User login
Can an antisense oligonucleotide benefit patients with SOD1-ALS?
PHILADELPHIA – In patients with amyotrophic lateral sclerosis (ALS) caused by gain-of-toxic function mutations in the SOD1 gene, according to phase 1/2 trial results presented at the annual meeting of the American Academy of Neurology. Exploratory analyses suggest that the drug, known as tofersen (also known as BIIB067), may lessen declines in function, respiratory function, and strength. The treatment generally was safe and well tolerated, researchers said.
Most ALS cases are sporadic, but about 10% are genetic, of which approximately 20% are caused by SOD1 mutations. “Although SOD1-ALS disease progression is heterogeneous, the underlying pathophysiology, attributable to mutant SOD1 toxicity, is thought to be consistent across SOD1 mutation types,” said Timothy M. Miller, MD, PhD, of Washington University, St. Louis, and colleagues. “As such, effective reduction of SOD1 protein, irrespective of mutation, has the potential to alter the disease course of people with SOD1-ALS.”
Tofersen is an antisense oligonucleotide ribonuclease H1-mediated inhibitor of SOD1 messenger RNA. To study its safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy in patients with SOD1-ALS, investigators conducted a double-blind study. The investigators randomized 50 patients with ALS with a SOD1 mutation to 20 mg (n = 10), 40 mg (n = 9), 60 mg (n = 9), or 100 mg (n = 10) of tofersen or placebo (n = 12).
Patients received tofersen by intrathecal bolus over 1-3 minutes. They received a loading regimen on days 1, 15, and 29 and maintenance dosing on days 57 and 85. After the dosing period, patients completed a 12-week follow-up period.
Adverse events
All patients received at least one dose of the study treatment, and 48 received all treatments. Three patients died during the study. One patient in the 20-mg group died of pulmonary embolism, and one patient in both the 60-mg group and placebo group died of respiratory failure. Investigators considered the deaths secondary to ALS or other conditions and not drug related.
Most adverse events were mild or moderate. The most common adverse events among tofersen-treated patients were headache (n = 16), procedural pain (n = 14), and post–lumbar puncture syndrome (n = 13). Five patients who received tofersen and two who received placebo experienced serious adverse events; no serious adverse events occurred in the 100-mg dose group.
“A reduction from baseline in CSF SOD1 concentrations was observed in the tofersen 40-, 60-, and 100-mg cohorts with the maximal reduction observed in the 100 mg–treated group [37% vs. no reduction in the placebo group; P less than 0.002] at day 85,” the investigators reported.
Possible efficacy
In exploratory analyses, the 100-mg dose slowed decline on the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ASLFRS-R), compared with placebo. Mean change in ASLFRS-R from baseline to day 85 was –1.1 in patients who received 100 mg of tofersen, compared with –5.3 for patients who received placebo. Declines on measures of respiratory function and muscle strength also slowed. “Across clinical measures, separation from placebo was most apparent in participants with fast progressing disease,” the researchers said.
“Lower concentrations of the protein in the spinal fluid suggest that there were also lower concentrations in the brain and spinal cord,” Dr. Miller said in a news release. “Such reductions could lead to preservation of motor neurons and slow progression of the disease, but more study is needed to examine this further.”
The study was sponsored by Biogen, which is developing tofersen. Some of the study authors are Biogen employees. Dr. Miller is on Biogen’s medical advisory board and receives clinical research support from Biogen. In addition, he consults, has licensing agreements with, and is a principal investigator for other companies.
PHILADELPHIA – In patients with amyotrophic lateral sclerosis (ALS) caused by gain-of-toxic function mutations in the SOD1 gene, according to phase 1/2 trial results presented at the annual meeting of the American Academy of Neurology. Exploratory analyses suggest that the drug, known as tofersen (also known as BIIB067), may lessen declines in function, respiratory function, and strength. The treatment generally was safe and well tolerated, researchers said.
Most ALS cases are sporadic, but about 10% are genetic, of which approximately 20% are caused by SOD1 mutations. “Although SOD1-ALS disease progression is heterogeneous, the underlying pathophysiology, attributable to mutant SOD1 toxicity, is thought to be consistent across SOD1 mutation types,” said Timothy M. Miller, MD, PhD, of Washington University, St. Louis, and colleagues. “As such, effective reduction of SOD1 protein, irrespective of mutation, has the potential to alter the disease course of people with SOD1-ALS.”
Tofersen is an antisense oligonucleotide ribonuclease H1-mediated inhibitor of SOD1 messenger RNA. To study its safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy in patients with SOD1-ALS, investigators conducted a double-blind study. The investigators randomized 50 patients with ALS with a SOD1 mutation to 20 mg (n = 10), 40 mg (n = 9), 60 mg (n = 9), or 100 mg (n = 10) of tofersen or placebo (n = 12).
Patients received tofersen by intrathecal bolus over 1-3 minutes. They received a loading regimen on days 1, 15, and 29 and maintenance dosing on days 57 and 85. After the dosing period, patients completed a 12-week follow-up period.
Adverse events
All patients received at least one dose of the study treatment, and 48 received all treatments. Three patients died during the study. One patient in the 20-mg group died of pulmonary embolism, and one patient in both the 60-mg group and placebo group died of respiratory failure. Investigators considered the deaths secondary to ALS or other conditions and not drug related.
Most adverse events were mild or moderate. The most common adverse events among tofersen-treated patients were headache (n = 16), procedural pain (n = 14), and post–lumbar puncture syndrome (n = 13). Five patients who received tofersen and two who received placebo experienced serious adverse events; no serious adverse events occurred in the 100-mg dose group.
“A reduction from baseline in CSF SOD1 concentrations was observed in the tofersen 40-, 60-, and 100-mg cohorts with the maximal reduction observed in the 100 mg–treated group [37% vs. no reduction in the placebo group; P less than 0.002] at day 85,” the investigators reported.
Possible efficacy
In exploratory analyses, the 100-mg dose slowed decline on the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ASLFRS-R), compared with placebo. Mean change in ASLFRS-R from baseline to day 85 was –1.1 in patients who received 100 mg of tofersen, compared with –5.3 for patients who received placebo. Declines on measures of respiratory function and muscle strength also slowed. “Across clinical measures, separation from placebo was most apparent in participants with fast progressing disease,” the researchers said.
“Lower concentrations of the protein in the spinal fluid suggest that there were also lower concentrations in the brain and spinal cord,” Dr. Miller said in a news release. “Such reductions could lead to preservation of motor neurons and slow progression of the disease, but more study is needed to examine this further.”
The study was sponsored by Biogen, which is developing tofersen. Some of the study authors are Biogen employees. Dr. Miller is on Biogen’s medical advisory board and receives clinical research support from Biogen. In addition, he consults, has licensing agreements with, and is a principal investigator for other companies.
PHILADELPHIA – In patients with amyotrophic lateral sclerosis (ALS) caused by gain-of-toxic function mutations in the SOD1 gene, according to phase 1/2 trial results presented at the annual meeting of the American Academy of Neurology. Exploratory analyses suggest that the drug, known as tofersen (also known as BIIB067), may lessen declines in function, respiratory function, and strength. The treatment generally was safe and well tolerated, researchers said.
Most ALS cases are sporadic, but about 10% are genetic, of which approximately 20% are caused by SOD1 mutations. “Although SOD1-ALS disease progression is heterogeneous, the underlying pathophysiology, attributable to mutant SOD1 toxicity, is thought to be consistent across SOD1 mutation types,” said Timothy M. Miller, MD, PhD, of Washington University, St. Louis, and colleagues. “As such, effective reduction of SOD1 protein, irrespective of mutation, has the potential to alter the disease course of people with SOD1-ALS.”
Tofersen is an antisense oligonucleotide ribonuclease H1-mediated inhibitor of SOD1 messenger RNA. To study its safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy in patients with SOD1-ALS, investigators conducted a double-blind study. The investigators randomized 50 patients with ALS with a SOD1 mutation to 20 mg (n = 10), 40 mg (n = 9), 60 mg (n = 9), or 100 mg (n = 10) of tofersen or placebo (n = 12).
Patients received tofersen by intrathecal bolus over 1-3 minutes. They received a loading regimen on days 1, 15, and 29 and maintenance dosing on days 57 and 85. After the dosing period, patients completed a 12-week follow-up period.
Adverse events
All patients received at least one dose of the study treatment, and 48 received all treatments. Three patients died during the study. One patient in the 20-mg group died of pulmonary embolism, and one patient in both the 60-mg group and placebo group died of respiratory failure. Investigators considered the deaths secondary to ALS or other conditions and not drug related.
Most adverse events were mild or moderate. The most common adverse events among tofersen-treated patients were headache (n = 16), procedural pain (n = 14), and post–lumbar puncture syndrome (n = 13). Five patients who received tofersen and two who received placebo experienced serious adverse events; no serious adverse events occurred in the 100-mg dose group.
“A reduction from baseline in CSF SOD1 concentrations was observed in the tofersen 40-, 60-, and 100-mg cohorts with the maximal reduction observed in the 100 mg–treated group [37% vs. no reduction in the placebo group; P less than 0.002] at day 85,” the investigators reported.
Possible efficacy
In exploratory analyses, the 100-mg dose slowed decline on the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ASLFRS-R), compared with placebo. Mean change in ASLFRS-R from baseline to day 85 was –1.1 in patients who received 100 mg of tofersen, compared with –5.3 for patients who received placebo. Declines on measures of respiratory function and muscle strength also slowed. “Across clinical measures, separation from placebo was most apparent in participants with fast progressing disease,” the researchers said.
“Lower concentrations of the protein in the spinal fluid suggest that there were also lower concentrations in the brain and spinal cord,” Dr. Miller said in a news release. “Such reductions could lead to preservation of motor neurons and slow progression of the disease, but more study is needed to examine this further.”
The study was sponsored by Biogen, which is developing tofersen. Some of the study authors are Biogen employees. Dr. Miller is on Biogen’s medical advisory board and receives clinical research support from Biogen. In addition, he consults, has licensing agreements with, and is a principal investigator for other companies.
REPORTING FROM AAN 2019
Researchers propose new risk groups for NK-AML
NEWPORT BEACH, CALIF. – New research suggests patients with normal karyotype acute myeloid leukemia (NK-AML) can be divided into four risk groups associated with overall survival.
Investigators used machine learning algorithms to study the association between mutations and overall survival in 1,352 patients with NK-AML. The analysis revealed combinations of mutations that could be used to classify NK-AML patients into favorable, intermediate-1, intermediate-2, and unfavorable risk groups.
For example, patients who had NPM1 mutations but wild-type FLT3-ITD and DNMT3A, had a median overall survival of 99.1 months and could be classified as favorable risk. Conversely, patients who had NPM1, FLT3-ITD, and DNMT3A mutations, had a median overall survival of 13.4 months and could be classified as unfavorable risk.
Aziz Nazha, MD, of the Cleveland Clinic, and his colleagues conducted this research and presented the findings at the Acute Leukemia Forum of Hemedicus.
The investigators looked at genomic and clinical data from 1,352 patients with NK-AML. The patients were a median age of 55 years and had a median white blood cell count of 21.3 x 109/L, a median hemoglobin of 9.1 g/dL, and a median platelet count of 61 x 109/L. More than half of patients (57.3%) were male.
The patients were screened for 35 genes that are commonly mutated in AML and other myeloid malignancies. The investigators used machine learning algorithms, including random survival forest and recommender system algorithms, to study the association between mutations and overall survival in an “unbiased” way.
Dr. Nazha said there were a median of three mutations per patient sample, and “there are some competing interests between those mutations to impact the prognosis of the patient.”
The investigators used the mutations and their associations with overall survival to classify patients into the risk groups outlined in the table below.
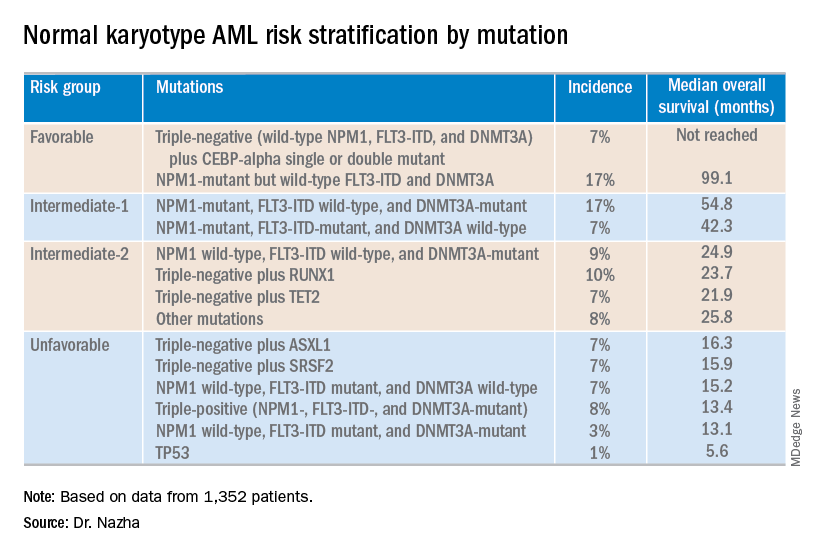
These findings can improve the risk stratification of NK-AML and may aid physicians in making treatment decisions, according to Dr. Nazha and his colleagues. To move this work forward, the investigators are attempting to develop a personalized model that can make predictions specific to an individual patient based on that patient’s mutation information.
