User login
What Are the Best Treatments for Bladder Dysfunction in MS?
LONDON—Many patients with multiple sclerosis (MS) suffer from bladder dysfunction, however, few of these patients have their symptoms managed, according to an overview presented at the 32nd Congress of the European Committee for Treatment and Research in MS.
If a patient reports bladder symptoms, most often those bladder symptoms will persist and increase, said Jalesh Panicker, MD, DM, Consultant Neurologist and Clinical Lead in Uroneurology at the University College London Institute of Neurology in Queen Square, London. Symptoms of urinary frequency, urgency, incontinence, and nocturia, which are collectively called overactive bladder syndrome, increase with increased duration of MS and tend to correlate with the neurologic disability, especially lower limb parameter weakness.

In a large survey of 3,000 patients with MS, 90% rated bladder complications as one of the top five common symptomatic problems. Seventy percent of the group reported that bladder issues had a moderate or high impact on them. Only one-third of patients surveyed reported improvement after starting disease-modifying therapies.
Bladder symptoms in MS occur, on average, six years into the illness. Lower urinary tract symptoms are present in 10% of patients at first diagnosis. To help address this issue, the Actionable eight-item questionnaire was developed to help make it easier for doctors to assess bladder problems, said Dr. Panicker.
Lesion location plays a significant role in urinary tract dysfunction. Patients with suprapontine lesions present predominantly with overactive bladder, whereas patients with spinal lesions tend to report voiding symptoms such as hesitancy and poor stream. Plaques in the cerebral cord can also alter the pattern of lower urinary tract dysfunction.
Measuring post-void residue is critical for bladder dysfunction management. Unlike in urgency and incontinence, patients tend to have difficulties expressing voiding and voiding dysfunction symptoms. A high post-void residue can cause urinary tract infections and lead to devastating effects on patients, Dr. Panicker said. Using a bladder scanner to detect high post-void residuals may help reduce urinary tract infections.
Oral Agents
The three most commonly used treatments for bladder dysfunction in patients with MS are oral agents, botulinum toxin, and neuromodulation. Oral antimuscarinics work through the muscarinic receptors of the bladder, reducing bladder sensation and intrusive pressures, inhibiting detrusor overactivity, and improving bladder capacity. Oxybutynin, for example, can cause dry mouth and dry eyes, whereas newer antimuscarinics such as tolteradine, solifenancin, or fesoteradine are less likely to cause dry eyes, dry mouth, or constipation, Dr. Panicker noted.
Anticholinergic burden refers to the cumulative effect of multiple medications with anticholinergic properties. The higher the anticholinergic burden, the greater the risk of cognitive impairment, impaired functional performance, mortality, and brain atrophy. The Anticholinergic Burden Scale (ACB) grades medications according to their anticholinergic burden—mild (ACB1), moderate (ACB2), and severe (ACB3). Total ACB score is the sum of each medication the patient is on; any ACB score over 3 is considered clinically relevant.
Beta 3 agonists are another class of oral agents used to control bladder symptoms. Beta 3 agonists inhibit intrusive muscle contractions and increase relaxation by acting on the beta 3 receptors present on the wall of the bladder. Unlike antimuscarinics, beta 3 agonists are devoid of side effects like dry mouth, dry eyes, and constipation; however, these oral agents are associated with cardiovascular side effects. Currently, there are pivotal phase III studies on the efficacy of mirabegron in the UK, however, there is limited evidence from neurologic patients and there is a need for more long-term data, Dr. Panicker said.
Botulinum Toxin
Botulinum toxin is effective for bladder management. Evidence shows that patients with MS experienced significant improvement in urinary incontinence, frequency, and urgency as early as four weeks after injections, and this beneficial effect is not lost with repeated injections. Three pivotal phase III studies have demonstrated the efficacy of onabotulinumtoxinA for improving incontinence. Studies suggest that onabotulinumtoxinA significantly improved incontinence at week 12. The median duration of retreatment was 42 weeks.
Neuromodulation
Stimulating the tibial nerve is beneficial in managing overactive bladder symptoms such as urinary frequency and incontinence. Percutaneous tibial nerve stimulation is one of the few treatments that does not tend to worsen voiding dysfunction. It is also potentially a treatment for patients who are retaining urine or experiencing incomplete bladder emptying.
“With several disease-modifying therapies over the last few years, it is clear that the number of MS relapses in patients is coming down. There is evidence to suggest that the neurologic disability that accumulates is also either halted or possibly even reversed. Whether the nonmotor symptoms of MS, such as urinary tract dysfunction, also would get halted or reversed with these treatments is an area for further research,” said Dr. Panicker.
—Erica Tricarico
Suggested Reading
Ginsberg D, Gousse A, Keppenne V, et al. Phase 3 efficacy and tolerability study of onabotulinumtoxinA for urinary incontinence from neurogenic detrusor overactivity. J Urol. 2012;187(6):2131-2139.
Jongen PJ, Blok BF, Heesakkers JP, et al. Simplified scoring of the Actionable 8-item screening questionnaire for neurogenic bladder overactivity in multiple sclerosis: a comparative analysis of test performance at different cut-off points. BMC Urol. 2015;15:106.
Wintner A, Kim MM, Bechis SK, Kreydin El. Voiding dysfunction in multiple sclerosis. Semin Neurol. 2016;36(1):34-40.
LONDON—Many patients with multiple sclerosis (MS) suffer from bladder dysfunction, however, few of these patients have their symptoms managed, according to an overview presented at the 32nd Congress of the European Committee for Treatment and Research in MS.
If a patient reports bladder symptoms, most often those bladder symptoms will persist and increase, said Jalesh Panicker, MD, DM, Consultant Neurologist and Clinical Lead in Uroneurology at the University College London Institute of Neurology in Queen Square, London. Symptoms of urinary frequency, urgency, incontinence, and nocturia, which are collectively called overactive bladder syndrome, increase with increased duration of MS and tend to correlate with the neurologic disability, especially lower limb parameter weakness.
In a large survey of 3,000 patients with MS, 90% rated bladder complications as one of the top five common symptomatic problems. Seventy percent of the group reported that bladder issues had a moderate or high impact on them. Only one-third of patients surveyed reported improvement after starting disease-modifying therapies.
Bladder symptoms in MS occur, on average, six years into the illness. Lower urinary tract symptoms are present in 10% of patients at first diagnosis. To help address this issue, the Actionable eight-item questionnaire was developed to help make it easier for doctors to assess bladder problems, said Dr. Panicker.
Lesion location plays a significant role in urinary tract dysfunction. Patients with suprapontine lesions present predominantly with overactive bladder, whereas patients with spinal lesions tend to report voiding symptoms such as hesitancy and poor stream. Plaques in the cerebral cord can also alter the pattern of lower urinary tract dysfunction.
Measuring post-void residue is critical for bladder dysfunction management. Unlike in urgency and incontinence, patients tend to have difficulties expressing voiding and voiding dysfunction symptoms. A high post-void residue can cause urinary tract infections and lead to devastating effects on patients, Dr. Panicker said. Using a bladder scanner to detect high post-void residuals may help reduce urinary tract infections.
Oral Agents
The three most commonly used treatments for bladder dysfunction in patients with MS are oral agents, botulinum toxin, and neuromodulation. Oral antimuscarinics work through the muscarinic receptors of the bladder, reducing bladder sensation and intrusive pressures, inhibiting detrusor overactivity, and improving bladder capacity. Oxybutynin, for example, can cause dry mouth and dry eyes, whereas newer antimuscarinics such as tolteradine, solifenancin, or fesoteradine are less likely to cause dry eyes, dry mouth, or constipation, Dr. Panicker noted.
Anticholinergic burden refers to the cumulative effect of multiple medications with anticholinergic properties. The higher the anticholinergic burden, the greater the risk of cognitive impairment, impaired functional performance, mortality, and brain atrophy. The Anticholinergic Burden Scale (ACB) grades medications according to their anticholinergic burden—mild (ACB1), moderate (ACB2), and severe (ACB3). Total ACB score is the sum of each medication the patient is on; any ACB score over 3 is considered clinically relevant.
Beta 3 agonists are another class of oral agents used to control bladder symptoms. Beta 3 agonists inhibit intrusive muscle contractions and increase relaxation by acting on the beta 3 receptors present on the wall of the bladder. Unlike antimuscarinics, beta 3 agonists are devoid of side effects like dry mouth, dry eyes, and constipation; however, these oral agents are associated with cardiovascular side effects. Currently, there are pivotal phase III studies on the efficacy of mirabegron in the UK, however, there is limited evidence from neurologic patients and there is a need for more long-term data, Dr. Panicker said.
Botulinum Toxin
Botulinum toxin is effective for bladder management. Evidence shows that patients with MS experienced significant improvement in urinary incontinence, frequency, and urgency as early as four weeks after injections, and this beneficial effect is not lost with repeated injections. Three pivotal phase III studies have demonstrated the efficacy of onabotulinumtoxinA for improving incontinence. Studies suggest that onabotulinumtoxinA significantly improved incontinence at week 12. The median duration of retreatment was 42 weeks.
Neuromodulation
Stimulating the tibial nerve is beneficial in managing overactive bladder symptoms such as urinary frequency and incontinence. Percutaneous tibial nerve stimulation is one of the few treatments that does not tend to worsen voiding dysfunction. It is also potentially a treatment for patients who are retaining urine or experiencing incomplete bladder emptying.
“With several disease-modifying therapies over the last few years, it is clear that the number of MS relapses in patients is coming down. There is evidence to suggest that the neurologic disability that accumulates is also either halted or possibly even reversed. Whether the nonmotor symptoms of MS, such as urinary tract dysfunction, also would get halted or reversed with these treatments is an area for further research,” said Dr. Panicker.
—Erica Tricarico
Suggested Reading
Ginsberg D, Gousse A, Keppenne V, et al. Phase 3 efficacy and tolerability study of onabotulinumtoxinA for urinary incontinence from neurogenic detrusor overactivity. J Urol. 2012;187(6):2131-2139.
Jongen PJ, Blok BF, Heesakkers JP, et al. Simplified scoring of the Actionable 8-item screening questionnaire for neurogenic bladder overactivity in multiple sclerosis: a comparative analysis of test performance at different cut-off points. BMC Urol. 2015;15:106.
Wintner A, Kim MM, Bechis SK, Kreydin El. Voiding dysfunction in multiple sclerosis. Semin Neurol. 2016;36(1):34-40.
LONDON—Many patients with multiple sclerosis (MS) suffer from bladder dysfunction, however, few of these patients have their symptoms managed, according to an overview presented at the 32nd Congress of the European Committee for Treatment and Research in MS.
If a patient reports bladder symptoms, most often those bladder symptoms will persist and increase, said Jalesh Panicker, MD, DM, Consultant Neurologist and Clinical Lead in Uroneurology at the University College London Institute of Neurology in Queen Square, London. Symptoms of urinary frequency, urgency, incontinence, and nocturia, which are collectively called overactive bladder syndrome, increase with increased duration of MS and tend to correlate with the neurologic disability, especially lower limb parameter weakness.
In a large survey of 3,000 patients with MS, 90% rated bladder complications as one of the top five common symptomatic problems. Seventy percent of the group reported that bladder issues had a moderate or high impact on them. Only one-third of patients surveyed reported improvement after starting disease-modifying therapies.
Bladder symptoms in MS occur, on average, six years into the illness. Lower urinary tract symptoms are present in 10% of patients at first diagnosis. To help address this issue, the Actionable eight-item questionnaire was developed to help make it easier for doctors to assess bladder problems, said Dr. Panicker.
Lesion location plays a significant role in urinary tract dysfunction. Patients with suprapontine lesions present predominantly with overactive bladder, whereas patients with spinal lesions tend to report voiding symptoms such as hesitancy and poor stream. Plaques in the cerebral cord can also alter the pattern of lower urinary tract dysfunction.
Measuring post-void residue is critical for bladder dysfunction management. Unlike in urgency and incontinence, patients tend to have difficulties expressing voiding and voiding dysfunction symptoms. A high post-void residue can cause urinary tract infections and lead to devastating effects on patients, Dr. Panicker said. Using a bladder scanner to detect high post-void residuals may help reduce urinary tract infections.
Oral Agents
The three most commonly used treatments for bladder dysfunction in patients with MS are oral agents, botulinum toxin, and neuromodulation. Oral antimuscarinics work through the muscarinic receptors of the bladder, reducing bladder sensation and intrusive pressures, inhibiting detrusor overactivity, and improving bladder capacity. Oxybutynin, for example, can cause dry mouth and dry eyes, whereas newer antimuscarinics such as tolteradine, solifenancin, or fesoteradine are less likely to cause dry eyes, dry mouth, or constipation, Dr. Panicker noted.
Anticholinergic burden refers to the cumulative effect of multiple medications with anticholinergic properties. The higher the anticholinergic burden, the greater the risk of cognitive impairment, impaired functional performance, mortality, and brain atrophy. The Anticholinergic Burden Scale (ACB) grades medications according to their anticholinergic burden—mild (ACB1), moderate (ACB2), and severe (ACB3). Total ACB score is the sum of each medication the patient is on; any ACB score over 3 is considered clinically relevant.
Beta 3 agonists are another class of oral agents used to control bladder symptoms. Beta 3 agonists inhibit intrusive muscle contractions and increase relaxation by acting on the beta 3 receptors present on the wall of the bladder. Unlike antimuscarinics, beta 3 agonists are devoid of side effects like dry mouth, dry eyes, and constipation; however, these oral agents are associated with cardiovascular side effects. Currently, there are pivotal phase III studies on the efficacy of mirabegron in the UK, however, there is limited evidence from neurologic patients and there is a need for more long-term data, Dr. Panicker said.
Botulinum Toxin
Botulinum toxin is effective for bladder management. Evidence shows that patients with MS experienced significant improvement in urinary incontinence, frequency, and urgency as early as four weeks after injections, and this beneficial effect is not lost with repeated injections. Three pivotal phase III studies have demonstrated the efficacy of onabotulinumtoxinA for improving incontinence. Studies suggest that onabotulinumtoxinA significantly improved incontinence at week 12. The median duration of retreatment was 42 weeks.
Neuromodulation
Stimulating the tibial nerve is beneficial in managing overactive bladder symptoms such as urinary frequency and incontinence. Percutaneous tibial nerve stimulation is one of the few treatments that does not tend to worsen voiding dysfunction. It is also potentially a treatment for patients who are retaining urine or experiencing incomplete bladder emptying.
“With several disease-modifying therapies over the last few years, it is clear that the number of MS relapses in patients is coming down. There is evidence to suggest that the neurologic disability that accumulates is also either halted or possibly even reversed. Whether the nonmotor symptoms of MS, such as urinary tract dysfunction, also would get halted or reversed with these treatments is an area for further research,” said Dr. Panicker.
—Erica Tricarico
Suggested Reading
Ginsberg D, Gousse A, Keppenne V, et al. Phase 3 efficacy and tolerability study of onabotulinumtoxinA for urinary incontinence from neurogenic detrusor overactivity. J Urol. 2012;187(6):2131-2139.
Jongen PJ, Blok BF, Heesakkers JP, et al. Simplified scoring of the Actionable 8-item screening questionnaire for neurogenic bladder overactivity in multiple sclerosis: a comparative analysis of test performance at different cut-off points. BMC Urol. 2015;15:106.
Wintner A, Kim MM, Bechis SK, Kreydin El. Voiding dysfunction in multiple sclerosis. Semin Neurol. 2016;36(1):34-40.
Potential Weaknesses in Major Trial Explored in Reevaluating CPAP Benefits
LAS VEGAS—Although continuous positive airway pressure (CPAP) failed to protect patients with obstructive sleep apnea from cardiovascular events in a recently completed large randomized trial, the results are not definitive, according to Alan Z. Segal, MD, Associate Professor of Clinical Neurology at Weill Cornell Medical College in New York City. In an update on sleep disorders at the American Academy of Neurology’s Fall 2016 Conference, Dr. Segal explained that the average adherence to CPAP in this trial, called SAVE, was less than that which has been widely accepted as adequate.

“Medicare has a requirement that CPAP be used for more than four hours on 70% of nights in a 30-day period for patients to keep their device,” observed Dr. Segal, providing one example of a way in which four hours or more of CPAP has been defined as a minimum duration. In the SAVE study by McEvoy et al, the mean duration of CPAP was 3.3 hours. Only 42% of patients met the four-hour definition of adherence, which was the duration the authors themselves had defined as good adherence.
“Our field was very much affected by this study,” Dr. Segal acknowledged. “It created quite a lot of controversy and criticisms over methodologic flaws. Like any study that does not produce the results we want, we start to pick at it to identify where it might have been inadequate, and the biggest inadequacy of this particular study is that patients in the CPAP arm may not have been adequately treated.”
Adherence Versus Effectiveness
In this study, 2,717 patients with moderate-to-severe obstructive sleep apnea and a history of coronary or cerebrovascular disease were randomized to CPAP plus usual care or usual care alone. The primary composite end point of the trial was death from cardiovascular causes, myocardial infarction, stroke, or hospitalization for unstable angina, heart failure, or transient ischemic attack.
After a mean follow-up of 3.7 years, an event had occurred in 17% of those in the CPAP group versus 15.4% of those in the usual-care group, producing a nonsignificantly increased hazard ratio (HR 1.10) for the primary end point in the CPAP arm. CPAP did reduce snoring and daytime sleepiness as well as improve quality of life and mood, all of which were secondary end points.
“The natural next step would be to look at the subgroup of patients who did have good adherence to CPAP, but the study was not powered to look at that,” Dr. Segal said. Although he reported that a post hoc analysis did show “a trend for benefit in those patients who actually adhered to the therapy,” these data are not sufficient to challenge the primary conclusion.
There was a reasonable expectation that CPAP, which has been an effective treatment in this and other trials for the symptoms of obstructive sleep apnea, would reduce cardiovascular events, according to the authors of the SAVE study. They cited strong evidence that obstructive sleep apnea increases the risk of stroke and other cardiovascular events, so control of the disorder had the potential to be protective. Moreover, CPAP has been specifically associated with improved endothelial function, improved insulin sensitivity, and reduced systolic blood pressure in patients with hypertension.
Study Flaw or Real-World Experience?
Yet judging CPAP on an intention-to-treat basis may be appropriate, because several other trials cited by the authors of SAVE, all of which were also negative for a cardiovascular benefit, have also found median adherence to be less than four hours. In SAVE, the mean adherence was 4.4 hours per night during the first 12 months of the trial, but it fell subsequently. As a reflection of what can be expected from a prescription of CPAP, the lack of protection from cardiovascular events in SAVE may not reflect a fundamental inability of CPAP to protect against vascular events, but the negative result is relevant if typical use means low adherence.
“CPAP adherence is a challenge,” Dr. Segal acknowledged. “In a dedicated sleep center such as ours, we work closely with patients to help them with adherence. One of the ways we do that is through mask fittings to find the style with which the patient is most comfortable.”
Mask styles range from minimalist devices that fit the nostrils to full “fighter-pilot” devices fitting over the entire face, according to Dr. Segal. He estimated that there are about 30 mask styles now available, and added that considerable time is devoted at his center to helping patients make the best choice.
Adherence may also be improved using modern devices, according to Dr. Segal. One example is software that ramps up pressure slowly so that the target pressure is not reached until the patient is already asleep, making use more acceptable. Another is technology that reduces pressure at each exhalation, facilitating a more natural breathing cycle and also increasing patient acceptance. Newer machines equipped with automated titration may also add convenience and ease of use.
Significant Benefit to Be Gained
The rewards of CPAP in patients who become comfortable with this therapy can be significant. The impairments in quality of sleep in patients with obstructive sleep apnea are often the impetus to seek care. “It is very important that patients get that subjective experience of feeling better the next day, and I would say about one-third of my patients get that next-day eureka feeling from initiating CPAP after sleeping so poorly for many years,” Dr. Segal reported.
These quality-of-life benefits were reflected in the SAVE study. For those in the CPAP arm, relative to those receiving usual care alone, there were highly significant improvements in objective anxiety and depression scales, as well as the physical and mental components of the 36-item Short Form (SF-36) quality of life tool. A reduction in daytime symptoms of sleep deprivation was also significant for CPAP on the Epworth Sleepiness Scale.
Relative to other tools to treat obstructive sleep apnea, CPAP is typically the most attractive option, according to Dr. Segal. For example, he is not an advocate of oral devices that advance the mandible, which he said have never been shown to be effective in moderate-to-severe apnea. He also suggested that surgery, which can be painful, does not uniformly provide benefit. However, he acknowledged that engaging patients in CPAP who do not experience a symptomatic benefit may now be more difficult after the results of the SAVE study. Although “the jury is still out” in regard to the ability of CPAP to reduce cardiovascular events in adherent patients, the evidence has now raised doubts for those already prescribed CPAP for the goal of reducing cardiovascular risk.“Those patients who are dedicated CPAP users will keep using this, but those who are perhaps looking for an excuse not to use CPAP may look at this data in a different way,” Dr. Segal said.
—Theodore Bosworth
Suggested Reading
McEvoy RD, Antic NA, Heeley E, et al. CPAP for prevention of cardiovascular events in obstructive sleep apnea. N Engl J Med. 2016;375(10):919-931.
LAS VEGAS—Although continuous positive airway pressure (CPAP) failed to protect patients with obstructive sleep apnea from cardiovascular events in a recently completed large randomized trial, the results are not definitive, according to Alan Z. Segal, MD, Associate Professor of Clinical Neurology at Weill Cornell Medical College in New York City. In an update on sleep disorders at the American Academy of Neurology’s Fall 2016 Conference, Dr. Segal explained that the average adherence to CPAP in this trial, called SAVE, was less than that which has been widely accepted as adequate.
“Medicare has a requirement that CPAP be used for more than four hours on 70% of nights in a 30-day period for patients to keep their device,” observed Dr. Segal, providing one example of a way in which four hours or more of CPAP has been defined as a minimum duration. In the SAVE study by McEvoy et al, the mean duration of CPAP was 3.3 hours. Only 42% of patients met the four-hour definition of adherence, which was the duration the authors themselves had defined as good adherence.
“Our field was very much affected by this study,” Dr. Segal acknowledged. “It created quite a lot of controversy and criticisms over methodologic flaws. Like any study that does not produce the results we want, we start to pick at it to identify where it might have been inadequate, and the biggest inadequacy of this particular study is that patients in the CPAP arm may not have been adequately treated.”
Adherence Versus Effectiveness
In this study, 2,717 patients with moderate-to-severe obstructive sleep apnea and a history of coronary or cerebrovascular disease were randomized to CPAP plus usual care or usual care alone. The primary composite end point of the trial was death from cardiovascular causes, myocardial infarction, stroke, or hospitalization for unstable angina, heart failure, or transient ischemic attack.
After a mean follow-up of 3.7 years, an event had occurred in 17% of those in the CPAP group versus 15.4% of those in the usual-care group, producing a nonsignificantly increased hazard ratio (HR 1.10) for the primary end point in the CPAP arm. CPAP did reduce snoring and daytime sleepiness as well as improve quality of life and mood, all of which were secondary end points.
