low level of HS education (13.0% to 20.9%), moderate level of HS education (7.0% to 12.9%), and highest level of HS education (≤ 7%). The 5- and 10-year survival probabilities were calculated using the number of months between the date of diagnosis and the date of death or last known contact.
Continuous variables are presented as median and interquartile range (IQR) whereas categorical variables are presented as frequencies and proportion. IBM SPSS version 25.0 was used to produce Kaplan-Meier survival curves and descriptive statistics. This study used Kaplan- Meier survival tables and log-rank tests to analyze both the 5- and 10-year OS rates for the 5 variables listed above. This study also used a multivariable Cox regression model that accommodated the correlative nature of outcomes within facilities to study the association of the treatment facility type and other socioeconomic factors, while controlling for age, race (which was collapsed into 3 categories), sex, primary site, tumor stage, and treatment approaches. The proportional hazards assumption was individually checked for all pertinent variables. Any patient records that were missing data were excluded from the multivariable Cox regression model, which was analyzed with SAS version 9.4 (Cary, NC). P < 0.05 was used to indicate statistical significance for all analyses.
Results
Table 1 provides descriptive analysis for demographic characteristics of the 3573 patients including age, sex, and race. The median age at diagnosis was 64 years. There were 1073 more men (65%) than women (35%) in this analysis. Whites were the predominant racial category, comprising 87.7% of the patient population, followed by African Americans (6.5%) and Asians (2.5%).
Socioeconomic Variables
The largest proportion of the patient population (45.5%) had private insurance (Table 2). Medicare came in a close second covering almost 42.2% of the population, followed by Medicaid (5.0%), uninsured (2.8%), and other government insurance (1.5%). About half (53.7%) of the patients were treated at academic or research facilities, while the fewest number of patients (5.2%) underwent treatment at community cancer facilities. The largest percentage (36.6%) of patients lived in zip codes with the highest level of median household income, while 26.0% and 22.3% had moderate and low levels of income, respectively. About 14% of patients lived within an area of the lowest level of income. Similarly, almost 15% of patients lived in an area of lowest level of HS education. The greatest percentage of the patient population (34.5%) lived in a zip code with moderate level of HS education. Surgery was the most common treatment modality with 90.8% of the cohort undergoing surgery, while 35.4% and 16.5% were treated with radiation and chemotherapy, respectively (some patients received more than one type of treatment modality).
Survival Data
Survival data were available for 3112 patients. Kaplan-Meier survival curves were used to analyze OS according to insurance status, racial background, treatment facility type, median family income, and percentage of adults with no high school education. Overall 5- and 10- year OS probabilities were 51.5% and 34.8%, respectively, while the median OS (SD) was 63.57 (2.8) months (Table 3).
Private insurance showed significantly higher 5- and 10-year OS probabilities and median OS: 5-year OS was 61.2%, 10-year OS was 47.2%, and median survival (SD) was 101.2 (8.2) months compared with that of all other insurance groups (Medicare, Medicaid, other government insurance, and uninsured) (Figure 1). These other insurance types were fairly similar in their 5-year and median OS, but surprisingly, patients with no insurance had the second longest 10-year OS. The difference between the 5-year OS probabilities of private insurance compared with an average of the other insurances was 15.1%, which had almost doubled to 28.5% at 10 years, with a median OS difference of almost 5 years (56 months; data not shown).
Using the Kaplan-Meier survival curve, Asian Indians had the longest 5-year OS probability of 77.9% and African Americans had the longest 10-year OS probability of 40.6%. However, Asians as a group demonstrated the longest median (SD) OS outcome with 119.8 (47.8) months (Figure 2).
Overall, academic/research programs had the longest median OS and 5-year OS probability (SD) of 66.6 (4.5) months and 52.6%, respectively (Figure 3). Comprehensive community cancer programs and integrated network cancer programs had nearly identical 10-year OS rates (35.2% vs 35.1%, respectively). Community cancer programs had the worst 5- and 10-year OS probabilities (41.1% and 21.8%, respectively).
The top 2 income quartiles combined to demonstrate the longest median, 5-year, and 10-year OS probabilities and were very similar. Patients living in a zip code with the highest income level had the longest 5-year OS rates of 54.3%, while patients living in zip codes with a moderate income level had the longest 10-year OS at 39.3% and the longest median OS of about 71 months. Patients with the lowest level of median household income had the worst 5-year OS rates (48.3%) and a median (SD) OS of 53.4 (5.4) months (Figure 4).
A Kaplan-Meier curve for percentage of adults without a HS degree is displayed in Figure 5. Zip codes with the highest level of education had the longest 5-year OS rates and median (SD) OS of 55.3% and 70.9 (4.8) months, respectively. The longest 10-year OS outcomes at 38.1% were found in patients who lived in areas of low-education levels. The worst 5- and 10- year OS outcomes and median OS were found in the least educated zip codes.
Results from the Cox regression model of OS are displayed in Table 4. Race and ethnicity, zip code-level median household income, and zip code-level education were not associated with OS. Patients with no insurance had an increased risk of death (hazard ratio [HR], 1.84; 95% CI, 1.17-2.88; P < .01) when compared with patients with private insurance. Patients with other government insurance also had an increased risk of death (HR, 2.12; 95% CI, 1.27-3.54; P < .01) when compared with patients with private insurance while controlling for all other variables. Patients with Medicare had a decreased risk of death when compared with patients with other government insurance and no insurance (HR, 0.53; 95% CI, 0.31-0.92; P = .02 and HR, 0.62; 95% CI, 0.38-0.99; P = .05, respectively). Patients treated at academic centers had better OS when compared with patients treated at comprehensive treatment centers (HR, 0.77; 95% CI, 0.65-0.92;P < .01) and community treatment centers (HR, 0.62; 95% CI, 0.44-0.86; P < .01).
Discussion
This study is the largest study to date that specifically studies the type of treatment facilities and socioeconomic factors, including insurance status, race, income, and education, and how they affect survival of DDLPS. The overall 5- and 10-year OS probabilities for DDLPS in this study were 51.5% and 34.8%, respectively, with median OS of 63.6 months. These results were more encouraging than previous reports, which found a 5-year survival probability of 36.5% and a median OS of 45 months.13,14
The largest age grouping was aged 61 to 80 years (48.9% of the cohort), and the median age at diagnosis was 64 years. DDLPSs most typically present between the ages of 50 and 70 years.15 Our cohort was 65% male. Previous studies have indicated that DDLPSs affect the sexes equally; however, another study showed a similar male predominance (68.8%) at the MD Anderson Cancer Center in Houston, Texas.13,16
In our study, approximately 88% of patients were white, 6.5% were African American, and 2.5% were Asian, which differed from a previous study of 84 patients that had a 78.6% white, 4.8% Asian, and 1.2% African American patient population.14
Asian Indian or Pakistani patients had the best 5-year OS probability at 77.9%, followed by African American (57.2%), Asian (51.6%), AI/AN (51.4%), and white patients (50.9%). This trend had disappeared by 10 years and Asian, AI/AN, African American, and Asian Indian or Pakistani groups all demonstrated longer median OS than did white patients. In fact, Asian patients had the longest median OS at 119.8 months, which was almost double that of white patients with the lowest median OS of 61.2 months. This finding is contrary to previous studies, which reported that racial minorities typically had worse OS outcomes when compared with white patients in different types of cancer.7,17 Notably, these findings were not statistically significant in our current study in the log-rank or multivariable analyses.
Private insurance was the most common form of insurance followed in decreasing order by Medicare, Medicaid, uninsured, and other government insurance. About 42% of the cohort had Medicare, which is a federally funded US insurance program designated for patients aged ≥ 65 years and certain younger patients with disabilities.
Patients with private insurance demonstrated the longest OS, essentially twice the median OS of all other insured groups at 101 months. Medicare had the worst 5-year OS probability and median OS of all groups. A previous study of 77 patients with DDLPS reported that patients aged > 65 years had reduced OS.13 Medicare patients in this study were older, with a mean and median age at DDLPS diagnosis of 71 and 72 years, respectively, while private insurance had a mean and median age at diagnosis of 56 and 57 years, respectively. Medicare inherently covers older patients and this age difference could account for the decrease in overall survival.
