User login
Less Invasive, Overlooked Option in Cardiac Surgery May Offer Benefit
Compared with traditional replacement valves, sutureless valves placed through minimally invasive cardiac surgery have less data supporting their use but offer unique features that might make them the preferred option for certain patients, reported specialists.
The sutureless device known as Perceval (Corcym) and a rapidly deployed device called Intuity (Edwards Lifesciences) are used as an alternative to surgical aortic valve replacement (SAVR) and transcatheter aortic valve replacement (TAVR). But despite being commercially available since 2016, the devices are still not being used much.
The devices are not discussed in substantial detail in either the joint guidelines from the American College of Cardiology and American Heart Association issued in 2020 or guidelines from the European Society of Cardiology issued in 2022.
Cristiano Spadaccio, MD, PhD, a cardiothoracic surgeon associated with Lancashire Cardiac Centre in Blackpool, England, and his colleagues reviewed the small number of studies evaluating the alternate approach to “make the cardiology world aware” of alternatives “that can relieve the surgical burden by minimizing the implantation time and length of the operation,” he said.
The comprehensive review is published in the Journal of the American College of Cardiology.
A Neglected Alternative
The sutureless Perceval device is held in place by a stent frame that self-expands. The Intuity device also relies primarily on its framework to anchor the valve in place but does involve three sutures. Both devices are still referred to as sutureless in the new review of them.
Only a small number of centers perform minimally invasive cardiac surgeries, and the main advantage of the devices — rapid deployment — has been eroded with the advent of automated knotting which has significantly reduced the time to implant and sutured valve.
The underuse of these devices is largely caused by the limited amount of comparative and prospective data, said Dr. Spadaccio. “The entire literature on sutureless aortic valve replacement with the exception of one randomized controlled trial is observational.”
That trial, PERSIST-AVR, found that the sutureless valves were just as good as conventional ones when it comes to major adverse cardiovascular events including all-cause death, myocardial infarction, stroke, or valve reintervention at 1 year.
In a subanalysis limited to patients who had isolated aortic valve replacement, the sutureless procedure was associated with lower adverse events (5.2% vs 10.8%) at the cost of a higher rate of pacemaker implantation (11% vs 1.6%).
There are also multiple retrospective studies and registries that have generated observational data comparing sutureless aortic valve replacement with SAVR and TAVR in various patient populations, said Dr. Spadaccio, and the review was based on more than a dozen studies published since 2015. Long-term follow-up data for sutureless aortic valve replacements, which now exceeds 10 years, suggest rates of structural valve deterioration and reintervention have been acceptably low.
The minimally invasive procedures have other advantages too. For example, relative to the greater trauma associated with open heart surgery, minimally invasive surgeries typically involve faster recovery, an advantage likely to appeal to many patients who are candidates for either.
Quicker Recovery
Collectively, these data suggest that sutureless aortic valve replacement might be a reasonable or even a more appropriate alternative to either SAVR or TAVR when considering specific patient characteristics and goals, according to the review, which included an algorithm identifying specifically where sutureless aortic valve replacement fits with SAVR and TAVR.
“The algorithm is based on different clinical scenarios and reflects current guidelines for SAVR,” said Dr. Spadaccio. For example, current guidelines identify SAVR as preferred in patients younger than 65 years and in older patients with a low Society of Thoracic Surgeons (STS) score, but there are many instances in which sutureless aortic valve replacement might be more attractive, such as in those also undergoing mitral valve repair, coronary artery bypass grafting, or another surgical procedure.
Dr. Spadaccio said that the STS score should not be considered in isolation when evaluating a patient for SAVR or TAVR. Other features such as mobility, frailty score, and comorbid liver or renal disease should also be considered when discussing the three options with patients. As a result, the algorithm emphasizes a detailed evaluation of patient characteristics in selecting one procedure over another.
“The treatment should be really tailored on the individual patient basis,” said Dr. Spadaccio.
Dr. Spadaccio acknowledged that there is a need for more comparative trials, particularly in regard to sutureless aortic valve replacement as an alternative to TAVR. “I really think that a 1:1 RCT on sutureless aortic valve replacement vs TAVR could give better answers to all of these interrogatives.”
But despite the limitations outlined in this review, Dr. Spadaccio and colleagues challenged the perception that current data are not sufficient to allow clinicians to consider sutureless aortic valve replacement in the mix of options.
A Viable Option
This comprehensive summary of what is known about sutureless aortic valve replacement compared with the other options addresses an important knowledge gap, said S. Chris Malaisrie, MD, a cardiac surgeon at Northwestern University Feinberg School of Medicine, Chicago, Illinois.
He said he agrees this option has unique qualities. “Minimally invasive surgery has been largely ignored by guideline writers, but patients certainly demand options that are less invasive than standard open heart surgery. Sutureless and rapid deployment valves facilitate minimally invasive surgery and offer an advantageous option for younger patients.”
Dr. Malaisrie said the review is generating discussion about a potentially valuable option within the cardiology community. And that is exactly what Dr. Spadaccio was hoping for. “This paper was meant to educate as much as possible on these details to assist and inform decision-making,” he said.
A version of this article first appeared on Medscape.com.
Compared with traditional replacement valves, sutureless valves placed through minimally invasive cardiac surgery have less data supporting their use but offer unique features that might make them the preferred option for certain patients, reported specialists.
The sutureless device known as Perceval (Corcym) and a rapidly deployed device called Intuity (Edwards Lifesciences) are used as an alternative to surgical aortic valve replacement (SAVR) and transcatheter aortic valve replacement (TAVR). But despite being commercially available since 2016, the devices are still not being used much.
The devices are not discussed in substantial detail in either the joint guidelines from the American College of Cardiology and American Heart Association issued in 2020 or guidelines from the European Society of Cardiology issued in 2022.
Cristiano Spadaccio, MD, PhD, a cardiothoracic surgeon associated with Lancashire Cardiac Centre in Blackpool, England, and his colleagues reviewed the small number of studies evaluating the alternate approach to “make the cardiology world aware” of alternatives “that can relieve the surgical burden by minimizing the implantation time and length of the operation,” he said.
The comprehensive review is published in the Journal of the American College of Cardiology.
A Neglected Alternative
The sutureless Perceval device is held in place by a stent frame that self-expands. The Intuity device also relies primarily on its framework to anchor the valve in place but does involve three sutures. Both devices are still referred to as sutureless in the new review of them.
Only a small number of centers perform minimally invasive cardiac surgeries, and the main advantage of the devices — rapid deployment — has been eroded with the advent of automated knotting which has significantly reduced the time to implant and sutured valve.
The underuse of these devices is largely caused by the limited amount of comparative and prospective data, said Dr. Spadaccio. “The entire literature on sutureless aortic valve replacement with the exception of one randomized controlled trial is observational.”
That trial, PERSIST-AVR, found that the sutureless valves were just as good as conventional ones when it comes to major adverse cardiovascular events including all-cause death, myocardial infarction, stroke, or valve reintervention at 1 year.
In a subanalysis limited to patients who had isolated aortic valve replacement, the sutureless procedure was associated with lower adverse events (5.2% vs 10.8%) at the cost of a higher rate of pacemaker implantation (11% vs 1.6%).
There are also multiple retrospective studies and registries that have generated observational data comparing sutureless aortic valve replacement with SAVR and TAVR in various patient populations, said Dr. Spadaccio, and the review was based on more than a dozen studies published since 2015. Long-term follow-up data for sutureless aortic valve replacements, which now exceeds 10 years, suggest rates of structural valve deterioration and reintervention have been acceptably low.
The minimally invasive procedures have other advantages too. For example, relative to the greater trauma associated with open heart surgery, minimally invasive surgeries typically involve faster recovery, an advantage likely to appeal to many patients who are candidates for either.
Quicker Recovery
Collectively, these data suggest that sutureless aortic valve replacement might be a reasonable or even a more appropriate alternative to either SAVR or TAVR when considering specific patient characteristics and goals, according to the review, which included an algorithm identifying specifically where sutureless aortic valve replacement fits with SAVR and TAVR.
“The algorithm is based on different clinical scenarios and reflects current guidelines for SAVR,” said Dr. Spadaccio. For example, current guidelines identify SAVR as preferred in patients younger than 65 years and in older patients with a low Society of Thoracic Surgeons (STS) score, but there are many instances in which sutureless aortic valve replacement might be more attractive, such as in those also undergoing mitral valve repair, coronary artery bypass grafting, or another surgical procedure.
Dr. Spadaccio said that the STS score should not be considered in isolation when evaluating a patient for SAVR or TAVR. Other features such as mobility, frailty score, and comorbid liver or renal disease should also be considered when discussing the three options with patients. As a result, the algorithm emphasizes a detailed evaluation of patient characteristics in selecting one procedure over another.
“The treatment should be really tailored on the individual patient basis,” said Dr. Spadaccio.
Dr. Spadaccio acknowledged that there is a need for more comparative trials, particularly in regard to sutureless aortic valve replacement as an alternative to TAVR. “I really think that a 1:1 RCT on sutureless aortic valve replacement vs TAVR could give better answers to all of these interrogatives.”
But despite the limitations outlined in this review, Dr. Spadaccio and colleagues challenged the perception that current data are not sufficient to allow clinicians to consider sutureless aortic valve replacement in the mix of options.
A Viable Option
This comprehensive summary of what is known about sutureless aortic valve replacement compared with the other options addresses an important knowledge gap, said S. Chris Malaisrie, MD, a cardiac surgeon at Northwestern University Feinberg School of Medicine, Chicago, Illinois.
He said he agrees this option has unique qualities. “Minimally invasive surgery has been largely ignored by guideline writers, but patients certainly demand options that are less invasive than standard open heart surgery. Sutureless and rapid deployment valves facilitate minimally invasive surgery and offer an advantageous option for younger patients.”
Dr. Malaisrie said the review is generating discussion about a potentially valuable option within the cardiology community. And that is exactly what Dr. Spadaccio was hoping for. “This paper was meant to educate as much as possible on these details to assist and inform decision-making,” he said.
A version of this article first appeared on Medscape.com.
Compared with traditional replacement valves, sutureless valves placed through minimally invasive cardiac surgery have less data supporting their use but offer unique features that might make them the preferred option for certain patients, reported specialists.
The sutureless device known as Perceval (Corcym) and a rapidly deployed device called Intuity (Edwards Lifesciences) are used as an alternative to surgical aortic valve replacement (SAVR) and transcatheter aortic valve replacement (TAVR). But despite being commercially available since 2016, the devices are still not being used much.
The devices are not discussed in substantial detail in either the joint guidelines from the American College of Cardiology and American Heart Association issued in 2020 or guidelines from the European Society of Cardiology issued in 2022.
Cristiano Spadaccio, MD, PhD, a cardiothoracic surgeon associated with Lancashire Cardiac Centre in Blackpool, England, and his colleagues reviewed the small number of studies evaluating the alternate approach to “make the cardiology world aware” of alternatives “that can relieve the surgical burden by minimizing the implantation time and length of the operation,” he said.
The comprehensive review is published in the Journal of the American College of Cardiology.
A Neglected Alternative
The sutureless Perceval device is held in place by a stent frame that self-expands. The Intuity device also relies primarily on its framework to anchor the valve in place but does involve three sutures. Both devices are still referred to as sutureless in the new review of them.
Only a small number of centers perform minimally invasive cardiac surgeries, and the main advantage of the devices — rapid deployment — has been eroded with the advent of automated knotting which has significantly reduced the time to implant and sutured valve.
The underuse of these devices is largely caused by the limited amount of comparative and prospective data, said Dr. Spadaccio. “The entire literature on sutureless aortic valve replacement with the exception of one randomized controlled trial is observational.”
That trial, PERSIST-AVR, found that the sutureless valves were just as good as conventional ones when it comes to major adverse cardiovascular events including all-cause death, myocardial infarction, stroke, or valve reintervention at 1 year.
In a subanalysis limited to patients who had isolated aortic valve replacement, the sutureless procedure was associated with lower adverse events (5.2% vs 10.8%) at the cost of a higher rate of pacemaker implantation (11% vs 1.6%).
There are also multiple retrospective studies and registries that have generated observational data comparing sutureless aortic valve replacement with SAVR and TAVR in various patient populations, said Dr. Spadaccio, and the review was based on more than a dozen studies published since 2015. Long-term follow-up data for sutureless aortic valve replacements, which now exceeds 10 years, suggest rates of structural valve deterioration and reintervention have been acceptably low.
The minimally invasive procedures have other advantages too. For example, relative to the greater trauma associated with open heart surgery, minimally invasive surgeries typically involve faster recovery, an advantage likely to appeal to many patients who are candidates for either.
Quicker Recovery
Collectively, these data suggest that sutureless aortic valve replacement might be a reasonable or even a more appropriate alternative to either SAVR or TAVR when considering specific patient characteristics and goals, according to the review, which included an algorithm identifying specifically where sutureless aortic valve replacement fits with SAVR and TAVR.
“The algorithm is based on different clinical scenarios and reflects current guidelines for SAVR,” said Dr. Spadaccio. For example, current guidelines identify SAVR as preferred in patients younger than 65 years and in older patients with a low Society of Thoracic Surgeons (STS) score, but there are many instances in which sutureless aortic valve replacement might be more attractive, such as in those also undergoing mitral valve repair, coronary artery bypass grafting, or another surgical procedure.
Dr. Spadaccio said that the STS score should not be considered in isolation when evaluating a patient for SAVR or TAVR. Other features such as mobility, frailty score, and comorbid liver or renal disease should also be considered when discussing the three options with patients. As a result, the algorithm emphasizes a detailed evaluation of patient characteristics in selecting one procedure over another.
“The treatment should be really tailored on the individual patient basis,” said Dr. Spadaccio.
Dr. Spadaccio acknowledged that there is a need for more comparative trials, particularly in regard to sutureless aortic valve replacement as an alternative to TAVR. “I really think that a 1:1 RCT on sutureless aortic valve replacement vs TAVR could give better answers to all of these interrogatives.”
But despite the limitations outlined in this review, Dr. Spadaccio and colleagues challenged the perception that current data are not sufficient to allow clinicians to consider sutureless aortic valve replacement in the mix of options.
A Viable Option
This comprehensive summary of what is known about sutureless aortic valve replacement compared with the other options addresses an important knowledge gap, said S. Chris Malaisrie, MD, a cardiac surgeon at Northwestern University Feinberg School of Medicine, Chicago, Illinois.
He said he agrees this option has unique qualities. “Minimally invasive surgery has been largely ignored by guideline writers, but patients certainly demand options that are less invasive than standard open heart surgery. Sutureless and rapid deployment valves facilitate minimally invasive surgery and offer an advantageous option for younger patients.”
Dr. Malaisrie said the review is generating discussion about a potentially valuable option within the cardiology community. And that is exactly what Dr. Spadaccio was hoping for. “This paper was meant to educate as much as possible on these details to assist and inform decision-making,” he said.
A version of this article first appeared on Medscape.com.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
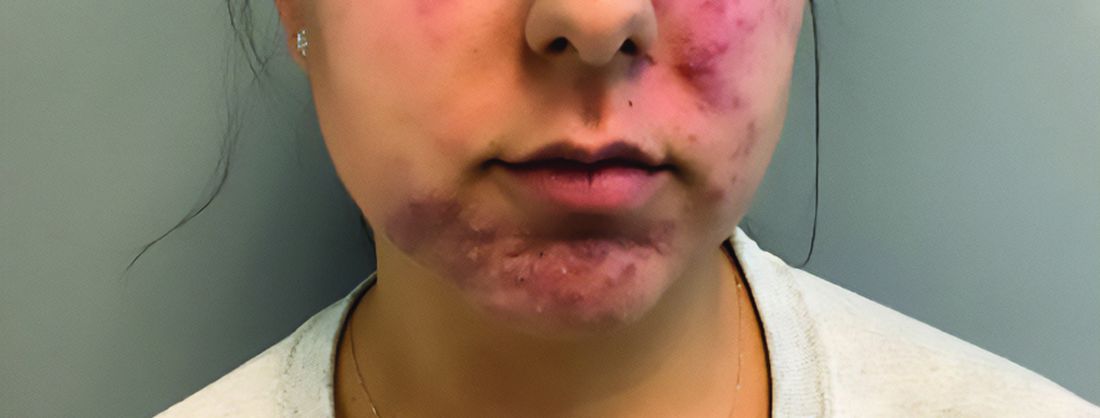
A young adult with a 1-year history of erythema, papules, and pustules on her cheeks and skin
. It typically presents with a sudden onset of papules, pustules, cysts, painful inflammatory nodules, and erythema on the centrofacial areas. The etiology is unknown but has been speculated to be hormone-related as it is more common in women and can be triggered by acute changes such as stress or medications.
Because of overlapping symptoms with other conditions, an accurate clinical assessment is crucial. Typically, there are no comedones and about half of the patients have a history of acne. Some cases have shown a possible link between pyoderma faciale with inflammatory bowel disease, thyroid disease and liver disease, highlighting the importance of considering these associations in treatment decisions.
