User login
Reserve thrombophilia testing for select subgroups
Clinicians should avoid routinely screening venous thromboembolism patients for thrombophilias, and should instead weigh the risks of recurrent thrombosis against the chances of bleeding with prolonged anticoagulation, according to a review article published in the April issue of the Journal of Vascular Surgery: Venous and Lymphatic Disorders.
“These laboratory tests are costly, and surprisingly, there is little evidence showing that testing leads to improved clinical outcomes,” said Dr. Elna Masuda at Straub Clinic and Hospital, Honolulu, and her associates. “Until data from well-designed, controlled trials are available comparing different durations of anticoagulation with specific thrombophilic states, treatment should be based on clinical risk factors and less on laboratory abnormalities.”
More than half of patients with an initial venous thromboembolism (VTE) episode had a positive thrombophilia screen in one study (Ann. Intern. Med. 2001;135:367-73), the reviewers noted. Testing, however, usually does not affect clinical management or prevent VTE recurrence, and it can cost more than $3,000 per patient, they said.
For these reasons, the American Society of Hematology, the National Institute for Health Care and Excellence, and the Society for Vascular Medicine discourage screening after an initial VTE episode if patients have a known cause or transient risk factor for thrombosis.
Testing also is unlikely to benefit patients with first-time unprovoked (or idiopathic) VTE, patients with a permanent risk factor for thrombosis such as cancer, or patients with arterial thrombosis or thrombosis of the retina or upper arm veins, Dr. Masuda and her associates said. And because recurrent VTE generally merits long-term anticoagulation, affected patients only should be screened if they are considering stopping treatment and test results could inform that decision, they added (J. Vasc. Surg. Venous Lymphat. Disord. 2015;3:228-35).
Some subgroups of patients, however, could benefit from targeted thrombophilia testing. The reviewers recommended antiphospholipid antibody testing if patients have a history of several unexplained pregnancy losses or another reason to suspect antiphospholipid syndrome. Patients with heparin resistance should be tested for antithrombin deficiency, and patients with warfarin necrosis or neonatal purpura fulminans should be tested for protein C and S deficiencies, they added.
Clinicians also should consider screening women with a personal or family history of VTE if they are pregnant and are considering anticoagulation or are considering oral contraceptives or hormone replacement therapy, Dr. Masuda and her associates said.
Screening such patients remains controversial, but it could help guide anticoagulation therapy before and after delivery or might help patients decide whether or not to take exogenous hormones. “In the subgroup of those pregnant or planning pregnancy, history of prior VTE and strong family history of thrombosis are two factors that appear to play a clinically important role in identifying those who may benefit from screening,” they concluded.
Patients who want to pursue testing need to understand that management is mainly based on clinical risk and that test results usually will not change treatment recommendations, the reviewers also emphasized. “If testing will change management, it may be appropriate to proceed,” they added. “If long-term anticoagulation is preferred on the basis of positive test results, the risk of bleeding should be considered.”
The researchers reported no funding sources. Dr. Masuda reported having served on the speakers bureau for Janssen Pharmaceuticals.
Clinical utility of thrombophilia testing is determined by the cost-benefit ratio to each patient. The extent of testing can be quite variable with the cost varying widely. Testing can range from factor V Leiden and homocystine levels to lupus anticoagulant and an isolated factor, or it can include panels of both fibrinolytic and thrombotic pathways as well as genetic testing. Duration of therapy and risk of recurrence can be influenced by the results. The real cost of underestimating the risk of recurrence is the sequela of recurrent thrombosis, such as the increased risk of valvular damage or obstruction, pulmonary embolism, and the development of the postthrombotic syndrome.
Even patients who have a provoked thrombus have been shown to have an increased incidence of thrombophilia. A positive test result can impact the patient’s treatment or potentially prevent events in families who have an unrecognized thrombophilic issue. Those outcomes matter to the patient and the family. In the past we ligated the saphenofemoral junction for patients with an isolated superficial vein thrombosis encroaching on the junction only to find out that many of these patients have an underlying undiagnosed thrombophilia, which had progressed to deep vein thrombosis.
Knowledge helps select appropriate therapies and potentially prevents life-threatening complications to the patient and their family members. Many people who have an underlying thrombophilia have a minor change in their baseline that then starts a cascade to promote a thrombotic event. Knowledge is power and testing to help identify risk is clinically warranted.
Treatments are based on risk factor assessment, which includes laboratory analysis, residual thrombus, and clinical risk. Understanding the fibrinolytic balance may further explain why some patients recanalize completely while other patients never recanalize and have a significant amount of residual thrombus.
Once a thrombophilia has been identified, family members can be tested for a specific defect, potentially avoiding any thrombotic events and preventing miscarriages in those of reproductive years. Further testing and identification of subgroups is needed to clearly define those who would benefit most. This will only be accomplished with further testing. Research can identify additional defects that will help treating physicians to further understand the thrombotic and fibrinolytic pathways. Management decisions need to be based on evidence. Some of these factors were unknown 20 years ago.
Dr. Joann Lohr is an associate program director at the Good Samaritan Hospital vascular surgery residency program and director of the John J. Cranley Vascular Laboratory at Good Samaritan Hospital, both in Cincinnati. She had no relevant financial disclosures.
Clinical utility of thrombophilia testing is determined by the cost-benefit ratio to each patient. The extent of testing can be quite variable with the cost varying widely. Testing can range from factor V Leiden and homocystine levels to lupus anticoagulant and an isolated factor, or it can include panels of both fibrinolytic and thrombotic pathways as well as genetic testing. Duration of therapy and risk of recurrence can be influenced by the results. The real cost of underestimating the risk of recurrence is the sequela of recurrent thrombosis, such as the increased risk of valvular damage or obstruction, pulmonary embolism, and the development of the postthrombotic syndrome.
Even patients who have a provoked thrombus have been shown to have an increased incidence of thrombophilia. A positive test result can impact the patient’s treatment or potentially prevent events in families who have an unrecognized thrombophilic issue. Those outcomes matter to the patient and the family. In the past we ligated the saphenofemoral junction for patients with an isolated superficial vein thrombosis encroaching on the junction only to find out that many of these patients have an underlying undiagnosed thrombophilia, which had progressed to deep vein thrombosis.
Knowledge helps select appropriate therapies and potentially prevents life-threatening complications to the patient and their family members. Many people who have an underlying thrombophilia have a minor change in their baseline that then starts a cascade to promote a thrombotic event. Knowledge is power and testing to help identify risk is clinically warranted.
Treatments are based on risk factor assessment, which includes laboratory analysis, residual thrombus, and clinical risk. Understanding the fibrinolytic balance may further explain why some patients recanalize completely while other patients never recanalize and have a significant amount of residual thrombus.
Once a thrombophilia has been identified, family members can be tested for a specific defect, potentially avoiding any thrombotic events and preventing miscarriages in those of reproductive years. Further testing and identification of subgroups is needed to clearly define those who would benefit most. This will only be accomplished with further testing. Research can identify additional defects that will help treating physicians to further understand the thrombotic and fibrinolytic pathways. Management decisions need to be based on evidence. Some of these factors were unknown 20 years ago.
Dr. Joann Lohr is an associate program director at the Good Samaritan Hospital vascular surgery residency program and director of the John J. Cranley Vascular Laboratory at Good Samaritan Hospital, both in Cincinnati. She had no relevant financial disclosures.
Clinical utility of thrombophilia testing is determined by the cost-benefit ratio to each patient. The extent of testing can be quite variable with the cost varying widely. Testing can range from factor V Leiden and homocystine levels to lupus anticoagulant and an isolated factor, or it can include panels of both fibrinolytic and thrombotic pathways as well as genetic testing. Duration of therapy and risk of recurrence can be influenced by the results. The real cost of underestimating the risk of recurrence is the sequela of recurrent thrombosis, such as the increased risk of valvular damage or obstruction, pulmonary embolism, and the development of the postthrombotic syndrome.
Even patients who have a provoked thrombus have been shown to have an increased incidence of thrombophilia. A positive test result can impact the patient’s treatment or potentially prevent events in families who have an unrecognized thrombophilic issue. Those outcomes matter to the patient and the family. In the past we ligated the saphenofemoral junction for patients with an isolated superficial vein thrombosis encroaching on the junction only to find out that many of these patients have an underlying undiagnosed thrombophilia, which had progressed to deep vein thrombosis.
Knowledge helps select appropriate therapies and potentially prevents life-threatening complications to the patient and their family members. Many people who have an underlying thrombophilia have a minor change in their baseline that then starts a cascade to promote a thrombotic event. Knowledge is power and testing to help identify risk is clinically warranted.
Treatments are based on risk factor assessment, which includes laboratory analysis, residual thrombus, and clinical risk. Understanding the fibrinolytic balance may further explain why some patients recanalize completely while other patients never recanalize and have a significant amount of residual thrombus.
Once a thrombophilia has been identified, family members can be tested for a specific defect, potentially avoiding any thrombotic events and preventing miscarriages in those of reproductive years. Further testing and identification of subgroups is needed to clearly define those who would benefit most. This will only be accomplished with further testing. Research can identify additional defects that will help treating physicians to further understand the thrombotic and fibrinolytic pathways. Management decisions need to be based on evidence. Some of these factors were unknown 20 years ago.
Dr. Joann Lohr is an associate program director at the Good Samaritan Hospital vascular surgery residency program and director of the John J. Cranley Vascular Laboratory at Good Samaritan Hospital, both in Cincinnati. She had no relevant financial disclosures.
Clinicians should avoid routinely screening venous thromboembolism patients for thrombophilias, and should instead weigh the risks of recurrent thrombosis against the chances of bleeding with prolonged anticoagulation, according to a review article published in the April issue of the Journal of Vascular Surgery: Venous and Lymphatic Disorders.
“These laboratory tests are costly, and surprisingly, there is little evidence showing that testing leads to improved clinical outcomes,” said Dr. Elna Masuda at Straub Clinic and Hospital, Honolulu, and her associates. “Until data from well-designed, controlled trials are available comparing different durations of anticoagulation with specific thrombophilic states, treatment should be based on clinical risk factors and less on laboratory abnormalities.”
More than half of patients with an initial venous thromboembolism (VTE) episode had a positive thrombophilia screen in one study (Ann. Intern. Med. 2001;135:367-73), the reviewers noted. Testing, however, usually does not affect clinical management or prevent VTE recurrence, and it can cost more than $3,000 per patient, they said.
For these reasons, the American Society of Hematology, the National Institute for Health Care and Excellence, and the Society for Vascular Medicine discourage screening after an initial VTE episode if patients have a known cause or transient risk factor for thrombosis.
