User login
Practical “pearls” to help improve your care

Although we reserve the term “PURL” for our popular feature, Priority Updates from the Research Literature, I’m proud to comment on the collection of articles in this issue of JFP, each of which contains important “pearls” of information for family physicians and other primary care clinicians.
Managing sport-related concussion. Revelations about serious head injuries in the National Football League have catalyzed important research regarding the management of sports-related head injuries, and the evidence for diagnosis and treatment is evolving. The article in this issue by Dr. Sprouse and colleagues provides some of the latest information regarding brain changes after concussion straight from the American Academy of Neurology’s 2016 Sports Concussion Conference held in Chicago in July, as well as valuable return-to-play recommendations.
Family medicine ultrasound. Because of advances in technology and reductions in the cost of portable machines, ultrasound use is rapidly moving into family medicine offices. Drs. Steinmetz and Oleskevich provide a no-nonsense review of the current uses of ultrasound in family medicine, leading me to wonder whether ultrasound might become the stethoscope of the future.
Shortness of breath. Although the diagnosis of shortness of breath is straightforward in many cases, misdiagnosis is not uncommon. Recently, I cared for a new patient who was diagnosed with asthma 15 years ago. Because of fine rales on exam, I suspected the patient’s diagnosis was incorrect. Indeed, he had pulmonary fibrosis, not asthma, and he is doing fine now without his asthma inhalers. Dr. Taggart outlines a thoughtful approach to the evaluation of shortness of breath, one that alerts you to when to suspect something beyond the usual culprits.
Cervical cancer screening. The days of yearly Pap smears for all women are over. Combined screening with cytology and human papillomavirus testing is now recommended at 5-year intervals for women 30 to 65 years of age who are at low risk for cervical cancer. In addition, Dr. Hofmeister reviews recent randomized trials that suggest HPV screening alone may be sufficient for low-risk women.
On-demand HIV prophylaxis. Our PURL for the month discusses an effective prevention strategy—other than condoms—that can be used as needed by people at high risk for human immunodeficiency virus.
We hope you enjoy this PURL—and the other “pearls”—this month. As diagnosis and treatments evolve, JFP will continue to bring you the information you need to provide the best possible care for your patients.
Although we reserve the term “PURL” for our popular feature, Priority Updates from the Research Literature, I’m proud to comment on the collection of articles in this issue of JFP, each of which contains important “pearls” of information for family physicians and other primary care clinicians.
Managing sport-related concussion. Revelations about serious head injuries in the National Football League have catalyzed important research regarding the management of sports-related head injuries, and the evidence for diagnosis and treatment is evolving. The article in this issue by Dr. Sprouse and colleagues provides some of the latest information regarding brain changes after concussion straight from the American Academy of Neurology’s 2016 Sports Concussion Conference held in Chicago in July, as well as valuable return-to-play recommendations.
Family medicine ultrasound. Because of advances in technology and reductions in the cost of portable machines, ultrasound use is rapidly moving into family medicine offices. Drs. Steinmetz and Oleskevich provide a no-nonsense review of the current uses of ultrasound in family medicine, leading me to wonder whether ultrasound might become the stethoscope of the future.
Shortness of breath. Although the diagnosis of shortness of breath is straightforward in many cases, misdiagnosis is not uncommon. Recently, I cared for a new patient who was diagnosed with asthma 15 years ago. Because of fine rales on exam, I suspected the patient’s diagnosis was incorrect. Indeed, he had pulmonary fibrosis, not asthma, and he is doing fine now without his asthma inhalers. Dr. Taggart outlines a thoughtful approach to the evaluation of shortness of breath, one that alerts you to when to suspect something beyond the usual culprits.
Cervical cancer screening. The days of yearly Pap smears for all women are over. Combined screening with cytology and human papillomavirus testing is now recommended at 5-year intervals for women 30 to 65 years of age who are at low risk for cervical cancer. In addition, Dr. Hofmeister reviews recent randomized trials that suggest HPV screening alone may be sufficient for low-risk women.
On-demand HIV prophylaxis. Our PURL for the month discusses an effective prevention strategy—other than condoms—that can be used as needed by people at high risk for human immunodeficiency virus.
We hope you enjoy this PURL—and the other “pearls”—this month. As diagnosis and treatments evolve, JFP will continue to bring you the information you need to provide the best possible care for your patients.
Although we reserve the term “PURL” for our popular feature, Priority Updates from the Research Literature, I’m proud to comment on the collection of articles in this issue of JFP, each of which contains important “pearls” of information for family physicians and other primary care clinicians.
Managing sport-related concussion. Revelations about serious head injuries in the National Football League have catalyzed important research regarding the management of sports-related head injuries, and the evidence for diagnosis and treatment is evolving. The article in this issue by Dr. Sprouse and colleagues provides some of the latest information regarding brain changes after concussion straight from the American Academy of Neurology’s 2016 Sports Concussion Conference held in Chicago in July, as well as valuable return-to-play recommendations.
Family medicine ultrasound. Because of advances in technology and reductions in the cost of portable machines, ultrasound use is rapidly moving into family medicine offices. Drs. Steinmetz and Oleskevich provide a no-nonsense review of the current uses of ultrasound in family medicine, leading me to wonder whether ultrasound might become the stethoscope of the future.
Shortness of breath. Although the diagnosis of shortness of breath is straightforward in many cases, misdiagnosis is not uncommon. Recently, I cared for a new patient who was diagnosed with asthma 15 years ago. Because of fine rales on exam, I suspected the patient’s diagnosis was incorrect. Indeed, he had pulmonary fibrosis, not asthma, and he is doing fine now without his asthma inhalers. Dr. Taggart outlines a thoughtful approach to the evaluation of shortness of breath, one that alerts you to when to suspect something beyond the usual culprits.
Cervical cancer screening. The days of yearly Pap smears for all women are over. Combined screening with cytology and human papillomavirus testing is now recommended at 5-year intervals for women 30 to 65 years of age who are at low risk for cervical cancer. In addition, Dr. Hofmeister reviews recent randomized trials that suggest HPV screening alone may be sufficient for low-risk women.
On-demand HIV prophylaxis. Our PURL for the month discusses an effective prevention strategy—other than condoms—that can be used as needed by people at high risk for human immunodeficiency virus.
We hope you enjoy this PURL—and the other “pearls”—this month. As diagnosis and treatments evolve, JFP will continue to bring you the information you need to provide the best possible care for your patients.
On-demand pill protocol protects against HIV
Offer patients at high risk for human immunodeficiency virus (HIV), particularly men who have sex with men, preexposure prophylaxis (PrEP) with a combination pill of tenofovir disoproxil fumarate and emtricitabine (TDF-FTC) on an on-demand basis to decrease HIV-1 infection rates.
Strength of recommendation
B: Based on one good quality randomized control trial.1
Molina JM, Capitant C, Spire B, et al. On-demand preexposure prophylaxis in men at high risk for HIV-1 infection. N Engl J Med. 2015;373:2237-2246.
ILLUSTRATIVE CASE
Your patient is a 31-year-old man who has sex with men. He is sexually active with several different partners. He asks you if there is anything he can do to decrease his risk of becoming infected with human immunodeficiency virus (HIV). Besides recommending condom use, what should you offer him?
In most high-income countries, including the United States, HIV-1 infection continues to occur in high-risk groups, especially among men who have sex with men (MSM).2 Without a vaccine, condom use has served as the primary method of preventing infection.
In 2014, the Centers for Disease Control and Prevention (CDC) began recommending daily use of tenofovir disoproxil fumarate and emtricitabine (TDF-FTC) in high-risk individuals, as a form of preexposure prophylaxis (PrEP).3-5 This recommendation is based primarily on the Preexposure Prophylaxis Initiative (iPrEx) trial, which showed a relative reduction of 44% (number needed to treat [NNT]=46 over 1.2 years) in the incidence of new HIV-1 infection among men and transgender women who have sex with men when TDF-FTC was used on a daily basis.6 However, the effectiveness of this strategy in the real world has not been as high as hoped, presumably because of the difficulty in getting patients to take the medication on a daily basis.7,8
While it would likely improve adherence rates, the use of prophylaxis in an on-demand manner is not currently recommended.5 That is because, until now, there have been no studies demonstrating the effectiveness of PrEP used episodically and taken only around the time of potential exposure.
STUDY SUMMARY
Fewer pills improves adherence, reduces HIV infection rates
The Intervention Preventive de l’Exposition aux Risques avec et pour les Gays (IPERGAY) study was a double-blind, multicenter study conducted in France and Canada that assessed the efficacy and safety of prophylaxis with TDF-FTC used in an on-demand fashion by MSM.1 The study hypothesis was that adherence would be higher if chemoprophylaxis was taken only around the time of intercourse, rather than daily, and that this would further reduce the risk of HIV infection.
The study randomized 414 participants who were considered to be at high risk for acquiring HIV-1 infection. The investigators defined high risk as having a history of unprotected anal sex with at least 2 partners in the previous 6 months. Other inclusion criteria included age ≥18 years, and male or transgender female sex. Exclusion criteria included current HIV infection, hepatitis B or C infection, creatinine clearance <60 mL/min, alanine aminotransferase level >2.5 times the upper limit of normal, and significant glycosuria or proteinuria.
The pill and visit schedule. After excluding those who withdrew consent, were lost to follow-up, or who acquired HIV-1 infection, the study participants (199 in the TDF-FTC group and 201 in the placebo group) were randomized to take TDF-FTC or placebo before and after sexual activity. The dose of TDF-FTC was fixed at 300 mg of TDF and 200 mg of FTC per pill. The participants were instructed to take a loading dose of 2 pills of TDF-FTC or placebo with food 2 to 24 hours prior to intercourse, followed by a third pill 24 hours after taking the first 2 pills, and a fourth pill 24 hours after the third pill. If there were multiple consecutive days with episodes of sexual intercourse, participants were to take one pill on each of the days of intercourse, and then the 2 post-exposure pills. If sexual activity resumed within a week of the prior episode, participants were instructed to take only one pill when resuming the preexposure prophylaxis; otherwise, they were to begin again with 2 pills 2 to 24 hours prior to intercourse and repeat the protocol.
Study coordinators followed participants 4 and 8 weeks after enrollment, and then every 8 weeks subsequently. The investigators tested the participants for HIV-1 and HIV-2 at each visit and assessed adherence by pill count and drug levels in plasma, as well as with an at-home, computer-assisted interview completed by each participant prior to each visit.