Treatment options for pyoderma faciale include isotretinoin, corticosteroids, dapsone, and antibiotics such as doxycycline. Isotretinoin is usually the first-line treatment, with dapsone reserved for cases where other methods have failed. Despite concerns about isotretinoin exacerbating inflammatory bowel disease (IBD), there has been at least one reported case where a patient with ulcerative colitis who had pyoderma faciale that was successfully treated with isotretinoin with no adverse effects.

Isotretinoin has been shown to be effective in treating pyoderma faciale by significantly reducing inflammation and scarring. This is imperative because the scarring from pyoderma faciale can be disfiguring and psychologically harmful for patients. Therefore, an early diagnosis and effective treatment method are essential in preventing these scars and improving patients’ confidence and overall dermatological care.

This patient’s initial bacterial culture was negative. She was treated with a course of low dose isotretinoin. Prednisone was initiated two weeks before starting isotretinoin and then was tapered off during the first month of isotretinoin treatment. The patient was also started on spironolactone. The course of isotretinoin was 9 months. She has remained clear and still takes oral contraceptive pills and low dose spironolactone.
This case and the photos were submitted by Ms. Towe, Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Florida, and Donna Bilu Martin, MD, of Premier Dermatology, MD, Aventura, Florida. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Angileri L et al. J Dermatolog Treat. 2021 Feb;32(1):110-3. doi: 10.1080/09546634.2019.1628175.
Coutinho JC et al. An Bras Dermatol. 2016 Sep-Oct;91(5 suppl 1):151-3. doi: 10.1590/abd1806-4841.20164943.
Rosen T and Unkefer RP. Cutis. 1999 Aug;64(2):107-9.
. It typically presents with a sudden onset of papules, pustules, cysts, painful inflammatory nodules, and erythema on the centrofacial areas. The etiology is unknown but has been speculated to be hormone-related as it is more common in women and can be triggered by acute changes such as stress or medications.
Because of overlapping symptoms with other conditions, an accurate clinical assessment is crucial. Typically, there are no comedones and about half of the patients have a history of acne. Some cases have shown a possible link between pyoderma faciale with inflammatory bowel disease, thyroid disease and liver disease, highlighting the importance of considering these associations in treatment decisions.
Treatment options for pyoderma faciale include isotretinoin, corticosteroids, dapsone, and antibiotics such as doxycycline. Isotretinoin is usually the first-line treatment, with dapsone reserved for cases where other methods have failed. Despite concerns about isotretinoin exacerbating inflammatory bowel disease (IBD), there has been at least one reported case where a patient with ulcerative colitis who had pyoderma faciale that was successfully treated with isotretinoin with no adverse effects.
Isotretinoin has been shown to be effective in treating pyoderma faciale by significantly reducing inflammation and scarring. This is imperative because the scarring from pyoderma faciale can be disfiguring and psychologically harmful for patients. Therefore, an early diagnosis and effective treatment method are essential in preventing these scars and improving patients’ confidence and overall dermatological care.
This patient’s initial bacterial culture was negative. She was treated with a course of low dose isotretinoin. Prednisone was initiated two weeks before starting isotretinoin and then was tapered off during the first month of isotretinoin treatment. The patient was also started on spironolactone. The course of isotretinoin was 9 months. She has remained clear and still takes oral contraceptive pills and low dose spironolactone.
This case and the photos were submitted by Ms. Towe, Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Florida, and Donna Bilu Martin, MD, of Premier Dermatology, MD, Aventura, Florida. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Angileri L et al. J Dermatolog Treat. 2021 Feb;32(1):110-3. doi: 10.1080/09546634.2019.1628175.
Coutinho JC et al. An Bras Dermatol. 2016 Sep-Oct;91(5 suppl 1):151-3. doi: 10.1590/abd1806-4841.20164943.
Rosen T and Unkefer RP. Cutis. 1999 Aug;64(2):107-9.
. It typically presents with a sudden onset of papules, pustules, cysts, painful inflammatory nodules, and erythema on the centrofacial areas. The etiology is unknown but has been speculated to be hormone-related as it is more common in women and can be triggered by acute changes such as stress or medications.
Because of overlapping symptoms with other conditions, an accurate clinical assessment is crucial. Typically, there are no comedones and about half of the patients have a history of acne. Some cases have shown a possible link between pyoderma faciale with inflammatory bowel disease, thyroid disease and liver disease, highlighting the importance of considering these associations in treatment decisions.
Treatment options for pyoderma faciale include isotretinoin, corticosteroids, dapsone, and antibiotics such as doxycycline. Isotretinoin is usually the first-line treatment, with dapsone reserved for cases where other methods have failed. Despite concerns about isotretinoin exacerbating inflammatory bowel disease (IBD), there has been at least one reported case where a patient with ulcerative colitis who had pyoderma faciale that was successfully treated with isotretinoin with no adverse effects.
Isotretinoin has been shown to be effective in treating pyoderma faciale by significantly reducing inflammation and scarring. This is imperative because the scarring from pyoderma faciale can be disfiguring and psychologically harmful for patients. Therefore, an early diagnosis and effective treatment method are essential in preventing these scars and improving patients’ confidence and overall dermatological care.
This patient’s initial bacterial culture was negative. She was treated with a course of low dose isotretinoin. Prednisone was initiated two weeks before starting isotretinoin and then was tapered off during the first month of isotretinoin treatment. The patient was also started on spironolactone. The course of isotretinoin was 9 months. She has remained clear and still takes oral contraceptive pills and low dose spironolactone.
This case and the photos were submitted by Ms. Towe, Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Florida, and Donna Bilu Martin, MD, of Premier Dermatology, MD, Aventura, Florida. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
Angileri L et al. J Dermatolog Treat. 2021 Feb;32(1):110-3. doi: 10.1080/09546634.2019.1628175.
Coutinho JC et al. An Bras Dermatol. 2016 Sep-Oct;91(5 suppl 1):151-3. doi: 10.1590/abd1806-4841.20164943.
Rosen T and Unkefer RP. Cutis. 1999 Aug;64(2):107-9.
Will Treating High Blood Pressure Curb Dementia Risk?
High blood pressure is an established risk factor for neurodegeneration and cognitive decline.
Valentin Fuster, MD, president of Mount Sinai Fuster Heart Hospital in New York City, told this news organization. “There is no question in the literature that untreated high blood pressure may lead to dementia,” he said. “The open question is whether treating blood pressure is sufficient to decrease or stop the progress of dementia.”
Studies are mixed, but recent research suggests that addressing hypertension does affect the risk for dementia. A secondary analysis of the China Rural Hypertension Control Project reported at the American Heart Association (AHA) Scientific Sessions in 2023 but not yet published showed that the 4-year blood pressure–lowering program in adults aged 40 or older significantly reduced the risk for all-cause dementia and cognitive impairment.
Similarly, a post hoc analysis of the SPRINT MIND trial found that participants aged 50 or older who underwent intensive (< 120 mm Hg) vs standard (< 140 mm Hg) blood pressure lowering had a lower rate of probable dementia or mild cognitive impairment.
Other studies pointing to a benefit included a pooled individual participant analysis of five randomized controlled trials, which found class I evidence to support antihypertensive treatment to reduce the risk for incident dementia, and an earlier systematic review and meta-analysis of the association of blood pressure lowering with newly diagnosed dementia or cognitive impairment.
How It Might Work
Some possible mechanisms underlying the connection have emerged.
“Vascular disease caused by hypertension is clearly implicated in one form of dementia, called vascular cognitive impairment and dementia,” Andrew Moran, MD, PhD, associate professor of medicine at Columbia University Vagelos College of Physicians and Surgeons in New York City, told this news organization. “This category includes dementia following a stroke caused by uncontrolled hypertension.”
“At the same time, we now know that hypertension and other vascular risk factors can also contribute, along with other factors, to developing Alzheimer dementia,” he said. “Even without causing clinically evident stroke, vascular disease from hypertension can lead to subtle damage to the brain via ischemia, microhemorrhage, and atrophy.”
“It is well known that hypertension affects the vasculature, and the vasculature of the brain is not spared,” agreed Eileen Handberg, PhD, ARNP, a member of the Hypertension Workgroup at the American College of Cardiology (ACC) and a professor of medicine and director of the Cardiovascular Clinical Trials Program in the University of Florida, Gainesville, Florida. “Combine this with other mechanisms like inflammation and endothelial dysfunction, and add amyloid accumulation, and there is a deterioration in vascular beds leading to decreased cerebral blood flow,” she said.
Treating hypertension likely helps lower dementia risk through “a combination of reduced risk of stroke and also benefits on blood flow, blood vessel health, and reduction in neurodegeneration,” suggested Mitchell S.V. Elkind, MD, chief clinical science officer and past president of the AHA and a professor of neurology and epidemiology at Columbia University Irving Medical Center in New York City. “Midlife blood pressure elevations are associated with deposition of amyloid in the brain, so controlling blood pressure may reduce amyloid deposits and neurodegeneration.”
Time in Range or Treat to Target?
With respect to dementia risk, does treating hypertension to a specific target make a difference, or is it the time spent in a healthy blood pressure range?
“Observational studies and a post hoc analysis of the SPRINT MIND trial suggest that more time spent in a healthy blood pressure range or more stable blood pressure are associated with lower dementia risk,” Dr. Moran said. Citing results of the CHRC program and SPRINT MIND trial, he suggested that while a dose-response effect (the lower the blood pressure, the lower the dementia risk) hasn’t been definitively demonstrated, it is likely the case.
In his practice, Dr. Moran follows ACC/AHA guidelines and prescribes antihypertensives to get blood pressure below 130/80 mm Hg in individuals with hypertension who have other high-risk factors (cardiovascular disease, diabetes, chronic kidney disease, or high risk for these conditions). “The treatment rule for people with hypertension without these other risk factors is less clear — lowering blood pressure below 140/90 mm Hg is a must; I will discuss with patients whether to go lower than that.”
“The relative contributions of time in range versus treating to a target for blood pressure require further study,” said Dr. Elkind. “It is likely that the cumulative effect of blood pressure over time has a big role to play — and it does seem clear that midlife blood pressure is even more important than blood pressure late in life.”
That said, he added, “In general and all things being equal, I would treat to a blood pressure of < 120/80 mmHg,” given the SPRINT trial findings of greater benefits when treating to this systolic blood pressure goal. “Of course, if patients have side effects such as lightheadedness or dizziness or other medical conditions that require a higher target, then one would need to adjust the treatment targets.”
According to Dr. Fuster, targets should not be the focus because they vary. For example, the ACC/AHA guidelines use < 130/80 mm Hg, whereas the European Society of Hypertension guidelines and those of the American Academy of Family Physicians specify < 140/90 mm Hg and include age-based criteria. Because there are no studies comparing the outcomes of one set of guidelines vs another, Dr. Fuster thinks the focus should be on starting treatment as early as possible to prevent hypertension leading to dementia.
He pointed to the ongoing PESA trial, which uses brain MRI and other tests to characterize longitudinal associations among cerebral glucose metabolism, subclinical atherosclerosis, and cardiovascular risk factors in asymptomatic individuals aged 40-54. Most did not have hypertension at baseline.
A recently published analysis of a subcohort of 370 PESA participants found that those with persistent high cardiovascular risk and subclinical carotid atherosclerosis already had signs of brain metabolic decline, “suggesting that maintenance of cardiovascular health during midlife could contribute to reductions in neurodegenerative disease burden later in life,” wrote the investigators.
Is It Ever Too Late?
If starting hypertension treatment in midlife can help reduce the risk for cognitive impairment later, can treating later in life also help? “It’s theoretically possible, but it has to be proven,” Dr. Fuster said. “There are no data on whether there’s less chance to prevent the development of dementia if you start treating hypertension at age 70, for example. And we have no idea whether hypertension treatment will prevent progression in those who already have dementia.”
“Treating high blood pressure in older adults could affect the course of further progressive cognitive decline by improving vascular health and preventing strokes, which likely exacerbate nonvascular dementia,” Dr. Elkind suggested. “Most people with dementia have a combination of vascular and nonvascular dementia, so treating reversible causes wherever possible makes a difference.”
Dr. Elkind treats older patients with this in mind, he said, “even though most of the evidence points to the fact that it is blood pressure in middle age, not older age, that seems to have the biggest impact on later-life cognitive decline and dementia.” Like Dr. Fuster, he said, “the best strategy is to identify and treat blood pressure in midlife, before damage to the brain has advanced.”
Dr. Moran noted, “The latest science on dementia causes suggests it is difficult to draw a border between vascular and nonvascular dementia. So, as a practical matter, healthcare providers should consider that hypertension treatment is one of the best ways to prevent any category of dementia. This dementia prevention is added to the well-known benefits of hypertension treatment to prevent heart attacks, strokes, and kidney disease: ‘Healthy heart, healthy brain.’ ”
“Our BP [blood pressure] control rates overall are still abysmal,” Dr. Handberg added. Currently around one in four US adults with hypertension have it under control. Studies have shown that blood pressure control rates of 70%-80% are achievable, she said. “We can’t let patient or provider inertia continue.”
Dr. Handberg, Dr. Elkind, Dr. Moran, and Dr. Fuster declared no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
High blood pressure is an established risk factor for neurodegeneration and cognitive decline.
Valentin Fuster, MD, president of Mount Sinai Fuster Heart Hospital in New York City, told this news organization. “There is no question in the literature that untreated high blood pressure may lead to dementia,” he said. “The open question is whether treating blood pressure is sufficient to decrease or stop the progress of dementia.”
Studies are mixed, but recent research suggests that addressing hypertension does affect the risk for dementia. A secondary analysis of the China Rural Hypertension Control Project reported at the American Heart Association (AHA) Scientific Sessions in 2023 but not yet published showed that the 4-year blood pressure–lowering program in adults aged 40 or older significantly reduced the risk for all-cause dementia and cognitive impairment.
Similarly, a post hoc analysis of the SPRINT MIND trial found that participants aged 50 or older who underwent intensive (< 120 mm Hg) vs standard (< 140 mm Hg) blood pressure lowering had a lower rate of probable dementia or mild cognitive impairment.
Other studies pointing to a benefit included a pooled individual participant analysis of five randomized controlled trials, which found class I evidence to support antihypertensive treatment to reduce the risk for incident dementia, and an earlier systematic review and meta-analysis of the association of blood pressure lowering with newly diagnosed dementia or cognitive impairment.
How It Might Work
Some possible mechanisms underlying the connection have emerged.
“Vascular disease caused by hypertension is clearly implicated in one form of dementia, called vascular cognitive impairment and dementia,” Andrew Moran, MD, PhD, associate professor of medicine at Columbia University Vagelos College of Physicians and Surgeons in New York City, told this news organization. “This category includes dementia following a stroke caused by uncontrolled hypertension.”
“At the same time, we now know that hypertension and other vascular risk factors can also contribute, along with other factors, to developing Alzheimer dementia,” he said. “Even without causing clinically evident stroke, vascular disease from hypertension can lead to subtle damage to the brain via ischemia, microhemorrhage, and atrophy.”
“It is well known that hypertension affects the vasculature, and the vasculature of the brain is not spared,” agreed Eileen Handberg, PhD, ARNP, a member of the Hypertension Workgroup at the American College of Cardiology (ACC) and a professor of medicine and director of the Cardiovascular Clinical Trials Program in the University of Florida, Gainesville, Florida. “Combine this with other mechanisms like inflammation and endothelial dysfunction, and add amyloid accumulation, and there is a deterioration in vascular beds leading to decreased cerebral blood flow,” she said.
Treating hypertension likely helps lower dementia risk through “a combination of reduced risk of stroke and also benefits on blood flow, blood vessel health, and reduction in neurodegeneration,” suggested Mitchell S.V. Elkind, MD, chief clinical science officer and past president of the AHA and a professor of neurology and epidemiology at Columbia University Irving Medical Center in New York City. “Midlife blood pressure elevations are associated with deposition of amyloid in the brain, so controlling blood pressure may reduce amyloid deposits and neurodegeneration.”
Time in Range or Treat to Target?
With respect to dementia risk, does treating hypertension to a specific target make a difference, or is it the time spent in a healthy blood pressure range?
“Observational studies and a post hoc analysis of the SPRINT MIND trial suggest that more time spent in a healthy blood pressure range or more stable blood pressure are associated with lower dementia risk,” Dr. Moran said. Citing results of the CHRC program and SPRINT MIND trial, he suggested that while a dose-response effect (the lower the blood pressure, the lower the dementia risk) hasn’t been definitively demonstrated, it is likely the case.
In his practice, Dr. Moran follows ACC/AHA guidelines and prescribes antihypertensives to get blood pressure below 130/80 mm Hg in individuals with hypertension who have other high-risk factors (cardiovascular disease, diabetes, chronic kidney disease, or high risk for these conditions). “The treatment rule for people with hypertension without these other risk factors is less clear — lowering blood pressure below 140/90 mm Hg is a must; I will discuss with patients whether to go lower than that.”
“The relative contributions of time in range versus treating to a target for blood pressure require further study,” said Dr. Elkind. “It is likely that the cumulative effect of blood pressure over time has a big role to play — and it does seem clear that midlife blood pressure is even more important than blood pressure late in life.”
That said, he added, “In general and all things being equal, I would treat to a blood pressure of < 120/80 mmHg,” given the SPRINT trial findings of greater benefits when treating to this systolic blood pressure goal. “Of course, if patients have side effects such as lightheadedness or dizziness or other medical conditions that require a higher target, then one would need to adjust the treatment targets.”
