User login
Long-term Cosmetic Use of Botulinum Toxin Type A
In the United States, the cosmetic use of botulinum toxin type A (BTX-A) has continued to grow over the last 15 years, according to multispecialty data recently released by the American Society for Aesthetic Plastic Surgery. During these years, many of our patients, if not ourselves, have undergone treatment faithfully every 3 to 6 months to combat the signs of aging. Subsequently, with the monitoring of adverse events (AEs), the US Food and Drug Administration has issued a black box warning that covers serious side effects—respiratory compromise and death—associated with treatment, yet most of what is listed in the black box warning pertains to medical use rather than cosmetic use. However, with the ever-growing indications for BTX-A, we must be cognizant of the fact that our patients may be receiving concomitant treatment with BTX-A for medical conditions (eg, migraines, hyperhidrosis, achalasia, dysphonia, dystonia, strabismus) by other specialists. Thus, there is a need to understand the potential side effects associated with BTX-A treatments and long-term consequences.
In a January 21 article published online in Pharmacology Yiannakopoulou looked at national monitoring programs through the US Food and Drug Administration and the Danish Medicines Agency. Many of the AEs reported were related to medical use of BTX-A, including anaphylaxis, death, generalized weakness, and dysphagia. Serious AEs related to the cosmetic use of BTX-A included thyroid eye disease, sarcoidal granuloma, pseudoaneurysm of the frontal branch of the superior temporal artery, and severe respiratory failure. Additionally, a patient receiving BTX-A for palmar and axillary hyperhidrosis developed botulinumlike generalized weakness. Upon review of epidemiological studies, the incidence and types of AEs were covered. The vast majority of these events were related to the medical use of BTX-A, which could stem from the lack of long-term studies on cosmetic patients and the lack of reporting of many AEs. The author summarized that minimizing potential AEs relies on proper injection technique, proper storage of the medication, proper dosing, and thorough knowledge of the anatomy.
What’s the issue?
Botulinum toxin type A remains one of the most gratifying treatments for both physicians and patients alike. However, with the potential for abuse, as in the case of the recent fake Botox Cosmetic that has shown up on the market in the United States), we must remain vigilant for AEs. Furthermore, we must continue to emphasize to patients that it is a medical treatment and deserves all the attention and respect we give to other medical interventions. However, as the Yiannakopoulou review has shown, proper injection techniques have a low rate of AEs in the cosmetic use of BTX-A. Have you seen an increase in the number of cosmetic BTX-A patients receiving concomitant treatments with BTX-A for medical conditions? If so, how do you manage them?
In the United States, the cosmetic use of botulinum toxin type A (BTX-A) has continued to grow over the last 15 years, according to multispecialty data recently released by the American Society for Aesthetic Plastic Surgery. During these years, many of our patients, if not ourselves, have undergone treatment faithfully every 3 to 6 months to combat the signs of aging. Subsequently, with the monitoring of adverse events (AEs), the US Food and Drug Administration has issued a black box warning that covers serious side effects—respiratory compromise and death—associated with treatment, yet most of what is listed in the black box warning pertains to medical use rather than cosmetic use. However, with the ever-growing indications for BTX-A, we must be cognizant of the fact that our patients may be receiving concomitant treatment with BTX-A for medical conditions (eg, migraines, hyperhidrosis, achalasia, dysphonia, dystonia, strabismus) by other specialists. Thus, there is a need to understand the potential side effects associated with BTX-A treatments and long-term consequences.
In a January 21 article published online in Pharmacology Yiannakopoulou looked at national monitoring programs through the US Food and Drug Administration and the Danish Medicines Agency. Many of the AEs reported were related to medical use of BTX-A, including anaphylaxis, death, generalized weakness, and dysphagia. Serious AEs related to the cosmetic use of BTX-A included thyroid eye disease, sarcoidal granuloma, pseudoaneurysm of the frontal branch of the superior temporal artery, and severe respiratory failure. Additionally, a patient receiving BTX-A for palmar and axillary hyperhidrosis developed botulinumlike generalized weakness. Upon review of epidemiological studies, the incidence and types of AEs were covered. The vast majority of these events were related to the medical use of BTX-A, which could stem from the lack of long-term studies on cosmetic patients and the lack of reporting of many AEs. The author summarized that minimizing potential AEs relies on proper injection technique, proper storage of the medication, proper dosing, and thorough knowledge of the anatomy.
What’s the issue?
Botulinum toxin type A remains one of the most gratifying treatments for both physicians and patients alike. However, with the potential for abuse, as in the case of the recent fake Botox Cosmetic that has shown up on the market in the United States), we must remain vigilant for AEs. Furthermore, we must continue to emphasize to patients that it is a medical treatment and deserves all the attention and respect we give to other medical interventions. However, as the Yiannakopoulou review has shown, proper injection techniques have a low rate of AEs in the cosmetic use of BTX-A. Have you seen an increase in the number of cosmetic BTX-A patients receiving concomitant treatments with BTX-A for medical conditions? If so, how do you manage them?
In the United States, the cosmetic use of botulinum toxin type A (BTX-A) has continued to grow over the last 15 years, according to multispecialty data recently released by the American Society for Aesthetic Plastic Surgery. During these years, many of our patients, if not ourselves, have undergone treatment faithfully every 3 to 6 months to combat the signs of aging. Subsequently, with the monitoring of adverse events (AEs), the US Food and Drug Administration has issued a black box warning that covers serious side effects—respiratory compromise and death—associated with treatment, yet most of what is listed in the black box warning pertains to medical use rather than cosmetic use. However, with the ever-growing indications for BTX-A, we must be cognizant of the fact that our patients may be receiving concomitant treatment with BTX-A for medical conditions (eg, migraines, hyperhidrosis, achalasia, dysphonia, dystonia, strabismus) by other specialists. Thus, there is a need to understand the potential side effects associated with BTX-A treatments and long-term consequences.
In a January 21 article published online in Pharmacology Yiannakopoulou looked at national monitoring programs through the US Food and Drug Administration and the Danish Medicines Agency. Many of the AEs reported were related to medical use of BTX-A, including anaphylaxis, death, generalized weakness, and dysphagia. Serious AEs related to the cosmetic use of BTX-A included thyroid eye disease, sarcoidal granuloma, pseudoaneurysm of the frontal branch of the superior temporal artery, and severe respiratory failure. Additionally, a patient receiving BTX-A for palmar and axillary hyperhidrosis developed botulinumlike generalized weakness. Upon review of epidemiological studies, the incidence and types of AEs were covered. The vast majority of these events were related to the medical use of BTX-A, which could stem from the lack of long-term studies on cosmetic patients and the lack of reporting of many AEs. The author summarized that minimizing potential AEs relies on proper injection technique, proper storage of the medication, proper dosing, and thorough knowledge of the anatomy.
What’s the issue?
Botulinum toxin type A remains one of the most gratifying treatments for both physicians and patients alike. However, with the potential for abuse, as in the case of the recent fake Botox Cosmetic that has shown up on the market in the United States), we must remain vigilant for AEs. Furthermore, we must continue to emphasize to patients that it is a medical treatment and deserves all the attention and respect we give to other medical interventions. However, as the Yiannakopoulou review has shown, proper injection techniques have a low rate of AEs in the cosmetic use of BTX-A. Have you seen an increase in the number of cosmetic BTX-A patients receiving concomitant treatments with BTX-A for medical conditions? If so, how do you manage them?

LISTEN NOW: SHM President Robert Harrington Jr., MD, SFHM, discusses hospital medicine, value of diversity and teamwork
New SHM President Robert Harrington Jr., MD, SFHM, talks about his views on hospital medicine, the society and the value of diversity and teamwork.
New SHM President Robert Harrington Jr., MD, SFHM, talks about his views on hospital medicine, the society and the value of diversity and teamwork.
New SHM President Robert Harrington Jr., MD, SFHM, talks about his views on hospital medicine, the society and the value of diversity and teamwork.
VIDEO: Updating the immune response to nonmelanoma skin cancer
ASHEVILLE, N.C. – Recent advances in basic science have shown how the local immune environment in tissue surrounding nonmelanoma skin cancer compares to adjacent normal tissue.
New Mexico Health Sciences Center’s Dr. Andrew Ondo reviewed the latest research in an interview at the annual meeting of the Noah Worcester Dermatological Society. “Each step along the way is a possible target for the treatment of squamous cell carcinoma,” said Dr. Ondo, who indicated that he had no financial conflicts to disclose.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ASHEVILLE, N.C. – Recent advances in basic science have shown how the local immune environment in tissue surrounding nonmelanoma skin cancer compares to adjacent normal tissue.
New Mexico Health Sciences Center’s Dr. Andrew Ondo reviewed the latest research in an interview at the annual meeting of the Noah Worcester Dermatological Society. “Each step along the way is a possible target for the treatment of squamous cell carcinoma,” said Dr. Ondo, who indicated that he had no financial conflicts to disclose.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ASHEVILLE, N.C. – Recent advances in basic science have shown how the local immune environment in tissue surrounding nonmelanoma skin cancer compares to adjacent normal tissue.
New Mexico Health Sciences Center’s Dr. Andrew Ondo reviewed the latest research in an interview at the annual meeting of the Noah Worcester Dermatological Society. “Each step along the way is a possible target for the treatment of squamous cell carcinoma,” said Dr. Ondo, who indicated that he had no financial conflicts to disclose.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
EXPERT ANALYSIS FROM NOAH 57

Prepping for the Boards? We can help

The results of the 2015 National Residency Match Program were announced on March 20. For family medicine, the glass was either half empty or half full, depending on your point of view.
On the plus side, 84 more family medicine positions were offered compared to 2014 (3216 vs 3132) and 60 more positions were filled, for a total of 3060 new family medicine residents in 2015.1 This was far more than matched in gloomy 2009, when only 2555 residents chose family medicine. On the negative side of the balance sheet, there will be 233 fewer family medicine residents this year than matched at the peak of medical student interest in family medicine in 1998.
I’m a glass half full kind of guy, so I am delighted that the trend of increased medical student interest in family medicine continues. According to Merritt Hawkins, a national recruitment firm, family medicine has been the top recruited specialty for several years. The firm reports that starting salaries for family physicians increased by nearly 12% from 2010/11 to 2013/14, which was a higher rate than that of most other specialties.2 So there is reason to be optimistic about the future of our specialty.
However, to be card-carrying family physicians, our new residents must take the American Board of Family Medicine certification exam, and not all pass on their first attempt. A 2013 study of family medicine residency graduates found that only 86% of graduates passed the board exam on their first try.3
We can help. In addition to the evidence-based reviews published in The Journal of Family Practice (JFP), we have launched a new feature called Residents’ Rapid Review (RRR) to provide an additional resource for residents.
