User login
Skin Examinations for Early Melanoma Detection


When the diagnosis is hard to swallow, take these management steps
CASE REPORTMr. C, age 72, reports a lack of desire to swallow food. He denies feeling a lump in his throat. Over the past 6 months, he lost >30 lb.
The patient had a similar episode 2 years ago, which resolved without intervention. The death of his wife recently has led to isolation and lack of desire to swallow food.
Testing with standard food samples to elicit eating behaviors is normal. Electromyography and video fluoroscopy test results show no abnormalities.
What is phagophobia?The case of Mr. C brings to light the condition known as phagophobia—a sensation of not being able to swallow. Phagophobia mimics oral apraxia; pharyngoesophageal and neurologic functions as well as the ability to speak remain intact, however.1
It is estimated that about 6% of the adult general population reports dysphagia.2 About 47% of patients with dysphagic complaints do not show motor-manometric or radiological abnormalities of the upper digestive tract. A number of psychiatric conditions, including panic disorder, obsessive-compulsive disorder, social phobia, anorexia nervosa, globus hystericus, hypersensitive gag reflex, and posttraumatic stress disorder can simulate this condition.3
When Barofsky and Fontaine4 compared phagophobia patients with other subjects—healthy controls, anorexia nervosa restrictors, dysphagic patients with esophageal obstruction, dysphagic patients with motility disturbance, and patients with non-motility non-obstructive dysphagia—they found that patients with psychogenic dysphagia did not appear to have an eating disorder. However, they did have a clinically significant level of psychological distress, particularly anxiety.
Diagnostic tools and management stepsThere are a number of approaches to assess your patient’s fear of swallowing (Table,5-7 page 68). Non-invasive assessment tools along with educational modalities usually are tried alone or together with psychopharmacological intervention. It is, however, imperative that you have an empathetic and understanding approach to such patients. When patients have confidence in the clinician they tend to respond more effectively with such approaches.

Investigations4 include questionnaires (swallow disorder history, Eating Disorder Inventory-2, and Symptom Checklist–90-R); weight assessment; testing with standardized food samples to elicit eating behaviors; self-reports; electromyography; and videofluoroscopy.
Education and reassurance includes individual demonstration of swallowing, combined with group therapy, exercises, and reassurance. Patients benefit from advice on how to maximize sensation within the oropharynx to increase taste, perception of temperature, and texture stimulation.8
Behavioral intervention involves practicing slow breathing and muscle relaxation techniques to gradually increase bite size and reduce the amount of time spent chewing each bite.
Introspection therapycomprises psychoeducation, cognitive restructuring, and in vivo and introspective exposure; helps patients replace anxiety-producing thoughts with probability estimation and decatastrophizing. Introspective exposure targets the fear of choking by having the patient create sensations of throat tightening by holding a swallow in mid-action and by rapid swallowing. In vivo exposure targets the fear of swallowing by having the patient practice feeding foods (such as semi-solid easy-to-swallow choices), in and outside of the session.6
Aversion therapy requires that you pinch the patient’s hand while he (she) chews, and release the hand when he swallows.
Psychopharmacotherapeutic intervention. A number of medications can be used to help, such as imipramine up to 150 mg; desipramine, up to 150 mg; or lorazepam, 0.25 mg, twice daily, to address anxiety or panic symptoms.
Acknowledgment
Duy Li, BS, and Yu Hsuan Liao, BS, contributed to the development of the manuscript of this article.
1. Evans IM, Pia P. Phagophobia: behavioral treatment of a complex case involving fear of fear. Clinical Case Studies. 2011;10(1):37-52.
2. Kim CH, Hsu JJ, Williams DE, et al. A prospective psychological evaluation of patients with dysphagia of various etiologies. Dysphagia. 1996;11(1):34-40.
3. McNally RJ. Choking phobia: a review of the literature. Compr Psychiatry. 1994;35(1):83-89.
4. Barofsky I, Fontaine KR. Do psychogenic dysphagia patients have an eating disorder? Dysphagia. 1998;13(1):24-27.
5. Bishop LC, Riley WT. The psychiatric management of the globus syndrome. Gen Hosp Psychiatry. 1988;10(3):214-219.
6. Ball SG, Otto MW. Cognitive-behavioral treatment of choking phobia: 3 case studies Psychother Psychosom. 1994;62(3-4):207-211.
7. Epstein SJ, Deyoub P. Hypnotherapy for fear of choking: treatment implications of a case report. Int J Clin Hypn. 1981;29(2):117-127.
8. Scemes S, Wielenska RC, Savoia MG, et al. Choking phobia: full remission following behavior therapy. Rev Bras Psiquiatr. 2009;31(3):257-260.
CASE REPORTMr. C, age 72, reports a lack of desire to swallow food. He denies feeling a lump in his throat. Over the past 6 months, he lost >30 lb.
The patient had a similar episode 2 years ago, which resolved without intervention. The death of his wife recently has led to isolation and lack of desire to swallow food.
Testing with standard food samples to elicit eating behaviors is normal. Electromyography and video fluoroscopy test results show no abnormalities.
What is phagophobia?The case of Mr. C brings to light the condition known as phagophobia—a sensation of not being able to swallow. Phagophobia mimics oral apraxia; pharyngoesophageal and neurologic functions as well as the ability to speak remain intact, however.1
It is estimated that about 6% of the adult general population reports dysphagia.2 About 47% of patients with dysphagic complaints do not show motor-manometric or radiological abnormalities of the upper digestive tract. A number of psychiatric conditions, including panic disorder, obsessive-compulsive disorder, social phobia, anorexia nervosa, globus hystericus, hypersensitive gag reflex, and posttraumatic stress disorder can simulate this condition.3
When Barofsky and Fontaine4 compared phagophobia patients with other subjects—healthy controls, anorexia nervosa restrictors, dysphagic patients with esophageal obstruction, dysphagic patients with motility disturbance, and patients with non-motility non-obstructive dysphagia—they found that patients with psychogenic dysphagia did not appear to have an eating disorder. However, they did have a clinically significant level of psychological distress, particularly anxiety.
Diagnostic tools and management stepsThere are a number of approaches to assess your patient’s fear of swallowing (Table,5-7 page 68). Non-invasive assessment tools along with educational modalities usually are tried alone or together with psychopharmacological intervention. It is, however, imperative that you have an empathetic and understanding approach to such patients. When patients have confidence in the clinician they tend to respond more effectively with such approaches.
Investigations4 include questionnaires (swallow disorder history, Eating Disorder Inventory-2, and Symptom Checklist–90-R); weight assessment; testing with standardized food samples to elicit eating behaviors; self-reports; electromyography; and videofluoroscopy.
Education and reassurance includes individual demonstration of swallowing, combined with group therapy, exercises, and reassurance. Patients benefit from advice on how to maximize sensation within the oropharynx to increase taste, perception of temperature, and texture stimulation.8
Behavioral intervention involves practicing slow breathing and muscle relaxation techniques to gradually increase bite size and reduce the amount of time spent chewing each bite.
Introspection therapycomprises psychoeducation, cognitive restructuring, and in vivo and introspective exposure; helps patients replace anxiety-producing thoughts with probability estimation and decatastrophizing. Introspective exposure targets the fear of choking by having the patient create sensations of throat tightening by holding a swallow in mid-action and by rapid swallowing. In vivo exposure targets the fear of swallowing by having the patient practice feeding foods (such as semi-solid easy-to-swallow choices), in and outside of the session.6
Aversion therapy requires that you pinch the patient’s hand while he (she) chews, and release the hand when he swallows.
Psychopharmacotherapeutic intervention. A number of medications can be used to help, such as imipramine up to 150 mg; desipramine, up to 150 mg; or lorazepam, 0.25 mg, twice daily, to address anxiety or panic symptoms.
Acknowledgment
Duy Li, BS, and Yu Hsuan Liao, BS, contributed to the development of the manuscript of this article.
CASE REPORTMr. C, age 72, reports a lack of desire to swallow food. He denies feeling a lump in his throat. Over the past 6 months, he lost >30 lb.
The patient had a similar episode 2 years ago, which resolved without intervention. The death of his wife recently has led to isolation and lack of desire to swallow food.
Testing with standard food samples to elicit eating behaviors is normal. Electromyography and video fluoroscopy test results show no abnormalities.
What is phagophobia?The case of Mr. C brings to light the condition known as phagophobia—a sensation of not being able to swallow. Phagophobia mimics oral apraxia; pharyngoesophageal and neurologic functions as well as the ability to speak remain intact, however.1
It is estimated that about 6% of the adult general population reports dysphagia.2 About 47% of patients with dysphagic complaints do not show motor-manometric or radiological abnormalities of the upper digestive tract. A number of psychiatric conditions, including panic disorder, obsessive-compulsive disorder, social phobia, anorexia nervosa, globus hystericus, hypersensitive gag reflex, and posttraumatic stress disorder can simulate this condition.3
When Barofsky and Fontaine4 compared phagophobia patients with other subjects—healthy controls, anorexia nervosa restrictors, dysphagic patients with esophageal obstruction, dysphagic patients with motility disturbance, and patients with non-motility non-obstructive dysphagia—they found that patients with psychogenic dysphagia did not appear to have an eating disorder. However, they did have a clinically significant level of psychological distress, particularly anxiety.
Diagnostic tools and management stepsThere are a number of approaches to assess your patient’s fear of swallowing (Table,5-7 page 68). Non-invasive assessment tools along with educational modalities usually are tried alone or together with psychopharmacological intervention. It is, however, imperative that you have an empathetic and understanding approach to such patients. When patients have confidence in the clinician they tend to respond more effectively with such approaches.
Investigations4 include questionnaires (swallow disorder history, Eating Disorder Inventory-2, and Symptom Checklist–90-R); weight assessment; testing with standardized food samples to elicit eating behaviors; self-reports; electromyography; and videofluoroscopy.
Education and reassurance includes individual demonstration of swallowing, combined with group therapy, exercises, and reassurance. Patients benefit from advice on how to maximize sensation within the oropharynx to increase taste, perception of temperature, and texture stimulation.8
Behavioral intervention involves practicing slow breathing and muscle relaxation techniques to gradually increase bite size and reduce the amount of time spent chewing each bite.
Introspection therapycomprises psychoeducation, cognitive restructuring, and in vivo and introspective exposure; helps patients replace anxiety-producing thoughts with probability estimation and decatastrophizing. Introspective exposure targets the fear of choking by having the patient create sensations of throat tightening by holding a swallow in mid-action and by rapid swallowing. In vivo exposure targets the fear of swallowing by having the patient practice feeding foods (such as semi-solid easy-to-swallow choices), in and outside of the session.6
Aversion therapy requires that you pinch the patient’s hand while he (she) chews, and release the hand when he swallows.
Psychopharmacotherapeutic intervention. A number of medications can be used to help, such as imipramine up to 150 mg; desipramine, up to 150 mg; or lorazepam, 0.25 mg, twice daily, to address anxiety or panic symptoms.
Acknowledgment
Duy Li, BS, and Yu Hsuan Liao, BS, contributed to the development of the manuscript of this article.
1. Evans IM, Pia P. Phagophobia: behavioral treatment of a complex case involving fear of fear. Clinical Case Studies. 2011;10(1):37-52.
2. Kim CH, Hsu JJ, Williams DE, et al. A prospective psychological evaluation of patients with dysphagia of various etiologies. Dysphagia. 1996;11(1):34-40.
3. McNally RJ. Choking phobia: a review of the literature. Compr Psychiatry. 1994;35(1):83-89.
4. Barofsky I, Fontaine KR. Do psychogenic dysphagia patients have an eating disorder? Dysphagia. 1998;13(1):24-27.
5. Bishop LC, Riley WT. The psychiatric management of the globus syndrome. Gen Hosp Psychiatry. 1988;10(3):214-219.
6. Ball SG, Otto MW. Cognitive-behavioral treatment of choking phobia: 3 case studies Psychother Psychosom. 1994;62(3-4):207-211.
7. Epstein SJ, Deyoub P. Hypnotherapy for fear of choking: treatment implications of a case report. Int J Clin Hypn. 1981;29(2):117-127.
8. Scemes S, Wielenska RC, Savoia MG, et al. Choking phobia: full remission following behavior therapy. Rev Bras Psiquiatr. 2009;31(3):257-260.
1. Evans IM, Pia P. Phagophobia: behavioral treatment of a complex case involving fear of fear. Clinical Case Studies. 2011;10(1):37-52.
2. Kim CH, Hsu JJ, Williams DE, et al. A prospective psychological evaluation of patients with dysphagia of various etiologies. Dysphagia. 1996;11(1):34-40.
3. McNally RJ. Choking phobia: a review of the literature. Compr Psychiatry. 1994;35(1):83-89.
4. Barofsky I, Fontaine KR. Do psychogenic dysphagia patients have an eating disorder? Dysphagia. 1998;13(1):24-27.
5. Bishop LC, Riley WT. The psychiatric management of the globus syndrome. Gen Hosp Psychiatry. 1988;10(3):214-219.
6. Ball SG, Otto MW. Cognitive-behavioral treatment of choking phobia: 3 case studies Psychother Psychosom. 1994;62(3-4):207-211.
7. Epstein SJ, Deyoub P. Hypnotherapy for fear of choking: treatment implications of a case report. Int J Clin Hypn. 1981;29(2):117-127.
8. Scemes S, Wielenska RC, Savoia MG, et al. Choking phobia: full remission following behavior therapy. Rev Bras Psiquiatr. 2009;31(3):257-260.
When to adjust the dosing of psychotropics in patients with renal impairment
Renal disease can play a large role in altering the pharmacokinetics of medications, especially in elimination or clearance and plasma protein binding. Specifically, renal impairment decreases the plasma protein binding secondary to decreased albumin and retention of urea, which competes with medications to bind to the protein.1
Electrolyte shifts—which could lead to a fatal arrhythmia—are common among patients with renal impairment. The risk can be further increased in this population if a patient is taking a medication that can induce arrhythmia. If a drug is primarily excreted by the kidneys, elimination could be significantly altered, especially if the medication has active metabolites.1
Normal renal function is defined as an estimated creatinine clearance (eCrCl) of >80 mL/min. Renal impairment is classified as:
- mild: eCrCl, 51 to 80 mL/min
- moderate: eCrCl, 31 to 50 mL/min
- severe: eCrCl, ≤30 mL/min
- end-stage renal disease (ESRD): eCrCl, <10 mL/min.2
Overall, there is minimal information about the effects of renal disease on psychotropic therapy; our goal here is to summarize available data. We have created quick reference tables highlighting psychotropics that have renal dosing recommendations based on manufacturers’ package inserts.
Antipsychotics
First-generation antipsychotics (FGAs). Dosage adjustments based on renal function are not required for any FGA, according to manufacturers’ package inserts. Some of these antipsychotics are excreted in urine, but typically as inactive metabolites.
Although there are no dosage recommendations based on renal function provided by the labeling, there has been concern about the use of some FGAs in patients with renal impairment. Specifically, concerns center around the piperidine phenothiazines (thioridazine and mesoridazine) because of the increased risk of electrocardiographic changes and medication-induced arrhythmias in renal disease due to electrolyte imbalances.3,4 Additionally, there is case evidence5 that phenothiazine antipsychotics could increase a patient’s risk for hypotension in chronic renal failure. Haloperidol is considered safe in renal disease because <1% of the medication is excreted unchanged through urine.6
Second-generation antipsychotics (SGAs). Overall, SGAs are considered safe in patients with renal disease. Most SGAs undergo extensive hepatic metabolism before excretion, allowing them to be used safely in patients with renal disease.
Sheehan et al7 analyzed the metabolism and excretion of SGAs, evaluating 8 antipsychotics divided into 4 groups: (1) excretion primarily as an unchanged drug in urine, (2) changed drug in urine, (3) changed drug in feces, (4) and unchanged drug in feces.
- Paliperidone was found to be primarily excreted as an unchanged drug in urine.
- Clozapine, iloperidone, olanzapine, quetiapine, and risperidone all were found to be primarily excreted as a changed drug in urine.
- Aripiprazole and ziprasidone were found to be primarily excreted as a changed drug in feces.
The manufacturers’ package inserts for clozapine, paliperidone, risperidone, and lurasidone have recommended dosage adjustments based on renal function (Table 1).8-11
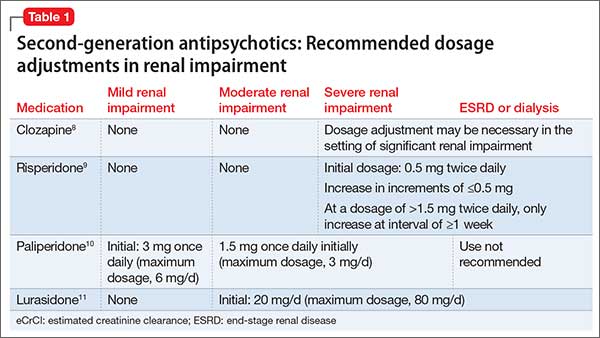
Ziprasidone. Although ziprasidone does not have a recommended renal dosage adjustment, caution is recommended because of the risk of electrocardiographic changes and potential for medication-induced arrhythmias in patients with electrolyte disturbances secondary to renal disease. A single-dosage study of ziprasidone by Aweeka et al12 demonstrated that the pharmacokinetics of ziprasidone are unchanged in patients with renal impairment.
Asenapine. A small study by Peeters et al13 evaluated the pharmacokinetics of asenapine in hepatic and renal impairment and found no clinically relevant changes in asenapine’s pharmacokinetics among patients with any level of renal impairment compared with patients with normal renal function.
Aripiprazole. Mallikaarjun et al14 completed a small study evaluating the pharmacokinetics of aripiprazole in patients with renal impairment. They found that the pharmacokinetics of aripiprazole in these patients is no different than it is in patients with normal renal function who are taking aripiprazole.
Quetiapine. Thyrum et al15 conducted a similar study with quetiapine, which showed no significant difference detected in the pharmacokinetics of quetiapine in patients with renal impairment. Additionally, quetiapine had no negative effect on patients’ creatinine clearance.
Lurasidone. During clinical trials of lurasidone in patients with mild, moderate, and severe renal impairment, the mean Cmax and area under the curve was higher compared with healthy patients, which led to recommended dosage adjustments in patients with renal impairment.11
As mentioned above, renal impairment decreases the protein binding percentage of medications. Hypothetically, the greater the protein binding, the lower the recommended dosage in patients with renal impairment because the free or unbound form correlates with efficacy and toxicity. Most FGAs and SGAs have the protein-binding characteristic of ≥90%.16 Although it seems this characteristic should result in recommendations to adjust dosage based on renal function, the various pharmacokinetic studies of antipsychotics have not shown this factor to play a role in the manufacturers’ recommendations.
Antidepressants
Comorbidity rates of depression in patients with renal disease range from 14% to 30%, making use of antidepressants in renal disease common.4 Antidepressants primarily are metabolized hepatically and excreted renally. Table 217-27 summarizes recommended dosing adjustments for antidepressants.
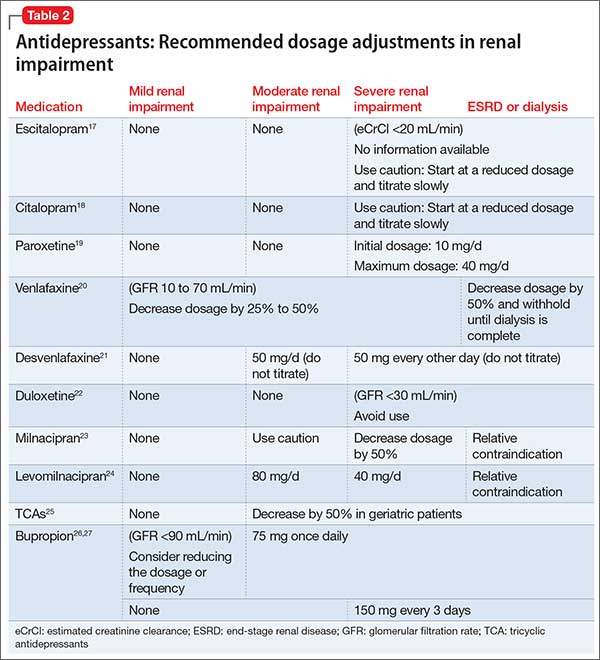
Selective serotonin reuptake inhibitors.Escitalopram is the (S)-enantiomer of the racemic antidepressant citalopram, both of which have been shown to decrease renal clearance in patients with mild or moderate renal impairment. However, according to the package insert, no dosage adjustments are needed.17 No extensive studies have been conducted on escitalopram or citalopram, but each should be initiated at a reduced dosage and the titration schedule should be prolonged in patients with severe renal impairment or ESRD.17,18
The plasma concentration of paroxetine has been noted to be elevated in patients with severe renal impairment, and the half-life can increase to nearly 50%.4 Paroxetine should be initiated at 10 mg/d, and then titrated slowly in patients with severe renal impairment.19,28
The pharmacokinetics of fluoxetine are unchanged in any stage of renal impairment. Patients in active renal dialysis report good tolerability and efficacy.4
Serotonin-norepinephrine reuptake inhibitors. Venlafaxine and its metabolite O-desmethylvenlafaxine (desvenlafaxine) are primarily excreted via renal elimination. Studies have shown that mild renal impairment can have an effect on plasma levels of the drug, and that moderate or severe impairment can increase the venlafaxine plasma concentration. According to the package insert, a dosage reduction of 50% is recommended for desvenlafaxine and venlafaxine.20,21
No significant pharmacokinetic changes with duloxetine have been noted in patients with mild or moderate renal impairment.22 However, duloxetine’s major metabolites, which are excreted renally, have been measured to be as much as 7 to 9 times higher in patients with ESRD compared with healthy subjects; therefore, it is recommended to avoid duloxetine in patients with severe renal disease.4,22 Our review of the literature produced limited recommendations on dosing milnacipran and its enantiomer levomilnacipran in renally impaired patients. The milnacipran package insert cautions its use in moderate renal impairment and recommends a 50% dosage reduction to 100 mg/d (50 mg twice daily) in patients with severe renal impairment.23 Dosage recommendations for levomilnacipran are 80 mg/d for moderate renal impairment and 40 mg/d for severe impairment. Both agents have relative contraindications for ESRD.23,24
Tricyclic antidepressants (TCAs) are predominantly metabolized hepatically, glucuronidated, and then eliminated renally. Desipramine, imipramine, and nortriptyline have nonspecific package insert recommendations for modified dosing in geriatric patients because of an age-related decrease in renal clearance.29-31 Review articles assert that elevated glucuronidated metabolites could increase patients’ sensitivity to side effects of TCAs. Because of concerns regarding elevated glucuronidated metabolites, it has been proposed to initiate TCAs at a low dosage, titrate slowly, and maintain the lowest effective dosage in patients with renal impairment.25
Monoamine oxidase inhibitors (MAOIs) and other antidepressants. The package inserts of the MAOIs isocarboxazid, phenelzine, selegiline, and tranylcypromine provide limited data and dosage recommendations for use in the context of renal impairment.32-36 Isocarboxazid should not be used in patients with severe renal impairment, according to the prescribing information.32 There are no dosing recommendations for transdermal selegiline in mild, moderate, or severe renal impairment.37 Extra vigilance is required when using MAOIs in patients with renal disease because of an increased risk of dialysis-induced hypotension (orthostatic hypotension is a common adverse effect of MAOIs).38
Bupropion is primarily metabolized hepatically to the active metabolite hydroxybupropion. Plasma levels of this metabolite at steady state are reported to be 10 times greater than bupropion’s concentration levels in healthy subjects; plasma levels are further increased in mild renal impairment.26 Hydroxybupropion is not dialyzable, which can increase the risk of toxicity with bupropion therapy in patients with renal impairment.3 If bupropion effectively treats depression in patients with declining renal function, specifically severe renal impairment and ESRD, then decreasing the dosage to 150 mg every 3 days is recommended to lessen the risk of toxicity. 27
Mood stabilizers
Lithium has the most published literature on dosing adjustments with renal impairment. Many providers are inclined to discontinue lithium use at the first sign of any change in renal function; however, monitoring, prevention, and treatment guidelines for lithium are well established after many years of research and clinical use.39 Lithium’s prescribing information recommends dosage adjustment in mild to moderate renal impairment and lists severe renal impairment and ESRD as relative contraindications.40
A recent study proposes more assertive use of lithium in patients with renal impairment of any severity. Rej et al41 compared continued lithium treatment to discontinuing treatment in geriatric patients with chronic renal failure, and reported (1) a statistically insignificant difference in renal function between groups at 2 years and (2) a “trending decrease” in renal function at 5 years in the lithium treatment group. With closely monitored plasma levels, lithium treatment is considered a workable treatment for patients with moderate renal impairment when mood stabilizer treatment has been effective.42
Lamotrigine and its main glucuronidated metabolite, lamotrigine-2N-glucuronide (L-2-N-G), are primarily excreted renally. In severe renal impairment and ESRD, the L-2-N-G levels are elevated but are not pharmacologically active and, therefore, do not affect plasma concentration or efficacy of lamotrigine.43 Although data are limited regarding the use of lamotrigine in severe renal impairment and ESRD, Kaufman44 reported a 17% to 20% decrease in concentration after dialysis—suggesting that post-dialysis titration might be needed in these patients.
Oxcarbazepine is metabolized by means of cytosolic enzymes in the liver to its primary pharmacologically active metabolite, 10-monohydroxy, which is further metabolized via glucuronidation and then renally excreted. There are no dosage adjustment recommendations for patients with an eCrCl >30 mL/min.45 Rouan et al46 suggest initiating oxcarbazepine at 50% of the recommended dosage and following a longer titration schedule in patients with an eCrCl 10 to 30 mL/min. No dosing suggestions for severe renal impairment and ESRD were provided because of study limitations; however, the general recommendation for psychotropic agents in patients in a severe stage of renal impairment is dosage reduction with close monitoring.46
Table 341,44,46 summarizes dosage adjustments for mood stabilizers in patients with renal impairment.
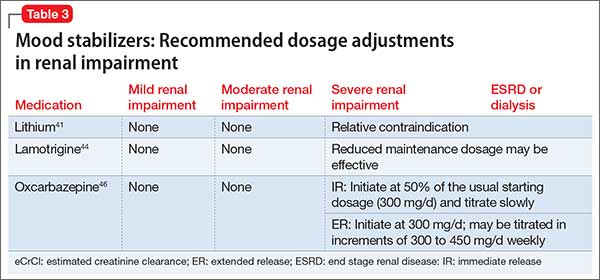
1. Levy G. Pharmacokinetics in renal disease. Am J Med. 1977;62(4):461-465.
2. Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 3. Effects of renal and hepatic impairment. J Psychiatr Pract. 2012;18(6):430-437.
3. Cohen LM, Tessier EG, Germain MJ, et al. Update on psychotropic medication use in renal disease. Psychosomatics. 2004;45(1):34-48.
4. Baghdady NT, Banik S, Swartz SA, et al. Psychotropic drugs and renal failure: translating the evidence for clinical practice. Adv Ther. 2009;26(4):404-424.
5. Sheehan J, White A, Wilson R. Hazards of phenothiazines in chronic renal failure. Ir Med J. 1982;75(9):335.
6. Haloperidol [monograph]. In: Micromedex Drugdex [online database]. Greenwood Village, CO: Truven Health Analytics. Accessed December 17, 2014.
7. Sheehan JJ, Sliwa JK, Amatniek JC, et al. Atypical antipsychotic metabolism and excretion. Curr Drug Metab. 2010;11(6):516-525.
8. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2014.
9. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
10. Invega [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
11. Latuda [package insert]. Fort Lee, NJ: Sunovion Pharmaceuticals; 2013.
12. Aweeka F, Jayesekara D, Horton M, et al. The pharmacokinetics of ziprasidone in subjects with normal and impaired renal function. Br J Clin Pharmacol. 2004;49(suppl 1):27S-33S.
13. Peeters P, Bockbrader H, Spaans E, et al. Asenapine pharmacokinetics in hepatic and renal impairment. Clin Pharmacol. 2011;50(7):471-481.
14. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533-542.
15. Thyrum PT, Wong YW, Yeh C. Single-dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521-533.
16. Lexi-Drugs. Lexicomp. Hudson, OH: Wolters Kluwer Health, Inc. http://online.lexi.com. Accessed May 28, 2015.
17. Lexapro [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
18. Celexa [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
19. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
20. Effexor [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2010.
21. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2014.
22. Cymbalta [package insert]. Indianapolis, IN: Lilly USA, LLC; 2014.
23. Savella [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2013.
24. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2014.
25. Kurella M, Bennett WM, Chertow GM. Analgesia in patients with ESRD: a review of available evidence. Am J Kidney Dis. 2003;42(2):217-228.
26. Wellbutrin [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
27. Worrall SP, Almond MK, Dhillon S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron Clin Pract. 2004;97(3):c83-c89.
28. Nagler EV, Webster AC, Vanholder R, et al. Antidepressants for depression in stage 3-5 chronic kidney disease: a systematic review of pharmacokinetics, efficacy and safety with recommendations by European Renal Best Practice (ERBP). Nephrol Dial Transplant. 2012;27(10):3736-3745.
29. Norpramin. [package insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; 2014.
30. Tofranil [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
31. Pamelor [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
32. Marplan [package insert]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012.
33. Nardil [package insert]. New York, NY: Parke-Davis Division of Pfizer Inc.; 2009.
34. EMSAM [package insert]. Morgantown, WV: Mylan Specialty, L.P.; 2014.
35. Eldepryl [package insert]. Morgantown, WV: Somerset Pharmaceuticals, Inc.; 2009.
36. Parnate [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
37. Culpepper L. Reducing the burden of difficult-to-treat major depressive disorder: revisiting monoamine oxidase inhibitor therapy. Prim Care Companion CNS Disord. 2013;15(5). doi: 10.4088/PCC.13r01515.
38. Tossani E, Cassano P, Fava M. Depression and renal disease. Semin Dial. 2005;18(2):73-81.
39. Young AH, Hammond JM. Lithium in mood disorders: increasing evidence base, declining use? Br J Psychiatry. 2007;191:474-476.
40. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
41. Rej S, Looper K, Segal M. The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging. 2013;30(6):409-415.
42. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211.
43. Lamictal [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
44. Kaufman KR. Lamotrigine and hemodialysis in bipolar disorder: case analysis of dosing strategy with literature review. Bipolar Disord. 2010;12(4):446-449.
45. Trileptal [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
46. Rouan MC, Lecaillon JB, Godbillon J, et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. 1994;47(2):161-167.
Renal disease can play a large role in altering the pharmacokinetics of medications, especially in elimination or clearance and plasma protein binding. Specifically, renal impairment decreases the plasma protein binding secondary to decreased albumin and retention of urea, which competes with medications to bind to the protein.1
Electrolyte shifts—which could lead to a fatal arrhythmia—are common among patients with renal impairment. The risk can be further increased in this population if a patient is taking a medication that can induce arrhythmia. If a drug is primarily excreted by the kidneys, elimination could be significantly altered, especially if the medication has active metabolites.1
Normal renal function is defined as an estimated creatinine clearance (eCrCl) of >80 mL/min. Renal impairment is classified as:
- mild: eCrCl, 51 to 80 mL/min
- moderate: eCrCl, 31 to 50 mL/min
- severe: eCrCl, ≤30 mL/min
- end-stage renal disease (ESRD): eCrCl, <10 mL/min.2
Overall, there is minimal information about the effects of renal disease on psychotropic therapy; our goal here is to summarize available data. We have created quick reference tables highlighting psychotropics that have renal dosing recommendations based on manufacturers’ package inserts.
Antipsychotics
First-generation antipsychotics (FGAs). Dosage adjustments based on renal function are not required for any FGA, according to manufacturers’ package inserts. Some of these antipsychotics are excreted in urine, but typically as inactive metabolites.
Although there are no dosage recommendations based on renal function provided by the labeling, there has been concern about the use of some FGAs in patients with renal impairment. Specifically, concerns center around the piperidine phenothiazines (thioridazine and mesoridazine) because of the increased risk of electrocardiographic changes and medication-induced arrhythmias in renal disease due to electrolyte imbalances.3,4 Additionally, there is case evidence5 that phenothiazine antipsychotics could increase a patient’s risk for hypotension in chronic renal failure. Haloperidol is considered safe in renal disease because <1% of the medication is excreted unchanged through urine.6
Second-generation antipsychotics (SGAs). Overall, SGAs are considered safe in patients with renal disease. Most SGAs undergo extensive hepatic metabolism before excretion, allowing them to be used safely in patients with renal disease.
Sheehan et al7 analyzed the metabolism and excretion of SGAs, evaluating 8 antipsychotics divided into 4 groups: (1) excretion primarily as an unchanged drug in urine, (2) changed drug in urine, (3) changed drug in feces, (4) and unchanged drug in feces.
- Paliperidone was found to be primarily excreted as an unchanged drug in urine.
- Clozapine, iloperidone, olanzapine, quetiapine, and risperidone all were found to be primarily excreted as a changed drug in urine.
- Aripiprazole and ziprasidone were found to be primarily excreted as a changed drug in feces.
The manufacturers’ package inserts for clozapine, paliperidone, risperidone, and lurasidone have recommended dosage adjustments based on renal function (Table 1).8-11
Ziprasidone. Although ziprasidone does not have a recommended renal dosage adjustment, caution is recommended because of the risk of electrocardiographic changes and potential for medication-induced arrhythmias in patients with electrolyte disturbances secondary to renal disease. A single-dosage study of ziprasidone by Aweeka et al12 demonstrated that the pharmacokinetics of ziprasidone are unchanged in patients with renal impairment.
Asenapine. A small study by Peeters et al13 evaluated the pharmacokinetics of asenapine in hepatic and renal impairment and found no clinically relevant changes in asenapine’s pharmacokinetics among patients with any level of renal impairment compared with patients with normal renal function.
Aripiprazole. Mallikaarjun et al14 completed a small study evaluating the pharmacokinetics of aripiprazole in patients with renal impairment. They found that the pharmacokinetics of aripiprazole in these patients is no different than it is in patients with normal renal function who are taking aripiprazole.
Quetiapine. Thyrum et al15 conducted a similar study with quetiapine, which showed no significant difference detected in the pharmacokinetics of quetiapine in patients with renal impairment. Additionally, quetiapine had no negative effect on patients’ creatinine clearance.
Lurasidone. During clinical trials of lurasidone in patients with mild, moderate, and severe renal impairment, the mean Cmax and area under the curve was higher compared with healthy patients, which led to recommended dosage adjustments in patients with renal impairment.11
As mentioned above, renal impairment decreases the protein binding percentage of medications. Hypothetically, the greater the protein binding, the lower the recommended dosage in patients with renal impairment because the free or unbound form correlates with efficacy and toxicity. Most FGAs and SGAs have the protein-binding characteristic of ≥90%.16 Although it seems this characteristic should result in recommendations to adjust dosage based on renal function, the various pharmacokinetic studies of antipsychotics have not shown this factor to play a role in the manufacturers’ recommendations.