Dr. Nazha reported having no financial disclosures relevant to this research. Other investigators reported relationships with the Munich Leukemia Laboratory.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – New research suggests patients with normal karyotype acute myeloid leukemia (NK-AML) can be divided into four risk groups associated with overall survival.
Investigators used machine learning algorithms to study the association between mutations and overall survival in 1,352 patients with NK-AML. The analysis revealed combinations of mutations that could be used to classify NK-AML patients into favorable, intermediate-1, intermediate-2, and unfavorable risk groups.
For example, patients who had NPM1 mutations but wild-type FLT3-ITD and DNMT3A, had a median overall survival of 99.1 months and could be classified as favorable risk. Conversely, patients who had NPM1, FLT3-ITD, and DNMT3A mutations, had a median overall survival of 13.4 months and could be classified as unfavorable risk.
Aziz Nazha, MD, of the Cleveland Clinic, and his colleagues conducted this research and presented the findings at the Acute Leukemia Forum of Hemedicus.
The investigators looked at genomic and clinical data from 1,352 patients with NK-AML. The patients were a median age of 55 years and had a median white blood cell count of 21.3 x 109/L, a median hemoglobin of 9.1 g/dL, and a median platelet count of 61 x 109/L. More than half of patients (57.3%) were male.
The patients were screened for 35 genes that are commonly mutated in AML and other myeloid malignancies. The investigators used machine learning algorithms, including random survival forest and recommender system algorithms, to study the association between mutations and overall survival in an “unbiased” way.
Dr. Nazha said there were a median of three mutations per patient sample, and “there are some competing interests between those mutations to impact the prognosis of the patient.”
The investigators used the mutations and their associations with overall survival to classify patients into the risk groups outlined in the table below.
These findings can improve the risk stratification of NK-AML and may aid physicians in making treatment decisions, according to Dr. Nazha and his colleagues. To move this work forward, the investigators are attempting to develop a personalized model that can make predictions specific to an individual patient based on that patient’s mutation information.
Dr. Nazha reported having no financial disclosures relevant to this research. Other investigators reported relationships with the Munich Leukemia Laboratory.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
NEWPORT BEACH, CALIF. – New research suggests patients with normal karyotype acute myeloid leukemia (NK-AML) can be divided into four risk groups associated with overall survival.
Investigators used machine learning algorithms to study the association between mutations and overall survival in 1,352 patients with NK-AML. The analysis revealed combinations of mutations that could be used to classify NK-AML patients into favorable, intermediate-1, intermediate-2, and unfavorable risk groups.
For example, patients who had NPM1 mutations but wild-type FLT3-ITD and DNMT3A, had a median overall survival of 99.1 months and could be classified as favorable risk. Conversely, patients who had NPM1, FLT3-ITD, and DNMT3A mutations, had a median overall survival of 13.4 months and could be classified as unfavorable risk.
Aziz Nazha, MD, of the Cleveland Clinic, and his colleagues conducted this research and presented the findings at the Acute Leukemia Forum of Hemedicus.
The investigators looked at genomic and clinical data from 1,352 patients with NK-AML. The patients were a median age of 55 years and had a median white blood cell count of 21.3 x 109/L, a median hemoglobin of 9.1 g/dL, and a median platelet count of 61 x 109/L. More than half of patients (57.3%) were male.
The patients were screened for 35 genes that are commonly mutated in AML and other myeloid malignancies. The investigators used machine learning algorithms, including random survival forest and recommender system algorithms, to study the association between mutations and overall survival in an “unbiased” way.
Dr. Nazha said there were a median of three mutations per patient sample, and “there are some competing interests between those mutations to impact the prognosis of the patient.”
The investigators used the mutations and their associations with overall survival to classify patients into the risk groups outlined in the table below.
These findings can improve the risk stratification of NK-AML and may aid physicians in making treatment decisions, according to Dr. Nazha and his colleagues. To move this work forward, the investigators are attempting to develop a personalized model that can make predictions specific to an individual patient based on that patient’s mutation information.
Dr. Nazha reported having no financial disclosures relevant to this research. Other investigators reported relationships with the Munich Leukemia Laboratory.
The Acute Leukemia Forum is held by Hemedicus, which is owned by the same company as this news organization.
REPORTING FROM ALF 2019
Ruzurgi approved for Lambert-Eaton myasthenic syndrome in patients under age 17
Amifampridine (Ruzurgi) has been approved for the treatment of Lambert-Eaton myasthenic syndrome (LEMS), a rare autoimmune neuromuscular disorder, in patients aged 6 to less than 17 years, according to a statement from the Food and Drug Administration.

The approval is the first for a LEMS treatment specifically for pediatric patients.
“This approval will provide a much-needed treatment option for pediatric patients with LEMS who have significant weakness and fatigue that can often cause great difficulties with daily activities,” Billy Dunn, MD, director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
The prevalence of LEMS in pediatric patients is not known, but the overall prevalence of LEMS is estimated to be three per million individuals worldwide, according to the FDA press release.
Use of amifampridine in patients 6 to less than 17 years of age is supported by pharmacokinetic data in adult patients, pharmacokinetic modeling and simulation to identify the dosing regimen in pediatric patients, and safety data from pediatric patients 6 to less than 17 years of age.
A randomized, double-blind, placebo-controlled withdrawal study enrolled 32 adult patients who had taken amifampridine for at least 3 months. The study compared patients continuing on amifampridine with patients switched to placebo. Effectiveness was measured by the degree of change in a test that assessed the time it took the patient to rise from a chair, walk three meters, and return to the chair for three consecutive laps without pause. The patients who continued on amifampridine experienced less impairment compared with those switched to placebo. Effectiveness was also measured with a self-assessment scale for LEMS-related weakness. The scores indicated greater perceived weakening in the patients switched to placebo.
The most common side effects among amifampridine users were paresthesia, abdominal pain, indigestion, dizziness, and nausea. Side effects reported in pediatric patients were similar to those seen in adult patients. Seizures have been observed in patients without a history of seizures. Signs of hypersensitivity reactions include rash, hives, itching, fever, swelling, or trouble breathing.
The FDA granted this application Priority Review and Fast Track designations. Amifampridine also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of amifampridine (Ruzurgi) to Jacobus Pharmaceutical Company.
Amifampridine (Ruzurgi) has been approved for the treatment of Lambert-Eaton myasthenic syndrome (LEMS), a rare autoimmune neuromuscular disorder, in patients aged 6 to less than 17 years, according to a statement from the Food and Drug Administration.
The approval is the first for a LEMS treatment specifically for pediatric patients.
“This approval will provide a much-needed treatment option for pediatric patients with LEMS who have significant weakness and fatigue that can often cause great difficulties with daily activities,” Billy Dunn, MD, director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
The prevalence of LEMS in pediatric patients is not known, but the overall prevalence of LEMS is estimated to be three per million individuals worldwide, according to the FDA press release.
Use of amifampridine in patients 6 to less than 17 years of age is supported by pharmacokinetic data in adult patients, pharmacokinetic modeling and simulation to identify the dosing regimen in pediatric patients, and safety data from pediatric patients 6 to less than 17 years of age.
A randomized, double-blind, placebo-controlled withdrawal study enrolled 32 adult patients who had taken amifampridine for at least 3 months. The study compared patients continuing on amifampridine with patients switched to placebo. Effectiveness was measured by the degree of change in a test that assessed the time it took the patient to rise from a chair, walk three meters, and return to the chair for three consecutive laps without pause. The patients who continued on amifampridine experienced less impairment compared with those switched to placebo. Effectiveness was also measured with a self-assessment scale for LEMS-related weakness. The scores indicated greater perceived weakening in the patients switched to placebo.
The most common side effects among amifampridine users were paresthesia, abdominal pain, indigestion, dizziness, and nausea. Side effects reported in pediatric patients were similar to those seen in adult patients. Seizures have been observed in patients without a history of seizures. Signs of hypersensitivity reactions include rash, hives, itching, fever, swelling, or trouble breathing.
The FDA granted this application Priority Review and Fast Track designations. Amifampridine also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of amifampridine (Ruzurgi) to Jacobus Pharmaceutical Company.
Amifampridine (Ruzurgi) has been approved for the treatment of Lambert-Eaton myasthenic syndrome (LEMS), a rare autoimmune neuromuscular disorder, in patients aged 6 to less than 17 years, according to a statement from the Food and Drug Administration.
The approval is the first for a LEMS treatment specifically for pediatric patients.
“This approval will provide a much-needed treatment option for pediatric patients with LEMS who have significant weakness and fatigue that can often cause great difficulties with daily activities,” Billy Dunn, MD, director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
The prevalence of LEMS in pediatric patients is not known, but the overall prevalence of LEMS is estimated to be three per million individuals worldwide, according to the FDA press release.
Use of amifampridine in patients 6 to less than 17 years of age is supported by pharmacokinetic data in adult patients, pharmacokinetic modeling and simulation to identify the dosing regimen in pediatric patients, and safety data from pediatric patients 6 to less than 17 years of age.
A randomized, double-blind, placebo-controlled withdrawal study enrolled 32 adult patients who had taken amifampridine for at least 3 months. The study compared patients continuing on amifampridine with patients switched to placebo. Effectiveness was measured by the degree of change in a test that assessed the time it took the patient to rise from a chair, walk three meters, and return to the chair for three consecutive laps without pause. The patients who continued on amifampridine experienced less impairment compared with those switched to placebo. Effectiveness was also measured with a self-assessment scale for LEMS-related weakness. The scores indicated greater perceived weakening in the patients switched to placebo.
The most common side effects among amifampridine users were paresthesia, abdominal pain, indigestion, dizziness, and nausea. Side effects reported in pediatric patients were similar to those seen in adult patients. Seizures have been observed in patients without a history of seizures. Signs of hypersensitivity reactions include rash, hives, itching, fever, swelling, or trouble breathing.
The FDA granted this application Priority Review and Fast Track designations. Amifampridine also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of amifampridine (Ruzurgi) to Jacobus Pharmaceutical Company.
Coalescing Papules on Bilateral Mastectomy Scars
The Diagnosis: Scar Sarcoidosis
Although scars on both breasts were involved, the decision was made to biopsy the right breast because the patient reported more pain on the left breast. Biopsy showed noncaseating granulomas consistent with scar sarcoidosis (Figure). Additional screening tests were performed to evaluate for any systemic involvement of sarcoidosis, including a complete blood cell count, comprehensive metabolic panel, angiotensin-converting enzyme level, tuberculosis serology screening, electrocardiogram, chest radiograph, and pulmonary function tests. She also was referred to rheumatology and ophthalmology for consultation. The results of all screenings were within reference range, and no sign of systemic sarcoidosis was found. She was treated with hydrocortisone ointment 2.5% for several weeks without notable improvement. She elected not to pursue any additional treatment and to monitor the symptoms with close follow-up only. One year after the initial visit, the skin lesions spontaneously and notably improved.