RRR is a monthly 5-question evidence-based quiz prepared by primary care faculty, including current and former residency program directors. After residents click on their answer, the system lets them know whether they’re right and provides the correct answer with an explanation and references. Monthly notifications are sent out to all jfponline.com registered users alerting them that a new quiz is available. (Not a registered user on the site? Sign up at jfponline.com/residents_reg.)
Getting ready for the recertification exam? The RRR quizzes can help you, too. Check out the latest quiz, today!
1. National Resident Matching Program. Advance data tables. 2015 main residency match. National Resident Matching Program Web site. Available at: http://www.nrmp.org/wp-content/uploads/2015/03/ADT2015_final.pdf. Accessed April 16, 2015.
2. Merritt Hawkins. 2014 review of physician and advanced practitioner recruiting incentives. Irving, TX: Merritt Hawkins; 2014:9.
3. Falcone JL, Middleton DB. Pass rates on the American Board of Family Medicine Certification Exam by residency location and size. J Am Board Fam Med. 2013;26:453-459.
The results of the 2015 National Residency Match Program were announced on March 20. For family medicine, the glass was either half empty or half full, depending on your point of view.
On the plus side, 84 more family medicine positions were offered compared to 2014 (3216 vs 3132) and 60 more positions were filled, for a total of 3060 new family medicine residents in 2015.1 This was far more than matched in gloomy 2009, when only 2555 residents chose family medicine. On the negative side of the balance sheet, there will be 233 fewer family medicine residents this year than matched at the peak of medical student interest in family medicine in 1998.
I’m a glass half full kind of guy, so I am delighted that the trend of increased medical student interest in family medicine continues. According to Merritt Hawkins, a national recruitment firm, family medicine has been the top recruited specialty for several years. The firm reports that starting salaries for family physicians increased by nearly 12% from 2010/11 to 2013/14, which was a higher rate than that of most other specialties.2 So there is reason to be optimistic about the future of our specialty.
However, to be card-carrying family physicians, our new residents must take the American Board of Family Medicine certification exam, and not all pass on their first attempt. A 2013 study of family medicine residency graduates found that only 86% of graduates passed the board exam on their first try.3
We can help. In addition to the evidence-based reviews published in The Journal of Family Practice (JFP), we have launched a new feature called Residents’ Rapid Review (RRR) to provide an additional resource for residents.
RRR is a monthly 5-question evidence-based quiz prepared by primary care faculty, including current and former residency program directors. After residents click on their answer, the system lets them know whether they’re right and provides the correct answer with an explanation and references. Monthly notifications are sent out to all jfponline.com registered users alerting them that a new quiz is available. (Not a registered user on the site? Sign up at jfponline.com/residents_reg.)
Getting ready for the recertification exam? The RRR quizzes can help you, too. Check out the latest quiz, today!
The results of the 2015 National Residency Match Program were announced on March 20. For family medicine, the glass was either half empty or half full, depending on your point of view.
On the plus side, 84 more family medicine positions were offered compared to 2014 (3216 vs 3132) and 60 more positions were filled, for a total of 3060 new family medicine residents in 2015.1 This was far more than matched in gloomy 2009, when only 2555 residents chose family medicine. On the negative side of the balance sheet, there will be 233 fewer family medicine residents this year than matched at the peak of medical student interest in family medicine in 1998.
I’m a glass half full kind of guy, so I am delighted that the trend of increased medical student interest in family medicine continues. According to Merritt Hawkins, a national recruitment firm, family medicine has been the top recruited specialty for several years. The firm reports that starting salaries for family physicians increased by nearly 12% from 2010/11 to 2013/14, which was a higher rate than that of most other specialties.2 So there is reason to be optimistic about the future of our specialty.
However, to be card-carrying family physicians, our new residents must take the American Board of Family Medicine certification exam, and not all pass on their first attempt. A 2013 study of family medicine residency graduates found that only 86% of graduates passed the board exam on their first try.3
We can help. In addition to the evidence-based reviews published in The Journal of Family Practice (JFP), we have launched a new feature called Residents’ Rapid Review (RRR) to provide an additional resource for residents.
RRR is a monthly 5-question evidence-based quiz prepared by primary care faculty, including current and former residency program directors. After residents click on their answer, the system lets them know whether they’re right and provides the correct answer with an explanation and references. Monthly notifications are sent out to all jfponline.com registered users alerting them that a new quiz is available. (Not a registered user on the site? Sign up at jfponline.com/residents_reg.)
Getting ready for the recertification exam? The RRR quizzes can help you, too. Check out the latest quiz, today!
1. National Resident Matching Program. Advance data tables. 2015 main residency match. National Resident Matching Program Web site. Available at: http://www.nrmp.org/wp-content/uploads/2015/03/ADT2015_final.pdf. Accessed April 16, 2015.
2. Merritt Hawkins. 2014 review of physician and advanced practitioner recruiting incentives. Irving, TX: Merritt Hawkins; 2014:9.
3. Falcone JL, Middleton DB. Pass rates on the American Board of Family Medicine Certification Exam by residency location and size. J Am Board Fam Med. 2013;26:453-459.
1. National Resident Matching Program. Advance data tables. 2015 main residency match. National Resident Matching Program Web site. Available at: http://www.nrmp.org/wp-content/uploads/2015/03/ADT2015_final.pdf. Accessed April 16, 2015.
2. Merritt Hawkins. 2014 review of physician and advanced practitioner recruiting incentives. Irving, TX: Merritt Hawkins; 2014:9.
3. Falcone JL, Middleton DB. Pass rates on the American Board of Family Medicine Certification Exam by residency location and size. J Am Board Fam Med. 2013;26:453-459.

Chest pain • shortness of breath • fever and nausea • Dx?
THE CASE
A 38-year-old Hispanic man was brought to the emergency department (ED) after losing consciousness and falling at home, striking his elbow, head, and neck. For the past week, he’d had palpitations, shortness of breath, mild swelling of the lower extremities, fever, nausea, and fatigue. He had also been experiencing squeezing chest pain that worsened with exertion and was only partially relieved by nitroglycerin.
The patient did not have any rashes and denied having contact with anyone who was sick. He said that he’d been bitten by mosquitos during recent outdoor activities. His medical history included hypertension, hemorrhagic basal ganglia stroke, hyperlipidemia, sleep apnea, metabolic syndrome, and gout. The patient denied smoking or using illicit drugs.
In the ED, his temperature was 101°F, heart rate was 112 beats/min, blood pressure was 175/100 mm Hg, and respiratory rate was 18 breaths/min. His head and neck exam was normal, with no neck stiffness. A lung exam revealed bilateral basal crackles, and a neurologic exam showed residual right-sided weakness due to the hemorrhagic stroke one year ago.
Lab test results revealed the following: white blood cell (WBC) count, 13,000/mm3 with relative monocytosis (14%); lymphocytosis (44%) with normal neutrophils and no bands; hemoglobin, 12 g/dL; hematocrit, 36/mm3; and platelets, 300,000/mm3. Liver function tests were within normal limits. Urinalysis was unremarkable. His troponin I level was elevated at 1.385 ng/dL. In addition to the tachycardia, his electrocardiogram (EKG) showed left axis deviation, left atrial enlargement, left anterior fascicular block, and diffuse nonspecific ST and T wave abnormalities. Chest x-ray was unremarkable except for cardiomegaly. A computed tomography (CT) scan of his head showed residual changes from the previous stroke.
The patient was admitted with a provisional diagnosis of systemic inflammatory response syndrome (SIRS), syncope, non–ST elevation myocardial infarction (NSTEMI), and acute heart failure. The patient had continuous EKG monitoring and serial assessments of his troponin levels. He was also given aspirin, metoprolol 25 mg BID, lisinopril 10 mg/d, furosemide 40 mg IV, isosorbide mononitrate 60 mg/d, and atorvastatin 40 mg/d.
The patient’s cardiac enzymes subsequently decreased. A left heart catheterization was performed, which showed minimum irregularities of the left anterior descending artery (< 20% narrowing) and an ejection fraction (EF) of 35%, without any evidence of obstructive coronary artery disease (CAD). An echocardiogram revealed systolic dysfunction, with an EF of 35% to 40% and global hypokinesis without any apical ballooning or pericardial effusion. (An echocardiogram performed 6 months earlier had shown normal systolic function, an EF of 60% to 65%, and no wall motion abnormalities.) Blood, urine, and fungal cultures were negative; stool studies for ova and parasites were also negative. A lower extremity venous Doppler was negative for deep vein thrombosis.
THE DIAGNOSIS
Because our patient had SIRS, troponinemia, acute systolic dysfunction, and global hypokinesis without any evidence of obstructive CAD, we considered a diagnosis of viral myocarditis. Serologic studies for echovirus, coxsackievirus B, parvovirus B19, adenovirus, and human herpesvirus 6 (HHV-6) all came back negative. However, an enzyme-linked immunosorbent assay (ELISA) for West Nile virus (WNV) was positive. WNV infection was confirmed with a positive plaque reduction neutralization test and a positive qualitative polymerase chain reaction (PCR) assay, which established a diagnosis of WNV myocarditis.
DISCUSSION
While most individuals infected with WNV are asymptomatic, 20% to 40% of patients will exhibit symptoms.1-4 Typical presentations of WNV infection include West Nile fever and neuroinvasive disease. West Nile fever is a self-limited illness characterized by a low-grade fever, headache, malaise, back pain, myalgia, and anorexia for 3 to 6 days.2 Neuroinvasive disease caused by WNV may present as encephalitis, meningitis, or flaccid paralysis.5 Atypical presentations of the virus include rhabdomyolysis,6 fatal hemorrhagic fever with multi-organ failure and palpable purpura,7 hepatitis,8 pancreatitis,9 central diabetes insipidus,10 and myocarditis.11
Although WNV has been linked to myocarditismin animals,12 few human cases of WNV myocarditis11,13 or cardiomyopathy14 have been reported. Viral myocarditis often leads to the development of dilated cardiomyopathy, and myocardial damage may result from direct virus-induced cytotoxicity, T cell-mediated immune response to the virus, or apoptosis.15 Some research suggests that immune-mediated mechanisms play a primary role in myocardial damage. Caforio et al16 found that anti-alpha myosin antibodies were present in 34% of myocarditis patients. In a follow-up study, these antibodies were shown to persist for up to 6 months, which far surpasses the viral cardiac replication timeline of 2 to 3 weeks,17 suggesting that damage occurring after that time is primarily an autoimmune process.
The differential diagnosis for WNV myocarditis includes myocardial stunning from demand ischemia related to SIRS, Takotsubo cardiomyopathy (stress cardiomyopathy), and Dressler’s syndrome. For our patient, myocardial stunning from demand ischemia was less likely because he had no obstructive coronary disease or focal hypokinesis. In addition, the persistence of left ventricular systolic dysfunction and global hypokinesis demonstrated in a repeat echocardiogram during follow-up 6 months later reinforced the likelihood of myocarditis.