Testing also is unlikely to benefit patients with first-time unprovoked (or idiopathic) VTE, patients with a permanent risk factor for thrombosis such as cancer, or patients with arterial thrombosis or thrombosis of the retina or upper arm veins, Dr. Masuda and her associates said. And because recurrent VTE generally merits long-term anticoagulation, affected patients only should be screened if they are considering stopping treatment and test results could inform that decision, they added (J. Vasc. Surg. Venous Lymphat. Disord. 2015;3:228-35).
Some subgroups of patients, however, could benefit from targeted thrombophilia testing. The reviewers recommended antiphospholipid antibody testing if patients have a history of several unexplained pregnancy losses or another reason to suspect antiphospholipid syndrome. Patients with heparin resistance should be tested for antithrombin deficiency, and patients with warfarin necrosis or neonatal purpura fulminans should be tested for protein C and S deficiencies, they added.
Clinicians also should consider screening women with a personal or family history of VTE if they are pregnant and are considering anticoagulation or are considering oral contraceptives or hormone replacement therapy, Dr. Masuda and her associates said.
Screening such patients remains controversial, but it could help guide anticoagulation therapy before and after delivery or might help patients decide whether or not to take exogenous hormones. “In the subgroup of those pregnant or planning pregnancy, history of prior VTE and strong family history of thrombosis are two factors that appear to play a clinically important role in identifying those who may benefit from screening,” they concluded.
Patients who want to pursue testing need to understand that management is mainly based on clinical risk and that test results usually will not change treatment recommendations, the reviewers also emphasized. “If testing will change management, it may be appropriate to proceed,” they added. “If long-term anticoagulation is preferred on the basis of positive test results, the risk of bleeding should be considered.”
The researchers reported no funding sources. Dr. Masuda reported having served on the speakers bureau for Janssen Pharmaceuticals.
Clinicians should avoid routinely screening venous thromboembolism patients for thrombophilias, and should instead weigh the risks of recurrent thrombosis against the chances of bleeding with prolonged anticoagulation, according to a review article published in the April issue of the Journal of Vascular Surgery: Venous and Lymphatic Disorders.
“These laboratory tests are costly, and surprisingly, there is little evidence showing that testing leads to improved clinical outcomes,” said Dr. Elna Masuda at Straub Clinic and Hospital, Honolulu, and her associates. “Until data from well-designed, controlled trials are available comparing different durations of anticoagulation with specific thrombophilic states, treatment should be based on clinical risk factors and less on laboratory abnormalities.”
More than half of patients with an initial venous thromboembolism (VTE) episode had a positive thrombophilia screen in one study (Ann. Intern. Med. 2001;135:367-73), the reviewers noted. Testing, however, usually does not affect clinical management or prevent VTE recurrence, and it can cost more than $3,000 per patient, they said.
For these reasons, the American Society of Hematology, the National Institute for Health Care and Excellence, and the Society for Vascular Medicine discourage screening after an initial VTE episode if patients have a known cause or transient risk factor for thrombosis.
Testing also is unlikely to benefit patients with first-time unprovoked (or idiopathic) VTE, patients with a permanent risk factor for thrombosis such as cancer, or patients with arterial thrombosis or thrombosis of the retina or upper arm veins, Dr. Masuda and her associates said. And because recurrent VTE generally merits long-term anticoagulation, affected patients only should be screened if they are considering stopping treatment and test results could inform that decision, they added (J. Vasc. Surg. Venous Lymphat. Disord. 2015;3:228-35).
Some subgroups of patients, however, could benefit from targeted thrombophilia testing. The reviewers recommended antiphospholipid antibody testing if patients have a history of several unexplained pregnancy losses or another reason to suspect antiphospholipid syndrome. Patients with heparin resistance should be tested for antithrombin deficiency, and patients with warfarin necrosis or neonatal purpura fulminans should be tested for protein C and S deficiencies, they added.
Clinicians also should consider screening women with a personal or family history of VTE if they are pregnant and are considering anticoagulation or are considering oral contraceptives or hormone replacement therapy, Dr. Masuda and her associates said.
Screening such patients remains controversial, but it could help guide anticoagulation therapy before and after delivery or might help patients decide whether or not to take exogenous hormones. “In the subgroup of those pregnant or planning pregnancy, history of prior VTE and strong family history of thrombosis are two factors that appear to play a clinically important role in identifying those who may benefit from screening,” they concluded.
Patients who want to pursue testing need to understand that management is mainly based on clinical risk and that test results usually will not change treatment recommendations, the reviewers also emphasized. “If testing will change management, it may be appropriate to proceed,” they added. “If long-term anticoagulation is preferred on the basis of positive test results, the risk of bleeding should be considered.”
The researchers reported no funding sources. Dr. Masuda reported having served on the speakers bureau for Janssen Pharmaceuticals.
FROM THE JOURNAL OF VASCULAR SURGERY: VENOUS AND LYMPHATIC DISORDERS
Key clinical point: Clinicians should reserve thrombophilia testing for select subgroups of patients with venous thromboembolism.
Major finding: Consider select thrombophilia testing for patients with suspected antiphospholipid syndrome, heparin resistance, warfarin necrosis, neonatal purpura fulminans, or mesenteric or cerebral deep venous thrombosis. Also consider screening women with a personal or family history of thrombosis who are or want to become pregnant or are considering oral contraceptives or hormone replacement therapy.
Data source: Review of 40 original research and review articles.
Disclosures: The researchers reported no funding sources. Dr. Masuda reported having served on the speakers bureau for Janssen Pharmaceuticals.
Shiitake Mushroom Dermatitis
To the Editor:
The shiitake mushroom (Lentinula edodes) is a popular Asian food and represents the second most consumed mushroom in the world. It is known for having a range of strong health benefits including antihypertensive, anti-inflammatory, and immunomodulatory effects. Especially in Asia, this mushroom has been used in patients with cancers of the gastrointestinal tract and also may be helpful in the treatment of human immunodeficiency virus.1,2 The source of these effects is lentinan, a polysaccharide in the mushroom. However, lentinan also can cause a toxic reaction of the skin when the mushrooms are eaten raw or undercooked. These reactions are mainly reported in Asia, but more cases have been published in the last decade in Europe and the United States, evidence that the incidence of this adverse effect has increased in the Western world.
A 65-year-old woman with no notable medical history presented to our outpatient practice with sudden onset of a pruritic, erythematous, papular eruption on the neck. The eruption began that morning. The diagnosis of eczematous dermatitis was made and hydrocortisone cream 2.5% was started. Three days later, she returned with spread of the rash to the trunk, arms, and legs despite the topical treatment. She denied fevers, chills, or constitutional symptoms. The patient also denied recent travel or bug bites. However, she reported that she recently had started using raw shiitake mushrooms in her salad; the first time was 3 days before the symptoms appeared. Physical examination revealed erythematous skin with long flagellate streaks composed of petechiae, papules, and vesicles involving the trunk, arms, and legs (Figure). Oral and nasal mucosae were uninvolved. Dermatographism was negative. The diagnosis of flagellate dermatitis from shiitake mushrooms was made given the patient’s history and the unique clinical findings of the skin. Blood work and a biopsy were not performed. Instead, the patient was advised to avoid shiitake mushrooms and use clobetasol propionate cream 0.05% twice daily for 2 weeks on the affected areas. The symptoms resolved within 10 days.
|
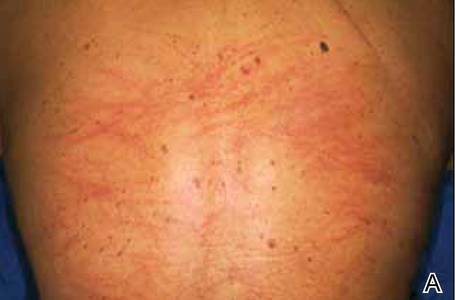

The first known case of toxicoderma to shiitake mushrooms was reported in Japan by Nakamura3 in 1977. Since this seminal report, numerous cases have followed. This disorder is mainly seen in Asia.
Patients usually present with linear groups of pruritic, papular, petechial, and vesicular lesions in a flagellate pattern, most commonly localized on the trunk, arms, and legs. Oral and nasal mucosae usually are not involved, and fever and malaise may be associated. All symptoms typically occur 1 to 2 days after ingestion of the mushrooms. The patient’s history and typical clinical findings lead to a diagnosis; however, blood tests may show inflammation with leukocytosis and elevated C-reactive protein levels. Biopsy of the skin shows lymphocytic dermal infiltrates with spongiosis and necrotic cells within the epidermis.4
Differential diagnoses include flagellate dermatitis associated with bleomycin, a glycopeptide antibiotic produced by the bacterium Streptomyces verticillus. Because it causes breaks in the DNA, bleomycin is commonly used as a chemotherapeutic agent in treating Hodgkin lymphoma and other malignancies. It presents with linear postinflammatory hyperpigmentation of the skin. However, unlike shiitake dermatitis, there is a lack of papules. Another differential diagnosis includes herpes zoster virus, which should be ruled out clinically.
All symptoms in shiitake dermatitis usually resolve within 1 to 8 weeks of avoidance of the culprit food. Topical steroids and antihistamines can be given.
The underlying pathology is a toxic reaction to the polysaccharide lentinan in the mushrooms, which is known as a thermolabile agent.5 Therefore, it may only cause a toxic reaction when the mushrooms are consumed raw or undercooked. Prick testing is usually negative in these patients, which suggests a toxic and not an immunologic reaction of the human body.6 Other forms of reaction to shiitake mushrooms include contact dermatitis after skin contact and allergic alveolitis after occupational exposure to mushroom spores, mainly in individuals cultivating shiitake mushrooms (mushroom worker’s lung). In these forms of the disease, prick testing may be positive.7,8
Flagellate dermatitis caused by shiitake mushrooms is still an uncommon dermatologic phenomenon in the Western world. Future studies and cases should be reported to increase the awareness of this disorder. Although the patients present with typical clinical findings, the diagnosis can be missed if history is not carefully considered.
1. Ng ML, Yap AT. Inhibition of human colon carcinoma development by lentinan from shiitake mushrooms (Lentinus edodes). J Altern Complement Med. 2002;8:581-589.
2. Gordon M, Bihari B, Goosby E, et al. A placebo-controlled trial of the immune modulator, lentinan, in HIV-positive patients: a phase I/II trial. J Med. 1998;29:305-330.
3. Nakamura T. Toxicoderma caused by shiitake. Jpn J Clin Dermatol. 1977;31:65-68.
4. Hanada K, Hashimoto I. Flagellate mushroom (shiitake) dermatitis and photosensitivity. Dermatology. 1998;197:255-257.
5. Lippert U, Martin V, Schwertfeger C, et al. Shiitake dermatitis. Br J Dermatol. 2003;148:178-179.
6. Nakamura T. Shiitake (Lentinus edodes) dermatitis. Contact Dermatitis. 1992;27:6570.
7. Ueda A, Obama K, Aoyama K, et al. Allergic contact dermatitis in shiitake (Lentinus edodes (Berk) Sing) growers. Contact Dermatitis. 1992;26:228-233.