Improved OS for privately insured patients was most notable compared with the uninsured or patients with other government insurance. Uninsured patients had an 83.7% increased risk of mortality when compared with patients with private insurance. When compared with patients with private insurance, patients with other government insurance had an 111.5% increased risk of mortality. Comparing patients with Medicare vs patients with no insurance or other government insurance, there was a decreased risk of mortality of 38.5% and 46.6%, respectively. This decreased OS in patients with other government insurance could be related to the choice of treatment facility, because only 31% of the patients with other government insurance went to academic or research centers when compared with the 58.4% and 50.8% of patients with private and Medicare insurance treated there (data not shown). Such centers often have access to more advanced technology and protocols that may not be available at other treatment facilities.
A little more than half of the patients in the cohort went to an academic or research center for treatment (53.7%); comprehensive community cancer programs were the second most common treatment facility at 28%. Patients treated at academic or research centers demonstrated the best outcomes with a 5-year OS of 52.6%, followed in decreasing order by comprehensive community cancer programs (49.7%), integrated network cancer programs (48.8%), and community cancer programs (41.1%). In our patient cocohort, patients treated at an academic/research center had slightly decreased 10-year OS rates compared with those patients treated at a comprehensive community cancer program, although the median OS for the academic/research centers were still the highest of all treatment facilities.
Treatment options varied significantly by facility, and the number of patients treated surgically followed a similar trend, with 92% undergoing surgery as the primary treatment at academic or research programs compared with 89% at comprehensive cancer programs and 82.7% at community cancer programs (data not shown). Another potential explaination for differing OS outcomes across facilities is the surgical margin outcome. Surgeries performed at community cancer programs or comprehensive cancer programs resulted with no residual tumor in 36% and 40% of cases, respectively, whereas cases performed at academic or research programs resulted with no residual tumor in 47% of cases (data not shown). Regardless, multivariate analysis demonstrated a marked decrease in the chance of mortality when comparing treatment received at academic facility centers with that received at comprehensive cancer centers (22.9%) and community cancer centers (38.3%) (data not shown).
A recent study demonstrated improved outcomes for patients with retroperitoneal or extremity STS treated at high-volume treatment centers.18 Patients treated at high-volume centers were found to have an 8% decreased risk of death compared with patients treated at low-volume centers. Notably, they found highvolume academic centers demonstrated the strongest improvement in survival, while highvolume community centers showed decreased survival.18 Similarly, we found that patients treated at academic/research institutions had improved 5-year OS and greater median OS than did patients treated at community cancer programs or comprehensive community cancer programs.
The top 2 income quartiles (≥ $48,000) combined to demonstrate the longest median, 5-year, and 10-year OS and were fairly similar between the quartiles. Patients living in zip codes with a median income of $38,000 to $47,999 had the worst 5-year OS and median OS. The log-rank analysis showed statistical evidence of differences in survival associated with income, but within the context of the multivariable analysis, there was no remaining evidence of a difference.
The longest 5-year OS outcomes were seen in patients living in zip codes with the highest level of education (55.3%). However, the difference in OS was not statistically significant using either the log-rank analysis or multivariate analysis.
Limitations
This study has certain inherent limitations in using a retrospective design and a large database such as the NCDB. Many different pathologists at CoC-accredited cancer programs perform the pathology that contributes to the data in the NCDB. There was no pathological review of these findings, which could potentially introduce error into the findings of this study. With the NCDB, potential selection bias is possible because patients in the database are added only from CoC-accredited cancer programs. This risk is minimized because NCDB contains data on most newly diagnosed cancer patients in the US. Further potential risks, which are unable to be controlled for, include potential interobserver error and data that may be incompletely, improperly, or inaccurately recorded from the patients’ charts. Without patient-specific information regarding income and education, it is challenging to utilize zip codes to estimate socioeconomic status and educational level. Even though a patient may live in a zip code identified with specific economic and educational characteristics, that patient may not share those characteristics. Furthermore, patients with Medicare tend to be older than patients with other forms of insurance, which limits the significance of comparisons across insurance groups. A future SEER (Surveillance, Epidemiology, and End Results) program study to confirm this study’s results and the effects of socioeconomic variables on DDLPS would be an excellent followup study.
Conclusion
This study used a large cohort of patients with DDLPS to study the effects of treatment facility, insurance status, and socioeconomic variables on survival outcomes. Although insurance status, median household income, and treatment facility were associated with differences in median OS and 5- and 10-year OS probabilities, evidence for a difference remained for only insurance status and facility type within the context of a multivariable analysis irrespective of age, race, sex, insurance status, education, and median income. Patients with private insurance and Medicaid had a decreased risk of mortality compared with other government insurance and no insurance. Patients receiving treatment at academic research programs had the highest median and 5-year OS of 66.6 months and 52.6%, respectively. Patients receiving treatment at academic centers had improved survival outcomes with a decrease in mortality of 23% and 38% compared to comprehensive or community cancer programs.
Approximately 17% to 25% of all softtissue sarcomas (STS) are liposarcomas, making liposarcoma the most common type of STS.1 The 2013 World Health Organization (WHO) classification separates liposarcoma into 4 histologic subtypes: atypical lipomatous tumor/well-differentiated (ALT/ WDLPS), dedifferentiated (DDLPS), myxoid, and pleomorphic.2 Each subtype has unique histology, morphology, and natural history. WDLPS and DDLPS are the most common histologic subtypes, comprising approximately 50% of all sarcomas that arise in the retroperitoneum.3 DDLPS represents 18% of all liposarcomas, making it the second most common subtype of liposarcoma.4
In 1979, DDLPS was first characterized.5 Most (90%) cases of DDLPS present de novo, whereas the other 10% transform from preexisting low-grade WDLPS.2 DDLPSs are formed by an amplification of 12q14-15 involving the MDM2 gene.4 These malignancies most commonly present in the retroperitoneum as a large painless mass, consisting of both fatty and nonfatty components.2 Primary site has been previously reported as a major prognostic factor for DDLPSs, with retroperitoneal DDLPSs demonstrating the worst prognosis.6 DDLPSs have a high risk of local recurrence, with some reports estimating recurrence rates approaching 40%.2 Overall mortality at 5 years for DDLPS is estimated to be between 30% and 40%.4
Previous literature has determined that median income, race, health insurance, and facility type are related to survival outcomes for patients with DDLPS.7-9 When comparing the most common types of cancers, residents of poorer US counties consistently had a higher risk of mortality than residents in affluent US counties, and all racial minorities showed worse survival outcomes when compared with white patients.7 Differences in survival outcomes have been reported in patients attending different treatment facilities for other cancers including pancreatic cancers, glioblastomas, and oral cancers, with multiple studies concluding that academic and research programs are associated with the longest survival outcomes.10-12 For many cancers, insurance status has been shown to be a significant prognostic factor, with private insurance typically resulting in the best prognosis.8,9
The goal of this retrospective study was to assess the prognostic effects of socioeconomic variables on the overall survival (OS) probabilities in a large cohort of DDLPS patients in order to inform clinicians about a potentially at-risk population.
Method
The National Cancer Database (NCDB) was created by the Commission on Cancer (CoC) of the American College of Surgeons and the American Cancer Society. The NCDB is the largest cancer database in the US and includes data on almost 70% of US patients with cancer. CoC-accredited cancer programs add data on patients with cancer to the NCDB. The authors accessed the NCDB data through the use of the NCDB Participant Use File program.
Patients’ data from 2004 through 2015 were abstracted. Only patients with the International Classification of Diseases for Oncology histology code 8858, corresponding to DDLPS, were analyzed. Patients with other comorbid malignant tumors were excluded to accurately capture the true survival rates for DDLPS. Variables analyzed included age, sex, race, insurance status, treatment facility type, median household income by zip code, and percentage of adults in the patient’s zip code with no high school (HS) education.
Median survival, 5- and 10-year OS probabilities, and Kaplan-Meier survival curves were calculated for multiple variables, specifically race, insurance status, treatment facility type, median family income, and percentage of adults without a HS degree. Both 5- and 10-year OS probabilities were determined by race with the patients separated into white, African American, Asian, American Indian/Alaska Native (AI/AN), and Asian Indian or Pakistani groups. Our study categorized Chinese, Japanese, Filipino, Hmong, Korean, Vietnamese, Thai, Guamanian, Asian not otherwise specified, and other Asian ethnicity patients together into one collective Asian group. Insurance status was classified into Medicare, Medicaid, other government insurance, and private insurance groups. Other government insurance consisted of US Department of Veterans Affairs, Indian Health Service, Public Health Service, and other government health care programs. Further analysis could not be performed into the distribution of the other government insurance variable.