The patient’s chest pain with syncope, elevated troponin level, and nonspecific EKG changes in the absence of obstructive CAD raised the possibility of Takotsubo cardiomyopathy. The characteristic echocardiogram finding in Takotsubo cardiomyopathy is transient apical ballooning with akinesis or hypokinesis in the apical and/or mid ventricular regions (typical variant) or isolated midventricular hypokinesis (apical sparing variant). Our patient’s echocardiogram did not show any of these focal wall motion abnormalities, but instead showed global hypokinesis. In addition, the persistence of systolic dysfunction during the repeat echocardiogram and the patient’s lack of psychological distress made the diagnosis of Takotsubo cardiomyopathy unlikely.
Dressler’s syndrome, which is also known as post-myocardial infarction (MI) syndrome, typically presents weeks to months after MI as pleuritic chest pain with a pericardial rub, elevated inflammatory markers, typical EKG changes (diffuse ST-segment elevation and PR-segment depression), and pericardial effusion. This did not fit our patient’s presentation.
Supportive care for heart failure is the mainstay of treatment
The standard treatment for WNV myocarditis is supportive care. Diuretics are used as needed for fluid overload, along with angiotensin-converting enzyme inhibitors and beta-blockers for cardiomyopathy with decreased EF.
Our patient’s dyspnea improved with treatment of furosemide 40 mg IV BID, and his blood pressure was controlled with metoprolol 25 mg BID and lisinopril 10 mg BID. His chest pain and fever resolved when his blood pressure improved. He was discharged home after 7 days on the furosemide, metoprolol, and lisinopril, in addition to isosorbide mononitrate 30 mg/d, atorvastatin 40 mg/d, and aspirin 325 mg/d. An echocardiogram performed 6 months later showed persistent systolic dysfunction, with an EF of 35% and global wall motion abnormalities.
THE TAKEAWAY
In addition to acute coronary syndrome, consider alternate etiologies in patients who present with chest pain and elevated cardiac biomarkers, particularly if diagnostic work-up is negative for obstructive coronary artery disease. WNV myocarditis should be considered as a diagnosis when a patient’s symptoms suggest acute coronary syndrome but are accompanied by fever, headache, and other constitutional symptoms, especially during mosquito season or a WNV outbreak.
1. Nash D, Mostashari F, Fine A, et al. The outbreak of West Nile virus infection in the New York City area in 1999. N Engl J Med. 2001;344:1807-1814.
2. Orton SL, Stramer SL, Dodd RY. Self-reported symptoms associated with West Nile virus infection in RNA-positive blood donors. Transfusion. 2006;46:272-277.
3. Brown JA, Factor DL, Tkachenko N, et al. West Nile viremic blood donors and risk factors for subsequent West Nile fever. Vector Borne Zoonotic Dis. 2007;7:479-488.
4. Zou S, Foster GA, Dodd RY, et al. West Nile fever characteristics among viremic persons identified through blood donor screening. J Infect Dis. 2010;202:1354-1361.
5. Davis LE, DeBiasi R, Goade DE, et al. West Nile virus neuroinvasive disease. Ann Neurol. 2006;60:286-300.
6. Montgomery SP, Chow CC, Smith SW, et al. Rhabdomyolysis in patients with west nile encephalitis and meningitis. Vector Borne Zoonotic Dis. 2005;5:252-257.
7. Paddock CD, Nicholson WL, Bhatnagar J, et al. Fatal hemorrhagic fever caused by West Nile virus in the United States. Clin Infect Dis. 2006;42:1527-1535.
8. Georges AJ, Lesbordes JL, Georges-Courbot MC, et al. Fatal hepatitis from West Nile virus. Ann Inst Pasteur Virol. 1988;138:237.
9. Perelman A, Stern J. Acute pancreatitis in West Nile Fever. Am J Trop Med Hyg. 1974;23:1150-1152.
10. Sherman-Weber S, Axelrod P. Central diabetes insipidus complicating West Nile encephalitis. Clin Infect Dis. 2004;38:1042-1043.
11. Pergam SA, DeLong CE, Echevarria L, et al. Myocarditis in West Nile virus infection. Am J Trop Med Hyg. 2006;75:1232-1233.
12. van der Meulen KM, Pensaert MB, Nauwynck HJ. West Nile virus in the vertebrate world. Arch Virol. 2005;150:637-657.
13. Kushawaha A, Jadonath S, Mobarakai N. West nile virus myocarditis causing a fatal arrhythmia: a case report. Cases J. 2009;2:7147.
14. Khouzam RN. Significant cardiomyopathy secondary to West Nile virus infection. South Med J. 2009;102:527-528.
15. Kawai C. From myocarditis to cardiomyopathy: mechanisms of inflammation and cell death: learning from the past for the future. Circulation. 1999;99:1091-1100.
16. Caforio AL, Goldman JH, Haven AJ, et al. Circulating cardiac-specific autoantibodies as markers of autoimmunity in clinical and biopsy-proven myocarditis. The Myocarditis Treatment Trial Investigators. Eur Heart J. 1997;18:270-275.
17. Lauer B, Schannwell M, Kühl U, et al. Antimyosin autoantibodies are associated with deterioration of systolic and diastolic left ventricular function in patients with chronic myocarditis. J Am Coll Cardiol. 2000;35:11-18.
THE CASE
A 38-year-old Hispanic man was brought to the emergency department (ED) after losing consciousness and falling at home, striking his elbow, head, and neck. For the past week, he’d had palpitations, shortness of breath, mild swelling of the lower extremities, fever, nausea, and fatigue. He had also been experiencing squeezing chest pain that worsened with exertion and was only partially relieved by nitroglycerin.
The patient did not have any rashes and denied having contact with anyone who was sick. He said that he’d been bitten by mosquitos during recent outdoor activities. His medical history included hypertension, hemorrhagic basal ganglia stroke, hyperlipidemia, sleep apnea, metabolic syndrome, and gout. The patient denied smoking or using illicit drugs.
In the ED, his temperature was 101°F, heart rate was 112 beats/min, blood pressure was 175/100 mm Hg, and respiratory rate was 18 breaths/min. His head and neck exam was normal, with no neck stiffness. A lung exam revealed bilateral basal crackles, and a neurologic exam showed residual right-sided weakness due to the hemorrhagic stroke one year ago.
Lab test results revealed the following: white blood cell (WBC) count, 13,000/mm3 with relative monocytosis (14%); lymphocytosis (44%) with normal neutrophils and no bands; hemoglobin, 12 g/dL; hematocrit, 36/mm3; and platelets, 300,000/mm3. Liver function tests were within normal limits. Urinalysis was unremarkable. His troponin I level was elevated at 1.385 ng/dL. In addition to the tachycardia, his electrocardiogram (EKG) showed left axis deviation, left atrial enlargement, left anterior fascicular block, and diffuse nonspecific ST and T wave abnormalities. Chest x-ray was unremarkable except for cardiomegaly. A computed tomography (CT) scan of his head showed residual changes from the previous stroke.
The patient was admitted with a provisional diagnosis of systemic inflammatory response syndrome (SIRS), syncope, non–ST elevation myocardial infarction (NSTEMI), and acute heart failure. The patient had continuous EKG monitoring and serial assessments of his troponin levels. He was also given aspirin, metoprolol 25 mg BID, lisinopril 10 mg/d, furosemide 40 mg IV, isosorbide mononitrate 60 mg/d, and atorvastatin 40 mg/d.
The patient’s cardiac enzymes subsequently decreased. A left heart catheterization was performed, which showed minimum irregularities of the left anterior descending artery (< 20% narrowing) and an ejection fraction (EF) of 35%, without any evidence of obstructive coronary artery disease (CAD). An echocardiogram revealed systolic dysfunction, with an EF of 35% to 40% and global hypokinesis without any apical ballooning or pericardial effusion. (An echocardiogram performed 6 months earlier had shown normal systolic function, an EF of 60% to 65%, and no wall motion abnormalities.) Blood, urine, and fungal cultures were negative; stool studies for ova and parasites were also negative. A lower extremity venous Doppler was negative for deep vein thrombosis.
THE DIAGNOSIS
Because our patient had SIRS, troponinemia, acute systolic dysfunction, and global hypokinesis without any evidence of obstructive CAD, we considered a diagnosis of viral myocarditis. Serologic studies for echovirus, coxsackievirus B, parvovirus B19, adenovirus, and human herpesvirus 6 (HHV-6) all came back negative. However, an enzyme-linked immunosorbent assay (ELISA) for West Nile virus (WNV) was positive. WNV infection was confirmed with a positive plaque reduction neutralization test and a positive qualitative polymerase chain reaction (PCR) assay, which established a diagnosis of WNV myocarditis.
DISCUSSION
While most individuals infected with WNV are asymptomatic, 20% to 40% of patients will exhibit symptoms.1-4 Typical presentations of WNV infection include West Nile fever and neuroinvasive disease. West Nile fever is a self-limited illness characterized by a low-grade fever, headache, malaise, back pain, myalgia, and anorexia for 3 to 6 days.2 Neuroinvasive disease caused by WNV may present as encephalitis, meningitis, or flaccid paralysis.5 Atypical presentations of the virus include rhabdomyolysis,6 fatal hemorrhagic fever with multi-organ failure and palpable purpura,7 hepatitis,8 pancreatitis,9 central diabetes insipidus,10 and myocarditis.11
Although WNV has been linked to myocarditismin animals,12 few human cases of WNV myocarditis11,13 or cardiomyopathy14 have been reported. Viral myocarditis often leads to the development of dilated cardiomyopathy, and myocardial damage may result from direct virus-induced cytotoxicity, T cell-mediated immune response to the virus, or apoptosis.15 Some research suggests that immune-mediated mechanisms play a primary role in myocardial damage. Caforio et al16 found that anti-alpha myosin antibodies were present in 34% of myocarditis patients. In a follow-up study, these antibodies were shown to persist for up to 6 months, which far surpasses the viral cardiac replication timeline of 2 to 3 weeks,17 suggesting that damage occurring after that time is primarily an autoimmune process.
The differential diagnosis for WNV myocarditis includes myocardial stunning from demand ischemia related to SIRS, Takotsubo cardiomyopathy (stress cardiomyopathy), and Dressler’s syndrome. For our patient, myocardial stunning from demand ischemia was less likely because he had no obstructive coronary disease or focal hypokinesis. In addition, the persistence of left ventricular systolic dysfunction and global hypokinesis demonstrated in a repeat echocardiogram during follow-up 6 months later reinforced the likelihood of myocarditis.