Antidepressants
Comorbidity rates of depression in patients with renal disease range from 14% to 30%, making use of antidepressants in renal disease common.4 Antidepressants primarily are metabolized hepatically and excreted renally. Table 217-27 summarizes recommended dosing adjustments for antidepressants.
Selective serotonin reuptake inhibitors.Escitalopram is the (S)-enantiomer of the racemic antidepressant citalopram, both of which have been shown to decrease renal clearance in patients with mild or moderate renal impairment. However, according to the package insert, no dosage adjustments are needed.17 No extensive studies have been conducted on escitalopram or citalopram, but each should be initiated at a reduced dosage and the titration schedule should be prolonged in patients with severe renal impairment or ESRD.17,18
The plasma concentration of paroxetine has been noted to be elevated in patients with severe renal impairment, and the half-life can increase to nearly 50%.4 Paroxetine should be initiated at 10 mg/d, and then titrated slowly in patients with severe renal impairment.19,28
The pharmacokinetics of fluoxetine are unchanged in any stage of renal impairment. Patients in active renal dialysis report good tolerability and efficacy.4
Serotonin-norepinephrine reuptake inhibitors. Venlafaxine and its metabolite O-desmethylvenlafaxine (desvenlafaxine) are primarily excreted via renal elimination. Studies have shown that mild renal impairment can have an effect on plasma levels of the drug, and that moderate or severe impairment can increase the venlafaxine plasma concentration. According to the package insert, a dosage reduction of 50% is recommended for desvenlafaxine and venlafaxine.20,21
No significant pharmacokinetic changes with duloxetine have been noted in patients with mild or moderate renal impairment.22 However, duloxetine’s major metabolites, which are excreted renally, have been measured to be as much as 7 to 9 times higher in patients with ESRD compared with healthy subjects; therefore, it is recommended to avoid duloxetine in patients with severe renal disease.4,22 Our review of the literature produced limited recommendations on dosing milnacipran and its enantiomer levomilnacipran in renally impaired patients. The milnacipran package insert cautions its use in moderate renal impairment and recommends a 50% dosage reduction to 100 mg/d (50 mg twice daily) in patients with severe renal impairment.23 Dosage recommendations for levomilnacipran are 80 mg/d for moderate renal impairment and 40 mg/d for severe impairment. Both agents have relative contraindications for ESRD.23,24
Tricyclic antidepressants (TCAs) are predominantly metabolized hepatically, glucuronidated, and then eliminated renally. Desipramine, imipramine, and nortriptyline have nonspecific package insert recommendations for modified dosing in geriatric patients because of an age-related decrease in renal clearance.29-31 Review articles assert that elevated glucuronidated metabolites could increase patients’ sensitivity to side effects of TCAs. Because of concerns regarding elevated glucuronidated metabolites, it has been proposed to initiate TCAs at a low dosage, titrate slowly, and maintain the lowest effective dosage in patients with renal impairment.25
Monoamine oxidase inhibitors (MAOIs) and other antidepressants. The package inserts of the MAOIs isocarboxazid, phenelzine, selegiline, and tranylcypromine provide limited data and dosage recommendations for use in the context of renal impairment.32-36 Isocarboxazid should not be used in patients with severe renal impairment, according to the prescribing information.32 There are no dosing recommendations for transdermal selegiline in mild, moderate, or severe renal impairment.37 Extra vigilance is required when using MAOIs in patients with renal disease because of an increased risk of dialysis-induced hypotension (orthostatic hypotension is a common adverse effect of MAOIs).38
Bupropion is primarily metabolized hepatically to the active metabolite hydroxybupropion. Plasma levels of this metabolite at steady state are reported to be 10 times greater than bupropion’s concentration levels in healthy subjects; plasma levels are further increased in mild renal impairment.26 Hydroxybupropion is not dialyzable, which can increase the risk of toxicity with bupropion therapy in patients with renal impairment.3 If bupropion effectively treats depression in patients with declining renal function, specifically severe renal impairment and ESRD, then decreasing the dosage to 150 mg every 3 days is recommended to lessen the risk of toxicity. 27
Mood stabilizers
Lithium has the most published literature on dosing adjustments with renal impairment. Many providers are inclined to discontinue lithium use at the first sign of any change in renal function; however, monitoring, prevention, and treatment guidelines for lithium are well established after many years of research and clinical use.39 Lithium’s prescribing information recommends dosage adjustment in mild to moderate renal impairment and lists severe renal impairment and ESRD as relative contraindications.40
A recent study proposes more assertive use of lithium in patients with renal impairment of any severity. Rej et al41 compared continued lithium treatment to discontinuing treatment in geriatric patients with chronic renal failure, and reported (1) a statistically insignificant difference in renal function between groups at 2 years and (2) a “trending decrease” in renal function at 5 years in the lithium treatment group. With closely monitored plasma levels, lithium treatment is considered a workable treatment for patients with moderate renal impairment when mood stabilizer treatment has been effective.42
Lamotrigine and its main glucuronidated metabolite, lamotrigine-2N-glucuronide (L-2-N-G), are primarily excreted renally. In severe renal impairment and ESRD, the L-2-N-G levels are elevated but are not pharmacologically active and, therefore, do not affect plasma concentration or efficacy of lamotrigine.43 Although data are limited regarding the use of lamotrigine in severe renal impairment and ESRD, Kaufman44 reported a 17% to 20% decrease in concentration after dialysis—suggesting that post-dialysis titration might be needed in these patients.
Oxcarbazepine is metabolized by means of cytosolic enzymes in the liver to its primary pharmacologically active metabolite, 10-monohydroxy, which is further metabolized via glucuronidation and then renally excreted. There are no dosage adjustment recommendations for patients with an eCrCl >30 mL/min.45 Rouan et al46 suggest initiating oxcarbazepine at 50% of the recommended dosage and following a longer titration schedule in patients with an eCrCl 10 to 30 mL/min. No dosing suggestions for severe renal impairment and ESRD were provided because of study limitations; however, the general recommendation for psychotropic agents in patients in a severe stage of renal impairment is dosage reduction with close monitoring.46
Table 341,44,46 summarizes dosage adjustments for mood stabilizers in patients with renal impairment.
Renal disease can play a large role in altering the pharmacokinetics of medications, especially in elimination or clearance and plasma protein binding. Specifically, renal impairment decreases the plasma protein binding secondary to decreased albumin and retention of urea, which competes with medications to bind to the protein.1
Electrolyte shifts—which could lead to a fatal arrhythmia—are common among patients with renal impairment. The risk can be further increased in this population if a patient is taking a medication that can induce arrhythmia. If a drug is primarily excreted by the kidneys, elimination could be significantly altered, especially if the medication has active metabolites.1
Normal renal function is defined as an estimated creatinine clearance (eCrCl) of >80 mL/min. Renal impairment is classified as:
- mild: eCrCl, 51 to 80 mL/min
- moderate: eCrCl, 31 to 50 mL/min
- severe: eCrCl, ≤30 mL/min
- end-stage renal disease (ESRD): eCrCl, <10 mL/min.2
Overall, there is minimal information about the effects of renal disease on psychotropic therapy; our goal here is to summarize available data. We have created quick reference tables highlighting psychotropics that have renal dosing recommendations based on manufacturers’ package inserts.
Antipsychotics
First-generation antipsychotics (FGAs). Dosage adjustments based on renal function are not required for any FGA, according to manufacturers’ package inserts. Some of these antipsychotics are excreted in urine, but typically as inactive metabolites.
Although there are no dosage recommendations based on renal function provided by the labeling, there has been concern about the use of some FGAs in patients with renal impairment. Specifically, concerns center around the piperidine phenothiazines (thioridazine and mesoridazine) because of the increased risk of electrocardiographic changes and medication-induced arrhythmias in renal disease due to electrolyte imbalances.3,4 Additionally, there is case evidence5 that phenothiazine antipsychotics could increase a patient’s risk for hypotension in chronic renal failure. Haloperidol is considered safe in renal disease because <1% of the medication is excreted unchanged through urine.6
Second-generation antipsychotics (SGAs). Overall, SGAs are considered safe in patients with renal disease. Most SGAs undergo extensive hepatic metabolism before excretion, allowing them to be used safely in patients with renal disease.
Sheehan et al7 analyzed the metabolism and excretion of SGAs, evaluating 8 antipsychotics divided into 4 groups: (1) excretion primarily as an unchanged drug in urine, (2) changed drug in urine, (3) changed drug in feces, (4) and unchanged drug in feces.
- Paliperidone was found to be primarily excreted as an unchanged drug in urine.
- Clozapine, iloperidone, olanzapine, quetiapine, and risperidone all were found to be primarily excreted as a changed drug in urine.
- Aripiprazole and ziprasidone were found to be primarily excreted as a changed drug in feces.
The manufacturers’ package inserts for clozapine, paliperidone, risperidone, and lurasidone have recommended dosage adjustments based on renal function (Table 1).8-11
Ziprasidone. Although ziprasidone does not have a recommended renal dosage adjustment, caution is recommended because of the risk of electrocardiographic changes and potential for medication-induced arrhythmias in patients with electrolyte disturbances secondary to renal disease. A single-dosage study of ziprasidone by Aweeka et al12 demonstrated that the pharmacokinetics of ziprasidone are unchanged in patients with renal impairment.
Asenapine. A small study by Peeters et al13 evaluated the pharmacokinetics of asenapine in hepatic and renal impairment and found no clinically relevant changes in asenapine’s pharmacokinetics among patients with any level of renal impairment compared with patients with normal renal function.
Aripiprazole. Mallikaarjun et al14 completed a small study evaluating the pharmacokinetics of aripiprazole in patients with renal impairment. They found that the pharmacokinetics of aripiprazole in these patients is no different than it is in patients with normal renal function who are taking aripiprazole.
Quetiapine. Thyrum et al15 conducted a similar study with quetiapine, which showed no significant difference detected in the pharmacokinetics of quetiapine in patients with renal impairment. Additionally, quetiapine had no negative effect on patients’ creatinine clearance.
Lurasidone. During clinical trials of lurasidone in patients with mild, moderate, and severe renal impairment, the mean Cmax and area under the curve was higher compared with healthy patients, which led to recommended dosage adjustments in patients with renal impairment.11
As mentioned above, renal impairment decreases the protein binding percentage of medications. Hypothetically, the greater the protein binding, the lower the recommended dosage in patients with renal impairment because the free or unbound form correlates with efficacy and toxicity. Most FGAs and SGAs have the protein-binding characteristic of ≥90%.16 Although it seems this characteristic should result in recommendations to adjust dosage based on renal function, the various pharmacokinetic studies of antipsychotics have not shown this factor to play a role in the manufacturers’ recommendations.
Antidepressants
Comorbidity rates of depression in patients with renal disease range from 14% to 30%, making use of antidepressants in renal disease common.4 Antidepressants primarily are metabolized hepatically and excreted renally. Table 217-27 summarizes recommended dosing adjustments for antidepressants.
Selective serotonin reuptake inhibitors.Escitalopram is the (S)-enantiomer of the racemic antidepressant citalopram, both of which have been shown to decrease renal clearance in patients with mild or moderate renal impairment. However, according to the package insert, no dosage adjustments are needed.17 No extensive studies have been conducted on escitalopram or citalopram, but each should be initiated at a reduced dosage and the titration schedule should be prolonged in patients with severe renal impairment or ESRD.17,18
The plasma concentration of paroxetine has been noted to be elevated in patients with severe renal impairment, and the half-life can increase to nearly 50%.4 Paroxetine should be initiated at 10 mg/d, and then titrated slowly in patients with severe renal impairment.19,28
The pharmacokinetics of fluoxetine are unchanged in any stage of renal impairment. Patients in active renal dialysis report good tolerability and efficacy.4
Serotonin-norepinephrine reuptake inhibitors. Venlafaxine and its metabolite O-desmethylvenlafaxine (desvenlafaxine) are primarily excreted via renal elimination. Studies have shown that mild renal impairment can have an effect on plasma levels of the drug, and that moderate or severe impairment can increase the venlafaxine plasma concentration. According to the package insert, a dosage reduction of 50% is recommended for desvenlafaxine and venlafaxine.20,21
No significant pharmacokinetic changes with duloxetine have been noted in patients with mild or moderate renal impairment.22 However, duloxetine’s major metabolites, which are excreted renally, have been measured to be as much as 7 to 9 times higher in patients with ESRD compared with healthy subjects; therefore, it is recommended to avoid duloxetine in patients with severe renal disease.4,22 Our review of the literature produced limited recommendations on dosing milnacipran and its enantiomer levomilnacipran in renally impaired patients. The milnacipran package insert cautions its use in moderate renal impairment and recommends a 50% dosage reduction to 100 mg/d (50 mg twice daily) in patients with severe renal impairment.23 Dosage recommendations for levomilnacipran are 80 mg/d for moderate renal impairment and 40 mg/d for severe impairment. Both agents have relative contraindications for ESRD.23,24
Tricyclic antidepressants (TCAs) are predominantly metabolized hepatically, glucuronidated, and then eliminated renally. Desipramine, imipramine, and nortriptyline have nonspecific package insert recommendations for modified dosing in geriatric patients because of an age-related decrease in renal clearance.29-31 Review articles assert that elevated glucuronidated metabolites could increase patients’ sensitivity to side effects of TCAs. Because of concerns regarding elevated glucuronidated metabolites, it has been proposed to initiate TCAs at a low dosage, titrate slowly, and maintain the lowest effective dosage in patients with renal impairment.25
Monoamine oxidase inhibitors (MAOIs) and other antidepressants. The package inserts of the MAOIs isocarboxazid, phenelzine, selegiline, and tranylcypromine provide limited data and dosage recommendations for use in the context of renal impairment.32-36 Isocarboxazid should not be used in patients with severe renal impairment, according to the prescribing information.32 There are no dosing recommendations for transdermal selegiline in mild, moderate, or severe renal impairment.37 Extra vigilance is required when using MAOIs in patients with renal disease because of an increased risk of dialysis-induced hypotension (orthostatic hypotension is a common adverse effect of MAOIs).38
Bupropion is primarily metabolized hepatically to the active metabolite hydroxybupropion. Plasma levels of this metabolite at steady state are reported to be 10 times greater than bupropion’s concentration levels in healthy subjects; plasma levels are further increased in mild renal impairment.26 Hydroxybupropion is not dialyzable, which can increase the risk of toxicity with bupropion therapy in patients with renal impairment.3 If bupropion effectively treats depression in patients with declining renal function, specifically severe renal impairment and ESRD, then decreasing the dosage to 150 mg every 3 days is recommended to lessen the risk of toxicity. 27
Mood stabilizers
Lithium has the most published literature on dosing adjustments with renal impairment. Many providers are inclined to discontinue lithium use at the first sign of any change in renal function; however, monitoring, prevention, and treatment guidelines for lithium are well established after many years of research and clinical use.39 Lithium’s prescribing information recommends dosage adjustment in mild to moderate renal impairment and lists severe renal impairment and ESRD as relative contraindications.40
A recent study proposes more assertive use of lithium in patients with renal impairment of any severity. Rej et al41 compared continued lithium treatment to discontinuing treatment in geriatric patients with chronic renal failure, and reported (1) a statistically insignificant difference in renal function between groups at 2 years and (2) a “trending decrease” in renal function at 5 years in the lithium treatment group. With closely monitored plasma levels, lithium treatment is considered a workable treatment for patients with moderate renal impairment when mood stabilizer treatment has been effective.42
Lamotrigine and its main glucuronidated metabolite, lamotrigine-2N-glucuronide (L-2-N-G), are primarily excreted renally. In severe renal impairment and ESRD, the L-2-N-G levels are elevated but are not pharmacologically active and, therefore, do not affect plasma concentration or efficacy of lamotrigine.43 Although data are limited regarding the use of lamotrigine in severe renal impairment and ESRD, Kaufman44 reported a 17% to 20% decrease in concentration after dialysis—suggesting that post-dialysis titration might be needed in these patients.
Oxcarbazepine is metabolized by means of cytosolic enzymes in the liver to its primary pharmacologically active metabolite, 10-monohydroxy, which is further metabolized via glucuronidation and then renally excreted. There are no dosage adjustment recommendations for patients with an eCrCl >30 mL/min.45 Rouan et al46 suggest initiating oxcarbazepine at 50% of the recommended dosage and following a longer titration schedule in patients with an eCrCl 10 to 30 mL/min. No dosing suggestions for severe renal impairment and ESRD were provided because of study limitations; however, the general recommendation for psychotropic agents in patients in a severe stage of renal impairment is dosage reduction with close monitoring.46
Table 341,44,46 summarizes dosage adjustments for mood stabilizers in patients with renal impairment.
1. Levy G. Pharmacokinetics in renal disease. Am J Med. 1977;62(4):461-465.
2. Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 3. Effects of renal and hepatic impairment. J Psychiatr Pract. 2012;18(6):430-437.
3. Cohen LM, Tessier EG, Germain MJ, et al. Update on psychotropic medication use in renal disease. Psychosomatics. 2004;45(1):34-48.
4. Baghdady NT, Banik S, Swartz SA, et al. Psychotropic drugs and renal failure: translating the evidence for clinical practice. Adv Ther. 2009;26(4):404-424.
5. Sheehan J, White A, Wilson R. Hazards of phenothiazines in chronic renal failure. Ir Med J. 1982;75(9):335.
6. Haloperidol [monograph]. In: Micromedex Drugdex [online database]. Greenwood Village, CO: Truven Health Analytics. Accessed December 17, 2014.
7. Sheehan JJ, Sliwa JK, Amatniek JC, et al. Atypical antipsychotic metabolism and excretion. Curr Drug Metab. 2010;11(6):516-525.
8. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2014.
9. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
10. Invega [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
11. Latuda [package insert]. Fort Lee, NJ: Sunovion Pharmaceuticals; 2013.
12. Aweeka F, Jayesekara D, Horton M, et al. The pharmacokinetics of ziprasidone in subjects with normal and impaired renal function. Br J Clin Pharmacol. 2004;49(suppl 1):27S-33S.
13. Peeters P, Bockbrader H, Spaans E, et al. Asenapine pharmacokinetics in hepatic and renal impairment. Clin Pharmacol. 2011;50(7):471-481.
14. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533-542.
15. Thyrum PT, Wong YW, Yeh C. Single-dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521-533.
16. Lexi-Drugs. Lexicomp. Hudson, OH: Wolters Kluwer Health, Inc. http://online.lexi.com. Accessed May 28, 2015.
17. Lexapro [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
18. Celexa [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
19. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
20. Effexor [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2010.
21. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2014.
22. Cymbalta [package insert]. Indianapolis, IN: Lilly USA, LLC; 2014.
23. Savella [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2013.
24. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2014.
25. Kurella M, Bennett WM, Chertow GM. Analgesia in patients with ESRD: a review of available evidence. Am J Kidney Dis. 2003;42(2):217-228.
26. Wellbutrin [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
27. Worrall SP, Almond MK, Dhillon S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron Clin Pract. 2004;97(3):c83-c89.
28. Nagler EV, Webster AC, Vanholder R, et al. Antidepressants for depression in stage 3-5 chronic kidney disease: a systematic review of pharmacokinetics, efficacy and safety with recommendations by European Renal Best Practice (ERBP). Nephrol Dial Transplant. 2012;27(10):3736-3745.
29. Norpramin. [package insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; 2014.
30. Tofranil [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
31. Pamelor [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
32. Marplan [package insert]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012.
33. Nardil [package insert]. New York, NY: Parke-Davis Division of Pfizer Inc.; 2009.
34. EMSAM [package insert]. Morgantown, WV: Mylan Specialty, L.P.; 2014.
35. Eldepryl [package insert]. Morgantown, WV: Somerset Pharmaceuticals, Inc.; 2009.
36. Parnate [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
37. Culpepper L. Reducing the burden of difficult-to-treat major depressive disorder: revisiting monoamine oxidase inhibitor therapy. Prim Care Companion CNS Disord. 2013;15(5). doi: 10.4088/PCC.13r01515.
38. Tossani E, Cassano P, Fava M. Depression and renal disease. Semin Dial. 2005;18(2):73-81.
39. Young AH, Hammond JM. Lithium in mood disorders: increasing evidence base, declining use? Br J Psychiatry. 2007;191:474-476.
40. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
41. Rej S, Looper K, Segal M. The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging. 2013;30(6):409-415.
42. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211.
43. Lamictal [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
44. Kaufman KR. Lamotrigine and hemodialysis in bipolar disorder: case analysis of dosing strategy with literature review. Bipolar Disord. 2010;12(4):446-449.
45. Trileptal [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
46. Rouan MC, Lecaillon JB, Godbillon J, et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. 1994;47(2):161-167.
1. Levy G. Pharmacokinetics in renal disease. Am J Med. 1977;62(4):461-465.
2. Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 3. Effects of renal and hepatic impairment. J Psychiatr Pract. 2012;18(6):430-437.
3. Cohen LM, Tessier EG, Germain MJ, et al. Update on psychotropic medication use in renal disease. Psychosomatics. 2004;45(1):34-48.
4. Baghdady NT, Banik S, Swartz SA, et al. Psychotropic drugs and renal failure: translating the evidence for clinical practice. Adv Ther. 2009;26(4):404-424.
5. Sheehan J, White A, Wilson R. Hazards of phenothiazines in chronic renal failure. Ir Med J. 1982;75(9):335.
6. Haloperidol [monograph]. In: Micromedex Drugdex [online database]. Greenwood Village, CO: Truven Health Analytics. Accessed December 17, 2014.
7. Sheehan JJ, Sliwa JK, Amatniek JC, et al. Atypical antipsychotic metabolism and excretion. Curr Drug Metab. 2010;11(6):516-525.
8. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2014.
9. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
10. Invega [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
11. Latuda [package insert]. Fort Lee, NJ: Sunovion Pharmaceuticals; 2013.
12. Aweeka F, Jayesekara D, Horton M, et al. The pharmacokinetics of ziprasidone in subjects with normal and impaired renal function. Br J Clin Pharmacol. 2004;49(suppl 1):27S-33S.
13. Peeters P, Bockbrader H, Spaans E, et al. Asenapine pharmacokinetics in hepatic and renal impairment. Clin Pharmacol. 2011;50(7):471-481.
14. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533-542.
15. Thyrum PT, Wong YW, Yeh C. Single-dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521-533.
16. Lexi-Drugs. Lexicomp. Hudson, OH: Wolters Kluwer Health, Inc. http://online.lexi.com. Accessed May 28, 2015.
17. Lexapro [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
18. Celexa [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
19. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
20. Effexor [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2010.
21. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2014.
22. Cymbalta [package insert]. Indianapolis, IN: Lilly USA, LLC; 2014.
23. Savella [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2013.
24. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2014.
25. Kurella M, Bennett WM, Chertow GM. Analgesia in patients with ESRD: a review of available evidence. Am J Kidney Dis. 2003;42(2):217-228.
26. Wellbutrin [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
27. Worrall SP, Almond MK, Dhillon S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron Clin Pract. 2004;97(3):c83-c89.
28. Nagler EV, Webster AC, Vanholder R, et al. Antidepressants for depression in stage 3-5 chronic kidney disease: a systematic review of pharmacokinetics, efficacy and safety with recommendations by European Renal Best Practice (ERBP). Nephrol Dial Transplant. 2012;27(10):3736-3745.
29. Norpramin. [package insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; 2014.
30. Tofranil [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
31. Pamelor [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
32. Marplan [package insert]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012.
33. Nardil [package insert]. New York, NY: Parke-Davis Division of Pfizer Inc.; 2009.
34. EMSAM [package insert]. Morgantown, WV: Mylan Specialty, L.P.; 2014.
35. Eldepryl [package insert]. Morgantown, WV: Somerset Pharmaceuticals, Inc.; 2009.
36. Parnate [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
37. Culpepper L. Reducing the burden of difficult-to-treat major depressive disorder: revisiting monoamine oxidase inhibitor therapy. Prim Care Companion CNS Disord. 2013;15(5). doi: 10.4088/PCC.13r01515.
38. Tossani E, Cassano P, Fava M. Depression and renal disease. Semin Dial. 2005;18(2):73-81.
39. Young AH, Hammond JM. Lithium in mood disorders: increasing evidence base, declining use? Br J Psychiatry. 2007;191:474-476.
40. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
41. Rej S, Looper K, Segal M. The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging. 2013;30(6):409-415.
42. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211.
43. Lamictal [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
44. Kaufman KR. Lamotrigine and hemodialysis in bipolar disorder: case analysis of dosing strategy with literature review. Bipolar Disord. 2010;12(4):446-449.
45. Trileptal [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
46. Rouan MC, Lecaillon JB, Godbillon J, et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. 1994;47(2):161-167.
Psychosis in treated neurosyphilis: Is now the time to stop his antipsychotic?
CASE Hallucinations, impaired memory
Mr. C is a 61-year-old African American man who visits the outpatient clinic for management of antipsychotic therapy for psychosis and depression. His most recent inpatient psychiatric hospitalization for auditory and visual hallucinations, paranoia, and agitation was more than 10 years ago. He has been taking chlorpromazine, 100 mg/d, for 11 years. Mr. C reports that he has had no psychotic symptoms in the past 3 years; he continues taking chlorpromazine, he says, because it helps him sleep.
How would you proceed with Mr. C’s care?
a) continue chlorpromazine because he has been symptom free
b) consider tapering and discontinuing chlorpromazine
c) obtain a more detailed history from Mr. C and perform additional tests
HISTORY Validation of diagnosis
Mr. C reports that, at age 48, he started hearing babies crying and started seeing dead infants crawling out of the incinerator at the hospital where he worked. He denies any psychiatric symptoms before that time. He stopped working 10 years ago because of his psychiatric symptoms and decline in cognition.
Subsequently, Mr. C had 3 inpatient psychiatric hospitalizations for auditory hallucinations; chlorpromazine, 100 mg/d, was prescribed for psychosis. Later efforts to discontinue chlorpromazine resulted in relapse of psychotic symptoms. Mr. C has no family history of psychiatric illness.
Mr. C’s medical history is significant for aortic regurgitation, congestive cardiac failure, hypertension, and left-sided sensorineural hearing loss. He has a history of cocaine abuse from age 21 to 45, but denies using any other substances, including alcohol and nicotine.
Urine toxicology and routine blood tests are within normal limits. The QTc is slightly prolonged over the past 2 years, recording 512, 520, and 505 milliseconds on serial electrocardiograms.
Mr. C is able to perform simple abstractions. He has a goal-directed thought process, devoid of any preoccupation, paranoia, and perceptual abnormalities. Cognitive screening reveals significant impairment of memory, registration, calculation, attention, and visuospatial skills.
Careful review of Mr. C’s history and medical records reveals a diagnosis of syphilis at age 48 after unprotected sexual intercourse. He recalls that he had a solitary genital lesion, which resolved over a few weeks. He then developed a slightly itchy, non-tender macular rash over his upper back, which he did not report to a physician. After a few months, he developed unsteady gait, blurry vision, and weakness of limbs, and had to crawl to the hospital. There, he was given a diagnosis of neurosyphilis. He also developed left-sided hearing loss during that time.
Mr. C was treated with aqueous penicillin G benzathine, 4 million units IV for 2 weeks. No follow-up cerebrospinal fluid (CSF) examination was documented after antibiotic treatment. He developed auditory and visual hallucinations and paranoia a few months after completing penicillin treatment. During the following year, he had 3 inpatient psychiatric hospitalizations for psychosis, agitation, and depressed mood.
How would you treat a patient with a history of neurosyphilis who presents with psychosis years after diagnosis?
a) repeat antibiotic treatment and stop the antipsychotic
b) repeat antibiotic treatment and continue the antipsychotic
c) attempt to discontinue the antipsychotic
d) continue the antipsychotic
The authors’ observations
Mr. C’s psychotic symptoms seem to be temporally related to his diagnosis of neurosyphilis at age 48. He and his family members deny that Mr. C had any history of psychosis or depression before the neurosyphilis diagnosis. All inpatient psychiatric hospitalizations were within 1 year of the neurosyphilis diagnosis.
Mr. C has been on a low dosage of chlorpromazine, which has significant antihistaminic action. Chlorpromazine also is known to cause QTc prolongation, especially in patients with heart disease.
TREATMENT Medication change
A serum rapid plasma reagin test is non-reactive, but Treponema pallidum particle agglutination is positive. MRI shows moderate atrophy suggestive of diffuse small-vessel disease.
Mr. C’s psychotic symptoms are considered to be sequelae of neurosyphilis, based on (1) the presence of positive antibody tests, (2) residual neurologic deficits, (3) other suggestive sequelae (aortic regurgitation, sensorineural deafness), and (4) age-inappropriate gradual cognitive decline in the absence of other psychiatric history.
Because we are concerned about the prolonged QTc, chlorpromazine is discontinued. Haloperidol, 5 mg at bedtime, is started. The neurology team does not recommend antibiotic treatment because symptoms have been stable for years. Mr. C refuses a lumbar puncture.
Mr. C returns to the outpatient clinic monthly. He is psychiatrically stable without any worsening of psychosis. Cognitive impairment remains stable over the next 6 months. Haloperidol is tapered to 2 mg at bedtime 6 months after initial evaluation. Mr. C remains psychiatrically stable on subsequent follow-up visits.
The authors’ observations
Mr. C’s psychotic symptoms persisted after standard antibiotic treatment of neurosyphilis and lapsed when he stopped taking antipsychotic medication 10 years after the initial treatment of neurosyphilis. He carried a diagnosis of schizophrenia for many years, even though his psychotic symptoms were atypical for the presentation of schizophrenia.
It is important to understand the natural course of syphilis, its implication on psychiatric symptom production, and long-term psychiatric prognosis.
Syphilis is a sexually transmitted infectious disease caused by T pallidum, a spirochete, that has varied clinical presentations. Osler called syphilis the “great imitator” for its array of system involvement, ranging from asymptomatic infection and afferent pupillary defect to depression, psychosis, and dementia. With wide use of penicillin, the rate of neurosyphilis declined steadily during the mid 1990s. By 1997, the overall rate reached its lowest point in the United States; in 1999 the Centers for Disease Control and Prevention released a national plan to eliminate syphilis.1 By 2004, however, prevalence had increased to 4.7/100,000. It is thought that this increase is mainly associated with substance use (especially crack cocaine) and HIV co-infection. Most cases were distributed in economically depressed geographical areas.
Psychiatric patients are at higher risk of acquiring the infection because of substance use, lack of education on safer sex practices, and impulsive behavior.
Stages of syphilis
Syphilis does not follow a step-wise progression. One-third of cases progress to the tertiary stage, even many years after initial infection, without adequate treatment.2
Almost 10% syphilis cases present with neurologic symptoms,3 and neurologic involvement can occur at any stage of disease progression. The most common symptoms of syphilis are presented in Table 1.
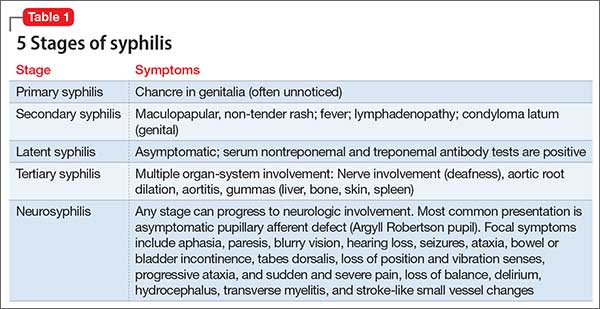
A range of psychiatric symptoms have been reported among patients with syphilis, including anhedonia, suicidality, mania, grandiosity, persecutory delusions, auditory and visual hallucinations, paranoia, and cognitive impairment. The incidence of psychiatric symptoms is not clearly described in literature.
Diagnosis and treatment
Neurosyphilis, at any disease stage, should be suspected if a patient:
- exhibits suggestive symptoms
- does not respond to antibiotic treatment
- has late latent syphilis
- is immunocompromised.
Lumbar puncture and examination of CSF is the most useful diagnostic test. Dark field microscopy to reveal T pallidum is definitive, but only is applicable during the primary stage. The role of dark field microscopy of the CSF sample to diagnose neurologic involvement has not been established. Tests and treatment protocol are described in Table 2.2-5
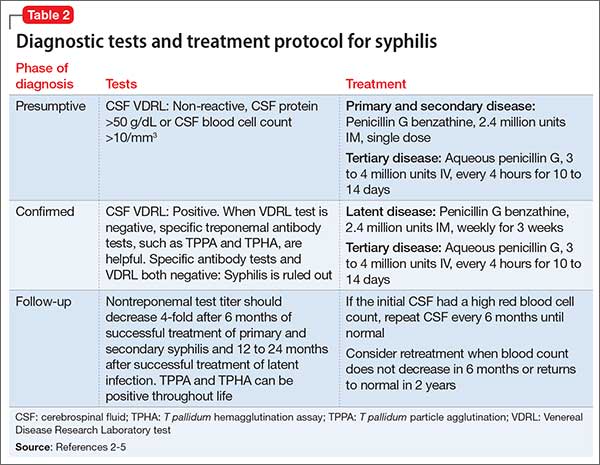
Treatment of psychiatric symptoms of neurosyphilis
There are inconsistent and limited data about the prevalence of psychiatric symptoms in neurosyphilis. A retrospective study6 of 161 patients with neurosyphilis in South Africa reported that 50.9% exhibited a complex spectrum of symptoms that included delirium and dementia. Of treated patients, 17% continued to have residual symptoms during follow-up.
A review of the literature did not reveal any widely accepted guideline for screening for neurosyphilis in general psychiatry practice or a treatment protocol for psychiatric symptoms. This lack of guidance could be attributed to the rarity of the disease, cost-benefit analyses, and low specificity of antibody tests. In the literature, syphilis screening is recommended as a routine protocol when evaluating and treating dementia.7
In most studies, a diagnosis of neurosyphilis was confirmed by CSF examination; however, many of these studies did not report a specific follow-up CSF examination protocol. Most of these patients were treated with an antipsychotic with partial improvement in symptoms, even after standard antibiotic protocol.8
First- and second-generation antipsychotics and mood stabilizers have been shown to be useful in the acute treatment of psychosis and agitation.8 In few instances, the psychotropic medication was continued beyond several months and the patient was placed in a long-term care facility. Psychiatric symptoms persisted for many years with or without residual neurosyphilis symptoms, possibly because of permanent neuronal loss.
Clinical considerations
It often is difficult to distinguish a preexisting psychiatric disorder made worse by neurosyphilis from a secondary psychiatric disorder caused by neurosyphilis. The 2 might coexist, or psychiatric symptoms could be wrongly attributed to schizophrenia because of a lack of careful clinical evaluation.
Often, the follow-up diagnostic protocol for neurosyphilis is not followed; as a result, the need for re-treatment remains unclear. Rarity of the disease makes it difficult to perform a prospective, randomized study to determine the duration and effect of long-term psychiatric treatment.