8. Ampere A, Delhaes L, Soots J, et al. Hypersensitivity pneumonitis induced by shiitake mushroom spores. Med Mycol. 2012;50:654-657.
To the Editor:
The shiitake mushroom (Lentinula edodes) is a popular Asian food and represents the second most consumed mushroom in the world. It is known for having a range of strong health benefits including antihypertensive, anti-inflammatory, and immunomodulatory effects. Especially in Asia, this mushroom has been used in patients with cancers of the gastrointestinal tract and also may be helpful in the treatment of human immunodeficiency virus.1,2 The source of these effects is lentinan, a polysaccharide in the mushroom. However, lentinan also can cause a toxic reaction of the skin when the mushrooms are eaten raw or undercooked. These reactions are mainly reported in Asia, but more cases have been published in the last decade in Europe and the United States, evidence that the incidence of this adverse effect has increased in the Western world.
A 65-year-old woman with no notable medical history presented to our outpatient practice with sudden onset of a pruritic, erythematous, papular eruption on the neck. The eruption began that morning. The diagnosis of eczematous dermatitis was made and hydrocortisone cream 2.5% was started. Three days later, she returned with spread of the rash to the trunk, arms, and legs despite the topical treatment. She denied fevers, chills, or constitutional symptoms. The patient also denied recent travel or bug bites. However, she reported that she recently had started using raw shiitake mushrooms in her salad; the first time was 3 days before the symptoms appeared. Physical examination revealed erythematous skin with long flagellate streaks composed of petechiae, papules, and vesicles involving the trunk, arms, and legs (Figure). Oral and nasal mucosae were uninvolved. Dermatographism was negative. The diagnosis of flagellate dermatitis from shiitake mushrooms was made given the patient’s history and the unique clinical findings of the skin. Blood work and a biopsy were not performed. Instead, the patient was advised to avoid shiitake mushrooms and use clobetasol propionate cream 0.05% twice daily for 2 weeks on the affected areas. The symptoms resolved within 10 days.
|
The first known case of toxicoderma to shiitake mushrooms was reported in Japan by Nakamura3 in 1977. Since this seminal report, numerous cases have followed. This disorder is mainly seen in Asia.
Patients usually present with linear groups of pruritic, papular, petechial, and vesicular lesions in a flagellate pattern, most commonly localized on the trunk, arms, and legs. Oral and nasal mucosae usually are not involved, and fever and malaise may be associated. All symptoms typically occur 1 to 2 days after ingestion of the mushrooms. The patient’s history and typical clinical findings lead to a diagnosis; however, blood tests may show inflammation with leukocytosis and elevated C-reactive protein levels. Biopsy of the skin shows lymphocytic dermal infiltrates with spongiosis and necrotic cells within the epidermis.4
Differential diagnoses include flagellate dermatitis associated with bleomycin, a glycopeptide antibiotic produced by the bacterium Streptomyces verticillus. Because it causes breaks in the DNA, bleomycin is commonly used as a chemotherapeutic agent in treating Hodgkin lymphoma and other malignancies. It presents with linear postinflammatory hyperpigmentation of the skin. However, unlike shiitake dermatitis, there is a lack of papules. Another differential diagnosis includes herpes zoster virus, which should be ruled out clinically.
All symptoms in shiitake dermatitis usually resolve within 1 to 8 weeks of avoidance of the culprit food. Topical steroids and antihistamines can be given.
The underlying pathology is a toxic reaction to the polysaccharide lentinan in the mushrooms, which is known as a thermolabile agent.5 Therefore, it may only cause a toxic reaction when the mushrooms are consumed raw or undercooked. Prick testing is usually negative in these patients, which suggests a toxic and not an immunologic reaction of the human body.6 Other forms of reaction to shiitake mushrooms include contact dermatitis after skin contact and allergic alveolitis after occupational exposure to mushroom spores, mainly in individuals cultivating shiitake mushrooms (mushroom worker’s lung). In these forms of the disease, prick testing may be positive.7,8
Flagellate dermatitis caused by shiitake mushrooms is still an uncommon dermatologic phenomenon in the Western world. Future studies and cases should be reported to increase the awareness of this disorder. Although the patients present with typical clinical findings, the diagnosis can be missed if history is not carefully considered.
To the Editor:
The shiitake mushroom (Lentinula edodes) is a popular Asian food and represents the second most consumed mushroom in the world. It is known for having a range of strong health benefits including antihypertensive, anti-inflammatory, and immunomodulatory effects. Especially in Asia, this mushroom has been used in patients with cancers of the gastrointestinal tract and also may be helpful in the treatment of human immunodeficiency virus.1,2 The source of these effects is lentinan, a polysaccharide in the mushroom. However, lentinan also can cause a toxic reaction of the skin when the mushrooms are eaten raw or undercooked. These reactions are mainly reported in Asia, but more cases have been published in the last decade in Europe and the United States, evidence that the incidence of this adverse effect has increased in the Western world.
A 65-year-old woman with no notable medical history presented to our outpatient practice with sudden onset of a pruritic, erythematous, papular eruption on the neck. The eruption began that morning. The diagnosis of eczematous dermatitis was made and hydrocortisone cream 2.5% was started. Three days later, she returned with spread of the rash to the trunk, arms, and legs despite the topical treatment. She denied fevers, chills, or constitutional symptoms. The patient also denied recent travel or bug bites. However, she reported that she recently had started using raw shiitake mushrooms in her salad; the first time was 3 days before the symptoms appeared. Physical examination revealed erythematous skin with long flagellate streaks composed of petechiae, papules, and vesicles involving the trunk, arms, and legs (Figure). Oral and nasal mucosae were uninvolved. Dermatographism was negative. The diagnosis of flagellate dermatitis from shiitake mushrooms was made given the patient’s history and the unique clinical findings of the skin. Blood work and a biopsy were not performed. Instead, the patient was advised to avoid shiitake mushrooms and use clobetasol propionate cream 0.05% twice daily for 2 weeks on the affected areas. The symptoms resolved within 10 days.
|
The first known case of toxicoderma to shiitake mushrooms was reported in Japan by Nakamura3 in 1977. Since this seminal report, numerous cases have followed. This disorder is mainly seen in Asia.
Patients usually present with linear groups of pruritic, papular, petechial, and vesicular lesions in a flagellate pattern, most commonly localized on the trunk, arms, and legs. Oral and nasal mucosae usually are not involved, and fever and malaise may be associated. All symptoms typically occur 1 to 2 days after ingestion of the mushrooms. The patient’s history and typical clinical findings lead to a diagnosis; however, blood tests may show inflammation with leukocytosis and elevated C-reactive protein levels. Biopsy of the skin shows lymphocytic dermal infiltrates with spongiosis and necrotic cells within the epidermis.4
Differential diagnoses include flagellate dermatitis associated with bleomycin, a glycopeptide antibiotic produced by the bacterium Streptomyces verticillus. Because it causes breaks in the DNA, bleomycin is commonly used as a chemotherapeutic agent in treating Hodgkin lymphoma and other malignancies. It presents with linear postinflammatory hyperpigmentation of the skin. However, unlike shiitake dermatitis, there is a lack of papules. Another differential diagnosis includes herpes zoster virus, which should be ruled out clinically.
All symptoms in shiitake dermatitis usually resolve within 1 to 8 weeks of avoidance of the culprit food. Topical steroids and antihistamines can be given.
The underlying pathology is a toxic reaction to the polysaccharide lentinan in the mushrooms, which is known as a thermolabile agent.5 Therefore, it may only cause a toxic reaction when the mushrooms are consumed raw or undercooked. Prick testing is usually negative in these patients, which suggests a toxic and not an immunologic reaction of the human body.6 Other forms of reaction to shiitake mushrooms include contact dermatitis after skin contact and allergic alveolitis after occupational exposure to mushroom spores, mainly in individuals cultivating shiitake mushrooms (mushroom worker’s lung). In these forms of the disease, prick testing may be positive.7,8
Flagellate dermatitis caused by shiitake mushrooms is still an uncommon dermatologic phenomenon in the Western world. Future studies and cases should be reported to increase the awareness of this disorder. Although the patients present with typical clinical findings, the diagnosis can be missed if history is not carefully considered.
1. Ng ML, Yap AT. Inhibition of human colon carcinoma development by lentinan from shiitake mushrooms (Lentinus edodes). J Altern Complement Med. 2002;8:581-589.
2. Gordon M, Bihari B, Goosby E, et al. A placebo-controlled trial of the immune modulator, lentinan, in HIV-positive patients: a phase I/II trial. J Med. 1998;29:305-330.
3. Nakamura T. Toxicoderma caused by shiitake. Jpn J Clin Dermatol. 1977;31:65-68.
4. Hanada K, Hashimoto I. Flagellate mushroom (shiitake) dermatitis and photosensitivity. Dermatology. 1998;197:255-257.
5. Lippert U, Martin V, Schwertfeger C, et al. Shiitake dermatitis. Br J Dermatol. 2003;148:178-179.
6. Nakamura T. Shiitake (Lentinus edodes) dermatitis. Contact Dermatitis. 1992;27:6570.
7. Ueda A, Obama K, Aoyama K, et al. Allergic contact dermatitis in shiitake (Lentinus edodes (Berk) Sing) growers. Contact Dermatitis. 1992;26:228-233.
8. Ampere A, Delhaes L, Soots J, et al. Hypersensitivity pneumonitis induced by shiitake mushroom spores. Med Mycol. 2012;50:654-657.
1. Ng ML, Yap AT. Inhibition of human colon carcinoma development by lentinan from shiitake mushrooms (Lentinus edodes). J Altern Complement Med. 2002;8:581-589.
2. Gordon M, Bihari B, Goosby E, et al. A placebo-controlled trial of the immune modulator, lentinan, in HIV-positive patients: a phase I/II trial. J Med. 1998;29:305-330.
3. Nakamura T. Toxicoderma caused by shiitake. Jpn J Clin Dermatol. 1977;31:65-68.
4. Hanada K, Hashimoto I. Flagellate mushroom (shiitake) dermatitis and photosensitivity. Dermatology. 1998;197:255-257.
5. Lippert U, Martin V, Schwertfeger C, et al. Shiitake dermatitis. Br J Dermatol. 2003;148:178-179.
6. Nakamura T. Shiitake (Lentinus edodes) dermatitis. Contact Dermatitis. 1992;27:6570.
7. Ueda A, Obama K, Aoyama K, et al. Allergic contact dermatitis in shiitake (Lentinus edodes (Berk) Sing) growers. Contact Dermatitis. 1992;26:228-233.
8. Ampere A, Delhaes L, Soots J, et al. Hypersensitivity pneumonitis induced by shiitake mushroom spores. Med Mycol. 2012;50:654-657.