Participants received counseling from a peer community member and were offered preventative services and testing for other sexually transmitted infections. They were given free condoms and gel at each visit, as well as enough pills (TDF-FTC or placebo) to cover daily use until their next visit.
Forty-three percent took the pills correctly. The participants were followed for a median of 9.3 months. Overall, 72% of the participants took the study drugs (TDF-FTC or placebo), although 29% took a suboptimal dose. There was no change in the sexual behavior of the participants during the study. The study was unblinded after 20 months and is continuing as an open-label study because of the discontinuation of another preexposure prophylaxis study in the United Kingdom, which showed an NNT of 13 to prevent one new HIV infection per year.3
An independent data and safety monitoring board recommended the unblinding because the placebo group was considered to be at significantly increased risk of contracting HIV without PrEP. The open-label part of the study, iPrex-OLE, completed enrollment and data gathering in November 2013, and the data analysis and results are presently pending.9
Eighty-six percent relative reduction in HIV. The primary end-point was the diagnosis of HIV-1 infection, and the results were based on an intention-to-treat analysis. HIV-1 infection was diagnosed in 19 study participants, with 3 of those new cases occurring between the time of randomization and enrollment. Fourteen of the cases were in the placebo group (6.6 infections per 100 person-years) and 2 of the new cases were in the TDF-FTC group (incidence 0.91 per 100 person-years). This translated to a relative reduction in the incidence of new HIV-1 seroconversion in the TDF-FTC group of 86% (95% confidence interval, 40%-98%; P=.002; NNT=17 over 9.3 months).
The 2 study participants in the TDF-FTC group diagnosed with new HIV-1 were found to be non-adherent to the prescribed prophylaxis, as they returned 58 and 60 of the 60 pills administered to them, and no study drugs were found in their plasma samples.
Adverse events included gastrointestinal symptoms of nausea, vomiting, diarrhea, and abdominal pain and were seen at a greater rate (14% vs 5%, P=.002; number needed to harm=11) in the treatment group than in the placebo group. There were also mild increases in serum creatinine level (seen in 18% of the TDF-FTC group), but only 2 participants had a transient decrease in creatinine clearance to <60 mL/min. None of the participants discontinued medications due to renal issues.
WHAT’S NEW
Risk reduction with on-demand use is nearly double that of daily use
This is the first study to look at on-demand preexposure prophylaxis with TDF-FTC to decrease the incidence of HIV-1 infection in high-risk MSM. The risk reduction in this study (86%) was much better than the 44% seen in the prior study that used daily PrEP in this population.6 We suspect the higher benefit of on-demand PrEP is likely due to increased compliance with medication use.
CAVEATS
Is fewer pills enough to maintain adherence over time?
The median length of follow-up in the study was 9.3 months. One concern is that adherence may wane over time, decreasing the efficacy of the prophylaxis. Continued efforts to improve compliance with this type of PrEP may be needed to ensure efficacy. Since the study was shortened and reported early, we will need to wait for the results of the open-label study to fully assess the risks of adverse events.
CHALLENGES TO IMPLEMENTATION
Efficacy and convenience come at a cost
The main challenge to implementation could be the cost of TDF-FTC, the retail price of which is about $50 per dose.10 Insurance coverage for the medication varies.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Molina JM, Capitant C, Spire B, et al. On-demand preexposure prophylaxis in men at high risk for HIV-1 infection. N Engl J Med. 2015;373:2237-2246.
2. Beyrer C, Sullivan P, Sanchez J, et al. The increase in global HIV epidemics in MSM. AIDS. 2013;27:2665-2678.
3. McCormack S, Dunn DT, Desai M, et al. Preexposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open-label randomised trial. Lancet. 2016;387:53-60.
4. Youle M, Wainberg MA. Could chemoprophylaxis be used as an HIV prevention strategy while we wait for an effective vaccine? AIDS. 2003;17:937-938.
5. US Public Health Service. Preexposure prophylaxis for the prevention of HIV infection in the United States – 2014. A clinical practice guideline. Available at: https://www.cdc.gov/hiv/pdf/prepguidelines2014.pdf. Accessed June 4, 2016.
6. Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363:2587-2599.
7. Marrazzo JM, Ramjee G, Richardson BA, et al. Tenofovir-based preexposure prophylaxis for HIV infection among African women. N Engl J Med. 2015;372:509-518.
8. Van Damme L, Corneli A, Ahmed K, et al. Preexposure prophylaxis for HIV infection among African women. N Engl J Med. 2012;367:411-422.
9. IPrEx open-label extension. Available at: http://www.iprexnews.com. Accessed July 13, 2016.
10. GoodRx. Truvada. Available at: https://www.goodrx.com/truvada. Accessed June 4, 2016.
Offer patients at high risk for human immunodeficiency virus (HIV), particularly men who have sex with men, preexposure prophylaxis (PrEP) with a combination pill of tenofovir disoproxil fumarate and emtricitabine (TDF-FTC) on an on-demand basis to decrease HIV-1 infection rates.
Strength of recommendation
B: Based on one good quality randomized control trial.1
Molina JM, Capitant C, Spire B, et al. On-demand preexposure prophylaxis in men at high risk for HIV-1 infection. N Engl J Med. 2015;373:2237-2246.
ILLUSTRATIVE CASE
Your patient is a 31-year-old man who has sex with men. He is sexually active with several different partners. He asks you if there is anything he can do to decrease his risk of becoming infected with human immunodeficiency virus (HIV). Besides recommending condom use, what should you offer him?
In most high-income countries, including the United States, HIV-1 infection continues to occur in high-risk groups, especially among men who have sex with men (MSM).2 Without a vaccine, condom use has served as the primary method of preventing infection.
In 2014, the Centers for Disease Control and Prevention (CDC) began recommending daily use of tenofovir disoproxil fumarate and emtricitabine (TDF-FTC) in high-risk individuals, as a form of preexposure prophylaxis (PrEP).3-5 This recommendation is based primarily on the Preexposure Prophylaxis Initiative (iPrEx) trial, which showed a relative reduction of 44% (number needed to treat [NNT]=46 over 1.2 years) in the incidence of new HIV-1 infection among men and transgender women who have sex with men when TDF-FTC was used on a daily basis.6 However, the effectiveness of this strategy in the real world has not been as high as hoped, presumably because of the difficulty in getting patients to take the medication on a daily basis.7,8
While it would likely improve adherence rates, the use of prophylaxis in an on-demand manner is not currently recommended.5 That is because, until now, there have been no studies demonstrating the effectiveness of PrEP used episodically and taken only around the time of potential exposure.
STUDY SUMMARY
Fewer pills improves adherence, reduces HIV infection rates
The Intervention Preventive de l’Exposition aux Risques avec et pour les Gays (IPERGAY) study was a double-blind, multicenter study conducted in France and Canada that assessed the efficacy and safety of prophylaxis with TDF-FTC used in an on-demand fashion by MSM.1 The study hypothesis was that adherence would be higher if chemoprophylaxis was taken only around the time of intercourse, rather than daily, and that this would further reduce the risk of HIV infection.
The study randomized 414 participants who were considered to be at high risk for acquiring HIV-1 infection. The investigators defined high risk as having a history of unprotected anal sex with at least 2 partners in the previous 6 months. Other inclusion criteria included age ≥18 years, and male or transgender female sex. Exclusion criteria included current HIV infection, hepatitis B or C infection, creatinine clearance <60 mL/min, alanine aminotransferase level >2.5 times the upper limit of normal, and significant glycosuria or proteinuria.
The pill and visit schedule. After excluding those who withdrew consent, were lost to follow-up, or who acquired HIV-1 infection, the study participants (199 in the TDF-FTC group and 201 in the placebo group) were randomized to take TDF-FTC or placebo before and after sexual activity. The dose of TDF-FTC was fixed at 300 mg of TDF and 200 mg of FTC per pill. The participants were instructed to take a loading dose of 2 pills of TDF-FTC or placebo with food 2 to 24 hours prior to intercourse, followed by a third pill 24 hours after taking the first 2 pills, and a fourth pill 24 hours after the third pill. If there were multiple consecutive days with episodes of sexual intercourse, participants were to take one pill on each of the days of intercourse, and then the 2 post-exposure pills. If sexual activity resumed within a week of the prior episode, participants were instructed to take only one pill when resuming the preexposure prophylaxis; otherwise, they were to begin again with 2 pills 2 to 24 hours prior to intercourse and repeat the protocol.
Study coordinators followed participants 4 and 8 weeks after enrollment, and then every 8 weeks subsequently. The investigators tested the participants for HIV-1 and HIV-2 at each visit and assessed adherence by pill count and drug levels in plasma, as well as with an at-home, computer-assisted interview completed by each participant prior to each visit.
Participants received counseling from a peer community member and were offered preventative services and testing for other sexually transmitted infections. They were given free condoms and gel at each visit, as well as enough pills (TDF-FTC or placebo) to cover daily use until their next visit.
Forty-three percent took the pills correctly. The participants were followed for a median of 9.3 months. Overall, 72% of the participants took the study drugs (TDF-FTC or placebo), although 29% took a suboptimal dose. There was no change in the sexual behavior of the participants during the study. The study was unblinded after 20 months and is continuing as an open-label study because of the discontinuation of another preexposure prophylaxis study in the United Kingdom, which showed an NNT of 13 to prevent one new HIV infection per year.3
An independent data and safety monitoring board recommended the unblinding because the placebo group was considered to be at significantly increased risk of contracting HIV without PrEP. The open-label part of the study, iPrex-OLE, completed enrollment and data gathering in November 2013, and the data analysis and results are presently pending.9
Eighty-six percent relative reduction in HIV. The primary end-point was the diagnosis of HIV-1 infection, and the results were based on an intention-to-treat analysis. HIV-1 infection was diagnosed in 19 study participants, with 3 of those new cases occurring between the time of randomization and enrollment. Fourteen of the cases were in the placebo group (6.6 infections per 100 person-years) and 2 of the new cases were in the TDF-FTC group (incidence 0.91 per 100 person-years). This translated to a relative reduction in the incidence of new HIV-1 seroconversion in the TDF-FTC group of 86% (95% confidence interval, 40%-98%; P=.002; NNT=17 over 9.3 months).
The 2 study participants in the TDF-FTC group diagnosed with new HIV-1 were found to be non-adherent to the prescribed prophylaxis, as they returned 58 and 60 of the 60 pills administered to them, and no study drugs were found in their plasma samples.
Adverse events included gastrointestinal symptoms of nausea, vomiting, diarrhea, and abdominal pain and were seen at a greater rate (14% vs 5%, P=.002; number needed to harm=11) in the treatment group than in the placebo group. There were also mild increases in serum creatinine level (seen in 18% of the TDF-FTC group), but only 2 participants had a transient decrease in creatinine clearance to <60 mL/min. None of the participants discontinued medications due to renal issues.
WHAT’S NEW