Close follow-up and consideration of the risk vs benefit of psychotropic medication is key. Because there are no proven guidelines for the length of treatment with antipsychotics, it is prudent to minimize their use until psychiatrically indicated. Side effects, such as (in Mr. C’s case) changes in the QTc interval, should warrant consideration of discontinuing psychotropic medication. Interdisciplinary collaboration with neurology and infectious disease will improve the overall outcome of a complex clinical presentation.
1. Centers for Disease Control and Prevention. National plan to eliminate syphilis from the United States. http://www.cdc.gov/stopsyphilis/plan.htm. Updated December 7, 2007. Accessed July 7, 2016.
2. Friedrich F, Aigner M, Fearns N, et al. Psychosis in neurosyphilis—clinical aspects and implications. Psychopathology. 2014;47(1):3-9.
3. Brown DL, Frank JE. Diagnosis and management of syphilis. Am Fam Physician. 2003;68(2):283-290.
4. Romanowski B, Sutherland R, Fick GH, et al. Serologic response to treatment of infectious syphilis. Ann Intern Med. 1991;114(12):1005-1009.
5. Centers for Disease Control and Prevention. 2015 Sexually transmitted diseases treatment guidelines. Syphilis. http://www.cdc.gov/std/tg2015/syphilis.htm. Updated June 4, 2015. Accessed July 13, 2016.
6. Timmermans M, Carr J. Neurosyphilis in the modern era. J Neurol Neurosurg Psychiatry. 2004;75(12):1727-1730.
7. Scott KR, Barrett AM. Dementia syndrome: evaluation and treatment. Expert Rev Neurother. 2007;7(4):407-422.
8. Sanchez FM, Zisselman MH. Treatment of psychiatric symptoms associated with neurosyphilis. Psychosomatics. 2007;48(5):440-445.
CASE Hallucinations, impaired memory
Mr. C is a 61-year-old African American man who visits the outpatient clinic for management of antipsychotic therapy for psychosis and depression. His most recent inpatient psychiatric hospitalization for auditory and visual hallucinations, paranoia, and agitation was more than 10 years ago. He has been taking chlorpromazine, 100 mg/d, for 11 years. Mr. C reports that he has had no psychotic symptoms in the past 3 years; he continues taking chlorpromazine, he says, because it helps him sleep.
How would you proceed with Mr. C’s care?
a) continue chlorpromazine because he has been symptom free
b) consider tapering and discontinuing chlorpromazine
c) obtain a more detailed history from Mr. C and perform additional tests
HISTORY Validation of diagnosis
Mr. C reports that, at age 48, he started hearing babies crying and started seeing dead infants crawling out of the incinerator at the hospital where he worked. He denies any psychiatric symptoms before that time. He stopped working 10 years ago because of his psychiatric symptoms and decline in cognition.
Subsequently, Mr. C had 3 inpatient psychiatric hospitalizations for auditory hallucinations; chlorpromazine, 100 mg/d, was prescribed for psychosis. Later efforts to discontinue chlorpromazine resulted in relapse of psychotic symptoms. Mr. C has no family history of psychiatric illness.
Mr. C’s medical history is significant for aortic regurgitation, congestive cardiac failure, hypertension, and left-sided sensorineural hearing loss. He has a history of cocaine abuse from age 21 to 45, but denies using any other substances, including alcohol and nicotine.
Urine toxicology and routine blood tests are within normal limits. The QTc is slightly prolonged over the past 2 years, recording 512, 520, and 505 milliseconds on serial electrocardiograms.
Mr. C is able to perform simple abstractions. He has a goal-directed thought process, devoid of any preoccupation, paranoia, and perceptual abnormalities. Cognitive screening reveals significant impairment of memory, registration, calculation, attention, and visuospatial skills.
Careful review of Mr. C’s history and medical records reveals a diagnosis of syphilis at age 48 after unprotected sexual intercourse. He recalls that he had a solitary genital lesion, which resolved over a few weeks. He then developed a slightly itchy, non-tender macular rash over his upper back, which he did not report to a physician. After a few months, he developed unsteady gait, blurry vision, and weakness of limbs, and had to crawl to the hospital. There, he was given a diagnosis of neurosyphilis. He also developed left-sided hearing loss during that time.
Mr. C was treated with aqueous penicillin G benzathine, 4 million units IV for 2 weeks. No follow-up cerebrospinal fluid (CSF) examination was documented after antibiotic treatment. He developed auditory and visual hallucinations and paranoia a few months after completing penicillin treatment. During the following year, he had 3 inpatient psychiatric hospitalizations for psychosis, agitation, and depressed mood.
How would you treat a patient with a history of neurosyphilis who presents with psychosis years after diagnosis?
a) repeat antibiotic treatment and stop the antipsychotic
b) repeat antibiotic treatment and continue the antipsychotic
c) attempt to discontinue the antipsychotic
d) continue the antipsychotic
The authors’ observations
Mr. C’s psychotic symptoms seem to be temporally related to his diagnosis of neurosyphilis at age 48. He and his family members deny that Mr. C had any history of psychosis or depression before the neurosyphilis diagnosis. All inpatient psychiatric hospitalizations were within 1 year of the neurosyphilis diagnosis.
Mr. C has been on a low dosage of chlorpromazine, which has significant antihistaminic action. Chlorpromazine also is known to cause QTc prolongation, especially in patients with heart disease.
TREATMENT Medication change
A serum rapid plasma reagin test is non-reactive, but Treponema pallidum particle agglutination is positive. MRI shows moderate atrophy suggestive of diffuse small-vessel disease.
Mr. C’s psychotic symptoms are considered to be sequelae of neurosyphilis, based on (1) the presence of positive antibody tests, (2) residual neurologic deficits, (3) other suggestive sequelae (aortic regurgitation, sensorineural deafness), and (4) age-inappropriate gradual cognitive decline in the absence of other psychiatric history.
Because we are concerned about the prolonged QTc, chlorpromazine is discontinued. Haloperidol, 5 mg at bedtime, is started. The neurology team does not recommend antibiotic treatment because symptoms have been stable for years. Mr. C refuses a lumbar puncture.
Mr. C returns to the outpatient clinic monthly. He is psychiatrically stable without any worsening of psychosis. Cognitive impairment remains stable over the next 6 months. Haloperidol is tapered to 2 mg at bedtime 6 months after initial evaluation. Mr. C remains psychiatrically stable on subsequent follow-up visits.
The authors’ observations
Mr. C’s psychotic symptoms persisted after standard antibiotic treatment of neurosyphilis and lapsed when he stopped taking antipsychotic medication 10 years after the initial treatment of neurosyphilis. He carried a diagnosis of schizophrenia for many years, even though his psychotic symptoms were atypical for the presentation of schizophrenia.
It is important to understand the natural course of syphilis, its implication on psychiatric symptom production, and long-term psychiatric prognosis.
Syphilis is a sexually transmitted infectious disease caused by T pallidum, a spirochete, that has varied clinical presentations. Osler called syphilis the “great imitator” for its array of system involvement, ranging from asymptomatic infection and afferent pupillary defect to depression, psychosis, and dementia. With wide use of penicillin, the rate of neurosyphilis declined steadily during the mid 1990s. By 1997, the overall rate reached its lowest point in the United States; in 1999 the Centers for Disease Control and Prevention released a national plan to eliminate syphilis.1 By 2004, however, prevalence had increased to 4.7/100,000. It is thought that this increase is mainly associated with substance use (especially crack cocaine) and HIV co-infection. Most cases were distributed in economically depressed geographical areas.
Psychiatric patients are at higher risk of acquiring the infection because of substance use, lack of education on safer sex practices, and impulsive behavior.
Stages of syphilis
Syphilis does not follow a step-wise progression. One-third of cases progress to the tertiary stage, even many years after initial infection, without adequate treatment.2
Almost 10% syphilis cases present with neurologic symptoms,3 and neurologic involvement can occur at any stage of disease progression. The most common symptoms of syphilis are presented in Table 1.
A range of psychiatric symptoms have been reported among patients with syphilis, including anhedonia, suicidality, mania, grandiosity, persecutory delusions, auditory and visual hallucinations, paranoia, and cognitive impairment. The incidence of psychiatric symptoms is not clearly described in literature.
Diagnosis and treatment
Neurosyphilis, at any disease stage, should be suspected if a patient:
- exhibits suggestive symptoms
- does not respond to antibiotic treatment
- has late latent syphilis
- is immunocompromised.
Lumbar puncture and examination of CSF is the most useful diagnostic test. Dark field microscopy to reveal T pallidum is definitive, but only is applicable during the primary stage. The role of dark field microscopy of the CSF sample to diagnose neurologic involvement has not been established. Tests and treatment protocol are described in Table 2.2-5
Treatment of psychiatric symptoms of neurosyphilis
There are inconsistent and limited data about the prevalence of psychiatric symptoms in neurosyphilis. A retrospective study6 of 161 patients with neurosyphilis in South Africa reported that 50.9% exhibited a complex spectrum of symptoms that included delirium and dementia. Of treated patients, 17% continued to have residual symptoms during follow-up.
A review of the literature did not reveal any widely accepted guideline for screening for neurosyphilis in general psychiatry practice or a treatment protocol for psychiatric symptoms. This lack of guidance could be attributed to the rarity of the disease, cost-benefit analyses, and low specificity of antibody tests. In the literature, syphilis screening is recommended as a routine protocol when evaluating and treating dementia.7
In most studies, a diagnosis of neurosyphilis was confirmed by CSF examination; however, many of these studies did not report a specific follow-up CSF examination protocol. Most of these patients were treated with an antipsychotic with partial improvement in symptoms, even after standard antibiotic protocol.8
First- and second-generation antipsychotics and mood stabilizers have been shown to be useful in the acute treatment of psychosis and agitation.8 In few instances, the psychotropic medication was continued beyond several months and the patient was placed in a long-term care facility. Psychiatric symptoms persisted for many years with or without residual neurosyphilis symptoms, possibly because of permanent neuronal loss.
Clinical considerations
It often is difficult to distinguish a preexisting psychiatric disorder made worse by neurosyphilis from a secondary psychiatric disorder caused by neurosyphilis. The 2 might coexist, or psychiatric symptoms could be wrongly attributed to schizophrenia because of a lack of careful clinical evaluation.
Often, the follow-up diagnostic protocol for neurosyphilis is not followed; as a result, the need for re-treatment remains unclear. Rarity of the disease makes it difficult to perform a prospective, randomized study to determine the duration and effect of long-term psychiatric treatment.
Close follow-up and consideration of the risk vs benefit of psychotropic medication is key. Because there are no proven guidelines for the length of treatment with antipsychotics, it is prudent to minimize their use until psychiatrically indicated. Side effects, such as (in Mr. C’s case) changes in the QTc interval, should warrant consideration of discontinuing psychotropic medication. Interdisciplinary collaboration with neurology and infectious disease will improve the overall outcome of a complex clinical presentation.
CASE Hallucinations, impaired memory
Mr. C is a 61-year-old African American man who visits the outpatient clinic for management of antipsychotic therapy for psychosis and depression. His most recent inpatient psychiatric hospitalization for auditory and visual hallucinations, paranoia, and agitation was more than 10 years ago. He has been taking chlorpromazine, 100 mg/d, for 11 years. Mr. C reports that he has had no psychotic symptoms in the past 3 years; he continues taking chlorpromazine, he says, because it helps him sleep.
How would you proceed with Mr. C’s care?
a) continue chlorpromazine because he has been symptom free
b) consider tapering and discontinuing chlorpromazine
c) obtain a more detailed history from Mr. C and perform additional tests
HISTORY Validation of diagnosis
Mr. C reports that, at age 48, he started hearing babies crying and started seeing dead infants crawling out of the incinerator at the hospital where he worked. He denies any psychiatric symptoms before that time. He stopped working 10 years ago because of his psychiatric symptoms and decline in cognition.
Subsequently, Mr. C had 3 inpatient psychiatric hospitalizations for auditory hallucinations; chlorpromazine, 100 mg/d, was prescribed for psychosis. Later efforts to discontinue chlorpromazine resulted in relapse of psychotic symptoms. Mr. C has no family history of psychiatric illness.
Mr. C’s medical history is significant for aortic regurgitation, congestive cardiac failure, hypertension, and left-sided sensorineural hearing loss. He has a history of cocaine abuse from age 21 to 45, but denies using any other substances, including alcohol and nicotine.
Urine toxicology and routine blood tests are within normal limits. The QTc is slightly prolonged over the past 2 years, recording 512, 520, and 505 milliseconds on serial electrocardiograms.
Mr. C is able to perform simple abstractions. He has a goal-directed thought process, devoid of any preoccupation, paranoia, and perceptual abnormalities. Cognitive screening reveals significant impairment of memory, registration, calculation, attention, and visuospatial skills.
Careful review of Mr. C’s history and medical records reveals a diagnosis of syphilis at age 48 after unprotected sexual intercourse. He recalls that he had a solitary genital lesion, which resolved over a few weeks. He then developed a slightly itchy, non-tender macular rash over his upper back, which he did not report to a physician. After a few months, he developed unsteady gait, blurry vision, and weakness of limbs, and had to crawl to the hospital. There, he was given a diagnosis of neurosyphilis. He also developed left-sided hearing loss during that time.
Mr. C was treated with aqueous penicillin G benzathine, 4 million units IV for 2 weeks. No follow-up cerebrospinal fluid (CSF) examination was documented after antibiotic treatment. He developed auditory and visual hallucinations and paranoia a few months after completing penicillin treatment. During the following year, he had 3 inpatient psychiatric hospitalizations for psychosis, agitation, and depressed mood.
How would you treat a patient with a history of neurosyphilis who presents with psychosis years after diagnosis?
a) repeat antibiotic treatment and stop the antipsychotic
b) repeat antibiotic treatment and continue the antipsychotic
c) attempt to discontinue the antipsychotic
d) continue the antipsychotic
The authors’ observations
Mr. C’s psychotic symptoms seem to be temporally related to his diagnosis of neurosyphilis at age 48. He and his family members deny that Mr. C had any history of psychosis or depression before the neurosyphilis diagnosis. All inpatient psychiatric hospitalizations were within 1 year of the neurosyphilis diagnosis.
Mr. C has been on a low dosage of chlorpromazine, which has significant antihistaminic action. Chlorpromazine also is known to cause QTc prolongation, especially in patients with heart disease.
TREATMENT Medication change
A serum rapid plasma reagin test is non-reactive, but Treponema pallidum particle agglutination is positive. MRI shows moderate atrophy suggestive of diffuse small-vessel disease.
Mr. C’s psychotic symptoms are considered to be sequelae of neurosyphilis, based on (1) the presence of positive antibody tests, (2) residual neurologic deficits, (3) other suggestive sequelae (aortic regurgitation, sensorineural deafness), and (4) age-inappropriate gradual cognitive decline in the absence of other psychiatric history.
Because we are concerned about the prolonged QTc, chlorpromazine is discontinued. Haloperidol, 5 mg at bedtime, is started. The neurology team does not recommend antibiotic treatment because symptoms have been stable for years. Mr. C refuses a lumbar puncture.
Mr. C returns to the outpatient clinic monthly. He is psychiatrically stable without any worsening of psychosis. Cognitive impairment remains stable over the next 6 months. Haloperidol is tapered to 2 mg at bedtime 6 months after initial evaluation. Mr. C remains psychiatrically stable on subsequent follow-up visits.
The authors’ observations
Mr. C’s psychotic symptoms persisted after standard antibiotic treatment of neurosyphilis and lapsed when he stopped taking antipsychotic medication 10 years after the initial treatment of neurosyphilis. He carried a diagnosis of schizophrenia for many years, even though his psychotic symptoms were atypical for the presentation of schizophrenia.
It is important to understand the natural course of syphilis, its implication on psychiatric symptom production, and long-term psychiatric prognosis.
Syphilis is a sexually transmitted infectious disease caused by T pallidum, a spirochete, that has varied clinical presentations. Osler called syphilis the “great imitator” for its array of system involvement, ranging from asymptomatic infection and afferent pupillary defect to depression, psychosis, and dementia. With wide use of penicillin, the rate of neurosyphilis declined steadily during the mid 1990s. By 1997, the overall rate reached its lowest point in the United States; in 1999 the Centers for Disease Control and Prevention released a national plan to eliminate syphilis.1 By 2004, however, prevalence had increased to 4.7/100,000. It is thought that this increase is mainly associated with substance use (especially crack cocaine) and HIV co-infection. Most cases were distributed in economically depressed geographical areas.
Psychiatric patients are at higher risk of acquiring the infection because of substance use, lack of education on safer sex practices, and impulsive behavior.
Stages of syphilis
Syphilis does not follow a step-wise progression. One-third of cases progress to the tertiary stage, even many years after initial infection, without adequate treatment.2
Almost 10% syphilis cases present with neurologic symptoms,3 and neurologic involvement can occur at any stage of disease progression. The most common symptoms of syphilis are presented in Table 1.
A range of psychiatric symptoms have been reported among patients with syphilis, including anhedonia, suicidality, mania, grandiosity, persecutory delusions, auditory and visual hallucinations, paranoia, and cognitive impairment. The incidence of psychiatric symptoms is not clearly described in literature.
Diagnosis and treatment
Neurosyphilis, at any disease stage, should be suspected if a patient:
- exhibits suggestive symptoms
- does not respond to antibiotic treatment
- has late latent syphilis
- is immunocompromised.
Lumbar puncture and examination of CSF is the most useful diagnostic test. Dark field microscopy to reveal T pallidum is definitive, but only is applicable during the primary stage. The role of dark field microscopy of the CSF sample to diagnose neurologic involvement has not been established. Tests and treatment protocol are described in Table 2.2-5
Treatment of psychiatric symptoms of neurosyphilis
There are inconsistent and limited data about the prevalence of psychiatric symptoms in neurosyphilis. A retrospective study6 of 161 patients with neurosyphilis in South Africa reported that 50.9% exhibited a complex spectrum of symptoms that included delirium and dementia. Of treated patients, 17% continued to have residual symptoms during follow-up.
A review of the literature did not reveal any widely accepted guideline for screening for neurosyphilis in general psychiatry practice or a treatment protocol for psychiatric symptoms. This lack of guidance could be attributed to the rarity of the disease, cost-benefit analyses, and low specificity of antibody tests. In the literature, syphilis screening is recommended as a routine protocol when evaluating and treating dementia.7
In most studies, a diagnosis of neurosyphilis was confirmed by CSF examination; however, many of these studies did not report a specific follow-up CSF examination protocol. Most of these patients were treated with an antipsychotic with partial improvement in symptoms, even after standard antibiotic protocol.8
First- and second-generation antipsychotics and mood stabilizers have been shown to be useful in the acute treatment of psychosis and agitation.8 In few instances, the psychotropic medication was continued beyond several months and the patient was placed in a long-term care facility. Psychiatric symptoms persisted for many years with or without residual neurosyphilis symptoms, possibly because of permanent neuronal loss.
Clinical considerations
It often is difficult to distinguish a preexisting psychiatric disorder made worse by neurosyphilis from a secondary psychiatric disorder caused by neurosyphilis. The 2 might coexist, or psychiatric symptoms could be wrongly attributed to schizophrenia because of a lack of careful clinical evaluation.
Often, the follow-up diagnostic protocol for neurosyphilis is not followed; as a result, the need for re-treatment remains unclear. Rarity of the disease makes it difficult to perform a prospective, randomized study to determine the duration and effect of long-term psychiatric treatment.
Close follow-up and consideration of the risk vs benefit of psychotropic medication is key. Because there are no proven guidelines for the length of treatment with antipsychotics, it is prudent to minimize their use until psychiatrically indicated. Side effects, such as (in Mr. C’s case) changes in the QTc interval, should warrant consideration of discontinuing psychotropic medication. Interdisciplinary collaboration with neurology and infectious disease will improve the overall outcome of a complex clinical presentation.
1. Centers for Disease Control and Prevention. National plan to eliminate syphilis from the United States. http://www.cdc.gov/stopsyphilis/plan.htm. Updated December 7, 2007. Accessed July 7, 2016.
2. Friedrich F, Aigner M, Fearns N, et al. Psychosis in neurosyphilis—clinical aspects and implications. Psychopathology. 2014;47(1):3-9.
3. Brown DL, Frank JE. Diagnosis and management of syphilis. Am Fam Physician. 2003;68(2):283-290.
4. Romanowski B, Sutherland R, Fick GH, et al. Serologic response to treatment of infectious syphilis. Ann Intern Med. 1991;114(12):1005-1009.
5. Centers for Disease Control and Prevention. 2015 Sexually transmitted diseases treatment guidelines. Syphilis. http://www.cdc.gov/std/tg2015/syphilis.htm. Updated June 4, 2015. Accessed July 13, 2016.
6. Timmermans M, Carr J. Neurosyphilis in the modern era. J Neurol Neurosurg Psychiatry. 2004;75(12):1727-1730.
7. Scott KR, Barrett AM. Dementia syndrome: evaluation and treatment. Expert Rev Neurother. 2007;7(4):407-422.
8. Sanchez FM, Zisselman MH. Treatment of psychiatric symptoms associated with neurosyphilis. Psychosomatics. 2007;48(5):440-445.
1. Centers for Disease Control and Prevention. National plan to eliminate syphilis from the United States. http://www.cdc.gov/stopsyphilis/plan.htm. Updated December 7, 2007. Accessed July 7, 2016.
2. Friedrich F, Aigner M, Fearns N, et al. Psychosis in neurosyphilis—clinical aspects and implications. Psychopathology. 2014;47(1):3-9.
3. Brown DL, Frank JE. Diagnosis and management of syphilis. Am Fam Physician. 2003;68(2):283-290.
4. Romanowski B, Sutherland R, Fick GH, et al. Serologic response to treatment of infectious syphilis. Ann Intern Med. 1991;114(12):1005-1009.
5. Centers for Disease Control and Prevention. 2015 Sexually transmitted diseases treatment guidelines. Syphilis. http://www.cdc.gov/std/tg2015/syphilis.htm. Updated June 4, 2015. Accessed July 13, 2016.
6. Timmermans M, Carr J. Neurosyphilis in the modern era. J Neurol Neurosurg Psychiatry. 2004;75(12):1727-1730.
7. Scott KR, Barrett AM. Dementia syndrome: evaluation and treatment. Expert Rev Neurother. 2007;7(4):407-422.
8. Sanchez FM, Zisselman MH. Treatment of psychiatric symptoms associated with neurosyphilis. Psychosomatics. 2007;48(5):440-445.
Rediscovering clozapine: Adverse effects develop—what should you do now?
Clozapine is a highly effective antipsychotic with superior efficacy in treatment-resistant schizophrenia, but its side effect profile is daunting (Figure 1).1 Adverse reactions lead to approximately 17% of patients who take clozapine eventually discontinuing the medication.1 As we noted in Part 1 of this 3-part series,2 clozapine remains the most efficacious, but most tedious, antipsychotic available to psychiatrists because of its monitoring requirements and potential side effects.
A powerful rationale for prescribing clozapine, despite its drawbacks, is its association with a reduced risk of all-cause mortality.3,4 People with serious mental illness, including schizophrenia, have a median 10-year shorter life expectancy than the general population.5
A recent cohort study6 examined electronic health records to test whether intensive monitoring or lower suicide risk might account for the reduced mortality with clozapine. The authors found that the reduced mortality rate was not directly related to clozapine’s clinical monitoring or other confounding factors. They did find an association between clozapine use and reduced risk of death from both natural and unnatural causes.
This second article in our series examines clozapine’s adverse effects from a systems perspective. Severe neutropenia, myocarditis, sedation, weight gain, orthostatic hypotension, and sialorrhea appear to be the most studied adverse effects, but myriad others can occur.7 We offer guidance to help the astute clinician continue this effective antipsychotic by monitoring carefully, treating side effects early, and managing potential drug interactions (Table 1).8
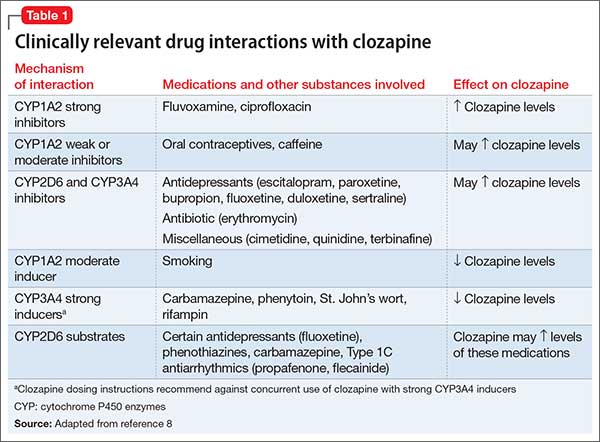
Hematologic eventsSevere neutropenia, defined as absolute neutrophil count (ANC) <500/µL, is a well-known adverse effect of clozapine that requires specific clinical monitoring, a requirement that was updated by the FDA in 2015.2 The incidence of severe neutropenia peaks in the first 2 months of clozapine therapy and tapers after 6 months, but some risk always remains.
Older efficacy studies in the United States gauged the 1-year cumulative incidence of severe clozapine-induced neutropenia to be 2%.9 A 1998 study of the effects of using a clozapine registry reported a lower incidence—0.38%—than the 2% noted above.10 Early recognition and recommended interventions can improve clinical outcomes.2
Drug interactions and neutropenia. A retrospective study of mental health inpatients taking clozapine concurrently with oseltamivir during an influenza outbreak found a statistically significant—but not clinically significant—change in ANC values.11 The authors noted that viral infection might lead to blood dyscrasia early in illness, and that oseltamivir has been associated with a small incidence of blood dyscrasia.11-13 This information might be useful when treating influenza in patients taking clozapine, although no specific change in management is recommended.
Similarly, concomitant treatment with clozapine and lithium can affect both white blood cell and ANC values.14,15 Lithium-treated patients often demonstrate increased circulating neutrophils via enhancement of granulocyte-colony stimulating factor.16 Case studies describe how initiating lithium treatment enabled some patients to continue clozapine after developing neutropenia.14,17 Leukocytosis can affect blood monitoring, possibly masking other blood dyscrasias, when lithium is used concurrently with clozapine.
Eosinophilia (blood eosinophil count >700/µL) occurs in approximately 1% of clozapine users, usually in the first 4 weeks of treatment.18 It can be benign and transient or a harbinger of a more rare adverse reaction such as myocarditis, pancreatitis, hepatitis, colitis, or nephritis.19 If a patient taking clozapine develops eosinophilia, clozapine’s package insert recommends that you:
- evaluate promptly for other systemic involvement (rash, other evidence of allergic reaction, myocarditis, other organ-specific disease)
- stop clozapine immediately if any of these are found.
If other causes of eosinophilia are identified (asthma, allergies, collagen vascular disease, parasitic infection, neoplasm), treat these and continue clozapine.
The manufacturer also mentions the occurrence of clozapine-related eosinophilia without organ involvement that can resolve without intervention, with careful monitoring over several weeks.8 In this scenario, there is flexibility to judge whether clozapine should be stopped or re-challenged, or if close monitoring is adequate. Consulting with an internal medicine or hematology specialist might be helpful.
Cardiovascular side effectsMost common events. Three of the 10 most common clozapine side effects are cardiac: tachycardia, hypotension, and hypertension (Figure 1).1 Orthostatic hypotension, bradycardia, and syncope also can occur, especially with rapid clozapine titration. Baseline electrocardiogram (ECG) can help differentiate whether abnormalities are clozapine-induced or related to a preexisting condition.
Reducing the dosage of clozapine or slowing titration could reverse cardiac side effects.8 If dosage reduction is not an option or is ineffective, first consider treating the side effect rather than discontinuing clozapine.20
Sinus tachycardia is one of the most common side effects of clozapine. First, rule out serious conditions—myocarditis, cardiomyopathy, neuroleptic malignant syndrome (NMS)—then consider waiting and monitoring for the first few months of clozapine treatment. If tachycardia continues, consider dosage reduction. Slower titration, or treatment with a cardio-selective beta blocker such as atenolol.21,22 Note that a recent Cochrane Review concluded that there is not enough randomized evidence to support any particular treatment for clozapine-induced tachycardia; the prescriber must therefore make a case-by-case clinical judgment.22
Similarly, orthostatic hypotension can be managed with a reduced dosage of clozapine or slower titration. Increased fluid intake, compression stockings, and, if necessary, fludrocortisone also can be initiated.20
Rare, potentially fatal events. Myocarditis, pericarditis, and cardiomyopathy are among the rare but potentially fatal adverse effects of clozapine. A recent study reported the incidence of myocarditis with clozapine at a range of 0.015% to 1.3%; cardiomyopathy was even more rare.23 Pulmonary embolism and deep venous thrombosis also are very rare possibilities; keep them in mind, however, when patients taking clozapine report new cardiovascular symptoms.
Patients with clozapine-induced cardiovascular effects most commonly report shortness of breath (60%), palpitations (36%), cough (16%), fatigue (16%), and chest pain (8%).7,24
Clozapine’s “black-box” warning specifically recommends discontinuing clozapine and consulting cardiology when myocarditis or cardiomyopathy is suspected. In 50% of cases, myocarditis symptoms present in the first few weeks of clozapine treatment.23 The manufacturer states that myocarditis usually presents in the first 2 months, and cardiomyopathy after 8 weeks of treatment; however, either can present at any time.8Figure 2 provides a clinical reference for monitoring a clozapine patient for cardiomyopathy.24
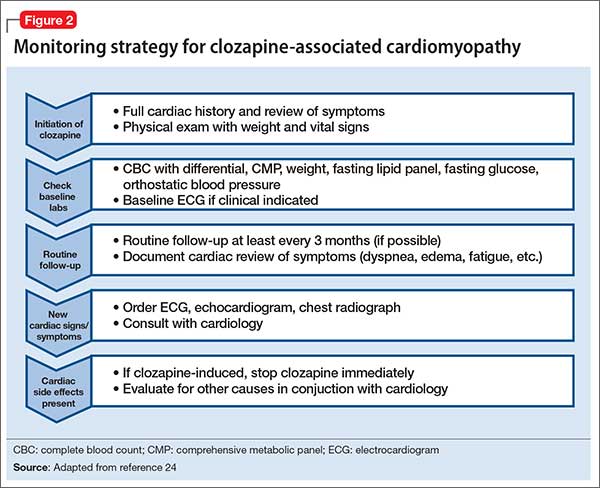
Laboratory findings that support a diagnosis of clozapine-related myocarditis include:
- elevated C-reactive protein
- elevated troponin I or T
- elevated creatine kinase-MB
- peripheral eosinophilia.8,25
ECG, echocardiography, and cardiac MRI can be helpful in diagnosis, in consultation with a cardiologist.
Neurologic side effectsSeizures are listed in the “black-box” warning for clozapine. Seizure incidence with clozapine is 5% per year, with higher incidence at dosages ≥600 mg/d.8 Because clozapine-induced seizures are dosage-dependent, slow titration can mitigate this risk. Tonic-clonic seizures are the most common type associated with clozapine.
The manufacturer recommends caution when using clozapine in patients with a known seizure disorder, alcohol use disorder, or other CNS pathology.8 Patients with a seizure disorder may be at increased risk of experiencing clozapine-induced seizures, but this is not an absolute contraindication.26 Smoking cessation increases clozapine blood levels by an average of 57.4%, further increasing seizure risk.26,27
Discontinuing clozapine is unnecessary when a patient experiences a seizure. Instead, you can:
- halve the dosage prescribed at the time of the seizure (or at least reduce to the last seizure-free dosage)
- consider any medications or medical problems that might have contributed to a lower seizure threshold
- consider prophylaxis with an antiepileptic medication (eg, valproic acid has efficacy for both myoclonic and tonic-clonic seizures).20,26
Sedation is the most common side effect of clozapine.1 Patients experiencing severe sedation should not drive or operate heavy machinery. To reduce sedation, consider instructing the patient to take all or most of the clozapine dosage at bedtime. A critical review of modafinil for sedation caused by antipsychotics in schizophrenia found only 1 open-label study that showed any positive effects; the authors concluded that further study is needed.28
Cognitive and motor slowing are possible neurologic side effects of clozapine. Caution patients about the risk of participating in activities that require cognitive or motor performance until the individual effects of clozapine are known.8
Tardive dyskinesia. Clozapine carries some risk of tardive dyskinesia, although that risk is lower than with other antipsychotics. Similarly, all antipsychotics including clozapine are associated with a risk of NMS. In the rare case of clozapine-induced NMS, stop clozapine immediately and initiate supportive therapy. Clozapine-induced NMS is not an absolute contraindication to re-challenging a patient with clozapine, however, if doing so is clinically appropriate.20
Cerebrovascular events. In older people with dementia, the use of antipsychotics—including clozapine—has been shown to increase the risk of cerebrovascular events. Because most antipsychotics are not FDA-approved for treating psychosis associated with dementia (only pimavanserin is FDA-approved for symptoms of psychosis in Parkinson’s disease), a risk-benefit analysis should be documented when prescribing any antipsychotic in this population. In practice, clozapine’s benefits may outweigh the mortality risks in specific situations.29,30
CASE Sialorrhea puts progress at risk
Ms. B, age 40, has a history of treatment-resistant schizophrenia and is starting clozapine because of residual psychosis during trials of other antipsychotics. She develops severe persistent drooling, mostly at night, during clozapine titration. Sugar-free candy, multiple bed pillows, and changing the dosing schedule do not significantly improve the sialorrhea.
As a result, Ms. B is embarrassed to continue her usual activities. She asks to stop clozapine, even though her psychotic symptoms have improved and she is functioning at her highest level in years.
Ms. B already is taking trihexyphenidyl, 5 mg, 3 times daily, to manage extrapyramidal symptoms related to haloperidol decanoate treatment. After discussing other medication options for sialorrhea, she agrees to a trial of glycopyrrolate, 1 mg, twice daily. She experiences significant improvement and continues taking clozapine.
Sialorrhea develops in 13% of patients taking clozapine.1 As in Ms. B’s case, this side effect can be embarrassing, can limit social or occupational functioning, and might lead patients to discontinue clozapine treatment despite efficacy. Nonpharmacotherapeutic options include covering the pillow with a towel, lowering the clozapine dosage or titrating slowly (or both), and using sugarless gum or candy to increase swallowing.