Hospital Medicine 2015 Photo Gallery - Day Three
Photographs from Hospital Medicine 2015, which took place March 29-April 1 at the Gaylord National Hotel and Conference Center in National Harbor, Md.
Photos by Manuel Noguera
[gallery ids="9476,9477,9479,9480,9481,9482,9483,9484,9485,9486,9487,9488,9489,9490,9491,9492,9493,9494,9495,9496,9497,9498,9499,9500,9501,9502,9503,9504,9505,9506,9507,9508,9509,9510,9511,9512,9516,9518,9519,9520,9521,9522,9523,9524,9525,9526,9528,9529,9530,9531,9532,9533,9534,9535,9536,9538,9540,9541,9542,9543,9545,9546,9548,9549,9551"]
Photographs from Hospital Medicine 2015, which took place March 29-April 1 at the Gaylord National Hotel and Conference Center in National Harbor, Md.
Photos by Manuel Noguera
[gallery ids="9476,9477,9479,9480,9481,9482,9483,9484,9485,9486,9487,9488,9489,9490,9491,9492,9493,9494,9495,9496,9497,9498,9499,9500,9501,9502,9503,9504,9505,9506,9507,9508,9509,9510,9511,9512,9516,9518,9519,9520,9521,9522,9523,9524,9525,9526,9528,9529,9530,9531,9532,9533,9534,9535,9536,9538,9540,9541,9542,9543,9545,9546,9548,9549,9551"]
Photographs from Hospital Medicine 2015, which took place March 29-April 1 at the Gaylord National Hotel and Conference Center in National Harbor, Md.
Photos by Manuel Noguera
[gallery ids="9476,9477,9479,9480,9481,9482,9483,9484,9485,9486,9487,9488,9489,9490,9491,9492,9493,9494,9495,9496,9497,9498,9499,9500,9501,9502,9503,9504,9505,9506,9507,9508,9509,9510,9511,9512,9516,9518,9519,9520,9521,9522,9523,9524,9525,9526,9528,9529,9530,9531,9532,9533,9534,9535,9536,9538,9540,9541,9542,9543,9545,9546,9548,9549,9551"]
Dr. Michael Krychman details new and in-the-pipeline treatment options for vulvovaginal atrophy
In an audiocast summarizing his Sunday Lunch Talk at the Annual Clinical Meeting of the American College of Obstetricians and Gynecologists (ACOG) on May 3, 2015, Dr. Michael L. Krychman discusses new treatment options for vulvar and vaginal atrophy (VVA), including over-the-counter and prescription products and procedures. He emphasizes that a better understanding of the physical and anatomic changes in menopause has led to these improved options.
Dr. Krychman also recommends the use of "genitourinary syndrome of menopause" (GSM), new terminology for VVA suggested by the International Society for the Study of Women's Sexual Health and the North American Menopause Society.1
Among the products Dr. Krychman details are neogyn® Feminine Soothing Cream (neogyn, inc., Switzerland); RepHresh™ Vaginal Gel (Church & Dwight Co., Inc., Princeton, New Jersey); Replens™ Long-Lasting Vaginal Moisturizer (Church & Dwight); silicone- and water-based lubricants (Replens™ Silky Smooth Lubricant [Church & Dwight]; JuvaGyn® Feminine Moisturizer [neogyn, inc.]); and ospemifene (Osphena®, Shionogi Inc., Florham Park, New Jersey).
Dr. Krychman is interested in a new laser procedure for VVA/GSM, but comments that more study is needed before he can recommend its general use. He also talks about other exciting alternatives in the pipeline.
- Portman DJ, Gass ML; Vulvovaginal Atrophy Terminology Consensus Conference Panel. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women’s Sexual Health and the North American Menopause Society. Menopause. 2014;11(12):2865–2872.
In an audiocast summarizing his Sunday Lunch Talk at the Annual Clinical Meeting of the American College of Obstetricians and Gynecologists (ACOG) on May 3, 2015, Dr. Michael L. Krychman discusses new treatment options for vulvar and vaginal atrophy (VVA), including over-the-counter and prescription products and procedures. He emphasizes that a better understanding of the physical and anatomic changes in menopause has led to these improved options.
Dr. Krychman also recommends the use of "genitourinary syndrome of menopause" (GSM), new terminology for VVA suggested by the International Society for the Study of Women's Sexual Health and the North American Menopause Society.1
Among the products Dr. Krychman details are neogyn® Feminine Soothing Cream (neogyn, inc., Switzerland); RepHresh™ Vaginal Gel (Church & Dwight Co., Inc., Princeton, New Jersey); Replens™ Long-Lasting Vaginal Moisturizer (Church & Dwight); silicone- and water-based lubricants (Replens™ Silky Smooth Lubricant [Church & Dwight]; JuvaGyn® Feminine Moisturizer [neogyn, inc.]); and ospemifene (Osphena®, Shionogi Inc., Florham Park, New Jersey).
Dr. Krychman is interested in a new laser procedure for VVA/GSM, but comments that more study is needed before he can recommend its general use. He also talks about other exciting alternatives in the pipeline.
In an audiocast summarizing his Sunday Lunch Talk at the Annual Clinical Meeting of the American College of Obstetricians and Gynecologists (ACOG) on May 3, 2015, Dr. Michael L. Krychman discusses new treatment options for vulvar and vaginal atrophy (VVA), including over-the-counter and prescription products and procedures. He emphasizes that a better understanding of the physical and anatomic changes in menopause has led to these improved options.
Dr. Krychman also recommends the use of "genitourinary syndrome of menopause" (GSM), new terminology for VVA suggested by the International Society for the Study of Women's Sexual Health and the North American Menopause Society.1
Among the products Dr. Krychman details are neogyn® Feminine Soothing Cream (neogyn, inc., Switzerland); RepHresh™ Vaginal Gel (Church & Dwight Co., Inc., Princeton, New Jersey); Replens™ Long-Lasting Vaginal Moisturizer (Church & Dwight); silicone- and water-based lubricants (Replens™ Silky Smooth Lubricant [Church & Dwight]; JuvaGyn® Feminine Moisturizer [neogyn, inc.]); and ospemifene (Osphena®, Shionogi Inc., Florham Park, New Jersey).
Dr. Krychman is interested in a new laser procedure for VVA/GSM, but comments that more study is needed before he can recommend its general use. He also talks about other exciting alternatives in the pipeline.
- Portman DJ, Gass ML; Vulvovaginal Atrophy Terminology Consensus Conference Panel. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women’s Sexual Health and the North American Menopause Society. Menopause. 2014;11(12):2865–2872.
- Portman DJ, Gass ML; Vulvovaginal Atrophy Terminology Consensus Conference Panel. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women’s Sexual Health and the North American Menopause Society. Menopause. 2014;11(12):2865–2872.
Hospital Medicine 2015 Photo Gallery - Day Two
Photographs from Hospital Medicine 2015, which took place March 29-April 1 at the Gaylord National Hotel and Conference Center in National Harbor, Md.
Photos by Manuel Noguera
[gallery ids="9426,9427,9428,9429,9430,9431,9432,9433,9434,9435,9436,9437,9439,9440,9441,9442,9444,9445,9447,9449,9450,9451,9452,9453,9454,9455,9457,9458,9459,9460,9461,9462,9463,9464,9465,9467,9468,9469,9471,9472,9474"]
tk
Photographs from Hospital Medicine 2015, which took place March 29-April 1 at the Gaylord National Hotel and Conference Center in National Harbor, Md.
Photos by Manuel Noguera
[gallery ids="9426,9427,9428,9429,9430,9431,9432,9433,9434,9435,9436,9437,9439,9440,9441,9442,9444,9445,9447,9449,9450,9451,9452,9453,9454,9455,9457,9458,9459,9460,9461,9462,9463,9464,9465,9467,9468,9469,9471,9472,9474"]
tk
Photographs from Hospital Medicine 2015, which took place March 29-April 1 at the Gaylord National Hotel and Conference Center in National Harbor, Md.
Photos by Manuel Noguera
[gallery ids="9426,9427,9428,9429,9430,9431,9432,9433,9434,9435,9436,9437,9439,9440,9441,9442,9444,9445,9447,9449,9450,9451,9452,9453,9454,9455,9457,9458,9459,9460,9461,9462,9463,9464,9465,9467,9468,9469,9471,9472,9474"]
tk
AAN: Facial nerve stimulator relieves cluster headaches
WASHINGTON – An implantable device that stimulates the sphenopalatine ganglion nerve bundle either reduced or eliminated pain in 68% of more than 5,000 cluster headaches, a 3-year study has determined.
The device, which is approved in Europe, was more effective in attacks of moderate severity, with a 78% rate of pain reduction or elimination, Dr. Jose Miguel Lainez reported at the annual meeting of the American Academy of Neurology.

The Pulsante System, manufactured by Autonomic Technologiesof Redwood City, Calif., consists of a neurostimulator about the size of an almond, and a lead with six electrodes. It’s inserted under local anesthetic via a small incision in the upper gum on the side in which the patient experiences symptoms. The electrodes are positioned along the sphenopalatine ganglion (SPG) nerve and the neurostimulator is affixed to the zygomatic process.
A hand-held remote controller placed against the cheek activates the device and controls the intensity of stimulation, which is thought to work by blocking signals to the postganglionic parasympathetic fibers. Those fibers innervate facial structures and the cerebral and meningeal blood vessels and are implicated in the pain and accompanying autonomic symptoms of a cluster headache attack.
Dr. Lainez, professor of neurology at Catholic University of Valencia (Spain), presented 3-year follow-up data from Pathway CH-1, a randomized, sham-controlled trial of 43 patients with cluster headache. Of these, 33 completed the 3-year follow-up period. Of the remaining 10, 1 was lost from observation, 5 violated protocol, 1 had the device implanted incorrectly, and 3 had the device explanted because of incorrect placement or lead migration.
Most of the patients were male. Mean age was 41 years. They had a mean disease duration of 10 years and averaged 17 cluster headaches per week but ranged from 4 to 70 attacks per week. Over the 3 years, 5,130 attacks were treated; the mean stimulation duration for these was 14 minutes with a mean response time of 11 minutes. Therapy was considered effective in 65% (3,354) of these attacks based on a clinically meaningful reduction in pain or pain elimination.
Dr. Lainez did not parse these results. However, in the initial 28-week phase of the Pathway CH-1 study, pain was reduced in 68% of attacks treated with the device and 7% of those treated with the sham control. Pain freedom by 15 minutes was achieved in 34% of attacks with full stimulation, compared with 1.5% of those treated with sham.
In the follow-up study, the device seemed most effective in attacks of moderate severity (78% response rate of pain reduction or elimination). The response rate was 59% in mild attacks and 51% in severe attacks. Most attacks treated with the device (77%) did not involve the use of abortive therapy.