Risk reduction with on-demand use is nearly double that of daily use
This is the first study to look at on-demand preexposure prophylaxis with TDF-FTC to decrease the incidence of HIV-1 infection in high-risk MSM. The risk reduction in this study (86%) was much better than the 44% seen in the prior study that used daily PrEP in this population.6 We suspect the higher benefit of on-demand PrEP is likely due to increased compliance with medication use.
CAVEATS
Is fewer pills enough to maintain adherence over time?
The median length of follow-up in the study was 9.3 months. One concern is that adherence may wane over time, decreasing the efficacy of the prophylaxis. Continued efforts to improve compliance with this type of PrEP may be needed to ensure efficacy. Since the study was shortened and reported early, we will need to wait for the results of the open-label study to fully assess the risks of adverse events.
CHALLENGES TO IMPLEMENTATION
Efficacy and convenience come at a cost
The main challenge to implementation could be the cost of TDF-FTC, the retail price of which is about $50 per dose.10 Insurance coverage for the medication varies.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Offer patients at high risk for human immunodeficiency virus (HIV), particularly men who have sex with men, preexposure prophylaxis (PrEP) with a combination pill of tenofovir disoproxil fumarate and emtricitabine (TDF-FTC) on an on-demand basis to decrease HIV-1 infection rates.
Strength of recommendation
B: Based on one good quality randomized control trial.1
Molina JM, Capitant C, Spire B, et al. On-demand preexposure prophylaxis in men at high risk for HIV-1 infection. N Engl J Med. 2015;373:2237-2246.
ILLUSTRATIVE CASE
Your patient is a 31-year-old man who has sex with men. He is sexually active with several different partners. He asks you if there is anything he can do to decrease his risk of becoming infected with human immunodeficiency virus (HIV). Besides recommending condom use, what should you offer him?
In most high-income countries, including the United States, HIV-1 infection continues to occur in high-risk groups, especially among men who have sex with men (MSM).2 Without a vaccine, condom use has served as the primary method of preventing infection.
In 2014, the Centers for Disease Control and Prevention (CDC) began recommending daily use of tenofovir disoproxil fumarate and emtricitabine (TDF-FTC) in high-risk individuals, as a form of preexposure prophylaxis (PrEP).3-5 This recommendation is based primarily on the Preexposure Prophylaxis Initiative (iPrEx) trial, which showed a relative reduction of 44% (number needed to treat [NNT]=46 over 1.2 years) in the incidence of new HIV-1 infection among men and transgender women who have sex with men when TDF-FTC was used on a daily basis.6 However, the effectiveness of this strategy in the real world has not been as high as hoped, presumably because of the difficulty in getting patients to take the medication on a daily basis.7,8
While it would likely improve adherence rates, the use of prophylaxis in an on-demand manner is not currently recommended.5 That is because, until now, there have been no studies demonstrating the effectiveness of PrEP used episodically and taken only around the time of potential exposure.
STUDY SUMMARY
Fewer pills improves adherence, reduces HIV infection rates
The Intervention Preventive de l’Exposition aux Risques avec et pour les Gays (IPERGAY) study was a double-blind, multicenter study conducted in France and Canada that assessed the efficacy and safety of prophylaxis with TDF-FTC used in an on-demand fashion by MSM.1 The study hypothesis was that adherence would be higher if chemoprophylaxis was taken only around the time of intercourse, rather than daily, and that this would further reduce the risk of HIV infection.
The study randomized 414 participants who were considered to be at high risk for acquiring HIV-1 infection. The investigators defined high risk as having a history of unprotected anal sex with at least 2 partners in the previous 6 months. Other inclusion criteria included age ≥18 years, and male or transgender female sex. Exclusion criteria included current HIV infection, hepatitis B or C infection, creatinine clearance <60 mL/min, alanine aminotransferase level >2.5 times the upper limit of normal, and significant glycosuria or proteinuria.
The pill and visit schedule. After excluding those who withdrew consent, were lost to follow-up, or who acquired HIV-1 infection, the study participants (199 in the TDF-FTC group and 201 in the placebo group) were randomized to take TDF-FTC or placebo before and after sexual activity. The dose of TDF-FTC was fixed at 300 mg of TDF and 200 mg of FTC per pill. The participants were instructed to take a loading dose of 2 pills of TDF-FTC or placebo with food 2 to 24 hours prior to intercourse, followed by a third pill 24 hours after taking the first 2 pills, and a fourth pill 24 hours after the third pill. If there were multiple consecutive days with episodes of sexual intercourse, participants were to take one pill on each of the days of intercourse, and then the 2 post-exposure pills. If sexual activity resumed within a week of the prior episode, participants were instructed to take only one pill when resuming the preexposure prophylaxis; otherwise, they were to begin again with 2 pills 2 to 24 hours prior to intercourse and repeat the protocol.
Study coordinators followed participants 4 and 8 weeks after enrollment, and then every 8 weeks subsequently. The investigators tested the participants for HIV-1 and HIV-2 at each visit and assessed adherence by pill count and drug levels in plasma, as well as with an at-home, computer-assisted interview completed by each participant prior to each visit.
Participants received counseling from a peer community member and were offered preventative services and testing for other sexually transmitted infections. They were given free condoms and gel at each visit, as well as enough pills (TDF-FTC or placebo) to cover daily use until their next visit.
Forty-three percent took the pills correctly. The participants were followed for a median of 9.3 months. Overall, 72% of the participants took the study drugs (TDF-FTC or placebo), although 29% took a suboptimal dose. There was no change in the sexual behavior of the participants during the study. The study was unblinded after 20 months and is continuing as an open-label study because of the discontinuation of another preexposure prophylaxis study in the United Kingdom, which showed an NNT of 13 to prevent one new HIV infection per year.3
An independent data and safety monitoring board recommended the unblinding because the placebo group was considered to be at significantly increased risk of contracting HIV without PrEP. The open-label part of the study, iPrex-OLE, completed enrollment and data gathering in November 2013, and the data analysis and results are presently pending.9
Eighty-six percent relative reduction in HIV. The primary end-point was the diagnosis of HIV-1 infection, and the results were based on an intention-to-treat analysis. HIV-1 infection was diagnosed in 19 study participants, with 3 of those new cases occurring between the time of randomization and enrollment. Fourteen of the cases were in the placebo group (6.6 infections per 100 person-years) and 2 of the new cases were in the TDF-FTC group (incidence 0.91 per 100 person-years). This translated to a relative reduction in the incidence of new HIV-1 seroconversion in the TDF-FTC group of 86% (95% confidence interval, 40%-98%; P=.002; NNT=17 over 9.3 months).
The 2 study participants in the TDF-FTC group diagnosed with new HIV-1 were found to be non-adherent to the prescribed prophylaxis, as they returned 58 and 60 of the 60 pills administered to them, and no study drugs were found in their plasma samples.
Adverse events included gastrointestinal symptoms of nausea, vomiting, diarrhea, and abdominal pain and were seen at a greater rate (14% vs 5%, P=.002; number needed to harm=11) in the treatment group than in the placebo group. There were also mild increases in serum creatinine level (seen in 18% of the TDF-FTC group), but only 2 participants had a transient decrease in creatinine clearance to <60 mL/min. None of the participants discontinued medications due to renal issues.
WHAT’S NEW
Risk reduction with on-demand use is nearly double that of daily use
This is the first study to look at on-demand preexposure prophylaxis with TDF-FTC to decrease the incidence of HIV-1 infection in high-risk MSM. The risk reduction in this study (86%) was much better than the 44% seen in the prior study that used daily PrEP in this population.6 We suspect the higher benefit of on-demand PrEP is likely due to increased compliance with medication use.
CAVEATS
Is fewer pills enough to maintain adherence over time?
The median length of follow-up in the study was 9.3 months. One concern is that adherence may wane over time, decreasing the efficacy of the prophylaxis. Continued efforts to improve compliance with this type of PrEP may be needed to ensure efficacy. Since the study was shortened and reported early, we will need to wait for the results of the open-label study to fully assess the risks of adverse events.
CHALLENGES TO IMPLEMENTATION
Efficacy and convenience come at a cost
The main challenge to implementation could be the cost of TDF-FTC, the retail price of which is about $50 per dose.10 Insurance coverage for the medication varies.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Molina JM, Capitant C, Spire B, et al. On-demand preexposure prophylaxis in men at high risk for HIV-1 infection. N Engl J Med. 2015;373:2237-2246.
2. Beyrer C, Sullivan P, Sanchez J, et al. The increase in global HIV epidemics in MSM. AIDS. 2013;27:2665-2678.
3. McCormack S, Dunn DT, Desai M, et al. Preexposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open-label randomised trial. Lancet. 2016;387:53-60.
4. Youle M, Wainberg MA. Could chemoprophylaxis be used as an HIV prevention strategy while we wait for an effective vaccine? AIDS. 2003;17:937-938.
5. US Public Health Service. Preexposure prophylaxis for the prevention of HIV infection in the United States – 2014. A clinical practice guideline. Available at: https://www.cdc.gov/hiv/pdf/prepguidelines2014.pdf. Accessed June 4, 2016.
6. Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363:2587-2599.
7. Marrazzo JM, Ramjee G, Richardson BA, et al. Tenofovir-based preexposure prophylaxis for HIV infection among African women. N Engl J Med. 2015;372:509-518.
8. Van Damme L, Corneli A, Ahmed K, et al. Preexposure prophylaxis for HIV infection among African women. N Engl J Med. 2012;367:411-422.
9. IPrEx open-label extension. Available at: http://www.iprexnews.com. Accessed July 13, 2016.
10. GoodRx. Truvada. Available at: https://www.goodrx.com/truvada. Accessed June 4, 2016.
1. Molina JM, Capitant C, Spire B, et al. On-demand preexposure prophylaxis in men at high risk for HIV-1 infection. N Engl J Med. 2015;373:2237-2246.
2. Beyrer C, Sullivan P, Sanchez J, et al. The increase in global HIV epidemics in MSM. AIDS. 2013;27:2665-2678.
3. McCormack S, Dunn DT, Desai M, et al. Preexposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open-label randomised trial. Lancet. 2016;387:53-60.
4. Youle M, Wainberg MA. Could chemoprophylaxis be used as an HIV prevention strategy while we wait for an effective vaccine? AIDS. 2003;17:937-938.
5. US Public Health Service. Preexposure prophylaxis for the prevention of HIV infection in the United States – 2014. A clinical practice guideline. Available at: https://www.cdc.gov/hiv/pdf/prepguidelines2014.pdf. Accessed June 4, 2016.
6. Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363:2587-2599.
7. Marrazzo JM, Ramjee G, Richardson BA, et al. Tenofovir-based preexposure prophylaxis for HIV infection among African women. N Engl J Med. 2015;372:509-518.
8. Van Damme L, Corneli A, Ahmed K, et al. Preexposure prophylaxis for HIV infection among African women. N Engl J Med. 2012;367:411-422.
9. IPrEx open-label extension. Available at: http://www.iprexnews.com. Accessed July 13, 2016.
10. GoodRx. Truvada. Available at: https://www.goodrx.com/truvada. Accessed June 4, 2016.
Copyright © 2016. The Family Physicians Inquiries Network. All rights reserved.