If the benefits of additional medications targeting side effects outweigh the risks, pharmacotherapeutic intervention may be appropriate. Options include the tricyclic antidepressant amitriptyline31; alpha-adrenergic agonists or antagonists (clonidine, terazosin); and anti-muscarinic medications (benztropine, atropine, trihexyphenidyl, glycopyrrolate) (Table 231). Scopolamine transdermal patch is another possible treatment strategy; however, the scopolamine patch was used for clozapine-induced sialorrhea in only a few case reports, and it is not considered a first-line treatment choice.30
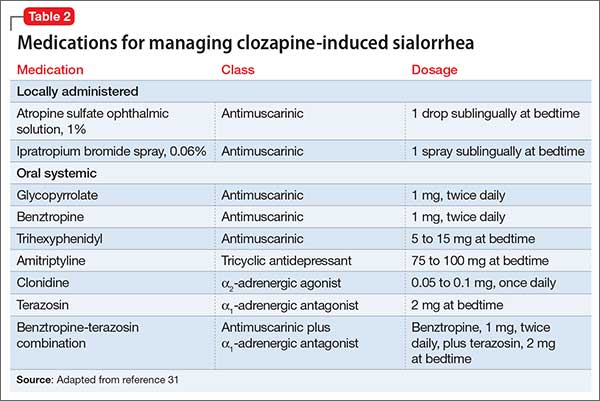
When prescribing, consider the possibility of combined side effects with clozapine and adjunct medications having antimuscarinic or alpha-adrenergic activity, or both. Even atropine ophthalmic drops, administered sublingually, are readily absorbed and cross the blood–brain barrier.31 Another antimuscarinic agent, glycopyrrolate, is less likely to cross the blood–brain barrier and therefore is less likely to cause cognitive side effects. Glycopyrrolate is 5 times more potent at blocking the muscarinic receptor than atropine.31,32 Ipratropium bromide, another nonselective muscarinic receptor antagonist, has less systemic absorption than atropine drops, with less anticholinergic side effects when administered sublingually.
Limited evidence supports the efficacy of alpha-adrenergic medications for managing clozapine-induced sialorrhea. Monitor blood pressure when prescribing terazosin or clonidine, which could potentiate clozapine’s hypotensive effects.
Endocrine side effectsAmong antipsychotics, clozapine is associated with the greatest weight gain—averaging nearly 10% of body weight.33,34 Similarly, the risk of new-onset diabetes mellitus is highest with clozapine in relation to other antipsychotics: 43% reported in a 10-year naturalistic study.35 The risk of hyperlipidemia also increases with clozapine treatment.36 These metabolic changes increase the risk of cardiovascular-related death, with a 10-year mortality rate from cardiovascular disease reported at 9% in clozapine-treated patients.35
Despite clozapine’s metabolic side effects, patients with schizophrenia who are treated with clozapine show a significant reduction in overall mortality compared with patients not treated with clozapine.6 Effective identification and management of metabolic side effects can prevent the need to discontinue clozapine.
Behavioral weight management and exercise are recommended as initial therapy.20 If, based on clinical judgment, these alone are insufficient, data support the use of pharmacotherapeutic interventions. Metformin demonstrates a positive effect on body weight, insulin resistance, and lipids, making it the first choice for adjunctive treatment of clozapine-induced metabolic side effects.37-39
Gastrointestinal side effectsClozapine’s anticholinergic activity can lead to serious gastrointestinal (GI) side effects, including constipation, intestinal obstruction, fecal impaction, and paralytic ileus.8 Ileus has produced more fatal adverse reactions with clozapine than has severe neutropenia.20,40 Co-administered anticholinergic medications could increase the risk of ileus. Obtaining a GI review of systems and monitoring bowel movements (in inpatient or residential facilities) can aid in early identification and limit morbidity and mortality from GI adverse events. A high-fiber diet, adequate hydration, bulk laxatives in patients who can reliably maintain hydration, and GI consultation (if needed) may help manage GI side effects.20
Constitutional side effectsFever can occur with clozapine, most often in the first month of treatment, but the incidence is quite variable (0.5% to 55%).20,41 Although benign fever is common, agranulocytosis with infection, NMS, and other systemic illness must be ruled out. The recommended workup when a patient develops fever while taking clozapine includes physical examination and relevant testing (urinalysis, measurement of ANC and serum creatine kinase, chest radiograph, ECG, and, possibly, blood cultures).41
If evidence supports a serious adverse reaction, stop clozapine immediately.20 If benign clozapine-related fever is suspected, acetaminophen or another antipyretic might provide symptomatic relief; discontinuing clozapine is then unnecessary.41
Pregnancy. When a patient with schizophrenia requires clozapine treatment during pregnancy, reliable clinical guidance is limited. The American College of Obstetricians and Gynecologists Practice Bulletin on the use of psychiatric medications during pregnancy and lactation can be a useful resource.42
Be aware that the FDA very recently made major changes to the format and content of pregnancy and lactation labeling, removing the letter categories that have been used for medications approved on or after June 30, 2001. The manufacturers of medications (such as clozapine) that were approved before June 30, 2001, have 3 years to comply with new requirements.43
The FDA had rated clozapine a pregnancy risk category B medication, meaning no evidence of risk in humans. In 2011, the FDA issued a general warning that antipsychotic use in pregnancy can cause extrapyramidal symptoms and discontinuation symptoms in newborns.44,45
A 2015 review of psychotropic medications and pregnancy noted that approximately 60% of women with schizophrenia became pregnant, with an increased incidence of unplanned pregnancy. A high risk of psychotic relapse (65%) during pregnancy and in the postpartum period may lead to insufficient prenatal care, drug use, and obstetric complications.45 Some data suggest low fetal birth weight and an increased rate of therapeutic abortions in women with schizophrenia.42,46
When treating a pregnant patient, weigh the benefits of clozapine against the risks of adverse events, and clearly document the analysis. Clozapine treatment is not recommended during breast-feeding because of the risk of side effects for newborns.8
We highly recommend keeping updated on the literature regarding pregnancy and lactation information with antipsychotics, including clozapine, because prescribing information will likely be updated in the near future to comply with recent FDA labeling changes.
Final installment: Using clozapine off-labelClozapine is FDA-approved for refractory schizophrenia and for reducing the risk of recurrent suicidal behavior in schizophrenia or schizoaffective disorder. In Part 3 of this series, we review off-label uses—such as managing bipolar disorder, borderline personality disorder, and aggressive behavior—that have varying degrees of scientific support.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Newman WJ, Newman BM. Rediscovering clozapine: after a turbulent history, current guidance on initiating and monitoring. Current Psychiatry. 2016;15(7):42-46,48-49.
3. Walker AM, Lanza LL, Arellano F, et al. Mortality in current and former users of clozapine. Epidemiology. 1997;8(6):671-677.
4. Tiihonen J, Lönnqvist J, Wahlbeck K, et al. 11-year follow-up of mortality in patients with schizophrenia: a population-based cohort study (FIN11 study). Lancet. 2009;374(9690):620-627.
5. Walker E, McGee RE, Druss BG. Mortality in mental disorders and global disease burden Implications: a systematic review and meta-analysis. JAMA Psychiatry. 2015;72(4):334-341.
6. Hayes RD, Downs J, Chang CK, et al. The effect of clozapine on premature mortality: an assessment of clinical monitoring and other potential confounders. Schizophr Bull. 2015;41(3):644-655.
7. De Fazio P, Gaetano R, Caroleo M, et al. Rare and very rare adverse effects of clozapine. Neuropsychiatr Dis Treat. 2015;11:1995-2003.
8. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 29, 2016.
9. Lieberman JA, Johns CA, Kane JM, et al. Clozapine-induced agranulocytosis: non-cross-reactivity with other psychotropic drugs. J Clin Psychiatry. 1988;49(7):271-277.
10. Honigfeld G, Arellano F, Sethi J, et al. Reducing clozapine-related morbidity and mortality: 5 years of experience with the Clozaril National Registry. J Clin Psychiatry. 1998;59(suppl 3):3-7.
11. Demler TL, Trigoboff E. Are clozapine patients at risk for blood dyscrasias with concomitant tamiflu use? Psychiatry (Edgmont). 2009;6(11):29-33.
12. Karalakulasingam R, Schacht RA, Lansing AM, et al. Influenza virus pneumonia after renal transplant. Postgrad Med. 1977;62(2):164-167.
13. Hoffman-La Roche Limited. Product monograph: Tamiflu. http://www.rochecanada.com/content/dam/roche_canada/en_CA/documents/Research/ClinicalTrialsForms/Products/ConsumerInformation/MonographsandPublicAdvisories/Tamiflu/Tamiflu_PM_E.pdf. Updated January 26, 2015. Accessed November 28, 2015.
14. Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
15. Palominao A, Kukoyi O, Xiong GL. Leukocytosis after lithium and clozapine combination therapy. Ann Clin Psychiatry. 2010;22(3):205-206.
16. Focosi D, Azzarà A, Kast RE, et al. Lithium and hematology: established and proposed uses. J Leukoc Biol. 2009;85(1):20-28.
17. Papetti F, Darcourt G, Giordana JY, et al. Treatment of clozapine-induced granulocytopenia with lithium (two observations) [in French]. Encephale. 2004;30(6):578-582.
18. Hummer M, Sperner-Unterweger B, Kemmler G, et al. Does eosinophilia predict clozapine induced neutropenia? Psychopharmacology (Berl). 1996;124(1-2):201-204.
19. Aneja J, Sharma N, Mahajan S, et al. Eosinophilia induced by clozapine: a report of two cases and review of the literature. J Family Med Prim Care. 2015;4(1):127-129.
20. Nielsen J, Correll CU, Manu P, et al. Termination of clozapine treatment due to medical reasons: when is it warranted and how can it be avoided? J Clin Psychiatry. 2013;74(6):603-613.
21. Stryjer R, Timinsky I, Reznik, I, et al. Beta-adrenergic antagonists for the treatment of clozapine-induced sinus tachycardia: a retrospective study. Clin Neuropharmacol. 2009;32(5):290-292.
22. Lally J, Docherty MJ, MacCabe JH. Pharmacological interventions for clozapine-induced sinus tachycardia. Cochrane Database Syst Rev. 2016;9(6):CD011566.
23. Kamphuis H, Arends J, Timmerman L, et al. Myocarditis and cardiomyopathy: underestimated complications resulting from clozapine therapy [in Dutch]. Tijdschr Psychiatr. 2010;52(4):223-233.
24. Alawami M, Wasywich C, Cicovic A, et al. A systematic review of clozapine induced cardiomyopathy. Int J Cardiol. 2014;176(2):315-320.
25. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
26. Williams AM, Park SH. Seizure associated with clozapine: incidence, etiology, and management. CNS Drugs. 2015;29(2):101-111.
27. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
28. Saavedra-Velez C, Yusim A, Anbarasan D, et al. Modafinil as an adjunctive treatment of sedation, negative symptoms, and cognition in schizophrenia: a critical review. J Clin Psychiatry. 2009;70(1):104-112.
29. Klein C, Gordon J, Pollak L, et al. Clozapine in Parkinson’s disease psychosis: 5-year follow-up review. Clin Neuropharmacol. 2003;26(1):8-11.
30. Lutz UC, Sirfy A, Wiatr G, et al. Clozapine serum concentrations in dopamimetic psychosis in Parkinson’s disease and related disorders. Eur J Clin Pharmacol. 2014;70(12):1471-1476.
31. Bird AM, Smith TL, Walton AE. Current treatment strategies for clozapine-induced sialorrhea. Ann Pharmacother. 2011;45(5):667-675.
32. Duggal HS. Glycopyrrolate for clozapine-induced sialorrhea. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(7):1546-1547.
33. Leadbetter R, Shutty M, Pavalonis D, et al. Clozapine-induced weight gain: prevalence and clinical relevance. Am J Psychiatry. 1992;149(1):68-72.
34. Lundblad W, Azzam PN, Gopalan, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;10(8):537-543.
35. Henderson DC, Nguyen DD, Copeland PM, et al. Clozapine, diabetes mellitus, hyperlipidemia, and cardiovascular risks and mortality: results of a 10-year naturalistic study. J Clin Psychiatry. 2005;66(9):1116-1121.
36. Stroup TS, Gerhard T, Crystal S, et al. Comparative effectiveness of clozapine and standard antipsychotic treatment in adults with schizophrenia. Am J Psychiatry. 2016;173(2):166-173.
37. Carrizo E, Fernández V, Connell L, et al. Extended release metformin for metabolic control assistance during prolonged clozapine administration: a 14 week, double-blind, parallel group, placebo-controlled study. Schizophr Res. 2009;113(1):19-26.
38. Chen CH, Huang MC, Kao CF, et al. Effects of adjunctive metformin on metabolic traits in nondiabetic clozapine-treated patients with schizophrenia and the effect of metformin discontinuation on body weight: a 24-week, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(5):e424-e430.
39. Mizuno Y, Suzuki T, Nakagawa A, et al. Pharmacological strategies to counteract antipsychotic-induced weight gain and metabolic adverse effects in schizophrenia: a systematic review and meta-analysis. Schizophr Bull. 2014;40(6):1385-1403.
40. Nielsen J, Meyer JM. Risk factors for ileus in patients with schizophrenia. Schizophr Bull. 2012;38(3):592-598.
41. Lowe CM, Grube RR, Scates AC. Characterization and clinical management of clozapine-induced fever. Ann Pharmacother. 2007;41(10):1700-1704.
42. ACOG Committee on Practice Bulletins–Obstetrics. ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces practice bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol. 2008;111(4):1001-1020.
43. U.S. Food and Drug Administration. Pregnancy and Lactation Labeling (Drugs) Final Rule. https://s3.amazonaws.com/public-inspection.federalregister.gov/2014-28241.pdf. Published December 4, 2014. Accessed July 6, 2016.
44. Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: a reference guide to fetal and neonatal risk. 9th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2011.
45. Larsen ER, Damkier P, Pedersen LH, et al; Danish Psychiatric Society; Danish Society of Obstetrics and Gynecology; Danish Paediatric Society; Danish Society of Clinical Pharmacology. Use of psychotropic drugs during pregnancy and breast-feeding. Acta Psychiatr Scand Suppl. 2015;(445):1-28.
46. McKenna K, Koren G, Tetelbaum M, et al. Pregnancy outcome of women using atypical antipsychotic drugs: a prospective comparative study. J Clin Psychiatry. 2005;66(4):444-449.
Clozapine is a highly effective antipsychotic with superior efficacy in treatment-resistant schizophrenia, but its side effect profile is daunting (Figure 1).1 Adverse reactions lead to approximately 17% of patients who take clozapine eventually discontinuing the medication.1 As we noted in Part 1 of this 3-part series,2 clozapine remains the most efficacious, but most tedious, antipsychotic available to psychiatrists because of its monitoring requirements and potential side effects.
A powerful rationale for prescribing clozapine, despite its drawbacks, is its association with a reduced risk of all-cause mortality.3,4 People with serious mental illness, including schizophrenia, have a median 10-year shorter life expectancy than the general population.5
A recent cohort study6 examined electronic health records to test whether intensive monitoring or lower suicide risk might account for the reduced mortality with clozapine. The authors found that the reduced mortality rate was not directly related to clozapine’s clinical monitoring or other confounding factors. They did find an association between clozapine use and reduced risk of death from both natural and unnatural causes.
This second article in our series examines clozapine’s adverse effects from a systems perspective. Severe neutropenia, myocarditis, sedation, weight gain, orthostatic hypotension, and sialorrhea appear to be the most studied adverse effects, but myriad others can occur.7 We offer guidance to help the astute clinician continue this effective antipsychotic by monitoring carefully, treating side effects early, and managing potential drug interactions (Table 1).8
Hematologic eventsSevere neutropenia, defined as absolute neutrophil count (ANC) <500/µL, is a well-known adverse effect of clozapine that requires specific clinical monitoring, a requirement that was updated by the FDA in 2015.2 The incidence of severe neutropenia peaks in the first 2 months of clozapine therapy and tapers after 6 months, but some risk always remains.
Older efficacy studies in the United States gauged the 1-year cumulative incidence of severe clozapine-induced neutropenia to be 2%.9 A 1998 study of the effects of using a clozapine registry reported a lower incidence—0.38%—than the 2% noted above.10 Early recognition and recommended interventions can improve clinical outcomes.2
Drug interactions and neutropenia. A retrospective study of mental health inpatients taking clozapine concurrently with oseltamivir during an influenza outbreak found a statistically significant—but not clinically significant—change in ANC values.11 The authors noted that viral infection might lead to blood dyscrasia early in illness, and that oseltamivir has been associated with a small incidence of blood dyscrasia.11-13 This information might be useful when treating influenza in patients taking clozapine, although no specific change in management is recommended.
Similarly, concomitant treatment with clozapine and lithium can affect both white blood cell and ANC values.14,15 Lithium-treated patients often demonstrate increased circulating neutrophils via enhancement of granulocyte-colony stimulating factor.16 Case studies describe how initiating lithium treatment enabled some patients to continue clozapine after developing neutropenia.14,17 Leukocytosis can affect blood monitoring, possibly masking other blood dyscrasias, when lithium is used concurrently with clozapine.
Eosinophilia (blood eosinophil count >700/µL) occurs in approximately 1% of clozapine users, usually in the first 4 weeks of treatment.18 It can be benign and transient or a harbinger of a more rare adverse reaction such as myocarditis, pancreatitis, hepatitis, colitis, or nephritis.19 If a patient taking clozapine develops eosinophilia, clozapine’s package insert recommends that you:
- evaluate promptly for other systemic involvement (rash, other evidence of allergic reaction, myocarditis, other organ-specific disease)
- stop clozapine immediately if any of these are found.
If other causes of eosinophilia are identified (asthma, allergies, collagen vascular disease, parasitic infection, neoplasm), treat these and continue clozapine.
The manufacturer also mentions the occurrence of clozapine-related eosinophilia without organ involvement that can resolve without intervention, with careful monitoring over several weeks.8 In this scenario, there is flexibility to judge whether clozapine should be stopped or re-challenged, or if close monitoring is adequate. Consulting with an internal medicine or hematology specialist might be helpful.
Cardiovascular side effectsMost common events. Three of the 10 most common clozapine side effects are cardiac: tachycardia, hypotension, and hypertension (Figure 1).1 Orthostatic hypotension, bradycardia, and syncope also can occur, especially with rapid clozapine titration. Baseline electrocardiogram (ECG) can help differentiate whether abnormalities are clozapine-induced or related to a preexisting condition.
Reducing the dosage of clozapine or slowing titration could reverse cardiac side effects.8 If dosage reduction is not an option or is ineffective, first consider treating the side effect rather than discontinuing clozapine.20
Sinus tachycardia is one of the most common side effects of clozapine. First, rule out serious conditions—myocarditis, cardiomyopathy, neuroleptic malignant syndrome (NMS)—then consider waiting and monitoring for the first few months of clozapine treatment. If tachycardia continues, consider dosage reduction. Slower titration, or treatment with a cardio-selective beta blocker such as atenolol.21,22 Note that a recent Cochrane Review concluded that there is not enough randomized evidence to support any particular treatment for clozapine-induced tachycardia; the prescriber must therefore make a case-by-case clinical judgment.22
Similarly, orthostatic hypotension can be managed with a reduced dosage of clozapine or slower titration. Increased fluid intake, compression stockings, and, if necessary, fludrocortisone also can be initiated.20
Rare, potentially fatal events. Myocarditis, pericarditis, and cardiomyopathy are among the rare but potentially fatal adverse effects of clozapine. A recent study reported the incidence of myocarditis with clozapine at a range of 0.015% to 1.3%; cardiomyopathy was even more rare.23 Pulmonary embolism and deep venous thrombosis also are very rare possibilities; keep them in mind, however, when patients taking clozapine report new cardiovascular symptoms.
Patients with clozapine-induced cardiovascular effects most commonly report shortness of breath (60%), palpitations (36%), cough (16%), fatigue (16%), and chest pain (8%).7,24
Clozapine’s “black-box” warning specifically recommends discontinuing clozapine and consulting cardiology when myocarditis or cardiomyopathy is suspected. In 50% of cases, myocarditis symptoms present in the first few weeks of clozapine treatment.23 The manufacturer states that myocarditis usually presents in the first 2 months, and cardiomyopathy after 8 weeks of treatment; however, either can present at any time.8Figure 2 provides a clinical reference for monitoring a clozapine patient for cardiomyopathy.24
Laboratory findings that support a diagnosis of clozapine-related myocarditis include:
- elevated C-reactive protein
- elevated troponin I or T
- elevated creatine kinase-MB
- peripheral eosinophilia.8,25
ECG, echocardiography, and cardiac MRI can be helpful in diagnosis, in consultation with a cardiologist.
Neurologic side effectsSeizures are listed in the “black-box” warning for clozapine. Seizure incidence with clozapine is 5% per year, with higher incidence at dosages ≥600 mg/d.8 Because clozapine-induced seizures are dosage-dependent, slow titration can mitigate this risk. Tonic-clonic seizures are the most common type associated with clozapine.
The manufacturer recommends caution when using clozapine in patients with a known seizure disorder, alcohol use disorder, or other CNS pathology.8 Patients with a seizure disorder may be at increased risk of experiencing clozapine-induced seizures, but this is not an absolute contraindication.26 Smoking cessation increases clozapine blood levels by an average of 57.4%, further increasing seizure risk.26,27
Discontinuing clozapine is unnecessary when a patient experiences a seizure. Instead, you can:
- halve the dosage prescribed at the time of the seizure (or at least reduce to the last seizure-free dosage)
- consider any medications or medical problems that might have contributed to a lower seizure threshold
- consider prophylaxis with an antiepileptic medication (eg, valproic acid has efficacy for both myoclonic and tonic-clonic seizures).20,26
Sedation is the most common side effect of clozapine.1 Patients experiencing severe sedation should not drive or operate heavy machinery. To reduce sedation, consider instructing the patient to take all or most of the clozapine dosage at bedtime. A critical review of modafinil for sedation caused by antipsychotics in schizophrenia found only 1 open-label study that showed any positive effects; the authors concluded that further study is needed.28
Cognitive and motor slowing are possible neurologic side effects of clozapine. Caution patients about the risk of participating in activities that require cognitive or motor performance until the individual effects of clozapine are known.8
Tardive dyskinesia. Clozapine carries some risk of tardive dyskinesia, although that risk is lower than with other antipsychotics. Similarly, all antipsychotics including clozapine are associated with a risk of NMS. In the rare case of clozapine-induced NMS, stop clozapine immediately and initiate supportive therapy. Clozapine-induced NMS is not an absolute contraindication to re-challenging a patient with clozapine, however, if doing so is clinically appropriate.20
Cerebrovascular events. In older people with dementia, the use of antipsychotics—including clozapine—has been shown to increase the risk of cerebrovascular events. Because most antipsychotics are not FDA-approved for treating psychosis associated with dementia (only pimavanserin is FDA-approved for symptoms of psychosis in Parkinson’s disease), a risk-benefit analysis should be documented when prescribing any antipsychotic in this population. In practice, clozapine’s benefits may outweigh the mortality risks in specific situations.29,30
CASE Sialorrhea puts progress at risk
Ms. B, age 40, has a history of treatment-resistant schizophrenia and is starting clozapine because of residual psychosis during trials of other antipsychotics. She develops severe persistent drooling, mostly at night, during clozapine titration. Sugar-free candy, multiple bed pillows, and changing the dosing schedule do not significantly improve the sialorrhea.
As a result, Ms. B is embarrassed to continue her usual activities. She asks to stop clozapine, even though her psychotic symptoms have improved and she is functioning at her highest level in years.
Ms. B already is taking trihexyphenidyl, 5 mg, 3 times daily, to manage extrapyramidal symptoms related to haloperidol decanoate treatment. After discussing other medication options for sialorrhea, she agrees to a trial of glycopyrrolate, 1 mg, twice daily. She experiences significant improvement and continues taking clozapine.
Sialorrhea develops in 13% of patients taking clozapine.1 As in Ms. B’s case, this side effect can be embarrassing, can limit social or occupational functioning, and might lead patients to discontinue clozapine treatment despite efficacy. Nonpharmacotherapeutic options include covering the pillow with a towel, lowering the clozapine dosage or titrating slowly (or both), and using sugarless gum or candy to increase swallowing.
If the benefits of additional medications targeting side effects outweigh the risks, pharmacotherapeutic intervention may be appropriate. Options include the tricyclic antidepressant amitriptyline31; alpha-adrenergic agonists or antagonists (clonidine, terazosin); and anti-muscarinic medications (benztropine, atropine, trihexyphenidyl, glycopyrrolate) (Table 231). Scopolamine transdermal patch is another possible treatment strategy; however, the scopolamine patch was used for clozapine-induced sialorrhea in only a few case reports, and it is not considered a first-line treatment choice.30
When prescribing, consider the possibility of combined side effects with clozapine and adjunct medications having antimuscarinic or alpha-adrenergic activity, or both. Even atropine ophthalmic drops, administered sublingually, are readily absorbed and cross the blood–brain barrier.31 Another antimuscarinic agent, glycopyrrolate, is less likely to cross the blood–brain barrier and therefore is less likely to cause cognitive side effects. Glycopyrrolate is 5 times more potent at blocking the muscarinic receptor than atropine.31,32 Ipratropium bromide, another nonselective muscarinic receptor antagonist, has less systemic absorption than atropine drops, with less anticholinergic side effects when administered sublingually.
Limited evidence supports the efficacy of alpha-adrenergic medications for managing clozapine-induced sialorrhea. Monitor blood pressure when prescribing terazosin or clonidine, which could potentiate clozapine’s hypotensive effects.
Endocrine side effectsAmong antipsychotics, clozapine is associated with the greatest weight gain—averaging nearly 10% of body weight.33,34 Similarly, the risk of new-onset diabetes mellitus is highest with clozapine in relation to other antipsychotics: 43% reported in a 10-year naturalistic study.35 The risk of hyperlipidemia also increases with clozapine treatment.36 These metabolic changes increase the risk of cardiovascular-related death, with a 10-year mortality rate from cardiovascular disease reported at 9% in clozapine-treated patients.35
Despite clozapine’s metabolic side effects, patients with schizophrenia who are treated with clozapine show a significant reduction in overall mortality compared with patients not treated with clozapine.6 Effective identification and management of metabolic side effects can prevent the need to discontinue clozapine.
Behavioral weight management and exercise are recommended as initial therapy.20 If, based on clinical judgment, these alone are insufficient, data support the use of pharmacotherapeutic interventions. Metformin demonstrates a positive effect on body weight, insulin resistance, and lipids, making it the first choice for adjunctive treatment of clozapine-induced metabolic side effects.37-39
Gastrointestinal side effectsClozapine’s anticholinergic activity can lead to serious gastrointestinal (GI) side effects, including constipation, intestinal obstruction, fecal impaction, and paralytic ileus.8 Ileus has produced more fatal adverse reactions with clozapine than has severe neutropenia.20,40 Co-administered anticholinergic medications could increase the risk of ileus. Obtaining a GI review of systems and monitoring bowel movements (in inpatient or residential facilities) can aid in early identification and limit morbidity and mortality from GI adverse events. A high-fiber diet, adequate hydration, bulk laxatives in patients who can reliably maintain hydration, and GI consultation (if needed) may help manage GI side effects.20
Constitutional side effectsFever can occur with clozapine, most often in the first month of treatment, but the incidence is quite variable (0.5% to 55%).20,41 Although benign fever is common, agranulocytosis with infection, NMS, and other systemic illness must be ruled out. The recommended workup when a patient develops fever while taking clozapine includes physical examination and relevant testing (urinalysis, measurement of ANC and serum creatine kinase, chest radiograph, ECG, and, possibly, blood cultures).41
If evidence supports a serious adverse reaction, stop clozapine immediately.20 If benign clozapine-related fever is suspected, acetaminophen or another antipyretic might provide symptomatic relief; discontinuing clozapine is then unnecessary.41
Pregnancy. When a patient with schizophrenia requires clozapine treatment during pregnancy, reliable clinical guidance is limited. The American College of Obstetricians and Gynecologists Practice Bulletin on the use of psychiatric medications during pregnancy and lactation can be a useful resource.42
Be aware that the FDA very recently made major changes to the format and content of pregnancy and lactation labeling, removing the letter categories that have been used for medications approved on or after June 30, 2001. The manufacturers of medications (such as clozapine) that were approved before June 30, 2001, have 3 years to comply with new requirements.43
The FDA had rated clozapine a pregnancy risk category B medication, meaning no evidence of risk in humans. In 2011, the FDA issued a general warning that antipsychotic use in pregnancy can cause extrapyramidal symptoms and discontinuation symptoms in newborns.44,45
A 2015 review of psychotropic medications and pregnancy noted that approximately 60% of women with schizophrenia became pregnant, with an increased incidence of unplanned pregnancy. A high risk of psychotic relapse (65%) during pregnancy and in the postpartum period may lead to insufficient prenatal care, drug use, and obstetric complications.45 Some data suggest low fetal birth weight and an increased rate of therapeutic abortions in women with schizophrenia.42,46
When treating a pregnant patient, weigh the benefits of clozapine against the risks of adverse events, and clearly document the analysis. Clozapine treatment is not recommended during breast-feeding because of the risk of side effects for newborns.8
We highly recommend keeping updated on the literature regarding pregnancy and lactation information with antipsychotics, including clozapine, because prescribing information will likely be updated in the near future to comply with recent FDA labeling changes.
Final installment: Using clozapine off-labelClozapine is FDA-approved for refractory schizophrenia and for reducing the risk of recurrent suicidal behavior in schizophrenia or schizoaffective disorder. In Part 3 of this series, we review off-label uses—such as managing bipolar disorder, borderline personality disorder, and aggressive behavior—that have varying degrees of scientific support.
Clozapine is a highly effective antipsychotic with superior efficacy in treatment-resistant schizophrenia, but its side effect profile is daunting (Figure 1).1 Adverse reactions lead to approximately 17% of patients who take clozapine eventually discontinuing the medication.1 As we noted in Part 1 of this 3-part series,2 clozapine remains the most efficacious, but most tedious, antipsychotic available to psychiatrists because of its monitoring requirements and potential side effects.
A powerful rationale for prescribing clozapine, despite its drawbacks, is its association with a reduced risk of all-cause mortality.3,4 People with serious mental illness, including schizophrenia, have a median 10-year shorter life expectancy than the general population.5
A recent cohort study6 examined electronic health records to test whether intensive monitoring or lower suicide risk might account for the reduced mortality with clozapine. The authors found that the reduced mortality rate was not directly related to clozapine’s clinical monitoring or other confounding factors. They did find an association between clozapine use and reduced risk of death from both natural and unnatural causes.
This second article in our series examines clozapine’s adverse effects from a systems perspective. Severe neutropenia, myocarditis, sedation, weight gain, orthostatic hypotension, and sialorrhea appear to be the most studied adverse effects, but myriad others can occur.7 We offer guidance to help the astute clinician continue this effective antipsychotic by monitoring carefully, treating side effects early, and managing potential drug interactions (Table 1).8
Hematologic eventsSevere neutropenia, defined as absolute neutrophil count (ANC) <500/µL, is a well-known adverse effect of clozapine that requires specific clinical monitoring, a requirement that was updated by the FDA in 2015.2 The incidence of severe neutropenia peaks in the first 2 months of clozapine therapy and tapers after 6 months, but some risk always remains.
Older efficacy studies in the United States gauged the 1-year cumulative incidence of severe clozapine-induced neutropenia to be 2%.9 A 1998 study of the effects of using a clozapine registry reported a lower incidence—0.38%—than the 2% noted above.10 Early recognition and recommended interventions can improve clinical outcomes.2
Drug interactions and neutropenia. A retrospective study of mental health inpatients taking clozapine concurrently with oseltamivir during an influenza outbreak found a statistically significant—but not clinically significant—change in ANC values.11 The authors noted that viral infection might lead to blood dyscrasia early in illness, and that oseltamivir has been associated with a small incidence of blood dyscrasia.11-13 This information might be useful when treating influenza in patients taking clozapine, although no specific change in management is recommended.
Similarly, concomitant treatment with clozapine and lithium can affect both white blood cell and ANC values.14,15 Lithium-treated patients often demonstrate increased circulating neutrophils via enhancement of granulocyte-colony stimulating factor.16 Case studies describe how initiating lithium treatment enabled some patients to continue clozapine after developing neutropenia.14,17 Leukocytosis can affect blood monitoring, possibly masking other blood dyscrasias, when lithium is used concurrently with clozapine.
Eosinophilia (blood eosinophil count >700/µL) occurs in approximately 1% of clozapine users, usually in the first 4 weeks of treatment.18 It can be benign and transient or a harbinger of a more rare adverse reaction such as myocarditis, pancreatitis, hepatitis, colitis, or nephritis.19 If a patient taking clozapine develops eosinophilia, clozapine’s package insert recommends that you:
- evaluate promptly for other systemic involvement (rash, other evidence of allergic reaction, myocarditis, other organ-specific disease)
- stop clozapine immediately if any of these are found.
If other causes of eosinophilia are identified (asthma, allergies, collagen vascular disease, parasitic infection, neoplasm), treat these and continue clozapine.
The manufacturer also mentions the occurrence of clozapine-related eosinophilia without organ involvement that can resolve without intervention, with careful monitoring over several weeks.8 In this scenario, there is flexibility to judge whether clozapine should be stopped or re-challenged, or if close monitoring is adequate. Consulting with an internal medicine or hematology specialist might be helpful.
Cardiovascular side effectsMost common events. Three of the 10 most common clozapine side effects are cardiac: tachycardia, hypotension, and hypertension (Figure 1).1 Orthostatic hypotension, bradycardia, and syncope also can occur, especially with rapid clozapine titration. Baseline electrocardiogram (ECG) can help differentiate whether abnormalities are clozapine-induced or related to a preexisting condition.
Reducing the dosage of clozapine or slowing titration could reverse cardiac side effects.8 If dosage reduction is not an option or is ineffective, first consider treating the side effect rather than discontinuing clozapine.20
Sinus tachycardia is one of the most common side effects of clozapine. First, rule out serious conditions—myocarditis, cardiomyopathy, neuroleptic malignant syndrome (NMS)—then consider waiting and monitoring for the first few months of clozapine treatment. If tachycardia continues, consider dosage reduction. Slower titration, or treatment with a cardio-selective beta blocker such as atenolol.21,22 Note that a recent Cochrane Review concluded that there is not enough randomized evidence to support any particular treatment for clozapine-induced tachycardia; the prescriber must therefore make a case-by-case clinical judgment.22
Similarly, orthostatic hypotension can be managed with a reduced dosage of clozapine or slower titration. Increased fluid intake, compression stockings, and, if necessary, fludrocortisone also can be initiated.20
Rare, potentially fatal events. Myocarditis, pericarditis, and cardiomyopathy are among the rare but potentially fatal adverse effects of clozapine. A recent study reported the incidence of myocarditis with clozapine at a range of 0.015% to 1.3%; cardiomyopathy was even more rare.23 Pulmonary embolism and deep venous thrombosis also are very rare possibilities; keep them in mind, however, when patients taking clozapine report new cardiovascular symptoms.
Patients with clozapine-induced cardiovascular effects most commonly report shortness of breath (60%), palpitations (36%), cough (16%), fatigue (16%), and chest pain (8%).7,24
Clozapine’s “black-box” warning specifically recommends discontinuing clozapine and consulting cardiology when myocarditis or cardiomyopathy is suspected. In 50% of cases, myocarditis symptoms present in the first few weeks of clozapine treatment.23 The manufacturer states that myocarditis usually presents in the first 2 months, and cardiomyopathy after 8 weeks of treatment; however, either can present at any time.8Figure 2 provides a clinical reference for monitoring a clozapine patient for cardiomyopathy.24
Laboratory findings that support a diagnosis of clozapine-related myocarditis include:
- elevated C-reactive protein
- elevated troponin I or T
- elevated creatine kinase-MB
- peripheral eosinophilia.8,25
ECG, echocardiography, and cardiac MRI can be helpful in diagnosis, in consultation with a cardiologist.