Facility types were divided into 4 groups: community, comprehensive community, academic/ research, and integrated network cancer treatment facilities. Median income quartiles and the percentage of adults with no high school degree were estimated by comparison of the patient’s zip code with US Census Bureau data. Median household income was separated into 4 groups, including lowest level of household income (< $38,000), low level of household income ($38,000 to $47,999), moderate level of household income ($48,000 to $62,999), and highest level of household income (≥ $63,000). The percentages of adults with no high school degree were divided into 4 groups: lowest level of HS education (≥ 21% ), low level of HS education (13.0% to 20.9%), moderate level of HS education (7.0% to 12.9%), and highest level of HS education (≤ 7%). The 5- and 10-year survival probabilities were calculated using the number of months between the date of diagnosis and the date of death or last known contact.
Continuous variables are presented as median and interquartile range (IQR) whereas categorical variables are presented as frequencies and proportion. IBM SPSS version 25.0 was used to produce Kaplan-Meier survival curves and descriptive statistics. This study used Kaplan- Meier survival tables and log-rank tests to analyze both the 5- and 10-year OS rates for the 5 variables listed above. This study also used a multivariable Cox regression model that accommodated the correlative nature of outcomes within facilities to study the association of the treatment facility type and other socioeconomic factors, while controlling for age, race (which was collapsed into 3 categories), sex, primary site, tumor stage, and treatment approaches. The proportional hazards assumption was individually checked for all pertinent variables. Any patient records that were missing data were excluded from the multivariable Cox regression model, which was analyzed with SAS version 9.4 (Cary, NC). P < 0.05 was used to indicate statistical significance for all analyses.
Results
Table 1 provides descriptive analysis for demographic characteristics of the 3573 patients including age, sex, and race. The median age at diagnosis was 64 years. There were 1073 more men (65%) than women (35%) in this analysis. Whites were the predominant racial category, comprising 87.7% of the patient population, followed by African Americans (6.5%) and Asians (2.5%).
Socioeconomic Variables
The largest proportion of the patient population (45.5%) had private insurance (Table 2). Medicare came in a close second covering almost 42.2% of the population, followed by Medicaid (5.0%), uninsured (2.8%), and other government insurance (1.5%). About half (53.7%) of the patients were treated at academic or research facilities, while the fewest number of patients (5.2%) underwent treatment at community cancer facilities. The largest percentage (36.6%) of patients lived in zip codes with the highest level of median household income, while 26.0% and 22.3% had moderate and low levels of income, respectively. About 14% of patients lived within an area of the lowest level of income. Similarly, almost 15% of patients lived in an area of lowest level of HS education. The greatest percentage of the patient population (34.5%) lived in a zip code with moderate level of HS education. Surgery was the most common treatment modality with 90.8% of the cohort undergoing surgery, while 35.4% and 16.5% were treated with radiation and chemotherapy, respectively (some patients received more than one type of treatment modality).
Survival Data
Survival data were available for 3112 patients. Kaplan-Meier survival curves were used to analyze OS according to insurance status, racial background, treatment facility type, median family income, and percentage of adults with no high school education. Overall 5- and 10- year OS probabilities were 51.5% and 34.8%, respectively, while the median OS (SD) was 63.57 (2.8) months (Table 3).
Private insurance showed significantly higher 5- and 10-year OS probabilities and median OS: 5-year OS was 61.2%, 10-year OS was 47.2%, and median survival (SD) was 101.2 (8.2) months compared with that of all other insurance groups (Medicare, Medicaid, other government insurance, and uninsured) (Figure 1). These other insurance types were fairly similar in their 5-year and median OS, but surprisingly, patients with no insurance had the second longest 10-year OS. The difference between the 5-year OS probabilities of private insurance compared with an average of the other insurances was 15.1%, which had almost doubled to 28.5% at 10 years, with a median OS difference of almost 5 years (56 months; data not shown).
Using the Kaplan-Meier survival curve, Asian Indians had the longest 5-year OS probability of 77.9% and African Americans had the longest 10-year OS probability of 40.6%. However, Asians as a group demonstrated the longest median (SD) OS outcome with 119.8 (47.8) months (Figure 2).
Overall, academic/research programs had the longest median OS and 5-year OS probability (SD) of 66.6 (4.5) months and 52.6%, respectively (Figure 3). Comprehensive community cancer programs and integrated network cancer programs had nearly identical 10-year OS rates (35.2% vs 35.1%, respectively). Community cancer programs had the worst 5- and 10-year OS probabilities (41.1% and 21.8%, respectively).
The top 2 income quartiles combined to demonstrate the longest median, 5-year, and 10-year OS probabilities and were very similar. Patients living in a zip code with the highest income level had the longest 5-year OS rates of 54.3%, while patients living in zip codes with a moderate income level had the longest 10-year OS at 39.3% and the longest median OS of about 71 months. Patients with the lowest level of median household income had the worst 5-year OS rates (48.3%) and a median (SD) OS of 53.4 (5.4) months (Figure 4).
A Kaplan-Meier curve for percentage of adults without a HS degree is displayed in Figure 5. Zip codes with the highest level of education had the longest 5-year OS rates and median (SD) OS of 55.3% and 70.9 (4.8) months, respectively. The longest 10-year OS outcomes at 38.1% were found in patients who lived in areas of low-education levels. The worst 5- and 10- year OS outcomes and median OS were found in the least educated zip codes.
Results from the Cox regression model of OS are displayed in Table 4. Race and ethnicity, zip code-level median household income, and zip code-level education were not associated with OS. Patients with no insurance had an increased risk of death (hazard ratio [HR], 1.84; 95% CI, 1.17-2.88; P < .01) when compared with patients with private insurance. Patients with other government insurance also had an increased risk of death (HR, 2.12; 95% CI, 1.27-3.54; P < .01) when compared with patients with private insurance while controlling for all other variables. Patients with Medicare had a decreased risk of death when compared with patients with other government insurance and no insurance (HR, 0.53; 95% CI, 0.31-0.92; P = .02 and HR, 0.62; 95% CI, 0.38-0.99; P = .05, respectively). Patients treated at academic centers had better OS when compared with patients treated at comprehensive treatment centers (HR, 0.77; 95% CI, 0.65-0.92;P < .01) and community treatment centers (HR, 0.62; 95% CI, 0.44-0.86; P < .01).
Discussion
This study is the largest study to date that specifically studies the type of treatment facilities and socioeconomic factors, including insurance status, race, income, and education, and how they affect survival of DDLPS. The overall 5- and 10-year OS probabilities for DDLPS in this study were 51.5% and 34.8%, respectively, with median OS of 63.6 months. These results were more encouraging than previous reports, which found a 5-year survival probability of 36.5% and a median OS of 45 months.13,14
The largest age grouping was aged 61 to 80 years (48.9% of the cohort), and the median age at diagnosis was 64 years. DDLPSs most typically present between the ages of 50 and 70 years.15 Our cohort was 65% male. Previous studies have indicated that DDLPSs affect the sexes equally; however, another study showed a similar male predominance (68.8%) at the MD Anderson Cancer Center in Houston, Texas.13,16
In our study, approximately 88% of patients were white, 6.5% were African American, and 2.5% were Asian, which differed from a previous study of 84 patients that had a 78.6% white, 4.8% Asian, and 1.2% African American patient population.14
Asian Indian or Pakistani patients had the best 5-year OS probability at 77.9%, followed by African American (57.2%), Asian (51.6%), AI/AN (51.4%), and white patients (50.9%). This trend had disappeared by 10 years and Asian, AI/AN, African American, and Asian Indian or Pakistani groups all demonstrated longer median OS than did white patients. In fact, Asian patients had the longest median OS at 119.8 months, which was almost double that of white patients with the lowest median OS of 61.2 months. This finding is contrary to previous studies, which reported that racial minorities typically had worse OS outcomes when compared with white patients in different types of cancer.7,17 Notably, these findings were not statistically significant in our current study in the log-rank or multivariable analyses.
Private insurance was the most common form of insurance followed in decreasing order by Medicare, Medicaid, uninsured, and other government insurance. About 42% of the cohort had Medicare, which is a federally funded US insurance program designated for patients aged ≥ 65 years and certain younger patients with disabilities.
Patients with private insurance demonstrated the longest OS, essentially twice the median OS of all other insured groups at 101 months. Medicare had the worst 5-year OS probability and median OS of all groups. A previous study of 77 patients with DDLPS reported that patients aged > 65 years had reduced OS.13 Medicare patients in this study were older, with a mean and median age at DDLPS diagnosis of 71 and 72 years, respectively, while private insurance had a mean and median age at diagnosis of 56 and 57 years, respectively. Medicare inherently covers older patients and this age difference could account for the decrease in overall survival.