The patient’s chest pain with syncope, elevated troponin level, and nonspecific EKG changes in the absence of obstructive CAD raised the possibility of Takotsubo cardiomyopathy. The characteristic echocardiogram finding in Takotsubo cardiomyopathy is transient apical ballooning with akinesis or hypokinesis in the apical and/or mid ventricular regions (typical variant) or isolated midventricular hypokinesis (apical sparing variant). Our patient’s echocardiogram did not show any of these focal wall motion abnormalities, but instead showed global hypokinesis. In addition, the persistence of systolic dysfunction during the repeat echocardiogram and the patient’s lack of psychological distress made the diagnosis of Takotsubo cardiomyopathy unlikely.
Dressler’s syndrome, which is also known as post-myocardial infarction (MI) syndrome, typically presents weeks to months after MI as pleuritic chest pain with a pericardial rub, elevated inflammatory markers, typical EKG changes (diffuse ST-segment elevation and PR-segment depression), and pericardial effusion. This did not fit our patient’s presentation.
Supportive care for heart failure is the mainstay of treatment
The standard treatment for WNV myocarditis is supportive care. Diuretics are used as needed for fluid overload, along with angiotensin-converting enzyme inhibitors and beta-blockers for cardiomyopathy with decreased EF.
Our patient’s dyspnea improved with treatment of furosemide 40 mg IV BID, and his blood pressure was controlled with metoprolol 25 mg BID and lisinopril 10 mg BID. His chest pain and fever resolved when his blood pressure improved. He was discharged home after 7 days on the furosemide, metoprolol, and lisinopril, in addition to isosorbide mononitrate 30 mg/d, atorvastatin 40 mg/d, and aspirin 325 mg/d. An echocardiogram performed 6 months later showed persistent systolic dysfunction, with an EF of 35% and global wall motion abnormalities.
THE TAKEAWAY
In addition to acute coronary syndrome, consider alternate etiologies in patients who present with chest pain and elevated cardiac biomarkers, particularly if diagnostic work-up is negative for obstructive coronary artery disease. WNV myocarditis should be considered as a diagnosis when a patient’s symptoms suggest acute coronary syndrome but are accompanied by fever, headache, and other constitutional symptoms, especially during mosquito season or a WNV outbreak.
THE CASE
A 38-year-old Hispanic man was brought to the emergency department (ED) after losing consciousness and falling at home, striking his elbow, head, and neck. For the past week, he’d had palpitations, shortness of breath, mild swelling of the lower extremities, fever, nausea, and fatigue. He had also been experiencing squeezing chest pain that worsened with exertion and was only partially relieved by nitroglycerin.
The patient did not have any rashes and denied having contact with anyone who was sick. He said that he’d been bitten by mosquitos during recent outdoor activities. His medical history included hypertension, hemorrhagic basal ganglia stroke, hyperlipidemia, sleep apnea, metabolic syndrome, and gout. The patient denied smoking or using illicit drugs.
In the ED, his temperature was 101°F, heart rate was 112 beats/min, blood pressure was 175/100 mm Hg, and respiratory rate was 18 breaths/min. His head and neck exam was normal, with no neck stiffness. A lung exam revealed bilateral basal crackles, and a neurologic exam showed residual right-sided weakness due to the hemorrhagic stroke one year ago.
Lab test results revealed the following: white blood cell (WBC) count, 13,000/mm3 with relative monocytosis (14%); lymphocytosis (44%) with normal neutrophils and no bands; hemoglobin, 12 g/dL; hematocrit, 36/mm3; and platelets, 300,000/mm3. Liver function tests were within normal limits. Urinalysis was unremarkable. His troponin I level was elevated at 1.385 ng/dL. In addition to the tachycardia, his electrocardiogram (EKG) showed left axis deviation, left atrial enlargement, left anterior fascicular block, and diffuse nonspecific ST and T wave abnormalities. Chest x-ray was unremarkable except for cardiomegaly. A computed tomography (CT) scan of his head showed residual changes from the previous stroke.
The patient was admitted with a provisional diagnosis of systemic inflammatory response syndrome (SIRS), syncope, non–ST elevation myocardial infarction (NSTEMI), and acute heart failure. The patient had continuous EKG monitoring and serial assessments of his troponin levels. He was also given aspirin, metoprolol 25 mg BID, lisinopril 10 mg/d, furosemide 40 mg IV, isosorbide mononitrate 60 mg/d, and atorvastatin 40 mg/d.
The patient’s cardiac enzymes subsequently decreased. A left heart catheterization was performed, which showed minimum irregularities of the left anterior descending artery (< 20% narrowing) and an ejection fraction (EF) of 35%, without any evidence of obstructive coronary artery disease (CAD). An echocardiogram revealed systolic dysfunction, with an EF of 35% to 40% and global hypokinesis without any apical ballooning or pericardial effusion. (An echocardiogram performed 6 months earlier had shown normal systolic function, an EF of 60% to 65%, and no wall motion abnormalities.) Blood, urine, and fungal cultures were negative; stool studies for ova and parasites were also negative. A lower extremity venous Doppler was negative for deep vein thrombosis.
THE DIAGNOSIS
Because our patient had SIRS, troponinemia, acute systolic dysfunction, and global hypokinesis without any evidence of obstructive CAD, we considered a diagnosis of viral myocarditis. Serologic studies for echovirus, coxsackievirus B, parvovirus B19, adenovirus, and human herpesvirus 6 (HHV-6) all came back negative. However, an enzyme-linked immunosorbent assay (ELISA) for West Nile virus (WNV) was positive. WNV infection was confirmed with a positive plaque reduction neutralization test and a positive qualitative polymerase chain reaction (PCR) assay, which established a diagnosis of WNV myocarditis.
DISCUSSION
While most individuals infected with WNV are asymptomatic, 20% to 40% of patients will exhibit symptoms.1-4 Typical presentations of WNV infection include West Nile fever and neuroinvasive disease. West Nile fever is a self-limited illness characterized by a low-grade fever, headache, malaise, back pain, myalgia, and anorexia for 3 to 6 days.2 Neuroinvasive disease caused by WNV may present as encephalitis, meningitis, or flaccid paralysis.5 Atypical presentations of the virus include rhabdomyolysis,6 fatal hemorrhagic fever with multi-organ failure and palpable purpura,7 hepatitis,8 pancreatitis,9 central diabetes insipidus,10 and myocarditis.11
Although WNV has been linked to myocarditismin animals,12 few human cases of WNV myocarditis11,13 or cardiomyopathy14 have been reported. Viral myocarditis often leads to the development of dilated cardiomyopathy, and myocardial damage may result from direct virus-induced cytotoxicity, T cell-mediated immune response to the virus, or apoptosis.15 Some research suggests that immune-mediated mechanisms play a primary role in myocardial damage. Caforio et al16 found that anti-alpha myosin antibodies were present in 34% of myocarditis patients. In a follow-up study, these antibodies were shown to persist for up to 6 months, which far surpasses the viral cardiac replication timeline of 2 to 3 weeks,17 suggesting that damage occurring after that time is primarily an autoimmune process.
The differential diagnosis for WNV myocarditis includes myocardial stunning from demand ischemia related to SIRS, Takotsubo cardiomyopathy (stress cardiomyopathy), and Dressler’s syndrome. For our patient, myocardial stunning from demand ischemia was less likely because he had no obstructive coronary disease or focal hypokinesis. In addition, the persistence of left ventricular systolic dysfunction and global hypokinesis demonstrated in a repeat echocardiogram during follow-up 6 months later reinforced the likelihood of myocarditis.
The patient’s chest pain with syncope, elevated troponin level, and nonspecific EKG changes in the absence of obstructive CAD raised the possibility of Takotsubo cardiomyopathy. The characteristic echocardiogram finding in Takotsubo cardiomyopathy is transient apical ballooning with akinesis or hypokinesis in the apical and/or mid ventricular regions (typical variant) or isolated midventricular hypokinesis (apical sparing variant). Our patient’s echocardiogram did not show any of these focal wall motion abnormalities, but instead showed global hypokinesis. In addition, the persistence of systolic dysfunction during the repeat echocardiogram and the patient’s lack of psychological distress made the diagnosis of Takotsubo cardiomyopathy unlikely.
Dressler’s syndrome, which is also known as post-myocardial infarction (MI) syndrome, typically presents weeks to months after MI as pleuritic chest pain with a pericardial rub, elevated inflammatory markers, typical EKG changes (diffuse ST-segment elevation and PR-segment depression), and pericardial effusion. This did not fit our patient’s presentation.
Supportive care for heart failure is the mainstay of treatment
The standard treatment for WNV myocarditis is supportive care. Diuretics are used as needed for fluid overload, along with angiotensin-converting enzyme inhibitors and beta-blockers for cardiomyopathy with decreased EF.
Our patient’s dyspnea improved with treatment of furosemide 40 mg IV BID, and his blood pressure was controlled with metoprolol 25 mg BID and lisinopril 10 mg BID. His chest pain and fever resolved when his blood pressure improved. He was discharged home after 7 days on the furosemide, metoprolol, and lisinopril, in addition to isosorbide mononitrate 30 mg/d, atorvastatin 40 mg/d, and aspirin 325 mg/d. An echocardiogram performed 6 months later showed persistent systolic dysfunction, with an EF of 35% and global wall motion abnormalities.
THE TAKEAWAY
In addition to acute coronary syndrome, consider alternate etiologies in patients who present with chest pain and elevated cardiac biomarkers, particularly if diagnostic work-up is negative for obstructive coronary artery disease. WNV myocarditis should be considered as a diagnosis when a patient’s symptoms suggest acute coronary syndrome but are accompanied by fever, headache, and other constitutional symptoms, especially during mosquito season or a WNV outbreak.
1. Nash D, Mostashari F, Fine A, et al. The outbreak of West Nile virus infection in the New York City area in 1999. N Engl J Med. 2001;344:1807-1814.
2. Orton SL, Stramer SL, Dodd RY. Self-reported symptoms associated with West Nile virus infection in RNA-positive blood donors. Transfusion. 2006;46:272-277.
3. Brown JA, Factor DL, Tkachenko N, et al. West Nile viremic blood donors and risk factors for subsequent West Nile fever. Vector Borne Zoonotic Dis. 2007;7:479-488.
4. Zou S, Foster GA, Dodd RY, et al. West Nile fever characteristics among viremic persons identified through blood donor screening. J Infect Dis. 2010;202:1354-1361.
5. Davis LE, DeBiasi R, Goade DE, et al. West Nile virus neuroinvasive disease. Ann Neurol. 2006;60:286-300.
6. Montgomery SP, Chow CC, Smith SW, et al. Rhabdomyolysis in patients with west nile encephalitis and meningitis. Vector Borne Zoonotic Dis. 2005;5:252-257.
7. Paddock CD, Nicholson WL, Bhatnagar J, et al. Fatal hemorrhagic fever caused by West Nile virus in the United States. Clin Infect Dis. 2006;42:1527-1535.
8. Georges AJ, Lesbordes JL, Georges-Courbot MC, et al. Fatal hepatitis from West Nile virus. Ann Inst Pasteur Virol. 1988;138:237.