Neurologic side effectsSeizures are listed in the “black-box” warning for clozapine. Seizure incidence with clozapine is 5% per year, with higher incidence at dosages ≥600 mg/d.8 Because clozapine-induced seizures are dosage-dependent, slow titration can mitigate this risk. Tonic-clonic seizures are the most common type associated with clozapine.
The manufacturer recommends caution when using clozapine in patients with a known seizure disorder, alcohol use disorder, or other CNS pathology.8 Patients with a seizure disorder may be at increased risk of experiencing clozapine-induced seizures, but this is not an absolute contraindication.26 Smoking cessation increases clozapine blood levels by an average of 57.4%, further increasing seizure risk.26,27
Discontinuing clozapine is unnecessary when a patient experiences a seizure. Instead, you can:
- halve the dosage prescribed at the time of the seizure (or at least reduce to the last seizure-free dosage)
- consider any medications or medical problems that might have contributed to a lower seizure threshold
- consider prophylaxis with an antiepileptic medication (eg, valproic acid has efficacy for both myoclonic and tonic-clonic seizures).20,26
Sedation is the most common side effect of clozapine.1 Patients experiencing severe sedation should not drive or operate heavy machinery. To reduce sedation, consider instructing the patient to take all or most of the clozapine dosage at bedtime. A critical review of modafinil for sedation caused by antipsychotics in schizophrenia found only 1 open-label study that showed any positive effects; the authors concluded that further study is needed.28
Cognitive and motor slowing are possible neurologic side effects of clozapine. Caution patients about the risk of participating in activities that require cognitive or motor performance until the individual effects of clozapine are known.8
Tardive dyskinesia. Clozapine carries some risk of tardive dyskinesia, although that risk is lower than with other antipsychotics. Similarly, all antipsychotics including clozapine are associated with a risk of NMS. In the rare case of clozapine-induced NMS, stop clozapine immediately and initiate supportive therapy. Clozapine-induced NMS is not an absolute contraindication to re-challenging a patient with clozapine, however, if doing so is clinically appropriate.20
Cerebrovascular events. In older people with dementia, the use of antipsychotics—including clozapine—has been shown to increase the risk of cerebrovascular events. Because most antipsychotics are not FDA-approved for treating psychosis associated with dementia (only pimavanserin is FDA-approved for symptoms of psychosis in Parkinson’s disease), a risk-benefit analysis should be documented when prescribing any antipsychotic in this population. In practice, clozapine’s benefits may outweigh the mortality risks in specific situations.29,30
CASE Sialorrhea puts progress at risk
Ms. B, age 40, has a history of treatment-resistant schizophrenia and is starting clozapine because of residual psychosis during trials of other antipsychotics. She develops severe persistent drooling, mostly at night, during clozapine titration. Sugar-free candy, multiple bed pillows, and changing the dosing schedule do not significantly improve the sialorrhea.
As a result, Ms. B is embarrassed to continue her usual activities. She asks to stop clozapine, even though her psychotic symptoms have improved and she is functioning at her highest level in years.
Ms. B already is taking trihexyphenidyl, 5 mg, 3 times daily, to manage extrapyramidal symptoms related to haloperidol decanoate treatment. After discussing other medication options for sialorrhea, she agrees to a trial of glycopyrrolate, 1 mg, twice daily. She experiences significant improvement and continues taking clozapine.
Sialorrhea develops in 13% of patients taking clozapine.1 As in Ms. B’s case, this side effect can be embarrassing, can limit social or occupational functioning, and might lead patients to discontinue clozapine treatment despite efficacy. Nonpharmacotherapeutic options include covering the pillow with a towel, lowering the clozapine dosage or titrating slowly (or both), and using sugarless gum or candy to increase swallowing.
If the benefits of additional medications targeting side effects outweigh the risks, pharmacotherapeutic intervention may be appropriate. Options include the tricyclic antidepressant amitriptyline31; alpha-adrenergic agonists or antagonists (clonidine, terazosin); and anti-muscarinic medications (benztropine, atropine, trihexyphenidyl, glycopyrrolate) (Table 231). Scopolamine transdermal patch is another possible treatment strategy; however, the scopolamine patch was used for clozapine-induced sialorrhea in only a few case reports, and it is not considered a first-line treatment choice.30
When prescribing, consider the possibility of combined side effects with clozapine and adjunct medications having antimuscarinic or alpha-adrenergic activity, or both. Even atropine ophthalmic drops, administered sublingually, are readily absorbed and cross the blood–brain barrier.31 Another antimuscarinic agent, glycopyrrolate, is less likely to cross the blood–brain barrier and therefore is less likely to cause cognitive side effects. Glycopyrrolate is 5 times more potent at blocking the muscarinic receptor than atropine.31,32 Ipratropium bromide, another nonselective muscarinic receptor antagonist, has less systemic absorption than atropine drops, with less anticholinergic side effects when administered sublingually.
Limited evidence supports the efficacy of alpha-adrenergic medications for managing clozapine-induced sialorrhea. Monitor blood pressure when prescribing terazosin or clonidine, which could potentiate clozapine’s hypotensive effects.
Endocrine side effectsAmong antipsychotics, clozapine is associated with the greatest weight gain—averaging nearly 10% of body weight.33,34 Similarly, the risk of new-onset diabetes mellitus is highest with clozapine in relation to other antipsychotics: 43% reported in a 10-year naturalistic study.35 The risk of hyperlipidemia also increases with clozapine treatment.36 These metabolic changes increase the risk of cardiovascular-related death, with a 10-year mortality rate from cardiovascular disease reported at 9% in clozapine-treated patients.35
Despite clozapine’s metabolic side effects, patients with schizophrenia who are treated with clozapine show a significant reduction in overall mortality compared with patients not treated with clozapine.6 Effective identification and management of metabolic side effects can prevent the need to discontinue clozapine.
Behavioral weight management and exercise are recommended as initial therapy.20 If, based on clinical judgment, these alone are insufficient, data support the use of pharmacotherapeutic interventions. Metformin demonstrates a positive effect on body weight, insulin resistance, and lipids, making it the first choice for adjunctive treatment of clozapine-induced metabolic side effects.37-39
Gastrointestinal side effectsClozapine’s anticholinergic activity can lead to serious gastrointestinal (GI) side effects, including constipation, intestinal obstruction, fecal impaction, and paralytic ileus.8 Ileus has produced more fatal adverse reactions with clozapine than has severe neutropenia.20,40 Co-administered anticholinergic medications could increase the risk of ileus. Obtaining a GI review of systems and monitoring bowel movements (in inpatient or residential facilities) can aid in early identification and limit morbidity and mortality from GI adverse events. A high-fiber diet, adequate hydration, bulk laxatives in patients who can reliably maintain hydration, and GI consultation (if needed) may help manage GI side effects.20
Constitutional side effectsFever can occur with clozapine, most often in the first month of treatment, but the incidence is quite variable (0.5% to 55%).20,41 Although benign fever is common, agranulocytosis with infection, NMS, and other systemic illness must be ruled out. The recommended workup when a patient develops fever while taking clozapine includes physical examination and relevant testing (urinalysis, measurement of ANC and serum creatine kinase, chest radiograph, ECG, and, possibly, blood cultures).41
If evidence supports a serious adverse reaction, stop clozapine immediately.20 If benign clozapine-related fever is suspected, acetaminophen or another antipyretic might provide symptomatic relief; discontinuing clozapine is then unnecessary.41
Pregnancy. When a patient with schizophrenia requires clozapine treatment during pregnancy, reliable clinical guidance is limited. The American College of Obstetricians and Gynecologists Practice Bulletin on the use of psychiatric medications during pregnancy and lactation can be a useful resource.42
Be aware that the FDA very recently made major changes to the format and content of pregnancy and lactation labeling, removing the letter categories that have been used for medications approved on or after June 30, 2001. The manufacturers of medications (such as clozapine) that were approved before June 30, 2001, have 3 years to comply with new requirements.43
The FDA had rated clozapine a pregnancy risk category B medication, meaning no evidence of risk in humans. In 2011, the FDA issued a general warning that antipsychotic use in pregnancy can cause extrapyramidal symptoms and discontinuation symptoms in newborns.44,45
A 2015 review of psychotropic medications and pregnancy noted that approximately 60% of women with schizophrenia became pregnant, with an increased incidence of unplanned pregnancy. A high risk of psychotic relapse (65%) during pregnancy and in the postpartum period may lead to insufficient prenatal care, drug use, and obstetric complications.45 Some data suggest low fetal birth weight and an increased rate of therapeutic abortions in women with schizophrenia.42,46
When treating a pregnant patient, weigh the benefits of clozapine against the risks of adverse events, and clearly document the analysis. Clozapine treatment is not recommended during breast-feeding because of the risk of side effects for newborns.8
We highly recommend keeping updated on the literature regarding pregnancy and lactation information with antipsychotics, including clozapine, because prescribing information will likely be updated in the near future to comply with recent FDA labeling changes.
Final installment: Using clozapine off-labelClozapine is FDA-approved for refractory schizophrenia and for reducing the risk of recurrent suicidal behavior in schizophrenia or schizoaffective disorder. In Part 3 of this series, we review off-label uses—such as managing bipolar disorder, borderline personality disorder, and aggressive behavior—that have varying degrees of scientific support.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Newman WJ, Newman BM. Rediscovering clozapine: after a turbulent history, current guidance on initiating and monitoring. Current Psychiatry. 2016;15(7):42-46,48-49.
3. Walker AM, Lanza LL, Arellano F, et al. Mortality in current and former users of clozapine. Epidemiology. 1997;8(6):671-677.
4. Tiihonen J, Lönnqvist J, Wahlbeck K, et al. 11-year follow-up of mortality in patients with schizophrenia: a population-based cohort study (FIN11 study). Lancet. 2009;374(9690):620-627.
5. Walker E, McGee RE, Druss BG. Mortality in mental disorders and global disease burden Implications: a systematic review and meta-analysis. JAMA Psychiatry. 2015;72(4):334-341.
6. Hayes RD, Downs J, Chang CK, et al. The effect of clozapine on premature mortality: an assessment of clinical monitoring and other potential confounders. Schizophr Bull. 2015;41(3):644-655.
7. De Fazio P, Gaetano R, Caroleo M, et al. Rare and very rare adverse effects of clozapine. Neuropsychiatr Dis Treat. 2015;11:1995-2003.
8. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 29, 2016.
9. Lieberman JA, Johns CA, Kane JM, et al. Clozapine-induced agranulocytosis: non-cross-reactivity with other psychotropic drugs. J Clin Psychiatry. 1988;49(7):271-277.
10. Honigfeld G, Arellano F, Sethi J, et al. Reducing clozapine-related morbidity and mortality: 5 years of experience with the Clozaril National Registry. J Clin Psychiatry. 1998;59(suppl 3):3-7.
11. Demler TL, Trigoboff E. Are clozapine patients at risk for blood dyscrasias with concomitant tamiflu use? Psychiatry (Edgmont). 2009;6(11):29-33.
12. Karalakulasingam R, Schacht RA, Lansing AM, et al. Influenza virus pneumonia after renal transplant. Postgrad Med. 1977;62(2):164-167.
13. Hoffman-La Roche Limited. Product monograph: Tamiflu. http://www.rochecanada.com/content/dam/roche_canada/en_CA/documents/Research/ClinicalTrialsForms/Products/ConsumerInformation/MonographsandPublicAdvisories/Tamiflu/Tamiflu_PM_E.pdf. Updated January 26, 2015. Accessed November 28, 2015.
14. Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
15. Palominao A, Kukoyi O, Xiong GL. Leukocytosis after lithium and clozapine combination therapy. Ann Clin Psychiatry. 2010;22(3):205-206.
16. Focosi D, Azzarà A, Kast RE, et al. Lithium and hematology: established and proposed uses. J Leukoc Biol. 2009;85(1):20-28.
17. Papetti F, Darcourt G, Giordana JY, et al. Treatment of clozapine-induced granulocytopenia with lithium (two observations) [in French]. Encephale. 2004;30(6):578-582.
18. Hummer M, Sperner-Unterweger B, Kemmler G, et al. Does eosinophilia predict clozapine induced neutropenia? Psychopharmacology (Berl). 1996;124(1-2):201-204.
19. Aneja J, Sharma N, Mahajan S, et al. Eosinophilia induced by clozapine: a report of two cases and review of the literature. J Family Med Prim Care. 2015;4(1):127-129.
20. Nielsen J, Correll CU, Manu P, et al. Termination of clozapine treatment due to medical reasons: when is it warranted and how can it be avoided? J Clin Psychiatry. 2013;74(6):603-613.
21. Stryjer R, Timinsky I, Reznik, I, et al. Beta-adrenergic antagonists for the treatment of clozapine-induced sinus tachycardia: a retrospective study. Clin Neuropharmacol. 2009;32(5):290-292.
22. Lally J, Docherty MJ, MacCabe JH. Pharmacological interventions for clozapine-induced sinus tachycardia. Cochrane Database Syst Rev. 2016;9(6):CD011566.
23. Kamphuis H, Arends J, Timmerman L, et al. Myocarditis and cardiomyopathy: underestimated complications resulting from clozapine therapy [in Dutch]. Tijdschr Psychiatr. 2010;52(4):223-233.
24. Alawami M, Wasywich C, Cicovic A, et al. A systematic review of clozapine induced cardiomyopathy. Int J Cardiol. 2014;176(2):315-320.
25. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
26. Williams AM, Park SH. Seizure associated with clozapine: incidence, etiology, and management. CNS Drugs. 2015;29(2):101-111.
27. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
28. Saavedra-Velez C, Yusim A, Anbarasan D, et al. Modafinil as an adjunctive treatment of sedation, negative symptoms, and cognition in schizophrenia: a critical review. J Clin Psychiatry. 2009;70(1):104-112.
29. Klein C, Gordon J, Pollak L, et al. Clozapine in Parkinson’s disease psychosis: 5-year follow-up review. Clin Neuropharmacol. 2003;26(1):8-11.
30. Lutz UC, Sirfy A, Wiatr G, et al. Clozapine serum concentrations in dopamimetic psychosis in Parkinson’s disease and related disorders. Eur J Clin Pharmacol. 2014;70(12):1471-1476.
31. Bird AM, Smith TL, Walton AE. Current treatment strategies for clozapine-induced sialorrhea. Ann Pharmacother. 2011;45(5):667-675.
32. Duggal HS. Glycopyrrolate for clozapine-induced sialorrhea. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(7):1546-1547.
33. Leadbetter R, Shutty M, Pavalonis D, et al. Clozapine-induced weight gain: prevalence and clinical relevance. Am J Psychiatry. 1992;149(1):68-72.
34. Lundblad W, Azzam PN, Gopalan, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;10(8):537-543.
35. Henderson DC, Nguyen DD, Copeland PM, et al. Clozapine, diabetes mellitus, hyperlipidemia, and cardiovascular risks and mortality: results of a 10-year naturalistic study. J Clin Psychiatry. 2005;66(9):1116-1121.
36. Stroup TS, Gerhard T, Crystal S, et al. Comparative effectiveness of clozapine and standard antipsychotic treatment in adults with schizophrenia. Am J Psychiatry. 2016;173(2):166-173.
37. Carrizo E, Fernández V, Connell L, et al. Extended release metformin for metabolic control assistance during prolonged clozapine administration: a 14 week, double-blind, parallel group, placebo-controlled study. Schizophr Res. 2009;113(1):19-26.
38. Chen CH, Huang MC, Kao CF, et al. Effects of adjunctive metformin on metabolic traits in nondiabetic clozapine-treated patients with schizophrenia and the effect of metformin discontinuation on body weight: a 24-week, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(5):e424-e430.
39. Mizuno Y, Suzuki T, Nakagawa A, et al. Pharmacological strategies to counteract antipsychotic-induced weight gain and metabolic adverse effects in schizophrenia: a systematic review and meta-analysis. Schizophr Bull. 2014;40(6):1385-1403.
40. Nielsen J, Meyer JM. Risk factors for ileus in patients with schizophrenia. Schizophr Bull. 2012;38(3):592-598.
41. Lowe CM, Grube RR, Scates AC. Characterization and clinical management of clozapine-induced fever. Ann Pharmacother. 2007;41(10):1700-1704.
42. ACOG Committee on Practice Bulletins–Obstetrics. ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces practice bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol. 2008;111(4):1001-1020.
43. U.S. Food and Drug Administration. Pregnancy and Lactation Labeling (Drugs) Final Rule. https://s3.amazonaws.com/public-inspection.federalregister.gov/2014-28241.pdf. Published December 4, 2014. Accessed July 6, 2016.
44. Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: a reference guide to fetal and neonatal risk. 9th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2011.
45. Larsen ER, Damkier P, Pedersen LH, et al; Danish Psychiatric Society; Danish Society of Obstetrics and Gynecology; Danish Paediatric Society; Danish Society of Clinical Pharmacology. Use of psychotropic drugs during pregnancy and breast-feeding. Acta Psychiatr Scand Suppl. 2015;(445):1-28.
46. McKenna K, Koren G, Tetelbaum M, et al. Pregnancy outcome of women using atypical antipsychotic drugs: a prospective comparative study. J Clin Psychiatry. 2005;66(4):444-449.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Newman WJ, Newman BM. Rediscovering clozapine: after a turbulent history, current guidance on initiating and monitoring. Current Psychiatry. 2016;15(7):42-46,48-49.
3. Walker AM, Lanza LL, Arellano F, et al. Mortality in current and former users of clozapine. Epidemiology. 1997;8(6):671-677.
4. Tiihonen J, Lönnqvist J, Wahlbeck K, et al. 11-year follow-up of mortality in patients with schizophrenia: a population-based cohort study (FIN11 study). Lancet. 2009;374(9690):620-627.
5. Walker E, McGee RE, Druss BG. Mortality in mental disorders and global disease burden Implications: a systematic review and meta-analysis. JAMA Psychiatry. 2015;72(4):334-341.
6. Hayes RD, Downs J, Chang CK, et al. The effect of clozapine on premature mortality: an assessment of clinical monitoring and other potential confounders. Schizophr Bull. 2015;41(3):644-655.
7. De Fazio P, Gaetano R, Caroleo M, et al. Rare and very rare adverse effects of clozapine. Neuropsychiatr Dis Treat. 2015;11:1995-2003.
8. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed June 29, 2016.
9. Lieberman JA, Johns CA, Kane JM, et al. Clozapine-induced agranulocytosis: non-cross-reactivity with other psychotropic drugs. J Clin Psychiatry. 1988;49(7):271-277.
10. Honigfeld G, Arellano F, Sethi J, et al. Reducing clozapine-related morbidity and mortality: 5 years of experience with the Clozaril National Registry. J Clin Psychiatry. 1998;59(suppl 3):3-7.
11. Demler TL, Trigoboff E. Are clozapine patients at risk for blood dyscrasias with concomitant tamiflu use? Psychiatry (Edgmont). 2009;6(11):29-33.
12. Karalakulasingam R, Schacht RA, Lansing AM, et al. Influenza virus pneumonia after renal transplant. Postgrad Med. 1977;62(2):164-167.
13. Hoffman-La Roche Limited. Product monograph: Tamiflu. http://www.rochecanada.com/content/dam/roche_canada/en_CA/documents/Research/ClinicalTrialsForms/Products/ConsumerInformation/MonographsandPublicAdvisories/Tamiflu/Tamiflu_PM_E.pdf. Updated January 26, 2015. Accessed November 28, 2015.
14. Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
15. Palominao A, Kukoyi O, Xiong GL. Leukocytosis after lithium and clozapine combination therapy. Ann Clin Psychiatry. 2010;22(3):205-206.
16. Focosi D, Azzarà A, Kast RE, et al. Lithium and hematology: established and proposed uses. J Leukoc Biol. 2009;85(1):20-28.
17. Papetti F, Darcourt G, Giordana JY, et al. Treatment of clozapine-induced granulocytopenia with lithium (two observations) [in French]. Encephale. 2004;30(6):578-582.
18. Hummer M, Sperner-Unterweger B, Kemmler G, et al. Does eosinophilia predict clozapine induced neutropenia? Psychopharmacology (Berl). 1996;124(1-2):201-204.
19. Aneja J, Sharma N, Mahajan S, et al. Eosinophilia induced by clozapine: a report of two cases and review of the literature. J Family Med Prim Care. 2015;4(1):127-129.
20. Nielsen J, Correll CU, Manu P, et al. Termination of clozapine treatment due to medical reasons: when is it warranted and how can it be avoided? J Clin Psychiatry. 2013;74(6):603-613.
21. Stryjer R, Timinsky I, Reznik, I, et al. Beta-adrenergic antagonists for the treatment of clozapine-induced sinus tachycardia: a retrospective study. Clin Neuropharmacol. 2009;32(5):290-292.
22. Lally J, Docherty MJ, MacCabe JH. Pharmacological interventions for clozapine-induced sinus tachycardia. Cochrane Database Syst Rev. 2016;9(6):CD011566.
23. Kamphuis H, Arends J, Timmerman L, et al. Myocarditis and cardiomyopathy: underestimated complications resulting from clozapine therapy [in Dutch]. Tijdschr Psychiatr. 2010;52(4):223-233.
24. Alawami M, Wasywich C, Cicovic A, et al. A systematic review of clozapine induced cardiomyopathy. Int J Cardiol. 2014;176(2):315-320.
25. Ronaldson KJ, Fitzgerald PB, Taylor AJ, et al. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust N Z J Psychiatry. 2011;45(6):458-465.
26. Williams AM, Park SH. Seizure associated with clozapine: incidence, etiology, and management. CNS Drugs. 2015;29(2):101-111.
27. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
28. Saavedra-Velez C, Yusim A, Anbarasan D, et al. Modafinil as an adjunctive treatment of sedation, negative symptoms, and cognition in schizophrenia: a critical review. J Clin Psychiatry. 2009;70(1):104-112.
29. Klein C, Gordon J, Pollak L, et al. Clozapine in Parkinson’s disease psychosis: 5-year follow-up review. Clin Neuropharmacol. 2003;26(1):8-11.
30. Lutz UC, Sirfy A, Wiatr G, et al. Clozapine serum concentrations in dopamimetic psychosis in Parkinson’s disease and related disorders. Eur J Clin Pharmacol. 2014;70(12):1471-1476.
31. Bird AM, Smith TL, Walton AE. Current treatment strategies for clozapine-induced sialorrhea. Ann Pharmacother. 2011;45(5):667-675.
32. Duggal HS. Glycopyrrolate for clozapine-induced sialorrhea. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(7):1546-1547.
33. Leadbetter R, Shutty M, Pavalonis D, et al. Clozapine-induced weight gain: prevalence and clinical relevance. Am J Psychiatry. 1992;149(1):68-72.
34. Lundblad W, Azzam PN, Gopalan, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;10(8):537-543.
35. Henderson DC, Nguyen DD, Copeland PM, et al. Clozapine, diabetes mellitus, hyperlipidemia, and cardiovascular risks and mortality: results of a 10-year naturalistic study. J Clin Psychiatry. 2005;66(9):1116-1121.
36. Stroup TS, Gerhard T, Crystal S, et al. Comparative effectiveness of clozapine and standard antipsychotic treatment in adults with schizophrenia. Am J Psychiatry. 2016;173(2):166-173.
37. Carrizo E, Fernández V, Connell L, et al. Extended release metformin for metabolic control assistance during prolonged clozapine administration: a 14 week, double-blind, parallel group, placebo-controlled study. Schizophr Res. 2009;113(1):19-26.
38. Chen CH, Huang MC, Kao CF, et al. Effects of adjunctive metformin on metabolic traits in nondiabetic clozapine-treated patients with schizophrenia and the effect of metformin discontinuation on body weight: a 24-week, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(5):e424-e430.
39. Mizuno Y, Suzuki T, Nakagawa A, et al. Pharmacological strategies to counteract antipsychotic-induced weight gain and metabolic adverse effects in schizophrenia: a systematic review and meta-analysis. Schizophr Bull. 2014;40(6):1385-1403.
40. Nielsen J, Meyer JM. Risk factors for ileus in patients with schizophrenia. Schizophr Bull. 2012;38(3):592-598.
41. Lowe CM, Grube RR, Scates AC. Characterization and clinical management of clozapine-induced fever. Ann Pharmacother. 2007;41(10):1700-1704.
42. ACOG Committee on Practice Bulletins–Obstetrics. ACOG Practice Bulletin: Clinical management guidelines for obstetrician-gynecologists number 92, April 2008 (replaces practice bulletin number 87, November 2007). Use of psychiatric medications during pregnancy and lactation. Obstet Gynecol. 2008;111(4):1001-1020.
43. U.S. Food and Drug Administration. Pregnancy and Lactation Labeling (Drugs) Final Rule. https://s3.amazonaws.com/public-inspection.federalregister.gov/2014-28241.pdf. Published December 4, 2014. Accessed July 6, 2016.
44. Briggs GG, Freeman RK, Yaffe SJ. Drugs in pregnancy and lactation: a reference guide to fetal and neonatal risk. 9th ed. Baltimore, MD: Lippincott Williams & Wilkins; 2011.
45. Larsen ER, Damkier P, Pedersen LH, et al; Danish Psychiatric Society; Danish Society of Obstetrics and Gynecology; Danish Paediatric Society; Danish Society of Clinical Pharmacology. Use of psychotropic drugs during pregnancy and breast-feeding. Acta Psychiatr Scand Suppl. 2015;(445):1-28.
46. McKenna K, Koren G, Tetelbaum M, et al. Pregnancy outcome of women using atypical antipsychotic drugs: a prospective comparative study. J Clin Psychiatry. 2005;66(4):444-449.

What clinicians need to know about treating opioid use disorder
Communities across the United States have experienced a near-epidemic of opioid abuse. Deaths from opioid overdose have doubled since 2000 and increased 14% from 2013 to 2014.1 Treatment strategies for opioid use disorder (OUD) target individual well-being with the goal of preventing relapse. Most treatment approaches help patients gain self-confidence and have been described by patients as “giving them their life back.”
Broadly, the major phases of treatment for OUD are similar to those for other substance use disorders, and involve acute detoxification followed by long-term maintenance of sobriety. There are 3 main phases of treatment for OUD:
- treatment engagement
- stabilization and harm reduction
- sustained abstinence.
Long-term maintenance of sobriety in OUD could involve medications that are FDA-approved for this indication: methadone, buprenorphine, and naltrexone. Psychosocial interventions can be used individually and in combination with pharmacotherapy. Because OUD is a chronic disorder typically characterized by intermittent relapse, patients could move back and forth between the different phases of treatment.
In this article, we highlight the medication and non-medication treatment options for the long-term management of OUD.
Defining abuseExogenous opioids are synthetic substances used for their analgesic and morphine-like properties. Opioids are indicated for the treatment of certain pain conditions and exist in varying potencies and delivery systems, which are tailored for specific types of pain.2 Opioids work through activity at opioid receptors found in the brain, spinal cord, gut, and other organs. Agonism of specific opioid receptors results in a decreased perception of pain and contributes to their abuse potential.
Because of their abuse potential, prescription of opioids is governed by the Controlled Substances Act3 of the United States. Despite these regulations, approximately 4.5% of U.S. adults from a nationally representative sample were found to be misusing prescription opioids.4 Another study used data from the National Survey on Drug Use and Health and showed an increasing prevalence of OUD involving prescription drugs and resulting in increased mortality.5 The mortality rate from prescription opioids was found to be higher than for all other illicit drugs combined in 2013.6 Recently, Congress passed the Comprehensive Addiction and Recovery Act, which is intended to address the opioid crisis by expanding access to treatments for opioid overdose and addiction treatment services.7
The term “opioid use disorder” is found in DSM-58 and replaces the previous diagnoses of opioid abuse and dependence in DSM-IV-TR.9 OUD is characterized by a strong desire to continue using opioids despite problems associated with their use. Patients with OUD often experience cravings for opioids, tolerance, repeated failures at cutting down or limiting use, and decreased involvement in social activities.
Maladaptive use of opioids can result in physiologic dependence and lead to withdrawal symptoms, including anxiety, drug craving, insomnia, rhinorrhea, lacrimation, diarrhea, and piloerection. However, physiologic dependence might not necessarily lead to development of OUD, which can be diagnosed when enough other diagnostic criteria present in the absence of physiologic dependence.
Misuse of prescription opioids is more prevalent among patients who meet criteria for other substance use disorders than among patients who do not.10 Misuse of prescription opioids often results in greater health care utilization, including the need for emergency services, hospitalization, and detoxification.
Assessing for OUDThe Addiction Severity Index was designed to classify and monitor misuse of opioids. The Index has poor sensitivity; it detects recent opioid use but fails to differentiate patients using prescribed opioids from those who are abusing opioids.11 By comparison, the Current Opioid Misuse Measure (COMM) was developed to monitor opioid use in chemical dependency treatment settings.12 The COMM has reliable internal consistency13 and validity and can be used to assess use of opioids outside of routine medical care. To gauge the severity of withdrawal symptoms, the Objective Opioid Withdrawal Scale or Clinical Opiate Withdrawal Scale14 can be used. Table 1 summarizes some standardized tools used to assess OUD.
Neurobiological considerationsOpioids work through activity at mu, kappa, and delta opioid receptors. These receptors are present in both the peripheral and the central nervous system. Exogenous opioids work through activity at G protein-coupled receptors and by activating specific neurotransmitter systems. The effect of a given opioid drug is dependent on the type and location of receptors it modulates and can range from CNS depression to euphoria.
For example, mu receptor activation produces sedation, euphoria, or analgesia depending upon the location, frequency, and duration of receptor occupancy. Activation of CNS mu receptors can cause miosis and respiratory depression, whereas mu receptor activation in the peripheral nervous system can cause constipation and cough suppression. Mu receptor stimulation through various G-proteins triggers the second messenger cascade, generating enzymes such as cyclic adenosine monophosphate.
Clinical considerationsTreating OUD is challenging because of the ease with which patients can obtain opioids and because sometimes OUD occurs iatrogenically. Engaging patients in treatment is an important step in recovery, but it does not necessarily lead to reduction in opioid use. The engagement stage can involve outreach workers to encourage further treatment. Developing a therapeutic alliance and appropriately incentivizing patients also promotes entry into treatment. Motivational interviewing is used often in substance use treatment programs and can help engage patients in treatment and evaluate their willingness to change problematic behaviors.
Managing acute withdrawal symptoms. Withdrawal symptoms usually are not life threatening, but can be in the context of other medical conditions, such as autonomic instability, hypertension, cardiovascular disease, and dehydration. Withdrawal symptoms also can be life-threatening during in utero exposure to a fetus. Pharmacotherapeutic options to treat opioid withdrawal symptoms include long-acting opioids, such as methadone and buprenorphine,15 which can be administered in an ambulatory setting. The combination of buprenorphine and naloxone also can be used to treat opioid withdrawal symptoms.
The alpha-2 agonist16 clonidine, although not FDA-approved for OUD or opioid withdrawal, could be used to shorten the duration of withdrawal symptoms. Clonidine also decreases methadone withdrawal and can be combined with naloxone to target naloxone-induced opioid withdrawal symptoms.17,18 Nalbuphine and butorphanol should be avoided during opioid withdrawal because they antagonize opioid receptors and can precipitate withdrawal symptoms.19,20
Maintenance phase involves long-term stabilization and relapse prevention. Treatment options include medication and non-medication interventions.21
Non-pharmacologic treatment options,22 principally psychosocial interventions, can be used on their own or in combination with medications for maintenance treatment of OUD. Psychosocial interventions include structured, professionally administered interventions such as cognitive-behavioral therapy (CBT), aversion therapy, and day-treatment programs. Interventions such as peer counseling and self-help groups also are considered psychosocial interventions, but do not require the same type of professional training.
Peer support groups such as Narcotics Anonymous (NA) help members achieve and maintain sobriety and often focus on a traditional 12-step format or on the more recent Matrix Model,23,24 which is an intensive outpatient treatment program based on components of relapse prevention, motivational interviewing, CBT, and psychoeducation.
In these peer support models, group members discuss patterns of substance use and help one another recognize and overcome problematic behaviors. Groups may vary in terms of their specific approach. For example, NA encourages group members to focus on addiction itself while Methadone Anonymous prefers participants also discuss pharmacologic treatment experiences. Additional services for finding housing and assisting with job placement are also part of some relapse prevention strategies.
Although studies on the use of abstinence-based treatments are limited, abstinence-based therapy is an option for patients wishing to undergo chemical dependency treatment without taking prescription medications to address cravings or withdrawal symptoms.25 However, abstinence-based treatments have been shown to be less effective in improving outcomes than medication-assisted treatment (MAT).26 MAT combines medications and behavioral therapies for treating substance use disorders.
Pharmacologically, OUD can be treated with opioid agonist and antagonist medications. As summarized in Tables 2-4,27-31 these medications differ based on their pharmacokinetic and pharmacodynamic profiles and intrinsic activity at mu opioid receptors. Opioid system agonists, such as methadone and buprenorphine, decrease cravings by mimicking the activity of exogenous opioids. The opioid antagonists naloxone and naltrexone reinforce abstinence by inhibiting the euphoric effects associated with opioid use. The medication of choice for a given patient depends on:
- treatment adherence
- clinical setting
- degree of withdrawal symptoms
- motivation.26
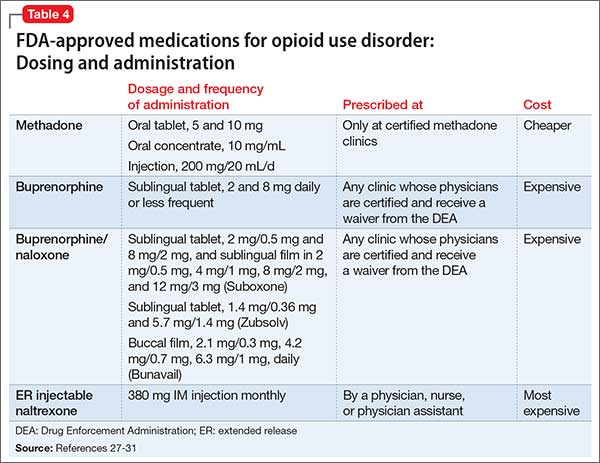
If a patient is actively seeking abstinence from opioids, either agonist or antagonist treatment can be used. In cases where a patient is not seeking abstinence, then preference should be given to opioid agonists to prevent overdose.26
Evidence suggests MAT can improve outcomes with OUD when compared with abstinence treatment alone. Several randomized, controlled, trials showed methadone and buprenorphine were more effective at treating OUD compared with treatment without medication. To date, 3 medications have been FDA-approved for treating OUD: methadone, buprenorphine, and naltrexone.26 All 3 medications differ in their pharmacokinetic and pharmacodynamic profiles and intrinsic activities at central mu-opioid receptor, as summarized in Tables 2-4.27-31
MethadoneMethadone reduces the euphoric effects of opioid use because it binds to and blocks opioid receptors. Methadone is an opioid replacement strategy; higher dosages are used for maintenance treatment to prevent additional dosages of opioids from causing euphoria. Methadone typically is administered once daily. However, in certain circumstances, such as rapid metabolism or pregnancy, it can be given as a twice-daily dosing regimen. Specific ABCB1 variants and DRD2 genetic polymorphisms (simultaneous occurrence of ≥2 genetically determined phenotypes) might determine the dosage requirements of methadone.32
Methadone during pregnancy. Methadone is the treatment of choice for opioid-dependent women during pregnancy33 and is listed as pregnancy category C because it can result in physiologic dependence of the newborn, although there are no documented controlled studies in humans to assess this risk. Methadone can be used while breast-feeding as long as patients are HIV-negative and not abusing other drugs.34,35 Because the methadone concentration in breast milk generally is low, the medication can be administered to nursing mothers after a careful consideration of risks and benefits.