Mobile App: VAM Info at Your Fingertips
At the Vascular Annual Meeting, there’s no need to cart around a lot of paper. Instead, the Mobile App puts all the information you need – abstracts, exhibitor map, schedules and more – within fingertip reach, to keep you more organized than ever.
Quick tips for using the Mobile App:

Create a personal schedule. Bookmark potential sessions in the program tab by clinking the small calendar icon the right for each session of interest. Reminders will pop up 10 minutes before the start time, including session details. The information will be pinned to the top of your activity feed to help you stay organized.
Review abstracts and take notes. The index contains all abstracts with author names and affiliations, presentation time, location, and a link to view the full abstract online. All index information is searchable. Take notes on each abstract within the app and export those notes to an email. Create a list of favorite abstracts by bookmarking those you want to refer to again.
Share. Share your thoughts in the app’s activity feed. Let all your friends know what you’re up to by linking your social media accounts to the app so you can post in the app and your social media feed at the same time!
The VAM Meeting App Is ...
Comprehensive: It includes all meeting content, including abstracts.
Searchable: Quickly locate sessions, abstracts, speakers and more.
Interactive: Network with colleagues, share photos and rate programs.
Visit vsweb.org/mobileapp to download the app.
At the Vascular Annual Meeting, there’s no need to cart around a lot of paper. Instead, the Mobile App puts all the information you need – abstracts, exhibitor map, schedules and more – within fingertip reach, to keep you more organized than ever.
Quick tips for using the Mobile App:
Create a personal schedule. Bookmark potential sessions in the program tab by clinking the small calendar icon the right for each session of interest. Reminders will pop up 10 minutes before the start time, including session details. The information will be pinned to the top of your activity feed to help you stay organized.
Review abstracts and take notes. The index contains all abstracts with author names and affiliations, presentation time, location, and a link to view the full abstract online. All index information is searchable. Take notes on each abstract within the app and export those notes to an email. Create a list of favorite abstracts by bookmarking those you want to refer to again.
Share. Share your thoughts in the app’s activity feed. Let all your friends know what you’re up to by linking your social media accounts to the app so you can post in the app and your social media feed at the same time!
The VAM Meeting App Is ...
Comprehensive: It includes all meeting content, including abstracts.
Searchable: Quickly locate sessions, abstracts, speakers and more.
Interactive: Network with colleagues, share photos and rate programs.
Visit vsweb.org/mobileapp to download the app.
At the Vascular Annual Meeting, there’s no need to cart around a lot of paper. Instead, the Mobile App puts all the information you need – abstracts, exhibitor map, schedules and more – within fingertip reach, to keep you more organized than ever.
Quick tips for using the Mobile App:
Create a personal schedule. Bookmark potential sessions in the program tab by clinking the small calendar icon the right for each session of interest. Reminders will pop up 10 minutes before the start time, including session details. The information will be pinned to the top of your activity feed to help you stay organized.
Review abstracts and take notes. The index contains all abstracts with author names and affiliations, presentation time, location, and a link to view the full abstract online. All index information is searchable. Take notes on each abstract within the app and export those notes to an email. Create a list of favorite abstracts by bookmarking those you want to refer to again.
Share. Share your thoughts in the app’s activity feed. Let all your friends know what you’re up to by linking your social media accounts to the app so you can post in the app and your social media feed at the same time!
The VAM Meeting App Is ...
Comprehensive: It includes all meeting content, including abstracts.
Searchable: Quickly locate sessions, abstracts, speakers and more.
Interactive: Network with colleagues, share photos and rate programs.
Visit vsweb.org/mobileapp to download the app.

Finding Food, Fun and History in DC Environs
National Harbor is a gateway port to Washington, D.C., situated just across the Potomac. For things to see and do while at the Vascular Annual Meeting, plan to vist the famous sights, monuments, museums, and cultural icons in the city (visit www.washington.org for a full rundown). Consider also the abundant lesser known tourist attractions unavailable anywhere else in the country.
There are many museums and cultural sites located away from the National Mall, and the DC’s Penn Quarter and Chinatown are home to most of them. There’s the Newseum, an interactive museum, devoted to the history of the news, and the International Spy Museum, developed to give a glimpse into the world of espionage. Nearby are the National Portrait Gallery and Smithsonian American Art Museum. And for an entertaining look at lifelike wax figures of the American presidents, stop in at Madame Tussauds.

Medical history enthusiasts may want to visit the National Musuem of American History, which houses collections of medical science and biotechnology artifacts. And those interested in the American Civil War with access to a car can visit the National Museum of Civil War Medicine in Frederick, Md., about an hour’s drive from National Harbor.
Cultural events abound during the annual meeting period. Theater goers can attend Shakespeare’s “Taming of the Shrew” or the musical “La Cage aux Folles.” You can attend the DC Jazz Festival or the National Symphony Orchestra’s performance of pieces by Bruckner and Mahler. For more cultural events, visit www.culturalcapitol.com.
For sports lovers, the baseball season will be in full swing. The Washington Nationals will be playing the Philadelphia Phillies on Saturday and Sunday at Nationals Park.
And family and visitor favorites are the various Potamac River boat tours and excursions, including a cruise to and tour of historic Mount Vernon, the faithfully restored home and plantation of George Washington.
Excellent dining options are available in an almost infinite variety, and the Washington Post offers a website dedicated to “eating your way” through the city: https://washington.org/eat-your-way-through-dcs-neighborhoods.
National Harbor, of course, has its own attractions. Hardest to miss is the Capital Wheel, a giant ferris wheel that soars 180 feet above the Potomac River waterfront, with views of the White House and Capitol, the National Mall, and all the surrounding DC metro area.
To plan dining, shopping, or other recreation at National Harbor, visit www.nationalharbor.com/consumer/entertainment.
National Harbor is a gateway port to Washington, D.C., situated just across the Potomac. For things to see and do while at the Vascular Annual Meeting, plan to vist the famous sights, monuments, museums, and cultural icons in the city (visit www.washington.org for a full rundown). Consider also the abundant lesser known tourist attractions unavailable anywhere else in the country.
There are many museums and cultural sites located away from the National Mall, and the DC’s Penn Quarter and Chinatown are home to most of them. There’s the Newseum, an interactive museum, devoted to the history of the news, and the International Spy Museum, developed to give a glimpse into the world of espionage. Nearby are the National Portrait Gallery and Smithsonian American Art Museum. And for an entertaining look at lifelike wax figures of the American presidents, stop in at Madame Tussauds.
Medical history enthusiasts may want to visit the National Musuem of American History, which houses collections of medical science and biotechnology artifacts. And those interested in the American Civil War with access to a car can visit the National Museum of Civil War Medicine in Frederick, Md., about an hour’s drive from National Harbor.
Cultural events abound during the annual meeting period. Theater goers can attend Shakespeare’s “Taming of the Shrew” or the musical “La Cage aux Folles.” You can attend the DC Jazz Festival or the National Symphony Orchestra’s performance of pieces by Bruckner and Mahler. For more cultural events, visit www.culturalcapitol.com.
For sports lovers, the baseball season will be in full swing. The Washington Nationals will be playing the Philadelphia Phillies on Saturday and Sunday at Nationals Park.
And family and visitor favorites are the various Potamac River boat tours and excursions, including a cruise to and tour of historic Mount Vernon, the faithfully restored home and plantation of George Washington.
Excellent dining options are available in an almost infinite variety, and the Washington Post offers a website dedicated to “eating your way” through the city: https://washington.org/eat-your-way-through-dcs-neighborhoods.
National Harbor, of course, has its own attractions. Hardest to miss is the Capital Wheel, a giant ferris wheel that soars 180 feet above the Potomac River waterfront, with views of the White House and Capitol, the National Mall, and all the surrounding DC metro area.
To plan dining, shopping, or other recreation at National Harbor, visit www.nationalharbor.com/consumer/entertainment.
National Harbor is a gateway port to Washington, D.C., situated just across the Potomac. For things to see and do while at the Vascular Annual Meeting, plan to vist the famous sights, monuments, museums, and cultural icons in the city (visit www.washington.org for a full rundown). Consider also the abundant lesser known tourist attractions unavailable anywhere else in the country.
There are many museums and cultural sites located away from the National Mall, and the DC’s Penn Quarter and Chinatown are home to most of them. There’s the Newseum, an interactive museum, devoted to the history of the news, and the International Spy Museum, developed to give a glimpse into the world of espionage. Nearby are the National Portrait Gallery and Smithsonian American Art Museum. And for an entertaining look at lifelike wax figures of the American presidents, stop in at Madame Tussauds.
Medical history enthusiasts may want to visit the National Musuem of American History, which houses collections of medical science and biotechnology artifacts. And those interested in the American Civil War with access to a car can visit the National Museum of Civil War Medicine in Frederick, Md., about an hour’s drive from National Harbor.
Cultural events abound during the annual meeting period. Theater goers can attend Shakespeare’s “Taming of the Shrew” or the musical “La Cage aux Folles.” You can attend the DC Jazz Festival or the National Symphony Orchestra’s performance of pieces by Bruckner and Mahler. For more cultural events, visit www.culturalcapitol.com.
For sports lovers, the baseball season will be in full swing. The Washington Nationals will be playing the Philadelphia Phillies on Saturday and Sunday at Nationals Park.
And family and visitor favorites are the various Potamac River boat tours and excursions, including a cruise to and tour of historic Mount Vernon, the faithfully restored home and plantation of George Washington.
Excellent dining options are available in an almost infinite variety, and the Washington Post offers a website dedicated to “eating your way” through the city: https://washington.org/eat-your-way-through-dcs-neighborhoods.
National Harbor, of course, has its own attractions. Hardest to miss is the Capital Wheel, a giant ferris wheel that soars 180 feet above the Potomac River waterfront, with views of the White House and Capitol, the National Mall, and all the surrounding DC metro area.
To plan dining, shopping, or other recreation at National Harbor, visit www.nationalharbor.com/consumer/entertainment.

Benvenuto, Bienvenue, Velkommen, Willkommen!
Welcome to the Capitol of the USA! More than 100 attendees at the 2016 Vascular Annual Meeting hail from overseas, coming to the nation’s capital from Argentina, China, Japan, the Netherlands, Poland, Sri Lanka, the Ukraine, the United Kingdom and more locales from around the globe.
Many special events for international guests take place Wednesday, including International Fast Talk.
International Fast Talk returns after its popular debut in 2015. It features 22 quick abstract presentations (three minutes) followed by discussion (two minutes). All presentations are scored and the top two scorers will win the event’s Young Surgeon Competition. To qualify for Fast Talk, presenters must be 35 or younger. The abstracts all are available on the meeting app or via the Journal of Vascular Surgery supplement.