9. Perelman A, Stern J. Acute pancreatitis in West Nile Fever. Am J Trop Med Hyg. 1974;23:1150-1152.
10. Sherman-Weber S, Axelrod P. Central diabetes insipidus complicating West Nile encephalitis. Clin Infect Dis. 2004;38:1042-1043.
11. Pergam SA, DeLong CE, Echevarria L, et al. Myocarditis in West Nile virus infection. Am J Trop Med Hyg. 2006;75:1232-1233.
12. van der Meulen KM, Pensaert MB, Nauwynck HJ. West Nile virus in the vertebrate world. Arch Virol. 2005;150:637-657.
13. Kushawaha A, Jadonath S, Mobarakai N. West nile virus myocarditis causing a fatal arrhythmia: a case report. Cases J. 2009;2:7147.
14. Khouzam RN. Significant cardiomyopathy secondary to West Nile virus infection. South Med J. 2009;102:527-528.
15. Kawai C. From myocarditis to cardiomyopathy: mechanisms of inflammation and cell death: learning from the past for the future. Circulation. 1999;99:1091-1100.
16. Caforio AL, Goldman JH, Haven AJ, et al. Circulating cardiac-specific autoantibodies as markers of autoimmunity in clinical and biopsy-proven myocarditis. The Myocarditis Treatment Trial Investigators. Eur Heart J. 1997;18:270-275.
17. Lauer B, Schannwell M, Kühl U, et al. Antimyosin autoantibodies are associated with deterioration of systolic and diastolic left ventricular function in patients with chronic myocarditis. J Am Coll Cardiol. 2000;35:11-18.
1. Nash D, Mostashari F, Fine A, et al. The outbreak of West Nile virus infection in the New York City area in 1999. N Engl J Med. 2001;344:1807-1814.
2. Orton SL, Stramer SL, Dodd RY. Self-reported symptoms associated with West Nile virus infection in RNA-positive blood donors. Transfusion. 2006;46:272-277.
3. Brown JA, Factor DL, Tkachenko N, et al. West Nile viremic blood donors and risk factors for subsequent West Nile fever. Vector Borne Zoonotic Dis. 2007;7:479-488.
4. Zou S, Foster GA, Dodd RY, et al. West Nile fever characteristics among viremic persons identified through blood donor screening. J Infect Dis. 2010;202:1354-1361.
5. Davis LE, DeBiasi R, Goade DE, et al. West Nile virus neuroinvasive disease. Ann Neurol. 2006;60:286-300.
6. Montgomery SP, Chow CC, Smith SW, et al. Rhabdomyolysis in patients with west nile encephalitis and meningitis. Vector Borne Zoonotic Dis. 2005;5:252-257.
7. Paddock CD, Nicholson WL, Bhatnagar J, et al. Fatal hemorrhagic fever caused by West Nile virus in the United States. Clin Infect Dis. 2006;42:1527-1535.
8. Georges AJ, Lesbordes JL, Georges-Courbot MC, et al. Fatal hepatitis from West Nile virus. Ann Inst Pasteur Virol. 1988;138:237.
9. Perelman A, Stern J. Acute pancreatitis in West Nile Fever. Am J Trop Med Hyg. 1974;23:1150-1152.
10. Sherman-Weber S, Axelrod P. Central diabetes insipidus complicating West Nile encephalitis. Clin Infect Dis. 2004;38:1042-1043.
11. Pergam SA, DeLong CE, Echevarria L, et al. Myocarditis in West Nile virus infection. Am J Trop Med Hyg. 2006;75:1232-1233.
12. van der Meulen KM, Pensaert MB, Nauwynck HJ. West Nile virus in the vertebrate world. Arch Virol. 2005;150:637-657.
13. Kushawaha A, Jadonath S, Mobarakai N. West nile virus myocarditis causing a fatal arrhythmia: a case report. Cases J. 2009;2:7147.
14. Khouzam RN. Significant cardiomyopathy secondary to West Nile virus infection. South Med J. 2009;102:527-528.
15. Kawai C. From myocarditis to cardiomyopathy: mechanisms of inflammation and cell death: learning from the past for the future. Circulation. 1999;99:1091-1100.
16. Caforio AL, Goldman JH, Haven AJ, et al. Circulating cardiac-specific autoantibodies as markers of autoimmunity in clinical and biopsy-proven myocarditis. The Myocarditis Treatment Trial Investigators. Eur Heart J. 1997;18:270-275.
17. Lauer B, Schannwell M, Kühl U, et al. Antimyosin autoantibodies are associated with deterioration of systolic and diastolic left ventricular function in patients with chronic myocarditis. J Am Coll Cardiol. 2000;35:11-18.

Seizure Prompts Man to Fall
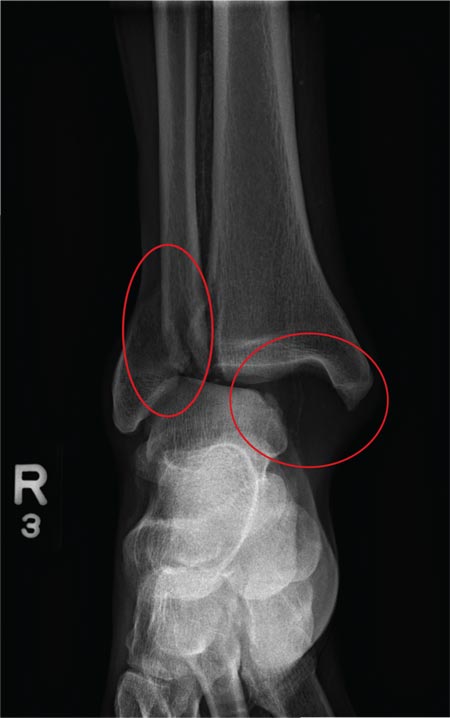
ANSWER
The radiograph shows a fracture dislocation of the ankle. The distal tibia is dislocated medially relative to the talus, as evidenced by the widened joint space. There is also an oblique fracture of the distal fibula.
Since the patient was experiencing neurovascular compromise, the dislocation was promptly reduced in the emergency department. Subsequently, he was taken to the operating room for open reduction and internal fixation of his fibula fracture.
ANSWER
The radiograph shows a fracture dislocation of the ankle. The distal tibia is dislocated medially relative to the talus, as evidenced by the widened joint space. There is also an oblique fracture of the distal fibula.
Since the patient was experiencing neurovascular compromise, the dislocation was promptly reduced in the emergency department. Subsequently, he was taken to the operating room for open reduction and internal fixation of his fibula fracture.
ANSWER
The radiograph shows a fracture dislocation of the ankle. The distal tibia is dislocated medially relative to the talus, as evidenced by the widened joint space. There is also an oblique fracture of the distal fibula.
Since the patient was experiencing neurovascular compromise, the dislocation was promptly reduced in the emergency department. Subsequently, he was taken to the operating room for open reduction and internal fixation of his fibula fracture.
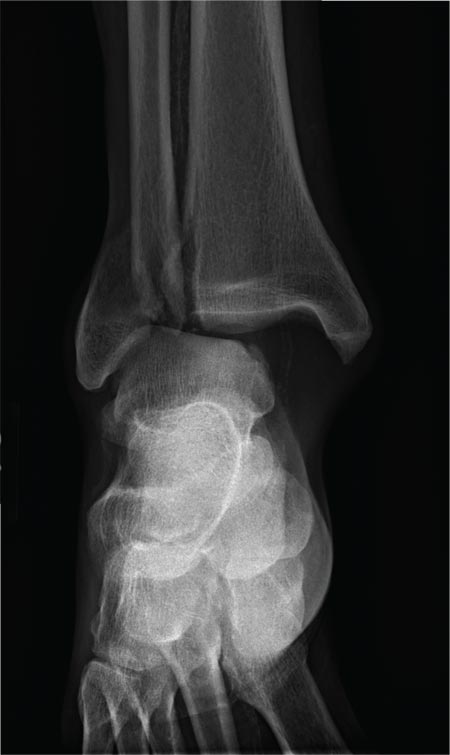
A 70-year-old man is brought to your facility by EMS following a new-onset, witnessed seizure. He reportedly fell down some steps. On arrival, he has returned to baseline but is complaining of left-sided weakness and right ankle pain. Medical history is significant for mild hypertension. Vital signs are stable. The patient exhibits slight confusion. He reports some mild weakness on his left side, especially in his lower extremity. There also appears to be moderate soft-tissue swelling of his right ankle, with a slight deformity noted. Dorsalis pedal pulse appears to be slightly diminished in that foot as well. You send the patient for noncontrast CT of the head, as well as a radiograph of the right ankle (the latter of which is shown). What is your impression?
When Wrong Test Is Ordered, “Wrongful Birth” Results
At a New Jersey hospital, a pregnant woman underwent an ultrasound examination with results suggesting a possible fetal abnormality. In response, DNA testing of the patient and her husband was ordered to investigate for a suspected hormonal disorder. But the wrong test was ordered, and the results of that test were negative.
A baby girl was born with congenital adrenal hyperplasia, a condition causing ambiguous genitalia due to exposure to high concentrations of androgens in utero. She underwent genital reconstructive surgery at age 4 months and is expected to require additional surgery, lifelong hormone replacement therapy, and lifelong monitoring.
The parents claimed that they would have elected to terminate the pregnancy if they had been properly informed of the child’s condition.
What was the outcome? >>
OUTCOME
A jury returned a ruling of 75% liability to the hospital and 25% liability to a hospital lab technician. The verdict was for $1 million, comprising $625,000 for the child and $375,000 for her parents.
COMMENT
The controversial legal theory of recovery in this case is known as “wrongful life” or “wrongful birth.” To prevail on these tort actions, one must prove that the defendant’s negligence led to the birth of an infant following a pregnancy that would have been terminated, had the parents been given all the prenatal screening information required by the standard of care.
The goal of any prenatal screening program should be to provide parents with information that is adequate, accurate, and timely. In this case, after the suspicious sonographic findings were encountered, the wrong test was ordered and the diagnosis was missed. Each practice providing prenatal screening should have a checklist to confirm that the correct test was ordered, completed, and documented—not to mention discussed with the patient in a timely manner.
In this case, the clinician ordered the wrong test, which left the patient with inadequate information. From the facts given, it is unclear if the ordering clinician became aware of this fact and what information, if any, the patient was given regarding the error. Importantly, information must also be given in a timely manner, leaving the patient adequate time to make an informed decision regarding termination—before fetal viability. But how is viability defined?
Although a detailed discussion of the constitutional principles of fetal viability is beyond the scope of this commentary, three US Supreme Court cases paved the way for successful wrongful life/wrongful birth actions. In Griswold v Connecticut (1965), the court held that decisions regarding birth control were protected by the right to privacy. In Roe v Wade (1973), the court held that a constitutionally protected right to privacy exists with regard to pregnancy terminations until the point of “viability,” originally defined as between 24 and 28 gestational weeks. Planned Parenthood v Casey (1991) held that advances in neonatal care required a revised definition of viability to a point “somewhat earlier,” without establishing a specific bright-line rule for viability.