Methadone administration. There are stringent eligibility criteria for methadone administration; not all physicians are authorized to prescribe methadone. Its use is federally regulated and only licensed treatment programs and licensed inpatient detoxification units can prescribe and dispense methadone in controlled settings and under the direct supervision of clinical personnel (Table 5).36 Patients meeting eligibility criteria can attend a specialized methadone clinic.

One of the challenges when using methadone for long-term management of OUD is tapering the dosage and attempting to discontinue the medication. Discontinuation of methadone leads to withdrawal symptoms and requires a carefully tailored tapering schedule. The literature on methadone tapering is limited. Tapering schedules could differ from practice to practice and, in many cases, are highly individualized based on the need and response of specific patients.
Dosage reduction schedules can last from 2 to 3 weeks to 6 months. Studies indicate rapid reduction worsens treatment outcomes and protracted tapering is associated with better outcomes. A suggested tapering schedule could involve decreasing the dosage by 20% to 25% until reaching a dosage of 30 mg/d, then decreasing by 5 mg/d every 3 to 5 days until reaching a dosage of 10 mg/d, before finally decreasing by 2.5 mg/d every 3 to 5 days.
Some randomized trials have shown better outcomes with long-term treatment. The goal of many programs is transitioning from maintenance treatment to abstinence. However, programs targeting maintenance rather than abstinence have been shown to be more effective.
The FDA has no defined limits for treatment duration with either methadone or buprenorphine. Therefore, the decision to taper or discontinue either medication should be made carefully case by case, using sound clinical judgment. Studies show that methadone treatment could reduce the spread of HIV,37,38 decrease criminal behaviors,39 and reduce overall mortality rates.40 A follow-up study comparing individuals randomly assigned to receive methadone or buprenorphine for OUD showed reduced risk of mortality overall40 in both groups.
Adverse events reported during treatment with methadone include decreased libido, erectile dysfunction, constipation, drowsiness, QTc prolongation, and torsade de pointes.41 Therefore, the FDA recommends obtaining a detailed medical history and baseline electrocardiogram (ECG), with a repeat ECG within the first month of treatment and then annually. Informing patients about the possibility of arrhythmias is part of the informed consent process before starting methadone.
Clinicians also should be vigilant when using methadone in combination with other medications that can prolong the QTc interval (eg, some antipsychotics). Methadone has a greater risk of fatal overdose then buprenorphine. A large-scale study of >16,000 patients reported a 4-fold increase in mortality resulting from methadone overdose compared with buprenorphine.42
BuprenorphineBuprenorphine is a partial opioid agonist at the mu opioid receptor. A full opioid agonist binds and fully activates the opioid receptors; an antagonist blocks the same. An opioid receptor partial agonist partially activates the receptor. Therefore, an opioid system partial agonist is a functional antagonist and, at lower dosages, has weak agonist effects; at higher dosages, a partial agonist antagonizes other endogenous and exogenous opioids that compete for binding at the same receptor.43 Because of the partial agonist effect, buprenorphine could result in less physical dependence and less withdrawal symptoms.
Administration. In contrast to methadone, buprenorphine can be prescribed by physicians for long-term management of OUD in the United States. Buprenorphine is available in 2 formulations: a sublingual form for daily use and a long-acting form that causes less withdrawal symptoms and cravings. In May 2016 the FDA approved the first buprenorphine implant for use in opioid dependence.44,45
To prevent withdrawal symptoms, a 24-hour period of opioid abstinence is recommended before starting buprenorphine or buprenorphine/naloxone treatment.46 Although lacking empirical evidence, catechol-O-methyltransferase (COMT) inhibitors, such as entacapone, have an anti-craving affect and are used by some clinicians to improve adherence with buprenorphine. This is because of their ability to balance dopamine, which is central to the reward pathway responsible for cravings. Although use of COMT inhibitors might make sense intuitively, such use is off-label and should be based on clinical judgment and a review of the available literature. A study showed that tapering buprenorphine for 4 weeks in combination with naltrexone improved the abstinence rate.47
Adverse effects. Some of the adverse events reported during treatment with buprenorphine include fever, back pain, nausea, cough, sedation, difficulty with urination, and constipation. Respiratory depression is a less common effect of buprenorphine, compared with full opioid agonists, because of the medication’s mechanism of action as a partial agonist.48 As a result, buprenorphine has been shown to have a lower risk of fatal overdose than methadone.49 Studies have shown buprenorphine to be more likely than methadone to reduce neonatal abstinence syndromes.50
NaltrexoneNaltrexone is an opioid antagonist and is an option to promote relapse prevention. Because of its antagonist properties, naltrexone treatment should always start after opioid detoxification because it can potentiate immediate withdrawal symptoms. Naltrexone is available in oral and long-acting formulations, the latter of which may be considered in patients who have difficulty with adherence.37
Oral naltrexone is taken as a single 50 mg-tablet once daily, whereas dosing for long-acting naltrexone in injectable and implantable formulations varies. These long-acting naltrexone formulations typically are administered monthly. Some of the adverse events reported during treatment with naltrexone are nausea, liver damage, and injection site pain.51
Buprenorphine/naloxoneBecause of buprenorphine’s agonist effects, it has a relatively high abuse potential compared with other opioids.52 Naloxone, on the other hand, is an opioid antagonist and is poorly absorbed when given orally and is associated with withdrawal symptoms if used intravenously. Therefore, naloxone is added to buprenorphine to decrease the likelihood of abuse when both are used as a combination product.53
Buprenorphine is combined with naloxone in a ratio of 4:1. Induction begins by using a 2 mg/0.5 mg tablet with dosage titration until symptoms abate. A combination of buprenorphine and naloxone also is available in film and tablet formulations. Patients must abstain from other opioids for at least 24 hours before initiating buprenorphine/naloxone treatment to prevent the precipitation of withdrawal symptoms.
1. Rudd RA, Aleshire N, Zibbell JE, et al. Increases in drug and opioid overdose deaths—United States, 2000-2014. MMWR Morb Mortal Wkly Rep. 2016;64(50-51):1378-1382.
2. Portenoy RK, Lesage P. Management of cancer pain. Lancet. 1999;353(9165):1695-1700.
3. Passik SD, Weinreb HJ. Managing chronic nonmalignant pain: overcoming obstacles to the use of opioids. Adv Ther. 2000;17(2):70-83.
4. Becker WC, Sullivan LE, Tetrault JM, et al. Non-medical use, abuse and dependence on prescription opioids among U.S. adults: psychiatric, medical and substance use correlates. Drug Alcohol Depend. 2008;94(1-3):38-47.
5. Han B, Compton WM, Jones CM, et al. Nonmedical prescription opioid use and use disorders among adults aged 18 through 64 years in the United States, 2003-2013. JAMA. 2015;314(14):1468-1478.
6. Centers for Disease Control and Prevention. National Center for Health Statistics, 2014. Multiple cause of death data. http://wonder.cdc.gov/mcd.html.
7. Twachtman G. Congress sends opioid legislation to the President. Clinical Psychiatry News. http://www.clinicalpsychiatrynews.com/?id=2407&tx_ttnews[tt_news]=524025&cHash=e93d5d1f86d20e53d3e2d8b07e9562b2. Published July 15, 2016. Accessed July 18, 2016.
8. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
9. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
10. McCabe SE, Cranford JA, West BT. Trends in prescription drug abuse and dependence, co-occurrence with other substance use disorders, and treatment utilization: results from two national surveys. Addict Behav. 2008;33(10):1297-1305.
11. Butler SF, Villapiano A, Malinow A. The effect of computer-mediated administration on self-disclosure of problems on the Addiction Severity Index. J Addict Med. 2009;3(4):194-203.
12. Meltzer EC, Rybin D, Saitz R, et al. Identifying prescription opioid use disorder in primary care: diagnostic characteristics of the Current Opioid Misuse Measure (COMM). Pain. 2011;152(2):397-402.
13. Butler SF, Budman SH, Fanciullo GJ, et al. Cross validation of the current opioid misuse measure to monitor chronic pain patients on opioid therapy. Clin J Pain. 2010;26(9):770-776.
14. Wesson DR, Ling W. The clinical opiate withdrawal scale (COWS). J Psychoactive Drugs. 2003;35(2):253-259.
15. Fiellin DA, O’Connor PG. Clinical practice. Office-based treatment of opioid-dependent patients. N Engl J Med. 2002;347(11):817-823.
16. Gowing LR, Farrell M, Ali RL, et al. α2‐Adrenergic agonists in opioid withdrawal. Addiction. 2002;97(1):49-58.
17. Loimer N, Hofmann P, Chaudhry H. Ultrashort noninvasive opiate detoxification. Am J Psychiatry. 1993;150(5):839.
18. Charney DS, Sternberg DE, Kleber HD, et al. The clinical use of clonidine in abrupt withdrawal from methadone. Effects on blood pressure and specific signs and symptoms. Arch Gen Psychiatry. 1981;38(11):1273-1277.
19. Preston KL, Bigelow GE, Liebson IA. Antagonist effects of nalbuphine in opioid-dependent human volunteers. J Pharmacol Exp Ther. 1989;248(3):929-937.
20. Preston KL, Bigelow GE, Liebson IA. Discrimination of butorphanol and nalbuphine in opioid-dependent humans. Pharmacol Biochem Behav. 1990;37(3):511-522.
21. Effective medical treatment of opiate addiction. National Consensus Development Panel on Effective Medical Treatment of Opiate Addiction. JAMA. 1998;280(22):1936-1943.
22. Amato L, Minozzi S, Davoli, M, et al. Psychosocial combined with agonist maintenance treatments versus agonist maintenance treatments alone for treatment of opioid dependence. Cochrane Database Syst Rev. 2011;(10):CD004147. doi: 10.1002/14651858.CD004147.pub4.
23. Obert JL, McCann MJ, Marinelli-Casey P, et al. The matrix model of outpatient stimulant abuse treatment: history and description. J Psychoactive Drugs. 2000;32(2):157-164.
24. Mayet S, Farrell M, Ferri M, et al. Psychosocial treatment for opiate abuse and dependence. Cochrane Database Syst Rev. 2005:CD004330.
25. McAuliffe WE. A randomized controlled trial of recovery training and self-help for opioid addicts in New England and Hong Kong. J Psychoactive Drugs. 1990;22(2):197-209.
26. Connery HS. Medication-assisted treatment of opioid use disorder: review of the evidence and future directions. Harv Rev Psychiatry. 2015;23(2):63-75.
27. Gibson AE, Degenhardt LJ. Mortality related to pharmacotherapies for opioid dependence: a comparative analysis of coronial records. Drug Alcohol Rev. 2007;26:405-410.
28. Clark L, Haram E, Johnson K, et al. Getting started with medication-assisted treatment with lessons from advancing recovery. Madison, WI: University of Wisconsin-Madison; 2010.
29. U.S. Food and Drug Administration. Vivitrol (naltrexone for extended-release injectable suspension): NDA 21-897C—Briefing document/background package. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharmacologic DrugsAdvisoryCommittee/UCM225664.pdf. Published September 16, 2010. Accessed July 11, 2016.
30. Providers Clinical Support System. PCSS Guidance. Buprenorphine induction. http://pcssmat.org/wp-content/uploads/2014/02/PCSS-MATGuidanceBuprenorphineInduction.Casadonte.pdf. Updated November 27, 2013. Accessed July 18, 2016.
31. An introduction to extended-release injectable naltrexone for the treatment of people with opioid dependence. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2012. HHS Publication No. (SMA) 12-4682.
32. Doehring A, von Hentig N, Graff J, et al. Genetic variants altering dopamine D2 receptor expression or function modulate the risk of opiate addiction and the dosage requirements of methadone substitution. Pharmacogenet Genomics. 2009;19(6):407-414.
33. Mitchell JL. Pregnant, substance-abusing women: treatment improvement protocol (TIP) Series 2. Rockville, MD: Substance Abuse and Mental Health Services Administration; 1993. DHHS Publication No. (SMA) 95-3056.
34. McCarthy JJ, Posey BL. Methadone levels in human milk. J Hum Lact. 2000;16(2):115-120.
35. Geraghty B, Graham EA, Logan B, et al. Methadone levels in breast milk. J Hum Lact. 1997;13(3):227-230.
36. Krambeer LL, von McKnelly W Jr, Gabrielli WF Jr, et al. Methadone therapy for opioid dependence. Am Fam Physician. 2001;63(12):2404-2410.
37. Novick DM, Joseph H, Croxson TS, et al. Absence of antibody to human immunodeficiency virus in long-term, socially rehabilitated methadone maintenance patients. Arch Intern Med. 1990;150(1):97-99.
38. Gowing LR, Farrell M, Bornemann R, et al. Brief report: methadone treatment of injecting opioid users for prevention of HIV infection. J Gen Intern Med. 2006;21(2):193-195.
39. Nurco DN, Ball JC, Shaffer JW, et al. The criminality of narcotic addicts. J Nerv Ment Dis. 1985;173(2):94-102.
40. Gibson A, Degenhardt L, Mattick RP, et al. Exposure to opioid maintenance treatment reduces long-term mortality. Addiction. 2008;103(3):462-468.
41. Pearson EC, Woosley RL. QT prolongation and torsades de pointes among methadone users: reports to the FDA spontaneous reporting system. Pharmacoepidemiol Drug Saf. 2005;14(11):747-753.
42. Bell JR, Butler B, Lawrance A, et al. Comparing overdose mortality associated with methadone and buprenorphine treatment. Drug Alcohol Depend. 2009;104(1-2):73-77.
43. Bickel WK, Amass L. Buprenorphine treatment of opioid dependence: a review. Experimental and Clinical Psychopharmacology. 1995;3(4):477-489.
44. U.S. Food and Drug Administration. FDA approves first buprenorphine implant for treatment of opioid dependence. http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm503719.htm. Published May 26, 2016. Accessed July 18, 2016.
45. The National Alliance of Advocates for Buprenorphine Treatment. https://www.naabt.org/index.cfm. Accessed July 18, 2016.
46. Buprenorphine: an alternative to methadone. Med Lett Drugs Ther. 2003;45(1150):13-15.
47. Sigmon SC, Dunn KE, Saulsgiver K, et al. A randomized, double-blind evaluation of buprenorphine taper duration in primary prescription opioid abusers. JAMA Psychiatry. 2013;70(12):1347-1354.
48. Dahan A, Yassen A, Bijl H, et al. Comparison of the respiratory effects of intravenous buprenorphine and fentanyl in humans and rats. Br J Anaesth. 2005;94(6):825-834.
49. Bell JR, Butler B, Lawrance A, et al. Comparing overdose mortality associated with methadone and buprenorphine treatment. Drug Alcohol Depend. 2009;104(1-2):73-77.
50. Kakko J, Heilig M, Sarman I. Buprenorphine and methadone treatment of opiate dependence during pregnancy: comparison of fetal growth and neonatal outcomes in two consecutive case series. Drug Alcohol Depend. 2008;96(1-2):69-78.
51. Stotts AL, Dodrill CL, Kosten TR. Opioid dependence treatment: options in pharmacotherapy. Expert Opin Pharmacother. 2009;10(11):1727-1740.
52. Robinson GM, Dukes PD, Robinson BJ, et al. The misuse of buprenorphine and a buprenorphine-naloxone combination in Wellington, New Zealand. Drug Alcohol Depend. 1993;33(1):81-86.
53. Fudala PJ, Bridge TP, Herbert S, et al; Buprenorphine/Naloxone Collaborative Study Group. Office-based treatment of opiate addiction with a sublingual-tablet formulation of buprenorphine and naloxone. N Engl J Med. 2003;349(10):949-958.
Communities across the United States have experienced a near-epidemic of opioid abuse. Deaths from opioid overdose have doubled since 2000 and increased 14% from 2013 to 2014.1 Treatment strategies for opioid use disorder (OUD) target individual well-being with the goal of preventing relapse. Most treatment approaches help patients gain self-confidence and have been described by patients as “giving them their life back.”
Broadly, the major phases of treatment for OUD are similar to those for other substance use disorders, and involve acute detoxification followed by long-term maintenance of sobriety. There are 3 main phases of treatment for OUD:
- treatment engagement
- stabilization and harm reduction
- sustained abstinence.
Long-term maintenance of sobriety in OUD could involve medications that are FDA-approved for this indication: methadone, buprenorphine, and naltrexone. Psychosocial interventions can be used individually and in combination with pharmacotherapy. Because OUD is a chronic disorder typically characterized by intermittent relapse, patients could move back and forth between the different phases of treatment.
In this article, we highlight the medication and non-medication treatment options for the long-term management of OUD.
Defining abuseExogenous opioids are synthetic substances used for their analgesic and morphine-like properties. Opioids are indicated for the treatment of certain pain conditions and exist in varying potencies and delivery systems, which are tailored for specific types of pain.2 Opioids work through activity at opioid receptors found in the brain, spinal cord, gut, and other organs. Agonism of specific opioid receptors results in a decreased perception of pain and contributes to their abuse potential.
Because of their abuse potential, prescription of opioids is governed by the Controlled Substances Act3 of the United States. Despite these regulations, approximately 4.5% of U.S. adults from a nationally representative sample were found to be misusing prescription opioids.4 Another study used data from the National Survey on Drug Use and Health and showed an increasing prevalence of OUD involving prescription drugs and resulting in increased mortality.5 The mortality rate from prescription opioids was found to be higher than for all other illicit drugs combined in 2013.6 Recently, Congress passed the Comprehensive Addiction and Recovery Act, which is intended to address the opioid crisis by expanding access to treatments for opioid overdose and addiction treatment services.7
The term “opioid use disorder” is found in DSM-58 and replaces the previous diagnoses of opioid abuse and dependence in DSM-IV-TR.9 OUD is characterized by a strong desire to continue using opioids despite problems associated with their use. Patients with OUD often experience cravings for opioids, tolerance, repeated failures at cutting down or limiting use, and decreased involvement in social activities.
Maladaptive use of opioids can result in physiologic dependence and lead to withdrawal symptoms, including anxiety, drug craving, insomnia, rhinorrhea, lacrimation, diarrhea, and piloerection. However, physiologic dependence might not necessarily lead to development of OUD, which can be diagnosed when enough other diagnostic criteria present in the absence of physiologic dependence.
Misuse of prescription opioids is more prevalent among patients who meet criteria for other substance use disorders than among patients who do not.10 Misuse of prescription opioids often results in greater health care utilization, including the need for emergency services, hospitalization, and detoxification.
Assessing for OUDThe Addiction Severity Index was designed to classify and monitor misuse of opioids. The Index has poor sensitivity; it detects recent opioid use but fails to differentiate patients using prescribed opioids from those who are abusing opioids.11 By comparison, the Current Opioid Misuse Measure (COMM) was developed to monitor opioid use in chemical dependency treatment settings.12 The COMM has reliable internal consistency13 and validity and can be used to assess use of opioids outside of routine medical care. To gauge the severity of withdrawal symptoms, the Objective Opioid Withdrawal Scale or Clinical Opiate Withdrawal Scale14 can be used. Table 1 summarizes some standardized tools used to assess OUD.
Neurobiological considerationsOpioids work through activity at mu, kappa, and delta opioid receptors. These receptors are present in both the peripheral and the central nervous system. Exogenous opioids work through activity at G protein-coupled receptors and by activating specific neurotransmitter systems. The effect of a given opioid drug is dependent on the type and location of receptors it modulates and can range from CNS depression to euphoria.
For example, mu receptor activation produces sedation, euphoria, or analgesia depending upon the location, frequency, and duration of receptor occupancy. Activation of CNS mu receptors can cause miosis and respiratory depression, whereas mu receptor activation in the peripheral nervous system can cause constipation and cough suppression. Mu receptor stimulation through various G-proteins triggers the second messenger cascade, generating enzymes such as cyclic adenosine monophosphate.
Clinical considerationsTreating OUD is challenging because of the ease with which patients can obtain opioids and because sometimes OUD occurs iatrogenically. Engaging patients in treatment is an important step in recovery, but it does not necessarily lead to reduction in opioid use. The engagement stage can involve outreach workers to encourage further treatment. Developing a therapeutic alliance and appropriately incentivizing patients also promotes entry into treatment. Motivational interviewing is used often in substance use treatment programs and can help engage patients in treatment and evaluate their willingness to change problematic behaviors.
Managing acute withdrawal symptoms. Withdrawal symptoms usually are not life threatening, but can be in the context of other medical conditions, such as autonomic instability, hypertension, cardiovascular disease, and dehydration. Withdrawal symptoms also can be life-threatening during in utero exposure to a fetus. Pharmacotherapeutic options to treat opioid withdrawal symptoms include long-acting opioids, such as methadone and buprenorphine,15 which can be administered in an ambulatory setting. The combination of buprenorphine and naloxone also can be used to treat opioid withdrawal symptoms.
The alpha-2 agonist16 clonidine, although not FDA-approved for OUD or opioid withdrawal, could be used to shorten the duration of withdrawal symptoms. Clonidine also decreases methadone withdrawal and can be combined with naloxone to target naloxone-induced opioid withdrawal symptoms.17,18 Nalbuphine and butorphanol should be avoided during opioid withdrawal because they antagonize opioid receptors and can precipitate withdrawal symptoms.19,20
Maintenance phase involves long-term stabilization and relapse prevention. Treatment options include medication and non-medication interventions.21
Non-pharmacologic treatment options,22 principally psychosocial interventions, can be used on their own or in combination with medications for maintenance treatment of OUD. Psychosocial interventions include structured, professionally administered interventions such as cognitive-behavioral therapy (CBT), aversion therapy, and day-treatment programs. Interventions such as peer counseling and self-help groups also are considered psychosocial interventions, but do not require the same type of professional training.
Peer support groups such as Narcotics Anonymous (NA) help members achieve and maintain sobriety and often focus on a traditional 12-step format or on the more recent Matrix Model,23,24 which is an intensive outpatient treatment program based on components of relapse prevention, motivational interviewing, CBT, and psychoeducation.
In these peer support models, group members discuss patterns of substance use and help one another recognize and overcome problematic behaviors. Groups may vary in terms of their specific approach. For example, NA encourages group members to focus on addiction itself while Methadone Anonymous prefers participants also discuss pharmacologic treatment experiences. Additional services for finding housing and assisting with job placement are also part of some relapse prevention strategies.
Although studies on the use of abstinence-based treatments are limited, abstinence-based therapy is an option for patients wishing to undergo chemical dependency treatment without taking prescription medications to address cravings or withdrawal symptoms.25 However, abstinence-based treatments have been shown to be less effective in improving outcomes than medication-assisted treatment (MAT).26 MAT combines medications and behavioral therapies for treating substance use disorders.
Pharmacologically, OUD can be treated with opioid agonist and antagonist medications. As summarized in Tables 2-4,27-31 these medications differ based on their pharmacokinetic and pharmacodynamic profiles and intrinsic activity at mu opioid receptors. Opioid system agonists, such as methadone and buprenorphine, decrease cravings by mimicking the activity of exogenous opioids. The opioid antagonists naloxone and naltrexone reinforce abstinence by inhibiting the euphoric effects associated with opioid use. The medication of choice for a given patient depends on:
- treatment adherence
- clinical setting
- degree of withdrawal symptoms
- motivation.26
If a patient is actively seeking abstinence from opioids, either agonist or antagonist treatment can be used. In cases where a patient is not seeking abstinence, then preference should be given to opioid agonists to prevent overdose.26
Evidence suggests MAT can improve outcomes with OUD when compared with abstinence treatment alone. Several randomized, controlled, trials showed methadone and buprenorphine were more effective at treating OUD compared with treatment without medication. To date, 3 medications have been FDA-approved for treating OUD: methadone, buprenorphine, and naltrexone.26 All 3 medications differ in their pharmacokinetic and pharmacodynamic profiles and intrinsic activities at central mu-opioid receptor, as summarized in Tables 2-4.27-31
MethadoneMethadone reduces the euphoric effects of opioid use because it binds to and blocks opioid receptors. Methadone is an opioid replacement strategy; higher dosages are used for maintenance treatment to prevent additional dosages of opioids from causing euphoria. Methadone typically is administered once daily. However, in certain circumstances, such as rapid metabolism or pregnancy, it can be given as a twice-daily dosing regimen. Specific ABCB1 variants and DRD2 genetic polymorphisms (simultaneous occurrence of ≥2 genetically determined phenotypes) might determine the dosage requirements of methadone.32
Methadone during pregnancy. Methadone is the treatment of choice for opioid-dependent women during pregnancy33 and is listed as pregnancy category C because it can result in physiologic dependence of the newborn, although there are no documented controlled studies in humans to assess this risk. Methadone can be used while breast-feeding as long as patients are HIV-negative and not abusing other drugs.34,35 Because the methadone concentration in breast milk generally is low, the medication can be administered to nursing mothers after a careful consideration of risks and benefits.
Methadone administration. There are stringent eligibility criteria for methadone administration; not all physicians are authorized to prescribe methadone. Its use is federally regulated and only licensed treatment programs and licensed inpatient detoxification units can prescribe and dispense methadone in controlled settings and under the direct supervision of clinical personnel (Table 5).36 Patients meeting eligibility criteria can attend a specialized methadone clinic.
One of the challenges when using methadone for long-term management of OUD is tapering the dosage and attempting to discontinue the medication. Discontinuation of methadone leads to withdrawal symptoms and requires a carefully tailored tapering schedule. The literature on methadone tapering is limited. Tapering schedules could differ from practice to practice and, in many cases, are highly individualized based on the need and response of specific patients.
Dosage reduction schedules can last from 2 to 3 weeks to 6 months. Studies indicate rapid reduction worsens treatment outcomes and protracted tapering is associated with better outcomes. A suggested tapering schedule could involve decreasing the dosage by 20% to 25% until reaching a dosage of 30 mg/d, then decreasing by 5 mg/d every 3 to 5 days until reaching a dosage of 10 mg/d, before finally decreasing by 2.5 mg/d every 3 to 5 days.
Some randomized trials have shown better outcomes with long-term treatment. The goal of many programs is transitioning from maintenance treatment to abstinence. However, programs targeting maintenance rather than abstinence have been shown to be more effective.
The FDA has no defined limits for treatment duration with either methadone or buprenorphine. Therefore, the decision to taper or discontinue either medication should be made carefully case by case, using sound clinical judgment. Studies show that methadone treatment could reduce the spread of HIV,37,38 decrease criminal behaviors,39 and reduce overall mortality rates.40 A follow-up study comparing individuals randomly assigned to receive methadone or buprenorphine for OUD showed reduced risk of mortality overall40 in both groups.
Adverse events reported during treatment with methadone include decreased libido, erectile dysfunction, constipation, drowsiness, QTc prolongation, and torsade de pointes.41 Therefore, the FDA recommends obtaining a detailed medical history and baseline electrocardiogram (ECG), with a repeat ECG within the first month of treatment and then annually. Informing patients about the possibility of arrhythmias is part of the informed consent process before starting methadone.
Clinicians also should be vigilant when using methadone in combination with other medications that can prolong the QTc interval (eg, some antipsychotics). Methadone has a greater risk of fatal overdose then buprenorphine. A large-scale study of >16,000 patients reported a 4-fold increase in mortality resulting from methadone overdose compared with buprenorphine.42
BuprenorphineBuprenorphine is a partial opioid agonist at the mu opioid receptor. A full opioid agonist binds and fully activates the opioid receptors; an antagonist blocks the same. An opioid receptor partial agonist partially activates the receptor. Therefore, an opioid system partial agonist is a functional antagonist and, at lower dosages, has weak agonist effects; at higher dosages, a partial agonist antagonizes other endogenous and exogenous opioids that compete for binding at the same receptor.43 Because of the partial agonist effect, buprenorphine could result in less physical dependence and less withdrawal symptoms.
Administration. In contrast to methadone, buprenorphine can be prescribed by physicians for long-term management of OUD in the United States. Buprenorphine is available in 2 formulations: a sublingual form for daily use and a long-acting form that causes less withdrawal symptoms and cravings. In May 2016 the FDA approved the first buprenorphine implant for use in opioid dependence.44,45
To prevent withdrawal symptoms, a 24-hour period of opioid abstinence is recommended before starting buprenorphine or buprenorphine/naloxone treatment.46 Although lacking empirical evidence, catechol-O-methyltransferase (COMT) inhibitors, such as entacapone, have an anti-craving affect and are used by some clinicians to improve adherence with buprenorphine. This is because of their ability to balance dopamine, which is central to the reward pathway responsible for cravings. Although use of COMT inhibitors might make sense intuitively, such use is off-label and should be based on clinical judgment and a review of the available literature. A study showed that tapering buprenorphine for 4 weeks in combination with naltrexone improved the abstinence rate.47
Adverse effects. Some of the adverse events reported during treatment with buprenorphine include fever, back pain, nausea, cough, sedation, difficulty with urination, and constipation. Respiratory depression is a less common effect of buprenorphine, compared with full opioid agonists, because of the medication’s mechanism of action as a partial agonist.48 As a result, buprenorphine has been shown to have a lower risk of fatal overdose than methadone.49 Studies have shown buprenorphine to be more likely than methadone to reduce neonatal abstinence syndromes.50
NaltrexoneNaltrexone is an opioid antagonist and is an option to promote relapse prevention. Because of its antagonist properties, naltrexone treatment should always start after opioid detoxification because it can potentiate immediate withdrawal symptoms. Naltrexone is available in oral and long-acting formulations, the latter of which may be considered in patients who have difficulty with adherence.37
Oral naltrexone is taken as a single 50 mg-tablet once daily, whereas dosing for long-acting naltrexone in injectable and implantable formulations varies. These long-acting naltrexone formulations typically are administered monthly. Some of the adverse events reported during treatment with naltrexone are nausea, liver damage, and injection site pain.51
Buprenorphine/naloxoneBecause of buprenorphine’s agonist effects, it has a relatively high abuse potential compared with other opioids.52 Naloxone, on the other hand, is an opioid antagonist and is poorly absorbed when given orally and is associated with withdrawal symptoms if used intravenously. Therefore, naloxone is added to buprenorphine to decrease the likelihood of abuse when both are used as a combination product.53
Buprenorphine is combined with naloxone in a ratio of 4:1. Induction begins by using a 2 mg/0.5 mg tablet with dosage titration until symptoms abate. A combination of buprenorphine and naloxone also is available in film and tablet formulations. Patients must abstain from other opioids for at least 24 hours before initiating buprenorphine/naloxone treatment to prevent the precipitation of withdrawal symptoms.
Communities across the United States have experienced a near-epidemic of opioid abuse. Deaths from opioid overdose have doubled since 2000 and increased 14% from 2013 to 2014.1 Treatment strategies for opioid use disorder (OUD) target individual well-being with the goal of preventing relapse. Most treatment approaches help patients gain self-confidence and have been described by patients as “giving them their life back.”
Broadly, the major phases of treatment for OUD are similar to those for other substance use disorders, and involve acute detoxification followed by long-term maintenance of sobriety. There are 3 main phases of treatment for OUD:
- treatment engagement
- stabilization and harm reduction
- sustained abstinence.
Long-term maintenance of sobriety in OUD could involve medications that are FDA-approved for this indication: methadone, buprenorphine, and naltrexone. Psychosocial interventions can be used individually and in combination with pharmacotherapy. Because OUD is a chronic disorder typically characterized by intermittent relapse, patients could move back and forth between the different phases of treatment.
In this article, we highlight the medication and non-medication treatment options for the long-term management of OUD.
Defining abuseExogenous opioids are synthetic substances used for their analgesic and morphine-like properties. Opioids are indicated for the treatment of certain pain conditions and exist in varying potencies and delivery systems, which are tailored for specific types of pain.2 Opioids work through activity at opioid receptors found in the brain, spinal cord, gut, and other organs. Agonism of specific opioid receptors results in a decreased perception of pain and contributes to their abuse potential.
Because of their abuse potential, prescription of opioids is governed by the Controlled Substances Act3 of the United States. Despite these regulations, approximately 4.5% of U.S. adults from a nationally representative sample were found to be misusing prescription opioids.4 Another study used data from the National Survey on Drug Use and Health and showed an increasing prevalence of OUD involving prescription drugs and resulting in increased mortality.5 The mortality rate from prescription opioids was found to be higher than for all other illicit drugs combined in 2013.6 Recently, Congress passed the Comprehensive Addiction and Recovery Act, which is intended to address the opioid crisis by expanding access to treatments for opioid overdose and addiction treatment services.7
The term “opioid use disorder” is found in DSM-58 and replaces the previous diagnoses of opioid abuse and dependence in DSM-IV-TR.9 OUD is characterized by a strong desire to continue using opioids despite problems associated with their use. Patients with OUD often experience cravings for opioids, tolerance, repeated failures at cutting down or limiting use, and decreased involvement in social activities.
Maladaptive use of opioids can result in physiologic dependence and lead to withdrawal symptoms, including anxiety, drug craving, insomnia, rhinorrhea, lacrimation, diarrhea, and piloerection. However, physiologic dependence might not necessarily lead to development of OUD, which can be diagnosed when enough other diagnostic criteria present in the absence of physiologic dependence.
Misuse of prescription opioids is more prevalent among patients who meet criteria for other substance use disorders than among patients who do not.10 Misuse of prescription opioids often results in greater health care utilization, including the need for emergency services, hospitalization, and detoxification.
Assessing for OUDThe Addiction Severity Index was designed to classify and monitor misuse of opioids. The Index has poor sensitivity; it detects recent opioid use but fails to differentiate patients using prescribed opioids from those who are abusing opioids.11 By comparison, the Current Opioid Misuse Measure (COMM) was developed to monitor opioid use in chemical dependency treatment settings.12 The COMM has reliable internal consistency13 and validity and can be used to assess use of opioids outside of routine medical care. To gauge the severity of withdrawal symptoms, the Objective Opioid Withdrawal Scale or Clinical Opiate Withdrawal Scale14 can be used. Table 1 summarizes some standardized tools used to assess OUD.