According to Dr. Fuster, targets should not be the focus because they vary. For example, the ACC/AHA guidelines use < 130/80 mm Hg, whereas the European Society of Hypertension guidelines and those of the American Academy of Family Physicians specify < 140/90 mm Hg and include age-based criteria. Because there are no studies comparing the outcomes of one set of guidelines vs another, Dr. Fuster thinks the focus should be on starting treatment as early as possible to prevent hypertension leading to dementia.
He pointed to the ongoing PESA trial, which uses brain MRI and other tests to characterize longitudinal associations among cerebral glucose metabolism, subclinical atherosclerosis, and cardiovascular risk factors in asymptomatic individuals aged 40-54. Most did not have hypertension at baseline.
A recently published analysis of a subcohort of 370 PESA participants found that those with persistent high cardiovascular risk and subclinical carotid atherosclerosis already had signs of brain metabolic decline, “suggesting that maintenance of cardiovascular health during midlife could contribute to reductions in neurodegenerative disease burden later in life,” wrote the investigators.
Is It Ever Too Late?
If starting hypertension treatment in midlife can help reduce the risk for cognitive impairment later, can treating later in life also help? “It’s theoretically possible, but it has to be proven,” Dr. Fuster said. “There are no data on whether there’s less chance to prevent the development of dementia if you start treating hypertension at age 70, for example. And we have no idea whether hypertension treatment will prevent progression in those who already have dementia.”
“Treating high blood pressure in older adults could affect the course of further progressive cognitive decline by improving vascular health and preventing strokes, which likely exacerbate nonvascular dementia,” Dr. Elkind suggested. “Most people with dementia have a combination of vascular and nonvascular dementia, so treating reversible causes wherever possible makes a difference.”
Dr. Elkind treats older patients with this in mind, he said, “even though most of the evidence points to the fact that it is blood pressure in middle age, not older age, that seems to have the biggest impact on later-life cognitive decline and dementia.” Like Dr. Fuster, he said, “the best strategy is to identify and treat blood pressure in midlife, before damage to the brain has advanced.”
Dr. Moran noted, “The latest science on dementia causes suggests it is difficult to draw a border between vascular and nonvascular dementia. So, as a practical matter, healthcare providers should consider that hypertension treatment is one of the best ways to prevent any category of dementia. This dementia prevention is added to the well-known benefits of hypertension treatment to prevent heart attacks, strokes, and kidney disease: ‘Healthy heart, healthy brain.’ ”
“Our BP [blood pressure] control rates overall are still abysmal,” Dr. Handberg added. Currently around one in four US adults with hypertension have it under control. Studies have shown that blood pressure control rates of 70%-80% are achievable, she said. “We can’t let patient or provider inertia continue.”
Dr. Handberg, Dr. Elkind, Dr. Moran, and Dr. Fuster declared no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
High blood pressure is an established risk factor for neurodegeneration and cognitive decline.
Valentin Fuster, MD, president of Mount Sinai Fuster Heart Hospital in New York City, told this news organization. “There is no question in the literature that untreated high blood pressure may lead to dementia,” he said. “The open question is whether treating blood pressure is sufficient to decrease or stop the progress of dementia.”
Studies are mixed, but recent research suggests that addressing hypertension does affect the risk for dementia. A secondary analysis of the China Rural Hypertension Control Project reported at the American Heart Association (AHA) Scientific Sessions in 2023 but not yet published showed that the 4-year blood pressure–lowering program in adults aged 40 or older significantly reduced the risk for all-cause dementia and cognitive impairment.
Similarly, a post hoc analysis of the SPRINT MIND trial found that participants aged 50 or older who underwent intensive (< 120 mm Hg) vs standard (< 140 mm Hg) blood pressure lowering had a lower rate of probable dementia or mild cognitive impairment.
Other studies pointing to a benefit included a pooled individual participant analysis of five randomized controlled trials, which found class I evidence to support antihypertensive treatment to reduce the risk for incident dementia, and an earlier systematic review and meta-analysis of the association of blood pressure lowering with newly diagnosed dementia or cognitive impairment.
How It Might Work
Some possible mechanisms underlying the connection have emerged.
“Vascular disease caused by hypertension is clearly implicated in one form of dementia, called vascular cognitive impairment and dementia,” Andrew Moran, MD, PhD, associate professor of medicine at Columbia University Vagelos College of Physicians and Surgeons in New York City, told this news organization. “This category includes dementia following a stroke caused by uncontrolled hypertension.”
“At the same time, we now know that hypertension and other vascular risk factors can also contribute, along with other factors, to developing Alzheimer dementia,” he said. “Even without causing clinically evident stroke, vascular disease from hypertension can lead to subtle damage to the brain via ischemia, microhemorrhage, and atrophy.”
“It is well known that hypertension affects the vasculature, and the vasculature of the brain is not spared,” agreed Eileen Handberg, PhD, ARNP, a member of the Hypertension Workgroup at the American College of Cardiology (ACC) and a professor of medicine and director of the Cardiovascular Clinical Trials Program in the University of Florida, Gainesville, Florida. “Combine this with other mechanisms like inflammation and endothelial dysfunction, and add amyloid accumulation, and there is a deterioration in vascular beds leading to decreased cerebral blood flow,” she said.
Treating hypertension likely helps lower dementia risk through “a combination of reduced risk of stroke and also benefits on blood flow, blood vessel health, and reduction in neurodegeneration,” suggested Mitchell S.V. Elkind, MD, chief clinical science officer and past president of the AHA and a professor of neurology and epidemiology at Columbia University Irving Medical Center in New York City. “Midlife blood pressure elevations are associated with deposition of amyloid in the brain, so controlling blood pressure may reduce amyloid deposits and neurodegeneration.”
Time in Range or Treat to Target?
With respect to dementia risk, does treating hypertension to a specific target make a difference, or is it the time spent in a healthy blood pressure range?
“Observational studies and a post hoc analysis of the SPRINT MIND trial suggest that more time spent in a healthy blood pressure range or more stable blood pressure are associated with lower dementia risk,” Dr. Moran said. Citing results of the CHRC program and SPRINT MIND trial, he suggested that while a dose-response effect (the lower the blood pressure, the lower the dementia risk) hasn’t been definitively demonstrated, it is likely the case.
In his practice, Dr. Moran follows ACC/AHA guidelines and prescribes antihypertensives to get blood pressure below 130/80 mm Hg in individuals with hypertension who have other high-risk factors (cardiovascular disease, diabetes, chronic kidney disease, or high risk for these conditions). “The treatment rule for people with hypertension without these other risk factors is less clear — lowering blood pressure below 140/90 mm Hg is a must; I will discuss with patients whether to go lower than that.”
“The relative contributions of time in range versus treating to a target for blood pressure require further study,” said Dr. Elkind. “It is likely that the cumulative effect of blood pressure over time has a big role to play — and it does seem clear that midlife blood pressure is even more important than blood pressure late in life.”
That said, he added, “In general and all things being equal, I would treat to a blood pressure of < 120/80 mmHg,” given the SPRINT trial findings of greater benefits when treating to this systolic blood pressure goal. “Of course, if patients have side effects such as lightheadedness or dizziness or other medical conditions that require a higher target, then one would need to adjust the treatment targets.”
According to Dr. Fuster, targets should not be the focus because they vary. For example, the ACC/AHA guidelines use < 130/80 mm Hg, whereas the European Society of Hypertension guidelines and those of the American Academy of Family Physicians specify < 140/90 mm Hg and include age-based criteria. Because there are no studies comparing the outcomes of one set of guidelines vs another, Dr. Fuster thinks the focus should be on starting treatment as early as possible to prevent hypertension leading to dementia.
He pointed to the ongoing PESA trial, which uses brain MRI and other tests to characterize longitudinal associations among cerebral glucose metabolism, subclinical atherosclerosis, and cardiovascular risk factors in asymptomatic individuals aged 40-54. Most did not have hypertension at baseline.
A recently published analysis of a subcohort of 370 PESA participants found that those with persistent high cardiovascular risk and subclinical carotid atherosclerosis already had signs of brain metabolic decline, “suggesting that maintenance of cardiovascular health during midlife could contribute to reductions in neurodegenerative disease burden later in life,” wrote the investigators.
Is It Ever Too Late?
If starting hypertension treatment in midlife can help reduce the risk for cognitive impairment later, can treating later in life also help? “It’s theoretically possible, but it has to be proven,” Dr. Fuster said. “There are no data on whether there’s less chance to prevent the development of dementia if you start treating hypertension at age 70, for example. And we have no idea whether hypertension treatment will prevent progression in those who already have dementia.”
“Treating high blood pressure in older adults could affect the course of further progressive cognitive decline by improving vascular health and preventing strokes, which likely exacerbate nonvascular dementia,” Dr. Elkind suggested. “Most people with dementia have a combination of vascular and nonvascular dementia, so treating reversible causes wherever possible makes a difference.”
Dr. Elkind treats older patients with this in mind, he said, “even though most of the evidence points to the fact that it is blood pressure in middle age, not older age, that seems to have the biggest impact on later-life cognitive decline and dementia.” Like Dr. Fuster, he said, “the best strategy is to identify and treat blood pressure in midlife, before damage to the brain has advanced.”
Dr. Moran noted, “The latest science on dementia causes suggests it is difficult to draw a border between vascular and nonvascular dementia. So, as a practical matter, healthcare providers should consider that hypertension treatment is one of the best ways to prevent any category of dementia. This dementia prevention is added to the well-known benefits of hypertension treatment to prevent heart attacks, strokes, and kidney disease: ‘Healthy heart, healthy brain.’ ”
“Our BP [blood pressure] control rates overall are still abysmal,” Dr. Handberg added. Currently around one in four US adults with hypertension have it under control. Studies have shown that blood pressure control rates of 70%-80% are achievable, she said. “We can’t let patient or provider inertia continue.”
Dr. Handberg, Dr. Elkind, Dr. Moran, and Dr. Fuster declared no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
Emergency Contraception Recommended for Teens on Isotretinoin
TORONTO —
That was one of the main messages from Andrea L. Zaenglein, MD, professor of dermatology and pediatrics, Penn State University, Hershey, who discussed hormonal therapies for pediatric acne at the annual meeting of the Society for Pediatric Dermatology.

Many doctors are reluctant to prescribe EC, which refers to contraceptive methods used to prevent unintended pregnancy after unprotected sexual intercourse or contraceptive failure, whether that’s from discomfort with EC or lack of training, Dr. Zaenglein said in an interview.
Isotretinoin, a retinoid marketed as Accutane and other brand names, is an effective treatment for acne but carries serious teratogenicity risks; the iPLEDGE Risk Evaluation and Mitigation Strategy is designed to manage this risk and minimize fetal exposure. Yet from 2011 to 2017, 210-310 pregnancies per year were reported to the Food and Drug Administration, according to a 2019 study.
There is a knowledge gap regarding EC among dermatologists who prescribe isotretinoin, which “is perpetuated by the iPLEDGE program because it is inadequate in guiding clinicians or educating patients about the use of EC,” Dr. Zaenglein and colleagues wrote in a recently published viewpoint on EC prescribing in patients on isotretinoin.
Types of EC include oral levonorgestrel (plan B), available over the counter; oral ulipristal acetate (ella), which requires a prescription; and the copper/hormonal intrauterine device.
Not all teens taking isotretinoin can be trusted to be sexually abstinent. Dr. Zaenglein cited research showing 39% of female high school students have had sexual relations. “In my opinion, these patients should have emergency contraception prescribed to them as a backup,” she said.
Dr. Zaenglein believes there’s a fair amount of “misunderstanding” about EC, with many people thinking it’s an abortion pill. “It’s a totally different medicine. This is contraception; if you’re pregnant, it’s not going to affect your fetus.”
Outgoing SPD President Sheilagh Maguiness, MD, professor of dermatology and pediatrics, University of Minnesota, Minneapolis, agreed that Dr. Zaenglein raised an important issue. “She has identified a practice gap and a knowledge gap that we need to address,” she said in an interview.
When discussing contraception with female patients taking isotretinoin, assume they’re sexually active or could be, Dr. Zaenglein told meeting attendees. Be explicit about the risks to the fetus and consider their past compliance.
Complex Disorder
During her presentation, Dr. Zaenglein described acne as a “very complex, multifactorial inflammatory disorder” of the skin. It involves four steps: Increased sebum production, hyperkeratinization, Cutibacterium acnes, and inflammation. External factors such as diet, genes, and the environment play a role.
“But at the heart of all of it is androgens; if you didn’t have androgens, you wouldn’t have acne.” That’s why some acne treatments block androgen receptors.
Clinicians are increasingly using one such therapy, spironolactone, to treat acne in female adolescents. Dr. Zaenglein referred to a Mayo Clinic study of 80 patients (mean age, 19 years), who had moderate to severe acne treated with a mean dose of 100 mg/day, that found 80% had improvement with a favorable side effect profile. This included nearly 23% who had a complete response (90% or more) and 36% who had a partial response (more than 50%); 20% had no response.
However, response rates are higher in adults, said Dr. Zaenglein, noting that spironolactone works “much better” in adult women.
Side effects of spironolactone can include menstrual disturbances, breast enlargement and tenderness, and premenstrual syndrome–like symptoms.
Dermatologists should also consider combined oral contraceptives (COCs) in their adolescent patients with acne. These have an estrogen component as well as a progestin component.
They have proven effectiveness for acne in adolescents, yet a US survey of 170 dermatology residents found only 60% felt comfortable prescribing them to healthy adolescents. The survey also found only 62% of respondents felt adequately trained on the efficacy of COCs, and 42% felt adequately trained on their safety.
Contraindications for COCs include thrombosis, migraine with aura, lupus, seizures, and hypertension. Complex valvular heart disease and liver tumors also need to be ruled out, said Dr. Zaenglein. One of the “newer concerns” with COCs is depression. “There’s biological plausibility because, obviously, hormones impact the brain.”
Preventing Drug Interactions
Before prescribing hormonal therapy, clinicians should carry out an acne assessment, aimed in part at preventing drug interactions. “The one we mostly have to watch out for is rifampin,” an antibiotic that could interact with COCs, said Dr. Zaenglein.
The herbal supplement St John’s Wort can reduce the efficacy of COCs. “You also want to make sure that they’re not on any medicines that will increase potassium, such as ACE inhibitors,” said Dr. Zaenglein. But tetracyclines, ampicillin, or metronidazole are usually “all okay” when combined with COCs.
It’s important to get baseline blood pressure levels and to check these along with weight on a regular basis, she added.
Always Consider PCOS
Before starting hormonal therapy, she advises dermatologists to “always consider” polycystic ovary syndrome (PCOS), a condition that’s “probably much underdiagnosed.” Acne is common in adolescents with PCOS. She suggests using a PCOS checklist, a reminder to ask about irregular periods, hirsutism, signs of insulin resistance such as increased body mass index, a history of premature adrenarche, and a family history of PCOS, said Dr. Zaenglein, noting that a person with a sibling who has PCOS has about a 40% chance of developing the condition.
“We play an important role in getting kids diagnosed at an early age so that we can make interventions because the impact of the metabolic syndrome can have lifelong effects on their cardiovascular system, as well as infertility.”
Dr. Zaenglein is a member of the American Academy of Dermatology (AAD) Acne Guidelines work group, the immediate past president of the American Acne and Rosacea Society, a member of the AAD iPLEDGE work group, co–editor in chief of Pediatric Dermatology, an advisory board member of Ortho Dermatologics, and a consultant for Church & Dwight. Dr. Maguiness had no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
TORONTO —
That was one of the main messages from Andrea L. Zaenglein, MD, professor of dermatology and pediatrics, Penn State University, Hershey, who discussed hormonal therapies for pediatric acne at the annual meeting of the Society for Pediatric Dermatology.
Many doctors are reluctant to prescribe EC, which refers to contraceptive methods used to prevent unintended pregnancy after unprotected sexual intercourse or contraceptive failure, whether that’s from discomfort with EC or lack of training, Dr. Zaenglein said in an interview.
Isotretinoin, a retinoid marketed as Accutane and other brand names, is an effective treatment for acne but carries serious teratogenicity risks; the iPLEDGE Risk Evaluation and Mitigation Strategy is designed to manage this risk and minimize fetal exposure. Yet from 2011 to 2017, 210-310 pregnancies per year were reported to the Food and Drug Administration, according to a 2019 study.
There is a knowledge gap regarding EC among dermatologists who prescribe isotretinoin, which “is perpetuated by the iPLEDGE program because it is inadequate in guiding clinicians or educating patients about the use of EC,” Dr. Zaenglein and colleagues wrote in a recently published viewpoint on EC prescribing in patients on isotretinoin.
Types of EC include oral levonorgestrel (plan B), available over the counter; oral ulipristal acetate (ella), which requires a prescription; and the copper/hormonal intrauterine device.
Not all teens taking isotretinoin can be trusted to be sexually abstinent. Dr. Zaenglein cited research showing 39% of female high school students have had sexual relations. “In my opinion, these patients should have emergency contraception prescribed to them as a backup,” she said.