Dr. Lainez did not mention adverse events related to the device. However, in the 28-week study, there were 92, including parasthesias and numbness; facial and tooth pain; and swelling. Others were considered mild and included dry eye, nose bleed, and facial asymmetry.
The device is currently being investigated in a U.S. study. The open-label Pathway-CH2 study aims to recruit 120 patients. For information on Pathway CH-2, contact Anthony Caparso.
The trial was sponsored by Autonomic Technologies Inc. Dr. Lainez had no financial ties with the company.
On Twitter @alz_gal
WASHINGTON – An implantable device that stimulates the sphenopalatine ganglion nerve bundle either reduced or eliminated pain in 68% of more than 5,000 cluster headaches, a 3-year study has determined.
The device, which is approved in Europe, was more effective in attacks of moderate severity, with a 78% rate of pain reduction or elimination, Dr. Jose Miguel Lainez reported at the annual meeting of the American Academy of Neurology.
The Pulsante System, manufactured by Autonomic Technologiesof Redwood City, Calif., consists of a neurostimulator about the size of an almond, and a lead with six electrodes. It’s inserted under local anesthetic via a small incision in the upper gum on the side in which the patient experiences symptoms. The electrodes are positioned along the sphenopalatine ganglion (SPG) nerve and the neurostimulator is affixed to the zygomatic process.
A hand-held remote controller placed against the cheek activates the device and controls the intensity of stimulation, which is thought to work by blocking signals to the postganglionic parasympathetic fibers. Those fibers innervate facial structures and the cerebral and meningeal blood vessels and are implicated in the pain and accompanying autonomic symptoms of a cluster headache attack.
Dr. Lainez, professor of neurology at Catholic University of Valencia (Spain), presented 3-year follow-up data from Pathway CH-1, a randomized, sham-controlled trial of 43 patients with cluster headache. Of these, 33 completed the 3-year follow-up period. Of the remaining 10, 1 was lost from observation, 5 violated protocol, 1 had the device implanted incorrectly, and 3 had the device explanted because of incorrect placement or lead migration.
Most of the patients were male. Mean age was 41 years. They had a mean disease duration of 10 years and averaged 17 cluster headaches per week but ranged from 4 to 70 attacks per week. Over the 3 years, 5,130 attacks were treated; the mean stimulation duration for these was 14 minutes with a mean response time of 11 minutes. Therapy was considered effective in 65% (3,354) of these attacks based on a clinically meaningful reduction in pain or pain elimination.
Dr. Lainez did not parse these results. However, in the initial 28-week phase of the Pathway CH-1 study, pain was reduced in 68% of attacks treated with the device and 7% of those treated with the sham control. Pain freedom by 15 minutes was achieved in 34% of attacks with full stimulation, compared with 1.5% of those treated with sham.
In the follow-up study, the device seemed most effective in attacks of moderate severity (78% response rate of pain reduction or elimination). The response rate was 59% in mild attacks and 51% in severe attacks. Most attacks treated with the device (77%) did not involve the use of abortive therapy.
Dr. Lainez did not mention adverse events related to the device. However, in the 28-week study, there were 92, including parasthesias and numbness; facial and tooth pain; and swelling. Others were considered mild and included dry eye, nose bleed, and facial asymmetry.
The device is currently being investigated in a U.S. study. The open-label Pathway-CH2 study aims to recruit 120 patients. For information on Pathway CH-2, contact Anthony Caparso.
The trial was sponsored by Autonomic Technologies Inc. Dr. Lainez had no financial ties with the company.
On Twitter @alz_gal
WASHINGTON – An implantable device that stimulates the sphenopalatine ganglion nerve bundle either reduced or eliminated pain in 68% of more than 5,000 cluster headaches, a 3-year study has determined.
The device, which is approved in Europe, was more effective in attacks of moderate severity, with a 78% rate of pain reduction or elimination, Dr. Jose Miguel Lainez reported at the annual meeting of the American Academy of Neurology.
The Pulsante System, manufactured by Autonomic Technologiesof Redwood City, Calif., consists of a neurostimulator about the size of an almond, and a lead with six electrodes. It’s inserted under local anesthetic via a small incision in the upper gum on the side in which the patient experiences symptoms. The electrodes are positioned along the sphenopalatine ganglion (SPG) nerve and the neurostimulator is affixed to the zygomatic process.
A hand-held remote controller placed against the cheek activates the device and controls the intensity of stimulation, which is thought to work by blocking signals to the postganglionic parasympathetic fibers. Those fibers innervate facial structures and the cerebral and meningeal blood vessels and are implicated in the pain and accompanying autonomic symptoms of a cluster headache attack.
Dr. Lainez, professor of neurology at Catholic University of Valencia (Spain), presented 3-year follow-up data from Pathway CH-1, a randomized, sham-controlled trial of 43 patients with cluster headache. Of these, 33 completed the 3-year follow-up period. Of the remaining 10, 1 was lost from observation, 5 violated protocol, 1 had the device implanted incorrectly, and 3 had the device explanted because of incorrect placement or lead migration.
Most of the patients were male. Mean age was 41 years. They had a mean disease duration of 10 years and averaged 17 cluster headaches per week but ranged from 4 to 70 attacks per week. Over the 3 years, 5,130 attacks were treated; the mean stimulation duration for these was 14 minutes with a mean response time of 11 minutes. Therapy was considered effective in 65% (3,354) of these attacks based on a clinically meaningful reduction in pain or pain elimination.
Dr. Lainez did not parse these results. However, in the initial 28-week phase of the Pathway CH-1 study, pain was reduced in 68% of attacks treated with the device and 7% of those treated with the sham control. Pain freedom by 15 minutes was achieved in 34% of attacks with full stimulation, compared with 1.5% of those treated with sham.
In the follow-up study, the device seemed most effective in attacks of moderate severity (78% response rate of pain reduction or elimination). The response rate was 59% in mild attacks and 51% in severe attacks. Most attacks treated with the device (77%) did not involve the use of abortive therapy.
Dr. Lainez did not mention adverse events related to the device. However, in the 28-week study, there were 92, including parasthesias and numbness; facial and tooth pain; and swelling. Others were considered mild and included dry eye, nose bleed, and facial asymmetry.
The device is currently being investigated in a U.S. study. The open-label Pathway-CH2 study aims to recruit 120 patients. For information on Pathway CH-2, contact Anthony Caparso.
The trial was sponsored by Autonomic Technologies Inc. Dr. Lainez had no financial ties with the company.
On Twitter @alz_gal
AT THE AAN 2015 ANNUAL MEETING
Key clinical point: An implantable device that stimulates the sphenopalatine ganglion nerve provided pain relief in cluster headaches.
Major finding: The device reduced or eliminated pain in 68% of more than 5,000 cluster headaches.
Data source: A 3-year follow-up study that examined response in more than 5,000 cluster headaches.
Disclosures: The trial was sponsored by Autonomic Technologies Inc., which makes the Pulsante System. Dr. Lainez had no financial disclosures.

Drug shows promise for lower-risk MDS

WASHINGTON, DC—An investigational drug can increase hemoglobin levels and eliminate transfusion dependence in patients with lower-risk myelodysplastic syndromes (MDS), results of a phase 2 trial suggest.
The drug, luspatercept, is a modified activin receptor type IIB fusion protein that acts as a ligand trap for members in the TGF-β superfamily involved in the late stages of erythropoiesis.
Luspatercept regulates late-stage erythrocyte precursor differentiation and maturation.
Uwe Platzbecker, MD, of the University Hospital in Dresden, Germany, presented results from an ongoing phase 2 study of luspatercept at the 13th International Symposium on Myelodysplastic Syndromes (abstract 53).
The trial is supported by Acceleron Pharma Inc. and Celgene Corporation, the companies developing luspatercept.
“We are excited by the results in lower-risk MDS patients, which confirm and extend our previous findings,” Dr Platzbecker said. “Luspatercept may be useful early in the treatment of lower-risk MDS patients, either as the initial treatment for anemia or in patients who do not respond or become refractory to treatment with erythropoiesis-stimulating agents.”
Patient and dosing details
The researchers enrolled 58 patients in this study. Twenty-seven have completed treatment as part of the dose-escalation cohort. These patients received luspatercept at 7 doses ranging from 0.125 mg/kg to 1.75 mg/kg.
Thirty-one patients are still receiving treatment in the expansion cohort. The starting dose in this cohort is 1.0 mg/kg, and patients are receiving individual dose titration up to 1.75 mg/kg. Seventeen patients from this cohort received at least 4 cycles of treatment or discontinued early and were included in the analysis presented at the meeting.
In all, Dr Platzbecker presented results in 44 patients. Their median age was 71 (range, 27-88), and 57% were male. The median time since diagnosis was 2.5 years (range, 0.2-13.6 years). Sixty-one percent of patients had received prior treatment with erythropoiesis-stimulating agents, and 21% had received lenalidomide.
Fifteen patients had a low transfusion burden (LTB), as they received less than 4 units of red blood cells (RBCs) over 8 weeks. For these patients, the median hemoglobin at baseline was 9.0 g/dL (range, 6.8-10.1), and the median number of RBCs transfused over 8 weeks was 2 (range, 2-2).
Twenty-nine patients had a high transfusion burden (HTB) and received 4 or more RBC units over 8 weeks. The median number of RBCs transfused in this group was 6 (range, 4-14).
Fifty percent of patients had low-risk MDS according to IPSS, 46% had intermediate-1-risk disease, and 4% had intermediate-2-risk MDS. Eighty-one percent of patients were positive for ring sideroblasts, and 58% had the SF3B1 splicing mutation.
Efficacy and safety
The study’s primary efficacy endpoint was an increase in hemoglobin and/or a reduction in transfusion use. For LTB patients, the endpoint was a hemoglobin increase of 1.5 g/dL or more for 2 weeks or longer. For HTB patients, it was decrease in transfusion of 4 or more RBC units or a 50% or greater reduction in transfusion over 8 weeks.
Among the 9 patients who received lower doses of luspatercept (0.125-0.5 mg/kg), 33% met the primary efficacy endpoint. And 63% of the 35 patients in the higher dose group (0.75-1.75 mg/kg) achieved the primary efficacy endpoint.
Twenty-two percent of patients in the lower dose group achieved the International Working Group (IWG) hematologic improvement-erythroid (HI-E) threshold of efficacy, as did 54% of patients in the higher dose group.
Fourteen percent of patients in the lower dose group achieved transfusion independence, as did 36% of patients in the higher dose group. In the higher dose group, this included 4 of 6 patients with LTB and 6 of 22 patients with HTB.
Among patients who were ring-sideroblast-positive and received higher doses of luspatercept, 39% achieved transfusion independence, and 63% achieved IWG HI-E.