“The natural next step would be to look at the subgroup of patients who did have good adherence to CPAP, but the study was not powered to look at that,” Dr. Segal said. Although he reported that a post hoc analysis did show “a trend for benefit in those patients who actually adhered to the therapy,” these data are not sufficient to challenge the primary conclusion.
There was a reasonable expectation that CPAP, which has been an effective treatment in this and other trials for the symptoms of obstructive sleep apnea, would reduce cardiovascular events, according to the authors of the SAVE study. They cited strong evidence that obstructive sleep apnea increases the risk of stroke and other cardiovascular events, so control of the disorder had the potential to be protective. Moreover, CPAP has been specifically associated with improved endothelial function, improved insulin sensitivity, and reduced systolic blood pressure in patients with hypertension.
Study Flaw or Real-World Experience?
Yet judging CPAP on an intention-to-treat basis may be appropriate, because several other trials cited by the authors of SAVE, all of which were also negative for a cardiovascular benefit, have also found median adherence to be less than four hours. In SAVE, the mean adherence was 4.4 hours per night during the first 12 months of the trial, but it fell subsequently. As a reflection of what can be expected from a prescription of CPAP, the lack of protection from cardiovascular events in SAVE may not reflect a fundamental inability of CPAP to protect against vascular events, but the negative result is relevant if typical use means low adherence.
“CPAP adherence is a challenge,” Dr. Segal acknowledged. “In a dedicated sleep center such as ours, we work closely with patients to help them with adherence. One of the ways we do that is through mask fittings to find the style with which the patient is most comfortable.”
Mask styles range from minimalist devices that fit the nostrils to full “fighter-pilot” devices fitting over the entire face, according to Dr. Segal. He estimated that there are about 30 mask styles now available, and added that considerable time is devoted at his center to helping patients make the best choice.
Adherence may also be improved using modern devices, according to Dr. Segal. One example is software that ramps up pressure slowly so that the target pressure is not reached until the patient is already asleep, making use more acceptable. Another is technology that reduces pressure at each exhalation, facilitating a more natural breathing cycle and also increasing patient acceptance. Newer machines equipped with automated titration may also add convenience and ease of use.
Significant Benefit to Be Gained
The rewards of CPAP in patients who become comfortable with this therapy can be significant. The impairments in quality of sleep in patients with obstructive sleep apnea are often the impetus to seek care. “It is very important that patients get that subjective experience of feeling better the next day, and I would say about one-third of my patients get that next-day eureka feeling from initiating CPAP after sleeping so poorly for many years,” Dr. Segal reported.
These quality-of-life benefits were reflected in the SAVE study. For those in the CPAP arm, relative to those receiving usual care alone, there were highly significant improvements in objective anxiety and depression scales, as well as the physical and mental components of the 36-item Short Form (SF-36) quality of life tool. A reduction in daytime symptoms of sleep deprivation was also significant for CPAP on the Epworth Sleepiness Scale.
Relative to other tools to treat obstructive sleep apnea, CPAP is typically the most attractive option, according to Dr. Segal. For example, he is not an advocate of oral devices that advance the mandible, which he said have never been shown to be effective in moderate-to-severe apnea. He also suggested that surgery, which can be painful, does not uniformly provide benefit. However, he acknowledged that engaging patients in CPAP who do not experience a symptomatic benefit may now be more difficult after the results of the SAVE study. Although “the jury is still out” in regard to the ability of CPAP to reduce cardiovascular events in adherent patients, the evidence has now raised doubts for those already prescribed CPAP for the goal of reducing cardiovascular risk.“Those patients who are dedicated CPAP users will keep using this, but those who are perhaps looking for an excuse not to use CPAP may look at this data in a different way,” Dr. Segal said.
—Theodore Bosworth
Suggested Reading
McEvoy RD, Antic NA, Heeley E, et al. CPAP for prevention of cardiovascular events in obstructive sleep apnea. N Engl J Med. 2016;375(10):919-931.
LAS VEGAS—Although continuous positive airway pressure (CPAP) failed to protect patients with obstructive sleep apnea from cardiovascular events in a recently completed large randomized trial, the results are not definitive, according to Alan Z. Segal, MD, Associate Professor of Clinical Neurology at Weill Cornell Medical College in New York City. In an update on sleep disorders at the American Academy of Neurology’s Fall 2016 Conference, Dr. Segal explained that the average adherence to CPAP in this trial, called SAVE, was less than that which has been widely accepted as adequate.
“Medicare has a requirement that CPAP be used for more than four hours on 70% of nights in a 30-day period for patients to keep their device,” observed Dr. Segal, providing one example of a way in which four hours or more of CPAP has been defined as a minimum duration. In the SAVE study by McEvoy et al, the mean duration of CPAP was 3.3 hours. Only 42% of patients met the four-hour definition of adherence, which was the duration the authors themselves had defined as good adherence.
“Our field was very much affected by this study,” Dr. Segal acknowledged. “It created quite a lot of controversy and criticisms over methodologic flaws. Like any study that does not produce the results we want, we start to pick at it to identify where it might have been inadequate, and the biggest inadequacy of this particular study is that patients in the CPAP arm may not have been adequately treated.”
Adherence Versus Effectiveness
In this study, 2,717 patients with moderate-to-severe obstructive sleep apnea and a history of coronary or cerebrovascular disease were randomized to CPAP plus usual care or usual care alone. The primary composite end point of the trial was death from cardiovascular causes, myocardial infarction, stroke, or hospitalization for unstable angina, heart failure, or transient ischemic attack.
After a mean follow-up of 3.7 years, an event had occurred in 17% of those in the CPAP group versus 15.4% of those in the usual-care group, producing a nonsignificantly increased hazard ratio (HR 1.10) for the primary end point in the CPAP arm. CPAP did reduce snoring and daytime sleepiness as well as improve quality of life and mood, all of which were secondary end points.
“The natural next step would be to look at the subgroup of patients who did have good adherence to CPAP, but the study was not powered to look at that,” Dr. Segal said. Although he reported that a post hoc analysis did show “a trend for benefit in those patients who actually adhered to the therapy,” these data are not sufficient to challenge the primary conclusion.
There was a reasonable expectation that CPAP, which has been an effective treatment in this and other trials for the symptoms of obstructive sleep apnea, would reduce cardiovascular events, according to the authors of the SAVE study. They cited strong evidence that obstructive sleep apnea increases the risk of stroke and other cardiovascular events, so control of the disorder had the potential to be protective. Moreover, CPAP has been specifically associated with improved endothelial function, improved insulin sensitivity, and reduced systolic blood pressure in patients with hypertension.
Study Flaw or Real-World Experience?
Yet judging CPAP on an intention-to-treat basis may be appropriate, because several other trials cited by the authors of SAVE, all of which were also negative for a cardiovascular benefit, have also found median adherence to be less than four hours. In SAVE, the mean adherence was 4.4 hours per night during the first 12 months of the trial, but it fell subsequently. As a reflection of what can be expected from a prescription of CPAP, the lack of protection from cardiovascular events in SAVE may not reflect a fundamental inability of CPAP to protect against vascular events, but the negative result is relevant if typical use means low adherence.
“CPAP adherence is a challenge,” Dr. Segal acknowledged. “In a dedicated sleep center such as ours, we work closely with patients to help them with adherence. One of the ways we do that is through mask fittings to find the style with which the patient is most comfortable.”
Mask styles range from minimalist devices that fit the nostrils to full “fighter-pilot” devices fitting over the entire face, according to Dr. Segal. He estimated that there are about 30 mask styles now available, and added that considerable time is devoted at his center to helping patients make the best choice.
Adherence may also be improved using modern devices, according to Dr. Segal. One example is software that ramps up pressure slowly so that the target pressure is not reached until the patient is already asleep, making use more acceptable. Another is technology that reduces pressure at each exhalation, facilitating a more natural breathing cycle and also increasing patient acceptance. Newer machines equipped with automated titration may also add convenience and ease of use.
Significant Benefit to Be Gained
The rewards of CPAP in patients who become comfortable with this therapy can be significant. The impairments in quality of sleep in patients with obstructive sleep apnea are often the impetus to seek care. “It is very important that patients get that subjective experience of feeling better the next day, and I would say about one-third of my patients get that next-day eureka feeling from initiating CPAP after sleeping so poorly for many years,” Dr. Segal reported.
These quality-of-life benefits were reflected in the SAVE study. For those in the CPAP arm, relative to those receiving usual care alone, there were highly significant improvements in objective anxiety and depression scales, as well as the physical and mental components of the 36-item Short Form (SF-36) quality of life tool. A reduction in daytime symptoms of sleep deprivation was also significant for CPAP on the Epworth Sleepiness Scale.
Relative to other tools to treat obstructive sleep apnea, CPAP is typically the most attractive option, according to Dr. Segal. For example, he is not an advocate of oral devices that advance the mandible, which he said have never been shown to be effective in moderate-to-severe apnea. He also suggested that surgery, which can be painful, does not uniformly provide benefit. However, he acknowledged that engaging patients in CPAP who do not experience a symptomatic benefit may now be more difficult after the results of the SAVE study. Although “the jury is still out” in regard to the ability of CPAP to reduce cardiovascular events in adherent patients, the evidence has now raised doubts for those already prescribed CPAP for the goal of reducing cardiovascular risk.“Those patients who are dedicated CPAP users will keep using this, but those who are perhaps looking for an excuse not to use CPAP may look at this data in a different way,” Dr. Segal said.
—Theodore Bosworth
Suggested Reading
McEvoy RD, Antic NA, Heeley E, et al. CPAP for prevention of cardiovascular events in obstructive sleep apnea. N Engl J Med. 2016;375(10):919-931.

Patient Satisfaction: Within Arm’s Reach, or Bending Over Backward?
In our December 2016 issue, we reported the results of our first annual survey on nonmonetary compensation (ie, the “perks”) and overall employment satisfaction (Clinician Reviews. 2016;26[12]:23-26). But the feedback I found most interesting came from the narrative responses—particularly those referencing patient satisfaction and the stress it creates for NPs and PAs.
Safety, quality, and affordability ha
Judging by the verbatim responses to our survey, NPs and PAs are concerned that quality measures don’t reflect the demands of our practice or focus on what matters to our patients.
One participant analogized, “Medicine is now like McDonalds or Burger King—patients want it their way, regardless of whether it’s in their best interest. I was fee-for-service for more than 10 years. As reimbursements have decreased significantly over time, I’m now employed by a hospital. I have become a waitress, considering my patients’ wishes—not for the benefit of their health, but to meet their more trivial ‘needs.’ These requirements can be as absurd as a specific brand of sweetener! If patients’ preferred sugar substitutes aren’t offered at my hospital, their ‘satisfaction’ may drop and I won’t get reimbursed as much. It’s a miserable experience.”
Perhaps the disparate views of what matters—Is it the softness of the pillows, or is it measurable improvement in the patient’s condition?—is the origin of the stress expressed by clinicians. This dissonance, in my opinion, exists among all involved—providers, patients, and payers. Today, patients see themselves as buyers of health services, and health care corporations have begun to function as a service industry. It may also explain why the concept of patient satisfaction has seemingly morphed into customer service, frustrating many of our colleagues.
Because it can affect clinical outcomes, patient retention, and medical malpractice claims, patient satisfaction is commonly used as a proxy for the success of doctors and hospitals.1 We know there is a correlation between higher patient satisfaction rates and improved outcomes—and conversely, research has demonstrated that unmet expectations significantly decrease satisfaction.2
However, there has been no explicit definition of patient satisfaction, nor systematic consideration of its determinants and consequences.3 As a result, measurement of “satisfaction” and its use as an indicator of quality of care remains controversial among health care providers. It can be a difficult concept to embrace.
Even setting aside the question of “amenities” and focusing on actual clinical care, satisfaction has different meanings for different people. For some, it is a positive, immediate improvement in the patient’s condition (recall my comments on pain management in my previous editorial).4 While that might be an unrealistic expectation, it is a factor in whether the patient and/or family express satisfaction with the care provided.
These high (if not unreasonable) expectations are fueled by the availability of information via the Internet. Patient attitudes and perceptions prior to receiving care also play a role. Instead of correlating with high-quality, appropriate, affordable care, a patient’s satisfaction might instead be based on the fulfillment of his or her predetermined ideas as to what treatment is needed!
The impetus for this change in perspective was the development of the patient-centered care model, which has patient satisfaction at its core.5 The model is intended to make patients partners in their health care; instead of depending solely on provider tools or standards, patients and providers discuss the options and preferences and develop a plan of care together. We all know that the relationship between patients and their providers greatly affects both treatment outcomes and patient satisfaction. But implementing a patient-centered care model means understanding and accepting from the start that patients will be asked to rate or judge their health care. It is therefore essential that there is agreement as to the standards that constitute “quality care” and congruence between these beliefs and the satisfaction ratings. You need to know what your patient expects to determine your likelihood of delivering it.
The patient-provider relationship has been a focus of the Consumer Assessment of Healthcare Providers and Systems (CAHPS®) Hospital Survey, which, since 2006, has measured patients’ perceptions of their hospital experiences.6 The CAHPS Clinician and Group Survey, initiated in 2011, is a standardized tool to measure patients’ perceptions of care in an office setting.7 Data from both surveys are used to improve performance and productivity in these settings. But while the information about quality of care has enabled consumers to make more informed decisions, the data are in many ways limited and subjective.
What cannot be measured by either survey alone is the health of patients, employees, and the community. This limitation is reflected in the feedback to our survey, which suggests a preponderance of NP and PA dissatisfaction with the current methods of evaluating the health care system. How much strain is incurred when evaluative measures fail to demonstrate that high-quality, safe, affordable care is being provided? That is difficult to ascertain, but it does give one pause. We know that providers who experience professional satisfaction have higher overall patient satisfaction scores.8 If we’re frustrated, are we able to provide the highest quality care? If not, our scores will suffer. If our scores drop … around we go again.
Currently, most data collection methods focus on physicians, making NPs and PAs “invisible” providers. That certainly won’t help our satisfaction! Only when the data gleaned from these measurement tools include all ambulatory settings, and all providers are recognized as valued contributors to patient health and satisfaction, will we have the information we need to improve satisfaction levels. That will benefit not only our patients, but also ourselves.
Please share your thoughts on patient satisfaction and “customer service” by emailing [email protected].
1. Prakash B. Patient satisfaction. J Cutan Aesthet Surg. 2010; 3(3):151-155.
2. Jackson JL,Chamberlin J, Kroenke K. Predictors of patient satisfaction. Soc Sci Med. 2001;52(4):609-620.
3. Linder-Pelz SU. Toward a theory of patient satisfaction. Soc Sci Med. 1982;16(5):577-582.
4. Onieal ME. The paradox of pain management. Clinician Reviews. 2016;26(11):12,16.
5. Rickert J. Measuring patient satisfaction: a bridge between patient and physician perceptions of care. http://healthaffairs.org/blog/2014/05/09/measuring-patient-satisfaction-a-bridge-between-patient-and-physician-perceptions-of-care. Accessed December 1, 2016.
6. Centers for Medicare & Medicaid Services. Hospital Consumer Assessment of Healthcare Providers and Systems CAHPS® Hospital Survey. www.hcahpsonline.org/home.aspx. Accessed December 1, 2016.
7. Agency for Healthcare Research and Quality. Consumer Assessment of Healthcare Providers and Systems Clinician and Group Survey. www.ahrq.gov/cahps/surveys-guidance/cg/index.html. Accessed December 1, 2016.
8. Haas JS, Cook EF, Puopolo AL, et al. Is the professional satisfaction of general internists associated with patient satisfaction? J Gen Intern Med. 2000;15(2):122-128.

In our December 2016 issue, we reported the results of our first annual survey on nonmonetary compensation (ie, the “perks”) and overall employment satisfaction (Clinician Reviews. 2016;26[12]:23-26). But the feedback I found most interesting came from the narrative responses—particularly those referencing patient satisfaction and the stress it creates for NPs and PAs.
Safety, quality, and affordability ha
Judging by the verbatim responses to our survey, NPs and PAs are concerned that quality measures don’t reflect the demands of our practice or focus on what matters to our patients.
One participant analogized, “Medicine is now like McDonalds or Burger King—patients want it their way, regardless of whether it’s in their best interest. I was fee-for-service for more than 10 years. As reimbursements have decreased significantly over time, I’m now employed by a hospital. I have become a waitress, considering my patients’ wishes—not for the benefit of their health, but to meet their more trivial ‘needs.’ These requirements can be as absurd as a specific brand of sweetener! If patients’ preferred sugar substitutes aren’t offered at my hospital, their ‘satisfaction’ may drop and I won’t get reimbursed as much. It’s a miserable experience.”
Perhaps the disparate views of what matters—Is it the softness of the pillows, or is it measurable improvement in the patient’s condition?—is the origin of the stress expressed by clinicians. This dissonance, in my opinion, exists among all involved—providers, patients, and payers. Today, patients see themselves as buyers of health services, and health care corporations have begun to function as a service industry. It may also explain why the concept of patient satisfaction has seemingly morphed into customer service, frustrating many of our colleagues.
Because it can affect clinical outcomes, patient retention, and medical malpractice claims, patient satisfaction is commonly used as a proxy for the success of doctors and hospitals.1 We know there is a correlation between higher patient satisfaction rates and improved outcomes—and conversely, research has demonstrated that unmet expectations significantly decrease satisfaction.2
However, there has been no explicit definition of patient satisfaction, nor systematic consideration of its determinants and consequences.3 As a result, measurement of “satisfaction” and its use as an indicator of quality of care remains controversial among health care providers. It can be a difficult concept to embrace.
Even setting aside the question of “amenities” and focusing on actual clinical care, satisfaction has different meanings for different people. For some, it is a positive, immediate improvement in the patient’s condition (recall my comments on pain management in my previous editorial).4 While that might be an unrealistic expectation, it is a factor in whether the patient and/or family express satisfaction with the care provided.
These high (if not unreasonable) expectations are fueled by the availability of information via the Internet. Patient attitudes and perceptions prior to receiving care also play a role. Instead of correlating with high-quality, appropriate, affordable care, a patient’s satisfaction might instead be based on the fulfillment of his or her predetermined ideas as to what treatment is needed!
The impetus for this change in perspective was the development of the patient-centered care model, which has patient satisfaction at its core.5 The model is intended to make patients partners in their health care; instead of depending solely on provider tools or standards, patients and providers discuss the options and preferences and develop a plan of care together. We all know that the relationship between patients and their providers greatly affects both treatment outcomes and patient satisfaction. But implementing a patient-centered care model means understanding and accepting from the start that patients will be asked to rate or judge their health care. It is therefore essential that there is agreement as to the standards that constitute “quality care” and congruence between these beliefs and the satisfaction ratings. You need to know what your patient expects to determine your likelihood of delivering it.
The patient-provider relationship has been a focus of the Consumer Assessment of Healthcare Providers and Systems (CAHPS®) Hospital Survey, which, since 2006, has measured patients’ perceptions of their hospital experiences.6 The CAHPS Clinician and Group Survey, initiated in 2011, is a standardized tool to measure patients’ perceptions of care in an office setting.7 Data from both surveys are used to improve performance and productivity in these settings. But while the information about quality of care has enabled consumers to make more informed decisions, the data are in many ways limited and subjective.
What cannot be measured by either survey alone is the health of patients, employees, and the community. This limitation is reflected in the feedback to our survey, which suggests a preponderance of NP and PA dissatisfaction with the current methods of evaluating the health care system. How much strain is incurred when evaluative measures fail to demonstrate that high-quality, safe, affordable care is being provided? That is difficult to ascertain, but it does give one pause. We know that providers who experience professional satisfaction have higher overall patient satisfaction scores.8 If we’re frustrated, are we able to provide the highest quality care? If not, our scores will suffer. If our scores drop … around we go again.
Currently, most data collection methods focus on physicians, making NPs and PAs “invisible” providers. That certainly won’t help our satisfaction! Only when the data gleaned from these measurement tools include all ambulatory settings, and all providers are recognized as valued contributors to patient health and satisfaction, will we have the information we need to improve satisfaction levels. That will benefit not only our patients, but also ourselves.
Please share your thoughts on patient satisfaction and “customer service” by emailing [email protected].
In our December 2016 issue, we reported the results of our first annual survey on nonmonetary compensation (ie, the “perks”) and overall employment satisfaction (Clinician Reviews. 2016;26[12]:23-26). But the feedback I found most interesting came from the narrative responses—particularly those referencing patient satisfaction and the stress it creates for NPs and PAs.
Safety, quality, and affordability ha
Judging by the verbatim responses to our survey, NPs and PAs are concerned that quality measures don’t reflect the demands of our practice or focus on what matters to our patients.
One participant analogized, “Medicine is now like McDonalds or Burger King—patients want it their way, regardless of whether it’s in their best interest. I was fee-for-service for more than 10 years. As reimbursements have decreased significantly over time, I’m now employed by a hospital. I have become a waitress, considering my patients’ wishes—not for the benefit of their health, but to meet their more trivial ‘needs.’ These requirements can be as absurd as a specific brand of sweetener! If patients’ preferred sugar substitutes aren’t offered at my hospital, their ‘satisfaction’ may drop and I won’t get reimbursed as much. It’s a miserable experience.”
Perhaps the disparate views of what matters—Is it the softness of the pillows, or is it measurable improvement in the patient’s condition?—is the origin of the stress expressed by clinicians. This dissonance, in my opinion, exists among all involved—providers, patients, and payers. Today, patients see themselves as buyers of health services, and health care corporations have begun to function as a service industry. It may also explain why the concept of patient satisfaction has seemingly morphed into customer service, frustrating many of our colleagues.
Because it can affect clinical outcomes, patient retention, and medical malpractice claims, patient satisfaction is commonly used as a proxy for the success of doctors and hospitals.1 We know there is a correlation between higher patient satisfaction rates and improved outcomes—and conversely, research has demonstrated that unmet expectations significantly decrease satisfaction.2
However, there has been no explicit definition of patient satisfaction, nor systematic consideration of its determinants and consequences.3 As a result, measurement of “satisfaction” and its use as an indicator of quality of care remains controversial among health care providers. It can be a difficult concept to embrace.
Even setting aside the question of “amenities” and focusing on actual clinical care, satisfaction has different meanings for different people. For some, it is a positive, immediate improvement in the patient’s condition (recall my comments on pain management in my previous editorial).4 While that might be an unrealistic expectation, it is a factor in whether the patient and/or family express satisfaction with the care provided.
These high (if not unreasonable) expectations are fueled by the availability of information via the Internet. Patient attitudes and perceptions prior to receiving care also play a role. Instead of correlating with high-quality, appropriate, affordable care, a patient’s satisfaction might instead be based on the fulfillment of his or her predetermined ideas as to what treatment is needed!
The impetus for this change in perspective was the development of the patient-centered care model, which has patient satisfaction at its core.5 The model is intended to make patients partners in their health care; instead of depending solely on provider tools or standards, patients and providers discuss the options and preferences and develop a plan of care together. We all know that the relationship between patients and their providers greatly affects both treatment outcomes and patient satisfaction. But implementing a patient-centered care model means understanding and accepting from the start that patients will be asked to rate or judge their health care. It is therefore essential that there is agreement as to the standards that constitute “quality care” and congruence between these beliefs and the satisfaction ratings. You need to know what your patient expects to determine your likelihood of delivering it.