International Fast Talk
12:30 – 2:45 p.m., Maryland Ballroom C
Moderators: Drs. Gustav Fraedrich, Kimihiro Komori, Peter F. Lawrence, Luca di Marzo, Gustavo S. Oderich, Kumud M. Rai and Wayne Zhang.
International Forum
3:00 – 6:00 p.m., Maryland Ballroom C
Moderators: Drs. E. Sebastian Debus, Fausto Miranda, Alberto Munoz, Carlo Setacci and Shenming Wang.
This session returns in its expanded three-hour time format. International Forum features 16 abstracts representing research from around the world. International authors have been selected to present their abstract selections, including video abstracts for the first time. Presentations last six minutes with an additional four minutes available for discussion. Only authors outside the United States and Canada may present.
International Reception
6:15 – 7:15 p.m., Maryland Ballroom 1-3
Mix and mingle with friends old and new, and see the International Scholar awards.
SVS vs. ESVS Debates (Friday, June 10)
3:30 – 5:00 p.m., Potomac Ballroom A/B
Moderators: Drs. E. Sebastian Debus, Glenn M. LaMuraglia, Alik Farber, Bruce Perler
“Combatants” take the gloves off during the annual – and always popular – debates between SVS and the European Society for Vascular Surgery. It’s one continent against another, with participants debating the hot topics of the day and scoring thought-provoking points amid witty repartee. The audience chooses the winners! Let the best side of the pond win.
Welcome to the Capitol of the USA! More than 100 attendees at the 2016 Vascular Annual Meeting hail from overseas, coming to the nation’s capital from Argentina, China, Japan, the Netherlands, Poland, Sri Lanka, the Ukraine, the United Kingdom and more locales from around the globe.
Many special events for international guests take place Wednesday, including International Fast Talk.
International Fast Talk returns after its popular debut in 2015. It features 22 quick abstract presentations (three minutes) followed by discussion (two minutes). All presentations are scored and the top two scorers will win the event’s Young Surgeon Competition. To qualify for Fast Talk, presenters must be 35 or younger. The abstracts all are available on the meeting app or via the Journal of Vascular Surgery supplement.
International Fast Talk
12:30 – 2:45 p.m., Maryland Ballroom C
Moderators: Drs. Gustav Fraedrich, Kimihiro Komori, Peter F. Lawrence, Luca di Marzo, Gustavo S. Oderich, Kumud M. Rai and Wayne Zhang.
International Forum
3:00 – 6:00 p.m., Maryland Ballroom C
Moderators: Drs. E. Sebastian Debus, Fausto Miranda, Alberto Munoz, Carlo Setacci and Shenming Wang.
This session returns in its expanded three-hour time format. International Forum features 16 abstracts representing research from around the world. International authors have been selected to present their abstract selections, including video abstracts for the first time. Presentations last six minutes with an additional four minutes available for discussion. Only authors outside the United States and Canada may present.
International Reception
6:15 – 7:15 p.m., Maryland Ballroom 1-3
Mix and mingle with friends old and new, and see the International Scholar awards.
SVS vs. ESVS Debates (Friday, June 10)
3:30 – 5:00 p.m., Potomac Ballroom A/B
Moderators: Drs. E. Sebastian Debus, Glenn M. LaMuraglia, Alik Farber, Bruce Perler
“Combatants” take the gloves off during the annual – and always popular – debates between SVS and the European Society for Vascular Surgery. It’s one continent against another, with participants debating the hot topics of the day and scoring thought-provoking points amid witty repartee. The audience chooses the winners! Let the best side of the pond win.
Welcome to the Capitol of the USA! More than 100 attendees at the 2016 Vascular Annual Meeting hail from overseas, coming to the nation’s capital from Argentina, China, Japan, the Netherlands, Poland, Sri Lanka, the Ukraine, the United Kingdom and more locales from around the globe.
Many special events for international guests take place Wednesday, including International Fast Talk.
International Fast Talk returns after its popular debut in 2015. It features 22 quick abstract presentations (three minutes) followed by discussion (two minutes). All presentations are scored and the top two scorers will win the event’s Young Surgeon Competition. To qualify for Fast Talk, presenters must be 35 or younger. The abstracts all are available on the meeting app or via the Journal of Vascular Surgery supplement.
International Fast Talk
12:30 – 2:45 p.m., Maryland Ballroom C
Moderators: Drs. Gustav Fraedrich, Kimihiro Komori, Peter F. Lawrence, Luca di Marzo, Gustavo S. Oderich, Kumud M. Rai and Wayne Zhang.
International Forum
3:00 – 6:00 p.m., Maryland Ballroom C
Moderators: Drs. E. Sebastian Debus, Fausto Miranda, Alberto Munoz, Carlo Setacci and Shenming Wang.
This session returns in its expanded three-hour time format. International Forum features 16 abstracts representing research from around the world. International authors have been selected to present their abstract selections, including video abstracts for the first time. Presentations last six minutes with an additional four minutes available for discussion. Only authors outside the United States and Canada may present.
International Reception
6:15 – 7:15 p.m., Maryland Ballroom 1-3
Mix and mingle with friends old and new, and see the International Scholar awards.
SVS vs. ESVS Debates (Friday, June 10)
3:30 – 5:00 p.m., Potomac Ballroom A/B
Moderators: Drs. E. Sebastian Debus, Glenn M. LaMuraglia, Alik Farber, Bruce Perler
“Combatants” take the gloves off during the annual – and always popular – debates between SVS and the European Society for Vascular Surgery. It’s one continent against another, with participants debating the hot topics of the day and scoring thought-provoking points amid witty repartee. The audience chooses the winners! Let the best side of the pond win.

Resident Research Prize: Seeking to Preserve AV Fistulae
Even though the use of arteriovenous fistulae (AVF) is considered optimal for hemodialysis access in patients with end-stage renal disease, outcomes for AVF are among the worst for any vascular procedure. Low rates of maturation result in up to 60% of fistulae failing to become suitable for dialysis. This year’s Resident Research Prize winner, Dr. Trenton Foster will report on research that he and his colleagues performed to examine the molecular biology of fistula development with the eventual goal of providing therapeutic targets and strategies to improve AVF maturation.

Working with his mentor, Dr. Alan Dardik, Dr. Foster and his colleagues at the Yale University School of Medicine, have built upon their earlier research showing that Eph (Erythropoietin-producing hepatocellular carcinoma)-B4 activity prevents adaptive remodeling in vein grafts. They hypothosized that Eph-B4 mediates AVF maturation.
Dr. Foster will present their research, which used a mouse model in which an infrarenal aorto-caval AVF was created, as well as a Wistar rat infrarenal patch model.
Human studies using high-resolution imaging have documented venous wall thickening and outward remodeling as consistent components of successful venous remodeling, according to the researchers. Venous remodeling is characterized by increased deposition of smooth muscle cells and changes in extracellular matrix component expression resulting in venous wall thickening and reduced compliance. However, it is not currently understood which aspects of remodeling are critical for successful venous adaptation to the arterial environment, according to Dr. Foster.
In control mice, by day 7, Eph-B4 protein expression increased a significant 2.3-fold in the fistula compared to sham veins. Similarly, venous patch neointimal thickening with a fistula showed a significant 3.8-fold increase in Eph-B4, compared to controls without fistulae. Stimulation of Eph-B4 with Ephrin-B2/Fc showed a sixfold significantly increased colocalization of Eph-B4 and p-Tyr (phosphotyrosine) immunoreactivity by day 21, and significantly reduced fistula diameter and reduced fistula wall thickness, according to Dr. Foster.
“To confirm an inhibitory role for Eph-B4 during AVF maturation, we used Eph-B4 heterozygous mice to determine whether AVF created in mice with less Eph-B4 activity would show altered outward remodeling or wall thickening,” said Dr. Foster. Eph-B4 heterozygous mice showed reduced Eph-B4 expression and AVF in Eph-B4 heterozygous mice had increased fistula wall thickening compared to AVF in wild-type mice, with a corresponding increase in proliferating cells and with no decrease in apoptotic cells. “These data confirm an inhibitory role of Eph-B4 on wall thickening during AVF maturation,” Dr. Foster said.
It is known that Eph-B4 activates the Akt (protein kinase B) pathway to promote cell migration and proliferation. Akt is a regulator of cellular metabolism affecting protein synthesis, cell growth, and promoting cell survival, all of which are essential functions critical to venous remodeling. Dr. Foster and his colleagues previously showed that Akt expression is upregulated during vein graft adaptation, suggesting a role for Akt during AVF maturation.
“Our study shows that Eph-B4 expression increases during AVF maturation and that Eph-B4 activity inhibits venous remodeling through an Akt-1-mediated mechanism. Eph-B4 is normally present in adult veins and may be a potential therapeutic target to reduce pathologic venous remodeling with failure of fistula maturation. These results suggest that strategies to alter Eph-B4 activity may improve AVF maturation,” concluded Dr. Foster and his colleagues.
Even though the use of arteriovenous fistulae (AVF) is considered optimal for hemodialysis access in patients with end-stage renal disease, outcomes for AVF are among the worst for any vascular procedure. Low rates of maturation result in up to 60% of fistulae failing to become suitable for dialysis. This year’s Resident Research Prize winner, Dr. Trenton Foster will report on research that he and his colleagues performed to examine the molecular biology of fistula development with the eventual goal of providing therapeutic targets and strategies to improve AVF maturation.
Working with his mentor, Dr. Alan Dardik, Dr. Foster and his colleagues at the Yale University School of Medicine, have built upon their earlier research showing that Eph (Erythropoietin-producing hepatocellular carcinoma)-B4 activity prevents adaptive remodeling in vein grafts. They hypothosized that Eph-B4 mediates AVF maturation.
Dr. Foster will present their research, which used a mouse model in which an infrarenal aorto-caval AVF was created, as well as a Wistar rat infrarenal patch model.
Human studies using high-resolution imaging have documented venous wall thickening and outward remodeling as consistent components of successful venous remodeling, according to the researchers. Venous remodeling is characterized by increased deposition of smooth muscle cells and changes in extracellular matrix component expression resulting in venous wall thickening and reduced compliance. However, it is not currently understood which aspects of remodeling are critical for successful venous adaptation to the arterial environment, according to Dr. Foster.
In control mice, by day 7, Eph-B4 protein expression increased a significant 2.3-fold in the fistula compared to sham veins. Similarly, venous patch neointimal thickening with a fistula showed a significant 3.8-fold increase in Eph-B4, compared to controls without fistulae. Stimulation of Eph-B4 with Ephrin-B2/Fc showed a sixfold significantly increased colocalization of Eph-B4 and p-Tyr (phosphotyrosine) immunoreactivity by day 21, and significantly reduced fistula diameter and reduced fistula wall thickness, according to Dr. Foster.
“To confirm an inhibitory role for Eph-B4 during AVF maturation, we used Eph-B4 heterozygous mice to determine whether AVF created in mice with less Eph-B4 activity would show altered outward remodeling or wall thickening,” said Dr. Foster. Eph-B4 heterozygous mice showed reduced Eph-B4 expression and AVF in Eph-B4 heterozygous mice had increased fistula wall thickening compared to AVF in wild-type mice, with a corresponding increase in proliferating cells and with no decrease in apoptotic cells. “These data confirm an inhibitory role of Eph-B4 on wall thickening during AVF maturation,” Dr. Foster said.
It is known that Eph-B4 activates the Akt (protein kinase B) pathway to promote cell migration and proliferation. Akt is a regulator of cellular metabolism affecting protein synthesis, cell growth, and promoting cell survival, all of which are essential functions critical to venous remodeling. Dr. Foster and his colleagues previously showed that Akt expression is upregulated during vein graft adaptation, suggesting a role for Akt during AVF maturation.
“Our study shows that Eph-B4 expression increases during AVF maturation and that Eph-B4 activity inhibits venous remodeling through an Akt-1-mediated mechanism. Eph-B4 is normally present in adult veins and may be a potential therapeutic target to reduce pathologic venous remodeling with failure of fistula maturation. These results suggest that strategies to alter Eph-B4 activity may improve AVF maturation,” concluded Dr. Foster and his colleagues.
Even though the use of arteriovenous fistulae (AVF) is considered optimal for hemodialysis access in patients with end-stage renal disease, outcomes for AVF are among the worst for any vascular procedure. Low rates of maturation result in up to 60% of fistulae failing to become suitable for dialysis. This year’s Resident Research Prize winner, Dr. Trenton Foster will report on research that he and his colleagues performed to examine the molecular biology of fistula development with the eventual goal of providing therapeutic targets and strategies to improve AVF maturation.
Working with his mentor, Dr. Alan Dardik, Dr. Foster and his colleagues at the Yale University School of Medicine, have built upon their earlier research showing that Eph (Erythropoietin-producing hepatocellular carcinoma)-B4 activity prevents adaptive remodeling in vein grafts. They hypothosized that Eph-B4 mediates AVF maturation.
Dr. Foster will present their research, which used a mouse model in which an infrarenal aorto-caval AVF was created, as well as a Wistar rat infrarenal patch model.
Human studies using high-resolution imaging have documented venous wall thickening and outward remodeling as consistent components of successful venous remodeling, according to the researchers. Venous remodeling is characterized by increased deposition of smooth muscle cells and changes in extracellular matrix component expression resulting in venous wall thickening and reduced compliance. However, it is not currently understood which aspects of remodeling are critical for successful venous adaptation to the arterial environment, according to Dr. Foster.
In control mice, by day 7, Eph-B4 protein expression increased a significant 2.3-fold in the fistula compared to sham veins. Similarly, venous patch neointimal thickening with a fistula showed a significant 3.8-fold increase in Eph-B4, compared to controls without fistulae. Stimulation of Eph-B4 with Ephrin-B2/Fc showed a sixfold significantly increased colocalization of Eph-B4 and p-Tyr (phosphotyrosine) immunoreactivity by day 21, and significantly reduced fistula diameter and reduced fistula wall thickness, according to Dr. Foster.
“To confirm an inhibitory role for Eph-B4 during AVF maturation, we used Eph-B4 heterozygous mice to determine whether AVF created in mice with less Eph-B4 activity would show altered outward remodeling or wall thickening,” said Dr. Foster. Eph-B4 heterozygous mice showed reduced Eph-B4 expression and AVF in Eph-B4 heterozygous mice had increased fistula wall thickening compared to AVF in wild-type mice, with a corresponding increase in proliferating cells and with no decrease in apoptotic cells. “These data confirm an inhibitory role of Eph-B4 on wall thickening during AVF maturation,” Dr. Foster said.
It is known that Eph-B4 activates the Akt (protein kinase B) pathway to promote cell migration and proliferation. Akt is a regulator of cellular metabolism affecting protein synthesis, cell growth, and promoting cell survival, all of which are essential functions critical to venous remodeling. Dr. Foster and his colleagues previously showed that Akt expression is upregulated during vein graft adaptation, suggesting a role for Akt during AVF maturation.
“Our study shows that Eph-B4 expression increases during AVF maturation and that Eph-B4 activity inhibits venous remodeling through an Akt-1-mediated mechanism. Eph-B4 is normally present in adult veins and may be a potential therapeutic target to reduce pathologic venous remodeling with failure of fistula maturation. These results suggest that strategies to alter Eph-B4 activity may improve AVF maturation,” concluded Dr. Foster and his colleagues.