The majority of adverse events (AEs) were mild to moderate (grade 1 or 2). AEs included nasopharyngitis (14%), diarrhea (14%), myalgia (11%), bone pain (9%), bronchitis (9%), headache (9%), and muscle spasms (9%).
There were 2 serious AEs—grade 3 muscle pain and grade 3 worsening of general condition—that were considered possibly related to treatment. One non-serious grade 3 AE of blast cell count increase was considered possibly treatment-related as well.
In closing, Dr Platzbecker said luspatercept was generally safe and well-tolerated, in addition to providing “robust hematologic improvement.” And these results support further study of the drug in patients with lower-risk MDS. ![]()
WASHINGTON, DC—An investigational drug can increase hemoglobin levels and eliminate transfusion dependence in patients with lower-risk myelodysplastic syndromes (MDS), results of a phase 2 trial suggest.
The drug, luspatercept, is a modified activin receptor type IIB fusion protein that acts as a ligand trap for members in the TGF-β superfamily involved in the late stages of erythropoiesis.
Luspatercept regulates late-stage erythrocyte precursor differentiation and maturation.
Uwe Platzbecker, MD, of the University Hospital in Dresden, Germany, presented results from an ongoing phase 2 study of luspatercept at the 13th International Symposium on Myelodysplastic Syndromes (abstract 53).
The trial is supported by Acceleron Pharma Inc. and Celgene Corporation, the companies developing luspatercept.
“We are excited by the results in lower-risk MDS patients, which confirm and extend our previous findings,” Dr Platzbecker said. “Luspatercept may be useful early in the treatment of lower-risk MDS patients, either as the initial treatment for anemia or in patients who do not respond or become refractory to treatment with erythropoiesis-stimulating agents.”
Patient and dosing details
The researchers enrolled 58 patients in this study. Twenty-seven have completed treatment as part of the dose-escalation cohort. These patients received luspatercept at 7 doses ranging from 0.125 mg/kg to 1.75 mg/kg.
Thirty-one patients are still receiving treatment in the expansion cohort. The starting dose in this cohort is 1.0 mg/kg, and patients are receiving individual dose titration up to 1.75 mg/kg. Seventeen patients from this cohort received at least 4 cycles of treatment or discontinued early and were included in the analysis presented at the meeting.
In all, Dr Platzbecker presented results in 44 patients. Their median age was 71 (range, 27-88), and 57% were male. The median time since diagnosis was 2.5 years (range, 0.2-13.6 years). Sixty-one percent of patients had received prior treatment with erythropoiesis-stimulating agents, and 21% had received lenalidomide.
Fifteen patients had a low transfusion burden (LTB), as they received less than 4 units of red blood cells (RBCs) over 8 weeks. For these patients, the median hemoglobin at baseline was 9.0 g/dL (range, 6.8-10.1), and the median number of RBCs transfused over 8 weeks was 2 (range, 2-2).
Twenty-nine patients had a high transfusion burden (HTB) and received 4 or more RBC units over 8 weeks. The median number of RBCs transfused in this group was 6 (range, 4-14).
Fifty percent of patients had low-risk MDS according to IPSS, 46% had intermediate-1-risk disease, and 4% had intermediate-2-risk MDS. Eighty-one percent of patients were positive for ring sideroblasts, and 58% had the SF3B1 splicing mutation.
Efficacy and safety
The study’s primary efficacy endpoint was an increase in hemoglobin and/or a reduction in transfusion use. For LTB patients, the endpoint was a hemoglobin increase of 1.5 g/dL or more for 2 weeks or longer. For HTB patients, it was decrease in transfusion of 4 or more RBC units or a 50% or greater reduction in transfusion over 8 weeks.
Among the 9 patients who received lower doses of luspatercept (0.125-0.5 mg/kg), 33% met the primary efficacy endpoint. And 63% of the 35 patients in the higher dose group (0.75-1.75 mg/kg) achieved the primary efficacy endpoint.
Twenty-two percent of patients in the lower dose group achieved the International Working Group (IWG) hematologic improvement-erythroid (HI-E) threshold of efficacy, as did 54% of patients in the higher dose group.
Fourteen percent of patients in the lower dose group achieved transfusion independence, as did 36% of patients in the higher dose group. In the higher dose group, this included 4 of 6 patients with LTB and 6 of 22 patients with HTB.
Among patients who were ring-sideroblast-positive and received higher doses of luspatercept, 39% achieved transfusion independence, and 63% achieved IWG HI-E.
The majority of adverse events (AEs) were mild to moderate (grade 1 or 2). AEs included nasopharyngitis (14%), diarrhea (14%), myalgia (11%), bone pain (9%), bronchitis (9%), headache (9%), and muscle spasms (9%).
There were 2 serious AEs—grade 3 muscle pain and grade 3 worsening of general condition—that were considered possibly related to treatment. One non-serious grade 3 AE of blast cell count increase was considered possibly treatment-related as well.
In closing, Dr Platzbecker said luspatercept was generally safe and well-tolerated, in addition to providing “robust hematologic improvement.” And these results support further study of the drug in patients with lower-risk MDS. ![]()
WASHINGTON, DC—An investigational drug can increase hemoglobin levels and eliminate transfusion dependence in patients with lower-risk myelodysplastic syndromes (MDS), results of a phase 2 trial suggest.
The drug, luspatercept, is a modified activin receptor type IIB fusion protein that acts as a ligand trap for members in the TGF-β superfamily involved in the late stages of erythropoiesis.
Luspatercept regulates late-stage erythrocyte precursor differentiation and maturation.
Uwe Platzbecker, MD, of the University Hospital in Dresden, Germany, presented results from an ongoing phase 2 study of luspatercept at the 13th International Symposium on Myelodysplastic Syndromes (abstract 53).
The trial is supported by Acceleron Pharma Inc. and Celgene Corporation, the companies developing luspatercept.
“We are excited by the results in lower-risk MDS patients, which confirm and extend our previous findings,” Dr Platzbecker said. “Luspatercept may be useful early in the treatment of lower-risk MDS patients, either as the initial treatment for anemia or in patients who do not respond or become refractory to treatment with erythropoiesis-stimulating agents.”
Patient and dosing details
The researchers enrolled 58 patients in this study. Twenty-seven have completed treatment as part of the dose-escalation cohort. These patients received luspatercept at 7 doses ranging from 0.125 mg/kg to 1.75 mg/kg.
Thirty-one patients are still receiving treatment in the expansion cohort. The starting dose in this cohort is 1.0 mg/kg, and patients are receiving individual dose titration up to 1.75 mg/kg. Seventeen patients from this cohort received at least 4 cycles of treatment or discontinued early and were included in the analysis presented at the meeting.
In all, Dr Platzbecker presented results in 44 patients. Their median age was 71 (range, 27-88), and 57% were male. The median time since diagnosis was 2.5 years (range, 0.2-13.6 years). Sixty-one percent of patients had received prior treatment with erythropoiesis-stimulating agents, and 21% had received lenalidomide.
Fifteen patients had a low transfusion burden (LTB), as they received less than 4 units of red blood cells (RBCs) over 8 weeks. For these patients, the median hemoglobin at baseline was 9.0 g/dL (range, 6.8-10.1), and the median number of RBCs transfused over 8 weeks was 2 (range, 2-2).
Twenty-nine patients had a high transfusion burden (HTB) and received 4 or more RBC units over 8 weeks. The median number of RBCs transfused in this group was 6 (range, 4-14).
Fifty percent of patients had low-risk MDS according to IPSS, 46% had intermediate-1-risk disease, and 4% had intermediate-2-risk MDS. Eighty-one percent of patients were positive for ring sideroblasts, and 58% had the SF3B1 splicing mutation.
Efficacy and safety
The study’s primary efficacy endpoint was an increase in hemoglobin and/or a reduction in transfusion use. For LTB patients, the endpoint was a hemoglobin increase of 1.5 g/dL or more for 2 weeks or longer. For HTB patients, it was decrease in transfusion of 4 or more RBC units or a 50% or greater reduction in transfusion over 8 weeks.
Among the 9 patients who received lower doses of luspatercept (0.125-0.5 mg/kg), 33% met the primary efficacy endpoint. And 63% of the 35 patients in the higher dose group (0.75-1.75 mg/kg) achieved the primary efficacy endpoint.
Twenty-two percent of patients in the lower dose group achieved the International Working Group (IWG) hematologic improvement-erythroid (HI-E) threshold of efficacy, as did 54% of patients in the higher dose group.
Fourteen percent of patients in the lower dose group achieved transfusion independence, as did 36% of patients in the higher dose group. In the higher dose group, this included 4 of 6 patients with LTB and 6 of 22 patients with HTB.
Among patients who were ring-sideroblast-positive and received higher doses of luspatercept, 39% achieved transfusion independence, and 63% achieved IWG HI-E.
The majority of adverse events (AEs) were mild to moderate (grade 1 or 2). AEs included nasopharyngitis (14%), diarrhea (14%), myalgia (11%), bone pain (9%), bronchitis (9%), headache (9%), and muscle spasms (9%).
There were 2 serious AEs—grade 3 muscle pain and grade 3 worsening of general condition—that were considered possibly related to treatment. One non-serious grade 3 AE of blast cell count increase was considered possibly treatment-related as well.
In closing, Dr Platzbecker said luspatercept was generally safe and well-tolerated, in addition to providing “robust hematologic improvement.” And these results support further study of the drug in patients with lower-risk MDS. ![]()
Whole blood better for cardiac surgery in young children

Photo by Elise Amendola
Using fresh whole blood (FWB) from single donors for cardiac procedures in children younger than 2 years of age is better than using component blood from multiple donors, researchers say.
FWB reduces the risk of getting transfusion-related illnesses by reducing donor exposures.
“Currently, whole blood is not generally made available to hospitals for use in pediatric heart surgery,” said David R. Jobes, MD, of The Children’s Hospital of Philadelphia in Pennsylvania.
“Blood centers separate donated blood into component parts, which are then stored for use in medical transfusions as needed.”
At The Children’s Hospital of Philadelphia, the standard preoperative blood order for elective pediatric heart surgery with cariopulmonary bypass is 2 units of FWB and 2 units of packed red blood cells. The FWB is to be used during and immediately after surgery and the components thereafter, if necessary.
The researchers set out to examine the effectiveness of this protocol. They conducted a retrospective study of patient records over a period of 15 years from a surgical registry and blood bank, comparing the cohort of 4111 patients to published reports.
The team defined donor exposures as transfusion requirements for the day of operation and the next postoperative day. All blood products issued were presumed to have been tranfused, and all aliquots from a single donor were counted as a single donor exposure.
Patients were a median age of 94 days and weighed a median of 4.4 kg.
Most (3836) patients received FWB, and 252 received components exclusively when no FWB was available. Twenty-three patients did not receive any blood products. A median of 2 whole blood units was transfused, for a total of 2 donor exposures for the entire cohort.