Neurobiological considerationsOpioids work through activity at mu, kappa, and delta opioid receptors. These receptors are present in both the peripheral and the central nervous system. Exogenous opioids work through activity at G protein-coupled receptors and by activating specific neurotransmitter systems. The effect of a given opioid drug is dependent on the type and location of receptors it modulates and can range from CNS depression to euphoria.
For example, mu receptor activation produces sedation, euphoria, or analgesia depending upon the location, frequency, and duration of receptor occupancy. Activation of CNS mu receptors can cause miosis and respiratory depression, whereas mu receptor activation in the peripheral nervous system can cause constipation and cough suppression. Mu receptor stimulation through various G-proteins triggers the second messenger cascade, generating enzymes such as cyclic adenosine monophosphate.
Clinical considerationsTreating OUD is challenging because of the ease with which patients can obtain opioids and because sometimes OUD occurs iatrogenically. Engaging patients in treatment is an important step in recovery, but it does not necessarily lead to reduction in opioid use. The engagement stage can involve outreach workers to encourage further treatment. Developing a therapeutic alliance and appropriately incentivizing patients also promotes entry into treatment. Motivational interviewing is used often in substance use treatment programs and can help engage patients in treatment and evaluate their willingness to change problematic behaviors.
Managing acute withdrawal symptoms. Withdrawal symptoms usually are not life threatening, but can be in the context of other medical conditions, such as autonomic instability, hypertension, cardiovascular disease, and dehydration. Withdrawal symptoms also can be life-threatening during in utero exposure to a fetus. Pharmacotherapeutic options to treat opioid withdrawal symptoms include long-acting opioids, such as methadone and buprenorphine,15 which can be administered in an ambulatory setting. The combination of buprenorphine and naloxone also can be used to treat opioid withdrawal symptoms.
The alpha-2 agonist16 clonidine, although not FDA-approved for OUD or opioid withdrawal, could be used to shorten the duration of withdrawal symptoms. Clonidine also decreases methadone withdrawal and can be combined with naloxone to target naloxone-induced opioid withdrawal symptoms.17,18 Nalbuphine and butorphanol should be avoided during opioid withdrawal because they antagonize opioid receptors and can precipitate withdrawal symptoms.19,20
Maintenance phase involves long-term stabilization and relapse prevention. Treatment options include medication and non-medication interventions.21
Non-pharmacologic treatment options,22 principally psychosocial interventions, can be used on their own or in combination with medications for maintenance treatment of OUD. Psychosocial interventions include structured, professionally administered interventions such as cognitive-behavioral therapy (CBT), aversion therapy, and day-treatment programs. Interventions such as peer counseling and self-help groups also are considered psychosocial interventions, but do not require the same type of professional training.
Peer support groups such as Narcotics Anonymous (NA) help members achieve and maintain sobriety and often focus on a traditional 12-step format or on the more recent Matrix Model,23,24 which is an intensive outpatient treatment program based on components of relapse prevention, motivational interviewing, CBT, and psychoeducation.
In these peer support models, group members discuss patterns of substance use and help one another recognize and overcome problematic behaviors. Groups may vary in terms of their specific approach. For example, NA encourages group members to focus on addiction itself while Methadone Anonymous prefers participants also discuss pharmacologic treatment experiences. Additional services for finding housing and assisting with job placement are also part of some relapse prevention strategies.
Although studies on the use of abstinence-based treatments are limited, abstinence-based therapy is an option for patients wishing to undergo chemical dependency treatment without taking prescription medications to address cravings or withdrawal symptoms.25 However, abstinence-based treatments have been shown to be less effective in improving outcomes than medication-assisted treatment (MAT).26 MAT combines medications and behavioral therapies for treating substance use disorders.
Pharmacologically, OUD can be treated with opioid agonist and antagonist medications. As summarized in Tables 2-4,27-31 these medications differ based on their pharmacokinetic and pharmacodynamic profiles and intrinsic activity at mu opioid receptors. Opioid system agonists, such as methadone and buprenorphine, decrease cravings by mimicking the activity of exogenous opioids. The opioid antagonists naloxone and naltrexone reinforce abstinence by inhibiting the euphoric effects associated with opioid use. The medication of choice for a given patient depends on:
- treatment adherence
- clinical setting
- degree of withdrawal symptoms
- motivation.26
If a patient is actively seeking abstinence from opioids, either agonist or antagonist treatment can be used. In cases where a patient is not seeking abstinence, then preference should be given to opioid agonists to prevent overdose.26
Evidence suggests MAT can improve outcomes with OUD when compared with abstinence treatment alone. Several randomized, controlled, trials showed methadone and buprenorphine were more effective at treating OUD compared with treatment without medication. To date, 3 medications have been FDA-approved for treating OUD: methadone, buprenorphine, and naltrexone.26 All 3 medications differ in their pharmacokinetic and pharmacodynamic profiles and intrinsic activities at central mu-opioid receptor, as summarized in Tables 2-4.27-31
MethadoneMethadone reduces the euphoric effects of opioid use because it binds to and blocks opioid receptors. Methadone is an opioid replacement strategy; higher dosages are used for maintenance treatment to prevent additional dosages of opioids from causing euphoria. Methadone typically is administered once daily. However, in certain circumstances, such as rapid metabolism or pregnancy, it can be given as a twice-daily dosing regimen. Specific ABCB1 variants and DRD2 genetic polymorphisms (simultaneous occurrence of ≥2 genetically determined phenotypes) might determine the dosage requirements of methadone.32
Methadone during pregnancy. Methadone is the treatment of choice for opioid-dependent women during pregnancy33 and is listed as pregnancy category C because it can result in physiologic dependence of the newborn, although there are no documented controlled studies in humans to assess this risk. Methadone can be used while breast-feeding as long as patients are HIV-negative and not abusing other drugs.34,35 Because the methadone concentration in breast milk generally is low, the medication can be administered to nursing mothers after a careful consideration of risks and benefits.
Methadone administration. There are stringent eligibility criteria for methadone administration; not all physicians are authorized to prescribe methadone. Its use is federally regulated and only licensed treatment programs and licensed inpatient detoxification units can prescribe and dispense methadone in controlled settings and under the direct supervision of clinical personnel (Table 5).36 Patients meeting eligibility criteria can attend a specialized methadone clinic.
One of the challenges when using methadone for long-term management of OUD is tapering the dosage and attempting to discontinue the medication. Discontinuation of methadone leads to withdrawal symptoms and requires a carefully tailored tapering schedule. The literature on methadone tapering is limited. Tapering schedules could differ from practice to practice and, in many cases, are highly individualized based on the need and response of specific patients.
Dosage reduction schedules can last from 2 to 3 weeks to 6 months. Studies indicate rapid reduction worsens treatment outcomes and protracted tapering is associated with better outcomes. A suggested tapering schedule could involve decreasing the dosage by 20% to 25% until reaching a dosage of 30 mg/d, then decreasing by 5 mg/d every 3 to 5 days until reaching a dosage of 10 mg/d, before finally decreasing by 2.5 mg/d every 3 to 5 days.
Some randomized trials have shown better outcomes with long-term treatment. The goal of many programs is transitioning from maintenance treatment to abstinence. However, programs targeting maintenance rather than abstinence have been shown to be more effective.
The FDA has no defined limits for treatment duration with either methadone or buprenorphine. Therefore, the decision to taper or discontinue either medication should be made carefully case by case, using sound clinical judgment. Studies show that methadone treatment could reduce the spread of HIV,37,38 decrease criminal behaviors,39 and reduce overall mortality rates.40 A follow-up study comparing individuals randomly assigned to receive methadone or buprenorphine for OUD showed reduced risk of mortality overall40 in both groups.
Adverse events reported during treatment with methadone include decreased libido, erectile dysfunction, constipation, drowsiness, QTc prolongation, and torsade de pointes.41 Therefore, the FDA recommends obtaining a detailed medical history and baseline electrocardiogram (ECG), with a repeat ECG within the first month of treatment and then annually. Informing patients about the possibility of arrhythmias is part of the informed consent process before starting methadone.
Clinicians also should be vigilant when using methadone in combination with other medications that can prolong the QTc interval (eg, some antipsychotics). Methadone has a greater risk of fatal overdose then buprenorphine. A large-scale study of >16,000 patients reported a 4-fold increase in mortality resulting from methadone overdose compared with buprenorphine.42
BuprenorphineBuprenorphine is a partial opioid agonist at the mu opioid receptor. A full opioid agonist binds and fully activates the opioid receptors; an antagonist blocks the same. An opioid receptor partial agonist partially activates the receptor. Therefore, an opioid system partial agonist is a functional antagonist and, at lower dosages, has weak agonist effects; at higher dosages, a partial agonist antagonizes other endogenous and exogenous opioids that compete for binding at the same receptor.43 Because of the partial agonist effect, buprenorphine could result in less physical dependence and less withdrawal symptoms.
Administration. In contrast to methadone, buprenorphine can be prescribed by physicians for long-term management of OUD in the United States. Buprenorphine is available in 2 formulations: a sublingual form for daily use and a long-acting form that causes less withdrawal symptoms and cravings. In May 2016 the FDA approved the first buprenorphine implant for use in opioid dependence.44,45
To prevent withdrawal symptoms, a 24-hour period of opioid abstinence is recommended before starting buprenorphine or buprenorphine/naloxone treatment.46 Although lacking empirical evidence, catechol-O-methyltransferase (COMT) inhibitors, such as entacapone, have an anti-craving affect and are used by some clinicians to improve adherence with buprenorphine. This is because of their ability to balance dopamine, which is central to the reward pathway responsible for cravings. Although use of COMT inhibitors might make sense intuitively, such use is off-label and should be based on clinical judgment and a review of the available literature. A study showed that tapering buprenorphine for 4 weeks in combination with naltrexone improved the abstinence rate.47
Adverse effects. Some of the adverse events reported during treatment with buprenorphine include fever, back pain, nausea, cough, sedation, difficulty with urination, and constipation. Respiratory depression is a less common effect of buprenorphine, compared with full opioid agonists, because of the medication’s mechanism of action as a partial agonist.48 As a result, buprenorphine has been shown to have a lower risk of fatal overdose than methadone.49 Studies have shown buprenorphine to be more likely than methadone to reduce neonatal abstinence syndromes.50
NaltrexoneNaltrexone is an opioid antagonist and is an option to promote relapse prevention. Because of its antagonist properties, naltrexone treatment should always start after opioid detoxification because it can potentiate immediate withdrawal symptoms. Naltrexone is available in oral and long-acting formulations, the latter of which may be considered in patients who have difficulty with adherence.37
Oral naltrexone is taken as a single 50 mg-tablet once daily, whereas dosing for long-acting naltrexone in injectable and implantable formulations varies. These long-acting naltrexone formulations typically are administered monthly. Some of the adverse events reported during treatment with naltrexone are nausea, liver damage, and injection site pain.51
Buprenorphine/naloxoneBecause of buprenorphine’s agonist effects, it has a relatively high abuse potential compared with other opioids.52 Naloxone, on the other hand, is an opioid antagonist and is poorly absorbed when given orally and is associated with withdrawal symptoms if used intravenously. Therefore, naloxone is added to buprenorphine to decrease the likelihood of abuse when both are used as a combination product.53
Buprenorphine is combined with naloxone in a ratio of 4:1. Induction begins by using a 2 mg/0.5 mg tablet with dosage titration until symptoms abate. A combination of buprenorphine and naloxone also is available in film and tablet formulations. Patients must abstain from other opioids for at least 24 hours before initiating buprenorphine/naloxone treatment to prevent the precipitation of withdrawal symptoms.
1. Rudd RA, Aleshire N, Zibbell JE, et al. Increases in drug and opioid overdose deaths—United States, 2000-2014. MMWR Morb Mortal Wkly Rep. 2016;64(50-51):1378-1382.
2. Portenoy RK, Lesage P. Management of cancer pain. Lancet. 1999;353(9165):1695-1700.
3. Passik SD, Weinreb HJ. Managing chronic nonmalignant pain: overcoming obstacles to the use of opioids. Adv Ther. 2000;17(2):70-83.
4. Becker WC, Sullivan LE, Tetrault JM, et al. Non-medical use, abuse and dependence on prescription opioids among U.S. adults: psychiatric, medical and substance use correlates. Drug Alcohol Depend. 2008;94(1-3):38-47.
5. Han B, Compton WM, Jones CM, et al. Nonmedical prescription opioid use and use disorders among adults aged 18 through 64 years in the United States, 2003-2013. JAMA. 2015;314(14):1468-1478.
6. Centers for Disease Control and Prevention. National Center for Health Statistics, 2014. Multiple cause of death data. http://wonder.cdc.gov/mcd.html.
7. Twachtman G. Congress sends opioid legislation to the President. Clinical Psychiatry News. http://www.clinicalpsychiatrynews.com/?id=2407&tx_ttnews[tt_news]=524025&cHash=e93d5d1f86d20e53d3e2d8b07e9562b2. Published July 15, 2016. Accessed July 18, 2016.
8. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
9. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
10. McCabe SE, Cranford JA, West BT. Trends in prescription drug abuse and dependence, co-occurrence with other substance use disorders, and treatment utilization: results from two national surveys. Addict Behav. 2008;33(10):1297-1305.
11. Butler SF, Villapiano A, Malinow A. The effect of computer-mediated administration on self-disclosure of problems on the Addiction Severity Index. J Addict Med. 2009;3(4):194-203.
12. Meltzer EC, Rybin D, Saitz R, et al. Identifying prescription opioid use disorder in primary care: diagnostic characteristics of the Current Opioid Misuse Measure (COMM). Pain. 2011;152(2):397-402.
13. Butler SF, Budman SH, Fanciullo GJ, et al. Cross validation of the current opioid misuse measure to monitor chronic pain patients on opioid therapy. Clin J Pain. 2010;26(9):770-776.
14. Wesson DR, Ling W. The clinical opiate withdrawal scale (COWS). J Psychoactive Drugs. 2003;35(2):253-259.
15. Fiellin DA, O’Connor PG. Clinical practice. Office-based treatment of opioid-dependent patients. N Engl J Med. 2002;347(11):817-823.
16. Gowing LR, Farrell M, Ali RL, et al. α2‐Adrenergic agonists in opioid withdrawal. Addiction. 2002;97(1):49-58.
17. Loimer N, Hofmann P, Chaudhry H. Ultrashort noninvasive opiate detoxification. Am J Psychiatry. 1993;150(5):839.
18. Charney DS, Sternberg DE, Kleber HD, et al. The clinical use of clonidine in abrupt withdrawal from methadone. Effects on blood pressure and specific signs and symptoms. Arch Gen Psychiatry. 1981;38(11):1273-1277.
19. Preston KL, Bigelow GE, Liebson IA. Antagonist effects of nalbuphine in opioid-dependent human volunteers. J Pharmacol Exp Ther. 1989;248(3):929-937.
20. Preston KL, Bigelow GE, Liebson IA. Discrimination of butorphanol and nalbuphine in opioid-dependent humans. Pharmacol Biochem Behav. 1990;37(3):511-522.
21. Effective medical treatment of opiate addiction. National Consensus Development Panel on Effective Medical Treatment of Opiate Addiction. JAMA. 1998;280(22):1936-1943.
22. Amato L, Minozzi S, Davoli, M, et al. Psychosocial combined with agonist maintenance treatments versus agonist maintenance treatments alone for treatment of opioid dependence. Cochrane Database Syst Rev. 2011;(10):CD004147. doi: 10.1002/14651858.CD004147.pub4.
23. Obert JL, McCann MJ, Marinelli-Casey P, et al. The matrix model of outpatient stimulant abuse treatment: history and description. J Psychoactive Drugs. 2000;32(2):157-164.
24. Mayet S, Farrell M, Ferri M, et al. Psychosocial treatment for opiate abuse and dependence. Cochrane Database Syst Rev. 2005:CD004330.
25. McAuliffe WE. A randomized controlled trial of recovery training and self-help for opioid addicts in New England and Hong Kong. J Psychoactive Drugs. 1990;22(2):197-209.
26. Connery HS. Medication-assisted treatment of opioid use disorder: review of the evidence and future directions. Harv Rev Psychiatry. 2015;23(2):63-75.
27. Gibson AE, Degenhardt LJ. Mortality related to pharmacotherapies for opioid dependence: a comparative analysis of coronial records. Drug Alcohol Rev. 2007;26:405-410.
28. Clark L, Haram E, Johnson K, et al. Getting started with medication-assisted treatment with lessons from advancing recovery. Madison, WI: University of Wisconsin-Madison; 2010.
29. U.S. Food and Drug Administration. Vivitrol (naltrexone for extended-release injectable suspension): NDA 21-897C—Briefing document/background package. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharmacologic DrugsAdvisoryCommittee/UCM225664.pdf. Published September 16, 2010. Accessed July 11, 2016.
30. Providers Clinical Support System. PCSS Guidance. Buprenorphine induction. http://pcssmat.org/wp-content/uploads/2014/02/PCSS-MATGuidanceBuprenorphineInduction.Casadonte.pdf. Updated November 27, 2013. Accessed July 18, 2016.
31. An introduction to extended-release injectable naltrexone for the treatment of people with opioid dependence. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2012. HHS Publication No. (SMA) 12-4682.
32. Doehring A, von Hentig N, Graff J, et al. Genetic variants altering dopamine D2 receptor expression or function modulate the risk of opiate addiction and the dosage requirements of methadone substitution. Pharmacogenet Genomics. 2009;19(6):407-414.
33. Mitchell JL. Pregnant, substance-abusing women: treatment improvement protocol (TIP) Series 2. Rockville, MD: Substance Abuse and Mental Health Services Administration; 1993. DHHS Publication No. (SMA) 95-3056.
34. McCarthy JJ, Posey BL. Methadone levels in human milk. J Hum Lact. 2000;16(2):115-120.
35. Geraghty B, Graham EA, Logan B, et al. Methadone levels in breast milk. J Hum Lact. 1997;13(3):227-230.
36. Krambeer LL, von McKnelly W Jr, Gabrielli WF Jr, et al. Methadone therapy for opioid dependence. Am Fam Physician. 2001;63(12):2404-2410.
37. Novick DM, Joseph H, Croxson TS, et al. Absence of antibody to human immunodeficiency virus in long-term, socially rehabilitated methadone maintenance patients. Arch Intern Med. 1990;150(1):97-99.
38. Gowing LR, Farrell M, Bornemann R, et al. Brief report: methadone treatment of injecting opioid users for prevention of HIV infection. J Gen Intern Med. 2006;21(2):193-195.
39. Nurco DN, Ball JC, Shaffer JW, et al. The criminality of narcotic addicts. J Nerv Ment Dis. 1985;173(2):94-102.
40. Gibson A, Degenhardt L, Mattick RP, et al. Exposure to opioid maintenance treatment reduces long-term mortality. Addiction. 2008;103(3):462-468.
41. Pearson EC, Woosley RL. QT prolongation and torsades de pointes among methadone users: reports to the FDA spontaneous reporting system. Pharmacoepidemiol Drug Saf. 2005;14(11):747-753.
42. Bell JR, Butler B, Lawrance A, et al. Comparing overdose mortality associated with methadone and buprenorphine treatment. Drug Alcohol Depend. 2009;104(1-2):73-77.
43. Bickel WK, Amass L. Buprenorphine treatment of opioid dependence: a review. Experimental and Clinical Psychopharmacology. 1995;3(4):477-489.
44. U.S. Food and Drug Administration. FDA approves first buprenorphine implant for treatment of opioid dependence. http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm503719.htm. Published May 26, 2016. Accessed July 18, 2016.
45. The National Alliance of Advocates for Buprenorphine Treatment. https://www.naabt.org/index.cfm. Accessed July 18, 2016.
46. Buprenorphine: an alternative to methadone. Med Lett Drugs Ther. 2003;45(1150):13-15.
47. Sigmon SC, Dunn KE, Saulsgiver K, et al. A randomized, double-blind evaluation of buprenorphine taper duration in primary prescription opioid abusers. JAMA Psychiatry. 2013;70(12):1347-1354.
48. Dahan A, Yassen A, Bijl H, et al. Comparison of the respiratory effects of intravenous buprenorphine and fentanyl in humans and rats. Br J Anaesth. 2005;94(6):825-834.
49. Bell JR, Butler B, Lawrance A, et al. Comparing overdose mortality associated with methadone and buprenorphine treatment. Drug Alcohol Depend. 2009;104(1-2):73-77.
50. Kakko J, Heilig M, Sarman I. Buprenorphine and methadone treatment of opiate dependence during pregnancy: comparison of fetal growth and neonatal outcomes in two consecutive case series. Drug Alcohol Depend. 2008;96(1-2):69-78.
51. Stotts AL, Dodrill CL, Kosten TR. Opioid dependence treatment: options in pharmacotherapy. Expert Opin Pharmacother. 2009;10(11):1727-1740.
52. Robinson GM, Dukes PD, Robinson BJ, et al. The misuse of buprenorphine and a buprenorphine-naloxone combination in Wellington, New Zealand. Drug Alcohol Depend. 1993;33(1):81-86.
53. Fudala PJ, Bridge TP, Herbert S, et al; Buprenorphine/Naloxone Collaborative Study Group. Office-based treatment of opiate addiction with a sublingual-tablet formulation of buprenorphine and naloxone. N Engl J Med. 2003;349(10):949-958.
1. Rudd RA, Aleshire N, Zibbell JE, et al. Increases in drug and opioid overdose deaths—United States, 2000-2014. MMWR Morb Mortal Wkly Rep. 2016;64(50-51):1378-1382.
2. Portenoy RK, Lesage P. Management of cancer pain. Lancet. 1999;353(9165):1695-1700.
3. Passik SD, Weinreb HJ. Managing chronic nonmalignant pain: overcoming obstacles to the use of opioids. Adv Ther. 2000;17(2):70-83.
4. Becker WC, Sullivan LE, Tetrault JM, et al. Non-medical use, abuse and dependence on prescription opioids among U.S. adults: psychiatric, medical and substance use correlates. Drug Alcohol Depend. 2008;94(1-3):38-47.
5. Han B, Compton WM, Jones CM, et al. Nonmedical prescription opioid use and use disorders among adults aged 18 through 64 years in the United States, 2003-2013. JAMA. 2015;314(14):1468-1478.
6. Centers for Disease Control and Prevention. National Center for Health Statistics, 2014. Multiple cause of death data. http://wonder.cdc.gov/mcd.html.
7. Twachtman G. Congress sends opioid legislation to the President. Clinical Psychiatry News. http://www.clinicalpsychiatrynews.com/?id=2407&tx_ttnews[tt_news]=524025&cHash=e93d5d1f86d20e53d3e2d8b07e9562b2. Published July 15, 2016. Accessed July 18, 2016.
8. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
9. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
10. McCabe SE, Cranford JA, West BT. Trends in prescription drug abuse and dependence, co-occurrence with other substance use disorders, and treatment utilization: results from two national surveys. Addict Behav. 2008;33(10):1297-1305.
11. Butler SF, Villapiano A, Malinow A. The effect of computer-mediated administration on self-disclosure of problems on the Addiction Severity Index. J Addict Med. 2009;3(4):194-203.
12. Meltzer EC, Rybin D, Saitz R, et al. Identifying prescription opioid use disorder in primary care: diagnostic characteristics of the Current Opioid Misuse Measure (COMM). Pain. 2011;152(2):397-402.
13. Butler SF, Budman SH, Fanciullo GJ, et al. Cross validation of the current opioid misuse measure to monitor chronic pain patients on opioid therapy. Clin J Pain. 2010;26(9):770-776.
14. Wesson DR, Ling W. The clinical opiate withdrawal scale (COWS). J Psychoactive Drugs. 2003;35(2):253-259.
15. Fiellin DA, O’Connor PG. Clinical practice. Office-based treatment of opioid-dependent patients. N Engl J Med. 2002;347(11):817-823.
16. Gowing LR, Farrell M, Ali RL, et al. α2‐Adrenergic agonists in opioid withdrawal. Addiction. 2002;97(1):49-58.
17. Loimer N, Hofmann P, Chaudhry H. Ultrashort noninvasive opiate detoxification. Am J Psychiatry. 1993;150(5):839.
18. Charney DS, Sternberg DE, Kleber HD, et al. The clinical use of clonidine in abrupt withdrawal from methadone. Effects on blood pressure and specific signs and symptoms. Arch Gen Psychiatry. 1981;38(11):1273-1277.
19. Preston KL, Bigelow GE, Liebson IA. Antagonist effects of nalbuphine in opioid-dependent human volunteers. J Pharmacol Exp Ther. 1989;248(3):929-937.
20. Preston KL, Bigelow GE, Liebson IA. Discrimination of butorphanol and nalbuphine in opioid-dependent humans. Pharmacol Biochem Behav. 1990;37(3):511-522.
21. Effective medical treatment of opiate addiction. National Consensus Development Panel on Effective Medical Treatment of Opiate Addiction. JAMA. 1998;280(22):1936-1943.
22. Amato L, Minozzi S, Davoli, M, et al. Psychosocial combined with agonist maintenance treatments versus agonist maintenance treatments alone for treatment of opioid dependence. Cochrane Database Syst Rev. 2011;(10):CD004147. doi: 10.1002/14651858.CD004147.pub4.
23. Obert JL, McCann MJ, Marinelli-Casey P, et al. The matrix model of outpatient stimulant abuse treatment: history and description. J Psychoactive Drugs. 2000;32(2):157-164.
24. Mayet S, Farrell M, Ferri M, et al. Psychosocial treatment for opiate abuse and dependence. Cochrane Database Syst Rev. 2005:CD004330.
25. McAuliffe WE. A randomized controlled trial of recovery training and self-help for opioid addicts in New England and Hong Kong. J Psychoactive Drugs. 1990;22(2):197-209.
26. Connery HS. Medication-assisted treatment of opioid use disorder: review of the evidence and future directions. Harv Rev Psychiatry. 2015;23(2):63-75.
27. Gibson AE, Degenhardt LJ. Mortality related to pharmacotherapies for opioid dependence: a comparative analysis of coronial records. Drug Alcohol Rev. 2007;26:405-410.
28. Clark L, Haram E, Johnson K, et al. Getting started with medication-assisted treatment with lessons from advancing recovery. Madison, WI: University of Wisconsin-Madison; 2010.
29. U.S. Food and Drug Administration. Vivitrol (naltrexone for extended-release injectable suspension): NDA 21-897C—Briefing document/background package. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Psychopharmacologic DrugsAdvisoryCommittee/UCM225664.pdf. Published September 16, 2010. Accessed July 11, 2016.
30. Providers Clinical Support System. PCSS Guidance. Buprenorphine induction. http://pcssmat.org/wp-content/uploads/2014/02/PCSS-MATGuidanceBuprenorphineInduction.Casadonte.pdf. Updated November 27, 2013. Accessed July 18, 2016.
31. An introduction to extended-release injectable naltrexone for the treatment of people with opioid dependence. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2012. HHS Publication No. (SMA) 12-4682.
32. Doehring A, von Hentig N, Graff J, et al. Genetic variants altering dopamine D2 receptor expression or function modulate the risk of opiate addiction and the dosage requirements of methadone substitution. Pharmacogenet Genomics. 2009;19(6):407-414.
33. Mitchell JL. Pregnant, substance-abusing women: treatment improvement protocol (TIP) Series 2. Rockville, MD: Substance Abuse and Mental Health Services Administration; 1993. DHHS Publication No. (SMA) 95-3056.
34. McCarthy JJ, Posey BL. Methadone levels in human milk. J Hum Lact. 2000;16(2):115-120.
35. Geraghty B, Graham EA, Logan B, et al. Methadone levels in breast milk. J Hum Lact. 1997;13(3):227-230.
36. Krambeer LL, von McKnelly W Jr, Gabrielli WF Jr, et al. Methadone therapy for opioid dependence. Am Fam Physician. 2001;63(12):2404-2410.
37. Novick DM, Joseph H, Croxson TS, et al. Absence of antibody to human immunodeficiency virus in long-term, socially rehabilitated methadone maintenance patients. Arch Intern Med. 1990;150(1):97-99.
38. Gowing LR, Farrell M, Bornemann R, et al. Brief report: methadone treatment of injecting opioid users for prevention of HIV infection. J Gen Intern Med. 2006;21(2):193-195.
39. Nurco DN, Ball JC, Shaffer JW, et al. The criminality of narcotic addicts. J Nerv Ment Dis. 1985;173(2):94-102.
40. Gibson A, Degenhardt L, Mattick RP, et al. Exposure to opioid maintenance treatment reduces long-term mortality. Addiction. 2008;103(3):462-468.
41. Pearson EC, Woosley RL. QT prolongation and torsades de pointes among methadone users: reports to the FDA spontaneous reporting system. Pharmacoepidemiol Drug Saf. 2005;14(11):747-753.
42. Bell JR, Butler B, Lawrance A, et al. Comparing overdose mortality associated with methadone and buprenorphine treatment. Drug Alcohol Depend. 2009;104(1-2):73-77.
43. Bickel WK, Amass L. Buprenorphine treatment of opioid dependence: a review. Experimental and Clinical Psychopharmacology. 1995;3(4):477-489.
44. U.S. Food and Drug Administration. FDA approves first buprenorphine implant for treatment of opioid dependence. http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm503719.htm. Published May 26, 2016. Accessed July 18, 2016.
45. The National Alliance of Advocates for Buprenorphine Treatment. https://www.naabt.org/index.cfm. Accessed July 18, 2016.
46. Buprenorphine: an alternative to methadone. Med Lett Drugs Ther. 2003;45(1150):13-15.
47. Sigmon SC, Dunn KE, Saulsgiver K, et al. A randomized, double-blind evaluation of buprenorphine taper duration in primary prescription opioid abusers. JAMA Psychiatry. 2013;70(12):1347-1354.
48. Dahan A, Yassen A, Bijl H, et al. Comparison of the respiratory effects of intravenous buprenorphine and fentanyl in humans and rats. Br J Anaesth. 2005;94(6):825-834.
49. Bell JR, Butler B, Lawrance A, et al. Comparing overdose mortality associated with methadone and buprenorphine treatment. Drug Alcohol Depend. 2009;104(1-2):73-77.
50. Kakko J, Heilig M, Sarman I. Buprenorphine and methadone treatment of opiate dependence during pregnancy: comparison of fetal growth and neonatal outcomes in two consecutive case series. Drug Alcohol Depend. 2008;96(1-2):69-78.
51. Stotts AL, Dodrill CL, Kosten TR. Opioid dependence treatment: options in pharmacotherapy. Expert Opin Pharmacother. 2009;10(11):1727-1740.
52. Robinson GM, Dukes PD, Robinson BJ, et al. The misuse of buprenorphine and a buprenorphine-naloxone combination in Wellington, New Zealand. Drug Alcohol Depend. 1993;33(1):81-86.
53. Fudala PJ, Bridge TP, Herbert S, et al; Buprenorphine/Naloxone Collaborative Study Group. Office-based treatment of opiate addiction with a sublingual-tablet formulation of buprenorphine and naloxone. N Engl J Med. 2003;349(10):949-958.

Unresolved questions about the specialty lurk in the cortex of psychiatrists
But many of our own questions await an answer. The fact is that psychiatrists have serious, nagging questions—in every cortical fold of their collective brain—about patients’ welfare, psychiatric practice, and professional matters. Their questions about frustrations of daily practice deserve an honest and convincing response, yet go begging—expressed so well in songwriter Bob Dylan’s lyric, “The answer is blowin’ in the wind.”
What follows are long-standing “Why?” questions whose answers are still blowin’ in the wind. (Dylan didn’t specify which wind is blowin’, so I’ve provided the names of 22 atmospheric movements of air molecules in the Box. Take your pick!)

Why is a jail OK for the mentally ill but an asylum is not? Why is it necessary to put armed guards in charge of psychiatric patients instead of a multidisciplinary team of psychiatrists, primary care providers, nurses, social workers, psychologists, and pharmacists? Why has a brain disease, such as psychosis or bipolar disorder, become a punishable felony instead of a treatable illness?
Why did the system of mental health care degenerate to the point that a severely depressed or suicidal, or acutely psychotic, patient can be hospitalized for only 4 or 5 days, then must be discharged before her (his) illness has been fully controlled? Why do health care insurers exhibit that atrocious combination of maximum greed and minimal compassion?
Why does a completely unjustified and hurtful stigma continue to plague mental brain disorders, patients who suffer from them, mental health professionals, and the very discipline of psychiatry?
Why do otherwise intelligent people show compassion toward people with a brain disorder such as stroke, Parkinson’s disease, multiple sclerosis, myasthenia gravis, or migraine, but express aversion and even disdain for psychiatric brain disorders such as schizophrenia, depression, obsessive-compulsive disorder, and panic disorder?
And why does this prejudice persist despite advances in psychiatric neuroscience that have used neurogenetics, neuroimaging, and molecular studies to establish, without a doubt, the neurobiological basis of all psychiatric disorders.
Why are there still no objective diagnostic criteria for psychiatric disorders? Why do we persist in using defining symptoms that have been volunteered by patients—symptoms that can be subject to distortion or malingering? Why aren’t the hundreds of established biomarkers being incorporated into the diagnostic formulation, to lessen subjectivity and improve reliability and validity?
Why is off-label prescribing, the judicious clinical repurposing of psychotropic medications, criticized and panned, even though there are no approved drugs for 88.5% psychiatric diagnoses?1 Why allow insurers to refuse to pay for a medication that can help a patient, just because the patient has not been given the “official” diagnosis for which the FDA approved that drug?
And why doesn’t the FDA solve this problem by revising its requirements that registration trials for new medications test their efficacy for a single symptom, rather than a diagnosis comprising multiple symptoms?
Why do people not accept the fact that all drugs have benefits and risks, and that it is impossible to have pure efficacy without side effects? Why empower lawyers to make clinical care adversarial? Why do lawyers refrain from suing oncologists or manufacturers of life-saving chemotherapy drugs because of terrible adverse effects, but pounce on other medications that might cause a serious side effect in a tiny percentage of patients that is clearly spelled out in the package insert?
Why do people demonize the pharmaceutical industry far more than other industries? No other entity discovers and develops life-saving medications.
Why don’t people realize that, without medications, massive numbers of patients would be hospitalized and the death rate would rise? Why can’t people weigh risks and benefits of having a pharmaceutical industry, just as they assess the risk-benefit ratio of everything in life?
Should the government impose a massive ($1 or $2 trillion) tax hike to establish infrastructure for drug research and development, for the benefit of psychiatry and all other medical specialties?
Why is there a severe shortage of psychiatrists but a glut of lawyers? Why doesn’t society rationally deploy its resources to meet urgent social needs and priorities? And why do lawyers bill us for every minute we talk to them, while we field telephone calls and e-mail messages from patients without compensation?