CMS seeks input on future of Open Payments program
The Centers for Medicare & Medicaid Services is seeking physician input on the Open Payments program.
The agency signaled its intent to gather information in its proposed Medicare Physician Fee Schedule for 2017 and will host a conference call for that purpose on August 2.

The agency already has released a slide presentation to be used during the call that highlights the information being requested, including whether the payment categories are inclusive enough; how many years of payment data is relevant; whether reporting entities should pre-vet data before reporting to the Open Payment system; the adequacy of the definition of a covered recipient teaching hospital; whether reporting entities should be able to submit data continuously throughout the calendar year; how mergers affect reporting; clarity on reporting of ownership and investment interests; clarity on the definition of physician-owned distributors; and clarity on ways to streamline the reporting process.
Details for participating in the conference call can be found here.
The Centers for Medicare & Medicaid Services is seeking physician input on the Open Payments program.
The agency signaled its intent to gather information in its proposed Medicare Physician Fee Schedule for 2017 and will host a conference call for that purpose on August 2.
The agency already has released a slide presentation to be used during the call that highlights the information being requested, including whether the payment categories are inclusive enough; how many years of payment data is relevant; whether reporting entities should pre-vet data before reporting to the Open Payment system; the adequacy of the definition of a covered recipient teaching hospital; whether reporting entities should be able to submit data continuously throughout the calendar year; how mergers affect reporting; clarity on reporting of ownership and investment interests; clarity on the definition of physician-owned distributors; and clarity on ways to streamline the reporting process.
Details for participating in the conference call can be found here.
The Centers for Medicare & Medicaid Services is seeking physician input on the Open Payments program.
The agency signaled its intent to gather information in its proposed Medicare Physician Fee Schedule for 2017 and will host a conference call for that purpose on August 2.
The agency already has released a slide presentation to be used during the call that highlights the information being requested, including whether the payment categories are inclusive enough; how many years of payment data is relevant; whether reporting entities should pre-vet data before reporting to the Open Payment system; the adequacy of the definition of a covered recipient teaching hospital; whether reporting entities should be able to submit data continuously throughout the calendar year; how mergers affect reporting; clarity on reporting of ownership and investment interests; clarity on the definition of physician-owned distributors; and clarity on ways to streamline the reporting process.
Details for participating in the conference call can be found here.

CDC reports three new cases of Zika-related birth defects
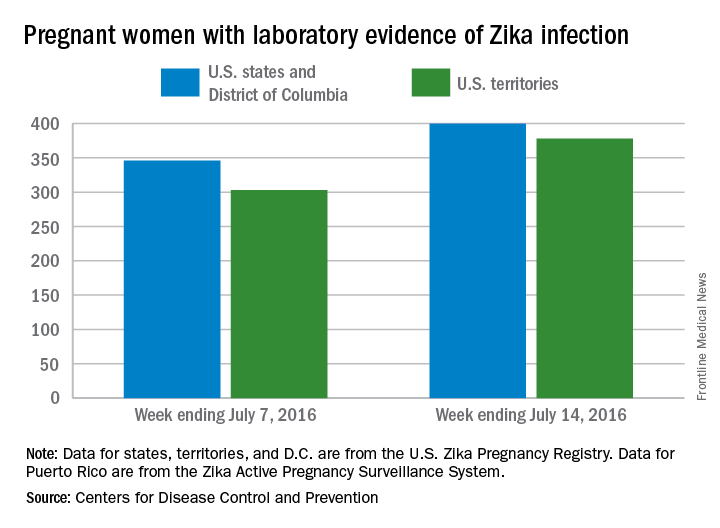
Three new cases of infants born with Zika virus–related birth defects were reported in the United States for the week ending July 14, 2016, along with 129 new infections in pregnant women, according to the Centers for Disease Control and Prevention.
The three infants were born in the 50 states and the District of Columbia, with no new pregnancy losses reported in the states or U.S. territories. Totals for the year are 12 infants with birth defects, all in the states, and seven pregnancy losses, of which six occurred in the states, the CDC reported July 21. State- or territorial-level data are not being reported to protect the privacy of affected women and children.
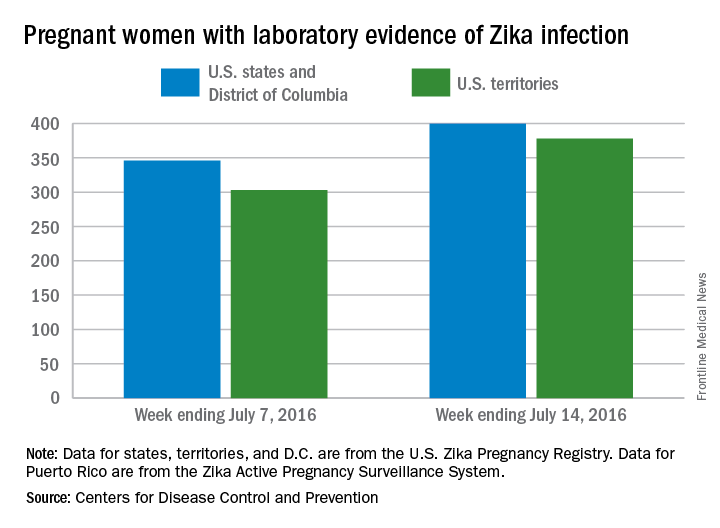
Of the 129 new infections in pregnant women for the week, 54 occurred in the states and 75 occurred in the U.S. territories. Those new cases bring the U.S. total to 778 for the year: 400 in the states and 378 in territories, the CDC also reported on July 21.
The figures for states, territories, and the District of Columbia reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
Three new cases of infants born with Zika virus–related birth defects were reported in the United States for the week ending July 14, 2016, along with 129 new infections in pregnant women, according to the Centers for Disease Control and Prevention.
The three infants were born in the 50 states and the District of Columbia, with no new pregnancy losses reported in the states or U.S. territories. Totals for the year are 12 infants with birth defects, all in the states, and seven pregnancy losses, of which six occurred in the states, the CDC reported July 21. State- or territorial-level data are not being reported to protect the privacy of affected women and children.
Of the 129 new infections in pregnant women for the week, 54 occurred in the states and 75 occurred in the U.S. territories. Those new cases bring the U.S. total to 778 for the year: 400 in the states and 378 in territories, the CDC also reported on July 21.
The figures for states, territories, and the District of Columbia reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
Three new cases of infants born with Zika virus–related birth defects were reported in the United States for the week ending July 14, 2016, along with 129 new infections in pregnant women, according to the Centers for Disease Control and Prevention.
The three infants were born in the 50 states and the District of Columbia, with no new pregnancy losses reported in the states or U.S. territories. Totals for the year are 12 infants with birth defects, all in the states, and seven pregnancy losses, of which six occurred in the states, the CDC reported July 21. State- or territorial-level data are not being reported to protect the privacy of affected women and children.
Of the 129 new infections in pregnant women for the week, 54 occurred in the states and 75 occurred in the U.S. territories. Those new cases bring the U.S. total to 778 for the year: 400 in the states and 378 in territories, the CDC also reported on July 21.
The figures for states, territories, and the District of Columbia reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
Intermediate alleles may confer mild, late-onset Huntington-like symptoms
Asymptomatic people who carry an intermediate number (27-35) of CAG repeats on the huntingtin gene – that is, the range just below the Huntington’s disease threshold of 36 CAG repeats – may be at increased risk for mild, late-onset symptoms, according to the first report to definitely associate intermediate alleles with such symptoms.
The prevalence of this distinct category of alleles for the huntingtin (HTT) gene, termed IA (for intermediate alleles), ranges from 1.5% to 5.8% both in the general population and in affected families. The clinical manifestations of Huntington’s disease associated with IAs have remain hidden until now in part because asymptomatic family members often decline to undergo genetic testing for the disorder and so are not identified as having IAs. Emerging evidence suggests that some people with IAs are more likely than those with a smaller number of CAG repeats to develop Huntington-like neuropathologic effects, but the data are sparse, and the question is controversial, said Esther Cubo, MD, PhD, of the department of neurology, Hospital Universitario Burgos (Spain) and her associates on behalf of the European Huntington’s Disease Network (Neurology. 2016 Jul 8. doi: 10.1212/WNL.0000000000002944).