Dr. Zaenglein believes there’s a fair amount of “misunderstanding” about EC, with many people thinking it’s an abortion pill. “It’s a totally different medicine. This is contraception; if you’re pregnant, it’s not going to affect your fetus.”
Outgoing SPD President Sheilagh Maguiness, MD, professor of dermatology and pediatrics, University of Minnesota, Minneapolis, agreed that Dr. Zaenglein raised an important issue. “She has identified a practice gap and a knowledge gap that we need to address,” she said in an interview.
When discussing contraception with female patients taking isotretinoin, assume they’re sexually active or could be, Dr. Zaenglein told meeting attendees. Be explicit about the risks to the fetus and consider their past compliance.
Complex Disorder
During her presentation, Dr. Zaenglein described acne as a “very complex, multifactorial inflammatory disorder” of the skin. It involves four steps: Increased sebum production, hyperkeratinization, Cutibacterium acnes, and inflammation. External factors such as diet, genes, and the environment play a role.
“But at the heart of all of it is androgens; if you didn’t have androgens, you wouldn’t have acne.” That’s why some acne treatments block androgen receptors.
Clinicians are increasingly using one such therapy, spironolactone, to treat acne in female adolescents. Dr. Zaenglein referred to a Mayo Clinic study of 80 patients (mean age, 19 years), who had moderate to severe acne treated with a mean dose of 100 mg/day, that found 80% had improvement with a favorable side effect profile. This included nearly 23% who had a complete response (90% or more) and 36% who had a partial response (more than 50%); 20% had no response.
However, response rates are higher in adults, said Dr. Zaenglein, noting that spironolactone works “much better” in adult women.
Side effects of spironolactone can include menstrual disturbances, breast enlargement and tenderness, and premenstrual syndrome–like symptoms.
Dermatologists should also consider combined oral contraceptives (COCs) in their adolescent patients with acne. These have an estrogen component as well as a progestin component.
They have proven effectiveness for acne in adolescents, yet a US survey of 170 dermatology residents found only 60% felt comfortable prescribing them to healthy adolescents. The survey also found only 62% of respondents felt adequately trained on the efficacy of COCs, and 42% felt adequately trained on their safety.
Contraindications for COCs include thrombosis, migraine with aura, lupus, seizures, and hypertension. Complex valvular heart disease and liver tumors also need to be ruled out, said Dr. Zaenglein. One of the “newer concerns” with COCs is depression. “There’s biological plausibility because, obviously, hormones impact the brain.”
Preventing Drug Interactions
Before prescribing hormonal therapy, clinicians should carry out an acne assessment, aimed in part at preventing drug interactions. “The one we mostly have to watch out for is rifampin,” an antibiotic that could interact with COCs, said Dr. Zaenglein.
The herbal supplement St John’s Wort can reduce the efficacy of COCs. “You also want to make sure that they’re not on any medicines that will increase potassium, such as ACE inhibitors,” said Dr. Zaenglein. But tetracyclines, ampicillin, or metronidazole are usually “all okay” when combined with COCs.
It’s important to get baseline blood pressure levels and to check these along with weight on a regular basis, she added.
Always Consider PCOS
Before starting hormonal therapy, she advises dermatologists to “always consider” polycystic ovary syndrome (PCOS), a condition that’s “probably much underdiagnosed.” Acne is common in adolescents with PCOS. She suggests using a PCOS checklist, a reminder to ask about irregular periods, hirsutism, signs of insulin resistance such as increased body mass index, a history of premature adrenarche, and a family history of PCOS, said Dr. Zaenglein, noting that a person with a sibling who has PCOS has about a 40% chance of developing the condition.
“We play an important role in getting kids diagnosed at an early age so that we can make interventions because the impact of the metabolic syndrome can have lifelong effects on their cardiovascular system, as well as infertility.”
Dr. Zaenglein is a member of the American Academy of Dermatology (AAD) Acne Guidelines work group, the immediate past president of the American Acne and Rosacea Society, a member of the AAD iPLEDGE work group, co–editor in chief of Pediatric Dermatology, an advisory board member of Ortho Dermatologics, and a consultant for Church & Dwight. Dr. Maguiness had no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
TORONTO —
That was one of the main messages from Andrea L. Zaenglein, MD, professor of dermatology and pediatrics, Penn State University, Hershey, who discussed hormonal therapies for pediatric acne at the annual meeting of the Society for Pediatric Dermatology.
Many doctors are reluctant to prescribe EC, which refers to contraceptive methods used to prevent unintended pregnancy after unprotected sexual intercourse or contraceptive failure, whether that’s from discomfort with EC or lack of training, Dr. Zaenglein said in an interview.
Isotretinoin, a retinoid marketed as Accutane and other brand names, is an effective treatment for acne but carries serious teratogenicity risks; the iPLEDGE Risk Evaluation and Mitigation Strategy is designed to manage this risk and minimize fetal exposure. Yet from 2011 to 2017, 210-310 pregnancies per year were reported to the Food and Drug Administration, according to a 2019 study.
There is a knowledge gap regarding EC among dermatologists who prescribe isotretinoin, which “is perpetuated by the iPLEDGE program because it is inadequate in guiding clinicians or educating patients about the use of EC,” Dr. Zaenglein and colleagues wrote in a recently published viewpoint on EC prescribing in patients on isotretinoin.
Types of EC include oral levonorgestrel (plan B), available over the counter; oral ulipristal acetate (ella), which requires a prescription; and the copper/hormonal intrauterine device.
Not all teens taking isotretinoin can be trusted to be sexually abstinent. Dr. Zaenglein cited research showing 39% of female high school students have had sexual relations. “In my opinion, these patients should have emergency contraception prescribed to them as a backup,” she said.
Dr. Zaenglein believes there’s a fair amount of “misunderstanding” about EC, with many people thinking it’s an abortion pill. “It’s a totally different medicine. This is contraception; if you’re pregnant, it’s not going to affect your fetus.”
Outgoing SPD President Sheilagh Maguiness, MD, professor of dermatology and pediatrics, University of Minnesota, Minneapolis, agreed that Dr. Zaenglein raised an important issue. “She has identified a practice gap and a knowledge gap that we need to address,” she said in an interview.
When discussing contraception with female patients taking isotretinoin, assume they’re sexually active or could be, Dr. Zaenglein told meeting attendees. Be explicit about the risks to the fetus and consider their past compliance.
Complex Disorder
During her presentation, Dr. Zaenglein described acne as a “very complex, multifactorial inflammatory disorder” of the skin. It involves four steps: Increased sebum production, hyperkeratinization, Cutibacterium acnes, and inflammation. External factors such as diet, genes, and the environment play a role.
“But at the heart of all of it is androgens; if you didn’t have androgens, you wouldn’t have acne.” That’s why some acne treatments block androgen receptors.
Clinicians are increasingly using one such therapy, spironolactone, to treat acne in female adolescents. Dr. Zaenglein referred to a Mayo Clinic study of 80 patients (mean age, 19 years), who had moderate to severe acne treated with a mean dose of 100 mg/day, that found 80% had improvement with a favorable side effect profile. This included nearly 23% who had a complete response (90% or more) and 36% who had a partial response (more than 50%); 20% had no response.
However, response rates are higher in adults, said Dr. Zaenglein, noting that spironolactone works “much better” in adult women.
Side effects of spironolactone can include menstrual disturbances, breast enlargement and tenderness, and premenstrual syndrome–like symptoms.
Dermatologists should also consider combined oral contraceptives (COCs) in their adolescent patients with acne. These have an estrogen component as well as a progestin component.
They have proven effectiveness for acne in adolescents, yet a US survey of 170 dermatology residents found only 60% felt comfortable prescribing them to healthy adolescents. The survey also found only 62% of respondents felt adequately trained on the efficacy of COCs, and 42% felt adequately trained on their safety.
Contraindications for COCs include thrombosis, migraine with aura, lupus, seizures, and hypertension. Complex valvular heart disease and liver tumors also need to be ruled out, said Dr. Zaenglein. One of the “newer concerns” with COCs is depression. “There’s biological plausibility because, obviously, hormones impact the brain.”
Preventing Drug Interactions
Before prescribing hormonal therapy, clinicians should carry out an acne assessment, aimed in part at preventing drug interactions. “The one we mostly have to watch out for is rifampin,” an antibiotic that could interact with COCs, said Dr. Zaenglein.
The herbal supplement St John’s Wort can reduce the efficacy of COCs. “You also want to make sure that they’re not on any medicines that will increase potassium, such as ACE inhibitors,” said Dr. Zaenglein. But tetracyclines, ampicillin, or metronidazole are usually “all okay” when combined with COCs.
It’s important to get baseline blood pressure levels and to check these along with weight on a regular basis, she added.
Always Consider PCOS
Before starting hormonal therapy, she advises dermatologists to “always consider” polycystic ovary syndrome (PCOS), a condition that’s “probably much underdiagnosed.” Acne is common in adolescents with PCOS. She suggests using a PCOS checklist, a reminder to ask about irregular periods, hirsutism, signs of insulin resistance such as increased body mass index, a history of premature adrenarche, and a family history of PCOS, said Dr. Zaenglein, noting that a person with a sibling who has PCOS has about a 40% chance of developing the condition.
“We play an important role in getting kids diagnosed at an early age so that we can make interventions because the impact of the metabolic syndrome can have lifelong effects on their cardiovascular system, as well as infertility.”
Dr. Zaenglein is a member of the American Academy of Dermatology (AAD) Acne Guidelines work group, the immediate past president of the American Acne and Rosacea Society, a member of the AAD iPLEDGE work group, co–editor in chief of Pediatric Dermatology, an advisory board member of Ortho Dermatologics, and a consultant for Church & Dwight. Dr. Maguiness had no relevant conflicts of interest.
A version of this article first appeared on Medscape.com.
FROM SPD 2024
Will Artificial Intelligence Replace Some Primary Care?
Within the next few years, patients will go to their primary care facility for a medical problem. They’ll be greeted by a nonhuman who speaks in the language of their choice. Based upon the initial interview, which will be taken in note form, the patient will be diagnosed, and a prescription called into the pharmacy. They’ll pay the robot at a reception kiosk, and their meds will be delivered via driverless car.
Or so suggests Allan Stewart, MD, medical director and chief of cardiothoracic surgery at HCA Florida Mercy Hospital in Miami.
The writing is on the wall. , he said.
If that sounds far too futuristic, buckle up. AI is already here and being used by most medical specialties. However, it’s primary care that stands to gain the most from this technology — right now — thanks to its ability to radically streamline patient care.
Seeing the Doctor and His or Her AI Assistant
AI is making doctors’ work lives easier, whether the technology helps with risk prevention and intervention or closing care gaps. It can also triage patient complaints, monitor patients remotely, or even perform digital health coaching to keep patients on track with their lifestyle regimens or monitor their health conditions.
Each of these AI components enables primary care physicians to reduce some of the paperwork requirements of their jobs and do what they were trained to do — listen and assess patients. Doctors currently spend 12 hours on average each week submitting prior authorization requests, according to an American Medical Association survey.
“Primary care can be overwhelming, especially today, with the advent of electronic records and data,” said Davin Lundquist, MD, a family medicine physician and chief medical officer at Augmedix, an automated medical documentation company that provides tools to reduce clinician burnout. “The amount of data we have to go through to try to get a complete and clear picture of our patients can be overwhelming on top of the referrals, administrative burdens, and regulatory requirements, which seem to be focused on the primary care space,” Dr. Lundquist said.
With an AI assist, primary care physicians can reduce their prep and pre-charting time, lessen the time needed for paperwork outside of clinic hours, and streamline information, including access to lab results, radiology reports, and consults.
“AI is already helping doctors manage their practices, make differential diagnoses, and input progress notes or histories,” said Dr. Stewart.
In Seattle, Ford Parsons, MD, chief of operational analytics at Providence Hospitals in Seattle, has been leading a generative AI project that recently developed a tool called Provaria to prioritize incoming messages from patients. The tool ensures that those with more urgent needs get immediate attention, and it supports the personnel who lead the responses.
The process begins with Provaria reviewing patient messages to ensure those with more urgent needs, such as a mental health crisis, get immediate attention instead of answering messages in the order they were received.
Provaria also provides resources to help responding staff craft a reply. If a patient’s message cites back pain, for example, the system might suggest a referral to a physical therapist, include a link to that department, and prompt the staff to ask about red flags that indicate a more urgent situation.
After an initial rollout, Providence recently deployed Provaria to manage the messages for all 4000 of its primary care, family medicine, and internal medicine providers. The system has reviewed and categorized more than 500,000 messages so far.
“This is another example where AI can increase the human connection in healthcare,” Dr. Parsons said. “That’s the opposite of what others are saying, but by using AI, you can automate the stuff that isn’t critical that doctors have wound up doing.”
AI Helps Foster Better Person-to-Person Communication
In recent years, the first thing most doctors do when they enter the exam room with a patient is log into the in-room computer and start to take notes — which can be off-putting to patients.
Now devices can ease this process, such as PLAUD, an AI voice recognition device that attaches to a cell phone. Just the size of a credit card, the device enables conversations to be easily recorded. It not only streamlines note-taking but also enables a physician to listen intently to a patient’s concerns instead of furiously jotting down notes.
“That device is already helping transcribe conversations into notes and then into a patient’s electronic medical record,” Dr. Stewart said. “This helps save doctors the work of having to input patient information.”
AI Can’t Be a Compassionate Human
The one thing AI can’t do is show compassion, at least not yet. The someday “vision” when a robot will gather intel about a patient’s symptoms and even offer a diagnosis does have some downsides. There is no replacement for human interaction, especially in the case of dire health news.
“If you have signs of a metastatic cancer and a nonhuman is delivering this news, there’s no way AI can share this news with compassion,” said Dr. Stewart.
For now, AI is becoming instrumental in helping reduce the number of extra demands on primary care doctors, as well as physicians in other specialties, so that they can continue focusing on what matters — healing patients.
A version of this article first appeared on Medscape.com.
Within the next few years, patients will go to their primary care facility for a medical problem. They’ll be greeted by a nonhuman who speaks in the language of their choice. Based upon the initial interview, which will be taken in note form, the patient will be diagnosed, and a prescription called into the pharmacy. They’ll pay the robot at a reception kiosk, and their meds will be delivered via driverless car.
Or so suggests Allan Stewart, MD, medical director and chief of cardiothoracic surgery at HCA Florida Mercy Hospital in Miami.
The writing is on the wall. , he said.
If that sounds far too futuristic, buckle up. AI is already here and being used by most medical specialties. However, it’s primary care that stands to gain the most from this technology — right now — thanks to its ability to radically streamline patient care.
Seeing the Doctor and His or Her AI Assistant
AI is making doctors’ work lives easier, whether the technology helps with risk prevention and intervention or closing care gaps. It can also triage patient complaints, monitor patients remotely, or even perform digital health coaching to keep patients on track with their lifestyle regimens or monitor their health conditions.
Each of these AI components enables primary care physicians to reduce some of the paperwork requirements of their jobs and do what they were trained to do — listen and assess patients. Doctors currently spend 12 hours on average each week submitting prior authorization requests, according to an American Medical Association survey.
“Primary care can be overwhelming, especially today, with the advent of electronic records and data,” said Davin Lundquist, MD, a family medicine physician and chief medical officer at Augmedix, an automated medical documentation company that provides tools to reduce clinician burnout. “The amount of data we have to go through to try to get a complete and clear picture of our patients can be overwhelming on top of the referrals, administrative burdens, and regulatory requirements, which seem to be focused on the primary care space,” Dr. Lundquist said.
With an AI assist, primary care physicians can reduce their prep and pre-charting time, lessen the time needed for paperwork outside of clinic hours, and streamline information, including access to lab results, radiology reports, and consults.
“AI is already helping doctors manage their practices, make differential diagnoses, and input progress notes or histories,” said Dr. Stewart.
In Seattle, Ford Parsons, MD, chief of operational analytics at Providence Hospitals in Seattle, has been leading a generative AI project that recently developed a tool called Provaria to prioritize incoming messages from patients. The tool ensures that those with more urgent needs get immediate attention, and it supports the personnel who lead the responses.
The process begins with Provaria reviewing patient messages to ensure those with more urgent needs, such as a mental health crisis, get immediate attention instead of answering messages in the order they were received.
Provaria also provides resources to help responding staff craft a reply. If a patient’s message cites back pain, for example, the system might suggest a referral to a physical therapist, include a link to that department, and prompt the staff to ask about red flags that indicate a more urgent situation.
After an initial rollout, Providence recently deployed Provaria to manage the messages for all 4000 of its primary care, family medicine, and internal medicine providers. The system has reviewed and categorized more than 500,000 messages so far.
“This is another example where AI can increase the human connection in healthcare,” Dr. Parsons said. “That’s the opposite of what others are saying, but by using AI, you can automate the stuff that isn’t critical that doctors have wound up doing.”
AI Helps Foster Better Person-to-Person Communication
In recent years, the first thing most doctors do when they enter the exam room with a patient is log into the in-room computer and start to take notes — which can be off-putting to patients.
Now devices can ease this process, such as PLAUD, an AI voice recognition device that attaches to a cell phone. Just the size of a credit card, the device enables conversations to be easily recorded. It not only streamlines note-taking but also enables a physician to listen intently to a patient’s concerns instead of furiously jotting down notes.