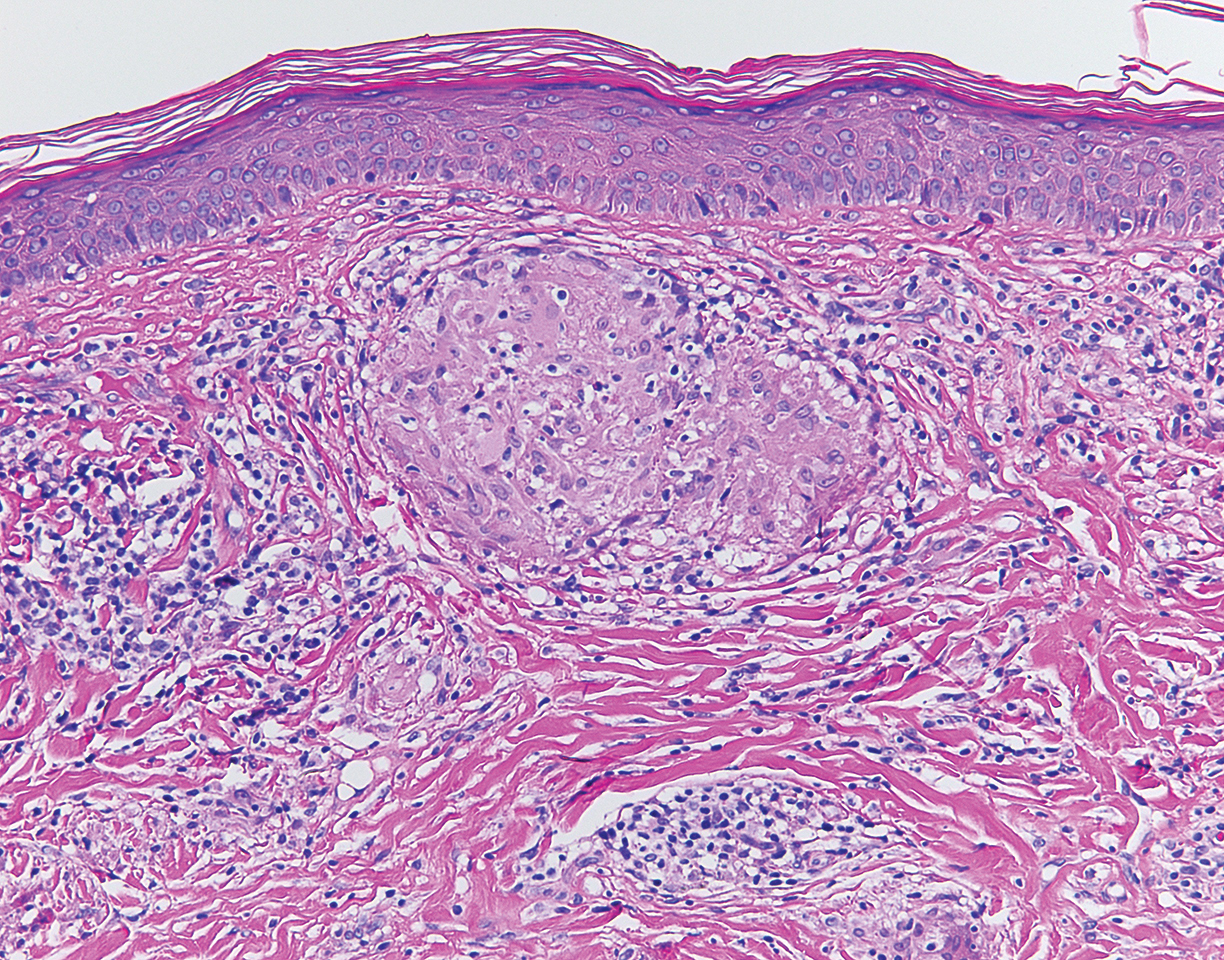
Sarcoidosis is a systemic granulomatous disorder of unknown etiology that most commonly affects the lungs. It also can involve the lymph nodes, liver, spleen, bones, gastrointestinal tract, eyes, and skin. Cutaneous sarcoidosis has been documented in the literature since the late 1800s and occurs in up to one-third of sarcoid patients.1 Cutaneous lesions developing within a preexisting scar is a well-known variant, occurring in 29% of patients with cutaneous sarcoidosis in one clinical study (N=818).2 There have been many reports describing scar sarcoidosis, with its development at prior sites of surgery, trauma, acne, or venipuncture.3 Other case reports have described variants of scar sarcoidosis developing at sites of hyaluronic acid injection, laser surgery, ritual scarification, tattoos, and desensitization injections, as well as prior herpes zoster infections.4-9
Cutaneous sarcoidosis has a wide range of clinical presentations. Lesions can be described as specific or nonspecific. Specific lesions demonstrate the typical sarcoid granuloma on histology and more often are seen in chronic disease, while nonspecific lesions more often are seen in acute disease.3,10 Scar sarcoidosis is an example of a specific lesion in which old scars become infiltrated with noncaseating granulomas. The granulomas typically are in the superficial dermis but may involve the full thickness of the dermis, extending into the subcutaneous tissue.11 The cause of granulomas developing in scars is unknown. Prior contamination of the scar with foreign material, possibly at the time of the trauma, is a possible underlying cause.12
Typical scar sarcoidosis presents as swollen, erythematous, indurated lesions with a purple-red hue that may become brown.3,12 Tenderness or pruritus also may be present.13 Interestingly, our patient's scar sarcoidosis presented with a yellow hue at both mastectomy sites.
Diagnosing scar sarcoidosis can be challenging. Patients are diagnosed with sarcoidosis when a compatible clinical or radiologic picture is present along with histologic evidence of a noncaseating granuloma and other potential causes are excluded.11 The differential includes an infectious etiology, other types of granulomatous dermatitis, hypertrophic scar, keloid, or foreign body granuloma.
Scar sarcoidosis can be isolated in occurrence. It also can precede or occur concomitantly or during a relapse of systemic sarcoidosis.10 Most commonly, patients with scar sarcoidosis also have systemic manifestations of sarcoidosis, and changing scars may be an indicator of disease exacerbation or relapse.10 For patients who only demonstrate specific skin lesions of cutaneous sarcoidosis, approximately 30% develop systemic involvement later in life.3 For this reason, close monitoring and regular follow-up are necessary.
Treatment of scar sarcoidosis is dependent on the extent of the disease and presence of systemic sarcoidosis. Topical and systemic corticosteroids, hydroxychloroquine, chloroquine phosphate, and methotrexate all have been shown to be helpful in treating cutaneous sarcoidosis.3 For scar sarcoidosis that is limited to only the scar site, as seen in our case, monitoring and close follow-up is acceptable. Topical steroids can be prescribed for symptomatic relief. Scar sarcoidosis can resolve slowly and spontaneously over time.10 Our patient notably improved 1 year after the initial presentation without treatment.
Scar sarcoidosis is a well-documented variant of cutaneous sarcoidosis that can have important implications for diagnosing systemic sarcoidosis. Although there are typical lesions that represent scar sarcoidosis, it is important to have a high degree of suspicion with any changing scar. Once diagnosed through biopsy, a thorough investigation for systemic signs of sarcoidosis needs to be performed to guide treatment.
- Bolognia JL, Jorizzo JL, Shaffer JV, eds. Dermatology. 3rd ed. Vol 2. Philadelphia, PA: Elsevier/Saunders; 2012.
- Neville E, Walker AN, James DG. Prognostic factors predicting the outcome of sarcoidosis: an analysis of 818 patients. Q J Med. 1983;52:525-533.
- Mañá J, Marcoval J, Graells J, et al. Cutaneous involvement in sarcoidosis: relationship to systemic disease. Arch Dermatol. 1997;133:882-888.
- Dal Sacco D, Cozzani E, Parodi A, et al. Scar sarcoidosis after hyaluronic acid injection. Int J Dermatol. 2005;44:411-412.
- Kormeili T, Neel V, Moy RL. Cutaneous sarcoidosis at sites of previous laser surgery. Cutis. 2004;73:53-55.
- Nayar M. Sarcoidosis on ritual scarification. Int J Dermatol. 1993;32:116-118.
- James WD, Elston DM, Berger TG, et al. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Elsevier/Saunders; 2011.
- Healsmith MF, Hutchinson PE. The development of scar sarcoidosis at the site of desensitization injections. Clin Exp Dermatol. 1992;17:369-370.
- Singal A, Vij A, Pandhi D. Post herpes-zoster scar sarcoidosis with pulmonary involvement. Indian Dermatol Online J. 2014;5:77-79.
- Chudomirova K, Velichkova L, Anavi B, et al. Recurrent sarcoidosis in skin scars accompanying systemic sarcoidosis. J Eur Acad Dermatol Venereol. 2003;17:360-361.
- Selim A, Ehrsam E, Atassi MB, et al. Scar sarcoidosis: a case report and brief review. Cutis. 2006;78:418-422.
- Singal A, Thami GP, Goraya JS. Scar sarcoidosis in childhood: case report and review of the literature. Clin Exp Dermatol. 2005;30:244-246.
- Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25:295-302.
The Diagnosis: Scar Sarcoidosis
Although scars on both breasts were involved, the decision was made to biopsy the right breast because the patient reported more pain on the left breast. Biopsy showed noncaseating granulomas consistent with scar sarcoidosis (Figure). Additional screening tests were performed to evaluate for any systemic involvement of sarcoidosis, including a complete blood cell count, comprehensive metabolic panel, angiotensin-converting enzyme level, tuberculosis serology screening, electrocardiogram, chest radiograph, and pulmonary function tests. She also was referred to rheumatology and ophthalmology for consultation. The results of all screenings were within reference range, and no sign of systemic sarcoidosis was found. She was treated with hydrocortisone ointment 2.5% for several weeks without notable improvement. She elected not to pursue any additional treatment and to monitor the symptoms with close follow-up only. One year after the initial visit, the skin lesions spontaneously and notably improved.
Sarcoidosis is a systemic granulomatous disorder of unknown etiology that most commonly affects the lungs. It also can involve the lymph nodes, liver, spleen, bones, gastrointestinal tract, eyes, and skin. Cutaneous sarcoidosis has been documented in the literature since the late 1800s and occurs in up to one-third of sarcoid patients.1 Cutaneous lesions developing within a preexisting scar is a well-known variant, occurring in 29% of patients with cutaneous sarcoidosis in one clinical study (N=818).2 There have been many reports describing scar sarcoidosis, with its development at prior sites of surgery, trauma, acne, or venipuncture.3 Other case reports have described variants of scar sarcoidosis developing at sites of hyaluronic acid injection, laser surgery, ritual scarification, tattoos, and desensitization injections, as well as prior herpes zoster infections.4-9
Cutaneous sarcoidosis has a wide range of clinical presentations. Lesions can be described as specific or nonspecific. Specific lesions demonstrate the typical sarcoid granuloma on histology and more often are seen in chronic disease, while nonspecific lesions more often are seen in acute disease.3,10 Scar sarcoidosis is an example of a specific lesion in which old scars become infiltrated with noncaseating granulomas. The granulomas typically are in the superficial dermis but may involve the full thickness of the dermis, extending into the subcutaneous tissue.11 The cause of granulomas developing in scars is unknown. Prior contamination of the scar with foreign material, possibly at the time of the trauma, is a possible underlying cause.12
Typical scar sarcoidosis presents as swollen, erythematous, indurated lesions with a purple-red hue that may become brown.3,12 Tenderness or pruritus also may be present.13 Interestingly, our patient's scar sarcoidosis presented with a yellow hue at both mastectomy sites.
Diagnosing scar sarcoidosis can be challenging. Patients are diagnosed with sarcoidosis when a compatible clinical or radiologic picture is present along with histologic evidence of a noncaseating granuloma and other potential causes are excluded.11 The differential includes an infectious etiology, other types of granulomatous dermatitis, hypertrophic scar, keloid, or foreign body granuloma.
Scar sarcoidosis can be isolated in occurrence. It also can precede or occur concomitantly or during a relapse of systemic sarcoidosis.10 Most commonly, patients with scar sarcoidosis also have systemic manifestations of sarcoidosis, and changing scars may be an indicator of disease exacerbation or relapse.10 For patients who only demonstrate specific skin lesions of cutaneous sarcoidosis, approximately 30% develop systemic involvement later in life.3 For this reason, close monitoring and regular follow-up are necessary.
Treatment of scar sarcoidosis is dependent on the extent of the disease and presence of systemic sarcoidosis. Topical and systemic corticosteroids, hydroxychloroquine, chloroquine phosphate, and methotrexate all have been shown to be helpful in treating cutaneous sarcoidosis.3 For scar sarcoidosis that is limited to only the scar site, as seen in our case, monitoring and close follow-up is acceptable. Topical steroids can be prescribed for symptomatic relief. Scar sarcoidosis can resolve slowly and spontaneously over time.10 Our patient notably improved 1 year after the initial presentation without treatment.
Scar sarcoidosis is a well-documented variant of cutaneous sarcoidosis that can have important implications for diagnosing systemic sarcoidosis. Although there are typical lesions that represent scar sarcoidosis, it is important to have a high degree of suspicion with any changing scar. Once diagnosed through biopsy, a thorough investigation for systemic signs of sarcoidosis needs to be performed to guide treatment.
The Diagnosis: Scar Sarcoidosis
Although scars on both breasts were involved, the decision was made to biopsy the right breast because the patient reported more pain on the left breast. Biopsy showed noncaseating granulomas consistent with scar sarcoidosis (Figure). Additional screening tests were performed to evaluate for any systemic involvement of sarcoidosis, including a complete blood cell count, comprehensive metabolic panel, angiotensin-converting enzyme level, tuberculosis serology screening, electrocardiogram, chest radiograph, and pulmonary function tests. She also was referred to rheumatology and ophthalmology for consultation. The results of all screenings were within reference range, and no sign of systemic sarcoidosis was found. She was treated with hydrocortisone ointment 2.5% for several weeks without notable improvement. She elected not to pursue any additional treatment and to monitor the symptoms with close follow-up only. One year after the initial visit, the skin lesions spontaneously and notably improved.