Why did the FDA allow the pharmaceutical industry to develop direct-to-consumer advertising? Why do they not realize how that decision has complicated the doctor–patient relationship, and how it preempts physicians’ evidence-based decision-making by encouraging consumers to demand a drug that they saw on television—a contorted version of prescribing by proxy?
Why (speaking of prescribing without a license), do politicians pass laws allowing people who do not have required medical training to take a short-cut to becoming a prescriber? Why not mandate that politicians, and their families, receive medical care exclusively from unqualified practitioners on whom they bestow prescribing privileges without requisite comprehensive medical training?Why do some psychiatrists resist changing their practice patterns despite continuous advances that update the care they provide? Why do reports of exciting therapeutic breakthroughs, published in top-tier journals, go unread by so many practitioners? Why do they say they are too busy to read journals or peruse PubMed?
Why don’t people realize that today’s research is tomorrow’s treatment? That research is not a luxury but an ongoing necessity? Why don’t more freshly minted, young psychiatrists pursue a career in research to accelerate the pace of progress about the biological causes and treatments of serious psychiatric disorders? Why aren’t there more incentives to grow the next generation of psychiatric discoverers and Nobel laureates? Why don’t clinicians support research by referring patients to clinical trials of medications or to National Institutes of Health-funded investigations of the neurobiology of psychiatric disorders?
Are these just rhetorical questions?
Some might sound that way. But they are not. These questions are brewing inside the hearts and minds of many psychiatrists, although only a few seem determined to relentlessly seek answers on which medical science and society can act.
We should collectively pose these “why” questions and not accept long-winded, hollow answers. We need to foster the winds of change—not resign ourselves to winds in which answers blow about but, ultimately, disappear.
1. Devulapalli KK, Nasrallah HA. An analysis of the high psychotropic off-label use in psychiatric disorders. Asian J Psychiatr. 2009;2(1):29-36.
But many of our own questions await an answer. The fact is that psychiatrists have serious, nagging questions—in every cortical fold of their collective brain—about patients’ welfare, psychiatric practice, and professional matters. Their questions about frustrations of daily practice deserve an honest and convincing response, yet go begging—expressed so well in songwriter Bob Dylan’s lyric, “The answer is blowin’ in the wind.”
What follows are long-standing “Why?” questions whose answers are still blowin’ in the wind. (Dylan didn’t specify which wind is blowin’, so I’ve provided the names of 22 atmospheric movements of air molecules in the Box. Take your pick!)
Why is a jail OK for the mentally ill but an asylum is not? Why is it necessary to put armed guards in charge of psychiatric patients instead of a multidisciplinary team of psychiatrists, primary care providers, nurses, social workers, psychologists, and pharmacists? Why has a brain disease, such as psychosis or bipolar disorder, become a punishable felony instead of a treatable illness?
Why did the system of mental health care degenerate to the point that a severely depressed or suicidal, or acutely psychotic, patient can be hospitalized for only 4 or 5 days, then must be discharged before her (his) illness has been fully controlled? Why do health care insurers exhibit that atrocious combination of maximum greed and minimal compassion?
Why does a completely unjustified and hurtful stigma continue to plague mental brain disorders, patients who suffer from them, mental health professionals, and the very discipline of psychiatry?
Why do otherwise intelligent people show compassion toward people with a brain disorder such as stroke, Parkinson’s disease, multiple sclerosis, myasthenia gravis, or migraine, but express aversion and even disdain for psychiatric brain disorders such as schizophrenia, depression, obsessive-compulsive disorder, and panic disorder?
And why does this prejudice persist despite advances in psychiatric neuroscience that have used neurogenetics, neuroimaging, and molecular studies to establish, without a doubt, the neurobiological basis of all psychiatric disorders.
Why are there still no objective diagnostic criteria for psychiatric disorders? Why do we persist in using defining symptoms that have been volunteered by patients—symptoms that can be subject to distortion or malingering? Why aren’t the hundreds of established biomarkers being incorporated into the diagnostic formulation, to lessen subjectivity and improve reliability and validity?
Why is off-label prescribing, the judicious clinical repurposing of psychotropic medications, criticized and panned, even though there are no approved drugs for 88.5% psychiatric diagnoses?1 Why allow insurers to refuse to pay for a medication that can help a patient, just because the patient has not been given the “official” diagnosis for which the FDA approved that drug?
And why doesn’t the FDA solve this problem by revising its requirements that registration trials for new medications test their efficacy for a single symptom, rather than a diagnosis comprising multiple symptoms?
Why do people not accept the fact that all drugs have benefits and risks, and that it is impossible to have pure efficacy without side effects? Why empower lawyers to make clinical care adversarial? Why do lawyers refrain from suing oncologists or manufacturers of life-saving chemotherapy drugs because of terrible adverse effects, but pounce on other medications that might cause a serious side effect in a tiny percentage of patients that is clearly spelled out in the package insert?
Why do people demonize the pharmaceutical industry far more than other industries? No other entity discovers and develops life-saving medications.
Why don’t people realize that, without medications, massive numbers of patients would be hospitalized and the death rate would rise? Why can’t people weigh risks and benefits of having a pharmaceutical industry, just as they assess the risk-benefit ratio of everything in life?
Should the government impose a massive ($1 or $2 trillion) tax hike to establish infrastructure for drug research and development, for the benefit of psychiatry and all other medical specialties?
Why is there a severe shortage of psychiatrists but a glut of lawyers? Why doesn’t society rationally deploy its resources to meet urgent social needs and priorities? And why do lawyers bill us for every minute we talk to them, while we field telephone calls and e-mail messages from patients without compensation?
Why did the FDA allow the pharmaceutical industry to develop direct-to-consumer advertising? Why do they not realize how that decision has complicated the doctor–patient relationship, and how it preempts physicians’ evidence-based decision-making by encouraging consumers to demand a drug that they saw on television—a contorted version of prescribing by proxy?
Why (speaking of prescribing without a license), do politicians pass laws allowing people who do not have required medical training to take a short-cut to becoming a prescriber? Why not mandate that politicians, and their families, receive medical care exclusively from unqualified practitioners on whom they bestow prescribing privileges without requisite comprehensive medical training?Why do some psychiatrists resist changing their practice patterns despite continuous advances that update the care they provide? Why do reports of exciting therapeutic breakthroughs, published in top-tier journals, go unread by so many practitioners? Why do they say they are too busy to read journals or peruse PubMed?
Why don’t people realize that today’s research is tomorrow’s treatment? That research is not a luxury but an ongoing necessity? Why don’t more freshly minted, young psychiatrists pursue a career in research to accelerate the pace of progress about the biological causes and treatments of serious psychiatric disorders? Why aren’t there more incentives to grow the next generation of psychiatric discoverers and Nobel laureates? Why don’t clinicians support research by referring patients to clinical trials of medications or to National Institutes of Health-funded investigations of the neurobiology of psychiatric disorders?
Are these just rhetorical questions?
Some might sound that way. But they are not. These questions are brewing inside the hearts and minds of many psychiatrists, although only a few seem determined to relentlessly seek answers on which medical science and society can act.
We should collectively pose these “why” questions and not accept long-winded, hollow answers. We need to foster the winds of change—not resign ourselves to winds in which answers blow about but, ultimately, disappear.
But many of our own questions await an answer. The fact is that psychiatrists have serious, nagging questions—in every cortical fold of their collective brain—about patients’ welfare, psychiatric practice, and professional matters. Their questions about frustrations of daily practice deserve an honest and convincing response, yet go begging—expressed so well in songwriter Bob Dylan’s lyric, “The answer is blowin’ in the wind.”
What follows are long-standing “Why?” questions whose answers are still blowin’ in the wind. (Dylan didn’t specify which wind is blowin’, so I’ve provided the names of 22 atmospheric movements of air molecules in the Box. Take your pick!)
Why is a jail OK for the mentally ill but an asylum is not? Why is it necessary to put armed guards in charge of psychiatric patients instead of a multidisciplinary team of psychiatrists, primary care providers, nurses, social workers, psychologists, and pharmacists? Why has a brain disease, such as psychosis or bipolar disorder, become a punishable felony instead of a treatable illness?
Why did the system of mental health care degenerate to the point that a severely depressed or suicidal, or acutely psychotic, patient can be hospitalized for only 4 or 5 days, then must be discharged before her (his) illness has been fully controlled? Why do health care insurers exhibit that atrocious combination of maximum greed and minimal compassion?
Why does a completely unjustified and hurtful stigma continue to plague mental brain disorders, patients who suffer from them, mental health professionals, and the very discipline of psychiatry?
Why do otherwise intelligent people show compassion toward people with a brain disorder such as stroke, Parkinson’s disease, multiple sclerosis, myasthenia gravis, or migraine, but express aversion and even disdain for psychiatric brain disorders such as schizophrenia, depression, obsessive-compulsive disorder, and panic disorder?
And why does this prejudice persist despite advances in psychiatric neuroscience that have used neurogenetics, neuroimaging, and molecular studies to establish, without a doubt, the neurobiological basis of all psychiatric disorders.
Why are there still no objective diagnostic criteria for psychiatric disorders? Why do we persist in using defining symptoms that have been volunteered by patients—symptoms that can be subject to distortion or malingering? Why aren’t the hundreds of established biomarkers being incorporated into the diagnostic formulation, to lessen subjectivity and improve reliability and validity?
Why is off-label prescribing, the judicious clinical repurposing of psychotropic medications, criticized and panned, even though there are no approved drugs for 88.5% psychiatric diagnoses?1 Why allow insurers to refuse to pay for a medication that can help a patient, just because the patient has not been given the “official” diagnosis for which the FDA approved that drug?
And why doesn’t the FDA solve this problem by revising its requirements that registration trials for new medications test their efficacy for a single symptom, rather than a diagnosis comprising multiple symptoms?
Why do people not accept the fact that all drugs have benefits and risks, and that it is impossible to have pure efficacy without side effects? Why empower lawyers to make clinical care adversarial? Why do lawyers refrain from suing oncologists or manufacturers of life-saving chemotherapy drugs because of terrible adverse effects, but pounce on other medications that might cause a serious side effect in a tiny percentage of patients that is clearly spelled out in the package insert?
Why do people demonize the pharmaceutical industry far more than other industries? No other entity discovers and develops life-saving medications.
Why don’t people realize that, without medications, massive numbers of patients would be hospitalized and the death rate would rise? Why can’t people weigh risks and benefits of having a pharmaceutical industry, just as they assess the risk-benefit ratio of everything in life?
Should the government impose a massive ($1 or $2 trillion) tax hike to establish infrastructure for drug research and development, for the benefit of psychiatry and all other medical specialties?
Why is there a severe shortage of psychiatrists but a glut of lawyers? Why doesn’t society rationally deploy its resources to meet urgent social needs and priorities? And why do lawyers bill us for every minute we talk to them, while we field telephone calls and e-mail messages from patients without compensation?
Why did the FDA allow the pharmaceutical industry to develop direct-to-consumer advertising? Why do they not realize how that decision has complicated the doctor–patient relationship, and how it preempts physicians’ evidence-based decision-making by encouraging consumers to demand a drug that they saw on television—a contorted version of prescribing by proxy?
Why (speaking of prescribing without a license), do politicians pass laws allowing people who do not have required medical training to take a short-cut to becoming a prescriber? Why not mandate that politicians, and their families, receive medical care exclusively from unqualified practitioners on whom they bestow prescribing privileges without requisite comprehensive medical training?Why do some psychiatrists resist changing their practice patterns despite continuous advances that update the care they provide? Why do reports of exciting therapeutic breakthroughs, published in top-tier journals, go unread by so many practitioners? Why do they say they are too busy to read journals or peruse PubMed?
Why don’t people realize that today’s research is tomorrow’s treatment? That research is not a luxury but an ongoing necessity? Why don’t more freshly minted, young psychiatrists pursue a career in research to accelerate the pace of progress about the biological causes and treatments of serious psychiatric disorders? Why aren’t there more incentives to grow the next generation of psychiatric discoverers and Nobel laureates? Why don’t clinicians support research by referring patients to clinical trials of medications or to National Institutes of Health-funded investigations of the neurobiology of psychiatric disorders?
Are these just rhetorical questions?
Some might sound that way. But they are not. These questions are brewing inside the hearts and minds of many psychiatrists, although only a few seem determined to relentlessly seek answers on which medical science and society can act.
We should collectively pose these “why” questions and not accept long-winded, hollow answers. We need to foster the winds of change—not resign ourselves to winds in which answers blow about but, ultimately, disappear.
1. Devulapalli KK, Nasrallah HA. An analysis of the high psychotropic off-label use in psychiatric disorders. Asian J Psychiatr. 2009;2(1):29-36.
1. Devulapalli KK, Nasrallah HA. An analysis of the high psychotropic off-label use in psychiatric disorders. Asian J Psychiatr. 2009;2(1):29-36.

CAR T-cell therapy eyed for CLL patients with residual disease
Four of eight patients with residual chronic lymphocytic leukemia (CLL) following initial chemotherapy had complete or partial responses to an outpatient therapy that used autologous T cells genetically targeted to the B cell–specific antigen CD19, Mark Blaine Geyer, MD, of Memorial Sloan Kettering Cancer Center, New York, reported at the annual meeting of the American Society of Clinical Oncology.
The therapy employing T cells genetically modified to express CD19-targeted 19-28z chimeric antigen receptors (CARs) was well tolerated but had limited observed efficacy, especially in patients with enlarged lymph nodes. The study goal was to find a safe dose of modified T cells for patients who have disease remaining after initial chemotherapy.
For the phase I dose escalation study (NCT01416974), Dr. Geyer and his associates enrolled eight CLL patients who had residual disease after upfront therapy consisting of six cycles of pentostatin, cyclophosphamide, and rituximab.
Five patients had clearly enlarged lymph nodes prior to T cell infusion.
Patients received cyclophosphamide 600 mg/m2 followed 2 days later by escalating doses of 19-28z T cells. Four of the five patients who received at least a 1 × 107 dose of 19-28z T cells/kg were admitted with fevers and mild cytokine release syndrome.
Maximal levels of CAR T cell persistence were detected at 8 weeks. With a median patient follow-up of 32 months, clinical complete response has been seen in two patients, partial response in two patients, and stable disease in one patient. Disease has progressed in three patients: one had a rising absolute lymphocyte count by the time of infusion and two had marrow response with progressive disease in lymph nodes. The median time to disease progression was 13.6 months, Dr. Geyer said.
Five of seven evaluable patients have received further CLL-directed therapy.
The researchers speculated that low-dose cyclophosphamide monotherapy, used before the CAR T-cell therapy, may be insufficient for lymphodepletion. Additionally, CAR T cell expansion and antitumor efficacy may be limited by a hostile CLL microenvironment. Strategies to enhance CAR T cell expansion and efficacy in patients with CLL are in preparation, Dr. Geyer reported.
Dr. Geyer had no financial disclosures. His colleagues reported various financial relationships with Juno Therapeutics, a developer of CAR technology.
On Twitter @maryjodales
Four of eight patients with residual chronic lymphocytic leukemia (CLL) following initial chemotherapy had complete or partial responses to an outpatient therapy that used autologous T cells genetically targeted to the B cell–specific antigen CD19, Mark Blaine Geyer, MD, of Memorial Sloan Kettering Cancer Center, New York, reported at the annual meeting of the American Society of Clinical Oncology.
The therapy employing T cells genetically modified to express CD19-targeted 19-28z chimeric antigen receptors (CARs) was well tolerated but had limited observed efficacy, especially in patients with enlarged lymph nodes. The study goal was to find a safe dose of modified T cells for patients who have disease remaining after initial chemotherapy.
For the phase I dose escalation study (NCT01416974), Dr. Geyer and his associates enrolled eight CLL patients who had residual disease after upfront therapy consisting of six cycles of pentostatin, cyclophosphamide, and rituximab.
Five patients had clearly enlarged lymph nodes prior to T cell infusion.
Patients received cyclophosphamide 600 mg/m2 followed 2 days later by escalating doses of 19-28z T cells. Four of the five patients who received at least a 1 × 107 dose of 19-28z T cells/kg were admitted with fevers and mild cytokine release syndrome.
Maximal levels of CAR T cell persistence were detected at 8 weeks. With a median patient follow-up of 32 months, clinical complete response has been seen in two patients, partial response in two patients, and stable disease in one patient. Disease has progressed in three patients: one had a rising absolute lymphocyte count by the time of infusion and two had marrow response with progressive disease in lymph nodes. The median time to disease progression was 13.6 months, Dr. Geyer said.
Five of seven evaluable patients have received further CLL-directed therapy.
The researchers speculated that low-dose cyclophosphamide monotherapy, used before the CAR T-cell therapy, may be insufficient for lymphodepletion. Additionally, CAR T cell expansion and antitumor efficacy may be limited by a hostile CLL microenvironment. Strategies to enhance CAR T cell expansion and efficacy in patients with CLL are in preparation, Dr. Geyer reported.
Dr. Geyer had no financial disclosures. His colleagues reported various financial relationships with Juno Therapeutics, a developer of CAR technology.
On Twitter @maryjodales
Four of eight patients with residual chronic lymphocytic leukemia (CLL) following initial chemotherapy had complete or partial responses to an outpatient therapy that used autologous T cells genetically targeted to the B cell–specific antigen CD19, Mark Blaine Geyer, MD, of Memorial Sloan Kettering Cancer Center, New York, reported at the annual meeting of the American Society of Clinical Oncology.
The therapy employing T cells genetically modified to express CD19-targeted 19-28z chimeric antigen receptors (CARs) was well tolerated but had limited observed efficacy, especially in patients with enlarged lymph nodes. The study goal was to find a safe dose of modified T cells for patients who have disease remaining after initial chemotherapy.
For the phase I dose escalation study (NCT01416974), Dr. Geyer and his associates enrolled eight CLL patients who had residual disease after upfront therapy consisting of six cycles of pentostatin, cyclophosphamide, and rituximab.
Five patients had clearly enlarged lymph nodes prior to T cell infusion.
Patients received cyclophosphamide 600 mg/m2 followed 2 days later by escalating doses of 19-28z T cells. Four of the five patients who received at least a 1 × 107 dose of 19-28z T cells/kg were admitted with fevers and mild cytokine release syndrome.
Maximal levels of CAR T cell persistence were detected at 8 weeks. With a median patient follow-up of 32 months, clinical complete response has been seen in two patients, partial response in two patients, and stable disease in one patient. Disease has progressed in three patients: one had a rising absolute lymphocyte count by the time of infusion and two had marrow response with progressive disease in lymph nodes. The median time to disease progression was 13.6 months, Dr. Geyer said.
Five of seven evaluable patients have received further CLL-directed therapy.
The researchers speculated that low-dose cyclophosphamide monotherapy, used before the CAR T-cell therapy, may be insufficient for lymphodepletion. Additionally, CAR T cell expansion and antitumor efficacy may be limited by a hostile CLL microenvironment. Strategies to enhance CAR T cell expansion and efficacy in patients with CLL are in preparation, Dr. Geyer reported.
Dr. Geyer had no financial disclosures. His colleagues reported various financial relationships with Juno Therapeutics, a developer of CAR technology.
On Twitter @maryjodales
FROM THE 2016 ASCO ANNUAL MEETING
Key clinical point: CAR T-cell therapy may be an option for chronic lymphocytic leukemia patients with residual disease after upfront therapy.
Major finding: Four of eight patients with residual CLL following initial chemotherapy had complete or partial responses to an outpatient therapy that used autologous T cells genetically targeted to the B cell–specific antigen CD19.
Data source: A phase I dose-finding and efficacy study in 8 patients with CLL.
Disclosures: Dr. Geyer had no financial disclosures. His colleagues reported various financial relationships with Juno Therapeutics, a developer of CAR technology.
Tips for Communicating with Empathy
Editor’s note: “Everything We Say and Do” is an informational series developed by SHM’s Patient Experience Committee to provide readers with thoughtful and actionable communication tactics that have great potential to positively impact patients’ experience of care. Each column will focus on how the contributor applies one of the “Key Communication” areas in practice.
View a chart outlining key communication tactics
What I Say and Do

In interactions with patients and families, I make sure I communicate with empathy. By communicating with empathy, I mean not only listening for and understanding a patient’s experiences, concerns, and perspective but also communicating this understanding with my intention to help.
Why I Do It
Time constraints, endless to-do lists, and racing minds can eclipse empathic, attentive, and personalized care. When empathy is missing from the patient-clinician relationship, patients and clinicians suffer. Patients feel disengaged from their clinician; they remain anxious and lose trust. And physicians miss out on the gratification of feeling connected with patients and on achieving the best possible patient engagement and outcomes.
Physician empathy is associated with not only higher levels of patient satisfaction and survey scores but also with patient engagement, adherence to care plans, and positive health outcomes as well as physician job satisfaction.1–3
How I Do It
I start with mindfulness. I sustain eye contact, sit eye to eye, and give the person my undivided attention, listening to their words and nonverbal behavior—without judgment.
Then I draw on several techniques that express empathy. My favorites are these:

On a Personal Note
Albert Schweitzer said, “At times, our own light goes out and is rekindled by a spark from another person. Each of us has cause to think with deep gratitude of those who have lighted the flame within us.” Communicating with empathy, to me, is the spark that rekindles the lights of patients, families, and colleagues in our relationships with them. TH
Wendy Leebov is founder and partner in Language of Caring, LLC, author of The Language of Caring Guide for Physicians, and developer of the Language of Caring for Physicians web-based learning program. Reach her at [email protected].
References
- Leebov W, Rotering C. The Language of Caring Guide for Physicians: Communication Essentials for Patient-Centered Care. 2nd ed. Language of Caring, LLC; 2015.
- Butterfield S. New research links empathy to outcomes. ACP Internist website. Available at: http://www.acpinternist.org/archives/2013/03/empathy.htm. Accessed July 8, 2016.
- Hojat M, Louis D, Maio V, Gonnella J. Empathy and health care quality. Am J Medical Quality. 2013;28(1):6-7.
Editor’s note: “Everything We Say and Do” is an informational series developed by SHM’s Patient Experience Committee to provide readers with thoughtful and actionable communication tactics that have great potential to positively impact patients’ experience of care. Each column will focus on how the contributor applies one of the “Key Communication” areas in practice.
View a chart outlining key communication tactics
What I Say and Do
In interactions with patients and families, I make sure I communicate with empathy. By communicating with empathy, I mean not only listening for and understanding a patient’s experiences, concerns, and perspective but also communicating this understanding with my intention to help.
Why I Do It
Time constraints, endless to-do lists, and racing minds can eclipse empathic, attentive, and personalized care. When empathy is missing from the patient-clinician relationship, patients and clinicians suffer. Patients feel disengaged from their clinician; they remain anxious and lose trust. And physicians miss out on the gratification of feeling connected with patients and on achieving the best possible patient engagement and outcomes.
Physician empathy is associated with not only higher levels of patient satisfaction and survey scores but also with patient engagement, adherence to care plans, and positive health outcomes as well as physician job satisfaction.1–3
How I Do It
I start with mindfulness. I sustain eye contact, sit eye to eye, and give the person my undivided attention, listening to their words and nonverbal behavior—without judgment.
Then I draw on several techniques that express empathy. My favorites are these:
On a Personal Note
Albert Schweitzer said, “At times, our own light goes out and is rekindled by a spark from another person. Each of us has cause to think with deep gratitude of those who have lighted the flame within us.” Communicating with empathy, to me, is the spark that rekindles the lights of patients, families, and colleagues in our relationships with them. TH
Wendy Leebov is founder and partner in Language of Caring, LLC, author of The Language of Caring Guide for Physicians, and developer of the Language of Caring for Physicians web-based learning program. Reach her at [email protected].
References
- Leebov W, Rotering C. The Language of Caring Guide for Physicians: Communication Essentials for Patient-Centered Care. 2nd ed. Language of Caring, LLC; 2015.
- Butterfield S. New research links empathy to outcomes. ACP Internist website. Available at: http://www.acpinternist.org/archives/2013/03/empathy.htm. Accessed July 8, 2016.
- Hojat M, Louis D, Maio V, Gonnella J. Empathy and health care quality. Am J Medical Quality. 2013;28(1):6-7.
Editor’s note: “Everything We Say and Do” is an informational series developed by SHM’s Patient Experience Committee to provide readers with thoughtful and actionable communication tactics that have great potential to positively impact patients’ experience of care. Each column will focus on how the contributor applies one of the “Key Communication” areas in practice.
View a chart outlining key communication tactics
What I Say and Do
In interactions with patients and families, I make sure I communicate with empathy. By communicating with empathy, I mean not only listening for and understanding a patient’s experiences, concerns, and perspective but also communicating this understanding with my intention to help.
Why I Do It
Time constraints, endless to-do lists, and racing minds can eclipse empathic, attentive, and personalized care. When empathy is missing from the patient-clinician relationship, patients and clinicians suffer. Patients feel disengaged from their clinician; they remain anxious and lose trust. And physicians miss out on the gratification of feeling connected with patients and on achieving the best possible patient engagement and outcomes.
Physician empathy is associated with not only higher levels of patient satisfaction and survey scores but also with patient engagement, adherence to care plans, and positive health outcomes as well as physician job satisfaction.1–3
How I Do It
I start with mindfulness. I sustain eye contact, sit eye to eye, and give the person my undivided attention, listening to their words and nonverbal behavior—without judgment.
Then I draw on several techniques that express empathy. My favorites are these:
On a Personal Note
Albert Schweitzer said, “At times, our own light goes out and is rekindled by a spark from another person. Each of us has cause to think with deep gratitude of those who have lighted the flame within us.” Communicating with empathy, to me, is the spark that rekindles the lights of patients, families, and colleagues in our relationships with them. TH
Wendy Leebov is founder and partner in Language of Caring, LLC, author of The Language of Caring Guide for Physicians, and developer of the Language of Caring for Physicians web-based learning program. Reach her at [email protected].
References
- Leebov W, Rotering C. The Language of Caring Guide for Physicians: Communication Essentials for Patient-Centered Care. 2nd ed. Language of Caring, LLC; 2015.
- Butterfield S. New research links empathy to outcomes. ACP Internist website. Available at: http://www.acpinternist.org/archives/2013/03/empathy.htm. Accessed July 8, 2016.
- Hojat M, Louis D, Maio V, Gonnella J. Empathy and health care quality. Am J Medical Quality. 2013;28(1):6-7.
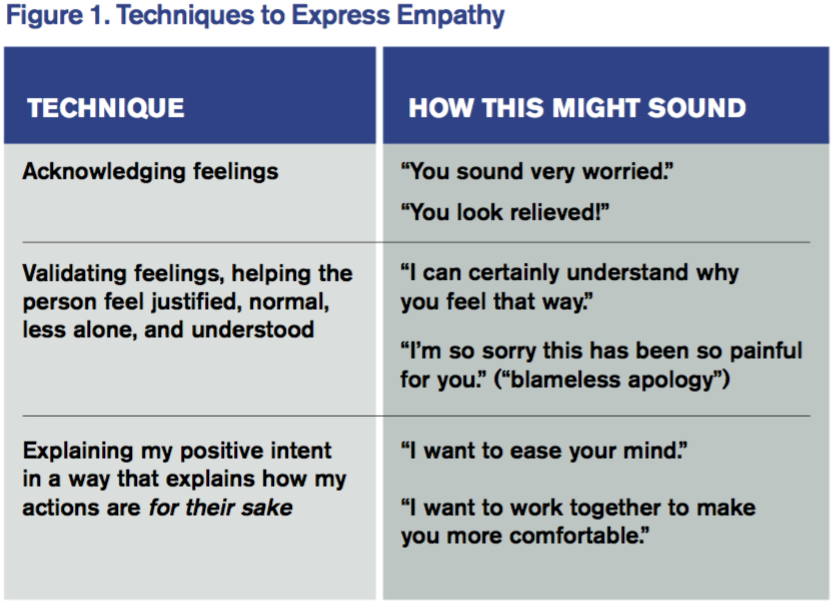
Patients can safely receive less FVIII, study suggests

ORLANDO—It may be possible for hemophilia patients to receive less factor VIII (FVIII) without increasing their risk of bleeding, according to a study presented at the World Federation of Hemophilia 2016 World Congress.
The study1 showed that hemophilia A patients who received prophylactic FVIII from the home infusion provider Option Care received 6 fewer units of FVIII per week than patients receiving prophylactic FVIII from specialty pharmacies.
This translated to a cost savings of more than $20,000 per patient each year.
The home infusion patients also had a lower annual bleed rate (ABR) than what is typically observed with intensive prophylaxis protocols in hemophilia, according to previous studies.
“By working with prescribers to closely monitor bleeds and collaborate on clinically appropriate optimization of treatment dose, Option Care’s utilization of factor VIII is less than the average with excellent outcomes,” said Joan Couden, RN, national program director for Option Care’s Bleeding Disorders Program.
“Our findings show we can save payers, including Medicare, Medicaid, and managed care insurers, significant costs without negatively impacting annual bleed rates.”
For this study, Couden and her colleagues conducted a retrospective analysis using dispensing data records from Option Care spanning the period from July 2015 through December 2015. The team compared these data to aggregate specialty pharmacy records from November 2013 through March 2014, which were analyzed in a previous study.2
In both data sets, patients receiving any FVIII product for prophylactic therapy were included. Patients being treated episodically or for immune tolerance induction were excluded, as were patients with extremely abnormal weights (40% below the 5th percentile or 40% above the 95th percentile based on weight-for-age charts from the US Centers for Disease Control and Prevention).
The researchers calculated a patient’s weekly dose of FVIII by multiplying the prescribed infusion dose by the dose frequency and dividing the product by the patient’s weight. Patients with an overall mean weekly dose greater than 2 standard deviations from the mean were excluded.
There were 77 home infusion patients and 520 specialty pharmacy patients.
The home infusion patients had a mean FVIII dose of 102 units/kg/week, compared to a mean of 108 units/kg/week for the specialty pharmacy patients.
This difference translates to savings of $21,166 per patient per year among the home infusion patients.
Couden and her colleagues could not compare the ABR between the 2 data sets because the ABR was not measured in the specialty pharmacy patients. However, they said the ABR in the home infusion patients was favorable when compared to ABRs in published studies.
The mean ABR for the home infusion patients was 1.70. And, according to a recent review of research on hemophilia treatment strategies, mean ABRs range from 2 to 5 for intensive treatment protocols.3![]()
ORLANDO—It may be possible for hemophilia patients to receive less factor VIII (FVIII) without increasing their risk of bleeding, according to a study presented at the World Federation of Hemophilia 2016 World Congress.
The study1 showed that hemophilia A patients who received prophylactic FVIII from the home infusion provider Option Care received 6 fewer units of FVIII per week than patients receiving prophylactic FVIII from specialty pharmacies.
This translated to a cost savings of more than $20,000 per patient each year.
The home infusion patients also had a lower annual bleed rate (ABR) than what is typically observed with intensive prophylaxis protocols in hemophilia, according to previous studies.
“By working with prescribers to closely monitor bleeds and collaborate on clinically appropriate optimization of treatment dose, Option Care’s utilization of factor VIII is less than the average with excellent outcomes,” said Joan Couden, RN, national program director for Option Care’s Bleeding Disorders Program.
“Our findings show we can save payers, including Medicare, Medicaid, and managed care insurers, significant costs without negatively impacting annual bleed rates.”
For this study, Couden and her colleagues conducted a retrospective analysis using dispensing data records from Option Care spanning the period from July 2015 through December 2015. The team compared these data to aggregate specialty pharmacy records from November 2013 through March 2014, which were analyzed in a previous study.2
In both data sets, patients receiving any FVIII product for prophylactic therapy were included. Patients being treated episodically or for immune tolerance induction were excluded, as were patients with extremely abnormal weights (40% below the 5th percentile or 40% above the 95th percentile based on weight-for-age charts from the US Centers for Disease Control and Prevention).
The researchers calculated a patient’s weekly dose of FVIII by multiplying the prescribed infusion dose by the dose frequency and dividing the product by the patient’s weight. Patients with an overall mean weekly dose greater than 2 standard deviations from the mean were excluded.
There were 77 home infusion patients and 520 specialty pharmacy patients.
The home infusion patients had a mean FVIII dose of 102 units/kg/week, compared to a mean of 108 units/kg/week for the specialty pharmacy patients.
This difference translates to savings of $21,166 per patient per year among the home infusion patients.
Couden and her colleagues could not compare the ABR between the 2 data sets because the ABR was not measured in the specialty pharmacy patients. However, they said the ABR in the home infusion patients was favorable when compared to ABRs in published studies.
The mean ABR for the home infusion patients was 1.70. And, according to a recent review of research on hemophilia treatment strategies, mean ABRs range from 2 to 5 for intensive treatment protocols.3![]()
ORLANDO—It may be possible for hemophilia patients to receive less factor VIII (FVIII) without increasing their risk of bleeding, according to a study presented at the World Federation of Hemophilia 2016 World Congress.
The study1 showed that hemophilia A patients who received prophylactic FVIII from the home infusion provider Option Care received 6 fewer units of FVIII per week than patients receiving prophylactic FVIII from specialty pharmacies.
This translated to a cost savings of more than $20,000 per patient each year.
The home infusion patients also had a lower annual bleed rate (ABR) than what is typically observed with intensive prophylaxis protocols in hemophilia, according to previous studies.
“By working with prescribers to closely monitor bleeds and collaborate on clinically appropriate optimization of treatment dose, Option Care’s utilization of factor VIII is less than the average with excellent outcomes,” said Joan Couden, RN, national program director for Option Care’s Bleeding Disorders Program.
“Our findings show we can save payers, including Medicare, Medicaid, and managed care insurers, significant costs without negatively impacting annual bleed rates.”
For this study, Couden and her colleagues conducted a retrospective analysis using dispensing data records from Option Care spanning the period from July 2015 through December 2015. The team compared these data to aggregate specialty pharmacy records from November 2013 through March 2014, which were analyzed in a previous study.2
In both data sets, patients receiving any FVIII product for prophylactic therapy were included. Patients being treated episodically or for immune tolerance induction were excluded, as were patients with extremely abnormal weights (40% below the 5th percentile or 40% above the 95th percentile based on weight-for-age charts from the US Centers for Disease Control and Prevention).
The researchers calculated a patient’s weekly dose of FVIII by multiplying the prescribed infusion dose by the dose frequency and dividing the product by the patient’s weight. Patients with an overall mean weekly dose greater than 2 standard deviations from the mean were excluded.
There were 77 home infusion patients and 520 specialty pharmacy patients.
The home infusion patients had a mean FVIII dose of 102 units/kg/week, compared to a mean of 108 units/kg/week for the specialty pharmacy patients.
This difference translates to savings of $21,166 per patient per year among the home infusion patients.
Couden and her colleagues could not compare the ABR between the 2 data sets because the ABR was not measured in the specialty pharmacy patients. However, they said the ABR in the home infusion patients was favorable when compared to ABRs in published studies.
The mean ABR for the home infusion patients was 1.70. And, according to a recent review of research on hemophilia treatment strategies, mean ABRs range from 2 to 5 for intensive treatment protocols.3![]()