“That device is already helping transcribe conversations into notes and then into a patient’s electronic medical record,” Dr. Stewart said. “This helps save doctors the work of having to input patient information.”
AI Can’t Be a Compassionate Human
The one thing AI can’t do is show compassion, at least not yet. The someday “vision” when a robot will gather intel about a patient’s symptoms and even offer a diagnosis does have some downsides. There is no replacement for human interaction, especially in the case of dire health news.
“If you have signs of a metastatic cancer and a nonhuman is delivering this news, there’s no way AI can share this news with compassion,” said Dr. Stewart.
For now, AI is becoming instrumental in helping reduce the number of extra demands on primary care doctors, as well as physicians in other specialties, so that they can continue focusing on what matters — healing patients.
A version of this article first appeared on Medscape.com.
Within the next few years, patients will go to their primary care facility for a medical problem. They’ll be greeted by a nonhuman who speaks in the language of their choice. Based upon the initial interview, which will be taken in note form, the patient will be diagnosed, and a prescription called into the pharmacy. They’ll pay the robot at a reception kiosk, and their meds will be delivered via driverless car.
Or so suggests Allan Stewart, MD, medical director and chief of cardiothoracic surgery at HCA Florida Mercy Hospital in Miami.
The writing is on the wall. , he said.
If that sounds far too futuristic, buckle up. AI is already here and being used by most medical specialties. However, it’s primary care that stands to gain the most from this technology — right now — thanks to its ability to radically streamline patient care.
Seeing the Doctor and His or Her AI Assistant
AI is making doctors’ work lives easier, whether the technology helps with risk prevention and intervention or closing care gaps. It can also triage patient complaints, monitor patients remotely, or even perform digital health coaching to keep patients on track with their lifestyle regimens or monitor their health conditions.
Each of these AI components enables primary care physicians to reduce some of the paperwork requirements of their jobs and do what they were trained to do — listen and assess patients. Doctors currently spend 12 hours on average each week submitting prior authorization requests, according to an American Medical Association survey.
“Primary care can be overwhelming, especially today, with the advent of electronic records and data,” said Davin Lundquist, MD, a family medicine physician and chief medical officer at Augmedix, an automated medical documentation company that provides tools to reduce clinician burnout. “The amount of data we have to go through to try to get a complete and clear picture of our patients can be overwhelming on top of the referrals, administrative burdens, and regulatory requirements, which seem to be focused on the primary care space,” Dr. Lundquist said.
With an AI assist, primary care physicians can reduce their prep and pre-charting time, lessen the time needed for paperwork outside of clinic hours, and streamline information, including access to lab results, radiology reports, and consults.
“AI is already helping doctors manage their practices, make differential diagnoses, and input progress notes or histories,” said Dr. Stewart.
In Seattle, Ford Parsons, MD, chief of operational analytics at Providence Hospitals in Seattle, has been leading a generative AI project that recently developed a tool called Provaria to prioritize incoming messages from patients. The tool ensures that those with more urgent needs get immediate attention, and it supports the personnel who lead the responses.
The process begins with Provaria reviewing patient messages to ensure those with more urgent needs, such as a mental health crisis, get immediate attention instead of answering messages in the order they were received.
Provaria also provides resources to help responding staff craft a reply. If a patient’s message cites back pain, for example, the system might suggest a referral to a physical therapist, include a link to that department, and prompt the staff to ask about red flags that indicate a more urgent situation.
After an initial rollout, Providence recently deployed Provaria to manage the messages for all 4000 of its primary care, family medicine, and internal medicine providers. The system has reviewed and categorized more than 500,000 messages so far.
“This is another example where AI can increase the human connection in healthcare,” Dr. Parsons said. “That’s the opposite of what others are saying, but by using AI, you can automate the stuff that isn’t critical that doctors have wound up doing.”
AI Helps Foster Better Person-to-Person Communication
In recent years, the first thing most doctors do when they enter the exam room with a patient is log into the in-room computer and start to take notes — which can be off-putting to patients.
Now devices can ease this process, such as PLAUD, an AI voice recognition device that attaches to a cell phone. Just the size of a credit card, the device enables conversations to be easily recorded. It not only streamlines note-taking but also enables a physician to listen intently to a patient’s concerns instead of furiously jotting down notes.
“That device is already helping transcribe conversations into notes and then into a patient’s electronic medical record,” Dr. Stewart said. “This helps save doctors the work of having to input patient information.”
AI Can’t Be a Compassionate Human
The one thing AI can’t do is show compassion, at least not yet. The someday “vision” when a robot will gather intel about a patient’s symptoms and even offer a diagnosis does have some downsides. There is no replacement for human interaction, especially in the case of dire health news.
“If you have signs of a metastatic cancer and a nonhuman is delivering this news, there’s no way AI can share this news with compassion,” said Dr. Stewart.
For now, AI is becoming instrumental in helping reduce the number of extra demands on primary care doctors, as well as physicians in other specialties, so that they can continue focusing on what matters — healing patients.
A version of this article first appeared on Medscape.com.
Several Skin Conditions More Likely in Children With Obesity
TORONTO — results of new research show.
The retrospective cohort study found markedly higher rates of skin infections, atopic dermatitis (AD), and acanthosis nigricans among children with overweight, compared with children with average weight.
“Many conditions associated with obesity are strong predictors of cardiovascular mortality as these children age, so doctors can play a key role in advocating for weight loss strategies in this population,” lead study author Samantha Epstein, third-year medical student at Case Western Reserve University, Cleveland, Ohio, said in an interview. The findings were presented at the annual meeting of the Society for Pediatric Dermatology.
Previous research has linked obesity, a chronic inflammatory condition, to psoriasis, AD, hidradenitis suppurativa (HS), acne vulgaris, infections, and rosacea in adults. However, there’s scant research exploring the connection between obesity and cutaneous conditions in children.
According to the Cleveland Clinic, childhood obesity is defined as a body mass index, which is weight in kg divided by the square of height in m2, at or above the 95th percentile for age and sex in children aged 2 years or older.
For the study, Ms. Epstein and coauthor Sonal D. Shah, MD, associate professor, Department of Dermatology, Case Western Reserve University, and a board-certified pediatric dermatologist accessed a large national research database and used diagnostic codes to identify over 1 million children (mean age, 8.5 years). Most (about 44%) were White; about one-quarter were Black. The groups were propensity matched, so there were about equal numbers of youngsters with and without obesity and of boys and girls.
They collected data on AD, HS, rosacea, psoriasis, and acanthosis nigricans (a thickened purplish discoloration typically found in body folds around the armpits, groin, and neck). They also gathered information on comorbidities.
Acanthosis nigricans, which is linked to metabolic syndrome, type 2 diabetes, and insulin resistance , was more prevalent among children with obesity (20,885 cases in the with-obesity group and 336 in the without-obesity group, for a relative risk [RR] of 62.16 and an odds ratio [OR] of 64.38).
Skin and subcutaneous tissue infections were also more common among those with obesity (14,795 cases) vs 4720 cases among those without obesity (RR, 3.14; OR, 3.2). As for AD, there were 11,892 cases in the with-obesity group and 2983 in the without-obesity group (RR, 3.99; OR, 4.06). There were 1166 cases of psoriasis among those with obesity and 408 among those without obesity (RR, 2.86; OR, 2.88).
HS (587 cases in the with-obesity group and 70 in the without-obesity group; RR, 8.39; OR, 8.39) and rosacea (351 in the with-obesity group and 138 in the without-obesity group; RR, 2.54; OR, 2.55) were the least common skin conditions.
Higher Comorbidity Rates
Compared with their average-weight counterparts, the children with obesity had higher rates of comorbidities, including type 2 diabetes. Ms. Epstein noted that children with diabetes and obesity had increased risks for every skin condition except for infections of the skin and subcutaneous tissue when compared with children without obesity.
Such infections were the most common skin conditions among children without obesity. “This was expected just due to the fact that children are outside, they’re playing in the grass and the dirt, and they get infected,” said Ms. Epstein. Still, these infections were three times more common in youngsters with obesity.
Although acanthosis nigricans is “highly correlated” with type 2 diabetes, “not as many children as we would expect in this population have developed type 2 diabetes,” said Ms. Epstein. This might make some sense, though, because these children are still quite young. “When dermatologists recognize this skin condition, they can advocate for weight loss management to try to prevent it.”
Other conditions seen more often in the overweight children with overweight included: hypertension, hyperlipidemia, obstructive sleep apnea, polycystic ovarian syndrome, attention-deficit/hyperactivity disorder, major depressive disorder, depressive episodes, and anxiety (all P < .001).
Commenting on the results, Sonia Havele, MD, a pediatrician and dermatology resident at Children’s Mercy Hospital, Kansas City, Missouri, said in an interview that the study reflects trends that she and her colleagues see in clinic: There are more common skin conditions in their patients with obesity.
She agreed that it offers an opening for education. “The results of this study highlight the opportunity we have as pediatric dermatologists to provide additional counseling on obesity and offer referrals to our colleagues in endocrinology, gastroenterology, and nutrition if needed.”
No conflicts of interest were reported.
TORONTO — results of new research show.
The retrospective cohort study found markedly higher rates of skin infections, atopic dermatitis (AD), and acanthosis nigricans among children with overweight, compared with children with average weight.
“Many conditions associated with obesity are strong predictors of cardiovascular mortality as these children age, so doctors can play a key role in advocating for weight loss strategies in this population,” lead study author Samantha Epstein, third-year medical student at Case Western Reserve University, Cleveland, Ohio, said in an interview. The findings were presented at the annual meeting of the Society for Pediatric Dermatology.
Previous research has linked obesity, a chronic inflammatory condition, to psoriasis, AD, hidradenitis suppurativa (HS), acne vulgaris, infections, and rosacea in adults. However, there’s scant research exploring the connection between obesity and cutaneous conditions in children.
According to the Cleveland Clinic, childhood obesity is defined as a body mass index, which is weight in kg divided by the square of height in m2, at or above the 95th percentile for age and sex in children aged 2 years or older.
For the study, Ms. Epstein and coauthor Sonal D. Shah, MD, associate professor, Department of Dermatology, Case Western Reserve University, and a board-certified pediatric dermatologist accessed a large national research database and used diagnostic codes to identify over 1 million children (mean age, 8.5 years). Most (about 44%) were White; about one-quarter were Black. The groups were propensity matched, so there were about equal numbers of youngsters with and without obesity and of boys and girls.
They collected data on AD, HS, rosacea, psoriasis, and acanthosis nigricans (a thickened purplish discoloration typically found in body folds around the armpits, groin, and neck). They also gathered information on comorbidities.
Acanthosis nigricans, which is linked to metabolic syndrome, type 2 diabetes, and insulin resistance , was more prevalent among children with obesity (20,885 cases in the with-obesity group and 336 in the without-obesity group, for a relative risk [RR] of 62.16 and an odds ratio [OR] of 64.38).
Skin and subcutaneous tissue infections were also more common among those with obesity (14,795 cases) vs 4720 cases among those without obesity (RR, 3.14; OR, 3.2). As for AD, there were 11,892 cases in the with-obesity group and 2983 in the without-obesity group (RR, 3.99; OR, 4.06). There were 1166 cases of psoriasis among those with obesity and 408 among those without obesity (RR, 2.86; OR, 2.88).
HS (587 cases in the with-obesity group and 70 in the without-obesity group; RR, 8.39; OR, 8.39) and rosacea (351 in the with-obesity group and 138 in the without-obesity group; RR, 2.54; OR, 2.55) were the least common skin conditions.
Higher Comorbidity Rates
Compared with their average-weight counterparts, the children with obesity had higher rates of comorbidities, including type 2 diabetes. Ms. Epstein noted that children with diabetes and obesity had increased risks for every skin condition except for infections of the skin and subcutaneous tissue when compared with children without obesity.
Such infections were the most common skin conditions among children without obesity. “This was expected just due to the fact that children are outside, they’re playing in the grass and the dirt, and they get infected,” said Ms. Epstein. Still, these infections were three times more common in youngsters with obesity.
Although acanthosis nigricans is “highly correlated” with type 2 diabetes, “not as many children as we would expect in this population have developed type 2 diabetes,” said Ms. Epstein. This might make some sense, though, because these children are still quite young. “When dermatologists recognize this skin condition, they can advocate for weight loss management to try to prevent it.”
Other conditions seen more often in the overweight children with overweight included: hypertension, hyperlipidemia, obstructive sleep apnea, polycystic ovarian syndrome, attention-deficit/hyperactivity disorder, major depressive disorder, depressive episodes, and anxiety (all P < .001).
Commenting on the results, Sonia Havele, MD, a pediatrician and dermatology resident at Children’s Mercy Hospital, Kansas City, Missouri, said in an interview that the study reflects trends that she and her colleagues see in clinic: There are more common skin conditions in their patients with obesity.
She agreed that it offers an opening for education. “The results of this study highlight the opportunity we have as pediatric dermatologists to provide additional counseling on obesity and offer referrals to our colleagues in endocrinology, gastroenterology, and nutrition if needed.”
No conflicts of interest were reported.
TORONTO — results of new research show.
The retrospective cohort study found markedly higher rates of skin infections, atopic dermatitis (AD), and acanthosis nigricans among children with overweight, compared with children with average weight.
“Many conditions associated with obesity are strong predictors of cardiovascular mortality as these children age, so doctors can play a key role in advocating for weight loss strategies in this population,” lead study author Samantha Epstein, third-year medical student at Case Western Reserve University, Cleveland, Ohio, said in an interview. The findings were presented at the annual meeting of the Society for Pediatric Dermatology.
Previous research has linked obesity, a chronic inflammatory condition, to psoriasis, AD, hidradenitis suppurativa (HS), acne vulgaris, infections, and rosacea in adults. However, there’s scant research exploring the connection between obesity and cutaneous conditions in children.
According to the Cleveland Clinic, childhood obesity is defined as a body mass index, which is weight in kg divided by the square of height in m2, at or above the 95th percentile for age and sex in children aged 2 years or older.
For the study, Ms. Epstein and coauthor Sonal D. Shah, MD, associate professor, Department of Dermatology, Case Western Reserve University, and a board-certified pediatric dermatologist accessed a large national research database and used diagnostic codes to identify over 1 million children (mean age, 8.5 years). Most (about 44%) were White; about one-quarter were Black. The groups were propensity matched, so there were about equal numbers of youngsters with and without obesity and of boys and girls.
They collected data on AD, HS, rosacea, psoriasis, and acanthosis nigricans (a thickened purplish discoloration typically found in body folds around the armpits, groin, and neck). They also gathered information on comorbidities.
Acanthosis nigricans, which is linked to metabolic syndrome, type 2 diabetes, and insulin resistance , was more prevalent among children with obesity (20,885 cases in the with-obesity group and 336 in the without-obesity group, for a relative risk [RR] of 62.16 and an odds ratio [OR] of 64.38).
Skin and subcutaneous tissue infections were also more common among those with obesity (14,795 cases) vs 4720 cases among those without obesity (RR, 3.14; OR, 3.2). As for AD, there were 11,892 cases in the with-obesity group and 2983 in the without-obesity group (RR, 3.99; OR, 4.06). There were 1166 cases of psoriasis among those with obesity and 408 among those without obesity (RR, 2.86; OR, 2.88).
HS (587 cases in the with-obesity group and 70 in the without-obesity group; RR, 8.39; OR, 8.39) and rosacea (351 in the with-obesity group and 138 in the without-obesity group; RR, 2.54; OR, 2.55) were the least common skin conditions.
Higher Comorbidity Rates
Compared with their average-weight counterparts, the children with obesity had higher rates of comorbidities, including type 2 diabetes. Ms. Epstein noted that children with diabetes and obesity had increased risks for every skin condition except for infections of the skin and subcutaneous tissue when compared with children without obesity.
Such infections were the most common skin conditions among children without obesity. “This was expected just due to the fact that children are outside, they’re playing in the grass and the dirt, and they get infected,” said Ms. Epstein. Still, these infections were three times more common in youngsters with obesity.
Although acanthosis nigricans is “highly correlated” with type 2 diabetes, “not as many children as we would expect in this population have developed type 2 diabetes,” said Ms. Epstein. This might make some sense, though, because these children are still quite young. “When dermatologists recognize this skin condition, they can advocate for weight loss management to try to prevent it.”
Other conditions seen more often in the overweight children with overweight included: hypertension, hyperlipidemia, obstructive sleep apnea, polycystic ovarian syndrome, attention-deficit/hyperactivity disorder, major depressive disorder, depressive episodes, and anxiety (all P < .001).
Commenting on the results, Sonia Havele, MD, a pediatrician and dermatology resident at Children’s Mercy Hospital, Kansas City, Missouri, said in an interview that the study reflects trends that she and her colleagues see in clinic: There are more common skin conditions in their patients with obesity.
She agreed that it offers an opening for education. “The results of this study highlight the opportunity we have as pediatric dermatologists to provide additional counseling on obesity and offer referrals to our colleagues in endocrinology, gastroenterology, and nutrition if needed.”
No conflicts of interest were reported.