Sarcoidosis is a systemic granulomatous disorder of unknown etiology that most commonly affects the lungs. It also can involve the lymph nodes, liver, spleen, bones, gastrointestinal tract, eyes, and skin. Cutaneous sarcoidosis has been documented in the literature since the late 1800s and occurs in up to one-third of sarcoid patients.1 Cutaneous lesions developing within a preexisting scar is a well-known variant, occurring in 29% of patients with cutaneous sarcoidosis in one clinical study (N=818).2 There have been many reports describing scar sarcoidosis, with its development at prior sites of surgery, trauma, acne, or venipuncture.3 Other case reports have described variants of scar sarcoidosis developing at sites of hyaluronic acid injection, laser surgery, ritual scarification, tattoos, and desensitization injections, as well as prior herpes zoster infections.4-9
Cutaneous sarcoidosis has a wide range of clinical presentations. Lesions can be described as specific or nonspecific. Specific lesions demonstrate the typical sarcoid granuloma on histology and more often are seen in chronic disease, while nonspecific lesions more often are seen in acute disease.3,10 Scar sarcoidosis is an example of a specific lesion in which old scars become infiltrated with noncaseating granulomas. The granulomas typically are in the superficial dermis but may involve the full thickness of the dermis, extending into the subcutaneous tissue.11 The cause of granulomas developing in scars is unknown. Prior contamination of the scar with foreign material, possibly at the time of the trauma, is a possible underlying cause.12
Typical scar sarcoidosis presents as swollen, erythematous, indurated lesions with a purple-red hue that may become brown.3,12 Tenderness or pruritus also may be present.13 Interestingly, our patient's scar sarcoidosis presented with a yellow hue at both mastectomy sites.
Diagnosing scar sarcoidosis can be challenging. Patients are diagnosed with sarcoidosis when a compatible clinical or radiologic picture is present along with histologic evidence of a noncaseating granuloma and other potential causes are excluded.11 The differential includes an infectious etiology, other types of granulomatous dermatitis, hypertrophic scar, keloid, or foreign body granuloma.
Scar sarcoidosis can be isolated in occurrence. It also can precede or occur concomitantly or during a relapse of systemic sarcoidosis.10 Most commonly, patients with scar sarcoidosis also have systemic manifestations of sarcoidosis, and changing scars may be an indicator of disease exacerbation or relapse.10 For patients who only demonstrate specific skin lesions of cutaneous sarcoidosis, approximately 30% develop systemic involvement later in life.3 For this reason, close monitoring and regular follow-up are necessary.
Treatment of scar sarcoidosis is dependent on the extent of the disease and presence of systemic sarcoidosis. Topical and systemic corticosteroids, hydroxychloroquine, chloroquine phosphate, and methotrexate all have been shown to be helpful in treating cutaneous sarcoidosis.3 For scar sarcoidosis that is limited to only the scar site, as seen in our case, monitoring and close follow-up is acceptable. Topical steroids can be prescribed for symptomatic relief. Scar sarcoidosis can resolve slowly and spontaneously over time.10 Our patient notably improved 1 year after the initial presentation without treatment.
Scar sarcoidosis is a well-documented variant of cutaneous sarcoidosis that can have important implications for diagnosing systemic sarcoidosis. Although there are typical lesions that represent scar sarcoidosis, it is important to have a high degree of suspicion with any changing scar. Once diagnosed through biopsy, a thorough investigation for systemic signs of sarcoidosis needs to be performed to guide treatment.
- Bolognia JL, Jorizzo JL, Shaffer JV, eds. Dermatology. 3rd ed. Vol 2. Philadelphia, PA: Elsevier/Saunders; 2012.
- Neville E, Walker AN, James DG. Prognostic factors predicting the outcome of sarcoidosis: an analysis of 818 patients. Q J Med. 1983;52:525-533.
- Mañá J, Marcoval J, Graells J, et al. Cutaneous involvement in sarcoidosis: relationship to systemic disease. Arch Dermatol. 1997;133:882-888.
- Dal Sacco D, Cozzani E, Parodi A, et al. Scar sarcoidosis after hyaluronic acid injection. Int J Dermatol. 2005;44:411-412.
- Kormeili T, Neel V, Moy RL. Cutaneous sarcoidosis at sites of previous laser surgery. Cutis. 2004;73:53-55.
- Nayar M. Sarcoidosis on ritual scarification. Int J Dermatol. 1993;32:116-118.
- James WD, Elston DM, Berger TG, et al. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Elsevier/Saunders; 2011.
- Healsmith MF, Hutchinson PE. The development of scar sarcoidosis at the site of desensitization injections. Clin Exp Dermatol. 1992;17:369-370.
- Singal A, Vij A, Pandhi D. Post herpes-zoster scar sarcoidosis with pulmonary involvement. Indian Dermatol Online J. 2014;5:77-79.
- Chudomirova K, Velichkova L, Anavi B, et al. Recurrent sarcoidosis in skin scars accompanying systemic sarcoidosis. J Eur Acad Dermatol Venereol. 2003;17:360-361.
- Selim A, Ehrsam E, Atassi MB, et al. Scar sarcoidosis: a case report and brief review. Cutis. 2006;78:418-422.
- Singal A, Thami GP, Goraya JS. Scar sarcoidosis in childhood: case report and review of the literature. Clin Exp Dermatol. 2005;30:244-246.
- Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25:295-302.
- Bolognia JL, Jorizzo JL, Shaffer JV, eds. Dermatology. 3rd ed. Vol 2. Philadelphia, PA: Elsevier/Saunders; 2012.
- Neville E, Walker AN, James DG. Prognostic factors predicting the outcome of sarcoidosis: an analysis of 818 patients. Q J Med. 1983;52:525-533.
- Mañá J, Marcoval J, Graells J, et al. Cutaneous involvement in sarcoidosis: relationship to systemic disease. Arch Dermatol. 1997;133:882-888.
- Dal Sacco D, Cozzani E, Parodi A, et al. Scar sarcoidosis after hyaluronic acid injection. Int J Dermatol. 2005;44:411-412.
- Kormeili T, Neel V, Moy RL. Cutaneous sarcoidosis at sites of previous laser surgery. Cutis. 2004;73:53-55.
- Nayar M. Sarcoidosis on ritual scarification. Int J Dermatol. 1993;32:116-118.
- James WD, Elston DM, Berger TG, et al. Andrews' Diseases of the Skin: Clinical Dermatology. 11th ed. Philadelphia, PA: Elsevier/Saunders; 2011.
- Healsmith MF, Hutchinson PE. The development of scar sarcoidosis at the site of desensitization injections. Clin Exp Dermatol. 1992;17:369-370.
- Singal A, Vij A, Pandhi D. Post herpes-zoster scar sarcoidosis with pulmonary involvement. Indian Dermatol Online J. 2014;5:77-79.
- Chudomirova K, Velichkova L, Anavi B, et al. Recurrent sarcoidosis in skin scars accompanying systemic sarcoidosis. J Eur Acad Dermatol Venereol. 2003;17:360-361.
- Selim A, Ehrsam E, Atassi MB, et al. Scar sarcoidosis: a case report and brief review. Cutis. 2006;78:418-422.
- Singal A, Thami GP, Goraya JS. Scar sarcoidosis in childhood: case report and review of the literature. Clin Exp Dermatol. 2005;30:244-246.
- Marchell RM, Judson MA. Chronic cutaneous lesions of sarcoidosis. Clin Dermatol. 2007;25:295-302.
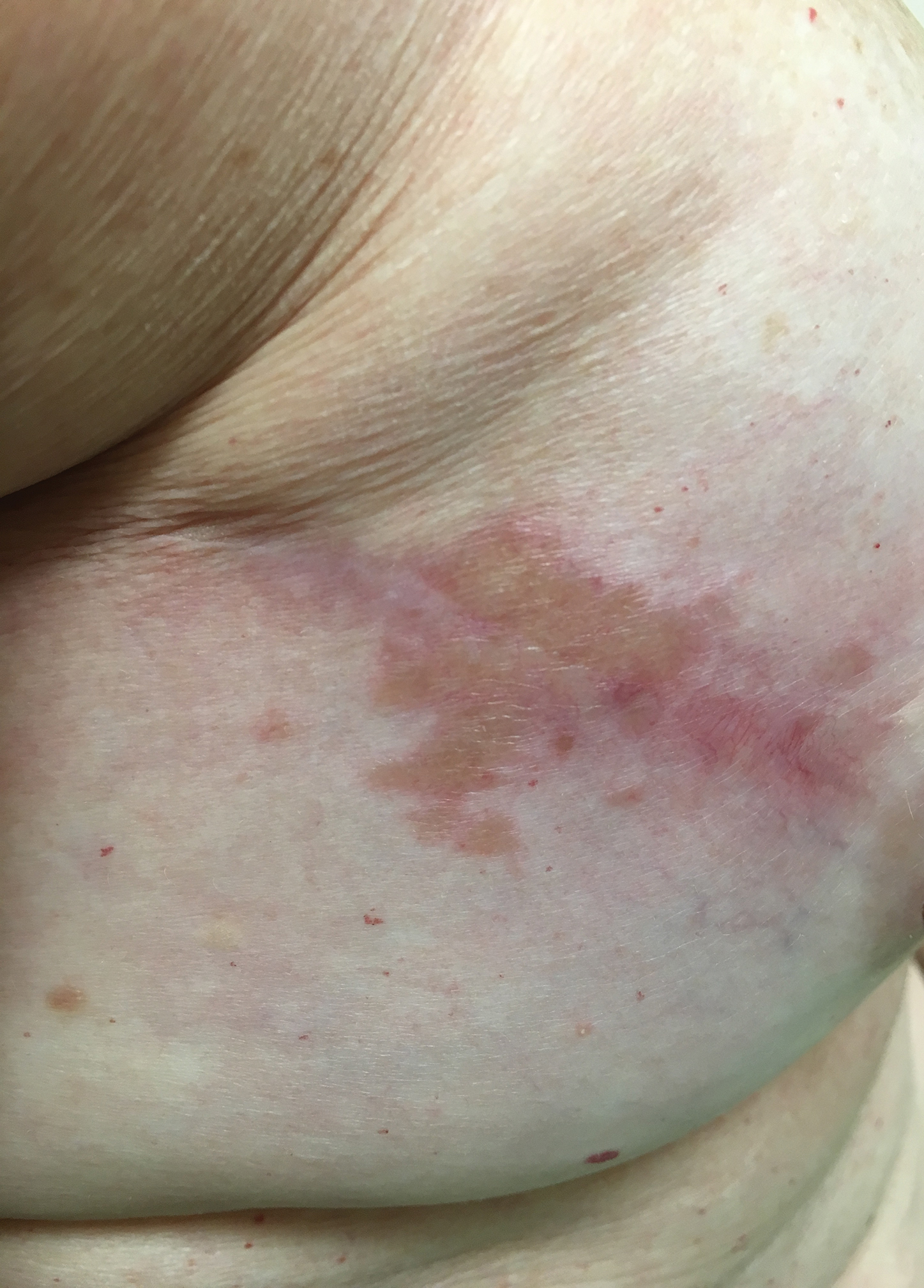
A 57-year-old woman with triple-negative ductal breast cancer presented with a mildly pruritic rash on bilateral mastectomy scars of 3 to 4 months' duration. More than a year prior to presentation, she was diagnosed with breast cancer and treated with a bilateral mastectomy and chemotherapy. On physical examination, faintly yellow, slightly indurated, coalescing papules with red rims were present on the bilateral mastectomy scars, with the scar on the left side appearing worse than the right. She previously had not sought treatment.
Angelman syndrome treatment safe, well-tolerated, and effective in exploratory analyses
Philadelphia – according to results of a study presented at the annual meeting of the American Academy of Neurology.
Clinician-rated clinical global impressions of improvement (CGI-I) were improved versus placebo in the randomized study, as were other outcomes in post hoc analyses, including measures of sleep and motor function, said Alexander Kolevzon, MD, professor of psychiatry and pediatrics at the Icahn School of Medicine at Mount Sinai, New York.
This study of OV101 was a genetics-driven trial for the rare genetic disorder, which is caused by mutations in UBE3A and characterized by seizures, speech impairments, profound intellectual disability, gait problems, and anxiety, Dr. Kolevzon said in a press conference.
“The only treatments that exist are really very symptomatically driven,” Dr. Kolevzon said. “Here, we are taking a genetics-first approach. Having identified the gene, there is some understanding of what the underlying biology is, and it seems to relate to deficits in tonic inhibition.”
OV101 is a delta-selective type A gamma-aminobutyric acid receptor agonist that may potentially normalize the tonic inhibition that is decreased in Angelman syndrome. “What we think this compound is doing is actually reversing the deficits of tonic inhibition, and sort of restoring that state to these patients,” Dr. Kolevzon said in the press conference.
A total of 78 patients completed the phase 2, randomized study, known as STARS, which had a primary endpoint of safety and tolerability over 12 weeks of treatment with OV101 once daily, twice daily, or placebo. The mean age of the 87 patients who enrolled and had at least one dose of study drug was 22.6 years.