To examine this issue in a large cohort of at-risk individuals, the investigators assessed clinical and sociodemographic factors for 657 people enrolled in the European Huntington’s Disease Registry, which tracks the natural course of the disease in patients and family members treated in 1998 through 2014 at 140 medical centers in 17 European and 3 other countries. They focused on individuals who were asymptomatic at baseline: 76 (11.6%) men and women who had IAs (cases) and 581 (88.4%) who had fewer than 27 CAG repeats (controls). The two groups were similar in age, body mass index, educational background, socioeconomic status, tobacco and alcohol use, and medication use, and they scored similarly on measures of health-related quality of life, functional capacity, and the Unified Huntington’s Disease Rating Scale (UHDRS) for motor, behavioral, and cognitive status.
IA status was not associated with any significant differences between cases and controls who were younger than age 60. However, after age 60, participants with IAs had higher median total UHDRS motor scores (13.0 vs. 2.0) and were more likely to show mild gait abnormalities, chorea, and bradykinesia. In the small subgroup of patients who were followed longitudinally for up to 2 years, those with IAs showed faster cognitive decline over time; however, longer follow-up of larger groups of participants is needed to confirm this trend, the investigators said.
In addition, study participants with IAs reported lower quality of life and a greater degree of apathy than those without IAs, but these differences did not reach statistical significance. Whether apathy and poor quality of life reflect Huntington’s disease or simply correlate with normal aging could not be determined.
This study was limited by the relatively small number of participants with IAs, but the findings suggest that IAs “could produce a mild phenotype” of Huntington’s disease in some carriers after age 60. Alternatively, “clinical manifestations of Huntington’s disease in patents with IA might also be potentially accelerated by medical conditions, treatments, and environmental factors” associated with normal aging, Dr. Cubo and her associates added.
This study was supported by the European Huntington’s Disease Registry. Dr. Cubo reported receiving consulting fees from UCB, Allergan, and AbbVie.
These findings are limited by the study’s small cohort size and short follow-up, but they suggest that younger IA carriers will develop few if any signs of Huntington’s disease while older IA carriers may develop a very mild phenotype late in life.
Young IA carriers and carriers who have a low number of CAG repeats (fewer than 27) on the longer allele have “not too much to worry about.” Older IA carriers may develop some mild motor symptoms and a slightly faster cognitive decline as they age. All carriers should remain as fit and healthy as possible, as they have always been advised, and should look to their family members’ experiences as a good yardstick of what might happen to them.
Patrick J. Morrison, MD, DSc, is in the department of genetic medicine at Belfast HSC Trust and the Centre for Cancer Research and Cell Biology at Queens University of Belfast (Northern Ireland). Julián Benito-León, MD, PhD, is in the department of neurology at University Hospital “12 de Octubre” and in the department of medicine at Complutense University, both in Madrid. They reported no targeted funding for this work. Dr. Morrison and Dr. Benito-León made these remarks in an editorial accompanying Dr. Cubo’s report (Neurology. 2016 Jul 8. doi: 10.1212/WNL.0000000000002958).
These findings are limited by the study’s small cohort size and short follow-up, but they suggest that younger IA carriers will develop few if any signs of Huntington’s disease while older IA carriers may develop a very mild phenotype late in life.
Young IA carriers and carriers who have a low number of CAG repeats (fewer than 27) on the longer allele have “not too much to worry about.” Older IA carriers may develop some mild motor symptoms and a slightly faster cognitive decline as they age. All carriers should remain as fit and healthy as possible, as they have always been advised, and should look to their family members’ experiences as a good yardstick of what might happen to them.
Patrick J. Morrison, MD, DSc, is in the department of genetic medicine at Belfast HSC Trust and the Centre for Cancer Research and Cell Biology at Queens University of Belfast (Northern Ireland). Julián Benito-León, MD, PhD, is in the department of neurology at University Hospital “12 de Octubre” and in the department of medicine at Complutense University, both in Madrid. They reported no targeted funding for this work. Dr. Morrison and Dr. Benito-León made these remarks in an editorial accompanying Dr. Cubo’s report (Neurology. 2016 Jul 8. doi: 10.1212/WNL.0000000000002958).
These findings are limited by the study’s small cohort size and short follow-up, but they suggest that younger IA carriers will develop few if any signs of Huntington’s disease while older IA carriers may develop a very mild phenotype late in life.
Young IA carriers and carriers who have a low number of CAG repeats (fewer than 27) on the longer allele have “not too much to worry about.” Older IA carriers may develop some mild motor symptoms and a slightly faster cognitive decline as they age. All carriers should remain as fit and healthy as possible, as they have always been advised, and should look to their family members’ experiences as a good yardstick of what might happen to them.
Patrick J. Morrison, MD, DSc, is in the department of genetic medicine at Belfast HSC Trust and the Centre for Cancer Research and Cell Biology at Queens University of Belfast (Northern Ireland). Julián Benito-León, MD, PhD, is in the department of neurology at University Hospital “12 de Octubre” and in the department of medicine at Complutense University, both in Madrid. They reported no targeted funding for this work. Dr. Morrison and Dr. Benito-León made these remarks in an editorial accompanying Dr. Cubo’s report (Neurology. 2016 Jul 8. doi: 10.1212/WNL.0000000000002958).
Asymptomatic people who carry an intermediate number (27-35) of CAG repeats on the huntingtin gene – that is, the range just below the Huntington’s disease threshold of 36 CAG repeats – may be at increased risk for mild, late-onset symptoms, according to the first report to definitely associate intermediate alleles with such symptoms.
The prevalence of this distinct category of alleles for the huntingtin (HTT) gene, termed IA (for intermediate alleles), ranges from 1.5% to 5.8% both in the general population and in affected families. The clinical manifestations of Huntington’s disease associated with IAs have remain hidden until now in part because asymptomatic family members often decline to undergo genetic testing for the disorder and so are not identified as having IAs. Emerging evidence suggests that some people with IAs are more likely than those with a smaller number of CAG repeats to develop Huntington-like neuropathologic effects, but the data are sparse, and the question is controversial, said Esther Cubo, MD, PhD, of the department of neurology, Hospital Universitario Burgos (Spain) and her associates on behalf of the European Huntington’s Disease Network (Neurology. 2016 Jul 8. doi: 10.1212/WNL.0000000000002944).
To examine this issue in a large cohort of at-risk individuals, the investigators assessed clinical and sociodemographic factors for 657 people enrolled in the European Huntington’s Disease Registry, which tracks the natural course of the disease in patients and family members treated in 1998 through 2014 at 140 medical centers in 17 European and 3 other countries. They focused on individuals who were asymptomatic at baseline: 76 (11.6%) men and women who had IAs (cases) and 581 (88.4%) who had fewer than 27 CAG repeats (controls). The two groups were similar in age, body mass index, educational background, socioeconomic status, tobacco and alcohol use, and medication use, and they scored similarly on measures of health-related quality of life, functional capacity, and the Unified Huntington’s Disease Rating Scale (UHDRS) for motor, behavioral, and cognitive status.
IA status was not associated with any significant differences between cases and controls who were younger than age 60. However, after age 60, participants with IAs had higher median total UHDRS motor scores (13.0 vs. 2.0) and were more likely to show mild gait abnormalities, chorea, and bradykinesia. In the small subgroup of patients who were followed longitudinally for up to 2 years, those with IAs showed faster cognitive decline over time; however, longer follow-up of larger groups of participants is needed to confirm this trend, the investigators said.
In addition, study participants with IAs reported lower quality of life and a greater degree of apathy than those without IAs, but these differences did not reach statistical significance. Whether apathy and poor quality of life reflect Huntington’s disease or simply correlate with normal aging could not be determined.
This study was limited by the relatively small number of participants with IAs, but the findings suggest that IAs “could produce a mild phenotype” of Huntington’s disease in some carriers after age 60. Alternatively, “clinical manifestations of Huntington’s disease in patents with IA might also be potentially accelerated by medical conditions, treatments, and environmental factors” associated with normal aging, Dr. Cubo and her associates added.
This study was supported by the European Huntington’s Disease Registry. Dr. Cubo reported receiving consulting fees from UCB, Allergan, and AbbVie.
Asymptomatic people who carry an intermediate number (27-35) of CAG repeats on the huntingtin gene – that is, the range just below the Huntington’s disease threshold of 36 CAG repeats – may be at increased risk for mild, late-onset symptoms, according to the first report to definitely associate intermediate alleles with such symptoms.
The prevalence of this distinct category of alleles for the huntingtin (HTT) gene, termed IA (for intermediate alleles), ranges from 1.5% to 5.8% both in the general population and in affected families. The clinical manifestations of Huntington’s disease associated with IAs have remain hidden until now in part because asymptomatic family members often decline to undergo genetic testing for the disorder and so are not identified as having IAs. Emerging evidence suggests that some people with IAs are more likely than those with a smaller number of CAG repeats to develop Huntington-like neuropathologic effects, but the data are sparse, and the question is controversial, said Esther Cubo, MD, PhD, of the department of neurology, Hospital Universitario Burgos (Spain) and her associates on behalf of the European Huntington’s Disease Network (Neurology. 2016 Jul 8. doi: 10.1212/WNL.0000000000002944).
To examine this issue in a large cohort of at-risk individuals, the investigators assessed clinical and sociodemographic factors for 657 people enrolled in the European Huntington’s Disease Registry, which tracks the natural course of the disease in patients and family members treated in 1998 through 2014 at 140 medical centers in 17 European and 3 other countries. They focused on individuals who were asymptomatic at baseline: 76 (11.6%) men and women who had IAs (cases) and 581 (88.4%) who had fewer than 27 CAG repeats (controls). The two groups were similar in age, body mass index, educational background, socioeconomic status, tobacco and alcohol use, and medication use, and they scored similarly on measures of health-related quality of life, functional capacity, and the Unified Huntington’s Disease Rating Scale (UHDRS) for motor, behavioral, and cognitive status.
IA status was not associated with any significant differences between cases and controls who were younger than age 60. However, after age 60, participants with IAs had higher median total UHDRS motor scores (13.0 vs. 2.0) and were more likely to show mild gait abnormalities, chorea, and bradykinesia. In the small subgroup of patients who were followed longitudinally for up to 2 years, those with IAs showed faster cognitive decline over time; however, longer follow-up of larger groups of participants is needed to confirm this trend, the investigators said.
In addition, study participants with IAs reported lower quality of life and a greater degree of apathy than those without IAs, but these differences did not reach statistical significance. Whether apathy and poor quality of life reflect Huntington’s disease or simply correlate with normal aging could not be determined.
This study was limited by the relatively small number of participants with IAs, but the findings suggest that IAs “could produce a mild phenotype” of Huntington’s disease in some carriers after age 60. Alternatively, “clinical manifestations of Huntington’s disease in patents with IA might also be potentially accelerated by medical conditions, treatments, and environmental factors” associated with normal aging, Dr. Cubo and her associates added.
This study was supported by the European Huntington’s Disease Registry. Dr. Cubo reported receiving consulting fees from UCB, Allergan, and AbbVie.
FROM NEUROLOGY
Key clinical point: Asymptomatic people who carry an intermediate number (27-35) of CAG repeats on the huntingtin gene may be at increased risk for mild symptoms after age 60.
Major finding: After age 60, people with intermediate alleles had higher median UHDRS motor scores (13.0) than did people with fewer CAG repeats (2.0).
Data source: A retrospective, international, observational study involving 76 asymptomatic adults with 27-35 CAG repeats and 581 with fewer than 27 CAG repeats.
Disclosures: This study was supported by the European Huntington’s Disease Registry. Dr. Cubo reported receiving consulting fees from UCB, Allergan, and AbbVie.

Feds sue to block mega-mergers by health insurers
The U.S. Department of Justice (DOJ) is suing to block two mega-mergers between four of the largest health insurers in the nation, claiming the alignments will harm competition and reduce patient choice.
The DOJ and a number of state attorneys general filed legal challenges July 21 in the U.S. District Court for the District of Columbia seeking to ban Anthem’s proposed acquisition of Cigna and Aetna’s proposed acquisition of Humana. The lawsuits allege the two mergers – valued at $54 billion and $37 billion respectively – would negatively affect doctors, patients, and employers, by limiting price competition, reducing benefits, decreasing incentives to provide innovative wellness programs, and lowering quality of care.