To complicate matters, in recent years, at least 14 states (Alabama, Arizona, Arkansas, Georgia, Idaho, Indiana, Kansas, Louisiana, Mississippi, Nebraska, North Carolina, North Dakota, Oklahoma, and Texas) have redefined viability and passed laws banning therapeutic abortion beyond week 20 (although some of these bans have been judicially blocked). In states with this type of legislation, whether a clinician could be held legally responsible for failing to provide information necessary to permit an informed decision prior to the 20-week mark is unclear.
Questions as to whether these state laws were in conflict with Roe v Wade led to a constitutional challenge. In 2013, the US Court of Appeals for the Ninth Circuit (the highest level before the Supreme Court) ruled that a 20-week cutoff was unconstitutional because it violated the “viability rule” established by Roe and Casey. The Supreme Court declined to review that decision.1
Damage awards in wrongful life/wrongful birth cases are often substantial. The verdict in this case was relatively restrained.
Without doubt, this is a sensitive issue, and respect for our fellow clinicians’ opinions is warranted. However, from a liability standpoint, the safest course of action is to provide patients with all the necessary information—including prenatal testing results—as soon as possible, allowing them to make an informed decision before viability (however that is defined in your state). —DML
REFERENCE
1. Isaacson v. Horne, 716 F.3d 1213, 1225 (9th Cir. 2013), cert denied, 134 S. Ct. 905 (2014).
At a New Jersey hospital, a pregnant woman underwent an ultrasound examination with results suggesting a possible fetal abnormality. In response, DNA testing of the patient and her husband was ordered to investigate for a suspected hormonal disorder. But the wrong test was ordered, and the results of that test were negative.
A baby girl was born with congenital adrenal hyperplasia, a condition causing ambiguous genitalia due to exposure to high concentrations of androgens in utero. She underwent genital reconstructive surgery at age 4 months and is expected to require additional surgery, lifelong hormone replacement therapy, and lifelong monitoring.
The parents claimed that they would have elected to terminate the pregnancy if they had been properly informed of the child’s condition.
What was the outcome? >>
OUTCOME
A jury returned a ruling of 75% liability to the hospital and 25% liability to a hospital lab technician. The verdict was for $1 million, comprising $625,000 for the child and $375,000 for her parents.
COMMENT
The controversial legal theory of recovery in this case is known as “wrongful life” or “wrongful birth.” To prevail on these tort actions, one must prove that the defendant’s negligence led to the birth of an infant following a pregnancy that would have been terminated, had the parents been given all the prenatal screening information required by the standard of care.
The goal of any prenatal screening program should be to provide parents with information that is adequate, accurate, and timely. In this case, after the suspicious sonographic findings were encountered, the wrong test was ordered and the diagnosis was missed. Each practice providing prenatal screening should have a checklist to confirm that the correct test was ordered, completed, and documented—not to mention discussed with the patient in a timely manner.
In this case, the clinician ordered the wrong test, which left the patient with inadequate information. From the facts given, it is unclear if the ordering clinician became aware of this fact and what information, if any, the patient was given regarding the error. Importantly, information must also be given in a timely manner, leaving the patient adequate time to make an informed decision regarding termination—before fetal viability. But how is viability defined?
Although a detailed discussion of the constitutional principles of fetal viability is beyond the scope of this commentary, three US Supreme Court cases paved the way for successful wrongful life/wrongful birth actions. In Griswold v Connecticut (1965), the court held that decisions regarding birth control were protected by the right to privacy. In Roe v Wade (1973), the court held that a constitutionally protected right to privacy exists with regard to pregnancy terminations until the point of “viability,” originally defined as between 24 and 28 gestational weeks. Planned Parenthood v Casey (1991) held that advances in neonatal care required a revised definition of viability to a point “somewhat earlier,” without establishing a specific bright-line rule for viability.
To complicate matters, in recent years, at least 14 states (Alabama, Arizona, Arkansas, Georgia, Idaho, Indiana, Kansas, Louisiana, Mississippi, Nebraska, North Carolina, North Dakota, Oklahoma, and Texas) have redefined viability and passed laws banning therapeutic abortion beyond week 20 (although some of these bans have been judicially blocked). In states with this type of legislation, whether a clinician could be held legally responsible for failing to provide information necessary to permit an informed decision prior to the 20-week mark is unclear.
Questions as to whether these state laws were in conflict with Roe v Wade led to a constitutional challenge. In 2013, the US Court of Appeals for the Ninth Circuit (the highest level before the Supreme Court) ruled that a 20-week cutoff was unconstitutional because it violated the “viability rule” established by Roe and Casey. The Supreme Court declined to review that decision.1
Damage awards in wrongful life/wrongful birth cases are often substantial. The verdict in this case was relatively restrained.
Without doubt, this is a sensitive issue, and respect for our fellow clinicians’ opinions is warranted. However, from a liability standpoint, the safest course of action is to provide patients with all the necessary information—including prenatal testing results—as soon as possible, allowing them to make an informed decision before viability (however that is defined in your state). —DML
REFERENCE
1. Isaacson v. Horne, 716 F.3d 1213, 1225 (9th Cir. 2013), cert denied, 134 S. Ct. 905 (2014).
At a New Jersey hospital, a pregnant woman underwent an ultrasound examination with results suggesting a possible fetal abnormality. In response, DNA testing of the patient and her husband was ordered to investigate for a suspected hormonal disorder. But the wrong test was ordered, and the results of that test were negative.
A baby girl was born with congenital adrenal hyperplasia, a condition causing ambiguous genitalia due to exposure to high concentrations of androgens in utero. She underwent genital reconstructive surgery at age 4 months and is expected to require additional surgery, lifelong hormone replacement therapy, and lifelong monitoring.
The parents claimed that they would have elected to terminate the pregnancy if they had been properly informed of the child’s condition.
What was the outcome? >>
OUTCOME
A jury returned a ruling of 75% liability to the hospital and 25% liability to a hospital lab technician. The verdict was for $1 million, comprising $625,000 for the child and $375,000 for her parents.
COMMENT
The controversial legal theory of recovery in this case is known as “wrongful life” or “wrongful birth.” To prevail on these tort actions, one must prove that the defendant’s negligence led to the birth of an infant following a pregnancy that would have been terminated, had the parents been given all the prenatal screening information required by the standard of care.
The goal of any prenatal screening program should be to provide parents with information that is adequate, accurate, and timely. In this case, after the suspicious sonographic findings were encountered, the wrong test was ordered and the diagnosis was missed. Each practice providing prenatal screening should have a checklist to confirm that the correct test was ordered, completed, and documented—not to mention discussed with the patient in a timely manner.
In this case, the clinician ordered the wrong test, which left the patient with inadequate information. From the facts given, it is unclear if the ordering clinician became aware of this fact and what information, if any, the patient was given regarding the error. Importantly, information must also be given in a timely manner, leaving the patient adequate time to make an informed decision regarding termination—before fetal viability. But how is viability defined?
Although a detailed discussion of the constitutional principles of fetal viability is beyond the scope of this commentary, three US Supreme Court cases paved the way for successful wrongful life/wrongful birth actions. In Griswold v Connecticut (1965), the court held that decisions regarding birth control were protected by the right to privacy. In Roe v Wade (1973), the court held that a constitutionally protected right to privacy exists with regard to pregnancy terminations until the point of “viability,” originally defined as between 24 and 28 gestational weeks. Planned Parenthood v Casey (1991) held that advances in neonatal care required a revised definition of viability to a point “somewhat earlier,” without establishing a specific bright-line rule for viability.
To complicate matters, in recent years, at least 14 states (Alabama, Arizona, Arkansas, Georgia, Idaho, Indiana, Kansas, Louisiana, Mississippi, Nebraska, North Carolina, North Dakota, Oklahoma, and Texas) have redefined viability and passed laws banning therapeutic abortion beyond week 20 (although some of these bans have been judicially blocked). In states with this type of legislation, whether a clinician could be held legally responsible for failing to provide information necessary to permit an informed decision prior to the 20-week mark is unclear.
Questions as to whether these state laws were in conflict with Roe v Wade led to a constitutional challenge. In 2013, the US Court of Appeals for the Ninth Circuit (the highest level before the Supreme Court) ruled that a 20-week cutoff was unconstitutional because it violated the “viability rule” established by Roe and Casey. The Supreme Court declined to review that decision.1
Damage awards in wrongful life/wrongful birth cases are often substantial. The verdict in this case was relatively restrained.
Without doubt, this is a sensitive issue, and respect for our fellow clinicians’ opinions is warranted. However, from a liability standpoint, the safest course of action is to provide patients with all the necessary information—including prenatal testing results—as soon as possible, allowing them to make an informed decision before viability (however that is defined in your state). —DML
REFERENCE
1. Isaacson v. Horne, 716 F.3d 1213, 1225 (9th Cir. 2013), cert denied, 134 S. Ct. 905 (2014).
Acute Kidney Injury: Prevalent in Sugarcane Harvesters
Q) I’ve heard a lot of talk about all the kidney problems that the sugarcane workers in Central America have. Does anyone know why this is happening?
The unusually high rates of chronic kidney disease (CKD) among sugarcane workers in Central America have been a subject of great interest since National Public Radio (NPR) aired a special on this topic.3 There has been a rising epidemic of CKD in otherwise healthy male farm workers (ages 20 to 50), particularly those who harvest sugarcane.4,5 It has been hypothesized that recurrent episodes of acute kidney injury (AKI)—related to dehydration, volume depletion, pollutants, and rhabdomyolysis with inflammatory stress—are the underlying cause.5
Sugarcane harvesters typically work nine-hour days, six days per week, in extremely high temperatures and while wearing heavy, hot clothing. Each worker cuts approximately 10 tons of sugarcane daily, since they are paid based on cutting volume. Workers drink between five and 10 L of water during their shifts.