Most adverse events were mild, and frequencies of specific adverse events were similar for OV101 and placebo treatment groups, according to Dr. Kolevzon and his coinvestigators.
Improvements in motor function, sleep, and behavior were seen in a series of exploratory analyses, including global improvement at week 12, as captured by CGI-I, which was significantly improved for daily OV101 versus placebo (P = .0006).
These phase 2 results have informed discussions of which specific endpoints might be incorporated into the design of a planned phase 3 trial in pediatric patients. The CGI-I may be especially useful to measure clinical improvement in Angelman syndrome, which is a very “heterogeneous” disorder, Dr. Kolevzon said in the press conference.
“Every child has a different composite of symptoms, so that is the big challenge,” Dr. Kolevzon said. “I do not think we are going to have one singular outcome. The idea is to have a global measure that really captures heterogeneity across each trial and allows for children to be compared to their baseline, and each as their own control, in essence, but with specific domains in mind.”
Funding for the study came from Ovid Therapeutics. Dr. Kolevzon reported disclosures related to Ovid Therapeutics, as well as Coronis Neurosciences, 5AM Ventures, SEMA4, LabCorp, and AMO Pharma.
Philadelphia – according to results of a study presented at the annual meeting of the American Academy of Neurology.
Clinician-rated clinical global impressions of improvement (CGI-I) were improved versus placebo in the randomized study, as were other outcomes in post hoc analyses, including measures of sleep and motor function, said Alexander Kolevzon, MD, professor of psychiatry and pediatrics at the Icahn School of Medicine at Mount Sinai, New York.
This study of OV101 was a genetics-driven trial for the rare genetic disorder, which is caused by mutations in UBE3A and characterized by seizures, speech impairments, profound intellectual disability, gait problems, and anxiety, Dr. Kolevzon said in a press conference.
“The only treatments that exist are really very symptomatically driven,” Dr. Kolevzon said. “Here, we are taking a genetics-first approach. Having identified the gene, there is some understanding of what the underlying biology is, and it seems to relate to deficits in tonic inhibition.”
OV101 is a delta-selective type A gamma-aminobutyric acid receptor agonist that may potentially normalize the tonic inhibition that is decreased in Angelman syndrome. “What we think this compound is doing is actually reversing the deficits of tonic inhibition, and sort of restoring that state to these patients,” Dr. Kolevzon said in the press conference.
A total of 78 patients completed the phase 2, randomized study, known as STARS, which had a primary endpoint of safety and tolerability over 12 weeks of treatment with OV101 once daily, twice daily, or placebo. The mean age of the 87 patients who enrolled and had at least one dose of study drug was 22.6 years.
Most adverse events were mild, and frequencies of specific adverse events were similar for OV101 and placebo treatment groups, according to Dr. Kolevzon and his coinvestigators.
Improvements in motor function, sleep, and behavior were seen in a series of exploratory analyses, including global improvement at week 12, as captured by CGI-I, which was significantly improved for daily OV101 versus placebo (P = .0006).
These phase 2 results have informed discussions of which specific endpoints might be incorporated into the design of a planned phase 3 trial in pediatric patients. The CGI-I may be especially useful to measure clinical improvement in Angelman syndrome, which is a very “heterogeneous” disorder, Dr. Kolevzon said in the press conference.
“Every child has a different composite of symptoms, so that is the big challenge,” Dr. Kolevzon said. “I do not think we are going to have one singular outcome. The idea is to have a global measure that really captures heterogeneity across each trial and allows for children to be compared to their baseline, and each as their own control, in essence, but with specific domains in mind.”
Funding for the study came from Ovid Therapeutics. Dr. Kolevzon reported disclosures related to Ovid Therapeutics, as well as Coronis Neurosciences, 5AM Ventures, SEMA4, LabCorp, and AMO Pharma.
Philadelphia – according to results of a study presented at the annual meeting of the American Academy of Neurology.
Clinician-rated clinical global impressions of improvement (CGI-I) were improved versus placebo in the randomized study, as were other outcomes in post hoc analyses, including measures of sleep and motor function, said Alexander Kolevzon, MD, professor of psychiatry and pediatrics at the Icahn School of Medicine at Mount Sinai, New York.
This study of OV101 was a genetics-driven trial for the rare genetic disorder, which is caused by mutations in UBE3A and characterized by seizures, speech impairments, profound intellectual disability, gait problems, and anxiety, Dr. Kolevzon said in a press conference.
“The only treatments that exist are really very symptomatically driven,” Dr. Kolevzon said. “Here, we are taking a genetics-first approach. Having identified the gene, there is some understanding of what the underlying biology is, and it seems to relate to deficits in tonic inhibition.”
OV101 is a delta-selective type A gamma-aminobutyric acid receptor agonist that may potentially normalize the tonic inhibition that is decreased in Angelman syndrome. “What we think this compound is doing is actually reversing the deficits of tonic inhibition, and sort of restoring that state to these patients,” Dr. Kolevzon said in the press conference.
A total of 78 patients completed the phase 2, randomized study, known as STARS, which had a primary endpoint of safety and tolerability over 12 weeks of treatment with OV101 once daily, twice daily, or placebo. The mean age of the 87 patients who enrolled and had at least one dose of study drug was 22.6 years.
Most adverse events were mild, and frequencies of specific adverse events were similar for OV101 and placebo treatment groups, according to Dr. Kolevzon and his coinvestigators.
Improvements in motor function, sleep, and behavior were seen in a series of exploratory analyses, including global improvement at week 12, as captured by CGI-I, which was significantly improved for daily OV101 versus placebo (P = .0006).
These phase 2 results have informed discussions of which specific endpoints might be incorporated into the design of a planned phase 3 trial in pediatric patients. The CGI-I may be especially useful to measure clinical improvement in Angelman syndrome, which is a very “heterogeneous” disorder, Dr. Kolevzon said in the press conference.
“Every child has a different composite of symptoms, so that is the big challenge,” Dr. Kolevzon said. “I do not think we are going to have one singular outcome. The idea is to have a global measure that really captures heterogeneity across each trial and allows for children to be compared to their baseline, and each as their own control, in essence, but with specific domains in mind.”
Funding for the study came from Ovid Therapeutics. Dr. Kolevzon reported disclosures related to Ovid Therapeutics, as well as Coronis Neurosciences, 5AM Ventures, SEMA4, LabCorp, and AMO Pharma.
REPORTING FROM AAN 2019
CSF and plasma biomarkers predict survival in sporadic Creutzfeldt-Jakob disease
, according to research published online ahead of print May 6 in JAMA Neurology. Levels of total tau in plasma and CSF are correlated, and plasma total tau is associated with survival time. These findings suggest that plasma total tau level could be a valid biomarker that guides clinical care for patients with sporadic Creutzfeldt-Jakob disease, said the investigators.

The accurate prediction of disease duration can assist clinicians and caregivers in clinical management, as well as influence the design of clinical trials. Previous studies have found that baseline protein levels in CSF and plasma are associated with disease duration in patients with sporadic Creutzfeldt-Jakob disease. To replicate these findings, Adam M. Staffaroni, PhD, of the department of neurology at the University of California, San Francisco, and colleagues conducted a longitudinal cohort study.
Evaluating fluid and nonfluid biomarkers
Dr. Staffaroni and colleagues recruited 193 participants with probable or definite sporadic Creutzfeldt-Jakob disease who had codon 129 genotyping and were referred to the UCSF Memory and Aging Center from March 2004 to January 2018. All participants underwent cognitive testing, informant measures, a neurologic examination, and CSF and blood sample collection. The researchers excluded from analysis five participants who had been placed on life-extending treatments. Participants were evaluated until death or censored at the time of statistical analysis.
Dr. Staffaroni and colleagues examined the following nonfluid biomarkers of survival: sex, age, codon 129 genotype, Barthel Index, and Medical Research Council (MRC) Prion Disease Rating Scale. In addition, they examined total tau level, phosphorylated tau level, total tau:phosphorylated tau ratio, neurofilament light (NfL) level, beta-amyloid 42 level, neuron-specific enolase level, 14-3-3 test result, and real-time quaking-induced conversion test in CSF as fluid biomarkers of survival. Finally, Dr. Staffaroni’s group analyzed total tau level, NfL level, and glial fibrillary acidic protein level in plasma as additional fluid biomarkers of survival.
The researchers fitted Cox proportional hazard models with time to event as the outcome. They log-transformed fluid biomarkers and ran models with and without nonfluid biomarkers of survival.
Plasma total tau was associated with survival
In all, 188 patients were included in the analysis. The population’s mean age was 63.8 years. Approximately 45% of participants were women. The diagnosis of sporadic Creutzfeldt-Jakob disease was pathologically confirmed for 78.2% of participants and probable for 21.8% of participants.
Dr. Staffaroni’s group observed strong correlations between plasma and CSF NfL concentrations and between plasma and CSF total tau concentrations. CSF total tau and CSF NfL concentrations were also correlated.
Among the nonfluid biomarkers, Barthel Index, MRC Scale, and codon 129 genotype were significantly associated with survival time. Lower level of function at baseline predicted a faster disease course.
Among the fluid biomarkers, greater levels of plasma total tau and NfL levels at baseline were associated with shorter survival. After the investigators controlled for Barthel Index and codon 129 genotype, the association of plasma total tau level with survival time remained significant. Plasma total tau level and Barthel Index (hazard ratio, 0.98) each independently predicted survival. Dr. Staffaroni and colleagues found that the hazard ratios for all CSF biomarkers were in the expected direction, and that those for total tau level, total tau:phosphorylated tau ratio, NfL level, and neuron-specific enolase level were statistically significant. Furthermore, positive results for 14-3-3 protein, neuron-specific enolase level, and total tau level were associated with a shorter time until death. Like the plasma biomarkers, CSF total tau level remained associated with survival after the investigators controlled for Barthel Index and codon 129 genotype. The same was true of CSF total tau:phosphorylated tau ratio, neuron-specific enolase level, and 14-3-3 result.
Plasma tau could be a diagnostic biomarker
“The hazard ratio associated with plasma total tau level was more than 40% higher than other fluid biomarkers of interest,” said the authors. “These findings further bolster the value of blood-based biomarkers, based on their minimally invasive and relatively inexpensive nature, and build on prior studies that suggested patients with sporadic Creutzfeldt-Jakob disease and controls can be discriminated with relatively high accuracy using blood-based assays.” When Dr. Staffaroni and colleagues modeled baseline functional status and plasma total tau levels together, they found that both were independent predictors of survival time. “This [finding] suggests that clinical measures and plasma total tau level could be combined to further improve prediction accuracy.”
Among the study’s limitations was its comparatively small subsample of patients for whom all plasma and CSF biomarkers were available. Disease duration and survival were longer in the study population than in the literature. “Another limitation is that the plasma biomarkers in this study, one of which showed great promise for predicting survival, were assayed using a research protocol,” said the researchers. “Widespread clinical use of these biomarkers will require well-validated commercial assays, development of which is underway.”
Before these biomarkers can be used in the clinic, these results will need to be replicated, and neurologists will need to develop consensus cutoffs for the biomarker levels. The researchers did not analyze plasma tau level as a diagnostic biomarker, but future studies should examine this potential, Dr. Staffaroni and colleagues concluded.
Grants from the National Institute on Aging and the National Institute of Allergy and Infectious Diseases supported the study.
SOURCE: Staffaroni AM et al. JAMA Neurol. 2019 May 6. doi: 10.1001/jamaneurol.2019.1071.
, according to research published online ahead of print May 6 in JAMA Neurology. Levels of total tau in plasma and CSF are correlated, and plasma total tau is associated with survival time. These findings suggest that plasma total tau level could be a valid biomarker that guides clinical care for patients with sporadic Creutzfeldt-Jakob disease, said the investigators.
The accurate prediction of disease duration can assist clinicians and caregivers in clinical management, as well as influence the design of clinical trials. Previous studies have found that baseline protein levels in CSF and plasma are associated with disease duration in patients with sporadic Creutzfeldt-Jakob disease. To replicate these findings, Adam M. Staffaroni, PhD, of the department of neurology at the University of California, San Francisco, and colleagues conducted a longitudinal cohort study.
Evaluating fluid and nonfluid biomarkers
Dr. Staffaroni and colleagues recruited 193 participants with probable or definite sporadic Creutzfeldt-Jakob disease who had codon 129 genotyping and were referred to the UCSF Memory and Aging Center from March 2004 to January 2018. All participants underwent cognitive testing, informant measures, a neurologic examination, and CSF and blood sample collection. The researchers excluded from analysis five participants who had been placed on life-extending treatments. Participants were evaluated until death or censored at the time of statistical analysis.