Improved OS for privately insured patients was most notable compared with the uninsured or patients with other government insurance. Uninsured patients had an 83.7% increased risk of mortality when compared with patients with private insurance. When compared with patients with private insurance, patients with other government insurance had an 111.5% increased risk of mortality. Comparing patients with Medicare vs patients with no insurance or other government insurance, there was a decreased risk of mortality of 38.5% and 46.6%, respectively. This decreased OS in patients with other government insurance could be related to the choice of treatment facility, because only 31% of the patients with other government insurance went to academic or research centers when compared with the 58.4% and 50.8% of patients with private and Medicare insurance treated there (data not shown). Such centers often have access to more advanced technology and protocols that may not be available at other treatment facilities.
A little more than half of the patients in the cohort went to an academic or research center for treatment (53.7%); comprehensive community cancer programs were the second most common treatment facility at 28%. Patients treated at academic or research centers demonstrated the best outcomes with a 5-year OS of 52.6%, followed in decreasing order by comprehensive community cancer programs (49.7%), integrated network cancer programs (48.8%), and community cancer programs (41.1%). In our patient cocohort, patients treated at an academic/research center had slightly decreased 10-year OS rates compared with those patients treated at a comprehensive community cancer program, although the median OS for the academic/research centers were still the highest of all treatment facilities.
Treatment options varied significantly by facility, and the number of patients treated surgically followed a similar trend, with 92% undergoing surgery as the primary treatment at academic or research programs compared with 89% at comprehensive cancer programs and 82.7% at community cancer programs (data not shown). Another potential explaination for differing OS outcomes across facilities is the surgical margin outcome. Surgeries performed at community cancer programs or comprehensive cancer programs resulted with no residual tumor in 36% and 40% of cases, respectively, whereas cases performed at academic or research programs resulted with no residual tumor in 47% of cases (data not shown). Regardless, multivariate analysis demonstrated a marked decrease in the chance of mortality when comparing treatment received at academic facility centers with that received at comprehensive cancer centers (22.9%) and community cancer centers (38.3%) (data not shown).
A recent study demonstrated improved outcomes for patients with retroperitoneal or extremity STS treated at high-volume treatment centers.18 Patients treated at high-volume centers were found to have an 8% decreased risk of death compared with patients treated at low-volume centers. Notably, they found highvolume academic centers demonstrated the strongest improvement in survival, while highvolume community centers showed decreased survival.18 Similarly, we found that patients treated at academic/research institutions had improved 5-year OS and greater median OS than did patients treated at community cancer programs or comprehensive community cancer programs.
The top 2 income quartiles (≥ $48,000) combined to demonstrate the longest median, 5-year, and 10-year OS and were fairly similar between the quartiles. Patients living in zip codes with a median income of $38,000 to $47,999 had the worst 5-year OS and median OS. The log-rank analysis showed statistical evidence of differences in survival associated with income, but within the context of the multivariable analysis, there was no remaining evidence of a difference.
The longest 5-year OS outcomes were seen in patients living in zip codes with the highest level of education (55.3%). However, the difference in OS was not statistically significant using either the log-rank analysis or multivariate analysis.
Limitations
This study has certain inherent limitations in using a retrospective design and a large database such as the NCDB. Many different pathologists at CoC-accredited cancer programs perform the pathology that contributes to the data in the NCDB. There was no pathological review of these findings, which could potentially introduce error into the findings of this study. With the NCDB, potential selection bias is possible because patients in the database are added only from CoC-accredited cancer programs. This risk is minimized because NCDB contains data on most newly diagnosed cancer patients in the US. Further potential risks, which are unable to be controlled for, include potential interobserver error and data that may be incompletely, improperly, or inaccurately recorded from the patients’ charts. Without patient-specific information regarding income and education, it is challenging to utilize zip codes to estimate socioeconomic status and educational level. Even though a patient may live in a zip code identified with specific economic and educational characteristics, that patient may not share those characteristics. Furthermore, patients with Medicare tend to be older than patients with other forms of insurance, which limits the significance of comparisons across insurance groups. A future SEER (Surveillance, Epidemiology, and End Results) program study to confirm this study’s results and the effects of socioeconomic variables on DDLPS would be an excellent followup study.
Conclusion
This study used a large cohort of patients with DDLPS to study the effects of treatment facility, insurance status, and socioeconomic variables on survival outcomes. Although insurance status, median household income, and treatment facility were associated with differences in median OS and 5- and 10-year OS probabilities, evidence for a difference remained for only insurance status and facility type within the context of a multivariable analysis irrespective of age, race, sex, insurance status, education, and median income. Patients with private insurance and Medicaid had a decreased risk of mortality compared with other government insurance and no insurance. Patients receiving treatment at academic research programs had the highest median and 5-year OS of 66.6 months and 52.6%, respectively. Patients receiving treatment at academic centers had improved survival outcomes with a decrease in mortality of 23% and 38% compared to comprehensive or community cancer programs.
1. Dodd LG. Update on liposarcoma: a review for cytopathologists. Diagn Cytopathol. 2012;40(12):1122-1131.
2. Mangham D. World Health Organisation classification of tumours: pathology and genetics of tumours of soft tissue and bone. J Bone Joint Surg Am. 2004;86(3):466.
3. Dalal KM, Kattan MW, Antonescu CR, Brennan MF, Singer S. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg. 2006;244(3):381-391.
4. Coindre JM, Pédeutour F, Aurias A. Well-differentiated and dedifferentiated liposarcomas. Virchows Arch. 2010;456(2):167-179.
5. Evans HL. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol. 1979;3(6):507-523.
6. Henricks WH, Chu YC, Goldblum JR, Weiss SW. Dedifferentiated liposarcoma: a clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol. 1997;21(3):271-281.
7. Ward E, Jemal A, Cokkinides V, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin. 2004;54(2):78-93.
8. Halpern MT, Ward EM, Pavluck AL, Schrag NM, Bian J, Chen AY. Association of insurance status and ethnicity with cancer stage at diagnosis for 12 cancer sites: a retrospective analysis. Lancet Oncol. 2008;9(3):222-231.
9. Niu X, Roche LM, Pawlish KS, Henry KA. Cancer survival disparities by health insurance status. Cancer Med. 2013;2(3):403-411.
10. Hauser A, Dutta SW, Showalter TN, Sheehan JP, Grover S, Trifiletti DM. Impact of academic facility type and volume on post-surgical outcomes following diagnosis of glioblastoma. J Clin Neurosci. 2018;47:103-110.
11. Chu Q, Medeiros K, Zhou M, et al. Effect of facility type on outcome following pancreatectomy for pancreatic adenocarcinoma: analysis of the National Cancer Data Base [Abstract FP26-02]. HPB (Oxford). 2016;18(suppl 1):E81-E82.
12. Rubin SJ, Cohen MB, Kirke DN, Qureshi MM, Truong MT, Jalisi S. Comparison of facility type outcomes for oral cavity cancer: analysis of the National Cancer Database. Laryngoscope. 2017;127(11):2551-2557.
13. Lahat G, Anaya DA, Wang X, Tuvin D, Lev D, Pollock RE. Resectable well-differentiated versus dedifferentiated liposarcomas: two different diseases possibly requiring different treatment approaches. Ann Surg Oncol. 2008;15(6):1585-1593.
14. Livingston JA, Bugano D, Barbo A, et al. Role of chemotherapy in dedifferentiated liposarcoma of the retroperitoneum: defining the benefit and challenges of the standard. Sci Rep. 2017;7(1):11836.
15. Brennan MF, Antonescu CR, Alektiar KM, Maki RG. Management of Soft Tissue Sarcoma. 2nd ed. New York, NY: Springer; 2016.
16. Goldblum JR, Folpe AL, Weiss SW. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Philadelphia, PA: Saunders; 2014.
17. White A, Djenaba J, Rim SH, Johnson CJ, Coleman MP, Allemani C. Colon cancer survival in the United States by race and stage (2001‐2009): findings from the CONCORD‐2 study. Cancer. 2017;123 (suppl 24):5014-5036.