The patient-provider relationship has been a focus of the Consumer Assessment of Healthcare Providers and Systems (CAHPS®) Hospital Survey, which, since 2006, has measured patients’ perceptions of their hospital experiences.6 The CAHPS Clinician and Group Survey, initiated in 2011, is a standardized tool to measure patients’ perceptions of care in an office setting.7 Data from both surveys are used to improve performance and productivity in these settings. But while the information about quality of care has enabled consumers to make more informed decisions, the data are in many ways limited and subjective.
What cannot be measured by either survey alone is the health of patients, employees, and the community. This limitation is reflected in the feedback to our survey, which suggests a preponderance of NP and PA dissatisfaction with the current methods of evaluating the health care system. How much strain is incurred when evaluative measures fail to demonstrate that high-quality, safe, affordable care is being provided? That is difficult to ascertain, but it does give one pause. We know that providers who experience professional satisfaction have higher overall patient satisfaction scores.8 If we’re frustrated, are we able to provide the highest quality care? If not, our scores will suffer. If our scores drop … around we go again.
Currently, most data collection methods focus on physicians, making NPs and PAs “invisible” providers. That certainly won’t help our satisfaction! Only when the data gleaned from these measurement tools include all ambulatory settings, and all providers are recognized as valued contributors to patient health and satisfaction, will we have the information we need to improve satisfaction levels. That will benefit not only our patients, but also ourselves.
Please share your thoughts on patient satisfaction and “customer service” by emailing [email protected].
1. Prakash B. Patient satisfaction. J Cutan Aesthet Surg. 2010; 3(3):151-155.
2. Jackson JL,Chamberlin J, Kroenke K. Predictors of patient satisfaction. Soc Sci Med. 2001;52(4):609-620.
3. Linder-Pelz SU. Toward a theory of patient satisfaction. Soc Sci Med. 1982;16(5):577-582.
4. Onieal ME. The paradox of pain management. Clinician Reviews. 2016;26(11):12,16.
5. Rickert J. Measuring patient satisfaction: a bridge between patient and physician perceptions of care. http://healthaffairs.org/blog/2014/05/09/measuring-patient-satisfaction-a-bridge-between-patient-and-physician-perceptions-of-care. Accessed December 1, 2016.
6. Centers for Medicare & Medicaid Services. Hospital Consumer Assessment of Healthcare Providers and Systems CAHPS® Hospital Survey. www.hcahpsonline.org/home.aspx. Accessed December 1, 2016.
7. Agency for Healthcare Research and Quality. Consumer Assessment of Healthcare Providers and Systems Clinician and Group Survey. www.ahrq.gov/cahps/surveys-guidance/cg/index.html. Accessed December 1, 2016.
8. Haas JS, Cook EF, Puopolo AL, et al. Is the professional satisfaction of general internists associated with patient satisfaction? J Gen Intern Med. 2000;15(2):122-128.
1. Prakash B. Patient satisfaction. J Cutan Aesthet Surg. 2010; 3(3):151-155.
2. Jackson JL,Chamberlin J, Kroenke K. Predictors of patient satisfaction. Soc Sci Med. 2001;52(4):609-620.
3. Linder-Pelz SU. Toward a theory of patient satisfaction. Soc Sci Med. 1982;16(5):577-582.
4. Onieal ME. The paradox of pain management. Clinician Reviews. 2016;26(11):12,16.
5. Rickert J. Measuring patient satisfaction: a bridge between patient and physician perceptions of care. http://healthaffairs.org/blog/2014/05/09/measuring-patient-satisfaction-a-bridge-between-patient-and-physician-perceptions-of-care. Accessed December 1, 2016.
6. Centers for Medicare & Medicaid Services. Hospital Consumer Assessment of Healthcare Providers and Systems CAHPS® Hospital Survey. www.hcahpsonline.org/home.aspx. Accessed December 1, 2016.
7. Agency for Healthcare Research and Quality. Consumer Assessment of Healthcare Providers and Systems Clinician and Group Survey. www.ahrq.gov/cahps/surveys-guidance/cg/index.html. Accessed December 1, 2016.
8. Haas JS, Cook EF, Puopolo AL, et al. Is the professional satisfaction of general internists associated with patient satisfaction? J Gen Intern Med. 2000;15(2):122-128.
Prepregnancy overweight boosts risk of depressive symptoms in pregnancy
NEW ORLEANS – Prepregnancy overweight and obesity are associated with increased incidence and severity of depressive symptoms during pregnancy, independent of preeclampsia and other hypertensive pregnancy disorders or gestational diabetes, Satu Kumpulainen reported at Obesity Week 2016.
The implications of this novel finding are clear: “Prepregnancy interventions targeting overweight and obesity and mental health will not only benefit the pregnant mother’s health but will also provide optimal odds for healthy development of the fetus as well,” said Ms. Kumpulainen, a doctoral student at the University of Helsinki Institute of Behavioral Sciences.
It’s well established that prepregnancy obesity is a risk factor for gestational diabetes, preeclampsia, and depression during pregnancy. This study was carried out to learn if a high prepregnancy BMI boosts the risk of prenatal depression independent of the cardiometabolic complications of pregnancy, Ms. Kumpulainen explained at the meeting, which was presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
This proved to be the case in the Finnish women, 67.3% of whom were normal weight before pregnancy; 19.1% were overweight and 13.6% obese. Gestational diabetes occurred in 10.6% of the PREDO participants, and hypertension-spectrum disorders of pregnancy occurred in 8.2%.
The women who were obese or overweight prepregnancy reported higher rates of clinically meaningful depressive symptoms throughout pregnancy, compared with women who were normal weight. Using a CES-D score of 16 or more to define clinically significant depressive symptoms, such symptoms were reported as early as gestational week 12 and on multiple occasions thereafter by 19.9% of the women who were normal weight before pregnancy, 23.3% of those who were overweight, and 27.4% of those who were obese. The differences were statistically significant.
The risk of clinically significant depressive symptoms during pregnancy was no higher in prepregnancy normal-weight women who developed gestational diabetes or preeclampsia than in those who did not, Ms. Kumpulainen reported.
“Our findings suggest that cardiometabolic pregnancy disorders per se don’t trigger higher levels of depressive symptoms, but women with prepregnancy overweight and obesity feel more depressed right from the beginning of pregnancy,” Ms. Kumpulainen said.
She reported having no financial conflicts of interest related to the study, which was supported by Finnish scientific research grants.
NEW ORLEANS – Prepregnancy overweight and obesity are associated with increased incidence and severity of depressive symptoms during pregnancy, independent of preeclampsia and other hypertensive pregnancy disorders or gestational diabetes, Satu Kumpulainen reported at Obesity Week 2016.
The implications of this novel finding are clear: “Prepregnancy interventions targeting overweight and obesity and mental health will not only benefit the pregnant mother’s health but will also provide optimal odds for healthy development of the fetus as well,” said Ms. Kumpulainen, a doctoral student at the University of Helsinki Institute of Behavioral Sciences.
It’s well established that prepregnancy obesity is a risk factor for gestational diabetes, preeclampsia, and depression during pregnancy. This study was carried out to learn if a high prepregnancy BMI boosts the risk of prenatal depression independent of the cardiometabolic complications of pregnancy, Ms. Kumpulainen explained at the meeting, which was presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
This proved to be the case in the Finnish women, 67.3% of whom were normal weight before pregnancy; 19.1% were overweight and 13.6% obese. Gestational diabetes occurred in 10.6% of the PREDO participants, and hypertension-spectrum disorders of pregnancy occurred in 8.2%.
The women who were obese or overweight prepregnancy reported higher rates of clinically meaningful depressive symptoms throughout pregnancy, compared with women who were normal weight. Using a CES-D score of 16 or more to define clinically significant depressive symptoms, such symptoms were reported as early as gestational week 12 and on multiple occasions thereafter by 19.9% of the women who were normal weight before pregnancy, 23.3% of those who were overweight, and 27.4% of those who were obese. The differences were statistically significant.
The risk of clinically significant depressive symptoms during pregnancy was no higher in prepregnancy normal-weight women who developed gestational diabetes or preeclampsia than in those who did not, Ms. Kumpulainen reported.
“Our findings suggest that cardiometabolic pregnancy disorders per se don’t trigger higher levels of depressive symptoms, but women with prepregnancy overweight and obesity feel more depressed right from the beginning of pregnancy,” Ms. Kumpulainen said.
She reported having no financial conflicts of interest related to the study, which was supported by Finnish scientific research grants.
NEW ORLEANS – Prepregnancy overweight and obesity are associated with increased incidence and severity of depressive symptoms during pregnancy, independent of preeclampsia and other hypertensive pregnancy disorders or gestational diabetes, Satu Kumpulainen reported at Obesity Week 2016.
The implications of this novel finding are clear: “Prepregnancy interventions targeting overweight and obesity and mental health will not only benefit the pregnant mother’s health but will also provide optimal odds for healthy development of the fetus as well,” said Ms. Kumpulainen, a doctoral student at the University of Helsinki Institute of Behavioral Sciences.
It’s well established that prepregnancy obesity is a risk factor for gestational diabetes, preeclampsia, and depression during pregnancy. This study was carried out to learn if a high prepregnancy BMI boosts the risk of prenatal depression independent of the cardiometabolic complications of pregnancy, Ms. Kumpulainen explained at the meeting, which was presented by the Obesity Society and the American Society for Metabolic and Bariatric Surgery.
This proved to be the case in the Finnish women, 67.3% of whom were normal weight before pregnancy; 19.1% were overweight and 13.6% obese. Gestational diabetes occurred in 10.6% of the PREDO participants, and hypertension-spectrum disorders of pregnancy occurred in 8.2%.
The women who were obese or overweight prepregnancy reported higher rates of clinically meaningful depressive symptoms throughout pregnancy, compared with women who were normal weight. Using a CES-D score of 16 or more to define clinically significant depressive symptoms, such symptoms were reported as early as gestational week 12 and on multiple occasions thereafter by 19.9% of the women who were normal weight before pregnancy, 23.3% of those who were overweight, and 27.4% of those who were obese. The differences were statistically significant.
The risk of clinically significant depressive symptoms during pregnancy was no higher in prepregnancy normal-weight women who developed gestational diabetes or preeclampsia than in those who did not, Ms. Kumpulainen reported.
“Our findings suggest that cardiometabolic pregnancy disorders per se don’t trigger higher levels of depressive symptoms, but women with prepregnancy overweight and obesity feel more depressed right from the beginning of pregnancy,” Ms. Kumpulainen said.
She reported having no financial conflicts of interest related to the study, which was supported by Finnish scientific research grants.
AT OBESITY WEEK 2016
Key clinical point:
Major finding: Women who were obese prior to pregnancy were over 50% more likely to experience clinically significant depressive symptoms throughout pregnancy, compared with women who were normal weight before pregnancy, independent of whether the women developed gestational diabetes or preeclampsia.
Data source: This was a secondary analysis from a prospective study of more than 3,000 pregnant Finnish women.
Disclosures: The study was supported by Finnish scientific research grants. The presenter reported having no financial conflicts of interest related to the study.
ADA: Empagliflozin and liraglutide reduce type 2 CV death
to reduce the risk of CV death, according to the American Diabetes Association 2017 Standards of Medical Care.
ADA updates it standards annually based on new information and research; like its predecessors, the 2017 guidance is comprehensive, addressing mental, social, and other challenges faced by patients with diabetes, along with clinical care (Diabetes Care. 2017 Jan;40(Suppl 1):S4-S5).
The 2017 guidance contains a great deal of new information. At 135 pages, there are 22 more pages than in 2016. “They did a really nice job. This guide is useful for anyone helping patients with diabetes,” including diabetologists, dietitians, educators, psychologists, and social workers, Richard Hellman, MD, a clinical endocrinologist in North Kansas City, Mo., said in an interview.

The empagliflozin and liraglutide recommendation applies to any patient with type 2 diabetes who has a history of stroke, heart attack, acute coronary syndrome, angina, or peripheral arterial disease. Data from recent trials have shown use of the drugs modestly reduces cardiovascular mortality in this population.
It’s unclear if the benefits are drug specific or group effects. “We anxiously await the results of several ongoing cardiovascular outcomes trials” to find out, said Helena Rodbard, MD, a clinical endocrinologist in Rockville, Md., who also commented on the new standards.
Basal insulin plus a GLP-1 receptor agonist, like liraglutide, are also now recommended for insulin-dependent type 2 disease. “This combination gives rise to a markedly reduced risk of hypoglycemia compared with basal insulin ... basal bolus insulin, or premixed insulins,” according to the ADA.
The newer drugs and insulins are expensive. To help doctors and patients negotiate the price hurdle, ADA added tables on how much the various options cost per month. It was a good move; “the cost of care is going up so fast” in diabetes “that many patients can no longer afford” what’s prescribed. “It’s a major problem,” said Dr. Hellman, clinical professor at the University of Missouri–Kansas City.
The ADA also set a blood glucose level of 54 mg/dL to trigger aggressive hypoglycemia treatment. “There has been confusion over when to treat aggressively. It was a good choice to land on 54 mg/dL” a safe, conservative number a bit higher than others have suggested, Dr. Hellman said.
Meanwhile, the group lowered its metabolic surgery cut point – the ADA has stopped using the term “bariatric surgery” – to type 2 patients with a body mass index of 30 kg/m2 when medications don’t work. The group also set a new hypertension treatment target of 120-160/80-105 mm Hg in pregnancy, and said that insulin is the treatment of choice for gestational diabetes, given concerns about metformin crossing the placenta and glyburide in cord blood.
The ADA expanded its list of diabetes comorbidities to include autoimmune disease, HIV, anxiety, depression, and disordered eating. In addition, doctors should ask patients how well they sleep – since sleep problems affect glycemic control – and should intervene when there’s a problem, according to the guidance.
The group updated its combination injection algorithm for type 2 diabetes “to reflect studies demonstrating the noninferiority of basal insulin plus” liraglutide and its class members “versus basal insulin plus rapid-acting insulin” or two daily injections of premixed insulin. The ADA added a section on the role of newly available biosimilar insulins, as well, and clarified that either basal insulin or basal plus bolus correctional insulin can be used to treat noncritical inpatients, but noted that “sole use of sliding scale insulin in the inpatient hospital setting is strongly discouraged.”
People on long-term metformin should have their vitamin B12 checked periodically, because of new evidence about the risk of B12 deficiency, the group said, and “due to the risk of malformations associated with unplanned pregnancies and poor metabolic control.” The group added “a new recommendation ... encouraging preconception counseling starting at puberty for all girls of childbearing potential.”
“Even though most of this information should be well known to practitioners treating patients, [it’s] a worthwhile read for everyone who treats people with diabetes,” Dr. Rodbard said.
The majority of the people on the ADA’s update committee had no disclosures, but a few reported ties to various companies, including Novo Nordisk, the maker of liraglutide, and Boehringer Ingelheim and Lilly, the companies that developed and/or marketed empagliflozin. Dr. Hellman had no conflicts. Dr. Rodbard is an adviser or researcher for AstraZeneca, Lilly, Janssen, Merck, Novo Nordisk, Sanofi, and Regeneron.
to reduce the risk of CV death, according to the American Diabetes Association 2017 Standards of Medical Care.
ADA updates it standards annually based on new information and research; like its predecessors, the 2017 guidance is comprehensive, addressing mental, social, and other challenges faced by patients with diabetes, along with clinical care (Diabetes Care. 2017 Jan;40(Suppl 1):S4-S5).
The 2017 guidance contains a great deal of new information. At 135 pages, there are 22 more pages than in 2016. “They did a really nice job. This guide is useful for anyone helping patients with diabetes,” including diabetologists, dietitians, educators, psychologists, and social workers, Richard Hellman, MD, a clinical endocrinologist in North Kansas City, Mo., said in an interview.
The empagliflozin and liraglutide recommendation applies to any patient with type 2 diabetes who has a history of stroke, heart attack, acute coronary syndrome, angina, or peripheral arterial disease. Data from recent trials have shown use of the drugs modestly reduces cardiovascular mortality in this population.
It’s unclear if the benefits are drug specific or group effects. “We anxiously await the results of several ongoing cardiovascular outcomes trials” to find out, said Helena Rodbard, MD, a clinical endocrinologist in Rockville, Md., who also commented on the new standards.
Basal insulin plus a GLP-1 receptor agonist, like liraglutide, are also now recommended for insulin-dependent type 2 disease. “This combination gives rise to a markedly reduced risk of hypoglycemia compared with basal insulin ... basal bolus insulin, or premixed insulins,” according to the ADA.
The newer drugs and insulins are expensive. To help doctors and patients negotiate the price hurdle, ADA added tables on how much the various options cost per month. It was a good move; “the cost of care is going up so fast” in diabetes “that many patients can no longer afford” what’s prescribed. “It’s a major problem,” said Dr. Hellman, clinical professor at the University of Missouri–Kansas City.
The ADA also set a blood glucose level of 54 mg/dL to trigger aggressive hypoglycemia treatment. “There has been confusion over when to treat aggressively. It was a good choice to land on 54 mg/dL” a safe, conservative number a bit higher than others have suggested, Dr. Hellman said.
Meanwhile, the group lowered its metabolic surgery cut point – the ADA has stopped using the term “bariatric surgery” – to type 2 patients with a body mass index of 30 kg/m2 when medications don’t work. The group also set a new hypertension treatment target of 120-160/80-105 mm Hg in pregnancy, and said that insulin is the treatment of choice for gestational diabetes, given concerns about metformin crossing the placenta and glyburide in cord blood.
The ADA expanded its list of diabetes comorbidities to include autoimmune disease, HIV, anxiety, depression, and disordered eating. In addition, doctors should ask patients how well they sleep – since sleep problems affect glycemic control – and should intervene when there’s a problem, according to the guidance.
The group updated its combination injection algorithm for type 2 diabetes “to reflect studies demonstrating the noninferiority of basal insulin plus” liraglutide and its class members “versus basal insulin plus rapid-acting insulin” or two daily injections of premixed insulin. The ADA added a section on the role of newly available biosimilar insulins, as well, and clarified that either basal insulin or basal plus bolus correctional insulin can be used to treat noncritical inpatients, but noted that “sole use of sliding scale insulin in the inpatient hospital setting is strongly discouraged.”
People on long-term metformin should have their vitamin B12 checked periodically, because of new evidence about the risk of B12 deficiency, the group said, and “due to the risk of malformations associated with unplanned pregnancies and poor metabolic control.” The group added “a new recommendation ... encouraging preconception counseling starting at puberty for all girls of childbearing potential.”
“Even though most of this information should be well known to practitioners treating patients, [it’s] a worthwhile read for everyone who treats people with diabetes,” Dr. Rodbard said.
The majority of the people on the ADA’s update committee had no disclosures, but a few reported ties to various companies, including Novo Nordisk, the maker of liraglutide, and Boehringer Ingelheim and Lilly, the companies that developed and/or marketed empagliflozin. Dr. Hellman had no conflicts. Dr. Rodbard is an adviser or researcher for AstraZeneca, Lilly, Janssen, Merck, Novo Nordisk, Sanofi, and Regeneron.
to reduce the risk of CV death, according to the American Diabetes Association 2017 Standards of Medical Care.
ADA updates it standards annually based on new information and research; like its predecessors, the 2017 guidance is comprehensive, addressing mental, social, and other challenges faced by patients with diabetes, along with clinical care (Diabetes Care. 2017 Jan;40(Suppl 1):S4-S5).
The 2017 guidance contains a great deal of new information. At 135 pages, there are 22 more pages than in 2016. “They did a really nice job. This guide is useful for anyone helping patients with diabetes,” including diabetologists, dietitians, educators, psychologists, and social workers, Richard Hellman, MD, a clinical endocrinologist in North Kansas City, Mo., said in an interview.
The empagliflozin and liraglutide recommendation applies to any patient with type 2 diabetes who has a history of stroke, heart attack, acute coronary syndrome, angina, or peripheral arterial disease. Data from recent trials have shown use of the drugs modestly reduces cardiovascular mortality in this population.
It’s unclear if the benefits are drug specific or group effects. “We anxiously await the results of several ongoing cardiovascular outcomes trials” to find out, said Helena Rodbard, MD, a clinical endocrinologist in Rockville, Md., who also commented on the new standards.
Basal insulin plus a GLP-1 receptor agonist, like liraglutide, are also now recommended for insulin-dependent type 2 disease. “This combination gives rise to a markedly reduced risk of hypoglycemia compared with basal insulin ... basal bolus insulin, or premixed insulins,” according to the ADA.
The newer drugs and insulins are expensive. To help doctors and patients negotiate the price hurdle, ADA added tables on how much the various options cost per month. It was a good move; “the cost of care is going up so fast” in diabetes “that many patients can no longer afford” what’s prescribed. “It’s a major problem,” said Dr. Hellman, clinical professor at the University of Missouri–Kansas City.
The ADA also set a blood glucose level of 54 mg/dL to trigger aggressive hypoglycemia treatment. “There has been confusion over when to treat aggressively. It was a good choice to land on 54 mg/dL” a safe, conservative number a bit higher than others have suggested, Dr. Hellman said.
Meanwhile, the group lowered its metabolic surgery cut point – the ADA has stopped using the term “bariatric surgery” – to type 2 patients with a body mass index of 30 kg/m2 when medications don’t work. The group also set a new hypertension treatment target of 120-160/80-105 mm Hg in pregnancy, and said that insulin is the treatment of choice for gestational diabetes, given concerns about metformin crossing the placenta and glyburide in cord blood.
The ADA expanded its list of diabetes comorbidities to include autoimmune disease, HIV, anxiety, depression, and disordered eating. In addition, doctors should ask patients how well they sleep – since sleep problems affect glycemic control – and should intervene when there’s a problem, according to the guidance.
The group updated its combination injection algorithm for type 2 diabetes “to reflect studies demonstrating the noninferiority of basal insulin plus” liraglutide and its class members “versus basal insulin plus rapid-acting insulin” or two daily injections of premixed insulin. The ADA added a section on the role of newly available biosimilar insulins, as well, and clarified that either basal insulin or basal plus bolus correctional insulin can be used to treat noncritical inpatients, but noted that “sole use of sliding scale insulin in the inpatient hospital setting is strongly discouraged.”
People on long-term metformin should have their vitamin B12 checked periodically, because of new evidence about the risk of B12 deficiency, the group said, and “due to the risk of malformations associated with unplanned pregnancies and poor metabolic control.” The group added “a new recommendation ... encouraging preconception counseling starting at puberty for all girls of childbearing potential.”
“Even though most of this information should be well known to practitioners treating patients, [it’s] a worthwhile read for everyone who treats people with diabetes,” Dr. Rodbard said.