Dr. Staffaroni and colleagues examined the following nonfluid biomarkers of survival: sex, age, codon 129 genotype, Barthel Index, and Medical Research Council (MRC) Prion Disease Rating Scale. In addition, they examined total tau level, phosphorylated tau level, total tau:phosphorylated tau ratio, neurofilament light (NfL) level, beta-amyloid 42 level, neuron-specific enolase level, 14-3-3 test result, and real-time quaking-induced conversion test in CSF as fluid biomarkers of survival. Finally, Dr. Staffaroni’s group analyzed total tau level, NfL level, and glial fibrillary acidic protein level in plasma as additional fluid biomarkers of survival.
The researchers fitted Cox proportional hazard models with time to event as the outcome. They log-transformed fluid biomarkers and ran models with and without nonfluid biomarkers of survival.
Plasma total tau was associated with survival
In all, 188 patients were included in the analysis. The population’s mean age was 63.8 years. Approximately 45% of participants were women. The diagnosis of sporadic Creutzfeldt-Jakob disease was pathologically confirmed for 78.2% of participants and probable for 21.8% of participants.
Dr. Staffaroni’s group observed strong correlations between plasma and CSF NfL concentrations and between plasma and CSF total tau concentrations. CSF total tau and CSF NfL concentrations were also correlated.
Among the nonfluid biomarkers, Barthel Index, MRC Scale, and codon 129 genotype were significantly associated with survival time. Lower level of function at baseline predicted a faster disease course.
Among the fluid biomarkers, greater levels of plasma total tau and NfL levels at baseline were associated with shorter survival. After the investigators controlled for Barthel Index and codon 129 genotype, the association of plasma total tau level with survival time remained significant. Plasma total tau level and Barthel Index (hazard ratio, 0.98) each independently predicted survival. Dr. Staffaroni and colleagues found that the hazard ratios for all CSF biomarkers were in the expected direction, and that those for total tau level, total tau:phosphorylated tau ratio, NfL level, and neuron-specific enolase level were statistically significant. Furthermore, positive results for 14-3-3 protein, neuron-specific enolase level, and total tau level were associated with a shorter time until death. Like the plasma biomarkers, CSF total tau level remained associated with survival after the investigators controlled for Barthel Index and codon 129 genotype. The same was true of CSF total tau:phosphorylated tau ratio, neuron-specific enolase level, and 14-3-3 result.
Plasma tau could be a diagnostic biomarker
“The hazard ratio associated with plasma total tau level was more than 40% higher than other fluid biomarkers of interest,” said the authors. “These findings further bolster the value of blood-based biomarkers, based on their minimally invasive and relatively inexpensive nature, and build on prior studies that suggested patients with sporadic Creutzfeldt-Jakob disease and controls can be discriminated with relatively high accuracy using blood-based assays.” When Dr. Staffaroni and colleagues modeled baseline functional status and plasma total tau levels together, they found that both were independent predictors of survival time. “This [finding] suggests that clinical measures and plasma total tau level could be combined to further improve prediction accuracy.”
Among the study’s limitations was its comparatively small subsample of patients for whom all plasma and CSF biomarkers were available. Disease duration and survival were longer in the study population than in the literature. “Another limitation is that the plasma biomarkers in this study, one of which showed great promise for predicting survival, were assayed using a research protocol,” said the researchers. “Widespread clinical use of these biomarkers will require well-validated commercial assays, development of which is underway.”
Before these biomarkers can be used in the clinic, these results will need to be replicated, and neurologists will need to develop consensus cutoffs for the biomarker levels. The researchers did not analyze plasma tau level as a diagnostic biomarker, but future studies should examine this potential, Dr. Staffaroni and colleagues concluded.
Grants from the National Institute on Aging and the National Institute of Allergy and Infectious Diseases supported the study.
SOURCE: Staffaroni AM et al. JAMA Neurol. 2019 May 6. doi: 10.1001/jamaneurol.2019.1071.
, according to research published online ahead of print May 6 in JAMA Neurology. Levels of total tau in plasma and CSF are correlated, and plasma total tau is associated with survival time. These findings suggest that plasma total tau level could be a valid biomarker that guides clinical care for patients with sporadic Creutzfeldt-Jakob disease, said the investigators.
The accurate prediction of disease duration can assist clinicians and caregivers in clinical management, as well as influence the design of clinical trials. Previous studies have found that baseline protein levels in CSF and plasma are associated with disease duration in patients with sporadic Creutzfeldt-Jakob disease. To replicate these findings, Adam M. Staffaroni, PhD, of the department of neurology at the University of California, San Francisco, and colleagues conducted a longitudinal cohort study.
Evaluating fluid and nonfluid biomarkers
Dr. Staffaroni and colleagues recruited 193 participants with probable or definite sporadic Creutzfeldt-Jakob disease who had codon 129 genotyping and were referred to the UCSF Memory and Aging Center from March 2004 to January 2018. All participants underwent cognitive testing, informant measures, a neurologic examination, and CSF and blood sample collection. The researchers excluded from analysis five participants who had been placed on life-extending treatments. Participants were evaluated until death or censored at the time of statistical analysis.
Dr. Staffaroni and colleagues examined the following nonfluid biomarkers of survival: sex, age, codon 129 genotype, Barthel Index, and Medical Research Council (MRC) Prion Disease Rating Scale. In addition, they examined total tau level, phosphorylated tau level, total tau:phosphorylated tau ratio, neurofilament light (NfL) level, beta-amyloid 42 level, neuron-specific enolase level, 14-3-3 test result, and real-time quaking-induced conversion test in CSF as fluid biomarkers of survival. Finally, Dr. Staffaroni’s group analyzed total tau level, NfL level, and glial fibrillary acidic protein level in plasma as additional fluid biomarkers of survival.
The researchers fitted Cox proportional hazard models with time to event as the outcome. They log-transformed fluid biomarkers and ran models with and without nonfluid biomarkers of survival.
Plasma total tau was associated with survival
In all, 188 patients were included in the analysis. The population’s mean age was 63.8 years. Approximately 45% of participants were women. The diagnosis of sporadic Creutzfeldt-Jakob disease was pathologically confirmed for 78.2% of participants and probable for 21.8% of participants.
Dr. Staffaroni’s group observed strong correlations between plasma and CSF NfL concentrations and between plasma and CSF total tau concentrations. CSF total tau and CSF NfL concentrations were also correlated.
Among the nonfluid biomarkers, Barthel Index, MRC Scale, and codon 129 genotype were significantly associated with survival time. Lower level of function at baseline predicted a faster disease course.
Among the fluid biomarkers, greater levels of plasma total tau and NfL levels at baseline were associated with shorter survival. After the investigators controlled for Barthel Index and codon 129 genotype, the association of plasma total tau level with survival time remained significant. Plasma total tau level and Barthel Index (hazard ratio, 0.98) each independently predicted survival. Dr. Staffaroni and colleagues found that the hazard ratios for all CSF biomarkers were in the expected direction, and that those for total tau level, total tau:phosphorylated tau ratio, NfL level, and neuron-specific enolase level were statistically significant. Furthermore, positive results for 14-3-3 protein, neuron-specific enolase level, and total tau level were associated with a shorter time until death. Like the plasma biomarkers, CSF total tau level remained associated with survival after the investigators controlled for Barthel Index and codon 129 genotype. The same was true of CSF total tau:phosphorylated tau ratio, neuron-specific enolase level, and 14-3-3 result.
Plasma tau could be a diagnostic biomarker
“The hazard ratio associated with plasma total tau level was more than 40% higher than other fluid biomarkers of interest,” said the authors. “These findings further bolster the value of blood-based biomarkers, based on their minimally invasive and relatively inexpensive nature, and build on prior studies that suggested patients with sporadic Creutzfeldt-Jakob disease and controls can be discriminated with relatively high accuracy using blood-based assays.” When Dr. Staffaroni and colleagues modeled baseline functional status and plasma total tau levels together, they found that both were independent predictors of survival time. “This [finding] suggests that clinical measures and plasma total tau level could be combined to further improve prediction accuracy.”
Among the study’s limitations was its comparatively small subsample of patients for whom all plasma and CSF biomarkers were available. Disease duration and survival were longer in the study population than in the literature. “Another limitation is that the plasma biomarkers in this study, one of which showed great promise for predicting survival, were assayed using a research protocol,” said the researchers. “Widespread clinical use of these biomarkers will require well-validated commercial assays, development of which is underway.”
Before these biomarkers can be used in the clinic, these results will need to be replicated, and neurologists will need to develop consensus cutoffs for the biomarker levels. The researchers did not analyze plasma tau level as a diagnostic biomarker, but future studies should examine this potential, Dr. Staffaroni and colleagues concluded.
Grants from the National Institute on Aging and the National Institute of Allergy and Infectious Diseases supported the study.
SOURCE: Staffaroni AM et al. JAMA Neurol. 2019 May 6. doi: 10.1001/jamaneurol.2019.1071.
FROM JAMA NEUROLOGY
Key clinical point: Invasive and minimally invasive biomarkers are associated with survival in sporadic Creutzfeldt-Jakob disease.
Major finding: The hazard ratio for plasma total tau level was more than 40% larger than any other biomarker.
Study details: A longitudinal study of 188 participants with sporadic Creutzfeldt-Jakob disease.
Disclosures: Grants from the National Institute on Aging and the National Institute of Allergy and Infectious Diseases supported the study.
Source: Staffaroni AM et al. JAMA Neurol. 2019 May 6. doi: 10.1001/jamaneurol.2019.1071.
FDA approves ivosidenib frontline for certain AML patients
The Food and Drug Administration has approved ivosidenib (Tibsovo) for newly diagnosed acute myeloid leukemia (AML) with a susceptible IDH1 mutation in patients who are at least 75 years old or have comorbidities preventing the use of intensive induction chemotherapy.
In July 2018, the FDA approved ivosidenib for adults with relapsed or refractory AML with a susceptible IDH1 mutation.
The latest approval was based on results from an open-label, single-arm, multicenter trial of patients with newly diagnosed AML with an IDH1 mutation. Patients were treated with 500 mg ivosidenib daily until disease progression, development of unacceptable toxicity, or hematopoietic stem cell transplantation; the median age of the 28 patients treated with ivosidenib was 77 years.
Of the 28 patients treated, 12 achieved complete remission or complete remission with partial hematologic recovery; 7 of the 17 transfusion-dependent patients achieved transfusion independence for at least 8 weeks.
The most common adverse events were diarrhea, fatigue, edema, decreased appetite, leukocytosis, nausea, arthralgia, abdominal pain, dyspnea, differentiation syndrome, and myalgia. The drug’s prescribing information includes a boxed warning on the risk of differentiation syndrome.
“The recommended ivosidenib dose is 500 mg orally once daily with or without food until disease progression or unacceptable toxicity. For patients without disease progression or unacceptable toxicity, treatment is recommended for a minimum of 6 months to allow time for clinical response,” the FDA noted.
Find the full press release on the FDA website.
The Food and Drug Administration has approved ivosidenib (Tibsovo) for newly diagnosed acute myeloid leukemia (AML) with a susceptible IDH1 mutation in patients who are at least 75 years old or have comorbidities preventing the use of intensive induction chemotherapy.
In July 2018, the FDA approved ivosidenib for adults with relapsed or refractory AML with a susceptible IDH1 mutation.
The latest approval was based on results from an open-label, single-arm, multicenter trial of patients with newly diagnosed AML with an IDH1 mutation. Patients were treated with 500 mg ivosidenib daily until disease progression, development of unacceptable toxicity, or hematopoietic stem cell transplantation; the median age of the 28 patients treated with ivosidenib was 77 years.
Of the 28 patients treated, 12 achieved complete remission or complete remission with partial hematologic recovery; 7 of the 17 transfusion-dependent patients achieved transfusion independence for at least 8 weeks.
The most common adverse events were diarrhea, fatigue, edema, decreased appetite, leukocytosis, nausea, arthralgia, abdominal pain, dyspnea, differentiation syndrome, and myalgia. The drug’s prescribing information includes a boxed warning on the risk of differentiation syndrome.
“The recommended ivosidenib dose is 500 mg orally once daily with or without food until disease progression or unacceptable toxicity. For patients without disease progression or unacceptable toxicity, treatment is recommended for a minimum of 6 months to allow time for clinical response,” the FDA noted.
Find the full press release on the FDA website.
The Food and Drug Administration has approved ivosidenib (Tibsovo) for newly diagnosed acute myeloid leukemia (AML) with a susceptible IDH1 mutation in patients who are at least 75 years old or have comorbidities preventing the use of intensive induction chemotherapy.
In July 2018, the FDA approved ivosidenib for adults with relapsed or refractory AML with a susceptible IDH1 mutation.
The latest approval was based on results from an open-label, single-arm, multicenter trial of patients with newly diagnosed AML with an IDH1 mutation. Patients were treated with 500 mg ivosidenib daily until disease progression, development of unacceptable toxicity, or hematopoietic stem cell transplantation; the median age of the 28 patients treated with ivosidenib was 77 years.