The researchers found that the youngest patients having complex procedures were exposed to the highest number of donors, while older patients having simpler procedures were exposed to fewer donors.
For example, 72 patients who were a median of 5 days old and underwent truncus arteriosus repair had a median of 4 donor exposures (range, 1-14). And 136 older patients who were a median of 610 days old and underwent fontan completion had a median of 1 donor exposure (range, 0-8).
The researchers concluded that the protocol resulted in fewer donor exposures compared with component use reported in the literature.
Dr Jobes said the risk for disease transmission in pediatric patients is essentially the same as the risk for adults, but it may be more costly for pediatric patients in the long run because infants and young children may live longer with chronic illness stemming from transfusion.
He added, “We hope that our research helps to re-examine current blood storage practice and make whole blood more readily available for pediatric patients.”
He and his colleagues described this research in The Annals of Thoracic Surgery. ![]()
Photo by Elise Amendola
Using fresh whole blood (FWB) from single donors for cardiac procedures in children younger than 2 years of age is better than using component blood from multiple donors, researchers say.
FWB reduces the risk of getting transfusion-related illnesses by reducing donor exposures.
“Currently, whole blood is not generally made available to hospitals for use in pediatric heart surgery,” said David R. Jobes, MD, of The Children’s Hospital of Philadelphia in Pennsylvania.
“Blood centers separate donated blood into component parts, which are then stored for use in medical transfusions as needed.”
At The Children’s Hospital of Philadelphia, the standard preoperative blood order for elective pediatric heart surgery with cariopulmonary bypass is 2 units of FWB and 2 units of packed red blood cells. The FWB is to be used during and immediately after surgery and the components thereafter, if necessary.
The researchers set out to examine the effectiveness of this protocol. They conducted a retrospective study of patient records over a period of 15 years from a surgical registry and blood bank, comparing the cohort of 4111 patients to published reports.
The team defined donor exposures as transfusion requirements for the day of operation and the next postoperative day. All blood products issued were presumed to have been tranfused, and all aliquots from a single donor were counted as a single donor exposure.
Patients were a median age of 94 days and weighed a median of 4.4 kg.
Most (3836) patients received FWB, and 252 received components exclusively when no FWB was available. Twenty-three patients did not receive any blood products. A median of 2 whole blood units was transfused, for a total of 2 donor exposures for the entire cohort.
The researchers found that the youngest patients having complex procedures were exposed to the highest number of donors, while older patients having simpler procedures were exposed to fewer donors.
For example, 72 patients who were a median of 5 days old and underwent truncus arteriosus repair had a median of 4 donor exposures (range, 1-14). And 136 older patients who were a median of 610 days old and underwent fontan completion had a median of 1 donor exposure (range, 0-8).
The researchers concluded that the protocol resulted in fewer donor exposures compared with component use reported in the literature.
Dr Jobes said the risk for disease transmission in pediatric patients is essentially the same as the risk for adults, but it may be more costly for pediatric patients in the long run because infants and young children may live longer with chronic illness stemming from transfusion.
He added, “We hope that our research helps to re-examine current blood storage practice and make whole blood more readily available for pediatric patients.”
He and his colleagues described this research in The Annals of Thoracic Surgery. ![]()
Photo by Elise Amendola
Using fresh whole blood (FWB) from single donors for cardiac procedures in children younger than 2 years of age is better than using component blood from multiple donors, researchers say.
FWB reduces the risk of getting transfusion-related illnesses by reducing donor exposures.
“Currently, whole blood is not generally made available to hospitals for use in pediatric heart surgery,” said David R. Jobes, MD, of The Children’s Hospital of Philadelphia in Pennsylvania.
“Blood centers separate donated blood into component parts, which are then stored for use in medical transfusions as needed.”
At The Children’s Hospital of Philadelphia, the standard preoperative blood order for elective pediatric heart surgery with cariopulmonary bypass is 2 units of FWB and 2 units of packed red blood cells. The FWB is to be used during and immediately after surgery and the components thereafter, if necessary.
The researchers set out to examine the effectiveness of this protocol. They conducted a retrospective study of patient records over a period of 15 years from a surgical registry and blood bank, comparing the cohort of 4111 patients to published reports.
The team defined donor exposures as transfusion requirements for the day of operation and the next postoperative day. All blood products issued were presumed to have been tranfused, and all aliquots from a single donor were counted as a single donor exposure.
Patients were a median age of 94 days and weighed a median of 4.4 kg.
Most (3836) patients received FWB, and 252 received components exclusively when no FWB was available. Twenty-three patients did not receive any blood products. A median of 2 whole blood units was transfused, for a total of 2 donor exposures for the entire cohort.
The researchers found that the youngest patients having complex procedures were exposed to the highest number of donors, while older patients having simpler procedures were exposed to fewer donors.
For example, 72 patients who were a median of 5 days old and underwent truncus arteriosus repair had a median of 4 donor exposures (range, 1-14). And 136 older patients who were a median of 610 days old and underwent fontan completion had a median of 1 donor exposure (range, 0-8).
The researchers concluded that the protocol resulted in fewer donor exposures compared with component use reported in the literature.
Dr Jobes said the risk for disease transmission in pediatric patients is essentially the same as the risk for adults, but it may be more costly for pediatric patients in the long run because infants and young children may live longer with chronic illness stemming from transfusion.
He added, “We hope that our research helps to re-examine current blood storage practice and make whole blood more readily available for pediatric patients.”
He and his colleagues described this research in The Annals of Thoracic Surgery. ![]()
Study shows importance of VTE screening

Image by Kevin MacKenzie
SEATTLE—Routine screening for venous thromboembolism (VTE) may decrease morbidity and mortality among patients undergoing pneumonectomy for benign or malignant indications, according to researchers.
The group conducted a study that showed the rate of VTE diagnosis was 3 times higher for patients who underwent routine VTE screening post-pneumonectomy than for patients who were tested for VTE only after they exhibited symptoms.
Siva Raja MD, PhD, of the Cleveland Clinic in Ohio, presented this finding at the 95th Annual Meeting of the American Association for Thoracic Surgery.
He and his colleagues analyzed 112 patients who underwent pneumonectomy for benign and malignant indications and were screened for VTE. The team compared the rate of VTE diagnosis in this group to the rate in a previously published group of 336 similar patients who did not undergo VTE screening.
The rate of in-hospital VTEs in the screened group was almost 3 times higher than the rate in patients who were not screened—8.9% and 3.0%, respectively (P=0.008).
Over the 30-day post-operative period, the rate of VTE for screened patients was more than double the rate for unscreened patients—13% and 5.1%, respectively (P=0.007).
In the screened group, 10 of 112 patients had VTE detected by screening just before discharge, and 4 additional patients developed symptomatic VTE within 30 days despite a negative pre-discharge screen. In all, 20 patients in this group developed a VTE.
In both the screened and unscreened cohorts, the risk of VTE peaked 6 days after surgery and plateaued after 30 days.
“We find that a large proportion (50%) of VTEs occurred prior to the time of discharge, and the risk of developing symptomatic VTE remained elevated for 30 days,” Dr Raja said. “It is possible that the prevalence of VTE may be even higher should a comprehensive serial screening program be initiated.”
Dr Raja also noted that VTEs are a particular problem after pneumonectomy, since these patients often have low pulmonary reserve to withstand the impact of pulmonary embolism.
Indeed, this study showed that post-pneumonectomy patients who developed VTE had worse long-term survival than patients who did not develop clots, although the difference was not statistically significant (hazard ratio=2.1, P=0.08).
Still, Dr Raja said these results suggest that patients undergoing pneumonectomy receive anticoagulants for a longer duration, as well as undergo repeat screening test for VTE even after hospital discharge. ![]()
Image by Kevin MacKenzie
SEATTLE—Routine screening for venous thromboembolism (VTE) may decrease morbidity and mortality among patients undergoing pneumonectomy for benign or malignant indications, according to researchers.
The group conducted a study that showed the rate of VTE diagnosis was 3 times higher for patients who underwent routine VTE screening post-pneumonectomy than for patients who were tested for VTE only after they exhibited symptoms.
Siva Raja MD, PhD, of the Cleveland Clinic in Ohio, presented this finding at the 95th Annual Meeting of the American Association for Thoracic Surgery.
He and his colleagues analyzed 112 patients who underwent pneumonectomy for benign and malignant indications and were screened for VTE. The team compared the rate of VTE diagnosis in this group to the rate in a previously published group of 336 similar patients who did not undergo VTE screening.
The rate of in-hospital VTEs in the screened group was almost 3 times higher than the rate in patients who were not screened—8.9% and 3.0%, respectively (P=0.008).
Over the 30-day post-operative period, the rate of VTE for screened patients was more than double the rate for unscreened patients—13% and 5.1%, respectively (P=0.007).
In the screened group, 10 of 112 patients had VTE detected by screening just before discharge, and 4 additional patients developed symptomatic VTE within 30 days despite a negative pre-discharge screen. In all, 20 patients in this group developed a VTE.
In both the screened and unscreened cohorts, the risk of VTE peaked 6 days after surgery and plateaued after 30 days.
“We find that a large proportion (50%) of VTEs occurred prior to the time of discharge, and the risk of developing symptomatic VTE remained elevated for 30 days,” Dr Raja said. “It is possible that the prevalence of VTE may be even higher should a comprehensive serial screening program be initiated.”
Dr Raja also noted that VTEs are a particular problem after pneumonectomy, since these patients often have low pulmonary reserve to withstand the impact of pulmonary embolism.
Indeed, this study showed that post-pneumonectomy patients who developed VTE had worse long-term survival than patients who did not develop clots, although the difference was not statistically significant (hazard ratio=2.1, P=0.08).
Still, Dr Raja said these results suggest that patients undergoing pneumonectomy receive anticoagulants for a longer duration, as well as undergo repeat screening test for VTE even after hospital discharge. ![]()
Image by Kevin MacKenzie
SEATTLE—Routine screening for venous thromboembolism (VTE) may decrease morbidity and mortality among patients undergoing pneumonectomy for benign or malignant indications, according to researchers.
The group conducted a study that showed the rate of VTE diagnosis was 3 times higher for patients who underwent routine VTE screening post-pneumonectomy than for patients who were tested for VTE only after they exhibited symptoms.
Siva Raja MD, PhD, of the Cleveland Clinic in Ohio, presented this finding at the 95th Annual Meeting of the American Association for Thoracic Surgery.
He and his colleagues analyzed 112 patients who underwent pneumonectomy for benign and malignant indications and were screened for VTE. The team compared the rate of VTE diagnosis in this group to the rate in a previously published group of 336 similar patients who did not undergo VTE screening.