18. Murphy JD, Padwal J, Guss ZD, Okamoto K, Sardar R. Impact of hospital volume on patterns of care and outcomes in soft tissue sarcoma [ASCO Abstract e23550]. J Clin Oncol. 2018;36(suppl 15):e23550
1. Dodd LG. Update on liposarcoma: a review for cytopathologists. Diagn Cytopathol. 2012;40(12):1122-1131.
2. Mangham D. World Health Organisation classification of tumours: pathology and genetics of tumours of soft tissue and bone. J Bone Joint Surg Am. 2004;86(3):466.
3. Dalal KM, Kattan MW, Antonescu CR, Brennan MF, Singer S. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg. 2006;244(3):381-391.
4. Coindre JM, Pédeutour F, Aurias A. Well-differentiated and dedifferentiated liposarcomas. Virchows Arch. 2010;456(2):167-179.
5. Evans HL. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol. 1979;3(6):507-523.
6. Henricks WH, Chu YC, Goldblum JR, Weiss SW. Dedifferentiated liposarcoma: a clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol. 1997;21(3):271-281.
7. Ward E, Jemal A, Cokkinides V, et al. Cancer disparities by race/ethnicity and socioeconomic status. CA Cancer J Clin. 2004;54(2):78-93.
8. Halpern MT, Ward EM, Pavluck AL, Schrag NM, Bian J, Chen AY. Association of insurance status and ethnicity with cancer stage at diagnosis for 12 cancer sites: a retrospective analysis. Lancet Oncol. 2008;9(3):222-231.
9. Niu X, Roche LM, Pawlish KS, Henry KA. Cancer survival disparities by health insurance status. Cancer Med. 2013;2(3):403-411.
10. Hauser A, Dutta SW, Showalter TN, Sheehan JP, Grover S, Trifiletti DM. Impact of academic facility type and volume on post-surgical outcomes following diagnosis of glioblastoma. J Clin Neurosci. 2018;47:103-110.
11. Chu Q, Medeiros K, Zhou M, et al. Effect of facility type on outcome following pancreatectomy for pancreatic adenocarcinoma: analysis of the National Cancer Data Base [Abstract FP26-02]. HPB (Oxford). 2016;18(suppl 1):E81-E82.
12. Rubin SJ, Cohen MB, Kirke DN, Qureshi MM, Truong MT, Jalisi S. Comparison of facility type outcomes for oral cavity cancer: analysis of the National Cancer Database. Laryngoscope. 2017;127(11):2551-2557.
13. Lahat G, Anaya DA, Wang X, Tuvin D, Lev D, Pollock RE. Resectable well-differentiated versus dedifferentiated liposarcomas: two different diseases possibly requiring different treatment approaches. Ann Surg Oncol. 2008;15(6):1585-1593.
14. Livingston JA, Bugano D, Barbo A, et al. Role of chemotherapy in dedifferentiated liposarcoma of the retroperitoneum: defining the benefit and challenges of the standard. Sci Rep. 2017;7(1):11836.
15. Brennan MF, Antonescu CR, Alektiar KM, Maki RG. Management of Soft Tissue Sarcoma. 2nd ed. New York, NY: Springer; 2016.
16. Goldblum JR, Folpe AL, Weiss SW. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Philadelphia, PA: Saunders; 2014.
17. White A, Djenaba J, Rim SH, Johnson CJ, Coleman MP, Allemani C. Colon cancer survival in the United States by race and stage (2001‐2009): findings from the CONCORD‐2 study. Cancer. 2017;123 (suppl 24):5014-5036.
18. Murphy JD, Padwal J, Guss ZD, Okamoto K, Sardar R. Impact of hospital volume on patterns of care and outcomes in soft tissue sarcoma [ASCO Abstract e23550]. J Clin Oncol. 2018;36(suppl 15):e23550
Hospital slashes S. aureus vancomycin resistance
LJUBLJANA, SLOVENIA – , Johannes Huebner, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
He presented a retrospective analysis of S. aureus isolates obtained from 540 patients at the Dr. von Hauner Children’s Hospital, Munich, from 2002 to 2017. All were either newly identified methicillin-resistant S. aureus (MRSA) or specimens from bacteremic children with invasive MRSA or methicillin-sensitive S. aureus (MSSA). The strains were tested for vancomycin resistance and minimum inhibitory concentration (MIC). The results from the 200 isolates obtained from 2002 to 2009 were then compared to the 340 specimens from 2010 to 2017, when antibiotic stewardship programs rose to the fore at the pediatric hospital.
All samples proved to be vancomycin sensitive. The further good news was there was absolutely no evidence of the worrisome vancomycin MIC creep that has been described at some centers. On the contrary, the MIC was significantly lower in the later samples, at 0.99 mcg/mL, compared with 1.11 mcg/mL in the earlier period. Moreover, the prevalence of heterogeneous glycopeptide-intermediate S. aureus (hGISA) – a phenotype that has been associated with increased rates of treatment failure – improved from 25% in the earlier period to 6% during the later period, reported Dr. Huebner, head of the division of pediatric infectious diseases at the children’s hospital, part of the University of Munich.
Vancomycin MICs weren’t significantly different between the MRSA and MSSA samples.
Based upon this favorable institutional experience, vancomycin remains the first-line treatment for suspected severe gram-positive cocci infections as well as proven infections involving MRSA at Dr. von Hauner Children’s Hospital.
These vancomycin MIC and hGISA data underscore the importance of periodically monitoring local S. aureus antimicrobial susceptibilities, which, as in this case, can differ from the broader global trends. The vancomycin MIC creep issue hadn’t been studied previously in German hospitals, according to Dr. Huebner.
He and his coworkers have published details of the elements of pediatric antibiotic stewardship programs they have found to be most effective (Infection. 2017 Aug;45[4]:493-504) as well as a systematic review of studies on the favorable economic impact of such programs (J Hosp Infect. 2019 Aug;102[4]:369-376).
Dr. Huebner reported having no financial conflicts regarding his study, which was conducted free of commercial support.
LJUBLJANA, SLOVENIA – , Johannes Huebner, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
He presented a retrospective analysis of S. aureus isolates obtained from 540 patients at the Dr. von Hauner Children’s Hospital, Munich, from 2002 to 2017. All were either newly identified methicillin-resistant S. aureus (MRSA) or specimens from bacteremic children with invasive MRSA or methicillin-sensitive S. aureus (MSSA). The strains were tested for vancomycin resistance and minimum inhibitory concentration (MIC). The results from the 200 isolates obtained from 2002 to 2009 were then compared to the 340 specimens from 2010 to 2017, when antibiotic stewardship programs rose to the fore at the pediatric hospital.
All samples proved to be vancomycin sensitive. The further good news was there was absolutely no evidence of the worrisome vancomycin MIC creep that has been described at some centers. On the contrary, the MIC was significantly lower in the later samples, at 0.99 mcg/mL, compared with 1.11 mcg/mL in the earlier period. Moreover, the prevalence of heterogeneous glycopeptide-intermediate S. aureus (hGISA) – a phenotype that has been associated with increased rates of treatment failure – improved from 25% in the earlier period to 6% during the later period, reported Dr. Huebner, head of the division of pediatric infectious diseases at the children’s hospital, part of the University of Munich.
Vancomycin MICs weren’t significantly different between the MRSA and MSSA samples.
Based upon this favorable institutional experience, vancomycin remains the first-line treatment for suspected severe gram-positive cocci infections as well as proven infections involving MRSA at Dr. von Hauner Children’s Hospital.
These vancomycin MIC and hGISA data underscore the importance of periodically monitoring local S. aureus antimicrobial susceptibilities, which, as in this case, can differ from the broader global trends. The vancomycin MIC creep issue hadn’t been studied previously in German hospitals, according to Dr. Huebner.
He and his coworkers have published details of the elements of pediatric antibiotic stewardship programs they have found to be most effective (Infection. 2017 Aug;45[4]:493-504) as well as a systematic review of studies on the favorable economic impact of such programs (J Hosp Infect. 2019 Aug;102[4]:369-376).
Dr. Huebner reported having no financial conflicts regarding his study, which was conducted free of commercial support.
LJUBLJANA, SLOVENIA – , Johannes Huebner, MD, reported at the annual meeting of the European Society for Paediatric Infectious Diseases.
He presented a retrospective analysis of S. aureus isolates obtained from 540 patients at the Dr. von Hauner Children’s Hospital, Munich, from 2002 to 2017. All were either newly identified methicillin-resistant S. aureus (MRSA) or specimens from bacteremic children with invasive MRSA or methicillin-sensitive S. aureus (MSSA). The strains were tested for vancomycin resistance and minimum inhibitory concentration (MIC). The results from the 200 isolates obtained from 2002 to 2009 were then compared to the 340 specimens from 2010 to 2017, when antibiotic stewardship programs rose to the fore at the pediatric hospital.