FROM SPD 2024
Topical Ruxolitinib: Analysis Finds Repigmentation Rates in Adolescents with Vitiligo
data showed.
“We consider repigmenting vitiligo a two-step process, where the overactive immune system needs to be calmed down and then the melanocytes need to repopulate to the white areas,” one of the study investigators, David Rosmarin, MD, chair of the Department of Dermatology at Indiana University School of Medicine, Indianapolis, said in an interview in advance of the annual meeting of the Society for Pediatric Dermatology, where the study results were presented during a poster session. “In younger patients, it may be that the melanocytes are more rapidly repigmenting the patches, which is why we see this effect.”

Ruxolitinib, 1.5% cream (Opzelura) is a Janus kinase inhibitor approved for the treatment of nonsegmental vitiligo in patients 12 years of age and older. Dr. Rosmarin and colleagues sought to evaluate differences in rates of complete or near-complete repigmentation and repigmentation by body region between adolescents 12-17 years of age and adults 18 years of age and older who applied ruxolitinib cream twice daily. The researchers evaluated patients who were initially randomized to ruxolitinib cream, 1.5% in the pivotal TRuE-V1 and TRuE-V2 studies and applied it for up to 104 weeks. Complete facial improvement was defined as 100% improvement on the Facial Vitiligo Area Scoring Index (F-VASI 100) from baseline, and near-total improvement was categorized as a ≥ 75% or ≥ 90% improvement from baseline on the Total body VASI (T-VASI). Responses for each of six body regions, excluding the face, were assessed by the proportion of patients who achieved at least a 50% improvement from baseline on the T-VASI.
Compared with adults, a greater proportion of adolescents achieved F-VASI 100 at week 24 (5.7% [3/53] vs 2.9% [10/341], respectively), but there were no differences between the two groups at week 52 (8.0% [4/50] vs 8.0% [24/300]). Response rates were greater among adolescents vs adults for T-VASI 75 at weeks 24 (13.2% [7/53] vs 5.6% [19/341]) and 52 (22.0% [11/50] vs 20.3% [61/300]), as well as T-VASI 90 at weeks 24 (3.8% [2/53] vs 0.3% [1/341]) and 52 (12.0% [6/50] vs 4.0% [12/300]).
The researchers observed that VASI 50 responses by body region were generally similar between adolescents and adults, but a greater proportion of adolescents achieved a VASI 50 in lower extremities (67.3% [33/49] vs 51.8% [118/228]) and feet (37.5% [12/32] vs 27.9% [51/183]) at week 52.
“Adolescents repigmented more rapidly than adults, so that at 24 weeks, more teens had complete facial repigmentation and T-VASI 75 and T-VASI 90 results,” Dr. Rosmarin said. “With continued use of ruxolitinib cream, both more adults and adolescents achieved greater repigmentation.” He acknowledged certain limitations of the study, including the fact that it was only vehicle controlled up through 24 weeks and that, after week 52, there were fewer patients who completed the long-term extension.
“The take-home message is that ruxolitinib cream can effectively and safely help many patients repigment, including adolescents,” he said.
The study was funded by topical ruxolitinib manufacturer Incyte. Dr. Rosmarin disclosed that he has consulted, spoken for, or conducted trials for AbbVie, Abcuro, Almirall, AltruBio, Amgen, Arena, Astria, Boehringer Ingelheim, Bristol Meyers Squibb, Celgene, Concert, CSL Behring, Dermavant Sciences, Dermira, Galderma, Incyte, Janssen, Kyowa Kirin, Lilly, Merck, Nektar, Novartis, Pfizer, RAPT, Regeneron, Recludix Pharma, Revolo Biotherapeutics, Sanofi, Sun Pharmaceuticals, UCB, Viela Bio, and Zura.
A version of this article first appeared on Medscape.com.
data showed.
“We consider repigmenting vitiligo a two-step process, where the overactive immune system needs to be calmed down and then the melanocytes need to repopulate to the white areas,” one of the study investigators, David Rosmarin, MD, chair of the Department of Dermatology at Indiana University School of Medicine, Indianapolis, said in an interview in advance of the annual meeting of the Society for Pediatric Dermatology, where the study results were presented during a poster session. “In younger patients, it may be that the melanocytes are more rapidly repigmenting the patches, which is why we see this effect.”
Ruxolitinib, 1.5% cream (Opzelura) is a Janus kinase inhibitor approved for the treatment of nonsegmental vitiligo in patients 12 years of age and older. Dr. Rosmarin and colleagues sought to evaluate differences in rates of complete or near-complete repigmentation and repigmentation by body region between adolescents 12-17 years of age and adults 18 years of age and older who applied ruxolitinib cream twice daily. The researchers evaluated patients who were initially randomized to ruxolitinib cream, 1.5% in the pivotal TRuE-V1 and TRuE-V2 studies and applied it for up to 104 weeks. Complete facial improvement was defined as 100% improvement on the Facial Vitiligo Area Scoring Index (F-VASI 100) from baseline, and near-total improvement was categorized as a ≥ 75% or ≥ 90% improvement from baseline on the Total body VASI (T-VASI). Responses for each of six body regions, excluding the face, were assessed by the proportion of patients who achieved at least a 50% improvement from baseline on the T-VASI.
Compared with adults, a greater proportion of adolescents achieved F-VASI 100 at week 24 (5.7% [3/53] vs 2.9% [10/341], respectively), but there were no differences between the two groups at week 52 (8.0% [4/50] vs 8.0% [24/300]). Response rates were greater among adolescents vs adults for T-VASI 75 at weeks 24 (13.2% [7/53] vs 5.6% [19/341]) and 52 (22.0% [11/50] vs 20.3% [61/300]), as well as T-VASI 90 at weeks 24 (3.8% [2/53] vs 0.3% [1/341]) and 52 (12.0% [6/50] vs 4.0% [12/300]).
The researchers observed that VASI 50 responses by body region were generally similar between adolescents and adults, but a greater proportion of adolescents achieved a VASI 50 in lower extremities (67.3% [33/49] vs 51.8% [118/228]) and feet (37.5% [12/32] vs 27.9% [51/183]) at week 52.
“Adolescents repigmented more rapidly than adults, so that at 24 weeks, more teens had complete facial repigmentation and T-VASI 75 and T-VASI 90 results,” Dr. Rosmarin said. “With continued use of ruxolitinib cream, both more adults and adolescents achieved greater repigmentation.” He acknowledged certain limitations of the study, including the fact that it was only vehicle controlled up through 24 weeks and that, after week 52, there were fewer patients who completed the long-term extension.
“The take-home message is that ruxolitinib cream can effectively and safely help many patients repigment, including adolescents,” he said.
The study was funded by topical ruxolitinib manufacturer Incyte. Dr. Rosmarin disclosed that he has consulted, spoken for, or conducted trials for AbbVie, Abcuro, Almirall, AltruBio, Amgen, Arena, Astria, Boehringer Ingelheim, Bristol Meyers Squibb, Celgene, Concert, CSL Behring, Dermavant Sciences, Dermira, Galderma, Incyte, Janssen, Kyowa Kirin, Lilly, Merck, Nektar, Novartis, Pfizer, RAPT, Regeneron, Recludix Pharma, Revolo Biotherapeutics, Sanofi, Sun Pharmaceuticals, UCB, Viela Bio, and Zura.
A version of this article first appeared on Medscape.com.
data showed.
“We consider repigmenting vitiligo a two-step process, where the overactive immune system needs to be calmed down and then the melanocytes need to repopulate to the white areas,” one of the study investigators, David Rosmarin, MD, chair of the Department of Dermatology at Indiana University School of Medicine, Indianapolis, said in an interview in advance of the annual meeting of the Society for Pediatric Dermatology, where the study results were presented during a poster session. “In younger patients, it may be that the melanocytes are more rapidly repigmenting the patches, which is why we see this effect.”
Ruxolitinib, 1.5% cream (Opzelura) is a Janus kinase inhibitor approved for the treatment of nonsegmental vitiligo in patients 12 years of age and older. Dr. Rosmarin and colleagues sought to evaluate differences in rates of complete or near-complete repigmentation and repigmentation by body region between adolescents 12-17 years of age and adults 18 years of age and older who applied ruxolitinib cream twice daily. The researchers evaluated patients who were initially randomized to ruxolitinib cream, 1.5% in the pivotal TRuE-V1 and TRuE-V2 studies and applied it for up to 104 weeks. Complete facial improvement was defined as 100% improvement on the Facial Vitiligo Area Scoring Index (F-VASI 100) from baseline, and near-total improvement was categorized as a ≥ 75% or ≥ 90% improvement from baseline on the Total body VASI (T-VASI). Responses for each of six body regions, excluding the face, were assessed by the proportion of patients who achieved at least a 50% improvement from baseline on the T-VASI.
Compared with adults, a greater proportion of adolescents achieved F-VASI 100 at week 24 (5.7% [3/53] vs 2.9% [10/341], respectively), but there were no differences between the two groups at week 52 (8.0% [4/50] vs 8.0% [24/300]). Response rates were greater among adolescents vs adults for T-VASI 75 at weeks 24 (13.2% [7/53] vs 5.6% [19/341]) and 52 (22.0% [11/50] vs 20.3% [61/300]), as well as T-VASI 90 at weeks 24 (3.8% [2/53] vs 0.3% [1/341]) and 52 (12.0% [6/50] vs 4.0% [12/300]).
The researchers observed that VASI 50 responses by body region were generally similar between adolescents and adults, but a greater proportion of adolescents achieved a VASI 50 in lower extremities (67.3% [33/49] vs 51.8% [118/228]) and feet (37.5% [12/32] vs 27.9% [51/183]) at week 52.
“Adolescents repigmented more rapidly than adults, so that at 24 weeks, more teens had complete facial repigmentation and T-VASI 75 and T-VASI 90 results,” Dr. Rosmarin said. “With continued use of ruxolitinib cream, both more adults and adolescents achieved greater repigmentation.” He acknowledged certain limitations of the study, including the fact that it was only vehicle controlled up through 24 weeks and that, after week 52, there were fewer patients who completed the long-term extension.
“The take-home message is that ruxolitinib cream can effectively and safely help many patients repigment, including adolescents,” he said.
The study was funded by topical ruxolitinib manufacturer Incyte. Dr. Rosmarin disclosed that he has consulted, spoken for, or conducted trials for AbbVie, Abcuro, Almirall, AltruBio, Amgen, Arena, Astria, Boehringer Ingelheim, Bristol Meyers Squibb, Celgene, Concert, CSL Behring, Dermavant Sciences, Dermira, Galderma, Incyte, Janssen, Kyowa Kirin, Lilly, Merck, Nektar, Novartis, Pfizer, RAPT, Regeneron, Recludix Pharma, Revolo Biotherapeutics, Sanofi, Sun Pharmaceuticals, UCB, Viela Bio, and Zura.
A version of this article first appeared on Medscape.com.
FROM SPD 2024
Mysteries Persist About Tissue Resident Memory T Cells in Psoriasis
SEATTLE — In fact, flare-ups often recur at the same site, a phenomenon that might be driven by these resident memory cells, according to Liv Eidsmo, MD, PhD.
This has led to their use as biomarkers in clinical trials for new therapies, but TRM T cells have a complex biology that is far from fully understood, Dr. Eidsmo said at the annual meeting of the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis. “With time, we’re understanding that the regulation of the functionality is more complicated than we thought, so following these cells as a positive outcome of a clinical trial is a little bit premature,” said Dr. Eidsmo, who is a consultant dermatologist at the University of Copenhagen, Copenhagen, Denmark.
Treatment strategies focus on inhibition of interleukin (IL)-23, which is an activator of TRM T cells and probably keeps them alive, according to Dr. Eidsmo. “The hope is that these cells can be silenced by IL-23 inhibition, which is a great idea, and it probably works. It’s just a matter of what is the readout of long-term remission, because the big challenge in the clinical world is when do we stop these expensive biological treatments? When can we feel secure that patients are in deep remission?” she asked.
TRM cells are also far from the only immune cells involved in psoriasis. Others include keratinocytes, Langerhans cells, and fibroblasts. Dr. Eidsmo referenced a recent spatial analysis that used single-cell and spatial RNA sequencing to identify the localization of specific cell populations and inflammatory pathways within psoriasis lesions and epidermal compartments as well as also suggested crosstalk links between cell types. Epigenetic changes in stem cells may also maintain a lower threshold for tissue inflammation.
Dr. Eidsmo advised caution in eliminating TRM T cells, which play a key role in protecting against melanoma and other cancers, especially later in life. “We don’t want to get rid of them. We want to have the right balance.”
She noted a study in her own lab that mapped TRM T cells in healthy epidermis and found that they could be renewed from both circulating precursors and cells within the epidermis. “So getting rid of the mature TRM T cells will most likely just lead to a new generation of the same subset.”
Other data show that there are a wide range of subsets of TRM T cells, and she recommended focusing on the functionality of TRM T cells rather than sheer numbers. “This is something we’re working on now: Can we change the functionality [of TRM T cells], rather than eradicate them and hope for the best in the next generation? Can we change the functionality of the T cells we already have in the skin?”
There is also epigenetic data in TRM T cells, keratinocytes, stem cells, and other cells thus suggesting complexity and plasticity in the system that remains poorly understood.
Taken together, the research is at too early of a stage to be clinically useful, said Dr. Eidsmo. “We need to go back to the drawing board and just realize what we need to measure, and with the new techniques coming out, maybe spatial [measurement] at a high resolution, we can find biomarkers that better dictate the future of this. Be a little bit wary when you read the outcomes from the clinical trials that are ongoing, because right now, it’s a bit of a race between different biologics. These cells are used as a readout of efficacy of the treatments, and we’re not quite there yet.”
During the Q&A session after the presentation, one audience member asked about the heterogeneity of cells found within the skin of patients with psoriasis and pointed out that many proinflammatory cells likely play a role in tumor control. Dr. Eidsmo responded that her group’s analysis of a large database of patients with metastatic melanoma found that a factor that is important to the development of TRM T cells was strongly correlated to survival in patients with metastatic melanoma receiving immune checkpoint blockade. “So we really don’t want to eradicate them,” she said.
Also during the Q&A, Iain McInnes, MD, PhD, commented about the need to understand the previous events that drove the creation of memory T cells. “For me, the question is about the hierarchy, the primacy of what really drives the memory. In the infectious world, we’re trained to think [that memory responses] are T cell driven memory, but I wonder whether you have an idea of whether the T cell is responding to other memories, particularly in the stroma. Because certainly in the arthropathies, we have really good evidence now of epigenetic change in the synovial stroma and subsets,” said Dr. McInnes, who is director of the Institute of Infection, Immunity, and Inflammation at the University of Glasgow, Glasgow, Scotland.
Dr. Eidsmo responded that she believes responses are different among different individuals. “We know too little about how these two systems interact with one another. I think the TRM T cells are very good at amplifying the stroma to recruit cells in. I think we need to think of two-step therapies. You need to normalize this [stromal] environment. How you can do that, I don’t know.”
Dr. McInnes agreed. “As a myeloid doctor, I strongly believe that perpetuators are innate and the adaptive is following on. But how do we test that? That’s really hard,” he said.
Dr. Eidsmo did not list any disclosures. Dr. McInnes has financial relationships with AbbVie, AstraZeneca, Bristol-Myers Squibb, Boehringer, Compugen, Cabaletta, Causeway, Dextera, Eli Lilly, Celgene, MoonLake, Pfizer, Novartis, Janssen, Roche, Versus Arthritis, MRC, and UCB.
SEATTLE — In fact, flare-ups often recur at the same site, a phenomenon that might be driven by these resident memory cells, according to Liv Eidsmo, MD, PhD.
This has led to their use as biomarkers in clinical trials for new therapies, but TRM T cells have a complex biology that is far from fully understood, Dr. Eidsmo said at the annual meeting of the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis. “With time, we’re understanding that the regulation of the functionality is more complicated than we thought, so following these cells as a positive outcome of a clinical trial is a little bit premature,” said Dr. Eidsmo, who is a consultant dermatologist at the University of Copenhagen, Copenhagen, Denmark.
Treatment strategies focus on inhibition of interleukin (IL)-23, which is an activator of TRM T cells and probably keeps them alive, according to Dr. Eidsmo. “The hope is that these cells can be silenced by IL-23 inhibition, which is a great idea, and it probably works. It’s just a matter of what is the readout of long-term remission, because the big challenge in the clinical world is when do we stop these expensive biological treatments? When can we feel secure that patients are in deep remission?” she asked.
TRM cells are also far from the only immune cells involved in psoriasis. Others include keratinocytes, Langerhans cells, and fibroblasts. Dr. Eidsmo referenced a recent spatial analysis that used single-cell and spatial RNA sequencing to identify the localization of specific cell populations and inflammatory pathways within psoriasis lesions and epidermal compartments as well as also suggested crosstalk links between cell types. Epigenetic changes in stem cells may also maintain a lower threshold for tissue inflammation.
Dr. Eidsmo advised caution in eliminating TRM T cells, which play a key role in protecting against melanoma and other cancers, especially later in life. “We don’t want to get rid of them. We want to have the right balance.”