The majority of the people on the ADA’s update committee had no disclosures, but a few reported ties to various companies, including Novo Nordisk, the maker of liraglutide, and Boehringer Ingelheim and Lilly, the companies that developed and/or marketed empagliflozin. Dr. Hellman had no conflicts. Dr. Rodbard is an adviser or researcher for AstraZeneca, Lilly, Janssen, Merck, Novo Nordisk, Sanofi, and Regeneron.

Antidepressant-associated purpura: A rare familial case presentation
Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) have been a welcome addition to the armamentarium for treating depressive disorders, neuropathic pain, and anxiety disorders. Despite the more favorable side-effect profile compared with tricyclic antidepressants and monoamine oxidase inhibitors, these serotonergic agents have been associated with bleeding disorders, purpura, thrombocytopenia, and, in extreme cases, death.1-4
We describe a case of purpura associated with different classes of antidepressants, including the non-serotonergic agent bupropion, as well as a family history of similar adverse effects to antidepressants.
CASE Purpura resolves when drug is stopped
Ms. R, age 70, presents with major depressive disorder and fibromyalgia and is receiving duloxetine, 20 mg/d, which is gradually increased to 60 mg. She also has a history of chronic obstructive pulmonary disease (COPD), for which she is taking albuterol and a steroid inhaler. Ms. R responds well to treatment; however, she develops blue–purple purpura on her arms each measuring 1 to 2 inches. Laboratory test results including platelet count and prothrombin time/international normalized ratio are within normal ranges. Duloxetine is stopped, and purpura resolves in 1 to 2 weeks. To avoid serotonergic antidepressants, Ms. R receives bupropion XL, 150 mg; however, similar purpura develops, then resolves when the medication is discontinued. She is lost to follow up for approximately 6 months, but returns requesting a rechallenge with duloxetine for her depression, which has worsened. Duloxetine is restarted with similar results and is then discontinued. Because she has developed neuropathy, Ms. R is started on nortriptyline, 25 mg/d, increased to 50 mg/d, but purpura develops again, which resolves when medication is discontinued. Ms. R’s daughter reports she also developed a similar reaction with several antidepressants, which resolved with medication discontinuation.
Bleeding risk with antidepressants
The role of serotonin reuptake inhibitors (SRIs) in inducing bleeding has emerged as a safety concern,5,6 which have been documented in case reports.7-11 Mechanisms of action that have been thought to affect platelet aggregation include:
- depletion of serotonin in platelets
- increase in capillary fragility
- modification of platelet plug formation
- responsiveness of peptide-induced activation of platelets through stimulation of the thrombin receptor.7,8,12,13
The severity of bleeding varies with patient-related factors, such as a history of gastritis, peptic ulcer disease, and heavy bleeding during menses; use of gastrotoxic drugs, particularly nonsteroidal anti-inflammatory drugs (NSAIDs), also have been shown to increase this risk.14,15 For patients taking SRIs and gastrotoxic drugs (eg, NSAIDs), use of acid-suppressing agents have been shown to limit the risk of bleeding.16
Studies evaluating relative bleeding risks among classes of antidepressants have not shown increased risk with tricyclic antidepressants compared with SSRIs.17 Adenosine triphosphate (ATP), adenosine diphosphate (ADP), and adenosine monophosphate (AMP) are signaling molecules in the vascular system and are important in the thromboregulatory system. Studies in rats reported significant inhibition of ATP, ADP, and AMP hydrolysis with chronic treatment with fluoxetine and nortriptyline, and suggested that both medications changed the nucleotide catabolism, which means that homeostasis of the vascular system can be altered by antidepressant treatments.18 This is one possible pathway in the role these medications play in the etiology of dysregulation of the thromboregulatory system.
We did not anticipate that our patient would develop similar purpura with bupropion because the bleeding risk associated with antidepressants has been attributed to the effect of serotonin on platelets. Studies observing the effect of SSRIs, SNRIs, and bupropion on platelets and bleeding have not shown significant risk with bupropion.19 Bleeding associated with bupropion is atypical and needs to be further studied. Although this medication is centrally selective in its action on dopamine receptors, it might have possible peripheral effect on other neurotransmitters, including serotonin.
Ms. R had no personal or family history of purpura or a bleeding disorder. Significant improvement in her physical signs after discontinuing medications and recurrence of pupura with rechallenge indicate that this reaction was triggered by 3 different classes of antidepressants. Family history of similar reaction further suggests a genetic predisposition to platelet dysfunction to antidepressant treatment in a select group of patients.
Limitations include the possibility of senile purpura, which cannot be ruled out despite strong indications that antidepressants were the cause. The possibility of drug interactions needs be considered as well. Ms. R was taking albuterol and a steroid inhaler for her COPD at the time of the initial medication trials, which did not interact with duloxetine or bupropion. During the trials with duloxetine and then nortriptyline, she was taking acetaminophen/hydrocodone in addition to her inhalers, and no significant interactions with the antidepressants were identified. Interactions with unreported or over-the-counter medications or supplements are a possibility.
Before prescribing an antidepressant, we suggest taking a careful history including a family history of bleeding disorders and adverse effects of antidepressants, especially in patients who have risk factors (eg, concomitant use of gastrotoxic medications). Use of gastric acid-suppressing medications could be considered if antidepressants are used. Further investigations into the incidence, risk factors, mechanism of action, and treatment of this adverse effect are indicated.
Drug Brand Names
Acetaminophen/hydrocodone • Lorcet, Norco, Vicodin
Albuterol • Proventil
Bupropion • Wellbutrin
Duloxetine • Cymbalta
Nortriptyline • Pamelor, Aventyl
1. Cymbalta [package insert]. Indianapolis, IN: Eli Lilly and Company; 2014.
2. Amitriptyline. Medscape. http://reference.medscape.com/drug/levate-amitriptyline-342936. Accessed December 19, 2016.
3. Wellbutrin [package insert]. Triangle Park, NC: GlaxoSmithKline; 2004.
4. Balhara Y, Sagar R, Varghese ST. Bleeding gums: duloxetine may be the cause. J Postgrad Med. 2007;53(1):44-45.
5. Paton C, Ferrier IN. SSRIs and gastrointestinal bleeding. BMJ. 2005;331(7516):529-530.
6. Turner MS, May DB, Arthur RR, et al. Clinical impact of selective serotonin reuptake inhibitors therapy with bleeding risks. J Intern Med. 2007;261(3):205-213.
7. Humphries JE, Wheby MS, VandenBerg SR. Fluoxetine and the bleeding time. Arch Pathol Lab Med. 1990;114(7):727-728.
8. Alderman CP, Moritz CK, Ben-Tovim DI. Abnormal platelet aggregation associated with fluoxetine therapy. Ann Pharmacother. 1992;26(12):1517-1519.
9. Calhoun JW, Calhoun DD. Prolonged bleeding time in a patient treated with sertraline. Am J Psychiatry. 1996;153(3):443.
10. Ottervanger JP, Stricker BH, Huls J, et al. Bleeding attributed to the intake of paroxetine. Am J Psychiatry. 1994;151(5):781-782.
11. de Abajo FJ, Rodríguez LA, Montero D, et al. Association between selective serotonin reuptake inhibitors and upper gastrointestinal bleeding: population based case-control study. BMJ. 1999;319(7217):1106-1109.
12. Nelva A, Guy C, Tardy-Poncet B, et al. Hemorrhagic syndromes related to selective serotonin reuptake inhibitor (SSRI) antidepressants: seven case reports and review of the literature [in French]. Rev Med Interne. 2000;21(2):152-160.
13. de Abajo FJ, Montero D, Rodríguez LA, et al. Antidepressants and risk of upper gastrointestinal bleeding. Basic Clin Pharmacol Toxicol. 2006;98(3):304-310.
14. Tata LJ, Fortun PJ, Hubbard RB, et al. Does concurrent prescription of selective serotonin reuptake inhibitors and non-steroidal anti-inflammatory drugs substantially increase the risk of upper gastrointestinal bleeding? Aliment Pharmacol Ther. 2005;22(3):175-181.
15. Yuan Y, Tsoi K, Hunt RH. Selective serotonin reuptake inhibitors and risk of upper GI bleeding: confusion or confounding? Am J Med. 2006;119(9):719-727.
16. de Abajo FJ, García-Rodríguez LA. Risk of upper gastrointestinal tract bleeding associated with selective serotonin reuptake inhibitors and venlafaxine therapy: interaction with nonsteroidal anti-inflammatory drugs and effect of acid-suppressing agents. Arch Gen Psychiatry. 2008;65(7):795-803.
17. Barbui C, Andretta M, De Vitis G, et al. Antidepressant drug prescription and risk of abnormal bleeding: a case-control study. J Clin Pharmacol. 2009;29(1):33-38.
18. Pedrazza EL, Senger MR, Rico EP, et al. Fluoxetine and nortriptyline affect NTPDase and 5’-nucleotidase activities in rat blood serum. Life Sci. 2007;81(15):1205-1210.
19. Song HR, Jung YE, Wang HR, et al. Platelet count alterations associated with escitalopram, venlafaxine and bupropion in depressive patients. Psychiatry Clin Neurosci. 2012;66(5):457-459.
Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) have been a welcome addition to the armamentarium for treating depressive disorders, neuropathic pain, and anxiety disorders. Despite the more favorable side-effect profile compared with tricyclic antidepressants and monoamine oxidase inhibitors, these serotonergic agents have been associated with bleeding disorders, purpura, thrombocytopenia, and, in extreme cases, death.1-4
We describe a case of purpura associated with different classes of antidepressants, including the non-serotonergic agent bupropion, as well as a family history of similar adverse effects to antidepressants.
CASE Purpura resolves when drug is stopped
Ms. R, age 70, presents with major depressive disorder and fibromyalgia and is receiving duloxetine, 20 mg/d, which is gradually increased to 60 mg. She also has a history of chronic obstructive pulmonary disease (COPD), for which she is taking albuterol and a steroid inhaler. Ms. R responds well to treatment; however, she develops blue–purple purpura on her arms each measuring 1 to 2 inches. Laboratory test results including platelet count and prothrombin time/international normalized ratio are within normal ranges. Duloxetine is stopped, and purpura resolves in 1 to 2 weeks. To avoid serotonergic antidepressants, Ms. R receives bupropion XL, 150 mg; however, similar purpura develops, then resolves when the medication is discontinued. She is lost to follow up for approximately 6 months, but returns requesting a rechallenge with duloxetine for her depression, which has worsened. Duloxetine is restarted with similar results and is then discontinued. Because she has developed neuropathy, Ms. R is started on nortriptyline, 25 mg/d, increased to 50 mg/d, but purpura develops again, which resolves when medication is discontinued. Ms. R’s daughter reports she also developed a similar reaction with several antidepressants, which resolved with medication discontinuation.
Bleeding risk with antidepressants
The role of serotonin reuptake inhibitors (SRIs) in inducing bleeding has emerged as a safety concern,5,6 which have been documented in case reports.7-11 Mechanisms of action that have been thought to affect platelet aggregation include:
- depletion of serotonin in platelets
- increase in capillary fragility
- modification of platelet plug formation
- responsiveness of peptide-induced activation of platelets through stimulation of the thrombin receptor.7,8,12,13
The severity of bleeding varies with patient-related factors, such as a history of gastritis, peptic ulcer disease, and heavy bleeding during menses; use of gastrotoxic drugs, particularly nonsteroidal anti-inflammatory drugs (NSAIDs), also have been shown to increase this risk.14,15 For patients taking SRIs and gastrotoxic drugs (eg, NSAIDs), use of acid-suppressing agents have been shown to limit the risk of bleeding.16
Studies evaluating relative bleeding risks among classes of antidepressants have not shown increased risk with tricyclic antidepressants compared with SSRIs.17 Adenosine triphosphate (ATP), adenosine diphosphate (ADP), and adenosine monophosphate (AMP) are signaling molecules in the vascular system and are important in the thromboregulatory system. Studies in rats reported significant inhibition of ATP, ADP, and AMP hydrolysis with chronic treatment with fluoxetine and nortriptyline, and suggested that both medications changed the nucleotide catabolism, which means that homeostasis of the vascular system can be altered by antidepressant treatments.18 This is one possible pathway in the role these medications play in the etiology of dysregulation of the thromboregulatory system.
We did not anticipate that our patient would develop similar purpura with bupropion because the bleeding risk associated with antidepressants has been attributed to the effect of serotonin on platelets. Studies observing the effect of SSRIs, SNRIs, and bupropion on platelets and bleeding have not shown significant risk with bupropion.19 Bleeding associated with bupropion is atypical and needs to be further studied. Although this medication is centrally selective in its action on dopamine receptors, it might have possible peripheral effect on other neurotransmitters, including serotonin.
Ms. R had no personal or family history of purpura or a bleeding disorder. Significant improvement in her physical signs after discontinuing medications and recurrence of pupura with rechallenge indicate that this reaction was triggered by 3 different classes of antidepressants. Family history of similar reaction further suggests a genetic predisposition to platelet dysfunction to antidepressant treatment in a select group of patients.
Limitations include the possibility of senile purpura, which cannot be ruled out despite strong indications that antidepressants were the cause. The possibility of drug interactions needs be considered as well. Ms. R was taking albuterol and a steroid inhaler for her COPD at the time of the initial medication trials, which did not interact with duloxetine or bupropion. During the trials with duloxetine and then nortriptyline, she was taking acetaminophen/hydrocodone in addition to her inhalers, and no significant interactions with the antidepressants were identified. Interactions with unreported or over-the-counter medications or supplements are a possibility.
Before prescribing an antidepressant, we suggest taking a careful history including a family history of bleeding disorders and adverse effects of antidepressants, especially in patients who have risk factors (eg, concomitant use of gastrotoxic medications). Use of gastric acid-suppressing medications could be considered if antidepressants are used. Further investigations into the incidence, risk factors, mechanism of action, and treatment of this adverse effect are indicated.
Drug Brand Names
Acetaminophen/hydrocodone • Lorcet, Norco, Vicodin
Albuterol • Proventil
Bupropion • Wellbutrin
Duloxetine • Cymbalta
Nortriptyline • Pamelor, Aventyl
Selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) have been a welcome addition to the armamentarium for treating depressive disorders, neuropathic pain, and anxiety disorders. Despite the more favorable side-effect profile compared with tricyclic antidepressants and monoamine oxidase inhibitors, these serotonergic agents have been associated with bleeding disorders, purpura, thrombocytopenia, and, in extreme cases, death.1-4
We describe a case of purpura associated with different classes of antidepressants, including the non-serotonergic agent bupropion, as well as a family history of similar adverse effects to antidepressants.
CASE Purpura resolves when drug is stopped
Ms. R, age 70, presents with major depressive disorder and fibromyalgia and is receiving duloxetine, 20 mg/d, which is gradually increased to 60 mg. She also has a history of chronic obstructive pulmonary disease (COPD), for which she is taking albuterol and a steroid inhaler. Ms. R responds well to treatment; however, she develops blue–purple purpura on her arms each measuring 1 to 2 inches. Laboratory test results including platelet count and prothrombin time/international normalized ratio are within normal ranges. Duloxetine is stopped, and purpura resolves in 1 to 2 weeks. To avoid serotonergic antidepressants, Ms. R receives bupropion XL, 150 mg; however, similar purpura develops, then resolves when the medication is discontinued. She is lost to follow up for approximately 6 months, but returns requesting a rechallenge with duloxetine for her depression, which has worsened. Duloxetine is restarted with similar results and is then discontinued. Because she has developed neuropathy, Ms. R is started on nortriptyline, 25 mg/d, increased to 50 mg/d, but purpura develops again, which resolves when medication is discontinued. Ms. R’s daughter reports she also developed a similar reaction with several antidepressants, which resolved with medication discontinuation.
Bleeding risk with antidepressants
The role of serotonin reuptake inhibitors (SRIs) in inducing bleeding has emerged as a safety concern,5,6 which have been documented in case reports.7-11 Mechanisms of action that have been thought to affect platelet aggregation include:
- depletion of serotonin in platelets
- increase in capillary fragility
- modification of platelet plug formation
- responsiveness of peptide-induced activation of platelets through stimulation of the thrombin receptor.7,8,12,13
The severity of bleeding varies with patient-related factors, such as a history of gastritis, peptic ulcer disease, and heavy bleeding during menses; use of gastrotoxic drugs, particularly nonsteroidal anti-inflammatory drugs (NSAIDs), also have been shown to increase this risk.14,15 For patients taking SRIs and gastrotoxic drugs (eg, NSAIDs), use of acid-suppressing agents have been shown to limit the risk of bleeding.16
Studies evaluating relative bleeding risks among classes of antidepressants have not shown increased risk with tricyclic antidepressants compared with SSRIs.17 Adenosine triphosphate (ATP), adenosine diphosphate (ADP), and adenosine monophosphate (AMP) are signaling molecules in the vascular system and are important in the thromboregulatory system. Studies in rats reported significant inhibition of ATP, ADP, and AMP hydrolysis with chronic treatment with fluoxetine and nortriptyline, and suggested that both medications changed the nucleotide catabolism, which means that homeostasis of the vascular system can be altered by antidepressant treatments.18 This is one possible pathway in the role these medications play in the etiology of dysregulation of the thromboregulatory system.
We did not anticipate that our patient would develop similar purpura with bupropion because the bleeding risk associated with antidepressants has been attributed to the effect of serotonin on platelets. Studies observing the effect of SSRIs, SNRIs, and bupropion on platelets and bleeding have not shown significant risk with bupropion.19 Bleeding associated with bupropion is atypical and needs to be further studied. Although this medication is centrally selective in its action on dopamine receptors, it might have possible peripheral effect on other neurotransmitters, including serotonin.
Ms. R had no personal or family history of purpura or a bleeding disorder. Significant improvement in her physical signs after discontinuing medications and recurrence of pupura with rechallenge indicate that this reaction was triggered by 3 different classes of antidepressants. Family history of similar reaction further suggests a genetic predisposition to platelet dysfunction to antidepressant treatment in a select group of patients.
Limitations include the possibility of senile purpura, which cannot be ruled out despite strong indications that antidepressants were the cause. The possibility of drug interactions needs be considered as well. Ms. R was taking albuterol and a steroid inhaler for her COPD at the time of the initial medication trials, which did not interact with duloxetine or bupropion. During the trials with duloxetine and then nortriptyline, she was taking acetaminophen/hydrocodone in addition to her inhalers, and no significant interactions with the antidepressants were identified. Interactions with unreported or over-the-counter medications or supplements are a possibility.
Before prescribing an antidepressant, we suggest taking a careful history including a family history of bleeding disorders and adverse effects of antidepressants, especially in patients who have risk factors (eg, concomitant use of gastrotoxic medications). Use of gastric acid-suppressing medications could be considered if antidepressants are used. Further investigations into the incidence, risk factors, mechanism of action, and treatment of this adverse effect are indicated.
Drug Brand Names
Acetaminophen/hydrocodone • Lorcet, Norco, Vicodin
Albuterol • Proventil
Bupropion • Wellbutrin
Duloxetine • Cymbalta
Nortriptyline • Pamelor, Aventyl
1. Cymbalta [package insert]. Indianapolis, IN: Eli Lilly and Company; 2014.
2. Amitriptyline. Medscape. http://reference.medscape.com/drug/levate-amitriptyline-342936. Accessed December 19, 2016.
3. Wellbutrin [package insert]. Triangle Park, NC: GlaxoSmithKline; 2004.
4. Balhara Y, Sagar R, Varghese ST. Bleeding gums: duloxetine may be the cause. J Postgrad Med. 2007;53(1):44-45.
5. Paton C, Ferrier IN. SSRIs and gastrointestinal bleeding. BMJ. 2005;331(7516):529-530.
6. Turner MS, May DB, Arthur RR, et al. Clinical impact of selective serotonin reuptake inhibitors therapy with bleeding risks. J Intern Med. 2007;261(3):205-213.
7. Humphries JE, Wheby MS, VandenBerg SR. Fluoxetine and the bleeding time. Arch Pathol Lab Med. 1990;114(7):727-728.
8. Alderman CP, Moritz CK, Ben-Tovim DI. Abnormal platelet aggregation associated with fluoxetine therapy. Ann Pharmacother. 1992;26(12):1517-1519.
9. Calhoun JW, Calhoun DD. Prolonged bleeding time in a patient treated with sertraline. Am J Psychiatry. 1996;153(3):443.
10. Ottervanger JP, Stricker BH, Huls J, et al. Bleeding attributed to the intake of paroxetine. Am J Psychiatry. 1994;151(5):781-782.
11. de Abajo FJ, Rodríguez LA, Montero D, et al. Association between selective serotonin reuptake inhibitors and upper gastrointestinal bleeding: population based case-control study. BMJ. 1999;319(7217):1106-1109.
12. Nelva A, Guy C, Tardy-Poncet B, et al. Hemorrhagic syndromes related to selective serotonin reuptake inhibitor (SSRI) antidepressants: seven case reports and review of the literature [in French]. Rev Med Interne. 2000;21(2):152-160.
13. de Abajo FJ, Montero D, Rodríguez LA, et al. Antidepressants and risk of upper gastrointestinal bleeding. Basic Clin Pharmacol Toxicol. 2006;98(3):304-310.
14. Tata LJ, Fortun PJ, Hubbard RB, et al. Does concurrent prescription of selective serotonin reuptake inhibitors and non-steroidal anti-inflammatory drugs substantially increase the risk of upper gastrointestinal bleeding? Aliment Pharmacol Ther. 2005;22(3):175-181.
15. Yuan Y, Tsoi K, Hunt RH. Selective serotonin reuptake inhibitors and risk of upper GI bleeding: confusion or confounding? Am J Med. 2006;119(9):719-727.
16. de Abajo FJ, García-Rodríguez LA. Risk of upper gastrointestinal tract bleeding associated with selective serotonin reuptake inhibitors and venlafaxine therapy: interaction with nonsteroidal anti-inflammatory drugs and effect of acid-suppressing agents. Arch Gen Psychiatry. 2008;65(7):795-803.
17. Barbui C, Andretta M, De Vitis G, et al. Antidepressant drug prescription and risk of abnormal bleeding: a case-control study. J Clin Pharmacol. 2009;29(1):33-38.
18. Pedrazza EL, Senger MR, Rico EP, et al. Fluoxetine and nortriptyline affect NTPDase and 5’-nucleotidase activities in rat blood serum. Life Sci. 2007;81(15):1205-1210.
19. Song HR, Jung YE, Wang HR, et al. Platelet count alterations associated with escitalopram, venlafaxine and bupropion in depressive patients. Psychiatry Clin Neurosci. 2012;66(5):457-459.
1. Cymbalta [package insert]. Indianapolis, IN: Eli Lilly and Company; 2014.
2. Amitriptyline. Medscape. http://reference.medscape.com/drug/levate-amitriptyline-342936. Accessed December 19, 2016.
3. Wellbutrin [package insert]. Triangle Park, NC: GlaxoSmithKline; 2004.
4. Balhara Y, Sagar R, Varghese ST. Bleeding gums: duloxetine may be the cause. J Postgrad Med. 2007;53(1):44-45.
5. Paton C, Ferrier IN. SSRIs and gastrointestinal bleeding. BMJ. 2005;331(7516):529-530.
6. Turner MS, May DB, Arthur RR, et al. Clinical impact of selective serotonin reuptake inhibitors therapy with bleeding risks. J Intern Med. 2007;261(3):205-213.