All samples proved to be vancomycin sensitive. The further good news was there was absolutely no evidence of the worrisome vancomycin MIC creep that has been described at some centers. On the contrary, the MIC was significantly lower in the later samples, at 0.99 mcg/mL, compared with 1.11 mcg/mL in the earlier period. Moreover, the prevalence of heterogeneous glycopeptide-intermediate S. aureus (hGISA) – a phenotype that has been associated with increased rates of treatment failure – improved from 25% in the earlier period to 6% during the later period, reported Dr. Huebner, head of the division of pediatric infectious diseases at the children’s hospital, part of the University of Munich.
Vancomycin MICs weren’t significantly different between the MRSA and MSSA samples.
Based upon this favorable institutional experience, vancomycin remains the first-line treatment for suspected severe gram-positive cocci infections as well as proven infections involving MRSA at Dr. von Hauner Children’s Hospital.
These vancomycin MIC and hGISA data underscore the importance of periodically monitoring local S. aureus antimicrobial susceptibilities, which, as in this case, can differ from the broader global trends. The vancomycin MIC creep issue hadn’t been studied previously in German hospitals, according to Dr. Huebner.
He and his coworkers have published details of the elements of pediatric antibiotic stewardship programs they have found to be most effective (Infection. 2017 Aug;45[4]:493-504) as well as a systematic review of studies on the favorable economic impact of such programs (J Hosp Infect. 2019 Aug;102[4]:369-376).
Dr. Huebner reported having no financial conflicts regarding his study, which was conducted free of commercial support.
REPORTING FROM ESPID 2019
Key clinical point: Staphylococcus aureus vancomycin MIC creep is reversible through dedicated antimicrobial stewardship.
Major finding: The prevalence of hGISA in MRSA and MSSA specimens improved from 25% during 2002-2009 to 6% during 2010-2017 at one German tertiary children’s hospital.
Study details: This was a retrospective single-center analysis of vancomycin resistance trends over time in 540 S. aureus specimens gathered in 2002-2017.
Disclosures: The presenter reported having no financial conflicts regarding this study, which was conducted free of commercial support.
FDA approves Turalio for symptomatic tenosynovial giant cell tumor
Turalio (pexidartinib) capsules have been approved for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) that is associated with severe morbidity or functional limitations not responsive to improvement with surgery, the U.S. Food and Drug Administration announced.

Turalio is the first therapy to be approved for the rare joint tumor and is available only through the Turalio Risk Evaluation and Mitigation Strategy (REMS) Program. The FDA granted the approval of Turalio to Daiichi Sankyo.
“TGCT can cause debilitating symptoms for patients such as pain, stiffness and limitation of movement,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a statement. “Surgery is the primary treatment option, but some patients are not eligible for surgery, and tumors can recur, even after the procedure.”
The approval was based on results of a study of 120 patients, 59 of whom received placebo. After 25 weeks of treatment, the overall response rate was 38% (15% complete responses and 23% partial responses) in those who received pexidartinib; no responses occurred in patients who received placebo. The response persisted in 22 of 23 responders who had been followed for a minimum of 6 months, and in 13 of 13 responders who had been followed for a minimum of 12 months.
Turalio comes with a Boxed Warning about the risk of serious and potentially fatal liver injury. Liver tests should be performed prior to beginning treatment and the results monitored at specified intervals during treatment. Patients who develop abnormal results may need to withhold therapy, reduce the dose, or discontinue therapy depending on the severity of the liver injury.
Common side effects for patients were increased levels of lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, and cholesterol. Loss of hair color also occurred in some patients.
Additional side effects included neutropenia, increased alkaline phosphatase levels, decreased lymphocytes, eye edema, decreased hemoglobin levels, rash, dysgeusia, and decreased phosphate levels.
Females of reproductive age and males with a female partner of reproductive potential should use effective contraception during treatment with pexidartinib. Pexidartinib may cause harm to a developing fetus or newborn baby.
Pexidartinib must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Turalio (pexidartinib) capsules have been approved for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) that is associated with severe morbidity or functional limitations not responsive to improvement with surgery, the U.S. Food and Drug Administration announced.
Turalio is the first therapy to be approved for the rare joint tumor and is available only through the Turalio Risk Evaluation and Mitigation Strategy (REMS) Program. The FDA granted the approval of Turalio to Daiichi Sankyo.
“TGCT can cause debilitating symptoms for patients such as pain, stiffness and limitation of movement,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a statement. “Surgery is the primary treatment option, but some patients are not eligible for surgery, and tumors can recur, even after the procedure.”
The approval was based on results of a study of 120 patients, 59 of whom received placebo. After 25 weeks of treatment, the overall response rate was 38% (15% complete responses and 23% partial responses) in those who received pexidartinib; no responses occurred in patients who received placebo. The response persisted in 22 of 23 responders who had been followed for a minimum of 6 months, and in 13 of 13 responders who had been followed for a minimum of 12 months.
Turalio comes with a Boxed Warning about the risk of serious and potentially fatal liver injury. Liver tests should be performed prior to beginning treatment and the results monitored at specified intervals during treatment. Patients who develop abnormal results may need to withhold therapy, reduce the dose, or discontinue therapy depending on the severity of the liver injury.
Common side effects for patients were increased levels of lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, and cholesterol. Loss of hair color also occurred in some patients.
Additional side effects included neutropenia, increased alkaline phosphatase levels, decreased lymphocytes, eye edema, decreased hemoglobin levels, rash, dysgeusia, and decreased phosphate levels.
Females of reproductive age and males with a female partner of reproductive potential should use effective contraception during treatment with pexidartinib. Pexidartinib may cause harm to a developing fetus or newborn baby.
Pexidartinib must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Turalio (pexidartinib) capsules have been approved for the treatment of adult patients with symptomatic tenosynovial giant cell tumor (TGCT) that is associated with severe morbidity or functional limitations not responsive to improvement with surgery, the U.S. Food and Drug Administration announced.
Turalio is the first therapy to be approved for the rare joint tumor and is available only through the Turalio Risk Evaluation and Mitigation Strategy (REMS) Program. The FDA granted the approval of Turalio to Daiichi Sankyo.
“TGCT can cause debilitating symptoms for patients such as pain, stiffness and limitation of movement,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a statement. “Surgery is the primary treatment option, but some patients are not eligible for surgery, and tumors can recur, even after the procedure.”
The approval was based on results of a study of 120 patients, 59 of whom received placebo. After 25 weeks of treatment, the overall response rate was 38% (15% complete responses and 23% partial responses) in those who received pexidartinib; no responses occurred in patients who received placebo. The response persisted in 22 of 23 responders who had been followed for a minimum of 6 months, and in 13 of 13 responders who had been followed for a minimum of 12 months.
Turalio comes with a Boxed Warning about the risk of serious and potentially fatal liver injury. Liver tests should be performed prior to beginning treatment and the results monitored at specified intervals during treatment. Patients who develop abnormal results may need to withhold therapy, reduce the dose, or discontinue therapy depending on the severity of the liver injury.
Common side effects for patients were increased levels of lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, and cholesterol. Loss of hair color also occurred in some patients.
Additional side effects included neutropenia, increased alkaline phosphatase levels, decreased lymphocytes, eye edema, decreased hemoglobin levels, rash, dysgeusia, and decreased phosphate levels.
Females of reproductive age and males with a female partner of reproductive potential should use effective contraception during treatment with pexidartinib. Pexidartinib may cause harm to a developing fetus or newborn baby.
Pexidartinib must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Lenabasum, a novel cannabinoid, shows promise in dermatomyositis
MILAN – according to Victoria Werth, MD, who presented early-stage findings at the World Congress of Dermatology.
Lenabasum – previously known as anabasum – is a synthetic cannabinoid that binds to the CB2 receptor present on a variety of cells, including lymphocytes, explained Dr. Werth, professor of medicine at the University of Pennsylvania, Philadelphia, and chief of dermatology at the Philadelphia Veterans Administration Hospital. The nonpsychoactive compound’s mechanism of action helps prevent tissue thickening and fibrosis. In addition to its potential for dermatomyositis treatment, it is also being investigated as a treatment for cystic fibrosis, scleroderma, and lupus.