The rate of in-hospital VTEs in the screened group was almost 3 times higher than the rate in patients who were not screened—8.9% and 3.0%, respectively (P=0.008).
Over the 30-day post-operative period, the rate of VTE for screened patients was more than double the rate for unscreened patients—13% and 5.1%, respectively (P=0.007).
In the screened group, 10 of 112 patients had VTE detected by screening just before discharge, and 4 additional patients developed symptomatic VTE within 30 days despite a negative pre-discharge screen. In all, 20 patients in this group developed a VTE.
In both the screened and unscreened cohorts, the risk of VTE peaked 6 days after surgery and plateaued after 30 days.
“We find that a large proportion (50%) of VTEs occurred prior to the time of discharge, and the risk of developing symptomatic VTE remained elevated for 30 days,” Dr Raja said. “It is possible that the prevalence of VTE may be even higher should a comprehensive serial screening program be initiated.”
Dr Raja also noted that VTEs are a particular problem after pneumonectomy, since these patients often have low pulmonary reserve to withstand the impact of pulmonary embolism.
Indeed, this study showed that post-pneumonectomy patients who developed VTE had worse long-term survival than patients who did not develop clots, although the difference was not statistically significant (hazard ratio=2.1, P=0.08).
Still, Dr Raja said these results suggest that patients undergoing pneumonectomy receive anticoagulants for a longer duration, as well as undergo repeat screening test for VTE even after hospital discharge. ![]()
FDA approves new drug for hemophilia B

The US Food and Drug Administration (FDA) has approved an intravenous, recombinant, human coagulation factor IX product (Ixinity) for use in patients
with hemophilia B.
The drug is intended to control and prevent bleeding episodes and for perioperative management in adults and children age 12 and older.
Concurrent with the FDA’s approval, Emergent Biosolutions (the company developing Ixinity) launched the Ixinity IXperience Concierge. This resource, which consumers can access by calling 1-855-IXINITY, provides information on the drug.
About Ixinity
Ixinity contains trenonacog alfa, a purified, single-chain glycoprotein derived from Chinese hamster ovary (CHO) cells that has an amino acid sequence comparable to the Thr148 allelic form of plasma-derived factor IX.
No human or animal proteins are added during any stage of manufacturing or formulation of Ixinity. The recombinant factor IX is purified by a chromatography purification process.
The process includes 3 validated steps for virus inactivation and removal. It also includes a validated manufacturing step to reduce the presence of CHO proteins in the final drug product.
Ixinity is contraindicated in patients who have known hypersensitivity to the drug or its excipients, including CHO protein. Hypersensitivity reactions, including anaphylaxis, may occur following treatment with Ixinity. Patients who receive Ixinity are also at risk of developing nephrotic syndrome and thromboembolism.
Trial data
The FDA approved Ixinity based on results from a phase 1/3 trial of the drug in previously treated adults and children (age 12 and older) with severe to moderately severe (factor IX level < 2%) hemophilia B.
Seventy-seven patients received at least 1 dose of Ixinity. The drug was given as routine prophylaxis or on-demand treatment for bleeding episodes.
Fifty-five patients received treatment for more than 50 exposure days, and 45 received the drug for more than 100 exposure days. The median duration of treatment on study was 16.2 months (range, 2.4-39.6 months) for the routine treatment regimen and 14.1 months (range, 2.3-36.9 months) for the on-demand treatment regimen.
A total of 508 bleeding episodes were treated with Ixinity—286 bleeds for patients on routine treatment and 222 for patients receiving on-demand treatment. A majority of the bleeds (84%) were resolved by 1 or 2 infusions of the drug.
Patients rated hemostatic efficacy at the resolution of a bleed as “excellent” or “good” in 84% of all treated bleeding episodes. “Excellent” was defined as a dramatic response with abrupt pain relief and clear reduction in joint or hemorrhage site size. “Good” was defined as pain relief or reduction in hemorrhage site size that may have required an additional infusion for resolution.
Ixinity also induced hemostasis in patients who underwent major surgical procedures.
The drug exhibited similar pharmacokinetic behavior as nonacog alfa, another licensed recombinant coagulation factor IX product. There was no significant reduction in steady-state factor IX levels or alteration in pharmacokinetic behavior over time with Ixinity.
There were 14 adverse events reported in 6 patients. The most common event, observed in 2.6% of patients, was headache. Other adverse events were asthenia, apathy, depression, dysgeusia, influenza, injection site discomfort, lethargy, and skin rash.
None of the patients developed inhibitors to Ixinity, and there were no reports of thrombotic events or allergic reactions.
For more details on this research, see the full prescribing information for Ixinity, available at www.IXINITY.com. ![]()
The US Food and Drug Administration (FDA) has approved an intravenous, recombinant, human coagulation factor IX product (Ixinity) for use in patients
with hemophilia B.
The drug is intended to control and prevent bleeding episodes and for perioperative management in adults and children age 12 and older.
Concurrent with the FDA’s approval, Emergent Biosolutions (the company developing Ixinity) launched the Ixinity IXperience Concierge. This resource, which consumers can access by calling 1-855-IXINITY, provides information on the drug.
About Ixinity
Ixinity contains trenonacog alfa, a purified, single-chain glycoprotein derived from Chinese hamster ovary (CHO) cells that has an amino acid sequence comparable to the Thr148 allelic form of plasma-derived factor IX.
No human or animal proteins are added during any stage of manufacturing or formulation of Ixinity. The recombinant factor IX is purified by a chromatography purification process.
The process includes 3 validated steps for virus inactivation and removal. It also includes a validated manufacturing step to reduce the presence of CHO proteins in the final drug product.
Ixinity is contraindicated in patients who have known hypersensitivity to the drug or its excipients, including CHO protein. Hypersensitivity reactions, including anaphylaxis, may occur following treatment with Ixinity. Patients who receive Ixinity are also at risk of developing nephrotic syndrome and thromboembolism.
Trial data
The FDA approved Ixinity based on results from a phase 1/3 trial of the drug in previously treated adults and children (age 12 and older) with severe to moderately severe (factor IX level < 2%) hemophilia B.
Seventy-seven patients received at least 1 dose of Ixinity. The drug was given as routine prophylaxis or on-demand treatment for bleeding episodes.
Fifty-five patients received treatment for more than 50 exposure days, and 45 received the drug for more than 100 exposure days. The median duration of treatment on study was 16.2 months (range, 2.4-39.6 months) for the routine treatment regimen and 14.1 months (range, 2.3-36.9 months) for the on-demand treatment regimen.
A total of 508 bleeding episodes were treated with Ixinity—286 bleeds for patients on routine treatment and 222 for patients receiving on-demand treatment. A majority of the bleeds (84%) were resolved by 1 or 2 infusions of the drug.
Patients rated hemostatic efficacy at the resolution of a bleed as “excellent” or “good” in 84% of all treated bleeding episodes. “Excellent” was defined as a dramatic response with abrupt pain relief and clear reduction in joint or hemorrhage site size. “Good” was defined as pain relief or reduction in hemorrhage site size that may have required an additional infusion for resolution.
Ixinity also induced hemostasis in patients who underwent major surgical procedures.
The drug exhibited similar pharmacokinetic behavior as nonacog alfa, another licensed recombinant coagulation factor IX product. There was no significant reduction in steady-state factor IX levels or alteration in pharmacokinetic behavior over time with Ixinity.
There were 14 adverse events reported in 6 patients. The most common event, observed in 2.6% of patients, was headache. Other adverse events were asthenia, apathy, depression, dysgeusia, influenza, injection site discomfort, lethargy, and skin rash.
None of the patients developed inhibitors to Ixinity, and there were no reports of thrombotic events or allergic reactions.
For more details on this research, see the full prescribing information for Ixinity, available at www.IXINITY.com. ![]()
The US Food and Drug Administration (FDA) has approved an intravenous, recombinant, human coagulation factor IX product (Ixinity) for use in patients
with hemophilia B.
The drug is intended to control and prevent bleeding episodes and for perioperative management in adults and children age 12 and older.
Concurrent with the FDA’s approval, Emergent Biosolutions (the company developing Ixinity) launched the Ixinity IXperience Concierge. This resource, which consumers can access by calling 1-855-IXINITY, provides information on the drug.
About Ixinity
Ixinity contains trenonacog alfa, a purified, single-chain glycoprotein derived from Chinese hamster ovary (CHO) cells that has an amino acid sequence comparable to the Thr148 allelic form of plasma-derived factor IX.
No human or animal proteins are added during any stage of manufacturing or formulation of Ixinity. The recombinant factor IX is purified by a chromatography purification process.
The process includes 3 validated steps for virus inactivation and removal. It also includes a validated manufacturing step to reduce the presence of CHO proteins in the final drug product.
Ixinity is contraindicated in patients who have known hypersensitivity to the drug or its excipients, including CHO protein. Hypersensitivity reactions, including anaphylaxis, may occur following treatment with Ixinity. Patients who receive Ixinity are also at risk of developing nephrotic syndrome and thromboembolism.
Trial data
The FDA approved Ixinity based on results from a phase 1/3 trial of the drug in previously treated adults and children (age 12 and older) with severe to moderately severe (factor IX level < 2%) hemophilia B.
Seventy-seven patients received at least 1 dose of Ixinity. The drug was given as routine prophylaxis or on-demand treatment for bleeding episodes.
Fifty-five patients received treatment for more than 50 exposure days, and 45 received the drug for more than 100 exposure days. The median duration of treatment on study was 16.2 months (range, 2.4-39.6 months) for the routine treatment regimen and 14.1 months (range, 2.3-36.9 months) for the on-demand treatment regimen.
A total of 508 bleeding episodes were treated with Ixinity—286 bleeds for patients on routine treatment and 222 for patients receiving on-demand treatment. A majority of the bleeds (84%) were resolved by 1 or 2 infusions of the drug.
Patients rated hemostatic efficacy at the resolution of a bleed as “excellent” or “good” in 84% of all treated bleeding episodes. “Excellent” was defined as a dramatic response with abrupt pain relief and clear reduction in joint or hemorrhage site size. “Good” was defined as pain relief or reduction in hemorrhage site size that may have required an additional infusion for resolution.
Ixinity also induced hemostasis in patients who underwent major surgical procedures.
The drug exhibited similar pharmacokinetic behavior as nonacog alfa, another licensed recombinant coagulation factor IX product. There was no significant reduction in steady-state factor IX levels or alteration in pharmacokinetic behavior over time with Ixinity.
There were 14 adverse events reported in 6 patients. The most common event, observed in 2.6% of patients, was headache. Other adverse events were asthenia, apathy, depression, dysgeusia, influenza, injection site discomfort, lethargy, and skin rash.
None of the patients developed inhibitors to Ixinity, and there were no reports of thrombotic events or allergic reactions.
For more details on this research, see the full prescribing information for Ixinity, available at www.IXINITY.com. ![]()