7. Humphries JE, Wheby MS, VandenBerg SR. Fluoxetine and the bleeding time. Arch Pathol Lab Med. 1990;114(7):727-728.
8. Alderman CP, Moritz CK, Ben-Tovim DI. Abnormal platelet aggregation associated with fluoxetine therapy. Ann Pharmacother. 1992;26(12):1517-1519.
9. Calhoun JW, Calhoun DD. Prolonged bleeding time in a patient treated with sertraline. Am J Psychiatry. 1996;153(3):443.
10. Ottervanger JP, Stricker BH, Huls J, et al. Bleeding attributed to the intake of paroxetine. Am J Psychiatry. 1994;151(5):781-782.
11. de Abajo FJ, Rodríguez LA, Montero D, et al. Association between selective serotonin reuptake inhibitors and upper gastrointestinal bleeding: population based case-control study. BMJ. 1999;319(7217):1106-1109.
12. Nelva A, Guy C, Tardy-Poncet B, et al. Hemorrhagic syndromes related to selective serotonin reuptake inhibitor (SSRI) antidepressants: seven case reports and review of the literature [in French]. Rev Med Interne. 2000;21(2):152-160.
13. de Abajo FJ, Montero D, Rodríguez LA, et al. Antidepressants and risk of upper gastrointestinal bleeding. Basic Clin Pharmacol Toxicol. 2006;98(3):304-310.
14. Tata LJ, Fortun PJ, Hubbard RB, et al. Does concurrent prescription of selective serotonin reuptake inhibitors and non-steroidal anti-inflammatory drugs substantially increase the risk of upper gastrointestinal bleeding? Aliment Pharmacol Ther. 2005;22(3):175-181.
15. Yuan Y, Tsoi K, Hunt RH. Selective serotonin reuptake inhibitors and risk of upper GI bleeding: confusion or confounding? Am J Med. 2006;119(9):719-727.
16. de Abajo FJ, García-Rodríguez LA. Risk of upper gastrointestinal tract bleeding associated with selective serotonin reuptake inhibitors and venlafaxine therapy: interaction with nonsteroidal anti-inflammatory drugs and effect of acid-suppressing agents. Arch Gen Psychiatry. 2008;65(7):795-803.
17. Barbui C, Andretta M, De Vitis G, et al. Antidepressant drug prescription and risk of abnormal bleeding: a case-control study. J Clin Pharmacol. 2009;29(1):33-38.
18. Pedrazza EL, Senger MR, Rico EP, et al. Fluoxetine and nortriptyline affect NTPDase and 5’-nucleotidase activities in rat blood serum. Life Sci. 2007;81(15):1205-1210.
19. Song HR, Jung YE, Wang HR, et al. Platelet count alterations associated with escitalopram, venlafaxine and bupropion in depressive patients. Psychiatry Clin Neurosci. 2012;66(5):457-459.

HHS Buys Growth Factor Products for Emergency Use
High doses of radiation are often followed by infection. HHS is preparing for emergencies by buying 2 colony-stimulating factor (CSF) products to reduce infection and risk of death in radiologic or nuclear incidents.
Related: Emergency Test for Absorbed Radiation
HHS is purchasing Neulasta (Amgen USA, Inc) and Leukine (Sanofi-Aventis US), under agreements totaling about $37.7 million and 37.6 million, respectively. Neulasta already is FDA approved to treat cancer patients exposed to high levels of radiation that damage bone marrow. Leukine is undergoing studies needed for approval.
The Biomedical Advanced Research and Development Authority had earlier sponsored advanced development and purchase of Neupogen, another leukocyte growth factor product approved for treating adults and children exposed to radiation that damages bone marrow.
The deal for Neulasta and Leukine thus increases the number of CSF factor doses available for use in an emergency. It also increases operational capability, HHS says, since treatments with Neulasta are given once weekly, whereas treatment with Neupogen is daily.
High doses of radiation are often followed by infection. HHS is preparing for emergencies by buying 2 colony-stimulating factor (CSF) products to reduce infection and risk of death in radiologic or nuclear incidents.
Related: Emergency Test for Absorbed Radiation
HHS is purchasing Neulasta (Amgen USA, Inc) and Leukine (Sanofi-Aventis US), under agreements totaling about $37.7 million and 37.6 million, respectively. Neulasta already is FDA approved to treat cancer patients exposed to high levels of radiation that damage bone marrow. Leukine is undergoing studies needed for approval.
The Biomedical Advanced Research and Development Authority had earlier sponsored advanced development and purchase of Neupogen, another leukocyte growth factor product approved for treating adults and children exposed to radiation that damages bone marrow.
The deal for Neulasta and Leukine thus increases the number of CSF factor doses available for use in an emergency. It also increases operational capability, HHS says, since treatments with Neulasta are given once weekly, whereas treatment with Neupogen is daily.
High doses of radiation are often followed by infection. HHS is preparing for emergencies by buying 2 colony-stimulating factor (CSF) products to reduce infection and risk of death in radiologic or nuclear incidents.
Related: Emergency Test for Absorbed Radiation
HHS is purchasing Neulasta (Amgen USA, Inc) and Leukine (Sanofi-Aventis US), under agreements totaling about $37.7 million and 37.6 million, respectively. Neulasta already is FDA approved to treat cancer patients exposed to high levels of radiation that damage bone marrow. Leukine is undergoing studies needed for approval.
The Biomedical Advanced Research and Development Authority had earlier sponsored advanced development and purchase of Neupogen, another leukocyte growth factor product approved for treating adults and children exposed to radiation that damages bone marrow.
The deal for Neulasta and Leukine thus increases the number of CSF factor doses available for use in an emergency. It also increases operational capability, HHS says, since treatments with Neulasta are given once weekly, whereas treatment with Neupogen is daily.
Old drug, new tricks possible in MM

SAN DIEGO—An antiretroviral drug used to treat the human immunodeficiency virus (HIV) may find a role in the treatment of multiple myeloma (MM) patients who are proteasome inhibitor (PI)-refractory.
According to investigators, nelfinavir may sensitize refractory patients so that PI-based treatments become an option for them.
In a phase 2 study of 34 patients, nelfinavir in combination with bortezomib and dexamethasone produced an objective response rate of 65%, which investigators called an “exceptional” response in this heavily pretreated, mostly dual-refractory patient population.
Christoph Driessen, MD, PhD, of Kantonsspital St Gallen in Switzerland, discussed the findings of this study, known as SAKK 39/13, at the 2016 ASH Annual Meeting as abstract 487.
Dr Driessen explained that downregulation of IRE1/XBP1 produces PI resistance, and this downregulation occurs in PI-refractory MM patients.
High expression of IRE1/XBP1 correlates with bortezomib sensitivity, and pharmacologic upregulation of IRE1/XBP1 re-sensitizes myeloma cells to PI treatment.
Nelfinavir, which overcomes PI resistance in vitro, is approved for oral HIV therapy.
“It’s an old drug, it’s a generic drug,” Dr Driessen said, and it’s approved at a dose of 2 x 1250 mg daily.
So the SAKK investigators undertook a phase 1 trial of nelfinavir in MM patients.
In an exploratory extension cohort, they found that 5 of 6 MM patients double-refractory to bortezomib and lenalidomide experienced clinical benefit from nelfinavir at the recommended phase 2 dose (2 x 2500 mg daily) in addition to standard treatment with bortezomib and dexamethasone.
Three patients achieved a partial response (PR) and 3 a minor response (MR).
The investigators’ objective in the phase 2 study was to determine whether the addition of nelfinavir to approved bortezomib-dexamethasone therapy is sufficiently active to merit further investigation in a randomized trial.
Study design
Patients in this prospective, single-arm, multicenter, open-label trial received the following treatment:
- Nelfinavir at 2 x 2500 mg orally on days 1–14
- Bortezomib at 1.3 mg/m2 intravenously or subcutaneously on days 1, 4, 8, and 11
- Dexamethasone at 20 mg orally on days 1-2, 4-5, 8-9, and 11-12 of each 21-day cycle.
Trial therapy lasted for a maximum of 6 cycles (18 weeks).
Dr Driessen explained that the trial “was a truly academic trial, without any finances from industry or drug support from industry. So we actually had to get a grant to buy commercial drugs for the study on the commercial drug market, and that limited the duration of treatment in this trial.”
The primary endpoint of the trial was response rate—best response of PR or better by IMWG criteria.
Investigators considered a 30% or higher response rate promising.
Secondary endpoints included adverse events, time to next new anti-myeloma therapy or death, progressive disease under trial treatment, duration of response, progression-free survival, and time to progression.
Patients were eligible to enroll if they had been exposed to or could not tolerate an immunomodulatory drug, were refractory to their most recent PI-containing regimen, had a performance status of 3 or less, had creatinine clearance of 15 mL/minute or greater, had a platelet count of 50,000/μL or more, and had a hemoglobin level of 8.0 g/dL or higher.
Patients were excluded if they had uncontrolled, clinically significant, active concurrent disease, concomitant additional systemic cancer treatment, concomitant radiotherapy, or significant neuropathy of grades 3-4 or grade 2 with pain.
Patient population
Thirty-four patients enrolled on the trial. They were a median age of 67 (range, 42–82), 62% were male, 91% had a performance status of 0 or 1, and 76% had a prior autologous stem cell transplant.
They had a median of 5 prior systemic therapies (range, 2–10), and 38% had poor-risk cytogenetics.
The time from last dose of prior therapy to enrollment on the study was a median of 27 days.
“So [it was] a truly progressive, highly refractory myeloma population,” Dr Driessen emphasized.
All 34 patients were refractory to bortezomib. All patients were also exposed to lenalidomide, and 79% were refractory to it.
Forty-four percent were refractory to pomalidomide, and 6% were refractory to carfilzomib. One patient was refractory to all 4 agents.
“Very few patients were exposed to carfilzomib because it wasn’t available in Switzerland at that time,” Dr Driessen explained.
Efficacy
Patients received a median of 4.5 cycles of therapy (range, 1–6), and the best response of PR or greater was achieved by 22 patients (65%).
Five patients (15%) achieved a very good partial response (VGPR), 17 (50%) PR, 3 (9%) MR, and 4 (12%) stable disease.
Twenty-five patients (74%) achieved a clinical benefit (VGPR+PR+MR).
Ten of the 13 patients (77%) with poor-risk cytogenetics achieved a best response of PR or greater.
Patients had a median of 16 weeks (range, 13–24) time to a new anti-myeloma therapy or death, and 13 patients (38%) had confirmed progressive disease while on trial therapy.
In 32 patients, all but 4 had a decrease from baseline in serum M protein or serum free light chain concentration.
Efficacy by prior therapy
Twenty-two of 34 patients (65%) refractory to bortezomib had a best response of PR or greater.
For patients refractory to bortezomib and lenalidomide, 70% achieved a best response of PR or greater.
For patients refractory to bortezomib, lenalidomide, and pomalidomide, 60% achieved a best response of PR or greater.
And for patients who were refractory to bortezomib, lenalidomide, and carfilzomib, 50% achieved a best response of PR or greater.
Adverse events
“The hematologic toxicity was essentially what you would expect from this heavily pretreated population,” Dr Driessen said.
“We did, however, experience 4 deaths on the trial therapy from infectious complications of sepsis and neutropenia, and we don’t know whether this is a true signal or whether this is due to the low numbers. We did not mandate antibiotic prophylaxis on the trial.”
Grade 3 or higher adverse events (AEs) occurring in 2 or more patients were anemia (n=10), febrile neutropenia (n=4, including 1 grade 5), thrombocytopenia (n=15), lung infection (n=8), sepsis (n=3, all grade 5), fatigue (n=5), peripheral sensory neuropathy (n=3), hypertension (n=6), increased creatinine (n=4), hyperglycemia (n=6) hypokalemia (n=3), and hyponatremia (n=5).
Dr Driessen indicated that with a future generic version of bortezomib, nelfinavir plus bortezomib and dexamethasone “has the potential to become a fully generic, affordable, active therapy option for PI-refractory patients.”
The investigators believe the results of their study call for further development of nelfinavir as a sensitizing drug for PI-based treatments and as a promising new agent for MM therapy. ![]()
SAN DIEGO—An antiretroviral drug used to treat the human immunodeficiency virus (HIV) may find a role in the treatment of multiple myeloma (MM) patients who are proteasome inhibitor (PI)-refractory.
According to investigators, nelfinavir may sensitize refractory patients so that PI-based treatments become an option for them.
In a phase 2 study of 34 patients, nelfinavir in combination with bortezomib and dexamethasone produced an objective response rate of 65%, which investigators called an “exceptional” response in this heavily pretreated, mostly dual-refractory patient population.
Christoph Driessen, MD, PhD, of Kantonsspital St Gallen in Switzerland, discussed the findings of this study, known as SAKK 39/13, at the 2016 ASH Annual Meeting as abstract 487.
Dr Driessen explained that downregulation of IRE1/XBP1 produces PI resistance, and this downregulation occurs in PI-refractory MM patients.
High expression of IRE1/XBP1 correlates with bortezomib sensitivity, and pharmacologic upregulation of IRE1/XBP1 re-sensitizes myeloma cells to PI treatment.
Nelfinavir, which overcomes PI resistance in vitro, is approved for oral HIV therapy.
“It’s an old drug, it’s a generic drug,” Dr Driessen said, and it’s approved at a dose of 2 x 1250 mg daily.
So the SAKK investigators undertook a phase 1 trial of nelfinavir in MM patients.
In an exploratory extension cohort, they found that 5 of 6 MM patients double-refractory to bortezomib and lenalidomide experienced clinical benefit from nelfinavir at the recommended phase 2 dose (2 x 2500 mg daily) in addition to standard treatment with bortezomib and dexamethasone.
Three patients achieved a partial response (PR) and 3 a minor response (MR).
The investigators’ objective in the phase 2 study was to determine whether the addition of nelfinavir to approved bortezomib-dexamethasone therapy is sufficiently active to merit further investigation in a randomized trial.
Study design
Patients in this prospective, single-arm, multicenter, open-label trial received the following treatment:
- Nelfinavir at 2 x 2500 mg orally on days 1–14
- Bortezomib at 1.3 mg/m2 intravenously or subcutaneously on days 1, 4, 8, and 11
- Dexamethasone at 20 mg orally on days 1-2, 4-5, 8-9, and 11-12 of each 21-day cycle.
Trial therapy lasted for a maximum of 6 cycles (18 weeks).
Dr Driessen explained that the trial “was a truly academic trial, without any finances from industry or drug support from industry. So we actually had to get a grant to buy commercial drugs for the study on the commercial drug market, and that limited the duration of treatment in this trial.”
The primary endpoint of the trial was response rate—best response of PR or better by IMWG criteria.
Investigators considered a 30% or higher response rate promising.
Secondary endpoints included adverse events, time to next new anti-myeloma therapy or death, progressive disease under trial treatment, duration of response, progression-free survival, and time to progression.
Patients were eligible to enroll if they had been exposed to or could not tolerate an immunomodulatory drug, were refractory to their most recent PI-containing regimen, had a performance status of 3 or less, had creatinine clearance of 15 mL/minute or greater, had a platelet count of 50,000/μL or more, and had a hemoglobin level of 8.0 g/dL or higher.
Patients were excluded if they had uncontrolled, clinically significant, active concurrent disease, concomitant additional systemic cancer treatment, concomitant radiotherapy, or significant neuropathy of grades 3-4 or grade 2 with pain.
Patient population
Thirty-four patients enrolled on the trial. They were a median age of 67 (range, 42–82), 62% were male, 91% had a performance status of 0 or 1, and 76% had a prior autologous stem cell transplant.
They had a median of 5 prior systemic therapies (range, 2–10), and 38% had poor-risk cytogenetics.
The time from last dose of prior therapy to enrollment on the study was a median of 27 days.
“So [it was] a truly progressive, highly refractory myeloma population,” Dr Driessen emphasized.
All 34 patients were refractory to bortezomib. All patients were also exposed to lenalidomide, and 79% were refractory to it.
Forty-four percent were refractory to pomalidomide, and 6% were refractory to carfilzomib. One patient was refractory to all 4 agents.
“Very few patients were exposed to carfilzomib because it wasn’t available in Switzerland at that time,” Dr Driessen explained.
Efficacy
Patients received a median of 4.5 cycles of therapy (range, 1–6), and the best response of PR or greater was achieved by 22 patients (65%).
Five patients (15%) achieved a very good partial response (VGPR), 17 (50%) PR, 3 (9%) MR, and 4 (12%) stable disease.
Twenty-five patients (74%) achieved a clinical benefit (VGPR+PR+MR).
Ten of the 13 patients (77%) with poor-risk cytogenetics achieved a best response of PR or greater.
Patients had a median of 16 weeks (range, 13–24) time to a new anti-myeloma therapy or death, and 13 patients (38%) had confirmed progressive disease while on trial therapy.
In 32 patients, all but 4 had a decrease from baseline in serum M protein or serum free light chain concentration.
Efficacy by prior therapy
Twenty-two of 34 patients (65%) refractory to bortezomib had a best response of PR or greater.
For patients refractory to bortezomib and lenalidomide, 70% achieved a best response of PR or greater.
For patients refractory to bortezomib, lenalidomide, and pomalidomide, 60% achieved a best response of PR or greater.
And for patients who were refractory to bortezomib, lenalidomide, and carfilzomib, 50% achieved a best response of PR or greater.
Adverse events
“The hematologic toxicity was essentially what you would expect from this heavily pretreated population,” Dr Driessen said.
“We did, however, experience 4 deaths on the trial therapy from infectious complications of sepsis and neutropenia, and we don’t know whether this is a true signal or whether this is due to the low numbers. We did not mandate antibiotic prophylaxis on the trial.”
Grade 3 or higher adverse events (AEs) occurring in 2 or more patients were anemia (n=10), febrile neutropenia (n=4, including 1 grade 5), thrombocytopenia (n=15), lung infection (n=8), sepsis (n=3, all grade 5), fatigue (n=5), peripheral sensory neuropathy (n=3), hypertension (n=6), increased creatinine (n=4), hyperglycemia (n=6) hypokalemia (n=3), and hyponatremia (n=5).
Dr Driessen indicated that with a future generic version of bortezomib, nelfinavir plus bortezomib and dexamethasone “has the potential to become a fully generic, affordable, active therapy option for PI-refractory patients.”
The investigators believe the results of their study call for further development of nelfinavir as a sensitizing drug for PI-based treatments and as a promising new agent for MM therapy. ![]()
SAN DIEGO—An antiretroviral drug used to treat the human immunodeficiency virus (HIV) may find a role in the treatment of multiple myeloma (MM) patients who are proteasome inhibitor (PI)-refractory.
According to investigators, nelfinavir may sensitize refractory patients so that PI-based treatments become an option for them.
In a phase 2 study of 34 patients, nelfinavir in combination with bortezomib and dexamethasone produced an objective response rate of 65%, which investigators called an “exceptional” response in this heavily pretreated, mostly dual-refractory patient population.
Christoph Driessen, MD, PhD, of Kantonsspital St Gallen in Switzerland, discussed the findings of this study, known as SAKK 39/13, at the 2016 ASH Annual Meeting as abstract 487.
Dr Driessen explained that downregulation of IRE1/XBP1 produces PI resistance, and this downregulation occurs in PI-refractory MM patients.
High expression of IRE1/XBP1 correlates with bortezomib sensitivity, and pharmacologic upregulation of IRE1/XBP1 re-sensitizes myeloma cells to PI treatment.
Nelfinavir, which overcomes PI resistance in vitro, is approved for oral HIV therapy.
“It’s an old drug, it’s a generic drug,” Dr Driessen said, and it’s approved at a dose of 2 x 1250 mg daily.
So the SAKK investigators undertook a phase 1 trial of nelfinavir in MM patients.
In an exploratory extension cohort, they found that 5 of 6 MM patients double-refractory to bortezomib and lenalidomide experienced clinical benefit from nelfinavir at the recommended phase 2 dose (2 x 2500 mg daily) in addition to standard treatment with bortezomib and dexamethasone.
Three patients achieved a partial response (PR) and 3 a minor response (MR).
The investigators’ objective in the phase 2 study was to determine whether the addition of nelfinavir to approved bortezomib-dexamethasone therapy is sufficiently active to merit further investigation in a randomized trial.
Study design
Patients in this prospective, single-arm, multicenter, open-label trial received the following treatment:
- Nelfinavir at 2 x 2500 mg orally on days 1–14
- Bortezomib at 1.3 mg/m2 intravenously or subcutaneously on days 1, 4, 8, and 11
- Dexamethasone at 20 mg orally on days 1-2, 4-5, 8-9, and 11-12 of each 21-day cycle.
Trial therapy lasted for a maximum of 6 cycles (18 weeks).
Dr Driessen explained that the trial “was a truly academic trial, without any finances from industry or drug support from industry. So we actually had to get a grant to buy commercial drugs for the study on the commercial drug market, and that limited the duration of treatment in this trial.”
The primary endpoint of the trial was response rate—best response of PR or better by IMWG criteria.
Investigators considered a 30% or higher response rate promising.
Secondary endpoints included adverse events, time to next new anti-myeloma therapy or death, progressive disease under trial treatment, duration of response, progression-free survival, and time to progression.
Patients were eligible to enroll if they had been exposed to or could not tolerate an immunomodulatory drug, were refractory to their most recent PI-containing regimen, had a performance status of 3 or less, had creatinine clearance of 15 mL/minute or greater, had a platelet count of 50,000/μL or more, and had a hemoglobin level of 8.0 g/dL or higher.
Patients were excluded if they had uncontrolled, clinically significant, active concurrent disease, concomitant additional systemic cancer treatment, concomitant radiotherapy, or significant neuropathy of grades 3-4 or grade 2 with pain.
Patient population
Thirty-four patients enrolled on the trial. They were a median age of 67 (range, 42–82), 62% were male, 91% had a performance status of 0 or 1, and 76% had a prior autologous stem cell transplant.
They had a median of 5 prior systemic therapies (range, 2–10), and 38% had poor-risk cytogenetics.
The time from last dose of prior therapy to enrollment on the study was a median of 27 days.
“So [it was] a truly progressive, highly refractory myeloma population,” Dr Driessen emphasized.
All 34 patients were refractory to bortezomib. All patients were also exposed to lenalidomide, and 79% were refractory to it.
Forty-four percent were refractory to pomalidomide, and 6% were refractory to carfilzomib. One patient was refractory to all 4 agents.
“Very few patients were exposed to carfilzomib because it wasn’t available in Switzerland at that time,” Dr Driessen explained.
Efficacy
Patients received a median of 4.5 cycles of therapy (range, 1–6), and the best response of PR or greater was achieved by 22 patients (65%).
Five patients (15%) achieved a very good partial response (VGPR), 17 (50%) PR, 3 (9%) MR, and 4 (12%) stable disease.
Twenty-five patients (74%) achieved a clinical benefit (VGPR+PR+MR).