Dr. Werth added that earlier in vitro work with peripheral blood mononuclear cells of patients with dermatomyositis showed that lenabasum markedly suppressed the cells’ secretion of tumor necrosis factor–alpha, interferon-alpha, and interferon-beta (J Invest Dermatol. 2017 Nov;137[11]:2445-7). A randomized, placebo-controlled trial tested lenabasum, known in the trial as JBT-101, in 22 patients with skin-predominant myositis. The study was structured so patients took a half dose of lenabasum for 1 month, followed by 2 months of taking a full dose, and finally 1 month with no lenabasum dosing. Compared with placebo, scores on the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI) were significantly improved with full dosing of lenabasum (P = .02), Dr. Werth said.
In this 16-week trial, the mean change in CDASI score dropped for participants in both the lenabasum and placebo groups for the first 4 weeks. After that, however, those taking lenabasum saw an improvement of about 8 points from baseline CDASI score at 8 weeks, while those taking placebo had a decline of 3 points (P = .05). The differences remained significant at trial’s end.
Patient-reported outcomes for pain were significantly better with lenabasum, with patients on lenabasum recording a decrease on the Patient-Reported Outcomes Measurement Information System–29 pain interference scale, while patients on placebo reported an increase in pain by the end of the study period (P = .026). Scores on two other patient-reported dermatomyositis scales were numerically improved for patients taking lenabasum, but the differences from the patients on placebo were not statistically significant, Dr. Werth said.
However, patients in the initial clinical trial were given the opportunity to continue in a long-term extension arm, and that group has seen the clinician-rated CDASI scores continue to fall, with a mean decrease of 22 points from baseline (about 12 further points from the mean for those on lenabasum in the initial trial) at the 68-week mark, she added.
Itch scores continued to drop as well, with a reduction of 3.7 points on the 5-D Itch scale by 68 weeks; at 16 weeks, the reduction for the lenabasum arm had been 1.3 points.
Skin samples taken from trial participants showed many fewer CD4 cells in those taking lenabasum, compared with placebo at week 12 of the initial study. Interferon-beta and -gamma levels also dropped in those taking lenabasum, but not in the placebo arm.
“Lenabasum has effects on cytokine signatures and inflammatory cells correlating with response to therapy,” Dr. Werth said, adding that the findings gave support for a planned global phase 3 clinical trial of lenabasum in dermatomyositis.
Dr. Werth reported receiving grants from Pfizer and Corbus, and consulting fees from Pfizer, Janssen, Neovacs, Idera Pharmaceuticals, Octapharma, CSL Behring, and Corbus Pharmaceuticals. The study was funded by Corbus, the National Institutes of Health, and the University of Pennsylvania.
Cannabinoids represent a broad class of chemical compounds originally comprised only phytocannabinoids – cannabinoids produced by the cannabis plant. The best-known and most-studied cannabinoids are tetrahydrocannabinol and cannabidiol, which are plant-derived, or “phyto”, cannabinoids. While tetrahydrocannabinol is best known for its mind-altering, munchy-causing properties, it was in fact the study of this illegal substance that led to the groundbreaking discovery of the human endocannabinoid system. (Meaning, we make our very own cannabinoids and receptors for them, which make up an extraordinary biological network that play a role in everything from sensations of pain and itch to mood, inflammation regulation, and wound healing.) Gold star for drugs?

It was this understanding that led to the development of synthetic cannabinoids, like ajulemic acid (aka lenabasum), all of which has created a flurry of investigative productivity and creativity to capitalize on these bioactive agents. That said, development, research and development, and even education in/on this area has been hindered for many years because of both regulatory limitations and negative public perceptions, given the Schedule 1 designation of every component of the cannabis plant (even nonpsychoactive actives) up until a few months ago with the passing of the Farm Bill (which made hemp legal).
However, the interest level among consumers, patients, and physicians is quickly growing regarding the effectiveness of cannabinoids in the treatment of a laundry list of skin conditions and symptoms, including psoriasis, atopic dermatitis, and wound healing. But there are just so many unanswered questions for anyone to be certain about the benefits. We recently published a study surveying 531 dermatologists to get a better idea about our community’s attitude and awareness on cannabinoids as therapeutics, and it turned out there’s a lot we all need to learn (J Drugs Dermatol. 2018 Dec 1;17[12]:1273-8). Some highlights of our findings:
- Dermatologists are being approached by their patients with questions on this subject matter, and this is more likely to occur in states where medical cannabis is legalized.
- While more than 90% of respondents agreed that this is an important area for research and development and 86% thought medical cannabinoids should be legal, more than 80% were not comfortable with their understanding or knowledge on this subject matter, which is not surprising given that 64% of respondents incorrectly responded that cannabidiol has psychoactive effects.
With the fast-tracking of lenabasum down the Food and Drug Administration approval pathway for not one but two diseases we as dermatologists manage, I am optimistic that this addition to our much needed armament will serve as further stimulus to expand the role of cannabinoids in the management of dermatologic diseases and spotlight the need for more education and research in this vastly underrecognized yet intriguing and promising space.
Adam Friedman, MD, is professor and interim chair of dermatology, director of translational research, and director of the supportive oncodermatology clinic at George Washington University, Washington, and is on the board of Dermatology News. Dr. Friedman is on the scientific advisory board for Corbus Pharmaceuticals.
Cannabinoids represent a broad class of chemical compounds originally comprised only phytocannabinoids – cannabinoids produced by the cannabis plant. The best-known and most-studied cannabinoids are tetrahydrocannabinol and cannabidiol, which are plant-derived, or “phyto”, cannabinoids. While tetrahydrocannabinol is best known for its mind-altering, munchy-causing properties, it was in fact the study of this illegal substance that led to the groundbreaking discovery of the human endocannabinoid system. (Meaning, we make our very own cannabinoids and receptors for them, which make up an extraordinary biological network that play a role in everything from sensations of pain and itch to mood, inflammation regulation, and wound healing.) Gold star for drugs?
It was this understanding that led to the development of synthetic cannabinoids, like ajulemic acid (aka lenabasum), all of which has created a flurry of investigative productivity and creativity to capitalize on these bioactive agents. That said, development, research and development, and even education in/on this area has been hindered for many years because of both regulatory limitations and negative public perceptions, given the Schedule 1 designation of every component of the cannabis plant (even nonpsychoactive actives) up until a few months ago with the passing of the Farm Bill (which made hemp legal).
However, the interest level among consumers, patients, and physicians is quickly growing regarding the effectiveness of cannabinoids in the treatment of a laundry list of skin conditions and symptoms, including psoriasis, atopic dermatitis, and wound healing. But there are just so many unanswered questions for anyone to be certain about the benefits. We recently published a study surveying 531 dermatologists to get a better idea about our community’s attitude and awareness on cannabinoids as therapeutics, and it turned out there’s a lot we all need to learn (J Drugs Dermatol. 2018 Dec 1;17[12]:1273-8). Some highlights of our findings:
- Dermatologists are being approached by their patients with questions on this subject matter, and this is more likely to occur in states where medical cannabis is legalized.
- While more than 90% of respondents agreed that this is an important area for research and development and 86% thought medical cannabinoids should be legal, more than 80% were not comfortable with their understanding or knowledge on this subject matter, which is not surprising given that 64% of respondents incorrectly responded that cannabidiol has psychoactive effects.
With the fast-tracking of lenabasum down the Food and Drug Administration approval pathway for not one but two diseases we as dermatologists manage, I am optimistic that this addition to our much needed armament will serve as further stimulus to expand the role of cannabinoids in the management of dermatologic diseases and spotlight the need for more education and research in this vastly underrecognized yet intriguing and promising space.
Adam Friedman, MD, is professor and interim chair of dermatology, director of translational research, and director of the supportive oncodermatology clinic at George Washington University, Washington, and is on the board of Dermatology News. Dr. Friedman is on the scientific advisory board for Corbus Pharmaceuticals.
Cannabinoids represent a broad class of chemical compounds originally comprised only phytocannabinoids – cannabinoids produced by the cannabis plant. The best-known and most-studied cannabinoids are tetrahydrocannabinol and cannabidiol, which are plant-derived, or “phyto”, cannabinoids. While tetrahydrocannabinol is best known for its mind-altering, munchy-causing properties, it was in fact the study of this illegal substance that led to the groundbreaking discovery of the human endocannabinoid system. (Meaning, we make our very own cannabinoids and receptors for them, which make up an extraordinary biological network that play a role in everything from sensations of pain and itch to mood, inflammation regulation, and wound healing.) Gold star for drugs?