“These mergers would restrict competition for health insurance products sold in markets across the country and would give tremendous power over the nation’s health insurance industry to just three large companies,” U.S. Attorney General Loretta E. Lynch said in a statement. “Our actions seek to preserve competition that keeps premiums down and drives insurers to collaborate with doctors and hospitals to provide better health care for all Americans.”
Anthem called the lawsuit “an unfortunate and misguided step backward for access to affordable health care.
“The DOJ’s action is based on a flawed analysis and misunderstanding of the dynamic, competitive, and highly regulated health care landscape and is inconsistent with the way that the DOJ has reviewed past health care transactions,” Anthem officials said in a statement. “Anthem has an unwavering commitment to enhancing access to affordable health care, and the benefits and efficiencies from its merger with Cigna is one way that Anthem will continue its mission of improving consumer choice, quality, and affordability.”
In a statement, Cigna officials said that company is evaluating its options given the nature of the concerns raised by the DOJ. Aetna and Humana meanwhile, vowed to vigorously defend their pending merger.
“A combined company will result in a broader choice of products, access to higher quality, and more affordable care, and a better overall experience for consumers,” according to a joint statement. “Aetna and Humana look forward to making this clear in court, where a judge will review the transaction based on its merits.”
Anthem’s proposed acquisition of Cigna would be the largest merger in the history of the health insurance industry, according to the DOJ. The companies began talks of a possible merger in early 2014 and Anthem agreed to acquire Cigna for $54 billion in 2015, according to court documents.
Meanwhile, Aetna began inquiring about a deal with Humana in March 2015, entering into a definitive agreement to acquire Humana for $37 billion later that year. The two proposed mergers have been closely watched by the DOJ and other regulatory agencies from the start.
The DOJ’s suit against Anthem and Cigna alleges the merger would substantially reduce competition for millions of patients who receive commercial health insurance coverage, from large-group employers in at least 35 metropolitan areas and from public exchanges created by the Affordable Care Act. The elimination of Cigna also threatens competition among commercial insurers for the purchase of health care services from hospitals, physicians, and other providers, the suit alleges. Eleven states and the District of Columbia joined the department’s challenge of the Cigna acquisition.
The government’s challenge against Aetna and Humana alleges the merger would greatly reduce Medicare Advantage competition in more than 350 counties in 21 states, affecting more than 1.5 million Medicare Advantage patients. The lawsuit also claims that Aetna’s purchase of Humana would substantially reduce competition to sell commercial health insurance to individuals and families on the public exchanges in 17 counties in Florida, Georgia, and Missouri. Eight states and the District of Columbia joined the department’s challenge of the Humana acquisition.
The American Medical Association voiced support for the lawsuit, condemning the proposed transactions as moves that will lessen competition and choice. In 2015, AMA issued special analyses showing that the combined impact of the proposed Anthem/Cigna and Aetna/Humana mergers and urged the federal government to block the transactions.

“The prospect of reducing five national health insurance carriers to just three is unacceptable,” AMA President Andrew W. Gurman, MD, said in a statement. “[The] action by the DOJ acknowledges the AMA’s concern that patients’ interests can be harmed when big insurers acquire rivals and develop strangleholds on local markets. Allowing commercial health insurers to become too big and exert control over the delivery of health care would be bad for patients and vitality of the nation’s health care system.”
On Twitter @legal_med
The U.S. Department of Justice (DOJ) is suing to block two mega-mergers between four of the largest health insurers in the nation, claiming the alignments will harm competition and reduce patient choice.
The DOJ and a number of state attorneys general filed legal challenges July 21 in the U.S. District Court for the District of Columbia seeking to ban Anthem’s proposed acquisition of Cigna and Aetna’s proposed acquisition of Humana. The lawsuits allege the two mergers – valued at $54 billion and $37 billion respectively – would negatively affect doctors, patients, and employers, by limiting price competition, reducing benefits, decreasing incentives to provide innovative wellness programs, and lowering quality of care.
“These mergers would restrict competition for health insurance products sold in markets across the country and would give tremendous power over the nation’s health insurance industry to just three large companies,” U.S. Attorney General Loretta E. Lynch said in a statement. “Our actions seek to preserve competition that keeps premiums down and drives insurers to collaborate with doctors and hospitals to provide better health care for all Americans.”
Anthem called the lawsuit “an unfortunate and misguided step backward for access to affordable health care.
“The DOJ’s action is based on a flawed analysis and misunderstanding of the dynamic, competitive, and highly regulated health care landscape and is inconsistent with the way that the DOJ has reviewed past health care transactions,” Anthem officials said in a statement. “Anthem has an unwavering commitment to enhancing access to affordable health care, and the benefits and efficiencies from its merger with Cigna is one way that Anthem will continue its mission of improving consumer choice, quality, and affordability.”
In a statement, Cigna officials said that company is evaluating its options given the nature of the concerns raised by the DOJ. Aetna and Humana meanwhile, vowed to vigorously defend their pending merger.
“A combined company will result in a broader choice of products, access to higher quality, and more affordable care, and a better overall experience for consumers,” according to a joint statement. “Aetna and Humana look forward to making this clear in court, where a judge will review the transaction based on its merits.”
Anthem’s proposed acquisition of Cigna would be the largest merger in the history of the health insurance industry, according to the DOJ. The companies began talks of a possible merger in early 2014 and Anthem agreed to acquire Cigna for $54 billion in 2015, according to court documents.
Meanwhile, Aetna began inquiring about a deal with Humana in March 2015, entering into a definitive agreement to acquire Humana for $37 billion later that year. The two proposed mergers have been closely watched by the DOJ and other regulatory agencies from the start.
The DOJ’s suit against Anthem and Cigna alleges the merger would substantially reduce competition for millions of patients who receive commercial health insurance coverage, from large-group employers in at least 35 metropolitan areas and from public exchanges created by the Affordable Care Act. The elimination of Cigna also threatens competition among commercial insurers for the purchase of health care services from hospitals, physicians, and other providers, the suit alleges. Eleven states and the District of Columbia joined the department’s challenge of the Cigna acquisition.
The government’s challenge against Aetna and Humana alleges the merger would greatly reduce Medicare Advantage competition in more than 350 counties in 21 states, affecting more than 1.5 million Medicare Advantage patients. The lawsuit also claims that Aetna’s purchase of Humana would substantially reduce competition to sell commercial health insurance to individuals and families on the public exchanges in 17 counties in Florida, Georgia, and Missouri. Eight states and the District of Columbia joined the department’s challenge of the Humana acquisition.
The American Medical Association voiced support for the lawsuit, condemning the proposed transactions as moves that will lessen competition and choice. In 2015, AMA issued special analyses showing that the combined impact of the proposed Anthem/Cigna and Aetna/Humana mergers and urged the federal government to block the transactions.
“The prospect of reducing five national health insurance carriers to just three is unacceptable,” AMA President Andrew W. Gurman, MD, said in a statement. “[The] action by the DOJ acknowledges the AMA’s concern that patients’ interests can be harmed when big insurers acquire rivals and develop strangleholds on local markets. Allowing commercial health insurers to become too big and exert control over the delivery of health care would be bad for patients and vitality of the nation’s health care system.”
On Twitter @legal_med
The U.S. Department of Justice (DOJ) is suing to block two mega-mergers between four of the largest health insurers in the nation, claiming the alignments will harm competition and reduce patient choice.
The DOJ and a number of state attorneys general filed legal challenges July 21 in the U.S. District Court for the District of Columbia seeking to ban Anthem’s proposed acquisition of Cigna and Aetna’s proposed acquisition of Humana. The lawsuits allege the two mergers – valued at $54 billion and $37 billion respectively – would negatively affect doctors, patients, and employers, by limiting price competition, reducing benefits, decreasing incentives to provide innovative wellness programs, and lowering quality of care.
“These mergers would restrict competition for health insurance products sold in markets across the country and would give tremendous power over the nation’s health insurance industry to just three large companies,” U.S. Attorney General Loretta E. Lynch said in a statement. “Our actions seek to preserve competition that keeps premiums down and drives insurers to collaborate with doctors and hospitals to provide better health care for all Americans.”
Anthem called the lawsuit “an unfortunate and misguided step backward for access to affordable health care.
“The DOJ’s action is based on a flawed analysis and misunderstanding of the dynamic, competitive, and highly regulated health care landscape and is inconsistent with the way that the DOJ has reviewed past health care transactions,” Anthem officials said in a statement. “Anthem has an unwavering commitment to enhancing access to affordable health care, and the benefits and efficiencies from its merger with Cigna is one way that Anthem will continue its mission of improving consumer choice, quality, and affordability.”
In a statement, Cigna officials said that company is evaluating its options given the nature of the concerns raised by the DOJ. Aetna and Humana meanwhile, vowed to vigorously defend their pending merger.
“A combined company will result in a broader choice of products, access to higher quality, and more affordable care, and a better overall experience for consumers,” according to a joint statement. “Aetna and Humana look forward to making this clear in court, where a judge will review the transaction based on its merits.”
Anthem’s proposed acquisition of Cigna would be the largest merger in the history of the health insurance industry, according to the DOJ. The companies began talks of a possible merger in early 2014 and Anthem agreed to acquire Cigna for $54 billion in 2015, according to court documents.
Meanwhile, Aetna began inquiring about a deal with Humana in March 2015, entering into a definitive agreement to acquire Humana for $37 billion later that year. The two proposed mergers have been closely watched by the DOJ and other regulatory agencies from the start.
The DOJ’s suit against Anthem and Cigna alleges the merger would substantially reduce competition for millions of patients who receive commercial health insurance coverage, from large-group employers in at least 35 metropolitan areas and from public exchanges created by the Affordable Care Act. The elimination of Cigna also threatens competition among commercial insurers for the purchase of health care services from hospitals, physicians, and other providers, the suit alleges. Eleven states and the District of Columbia joined the department’s challenge of the Cigna acquisition.
The government’s challenge against Aetna and Humana alleges the merger would greatly reduce Medicare Advantage competition in more than 350 counties in 21 states, affecting more than 1.5 million Medicare Advantage patients. The lawsuit also claims that Aetna’s purchase of Humana would substantially reduce competition to sell commercial health insurance to individuals and families on the public exchanges in 17 counties in Florida, Georgia, and Missouri. Eight states and the District of Columbia joined the department’s challenge of the Humana acquisition.
The American Medical Association voiced support for the lawsuit, condemning the proposed transactions as moves that will lessen competition and choice. In 2015, AMA issued special analyses showing that the combined impact of the proposed Anthem/Cigna and Aetna/Humana mergers and urged the federal government to block the transactions.
“The prospect of reducing five national health insurance carriers to just three is unacceptable,” AMA President Andrew W. Gurman, MD, said in a statement. “[The] action by the DOJ acknowledges the AMA’s concern that patients’ interests can be harmed when big insurers acquire rivals and develop strangleholds on local markets. Allowing commercial health insurers to become too big and exert control over the delivery of health care would be bad for patients and vitality of the nation’s health care system.”
On Twitter @legal_med