Ten of the 13 patients (77%) with poor-risk cytogenetics achieved a best response of PR or greater.
Patients had a median of 16 weeks (range, 13–24) time to a new anti-myeloma therapy or death, and 13 patients (38%) had confirmed progressive disease while on trial therapy.
In 32 patients, all but 4 had a decrease from baseline in serum M protein or serum free light chain concentration.
Efficacy by prior therapy
Twenty-two of 34 patients (65%) refractory to bortezomib had a best response of PR or greater.
For patients refractory to bortezomib and lenalidomide, 70% achieved a best response of PR or greater.
For patients refractory to bortezomib, lenalidomide, and pomalidomide, 60% achieved a best response of PR or greater.
And for patients who were refractory to bortezomib, lenalidomide, and carfilzomib, 50% achieved a best response of PR or greater.
Adverse events
“The hematologic toxicity was essentially what you would expect from this heavily pretreated population,” Dr Driessen said.
“We did, however, experience 4 deaths on the trial therapy from infectious complications of sepsis and neutropenia, and we don’t know whether this is a true signal or whether this is due to the low numbers. We did not mandate antibiotic prophylaxis on the trial.”
Grade 3 or higher adverse events (AEs) occurring in 2 or more patients were anemia (n=10), febrile neutropenia (n=4, including 1 grade 5), thrombocytopenia (n=15), lung infection (n=8), sepsis (n=3, all grade 5), fatigue (n=5), peripheral sensory neuropathy (n=3), hypertension (n=6), increased creatinine (n=4), hyperglycemia (n=6) hypokalemia (n=3), and hyponatremia (n=5).
Dr Driessen indicated that with a future generic version of bortezomib, nelfinavir plus bortezomib and dexamethasone “has the potential to become a fully generic, affordable, active therapy option for PI-refractory patients.”
The investigators believe the results of their study call for further development of nelfinavir as a sensitizing drug for PI-based treatments and as a promising new agent for MM therapy. ![]()
A Primary Care Approach to Managing Chronic Noncancer Pain
The Primary Care Chronic Pain Program (PC-CPP) of the Women’s Primary Care Clinics at the VA Salt Lake City Health Care System (VASLCHCS) in Utah was the first VA primary care clinical service to incorporate patient participation in obtaining chronic opioid medications in the treatment of chronic noncancer pain. In addition, the program used a multimodality approach for chronic pain treatment and veteran education about the relationship between physical and mental health issues.
Treatment Complexity
Chronic, noncancer pain is a complex issue in the primary care setting. Diagnosis is difficult, patient education is time consuming, goals and expectations are often unclear, and the experience can be unsatisfying for the patient and the provider.1 These issues, combined with an estimated prevalence rate of 71% for moderate pain among veterans seen in primary care, present a unique challenge for the primary care provider (PCP), given the limited time available to spend with these complex patients.2 Comorbidity rates with mental health issues (eg, depression, anxiety, substance use disorders, etc), which range from 18% to 44%, add to the management challenges for PCPs.3
Veterans also pose unique challenges in pain care as they have a 2-fold greater risk of death from opioid overdose compared with that of the general population, and Utah has been shown to have the highest rate of veteran overdoses.4 Developing programs to help PCPs efficiently manage patients with chronic noncancer pain and mental health comorbidities was vital at VASLCHCS.
Before VASLCHCS established the PC-CPP, the treatment for chronic noncancer pain and related mental health comorbidities followed a biomedical model that separated physical and mental health with the treatment focus on pharmacologic management of symptoms by separate services. Consistent with the biomedical model, management of chronic noncancer pain commonly included long-term use of opioids.
Over the past 2 decades, the use of opioids for treating chronic noncancer pain has significantly increased, with more than 62 million opioid prescriptions dispensed in 2012.5 There are no longitudinal follow-up studies, however, beyond 16 weeks on the use of opioids.6 Further, patients who are prescribed increased opioids continue to report high levels of pain, poor quality of life, and functional disability.7 High-dose opioids also are associated with overdose deaths.
Likewise, PCPs in the Women’s Primary Care Clinics at the VASLCHCS struggled with decreasing opioid use, often because other interventions for managing pain and related mental health conditions in primary care were not readily available. Although the VASLCHCS has an effective specialty pain service caring for patients with complex pain issues, opioid morphine equivalent doses > 200 mg/d, and palliative care, patients with chronic noncancer pain treated in the primary care setting did not have a consistent treatment approach.
A chart review of women veterans seen in Women’s Primary Care Clinic (N = 122) revealed that the majority of patients lacked timely urine drug screening, state database queries, signed medication management agreements, and documentation consistent with state and national guidelines. Additionally, many patients lacked provider follow-through regarding alternative and adjunctive therapy consults, which were often discontinued after failed contact attempts or no-shows to scheduled appointments.
There also was a general consensus among the Women’s Primary Care Clinic PCPs that caring for patients with chronic noncancer pain was exhausting, time consuming, ineffective, and often straining on the patient-provider relationship, as evidenced in many patients’ request to change providers secondary to pain management. The PC-CPP was developed to help systematically facilitate safe opioid prescribing, manage chronic pain issues, and document evidence-based care among women veterans receiving treatment for chronic noncancer pain at the Women’s Primary Care Clinics at VASLCHCS while coordinating and following through with nonpharmacologic interventions.
Program Development
National, state, VA, and professional licensure guidelines for chronic noncancer pain treatment standards were reviewed with the goal of creating a program that was evidence based, would benefit the patient in terms of opioid prescribing and pain control, and improve function while identifying key elements of care and documentation that adequately covered the prescriber of retribution.1,8-10
Concurrent to a review of the guidelines was a review of the literature with the goal of identifying useful patient education and alternative interventions and chronic pain programs that were already established and might meet the clinic’s needs.10,11 These reviews provided direction for a generalized approach to caring for patients with chronic nonmalignant pain. They also clarified that although pain education programs existed nationally, a program that offered a holistic, reproducible, adherence-driven yet patient-centered approach to the patient prescribed opioids chronically in a primary care setting was lacking.
Guideline recommendations included but were not limited to the following1,8-10:
- Patient education about chronic pain and opioids
- Evaluation of pain, function, opioid misuse risk at least twice yearly
- Patient-centered and driven treatment plans
- A holistic approach to chronic pain interventions
- Review of treatment plan efficacy at least twice yearly
- Enzyme multiplied immunoassay technique urine drug screening (UDS) 2 times per year
- State prescription monitoring program query annually
- Signed iMedConsent for treatment of chronic pain
- Plan for safe discontinuation of opioids
- Documentation that the above has been performed with patient understanding
The literature suggested a multimodality approach to chronic nonmalignant pain by minimizing the use of opioids over time while emphasizing nonpharmacologic therapies, such as cognitive behavioral therapy (CBT), mindfulness, meditation, yoga, and spiritual growth, to name a few.10,11 These findings are based on several studies, which suggested that passive coping strategies (eg, use of medication for immediate relief, depending on others, restricting medications) result in an increase in subjective pain among chronic nonmalignant pain patients.12 Helping patients reduce frequent use of passive coping strategies is believed to decrease pain.12 Active coping strategies (eg, engaging in therapies, staying busy or active, distracting attention from pain) have been found to decrease pain.12 The PC-CPP program shifted health care outcomes and responsibilities away from the hierarchal PCP-patient relationship toward a collaborative relationship that encourages patient-driven, patient-centered care outcomes and shared responsibilities.
Program Overview
The PC-CPP was shaped by the following hypotheses: (1) Transparent expectations and consequences would increase functional scores and decrease chronic opioid doses; (2) Treatment plans consisting of chronic opioid prescriptions linked with interactive nonpharmacologic interventions led to decreased pain and increased functional scores; (3) Transparent expectations combined with a streamlined approach to the chronic nonmalignant pain patient would improve patient and PCP satisfaction scores.
The PC-CPP was developed to provide an efficient, effective, and evidence-based approach to managing chronic nonmalignant pain and opioid therapy issues in the primary care setting. Referred patients attend 1 shared medical appointment (SMA) every 6 months with up to 19 other female veterans also referred to the PC-CPP. The group was composed of only female veterans as the pilot study for this SMA occurred in the Women’s Clinic. At each 6-month SMA, patients received education from the Taking Opioids Responsibly for Your Safety and the Safety of Others (TORYSSO) guide13 and signed the corresponding long-term opioid therapy for pain informed consent form (iMedConsent).
The patient and a staff member developed a treatment plan that was patient driven and included at least 1 nonpharmacologic treatment option. The 1-hour nonpharmacologic sessions were either group or individual and occurred weekly for 6 to 8 weeks. These options included CBT for chronic pain, Living Well With Chronic Conditions, trauma-sensitive yoga, smoking cessation, mindfulness for stress and anxiety, MOVE! weight management, Walk With Ease, and a self-help option (VA-issued Manage Stress Workbook, 2014). The workbook was included as an option for those who lived far away, were limited by work schedules, or were unable to afford the copays for a 6- to 8-session program.
Inclusion and Exclusion Criteria
Any female veteran patient enrolled in the VASLCHCS with a chronic nonmalignant pain diagnosis who received daily opioids for 3 or more consecutive months from a PCP was included. Excluded individuals were those with cognitive decline/dementia, serious mental illness, psychosis, active suicidality, disruptive behavior flag, or those excluded by PCP discretion if it was determined that the patient would do better in a one-on-one setting with the PCP (Table 1). Patients taking > 200 MED/d of opioids who were seen in the VASLCHCS specialty pain clinic were also excluded.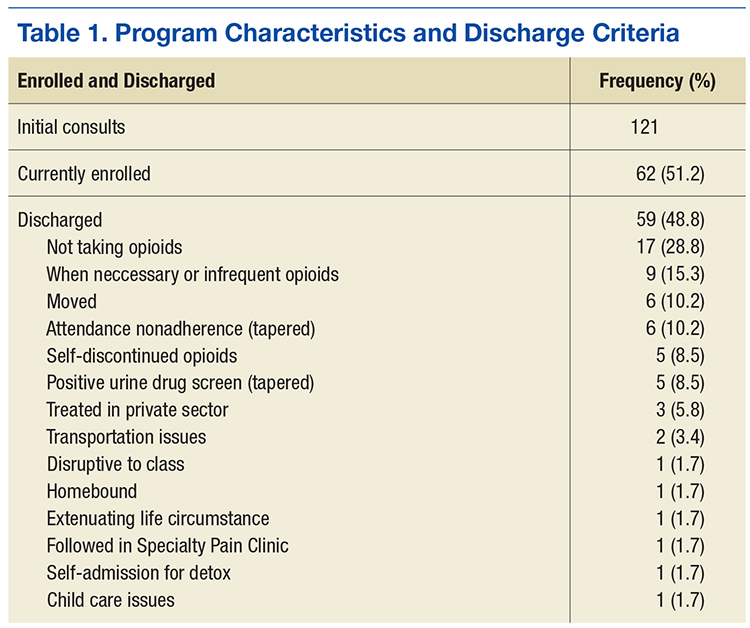
Patient and PCP Responsibilities
The patient was responsible for timely attendance and full participation in all SMA group classes as determined in the veteran’s Treatment Plan Agreement (TPA). In addition, the patient had to provide UDS when requested (a minimum of twice yearly) and communicate with the PCP if having a procedure requiring additional opioids. This was in line with the current standards set forth by the VA Opioid Safety Initiative (OSI) Taskforce.12
The PC-CPP provided education, evaluation, documentation, and referral and follow-up with the nonpharmacologic treatment options discussed but did not provide prescription medications. The PCP reviewed the medical documents completed in the PC-CPP, and the PCP was strongly encouraged to follow its recommendations. The expectation was that the PCP would support the PC-CPP when the care recommendation was for a pharmacist-guided opioid taper.
Lack of attendance was defined as a no-show or a reschedule. Patients were considered adherent if they missed fewer than 2 SMA appointments and 2 nonpharmacologic treatment appointments every 6 months. The patient was required to attend the SMA and nonpharmacologic treatment on the third appointment to remain adherent with PC-CPP expectations and agreements. Adherence was acknowledged after 12 and 24 months by a reduction in PC-CPP requirements.
Shared Medical Appointment
Patients referred to the program and who met inclusion criteria received letters explaining the importance of SMA attendance and follow-up reminder calls. At least 30 minutes before the SMA, the patient provided an UDS sample at the laboratory. Next, the patient received an individualized program packet that included the TORYSSO guide, iMedConsent, a TPA specific to the program, a brief pain inventory (BPI) and opioid risk tool, a list of medication disposal sites, and short descriptions of available nonpharmacologic therapies.
Each SMA began with a presentation delivered by a pharmacist, a psychologist, and a medical provider, discussing TORYSSO, program expectations, and holistic approaches to pain. Each SMA also included a rotating chronic pain information topic (eg, nutrition and pain, the physiology of addiction, and the value of multiple modalities in pain treatment). Together, the staff and patients reviewed and completed the blank forms enclosed in the individualized 
Each visit was entered into a Computerized Patient Record System (CPRS) template, which included a pain diagnosis, Opioid Risk Tool score, pain and functional scores, opioid fill history, last comprehensive metabolic panel, last electrocardiogram if on methadone, dates of signed agreements, patient adherence with SMA and optional therapies, and follow-up (eFigure). 
Every patient enrolled in the PC-CPP had to attend a SMA every 6 months. Patients continued this indefinitely while receiving opioids, and requirements were lessened for patients who had a history of meeting program requirements. For those fully adherent after the first year, only 1 nonpharmacologic intervention was needed (instead of 1 every 6 months) yearly. After 2 years of full adherence, nonpharmacologic interventions were no longer necessary as the expectation was that the patient would continue to use the strategies that they had learned over the previous 2 years. Patients left the PC-CPP if they chose to discontinue opioids, met any of the exclusion criteria, or were nonadherent. Tapering opioid medication was recommended for patients who missed a SMA meeting or 2 nonpharmacologic treatment meetings in a 6-month period; received opioids from more than 1 provider; test positive on a UDS for substances that should not be present; consistently testing negative on a UDS for substances that should be present (indicating diversion); or exhibiting other aberrant behavior (frequent requests for early refills, medications often lost/stolen, etc).
Program Barriers
The PC-CPP took about 2 years to set up, and several barriers were encountered. A thorough understanding of the following factors is necessary for establishing a similar program.
Initially, consults were placed by a designee (someone other than the PCP currently caring for the patient). The designee was usually a member of the PC-CPP who placed consults for all patients who had opioids listed on the CPRS profile. Further, patients who had any opioids within the past 3 months were initially included as were patients who wanted pain education but were not taking opioids. After 12 months, it became apparent that the focus of the PC-CPP should center on patients taking opioids for a minimum of 3 months consecutively. Patients who wanted only education could attend other hospital education opportunities, which helped keep the patient load manageable for PC-CPP staff. Further, to lessen patient confusion and improve adherence, the PCP placed the consult and discussed the program with the patient. Class sizes of 5 to 10 patients seemed to be ideal for patient participation and provider workload.
Patient Education
Initially, the SMA did not follow a standard curriculum, but the current format is more consistent, reproducible, streamlined, and organized. This adjustment improved SMA attendance as well as patient satisfaction, as the class started and finished on time. The SMA also started with numerous handouts, including brochures for nonpharmacologic programs offered at this facility. This led to patients feeling overwhelmed, missing the important forms, and wasted paper. Handouts were simplified to 2 color-coded forms (TPA and BPI).
The take-home assessment was streamlined to a single general assessment. This assessment consisted of 2 questions that asked patients to write a summary of what they learned and then write a summary of how they applied what they learned to their pain management. The VA Manage Stress Workbook also was added to the take-home materials. There are currently 5 different take-home options, which are necessary for those who live more than 50 miles from any VA facility or for those who have transportation issues.
Patient Distress
The SMA could be stressful for patients who felt they were being “punished” or who showed up more than 15 minutes late and had to reschedule the SMA. Having a mental health provider available was crucial for these situations.
Therapeutic Option Development
A cornerstone of the program was getting patients to participate in nonpharmacologic treatment options, which required a robust selection of programs. The VASLCHCS was fortunate to have many programs already available (Table 2), but this was not always the case for the VA community-based outpatient clinics (CBOCs).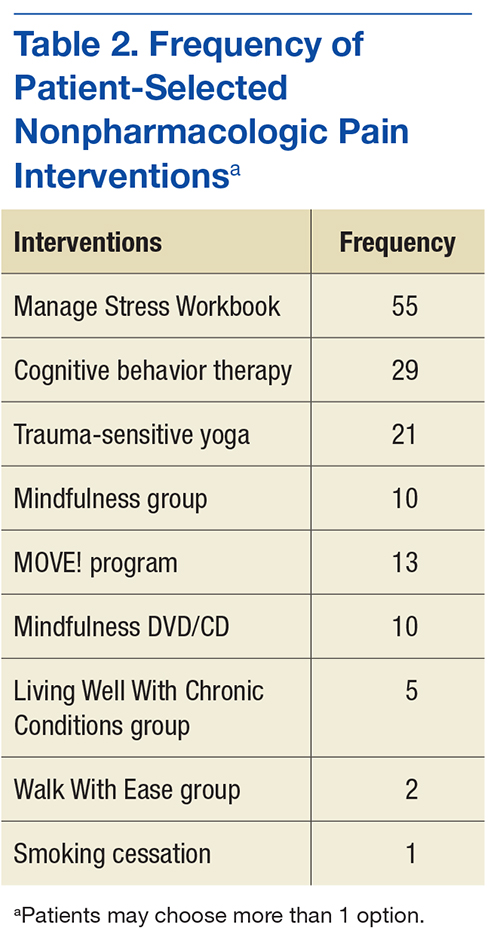
Stakeholder Support
Before its start, PC-CPP was presented to the Pentad (a group of 5 individuals in the local facility who hold executive leadership positions) for approval. Tapering opioids can lead to feelings of hostility, frustration, or sadness for patients, so having the Pentad support for the program was crucial to address complaints made to patient advocates or senators. Provider support also was important to reinforce program rules. The PC-CPP inclusion criteria included only those patients whose PCP was agreeable to a taper when the patient did not comply with program expectations. This strategy helped to improve patient adherence with the PC-CPP and decrease patient arguments with clinic staff, as all patients are held to the same standards.
Staff
Finding willing staff can be a challenge. It is estimated that each site needed a program leader who can champion the program objectives and drive organization of staff, space, documentation, and consistency for the patients consulted to the PC-CPP. The goal is that the consistent, reproducible expectations for both the PCP and the patient will reduce overall workload for a clinic. Patients may test the firmness and conviction of the staff to the PC-CPP. Having staff who are able and willing to be firm on relaying information for adherence to the patient is vital.
Administrative Support
At a minimum, a medical support assistant was required to help with scheduling, reminder calls and letters, CPRS check-in/check-out, ensuring necessary forms are ready for the SMA, tracking adherence, and following-up on no-shows and rescheduling.
Documentation
The CPRS consult and note template titles required the approval of the template committee. Although the template is helpful, there is still a great deal that needs to be manually entered in the note, such as BPI scores, opioid risk scores, and chosen nonpharmacologic interventions scores of pain, function, and opioid risk as well individual comorbidities, diagnosis, and follow-up dates. Documentation is geared toward easy review for the PCP who should scan the document prior to renewing opioid medications. The PC-CPP consult became a message board. Once the patient attends the SMA, the designated staff will add a comment to the message board, identifying all dates attended, complete history of the patient’s intervention choices and rate of adherence, as well a follow-up SMA date and whether the patient should bring materials such as take-home tests.
Time Commitment
Program development carries a heavy time burden. One full-time equivalent clinician for 6 weeks for program development is needed. Time allotment is estimated to be the following:
- Medical provider—30 minutes per patient (chart review, documentation, consult resolution). With training, these duties could be completed by support staff
- Pharmacist—30 minutes per patient (chart review, UDS, Utah Division of Occupational and Professional Licensing, fill history). Additional time is needed for writing opioid tapers for qualifying patients
- Primary care mental health integration—a PhD spent 1 to 2 hours per SMA visit assisting patients who became distressed during the visit. Only once has a patient needed to be escorted to the emergency department for active suicidality. A PhD also spent 10+ hours per week running and managing the CBT for Chronic Pain Group
- Support staff—a registered nurse spent 4 hours each month preparing for the SMA (entering consults, ordering EMITs, purchasing snacks)
Conclusion
In this descriptive report, the authors presented an overview of a newly developed program to manage chronic nonmalignant pain and safe opioid prescribing in a primary care setting. A final report is pending. The intent with this interim report was to describe the PC-CPP at the VASLCHCS, its methods and protocols, and logistic considerations for other providers who are working with patients with chronic pain in a primary care model. Standard operating procedure and inclusion/exclusion criteria were included to help with clinical decision making for patients chronic pain for whom aberrant opioid-related behavior presents a problem.
The authors expect that the PC-CPP will provide more comprehensive assisted care, lending to decreased complications associated with accidental overdose, because since patients have been educated about risks for accidental overdose from chronic opioids and have the responsibility for their outcomes. The authors also anticipated that functional scores (as measured by the BPI) will increase despite lowering opioid doses because patients will use ancillary treatments for pain. The desired outcome is that patients will come to understand that pain control is best approached holistically rather than through opioid monotherapy.
There have been several recent initiatives within the VA to decrease opioid prescribing and increase patient safety. With this in mind, continued expansion of this program to CBOCs and male patients could be useful to providers. Also, this program was conducted in a small setting (Women’s Clinic), and there are many challenges with rolling out such a program in a larger clinic (eg, greater chance for provider disagreement, greater need for administrative staff support). Nonetheless, the benefits of close monitoring of prescription opioids and active encouragement to engage in nonpharmacologic therapies are substantial and deserve further advancement.
1. Federation of State Medical Boards. Model policy on the use of opioid analgesics in the treatment of chronic pain. http://www.fsmb.org/Media/Default /PDF/FSMB/Advocacy/pain_policy_july2013.pdf. Accessed November 4, 2016.
2. Buse D, Loder E, McAlary P. Chronic pain rehabilitation. Pain Management Rounds. 2005;6:355-360.
3. Reid MC, Engles-Horton LL, Weber MB, Kerns RD, Rogers EL, O’Conner PG. Use of opioid medications for chronic noncancer pain syndromes in primary care. J Gen Intern Med. 2002;17(3):173-179.
4. Dart RC, Surratt HL, Cicero TJ, et al. Trends in opioid analgesic abuse and mortality in the United States. N Engl J Med. 2015;372(3):241-248.
5. Bohnert AS, Ilgen MA, Trafton JA, et al. Trends in regional variation in opioid overdose mortality among Veterans Health Administration patients, fiscal year 2001 to 2009. Clin J Pain. 2014;30(7):605-6012.
6. Busse JW, Guyatt GH. Optimizing the use of patient data to improve outcomes for patients: narcotics for chronic noncancer pain. Expert Rev Pharmacoecon Outcomes Res. 2009;9(2):171-179.
7. Eriksen J, Sjøgren P, Bruera E, Ekholm O, Rasmussen NK. Critical issues on opioids in chronic non-cancer pain: an epidemiological study. Pain. 2006;125(1-2):172-179.