Of the 28 patients treated, 12 achieved complete remission or complete remission with partial hematologic recovery; 7 of the 17 transfusion-dependent patients achieved transfusion independence for at least 8 weeks.
The most common adverse events were diarrhea, fatigue, edema, decreased appetite, leukocytosis, nausea, arthralgia, abdominal pain, dyspnea, differentiation syndrome, and myalgia. The drug’s prescribing information includes a boxed warning on the risk of differentiation syndrome.
“The recommended ivosidenib dose is 500 mg orally once daily with or without food until disease progression or unacceptable toxicity. For patients without disease progression or unacceptable toxicity, treatment is recommended for a minimum of 6 months to allow time for clinical response,” the FDA noted.
Find the full press release on the FDA website.
Erythema on abdomen

The FP was puzzled by the lack of response to treatment and decided to perform a 4-mm punch biopsy at the edge of the nonhealing ulcer. (Note that the correct location for a biopsy of an ulcer is on the edge, not in the middle). His differential diagnosis included pyoderma gangrenosum and a deep fungal infection. The pathologist called a week later FP with a surprising result: anaplastic large cell cutaneous T-cell lymphoma. (See the Watch & Learn video on “Punch biopsy.”)
Anaplastic large cell cutaneous T-cell lymphoma is a rare diagnosis—especially in a teenager—and it can’t be determined by appearance only. On follow-up, the FP explained the diagnosis to the patient and her mother. He called Hematology/Oncology to facilitate the referral.
The patient was treated by the specialist with weekly oral methotrexate and her skin cleared up completely. Although she would likely need treatment for years, the prognosis was good. This case is a reminder that when a treatment is not working for an expected diagnosis, it’s time to reconsider the diagnosis and do further testing to identify the correct diagnosis.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Chacon G, Nayar A, Usatine R, Smith M. Cutaneous T-cell lymphoma. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas and Synopsis of Family Medicine. 3rd ed. New York, NY: McGraw-Hill; 2019:1124-1131.
To learn more about the newest 3rd edition of the Color Atlas and Synopsis of Family Medicine, see: https://www.amazon.com/Color-Atlas-Synopsis-Family-Medicine/dp/1259862046/
You can get the Color Atlas of Family Medicine app by clicking on this link: usatinemedia.com
The FP was puzzled by the lack of response to treatment and decided to perform a 4-mm punch biopsy at the edge of the nonhealing ulcer. (Note that the correct location for a biopsy of an ulcer is on the edge, not in the middle). His differential diagnosis included pyoderma gangrenosum and a deep fungal infection. The pathologist called a week later FP with a surprising result: anaplastic large cell cutaneous T-cell lymphoma. (See the Watch & Learn video on “Punch biopsy.”)
Anaplastic large cell cutaneous T-cell lymphoma is a rare diagnosis—especially in a teenager—and it can’t be determined by appearance only. On follow-up, the FP explained the diagnosis to the patient and her mother. He called Hematology/Oncology to facilitate the referral.
The patient was treated by the specialist with weekly oral methotrexate and her skin cleared up completely. Although she would likely need treatment for years, the prognosis was good. This case is a reminder that when a treatment is not working for an expected diagnosis, it’s time to reconsider the diagnosis and do further testing to identify the correct diagnosis.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Chacon G, Nayar A, Usatine R, Smith M. Cutaneous T-cell lymphoma. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas and Synopsis of Family Medicine. 3rd ed. New York, NY: McGraw-Hill; 2019:1124-1131.
To learn more about the newest 3rd edition of the Color Atlas and Synopsis of Family Medicine, see: https://www.amazon.com/Color-Atlas-Synopsis-Family-Medicine/dp/1259862046/
You can get the Color Atlas of Family Medicine app by clicking on this link: usatinemedia.com
The FP was puzzled by the lack of response to treatment and decided to perform a 4-mm punch biopsy at the edge of the nonhealing ulcer. (Note that the correct location for a biopsy of an ulcer is on the edge, not in the middle). His differential diagnosis included pyoderma gangrenosum and a deep fungal infection. The pathologist called a week later FP with a surprising result: anaplastic large cell cutaneous T-cell lymphoma. (See the Watch & Learn video on “Punch biopsy.”)
Anaplastic large cell cutaneous T-cell lymphoma is a rare diagnosis—especially in a teenager—and it can’t be determined by appearance only. On follow-up, the FP explained the diagnosis to the patient and her mother. He called Hematology/Oncology to facilitate the referral.
The patient was treated by the specialist with weekly oral methotrexate and her skin cleared up completely. Although she would likely need treatment for years, the prognosis was good. This case is a reminder that when a treatment is not working for an expected diagnosis, it’s time to reconsider the diagnosis and do further testing to identify the correct diagnosis.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Chacon G, Nayar A, Usatine R, Smith M. Cutaneous T-cell lymphoma. In: Usatine R, Smith M, Mayeaux EJ, et al. Color Atlas and Synopsis of Family Medicine. 3rd ed. New York, NY: McGraw-Hill; 2019:1124-1131.
To learn more about the newest 3rd edition of the Color Atlas and Synopsis of Family Medicine, see: https://www.amazon.com/Color-Atlas-Synopsis-Family-Medicine/dp/1259862046/
You can get the Color Atlas of Family Medicine app by clicking on this link: usatinemedia.com

Report card may foretell achalasia surgery outcomes
BALTIMORE – The Eckardt score has been established as a tool to evaluate outcomes of surgery for achalasia, but researchers have developed a report card that uses multiple variables that may provide a more accurate picture of surgical outcomes, according to results of study reported at the annual meeting of the Society of American Gastrointestinal Endoscopic Surgeons.
“The use of an accurate score to assess outcomes after achalasia surgery shows outstanding results,” said Ealaf Shemmeri, MD, of Swedish Medical Center in Seattle. “Using patient-reported symptoms, objective measures, and rates of reinterventions organized into a report card provides a more comprehensive and informative view.”
The Eckardt score evaluates four symptoms to evaluate outcomes of surgery to treat achalasia: weight loss, retrosternal pain, regurgitation, and dysphagia. “However, it does not address the other changes that can occur after myotomy, including the quality of swallowing and the onset of reflux disease,” she said. “Thus, there is a need for a more comprehensive assessment of quality after achalasia treatment.”
So the Swedish investigators set out to devise a report card that provides “a comprehensive and informative assessment” of surgical myotomy outcomes, she said. This involved a retrospective, single-center chart review of 185 patients who had surgical myotomy for primary achalasia from 2005 to 2017.
To determine patient-reported outcomes, the report card defines success as an Eckardt score below 3, Dakkak dysphagia score above 40, and GERD-HRQL (health-related quality of life) score below 10. The objective measures consisted of DeMeester (pH) score below 14.72, no column at 5 minutes on timed barium swallow, normalized integrated relaxation pressure less than 15 on manometry, and absence of esophagitis on endoscopy. For the third pillar of the report card, no reintervention was recorded as a success, Dr. Shemmeri said.
Regarding the etiology of achalasia in the study population, 42 had type 1, 109 had type 2, and 34 had type 3. A total of 71 patients had per oral endoscopic myotomy and 114 had Heller myotomy, 92 with Dor fundoplication and 20 with Toupet. Major perioperative complications included four per oral endoscopic myotomy patients who developed a leak requiring intervention. Six patients required return to the operating room for persistent dysphagia, Dr. Shemmeri said.
After the procedures, 93% of study patients reported an Eckardt score less than 3. However, only 45% have a Dakkak dysphagia score greater than 40 and 71% had a GERD-HRQL score less than 10, Dr. Shemmeri said. The objective measures told a similar story: Integrated relaxation pressure normalized in 80%, barium clearance was achieved in 61%, normal esophageal mucosa was recorded in 71%, “but pH testing was normal only 50% of the time,” Dr. Shemmeri said.
“The final success of not needing intervention is 79%,” she said. At this point in the study, 139 patients were available for follow-up. Among the 29 who needed reintervention, 19 had dilation below 20 mm Hg, 3 underwent pneumatic dilation, and 2 had botulinum toxin. Two patients required a redo myotomy, two had antireflux surgery, and one had an esophagectomy.
“When you only focus on a singular outcome, you can miss the whole story that occurs after myotomy,” Dr. Shemmeri said. “Providing a comprehensive tool gives you the ability to identify areas for improvement in your achalasia practice. Its simplicity allows it to be applied in various settings.” In the academic setting, it can be a tool for evaluating technologies and approaches for postmyotomy management. In hospitals and surgeons’ practices, it can aid in quality improvement, comparative outcomes research, and in evaluating operative approaches to myotomy.
The outcomes highlight the high prevalence of GERD, thus stressing the importance of pH testing after myotomy, Dr. Shemmeri said. Her study team recommends pH testing at 6-month follow-up because patients may not always self-report the extent of esophagitis present. Coauthor Brian Louie, MD, also of Swedish Medical Center, added during the discussion that ongoing follow-up of achalasia patients is necessary to address issues patients encounter with their swallowing over time.
Dr. Shemmeri had no relevant financial relationships to disclose. Dr. Louie reported relationships with Boston Scientific, ERBE, and Olympus.
SOURCE: Shemmeri E et al. SAGES 2019, Presentation S085.
BALTIMORE – The Eckardt score has been established as a tool to evaluate outcomes of surgery for achalasia, but researchers have developed a report card that uses multiple variables that may provide a more accurate picture of surgical outcomes, according to results of study reported at the annual meeting of the Society of American Gastrointestinal Endoscopic Surgeons.
“The use of an accurate score to assess outcomes after achalasia surgery shows outstanding results,” said Ealaf Shemmeri, MD, of Swedish Medical Center in Seattle. “Using patient-reported symptoms, objective measures, and rates of reinterventions organized into a report card provides a more comprehensive and informative view.”
The Eckardt score evaluates four symptoms to evaluate outcomes of surgery to treat achalasia: weight loss, retrosternal pain, regurgitation, and dysphagia. “However, it does not address the other changes that can occur after myotomy, including the quality of swallowing and the onset of reflux disease,” she said. “Thus, there is a need for a more comprehensive assessment of quality after achalasia treatment.”
So the Swedish investigators set out to devise a report card that provides “a comprehensive and informative assessment” of surgical myotomy outcomes, she said. This involved a retrospective, single-center chart review of 185 patients who had surgical myotomy for primary achalasia from 2005 to 2017.
To determine patient-reported outcomes, the report card defines success as an Eckardt score below 3, Dakkak dysphagia score above 40, and GERD-HRQL (health-related quality of life) score below 10. The objective measures consisted of DeMeester (pH) score below 14.72, no column at 5 minutes on timed barium swallow, normalized integrated relaxation pressure less than 15 on manometry, and absence of esophagitis on endoscopy. For the third pillar of the report card, no reintervention was recorded as a success, Dr. Shemmeri said.
Regarding the etiology of achalasia in the study population, 42 had type 1, 109 had type 2, and 34 had type 3. A total of 71 patients had per oral endoscopic myotomy and 114 had Heller myotomy, 92 with Dor fundoplication and 20 with Toupet. Major perioperative complications included four per oral endoscopic myotomy patients who developed a leak requiring intervention. Six patients required return to the operating room for persistent dysphagia, Dr. Shemmeri said.
After the procedures, 93% of study patients reported an Eckardt score less than 3. However, only 45% have a Dakkak dysphagia score greater than 40 and 71% had a GERD-HRQL score less than 10, Dr. Shemmeri said. The objective measures told a similar story: Integrated relaxation pressure normalized in 80%, barium clearance was achieved in 61%, normal esophageal mucosa was recorded in 71%, “but pH testing was normal only 50% of the time,” Dr. Shemmeri said.
“The final success of not needing intervention is 79%,” she said. At this point in the study, 139 patients were available for follow-up. Among the 29 who needed reintervention, 19 had dilation below 20 mm Hg, 3 underwent pneumatic dilation, and 2 had botulinum toxin. Two patients required a redo myotomy, two had antireflux surgery, and one had an esophagectomy.
“When you only focus on a singular outcome, you can miss the whole story that occurs after myotomy,” Dr. Shemmeri said. “Providing a comprehensive tool gives you the ability to identify areas for improvement in your achalasia practice. Its simplicity allows it to be applied in various settings.” In the academic setting, it can be a tool for evaluating technologies and approaches for postmyotomy management. In hospitals and surgeons’ practices, it can aid in quality improvement, comparative outcomes research, and in evaluating operative approaches to myotomy.
The outcomes highlight the high prevalence of GERD, thus stressing the importance of pH testing after myotomy, Dr. Shemmeri said. Her study team recommends pH testing at 6-month follow-up because patients may not always self-report the extent of esophagitis present. Coauthor Brian Louie, MD, also of Swedish Medical Center, added during the discussion that ongoing follow-up of achalasia patients is necessary to address issues patients encounter with their swallowing over time.
Dr. Shemmeri had no relevant financial relationships to disclose. Dr. Louie reported relationships with Boston Scientific, ERBE, and Olympus.