Santos et al designed a study to prospectively examine the effects of burnt sugarcane harvesting on renal function in healthy male farm workers. Twenty-eight men (ages 19 to 39) with no CKD risk factors (diabetes, smoking, obesity, hypertension, illicit drug or alcohol use) were followed for eight months from preharvest to postharvest. Blood samples were collected at the beginning and at the end of the workday and preharvest and postharvest season.5
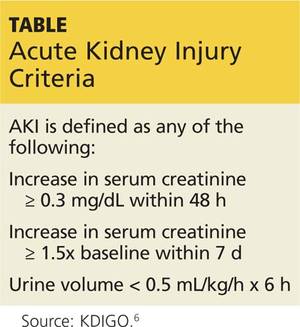
Preseason lab values were normal in all 28 men. But postseason, all workers had elevated creatinine levels, with five meeting the criteria for AKI (see Table at left).5,6
Santos and colleagues identified potential causes for AKI in this population. These included
• Dehydration and volume depletion (episodes of tachycardia, increased urine density, lower urinary/serum sodium, higher hematocrit)
• Rhabdomyolysis (increased creatine kinase at the end of each workday)
• Systemic inflammation (increased white blood count, neutrophils, lymphocytes, and monocytes during the workday—possibly indicative of an inflammatory burst)
• Other factors (burning of the sugarcane releasing unknown nephrotoxic substances; unreported NSAIDs use)5
Compared to workers who showed early signs of CKD, those who developed frank AKI were more likely to have hyponatremia. Recommendations to reduce the problem include consumption of water/salt hydrating drinks, use of appropriate clothing, work-hour limitations, and changes to payment structures (ie, from a volume system to an hourly or daily system). Furthermore, education on the need to avoid alcohol, illicit drugs, and NSAIDs during the harvest season should help to decrease incidence of AKI among these workers.
Elizabeth C. Evans, RN, MSN, CNP, DNP
Renal Medicine Associates, Albuquerque, New Mexico
REFERENCES
1. Ayuk J, Gittoes N. Contemporary view of the clinical relevance of magnesium homeostasis. Ann Clin Biochem. 2014;51(Pt 2):179-188.
2. Firouzi A, Maadani M, Kiani R, et al. Intravenous magnesium sulfate: new method in prevention of contrast-induced nephropathy in primary percutaneous coronary intervention. Int Urol Nephrol. 2015;47(3):521-525.
3. Beaubien J. Mysterious kidney disease slays farm workers in central America. National Public Radio; 2014. www.npr.org/blogs/health/2014/04/30/306907097/mysterious-kidney-disease-slays-farmworkers-in-central-america. Accessed April 1, 2015.
4. Almaguer M, Herrera R, Orantes CM. Chronic kidney disease of unknown etiology in agricultural communities. MEDICC Rev. 2014;16(2):9-15.
5. Santos UP, Zanetta DMT, Burdmann EA. Burnt sugarcane harvesting is associated with acute renal dysfunction. Kidney Int. 2015;87(4):792-799.
6. Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2:1-138.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Debra L. Coplon, DNP, DCC, who practices at the City of Memphis Wellness Clinic in Tennessee, and Elizabeth C. Evans, RN, MSN, CNP, DNP, who practices with Renal Medicine Associates in Albuquerque.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Debra L. Coplon, DNP, DCC, who practices at the City of Memphis Wellness Clinic in Tennessee, and Elizabeth C. Evans, RN, MSN, CNP, DNP, who practices with Renal Medicine Associates in Albuquerque.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Debra L. Coplon, DNP, DCC, who practices at the City of Memphis Wellness Clinic in Tennessee, and Elizabeth C. Evans, RN, MSN, CNP, DNP, who practices with Renal Medicine Associates in Albuquerque.
Q) I’ve heard a lot of talk about all the kidney problems that the sugarcane workers in Central America have. Does anyone know why this is happening?
The unusually high rates of chronic kidney disease (CKD) among sugarcane workers in Central America have been a subject of great interest since National Public Radio (NPR) aired a special on this topic.3 There has been a rising epidemic of CKD in otherwise healthy male farm workers (ages 20 to 50), particularly those who harvest sugarcane.4,5 It has been hypothesized that recurrent episodes of acute kidney injury (AKI)—related to dehydration, volume depletion, pollutants, and rhabdomyolysis with inflammatory stress—are the underlying cause.5
Sugarcane harvesters typically work nine-hour days, six days per week, in extremely high temperatures and while wearing heavy, hot clothing. Each worker cuts approximately 10 tons of sugarcane daily, since they are paid based on cutting volume. Workers drink between five and 10 L of water during their shifts.
Santos et al designed a study to prospectively examine the effects of burnt sugarcane harvesting on renal function in healthy male farm workers. Twenty-eight men (ages 19 to 39) with no CKD risk factors (diabetes, smoking, obesity, hypertension, illicit drug or alcohol use) were followed for eight months from preharvest to postharvest. Blood samples were collected at the beginning and at the end of the workday and preharvest and postharvest season.5
Preseason lab values were normal in all 28 men. But postseason, all workers had elevated creatinine levels, with five meeting the criteria for AKI (see Table at left).5,6
Santos and colleagues identified potential causes for AKI in this population. These included
• Dehydration and volume depletion (episodes of tachycardia, increased urine density, lower urinary/serum sodium, higher hematocrit)
• Rhabdomyolysis (increased creatine kinase at the end of each workday)
• Systemic inflammation (increased white blood count, neutrophils, lymphocytes, and monocytes during the workday—possibly indicative of an inflammatory burst)
• Other factors (burning of the sugarcane releasing unknown nephrotoxic substances; unreported NSAIDs use)5
Compared to workers who showed early signs of CKD, those who developed frank AKI were more likely to have hyponatremia. Recommendations to reduce the problem include consumption of water/salt hydrating drinks, use of appropriate clothing, work-hour limitations, and changes to payment structures (ie, from a volume system to an hourly or daily system). Furthermore, education on the need to avoid alcohol, illicit drugs, and NSAIDs during the harvest season should help to decrease incidence of AKI among these workers.
Elizabeth C. Evans, RN, MSN, CNP, DNP
Renal Medicine Associates, Albuquerque, New Mexico
REFERENCES
1. Ayuk J, Gittoes N. Contemporary view of the clinical relevance of magnesium homeostasis. Ann Clin Biochem. 2014;51(Pt 2):179-188.
2. Firouzi A, Maadani M, Kiani R, et al. Intravenous magnesium sulfate: new method in prevention of contrast-induced nephropathy in primary percutaneous coronary intervention. Int Urol Nephrol. 2015;47(3):521-525.
3. Beaubien J. Mysterious kidney disease slays farm workers in central America. National Public Radio; 2014. www.npr.org/blogs/health/2014/04/30/306907097/mysterious-kidney-disease-slays-farmworkers-in-central-america. Accessed April 1, 2015.
4. Almaguer M, Herrera R, Orantes CM. Chronic kidney disease of unknown etiology in agricultural communities. MEDICC Rev. 2014;16(2):9-15.
5. Santos UP, Zanetta DMT, Burdmann EA. Burnt sugarcane harvesting is associated with acute renal dysfunction. Kidney Int. 2015;87(4):792-799.
6. Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2:1-138.
Q) I’ve heard a lot of talk about all the kidney problems that the sugarcane workers in Central America have. Does anyone know why this is happening?
The unusually high rates of chronic kidney disease (CKD) among sugarcane workers in Central America have been a subject of great interest since National Public Radio (NPR) aired a special on this topic.3 There has been a rising epidemic of CKD in otherwise healthy male farm workers (ages 20 to 50), particularly those who harvest sugarcane.4,5 It has been hypothesized that recurrent episodes of acute kidney injury (AKI)—related to dehydration, volume depletion, pollutants, and rhabdomyolysis with inflammatory stress—are the underlying cause.5
Sugarcane harvesters typically work nine-hour days, six days per week, in extremely high temperatures and while wearing heavy, hot clothing. Each worker cuts approximately 10 tons of sugarcane daily, since they are paid based on cutting volume. Workers drink between five and 10 L of water during their shifts.
Santos et al designed a study to prospectively examine the effects of burnt sugarcane harvesting on renal function in healthy male farm workers. Twenty-eight men (ages 19 to 39) with no CKD risk factors (diabetes, smoking, obesity, hypertension, illicit drug or alcohol use) were followed for eight months from preharvest to postharvest. Blood samples were collected at the beginning and at the end of the workday and preharvest and postharvest season.5
Preseason lab values were normal in all 28 men. But postseason, all workers had elevated creatinine levels, with five meeting the criteria for AKI (see Table at left).5,6
Santos and colleagues identified potential causes for AKI in this population. These included
• Dehydration and volume depletion (episodes of tachycardia, increased urine density, lower urinary/serum sodium, higher hematocrit)
• Rhabdomyolysis (increased creatine kinase at the end of each workday)
• Systemic inflammation (increased white blood count, neutrophils, lymphocytes, and monocytes during the workday—possibly indicative of an inflammatory burst)
• Other factors (burning of the sugarcane releasing unknown nephrotoxic substances; unreported NSAIDs use)5
Compared to workers who showed early signs of CKD, those who developed frank AKI were more likely to have hyponatremia. Recommendations to reduce the problem include consumption of water/salt hydrating drinks, use of appropriate clothing, work-hour limitations, and changes to payment structures (ie, from a volume system to an hourly or daily system). Furthermore, education on the need to avoid alcohol, illicit drugs, and NSAIDs during the harvest season should help to decrease incidence of AKI among these workers.
Elizabeth C. Evans, RN, MSN, CNP, DNP
Renal Medicine Associates, Albuquerque, New Mexico
REFERENCES
1. Ayuk J, Gittoes N. Contemporary view of the clinical relevance of magnesium homeostasis. Ann Clin Biochem. 2014;51(Pt 2):179-188.
2. Firouzi A, Maadani M, Kiani R, et al. Intravenous magnesium sulfate: new method in prevention of contrast-induced nephropathy in primary percutaneous coronary intervention. Int Urol Nephrol. 2015;47(3):521-525.
3. Beaubien J. Mysterious kidney disease slays farm workers in central America. National Public Radio; 2014. www.npr.org/blogs/health/2014/04/30/306907097/mysterious-kidney-disease-slays-farmworkers-in-central-america. Accessed April 1, 2015.
4. Almaguer M, Herrera R, Orantes CM. Chronic kidney disease of unknown etiology in agricultural communities. MEDICC Rev. 2014;16(2):9-15.
5. Santos UP, Zanetta DMT, Burdmann EA. Burnt sugarcane harvesting is associated with acute renal dysfunction. Kidney Int. 2015;87(4):792-799.
6. Kidney Disease: Improving Global Outcomes (KDIGO) Acute Kidney Injury Work Group. KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int Suppl. 2012;2:1-138.
Marathon Runner Has History of A-fib
ANSWER
This ECG shows marked sinus bradycardia with a first-degree atrioventricular block and nonspecific T-wave abnormality. The QT interval of 524 ms is consistent with prolonged QT interval but is normal when corrected for rate.
These findings were consistent with previous ECGs. Since the patient’s bradycardia is asymptomatic, no intervention (ie, placement of a permanent pacemaker) is indicated.
ANSWER
This ECG shows marked sinus bradycardia with a first-degree atrioventricular block and nonspecific T-wave abnormality. The QT interval of 524 ms is consistent with prolonged QT interval but is normal when corrected for rate.
These findings were consistent with previous ECGs. Since the patient’s bradycardia is asymptomatic, no intervention (ie, placement of a permanent pacemaker) is indicated.