8. Agency Medical Directors Group. Interagency guideline on opioid dosing for chronic non-cancer pain: an educational aid to improve care and safety with opioid therapy 2010 update. http://www.agen cymeddirectors.wa.gov/files/opioidgdline.pdf. Accessed November 4, 2016.
9. Utah Department of Health. Utah clinical guidelines on prescribing opioids for treatment of pain. http://health.utah.gov/prescription/pdf/guidelines/final.04.09opioidGuidlines.pdf. Published February 2009. Accessed November 4, 2016.
10. U.S. Department of Veterans Affairs, VA Academic Detailing Service. Pain management, opioid safety. VA educational guide (2014).http://www.va.gov/PAINMANAGEMENT/docs/OSI_1 _Tookit_Provider_AD_Educational_Guide_7_17.pdf. Published July 2014. Accessed November 2016.
11. Dobscha SK, Corson K, Leibowitz RQ, Sullivan MD, Gerrity MS. Rational, design, and baseline findings from a randomized trial of collaborative care for chronic musculoskeletal pain in primary care. Pain Med. 2008;9(8):1050-1064.
12. Mercado AC, Carroll LJ, Cassidy JD, Côté P. Passive coping is a risk factor for disabling neck or low back pain. Pain. 2005;117(1-2):51-57.
13. U.S. Department of Veterans Affairs, VA National Pain Management Program. Taking opioid responsibly for your safety and the safety of others: patient information guide on long-term opioid therapy for pain. http://www.veteranshealthlibrary.org/DiseasesConditions/ChronicPain/142,OpioidsIntro_VA. Updated December 9, 2015. Accessed November 17, 2016.
The Primary Care Chronic Pain Program (PC-CPP) of the Women’s Primary Care Clinics at the VA Salt Lake City Health Care System (VASLCHCS) in Utah was the first VA primary care clinical service to incorporate patient participation in obtaining chronic opioid medications in the treatment of chronic noncancer pain. In addition, the program used a multimodality approach for chronic pain treatment and veteran education about the relationship between physical and mental health issues.
Treatment Complexity
Chronic, noncancer pain is a complex issue in the primary care setting. Diagnosis is difficult, patient education is time consuming, goals and expectations are often unclear, and the experience can be unsatisfying for the patient and the provider.1 These issues, combined with an estimated prevalence rate of 71% for moderate pain among veterans seen in primary care, present a unique challenge for the primary care provider (PCP), given the limited time available to spend with these complex patients.2 Comorbidity rates with mental health issues (eg, depression, anxiety, substance use disorders, etc), which range from 18% to 44%, add to the management challenges for PCPs.3
Veterans also pose unique challenges in pain care as they have a 2-fold greater risk of death from opioid overdose compared with that of the general population, and Utah has been shown to have the highest rate of veteran overdoses.4 Developing programs to help PCPs efficiently manage patients with chronic noncancer pain and mental health comorbidities was vital at VASLCHCS.
Before VASLCHCS established the PC-CPP, the treatment for chronic noncancer pain and related mental health comorbidities followed a biomedical model that separated physical and mental health with the treatment focus on pharmacologic management of symptoms by separate services. Consistent with the biomedical model, management of chronic noncancer pain commonly included long-term use of opioids.
Over the past 2 decades, the use of opioids for treating chronic noncancer pain has significantly increased, with more than 62 million opioid prescriptions dispensed in 2012.5 There are no longitudinal follow-up studies, however, beyond 16 weeks on the use of opioids.6 Further, patients who are prescribed increased opioids continue to report high levels of pain, poor quality of life, and functional disability.7 High-dose opioids also are associated with overdose deaths.
Likewise, PCPs in the Women’s Primary Care Clinics at the VASLCHCS struggled with decreasing opioid use, often because other interventions for managing pain and related mental health conditions in primary care were not readily available. Although the VASLCHCS has an effective specialty pain service caring for patients with complex pain issues, opioid morphine equivalent doses > 200 mg/d, and palliative care, patients with chronic noncancer pain treated in the primary care setting did not have a consistent treatment approach.
A chart review of women veterans seen in Women’s Primary Care Clinic (N = 122) revealed that the majority of patients lacked timely urine drug screening, state database queries, signed medication management agreements, and documentation consistent with state and national guidelines. Additionally, many patients lacked provider follow-through regarding alternative and adjunctive therapy consults, which were often discontinued after failed contact attempts or no-shows to scheduled appointments.
There also was a general consensus among the Women’s Primary Care Clinic PCPs that caring for patients with chronic noncancer pain was exhausting, time consuming, ineffective, and often straining on the patient-provider relationship, as evidenced in many patients’ request to change providers secondary to pain management. The PC-CPP was developed to help systematically facilitate safe opioid prescribing, manage chronic pain issues, and document evidence-based care among women veterans receiving treatment for chronic noncancer pain at the Women’s Primary Care Clinics at VASLCHCS while coordinating and following through with nonpharmacologic interventions.
Program Development
National, state, VA, and professional licensure guidelines for chronic noncancer pain treatment standards were reviewed with the goal of creating a program that was evidence based, would benefit the patient in terms of opioid prescribing and pain control, and improve function while identifying key elements of care and documentation that adequately covered the prescriber of retribution.1,8-10
Concurrent to a review of the guidelines was a review of the literature with the goal of identifying useful patient education and alternative interventions and chronic pain programs that were already established and might meet the clinic’s needs.10,11 These reviews provided direction for a generalized approach to caring for patients with chronic nonmalignant pain. They also clarified that although pain education programs existed nationally, a program that offered a holistic, reproducible, adherence-driven yet patient-centered approach to the patient prescribed opioids chronically in a primary care setting was lacking.
Guideline recommendations included but were not limited to the following1,8-10:
- Patient education about chronic pain and opioids
- Evaluation of pain, function, opioid misuse risk at least twice yearly
- Patient-centered and driven treatment plans
- A holistic approach to chronic pain interventions
- Review of treatment plan efficacy at least twice yearly
- Enzyme multiplied immunoassay technique urine drug screening (UDS) 2 times per year
- State prescription monitoring program query annually
- Signed iMedConsent for treatment of chronic pain
- Plan for safe discontinuation of opioids
- Documentation that the above has been performed with patient understanding
The literature suggested a multimodality approach to chronic nonmalignant pain by minimizing the use of opioids over time while emphasizing nonpharmacologic therapies, such as cognitive behavioral therapy (CBT), mindfulness, meditation, yoga, and spiritual growth, to name a few.10,11 These findings are based on several studies, which suggested that passive coping strategies (eg, use of medication for immediate relief, depending on others, restricting medications) result in an increase in subjective pain among chronic nonmalignant pain patients.12 Helping patients reduce frequent use of passive coping strategies is believed to decrease pain.12 Active coping strategies (eg, engaging in therapies, staying busy or active, distracting attention from pain) have been found to decrease pain.12 The PC-CPP program shifted health care outcomes and responsibilities away from the hierarchal PCP-patient relationship toward a collaborative relationship that encourages patient-driven, patient-centered care outcomes and shared responsibilities.
Program Overview
The PC-CPP was shaped by the following hypotheses: (1) Transparent expectations and consequences would increase functional scores and decrease chronic opioid doses; (2) Treatment plans consisting of chronic opioid prescriptions linked with interactive nonpharmacologic interventions led to decreased pain and increased functional scores; (3) Transparent expectations combined with a streamlined approach to the chronic nonmalignant pain patient would improve patient and PCP satisfaction scores.
The PC-CPP was developed to provide an efficient, effective, and evidence-based approach to managing chronic nonmalignant pain and opioid therapy issues in the primary care setting. Referred patients attend 1 shared medical appointment (SMA) every 6 months with up to 19 other female veterans also referred to the PC-CPP. The group was composed of only female veterans as the pilot study for this SMA occurred in the Women’s Clinic. At each 6-month SMA, patients received education from the Taking Opioids Responsibly for Your Safety and the Safety of Others (TORYSSO) guide13 and signed the corresponding long-term opioid therapy for pain informed consent form (iMedConsent).
The patient and a staff member developed a treatment plan that was patient driven and included at least 1 nonpharmacologic treatment option. The 1-hour nonpharmacologic sessions were either group or individual and occurred weekly for 6 to 8 weeks. These options included CBT for chronic pain, Living Well With Chronic Conditions, trauma-sensitive yoga, smoking cessation, mindfulness for stress and anxiety, MOVE! weight management, Walk With Ease, and a self-help option (VA-issued Manage Stress Workbook, 2014). The workbook was included as an option for those who lived far away, were limited by work schedules, or were unable to afford the copays for a 6- to 8-session program.
Inclusion and Exclusion Criteria
Any female veteran patient enrolled in the VASLCHCS with a chronic nonmalignant pain diagnosis who received daily opioids for 3 or more consecutive months from a PCP was included. Excluded individuals were those with cognitive decline/dementia, serious mental illness, psychosis, active suicidality, disruptive behavior flag, or those excluded by PCP discretion if it was determined that the patient would do better in a one-on-one setting with the PCP (Table 1). Patients taking > 200 MED/d of opioids who were seen in the VASLCHCS specialty pain clinic were also excluded.
Patient and PCP Responsibilities
The patient was responsible for timely attendance and full participation in all SMA group classes as determined in the veteran’s Treatment Plan Agreement (TPA). In addition, the patient had to provide UDS when requested (a minimum of twice yearly) and communicate with the PCP if having a procedure requiring additional opioids. This was in line with the current standards set forth by the VA Opioid Safety Initiative (OSI) Taskforce.12
The PC-CPP provided education, evaluation, documentation, and referral and follow-up with the nonpharmacologic treatment options discussed but did not provide prescription medications. The PCP reviewed the medical documents completed in the PC-CPP, and the PCP was strongly encouraged to follow its recommendations. The expectation was that the PCP would support the PC-CPP when the care recommendation was for a pharmacist-guided opioid taper.
Lack of attendance was defined as a no-show or a reschedule. Patients were considered adherent if they missed fewer than 2 SMA appointments and 2 nonpharmacologic treatment appointments every 6 months. The patient was required to attend the SMA and nonpharmacologic treatment on the third appointment to remain adherent with PC-CPP expectations and agreements. Adherence was acknowledged after 12 and 24 months by a reduction in PC-CPP requirements.
Shared Medical Appointment
Patients referred to the program and who met inclusion criteria received letters explaining the importance of SMA attendance and follow-up reminder calls. At least 30 minutes before the SMA, the patient provided an UDS sample at the laboratory. Next, the patient received an individualized program packet that included the TORYSSO guide, iMedConsent, a TPA specific to the program, a brief pain inventory (BPI) and opioid risk tool, a list of medication disposal sites, and short descriptions of available nonpharmacologic therapies.
Each SMA began with a presentation delivered by a pharmacist, a psychologist, and a medical provider, discussing TORYSSO, program expectations, and holistic approaches to pain. Each SMA also included a rotating chronic pain information topic (eg, nutrition and pain, the physiology of addiction, and the value of multiple modalities in pain treatment). Together, the staff and patients reviewed and completed the blank forms enclosed in the individualized
Each visit was entered into a Computerized Patient Record System (CPRS) template, which included a pain diagnosis, Opioid Risk Tool score, pain and functional scores, opioid fill history, last comprehensive metabolic panel, last electrocardiogram if on methadone, dates of signed agreements, patient adherence with SMA and optional therapies, and follow-up (eFigure).
Every patient enrolled in the PC-CPP had to attend a SMA every 6 months. Patients continued this indefinitely while receiving opioids, and requirements were lessened for patients who had a history of meeting program requirements. For those fully adherent after the first year, only 1 nonpharmacologic intervention was needed (instead of 1 every 6 months) yearly. After 2 years of full adherence, nonpharmacologic interventions were no longer necessary as the expectation was that the patient would continue to use the strategies that they had learned over the previous 2 years. Patients left the PC-CPP if they chose to discontinue opioids, met any of the exclusion criteria, or were nonadherent. Tapering opioid medication was recommended for patients who missed a SMA meeting or 2 nonpharmacologic treatment meetings in a 6-month period; received opioids from more than 1 provider; test positive on a UDS for substances that should not be present; consistently testing negative on a UDS for substances that should be present (indicating diversion); or exhibiting other aberrant behavior (frequent requests for early refills, medications often lost/stolen, etc).
Program Barriers
The PC-CPP took about 2 years to set up, and several barriers were encountered. A thorough understanding of the following factors is necessary for establishing a similar program.
Initially, consults were placed by a designee (someone other than the PCP currently caring for the patient). The designee was usually a member of the PC-CPP who placed consults for all patients who had opioids listed on the CPRS profile. Further, patients who had any opioids within the past 3 months were initially included as were patients who wanted pain education but were not taking opioids. After 12 months, it became apparent that the focus of the PC-CPP should center on patients taking opioids for a minimum of 3 months consecutively. Patients who wanted only education could attend other hospital education opportunities, which helped keep the patient load manageable for PC-CPP staff. Further, to lessen patient confusion and improve adherence, the PCP placed the consult and discussed the program with the patient. Class sizes of 5 to 10 patients seemed to be ideal for patient participation and provider workload.
Patient Education
Initially, the SMA did not follow a standard curriculum, but the current format is more consistent, reproducible, streamlined, and organized. This adjustment improved SMA attendance as well as patient satisfaction, as the class started and finished on time. The SMA also started with numerous handouts, including brochures for nonpharmacologic programs offered at this facility. This led to patients feeling overwhelmed, missing the important forms, and wasted paper. Handouts were simplified to 2 color-coded forms (TPA and BPI).
The take-home assessment was streamlined to a single general assessment. This assessment consisted of 2 questions that asked patients to write a summary of what they learned and then write a summary of how they applied what they learned to their pain management. The VA Manage Stress Workbook also was added to the take-home materials. There are currently 5 different take-home options, which are necessary for those who live more than 50 miles from any VA facility or for those who have transportation issues.
Patient Distress
The SMA could be stressful for patients who felt they were being “punished” or who showed up more than 15 minutes late and had to reschedule the SMA. Having a mental health provider available was crucial for these situations.
Therapeutic Option Development
A cornerstone of the program was getting patients to participate in nonpharmacologic treatment options, which required a robust selection of programs. The VASLCHCS was fortunate to have many programs already available (Table 2), but this was not always the case for the VA community-based outpatient clinics (CBOCs).
Stakeholder Support
Before its start, PC-CPP was presented to the Pentad (a group of 5 individuals in the local facility who hold executive leadership positions) for approval. Tapering opioids can lead to feelings of hostility, frustration, or sadness for patients, so having the Pentad support for the program was crucial to address complaints made to patient advocates or senators. Provider support also was important to reinforce program rules. The PC-CPP inclusion criteria included only those patients whose PCP was agreeable to a taper when the patient did not comply with program expectations. This strategy helped to improve patient adherence with the PC-CPP and decrease patient arguments with clinic staff, as all patients are held to the same standards.
Staff
Finding willing staff can be a challenge. It is estimated that each site needed a program leader who can champion the program objectives and drive organization of staff, space, documentation, and consistency for the patients consulted to the PC-CPP. The goal is that the consistent, reproducible expectations for both the PCP and the patient will reduce overall workload for a clinic. Patients may test the firmness and conviction of the staff to the PC-CPP. Having staff who are able and willing to be firm on relaying information for adherence to the patient is vital.
Administrative Support
At a minimum, a medical support assistant was required to help with scheduling, reminder calls and letters, CPRS check-in/check-out, ensuring necessary forms are ready for the SMA, tracking adherence, and following-up on no-shows and rescheduling.
Documentation
The CPRS consult and note template titles required the approval of the template committee. Although the template is helpful, there is still a great deal that needs to be manually entered in the note, such as BPI scores, opioid risk scores, and chosen nonpharmacologic interventions scores of pain, function, and opioid risk as well individual comorbidities, diagnosis, and follow-up dates. Documentation is geared toward easy review for the PCP who should scan the document prior to renewing opioid medications. The PC-CPP consult became a message board. Once the patient attends the SMA, the designated staff will add a comment to the message board, identifying all dates attended, complete history of the patient’s intervention choices and rate of adherence, as well a follow-up SMA date and whether the patient should bring materials such as take-home tests.
Time Commitment
Program development carries a heavy time burden. One full-time equivalent clinician for 6 weeks for program development is needed. Time allotment is estimated to be the following:
- Medical provider—30 minutes per patient (chart review, documentation, consult resolution). With training, these duties could be completed by support staff
- Pharmacist—30 minutes per patient (chart review, UDS, Utah Division of Occupational and Professional Licensing, fill history). Additional time is needed for writing opioid tapers for qualifying patients
- Primary care mental health integration—a PhD spent 1 to 2 hours per SMA visit assisting patients who became distressed during the visit. Only once has a patient needed to be escorted to the emergency department for active suicidality. A PhD also spent 10+ hours per week running and managing the CBT for Chronic Pain Group
- Support staff—a registered nurse spent 4 hours each month preparing for the SMA (entering consults, ordering EMITs, purchasing snacks)
Conclusion
In this descriptive report, the authors presented an overview of a newly developed program to manage chronic nonmalignant pain and safe opioid prescribing in a primary care setting. A final report is pending. The intent with this interim report was to describe the PC-CPP at the VASLCHCS, its methods and protocols, and logistic considerations for other providers who are working with patients with chronic pain in a primary care model. Standard operating procedure and inclusion/exclusion criteria were included to help with clinical decision making for patients chronic pain for whom aberrant opioid-related behavior presents a problem.
The authors expect that the PC-CPP will provide more comprehensive assisted care, lending to decreased complications associated with accidental overdose, because since patients have been educated about risks for accidental overdose from chronic opioids and have the responsibility for their outcomes. The authors also anticipated that functional scores (as measured by the BPI) will increase despite lowering opioid doses because patients will use ancillary treatments for pain. The desired outcome is that patients will come to understand that pain control is best approached holistically rather than through opioid monotherapy.
There have been several recent initiatives within the VA to decrease opioid prescribing and increase patient safety. With this in mind, continued expansion of this program to CBOCs and male patients could be useful to providers. Also, this program was conducted in a small setting (Women’s Clinic), and there are many challenges with rolling out such a program in a larger clinic (eg, greater chance for provider disagreement, greater need for administrative staff support). Nonetheless, the benefits of close monitoring of prescription opioids and active encouragement to engage in nonpharmacologic therapies are substantial and deserve further advancement.
The Primary Care Chronic Pain Program (PC-CPP) of the Women’s Primary Care Clinics at the VA Salt Lake City Health Care System (VASLCHCS) in Utah was the first VA primary care clinical service to incorporate patient participation in obtaining chronic opioid medications in the treatment of chronic noncancer pain. In addition, the program used a multimodality approach for chronic pain treatment and veteran education about the relationship between physical and mental health issues.
Treatment Complexity
Chronic, noncancer pain is a complex issue in the primary care setting. Diagnosis is difficult, patient education is time consuming, goals and expectations are often unclear, and the experience can be unsatisfying for the patient and the provider.1 These issues, combined with an estimated prevalence rate of 71% for moderate pain among veterans seen in primary care, present a unique challenge for the primary care provider (PCP), given the limited time available to spend with these complex patients.2 Comorbidity rates with mental health issues (eg, depression, anxiety, substance use disorders, etc), which range from 18% to 44%, add to the management challenges for PCPs.3
Veterans also pose unique challenges in pain care as they have a 2-fold greater risk of death from opioid overdose compared with that of the general population, and Utah has been shown to have the highest rate of veteran overdoses.4 Developing programs to help PCPs efficiently manage patients with chronic noncancer pain and mental health comorbidities was vital at VASLCHCS.
Before VASLCHCS established the PC-CPP, the treatment for chronic noncancer pain and related mental health comorbidities followed a biomedical model that separated physical and mental health with the treatment focus on pharmacologic management of symptoms by separate services. Consistent with the biomedical model, management of chronic noncancer pain commonly included long-term use of opioids.
Over the past 2 decades, the use of opioids for treating chronic noncancer pain has significantly increased, with more than 62 million opioid prescriptions dispensed in 2012.5 There are no longitudinal follow-up studies, however, beyond 16 weeks on the use of opioids.6 Further, patients who are prescribed increased opioids continue to report high levels of pain, poor quality of life, and functional disability.7 High-dose opioids also are associated with overdose deaths.
Likewise, PCPs in the Women’s Primary Care Clinics at the VASLCHCS struggled with decreasing opioid use, often because other interventions for managing pain and related mental health conditions in primary care were not readily available. Although the VASLCHCS has an effective specialty pain service caring for patients with complex pain issues, opioid morphine equivalent doses > 200 mg/d, and palliative care, patients with chronic noncancer pain treated in the primary care setting did not have a consistent treatment approach.
A chart review of women veterans seen in Women’s Primary Care Clinic (N = 122) revealed that the majority of patients lacked timely urine drug screening, state database queries, signed medication management agreements, and documentation consistent with state and national guidelines. Additionally, many patients lacked provider follow-through regarding alternative and adjunctive therapy consults, which were often discontinued after failed contact attempts or no-shows to scheduled appointments.
There also was a general consensus among the Women’s Primary Care Clinic PCPs that caring for patients with chronic noncancer pain was exhausting, time consuming, ineffective, and often straining on the patient-provider relationship, as evidenced in many patients’ request to change providers secondary to pain management. The PC-CPP was developed to help systematically facilitate safe opioid prescribing, manage chronic pain issues, and document evidence-based care among women veterans receiving treatment for chronic noncancer pain at the Women’s Primary Care Clinics at VASLCHCS while coordinating and following through with nonpharmacologic interventions.
Program Development
National, state, VA, and professional licensure guidelines for chronic noncancer pain treatment standards were reviewed with the goal of creating a program that was evidence based, would benefit the patient in terms of opioid prescribing and pain control, and improve function while identifying key elements of care and documentation that adequately covered the prescriber of retribution.1,8-10
Concurrent to a review of the guidelines was a review of the literature with the goal of identifying useful patient education and alternative interventions and chronic pain programs that were already established and might meet the clinic’s needs.10,11 These reviews provided direction for a generalized approach to caring for patients with chronic nonmalignant pain. They also clarified that although pain education programs existed nationally, a program that offered a holistic, reproducible, adherence-driven yet patient-centered approach to the patient prescribed opioids chronically in a primary care setting was lacking.
Guideline recommendations included but were not limited to the following1,8-10:
- Patient education about chronic pain and opioids
- Evaluation of pain, function, opioid misuse risk at least twice yearly
- Patient-centered and driven treatment plans
- A holistic approach to chronic pain interventions
- Review of treatment plan efficacy at least twice yearly
- Enzyme multiplied immunoassay technique urine drug screening (UDS) 2 times per year
- State prescription monitoring program query annually
- Signed iMedConsent for treatment of chronic pain
- Plan for safe discontinuation of opioids
- Documentation that the above has been performed with patient understanding
The literature suggested a multimodality approach to chronic nonmalignant pain by minimizing the use of opioids over time while emphasizing nonpharmacologic therapies, such as cognitive behavioral therapy (CBT), mindfulness, meditation, yoga, and spiritual growth, to name a few.10,11 These findings are based on several studies, which suggested that passive coping strategies (eg, use of medication for immediate relief, depending on others, restricting medications) result in an increase in subjective pain among chronic nonmalignant pain patients.12 Helping patients reduce frequent use of passive coping strategies is believed to decrease pain.12 Active coping strategies (eg, engaging in therapies, staying busy or active, distracting attention from pain) have been found to decrease pain.12 The PC-CPP program shifted health care outcomes and responsibilities away from the hierarchal PCP-patient relationship toward a collaborative relationship that encourages patient-driven, patient-centered care outcomes and shared responsibilities.