She noted a study in her own lab that mapped TRM T cells in healthy epidermis and found that they could be renewed from both circulating precursors and cells within the epidermis. “So getting rid of the mature TRM T cells will most likely just lead to a new generation of the same subset.”
Other data show that there are a wide range of subsets of TRM T cells, and she recommended focusing on the functionality of TRM T cells rather than sheer numbers. “This is something we’re working on now: Can we change the functionality [of TRM T cells], rather than eradicate them and hope for the best in the next generation? Can we change the functionality of the T cells we already have in the skin?”
There is also epigenetic data in TRM T cells, keratinocytes, stem cells, and other cells thus suggesting complexity and plasticity in the system that remains poorly understood.
Taken together, the research is at too early of a stage to be clinically useful, said Dr. Eidsmo. “We need to go back to the drawing board and just realize what we need to measure, and with the new techniques coming out, maybe spatial [measurement] at a high resolution, we can find biomarkers that better dictate the future of this. Be a little bit wary when you read the outcomes from the clinical trials that are ongoing, because right now, it’s a bit of a race between different biologics. These cells are used as a readout of efficacy of the treatments, and we’re not quite there yet.”
During the Q&A session after the presentation, one audience member asked about the heterogeneity of cells found within the skin of patients with psoriasis and pointed out that many proinflammatory cells likely play a role in tumor control. Dr. Eidsmo responded that her group’s analysis of a large database of patients with metastatic melanoma found that a factor that is important to the development of TRM T cells was strongly correlated to survival in patients with metastatic melanoma receiving immune checkpoint blockade. “So we really don’t want to eradicate them,” she said.
Also during the Q&A, Iain McInnes, MD, PhD, commented about the need to understand the previous events that drove the creation of memory T cells. “For me, the question is about the hierarchy, the primacy of what really drives the memory. In the infectious world, we’re trained to think [that memory responses] are T cell driven memory, but I wonder whether you have an idea of whether the T cell is responding to other memories, particularly in the stroma. Because certainly in the arthropathies, we have really good evidence now of epigenetic change in the synovial stroma and subsets,” said Dr. McInnes, who is director of the Institute of Infection, Immunity, and Inflammation at the University of Glasgow, Glasgow, Scotland.
Dr. Eidsmo responded that she believes responses are different among different individuals. “We know too little about how these two systems interact with one another. I think the TRM T cells are very good at amplifying the stroma to recruit cells in. I think we need to think of two-step therapies. You need to normalize this [stromal] environment. How you can do that, I don’t know.”
Dr. McInnes agreed. “As a myeloid doctor, I strongly believe that perpetuators are innate and the adaptive is following on. But how do we test that? That’s really hard,” he said.
Dr. Eidsmo did not list any disclosures. Dr. McInnes has financial relationships with AbbVie, AstraZeneca, Bristol-Myers Squibb, Boehringer, Compugen, Cabaletta, Causeway, Dextera, Eli Lilly, Celgene, MoonLake, Pfizer, Novartis, Janssen, Roche, Versus Arthritis, MRC, and UCB.
SEATTLE — In fact, flare-ups often recur at the same site, a phenomenon that might be driven by these resident memory cells, according to Liv Eidsmo, MD, PhD.
This has led to their use as biomarkers in clinical trials for new therapies, but TRM T cells have a complex biology that is far from fully understood, Dr. Eidsmo said at the annual meeting of the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis. “With time, we’re understanding that the regulation of the functionality is more complicated than we thought, so following these cells as a positive outcome of a clinical trial is a little bit premature,” said Dr. Eidsmo, who is a consultant dermatologist at the University of Copenhagen, Copenhagen, Denmark.
Treatment strategies focus on inhibition of interleukin (IL)-23, which is an activator of TRM T cells and probably keeps them alive, according to Dr. Eidsmo. “The hope is that these cells can be silenced by IL-23 inhibition, which is a great idea, and it probably works. It’s just a matter of what is the readout of long-term remission, because the big challenge in the clinical world is when do we stop these expensive biological treatments? When can we feel secure that patients are in deep remission?” she asked.
TRM cells are also far from the only immune cells involved in psoriasis. Others include keratinocytes, Langerhans cells, and fibroblasts. Dr. Eidsmo referenced a recent spatial analysis that used single-cell and spatial RNA sequencing to identify the localization of specific cell populations and inflammatory pathways within psoriasis lesions and epidermal compartments as well as also suggested crosstalk links between cell types. Epigenetic changes in stem cells may also maintain a lower threshold for tissue inflammation.
Dr. Eidsmo advised caution in eliminating TRM T cells, which play a key role in protecting against melanoma and other cancers, especially later in life. “We don’t want to get rid of them. We want to have the right balance.”
She noted a study in her own lab that mapped TRM T cells in healthy epidermis and found that they could be renewed from both circulating precursors and cells within the epidermis. “So getting rid of the mature TRM T cells will most likely just lead to a new generation of the same subset.”
Other data show that there are a wide range of subsets of TRM T cells, and she recommended focusing on the functionality of TRM T cells rather than sheer numbers. “This is something we’re working on now: Can we change the functionality [of TRM T cells], rather than eradicate them and hope for the best in the next generation? Can we change the functionality of the T cells we already have in the skin?”
There is also epigenetic data in TRM T cells, keratinocytes, stem cells, and other cells thus suggesting complexity and plasticity in the system that remains poorly understood.
Taken together, the research is at too early of a stage to be clinically useful, said Dr. Eidsmo. “We need to go back to the drawing board and just realize what we need to measure, and with the new techniques coming out, maybe spatial [measurement] at a high resolution, we can find biomarkers that better dictate the future of this. Be a little bit wary when you read the outcomes from the clinical trials that are ongoing, because right now, it’s a bit of a race between different biologics. These cells are used as a readout of efficacy of the treatments, and we’re not quite there yet.”
During the Q&A session after the presentation, one audience member asked about the heterogeneity of cells found within the skin of patients with psoriasis and pointed out that many proinflammatory cells likely play a role in tumor control. Dr. Eidsmo responded that her group’s analysis of a large database of patients with metastatic melanoma found that a factor that is important to the development of TRM T cells was strongly correlated to survival in patients with metastatic melanoma receiving immune checkpoint blockade. “So we really don’t want to eradicate them,” she said.
Also during the Q&A, Iain McInnes, MD, PhD, commented about the need to understand the previous events that drove the creation of memory T cells. “For me, the question is about the hierarchy, the primacy of what really drives the memory. In the infectious world, we’re trained to think [that memory responses] are T cell driven memory, but I wonder whether you have an idea of whether the T cell is responding to other memories, particularly in the stroma. Because certainly in the arthropathies, we have really good evidence now of epigenetic change in the synovial stroma and subsets,” said Dr. McInnes, who is director of the Institute of Infection, Immunity, and Inflammation at the University of Glasgow, Glasgow, Scotland.
Dr. Eidsmo responded that she believes responses are different among different individuals. “We know too little about how these two systems interact with one another. I think the TRM T cells are very good at amplifying the stroma to recruit cells in. I think we need to think of two-step therapies. You need to normalize this [stromal] environment. How you can do that, I don’t know.”
Dr. McInnes agreed. “As a myeloid doctor, I strongly believe that perpetuators are innate and the adaptive is following on. But how do we test that? That’s really hard,” he said.
Dr. Eidsmo did not list any disclosures. Dr. McInnes has financial relationships with AbbVie, AstraZeneca, Bristol-Myers Squibb, Boehringer, Compugen, Cabaletta, Causeway, Dextera, Eli Lilly, Celgene, MoonLake, Pfizer, Novartis, Janssen, Roche, Versus Arthritis, MRC, and UCB.
FROM GRAPPA 2024
Risk Stratification May Work Well for FIT-Based CRC Screening in Elderly
WASHINGTON — , according to a study presented at the annual Digestive Disease Week® (DDW).
In particular, interval CRC risk can vary substantially based on the fecal hemoglobin (f-Hb) concentration in the patient’s last fecal immunochemical test (FIT), as well as the number of prior screening rounds.
“Less is known about what happens after the upper age limit has been reached and individuals are not invited to participate in more screening rounds. This is important as life expectancy is increasing, and it is increasingly important to consider the most efficient way of screening the elderly,” said lead author Brenda van Stigt, a PhD candidate focused on cancer screening at Erasmus University Medical Center in Rotterdam, the Netherlands.
In the Netherlands, adults between ages 55 and 75 are invited to participate in stool-based CRC screening every 2 years. Based on a fecal immunochemical testing (FIT) threshold of 47 μg Hb/g, those who test positive are referred to colonoscopy, and those who test negative are invited to participate again after a 2-year period.
FIT can play a major role in risk stratification, Ms. van Stigt noted, along with other factors that influence CRC risk, such as age, sex, and CRC screening history. Although this is documented for ages 55-75, she and colleagues wanted to know more about what happens after age 75.
Ms. Van Stigt and colleagues conducted a population-based study by analyzing Dutch national cancer registry data and FIT results around the final screening at age 75, looking at those who were diagnosed with CRC within 24 months of their last negative FIT. The researchers assessed interval CRC risk and cancer stage, accounting for sex, last f-Hb concentration, and the number of screening rounds.
Among 305,761 people with a complete 24-month follow-up after a negative FIT, 661 patients were diagnosed with interval CRC, indicating an overall interval CRC risk of 21.6 per 10,000 individuals with a negative FIT. There were no significant differences by sex.
However, there were differences by screening rounds, with those who had participated in three or four screening rounds having a lower risk than those who participated only once (HR, .49).
In addition, those with detectable f-Hb (>0 μg Hb/g) in their last screening round had a much higher interval CRC risk (HR, 4.87), at 65.8 per 10,000 negative FITs, compared with 13.8 per 10,000 among those without detectable f-Hb. Interval CRC risk also increased over time for those with detectable f-Hb.
About 15% of the total population had detectable f-Hb, whereas 46% of those with interval CRC had detectable f-Hb, Ms. van Stigt said, meaning that nearly half of patients who were diagnosed with interval CRC already had detectable f-Hb in their prior FIT.
In a survival analysis, there was no association between interval CRC risk and sex. However, those who participated in three or four screening rounds were half as likely to be diagnosed than those who participated once or twice, and those with detectable f-Hb were five times as likely to be diagnosed.
For late-stage CRC, there was no association with sex or the number of screening rounds. Detectable f-Hb was associated with not only a higher risk of interval CRC but also a late-stage diagnosis.
“These findings indicate that one uniform age to stop screening is suboptimal,” Ms. van Stigt said. “Personalized screening strategies should, therefore, also ideally incorporate a risk-stratified age to stop screening.”
The US Preventive Services Task Force recommends that clinicians personalize screening for ages 76-85, accounting for overall health, prior screening history, and patient preferences.
“But we have no clear guidance on how to quantify or weigh these factors. This interesting study highlights how one of these factors (prior screening history) and fecal hemoglobin level (an emerging factor) are powerful stratifiers of subsequent colorectal cancer risk,” said Sameer D. Saini, MD, AGAF, director and research investigator at the VA Ann Arbor Healthcare System’s Center for Clinical Management Research. Dr. Saini wasn’t involved with the study.

At the clinical level, Dr. Saini said, sophisticated modeling is needed to understand the interaction with competing risks and identify the optimal screening strategies for patients at varying levels of cancer risk and life expectancy. Models could also help to quantify the population benefits and cost-effectiveness of personalized screening.
“Finally, it is important to note that, in many health systems, access to quantitative FIT may be limited,” he said. “These data may be less informative if colonoscopy is the primary mode of screening.”
Ms. van Stigt and Dr. Saini reported no relevant disclosures.
WASHINGTON — , according to a study presented at the annual Digestive Disease Week® (DDW).
In particular, interval CRC risk can vary substantially based on the fecal hemoglobin (f-Hb) concentration in the patient’s last fecal immunochemical test (FIT), as well as the number of prior screening rounds.
“Less is known about what happens after the upper age limit has been reached and individuals are not invited to participate in more screening rounds. This is important as life expectancy is increasing, and it is increasingly important to consider the most efficient way of screening the elderly,” said lead author Brenda van Stigt, a PhD candidate focused on cancer screening at Erasmus University Medical Center in Rotterdam, the Netherlands.
In the Netherlands, adults between ages 55 and 75 are invited to participate in stool-based CRC screening every 2 years. Based on a fecal immunochemical testing (FIT) threshold of 47 μg Hb/g, those who test positive are referred to colonoscopy, and those who test negative are invited to participate again after a 2-year period.
FIT can play a major role in risk stratification, Ms. van Stigt noted, along with other factors that influence CRC risk, such as age, sex, and CRC screening history. Although this is documented for ages 55-75, she and colleagues wanted to know more about what happens after age 75.
Ms. Van Stigt and colleagues conducted a population-based study by analyzing Dutch national cancer registry data and FIT results around the final screening at age 75, looking at those who were diagnosed with CRC within 24 months of their last negative FIT. The researchers assessed interval CRC risk and cancer stage, accounting for sex, last f-Hb concentration, and the number of screening rounds.
Among 305,761 people with a complete 24-month follow-up after a negative FIT, 661 patients were diagnosed with interval CRC, indicating an overall interval CRC risk of 21.6 per 10,000 individuals with a negative FIT. There were no significant differences by sex.
However, there were differences by screening rounds, with those who had participated in three or four screening rounds having a lower risk than those who participated only once (HR, .49).
In addition, those with detectable f-Hb (>0 μg Hb/g) in their last screening round had a much higher interval CRC risk (HR, 4.87), at 65.8 per 10,000 negative FITs, compared with 13.8 per 10,000 among those without detectable f-Hb. Interval CRC risk also increased over time for those with detectable f-Hb.
About 15% of the total population had detectable f-Hb, whereas 46% of those with interval CRC had detectable f-Hb, Ms. van Stigt said, meaning that nearly half of patients who were diagnosed with interval CRC already had detectable f-Hb in their prior FIT.
In a survival analysis, there was no association between interval CRC risk and sex. However, those who participated in three or four screening rounds were half as likely to be diagnosed than those who participated once or twice, and those with detectable f-Hb were five times as likely to be diagnosed.
For late-stage CRC, there was no association with sex or the number of screening rounds. Detectable f-Hb was associated with not only a higher risk of interval CRC but also a late-stage diagnosis.
“These findings indicate that one uniform age to stop screening is suboptimal,” Ms. van Stigt said. “Personalized screening strategies should, therefore, also ideally incorporate a risk-stratified age to stop screening.”
The US Preventive Services Task Force recommends that clinicians personalize screening for ages 76-85, accounting for overall health, prior screening history, and patient preferences.
“But we have no clear guidance on how to quantify or weigh these factors. This interesting study highlights how one of these factors (prior screening history) and fecal hemoglobin level (an emerging factor) are powerful stratifiers of subsequent colorectal cancer risk,” said Sameer D. Saini, MD, AGAF, director and research investigator at the VA Ann Arbor Healthcare System’s Center for Clinical Management Research. Dr. Saini wasn’t involved with the study.
At the clinical level, Dr. Saini said, sophisticated modeling is needed to understand the interaction with competing risks and identify the optimal screening strategies for patients at varying levels of cancer risk and life expectancy. Models could also help to quantify the population benefits and cost-effectiveness of personalized screening.
“Finally, it is important to note that, in many health systems, access to quantitative FIT may be limited,” he said. “These data may be less informative if colonoscopy is the primary mode of screening.”
Ms. van Stigt and Dr. Saini reported no relevant disclosures.
WASHINGTON — , according to a study presented at the annual Digestive Disease Week® (DDW).
In particular, interval CRC risk can vary substantially based on the fecal hemoglobin (f-Hb) concentration in the patient’s last fecal immunochemical test (FIT), as well as the number of prior screening rounds.
“Less is known about what happens after the upper age limit has been reached and individuals are not invited to participate in more screening rounds. This is important as life expectancy is increasing, and it is increasingly important to consider the most efficient way of screening the elderly,” said lead author Brenda van Stigt, a PhD candidate focused on cancer screening at Erasmus University Medical Center in Rotterdam, the Netherlands.
In the Netherlands, adults between ages 55 and 75 are invited to participate in stool-based CRC screening every 2 years. Based on a fecal immunochemical testing (FIT) threshold of 47 μg Hb/g, those who test positive are referred to colonoscopy, and those who test negative are invited to participate again after a 2-year period.
FIT can play a major role in risk stratification, Ms. van Stigt noted, along with other factors that influence CRC risk, such as age, sex, and CRC screening history. Although this is documented for ages 55-75, she and colleagues wanted to know more about what happens after age 75.
Ms. Van Stigt and colleagues conducted a population-based study by analyzing Dutch national cancer registry data and FIT results around the final screening at age 75, looking at those who were diagnosed with CRC within 24 months of their last negative FIT. The researchers assessed interval CRC risk and cancer stage, accounting for sex, last f-Hb concentration, and the number of screening rounds.
Among 305,761 people with a complete 24-month follow-up after a negative FIT, 661 patients were diagnosed with interval CRC, indicating an overall interval CRC risk of 21.6 per 10,000 individuals with a negative FIT. There were no significant differences by sex.
However, there were differences by screening rounds, with those who had participated in three or four screening rounds having a lower risk than those who participated only once (HR, .49).