It was this understanding that led to the development of synthetic cannabinoids, like ajulemic acid (aka lenabasum), all of which has created a flurry of investigative productivity and creativity to capitalize on these bioactive agents. That said, development, research and development, and even education in/on this area has been hindered for many years because of both regulatory limitations and negative public perceptions, given the Schedule 1 designation of every component of the cannabis plant (even nonpsychoactive actives) up until a few months ago with the passing of the Farm Bill (which made hemp legal).
However, the interest level among consumers, patients, and physicians is quickly growing regarding the effectiveness of cannabinoids in the treatment of a laundry list of skin conditions and symptoms, including psoriasis, atopic dermatitis, and wound healing. But there are just so many unanswered questions for anyone to be certain about the benefits. We recently published a study surveying 531 dermatologists to get a better idea about our community’s attitude and awareness on cannabinoids as therapeutics, and it turned out there’s a lot we all need to learn (J Drugs Dermatol. 2018 Dec 1;17[12]:1273-8). Some highlights of our findings:
- Dermatologists are being approached by their patients with questions on this subject matter, and this is more likely to occur in states where medical cannabis is legalized.
- While more than 90% of respondents agreed that this is an important area for research and development and 86% thought medical cannabinoids should be legal, more than 80% were not comfortable with their understanding or knowledge on this subject matter, which is not surprising given that 64% of respondents incorrectly responded that cannabidiol has psychoactive effects.
With the fast-tracking of lenabasum down the Food and Drug Administration approval pathway for not one but two diseases we as dermatologists manage, I am optimistic that this addition to our much needed armament will serve as further stimulus to expand the role of cannabinoids in the management of dermatologic diseases and spotlight the need for more education and research in this vastly underrecognized yet intriguing and promising space.
Adam Friedman, MD, is professor and interim chair of dermatology, director of translational research, and director of the supportive oncodermatology clinic at George Washington University, Washington, and is on the board of Dermatology News. Dr. Friedman is on the scientific advisory board for Corbus Pharmaceuticals.
MILAN – according to Victoria Werth, MD, who presented early-stage findings at the World Congress of Dermatology.
Lenabasum – previously known as anabasum – is a synthetic cannabinoid that binds to the CB2 receptor present on a variety of cells, including lymphocytes, explained Dr. Werth, professor of medicine at the University of Pennsylvania, Philadelphia, and chief of dermatology at the Philadelphia Veterans Administration Hospital. The nonpsychoactive compound’s mechanism of action helps prevent tissue thickening and fibrosis. In addition to its potential for dermatomyositis treatment, it is also being investigated as a treatment for cystic fibrosis, scleroderma, and lupus.
Dr. Werth added that earlier in vitro work with peripheral blood mononuclear cells of patients with dermatomyositis showed that lenabasum markedly suppressed the cells’ secretion of tumor necrosis factor–alpha, interferon-alpha, and interferon-beta (J Invest Dermatol. 2017 Nov;137[11]:2445-7). A randomized, placebo-controlled trial tested lenabasum, known in the trial as JBT-101, in 22 patients with skin-predominant myositis. The study was structured so patients took a half dose of lenabasum for 1 month, followed by 2 months of taking a full dose, and finally 1 month with no lenabasum dosing. Compared with placebo, scores on the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI) were significantly improved with full dosing of lenabasum (P = .02), Dr. Werth said.
In this 16-week trial, the mean change in CDASI score dropped for participants in both the lenabasum and placebo groups for the first 4 weeks. After that, however, those taking lenabasum saw an improvement of about 8 points from baseline CDASI score at 8 weeks, while those taking placebo had a decline of 3 points (P = .05). The differences remained significant at trial’s end.
Patient-reported outcomes for pain were significantly better with lenabasum, with patients on lenabasum recording a decrease on the Patient-Reported Outcomes Measurement Information System–29 pain interference scale, while patients on placebo reported an increase in pain by the end of the study period (P = .026). Scores on two other patient-reported dermatomyositis scales were numerically improved for patients taking lenabasum, but the differences from the patients on placebo were not statistically significant, Dr. Werth said.
However, patients in the initial clinical trial were given the opportunity to continue in a long-term extension arm, and that group has seen the clinician-rated CDASI scores continue to fall, with a mean decrease of 22 points from baseline (about 12 further points from the mean for those on lenabasum in the initial trial) at the 68-week mark, she added.
Itch scores continued to drop as well, with a reduction of 3.7 points on the 5-D Itch scale by 68 weeks; at 16 weeks, the reduction for the lenabasum arm had been 1.3 points.
Skin samples taken from trial participants showed many fewer CD4 cells in those taking lenabasum, compared with placebo at week 12 of the initial study. Interferon-beta and -gamma levels also dropped in those taking lenabasum, but not in the placebo arm.
“Lenabasum has effects on cytokine signatures and inflammatory cells correlating with response to therapy,” Dr. Werth said, adding that the findings gave support for a planned global phase 3 clinical trial of lenabasum in dermatomyositis.
Dr. Werth reported receiving grants from Pfizer and Corbus, and consulting fees from Pfizer, Janssen, Neovacs, Idera Pharmaceuticals, Octapharma, CSL Behring, and Corbus Pharmaceuticals. The study was funded by Corbus, the National Institutes of Health, and the University of Pennsylvania.
MILAN – according to Victoria Werth, MD, who presented early-stage findings at the World Congress of Dermatology.
Lenabasum – previously known as anabasum – is a synthetic cannabinoid that binds to the CB2 receptor present on a variety of cells, including lymphocytes, explained Dr. Werth, professor of medicine at the University of Pennsylvania, Philadelphia, and chief of dermatology at the Philadelphia Veterans Administration Hospital. The nonpsychoactive compound’s mechanism of action helps prevent tissue thickening and fibrosis. In addition to its potential for dermatomyositis treatment, it is also being investigated as a treatment for cystic fibrosis, scleroderma, and lupus.
Dr. Werth added that earlier in vitro work with peripheral blood mononuclear cells of patients with dermatomyositis showed that lenabasum markedly suppressed the cells’ secretion of tumor necrosis factor–alpha, interferon-alpha, and interferon-beta (J Invest Dermatol. 2017 Nov;137[11]:2445-7). A randomized, placebo-controlled trial tested lenabasum, known in the trial as JBT-101, in 22 patients with skin-predominant myositis. The study was structured so patients took a half dose of lenabasum for 1 month, followed by 2 months of taking a full dose, and finally 1 month with no lenabasum dosing. Compared with placebo, scores on the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI) were significantly improved with full dosing of lenabasum (P = .02), Dr. Werth said.
In this 16-week trial, the mean change in CDASI score dropped for participants in both the lenabasum and placebo groups for the first 4 weeks. After that, however, those taking lenabasum saw an improvement of about 8 points from baseline CDASI score at 8 weeks, while those taking placebo had a decline of 3 points (P = .05). The differences remained significant at trial’s end.
Patient-reported outcomes for pain were significantly better with lenabasum, with patients on lenabasum recording a decrease on the Patient-Reported Outcomes Measurement Information System–29 pain interference scale, while patients on placebo reported an increase in pain by the end of the study period (P = .026). Scores on two other patient-reported dermatomyositis scales were numerically improved for patients taking lenabasum, but the differences from the patients on placebo were not statistically significant, Dr. Werth said.
However, patients in the initial clinical trial were given the opportunity to continue in a long-term extension arm, and that group has seen the clinician-rated CDASI scores continue to fall, with a mean decrease of 22 points from baseline (about 12 further points from the mean for those on lenabasum in the initial trial) at the 68-week mark, she added.
Itch scores continued to drop as well, with a reduction of 3.7 points on the 5-D Itch scale by 68 weeks; at 16 weeks, the reduction for the lenabasum arm had been 1.3 points.
Skin samples taken from trial participants showed many fewer CD4 cells in those taking lenabasum, compared with placebo at week 12 of the initial study. Interferon-beta and -gamma levels also dropped in those taking lenabasum, but not in the placebo arm.
“Lenabasum has effects on cytokine signatures and inflammatory cells correlating with response to therapy,” Dr. Werth said, adding that the findings gave support for a planned global phase 3 clinical trial of lenabasum in dermatomyositis.
Dr. Werth reported receiving grants from Pfizer and Corbus, and consulting fees from Pfizer, Janssen, Neovacs, Idera Pharmaceuticals, Octapharma, CSL Behring, and Corbus Pharmaceuticals. The study was funded by Corbus, the National Institutes of Health, and the University of Pennsylvania.
EXPERT ANALYSIS FROM WCD2019