SOURCE: Shemmeri E et al. SAGES 2019, Presentation S085.
BALTIMORE – The Eckardt score has been established as a tool to evaluate outcomes of surgery for achalasia, but researchers have developed a report card that uses multiple variables that may provide a more accurate picture of surgical outcomes, according to results of study reported at the annual meeting of the Society of American Gastrointestinal Endoscopic Surgeons.
“The use of an accurate score to assess outcomes after achalasia surgery shows outstanding results,” said Ealaf Shemmeri, MD, of Swedish Medical Center in Seattle. “Using patient-reported symptoms, objective measures, and rates of reinterventions organized into a report card provides a more comprehensive and informative view.”
The Eckardt score evaluates four symptoms to evaluate outcomes of surgery to treat achalasia: weight loss, retrosternal pain, regurgitation, and dysphagia. “However, it does not address the other changes that can occur after myotomy, including the quality of swallowing and the onset of reflux disease,” she said. “Thus, there is a need for a more comprehensive assessment of quality after achalasia treatment.”
So the Swedish investigators set out to devise a report card that provides “a comprehensive and informative assessment” of surgical myotomy outcomes, she said. This involved a retrospective, single-center chart review of 185 patients who had surgical myotomy for primary achalasia from 2005 to 2017.
To determine patient-reported outcomes, the report card defines success as an Eckardt score below 3, Dakkak dysphagia score above 40, and GERD-HRQL (health-related quality of life) score below 10. The objective measures consisted of DeMeester (pH) score below 14.72, no column at 5 minutes on timed barium swallow, normalized integrated relaxation pressure less than 15 on manometry, and absence of esophagitis on endoscopy. For the third pillar of the report card, no reintervention was recorded as a success, Dr. Shemmeri said.
Regarding the etiology of achalasia in the study population, 42 had type 1, 109 had type 2, and 34 had type 3. A total of 71 patients had per oral endoscopic myotomy and 114 had Heller myotomy, 92 with Dor fundoplication and 20 with Toupet. Major perioperative complications included four per oral endoscopic myotomy patients who developed a leak requiring intervention. Six patients required return to the operating room for persistent dysphagia, Dr. Shemmeri said.
After the procedures, 93% of study patients reported an Eckardt score less than 3. However, only 45% have a Dakkak dysphagia score greater than 40 and 71% had a GERD-HRQL score less than 10, Dr. Shemmeri said. The objective measures told a similar story: Integrated relaxation pressure normalized in 80%, barium clearance was achieved in 61%, normal esophageal mucosa was recorded in 71%, “but pH testing was normal only 50% of the time,” Dr. Shemmeri said.
“The final success of not needing intervention is 79%,” she said. At this point in the study, 139 patients were available for follow-up. Among the 29 who needed reintervention, 19 had dilation below 20 mm Hg, 3 underwent pneumatic dilation, and 2 had botulinum toxin. Two patients required a redo myotomy, two had antireflux surgery, and one had an esophagectomy.
“When you only focus on a singular outcome, you can miss the whole story that occurs after myotomy,” Dr. Shemmeri said. “Providing a comprehensive tool gives you the ability to identify areas for improvement in your achalasia practice. Its simplicity allows it to be applied in various settings.” In the academic setting, it can be a tool for evaluating technologies and approaches for postmyotomy management. In hospitals and surgeons’ practices, it can aid in quality improvement, comparative outcomes research, and in evaluating operative approaches to myotomy.
The outcomes highlight the high prevalence of GERD, thus stressing the importance of pH testing after myotomy, Dr. Shemmeri said. Her study team recommends pH testing at 6-month follow-up because patients may not always self-report the extent of esophagitis present. Coauthor Brian Louie, MD, also of Swedish Medical Center, added during the discussion that ongoing follow-up of achalasia patients is necessary to address issues patients encounter with their swallowing over time.
Dr. Shemmeri had no relevant financial relationships to disclose. Dr. Louie reported relationships with Boston Scientific, ERBE, and Olympus.
SOURCE: Shemmeri E et al. SAGES 2019, Presentation S085.
REPORTING FROM SAGES 2019
Neurodevelopmental concerns may emerge later in Zika-exposed infants
BALTIMORE – Most infants prenatally exposed to Zika showed relatively normal neurodevelopment if their fetal MRI and birth head circumference were normal, but others with similarly initial normal measures appeared to struggle with social cognition and mobility as they got older, according to a new study.

“I think we need to be cautious with saying that these children are normal when these normal-appearing children may not be doing as well as we think,” lead author Sarah Mulkey, MD, of Children’s National Health System and George Washington University, Washington, said in an interview. “While most children are showing fairly normal development, there are some children who are … becoming more abnormal over time.”
Dr. Mulkey shared her findings at the Pediatric Academic Societies annual meeting. She and her colleagues had previously published a prospective study of 82 Zika-exposed infants’ fetal brain MRIs. In their new study, they followed up with the 78 Colombian infants from that study whose fetal neuroimaging and birth head circumstance had been normal.
The researchers used the Alberta Infant Motor Scale (AIMS) and the Warner Initial Developmental Evaluation of Adaptive and Functional Skills (WIDEA) to evaluate 72 of the children, 34 of whom underwent assessment twice. Forty of the children were an average 5.7 months old when evaluated, and 66 were an average 13.5 months old.
As the children got older, their overall WIDEA z-score and their subscores in the social cognition domain and especially in the mobility domain trended downward. Three of the children had AIMS scores two standard deviations below normal, but the rest fell within the normal range.
Their WIDEA communication z-score hovered relatively close to the norm, but self-care also showed a very slight slope downward, albeit not as substantially as in the social cognition and mobility domains.
The younger a child is, the fewer skills they generally show related to neurocognitive development, Dr. Mulkey explained. But as they grow older and are expected to show more skills, it becomes more apparent where gaps and delays might exist.
“We can see that there are a lot of kids doing well, but some of these kids certainly are not,” she said. “Until children have a long time to develop, you really can’t see these changes unless you follow them long-term.”
The researchers also looked separately at a subgroup of 19 children (26%) whose cranial ultrasounds showed mild nonspecific findings. These findings – such as lenticulostriate vasculopathy, choroid plexus cysts, subependymal cysts and calcifications – do not usually indicate any problems, but they appeared in a quarter of this population, considerably more than the approximately 5% typically seen in the general population, Dr. Mulkey said.
Though the findings did not reach significance, infants in this subgroup tended to have a lower WIDEA mobility z-scores (P = .054) and lower AIMS scores (P = .26) than the Zika-exposed infants with normal cranial ultrasounds.
“Mild nonspecific cranial ultrasound findings may represent a mild injury” related to exposure to their mother’s Zika infection during pregnancy, the researchers suggested. “It may be a risk factor for the lower mobility outcome,” Dr. Mulkey said.
The researchers hope to continue later follow-ups as the children age.
The research was funded by the Thrasher Research Fund. Dr. Mulkey had no conflicts of interest.
BALTIMORE – Most infants prenatally exposed to Zika showed relatively normal neurodevelopment if their fetal MRI and birth head circumference were normal, but others with similarly initial normal measures appeared to struggle with social cognition and mobility as they got older, according to a new study.
“I think we need to be cautious with saying that these children are normal when these normal-appearing children may not be doing as well as we think,” lead author Sarah Mulkey, MD, of Children’s National Health System and George Washington University, Washington, said in an interview. “While most children are showing fairly normal development, there are some children who are … becoming more abnormal over time.”
Dr. Mulkey shared her findings at the Pediatric Academic Societies annual meeting. She and her colleagues had previously published a prospective study of 82 Zika-exposed infants’ fetal brain MRIs. In their new study, they followed up with the 78 Colombian infants from that study whose fetal neuroimaging and birth head circumstance had been normal.
The researchers used the Alberta Infant Motor Scale (AIMS) and the Warner Initial Developmental Evaluation of Adaptive and Functional Skills (WIDEA) to evaluate 72 of the children, 34 of whom underwent assessment twice. Forty of the children were an average 5.7 months old when evaluated, and 66 were an average 13.5 months old.
As the children got older, their overall WIDEA z-score and their subscores in the social cognition domain and especially in the mobility domain trended downward. Three of the children had AIMS scores two standard deviations below normal, but the rest fell within the normal range.
Their WIDEA communication z-score hovered relatively close to the norm, but self-care also showed a very slight slope downward, albeit not as substantially as in the social cognition and mobility domains.
The younger a child is, the fewer skills they generally show related to neurocognitive development, Dr. Mulkey explained. But as they grow older and are expected to show more skills, it becomes more apparent where gaps and delays might exist.
“We can see that there are a lot of kids doing well, but some of these kids certainly are not,” she said. “Until children have a long time to develop, you really can’t see these changes unless you follow them long-term.”
The researchers also looked separately at a subgroup of 19 children (26%) whose cranial ultrasounds showed mild nonspecific findings. These findings – such as lenticulostriate vasculopathy, choroid plexus cysts, subependymal cysts and calcifications – do not usually indicate any problems, but they appeared in a quarter of this population, considerably more than the approximately 5% typically seen in the general population, Dr. Mulkey said.
Though the findings did not reach significance, infants in this subgroup tended to have a lower WIDEA mobility z-scores (P = .054) and lower AIMS scores (P = .26) than the Zika-exposed infants with normal cranial ultrasounds.
“Mild nonspecific cranial ultrasound findings may represent a mild injury” related to exposure to their mother’s Zika infection during pregnancy, the researchers suggested. “It may be a risk factor for the lower mobility outcome,” Dr. Mulkey said.
The researchers hope to continue later follow-ups as the children age.
The research was funded by the Thrasher Research Fund. Dr. Mulkey had no conflicts of interest.
BALTIMORE – Most infants prenatally exposed to Zika showed relatively normal neurodevelopment if their fetal MRI and birth head circumference were normal, but others with similarly initial normal measures appeared to struggle with social cognition and mobility as they got older, according to a new study.
“I think we need to be cautious with saying that these children are normal when these normal-appearing children may not be doing as well as we think,” lead author Sarah Mulkey, MD, of Children’s National Health System and George Washington University, Washington, said in an interview. “While most children are showing fairly normal development, there are some children who are … becoming more abnormal over time.”
Dr. Mulkey shared her findings at the Pediatric Academic Societies annual meeting. She and her colleagues had previously published a prospective study of 82 Zika-exposed infants’ fetal brain MRIs. In their new study, they followed up with the 78 Colombian infants from that study whose fetal neuroimaging and birth head circumstance had been normal.
The researchers used the Alberta Infant Motor Scale (AIMS) and the Warner Initial Developmental Evaluation of Adaptive and Functional Skills (WIDEA) to evaluate 72 of the children, 34 of whom underwent assessment twice. Forty of the children were an average 5.7 months old when evaluated, and 66 were an average 13.5 months old.
As the children got older, their overall WIDEA z-score and their subscores in the social cognition domain and especially in the mobility domain trended downward. Three of the children had AIMS scores two standard deviations below normal, but the rest fell within the normal range.
Their WIDEA communication z-score hovered relatively close to the norm, but self-care also showed a very slight slope downward, albeit not as substantially as in the social cognition and mobility domains.
The younger a child is, the fewer skills they generally show related to neurocognitive development, Dr. Mulkey explained. But as they grow older and are expected to show more skills, it becomes more apparent where gaps and delays might exist.
“We can see that there are a lot of kids doing well, but some of these kids certainly are not,” she said. “Until children have a long time to develop, you really can’t see these changes unless you follow them long-term.”
The researchers also looked separately at a subgroup of 19 children (26%) whose cranial ultrasounds showed mild nonspecific findings. These findings – such as lenticulostriate vasculopathy, choroid plexus cysts, subependymal cysts and calcifications – do not usually indicate any problems, but they appeared in a quarter of this population, considerably more than the approximately 5% typically seen in the general population, Dr. Mulkey said.
Though the findings did not reach significance, infants in this subgroup tended to have a lower WIDEA mobility z-scores (P = .054) and lower AIMS scores (P = .26) than the Zika-exposed infants with normal cranial ultrasounds.
“Mild nonspecific cranial ultrasound findings may represent a mild injury” related to exposure to their mother’s Zika infection during pregnancy, the researchers suggested. “It may be a risk factor for the lower mobility outcome,” Dr. Mulkey said.
The researchers hope to continue later follow-ups as the children age.
The research was funded by the Thrasher Research Fund. Dr. Mulkey had no conflicts of interest.
REPORTING FROM PAS 2019
Key clinical point: .
Major finding: Zika-exposed infants with normal fetal MRI neuroimaging showed increasingly lower mobility and social cognition skills as they approached their first birthday.
Study details: The findings are based on neurodevelopmental assessments of 72 Zika-exposed Colombian children at 4-18 months old.
Disclosures: The research was funded by the Thrasher Research Fund. Dr. Mulkey had no conflicts of interest.