In addition, those with detectable f-Hb (>0 μg Hb/g) in their last screening round had a much higher interval CRC risk (HR, 4.87), at 65.8 per 10,000 negative FITs, compared with 13.8 per 10,000 among those without detectable f-Hb. Interval CRC risk also increased over time for those with detectable f-Hb.
About 15% of the total population had detectable f-Hb, whereas 46% of those with interval CRC had detectable f-Hb, Ms. van Stigt said, meaning that nearly half of patients who were diagnosed with interval CRC already had detectable f-Hb in their prior FIT.
In a survival analysis, there was no association between interval CRC risk and sex. However, those who participated in three or four screening rounds were half as likely to be diagnosed than those who participated once or twice, and those with detectable f-Hb were five times as likely to be diagnosed.
For late-stage CRC, there was no association with sex or the number of screening rounds. Detectable f-Hb was associated with not only a higher risk of interval CRC but also a late-stage diagnosis.
“These findings indicate that one uniform age to stop screening is suboptimal,” Ms. van Stigt said. “Personalized screening strategies should, therefore, also ideally incorporate a risk-stratified age to stop screening.”
The US Preventive Services Task Force recommends that clinicians personalize screening for ages 76-85, accounting for overall health, prior screening history, and patient preferences.
“But we have no clear guidance on how to quantify or weigh these factors. This interesting study highlights how one of these factors (prior screening history) and fecal hemoglobin level (an emerging factor) are powerful stratifiers of subsequent colorectal cancer risk,” said Sameer D. Saini, MD, AGAF, director and research investigator at the VA Ann Arbor Healthcare System’s Center for Clinical Management Research. Dr. Saini wasn’t involved with the study.
At the clinical level, Dr. Saini said, sophisticated modeling is needed to understand the interaction with competing risks and identify the optimal screening strategies for patients at varying levels of cancer risk and life expectancy. Models could also help to quantify the population benefits and cost-effectiveness of personalized screening.
“Finally, it is important to note that, in many health systems, access to quantitative FIT may be limited,” he said. “These data may be less informative if colonoscopy is the primary mode of screening.”
Ms. van Stigt and Dr. Saini reported no relevant disclosures.
FROM DDW 2024

Statins, Vitamin D, and Exercise in Older Adults
In this article, I will review several recently published articles and guidelines relevant to the care of older adults in primary care. The articles of interest address statins for primary prevention, vitamin D supplementation and testing, and physical activity for healthy aging.
Statins for Primary Prevention of Cardiovascular Disease
A common conundrum in primary care is whether an older adult should be on a statin for primary prevention. This question has been difficult to answer because of the underrepresentation of older adults in clinical trials that examine the effect of statins for primary prevention. A recent study by Xu et al. published in Annals of Internal Medicine sought to address this gap in knowledge, investigating the risks and benefits of statins for primary prevention for older adults.1
This study stratified participants by “old” (aged 75-84 years) and “very old” (85 years or older). In this study, older adults who had an indication for statins were initiated on statins and studied over a 5-year period and compared with age-matched cohorts not initiated on statin therapy. Participants with known cardiovascular disease at baseline were excluded. The outcomes of interest were major cardiovascular disease (CVD) (a composite of myocardial infarction, stroke, or heart failure), all-cause mortality, and adverse effect of drug therapy (myopathy or liver dysfunction).
The study found that among older adults aged 75-84, initiation of statin therapy led to a 1.2% risk reduction in major CVD over a 5-year period. For older adults aged 85 and greater, initiation of statins had an even larger impact, leading to a 4.4% risk reduction in major CVD over a 5-year period. The study found that there was no significant difference in adverse effects including myopathy or liver dysfunction in both age groups.
Statins, the study suggests, are appropriate and safe to initiate for primary prevention in older adults and can lead to substantial benefits in reduction of CVD. While time to benefit was not explicitly examined in this study, a prior study by Yourman et al. suggested that the time to benefit for statins for primary prevention in adults aged 50-75 would be least 2.5 years.2
My takeaway from these findings is to discuss statin initiation for primary prevention for older patients who are focused on longevity, have good functional status (often used in geriatrics as a proxy for prognosis), and are willing to accept more medications.
Empiric Vitamin D Supplementation in Adults over 75 Years
Vitamin D is one of the most common supplements taken by older adults but evidence supporting vitamin D supplementation is variable in published literature, as most data comes from observational trials. New guidelines from the Endocrine Society focused on developing recommendations for healthy individuals with data obtained from randomized controlled trials (RCTs) and large longitudinal observational trials with comparison groups if RCTs were not available. These guidelines recommend against empiric supplementation of vitamin D for healthy adults aged 18-74, excluding pregnant women and patients with high-risk diabetes.3
For older adults aged 75 or greater, empiric vitamin D supplementation is recommended because of the possible reduction of risk in all-cause mortality in this population. Of note, this was a grade 2 recommendation by the panel, indicating that the benefits of the treatment probably outweigh the risks. The panel stated that vitamin D supplementation could be delivered through fortified foods, multivitamins with vitamin D, or as a separate vitamin D supplement.
The dosage should remain within the recommended daily allowance outlined by the Institute of Medicine of 800 IU daily for adults over 70, and the panel recommends low-dose daily vitamin D supplementation over high-dose interval supplementation. The panel noted that routine screening of vitamin D levels should not be used to guide decision-making on whether to start supplementation, but vitamin D levels should be obtained for patients who have an indication for evaluation.
The reviewers highlight that these guidelines were developed for healthy individuals and are not applicable to those with conditions that warrant vitamin D evaluation. In my clinical practice, many of my patients have bone-mineral conditions and cognitive impairment that warrant evaluation. Based on these guidelines, I will consider empiric vitamin D supplementation more often for healthy patients aged 75 and older.
Sedentary Behaviors and Healthy Aging
Engaging inactive older adults in regular physical activity can be challenging, particularly as the pandemic has led to more pervasive social isolation and affected the availability of in-person exercise activities in the community. Physical activity is a key component of healthy aging and cognition, and its benefits should be a part of routine counseling for older adults.
An interesting recent study published in JAMA Network Open by Shi et al. evaluated the association of health behaviors and aging in female US nurses over a 20-year period.4 Surveys were administered to capture time spent in each behavior, such as being sedentary (TV watching, sitting at home or at work), light activity (walking around the house or at work), and moderate to vigorous activity (walking for exercise, lawn mowing). “Healthy aging” was defined by the absence of chronic conditions such as heart failure, and lack of physical, mental, and cognitive impairment.
The study found that participants who were more sedentary were less likely to age healthfully, with each additional 2 hours of TV watching per day associated with a 12% reduction in likelihood of healthy aging. Light physical activity was associated with a significant increase in healthy aging, with a 6% increase in the likelihood of healthy aging for each additional 2 hours of light activity. Each additional 1 hour of moderate to vigorous activity was associated with a 14% increase in the likelihood of healthy aging. These findings support discussions with patients that behavior change, even in small increments, can be beneficial in healthy aging.
References
1. Xu W et al. Ann Intern Med. 2024 Jun;177(6):701-10.
2. Yourman LC et al. JAMA Intern Med. 2021;181:179-85.
3. Demay MB et al. J Clin Endocrinol Metab. August 2024;109(8):1907-47.
4. Shi H et al. JAMA Netw Open. 2024;7(6):e2416300.
In this article, I will review several recently published articles and guidelines relevant to the care of older adults in primary care. The articles of interest address statins for primary prevention, vitamin D supplementation and testing, and physical activity for healthy aging.
Statins for Primary Prevention of Cardiovascular Disease
A common conundrum in primary care is whether an older adult should be on a statin for primary prevention. This question has been difficult to answer because of the underrepresentation of older adults in clinical trials that examine the effect of statins for primary prevention. A recent study by Xu et al. published in Annals of Internal Medicine sought to address this gap in knowledge, investigating the risks and benefits of statins for primary prevention for older adults.1
This study stratified participants by “old” (aged 75-84 years) and “very old” (85 years or older). In this study, older adults who had an indication for statins were initiated on statins and studied over a 5-year period and compared with age-matched cohorts not initiated on statin therapy. Participants with known cardiovascular disease at baseline were excluded. The outcomes of interest were major cardiovascular disease (CVD) (a composite of myocardial infarction, stroke, or heart failure), all-cause mortality, and adverse effect of drug therapy (myopathy or liver dysfunction).
The study found that among older adults aged 75-84, initiation of statin therapy led to a 1.2% risk reduction in major CVD over a 5-year period. For older adults aged 85 and greater, initiation of statins had an even larger impact, leading to a 4.4% risk reduction in major CVD over a 5-year period. The study found that there was no significant difference in adverse effects including myopathy or liver dysfunction in both age groups.
Statins, the study suggests, are appropriate and safe to initiate for primary prevention in older adults and can lead to substantial benefits in reduction of CVD. While time to benefit was not explicitly examined in this study, a prior study by Yourman et al. suggested that the time to benefit for statins for primary prevention in adults aged 50-75 would be least 2.5 years.2
My takeaway from these findings is to discuss statin initiation for primary prevention for older patients who are focused on longevity, have good functional status (often used in geriatrics as a proxy for prognosis), and are willing to accept more medications.
Empiric Vitamin D Supplementation in Adults over 75 Years
Vitamin D is one of the most common supplements taken by older adults but evidence supporting vitamin D supplementation is variable in published literature, as most data comes from observational trials. New guidelines from the Endocrine Society focused on developing recommendations for healthy individuals with data obtained from randomized controlled trials (RCTs) and large longitudinal observational trials with comparison groups if RCTs were not available. These guidelines recommend against empiric supplementation of vitamin D for healthy adults aged 18-74, excluding pregnant women and patients with high-risk diabetes.3
For older adults aged 75 or greater, empiric vitamin D supplementation is recommended because of the possible reduction of risk in all-cause mortality in this population. Of note, this was a grade 2 recommendation by the panel, indicating that the benefits of the treatment probably outweigh the risks. The panel stated that vitamin D supplementation could be delivered through fortified foods, multivitamins with vitamin D, or as a separate vitamin D supplement.
The dosage should remain within the recommended daily allowance outlined by the Institute of Medicine of 800 IU daily for adults over 70, and the panel recommends low-dose daily vitamin D supplementation over high-dose interval supplementation. The panel noted that routine screening of vitamin D levels should not be used to guide decision-making on whether to start supplementation, but vitamin D levels should be obtained for patients who have an indication for evaluation.
The reviewers highlight that these guidelines were developed for healthy individuals and are not applicable to those with conditions that warrant vitamin D evaluation. In my clinical practice, many of my patients have bone-mineral conditions and cognitive impairment that warrant evaluation. Based on these guidelines, I will consider empiric vitamin D supplementation more often for healthy patients aged 75 and older.
Sedentary Behaviors and Healthy Aging
Engaging inactive older adults in regular physical activity can be challenging, particularly as the pandemic has led to more pervasive social isolation and affected the availability of in-person exercise activities in the community. Physical activity is a key component of healthy aging and cognition, and its benefits should be a part of routine counseling for older adults.
An interesting recent study published in JAMA Network Open by Shi et al. evaluated the association of health behaviors and aging in female US nurses over a 20-year period.4 Surveys were administered to capture time spent in each behavior, such as being sedentary (TV watching, sitting at home or at work), light activity (walking around the house or at work), and moderate to vigorous activity (walking for exercise, lawn mowing). “Healthy aging” was defined by the absence of chronic conditions such as heart failure, and lack of physical, mental, and cognitive impairment.
The study found that participants who were more sedentary were less likely to age healthfully, with each additional 2 hours of TV watching per day associated with a 12% reduction in likelihood of healthy aging. Light physical activity was associated with a significant increase in healthy aging, with a 6% increase in the likelihood of healthy aging for each additional 2 hours of light activity. Each additional 1 hour of moderate to vigorous activity was associated with a 14% increase in the likelihood of healthy aging. These findings support discussions with patients that behavior change, even in small increments, can be beneficial in healthy aging.
References
1. Xu W et al. Ann Intern Med. 2024 Jun;177(6):701-10.
2. Yourman LC et al. JAMA Intern Med. 2021;181:179-85.
3. Demay MB et al. J Clin Endocrinol Metab. August 2024;109(8):1907-47.
4. Shi H et al. JAMA Netw Open. 2024;7(6):e2416300.
In this article, I will review several recently published articles and guidelines relevant to the care of older adults in primary care. The articles of interest address statins for primary prevention, vitamin D supplementation and testing, and physical activity for healthy aging.
Statins for Primary Prevention of Cardiovascular Disease
A common conundrum in primary care is whether an older adult should be on a statin for primary prevention. This question has been difficult to answer because of the underrepresentation of older adults in clinical trials that examine the effect of statins for primary prevention. A recent study by Xu et al. published in Annals of Internal Medicine sought to address this gap in knowledge, investigating the risks and benefits of statins for primary prevention for older adults.1
This study stratified participants by “old” (aged 75-84 years) and “very old” (85 years or older). In this study, older adults who had an indication for statins were initiated on statins and studied over a 5-year period and compared with age-matched cohorts not initiated on statin therapy. Participants with known cardiovascular disease at baseline were excluded. The outcomes of interest were major cardiovascular disease (CVD) (a composite of myocardial infarction, stroke, or heart failure), all-cause mortality, and adverse effect of drug therapy (myopathy or liver dysfunction).
The study found that among older adults aged 75-84, initiation of statin therapy led to a 1.2% risk reduction in major CVD over a 5-year period. For older adults aged 85 and greater, initiation of statins had an even larger impact, leading to a 4.4% risk reduction in major CVD over a 5-year period. The study found that there was no significant difference in adverse effects including myopathy or liver dysfunction in both age groups.
Statins, the study suggests, are appropriate and safe to initiate for primary prevention in older adults and can lead to substantial benefits in reduction of CVD. While time to benefit was not explicitly examined in this study, a prior study by Yourman et al. suggested that the time to benefit for statins for primary prevention in adults aged 50-75 would be least 2.5 years.2
My takeaway from these findings is to discuss statin initiation for primary prevention for older patients who are focused on longevity, have good functional status (often used in geriatrics as a proxy for prognosis), and are willing to accept more medications.
Empiric Vitamin D Supplementation in Adults over 75 Years
Vitamin D is one of the most common supplements taken by older adults but evidence supporting vitamin D supplementation is variable in published literature, as most data comes from observational trials. New guidelines from the Endocrine Society focused on developing recommendations for healthy individuals with data obtained from randomized controlled trials (RCTs) and large longitudinal observational trials with comparison groups if RCTs were not available. These guidelines recommend against empiric supplementation of vitamin D for healthy adults aged 18-74, excluding pregnant women and patients with high-risk diabetes.3
For older adults aged 75 or greater, empiric vitamin D supplementation is recommended because of the possible reduction of risk in all-cause mortality in this population. Of note, this was a grade 2 recommendation by the panel, indicating that the benefits of the treatment probably outweigh the risks. The panel stated that vitamin D supplementation could be delivered through fortified foods, multivitamins with vitamin D, or as a separate vitamin D supplement.
The dosage should remain within the recommended daily allowance outlined by the Institute of Medicine of 800 IU daily for adults over 70, and the panel recommends low-dose daily vitamin D supplementation over high-dose interval supplementation. The panel noted that routine screening of vitamin D levels should not be used to guide decision-making on whether to start supplementation, but vitamin D levels should be obtained for patients who have an indication for evaluation.
The reviewers highlight that these guidelines were developed for healthy individuals and are not applicable to those with conditions that warrant vitamin D evaluation. In my clinical practice, many of my patients have bone-mineral conditions and cognitive impairment that warrant evaluation. Based on these guidelines, I will consider empiric vitamin D supplementation more often for healthy patients aged 75 and older.
Sedentary Behaviors and Healthy Aging
Engaging inactive older adults in regular physical activity can be challenging, particularly as the pandemic has led to more pervasive social isolation and affected the availability of in-person exercise activities in the community. Physical activity is a key component of healthy aging and cognition, and its benefits should be a part of routine counseling for older adults.
An interesting recent study published in JAMA Network Open by Shi et al. evaluated the association of health behaviors and aging in female US nurses over a 20-year period.4 Surveys were administered to capture time spent in each behavior, such as being sedentary (TV watching, sitting at home or at work), light activity (walking around the house or at work), and moderate to vigorous activity (walking for exercise, lawn mowing). “Healthy aging” was defined by the absence of chronic conditions such as heart failure, and lack of physical, mental, and cognitive impairment.
The study found that participants who were more sedentary were less likely to age healthfully, with each additional 2 hours of TV watching per day associated with a 12% reduction in likelihood of healthy aging. Light physical activity was associated with a significant increase in healthy aging, with a 6% increase in the likelihood of healthy aging for each additional 2 hours of light activity. Each additional 1 hour of moderate to vigorous activity was associated with a 14% increase in the likelihood of healthy aging. These findings support discussions with patients that behavior change, even in small increments, can be beneficial in healthy aging.
References
1. Xu W et al. Ann Intern Med. 2024 Jun;177(6):701-10.
2. Yourman LC et al. JAMA Intern Med. 2021;181:179-85.
3. Demay MB et al. J Clin Endocrinol Metab. August 2024;109(8):1907-47.
4. Shi H et al. JAMA Netw Open. 2024;7(6):e2416300.