ANSWER
This ECG shows marked sinus bradycardia with a first-degree atrioventricular block and nonspecific T-wave abnormality. The QT interval of 524 ms is consistent with prolonged QT interval but is normal when corrected for rate.
These findings were consistent with previous ECGs. Since the patient’s bradycardia is asymptomatic, no intervention (ie, placement of a permanent pacemaker) is indicated.
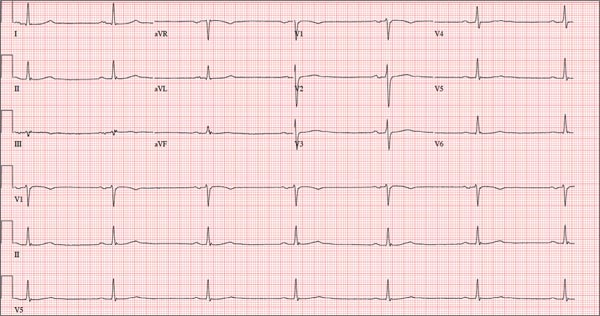
A 52-year-old man has a cardiac diagnosis of paroxysmal atrial fibrillation (A-fib). An echocardiogram demonstrates no valvular heart disease and a left ventricular ejection fraction of 62%. There are no symptoms to suggest left ventricular dysfunction or volume overload. He denies exertional angina or dyspnea and says he has had no palpitations or recurrences of A-fib since you saw him six months ago. The patient is very active: In the past year, he has completed two half marathons and one full marathon. In addition to his running schedule, he also swims 30 min/d and trains on an elliptical machine for 1 h/d. His only complaint today is that he recently lost the toenails off each big toe, which he attributes to his running, adding that this isn’t the first time it’s happened. Medical history is remarkable for two episodes of A-fib that manifested with palpitations and a rapid heart rate, which caused dyspnea. The last episode was approximately eight months ago. Both were treated with cardioversion in the emergency department of your institution. He was not started on an anticoagulant or antiarrhythmic medication after either occurrence. The patient currently takes no medications except the occasional ibuprofen for muscle soreness related to training. He has no known drug allergies and does not use naturopathic medications or illicit drugs. He has never smoked, and he only drinks wine socially, usually on weekends. The patient works as a certified public accountant for a large corporation. He is married with two teenage children. A 12-point review of systems is remarkable only for an inguinal rash and the aforementioned missing toenails. On physical exam, the vital signs include a blood pressure of 107/60 mm Hg; pulse, 46 beats/min; respiratory rate, 12 breaths/min-1; and temperature, 97.8°F. His height is 74 in and his weight, 172 lb. The patient is in no distress. The neck veins are flat, the lungs are clear, and the cardiac exam reveals no murmurs or gallops. The abdominal exam is unremarkable. There is no edema in the peripheral extremities, and pulses are strong bilaterally. Both feet reveal multiple callouses, and the two great toes have missing nails but healthy nail beds. The neurologic exam is intact. As part of his clinic visit, a 12-lead ECG is obtained. It reveals a ventricular rate of 38 beats/min; PR interval, 222 ms; QRS duration, 112 ms; QT/QTc interval, 524/416 ms; P axis, 20°; R axis, 26°; and T axis, 33°. What is your interpretation of this ECG?
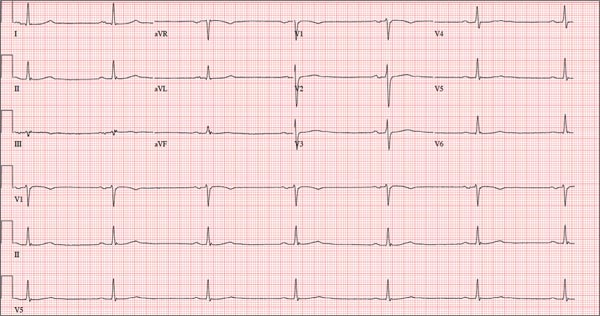
Stomach pain chalked up to flu; patient suffers fatal cardiac event ... More
Stomach pain chalked up to flu; patient suffers fatal cardiac event
A 40-YEAR-OLD MAN went to the emergency department (ED) after 2 days of stomach discomfort. The ED physician who evaluated him released him after 4 or 5 hours without testing for levels of troponin or other cardiac enzymes. The patient’s discomfort continued, and about 3 days later, he told his wife to call 911. He was transported to the ED but did not survive.
PLAINTIFF’S CLAIM The decedent had been suffering from an acute cardiac event during the first ED visit. Testing to rule out cardiac problems should have been performed.
THE DEFENSE The patient had been suffering from a stomach flu during his initial ED visit. Any testing performed at that time would have been normal. The patient’s death was unrelated to the symptoms he was experiencing when he was first seen.
VERDICT $4 million Alabama verdict.
COMMENT Many questions come to mind with this case: How careful was the history? Did the patient’s discomfort get worse with activity? What were the characteristics of his pain? What were the patient’s cardiac risk factors? A colleague of mine missed a very similar case several years ago in a 67-year-old. The patient even had vomiting and diarrhea, but clearly had a myocardial infarction when diagnosed a few days later.
Follow-up failure on PSA results costs patient valuable Tx time
A PATIENT AT A GROUP PRACTICE underwent prostate specific antigen (PSA) screening, which revealed an abnormal result (4.1 ng/mL). The physician circled this value on the lab report, wrote, “Discuss next visit,” and placed the report in the patient’s chart. However, the patient switched to another physician in the group and was not told of the abnormal result for more than 2 years. When the patient went to a medical center for back pain, magnetic resonance imaging of his spine revealed the presence of cancer in his spine, shoulder blades, pelvis, and ribs. A PSA test performed at that time came back at 100 ng/mL. Two days later, a biopsy confirmed the diagnosis of prostate cancer (Gleason score, 9).
PLAINTIFF’S CLAIM In addition to failing to inform the patient of his abnormal PSA test result, the physician did not perform digital rectal exams.
THE DEFENSE Earlier treatment would not have made a difference in the outcome.
VERDICT $934,000 Florida verdict.
COMMENT If you order a PSA, you must follow up on it. When a patient transfers to your care, be sure to obtain and review past testing and provide follow-up on abnormal results. We now send all test results directly to patients so they can serve as a safety check for their own care. Despite fears of being inundated with calls, most organizations that have instituted such a policy have not turned back.
Stomach pain chalked up to flu; patient suffers fatal cardiac event
A 40-YEAR-OLD MAN went to the emergency department (ED) after 2 days of stomach discomfort. The ED physician who evaluated him released him after 4 or 5 hours without testing for levels of troponin or other cardiac enzymes. The patient’s discomfort continued, and about 3 days later, he told his wife to call 911. He was transported to the ED but did not survive.
PLAINTIFF’S CLAIM The decedent had been suffering from an acute cardiac event during the first ED visit. Testing to rule out cardiac problems should have been performed.
THE DEFENSE The patient had been suffering from a stomach flu during his initial ED visit. Any testing performed at that time would have been normal. The patient’s death was unrelated to the symptoms he was experiencing when he was first seen.
VERDICT $4 million Alabama verdict.
COMMENT Many questions come to mind with this case: How careful was the history? Did the patient’s discomfort get worse with activity? What were the characteristics of his pain? What were the patient’s cardiac risk factors? A colleague of mine missed a very similar case several years ago in a 67-year-old. The patient even had vomiting and diarrhea, but clearly had a myocardial infarction when diagnosed a few days later.
Follow-up failure on PSA results costs patient valuable Tx time
A PATIENT AT A GROUP PRACTICE underwent prostate specific antigen (PSA) screening, which revealed an abnormal result (4.1 ng/mL). The physician circled this value on the lab report, wrote, “Discuss next visit,” and placed the report in the patient’s chart. However, the patient switched to another physician in the group and was not told of the abnormal result for more than 2 years. When the patient went to a medical center for back pain, magnetic resonance imaging of his spine revealed the presence of cancer in his spine, shoulder blades, pelvis, and ribs. A PSA test performed at that time came back at 100 ng/mL. Two days later, a biopsy confirmed the diagnosis of prostate cancer (Gleason score, 9).
PLAINTIFF’S CLAIM In addition to failing to inform the patient of his abnormal PSA test result, the physician did not perform digital rectal exams.
THE DEFENSE Earlier treatment would not have made a difference in the outcome.
VERDICT $934,000 Florida verdict.
COMMENT If you order a PSA, you must follow up on it. When a patient transfers to your care, be sure to obtain and review past testing and provide follow-up on abnormal results. We now send all test results directly to patients so they can serve as a safety check for their own care. Despite fears of being inundated with calls, most organizations that have instituted such a policy have not turned back.
Stomach pain chalked up to flu; patient suffers fatal cardiac event
A 40-YEAR-OLD MAN went to the emergency department (ED) after 2 days of stomach discomfort. The ED physician who evaluated him released him after 4 or 5 hours without testing for levels of troponin or other cardiac enzymes. The patient’s discomfort continued, and about 3 days later, he told his wife to call 911. He was transported to the ED but did not survive.
PLAINTIFF’S CLAIM The decedent had been suffering from an acute cardiac event during the first ED visit. Testing to rule out cardiac problems should have been performed.
THE DEFENSE The patient had been suffering from a stomach flu during his initial ED visit. Any testing performed at that time would have been normal. The patient’s death was unrelated to the symptoms he was experiencing when he was first seen.
VERDICT $4 million Alabama verdict.
COMMENT Many questions come to mind with this case: How careful was the history? Did the patient’s discomfort get worse with activity? What were the characteristics of his pain? What were the patient’s cardiac risk factors? A colleague of mine missed a very similar case several years ago in a 67-year-old. The patient even had vomiting and diarrhea, but clearly had a myocardial infarction when diagnosed a few days later.
Follow-up failure on PSA results costs patient valuable Tx time
A PATIENT AT A GROUP PRACTICE underwent prostate specific antigen (PSA) screening, which revealed an abnormal result (4.1 ng/mL). The physician circled this value on the lab report, wrote, “Discuss next visit,” and placed the report in the patient’s chart. However, the patient switched to another physician in the group and was not told of the abnormal result for more than 2 years. When the patient went to a medical center for back pain, magnetic resonance imaging of his spine revealed the presence of cancer in his spine, shoulder blades, pelvis, and ribs. A PSA test performed at that time came back at 100 ng/mL. Two days later, a biopsy confirmed the diagnosis of prostate cancer (Gleason score, 9).
PLAINTIFF’S CLAIM In addition to failing to inform the patient of his abnormal PSA test result, the physician did not perform digital rectal exams.
THE DEFENSE Earlier treatment would not have made a difference in the outcome.
VERDICT $934,000 Florida verdict.
COMMENT If you order a PSA, you must follow up on it. When a patient transfers to your care, be sure to obtain and review past testing and provide follow-up on abnormal results. We now send all test results directly to patients so they can serve as a safety check for their own care. Despite fears of being inundated with calls, most organizations that have instituted such a policy have not turned back.