Program Overview
The PC-CPP was shaped by the following hypotheses: (1) Transparent expectations and consequences would increase functional scores and decrease chronic opioid doses; (2) Treatment plans consisting of chronic opioid prescriptions linked with interactive nonpharmacologic interventions led to decreased pain and increased functional scores; (3) Transparent expectations combined with a streamlined approach to the chronic nonmalignant pain patient would improve patient and PCP satisfaction scores.
The PC-CPP was developed to provide an efficient, effective, and evidence-based approach to managing chronic nonmalignant pain and opioid therapy issues in the primary care setting. Referred patients attend 1 shared medical appointment (SMA) every 6 months with up to 19 other female veterans also referred to the PC-CPP. The group was composed of only female veterans as the pilot study for this SMA occurred in the Women’s Clinic. At each 6-month SMA, patients received education from the Taking Opioids Responsibly for Your Safety and the Safety of Others (TORYSSO) guide13 and signed the corresponding long-term opioid therapy for pain informed consent form (iMedConsent).
The patient and a staff member developed a treatment plan that was patient driven and included at least 1 nonpharmacologic treatment option. The 1-hour nonpharmacologic sessions were either group or individual and occurred weekly for 6 to 8 weeks. These options included CBT for chronic pain, Living Well With Chronic Conditions, trauma-sensitive yoga, smoking cessation, mindfulness for stress and anxiety, MOVE! weight management, Walk With Ease, and a self-help option (VA-issued Manage Stress Workbook, 2014). The workbook was included as an option for those who lived far away, were limited by work schedules, or were unable to afford the copays for a 6- to 8-session program.
Inclusion and Exclusion Criteria
Any female veteran patient enrolled in the VASLCHCS with a chronic nonmalignant pain diagnosis who received daily opioids for 3 or more consecutive months from a PCP was included. Excluded individuals were those with cognitive decline/dementia, serious mental illness, psychosis, active suicidality, disruptive behavior flag, or those excluded by PCP discretion if it was determined that the patient would do better in a one-on-one setting with the PCP (Table 1). Patients taking > 200 MED/d of opioids who were seen in the VASLCHCS specialty pain clinic were also excluded.
Patient and PCP Responsibilities
The patient was responsible for timely attendance and full participation in all SMA group classes as determined in the veteran’s Treatment Plan Agreement (TPA). In addition, the patient had to provide UDS when requested (a minimum of twice yearly) and communicate with the PCP if having a procedure requiring additional opioids. This was in line with the current standards set forth by the VA Opioid Safety Initiative (OSI) Taskforce.12
The PC-CPP provided education, evaluation, documentation, and referral and follow-up with the nonpharmacologic treatment options discussed but did not provide prescription medications. The PCP reviewed the medical documents completed in the PC-CPP, and the PCP was strongly encouraged to follow its recommendations. The expectation was that the PCP would support the PC-CPP when the care recommendation was for a pharmacist-guided opioid taper.
Lack of attendance was defined as a no-show or a reschedule. Patients were considered adherent if they missed fewer than 2 SMA appointments and 2 nonpharmacologic treatment appointments every 6 months. The patient was required to attend the SMA and nonpharmacologic treatment on the third appointment to remain adherent with PC-CPP expectations and agreements. Adherence was acknowledged after 12 and 24 months by a reduction in PC-CPP requirements.
Shared Medical Appointment
Patients referred to the program and who met inclusion criteria received letters explaining the importance of SMA attendance and follow-up reminder calls. At least 30 minutes before the SMA, the patient provided an UDS sample at the laboratory. Next, the patient received an individualized program packet that included the TORYSSO guide, iMedConsent, a TPA specific to the program, a brief pain inventory (BPI) and opioid risk tool, a list of medication disposal sites, and short descriptions of available nonpharmacologic therapies.
Each SMA began with a presentation delivered by a pharmacist, a psychologist, and a medical provider, discussing TORYSSO, program expectations, and holistic approaches to pain. Each SMA also included a rotating chronic pain information topic (eg, nutrition and pain, the physiology of addiction, and the value of multiple modalities in pain treatment). Together, the staff and patients reviewed and completed the blank forms enclosed in the individualized
Each visit was entered into a Computerized Patient Record System (CPRS) template, which included a pain diagnosis, Opioid Risk Tool score, pain and functional scores, opioid fill history, last comprehensive metabolic panel, last electrocardiogram if on methadone, dates of signed agreements, patient adherence with SMA and optional therapies, and follow-up (eFigure).
Every patient enrolled in the PC-CPP had to attend a SMA every 6 months. Patients continued this indefinitely while receiving opioids, and requirements were lessened for patients who had a history of meeting program requirements. For those fully adherent after the first year, only 1 nonpharmacologic intervention was needed (instead of 1 every 6 months) yearly. After 2 years of full adherence, nonpharmacologic interventions were no longer necessary as the expectation was that the patient would continue to use the strategies that they had learned over the previous 2 years. Patients left the PC-CPP if they chose to discontinue opioids, met any of the exclusion criteria, or were nonadherent. Tapering opioid medication was recommended for patients who missed a SMA meeting or 2 nonpharmacologic treatment meetings in a 6-month period; received opioids from more than 1 provider; test positive on a UDS for substances that should not be present; consistently testing negative on a UDS for substances that should be present (indicating diversion); or exhibiting other aberrant behavior (frequent requests for early refills, medications often lost/stolen, etc).
Program Barriers
The PC-CPP took about 2 years to set up, and several barriers were encountered. A thorough understanding of the following factors is necessary for establishing a similar program.
Initially, consults were placed by a designee (someone other than the PCP currently caring for the patient). The designee was usually a member of the PC-CPP who placed consults for all patients who had opioids listed on the CPRS profile. Further, patients who had any opioids within the past 3 months were initially included as were patients who wanted pain education but were not taking opioids. After 12 months, it became apparent that the focus of the PC-CPP should center on patients taking opioids for a minimum of 3 months consecutively. Patients who wanted only education could attend other hospital education opportunities, which helped keep the patient load manageable for PC-CPP staff. Further, to lessen patient confusion and improve adherence, the PCP placed the consult and discussed the program with the patient. Class sizes of 5 to 10 patients seemed to be ideal for patient participation and provider workload.
Patient Education
Initially, the SMA did not follow a standard curriculum, but the current format is more consistent, reproducible, streamlined, and organized. This adjustment improved SMA attendance as well as patient satisfaction, as the class started and finished on time. The SMA also started with numerous handouts, including brochures for nonpharmacologic programs offered at this facility. This led to patients feeling overwhelmed, missing the important forms, and wasted paper. Handouts were simplified to 2 color-coded forms (TPA and BPI).
The take-home assessment was streamlined to a single general assessment. This assessment consisted of 2 questions that asked patients to write a summary of what they learned and then write a summary of how they applied what they learned to their pain management. The VA Manage Stress Workbook also was added to the take-home materials. There are currently 5 different take-home options, which are necessary for those who live more than 50 miles from any VA facility or for those who have transportation issues.
Patient Distress
The SMA could be stressful for patients who felt they were being “punished” or who showed up more than 15 minutes late and had to reschedule the SMA. Having a mental health provider available was crucial for these situations.
Therapeutic Option Development
A cornerstone of the program was getting patients to participate in nonpharmacologic treatment options, which required a robust selection of programs. The VASLCHCS was fortunate to have many programs already available (Table 2), but this was not always the case for the VA community-based outpatient clinics (CBOCs).
Stakeholder Support
Before its start, PC-CPP was presented to the Pentad (a group of 5 individuals in the local facility who hold executive leadership positions) for approval. Tapering opioids can lead to feelings of hostility, frustration, or sadness for patients, so having the Pentad support for the program was crucial to address complaints made to patient advocates or senators. Provider support also was important to reinforce program rules. The PC-CPP inclusion criteria included only those patients whose PCP was agreeable to a taper when the patient did not comply with program expectations. This strategy helped to improve patient adherence with the PC-CPP and decrease patient arguments with clinic staff, as all patients are held to the same standards.
Staff
Finding willing staff can be a challenge. It is estimated that each site needed a program leader who can champion the program objectives and drive organization of staff, space, documentation, and consistency for the patients consulted to the PC-CPP. The goal is that the consistent, reproducible expectations for both the PCP and the patient will reduce overall workload for a clinic. Patients may test the firmness and conviction of the staff to the PC-CPP. Having staff who are able and willing to be firm on relaying information for adherence to the patient is vital.
Administrative Support
At a minimum, a medical support assistant was required to help with scheduling, reminder calls and letters, CPRS check-in/check-out, ensuring necessary forms are ready for the SMA, tracking adherence, and following-up on no-shows and rescheduling.
Documentation
The CPRS consult and note template titles required the approval of the template committee. Although the template is helpful, there is still a great deal that needs to be manually entered in the note, such as BPI scores, opioid risk scores, and chosen nonpharmacologic interventions scores of pain, function, and opioid risk as well individual comorbidities, diagnosis, and follow-up dates. Documentation is geared toward easy review for the PCP who should scan the document prior to renewing opioid medications. The PC-CPP consult became a message board. Once the patient attends the SMA, the designated staff will add a comment to the message board, identifying all dates attended, complete history of the patient’s intervention choices and rate of adherence, as well a follow-up SMA date and whether the patient should bring materials such as take-home tests.
Time Commitment
Program development carries a heavy time burden. One full-time equivalent clinician for 6 weeks for program development is needed. Time allotment is estimated to be the following:
- Medical provider—30 minutes per patient (chart review, documentation, consult resolution). With training, these duties could be completed by support staff
- Pharmacist—30 minutes per patient (chart review, UDS, Utah Division of Occupational and Professional Licensing, fill history). Additional time is needed for writing opioid tapers for qualifying patients
- Primary care mental health integration—a PhD spent 1 to 2 hours per SMA visit assisting patients who became distressed during the visit. Only once has a patient needed to be escorted to the emergency department for active suicidality. A PhD also spent 10+ hours per week running and managing the CBT for Chronic Pain Group
- Support staff—a registered nurse spent 4 hours each month preparing for the SMA (entering consults, ordering EMITs, purchasing snacks)
Conclusion
In this descriptive report, the authors presented an overview of a newly developed program to manage chronic nonmalignant pain and safe opioid prescribing in a primary care setting. A final report is pending. The intent with this interim report was to describe the PC-CPP at the VASLCHCS, its methods and protocols, and logistic considerations for other providers who are working with patients with chronic pain in a primary care model. Standard operating procedure and inclusion/exclusion criteria were included to help with clinical decision making for patients chronic pain for whom aberrant opioid-related behavior presents a problem.
The authors expect that the PC-CPP will provide more comprehensive assisted care, lending to decreased complications associated with accidental overdose, because since patients have been educated about risks for accidental overdose from chronic opioids and have the responsibility for their outcomes. The authors also anticipated that functional scores (as measured by the BPI) will increase despite lowering opioid doses because patients will use ancillary treatments for pain. The desired outcome is that patients will come to understand that pain control is best approached holistically rather than through opioid monotherapy.
There have been several recent initiatives within the VA to decrease opioid prescribing and increase patient safety. With this in mind, continued expansion of this program to CBOCs and male patients could be useful to providers. Also, this program was conducted in a small setting (Women’s Clinic), and there are many challenges with rolling out such a program in a larger clinic (eg, greater chance for provider disagreement, greater need for administrative staff support). Nonetheless, the benefits of close monitoring of prescription opioids and active encouragement to engage in nonpharmacologic therapies are substantial and deserve further advancement.
1. Federation of State Medical Boards. Model policy on the use of opioid analgesics in the treatment of chronic pain. http://www.fsmb.org/Media/Default /PDF/FSMB/Advocacy/pain_policy_july2013.pdf. Accessed November 4, 2016.
2. Buse D, Loder E, McAlary P. Chronic pain rehabilitation. Pain Management Rounds. 2005;6:355-360.
3. Reid MC, Engles-Horton LL, Weber MB, Kerns RD, Rogers EL, O’Conner PG. Use of opioid medications for chronic noncancer pain syndromes in primary care. J Gen Intern Med. 2002;17(3):173-179.
4. Dart RC, Surratt HL, Cicero TJ, et al. Trends in opioid analgesic abuse and mortality in the United States. N Engl J Med. 2015;372(3):241-248.
5. Bohnert AS, Ilgen MA, Trafton JA, et al. Trends in regional variation in opioid overdose mortality among Veterans Health Administration patients, fiscal year 2001 to 2009. Clin J Pain. 2014;30(7):605-6012.
6. Busse JW, Guyatt GH. Optimizing the use of patient data to improve outcomes for patients: narcotics for chronic noncancer pain. Expert Rev Pharmacoecon Outcomes Res. 2009;9(2):171-179.
7. Eriksen J, Sjøgren P, Bruera E, Ekholm O, Rasmussen NK. Critical issues on opioids in chronic non-cancer pain: an epidemiological study. Pain. 2006;125(1-2):172-179.
8. Agency Medical Directors Group. Interagency guideline on opioid dosing for chronic non-cancer pain: an educational aid to improve care and safety with opioid therapy 2010 update. http://www.agen cymeddirectors.wa.gov/files/opioidgdline.pdf. Accessed November 4, 2016.
9. Utah Department of Health. Utah clinical guidelines on prescribing opioids for treatment of pain. http://health.utah.gov/prescription/pdf/guidelines/final.04.09opioidGuidlines.pdf. Published February 2009. Accessed November 4, 2016.
10. U.S. Department of Veterans Affairs, VA Academic Detailing Service. Pain management, opioid safety. VA educational guide (2014).http://www.va.gov/PAINMANAGEMENT/docs/OSI_1 _Tookit_Provider_AD_Educational_Guide_7_17.pdf. Published July 2014. Accessed November 2016.
11. Dobscha SK, Corson K, Leibowitz RQ, Sullivan MD, Gerrity MS. Rational, design, and baseline findings from a randomized trial of collaborative care for chronic musculoskeletal pain in primary care. Pain Med. 2008;9(8):1050-1064.
12. Mercado AC, Carroll LJ, Cassidy JD, Côté P. Passive coping is a risk factor for disabling neck or low back pain. Pain. 2005;117(1-2):51-57.
13. U.S. Department of Veterans Affairs, VA National Pain Management Program. Taking opioid responsibly for your safety and the safety of others: patient information guide on long-term opioid therapy for pain. http://www.veteranshealthlibrary.org/DiseasesConditions/ChronicPain/142,OpioidsIntro_VA. Updated December 9, 2015. Accessed November 17, 2016.
1. Federation of State Medical Boards. Model policy on the use of opioid analgesics in the treatment of chronic pain. http://www.fsmb.org/Media/Default /PDF/FSMB/Advocacy/pain_policy_july2013.pdf. Accessed November 4, 2016.
2. Buse D, Loder E, McAlary P. Chronic pain rehabilitation. Pain Management Rounds. 2005;6:355-360.
3. Reid MC, Engles-Horton LL, Weber MB, Kerns RD, Rogers EL, O’Conner PG. Use of opioid medications for chronic noncancer pain syndromes in primary care. J Gen Intern Med. 2002;17(3):173-179.
4. Dart RC, Surratt HL, Cicero TJ, et al. Trends in opioid analgesic abuse and mortality in the United States. N Engl J Med. 2015;372(3):241-248.
5. Bohnert AS, Ilgen MA, Trafton JA, et al. Trends in regional variation in opioid overdose mortality among Veterans Health Administration patients, fiscal year 2001 to 2009. Clin J Pain. 2014;30(7):605-6012.
6. Busse JW, Guyatt GH. Optimizing the use of patient data to improve outcomes for patients: narcotics for chronic noncancer pain. Expert Rev Pharmacoecon Outcomes Res. 2009;9(2):171-179.
7. Eriksen J, Sjøgren P, Bruera E, Ekholm O, Rasmussen NK. Critical issues on opioids in chronic non-cancer pain: an epidemiological study. Pain. 2006;125(1-2):172-179.
8. Agency Medical Directors Group. Interagency guideline on opioid dosing for chronic non-cancer pain: an educational aid to improve care and safety with opioid therapy 2010 update. http://www.agen cymeddirectors.wa.gov/files/opioidgdline.pdf. Accessed November 4, 2016.
9. Utah Department of Health. Utah clinical guidelines on prescribing opioids for treatment of pain. http://health.utah.gov/prescription/pdf/guidelines/final.04.09opioidGuidlines.pdf. Published February 2009. Accessed November 4, 2016.
10. U.S. Department of Veterans Affairs, VA Academic Detailing Service. Pain management, opioid safety. VA educational guide (2014).http://www.va.gov/PAINMANAGEMENT/docs/OSI_1 _Tookit_Provider_AD_Educational_Guide_7_17.pdf. Published July 2014. Accessed November 2016.
11. Dobscha SK, Corson K, Leibowitz RQ, Sullivan MD, Gerrity MS. Rational, design, and baseline findings from a randomized trial of collaborative care for chronic musculoskeletal pain in primary care. Pain Med. 2008;9(8):1050-1064.
12. Mercado AC, Carroll LJ, Cassidy JD, Côté P. Passive coping is a risk factor for disabling neck or low back pain. Pain. 2005;117(1-2):51-57.
13. U.S. Department of Veterans Affairs, VA National Pain Management Program. Taking opioid responsibly for your safety and the safety of others: patient information guide on long-term opioid therapy for pain. http://www.veteranshealthlibrary.org/DiseasesConditions/ChronicPain/142,OpioidsIntro_VA. Updated December 9, 2015. Accessed November 17, 2016.
Anticoagulant receives priority review

Image by Andre E.X. Brown
The US Food and Drug Administration (FDA) has granted priority review to the new drug application (NDA) for betrixaban, an oral factor Xa inhibitor, for extended-duration prophylaxis of venous thromboembolism (VTE) in acute medically ill patients with risk factors for VTE.
A priority review shortens the FDA review timeline to 6 months from the standard review period of 10 months.
The application for betrixaban has been given a Prescription Drug User Fee Act action date of June 24, 2017. (Betrixaban also has fast track designation from the FDA.)
Meanwhile, the European Medicines Agency (EMA) is reviewing a marketing authorization application (MAA) for betrixaban for extended-duration prophylaxis of VTE in adults with acute medical illness and risk factors for VTE.
However, the EMA’s Committee for Medicinal Products for Human Use is reviewing the application under a standard 210-day review period.
“With the filing of the betrixaban NDA and the MAA validation, we now look forward to working with the FDA and EMA to bring this drug to market,” said Bill Lis, chief executive officer of Portola Pharmaceuticals, Inc., the company developing betrixaban.
The NDA and MAA for betrixaban are supported by data from Portola’s phase 3 APEX study, which enrolled 7513 patients at more than 450 clinical sites worldwide.
In this study, researchers compared extended-duration anticoagulation with oral betrixaban for 35-42 days to standard-duration enoxaparin for 10±4 days in preventing VTE in high-risk acute medically ill patients.
Patients who received betrixaban had a significantly lower incidence of VTE than those who received enoxaparin, and there was no significant difference in major bleeding between the treatment groups.
Full results from this study were presented at the 62nd Annual International Society on Thrombosis and Haemostasis Scientific and Standardization Committee Meeting and published in NEJM in May 2016. ![]()
Image by Andre E.X. Brown
The US Food and Drug Administration (FDA) has granted priority review to the new drug application (NDA) for betrixaban, an oral factor Xa inhibitor, for extended-duration prophylaxis of venous thromboembolism (VTE) in acute medically ill patients with risk factors for VTE.
A priority review shortens the FDA review timeline to 6 months from the standard review period of 10 months.
The application for betrixaban has been given a Prescription Drug User Fee Act action date of June 24, 2017. (Betrixaban also has fast track designation from the FDA.)
Meanwhile, the European Medicines Agency (EMA) is reviewing a marketing authorization application (MAA) for betrixaban for extended-duration prophylaxis of VTE in adults with acute medical illness and risk factors for VTE.
However, the EMA’s Committee for Medicinal Products for Human Use is reviewing the application under a standard 210-day review period.
“With the filing of the betrixaban NDA and the MAA validation, we now look forward to working with the FDA and EMA to bring this drug to market,” said Bill Lis, chief executive officer of Portola Pharmaceuticals, Inc., the company developing betrixaban.
The NDA and MAA for betrixaban are supported by data from Portola’s phase 3 APEX study, which enrolled 7513 patients at more than 450 clinical sites worldwide.
In this study, researchers compared extended-duration anticoagulation with oral betrixaban for 35-42 days to standard-duration enoxaparin for 10±4 days in preventing VTE in high-risk acute medically ill patients.
Patients who received betrixaban had a significantly lower incidence of VTE than those who received enoxaparin, and there was no significant difference in major bleeding between the treatment groups.
Full results from this study were presented at the 62nd Annual International Society on Thrombosis and Haemostasis Scientific and Standardization Committee Meeting and published in NEJM in May 2016. ![]()
Image by Andre E.X. Brown
The US Food and Drug Administration (FDA) has granted priority review to the new drug application (NDA) for betrixaban, an oral factor Xa inhibitor, for extended-duration prophylaxis of venous thromboembolism (VTE) in acute medically ill patients with risk factors for VTE.
A priority review shortens the FDA review timeline to 6 months from the standard review period of 10 months.
The application for betrixaban has been given a Prescription Drug User Fee Act action date of June 24, 2017. (Betrixaban also has fast track designation from the FDA.)
Meanwhile, the European Medicines Agency (EMA) is reviewing a marketing authorization application (MAA) for betrixaban for extended-duration prophylaxis of VTE in adults with acute medical illness and risk factors for VTE.
However, the EMA’s Committee for Medicinal Products for Human Use is reviewing the application under a standard 210-day review period.
“With the filing of the betrixaban NDA and the MAA validation, we now look forward to working with the FDA and EMA to bring this drug to market,” said Bill Lis, chief executive officer of Portola Pharmaceuticals, Inc., the company developing betrixaban.
The NDA and MAA for betrixaban are supported by data from Portola’s phase 3 APEX study, which enrolled 7513 patients at more than 450 clinical sites worldwide.
In this study, researchers compared extended-duration anticoagulation with oral betrixaban for 35-42 days to standard-duration enoxaparin for 10±4 days in preventing VTE in high-risk acute medically ill patients.
Patients who received betrixaban had a significantly lower incidence of VTE than those who received enoxaparin, and there was no significant difference in major bleeding between the treatment groups.
Full results from this study were presented at the 62nd Annual International Society on Thrombosis and Haemostasis Scientific and Standardization Committee Meeting and published in NEJM in May 2016. ![]()